Способы и составы для лечения поражения центральной нервной системы (ЦНС), вызванного ВИЧ, в частности СПИД-обусловленной деменции.
Вирус иммунодефицита человека (ВИЧ) является каузальным агентом синдрома приобретенного иммунодефицита (СПИД) - заболевания, характеризующегося разрушением иммунной системы, в частности, CD4+ Т-клеток, с сопутствующей подверженностью условно-патогенным инфекциям - и его предшественника СПИД-обусловленного комплекса (AIDS-related complex, "ARC") - синдрома, характеризующегося такими симптомами, как персистирующая генерализованная лимфаденопатия, лихорадка и потеря веса.
Как и в случае нескольких других ретровирусов, ВИЧ кодирует продуцирование протеазы, которая осуществляет посттрансляционное дробление предшественника полипептидов в процессе, необходимом для образования инфекционных вирионов (S. Crawford et al., "A Deletion Mutation in the 5' Part of the pol Gene of Moloney Murine Leikemia Virus Blocks Proteolytic Processing of the gag and pol Polyproteins", J.Virol., 53, p.899 (1985). Эти генные продукты включают в себя pol, который кодирует РНК-зависимую ДНК полимеразу вириона (обратную транскриптазу), эндонуклеазу, протеазу ВИЧ, и gag, который кодирует коровые белки вириона (Н. Toh et al., "Close Structural Resemblance Between Putative Polymerase of a Drosophila Transposable Genetic Element 17.6 and pol gene product of Moloney Murine Leukemia Virus", EMBO J., 4, p. 1267 (1985); L.H.Pearl et al., "A Structural Model for the Retroviral Proteases", Nature, pp. 329-351 (1987); M. D. Power et al., "Nucleotide Sequence of SRV-1, a Type D Simian Acquired Immune Deficiency Syndrome Retrovirus", Science, 231, p.1567 (1986).
Был разработан ряд синтетических антивирусных агентов, нацеленных на различные стадии репликационного цикла ВИЧ. Эти агенты включают в себя соединения, которые блокируют связь вируса с CD4+ Т-лимфоцитами (например, растворимый CD4), и соединения, которые вмешиваются в репликацию вируса путем ингибирования вирусной обратной транскриптазы (например, диданозин и зидовудин (AZT) и ингибируют интеграцию вирусной ДНК в клеточную ДНК (M.S. Hirsh and R. T.D'Aqulia, "Therapy for Human Immunodeficiency Virus Infection", N. Eng. J. Med. , 328, p. 1686 (1993)). Однако такие агенты, которые направлены первично на ранние стадии вирусной репликации, не предотвращают продуцирование инфекционных вирионов в хронически инфицированных клетках. Более того, назначение некоторых их этих агентов в эффективных количествах привело к клеточной токсичности и нежелательным побочным эффектам, таким как анемия и супрессия костного мозга.
В самое последнее время фокусом разработки антивирусных препаратов было создание соединений, которые ингибируют образование инфекционных вирионов путем вмешательства в обработку вирусных полипротеиновых предшественников. Обработка этих предшественников белков требует действия вирус-закодированных протеаз, которые существенно важны для репликации (Kohl, N.E., et al., "Active HIV Protease is Required for Viral Infectivity" Proc. Natl. Acad. Sci. USA, 85, h.4686 (1988)). Антивирусный потенциал ингибирования ВИЧ-протеазы продемонстрирован применением пептидных ингибиторов. Такие пептидные соединения, однако, обычно представляют собой большие и сложные молекулы, которые имеют тенденцию к проявлению слабой биодоступности и в большинстве своем несовместимы с пероральным введением. Соответственно по-прежнему существует необходимость в соединениях, которые могут эффективно ингибировать действие вирусных протеаз, для применения в качестве агентов для предупреждения и лечения хронических и острых вирусных инфекций.
СПИД и другие ВИЧ-обусловленные заболевания нередко содержат компоненты поражения ЦНС. Одним из таких компонентов является ВИЧ-обусловленная деменция.
В то время как имеется растущее число видов лечения ВИЧ и связанных с ним заболеваний, например СПИД и ARC, такие виды лечения мало оказывают или не оказывают влияния на эффекты поражения ЦНС при ВИЧ-инфекции.
Причиной того, что эти виды лечения не так эффективны против эффектов поражения ЦНС при ВИЧ-инфекции, является то, что фармацевтические составы, которые характеризуют их, не способны преодолевать гематоэнцефалический барьер в количестве, достаточном для эффекта, и затормозить ВИЧ-инфекцию в ЦНС.
AZT, наиболее известный из видов лечения ВИЧ, например, имеет распределение мозг/кровь лишь около 0,3. А через 60 минут AZT с мозговой ткани не обнаруживается совсем. Другие ВИЧ-нуклеозиды, ddC, DDI и d4T, имеют еще худшие распределительные профили в ЦНС.
Ингибиторы ВИЧ-протеазы также не проникают в ЦНС на полезных уровнях. Препарат Abbot's ABT 538, например, показывает очень ограниченное проникновение в ЦНС. Ингибитор Searle's inhibitor имеет распределение мозг/кровь от 0,2 до 0,3. Препарат Merck's L-535524 имеет то же распределение.
Таким образом, существующие в настоящее время виды терапии, основанные на применении ВИЧ-нуклеозидов и протеаз, оказывают менее чем желаемые эффекты на ВИЧ-обусловленные компоненты ЦНС.
Настоящее изобретение обеспечивает способ и состав для лечения ВИЧ-компонентов ЦНС, в частности, обусловленной СПИДом деменции.
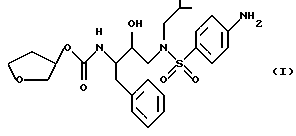
Способ и состав этого изобретения характеризуются ингибитором ВИЧ-протеазы Формулы I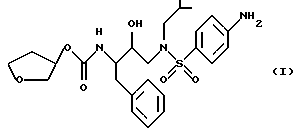
Соединение Формулы I является ингибитором ВИЧ-протеазы. Однако в отличие от других протеазных ингибиторов он имеет распределение мозг/кровь более 1,0. Это означает, что он очень эффективен в преодолении гематоэнцефалитического барьера. Действительно, он присутствует в мозге примерно с тем же уровнем, что и в крови. Кроме того, соединение Формулы I неожиданно обладает долгим полупериодом жизни в мозгу. Оба эти свойства дают в результате то, что соединение Формулы I неожиданно оказывается пригодным для лечения ВИЧ-обусловленных эффектов поражения ЦНС, в частности обусловленной СПИДом деменции.
Соединение с Формулой I может быть получено из доступных исходных материалов при использовании любого из нескольких хорошо известных методов синтеза. Примеры такого синтеза включают способы, описанные в публикации Международной патентной заявки WO 94/05639, которая включена здесь как ссылка.
В целом, сульфонамиды Формулы I легко получают из производных α-аминокислот, имеющих общую формулу P-N(G)-CH(D)-COOH, где Р определено как THF-O-C(О)- или аминокислотная защитная группа, D определено как бензил, a G представляет собой Н или бензил. Подходящие аминокислотные защитные группы описаны во многих публикациях, в том числе T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2d Ed., John Wiley and Sons (1991). Примеры таких аминокислотных защитных групп включают в себя, хотя не ограничиваются ими, карбаматсодержащие группы, такие как Воc, Cbz или Alloc, либо альтернативно, амин может быть защищен как производное алкила, такого как N, N-дибензил или тритил. Такие производные α-аминокислот нередко коммерчески доступны либо могут быть традиционно получены из коммерчески доступных производных α-аминокислот с помощью известных технологических методов. Хотя данное изобретение предполагает использование рацемических смесей таких исходных материалов, предпочтительным является одиночный энантиомер S-образной конфигурации.
С использованием известных технологических методов производное α-аминокислоты с общей формулой P-N(G)-CH(D)-COOH может быть легко конвертировано в производное аминокетона с общей формулой P-N(G) -CH(D)-CO-CH2-X, где Х является уходящей группой, которая активирует α-углерод (т.е. повышает восприимчивость метилена к нуклеофильной атаке).
Приемлемые уходящие группы хорошо известны в практике и включают галогениды и сульфонаты, такие как метансульфонат, трифторметансульфонат или 4-толуолсульфонат. Х также может быть гидроксилом, который конвертируется на месте (in situ) в уходящую группу (например, путем обработки с помощью триалкил- или триарилфосфина в присутствии диалкилазодикарбоксилата). Способы для образования таких аминокетонных производных хорошо известны специалистам в данной области (см. , например, S.J.Fittkau, J. Prakt. Chem., 315, р.1037 (1973)). Альтернативно, определенные аминокетоновые производные являются коммерчески доступными (например, от фирмы Bachem Biosciences, Inc., Philadelphia, Pensylvania).
Производное аминокетона может затем быть восстановлено в соответствующий аминоспирт, представленный формулой P-N(G)-CH(D)-СО-СН2-Х. Альтернативно, производное аминокетона может потом быть восстановлено по схеме синтеза. Многие технологические методы для восстановления производных аминокетонов, таких как P-N(G)-СН(D)-СО-СН2-Х, хорошо известны практическим специалистам (Larock, R. С."Comprehensive Organic Transformations", pp.527-547, VCH Publishers, Inc.© 1989 и ссылки, приведенные там). Предпочтительным восстановительным агентом является борогидрид натрия. Реакция восстановления проводится при температуре приблизительно от -40oС до 40oС (предпочтительнее от около -10oС до приблизительно 20oС) в подходящей системе растворителей, таких как, например, водный или чистый тетрагидрофуран или более низкий спирт, такой как метанол или этанол. Хотя данное изобретение предполагает как стереоспецифическое, так и нестереоспецифическое восстановление аминокетонового производного P-N(G)-СН(D)-СО-СН2-Х, предпочтительным является стереоселективное восстановление. Стереоселективное восстановление может совершаться с использованием хиральных реагентов, известных в практике. В настоящем изобретении стереоселективное восстановление может быть легко достигнуто, например, в нехелатных восстановительных условиях, где хиральная индукция новообразованной гидроксильной группы устанавливается стереохимией D-группы (т. е. добавление гидрида по Felkin-Ahn). В частности, предпочтительны стереоселективные восстановления, где возникающий гидроксил представляет собой syn к D. Установлено, что когда гидроксильная группа является syn к D, конечным продуктом сульфонамида является ингибитор ВИЧ-протеазы более высокой потенции, чем анти-диастереомер.
Гидроксильная группа аминоспирта может необязательно быть защищена любой из известных защитных групп кислорода (такой как триалкилсилил, бензил или алкилоксиметил), давая выход защищенного аминоспирта, имеющего формулу P-N(G)-CH(D)-С(OR7)-СН2-Х, где R7 представляет собой Н или любую подходящую гидроксизащитную группу. Несколько приемлемых защитных групп описано в T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2d Ed., John Wiley and Sons (1991).
Этот защищенный аминоспирт может быть затем подвергнут реакции с нуклеофильным аминосоединением с образованием промежуточного продукта Формулы III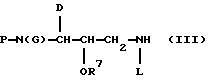
где Р определено как THF-O-C(О)- или аминокислотная защитная группа, D-бензил, R7 - как описано выше, а L - изобутил либо водород.
Альтернативно, должным образом защищенное и активированное производное аминокислоты можно подвергнуть реагированию с нуклеофильным нитросоединением (например, анионом нитрометана или его производным), которое после сочетания можно восстанавливать и получать выход промежуточного продукта Формулы III.
По особенно выгодной схеме синтеза одновременная активация метилена и защита спирта могут осуществляться за счет образования N-защищенного амино-эпоксида из кислорода и смежного с ним метилена, с получением промежуточного продукта Формулы II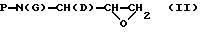
где Р, D и G определены выше. Подходящие системы растворителей для получения N-защищенного амино-эпоксида включают в себя безводные или водные органические растворители, такие как этанол, метанол, изопропанол, тетрагидрофуран, диоксан, диметилформамид и подобные (включая их смеси). Подходящие основания для производства эпоксида включают в себя гидроокиси щелочных металлов, t-бутоксид калия, DBU и подобные. Предпочтительным основанием является гидроокись калия.
Предпочтительнее, чтобы соединение Формулы I создавалось путем получения N-защищенного амино-эпоксида посредством реагирования дианиона производного уксусной кислоты, содержащего потенциальную уходящую группу на α-углероде, с циклическим N-карбоксиангидридом защищенной α-аминокислоты (такой как BOC-Phe-NCA, доступной из Пропептида) или другим соответствующим образом защищенным и активированным производным аминокислоты. Этот способ включает в себя использование галоуксусных кислот или, главным образом, гетероатом-замещенных уксусных кислот, где гетероатом может быть конвертирован в уходящую группу. Предпочтительным дианионом уксусной кислоты является дианион (метилтио) уксусной кислоты. Возникающий в результате кетон может быть затем восстановлен (например, с помощью натрия борогидрата). В том случае, когда дианионом метилтиоуксусной кислоты является нуклеофил, возникающий в результате аминоспирт легко конвертируется в амино-эпоксид путем алкилирования (например, метил-йодидом) с последующим замыканием кольца (с использованием, например, гидрата натрия).
Реакция N-защищенного амино-эпоксида (или другого подходящим образом активированного промежуточного продукта) с амином осуществляется в чистом виде, т. е. в отсутствие растворителя, либо в присутствии полярного растворителя, такого как низкомолекулярные спирты, вода, диметилформамид или диметилсульфоксид. Реакция может быть осуществлена в интервале температур приблизительно -30oС и 120oС, предпочтительно между приблизительно -5oС и 100oС. Альтернативно, реакция может осуществляться в присутствии активирующего агента, такого как активированные квасцы или инертный растворитель, предпочтительно эфир, такой как диэтиловый эфир, тетрагидрофуран, диоксан или трет-бутил метил эфир, при температуре от близкой к комнатной до примерно 110oС, как это описано Posner and Rogers, J. Am. Chem. Soc., 99, p.8208 (1977). Другие активирующие реагенты включают в себя низкомолекулярные триалкилалюминиевые ряды, такие как триэтилалюминий, либо диалкилалюминиевые-галогенидные ряды, такие как диэтилалюминиевый хлорид (Overman and Flippin, Tetrahedron Letters, p.195 (1981)). Реакции, использующие эти ряды, удобно осуществимы в инертных растворителях, таких как дихлорметан, 1,2-дихлорэтан, толуол или ацетонитрил, при температуре между приблизительно 0oС и приблизительно 110oС. Известны также и другие способы перемещения уходящих групп, либо открытия эпоксидов амина, либо их эквивалентами, такими как азиды или тиметилсилил цианид (Gassman and Guggenheim, J. Am. Chem. Soc., 104, p.5849 (1982)), которым владеют практические специалисты в данной области.
Соединения Формул II и III и их функционально защищенные производные применимы как промежуточные продукты для получения соединения Формулы I. Когда L представляет изобутил, соединения Формулы III могут быть конвертированы в соединение Формулы I и путем реакции с сульфонил-активированными рядами и образования сульфонамида. Способы получения таких сульфонил-активированных рядов хорошо освоены в практической работе специалистов данной области. Как правило, сульфонил-галогениды используются для получения сульфонамидов. Многие сульфонил-галогениды являются коммерчески доступными; другие можно легко получить с применением традиционных методов синтеза (Gilbert, E.E. "Recent Developments in Preparative Sulfonation and. Sulfation" Synthesis 1969: 3 (1969) и приведенные там ссылки; Hoffman, R.V. "М-Trifluoromethylbenzenesulfonyl Chloride" Org. Sunth. Coll. Vol. VII, John Wiley Sons (1990); Hartman, G.D. et al. "4-Substituted Thiophene-and Furan-2-sulfonamides as Topical Carbonic Anhydrase Inhibitors", J. Med. Chem., 35, p.3822 (1992)) и приведенные там ссылки).
В случае соединений Формулы III, где L - водород, конверсия возникающего в результате первичного амина во вторичный амин может осуществляться известными методами. Такие методы включают в себя реакцию с алкил-галогенидом или алкил-сульфонатом, либо путем восстановительного алкилирования альдегидом с использованием, например, каталитической гидрогенизации или цианоборогидрида натрия (Borch et al., J. Am. Chem. Soc., 93, p.2897 (1971)). Альтернативно, первичный амин может быть ацилирован с последующим восстановлением с помощью борана или другого подходящего восстанавливающего реагента, например, как описано Gushman et al., J. Org. Chem., 56, р.4161 (1991). Этот метод особенно применим для соединений Формулы III, где Р представляет собой защитную группу, такую как трет-бутоксикарбонил (Воc) или бензилоксикарбонил (Cbz) и G - водород, либо где и Р и G оба являются бензилом.
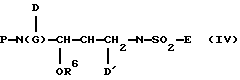
Если переменные Р и G конкретного соединения Формулы IV представляют собой подвижные защитные группы, удаление одной из них или обеих групп с последующей реакцией возникающего в результате амина с соответствующим активированным реагентом будут выгодно давать выход иного соединения Формулы IV. Например, могут быть получены карбаматы путем реакции с хлоркарбонатами или с карбонатами, эстерифицированными уходящими группами, такими как 1-гидроксибензотриазол (НОВТ) или HOSu, либо 4-нитрофенол (протонизированный ряд). Примером такого карбоната является N-сукцинимидил-(3S)-тетрагидрофуран-3-ил карбонат. Нетрудно признать, что для того, чтобы обеспечить возможность специфических реакций, может потребоваться защита одной или более потенциально реактивных групп с дальнейшим последовательным удалением этой группы. Такая модификация в схемах реакции, обрисованных выше, вполне в пределах практических возможностей технологий данной области.
Как должно быть понятно специалисту в данной области, приведенные выше схемы синтеза не претендуют на то, чтобы рассматривать как исчерпывающий перечень всех возможных средств, с помощью которых соединения, описанные и заявленные в настоящей заявке, могут быть синтезированы. Для специалистов будут очевидны и другие способы.
Соединения данного изобретения могут модифицироваться добавлением соответствующих функциональностей для усиления селективных биологических свойств. Такие модификации известны специалистам в данной области и включают в себя те, которые повышают биологическую пенетрацию в соответствующий биологический отдел (например, кровь, лимфатическая система, центральная нервная система), повышают пероральную доступность, повышают растворимость для обеспечения возможности введения посредством инъекции, изменяют метаболизм и изменяют интенсивность экскреции.
Соединение настоящего изобретения представляет собой превосходный лиганд для аспартил-протеаз, в частности ВИЧ-1 и ВИЧ-2 протеазы. Соответственно соединение способно избирать целью и ингибировать события поздней стадии в ВИЧ-репликации, т. е. обрабатывать вирусные полипротеины с помощью ВИЧ-закодированных протеаз. Соединение ингибирует протеолитическую обработку предшественников вирусных полипротеинов путем ингибирования аспартил-протеазы. Поскольку аспартил-протеаза существенно важна для продуцирования зрелых вирионов, ингибирование этой обработки эффективно блокирует распространение вируса путем подавления продуцирования инфекционных вирионов, в частности, из хронически инфицированных клеток. Соединение согласно данному изобретению выгодно ингибирует способность вируса ВИЧ-1 инфицировать иммортализированные человеческие Т-клетки за некоторый период времени, как установлено исследованием внеклеточного р24 антигена - специфического маркера вирусной репликации. Другие антивирусные исследования подтвердили действенность данного соединения.
Соединение данного изобретения может применяться традиционным способом для воздействия на вирусы, такие как ВИЧ и HTLV, которые зависят от аспартил-протеазы для обязательных событий своего жизненного цикла. Такие способы воздействия, уровни доз и необходимые условия могут быть избраны практическими специалистами в этой области из числа доступных способов и технологий. Например, соединение данного изобретения может комбинироваться с фармацевтически приемлемым вспомогательным средством для введения вирус-инфицированному пациенту фармацевтически приемлемым способом и в количестве, достаточно эффективном для уменьшения тяжести вирусной инфекции или смягчения патологических эффектов, связанных с ВИЧ- инфекцией.
Альтернативно, соединение данного изобретения может быть использовано в профилактике и в способах для защиты индивидуумов от вирусной инфекции во время специфического события, такого как роды, либо для приема в течение продолжительного периода времени. Соединение может быть использовано для такой профилактики либо изолированно, либо вместе с другими антивирусными агентами в целях повышения эффективности каждого из агентов. Как таковой, новый ингибитор протеазы данного изобретения может быть назначен для приема в качестве агентов для лечения или предупреждения ВИЧ-инфекции у млекопитающих.
Соединение Формулы I может легко абсорбироваться в кровоток млекопитающих после перорального введения. Соединение Формулы I, имеющее молекулярную массу менее чем приблизительно 600 г/моль и растворимость в воде более чем или равную 0,1 мг/мл, по всем данным обнаруживает высокую и устойчивую пригодность для перорального применения. Эта неожиданно впечатляющая пероральная пригодность делает соединение отличным агентом для способов перорального приема при назначении в лечении и профилактике ВИЧ-инфекции.
Дополнительно к своей пероральной биодоступности соединение данного изобретения также обладает впечатляюще высоким терапевтическим индексом (которым измеряется токсичность в сравнении с антивирусным эффектом). Соответственно соединение данного изобретения обладает эффективностью на более низких уровнях дозировок, чем многие ранее описанные традиционные антиретровирусные агенты, и это позволяет избежать многих тяжелых токсических эффектов, связанных с этими препаратами. Потенциальная возможность данного соединения поставляться в дозах, намного превышающих его эффективный антивирусный уровень, выгодна для замедления или предотвращения возможности развития резистентных вариантов.
Соединение данного изобретения может назначаться здоровому или ВИЧ-инфицированному пациенту либо в виде изолированного агента, либо в комбинации с другими антивирусными агентами, которые вмешиваются в репликационный цикл ВИЧ. Назначением вещества данного изобретения с другими антивирусными агентами, направленными на другие события жизненного цикла вируса, потенциируется терапевтический эффект этих соединений. Например, такой совместно вводимый антивирусный агент может относиться к числу направленных на ранние события жизненного цикла вируса, такие как проникновение в клетку, обратную транскрипцию и ДНК-интеграцию вируса в клеточную ДНК. Анти-Вич агенты, нацеленные на такие ранние события жизненного цикла, включают в себя: диданозин (ddl), дидеоксицитидин (ddC), d4T, зидовудин (AZT), ЗТС, 935U83, 1592U89, 524W91, полисульфатные полисахариды, sT4 (растворимый CD4), ганикловир, тринатрий-фосфоноформат эфлорнитин, рибавирин, ацикловир, альфа-интерферон и трименотрексат. Дополнительно, ненуклеозидные ингибиторы обратной транскриптазы, такие как TIBO, делавирдин (U90) или невирапин, могут применяться для потенциирования эффекта соединений данного изобретения, как и ингибиторы раздевания вируса, ингибиторы трансактивации протеинов, такие как tat или rev, либо ингибиторы вирусной интегразы.
Комбинационные виды терапии соответственно данному изобретению обеспечивают аддитивный или синергический эффект в ингибировании ВИЧ-репликации, потому что каждый составляющий агент комбинации действует на другой участок ВИЧ-репликации. Использование таких комбинационных видов терапии также выгодно снижает дозу взятого традиционного антиретровирусного агента, которая потребовалась бы в целях желаемого терапевтического или профилактического эффекта, в сравнении с тем, что достигается при назначении монотерапии. Такие комбинации могут снижать или устранять побочные эффекты традиционных видов терапии изолированными антиретровирусными агентами, не нарушая антиретровирусную активность этих агентов. Эти комбинации снижают потенциал резистентности к видам лечения изолированными агентами, минимизируя в то же время какую-либо ассоциированную токсичность. Эти комбинации могут также увеличивать эффективность традиционных агентов без увеличения связанной с ними токсичности. В частности, было открыто, что в комбинации с другими анти-ВИЧ агентами, соединение данного изобретения действует аддитивным или синергическим образом в предупреждении репликации ВИЧ в Т-клетках человека. Предпочтительные терапевтические комбинации включают введение соединения данного изобретения с AZT, ddI, ddC, d4T, 3TC, 935U83, 1592U89, 524W91 или их комбинациями.
Альтернативно, соединение данного изобретения может также быть совместно введено с другими ингибиторами ВИЧ-протеаз, такими как saquinavir (Ro 31-8959, Roche), L-735,524 (Merck), ABT 538 (A-80538, Abbott), AG 1341 (Agouron), XM 412 (DuPont Merck), XM 450 (DuPont Merck), BMS 186318 (Bristol-Meyers Squibb) and CPG 53,437 (Ciba Geigy), или пропрепаратами этих или родственных им соединений для повышения эффекта терапии или профилактики против различных вирусных мутантов или членов квази-ВИЧ ряда.
Предпочтительно введение соединения данного изобретения в качестве изолированного агента либо в комбинации с ингибиторами обратной ретровирусной транскриптазы, такими как производные AZT или другие ингибиторы ВИЧ-аспартил-протеазы, включая множественные комбинации, состоящие из 3-5 агентов. Предполагается, что совместное введение соединения данного изобретения обратной ретровирусной транскриптазы или ингибиторами ВИЧ-аспартил-протеазы может обеспечивать существенный аддитивный или синергический эффект и таким образом предотвращать, существенно снижать или полностью устранять репликацию вируса или инфицирование им, либо и то, и другое, а также и связанные с этим симптомы.
Соединение данного изобретения также может вводиться в комбинации с иммуномодуляторами и иммуностимуляторами (например, бропиримином, античеловеческим антителом альфа-интерферона, IL-2, GM-CSF, интерфероном альфа, диэтилдитиокарбаматом, фактором опухолевого некроза, налтрексоном, тускаразолом и rEPO) и антибиотиками (например, пентамидином изетиоратом) для предупреждения или борьбы с инфекцией и заболеваниями, связанными с ВИЧ-инфекциями, такими как СПИД и ARC.
Когда соединение данного изобретения назначается в комбинационных видах терапии вместе с другими агентами, они могут вводиться пациенту последовательно или одновременно. Альтернативно, фармацевтические рецептуры в соответствии с данным изобретением могут содержать комбинацию ингибитора аспартил-протеазы данного изобретения и другого терапевтического или профилактического агента.
Хотя данное изобретение сфокусировано на применение раскрываемого в нем соединения для предупреждения и лечения ВИЧ-инфекции, соединение данного изобретения может также быть использовано в качестве ингибиторного агента для других вирусов, которые зависят от подобных аспартил-протеаз для обязательных событий в их жизненном цикле. Эти вирусы включают другие СПИД-подобные заболевания, вызываемые ретровирусами, такими как вирус иммунодефицита обезьян, HTLV-I и HTLV-II. Кроме того, соединение данного изобретения может также применяться для ингибирования других аспартил-протеаз и, в частности, других аспартил-протеаз человека, включая ренин и аспартил-протеазы, которые подвергают обработке предшественник эндотелина.
Составы данного изобретения, как правило, принимаются перорально. Они содержат некоторое количество соединения Формулы I, которое эффективно в ингибировании репликации ВИЧ путем ингибирования его ВИЧ-протеазы в ЦНС.
Соединение Формулы I используется в способе и составе данного изобретения в комбинации с фармацевтически приемлемым носителем. Как правило, он также применяется в комбинации с другими видами лечения СПИДа, в частности AZT и ЗТС.
Фармацевтические составы данного изобретения содержат соединение настоящего изобретения и его фармацевтически приемлемые соли, с любым фармацевтически приемлемым носителем, вспомогательным средством или растворителем. Фармацевтически приемлемые носители, вспомогательные средства или растворители, которые могут быть использованы в фармацевтических составах настоящего изобретения, включают в себя, хотя и не ограничиваются только ими; ионообменники, квасцы, стеарат алюминия, лецитин, системы доставки самоэмульгирующихся препаратов SEDDS (self-emulsifying drug delivery systems), такие как dα-токоферол полиэтиленгликоль 1000 сукцинат, сывороточные белки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, парциальные глицеридные смеси насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протамин сульфат, двунатриевый гидрофосфат, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный кремнезем, магния трисиликат, поливинил пирролидон, основанные на клетчатке вещества, полиэтиленгликоль, натрий карбоксиметилцеллюлоза, полиакрилаты, воски, полиэтилен-полиоксипропилен-блок полимеры, полиэтиленгликоль и ланолин. Циклодекстрины, такие как α-, β-, γ- циклодекстрин, или химически видоизмененные производные, такие как гидроксиалкил циклодекстрины, включая 2-3-гидроксипропил-β-циклодекстрины, либо другие переведенные в растворимую форму производные также могут быть выгодно использованы для доставки соединения Формулы I.
Фармацевтические составы данного изобретения могут вводиться перорально, парентерально, с помощью ингаляционного спрея, местно, ректально, назально, буккально, вагинально или путем имплантированного резервуара. Предпочтительно пероральное введение или введение путем инъекций. Фармацевтические составы данного изобретения могут содержать любые традиционные нетоксические фармацевтически приемлемые носители, вспомогательные вещества или растворители. В ряде случаев, рН рецептуры может быть доведено с помощью фармацевтически приемлемых кислот, оснований или буферов для повышения стабильности рецептур соединения в его отпускной форме. Термин "парентеральный" в том виде, в каком он используется здесь, включает в себя подкожные, внутрикожные, внутривенные, внутримышечные, внутрисуставные, внутрисиновиальные, интрастернальные, интратекальные, интралезиональные и интракраниальные инъекционные и инфузионные методики.
Фармацевтические составы могут быть в форме стерильного инъекционного препарата, например в виде стерильной водной или масляной суспензии. Эта суспензия может иметь рецептуру соответственно методике, известной в практике, при использовании подходящих диспергирующих или увлажняющих агентов (таких, например, как Tween 80) и суспендирующих агентов. Стерильный инъекционный препарат может также быть в виде стерильного инъекционного раствора или суспензии в нетоксичном парентерально-приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые могут применяться, можно назвать маннит, воду, раствор Рингера (Ringer's solution) и изотонический раствор натрия хлорида. Кроме того, традиционно применяются в качестве растворителей или суспендирующей среды стерильные, фиксированные масла. В этих целях может применяться любое мягкое фиксированное масло, включая синтетические моно- или диглицериды. Пригодны в получении инъекционных препаратов жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, так же как и натуральные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных вариантах. Эти масляные растворы или суспензии могут также содержать спирт с длинной цепочкой как разбавитель или диспергент, такой как Ph. Hely или подобный спирт.
Фармацевтические составы данного изобретения могут назначаться перорально в любой перорально приемлемой дозированной форме, включая капсулы, таблетки и водные суспензии и растворы, но не ограничиваясь ими. В случае таблеток для перорального применения носителями, которые обычно используются, являются лактоза и кукурузный крахмал. Смазывающие агенты, такие как стеарат магния, тоже добавляются, как правило. Для перорального применения в форме капсул пригодны разбавители, включая лактозу и сушеный кукурузный крахмал. Когда водные суспензии назначаются перорально, активный ингредиент комбинируется с эмульгирующими и суспендирующими агентами. Если это необходимо, могут добавляться определенные подслащающие, ароматизирующие и/или красящие агенты.
Фармацевтические составы данного изобретения могут назначаться в форме суппозиториев для ректального введения. Эти составы можно получать путем смешивания соединения данного изобретения с подходящим нераздражающим наполнителем, который является твердым веществом при комнатной температуре, но жидким при ректальной температуре и поэтому будет таять в прямой кишке и высвобождать активные компоненты. Такие материалы включают в себя масло какао, пчелиный воск и полиэтиленгликоли, но не ограничиваются только ими.
Местное применение фармацевтических составов данного изобретения особенно целесообразно, когда необходимое лечение должно охватывать участки или органы, легко доступные для местных аппликаций. Для аппликаций местно на кожу фармацевтические составы должны обладать рецептурой с включением подходящей мази, содержащей активные компоненты, взвешенные или растворенные в носителе. Носители для соединений данного изобретения при местном их применении включают в себя минеральное масло, жидкую нефть, вазелин, пропиленгликоль, полиоксиэтилен полиоксипропиленовое соединение, эмульгирующий воск и воду, но не ограничиваются только ими. Альтернативно, фармацевтические составы могут иметь рецептуру с подходящим лосьоном или кремом, содержащим активное соединение, взвешенное или растворимое в носителе. Подходящие носители включают в себя минеральное масло, сорбитан моностеарат, полисорбат 60, цетиловые эфиры, воск, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду, но не ограничиваются только ими. Фармацевтические составы данного изобретения могут также применяться местно в области нижнего отдела кишечника с помощью рецептур для ректальных свечей или подходящих рецептур для клизм. Местные наклейки чрескожного действия также включены в данное изобретение.
Фармацевтические составы данного изобретения могут назначаться в форме назального аэрозоля или ингаляции. Такие составы получают в соответствии с методиками, хорошо известными в практике фармацевтической рецептуры, и могут приготавливаться в виде растворов в физиологическом растворе, с использованием бензилового спирта или других подходящих консервантов, абсорбционных промоторов для повышения биодоступности, фторуглеродоводородов и/или других растворяющих или диспергирующих агентов, известных в практике.
Для предупреждения и лечения вирусных инфекций, включая ВИЧ-инфекцию, пригодны уровни дозировок предпочтительно от 0,01 до 100 мг/кг веса тела в день, предпочтительно приблизительно 0,5-75 мг/кг веса тела в день активного ингредиента соединения. Как правило, фармацевтические составы данного изобретения должны назначаться на прием приблизительно от 1 до 5 раз в день или, альтернативно, в виде непрерывной инфузии. Такое введение может применяться в порядке экстренной или долговременной терапии. Количество активного ингредиента, которое может комбинироваться с материалами-носителями для получения формы разовой дозировки, должно варьировать в зависимости от хозяина, подвергаемого лечению, и конкретного способа введения. Типичный препарат должен содержать приблизительно от 5% до 95% (мас./мас.) активного соединения. Предпочтительно, чтобы такие препараты содержали от приблизительно 20% до приблизительно 80% активного соединения.
По улучшении состояния пациента может быть назначена поддерживающая доза соединения, состава или комбинации данного изобретения, если это необходимо. В последующем дозировка или частота приема, либо и то, и другое, будучи рассмотрены как функция зависимости от симптоматики, могут быть снижены до уровня, на котором сохраняется улучшение состояния; когда симптоматика улучшилась до желаемого уровня, лечение прекращается. Пациенты могут, однако, нуждаться в прерывистом лечении на долговременной основе в связи с каким-нибудь рецидивом симптомов болезни.
Как это будет понятно практическому специалисту, могут потребоваться дозы, более низкие или более высокие, чем те, которые приведены выше. Специфическое определение доз и схемы лечения для какого-нибудь конкретного пациента будут зависеть от разнообразных факторов, включая специфическую активность используемого соединения, возраст, вес тела, общее состояние здоровья, пол, режим питания, время приема, интенсивность выделения, комбинацию препаратов, тяжесть и течение инфекции, предрасположенность пациента к инфекции и мнение лечащего врача.
В целях более полного понимания данного изобретения предложены для рассмотрения примеры, приведенные ниже. Эти примеры служат лишь целям иллюстрирования и не должны никоим образом истолковываться как ограничивающие объем изобретения.
Пример 1
Предшественник А. Раствор 102 мг N-((2 syn, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламинобутиламина в 4:1 CH2Cl2/насыщенный водный NaHCO3 обрабатывали последовательно при температуре окружающей среды под атмосферой азота, с помощью 65 мг р-нитробензолсульфонила хлорида и 51 мг бикарбоната натрия. Смесь перемешивали в течение 14 ч, разбавляли с помощью CH2Cl2, промывали насыщенным NaCl, затем сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали селикогельной хроматографией низкого давления с использованием 20% диэтилового эфира/ CH2Cl2 в качестве элюента, получая 124 мг заглавного продукта в виде белого твердого вещества. TLC (тонкослойная хроматография): Rf= 0,36, 20% диэтиловый эфир/ CH2Cl2. HPLC: Rt=15,15 мин. (1Н) - NMR (CDCl3) совместимый со структурой.
Пример 2
Соединение I. Раствор 124 мг вещества, полученного в Примере 1 в этил-ацетате, обрабатывали при температуре окружающей среды с помощью 13 мг 10% палладия на угле. Смесь перемешивали в течение 14 ч под атмосферой водорода, фильтровали через прокладку фильтровального агента Celite и концентрировали в вакууме. Остаток подвергали препаративной высокоэффективной жидкостной хроматографии и получали 82 мг заглавного продукта в виде белого твердого вещества. TLC: Rf= 0,10, 20% эфир/ CH2Cl2. HPLC: Rf=13,16 мин. (1Н) - NMR (CDCl3), совместимый со структурой.
Безусловно, несмотря на то, что выше приведен ряд воплощений данного изобретения, основные построения могут подвергаться изменениям для обеспечения еще и других воплощений, использующих продукты и способы данного изобретения. Следовательно, должно быть понятно, что объем настоящего изобретения следует определять прилагаемыми пунктами формулы изобретения, а не теми специфическими воплощениями, которые представлены с помощью примеров.
Изобретение относится к медицине, и может быть использовано для лечения поражения центральной нервной системы (ЦНС), вызванного ВИЧ, в частности СПИД-обусловленной деменции. Изобретение представляет собой применение ингибитора ВИЧ-протеазы для лечения поражений ЦНС, вызванной ВИЧ. Предложенное изобретение позволяет повысить эффективность лечения за счет преодоления заявленными соединениями гематоэнцефалического барьера. 4 с. и 2 з.п. ф-лы.
в качестве средства для лечения млекопитающего, страдающего поражением центральной нервной системы (ЦНС), обусловленным вирусом, который зависит от аспартил-протеазы для протеолитической обработки предшественников вирусных полипротеинов.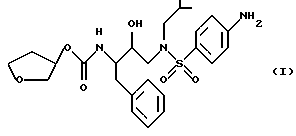
в качестве средства для ингибирования в млекопитающем продуцирования инфекционных ВИЧ-вирионов в ЦНС.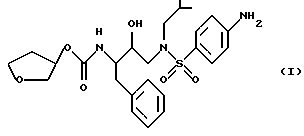
в качестве средства для ингибирования энзиматической активности аспартил-протеазы в ЦНС млекопитающего.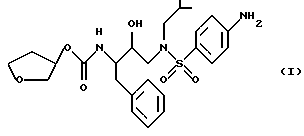
в качестве средства для редуцирования вирусной инфекции в ЦНС млекопитающего, где упомянутый вирус требует аспартил-протеазы для протеолитической обработки предшественников вирусных полипротеинов.
| СПОСОБ ТОРМОЖЕНИЯ РЕТРОВИРУСНЫХ ПРОТЕАЗ | 1991 |
|
RU2028155C1 |
| Livington DJ et al | |||
| Двухколесный автомобиль для формовки кирпичей из разлитой по полю сушки торфяной массы | 1923 |
|
SU478A1 |
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |
| Partaledis JA et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| J Virol | |||
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |
| ШУВАЛОВА Е.П | |||
| Инфекционные болезни | |||
| - М.: Медицина, 1990, с.537 и 539. | |||

