Область изобретения
Настоящее изобретение относится к способу получения 3S-3-амино-3-арилпропионовой кислоты и ее производных
Предпосылки изобретения
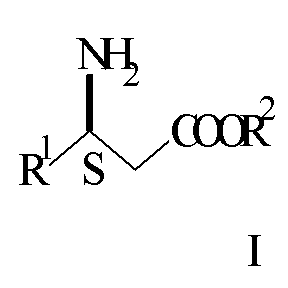
Производные 3S-3-амино-3-арилпропионовой кислоты формулы I
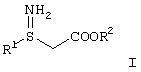
где
R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил, и R2 представляет водород, алкил или аралкил, или их кислотно-аддитивные соли могут использоваться в качестве промежуточных продуктов при синтезе соединений, описанных в WO 97/41102, включенной в. настоящее описание в качестве ссылки. Соединения, описанные в WO 97/41102, представляют собой антагонисты тромбоцитного фибриногенового рецептора (gp IIb/IIIa антагонист) и таким образом могут использоваться для лечения тромбоцит-опосредованных тромботических заболеваний, таких как артериальный и венозный тромбоз, острый инфаркт миокарда, повторное сужение сосудов после тромболитической терапии и ангиопластики, воспаление, нестабильная стенокардия и заболеваний, связанных с закупоркой сосудов.
Известные способы получения соединений формулы I включают асимметрическое присоединение по Михаэлю N-(триметилсилил)-(R)-фенетиламида лития к этил-3-пиридилакрилату с получением сложного этил-β-аминоэфира [Rico J.G., Lindmark R.J., Rogers Т.Е., Bovy P.P., J. Org. Chem., 1993, 58, 7948]. Данный способ приводит к неэффективному образованию амида лития и трудному удалению N-метилбензильной группы.
В J. Org. Chem., vol. 61, p. 2222 (1996) описан способ, в котором литиевый енолят этилацетата добавляют к энантиомерно чистому сульфинимину, продукт взаимодействия очищают хроматографией и снимают защитную группу в кислых условиях, получая сложный β-аминоэфир с более чем 90% ее (энантиомерным избытком). Необходимость хроматографии делает данный способ непривлекательным для крупномасштабного производства. Аналогично, в J. Org. Chem., vol. 64, p. 12 (1999) описан способ, в котором титановый енолят метилацетата добавляют к энантиомерно чистому трет-бутилсульфинимину, получая сложный β-аминоэфир с примерно 90% ее.
В WO 98/02410 описан способ стереоселективного присоединения реактива Реформатского, полученного из трет-бутилбромацетата, к энантиомерно чистому имину, полученному из 3-пиридинкарбоксальдегида и (R)-2-фенилглицинола. Окислительное расщепление N-(1-фенил-2-гидроксиэтильной) группы NaIO4 в этаноле с последующим кислотным гидролизом дает энантиомерно чистый трет-бутиловый сложный β-аминоэфир. Применение окисляющих агентов делает данный способ непривлекательным для крупномасштабного производства.
В WO 97/41102 описано ферментативное разделение (±)β-фенилацетамидо кислоты с использованием пенициллинамидазы с получением S-кислоты. Данный способ, в котором используют ферменты, не эффективен и не практичен для крупномасштабного производства.
Таким образом, существует необходимость в способе, который был бы совместим с потребностями крупномасштабного производства и при котором достигался бы приемлемый уровень чистоты и выхода.
Краткое изложение сущности изобретения
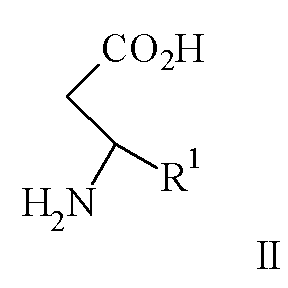
Изобретение относится к способу получения соединения формулы I, как описано выше, включающему взаимодействие соединения формулы II
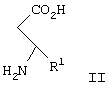
где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил,
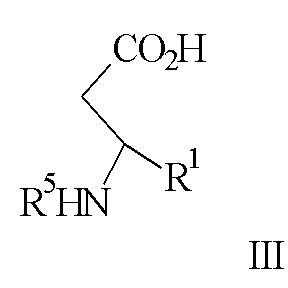
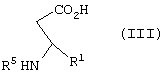
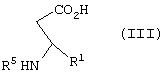
в диапазоне рН между примерно 7 и примерно 11, с образованием соединения формулы III
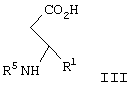
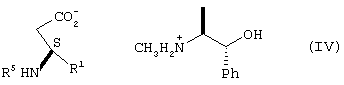
где R5 представляет N-трет-бутоксикарбонил, взаимодействие соединения формулы III с по крайней мере 0,5 эквивалентами (1R,2S)-(-)эфедрина в алкилацетатном растворителе с образованием соли формулы IV
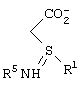
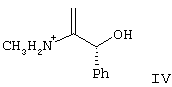
где Ph представляет фенил,
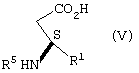
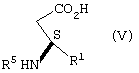
взаимодействие соли формулы IV с неорганическим основанием в воде с образованием карбоксилатной соли соединения формулы V, подкисление карбоксилатной соли соединения формулы V кислотой, рКа которой меньше чем или равно трем, до рН в диапазоне между примерно 3,5 и примерно 6,5, с образованием соединения формулы V

взаимодействие соединения формулы V при температуре меньше примерно 25°С с образованием соединения формулы I.
В другом аспекте, изобретение относится к новой кристаллической форме (3S)-3-[(трет-бутокси)карбонил]амино-3-[3’-пиридил]пропионовой кислоты, промежуточному соединению формулы Va

где Вос представляет собой (трет-бутокси)карбонил, обозначенному как форма 2 и охарактеризованному порошковыми рентгенограммами.
Подробное описание изобретения
Как использовано в данном описании, если не указано другого, “алкил” используется или отдельно, или как часть замещающей группы, будет включать линейные и разветвленные цепи, содержащие от 1 до 10 атомов углерода. Например, алкильные радикалы включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил и тому подобное.
Как использовано в данном описании, если не указано другого, “алкокси” будет означать простой эфирный радикал с атомом кислорода описанных выше линейных или разветвленных алкильных групп. Например, метокси, этокси, н-пропокси, втор-бутокси, трет-бутокси, н-гексилокси и тому подобное.
Как использовано в данном описании, если не указано другого, “арил” будет относиться к незамещенным ароматическим группам, таким как фенил, нафтил и тому подобно. Арильная группа может быть замещена одним или двумя заместителями. Подходящие заместители арильной группы выбирают независимо из группы, состоящей из галогена, алкила, алкокси, аралкила, -NR
Как использовано в данном описании, если не указано другого, “гетероарил” будет означать любую пяти- или шестичленную моноциклическую кольцевую структуру, содержащую по крайней мере один гетероатом, выбранный из О, N и S, или бициклическую кольцевую систему, где гетероарил сконденсирован с арильной группой. Примеры подходящих гетероарильных групп включают, но не ограничиваются ими, пирролил, пиридил, пиразинил, пиримидинил, пиразолил, пиридазинил, фуранил, пиранил, имидазолил, тиофенил, оксазолил, изотиазолил, изоксазолил, фуразанил, бензотиенил, бензофуранил, индолил, изоиндолил, индолизинил, индазолил, пуринил, изохинолил, хинолил, изотиазолил и тому подобно. Гетероарил может быть замещен одним или двумя заместителями. Подходящие заместители гетероарильной группы выбирают независимо из группы, состоящей из галогена, алкила, алкокси, аралкила, -NR
Предпочтительно гетероарил представляет пиридил. Предпочтительный гетероарил может быть замещен одним или двумя заместителями, как описано выше. Наиболее предпочтительно пиридил является незамещенным.
Как использовано в данном описании, если не указано другого, “аралкил” будет означать любую алкильную группу, замещенную арильной группой, такой как фенил, нафтил и тому подобное.
Как использовано в данном описании, если не указано другого, “галоген” будет означать хлор, бром, фтор и иод.
Как использовано в данном описании, если не указано другого, “кислота, рКа которой меньше чем или равно трем” включает монохлоруксусную, дихлоруксусную, трихлоруксусную, хлористоводородную, бромистоводородную, иодистоводородную, перхлорную, пикриновую, азотную, серную, фосфорную, метансульфоновую, толуолсульфоновую, трифторметансульфоновую, трифторуксусную, бисульфат калия, бисульфат натрия, лимонную кислоту и тому подобное.
Как использовано в данном описании, если не указано другого, “неорганическое основание” будет означать основание, имеющее одновалентный катионный компонент, такое как карбонат лития, карбонат натрия, карбонат калия, гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид тетрабутиламмония, гидроксид триметилбензиламмония и тому подобное.
Как использовано в данном описании, если не указано другого, “алкиловый спирт” будет означать гидроксильное производное вышеописанных линейных или разветвленных алкильных групп. Например, метанол, этанол, N-пропанол, изопропанол, изобутанол, трет-бутанол и тому подобное.
Как использовано в данном описании, обозначение ″S″ и ″R″ будет означать присутствие стереогенного центра, имеющего S или R конфигурацию.
В предпочтительном воплощении изобретения в соединении формулы I R1 представляет незамещенный фенил, замещенный фенил, незамещенный пиримидил, замещенный пиримидил, незамещенный пиридил, замещенный пиридил, незамещенный нафтил или замещенный нафтил. Подходящие заместители выбирают независимо из группы, состоящей из галогена, алкила, алкокси, аралкила, -NR
В воплощении настоящего изобретения, где соединение формулы I присутствует в виде кислотно-аддитивной соли, кислота представляет собой кислоту, рКа которой меньше чем или равно трем, как определено выше, отличной от бисульфата калия, бисульфата натрия и лимонной кислоты. Предпочтительно соединение формулы I представляет аддитивную соль хлористоводородной кислоты.
Настоящее изобретение относится к способу получения соединения формулы I
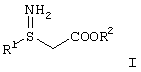
где R1 и R2 являются такими, как описано выше, или его кислотно-аддитивной соли, который включает взаимодействие соединения формулы II
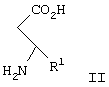
с ди-трет-бутил бикарбонатом с образованием соединения формулы III
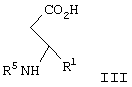
где R5 представляет N-трет-бутоксикарбонил,
взаимодействие соединения формулы III с образованием соли формулы IV
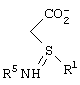
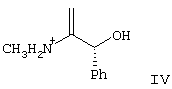
где Ph представляет фенил,
взаимодействие соли формулы IV с образованием соединения формулы V
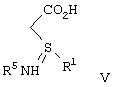
взаимодействие соединения формулы V с кислотой, рКа которой меньше чем или равно 3, отличной от бисульфата калия, бисульфата натрия и уксусной кислоты, с образованием соединения формулы I в виде кислоты или превращением ее, в случае необходимости, в эфир.
Согласно изобретению соединение формулы II, являющееся известным соединением или соединением, полученным известными способами [Profft V.E., Becker F.J., J. Prakt. Chem. 1965, 30(1-2), 18], подвергают взаимодействию с ди-трет-бутилдикарбонатом в органическом растворителе, таком как 1,4-диоксан, трет-бутанол или тетрагидрофуран, предпочтительно тетрагидрофуран, с добавлением водного раствора неорганического основания, как определено ранее, предпочтительно гидроксида натрия, при температуре в диапазоне от примерно 0°С до примерно 100°С, предпочтительно при температуре от примерно 0°С до примерно 35°С, при рН в диапазоне от примерно 7 до примерно 11, предпочтительно при рН между примерно 9,9 и примерно 10,2, с образованием соединения формулы III.
Предпочтительно целевое соединение формулы III выделяют, удаляя органический растворитель упариванием при пониженном давлении с последующим подкислением оставшегося водного раствора добавлением кислоты, имеющей рКа меньше чем или равное трем, как определено выше, предпочтительно бисульфатом натрия или лимонной кислотой, до рН в диапазоне от примерно 3,5 до примерно 6,5, предпочтительно до рН примерно 3,8, фильтрованием или экстракцией органическим растворителем, таким как хлористый метилен, 1,2-дихлорэтан, хлороформ, диоксан, толуол, алкилацетат, такой как этилацетат, или их смесями, предпочтительно этилацетатом, и необязательно удалением органического растворителя выпариванием при пониженном давлении.
Соединение формулы III подвергают взаимодействию с по крайней мере 0,5 эквивалента (1R,2S)-(-)эфедрина, предпочтительно 0,5-1,0 эквивалента (1R,2S)-(-)эфедрина, в алкилацетатном растворителе, предпочтительно в этилацетате, при температуре от примерно 25°С до примерно 78°С с образованием соли формулы IV.
Соль формулы IV подвергают взаимодействию с неорганическим основанием, как определено ранее, предпочтительно гидроксидом натрия, в воде с образованием карбоксилатной соли соединения формулы V (растворенной в водном растворе).
Целевое соединение формулы V выделяют удалением (1R,2S)-(-)эфедрина экстракцией органическим растворителем, который в значительной степени не смешивается с водой, таким как хлористый метилен, 1,2-дихлорэтан, алкилацетат или ароматический углеводород, такой как толуол, или кетон, такой как метил-изобутилкетон, с последующим подкислением оставшегося водного раствора добавлением кислоты, имеющей рК меньше чем или равное трем, как определено выше, предпочтительно бисульфата натрия или серной кислоты, до рН в диапазоне от примерно 3,5 до примерно 6,5, предпочтительно до рН примерно 3,8, и фильтрованием для получения соединения формулы V. Когда органический растворитель, который в значительной степени не смешивается с водой, представляет собой толуол, предпочтительно водный раствор, содержащий соединение формулы V, нагревают до температуры в диапазоне примерно 70-80°С перед экстракцией толуолом и охлаждают до примерно комнатной температуры после экстракции толуолом и перед подкислением.
Соединение формулы V подвергают взаимодействию с кислотой, рКа которой меньше чем или равно трем, как определено выше, отличной от бисульфата калия, бисульфата натрия и лимонной кислоты, предпочтительно с хлористоводородной кислотой, в C1-С10 алкиловом спирте, предпочтительно метаноле, при температуре меньше чем примерно 25°С с образованием соответствующего соединения формулы I, которое выделяют обычными способами, такими как фильтрование.
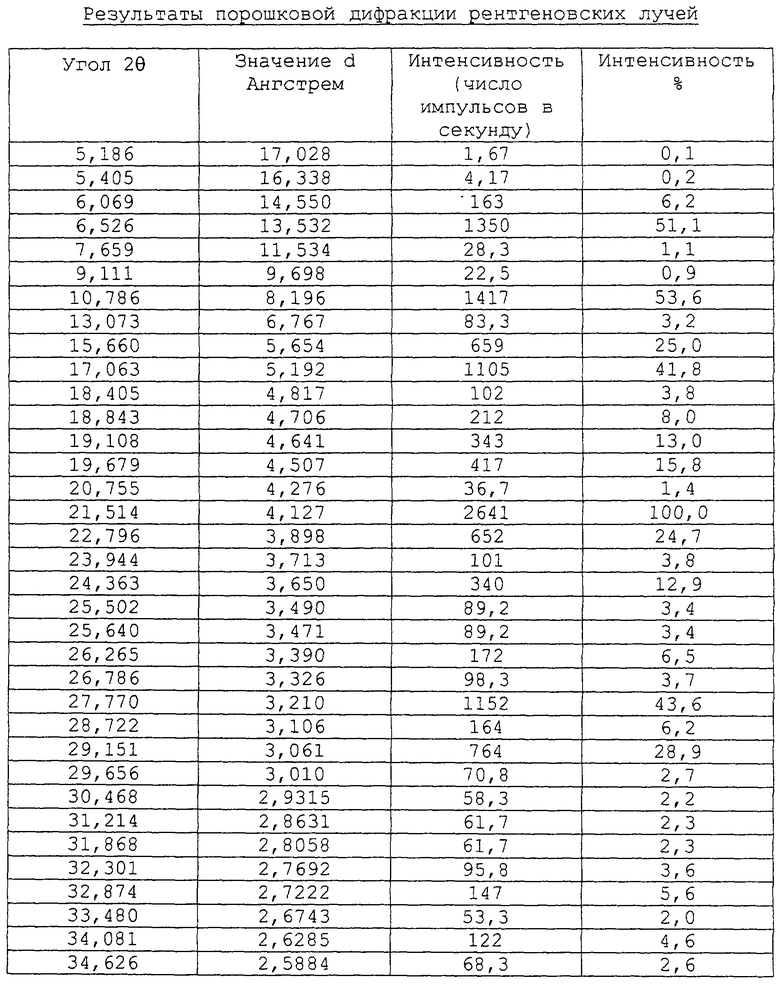
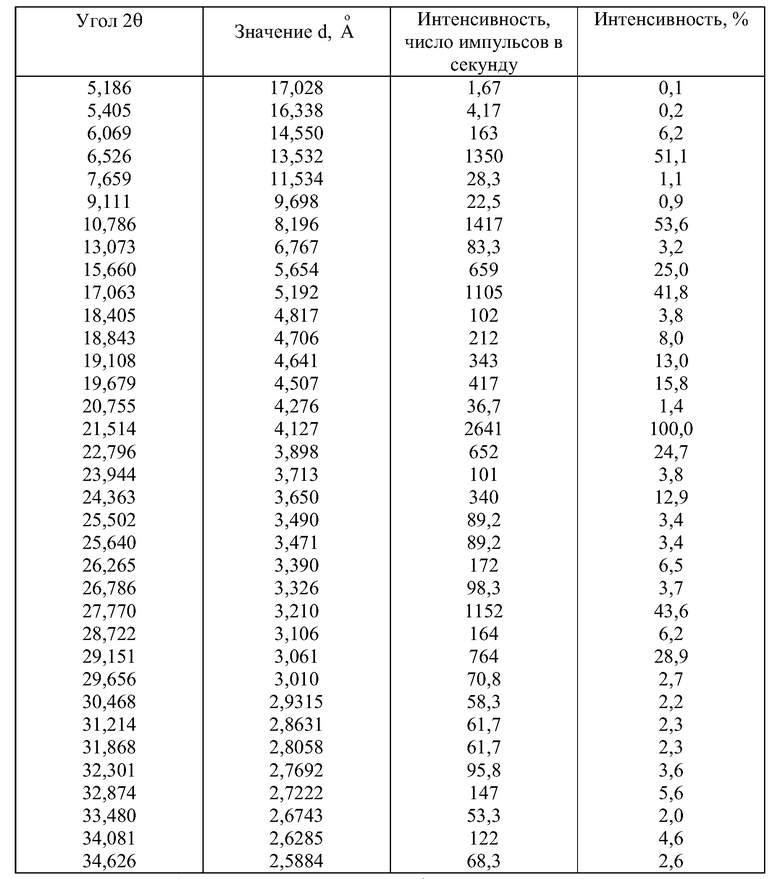
Форма 2 соединения формулы Va может быть охарактеризована его порошковой рентгенограммой, полученной с использованием дифрактометра Siemens D5000 и при следующих системных условиях:
- а) СuКα облучение, 35 мА, 40 кВ,
- b) Оптика,
1 мм щель, зеркала Goebel, 0,6 мм щель и вертикальные соллерные щели между трубкой и образцом,
LiF монохроматор между образцом и детектором,
- с) Скан от 5 до 35°2θ при размере шага 0,02 со скоростью 1°2θ/минуту,
- d) Нулевой фон держателя образца.
Результаты порошковой дифракции рентгеновских лучей приведены в таблице.
Следующие примеры более подробно описывают изобретение и предназначены только для иллюстрации изобретения и никоим образом не для его ограничения.
Пример 1
3-Амино-3-(3’-пиридил)пропионовая кислота
Часть А
В реакционную колбу загружали 300 г (264,3 мл) (2,8 моль) охлажденного 3-пиридинкарбоксальдегида и 60 мл абсолютного этанола. Температура реакции составила 13°С. При хорошем перемешивании добавляли 291,36 г (2,8 моль) малоновой кислоты в виде одной порции (добавление твердого вещества) в течение примерно 2 мин с последующим добавлением 90 мл этанола. Сразу же после такого добавления в течение примерно 10 мин добавили 323,73 г (4,2 моля) ацетата аммония (добавление твердого вещества) с последующим добавлением 250 мл этанола. Во время такого добавления реакционную смесь охлаждали до примерно 38°С. Ярко-оранжевую смесь нагревали и выдерживали при легком кипячении с обратным холодильником в течение 5 ч. Нагревающий кожух удаляли и смесь оставляли охлаждаться до температуры окружающей среды в течение ночи. Твердое вещество собирали фильтрованием с отсосом (50 мин) и промывали 400 мл метанола (30 мин). Фильтровальную лепешку промывали второй порцией 200 мл метанола (10 мин). Фильтровальную лепешку частично высушивали при отсасывании в течение примерно 60 мин. Твердые вещества сушили в вакууме при 35-40°С до постоянного веса (38 ч), получая 303,38 г (65,2%) 3-амино-3-(3’-пиридил)пропионовой кислоты в виде белого порошка.
Часть В
В реакционную колбу загружали 594,52 г смеси 3-амино-3-(3’-пиридил)пропионовой кислоты и транс-3-(3’-пиридил)акриловой кислоты и 1600 мл метанола. При хорошем перемешивании суспензию нагревали и выдерживали при легком кипячении с обратным холодильником в течение 1,25 ч. Суспензию фильтровали горячей и промывали 2×80 мл, затем 160 мл теплого (>50°С) метанола (30 мин). Фильтровальную лепешку частично высушивали при отсасывании в течение примерно 1,25 ч. Дополнительная сушка в вакууме при 35-40°С в течение 21,5 ч давала 540,22 г (90,9% выделения по весу) продукта в виде белого порошка.
В реакционную колбу загружали 540,12 г смеси 3-амино-3-(3’-пиридил)пропионовой кислоты и транс-3-(3’-пиридил)акриловой кислоты и 1600 мл метанола. При хорошем перемешивании суспензию нагревали и выдерживали при легком кипячении с обратным холодильником в течение 5-6 ч. Суспензию фильтровали горячей и промывали 3×160 мл теплого (>50°С) метанола (35 мин). Фильтровальную лепешку частично высушивали при отсасывании в течение примерно 1,75 ч, получая 814,45 г (>100% выделения по весу) продукта в виде белого твердого вещества (влажная лепешка).
В реакционную колбу загружали 814,35 г смеси 3-амино-3-(3’-пиридил)пропионовой кислоты и транс-3-(3’-пиридил)акриловой кислоты (влажная лепешка) и 1600 мл метанола. При хорошем перемешивании суспензию нагревали и выдерживали при легком кипячении с обратным холодильником в течение 3,75 ч. Суспензию фильтровали горячей и промывали 3×160 мл теплого (>50°С) метанола (20 мин). Фильтровальную лепешку частично высушивали при отсасывании в течение примерно 17,25 ч. Дополнительная сушка в вакууме при 40-45°С в течение 20 часов давала 390,69 г (65,7% выделения по весу от первоначальной загрузки) продукта в виде белого порошка.
Пример 2
3-[(трет-Бутокси)карбонил]амино-3-(3’-пиридил)пропионовая кислота
В реакционную колбу загружали 200 мл тетрагидрофурана и 170,88 г (1,03 моля) 3-амино-3-(3’-пиридил)пропионовой кислоты и 614 мл тетрагидрофурана при хорошем перемешивании. При температуре окружающей среды (22°С) добавляли 514 мл (1,03 моля) 2М раствора гидроксида натрия, медленно выливая его в течение примерно 2 мин. Во время такого добавления реакционная температура поднималась до 26°С, и большая часть твердого вещества растворялась. После перемешивания дополнительно в течение 45 мин при температуре окружающей среды получали слегка замутненный бесцветный раствор; рН 10,57 при 24°С. В капельную воронку постоянного давления емкостью 1 л загружали 407 мл тетрагидрофурана (ТГФ) и 365,3 мл (1,54 моля) ди-трет-бутилдикарбоната. Во вторую 1 л капельную воронку постоянного давления загружали 771 мл (1,54 моля) 2М раствора гидроксида натрия. Раствор ди-трет-бутилдикарбоната в тетрагидрофуране медленно добавляли струей в течение примерно 25 мин при рН-мониторинге. Как только рН достигало значения примерно 9,90, начинали сопутствующее добавление 2М раствора гидроксида натрия. Для поддержания реакционной температуры при <35°С применяли внешнее охлаждение. Реакционную смесь поддерживали при рН 9,9-10,2 во время добавления раствора тетрагидрофуран/ди-трет-бутилдикарбонат. После его добавления капельную воронку, которая содержала раствор дикарбонат/тетрагидрофуран, споласкивали затем с использованием 81 мл свежего тетрагидрофурана. Добавление оставшегося количества 2М раствора гидроксида натрия продолжали так, чтобы поддерживать рН в диапазоне 9,9-10,2. После добавления оставшегося количества 2М раствора гидроксида натрия рН зонд удаляли и реакционную смесь перемешивали при температуре окружающей среды в течение 17 ч. При хорошем перемешивании рН понижали от 7,68 при 21°С до 3,87 при 27°С с помощью порционного добавления 393,0 г (2,85 моля) моногидрата бисульфата натрия в течение примерно 3 ч. Гетерогенную смесь охлаждали в течение 75 мин на бане со смесью лед-вода при хорошем перемешивании. Объем воды, равный 1,285 мл, отмечали снаружи на 5 л широкогорлой круглодонной колбе, установив ее на роторный испаритель. Колбу освобождали, затем повторно наполняли реакционной суспензией. Реакционную колбу споласкивали затем 2×250 мл раствора 98/2 (об/об) тетрагидрофуран-вода. Суспензию концентрировали при 25°С в течение примерно 3 ч до объема примерно 1,285 мл. Концентрированную смесь экстрагировали 4×650 мл хлористого метилена. Объединенные экстракты сушили в течение 45 мин над 23,5 г (0,20 моля) безводного сульфата магния при хорошем перемешивании. После фильтрования прозрачный бесцветный фильтрат концентрировали в течение 90 мин при 25°С до получения подвижного сиропа. Его осторожно дополнительно концентрировали в течение 45 мин при 35°С и при <5 мм Нg до густого сиропа. Дополнительная сушка в вакууме при температуре окружающей среды в течение 60 ч давала 227,24 г (83,0%) продукта в виде прозрачного неподвижного стекла. По данным 1H ЯМР спектроскопии чистота данного вещества составила 89,8 вес./вес.%.
Пример 3
(1R,2S)-(-)Эфедриновая соль (3S)-3-[(трет-бутокси)карбонил)амино-3-(3’-пиридил)пропионовой кислоты
Часть А
В реакционную колбу загружали 2,750 мл этилацетата и 227,23 г (0,77 моля) 3-[(трет-бутокси)карбонил]амино-3-(3’-пиридил)пропионовой кислоты. При хорошем перемешивании раствор нагревали до реакционной температуры 62°С. Свежеприготовленный раствор 126,9 г (0,77 моля) (1R,2S)-(-)-эфедрина в 350 мл этилацетата добавляли медленно струей в течение примерно 9 минут при 64°С. Капельную воронку, которая содержала раствор эфедрина, споласкивали затем 50 мл этилацетата. В прозрачный бесцветный раствор (63°С) вносили затравку. В течение 1-2 мин начиналось интенсивное осаждение продукта. (Во время кристаллизации скорость перемешивания увеличивали для поддержания хорошего перемешивания суспензии). Суспензию медленно охлаждали до температуры окружающей среды. После примерно 18 ч твердое вещество собирали фильтрованием с отсосом и промывали 2×125 мл свежего этилацетата (15 мин). Фильтровальную лепешку частично высушивали при отсасывании в течение 35 мин. Дополнительная сушка в вакууме при 40-45°С в течение 21 ч давала 153,04 г (46,3%) продукта в виде рыхлого белого твердого вещества.
Часть В
В реакционную колбу загружали 415,33 г (0,96 моля) соли (1R,2S)-(-)-эфедрина (3S)-3-[(трет-бутокси)карбонил]амино-3-(3’-пиридил)пропионовой кислоты и 9 л этилацетата. Полученную суспензию нагревали и выдерживали при легком кипячении с обратным холодильником в течение 30 мин при хорошем перемешивании. Суспензию оставляли медленно охлаждаться до температуры окружающей среды при непрерывном хорошем перемешивании. Через 15 ч для восстановления хорошего перемешивания добавляли 250 мл свежего этилацетата. Еще через 2 ч твердое вещество собирали фильтрованием с отсосом и промывали 2×450 мл свежего этилацетата (10 мин). Фильтровальную лепешку частично высушивали при отсасывании в течение 75 мин. Дополнительная сушка в вакууме при 50-55°С в течение 44 ч давала 311,25 г (74,9%) продукта в виде рыхлого белого твердого вещества.
Пример 4
(3S)-3-[(трет-Бутокси)карбонил)амино-3-(3’-пиридил)-пропионовая кислота
В реакционный химический стакан загружали 303,47 г (0,67 моля) соли (1R,2S)-(-)-эфедрина (3S)-3-[(трет-бутокси)-карбонил]амино-3-(3’-пиридил)пропионовой кислоты и 792 мл милликвоты воды. После перемешивания в течение 5 мин рН суспензии составляло 7,32 при 21°С. Добавляли всего 595 мл (0,60 моль) 1М раствора гидроксида натрия, медленно выливая его в течение примерно 2 мин при хорошем перемешивании, до рН 11,00 при 23°С. Еще через 5 мин (рН 10,86) добавляли 20 мл (0,02 моль) 1М раствора гидроксида натрия при непрерывном хорошем перемешивании до рН 11,00 при 23°С. После перемешивания в течение дополнительных 65 мин (рН 10,73 при 22°С) добавляли 50 мл (0,05 моль) 1М раствора гидроксида натрия до рН 11,07 при 22°С. Слегка замутненный бесцветный раствор промывали 12×880 мл хлористого метилена. После последнего промывания рН составляло 7,76 при 20°С. При хорошем перемешивании добавляли порциями 47,89 г (0,35 моль) моногидрата бисульфата натрия в течение 10 мин до рН 5,18 при 21°С. В мутный раствор вносили затравку и образовавшуюся суспензию перемешивали в течение 5 мин. Во время этой кристаллизации рН увеличивалось до 5,76 при 21°С. Затем добавляли 47,82 г (0,34 моля) моногидрата бисульфата натрия в течение 25 мин до рН 3,79 при 22°С. Суспензию охлаждали в течение 40 мин на ледяной водной бане, фильтровали (5 мин) и частично сушили под отсосом в течение 20 мин. Фильтровальную лепешку сушили в вакууме при 50-55°С в течение 17 ч, получая 151,72 г (85,1%) продукта в виде бесцветного кристаллического твердого вещества.
Пример 5
Дигидрохлорид метил (3S)-3-амино-3-(3’-пиридил)пропионата
В круглодонную колбу загружали 142,01 г (0,53 моля) (3S)-3-[(трет-бутокси)карбонил)амино-3-(3’-пиридил)пропионовой кислоты и 1350 мл метанола. Смесь центрифугировали без вакуума, с использованием роторного испарителя при 25°С в течение 2 ч, затем осторожно концентрировали до получения бесцветного подвижного сиропа. Данный сироп переносили в реакционную колбу с использованием 1350 мл метанола. Непрозрачный раствор охлаждали на ледяной водной бане до 0-5°С. Затем примерно 194,4 г (5,33 моля) хлористого водорода барботировали в раствор в течение примерно 3 ч. Во время такого добавления температуру реакции поддерживали при <15°С. После внесения затравки суспензию нагревали до температуры окружающей среды и перемешивали в течение 2 ч. Суспензию охлаждали в течение 30 мин на ледяной водной бане. Твердое вещество собирали фильтрованием с отсосом и промывали 2×65 мл холодным (0-5°С) метанолом (5 мин). Фильтровальную лепешку частично высушивали отсасывая в течение примерно 20 мин.
Дополнительная сушка в вакууме при температуре окружающей среды в течение 14 ч давала 105,21 г (77,9%) продукта в виде белого кристаллического порошка.
Пример 6
3-Амино-3-[3’-(6-метил)пиридил]пропионовая кислота
К смеси 6-метилпиридин-3-карбоксальдегида (23,5 г, 0,149 моля), ацетата аммония (22,9 г, 1,5 экв.) и этанола (350 мл) добавляли малоновую кислоту (20,5 г, 1 экв.). Смесь нагревали с обратным холодильником в течение 6 ч, охлаждали до комнатной температуры и фильтровали. Данное отфильтрованное твердое вещество промывали этанолом (2×30 мл) и диэтиловым эфиром (2×100 мл) и сушили, получая продукт в виде белого твердого вещества (23,2 г, выход 66%).
Пример 7
3-Амино-3-[3’-(5-бром)пиридил]пропионовая кислота
К смеси 5-бромпиридин-3-карбоксальдегида (40,4 г, 0,21 моль), ацетата аммония (24,3 г, 1,5 экв.) и абсолютного этанола (650 мл) добавляли малоновую кислоту (21,8 г, 1 экв.). Смесь нагревали с обратным холодильником в течение 6,5 ч, охлаждали до комнатной температуры и фильтровали. Такое отфильтрованное твердое вещество промывали этанолом и диэтиловым эфиром и сушили, получая неочищенный продукт (28,0 г, выход 55%). Данное твердое вещество нагревали с обратным холодильником в течение 30 мин в метаноле (500 мл), фильтровали горячим, промывали горячим метанолом и сушили, получая продукт в виде белого твердого вещества (17,4 г, 34%).
Пример 8
3-Амино-3-[3’-(6-хлор)пиридил]пропионовая кислота
К смеси 6-хлорпиридин-3-карбоксальдегида (32,6 г, 0,229 моль), ацетата аммония (26,5 г, 1,5 экв.) и абсолютного этанола (300 мл) добавляли малоновую кислоту (23,8 г, 1 экв.). Смесь нагревали с обратным холодильником в течение 2,5 ч, охлаждали до комнатной температуры и фильтровали. Такое отфильтрованное твердое вещество промывали этанолом и диэтиловым эфиром и сушили, получая неочищенный продукт (30,6 г, выход 66%). Данное твердое вещество нагревали с обратным холодильником в метаноле (500 мл), фильтровали горячим, промывали горячим метанолом (3×150 мл) и диэтиловым эфиром и сушили, получая указанное в заголовке соединение (24,5 г, 53%). Это твердое вещество нагревали с обратным холодильником в метаноле (500 мл), фильтровали горячим, промывали горячим метанолом и диэтиловым эфиром и сушили, получая продукт в виде белого твердого вещества (21,0 г; 46%).
Пример 9
3-Амино-3-[3’-[5,6-дихлор)пиридил]пропионовая кислота
К смеси 5,6-дихлорпиридин-3-карбоксальдегида (9,4 г, 0,053 моль), ацетата аммония (6,2 г, 1,5 экв.) и абсолютного этанола (100 мл) добавляли малоновую кислоту (5,6 г, 1 экв.). Смесь нагревали с обратным холодильником в течение 8 ч, охлаждали до комнатной температуры, перемешивали в течение ночи и фильтровали. Такое отфильтрованное твердое вещество промывали этанолом и диэтиловым эфиром и сушили, получая неочищенный продукт (7,0 г, выход 56%). Данное твердое вещество нагревали с обратным холодильником в метаноле (125 мл) в течение 15 мин, фильтровали горячим, промывали горячим метанолом (50 мл) и сушили, получая продукт в виде белого твердого вещества (4,4 г, 35%).
Пример 10
3-[(трет-Бутокси)карбонил]амино-3-[3’-(6-метил)пиридил]-пропионовая кислота
К охлажденному (5°С) раствору 3-амино-3-[3’-(6-метил)пиридил]пропионовой кислоты (23,2 г, 0,128 моля), 1 н. гидроксида натрия (256 мл, 2 экв.) и диоксана (150 мл) добавляли ди-трет-бутилдикарбонат (27,9 г, 1 экв.). Данную смесь перемешивали в течение 1,5 ч, ледяную баню убирали и перемешивание продолжали в течение 16 ч. Диоксан удаляли в вакууме и полученную водную смесь доводили до рН 4 10%-ной лимонной кислотой (250 мл). Данный раствор охлаждали на ледяной бане, перемешивали в течение 30 мин и фильтровали, получая 7,7 г белого порошка. Фильтрат уменьшали в объеме наполовину в вакууме, обрабатывали хлоридом натрия (30 г), охлаждали в течение 70 ч и фильтровали, получая вторую порцию (22,4 г) продукта. Порции объединяли, получая продукт в виде белого твердого вещества (30,1 г, выход 84%).
Пример 11
3-[(трет-Бутокси)карбонил]амино-3-[3’-(5-бром)пиридил]-пропионовая кислота
К охлажденному (5°С) раствору 3-амино-3-[3’-(5-бром)пиридил]пропионовой кислоты (17,4 г, 0,071 моля), 1 н. гидроксида натрия (142 мл, 2 экв.) и диоксана (100 мл) добавляли ди-трет-бутилдикарбонат (15,5 г, 1 экв.). Данную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение ночи. Диоксан удаляли в вакууме и полученную водную смесь доводили до рН 3-4 10%-ной лимонной кислотой. Данный раствор экстрагировали смесью 9/1 (об/об) хлороформ-диоксан (3×150 мл) и объединенные экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали, получая продукт в виде белого твердого вещества (21,1 г, 86%).
Пример 12
3-[(трет-Бутокси)карбонил]амино-3-[3’-(6-хлор)пиридил]-пропионовая кислота
К охлажденному (5°С) раствору 3-амино-3-[3’-(6-хлор)пиридил]пропионовой кислоты (21,0 г, 0,105 моля), 1 н. гидроксида натрия (210 мл, 2 экв.) и диоксана (150 мл) добавляли ди-трет-бутилдикарбонат (22,89 г, 1 экв.). Данную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение ночи. Диоксан удаляли в вакууме и полученную водную смесь доводили до рН 3-4 10%-ной лимонной кислотой. Смесь фильтровали и твердые вещества промывали водой и сушили на воздухе в течение ночи, получая продукт в виде белого твердого вещества (21,5 г, 68%).
Пример 13
3-[(трет-Бутокси)карбонил]амино-3-[3’-5,6-дихлор)-пиридил]пропионовая кислота
К охлажденному (5°С) раствору 3-амино-3-[3’-(5,6-дихлор)пиридил]пропионовой кислоты (4,4 г, 0,019 моля), 1 н. гидроксида натрия (37,6 мл, 2 экв.) и диоксана (30 мл) добавляли ди-трет-бутилдикарбонат (4,1 г, 1 экв.). Данную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение ночи. Диоксан удаляли в вакууме и полученную водную смесь доводили до рН 3-4 10%-ной лимонной кислотой. После дополнительного перемешивания при температуре окружающей среды смесь фильтровали и твердые вещества промывали водой и сушили на воздухе, получая продукт в виде белого твердого вещества (5,0 г, 79%).
Пример 14
(1R,2S)-(-)-Эфедриновая соль (3S)-3-[(трет-бутокси)-карбонил]амино-3-[3’-(6-метил)пиридил]пропионовой кислоты
К суспензии 3-[(трет-бутокси)карбонил]амино-3-[3’-(6-метил)пиридил]пропионовой кислоты (3,85 г, 0,0137 моля) и теплого этилацетата (170 мл) добавляли раствор (1R,2S)-(-)эфедрина (2,31 г, 1 экв.) в этилацетате (60 мл). Данную смесь нагревали с обратным холодильником, получая прозрачный раствор. Через 5 мин при условиях окружающей среды в теплый раствор вносили затравку и оставляли охлаждаться до комнатной температуры. Кристаллизация соли начиналась, когда раствор достигал температуры 30°С. После кристаллизации в течение 15 мин смесь фильтровали и твердый продукт промывали этилацетатом (50 мл) и диэтиловым эфиром (50 мл) и сушили, получая белый порошок (1,9 г, выход 31%).
Вторую порцию выделяли из маточных растворов после выдерживания в течение 16 ч при условиях окружающей среды (0,33 г, выход 5,4%). Две порции объединяли, суспендировали в теплом этилацетате (60 мл) и фильтровали, получая 2,2 г (36%) продукта в виде белого твердого вещества.
Пример 15
(1R,2S)-(-)-Эфедриновая соль (3S)-3-[(трет-бутокси)-карбонил]амино-3-[3’-(5-бром)пиридил]пропионовой кислоты
К раствору 3-[(трет-бутокси)карбонил]амино-3-[3’-(5-бром)пиридил]пропионовой кислоты (21,0 г, 0,061 моля) и этилацетата (125 мл) добавляли (1R,2S)-(-)эфедрин (10,1 г, 1 экв.), получая прозрачный раствор. После внесения затравки и выдерживания в течение ночи при температуре окружающей среды смесь фильтровали, твердый продукт промывали холодным этилацетатом и диэтиловым эфиром и сушили, получая продукт в виде белого порошка (8,3 г, выход 27%).
Пример 16
(1R,2S)-(-)-Эфедриновая соль (3S)-3-[(трет-бутокси)-карбонил]амино-3-[3’-(6-хлор)пиридил]пропионовой кислоты
К суспензии 3-[(трет-бутокси)карбонил]амино-3-[3’-(6-хлор)пиридил]пропионовой кислоты (21,4 г, 0,071 моля) и этилацетата (250 мл) добавляли (1R,2S)-(-)эфедрин (11,79 г, 1 экв.). Данную смесь нагревали с обратным холодильником, получая прозрачный раствор. Раствор охлаждали до комнатной температуры и вносили затравку. После кристаллизации, добавляли дополнительное количество этилацетата (700 мл), суспензию нагревали до кипения и затем медленно охлаждали до температуры окружающей среды. После перемешивания в течение ночи смесь фильтровали, твердый продукт промывали этилацетатом и диэтиловым эфиром и сушили, получая продукт в виде рыхлого белого твердого вещества (15,36 г, выход 46%).
Пример 17
(1R,2S)-(-)-Эфедриновая соль (3S)-3-[(трет-бутокси)-карбонил]амино-3-[3’-(5,6-дихлор)пиридил]пропионовой кислоты
К суспензии 3-[(трет-бутокси)карбонил]амино-3-[3’-(5,6-дихлор)пиридил]пропионовой кислоты (4,9 г, 0,015 моля) и этилацетата (150 мл) добавляли (1R,2S)-(-)эфедрин (2,42 г, 1 экв.). Данную смесь нагревали до образования флегмы и фильтровали для удаления небольшого количества нерастворившегося вещества. Прозрачный фильтрат медленно охлаждали до температуры окружающей среды. Смесь фильтровали, твердый продукт промывали этилацетатом и диэтиловым эфиром и сушили, получая сырой продукт (3,1 г). Перекристаллизация из кипящего этилацетата (75 мл) давала продукт в виде белого твердого вещества (2,8 г, 38%).
Пример 18
3-Амино-3-(3’-пиридил)пропионовая кислота
Ацетат аммония (194,28 г, 2,52 моля) суспендировали в этаноле (285 г). К суспензии в течение 10 мин при 15-20°С добавляли раствор пиридин-3-карбальдегида (62,9 г, 1,68 моля) в этаноле (80 г). Реакционную смесь перемешивали в течение 1 ч до образования прозрачного желтоватого раствора. Затем добавляли суспензию малоновой кислоты (174,86 г, 1,68 моля) в этаноле (235 г) (в течение 30 мин). После перемешивания в течение 30 мин при температуре окружающей среды реакционную смесь нагревали в течение 5 ч до образования флегмы (78°С). Наблюдалось выделение газа. Через 3-4 ч начинало осаждаться белое твердое вещество. Оранжевую окрашенную суспензию охлаждали до 15-20°С и получали густую суспензию. К данной суспензии добавляли метанол (80 г) и реакционную смесь опять нагревали до образования флегмы (65°С). Суспензию фильтровали горячей и фильтровальную лепешку промывали тремя порциями горячего метанола (120 г) (при температуре 55-65°С). Влажный продукт сушили в вакууме при 70-80°С, получая продукт в виде бесцветного твердого вещества (137,73 г, 49,3%). Чистота по ЖХ 95,8%.
Пример 19
3-[(трет-Бутокси)карбонил]амино-3-(3’-пиридил)пропионовая кислота
3-Амино-3-(3’-пиридил)пропионовую кислоту (73,00 г, 0,439 моля) суспендировали в ТГФ (239,5 г) при 15-25°С. В течение 10-20 мин добавляли раствор гидроксида натрия (43,93 г, 1,10 моля) в очищенной воде (395,4 г), получая слегка желтоватый раствор. В течение 2-3 ч добавляли раствор ВОС-ангидрида (143,82 г, 0,659 моля) в ТГФ (124,2 г), поддерживая температуру раствора при не выше 30°С. Раствор перемешивали в течение ночи (примерно 17 ч) при 15-25°С. В течение 1-2 ч добавляли раствор, моногидрата бисульфата натрия (167,9 г, 1,22 моля) в воде (160 г) для доведения рН до примерно 3,8-3,9. Наблюдалось осаждение некоторого количества твердых веществ при сильном выделении газа. Суспензию охлаждали до 5-10°С, фильтровали и промывали ТГФ (50 г). Объединенные фильтраты уменьшали отгонкой до одной трети их первоначального объема. Затем добавляли этилацетат (102,9 г), равный объем отгоняли, опять добавляли этилацетат (99,4 г) и такой же объем отгоняли. К полученной водной эмульсии добавляли хлорид натрия (64,6 г) и этилацетат (100,5 г). Фазы отделяли и водную фазу промывали этилацетатом (3×20 г). Объединенные органические фазы сушили над безводным сульфатом натрия (20 г) и осушающий агент отфильтровывали и фильтровальную лепешку промывали этилацетатом (5 г). Полученный раствор (примерно 250 г) использовали непосредственно на следующей стадии.
Пример 20
(1R,2S)-(-)-Эфедриновая соль (3S)-3-[(трет-бутокси)-карбонил]амино-3-(3’-пиридил)пропионовой кислоты
Раствор 3-[(трет-бутокси)карбонил]амино-3-(3’-пиридил)-пропионовой кислоты (250 г) в этилацетате, содержащий 3-[(трет-бутокси)карбонил]амино-3-(3’-пиридил)пропионовую кислоту (71,5 г, 0,269 ммоль) и этилацетат (178,5 г), нагревали до 60-70°С. В течение 10 мин добавляли раствор (-)-эфедрина (48,80 г, 0,295 моля) в этилацетате (90,0 г). Прозрачный раствор охлаждали до 20-30°С, в то время как продукт кристаллизовался в виде объемного белого твердого вещества. После начала кристаллизации для поддержания возможности перемешивания смеси добавляли этилацетат (270 г). Суспензию охлаждали до 15-25°С, перемешивали в течение 3-5 ч и твердые вещества собирали фильтрованием. Фильтровальную лепешку промывали этилацетатом (90 г) двумя порциями и сушили в вакууме при 70-80°C, получая продукт в виде бесцветного твердого вещества (52,72 г, 28%). Чистота по ЖХ >99%.
Пример 21
(3S)-3-[(трет-Бутокси)карбонил]амино-3-(3’-пиридил)-пропионовая кислота
(1R,2S)-(-)-Эфедриновую соль (3S)-3-[(трет-бутокси)-карбонил]амино-3-(3’-пиридил)пропионовой кислоты (25,11 г, 0,058 моля) растворяли в воде (50,0 г) при 15-25°С. В течение 10-20 минут добавляли раствор гидроксида натрия (2,63 г, 0,066 моля) в воде (23,3 г), получая прозрачный раствор. При хорошем перемешивании добавляли толуол (17,4 г) и полученную эмульсию нагревали до 70-80°С. После прекращения перемешивания прозрачную органическую фазу отделяли от слегка мутной водной фазы. Водную фазу экстрагировали толуолом (4×17,4 г) при 7-80°С. Затем водную фазу охлаждали до 15-25°C, фильтровали и фильтр промывали водой (2,5 г). К объединенным фильтратам добавляли раствор моногидрата бисульфата натрия (9,50 г, 0,069 моля) в воде (12,1 г), что приводило к рН 3,6-3,9 и закристаллизованному продукту. Реакционную смесь охлаждали до 0-5°С, перемешивали дополнительно в течение 1 ч и продукт собирали фильтрованием, промывали водой (5 г) двумя порциями, сушили в вакууме при 40-50°С, получая продукт в виде бесцветного твердого вещества (11,08 г, 72%). Чистота по ЖХ>95%.
Пример 22
Дигидрохлорид метил (3S)-3-амино-3-(3’-пиридил)пропионата
(3S)-3-[(трет-Бутокси)карбонил]амино-3-(3’-пиридил)-пропионовую кислоту (9,93 г, 0,037 моля) растворяли в метаноле (45,8 г) при 0-5°С. В течение 2-3 ч через раствор барботировали хлористый водород (34,2 г, 0,938 моля), поддерживая температуру раствора ниже примерно 15°С. После завершения добавления хлористого водорода реакционную смесь перемешивали в течение, по крайней мере, 2 ч при 20-25°С и затем охлаждали до 0-5°С. После перемешивания в течение 30 мин при 0-5°С твердые вещества собирали фильтрованием, промывали холодным метанолом (14,2 г) (при температуре примерно 0-5°С) двумя порциями и сушили в вакууме при 20-30°С, получая продукт в виде бесцветного твердого вещества (7,86 г, 83%). Чистота по ЖХ >95%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СУЛЬФОНИЛАЛКАНОИЛАМИНО-ГИДРОКСИЭТИЛАМИНО-СУЛЬФОНАМИДНОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ И ИНГИБИРОВАНИЯ РЕТРОВИРУСНЫХ ПРОТЕАЗ | 1993 |
|
RU2146668C1 |
| СИНТЕЗ 3-АМИНО-3-АРИЛПРОПАНОАТОВ | 2000 |
|
RU2225859C2 |
| ПЕПТИДНЫЕ МАКРОЦИКЛЫ ПРОТИВ ACINETOBACTER BAUMANNII | 2019 |
|
RU2795374C2 |
| ПРОТИВОГЕЛЬМИНТНЫЕ ДЕПСИПЕПТИДНЫЕ СОЕДИНЕНИЯ | 2016 |
|
RU2707298C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩИЕ СВОЙСТВАМИ ВЫСВОБОЖДЕНИЯ ГОРМОНА РОСТА, СПОСОБ СТИМУЛЯЦИИ ВЫДЕЛЕНИЯ ГОРМОНА РОСТА ИЗ ГИПОФИЗА МЛЕКОПИТАЮЩЕГО | 1999 |
|
RU2298547C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2833354C1 |
| 3-КАРБОНИЛАМИНО-5-ЦИКЛОПЕНТИЛ-1H-ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ИНГИБИТОРОВ CDK2 | 2020 |
|
RU2797889C2 |
| КОНЪЮГАТЫ АНТАГОНИСТОВ ИНТЕГРИНА ДЛЯ НАЦЕЛЕННОЙ ДОСТАВКИ К КЛЕТКАМ, ЭКСПРЕССИРУЮЩИМ АЛЬФА-V-БЕТА-3 | 2013 |
|
RU2623441C2 |
| СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ КОНДЕНСИРОВАННОЕ БИЦИКЛИЧЕСКОЕ ЯДРО, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ ПРЕВРАЩЕНИЯ ФЕРМЕНТА АНГИОТЕНЗИНА И НЕЙТРАЛЬНОЙ ЭНДОПЕПТИДАЗЫ | 1994 |
|
RU2125056C1 |
| КОНДЕНСИРОВАННОЕ ХИНОЛИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2384571C2 |
Изобретение относится к способу получения 3S-3-амино-3-пиридилпропионовой кислоты или ее производных формулы (I), где R1 представляет пиридил или замещенный пиридил, R2 представляет водород, алкил или аралкил, включающему взаимодействие соединения формулы (II) при рН 7-11 с ди-трет-бутилбикарбонатом с образованием соединения формулы (III), где R5 представляет N-трет-бутоксикарбонил, взаимодействие соединения формулы (III) c по крайней мере 0,5 эквивалентами (1R,2S)-(-)эфедрина в алкилацетатном растворителе с образованием соли формулы (IV), где Ph представляет фенил, взаимодействие соли формулы (IV) с неорганическим основанием в воде с образованием карбоксилатной соли соединения формулы (V), подкисление карбоксилатной соли соединения формулы (V) кислотой, pKa которой меньше чем или равно трем, до рН примерно 3,5 - 6,5, с образованием соединения формулы (V), взаимодействие соединения формулы (V) с кислотой, pKaкоторой меньше чем или равно 3, отличной от бисульфата калия, бисульфата натрия и уксусной кислоты, при температуре меньше примерно 25оС, с образованием соединения формулы (I) в виде кислоты или превращением ее, в случае необходимости, в эфир. Предлагаемый способ характеризуется доступностью исходных реагентов и позволяет получать целевые продукты с хорошими выходом и чистотой. Описывается также промежуточное соединение формулы (Va). 4 с. и 22 з.п. ф-лы, 1 табл.


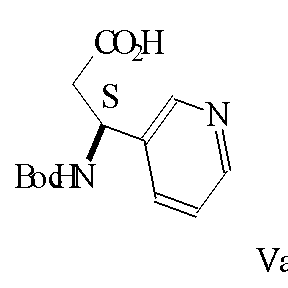
Форма 2 соединения формулы Vа
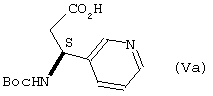
где Boc представляет трет-бутоксикарбонил,
характеризующаяся следующей порошковой рентгенограммой:
Результаты порошковой дифракции рентгеновских лучей
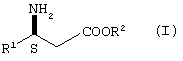
где R1 представляет пиридил или замещенный пиридил;
R2 представляет водород, алкил или аралкил,
или его кислотно-аддитивной соли, включающий взаимодействие соединения формулы II при рН примерно 7 - 11
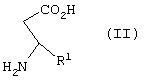
с ди-трет-бутил бикарбонатом с образованием соединения формулы III
где R5 представляет N-трет-бутоксикарбонил,
взаимодействие соединения формулы III c по крайней мере 0,5 эквивалентами (1R,2S)-(-)эфедрина в алкилацетатном растворителе с образованием соли формулы IV

где Ph представляет фенил,
взаимодействие соли формулы IV с неорганическим основанием в воде с образованием карбоксилатной соли соединения формулы V, подкисление карбоксилатной соли соединения формулы V кислотой с рКa ≤ 3 до рН примерно 3,5 - 6,5 с образованием соединения формулы V
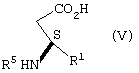
взаимодействие соединения формулы V с кислотой, pKa которой ≤ 3, отличной от бисульфата калия, бисульфата натрия и уксусной кислоты, при температуре меньше примерно 25°С с образованием соединения формулы I в виде кислоты или превращением ее в случае необходимости в эфир.
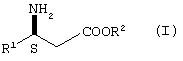
где R1 представляет пиридил или замещенный пиридил;
R2 представляет водород, алкил или аралкил, или его кислотно-аддитивной соли,
включающий взаимодействие соединения формулы III
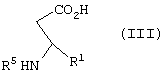
где R5 представляет N-трет-бутоксикарбонил,
c по крайней мере 0,5 эквивалентами (1R,2S)-(-)эфедрина в алкилацетатном растворителе с образованием соли формулы IV
где Ph представляет фенил,
взаимодействие соли формулы IV с неорганическим основанием в воде с образованием карбоксилатной соли соединения формулы V, подкисление карбоксилатной соли соединения формулы V кислотой с рКa ≤ 3 до рН примерно 3,5 - 6,5 с образованием соединения формулы V
взаимодействие соединения формулы V с кислотой, pKa которой ≤ 3, отличной от бисульфата калия, бисульфата натрия и уксусной кислоты, при температуре меньше примерно 25°С с образованием соединения формулы I в виде кислоты или превращением ее в случае необходимости в эфир.
где R1 представляет пиридил или замещенный пиридил;
R5 представляет N-трет-бутоксикарбонил,
включающий взаимодействие соединения формулы III
с по крайней мере 0,5 эквивалента (1R,2S)-(-)эфедрина в алкилацетатном растворителе с образованием соли формулы IV

где Ph представляет фенил,
взаимодействие соли формулы IV с неорганическим основанием в воде с образованием карбоксилатной соли соединения формулы V, подкисление карбоксилатной соли соединения формулы V кислотой с рКа ≤ 3 до рН от примерно 3,5 до примерно 6,5.
| ПРОИЗВОДНЫЕ ПИРИДИЛА, ИХ ЭНАНТИОМЕРЫ, ЦИС- ИЛИ ТРАНС-ИЗОМЕРЫ И ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2119915C1 |

