Изобретение относится к новому способу получения бифенилкарбонитрилов, которые пригодны, например, для использования в качестве промежуточных продуктов при получении как новых, так и известных производных хинолина и имидазола, ингибирующих действия ангиобензина П (АП), и которые могут быть использованы при лечении таких заболеваний, как гипертония или хроническая сердечная недостаточность.
Известен способ получения бифенилкарбонитрилов, например 4-метилбифенил-2-карбонитрила, которые используются для получения замещенных производных имидазола, используемых в качестве ингибитора ангиобензина АП. Указанные карбонитрилы получают в результате многостадийного синтеза, который требует использования 4-метилбифенил-2-кар- боновой кислоты с последующим превращением карбоксильной группы [1].
Исходную кислоту 4-метилбифенил-2-карбоновую кислоту получают по реакции сочетания Ульмана при высокой температуре с получением соответствующего эфира, который гидролизуют, или гидролизом 1-(4-метилбифенил-2-ил)-4,4-диметилоксазоли-на. Последний получают путем многостадийного синтеза (1).
Наиболее близким к предложенному способу является способ получения 4-метил-6-цианобифенил-2-карбонитрила, заключающийся в том, что галоидбензонитрил подвергают взаимодействию с 4-метилфенилцинком с получением целевого продукта (2).
Недостатком этого способа является многостадийность синтеза и необходимость использования соответствующего магнийгалогенида для получения цинкового соединения.
Предложенный способ получения бифенилкарбонитрила формулы 1 является более простым и удобным по сравнению с прототипом.
Способ получения бифенилкарбонитрила формулы 1: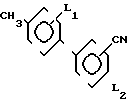 где L1 и L2 независимо друг от друга могут быть выбраны из группы водород, С1-С4-алкил, С1-С4 алкокси, трифторметил, циано, нитро, отличительная особенность которого состоит в том, что соединение бора формулы II
где L1 и L2 независимо друг от друга могут быть выбраны из группы водород, С1-С4-алкил, С1-С4 алкокси, трифторметил, циано, нитро, отличительная особенность которого состоит в том, что соединение бора формулы II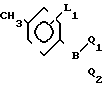 где L1 имеет указанное значение Q1 и Q2 - гидроксигруппа, или Q1 и Q2вместе с атомом бора образуют бороксиновое кольцо, или их смесь, подвергают взаимодействию с галоидбензолом общей формулы III
где L1 имеет указанное значение Q1 и Q2 - гидроксигруппа, или Q1 и Q2вместе с атомом бора образуют бороксиновое кольцо, или их смесь, подвергают взаимодействию с галоидбензолом общей формулы III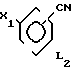 где Х1 - бром, йод или трифторметансульфонилоксигруппа,
где Х1 - бром, йод или трифторметансульфонилоксигруппа,
L2 имеет указанные значения, в присутствии основания и палладиевого (О) и (П) катализатора в среде растворителя.
Соединения формулы II являются известными и могут быть получены например взаимодействием тиоалкилбороната Р(OR3), где R - С1-С6 алкил, с реактивом Гриньяра, или соединением фениллития, полученным с помощью 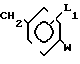 известного способа из соединения формулы IV где L1 имеет указанное значение; E - галоген такой, как хлор, бром, йод,
известного способа из соединения формулы IV где L1 имеет указанное значение; E - галоген такой, как хлор, бром, йод,
Реакцию проводят обычно в растворителе, таком как тетрагидрофуран или эфир, или их смеси при температуре (-78)-(25)оС.
Последующий кислотный гидролиз в обычных условиях приводит к образованию соединения формулы II, где Q1 и Q2 гидроксигруппа, из которых дегидратацией получают соединения формулы II, где Q1 и Q2 - образуют бороксиновое кольцо.
Кроме того, реактив Гриньяра или соединения лития могут взаимодействовать с бораном формулы B(Q1)(Q2)Hal, где Q1 и Q2представляет алкил, или возможно замещенный фенил; Hal - галоген, такой как хлор, бром, йод.
В предложенном способе подходящим основанием является алкоксид или гидроксид щелочного металла, карбонат щелочного металла, а также триалкиламин. В качестве растворителя могут быть использованы углеводороды, низший алифатический спирт, циклический эфир, такой как тетрагидрофуран или слив указанных соединений, температура реакции колеблется в диапазоне 50-150оС, предпочтительной температурой является температура кипения растворителя.
Биологическая активность полученных соединений, в частности антагонизм по отношению к одному или более действиям АII, а в частности антагонизм по отношению к взаимодействию АII с рецепторами, проводящими его влияние на ткань-мишень, может быть обнаружен путем использования одной или нескольких рутинных лабораторных процедур.
За исключением 4-метилбифенил-2-карбонитрила и 4-метил-6-цианобифенил-2-карбонитрила формулы I представляют собой новые соединения.
П р и м е р 1. 1,2 г (0,012 ммоля) 4-метилфенилборной кислоты добавили к раствору, содержащему 1,82 г (0,01 моля) 2-бромбензонитрила, 0,35 г (3 мол. % ) /тетракис/трифенилфосфин/палладия (0) и 10 мл 2М водного раствора карбоната натрия в 20 мл толуола, и смесь нагревают в течение 6 ч при 80оС. Смеси дали охладиться и добавили пероксид водорода (0,5 мл 30 мас.% в воде). Смесь перемешивали в течение 20 мин, после чего проэкстрагировали эфиром и высушили над безводным сульфатом магния. Растворитель упарили, а полученный маслянистый остаток очистили, пропустив через колонку с силикагелем, используя в качестве элюирующего растворителя 15%-ный раствор этилацетата в гексана, в результате получили 4-метилбифенил- 2-карбонитрил - в виде твердого продукта (1,65 г), т.пл. 44-46оС.
Параметры ЯМР : (ДМСО - d6)s, 2,40 (s, 3H), 7,30 (d, 2H)m, 7,35-7,55 (m, 4H), 7,60-7,65 (m, 1Н), 7,75 (d, 1Н).
П р и м е р 2. При 5оС перемешиваемой смеси, содержащей 30 г 4-метилфенилборной кислоты, 36,4 г 2-бромбензонитрила, 0,4 г хлорида палладия (II) 200 мл метанола и 200 мл толуола, добавили 200 мл 2М раствора карбоната натрия. Температура поднялась примерно до 20оС и выпал осадок. После этого смесь нагревали с обратным холодильником в течение 2 ч. Реакционной смеси дали охладиться и добавили 100 мл воды вслед за этим - 5 г диатомовой земли. Смесь перемешивали в течение 15 мин, затем отфильтровали через диатомовую землю. Отделили органический слой фильтра и промыли водным 2М раствором карбоната натрия, а затем водой. Органический слой затем отфильтровали из петролейного эфира (температура кипения 110-120оС) и получили в результате с 80% выходом 4-метилбифенил-2-карбонитрил, идентичный продукту полученному в примере 1.
П р и м е р 3. К 7,5 М раствору хлористого водорода в 10 мл диоксана и 1 мл воды добавили 1,0 г 2-бутил-4-хлор-5-гидроксиметил-1- [2-трифенилметил-2Н-тетразол-5-ил(бифенил-4-ил)метил] имидазола и смесь перемешивали в течение часа при комнатной температуре. Летучий продукт отогнали при упаривании к остатку добавили избыток водного раствора карбоната натрия, и смесь промыли эфиром (2 х 10 мл). Водный слой подкислили до рН 3, добавляя 2М раствор хлористоводородной кислоты, и проэкстрагировали дихлорметаном. Экстракты высушили над безводным сульфатом магния и растворитель отогнали при упаривании. Оставшуюся пену белого цвета растерли с эфиром, полученный твердый продукт отфильтровали и получили в результате 0,37 г 2-бутил-2-хлоро-5-гидроксиметил-1-2'-(1Н-тетразол-5-ил)бифенил- 4-ил(метил) имидазола, т.пл. 179-180оС.
Параметры ЯМР (COCl3): 0,9 (t, 3H), 1,2-1,4 (m, 1H), 1,5-1,7 (m, 2H), 2,6 (t, 2H), 4,5 (s, 2H), 5,2 (s, 2H), 6,95 (d, 2H), 7,15 (d, 2H), 7,35-7,55 (m, 3H), 7,80-7,90 (m, 2H).
Исходный материал (А) был получен следующим образом.
Раствор, содержащий 0,67 г 4-метилбифенил-2-карбонитрила и 9,3 г азида трибутилолова в 20 мл толуола, нагревали с обратным холодильником в течение 46 ч. Охлажденную реакционную смесь подкислили, насыщением ее газообразным хлористым водородом и затем охладили на ледяной бане. Суспендированный твердый продукт отфильтровали и растерли с толуолом, получили в результате с 90% выходом 5-(2-(4-метилбифенилил-2Н-тетразол) (В), т.пл. 149-150оС.
Параметры ЯМР спектра: (СOCl3) ДМСО - d6) 2,25 (s, 3H), 6,95 (d, 2H), 7,10 (d, 2H), 7,50-7,70 (m, 4H).
К перемешиваемому раствору соединения (В) в 150 мл дихлорметана при комнатной температуре добавили 18,73 г трифенилметилхлорида. Затем добавили 10,2 мл триэтиламина и смесь нагревали с обратным холодильником в течение 2,5 ч. Реакционной смеси дали охладиться, промыли водой и высушили над безводным сульфатом магния.
Растворитель отогнали при упаривании и получили в результате 26,7 г 5-(2-(4-метилбифенилил-2-трифенилметил-2Н-тетразол) (С), т.пл. 16-168оС.
Параметры ЯМР спектра (СDCl3) 2,25 (s, 3H)- 6,90-7,00 (m, 10H), 7,20-7,45 (m, 12H), 7,85-7,90 (m, 1H).
Смесь, содержащую 0,54 г соединения (С), 0,20 г N-бромосукцинимида и 18 молей азо(бисизобутиронитрила) в 10 мл четыреххлористого углерода, нагревали с обратным холодильником в течение 3 ч. Нерастворимый продукт отфильтровали и фильтрат сконцентрировали. Остаток растворили в этилацетате, промыли водой и высушили над безводным сульфатом магния. Растворитель удалили при выпаривании, и остаток растерли с эфиром, в результате получили в виде твердого белого продукта с 92% выходом 4-(2-(4-бромометилбифенилил)-2-трифенилметил-2Н, тетразол (D), т.пл. 136-138оС.
Параметры ЯМР: 4,4 (s, 2H), 6,85-7,10 (m, 10H), (m, 10H), 7,20-7,45 (m, 12H), 7,95-8,00 (m, 1H).
К перемешиваемому раствору 1,87 г 2-бутил-4-хлор-5-гидроксиметилимидазола в диметилформамиде (25 мл) добавили соединение D, оставили перемешиваться при комнатной температуре на 72 ч. Растворитель удалили при выпаривании, а остаток растворили в этилацетате и промыли водой. Органический слой высушили над безводным сульфатом магния, растворитель удалили при выпаривании, полученный остаток очистили, используя хроматографию на силикагале с элюированием смесью этилацетата (гексан 1:1) объемное отношение. В результате получили в виде твердого продукта 1,8 г 2-бутил-4-хлоро-5-гидроксиметил-1-(2-трифенилметил-2Н-тетразол-5- ил)бифенил-4-ил(метил)имидазола (А), т.пл. 89-93оС.
Параметры ЯМР спектра СDCl3: 0,85 (t, 3H), 1.25 (синстет, 2Н), 1,60-1,75 (m, 2H), 2,50 (t, 2H), 4,3 (s, 2H), 5,0 (s, 2H), 6,75 (d, 2H), 6,90-7,00 (m, 6H), 7,10 (d, 2H), 7,20-7,50 (m, 12H), 7,90-7,95 (m, 1H).
П р и м е р 4. Смесь, содержащую, 890 мг 2-метил-4-[(2-(2-три-фенилметил-2H-тетразол-5-ил)бифенил-4-ил(метокси)] хинол ина (А) и раствор 7,5 М хлористого водорода в диоксане (10 мл) и воде (1 мл), оставили на 72 ч. Летучий продукт удалили при выпаривании, а остаток растерли с эфиром (2 х 50 мл). Эфир декантировали и твердый остаток перекристаллизовали из изопропанола, в результате получили 370 мг гидрохлорида 2-метил-4-[(2-(1Н-тетразол-5-ил)бифенил-4-ил(метокси)] хинолина, в виде твердого продукта белого цвета, с т.пл. 188-190оС.
Параметры ЯМР спектра (ДМСО - d6): 2,92 (s, 3H), 5,63 (s, 2H), 7,21 (d, 2H), 7,56-7,87 (m, 8H), 8,07 (dt, 1H), 8,28 (dd, 1H), 8,32 (dd, 1H).
Масс-спектр [ve FAB, ДМСО].
[NBA] 392 (М-Н)- 158. Данные микроанализа:
Найдено, %: C 66,0; H 4,6; N 15,5.
С 24Н19N5O.HCl.0,5.H2O
Вычислено, %: C 76,7; H 4,7; N 16,0.
Исходный продукт (А) был получен следующим образом.
90 мг 60%-ной дисперсии в минеральном масле (гидрид натрия добавили к перемешиваемому раствору 2-метил-4-хи- нолина, полученному в соответствии с методикой, приведенной в Org. Syn. 1955. Coll. vol III, с.374, и с.593) 340 мг в 10 мл диметилформамида. Смесь перемешивали до полного выделения Н2, после чего добавили раствор 1,2 г 5-(2-(4-бромметилбифенилил/2-трифенилметил-2Н-тетразола в 5 мл ДМФА.
Полученную смесь перемешивали в течение 16 ч. Растворитель удаляют при выпаривании, а остаток разделяют на части между 20 мл воды и двумя порциями по 10 мл дихлорметана. Органический слой промыли насыщенным раствором хлорида натрия (5 мл) и высушили над безводным сульфатом магния. Растворитель удалили при выпаривании и полученное масло очистили методом колоночной хроматографии, используя в качестве элюирующего растворителя смесь метанол/дихлорметан объемное соотношение 1:99, получили в результате 890 мг 2-метил-4-[(2-трифенилметил-2Н-тетразол-5-ил) бифенил-4-илметокси)] -хинолина (А) в виде твердого продукта белого цвета, с т.пл. 168-170оС (разложение).
Параметры ЯМР спектра: 2,7 (s, 3H), 5,14 (s, 2H), 6,7 (s, 1H), 6,9 (dd, 6H), 7,15-7,55 (сложный m, 17H), 7,65 (dt, 1H), 7,95 (m, 2H), 8,15 (dd, 1H).
П р и м е р 5. Используя процедуру, аналогичную описанной в примере 4, но в качестве исходного продукта - 2-этил-4-[2-(2-трифенилметил-2Н-тетразол-5-ил)бифе-нил-4-ил (метокси)]хинолин получили с 70% выходом гидрохлорид 2-этил-4- (2-1Н)тетразол -5-ил)бифенил-4-ил(метокси)хинолина, с т.пл. 178-181оС (разл.).
Параметр ЯМР спектра (ДМСО - d6): 1,48 (t, 3Н), 3,22 (q, 2H),m 5,68 (s, 2H), 7,23 (d, 2H), 7,5-7,8 (m, 7H), 7,83 (t, 1H), 8,08 (t, 1H), 8,32 (t, 2H).
Исходный материал А был получен согласно процедуре, описанной в примере 4, но с использованием 2-этил-4-хинолина, который был сам получен с использованием методики получения 2-метил-4-хинолина, с использованием в качестве исходных реагентов аналина и этилпропионилацетата.
П р и м е р 6. 4-Метил-3-нитро-фенилборную кислоту (полученную согласно описанию JA. C.S, 1932, 54, 4415 в количестве 3 г добавили к раствору, содержащему 2,78 г 2-бромбензнитрила и 0,525 г тетракис(трифенилфосфин)палладия (0) в смеси 15 мл водного 2М раствора карбоната натрия и 38 мл толуола. Смесь нагревали при 100оС в течение 16 ч, затем оставили охлаждаться. Смесь проэкстрагировали этилацетатом и экстракты промыли насыщенным раствором хлорида натрия и высушили. Растворитель удалили при упаривании, полученный твердый продукт светло-коричневого цвета перекристаллизовали из этилацетата, получив в результате 4-метил-3-нитробифенил-2-карбонитрил 33,13 г, в виде твердого продукта с т.пл. 155-156оС.
ЯМР (CDCl3): 2,88 (s, 3H), 7,44-7,86 (сложный m, 6H), 8,14 (d, 1H).
Найдено, %: C 70,3; H 4,0; N 11,38.
C14H10N2O2.
Вычислено, %: C 70,6; Н 4,2; N 11,8.
Антагонизм по отношению к одному или более действиям АII и, в частности, антагонизм по отношению к взаимодействию AII с рецепторами, проводящим его влияние на ткань-мишень, может быть обнаружен путем использования одной или нескольких рутинных лабораторных процедур.
Тест А. Этот тест in vitro включает инкубацию тестируемого соединения вначале при концентрации 100 мкмолей (или менее) в буферной смеси, содержащей фиксирование концентрации меченого радиоактивной меткой AII и фракции поверхностной мембраны клетки, полученной из подходящей ангиобензину ткани-мишени. В этом тесте источником поверхностной мембраны клетки является надпочечник морской свинки, который, как известно, дает ответную реакцию на действие AII. Взаимодействие меченого AII с его рецепторами (оценивается по количеству радиоактивной метки, связавшейся с частицами выделенной фракции мембраны, вслед за удалением несвязанной метки путем быстрой фильтрации традиционно используемой в таких исследованиях (оказывается под влиянием соединений, которые также способны образовывать связь с рецепторными участками мембраны, и степень антагонизма (наблюдаемого в исследовании по исчезновению мембран-связанной радиоактивности) определяется с легкостью путем сравнения рецептор-связанной радиоактивности в присутствии тестируемого соединения при специфической тестируемой концентрации с контрольным значением радиоактивности, определяемой в отсутствии тестируемого соединения. Используя такую процедуру, соединения, показавшие по крайней мере 50%-ное замещение радиоактивного меченого АII, при концентрации, равной 10-4 М, были подвергнуты тестированию вновь при более низкой концентрации для определения их эффективности.
Для определения ИК50 (концентрация, при которой происходит 50%-ное замещение связанного радиоактивномеченого АII) концентрации тестируемого соединения выбирают таким образом, чтобы иметь возможность проверить по крайней мере четыре значения величины концентрирующих около примерно определенной по предварительным данным ИК50, которая затем определяется из графика зависимости процента замещения от концентрации тестируемого соединения.
Как правило соединения формулы VI, определенные выше, проявляют значительное ингибирующее действие в тесте А при концентрации 50 мкмолей, или существенно меньшей концентрации.
Тест В. Этот тест in vitro включает определение величины антагонистического действия тестируемого соединения на сокращения изолированной аорты кролика, помещенной в физиологический солевой раствор при 37оС, индуцированные AII. Для того, чтобы быть уверенным в том, что действие соединения специфично именно в отношении AII, можно провести исследование действия тестируемого соединения на сокращения, индуцированные норадреналином в том же самом объекте.
Как правило, соединения формулы VI, определенные выше, проявляют значительное ингибирующее действие в тесте В при конечной концентрации 50 мкмолей или существенно меньшей концентрации.
Тест С. Этот тест in vivo включает использование крыс, находящихся в сознании или с поверхностным обезболиванием, которым в условиях анестезии имплантировали артериальный катетер для измерения изменения кровяного давления. Антагонистическое действие тестируемого соединения после его перорального или парентерального введения оценивают по величине снижения вазопрессорного действия, индуцированного AII. Для того, чтобы быть уверенным в том, что действие тестируемого соединения специально именно в отношении AII, на этом же объекте можно проверить его влияние на вазопрессорное действие, индуцированное вазопрессином.
Как правило, соединения формулы VI проявляют специфическое антагонистическое действие в отношении AII в тесте С при концентрации 50 мг/кг массы тела или существенно меньшей концентрации, при этом отсутствует какое-либо явное токсикологическое или другое неблагоприятное фармацевтическое действие.
Тест Д. Этот тест in vivo включает стимулирование эндогенного биосинтеза АII у различных особей, включая крыс, обезьян и собак, путем содержания последних на диете с низким содержанием натрия и назначения им соответствующей ежесуточной дозы салуретика, известного под названием фуросемид. После этого тестируемое соединение вводят животным перорально или парэнтерально, предварительно этим животным в условиях анестезии имплантируют артериальный катетер для определения изменений кровяного давления.
Как правило, соединения формулы VI могут оказывать антагонистическое действие в отношении AII в тесте Д, что продемонстрировано на примере резкого снижения кровяного давления при дозе 50 мг/кг массы тела или существенно меньшей концентрации, при этом отсутствует какое-либо явное токсикологическое или другое неблагоприятное фармакологическое действие.
В качестве иллюстрации свойств соединений формулы VI, свидетельствующих об ингибировании действия антиотензина II, приводятся следующие результаты, полученные с использованием гидрохлорида 2-метил-4-[(2-(1Н-тетразол-5-ил)бифенил-4-илметокси)]хинолина в тестах А, В, и С, описанных выше.
В тесте А: среднее значение ИК50 при концентрации 1,7х10-8С, в тесте В: среднее значение рА2 при 8,95, в тесте С: ЕД50, при концентрации 0,5 кг/га массы тела (внутривенное введение).
Использование: в медицине, так как продукт обладает фармакологической активностью. Сущность: усовершенствованный способ получения бифенилкарбонитрила общей формулы I, где L1, L2 водород, C1-C4 алкил, C1-C4 - алкокси, трифторметил, циано, нитро. Реагенты 1: соединение общей формулы II, где X1 - бром, йод или трифторметансульфонилоксигруппа; L2 имеет указанные значения. Реагент 2: соединение общей формулы III, в которой L1 - имеет указанное значение Q1 и Q2 гидроксигруппа или Q1 и Q2 вместе с атомом бора образуют бороксиновое кольцо, или их смесь, в присутствии основания и палладиевого /О/ или 3 /II/ катализатора в среде растворителя.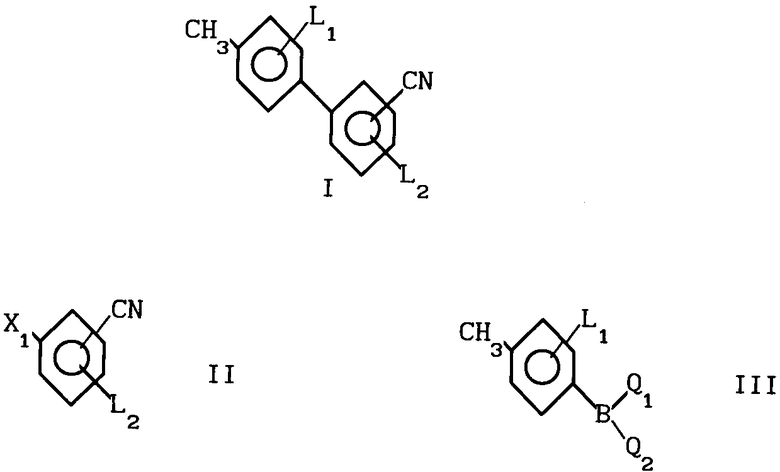
СПОСОБ ПОЛУЧЕНИЯ БИФЕНИЛКАРБОНИТРИЛОВ общей формулы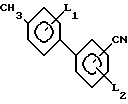
где L1 и L2 - независимо друг от друга могут быть выбраны из группы: водород, С1 - С4-алкил, С1 - С4-алкокси, трифторметил, циано, нитро, с использованием галоидианобензола, отличающийся тем, что в качестве галоидцианобензола используют соединение общей формулы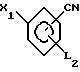
где Х - бром, йод или трифторметаносульфонилоксигруппа, L2 имеет указанные значения,
которое подвергают взаимодействию с соединением бора общей формулы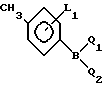
в котором L1 имеет указанное значение, Q1 и Q2 - гидроксигруппа или Q1 и Q2 вместе с атомом бора образуют бороксиновое кольцо, или их смесь, в присутствии основания и палладиевого (О) или (П) катализатора в среде растворителя.
| УСТРОЙСТВО ДЛЯ ОТБОРА КЕРНА | 0 |
|
SU324377A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
