Настоящее изобретение относится к новым продуктам, имеющим противовоспалительную, анальгезирующую и антитромботическую активность.
В частности оно относится к ингибиторам циклооксигеназы (СОХ).
Известно, что противовоспалительная и антитромботическая эффективность NSAIDs (нестероидные противовоспалительные лекарственные средства), также известных как FANS (нестероидные противовоспалительные лекарственные средства), и в особенности их толерантность, по-видимому, значительно влияют на их ингибиторную активность в отношении циклооксигеназы (СОХ) как в участке воспаления, так и в здоровой ткани. Смотри, например, FASEB Journal 1, 89, 1987; Bioch. Biophys. Acta 1083, 1, 1991. Недостаток этих продуктов заключается в том, что они токсичны, как уже описано в патенте США 5861426.
Также известны нитропроизводные, описанные в вышеупомянутом патенте, имеющие высокую эффективность в ингибировании циклооксигеназы и низкую токсичность. Однако эти соединения имеют некоторые недостатки, связанные с химико-физическими и структурными характеристиками их молекул, так как последние высоколипофильны и, следовательно, имеют низкую растворимость в воде. Хорошо известно, что процесс растворения имеет значение для абсорбции и взаимодействия с эффектором. Как правило, плохая растворимость приводит к переменному и непредсказуемому действию, в результате чего трудно установить правильную позологию. На практике необходимо вводить более высокие дозы, для того чтобы учитывать вышеупомянутые изменения. Недостатком является опасность более высокой степени побочных эффектов. Другое неудобство связано с низкой растворимостью нитропроизводных вышеупомянутой патентной заявки, так как это приводит к трудностям при изготовлении лекарственных средств. Хорошо известно, что растворимость в воде молекулы является одним из самых важных свойств, влияющих на процессы фармакокинетики и фармакодинамики. Например, для парентерального введения, в частности для внутривенного введения, лекарственные средства должны быть приготовлены в виде растворов. Для того чтобы увеличить растворимость, когда она является неудовлетворительной для этих применений, выбор подходящих растворителей и/или наполнителей является критичным, например, среди последних могут быть упомянуты поверхностно-активные вещества и т.д. Это может привести к недостаткам с токсикологической точки зрения, связанным с толерантностью наполнителя; кроме того, существуют другие недостатки, например, во внутривенной форме, которая, как хорошо известно, не должна вызывать гемолиз или несовместимость с компонентами крови. Кроме того, необходимо отметить, что, как это хорошо известно, поверхностно-активные вещества и неполярные растворители могут быть раздражающими. Смотри, например, J. Pharm. Science 72, 1014, 1983.
Эксперименты, проводимые заявителем, где использовались 0/1% Tween 80 и 1% диметилсульфоксид для суспендирования нитроксипроизводных противовоспалительных соединении, описанных в заявке на патент WO 95/30641, показали, что эти вещества являются раздражающими по отношению к слизистой оболочке желудка.
Неожиданно было обнаружено, что производные согласно настоящему изобретению в отличие от вышеупомянутых соединений предшествующего уровня техники могут быть растворимы без использования веществ, обычно применяемых в фармацевтической практике для получения растворов или суспензий, при этом сохраняя или даже улучшая активность нитроксипризводных предшествующего уровня техники. Следующее преимущество соединений согласно настоящему изобретению состоит в том, что можно избежать добавления наполнителей к форме лекарственного средства, таких как, например, вышеупомянутые, которые вызывают или могут вызывать раздражающие эффекты.
Противовоспалительные продукты, описанные в настоящей заявке, имеют высокую ингибирующую циклооксигеназу активность, объединенную с низкой токсичностью и хорошими фармакокинетическими откликами, и, кроме того, улучшенную общую степень абсорбции.
Это действительно удивительно и неожиданно, так как факторы, влияющие на противовоспалительную и антитромботическую активность FANS, зависят от различных параметров, в результате чего невозможно предсказать априори фармакокинетику, например, абсорбированной фракции продукта, фармакодинамическую активность, токсичность и ингибирующие СОХ свойства, и для большинства из них не могут быть сделаны предположения, чтобы предсказать или ограничить разнообразие ответа.
Объектом настоящего изобретения являются соединения или органические или неорганические соли соединений общей формулы
A-X1-N(O)z
для использования в качестве лекарственных средств, в частности, как противовоспалительных и антитромботических агентов, где:
z является целым числом и означает 1 или 2;
А=R(COXu)t, где t и и являются целым числом и означают 1; X=O,
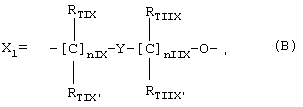
где nIX означает целое число между 0 и 3, предпочтительно 1;
nIIX означает целое число между 1 и 3, предпочтительно 1;
RTIX, RTIX', RTIIX, RTIIX' равны или отличны друг от друга, означают Н или линейный или разветвленный C1-C4 алкил;
Y означает гетероциклическое кольцо, насыщенное или ненасыщенное или ароматическое, имеющее 6 атомов и содержащее один или два атома азота;
R выбран из следующих групп:
Группа I)
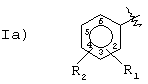
R1 означает ОСОR3 группу; где R3 означает метил, этил или линейный или разветвленный С3-C5 алкил;
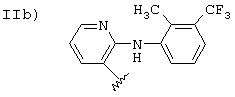
Ib)
Ic)
Группа II)

где RII1, RII2 и RII3 означают водород, линейный или разветвленный C1-C6 алкил или Cl, F, Вr;
RII4 означает RII1,
где в формуле IIа, когда RII2 и RII3 означают хлор в ортоположении по отношению к NH,
RII1, RII4, RII5 и RII6 означают водород, R означает остаток диклофенака.
Группа III)
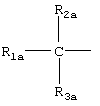
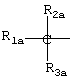
где R2a и R3a означают водород, линейный или разветвленный, C1-C12 алкил;
R1a выбран из
IIID) R1a соответствует следующим формулам:
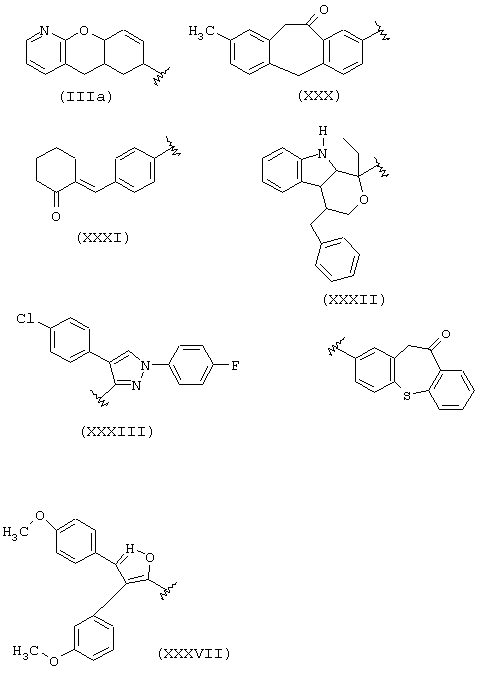
где в группе III значения следующие:
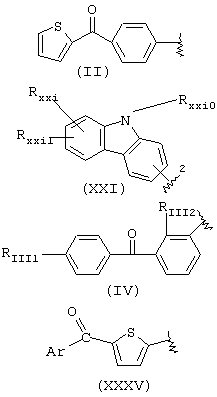
когда R1a такой, как определено в формуле (IV), RIII1 и RIII2 означают водород, R2a означает метил, R3a=Н, R означает остаток кетопрофена;
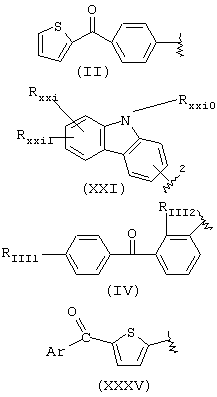
когда R1a такой, как определено в формуле (XXI), остаток карпрофена:
Rxxio и Rxxi означают Н, Rxxi1 означает Сl и находится в пара-положении по отношению к азоту, связующий мостик находится в положении 2, R2a означает метил, R3a=Н, R означает остаток карпрофена,
когда R1a такой как определено в формуле (XXXV), Аr означает фенил, R2a означает метил, R3a=Н, R означает остаток тиапрофеновой кислоты;
когда R1a такой, как определено в формуле (II), R2a означает метил, R3a=Н, R означает остаток супрофена,
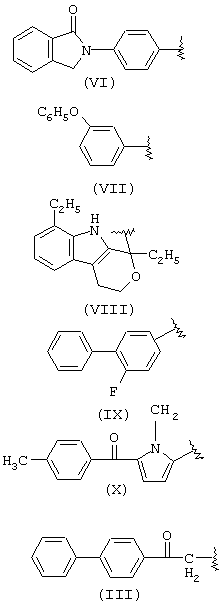
когда R1a такой, как определено в формуле (VI), R2a=Н и R3a=СН3, R означает остаток индопрофена;
когда R1a такой, как определено в формуле (VI), R2a=Н, R3a=C2H2, R означает остаток инобуфена;
когда R1a такой, как определено в формуле (VIII), R2a=R3a=Н, R означает остаток этодолака;
когда R1a такой, как определено в формуле (VII), R3a=Н, R2a=СН3, R означает остаток фенопрофена;
когда R1a такой, как определено в формуле (III), R2a=R3a=Н, R означает остаток фенбуфена;
когда R1a такой, как определено в формуле (IX), R3a=Н, R2a=СН3, R означает остаток флурбипрофена;
когда R1a такой, как определено в формуле (X), R2a=R3a=Н, R означает остаток толметина.
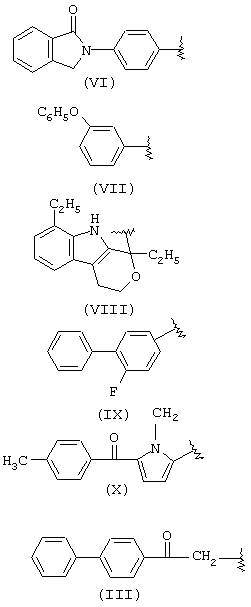
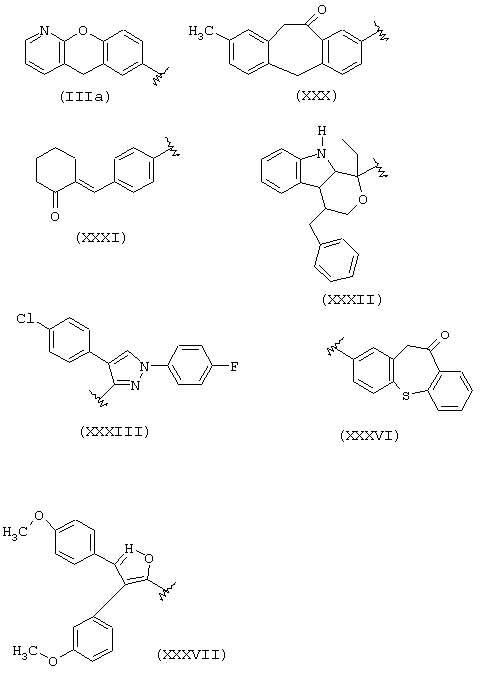
В группе IIID, когда R1a такой, как определено в формуле IIIa, R2a=Н, R3a=СН3, R означает остаток пранопрофена;
когда R1a такой, как определено в формуле (XXX), R2a=Н, R3a=СН3, R означает остаток бермопрофена;
когда R1a такой, как определено в формуле (XXXI), R2a=Н, R3a=СН3, R означает радикал соединения CS-670: 2-[4-(2-оксо-1-циклогексилиденметил)фенил]пропионовая кислота;
когда R1a такой, как определено в формуле (XXXII), R2a=R3a=Н, R означает остаток пемедолака;
когда R1a такой, как определено в формуле (XXXIII), R2a=R3a=H, R означает остаток пиразолака;
когда R1a такой, как определено в формуле (XXXVI), R2a=Н, R3a=СН3, R означает остаток залтопрофена;
когда R1a такой, как определено в формуле (XXXVII), R2a=R3a=Н, R означает остаток мофезолака: 3,4-ди(п-метоксифенил)изоксазол-5-уксусная кислота, когда остаток означает СН2-СООН; в предпочтительных соединениях R2a=R3a=Н, t=1 и Х=0;
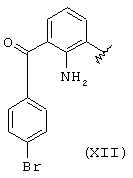
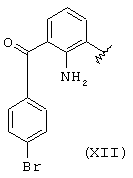
когда R1a такой, как определено в формуле (XII), R2a=R3a=Н, R означает остаток бромфенака.
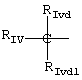
В группе IV R означает
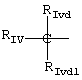
где RIvd и RIvd1 означают, по крайней мере, один Н и другой алкил от C1 до С6,
Riv имеет следующее значение:
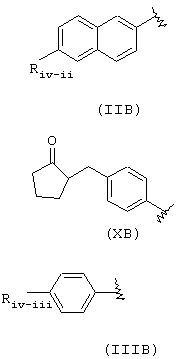
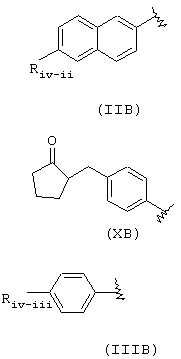
где Riv, как определено в формуле (IIB), Riv-ii означает СН3-O-, Rivd означает Н, Rivd1 означает СН3, R означает остаток напроксена;
Riv, как определено в формуле (ХВ), Rivd=Н, RIvd1=СН3, R означает остаток локсопрофена;
Riv, как определено в формуле (IIIB), Riv-iii означает
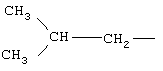
и RIvd=Н, RIvd1 означает СН3, R означает остаток ибупрофена.
Группа V, где R означает:
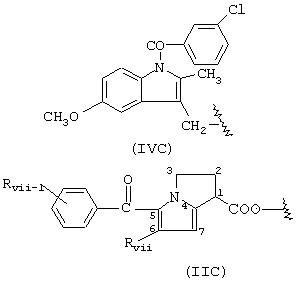
В группе V соединения имеют следующие значения:
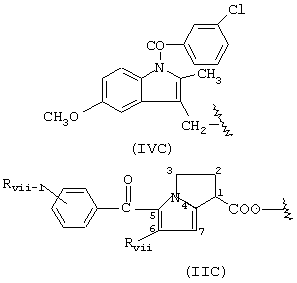
когда R означает формулу (IIC),
Rvii и Rvii-1 означают Н, R означает остаток кетополака; когда R имеет формулу (IVC), R означает остаток индометасина.
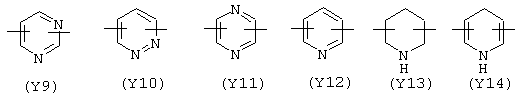
Y в вышеупомянутой X1 формуле содержит один или два атома азота в кольце и предпочтительно выбран из:
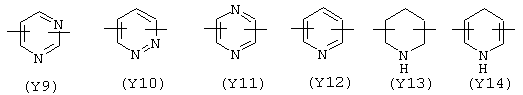
Предпочтительным Y является Y12 (пиридил), замещенный в положении 2 и 6. Связи могут быть также в не симметричном положении, например Y12 (пиридил) может быть замещен в положении 2 и 3;
Предшественники X1, где свободная валентность кислорода насыщена Н и свободная валентность концевого углерода насыщена карбоксильной или гидроксильной группой, являются коммерчески доступными продуктами или их получают способами, известными в предшествующем уровне техники.
Соединения, содержащие группу I типа Iа, описаны в патенте WO 92/01668, где также описаны способы получения. Этот патент приведен здесь путем ссылки. Соединения типа Ib получают, например, используя способ, показанный в Merck Index, XI изд., 1989, стр. 16, №95, для остатка ацетилсалицилсалициловой кислоты. Варианты соединений формулы Ib могут быт получены при применении процессов, упомянутых в патенте WO 92/01668.
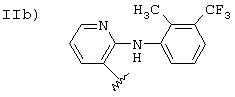
Соединения, где R означает группу II, описаны в патенте WO 94/04484 и патенте США 3558690, где также описаны способы получения. Эти патенты приведены здесь путем ссылки.
Исходное соединение IIb, когда валентность насыщена -СООН (флуниксин), получают согласно патентам США 3337570 и 3689653, приведенным здесь путем ссылки. Соединения, содержащие заместители, упомянутые в предыдущих патентах, эквивалентны флуниксину.
Соединения, где R означает группу III), описаны и получены способами, упомянутыми в следующих патентах:
патентная заявка РСТ/ЕР 9303193; для соединений формулы (IV) смотри также патент США 3641127; для соединений формулы (XXI) смотри также патент США 3896145; для соединений формулы (IX), остаток фторбипрофена, смотри также патент США 3755427; для соединений формулы (II) смотри также патент США 4035376; для соединений формулы (VI) смотри также патент США 3997669; для соединений формулы (VIII) смотри также патент США 3843681; для соединений формулы (VII) смотри также патент США 3600437; для соединений формулы (III) смотри также патент США 3784701. Все упомянутые патенты приведены здесь путем ссылки.
Способами для получения соединений класса II ID) являются следующие: остаток IIIa) получают приготовлением соединения кислоты согласно патенту США 3931205, валентность насыщают -СН(СН3)-СООН. Соединения, содержащие заместители, упомянутые в вышеуказанном патенте, эквивалентны пранопрофену. Остаток (XXX) получают через соединение с -СН(СН3)-СООН группой (бермопрофен) согласно патенту США 4238620, приведенному здесь путем ссылки. Другие эквивалентные продукты описаны в вышеупомянутом патенте.
Остаток (XXXI) получают начиная с соответствующей кислоты -СН (СН3)-СООН согласно патенту США 4254274. Эквивалентные соединения описаны в том же патенте.
Остаток (XXXII) получают согласно ЕР 238226, приведенному здесь путем ссылки, когда валентность насыщают –СН2-СООН. Эквивалентные продукты приведены в вышеупомянутых патентах как замещенные 1, 3, 4, 9 тетрагидрофуран [3,4-b]индол-1-уксусные кислоты.
Остаток (XXXIII) получают из пиразолака и валентность насыщают –СН2-СООН как упомянуто в ЕР 54812, приведенном здесь путем ссылки. Эквивалентные продукты описаны в вышеупомянутом патенте.
Остаток (XXXVI) получают согласно UK 2035311, приведенному здесь путем ссылки, начиная с залтопрофена и при наличии -СН(СН3)-СООН концевой группы. Эквивалентные продукты описаны в вышеупомянутом патенте.
Способ получения остатка (XXXVII) начинают с мофезолака и получают согласно ЕР 26928. Эквивалентные продукты приведены в том же патенте.
Соединения, в которых R означает группу IV, описаны в английской патентной заявке 2283238, где также указаны способы получения; этот патент приведен здесь путем ссылки.
В группе IV соединения могут быть также получены: для соединений формулы (II), используя патент США 3904682; соединения формулы (Х) получают согласно патенту США 4161538, соединения формулы (III) согласно патенту США 3228831. Эти патенты приведены здесь путем ссылки.
В группе V могут быть также получены соединения: для соединений формулы (II), используя патент США 4089969, приведенный здесь путем ссылки; соединение формулы (V) может быть получено согласно патенту США 4556672, приведенному здесь путем ссылки.
Остаток (X) получают согласно немецкому патенту 2756113. Эквивалентные продукты описаны в вышеупомянутом патенте.
Остаток (XI) получают согласно ЕР 147177, начиная с ампироксикама, имеющего -СН(СН3)ОСООС2Н5 концевую группу. Эквивалентные продукты описаны в вышеупомянутом патенте.
Остаток (XII) получают согласно J. Med. Chem., т.27, №11, ноябрь, 1984, Walsh et al. "Anti-inflammatory Agents. 3. Synthesis and Pharmacological Evaluation of 2-amino-3-benzoylphenylacetic Acid and Analogues". Эквивалентные продукты описаны в вышеупомянутой публикации.
Остаток (XIII) получают исходя из лорноксикама, где валентность насыщена водородом. Его получают согласно GB 2033877. Эквивалентные продукты описаны в вышеуказанном патенте.
Как правило, связь между А и X1, как видно, эфирного или амидного типа (NH или NR1c, как определено в X), когда R означает группы I, II, III, IV и V. Все хорошо известные пути синтеза формирования таких связей могут быть использованы для образования вышеупомянутой связи.
В случае сложных эфиров групп I, II, III и IV и для соединений группы V, заканчивающихся карбоксильной функцией, наиболее прямой синтетический путь получения соответствующих нитропроизводных настоящего изобретения включает:
a) реакцию ацилхлоридов R-CO-Cl с галогенспиртами типа HO-X1z-Cl, HO-X1z-Br, HO-X1z-I, где X1z означает X1, как определено выше без атома кислорода, в экспериментальных условиях предшествующего уровня техники и выделение соединений формулы R-CO-O-X1z-Cl (Вr, I). Вышеупомянутые продукты могут быть также получены реакцией солей натрия или калия вышеупомянутых R-CO-OH кислот с дигалогеновыми производными общей формулы X1zCl2, X1zBr2 или X1zI2.
b) Вышеупомянутые продукты трансформируют в конечные продукты реакцией АgNО3 в ацетонитриле согласно тому способу, как это известно из предшествующего уровня техники.
Общая схема является следующей:
R-CO-Cl+HO-X1z-Br→ R-CO-O-X1z-Br+AgNO3→A-X1NO2,
где X1=X1zO.
R-CO-ONa+Br2X1z→R-CO-O-X1z-Br+AgNO3→A-X1NO2,
где X1=X1zO.
В случае амидов последовательность синтеза включает реакцию таких же ацилхлоридов RCOC1 с аминоспиртами общей формулы NH2-X1z-OH, NHR1c-X1z-OH с получением амидов общей формулы:
R-CO-NH-X1z-ОН и R-CO-NR1c-X1z-OH
согласно известным способам.
Реакция вышеуказанных амидов с галогенирующими агентами, такими как, например, PCl5, РВr3, SOCl2 и т.д., приводит к галогенпроизводным общей формулы:
R-CO-NH-X1z-Br(Cl) и R-CO-NR1c-X1z-Br(Cl).
Последнее путем реакции с АgNО3 в ацетонитриле согласно известным способам в предшествующем уровне техники приводит к конечным продуктам A-X1-NO2.
Схема синтеза является следующей:
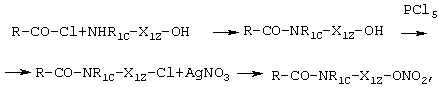
где X1zO означает X1.
с) Альтернативный путь: синтез через вышеуказанные стадии а) и b) является реакцией натриевых или калиевых солей кислот с эфирами азотной кислоты галогеноспиртов общей формулы
NO2-O-X1z-Cl(Br, I)
для получения непосредственно нитроксипроизводных настоящего изобретения.
Реакционная схема является следующей:
R-CO-ONa+Br-X1z-ONO2→R-CO-O-X1z-ONO2
где X1zO является X1.
Подобные вышеописанным пути синтеза используются для продуктов группы V, например теноксикама и пироксикама, где дигалогеновые производные формулы Br2X1z реагируют с соответствующими энолятами. Полученные продукты затем превращаются в соединения изобретения реакцией с АgNО3 в ацетонитриле согласно вышеуказанной схеме реакции.
Здесь приведена схема для пироксикама формулы (IX) группы V.
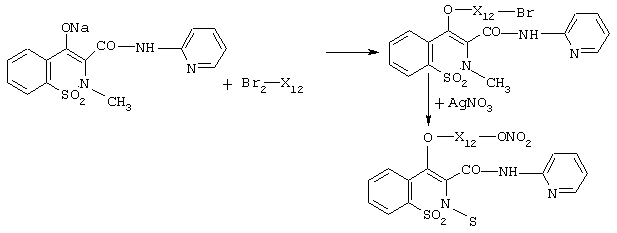
Продукты V группы, такие как теноксикам и пироксикам, где противовоспалительной реакционной функцией является гидроксил, могут также реагировать с ацилхлоридом формулы ClCO-X1z-Q1, где Q1 является Сl, Вr, I, ОН. Когда Q1=ОН, гидроксил замещают галогеном как описано выше, перед конечной реакцией нитрования с АgNО3.
Нитрование проводят, как описано выше.
Для того чтобы получить соединения формулы A-X1-NO, ацилхлориды формулы R-COC1 вступают в реакцию с HX-X1z-OH, где R, Х и X1z имеют вышеупомянутые значения, в экспериментальных условиях, описанных в предшествующем уровне техники. Полученные спирты реагируют с нитритом натрия в растворителе, например, представляющем собой смесь воды с тетрагидрофураном в присутствии соляной кислоты. Эта реакция описана в предшествующем уровне техники. Общая схема является следующей:
R-COCl+HX-X1z-OH→ R-CO-X-X1z-ОН+NaNO3→A-X1-NO
Соединения согласно настоящему изобретению превращают в соответствующие соли реакцией в органическом растворителе, таком как, например, ацетонитрил и тетрагидрофуран, с эквимолекулярным количеством соответствующей органической или неорганической кислоты.
Примеры подходящих органических кислот являются следующими; щавелевая кислота, винная кислота, малеиновая кислота, янтарная кислота, лимонная кислота.
Примеры подходящих неорганических кислот являются следующими: азотная кислота, соляная кислота, серная кислота, фосфорная кислота.
Другой объект изобретения заключается в том, что, как это было с удивлением обнаружено, продукты изобретения, содержащие ON-(O)z группы, также способны оказывать ингибирующее действие на воспаление, вызванное липосахаридом (ЛПС) и, следовательно, могут использоваться при септическом шоке.
Это является удивительным, так как хорошо известно, что, как правило, противовоспалительные агенты не изменяют значительно нитросинтетазную активность, вызванную липополисахаридами в крысе, и следовательно, они не могут использоваться при септическом шоке.
Соединения настоящего изобретения могут быть использованы как противовоспалительные лекарственные средства или для терапии и профилактики сердечно-сосудистых заболевания или тех патологий, где клеточная гиперпролиферация играет важную патогенетическую роль.
Должно быть понятно, что когда соединения различных групп содержат, по крайней мере, один асимметрический углерод, продукты могут быть использованы в рацемической форме или как отдельные изомеры. Действительно, хорошо известно, что в терапевтических применениях изобретения, как правило, одна изомерная форма активнее, чем другие. Когда соединение представлено цис/транс изомерами, они могут быть использованы в этой отдельной форме или в смеси.
Фармацевтические составы соединений согласно настоящему изобретению содержат такую же дозу противовоспалительных продуктов предшественника или ниже.
Фармацевтические составы могут приниматься орально или парентерально и могут быть получены согласно хорошо известным процессам в предшествующем уровне техники. Смотри книгу "Remington's Pharmaceutical Sciences".
Следующие примеры даны в иллюстративных целях и не ограничивают настоящего изобретения.
Пример 1
Синтез хлоргидрата 6-(нитроксиметил)-2-пиридинилметил эфира 2-ацетилоксибензойной кислоты (NCX 4050) формулы
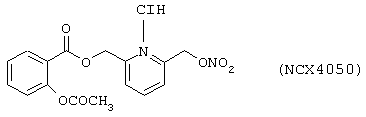
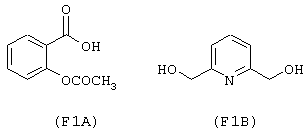
исходя из ацетилсалициловой кислоты (формула (F1A)) и 2,6-бис-(гидроксиметил)пиридина (формула (F1B))
А) Синтез 2,6-бис-(хлорметил)пиридина
К тионилхлориду (11,6 мл, 158 моль), охлажденному при 0° С, очень медленно добавляют 2,6-бис-(гидроксиметил)пиридин (4 г, 28 ммоль). Полученный раствор оставляют перемешиваться в течение 2 часов при комнатной температуре, затем избыток тионилхлорида выпаривают при пониженном давлении. Полученный остаток обрабатывают хлороформом и выпаривают снова при пониженном давлении для удаления остатков тионилхлорида. Сырой продукт обрабатывают хлороформом и промывают водой. Органическую фазу обезвоживают сульфатом натрия и высушивают, получая 4,81 г продукта в виде белого твердого вещества с темп. пл.=76-78° С.
B) Синтез 6-(хлорметил)-2-метилпиридинил эфира 2-ацетилоксибензойной кислоты.
К раствору салициловой кислоты (1,6 г, 8,88 ммоль) в N,N'-диметилформамиде (20 мл) и при перемешивании добавляют этилат натрия (0,64 г, 8,88 ммоль). Через 30 минут полученный раствор добавляют к раствору 2,6-бис-(хлорметил)пиридина (4,72 г, 26,81 ммоль) в N,N'-диметилформамиде (20 мл). Раствор оставляют при комнатной температуре на 7 дней при перемешивании, затем разбавляют этиловым эфиром и промывают водой. Отобранные органические фазы обезвоживают сульфатом натрия и растворитель выпаривают при пониженном давлении. Сырой продукт реакции очищают хроматографией на силикагеле, элюируя н-гексаном/этилацетатом 7/3. Получают 1,7 г продукта в виде желтого масла.
1Н-ЯМР (200 МГц) (CDC13): 8,10 (1Н, d); 7,74 (1H, t); 7,57 (1H, t); 7,42 (1H, d); 7,33 (2H, m); 7,11 (1H, d); 5,42 (2H, s); 4,67 (2H, s); 2,41 (3H, s).
C) Синтез 6-(нитроксиметил)-2-метилпиридинилового эфира 2-ацетилоксибензойной кислоты.
К раствору 6-(хлорметил)-2-метилпиридинил эфира 2-ацетилоксибензойной кислоты (1,5 г, 4,7 ммоль) в ацетонитриле (20 мл), поддерживаемому при перемешивании, добавляют нитрат серебра (1,3 г, 7,65 ммоль). Раствор нагревают до 80° С, пряча от света, при перемешивании в течение 30 часов. Образовавшийся хлорид серебра фильтруют, растворитель выпаривают. Сырой продукт реакции очищают хроматографией на силикагеле, элюируя н-гексаном/этилацетатом 7/3. Получают 1,2 г продукта в виде желтого масла.
1H-ЯМР (200 МГц) (CDC13): 8,10 (1H, d); 7,74 (1H, t); 7,57 (1H, t); 7,42 (1H, d); 7,33 (2H, m); 7,11 (1H, d); 5,60 (2H, s); 5,42 (2H, s); 2,41 (3H, s).
D) Синтез гидрохлорида 6-(нитроксиметил)-2-метилпиридинил эфира 2-ацетилоксибензойной кислоты.
К раствору 6-(нитроксиметил)-2-метилпиридинил эфира 2-ацетилоксибензойной кислоты (1 г, 2,88 ммоль) в этилацетате (20 мл), охлажденному до 0° С, добавляют по каплям при перемешивании раствор этилацетата/НС1 5 М. Раствор оставляют на один час при 0° С, затем позволяют температуре подняться до комнатных значений. Образовавшийся осадок фильтруют и промывают этиловым эфиром. Получают 900 мг твердого продукта.
Элементный анализ
Рассчитано: C 50,21%; H 3,95%; N 7,31%; Cl 9,26%.
Найдено: C 50,23%; H 3,97%; N 7,29%; Cl 9,20%.
Пример 2
Синтез нитрата 6-(нитроксиметил)-2-пиридинилметил эфира 2-ацетилоксибензойной кислоты (NCX 4051) формулы
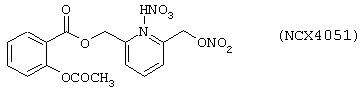
исходя из 6-(нитроксиметил)-2-метилпиридинил эфира 2-ацетилоксибензойной кислоты, выделенного на стадии С) предыдущего примера 1.
Синтез нитрата 6-(нитроксиметил)-2-метилпиридинил эфира 2-ацетилоксибензойной кислоты
К раствору 6-(нитроксиметил)-2-метилпиридинил эфира 2-ацетилоксибензойной кислоты (1 г, 2,88 ммоль) в ацетонитриле (10 мл), охлажденному до 0° С, добавляют по каплям при перемешивании раствор 65% азотной кислоты (0,2 мл) в ацетонитриле (2 мл). Оставляют на два часа при 0° С, затем позволяют повыситься температуре до комнатных величин. Сформировавшийся осадок фильтруют и промывают этиловым эфиром. Получают 1 г твердого продукта.
Элементный анализ
Рассчитано: С 46,95%; Н 3,69%; N 10,26%.
Найдено: С 46,99%; Н 3,72%; N 10,22%.
Пример 3
Синтез гидрохлорида 6-(нитроксиметил)-2-пиридинилметил эфира (S)-6-метокси-α -метилнафталинуксусной кислоты формулы
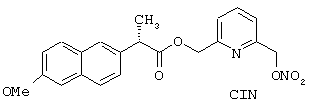
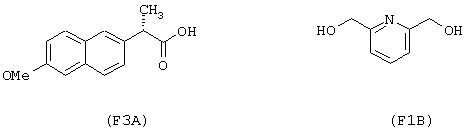
исходя из напроксена (формула (F3A)) и 2,6-бис-(гидроксиметил)пиридина (формула (F1B))
Соединение синтезируют, следуя процедуре, приведенной в примере 1. Выход 38%.
Элементный анализ
Рассчитано: С 58,25%; Н 4,88%; N 6,47%; Cl 8,19%.
Найдено: С 58,29%; Н 5,00%; N 6,44%; Cl 8,11%.
Пример 4
Синтез нитрата 6-(нитроксиметил)-2-пиридинилметил эфира (S)-6-метокси-α -метилнафталинуксусной кислоты формулы
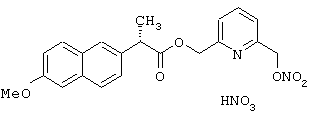
Соединение синтезируют, следуя процедуре, приведенной в примере 2.
Выход 42%.
Элементный анализ
Рассчитано: С 54,88%; H 4,60%; N 9,15%.
Найдено: С 54,91%; H 4,65%; N 9,10%.
Пример 5
Синтез гидрохлорида 6-(нитроксиметил)-2-пиридинилметил эфира 2-фтор-α -метил-(1,1’-бифенил)-4-уксусной кислоты формулы
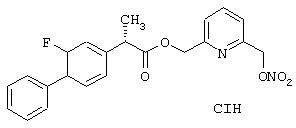
исходя из флурбипрофена (формула (F5A)) и 2,6-бис-(гидроксиметил)пиридина (формула (F1B))
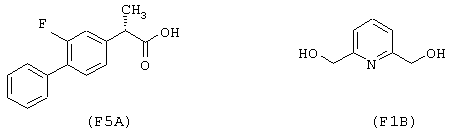
Соединение синтезируют, следуя процедуре, приведенной в примере 1. Выход 35%.
Элементный анализ
Рассчитано: C 59,12%; H 4,51%; N 6,29%; Cl 7,93%; F 4,25%.
Найдено: C 59,17%; H 4,55%; N 6,21%; Cl 7,91%; F 4,22%.
Пример 6
Синтез нитрата 6-(нитроксиметил)-2-пиридинметил эфира 2-фтор-α -метил-(1,1-бифенил)-4-уксусной кислоты формулы
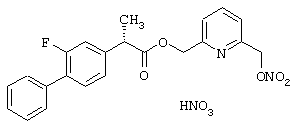
Соединение синтезируют, следуя процедуре, приведенной в примере 2. Выход 39%.
Элементный анализ
Рассчитано: C 55,79%; H 4,26%; N 8,91%; F 4,01%.
Найдено: C 55,83%; H 4,30%; N 8,88%; F 4,00%.
Пример 7
Синтез гидрохлорида 6-(нитроксиметил)-2-пиридинилметил эфира 1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-уксусной кислоты формулы
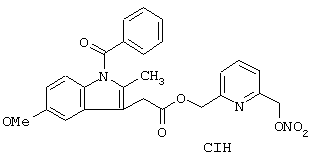
исходя из индометацина (формула (F7A)) и 2,6-бис(гидроксиметил)пиридина (формула (F1B))
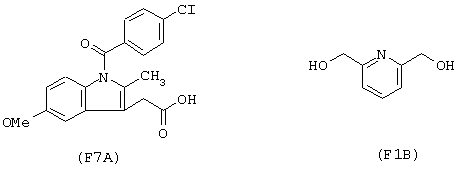
Соединение синтезируют, следуя процедуре, приведенной в примере 1. Выход 41%.
Элементный анализ
Рассчитано: C 55,71%; H 4,13%; N 7,53%; Cl 7,93%.
Найдено: C 55,73%; H 4,16%; N 7,49%; Cl 7,91%.
Пример 8
Синтез нитрата 6-(нитроксиметил)-2-пиридинилметил эфира 1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-уксусной кислоты формулы
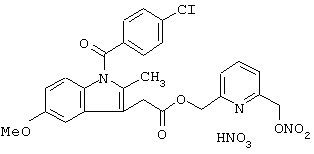
Соединение синтезируют, следуя процедуре, приведенной в примере 2. Выход 35%.
Элементный анализ
Рассчитано: C 53,18%; H 3,95%; N 9,58%; Cl 6,04%.
Найдено: C 53,20%; H 4,41%; N 9,56%; Cl 6,01%.
Пример 13
Синтез гидрохлорида 6-(нитроксиметил)-2-пиридинилметил эфира 2-[(2,6-дихлорфенил)амино]бензолуксусной кислоты формулы
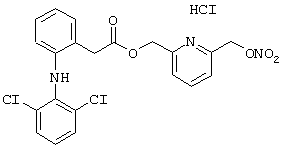
исходя из натриевой соли 2-[(2,6-дихлорфенил) амино]бензолуксусной кислоты (формула) и 2,6-бис-(гидроксиметил)пиридина
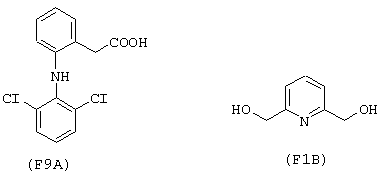
A) Синтез 6-(хлорметил)-2-метилпирилинил эфира 2-[(2,6-дихлорфенил)амино]бензолуксусной кислоты
К раствору 2,6-бис-(хлорметил)пиридина (3,83 г, 21,75 ммоль), полученному, как описано в примере 1А, в N,N'-диметилформамиде (20 мл) при перемешивании добавляют по каплям раствор натриевой соли 2-[(2,6-дихлорфенил)амино]бензолуксусной кислоты (3,04 г, 9,54 ммоль) в N,N'-диметилформамиде (25 мл). Раствор перемешивают при комнатной температуре в течение дня, затем его разбавляют этилацетатом и промывают водой. Отбирают органические фазы и обезвоживают их сульфатом натрия. Растворитель выпаривают при пониженном давлении. Сырой продукт реакции очищают хроматографией на колонке с силикагелем, элюируя н-гексаном/этилацетатом 8/2. Получают 2,88 г продукта в виде белого твердого вещества. Выход 69%.
1H-ЯМР (200 МГц) (CDC13): 7,66 (1Н, t); 7,41 (1Н, d); 7,33 (1Н, d); 7,27 (1Н, d); 7,18 (2H, m); 6,97 (2H, dd); 6,81 (1Н, s); 6,57 (1Н, d); 5,3 (2H, s); 4,62 (2H, s); 3,93 (2H, s).
B) Синтез 6-(нитроксиметил)-2-метилпиридинил эфира 2-[(2,6-дихлорфенил) амино]бензолуксусной кислоты.
К перемешиваемому раствору 6-(хлорметил)-2-метилпиридинил эфира 2-[(2,6-дихлорфенил)амино]бензолуксусной кислоты (2,438 г, 5,59 ммоль) в 90 мл ацетонитрила добавляют нитрат серебра (2,19 г, 12/89 ммоль). Далее раствор перемешивают в течение 30 часов при 80° С, пряча от света. Образовавшийся хлорид серебра фильтруют и растворитель выпаривают. Сырой продукт реакции очищают хроматографией на колонке с силикагелем, элюируя н-гексаном/этилацетатом 7/3. Получают 1,2 г продукта в виде желтого масла. Выход 46%.
1H ЯМР (200 МГц) (CDC13): 7,69 (1Н, dd); 7,33 (1H, d); 7,25 (1H, m); 7,23 (2H, m); 7,16 (1H, dd); 6,98 (2H, m); 6,82 (1H, s); 6,57 (1H, d); 5,49 (2H, s); 5,31 (2H, s); 3,94 (2H, s).
С) Синтез гидрохлорида 6-(нитроксиметил)-2-метилпиридинил эфира 2-[(2,6-дихлорфенил)амино]бензолуксусной кислоты
К раствору 6-(нитроксиметил)-2-метилпиридинил эфира 2-[(2,6-дихлорфенил)амино]бензолуксусной кислоты (0,400 г, 0,86 ммоль) в этилацетате (6 мл), охлажденному при 0° С, добавляют по каплям раствор HCl/этилацетата 3 М (0,6 мл) при перемешивании. Реакционную смесь перемешивают в течение одного часа при 0° С, затем нагревают до комнатной температуры.
Образовавшийся осадок фильтруют и промывают этиловым эфиром. Получают 0,310 г твердого продукта. Выход 73%.
Элементный анализ
Рассчитано: C 50,58%; H 3,63%; N 8,42%; Cl 21,32%.
Найдено: C 50,62%; H 3,66%; N 8,40%; Cl 21,20%.
Пример 14
Синтез нитрата 6-(нитроксиметил)-2-метилпиридинил эфира 2-[(2,6-дихлорфенил) амино]бензолуксусной кислоты формулы
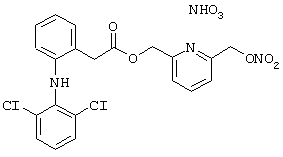
исходя из 6-(нитроксиметил)-2-метилпиридинил эфира 2-[(2,6-дихлорфенил)амино]бензолуксусной кислоты, полученного на стадии В предыдущего примера 13.
Синтез нитрата 6-(нитроксиметил)-2-метилпиридинил эфира 2-[(2,6-дихлорфенил) амино]бензолуксусной кислоты
К раствору 6-(нитроксиметил)-2-метилпиридинилэфира 2-[(2,6-дихлорфенил)аминорензолуксусной кислоты (0,760 г, 1,65 ммоль) в ацетонитриле (6 мл), охлажденному до 0° С, добавляют по каплям при перемешивании раствор азотной кислоты (65%) (0,150 мл) в ацетонитриле (2 мл). Реакционную смесь перемешивают один час при 0° С, затем нагревают до комнатной температуры. Сформировавшийся осадок фильтруют и промывают этиловым эфиром. Получают 0,600 г продукта в форме твердого вещества. Выход 70%.
Элементный анализ
Рассчитано: C 48,02%; H 3,45%; N 10,67%; Cl 13,50%.
Найдено: C 48,06%; H 3,47%; N 10,66%; Cl 13,60%.
Пример 15
Изучение ингибирующего воздействия на сокращение гладких мышц и пролиферацию гладко мышечных клеток.
Как известно, сокращение и/или клеточная пролиферация гладкой мышцы являются важными стадиями в процессе воспаления. Сокращение гладких мышц.
Новозеландских белых кроликов (2,0-2,5 кг) убивают цервикальным смещением, удаляют кавернозную ткань (кавернозные тела) и аорту.
Ткань помещают в баню для органов для регистрации изометрического натяжения согласно способу, описанному Khan MA и соавт. (BJU Int. 1999 84(6); 720-4). Ткани предварительно сокращают фенилэфрином (10 мкМ) и оценивают релаксационные отклики на карбакол в присутствии изучаемого соединения.
Соединение изобретения, используемое в анализе, является гидрохлоридом 6-(нитроксиметил)-2-пиридинилметил эфира 2-ацетилоксибензойной кислоты (NCX 4050), синтез которого описан в предшествующем примере 1.
Сравнительным соединением является (3-нитроксиметил)фенил эфир 2-ацетоксибензойной кислоты формулы
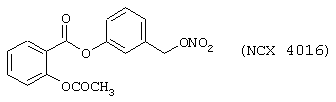
Синтез которого описан в примере 3 заявки PCT WO 97/16405, поданной на имя заявителя.
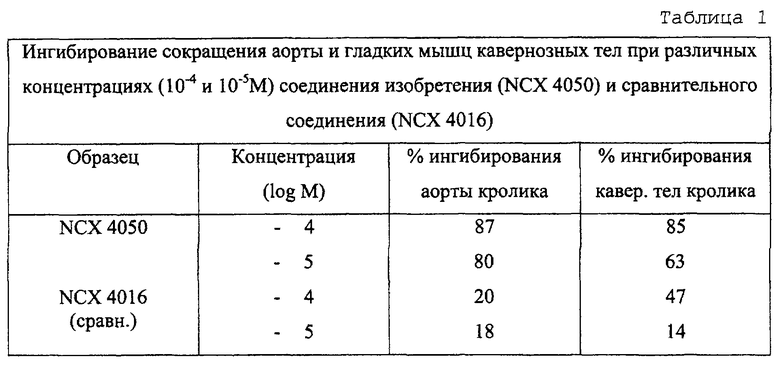
Результаты представлены в таблице 1, которая показывает, что соединение по изобретению является более активным, чем сравнительное соединение при ингибировании сокращения гладких мышц.
Клеточная пролиферация гладких мышц.
Подкожные вены ноги человека культивировали стандартными способами эксплантации (J. Cardiovasc. Pharmacol. 1999, 33(2), 204-11). Ткани собирали в стерильные сосуды, содержащие PBS (физиологический раствор с фосфатным буфером), пенициллин и стрептомицин. В условиях для стерильных тканевых культур ткани разрезают на маленькие кусочки (приблизительно массой 1 мг) и помещают в стандартную среду для культивирования, содержащую 20% эмбриональной телячьей сыворотки (FCS), на несколько дней (среду меняют каждые 2-4 дня). 3H-тимидин измеряют в ДНК фракции клеток, культивированных в 48-луночных планшетах. Клетки культивируют до слияния в среде, содержащей 10% FCS. Клетки лишены сыворотки в течение 24 часов перед добавлением 10% FCS вместе с различной концентрацией соединений. Через 24 часа 3H-тимидин добавляют к клеткам в течение 4 часов. Клетки промывают физиологическим раствором с фосфатным буфером и этанолом. ДНК экстрагируют раствором гидроксида натрия и 3H материал подсчитывают сцинтилляцией. Данные представляют наблюдения, сделанные в лунках в трех экземплярах.
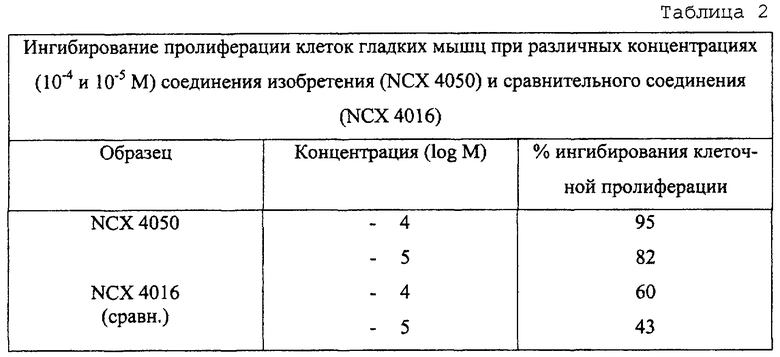
Таблица 2 представляет результаты, полученные для ингибирующего воздействия исследуемых соединений на пролиферацию клеток гладких мышц сосудов человека.
Таблица 2 показывает, что соединения изобретения гораздо более активны, чем сравнительные соединения.
Таблицы 1 и 2 демонстрируют, что противовоспалительная активность соединения изобретения выше, чем активность у сравниваемого соединения
А). Синтез дигидрохлорида 3-[4-(3-нитроксипропил)-1-пиперацинил]-пропилового эфира-(S)-6-метокси-альфа-метил-2-нафталенуксусной кислоты.
1а) 3-Бромпропиловый эфир-(S)-6-метокси-альфа-метил-2-нафталенуксусной кислоты.
К раствору 1,3-дибромпропана (2,42 мл) в DMF (50 мл) добавляли суспензию небольшими порциями и полученную суспензию перемешивали при комнатной температуре в течение 24 часов. Затем добавляли воды (200 мл) и органическую фазу извлекали с этиловым эфиром (100 мл × 3). Комбинированные органические фазы просушивали над Na2SО4 и раствор выпаривали и остаток очищали хроматографией на силикагеле, элюируя 9/1 гексан/этилацетатом, получая названное соединение (2,15 г).
2а) 3-[4-(3-хлорпропил)-1-пиперацинил]пропиловый эфир-(S)-6-метокси-альфа-метил-2-нафталенуксусной кислоты.
К раствору 3-бромпропилового эфира-(S)-6-метокси-альфа-метил-2-нафталенуксусной кислоты (1,5 г, 4,3 ммоль) в THF (20 мл) при температуре 40° С добавляли раствор дигидрохлорида N-(3-хлорпропил)пиперацина (1 г, 4,3 ммоль) в THF (25 мл), DMF (20 мл) и тритиламина (TEA) (2 мл). Полученный раствор нагревали до 55° С в течение 24 часов. Раствор охлаждали и экстрагировали с этиловым эфиром. Комбинированные органические фазы промывали водой, просушивали над Na2SО4 и фильтровали. Раствор выпаривали и остаток очищали хроматографией на силикагеле, элюируя 10:0,4 этилацетатом : TEA, получая 3-[4-(3-хлорпропил)-1-пиперацинил] пропиловый эфир-(S)-6-метокси-альфа-метил-2-нафталенуксусной кислоты (0,725 г).
1H NMR (CDC13): 7,67 (ЗН, m); 7,39 (1H, dd); 7,11 (2H, m); 4,11 (2H, m); 3,9 (3Н, s); 3,85 (1H, q); 3,56 (2H, t); 2,42-2,21 (12H, m); 1,90 (2H, m); 1,70 (2H, m); 1,56 (3Н, d).
3а) 3-[4-(3-нитроксипропил)-1-пиперацинил]-пропиловый эфир-(S)-6-метокси-альфа-метил-2-нафталенуксусной кислоты.
К раствору 3-[4-(3-хлорпропил)-1-пиперацинил]пропилового эфира-(S)-6-метокси-альфа-метил-2-нафталенуксусной кислоты (0,7 г, 1,6 ммоль) в ацетонитриле (50 мл) добавляли АgNО3 (0,545 г, 3,2 ммоль) и раствор нагревали до 60° С в течение 24 часов в темноте. Соли фильтровали, раствор выпаривали и остаток очищали хроматографией на силикагеле, элюируя 10:0,4 этилацетатом : ТЕА, получая 3-[4-(3-нитроксипропил)-1-пиперацинил]пропиловый эфир-(S)-6-метокси-альфа-метил-2-нафталенуксусной кислоты (0,1 г).
1H NMR (CDC13): 7,67 (3Н, m); 7,39 (1H, dd); 7,11 (2H, m); 4,5 (2H, t); 4,11 (2H, m); 3,9 (3Н, s); 3,85 (1H, q); 3,56 (2H, t); 2,42-2,21 (12H, m); 1,90 (2H, m); 1,70 (2H, m); 1,56 (3Н, d).
4a) Дигидрохлорид-3-[4-(3-нитроксипропил)-1-пиперацинил] пропилового эфира-(S)-6-метокси-альфа-метил-2-нафталенуксусной кислоты.
К раствору 3-[4-(3-нитроксипропил)-1-пиперацинил] пропилового эфира-(S)-6-метокси-альфа-метил-2-нафталенуксусной кислоты (0,1 г, 0,22 ммоль) в этилацетате, охлажденному в ледяной ванне, капали HCl/этилацетат (0,2 мл, 2,5 N), через 1 час температуру суспензии понижали до комнатной температуры. Продукт фильтровали, промывали Et2О и просушивали в условиях очень разреженного вакуума.
В) Синтез N-(3-хлорпропил)-пиперацин дигидрохлорида. К раствору N-Boc-пиперацин (2 г) в СН2Сl2 (40 мл) и TEA (1,8 мл), охлажденному до 0° С, добавляли 3-хлор-1-бромпропан (1,3 мл) и раствор нагревали до 50° С в течение 3 часов. Раствор выпаривали и остаток растворяли в CH2Cl2 и промывали водой. Органическую фазу просушивали над Nа2SO4 и фильтровали, раствор выпаривали и сырой продукт очищали хроматографией на силикагеле, элюируя 8:2 этилацетатом : гексан, получая N'-(3-хлорпропил)-N-пиперацин (1,1 г). Продукт растворяли в HCl/этилацетате (10 мл), раствор охлаждали до 0° С в ледяной ванне и перемешивали в течение 1 часа, затем нагревали до комнатной температуры. Раствор удаляли и остаток обрабатывали диэтиловым эфиром и полученный монолитный продукт фильтровали и применяли без последующей очистки.
Изучение эффекта ингибирования хлоргидрата 6-(нитроксиметил)- 2-пиридинилметипового эфира-2-ацетилбензойной кислоты.
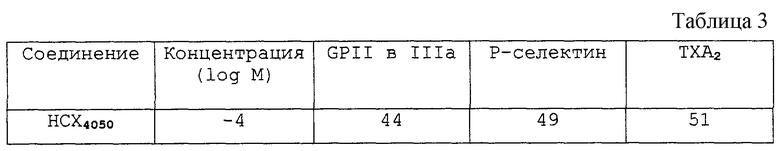
У нормального человека бралась проба крови, стимулировалась 100 μ М ADP и Р-селектином и значение GPII в IIIa измерялось с использованием потока ситометрии. Пластинка, обогащенная плазмой (PRP), также готовилась и эффект соединения проверялся на кальцийионофоре А 23187 (10 μ М) - стимулированный тромбоксан А2 (TXA2), определенный радиоиммунным испытанием (Jeremy et al., Eur J. Pharmacol., 1993, 15, 245 (1); 67-73).
В таблице 3 приведены результаты, относящиеся к эффекту ингибирования тестуемого соединения на активность на человеке.
| название | год | авторы | номер документа |
|---|---|---|---|
| НИТРОКСИСОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ИМЕЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНУЮ, АНАЛЬГЕТИЧЕСКУЮ И АНТИТРОМБОЦИТАРНУЮ АКТИВНОСТИ | 1995 |
|
RU2145595C1 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ ОПУХОЛЕЙ | 1997 |
|
RU2269510C2 |
| СТЕРОИДЫ, ВЫСВОБОЖДАЮЩИЕ ОКСИД АЗОТА | 2008 |
|
RU2442790C2 |
| ПРОИЗВОДНЫЕ НИТРОЭФИРОВ И КОМПОЗИЦИИ НА ИХ ОСНОВЕ ДЛЯ ИСПОЛЬЗОВАНИЯ ПРИ КОНТРОЛИРОВАНИИ НЕДЕРЖАНИЯ МОЧИ | 1997 |
|
RU2210563C2 |
| НИТРОСОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ И АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЯМИ | 1996 |
|
RU2165921C2 |
| СПОСОБ СИНТЕЗА НИТРОКСИМЕТИЛФЕНИЛОВЫХ ЭФИРОВ АСПИРИНА И ЕГО ПРОИЗВОДНЫХ | 2000 |
|
RU2232747C2 |
| ИНГИБИТОРЫ MMPL3, КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2795229C2 |
| ПРОЛЕКАРСТВА НА ОСНОВЕ ГИДРОГЕЛЯ | 2013 |
|
RU2647729C2 |
| ДИАГНОСТИКА, ПРОФИЛАКТИКА И ЛЕЧЕНИЕ БОЛЕЗНЕЙ СУСТАВОВ | 2013 |
|
RU2682676C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ - 759 | 2008 |
|
RU2481348C2 |
Изобретение относится к органической химии и может найти применение в медицине. Описываются органические или неорганические соли соединений общей формулы A-X1-N(O)Z,
где z является целым числом и означает 1 или 2; A=R(COXu)t, где t и u являются целым числом и означают 1; Х=0;
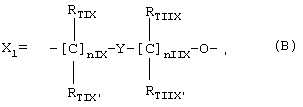
где nIX означает целое число между 0 и 3; nIIX означает целое число между 1 и 3; RTIX, RTIX', RTIIX, RTIIX' равны или отличны друг от друга, означают Н или линейный или разветвленный алкил; Y означает гетероциклическое кольцо, насыщенное или ненасыщенное или ароматическое, имеющее 6 атомов и содержащее один или два атома азота; R выбран из групп раскрытых в формуле изобретения. Также описываются фармацевтические составы для орального и парентерального применения, содержащие в качестве активных компонентов соли, описанные выше и обладающие ингибирующим воздействием на сокращение гладких мышц и клеточную пролиферацию. Технический результат - получены новые соли соединений, обладающие полезными биологическими свойствами. 2 с. и 9 з.п. ф-лы, 3 табл.
A-X1-N(O)Z
где z является целым числом и означает 1 или 2;
А=R(COXu)t, где t и u являются целым числом и означают 1;
X=O;
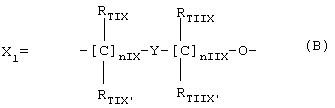
где nIX означает целое число между 0 и 3;
nIIX означает целое число между 1 и 3;
rtix, rtix’, rtiix, rtiix’ равны или отличны друг от друга, означают Н или линейный или разветвленный C1-C4 алкил;
Y означает гетероциклическое кольцо, насыщенное или ненасыщенное или ароматическое, имеющее 6 атомов и содержащее один или два атома азота;
R выбран из следующих групп:
группа I)
где R1 означает OCOR3 группу, где R3 означает метил, этил или линейный или разветвленный С3-С5 алкил;
Ib)
Ic)
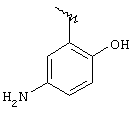
группа II)
rii1, rii2 и RII3 означают водород, линейный или разветвленный C1-C4 алкил или Cl, F, Вr;
Rii4 означает RII1;
где в формуле IIа, когда RII2 и RII3 означают Cl в орто-положении по отношению к NH,
RII1, Rii4, RII5 и RII6 означают водород, R означает остаток диклофенака.
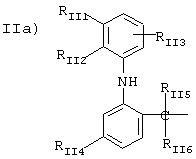
группа III)
где R2a и R3a означают водород, линейный или разветвленный, C1-C12 алкил;
R1a выбран из
IIID) R1a соответствует следующим формулам
где в группе III значения следующие:
когда R1a такой, как определено в формуле (IV), RIII1 и RIII2 означают водород, R2a означает метил, R3a=Н, R означает остаток кетопрофена:
когда R1a такой, как определено в формуле (XXI), остаток карпрофена:
Rxxio и Rxxi означают Н, Rxxil означает Сl и находится в пара-положении по отношению к азоту, связующий мостик находится в положении 2,
R2a означает метил, R3a=Н, R означает остаток карпрофена,
когда R1a такой, как определено в формуле (XXXV),
Аr означает фенил, R2a означает метил, R3a=Н, R означает остаток тиапрофеновой кислоты;
когда R1a такой, как определено в формуле (II), R2a означает метил, R3a=Н, R означает остаток супрофена,
когда R1a такой, как определено в формуле (VI), R2a=Н и R3a=СН3, R означает остаток индопрофена;
когда R1a такой, как определено в формуле (VI), R2a=Н, R3a=С2Н2, R означает остаток индобуфена;
когда R1a такой, как определено в формуле (VIII), R2a=R3a=Н, R означает остаток этодолака;
когда R1a такой, как определено в формуле (VII), R3a=Н, R2a=СН3, R означает остаток фенопрофена;
когда R1a такой, как определено в формуле (III), R2a=R3a=Н, R означает остаток фенбуфена;
когда R1a такой, как определено в формуле (IX), R3a=Н, R2a=СН3, R означает остаток флурбипрофена;
когда R1a такой, как определено в формуле (X), R2a=R3a=Н, R означает остаток толметина;
в группе IIID):
когда R1a, такой, как определено в формуле:
III а), R2a=H, R3a=СН3, R означает остаток пранопрофена;
когда R1a такой, как определено в формуле (XXX), R2а=Н, R3a=СН3, R означает остаток бермопрофена;
когда R1a такой, как определено в формуле (XXXI), R2a=Н, R3a=СН3, R означает радикал соединения CS-670; 2-[4-(2-оксо-1-циклогексилиденметил)фенил]пропионовая кислота;
когда R1a такой, как определено в формуле (XXXII), R2a=R3a=Н, R означает остаток пемедолака;
когда R1a такой, как определено в формуле (XXXIII), R2a=R3a=Н, R означает остаток пиразолака;
когда R1a такой, как определено в формуле (XXXVI), R2a=Н, R3a=СН3, R означает остаток залтопрофена;
когда R1a такой, как определено в формуле (XXXVII), R2a=R3a=H, R означает остаток мофезолака; 3,4-ди (п-метоксифенил)изоксазол-5-уксусная кислота, когда остаток означает CH2-СООН; в предпочтительных соединениях R2a=R3a=H, t=1 и Х=0;
когда R1a такой, как определено в формуле (XII), R2a=R3a=Н, R означает остаток бромфенака;
в группе IV) R означает
где RIvd и RIvd1 означают, по крайней мере, один Н и другой алкил от C1 до С6;
riv имеет следующее значение:
где riv как определено в формуле (IIВ), Riv-ii означает СН3-O-,
Rivd=Н, Rivd1=СН3, R означает остаток напроксена;
riv как определено в формуле (ХВ), Rivd=Н,
Rivd1=СН3, R означает остаток локсопрофена;
Riv как определено в формуле (IIIB), Riv-iii означает
и Rivd=Н, Rivd1 означает СН3, R означает остаток ибупрофена;
группа V), где R означает:
в группе V) имеются следующие значения:
когда R имеет формулу (IIC),
Rvii и Rvii-1 означают Н, R означает остаток кетополака; когда R имеет формулу (IVC) R означает остаток индометасина.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |

