Изобретение относится к способам низкотемпературного окисления органических соединений, в частности, к уничтожению химических боевых отравляющих веществ (OВ), продуктов их детоксикации, а также отходов химических производств и пестицидов.
В настоящее время существует серьезная необходимость в безопасном уничтожении химических боевых отравляющих веществ как в России, так и за рубежом. Технология уничтожения OВ должна обеспечивать безопасность для окружающей среды, процесс должен проводиться быстро и по возможности с меньшими затратами, причем должна быть достигнута оптимальная комбинация всех этих факторов. В решении проблемы уничтожения отравляющих веществ важное значение имеет вопрос о возможности дальнейшего использования получаемых продуктов. В основном боевые отравляющие вещества являются органическими соединениями, принадлежащими к разным классам химических веществ. Для их уничтожения применяют различные методы - термические, химические, биологические.
Известен способ уничтожения отравляющих веществ путем окисления их кислородом воздуха при температурах 1000-1400°С с последующей нейтрализацией абгазов гидроксидами, оксидами щелочных и щелочноземельных металлов (Российский химический журнал. 1993, т.37, №3, с.29-33. Российский химический журнал. 1995, т.39, №4, с.31-36). К недостаткам известного способа следует отнести большой расход топлива (до 4 т/т OВ), используемого для поддержания заданной температуры в зоне реакции и значительные объемы абгазов (до 100 000 м3/т OВ), подлежащих очистке щелочными растворами.
С целью повышения интенсивности процесса окисления и снижения объема абгазов, сбрасываемых в атмосферу, предложен способ окисления отравляющих веществ кислородом при температурах 2200-3200 К и подаче отравляющих веществ в зону реакции в виде раствора в керосине в соотношении 1:4, нейтрализацию абгазов предложено проводить 5-10%-ным раствором ацетата кальция (RU 2022590, А 62 D 3/00, 15.11.94). Расход раствора ацетата кальция на нейтрализацию кислых газов, содержащих хлористоводородную, фтористоводородную и фосфорную кислоты составил 60-65 т/т уничтожаемого OВ. Использование кислорода в качестве окислителя позволяет в 2-3 раза уменьшить объем абгазов, сбрасываемых в атмосферу.
К недостаткам способа следует отнести высокий уровень капитальных затрат на создание установки, обусловленный необходимостью эксплуатации оборудования в условиях коррозионно-агрессивных сред и высоких температур. Кроме того, дополнительные меры безопасности должны быть предусмотрены на периоды запуска и остановки процесса окисления, так как в этих ситуациях не исключается вероятность выброса аэрозоля OВ в керосине в систему нейтрализации абгазов. Использование ацетата кальция в качестве вещества, нейтрализующего кислые компоненты абгазов, по нашему мнению, также не вполне оправдано, так как проведение процесса нейтрализации при температурах 700-1000 К, т.е. в условиях пирогидролитического разложения ацетата кальция, неизбежно приводит к необходимости окисления уксусной кислоты, образующейся в процессе гидролиза, и дополнительному контролю за ее содержанием в абгазах.
С целью уменьшения расхода топлива и соответственно абгазов, сбрасываемых в атмосферу, предложен способ уничтожения высокотоксичных органических соединений путем пиролиза OВ без доступа кислородсодержащих газов с последующей очисткой полученного пиролизного газа твердым щелочным адсорбентом (RU 2113874, А 62 D 3/00, 27.06.98).
Способ предусматривает проведение процесса пиролиза в циклическом режиме с использованием неподвижной теплоаккумулирующей насадки, нагретой до температуры 660-800°С. Нагрев насадки осуществляется топочными газами с температурой 1000-1100°С, образующимися при сжигании предварительно очищенного пиролизного газа. Очистку газов процесса пиролиза проводят с помощью твердого щелочного адсорбента при 250-400°С и органического растворителя (солярового масла) при 35-50°С.
Несмотря на кажущуюся простоту технологического оборудования, предложенного для реализации метода, постоянные циклические колебания температуры в реакторе пиролиза от 650 до 1100°С (при подаче жидкого OВ в зону пиролиза перепад температур будет значительно больше) в условиях высокой коррозионной активности газов пиролиза, обусловленной присутствием кислот НСl, HF, Н3РO4, СН3РО(ОН)2 и водяного пара, могут существенно изменить прочностные характеристики конструкционных материалов реактора и срок его службы. Кроме того, наличие в реальных OВ продуктов осмоления и коррозии неизбежно приведет к забивке теплоаккумулирующей насадки реактора пиролиза, изменению ее характеристик по теплопроводности и удельной поверхности. Рекомендуемое авторами удаление из реактора пиролиза продуктов коррозии и углеродных шлаков с помощью очищенного пиролизного газа, либо продуктов его окисления, вряд ли способно исключить забивки насадки. Использование для очистки пиролизного газа кусковой негашеной извести, которая достаточно неоднородна по составу и адсорбционным свойствам, вряд ли можно считать удачным. В дополнение к перечисленному следует отметить пожаровзрывоопасность технологии при проведении в одном реакторе в циклическом режиме и процесса пиролиза и сжигания пиролизного газа.
Известен способ утилизации отравляющего вещества типа Vx, заключающийся в детоксикации OВ методом гидролиза, последующей нейтрализации образовавшихся реакционных масс 10-20%-ным водным раствором гидроксида калия в количестве, обеспечивающем мольное соотношение OВ:КОН, равное 1:2, и добавлением к смеси нитрата калия в количестве, достаточном для обеспечения 10%-ного избытка активного кислорода по отношению к сумме окисляемых в смеси элементов - фосфору, водороду, углероду, сере и азоту. Полученную смесь упаривают и нагревают до 390-400°С до начала самоокисления смеси (RU 2123368, А 62 D 3/00, 20.12.98, прототип).
В соответствии с приведенными примерами реализации способа расход гидроксида калия составит 3 т 20%-ного или 6 т 10%-ного раствора, а нитрата калия 5,9-8,9 т на 1 т OВ типа Vx. Достаточно мягкие условия практически полного окисления фосфорсодержащих эфиров по сравнению с сжиганием могут значительно снизить капитальные затраты на создание производства по уничтожению отравляющих веществ, однако при практической реализации способа возникают серьезные технологические сложности, обусловленные следующими факторами: большой расход нитрата калия как окислителя; сложное изменение фазового состава системы от раствора реакционных масс, суспензии нитрата калия, спека до солевого расплава с температурой 390-400°С и обусловленное этим обстоятельством сложное аппаратурное оформление процесса; большие затраты тепловой энергии на упарку нейтрализованных 10-20%-ным водным раствором гидроксида калия продуктов гидролиза Vx, так как необходимо упарить не менее 3 т воды на 1 т утилизируемого Vx; значительные количества 6-9 т/т Vx плава 75-80%-ного нитрата калия, подлежащего переработке в минеральные удобрения.
Наиболее близким техническим решением является способ уничтожения отравляющих веществ, продуктов их детоксикации и пестицидов по патенту RU 2156631 С1, кл. А 62 D 3/00, 27.09.2000.
Задачей изобретения является упрощение технологического процесса уничтожения отравляющих веществ и продуктов их детоксикации, а также снижение сырьевых и энергетических затрат.
Поставленная задача достигается путем смешения отравляющего вещества или продукта его детоксикации с раствором нитрата щелочноземельного металла в аминоспирте и нагревании полученной смеси до 300°С.
Предлагаемый способ позволяет уничтожать отравляющие вещества класса фосфорорганических OВ, таких как зарин, зоман, Vx, иприт и продукты их детоксикации, имеющие в своем составе аминоэфир соответствующего OВ, содержащие в своем составе, по меньшей мере, одну аминогруппу, а также пестициды, содержащие, по меньшей мере, одну аминогруппу. При этом обеспечивается степень деструкции исходных соединений более 99,9%.
Для осуществления способа вышеуказанное исходное соединение смешивают с раствором нитрата щелочноземельного металла в аминоспирте.
Мольное соотношение нитрат : аминоспирт зависит от содержания аминогрупп в исходном соединении и должно быть достаточным для окисления аминоэфира и углеродсодержащих компонентов исходного сырья. Предпочтительное мольное соотношение нитрата к аминоспирту составляет 2:1. Этот состав обеспечивает минимальное содержание оксидов азота в абгазах и существенно упрощает их последующую очистку от оксидов азота.
В качестве нитрата щелочноземельного металла используют Са(NО3)2, Mg(NO3)2 и др. Технологически удобно и экономически целесообразно применять промышленно выпускаемый тетрагидрат нитрата кальция или его водный раствор, предпочтительно, 50%-ный водный раствор Са(NО3)2. Может быть также использован безводный нитрат щелочноземельного металла или его спиртовый раствор. Опыты, представленные в таблице, проведены главным образом, с использованием 50%-ного водного раствора щелочноземельного металла.
В качестве аминоспирта могут быть использованы, например, моноэтаноламин, диэтаноламин, триэтаноламин, моноизопропаноламин, диизопропаноламин, триизопропаноламин, 3-амино-1-пропанол, 4-амино-1-бутанол, 2-амино-1-бутанол и другие, но экономически целесообразно использовать моноэтаноламин.
Для проведения процесса исходный раствор нитрата в аминоспирте используют в стехиометрическом количестве или в избытке, что зависит от вида OВ, его вязкости, загрязнения продуктами коррозии и степени полимеризации.
В предлагаемом способе OВ смешивают с заранее приготовленным раствором нитрата щелочноземельного металла в аминоспирте. Процесс можно также осуществлять и последовательным смешением OВ с аминоспиртом и нитратом.
Полученную смесь перемешивают при комнатной температуре в течение суток, а затем постепенно нагревают до 300°С.
При повышении температуры до 180°С протекает преимущественно реакция взаимодействия OВ с аминоспиртом, которая приводит к образованию аминоэфиров:
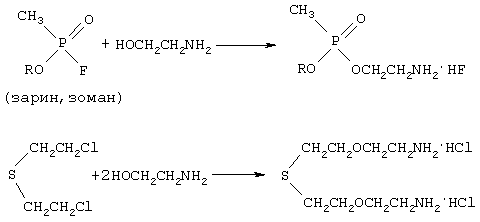
При дальнейшем повышении температуры до 300°С происходит окисление аминоэфиров нитратом щелочноземельного металла согласно следующей схеме:
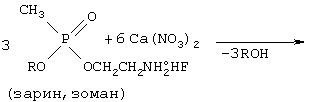
Ca5(PO4)3F+CaF2+7,5N2+12H2O+12H2O+7,5CO2+1,5CO
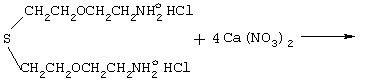
CaSO4+CaCl2+2CaCO3+2CO2+3CO+9H2O+CH4+5N2
Нагрев реакционной смеси до 300°С является достаточным для полного уничтожения OВ.
Полученные в процессе водно-спиртовые конденсата подвергают ректификации с выделением чистого спирта с выходом 50-60%. Водный остаток используют для приготовления исходного раствора нитрат - аминоспирт.
Твердые продукты реакции представляют собой фторфосфатный или хлорсульфатный шлам и соответствуют по своему составу природному минеральному сырью. ИК-спектроскопическим анализом шлама установлено, что содержание в нем соединений с С-Р и C-S связями составляет не более 1 ppm. Проведенная токсикологическая экспертиза показала, что фторфосфатный шлам имеет ЛД 50 более 10000 мг/кг и по ГОСТ 12.1.007-76 может быть отнесен к 4 классу опасности. Поэтому его наиболее целесообразно использовать в качестве фторсодержащего минерализатора в производстве строительных материалов.
Абгазы, содержащие N2, CO2, CO, N2O, CH4, SO2, после щелочной очистки подвергают доокислению кислородом с использованием тепла сгорания в стандартных системах водяного (парового) отопления.
Газообразные соединения, подлежащие выбросу, не отличаются по составу от содержащихся в атмосфере.
Таким образом, разработан универсальный, экологически безопасный метод уничтожения OВ, продуктов их детоксикации, а также пестицидов, базирующийся на их окислении дешевым и доступным окислителем с образованием нетоксичных газообразных, жидких и твердых продуктов.
Предлагаемый способ позволяет упростить технологический процесс уничтожения OВ по сравнению с прототипом, снизить расход химических реагентов до 5 т на 1 т OВ ( в прототипе 9-15 т на 1 т OВ), а также существенно уменьшить энергозатраты.
Данный способ прост в технологическом и аппаратурном оформлении и является перспективным для промышленной реализации.
Следующие примеры иллюстрируют предлагаемое изобретение, но не ограничивают его.
Пример 1.
В металлический контейнер емкостью 4,8 л, снабженный термопарой, технологическими штуцерами загружают 100 г изопропилметилфосфоната и 520 г раствора нитрата кальция в моноэтаноламине. Мольное соотношение Са(NО3)2 к моноэтаноламину равно 2:1. Полученную смесь подвергают перемешиванию на встряхивающей установке в течение 24 часов при комнатной температуре.
Затем контейнер помещают в печь, соединяют с сепаратором, холодильником, охлаждаемым водой, приемником конденсата и газгольдером. Реакционную массу нагревают, постепенно поднимая температуру до 180°С в течение 4-5 часов. Скорость нагрева реакционной массы начиная с температуры 100°С желательно чтобы не превышала 1 градус в минуту, так как происходит сильное вспенивание массы.
При достижении температуры 180°С из приемника извлекают 247,5 г конденсата следующего состава, мас.%: 16,1 изопропанола, 0,5 моноэтаноламина и 83,4 воды. Содержание в конденсате изопропанола и моноэтаноламина определяют хроматографическим методом, а воду методом Фишера. Наличие соединений с С-Р и Р-O связями в конденсате, определяемое ИК-спектрометрическим методом анализа, не выявлено. Из полученного конденсата ректификацией выделяют изопропанол.
Оставшийся в контейнере кубовый остаток нагревают до 300°С и выдерживают при этой температуре в течение часа для наиболее полного удаления газообразных продуктов.
После окончания реакции окисления из приемника извлекают 96,3 г конденсата следующего состава, мас.%: 0,9 изопропанола, 1,2 моноэтаноламина, 0,4 пиперидина, 2,1 аммиака и 95,4 воды. Содержание аминоэфиров метилфосфоновой кислоты и их солей в конденсате не превышает 0,3 мас.% в пересчете на P2O
Абгазы процесса окисления, прошедшие через сепаратор и холодильник, собирают в газгольдере. Получено 82 л абгазов состава, об.%: 43,4 N2; 42,0 СO2; 9,1 СО; 0,6 N2O и 4,9 CH4.
После охлаждения контейнера из него извлекают 151,2 г фторфосфатного шлама в виде спека серого цвета. Элементным анализом установлено, что содержание углерода в нем не превышает 0,4 мас.%, а содержание азота не превышает 1,1 мас.%. Рентгенофазовый анализ показал, что шлам включает до 10 мас.% фторида кальция и около 70% фторапатита.
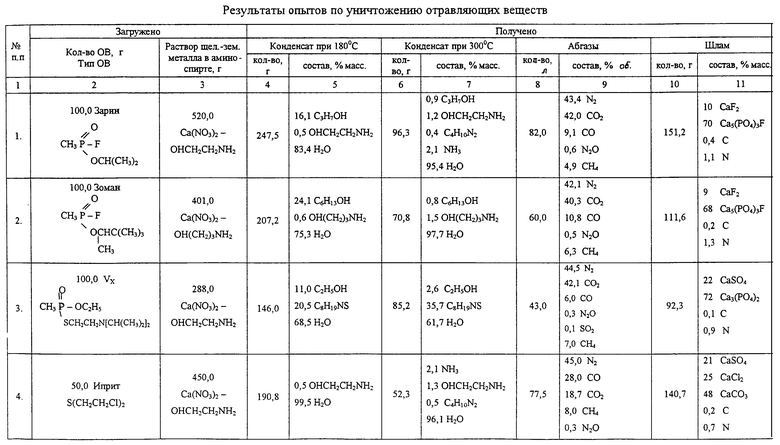
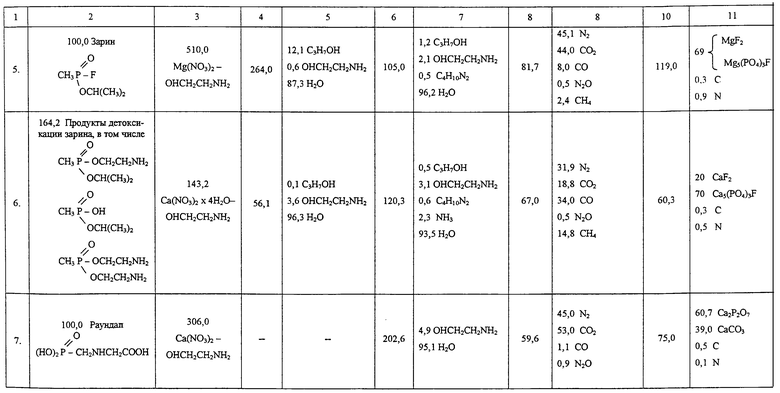
Последующие опыты проверены аналогично примеру 1. Условия и полученные результаты приведены в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ УНИЧТОЖЕНИЯ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ | 2004 |
|
RU2286822C2 |
| НЕПРЕРЫВНЫЙ СПОСОБ ДЕТОКСИКАЦИИ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ И ТОКСИЧНЫХ ХИМИЧЕСКИХ СОЕДИНЕНИЙ | 2001 |
|
RU2182505C1 |
| СПОСОБ ОБЕЗЗАРАЖИВАНИЯ ВНУТРЕННЕЙ ПОВЕРХНОСТИ ХИМИЧЕСКИХ БОЕПРИПАСОВ ОТ ОСТАТКОВ ФОСФОРОРГАНИЧЕСКИХ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ | 2005 |
|
RU2302891C2 |
| СПОСОБ УТИЛИЗАЦИИ ОТРАВЛЯЮЩЕГО ВЕЩЕСТВА ТИПА V | 1997 |
|
RU2123368C1 |
| СПОСОБ ФЕРМЕНТАТИВНОГО ГИДРОЛИЗА ФОСФОРОРГАНИЧЕСКИХ БОЕВЫХ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ | 2005 |
|
RU2296164C1 |
| СПОСОБ ОЧИСТКИ ГАЗООБРАЗНОГО ТРИФТОРИДА АЗОТА | 2002 |
|
RU2206499C1 |
| СПОСОБ ОБЕЗЗАРАЖИВАНИЯ ВНУТРЕННЕЙ ПОВЕРХНОСТИ ХИМИЧЕСКИХ БОЕПРИПАСОВ ОТ ОСТАТКОВ ФОСФОРОРГАНИЧЕСКИХ ОВ ТИПА "ЗАРИН" И "ЗОМАН" | 2001 |
|
RU2200046C1 |
| СПОСОБ УНИЧТОЖЕНИЯ О-ИЗОБУТИЛ-S-2-(N, N-ДИЭТИЛАМИНО)-ЭТИЛМЕТИЛТИОЛФОСФОНАТА (ВЕЩЕСТВА ТИПА Vx) | 2006 |
|
RU2355452C2 |
| ДЕГАЗИРУЮЩАЯ РЕЦЕПТУРА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2288016C1 |
| СПОСОБ ФОТОХИМИЧЕСКОГО ОКИСЛЕНИЯ О-ИЗОБУТИЛ-S-2-(N,N-ДИЭТИЛАМИНО)ЭТИЛМЕТИЛТИОФОСФОНАТА (ВЕЩЕСТВА ТИПА Vx) В ПРИСУТСТВИИ ПЕРХЛОРАТА 2,4,6-ТРИФЕНИЛСЕЛЕНОПИРИЛИЯ И ХЛОРОФОРМА | 2010 |
|
RU2494782C2 |
Изобретение относится к области уничтожения химических боевых отравляющих веществ, продуктов их детоксикации и пестицидов. Способ осуществляют путем смешения отравляющего вещества или продукта его детоксикации или пестицида с раствором нитрата щелочно-земельного металла в аминоспирте и нагревании полученной смеси до 300°С. В качестве продуктов детоксикации или пестицидов используют соединения, содержащие в своем составе, по меньшей мере, одну аминогруппу. Изобретение позволяет упростить технологический процесс уничтожения отравляющих веществ и продуктов их детоксикации, снизить сырьевые и энергетические затраты. 1 з.п.ф-лы, 1 табл.
| СПОСОБ ОБРАБОТКИ ХИМИЧЕСКИХ БОЕВЫХ ВЕЩЕСТВ | 1995 |
|
RU2156631C2 |
| СПОСОБ УТИЛИЗАЦИИ ОТРАВЛЯЮЩЕГО ВЕЩЕСТВА ТИПА V | 1997 |
|
RU2123368C1 |
| СПОСОБ ОКИСЛИТЕЛЬНОГО ЖИДКОФАЗНОГО ОБЕЗВРЕЖИВАНИЯ ПЕСТИЦИДОВ ФЕНОКСИЛЬНОГО РЯДА | 1999 |
|
RU2163158C1 |
| GB 1543685 А1, 04.04.1979 | |||
| DE 4301639 А1, 04.08.1984 | |||
| US 4497782 А1, 05.02.1985 | |||
| US 5663479 А1, 02.09.1997 | |||
| Устройство для укладки теста в хлебные формы люлечно-конвейерных печей | 1960 |
|
SU135043A1 |

