Изобретение относится к фармацевтической промышленности и касается способа получения лекарственных средств с высокой детоксицирующей способностью.
Использование атомной энергии, тяжелых металлов создало проблемы предупреждения всасывания радионуклидов и способов выведения их из организма. В результате исследования значительного количества химических соединений синтетического и природного генеза было установлено, что даже при относительной эффективности отдельных веществ в отношении замедления всасывания радионуклидов, они являются далеко небезразличными для организма, оказывая токсическое действие на функцию (и структуру) различных органов и систем и, в первую очередь, на почки, ретикулоэндотелий, костный мозг. Деградированные производные альгиновой кислоты, характеризующиеся повышенным содержанием остатков α-L-гулуроновой кислоты, полностью освобожденные от катионов тяжелых металлов, способны избирательно ингибировать всасывание радионуклидов, практически не влияя на абсорбцию ионов кальция. Наряду с этим альгинаты даже при назначении людям в дозах до 45 г в день не проявляют нежелательного действия местно и резобтивно (Особенности действия и перспективы применения в медицине деградированных альгинатов /И.С. Ажгихин, А.И. Аразашвили, Н.Н. Аракелова и др.//Фармация. - М.: Медицина, 1988. - 1. -C. 77-85).
Альгиновая кислота - сополимер остатков Д-маннуроновой кислоты и остатков L-гулуроновой кислоты, связана в тканях бурых водорослей с одно-, двух- и поливалентными металлами. Известно, что в процессе биосинтеза альгиновой кислоты водорослями содержание в ней L-гулуроновой кислоты увеличивается. Это приводит к предпочтительному связыванию двух- и поливалентных металлов. Альгинаты, обогащенные L-гулуроновой кислотой, содержатся в основном в клеточных стенках водорослей, и при экстрагировании их очень трудно перевести в раствор (Подкорытова А.В. Зависимость эффективности экстрагирования альгината натрия из ламинарии японской от условий обработки. /Рыбное хозяйство. - 1987. - 2. - С. 64-68).
Весьма эффективным представляется применение альгинатов, обогащенных фрагментами полигулуронида, в терапии отравлений тяжелыми металлами. При сравнении детоксицирующего действия импортных и отечественных образцов альгината был установлен преимущественный эффект первых. При изучении состава этих образцов оказалось, что импортные образцы обогащены остатками L-гулуроновой кислоты и максимально освобождены от катионов двух- и трехвалентных металлов, в то время как отечественные образцы характеризовались минимальным содержанием гулуроновой кислоты и высоким содержанием маннуроновой кислоты при повышенном содержании катионов поливалентных металлов. А ведь именно полигулурониды гораздо эффективнее для детоксикации металлов, чем полиманнурониды (Особенности действия и перспективы применения в медицине деградированных альгинатов /И.С. Ажгихин, А.И. Аразашвили, Н.Н. Аракелова и др. //Фармация. - М.: Медицина, 1988. - 1. -С. 77-85). Таким образом, для характеристики антидотного действия альгинатов наиболее важным вопросом является содержание и соотношение уроновых кислот (маннуронид/гулуронид М/G) в препарате, а также содержание катионов поливалентных металлов.
Для альгиновой кислоты, полученной из беломорских водорослей, характерна повышенная сорбционность в отношении ионов железа (Ш) и других поливалентных катионов (Особенности действия и перспективы применения в медицине деградированных альгинатов /И.С. Ажгихин, А.И. Аразашвили, Н.Н. Аракелова и др. //Фармация. - М.: Медицина, 1988. - 1. -С. 77-85), поэтому в процессе выделения альгината натрия необходимо максимально отделить альгинат от ионов тяжелых металлов и, в первую очередь, от катионов железа (Ш) (коэффициент накопления ионов железа равен 77, а стронция 44).
Описан способ получения альгината натрия (Ажгихин И.С. Шпаков Ю.Н., Кипиани Р. Е. , Гандель В.Г. Морская фармация (Теория и практика нового направления в фармацевтической науке). - Кишинев: Штиинца, 1982. - 260с.) из бурых водорослей. Способ заключается в том, что водоросли помещают в 0,35-0,4%-й раствор серной кислоты, нагретый до 45-50oС, и тщательно перемешивают в течение 1 ч. Затем температуру экстракционной среды повышают, избыток кальцинированной соды загружают в реактор. Процесс варки продолжается 1,5-2 ч. По окончании горячей экстракции вытяжку перекачивают в отстойник, заполняя оставшийся объем водой. Раствор отстаивается 4-5 ч. Очистку растворов альгината натрия осуществляют декантацией и фильтрованием. Осветленный раствор затем обрабатывают концентрированной серной кислотой, образующаяся альгиновая кислота освобождается от маточника. Затем ее промывают проточной водой и загружают в смесители вместе с рассчитанным количеством безводного карбоната натрия, перемешивая до завершения процесса растворения и образования альгината натрия. Реакция продолжается около 1 ч. Полученный раствор альгината натрия высушивают на барабанной сушилке при температуре 120oС и растирают в крупный порошок. Полученный альгинат натрия - пластинки коричневого цвета, без запаха. Содержание альгиновой кислоты 60%.
Недостатками описанного способа являются: а) невысокое количественное содержание альгиновой кислоты (60%) в препарате, б) низкое содержание гулуроновой кислоты (соотношение М/G=5,15) и, как следствие, комплексообразующей способности; в) высокое содержание общей золы (20,8%), г) использование высокой температуры сушки (120oС) растворов альгината натрия ведет к ухудшению растворимости препарата. Такой препарат для детоксикации тяжелых металлов с целью применения в медицине не представляет интереса.
По другому методу, разработанному в ТИНРО (Ажгихин И.C., Шпаков Ю.Н., Кипиани Р.Е., Гандель В.Г. Морская фармация (Теория и практика нового направления в фармацевтической науке). - Кишинев: Штиинца, 1982. - 260 с.), для получения альгината натрия водоросли измельчают, промывают в воде и мацерируют 2-3 ч в 0,5% растворе хлористоводородной кислоты. Предварительно обработанные водоросли мацерируют нагретым раствором гидроксида натрия и образующийся щелочной раствор отфильтровывают. Далее щелочной раствор альгината натрия разбавляют 15% раствором хлорида кальция, получая таким образом кальциевую соль альгиновой кислоты. Альгинат кальция обесцвечивают с помощью 1% раствора гипохлорита натрия. Альгиновую кислоту получают при обработке отбеленного альгината кальция 5% раствором хлористоводородной кислоты, переводимой в свою очередь в легко образуемый при взаимодействии альгиновой кислоты с карбонатом натрия альгинат натрия, который после предварительного обезвоживания с помощью метанола сушат при температуре 40-50oС. Образующийся альгинат натрия представляет собою продукт белого цвета, без вкуса и запаха, с содержанием альгиновой кислоты до 85%. Несмотря на более высокое содержание альгиновой кислоты и относительно низкую температуру сушки (40-50oC), данный способ имеет практически те жа недостатки, что и предыдущий и, кроме того, по-видимому, применение для отбеливания препарата очень сильного окислителя - гипохлорита натрия способствует разложению уроновых кислот, прежде всего гулуроновой, т.к. соотношение составляет 4,70. Использование очень токсичного реактива - метанола для обезвоживания альгината натрия также ограничивает применение описанного способа.
Известен метод получения очищенных образцов альгината натрия (Ажгихин И. C. , Шпаков Ю. Н. , Кипиани Р.Е., Гандель В.Г., Морская фармация (Теория и практика нового направления в фармацевтической науке). - Кишинев:иШтиинца, 1982. - 260с. ), который заключается в следующем. Заготовленные водоросли (Laminaria Ligitata) освобождают от механических загрязнений, тщательно промывают проточной водой и измельчают. Тонкий порошок водорослей заливают 0,2 н. раствором серной кислоты в соотношении 1:50 и при постоянном перемешивании мацерируют в течение 12-15 ч. По окончании экстрагирования смесь фильтруют через бумажный фильтр, на фильтре водорослевый порошок промывают водой, отбрасывая фильтрат. Оставшиеся водоросли экстрагируют 1% раствором карбоната натрия в соотношении 1:50 в течение 8-12 ч при постоянном перемешивании смеси. Затем разбавляют экстракционную вязкую массу пятикратным количеством воды и после тщательного перемешивания фильтруют через крупнопористый бумажный фильтр. Полученный фильтрат смешивают с равным объемом этанола, перемешивая до образования клейкого осадка, который центрифугируют. Осадок промывают последовательно этанолом и эфиром и высушивают при температуре 30-40oС. Для получения особо чистого препарата альгинат натрия растворяют в воде и повторно осаждают этанолом. Осаждение полисахарида ускоряется в присутствии хлорида натрия уже в концентрации 0,1-0,2%. Данный способ получения альгината натрии близок к предлагаемому и выбран за прототип.
Недостатками указанного способа являются: а) относительно невысокое содержание альгинатов в препарате (92%), б) низкое содержание гулуроновой кислоты (22,47%, или соотношение М/G составляет 3,45) и, как следствие, невысокая комплексообразующая способность 1320 мг Рв2+/г, в) относительно высокое содержание сульфатной золы (16,5%), г) высокое содержание железа (0,09% к препарату), д)низкая относительная вязкость альгината натрия (примерно 20 для 1% водного раствора).
Поскольку полученный препарат альгината натрия характеризуется невысоким содержанием полигулуронида и высокой зольностью, то он имеет ограниченное применение для медицинских целей в качестве детоксиканта тяжелых металлов. Поэтому совершенствование технологии получения альгината натрия, обогащенного полигулуронидами и освобожденного от катионов поливалентных металлов, для усиления его сорбционных свойств позволит получить альгинат натрия для медицинского применения в качестве антидота при отравлении тяжелыми металлами.
Цель изобретения - повышение количественного содержания альгината натрия в препарате, повышение содержания полигулуронидной составляющей в препарате, повышение степени чистоты для возможности применения альгината натрия в медицине в качестве детоксиканта тяжелых металлов.
Поставленная цель достигается тем, что способ получения медицинского очищенного альгината натрия из ламинарии сахаристой осуществляют путем освобождения водорослей от механических загрязнений и измельчения. Далее сырье заливают 0,1 моль/л раствором серной кислоты в массообъемном соотношении сырье: раствор кислоты 1: 50 и экстрагируют в течение 15 ч при медленном встряхивании на качалке. Затем смесь фильтруют через плотный бумажный фильтр ("синяя лента"); остаток на фильтре промывают водой и фильтрат отбрасывают. Оставшиеся водоросли экстрагируют 1% раствором карбоната натрия в массообъемном соотношении сырье:экстрагент 1:50 в течение 12 ч при встряхивании на качалке. Далее вязкую экстракционную массу обрабатывают 1% раствором оксалата аммония при объемном соотношении экстракт:1% раствор оксалата аммония 1: 4, чтобы доосадить катионы поливалентных металлов, которые не осадились в виде сульфатов, а также чтобы сделать фильтрование возможным. Смесь фильтруют через крупнопористый бумажный фильтр ("красная лента"). Остаток отбрасывают. Полученный фильтрат при определенной ионной силе, создаваемой с помощью 0,1 моль/л раствора хлорида натрия, диализуют через целлофановую мембрану при толщине мембраны 8 нм с диаметром пор 0,4 нм и времени диализа 24 ч. Полученный диализат упаривают под вакуумом примерно до 1/25 от объема экстракта, смешивают с равным объемом 96 мас.% этанола и перемешивают стеклянной палочкой. Обычно клейкий осадок альгината натрия прилипает практически полностью к палочке и его можно выделить из смеси таким путем без фильтрования. Осадок промывают спиртом и эфиром по два раза. Затем осадок растворяют в воде в соотношении альгинат натрия:вода 1:200, полученный раствор смешивают с равным объемом насыщенного раствора хлорида калия; смесь центрифугируют в течение 1 ч на препаративной ультрацентрифуге при 30000 обор./мин. Прозрачную жидкость количественно отделяют от осадка пипеткой и прибавляют к ней равный объем спирта; выпавший осадок снова растворяют в воде и еще раз осаждают спиртом. Образовавшийся осадок суспендируют в воде при соотношении 1: 200 и смешивают с раствором 0,09 моль/л по сульфату марганца (II) и 0,09 моль/л по хлориду калия в объемном соотношении суспензия:раствор солей 2:1. После центрифугирования смеси в течение 1,5 ч при 30000 обор./мин прозрачный раствор отделяют от рыхлого студенистого осадка пипеткой и отбрасывают. Осадок вымывают из центрифужной пробирки водой, осаждают равным объемом спирта и суспендируют в воде. К суспензии добавляют 0,1 моль/л раствора этилендиаминтетрауксусной кислоты (в объемном соотношении 3:1), 1% раствор карбоната натрия (объемное соотношение 10:1) и 1% раствор гидрокарбоната натрия (объемное соотношение 10:1). Когда осадок полностью растворится, альгинат снова осаждают равным объемом спирта, промывают спиртом и эфиром и сушат при температуре 30-40oС.
В предлагаемом способе, как и в способе прототипе, проводится длительный кислотный гидролиз. Это связано с различной растворимостью уроновых кислот в кислой среде: полиманнурониды хорошо растворимы в кислоте, а полигулурониды - мало растворимы. Таким образом, стадией кислотного гидролиза проводится разделение полигулуронидов и полиманнуронидов; фильтрат, содержащий полиманнурониды, отделяется.
В предлагаемом способе щелочной раствор альгината натрия обрабатывают 1% раствором оксалата аммония при объемном соотношении 1:4. Это позволяет осадить катионы поливалентных металлов в виде оксалатов, которые характеризуются высокими значениями констант устойчивости:(lgК):оксалат железа (III) 20,2; оксалат магния 4,38; оксалат алюминия 16,3 или малыми значениями произведений растворимости (рПР): оксалат кальция 8,64; оксалат стронция 6,80 (Лурье Ю.Ю. Справочник по аналитической химии. - М.: Химия, 1989. -С. 69-82, 322-323), т. к. указанные поливалентные катионы содержатся в сырье. Оксалат аммония хелатообразно связывает катионы металлов, сопутствующие уроновым кислотам (особенно полигулуронидам) в альгинате. При гидролизе солей уроновых кислот, связанных с поливалентными катионами, эти катионы освобождаются и затем они с помощью оксалата аммония переводятся в нерастворимые соединения. При этом натрий и аммоний, связанные с уроновыми кислотами, легко обмениваются на поливалентные катионы при терапии интоксикаций тяжелыми металлами. Выделенные оксалаты металлов отделяются от альгината фильтрацией.
Стадия диализа фильтрата позволяет также очистить альгинат натрия. Это можно подтвердить записью УФ-спектра альгината натрия до и после диализа (фиг. 1). Как видно из фиг.1, после диализа (кривая 2), спектр имеет отчетливый максимум поглощения при длине волны 225 нм с большей величиной оптической плотности, чем до диализа (кривая 1).
Диализ растворов альгинатов в предлагаемом способе производится при постоянной ионной силе, создаваемой с помощью хлорида натрия. Присутствие сильного электролита разрывает водородные связи между цепями полиуроновых кислот в макромолекуле альгината натрия, что способствует их лучшему прохождению через целлофановую мембрану.
Для оценки эффективности связывания полисахаридами ионов металлов используется показатель комплексообразующая способность (КС), который характеризует число мг ионов металла, связанное 1 г полисахарида (Давидюк Л.П., Вшивкова Г.Ф. Сравнительное изучение комплексообразующей способности пектинов разного происхождения. /Бюллетень государ. Никитского ботанического сада. - Ялта: ГНБС, 1981. -Вып. 3 (46). -С. 98-101).
Как указывалось выше, образцы альгината натрия, значительно освобожденные от ионов тяжелых металлов, но обедненные полигулуронидами, по способности ингибировать всасывание тяжелых металлов уступают образцам с высоким содержанием L-гулуроновой кислоты. В способе, принятом за прототип, осуществляются стадии деминерализации и разделения полигулуронидов от полиманнуронидов (за счет кислотного гидролиза), но не проводится стадия фракционирования гулуронидов. Эта стадия осуществляется в заявляемом способе, она основана на различном сродстве гулуронидов к ионам калия и марганца (II): образец альгината натрия, растворимый в насыщенном растворе хлорида калия и нерастворимый в растворе сульфата марганца (II), характеризуется высоким содержанием гулуронидов.
Таким образом, предлагаемый способ получения альгината натрия, основанный на сочетанном использовании частичного гидролиза, деминерализации и фракционирования гулуронидов, позволяет получить медицинский альгинат натрия, т.е. альгинат натрия, обогащенный гулуронидами (стадии гидролиза, фракционирования) и освобожденный от катионов поливалентных металлов (стадии осаждения в виде сульфатов и оксалатов).
Для оценки качества препаратов альгината натрия определяли:
1) количественное содержание альгината натрия - методом спектрофотометрии по реакции с карбазолом (ВФС 42-1680-87. Натрия альгинат);
2) относительное содержание Д-маннуроновой и L-гулуроновой кислот - методами спектрофотометрии (Knutson, (Geanes, 1968), ГЖХ (Lehrfeld, 1981). Образцы альгината натрия подвергали гидролизу в соответствии с методикой Lehrfeld и затем в виде ацетатов полиолов подвергали хроматографическому разделению на приборе Pye Series 104 (Подкорытова А.В. Разработка технологии получения высокомолекулярного альгината натрия из культивируемой ламинарии японской /Автореферат дис. ... канд. техн.наук.-М., 1986. - 24с.). По высотам пиков соответствующих ацетатов полиолов подсчитывали соотношение уроновых кислот (М/G);
3) содержание сульфатной золы (ГФ XI. - М.: Медицина, 1990. - Вып. 2. - С. 25);
4) содержание железа - методом спектрофотометрии по реакции с 1,10-фенантролин сульфатом и калия иодидом (ВФС 42-2604-95. Альгисорб);
5) относительную вязкость 1% водного раствора - по скорости истечения раствора на вискозиметре ВПЖ-1 при 20oС (ГФ XI. - М.: Медицина, 1987. -Вып. 1. - С. 89).
6) КС - методом комплексонометрии (по числу мг свинца, связанного 1 г полиуронида) (Компанцев В.А., Кайшева Н.Ш., Гокжаева Л.П., Сизова Н.М. Определение комплексообразующей способности пектинов и пектиносодержащих препаратов/Охрана окружающей среды. - М.: НИИТЭХИМ, 1991. - С. 25-27).
Заявляемый способ обеспечивает получение образца альгината натрия с высоким количественным содержанием альгината натрия (99,95%); с высоким содержанием гулуроновой кислоты (соотношение М/G- составляет 0,75 или содержание гулуроновой кислоты 57%), с низким содержанием сульфатной золы (5,2%), не содержащим железо, с высокой относительной вязкостью альгината натрия (22,4), с высокой КС (2875 мг Рв2+/г).
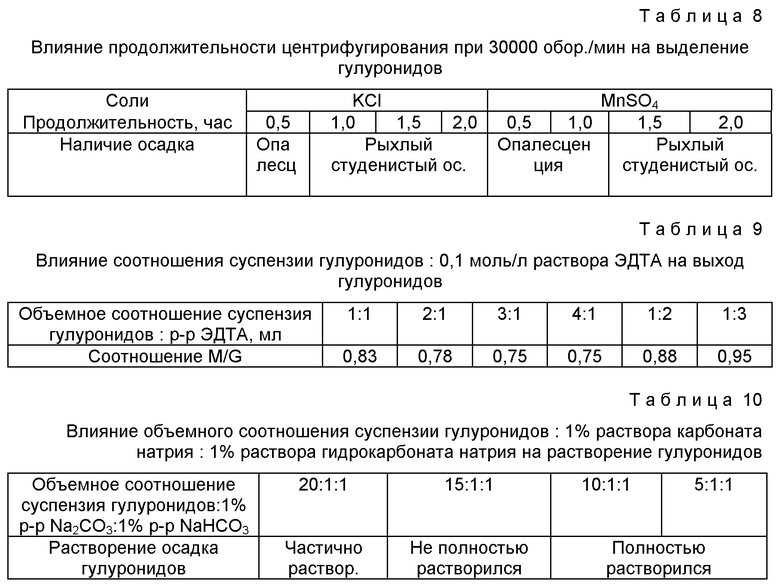
В результате исследований подобраны следующие оптимальные условия выделения очищенного альгината натрия (таблицы 1-10, фиг.1, 2).
Из таблицы 1 видно, что применение оксалата аммония для осаждения поливалентных катионов в щелочном растворе обеспечивает наиболее высокое количественное содержание альгината натрия в препарате.
Из таблицы 2 следует, что оптимальной концентрацией раствора оксалата аммония, обеспечивающей наибольшее количественное содержание альгината натрия, является 1,0%. Дальнейшее повышение концентрации раствора оксалата аммония не приводит к заметным изменениям содержания альгината натрия в препарате.
Из таблицы 3 видно, что оптимальным объемным соотношением (в мл) растворов альгината натрия и 1% раствора оксалата аммония, при котором количественное содержание альгината натрия максимальное, является соотношение 1:4. Хотя при соотношении 1:5 достигается аналогичное количественное содержание действующих веществ в препарате, но в этом случае происходит очень сильное разбавление раствора альгината натрия раствором оксалата аммония, что в дальнейшем увеличивает продолжительность упаривания экстрактов.
Влияние диализа альгинатных растворов через целлофановую мембрану (ее стандартные параметры: толщина 8 нм, диаметр пор 0,4 нм) показано на фиг.1.
Как видно из табл.4, оптимальная продолжительность диализа для альгинатных растворов составляет 24 ч; более продолжительный диализ не приводит к существенному изменению оптической плотности растворов.
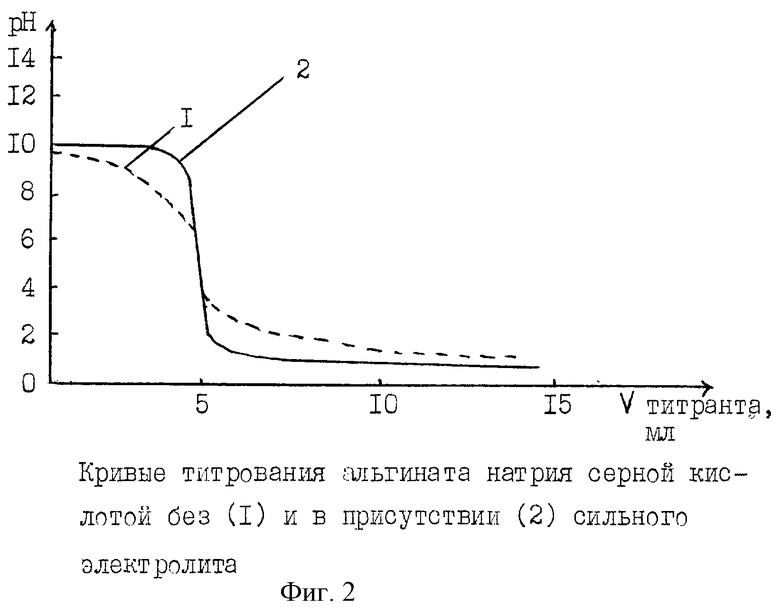
Влияние сильного электролита на характер кривой титрования альгината натрия серной кислотой показано на фиг.2.
Влияние электролита (0,1 моль/л раствора хлорида натрия) на характер кривой потенциометрического титрования альгината натрия с помощью 0,1 моль/л раствора серной кислоты очевидно: если раствор альгината натрия имеет рН 9,58, то после добавления к нему 0,1 моль/л раствора хлорида натрия рН раствора становится 9,16. Кривая потенциометрического титрования альгината натрия в присутствии хлорида натрия (кривая 2) более вертикальна, чем без электролита (кривая 1), т. е. в этом случае скачок титрования больше. Хлорид натрия разрушает водородные связи в макромолекуле альгината между цепями уроновых кислот, уменьшая их "сцепляемость", это приводит к разобщению полиуронидных цепочек и улучшению их проходимости через мембрану.
Как видно из табл.5, осаждение хлоридом калия (нерастворимая фракция) и растворение в сульфате марганца (II) (растворимая фракция) ведут к накоплению молекул с высоким содержанием остатков Д-маннуроновой кислоты, и, наоборот, растворение в хлориде калия (растворимая фракция) и осаждение сульфата марганца (II) приводят к накоплению молекул с высоким содержанием остатков L-гулуроновой кислоты. Сочетанное применение осаждения хлоридом калия и растворение в сульфате марганца (II) ведет к накоплению молекул, обогащенных Д-маннуроновой кислотой, а применение растворения в хлориде калия и осаждения. сульфатом марганца (II) - к накоплению L -гулуроновой кислоты. Поэтому последнее сочетание нами использовано в предлагаемом способе.
Как следует из табл. 6, наиболее оптимальной концентрацией обеих солей, обеспечивающей наибольшее содержание гулуроновой кислоты, является 0,09 моль/л.
Как видно из табл. 7, при соотношении раствор альгината натрия: растворы солей (в концентрации по 0,09 моль/л) 2:1 достигается наиболее высокое содержание гулуронидов.
Из табл. 8 видно, что при выделении гулуронидов разделение осадка и надосадочной жидкости с помощью хлорида калия четко наблюдается при центрифугировании в течение 1,0 ч, а при разделении сульфатом марганца (II) - при центрифугировании в течение 1,5 ч.
Добавление этилендиаминтетрауксусной кислоты (ЭДТА) при растворении гулуронидов связано с необходимостью устранения ионов кальция, которые, будучи связаны с гулуронидами в виде нерастворимых соединений, снижают выход гулуронидов.
Как следует из табл. 9, наиболее высокое содержание гулуронидов (0,75) достигается при объемном соотношении суспензии гулуронидов: раствора ЭДТА 3: 1, дальнейшее увеличение объема раствора ЭДТА не влияет на показатель М/G.
Поскольку гулурониды в воде нерастворимы, то для перевода их в растворимое состояние необходимо создание щелочной среды. Использование для этих целей растворов карбоната и гидрокарбоната натрия позволяет растворить полностью целевой продукт.
Из таблицы 10 видно, что при соотношении растворов (мл) - суспензия:1% раствор карбоната натрия: 1% раствор гидрокарбоната натрия 10:1:1 осадок гулуронидов полностью растворяется, т.е. образуется истинный раствор.
Предлагаемый способ получения медицинского очищенного альгината натрия поясняется следующими примерами конкретного выполнения.
ПРИМЕР 1. Беломорские водоросли Северного бассейна (Laminaria saceharina L. ) высушивают при температуре 50oС в сушильном шкафу, затем освобождают их от механических примесей, моллюсков и измельчают на лабораторном измельчителе. Отвешивают 10 г водорослей, помещают в колбу вместимостью 1 л, приливают 500 мл 0,1 моль/л раствора серной кислоты и экстрагируют в течение 15 ч при медленном встряхивании на качалке. Затем смесь фильтруют через плотный бумажный фильтр ("синяя лента"); остаток на фильтре промывают 50-100 мл воды и фильтрат отбрасывают. Оставшиеся водоросли вновь помещают в колбу вместимостью 1 л, приливают 500 мл 1% раствора карбоната натрия и экстрагируют в течение 12 ч при встряхивании на качалке. Образуется вязкая экстракционная масса, к которой добавляют 2 л 1% раствора оксалата аммония, перемешивают и фильтруют через бумажный фильтр "красная лента". Oстаток на фильтре отбрасывают. К полученному фильтрату добавляют хлорид натрия из расчета его концентрации в конечном растворе 0,1 моль/л (т.е. на 2,5 л экстракта необходимо добавить 14,625 г хлорида натрия), экстракт перемешивают до растворения хлорида натрия, после чего проводят диализ экстракта в течение 24 ч через целлофановую мембрану толщиной 8 нм и диаметром пор 0,4 нм. Для диализа используют горизонтальный прибор, в котором имеется два отсека, разделенных между собой целлофановой мембраной, края которой закреплены на внутренней стороне корпуса с помощью водостойкого клея. В один отсек через отверстие прибора сверху помещается диализуемый экстракт, который диализуется через мембрану в другой отсек (диализатор). Затем полученный диализат упаривают под вакуумом примерно до 1/25 от объема экстракта, смешивают с равным объемом 96% этанола и перемешивают стеклянной палочкой. Клейкий осадок альгината натрия прилипает практически полностью к палочке и его можно выделить из смеси таким путем без фильтрования. Осадок промывают два раза 96% спиртом и два раза эфиром. Затем осадок растворяют в 400 мл воды; образующийся прозрачный гомогенный раствор смешивают с 400 мл насыщенного раствора хлорида калия (при 20oС в 400 мл воды растворяют 136 г хлорида калия); смесь центрифугируют на препаративной ультрацентрифуге при 30000 обор./мин в течение 1 ч. Прозрачную жидкость (в основном гулурониды) тщательно отделяют от рыхлого студенистого осадка (в основном маннурониды) пипеткой; к жидкости приливают равный объем 96% спирта; выпавший осадок снова растворяют в таком количестве воды, чтобы получить 0,5%-й раствор альгината, и еще раз осаждают равным объемом 96% спирта, отделяют осадок центрифугированием. Осадок суспендируют в 400 мл воды и смешивают с 200 мл раствора 0,09 моль/л по сульфату марганца (II) и 0,09 моль/л по хлориду калия. После центрифугирования смеси при 30000 сбор. /мин в течение 1,5 ч прозрачный раствор (преимущественно маннурониды) отделяют от рыхлого студенистого осадка (преимущественно гулурониды) пипеткой. Полученный осадок вымывают из центрифужной пробирки водой, осаждают равным объемом 96% спирта и суспендируют в 100 мл воды. К суспензии добавляют 30 мл 0,1 моль/л раствора ЭДТА и по 10 мл 1% раствора карбоната натрия и 1% раствора гидрокарбоната натрия. Когда осадок полностью растворится, альгинат снова осаждают равным объемом 96% спирта, осадок отделяют центрифугированием, промывают спиртом и эфиром и сушат при температуре 30-40oС.
Целевой продукт - альгинат натрия, обогащенный гулуронидами, представляет собой тонкие пластинки белого цвета, без запаха, слизистого вкуса. Количественное содержание альгината натрия в препарате 99,95%. Соотношение М/G составляет 0,75 (или содержание гулуроновой кислоты 57%). Содержание сульфатной золы 5,2%. В препарате отсутствует железо. Относительная вязкость 1% водного раствора альгината натрия 22,4. КС препарата 2875 мг Рв2+/г.
Маточные спиртовые растворы подвергают обработке с целью регенерации из них спирта.
ПРИМЕР 2. Извлечение альгината натрия проводят аналогично примеру 1. К вязкому щелочному экстракту (после добавления 1% раствора карбоната натрия) добавляют 2,5 л 1,5% раствора оксалата аммония и также фильтруют. Полученный фильтрат при ионной силе, создаваемой 0,1 моль/л раствором хлорида натрия, диализуют в течение 48 ч через целлофановую мембрану толщиной 8 нм и диаметром пор 0,4 нм. Выделение из диализата альгината натрия, растворение его в воде и осаждение хлоридом калия проводят аналогично примеру 1. Смесь центрифугируют при 30000 обор. /мин в течение 1,5 ч. Из жидкости, содержащей преимущественно гулурониды, выделяют альгинат натрия, как описано в примере 1. Осадок суспендируют в 400 мл воды и смешивают с 800 мл раствора 0,10 моль/л по сульфату марганца (II) и 0,10 моль/л по хлориду калия. Смесь центрифугируют при 30000 обор./мин в течение 2,0 ч. Выделение фракции гулуронидов проводят аналогично примеру 1. К суспензии добавляют 25 мл 0,1 моль/л раствора ЭДТА и по 20 мл 1% раствора карбоната натрия и 1% раствора гидрокарбоната натрия. Далее выделяют альгинат натрия, как описано в примере 1.
Целевой продукт имеет следующие характеристики: количественное содержание альгината натрия в препарате 99,33%; соотношение М/G 0,75 (содержание гулуроновой кислоты 57%); содержание сульфатной золы 5,4%; в препарате отсутствует железо. Относительная вязкость 1% водного раствора альгината натрия 23,8. КС препарата 2860 мг Рв2+/г.
ПРИМЕР 3. Извлечение альгината натрия проводят аналогично примеру 1. К вязкому щелочному экстракту после добавления 1% раствора карбоната натрия добавляют 1,5 л 0,5% раствора оксалата аммония и также фильтруют. Полученный фильтрат при ионной силе, создаваемой 0,1 моль/л раствором хлорида натрия, диализуют в течение 10 ч через целлофановую мембрану толщиной 8 нм и диаметром пор 0,4 нм. Выделение из диализата альгината натрия, растворение его в воде и осаждение хлоридом калия проводят аналогично примеру 1. Смесь центрифугируют при 30000 обор./мин в течение 0,5 ч. Из раствора, содержащего преимущественно гулурониды, выделяют альгинат натрия, как описано в примере 1. Осадок суспендируют в 400 мл воды и смешивают с 400 мл раствора 0,08 моль/л по сульфату марганца (II) и 0,08 моль/л по хлориду калия. Смесь центрифугируют при 30000 обор./мин в течение 1,0 ч. Выделение фракции гулуронидов проводят аналогично примеру 1. К суспензии добавляют 50 мл 0,1 моль/л раствора ЭДТА и по 7,5 мл 1% раствора карбоната натрия и 1% раствора гидрокарбоната натрия. Далее выделяют альгинат натрия, как описано в примере 1.
Целевой продукт имеет следующие характеристики: количественное содержание альгината натрия в препарате 98,4%; соотношение M/G 0,93 (содержание гулуроновой кислоты 51,8%); содержание сульфатной золы 6,2%; в препарате отсутствует железо. Относительная вязкость 1% водного раствора альгината натрия 18,9. КС препарата 2110 мг Рв2+/г.
Таким образом, предлагаемый способ получения медицинского очищенного альгината натрия обеспечивает следующий положительный эффект:
1. Повышение количественного содержания альгината натрия в препарате: так, если по способу, принятому за прототип, содержание альгината натрия составляет 92%, то по заявляемому способу оно достигает 99,95%, т.е. превышение содержания составляет 1,1 раза. Этот эффект обусловлен следующими причинами: а) по способу-прототипу водоросли тщательно промывают проточной водой, по заявляемому - промывка не предусматривается. При промывке водой водоросли попадают в среду, отличную от среды водоемов как по концентрации, так и по составу солей. Поэтому между водорослями и растворителем наступает обмен компонентами. Если водоросли промываются водой, то происходит экстракция всех водорастворимых компонентов, в т.ч. и солей. Соли создавали слабокислую среду, и удаление их равносильно нейтрализации одноосновным катионом. Вследствие этого в равновесной системе: полиуроновая кислота ⇄ ангидрид полиуроновой кислоты происходит сдвиг в сторону образования кислоты, а это приводит к потерям целевого продукта с промывными водами (Евтушенко В.А. Химические основы технологии альгиновой кислоты /Рыбное хозяйство. - М.: Пищепромиздат, 1949. - 7. - С. 44-48); б) добавление оксалата аммония к раствору альгината натрия способствует переведению оставшихся (после гидролиза и осаждения серной кислотой) поливалентных катионов в нерастворимые соединения. При этом натрий и аммоний, связанные с уроновыми кислотами, легко обмениваются на поливалентные катионы при дальнейшем использовании альгината в качестве детоксиканта тяжелых металлов. Деминерализация уроновых кислот альгината способствует снижению потерь уронидов за счет перевода их в растворимое состояние.
2. Повышение содержания полигулуронидной составляющей в полученном препарате альгината натрия: если в препарате, полученном по способу-прототипу, соотношение М/G составляет 3,45 (или содержание гулуронидов 22,47% от уроновых кислот), то по заявляемому способу это соотношение составляет 0,75 (или содержание гулуронидов 57%), т.е. превышение содержания гулуронидов составляет 2,5 раза. Это особенно важно, т.к. применение альгината натрия в качестве медицинского препарата основано на его способности к селективному ионообмену, который зависит от содержания L-гулуроновой кислоты в полимере.
Этот положительный эффект обусловлен не только кислотным гидролизом (осуществляемым в обоих способах), но и стадией фракционирования уронидов (заявляемый способ), основанной на различном сродстве к ионам марганца (II) и калия. Именно сочетанное использование частичного гидролиза, деминерализации и фракционирования позволяет получить альгинат натрия, обогащенный гулуронидами.
3. Повышение КС альгината натрия: по предлагаемому способу она составляет 2875 мг Рв 2+/г, по прототипу - 1320 мг Рв 2+/г (превышение в 2,2 раза). Увеличение КС альгината натрия является следствием повышения содержания гулуронидов в препарате.
4. Повышение степени чистоты альгината натрия. Высокая степень чистоты альгината натрия, полученного по заявляемому способу, подтверждается следующими физико-химическими характеристиками: низкое содержание сульфатной золы 5,2% (по прототипу 16,5%; снижение зольности составляет 3,2 раза); отсутствие ионов железа (III) (по прототипу содержание ионов железа (III) 0,09%); высокое количественное содержание альгината натрия (превышение в 1,1 раза). Высокая степень чистоты объясняется деминерализацией экстракта оксалатом аммония, диализом раствора в присутствии электролита через целлофановую мембрану; дополнительной очисткой спиртом и эфиром в ходе фракционирования уронидов. Препарат альгината натрия с полученными характеристиками вполне может быть использован как лекарственный препарат перорального и парентерального применения.
5. Повышение вязкости раствора альгината натрия: по предлагаемому способу относительная вязкость 1% раствора альгината натрия составляет 22,4, а по способу-прототипу 20 (превышение в 1,1 раза).


Изобретение относится к фармацевтической промышленности. Проводят освобождение водорослей ламинарии сахаристой от механических загрязнений и измельчение. Затем заливают 0,1 моль/л раствором серной кислоты в массообъемном соотношении сырье: раствор кислоты 1:50 и экстрагируют в течение 15 ч при медленном встряхивании. Затем смесь фильтруют, остаток на фильтре промывают водой и фильтрат отбрасывают. Оставшиеся водоросли экстрагируют 1% раствором карбоната натрия в соотношении сырье: экстрагент 1:50 в течение 12 ч при перемешивании. Далее экстракционную массу обрабатывают 1% раствором оксалата аммония при соотношении 1:4. Смесь фильтруют. Полученный фильтрат диализуют через целлофановую мембрану в течение 24 ч. Диализат упаривают, смешивают с равным объемом 96% этанола. Полученный осадок промывают спиртом и эфиром. Затем растворяют в воде, смешивают с равным объемом насыщенного раствора хлорида калия, смесь центрифугируют. Жидкость отделяют от осадка и прибавляют к ней равный объем спирта. Выпавший осадок снова растворяют в воде и еще раз осаждают спиртом. Осадок суспендируют в воде и смешивают с раствором 0,09 моль/л по сульфату марганца (II) и 0,09 моль/л по хлориду калия в объемном соотношении суспензия:раствор солей 2:1. После центрифугирования смеси раствор отделяют от осадка. Осадок вымывают водой, осаждают равным объемом спирта и суспендируют в воде. К суспензии добавляют 0,1 моль/л раствора ЭДТА в объемном соотношении 3:1, 1% раствор карбоната натрия в объемном соотношении 10: 1 и 1% раствор гидрокарбоната натрия в объемном соотношении 10:1. Когда осадок полностью растворится, альгинат снова осаждают равным объемом спирта, промывают спиртом и эфиром и сушат. Изобретение позволяет повысить чистоту и выход продукта. 10 табл., 2 ил.
Способ получения медицинского очищенного альгината натрия, включающий обработку ламинарии раствором серной кислоты, удаление фильтрата, обработку остатка раствором карбоната натрия, выделение и сушку продукта, отличающийся тем, что используют ламинарию сахаристую, после обработки раствором карбоната натрия щелочную экстракционную массу обрабатывают 1%-ным раствором оксалата аммония при объемном соотношении экстракт: 1%-ный раствор оксалата аммония 1: 4, полученный раствор альгината натрия при постоянной ионной силе, создаваемой 0,1 моль/л раствором хлорида натрия, диализуют через целлофановую мембрану в течение 24 ч, затем альгинат натрия растворяют в воде и фракционируют путем смешивания с равным объемом насыщенного раствора хлорида калия, проводят операции центрифугирования в течение 1 ч, смешивания центрифугата с равным объемом спирта, отделения осадка, растворения его в воде и повторного осаждения спиртом, отделения осадка, суспендирования его в воде, смешивания полученной суспензии с раствором 0,09 моль/л по сульфату марганца (II) и 0,09 моль/л по хлориду калия в объемном соотношении суспензия: раствор солей 2: 1, центрифугирования смеси в течение 1,5 ч, вымывания осадка водой и осаждения его равным объемом спирта, отделения осадка, повторного суспендирования в воде, смешивания с раствором ЭДТА в объемном соотношении суспензия: раствор ЭДТФ 3: 1, добавления растворов карбоната натрия и гидрокарбоната натрия при объемном соотношении суспензия: раствор карбоната натрия: раствор гидрокарбоната натрия 10: 1: 1.
| АЖГИХИН И.С | |||
| и др | |||
| Морская фармация, теория и практика нового направления в фармацевтической науке | |||
| - Кишинев, Штинца, 1982, с.260 | |||
| СПОСОБ ПЕРЕРАБОТКИ БУРЫХ ВОДОРОСЛЕЙ | 1998 |
|
RU2132622C1 |
| СПОСОБ КОМПЛЕКСНОЙ ПЕРЕРАБОТКИ СУХОГО СЫРЬЯ ВОДОРОСЛЕЙ | 1998 |
|
RU2142812C1 |
| СПОСОБ ПЕРЕРАБОТКИ ВОДОРОСЛЕЙ С ПОЛУЧЕНИЕМ ПРОДУКТА, СОДЕРЖАЩЕГО АЛЬГИНАТ НАТРИЯ | 1991 |
|
RU2019981C1 |

