Изобретение относится к антагонистам LHPH и к системной и пероральной "доставке" этих антагонистов путем введения комплекса, содержащего антагонисты, связанные с витамином B12 (VB12) или его аналогом. В частности, настоящее изобретение относится к способам синтеза указанных комплексов.
Гипоталамический фактор выделения гонадотропина (или гонадотропин-рилизинг-фактор, GnRH, известный также как фактор, высвобождающий лютеинизирующий гормон, ЛГ-рилизинг-фактор LHRH) опосредует регуляцию синтеза и секреции гипофизарного гонадотропина. С тех пор, как Schally и др. (1971) впервые выделили LHRH, было синтезировано большое количество аналогов, которые могут быть классифицированы по своему сильному воздействию на высвобождение гонадотропина, либо как антагонисты (ингибирующие высвобождение). Агонисты характеризуются увеличением аффинности связывания с рецепторами гипофизарного LHRH (Loumay и др., 1982, Perrin и др., 1980) и повышенной резистентностью к протеолитической деградации, что способствует быстрому выведению нативного LHRH.
В результате наблюдений, которые показали, что указанные сильные аналоги могут индуцировать медикоментозную кастрацию, был разработан новый способ лечения различных гонадотропинзависимых расстройств, а в частности гормонозависимый простатит и рак молочной железы. При индуцировании кастрации имеют место две различные фазы. Сначала аналог стимулирует систему гипофиз - гонада, вызывая кратковременное увеличение секреции гонадотропина и половых стероидных гормонов в первые две недели или около того. По истечении этого времени происходит снижение регуляции рецепторов гипофизарного LHRH с последующим уменьшением секреции гонадотропина и стероидных половых гормонов. Эта первоначальная стимуляция является главным недостатком использования агонистов в лечении злокачественных опухолей представленной железы, поскольку первоначальная стимуляция тестостерона может стимулировать внезапное и тяжелое обострение болезни с последующими неблагоприятными клиническими эффектами (Eisenberger & Abrams, 1988, Crawford & Davis, 1988). В противоположность этому, первый успешный синтез антагонистов должен дать положительный результат по предотвращению начального обострения болезни. Первые попытки продуцирования антагонистов привели к получению соединений, отличающихся нежелательной способностью к высвобождению гистамина (Karten & Rivier, 1986, Schmidt et al., 1984; Phillips et al., 1988). Однако затем были разработаны новые антагонисты, обладающие повышенной эффективностью и меньшей склонностью к высвобождению гистамина (например, Karten & Rivier, 1986). Одним из наиболее сильных антагонистов, описанных до настоящего времени, является аналог N-Ac-D-NaI (2), D-Phe (pCI), D-PaI (3), Ser, Lys (Nic), D-Lys (Nic), Leu, Lys (iPr), Pro, D-AIa-NH2 (ANTIDE), который был синтезирован Folkers & Bowers. Этот аналог обладает высокой эффективностью в ингибировании овуляции у крыс (Ljundquist и др. , 1988), а одноразовые дозы указанного аналога оказывают сильное и продолжительное ингибирующее действие на концентрацию LH в сыворотке кастрированных самок собакоподобных обезьян (Leal et al., 1988). Было также показано, что данный аналог обладает способностью индуцировать продолжительную химическую кастрацию у интактных половозрелых крыс самцов и собакоподобных обезьян и оказывает ингибирующее действие на опухолевый рост в модели карциномы простаты Dunning R3327, аналогично тому, как это имеет место при кастрации (Habennicht et al., 1990). Поэтому новый антагонист должен представлять особый клинический интерес во всех случаях, при которых лекарственная кастрация является весьма желательной, особенно при лечении рака представленной железы и различных гинекологических расстройств (MeLachlan et al., 1986). Применяемый в настоящее время способ введения антагониста ограничивается парентеральным введением. Однако при этом способе введения доза аналога, которая может быть "доставлена" к нужному участку организма, ограничена из-за плохой растворимости указанного антагониста. Так, например, при клинических испытаниях продолжительности действия ANTIDE, дозы были снижены до 2,5 мг или менее.
Пероральное введение пептидов, таких как LHRH и их аналоги в качестве фармацевтических средств для лечения системных заболеваний, оказалось малоуспешным. Другими словами, количество пептида, необходимое для успешного перорального введения, в 100 - 1000 раз превышает количество, требуемое для парентеральной "доставки" этих агентов, что делает их применение путем перорального введения недопустимо дорогостоящим. Неудача перорального введения была обусловлена двумя основными причинами. Во-первых, среда в кишечнике обладает высокой степенью протеолитической активности, способствующей быстрой деградации большинства пептидов. И во-вторых, хотя механизмы поглощения отдельных аминокислот и депептидов хорошо известны, однако, общий механизм транспорта полипептидов через мембрану эпителия слизистой в кровоток пока не ясен. Эта мембрана скорее всего является основным барьером, препятствующим поглощению многочисленных чужеродных белков, присутствующих в окружающей среде. Поэтому, хотя для защиты от ферментативного расщепления, происходящего в кишечнике, пептид и может быть модифицирован, однако такая модификация будет малоэффективной, если данный пептид не сможет затем преодолеть барьер слизистой оболочки и войти в большой круг кровообращения.
Однако в последней работе автора настоящей заявки (PCT/AU86/00299) был разработан метод, позволяющий преодолеть барьер слизистой оболочки. В этом методе эффективно используется механизм опосредованного нативным внутренним фактором (IF) поглощения витамина B12 (VB12). VB12 представляет собой природную поступающую в организм вместе с пищей молекулу, которая активно поглощается организмом из кишечника, в процессе чего, указанная молекула сначала связывается с IF в верхней тонкой кишке. После этого комплекс "VB12-IF" проходит по тонким кишкам и связывается с IF-рецептором, локализованным на поверхности эпителия подвздошной кишки. Затем, посредством рецепторопосредованного эндоцитоза, происходит интернационализация всего комплекса "VB12-IF-Рецептор", после чего через некоторое время VB-12 появляется в сыворотке.
В PCT-заявке PCT/AU86/00299 (087/02251) описаны способы химической модификации VB12, осуществляемые в целях введения соответствующих функциональных групп для конъюгации VB12 с различными лекарственными средствами и пептидными/протеиновыми фармацевтическими средствами. Если комплекс "VB12-фармацевтическое средство" вводят перорально, то для доставки фармацевтического средства в кровоток может быть использована система поглощения VB12, опосредованного натуральным IF.
Поскольку пероральный способ введения является наиболее предпочтительным способом доставки фармацевтически активного агента, то любой способ, обеспечивающий пероральную доставку антагонистов LHRH в организм человека, будет пользоваться большим спросом на имеющемся рынке предложений. Такой способ может быть осуществлен путем образования комплекса между VB12 или его аналогами и антагонистами LHRH.
В своем первом аспекте настоящее изобретение относится к аналогам ANTIDE.
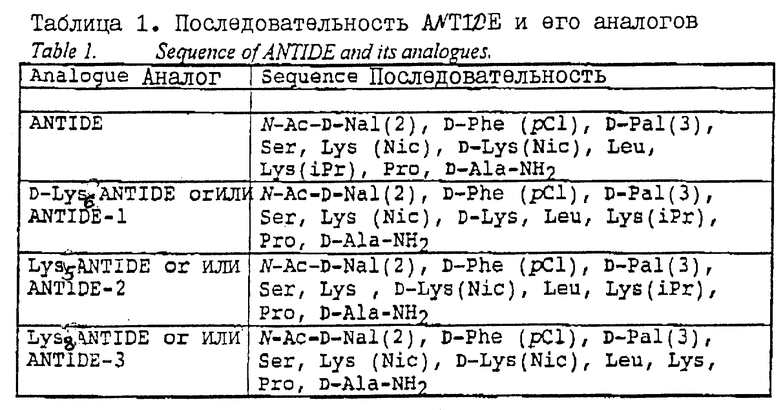
ANTIDE представляет собой молекулу, которая является особенно подходящей для перорального введения, поскольку она содержит очень небольшое количество природных аминокислот и является в высокой степени резистентной к действию пепсина, трипсина или химотрипсина. Однако саму молекулу ANTIDE необходимо слегка модифицировать в целях сообщения ей подходящего сайта для конъюгации с VB12. Заявителем было обнаружено, что могут быть синтезированы три аналога ANTIDE, которые являются подходящими для конъюгации с VB12, а именно N-Ac-D-Nal(2), D-Shc(pCl), D-Pal(3), Ser, Lys (Nic), D-Lys, Leu, Lus (iPr), Pro, D-Ala-NH2(D-Lus6 ANTIDE или ANTIDE-1), N-Ac-D-Nal(2), D-Phe(pCl), D-Pal(3), Ser, Lys, D-Lys(Nic), Leu Lys (iPr), Pro, D-Ala-NH2 (Lys5 ANTIDE или ANTIDE-2), и N-Ac-D- Nal(2), D-Phe(pCl), D-Pal(3), Ser, Lys(Nic), D-Lys(Nic), Leu, Lys, Pro, D-Ala-NH2(Lys8 ANTIDE или ANTIDE-3).
Во втором варианте настоящее изобретение относится к комплексу между VB12 или аналогом VB12 и антагонистами LHRH, рассматриваемыми в первом аспекте изобретения, где для поглощения и транспорта VB12 в хозяине-позвоночном необходимо, чтобы указанный VB12 или его аналог обладал способностью вступать в реакцию связывания, и где активность антагониста I HPH сохраняется в основном на постоянном уровне.
Аналоги VB12 представляют собой любой вариант или любое производное VB12, которые обладают способностью к связыванию с внутренним фактором (IF). Предпочтительными аналогами VB12 являются цианокобаламин (CN-CbI), аквокобаламин, аденозилкобаламин, метилкобаламин, гидроксикобаламин, цианокобаламина карбаналид, 5-метоксибензилкобаламин [(5-MeOBz)CN-CbI], а также десдиметиловые, моноэтиламидные и метиламидные аналоги всех указанных выше соединений. Другими аналогами являются кофермент B12, 5'-дезоксиаденозилкобаламин, хлорокобаламин, сульфитокобаламин, нитрокобаламин, тиоцианатокобаламин, производные бензимидазола, такие как 5,6-дихлоробензимидазол, 5-гидроксибензимидазол, триметилбензимидазол, а также аденозилцианокобаламин [(Ade)CN-CbI] , кобаламинлактон, кобаламинлактам и анилидные, этиламидные, монокарбоновокислотные и дикарбоновокислотные производные VB12 или его аналоги.
Предпочтительными производными VB12 также являются моно- ди- и трикарбоновокислотные производные пропионамидных производных VB12. Другими производными являются аналоги VB12, в которых вместо кобальта присутствует цинк или никель. Корриновое кольцо витамина B12 или его аналогов может быть также замещено любым заместителем, не оказывающим какого-либо влияния на связывание VB12 IF, причем эти производные VB12 или его аналоги также являются частью настоящего изобретения. Другие производные и аналоги витамина B12 обсуждаются в работе Schneider Z. и Stroinski A. (Walter De Gruyter, Berlin, NY; 1987), и это обсуждение вводится в настоящее описание посредством ссылки.
Подходящими для конъюгации протяженными спейсерами являются дисукцинимидилсуберат (DSS), бис(сульфосукцинимидил)суберат (BSS), этиленгликольбис(сукцинимидилсукцинат)(EGS), этиленгликольбис(сульфосукцинимидилсукцинат) (сульфо-EGS), n-аминофенилуксусная кислота, дитиобис(сукцинимидилпропионат) (DSP), 3,3'-дитиобис(сульфосукцинимидилпропионат) (DTSSP), дисукцинимидилтартарат (DST), дисульфосукцинимидилтартарат (сульфо-DST) бис[2-(сукцинимидооксикарбонилокси) -этилен] сульфон (BSOCOES), бис[2-сульфосукцинимидооксикарбонилокси)-этилен] сульфон (сульфо-BSOCOES), диметиладипимидат, 2HCl (DMA), диметилпимелимидат, 2HCl (DMP), диметилсуберимидат, 2HCl (DMS).
Перекрестносшивающими агентами, подходящими для использования в получении расщепляемых тиолом биологически разлагаемых линкеров, являются N-сукцинимидил 3-(2-пиридилдитио)пропионат (SPDP), иминотиолан, сульфосукцинимидил 6-[3-(2-пиридилдитио) пропионамидо] гексаноат (Сульфо-IC-SPDP), сукцинимидил 6-[3-(2- пиридилдитио)пропионамидо]гексаноат (IC-SPDP), сульфосукцинимидил 6-[ α -метил- α -(2-пиридилдитио)толуамидо]гексаноат (Сульфо-IC-SMPT), 1,4-ди-[3'-(2'-пиридилдитио)пропионамидо] бутан (DPDPB), 4-сукцинимидилоксикарбонил- α метил- α -(2-пиридилдитио)-толуол (SMPT), диметил 3,3'-дитиобиспропионимидат, 2 HCl (DTBP).
В другом аспекте настоящее изобретение относится к способу получения биологически расщепляемых комплексов между VB12 и антагонистами LHRH, такими как аналоги ANTIDE, описанные при обсуждении первого изобретения, причем указанный комплекс получают таким образом, чтобы VB12 отщеплялся от антагониста IHRH in vivo.
В следующем аспекте настоящее изобретение относится к комплексу, содержащему аналог ANTIDE первого аспекта, связанный с носителем VB12 или его аналогом, где с указанным носиетлем связывается более, чем один аналог ANTIDE.
В другом аспекте настоящее изобретение относится к способу пероральной "доставки" антагониста IHRH, в соответствии с которой антагонист LHRH подвергается трансэпителиальному транспорту из кишечника в кровоток хозяина-позвоночного.
В еще одном аспекте настоящего изобретения рассматривается новый класс аналогов ANTIDE, которые обладают повышенной растворимостью в физиологических растворах. Эти аналоги могут быть введены системно.
Для лучшего понимания настоящего изобретения ниже приводится описание его предпочтительных вариантов со ссылками на нижеследующие примеры и чертежи, где:
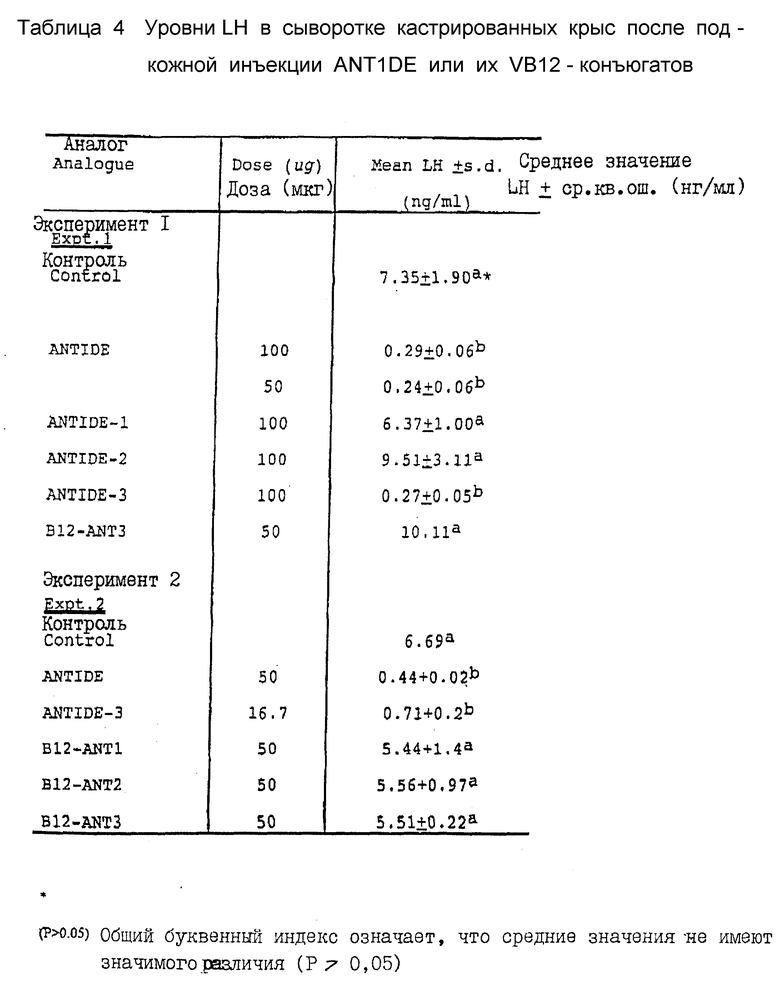
На фиг. 1 представлен график, иллюстрирующий средние ингибирующие концентрации (IC50) ANTIDE, их аналогов и VB12-конъюгатов, определенные с помощью in vitro - биоанализа на блокирование IHPH-стимулированного высвобождения IH из клеток гипофиза крыс. Средние ингибирующие концентрации ANTIDE, аналога ANTIDE и конъюгатов VB12-ANTIDE для подавления LHRH-стимулированного высвобождения LH из культур клеток передней доли гипофиза крыс определяли после 4-часового культивирования. Результаты для конъюгатов даны в нг введенного аналога/мл (конечная концентрация в лунке с культурой). Результаты, которые не отличались друг от друга (т.е. P>0,05), сгруппированы под общей строчной буквой.
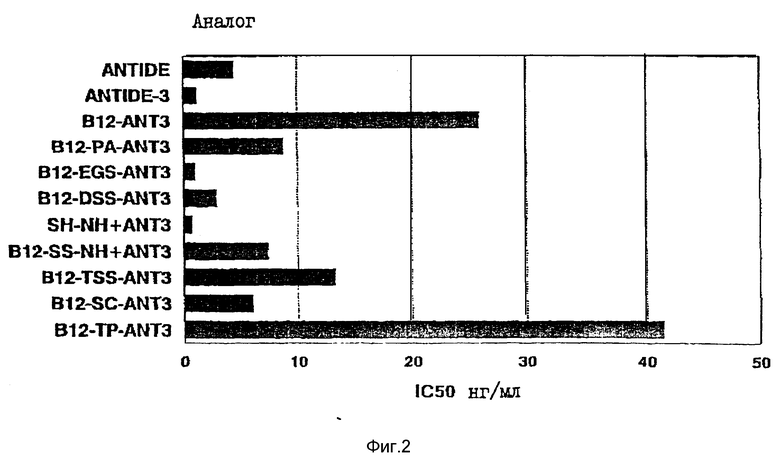
На фиг. 2 представлен график, иллюстрирующий in vitro - биоанализ на блокирование LHRH-стимулированного высвобождения из клеток гипофиза крыс ANTIDE, ANTIDE-3- и их VB12-конъюгатами. Анализ проводили в условиях, аналогичных условиям, описанным на фиг. 1.
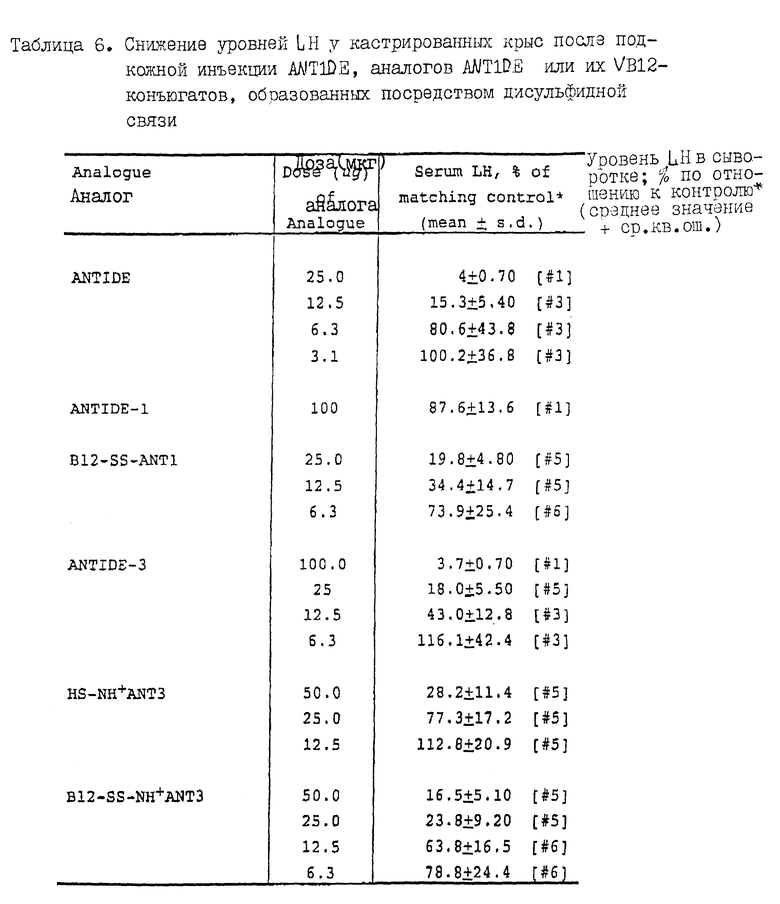
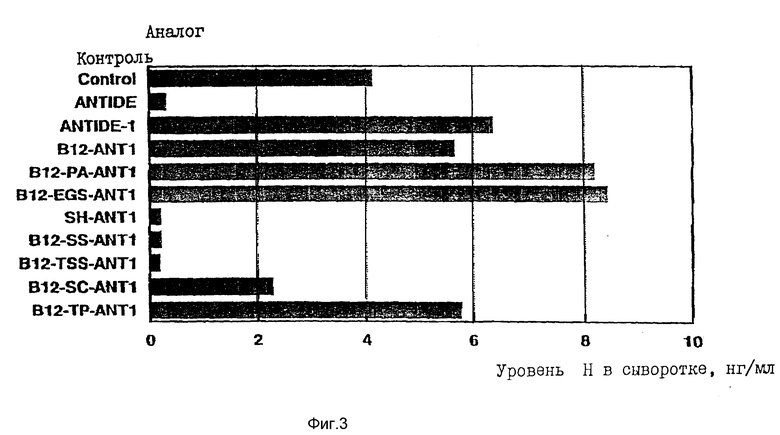
На фиг. 3 представлен график, иллюстрирующий снижение уровня LH в сыворотке кастрированных крыс после инъекции ANTIDE, ANTIDE-1- или конъюгатов ANTIDE-1-VB12. Действие подкожных инъекций ANTIDE или конъюгатов VB12-ANTIDE оценивают с использованием 6-недельных кастрированных крыс. Через 24 часа после инъекции 100 мкг-дозы различных аналогов ANTIDE крыс умерщвляют, а кровь собирают для проведения РИА-анализа (см. Puente & Catt, 1986) на уровни LH в сыворотке.
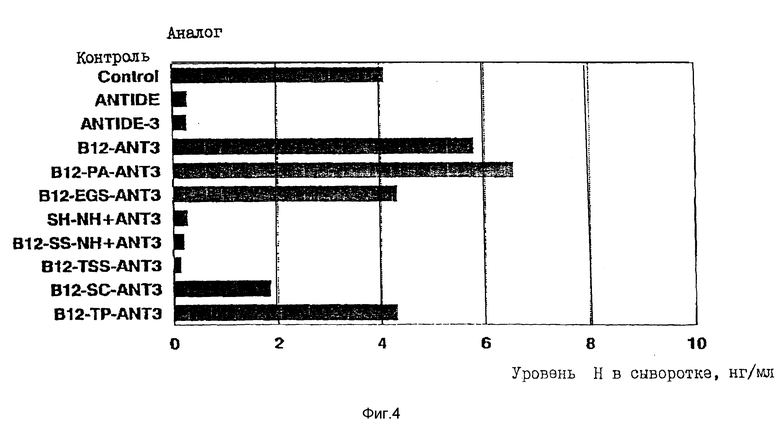
На фиг. 4 представлен график, иллюстрирующий снижение уровней IH в сыворотке кастрированных крыс после подкожной инъекции 100 мкг-доз ANTIDE, ANTIDE-3- или ANTIDE-3-VB12-конъюгатов.
Материалы.
Анализ клеток гипофиза на высвобождение IH.
Культуры клеток получают от передней доли гипофиза 4-месячных крыс Sprague-Dawley. Клетки диспергируют с получением суспензии одиночных клеток, содержащей 250 000 жизнеспособных клеток на 1 мл среды Игла, модифицированной по способу Дульбекко, в которую были добавлены 10% фетальная телячья сыворотка и антибиотики. Затем 0,5 мл-аликвоты вносят в многолуночные планшеты для культивирования тканей и выдерживают при 30oC в закрытом виде во влажной атмосфере 5% CO2 в воздухе. Клетки культивируют в течение 5 дней, после чего среду удаляют. Эту среду заменяют свежей средой, содержащей различные дозы ANTIDE, аналогов ANTIDE или конъюгатов VB12-ANTIDE. Через 4 часа к каждой лунке добавляют 10 нМ LHRH. Затем клетки инкубируют еще 4 часа, после чего среду удаляют для определения высвобожденного в среду количества FSH и LH с помощью радиоиммуноанализа (РИА).
Модель кастрированных крыс.
Взрослых особей крыс Sprague-Dawley кастрируют и за неделю до введения испытуемых соединений выдерживают в специальном помещении для животных. Испытуемые ANTIDE или его аналоги вводят кастрированным крысам путем подкожной инъекции. Группы из 5 крыс используют в качестве контроля, и для сбора проб крови, которые берут из наружной яремной вены через 24 часа после ввода дозы, определение уровня LH осуществляют с помощью стандартного радиоиммуноанализа.
Анализ на IF.
Аффинность различных конъюгатов VB12 ANTIDE по отношению к внутреннему фактору (IF) определяют с помощью анализа на конкурентное связывание (Russel-Jones 1994). Для этого разведения немеченного B12 или аналога VB12 или конъюгата смешивают с 1 нг 57Co VB12 (Amersham). Затем к смеси добавляют IIU IF (единица IF представляет собой количество IF, необходимое для связывания 1 нг VB12), и полученную смесь инкубируют при комнатной температуре (РТ) в течение 20 минут, после чего добавляют раствор 5% активированного угля в 0,1% BSA (несодержащий IF и VB12, Sigma). Пробы центрифугируют и проводят подсчеты для супернатанта (связанное IF) и для осадка (свободного 57Co VB12), которые используют для определения относительной аффинности IF по отношению к испытуемому материалу.
Синтез аналогов ANTIDE.
Три аналога ANTIDE, содержащие отдельные, немодифицированные лизины, а именно D-Lys6 ANTIDE или ANTIDE-1; Lys5 ANTIDE или ANTIDE-2; и LYS8 ANTIDE или ANTIDE-3 (см. таблицу 1) синтезируют на пептидном синтезаторе Applied Biosystems. Пептиды отщепляют от смолы и очищают с помощью обращенно-фазовой ВЭЖХ, используя градиент 5 - 100% ацетонитрила в 0,1% TFA.
Два аналога - ANTIDE-1 и ANTIDE-3 обладают значительной in vitro - активностью, а аналог ANTIDE-3 имел активность, сравнимую с in vivo - активностью нативного ANTIDE. Было установлено, что прямое ковалентное связывание "e"-изомерного карбоксилата VB12 (eV VB12) с ANTIDE-1, ANTIDE-2 или ANTIDE-3 приводит к снижению биоактивности аналога ANTIDE до уровней в основном ниже, чем уровень самого ANTIDE, как при in vitro, так и при in vivo-испытаниях.
Прямое связывание аналога ANTIDE с монокарбокси - VB12.
В целях получения функциональной группы, подходящей для конъюгирования VB12 с ANTIDE, цианокобаламин (VB12) сначала гидролизуют в 0,4 M HCl в течение 72 часов. "e"-Изомер монокарбокси- VB12 (e VB12, e-изомер), который, как было предварительно установлено, из всех трех изомеров обладает наиболее высокой аффинностью по отношению к IF, отделяют от b- и d-изомеров (образуемых также в процессе кислотного гидролиза цианокобаламина) с помощью комбинации Dowex IX2-хроматографии и полупрепаративной обращенно-фазовой (С-18) ВЭЖХ с использованием градиента 5 - 100% ацетонитрила в 0,1% TFA.
Аминоэтил-e VB12 получают посредством реакции e VB12-изомера с 1,2-диаминоэтаном при pH 6,5 с использованием 20-кратного молярного избытка диамина по сравнению с e-изомером и 20-кратного молярного избытка 1-этил-3-(4-диметил-аминопропил)-карбодиимида (EDAC, Biorad, Richmond, CA). Аминоэтиловое производное очищают с помощью обращенно-фазовых ВЭЖХ на полупрепаративной C-4-колонке с использованием градиента 5 - 100% ацетонитрила в 0,1% TFA. Элюированный материал затем очищают с помощью хроматографии на S-Сефарозе. Аминопроизводное элюируют 0,1 M HCl и экстрагируют в фенол, а затем после добавления в феноловую фазу дихлорометана, подвергают обратной экстракции в воду. После этого аминоэтил-e VB12 выделяют из водной фазы путем лиофилизации.
Три аналога ANTIDE подвергают конъюгации с e VB12, используя карбодиимид. EDAC и полученные продукты (B12-ANT1, B12-ANT2 и B12-ANT3 в зависимости от того, был ли данный комплекс образован между ANTIDE-1, ANTIDE-2 или ANTIDE-3 соответственно) очищают с помощью хроматографии на Сефадексе G-25 в 10% уксусной кислоте, а затем с помощью обращенно-фазовой ВЭЖХ на полупрепаративной C-4 колонке, как описано выше.
Конъюгирование VB12 с ANTIDE с использованием перекрестносшивающего агента, содержащего пространственно затрудненный тиол.
Аминоэтил-e VB12 подвергают реакции с 4-сукцинимидилоксикарбонил- α -метил- α -(2 пиридилдитио)толуолом (SMPT; Pierce). Иминотиолированные ANTIDE-1 и ANTIDE-3 получают, как описано выше. Затем SNPT-производное аминоэтил-e VB12 подвергают реакции с тиолированными производными ANTIDE-1- и -3, как описано выше, с последующей очисткой B12-TSS -ANTI и B12-TSS - ANT3 стандартным методом (таблица 2).
Образование нерасщепляемой тиоэфирной связи между VB12 и ANTIDE.
6-(Иодоацетамидо)гексиламидо-e VB12 в диизопропилэтиламиде/диметилформамиде (ДМФ/DTEA, 1:20 об./об.) подвергают дезоксигенированию с использованием аргона в течение 10 минут и к размешанному раствору по капле добавляют раствор иминотиолированного ANTIDE-1 или -3 в ДМФ. Каждую реакционную смесь перемешивают в течение 30 минут при комнатной температуре, после чего разводят водой, фильтруют, а продукты B12- SC- ANTI и B12-SC-ANT-3 очищают с помощью препаративной обращенно-фазовой ВЭЖХ (таблица 2).
Конъюгирование VB12 с ANTIDE с использованием связи, расщепляемой трансглутаминазой.
ANTIDE-1 и -3 растворяли в 5% DIEA/ДМФ, а затем добавляли N- сукцинимидилэфирное производное e VB12-GGEA-OMe (e VB12-GlyGlyGly Ala)в ДМФ (100 мкл). Каждую реакционную смесь размешивали в течение 30 минут при комнатной температуре, затем разводили 2% уксусной кислотой и фильтровали. Продукты B12-TP-ANTI и B12-TP-ANT3 очищали с помощью обращенно-фазовой ВЭЖХ, лиофилизовали и перед использованием хранили под осушителем (таблица 2).
Статистический анализ.
Данные, полученные от дублированных индивидуумов (in vivo - испытания) и из дублированных экспериментов (in vitro испытания), подвергают одностороннему анализу дисперсии, и для попарного сравнения средних значений, которые, как предполагается, значительно отличаются по степени вероятности p<0,05, применяют post hoc - критерии HSD Тьюки.
Аффинность конъюгатов VB12-ANTIDE по отношению к внутреннему фактору.
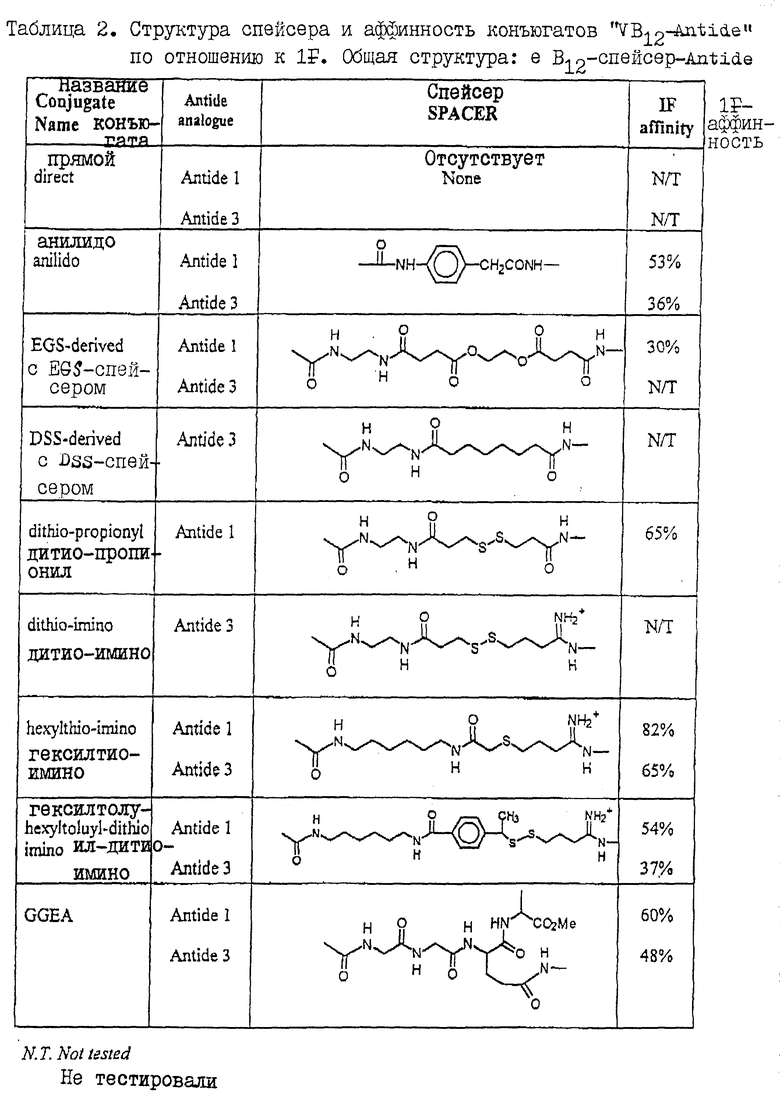
Все испытанные конъюгаты VB12-ANTIDE показывают хорошую аффинность по отношению к внутреннему фактору (таблица 2). Действительно, большинство конъюгатов имеет относительную аффинность, которая превышает аффинность изомера e B12, использованного для получения конъюгата (обычно около 35%).
Пример 1. Образование конъюгатов "VB12-аналог ANTIDE" с использованием нерасщепляемых гомо-бифункциональных перекрестносшивающих агентов.
Путем прямого связывания VB12 с ANTIDE-1 и ANTIDE-3 получают конъюгаты с гораздо меньшей биоактивностью по сравнению с ANTIDE. Тесная пространственная близость VB12 c ANTIDE, обеспечиваемая описанной методикой конъюгирования, стерически препятствует связыванию ANTIDE с LHRH-рецептором. Для снижения стерического эффекта, связанного очевидно с прямым конъюгированием, используют нерасщепляемые линкеры для получения ковалентных комплексов между аминоэтил-e VB12 и ANTIDE-1 и ANTIDE-3. Для этого ANTIDE-1 и -3 подвергают реакции с 1,5-молярным избытком дисукцинимидилсуберата (DSS) или этиленгликольбис (сукцинимидилсукцинат) (EGS) в течение 10 минут при комнатной температуре (RT). Затем добавляют аминоэтил -"e" VB12, и смесь оставляют на ночь для прохождения реакции. Конъюгированный материал очищают с помощью хроматографии на Сефадексе G-25 в 10% уксусной кислоте, а затем с помощью обращенно-фазовой ВЭЖХ (ОФ-ВЭЖХ). Конъюгаты VB12-анилидо- ANTIDE-1 и ANTIDE-3 получают посредством реакции аминоэтил- "e" VB12 с n-аминофенилукусуной кислоты с использованием EDAC-NHS. Затем VB12-анилид в свою очередь подвергают конъюгированию c ANTIDE-1 и ANTIDE-3 с использованием EDAC/NHS. Конъюгированный материал очищают с помощью хроматографии на Сефадексе G-25 в 10% уксусной кислоте, а затем с помощью ОФ-ВЭЖХ.
Пример 2. Образование конъюгатов "VB12-аналог ANTIDE" с использованием спейсеров.
Получение конъюгатов между VB12 и ANTIDE-1 и ANTIDE-3 с использованием либо гидрофильных спейсеров, таких как EGS (16,1  ), либо гидрофобных спейсеров, таких как DSS (11,4
), либо гидрофобных спейсеров, таких как DSS (11,4  ), либо более короткой объемной анилидогруппы, позволяет значительно повысить in vitro - биологическую активность конъюгатов до уровней, сравнимых с уровнем активности исходного аналога (фиг. 1 и 2). Так, например, все B12-PA-ANTI, B12-EGS-ANT3 и B12-DSS-ANT3 имеют биоактивности, сравнимые с активностью нативного ANTIDE (фиг. 1 и 2). В противоположность этому, биологическая in vivo-активность указанных конъюгатов, введенных подкожно крысам, оказалась очень низкой, о чем свидетельствовала неспособность этих конъюгатов вызывать заметное снижение уровней LH в сыворотке кастрированных крыс даже при введении аналога в дозах 100 мкг. С другой стороны ANTIDE и ANTIDE-3 обладают активностью уже при дозах 12,5 мкг (фиг. 3 и 4). Поэтому очевидно, что помимо пространственного затруднения, на in vivo - активность указанных конъюгатов VB12- ANTIDE оказывают влияние и другие факторы (ср. фиг. 1 с фиг. 3 и фиг. 2 с фиг. 4).
), либо более короткой объемной анилидогруппы, позволяет значительно повысить in vitro - биологическую активность конъюгатов до уровней, сравнимых с уровнем активности исходного аналога (фиг. 1 и 2). Так, например, все B12-PA-ANTI, B12-EGS-ANT3 и B12-DSS-ANT3 имеют биоактивности, сравнимые с активностью нативного ANTIDE (фиг. 1 и 2). В противоположность этому, биологическая in vivo-активность указанных конъюгатов, введенных подкожно крысам, оказалась очень низкой, о чем свидетельствовала неспособность этих конъюгатов вызывать заметное снижение уровней LH в сыворотке кастрированных крыс даже при введении аналога в дозах 100 мкг. С другой стороны ANTIDE и ANTIDE-3 обладают активностью уже при дозах 12,5 мкг (фиг. 3 и 4). Поэтому очевидно, что помимо пространственного затруднения, на in vivo - активность указанных конъюгатов VB12- ANTIDE оказывают влияние и другие факторы (ср. фиг. 1 с фиг. 3 и фиг. 2 с фиг. 4).
Пример 3. Образование конъюгатов "VB12 ANTIDE" с использованием расщепляемых тиолом перекрестносшивающих агентов.
Посредством связывания VB12 с аналогами ANTIDE с использованием протяженных гидрофильных или гидрофобных спейсеров получают конъюгаты, которые обладают in vitro-активностью, аналогичной активности нативного ANTIDE. Однако парентеральное введение этих конъюгатов дает незначительный эффект в снижении уровней LH в сыворотке кастрированных крыс в пределах испытанных доз. Эти результаты дают основание предложить, что, хотя общая стратегия повышения биоактивности конъюгатов "VB12-ANTIDE" путем введения протяженных спейсеров является правильной, однако образованный таким образом конъюгат имеет, тем не менее пониженную in vivo-активность, что вероятно обусловлено присутствием в конъюгате молекулы VB12. Поэтому были получены конъюгаты, в которых ковалентный линкер (сшивающий агент) содержал биологически расщепляемую дисульфидную связь, которая должна быть восстановлена in vivo, вероятно с помощью глутатиона, присутствующего в сыворотке. Для этого аминоэтил-e VB12 подвергают реакции с N-сукцинимидил 3-(2-пиридилдитио)пропионатом (SPDP). Полученный продукт дитиопиридил-аминоэтил "e" VB12 (DTP "e" VB12) очищают с помощью обращенно-фазовых ВЭЖХ (ОФ-ВЭЖХ). Свободный тиол вводят в ANTIDE-1 посредством реакции с SPDP. Дитиопиридильную группу затем восстанавливают с использованием меркапто-этанола, после чего продукт очищают с помощью ОФ-ВЭЖХ. Аналогичным образом свободный тиол вводят в ANTIDE-3 посредством реакции с иминотиоланом. Тиолированный продукт (SH-NH+ ANTIDE-3) очищают с помощью ОФ-ВЭЖХ. Образование связанных посредством дисульфидной связи конъюгатов "VB12-ANTIDE-1" и "VB12-ANTIDE-2" осуществляют с помощью реакции тиолированного производного ANTIDE с DTP "e" VB12 в 2,5% уксусной кислоте в течение 24 часов. Конъюгированный материал очищают с помощью хроматографии на Сефадексе G-25, а затем с помощью ОФ-ВЭЖХ.
Пример 4. Образование конъюгатов "VB12-аналог ANTIDE" с использованием биолологически разлагаемых линкеров.
Для снижения вероятности удаления конъюгатов "VB12-ANTIDE" из кровотока после их инъекции были получены конъюгаты "VB12-ANTIDE" с использованием расщепляемого тиолом дисульфидсодержащего спейсера. Этот спейсер потенциально должен обеспечивать отщепление ANTIDE от VB12 при низких уровнях восстановленного глутатиона в сыворотке. Для этого получают конъюгаты между ANTIDE-1 и ANTIDE-3 с использованием расщепляемого тиолом спейсера (SS) и пространственно затрудненной тиоловой группы (TSS). Присутствие пространственно затрудненного тиола в указанном спейсере должно гарантировать более медленное его расщепление под действием сывороточного глутатиона. Был получен также конъюгат, содержащий нерасщепляемый тиоэфирный линкер с аналогичной длиной (SC).
Было установлено, что расщепляемые тиолом конъюгаты B12-SS-ANTI и B12-SS-NH+ANT3 имеют in vitro - биоактивность, аналогичную биоактивности конъюгатов с протяженным спейсером, описанных выше (фиг. 1 и 2; таблицы 5 и 6). Парентеральное введение B12-SS-NH+ ANT3 кастрированным крысам приводило к снижению уровня LH в сыворотке аналогично тому, которое наблюдалось в случае немодифицированного ANTIDE-3, хотя B12-SS-ANTI имеет такую же биоактивность in vivo, что и нативный ANTIDE (таблица 6). Таким образом, использование расщепляемого тиолом спейсера (вместо нерасщепляемого спейсера) для получения VB12-ANTIDE-конъгатов, хотя и оказывает небольшое влияние на IC50 конъюгатов "VB12-спейсер-ANTIDE" в in vitro - анализе, но при этом оно значительно повышает активность конъюгатов in vivo до уровней, аналогичных уровню ANTIDE (фиг. 3 и 4, таблицы 5 и 6). Введение толильной группы или пространственно затрудненного тиола не оказывает значительного влияния ни на in vitro, ни на in vivo - активность любого аналога по сравнению с соответствующим аналогом, содержащим пространственно незатрудненный тиол. Было установлено, что нерасщепляемые конъюгаты, образованные посредством тиоэфирной связи, имели при испытаниях in vitro активность, аналогичную активности тиолового аналога однако при испытании in vivo, этот аналог обнаруживал гораздо более низкую активность.
Прим. к таблице 3:
Среднее ингибирующие концентрации (IC50) ABTIDE, аналогов ANTIDE и BV12-ANTIDE-конъюгатов для ингибирования LHRH-стимулированного высвобождения LH из культур клеток передней доли гипофиза крысы определяют после 4-часового культивирования. Результаты для конъюгатов (среднее значение +/- ср.кв. ош.) даны в "нг" введенного аналога ANTIDE/мл (конечная концентрация в лунке с культурой) по n независимым испытаниям. Результаты, которые не имели значимого отличия друг от друга (т.е. P>0,05), сгруппированы под общей строчной буквой.
Пример 5. Образование конъюгатов "VB12-аналог ANTIDE" с использованием линкера, расщепляемого трансглутаминазой.
Было установлено, что конъгаты между VB12 и ANTIDE-1 и ANTIDE-3, образованные с использованием расщепляемой трансглутаминазой γ-глутамил-ε-лизиновой связи, (B12-TP-ANTI и B12-TP-ANT3) имеют плохую активность in vitro, и ничтожно малую активность in vivo при испытуемых дозах, что дает основание предположить, что эта связь не расщепляется ни in vitro, ни in vivo. Возможно, стереоспецифичность ингибирует расщепление ANTIDE-1-конъюгатов (благодаря присутствию D-лизиновой группы), тогда как пространственное затруднение, возможно, препятствует расщеплению ANTIDE-1- и ANTIDE-3-конъюгатов трансглутаминазой.
Пример 6. Образование конъюгатов "VB12-ANTIDE-аналог-лизилполиглутамат" с использованием перекрестносшивающих агентов, расщепляемых тиолом.
Желательно увеличить количество лекарственного средства или его аналогов, которое может быть доставлено VB12-транспортной системой, посредством присоединения множества копий лекарственного средства к полимерному остову, с которым связана одна или несколько молекул VB12. Ниже описан способ получения таких VB12-лекарственное средство-полимерных комплексов.
a) Образование DTP-лизил-полиглутамата.
Полиглутамат (100 мг) (MW 64600 - 70000; Sigma) подвергают реакции с EDAC (100 мг) и NHS (50 мг в ацетоне) в течение 10 минут при комнатной температуре. Затем добавляют лизин (400 мг в 4 мл 1 % NaHCO3) и реакционную смесь оставляют на ночь для протекания реакции (O/N). Продукт лизил-полиглутамат (LPG) очищают путем экстензивного диализа против DW, а затем лиофилизуют.
Дитиопиридиловое производное LPT получают с помощью реакции дитиопиридил-пропионовой кислоты (50 мг) с тетрафтороборатом O-(N- сукцинимидил)-N,N, N',N'-тетраметилилурония (TSTU) (100 мг) и N- этилдиизопропиламином (100 мг; DIEA) в ДМФ в течение 1 часа. Полученный таким образом сукцинимидиловый сложный эфир вносят непосредственно в 100 мг LPG, растворенного в 4 мл 2% NaHCO3. Реакционную смесь оставляют на ночь для протекания реакции, после чего продукт (DTP-IPG) очищают путем исчерпывающего диализа против DW, а затем лиофилизуют.
b) Получение иминотиолированного ANTIDE-1.
ANTIDE-1 (20 мг) растворяют в 300 мкл 5% DIEA в ДМФ, содержащем 5 мг EDTA и 5 мг DTT, который был дегазирован в присутствии аргона. Затем добавляют иминотиолан (20 мг в 50 мкл DIEA/ДФМ и 50 мкл боратного буфера, 100 мМ, pH 8,2) и проводят реакцию в течение 60 минут, после чего продукт очищают с помощью обращенно-фазовой ВЭЖХ и лиофилизуют.
c) Получение иминотиолированного аминоэтил- VB12.
Аминоэтил "e" VB12 (20 мг) растворяют в 300 мкл 5% DIEA в ДМФ, содержащем 5 мг EDTA и 5 мг DTT, который был дегазирован в присутствии аргона. Затем добавляют иминотиолан (20 мг в 50 мкл DIEA/ДМФ и 50 мкл боратного буфера, 100 мМ, pH 8,2) и проводят реакцию в течение 60 минут, после чего продукт очищают с помощью обращенно-фазовой ВЭЖХ и лиофилизуют.
d) Образование VB12-ANTIDE-лизил-полиглутамата.
DTP-IPG растворяют в DW при 20 мг/мл. Иминотиолированный аминоэтил-VB12 растворяют в DW при 5мг/мл и добавляют к DTP-IPG (1:20 мас./мас.), оставляют на 20 минут при комнатной температуре для прохождения реакции, после чего, размешивая, по капле добавляют иминотиолированный ANTIDE-1 (50 мг/мл в DW). Реакционную смесь выдерживают при pH 6,5 - 7,0 путем добавления Трис-HCl, pH 7,0 и ацетата натрия, pH 5,5. Реакция протекает в течение ночи, после чего продукт очищают путем диализа, а затем лиофилизуют. Состав продукта определяют с помощью аминокислотного анализа, который показывает, что полученный продукт имеет следующий состав: 1:5:21=ANTIDE-1:лизин:глутамат или по грубой оценке этот продукт содержит 25 мас.% ANTIDE.
Ранее получение антагониста LHRH, подходящего для использования в целях лечения гормонозависимого рака молочной железы и простаты у человека, было ограничено низкой активностью этих молекул, их плохой растворимостью в физиологических буферах и плохой биологической усвоямосью при пероральном введении. В попытке разрешить эти проблемы автором настоящего изобретения была рассмотрена возможность совмещения относительно высокой биологической активности сильного антагониста LHRH, а именно ANTIDE, с высокой биологической усвояемостью при пероральном введении витамина B12. ANTIDE является особенно подходящим для пероральной "доставки", поскольку он состоит из многих не встречающихся в природе аминокислот, кроме того, он является резистентным к действию трипсина или химотрипсина. Однако указанный ANTIDE должен быть модифицирован таким образом, чтобы он содержал сайт, необходимый для конъюгации с VB12, но при этом он должен полностью сохранять свою биологическую активность. Обычно ANTIDE имеет модифицированные лизиновые остатки в положениях 5, 6 и 8, а именно: Lys (Nic), D-Lys (Nic) и Lys (iPr) соответственно. Удаление этих модифицикаций в любом из трех положений дает свободный амин, подходящий для конъюгации с VB12. Были синтезированы два производных ANTIDE, а именно: D-Lys6-ANTIDE (ANTIDE-1) и Lys8-ANTIDE (ANTIDE-3), подходящих для конъюгации с VB12, и при этом было установлено, что они обладают высокой степенью активности в in vitro - анализе клеток гипофиза. Было также обнаружено, что ANTIDE-3 обладает высокой активностью при подкожном введении крысам, хотя ANTIDE-1, как было обнаружено, обладает ничтожно малой антагонистической активностью, что вероятно обусловлено изменением скорости его выведения из сыворотки. Однако in vitro- и in vivo -активность любого их этих аналогов значительно снижается при прямом конъюгировании с VB12 (таблицы 3 и 4), что вероятно обусловлено стерическим препятствием связыванию конъюгата с гипофизарным рецептором для LHRH посредством объемной молекулы VB12, находящейся в непосредственной близости с ANTIDE.
Для уменьшения стерического эффекта, наблюдающегося при прямом конъюгировании VB12 с ANTIDE-1 и ANTIDE-3, продуцируют ковалентные комплексы между аминоэтил-e VB12 и ANTIDE-1 и -3 с использованием нерасщепляемых линкеров, которые отделяют объемную группу VB12 от аналога ANTIDE. Все продуцированные конъюгаты, а именно конъюгаты VB12-ANTIDE-1, образованные с помощью либо анилидо (PA)-, либо EGS - спейсера, или конъюгаты VB12-ANTIDE-3, образованные с помощью анилидо-, EGS- или DSS - спейсера, обнаруживали in vitro - активность, аналогичную активности нативного ANTIDE (фиг. 1), что дает основание предположить, что размещение B12-группы на некотором расстоянии от ANTIDE позволяет сохранить аффинность ANTIDE по отношению к его рецептору. Эти конъюгаты имели очень низкую активность in vivo (фиг. 2), что вероятно обусловлено удалением указанных конъюгатов из кровотока VB12-связывающими белками, такими как транскобаламин I или транскобаламин II, которые в значительной степени могут снижать их in vivo-активность.
В попытке уменьшить возможность стерического затруднения минимизировать возможность снижения системной биологической доступности конъюгатов, обусловленного выведением их из кровотока VB12-связывающими белками, были получены VB12- ANTIDE - конъюгаты с использованием линкеров, которые повержены биолоческому расщеплению in vivo. Для этого была вабрана дисульфтдная связь, поскольку эта связь наиболее легко расщепляется in vivo и, кроме того, она присутствует в биологических конъюгатах, таких как A-SS-B субъединичные токсины, столбнячный токсин и дифтерийный токсин (Sabdvig & Olsens, 1981; Matsuda & Yoneda, 1974). Было показано, что дисульфидные связи расщепляются при низких уровнях глутатиона, присутствующего в сыворотке, либо в цитоплазме клеток (Anderson & Meister, 1980).
Было установлено, что образованные с помощью дисульфидной связи конъюгаты VB12-ANTIDE-1 и VB12-ANTIDE-3 имеют немного меньшую in vitro - активность по сравнению с ANTIDE или по сравнению с конъюгатами VB12-ANTIDE-1 и ANTIDE-3, полученными с использованием анилидо- и EGS-спейсеров. В противоположность этому, конъюгаты с дисульфидной связью обнаруживают резкое увеличение своей in vivo - активности по сравнению с конъюгатами, полученными с использованием нерасщепляемого протяженного спейсера (фиг. 3 и 4, таблицы 5 и 6). Что касается B12-SS-NH+-ANT3-конъюгата, то его in vivo - биоактивность слегка превышает активность NS-NH+-ANT3-аналога. Использование спейсера аналогичной длины, содержащего нерасщепляемую тиоэфирную связь вместо дисульфидной связи, приводит к значительному снижению in vivo - активности, что дает основание предположить, что увеличение активности конъюгата с дисульфидной связью обусловлено его способностью расщепляться in vivo. Было установлено, что все конъюгаты, описанные выше, независимо от того, были ли они получены путем прямого связывания, либо путем связывания посредством расщепляемого или нерасщепляемого спейсера, имеют гораздо более высокую растворимость в физиологических буферах, таких как PBS или физиологический раствор, чем нативная молекула ANTIDE (результаты не показаны).
Были получены также конъюгаты между VB12 и ANTIDE-1 и -3, образованные спейсером, содержащим γ -глутамил- ε -лизиновую связь, которая может расщепляться сывороточными трансглутаминазами. Несмотря на высокую in vitro - активность этих конъюгатов, их in vivo - активность оказалась очень низкой, что объясняется очевидно тем, что указанный спейсер не расщепляется in vivo.
Было установлено, что аффинность конъюгатов VB12-ANTIDE-1 и -3 в отношении IF равна или превышает аффинность e VB12-изомера, из которого эти конъюгаты были получены. Аналогичные наблюдения были сделаны и в отношении других конъюгатов e VB12-изомера (Russell - Jones, 1994).
Эти исследования показали, что две молекулы, имеющие в принципе различные биологические активности, могут быть связаны друг с другом и при этом может быть в основном сохранена биологическая активность каждого отдельного компонента. Однако в процессе конъюгирования необходимо следить за тем, чтобы сохранялась активность связывания "рецептор-лиганд" для обеих молекул, а также сохранялась биологическая доступность фармакологического агента. Так, например, оказалось вполне возможным получить несколько комплексов между VB12 и ANTIDE с использованием спейсеров, которые обнаруживали не только высокую in vitro - активность в анализе клеток гипофиза, но и высокую относительную аффинность по отношению к IF, однако биологическая активность in vivo этих конъюгатов была значительно более низкой. В этих случаях положительные результаты дает альтернативный способ конъюгирования двух молекул, предусматривающий использование биологически расщепляемых спейсеров.
Необходимо отметить, что любой специалист может внести в данное изобретение, проиллюстрированное выше конкретными примерами, различные изменения и/или модификации, не выходящие однако за рамки объема и существа этого изобретения. Поэтому представленные примеры осуществления изобретения должны рассматриваться как иллюстративные и не ограничивающие его объема.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМПЛИФИКАЦИЯ СИСТЕМЫ ПОГЛОЩЕНИЯ ВИТАМИНА B ПРИ ПОМОЩИ ПОЛИМЕРОВ | 1994 |
|
RU2139732C1 |
| RGD-СОДЕРЖАЩИЕ ПЕПТИДОМИМЕТИКИ И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2519736C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), ПРЕПАРАТ И СПОСОБ ЛЕЧЕНИЯ СОСТОЯНИЯ, ПОДДАЮЩЕГОСЯ ВОЗДЕЙСТВИЮ АНАЛОГА LHRH, ШПРИЦ | 1997 |
|
RU2202371C2 |
| АНТАГОНИСТЫ GNRH, МОДИФИЦИРОВАННЫЕ В ПОЛОЖЕНИЯХ 5 И 6 | 1998 |
|
RU2199549C2 |
| МОЛЕКУЛЯРНЫЙ КОНЪЮГАТ НА ОСНОВЕ СИНТЕТИЧЕСКИХ АНАЛОГОВ ЛЮЛИБЕРИНА И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ СРЕДСТВА ДОСТАВКИ ДНК В КЛЕТКИ ГОРМОН-ЧУВСТВИТЕЛЬНЫХ ОПУХОЛЕЙ (ВАРИАНТЫ) | 2007 |
|
RU2377247C2 |
| ЛЕЧЕНИЕ ИЛИ ПРОФИЛАКТИКА ИНФЕКЦИИ | 2011 |
|
RU2617399C2 |
| УСОВЕРШЕНСТВОВАННЫЕ КОНЪЮГАТЫ N4 ХЕЛАТООБРАЗУЮЩИХ АГЕНТОВ | 2005 |
|
RU2360701C2 |
| БИЦИКЛИЧЕСКИЕ ПЕПТИДНЫЕ ЛИГАНДЫ, СПЕЦИФИЧНЫЕ ДЛЯ МТ1-ММР | 2015 |
|
RU2708459C2 |
| БИЦИКЛИЧЕСКИЕ ПЕПТИДНЫЕ ЛИГАНДЫ, СПЕЦИФИЧНЫЕ ДЛЯ MT1-MMP | 2015 |
|
RU2824959C2 |
| МАТЕРИАЛЫ И СПОСОБЫ, СВЯЗАННЫЕ С ЛИНКЕРАМИ ДЛЯ ПРИМЕНЕНИЯ В КОНЪЮГАТАХ ЛЕКАРСТВЕННОГО СРЕДСТВА И БЕЛКА | 2015 |
|
RU2737553C2 |


Изобретение относится к новым антагонистам LHRH и комплексам между этими антагонистами и VВ12. В частности, в настоящем изобретении предусматривается пероральное введение антагонистов LHRH. Антагонисты настоящего изобретения выбирают из группы, состоящей из ANTIDE-1, ANTIDE-2 и ANTIDE-3, которые являются подходящими для конъюгации с VB12, а именно: N-Ac-D-Nal (2), D-Phe(pCl), D-Pal (3), ser, Lys (Nic), D-Lys, Leu, Lys (iPr), Pro, O-Ala-NH2 (D-Lys6ANTIDE) или (ANTIDE-1), N-Ac-D-Nal (2), D-Phe (pCl), D-Pal (3), Ser, Lys, (Nic), D-Lys (Nic), Leu, Lys, Pro, D-Ala-NH2 (Lys8ANTIDE или ANTIDE-3). Во втором аспекте изобретение относится к комплексу между VВ12 или аналогом VB12 и антагонистами LHRH, рассматриваемыми в первом аспекте изобретения, где для поглощения и транспорта VВ12 в хозяине-позвоночном необходимо, чтобы указанный VB12 или его аналог обладал способностью вступать в реакцию связывания, и где активность антагониста LHRН сохраняется в основном на постоянном уровне. Описывается также способ химической кастрации и/или лечения гонадотропинзависимых расстройств индивидуума, заключающийся во введении вышеуказанных соединений. 3 с. и 6 з.п. ф-лы, 4 ил., 6 табл.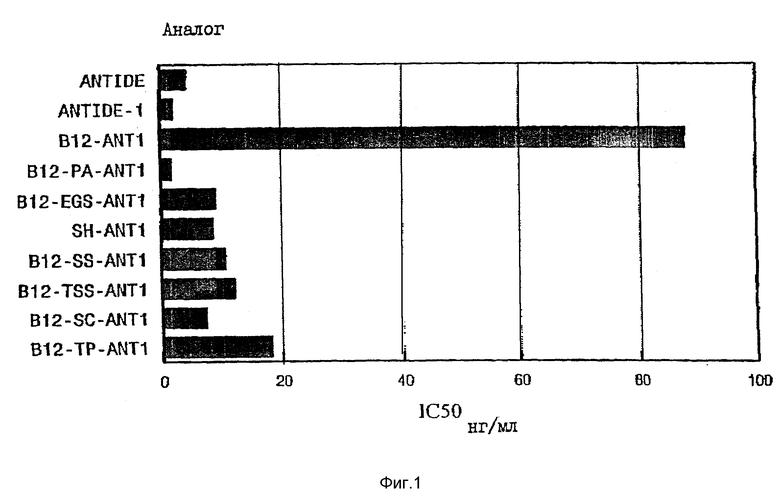
| ПРОИЗВОДНЫЕ ПЕПТИДОВ И КОМПОЗИЦИЯ, ВЫЗЫВАЮЩАЯ ИММУННЫЙ ОТВЕТ НА LHRH | 1990 |
|
RU2078770C1 |
| Торфодобывающая машина с вращающимся измельчающим орудием | 1922 |
|
SU87A1 |
| Устройство для контроля запальных свеч | 1940 |
|
SU65289A1 |
| Наконечник для электромедицинских аппаратов | 1932 |
|
SU32637A1 |
| Пружинный динамометр | 1939 |
|
SU57178A1 |
