Изобретение относится к области органической химии и может быть использовано для получения тетрафторэтилена.
Тетрафторэтилен в промышленности получают пиролизом дифторхлорметана. Технологический процесс состоит из следующих основных стадий: пиролиз, закалка, нейтрализация, компримирование и конденсация пиролизата, выделение товарных продуктов ректификацией [Промышленные фторорганические продукты. Справочник. Максимов Б.Н., Барабанов В.Г., Серушкин И.Л. и др. - С.-Петербург: Химия, 1996, с.314]. При этом пиролиз дифторхлорметана сопровождается образованием побочных продуктов, снижающих коэффициент использования сырья.
Известен способ получения тетрафторэтилена, который решает большинство проблем по его производству, улучшает экологические параметры, повышает экономичность производства тетрафторэтилена за счет утилизации ряда побочных продуктов пиролиза [пат. РФ №2061672, МПК С 07 С 21/185, 17/00, опубл.10.06.96]. Этот способ включает пиролиз дифторхлорметана, закалку продуктов пиролиза, отделение побочного хлористого водорода с получением соляной кислоты, нейтрализацию, сушку, компримирование, конденсацию продуктов пиролиза с выделением целевого продукта из газов пиролиза, при этом пиролиз дифторхлорметана проводят в присутствии водяного пара и добавки октафторциклобутана и 1,1,2,2-тетрафторхлорэтана в виде азеотропнокипящей смеси в количестве 2-15 мас.% по отношению к дифторхлорметану, далее азеотропнокипящую смесь путем ректификации выделяют из кубовой фракции, полученной в процессе многоступенчатой ректификации продуктов пиролиза после выделения из них целевого продукта и непрореагировавшего дифторхлорметана. Недостаток этого способа состоит в том, что в результате последовательной многоступенчатой ректификации продуктов пиролиза в кубовых фракциях, особенно после отделения азеотропной смеси октафторциклобутана и 1,1,2,2-тетрафторхлорэтана, накапливаются токсичные непредельные соединения, в частности перфторизобутилен и пентафторхлорпропилен.
Известен способ получения тетрафторэтилена, наиболее близкий к предлагаемому по совокупности существенных признаков [пат. РФ №2162835, МПК С 07 С 21/185, 17/00, опубл. 10.02.2001]. Известный способ включает пиролиз дифторхлорметана с добавками октафторциклобутана и 1,1,2,2-тетрафторхлорэтана в виде азеотропной смеси в присутствии водяного пара, закалку продуктов пиролиза, отделение побочного хлористого водорода с получением соляной кислоты, нейтрализацию, компримирование и конденсацию продуктов пиролиза с выделением из них целевого продукта, непрореагировавшего дифторхлорметана и азеотропной смеси октафторциклобутана и 1,1,2,2-тетрафторхлорэтана многоступенчатой ректификацией, причем для повышения безопасности процесса, уменьшения возможности поражения токсичными непредельными соединениями за счет их перевода в малолетучие соединения процесс ведут с добавлением диэтиламина либо в куб ректификационной колонны, предназначенной для выделения дифторхлорметана, либо в куб ректификационной колонны, предназначенной для выделения азеотропной смеси. Диэтиламин используют либо в чистом виде, либо в виде смеси с водным раствором гидроксида натрия. Кубовую фракцию после выделения азеотропной смеси октафторциклобутана и 1,1,2,2-тетрафторхлорэтана можно подвергнуть дальнейшей ректификации с выделением теломерных продуктов общей формулы Н(СF2)nСl, где n более 2, являющихся ценными гидрофторхлоруглеродами, использование которых разрешено Монреальским протоколом по защите озонового слоя вплоть до 2020 г. Содержание непредельных соединений в выделяемых азеотропной смеси и указанных гидрофторхлоруглеродах не превышает 0,05 мол.%.
Существенный недостаток известного способа - образование высококипящих смолообразных продуктов взаимодействия диэтиламина и непредельных соединений, высокая коррозионная активность этих продуктов в условиях проведения ректификации, что может привести к выходу из строя дорогостоящего ректификационного оборудования. Кроме того, выделяемые по известному способу теломерные продукты общей формулы H(CF2)nCl, где n=3-5, имеют температуру кипения 21-77°C, а добавляемый диэтиламин 57oС, поэтому после ректификационного разделения требуется специальная очистка от диэтиламина. Необходимо отметить, что диэтиламин характеризуется также и высокой стоимостью.
Техническая задача настоящего изобретения состоит в исключении использования диэтиламина - дорогого реагента, загрязняющего получаемые продукты и создающего коррозионную среду на стадии ректификации, при сохранении высокой степени очистки от непредельных соединений.
Поставленная техническая задача решается тем, что в способе получения тетрафторэтилена, включающем пиролиз дифторхлорметана с добавками октафторциклобутана и 1,1,2,2-тетрафторхлорэтана в виде азеотропной смеси в присутствии водяного пара, закалку продуктов пиролиза, отделение побочного хлористого водорода с получением соляной кислоты, нейтрализацию, компримирование, конденсацию, многоступенчатую ректификацию продуктов пиролиза с выделением целевого продукта, непрореагировавшего дифторхлорметана, азеотропной смеси октафторциклобутана и 1,1,2,2-тетрафторхлорэтана, а также теломерных продуктов общей формулы Н(СF2)nСl, где n более 2, кубовую фракцию после выделения дифторхлорметана или смеси октафторциклобутана и 1,1,2,2-тетрафторхлорэтана очищают от токсичных примесей путем контактирования с реагентом в газовой фазе при повышенной температуре на катализаторе, содержащем активированный уголь.
В качестве реагента могут использовать фтористый водород. В этом случае процесс очистки от токсичных примесей проводят при температуре 300-400oC и при мольном отношении фтористого водорода к сумме токсичных примесей, равном (2-10):1, а в качестве катализатора используют активированный уголь, модифицированный гидроксидом калия, взятым в количестве 3% от массы угля.
В качестве реагента могут использовать хлор. В этом случае процесс очистки от токсичных примесей проводят при температуре 100-250°C и при мольном отношении хлора к сумме токсичных примесей, равном (1-2):1.
В качестве реагента могут использовать водяной пар. В этом случае очистку от токсичных примесей проводят при температуре 400-500°С и при мольном отношении водяного пара к сумме токсичных примесей, равном (20-80):1.
После очистки от токсичных примесей продукт отмывают от непрореагировавшего реагента, конденсируют и направляют на очередную ступень ректификации.
Токсичный пентафторхлорпропилен превращается в процессе очистки в нетоксичные насыщенные соединения: в 1,1,1,3,3,3-гексафтор-2-хлорпропан при использовании в качестве реагента фтористого водорода, в 1,1,3,3,3-пентафтортрихлорпропан при использовании хлора, в диоксид углерода и оксид углерода при использовании в качестве реагента водяного пара.
Пример 1
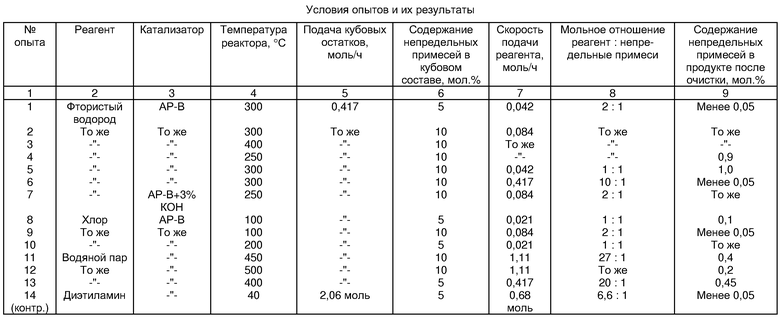
Тетрафторэтилен получают на промышленной установке путем пиролиза дифторхлорметана с добавками октафторциклобутана и 1,1,2,2-тетрафторхлорэтана в виде азеотропной смеси в присутствии водяного пара. Продукты пиролиза подвергают затем закалке, освобождают от непрореагировавшего хлористого водорода с получением соляной кислоты, нейтрализуют, проводят компримирование и конденсацию нейтральных продуктов пиролиза. Многоступенчатой ректификацией конденсата последовательно выделяют тетрафторэтилен и непрореагировавший дифторхлорметан. Кубовую фракцию после выделения дифторхлорметана анализируют на содержание непредельных примесей методом ИК-спектроскопии (определяют оптическую плотность полосы спектра с частотой 1790-1800 см-1). Количество непредельных примесей составляет 5 мол.%.
Катализатор для очистки кубовой фракции от токсичных примесей готовят следующим образом. В цилиндрический реактор диаметром 25 мм и длиной 300 мм, изготовленный из стали 12Х18Н10Т, помещают 100 см3 активированного угля марки АР-В с размером гранул 3-5 мм. Реактор помещают в трубчатый электрообогреватель и проводят дегидратацию угля в токе азота при температуре 160oС в течение 2 ч и при температуре 300oС в течение 4 ч. Не прекращая подачи азота, проводят насыщение катализатора фтористым водородом при температуре 300oС. Скорость подачи фтористого водорода 5 г/ч, насыщение заканчивают при выравнивании скоростей фтористого водорода на входе и выходе из реактора. Через слой приготовленного катализатора в реакторе при температуре 300oС в течение 5 ч совместно пропускают в испаренном виде кубовую фракцию со скоростью 0,417 моль/ч (66,7 г/ч) и фтористый водород со скоростью 0,042 моль/ч (0,84 г/ч). Газы после реактора отмывают от непрореагировавшего фтористого водорода, сушат дегидратированным хлоридом кальция и конденсируют в баллоне, охлаждаемом жидким азотом. Сконденсированный продукт подвергают низкотемпературной ректификации на лабораторной колонке эффективностью 40 теоретических тарелок.
За опыт подано 333,3 г кубовой фракции после выделения дифторхлорметана и 4,2 г фтористого водорода. Получено 335 г очищенной кубовой фракции, содержание непредельных примесей в которой, определяемое методом ИК-спектроскопии, составляет менее 0,05 мол.%.
Из очищенной фракции ректификацией выделено, г:
Азеотропная смесь
октафторциклобутана
с 1,1,2,2-тетрафторхлорэтаном 197
H(CF2)3Cl 101
H(CF2)4Cl 17
Н(СF2)5Сl 5
H(CF2)6Cl 5
Более высококипящие продукты 10
Пример 2
Получение тетрафторэтилена проводят аналогично описанному в примере 1. Кубовую фракцию после выделения дифторхлорметана подвергают дальнейшей ректификации на промышленной колонне с выделением азеотропной смеси октафторциклобутана с 1,1,2,2-тетрафторхлорэтаном. Полученную при этом кубовую фракцию анализируют на содержание непредельных примесей методом ИК-спектроскопии. Результат анализа 10 мол.%. Эту кубовую фракцию подвергают очистке на том же катализаторе и в тех же условиях, как описано в примере 1, но при скорости подачи фтористого водорода 0,084 моль/ч.
Примеры 3-4
Опыты проводят, как описано в примере 2, но при других температурах очистки. Условия опытов и результаты приведены в таблице.
Пример 5
Опыт проводят, как описано в примере 2, но при скорости подачи фтористого водорода 0,042 моль/ч. Результаты приведены в таблице.
Пример 6
Опыт проводят, как описано в примере 2, но при скорости подачи фтористого водорода 0,417 моль/ч. Результаты приведены в таблице.
Пример 7
Опыт проводят, как описано в примере 4, но для очистки используют катализатор, который готовят путем пропитки предварительно дегидратированного активированного угля марки АР-В объемом 100 см3 водным раствором объемом 120 см3, содержащим 1,8 г гидроксида калия. Катализатор после пропитки сушат на воздухе, а затем загружают в реактор (см. пример 1) и дегидратируют, как описано выше. Результаты очистки представлены в таблице.
Пример 8
Очистке подвергают кубовую фракцию после отделения дифторхлорметана, полученную аналогично описанному в примере 1.
Катализатор готовят, как описано в примере 1, но, в отличие от описанного, по окончании дегидратации катализатора реактор охлаждают в токе азота до 50oС и, не прекращая подачи азота, проводят насыщение катализатора хлором в токе хлора и азота при температуре 50-100oC до выравнивания скоростей хлора на входе и выходе из реактора, после чего отключают подачу азота и включают подачу кубовой фракции после отделения дифторхлорметана в испаренном виде. Эту кубовую фракцию со скоростью 0,417 моль/ч (66,7 г/ч) и хлор со скоростью 0,021 моль/ч (1,5 г/ч) пропускают через слой катализатора при температуре 100oC в течение 5 ч. Газы после реактора отмывают от непрореагировавшего хлора раствором гидроксида натрия и тиосульфата натрия в промывателе, сушат дегидратированным хлоридом кальция и конденсируют в баллоне, охлаждаемом жидким азотом. Продукт подвергают низкотемпературной ректификации на лабораторной колонке эффективностью 40 теоретических тарелок.
За опыт подано 333,3 г вышеуказанной кубовой фракции и 7,5 г хлора. Получено 351 г очищенной кубовой фракции, содержание непредельных примесей в которой, определяемое методом ИК-спектроскопии, составляет 0,1 мол.%.
Из очищенной фракции ректификацией выделено, г:
Азеотропная смесь
октафторциклобутана с
1,1,2,2-тетрафторхлорэтаном 197
Н(СF2)3Сl 101
H(CF2)4Cl 17
H(CF2)5Cl 5
Н(СF2)6Сl 5
CF2ClCFCl2CF3 –
1,2,2-трихлорпентафторпропан
(продукт присоединения
хлора к пентафторхлорпропилену) 7
Более высококипящие продукты 19
Пример 9
Для очистки используют кубовую фракцию после отделения азеотропной смеси октафторциклобутана с 1,1,2,2-тетрафторхлорэтаном, полученную аналогично описанному в примере 2.
Подготовку катализатора и очистку проводят, как в примере 8, но при скорости подачи хлора 0,084 моль/ч. Результаты очистки представлены в таблице.
Пример 10
Опыт проводят, как описано в примере 8, но при температуре очистки 200oC. Результаты очистки представлены в таблице.
Пример 11
Для очистки используют кубовую фракцию после отделения азеотропной смеси октафторциклобутана с 1,1,2,2-тетрафторхлорэтаном, полученную аналогично описанному в примере 2.
Катализатор для очистки кубовой фракции от токсичных примесей готовят следующим образом. В цилиндрический реактор диаметром 40 мм и длиной 390 мм, изготовленный из стали 12Х18Н10Т, помещают 490 см3 активированного угля марки АР-В с размером гранул 3-5 мм. Реактор помещают в трубчатый электрообогреватель и проводят активацию угля перегретым водяным паром при температуре 450oC в течение 1 ч. Перегретый водяной пар получают испарением воды в пароперегревателе, представляющем собой кварцевую трубку диаметром 6 мм и длиной 300 мм, помещенную в электрообогреватель. Подача воды на активацию угля составляет 20 г/ч (1,11 моль/ч). Не прекращая подачи воды, включают подачу вышеуказанной кубовой фракции со скоростью 0,417 моль/ч (72 г/ч).
Газы после реактора освобождают от непрореагировавшего водяного пара и продуктов гидролиза путем промывки водой, сушат дегидратированным хлоридом кальция и конденсируют в баллоне, охлаждаемом жидким азотом. Продукт подвергают низкотемпературной ректификации на лабораторной колонке эффективностью 40 теоретических тарелок.
За 5 ч подано 365 г вышеуказанной кубовой фракции и 100 г водяного пара. Получено 320 г очищенной кубовой фракции, содержание непредельных примесей в которой, определяемое методом ИК-спектроскопии, 0,4 мол.%.
Пример 12
Опыт проводят, как описано в примере 11, но при температуре очистки 500oC. Результаты приведены в таблице.
Пример 13
Для очистки используют кубовую фракцию после отделения дифторхлорметана, полученную аналогично описанному в примере 1.
Подготовку катализатора и очистку проводят, как в примере 11, но, в отличие от указанного примера, температура очистки составляет 400oC, а скорость подачи реагента (водяного пара) 0,417 моль/ч (7,5 г/ч). Результаты приведены в таблице.
Пример 14 (по прототипу)
Для очистки используют кубовую фракцию после отделения дифторхлорметана, полученную аналогично описанному в примере 1. Указанную кубовую фракцию с содержанием непредельных примесей 5 мол.% в количестве 330 г (2,06 моль) смешивают с 50 г (0,68 моль) диэтиламина в кубе стеклянной лабораторной ректификационной колонки эффективностью около 40 теоретических тарелок. Колонку вводят в режим ректификации и в легкую фракцию выделяют 180 г азеотропной смеси октафторциклобутана с 1,1,2,2-тетрафторхлорэтаном с содержанием непредельных примесей менее 0,05 мол.%.
Ректификацией кубового остатка получены фракции, г:
Н(СF2)3Сl 100
Н(СF2)4Сl 15
Н(СF2)5Сl 7
Более высококипящие
продукты и продукты
взаимодействия диэтиламина с
непредельными примесями 78
Полученные продукты отмывают водой от примеси диэтиламина, при этом рН водных вытяжек составляет:
H(CF2)3Cl 7,4
H(CF2)4Cl 8,2
H(CF2)5Cl 9,6
После очистки полученных фракций на катионообменной смоле КУ-2 рН водной вытяжки всех фракций не превышает 6,0. Удельный расход катионообменной смолы на очистку 0,1 кг/кг продукта.
Из примеров видно, что контактирование с фтористым водородом, хлором или водяным паром кубовой фракции после отделения непрореагировавшего дифторхлорметана или кубовой фракции после отделения азеотропной смеси октафторциклобутана и 1,1,2,2-тетрафторхлорэтана в слое катализатора, содержащего активированный уголь, приводит к очистке от пентафторхлорпропилена и других токсичных непредельных примесей на 90-99,5%. При этом наилучшие результаты достигаются в условиях, указанных в зависимых пунктах формулы изобретения. В процессе гидрофторирования использование фтористого водорода в мольном отношении к непредельным примесям 1:1 характеризуется минимальной степенью очистки, а в мольном отношении 10:1 - большими потерями фтористого водорода на стадии нейтрализации. Использование в качестве катализатора активированного угля, пропитанного гидроксидом калия в количестве 3 мас.%, способствует снижению температуры очистки. В процессе хлорирования достигается высокая степень очистки от токсичных примесей уже при 100-200°С и мольном отношении хлора к непредельным примесям 1:1. Процесс очистки от токсичных примесей водяным паром характеризуется использованием дешевого реагента.
Благодаря предлагаемому способу очистки исключается использование в качестве реагента дорогостоящего диэтиламина, исключается смолообразование, что позволяет организовать работу узла очистки в непрерывном режиме. Кроме того, предотвращается возможность коррозии дорогостоящего ректификационного оборудования. Вместе с тем отпадает необходимость стадии очистки теломерных продуктов общей формулы H(CF2)nCl, где n=3-5, после ректификационного разделения от диэтиламина дорогостоящими ионообменными смолами.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРЭТИЛЕНА | 1998 |
|
RU2162835C2 |
| СПОСОБ КОМПЛЕКСНОГО ПОЛУЧЕНИЯ ФТОРУГЛЕРОДОВ | 2001 |
|
RU2188814C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРЭТИЛЕНА | 1997 |
|
RU2136652C1 |
| СПОСОБ КОМПЛЕКСНОГО ПОЛУЧЕНИЯ ФТОРУГЛЕРОДОВ | 1999 |
|
RU2150456C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРЭТИЛЕНА | 1994 |
|
RU2061672C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРЭТИЛЕНА | 2007 |
|
RU2339607C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКТАФТОРЦИКЛОБУТАНА И ГЕКСАФТОРПРОПИЛЕНА | 2001 |
|
RU2186052C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕНТАФТОРЭТАНА | 1993 |
|
RU2049085C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРЭТИЛЕНА | 2000 |
|
RU2167847C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРЭТИЛЕНА | 1991 |
|
RU2097370C1 |
Изобретение относится к области органической химии, а именно к получению тетрафторэтилена. Способ осуществляют путем пиролиза дифторхлорметана с добавками октафторциклобутана и 1,1,2,2-тетрафторхлорэтана в виде азеотропной смеси в присутствии водяного пара. Закаляют продукты пиролиза, отделяют побочный хлористый водород с получением соляной кислоты. Нейтрализуют, компримируют, конденсируют, проводят многоступенчатую ректификацию продуктов пиролиза с выделением целевого продукта, непрореагировавшего дифторхлорметана, а также теломерных продуктов общей формулы Н(CF2)nCl, где n более 2. Кубовую фракцию после выделения дифторхлорметана очищают от токсичных примесей путем контактирования с реагентом, в качестве которого используют фтористый водород, хлор или водяной пар на катализаторе, содержащем активированный уголь. Технический результат - снижение экономических показателей процесса. 11 з.п. ф-лы, 1 табл.
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРЭТИЛЕНА | 1998 |
|
RU2162835C2 |

