Настоящее изобретение относится к области органической химии, в частности к синтезу йодсодержащих контрастных веществ, более определенно, к применению оксикислот бора и их производных в качестве защитных групп. Настоящее изобретение также обеспечивает соединения, пригодные в качестве промежуточных соединений в вышеуказанном синтезе.
УРОВЕНЬ ТЕХНИКИ
Контрастные вещества или контрастные средства представляют собой вещества, которые могут изменить способ, в котором анализируют область при медицинской визуализации. В частности, они способны изменить контрастность органа, повреждения или любой другой окружающей структуры, чтобы сделать видимыми такие детали, которые другим способом было бы трудно обнаружить или оценить.
Контрастные вещества в основном используются в области радиологии или ядерно-магнитно-резонансной диагностики. В зависимости от области применения данные производные представляют структурные особенности, такие как в случае молекул, пригодных в качестве контрастных веществ для рентгеноструктурного анализа, присутствие одного или более атомов с большим атомным номером (например, йода или бария).
Йопамидол (N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-5-[(2S)(2-гидрокси-1-оксопропил)амино]-2,4,6-трийодо-1,3-бензолдикарбоксамид) (II), структурная формула которого приведена ниже, представляет собой одно из многочисленных трийодзамещенных диагностических средств, коммерчески доступных и широко используемых для этой цели:
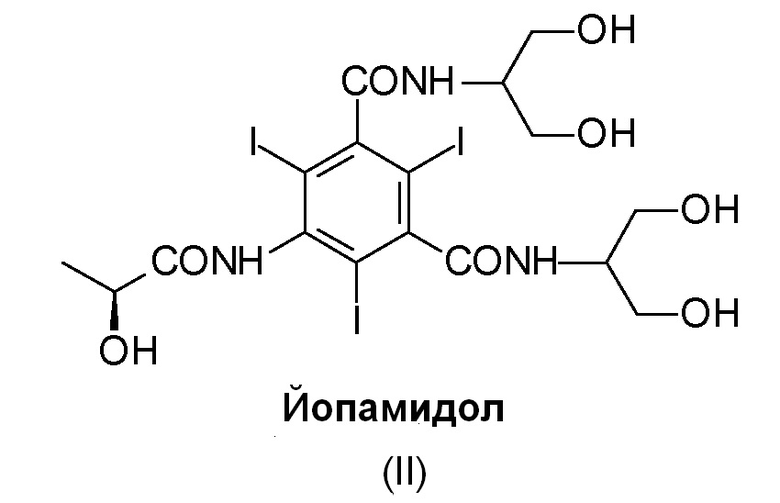
Широкое применение данного соединения в диагностике вызывает необходимость для производителей рассмотреть простой и удобный синтез в промышленном масштабе.
Йопамидол и его синтез впервые были раскрыты в GB1472050.
Несколько синтетических подходов были описаны за прошедшее время: они в основном характеризуются превращением ароматических аминопроизводных в соответствующие карбоксамиды с помощью взаимодействия с подходящим производным α-гидроксикислоты, смотри, например: WO 02/44132, WO 02/44125, WO 96/37459, WO 96/37460, патент США 5,362,905, WO 97/47590, WO 98/24757, WO 98/28259 и WO 99/58494.
5-Амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-1,3-бензолдикарбоксамид (V) представляет собой ключевое промежуточное соединение в синтезе Йопамидола. Как показано на Схеме 1 ниже, где был обобщен синтез предшествующего уровня техники, его йодирование обеспечивает промежуточное соединение 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV), который может дополнительно взаимодействовать с подходящими ацилирующими агентами, такими как уксусный ангидрид с целью защиты гидроксильных групп (как описано, т.е. в WO 02/44132 или в WO 00/050385) и предотвращения их взаимодействия с N-(S)-2-(ацетилокси)пропаноилхлоридом (2-ацетилоксипропаноилхлоридом) в последующей реакции. С помощью ацетилирования более реакционно-способных карбоксамидо-гидроксигрупп предотвращается применение избытка хлорида 2-ацетилоксипропаноила. Тем не менее, защитная группа после окончательного снятия защитной группы с помощью NaOH теряется и не может быть повторно использована.
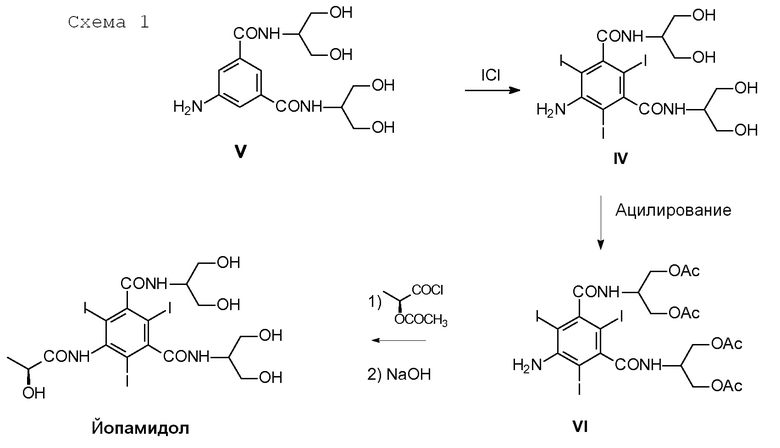
Кроме того, чтобы защитить гидроксильные группы, требуется избыток уксусного ангидрида, и его присутствие в смеси является несовместимым в следующей стадии реакции. В дальнейшем, необходимы дополнительные стадии осаждения и кристаллизации.
Основной недостаток данного подхода связан с выделением промежуточного соединения (VI) для получения твердого вещества в подходящей кристаллической форме. Данная методика может привести к потере 10% выхода.
Существует необходимость экономически целесообразного синтеза Йопамидола, в частности синтеза, обеспечивающего восстановление и повторное применение реагирующего вещества, используемого в качестве гидроксильной защитной группы.
Существует также необходимость в обеспечении синтеза, который, по меньшей мере, на последних стадиях предоставляет одно- или многостадийную реакцию, которую проводят в одном реакционном сосуде, чтобы избежать выделения промежуточных соединений и повысить общий выход. Кроме того, восстановление реагирующих веществ вместе с уменьшенным образованием и удалением отходов представляют собой крайне желательные задачи, принимая во внимание положительный конечный баланс реакции.
Производные бора известны в качестве защитных агентов в химическом синтезе.
GB2331098 и HR Bjørsvik, H Priebe, J Cervenka, AW Aabye, T Gulbrandsen и AC Bryde (A Selective Process for N-Alkylation in Competition with O-Alkylation: Boric Acid, Borax, and Metaborate as a Cheap and Effective Protecting Group Applicable for Industrial-Scale Synthetic Processes; Organic Process Research and Development 2001, 5, 472-478) раскрывают способ N-алкилирования соединений, содержащих 1,2 и/или 1,3-диольные структуры. Йодсодержащие контрастные вещества раскрыты как определенный вариант осуществления. Для предотвращения конкуренции O-алкилирования в данном документе описано применение оксикислоты бора в качестве диольных защитных агентов. Также могут быть использованы соли и сложные эфиры. Реакцию, включающую диольную защиту с помощью оксикислоты бора, проводят в воде. После завершения реакции N-алкилирования проводят снятие защитной группы диола.
Другое характерное использование оксикислот бора применительно к йодсодержащим контрастным средствам раскрыто в Journal of Hazardous Materials 205-206 (2012) 10-16 (I Rustighia, I Donatia, M Ferluga, C Campa, AE Pasqua, M Rossi, S Paoletti; Borate complexes of X-ray iodinated contrast agents: Characterization and sorption studies for their removal from aqueous media). Авторы показывают эффективное применение оксикислот бора в качестве средств для удаления йодсодержащих контрастных средств из сточных вод. Данный аддукт имеет хорошую стабильность при щелочном рН и адсорбируется на ионообменной смоле Dowex 1Х4, из которой он десорбируется с помощью использования ряда десорбирующих агентов, главным образом солей.
Сущность изобретения
В настоящее время были обнаружены борсодержащие защитные группы, которые являются универсальными и пригодными для эффективного повторного использования. Новые защитные борсодержащие функциональные группы обеспечивают одно- или многостадийный синтез, который проводят в одном реакционном сосуде без выделения промежуточных соединений, предоставляя восстановление и повторное использование защитных функциональных групп и значительное увеличение общих выходов способа.
Кроме того, данные защитные группы могут быть восстановлены и повторно использованы в способе, и это представляет собой значительное преимущество с точки зрения экономической эффективности всего способа и с учетом экологических проблем.
Задача настоящего изобретения состоит в обеспечении промежуточного соединения, представляющего собой Соединение формулы (I)
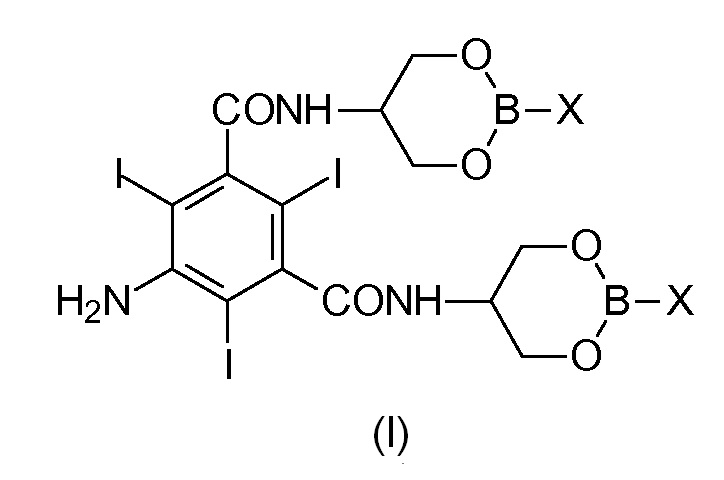
в которой Х представляет собой OR2 или R3, и в которой R2 и R3 представляют собой С1-С6линейный или разветвленный алкил, С3-С6циклоалкил, C6арил, необязательно замещенный группой, выбранной из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, втор-бутила, трет-бутила и фенила.
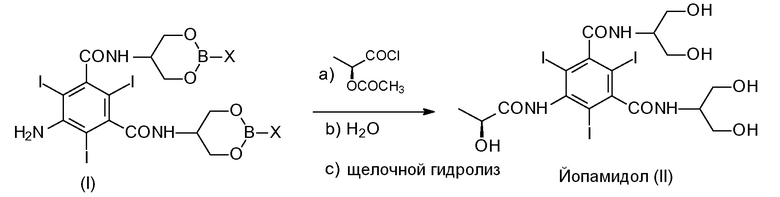
Другой целью настоящего изобретения является способ получения Йопамидола формулы (II),
включающий следующую реакцию, обобщенную на Схеме 2:
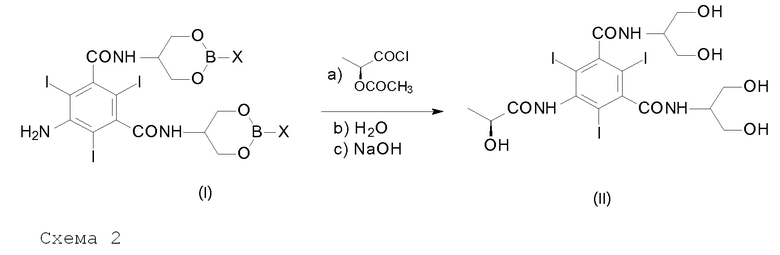
в которой различные группы представляют собой, как определено выше, и включающий следующие стадии:
a) взаимодействие Соединения (I) с ацилирующим агентом (S)-2-(ацетилокси)пропаноилхлоридом в реакционной среде для получения N-(S)-2-(ацетилокси)пропаноильного производного Соединения (I);
b) гидролиз промежуточного соединения со стадии а) с водным раствором при рН в диапазоне от 0 до 7, предпочтительно от 6 до 7 с помощью добавления воды или разбавленного щелочного раствора, такого как гидроксид натрия или гидроксид калия, освобождение гидроксилов от борсодержащих защитных групп, получение ацетилоксипроизводного Соединения (II) и необязательно восстановление борпроизводного;
c) щелочной гидролиз ацетилоксипроизводного Соединения (II), восстановление (S)-2-(гидрокси)пропаноильной группы для получения Йопамидола (II).
В первом предпочтительном варианте осуществления в соединении формулы (I) Х представляет собой OR2, в которой R2 представляет собой, как определено выше, и предпочтительно выбран из группы, состоящей из: этила, н-пропила и н-бутила.
Во втором предпочтительном варианте осуществления в Соединении формулы (I) Х представляет собой R3, в которой R3 представляет собой, как определено выше, и предпочтительно выбран из группы, состоящей из: бутила, изобутила, изопентила, н-пентила, н-гексила, циклопентила, циклогексила или фенила, необязательно замещенного метилом, этилом, н-пропилом, изопропилом, н-бутилом, втор-бутилом, трет-бутилом или фенилом.
На стадии а) вышеупомянутого способа указанная реакционная среда представляет собой органический растворитель, выбранный из группы, состоящей из N,N-диметилформамида, N,N-диметилацетамида, N,N-диэтилацетамида, N,N-диметилпропионамида, N-метилпирролидона, N-этилпирролидона, тетраметилмочевины, N,N'-диметилэтиленмочевины (DMEU), N,N'-диметилпропиленмочевины (DMPU), необязательно в смеси с несмешивающимся с водой органическим растворителем, в настоящем описании определенным, как сорастворитель.
На стадии b) после гидролиза борзащитных групп восстановление борпроизводного может быть проведено с помощью хроматографии или с помощью экстракции растворителем. В случае использования хроматографии можно применить соответствующую специальную смолу для удаления бора. Например, подходящая смола содержит диольные группы и предназначена для комплексообразования с борной или бороновой кислотой и последующего удаления. Предпочтительная смола представляет собой смолу, содержащую функциональные группы N-метил(полигидроксигексил)амина, также называемую метилглюкамин. Коммерчески доступным примером такой смолы является Amberlite® IRA743. Тем не менее, другие смолы могут быть выбраны из коммерчески доступных смол, например, или эквивалентные, или аналогичные колонки, такие как Duolite ES-371, Diaion CRB 02, Dowex BSR 1, Purolite S 108 и Purolite S110. Дополнительная информация представлена в Подробном Описании ниже. Гидролиз борсодержащих защитных групп проводится с помощью добавления воды.
Тем не менее, восстановление борпроизводных предпочтительно проводят с помощью экстракции растворителем, в частности, когда в соответствии со Схемой 3 и реагентом 3 R3 представляет собой бутил, фенил или метилзамещенный фенил (толильную группу), бутил или когда используют бороксин Формулы III, и R3 имеет то же значение, как и в случае с реагентом 3.
В целом, способ получения Йопамидола в соответствии с настоящим изобретением представлен на следующей Схеме 3:
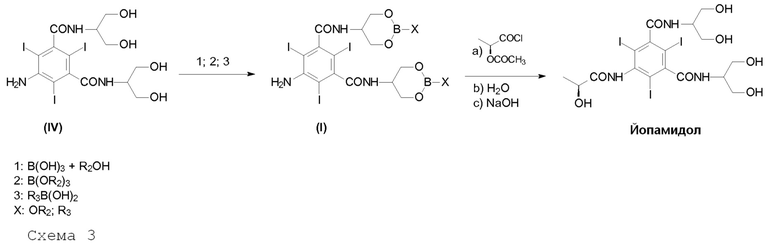
На Схеме 3, которая описывает синтез Йопамидола (II) из 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамида (IV), Х представляет собой, как определено выше, и на первой стадии реакции числа 1; 2; 3 представляют собой реагенты в отдельных, альтернативных вариантах осуществления. На схеме не показано, но частью настоящего изобретения является также альтернативный реагент бороксин Формулы III.
В соответствии с настоящим изобретением термины «борпроизводное», «борпроизводные» или «борсодержащая защитная группа» означают соединения бора, которые используют в соответствии со Схемой 3 выше, с исходным Соединением (IV) для получения Соединения (I); также как соединения, полученные в результате гидролиза промежуточного соединения (I) и последующего освобождения гидроксилов от борсодержащих защитных групп. Данные борпроизводные могут быть необязательно восстановлены в вышеуказанной стадии с) и повторно использованы в способе. Термины «борпроизводное» или «борпроизводные» обычно содержат оксикислоты бора (такие как борная кислота и бороновая кислота), их сложные эфиры и бороксин.
В соответствии с первым предпочтительным вариантом осуществления настоящего изобретения и ссылаясь на Схему 3, промежуточное Соединение (I), в котором Х представляет собой OR2, получают с помощью взаимодействия Соединения (IV) с одной из борных кислот в спирте R2OH или борным эфиром В(OR2)3, в которой R2 представляет собой, как определено выше.
В соответствии со вторым предпочтительным вариантом осуществления настоящего изобретения и также ссылаясь на Схему 3, промежуточное Соединение (I), в котором Х представляет собой R3, получают с помощью взаимодействия Соединения (IV) с бороновой кислотой R3-B(OH)2, в которой R3 представляет собой, как определено выше. Альтернативно данный второй предпочтительный вариант осуществления, не показанный на Схеме 3, но часть настоящего изобретения достигается с помощью взаимодействия Соединения (IV) с бороксином формулы (III)
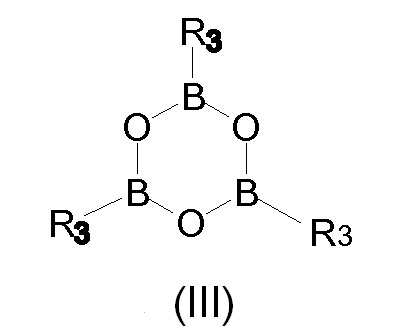
в которой R3 представляет собой, как определено выше.
В одном варианте осуществления настоящего изобретения указанный способ получения Йопамидола (II) включает:
х) взаимодействие Соединения формулы (IV) с эфиром борной кислоты B(OR2)3, в которой R2 представляет собой, как определено выше (см. Схему 3 выше, реагент 2), для получения промежуточного соединения формулы (I), раскрытого выше;
а) обработку указанного промежуточного соединения Соединения (I) (S)-2-(ацетилокси)пропаноилхлоридом для получения N-(S)-2-(ацетилокси)пропаноильного производного Соединения (I)
b) высвобождение борной кислоты; и
c) восстановление (S)-2-(гидрокси)пропаноильной группы для получения Йопамидола (II) с помощью щелочного гидролиза.
В указанном способе эфир борной кислоты может быть восстановлен для последующего применения. В связи с этим борную кислоту, полученную на конечной стадии, восстанавливают, подвергают взаимодействию со спиртом R2-OH, в которой R2 представляет собой, как определено выше, и повторно используют в новом способе.
В варианте осуществления стадии восстановления указанную борную кислоту обрабатывают подходящей смолой, такой как смола, специфичная для борной кислоты, например, коммерчески доступная AmberliteTM IRA743, подразумевая, что специалист в данной области техники сможет выбрать соответствующий способ для восстановления борной кислоты, обращаясь к общеизвестным знаниям в данном вопросе.
В другом варианте осуществления настоящего изобретения указанный способ получения Йопамидола (II) включает:
х') взаимодействие Соединения (IV) с борной кислотой со спиртом R2OH, в которой R2 представляет собой, как определено выше (см. Схему 3 выше, реагент 1), для получения промежуточного соединения формулы (I), раскрытого выше;
а) обработку указанного промежуточного соединения (I) (S)-2-(ацетилокси)пропаноилхлоридом для получения N-(S)-2-(ацетилокси)пропаноильного производного Соединения (I)
b) высвобождение борной кислоты; и
c) восстановление (S)-2-(гидрокси)пропаноильной группы для получения Йопамидола (II) с помощью щелочного гидролиза.
В указанном способе борная кислота и спирт образуют соответствующий эфир борной кислоты in situ, и способ может быть проведен, как и в случае объясненного выше, с использованием эфира борной кислоты. Эфир борной кислоты затем восстанавливают для последующего применения, как показано выше.
Способ может быть проведен в периодическом режиме или в целях удобства в непрерывном режиме.
В другом варианте осуществления настоящего изобретения в Соединении формулы (I) Х представляет собой R3, как определено выше, и представляет собой предпочтительно фенильную, метилзамещенную фенильную, метильную или бутильную группу.
В соответствии с данным вариантом осуществления примерный способ получения Йопамидола (II) продемонстрирован на вышеприведенной Схеме 3, реагент 3.
Указанный способ включает:
х'') взаимодействие Соединения формулы (IV) с бороновой кислотой R3-B(OH)2 или бороксином (III), в которой R3 представляет собой, как определено выше, и предпочтительно выбран из фенила, метилзамещенного фенила, метила и бутила, для получения промежуточного соединения формулы (I);
а) обработку указанного промежуточного соединения (I) (S)-2-(ацетилокси)пропаноилхлоридом для получения ацетилоксипроизводного Соединения (I)
b) высвобождение бороновой кислоты;
c) восстановление (S)-2-(гидрокси)пропаноильной группы для получения Йопамидола (II) с помощью щелочного гидролиза.
В указанном способе бороновая кислота может быть восстановлена для последующего применения. В связи с этим бороновую кислоту, полученную на конечной стадии, восстанавливают с помощью двух возможных подходов: с помощью экстракции органическим несмешивающимся с водой растворителем, например, 4-метил-2-пентаноном, 2-пентаноном, 3-пентаноном, дибутиловым эфиром, 2-метилтетрагидрофураном, циклопентилметиловым эфиром, метилизопропилкетоном, метилизопентилкетоном, этилацетатом, бутилацетатом, пентилацетатом, изопентилацетатом, изопропилацетатом, удаления растворителя и повторного использования восстановленной бороновой кислоты в способе, или в качестве альтернативы, как описано выше, для комплексообразования с борной или бороновой кислотой, то есть с помощью обработки конечной реакционной смеси смолой, подходящей для удаления бора, такой как упомянутые на стадии с) выше, среди которых, например, Amberlite® IRA 743.
Способ восстановления может быть проведен в периодическом режиме или в целях удобства в непрерывном режиме. В предпочтительном варианте осуществления фенилбороновую, п-толилбороновую или н-бутилбороновую кислоты используют и повторно используют.
В другом варианте осуществления в Соединении формулы (I) Х представляет собой R3, как определено выше, предпочтительно фенил, метилзамещенный фенил, метил или бутил. Более предпочтительно в Соединении формулы (I) R3 представляет собой фенил.
Получение соединения формулы (I) в качестве промежуточного соединения при получении Йопамидола (II) также может быть проведено, как раскрыто в предшествующем варианте осуществления, но с использованием бороксина формулы (III) вместо бороновой кислоты. Трифенилбороксин и триметилбороксин являются предпочтительными бороксинами, и R3 в соединении (I) представляет собой предпочтительно фенил или метил. В соответствии с данным вариантом осуществления фенилбороновая кислота или метилбороновая кислота высвобождаются при гидролизе гидроксильных групп, освобожденных от борсодержащего защитного фрагмента, и бороновые кислоты могут быть восстановлены для последующего применения, как раскрыто выше.
Соединение (I) может быть выделено и охарактеризовано, как будет описано более подробно ниже. Соответственно, соединение формулы (I) представляет собой дополнительную цель настоящего изобретения, также как его применение в качестве промежуточного соединения в синтезе Йопамидола (II).
После завершения защиты ОН, которая предпочтительно достигается с помощью дистилляции воды, проводят стадию ацетилирования на Соединении (I), предпочтительно в растворителе, выбранном из группы, состоящей из: N,N-диметилформамида, N,N-диметилацетамида (DMAC), N,N-диэтилацетамида, N,N-диметилпропионамида, 1-метил-2-пирролидона, 1-этил-2-пирролидона, тетраметилмочевины, N,N'-диметилэтиленмочевины (DMEU), N,N'-диметилпропиленмочевины (DMPU). Предпочтительно, используют N,N-диметилацетамид, и, более предпочтительно, N,N-диметилацетамид имеет очень низкое содержание воды или является безводным. Растворитель может также содержать сорастворитель, который представляет собой органический несмешивающийся с водой растворитель, выбранный из: 4-метил-2-пентанона, 2-пентанона, 3-пентанона, дибутилового эфира, 2-метилтетрагидрофурана, циклопентилметилового эфира, метилизопропилкетона, метилизопентилкетона, этилацетата, бутилацетата, пентилацетата, изопентилацетата, изопропилацетата. Предпочтительная смесь растворителя/сорастворителя представлена DMAC и 4-метил-2-пентаноном, 3-пентаноном или 2-пентаноном.
Присутствие сорастворителя на стадии а) является наиболее предпочтительным, когда борзащитные группы должны быть восстановлены позднее с помощью экстракции сорастворителем.
В другом варианте осуществления настоящего изобретения способ может быть проведен исходя из соответствующего Соединения (V).
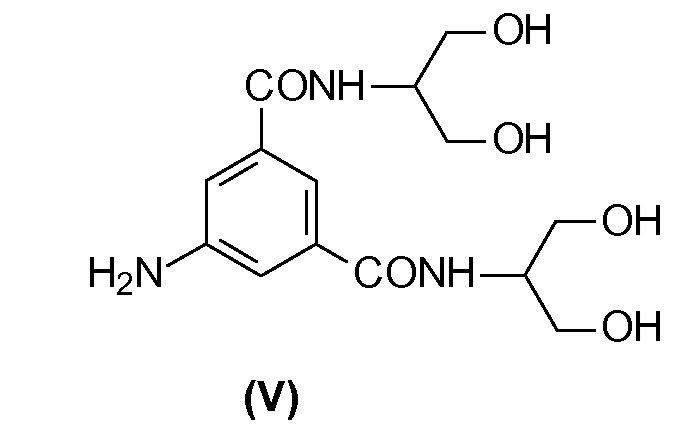
В соответствии с данным вариантом осуществления целью настоящего изобретения является способ в соответствии со следующей Схемой 4:
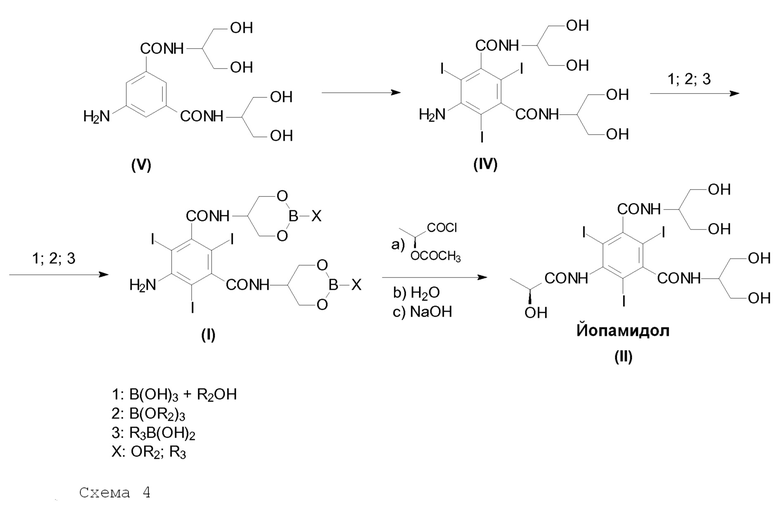
Схема 4 описывает синтез Йопамидола (II) из 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-1,3-бензолдикарбоксамида (V); Х представляет собой, как определено выше, и на первой стадии реакции числа 1; 2; 3 представляют собой реагенты в отдельных, альтернативных вариантах осуществления. На схеме не показано, но частью настоящего изобретения является также альтернативный реагент бороксин.
Соединение (V) также могут получить, как описано в WO02/44125 или WO00/029372.
Йодирование ароматического кольца также проводят в соответствии со способами, раскрытыми в многочисленной литературе по синтезу Йопамидола, например, в WO96/037458, WO2009/103666, WO2010/121904, WO2011/154500 и WO2011/003894.
В определенном аспекте и цели настоящего изобретения Соединение (V) получают в соответствии со следующей Схемой 5:
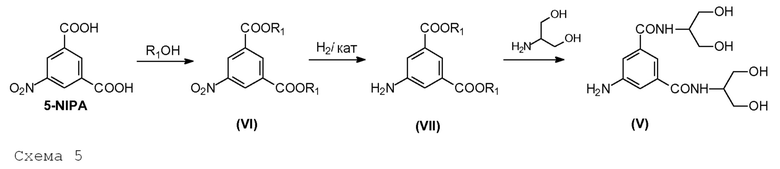
Схема 5 описывает синтез 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-1,3-бензолдикарбоксамида (V) из 5-нитроизофталевой кислоты (5-NIPA), в котором:
i) 5-нитроизофталевую кислоту обрабатывают спиртом R1OH, в которой R1 представляет собой линейный или разветвленный С1-С4алкил, для получения соответствующего диэфира (VI);
ii) 5-нитрогруппу восстанавливают до соответствующей 5-аминогруппы в Соединении (VII);
iii) диэфир взаимодействует с 2-амино-1,3-пропандиолом для получения Соединения (V).
Другой целью настоящего изобретения является способ получения Йопамидола в соответствии со следующей Схемой 6:
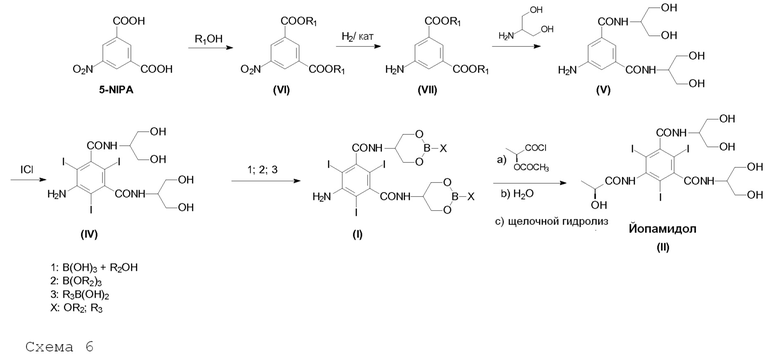
Схема 6 описывает синтез Йопамидола (II) из 5-нитроизофталевой кислоты, в котором:
i) 5-нитроизофталевую кислоту обрабатывают спиртом R1OH, в которой R1 представляет собой линейный или разветвленный С1-С4алкил, предпочтительно бутил, для получения диэфира (VI);
ii) 5-нитрогруппу восстанавливают для получения Соединения (VII);
iii) диэфир взаимодействует с 2-амино-1,3-пропандиолом для получения Соединения (V);
iv) Соединение (V) йодируют в положениях 2,4,6 для получения Соединения (IV);
v) Соединение (IV) обрабатывают борной кислотой или ее производным в соответствии с настоящим изобретением для получения Соединения формулы (I) в соответствии с настоящим изобретением;
vi) Соединение формулы (I) окончательно превращают в Йопамидол (II), как описано выше.
Вышеуказанные цели настоящего изобретения и другие варианты осуществления будут подробно раскрыты в следующем описании.
Подробное описание изобретения
Настоящее изобретение раскрывает способ получения Йопамидола (II), включающий следующую реакцию:
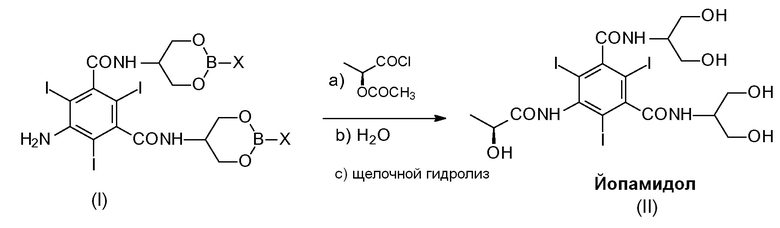
в которой Х представляет собой OR2 или R3, и в которой R2 и R3 представляют собой С1-С6линейный или разветвленный алкил, С3-С6циклоалкил, C6арил, необязательно замещенный группой, выбранной из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, втор-бутила, трет-бутила и фенила, более предпочтительно фенила, метилзамещенного фенила, метила и бутила,
и включающий следующие стадии:
a) взаимодействие Соединения (I) с ацилирующим агентом (S)-2-(ацетилокси)пропаноилхлоридом в реакционной среде для получения N-(S)-2-(ацетилокси)пропаноильного производного Соединения (I);
b) гидролиз промежуточное соединение со стадии а) с водным раствором при рН в диапазоне от 0 до 7, предпочтительно от 6 до 7 с помощью добавления воды или разбавленного щелочного раствора, такого как гидроксид натрия или гидроксид калия, освобождение гидроксилов от борсодержащих защитных групп, получение ацетилоксипроизводного Соединения (II) и необязательное восстановление борпроизводного;
c) щелочной гидролиз ацетилоксипроизводного Соединения (II), восстановление (S)-2-(гидрокси)пропаноильной группы для получения Йопамидола (II).
В соответствии со стадией а) реакционная среда представляет собой предпочтительно реакционную среду, имеющую минимальное содержание воды, совместимое с реакцией, более предпочтительно безводную реакционную среду.
Реакционную среду обычно выбирают с помощью специалиста в данной области техники, основываясь на общеизвестных знаниях для такого типа реакции. Среда обычно представляет собой органический растворитель, способный растворять Соединение формулы (I) и не затрагивать ацилирующий реагент (S)-2-(ацетилокси)пропаноилхлорид. Предпочтительные примеры органического растворителя, пригодного для использования на данной стадии, представляют собой инертные диполярные апротонные растворители, такие как, например, N,N-диметилформамид, N,N-диметилацетамид, N,N-диэтилацетамид, N,N-диметилпропионамид, 1-метил-2-пирролидон, 1-этил-2-пирролидон, тетраметилмочевина, N,N'-диметилэтиленмочевина (DMEU), N,N'-диметилпропиленмочевина (DMPU).
N,N-Диметилацетамид (DMAC) с низким содержанием воды или безводный является предпочтительным. Органический растворитель, используемый на данной стадии, также может быть в смеси с сорастворителем. Предпочтительные сорастворители выбраны из органических и несмешивающихся с водой сорастворителей, таких как: 4-метил-2-пентанон, 2-пентанон, 3-пентанон, дибутиловый эфир, 2-метилтетрагидрофуран, циклопентилметиловый эфир, метилизопропилкетон, метилизопентилкетон, этилацетат, бутилацетат, пентилацетат, изопентилацетат, изопропилацетат. Предпочтительная смесь растворителя/сорастворителя представлена DMAC и 4-метил-2-пентаноном, 3-пентаноном или 2-пентаноном.
На стадии а) стехиометрическое соотношение между Соединением формулы (I) и хиральным ацилирующим агентом (S)-2-(ацетилокси)пропаноилхлоридом представляет собой соотношение, обычно используемое в синтезе Йопамидола (см., например, GB1472050 Пример 1b). Стехиометрический избыток предпочитается в зависимости от условий реакции.
Влажность воздуха должна быть проконтролирована в окружающей среде реакции, следовательно, предпочтительно проводить реакцию в инертной атмосфере, например, в атмосфере сухого азота или аргона.
Температура реакции обычно представляет собой приблизительно комнатную температуру, даже если более высокие или более низкие температуры могут быть использованы совместимо со стабильностью реагентов и конечного продукта.
Реакцию проводят в течение времени, проходящего от нескольких минут до нескольких дней, обычно от 8 до 30 часов, более удобно от 12 до 30 часов, например, в течение 18 часов. Время реакции зависит от условий реакции: используемого растворителя, соотношения и чистоты реагентов, температуры. Специалист в данной области техники может найти оптимальные условия, обращаясь к своим собственным знаниям и опыту.
Завершение реакции можно обнаружить с помощью стандартных аналитических средств, используемых в органической химии, например, спектрометрического оборудования, например, 1H-ЯМР, ИК; хроматографического оборудования, например, ТСХ, ВЭЖХ, ГЖХ.
С этой целью и ссылаясь на Схему 2, реакционную смесь, полученную на стадии а) способа настоящего изобретения, переносят в водную среду (стадия b). В целях удобства воду (или разбавленный щелочной раствор, такой как раствор NaOH или KOH) добавляют в тот же реакционный сосуд, где была проведена стадия ацилирования а). Перенос органической фазы в другой сосуд, содержащий воду, также может быть осуществлен. Как правило, количество объема или массы воды представляет собой, по меньшей мере, такое же количество органической фазы, предпочтительно выше, например, в 2-3 раза превышающее объем органической фазы, совместимо с последующими действиями и не превышая в разбавлении. Ацетил-Йопамидол защищенные гидроксильные группы затем освобождают от борсодержащих защитных групп с помощью гидролиза, добавляя воду или разбавленные щелочные растворы, такие как разбавленный NaOH или KOH к кислой реакционной смеси.
Восстановление борсодержащих защитных групп после их гидролиза на ацетилоксипроизводном Соединения Формулы (I) на стадии b) может быть проведено с помощью обработки реакционной смеси ионообменной смолой, обычно анионообменной смолой, предпочтительно специфичной для удаления бора, такой как смола с диольными функциональными группами, более предпочтительно с функциональными группами, выбранными из группы, состоящей из: метилглюкамина, диэтаноламинометила предпочтительно на полистирольной матрице, глицидила предпочтительно на метакрилатной матрице, иминодипропиленгликоля, амино-бис(пропан-цис-2,3-диола), гидроксиэтиламинопропиленгликоля. Некоторые смолы, такие как с метилглюкаминовыми функциональными группами являются также коммерчески доступными и могут быть выбраны из каталогов производителей, например, Resindion of Mitsubishi Chemical, Dow Chemical, etc. Типичным примером является Duolite ES-371, предпочтительным примером является AmberliteTM IRA743 от Dow Chemical Company или других поставщиков. Данный вариант осуществления предпочтительно относится к производным борной кислоты: в целях удобства смолу загружают в колонку и фазу элюируют через нее.
В качестве альтернативы и в соответствии с предпочтительным вариантом осуществления, описанным ниже, когда бороновую кислоту, такую как фенилбороновая, п-толилбороновая или бутилбороновая кислота или бороксин (например, фенилбороксин или метилбороксин) используют в качестве борсодержащих защитных групп, восстановление борсодержащих защитных групп проводят с помощью экстракции органическим несмешивающимся с водой растворителем, выбранным из группы, состоящей из: 4-метил-2-пентанона, 3-пентанона, 2-пентанона, дибутилового эфира, 2-метилтетрагидрофурана, циклопентилметилового эфира, метилизопропилкетона, метилизопентилкетона, этилацетата, бутилацетата, пентилацетата, изопентилацетата, изопропилацетата. Предпочтительными экстракционными растворителями являются 4-метил-2-пентанон (МИБК), 3-пентанон или 2-пентанон.
В соответствии с данным вариантом осуществления и Схемой 3 Соединение I предпочтительно получают непосредственно в полярном растворителе в смеси с несмешивающимся с водой органическим растворителем (сорастворителем), пригодным для экстракции борсодержащих защитных групп, как описано ниже. Предпочтительная смесь растворителя/сорастворителя представлена DMAC и 4-метил-2-пентаноном (МИБК), 3-пентаноном или 2-пентаноном (т.е. в соотношении, составляющем от 1:10 до 1:4 масс./масс.).
Бороновую кислоту или бороксин добавляют в незначительном молярном избытке по сравнению с трийодобензолкарбоксамидом (Соединение IV). Суспензию перемешивают и нагревают до 90-95°С, и воду предпочтительно отгоняют для завершения реакции защиты. Образование Соединения I может быть определено, т.е. с помощью 1H-ЯМР.
В соответствии с наиболее предпочтительным вариантом осуществления стадии а)-с) могут затем проводить в одном реакционном сосуде, например, следующим образом: (S)-2-(ацетилокси)пропаноилхлорид добавляют к смеси в атмосфере азота и перемешивают в течение нескольких часов, чтобы получить ацетилоксипропаноильное производное Соединения I. Высвобождение борзащитных групп с помощью гидролиза ацетилоксипропаноильного производного Соединения I обычно получают с (водой или) разбавленным щелочным раствором до нейтрального рН (т.е. составляющего от 5-8), предпочтительно составляющего от 6-7, который обеспечивает хорошую селективность в дополнительном восстановлении борзащитной группы с несмешивающимся с водой растворителем.
Восстановление борзащитной группы, которое обычно является количественным, могут проводить в периодическом или непрерывном режиме. В обоих способах количество органического несмешивающегося с водой растворителя поддерживается в соотношении с соединением бороновой кислоты, составляющем от 1:10 до 1:20, предпочтительно от 1:13 до 1:16, более предпочтительно приблизительно 1:15. В периодическом способе данное количество может быть добавлено за одну или более аликвот.
Полученная таким образом двухфазная смесь содержит водную фазу с ацетил-Йопамидолом, которую восстанавливают для очистки с последующим гидролизом ацетильной группы для получения Йопамидола, в то время как бороновую кислоту отделяют в органической фазе, которую восстанавливают, необязательно предпочтительно дистиллируют для концентрирования борсодержащих защитных групп и повторно используют.
Повторное использование раствора бороновой кислоты может быть осуществлено после добавления предпочтительного растворителя для реакции, т.е. DMAC (5-10% органического раствора) и сорастворителя дистилляции, предпочтительно под вакуумом и при температуре ниже 40°C для достижения концентрации бороновой кислоты приблизительно 10%. Повторно использованный раствор затем может быть применен неограниченно предпочтительно с помощью добавления небольшого количества (т.е. соответствующего 5-20% повторно применяемой бороновой кислоты, присутствующей в органическом растворе) свежей бороновой кислоты.
В качестве следующей стадии или водный раствор, элюированный из колонки, специфичной для удаления бороновой кислоты, или в качестве альтернативы поступающий с экстракции с помощью использования органического растворителя и содержащий N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-5-[(2S)(2-ацетокси-1-оксопропил)амино]-2,4,6-трийодо-1,3-бензолдикарбоксамид, также известный как ацетил-Йопамидол, затем обессоливают от органических и неорганических солей с помощью использования ионообменных смол.
На стадии с) полученный нейтральный раствор загружают на сильноосновную анионообменную смолу, предпочтительно смолу с триметиламиновыми функциональными группами, связанными с полимерным матричным материалом, таким как Relite® 3ASFB, для высвобождения неочищенного Йопамидола, по существу, как описано в патенте США 5550287 за исключением того, что Йопамидол высвобождается с разбавленным водным раствором сильной кислоты, такой как HCl, H2SO4, причем хлористоводородная кислота является предпочтительной. В качестве альтернативы, высвобождение Йопамидола может быть осуществлено в периодическом режиме при основных условиях, как раскрыто, например, в WO97/30735, Пример 1, т.е. с помощью обработки ацетил-Йопамидол-содержащей смеси в сильнощелочных условиях рН с последующим удалением соли, предпочтительно с помощью хроматографии на ионообменных смолах, предпочтительно сначала на сильнокислотной катионообменной смоле с последующей слабоосновной анионообменной смолой.
Гидролиз ацетил-Йопамидола на сильноосновной анионообменной смоле является более эффективным и, следовательно, является предпочтительным. В соответствии с данным вариантом осуществления способ становится более простым, так как очистку и гидролиз, на практике, проводят в одну стадию.
Продукт дополнительно очищают стандартным способом, например, как описано более подробно в Экспериментальной Части, с помощью кристаллизации с 2-бутанол-водой для фармацевтической степени чистоты в соответствии с национальной или Европейской фармакопеей.
Борную кислоту или ее производное, используемые в вышеуказанных вариантах осуществления способа, восстанавливают, как описано выше, и предпочтительно повторно применяют в способе.
Новое Соединение формулы (I) могут получить в соответствии с различными вариантами осуществления. Например, исходя из Соединения (IV), как описано ниже, или другими путями синтеза, раскрытыми в настоящем описании.
Хотя в способе получения Йопамидола (II) Соединение (I) необязательно выделяют, оно может быть выделено, например, после взаимодействия с Соединением (IV), как показано на вышеприведенной Схеме 3, в целях характеристики, то есть с помощью 1H-ЯМР. Выделение Соединения (I) может быть проведено в соответствии со стандартными способами обработки, хорошо известными в данной области техники, такими как, например, экстракция, осаждение, хроматографическое разделение. Один примерный способ для характеристики Соединения (I) представляет собой растворение его в подходящем растворителе, например, в диметилацетамиде, добавляя растворитель осаждения, например, толуол и выделяя образовавшийся осадок. В целях удобства осадок могут повторно растворить, например, нагревание раствора осадка и последующее охлаждение. Полученное белое твердое вещество выделяют, например, с помощью фильтрования. Аналитическое исследование может быть сделано в соответствии с хорошо известными способами, например, элементного анализа, температуры плавления, спектроскопии (такой как ЯМР, ИК), как лучше подробно описано в Экспериментальной Части.
В другом аспекте настоящего изобретения Йопамидол (II) получают исходя из вышеуказанного Соединения формулы (IV).
Данное соединение представляет собой хорошо известный интермедиат, так как его получение изложено в предшествующем уровне техники синтезов Йопамидола (II), смотри, например, WO0244125 и противопоставленные там материалы.
В соответствии с настоящим изобретением данное Соединение (IV) обрабатывают борной кислотой или ее производным для получения Соединения формулы (I), как раскрыто выше.
Борная кислота и ее производные, используемые в настоящем изобретении, являются хорошо известными соединениями, которые являются коммерчески доступными или могут быть получены в соответствии с описанными в литературе способами. Например, эфиры борной кислоты B(OR2)3, в которой R2 представляет собой, как определено выше, бороновые кислоты R3-В(ОН)2 и бороксин формулы (III),
в которой R3 имеет одинаковое значение, и предпочтительные варианты осуществления, как определено выше, описаны вместе с их получением в литературе общего содержания, такой как, например, Ullmann’s Encyclopedia of Industrial Chemistry, VCH, последнее издание; Dennis G. Hall (ed.) Boronic Acids, Wiley VCH, последнее изд.; March’s Advanced Organic Chemistry, Wiley, последнее изд.; Kirk Othmer Encyclopedia of Chemical Technology, Wiley, последнее изд.; Lawrence Barton et al. (eds.) Boron Compounds, Springer Verlag, 1977.
Только для иллюстративного примера эфиры борной кислоты получают в соответствии с общей реакцией:
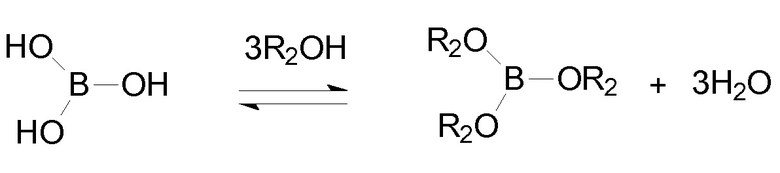
в которой R2 представляет собой, как определено выше.
В одном варианте осуществления настоящего изобретения Соединение (IV) обрабатывают борной кислотой и спиртом R2OH в реакционной среде. Реакционная среда представляет собой растворитель, совместимый с условиями реакции, реагентами и конечным продуктом. В целях удобства реакционная среда представляет собой тот же органический растворитель, используемый в реакции из Соединения (I) в Йопамидол (II) (стадия а) (см. Схему 2). В предпочтительном варианте осуществления N,N-диметилацетамид является растворителем. Реакцию проводят при температуре, составляющей от 60°С до 100°С или выше в течение времени, достаточного для завершения. Проверку завершения реакции, то есть пока содержание воды не станет минимальным или очень низким, например, <0,5%, проводят с помощью стандартных способов, например, с помощью 1H-ЯМР или с помощью определения содержания воды в реакционной смеси, то есть с помощью титрования по Карлу Фишеру. Полученное Соединение (I) могут выделить или непосредственно обработать хиральным ацилирующим агентом (S)-2-(ацетилокси)пропаноилхлоридом в том же растворителе реакции, обеспечивая таким образом способ «в одном реакционном сосуде». Специалист в данной области техники знает значение термина способ «в одном реакционном сосуде», и никаких дополнительных объяснений не требуется.
В другом варианте осуществления настоящего изобретения Соединение (IV) непосредственно обрабатывают эфиром борной кислоты B(OR2)3 в реакционной среде. Реакция может быть проведена как и в случае с вышеприведенным вариантом осуществления борной кислоты и спирта. Соединение (I) может быть или выделено для последующей реакции, или использовано в способе «в одном реакционном сосуде».
Предпочтительные бораты выбраны из группы, состоящей из трет-бутил-, н-пропил- и этилбората. Также могут быть использованы эфиры с различными алкильными группами. Реакция может быть проведена, как и в случае с вышеприведенным вариантом осуществления борной кислоты и спирта. Соединение (I) может быть или выделено для последующей реакции, или использовано в способе «в одном реакционном сосуде».
В другом варианте осуществления настоящего изобретения Соединение (IV) обрабатывают ангидридом бороновой кислоты или бороксином формулы (III) выше. Предпочтительные бороксины представляют собой три(фенил)бороксин и три(метил)бороксин. Реакция может быть проведена как и в случае с вышеприведенным вариантом осуществления бороновой кислоты. Соединение (I) может быть или выделено для последующей реакции, или использовано в способе «в одном реакционном сосуде».
В предпочтительном варианте осуществления реакцию между Соединением (IV) и борной кислотой или ее производным проводят в течение определенного времени и до перехода к следующей стадии N-ацилирования до Йопамидола (II), целесообразно удалить часть растворителя, например, с помощью перегонки, даже лучше с помощью вакуумной перегонки, чтобы контролировать содержание воды, предпочтительно пока содержание воды не станет минимальным или, по меньшей мере, очень низким, например, <0,5% (определено с помощью титрования по Карлу Фишеру).
Достойный внимания подход диольной защиты с помощью борпроизводных, используемый в соответствии с настоящим изобретением, может быть использован также для получения других йодсодержащих рентгеноконтрастных веществ, кроме Йопамидола, таких как, например, Йомепрол, Йодиксанол, Йоверсол, Йогексол, Йопромид и т.д.
В другом аспекте настоящее изобретение обеспечивает способ получения Йопамидола (II), как показано на вышеприведенной Схеме 4. 5-Амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-1,3-бензолдикарбоксамид (Соединение (V)) является хорошо известным соединением и одно из его получений раскрыто, например, в WO0244125. В соответствии с настоящим изобретением данное Соединение подвергают йодированию бензольного кольца с помощью способа, известного в данной области техники, например, как раскрыто в том же WO0244125 и противопоставленных там материалах, для получения 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамида (Соединение (IV)).
В другом аспекте настоящее изобретение обеспечивает способ получения Йопамидола (II), как показано на вышеприведенной Схеме 5. Исходя из 5-нитро-1,3-бензолдикарбоновой кислоты (5-нитроизофталевой кислоты или 5-NIPA) получают, например, 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-1,3-бензолдикарбоксамид (V), как раскрыто в WO0244125 и WO0029372.
В соответствии с настоящим изобретением данное Соединение (V) подвергают йодированию бензольного кольца, как указано выше, для получения 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамида (Соединение (IV)).
Соединение (IV) затем обрабатывают в соответствии с настоящим изобретением до Йопамидола (II) через интермедиат (I).
Предпочтительно, может быть проведен способ «в одном реакционном сосуде».
Предпочтительный путь синтеза Соединения (V) описан на Схеме 6 ниже, показанный для одного примерного варианта осуществления:
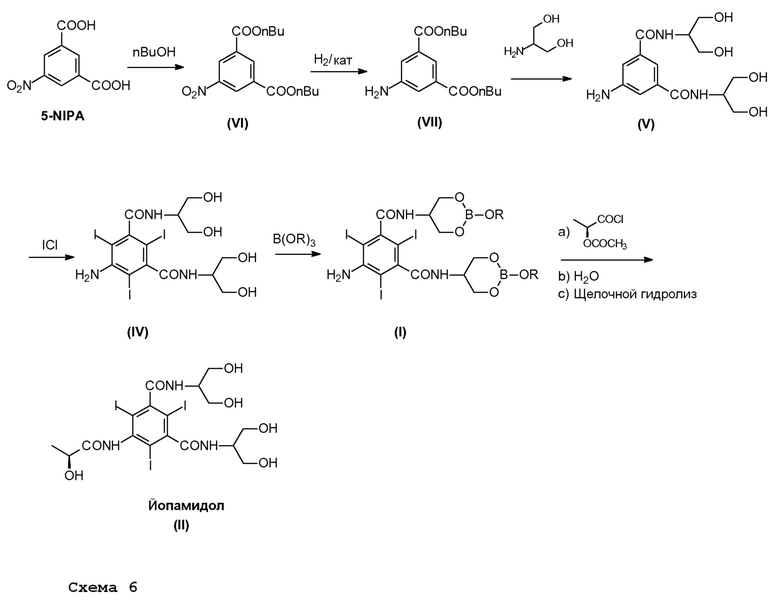
Схема 6 представляет собой путь синтеза Йопамидола (II) из 5-NIPA в соответствии с примерным вариантом осуществления.
Первая часть синтеза из NIPA в Соединение (V) представляет собой синтез «в одном реакционном сосуде» без выделения промежуточного соединения. Он состоит из трех стадий (этерификация, гидрирование, амидирование), проводимых в подходящем растворителе, например, н-бутанол в случае н-бутилового эфира является предпочтительным. Этерификацию проводят в присутствии хорошо известного катализатора этерификации, например, кислотного катализатора, такого как пара-толуолсульфоновая кислота, метансульфоновая кислота, серная кислота, предпочтительно метансульфоновая кислота. В дальнейшем проводят гидрирование, как известно специалисту в данной области техники, например, как описано в EP1337505 и предпочтительно с помощью каталитического гидрирования в присутствии 5% Pd/C или другого эквивалентного катализатора. В данном случае после удаления катализатора смесь концентрируют для следующей стадии амидирования, которую проводят по существу с помощью двух альтернативных способов:
- Амидирование без примесей, без растворителя и с соответствующим избытком серинола, который восстанавливают с помощью анионообменной смолы и повторно используют в реакции. Соединение (V) не выделяют, а непосредственно переносят в новый реакционный сосуд для стадии йодирования.
- Амидирование в присутствии органического растворителя и сорастворителя, как описано в EP1337505, предпочтительно в метаноле в присутствии основного катализатора. В ходе реакции происходит осаждение и полученное Соединение (V) отфильтровывают. Твердое вещество непосредственно повторно растворяют в воде и переносят в следующий реакционный сосуд для стадии йодирования.
Предпочтительный вариант осуществления представлен амидированием в присутствии органического растворителя и сорастворителя.
Реакцию йодирования проводят в водном растворе, содержащем Соединение (V), синтезированное в результате одного из двух вышеупомянутых подходов. Методику йодирования проводят в соответствии с хорошо известными способами, смотрите выше для родственных ссылок, с получением промежуточного соединения (IV). Затем способ протекает, как раскрыто выше, в соответствии со стадиями а)-с).
В соответствии с дополнительным вариантом осуществления изобретение относится к способу восстановления бороновых кислот из реакционной смеси, где они используются в качестве диольных защитных групп. Это обеспечивает их повторное использование для той же цели в новом синтезе. Даже если данное восстановление и повторное использование предпочтительно проводят в способе получения Йопамидола в соответствии с настоящим изобретением и как описано выше, восстановление представляет собой более общий вариант осуществления для повторно используемых бороновых кислот, так как оно обеспечивает количественные выходы (обычно >90%, предпочтительно >95% и более предпочтительно, по меньшей мере, 99%) данных обычно дорогих реагентов, таким образом представляя собой значительное экономическое преимущество для крупномасштабного развития промышленности.
Еще большее преимущество можно ожидать, когда данный способ проводят в непрерывном режиме и в промышленных способах, в которых восстановление и повторное использование могут быть оптимизированы.
В соответствии с данным вариантом осуществления водную реакционную смесь, полученную после гидролиза диольных защитных групп, которая включает бороновую кислоту, используемую для диольной защиты, или в которой бороновая кислота образуется после гидролиза (например, когда бороксин Формулы III используется для диольной защиты) и которая необязательно содержит полярный растворитель, такой как: N,N-диметилформамид, N,N-диметилацетамид (DMAC), N,N-диэтилацетамид, N,N-диметилпропионамид, N-метилпирролидон, N-этилпирролидон, тетраметилмочевина, N,N'-диметилэтиленмочевина (DMEU), N,N'-диметилпропиленмочевина (DMPU), добавляют с органическим несмешивающимся с водой растворителем.
Органический несмешивающийся с водой растворитель выбран из: 4-метил-2-пентанона (МИБК), 3-пентанона, 2-пентанона, дибутилового эфира, 2-метилтетрагидрофурана, циклопентилметилового эфира, метилизопропилкетона, метилизопентилкетона, этилацетата, бутилацетата, пентилацетата, изопентилацетата, изопропилацетата.
Предпочтительное соотношение между бороновой кислотой и несмешивающимся с водой растворителем (экстракционный растворитель бороновой кислоты) составляет от 1:10-1:20 (масс./масс.), более предпочтительно составляет от 1:13-1:16 и еще более предпочтительно приблизительно 1:15.
Бороновая кислота, используемая в качестве борсодержащей диольной защитной группы, восстановленная в соответствии с данным вариантом осуществления, представляет собой предпочтительно фенилбороновую, п-толилбороновую или бутилбороновую кислоту, или когда используют фенилбороксин или метилбороксин для диольной защиты, фенилбороновая кислота или метилбороновая кислота может быть восстановлена после гидролиза.
Несмешивающийся с водой органический растворитель может быть дополнительно повторно экстрагирован с помощью аликвоты водного раствора с рН, составляющем от 0-7 или предпочтительно близким к нейтральному при рН, составляющем от 6 до 7, когда восстановление проводят после гидролиза Соединения I и в соответствии с настоящим изобретением, чтобы улучшить селективность восстановления бороновой кислоты.
Отделение бороновой кислоты в органической фазе для ее восстановления может быть достигнуто или в периодическом, или в непрерывном режиме с помощью методов оптимизации, известных специалисту в данной области техники.
Следующие примеры дополнительно иллюстрируют изобретение более подробно.
Пример 1: Получение Йопамидола (II) исходя из Соединения (IV) и с использованием бороновой кислоты
Ссылаясь на Схему 3, 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV) (1 кг; 1,42 моль) и фенилбороновую кислоту (Х=Ph) (363 г; 2,98 моль) смешивали в N,N-диметилацетамиде (4 кг). Суспензию перемешивали и нагревали при 90-95°С. Полученный раствор нагревали при 90-95°С в течение 1 ч, затем N,N-диметилацетамид (приблизительно 3 кг) отгоняли под вакуумом и доводили до содержания воды менее 0,5%, оцененного с помощью титрования по Карлу Фишеру. В этот момент завершали образование промежуточного соединения (I) (оцененное по данным 1H-ЯМР). Остаток охлаждали до 30-35°С и в атмосфере азота медленно добавляли (S)-2-(ацетилокси)пропаноилхлорид (380 г; 2,52 моль). Смесь перемешивали в течение 18 ч при комнатной температуре и в атмосфере азота, затем добавляли воду для получения разбавленного раствора, пригодного для хроматографической очистки. После 1 ч перемешивания раствор загружали в колонку Amberlite® IRA743 (17 л) (Dow Chemical Company) и элюировали водой (3 объема слоя). Раствор загружали в колонку Relite® 3ASFB (анионообменная смола; 4 л) и элюат выливали. Колонку Relite 3ASFB затем последовательно элюировали 2 объемами слоя водного кислотного раствора (разбавленной хлористоводородной кислоты) и промывали 3-4 ОС (объемами слоя) воды, количественно восстанавливая субстрат. Полученный раствор нейтрализовали до рН 7, концентрировали с помощью вакуумной дистилляции более 2 часов. Раствор загружали в колонку Amberlite® XAD 1600 (3,6 л) (Dow Chemical Company) и элюировали 4 ОС сильно разбавленного раствора гидроксида натрия. Раствор загружали в две колонки с ионообменной смолой (катионообменная Dowex® C350, 4,7 л; анионообменная Relite® MG1/P 2 л, Dow Chemical Company). Элюат концентрировали и сухой остаток кристаллизовали из 2-бутанола с получением Йопамидола (II) (904 г; 1,16 моль) в виде чистого белого твердого вещества с выходом 82%.
1H-ЯМР, 13С-ЯМР, ИК и МС соответствуют указанной структуре.
Фенилбороновую кислоту восстанавливали с выходом >90%.
Идентичную методику использовали с н-бутилбороновой кислотой (X=n-Bu) и Йопамидол (II) выделяли с выходом 80%.
Пример 2: Получение Йопамидола (II) исходя из Соединения (IV) и с использованием бороновой кислоты
Ссылаясь на Схему 3, 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV) (1 кг; 1,42 моль) и фенилбороновую кислоту (Х=Ph) (363 г; 2,98 моль) смешивали в N,N-диметилацетамиде (4 кг). Суспензию перемешивали и нагревали при 90-95°С. Полученный раствор нагревали при 90-95°С в течение 1 ч, затем N,N-диметилацетамид (приблизительно 3 кг) отгоняли под вакуумом. Остаток должен был иметь содержание воды менее 0,5%, оцененное с помощью титрования по Карлу Фишеру. В этот момент завершали образование промежуточного соединения (I), как определено с помощью 1H-ЯМР на аликвоте реакционной смеси, высушивали и остаток обрабатывали, как описано ниже. Остаток охлаждали до 30-35°С и в атмосфере азота медленно добавляли (S)-2-(ацетилокси)пропаноилхлорид (384 г; 2,55 моль). Смесь перемешивали в течение 18 ч при комнатной температуре и в атмосфере азота, затем добавляли воду (9 кг). После 1 ч перемешивания раствор загружали в колонку Relite MG1/P (2,2 л) и элюировали водой (15 л). Раствор экстрагировали 4-метил-2-пентаноном (3×4 л) и водную фазу загружали в колонку Relite 3ASFB для гидролиза, последовательно элюировали 2 объемами слоя водного кислотного раствора (разбавленной хлористоводородной кислоты) и промывали 3-4 объемами слоя воды, количественно восстанавливая субстрат. Полученный раствор нейтрализовали до рН 7, концентрировали с помощью вакуумной дистилляции более 2 часов. Раствор загружали в колонку Amberlite® XAD 1600 (3,6 л) и элюировали 4 ОС сильно разбавленного раствора гидроксида натрия. Раствор загружали в две колонки с ионообменной смолой (катионообменная Dowex® C350, 4,7 л; анионообменная Relite® MG1/P, 2 л). Элюат концентрировали и сухой остаток кристаллизовали из 2-бутанола с получением Йопамидола (II) (959 г; 1,23 моль) в виде чистого белого твердого вещества с выходом 87%. 1H-ЯМР, 13С-ЯМР, ИК и МС соответствуют указанной структуре.
Фенилбороновую кислоту восстанавливали с выходом >95%. Растворитель отгоняли и концентрированный раствор непосредственно повторно применяли в синтезе.
Фенилбороновую кислоту также экстрагировали 3-пентаноном с сопоставимыми результатами.
Методика выделения Соединения (I):
Защищенный интермедиат (12,4 г) повторно растворяли в диметилацетамиде (10 г) и добавляли толуол (100 мл) для образования осадка. Раствор нагревали при 60°С более 30 минут и осадок повторно растворяли; раствор охлаждали до 5°С более 2 ч и полученное твердое вещество отфильтровывали, получая белое твердое вещество. Аналитическое исследование находится в соответствии с предложенной структурой.
Температура плавления = 180-185°C.
1H-ЯМР (ДМСО-d6) (м.д.): 4,07 (дд, 1H, 7), 4,31 (дд, 1H, 7), 4,38 (м, 1H, 6), 7,35 (т, 1H, 10), 7,43 (т, 1H, 11), 7,70 (д, 1H, 9), 9,12 (д, 1H, CONH).
13С-ЯМР (ДМСО-d6) (м.д.): 45,35 (С6), 64,05 (С7), 74,45 (C4), 80,30 (C2), 127,94 (С10), 131,06 (С11), 133,00 (С8), 134,05 (С9), 147,88 (С1), 148,82 (С3), 170,20 (С5).
Пример 3: Получение Йопамидола (II) исходя из Соединения (IV) и с использованием бороновой кислоты
Ссылаясь на Схему 3, 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV) (1 кг; 1,42 моль) и фенилбороновую кислоту (Х=Ph) (360 г; 2,95 моль) смешивали в N,N-диметилацетамиде (0,76 кг) и метилизобутилкетоне (МИБК) (3,24 кг). Суспензию перемешивали и нагревали при 90-95°С, затем смесь МИБК/вода (2,8 кг) отгоняли под вакуумом и доводили до содержания воды менее 0,5%, оцененного с помощью титрования по Карлу Фишеру, получая прозрачный желтый раствор. В этот момент завершали образование промежуточного соединения (I) (оцененное по данным 1H-ЯМР). Остаток охлаждали до 30-35°С и в атмосфере азота медленно добавляли (S)-2-(ацетилокси)пропаноилхлорид (380 г; 2,52 моль). Смесь перемешивали в течение 18 ч при 30-35°С и в атмосфере азота, затем добавляли разбавленный раствор NaOH до нейтрального рН. Другую часть метилизобутилкетона (4-5 кг) добавляли к двухфазной смеси и фенилбороновую кислоту экстрагировали. Водную фазу загружали в две колонки с ионообменной смолой (катионообменная смола, Dowex® C350, 2 л; анионообменная смола Relite® MG1/P, 2,6 л). Колонки элюировали 2 ОС воды. Полученный раствор загружали в колонку Relite 3ASFB для гидролиза: Йопамидол (II) восстанавливали с помощью элюирования 2 ОС водного кислотного раствора (разбавленной хлористоводородной кислоты) и промывали 3-4 объемами слоя воды. Полученный раствор нейтрализовали до рН 7, концентрировали с помощью вакуумной дистилляции более 2 часов. Раствор загружали в колонку Amberlite® XAD 1600 (3,6 л) и элюировали 4 ОС сильно разбавленного раствора гидроксида натрия. Раствор загружали в две колонки с ионообменной смолой (катионообменная Dowex® C350, 4,7 л; анионообменная Relite® MG1/P, 2 л). Элюат концентрировали и сухой остаток кристаллизовали из 2-бутанола с получением Йопамидола (II) (992 г; 1,28 моль) в виде белого твердого вещества. Выход 90%.
PBA восстанавливали с выходом 95%.
Пример 4: Получение Йопамидола (II) исходя из Соединения (IV) и с использованием бороновой кислоты
Синтез проводили по существу, как описано в Примере 3. Кратко и ссылаясь на Схему 3, 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV) (1 кг; 1,42 моль) и фенилбороновую кислоту (Х=Ph) (360 г; 2,95 моль) смешивали в N,N-диметилацетамиде (0,76 кг) и метилизобутилкетоне (МИБК) (3,24 кг). Суспензию перемешивали и нагревали при 90-95°С, затем смесь МИБК/вода (2,8 кг) отгоняли под вакуумом и доводили до содержания воды менее 0,5%, оцененного с помощью титрования по Карлу Фишеру, получая прозрачный желтый раствор. В этот момент завершали образование промежуточного соединения (I), как оценивали с помощью 1Н-ЯМР. Остаток охлаждали до 30-35°С и в атмосфере азота медленно добавляли (S)-2-(ацетилокси)пропаноилхлорид (380 г; 2,52 моль). Смесь перемешивали в течение 18 ч при 30-35°С и в атмосфере азота, затем добавляли разбавленный раствор NaOH (9 кг) до нейтрального рН. РВА экстрагировали тремя порциями метилизобутилкетона (3×3,2 кг). Водную фазу загружали в две колонки с ионообменной смолой (катионообменная смола Dowex® C350, 2 л; анионообменная смола Relite® MG1/P, 2,6 л). Колонки элюировали водой. Полученный раствор загружали в колонку Relite 3ASFB для гидролиза: Йопамидол (II) восстанавливали с помощью элюирования водным кислотным раствором (разбавленной хлористоводородной кислотой) и промывали водой. Полученный раствор загружали в колонку Relite® MG1/P (1,8 л), Amberlite® XAD 1600 (3,6 л) и катионообменную Dowex® С350 (0,2 л) и элюировали 4 ОС сильно разбавленного раствора гидроксида натрия. Элюат концентрировали и твердый остаток кристаллизовали из 2-бутанола с получением Йопамидола (II) (1003 г; 1,29 моль) в виде белого твердого вещества. Выход 91%.
PBA восстанавливали с выходом 95%.
Пример 5: Получение Йопамидола (II) исходя из Соединения (IV) и с использованием повторно применяемой фенилбороновой кислоты (РВА)
Было исследовано, могла ли восстановленная фенилбороновая кислота быть повторно использована с помощью непосредственного добавления к реакционной смеси.
Ссылаясь на Схему 3, органическую фазу (МИБК-содержащей фенилбороновой кислоты), полученную из экстракции фенилбороновой кислоты и восстановленную из реакционной смеси, полученной в примере 4, использовали для проведения синтеза новой партии Йопамидола. DMAC (0,6 кг) сначала добавляли к органической смеси и раствор отгоняли под вакуумом при <40°C, концентрируя РВА, чтобы достичь подходящую концентрацию. В дальнейшем органический раствор, содержащий 95% требуемой восстановленной фенилбороновой кислоты, смешивали с 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамидом (IV) (1 кг; 1,42 моль) и добавляли свежую фенилбороновую кислоту (Х=Ph) (18 г; 0,15 моль), что соответствует приблизительно 5% общего требуемого количества. Суспензию перемешивали и нагревали при 90-95°С, и реакция продолжалась, как описано в Примере 4, обеспечивая выход Йопамидола, сопоставимый с выходом, полученным с твердой (свежей) РВА.
Пример 6. Получение Йопамидола (II) исходя из Соединения (IV) с фенилбороновой кислотой (РВА) в DMAC и сорастворителе. Восстановление PBA.
Синтез проводили по существу, как описано в Примере 4, но с использованием восстановленных объемов. Кратко: 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV) (1 кг; 1,42 моль) и фенилбороновую кислоту (Х=Ph) (360 г; 2,95 моль) смешивали в N,N-диметилацетамиде (0,76 кг) и метилизобутилкетоне (МИБК) (3,24 кг). Суспензию перемешивали и нагревали при 90-95°С, затем смесь МИБК/вода (2,8 кг) отгоняли под вакуумом и доводили до содержания воды менее 0,5%, оцененного с помощью титрования по Карлу Фишеру, получая прозрачный желтый раствор. В этот момент завершали образование промежуточного соединения (I), как определено с помощью 1H-ЯМР. Остаток охлаждали до 30-35°С и в атмосфере азота медленно добавляли (S)-2-(ацетилокси)пропаноилхлорид (380 г; 2,52 моль). Смесь перемешивали в течение 18 ч при 30-35°С и в атмосфере азота, затем добавляли NaOH (2,7 кг) до нейтрального рН. Другую часть метилизобутилкетона (2 кг) добавляли к двухфазной смеси и экстрагировали фенилбороновую кислоту. Органическую фазу промывали водой (0,9 кг) и водные фазы собирали и экстрагировали двумя другими частями МИБК (2 кг). Менее 0,8% ацетил-Йопамидола переходило в органическую фазу. Водную фазу загружали в две колонки с ионообменной смолой и продолжали обработку, как описано выше.
Органическую фазу (МИБК-содержащей фенилбороновой кислоты), полученную из экстракции фенилбороновой кислоты и восстановленную из реакционной смеси, использовали для проведения синтеза новой партии Йопамидола, по существу, как описано в Примере 5. Кратко, DMAC (0,6 кг) сначала добавляли к органической смеси и раствор отгоняли под вакуумом при <40°C, удаляя 2-2,3 кг МИБК, чтобы получить конечное количество 3,65 кг МИБК. В дальнейшем органический раствор, содержащий 95% требуемой восстановленной фенилбороновой кислоты, смешивали с 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамидом (IV) (1 кг; 1,42 моль) и добавляли свежую фенилбороновую кислоту (Х=Ph) (18 г; 0,15 моль), что соответствует приблизительно 5% общего требуемого количества. Суспензию перемешивали и нагревали при 90-95°С, и реакция продолжалась, как описано предварительно, до получения Йопамидола.
Пример 7. Получение Йопамидола (II) исходя из Соединения (IV) с фенилбороновой кислотой (РВА) в DMAC и сорастворителе. Восстановление PBA с помощью 2-пентанона.
Способ проводили по существу, как описано в Примере 4, с использованием 2-пентанона вместо МИБК. Кратко: 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV) (1 кг; 1,42 моль) и фенилбороновую кислоту (Х=Ph) (360 г; 2,95 моль) смешивали в N,N-диметилацетамиде (0,76 кг) и 2-пентаноне (3,24 кг). Суспензию перемешивали и нагревали при 90-95°С, затем смесь 2-пентанон/вода (2,8 кг) отгоняли под легким вакуумом и доводили до содержания воды менее 0,5%, оцененного с помощью титрования по Карлу Фишеру, получая прозрачный желтый раствор. В этот момент завершали образование промежуточного соединения (I), как определено с помощью 1H-ЯМР. Остаток охлаждали до 30-35°С и в атмосфере азота медленно добавляли (S)-2-(ацетилокси)пропаноилхлорид (380 г; 2,52 моль). Смесь перемешивали в течение 18 ч при 30-35°С и в атмосфере азота, затем добавляли раствор NaOH (4,4 кг) до нейтрального рН. Другую часть 2-пентанона (2,4 кг) добавляли к двухфазной смеси и экстрагировали фенилбороновую кислоту. Экстракцию повторяли еще 2 раза 2-пентаноном (1,6 кг×2). Количественное восстановление РВА достигали, но приблизительно 4% ацетил-Йопамидола было обнаружено в органической фазе.
Методику повторяли с помощью промывания органической фазы (2-пентанона) водой, таким образом восстанавливая большее количество продукта и обеспечивая эквивалентное восстановление РВА.
Пример 8. Восстановление РВА с помощью использования разбавленного раствора NaOH и увеличенных объемов растворителя.
Способ проводили по существу, как описано в Примерах 6 и 7, но с использованием увеличенных объемов более разбавленного раствора NaOH. В данном случае водную фазу повторно экстрагировали приблизительно в два раза увеличенным объемом растворителя для достижения такого же восстановления ацетил-Йопамидола и РВА.
Подобную обработку проводили с 3-пентаноном, 2-пентаноном, метилизопропилкетоном, метилизопентилкетоном и циклопентилметиловым эфиром, что обеспечивало сопоставимые выходы синтеза Йопамидола, также как выходы восстановления одновременно Йопамидола и РВА.
Пример 9. Получение Йопамидола (II) исходя из Соединения (IV) и с использованием п-толилбороновой кислоты
Ссылаясь на Схему 3, 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV) (1 кг; 1,42 моль) и п-толилбороновую кислоту (405 г; 2,98 моль) смешивали в N,N-диметилацетамиде (0,75 кг) и метилизобутилкетоне (МИБК) (3,25 кг). Суспензию перемешивали и нагревали при 90-95°С, затем смесь МИБК/вода (2,8 кг) отгоняли под вакуумом и доводили до содержания воды менее 0,5%, оцененного с помощью титрования по Карлу Фишеру, получая прозрачный желтый раствор. В этот момент завершали образование промежуточного соединения (I), как определено с помощью 1H-ЯМР. Остаток охлаждали до 30-35°С и в атмосфере азота медленно добавляли (S)-2-(ацетилокси)пропаноилхлорид (380 г; 2,52 моль). Смесь перемешивали в течение 18 ч при 30-35°С и в атмосфере азота, затем добавляли разбавленный раствор NaOH до нейтрального рН. Другую часть метилизобутилкетона (4 кг) добавляли к двухфазной смеси и экстрагировали п-толилбороновую кислоту. Водную фазу загружали в две колонки с ионообменной смолой (катионообменная смола Dowex® C350, 2 л; анионообменная смола Relite® MG1/P, 2,6 л) и продолжали обработку, как описано в Примере 4, с получением Йопамидола (II) (992 г; 1,28 моль) в виде белого твердого вещества. Выход 90%.
Пример 10: Получение Йопамидола (II) исходя из Соединения (IV) и с использованием бутилбороновой кислоты
Ссылаясь на Схему 3, 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV) (1 кг; 1,42 моль) и бутилбороновую кислоту (303,6 г; 2,98 моль) смешивали в N,N-диметилацетамиде (4,0 кг). Суспензию перемешивали и нагревали при 90-95°С, затем смесь DMA/вода (2,8 кг) отгоняли под вакуумом и доводили до содержания воды менее 0,5%, оцененного с помощью титрования по Карлу Фишеру, получая прозрачный желтый раствор. В этот момент завершали образование промежуточного соединения (I), как определено с помощью 1H-ЯМР. Остаток охлаждали до 25°С и в атмосфере азота медленно добавляли (S)-2-(ацетилокси)пропаноилхлорид (380 г; 2,52 моль). Смесь перемешивали в течение 18 ч при КТ и в атмосфере азота, затем добавляли разбавленный раствор NaOH до нейтрального рН. Часть метилизобутилкетона (5 кг) добавляли к двухфазной смеси и экстрагировали бутилбороновую кислоту. Водную фазу загружали в две колонки с ионообменной смолой (катионообменная смола Dowex® C350, 2 л; анионообменная смола Relite® MG1/P, 2,6 л) и продолжали обработку, как описано в Примере 4, с получением Йопамидола (II) (992 г; 1,28 моль) в виде белого твердого вещества. Выход 90%.
Пример 11: Получение Йопамидола (II) исходя из Соединения (IV) и с использованием бороксина
Ссылаясь на Схему 3, 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV) (255 г; 0,362 моль) и трифенилбороксин (Соединение (III), R3 = Ph) (82,3 г; 0,264 моль) смешивали в N,N-диметилацетамиде (1 кг). Суспензию перемешивали и нагревали при 90-95°С. Полученный раствор нагревали при 90-95°С в течение 1 ч, затем N,N-диметилацетамид (приблизительно 700 г) отгоняли под вакуумом. Остаток должен был иметь содержание воды менее 0,5%, оцененное с помощью титрования по Карлу Фишеру. В этот момент завершали образование промежуточного соединения (I) (оцененное по данным 1H-ЯМР). Остаток охлаждали до 30-35°С и в атмосфере азота медленно добавляли (S)-2-(ацетилокси)пропаноилхлорид (98 г; 0,651 моль). Смесь перемешивали в течение 18 ч при комнатной температуре и в атмосфере азота, затем добавляли воду (190 г). После 1 ч перемешивания раствор очищали с помощью элюирования на серии колонок, как описано в Примере 4, с получением Йопамидола (II) (219 г; 0,282 моль) в виде белого твердого вещества.
Выход: 78%. 1H-ЯМР, 13С-ЯМР, ИК и МС соответствуют указанной структуре.
Идентичную методику использовали с триметилбороксином (III) (R3 = Me); выход Йопамидола (II) представлял собой 75%.
Пример 12: Получение Йопамидола (II) исходя из Соединения (IV) и с использованием эфира борной кислоты
Ссылаясь на Схему 3, 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV) (200 г; 0,284 моль) в N,N-диметилацетамиде (800 г) нагревали при 60°С с получением раствора, затем добавляли три-н-бутилборат (X=OR2, R2 = n-Bu) (137,2 г; 0,596 моль). Раствор перемешивали и нагревали при 105°С в течение 2 ч, затем N,N-диметилацетамид и н-бутанол отгоняли под вакуумом, собирая приблизительно 730 г дистиллята. Большее количество N,N-диметилацетамида (95 г) добавляли к реакционной смеси и отгоняли под вакуумом. В этот момент завершали образование промежуточного соединения (I) (оцененное по данным 1H-ЯМР). Остаток охлаждали до комнатной температуры и в атмосфере азота медленно добавляли (S)-2-(ацетилокси)пропаноилхлорид (85,5 г; 0,568 моль). Смесь перемешивали в течение 18 ч при комнатной температуре и в атмосфере азота, затем добавляли воду (1,5 кг), снимая защиту с гидроксильных групп и получая разбавленный раствор, пригодный для хроматографической очистки. После 1 ч перемешивания раствор загружали в колонку XAD® 1600 (4 л), смолу промывали водой (3 ОС) и элюат, содержащий борную кислоту, N,N-диметилацетамид и бутанол, загружали в колонку IRA743 (4,1 л) для восстановления DMAC, бутанола и борной кислоты. Ацетил-Йопамидол элюировали из XAD 1600 с помощью NaOH (0,20% масс./масс.; 5 ОС), концентрировали под вакуумом до конечного объема 2 л и гидролизовали в периодическом режиме с помощью NaOH при рН 12 при 35°С более 20-24 часов. Раствор загружали в две колонки с ионообменной смолой (катионообменная Amberjet® 1200, 0,9 л; анионообменная Relite® MG1, 0,8 л). Элюат концентрировали и твердый остаток кристаллизовали из 2-бутанола с получением Йопамидола (II) (159 г; 0,205 моль) в виде белого твердого вещества.
Выход: 72%. 1H-ЯМР, 13С-ЯМР, ИК и МС соответствуют указанной структуре.
Идентичную методику использовали с триэтилборатом и три-н-пропилборатом.
Пример 13: Получение Йопамидола (II) исходя из Соединения (IV) и с использованием борной кислоты и спирта.
Ссылаясь на Схему 3, 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамид (IV) (200 г; 0,284 моль), n-BuOH (600 г, 8,10 моль) и борную кислоту (36,8 г, 0,60 моль) суспендировали в N,N-диметилацетамиде (720 г) и нагревали при 60°С с получением однородного раствора. Раствор перемешивали и нагревали при 90°С в течение 1 ч, затем N,N-диметилацетамид и н-бутанол отгоняли под вакуумом более 4 ч, собирая приблизительно 1,1 кг дистиллята. В этот момент завершали образование промежуточного соединения (I) (оцененное по данным 1H-ЯМР). Остаток охлаждали до комнатной температуры и в атмосфере азота медленно добавляли (S)-2-(ацетилокси)пропаноилхлорид (85,28 г; 0,57 моль). Смесь перемешивали более 18 ч при комнатной температуре в атмосфере азота, затем добавляли воду (1,5 кг). После 1 ч перемешивания раствор загружали в колонку XAD® 1600 (4 л), смолу промывали водой (3 ОС) и элюат, содержащий борную кислоту, DMAC и n-BuOH, загружали в колонку IRA743 (4,1 л) для восстановления DMAC, n-BuOH и борной кислоты. Ацетил-Йопамидол элюировали из XAD 1600 с помощью NaOH (0,20% масс./масс.; 5 объемов слоя), концентрировали под вакуумом до конечного объема 2 л и гидролизовали в периодическом режиме с помощью NaOH при рН 12 и при 35°С. Раствор загружали в две колонки с ионообменной смолой (катионообменная Amberjet® 1200, 0,9 л; анионообменная Relite® MG1, 0,8 л). Элюат концентрировали и твердый остаток кристаллизовали из 2-бутанола с получением Йопамидола (II) (170 г; 0,218 моль) в виде белого твердого вещества. Выход: 77%.
Пример 14: Методика: Синтез в одном реакционном сосуде исходя из 5-нитроизофталевой кислоты в Соединение (V).
Ссылаясь на Схему 6, 5-нитроизофталевую кислоту (NIPA; 100 г; 0,47 моль) растворяли в бутаноле (600 г) в присутствии каталитического количества моногидратированной п-толуолсульфоновой кислоты (9,01 г; 0,047 моль). Смесь нагревали при 125°С и воду удаляли с помощью азеотропной дистилляции. Промежуточное соединение (VI) получали с количественным превращением (>98%). Однородный раствор гидрировали без выделения в присутствии 5% Pd/C (3,0 г) в качестве катализатора. Полученную суспензию поддерживали при механическом перемешивании и продували азотом, в конце которого проводили реакцию гидрирования при температуре, находящейся в диапазоне от 50 до 70°С. Реакцию завершали через 4-8 часов (132,76 г; 0,453 моль). Поток азота пропускали через реакционный сосуд, чтобы промыть газ водород, катализатор отфильтровывали и полученный раствор переносили в новый реактор.
В отношении амидирования реакцию проводили с помощью двух альтернативных способов.
i) Амидирование с использованием метанола в качестве сорастворителя:
Серинол в незначительном избытке (94,84 г; 1,04 моль) загружали в гидрированную смесь, содержащую 132,76 г (VII). Раствор концентрировали, удаляя воду, полученную на предыдущей стадии, и большую часть бутанола.
Смесь охлаждали, добавляли метанол (524 г) и температуру повышали до 55-60°С. Раствор метилата натрия (21,19 г; 0,118 моль) в метаноле добавляли по каплям и поддерживали данную температуру до полного превращения (7-10 часов). Смесь охлаждали до 15°С, и выдерживали в течение 3 ч, и затем твердое вещество отфильтровывали, получая белое твердое вещество (V), которое промывали метанолом. Полученное твердое вещество непосредственно повторно растворяли в воде и переносили в реактор для следующей реакции йодирования. Выход в расчете на сухое твердое вещество = 95%
ii) Амидирование без примесей с избытком серинола:
Гидрированную смесь, содержащую (VII) (132,76 г; 0,45 моль), охлаждали до комнатной температуры и добавляли избыток серинола (247,40 г; 2,72 моль). Раствор концентрировали, азеотропно удаляя смесь вода/бутанол при 100°С под вакуумом. Смесь нагревали при 125°С в течение периода 4-6 часов, затем охлаждали при 70-80°С. Воду (929,3 г) загружали в реактор. Разбавленный раствор, полученный таким образом, загружали в серию из двух колонок, первая представляла собой колонку со слабокислотной смолой (карбоксильная, 700 мл), чтобы выборочно восстановить и повторно использовать серинол, вторая представляла собой колонку с анионной смолой (третичный амин, 50 мл), чтобы очистить раствор (V).
Методика йодирования: с (V), полученным из методики сорастворителя:
iа) Влажное твердое вещество (735 г; 2,05 моль) повторно растворяли в воде (7 л), остаточный метанол отгоняли, нагревая при 70-75°С, затем раствор нагревали при 70-90°С и загружали серную кислоту (106 г; 1,03 моль). ICl (1919 г; 6,65 моль) добавляли по каплям более 1,5 ч. (IV) стало осаждаться, и суспензию нагревали более 6-8 ч. Суспензию охлаждали до комнатной температуры и осадок отфильтровывали, обеспечивая белое твердое вещество. Выход = 92%
iiа) Йодирование с (V), полученным из методики без примесей:
Раствор, полученный из амидирования, концентрировали, нагревали при 70-90°С и загружали серную кислоту. Методика представляла собой, как сообщалось выше.
Синтез Йопамидола (II) затем проводили в соответствии со схемой 3 или по любому одному из предшествующих Примеров.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЙОДИРОВАННОГО КОНТРАСТНОГО АГЕНТА | 2009 |
|
RU2493146C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОНТРАСТНЫХ АГЕНТОВ | 2011 |
|
RU2566823C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 5-АМИНО-2,4,6-ТРИИОДО- 1,3-БЕНЗОЛКАРБОНОВОЙ КИСЛОТЫ | 1990 |
|
RU2046795C1 |
| НЕБЕЛКОВЫЕ КОНЪЮГАТНЫЕ СОЕДИНЕНИЯ БОРОНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2076127C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ БОРОНОВОЙ КИСЛОТЫ | 2014 |
|
RU2658915C2 |
| СПОСОБ ЙОДИРОВАНИЯ ПРОИЗВОДНЫХ ФЕНОЛА | 2011 |
|
RU2563645C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАСПОФУНГИНА И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2009 |
|
RU2489442C2 |
| НОВЫЙ СПОСОБ СИНТЕЗА ПРОИЗВОДНЫХ ПИПЕРАЗИНИЛ-ЭТОКСИ-БРОМФЕНИЛА И ИХ ПРИМЕНЕНИЕ В ПОЛУЧЕНИИ СОДЕРЖАЩИХ ИХ СОЕДИНЕНИЙ | 2019 |
|
RU2804353C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИЙОДИРОВАННЫХ КАРБОКСИЛЬНЫХ АРОМАТИЧЕСКИХ ПРОИЗВОДНЫХ | 2010 |
|
RU2512358C2 |
| СПОСОБ ПРОСТОГО И УДОБНОГО ПОЛУЧЕНИЯ ВАБОРБАКТАМА | 2019 |
|
RU2770434C1 |
Изобретение относится к способам синтеза йодсодержащих контрастных веществ, конкретно к способу получения Йопамидола (II), который включает указанную ниже реакцию, где Х представляет собой OR2 или R3, и R2 и R3 представляют собой С1-С6линейный или разветвленный алкил, С3-С6циклоалкил, C6арил, необязательно замещенный группой, выбранной из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, втор-бутила, трет-бутила и фенила. Способ включает стадии a)-c). На стадии a) осуществляют взаимодействие соединения (I) с ацилирующим агентом (S)-2-(ацетилокси)пропаноилхлоридом в реакционной среде для получения N-(S)-2-(ацетилокси)пропаноильного производного соединения (I). На стадии b) осуществляют гидролиз промежуточного соединения со стадии а) с водным раствором при рН, составляющем от 0 до 7, с помощью добавления воды или разбавленного щелочного раствора, освобождение гидроксилов от борсодержащих защитных групп, получение ацетилоксипроизводного соединения (II) и необязательное восстановление борпроизводного. На стадии c) осуществляют щелочной гидролиз ацетилоксипроизводного соединения (II), восстановление (S)-2-(гидрокси)пропаноильной группы для получения Йопамидола (II). Предлагаемый способ позволяет проводить реакцию в одном реакционном сосуде и повысить общий выход Йопамидола (II), а также обеспечивает восстановление и повторное применение реагирующего вещества, используемого в качестве гидроксильной защитной группы. Изобретение относится также к способу получения Йопамидола (II) исходя из 5-нитроизофталевой кислоты (5-NIPA), соединению формулы (I) и его применению в синтезе Йопамидола (II). 4 н. и 25 з.п. ф-лы, 14 пр.
1. Способ получения Йопамидола (II), включающий следующую реакцию:
 ,
,
в которой Х представляет собой OR2 или R3, и в которой R2 и R3 представляют собой С1-С6линейный или разветвленный алкил, С3-С6циклоалкил, C6арил, необязательно замещенный группой, выбранной из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, втор-бутила, трет-бутила и фенила;
и включающий следующие стадии:
a) взаимодействие соединения (I) с ацилирующим агентом (S)-2-(ацетилокси)пропаноилхлоридом в реакционной среде для получения N-(S)-2-(ацетилокси)пропаноильного производного соединения (I);
b) гидролиз промежуточного соединения со стадии а) с водным раствором при рН, составляющем от 0 до 7, с помощью добавления воды или разбавленного щелочного раствора, освобождение гидроксилов от борсодержащих защитных групп, получение ацетилоксипроизводного соединения (II) и необязательное восстановление борпроизводного;
c) щелочной гидролиз ацетилоксипроизводного соединения (II), восстановление (S)-2-(гидрокси)пропаноильной группы для получения Йопамидола (II).
2. Способ по п.1, в котором Х представляет собой OR2.
3. Способ по п.1, в котором Х представляет собой R3.
4. Способ по п.1, в котором указанная реакционная среда на стадии а) представляет собой безводный органический растворитель.
5. Способ по п.1, в котором указанная реакционная среда на стадии а) выбрана из группы, состоящей из N,N-диметилформамида, N,N-диметилацетамида, N,N-диэтилацетамида, N,N-диметилпропионамида, N-метилпирролидона, N-этилпирролидона, тетраметилмочевины, N,N'-диметилэтиленмочевины (DMEU) и N,N'-диметилпропиленмочевины (DMPU), необязательно в смеси с сорастворителем.
6. Способ по п.1, в котором на указанной стадии b) восстановление борпроизводного проводят с помощью хроматографии или с помощью экстракции сорастворителем.
7. Способ по любому одному из пп.1-6, в котором указанное соединение формулы (I) получают исходя из соединения формулы (IV) в соответствии со следующей реакцией:
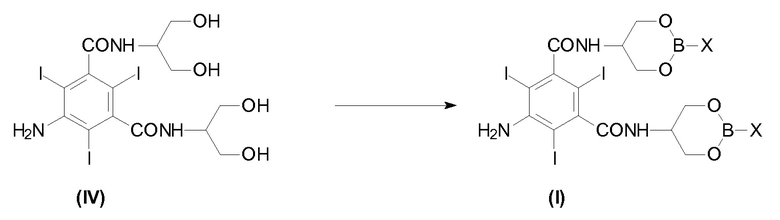 ,
,
в которой Х представляет собой OR2 или R3, и в которой R2 и R3 представляют собой С1-С6линейный или разветвленный алкил, С3-С6циклоалкил, C6арил, необязательно замещенный группой, выбранной из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, втор-бутила, трет-бутила и фенила; и включающий:
взаимодействие соединения формулы (IV) с одной из борных кислот в спирте R2OH или с эфиром борной кислоты B(OR2)3, где R2 представляет собой, как определено выше, для получения соединения формулы (I), в которой Х представляет собой OR2; или
взаимодействие соединения формулы (IV) с одной из бороновых кислот R3-B(OH)2 или бороксином формулы (III)
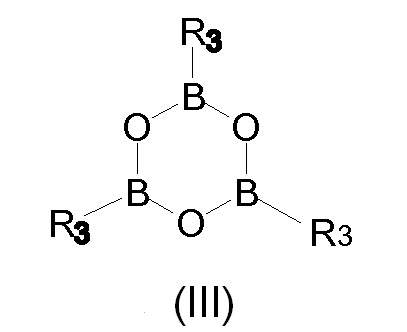
для получения соединения формулы (I), в которой Х представляет собой R3.
8. Способ по п.7, в котором используют эфир борной кислоты B(OR2)3.
9. Способ по п.8, в котором эфир борной кислоты выбран из группы, состоящей из три-н-бутилбората, три-н-пропилбората и триэтилбората.
10. Способ по п.7, в котором используют бороновую кислоту R3B(OH)2 или бороксин (III).
11. Способ по п.10, в котором бороновая кислота выбрана из группы, состоящей из фенилбороновой кислоты, толилбороновой кислоты и бутилбороновой кислоты, или бороксин (III) выбран из группы, состоящей из трифенилбороксина и триметилбороксина.
12. Способ по любому одному из пп.10 или 11, который включает восстановление бороновой кислоты с помощью экстракции сорастворителем, в котором указанный сорастворитель представляет собой органический несмешивающийся с водой растворитель, выбранный из группы, состоящей из 4-метил-2-пентанона, 2-пентанона, 3-пентанона, дибутилового эфира, 2-метилтетрагидрофурана, циклопентилметилового эфира, метилизопропилкетона, метилизопентилкетона, этилацетата, бутилацетата, пентилацетата, изопентилацетата, изопропилацетата.
13. Способ по п.1, который представляет собой способ, проводящийся в одном реакционном сосуде.
14. Способ по п.7, в котором указанное соединение формулы (IV) получают, подвергая йодированию следующее соединение (V):
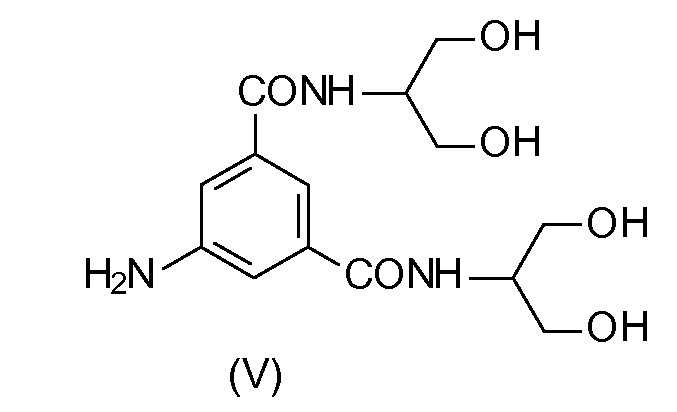
15. Способ по п.14, в котором указанное соединение (V) получают в соответствии со следующей схемой реакции:
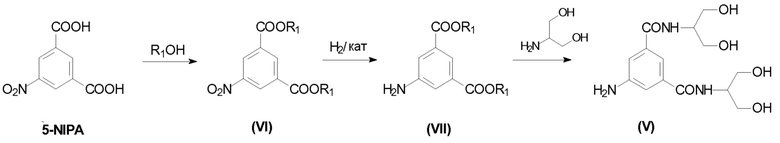
в котором:
i) 5-нитроизофталевую кислоту (5-NIPA) обрабатывают спиртом R1OH, в котором R1 представляет собой линейный или разветвленный С1-С4алкил, для получения соответствующего диэфира;
ii) 5-нитрогруппу восстанавливают до соответствующей 5-аминогруппы для получения соединения (VII);
iii) диэфир взаимодействует с 2-амино-1,3-пропандиолом для получения соединения (V).
16. Способ получения Йопамидола (II) в соответствии со следующей схемой реакции:
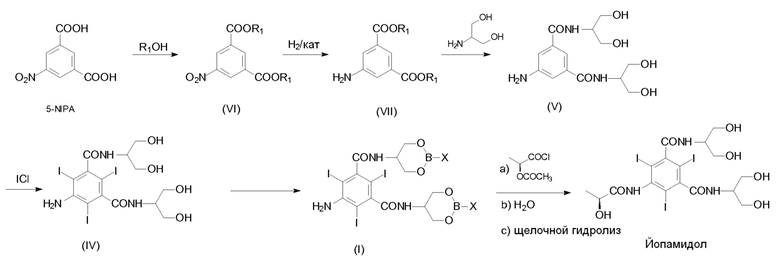
в котором:
i) 5-нитроизофталевую кислоту (5-NIPA) обрабатывают спиртом R1OH, в котором R1 представляет собой линейный или разветвленный С1-С4алкил, для получения диэфира (VI);
ii) 5-нитрогруппу восстанавливают для получения соединения (VII);
iii) диэфир взаимодействует с 2-амино-1,3-пропандиолом для получения 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-1,3-бензолдикарбоксамида (V);
iv) соединение (V) йодируют в положениях 2,4,6 для получения 5-амино-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-2,4,6-трийодо-1,3-бензолдикарбоксамида (IV);
v) соединение (IV) обрабатывают борной кислотой или ее производным по любому одному из пп.8-12 для получения соединения формулы (I);
vi) соединение формулы (I) превращают в Йопамидол (II) по любому одному из пп.1-7 или 14.
17. Способ по п.1, дополнительно включающий очистку и выделение Йопамидола (II).
18. Способ по п.16, дополнительно включающий очистку и выделение Йопамидола (II).
19. Способ по п.17 или 18, в котором указанная очистка должна быть фармацевтической категории.
20. Соединение формулы (I)
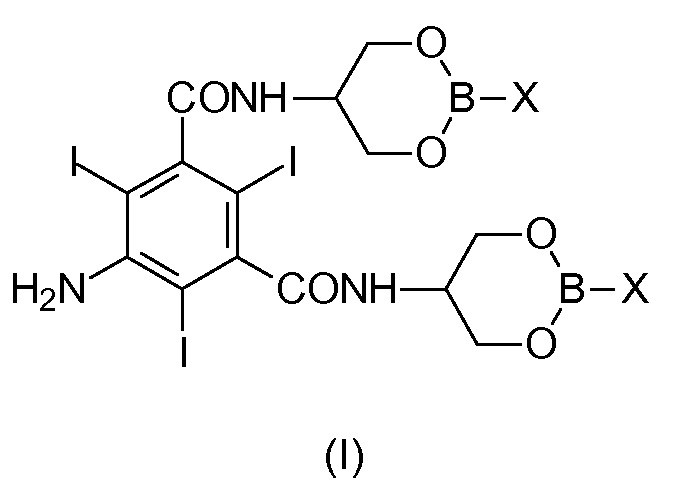 ,
,
в которой Х представляет собой OR2 или R3, и в которой R2 и R3 представляют собой С1-С6линейный или разветвленный алкил, С3-С6циклоалкил, C6арил, необязательно замещенный группой, выбранной из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, втор-бутила, трет-бутила и фенила.
21. Соединение по п.20, в котором Х выбран из группы, состоящей из фенила, метилзамещенного фенила, метила и бутила.
22. Соединение по п.21, в котором Х представляет собой фенил.
23. Применение соединения формулы (I) по п.21 или 22 и его ацетилоксипроизводного в качестве промежуточного соединения в синтезе Йопамидола (II).
24. Способ по п.1, в котором восстановление бороновой кислоты формулы
R3-B(OH)2,
в которой R3 выбран из группы, состоящей из C1-C6линейного или разветвленного алкила, С3-С6циклоалкила и C6арила, необязательно замещенного группой, выбранной из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, втор-бутила, трет-бутила и фенила,
из водной смеси стадии b), необязательно содержащей полярный растворитель, включает следующие стадии:
- перемешивание водной смеси с несмешивающимся с водой органическим растворителем, где массовое соотношение между бороновой кислотой и несмешивающимся с водой растворителем составляет от 1:10 до 1:20 (мас./мас.),
- добавление водного раствора до получения конечного рН, составляющего от 0 до 7, и
- отделение бороновой кислоты в органической несмешивающейся с водой фазе и восстановление органической несмешивающейся с водой фазы.
25. Способ по п.24, в котором бороновая кислота Формулы R3-B(OH)2 используется для диольной защиты или образуется после гидролиза.
26. Способ по п.24, в котором органический несмешивающийся с водой растворитель выбран из группы, состоящей из 4-метил-2-пентанона (МИБК), 3-пентанона, 2-пентанона, дибутилового эфира, 2-метилтетрагидрофурана, циклопентилметилового эфира, метилизопропилкетона, метилизопентилкетона, этилацетата, бутилацетата, пентилацетата, изопентилацетата и изопропилацетата.
27. Способ по п.24, в котором соотношение между бороновой кислотой и несмешивающимся с водой растворителем (экстракционным растворителем) составляет от 1:13 до 1:16 (мас./мас.).
28. Способ по п.24, в котором R3 представляет собой метил, бутил и фенил, необязательно замещенный метилом.
29. Способ по п.24, в котором отделение бороновой кислоты в органической фазе проводят в периодическом или непрерывном режиме.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| СПОСОБ ПОЛУЧЕНИЯ ЙОДИРОВАННОГО КОНТРАСТНОГО АГЕНТА | 2009 |
|
RU2493146C2 |

