Изобретение относится к области электрохимических методов анализа, в частности к анализу состава раствора, и может использоваться в химической, металлургической, пищевой промышленности, медицине, экологии и, в частности, для контроля состава природных, сточных вод, биологических объектов, пищевых продуктов, диагностики заболеваний.
Известен способ изготовления электродов для электрохимического анализа, включающий осаждение ртутной пленки на электроды из графитовых материалов или иридия (Патент США 5942103, G 01 N 27/26). Способ имеет следующие недостатки: необходимость дополнительной стадии осаждения ртутной пленки перед анализом, высокая токсичность растворов солей ртути и самой ртутной пленки, полученной электроосаждением.
Известен способ, включающий нанесение коллоидного золота, смешанного с катионообменным полимером, на поверхность углеродного электрода, изготовленного методом трафаретной печати, либо стеклоуглеродного электрода (Патент США 5468366, G 01 N 27/26).
Использование известного электрода позволяет определять лишь ионы свинца. В данном способе высокий предел обнаружения - 10 мкг/л.
Известен способ изготовления электродов для электрохимического определения тяжелых металлов в различных объектах (сточные и природные воды, биологические объекты, продукты питания, воздух). Для определения используют электрод, изготовленный нанесением на углеродную поверхность полимера, содержащего ртуть, соединения ртути или соли ртути (Патент США 5672257, G 01 N 27/26). Однако известный способ имеет следующие недостатки: высокий предел обнаружения - 50 мкг/л. Многие объекты, такие как питьевая и природная вода содержат тяжелые металлы в малом количестве на уровне 1-10 мкг/л, поэтому определение свинца в этих объектах с использованием предложенного электрода невозможно. Также недостатком является использование в процессе изготовления электрода и в анализе объектов ртути и растворов с высоким содержанием растворимых солей ртути (12 г/л).
Известен способ изготовления электродов, в котором изготавливают графитсодержащие электроды, модифицированные оксидом ртути HgO (WO 98/58249 А1, G 01 N 27/34). Однако предложенная в способе технология модифицирования оксидом ртути очень сложна. Хлорид ртути растворяют в этаноле, смешивают с угольным порошком, тщательно перемешивают. К этой смеси добавляют 1,5 М NaOH для осаждения на графитовых частицах оксида ртути. Последняя стадия трудновоспроизводимая, поскольку реакция должна проходить при высокой степени адсорбции оксида ртути на графитовых частицах. В тех случаях, когда смесь имеет желтый оттенок оксида ртути, осадок растворяют добавлением кислого раствора, затем переосаждают. Полученную смесь фильтруют и высушивают. Полученный порошок с адсорбированными частицами оксида ртути смешивают с полимерной матрицей, изготавливают электрод и полимеризуют. Для снижения предела обнаружения изготовленных таким образом электродов необходима полировка рабочей поверхности. К тому же оксид ртути растворим в кислых средах, что приводит к загрязнению отработанных растворов ионами ртути.
Наиболее близким техническим решением служит способ изготовления электрода для электрохимического анализа, включающий нанесение на основу графитсодержащего слоя и его модифицирование растворами химических реагентов (Патент РФ 2124720. Бюл. 1, 1999).
Недостатками такого способа являются сложность автоматизации замены рабочего электрода в случае использования толстопленочных электродов из-за недостаточной гибкости электрода и нарушения целостности покрытия при изгибе электрода в процессе его замены. В случае использования графитовой нити такая автоматизация возможна, однако процесс модифицирования графитовой нити сложен и длителен (последовательное вымачивание в разных растворах в течение 5 часов, затем сушка), а при анализе графитовая нить быстро намокает и анализируемый раствор попадает в контактные механизмы, что приводит к снижению чувствительности и искажению полученных результатов. При использовании модифицированной графитовой нити круг определяемых элементов ограничен (свинец и кадмий).
Изобретение направлено на упрощение технологии изготовления электродов, расширение спектра определяемых веществ (возможность определения ионов металлов: меди, свинца, олова, кадмия, цинка, никеля, хрома, марганца, молибдена, вольфрама, селена, сурьмы, ртути, мышьяка, урана; анионов: сульфидов, хлоридов и др. ; органических веществ, таких как мочевина, фонолы, фенотиазины и др. , снижение стоимости электрода, снижение предела обнаружения, улучшение метрологических характеристик электродов, увеличение продолжительности жизни и срока их хранения, расширение возможностей использования.
Данная техническая задача решается тем, что в качестве основы используют полимерные пленочные материалы различной толщины, а перед модифицированием в один или каждый раствор химического реагента дополнительно вводят органический растворитель для образования мелкокристаллической структуры осадка, а также тем, что в один из растворов химических реагентов дополнительно вводят ионнообменный полимер; тем, что в состав графитсодержащего слоя вводят отверждающее связующее.
По другому варианту исполнения задача решается тем, что в качестве основы используют полимерные пленочные материалы различной толщины, а модифицирование графитсодержащего слоя осуществляют до его нанесения на основу путем введения в объем графитсодержащей композиции органического раствора, содержащего водонерастворимый модификатор, полученный экстракцией или растворением в органическом растворителе.
Указанные отличия существенны. Применение в качестве основы полимерных материалов в совокупности с модифицированием графитсодержащего покрытия, приводящего к образованию мелкокристаллической структуры, повышает гибкость, прочность и чувствительность электрода, что позволяет использовать такой электрод в автоматических устройствах с системой перемотки электрода. Кроме того, мелкокристаллическая структура снижает предел обнаружения.
На фиг.1 представлен общий вид электрода, где
1 - гибкая полимерная пленка;
2 - гибкий графитсодержащий слой;
3 - рабочие зоны долгоживущего электрода;
4 - изолированные участки.
На фиг.2 - вариант изготовления электрода, где
1 - основа;
2 - графитсодержащий слой;
3 - слой изолятора;
4 - рабочая поверхность электрода.
На фиг. 3 приведены дифференциально-импульсные (DP) вольтамперограммы свинца и кадмия.
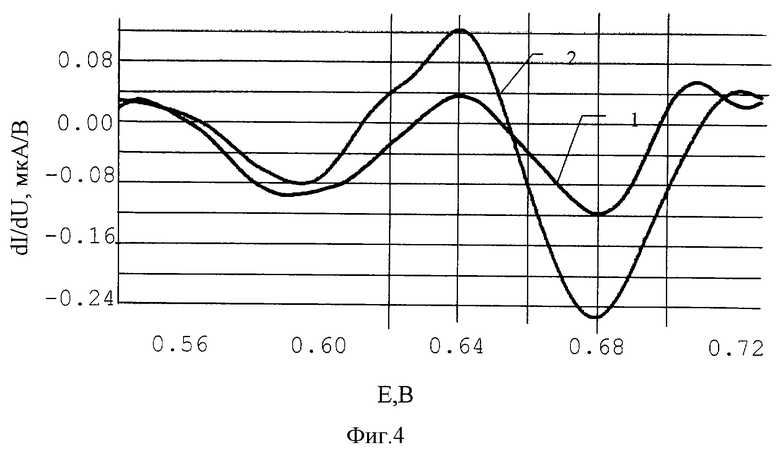
На фиг.4 представлены дифференциально-импульсные (DP) вольтамперограммы ртути.
На фиг.5 приведены хроноамперограммы мочевины.
На фиг.6 приведены производные вольтамперограммы свинца и кадмия.
На фиг. 7 приведены дифференциально-импульсные (DP) вольтамперограммы свинца.
На фиг.8 приведены производные вольтамперограммы цинка.
Способ иллюстрируется следующими примерами.
Пример 1. На длинную гибкую пленку полиэстра толщиной 0.1 мм наносят графитсодержащие чернила в виде сплошной дорожки. После отверждения нерабочие зоны электрода изолируют в соответствии с фиг.1. На рабочие зоны наносят последовательно микропипеткой 5 мкл 0.2% водного раствора диэтилдитиокарбомината натрия и 5 мкл водно-спиртового раствора, содержащего 1 г/л ионов ртути (II). Дают высохнуть при комнатной температуре. В результате последовательного нанесения двух растворов на поверхности электрода образуется мелкокристаллическое водно-нерастворимое соединение - диэтилдитиокарбаминат ртути.
Анализ раствора проводят методом анодной инверсионной вольтамперометрии в дифференциально-импульсном режиме. Электрод можно использовать для определения концентрации ионов меди, свинца, кадмия, цинка. После проведения 8-10 анализов электрод прокручивается устройством с целью замены рабочей поверхности.
Дифференциально-импульсные (DP) вольтамперограммы свинца и кадмия приведены на фиг.3, где
1 - питьевая вода,
2 - питьевая вода + 2 мкг/л кадмия + 5 мкг/л свинца,
3 - питьевая вода + 4 мкг/л кадмия + 10 мкг/л свинца.
В результате анализа в питьевой воде найдено 1.73 мкг/л кадмия с ошибкой определения 2.59% и 5.27 мкг/л свинца с ошибкой определения 3.31%.
Пример 2. Готовят смесь графитового порошка 70% и эпоксидной смолы 30%. Смесь наносят на основу из стеклотекстолита толщиной 0.5 мм. После отверждения графито-эпоксидной пасты нерабочую зону электрода изолируют согласно фиг. 2. На рабочую зону, которая уже представляет собой графито-эпоксидную композицию, наносят последовательно микропипеткой 5 мкл 0.001% водного раствора пирролидиндитиокарбомината аммония и 5 мкл водно-спиртовый раствора, содержащего 0.1 г/л ионов золота (III). Дают высохнуть при комнатной температуре. В результате последовательного нанесения двух растворов на поверхности электрода образуется мелкокристаллическое воднонерастворимое соединение - пирролидиндитиокарбоминат золота. Поверхность модифицированного электрода перед анализом раствора дополнительно подвергают электрохимической подготовке. После чего проводят анализ раствора методом анодной инверсионной вольтамперометрии в дифференциальноимпульсном режиме. Электрод можно использовать для определения концентрации ионов меди, свинца, ртути, мышьяка, селена. Дифференциально-импульсные (DP) вольтамперограммы ртути приведены на фиг.4, где
1 - морская вода
2 - речная вода + 0.02 мкг/л ртути.
В результате анализа в морской воде найдено 0.016 мкг/л ртути.
Пример 3. Готовят смесь графитового порошка 70% и эпоксидной смолы 30%. Пасту наносят на основу из мелинекса. После отверждения графито-эпоксидной пасты нерабочую зону изолируют согласно фиг.2. На рабочую зону, которая уже представляет собой графито-эпоксидную композицию, наносят последовательно микропипеткой 5 мкл 0.14% водного раствора гидроксида натрия и 5 мкл 0.7% спиртового раствора никеля (II), содержащего 0.3% ионообменного полимера Nafion. Дают высохнуть при комнатной температуре. В результате последовательного нанесения двух растворов на поверхности электрода образуется мелкокристаллическое воднонерастворимое соединение - гидроксид никеля (II) (Ni(ОН)2).
Поверхность модифицированного электрода перед анализом раствора дополнительно подвергают электрохимической подготовке. После чего проводят анализ раствора методом хроноамперометрии.
Электрод можно использовать для определения концентрации мочевины, органических тиолов.
На фиг.5 приведены хроноамперограммы мочевины, где
1 - фоновый раствор;
2 - 10-4 М мочевины;
3 - 2•10-4 М мочевины.
Пример 4. Готовят модифицированные графитовые чернила. Для этого берут метилизобутилкетон (МИБК), растворяют в нем пирролидиндитиокарбамат аммония. 0.1% раствор пирролидиндитиокарбамата аммония в МИБК смешивают с водным раствором, содержащим 10 г/л ионов ртути, и встряхивают в течение 10 минут. После этого отделяют органическую фазу и смешивают ее с графитсодержащими чернилами. Дают испариться растворителю и полученные модифицированные чернила наносят на полиэстер толщиной 0.25 мм. Отверждают 1 час при температуре 70oС. После этого нерабочую зону электрода изолируют согласно фиг.2. В результате этих операций рабочая зона содержит мелкокристаллическое воднонерастворимое соединение - пирролидиндитиокарбаминат ртути.
Поверхность модифицированного электрода перед анализом раствора дополнительно подвергают электрохимической подготовке.
После чего проводят анализ раствора методом анодной инверсионной вольтамперометрии. Электрод можно использовать для определения концентрации ионов меди, свинца, кадмия, цинка.
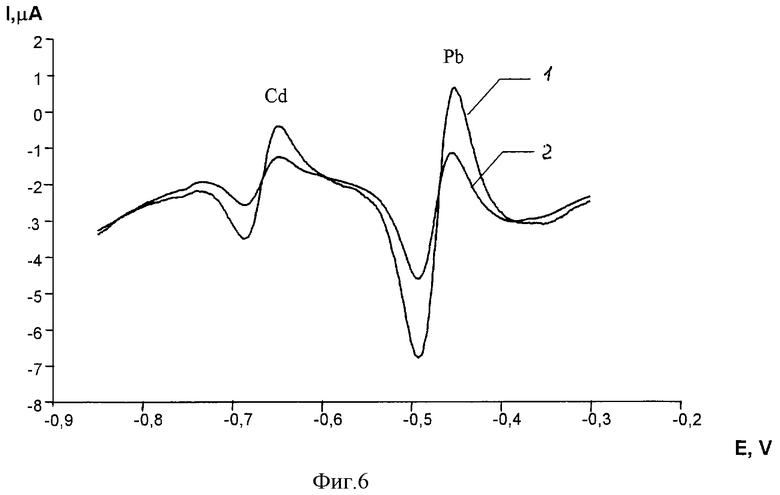
Производные вольтамперограммы свинца и кадмия приведены на фиг.6, где
1 - питьевая вода + 2 мкг/л кадмия + 4 мкг/л свинца
2 - питьевая вода + 4 мкг/л кадмия + 8 мкг/л свинца.
В результате анализа в питьевой воде найдено 1.4 мкг/л и 3.2 мкг/л свинца.
Пример 5. Готовят модифицированные графитовые чернила. Для этого берут метилизобутилкетон (МИБК), растворяют в нем пирролидиндитиокарбамат аммония. 0.1% раствор пирролидиндитиокарбамата аммония в МИБК смешивают с водным раствором, содержащим 10 г/л ионов цинка, и встряхивают в течение 10 минут. После этого отделяют органическую фазу и смешивают ее с графитсодержащими чернилами. Дают испариться растворителю и полученные модифицированные чернила наносят на полимерную основу из рулонного электроизоляционного материала толщиной 0.35 мм. Отверждают 1 час при температуре 70oС. После этого нерабочую зону электрода изолируют согласно фиг.2. В результате этих операций рабочая зона содержит мелкокристаллическое водно-нерастворимое соединение - пирролидиндитиокарбаминат цинка.
После чего проводят анализ раствора методом анодной инверсионной вольтамперометрии. Электрод можно использовать для определения концентрации ионов меди, свинца, кадмия.
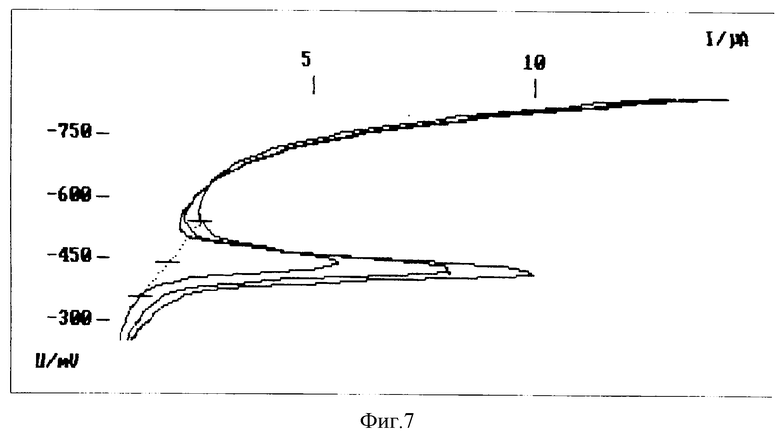
Дифференциально-импульсные (DP) вольтамперограммы свинца приведены на фиг.7, где
1 - питьевая вода;
2 - питьевая вода + 2 мкг/л свинца;
3 - питьевая вода + 4 мкг/л свинца.
В результате анализа в питьевой воде найдено 2.68 мкг/л свинца с ошибкой 14,5%.
Пример 6. Готовят модифицированные графитовые чернила. Для этого готовят 10% раствор 1-(о-хлорфенил)-3-фенил-5-(6-метил-4-оксо-пиримидинил-2)формазана в хлороформе и смешивают его с графитсодержащими чернилами. Полученные модифицированные чернила наносят на полимерную основу из гибкого целлулоида толщиной 0.1 мм, дают испариться растворителю, затем отверждают 1 час при температуре 70oС. После этого нерабочую зону электрода изолируют согласно фиг.2. В результате этих операций рабочая зона содержит мелкокристаллическое водно-нерастворимое соединение - 1-(о-хлорфенил)-3-фенил-5-(6-метил-4-оксо-пиримидинил-2)формазан.
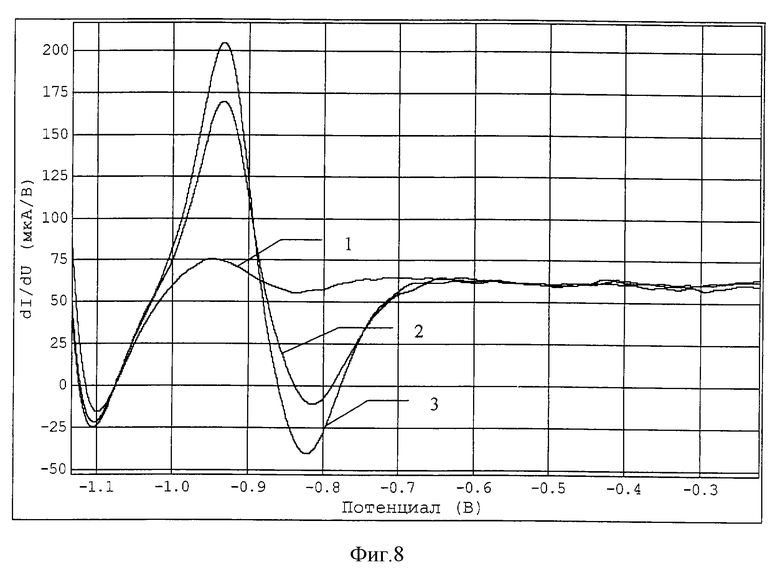
После чего проводят анализ раствора методом анодной инверсионной вольтамперометрии. Электрод можно использовать для определения концентрации ионов меди, свинца, кадмия, цинка. Производные вольтамперограммы цинка приведены на фиг.8, где
1 - фоновый раствор;
1 - фоновый раствор + водопроводная вода;
2 - фоновый раствор + водопроводная вода + 20 мкг/л цинка.
В результате анализа в водопроводной воде найдено 399 мкг/л цинка.
Преимущество заявляемого способа от известных состоит в том, что изготовленный таким способом электрод позволяет использовать его в автоматизированных системах анализа водных и других сред и упростить автоматизацию анализа.
Использование различных способов иммобилизации модификатора на поверхности или в объеме электрода позволяет расширить спектр определяемых элементов, а также обеспечить экологическую безопасность отработанных растворов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ИЗГОТОВЛЕНИЯ МОДИФИЦИРОВАННОГО ЭЛЕКТРОДА ДЛЯ ИНВЕРСИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО МЕТОДА ОПРЕДЕЛЕНИЯ СЛЕДОВ ТЯЖЕЛЫХ И ТОКСИЧНЫХ МЕТАЛЛОВ | 1997 |
|
RU2124720C1 |
| УСТРОЙСТВО ДЛЯ ЭЛЕКТРОХИМИЧЕСКИХ ИЗМЕРЕНИЙ | 2001 |
|
RU2192002C1 |
| УСТРОЙСТВО ДЛЯ ЭЛЕКТРОХИМИЧЕСКИХ ИЗМЕРЕНИЙ | 1998 |
|
RU2150108C1 |
| СПОСОБ ЭЛЕКТРОХИМИЧЕСКОГО ОПРЕДЕЛЕНИЯ СПЕЦИФИЧЕСКИХ АНТИТЕЛ В СЫВОРОТКЕ КРОВИ С ПОМОЩЬЮ БЕЛКОВ, МЕЧЕННЫХ МЕТАЛЛОМ | 2002 |
|
RU2249217C2 |
| СПОСОБ ИЗГОТОВЛЕНИЯ МОДИФИЦИРОВАННОГО ЭЛЕКТРОДА ДЛЯ ЭЛЕКТРОХИМИЧЕСКОГО АНАЛИЗА (ВАРИАНТЫ) | 2012 |
|
RU2507512C2 |
| СПОСОБ ИЗГОТОВЛЕНИЯ МОДИФИЦИРОВАННОГО ЭЛЕКТРОДА ДЛЯ ОДНОВРЕМЕННОГО ИНВЕРСИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ СЛЕДОВ ТЯЖЕЛЫХ МЕТАЛЛОВ И ИОДИД-ИОНОВ | 2003 |
|
RU2237888C1 |
| ЭЛЕКТРОХИМИЧЕСКИЙ ДАТЧИК | 1999 |
|
RU2166752C1 |
| ЭКСТРАКЦИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ ОПРЕДЕЛЕНИЯ ЦИНКА, КАДМИЯ, СВИНЦА И МЕДИ | 2011 |
|
RU2476853C1 |
| ЭКСТРАКЦИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ ОПРЕДЕЛЕНИЯ ЦИНКА, КАДМИЯ, СВИНЦА И МЕДИ В ПРИРОДНЫХ ВОДАХ | 2008 |
|
RU2383014C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОКСИДАНТНОЙ/АНТИОКСИДАНТНОЙ АКТИВНОСТИ РАСТВОРОВ | 2002 |
|
RU2235998C2 |



Изобретение относится к области электрохимических методов анализа. Сущность: способ изготовления включает изготовление основы из полимерного материала и нанесение на основу графитсодержащего материала, модифицированного химическими реагентами для придания ему водно-нерастворимой мелкокристаллической структуры. Предложены 2 варианта способа. Технический результат изобретения заключается в упрощении технологии изготовления электродов, в расширении спектра определяемых веществ, в том числе большого числа ионов металлов, анионов и ряда органических веществ, а также улучшению метрологических характеристик электродов, увеличению продолжительности жизни и срока хранения, расширению возможностей использования. 2 с. и 3 з.п. ф-лы, 8 ил.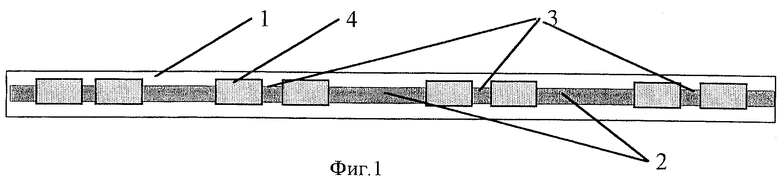
| СПОСОБ ИЗГОТОВЛЕНИЯ МОДИФИЦИРОВАННОГО ЭЛЕКТРОДА ДЛЯ ИНВЕРСИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО МЕТОДА ОПРЕДЕЛЕНИЯ СЛЕДОВ ТЯЖЕЛЫХ И ТОКСИЧНЫХ МЕТАЛЛОВ | 1997 |
|
RU2124720C1 |
| US 6019880 A, 01.02.2000 | |||
| US 5002651 A, 26.03.1991. | |||

