Изобретение относится к области аналитической химии, в частности может быть использовано для одновременного определения неорганических веществ методом инверсионной вольтамперометрии.
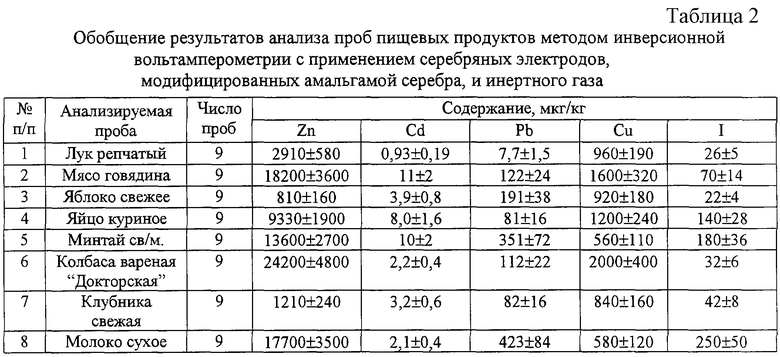
Определение иодид-ионов методом инверсионной вольтамперометрии проводилось в основном на ртутных и серебряных электродах. Сущность метода заключается в предварительном электроконцентрировании иодид-ионов на поверхности электрода при постоянном потенциале в виде малорастворимого соединения (иодида ртути или иодида серебра) и последующем электрорастворении осадка с поверхности электрода. Результаты работ по этому вопросу обобщены [Гунцов А.В., Захаров М.С., Захарова О.М., Ларина Н.С. Катодная инверсионная вольтамперометрия галогенид-ионов и некоторых органических веществ. - Тюмень: ТюмГНГУ, 2001, 95 с.]. Обеспечить удовлетворительную воспроизводимость результатов на серебряных электродах удается только при сложной регенерации поверхности, которая обычно включает стадии механической, химической и электрохимической обработки. Недостатком ртутных электродов является токсичность ртути.
В одной из первых работ, посвященных определению иодид-ионов на серебряных электродах [Shain I., Perone S.P. Anal. Chem. 33, 325 (1961)], достигнут достаточно низкий предел обнаружения - 4·10-8 M. При этом в качестве фонового электролита использовали 0,1 М раствор ацетата натрия и 0,1 М раствор муравьиной кислоты, накопление проводили при потенциале +0,18 В. В более поздней работе [Райкова Н.С., Захаров М.С., Гунцов А.В. Определение галогенид-ионов при их совместном присутствии в растворе методом инверсионной вольтамперометрии с серебряньм электродом// Журн. аналит. химии, 1988, Т.43, №4, С.666-672] при определении иодид-ионов на серебряном электроде на фоне 0,1 М раствора ацетата натрия предел обнаружения составил 4·10-6 M. При этом для получения воспроизводимых результатов перед проведением измерений индикаторный электрод шлифовали оксидом хрома и обезжиривали азотной кислотой.
Предложено определение иодид-ионов на серебряных электродах, обновляемых срезанием поверхностного слоя субмикронной толщины на фоне 0,05 М серной кислоты при потенциале электролиза -0,1 В. Интервал определяемых концентраций составил 10-1-10-4 М [Клетеник Ю.Б., Тарасова В.А., Александрова Т.П., Скворцова Л.И., Кирюшов В.И. Анализ питьевой воды методом инверсионной вольтамперометрии на обновляемых твердых электродах. Опыт и совершенствование. //Химия в интересах устойчивого развития. - 1997, Т.5, №4, С.401-406].
Описано определение иодид-ионов методом инверсионной вольтамперометрии в минеральных водах на стационарном ртутном электроде на фонах 0,06-0,10 М водных растворов нитрата калия [Хелашвили Н.В. Определение иодидов в минеральных водах методом инверсионной вольтамперометрии со стационарным ртутным электродом // Сообщение АН СССР, 1972, Т.65, №3, С.609-612]. Нижний предел обнаружения иодид-иона составил 5·10-8M. Определению не мешает 5000-кратный избыток хлорид-ионов или 60-кратный избыток бромид-ионов.
Определение иодид-ионов предложено проводить, концентрируя его на графитовом электроде в виде малорастворимого соединения с трифенилметановыми красителями - кристаллическим фиолетовым, метиловым фиолетовым и малахитовым зеленым. [Брайнина Х.З., Чернышева Л.В. Определение концентрации иодид-ионов и йода в природных водах методом инверсионной вольтамперометрии твердых фаз//Гидрохимические материалы. - Л.: Гидрометеоиздат, 1975, Т.62, С.119-123]. Нижний предел обнаружения составляет 4·10-9 M при продолжительности концентрирования 30 минут.
Использование в качестве материала электрода для ИВА сплавов системы AgBr-Ag2S-As2S3 дает возможность определять иодид-ионы на уровне 1·10-7 М [Кустова О.В., Никоноров В.В., Москвин Л.Н. Инверсионно-вольтмаперометрическое определение галогенид-ионов с использованием датчиков на основе сплавов AgBr-Ag2S-As2S3//Журн. ана-лит. химии, 1994, Т.49, №6, С.648-650]. Однако при совместном присутствии нескольких галогенид-ионов получается единый результирующий аналитический сигнал с потенциалом, характерным для более тяжелого галогена. Для устранения мешающего влияния хлорид- и бромид-ионов необходим тщательный выбор потенциала электролиза.
Определение ионов цинка, кадмия, свинца и меди методом инверсионной вольтамперометрии широко распространено. В качестве индикаторного электрода чаще всего используют ртутно-пленочные электроды, которые готовят путем нанесения пленки ртути на инертную электропроводящую подложку. Используют химические или электрохимические способы нанесения ртути. Формирование пленки проводят предварительно или в режиме in situ - одновременно с электроосаждением определяемых элементов. Для этого в анализируемый раствор добавляют раствор ртути (II). В качестве материала подложки используют серебро, непористые модификации графита - стеклоуглерод, пирографит и углеситал, а также графит, пропитанный полимерными материалами с целью устранения пористости. Тяжелые металлы концентрируют на индикаторном электроде в виде амальгамы, после чего регистрируют анодный ток окисления металлов из амальгамы [Будников Г.К., Майстренко В.Н., Вяселев М.Р. Основы современного электрохимического анализа - М.: Мир: Бином Л3, 2003. - 592 с.].
Наиболее распространенные способы определения тяжелых металлов описаны в [ГОСТ Р 51301-99. Продукты пищевые и продовольственное сырье. Инверсионно-вольтампрометрические методы определения содержания токсичных элементов (кадмия, свинца, меди и цинка)], Цинк, кадмий, свинец и медь определяют на фоне муравьиной кислоты или солянокислого раствора хлорида натрия, применяя в качестве индикаторных электродов серебряные, графитсодержащие и вращающийся дисковый стеклоуглеродный электроды. Пленку ртути формируют предварительно при использовании серебряных электродов и в режиме in situ, добавляя в фоновый электролит раствор азотнокислой ртути - при использовании графитсодержащих или стеклоуглеродных электродов. Нижние пределы обнаружения цинка и меди в растворе подготовленной пробы составляют 0,0005 мг/л, кадмия и свинца 0,0001 мг/л. Недостатком применения ртутно-пленочных электродов является необходимость тщательной механической подготовки и регенерации их поверхности в процессе эксплуатации для обеспечения необходимых метрологических характеристик анализа.
Наиболее близким является способ изготовления модифицированного электрода для инверсионно-вольтамперометрического метода определения следов тяжелых и токсичных металлов, включающий нанесение модификатора на рабочую поверхность разового электрода, отличающийся тем, что берут разовый толстопленочный графитовый электрод с плоской поверхностью или гибкий электрод в виде графитовой нити, модифицированные методом адсорбции органического вещества, избирательного на конкретный металл, с последующим образованием нерастворимого соединения этого металла, полученного при нанесении на поверхность электрода органического вещества в виде водного раствора концентрации 0,04-0,2% и соли металла в виде водного раствора концентрации 0,2-10 г/л последовательно, причем поверхность модифицированного электрода перед анализом дополнительно подвергают электрохимической подготовке, заключающейся в чередовании наложения импульсов потенциалов в интервале -1,4...+1,3 В и постоянного потенциала -1,4...-0,8 В [Брайнина Х.З., Иванова А.В., Сараева С.Ю., Стожко Н.Ю., Малахова Н.А. Патент РФ №2124720, БИ №1, 1999]. Пределы обнаружения ионов металлов при использовании таких электродов составляют 0,1-2 мкг/л в зависимости от определяемого элемента. Однако известный способ имеет следующие недостатки: ограниченный срок службы электрода - 8-10 часов непрерывной работы, сложность изготовления электрода, необходимость дополнительной электрохимической подготовки, использование ограниченного числа вольтамперометрических анализаторов.
Задачами заявляемого изобретения являются: изготовление твердых электродов, не нуждающихся в механической и электрохимической регенерации поверхности, совместимых с большинством вольтамперометрических анализаторов; отказ от использования растворов ртути и металлической ртути при определении тяжелых металлов; повышение чувствительности определения иодид-ионов; совместное определение ионов цинка(II), кадмия(II), свинца(II), меди(II) и иодид-ионов из одного раствора, что позволит сократить время анализа.
Поставленные задачи достигаются модифицированием индикаторного электрода амальгамой серебра путем последовательного электрохимического нанесения ртути и серебра. Для этого рабочую поверхность электрода опускают на одну-две секунды в концентрированную азотную кислоту и хорошо промывают бидистиллированной водой. Проводят электролиз ртути на рабочую поверхность электрода из раствора ртути при постоянном токе 1-1,5 мА в течение 600-800 секунд при слабом перемешивании раствора. Промывают полученный ртутно-пленочный электрод бидистиллированной водой, опускают в раствор нитрата серебра и проводят электролиз при постоянном токе 0,8-1,0 мА и слабом перемешивании раствора до исчезновения металлического блеска рабочей поверхности электрода. Готовый электрод выдерживают в бидистиллированной воде не менее 12 часов, после чего хранят на воздухе. Электрод позволяет одновременно регистрировать анодные вольтамперограммы для измерения токов пиков цинка, кадмия, свинца и меди и катодные - для регистрации тока пика иодид-ионов, что позволяет проводить определение содержания катионов и анионов из одного раствора.
При проведении измерений в 10-12 мл анализируемого раствора добавляют 0,1-0,4 мл концентрированной муравьиной кислоты, проводят УФ-облучение полученного раствора при перемешивании или продувают раствор инертным газом в течение 120-300 секунд для дезактивации растворенного кислорода. Проводят электронакопление тяжелых металлов при потенциале накопления минус(1,4-1,5) В в течение 10-120 секунд при продувании раствора инертным газом или УФ-облучении. По окончании накопления отключают перемешивание и УФ-лампу или инертный газ, через 5 секунд регистрируют анодную вольтамперограмму в диапазоне потенциалов от минус 1,2 до плюс 0,1 В при постоянно-токовой или дифференциально-импульсной развертке потенциала со скоростью 80-100 мВ/с. По окончании анодной развертки потенциала проводят электронакопление иодид-ионов при потенциале минус 0,05 - плюс 0,05 В в течение 5-120 секунд при барботаже инертным газом или УФ-облучении и перемешивании раствора. По окончании накопления отключают перемешивание и УФ-лампу или инертный газ и через 2 секунды регистрируют катодную вольтамперограмму от плюс 0,1 до минус 1,0 В при скорости развертки 80-100 мВ/с в постоянно-токовом или дифференциально-импульсном режиме. В качестве электрода сравнения используют 1 М хлорсеребряный электрод, в качестве вспомогательного - стеклоуглеродный электрод. Измерение концентраций определяемых ионов проводят методом добавок аттестованных смесей. Нижние пределы обнаружения цинка и меди в анализируемом растворе составляют 0,5 мкг/л, кадмия и свинца - 0,05 мкг/л, иодид-ионов - 0,2 мкг/л (4·10-9 M) при постоянно-токовой форме поляризующего напряжения и 0,1 мкг/л (1,6·10-9 M) при дифференциально-импульсной. Для иодид-ионов это на порядок меньше предела обнаружения, достигнутого методом инверсионной вольтамперометрии и описанного в литературе. Определению иодид-ионов не мешают 500-кратный избыток цинка, кадмия, свинца, 200-кратный избыток меди, 20000-кратный избыток хлоридов и 2000-кратный избыток бромидов.
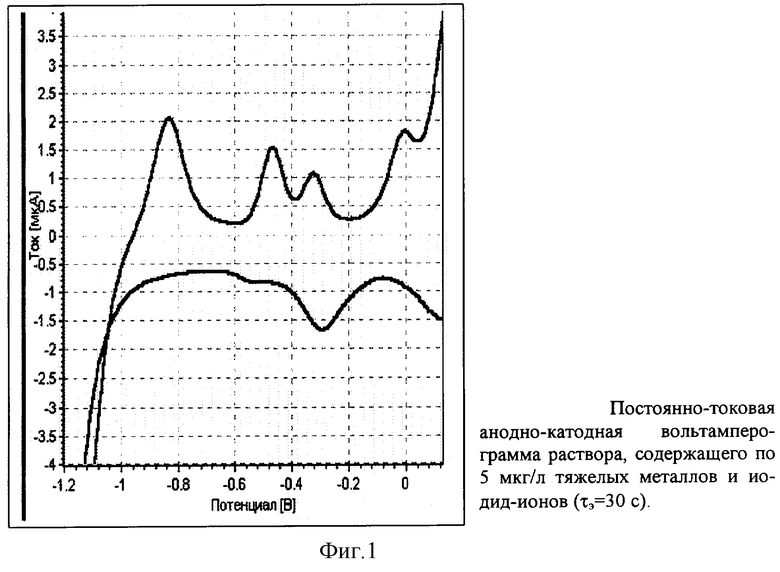
На фиг.1 представлена постоянно-токовая анодно-катодная вольтамперограмма раствора, содержащего по 5 мкг/л тяжелых металлов и иодид-ионов.
На фиг.2 представлена дифференциально-импульсная анодно-катодная вольтамперограмма раствора, содержащего по 2 мкг/л тяжелых металлов и иодид-ионов.
Изобретение характеризуется следующими примерами.
Пример 1. Определение цинка, кадмия, свинца, меди и йода в минеральной и питьевой воде с использованием серебряного электрода, модифицированного амальгамой серебра
Серебряную проволоку, запрессованную в полимерный стержень, опускают на одну-две секунды в концентрированную азотную кислоту и хорошо промывают бидистиллированной водой. Проводят электролиз ртути на серебряную проволоку из насыщенного раствора Hg2(NO3)2 при постоянном токе 1,5 мА в течение 600 секунд при слабом перемешивании раствора. Промывают полученный ртутно-пленочный электрод бидистиллированной водой, опускают в 0,01 М раствор нитрата серебра и проводят электролиз при постоянном токе 1,0 мА и слабом перемешивании раствора в течение 40-50 секунд до исчезновения металлического блеска рабочей поверхности электрода. Готовый электрод выдерживают в бидистиллированной воде не менее 12 часов, после чего хранят на воздухе.
В кварцевый стаканчик объемом 20-30 мл наливают 8-10 мл бидистиллированной воды, 0,2-0,3 мл концентрированной муравьиной кислоты и 1-3 мл анализируемой воды. Стаканчик, серебряный электрод, модифицированный амальгамой серебра, вспомогательный стеклоуглеродный электрод и хлорсеребряный электрод сравнения устанавливают в вольтамперометрический анализатор с встроенной УФ-лампой. Проводят УФ-облучение раствора в течение 300 секунд при перемешивании. Регистрируют анодную вольтамперограмму в постоянно-токовом режиме от минус 1,2 до плюс 0,1 В при условиях: потенциал электролиза Еэ=-1,5 В; время электролиза τэ=40-120 секунд; скорость развертки потенциала W=100 мВ/с. Проводят электронакопление иодид-ионов при потенциале -0,05 В в течение 5-40 секунд. Регистрируют катодную вольтамперограмму в постоянно-токовом режиме от плюс 0,1 В до минус 1,0 В со скоростью развертки потенциала 100 мВ/с. Содержание определяемых элементов оценивают методом добавок аттестованных смесей. Время анализа одной пробы не превышает 40 минут. Результаты анализа различных вод представлены в табл.1.
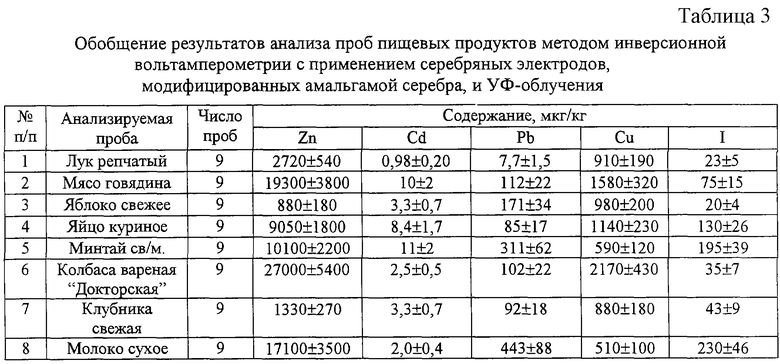
Пример 2. Определение цинка, кадмия, свинца, меди и йода в пищевых продуктах, овощах и фруктах с использованием серебряного электрода, модифицированного амальгамой серебра
Серебряную проволоку, запрессованную в полимерный стержень, опускают на одну-две секунды в концентрированную азотную кислоту и хорошо промывают бидистиллированной водой. Проводят электролиз ртути на серебряную проволоку из насыщенного раствора Hg2(NO3)2 при постоянном токе 1,5 мА в течение 600 секунд при слабом перемешивании раствора. Промывают полученный ртутно-пленочный электрод бидистиллированной водой, опускают в 0,01 М раствор нитрата серебра и проводят электролиз при постоянном токе 1,0 мА и слабом перемешивании раствора в течение 40-50 секунд до исчезновения металлического блеска рабочей поверхности электрода. Готовый электрод выдерживают в бидистиллированной воде не менее 12 часов, после чего хранят на воздухе.
Пробу навеской 0,2-0,5 г помещают в кварцевый стаканчик и добавляют 1 мл 10% раствора гидроксида калия. Стаканчик накрывают фильтровальной бумагой и дают постоять 30 минут. После чего тщательно перемешивают и высушивают при температуре 100-130°С. Стаканчик помещают в муфельную печь и выдерживают 15 минут при температуре 460-480°С. Стаканчики охлаждают, добавляют 1 мл 1 М калия азотнокислого и 1 мл бидистиллированной воды. Раствор упаривают досуха, постепенно повышая температуру от 100 до 350°С. Стаканчики вновь помещают в муфельную печь при температуре 460-480°С и выдерживают 15 минут. Стаканчик охлаждают, еще раз добавляют 0,5 см3 1 М раствора калия азотнокислого, упаривают, постепенно поднимая температуру от 100 до 350°С, и выдерживают в муфеле при 460-480°С в течение 15 минут. Полученная зола должна быть без угольных включений. Стаканчик охлаждают до комнатной температуры, добавляют 1 мл концентрированной муравьиной кислоты и 4 см3 бидистиллированной воды. Раствор тщательно перемешивают стеклянной палочкой до растворения золы.
В кварцевый стаканчик объемом 20-30 мл наливают 10-12 мл бидистиллированной воды, 0,2 мл концентрированной муравьиной кислоты и 0,5-1,0 мл раствора подготовленной пробы. Если удаление кислорода из раствора проводится с помощью инертного газа в полученный раствор добавляют 30-50 мг аскорбиновой кислоты. Стаканчик, серебряный электрод, модифицированный амальгамой серебра, вспомогательный стеклоуглеродный электрод и хлорсеребряный электрод сравнения устанавливают в вольтамперометрический анализатор. Полученный раствор подвергают УФ-облучению при перемешивании или продувают инертным газом в течение 120-300 секунд для дезактивации растворенного кислорода. Проводят электронакопление тяжелых металлов при потенциале накопления - 1,4 В в течение 10-60 секунд при барботаже раствора инертным газом или УФ-облучении. По окончании накопления отключают перемешивание и УФ-лампу или инертный газ, через 5 секунд регистрируют анодную вольтамперограмму в диапазоне потенциалов от минус 1,2 до плюс 0,1 В при дифференциально-импульсной развертке потенциала со скоростью 80 мВ/с. По окончании анодной развертки потенциала проводят электронакопление иодид-ионов при потенциале +0,05 В в течение 20-120 секунд при барботаже инертным газом или УФ-облучении и перемешивании раствора. По окончании накопления отключают перемешивание и УФ-лампу или инертный газ и через 2 секунды регистрируют катодную вольтамперограмму от плюс 0,1 до минус 1,0 В при скорости развертки 80 мВ/с в дифференциально-импульсном режиме. Содержание определяемых элементов оценивают методом добавок аттестованных смесей. Время анализа раствора подготовленной пробы не превышает 30 минут. Результаты анализа различных проб представлены в табл.2 и 3.
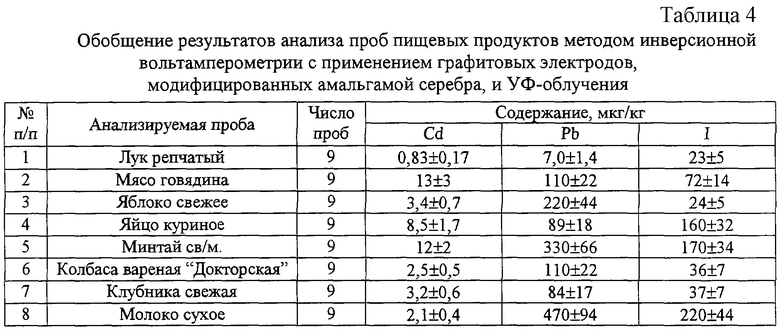
Пример 3. Определение кадмия, свинца и йода в пищевых продуктах, овощах и фруктах с использованием графитового электрода, модифицированного амальгамой серебра
Проводят электролиз ртути на рабочую поверхность графитового электрода, изготовленного из электропроводящей смеси полиэтилена с сажей по технологии “литье под давлением” (производство ООО НПП “Томьаналит”, Томск, Россия) из 0,1 г/л раствора нитрата одновалентной ртути при постоянном токе 1 мА в течение 800 секунд при слабом перемешивании раствора. Промывают электрод бидистиллированной водой, опускают в 0,01 М раствор нитрата серебра и проводят электролиз при постоянном токе 0,8 мА и слабом перемешивании раствора в течение 40-50 секунд до исчезновения металлического блеска рабочей поверхности электрода. Готовый электрод хранят на воздухе.
Пробу навеской 0,2-0,5 г помещают в кварцевый стаканчик и добавляют 1 мл 10% раствора гидроксида калия. Стаканчик накрывают фильтровальной бумагой и дают постоять 30 минут. После чего тщательно перемешивают и высушивают при температуре 100-130°С. Стаканчик помещают в муфельную печь и выдерживают 15 минут при температуре 460-480°С. Стаканчики охлаждают, добавляют 1 мл 1 М калия азотнокислого и 1 мл бидистиллированной воды. Раствор упаривают досуха, постепенно повышая температуру от 100 до 350°С. Стаканчики вновь помещают в муфельную печь при температуре 460-480°С и выдержавают 15 минут. Стаканчик охлаждают, еще раз добавляют 0,5 см3 1 М раствора калия азотнокислого, упаривают, постепенно поднимая температуру от 100 до 350°С, и выдерживают в муфеле при 460-480°С в течение 15 минут. Полученная зола должна быть без угольных включений. Стаканчик охлаждают до комнатной температуры, добавляют 1 мл концентрированной муравьиной кислоты и 4 см3 бидистиллированной воды. Раствор тщательно перемешивают стеклянной палочкой до растворения золы.
В кварцевый стаканчик объемом 20-30 мл наливают 10-12 мл бидистиллированной воды, 0,2 мл концентрированной муравьиной кислоты и 0,5-1,0 мл раствора подготовленной пробы. Если удаление кислорода из раствора проводится с помощью инертного газа, в полученный раствор добавляют 30-50 мг аскорбиновой кислоты. Стаканчик, серебряный электрод, модифицированный амальгамой серебра, вспомогательный стеклоуглеродный электрод и хлорсеребряный электрод сравнения устанавливают в вольтамперометрический анализатор. Полученный раствор подвергают УФ-облучению при перемешивании или продувают инертным газом в течение 120-300 секунд для дезактивации растворенного кислорода. Проводят электронакопление тяжелых металлов при потенциале накопления - 1,4 В в течение 30-90 секунд при барботаже раствора инертным газом или УФ-облучении. По окончании накопления отключают перемешивание и УФ-лампу или инертный газ, через 5 секунд регистрируют анодную вольтамперограмму в диапазоне потенциалов от минус 1,2 до плюс 0,1 В при дифференциально-импульсной развертке потенциала со скоростью 80 мВ/с. По окончании анодной развертки потенциала проводят электронакопление иодид-ионов при потенциале +0,05 В в течение 40-120 секунд при барботаже инертным газом или УФ-облучении и перемешивании раствора. По окончании накопления отключают перемешивание и УФ-лампу или инертный газ и через 2 секунды регистрируют катодную вольтамперограмму от плюс 0,1 до минус 1,0 В при скорости развертки 80 мВ/с в дифференциально-импульсном режиме. Содержание определяемых элементов оценивают методом добавок аттестованных смесей. Время анализа раствора подготовленной пробы не превышает 30 минут. Результаты анализа различных проб представлены в табл.4.
В примере 1 для дезактивация мешающего влияния кислорода пробу подвергают УФ-облучению в течение 120 секунд. При этом происходит одновременное восстановление иодат-ионов до иодид-ионов и устранение мешающего влияния растворенных органических веществ, адсорбирующихся на поверхности электрода. В настоящее время в патентной и научно-технической литературе не описан инверсионно-вольтамперометрический способ определения йода в воде с применением УФ-облучения для дезактивации растворенного кислорода и устранения мешающего влияния растворенных органических веществ.
В примерах 2 и 3 описан анализ раствора минерализованной пробы, поэтому для удаления кислорода из анализируемого раствора можно применять и УФ-облучение, и инертный газ. В случае барботажа раствора инертным газом для восстановления иодат-ионов до иодид-ионов в анализируемый раствор добавляют аскорбиновую кислоту. Как видно из табл.2 и 3, применение различных способов удаления кислорода не оказывает существенного влияния на результаты анализа.
В примерах 1 и 2 в качестве подложки используют серебро, в примере 3 - графит. Результаты, полученные с использованием электродов с различной подложкой и представленные в табл.3 и 4, достаточно близки. Это служит подтверждением возможности определения следов тяжелых металлов и иодид-ионов методом инверсионной вольтамперометрии с применением электродов, модифицированных амальгамой серебра.
Преимущество заявляемого способа от известных заключается в том, что использование предложенного модифицированного электрода позволяет проводить измерения без регенерации поверхности индикаторного электрода, снизить предел обнаружения иодид-ионов, уменьшить время анализа одной пробы за счет одновременного определения содержания катионов и анионов.

| название | год | авторы | номер документа |
|---|---|---|---|
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ ОДНОВРЕМЕННОГО ОПРЕДЕЛЕНИЯ СЕЛЕНА И ЙОДА | 2009 |
|
RU2415411C1 |
| СПОСОБ ОДНОВРЕМЕННОГО ОПРЕДЕЛЕНИЯ ИОДИД- И ИОДАТ-ИОНОВ МЕТОДОМ ВОЛЬТАМПЕРОМЕТРИИ | 2004 |
|
RU2257570C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЙОДА МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2011 |
|
RU2459199C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ НИКЕЛЯ МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ НА ОРГАНО-МОДИФИЦИРОВАННОМ ЭЛЕКТРОДЕ | 2012 |
|
RU2504761C1 |
| Экстракционно-вольтамперометрический способ определения ионов цинка, кадмия, свинца и меди в поверхностных водах | 2018 |
|
RU2680075C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СУРЬМЫ, ВИСМУТА, МЕДИ В ВОДНЫХ РАСТВОРАХ МЕТОДОМ АНОДНО-КАТОДНОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2010 |
|
RU2419786C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ СМЕСИ АФЛАТОКСИНОВ B1, B2, G1, G2 МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2015 |
|
RU2592049C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ВИСМУТА В РУДАХ И РУДНЫХ КОНЦЕНТРАТАХ МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2008 |
|
RU2367937C1 |
| Инверсионный вольтамперометрический способ определения тиоглюкозы в водных растворах | 1989 |
|
SU1670567A1 |
| Способ определения иодид-ионов катодной вольтамперометрией | 2016 |
|
RU2645003C2 |

Изобретение относится к области аналитической химии, в частности может быть использовано для одновременного определения неорганических веществ методом инверсионной вольтамперометрии. Сущность: рабочую поверхность индикаторного электрода модифицируют амальгамой серебра путем последовательного электрохимического нанесения ртути и серебра. Для этого проводят электролиз ртути на рабочую поверхность электрода из раствора ртути при постоянном токе 1-1,5 мА в течение 600-800 секунд при слабом перемешивании раствора. Промывают полученный ртутно-пленочный электрод бидистиллированной водой, опускают в раствор нитрата серебра и проводят электролиз при постоянном токе 0,8-1,0 мА и слабом перемешивании раствора до исчезновения металлического блеска рабочей поверхности электрода. Технический результат изобретения: повышение чувствительности, уменьшение времени анализа одной пробы за счет одновременного определения содержания катионов и анионов. 4 табл., 2 ил.
Способ изготовления модифицированного электрода для одновременного инверсионно-вольтамперометрического определения следов тяжелых металлов и иодид-ионов, включающий нанесение модификатора на рабочую поверхность электрода, отличающийся тем, что последовательно проводят электрохимическое восстановление ртути из раствора при постоянном токе 1-1,5 мА в течение 600-800 с, затем электрод опускают в раствор нитрата серебра и проводят электролиз при постоянном токе 0,8-1 мА и слабом перемешивании раствора до исчезновения металлического блеска рабочей поверхности электрода.
| СПОСОБ ИЗГОТОВЛЕНИЯ МОДИФИЦИРОВАННОГО ЭЛЕКТРОДА ДЛЯ ИНВЕРСИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО МЕТОДА ОПРЕДЕЛЕНИЯ СЛЕДОВ ТЯЖЕЛЫХ И ТОКСИЧНЫХ МЕТАЛЛОВ | 1997 |
|
RU2124720C1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ ЭЛЕКТРОДА ДЛЯ ВОЛЬТАМПЕРОМЕТРИЧЕСКИХ ИЗМЕРЕНИЙ | 1991 |
|
RU2039975C1 |
| US 6019880 А, 01.02.2000 | |||
| US 5002651 А, 26.03.1991. | |||

