Настоящее изобретение испрашивает приоритет по заявке на патент Китая № CN201710737513.4, поданной 24 августа 2017 г., содержание которой включено в данный документ посредством ссылки во всей своей полноте.
Область техники, к которой относится изобретение
Настоящее изобретение относится к кристаллической форме ингибитора PARP-1 и способу ее получения.
Предшествующий уровень техники
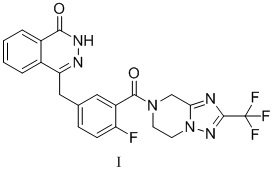
Поли(АДФ-рибоза)полимеразы (PARP), которые характеризуются осуществлением полиаденозиндифосфатрибозилирования, составляют суперсемейство из 18 ферментов клеточного ядра и цитоплазматических ферментов. PARP-1 является одним из важных представителей семейства PARP и рассматривается в качестве перспективной мишени для исследования новых средств лечения рака. В ZL201180003990.9 раскрыт новый ингибитор PARP, который представляет собой 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинон (формула I). Данное соединение может в значительной степени ингибировать активность фермента PARP in vitro, и оно может в значительной степени подавлять рост опухоли в модели на основе голой мыши с трансплантированной опухолью. В то же время, токсикологические данные в отношении крыс и собак также подтвердили, что соединение характеризуется соответствующей безопасностью. Конкретная структура является следующей:
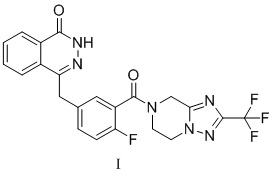 .
.
Полиморфизм относится к явлению, при котором твердые вещества существуют в двух или более отличающихся пространственных расположениях атомов, которые характеризуются разными физическими и химическими свойствами. Биологическая доступность одного и того же фармацевтического препарата может также различаться среди разных кристаллических форм ввиду отличающихся расположений атомов. Принимая во внимание важное значение кристаллических форм и их стабильность как твердых лекарственных средств, применяемых в клиническом лечении, для исследователей лекарственных средств необходимо проведение исследований нескольких кристаллических форм 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I).
Содержание настоящего изобретения
В настоящем изобретении представлена кристаллическая форма A 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I), которая характеризуется порошковой рентгеновской дифрактограммой, полученной с применением Cu-Kα-излучения, на которой представлены характеристические пики при значениях угла дифракции 2θ 9,58, 15,25, 17,09, 18,63, 21,11, 22,79, 23,99, 24,23, 27,26, 28,97,
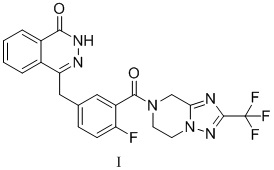 .
.
Кроме того, в неограничивающем примере кристаллическая форма A соединения формулы I характеризуется порошковой рентгеновской дифрактограммой, полученной с применением Cu-Kα-излучения, на которой представлены характеристические пики при значениях угла дифракции 2θ 9,58, 15,25, 17,09, 18,29, 18,63, 19,18, 21,11, 22,79, 23,99, 24,23, 27,26, 28,97.
Кроме того, кристаллическая форма A соединения формулы I характеризуется порошковой рентгеновской дифрактограммой, полученной с применением Cu-Kα-излучения, на которой представлены характеристические пики при значениях угла дифракции 2θ 9,58, 10,22, 13,00, 15,25, 17,09, 18,29, 18,63, 19,18, 21,11, 22,79, 23,99, 24,23, 27,26, 28,97.
В предпочтительном варианте осуществления кристаллическая форма A соединения формулы I характеризуется порошковой рентгеновской дифрактограммой, полученной с применением Cu-Kα-излучения, на которой представлены характеристические пики при значениях угла дифракции 2θ 9,58, 10,22, 12,76, 13,00, 15,25, 15,82, 16,11, 16,90, 17,09, 18,29, 18,63, 19,18, 20,65, 21,11, 22,79, 23,99, 24,23, 27,26, 28,97. Кроме того, порошковая рентгеновская дифрактограмма кристаллической формы A представлена на фигуре 1.
В настоящем изобретении также представлен способ получения кристаллической формы A соединения формулы I, который включает:
(a) добавление 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в растворитель (I), растворение путем перемешивания или нагревания, фильтрование, концентрирование до сухого состояния, где растворитель (I) выбран из по меньшей мере одного из бутанона, дихлорметана, этилацетата и тетрагидрофурана, причем применяемый объем в 20–200 раз превышает значение веса формулы I, предпочтительно в 50–100 раз, и может превышать его в 50, 55, 60, 65, 70, 75, 80, 85, 90, 95 и 100 раз в неограничивающем варианте осуществления;
(b) добавление растворителя (II), растворение вышеуказанного твердого вещества путем перемешивания или нагревания и перемешивание с кристаллизацией; или добавление растворителя (II), нагревание с обратным холодильником со взбиванием, перемешивание и охлаждение;
(c) фильтрование с получением кристаллов соединения формулы I.
Растворитель (II), описанный в данном способе, может быть выбран без ограничения из по меньшей мере одного из бутанона, тетрагидрофурана, ацетона, метанола, этанола, воды, ацетонитрила и этилацетата, предпочтительно из бутанона, тетрагидрофурана, ацетона, метанола, этанола/воды, тетрагидрофурана/воды, ацетона/воды, ацетонитрила, ацетонитрила/воды, этилацетата, бутанона/воды; причем применяемый объем растворителя (II) может в 1–100 раз превышать значение веса формулы I, предпочтительно в 5–70 раз, и может превышать его в 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70 раз в неограничивающем варианте осуществления.
Температура кристаллизации, описанной в данном способе, может составлять 0–40°C, и в неограничивающем варианте осуществления температура кристаллизации может составлять 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40°C, предпочтительно 10–30°C.
В настоящем изобретении также представлена фармацевтическая композиция, которая получена из кристаллической формы A вышеуказанного соединения формулы I. Фармацевтическая композиция может также содержать одно или более фармацевтически приемлемых вспомогательных веществ.
В неограничивающем варианте осуществления фармацевтическая композиция согласно настоящему изобретению может быть дополнительно получена в виде раствора для инъекций или твердого препарата с использованием промежуточного препарата, и твердый препарат выбран без ограничения из таблеток, пилюль, гранул, инъекций на основе лиофилизированного порошка или капсул.
Кроме того, вспомогательное вещество в твердом препарате является хорошо известным специалисту в данной области техники или может быть определено им и выбрано без ограничения из по меньшей мере одного из разрыхлителя, наполнителя, связующего и смазывающего средства; при этом вспомогательное вещество в растворе для инъекций выбрано без ограничения из по меньшей мере одного из нетоксичного физиологически приемлемого жидкого носителя, такого как физиологический солевой раствор, вода для инъекций, 5% раствор глюкозы для инъекций, раствор глюкозы с хлоридом натрия для инъекций, регулятор pH или консервант.
В настоящем изобретении также представлено применение кристаллической формы A соединения формулы I или вышеуказанной фармацевтической композиции в получении лекарственного препарата для ингибирования PARP или применение в получении лекарственного препарата, который используется в качестве вспомогательного средства или для придания опухолевым клеткам восприимчивости к ионизирующему излучению или химиотерапии при лечении рака, причем рак выбран из рака молочной железы, рака яичника, рака поджелудочной железы, рака предстательной железы, рака прямой кишки, рака печени или рака толстой кишки.
Кроме того, вышеуказанный лекарственный препарат можно применять в комбинации с терапевтически эффективной дозой фармацевтических препаратов, выбранных из темозоломида, доксорубицина, цисплатина, карбоплатина или дакарбазина.
«Порошковая рентгеновская дифрактограмма или XRPD» в настоящем изобретении относится к тому, что в соответствии с уравнением Брэгга 2dsinθ = nλ (где λ представляет собой длину волны рентгеновского излучения, λ = 1,54056Å, и порядок дифракции n представляет собой любое положительное целое число, при этом, как правило, регистрируют дифракционный пик первого порядка, n = 1), если рентгеновское излучение падает при угле скольжения (угле, комплементарном углу падения, также известному как угол Брэгга) на атомную плоскость кристалла или часть образца кристалла с расстоянием между плоскостями решетки d, то могут быть удовлетворены условия уравнения Брэгга с получением, таким образом, порошковой рентгеновской дифрактограммы.
«Дифференциальная сканирующая калориметрия или DSC» в настоящем изобретении относится к измерению температурной разницы и разницы теплового потока между образцом и эталоном в ходе обработки образца с помощью нагревания или постоянной температуры, выполняемому для характеристики всех физических изменений и химических изменений, связанных с температурными эффектами, чтобы обеспечить получение информации в отношении фазового перехода образца.
«2θ или угол 2θ» в настоящем изобретении относится к углу дифракции, причем θ представляет собой угол Брэгга, при этом единицей измерения является ° или градус.
Диапазон погрешности 2θ в настоящем изобретении может составлять ±0,5, а также может составлять ±0,1, ±0,2, ±0,3, ±0,4, ±0,5.
«Взбивание», описанное в настоящем изобретении, относится к способу осуществления очистки посредством применения свойств плохой растворимости вещества в растворителе, но хорошей растворимости примесей в растворителе. Очистка взбиванием может обеспечивать устранение окрашивания, изменение кристаллической формы или удаление небольшого количества примесей.
Температура высушивания в настоящем изобретении, как правило, составляет 20–100°C, предпочтительно 30–70°C, и высушивание можно проводить при атмосферном давлении или при пониженном давлении. Предпочтительно высушивание проводят при пониженном давлении.
Применяемый в настоящем изобретении 4-[[3-[[2-(Трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинон (формула I) может быть приобретен коммерчески или получен в соответствии со способом, описанным в ZL2011800039909. Другие химические реагенты или растворители, применяемые в настоящем изобретении, могут быть приобретены коммерчески.
«Межплоскостное расстояние кристалла или межплоскостное расстояние (значение d)» в настоящем изобретении относится к тому, что в пространственной решетке выбраны три непараллельных единичных вектора a, b, c, каждый из которых соединяет две смежные точки решетки, которые разделяют решетку на размещенные рядом единицы в форме параллелепипедов, называемые межплоскостным расстоянием кристалла. Пространственная решетка разделена с помощью соединенных линий между установленными единицами в форме параллелепипеда с получением группы линейных сеток, которые называют пространственной решеткой или кристаллической решеткой. Решетка и кристаллическая решетка отражают регулярность кристаллической структуры посредством геометрических точек и линий соответственно. Плоскости разных кристаллов характеризуются разными значениями межплоскостного расстояния (то есть расстоянием между двумя смежными параллельными плоскостями кристалла); при этом единицей измерения является Å или ангстрем.
Условия проведения испытаний для прибора, применяемого в экспериментах настоящего изобретения
1. Дифференциальный сканирующий калориметр, DSC
Модель прибора: система термического анализа Perkin-Elmer Pyris 7 Series.
Продувочный газ: азот
Скорость нагревания: 10,0°C/мин.
Диапазон температур: 50–250°C.
2. Порошковая рентгеновская дифракция, XRPD
(1) Модель прибора: порошковый рентгеновский дифрактометр Bruker D8 Discover A25.
Излучение: монохромное Cu-Kα-излучение (λ = 1,5418Å).
Способ сканирования: θ/2θ, диапазон сканирования: 8–35°.
Напряжение: 40 КВ, температурный диапазон: 294 K.
Краткое описание графических материалов
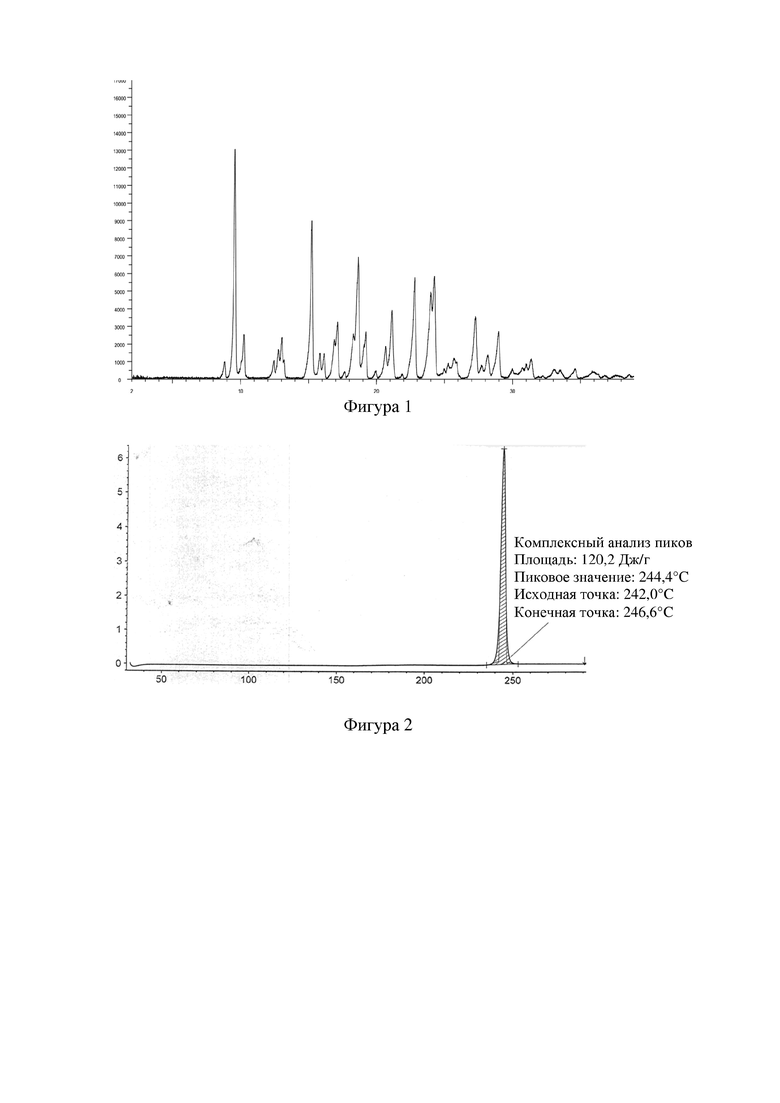
Фигура 1: XRPD-дифрактограмма кристаллической формы A соединения формулы I.
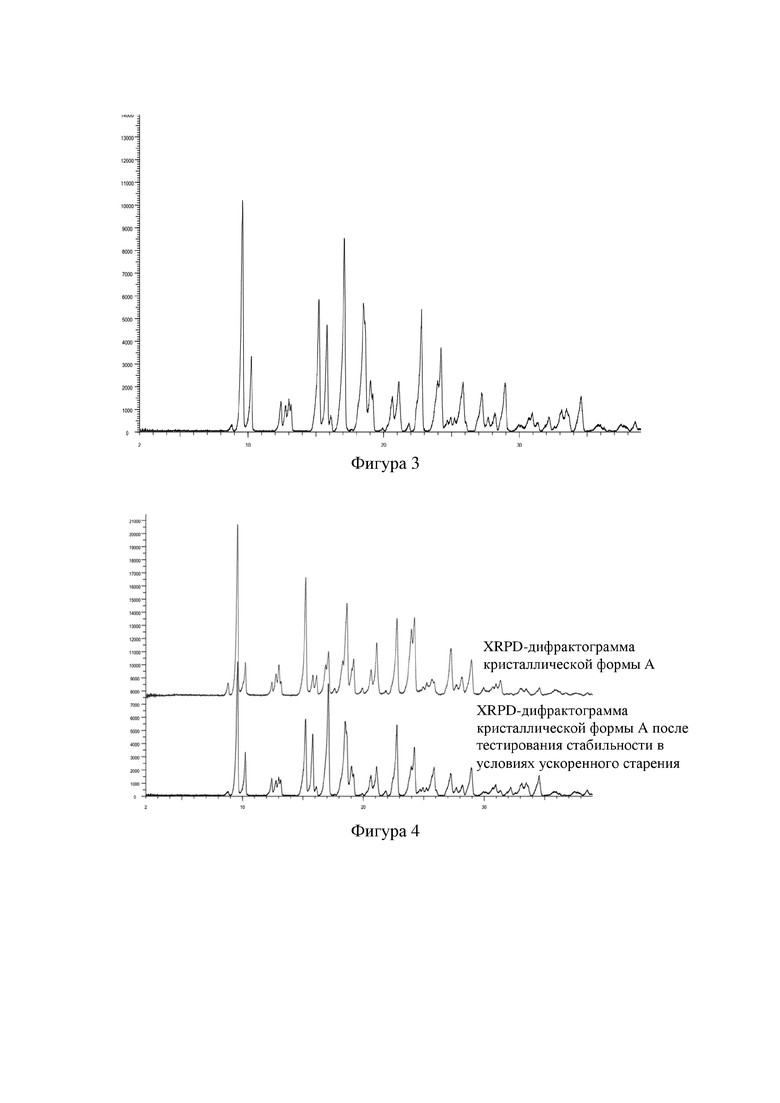
Фигура 2: DSC-спектр кристаллической формы A соединения формулы I.
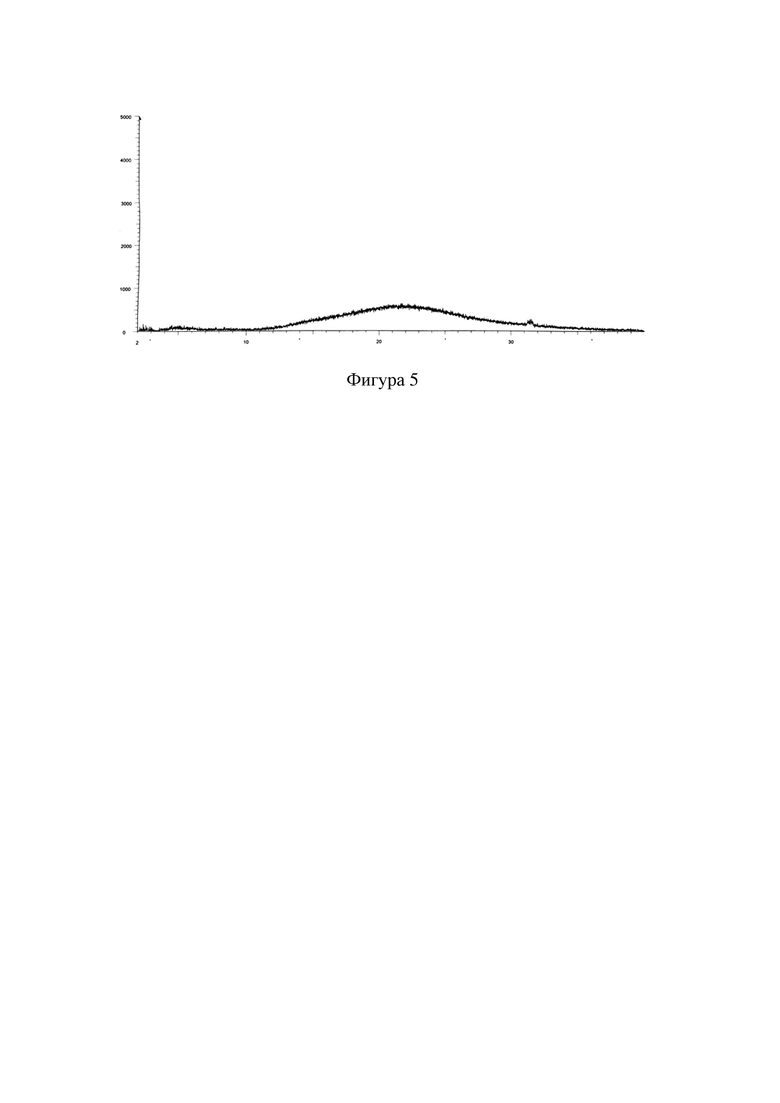
Фигура 3: XRPD-дифрактограмма кристаллической формы A соединения формулы I через 6 месяцев тестирования стабильности в условиях ускоренного старения (40°C, относительная влажность 75%).
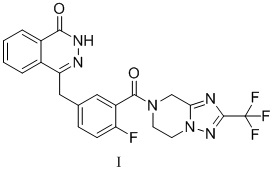
Фигура 4: сравнительная XRPD-дифрактограмма кристаллической формы A соединения формулы I после тестирования стабильности в условиях ускоренного старения.
Фигура 5: XRPD-дифрактограмма аморфной формы соединения формулы I.
Подробное описание проиллюстрированных вариантов осуществления
Далее в данном документе настоящее изобретение будет поясняться более подробно в сочетании с примерами или экспериментальными примерами. Примеры или экспериментальные примеры настоящего изобретения применяются лишь для иллюстрации технического решения согласно настоящему изобретению и не ограничивают сущность и объем настоящего изобретения.
Пример 1. Получение кристаллической формы A
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в 160 мл бутанона, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния, затем растворяли в 20 мл бутанона, перемешивали до кристаллизации при комнатной температуре, фильтровали и высушивали, получали 1,6 г белого твердого вещества. XRPD-дифрактограмма данного кристаллического образца представлена на фигуре 1, и его DSC-спектр представлен на фигуре 2. Измеренный с помощью DSC пик плавления образца соответствовал около 244,4°C, и температура начала плавления составила 242,0°C. Положения характеристических пиков представлены в таблице 1 ниже.
Таблица 1
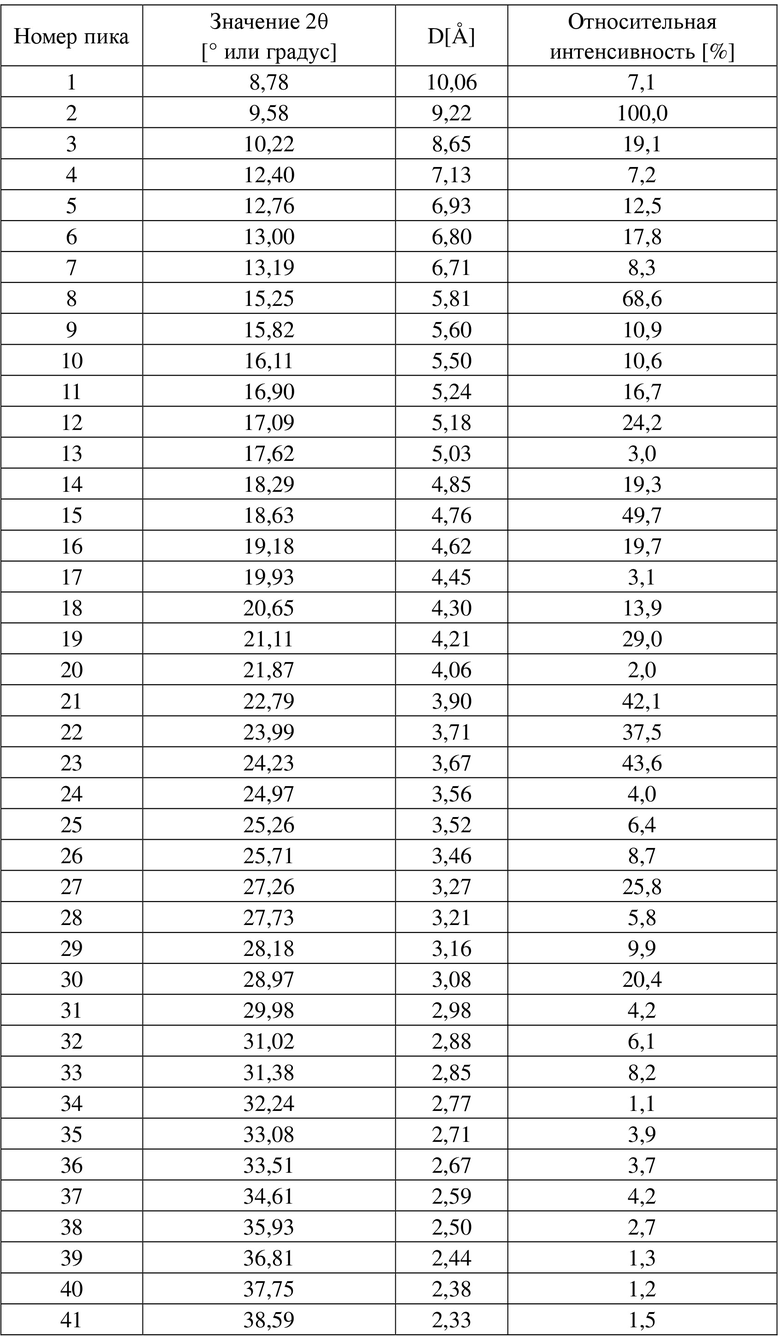
Пример 2. Получение кристаллической формы A
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в 160 мл бутанона, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния, затем растворяли в 20 мл тетрагидрофурана, перемешивали до кристаллизации при комнатной температуре, фильтровали и высушивали, получали 0,7 г белого твердого вещества. Измеренный с помощью DSC пик плавления образца соответствовал около 244,3°C, и температура начала плавления составила 242,1°C.
Пример 3. Получение кристаллической формы A
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в 160 мл бутанона, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния, затем растворяли в 20 мл ацетона, перемешивали до кристаллизации при комнатной температуре, фильтровали и высушивали, получали 1,4 г белого твердого вещества. Измеренный с помощью DSC пик плавления образца соответствовал около 244,2°C, и температура начала плавления составила 242,1°C.
Пример 4. Получение кристаллической формы A
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в 160 мл бутанона, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния, затем растворяли в 20 мл метанола, перемешивали до кристаллизации при комнатной температуре, фильтровали и высушивали, получали 1,3 г белого твердого вещества. Измеренный с помощью DSC пик плавления образца соответствовал около 244,3°C, и температура начала плавления составила 242,2°C.
Пример 5. Получение кристаллической формы A
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в 160 мл бутанона, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния, затем растворяли в 20 мл этанола, перемешивали до кристаллизации при комнатной температуре, фильтровали и высушивали, получали 1,8 г белого твердого вещества. Измеренный с помощью DSC пик плавления образца соответствовал около 244,1°C, и температура начала плавления составила 241,9°C.
Пример 6. Получение кристаллической формы A
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в 160 мл бутанона, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния, затем добавляли в 20 мл тетрагидрофурана, 100 мл воды добавляли по каплям при нагревании с обратным холодильником, перемешивали до кристаллизации при комнатной температуре, фильтровали и высушивали, получали 1,9 г белого твердого вещества. Измеренный с помощью DSC пик плавления образца соответствовал около 244,1°C, и температура начала плавления составила 241,9°C.
Пример 7. Получение кристаллической формы A
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) добавляли в 160 мл бутанона, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния, затем в 20 мл ацетона, 100 мл воды добавляли по каплям при нагревании с обратным холодильником, перемешивали до кристаллизации при комнатной температуре, фильтровали и высушивали, получали 1,6 г белого твердого вещества. Измеренный с помощью DSC пик плавления образца соответствовал около 244,1°C, и температура начала плавления составила 241,8°C.
Пример 8. Получение кристаллической формы A
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в 160 мл бутанона, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния, затем добавляли в 20 мл ацетонитрила, 100 мл воды добавляли по каплям при нагревании с обратным холодильником, перемешивали до кристаллизации при комнатной температуре, фильтровали и высушивали, получали 1,9 г белого твердого вещества. Измеренный с помощью DSC пик плавления образца соответствовал около 243,7°C, и температура начала плавления составила 241,6°C.
Пример 9. Получение кристаллической формы A
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в 160 мл бутанона, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния, затем добавляли в 20 мл ацетонитрила, перемешивали до кристаллизации при комнатной температуре, фильтровали и высушивали, получали 1,8 г белого твердого вещества. Измеренный с помощью DSC пик плавления образца соответствовал около 244,3°C, и температура начала плавления составила 242,1°C.
Пример 10. Получение кристаллической формы A
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в 160 мл бутанона, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния, затем растворяли в 20 мл этилацетата, перемешивали до кристаллизации при комнатной температуре, фильтровали и высушивали, получали 1,8 г белого твердого вещества. Измеренный с помощью DSC пик плавления образца соответствовал около 244,0°C, и температура начала плавления составила 241,6°C.
Пример 11. Получение кристаллической формы A
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в 160 мл бутанона, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния, затем добавляли в 21 мл бутанона, 80 мл воды добавляли по каплям при нагревании с обратным холодильником, проводили нагревание с обратным холодильником со взбиванием в течение 10 ч., охлаждали, фильтровали и высушивали, получали 1,2 г белого твердого вещества. Измеренный с помощью DSC пик плавления образца соответствовал около 243,9°C, и температура начала плавления составила 241,7°C.
Комплексный анализ данных порошковой рентгеновской дифракции и DSC показал, что твердые кристаллические формы, полученные путем кристаллизации в условиях вышеуказанных систем растворителей, являются полностью сопоставимыми и все представляют собой кристаллическую форму A.
Пример 12. Исследование стабильности кристаллов
Образец примера 1 подвергали тестированию стабильности в условиях ускоренного старения (40°C, относительная влажность 75%) в течение 6 месяцев, а затем отправляли на XRD для тестирования и сравнивали с исходными данными для подтверждения того, подверглась ли кристаллическая форма изменениям. Результаты показаны в таблице 2.
Среди них XRPD-дифрактограмма кристаллической формы A соединения формулы I через 6 месяцев тестирования стабильности в условиях ускоренного старения (40°C, относительная влажность 75%) представлена на фигуре 3. Сравнительная XRPD-дифрактограмма кристаллической формы A соединения формулы I после тестирования стабильности в условиях ускоренного старения представлена на фигуре 4.
Таблица 2. Сравнение данных XRD для образца и образца через 6 месяцев тестирования стабильности в условиях ускоренного старения
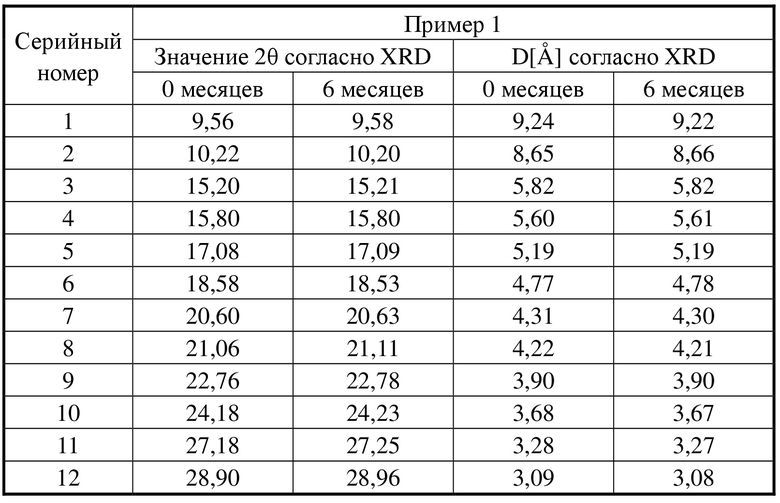
Заключение
Кристаллическая форма образца не подвергалась изменению после помещения для тестирования стабильности в условиях ускоренного старения (40°C, относительная влажность 75%) в течение 6 месяцев, что указывает на то, что кристаллическая форма является устойчивой и подходящей для разработки лекарственных средств.
Пример 13.
Растворяли 2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1a (780 мг, 2,65 ммоль) в 15 мл N,N-диметилформамида, затем добавляли бензотриазол-N,N,N',N'-тетраметилмочевины гексафторфосфат (1,80 г, 4,77 ммоль), 2-(трифторметил)-5,6,7,8-тетраводород-[1,2,4]триазоло[1,5-α]пиразин (560 мг, 2,92 ммоль, полученный с помощью хорошо известного способа, представленного в патенте WO 2009025784) и N,N-диизопропилэтиламин (1,4 мл, 7,95 ммоль) и осуществляли реакцию в течение 12 часов. Концентрировали при пониженном давлении, добавляли 30 мл воды, экстрагировали с помощью этилацетата (30 мл × 3), органические фазы объединяли, промывали с помощью насыщенного раствора хлорида натрия (20 мл), высушивали посредством безводного сульфата натрия, фильтровали и фильтрат концентрировали при пониженном давлении, остатки элюировали и очищали с использованием метанола/дихлорметана путем тонкослойной хроматографии, затем получали 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинон (210 мг, бледно-желтое твердое вещество). Как показано на фигуре 5, каких-либо значимых характеристических пиков посредством XRPD выявлено не было.
Пример 14.
Добавляли 2,0 г 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в дихлорметан, растворяли путем нагревания, фильтровали, концентрировали до сухого состояния и получали твердое вещество. Каких-либо значимых характеристических пиков посредством XRPD выявлено не было.
Изобретение относится к способу получения кристаллической формы A соединения формулы I, где на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения, представлены характеристические пики при значениях угла дифракции 2θ 9,58, 15,25, 17,09, 18,63, 21,11, 22,79, 23,99, 24,23, 27,26, 28,97, который включает: (a) добавление 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в растворитель (I), растворение, фильтрование, концентрирование до сухого состояния, где растворитель (I) представляет собой бутанон; (b) добавление растворителя (II), растворение вышеуказанного твердого вещества путем перемешивания или нагревания и перемешивание с кристаллизацией или добавление растворителя (II), нагревание с обратным холодильником со взбиванием, перемешивание и охлаждение; (c) фильтрование с получением кристаллов соединения формулы I; при этом растворитель (II) выбран из бутанона, тетрагидрофурана, ацетона, метанола, этанола, тетрагидрофурана/воды, ацетона/воды, ацетонитрила, ацетонитрила/воды, этилацетата и бутанона/воды. Технический результат – разработан способ получения кристаллической формы А соединения формулы I, которая является стабильной и может найти применение в медицине для получения лекарственных средств, ингибирующих PARP-1. 4 з.п. ф-лы, 5 ил., 2 табл., 14 пр.
1. Способ получения кристаллической формы A соединения формулы I, который включает:
(a) добавление 4-[[3-[[2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-α]пиразин-7-ил]карбонил]-4-фторфенил]метил-1(2H)-фталазинона (формулы I) в растворитель (I), растворение, фильтрование, концентрирование до сухого состояния, где растворитель (I) представляет собой бутанон;
(b) добавление растворителя (II), растворение вышеуказанного твердого вещества путем перемешивания или нагревания и перемешивание с кристаллизацией; или добавление растворителя (II), нагревание с обратным холодильником со взбиванием, перемешивание и охлаждение;
(c) фильтрование с получением кристаллов соединения формулы I;
при этом растворитель (II) выбран из бутанона, тетрагидрофурана, ацетона, метанола, этанола, тетрагидрофурана/воды, ацетона/воды, ацетонитрила, ацетонитрила/воды, этилацетата и бутанона/воды;
где на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения, представлены характеристические пики при значениях угла дифракции 2θ 9,58, 15,25, 17,09, 18,63, 21,11, 22,79, 23,99, 24,23, 27,26, 28,97,
 .
.
2. Способ по п. 1, где на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения, представлены характеристические пики при значениях угла дифракции 2θ 9,58, 15,25, 17,09, 18,29, 18,63, 19,18, 21,11, 22,79, 23,99, 24,23, 27,26, 28,97.
3. Способ по п. 1 или 2, где на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения, представлены характеристические пики при значениях угла дифракции 2θ 9,58, 10,22, 13,00, 15,25, 17,09, 18,29, 18,63, 19,18, 21,11, 22,79, 23,99, 24,23, 27,26, 28,97.
4. Способ по п. 1 или 2, где на порошковой рентгеновской дифрактограмме, полученной с применением Cu-Kα-излучения, представлены характеристические пики при значениях угла дифракции 2θ 9,58, 10,22, 12,76, 13,00, 15,25, 15,82, 16,11, 16,90, 17,09, 18,29, 18,63, 19,18, 20,65, 21,11, 22,79, 23,99, 24,23, 27,26, 28,97.
5. Способ по п. 1, где порошковая рентгеновская дифрактограмма, полученная с применением Cu-Kα-излучения, представлена на фигуре 1.
| ПРОИЗВОДНОЕ ФТАЛАЗИНОНКЕТОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2564527C2 |
| WO 2012019426 A1, 16.02.2012 | |||
| US 20080200469 A1, 21.08.2008 | |||
| US 20080161280 A1, 03.07.2008 | |||
| WO 2007138351 A2, 06.12.2007. | |||

