Изобретение относится к улучшенному синтезу для получения (нитроксиметил)фениловых эфиров аспирина и его производных.
Эти сложные эфиры обладают интересными фармакологическими и терапевтическими свойствами; в частности они показывают улучшенную общую и местную толерантность на уровне слизистой оболочки желудка (WO 95/030641) и они являются более эффективными как антитромботические лекарственные средства (WO 97/16405).
Из предшествующего уровня техники известно, что (нитроксиметил)фениловые эфиры аспирина и его производных получают реакцией (нитроксиметил)фенола с аспирином и его производным в кислой форме (WO 97/16405).
В частности, получение (нитроксиметил)фенола проводят, начиная с (гидроксиметил)фенола через следующие стадии:
- реакция фенола с НВr в органическом растворителе для получения (бромметил)фенола;
- реакция (бромметил)фенола в органическом растворителе с AgNO3 для получения (нитроксиметил)фенола.
Синтез (нитроксиметил)фенольного промежуточного соединения имеет следующие недостатки. (Бромметил)фенол является химически нестабильным и раздражающим соединением. Нитроксипроизводное, полученное из бромметилфенола, все еще является нестабильным соединением, которое должно быть очищено перед реакцией с кислым хлоридом. (Нитроксиметил)фенол может далее разлагаться неконтролируемым путем, поэтому чтобы получить в промышленном масштабе это соединение с требуемой чистотой для конечной стадии эстерификации процессы очистки, обычно используемые в лабораторных органических синтезах, не могут быть использованы.
Следовательно, использование (нитроксиметил)фенола для синтеза (нитроксиметил)фениловых эфиров аспирина и его производных промышленно неосуществимо.
Неожиданно и с удивлением заявитель обнаружил, что синтезировать (нитроксиметил)фениловые эфиры аспирина и его производных, в частности (нитроксиметил)фениловые эфиры N-ацетилсалициловой кислоты, можно посредством синтетических реакций, в которых можно избежать использования вышеупомянутых производных фенола и таким образом стадий очистки промежуточных соединений, получая конечные продукты с хорошими выходами. Следовательно, этот новый процесс более благоприятный по сравнению с процессами предшествующего уровня техники.
Следовательно, объектом настоящего изобретения является новый способ получения (нитроксиметил)фениловых эфиров аспирина и его производных с формулой R-СООН с типами радикалов, имеющих следующую формулу:

где:
R1 означает группу ОСОR3; где R3 означает метил, этил или алкил С3-C5, линейный или разветвленный,
R2 означает водород;
nI означает 0;
предпочтительно в 1a) R1 означает ацетокси, предпочтительно в орто-положении по отношению к –СО-группе, R2 означает водород;
предпочтительно в 1b) R3=СН3, nI=0;
предпочтительно R-COOH означает ацетилсалициловую кислоту;
причем вышеупомянутый способ содержит следующие стадии, в основном проводимые в присутствии растворителя, инертного к реакционным условиям;
(1) реакцию между галогенангидридом R-C(O)-X1, где:
X1 означает галоген, выбранный между Сl и Вr,
R означает радикал как определено выше,
в присутствии основания с изомером гидроксибензальдегида, т.е. где гидроксильная группа может быть в орто, мета или пара положении, с образованием (карбонил)фенилового эфира (I):
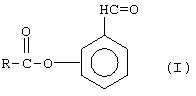
(2) селективное восстановление альдегидной группы соединения (I) с образованием (гидроксиметил)фенилового эфира (II):

(3) реакцию между (гидроксиметил)фениловым эфиром формулы (II) с:
a) SOX2, причем Х означает галоген, выбранный между Сl и Вr, с образованием (галогенометил)фенилового эфира формулы (III), где Х = галоген,
или
b) с тозилхлоридом или мезилхлоридом с образованием (тозилоксиметил)или (мезилокси)фенилэфир, причем Х означает O-тозил или O-мезил в формуле (III):
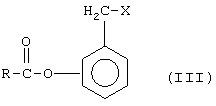
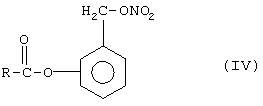
(4) реакцию между соединением формулы (III) с неорганической нитратной солью, катион металла которой принадлежит группе IB или IIВ, с образованием соответствующего (нитроксиметил)фенилового эфира
Образование (карбонил)фенилового эфира на стадии (1) может альтернативно достигаться другими реакциями. Например, реакцией аспирина и его производного общей формулы R-COOH с дегидратирующим агентом, таким как, например, N,N’-дициклогексилкарбодиимидом, в присутствии аминопиридинового производного, N,N-дизамещенного алкильными радикалами C1-C4 (стадия (I1)), или с C1-C4 алкилхлорформиатом в присутствии основания, растворимого или нерастворимого в реакционной среде, как определено ниже (стадия (III)), или с N,N’-карбонилдиимидазол (стадия (IIII)).
Процесс, являющийся объектом настоящего изобретения, позволяет получать продукты с требуемой степенью чистоты. Таким образом, нет необходимости очищать соединения, полученные после каждой стадии. Общие выходы являются хорошими (50-70%).
На стадии (1) ацилхлориду или бромиду аспирина и его производного, полученному из соответствующего соединения в кислой форме посредством использования известных реагентов (например, тионилхлорида, тионилбромида, оксалилхлорида, оксалилбромида, РСl3, РВr3) позволяют реагировать в инертных растворителях (например, галогенированных углеводородах, таких как дихлорметан, трихлорметан; простых эфирах, таких как этилацетат, пропиловый эфир, изопропиловый эфир, диоксан; сложных эфирах, таких как этилацетат, пропилацетат, бутилацетат) в присутствии органического или неорганического основания с изомером гидроксибензальдегида как определено выше. Вышеупомянутое основание может быть растворимо в растворителе реакции, как в случае третичных алифатических аминов формулы N(RN)3, где RN означает алкильную группу C1-C4 такую как, например, трибутиламин, триэтиламин, диэтилметиламин, триметиламин; или вышеуказанное основание может являться или нерастворимым в растворителе, таким как, например, в случае щелочных неорганических солей, например карбоната калия, карбоната натрия, или основаниями щелочных металлов, такими как NaOH и КОН.
Когда стадию 1) заменяют стадией (1I), как определено выше, аминопиридиновое производное, N,N-дизамещенное алкильными радикалами C1-C4, использованное в сочетании с дегидратирующим агентом, предпочтительно выбирают, например, из диметиламинопиридина и дибутиламинопиридина; когда вместо стадии 1) используют стадию (1II), соединение C1-C4 алкилхлорформиат предпочтительно выбирают между этилхлорформиатом и изобутилхлорформиатом.
Реакцию (2) селективного восстановления альдегидной группы до спирта можно проводить путем гидрирования газообразньм водородом, используя обычные катализаторы на носителе из угля, такие как, например, палладий, в растворе соединения формулы (I) в инертном растворителе. Температура реакции находится в диапазоне 0-40°С, давление газа может находиться в пределах от 1 до 3 атм.
Альтернативно гидрированию газообразным водородом восстановление соединения (II) можно проводить также с помощью других восстанавливающих агентов, например неорганических смешанных гидридов, таких как, например, NaBH4, в условиях, хорошо известных специалисту в данной области техники.
Стадию (3) проводят в инертном органическом растворителе при температуре в диапазоне 0-40°С.
Альтернативную реакцию между спиртом и тозилхлоридом или мезилхлоридом проводят согласно способам, известным из предшествующего уровня техники.
Стадию (4) проводят добавлением неорганической нитратной соли, катион которой выбран из металлов, принадлежащих группам IB и IIВ, к раствору соединения формулы (III), где Х означает галоген, как определено выше, или O-тозил или O-мезил, в органическом растворителе, в котором вышеупомянутой нитратной соли следует быть растворимой, таком как, например, ацетонитрил, тетрагидрофуран. Катион соли может быть цинком, серебром или ртутью. Предпочтительно соль является нитратом серебра. Температура реакции может находиться в диапазоне между 20° и 90°С.
Очевидно, что синтез является специфическим:
когда в процессе согласно настоящему изобретению используются в качестве исходных соединений другие терапевтически активные молекулы, имеющие реакционную карбоксильную функцию, обнаружено, что соответствующие нитроксиметилфениловые эфиры получаются с более низкими выходами, как это показано в примерах.
Следующие примеры представлены только в целях иллюстрации настоящего изобретения и они не ограничивают его.
Пример 1
Получение 3-(нитроксиметил)фенилового эфира 2-(ацетилокси)бензойной кислоты.
Пример 1а
Получение 3-(формил)фенилового эфира 2-(ацетилокси)бензойной кислоты.
Смесь 3-гидроксибензальдегида (830 г) и триэтиламина (8,24 г) в метиленхлориде (12,6 л) выдерживают при перемешивании в инертной атмосфере азота, охлаждая до температуры между -5°С и 0°С. Салицилоилхлорид (1650 г) добавляют маленькими порциями в течение часа. Смесь выдерживают при перемешивании еще в течение 15 мин, затем добавляют воду (10 л) и разделяют фазы. Водную фазу отбирают и отдельно экстрагируют метиленхлоридом (3 л). Органические фазы объединяют вместе, промывают 5% раствором Nа2СО3 (5 л × 2 раза) и затем водой (5 л × 2 раза). Органическую фазу высушивают сульфатом магния (2 кг) в присутствии обесцвечивающего угля (300 г). Фильтруют под вакуумом и растворитель выпаривают при пониженном давлении при температуре бани ниже, чем 40°С, получая в конечном счете 1929 г 3-(формил)фенилового эфира 2-(ацетокси)бензойной кислоты (количественный выход) с темп. плав. 80-84°С. Чистота соединения, определяемая ВЭЖХ, при использовании колонки LiChrospher® 100 RP 8 и элюента в виде фосфатного буфера рН 8/ацетонитрила 55/45, составляет 98,5%.
Пример 1b
Получение 3-(гидроксиметил)фенилового эфира 2-(ацетилокси)бензойной кислоты.
3-(Формил)фениловый эфир 2-(ацетилокси)бензойной кислоты (1929 г) растворяют в этилацетате (11 л) в присутствии 5% палладия на угле (290 г) с 50% влажностью.
Смесь гидрируют при комнатной температуре и давлении водорода примерно 2,5 атм при перемешивании. Реакция в течение первого часа является слегка экзотермичной и температура в реакторе повышается до 35°С. Через восемь часов добавляют свежий катализатор (100 г) для завершения реакции. Через 12 часов реактор разгружают, катализатор удаляют фильтрацией под вакуумом в атмосфере азота, промывая перегородку этилацетатом (2 л). Органические фазы объединяют вместе и промывают 5% раствором бикарбоната натрия (3 л × 2) и водой (3 л × 2). Органическую фазу высушивают сульфатом магния (2 кг) в присутствии обесцвечивающего угля (100 г). Затем ее фильтруют под вакуумом и выпаривают при пониженном давлении при температуре бани ниже, чем 40°С, получая 1850 г 3-(гидроксиметил)фенилового эфира 2-(ацетилокси)бензойной кислоты с выходом 95,2%, темп. пл. 77-79°С. Чистота соединения, определенная ВЭЖХ при использовании колонки LiChrospher® 100 RP 8 и элюента в виде фосфатного буфера рН 8/ацетонитрила 55/45, составляет 99,0%.
Пример 1с
Получение 3-(хлорметил)фенилового эфира 2-(ацетилокси)бензойной кислоты. К смеси, состоящей из 3-(гидроксиметил)фенилового эфира 2-(ацетилокси)бензойной кислоты (1850 г) и тионилхлорида (5,5 л), поддерживаемой при перемешивании, добавляют диметилформамид (5 мл) при комнатной температуре и оставляют при перемешивании в течение одного часа. По окончанию тионил-хлорид выпаривают при пониженном давлении при температуре бани ниже, чем 40°С. Остаточные следы тионилхлорида в соединении устраняют обработкой твердого вещества толуолом (2 л × 2), который затем удаляют выпариванием при пониженном давлении при температуре бани ниже, чем 40°С. Полученное таким образом неочищенное твердое вещество очищают кристаллизацией с изопропиловым эфиром (30 л), удаляя фильтрацией осадок, который остается нерастворимым в кристаллизационном растворе, доведенном до температуры кипения.
После охлаждения и фильтрации при пониженном давлении выделяют твердое вещество, которое высушивают под вакуумом при комнатной температуре, получая 1367 г (выход 69,4%) 3-(хлорметил)фенилового эфира 2-(ацетилокси)бензойной кислоты, темп. пл. 71-73°С. Чистота соединения, определенная ВЭЖХ при использовании колонки LiChrospher® 100 RP 8 и элюента в виде фосфатного буфера рН 8/ацетонитрила 40/60, составляет 99,0%.
Пример 1d
Получение 3-(нитроксиметил)фенилового эфира 2-(ацетилокси)бензойной кислоты Раствор 3-(хлорметил)фенилового эфира 2-(ацетилокси)бензойной кислоты (1367 г) в ацетонитриле (8 л) обрабатывают при перемешивании AgNO3 (915 г) при комнатной температуре, защищая от света. Раствор нагревают с обратным холодильником в течение двух часов и затем охлаждают при комнатной температуре и добавляют AgNO3 (115 г). Раствор вновь нагревают с обратным холодильником и через 4 часа охлаждают до 10°С; осадок (соли серебра) отфильтровывают под вакуумом и промывают ацетонитрилом (1 л × 2). Органические фазы объединяют вместе, и растворитель выпаривают под вакуумом при температуре бани ниже чем 40°С. Остаток растворяют в хлороформе (4 л), добавляют обесцвечивающий уголь (100 г), перемешивают и органическую фазу фильтруют через стеклянную перегородку (2,5 кг). Стеклянную перегородку промывают хлороформом (10 л).
Органические фазы объединяют вместе и концентрируют до маленького объема при пониженном давлении и температуре ниже, чем 40°С до тех пор, пока в растворе будет замечено образование осадка (примерно 3 л по объему). Объем раствора поддерживают непрерывным добавлением изопропилового эфира (6 л), продолжая выпаривание хлороформа при пониженном давлении до полного его удаления из органической фазы. Органическую фазу оставляют при перемешивании в течение двух часов при температуре примерно 20°С. Затем ее фильтруют под вакуумом, промывая изопропиловым эфиром (1,5 л) твердое вещество на фильтре. После высушивания под вакуумом при комнатной температуре получают 1200 г 3-(нитроксиметил)фенилового эфира 2-(ацетилокси)бензойной кислоты (выход 80,7%), темп. плавл. 63,5-64°С. Чистота соединения, определенная ВЭЖХ при использовании колонки LiChrospher® 100 RP 8 и элюента в виде фосфатного буфера рН 8/ ацетонитрила 50/50, составляет 99,75%. Структура конечного продукта подтверждена 1Н-ЯМР (CDCl3): 8,22 (1Н, dd), 7,66 (1H, td), 7,47 (1H, t), 7,40 (1H, td), 7,32 (1H, d), 7,24-7,21 (2H, m), 7,18 (1H, dd), 5,44 (2H, s), 2,30 (3H, s). Общий выход процесса составляет 53,3%.
Пример 2
Получение 2-(нитроксиметил)фенилового эфира 2-(ацетилокси)бензойной кислоты Продукт получают согласно процедуре, описанной в Примере 1, используя в качестве спирта 2-гидроксибензальдегид. При анализе конечного соединения, полученного хроматографией на тонком слое силикогеля, используя как элюент гексан/этилацетат 7/3, получают единичное пятно. Структуру конечного продукта подтверждают 1Н-ЯМР (CDCl3): 8,22 (1H, dd), 7,68 (1H, dt), 7,35 (6Н, m), 5,40 (2H, s), 2,30 (3H, s). Общий выход процесса составляет 67,8%.
Пример 3
Получение 4-(нитроксиметил)фенилового эфира 2-(ацетилокси)бензойной кислоты. Продукт получают согласно процедуре, описанной в Примере 1. Используемый ароматический гидроксиальдегид представляет собой 4-гидроксибензальдегид. На тонком слое силикагеля, используя как элюент гексан/этилацетат 7/3, получают единичное пятно. Структуру конечного продукта подтверждают 1Н-ЯМР (СDСl3): 8,21 (1Н, dd), 7,66 (1Н, dt), 7,42 (6Н, m), 5,40 (2Н, s), 2,25 (3Н, s). Общий выход процесса составляет 57,5%.
Изобретение описывает способ синтеза нитроксиметилфениловых эфиров аспирина и его производных. Описывается способ получения (нитроксиметил)фениловых эфиров аспирина и его производных формулы R-COOH с типами радикалов, имеющих следующую формулу:
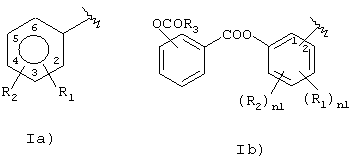
где R1 означает группу OCOR3; где R3 означает метил, этил или алкил С3-С5, линейный или разветвленный; R2 означает водород; nI означает 0; причем вышеупомянутый способ синтеза включает следующие стадии: (1) реакцию между галогенидом R-С(O)-Х1А), где X1 означает Cl, Br, и R означает радикал как определено выше, с изомером гидроксибензальдегида, в присутствии основания, с образованием (карбонил)фенилового эфира (I); (2) восстановление альдегидной группы (карбонил)фенилового эфира с образованием (гидроксиметил)фенилового эфира; (3) реакцию между (гидроксиметил)фениловым эфиром формулы (II) с: a) SOX2, причем Х означает галоген, выбранный между Cl и Br, или b) с тозилхлоридом или мезилхлоридом; (4) реакцию между сложным эфиром, полученным на предыдущей стадии, с неорганической нитратной солью, катион металла которой принадлежит группе IB или IIB, с образованием (нитроксиметил)фенилового эфира. Также описываются (гидроксиметил)фениловый эфир производных аспирина формулы R-COOH, 2-(нитроксиметил)фениловый эфир 2-(ацтилокси)бензойной кислоты и 4-(нитроксиметил)фениловый эфир 2-(ацетилокси)бензойной кислоты. Технический результат – упрошение процесса получения конечных продуктов и получение их с хорошими выходами. 4 н. и 3 з.п. ф-лы.
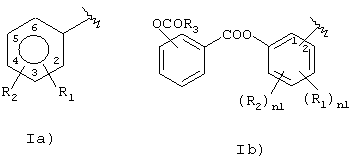
где R1 означает группу ОСОR3, где R3 означает метил, этил или алкил С3-С5, линейный или разветвленный;
R2 означает водород;
n1 = 0,
причем вышеупомянутый способ синтеза включает следующие стадии: (1) реакцию между галогенидом R-С(O)-Х1А), где X1 означает Сl, Вr и R означает радикал, как определено выше, с изомером гидроксибензальдегида в присутствии основания с образованием (карбонил)фенилового эфира (I); (2) восстановление альдегидной группы (карбонил)фенилового эфира с образованием (гидроксиметил)фенилового эфира; (3) реакцию между (гидроксиметил)фениловым эфиром формулы (II) с a) SOX2, причем Х означает галоген, выбранный между Сl и Вr, или b) с тозилхлоридом или мезилхлоридом; (4) реакцию между сложным эфиром, полученным на предыдущей стадии, с неорганической нитратной солью, катион металла которой принадлежит группе IВ или IIВ, с образованием (нитроксиметил)фенилового эфира.
| BOWDEN K | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Formylphenyl esters of aspirin | |||
| European Journal of Medicinal Chemistry | |||
| Chimica Therapeutica | |||
| Электрическое сопротивление для нагревательных приборов и нагревательный элемент для этих приборов | 1922 |
|
SU1997A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| ПРОИЗВОДНЫЕ ЯНТАРНОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ СИНТЕЗА И СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 1997 |
|
RU2125040C1 |

