Изобретение относится к новым соединениям, содержащим конденсированное бициклическое ядро, которые используются в качестве ангистензина ингибиторов превращения фермента. Некоторые из этих соединений обладают ингибирующей активностью по отношению к нейтральной эндопептидазе. Это изобретение также относится к фармацевтическим композициям, содержащим такие селективные или двойственного действия ингибиторы, и способу использования таких композиций. Это изобретение также относится к способу получения таких новых соединений, новых промежуточных соединений и способам получения таких промежуточных соединений.
Новые конденсированные бициклические ингибиторы этого изобретения включают соединения формулы I
и их фармацевтически приемлемые соли:
где
A представляет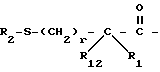

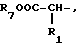
или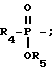
X представляет O, или S -(O)t;
R1 и R12 независимо выбраны из водорода, алкила, алкенила, циклоалкила, замещенного алкила, замещенного алкенила, арила, замещенного арила, гетероарила, циклоалкилалкилена-, арил-алкилена, замещенного арил-алкилена, и гетероарил-алкилена или R1 и R12 взятые вместе с атомом углерода, к которому они присоединяются, образуют циклоалкильное кольцо или конденсированное бензоциклоалкильное кольцо;
R2 представляет водород,  или R11 -S-;
или R11 -S-;
R3, R5 и R7 независимо выбраны из водорода, алкила, замещенного алкила, арила-(CH2)p-, замещенного арила-CH2)p-, гетероарила-(CH2)p-,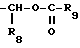
и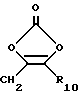
R4 представляет алкил, циклоалкил-(CH2)p-, замещенный алкил, арил-(CH2)p-, замещенный арил-(CH2)p- или гетероарил-(CH2)p-;
R6 представляет алкил, замещенный алкил, циклоалкил-(CH2)p-, арил-(CH2)p-, замещенный арил-(CH2)p-, или гетероарил-(CH2)p;
R8 представляет водород, низший алкил, циклоалкил или фенил;
R9 представляет водород, низший алкил, низший алкокси или фенил;
R10 представляет низший алкил или арил-(CH2)p-;
R11 представляет водород, алкил, замещенный алкил, циклоалкил-(CH2)p-, арил-(CH2)p-, замещенный арил-(CH2)p-, гетероарил-(CH2)p- или -S-R11 образует симметричный дисульфид, где R11 представляет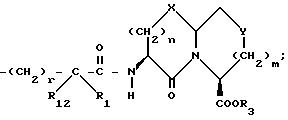
m равно 0 или 1;
Y представляет CH2, S(O)t или O при условии, что Y представляет S-(O)t или O только если m равно 1;
n равно 1 или 2;
p равно 0 или целому от 1 до 6;
q равно 0 или целому от 1 до 3;
r равно 0 или 1; и
t равно 0, 1 или 1.
Термин "алкил" относится к радикалам с неразветвленной или разветвленной цепью, содержащим вплоть до 7 атомов углерода. Термин "низший алкил" относится к радикалам с неразветвленной или разветвленной цепью, содержащим вплоть до 4 атомов углерода, и является предпочтительной подгруппой для термина алкил.
Термин "замещенный алкил" относится к таким радикалам с неразветвленной или разветвленной цепью с 1 - 7 атомами углерода, где один или более, предпочтительно один, два или три атома водорода заменены гидрокси, амино, циано, галоид, трифторметильной, -NH(низшей алкильной), -N-(низшей алкильной)2, низшей алкокси, низшей алкилтио, или карбокси группой.
Термины "низший алкокси" и "низший алкилтио" относятся к таким низшим алкильным группам, которые, как определенные выше, присоединяются к атому кислорода или серы.
Термин "циклоалкил" относится к насыщенным кольцам с 3 - 7 атомами углерода с циклопропилом, циклобутилом, циклопентилом и циклогексилом, являющимся наиболее предпочтительным.
Термин "алкенил" относится к радикалам с неразветвленной и разветвленной цепью с 3 - 7 атомами углерода, содержащим одну или две двойных связи. Предпочтительно "алкенильные" группы представляют радикалы с неразветвленной цепью с 3 - 5 атомами углерода, содержащие одну двойную связь.
Термин "замещенный алкенил" относится к таким радикалам с неразветвленной или разветвленной цепью с 3 - 7 атомами углерода, содержащим одну или две двойные связи, где водород замещен гидрокси, амино, галоидной, трифторметильной, циано, -NH(низшей алкильной), -N(низшей алкильной)2, низшей алкокси, низшей алкилтио или карбокси группой.
Термин "алкилен" относится к радикалам с неразветвленной или разветвленной цепью, содержащим вплоть до 7 атомов углерода, т.е. -CH2-, -(CH2)2-, -(CH2)3-, -(CH2)4-, 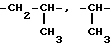 и т.д.
и т.д.
Термин "арил" относится к фенилу, 1-нафтилу и 2-нафтилу. Термин "замещенный арил" относится к фенилу, 1-нафтилу и 2-нафтилу содержащим заместитель, выбранный из низшего алкила, низшего алкокси, низшего алкилтио, галоида, гидрокси, трифторметила, амино, -NH(низшего алкила) или -N(низшего алкила)2, и ди- или три-замещенного фенила, 1-нафтила или 2-нафтила, где указанные заместители выбраны из метила, метокси, метилтио, галоида, гидрокси и амино.
Термин "гетероарил" относится к ненасыщенным кольцам с 5 или 6 атомами углерода, содержащим один или два атома O и S и/или 1 - 4 атома азота при условии, что общее число гетероатомов в кольце равно 4 или меньше. Гетероарильное кольцо присоединяется с помощью имеющегося атома углерода или атома азота. Предпочтительные гетероарильные группы включают 2-, 3- или 4-пиридил, 4-имидазолил, 4-тиазолил, 2- и 3-тиенил, и 2- и 3-фурил. Термин "гетероарил" также включает бициклические кольца, где 5- или 6-членное кольцо, содержащее атомы O, S и N, как оно определено выше, представляется конденсированным с бензольным или пиридильным кольцом. Предпочтительные бициклические ядра представляют 2- или 3-индоил и 4- и 5-хинолинил. Моно- или бициклическое гетероарильное кольцо может быть также дополнительно замещено по имеющемуся атому углерода низшим алкилом, галоидом, гидрокси, бензилом или циклогексилметилом. Кроме того, если моно- или бициклическое ядро имеет N-атом, то такой N-атом также может быть замещен N-защищающей группой, такой как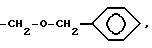

2,4-динитрофенил, низший алкил, бензил или бензгидрил.
Соединения формулы I, где A представляет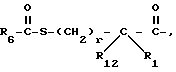
X представляет O или S, и
Y представляет CH2, O или S, могут быть получены реакцией сочетания соединения с арилмеркапто боковой цепью формулы II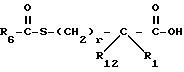
с соединением с конденсированным бициклическим кольцом формулы III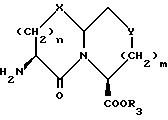
с получением продукта формулы IV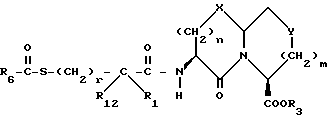
где
R3 представляет водород или кислую защищающую группу, такую как метил, этил, т-бутил или бензил.
Вышеприведенная реакция может быть проведена в органическом растворителе, таком как метиленхлорид, и в присутствии агента сочетания, такого как 1-этил-3-(3-диметиламинопропил)карбодиимид, дициклогексилкарбодиимид, бензолтриазол-1-илокситрис(диметиламино)-фосфонийгексафторфосфат, или карбонилдиимидазол. Или же ацилмеркаптокарбоновая кислота формулы II может быть превращена в активированную форму до реакции сочетания, такую как хлорид кислоты, смешанный ангидрид, симметричный ангидрид, активированный эфир и т.д.
Продукт формулы IV может быть превращен в меркаптановый продукт формулы I, где R2 представляет водород и R3 представляет водород, способом известным в литературе. Например, если R6 представляет метил и R3 представляет этил, обработка метанольным раствором гидроокиси натрия с последующей обработкой водным раствором кислоты дает продукты, где R2 и R3 представляют водород.
Продукт формулы I, где R2 представляет водород, может быть ацилирован ацилгалоидом формулы V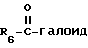
где
галоид представляет F, Cl или Br,
или ацилирован ангидридом формулы VI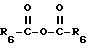
с получением других продуктов формулы I, где R2 представляет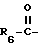
Продукты формулы I, где R2 представляет -S-R11 и R11 представляет алкил, замещенный алкил, циклоалкил-(CH2)p-, арил-(CH2)p-, замещенный арил-(CH2)p-, или гетероарил-(CH2)p-, могут быть получены взаимодействием продуктов формулы I, где R2 представляет водород, с сульфонильным соединением формулы VII
H3C-SO2-S-R11
в водном спиртовом растворителе, с получением желаемых продуктов. Соединения формулы VII являются известными в литературе или могут быть получены известными способами, например Smith и др., Biochemistry, 14, с. 766-771 (1975).
Продукт формулы I, где R2 представляет SH, может быть получен взаимодействием продукта формулы I, где R2 представляет водород, с соединением формулы VII, где R11 представляет трифенилметил или триарилсилил, с последующим удалением трифенилметильной или триалкилсилильной группы в кислых условиях.
Симметричные дисульфидные продукты формулы I могут быть получены прямым окислением продукта формулы I, где R2 представляет водород, иодом согласно известным способам, например см. Ondetti и др., патент США 4105776.
Соединения с боковой арилмеркаптоцепью формулы II, где R12 представляет водород, описаны в литературе (см. , например Ondetti и др. патент США 4105776 и 4339600, Haslanger и др. патент США 4801609, Delaney и др. патент США 4722810 и т.д.).
Соединения с боковой арилмеркаптоцепью формулы II, где R1 и R12 оба другие, чем водород, и r равно 0, могут быть получены взаимодействием замещенной карбоновой кислоты формулы VIII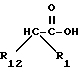
с бис///(4-метокси)фенил/метилдисульфидом в присутствии литийдиизопропиламида с получением соединения формулы IX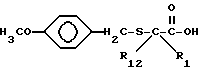
Обработка соединения формулы IX сильной кислотой, такой как трифторметансульфоновая кислота удаляет метоксибензильную защищающую группу и последующее ацилирование ацилгалоидом формулы V или ангидридом формулы VI дает соединение формулы II, где R1 и R12 - оба водород и r равно 0.
Или же замещенная карбоновая кислота формулы VIII может взаимодействовать с литийдиизопропиламидом и серой, давая меркаптан формулы X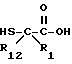
Меркаптан формулы X может быть затем ацилирован ацилгалоидом формулы V или ангидридом формулы VI, давая соединение формулы II, где R1 и R12 оба другие, чем водород, и r равно 0.
Соединения с боковой ацилмеркаптоцепью формулы II, где R1 и R12 оба другие, чем водород, и r равно 1, могут быть получены взаимодействием замещенной карбоновой кислоты формулы XI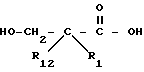
с пара-толуолсулфонилхлоридом в пиридине с получением лактама формулы XII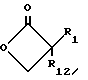
Обработка лактама формулы XII цезийтиокислотой формулы XIII
в присутствии диметилформамида дает желаемое соединение с арилмеркаптобоковой цепью формулы II, где R1 и R12 оба другие, чем водород, и r равно 1.
Соединения формулы I, где A представляет \\\ 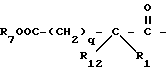
X представляет O или S и Y представляет CH2, O или S, могут быть получены реакцией сочетания кислоты формулы XIV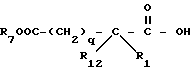
где
R7 представляет кислую защищающую группу,
с соединением с конденсированным бициклическим ядром формулы III в присутствии агента сочетания, как он определен выше, с получением продукта формулы XV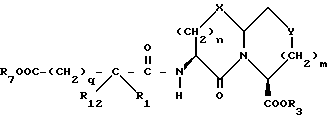
Или же кислота формулы XIV может быть превращена в активированную форму, такую как хлорид кислоты, до реакции сочетания.
Кислоты формулы XIV описаны Warshawsky и др. в Европейской патентной заявке 534396 и 534492.
Соединения формулы I, где A представляет
X представляет O или S и Y представляет CH2, O или S, могут быть получены взаимодействием кетокислоты или эфира формулы XVI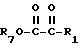
с соединением с конденсированным бициклическим ядром формулы III в условиях восстановления с получением продукта формулы XVII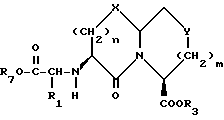
Кетокислоты и эфиры формулы XVI описаны в литературе (см., например, Ruyle патент США 4584294 и Parsons и др. патент США 4873235).
Или же соединение с конденсированным бициклическим ядром формулы III может взаимодействовать с трифталатом формулы XVIII
давая продукт формулы XVII.
Соединения формулы I, где A представляет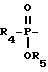
X представляет O или S и Y представляет CH2, O или S, могут быть получены реакцией сочетания фосфонхлоридата формулы XIX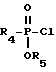
где
R5 представляет низший алкил или бензил,
с соединением с конденсированным бициклическим ядром формулы III, с получением продукта формулы XX
Предпочтительно R3 в соединении формулы III представляет низший алкил или бензил. R3 и R5 кислые защищающие группы могут быть затем удалены, например, гидрированием с получением соответствующих продуктов формулы I, где R3 и R5 представляют водород.
Фосфонохлоридаты формулы XIX известны в литературе (см., например, Karanewsky и др. патент США 4432971 и 4432972 и Karanewsky патент США 4460579).
Продукты формулы I, где X или Y или оба представляют S-(O)t и t равно 1 или 2, могут быть получены окислением соединений формул IV, XV, XVII или XX известными окисляющими агентами, такими как мета-хлорбензойная кислота, перуксусная кислота, монопероксифталевая кислота, гексагидратная соль магния и т. д. Контролированием количества окисляющего агента и времени реакции получают продукты, где t равно 1 или 2.
Продукты формулы I в эфирной форме, где R5 или R7 представляет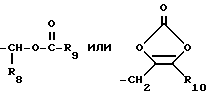
могут быть получены обработкой соответствующих соединений формулы I, где R5 или R7 представляет водород и R3 представляет кислую защищающую группу, соединением формулы XXI
где
L представляет удаляемую группу, такую как хлор, бром или толилсульфонилокси,
с последующим удалением кислой защищающей группы R3.
Эфирные продукты формулы I, где R3 представляет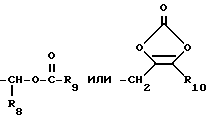
могут быть получены обработкой соответствующих соединений формулы I, где R3 представляет водород и R2 представляет 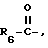 соединением формулы XXI.
соединением формулы XXI.
Соединения с конденсированными бициклическими ядрами формулы III могут быть получены согласно следующим способам, которые также являются частью этого изобретения. Например, если Y представляет CH2, то N-защищенная аминокислота формулы XXII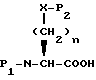
может быть подвергнута реакции сочетания с эфиром аминокислоты формулы XXIII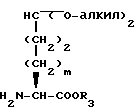
с получением дипептида формулы XXIV
где
P1 представляет аминозащищающую группу, такую, как бензилоксикарбонил или т-бутилоксикарбонил, или группу, которая вместе с N-атомом образует защищающую группу, такую как фталимидо;
P2 представляет гидрокси или меркаптозащищающую группу;
R3 представляет легко удаляемую эфирную защищающую группу.
Предпочтительными P2 защищающими группами, если X представляет S, являются ацильные группы, такие как ацетильная или бензоильная, особенно ацетильная. Предпочтительными P2 защищающими группами, если X представляет O, являются ацильные группы, тетрагидропираны, затрудненные силильные группы и тритилы, особенно трифенилметил и 1,1-диметилэтилдиметилсилил. Эту реакцию сочетания проводят предпочтительно в присутствии агента сочетания, такого как бензотриазол-1-илокситрис-(диметиламино)фосфонийгексафторфосфат, этил-3-(3-диметиламино)пропилкарбодиимид или метансульфонилоксибензотриазол.
P2 защищающая группа может быть селективно удалена из промежуточного соединения формулы XXIV, например, обработкой метоксидом натрия в метаноле, если P2 представляет ацетил или бензоил, или обработкой кислотой, такой как п-толуолсульфоновая кислота в метаноле, если P2 представляет ацетил, бензоил, тритил, тетрагидропиранил, или 1,1-диметилэтилдиметилсилил. Полученный продукт затем подвергают реакции циклизации катализируемой кислотой, предпочтительно обработкой сильной кислотой, такой как трифторуксусная кислота, пара-толуолсульфоновая кислота или коммерчески доступным полистиролсульфонатным полимером типа ионообменной смолы, такой как Амберлит 15®. Эта реакция циклизации может быть выполнена в непротонном растворителе, таком как метиленхлорид или хлороформ, с получением промежуточного соединения формулы XXV
Соединения формулы XXIV, после удаления P2 защищающей группы и до реакции циклизации, где X представляет O, могут быть превращены в соответствующие соединения, где X представляет S. Это может быть проведено различными способами. Например, соединение формулы XXIV, после удаления P2 группы, может быть обработано трифенилфосфином, диизопропилазадикарбоксилатом и тиоуксусной кислотой. Полученный тиоацетат затем обрабатывают метоксидом натрия в метаноле, получая соответствующий меркаптан, который может быть затем подвергнут описанной выше реакции циклизации.
В другом способе, соединение формулы XXIV, после удаления P2 группы, обрабатывают известными способами, получая соединение формулы XXVI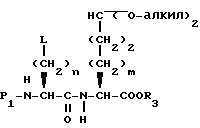
где
L представляет удаляемую группу, такую как метансульфонилокси, пара-толуолсульфонилокси, йод или бром. Например, обработка соединения формулы XXIV, после удаления P2 защищающей группы метансульфонилхлоридом, дает соединение формулы XXVI, где L представляет метансульфонилокси.
Соединение формулы XXVI затем обрабатывают цезийтиоацетатом, получая соответствующий тиоацетат. Обработка метоксидом натрия в метаноле дает соответствующий меркаптан, который может быть затем подвергнут реакции циклизации, описанной выше.
Или же соединение формулы XXIV, где X представляет O, может быть непосредственно превращено в промежуточное соединение формулы XXV обработкой сильной кислотой, такой как трифторуксусная кислота, пара-толуолсульфоновая кислота, или коммерчески доступным полистиролсульфонатным полимером типа ионообменной смолы, такой как Амберлит 15®, в соответствующем растворителе, таком как метиленхлорид или хлороформ.
N-Защищающую группу затем удаляют из соединения формулы XXV, например, обработкой гидразинмоногидратом, если R1 образует вместе с атомом N фталимидо группу, или обработкой иодтриметилсиланом или палладием на угле и аммониевой солью муравьиной кислоты или водородом, если P1 представляет бензилоксикарбонил, или обработкой хлористоводородной кислотой в диоксане или другой сильной кислоте, если P1 представляет т-бутоксикарбонил, получая соединение формулы III с конденсированным бициклическим ядром.
Еще в другом способе, если Y представляет CH2, N-защищенная аминокислота формулы XXII может быть подвергнута реакции сочетания с эфиром гидроксиаминовой кислоты формулы XXVII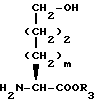
с получением дипептида формулы XXVIII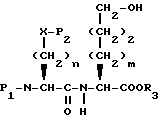
где
P1 и P2 имеют значения, как они определены выше.
Эту реакцию сочетания предпочтительно проводят в присутствии агента сочетания, такого как метансульфонилоксибензотриазол или этил-3-(диметиламино)карбодиимид.
Гидроксисоединение XXVIII затем окисляют до альдегида формулы XXIX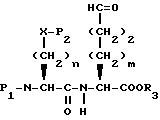
обработкой оксалилхлоридом/диметилсульфоксидом с последующей обработкой третичным амином в непротонном растворителе, таком как метиленхлорид. Альдегид формулы XXIX затем обрабатывают, как описано выше, для удаления P2 защищающей группы и затем подвергают реакции циклизации катализируемой кислотой, как описано выше, давая промежуточное соединение формулы XXV.
Исходное вещество формулы XXIII, где m равно 1, может быть получено селективной защитой N-атома L-ε-гидроксинорлейцина, что дает соединение формулы XXX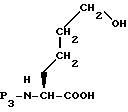
где
P3 представляет N-защищающую группу. Например, R3 и N-атом могут образовывать фталимидный остаток. N-защищенный L -ε- гидроксинорлейцин формулы XXX затем обрабатывают для введения P3 кислой защищающей группы метилиодидом в присутствии основания или обрабатывают сильной кислотой в метаноле, где R3 представляет метил. Этот эфир затем окисляют, получая альдегид формулы XXXI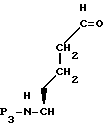
Альдегид формулы XXXI затем обрабатывают орто-муравьиным эфиром формулы XXXII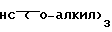
в присутствии в качестве катализатора сильной кислоты и соответствующего спирта, т. е. OH-алкила, где алкил представляет такой же алкил как в орто-муравьином эфире формулы XXXII, получая соединение формулы XXXIII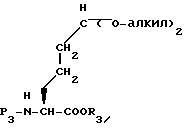
Удаление N-защищающей группы R3, например, обработкой гидразингидратом, если R3 и N-атом образуют фталимидный остаток, дает исходное вещество формулы XXIII, где m равно 1.
Исходное вещество формулы XXIII, где m равно 0, может быть получено путем защиты N-атома γ - бензилглутамата с получением соединения формулы XXXIV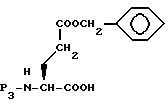
где
P3 представляет N-защищающую группу, такую как т-бутилоксикарбонил, или где P3 и N-атом могут образовать фталимидный остаток.
N-защищенную глутаминовую кислоту формулы XXXIV затем обрабатывают для введения R3 кислой защищающей группы, как описано выше, с получением XXXV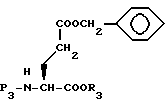
Гидролизом, если R3 представляет низший алкил, удаляют бензильную эфирную группу из соединения XXXV с получением XXXVI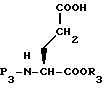
Селективное восстановление соединения XXXVI, например, обработкой этантиолом, этил-3-(3-диметиламино)пропил карбодиимидом и диметиламинопиридином с последующей обработкой триэтилсиланом, палладием на угле и ацетонитрилом дает альдегид формулы XXXVII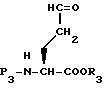
Альдегид формулы XXXVII затем обрабатывают орто-эфиром муравьиной кислоты, как описано выше, и N-защищающую группу P3 удаляют, как описано выше, с получением исходного вещества формулы XXIII, где m равно 0.
Гидроксисодержащий эфир аминокислоты исходного вещества формулы XXVII может быть получен взаимодействием раствора диэтилацетамидомалоната с суспензией гидрида натрия при перемешивании с последующей реакцией с галоидалкилацетатом формулы XXXVIII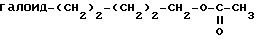
где галоид представляет Br, I или Cl,
с получением соединения формулы XXXIX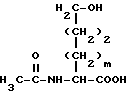
Раствор диэтилового эфира формулы XXXIX обрабатывают гидроокисью натрия и нагревают и затем подкисляют и нагревают вновь с получением гидроксиаминокислоты формулы XL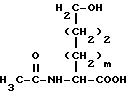
Гидроксиаминокислоту формулы XL затем обрабатывают ацилазой свиной почки или другим соответствующим гидролизующим ферментом с получением гидроксиаминокислоты формулы XLI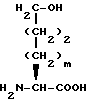
Гидроксиаминокислоту формулы XLI затем превращают в эфир формулы XXVII обычным способом. Например, гидроксиаминокислота формулы XLI может быть обработана триметилсилилхлоридом в метаноле с получением гидрохлоридной соли метилового эфира соединения формулы XXVII.
Исходные вещества формулы XXII могут быть получены следующим образом. Если X представляет O, гидрокси α- аминокислоту формулы XLII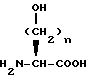
подвергают взаимодействию для введения P1 и P2 защищающих групп. Например, обработка кислоты формулы XLII N-карбетоксифталимидом в присутствии карбоната натрия с последующей обработкой хлортрифенилметаном и триэтиламином дает исходное вещество формулы XXII, где X представляет O, R1 вместе с атомом N образует фталимидо и P2 представляет тритил. Или же обработка кислоты формулы XLII N-(бензилоксикарбонилокси)сукцинимидом в водном растворе карбоната натрия и ацетона с последующей обработкой т-бутилдиметилсилилхлоридом или ацилирующим агентом формулы V или VI дает исходное вещество формулы XXII, где X представляет O, P1 представляет бензилоксикарбонил и P2 представляет т-бутилдиметилсилил или ацильную группу, такую как ацетильная.
Если X представляет S и n равно 1, N,N'-бис[(фенилметокси)карбонил]-L-цистеин может быть обработан цинковой пылью и водным раствором серной кислоты с получением меркаптана формулы XLIII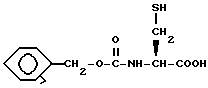
Меркаптан формулы XLIII затем обрабатывают для введения P2 защищающей группы. Например, обработка меркаптана формулы XLIII уксусным ангидридом дает исходное вещество формулы XXII, где X представляет S, n равно 1, P2 представляет ацетил и P1 представляет бензилоксикарбонил.
Если X представляет S и n равно 2, L-метионин может быть защищен по N-атому. Например, реакция с бензилхлорформатом или N-(бензилоксикарбонилокси)сукцинимидом дает N-[(фенилметокси)карбонил]-L-метионин, который затем этерифицируют обработкой спиртом, алкил-OH, в присутствии в качестве катализатора кислоты, такой как п-толуолсульфоновая кислота. Обработка окисляющим агентом, таким как N-хлорсукцинимид, в водном растворе дает сульфоксид формулы XLIV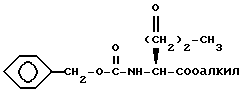
Сульфоксид формулы XLIV затем обрабатывают ангидридом кислоты, таким как уксусный ангидрид, с получением соединения формулы XLV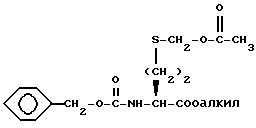
Обработка гидроокисью щелочного металла, с последующим удалением формальдегида, например, обработкой восстанавливающим агентом, например боргидридом натрия, с последующей обработкой ангидридом кислоты, таким как уксусный ангидрид, дает исходное вещество формулы XXII, где X представляет S, n равно 2, R2 представляет ацетил и P1 представляет бензилоксикарбонил.
Соединения с бициклическим конденсированным ядром формулы III, где Y представляет S или O и m равно 1, могут быть получены реакцией сочетания N-защищенной аминокислоты формулы XXII с эфиром аминокислоты формулы XLVI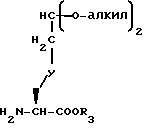
давая дипептид формулы XLVII
где
P1 и P2 имеют значения, как они определены ранее;
R3 представляет кислую защищающую группу.
Эту реакцию сочетания предпочтительно проводят в присутствии агента сочетания, такого как бензотриазол-1-илокситрис(диметиламино)фосфонийгексафторфосфата или этил-3-(3-диметиламино)пропил карбодиимида.
P2 защищающая группа может быть селективно удалена из промежуточного соединения формулы XLVII, например, обработкой метоксидом натрия в метаноле, если P2 представляет ацильную группу такую как ацетил или бензоил, и обработкой кислотой, такой как п-толуолсульфоновая кислота в метаноле, если P2 представляет тритил, тетрагидропиранил, или затрудненную силильную группу. Полученный продукт затем подвергают реакции циклизации катализируемой кислотой, как описано выше, с получением промежуточного соединения формулы XLVIII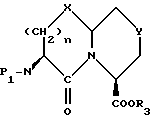
Промежуточное соединение формулы XLVIII, где X представляет S и n равно 2, может быть получено обработкой соединения формулы XLVII, где X представляет O и n равно 2, с селективным удалением группы P2 и превращением гидрокси производного в меркаптан, как описано выше, с последующей реакцией циклизации катализируемой кислотой.
N-Защищающую группу затем удаляют из соединения формулы XLVIII, например обработкой гидразингидратом, если P1 вместе с N-атомом образуют фталимидо группу, или обработкой иодтриметилсиланом или палладием на угле и аммониевой солью муравьиной кислоты или водородом, если P1 представляет бензилоксикарбонил, давая соединения формулы III с конденсированным ядром.
Исходное вещество формулы XLVI, где Y представляет O, может быть получено взаимодействием N-фталимино защищенного эфира аминокислоты формулы XLIX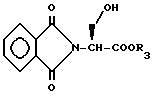
с аллилтрихлорацетимидатом в присутствии трифторметансульфоновой кислоты с получением соединения формулы L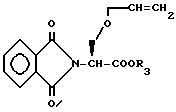
Обработка соединения формулы L озоном в метаноле, затем диметилсульфидом с последующей обработкой орто-формиатом формулы XXXII в присутствии п-толуолсульфоновой кислоты дает защищенное соединение формулы LI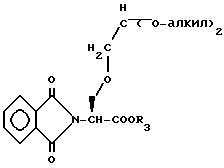
Удаление N-защищающей группы, например, обработкой гидразингидратом, дает исходное вещество формулы XLVI, где Y представляет O.
Исходное вещество формулы XLVI, где Y представляет S, может быть получено взаимодействием цистеинового эфира формулы LII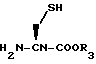
с бромацеталем формулы LIII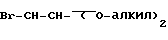
в присутствии гидрида натрия и иодида калия с получением эфира аминокислоты формулы LIV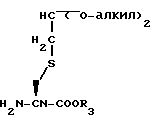
Соединения формулы I содержат три асимметричных центра в конденсированной бициклической части структуры с возможными дополнительными центрами в боковой цепи. Ввиду того, что оптически чистая форма конденсированных бициклических продуктов является предпочтительной, все такие формы находятся в пределах объема этого изобретения. Вышеописанные способы могут использовать рацематы, энантиомеры или диастереомеры в качестве исходных веществ. Если получают диастереомерные соединения, они могут быть разделены обычными хроматографическими способами или фракционной кристаллизацией. Предпочтительно, водород присоединяется к главному мостиковому атому углерода в ориентации, показанной ниже
Соединения формулы I, где R3, R5 и/или R7 представляют водород, могут быть выделены в форме фармацевтически приемлемой соли. Соответствующие соли для этой цели представляют соли щелочных металлов, таких как натрий и калий, соли щелочноземельных металлов, таких как кальций и магний, соли полученные из аминокислот, таких как аргинин, лизин и т.д. и соли полученные из аминов, таких как алкиламины, например т-бутиламин, т-амиламин и т.д., замещенных алкиламинов, например бензиламин, диалкиламинов, замещенных диалкиламинов, например N-метилглюкамин, триалкиламинов, замещенных триалкиламинов и четвертичных аммониевых солей. Эти соли могут быть получены взаимодействием кислой формы соединения с основанием, обеспечивающим нужный ион, в среде, в которой осаждается соль, или в водной среде, затем лиофилизацией.
Предпочтительными соединениями этого изобретения являются те соединения, где
A представляет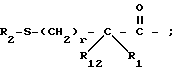
R2 представляет водород, 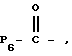 или R11 -S-; R3 представляет водород или низший алкил с 1-4 атомами углерода; r равно 0 или 1; R11 представляет низший алкил с 1-4 атомами углерода; R1 представляет арил-CH2-, замещенный арил-CH2-; гетероарил-CH2-; циклоалкил-CH2-, где циклоалкил содержит 3-7 атомов углерода, или неразветвленную или разветвленную алкильную цепь с 1-7 атома углерода и R12 представляет водород; или R1 и R12 взятые вместе с атомом углерода, к которому они присоединяются, дают циклоалкильное кольцо с 5-7 атомами углерода; R6 представляет низший алкил с 1-4 атомами углерода или фенил; n равно 1 или 2; m равно 0 или 2; X представляет O или S и Y представляет CH2, O или S при условии, что Y представляет O или S, только если m равно 1.
или R11 -S-; R3 представляет водород или низший алкил с 1-4 атомами углерода; r равно 0 или 1; R11 представляет низший алкил с 1-4 атомами углерода; R1 представляет арил-CH2-, замещенный арил-CH2-; гетероарил-CH2-; циклоалкил-CH2-, где циклоалкил содержит 3-7 атомов углерода, или неразветвленную или разветвленную алкильную цепь с 1-7 атома углерода и R12 представляет водород; или R1 и R12 взятые вместе с атомом углерода, к которому они присоединяются, дают циклоалкильное кольцо с 5-7 атомами углерода; R6 представляет низший алкил с 1-4 атомами углерода или фенил; n равно 1 или 2; m равно 0 или 2; X представляет O или S и Y представляет CH2, O или S при условии, что Y представляет O или S, только если m равно 1.
Наиболее предпочтительными являются соединения, приведенные выше, где R2 представляет водород или 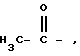 особенно водород; R3 представляет водород; r равно 0 или 1, особенно 1; R1 представляет бензил, циклопропилметил, или неразветвленную или разветвленную алкильную цепь с 3-5 атомами углерода, особенно бензил; R12 представляет водород; n равно 1 или 2; m равно 0 или 1; X представляет O или S и Y представляет CH2, O или S при условии, что Y представляет O или S, только если m равно 1.
особенно водород; R3 представляет водород; r равно 0 или 1, особенно 1; R1 представляет бензил, циклопропилметил, или неразветвленную или разветвленную алкильную цепь с 3-5 атомами углерода, особенно бензил; R12 представляет водород; n равно 1 или 2; m равно 0 или 1; X представляет O или S и Y представляет CH2, O или S при условии, что Y представляет O или S, только если m равно 1.
Единственное наиболее предпочтительное соединение представляет /4S-/4 α (r*), 7 α , 10a β/ //-октагидро-4-/(2-меркапто-1-оксо-3-фенилпропил)амино/-5-оксо-7H- пиридо[2,1-b]-/1,3/тиазепин-7-карбоновую кислоту, т.е. соединение формулы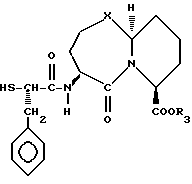
Соединения формулы I, где A представляет
являются ингибиторами двойного действия, обладающими способностью ингибировать превращение фермента ангиотензина и нейтральной эндопептидазы. Соединения формулы I, где A представляет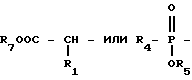
являются селективными ингибиторами, обладающими способностью ингибировать превращение фермента ангиотензина. Таким образом, соединения формулы I, включающие их фармацевтически приемлемые соли, являются полезными при лечении физиологических состояний, в которых ингибиторы превращения фермента ангиотензина были показаны как полезные. Такие состояния включают болезненные состояния, характеризующиеся нарушениями давления крови, внутриглазного давления, включая сердечно-сосудистые заболевания, в частности гипертензию и застойную сердечную недостаточность, глаукому и ренальные болезни, такие как почечная недостаточность, диабетическая нефропания, и почечная недостаточность (renal impairment) с последующим лечением циклоспорином или другими иммунодепрессантами. Другие состояния, в которых ингибиторы превращения фермента ангиотензина, как сообщалось, являются полезными, включают цирроз печени, ингибирование прогрессирования атеросклероза, предупреждение или лечение гипотензивной или диабетической ретинопатии, улучшение сердечной дисфункции или последующие перебои сердечной деятельности и предупреждение рестинозиса после пластической операции на сосудах. Ингибиторы двойного действия являются также полезными при лечении физиологических состояний, в которых, как показано, будут полезными ингибиторы нейтральной эндопептидазы. Такие состояния также включают сердечно-сосудистые заболевания, в частности гипертензию, гиперальдостеромию, почечные болезни, глаукому, а также ослабление острой и хронической боли. Таким образом, соединения формулы I являются полезными для снижения кровяного давления, а ингибиторы формулы I двойного действия являются, кроме того, полезными для этой цели благодаря их диурезным и натриурезным свойствам. Ингибиторы двойного действия являются особенно полезными для лечения застойных сердечных нарушений.
Соединения формулы I, включающие их фармацевтически приемлемые соли, могут быть введены для этих целей в количествах, аналогичных тем, которые применяли ранее для ингибиторов превращения фермента ангиотензина. Например, соединения формулы I могут быть введены млекопитающему, например человеку, дозой от около 0,1 мг до около 100 мг на кг веса тела в день, предпочтительно от около 0,5 мг до около 25 мг на кг веса тела в день. Соединения формулы I предпочтительно вводятся орально, но парентеральные пути, такие как подкожно, внутримышечно и внутривенно, также могут быть использованы, как могут быть использованы и локальные пути введения. Дневная доза может быть введена в один прием или может быть разделена на 2 - 4 дозы введения в течение дня.
Ингибиторы формулы I могут быть введены в комбинации с ANF 99 - 126 человека. Такая комбинация будет содержать ингибитор формулы I от около 1 до около 100 мг на 1 кг веса тела и ANF 99-126 человека от около 0,001 до около 0,1 мг на 1 кг веса тела.
Ингибиторы формулы I могут быть введены в комбинации с другими типами фармацевтически активных соединений. Например, диуретических, блокаторов кальциевых каналов, активаторов калиевых каналов, восстанавливающих агентов холестерина, β- блокаторов и т.д.
Ингибиторы формулы I или их фармацевтически приемлемые соли и другие фармацевтически приемлемые ингредиенты могут быть включены в композицию для вышеописанного фармацевтического использования. Соответствующие композиции для орального введения включают таблетки, капсулы и эликсиры, и соответствующие композиции для парентерального введения включают стерильные растворы и суспензии. Соответствующие композиции для лечения глаукомы также включают локальные композиции, такие как растворы, мази и твердые включения, как описано в патенте США 4442089. От около 10 до 500 мг активного ингредиента включается вместе с физиологически приемлемым связующим, носителем, наполнителем, консервантом, стабилизатором, ароматическим веществом и т.д. в форме единичной дозы, как отмечено, для приемлемой фармацевтической практики.
Следующие примеры иллюстрируют изобретение. Температуры даны в градусах. Тонкослойную хроматографию выполняли на силикагеле, если не оговорено особо.
Пример 1
/4S-/ 4α (R*), 7α 10a β //-октагидро-4-//2- меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-7H-пиридо/2,1-b//1,3/ оксазепин-7-карбоновая кислота
a) (S)-2-фталимидо-4-гидроксибутановая кислота, соль триэтиламина
Раствор L-гомосерина (3,0 г, 25,2 ммоля) и карбоната натрия (2,670 г, 25,2 ммоля) в воде (60 мл) обрабатывали N-карбетоксифталимидом (5,570 г, 25,4 ммоля). После перемешивания при комнатной температуре в течение 2 часов раствор подкисляли 6N соляной кислотой и экстрагировали этилацетатом. Этилацетатный экстракт промывали рассолом, сушили (сульфатом натрия) и фильтровали в раствор триэтиламина (4,0 мл) в метиленхлориде (40 мл). Мутный раствор концентрировали и растирали с этилацетатом и этиловым эфиром с получением 5,11 г указанного в названии соединения в виде белого твердого вещества: т.пл. 142 - 144oC, TLC (5% уксусная кислота в этилацетате) Rf = 0.36;
[α]D= -6,2o (c = 0,8 хлороформ).
Данные анализа для C18H26N2O5:
Вычислено, %: С 61,70; Н 7,48; N 7,99
Найдено, %: С 61,45; Н 7,47; N 7,84
b) (S)-2-фталимидо-4-/трифенилметокси/бутановая кислота, соль триэтиламина
Гомогенный раствор продукта стадии (a) (1,890 г, 5,4 ммоля) в хлороформе (20 мл) обрабатывали триэтиламином (80 мкл) с последующей обработкой твердым хлортрифенилметаном (1,590 г, 5,70 ммоля). После перемешивания при комнатной температуре в течение 2,5 часов раствор разделялся между этилацетатом и 0,1 N соляной кислотой (150 мл). Органический слой промывали водой и рассолом, затем сушили (сульфатом натрия) и фильтровали в раствор триэтиламина (1,0 мл) в метиленхлориде (30 мл). Раствор концентрировали до масла, вновь растворяли в небольшом количестве метиленхлорида и этилацетата и растирали с этилацетатом до тех пор, пока раствор не помутнеет. В смесь помещали затравку и оставляли при комнатной температуре. Образующийся осадок собирали фильтрованием, промывали этилацетатом и этиловым эфиром и сушили в вакууме с получением 2,538 г указанного в названии соединения в виде белого твердого вещества: т. пл. 165 - 170oC (разлож.) TLC (10% метанола в хлороформе) Rf = 0,23;
[α]D= +7,0o (c = 1,2, хлороформ).
c) (S)-2-фталимидо-6-гидроксигексановая кислота
Раствор (+)-L-ε-гидроксинорлейцина (полученного согласно способу Bodanszky и др. , J. Med. Chem. 1978, 21, 1030-1035) (1,030 г, 7,0 ммоля) и карбоната натрия (745 мг, 7,0 ммоля) в воде (12 мл) обрабатывали N-карбетоксифталимидом (1,495 г, 7,0 ммоля) и смесь перемешивали при комнатной температуре в течение 2 часов. Раствор фильтровали, охлаждали до 0oC и подкисляли 6 N соляной кислотой с получением белого осадка. Твердое вещество собирали фильтрованием и сушили в вакууме при 80oC в течение 1 часа с получением 1,297 г указанного в названии соединения: т.пл. 162 - 163oC; [α]D= -35,7o (c = 1,3, метанол).
d) (S)-2-фталимидо-6,6-диметоксигексановая кислота, метиловый эфир
Сырой продукт из стадии (c) (3,752 г, 13,5 ммоля) и карбонат цезия (2,178 г, 6,7 ммоля) в диметилформамиде (44 мл) обрабатывали метилиодидом (3,0 мл, 6,84 г, 48,2 ммоля). После перемешивания при комнатной температуре в течение 2 часов смесь разбавляли этилацетатом и промывали последовательно водой, содержащей небольшое количество бисульфита натрия, водой, 50% насыщенным раствором бикарбоната натрия и рассолом, затем сушили (сульфатом натрия), фильтровали и удаляли с получением промежуточного эфира в виде бесцветного масла (3,825 г). Масло было гомогенным по данным TLC (1:1 - ацетон:гексан) Rf = 0,37.
Раствор оксалилхлорида при -78oC (1,37 мл, 2,00 г, 15,7 ммоля) в сухом метиленхлориде (58 мл) обрабатывали по каплям раствором сухого диметилсульфоксида (2,24 мл, 2,47 г, 31,6 ммоля) в метиленхлориде (2 мл). Через 10 минут добавляли раствор приведенной выше спирт-эфирной смеси, (3,825 г, 13,1 ммоля) в метилехлориде (10 мл). Затем через 15 минут добавляли триэтиламин (8,0 мл) и смесь перемешивали при -78oC в течение 5 минут, затем нагревали до 0oC. Смесь разбавляли этилацетатом/этиловым эфиром и последовательно промывали 1 N соляной кислотой, водой и рассолом, затем сушили (сульфатом натрия), фильтровали и удаляли с получением неочищенного желаемого альдегида. Масло было гомогенным по данным TLC (1:1 ацетон:гексан) Rf = 1,48.
Раствор приведенного выше альдегида в метаноле (17 мл) и метиленхлорида (17 мл) обрабатывали триметиловым эфиром орто-муравьиной кислоты (1,7 мл) с последующей обработкой моногидратом п-толуолсульфоновой кислоты (180 мг). Смесь перемешивали при комнатной температуре в течение 1,5 часов, затем разделяли между этилацетатом и 50% насыщенным раствором бикарбоната натрия. Органический слой промывали водой и рассолом, затем сушили (сульфатом натрия), фильтровали и удаляли. Остаток подвергали флеш-хроматографии (Мерк силикагель, 1: 1 этилацетат:гексан) и чистили фракции продукта кристаллизацией из смеси этилацетат/гексан, получая аналитически чистый указанный в названии продукт (3,452 г, первый отбор и 215 мг второй отбор) в виде белого игольчатого вещества: т. пл. 69-70oC, TLC (1:1 - этилацетат:гексан) Rf = 0,35; [α]D= -27,4o (c = 1,5, хлороформ).
Данные для анализа для C17H21NO6:
Вычислено, %: С 60,89; Н 6,31; N 4,18
Найдено, %: С 60,80; Н 6,32; N 4,16.
e) /S/-/R*R*//-2//2-фталимидо-4-/трифенилметокси/- 1-оксобутил/амино/-6,6-диметоксигексановая кислота, метиловый эфир
Неочищенный продукт из части (d) (2,540 г, 7,57 ммоля) в метаноле (18 мл) обрабатывали гидразинмоногидратом (378 мкл, 390 мг, 7,80 ммоля). Смесь становилась гомогенной в течение 10 минут. После перемешивания при комнатной температуре в течение 3 дней полученный шлам фильтровали, удаляли, смешивали с метиленхлоридом, фильтровали и вновь удаляли, получая неочищенный промежуточный амин в виде бесцветного масла. В то же время раствор соли триэтиламина продукта из части (b) (4,622 г, 7,80 ммоля) в метиленхлориде (50 мл) при 0oC обрабатывали бензотриазол-1-илокситрис/диметиламино/фосфоний гексафторфосфатом (3,519 г, 7,95 ммоля). Смесь перемешивали в течение 35 минут, затем обрабатывали раствором приведенного выше амина в метиленхлориде (15 мл). Через 10 минут реакции при 0oC и 2 часов при комнатной температуре, раствор разделялся между этиловым эфиром и водой. Органический слой промывали 50% насыщенным раствором бикарбоната натрия и рассолом, затем сушили (сульфатом натрия), фильтровали и удаляли. Остаток подвергали флеш-хроматографированию (Мерк силикагель, 6:4 этилацетат:гексан), получая 3,580 г чистого указанного в названии соединения в виде белой пены. TLC (6:4 этилацетат:гексан) Rf = 0,32; [α]D= +26,2o (c = 0,6, хлороформ).
f) /S-/R*, R*//-2//2-фталимидо-4-гидрокси-1-оксо- бутил/амино/-6,6-диметоксигексановая кислота, метиловый эфир
Раствор продукта из части (e) (5,420 г, 8,0 ммоля) в метаноле (60 мл) обрабатывали моногидратом п-толуолсульфоновой кислоты (520 мг). После перемешивания при комнатной температуре в течение 1,5 часов смесь разделялась между этилацетатом и разбавленным раствором бикарбоната натрия. Фазы разделяли и водный слой вновь экстрагировали этилацетатом. Собранные органические экстракты промывали рассолом, сушили (сульфатом натрия), фильтровали и удаляли. Остаток подвергали флеш-хроматографированию (Мерк силикагель 8:2 - этилацетат: гексан с последующим 5% метанола в этилацетате) с получением 2,860 г указанного в названии продукта в виде бесцветного масла. TLC (7:3 этилацетат:гексан) Rf = 0,26; [α]D= +18,7o (c = 1,3, хлороформ).
g) /4S- 4α,7α,10aβ //-Октагидро-4-фталимидо-5-оксо-7H-пиридо[2,1-b] - /1,3/оксазепин-7-карбоновая кислота, метиловый эфир
Раствор продукта из части (f) (2,10 г, 4,95 ммоля) в метиленхлориде (100 мл) обрабатывали Амберлитом® 15 - ионообменной смолой (240 мг, предварительно последовательно промытой 6N соляной кислотой, водой, тетрагидрофураном, затем метиленхлоридом). После перемешивания при комнатной температуре в течение 2,5 часов раствор фильтровали, удаляли и подвергали флеш-хроматографированию (Мерк силикагель, 6:4 - этилацетат:гексан с последующим 100% этилацетатом), получая 1,40 г указанного в названии продукта в виде белой пены.
h) /S/-2-/Ацетилтио/бензолпропановая кислота
Раствор нитрита (10,3 г, 280 ммолей) добавляли к раствору D-фенилаланина (30,0 г, 181 ммоля) и бромида калия (73,5 г) в серной кислоте (2,5 N, 365 мл) в течение 1 часа, поддерживая одновременно температуру реакции при 0oC. Смесь дополнительно перемешивали в течение 1 часа при 0oC и затем в течение 1 часа при комнатной температуре. Реакционный раствор экстрагировали эфиром, эфир обратно экстрагировали водой и эфирный слой сушили над сульфатом натрия. Эфир удаляли в вакууме - отгоняли маслообразный остаток, получая 25,7 г /R/-2-бром-3-бензолпропановой кислоты: т. кип. 141oC (0,55 мм Hg); [α]D= + 14,4o(c = 2,4, хлороформ).
Смесь тиоуксусной кислоты (7 мл, 97,9 ммоля) и гидроокиси калия (5,48 г, 97,9 ммоля) в ацетонитриле (180,5 мл) перемешивали в атмосфере аргона при комнатной температуре в течение 1 и 3/4 часа. Смесь охлаждали в бане со льдом и раствор /R/-2-бром-3-бензолпропановой кислоты (20,4 г, 89 ммоля) в ацетонитриле (20 мл) добавляли в течение 10 минут. Реакционную смесь перемешивали в атмосфере аргона при комнатной температуре в течение 5 часов, фильтровали и удаляли ацетонитрил в вакууме. Маслообразный остаток вновь растворяли в этилацетате и промывали 10% раствором бисульфата калия и водой. Этилацетат удаляли в вакууме, получая 19,6 г неочищенного продукта. Неочищенный продукт чистили через получение его дициклогексиламиновой соли, используя изопропиловый эфир в качестве растворителя для кристаллизации. Аналитически чистый образец /S/-2-/ацетилтио/бензолпропановой кислоты, дициклогексиламиновой соли получали перекристаллизацией из этилацетата: т.пл. 146 - 147oC; [α]D= - 39,6oC (c = 1,39, хлороформ).
Данные анализа для C11H12O3S • C12H23N:
Вычислено, %: С 68,11; Н 8,70; N 3,45, S 7,91
Найдено, %: С 67,93; Н 8,71; N 3,37, S 7,94.
Свободную кислоту регенерировали разделением дициклогексиламиновой соли между 5% раствором бисульфата калия и этилацетатом, получая /S/-2-/ацетилтио/бензолпропановую кислоту; [α]D= -70,1oC (c= 1,91, хлороформ).
Данные анализа для C11H12O3S:
Вычислено, %: С 58,91; Н 5,39; S 14,30
Найдено, %: С 58,73; Н 5,41; S 14,53.
i/ Метиловый эфир /4S-/4 α /R*/, 7 α 10a β //-октагидро-4-//2-/ацетилтио/-1-оксо-3-фенилпропил/амино/-5-оксо-7H- [2,1-b] /1,3/оксазепин-7-карбоновой кислоты
Продукт из части (g) (620 мг, 166 ммоля) в метаноле (10 мл) обрабатывали гидразинмоногидратом (85 мкл, 88 мг, 1,75 ммоля) и раствор перемешивали при комнатной температуре в течение 44 часов. Смесь фильтровали и твердое вещество промывали метанолом. Фильтрат удаляли, растирали с метиленхлоридом, снова фильтровали и удаляли, получая неочищенный амин в виде мутного масла (около 400 мг).
Холодный раствор (0oC) /S/-2-/ацетилтио/бензолпропановой кислоты (410 мг, 1,83 ммоля) и триэтиламина (250 мкл, 182 мг, 1,80 ммоля) в метиленхлориде (10 мл) обрабатывали вышеуказанным амином (в виде раствора в 8 мл метилхлорида) с последующей обработкой бензтриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфатом (808 мг, 1,83 ммоля). Прозрачный, практически бесцветный раствор перемешивали при 0oC в течение 40 минут и затем при комнатной температуре в течение 2 часов. Смесь разделялась между этилацетатом /этиловым эфиром и водой. Органический слой промывали последовательно 50% раствором насыщенного бикарбоната натрия и рассолом, затем сушили (сульфатом натрия), фильтровали и удаляли. Остаток подвергали флэш хроматографированию (Мерк силикагель 60-70% этилацетата в гексане), получая 602 мг чистого, указанного в названии продукта в виде белой пены: TLC (6:4 - этилацетат:гексан) Rf = 0.27.
j) /4S -/4α/R*/, 7α, 10aβ/- октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5 -оксо-7H-пиридо[2,1-b] /1,3/оксазепин-7-карбоновая кислота
Раствор продукта из части (i) при 0oC (590 мг, 1,32 ммоля) в метаноле (10 мл, обескислороженный пробулькиванием аргона) обрабатывали 1 N раствором гидроокиси натрия (7 мл, обескислороженным пробулькиванием аргона). После перемешивания в течение 15 минут раствор нагревали до комнатной температуры и продолжали перемешивание в атмосфере аргона дополнительно 4,5 часа. Смесь подкисляли 5% бисульфатом калия, разбавляли водой и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой и рассолом, затем сушили (сульфатом натрия), фильтровали и концентрировали до приблизительно 3 мл. Остаток смешивали с этилацетатом и небольшим количеством гексана и полученное твердое вещество собирали фильтрованием и сушили в вакууме, получая 413 мг указанного в названии продукта: т.пл. 180,5oC (разложен.). TLC (2% уксусной кислоты в этилацетате) Rf = 0,39;
[α]D= -37,6o /c = 0,36, метанол/.
HPLC: YMC S30DS колонка (6,0 х 150 мм); элюируют 40% A:90% воды - 10% метанола - 0,2% фосфоновой кислоты и 60% B:10% воды - 90% метанола - 0,2% фосфоновой кислоты; скорость потока 1,5 мл/мин с детектированием при 220 нм; t = 6,73 мин (95,7%).
Данные анализа для C20H28N2O4S • 0,12 этилацетат:
Вычислено, %: С 58,05; Н 6,24; N 6,95; S 7,96
Найдено, %: С 58,23; Н 6,34; N 6,83; S 7,81.
Пример 2
/3R /3α/S*/, 6α, 9aβ//- гексагидро-3-//2-меркапто-1-оксо-3-фенилпропил/амино/-4-оксо-2H, 6H-пиридо [21-b]/1,3-тиазин-6-карбоновая кислота
a) N-//фенилметокси/карбонил/-L-цистеин
Раствор N, N'-бис//фенилметокси/карбонил/-L-цистеина (4,658 г, 9,16 ммоля) в метаноле (35 мл) обрабатывали 2N раствором серной кислоты (23 мл) с последующей обработкой порциями цинковой пылью (2,442 г, 37,3 ммоля). Смесь нагревали при 70oC в течение 1,5 часов, фильтровали пока раствор остается еще теплым и концентрировали на роторном испарителе. Оставшийся раствор экстрагировали этиловым эфиром и эфирный экстракт промывали водой и рассолом и сушили (сульфатом натрия), фильтровали и удаляли. Остаток (масло) растворяли в четыреххлористом углероде, охлаждали до 0oC и вводили затравку до медленного образования осадка. Твердое вещество собирали фильтрованием и промывали холодным четыреххлористым углеродом и подвергали флеш-хроматографированию (Мерк силикагель, этилацетат с последующей обработкой 4% уксусной кислоты в этилацетате), получая дополнительный продукт после кристаллизации (246 мг). Полный выход продукта 2,894 г. TLC (5% уксусная кислота в этилацетате) Rf = 0,58.
b) S-Ацетил-N-//фенилметокси/карбонил/-L-цистеин
Гомогенный раствор продукта из части (a) (2,70 г, 10,6 ммоля) в воде (30 мл, обескислороженный пробулькиванием аргона), содержащий бикарбонат калия (2,140 г, 21,4 ммоля), обрабатывали уксусным ангидридом (8,0 мл, 8,66 г, 84,8 ммоля). Через 10 минут при комнатной температуре смесь подкисляли 10% соляной кислотой и экстрагировали этиловым эфиром. Эфирный экстракт промывали дважды водой и рассолом и сушили (сульфатом натрия), фильтровали и удаляли, получая масло. Остаток подвергали азеотропной разгонке три раза с толуолом и дважды со смесью этиловый эфир/гексан, после чего масло кристаллизовалось. Остаток растирали со смесью этиловый эфир/гексан и твердое вещество собирали фильтрованием, получая 2,19 г чистого указанного в названии продукта. TLC (5% уксусная кислота в этилацетате) Rf = 0,56.
c) Метиловый эфир /S/-2-//N-фенилметокси/карбонил-S-ацетил-L-цистеинил/амино/-6,6- диметоксигексановой кислоты
Шлам метилового эфира /S/-2-фталимидо-6,6-диметокси-гексановой кислоты (полученный как описано в примере 1 (d), 1,158 г, 3,45 ммоля) в метаноле (12 мл) обрабатывали гидразинмоногидратом (176 мкл, 182 мг, 3,63 ммоля). Смесь становилась гомогенной в течение 10 минут. После перемешивания при комнатной температуре 67 часов полученный шлам фильтровали, удаляли, смешивали с метиленхлоридом, фильтровали и вновь удаляли, получая неочищенный промежуточный амин в виде бесцветного масла.
В то же время часть шлама продукта из части (b) (1,185 г, 3,98 ммоля) в метиленхлориде (14 мл) обрабатывали триэтиламином (555 мкл, 403 мг, 3,98 ммоля). Затем гомогенный раствор охлаждали до 0oC, обрабатывали указанным выше амином в виде раствора в метиленхлориде (7 мл), затем обрабатывали бензотриазол-1-илокситрис/диметиламино/фосфоний-гексафторфосфатом (1,762 г, 3,98 ммоля). Смесь перемешивали при 0oC в течение 2,5 часов, затем при комнатной температуре в течение 45 минут. Растворитель удаляли и остаток разделяли между этилацетатом и водой. Органический слой промывали 50% насыщенным раствором бикарбоната натрия и рассолом, затем сушили (сульфатом натрия), фильтровали и удаляли. Остаток подвергали флеш-хроматографированию (Мерк силикагель, 65: 35 этилацетат:гексан), получая 1,15 г чистого указанного в названии продукта в виде белой пены. TLC (75:25 - этилацетат:гексан) Rf = 0,42.
Данные анализа для C22H32N2O8S:
Вычислено, %: С 54,53; Н 6,66; N 5,78; S 6,62
Найдено, %: С 54,79; Н 6,72; N 5,77; S 6,95.
d) Метиловый эфир /3R- /3α, 6α, 9aβ//- гексагидро-3-//-фенилметокси/карбонил/амино/-4-оксо-2H, 6H-пиридо [2,1-b] /1,3/-тиазин-6-карбоновой кислоты
Обескислороженный раствор (пробулькивание аргоном) продукта из части (c) (1,040 г, 2,15 ммоля) в метаноле (12 мл) при 0oC обрабатывали метоксидом натрия (25 вес. % в метаноле, 490 мкл, 463 мг, 2,14 ммоля). Через 20 минут смесь гасили насыщенным раствором хлористого аммония, разбавляли водой и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой и рассолом и сушили (сульфатом натрия), фильтровали и удаляли. Остаток вновь растворяли в метиленхлориде (200 мл) и перемешивали при комнатной температуре с Амберлитом® - ионообменной смолой 820 мг, предварительно промытой последовательно 6N соляной кислотой, водой, тетрагидрофураном, затем метиленхлоридом. Через 3 часа раствор фильтровали, удаляли и подвергали флеш-хроматографированию (Мерк силикагель, 65:35 - этилацетат:гексан), получая 757 мг указанного в названии продукта в виде бесцветного масла. TLC (75:25 - этилацетат:гексан/Rf = 0.58.
e/ Метиловый эфир /3R /3α, 6α, 9aβ//- -гексагидро-3-амино-4-оксо-2H, 6H-пиридо [2,1-b]/1,3/тиазин-6-карбоновой кислоты
Раствор продукта из части (d) (752 мг, 1,99 ммоля) в сухом метиленхлориде (15 мл) обрабатывали при комнатной температуре иодтриметилсиланом (620 мкл, 872 мг, 4,36 ммоля). После перемешивания в течение 3 часов смесь гасили водой, обрабатывали небольшим количеством 10% соляной кислоты и экстрагировали этиловым эфиром. Слои разделяли и эфирный слой вновь экстрагировали водой. Собранные водные слои подщелачивали (pH 13) 10% раствором гидроокиси натрия и дважды экстрагировали метиленхлоридом. Собранные метиленхлоридные экстракты (сульфатом натрия) фильтровали и удаляли, получая 290 мг неочищенного указанного в названии продукта в виде бесцветного масла. TCL (10% метанола в метиленхлориде) Rf = 0,38.
f) Метиловый эфир /3R /3α/ S*/6α 9aβ//- гексагидро-3-//2-/ацетилтио/1-оксо-3-фенилпропил/амино/-4-оксо-2H, 6H-пиридо [2,1-b] -/1,3/тиазин-6-карбоновой кислоты
Холодный раствор (0oC) /S/-2-ацетилтио/бензолпропановой кислоты (294 мг, 1,31 ммоля) и триэтиламина (180 мкл, 131 мг, 1,29 ммоля) в метиленхлориде (8 мл) обрабатывали продуктом из части (e) (287 мг, 1,17 ммоля) в виде раствора в 6 мл метиленхлорида. Затем добавляли бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторофосфат (575 мг, 1,30 ммоля). Прозрачный, практически бесцветный раствор перемешивали при 0oC в течение 1 часа и затем при комнатной температуре в течение 1 часа. Растворитель удаляли на роторном испарителе и остаток разделяли между этилацетатом и 5% раствором бисульфата калия. Органический слой последовательно промывали водой, 50% насыщенным раствором бикарбоната натрия и рассолом, затем сушили (сульфатом натрия), фильтровали и удаляли. Остаток подвергали флеш-хроматографированию (Мерк силикагель; 1: 1 - этилацета:гексан), получая 412 мг чистого указанного в названии продукта в виде белой пены. TLС (1:1 - этилацетат:гексан) Rf = 0,27;  -107,0oC (с = 0,6, хлороформ).
-107,0oC (с = 0,6, хлороформ).
g) /3R-/3α/ S*,/6α 9aβ//- гексагидро-3-//-2-меркапто-1-оксо-3-фенилпропил/амино/-4-оксо-2H, 6H-пиридо[2,1-b]-/1,3/тиазин-6-карбоновая кислота
Раствор продукта из части (f) при 0oC (406 мг, 0,90 ммоля) в метаноле (5 мл, обескислороженной пробулькиванием аргона) обрабатывали 1 N раствором гидроокиси натрия (5 мл, обескислороженным пробулькиванием аргона). После перемешивания в течение 1 часа раствор нагревали до комнатной температуры и перемешивание в атмосфере аргона продолжали дополнительно 1,25 часа. Смесь подкисляли 5% раствором бисульфата калия, разбавляли водой и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой и рассолом, затем сушили (сульфатом натрия), фильтровали и удаляли. Остаток дважды подвергали флеш-хроматографированию (Мерк силикагель, 2% уксусная кислота в этилацетате).
Фракции продукта контролировали по данным HPLC. Желаемые фракции собирали, удаляли и дважды подвергали азеотропной разгонке с этилацетатом. Остаток помещали в небольшое количество этилацетата и растирали с гексаном. Растворитель удаляли и остаток с гексаном, удаляли и сушили в вакууме, получая 98,3 мг указанного в названии продукта в виде твердой белой пены. TLC (2% уксусная кислота в этилацетате) Rf = 0,46; [α]D= -57,0oC (с = 0,4, хлороформ).
Данные HPLC (гель-проникающей хроматографии) : YMC S 3 ODS колонка (6,0 • 150 мм); элюированная 40% A:90% воды-10 метанола -0,2% фосфорной кислоты и 60% B : 10% воды - 90% метанола - 0,2% фосфорной кислоты; скорость потока 1,5 мл/мин детектирование при 220 нм; tR = 8,33 мин (95,0%).
Данные анализа для C18H22N2O4S2 • 0,2 этилацетат:
Вычислено, %: С 54,79; Н 5,77; N 6,80; S 15,56
Найдено, %: С 54,59; Н 6,04; N 6,59; S 15,16.
Пример 3
/4S -/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-7H- пиридо[2,1-b]/1,3/тиазепин-7-карбоновая кислота
a) Метиловый эфир /S-/R*, R*//-2-//2-фталимидо-4-/ацетилтио/-1-оксобутил/амино-6,6- диметоксигексановой кислоты
Холодный раствор (0oC) трифенилфосфина (1,143 г, 4,36 ммоля) в тетрагидрофуране (20 мл) обрабатывали диизопропилазидодикарбоксилатом (860 мкл, 883 мг, 4,37 ммоля). В течение 5 минут появлялся белый шлам. Через 30 минут добавляли раствор метилового эфира /S-/R*, R*/-2-//2-фталимидо-4-гидрокси-1-оксобутил/амино/-6,6- диметоксигексановой кислоты (полученный как описано в примере 1 (f), 928 мг, 2,19 ммоля) в тетрагидрофуране (8 мл) с последующим добавлением слабой тиоуксусной кислоты (312 мкл, 332 мг, 4,36 ммоля). Смесь перемешивали при 0oC в течение 1,25 часа, затем разделяли между 50% раствором насыщенного бикарбоната натрия и этилацетатом. Этилацетатный экстракт промывали рассолом, сушили (сульфатом натрия), фильтровали и удаляли. Остаток вновь растворяли в этилацетате и обрабатывали небольшим количеством гексана для осаждения трифенилфосфиноксида. Смесь фильтровали и фильтрат подвергали флеш-хроматографированию (Мерк силикагель, 65:35 - этилацетат:гексан), получая 894 мг указанного в названии продукта в виде бесцветного масла. TLC (75:25 - этилацетат:гексан) Rf = 0,43.
b) Метиловый эфир /4S-/4α, 7α, 10aβ//- октагидро-4-фталимидо-5-оксо-7H-пиридо[2,1-b]/1,3/тиазепин- 7-карбоновой кислоты
Обескислороженный (пробулькиванием аргона) продукт из части (a) (814 мг, 1,65 ммоля) в метаноле (15 мл) при 0oC обрабатывали метоксидом натрия (25 вес. % в метаноле, 1,05 мл, 4,6 ммоля). Через 5 минут смесь гасили раствором насыщенного хлористого аммония, разбавляли водой и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой и рассолом, затем сушили (сульфатом натрия), фильтровали и удаляли. Остаток вновь растворяли в метиленхлориде (180 мл) и перемешивали при комнатной температуре с ионнообменной смолой Амберлит®15 (285 мг, предварительно промытой последовательно 6 N соляной кислотой, водой, тетрагидрофураном, затем метиленхлоридом). Через 46 часов раствор фильтровали, удаляли и подвергали флеш-хроматографированию (Мерк силикагель, 1:1 - этилацетат:гексан), получая 314 мг указанного в названии продукта в виде белой пены. Растирание пены с этилацетатом дало указанный в названии продукт в виде твердого белого вещества. Т.пл. = 147 - 148oC. TLC (75: 25 этилацетат: гексан) Rf = 0,56; [α]D= -143,2o (с = 0,6, хлороформ).
c) Метиловый эфир /4S-/4α/R*/, 7α, 10aβ//- октагидро-4-//2-/ацетилтио/-1-оксо-3-фенилпропил /амино/-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Продукт из части (b) (280 мг, 0,72 ммоля) в метаноле (8 мл) обрабатывали гидразинмоногидратом (42 мкл, 43,3 мг, 0,86 ммоля) и раствор перемешивали при комнатной температуре в течение 67 часов. Смесь фильтровали и твердое вещество промывали метанолом. Фильтрат удаляли, растирали с метиленхлоридом, вновь фильтровали и удаляли, получая неочищенный амин в виде желтого масла (около 205 мг).
Холодный раствор (0oC) /S/-2-ацетилтио/бензопропановой кислоты (178 мг, 0,79 ммоля) и триэтиламина (111 мкл, 80 мг, 0,80 ммоля) в метиленхлориде (3 мл) обрабатывали указанным выше амином (в виде раствора в 7 мл метиленхлорида) с последующей обработкой бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфатом (353 мг, 0,80 ммоля). Раствор перемешивали при 0oC в течение 1 часа и затем при комнатной температуре в течение 2 часов. Растворитель удаляли и остаток разделяли между этилацетатом и 5% раствором бисульфата калия. Органический слой последовательно промывали водой, 50% насыщенным раствором бикарбоната натрия и рассолом и затем сушили (сульфатом натрия), фильтровали и удаляли. Остаток подвергали флеш-хроматографированию (Мерк силикагель, 1:1 - этилацетат:гексан), получая 272 мг чистого указанного в названии продукта в виде белой пены.
d) /4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5- оксо-7H-пиридо[2,1-b] /1,3/-тиазепин-7-карбоновая кислота
Раствор продукта из части (c) (227 мг, 0,49 ммоля) в метаноле (5 мл, обескислороженный пробулькиванием аргона) обрабатывали при комнатной температуре 1 N раствором гидроокиси натрия (8 мл, обескислороженным пробулькиванием аргона). После перемешивания в течение 1 часа смесь подкисляли 10% соляной кислотой, разбавляли водой и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой и рассолом и затем сушили (сульфатом натрия), фильтровали и концентрировали. Полученное твердое вещество смешивали с этилацетатом и собирали фильтрованием. Фильтрат подвергали флеш-хроматографированию (Мерк силикагель, 1% уксусная кислота в этилацетате) и желаемую фракцию собирали, удаляли и растирали со смесью этилацетат/этиловый эфир, получая дополнительно твердое вещество. Твердое вещество собирали, получая общее количество 150 мг указанного в названии продукта, т. пл. 216 - 217oC (разложение) TLC (2% уксусная кислота в этилацетате) Rf = 0,56; [α]D= = -72,6oC (c = 0,28, диметилформамид).
HPLC YMC S 3 ODS колонка (6,0 • 150 мм) : элюируют 40% A: 90% воды - 10% метанола - 0,2% фосфорной кислоты и 60% B : 10% воды - 90% метанола - 0,2% фосфорной кислоты, скорость потока 1,5 мл/мин, детектирование при 220 нм, tR = 9,48 мин (97,4%).
Данные анализа для C19H24N2O4S2 • 0,14 этилацетат:
Вычислено, %: С 55,82; Н 6,02; N 6,66; S 15,24
Найдено, %: С 55,53; Н 6,01; N 6,63; S 14,91.
Пример 4
/4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксопирроло [2,1-b]/1,3/оксазепин-7-карбоновая кислота
a) /S/-2-фталимидо-5-оксо-5-/фенилметокси/пентановая кислота
К раствору γ-бензил-L-глутамата (17,49 г, 73,70 ммоля) в водной среде (180 мл) карбоната натрия (7,81 г, 73,70 ммоля) и диоксана (120 мл) добавляли N-карбетоксифталимид (16,50 г, 75,27 ммоля, 1,02 экв). После перемешивания при комнатной температуре в течение 4,5 часов реакционную смесь подкисляли 6 N соляной кислотой (30 мл) и экстрагировали этилацетатом (2 • 400 мл). Объединенные этилацетатные экстракты промыли 50% рассолом (200 мл) и рассолом (200 мл), сушили над сульфатом натрия, фильтровали, концентрировали и сушили в вакууме, получая неочищенное масло (41,4 г). К раствору неочищенного остатка в этилацетате (100 мл) добавляли дициклогексиламин (14 мл). После пребывания в холодильнике в течение ночи этиловый эфир удаляли на роторном испарителе и маслообразный остаток кристаллизовали из смеси этилацетат/гексан. Полученный осадок собирали фильтрованием, промывали гексаном и сушили в вакууме, получая 21,21 г указанного в названии продукта в виде дициклогексиламиновой соли. Суспензию этой дициклогексиламиновой соли в этилацетате (200 мл) промывали 5% раствором бисульфата натрия (3•50 мл), рассолом (50 мл) и сушили над сульфатом магния, фильтровали и концентрировали, получая 13,5 г указанного в названии продукта в виде белой пены. TLC (3% уксусная кислота в 9:1 этилацетат:гептане) Rf = 0,30.
b) Этиловый эфир /S/-2-фталимидо-5-оксо-5-/фенилметокси/- пентановой кислоты
К раствору продукта из части (a) (13,22 г, 36,0 ммоля) и карбоната цезия (5,86 г, 18,0 ммоля) в диметилформамиде (100 мл) добавляли иодметан (8,1 мл, 129,6 ммоля, 3,6 экв.). Желтый раствор перемешивали в течение 2,5 часов и затем разделяли между этилацетатом (300 мл) и водой (250 мл). Этилацетатный экстракт промывали 5% раствором бикарбоната натрия и рассолом, сушили на сульфатом магния, фильтровали и концентрировали, получая 13,68 г желтого масла. Остаток чистили хроматографически на колонке с силикагелем (5•20 см), элюируя 30% смесью этилацетат/гексан. Желаемые фракции объединяли и концентрировали, получая 10,0 г указанного в названии продукта. TLC (1:1 этилацетат:гексан) Rf = 0,45.
c) Метиловый эфир /S/-2-фталимидо-4-/карбокси/бутановой кислоты
К раствору продукта из части (b) (10,0 г, 26,22 ммоля) в этилацетате (115 мл) добавляли 20% гидроокиси палладия на углероде в качестве катализатора (1,90 г) и полученную суспензию перемешивали в атмосфере водорода (баллон) в течение 2,5 часов. Смесь фильтровали, интенсивно промывали этилацетатом, концентрировали и сушили в вакууме с выходом 7,29 г неочищенного указанного в названии продукта в виде белого твердого вещества, т. пл. 137 - 138oC. TLC (10 % метанол/метилен хлорид) Rf = 0.43.
d) Метиловый эфир /S/-2-фталимидо-5-оксо-5-/этилтио/-пентановой кислоты
К раствору продукта из части (c) (7,27 г, 24,95 ммоля) в метиленхлориде (125 мл) при 0oC в атмосфере аргона добавляли этантиол (4,81 мл, 64,92 ммоля, 2,6 экв.), 4-диметил-аминопиридин (609 мг, 4,99 ммоля, 0,2 экв.) и этил-3-/3-ди-метиламино/ пропилкарбодиимид гидрохлоридной соли (5,27 г, 27,47 ммоля, 1,1 экв.). После перемешивания при 0oC в течение 2 часов и при комнатной температуре в течение 1 часа реакционную смесь концентрировали, разбавляли этилацетатом (400 мл) и промывали 5% бисульфатом калия (200 мл) и рассолом (200 мл), сушили над сульфатом натрия, фильтровали, концентрировали и сушили в вакууме с выходом 8,30 г указанного в названии продукта в виде неочищенного масла. TLC (1:1, этилацетат:гексан) Rf = 0,47.
e) Метиловый эфир /S/-2-фталимидо-5-оксопентановой кислоты
Суспензию продукта из части (d) (8,30 г, 24,75 ммоля) и 10% палладия на угле (1,24 г) в ацетонитриле (150 мл) в атмосфере аргона обрабатывали по каплям триэтилсиланом (7,91 мл, 49,5 ммоля, 2 экв.). После перемешивания при комнатной температуре в течение 45 минут смесь фильтровали, концентрировали и сушили в вакууме. Неочищенный остаток чистили хроматографически на колонке (5•25 см) с силикагелем, элюируя 25% смесью этилацетат/гексан (4 л) с последующим элюированием 35% смесью этилацетат/гексан (2 л). Желаемые фракции объединяли, получая 5,60 г указанного в названии продукта. TLC (1:1 этилацетат:гексан) Rf = 0,32.
f) Метиловый эфир /S/-2-фталимидо-5,5-диметоксипентановой кислоты
Раствор продукта из части (e) (5,60 г, 20,34 ммоля) в метаноле (60 мл) и метиленхлориде (40 мл) обрабатывали триметиловым эфиром орто-муравьиной кислоты (3,8 мл, 34,59 ммоля, 1,7 экв.) и моногидратом n-толуолсульфоновой кислоты (280 мг). После перемешивания при комнатной температуре в течение 1,5 часа реакцию гасили 2 мл насыщенного раствора бикарбоната натрия, концентрировали и разделяли между этилацетатом (400 мл) и водой (100 мл). Этилацетатный экстракт промывали насыщенным раствором бикарбоната натрия (100 мл), рассолом (100 мл), сушили над сульфатом магния, фильтровали и концентрировали до неочищенного масла. Неочищенный остаток чистили хроматографически на колонке 5•20 см с силикагелем, элюируя 30% смесью этилацетат/гексан (2 л). Желаемые фракции объединяли, концентрировали и сушили в вакууме, получая 6,20 указанного в названии продукта. TLC (1:1 этилацетат/гексан) Rf = 0,40.
g) метиловый эфир /S/-2-амино-5,5-диметоксипентановой кислоты
Раствор продукта из части (f) (6,16 г, 19,18 ммоля) в метаноле /125 мл/ обрабатывали гидразинмоногидратом (0,98 мл, 20,14 ммоля, 1,05 экв.). После перемешивания при комнатной температуре в течение 6 дней полученный шлам фильтровали, концентрировали, растирали в метиленхлориде, фильтровали, концентрировали и сушили в вакууме с получением 3,57 г указанного в названии продукта в виде мутного масла. TLC (10% метанола в метиленхлориде) Rf = 0,41.
h) Метиловый эфир /S -/R*, R*//-2-//2-фталимидо-4- /трифенилметокси/-1-оксобутил/амино-5,5-диметоксипентановой кислоты
Раствор триэтиламиновой соли /S/-2-фталимидо-4-/трифенилметокси/ бутановой кислоты, полученной, как описано в примере 1 (b), (11,62 г, 19,60 ммоля, 1,05 экв. ) в метиленхлориде (100 мл) при 0oC обрабатывали бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфатным реагентом (8,67 г, 19,60 ммоля, 1,05 экв.). Смесь перемешивали в течение 45 минут при 0oC, затем обрабатывали раствором продукта из части (g) (3,57 г, 18,67 ммоля) в метиленхлориде (50 мл). Через 10 минут реакции при 0oC и 2 часов при комнатной температуре, раствор разделяли между этилацетатом (300 мл) и водой (100 мл). Этилацетатный слой промывали 50% насыщенным раствором бикарбоната натрия (100 мл) и рассолом (100 мл), сушили над сульфатом магния, фильтровали и концентрировали. Остаток чистили хроматографически на колонке 5•25 см с силикагелем, элюируя смесь 1:1 этилацетат/гексан, с получением 8,58 г указанного в названии продукта. TLC (1:1 этилацетат/гексан) Rf = 0,20.
i) Метиловый эфир /S-/R*, R*//-2-//2-фталимидо-4- гидрокси-1-оксобутил/амино/-5,5-диметоксипентановой кислоты
Раствор продукта из части (h) (8,58 г, 12,91 ммоля) в метаноле (100 мл) обрабатывали моногидратом n-толуолсульфоновой кислоты (850 мг). После перемешивания при комнатной температуре в течение 3,5 часов смесь разделяли между этилацетатом (200 мл) и 10% насыщенным раствором бикарбоната натрия (100 мл). Фазы разделяли и водный слой экстрагировали вновь этилацетатом (100 мл). Объединенные этилацетатные экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. Остаток чистили хроматографически на колонке 5•20 см с силикагелем, элюируя 8:2 смесью этилацетат: гексан (1 л) с последующим элюированием 1% метанолом в этилацетате (2 л). Желаемые фракции объединяли, концентрировали и сушили в вакууме, получая 4,33 г указанного в названии соединения. TLC (8:2 этилацетат:гексан) Rf = 0,21.
j) Метиловый эфир /4S-/4α, 7α, 9aβ/- октагидро-4-фталимидо-5-оксопирроло [2.1-b] /1,3/оксазепин-7-карбоновой кислоты
Раствор продукта из части (i) (1,89 г, 4,48 ммоля) в метиленхлориде (90 мл) обрабатывали ионообменной смолой - Амберлит®15 (400 мг, предварительно последовательно промытой 6 N соляной кислотой, водой, тетрагидрофураном и метиленхлоридом). После перемешивания при комнатной температуре в течение 3 часов раствор фильтровали, концентрировали и подвергали флеш-хроматографированию на колонке 5• 15 см с силикагелем, элюируя смесью 6:4 этилацетат/гексан, получая 1,51 г указанного в названии продукта в виде белой пены. TLC (8:2 этилацетат - гексан) Rf = 0,32.
k) Метиловый эфир /4S-/4α, 7α, 9aβ/- -4-амино-оксагидро-5-оксопирроло [2,1 - b] /1,3/оксазепин-7-карбоновой кислоты
Продукт из части (j) (764 г, 2,13 ммоля) в метаноле (15 мл) обрабатывали гидразинмоногидратом (109 мкл, 2,24 ммоля, 1,05 экв.) и раствор перемешивали при комнатной температуре в течение 4 дней. Смесь фильтровали и твердое вещество промывали метанолом. Фильтрат концентрировали, растирали в метиленхлоридом, вновь фильтровали и концентрировали. Остаток чистили хроматографически на колонке 2•15 см с силикагелем, элюируя 3% метанолом в метиленхлориде (3 л). Желаемые фракции объединяли и концентрировали с получением 451 мг указанного в названии продукта в виде масла. TLC (10% метанола в метиленхлориде) Rf = 0,18.
l) Метиловый эфир /4S-/4α/R*/, 7α, 9aβ//- октагидро-4-//2-/ацетилтио/-1-оксо-3- фенилпропил/амино/-5-оксопирроло [2,1-b]/1,3/-оксазепин-7-карбоновой кислоты
Суспензию дициклогексиламиновой соли /S/-2-ацетилтио-3- бензолпропановой кислоты /полученной, как описано в примере 1 (h), 870 мг, 2,14 ммоля, 1,14 экв. / в этилацетате (70 мл) промывали 5% раствором бисульфата калия (5•20 мл), 50% рассолом (20 мл) и рассолом (20 мл), сушили (безводным сульфатом натрия). Фильтровали, концентрировали и сушили в вакууме в течение ночи, получая /S/-2-/ацетилтио/бензолпропановую кислоту.
Эту свободную кислоту растворяли в сухом метиленхлориде (10 мл), охлаждали до 0oC (в бане лед с солью) и обрабатывали триэтиламином (298 мкл, 2,14 ммоля) с последующей обработкой раствором продукта из части (k) (430 мг, 1,88 ммоля) в метиленхлориде (10 мл) и бензотриазол-1-илокситрис/-диметиламино/ фосфонийгексафторфосфатом (947 мг, 2,14 ммоля, 1,14 экв.). Полученный раствор перемешивали при 0oC в течение 50 минут и затем при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали, разбавляли этилацетатом (150 мл), промывали 0,5 N соляной кислотой (50 мл), водой (50 мл), насыщенным раствором бикарбоната натрия (50 мл), водой (50 мл) и рассолом (50 мл), сушили (безводным сульфатом магния), фильтровали и выпаривали досуха. Неочищенный продукт адсорбировали на целите (CeliteR) и хроматографировали на колонке с силикагелем (5• 10 см), элюируя 60% смесью этилацетат/гексан (3 л). Желаемые фракции объединяли и концентрировали, получая 779 мг чистого указанного в названии продукта. TLC (6:4 этилацетат:гексан) Rf = 0,17.
m/ /4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5- оксопирроло[2,1-b]/1,3/оксазепин-7-карбоновая кислота
Раствор продукта из части (1) (754 мг, 1,74 ммоля) в метаноле (15 мл) продували аргоном в течение 30 минут, охлаждали до 0oC (баня лед с солью), затем по каплям обрабатывали предварительно продутым (аргоном, в течение 30 минут) раствором 1,0 N гидроокиси натрия (12 мл), продолжая пробулькивание аргона в течение дополнительного времени протекания реакции. Реакционную смесь перемешивали при 0oC в течение 3 часов, подкисляли при 0oC 5% раствором бисульфата калия до pH 1, затем экстрагировали этилацетатом (3 x 100 мл). Объединенные органические экстракты промывали 50% рассолом (100 мл), рассолом (100 мл), сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме, получая белую пену. Остаток чистили хроматографически на колонке 2,5 • 15 см с силикагелем, элюируя этилацетатом (500 мл) и 0,3% уксусной кислотой в этилацетате (1 л). Желаемые фракции концентрировали, удаляли хлороформом и сушили в вакууме в течение ночи при 50oC над пятиокисью фосфора, получая указанный в названии продукт в виде белой пены, т. пл. 88 - 92oC, [α]D= (c = 1,0, метанол). TLC (1% уксусная кислота в этилацетате) Rf = 0,24.
1H-NMR: 400 MHz; CDCl3: δ 1,80 - 2,31 (m's, 7H), 3,10 (m, 1H), 3, 27 (m, 1H), 3,63 (m, 1H), 4,0 (m, 1H), 4,20 (m, 1H), 4,49 (m, 1H), 4,75 (m, 1H), 5,23 (m, 1H), 7,19 - 7,30 (m's, 5H), 7,52 (d, 1H, J = 6 Hz).
13C-NMR: 100 MHz; CDCl3: δ 26,4, 32,0, 32,6, 41,2, 44,2, 53,0, 59,4, 70,6, 89,47, 126,9, 128,4, 129,3, 137,4, 171,2, 171,6, 174,8.
Данные анализа для C18H22N2O5S • 0,85 H2O
Вычислено, %: С 54,91; Н 6,07; N 7,12; S 8,14
Найдено, %: С 54,85; Н 5,68; N 7,18; S 8,14.
HPLC: tR = 13,5 мин (96,7%, УФ 220): YMCS - 3ODS (C-18) 6,0 • 150 мм 30% B: A - 100% B:A, 25 мин линейный градиент /A = 90% вода/метанол - 0,2% фосфорной кислоты B = 90% метанол/вода + 0,2% фосфорной кислоты/скорость потока 1,5 мл/мин.
Пример 5
/4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5- оксопирроло[2,1-b]/1,3/тиазепин-7-карбоновая кислота
a) Метиловый эфир/S-/R*, R*//-2-//2-фталимидо-4- /ацетилтио/-1-оксобутил/амино/-5,5-диметоксипентановой кислоты
Раствор трифенилфосфина (1,26 г, 4,79 ммоля) при 0oC в сухом тетрагидрофуране (15 мл) обрабатывали диизопропил азодикарбоксилатом (943 мкл, 4,79 ммоля). Полученный белый шлам перемешивали в течение 30 минут и затем обрабатывали раствором метилового эфира /S-/R*,R*//-2-//2-фталимидо-4- гидрокси-1-оксобутил/амино/-5,5-диметоксипентановой кислоты (полученной как описано в примере 4 (i), 1,35 г, 3,20 ммоля) в сухом тетрагидрофуране (15 мл) с последующей обработкой слабой тиолуксусной кислотой (343 мкл, 4,79 ммоля). Смесь перемешивали при 0oC в течение 1,5 часов и затем разделяли между этилацетатом (150 мл) и 50% раствором бикарбоната натрия (100 мл). Этилацетатный слой промывали рассолом, сушили над сульфатом магния, фильтровали, концентрировали, поглощали на целит и сушили в вакууме. Неочищенное вещество чистили хроматографически на колонке 2,5 • 15 см с силикагелем, элюируя 1:1 смесью этилацетат:гексан (1 л) и 6:4 этилацетат:гексан (1 л). Желаемые фракции объединяли, концентрировали и сушили в вакууме, получая 1,35 г указанного в названии продукта в виде масла. TLC (8:2 этилацетат : гексан) Rf = 0,42.
b) Метиловый эфир /S-/R*, R*//-2-//2-фталимидо-4- меркапто-1-оксобутил/амино/-5,5-диметоксипентановой кислоты
Обескислороженный (пробулькиванием аргона) раствор продукта из части (a) (1,33 г, 2,76 ммоля) в метаноле (25 мл) при 0oC обрабатывали метоксидом натрия (25 вес. % в метаноле), 1,52 мл, 6,63 ммоля, 2.4 экв./. Через 3 минуты смесь гасили насыщенным раствором хлористого аммония (3 мл), разбавляли водой и экстрагировали этилацетатом (100 мл). Этилацетатный экстракт промывали водой (50 мл) и рассолом (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток чистили хроматографически на колонке 5 • 15 см с силикагелем, элюируя 1:1 (3 л), с последующим элюированием 8:2 /2 л/ смесью этилацетат:гексан. Фракции, содержащие желаемый продукт, объединяли, концентрировали, получая 853 мг указанного в названии соединения в виде масла. TLC (8:2 этилацетат:гексан) Rf = 0,43.
c) Метиловый эфир /4S-/4α, 7α, 9aβ//- октагидро-4-фталимидо-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Раствор продукта из части (b) (847 мг, 1,93 ммоля) в метиленхлориде (20 мл) обрабатывали ионообменной смолой - Амберлит®15 (700 мг, предварительно последовательно промытой 6 N соляной кислотой, водой, тетрагидрофураном и метиленхлоридом). После перемешивания при комнатной температуре в течение 17 часов раствор фильтровали, концентрировали и подвергали флеш-хроматографированию на колонке 2,5 • 15 см с силикагелем, элюируя смесь 1:1 этилацетат: гексан, получая 691 мг указанного в названии продукта в виде белой пены. TLC (8:2 этилацетат:гексан) Rf = 0,48.
d) Метиловый эфир /4S-/4α, 7α, 9aβ//- 4-амино-октагидро-5-оксопирроло[2,1-b]/1,3/тиазепин-7-карбоновой кислоты
Продукт из части (c) (899 мг, 2,40 ммоля) в метаноле (17 мл) обрабатывали гидразинмоногидратом (122 мкл, 2,52 ммоля, 1,05 экв.) и раствор перемешивали при комнатной температуре в течение 3 дней. Смесь фильтровали, и твердое вещество промывали метанолом. Фильтрат концентрировали, растирали с метиленхлоридом, вновь фильтровали, концентрировали и сушили в вакууме, получая 572 мг указанного в названии продукта в виде мутного масла. TLC (10% метанол в метиленхлориде) Rf = 0,13.
e) Метиловый эфир /4S-/4α/R*/, 7α, 9aβ/- октагидро-4-//2-/ацетилтио/-1-оксо-3- фенилпропил/амино/-5-оксопирроло[2,1-b]/1,3/тиазепин-7-карбоновой кислоты
Суспензию дициклогексиламиновой соли /S/-2-/ацетилтио/- бензолпропановой кислоты (полученной как описано в примере 1 (h), (1,045 г, 2,58 ммоля, 1,1 экв.) в этилацетате (100 мл) промывали 5% раствором бисульфата калия (5 • 25 мл), 50% рассолом (25 мл) и рассолом (25 мл), сушили (безводным сульфатом натрия), фильтровали, концентрировали и сушили в вакууме в течение 1 часа, получая /S/-2-/ацетилтио/-бензолпропановую кислоту.
Эту свободную кислоту растворяли в сухом метиленхлориде (10 мл), охлаждали до 0oC (в бане лед с солью) и обрабатывали триэтиламином (360 мкл, 2,58 ммоля), бензотриазол-1-илокситрис/ диметиламино/фосфонийгексафторфосфатом (1,141 г, 2,58 ммоля) и затем раствором продукта из части (d) (572 мг, 2,34 ммоля) в метиленхлориде (10 мл). Полученный раствор перемешивали при 0oC в течение 30 минут и затем при комнатной температуре в течение 2,5 часов. Реакционную смесь концентрировали, разбавляли этилацетатом (100 мл), промывали 0,5 N соляной кислотой (50 мл), водой (50 мл), насыщенным раствором бикарбоната натрия (50 мл), водой (50 мл) и рассолом (50 мл), сушили (безводным сульфатом магния), фильтровали и выпаривали досуха. Неочищенный продукт адсорбировали на целите и подвергали хроматографированию на колонке (5 • 10 см) с силикагелем, элюируя 25% (5 л), 30% (2 л) и 40% (2 л) смесью этилацетат/гексан. Смешанные фракции объединяли и повторно хроматографировали, элюируя с тем же градиентом. Желаемые фракции объединяли и концентрировали, получая 490 мг чистого указанного в названии продукта. TLC (1:1 этилацетат:гексан) Rf = 0,16.
f) /4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//-2-меркапто-1-оксо-3-фенилпропил/амино/-5- оксопирроло[2,1-b]/1,3/тиазепин-7-карбоновая кислота
Раствор продукта из части (e) (490 мг, 1,09 ммоль) в смеси метанол:тетрагидрофуран (8 мл : 4 мл) продували аргоном в течение 30 минут, охлаждали до 0oC (в бане лед с солью) и затем обрабатывали по каплям предварительно продутым (аргоном в течение 30 минут) раствором 1,0 N гидроокиси натрия (10 мл), пробулькивая аргон в течение добавления и протекания реакции. Реакционную смесь перемешивали при 0oC в течение 3 часов, подкисляли при 0oC 5% раствором бисульфата калия до pH 2 и затем экстрагировали этилацетатом (3•75 мл). Объединенные органические экстракты промывали рассолом (75 мл), сушили (безводным сульфатом магния), фильтровали, выпаривали досуха и сушили в вакууме, получая белую пену (489 мг). Остаток чистили хроматографически на колонке (2,5 • 15 см) с силикагелем, элюируя смесью этилацетат:гептан 9:1 (400 мл) и 0,5% уксусной кислотой в 9:1 смеси этилацетат:гептан (1 л). Желаемые фракции концентрировали, удаляли смесью метиленхлорид/гептан и сушили в вакууме, получая 428 мг продукта. Чистое вещество перекристаллизовывали из смеси этилацетат/метанол/гексан. Кристаллы собирали фильтрованием, интенсивно промывали этиловым эфиром и сушили в вакууме в течение ночи при 40oC над пятиокисью фосфора, получая 305 мг указанного в названии продукта в виде белых кристаллов, т.пл. 206 - 208oC, [α]D= -96,3o (c = 1,0 метанол). TLC (5% уксусная кислота в 9:1 смеси этилацетат:гептан) Rf = 0,29.
1H-NMR: 400 Hz; CDCl3 w/2 drops CD3OD: δ 1,94 (m, 1H), 2,02 (d, 1H, J = 9 Hz), 2,08 (m, 1H), 2,20 - 2, 55 (m's, 4H), 2,95 (m, 1H), 3,08 (m, 1H), 3,23 (m, 1H), 3,27 (m, 1H), 3,59 (m, 1H), 4,54 (t, 1H, J = 7,3 Hz), 4,60 (m, 1H), 5,23 (m, 1H), 7,18 - 7,34 (m's, 5H), 7,63 (d, 1H, J = 6 Hz).
13C - NMR: 100 MHz; CDCl3 w/2 drops CD3OD: δ 27,6, 31,1, 32,1, 32,9, 41,1, 44,0, 52,8, 60,4, 62,2, 126,77, 128,3, 129,0, 137,4, 170,2, 171,4, 172,6.
Данные анализа для C18H22N2O4S2 • 0,08 H2O:
Вычислено, %: С 54,60; Н 5,64; N 7,07; S 16,19
Найдено, %: С 54,65; Н 5,54; N 7,02; S 15,80.
HPLC: tR = 13,0 мин (98,8%, УФ 220): YMC S-3 ODS (C-18) 6,0 • 150 мм; 40% B:A - 100% B:A, 25 минут линейный градиент (A = 90% вода/метанол + 0,2% фосфорной кислоты; B = 90% метанол/вода + 0,2% фосфорной кислоты) скорость потока 1,5 мл/мин.
Пример 6
/4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновая кислота
Продукт примера 5 был также приготовлен как показано далее:
a/N-//Фенилметокси/карбонил/-L-гомосерин
N-/Бензилоксикарбонилокси/сукцинимид /23,57 г, 94,58 ммоля/ добавляли в раствору L-гомосерина (10,24 г, 85,98 ммоля) и бикарбоната натрия (7,95 г, 94,58 ммоля, 1,1 экв.). В смеси воды (100 мл) и ацетона (100 мл). Смесь перемешивали при комнатной температуре в течение ночи. Ацетон удаляли при пониженном давлении (на роторном испарителе) и водный раствор промывали метиленхлоридом (2•75 мл). Затем водный слой подкисляли до pH 2 добавлением 6 N соляной кислоты и экстрагировали этилацетатом (2•25 мл). Объединенные этилацетатные слои промывали водой (2•100 мл) и рассолом, сушили над сульфатом натрия, фильтровали, концентрировали и сушили в вакууме, получая 19,54 г указанного в названии продукта в виде белого твердого вещества. TLC (этилацетат: н-бутанол:уксусная кислота: вода 2:1:1:1) Rf = 0,74.
b) N-//Фенилметокси/карбонил/-O-/трифенилметил-L-гомосерин
К суспензии продукта из части (a) (19,54 г, 77,04 ммоля) в хлороформе (250 мл) добавляли триэтиламин (12,35 мл, 88,59 ммоля, 1,15 экв.). Гомогенную смесь обрабатывали трифенилметилхлоридом (24,70 г, 88,59 ммоля) и реакционную смесь перемешивали в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении (на роторном испарителе), разделяли между этилацетатом (400 мл) и 5% раствором бисульфата калия (200 мл). Этилацетатный слой промывали 5% раствором бисульфата калия (200 мл), водой (2•200 мл), и рассолом (200 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая 45,4 г вещества. Остаток хроматографировали на колонке 10 • 30 см с силикагелем, элюируя смесью 6:4 этилацетат:гексан (2 л) с последующим элюированием 1% уксусной кислотой в смеси 8:2 этилацетат:гексан, получая 8,76 г чистого указанного в названии продукта.
c) Метиловый эфир /S/-2-амино-5,5-диметоксипентановой кислоты
Метиловый эфир /S/-2-фталимидо-5,5-диметоксипентановой кислоты (полученный как описано в примере 4 (f), 3,35 г 10,43 ммоля) в метаноле (70 мл) обрабатывали гидразинмоногидратом (531 мкл, 10,95 ммоля, 1,05 экв.) и раствор перемешивали при комнатной температуре в течение 6 дней. Смесь фильтровали и твердое вещество промывали метанолом. Фильтрат концентрировали, растирали с метиленхлоридом, вновь фильтровали, концентрировали и сушили в вакууме, получая 1,89 г указанного в названии продукта в виде мутного масла. TLC (10% метанол в метиленхлориде) Rf = 0,39.
d) Метиловый эфир /S-/R*, R*//-2-//2-///фенилметокси/карбонил/амино/-4-/трифенилметокси/ -1-оксобутил/амино/-5,5-диметоксипентановой кислоты
Раствор продукта из части (b) (5,28 г. 10,65 ммоля, 1,1 экв.) в сухом метиленхлориде (50 мл) обрабатывали при 0oC триэтиламином (1,48 мл, 10,65 ммоля) с последующей обработкой продуктом из части (с) (1,85 г, 9,68 ммоля) в сухом метиленхлориде (30 мл) в бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфатом (4,71 г, 10,65 ммоля, 1,1 экв.). Смесь перешивали в течение 1 часа при 0oC и затем перешивали при комнатной температуре в течение 2 часов. Реакционную смесь разделяли между этилацетатом (300 мл) и водой (150 мл). Этилацетатный слой промывали 50% насыщенным раствором бикарбоната натрия (200 мл) и рассолом (2•200 мл), сушили над сульфатом магния, фильтровали, концентрировали, адсорбировали на целите и чистили на колонке 7•20 см с силикагелем, элюируя 40% смесью этилацетат/гексан (3 л) с последующим элюированием 50% (2 л) смесью этилацетат/гексан, получая 4,84 г указанного в названии продукта. TLC (этилацетат/гексан 1:1) Rf = 0,22.
e) Метиловый эфир /S-/R*, R*//-2-//2-///фенилметокси/карбонил/амино/-4-гидрокси-1 -оксобутил/амино/-5,5-диметоксипентановой кислоты
Раствор продукта из части (d) (4,80 г, 7,18 ммоля) в метаноле (70 мл) обрабатывали моногидратом п-толуолсульфоновой кислоты (300 мг). После перемешивания при комнатной температуре в течение 2 часов смесь разделяли между этилацетоном (400 мл) и 25% насыщенным раствором бикарбоната натрия (200 мл). Фазы разделяли и водный слой вновь экстрагировали этилацетатом (100 мл). Объединенные этилацетатные экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. Остаток чистили хроматографически на колонке 5•20 см с силикагелем, элюируя 7:3 (1 л), 8:2 (1 л) смесью этилацетат:гексан с последующим элюированием 10% метанолом в этилацетате (2 л). Желаемые фракции объединяли, концентрировали и сушили в вакууме, получая 2,92 г указанного в названии продукта. TLC (этилацетат:гексан 8:2) Rf = 0,09.
f) Метиловый эфир /S-/R*, R* //-2-//2-///фенилметокси/карбонил/амино-4-/ацетилтио/-1-оксобутил/ амино-5,5-диметоксипентановой кислоты
Раствор трифенилфосфина (3,06 г, 11,65 ммоля, 1,7 экв.) в сухом тетрагидрофуране (40 мл) при 0oC обрабатывали диизопропилазадикарбоксилатом (2,29 мл, 11,65 ммоля). Полученный белый шлам перемешивали в течение 30 минут и затем обрабатывали раствором продукта из части (е) (2,92 г, 6,85 ммоля) в сухом тетрагидрофуране с последующей обработкой слабой тиолуксусной кислотой (833 мкл, 11,65 ммоля). Смесь перемешивали при 0oC в течение 2 часов и затем разделяли между этилацетатом (300 мл) и 50% раствором бикарбоната натрия (200 мл). Этилацетатный слой промывали рассолом, сушили над сульфатом магния, фильтровали, концентрировали, адсорбировали на целит и сушили в вакууме. Неочищенное вещество чистили хроматографически на колонке 5•20 см с силикагелем, элюируя смесью этилацетат:гексан 1:1 (3 л). Желаемые фракции объединяли, концентрировали и сушили в вакууме, получая 2,58 г указанного в названии продукта в виде сверхбелого твердого продукта. TLC (этилацетат:гексан, 8:2) Rf = 0,40.
g) Метиловый эфир /S-/R*, R* //-2-//2-///фенилметокси/карбонил/амино/-4-меркапто-1-оксобутил/амино/-5,5- диметоксипентановой кислоты
Обескислороженный (пробулькиванием аргона) раствор продукта из части (f) (2,56 г, 5,28 ммоля) в метаноле (50 мл) при 0oC обрабатывали метоксидом натрия (25 вес.% в метаноле, 3,62 мл, 15,84 ммоля, 3 экв.). Через 10 минут смесь гасили насыщенным раствором хлористого аммония (40 мл), разбавляли водой (100 мл) и экстрагировали этилацетатом (300 мл). Этилацетатный экстракт промывали водой (100 мл) и рассолом (150 мл), сушили над сульфатом магния, фильтровали и концентрировали. Остаток чистили хроматографически на колонке 5•20 см с силикагелем, элюируя 1:1 (3 л) смесью этилацетат:гексан. Желаемое соединение объединяли и концентрировали, получая 1,99 г указанного в названии продукта в виде масла. TLC (этилацетат:гексан, 8:1) Rf = 0,43.
h) Метиловый эфир /4S-/4α, 7α, 9aβ//- октагидро-4-///фенилметоксикарбонил/амино/-5-оксопирроло [2,1-b] /1,3/-тиазепин-7-карбоновой кислоты
Раствор продукта из части (g) (2,16 г, 4,88 ммоля) в метиленхлориде (50 мл) обрабатывали ионообменной смолой - Амберлитом® 15 (620 мг, предварительно последовательно промытой 6 N соляной кислотой водой, тетрагидрофураном и метиленхлоридом). После перемешивания при комнатной температуре в течение 3 часов раствор фильтровали, концентрировали и подвергали флеш-хроматографированию на колонке 5•20 см с силикагелем, элюируя 6:4 смесью этилацетат: гексан, получая 1,34 г указанного в названии продукта в виде белой пены. TLC (этилацетат:гексан, 8:2) Rf = 0,51.
i) Метиловый эфир /4S-/2α, 7α, 9aβ//- 4-амино-октагидро-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Раствор продукта из части (h) (1,20 г, 3,17 ммоля, удаленный толуолом три раза и высушенный в вакууме в течение ночи) в сухом метиленхлориде (40 мл) обрабатывали иодтриметилсиланом (632 мкл, 4,44 ммоля, 1,4 экв.) и перемешивали при комнатной температуре в атмосфере аргона в течение 1,5 часов. Смесь гасили водой (50 мл), обрабатывали 10% соляной кислотой (5 мл, pH 1) и промывали этилацетатом (50 мл). Водную фазу обрабатывали 10% раствором гидроокиси натрия и экстрагировали метиленхлоридом (три раза). Собранные экстракты сушили над сульфатом натрия, фильтровали, концентрировали и сушили в вакууме, получая 396 мг указанного в названии продукта в виде прозрачного масла. TLC (10% метанол в метиленхлориде) Rf = 0,10.
j) /4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксопирроло [2,1 - b] /1,3/-тиазепин-7-карбоновая кислота
Раствор /S/-2-/ацетилтио/бензолпропановой кислоты в сухом метиленхлориде обрабатывали триэтиламином. Затем добавляли раствор продукта из части (i) в метиленхлориде с последующим добавлением бензотриазол-1-илокситрис-/диметиламино/ фосфонийгексафторфосфата. Полученный раствор обрабатывали, как описано в примере 5 (e), получая метиловый эфир /4S-/4α/R*/, 7α, 9aβ//- окстагидро-4-//2- ацетилтио/-1-оксо-3-фенилпропил/амино/-5-оксопирроло[2.1-b]/1,3/- тиазепин-7-карбоновой кислоты.
Суспензию этого метилового эфира в смеси метанол: тетрагидрофуран продували аргоном, охлаждали до 0oC и обрабатывали предварительно продутым 1,0 N раствором гидроокиси натрия. Обрабатывали таким образом, как описано в примере 5 (f), и получали указанный в названии продукт.
Пример 7
/4S-/4α/R*/, 7α, 9aβ//- Окстагидро-4-//3-циклогексил-2-меркапто-1-оксопропил/амино/-5 -оксопирроло[2,1-b]/1,3/-тиазепин-7-карбоновая кислота
a/ Соль метилового эфира /4S-/4α, 7α, 9aβ//- 4-аминооксагидро-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты и п-толуолсульфоновой кислоты.
Раствор метилового эфира /4S-/4α, 7α, 9aβ//- октагидро-4-///фенилметокси/карбонил/амино/-5-оксопирроло[2,1-b] -/1,3/ -тиазепин-7-карбоновой кислоты (полученный, как описано в примере 6 (h), 738 мг, 1,95 ммоля, удаленный три раза толуолом и высушенный в вакууме в течение ночи) в сухом метиленхлориде (25 мл) обрабатывали иодтриметилсиланом (389 мкл, 2,73 ммоля, 1,4 экв.) и перемешивали при комнатной температуре в атмосфере аогона. Через 2 часа реакционную смесь обрабатывали дополнительным количеством иодтриметилсилана (40 мкл) и перемешивали в течение 30 минут. Смесь гачили 0,4 M соляной кислотой в растворе метанол:диоксан (9:1, 9,7 мл) и перемешивали в течение 5 минут. Летучие удаляли в вакууме (на роторном испарителе) и остаток разделяли между водой и этилацетатом. Отделенный этилацетатный слой промывали водой и объединенные водные фазы промывали этилацетатом. Водные фазы охлаждали до 0oC и доводили pH до 10,3 (по показаниям pH-метра) 1,0 N раствором гидроокиси натрия. Водную фазу экстрагировали метиленхлоридом (три раза) и затем водную фазу насыщали солью и вновь экстрагировали метиленхлоридом (три раза). Собранные экстракты сушили над сульфатом натрия, фильтровали, концентрировали и сушили в вакууме, получая 455 мг свободного амина в виде прозрачного масла, TLC (10% метанола в метиленхлориде) Rf = 0,28.
Этот свободный амин растворяли в этилацетате (5 мл) и обрабатывали раствором моногидрата п-толуолсульфоновой кислоты (354 мг, 1 экв) в этилацетате (1 мл). Немедленно образовывались белые кристаллы. Кристаллы держали в холодильнике (5oC) в течение 30 минут и затем собирали фильтрованием, хорошо промывая этилацетатом, и высушивали в течение ночи в ваккууме с получением 639 мг указанного в названии продукта в виде белого твердого вещества.
b) Дициклогексиламиновая соль /S/-2-/ацетилтио/-3-циклогексилпропионовой кислоты
Раствор D-фенилаланина (5,20 г, 31,5 ммоля) в 2 M растворе соляной кислоты (75 мл) в 500 мл сосуде для гидрирования (Parr) продували газообразным азотом и обрабатывали оксидом платины (640 мг, 2,82 ммоля). Гидрирование начинали при давлении P0 ≈ 3 кг/см2 в откаченной склянке, наполняя вновь, если необходимо. Полное количество поглощенного водорода было около 5,8 кг/см2 (теоретически 5,86 кг/см2) в течение 6 часов. Реакционную смесь продували газообразным азотом и фильтровали через целит, промывая остаток на фильтре горячей водой. Фильтрат концентрировали до около 40 мл и хранили в течение ночи при 5oC. Полученные твердые вещества собирали, промывали небольшим количеством холодной воды и сушили в вакууме при 60oC, получая 5,46 г гидрохлоридной соли /R/-2-амино-3-циклогексилпропановой кислоты.
К перемешиваемому раствору этой гидрохлоридной соли (2,81 г, 13,5 ммоля) в 2,5 N серной кислоте (32 мл) при комнатной температуре добавляли бромид калия (10,0 г, 84 ммоля). Реакционную смесь охлаждали до -4oC и добавляли порциями в течение 1 часа твердый нитрит натрия (1,75 г, 25,4 ммоля), поддерживая температуру ниже 0oC. Реакционная смесь вспенивалась и начинало образовываться масло. После окончания добавления реакционную смесь перемешивали в течение 1 часа и затем нагревали до комнатной температуры и перемешивали еще 1 час. Реакционную смесь затем дважды экстрагировали эфиром, экстракты сушили (сульфатом магния), фильтровали и выпаривали, получая 2,3 г /R/-2-бром-3-циклогексилпропионовой кислоты в виде бесцветного масла.
К перемешиваемому шламу тиоацетата калия (1,07 г, 9,36 ммоля) в сухом ацетонитриле (15 мл) при 0oC в атмосфере аргона добавляли раствор /R/-2-бром-3-циклогексилпропановой кислоты (2,20 г, 9,36 ммоля) в ацетонитриле (3 мл) в течение 10 минут. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. Полученный шлам фильтровали и выпаривали. Остаток вновь растворяли в этилацетате, промывали один раз 5% раствором бисульфата калия, сушили (сульфатом натрия) и выпаривали. Маслообразный желтый остаток (2,21 г) растворяли в эфире и обрабатывали раствором дициклогексиламина (1,8 мл, 9,0 ммоля) в 5 мл эфира. Царапание поверхности склянки стеклянным стержнем давало 2,38 г белых кристаллов указанного в названии продукта, т.пл. 159 - 161oC, [α]D= -41,2o (с=1,0, хлороформ).
Данные анализа для C23H41NSO3:
Вычислено, %: С 67,11; Н 10,04; N 3,40; S 7,79
Найдено, %: С 66,95; Н 10,12; N 3,25; S 7,89.
c) Метиловый эфир /4S-/4α/R*/, 7α, 9aβ/- окстагидро-4-//2-/ацетилтио/-3-циклогексил-1-оксопропил/амино/-5- оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Суспензию дициклогексиламиновой соли /S/-2-/-ацетилтио/-3- циклогексилпропановой кислоты (285 г, 0,69 ммоля, 1,05 экв) в этилацетате (15 мл) промывали 5% раствором бисульфата калия (3•10 мл), 50% рассолом (10 мл) и рассолом (10 мл), сушили (безводным сульфатом магния), фильтровали, концентрировали, удаляли метиленхлоридом (дважды) и сушили в вакууме в течение 1 часа, получая /S/-2-/-3-циклогексилпропановую кислоту в виде масла.
Эту свободную кислоту растворяли в сухом метиленхлориде (5 мл), охлаждали 0oC (баня со льдом) и обрабатывали триэтиламином (96 мкл, 0,69 ммоля, 1,05 экв.), затем продуктом из части (a) (275 мг, 0,66 ммоля), триэтиламином (92 мкл, 0,66 ммоля) и наконец бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфатом/ 305 мг, 0,69 ммоля. Полученный раствор перемешивали при 0oC в течение 1 часа и затем при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали, разбавляли этилацетатом, промывали 5% раствором бисульфатом калия (20 мл), 50% насыщенным раствором бикарбоната натрия (20 мл), рассолом (20 мл), сушили (безводным сульфатом магния), фильтровали и выпаривали досуха. Неочищенный продукт адсорбировали на целит и хроматографировали на колонке с силикагелем (2,5•10 см), элюируя 40% (1 л) смесью этилацетат/гексан. Желаемые фракции объединяли и концентрировали, получая 265 мг чистого указанного в названии продукта. TLC (этилацетат:гексан, 8:2) Rf=0,53.
d) /4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//3-циклогексил-2-меркапто-1-оксопропил/амино/ -5-оксопирроло[2,1-b]/1,3/тиазепин-7-карбоновая кислота
Раствор продукта из части (с) (245 мг, 0,54 ммоля) в метаноле (6 мл), продутый аргоном в течение 30 минут, охлажденный до 0oC, обрабатывали по каплям предварительно продутым (аргоном в течение 30 минут) раствором 1,0 N гидроокиси натрия (5 мл), продолжая пробулькивание аргона в течение добавления раствора и протекания реакции. Реакционную смесь перемешивали при 0oC в течение 2 часов, подкисляли при 0oC 5% раствором бисульфита калия до pH 2, затем экстрагировали этилацетатом (3•20 мл). Объединенные органические экстракты промывали 50% рассолом (20 мл) и рассолом (20 мл), сушили (безводным сульфатом магния), фильтровали и выпаривали досуха. Остаток чистили хроматографически на колонке (2,5•10 см) с силикагелем, элюируя 7:3 смесью этилацетат: гептан (300 мл) и 1% уксусной кислотой в 7:3 смеси этилацетата/гептана (500 мл). Желаемые фракции концентрировали, удаляли метиленхлоридом и сушили в вакууме, получая 172 мг указанного в названии продукта в виде белой пены; [α]D= -116,9o (c=0,5 метанол). TLC (1% уксусная кислота в этилацетате) Rf=0,35.
1H-NMR: 400 MHz; CDCl3: δ 0,91 (m, 2H), 1,22 (m, 3H), 1,44 (m, 1H), 1,55 (m, 1H), 1,68 (m'S, 5H), 1,83 (m, 1H), 1,97 (m'S, 2H), 2,12 (m, 1H), 2,19 - 2,40 (m' S, 3H), 2,53 (m, 1), 2,96 (m, 1H), 3,38 (m'S, 2H), 4,62 (t, 1H, J= 6,8Hz), 4,70(m, 1H), 5,25 (m, 1H), 7..55(d, 1H, J=6,4 Hz).
13C-NMR: 100 MHz; CDCl3: δ/ 26,0, 26,1, 27,5, 31,5, 32,3, 33,0, 33,3, 35,2, 40,7, 43,0, 52,9, 60,6, 62,5, 170,9, 172,7, 175,3.
Данные анализа для C18H28N2O4S2:
Вычислено, %: С 53,98; Н 7,05; N 6,99; S 16,01
Найдено, %: С 53,97; Н 7,18; N 6,84; S 15,75.
HPLC: tR= 16 мин/>99%, УФ 217/; YMCS-3ODS /C-18/ 6,0• 150 мм; 50% B:A - 100% B:A, 25 минут линейный градиент /A=90% вода/метанол+0,2 фосфорной кислоты; B: 90% метанол/вода+0,2% фосфорной кислоты/; скорость потока при 1,5 мл/мин.
Пример 8
/4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксогексил/амино/-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновая кислота
a) /S/-2-Бромгексановая кислота
Бромид калия (15,9 г, 133 ммоля) добавляли к перемешиваемому раствору D-норлейцина (5,0 г, 38 ммоля) в 2,5 N серной кислоте (77 мл) при комнатной температуре. Реакционную смесь охлаждали до -10oC и добавляли твердый нитрит натрия (3,94 г, 57 ммоля) порциями, поддерживая температуру между -10o и -5oC. После окончания добавления пенистую реакционную массу перемешивали в течение 1 часа и затем нагревали до комнатной температуры и перемешивали еще 1 час. Затем реакционную смесь экстрагировали дважды эфиром, эфирные экстракты промывали один раз водой и сушили (сульфатом магния), фильтровали и выпаривали, получая 3,3 г неочищенного указанного в названии продукта.
b) Дициклогексиламиновая соль /S/-2-/ацетилтио/гексановой кислоты
К перемешиваемому шламу тиоцетата калия (2,11 г, 18,5 ммоля) в 50 мл сухого ацетонитрила при комнатной температуре в атмосфере аргона добавляли раствор продукта из части (a) (3,27 г, 16,8 ммоля) в 26 мл ацетонитрила. Реакционную смесь перемешивали в течение 5 часов. Полученный шлам фильтровали и выпаривали. Остаток вновь растворяли в этиловом эфире, промывали один раз 5% раствором бисульфата калия и один раз рассолом, сушили (сульфатом магния) и выпаривали. Остаток растворяли в эфире (64 мл) и обрабатывали дициклогексиламином (3,4 мл, 16,8 ммоля/. Эфирный раствор концентрировали в вакууме и растирали с гексаном, получая белое твердое вещество, которое перекристаллизовывали из смеси этиловый эфир/гексан, получая указанный в названии продукт. Маточный раствор концентрировали и перекристаллизовывали дважды, получая полный выход 2,2 г указанного в названии продукта; т.пл. 145 - 147oC; [α]D= -33,8o (с = 1,08, хлороформ).
c) Метиловый эфир /4S-/4α/R*/, 7α, 9aβ//- октагидро-4-//2-/ацетилтио/-1-оксогексил/амино/-5-оксопирроло- [2,1-b] /3,3/тиазепин-7-карбоновой кислоты
Суспензию дициклогексиламиновой соли из части (b) (255 мг, 9,69 ммоля, 1,05 экв.) в этилацетате (15 мл) промывали 5% раствором бисульфата калия (3 • 5 мл) и рассолом (10 мл), сушили (безводным сульфатом магния), фильтровали, концентрировали, удаляли метиленхлоридом (дважды) и сушили в вакууме в течение 1 часа, получая свободную кислоту в виде масла.
Это масло растворяли в сухом метиленхлориде (6 мл), охлаждали до 0oC (баня со льдом) и обрабатывали триэтиламином (96 мкл, 0,69 ммоля, 1,05 экв. ), затем солью метилового эфира /4S-/4α, 7α, 9aβ//- 4-аминооктагидро-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты и п-толуолсульфоновой кислоты, полученной как описано в примере 7 (a), (275 мг, 0,66 ммоля), триэтиламином (92 мкл, 0,66 ммоля) и наконец, бензотриазол-1-илокситрис/диметиламино/ фосфонийгексафторфосфатом (305 мг, 0,69 ммоля). Полученный раствор перемешивали при 0oC в течение 1 часа, затем при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали, разбавляли этилацетатом, промывали 5% раствором бисульфата калия (20 мл), 50% насыщенным раствором бикарбоната натрия (20 мл), рассолом (20 мл), сушили (безводным сульфатом магния), фильтровали и выпаривали досуха. Неочищенный продукт адсорбировали на целите и хроматографировали на колонке (2,5 • 10 см) с силикагелем, элюируя 40% (1 л) смесью этилацетат/гексан. Желаемые фракции объединяли и концентрировали, получая 258 мг чистого указанного в названии продукта. TLC (этилацетат:гексан 8:2) Rf = 0,54.
d) /4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксогексил/-амино/-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновая кислота
Раствор продукта из части (c) (255 мг, 0,54 ммоля) в метаноле (5 мл), продутый аргоном в течение 30 минут, охлажденный до 0oC, обрабатывали по каплям предварительно продутым (аргоном в течение 30 минут) раствором 1,0 N гидроокиси натрия (5 мл), поддерживая условия пробулькивания аргона в течение периода добавления и протекания реакции. Реакционную смесь перемешивали при 0oC в течение 2 часов, подкисляли при 0oC 5% раствором бисульфата калия до pH 2, затем экстрагировали этилацетатом (3 • 20 мл). Объединенные органические экстракты промывали 50% рассолом (20 мл) и рассолом (20 мл), сушили (безводным сульфатом магния), фильтровали и выпаривали досуха. Остаток чистили хроматографически на колонке 2,5 • 10 см с силикагелем, элюируя 7:3 смесью этилацетат:гептан (300 мл) и 1% уксусной кислотой в 7:3 смеси этилацетат: гептан (500 мл). Желаемые фракции концентрировали, удаляли метиленхлоридом и сушили в вакууме, получая 170 мг указанного в названии продукта в виде белой пены; [α]D= -135,1o (с = 0,5, метанола). TLC (1% уксусная кислота в этилацетате) Rf = 0,32.
1H NMR: 400 MHz; CDCl3: δ 0,89 (t, 3H, J=7Hz), 1,32(m,4H), 1,73(m,1H), 1,96(m, 2H), 2,00(d, 1H), J=8,6 Hz), 2,11(m, 1H), 2,32(m's, 3H), 2,52(m, 1H), 2,98(m, 1H), 3,32(m's, 2H), 4,61(t, 1H, J=7,1 Hz), 4,72(m, 1H), 5,25(m, 1H), 7,63(d, 1H, J=6,4 Hz).
13C - NMR: 100 MHz; CDCl3: δ 13,8, 22,2, 27,6, 29,2, 31,4, 32,6, 33,1, 35,3, 43,0, 52,8, 60,6, 62,42, 170,7, 172,5, 174,0.
Данные анализа для C15H24N4O4S2O • 0,08 H2O:
Вычислено, %: С 49,79; Н 6,73; N 7,74; S 17,72
Найдено, %: С 49,90; Н 6,92; N 7,63; S 17,57.
HPLC: tR = 9,4 мин (> 9%, УФ 220); YMC S - 3 ODS (C-18) 6,0 • 150 мм; 50% B: A-100% B: A, 25 минут линейный градиент /A=90% вода/метанол + 0,2% фосфорной кислоты/; B= 90% метанол/вода + 0,2% фосфорной кислоты/ скорость потока при 1,5 мл/мин.
Пример 9
/4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1- оксо-4-метилфенил/амино/-5-оксопирроло[2,1-b] /1,3/тиазепин-7- карбоновая кислота
a) /S/-2-Бром-4-метилпентановая кислота
Бромид калия (9,5 г, 80 ммоля) добавляли к перемешиваемому раствору D-лейцина (3,0 г, 23 ммоля) в 2,5 N серной кислоте (47 мл) при комнатной температуре. Реакционную смесь охлаждали до -10oC и порциями добавляли твердый нитрит натрия (2,4 г, 34 ммоля), поддерживая температуру между -10o и -5oC. После окончания добавления, реакционную смесь перемешивали в течение 1 часа и затем нагревали до комнатной температуры и перемешивали еще один час. Затем реакционную смесь дважды экстрагировали эфиром, эфирные экстракты промывали один раз водой, сушили (сульфатом магния), фильтровали и выпаривали, получая 2,7 г неочищенного указанного в названии продукта.
b) Дициклогексиламиновая соль /S/-2-/ацетилтио/-4-метиленпентановой кислоты
К перемешиваемому шламу тиоацетата калия (1,7 г, 15,0 ммоля) в 50 мл сухого ацетонитрила при комнатной температуре аргона добавляли раствор продукта из части (a) (2,6 г, 13 ммоля) в 17 мл ацетонитрила. Реакционную смесь перемешивали в течение 4 часов. Полученный шлам фильтровали и выпаривали. Остаток вновь растворяли в этиловом эфире, промывали один раз 5% раствором гидросульфата калия и один раз рассолом, сушили (сульфатом магния) и выпаривали. Остаток растворяли в эфире (64 мл) и обрабатывали дициклогексиламином (2,7 мл, 1,4 ммоля). Из раствора немедленно начинало выпадать белое твердое вещество. Раствор фильтровали и собирали белое твердое вещество, получая 2,0 г указанного в названии продукта, т.пл. 153 - 158oC; [α]D= -54,5oC /с = 0,61, хлороформ/.
c) Метиловый эфир /4S-/4α/R*/, 7α, 9aβ//- октагидро-4-//2-/ацетилтио/-1-оксо-4-метилпентил/амино/-5-оксопирроло [2,1-b]/1,3/тиазепин-7-карбоновой кислоты
Перемешиваемую суспензию дициклогексиламиновой соли продукта из части (b) (234 мг, 0,63 ммоля) в этилацетате (15 мл) промывали 5% водным раствором бисульфата калия (3 • 5 мл). Органический экстракт сушили (безводным сульфатом магния), фильтровали и дважды выпаривали из гексана. Полученное масло растворяли в метиленхлориде (6 мл) и перемешивали в атмосфере азота при 0oC. К этому раствору добавляли триэтиламин (88 мкл, 0,63 ммоля), затем соль метилового эфира /4S-/4α, 7α, 9aβ//- 4-амино-октагидро-5-оксопирроло[2,1-b] /1,3/-тиазепин-7-карбоновой кислоты и п-толуолсульфоновой кислоты (полученной как описано в примере 7 (a), 249 мг, 0,6 ммоля), дополнительное количество триэтиламина (84 мкл, 0,609 ммоля) и через 10 минут бензотриазол-1-илокситрис-/диметиламино/- фосфоний гексафторфосфата (279 мг, 0,63 ммоля). Через 1 час реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Полученный бесцветный раствор выпаривали при температуре ниже 30oC и маслообразный остаток вновь растворяли в этилацетате. Раствор промывали один раз 5% раствором бисульфата калия, один раз насыщенным раствором бикарбоната натрия и один раз рассолом. Органический слой сушили (сульфатом магния), фильтровали и выпаривали на 5 г силикагеля. Очистка флеш-хроматографированием на колонке (2,5•15 см, элюирование смесью 1:1 этилацетат/гексан) давала 203 мг указанного в названии продукта в виде белого твердого вещества, т.пл. 101-103oC, [α]D= -157,5oC (c = 1,04, хлороформ). TLC (этилацетат:гексан, 1:1) Rf = 0,19.
d) /4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксо-4-метилпентил/амино/-5-оксопирроло [2,1-b]/1,3/-тиазепин-7-карбоновая кислота
Раствор продукта из части (c) (184 мг, 0,44 ммоля) в 5 мл метанола продували азотом в течение 10 минут и охлаждали до 0oC. К этому раствору по каплям добавляли 5 мл продутого азотом раствора 1 M гидроокиси натрия. Азот медленно пробулькивали через раствор в процессе реакции. Через 2 часа реакционную смесь подкисляли 2 мл 6M соляной кислоты, экстрагировали дважды этилацетатом и экстракты объединяли, сушили (сульфатом магния) и выпаривали. Повторное выпаривание из гексана и растирание остатка в смеси метанол/вода давало 132 мг указанного в названии продукта в виде твердого кристаллического вещества, т. пл. 94-96oC, [α]D= -158,6oC (c = 0,42, метанол). TLC (этилацетат:уксусная кислота 4:4:0,1), Rf = 0,13.
Данные анализа для C15H24N2S2O4 • 0,75 H2O:
Вычислено, %: С 48,17; Н 6,87; N 7,49; S 17,15
Найдено, %: С 48,33; Н 6,51; N 7,37; S 16,82.
HPLC: Rf = 17,6 мин; (99,2 %) YMC S-3-ODS(C-18), 6,0•150 мм; 0% до 100% B:A, 25 мин линейный градиент и 15 мин выдерживания 1,5 мл/мин.
A = 90% вода/метанол + 0,2% фосфорной кислоты;
B = 90% метанол/вода + 0,2 фосфорной кислоты;
220 нм.
Пример 10
/4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-/1,4/оксазино [3,4-b]/1,3/оксазепин-7-карбоновая кислота
a) 2-Пропениловый эфир 2,2,2-трихлорацетимидовой кислоты
Суспензия 80% гидрида натрия (945 мг, 31,5 ммоля, дважды промытую 25 мл гексана) в сухом эфире (30 мл) обрабатывали по каплям раствором 2-пропен-1-олом (21,4 мл, 18,3 г, 315 ммоля) в сухом эфире (45 мл), перемешивали в течение 20 минут при комнатной температуре в атмосфере аргона и затем охлаждали до 0oC (в бане лед с солью). Трихлорацетонитрил (30 мл или 42,3 г, 0,30 моля) добавляли в течение 15 минут и коричневый раствор перемешивали при 0oC в течение 40 минут, при 10oC в течение 10 минут и при комнатной температуре в течение 10 минут. Реакционную смесь концентрировали до сиропа, обрабатывали раствором метанола (1,2 мл) в пентане (30 мл) и интенсивно перемешивали в течение 5,0 минут. Светло-коричневый осадок отфильтровывали, промывали пентаном (2•30 мл) и объединенные фильтраты концентрировали до светло-коричневой жидкости. Жидкость повторно растворяли в пентане (30 мл) перемешивали в течение нескольких минут и полученную суспензию отфильтровывали и полученные осадки промывали пентаном (30 мл), повторяли эту процедуру по крайней мере еще не менее одного раза. Чистый фильтрат концентрировали и сушили в вакууме, получая 54,0 г указанного в названии соединения в виде светло-красной окрашенной жидкости. Это вещество хранили в виде раствора в гексане при 10oC.
b) Метиловый эфир N-фталоил-L-серина
Суспензию гидрохлорида метилового эфира L-серина (25 г, 161 ммоля) в воде (350 мл) разбавляли диоксаном (250 мл) и полученный прозрачный раствор обрабатывали твердым карбонатом натрия (17,0 г, 1,0 экв.) с последующей обработкой N-карбетоксифталимидом (37 г, 1,05 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2,5 часов в атмосфере аргона. Смесь экстрагировали этилацетатом (3•500 мл) и объединенные органические экстракты промывали последовательно 5% раствором бикарбоната натрия (250 мл), 5% бисульфатом калия (250 мл) и рассолом (250 мл), сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме. Продукт смеси хроматографировали на колонке с силикагелем (Мерк), элюируя смесью этилацетат: гексан (1: 3, 1: 2), и желаемые фракции объединяли, выпаривали досуха и сушили в вакууме, получая 31 г указанного в названии соединения в виде вязкого сиропа. TLC/этилацетат:гексан, 1:1/Rf = 0,52.
c) Метиловый эфир N-фталоил-1-/2-пропенил/-L-серина
Раствор продукта из части (b) (7,37 г, 29,5 ммоля) в сухом метиленхлориде (30 мл) обрабатывали раствором продукта из части (a) (11,97 г, 59,1 ммоля, 2 экв.) в циклогексане (60 мл) с последующей обработкой трифторметансульфоновой кислотой (0,37 мл) и реакционную смесь перемешивали при комнатной температуре в течение 20 часов в атмосфере аргона. Осадки отфильтровывали, промывали минимальным количеством метиленхлорида и объединенные фильтраты промывали 5% раствором бикарбоната натрия (30 мл) и водой (30 мл), сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме. Неочищенную смесь хроматографировали на колонке с силикагелем (Мерк), элюируя смесью этилацетат:гексан (1:9). Желаемые фракции объединяли, выпаривали досуха и сушили в вакууме, получая 7,56 г указанного в названии соединения в виде прозрачного вязкого сиропа.
TLC (этилацетат:гексан, 1:1), Rf = 0,70.
d) Метиловый эфир N-фталоил-0-/ацетальдегид/-L-серина
Раствор продукта из части (c) (2,5 г, 8,64 ммоля) в смеси сухого метиленхлорида (46,4 мл) и метанола (4,6 мл) охлаждали до -78oC (в бане сухой лед-ацетон) и обрабатывали озоном до устойчивой голубой окраски (около 15 минут). Затем смесь продували азотом в течение 10 минут (до исчезновения голубой окраски), обрабатывали диметилсульфидом (14,0 мл, 0,19 моля, 22,1 экв.), нагревали до комнатной температуры и перемешивали в течение 2,5 часов в атмосфере азота. Реакционную смесь выпаривали досуха и остаток в виде сиропа растворяли в этилацетате (50 мл), промывали водой (15 мл) и рассолом (15 мл), сушили (безводным сульфатом магния), фильтровали, выпаривали досуха и сушили в вакууме.
Неочищенный продукт хроматографировали на колонке с силикагелем (Мерк), элюируя смесью этилацетат: гексан (1:9; 1:4; 1:2) с получением 1,54 г указанного в названии соединения. TLC (этилацетат:гексан, 1:1) Rf = 0,33.
e) Метиловый эфир N-фталоил-0-/2,2-диметоксиэтил/-L-серина
Раствор продукта из части (d) (1,54 г, 5,29 ммоля) в смеси сухого метиленхлорида (8,3 мл) и сухого метанола (8,3 мл) обрабатывали триметиловым эфиром орто-муравьиной кислоты (0,84 мл, 7,68 ммоля, 1,45 экв.) и моногидратом п-толуолсульфоновой кислоты (92 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение 2,5 часов, затем разделяли между этилацетатом (50 мл) и насыщенным раствором бикарбоната натрия (15 мл). Органическую фазу промывали водой (15 мл) и рассолом (15 мл), сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Мерк), элюируя смесью этилацетат:гексан (1:4), получая 1,35 г указанного в названии продукта в виде вязкого прозрачного сиропа. TLC (этилацетат:гексан, 1:1) Rf = 0,58.
f) Метиловый эфир О-/2,2-диметоксиэтил/-L-серина
Раствор продукта из части (e) (2,0 г, 5,93 ммоля) в сухом метаноле (14 мл) обрабатывали гидразингидратом (0,30 мл, 6,1 ммоля) и перемешивали при комнатной температуре в течение 4 дней в атмосфере аргона. Полученную суспензию фильтровали, промывали осадок метанолом (2•14 мл) и фильтрат концентрировали досуха. Сироп повторно растворяли в метиленхлориде и фильтровали более двух раз до тех пор, пока осадка уже больше не получалось. Прозрачный фильтрат концентрировали, получая 1,17 г указанного в названии продукта в виде светло-желтого сиропа. TLC (метиленхлорид:метанол, 9:1) Rf = 0,54.
g) N-//фенилметокси/карбонил/-О-//1,1-диметилэтил/-диметилсилил/-L- гомосерин
Раствор N-//фенилметокси/карбонил/-L-гомосерина /полученного как описано в примере 6(a), (3,0 г, 11,85 ммоля) в сухом диметилформамиде /65 мл/ обрабатывали //1,1-диметилэтил/-диметилсилил хлоридом (10,72 г, 71,1 ммоля) и имидазолом (9,65 г, 0,14 моля) и перемешивали при комнатной температуре в атмосфере аргона в течение 24 часов. Реакционную смесь разбавляли метанолом (207 мл), перемешивали еще 24 часа при комнатной температуре и затем концентрировали до сиропа. Оставшийся сироп растворяли в этилацетате (200 мл), промывали 10% лимонной кислотой (2•75 мл) и рассолом, сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме. Неочищенную смесь хроматографировали на колонке с силикагелем (Мерк), элюируя смесью этилацетат: гексан (1:1) с последующим элюированием смесью этилацетат: уксусная кислота (99,5: 0,5). Желаемые фракции объединяли, концентрировали и выпаривали несколько раз из толуола, получая 3,56 г указанного в названии соединения в виде воскообразного твердого вещества. TLC (этилацетат:уксусная кислота, 95:5) Rf = 0,82.
h) Метиловый эфир N-/O-//1,1-диметилэтил/ диметилсилил/-N-//фенилметокси/ карбонил/-L-гомосерил/ -O-/2,2-диметоксиэтил/-L-серина
Раствор продукта из части (g) (2,18 г, 5,93 ммоля) в сухом метиленхлориде до 0oC (в бане лед с солью) и обрабатывали последовательно раствором продукта из части (f) (1,71 г, 5,65 ммоля) в сухом метиленхлориде (5 мл), триэтиламином (0,78 мл, 5,65 ммоля) и бензотриазол-1-илокситрис-/диметиламино/фосфонийгексафторфосфатом (2,63 г, 6,0 ммоля). Реакционную смесь перемешивали при 0oC в течение 30 минут и при комнатной температуре в течение 1 часа и 45 минут. Реакционную смесь разделяли между этиловым эфиром (2•100 мл) и водой (30 мл) и объединенные органические экстракты промывали 50% насыщенным раствором бикарбоната натрия (20 мл) и рассолом (25 мл), сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме. Неочищенную смесь хроматографировали на колонке с силикагелем (Мерк), элюируя смесью этилацетат: гексан (1:4, 1:3, 1:1), получая 2,38 г указанного в названии продукта, TLC (этилацетат:гексан, 1:1) Rf = 0,40,
i) Метиловый эфир /4S-/4α, 7α, 10aβ//- октагидро-4-//фенилметокси/-карбонил/амино-5-оксо-/1,4/оксазино[3,4-b] /1,3/ оксазепин-7-карбоновой кислоты
Раствор продукта из части (h) (1,0 г, 1,8 ммоля) в сухом метиленхлориде (50 мл) обрабатывали ионообменной смолой - Амберлит®15 (кислая форма) и метанолом (0,1 мл) и полученную смесь перемешивали при комнатной температуре в атмосфере аргона в течение трех дней. Смолу отфильтровывали, промывали небольшим количеством метиленхлорида и фильтрат концентрировали до сиропа. Неочищенную смесь хроматографировали на колонке с силикагелем, элюируя смесью этилацетат: гексан (1:1), получая 339 мг указанного в названии продукта. TLC (этилацетат: гексан, 3:1) Rf = 0,53.
j) Метиловый эфир /4S-/4α, 7α, 10aβ/- октагидро-4-амино-5-оксо/1,4/оксазино[3,4-b]/1,3/оксазепин-7-карбоновой кислоты
Раствор продукта, полученного, как описано в части (i) (771 мг, 2,04 ммоля), в сухом метаноле (25 мл) обрабатывали 10% палладием на угле в качестве катализатора (125 мг) и гидрировали (из баллона) при комнатной температуре в течение 16 часов. Реакционную смесь фильтровали через целитную подложку и подложку промывали метанолом (2 •25 мл). Прозрачный фильтрат выпаривали досуха и сушили в вакууме, получая 448 мг указанного в названии продукта в виде сиропа. TLC (метиленхлорид:метанол, 9:1) Rf = 0,22.
k) Метиловый эфир /4S-/4α/R*/, 7α, 10aβ/- октагидро-4-//2-/ацетилтио/-1-оксо-3-фенилпропил/амино/-5-оксо/1,4/оксазино [3,4-b] /1,3/оксазепин-7-карбоновой кислоты
Дициклогексиламиновую соль /S/-2-/ацетилтио/бензолпропановой кислоты (813 мг, 2,01 ммоля) суспендировали в этилацетате (70 мл), промывали 5% раствором бисульфата калия (5•9,3 мл) и рассолом (9,3 мл), сушили (безводным сульфатом магния), фильтровали, выпаривали досуха и сушили в вакууме.
Эту свободную кислоту растворяли в сухом метиленхлориде (12 мл), охлаждали до 0oC (в бане лед с солью) и обрабатывали последовательно раствором продукта из части (j) (448 мг, 1,84 ммоля) в сухом метиленхлориде (4,0 мл), триэтиламином (0,25 мл, 1,80 ммоля) и бензотриазол-1-илокситрис/ диметиламино/фосфонийгексафторфосфатом (823 мг, 1,86 ммоля). Реакционную смесь перемешивали при 0oC в течение 1 часа и при комнатной температуре в течение 2 часов в атмосфере аргона. Реакционную смесь отгоняли досуха и полученный сироп вновь растворяли в этилацетате (60 мл), промывали 0,5 N соляной кислотой (2•11 мл), водой (11 мл) и рассолом (11 мл), сушили (безводный сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме. Неочищенную смесь хроматографировали дважды на колонке с силикагелем (Мерк), элюируя каждый раз смесью этилацетат:гексан (1:1, 1:1), получая 665 мг указанного в названии продукта в виде сиропа. TLC (этилацетат:гексан, 3:1) Rf = 0,30.
i) /4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//меркапто-1-оксо-3-фенилпропил/амино/5-оксо /1,4/оксазино[3,4-b]/1,3/оксазепин-7-карбоновая кислота
Раствор продукта из части (k) (650 мг, 1,44 ммоля) в метаноле (13 мл) продували аргоном в течение 30 минут, охлаждали до 0oC (в бане лед с солью) и обрабатывали по каплям раствором 1,0 N гидроокиси натрия (5,84 мл, предварительно продутым аргоном в течение 30 минут), продолжая пробулькивание аргона в процессе добавления и протекания реакции. Реакционную смесь перемешивали при 0oC в течение 5,0 часов и гасили при 0oC 5% раствором бисульфата калия (25,4 мл). Смесь нагревали до комнатной температуры, экстрагировали этилацетатом (3•50 мл) и объединенные органические экстракты промывали рассолом (15 мл), сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме. Неочищенный продукт растирали со смесью гексан: метиленхлорид (130: 7) и полученное твердое вещество хроматографировали на колонке с силикагелем (Мекр), элюируя смесью этилацетат (гексан) 1: 2, 1:1 (с последующим элюированием смесью метиленхлорид: метанол:уксусная кислота (100:4:0,2). Желаемые фракции объединяли, выпаривали досуха и выпариванием несколько раз из толуола получали 364 мг указанного в названии продукта, который сушили в вакууме в течение 9 часов. Полученный продукт затем растирали со смесью метиленхлорид: гексан (1:10), гексаном (50 мл) и пентаном (2•50 мл), перемешивая с первыми 50 мл в течение 4 часов и со следующими 50 мл пентана в течение ночи в атмосфере аргона. Растворитель декантировали и твердое вещество сушили в вакууме в течение 6,0 часов, получая указанный в названии продукт в виде твердой аморфной пены; [α]D= -49,1o (с = 0,48, метанол). TLC (толуол:уксусная кислота, 5:1) Rf = 0,17.
Данные анализа для C18H22N2O6S • 0,56 H2O:
Вычислено, %: С 53,45; Н 5,76; N 6,98; S 7,92
Найдено, %: С 53,45; Н 5,53; N 6,75; S 7,48.
HPL C: Rt = 10,45 мин; (98,3%); YMC S - 3 ODS(C=18) 6,0• 150 мм; 44% (10% воды - 90 метанола - 0,2% фосфорной кислоты) (56%/ 90% воды - 10% метанола - 0,2% фосфорной кислоты), изократный; 1,5 мл/мин.
Пример 11
/4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-7H-пиридо [2,1-b] /1,3/ тиазепин-7-карбоновая кислота
Продукт примера 3 получали также следующим образом:
а) N-//Фенилметокси/карбонил/-O-//1,1-диметилэтил/-диметилсилил/-L-гомосерин
//1,1-Диметилэтил/диметилсилил/хлорид /37,5 г, 249 ммоля/ добавляли к раствору N-//Фенилметокси/ карбонил/-L-гомосерина (полученному, как описано в примере 6 (a), 41,56 ммоля) в диметилформамиде (125 мл) с последующим добавлением имидазола (33,95 г, 498 ммоля). Полученный светло-желтый раствор перемешивали при комнатной температуре в течение 22 часов. Добавляли метанол (500 мл), реакционную смесь дополнительно перемешивали в течение 6 часов и затем метанол и большую часть диметилформамида удаляли в вакууме. Оставшийся остаток помещали в этилацетат (800 мл), промывали 10% лимонной кислотой (2•300 мл) и объединенные водные фазы экстрагировали этилацетатом (300 мл). Объединенные этилацетатные фазы промывали водой и рассолом, сушили (сульфатом натрия), концентрировали и остаток выпаривали с гексаном с получением белого порошка. Этот порошок сушили в вакууме, получая 12,942 г указанного в названии продукта.
b) Метиловый эфир /S-/R*, R* //-2-//4-//1,1-диметилэтил/окси-1-оксо-2-//фенилметокси/карбонил/-амино/ бутил/амино/-6,6-диметоксигексановой кислоты
К раствору продукта из части (а) (22,78 г, 61,97 ммоля) в метиленхлориде (100 мл), охлажденному до 0oC, добавляли N-метилморфолин (6,81 мл, 61,97 ммоля) с последующим добавлением гидроксибензотриазола (8,37 г, 61,97 ммоля), метилового эфира /S/-2-амино-6,6-диметоксигенсановой кислоты /полученного как описано в примере 1 (e), 10,6 г, 51,64 ммоля/ в метиленхлориде (50 мл) и затем 1-этил-3-/3-диметиламинопропил/карбодиимида (11,88 г, 61,97 ммоля). Реакционную смесь перемешивали при 0oC в течение 1 часа и затем при комнатной температуре в течение ночи. Реакционную смесь концентрировали в вакууме и остаток разбавляли этилацетатом (600 мл), промывали 5% раствором бисульфата калия (200 мл), 0,5 N раствором гидроокиси натрия (200 мл), водой и рассолом, и сушили (сульфатом натрия). Фильтрат концентрировали и остаток помещали в этиловый эфир (150 мл). Полученную суспензию фильтровали и собранное твердое вещество интенсивно промывали этиловым эфиром. Фильтрат концентрировали в вакууме досуха, получая 30 г неочищенного указанного в названии продукта в виде маслообразного соединения, которое использовали в следующей реакции без очистки. TLC (этилацетат: гексан, 8:2) Rf = 0,55.
c) Метиловый эфир /S-/R*,R*//-2-//4-гидрокси-1- оксо-2-//фенилметокси/карбонил/амино/бутил/амино/-6,6- диметоксигексановой кислоты
К раствору продукта из части (b) (30 г) в метаноле (150 мл), охлажденному до 0oC, добавляли моногидрат п-толуолсульфоновой кислоты (1,96 г). Реакционную смесь перемешивали при 0oC в течение 2 часов до тушения ее водным раствором бикарбоната натрия (1,3 г, бикарбоната натрия в 100 мл воды). Смесь концентрировали в вакууме и остаток разделяли между этилацетатом (400 мл) и водой (150 мл). Отделенную водную фазу экстрагировали этилацетатом (2 • 150 мл). Объединенные этилацетатные слои промывали 10% раствором бикарбоната натрия, рассолом (2 раза), сушили (сульфатом натрия), фильтровали и выпаривали досуха. Остаток подвергали флеш-хроматографированию на колонке 10 • 25 см с силикагелем, элюируя 80% этилацетатом в гексане (5 л), этилацетатом (3 л) и 2% метанолом в этилацетате (5 л). Желаемые фракции объединяли и концентрировали и сушили в вакууме, получая 17,45 г указанного в названии продукта в виде бледно-желтого масла. TLC (8:2, этилацетат:гексан) Rf = 0,17.
d) Метиловый эфир /S-/R*,R*/-2-//4-/метансульфонил/окси-1-оксо-2-// фенилметокси/карбонил/амино/бутил/-амино/-6,6-диметоксигексановой кислоты
К раствору продукта из части (c) (17,40 г, 39,50 ммоля) (удаленному с толуолом три раза и высушенному в вакууме в течение ночи) в сухом метиленхлориде (250 мл), охлажденному при -15oC (лед/ацетон), добавляли триэтиламин (8,26 мл, 59,28 ммоля, свежеперегнанный) с последующим добавлением метансульфонилхлорида (3,67 мл, 47,4 ммоля) по каплям. Реакционную смесь перемешивали при -15oC в течение 30 минут, затем гасили насыщенным раствором хлористого аммония (100 мл). После перемешивания в течение 5 минут смесь разбавляли этилацетатом (600 мл) и промывали 5% раствором бисульфата калия, рассолом, сушили (сульфатом натрия), фильтровали и выпаривали досуха. Остаток сушили в вакууме, получая 20,40 г указанного в названии соединения в виде желтого масла, которое использовали в следующей реакции без очистки. TLC (8:2, этилацетат:гексан) Rf = 0,35.
e) Метиловый эфир /S-/R*,R*//-2-//4-ацетилтио/-1- оксо-2-///фенилметокси/карбонил/амино/бутил/амино/-6,6- диметоксигексановой кислоты
К раствору тиоуксусной кислоты (5,09 г, 71,10 ммоля) в метаноле (100 мл) добавляли карбонат цезия (10,81 г, 33,18 ммоля). Полученный раствор концентрировали в вакууме. Твердое вещество растирали с сухим ацетоном (3 раза) и затем сушили в вакууме над пятиокисью фосфора в течение ночи, получая тиоацетат цезия.
Раствор продукта из части (d) (20,40 г, 39,50 ммоля) в сухом диметилформамиде (150 мл) добавляли через канюлю к суспензии тиоацетата цезия (10,576 г, 50,85 ммоля) в диметилформамиде (50 мл). Полученный желтый раствор перемешивали в атмосфере аргона при комнатной температуре в течение ночи, затем концентрировали при высоком вакууме для удаления большей части диметилформамида. Остаток помещали в этилацетат (1 л), промывали 10% раствором бикарбоната натрия (200 мл), водой (4 • 200 мл), рассолом (400 мл) и сушили (сульфатом натрия). Фильтрат концентрировали и остаток выпаривали с толуолом (3 раза), затем сушили в вакууме, получая 20 г указанного в названии соединения в виде светло-желтого твердого вещества, которое использовали для следующей реакции без очистки. TLC (8:2, этилацетат:гексан) Rf = 0,47.
f) Метиловый эфир /S-/R*,R*//-2-//4-меркапто-1- оксо-2-//фенилметоксикарбонил/амино/бутил/амино/-6,6- диметоксигексановой кислоты
Раствор продукта из части (e) (20 г, 39,50 ммоля) в метаноле (250 мл) при 0oC продували аргоном в течение 15 минут. По каплям добавляли раствор 25% (вес/вес, плотность = 0,945) метоксида натрия в метаноле (9,17 мл, 40 ммоля) при непрерывной продувке аргона. После перемешивания в течение 5 минут реакцию гасили насыщенным раствором хлористого аммония (200 мл) и смесь разделяли между этилацетатом (1 л) и водой (200 мл). Водную фазу экстрагировали этилацетатом (200 мл). Объединенный этилацетатный экстракт промывали насыщенным раствором хлористого аммония (400 мл), рассолом (400 мл), сушили (сульфатом натрия), фильтровали и концентрировали в вакууме, получая 17,5 г указанного в названии продукта в виде желтого масла, которое использовали для следующей реакции без очистки. TLC (8:2, этилацетат:гексан) Rf = 0,45.
g) Метиловый эфир /4S-/4α, 7α, 10aβ/- октагидро-4-///фенилметокси/карбонил/амино/-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
К раствору продукта из части (f) (17,5 г, 38,3 ммоля) в метиленхлориде (600 мл) добавляли ионообменную смолу - Амберлит®15 (6 г, предварительно обработанную 6 N соляной кислотой, водой, тетрагидрофураном и метиленхлоридом и высушенную). Суспензию перемешивали в атмосфере аргона при комнатной температуре в течение 18 часов и фильтровали. Фильтрат концентрировали и остаток адсорбировали на целите, очищали на колонке с силикагелем (10 • 30 см), элюируя 20 - 30% этилацетатом в гексане. Желаемые фракции объединяли и выпаривали в вакууме досуха, получая 9,18 г указанного в названии продукта в виде желтого масла. TLC (1:1, этилацетат:гексан) Rf = 0,32.
h) Метиловый эфир /4S-/4α, 7α, 10aβ//- октагидро-4-амино-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
К раствору продукта из части (g) (9,1 г, 23,19 ммоля, выпаренному с толуолом три раза и высушенному в вакууме в течение ночи/ в сухом метиленхлориде (150 мл) добавляли иодтриметилсилан (4,95 г, 34,78 ммоля) по каплям. Полученный желтый раствор перемешивали в атмосфере аргона при комнатной температуре в течение 1,5 часов, затем гасили 0,4 N соляной кислотой в смеси метанол/диоксан (120 мл). Летучие удаляли в вакууме и остаток разделяли между эфиром (500 мл) и водой (700 мл). Органическую фазу экстрагировали 0,1 N соляной кислотой (150 мл) и объединенный кислый водный экстракт охлаждали до 0oC, доводили до щелочной реакции pH 10,5 раствором 1,0 N гидроокиси натрия (по показаниям pH-метра), затем экстрагировали метиленхлоридом (4 • 400 мл). Объединенные органические экстракты промывали рассолом, сушили (сульфатом натрия), фильтровали и концентрировали в вакууме, получая 6,45 г указанного в названии продукта в виде желтого масла, которое использовали для следующей реакции без дальнейшей очистки. TLC (1:9, метанол:метиленхлорид) Rf = 0,20.
i) /4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5- оксо-7H-пиридо[2,1-b]/1,3/тиазепин-7-карбоновая кислота
Холодный (0oC) раствор /S/-/2-ацетилтио/бензолпропановой кислоты и триэтиламина в метиленхлориде обрабатывали раствором продукта из части (h) в метиленхлориде с последующей обработкой бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфатом. Реакцию проводили согласно процедуре, описанной в примере 3 (c), получая метиловый эфир /4S-/4α/R*/, 7α, 10aβ//- октагидро-4-//2-/ацетилтио/-1-оксо-3- фенилпропил/амино/-5-оксо-7H-пиридо[2,1-b]/1,3/тиазепин-7-карбоновой кислоты.
Раствор этого метилэфирного продукта в обескислороженном метаноле обрабатывали 1 N раствором гидроокиси натрия согласно процедуре примера 3 (d), получая указанный в названии продукт.
Пример 12
/4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркаптометил/-1-оксо-3-фенилпропил/амино/-5- оксо-7H-пиридо[2,1-b]/1,3/тиазепин-7-карбоновая кислота
a) Эфедриновая соль /S/-2-//ацетилтио/метил/бензолпропановой кислоты
Раствор /1R,2S/-/-/-эфедрина (17,3 г, 105 ммоля) в диэтиловом эфире (175 мл) добавляли в одну порцию к раствору 2-//ацетилтио/метил/бензолпропановой кислоты (50,0 г, 210 ммоля) в диэтиловом эфире (175 мл). После пребывания при комнатной температуре в течение 16 часов кристаллизованную соль эфедрина собирали фильтрованием (19, г); т. пл. 114 - 125oC; [α]D= -40,6o (c = 1, метанол). Дополнительно количество твердого вещества (8,9 г); т. пл. 121 - 126o; [α]D= -47,2o (c = 1, метанол) выделяли из фильтрата после пребывания при комнатной температуре в течение 20 часов. Твердое вещество объединяли и перекристаллизовывали из ацетонитрила (1500 мл). Через 16 часов при комнатной температуре собирали 20,8 г твердого вещества, т. пл. 125 - 130oC; [α]D= -48,9o (c = 1, метанол). Это вещество перекристаллизовывали таким же образом из ацетонитрила (300 мл), получая 18,7 г. т. пл. 128 - 130o; [α]D= -48,9o (c = 1, метанол).
Третья перекристаллизация из ацетонитрила (225 мл) давала 17,4 г твердого /S/-2-//ацетилтио/метил/бензолпропановой кислоты, эфедриновой соли, т. пл. 128 - 129oC; [α]D= -50,1o (c = 1, метанол).
Данные анализа для C12H14O3S • C10H15NO:
Вычислено, %: С 65,48; Н 7,24; N 3,47; S 7,95
Найдено, %: С 65,46; Н 7,34; N 3,21; S 8,00.
b) Метиловый эфир /4S-/4α/R*/, 7α, 10aβ//- октагидро-4-//2-//ацетилтио/метил/-1-оксо- 3-фенилпропил/амино/-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7 -карбоновой кислоты
Перемешанную суспензию эфедриновой соли из части (a) (333,1 г, 0,822 ммоля) в этилацетате (5 мл) промывали три раза порциями по 5 мл 1N раствором соляной кислоты. Органические экстракты объединяли, промывали рассолом, сушили (сульфатом магния), фильтровали, концентрировали и сушили в вакууме в течение 30 минут. Полученное масло растворяли в метиленхлориде (2 мл) и перемешивали в атмосфере азота при 0oC. К этому раствору добавляли раствор метилового эфира /4S-/4α, 7α, 10aβ//- октагидро-4-амино-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7- карбоновой кислоты (200,0 мг, 0,774 ммоля, полученный как описано в примере 3 (c)) в метиленхлориде (6 мл), затем триэтил (0,113 мл, 0,813 ммоля) и, наконец, бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфат (360,0 мг, 0,813 ммоля). Реакционную смесь перемешивали при 0oC и позволяли медленно нагреваться до комнатной температуры. Через 19 часов реакционную смесь концентрировали в вакууме и остаток растворяли в этилацетате. Раствор один раз промывали 5% раствором бисульфата калия (20 мл), один раз насыщенным раствором бикарбоната натрия (20 мл) и один раз рассолом. Органический слой сушили (сульфатом магния), фильтровали и концентрировали до желтой пены. Очистка флеш-хроматографией (силикагель, 230 - 400 меш при давлении азота 0,7 - 1,4 кг/см2) с элюированием смесью 4:3 этилацетат/гексан давала 303 мг продукта в виде прозрачного масла.
c) /4S-/4α/R*/, 7α, 10aβ//- Октагидро-4//2-/меркаптометил/-1-оксо-3-фенилпропил/амино/-5-оксо- 7H-пиридо[2,1-b]/1,3/тиазепин-7-карбоновая кислота
Раствор продукта из части (b) (303,1 мг, 0,635 ммоля) в метаноле (6,5 мл, обескислороженный пробулькиванием азота) охлаждали до 0oC и обрабатывали 1N раствором гидроокиси натрия (6,5 мл, обескислороженным пробулькиванием азота). После перемешивания в течение 1 часа при 0oC с непрерывной продувкой азотом реакционную смесь нагревали до комнатной температуры. Через полных три часа реакционную смесь подкисляли до pH 1 5% раствором бисульфата калия и экстрагировали этилацетатом. Органические слои объединяли, промывали водой, рассолом, сушили /сульфатом натрия/, фильтровали и концентрировали в вакууме, получая 219 мг указанного в названии продукта в виде белого твердого вещества; т. пл. 200oC (разложение). TLC (6: 0,01: 3,99 этилацетат/уксусная кислота/гексан) Rf = 0,15.
HPL C: tR =26,3 мин. примеси при 27,0 мин.; YMC. S-3 ODS (C-18) 6,0 • 150 мм 0% до 100% B:A, 30 мин. линейный градиент и 10 мин. выдерживан. 1,5 мл/мин; A = 90% вода:метанол + 0,2% фосфорная кислота, B = 90% метанол:вода + 0,2% фосфорная кислота; 220 нм.
Данные анализа для C20H26O4N2S2 • 0,11 C4H8O2 • 0,07 CH2Cl2:
Вычислено, %: С 56,22; Н 6,21; N 6,39; S 14,63
Найдено, %: С 56,46; Н 6,28; N 6,31; S 14,59.
Пример 13
/4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-4-метил-1-оксопентил/амино/-5- оксо-7H-пиридо[2,1-b]/1,3/-тиазепин-7-карбоновая кислота
a) /R/-2-Бром-4-метилпентановая кислота
Бромид калия (9,5 г, 80 ммоля) добавляли к перемешиваемому раствору D-лейцина (3,0 г, 23 ммоля) в 2,5 N серной кислоте (47 мл) при комнатной температуре. Реакционную смесь охлаждали до -10oC и твердый нитрит натрия (2,4 г, 34 ммоля) добавляли порциями, поддерживая температуру между -10o и -5oC. После окончания добавления, реакционную смесь перемешивали в течение 1 часа и затем нагревали до комнатной температуры и перемешивали еще один час. Затем реакционную смесь дважды экстрагировали эфиром, эфирные экстракты промывали один раз водой, сушили сульфатом магния, фильтровали и выпаривали, получая 2,7 г неочищенного продукта указанного в названии.
b) Дициклогексиламиновая соль /S/-2-/ацетилтио/-4-/метилпентановой кислоты
К перемешиваемому шламу тиоацетата калия (1,7 г, 15,0 ммоля) в 50 мл сухого ацетонитрила при комнатной температуре в атмосфере аргона добавляли раствор продукта из части (a) (2,6 г, 13 ммоля) в 17 мл ацетонитрила. Реакционную смесь перемешивали в течение 4 часов. Полученный шлам отфильтровывали и выпаривали. Остаток повторно растворяли в этиловом эфире, промывали один раз 5% раствором бисульфата калия и один раз рассолом, сушили (сульфатом магния) и выпаривали. Остаток растворяли в эфире (64 мл) и обрабатывали дициклогексиламином (2,7 мл, 14 ммоля). Немедленно начиналось осаждение белого твердого вещества из раствора. Раствор фильтровали и собирали белое твердое вещество, получая 2,0 г указанного в названии продукта, т.пл. 153 - 158oC; [α]D= -54,5o (c = 0,61, хлороформ).
c) Метиловый эфир /4S-/4α/R*/, 7α, 10aβ//- октагидро-//2-ацетилтио/-4-метил-1-оксопентил/амино/-5-оксо-7H- пиридо[2,1-b]/1,3/тиазепин-7-карбоновой кислоты
Перемешанную суспензию продукта из части (b) (403,3 мг, 1,09 ммоля) в этилацетате (5 мл) промывали три раза порциями по 5 мл 5% раствором бисульфата калия. Органические экстракты объединяли, промывали рассолом, сушили (сульфатом натрия), фильтровали, концентрировали и сушили в вакууме в течение 30 минут. Полученное масло (179,4 мг, 0,943 ммоля) растворяли в метиленхлориде (2 мл) и перемешивали в атмосфере азота при 0oC. К этому раствору добавляли раствор метилового эфира /4S-/4α, 7α, 10aβ//-/ октагидро-4-амино-5-оксо-7H- пиридо[2,1-b]/1,3/тиазепин-7-карбоновой кислоты (232,0 мг, 0,898 ммоля, полученного, как описано в примере (c)) в метиленхлориде (6 мл), затем триэтиламин (0,131 мл, 0,943 ммоля) и, наконец, бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфат (417,1 мг 0,943 ммоля). Реакционную смесь перемешивали при 0oC в течение 1 часа и 3,5 часа при комнатной температуре. Через полных 4,5 часа реакционную смесь концентрировали в вакууме и остаток растворяли в этилацетате. Раствор промывали один раз 5% раствором бисульфата калия (20 мл), один раз насыщенным раствором бикарбоната натрия (20 мл) и один раз рассолом. Органический слой сушили (сульфатом магния), фильтровали и концентрировали до желтой пены. Очистка флеш-хроматографией (силикагель 230 - 400 меш, при 0,7 - 1,4 кг/см2 давлении азота) с элюированием 2: 3 смесью этилацетат: гексан давала 209,4 мг указанного в названии продукта в виде прозрачного масла.
d) /4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-4-метил-1-оксопентил/амино/-5- оксо-7H-пиридо[2,1-b]/1,3/тиазепин-7-карбоновая кислота
Раствор продукта из части (c) (209,4 мг, 0,486 ммоля) в метаноле (5 мл, обескислороженный пробулькиванием азота) охлаждали до 0oC и обрабатывали 1N раствором гидроокиси натрия (5 мл, обескислороженным пробулькиванием азота).
После перемешивания в течение 1 часа при 0oC с непрерывной продувкой азота, реакционную смесь нагревали до комнатной температуры. Через полных 2,5 часа, реакционную смесь подкисляли до pH 1 5% раствором бисульфата калия и экстрагировали этилацетатом. Органические слои объединяли, промывали водой, рассолом, сушили (сульфатом натрия), фильтровали и концентрировали в вакууме. Очистка флеш-хроматографией (силикагель, 230 - 400 меш, при давлении азота 0,7 - 1,4 кг/см2) и элюирование с пропусканием азота смесью 6:0,01: 3,99 этилацетат/уксусной кислоты/гексан давали 142,0 мг указанного в названии продукта в виде белого твердого вещества. TLC (6:0,1:3,9 этилацетат/уксусная кислота/гексан) Rf=0,20 [α]D= (c=0,43, хлороформ).
HPLC: tR= 26,7 мин. ; YMC S-3ODS (С-18) 6,0•150 мм; 0% до 100% B:A, 30 мин. линейный градиент и 10 мин. выдержив. 1,5 мл/мин, A=90% вода:метанол+0,2% фосфорной кислоты, B=90% метанол:вода+0,2% фосфорной кислоты; 220 нм.
Данные анализа для C16H26O4N2S2 • 0,19 C4H8O2 • 0,18 C7H16 • 0,9 H2O: Вычислено, %: С 50,87; Н 7,63; N 6,58; S 15,07
Найдено, %: С 50,57; Н 7,20; N 6,83; S 14,75.
Пример 14
/4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксобутил/амино/-5-оксопирроло[2,1-b]/ 1,3/тиазепин-7-карбоновой кислоты
a) /R/-2-Бромбутановая кислота
Бромид калия (7,85 г, 65,94 ммоля) добавляли к раствору /R/-2-аминобутановой кислоты (2,0 г, 19,40 ммоля) в 2,5 N серной кислоте (25 мл) и охлаждали до 0oC. Нитрит натрия (2,06 г, 29,87 ммоля) медленно добавляли в несколько порций. Температуру сохраняли по крайней мере ниже 2oC в течение этого процесса добавления. Реакционную смесь перемешивали при 0oC в течение 1 часа и при комнатной температуре в течение 16 часов, затем экстрагировали 3 порциями по 50 мл этилацетата. Объединенные этилацетатные слои промывали 2 порциями по 50 мл воды и одной порцией 50 мл рассола, сушили (сульфатом магния) и концентрировали в вакууме, получая 2,86 г указанного в названии продукта в виде неочищенного масла.
b) Дициклогексиламиновая соль /S/-2-/ацетилтио/бутановой кислоты
К шламу 2,14 г (18,77 ммоля) тиоацетата калия в 15 мл ацетонитрила, перемешанному при комнатной температуре, добавляли по каплям в течение 15 минут раствор /R/-2-бромбутановой кислоты (2,83 г, 17,07 ммоля) в 15 мл ацетонитрила. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, затем фильтровали. Фильтрат концентрировали в вакууме и полученное масло растворяли в 30 мл этилового эфира. Эфирный слой промывали 2 порциями по 20 мл 5% раствора бисульфата калия, 2 порциями по 20 мл воды, 2 порциями по 20 мл рассола, сушили (сульфатом магния) и фильтровали. К фильтрату добавляли 3,4 мл (17,07 ммоля) дициклогексиламина. После перемешивания при комнатной температуре в течение 2 часов шлам фильтровали, получая 1,24 г дициклогексиламиновой соли продукта. Вторую порцию дициклогексиламиновой соли продукта 724 мг получали из фильтрата.
c) Метиловый эфир /4S-/4α/R*/, 7α, 9aβ//-октагидро-4-//2-/ацетилтио/-1-оксобутил/амино/-5-оксопирроло[2,1-b] 1,3/тиазепин-7-карбоновой кислоты
Дициклогексиламиновую соль продукта из части (b) (227 мг, 0,66 ммоля) растворяли в 15 мл этилацетата и промывали три раза порциями по 10 мл бисульфатом калия, одной порцией 10 мл рассола, сушили (сульфатом магния) и концентрировали в вакууме, получая 108 мг /S/-2-/ацетилтио/бутановой кислоты в виде прозрачного масла.
К раствору этой свободной кислоты (108 мг, 0,66 ммоля) в 5 мл сухого метиленхлорида, охлажденного до 0oC, добавляли триэтиламин (90 мкл, 0,66 ммоля) с последующим добавлением соли метилового эфира /4S-/4α, 7α, 9aβ//- 4-амино-октагидро-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты и п-толуолсульфоновой кислоты (250 мг, 0,66 ммоля, полученной из вещества описанного в примере 5 (d)) и второй порции триэтиламина (90 мкл, 0,66 ммоля). Реакционную смесь перемешивали при 0oC в течение 20 минут, затем в одну порцию добавляли бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфат (292 мг, 0,66 ммоля). Реакционную смесь перемешивали при 0oC в течение 1 часа, оставляли в холодильнике на 56 часов, затем перемешивали при комнатной температуре в течение 3 часов. Затем реакционную смесь концентрировали в вакууме, вновь растворяли в 30 мл этилацетата и промывали 20 мл 5% раствора бисульфата калия, 20 мл насыщенного раствора бикарбоната натрия, 20 мл рассола, сушили (сульфатом магния) и концентрировали в вакууме, получая неочищенное масло. Неочищенное масло флеш-хроматографировали (Мерк силикагель, 25•100 мм, 2:3 этилацетат/гексан, получая 208 мг указанного в названии продукта в виде белой пены.
d) /4S-/4α/R*/, 7α, 9aβ//- октагидро-4-//2-меркапто-1-оксобутил/амино/-5-оксопирроло[2,1-b]/1,3 тиазепин-7-карбоновая кислота
Раствор продукта из части (c) (225 мг, 0,559 ммоля) в метаноле (5 мл) продували аргоном в течение 30 минут и охлаждали до 0oC. К этому раствору по каплям добавляли 5 мл 1 M раствора гидроокиси натрия, также продували аргоном в течение 30 минут и охлаждали до 0oC. Реакционную смесь перемешивали при 0oC в течение 3 часов с непрерывной продувкой аргоном, затем подкисляли до pH 2 5% раствором бисульфата калия. Смесь экстрагировали 3 порциями по 40 мл этилацетата и объединенные этилацетатные слои сушили (сульфатом магния) и концентрировали в вакууме, получая неочищенную пену. Неочищенный продукт флеш-хроматографировали (Мерк силикагель, 25•180 мм, 3% уксусная кислота/этилацетат), получая белую пену, которую растворяли в метиленхлориде и растирали с гексаном, получая 176 мг указанного в названии продукта в виде компактной белой пены, [α]D= -125,4o (c=1,0, хлороформ). TLC (метанол/метиленхлорид 1:9) Rf=0,16.
HPLC:tR=15,5 мин. (97% полная площадь, УФ 220 нм); линейный 25 мин. градиент (A= 90% вода/метанол+0,2% фосфорной кислоты) B=90% метанол/вода+0,2% фосфорной кислоты; скорость потока 1,5 мл/мин.
Данные анализа для C13H20N2S2O4 • 0,8 H2O • 0,2 C6H14 0,1 • CH2Cl2: Вычислено, %: С 46,10; Н 6,66; N 7,52; S 17,21
Найдено, %: С 46,00; Н 6,17; N 7,52; S 16,82.
Пример 15
/4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксопентил/амино-5-оксопирроло [2,1-b]/1,3/тиазепин-7-карбоновая кислота
a) /R/-2-Бромпентановая кислота
Следуя процедуре примера 14 (a), но используя D-норвалин вместо /R/-2-аминобутановой кислоты, получали /R/-2-бромпентановую кислоту в виде прозрачной жидкости.
b) Дициклогексиламиновая соль /S/-2-ацетилтио/пентановой кислоты
Взаимодействием /R/-2-бромпентановой кислоты с тиоацетатом калия в ацетонитриле с последующей обработкой продукта дициклогексиламином согласно процедуре примера 14 (b) получали дициклогексиламиновую соль /S/-2-/ацетилтио/пентановой кислоты в виде твердого вещества.
c) Метиловый эфир /4S-/4α/R*/, 7α, 9aβ//- октагидро-4-//2-ацетилтио/-1-оксопентил/амино/-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Взаимодействием свободной кислоты дициклогексиламиновой соли из части (b) с метиловым эфиром /4S-/4α, 7α, 9aβ//- -4-амино-октагидро-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты, согласно процедуре примера 14 (c), получали указанный в названии продукт в виде белой пены.
d) /4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксопентил/амино-5- оксопирроло[2,1-b]/1,3/тиазепин-7-карбоновой кислоты
Раствор продукта из части (c) в метаноле обрабатывали 1 M раствором гидроокиси натрия, согласно процедуре примера 14 (d), получая указанный в названии продукт в виде компактной белой пены; [α]D= -122,8o (c=1,0, CDCl3). TLC (метанол/метиленхлорид, 1:9), Rf=0,15.
HPLC: tr=20,5 мин; (97% общая площадь, УФ 220 нм), YMC S-3ODS (C-18, 120 A) 6•150 мм; 0% B:A = 100% B:A, линейный 25 мин. градиент (A=90% вода:метанол+0,2% фосфорной кислоты); B=90% метанол:вода+0,2% фосфорной кислоты; скорость потока=1,5 мл/мин.
Данные анализа для C14H22N2S2O4 • 0,65 H2O • 0,20 C6H14:
Вычислено, %: С 48,63; Н 7,01; N 7,46; S 17,08
Найдено, %: С 48,86; Н 6,70; N 7,24; S 16,74.
Пример 16
/4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто- 4,4-диметил-1-оксопентил// амино/-5- оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновая кислота
a//R-2-Бром-4,4-диметилпентановая кислота
Раствор /R/-2-амино-4,4-диметилпентановой кислоты (950 мг, 6,55 ммоля) в 2,5 N растворе серной кислоты (13 мл, 33 ммоля) охлаждали до -5oC и обрабатывали бромидом калия (2,72 г, 22,9 ммоля) одной порцией. Бесцветный раствор обрабатывали нитритом натрия (680 мг, 9,86 ммоля) порциями, сохраняя температуру между 0 и 3oC в течение 25 минут. Реакционную смесь перемешивали при 0oC в течение одного часа и при комнатной температуре в течение 1,5 часа. Реакционную смесь выливали в воду (10 мл). Продукт экстрагировали эфиром (60 мл), промывали водой (20 мл) и рассолом (20 мл), сушили (сульфатом магния) и концентрировали в вакууме, получая указанный в названии продукт в виде бесцветной жидкости.
b) /S/-2-Ацетилтио/-4,4-диметилпентановая кислота
Шлам тиоацетата калия (750 мг, 6,58 ммоля) и ацетонитрила (10 мл, высушенный над молекулярными ситами) охлаждали до 0oC и обрабатывали раствором /R/-2-бром-4,4-диметилпентановой кислоты из части (a) в ацетонитриде (2 мл) в течение 5 минут. Реакционной смеси позволяли нагреваться до комнатной температуры и перемешивали в течение 2,5 часов. Шлам отфильтровывали. Фильтрат концентрировали в вакууме. Остаток разбавляли эфиром (50 мл), промывали двумя порциями 5% водного раствора тиолсульфата натрия (50 мл) и рассолом (25 мл), сушили (сульфатом магния) и концентрировали в вакууме. Бледно-желтое масло чистили флеш-хроматографией (Мерк силикагель, 12•3 см, 10% метанол/метиленхлорид), получая 575 мг указанного в названии продукта в виде бледно-желтого масла.
с) Метиловый эфир /4S-/4α/R*/, 7α, 9aβ//- октагидро-4-//2-ацетилтио/-4,4-диметил-1- оксопентил/амино/-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Прозрачный раствор продукта из части (b) (148 мг, 0,72 ммоля) в метиленхлориде (5 мл, разогнанный над гидридом кальция) охлаждали 0oC и обрабатывали раствором соли метилового эфира /4S-/4α, 7α, 9aβ//- -4-амино-октагидро -5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты и п-толуолсульфоновой кислоты (250,0 мг, 0,60 ммоля, полученной из вещества описанного в примере 5 (d)) в метиленхлориде (3 мл, разогнанным над гидридом кальция), триэтиламином (122 мг, 1,2 ммоля), с последующей обработкой бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфатом (319 мл, 0,72 ммоля). Реакционную смесь перемешивали при 0oC в течение 24 часов и при комнатной температуре в течение 5 часов. Неочищенную реакционную смесь концентрировали в вакууме. Остаток разбавляли этилацетатом (50 мл), промывали 5% водным раствором бисульфатом калия (50 мл), 50% насыщенным водным раствором бикарбоната натрия (50 мл) и рассолом (50 мл), сушили (сульфатом натрия) и концентрировали в вакууме. Неочищенный продукт флеш-хроматографировали (Мерк, силикагель 20 г, этилацетат), получая 244 мг указанного в названии продукта в виде белой пены.
d) /4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-4,4-диметил-1-оксопентил/амино/-5- оксопирроло[2,1-b]/1,3/тиазепин-7-карбоновая кислота
Прозрачный раствор продукта из части (c) (240 мг, 0,56 ммоля) в метаноле (2 мл, продутый азотом) охлаждали до 0oC и обрабатывали по каплям 1 N раствором гидроокиси натрия (2,27 мл, 2,24 ммоля, продутым аргоном) с непрерывным барботированием при 0oC. Смеси позволяли перемешиваться в течение 3 часов при 0oC и при комнатной температуре в течение 3 часов. Смесь подкисляли до pH 15% водным раствором бисульфата калия (барботированным аргоном). Продукт экстрагировали метиленхлоридом (50 мл, барбортировали азотом), промывали рассолом, сушили (сульфатом натрия) и концентрировали в вакууме. Неочищенный продукт перекристаллизовывали из смеси метиленхлорид/гексан, получая 75 мг указанного в названии продукта в виде белого твердого вещества; т. пл. 124 - 126oC; [α]D= -175oC (c=0,25, метанол). TLC (уксусная кислота/метанол/метиленхлорид 1:5:94) Rf = 0,65.
HPLC: tR (YMC, S-3 ODS (C-18) 6,0•150 мм; 1,5 мл/мин: линейный градиент 0 - 100% B в течение 30 минут, Буфер A = метанол/вода/фосфорная кислота (10: 90: 02), Буфер B = метанол/вода/фосфорная кислота (90:10:0,2) = 2,4 мин, 95% общей площади пика при 254 нм.
Данные анализа для C16H26N2O4S2 • 0,13 C6H14:
Вычислено, %: С 52,24; Н 7,28; N 7,26; S 16,62
Найдено, %: С 51,97; Н 7,26; N 7,09; S 16,23.
Пример 17.
/4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксопропил/амино/-5-оксопирроло[2,1-b]/1,3/ тиазепин-7-карбоновая кислота
a) /R-2-Бромпропановая кислота
Следуя процедуре примера 14 (a), но применяя D-аланин вместо /R/-аминобутановой кислоты, получали /R/-бромпропановую кислоту в виде светло-желтого масла.
b) /S/-2-/Ацетилтио/пропановая кислота
К светло-зеленому раствору тиоацетата калия (3,94 г, 34,5 ммоля) в ацетонитриле (150 мл) добавляли раствор /R/-2-бромпропановой кислоты (4,8 г, 31 ммоля) в ацетонитриле (12 мл) при комнатной температуре в атмосфере аргона. Полученный белый шлам перемешивали при комнатной температуре в течение 2 часов, затем фильтровали. Фильтрат концентрировали в вакууме. Остаток разбавляли этилацетатом (100 мл), промывали 10% водным раствором бисульфата калия (50 мл) и рассолом, сушили (сульфатом натрия) и концентрировали в вакууме. Неочищенный продукт (4,61 г), чистили флеш-хроматографией (60 г, Мерк силикагель, 1: 45:54 уксусная кислота/ этилацетат/гексан/, получая 3,7 г указанного в названии продукта в виде светло-желтого масла: [α]D= -114o (c= 0,50, метанол).
c) Метиловый эфир /4S-/4α/R*/, 7α, 9aβ//- октагидро-4-//2-ацетилтио/-1-оксопропил/амино/-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Прозрачный раствор /S/-2-/ацетилтио/пропановой кислоты (86 мг, 0,58 ммоля) в метиленхлориде (5 мл, разогнанным над гидридом кальция) охлаждали до 0oC и обрабатывали раствором соли метилового эфира /4S-/4α, 7α, 9aβ//- 4-аминооктагидро-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты и п-толуолсульфоновой кислоты (200,0 мг, 0,48 ммоля, полученной из вещества, описанного в примере 5 (d)) в метиленхлориде (5 мл, разогнанном над гидридом кальция), триэтиламином (98 мг, 0,97 ммоля), с последующей обработкой бензотриазол-1-илокситрис/диметиламино/фосфорнийгексафторфосфатом (255 мг, 0,58 ммоля). Реакционную смесь перемешивали при 0oC в течение 22 часов и при комнатной температуре в течение 2 часов. Неочищенную реакционную смесь концентрировали в вакууме. Остаток разбавляли этилацетатом (100 мл), промывали 5% водным раствором бисульфата калия (30 мл), 50% насыщенным водным раствором бикарбоната натрия и рассолом, сушили (сульфатом натрия) и концентрировали в вакууме. Неочищенный продукт очищали флеш-хроматографированием (40 г, Мерк силикагель, этилацетат), получая 180 мг указанного в названии продукта в виде белого твердого вещества; т. пл. 143 - 145oC.
d) /4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксопропил/амино/-5- оксопирроло [2,1-b]/1,3/тиазепин-7-карбоновая кислота
Прозрачный раствор продукта из части (c) (180 мг, 0,48 ммоля) в метаноле (2 мл) в атмосфере аргона охлаждали до -10oC и по каплям обрабатывали с барботированием аргоном 1 N раствором гидроокиси натрия (1,95 мл, 1,95 ммоля), сохраняя температуру ниже 0oC. Смеси позволяли перемешиваться с барботированием аргоном при 0oC в течение 3 часов. Смесь подкисляли до pH 1 5% водным раствором бисульфата калия в атмосфере аргона. Продукт экстрагировали этилацетатом с барботированием азота (100 мл), промывали рассолом, сушили (сульфатом натрия) и концентрировали в вакууме. Неочищенный продукт чистили флеш-хроматографированием (40 г, Мерк силикагель, 1:5:94 уксусная кислота/метанол/метиленхлорид), получая 154 мг указанного в названии продукта в виде белого твердого вещества, т. пл. 150 - 152oC, [α]D= 156o (c = 0,50, метанол). TLC (1:5;94 уксусная кислота/метанол/метиленхлорид) Rf = 0,28.
HPL C: tR (YMC, S - 3 ODS (C-18) 6,0•150 мм; 1,5 мл/мин, линейный градиент 0 - 100 % B в течение 30 минут. Буфер A = метанол/вода/фосфорная кислота (10: 90: 0,2), Буфер B = метанол/вода/фосфорная кислота (90:10:0,2) = 14,69 мин, более 95% общей площади пика при 254 нм.
Данные анализа для C12H18N2O4S2 • 0,75 CH3CO2H:
Вычислено, %: С 44,61; Н 5,82; N 7,71; S 17,64
Найдено, %: С 44,76; Н 5,71; N 7,81; S 17,76.
Пример 18
/4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-/3-циклопропил-2-меркапто-1-оксопропил/амино/- 5-оксопирроло[2,1-b]/1,3/-тиазепин-7-карбоновая кислота
a) /R/-2-///Фенилметокси/карбонил/амино/-4-пентановая кислота
Смесь D-аллилглицина (2,8 г, 24,3 ммоля), 1 M водного раствора гидроокиси натрия (25 мл) и тетргидрофурана (10 мл, отогнанный от кетила) перемешивали при комнатной температуре до получения гомогенного раствора, затем охлаждали в бане со льдом. К полученному быстроперемешиваемому раствору добавляли около 5 мл 1,0 M водного раствора гидроокиси натрия, затем по каплям около 1 г бензилхлорформиата. Эту операцию повторяли дополнительно 4 раза до тех пор, пока было добавлено общее количество 28 мл 1,0 M водного раствора гидроокиси натрия и 4,80 г (95%, 27 ммоля) бензилхлорформиата. Реакционную смесь перемешивали в течение 15 минут при 0oC, затем 30 минут при комнатной температуре и затем экстрагировали 50 мл эфира. Водный слой подкисляли (pH - 1,5) добавлением 6 N соляной кислоты (около 10 мл), затем экстрагировали 3-я порциями по 50 мл эфира. Три эфирных экстракта объединяли, сушили (сульфатом магния) и концентрировали в вакууме, получая 6,01 г указанного в названии продукта в виде бесцветного масла.
b) Фенилметиловый эфир /R/-2-///фенилметокси/карбонил/амино/-4- пентановой кислоты
Карбонат цезия (4,28 г, 13,1 ммоля) добавляли к раствору продукта из части (a) (5,96 г, 23,9 ммоля) в безводном диметилформамиде (25 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 20 минут, затем быстро добавляли бензилбромид (4,5 г, 26,3 ммоля, слабо экзотермическая). Смесь перемешивали в течение 30 минут, затем разделяли между 100 мл воды и 100 мл эфира. Органический слой отделяли, промывали тремя порциями по 100 мл воды, 50 мл рассола, сушили (сульфатом магния) и концентрировали в вакууме с получением масла. Неочищенное вещество чистили флеш-хроматографированием (Мерк силикагель, 24•5,0 см, 1:10 этилацетат: гексан, затем 1:4 этилацетат/гексан), получая 6,13 г указанного в названии продукта в виде бесцветного масла.
c) Фенилметиловый эфир /R/ -α- ///фенилметокси/карбонил/амино/ циклопропанпропановой кислоты
Палладий ацетат /II/ (65 мг, 0,29 ммоля) добавляли к раствору продукта из части (b) (5,78 г, 17,1 ммоля) в безводном эфире (60 мл) и перемешивали в течение 10 минут. Полученную смесь охлаждали до 0oC, добавляли избыток эфирного диазометана (полученного из 12 г N-метил-N'-нитро-II-нитрозогуанидина (120 мл эфира) порциями в течение 15 минут. Реакционную смесь перемешивали в течение 15 минут, затем гасили добавлением 1 мл ледяной уксусной кислоты. Раствор переносили в делительную воронку, промывали в 100 мл насыщенного водного раствора бикарбоната натрия, 50 мл рассола, сушили (сульфатом магния) и концентрировали в вакууме, получая желтое масло. Неочищенное вещество чистили флеш-хроматографированием (Мерк силикагель, 20•5,0 см, 1:4 этилацетат/гексан), получая 5,74 г указанного в названии продукта в виде бесцветного масла.
d) /R/ -α- Амино/циклопропанпропановая кислота
Катализатор - палладий на угле (10%, 1,14 г) добавляли к раствору продукта из части (c) (5,71 г, 16,2 ммоля) в метаноле (75 мл) и перемешивали в атмосфере водорода (баллон) при комнатной температуре в течение 48 часов. Реакционную смесь фильтровали для удаления катализатора и катализатор промывали теплой водой. Затем фильтрат пропускали через поликарбонатный мембранный фильтр 0,5 мкМ. Фильтрат концентрировали в вакууме, получая 2,03 г указанного в названии продукта в виде белого твердого вещества.
e) /S/ -α- /Ацетилтио/циклопропанпропановая кислота
Смесь продукта из части (d) (2,00 г, 15,5 ммоля) в 2,5 N водном растворе серной кислоты (30 мл) перемешивали при комнатной температуре до получения гомогенного раствора и затем охлаждали до -5oC. К этому раствору порциями добавляли бромид калия (6,50 г, 54,6 ммоля). Смесь перемешивали до получения гомогенного раствора. Затем небольшими порциями добавляли нитрит натрия (1,60 г, 23,2 ммоля) в течение 25 минут, поддерживая температуру реакции ниже 0oC. Реакционную смесь перемешивали дополнительно в течение 1 часа при 0oC, затем при комнатной температуре в течение 1,5 часов. Полученную смесь разбавляли 30 мл воды и экстрагировали тремя порциями по 30 мл эфира. Объединенные эфирные экстракты промывали 25 мл рассола, сушили (сульфатом магния) и концентрировали в вакууме, получая 2,82 г неочищенной /R/-α-бром/циклопропанпропановой кислоты в виде бледно-желтого масла.
Раствор этой неочищенной /R/-α- бром/циклопропанпропановой кислоты в ацетонитриле (10 мл) добавляли в течение 5 минут к перемешиваемому шламу тиоацетата калия (1,83 г, 16,1 ммоля) в ацетонитриле (20 мл) при охлаждении в бане со льдом. Реакционную смесь перемешивали при 0oC в течение 1 часа, затем при комнатной температуре в течение 16 часов. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме, получая масло. Масло разделяли между 50 мл эфира и 50 мл 5% водного раствора тиосульфата натрия. Органический слой отделяли, промывали 25 мл рассола, сушили (сульфатом магния) и концентрировали в вакууме с получением желтого масла. Неочищенное вещество чистили флеш-хроматографированием (Мерк силикагель, 12•5,0 см, 1:19 метанол/метиленхлорид), получая 1,52 г указанного в названии продукта в виде желтого масла.
f) Метиловый эфир /4S-/4α/R*/, 7α, 9aβ//- октагидро-4-//2-/ацетилтио/-3-циклопропил- 1-оксопропил/амино/-5-оксопирроло [2,1 - b] /1,3/тиазепин-7-карбоновой кислоты
Бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфат (292 мг, 0,66 ммоля) добавляли одной порцией к смеси соли метилового эфира /4S-/4α, 7α, 9aβ//- октагидро-4-амино-5-оксопирроло [2,1 - b] /1,3/тиазепин-7-карбоновой кислоты и n-толуолсульфоновой кислоты (250 мг, 0,60 ммоля, полученной из вещества, описанного в примере 5 (d)) в метиленхлориде (3 мл, разогнанном над гидридом кальция), триэтиламина (121 мг, 1,20 ммоля) и продукта из части (e) (122 мг, 0,65 ммоля) в метиленхлориде (3 мл, разогнанном над гидридом кальция). Реакционную смесь перемешивали при 0oC в течение 1 часа, затем при комнатной температуре в течение 1,5 часов. Полученную смесь разделяли между 20 мл этилацетата и 20 мл 1 M водного раствора бисульфата калия. Органический слой отделяли, промывали 20 мл 5% водного раствора бикарбоната натрия, 20 мл рассола, сушили (сульфатом магния) и концентрировали в вакууме, получая масло. Неочищенное вещество чистили флеш-хроматографированием (Мерк силикагель, 12•3,0 см, 1:1 этилацетат/гексан), получая 191 мг указанного в названии продукта в виде твердой белой пены.
g) /4S-/4α/R*/, 7α, 9aβ//- Октагидро-4-//3-цикло-пропил-2-меркапто-1-оксопропил/ амино/-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновая кислота
Раствор продукта из части (f) (185 мг, 0,45 ммоля) в метаноле (3 мл) барботировали аргоном в течение 10 минут при 0oC и затем добавляли 1 М водный раствор гидроокиси натрия (3 мл, свежебарботированный аргоном в течение 10 минут). Реакционную смесь перемешивали в течение 2,5 часов при комнатной температуре с непрерывным барботированием аргоном, затем подкисляли добавлением 20 мл 1 М раствора бисульфата калия и экстрагировали 20 мл этилацетата. Органический экстракт промывали 20 мл рассола, сушили (сульфатом натрия) и концентрировали в вакууме, получая смолу. Смолу промывали безводным эфиром, затем концентрировали в вакууме, получая 121 мг указанного в названии продукта в виде белой пены; [α]D= -103o (с=0,23, метанол). TLC (1:10:90, уксусная кислота/метанол/метиленхлорид) Rf = 0,46.
HPLC: tR (YMC S-3 ODS 6,0 •150 мм, 1,5 мл/мин линейный градиент 0-100% B в течение 30 минут, Буфер A = метанол/вода/фосфорная кислота (10:90:0,2), Буфер B = метанол/вода/фосфорная кислота (90:10:0,2) = 20,7 минут больше 97% общей площади пика при 254 нм).
Данные анализа для C15H22N2O4S2 • 0,20 H2O:
Вычислено, %: С 49,77; Н 6,23; N 7,74; S 17,71
Найдено, %: С 50,01; Н 6,27; N 7,50; S 17,40.
Пример 19
/4S-/4α, 7α, 9aβ//- Октагидро-4-///1-мерокаптоциклопентил/карбонил/амино/-5- оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновая кислота
a) 1-Меркаптоциклопентанкарбоновая кислота
Раствор литийциизопропиламида получали в атмосфере азота из циизопропиламина (5,4 мл, 38,5 ммоля) и н-бутиллития (2,5 М в гексане, 15,4 мл, 38,5 ммоля) в тетрагидрофуране (17,6 мл), поддерживая температуру между -3oC и 0oC. После перемешивания в течение 15 минут добавляли циклопентанкарбоновую кислоту (2,0 г, 17,5 ммоля) в тетрагидрофуране (2 мл) при 0 - 3oC в течение 25 минут. После проведения реакции в течение 15 минут при 0oC баню удаляли и реакцию перемешивали более 15 минут, вызывая подъем температуры до 15oC. Молочно-белый раствор охлаждали до -78oC и добавляли серу в виде твердого вещества (S8, 618,0 мг, 19,3 ммоля), поддерживая температуру при -78oC. Реакционной смеси позволяли нагреться до комнатной температуры in situ. После 70 часов реакционную смесь охлаждали до 0oC, гасили водой (pH 8-9) и быстро подкисляли до pH 1 6 N соляной кислотой. Водный раствор экстрагировали этилацетатом (3 • 30 мл), промывали рассолом, сушили (сульфатом магния), фильтровали и концентрировали, получая 2,62 г указанного в названии продукта в виде желтого масла.
b) 1-/Ацетилтио/циклопентанкарбоновая кислота
К раствору продукта из части (a) (1,44 г, 9,89 ммоля) в барботируемом азотом растворе 1N гидроокиси натрия (20 мл, 19,7 ммоля) при 0oC добавляли уксусный ангидрид (0,93 мл, 9,89 ммоля). Добавляли тетрагидрофуран (13 мл) для того, чтобы солюбилизировать масло, которое образовалось после перемешивания в течение 1 часа при 0oC (pH 7), реакционную смесь нагревали до комнатной температуры и добавляли дополнительно уксусный ангидрид (0,47 мл, 4,9 ммоля), а также карбонат калия (2,04 г, 14,8 ммоля) до pH 10 и тетрагидрофуран (4 мл). После перемешивания в течение ночи при комнатной температуре реакционную смесь подкисляли до pH 1 1 N соляной кислотой и экстрагировали этилацетатом. Этилацетатные экстракты объединяли, промывали рассолом, сушили /сульфатом магния/, фильтровали и концентрировали в вакууме, получая желтое твердое вещество (1,61 г). Твердое вещество перекристаллизовывали дважды из смеси этилацетат/гексан, получая 614 мг указанного в названии продукта в виде светло-коричневого вещества, т.пл. 119 - 121, 5oC.
c) Метиловый эфир /4S-/4α, 7α, 9aβ//- октагидро-4-//ацетилтио/циклопентил/карбонил/амино-5-/оксопирроло [2,1-b] /1,3/тиазепин-7-карбоновой кислоты
К раствору продукта из части (b) (94,5, 0,502 ммоля) в метиленхлориде (3,6 мл) при 0oC в атмосфере азота добавляли триэтиламин (70 мкл, 0,502 ммоля) с последующим добавлением соли метилового эфира /4S-/4α, 7α, 9aβ//- 4-амино-октагидро-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты и п-толуолсульфоновой кислоты /полученной из вещества, описанного в примере 5 (d), 198 мг, 0,478 ммоля/ одной порцией, с последующим добавлением триэтиламина (66,6 мкл, 0,478 ммоля). Реакцию перемешивали в течение 5 минут при 0oC. Затем добавляли бензотриазол-1-иолокситрис/диметиламино/фосфонийгексафторфосфат (222,0 мг, 0,502 ммоля) в виде твердого вещества. Реакцию перемешивали при 0oC в течение 1 часа и затем при комнатной температуре в течение 2,25 часа. Реакционную смесь концентрировали в вакууме и остаток разделяли между этилацетатом и 5% водным раствором бисульфата калия (20 мл). Органический слой промывали полунасыщенным раствором бикарбоната натрия и рассолом, сушили (сульфатом магния), фильтровали и концентрировали до прозрачного масла. Очистка флеш-хроматографированием, элюирование смесью 11:9 этилацетат:гексан давала 169,3 мг указанного в названии продукта в виде прозрачного масла.
d) /4S-/4α, 7α, 9aβ//- Октагидро-4-//1-меркаптоциклопентил/карбонил/амино/-5- оксопирроло[2,1-b]/1,3/тиазепин-7-карбоновая кислота.
Раствор продукта из части (c) (167,3 мг, 0,404 ммоля) в метаноле (4 мл, обескислороженный пробулькиванием азота) охлаждали до 0oC и обрабатывали 1 N раствором гидроокиси натрия (4 мл, обескислороженным пробулькиванием азота). После перемешивания в течение 1,5 часа при 0oC и непрерывном продувании азотом реакцию нагревали до комнатной температуры. Через три часа реакционную смесь подкисляли до pH 1 5% раствором бисульфата калия и экстрагировали этилацетататом. Органические слои объединяли, промывали водой (20 мл), рассолом, сушили (сульфатом натрия), фильтровали, концентрировали в вакууме и повторно выпаривали из гексана, получая белое твердое вещество. Соединение растворяли в диоксане (безводном) и лиофилизовали, получая 110 мг указанного в названии продукта в виде белого твердого вещества; [α]D= - 106,5o (с = 0,68, хлороформ). TLC (7:2,9:0,1, этилацетат/гексан/уксусная кислота) Rf = 0,12.
HPLC : tR = 21,5 мин.; YMC S - 3 ODS (C-18) 6,0 • 150 мм; O - 100% B:A, 30 минут линейный градиент и 10 минут выдержки, 1,5 мл/мин, A = 90% воды/10% метанола + 0,2% фосфорной кислоты, B = 90% метанола/10% воды + 0,2% фосфорной кислоты; 220 нм.
Данные анализа для C15H22N2O4S2 • 0,15 C4H8O2 • 0,7 H2O • 0,08 C6H14: Вычислено, %: С 49,37; Н 6,63; N 7,16; S 16,39
Найдено, %: С 49,03; Н 6,37; N 7,21; S 16,65.
Пример 20
/4S-/4α, 7α, 10aβ//- -4-//2-Карбоксил-1-оксо-3-фенилпропил/амино/октагидро-5- оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновая кислота.
a) Моноэтиловый эфир 3-/фенилметил/пропандионовой кислота
Диэтиловый эфир 3-/фенилметил/пропандиоловой кислоты (2,5 г, 10 ммоль) в 10 мл тетрагидрофурана перемешивали в течение ночи с 10 мл 1 N раствора гидроокиси лития. Реакционную смесь подкисляли 11 мл 1 N соляной кислоты и экстрагировали двумя порциями по 50 мл этилацетата. Этилацетатные экстракты промывали рассолом, сушили (сульфатом магния) и концентрировали в вакууме. Концентрат хроматографировали на колонке с силикагелем (80 г), используя систему 5% метанол:хлороформ. Соответствующие фракции объединяли, концентрировали, получая 1,23 г указанного в названии продукта.
b) Метиловый эфир /4S-/4α, 7α, 10aβ//- -4-//2-/этоксикарбонил/-1-оксо-3-фенилпропил/амино/октагидро- 5-оксо-7H-пиридо[2,1-b] /1,3/триазепин-7-карбоновой кислоты
Продукт из части (a) (0,222 г, 1 ммоль) и метиловый эфир /4S-/4α, 7α, 10aβ//- октагидро-4-амино-4-амино-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты (0,258 г, 1 моль, полученный как описано в примере 3 (c)) растворяли в метиленхлориде (5 мл) и охлаждали до 0oC. Добавляли триэтиламин (0,14 мл, 1 ммоль) и реакционную смесь перемешивали в течение 1 часа. Добавляли бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфат (0,442 г, 1 ммоль) и раствор перемешивали при 0oC в течение 1 часа при комнатной температуре в течение 2,5 часов. Реакционную смесь разбавляли 50 мл метиленхлорида и промывали водой, 10% раствором бисульфата натрия, насыщенным водным раствором бикарбоната натрия, сушили (сульфатом натрия), фильтровали и концентрировали в вакууме. Остаток хроматографировали на силикагеле, используя 30% этилацетата в гексане. Соответствующие фракции объединяли и концентрировали в вакууме, получая 0,22 г указанного в названии продукта.
c) /4S-/4α, 7α, 10aβ//- 4-//2-Карбокси-1-оксо-3-фенилпропил/амино/октагидро-5-/оксо-7H- пиридо[2,1-b] /1,3/тиазепин-7-карбоновая кислота
Продукт из части (b) (0,22 г, 0,476 ммоля) перемешивали с 1 N раствором гидроокиси лития (5 мл) в тетрагидрофуране (5 мл) при комнатной температуре в течение 3 часов. Реакционную смесь подкисляли до pH 2 1 N соляной кислотой и концентрировали в вакууме. Остаток растворяли в этилацетате и промывали водой, рассолом, сушили (сульфатом натрия) и концентрировали в вакууме до 3 мл, до состояния, при котором продукт кристаллизовался. После пребывания при 0oC в течение ночи твердое вещество отфильтровывали и сушили, получая 0,16 г указанного в названии продукта в виде твердого белого вещества, т.пл. 159 - 162oC;  - 84,92o (с = 0,7, метанол) TLC/хлороформ:метанол, 9:1/Rf = 0,23; 0,28.
- 84,92o (с = 0,7, метанол) TLC/хлороформ:метанол, 9:1/Rf = 0,23; 0,28.
HPLC : tR = 16,15, 16,35 мин.; (УФ-254 нм); YMC S-3 ODS (C-18) 6,0•150 мм, 3 мк колонка с заглушенным концом, линейный градиент 50 - 90% метанол, содержащий 0,2% фосфорной кислоты, 20 мин.; 1,5 мл/мин (44,9%, 55,1% изомерная смесь).
Данные анализа для C20H24N2SO6 • 0,1 H2O:
Вычислено, %: С 56,89; Н 5,78; N 6,66; S 7,59
Найдено, %: С 56,98; Н 5,68; N 6,58; S 7,15.
Пример 21.
/4S-/4α,R*7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-/1,4/оксазино [3,4-b]/1,3/-тиазепин-7-карбоновая кислота
a) Метиловый эфир O-/2,2-диметоксиэтил/-N-/N//фенилметокси/карбонил/-L-гомосерил/L-серина
Раствор метилового эфира N-/O-//1,1-диметилэтил/диметилсилил/-N-//фенилметокси/карбонил/-L-гомосерил/ -O-/2,2-диметоксиэтил/-L-серина /5,56 г, 10 ммоля, полученный, как описано в примере 10 (h)/ в метаноле (65 мл) охлаждали до 0oC (баня лед с солью), обрабатывали моногидратом п-толуол-сульфоновой кислоты (386 мг, 2,0 ммоля) и перемешивали при 0oC в течение 1,5 часа. Реакцию гасили раствором бикарбоната натрия (198 мг, 20 мл воды), перемешивали в течение 5 минут, затем выпаривали для удаления метанола. Водную фазу экстрагировали этилацетатом (2•200 мл) и объединенный органический экстракт промывали водой (110 мл), 5% раствором бикарбоната натрия (80 мл) и рассолом (80 мл), сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Мерк), элюируя смесью этилацетат:гексан (2:1) и смесью этилацетат:метанол (98:2), получая 3,975 г указанного в названии продукта в виде сиропа. TLC (этилацетат:гексан, 4:1) Rf = 0,17.
b) Метиловый эфир 0 - /2,2-диметилоксиэтил/-N-/O-/метилсульфонил/-N-//фенилметокси/карбонил/-L- гомосерил/-L-серина
Раствор продукта из части (a) (3,975 г, 8,98 ммоля) в сухом метиленхлориде (52 мл) охлаждали до -15oC, обрабатывали триэтиламином (1,82 мл, 13,1 ммоля) и метансульфонилхлоридом (0,82 мл, 10,6 ммоля) и перемешивали при -15oC в течение 30 минут. Реакционную смесь гасили 25% раствором хлористого аммония (19 мл), нагревали до комнатной температуры и разбавляли этилацетатом (750 мл). Органическую фазу промывали 5% раствором бисульфата калия (100 мл), 50% насыщенным рассолом (100 мл) и насыщенным рассолом (100 мл), сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме, получая 4,9 г указанного в названии продукта в виде воскообразного твердого вещества. TLC (этилацетат:гексан, 4:1) Rf = 0,32.
c) Метиловый эфир N-/S-ацетил-N-//фенилметокси/-карбонил/L-гомоцистеинил/-О- /2,2-диметоксиэтил/-L-серина
Карбонат цезия (5,56 г, 17,04 ммоля) добавляли к раствору тиоуксусной кислоты (2,6 мл) в сухом метаноле (40 мл), перемешивали в течение 10 минут, затем выпаривали досуха. Полученное твердое вещество растирали с ацетоном (7•8 мл) и полученное сверхбелое твердое вещество сушили в вакууме, получая 4,39 г цезиевой соли тиоуксусной кислоты.
Суспензию цезиевой соли тиоуксусной кислоты (2,43 г, 1,3 экв.) в сухом диметилформамиде (8,0 мл) обрабатывали раствором продукта из части (b) (4,9 г, 8,98 ммоля) в сухом диметилформамиде (24 мл) и перемешивали в течение 16 часов при комнатной температуре в атмосфере аргона. Смесь разбавляли этилацетатом (1,0 л), промывали последовательно 5% раствором бикарбоната натрия (2•150 мл), водой (2•150 мл) и рассолом (150 мл), сушили (безводным сульфатом магния), фильтровали, выпаривали, досуха и сушили в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Мерк), элюируя смесями этилацетат: гексан (1:1, 2:1), получая 3,93 г указанного в названии продукта в виде воскообразного твердого вещества. TLC/этилацетат:гексан, 4: 1/Rf = 0,63.
d) Метиловый эфир O-/2,2-диметоксиэтил/-N-/N-//фенилметокси/карбонил-L-гомоцистеинил/-L-серина
Раствор продукта из части (c) (200 мг, 0,49 ммоля) в метаноле (8,0 мл) продували аргоном в течение 30 минут, охлаждали до 0oC (баня лед с солью) и обрабатывали 25% раствором метоксида натрия в метаноле (0,11 мл, 0,5 ммоля), продолжая пробулькивание аргона в процессе добавления и протекания реакции. После 5 минут при 0oC смесь гасили 25% раствором хлористого аммония (2,3 мл), и разделяли между этилацетатом (2•12 мл) и водой (2,3 мл). Объединенные органические экстракты промывали 25% раствором хлористого аммония (4,6 мл) и рассолом (4,6 мл), сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме, получая 183,2 мг указанного в названии продукта в виде белого твердого вещества. TLC (этилацетат:гексан, 4:1) Rf = 0,62.
e) /4S-/4α, 7α, 10aβ//- Октагидро-5-оксо-4-///фенил-метокси/карбонил/амино//1,4/оксазино [3,4-b]/1,3/тиазепин-7-карбоновая кислота
Раствор продукта из части (d) (150 мг, 0,11 ммоля) в сухом метиленхлориде (2,0 мл) обрабатывали смолой - Амберлит®15 (в H+ форме, 13 мг), перемешивали в течение 3 дней при комнатной температуре в атмосфере аргона, обрабатывали еще смолой Амберлитa®15 (13 мг) и перемешивали дополнительно в течение 3 дней. Раствор декантировали и хроматографировали на колонке с силикагелем (Мерк), элюируя смесями эилацетат:гексан (1:3, 1:1), получая 21,1 мг указанного в названии продукта в виде сиропа. TLC/этилацетат:гексан, 4:1/Rf = 0,70
f) Метиловый эфир /4S-/4α, 7α, 10aβ//- -октагидро-4-амино-5-оксо-/1.4/-оксазино [3,4-b]/1,3/тиазепин-7-карбоновой кислоты
Раствор продукта из части (e) (421 мг, 1,01 ммоля) в сухом метиленхлориде (25 мл) обрабатывали триметилсилилиодидом (0,72 мл, 5,06 ммоля) и перемешивали при комнатной температуре в атмосфере аргона в течение 1,75 часа. Смесь выпаривали досуха и полученный сироп разделяли между этиловым эфиром (50 мл) и 0,2 N соляной кислотой (2•25 мл). Водную фазу доводили до pH 10 насыщенным раствором бикарбоната натрия (25 мл), обрабатывали твердым хлористым натрием (2,0 г) и экстрагировали метиленхлоридом (3•75 мл), получая 219 мг указанного в названии продукта в виде сиропа. Вторая обработка водной фазы хлористым натрием (2,0 г) и повторная экстракция метиленхлоридом (2•100 мл) давала дополнительно 37 мг указанного в названии продукта. TLC (метиленхлорид:метанол, 9:1) Rf = 0,23.
g) Метиловый эфир /4S-/4α, 7α, 10aβ//- -октагидро-4-//2-ацетилтио/-1-оксо-3-фенилпропил/амино/-5-оксо-/1,4/- оксазино [3,4-b] /1,3/тиазепин-7-карбоновой кислоты.
Дициклогексиламиновую соль /S/-2-/ацетилтио/бензол-пропановой кислоты (516 мг, 1,32 ммоля, 1,2 экв.) суспендировали в этилацетате (42 мл), промывали 5% раствором бисульфатом калия (5•6,0 мл) и рассолом (6,0 мл), сушили (безводным сульфатом магния), фильтровали, выпаривали досуха и сушили в вакууме.
Свободную кислоту растворяли в сухом метиленхлориде (9,5 мл), охлаждали до 0oC (в бане лед с солью) и обрабатывали последовательно раствором продукта из части (f) (285,5 мг, 1,09 ммоля) в сухом метиленхлориде (4,2 мл), триэтиламино (0,14 мл, 1,15 ммоля) и бензотриазол-1-илокситрис/-диметиламино/фосфонийгексафторфосфатом (484 мг, 1,09 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и при комнатной температуре в течение 2,0 часов в атмосфере аргона, затем отгоняли досуха. Остаточный сироп растворяли в этилацетате (40 мл), промывали 0,5 N соляной кислотой (2 •6,6 мл), водой (6,6 мл) и рассолом (6,6 мл), сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Мерк), элюируя смесями этилацетат:гексан (1:2, 1:1), получая 382,9 мг указанного в названии продукта в виде сиропа. TLC (этилацетат:гексан, 1:1) Rf = 0,28.
h) /4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо/1,4/оксазино [3,4-b] /1,3/тиазепин-7-карбоновая кислота
Раствор продукта из части (g) (382,9 мг, 0,82 ммоля) в метаноле (9,0 мл) продували аргоном в течение 30 минут, охлаждали до 0oC (баня лед с солью), затем обрабатывали 1,0 N раствором гидроокиси натрия (3,32 мл, 4,0 экв., предварительно продутым аргоном в течение 30 минут), продолжая пробулькивание в процессе добавления и в течение реакции. Реакционную смесь перемешивали при 0oC в течение 5,0 часов и при комнатной температуре в течение 1,0 часа, доводили до PH 2,0 5% раствором бисульфата калия (14,5 мл), нагревали до комнатной температуры и экстрагировали этилацетатом (2•50 мл). Объединенные органические экстракты промывали рассолом (10 мл), сушили (безводным сульфатом натрия), фильтровали, выпаривали досуха и сушили в вакууме. Полученный сироп дважды выпаривали из гексана (25 мл) и полученное твердое вещество в виде пены хроматографировали на колонке с силикагелем (Мерк), элюируя смесями толуол: уксусная кислота (100:1, 50:1, 20:1), получая 212 мг указанного в названии продукта в виде твердого вещества, и.пл. 224 - 226oC; [α]D= -50,2o (с = 0,45, метанол). TLC (толуол: уксусная кислота, 5:1) Rf = 0,28.
HPLC: tR = 5,37 мин (УФ 220 нм) (98,9%); YMC S - 3 ODS (C-18) 6,0•150 мм; 60% (10% воды - 90% метанола - 2,0% фосфорной кислоты) (40% 90% воды - 10% метанола - 0,2% фосфорной кислоты), изократное.
Данные анализа для C18H22N2O5S2 • 0,14 H2O:
Вычислено, %: С 52,34; Н 5,44; N 6,78; S 15,53
Найдено, %: С 52,58; Н 5,57; N 6,44; S 15,16.
Пример 22
/4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-7H-пиридо [2,2-b]/1,3/-тиазепин-7-карбоновая кислота
Продукт примеров 3 и 11 получали также следующим образом:
а) Соль метилового эфира /4S-/4α, 7α, 10aβ//- октагидро-4-амино-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты и п-толуосульфоновой кислоты
Метиловый эфир /4S-/4α, 7α, 10aβ//- октагидро-4-амино-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты (6,11 г) растворяли в этилацетате (около 100 мл) и обрабатывали раствором моногидрата п-толуосульфоновой кислоты (4,52 г) в метаноле (3 мл) и этилацетата (20 мл). Немедленно образовывался осадок. Смесь дополнительно разбавляли этилацетатом и твердое вещество собирали фильтрованием. Твердое вещество промывали этиловым эфиром и сушили в вакууме, получая 7,908 г указанного в названии продукта в виде бледно-желтого твердого вещества с 98% чистотой; т.пл. 179 - 181oC (разложение).
b) Метиловый эфир /4S-/4α/R*/, 7α, 10aβ//- октагидро-4-//2-/ацетилтио/-1-оксо-3-фенилпропил/амино/-5-оксо- 7H-пиридо[2,1-b] /1,3/тиазепин-7- карбоновой кислоты
Неочищенный продукт из части (a) (636 мг, 1,48 ммоля) в метиленхлориде (5 мл) и диметилформамиде (1 мл) обрабатывали N-метилморфолином (163 мкл, 150 мг, 1,48 ммоль) с последующей обработкой 1-гидрокси-7-азабензотриазолом (208 мг, 1,52 ммоля). Ярко желтый раствор затем обрабатывали /S/-2-/ацетилтио/бензолпропановой кислотой (333 мг, 1,48 ммоля) в метиленхлориде (5 мл) и охлаждали в бане со льдом. Добавляли этил-3-/диметиламино/пропилкарбодиимид, гидрохлоридную соль (2,88 мг, 1,50 ммоля) и смесь перемешивали при 0oC в течение 1 часа и при комнатной температуре в течение 1,5 часа. Растворитель удаляли на роторном испарителе и остаток разделяли между этилацетатом и 0,5 N соляной кислотой. Этилацетатный экстракт промывали последовательно водой (дважды), 50% насыщенным раствором бикарбоната натрия и рассолом, затем сушили (сульфатом натрия), фильтровали и отгоняли, получая 651,2 мг указанного в названии продукта в виде белой пены.
c) /4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенил-пропил/амино/-5-оксо-7H-пиридо[2,1-b] /1,3/-тиазепин-7-карбоновая кислота
Раствор метилового эфира продукта части (b) в обескислороженном метаноле обрабатывали 1 N раствором гидроокиси натрия согласно процедуре примера 3(d), получая указанное в названии соединение.
Пример 23
/4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-7H-пиридо[2,1-b] /1,3/-тиазепин-7-карбоновая кислота
Продукт примеров 3, 11 и 22 получали также следующим образом:
a) N-/Фенилметокси/карбонил/-L-метионин
В 2 л склянке, оборудованной механической мешалкой и внутренним термометром, растворяли гидроокись натрия (61,65 г, 1,541 моль) в дистиллированной воде (1000 мл). К этому раствору добавляли L-метионин (100,0 г, 0,670 ммоля) при комнатной температуре. Раствор охлаждали в бане со льдом (внутренняя температуре 3oC) и добавляли бензилхлорформиат (110 мл, 0,737 ммоля) в течение 10 минут. После 15 минут индукционного периода внутренняя температура поднималась от 3oC до 12oC в течение 30 минут и затем падала до 0oC в течение 15 минут. Реакционную смесь перемешивали при 0oC в течение 2 часов, время в течение, которого первоначально мутная реакционная смесь становилась гомогенной. Баню со льдом удаляли и реакционной смеси позволяли нагреться до комнатной температуры в течение 1 часа. Реакционную смесь переносили в делительную воронку и промывали гексаном (2•300 мл). Водный слой подкисляли 6 N соляной кислотой до рH 5 и разбавляли этилацетатом (600 мл). Смесь далее подкисляли до pH 2. Органический слой отделяли и водный слой экстрагировали этилацетатом (3•600 мл). Органические экстракты объединяли, промывали рассолом (750 мл), сушили (сульфатом натрия), фильтровали и концентрировали в вакууме, получая светло-желтое масло. Неочищенный продукт (масло) растворяли в толуоле (1500 мл) и раствор концентрировали до половины его объема. Добавляли вторую порцию толуола (750 мл) и вновь концентрировали таким образом, что оставалось 650 мл толуола. Этот раствор хранили в течение ночи при 5oC, время, в течение которого некоторая часть продукта выкристаллизовывалась из раствора. Твердое вещество вновь растворяли нагреванием до комнатной температуры. Затем добавляли толуол (134 мл) (оставалось несколько кристаллов). Добавляли гептан при механическом перемешивании (500 мл) порциями по 30 мл с интервалом в 10 минут (приблизительно 3 часа - общее время добавления). В это момент продукт начинал кристаллизоваться из раствора. Добавляли дополнительную порцию гептана (1020 мл) в течение 1,5 часов и полученный шлам перемешивали в течение 2 часов. Продукт собирали вакуумным фильтрованием, промывали смесью 1:1 толуол: гептан (3•150 мл) и гептаном (3•500 мл) и сушили на воздухе, получая 158,6 г указанного в названии продукта в виде белого твердого вещества; и.пл. 66oC; [α]D= -1,5o (c = 1,95% этанол), TLC (этанол/вода, 3:1) Rf = 0,78.
Данные анализа для C13H17NO4:
Вычислено, %: С 55,11; Н 6,05; N 4,94
Найдено, %: С 54,96; Н 6,20; N 4,83.
b) Метиловый эфир N-//фенилметокси/карбонил/-L-метионина
В 3 л склянке, оборудованной механической мешалкой и вводом для продувки аргоном, растворяли продукт из части (a) (100,0 г, 0,353 ммоль) в метаноле (2 л) и добавляли моногидрат п-толуолсульфоновой кислоты (6,71 г, 0,035 моля). Реакционную смесь перемешивали в атмосфере аргона в течение 21 часа. Добавляли триэтиламин /4,9 г, 0,035 моля/ и реакционную смесь дополнительно перемешивали в течение 15 минут. Реакционную смесь концентрировали в вакууме до бледно-желтого масла. Масло растворяли в этилацетате (900 мл) и раствор промывали 1 N раствором соляной кислоты (740 мл), насыщенным раствором бикарбоната натрия (2•740 мл) и рассолом (740 мл). Органический слой сушили (сульфатом магния), фильтровали и концентрировали в вакууме до светло-желтого масла. Масло концентрировали из гексана (2•100 мл), получая 98,22 г указанного в названии продукта в виде белого твердого вещества.
c) Метиловый эфир N-//фенилметокси/карбонил/-L-метионинсульфоксида
В 3 л склянке, оборудованной механической мешалкой растворяли продукт из части (b) (97,95 г, 0,329 моля) в метаноле (1675 мл) и дистиллированной воде (215 мл). Раствор охлаждали в бане со льдом и добавляли бикарбонат натрия (28,5 г, 0,339 моля). Небольшими порциями добавляли N-хлорсукцинимид (44,0 г, 0,29 моля) в течение 25 минут таким образом, чтобы внутренняя температура не превышала 7oC. Смесь перемешивали в бане со льдом в течение 1 часа и затем позволяли нагреваться до комнатной температуры в течение 1 часа. Смесь концентрировали в вакууме с удалением около 75% метанола, разбавляли этилацетатом (1000 мл) и промывали рассолом (500 мл). Слой рассола повторно экстрагировали этилацетатом (2•200 мл). Органические экстракты объединяли, сушили (сульфатом магния), фильтровали и концентрировали в вакууме до прозрачного вязкого масла. Масло концентрировали из толуола (3•100 мл) и остаточный растворитель удаляли в условиях высокого вакуума, получая неочищенное указанное в названии вещество в виде прозрачного масла, которое превращалось в белое твердое вещество (131,4 г). Неочищенный продукт содержал 12 весовых % сукцинимида и 9 весовых толуола по данным ЯМР. Продукт являлся смесью диастереомеров сульфоксида.
d) Метиловый эфир S-//ацетилокси/метил/-N-//фенилметокси/карбонил-L-гомоцистеина
В 1 л склянку, содержащую продукт из части (c) (102,8 г, поправленный вес, 0,328 моля), добавляли толуол (480 мл), ацетат натрия (32,3 г, 0,394 моля) и уксусный ангидрид (186 мл, 1,970 моля). Полученную смесь кипятили с обратным холодильником в атмосфере аргона (118oC) в течение 18 часов. Темно-коричневой реакционной смеси позволяли охладиться до комнатной температуры. Через 1 час при комнатной температуре реакционная смесь становилась очень вязкой с твердыми включениями. Твердые вещества растворяли этилацетатом (100 мл) и смесь частично концентрировали в вакууме до вязкого коричневого остатка. Остаток концентрировали из толуола (240 мл) с удалением уксусного ангидрида, разбавляли этилацетатом (1000 мл) и осторожно промывали насыщенным раствором бикарбоната натрия (4•680 мл). Органический слой промывали рассолом (450 мл), сушили (сульфатом магния), фильтровали и концентрировали в вакууме. Остаточный растворитель удаляли при высоком вакууме с получением светло-коричневого твердого вещества. Неочищенный продукт растворяли в н-бутилацетате (450 мл) с нагреванием (35oC) и перемешиванием. После охлаждения до комнатной температуры медленно добавляли гексан (200 мл) к раствору при перемешивании в течение 15 минут. В этот момент продукт кристаллизовался из раствора. Добавляли дополнительную порцию гексана (700 мл) в течение 30 минут и полученный шлам перемешивали в течение 3 часов. Продукт собирали фильтрованием, промывали смесью 1:2 н-бутилацетат:гексан (200 мл), 1:4 н-бутилацетат: гексан (2 •240 мл) и гексаном (2 • 250 мл). Продукт сушили на воздухе, затем сушили в условиях высокого вакуума, получая 87,7 г указанного в названии продукта в виде бледно-коричневого твердого вещества, т.пл. 73oC; [α]D= -1,6o (c = 1,95% этанол). TLC (5% метанол/метиленхлорид) Rf = 0,80.
Данные анализа для C16H21NO6S:
Вычислено, %: С 54,07; Н 5,95; N 3,94
Найдено, %: С 53,48; Н 5,74; N 3,82.
e) S-Ацетил-N-//Фенилметокси/карбонил/-L-гомоцистеин
В 1 л склянке раствор продукта из части (d) (83,0 г, 0,233 моля) в тетрагидрофуране (415 мл) продували аргоном в течение 30 минут. В отдельной 2 л склянке, оборудованной механической мешалкой и вводом для аргона, 86,8% раствор гидроокиси калия (62,7 г, 0,969 моля) в дистиллированной воде продували аргоном в течение 15 минут.
Тетрагидрофурановый раствор быстро добавляли к раствору гидроокиси калия (внутренняя температура 20oC) через канюлю с интенсивным перемешиванием в атмосфере аргона. Склянку, содержащую продукт из части (d), промывали 20 мл тетрагидрофурана (продутого аргоном в течение 15 минут) и смыв добавляли к реакционной смеси. Через 30 минут реакционная смесь была прозрачной и разделенной на две фазы и имела место изотермическая реакция до 28oC.
Еще через 2 часа реакционную смесь охлаждали до 1oC (внутренняя) и добавляли одной порцией гидроокись натрия (2,75 г, 0,073 моля) (экзотермическая реакция до 6,8oC). Реакционную смесь дополнительно перемешивали в течение 20 минут при 0oC и затем позволяли нагреваться до 11oC в течение 30 минут. Реакционную смесь охлаждали до 1oC и в течение 10 минут добавляли уксусный ангидрид (68,6 г, 0,727 моля). В процессе добавления имела место экзотермическая реакция до 10oC. Внутренняя температура вновь падала до 4oC до окончания добавления. Охлажденную баню удаляли и реакционную смесь перемешивали при температуре окружающего воздуха в течение 45 минут.
Реакционную смесь концентрировали в вакууме приблизительно на половину ее объема, подкисляли до pH 2 6 N раствором соляной кислоты (175 мл) и экстрагировали этилацетатом (2 • 1,1 л). Объединенные органические экстракты промывали рассолом (560 мл). Органический слой обрабатывали активированным углем и безводным сульфатом магния, фильтровали и концентрировали в вакууме до желтого масла. Добавляли н-бутилацетат (380 мл) и раствор концентрировали в вакууме (45oC) на половину его объема. Добавляли вторую порцию н-бутилацетата (190 мл/ и вновь концентрировали таким образом, чтобы осталось 190 мл н-бутилацетата. Медленно добавляли гептан (300 мл) при перемешивании до появления вуали и добавляли в качестве затравки кристаллы. Через 15 минут из раствора кристаллизовалось белое твердое вещество. Медленно добавляли вторую порцию гептана (570 мл) в течение 30 минут и полученный шлам перемешивали при комнатной температуре в течение ночи. Продукт собирали фильтрованием, промывали смесью 1:3 н-бутилацетата:гептана (2 • 275 мл) и гексаном (2 • 275 мл), сушили на воздухе и затем сушили в условиях высокого вакуума, получая 50,1 г указанного в названии продукта в виде белого твердого вещества; т.пл. 73 - 74oC; [α]D= -1,3o (c = 1,95% этанол). TLC (этанол/вода, 3:1) Rf = 0,83.
Данные анализа для C14H17NO5S:
Вычислено, %: С 54,01; Н 5,50; N 4,50
Найдено, %: С 53,88; Н 5,45; N 4,44.
Фильтрат концентрировали таким образом, чтобы осталось 100 мл н-бутилацетата. Этот раствор обрабатывали 310 мл гептана, как описано выше, получая вторую часть 8,4 г указанного в названии продукта в виде белого твердого вещества с общим выходом 58,5 г.
f) Метиловый эфир /S-/R*,R*/-2-//4-/ацетилтио/-1- оксо-2-///фенилметокси/карбонил/амино/бутил/амино/-6,6- диметоксигексановой кислоты
S-Ацетил-N-//фенилметокси/карбонил/-L гомоцистеин (0,456 моля) растворяли в смеси метиленхлорида (600 мл) и диметилформамида (90 мл) и добавляли гидроксибензотриазол гидрат (64,72 г, 0,479 моля). Смесь охлаждали в бане со льдом и добавляли раствор метилового эфира /S/-2-амино-6,6-диметоксигексановой кислоты (полученного как описано в примере 1 (e), 93,7 г, 0,456 моля), растворенного в метиленхлориде (600 мл). Наконец, добавляли гидрохлоридную соль этил-3-/диметиламино/пропилкарбодиимида (91,83 г, 0,479 моля) и реакционную смесь перемешивали в течение 1 часа при 0oC, затем при комнатной температуре в течение 2 часов. По окончании этого времени реакционную смесь концентрировали в вакууме и остаток разделяли между этилацетатом (3 л) и насыщенным водным раствором бикарбоната натрия (1 л). Органический экстракт промывали водой (1 л), 5% раствором бисульфата калия (1 л), водой (1 л) и рассолом (1 л), затем сушили (сульфатом натрия) и концентрировали в вакууме до 238 г неочищенного продукта. Неочищенный продукт растворяли в смеси этилацетат: метиленхлорид (1:1, 300 мл) и переносили на подложку колонки с силикагелем (Мерк) 10 • 15 см. Элюирование смесью 8:2 этилацетат:гексан (7 л) с последующим элюированием этилацетатом (4 л) давало 205,28 г указанного в названии продукта.
g) Метиловый эфир /4S-/4α, 7α, 10aβ//- октагидро-4-///фенилметокси/карбонил/амино/-5-оксо-7H- пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Раствор продукта из части (f) (205,28 г, 0,412 моля), высушенный выпариванием со смесью метиленхлорид/толуол/ в метаноле (2 л), охлаждали до 0oC (баня со льдом) и продували аргоном в течение 30 минут. Быстро добавляли 25 вес.% раствором метоксида натрия в метаноле (95,1 мл, 1,01 экв.) с непрерывной продувкой аргона, и реакционную смесь перемешивали более 10 минут, затем гасили добавлением 1 л насыщенного раствора хлористого аммония, разбавляли 0,5 л воды и обрабатывали 3 л этилацетата. Полученную смесь разделяли на две порции, каждую из которых отдельно концентрировали в вакууме для удаления органики (этилацетата и метанола). Концентрированные остатки объединяли и обрабатывали 1 л этилацетата. Органический слой отделяли и промывали насыщенным раствором хлористого аммония (0,5 л). Объединенные водные растворы повторно экстрагировали этилацетатом 1 л. Органические экстракты объединяли и промывали 1 л воды и двумя порциями по 1 л рассола, сушили (сульфатом натрия), фильтровали и концентрировали. Далее остаток выпаривали с метиленхлоридом и сушили в вакууме, получая 182,65 г свободного сульфогидрила продукта из части (f).
Этот свободный промежуточный сульфогидрильный продукт (0,400 моля) растворяли в метиленхлориде (4 л) и обрабатывали 30,8 мл (0,400 моля) трифторуксусной кислоты. Реакционную смесь кипятили с обратным холодильником в течение 16 часов, затем охлаждали и концентрировали в вакууме. Полученный остаток растворяли в 2 л этилацетата, затем промывали 400 мл 0,1 N раствора соляной кислоты, 1 л воды, 1 л насыщенного раствора бикарбоната натрия, 1 л воды и 1 л рассола, сушили (сульфатом натрия), фильтровали и концентрировали. Остаток выпаривали с метиленхлоридом и сушили в вакууме, получая 166,24 г указанного в названии продукта.
h) Метиловый эфир /4S-/4α, 7α, 10aβ//- октагидро-4-амино-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Иодтриметилсилан (76,6 мл, 0,538 моля) добавляли к раствору под аргоном, содержащему продукт из части (g) (162,43 г, 0,414 моля), растворенного в метиленхлориде (1,5 л). После перемешивания реакционной смеси в течение 1,5 часов смесь концентрировали в вакууме и разделяли между 1 л этилацетата и 700 мл 1N соляной кислоты (имело место выделение CO2, pH 1,2). Этилацетатный слой отделяли и экстрагировали 300 мл 1 N соляной кислоты. Объединенные кислые водные экстракты промывали далее 1 л этилацетата, затем охлаждали до 0oC и доводили до pH 10,0 4 N раствором гидроокиси натрия (около 275 мл). Водный слой насыщали твердым хлористым натрием, затем экстрагировали 5 порциями по 1 л метиленхлорида. Объединенные органические экстракты сушили (сульфатом натрия), фильтровали и концентрировали в вакууме. Остаток повторно растворяли в 1 л метиленхлорида и промывали 0,5 л рассола, сушили (сульфатом натрия), фильтровали и концентрировали, получая 98,8 г указанного в названии продукта.
i) Метиловый эфир /4S-/4α/R*/, 7α, 10aβ//- октагидро-4-//2-/ацетилтио/-1-оксо-3-фенилпропил-амино/-5-оксо-7H- пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Дициклогексиламиновую соль /S/-2-/ацетилтио/бензолпропановой кислоты (173,1 г, 0,427 моля) разделяли между этиленцетатом (1 л) и 10% раствором бисульфата калия (800 мл). Органический слой отделяли и промывали 5% водным раствором бисульфата калия (1 л), 50% рассолом (1 л) и рассолом (1 л), сушили (сульфатом натрия), фильтровали и концентрировали в вакууме. Остаток несколько раз выпаривали с метиленхлоридом, затем сушили в течение ночи в вакууме, получая 97,8 г неочищенной /S/-2-/ацетилтио/бензолпропановой кислоты.
Раствор этой /S/-2-/ацетилтио/бензолпропановой кислоты (0,4 г, 27 ммоля), растворенный в метиленхлориде (900 мл), охлаждали в бане со льдом и обрабатывали раствором продукта из части (h) (100,28 г, 0,388 моля) в метиленхлориде (600 мл), триэтиламином (154,2 мл, 0,388 ммоля), и, наконец, добавляли в одну порцию бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфат (188,9 г, 0,427 моля). После 1 часа при 0oC и 2 часов при комнатной температуре реакционную смесь концентрировали в вакууме и растворяли в 2 л этилацетата. Органический раствор промывали 0,5 л рассола, 1 л 0,5 N соляной кислоты, 1 л воды, 2 л насыщенного раствора бикарбоната натрия, 1 л воды и 1 л рассола, сушили (сульфатом натрия), фильтровали и концентрировали. К этому моменту, ту промывную воду, которая содержала продукт (TLC определение), повторно экстрагировали этилацетатом. Этилацетатные экстракты обрабатывали обычным образом и все объединяли, получая неочищенный желтый маслообразный продукт. Желтое масло, приготовленное в смеси 1:1 этилацетат:гексан, подвергали обработке на колонке с силикагелем 15• 15 см и элюировали 7 мл смеси этилацетат:гексан 1:1 с последующим элюированием 4 л смеси 6:4 этилацетат:гексан, и, наконец, 2 л смеси 7:3 этилацетат: гексан. Фильтраты, содержащие желаемый продукт, концентрировали, получая 123,57 г указанного в названии продукта.
j) /4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/-5-оксо-7H-пиридо[2,1-b]- /13,/тиазепин-7-карбоновая кислота
В 12 л трехгорлую склянку, оборудованную делительной воронкой и механической мешалкой, помещали раствор продукта из части (i) (96,0 г, 0,207 ммоль) в метаноле (1,1 л). Раствор продували в течение 30 минут, затем охлаждали в бане со льдом до достижения внутренней температуры +7oC. В течение 1 часа было добавлено общее количество 1,45 л 1 N раствора гидроокиси натрия (предпочтительно продутого аргоном в течение 30 минут). Реакционную смесь непрерывно продували аргоном в процессе добавления. Температура реакции поднималась до +12oC и поддерживалась в процессе добавления. Реакционную смесь дополнительно перемешивали в течение 30 минут, затем нагревали до комнатной температуры водяной баней с температурой окружающей среды и перемешивали с барбортированием с течение 2,5 часов. Около 250 мл 6 N соляной кислоты добавляли по каплям в течение 15 - 250 минут до достижения pH 2. В процессе подкисления образовался смолообразный осадок. После непрерывного перемешивания дополнительно в течение 2 часов осадок изменялся до белого тонкодисперсного твердого вещества с наличием более крупных комков твердого вещества. Продукт собирали на 600 мл фильтре Шотта. Промывка собранного твердого вещества 1 л воды с последующей промывкой 2 л безводного эфира и, наконец, сушка в вакууме давали 70,3 г указанного в названии продукта в виде тонкодисперсного твердого вещества, т.пл. 218 - 220oC (разложение). TLC (1:99 уксусная кислота/этилацетат) Rf=0,48
HPLC: tR (YMC S-3 ODS 6,0•150 мм, 1,5 мл/мин, изократный 60% B, Буфер A = метанол/вода/фосфорная кислота (10:90:0,2), Буфер B = метанол/вода/фосфорная кислота (90:10:0,2) = 9,33 мин, 99,3% общей площади пика при 220 нм.
Данные анализа для C19H24N2O4S2:
Вычислено, %: С 55,86; Н 5,92; N 6,86; S 15,70
Найдено, %: С 55,83; Н 5,83; N 6,96; S 15,70.
Пример 24
Метиловый эфир /4S-/4α/R*/, 7α, 10aβ//- октагидро-4-//2-ацетилтио/-1-оксо-3- фенилпропил/амино/-5-оксо-7H-пиридо[2,1-b]/1,3/тиазепин-7-карбоновой кислоты
Реакцию сочетания, описанную в примерах 3 (c), 11 (i), 22 (b), и 23 (i), проводили также следующим образом.
Раствор /S/-2-/ацетилтио/бензолпропановой кислоты (1,83 г, 8,14 ммоля) и метилового эфира /4S-/4α, 7α, 10aβ//- -октагидро-4-амино-5-оксо-7H-пиридо [2,1-b]/1,3/тиазепин-7-карбоновой кислоты (2,11 г, 8,17 ммоля) в сухом метиленхлориде (20 мл) охлаждали до 0oC и добавляли в одну порцию гидрохлоридную соль этил-3-/диметиламино/пропил/карбодиимида (1,77 г, 9,32 ммоля). Реакционную смесь перемешивали при 0oC в течение 6 часов и затем концентрировали до маслообразной пены. Затем остаток разделяли между этилацетатом (100 мл) и 1 N раствором соляной кислоты (50 мл). Органическую фазу промывали 1 N соляной кислотой (50 мл), насыщенным водным раствором бикарбоната натрия (2•50 мл) и насыщенным водным раствором хлористого натрия (50 мл), сушили (безводным сульфатом натрия), фильтровали и концентрировали в вакууме, получая 3,43 г указанного в названии продукта в виде белой пены.
Пример 25
1,1-Диметилэтиламиновая соль /4S-/4α/R*/, 7α, 10aβ//- октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо -7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
15 мл 3-горлую склянку, оборудованную обратным холодильником, вакуумировали и повторно заполняли три раза аргоном. В склянку загружали /4S-/4α/R*/, 7α, 10aβ//- октагидро-4-//2-меркапто-1-окксо-3-фенилпропил/амино/- 5-оксо-7H-пиридо[2,1-b]/1,3/тиазепин-7-карбоновую кислоту (0,20 г) и 1:1 раствор обезгаженного абсолютного этанола и ацетонитрила (1,0 мл). Как только гетерогенную смесь перемешивали, добавляли по каплям трет-бутиламин (63,0 мкл, 1,03 экв). Раствор становился гомогенным через 3 минуты после окончания добавления амина. Раствор (внутренняя температура около 20oC) медленно по каплям разбавляли добавлением ацетонитрила с конечным объемом 10 мл. После дополнительного перемешивания в течение 2 часов твердое вещество отфильтровывали, промывали один раз 100% ацетонитрилом (5 мл), сушили на воздухе и помещали под высокий вакуум на 2 часа для удаления остаточных растворителей, получая 20,2 г указанного в названии продукта в виде белого кристаллического твердого вещества.
Вышеуказанное твердое вещество объединяли с веществом из других опытов и перекристаллизовывали следующим образом. 25 мл 3-горлую склянку, оборудованную обратным холодильником, магнитной мешалкой и капельной воронкой, вакуумировали и трижды заполняли аргоном. В склянку добавляли загрузки 1,1-диметиламиновой соли (0,37 г) и 59% ацетонитрил/метанол (2,28 мл). Склянку и содержимое нагревали до 29 - 32oC для растворения твердых веществ. Раствор разбавляли ацетонитрилом (27 мл). Баню для нагревания удаляли и склянке позволяли охладиться до комнатной температуры (20oC). Через час дополнительного перемешивания смесь фильтровали и твердые вещества промывали один раз ацетонитрилом (10 мл), сушили на воздухе, и помещали в условия высокого вакуума для удаления остаточных растворителей, получая 0,29 г указанного в названии продукта в виде белого кристаллического твердого вещества: т.пл. с усадкой 160oC и медленное плавление и разложение при температуре, увеличившейся до 190 - 191oC (при 190 - 191oC оставшееся блестящее вещество быстро плавится).
Пример 26
Метиловый эфир /4S-/4α, 7α, 10aβ//- октагидро-4-амино-5-оксо-7H-пиридо[2,1-b]/1,3/тиазепин-7-карбоновой кислоты
Это промежуточное соединение примеров 3 (c) и 11 (h) и 23 (h) получали также следующим образом:
a) Диэтиловый эфир 2-/ацетиламино/-2-/4-/ацетилокси/бутил/пропандионовой кислоты
Перемешиваемую суспензию 95% гидрида натрия (60,8 г, 2,532 моля) в безводном диметилформамиде (500 мл) в атмосфере аргона охлаждали до 0oC (в бане со льдом). Раствор диэтилацетамидомалоната (500 г, 2,302 ммоля) в безводном диметилформамиде (1,2 л) добавляли в течение 45 минут, в то же время сохраняя температуру реакции ниже 18oC. После окончания добавления мутный раствор постепенно нагревали до комнатной температуры. После перемешивания в течение 1 часа при комнатной температуре добавляли 4-бромбутилацетат (471,5 г, 2,417 моля). Затем смесь перемешивали при 59 - 60oC в течение 18 часов. Полученный шлам охлаждали до комнатной температуры, гасили абсолютным этанолом (40 мл) и ледяной уксусной кислотой (4 мл), перемешивали около 15 минут, выливали в 10% раствор хлористого лития и экстрагировали этилацетатом (2•3 л). Объединенные этилацетатные экстракты промывали 10% раствором хлористого лития (3•3 л), сушили безводным сульфатом натрия) и выпаривали в вакууме, получая 750 г указанного в названии продукта в виде масла.
b) 2-/Ацетиламино/-6-гидроксигексановая кислота
Продукт из части (a) (730 г, 2,2 моля) взвешивали в 5 л 3-горлую склянку (оборудованную термометром, магнитной мешалкой и воздушным холодильником) и разбавляли абсолютным этанолом (300 мл) с последующим добавлением водного раствора 6 N гидроксида натрия (1,6 л, 9,6 моля). Реакционную смесь нагревали при 68 - 70oC в течение 5 часов и получали гомогенный раствор. Реакцию охлаждали до комнатной температуры и медленно добавляли 6 N соляную кислоту (1,32 л) до pH 1,3. Склянку оборудовали коротким дефлегматором для отгонки этанола, когда температура медленно поднималась до 87 - 90oC, и эта температура поддерживалась в течение 8,5 часов. Наблюдалось медленное выделение двуокиси углерода. Полный объем дистиллата составлял 600 мл. pH конечного раствора была 3,0. Реакционную смесь концентрировали в вакууме до полного выпаривания воды и затем концентрировали из толуола (2•500 мл). Полутвердую массу растирали с абсолютным этанолом (1 л), фильтровали и промывали дополнительным количеством абсолютного этанола (500 мл). Фильтрат концентрировали в вакууме, получая 509 г неочищенного масла (82% чистоты), которое содержало этанол и толуол.
c) /S/-2-Амино-6-гидроксигексановая кислота
Неочищенный продукт из части (b) (443 г, включая некоторое количество этанола и толуола (установленный вес исходного вещества был 394 г), растворяли в воде (3,3 л) и добавляли 1N раствор гидроокиси лития до pH 7,5 (потребовалось 1,53 л). Смесь нагревали до 35oC, добавляли ацилазу (марки I из свиной почки, 0,4 г) и реакционную смесь перемешивали в течение 24 часов. По окончании этого времени pH было 7,33. Доводили pH 7,5 1 N раствором гидроокиси лития (около 2 мл), добавляли дополнительное количество ацилазы (0,4 г) и реакцию перемешивали более 17 часов (pH 7,3). Доводили pH раствора до 5,9 уксусной кислотой. Добавляли целит (20 г) и уголь (20 г) и реакцию нагревали до 92oC и поддерживали температуру в течение 5 минут. Реакционную смесь фильтровали через целитовую подложку и концентрировали в вакууме до полупастообразного состояния (441 г). Это вещество растирали с 900 мл смеси 1: 5:10 вода: этанол:диметилформамид. Требовалось некоторое подогревание для разрушения первоначального комка. Реакционную смесь выдерживали в холодильнике в течение ночи, фильтровали и промывали 200 мл приведенной выше смеси растворителей, получая 214 г неочищенного вещества (около 40% N-ацетильного вещества). Это вещество суспендировали в метаноле (500 мл), нагревали на паровой бане, выдерживали в течение 2 часов и фильтровали. Эту процедуру повторяли два раза, получая 108 г указанного в названии продукта; [α]D= +22o (c=1,44, 6 N соляная кислота).
Или же стадии (b) и (c) также проводили следующим образом:
b) 2-/Ацетиламино/-6-гидроксигексановая кислота
5 л трехгорлую склянку, оборудованную механической мешалкой и термометром с термопарой, загружали продуктом из части (a) (631 г, 1,933 моля) и тетрагидрофураном (259 мл). К перемешиваемому раствору по каплям добавляли 6 N раствор гидроокиси натрия (1385 мл, 8,31 ммоля) в течение 40 минут. Имела место сильная экзотермическая реакция и было необходимо для охлаждения реакционной смеси помещать реактор в баню со льдом для поддерживания температуры ниже 60oC. Реакционную смесь затем нагревали до температуры, близкой к температуре кипения, с обратным холодильником (температура реактора 67 - 68o) в течение 5,5 часов.
Смесь перемешивали при комнатной температуре в течение ночи (16,5 часов). Доводили pH от 12,75 до 1,3 постепенным добавлением 6 N соляной кислоты (1150 мл, 6,9 ммоля), поддерживая температуру около 25oC. Смесь постепенно нагревали с коротким дефлегматором до отгонки и происходило выделение газа (двуокиси углерода) (температура реактора 72,3o, температура дефлегматора 70oC) до тех пор, пока не прекращалась отгонка и выделение газа становилось очень медленным (94,1oC температура реактора, 50o температура дефлегматора). Полный объем собранного дистиллата составлял 410 мл и остаток в реакторе имел pH 3,9. Нагревание продолжали еще в течение 10 минут без дополнительного выделения газа. Полное время нагревания от начала отгонки составляло 7,5 часов.
После перемешивания при комнатной температуре в течение ночи, прозрачную реакционную смесь (pH 3,50) отгоняли на роторном испарителе в вакууме при 60o температуре бани и пастообразный остаток перемешивали с абсолютным этанолом (750 мл). Полученную суспензию с кристаллами отгоняли на роторном испарителе (вакуумный насос, 60o баня) и пастообразный остаток отделяли абсолютным этанолом (2•750 мл). К конечному остатку добавляли абсолютный этанол (1500 мл) и смесь перемешивали при температуре бани 60o до тех пор, пока она не превращалась в суспензию с тонкокристаллическими образованиями, в течение 20 минут, и затем перемешивали при комнатной температуре в течение 20 минут. Суспензию фильтровали и остаток промывали абсолютным этанолом (2•300 мл). Фильтраты имели заметную мутность и в дальнейшем очищались фильтрованием через целитовую подложку. Новый прозрачный фильтрат отгоняли на роторном испарителе, получая 434,6 г указанного в названии продукта в виде густого сиропа янтарного цвета. TLC (10:1:1, метанол:уксусная кислота:вода) Rf=0,59.
c) /S/-2-Амино-6-гидроксигексановая кислота
5 л трехгорлую склянку, оборудованную механической мешалкой и термометром, загружали продуктом из части (b) (434 г, 1,93 моля) и водой (3 л). pH мутного раствора доводили от 4,05 до 7,50 добавлением 1 N раствора гидроокиси лития (705 мл). Раствор нагревали до 36o и добавляли ацилазу из свиной почки I (0,710 г). Смесь перемешивали при температуре 35 - 36oC в течение 23,5 часов. Реакционную смесь охлаждали до комнатной температуры и доводили pH от 7,0 до 5,9 добавлением ледяной уксусной кислоты (4,4 мл). Добавляли целит (29 г) и уголь (29 г) и поднимали температуру до 91oC при перемешивании. Нагревание прекращали и смеси позволяли охладиться до комнатной температуры. Суспензию фильтровали через бумажный фильтр 18,5 см и полученный остаток интенсивно промывали водой. Бесцветный фильтрат (около 3,9 л) концентрировали на роторном испарителе при 60oC, получая 476 г прозрачного вязкого масла. Добавляли абсолютный этанол (7250 мл) и смесь перемешивали до тех пор, пока она не становилась гомогенной суспензией, содержащей кристаллы. Вновь отгоняли растворитель и добавляли абсолютный этанол (1584 мл) к белому твердому остатку. Суспензию крутили в роторном испарителе в течение ночи (15 часов) при комнатной температуре и фильтровали через бумажный фильтр 18,5 см. Остаток промывали абсолютным этанолом (7 • 100 мл) и сушили до постоянного веса в вакууме, получая 85,8 г белого кристаллического указанного в названии продукта. TLC (метанол:вода:уксусная кислота 10:1:1) Rf=0,62.
Данные анализа для C6H13NO3:
Вычислено, %: С 48,25; Н 8,94; N 9,38
Найдено, %: С 48,66; Н 8,77; N 9,43
d) Метиловый эфир /S-/R*, R*/-2-/4-/ацетилтио/-1-оксо-2-///фенилметокси/карбонил/ амино/бутил/амино/-6-гидроксигексановой кислоты
Шлам /S/-2-амино-6-гидроксигексановой кислоты (1,0 г, 6,8 ммоля) в метаноле (20 мл) перемешивали при комнатной температуре в атмосфере аргона и обрабатывали триметилсилилхлоридом (1,9 мл, 15 ммоля). Полученный раствор перемешивали при комнатной температуре в течение 18 часов. Объем растворителя снижали до 3,5 мл при пониженном давлении. Добавляли ацетонитрил (5 мл) и раствор охлаждали до -10oC. Затем добавляли N,N-диизопропилэтиламин /84,15 мл, 23,8 ммоля/ и раствор охлаждали до -40oC, получая раствор, содержащий метиловый эфир /S/-2-амино-6-гидроксигексановой кислоты.
В отдельной склянке раствор S-ацетил-N-//фенилметокси/карбонил/гомоцистеина (полученный как описано в примере 23 (e), 2,117 г, 6,8 ммоля) в ацетонитриле (5 мл) обрабатывали при 0oC N,N-диизопропилэтиламином (1,20 мл, 6,8 ммоля). В другой склянке гидроксибензотриазол гидрат (0,104 г, 0,68 ммоля) и метансульфонилгидроксибензотриазол (1,450 г, 6,8 ммоля) растворяли в ацетонитриле и охлаждали до -18oC. Предварительно полученный S-ацетил -N-//фенилметокси/карбонил/гомоцистеин добавляли затем по каплям к этому раствору, поддерживая при этом внутреннюю температуру ниже -10oC. После перемешивания при температуре от -18o до -12oC в течение 3 часов полученный раствор по каплям добавляли к раствору, приведенному выше, содержащему метиловый эфир /S/-2-амино-6-гидроксигексановой кислоты при -40oC. Смеси позволяли медленно нагреваться до 16oC в течение 18 часов. Затем реакционную смесь выливали в этилацетат (50 мл) и 1 N соляную кислоту (50 мл). Смесь переносили в делительную воронку и разделяли слои. Водный слой экстрагировали этилацетатом (2 х 50 мл). Органические слои объединяли и затем промывали 1 N соляной кислотой (100 мл), насыщенным раствором бикарбоната натрия (100 мл). Раствор сушили над сульфатом магния, фильтровали и концентрировали до белого твердого вещества. К этому твердому веществу добавляли трет-бутилметиловый эфир (20 мл) и полученный шлам перемешивали при комнатной температуре в течение 4 часов и фильтровали. Продукт промывали трет-бутилметиловым эфиром и сушили, получая 2,087 г указанного в названии продукта, т. пл. 89 - 90oC.
e) Метиловый эфир /S-/R*, R*//-2-//4-ацетилтио/-1- оксо-2-//фенилметокси/карбонил/ амино/бутил/амино/-6- оксогексановой кислоты
К раствору оксалилхлорида (546 мкл, 6,27 ммоля) в сухом метиленхлориде (16 мл) при -65oC (внутренняя температура) по каплям добавляли раствор диметилсульфоксида (905 мкл, 12,54 ммоля) в метиленхлориде (13 мл) в течение 12 минут, поддерживая при этом внутреннюю температуру между -65oC и 60oC. Раствор продукта из части (d) (1,90 г, 4,18 ммоля) в метиленхлориде (7 мл) добавляли в реакционную склянку в течение 20 минут, делая смесь мутной. Для окончательного перевода спирта в реакционную склянку использовали дополнительное количество (1 мл) метиленхлорида и реакционной смеси позволяли перемешиваться при -65oC в течение 40 минут. Затем добавляли N,N-диизопропилэтиламин (3,7 мл, 20,90 ммоля), таким образом делая раствор прозрачным. После дополнительного перемешивания в течение 90 минут при -65oC реакции позволяли нагреться до -18oC в течение 2 часов. Реакцию гасили 10% водным раствором бисульфата калия (30 мл) и затем нагревали до комнатной температуры. Реакционную смесь разбавляли 25 мл воды, перемешивали и фазы разделяли. Водную фракцию обратно экстрагировали метиленхлоридом (2х35 мл). Объединенные органические экстракты промывали 10% водным раствором бисульфата калия (25 мл), насыщенным водным раствором бикарбоната натрия (2х25 мл), рассолом (25 мл), сушили (сульфатом магния), фильтровали и концентрировали в вакууме, получая 1,84 г указанного в названии продукта в виде белого твердого вещества.
f) Метиловый эфир /4S-/4α, 7α, 10aβ//- октагидро-4-///фенилметокси/карбонил/амино/-5-оксо-7H-пиридо[2,1-b] /1,3/- тиазепин-7-карбоновой кислоты
Сухую склянку под аргоном загружали продуктом из части (е) (1,76 г, 3,89 ммоля) и метанолом (17 мл). Раствор охлаждали до 0oC и барботировали аргоном в течение 25 минут. Раствор метоксида натрия (25 вес.% в метаноле, 983 мкл, 4,28 ммоля) добавляли к реакционной смеси в течение 15 секунд. Через час реакцию гасили 1 N соляной кислотой (20 мл) и затем позволяли нагреться до комнатной температуры. Добавляли этилацетат (35 мл) и после перемешивания разделяли слои. Водную фракцию обратно экстрагировали этилацетатом (2х15 мл). Объединенные органические фракции промывали 1 N соляной кислотой (15 мл), рассолом, сушили (сульфатом магния), фильтровали и концентрировали в вакууме,получая1,69гметиловогоэфира/S-/R*,R*//2-//4-меркапто-1-оксо///фенилметокси/карбонил/амино/ бутил/амино/-6-оксогексановой кислоты в виде белой пены.
Раствор этой белой пены и трифторуксусной кислоты (305 мкл, 3,95 ммоля) в метиленхлориде (17 мл) кипятили с обратным холодильником в течение 2,25 часа. После охлаждения до комнатной температуры реакционную смесь концентрировали и остаток растворяли в этилацетате (25 мл), промывали насыщенным раствором бикарбоната натрия (2х20 мл), рассолом, сушили (сульфатом магния), фильтровали и концентрировали в вакууме, получая 1,50 г указанного в названии продукта в виде белой пены.
g) Метиловый эфир /4S-/4α, 7α, 10aβ//- октагидро-4-амино-5-оксо-7H-пиридо[2,1-b]/1,3/ тиазепин-7-карбоновой кислоты.
Обработка продукта из части (f) иодтриметилсиланом, согласно процедуре примера 23 (h) или 11 (h), удаляла N-защищающую группу и давала желаемый 4-амино продукт.
Пример 27
/S/-2-фталимидо-6-гидроксигексановая кислота
Это промежуточное соединение примера 1 (с) получали также следующим образом.
К раствору диэтилового эфира 2-/ацетиламино/-2-/4-/ацетилокси/бутил/пропандионовой кислоты (730 г, 2,2 ммоля), полученной как описано в примере 24 (а), в абсолютном этаноле (300 мл) добавляли 6 N раствор гидроокиси натрия (1,6 л). Реакцию нагревали при 70 - 75oC в течение 5 часов, затем при 90 - 95oC для отгонки большей части этанола. Реакцию охлаждали и подкисляли до pH 1,3, используя 6 N соляную кислоту (около 1,3 л), затем нагревали при 90 - 100oC для достижения условий декарбоксилирования. По окончании неочищенную реакционную смесь охлаждали до комнатной температуры.
Вышеприведенную неочищенную реакционную смесь нагревали до 35oC и обрабатывали около 600 мл 6 N гидроокиси натрия с последующей обработкой 1 N раствором гидроокиси натрия для достижения pH 7,5 (общий объем был около 5,3 л). К этой смеси добавляли 0,6 г ацилазы 1 из свиной почки. После перемешивания при 35oC в течение ночи pH было 7,25. pH доводили до 7.5 и добавляли дополнительное количество ацилазы (300 мг). После перемешивания в течение ночи реакция заканчивалась на около 90%. Реакционную смесь затем обрабатывали 20 г угля и 20 г целита, затем нагревали до 85oC и поддерживали при этой температуре в течение 10 минут, затем охлаждали до 50oC и фильтровали. В этот момент полный объем фильтрата составлял около 4,9 л. Фильтрат охлаждали до 5oC и добавляли твердый карбонат натрия для доведения pH до 9,3. Добавляли N-карбетоксифталимид (263,04 г, 1,2 моля) в одну порцию и добавляли карбонат натрия, так как необходимо было сохранить pH при 9,3. Через 2 часа при 5oC с последующими 3 часами при комнатной температуре, pH падало до 8,5 и большая часть реагентов растворялась. Реакционную смесь фильтровали, охлаждали до 5oC и подкисляли до pH 2,3 6 N соляной кислотой. Выпавшее твердое вещество собирали фильтрованием и промывали 200 мл холодной воды, затем сушили в вакууме, получая 220 г указанного в названии продукта.
Пример 28
/4S-/4α, 7α, 10aβ//- Октагидро-4/ /2-меркапто-3/1-нафталенил/-1-оксопропил/ амино/-5-оксо-7H-пиридо [2,1-b]/1,3/ тиазепин-7-карбоновая кислота
а) Диэтиловый эфир/ацетиламино//1-нафталенилметил/-пропандионовой кислоты
К раствору этоксида натрия (21% в этаноле, 4,613 г, 67,8 ммоля) в этаноле (100 мл) добавляли диэтилацетамидомалонат (14,74 г, 67,8 ммоля), затем 1-бромметил/нафталин (10,0 г, 45,2 ммоля). Раствор перемешивали при комнатной температуре в течение одного часа. Реакционную смесь концентрирования до оранжевого масла. Масло растворяли в этилацетате и промывали 50% насыщенным раствором хлористого аммония, водой и рассолом, затем сушили над сульфатом натрия, фильтровали и концентрировали, получая оранжевое твердое вещество. Твердое вещество перекристаллизовывали из этилацетата и гексана, получая бежевые кристаллы, загрязненные диэтилацетамидомалонатом. Твердое вещество растворяли в 50% этилацетате в гексане и чистили флеш-хроматографированием на силикагеле (Мерк) в 50% этилацетате в гексане. Фракции, содержащие чистый продукт, объединяли и концентрировали, получая 10,225 г продукта в виде белого твердого вещества, т.пл. 105 - 108oC = 0,57 (50% этилацетат в гексане).
b) α- Амино-1-нафталинпропановая кислота
Раствор продукта из части (a) (16,182 г, 47,5 ммоля) суспендировали в 48% гидробромиде (100 мл) и кипятили с обратным холодильником под аргоном в течение 14 часов. Гидробромидную соль продукта отфильтровывали из раствора в виде белого твердого вещества, затем помещали в горячую воду (50oC) (500 мл) и раствор нейтрализовывали концентрированной гидроокисью аммония. Продукт осаждался из раствора в виде тонкодисперсного твердого вещества. При фильтровании в сушке в высоком вакууме в течение ночи (18 часов) получали 8,335 г продукта в виде пушистого белого твердого вещества, т.пл. 264oC.
c) α- Бром-1-нафталинпропановая кислота
К раствору продукта из части (b) (4,000 г, 18,6 ммоля) и бромида калия (7,63 г, 63,2 ммоля) в 2,5 N серной кислоте (35 мл), сохраняемому при 0oC, добавляли нитрит натрия (1,92 г, 27,8 ммоля) в течение 1 часа. Смесь дополнительно перемешивали в течение часа при 0oC, затем нагревали до комнатной температуры и перемешивали в течение 2,5 часов. Реакционную смесь затем экстрагировали эфиром (трижды). Эфирные слои объединяли и промывали водой и рассолом, затем сушили над сульфатом натрия, фильтровали и концентрировали, получая оранжевое масло. Масло чистили флеш-хроматографированием на Мерк силикагеле в 70% этилацетате в гексане с добавлением 1% уксусной кислоты для снижения хвоста. Эти фракции, содержащие бромиды, объединяли и концентрировали, получая слегка загрязненный продукт в виде оранжевого масла, которое становилось твердым при сохранении в течение ночи. Rf = 0,40 (40% этилацетат в гексане с 1% уксусной кислоты).
d) α- /Ацетилтио/-1-нафталинпропановая кислота
К шламу тиацетата калия (0,912 г, 8,00 ммоля) в ацетонитриле (300 мл) при 0oC добавляли продукт из части (c) (2,030 г, 7,27 ммоля) в виде раствора в ацетонитриле (3 мл). Раствор перемешивали в течение часа при 0oC, затем нагревали до комнатной температуры и перемешивали в течение 15 часов. Затем бромид калия отфильтровывали из реакционной смеси и фильтрат концентрировали с получением оранжевого масла. Масло растворяли в этилацетате и промывали 10% раствором бисульфата калия и рассолом, затем сушили над сульфатом натрия, фильтровали и концентрировали, получая оранжевое масло. Масло чистили флеш-хроматографированием на силикагеле Мерк в 50% этилацетате в гексане с добавлением 1% уксусной кислоты для снижения хвоста. Все фракции содержащие продукт были загрязнены соединением с Rf = 0,43.
Эти фракции отстаивали и концентрировали, получая оранжевое масло. Неочищенный продукт чистили через дициклогексиламиновую соль растворением оранжевого масла в эфире и добавлением эквивалентного количества дициклогексиламина (1,32 г, 7,27 ммоля) к раствору. Получали дициклогексиламиновую соль в виде двух типов коричневых кристаллов (1,450 г), еще слегка загрязненную примесями. Кристаллы суспендировали в этилацетате и трясли с раствором 10% бисульфата калия (трижды). Затем органический слой промывали водой и рассолом, затем сушили над сульфатом натрия, фильтровали и концентрировали, получая 875 мг продукта в виде желтого масла; Rf = 0,40 (40% этилацетата в гексане с 1% уксусной кислоты).
e) Метиловый эфир /4S-/4α, 7α, 10aβ//- октагидро-4-//2-/ацетилтио/-3-/1-нафталенил/-1-оксопропил/ амино/-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Раствор рацемической кислоты продукта из части (d) в метиленхлориде и раствор метилового эфира /4S-/4α, 7α, 10aβ//- //-октагидро-4-амино-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7- карбоновой кислоты в метиленхлориде реагировали в присутствии триэтиламина и бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфата согласно процедуре примера 23 (i), давая указанный в названии продукт.
f) /4S-/4α, 7α, 10aβ//- Октагидро-4-//2-меркапто-3-/1-нафталенил/-1-оксопропил/амино/-5-оксо- 7H-пиридо[2,10-b]/1,3/тиазепин-7-карбоновая кислота
Раствор продукта из части (e) в метаноле обрабатывали 1 N раствором гидроокиси натрия, согласно процедуре примера 23 (j), получая указанный в названии продукт.
Пример 29
/4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-/2-тиенил/пропил/ амино/-5-оксо-7H-пиридо [2,1-b]/1,3/-тиазепин-7-карборновая кислота
a) /S/ -α- Ацетилтио/-2-/тиофенилпропановая кислота
Хлорид калия (3,0 г, 40,1 ммоля) добавляли к раствору β- /2-тиенил/-D-аланина (1,37 г, 8,03 ммоля) в 2,5 N соляной кислоте (25 мл) при комнатной температуре под аргоном. После перемешивания в течение 10 минут полученную смесь охлаждали до 0oC и обрабатывали нитритом натрия (720 мг, 10,44 ммоля). Через 2,5 часа реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Смесь разделяли между водой и этилацетатом и органический слой сушили (сульфатом натрия), фильтровали и концентрировали. Остаток подвергали флеш-хроматографированию (Мерк силикагель), элюируя 1% уксусной кислотой в 3: 1 смеси гексан/этилацетат, получая 760 мг /R/ -α- хлор-2-тиофенилкарбоновую кислоту в виде желтого масла.
Тиоацетат цезия (2,95 г, 14,19 ммоля) добавляли к раствору, содержащему вышеприведенный хлорид (750 мг, 4,73 ммоля) в диметилформамиде (15 мл) при комнатной температуре под аргоном. После перемешивания в течение 2 часов реакционную смесь разделяли между 10% бисульфатом калия и этилацетатом. Органический слой промывали рассолом, сушили (сульфатом натрия), фильтровали и концентрировали и остаток подвергали флеш-хроматографированию (Мерк силикагель), элюируя 1% уксусной кислотой в смеси 4:1 гексан/этилацетат, получая 500 мг указанного в названии продукта в виде масла. TLC (2% уксусная кислота в смеси 3:1 этилацетат/гексан) Rf = 0,73.
b) Метиловый эфир /4S-/4α/R*/, 7α, 10aβ//- октагидро-4-//2-/ацетилтио/-1-оксо-3-/2-тиенил/пропил/амино/- 5-оксо-7H-пиридо[2,1-b] 1,3/тиазепин-7-карбоновой кислоты
Раствор кислоты продукта из части (a) в метиленхлориде метилового эфира /4S-/4α, 7α, 10aβ//- октагидро-4-амино-5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты в метиленхлориде взаимодействовали в присутствии триэтиламина и бензотриазол-1-илокситрис/диметиламино/-фосфонийгексафторфосфата согласно процедуре примера 23 (i), давая указанный в названии продукт.
c) /4S-/4α/R*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-/2-тиенил/пропил/амино -5-оксо-7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновая кислота
Раствор продукта из части (b) в метаноле обрабатывали 1 N раствором гидроокиси натрия согласно процедуре примера 23 (j), получая указанный в названии продукт.
Пример 30
/4S-/4α/S*/, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/ -5-оксо-7H-пиридо[2,1-b] /1,3/-тиазепин-7-карбоновая кислота
a) Дициклогексиламиновая соль /R/-2-/ацетилтио/бензолпропановая кислота
Следуя процедуре примера 1 (h), но заменяя L-фенилаланин на D - фенилаланин, получали дициклогексиламиновую соль /R/-2-/ацетилтио/бензопропановой кислоты.
b) Метиловый эфир /4S-/4α/S*/, 7α, 10aβ//-/ окстагидро-4-//2-/ацетилтио/-1-оксо -7H-пиридо[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Перемешиваемую суспензию дициклогексиламиновой соли /R/-2-/ацетилтио/бензопропановой кислоты (353,5 мг, 0,872 ммоля) в этилацетате (5 мл) промывали 5% раствором бисульфата калия (3 • 5 мл). Органические экстракты объединяли, промывали рассолом, сушили (сульфатом магния), фильтровали, концентрировали, сушили в вакууме и отгоняли дважды из гексана с получением /R/-2-/ацетилтио/бензопропановой кислоты в виде масла.
Полученную свободную кислоту (181, 4 мг, 0,809 ммоля) растворяли в метиленхлориде (2 мл) и перемешивали под азотом при 0oC. К этому раствору добавляли раствор метилового эфира /4S-/4α, 7α, 10aβ//- октагидро-4-амино-5-оксо-7H-пиридо[2,1-b]/1,3/тиазепин-7-карбоновой кислоты (200 мг, 0,774 ммоля) в метиленхлориде (6 мл), затем триэтиламин (0,113 мл, 0,813 ммоля) и, наконец, бензотриазол-1-илокситрис/диметиламино/фосфонийгексафторфосфат (360 мг, 0,813 ммоля). Реакцию перемешивали при 0oC и затем медленно позволяли нагреться до комнатной температуры. Через полных 20 часов реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате и раствор промывали 5% раствором бисульфата калия, насыщенным раствором бикарбоната натрия и рассолом. Органический слой сушили (сульфатом магния), фильтровали и концентрировали до желтого твердого вещества. Очистка флеш-хроматографированием (элюирование 2:3 смесью этилацетат:гексан) давала 261,7 мг указанного в названии продукта в виде прозрачного масла.
c) /4S-/4α/S*/, 7α, 10aβ//- октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-7H-пиридо [2,1-b] /1,3/-тиазепин-7-карбоновая кислота
Раствор продукта из части (b) (261,1 мг, 0,562 ммоля) в метаноле (6 мл, обескислороженном пробулькиванием азота) охлаждали до 0oC и обрабатывали 1 N раствором гидроокиси натрия (6 мл, обескислороженным пробулькиванием азота). После перемешивания в течение 1 часа при 0oC при непрерывном продувании азота реакцию нагревали до комнатной температуры. После перемешивания в течение 30 минут при комнатной температуре получали прозрачный раствор. Через 5,5 часов реакцию подкисляли до pH 1 5% раствором бисульфата калия и экстрагировали этилацетатом. Органические слои объединяли, промывали водой и рассолом, сушили (сульфатом натрия), фильтровали и концентрировали в вакууме. Очистка флеш-хроматографированием (6:0,01:3,99 этилацетат:уксусная кислота:гексан) давала 190 мг указанного в названии продукта в виде белого твердого вещества; [α]D= -87,5o (с = 0,51, хлороформ). TLC (6:0,01:3,99, этилацетат: уксусная кислота:гексан) Rf = 0,20.
HPLC: tR = 25,3 мин; YМС S-3 ODS (C-18) 6,0 х 150 мм, 0% до 100% B: A, 30 мин линейный градиент и 10 минут выдерживали, 1,5 мл/мин: A = 90% вода: метанол + 0,2% фосфорной кислоты, B = 90% метанол:вода + 0,2% фосфорной кислоты, 220 нм.
Данные анализа для C19H24O4N2S2 • 0,15 C4H8O2 • 0,15 C7H16 • 0,39 H2O:
Вычислено, %: С 55,89; Н 6,45; N 6,31; S 14,45
Найдено, %: С 56,19; Н 6,50; N 6,71; S 13,96.
Пример 31
/4S-/4α, 7α, 10aβ//- Октагидро-4-//2-меркапто-1-оксо-3-/4-тиазолил/пропил/амино/- 5-оксопирроло [2,1-b]/1,3/ тиазепин-7-карбоновая кислота
a) /R/-2-амино-3-/4-тиазолил/пропановая кислота
Раствор 4N соляной кислоты в диоксане (10 мл) добавляли к раствору /R/-2-//1,1-диметилэтокси/карбонил/амино-3-/4-тиазолил/пропановой кислоты (2,0 г, 7,3 ммоля) в диоксане (2 мл. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, концентрировали в вакууме и остаток растворяли в воде (3 мл). pH доводили до 6,5 1N раствором гидроокиси натрия и этот раствор пропускали через 20 мл смолы Дауэкс®AG 50 (H+). Колонку элюировали водой (250 мл) с последующим элюированием 2% пиридином в воде (300 мл). Фракции, содержащие продукт, концентрировали в вакууме, получая 0,94 г указанного в названии продукта.
b) /R/-2-Бром-3-/4-тиазолил/пропановая кислота
Раствор продукта из части (a) (0,516 г, 3 ммоля) и бромида калия (1,19 г, 10,1 ммоля) в воде (5,94 мл) и серной кислоты (0,43 мл) перемешивали при -10oC в течение 10 минут с последующим добавлением порциями нитрита натрия (0,318 г, 4,61 ммоля) в течение 10 минут. Реакционную смесь дополнительно перемешивали в течение 10 минут при 0oC и при комнатной температуре в течение одного часа и затем экстрагировали эфиром (3 х 100 мл). Эфирные экстракты промывали рассолом (2 х 20 мл), сушили (сульфатом натрия), фильтровали и концентрировали в вакууме, получая 0,37 г указанного в названии продукта, [α]D= + 37,35o (c = 0,7,метанол). Второй опыт проводили исходя из 2,67 ммолей продукта из части (a), используя ту же самую процедуру, и получали дополнительно 0,35 г указанного в названии продукта.
c) /S/-2-/Ацетилтио/-3-/4-тиазолил/пропановая кислота
Продукт из части (b) (0,72 г, 3,05 ммоля) и тиоацетат калия (0,35 г, 3,05 ммоля) перемешивали в ацетонитриле (9 мл) в течение ночи при комнатной температуре и при 30oC в течение одного часа. Реакционную смесь разбавляли этилацетатом (100 мл) и фильтровали. Фильтрат концентрировали в вакууме. Остаток вновь растворяли в этилацетате (100 мл), промывали водой (2 х 50 мл) и рассолом (20 мл), сушили (сульфатом натрия), фильтровали и концентрировали в вакууме, получая 0,52 г указанного в названии продукта; [α]D= - 15,89o (c = 0,6: метанол).
d) Метиловый эфир /4S-/4α, 7α, 9aβ//- октагидро- 4-//2-/ацетилтио/- 1-оксо-3-/4-тиазолил/ пропил/амино/-5- оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты
Соль метилового эфира /4S-/4α, 7α, 9aβ//- 4-аминооктагидро-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновой кислоты и п-толуолсульфоновой кислоты (0,367 г, 0,882 ммоля), полученная из вещества, описанного в примере 5 (d), растворяли в метиленхлориде (5 мл) 0oC с последующим добавлением триэтиламина (0,12 мл, 0,868 ммоля). Продукт из части (c) (0,2 г, 0,865 ммоля) добавляли к этому раствору с последующим добавлением второй порции триэтиламина (0,12 мл, 0,865 ммоля). Добавляли бензотриазол-1-илокситрис-/диметиламино/фосфонийгексафторофосфат (0,383 г, 0,865 ммоля) и реакционную смесь перемешивали при 0oC в течение одного часа и при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали в вакууме и остаток растворяли в этилацетате (60 мл). Органический экстракт промывали 5% водным раствором бисульфата калия (10 мл) и рассолом (2 х 10 мл), сушили (сульфатом натрия), фильтровали и концентрировали в вакууме. Это неочищенное вещество хроматографировали на силикагеле Мерк 100 г используя 0,2% метанола в этилацетате. Фракции, обогащенные медленновращающим изомером, концентрировали в вакууме, получая 0,134 г указанного в названии продукта.
e) /4S-/4α, 7α, 9aβ//- Октагидро-4-//2-меркапто-1-оксо-3-/4-тиазолил/пропил/амино/-5-оксопирроло [2,1-b]/1,3/-тиазепин-7-карбоновая кислота
Продукт из части (d) (0,135 г, 0,29 ммоля) растворяли в метаноле и в раствор пробулькивали аргон в течение 30 минут при 0oC. 1N раствор гидроокиси натрия (1,32 мл), также продутый аргоном, добавляли к упомянутому выше раствору и реакционную смесь перемешивали при 0oC с пробулькиванием аргона через раствор в течение 1 часа и при комнатной температуре в течение 2 часов. Реакцию гасили добавлением 5% водного раствора бисульфата калия (20 мл) и органические соли экстрагировали этилацетатом (3 х 50 мл). Этилацетатный раствор промывали рассолом, сушили (сульфатом натрия), фильтровали и концентрировали в вакууме. Концентрат хроматографировали на силикагеле Мерк 40 г, используя хлороформ, содержащий 5% метанола и 0,5% уксусной кислоты. Соответствующие фракции объединяли, концентрировали и разделяли между 20 мл этилацетата и 5% водным раствором бисульфата калия. Этилацетатный раствор промывали водой и рассолом, сушили (сульфатом натрия) и концентрировали в вакууме. Остаток лиофилизовали из диоксана (4 мл), получая 36 мг указанного в названии продукта в виде 70:30 смеси изомеров, т.пл. 95 - 115oC; [α]D= -191,7o (c = 0,06, хлороформ) TLC (хлороформ: метанол:уксусная кислота, 8:2: 0,2) Rf = 0,59.
HPLC: tR = 3,06 мин, YМС S-3 ODS (C-18) 6,0 х 150 мм, 3 мк колонка с заглушенным концом, изократич. 60% водный метанол, содержащий 0,2% фосфорной кислоты, 25 мин, 1,5 мл/мин (95,4%).
Данные анализа для C15H19N3O4S3 • 0,2 C4H8O2 • 0,9 H2O:
Вычислено, %: С 43,59; Н 5,19; N 9,65; S 22,09
Найдено, %: С 43,54; Н 4,89; N 9,44; S 21,90.
Пример 32
1000 таблеток, каждая из которых содержит следующие ингредиенты:
/4S-/4α/R*/, 7α, 10aβ//- октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-7H-пиридо [2,1-b]/1,3/тиазепин-7- карбоновая кислота - 100 мг
Кукурузный крахмал - 100 мг
Желатин - 20 мг
Авицел (микрокристаллическая целлюлоза) - 50 мг
Стеарат магния - 5 мг - 275 мг
получают из достаточного количества массы смешением продукта примера 3 и кукурузного крахмала с водным раствором желатины. Смесь сушат и измельчают до тонкодисперсного порошка. Авицел и затем стеарат магния смешивают с гранулированием. Затем смесь прессуют в прессе для таблеток с образованием 1000 таблеток, каждая содержит 100 мг активного ингредиента.
Аналогичным способом могут быть получены таблетки, содержащие 200 мг продукта примеров 1, 2, 4 - 23, 25 и 28 - 31.
Подобная процедура может быть использована для получения таблеток или капсул, содержащих от 10 мг до 500 мг активного ингредиента.
Данные и методика биологических испытаний
Активность in vitro соединений по примерам определяли следующими методами.
Ингибирующую активность фермента превращения ангиотензина ((ACE) определяли по методу Cushman et al., Biochem. Pharmacol., vol. 20, p. 1637-1648 (1971).
Экстракт легкого кролика использовали как источник ACE и прослеживали скорость образования гиппуриновой кислоты из субстрата гиппурил-L-гистидил-L-лейцина (ГГЛ). После 30 минут инкубации испытуемого соединения с ферментом и субстратом реакцию прекращали добавлением HCl. Добавляли этилацетат. Этилацетатный слой отделяли и выпаривали до сухого остатка; образовавшуюся гиппуровую кислоту восстанавливали в воде и определяли ее концентрацию в испытуемом соединении, замедляющую реакцию на 50% (IC50).
Активность почечной нейтральной эндопептидазы (ПНЭП) определяли относительно очищенной нейтральной эндопептидазы почек крыс, используя метод флуорометрического анализа. Анализ основан на разложении флуоресцирующего дансилат трипептида Dns-Gly-Phe-Arg с образованием Dns-Gly; последний экстрагировали из подкисленной смеси этилацетатом и он сильно флуоресцировал. Испытуемое соединение предварительно инкубировали в течение 10 минут с препаратом фермента перед тем как инициировать реакцию добавлением субстрата. После 30 минут инкубации реакцию прерывали добавлением HCl. Dns-Gly экстрагировали этилацетатом. Относительную интенсивность флуоресценции измеряли с использованием спектрометра. Определяли концентрацию испытуемого соединения, вызывающую замедление реакции на 50% (IC50). Результаты испытаний представлены в таблице.
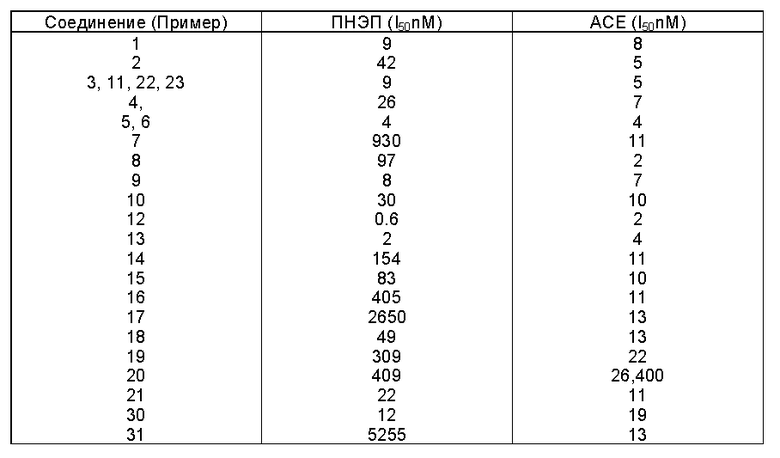
Соединения, содержащие конденсированное бициклическое ядро формулы I, где A представляет собой остаток формулы (а) или (б), X представляет O или S; R1 и R12 - водород, C1-5-алкил, фенил-(низший)алкелен, нафтил-(низший)алкилен, C3-7-циклоалкил-(низший)алкилен, тиенил-(низший)алкилен, или R1 и R12 взятые вместе с атомом углерода, к которому они присоединяются, замыкают C5 - C6 -циклоалкильное кольцо; R2 - водород или C = O, R3 и R7 - водород или низший алкил; R6 - низший алкил; m и r = 0 или 1; Y - CH2 или O; n = 1 или 2, или их фармацевтически приемлемые соли являются ингибиторами двойного действия, обладающими способностью ингибировать превращение фермента ангиотензина и нейтральной эндопептидазы.
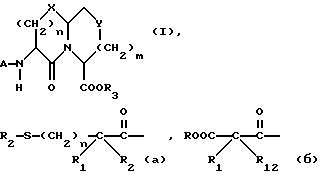
9 с. и 4 з.п. ф-лы, 1 табл.
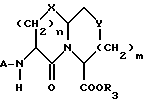
или их фармацевтически приемлемая соль,
где A представляет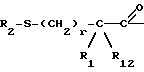
или
X представляет O или S;
R1 и R12 независимо выбраны из группы, состоящей из водорода, C1 - C5-алкила с прямой или разветвленной цепью, фенил-(низшего)алкилена, нафтил-(низшего)алкилена, C3 - C7-циклоалкил-(низшего)алкилена, тиенил-(низшего)алкилена, или R1 и R12 взятые вместе с атомом углерода, к которому они присоединяются, замыкают C5 - C6-циклоалкильное кольцо;
R2 представляет водород или 
R3 и R7 независимо выбраны из группы, состоящей из водорода или низшего алкила;
R6 представляет низший алкил;
m равно 0 или 1;
Y представляет CH2 или 0, при условии, что Y представляет 0 только, если m равно 1;
n равно 1 или 2;
r равно 0 или 1.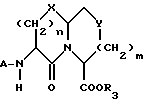
где A представляет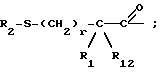
R2 представляет водород или 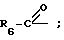
R3 представляет водород или низший алкил с 1 - 4 атомами углерода;
r равно 0 или 1;
R1 - C1 - C5-алкил с прямой или разветвленной цепью, фенил - (CH2)-, нафтил - (CH2)-, тиенил - (CH2)-, (C3 - C7)-циклоалкил - (CH2)-;
R12 - водород или R1 и R12 взятые вместе с атомом углерода, к которому они присоединяются, замыкают C5 - C6-циклоалкильное кольцо;
R6 представляет низший алкил с 1 - 4 атомами углерода;
n равно 1 или 2;
m равно 0 или 1;
X представляет O или S;
Y представляет O или CH2 при условии, что Y представляет O только, если m равно 1. R3 представляет водород; r равно 0 или 1; R1 - C3 - C5-алкил с прямой или разветвленной цепью; R12 представляет водород; n равно 1 или 2; m равно 0 или 1; X представляет O или S; Y представляет O или CH2, при условии, что Y представляет O только, если m равно 1.
R3 представляет водород; r равно 0 или 1; R1 - C3 - C5-алкил с прямой или разветвленной цепью; R12 представляет водород; n равно 1 или 2; m равно 0 или 1; X представляет O или S; Y представляет O или CH2, при условии, что Y представляет O только, если m равно 1.
1,1-диметилэтиламиновая соль /4S-/4α/R*/,7α,10aβ//-октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-7Н-пиридо[2,1-b] /1,3/тиазепин-7-карбоновую кислоту;
/4S-/4α/S*/,7α,10aβ//-октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-7Н-пиридо[2,1-b] /1,3/-тиазепин-7-карбоновую кислоту;
/4S-/4α/R*/,7α,10aβ//-октагидро-4-//2-меркаптометил/-1-оксо-3-фенилпропил/амино/-5-оксо-7Н-пиридо[2,1-b] /1,3/тиазепин-7-карбоновую кислоту;
/4S-/4α/R*/,7α,10aβ//-октагидро-4-//2-меркапто-4-метил-1-оксопентил/-амино/-5-оксо-7Н-пиридо[2,1-b]/1,3/тиазепин-7-карбоновую кислоту;
/3R-/3α/S*/,6α,9aβ//-гексагидро-3-//2-меркапто-1-оксо-3-фенилпропил/амино/-4-оксо-2Н,6Н-пиридо[2,1-b]/1,3/тиазепин-6-карбоновую кислоту;
/4S-/4α/R*/,7α,9aβ//-октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксопирроло[2,1-b] /1,3/-тиазепин-7-карбоновую кислоту;
/4S-/4α/R*/,7α,9aβ//-октагидро-4-//3-циклопропил-2-меркапто-1-оксопропил/амино/-5-оксопирроло[2,1-b] /1,3/-тиазепин-7-карбоновую кислоту;
/4S-/4α/R*/,7α,9aβ//-октагидро-4-//2-меркапто-1-оксогексил/амино/-5-оксопирроло[2,1-b] /1,3/-тиазепин-7-карбоновую кислоту;
/4S-/4α/R*/,7α,9aβ//-октагидро-4-//2-меркапто-1-оксо-4-метилпентил/амино/-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновую кислоту;
/4S-/4α/R*/,7α,9aβ//-октагидро-4-//2-меркапто-1-оксопентил/амино/-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновую кислоту;
/4S-/4α/R*/,7α,9aβ//-октагидро-4-//2-меркапто-4,4-диметил-1-оксопентил/амино-5-оксопирроло[2,1-b] /1,3/тиазепин-7-карбоновую кислоту;
/4S-/4α/R*/,7α,10aβ//-октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-7Н-пиридо[2,1-b] /1,3/-оксазепин-7-карбоновую кислоту;
/4S-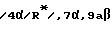 //-октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино-5-оксопирроло[2,1-b] /1,3/оксазепин-7-карбоновую кислоту;
//-октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино-5-оксопирроло[2,1-b] /1,3/оксазепин-7-карбоновую кислоту;
/4S-/4α/R*/,7α,10aβ//-октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-/1,4/оксазино[3,4-b] /1,3/оксазепин-7-карбоновую кислоту, или
/4S-/4α/R*/,7α,10aβ//-октагидро-4-//2-меркапто-1-оксо-3-фенилпропил/амино/-5-оксо-/1,4/оксазино[3,4-b] /1,3/ тиазепин-7-карбоновую кислоту.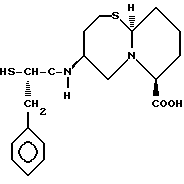
или его фармацевтически приемлемая соль.
или его фармацевтически приемлемой соли,
где A, X, Y, n, m и R3 имеют значения, как они определены по п.1.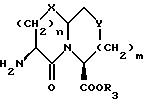
или его фармацевтически приемлемая соль,
где X представляет O или S,
n равно 1 или 2;
m равно 0 или 1;
Y представляет CH2 или O, при условии, что
Y представляет O, только если m равно 1;
R3 представляет водород или низший алкил, при условии, что когда X представляет O, n равно 1, Y - группа CH2, то m должно быть равно 1.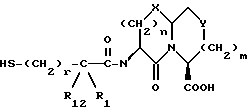
отличающийся тем, что включает
a) реакцию сочетания ацилмеркаптогруппы боковой цепи формулы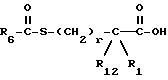
или ее активированной формы с амином формулы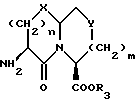
в присутствии реагента сочетания,
где X, Y, m, n, R6, r, R1 и R12 определены в п.1;
R3 представляет низший алкил, защищающий кислотную группу,
с получением соединения формулы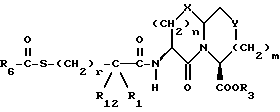
b) обработку продукта со стадии a) для удаления ацильной группы  и кислотнозащитной группы R3 с получением целевого продукта, который необязательно превращают в его фармацевтически приемлемую соль.
и кислотнозащитной группы R3 с получением целевого продукта, который необязательно превращают в его фармацевтически приемлемую соль.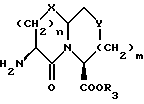
где X представляет O или S;
n равно 1 или 2;
n равно 0 или 1;
Y представляет CH2 или O, при условии, что Y представляет O, только если m равно 1;
R3 представляет кислотную защищающую группу,
отличающийся тем, что включает:
a) сочетание аминокислоты формулы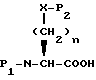
с эфиром аминокислоты формулы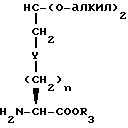
с образованием дипептида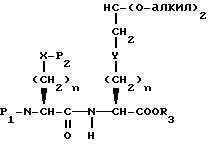
где P1 представляет аминозащитную группу или группу, которая вместе с N-атомом образует защищающую группу;
P2 представляет гидрокси- или меркаптозащищающую группу;
b) селективное удаление P2 защищающей группы из продукта части (a);
c) циклизацию продукта части (b) с получением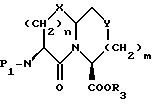
d) удаление защитной группы P1 продукта части (c) с получением желаемого продукта.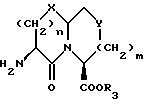
где X представляет S;
n равно 1 или 2;
Y представляет CH2;
m равно 0 или 1,
отличающийся тем, что включает:
a) сочетание аминокислоты формулы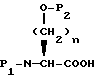
c эфиром аминокислоты формулы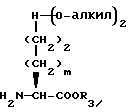
с образованием дипептида формулы
где P1 представляет аминозащищающую группу или группу, которая вместе с N-атомом образует защищающую группу;
P2 представляет гидрокси защищающую группу;
b) селективное удаление P2 защитной группы из продукта части (a) с образованием соответствующего гидроксисоединения;
c) превращение гидрокси продукта из части (b) в меркаптан формулы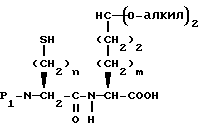
d) циклизацию меркаптанового продукта части (с),
с образованием соединения формулы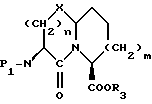
e) удаление P1 защитной группы из продукта части (d) с образованием желаемого продукта.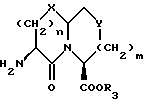
где X представляет S;
n равно 2;
Y представляет O; и
m равно 1;
отличающийся тем, что включает:
a) сочетание аминокислоты формулы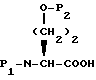
с эфиром аминокислоты формулы
с образованием дипептида формулы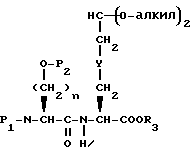
где P1 представляет аминозащитную группу или группу, которая вместе с N-атомом образует защищающую группу;
P2 представляет гидроксизащитную группу;
b) селективное удаление P2 защитной группы из продукта части (a) с образованием соответствующего гидрокси соединения;
c) превращение гидрокси продукта из части (b) в меркаптан формулы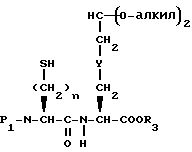
d) циклизацию меркаптанового продукта части (c),
с образованием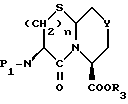
e) удаление защитной группы P1 из продукта части (d), с образованием желаемого продукта.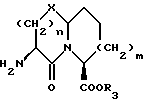
где X представляет O или S;
n равно 1 или 2;
m равно 0 или 1;
R3 представляет кислотную защитную группу,
отличающийся тем, что включает:
a) сочетание аминокислоты формулы
с эфиром гидроксиаминокислоты формулы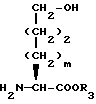
с образованием дипептида формулы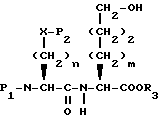
где P1 представляет аминозащитную группу или группу, которая вместе с N-атомом образует защитную группу;
P2 представляет гидрокси или меркаптозащитную группу:
b) окисление гидрокси продукта части (a) до альдегида формулы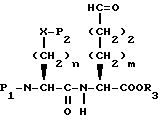
c) селективное удаление P2 защитной группы из альдегидного продукта части (b);
d) циклизацию продукта части (c),
с образованием соединения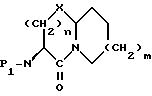
e) удаление защитной группы P1 продукта части (d),
с образованием желаемого продукта.
| Способ получения карбоксиалкенамидоцефалоспоринов, или их сложных эфиров, или их солей с щелочными металлами | 1984 |
|
SU1500163A3 |
| Способ получения производных 2-тиацефемов | 1984 |
|
SU1340591A3 |
| Способ получения производных 1-детиа-2-тиацефалоспорановой кислоты или их солей с йодистоводородной или фармацевтически приемлемой органической кислотой | 1986 |
|
SU1648252A3 |
| SU 1287756 A3, 1987 | |||
| Способ приготовления шампанского | 1939 |
|
SU61187A1 |
| Радиатор для насосно-водяных систем центрального отопления | 1936 |
|
SU50671A1 |
| ЛЕНТОПРОТЯЖНЫЙ МЕХАНИЗМ С ПРЕРЫВИСТЫМ ПЕРЕМЕЩЕНИЕМ НОСИТЕЛЯ | 0 |
|
SU272594A1 |
| US 4880804 A, 1989. | |||

