Область изобретения
Настоящее изобретение относится к кристаллическим формам 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида, которые обладают гипогликемической активностью или эффектом ингибирования PDE5, и к способу их получения.
Уровень техники
Когда соединение, содержащее кристаллические полиморфные формы, используют в качестве лекарственного средства, часто оказывается необходимым получить лекарственное вещество, имеющее конкретную кристаллическую форму, чтобы гарантировать постоянство физико-химических и биологических свойств соединения. Кроме того, в способе получения лекарственного вещества часто бывает важно выделить конкретную форму кристаллов в процессе кристаллизации для того, чтобы поддерживать определенные уровни выхода и эффективности очистки.
3-(2,4-Дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамид был раскрыт в WO 97/24334 как соединение бензимидазола, обладающее гипогликемической активностью или эффектом ингибирования PDE5 (см. пример 251). Однако существование кристаллических полиморфных форм этого соединения до сих пор не было установлено, и не были получены по существу и кристаллографически чистые кристаллы этого соединения, обладающие определенной формой.
Описание изобретения
Целью настоящего изобретения является обеспечение по существу и кристаллографически чистой кристаллической формы 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида, которая полезна в качестве медицинского средства, способа ее получения и лекарственной композиции, ее содержащей.
Авторы настоящего изобретения изучали различные условия кристаллизации -(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида. В результате они обнаружили три формы кристаллов этого соединения. Здесь эти три кристаллические формы называются кристаллическими формами А, В и С. Авторы обнаружили также, что кристаллические формы А и В обладают преимуществами по сравнению с другими кристаллическими формами соответственно. Так, кристаллическая форма А является кристаллографически более стабильной нежели кристаллические формы В и С, хотя кристаллы этой формы отличаются меньшими размерами. Поэтому ее легче получать в виде по существу и кристаллографически чистых кристаллов, что благоприятно для сохранения качества фармацевтического препарата как лекарственного средства. С другой стороны, кристаллическая форма В, хотя и не столь кристаллографически стабильная, как кристаллическая форма А, образует более крупные кристаллы, чем кристаллическая форма А, так что ее можно значительно легче выделить фильтрованием и эффективно очистить с помощью кристаллизации.
Кроме того, авторы настоящего изобретения обнаружили, что каждую кристаллическую форму можно получить в по существу чистой кристаллографической форме и промышленно стабильным способом, используя метод кристаллизации, предпочтительный для соответствующей формы, и тем самым осуществили настоящее изобретение.
Таким образом, настоящее изобретение относится к (1) по существу и кристаллографически чистой кристаллической форме 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида, имеющей следующие значения порошковой дифракции рентгеновских лучей (2θ), с ошибкой в интервале ±0,2, в порошковом рентгеноструктурном анализе с использованием СuKα-излучения в качестве характеристического рентгеновского излучения: угол 2θ (°): около 4,7, около 9,5, около 10,5, около 15,6 и около 18,4, и
(2) по существу и кристаллографически чистой кристаллической форме 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида, имеющей следующие значения порошковой дифракции рентгеновских лучей (2θ), с ошибкой в интервале ±0,2, в порошковом рентгеноструктурном анализе с использованием СuKα-излучения в качестве характеристического рентгеновского излучения: угол 2θ (°): около 4,4, около 8,9 и около 13,4.
Настоящее изобретение относится также к фармацевтической композиции, содержащей в качестве активного ингредиента кристаллическую форму 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида, определенную в (1). Настоящее изобретение относится также к фармацевтической композиции, содержащей в качестве активного ингредиента кристаллическую форму 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида, определенную в (2).
Настоящее изобретение относится далее к способу получения кристаллической формы (1), определенной выше, который включает кристаллизацию 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида из органического растворителя или смеси органического растворителя с водой. Настоящее изобретение относится также к способу получения кристаллической формы (2), определенной выше, который включает кристаллизацию 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида путем добавления кислоты к раствору 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида в органическом растворителе или в смеси органического растворителя с водой в присутствии щелочи.
Как используется в описании, термин “по существу и кристаллографически чистый” означает, что другие кристаллические формы аналитически не определяются. В этом контексте анализ относится к, по крайней мере, одному из анализов: порошковая дифракция рентгеновских лучей, инфракрасная спектрометрия (ИК) и термогравиметрический/дифференциальный термический анализ (TG/DTA), которые раскрыты далее.
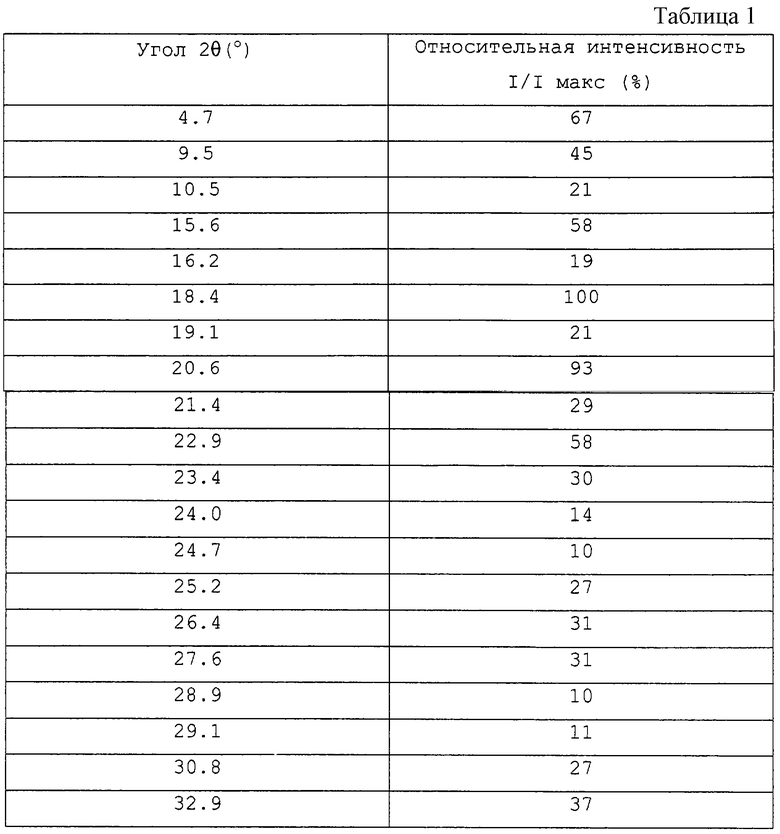
Кристаллическая форма А 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида, часто обозначаемая далее как соединение (I), характеризуется как имеющая следующие величины порошковой дифракции рентгеновских лучей (2θ), с ошибкой в интервале ±0,2, в порошковом рентгеноструктурном анализе с использованием СuKα-излучения в качестве характеристического рентгеновского излучения: угол 2θ (°): около 4,7, около 9,5, около 10,5, около 15,6 и около 18,4.
Более конкретно, например, кристаллическая форма А демонстрирует дифракционные значения, приведенные в таблице 1.
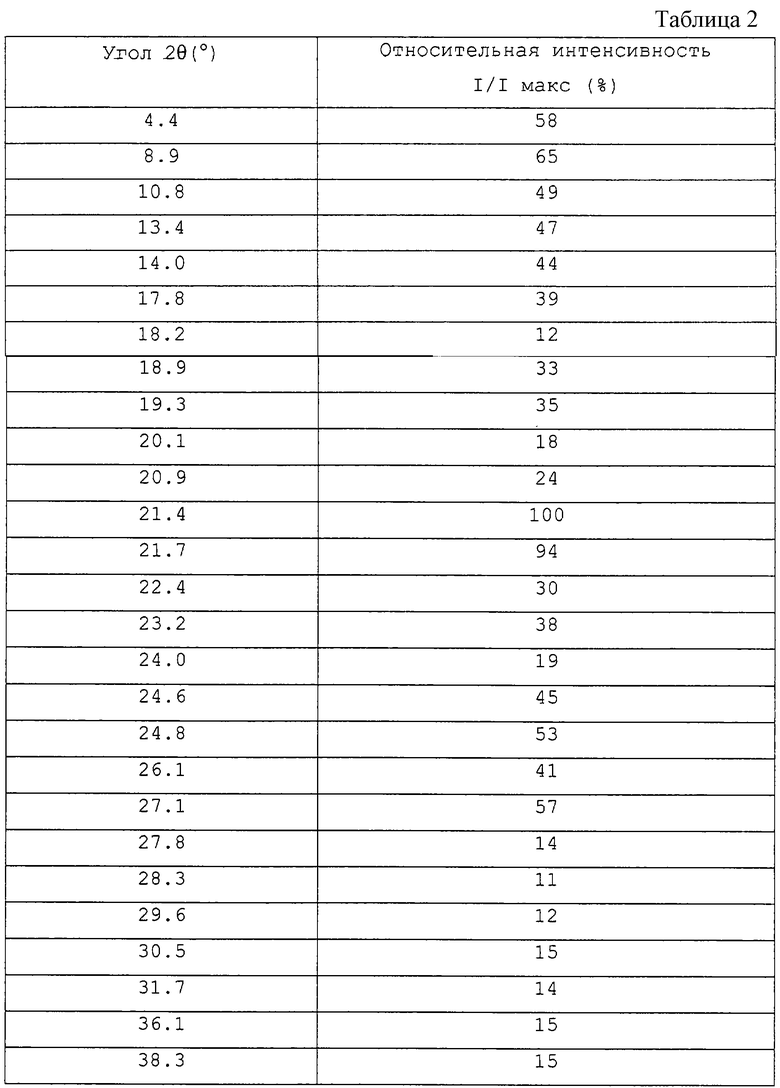
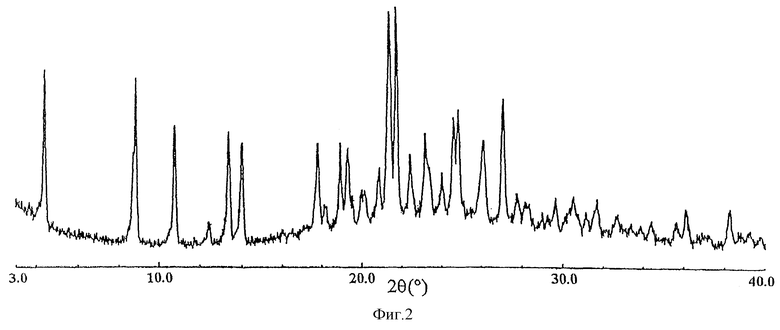
Кристаллическая форма В соединения (I) характеризуется как имеющая следующие значения порошковой дифракции рентгеновских лучей 2θ, с ошибкой в интервале ±0,2, в порошковом рентгеноструктурном анализе с использованием СuKα-излучения в качестве характеристического рентгеновского излучения: угол 2θ (°): около 4,4, около 8,9 и около 13,4.
Более конкретно, например, кристаллическая форма В демонстрирует дифракционные значения, приведенные в таблице 2.
Кроме того, кристаллическая форма С соединения (I) имеет следующие значения порошковой дифракции рентгеновских лучей 2θ, с ошибкой в интервале ±0/2, в порошковом рентгеноструктурном анализе с использованием СuKα-излучения в качестве характеристического рентгеновского излучения, приведенные в таблице 3.

Значения порошковой дифракции рентгеновских лучей (2θ), указанные выше, определяют, используя следующий прибор и условия:
Прибор: Rigaku RINT-1500 (Rigaku Denki Kogyo Inc.);
Характеристическое рентгеновское излучение: CuKα-излучение (с использованием монохроматора);
Соотношение электрический ток/напряжение в электронной трубке: 40 кV/30 mA;
Детектор: пропорциональный счетчик;
Скорость сканирования: 2θ=3-40° и
Щелевая система: отклоняющая щель 1°, рассеивающая щель 1°, щель приемника 0,3 мм.
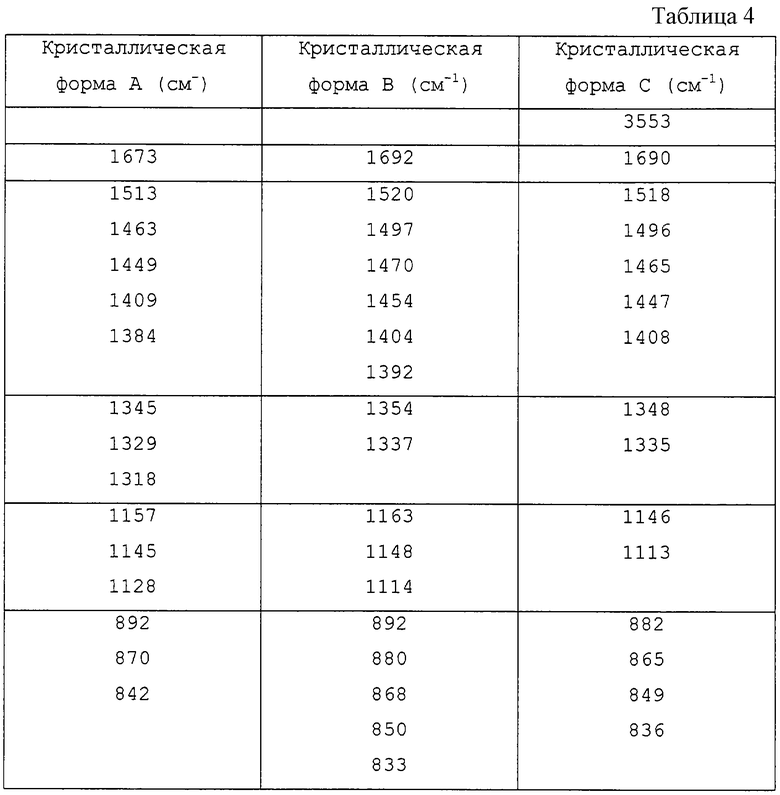
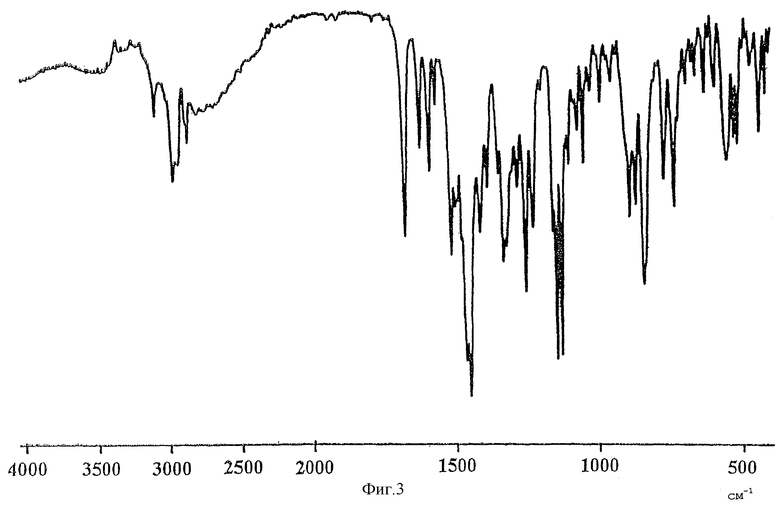
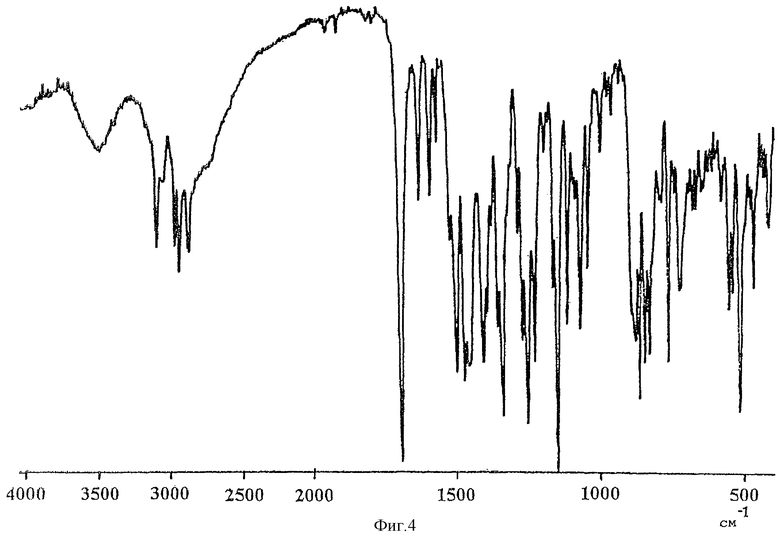
Кристаллические формы А, В и С соединения (I) можно также различить с помощью ИК-спектров. Значительно отличающиеся пики в картине поглощения для каждой кристаллической формы в ИК-спектрах (КВr), определенные с помощью инфракрасного спектрофотометрического анализа (метод с использованием таблеток КВr), представлены в таблице 4.
Представленные выше ИК-спектры получают с использованием следующего прибора и условий:
Прибор: PERKIN ELMER 1650 FT-1R (Perkin-Elmer, Japan);
Способ измерения: метод с использованием таблеток КВr и
Таблетки с диаметром 3 мм.
Данные ИК-спектров для каждой кристаллической формы, определенные методом Nujol, а также данные для соединения (I), полученного и очищенного обычным способом (WO 97/24334), представлены в таблице 5.

Кроме того, кристаллические формы А, В и С соединения (I) можно различить с помощью термогравиметрического/дифференциального термического анализа (TG/DTA), как раскрыто далее.
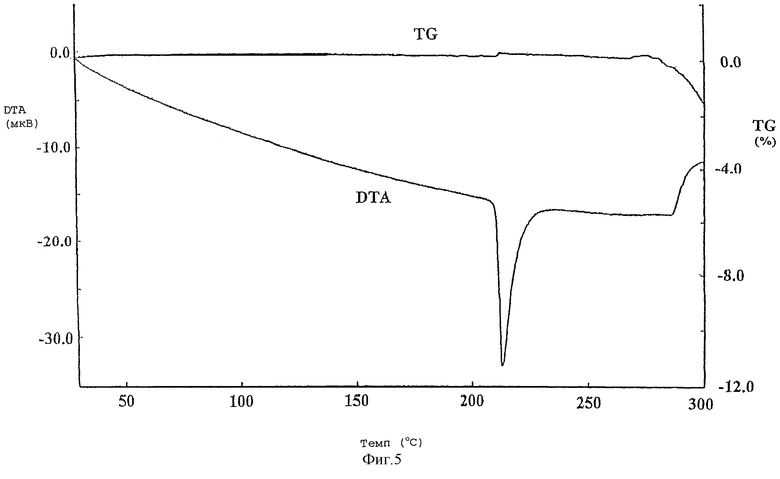
Кристаллическая форма А: максимум эндотермы плавления наблюдается при экстраполяции температуры инициирования примерно 211°С.
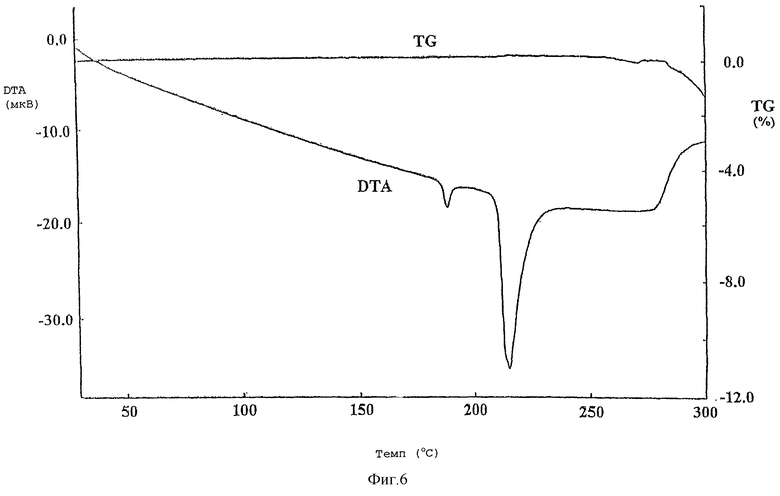
Кристаллическая форма В: максимум кратковременной эндотермы наблюдается при экстраполяции температуры инициирования примерно 186°С и максимум эндотермы плавления наблюдается при экстраполяции температуры инициирования примерно 211°С.
Кристаллическая форма С: максимум эндотермы плавления наблюдается при экстраполяции температуры инициирования примерно 102°С и последующий максимум экзотермы, максимум эндотермы плавления при экстраполяции температуры инициирования примерно 211°С, и 1-2% потери веса около первого максимума эндотермы плавления.
Вышеуказанные результаты термогравиметрического/дифференциального термического анализа (TG/DTA) получают, используя следующий прибор и условия:
Прибор: SII TG/DTA 6300 (Seiko Instruments Inc.);
Температурные условия: 30°С (0 мин) → 10°С, мин → 350°С;
Контейнер для образца: А1 запаянный контейнер;
Атмосфера: N2, 300 мл/мин;
Время отбора: 0,5 сек.
По существу и кристаллографически чистую кристаллическую форму А соединения (I) можно получить стабильным способом, растворяя соединение (I) в органическом растворителе или в смеси органического растворителя и воды, нагревая раствор, и затем кристаллизуя этот раствор в процессе нагревания. В качестве растворителя предпочтительно использовать смесь воды и органического растворителя, включая, но не ограничиваясь ими, амиды (например, N,N-диметилформамид, N,N-диметилацетамид и т.д.), спирты (например, метанол, этанол, 1-пропанол, 2-пропанол и т.д.) и кетоны (например, ацетон, метилэтилкетон и т.д.). Один или более из этих растворителей можно объединить в смешанный растворитель. Среди смесей водный/органический растворитель, наиболее предпочтителен смешанный растворитель состоящий из кетона и воды, и смешанный растворитель, состоящий из ацетона и воды, еще более предпочтителен.
Так как растворимость меняется в зависимости от типа и состава используемого растворителя, не существует конкретных ограничений на соотношение органического растворителя и воды. При растворении соединения (I) более высокое отношение органического растворителя к воде является преимуществом, обеспечивая повышенную растворимость; отношение органического растворителя к воде предпочтительно составляет от 100:1 до 50:50 (мас/мас) и более предпочтительно от 100:0 до 70:30 (мас/мас). После завершения кристаллизации соединения (I) выгодно уменьшать количество органического растворителя соответствующим образом для получения достаточного выхода; отношение органического растворителя к воде предпочтительно снижают от 95:5 до 5:95 (мас/мас) и более предпочтительно от 90:10 до 30:70 (мас/мас).
Для осаждения кристаллов во время нагревания слабый растворитель, такой как вода, добавляют к раствору соединения (I) в вышеуказанном растворителе во время нагревания; альтернативно органический растворитель выпаривают. Кроме того, если соединение (I) поддерживают при высокой температуре, осадить кристаллы можно с помощью охлаждения.
В любом из описанных выше способов для получения кристаллической формы А желательно инициировать кристаллизацию при 30°С или выше. Не существует определенного верхнего предела температуры инициирования, так как растворимость меняется в зависимости от типа или состава используемого растворителя. Температура инициирования может находиться между 30°С и температурой кипения используемого растворителя, и ниже растворимости кристаллической формы А. Предпочтительно кристаллизацию инициируют при температуре выше 40°С для получения более стабильной кристаллической формы А. В контексте настоящего изобретения термин “инициирование кристаллизации” относится ко времени, когда любые кристаллы начинают осаждаться, если не добавляют затравочные кристаллы, или ко времени, когда любые кристаллы, отличные от затравочных кристаллов, начинают осаждаться в случае, если добавляют затравочные кристаллы.
Кристаллическую форму А соединения (I) можно также получить, поддерживая суспензию, содержащую соединение (I), в любой кристаллической форме или в аморфной форме, или в их смеси, в растворителе при нагревании, чтобы вызвать переход кристаллической формы в суспендированное состояние.
В этом случае температура нагревания не ограничивается до определенного интервала, до тех пор, пока она обеспечивает такой переход; однако желательно сохранять температуру 30°С или выше для получения кристаллической формы А стабильным образом. И снова, не существует определенного верхнего предела удерживания температуры, так как растворимость меняется в зависимости от типа или состава используемого растворителя. Температура удерживания может быть от 30°С до температуры кипения используемого растворителя, и также ниже растворимости кристаллической формы А. Температура удерживания 40°С или выше является предпочтительной для получения кристаллической формы А более стабильным способом.
Не существует конкретных ограничений в отношении времени удерживания, пока оно может обеспечивать переход; однако предпочтительно, чтобы оно составляло, по крайней мере, 5 минут, более предпочтительно, по крайней мере, один час. Не существует определенного верхнего предела относительно времени удерживания; однако, с экономической точки зрения, предпочтительно, чтобы оно составляло три дня или менее, более предпочтительно один день или менее.
Кроме того, можно комбинировать раскрытые выше методы кристаллизации и перехода, хотя каждый из них можно использовать отдельно, для получения кристаллической формы А. Для уменьшения возможности кристаллизации кристаллических форм, которые отличаются от нужной кристаллической формы соединения (I) в этих методах кристаллизации и перехода, может оказаться эффективным добавлять к раствору небольшие количества затравочных кристаллов формы А, например, перед инициированием кристаллизации.
Для увеличения выхода кристаллы можно выращивать, используя уже закристаллизовавшиеся затравочные кристаллы формы А, путем дополнительного добавления слабого растворителя, такого как вода, или охлаждая раствор после того, как выпадут кристаллы. После кристаллизации фильтрат удаляют обычным способом, например центрифугированием, фильтрацией и т.п., и кристаллы сушат обычными способами сушки, такими как вакуумная сушка или сушка горячим воздухом и т.п., для получения требуемой кристаллической формы А.
По существу кристаллографически чистую кристаллическую форму В соединения (I) можно стабильно получать путем кристаллизации свободного соединения (I), добавляя кислоту к солевому раствору соединения (I) и основания.
Основания, которые используют для получения солей соединения (I), включают, но ими не ограничиваются, неорганические основания (например, гидроксид натрия, гидроксид калия, карбонат натрия, карбонат калия, метилат натрия и т.д.) и органические основания (например, 4-N,N-диметиламинопиридин, 1,8-диазабицикло[5.4.1]-7-ундецен, 1,5-диазабицикло[4.3.0]-5-нонен, триэтиламин, имидазол и т.д.). Эти основания можно использовать отдельно или в комбинации в виде смеси.
Нет необходимости выделять солевую форму соединения (I) с основанием заблаговременно. Солевой раствор соединения (I) и основания можно получить, добавляя основание в количестве, достаточном для растворения суспензии свободного соединения (I) в системе растворителя, которую используют для кристаллизации, или используя основание, используемое в реакции конденсации, в качестве основания для получения соли соединения (I).
Не существует ограничений относительно количества основания до тех пор, пока оно достаточно для полного растворения соединения (I) в растворителе, используемом для соединения (I) в присутствии основания. Однако обычно общее количество используемого основания предпочтительно составляет от половины до 10-кратного от эквивалентного количества соединения (I). Более предпочтительно использовать от эквивалентного количества до 4-кратного количества основания.
Кислоты, которые используются для нейтрализации, включают, но ими не ограничиваются, неорганические кислоты (например, хлористоводородная кислота, серная кислота, фосфорная кислота и т.д., и органические кислоты (например, уксусная кислота, пропионовая кислота, метансульфоновая кислота и т.д.). Эти кислоты можно использовать отдельно или в сочетании, в виде смеси.
Не существует ограничений относительно количества кислоты, пока оно достаточно для получения свободного соединения (I) путем нейтрализации основания, используемого для растворения соединения (I), в результате осаждая кристаллическую форму В. Обычно общее количество используемой кислоты составляет предпочтительно от 0,1- до 10-кратного и более предпочтительно от 0,2- до 2-кратного количества основания, которое используют для растворения соединения (I).
Не существует конкретных ограничений в отношении типов растворителей, которые используют для растворения; можно использовать органические растворители, воду или их смеси. Не существует определенных ограничений в отношении типов органических растворителей; однако с точки зрения растворимости и оперативности примерами могут служить амиды (например, N,N-диметилформамид, N,N-диметилацетамид и т.д.), спирты (например, метанол, этанол, 1-пропанол, 2-пропанол и т.д.) и кетоны (например, ацетон, метилэтилкетон и т.д.). Эти растворители можно использовать отдельно или в комбинации, в любых соотношениях в смеси. Из органических растворителей предпочтительны спирты и особенно предпочтителен метанол. Кроме того, в том случае, если используют неорганическое основание или кислоту, предпочтительно включать воду в качестве растворителя для удаления образующихся солей.
Для обеспечения кристаллизации и выхода кристаллической формы В соединения (I) в процессе кристаллизации, к раствору можно добавлять небольшое количество затравочных кристаллов формы В.
Обычно температура кристаллизации бывает ниже точки кипения используемого растворителя; однако температура кристаллизации меняется в зависимости от используемой системы растворителей и их растворяющей способности, и поэтому конкретно не определена. Однако необходимо, чтобы эта температура находилась в интервале значений, которые могут предотвратить переход в кристаллическую форму А соединения (I) после того, как закристаллизовалась кристаллическая форма В.
Если соединение (I) растворено в присутствии основания, а затем к осажденной кристаллической форме В добавляют кислоту, эта форма будет существовать как относительно стабильная форма, даже если она была суспендирована в растворителе. Однако переход в кристаллическую форму А происходит, если температура становится слишком высокой или время удерживания длится слишком долго. Таким образом, необходимо предотвратить этот переход, чтобы получить кристаллическую форму В. Температура, при которой кристаллическая форма В переходит в кристаллическую форму А, и время удерживания, необходимое для завершения выделения, меняются в зависимости от типов и составов, используемых в процессе, а также от взаимодействия температуры и времени удерживания. Поэтому температура и время удерживания конкретно не определены. Однако благоприятны более низкие температуры и более короткие времена удерживания. Обычно температура удерживания предпочтительно ниже точки кипения растворителя, или 60°С или ниже, и более предпочтительно 55°С или ниже. Кроме того, время удерживания до завершения выделения составляет предпочтительно 5 дней или менее и более предпочтительно 2 дня или менее при температуре, например, 50°С.
После кристаллизации фильтрат удаляют обычным способом, например центрифугированием, фильтрованием и т.п., и кристаллы сушат, используя обычные методы сушки, такие как вакуумная сушка, сушка горячим воздухом и т.п., до получения требуемой кристаллической формы В.
Кроме того, настоящее изобретение предлагает фармацевтическую композицию, содержащую в качестве активного ингредиента кристаллическую форму А соединения (I), полученную описанным выше способом.
Соединение (I) можно использовать для профилактики или лечения различных заболеваний на основании его гипогликемической активности или эффекта ингибирования PDE5. Оно полезно для профилактики или лечения заболеваний, включая, но ими не ограничиваясь, синдром поликистоза яичников, гестационный диабет, осложнения, связанные с диабетом (диабетическое нарушение остеогенеза, остеопороз), автоиммунные заболевания, панкреатит, кахексию (прогрессирующую потерю веса из-за липолиза, миолиза, анемии, отеков, анорексией и т.п.) и хронические заболевания, такие как рак, туберкулез, эндокринные заболевания и СПИД).
Фармацевтическую композицию по настоящему изобретению можно получить путем смешивания кристаллической формы А соединения (I) с фармацевтически приемлемым носителем, таким как органический или неорганический наполнитель, в твердом, полутвердом или жидком состоянии, пригодном для перорального, парэнтерального введения и наружного применения (локальное применение). Фармацевтическая композиция может быть в твердой форме, такой как таблетки, гранулы, порошки, капсулы, драже или суппозитории; в жидкой форме, такой как суспензия, молочко сироп, эмульсия, лимонад, лосьон и т.д.; в форме мази и геля. Вышеуказанные препараты могут содержать, при необходимости, вспомогательные агенты, стабилизаторы, смачивающие агенты, эмульгаторы, буферы и обычные добавки, такие как лактоза, цитрат, тартрат, стеарат, стеарат магния, глина, сахароза, кукурузный крахмал, тальк, желатин, агар, пектин, арахисовое масло, оливковое масло, масло какао, этиленгликоль и т.д.
Дозу соединения (I) можно определить обычным способом в зависимости от возраста и состояния пациента, типа и степени тяжести заболевания. Обычно доза составляет 1-100 мг/кг соединения (I) для перорального введения и 0,1-10 мг/кг для внутримышечных или внутривенных инъекций, вводимых от одного до четырех раз в день.
Краткое описание рисунков
Фиг.1 представляет порошковую рентгенограмму кристаллической формы А соединения (I).
Фиг.2 представляет порошковую рентгенограмму кристаллической формы В соединения (I).
Фиг.3 представляет ИК-спектр (метод с таблеткой КВr) кристаллической формы А соединения (I).
Фиг.4 представляет ИК-спектр (метод с таблеткой КВr) кристаллической формы В соединения (I).
Фиг.5 представляет кривую TG/DTA, полученную с помощью термогравиметрического/дифференциального термического анализа кристаллической формы А соединения (I).
Фиг.6 представляет кривую TG/DTA, полученную с помощью термогравиметрического/дифференциального термического анализа кристаллической формы В соединения (I).
Наилучший способ осуществления изобретения
Далее настоящее изобретение будет раскрыто более подробно со ссылкой на примеры, которые не следует рассматривать как ограничивающие изобретение.
Пример 1. Получение кристаллической формы В 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида.
3-(2,4-Дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамид (110 г, 0,235 моль) суспендируют в смеси растворителя из метанола (1100 г) и воды (1100 г), в которую добавляют водный раствор 25% гидроксида натрия (38 г), и затем полученную смесь нагревают до 40°С для растворения соединения. После удаления нерастворившегося материала фильтрованием полученный фильтрат нагревают вплоть до 50°С и по каплям добавляют 10% водный раствор (17 г) хлористоводородной кислоты в течение 2 часов для осаждения кристаллов. Суспензию кристаллов перемешивают при 50°С в течение 1 часа для созревания, а затем добавляют по каплям еще 10% водный раствор хлористоводородной кислоты (68 г) в течение 4 часов для роста кристаллов. После завершения добавления по каплям суспензию охлаждают до 25°С и кристаллы собирают фильтрованием. Влажные кристаллы промывают смешанным растворителем из метанола (550 г) и воды (550 г), а затем сушат в вакууме, получая кристаллическую форму В 3-(2,4-дихлобензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида.
Выход составляет 102 г. ИК-спектр этого продукта согласуется со спектром известной кристаллической формы В 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида.
Пример 2. Получение 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида и его кристаллической формы В.
Смесь 3-(2,4-дихлорбензил)-2-метил-3Н-бензимидазол-5-карбоновой кислоты (34,1 г, 102 ммоль), N,N’-карбонилдиимидазола (21,4 г) и N,N-диметилформамида (122,7 г) перемешивают в течение одного часа при 35°С, затем к этому добавляют 1-пентансульфонамид (20,0 г) и 1,8-диазабицикло[5.4.0]-7-унден (20,1 г) и перемешивают в течение 6 часов при 40°С. После реакции порцию N,N-диметилформамида (62,9 г) отгоняют в вакууме и к остатку добавляют метанол (227, 8 г) и воду (227,8 г). Этот раствор содержит 1,8-диазабицикло[5.4.0]-7-ундецен и имидазол, образовавшийся из N,N-карбонилдиимидазола в качестве основания. Этот раствор фильтруют для удаления нерастворимого материала, полученный фильтрат нагревают до 50°С и по каплям добавляют концентрированную хлористоводородную кислоту (20,4 г) в течение одного часа. После добавления по каплям кислоты в качестве затравочных кристаллов добавляют кристаллическую форму В 3-(3,2-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида (119 мг) и полученную смесь оставляют созревать в течение 2 часов. После созревания, в течение 2 часов добавляют по каплям концентрированную хлористоводородную кислоту (15,5 г). Суспензия созревает в течение одного часа при 50°С и ее охлаждают до 30°С, а затем кристаллы собирают фильтрованием. Кристаллы промывают смешанным растворителем, состоящим из метанола (120 г) и воды (120 г), сушат в вакууме и получают кристаллическую форму В 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида.
Выход составляет 41,7 г. ИК-спектр этого продукта согласуется со спектром известной кристаллической формы В 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида.
Пример 3. Получение кристаллической формы А путем перехода кристаллической формы В 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида.
Кристаллическую форму В 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида (5,0 г) суспендируют в смешанном растворителе, состоящем из ацетона (51,2 г) и воды (17,1 г), и полученную суспензию перемешивают при нагревании при 40°С. Спустя примерно 4 часа кристаллы в суспензии полностью переходят в кристаллы, ИК-спектр которых соответствует спектру кристаллической формы А 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида.
Пример 4. Получение кристаллической формы А 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида.
Смешанный растворитель, содержащий ацетон (332,5 г) и воду (42,0 г), добавляют к 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамиду (35,0 г) и полученную смесь кипятят с обратным холодильником для растворения кристаллов. Этот раствор фильтруют, пока он горячий, и полученный фильтрат снова нагревают при кипении с обратным холодильником, при этом в него по каплям добавляют воду (238 г) в течение 1 часа. По мере добавления по каплям воды начинают осаждаться кристаллы. После завершения добавления воды полученную суспензию охлаждают до 25°С и кристаллы собирают фильтрованием. Кристаллы промывают смешанным растворителем, состоящим из ацетона (87,2 г) и воды (73, 2 г), и затем сушат в вакууме, получая кристаллическую форму А 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида.
Выход составляет 24,2 г. ИК-спектр этого продукта соответствует спектру известной кристаллической формы А 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида.
Промышленная применимость
Кристаллическая форма А 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида, соединение, котрое обладает гипогликемической активностью или эффектом ингибирования PDE5, является по существу и кристаллографически стабильной и полезной в качестве лекарственного вещества для медицинских целей. С другой стороны, кристаллическую форму В 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида можно эффективно очистить путем кристаллизации, так как эта форма образует более крупные кристаллы и может быть очень легко выделена фильтрованием. Таким образом, кристаллическая форма В полезна для очистки 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида.
Изобретение относится к кристаллическим формам 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида, которые различаются по их данным порошковой дифракции рентгеновских лучей. Также описаны фармацевтические композиции, обладающие гипогликемической активностью или эффектом ингибирования PDE5, на основе этих кристаллических форм. Технический результат – получены новые формы 3-(2,4-дихлорбензил)-2-метил-N-(пентилсульфонил)-3Н-бензимидазол-5-карбоксамида, которые проявляют постоянство физикохимических и биологических свойств. 6 с. и 4 з.п. ф-лы, 6 ил., 5 табл.
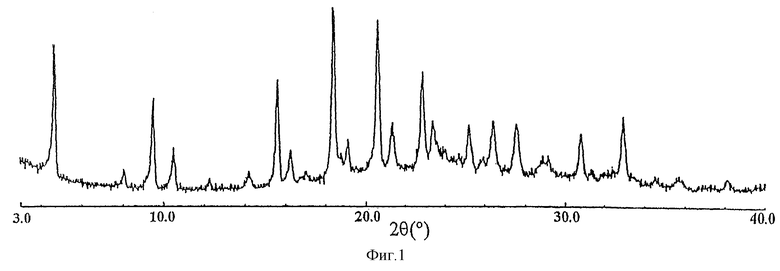
Угол 2θ(°): около 4,7, около 9,5, около 10,5, около 15,6 и около 18,4.
Угол 2θ(°): около 4,4, около 8,9 и около 13,4.
(a) растворения соединения (I) в растворителе при нагревании указанного растворителя,
(b) получения раствора,
(c) инициирования осаждения из раствора при 30°С или выше, и
(d) получения по существу и кристаллографически чистой кристаллической формы соединения (I), где значения порошковой дифракции рентгеновских лучей (2θ) указанной кристаллической формы соединения (I) в порошковом рентгеноструктурном анализе с использованием СuКα-излучения в качестве характеристического рентгеновского излучения и с ошибкой в интервале ±0,2 следующие:
Угол 2θ (°): около 4,7, около 9,5, около 10,5, около 15,6 и около 18,4.
(a) растворения соединения (I) в растворителе в присутствии основания и получения раствора,
(b) добавления к раствору кислоты,
(c) инициирования осаждения из раствора и
(d) получения по существу и кристаллографически чистой кристаллической формы соединения (I), где значения порошковой дифракции рентгеновских лучей (2θ) указанной кристаллической формы соединения (I) в порошковом рентгеноструктурном анализе с использованием СuКα-излучения в качестве характеристического рентгеновского излучения и с ошибкой в интервале ±0,2 следующие:
Угол 2θ (°): около 4,4, около 8,9 и около 13,4.
| WO 9724334 A1, 10.07.1997 | |||
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ НА ИХ ОСНОВЕ | 1991 |
|
RU2057126C1 |
| RU 2052455 C1, 20.01.1996. | |||

