Изобретение относится к области органической химии - способу получения о-арилгликолевых кислот [1].
Известен способ получения о-арилгликолевых кислот (I) путем конденсации эфира галоидуксусной кислоты с фенолами под действием основания (этилат натрия, углекислый калий, иодид натрия и т.п.), с последующим выделением синтезированных эфиров о-арилгликолевых кислот (II) и омылением их до о-арилгликолевых кислот (I) с помощью избытка водного раствора щелочи при нагревании в водно-спиртовой среде (схема 1) [1, 2], (приведена в конце описания).
Таким образом, например, синтезируют о-арилгликолевую кислоту (Iа) исходя из эстрадиола (III) [1].
Однако распространить этот метод на получение о-арилгликолевой кислоты (Iб), содержащей оксиацетиленовую группировку, затруднительно.
Известно, что соединения, содержащие подобную группировку, легко гидролизуются в присутствии водных растворов оснований, а в присутствии спиртов присоединяют их по тройной связи (схема 2) [3] (приведена в конце описания).
С целью устранения указанных недостатков нами был разработан новый способ получения о-арилгликолевых кислот (I), отличающийся тем, что о-арилгликолевые кислоты получают в одну стадию и омыление образующихся в процессе реакции эфиров о-арилгликолевых кислот (II) проводят при 10-30oС в среде апротонного биполярного растворителя эквивалентным количеством водного раствора щелочи, доводя постепенно рН смеси по окончании реакции до 9-10 единиц.
По существу процесс омыления эфиров о-арилгликолевых кислот (II) сводится к процессу их титрования при комнатной температуре.
Высокая скорость гидролиза эфиров о-арилгликолевых кислот (II) обусловлена применением высокополярных растворителей и значительно более высокой степенью диссоциации о-арилгликолевых кислот (I) по сравнению с соответствующими бензойными кислотами.
Подобранные мягкие условия омыления эфиров (II) позволяют получить о-арилгликолевую кислоту (Iб), содержащую лабильную оксиацетиленовую группировку, с высоким выходом.
Используя разработанный метод омыления эфиров (II), удается заменить двухстадийный метод получения о-арилгликолевых кислот (I) одностадийным. Новый способ получения о-арилгликолевых кислот (I) отличается тем, что конденсацию эфиров галоидуксусных кислот с фенолами и последующее превращение эфиров о-арилгликолевых кислот (II) в целевой продукт осуществляют в одну стадию.
Так, о-арилгликолевую кислоту (Iа) получают с хорошим выходом путем обработки раствора метилового эфира хлоруксусной кислоты и эстрадиола (III) в диметилсульфоксиде эквивалентным количеством водного раствора едкого кали при 20oС.
Предложенный способ имеет общее значение и может быть распространен на получение различных о-арилгликолевых кислот (I). Например, n-хлорфенилгликолевую кислоту (Iс) получают с выходом до 96%. Реакция идет уже при температуре 10oС.
Изобретение иллюстрируется следующими примерами
Пример 1.
Получают о-[19-нор-17α-этинил-17β-гидроксипрегна-1,3,5(10)-триен-3-ил] гликолевую кислоту (Iб)
Раствор 29,6 мг (0,1 ммоль) этинилэстрадиола (IV) в 0,4 см3 диметилсульфоксида прибавляют при перемешивании к суспензии 22 мг (0,4 ммоль) порошкообразного едкого кали в 0,4 см3 диметилсульфоксида. Через 30 минут прибавляют раствор 22 мг (0,2 ммоль) метилового эфира хлоруксусной кислоты в 0,1 см3 диметилсульфоксида, перемешивают 1 час, после чего прибавляют 0,1 см воды и продолжают перемешивание в течение 30 минут. Реакционную массу разбавляют 5 см3 холодной воды (рН 9,0-9,5), переносят в делительную воронку, подкисляют 5% соляной кислотой и экстрагируют последовательно 2, 1 и 1 см3 этилацетата. Объединенные этилацетатные вытяжки промывают 2 порциями по 5 см3 воды, встряхивают с 0,5 см3 насыщенного раствора соли, сушат над сульфатом натрия, осветляют активированным углем и упаривают в вакууме на водяной бане при 100oС. Маслянистый остаток при охлаждении кристаллизуется.
Выделяют 297 мг (84%) о-арилгликолевой кислоты (Iб), т.пл. 180-181oC.
ИК-спектр (КВr), см-1: 1736с, 1215с (СООН), 1610ср(Аr), 1500ср, 1160ср, 3280с (ОН, С=СН).
ПМР-спектр (СDСl3), м.д.: 0,88 (3H, СН3), 2,62с (1Н, С=СН), 2,86с (2Н, СН2), 4,65с (2Н, ОСН2), 6,67с (1Н, Аr), 6,72д (1Н, Аr), 7,28м (1Н, Аr).
Пример 2.
Получают о-(п-хлорфенил)гликолевую кислоту (Iс).
Раствор 130 мг (1 ммоль) п-хлорфенола и 214 мг (1 ммоль) этилового эфира иодуксусной кислоты в 3 см3 диметилсульфоксида обрабатывают при охлаждении (8-10oС) 112 мг (2 ммоль) едкого кали в 0,2 см3 воды, размешивают 2 часа при 10oС и упаривают в вакууме. Остаток разбавляют водой, встряхивают с этилацетатом и подкисляют 10% соляной кислотой до рН 2.
Выпавшую о-(п-хлорфенил)гликолевую кислоту(Iс) отделяют фильтрованием.
Получают 67 мг (36%) кислоты (Iс), т.пл. 155-156oС (вода-метанол). Литературные данные: т.пл. l55-156oC [4].
Пример 3.
Получают о-(п-хлорфенил)гликолевую кислоту (Iс)
Раствор 130 мг (1 ммоль) п-хлорфенола и 428 мг (2 ммоль) этилового эфира иодуксусной кислоты в 3 см3 диметилсульфоксида обрабатывают при 8-10oС 224 мг (4 ммоль) едкого кали в 0,4 см3 воды. Через несколько часов избыток диметилсульфоксида отгоняют в вакууме, а смесь разбавляют водой и подкисляют до рН 2.
Выход кислоты (Iс) 110 мг (59%), т.пл. 154-155oС.
Пример 4.
Получают о-(п-хлорфенил)гликолевую кислоту (Iс).
Раствор 215 мг (1 ммоль) этилового эфира п-хлорфеноксиуксусной кислоты в 4 см3 диметилсульфоксида обрабатывают при перемешивании раствором 40 мг (1 ммоль) едкого натра в 0,3 см3 воды при температуре 30oС, выдерживают 30 минут (рН 10) и обрабатывают так, как указано в примере 3.
Выход о-(п-хлорфенил)гликолевой кислоты (Iс) 185 мг (86%), т.пл. 155-156oС.
Пример 5.
Получают о-[17β-гидрокси-эстра-1,3,5(10)триен-3-ил] гликолевую кислоту (Iа)
К раствору 272 мг (1 ммоль) эстрадиола (III) и 0,22 см3 (334 мг, 2 ммоль) этилового эфира бромуксусной кислоты в 8 см3 диметилсульфоксида прибавляют при интенсивном перемешивании по каплям раствор 224 мг (4 ммоль) едкого кали в 0,7 см3 воды в течение часа, выдерживают 30 минут при 20oС (рН 9,5-10,0), разбавляют 60 см3 воды и экстрагируют 3 порциями по 15 см3 этилацетата. Дальнейшая обработка проводится так же, как указано в примере 1.
Получают о-арилгликолевую кислоту (Iа) с выходом 95%, т.пл. 186-187oС. Литературные данные: т.пл. 187-188oС [1].
Пример 6.
Получают о-[19-нор-17α-этинил-17β-гидроксипрегна-1,3,5(10)-триен-3-ил] гликолевую кислоту (Iб)
Раствор 382 мг (1 ммоль) этилового эфира (IIб) в 6 см3 диметилсульфоксида обрабатывают при 20oC по каплям дважды по 220 мг 10% раствора едкого натра (1,1 ммоль) при интенсивном перемешивании. Содержимое колбы постепенно загустевает. Через два часа реакционную массу разбавляют 50 см3 холодной воды (рН 9,0-9,5) и обрабатывают, как указано в примере 1. Получают 326 мг (92%) о-арилгликолевой кислоты (Iб), т.пл. 181-182oС (этилацетат-гексан).
Пример 7.
Получают о-[19-нор-17α-этинил-17β-гидроксипрегна-1,3,5(10)-триен-3-ил] гликолевую кислоту (Iб)
Раствор 38,2 мг (0,1 ммоль) этилового эфира (IIб) в 1 см3 диметилацетамида обрабатывают дважды 22 мг (0,11 ммоль) раствора едкого натра в 0,05 см3 воды при интенсивном перемешивании, выдерживают 1 час при 30oС и разбавляют водой (рН 9,5-10,0). Реакционную смесь обрабатывают так же, как изложено в примере 2.
Получают 78% о-арилгликолевой кислоты (Iб), т.пл. 179-180oС.
Пример 8.
Получают этиловый эфир о-[19-нор-17α-этинил-17β-гидрокси-прегна-1,3,5-(10)триен-3-ил]гликолевой кислоты (IIб)
Смесь 296 мг (1 ммоль) этинилэстрадиола (IV), 200 мг тонкорастертого углекислого калия, 10 мг бензилтриэтиламмоний бромида, 0,22 см3 (334 мг, 2 ммоль) этилового эфира бромуксусной кислоты и 4 см3 ацетона кипятят несколько часов на водяной бане.
Ацетон отгоняют, а маслообразный остаток растирают с 10 см3 гексана. Выпавшие кристаллы отделяют фильтрованием. Выход этилового эфира (IIб) 350 мг (94%), т. пл. 95-96oС (метанол).
ИК-спектр (КВr), см: 1760с, 1225с (COOC2H5), 1610cp (Аr), 3330ш, 3390 с. ш. (ОН, С≡СН).
ПМР-спектр (CDCl3,), м.д.: 0,88с (3Н, СН3), 1,38т (3Н, СН3), 2,62с (1Н, С≡СН), 2,83с (2Н, СН2Аr), 4,25кв (2Н, ОСН2), 4,62с (2Н, OCH2COOEt), 6,63с (1Н, Аr), 6,72д (1Н, Аr), 7,26м (1Н, Аr).
О-арилгликолевые кислоты (I) имеют большое практическое значение. Например, о-арилгликолевую кислоту (Iа), используют в качестве гаптена при получении антисывороток для определения анаболического стероида эстрадиола (III) в физиологических жидкостях и тканях животных и человека иммунохимическими методами [1].
Аналогичным образом может быть применена и о-арилгликолевая кислота(Iб) для определения анаболического стероида - этинилэстрадиола(IV), незаконно применяемого в животноводстве.
Источники информации
1. Патент США 3966744, кл. 546-125, 1976.
2. С.А. 72, р. 66698 к.
3. Волков А.Н., Скворцов Ю.М. и др. - Органическая химия, 1970, т.6, 5, с. 897-902.
4. Справочник химреактивов Аldrich, 1990, c. 311.
Изобретение относится к улучшенному способу получения о-арилгликолевых кислот формулы ArOCH2СООН. Способ включает конденсацию эфиров галоидуксусных кислот с фенолами в присутствии оснований и омыление синтезированных эфиров о-арилгликолевых кислот в целевой продукт при 10-30oС в среде апротонного биполярного растворителя путем обработки эквивалентным количеством водного раствора щелочи до рН 9-10. Способ позволяет совместить две стадии получения о-арилгликолевых кислот в одну. Способ позволяет осуществлять процесс в более мягких условиях с повышением выхода, а также позволяет синтезировать о-арилгликолевые кислоты, содержащие лабильные группировки. Некоторые из синтезированных о-арилгликолевых кислот могут быть использованы для получения антисывороток при разработке иммунохимических методов определения анаболических стероидов.
Способ получения о-арилгликолевых кислот общей формулы:
АrОСН2СООН,
где Аr - замещенный фенил или стероидный радикал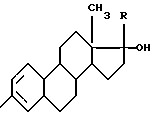
где R = H или C≡CH,
путем конденсации эфиров галоидуксусных кислот с фенолами в присутствии щелочных агентов и последующего омыления выделенных эфиров о-арилгликолевых кислот водным или водно-спиртовым раствором щелочи при нагревании и подкислении, отличающийся тем, что о-арилгликолевые кислоты получают в одну стадию и омыление образующихся в процессе реакции эфиров о-арилгликолевых кислот проводят при 10-30oС в среде апротонного биполярного растворителя эквивалентным количеством водного раствора щелочи таким образом, чтобы рН смеси по окончании реакции не превышал 9-10 единиц.
| US 3966744, 29.06.1976 | |||
| FR 2000174, 20.03.1973 | |||
| Волков А.Н., Скворцов Ю.М | |||
| и др | |||
| - Органическая химия, 1970, т.6, №5, с.897-902. |

