Предпосылки изобретения
Область изобретения
Настоящее изобретение в целом относится к области органической химии и к соединениям с антипролиферативными и проапоптотическими свойствами. Более конкретно, настоящее изобретение относится к соединениям на основе хромана и его производным и аналогам и их применению в качестве антипролиферативных, проапоптотических, иммуномодулирующих и противовирусных средств.
Область техники
Биология клеточной пролиферации и клеточной гибели (апоптоза) является крайне сложной, включающей множество межклеточных путей передачи сигнала и множество продуктов взаимодействия генов. Раковые клетки могут проявлять разнообразные дефекты нормального контроля регуляции клеточной пролиферации, которые позволяют им увеличивать свою численность. Кроме того, раковые клетки проявляют дефекты механизмов, участвующих в элиминации аномальных клеток с помощью многостадийного процесса, рассматриваемого как запрограммированная клеточная гибель или апоптоз. Таким образом, сочетание нерегулируемой клеточной пролиферации и супрессии гибели, затрагивающей пути передачи сигнала, способствует как росту раковых клеток, так и их жизнеспособности.
Происходит ли увеличение численности клеток или не происходит, зависит от баланса экспрессии отрицательно и положительно регулирующих рост продуктов регуляторных генов и наличия и отсутствия функциональных путей передачи сигнала клеточной гибели. Отрицательно регулирующие рост регуляторные гены приводят к блокированию клеток в ходе клеточного цикла. Положительно регулирующие рост регуляторные гены стимулируют клетки к прохождению клеточного цикла. Гены, участвующие в апоптозе, могут быть как проапоптотического, так и противоапоптотического характера, и динамический баланс между ними определяет выбор роста клетки или ее гибели.
Раковые клетки для выживания и увеличения своей численности проходят ряд мутаций в течение времени, которые устраняют регуляторный контроль, что дает им способность к неконтролируемому росту и жизнеспособности даже в присутствии проапоптотических сигналов и придает свойства, позволяющие ускользать от обнаружения и удаления с помощью защитной реакции иммунной системы. Злокачественные опухоли могут быть причиной смерти пациентов при отсутствии удаления хирургическим путем или эффективного лечения с помощью лекарственных препаратов.
Существует большое разнообразие патологических состояний пролиферации клетки, которые нуждаются в новых терапевтических стратегиях и препаратах для достижения терапевтического успеха. Данные патологические состояния могут наблюдаться почти во всех типах клеток, способных к аномальной клеточной пролиферации или аномальной быстроте реагирования на сигналы клеточной гибели. В числе типов клеток, проявляющих патологические или аномальные характеристики роста и гибели, находятся (1) фибробласты, (2) эндотелиальные клетки сосудов и (3) эпителиальные клетки. Таким образом, необходимы новые способы лечения локальных или распространенных патологических состояний во всех или почти во всех системах органов и тканях человека.
Большинство злокачественных опухолей, являющихся специфическими для мужчин, такие как опухоли предстательной железы или яичек, или специфическими для женщин, такие как опухоли молочной железы, яичников или шейки матки, или поражающих мужчин и женщин в равной степени, такие как опухоли печени, кожи или легких, со временем претерпевают повышенное количество генетических повреждений и эпигенетических воздействий и становятся в равной степени высоко метастатическими и плохо поддающимися лечению. Хирургическое удаление обособленных злокачественных опухолей является оправданно эффективным только в случае, если злокачественная опухоль не распространилась далеко за пределы первичного очага поражения. Как только злокачественная опухоль распространяется на другие ткани и органы, хирургические процедуры должны дополняться другими более специфическими процедурами для уничтожения нездоровых или малигнизированных клеток. Большинство из обычно применяемых дополнительных процедур удаления нездоровых или малигнизированных клеток, такие как химиотерапия или биооблучение, не являются локальными по отношению к расположению опухолевых клеток и, хотя они оказывают пропорционально большее разрушающее действие на малигнизированные клетки, часто до некоторой степени затрагивают нормальные клетки.
Некоторые производные токоферолов, токотриенолов и витамин Е применяются в качестве проапоптотических и ингибирующих синтез ДНК веществ. Структурно витамин Е состоит из хроманольной группы и алкильной боковой цепи. Существует восемь основных форм витамина Е, встречающихся в природе: альфа (α), бета (β), гамма (γ) и дельта (δ) токоферолы и α, β, γ и δ токотриенолы. Токоферолы отличаются от токотриенолов наличием насыщенной фитильной боковой цепи в отличие от ненасыщенной изопренильной боковой цепи. Четыре формы токоферолов и токотриенолов отличаются числом метильных групп у хроманольной группы (α содержит три, β и γ содержат две и 8 содержит одну группу).
RRR-α-токоферилсукцинат представляет собой производное RRR-α-токоферола, которое структурно модифицировано с помощью сложноэфирной связи для присоединения сукцинильной группы взамен гидроксильной группы по 6-му положению в хромановой группе. Данный сложный эфир RRR-α-токоферола, содержащий сукцинатную составляющую, представляет наиболее эффективную форму витамина Е, оказывающую биологическое действие в виде запуска апоптоза и ингибирования синтеза ДНК. Данная форма витамина Е индуцирует в опухолевых клетках переход в апоптоз, не оказывая индуцирующего апоптоз влияния на нормальные клетки. Основное преимущество данной формы витамина Е в качестве противоопухолевого вещества заключается в том, что многие малигнизированные клетки либо экспрессируют низкие уровни эстеразы, либо вовсе не экспрессируют эстеразы, которые могут отщепить сукцинатную группу, преобразуя RRR-α-токоферол из сукцинатной формы в свободный RRR-α-токоферол. RRR-α-токоферол не обладает ни высокой антипролиферативной, ни запускающей апоптоз биологической активностью. Однако сложноэфирный сукцинат витамина Е не эффективен в vivo, поскольку естественные эстеразы хозяина отщепляют сукцинатную группу, преобразуя в неэффективное противораковое средство RRR-α-токоферол.
Данные предшествующего уровня техники недостаточны из-за отсутствия эффективных способов ингибирования нежелательной или неконтролируемой клеточной пролиферации при большом ряде патофизиологических состояний, характеризующихся отсутствием или небольшим воздействием на нормальные клетки. Настоящим изобретением реализована данная давно существующая необходимость и потребность в данной области.
Сущность изобретения
Согласно одному варианту осуществления настоящего изобретения предложено соединение, имеющее структурную формулу
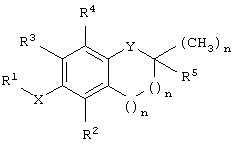
где Х представляет собой кислород, азот или серу; Y выбран из группы, включающей кислород, азот и серу, при этом в случае, когда Y представляет собой кислород или азот, n равно 1, и когда Y представляет собой серу, n равно 0; R1 выбран из группы, включающей алкил, алкенил, алкинил, арил, гетероарил, карбоновую кислоту, карбоксилат, карбоксамид, сложный эфир, тиоамид, сложный тиолэфир, тиолкислоту, сахарид, алкоксисвязанный сахарид, амин, сульфонат, сульфат, фосфат, спирт, эфир и нитрил; R2 выбран из группы, включающей водород, метил, бензилкарбоновую кислоту, бензилкарбоксилат, бензилкарбоксамид, сложный бензиловый эфир, сахарид и амин; R3 выбран из группы, включающей водород, метил, бензилкарбоновую кислоту, бензилкарбоксилат, бензилкарбоксамид, сложный бензиловый эфир, сахарид и амин; R4 выбран из группы, включающей метил, бензилкарбоновую кислоту, бензилкарбоксилат, бензилкарбоксамид, сложный бензиловый эфир, сахарид и амин, и R5 выбран из группы, включающей алкил, алкенил, алкинил, арил, гетероарил, карбоксил, амин и сложный эфир, где, когда Y представляет собой азот, указанный азот замещен R6, где R6 выбран из группы, включающей водород и метил; где, когда Х представляет собой кислород, R2 представляет собой метил, R3 представляет собой метил, R4 представляет собой метил и R5 представляет собой фитил, тогда R1 не является бутановой кислотой.
Согласно другому варианту осуществления настоящего изобретения предложен способ лечения клеточно-пролиферативных заболеваний, включающий введение животному фармакологически эффективной дозы соединения, имеющего структурную формулу
где Х представляет собой кислород, азот или серу; Y выбран из группы, включающей кислород, азот и серу, при этом, когда Y представляет собой кислород или азот, n равно 1, и, когда Y представляет собой серу, n равно 0; R1 выбран из группы, включающей алкил, алкенил, алкинил, арил, гетероарил, карбоновую кислоту, карбоксилат, карбоксамид, сложный эфир, тиоамид, сложный тиолэфир, тиолкислоту, сахарид, алкоксисвязанный сахарид, амин, сульфонат, сульфат, фосфат, спирт, эфир и нитрил; R2 выбран из группы, включающей водород, метил, бензилкарбоновую кислоту, бензилкарбоксилат, бензилкарбоксамид, сложный бензиловый эфир, сахарид и амин; R3 выбран из группы, включающей водород, метил, бензилкарбоновую кислоту, бензилкарбоксилат, бензилкарбоксамид, сложный бензиловый эфир, сахарид и амин; R4 выбран из группы, включающей метил, бензилкарбоновую кислоту, бензилкарбоксилат, бензилкарбоксамид, сложный бензиловый эфир, сахарид и амин, и R5 выбран из группы, включающей алкил, алкенил, алкинил, арил, гетероарил, карбоксил, амин и сложный эфир, где, когда Y представляет собой азот, указанный азот замещен с помощью R6, где R6 выбран из группы, включающей водород и метил.
Согласно еще одному варианту осуществления настоящего изобретения предложена фармацевтическая композиция, содержащая соединение, представленное здесь, и фармацевтически приемлемый носитель.
Согласно еще одному варианту осуществления настоящего изобретения предложен способ индуцирования апоптоза клетки, включающий стадию контактирования указанной клетки с фармакологически эффективной дозой соединения согласно настоящему изобретению.
Другие и дополнительные аспекты, признаки, выгоды и преимущества настоящего изобретения будут понятны из последующего описания предпочтительных воплощений изобретения, представленных с целью иллюстрации изобретения.
Краткое описание чертежей
Для того чтобы вышеперечисленные признаки, преимущества и объекты изобретения, а также остальное были бы ясны в деталях, далее следуют более конкретные описания с отсылкой к некоторым вариантам осуществления, которые проиллюстрированы прилагаемыми чертежами. Данные чертежи образуют часть описания. Следует отметить, однако, что прилагаемые чертежи иллюстрируют предпочтительные варианты осуществления изобретения и поэтому не должны рассматриваться как ограничивающие его объем.
На фиг.1 представлена общая структура токоферола, токотриенола и других соединений на основе хромана.
На фиг.2 представлены основные соединения 1-29 на основе токоферола, синтезированные и исследованные здесь.
На фиг.3А, 3В и 3С представлены основные способы органического синтеза для химической модификации хроманольных соединений по положению R1.
На фиг.4 представлены основные способы органического синтеза для химической модификации хроманольных соединений по положению R2.
На фиг.5 представлены основные способы органического синтеза для химической модификации хроманольных соединений по положению R3 и R4.
На фиг.6 представлены основные способы органического синтеза для химической модификации хроманольных соединений по положению R5.
На фиг.7 представлены основные способы органического синтеза всех рацемических аналогов 1-аза-α-токоферола. На фиг.7А представлена схема синтеза соединений 31-38 и на фиг.7В представлена схема синтеза соединений 39-43.
На фиг.8 представлены средние значения массы тела самок мышей безопухолевой линии Balb/c на 11-е сутки и на 23-и сутки изучения максимально переносимой дозы.
На фиг.9 представлено сравнение среднего процентного значения массы опухоли после лечения в раковых клетках молочной железы человека MDA МВ-435 (фиг.9А), раковых клетках предстательной железы человека DU-145 (фиг.9В) и раковых клетках толстого кишечника человека НТ-29 (фиг.9С).
Подробное описание изобретения
Следующие определения даны с целью облегчения понимания описываемого здесь изобретения. Любые термины, не определенные конкретным образом, должны интерпретироваться согласно обычным значениям терминов в данной области.
Применяемый здесь термин "индивидуум" относится к животным и человеку.
Применяемый здесь термин "биологическое ингибирование" или "ингибирование" роста пролиферирующих клеток должен включать частичное или полное ингибирование роста и также подразумевает снижение скорости пролиферации или роста клеток. Биологически ингибирующая доза композиции согласно настоящему изобретению может быть определена с помощью оценки действия исследуемого соединения на рост малигнизированных или аномально пролиферирующих клеток в культуре ткани, на рост опухоли у животных и в клеточной культуре или любым другим способом, известным специалисту в данной области.
Применяемый здесь термин "индукция запрограммированной клеточной гибели или апоптоза" включает частичную или полную гибель клетки с проявлением клетками апоптотических характеристик, определяемых морфологически и биохимически. Доза композиции согласно настоящему изобретению, индуцирующая апоптоз, может быть определена с помощью оценки эффектов исследуемого соединения, действующего на рост малигнизированных или аномально пролиферирующих клеток в культуре ткани, на рост опухоли у животных и в клеточной культуре или любым другим способом, известным специалисту в данной области.
Применяемый здесь термин "индукция остановки клеточного цикла" должен включать остановку роста в результате задержки обработанных клеток на стадии GO/G1 или G2/M клеточного цикла. Доза композиции согласно настоящему изобретению, индуцирующая остановку клеточного цикла, может быть определена с помощью оценки действия исследуемого соединения на рост малигнизированных или аномально пролиферирующих клеток в культуре ткани, на рост опухоли у животных и в клеточной культуре или любым другим способом, известным специалисту в данной области.
Применяемый здесь термин "индукция клеточной дифференцировки" должен включать остановку роста в результате индуцирования прохождения обработанными клетками стадии клеточной дифференцировки, в ходе которой не происходит пролиферации клеток. Доза композиции согласно настоящему изобретению, индуцирующая дифференцировку клеток, может быть определена с помощью оценки действия исследуемого соединения на рост малигнизированных или аномально пролиферирующих клеток в культуре ткани, на рост опухоли у животных и в клеточной культуре, или любым другим способом, известным специалисту в данной области.
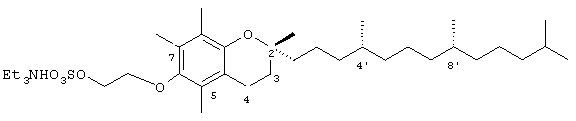
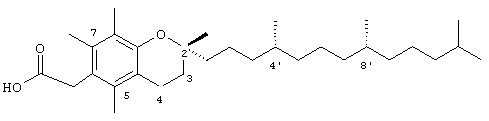
Настоящее изобретение относится к токоферолам, токотриенолам и другим хромановым производным вместе с производными по насыщенной фитильной или ненасыщенной изопренильной боковым цепям или без них, и их аналогам, например азо- и тиоловым аналогам. Применяя эфирные и некоторые другие химические связи для присоединения различных последовательностей к токоферолу, токотриенолу и другим хромановым производным, получали новые противоопухолевые соединения для применения в vivo. Основные структуры новых соединений согласно настоящему изобретению представлены на фиг.1, предпочтительные соединения представлены на фиг.2, и возможные пути их синтеза представлены на фиг.3-7. Новые свойства данных молекул включают введение функциональных групп в хромановую структуру по положениям R1-R5 и введение функциональных групп в фитильную и изопренильную боковые цепи, в частности в соединения на основе токоферолов и токотриенолов (фиг.1). Кроме того, представлены соединения с гетероатомными заместителями (N или S) кислорода хроманового кольца (фиг.7). Особенно предпочтительные соединения включают 2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту (1), 2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)пропионовую кислоту (2), 2,5,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислому (7), 2,7,8-триметил(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту (8), 2,8-диметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту (9), 2-(N,N-(карбоксиметил)-2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту (12), 2,5,7,8-тетраметил-(2RS-(4RS,8RS,12-триметилтридецил)хроман-6-илокси)уксусную кислоту (15), 2,5,7,8-тетраметил-2R-(2RS,6RS,10-триметилундецил)хроман-6-илокси)уксусную кислоту (17), хлорид 3-(2,5,7,8-тетраметил-(2R-(4R,8,12-триметилтридецил)хроман-6-илокси)пропил-1-аммония (19), 2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-3-ен-6-илокси)уксусную кислоту (20), сульфат 2-(2,5,7,8-тетраметил-(2R-(4R,8,12-триметилтридецил) хроман-6-илокси)триэтиламмония (21), 6-(2,5,7,8-тетраметил-(2R-(4R,8,12-триметилтридецил)хроман)уксусную кислоту (22), 2,5,7,8-тетраметил-(2R-(гептадецил)хроман-6-илокси)уксусную кислоту (25), 2,5,7,8-тетраметил-2R-(4,8-диметил-1,3,7-EZ-нонотриен)хроман-6-илокси)уксусную кислоту (26), E,Z,RS,RS-(фитилтриметилбензолтиол-6-илокси)уксусную кислоту (27), 1-аза-α-токоферол-6-илоксилуксусную кислоту (39), 1-аза-α-токоферол-6-илоксилметилацетат (40), 1-аза-N-метил-α-токоферол-6-илоксилметилацетат (41), 1-аза-N-метил-α-токоферол-6-илоксилуксусную кислоту (42), 6-(2,4-динитрофенил)азо-(2,5,7,8-тетраметил)-2-(4,8,12-триметилтридецил)-1,2,3,4-тетрагидрохинолин (43).
Фармакодинамически полученные соединения согласно настоящему изобретению обладают улучшенным терапевтическим индексом и являются эффективными ингибиторами роста раковых клеток, т.е. они проявляют высокую противоопухолевую активность с минимальными побочными эффектами. Указанные соединения, которые не могут легко разлагаться, поскольку у млекопитающих отсутствуют известные эстеразы, могут применяться при лечении злокачественных опухолей и расстройств, характеризующихся избытком клеточной пролиферации, а также в отношении клеток, увеличивающихся в количестве в результате супрессии механизмов гибели клеток, с минимальными побочными эффектами. Соединения согласно настоящему изобретению ингибируют рост раковых клеток с помощью индукции апоптоза и остановки синтеза ДНК. Индукция апоптоза данными соединениями опосредуется активацией TGF-β, стресс-киназы и путей передачи сигнала лиганда Fas/Fas. He исключается индукция апоптоза другими путями, например с помощью продукции церамида. Данные свойства ингибирования роста позволяют применять данные соединения для лечения пролиферативных заболеваний, включая злокачественные опухоли различных клеточных типов и происхождения, ненеопластических гиперпролиферативных заболеваний и расстройств с дефектами путей передачи сигнала к апоптозу. Несколько соединений согласно настоящему изобретению являются как сильными индукторами апоптоза, так и сильными ингибиторами синтеза ДНК в малигнизированных клетках, представляющих собой клетки различного происхождения.
Проиллюстрировано терапевтическое применение соединений согласно настоящему изобретению для лечения злокачественных опухолей и других заболеваний и расстройств, характеризующихся избытком клеточной пролиферации или неспособностью клеток к гибели. Новые производные (таблицы 1 и 2), показанные при концентрации ЕС50, индуцируют апоптоз раковых клеток молочной железы человека (раковых клеток молочной железы MDA MB 435, MDA MB 231 и MCF-7), раковых клеток предстательной железы человека (PC-3, DU-145 и LnCaP), раковых клеток яичника человека (С-170), раковых клеток шейки матки человека (МЕ-180), эндометриальных клеток человека (RL-95-2), лимфоидных клеток человека (миеломы, Raji, Ramos, Jurkat и HL-60), раковых клеток толстого кишечника (НТ29 и DLD-1) и раковых клеток легких (А-549). Новые производные не демонстрируют индукцию апоптоза нормальных эпителиальных клеток молочной железы человека (НМЕС) и иммортализованных, но не онкогенных, клеток молочной железы MCF-10A.
Данные новые соединения и способы согласно настоящему изобретению могут применяться для лечения неопластических заболеваний и ненеопластических заболеваний. Типичными примерами неопластических заболеваний являются рак яичника, рак шейки матки, рак эндометрия, рак мочевого пузыря, рак легких, рак молочной железы, рак предстательной железы, рак яичек, глиомы, фибросаркомы, ретинобластомы, меланомы, саркомы мягких тканей, остеосаркомы, рак толстого кишечника, карцинома почки, рак поджелудочной железы, базально-клеточная карцинома и плоскоклеточная карцинома. Типичные примеры ненеопластичеких заболеваний выбраны из группы, включающей псориаз, доброкачественные пролиферативные заболевания кожи, ихтиоз, папиллому, рестеноз, склеродермию и гемангиому.
Соединения и способы согласно настоящему изобретению могут применяться для лечения ненеопластических заболеваний, развивающихся в результате неспособности отдельных клеток нормально пройти запрограммированную клеточную гибель или апоптоз. Типичными примерами заболеваний и расстройств, возникающих в результате неспособности клеток к гибели, являются аутоиммунные заболевания. Аутоиммунные заболевания характеризуются повреждением собственных клеток, тканей и органов клетками иммунной системы. Типичная группа аутоиммунных заболеваний включает аутоиммунный тиреоидит, рассеянный склероз, тяжелую миастению, системную красную волчанку, герпетиформный дерматит, целиакию и ревматоидный артрит. Данное изобретение не ограничивается аутоиммунными заболеваниями, а включает все расстройства, имеющие иммунный компонент, такие как воспалительный процесс при образовании сердечно-сосудистых бляшек или кожные повреждения, вызванные ультрафиолетовым облучением.
Соединения и способы согласно настоящему изобретению могут применяться для лечения расстройств и заболеваний, развивающихся в результате вирусной инфекции. Типичными примерами заболеваний и расстройств, возникающих в результате вирусных инфекций, являются вирусы иммунодефицита человека (ВИЧ). Поскольку данные соединения участвуют в работе внутриклеточных путей передачи сигнала, они способны влиять на наружный клеточный сигнал любого типа, такой как цитокины, вирусы, бактерии, токсины, тяжелые металлы и т.п.
Способы согласно настоящему изобретению могут применяться для лечения любых животных. Более предпочтительно, способы согласно настоящему изобретению применимы для человека.
В основном для фармакологически эффективного достижения клеточной гибели и антипролиферативного эффекта данные соединения и их аналоги могут вводиться в любой терапевтически эффективной дозе. Предпочтительно структурно-модифицированные токоферолы и токотриенолы и аналоги вводят в дозе приблизительно от 0,1 до 100 мг/кг. Более предпочтительно структурно модифицированные токоферолы и токотриенолы и аналоги вводят в дозе приблизительно от 0,1 до 10 мг/кг.
Введение композиций согласно настоящему изобретению может проводиться местным, внутриглазным, парентеральным, пероральным, интраназальным, внутривенным, внутримышечным, подкожным или любым другим подходящим путем. Вводимая доза зависит от возраста, клинической стадии и тяжести заболевания или генетической предрасположенности индивидуума, расположения, массы тела, вида сопутствующего лечения, если оно имеет место, и природы патологического или злокачественного состояния. Эффективная система доставки, пригодная для способа согласно настоящему изобретению, может применяться в таких формах как капсулы, таблетки, жидкие растворы, суспензии или эликсиры для перорального применения или стерильные жидкие формы, такие как растворы, суспензии или эмульсии. Для местного применения могут использоваться такие формы, как мази, кремы или спреи. Предпочтительно применяется любой инертный носитель в комбинации с подходящими растворителями, такими как солевой раствор или фосфатный буферный солевой раствор, или любой носитель, такой как этанол, ацетон или ДМСО, в котором соединения, применяемые в данном способе согласно настоящему изобретению, обладают подходящими свойствами растворимости.
Существует большое количество патологических злокачественных и незлокачественных клеточно-пролиферативных состояний и накопления клеток в результате отсутствия нормального процесса клеточной гибели, для которых композиции и способы согласно настоящему изобретению будут обеспечивать терапевтический успех. Данные патологические состояния могут наблюдаться почти во всех типах клеток, способных к аномальной клеточной пролиферации или дефектных в отношении механизмов запрограммированной клеточной гибели. Среди типов клеток, которые проявляют способность к патологическому или аномальному росту или аномальной гибели, представлены (1) фибробласты, (2) эндотелиальные клетки сосудов и (3) эпителиальные клетки. Из вышесказанного можно увидеть, что способы согласно настоящему изобретению пригодны для лечения местных или системных патологических состояний во всех или почти во всех системах органов и тканей индивидуумов.
Специально предусмотрено, что фармацевтические композиции могут быть получены с помощью новых соединений на основе хромана и их производных и аналогов согласно настоящему изобретению. В таком случае фармацевтическая композиция содержит новые соединения согласно настоящему изобретению и фармацевтически приемлемый носитель. Специалисту в данной области будет легко определить без большого числа экспериментов соответствующие дозы и пути введения соединений и аналогов согласно настоящему изобретению.
Таким образом, настоящее изобретение направлено на создание и эффективное применение новых соединений, которые могут специфически воздействовать на раковые клетки и также отрицательно регулировать сигналы, стимулирующие рост, положительно регулировать сигналы, ингибирующие рост, отрицательно регулировать сигналы, способствующие выживаемости, и/или положительно регулировать сигналы, приводящие к гибели. Более конкретно, в данном изобретении разработаны и описаны новые соединения, активирующие факторы ингибирования роста, запускающие пути передачи сигнала к гибели и ингибирующие синтез ДНК.
Следующие примеры представлены с целью иллюстрации различных вариантов осуществления изобретения и не ограничивают изобретение каким-либо образом.
Пример 1
Методы органического синтеза
Синтез ряда токоферола, токотриенола и других хромановых производных, содержащих или не содержащих производных по насыщенной фитильной или ненасыщенной изопренильной боковым цепям, и их аналогов возможен с помощью структурных модификаций хромановой кольцевой системы (фиг.3-6) и гетероатомных заместителей (N или S) кислорода хроманового кольца (фиг.7А и 7В). Структурные переменные R1, R2, R3, R4, R5 и Х представляют группы на хромановой системе, которые изменяемы, и Y представляет либо кислород, либо гетероатомные заместители (N или S) кислорода хроманового кольца. Используя химические реакции алкилирования, можно синтезировать большое количество соединений, содержащих различные группы R1, особенно когда Х представляет собой кислород. После алкилирования дальнейшая химическая модификация групп R1 позволяет синтезировать большой диапазон новых соединений. Бромирование метильных групп бензила хроманового кольца обеспечивает промежуточные соединения, которые допускают варьирование групп R2, R3 и R4. Также возможно варьирование группы R5, особенно исходя из коммерчески доступной 6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоновой кислоты. В случае присутствия азотного гетероатомного заместителя кислорода хроманового кольца, азот может быть замещен с помощью R6, который представляет собой водород или метил. Замена Х на группы, отличные от кислорода, которые идентичны Х в токоферолах и токотриенолах, может выполняться с помощью химических реакций с палладием (для Х=СН2) и нуклеофильного ароматического замещения (для X=N или S). Другие возможные модификации хромановой структуры включают ненасыщенное состояние по положениям 3-4 и сужение цикла для получения пятичленного фуранильного кольца.
Используемые реагенты могут быть как коммерчески доступными, так и полученными согласно известным способам. Безводный СН2Cl2 и ТГФ получали с помощью дистилляции. Все другие применяемые растворители представляли собой реагенты. Безводные условия реакции поддерживали в слабо положительной атмосфере аргона в высушенной в печи стеклянной посуде. Хроматографию на силикагеле проводили через силикагель с 230-400 меш, приобретенный в ЕМ Science. Обычные спектры 1Н- и 13С-ЯМР получали на спектрометре Varian Unity при частотах 300,132 МГц и 75,033 МГц соответственно. Спектр ЯМР регистрировали с контролем с ТМС (0 м.д.) или с пиком CDCl3 с изотопными примесями (7,26 и 77,0 м.д. для 1H и 13С соответственно). Высокоразрешающую электронную масс-спектроскопию с ударной ионизацией проводили в Mass Spectrometry Center в University of Texas at Austin.
Пример 2
Синтез и характеристики нового соединения токоферола-2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусной кислоты (1)
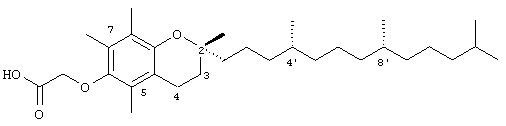
Раствор R,R,R-α-токоферола (0,5 г, 1,16 ммоль) в N.N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли с помощью 5 н. HCl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали с помощью H2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Раствор эфира концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 19 об.% EtOAc и 2% уксусной кислотой в гексане. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтой маслянистой жидкости и сушили в вакууме в течение 48 ч. В результате получали соединение 1 в виде воскообразного не совсем белого твердого вещества (0,50 г, 88%). 1H-ЯМР (CDCl3/ТМС, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-,11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,07, 2,14, 2,16 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 4,34 (с, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 69,2 (ОСН2), 75,0 (2-С), 117,8, 123,2, 125,4, 127,3 (арил С), 147,0, 148,5 (арил С-O), 173,7 (СООН); HRMS (CI, m/z): 489,394374 (М+Н+), вычисл. для С31Н53O4 489, 394386). Все определения были подтверждены с помощью HMQC, DEPT-135 и 1H-NOSEY.
Соединения 2-6 синтезировали способом, индентичным синтезу соединения 1, используя соответствующие бромалкановые кислоты.
2,5,7,8-Тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман)-6-илокси)пропионовая кислота (2)
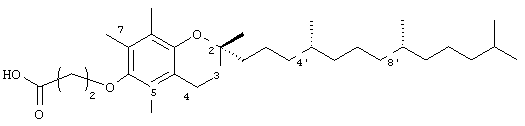
(выход 89%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,09, 2,14, 2,19 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 2,85 (т, J=6,4 Гц, 2Н, СН2СООН), 3,96 (т, J=6,4 Гц, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 35,1, 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 67,5 (ОСН2), 74,8 (2-С), 117,5, 122,9, 125,8, 127,8 (арил С), 147,6, 148,0 (арил С-O), 177,1 (СООН); HRMS (CI, m/z): 503, 408610 (М+Н)+, вычисл. для С32Н55O4 503,410036.
2,5,7,8-Тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)бутановая кислота (3)

(выход 85%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 26Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,14, 2,17, 2,21 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,62 (т, J=6,6 Гц, 2Н, 4-СН2), 2,72 (т, J=7,2 Гц, 2Н, СН2СООН), 3,74 (т, J=6,1 Гц, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,9 (2а-СН3), 24,4, 24,8, 25,3 (СН2), 28,0 (СН), 30,9, 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 71,3 (ОСН2), 74,8 (2-С), 117,5, 122,9, 125,7, 127,7 (арил С), 147,8, 147,9 (арил С-O), 178,9 (СООН); HRMS (CI, m/z): 516,424374 (М+Н)+, вычисл. для С33Н57O4 516,424386.
2,5,7,8-Тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)валериановая кислота (4)
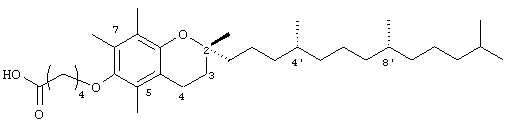
(выход 90%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 28Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 1'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,09, 2,14, 2,18 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,49 (т, J=6,8 Гц, 2Н, СН2СООН), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 3,68 (т, J=5,5 Гц, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0, 21,4 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 30,0 (СН2), 31,2 (3-СН2), 32,7, 32,8 (СН), 35,8, 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 72,2 (ОСН2), 74,9 (2-С), 117,8, 123,2, 125,4, 127,3 (арил С), 147,6, 148,3 (арил С-O), 178,7 (СООН); HRMS (CI, m/z): 530,433514 (М+Н)+, вычисл. для С34Н59O4 530, 433516.
2,5,7,8-Тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)гексановая кислота (5)
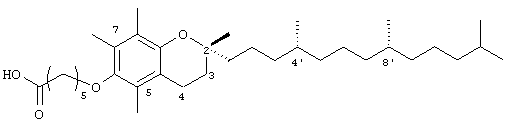
(выход 77%). 1H-ЯМР (CDCl3/ТМС, м.д.): 0,87 (м, 12Н, 4а'-,8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 30Н, 4'-, 8'-, 12'-СН, 1'-, 2', 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,08, 2,12, 2,16 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,32 (т, J=6,5 Гц, 2Н, CH2COOH), 2,57 (т, J=6,6 Гц, 2Н, 4-СН2), 3,64 (т, J=5,5 Гц, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,8, 11,9, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (CH2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,6, 24,8, 25,7 (СН2), 28,0 (СН), 30,0 (СН2), 31,3 (3-СН3), 32,7, 32,8 (СН), 34,0, 37,3, 37,3, 37,4, 39,3, 40,0 (СН2), 72,6 (ОСН2), 74,7 (2-С), 117,4, 122,7, 125,4, 127,8 (арил С), 147,6, 148,2 (арил С-O), 179,6 (СООН); HRMS (CI, m/z): 545, 457026 (М+H)+,вычисл. для С35Н61O4 545, 456986.
2,5,7,8-Тетраметил-2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)октановая кислота (6)
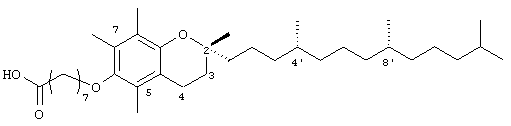
(выход 91%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 34Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,08, 2,11, 2,16 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,36 (м, 2Н, СН2СООН), 2,58 (т, J=6,6 Гц, 2Н, 4-СН2), 3,62 (т, J=5,5 Гц, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,6, 24,8, 25,1, 25,7, 26,6 (CH2), 28,0 (СН), 30,0 (СН2), 31,3 (3-СН2), 32,7, 32,8 (СН), 34,0, 37,3, 37,3, 37,4, 39,3, 40,0 (СН2), 72,7 (ОСН2), 74,6 (2-С), 117,6, 122,8, 125,5, 127,6 (арил С), 147,5, 148,3 (арил С-O), 179,4 (СООН); HRMS (CI, m/z): 573,484396 (М+Н)+, вычисл. для С37Н65O4 573, 488286.
2,5,8-Триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусная кислота (7)
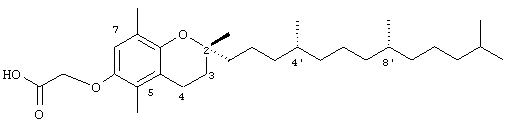
Раствор R,R,R-α-токоферола (75 мг, 0,18 ммоль) в N,N-диметилформамиде (2 мл) обрабатывали метилбромацетатом (0,4 г, 2,8 ммоль) и избытком порошкообразного NaOH (0,5 г, 12,5 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли с помощью 5 н. HCl и экстрагировали диэтиловым эфиром (3×10 мл). Объединенные эфирные фазы промывали Н2О (3×10 мл) и солевым раствором (1×10 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 19 об.% EtOAc и 2% уксусной кислотой в гексане. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2O (3×10 мл) и солевым раствором (1×10 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтой маслянистой жидкости и сушили в вакууме в течение 48 ч. В результате получали 7 в виде воскообразного не совсем белого твердого вещества (80 мг, 97%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,12, 2,14 (2 х с, 6Н, 5а-, 8а-СН3), 2,61 (т, J=6,6 Гц, 2Н, 4-СН2), 4,59 (с, 2Н, ОСН2), 6,53 (с, 1Н, арил СН); 13С-ЯМР (CDCl3, м.д.): 11,2, 16,1 (5а-, 8а-СН3), 19,6, 19,7 (СН3), 20,7, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 27,9 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,2, 37,4, 37,5, 39,4, 40,0 (СН2), 66,8 (ОСН2), 74,8 (2-С), 113,8, 120,7, 123,1, 127,3 (арил С), 147,1, 148,2 (арил С-O), 175,3 (СООН); HRMS (CI, m/z): 475,377840 (М+Н)+, вычисл. для С30H51О4 475,378736.
2,7,8-Триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусная кислота (8)
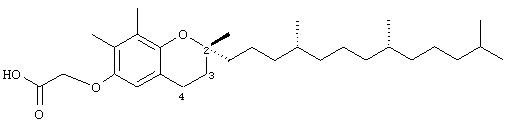
Раствор R,R,R-α-токоферола (100 мг, 0,24 ммоль) в N,N-диметилформамиде (5 мл) обрабатывали метилбромацетатом (1,1 г, 7,4 ммоль) и избытком порошкообразного NaOH (1,0 г, 25 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли с помощью 5 н. HCl и экстрагировали диэтиловым эфиром (3×10 мл). Объединенные эфирные фазы промывали Н2O (3×10 мл) и солевым раствором (1×10 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 19 об.% EtOAc и 2% уксусной кислотой в гексане. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2О (3×10 мл) и солевым раствором (1×10 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтой маслянистой жидкости и сушили в вакууме в течение 48 ч. В результате получали 8 в виде воскообразного, не совсем белого твердого вещества (110 мг, 97%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-,2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,12, 2,19 (2 х с, 6Н, 7а-, 8а-СН3), 2,61 (т, J=6,6 Гц, 2Н, 4-СН2), 4,59 (с, 2Н, ОСН2), 6,39 (с, 1Н, арил СН); 13С-ЯМР (CDCl3, м.д.): 11,9, 12,0 (7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,7, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (CH2), 27,9 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,2, 37,4, 37,5, 39,4, 40,0 (CH2), 66,6 (ОСН2), 75,7 (2-С), 110,6, 117,7, 125,0, 126,3 (арил С), 146,9, 148,7 (арил С-O), 175,0 (СООН); HRMS (CI, m/z): 475,377962 (М+Н)+, вычисл. для С30Н51O4 475,378736.
2,8-Диметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусная кислота (9)
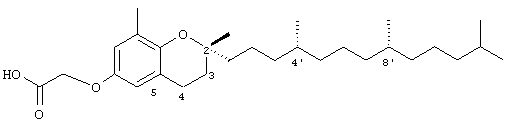
Раствор R,R,R-α-токоферола (100 мг, 0,25 ммоль) в N,N-диметилформамиде (5 мл) обрабатывали метилбромацетатом (1,1 г, 7,4 ммоль) и избытком порошкообразного NaOH (1,0 г, 25 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли с помощью 5 н. HCl и экстрагировали диэтиловым эфиром (3×10 мл). Объединенные эфирные фазы промывали Н2O (3×10 мл) и солевым раствором (1×10 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 19 об.% EtOAc и 2% уксусной кислотой в гексане. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2О (3×10 мл) и солевым раствором (1×10 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтой маслянистой жидкости и сушили в вакууме в течение 48 ч. В результате получали 9 в виде воскообразного, не совсем белого твердого вещества (111 мг, 98%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,15 (с, 3Н, 8а-СН3), 2,71 (т, J=6,6 Гц, 2Н, 4-СН2), 4,59 (с, 2Н, ОСН2), 6,48 (д, J=3,0 Гц, 1Н, арил СН), 6,61 (д, J=3,0 Гц, 1H, арил СН); 13С-ЯМР (CDCl3, м.д.): 16,2 (8а-СН3), 19,6, 19,7 (СН3), 21,0 (СН2), 22,6, 22,7 (СН3), 24,0 (2а-СН3), 24,4, 24,8 (СН2), 27,9 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,2, 37,4, 37,5, 39,4, 40,0 (СН2), 65,7 (ОСН2), 75,8 (2-С), 112,3, 115,6, 121,1, 127,5 (арил С), 147,2, 149,9 (арил С-O), 174,8 (СООН); HRMS (CI, m/z): 460,3552022 (М+Н)+, вычисл. для С30Н51O4 460,355262.
2,5,7,8-Тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетамид (10)
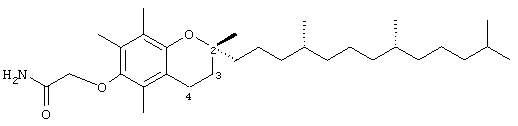
Раствор соединения 1 (0,1 г, 0,2 ммоль) в СН2Cl2 (5 мл) обрабатывали с помощью N-гидроксисукцинимида (26 мг, 0,23 ммоль) и дициклогексилкарбодиимида (46 мг, 0,23 ммоль). Через 2 мин образовывался белый осадок. Полученную суспензию перемешивали в течение 2 ч. Реакционную смесь перемешивали дополнительно в течение 6 ч. Реакционную смесь охлаждали до -30°С и фильтровали. Фильтрат концентрировали и полученную бесцветную маслянистую жидкость очищали с помощью хроматографии на силикагеле, проводя элюирование EtOAc (35%, об/об) в гексане. В результате получали белое твердое вещество (75 мг, 76%). 1H-ЯМР (CDCl3/ТМС, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-CH2), 2,10, 2,12, 2,16 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 4,19 (с, 2Н, ОСН2), 6,36, 6,92 (2 х ушир., 2Н, NH); 13С-ЯМР, (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 70,9 (ОСН2), 74,9 (2-С), 117,8, 123,3, 125,4, 127,3 (арил С), 146,5, 148,4 (арил С-O), 172,1 (СООН); HRMS (CI, m/z): 488, 409341 (М+H)+, вычисл. для C31H54NO3 488, 410370.
Метил-2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетат (11)
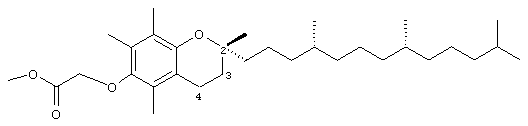
Раствор соединения 1 (0,1 г, 0,2 ммоль) в СН2Cl2 (5 мл) обрабатывали N,N-диметиламинопиридином (26 мг, 0,23 ммоль), метанолом (1 мл) и дициклогексилкарбодиимидом (46 мг, 0,23 ммоль). Через 2 мин образовывался белый осадок. Полученную суспензию перемешивали в течение 6 ч. Реакционную смесь охлаждали до -30°С и фильтровали. Фильтрат концентрировали и полученную бесцветную маслянистую жидкость очищали с помощью хроматографии на силикагеле, проводя элюирование EtOAc (40%, об/об) в гексане. В результате получали белое твердое вещество (82 мг, 80%.). 1H-ЯМР (CDCl3/ТМС, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,10, 2,16, 2,20 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 3,85 (с, 3Н, ОСН3), 4,32 (с, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 50,2 (ОСН3), 69,8 (ОСН2), 74,9 (2-С), 117,6, 123,0, 125,6, 127,5 (арил С), 147,6, 148,2 (арил С-O), 169,8 (СООН); HRMS (CI, m/z): 503, 408411 (М+Н)+, вычисл. для С32Н55O4 503, 410036.
2-(N,N-(Карбоксиметил)-2(2,5,7,8-тетраметил-(2R(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусная кислота (12)
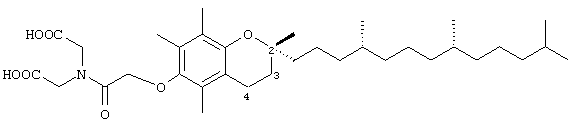
Раствор соединения 1 (0,2 г, 0,4 ммоль) в СН2Cl2 (5 мл) обрабатывали диэтилиминодиацетатом (77 мг, 0,4 ммоль) и гексафторфосфатом O-7-азабензотриазол-1-ил-N,N,N',N' -тетраметилурония (HATU) (46 мг, 0,23 ммоль). Через 12 ч реакционную смесь концентрировали до пастообразного состояния и затем очищали с помощью хроматографии на силикагеле, проводя элюирование EtOAc (30%, об/об) в гексане. В результате получали желаемый сложноэфирный промежуточный продукт в виде бесцветной маслянистой жидкости (150 мг, 55%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 30Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11-СН2, 2а-СН3), 1,78 (м, 2Н, 3-СН2), 2,08, 2,13, 2,17 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,58 (т, J=6,8 Гц, 2Н, 4-СН2), 4,19, 4,22 (кв., J=7,4 Гц, 4Н, ОСН2), 4,30, 4,33, 4,42 (3 х с, 6Н, 2 х NCH2, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 14,0 (СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (CH2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (CH2), 48,1, 49,4 (NCH2), 61,2, 61,5 ОСН2), 71,8 (ОСН2), 74,8 (2-С), 117,5, 122,9, 125,6, 127,4 (арил С), 148,0, 148,1 (арил С-O), 168,8, 169,0 (СО); MS (CI, m/z): 660 (М+Н)+, вычисл. для С39Н65NO7 659, 47610.
Раствор сложноэфирного промежуточного продукта (0,15 г, 0,23 ммоль) в этаноле (4 мл) обрабатывали 1 н. NaOH (1 мл). Полученную мутную смесь перемешивали при 70°С в течение 15 ч. Реакционную смесь подкисляли 1 н. HCl и удаляли этанол в вакууме. Полученный водный раствор экстрагировали с помощью CHCl3 (5×20 мл) и объединенные органические фазы сушили над Na2SO4. В результате получали 12 (0,13 г, 52%) в виде белого твердого вещества. 1H-ЯМР (CDCl3/ТМС, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11-СН2, 2а-СН3), 1,70 (м, 2Н, 3-СН2), 2,01, 2,05, 2,08 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,47 (м, 2Н, 4-СН2), 4,18 (м, 4Н, 2 х NCH2), 4,31 (м, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,5, 11,6, 12,4 (5а-, 7a-, 8а-СН3), 19,4, 19,5 (СН3), 20,6, 21,0 (СН3), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (CH2), 28,0 (СН), 31,2 (3-СН2), 32,4, 32,5 (СН), 37,0, 37,2, 37,59 39,1, 40,0 (СН2), 48,1, 49,4 (NCH2), 71,1 (ОСН2), 74,8 (2-С), 117,5, 122,9, 125,4, 127,2 (арил С), 147,8, 148,1 (арил С-O), 168,8, 169,0 (СО); HRMS (CI, m/z): 604, 420882 (М+Н)+, вычисл. для С35Н58NO7 604, 421329.
2-(2,5,7,8-Тетраметил-(2R-(4R,8R,12-триметилтридецилхроман-6-илокси))этан-1-ол (13)
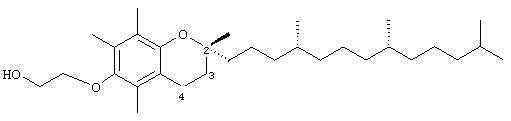
Раствор R,R,R-α-токоферола (0,5 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали иодэтанолом (1,7 г, 10 ммоль) и избытком порошкообразного NaOH (2,5 г, 63 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли с помощью 5 н. HCl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 30 об.% EtOAc и 2% уксусной кислотой в гексане. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2О (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтой маслянистой жидкости и сушили в вакууме в течение 48 ч. В результате получали 13 в виде желтой маслянистой жидкости (0,40 г, 73%). 1H-ЯМР (CDCl3/ТМС, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,07, 2,14, 2,16 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 3,79 (м, 2Н, ОСН2), 3,94 (м, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 63,1, 69,2 (ОСН2), 75,0 (2С), 117,8, 123,4, 126,4, 128,3 (арил С), 149,2, 149,5 (арил С-O); MS (CI, m/z): 475 (М+Н)+, вычисл. для С31Н54O3 474, 40729.
2-(2,5,7,8-Пентаметилхроман-6-илокси)уксусная кислота (14)
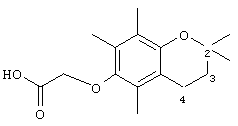
Раствор 2,2,5,7,8-пентаметил-6-хроманола (0,3 г, 1,36 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (0,8 г, 5,3 ммоль) и избытком порошкообразного NaOH (0,7 г, 18 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли с помощью 5 н. HCl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали Н2О (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 30 об.% EtOAc и 2% уксусной кислотой в гексане. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтой маслянистой жидкости и сушили в вакууме в течение 48 ч. В результате получали 14 в виде белого твердого вещества (0,31 г, 82%). 1H-ЯМР (CDCl3/TMC, м.д.): 1,31 (с, 6Н, СН3), 1,81 (т, J=7,8 Гц, 3-СН2), 2,10, 2,16, 2,19 (3 х с, 9Н, 5а-, 7а- 8а-СН3), 2,61 (т, J=7,8 Гц, 2Н, 4-СН2), 4,39 (с, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 11,8, 12,7 (5а-, 7а-, 8а-СН3), 20,9, 26,8, 32,7 (алкил), 69,1, (ОСН2), 72,9 (2-С), 117,5, 123,2, 125,5, 127,3 (арил), 147,0, 148,6 (O-арил), 173,8 (СООН); HRMS (CI, m/z): 279, 159238 (М+Н)+, вычисл. для С1бН23O4 279, 159634.
2,5,7,8-Тетраметил-(2RS-(4RS,8RS,12-триметилтридецил)хроман-6-илокси)уксусная кислота (15)
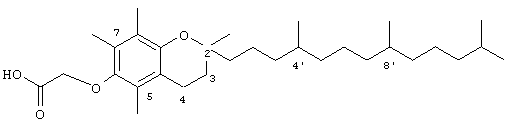
Раствор всех рацемических α-токоферолов (0,5 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли с помощью 5 н. HCl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали Н2О (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 19 об.% EtOAc и 2% уксусной кислотой в гексане. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали H2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтой маслянистой жидкости и сушили в вакууме в течение 48 ч. В результате получали 15 в виде воскообразного, не совсем белого твердого вещества (80%). 1H-ЯМР (CDCl3/ТМС, м.д.): 0,88 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,84 (м, 2Н, 3-СН2), 2,07, 2,14, 2,16 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,61 (т, J=6,6 Гц, 2Н, 4-СН2), 4,34 (с, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,5, 11,7, 12,6 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,3 (СН2), 22,6, 22,8 (СН3), 23,8 (2а-СН3), 24,5, 24,9 (CH2), 29,0 (СН), 31,6 (3-СН2), 32,6, 32,8 (СН), 37,5, 37,8, 37,9, 39,5, 41,0 (СН2), 69,3 (ОСН2), 75,1 (2-С), 117,9, 123,3, 125,5, 127,3 (арил С), 147,0, 148,0 (арил С-O), 173,9 (СООН); HRMS (CI, m/z): 489, 394375 (М+Н+), вычисл. для С31Н53O4 489, 394383.
2,5,7,8-Тетраметил-(2R-(карбокси)хроман-6-илокси)уксусная кислота (16)
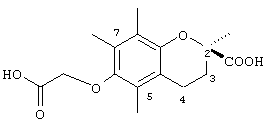
Раствор (-)-(R)-6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоновой кислоты (0,34 г, 1,36 ммоль) в N,N-диметилфомамиде (20 мл) обрабатывали метилбромацетатом (0,8 г, 5,3 ммоль) и избытком порошкообразного NaOH (0,7 г, 18 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли с помощью 5 н. HCl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 30 об.% EtOAc и 2% уксусной кислотой в гексане. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали H2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтой маслянистой жидкости и сушили в вакууме в течение 48 ч. В результате получали 16 в виде белого твердого вещества (0,33 г, 80%). 1H-ЯМР (CDCl3/ТМС, м.д.): 1,52 (с, 3Н, 2а-СН3), 2,10 (м, 2Н, 3-СН2), 2,12, 2,16, 2,19 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,56 (т, J=6,5 Гц, 2Н, 4-СН2), 4,36 (с, 2Н, ОСН2).
2,5,7,8-Тетраметил-2R-(2RS,6RS,10-триметилундецил)хроман-6-илокси)уксусная кислота (17)
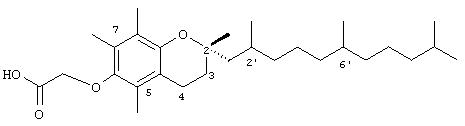
Раствор 10 г (40 ммоль) (-)-(S)-6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоновой кислоты и 0,5 г моногидрата пара-толуолсульфоновой кислоты в 200 мл метанола перемешивали и нагревали с обратным холодильником в течение 4 ч. После охлаждения раствор разбавляли водой и экстрагировали диэтиловым эфиром. Объединенные эфирные фазы промывали насыщенным водным раствором бикарбоната натрия, Н2О и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали и сушили в вакууме в течение 48 ч. В результате получали 10 г (95%) метил-(-)-(S)-6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоксилата в виде бесцветного твердого вещества, которое применяли без дальнейшей очистки. 1H-ЯМР (CDCl3/TMC, м.д.): 1,52 (с, 3Н, 2а-СН3), 2,10 (м, 2Н, 3-СН2), 2,12, 2,16, 2,19 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,56 (т, J=6,5 Гц, 2Н, 4-СН2), 3,55 (с, 3Н, ОСН3); MS (CI, m/z): 264,422 (М+Н)+, вычисл. для C15H20O4 265,3224.
К раствору 2 г (7,58 ммоль) данного сложного эфира в 7,5 мл N,N-диметилформамида (ДМФ) добавляли 2,6 г (18,8 ммоль) безводного гранулированного карбоната калия с последующим добавлением 2,3 мл (20 ммоль) бензилхлорида. Полученную смесь перемешивали при комнатной температуре в течение 41 ч, далее вносили в 50 мл воды и обрабатывали эфиром обычным образом.
Продукт освобождали от избытка бензилхлорида при 50°С в глубоком вакууме. В результате получали 2,69 г (100%) чистого (ТСХ) метилового эфира (-)-(S)-6-бензилокси-2,5,7,8-тетраметилхроман-2-карбоновой кислоты в виде желтого твердого вещества, точка плавления 102-106°С. Аналитический образец данного вещества получали в виде бесцветного твердого вещества, точка плавления 108-109°С (из эфира/метанола). 1H-ЯМР (CDCl3/TMC, м.д.): 1,54 (с, 3Н, 2а-СН3), 2,01 (м, 2Н, 3-СН2), 2,14, 2,17, 2,19 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,51 (т, J=6,7 Гц, 2Н, 4-СН2), 3,64 (с, 3Н, ОСН3), 5,12 (с, 2Н, 6-ОСН2), 7,15 (м, 5Н, ArH); MS (CI, m/z): 355,232 (М+Н)+, вычисл. для С22Н25O4 354,448.
Раствор 3,54 г (10 ммоль) вышеописанного сложного эфира в 20 мл толуола и 10 мл СН2Cl2 перемешивали при охлаждении на сухой бане лед/ацетон, при этом добавляя по капле 12 мл (18 ммоль) 25% гидрида диизобутилалюминия в толуоле (Texas AlkyIs) в течение 10 мин. После перемешивания при около -70°С в течение 30 мин реакционную смесь осторожно разлагали (-70°С) с помощью 10 мл МеОН. После добавления 50 мл воды и 50 мл 1 н. водного раствора H2SO4 смесь нагревали до комнатной температуры и обрабатывали эфиром обычным способом с получением 3,2 г (100%) неочищенного альдегида ((+)-S-6-бензилокси-2,5,7,8-тетраметилхроман-2-карбальдегид) в виде вязкой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 19 об.% EtOAc в гексане. 1H-ЯМР (CDCl3/ТМС, м.д.): 1,53 (с, 3Н, 2а-СН3), 2,11 (м, 2Н, 3-СН2), 2,24, 2,27, 2,29 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,481 (т, J=6,7 Гц, 2Н, 4-СН2), 5,19 (с, 2Н, 6-OCH2), 7,20 (м, 5Н, ArH), 9,6 (с, 1Н, СНО); MS (CI, m/z): 325,332 (М+Н)+, вычисл. для С21Н24O3 324,422.
Раствор 9,6 г псевдоионона растворяли в 100 мл 95% этанола; после добавления 0,68 г борогидрида натрия в этаноле при комнатной температуре смесь перемешивали в течение 2 ч и затем оставляли на ночь. Смесь добавляли к раствору 2 г гидроксида натрия в 500 мл воды. Смесь экстрагировали эфиром и эфирный экстракт промывали водой, сушили и концентрировали. Проводили дистилляцию полученной маслянистой жидкости в вакууме с получением бесцветной маслянистой жидкости (псевдоионол); точка кипения 112-120°С/5 мм рт.ст. 7,7 г (80%).
К раствору 2,97 г псевдоионола в 10 мл ацетонитрила добавляли при перемешивании и температуре ниже 30° С 4,53 г гидрохлорида трифенилфосфина, полученного пропусканием безводного хлористого водорода через раствор трифенилфосфина в безводном эфире. После оставления смеси на ночь при комнатной температуре удаляли ацетонитрил при пониженном давлении ниже 50° С. К остатку добавляли 4,47 г (+)-S-6-бензилокси-2,5,7,8-тетраметилхроман-2-карбальдегида в 15 мл диметилформамида, и смесь перемешивали. После получения прозрачного раствора вносили при перемешивании по капле метоксид натрия, полученный из 0,352 г натрия и 7 мл безводного метанола, при температуре ниже 15°С. Реакционная смесь становилась красной при образовании илида. После завершения добавления продолжали перемешивание в течение в течение 30 мин при 10°С; далее смесь постепенно нагревали до 80°С до исчезновения красной окраски. Продукт вносили в 200 мл 50% водного раствора метанола, обезвоживали и концентрировали в вакууме. Оставшуюся маслянистую жидкость растворяли в 20 мл эфира и добавляли раствор хлорида ртути в эфире до прекращения образования осадка. После фильтрования осадка фильтрат промывали водой, сушили и концентрировали с получением 4,7 г желтой маслянистой жидкости. Неочищенную смесь цис- и транс-изомеров алкена (MS (CI, m/z): 485,22, М+Н+, рассчит. для С34Н44O2 484,7255) растворяли в 30 мл этилацетата и добавляли 0,80 г 5% палладия на углероде, смесь встряхивали при давлении H2 40 фунтов/кв.дюйм в течение 30 ч и далее фильтровали через целит и хорошо промывали этилацетатом. Фильтрат концентрировали и очищали с помощью хроматографии на силикагеле, проводя элюирование EtOAc в гексане (1:9), с получением 2,5,7,8-тетраметил-(2R-(2RS,6RS,10-триметилундецил))-6-хроманола (выход 60%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,97 (м, 12Н, 2а'-, 6а'-, 10а'-, 11'-СН3), 1,1-1,7 (м, 20Н, 2'-, 6'-, 10'- CH, 1'-, 3'-, 4'-, 5'-, 7'-, 8'-, 9'-СН2, 2а-СН3), 1,88 (м, 2Н, 3-СН2), 2,17, 2,19, 2,20 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,63 (т, J=6,7 Гц, 2Н, 4-СН2); MS (CI, m/z): 403,27 (М+H)+, вычисл. для С27Н46O2 402,6632.
Раствор 2,5,7,8-тетраметил-(2R-(2RS,6RS,10-триметилундецил))-6-хроманола (0,466 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли с помощью 5 н. HCl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 19 об.% EtOAc и 2% уксусной кислотой в гексане. В результате получали соединение 17 с выходом 76%. 1H-ЯМР (CDCl3/TMC, м.д.): 0,97 (м, 12Н, 2а'-, 6а'-,10а'-, 11'-СН3), 1,2-1,7 (м, 20Н, 2'-, 6'-,10'-CH, 1'-, 3'-, 4'-, 5'-, 7'-, 8'-, 9'-СН2, 2а-СН3), 1,92 (м, 2Н, 3-СН2), 2,18, 2,20, 2,23 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,68 (т, J=6,8 Гц, 2Н, 4-СН2), 4,48 (с, 2Н, ОСН2); MS (CI, m/z): 461,44 (М+Н)+, вычисл. для С29Н48O4 460,700.
2,5,7,8-Тетраметил-2R-(2,6,10-триметил-1,3,5,9-EZ-декатетраен)хроман-6-илокси)уксусная кислота' (18)
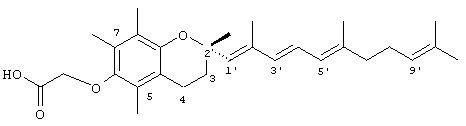
К раствору метил-(-)-(S)-6-гидрокси-2,5,7,8-тетраметилхроман-2-карбоксилата (20 г, 0,075 моль) в 50 мл безводного ДМФ добавляли имидазол (13 г, 0,1911 моль) и трет-бутилдиметилсилилхлорид (14 г, 0,0933 моль). Смесь перемешивали при 23°С в течение 24 ч и далее обрабатывали эфиром и выливали в 1 н. HCl. Органические экстракты сушили (солевой раствор, Na2SO4) и концентрировали в вакууме. Неочищенный продукт очищали с помощью тонкослойной хроматографии (гексан:этилацетат 9:1) с получением 6-(диметил(1,1-диметилэтил)силил)-2,5,7,8-тетраметилхроман-2-карбоксилата (сложный метиловый эфир, защищенный трет-бутилстиролом). 1H-ЯМР (CDCl3/TMC, м.д.): 0,12 (с, 6Н), 1,102 (с, 9Н), 1,18 (с, 3Н), 1,48 (с, 3Н), 1,645 (с, 3Н), 2,07 (с, 3Н), 2,2 (т, J=6,5 Гц, 2Н), 2,48-2,7 (м, 2Н) и 3,72 (с, 3Н, ОСН3); MS (CI, m/z): 379,32 (М+HN)+, вычисл. для С21Н34O4 378,586.
Раствор 3,78 г (10 ммоль) вышеуказанного бифункционального соединения с простой и сложной эфирной связью (ether ester) в 20 мл толуола и 10 мл CH2Cl2 перемешивали при охлаждении на бане сухой лед/ацетон, при этом добавляя по капле 12 мл (18 ммоль) 25% гидрида диизобутилалюминия в толуоле (Texas Alkyls) в течение 10 мин. После перемешивания при около -70°С в течение 30 мин реакционную смесь осторожно разлагали (-70°С с помощью 10 мл МеОН. После добавления 50 мл воды и 50 мл 1 н. водного раствора H2SO4 смесь нагревали до комнатной температуры и обрабатывали эфиром обычным способом с получением 3,2 г (90%) неочищенного альдегида ((+)-S-6-(диметил(1,1-диметилэтил)силил)-2,5,7,8-тетраметилхроман-2-карбальдегид) в виде вязкой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 19 об.% EtOAc в гексане. Концентрировали раствор с последующим высушиванием в вакууме в течение 48 ч с получением альдегида TBDS (78%) в виде твердого вещества с точкой плавления 66-68° С. 1H-ЯМР (CDCl3/TMC, м.д.): 0,12 (с, 6Н), 1,1 (с, 9Н), 1,38 (с, 3Н), 1,64 (с, 3Н), 2,12 (с, 3Н), 2,16 (с, 3Н), 2,3-2,2 (м, 2Н), 2,53 (м, 2Н) и 9,82 (д, J=1,4 Гц, 1Н); MS (CI, m/z): 349,40 (М+Н)+, вычисл. для С20Н32SiO3 348,560.
К раствору 2,97 г псевдоионола в 10 мл ацетонитрила добавляли при перемешивании и поддерживании температуры ниже 30°С 4,53 г гидрохлорида трифенилфосфина, который получали пропусканием безводного хлористого водорода через раствор трифенилфосфина в безводном эфире. После оставления смеси на ночь при комнатной температуре удаляли ацетонитрил при пониженном давлении и температуре ниже 50°С. К остатку добавляли 4,80 г ((+)-S-6-(диметил(1,1-диметилэтил)силил)-2,5,7,8-тетраметилхроман-2-карбальдегида) в 15 мл диметилформамида и смесь перемешивали. После получения прозрачного раствора вносили при перемешивании по капле метоксид натрия, полученный из 0,352 г натрия и 7 мл безводного метанола, при температуре ниже 15° С. Реакционная смесь становилась красной при образовании илида. После завершения добавления продолжали перемешивание в течение 30 мин при 10°С; далее смесь постепенно нагревали до 80°С до исчезновения красной окраски. Продукт вносили в 200 мл 50% водного раствора метанола, сушили и концентрировали в вакууме. Оставшуюся маслянистую жидкость растворяли в 20 мл эфира и добавляли раствор хлорида ртути в эфире до прекращения образования осадка. После фильтрования осадка фильтрат промывали водой, сушили и концентрировали с получением 4,7 г желтой маслянистой жидкости. Неочищенную силилэфирную смесь цис- и транс-изомеров алкена растворяли в ТГФ и добавляли фторид тетра-н-бутиламмония (0,031 моль). После перемешивания при 23°С в течение 40 минут смесь вносили в воду и экстрагировали эфиром. Эфирный экстракт сушили, концентрировали и очищали с помощью хроматографии на силикагеле, проводя элюирование EtOAc в гексане (1:9), с получением 2,5,7,8-тетраметил-2R-(2,6,10-триметил-1,3,5,9-Е:Z-декатетраен)-6-хроманола (выход 68%). 1H-ЯМР (CDCl3/TMC, м.д.); 1,28 (с, 3Н, 2аСН3), 1,65 (с, 3Н), 1,70 (с, 6Н), 1,72 (с, 3Н), 1,9 (м, 6Н), 2,18 (с, 3Н), 2,35 (с, 6Н), 2,53 (т, J=6,6 Гц, 2H, 4CH2), 5,13-5,27 (м, 3Н) и 6,44 (м, 2H); MS (CI, m/z): 395,17 (М+Н)+, вычисл. для С27Н38O2 394,60.
Раствор 2,5,7,8-тетраметил-2R-(2, 6, 10-триметил-1, 3, 5, 9-Е:Z-декатетраен)-6-хроманола (0,457 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли с помощью 5 н. HCl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали Н2О (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 19 об.% EtOAc и 2% уксусной кислотой в гексане. Полученную жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали и сушили в вакууме в течение 48 ч. В результате получали соединение 18 с выходом 67%. 1Н-ЯМР (CDCl3/TMC, м.д.): 1,24 (с, 3Н, 2аСН3), 1,63 (с, 3Н), 1,72 (с, 6Н) 1,74 (с, 3Н), 1,92 (м, 6Н), 2,18 (с, 3Н), 2,29 (с, 6Н), 2,43 (т, J=6,6 Гц, 2H, 4СН2), 4,68 (с, 2Н, ОСН2), 5,10-5,27 (м, 3Н) и 6,34 (м, 2H); MS (CI, m/z): 452,24 (М-Н)+, вычисл. для С27Н38O2 452,63.
Хлорид 3-(2,5,7,8-тетраметил-(2R-(4R,8,12-триметилтридецил)хроман-6-илокси)пропил-1-аммония (19)
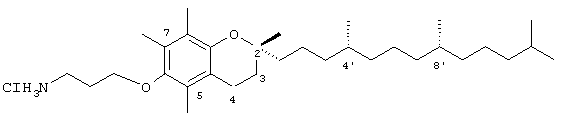
Раствор- 3-бромпропиламингидробромида (1,0 г, 4,6 ммоль) в диоксане/Н2O 2:1 (45 мл) охлаждали до 0°С и обрабатывали К2СО3 (6,22 г, 45 ммоль) и ди-трет-бутилдикарбонатом (1,5 г, 6,9 ммоль). Реакционную смесь перемешивали в течение 15 ч, при этом нагревая до комнатной температуры. Диоксан удаляли в вакууме и оставшуюся водную смесь подкисляли 5 н. HCl и экстрагировали этилацетатом (5×25 мл). Объединенные органические фазы сушили с помощью MgSO4 и получали 3-бром-N-(трет-бутоксикарбонил)пропиламин в виде бесцветной маслянистой жидкости (0,93 г, 93%). 1H-ЯМР (CDCl3/ТМС, м.д.): 1,41 (s, 9Н, СН3), 2,02 (квинтет, J=6,4 Гц, 2Н, СН2), 3,23 (м, 2Н, NCH2), 3,41 (т, J=6,6 Гц, CH2Br), 4,8 (ушир., 1Н, NH); 13С-ЯМР (CDCl3, м.д.): 28,3 (СН3), 30,7, 32,6, 38,9 (СН2), 79,3 (четвертичный С), 155,9 (СО); MS (CI, m/z): 239,241 (М+H)+,вычисл. для C8H16BrNO2 237,03644.
Раствор R,R,R-α-токоферола (0,5 г, 1,16 ммоль) в N,N-диметилформамиде (15 мл) обрабатывали 3-бром-N-(трет-бутоксикарбонил)пропиламином (0,9 г, 3,8 ммоль) и избытком порошкообразного NaOH (0,32 г, 8 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли с помощью 5 н. HCl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали Н2О (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование EtOAc (10 об.%) в гексане. В результате получали желаемый эфир в виде бесцветной маслянистой жидкости (0,45 г, 66%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 33Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 1,99 (квинтет, J=6,2 Гц, 2Н, CH2), 2,07, 2,14, 2,16 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,59 (т, J=6,6 Гц, 2Н, 4-СН2), 3,43 (м, 2Н, NCH2), 3,73 (т, J=5,7 Гц, 2Н, ОСН2), 4,34 (с, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 11,7, 12,0, 12,9 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,7 (2а-СН3), 24,4, 24,8 (СН2), 27,9 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,2, 37,4, 37,5, 39,3, 40,1 (СН2), 70,2 (ОСН2), 74,8 (2-С), 117,5, 122,9, 125,5, 127,5 (арил С), 147,5, 148,0 (арил С-O), 156,0 (CO); MS (CI, m/z): 589 (М+H)+,вычисл. для С37Нб5NO4 587,49136.
Вышеуказанный N-защищенный эфир (0,1 г, 0,17 ммоль) растворяли в 4 н. HCl в диоксане (1 мл, 4 ммоль) и перемешивали в течение 4 ч. Диоксан удаляли пропусканием струи аргона через реакционную смесь. Полученное вещество сушили в вакууме в течение 8 ч с получением 19 в виде белого твердого вещества (82 мг, 99%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 33Н, 4'-, 8'-, 12'-СН, 1'-, 2'-,'3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'- СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 1,99 (квинтет, J=6,2 Гц, 2Н, CH2), 2,07, 2,11, 2,15 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,29 (м, 2Н, СН2), 2,59 (т, J=6,6 Гц, 2Н, 4-CH2), 3,43 (м, 2Н, NCH2), 3,79 (м, 2Н, OCH2); 13С-ЯМР (CDCl3, м.д.): 11,8, 11,9, 12,7 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (CH2), 22,6, 22,7 (СН3), 23,9 (2а-СН3), 24,4, 24,8 (CH2), 28,0 (СН), 28,4 (СН3), 31,2 (3-CH2), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (CH2), 74,8 (OCH2), 75,0 (2-С), 117,5, 122,9, 126,0, 127,3 (арил С), 147,8, 148,0 (арил С-0); HRMS (CI, m/z): 487,438887 (М+H)+ ,вычисл. для С32Н57NO2 487,438935.
2,5,7,8-Тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-3-ен-6-илокси)уксусная кислота (20)
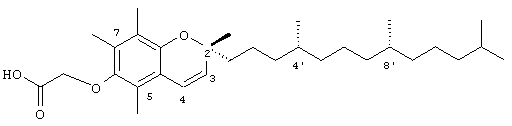
Раствор ацетата R,R,R-α-токоферола (2 г, 4,2 ммоль) в безводном толуоле (150 мл) нагревали с обратным холодильником и затем обрабатывали 2,3-дихлор-5,6-дициано-1,4-бензохиноном (0,96 г, 4,2 ммоль) 4 порциями с интервалом в 1 ч. Реакционную смесь нагревали с обратным холодильником в течение 24 ч. В течение этого времени реакционная смесь становилась темно-красного цвета, и далее образовывался осадок в виде слабоокрашенного твердого вещества. Реакционную смесь охлаждали до комнатной температуры, фильтровали и фильтрат концентрировали. Полученную темноокрашенную маслянистую жидкость очищали с помощью хроматографии на силикагеле, проводя элюирование этилацетатом (10 об.%) в гексане. В результате получали желаемый хроменацетат в виде бесцветной маслянистой жидкости (1,74 г, 88%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 2,07, 2,13, 2,18 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,35 (с, 3Н, СН3СО-), 5,61, 6,52 (2 х д, J=10,0 Гц, 2Н, СН); 13С-ЯМР (CDCl3, м.д.): 11,5, 11,6, 13,1 (5а-, 7а-, 8а-СН3), 14,1 (СН3), 19,6, 19,7 (СН2), 20,4, 21,4 (СН2), 22,6, 22,7 (СН3), 24,4, 24,8 (CH2), 25,8 (2а-СН2), 27,9 (СН), 30,8 (3-СН2), 32,7, 32,8 (СН), 37,2, 37,4, 39,4, 41,0 (СН2), 60,3 (2-С), 117,6, 119,7, 122,3, 122,6, 128,9, 129,6 (арил и винил С), 141,2, 148,4 (арил С-O), 169,4 (СО); HRMS (CI, m/z): 471,375799 (М+HN)+, вычисл. для С31H50O3 470,375996.
Раствор хроменацетата (1,0 г, 2,13 ммоль) в этаноле (20 мл) обрабатывали 2 н. NaOH (20 мл) и перемешивали при 60°С в течение 90 мин. Реакционную смесь охлаждали, подкисляли с помощью 5 н. HCl и удаляли этанол в ваууме. Полученный водный раствор экстрагировали эфиром и концентрировали с получением светло-желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование этилацетатом (15 об.%) в гексане. В результате получали желаемый промежуточный продукт хромен-6-ол в виде бесцветной маслянистой жидкости (0,92 г, 98%). 1H-ЯМР (CDCl3/ТМС, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'- СН2, 2а-СН3), 2,14, 2,18, 2,19 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 5,63, 6,55 (2 х д, J=10,0 Гц, 2Н, СН); 13С-ЯМР (CDCl3, м.д.): 10,8, 11,6, 12,4 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 21,3 (CH2), 22,6, 22,7 (СН3), 24,4, 24,8 (СН2), 25,2 (2а-СН3), 27,9 (СН), 30,9 (3СН2), 32,7, 32,8 (СН), 37,2, 37,4, 37,5, 39,3, 40,5 (СН2), 50,8 (2-С), 116,2, 117,8, 120,1, 122,3, 123,0, 130,0 (арил и винил С), 144,6, 145,3 (арил С-0), 169,4 (СО); HRMS (CI, m/z): 428,365275 (М+Н)+, вычисл. для С29Н48O2 428,365431.
Раствор промежуточного продукта хромен-6-ола (0,9 г, 2,1 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли 5 н. HCl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование EtOAc 19 об.% и 2% уксусной кислотой в гексане. Полученную желтую жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали до светло-желтой маслянистой жидкости и сушили в вакууме в течение 48 ч. В результате получали 19 в виде бесцветного масла (0,90 г, 88%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-CH2, 2а-СН3), 2,07, 2,10, 2,19 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 4,37 (с, 2Н, ОСН3), 5,62, 6,50 (2 х д, J=10,0 Гц, 2Н, СН); 13С-ЯМР (CDCl3, м.д.): 11,3, 11,5, 12,9 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 21,3 (СН2), 22,6, 22,7 (СН3), 24,4, 24,8 (CH2), 25,6 (2а-СН3), 27,9 (СН), 30,9 (3-СН2), 32,7, 32,8 (СН), 37,2, 37,4, 37,5, 39,3, 40,9 (СН2), 60,5 (ОСН2), 69,1 (2-С), 118,0, 1198, 122,8, 122,9, 129,6, 19,8 (арил и винил С), 147,5, 147,8 (арил С-O), 173,4 (СО); HRMS (CI, m/z): 487,378731 (М+Н)+, вычисл. для C31H51O4 487,378736.
Сульфат 2-(2,5,7,8-тетраметил-(2R-(4R,8,12-триметилтридецил)хроман-6-илокси)триэтиламмония (21)
Раствор 2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси))этан-1-ола (13) (0,1 г, 0,21 ммоль) в безводном ДМФ (2 мл) и пиридине (0,6 мл) обрабатывали комплексом серы и триоксид-N,N-диметилформамида (0,16 г, 1,0 ммоль), и полученный раствор перемешивали в течение 24 ч. Реакцию гасили 1 н. HCl и затем экстрагировали с помощью СН2Cl2 (5×5 мл). Газообразный аммиак барботировали через раствор СН2Cl2 в течение 10 мин. Полученный раствор концентрировали до получения желтой пасты и очищали с помощью хроматографии на силикагеле, проводя элюирование МеОН (10 об.%) и триэтиламином (2%) в CHCl3. В результате получали 21 в виде желтого полутвердого вещества (92 мг, 77%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 33Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 1,95, 2,01, 2,05 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,45 (т, J=6,6 Гц, 2Н, 4-СН2), 3,05 (м, 6Н, NCH2), 3,79 (м, 2Н, ОСН2), 4,21 (м, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 9,46 (СН3), 12,4, 12,6, 13,5 (5а, 7а-, 8а-СН3), 20,3, 20,4 (СН3), 21,3, 21,7 (СН2), 23,3, 23,4 (СН3), 24,5 (2а-СН3), 25,1, 25,5 (СН2), 28,6 (СН), 31,9 (3-СН2), 33,3, 33,4 (СН), 37,9, 38,1, 40,0, 40,8 (СН2), 46,9 (NCH2), 67,4, 71,9 (ОСН2), 75,5 (2-С), 118,3, 123,5, 126,5, 128,3 (арил С), 148,5 (арил С-O); HRMS (CI, m/z): 554,364102 (М-NH3), вычисл. для С31Н54O6S 554,364119.
6-(2,5,7,8-Тетраметил-(2R-(4R,8,12-триметилтридецил)хроман)уксусная кислота (22)
Раствор R,R,R-α-токоферола (1,0 г, 2,3 ммоль) в безводном СН2Cl2 (25 мл) охлаждали до 0°С. Добавляли диизопропилэтиламин (2 мл, 11,6 ммоль) с последующим добавлением по капле трифторметилсульфонового ангидрида (5,0 г, 17,7 ммоль). Раствор немедленно приобретал темную окраску, и его оставляли нагреваться до комнатной температуры при перемешивании в течение 24 ч. Реакцию гасили H2O и затем экстрагировали диэтиловым эфиром (2×100 мл). Объединенные эфирные фазы промывали 1 н. HCl (50 мл), Н2О (50 мл), солевым раствором (50 мл) и затем сушили MgSO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, очищали с помощью хроматографии на силикагеле, проводя элюирование этилацетатом (3 об.%) в гексане. В результате получали желаемый промежуточный продукт трифлат в виде желтой маслянистой жидкости (1,3 г, количественный выход). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-,3'-,5'-,6'-,7'-,9'-,10'-,11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,07, 2,13, 2,21 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,62 (т, J=6,6 Гц, 2Н, 4-СН2); 13С-ЯМР (CDCl3, м.д.): 11,9, 13,2, 14,0 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (CH2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН3), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 75,6 (2-С), 118,4, 124,4, 126,7, 128,1 (арил С), 139,6, 150,9 (арил С-O); 19F-ЯМР (CDCl3, м.д.): -73,52; HRMS (CI, m/z): 563,337803 (М+Н)+, вычисл. для С30Н50С4F3S 563,338192.
Раствор трифлата (1,3 г, 2,31 ммоль) в безводном ДМФ (23 мл) обрабатывали с помощью LiCl (0,98 г, 4,62 ммоль), трифенилфосфина (0,37 г, 1,4 ммоль), 2,6-ди-трет-бутил-4-метилфенола (2-3 кристалла), трибутил(винил)олова (0,73 г, 2,31 ммоль) и дихлорбис(трифенилфосфин)палладия (II) (0,24 г, 0,35 ммоль). Данную смесь нагревали до 120°С и перемешивали. Через 2 ч добавляли дополнительно трибутил(винил)олово (0,73 г, 2,31 ммоль). Через 8 ч реакционную смесь охлаждали до комнатной температуры и добавляли смесь Н2O (50 мл) и диэтилового эфира (50 мл). Эфирную фазу промывали 1 н. HCl (6×30 мл) и насыщенным раствором KF (6×30 мл). Эфирный раствор сушили над Na2SO4 и затем концентрировали до темной маслянистой жидкости. Данное вещество очищали с помощью хроматографии на силикагеле, проводя элюирование этилацетатом (3 об.%) в гексане, с получением промежуточного продукта 6-винилхромана в виде прозрачной маслянистой жидкости (0,38 г, 38%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0- 1,6 (м, 24Н, 4'-, 8'-, 12'-CH, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'- СН2, 2а-СН3), 1,86 (м, 2Н, 3-СН2), 2,20, 2,24, 2,28 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,62 (т, J=6,8 Гц, 2Н, 4-СН2), 5,18, 5,56 (2 х дд, Jgem=2,3 Гц, Jcis=11,2 Гц, Jtrans=18,7 Гц, 2Н, =СН2), 6,77 (dd, J=18,7, 11,2 Гц, 1Н, СН); 13С-ЯМР (CDCl3, м.д.): 11,9, 16,3, 17,2 (5а-, 7а-, 8а-СН3),'19,7, 19,8 (СН3), 20,8, 21,1 (СН2), 22,6, 22,7 (СН3), 23,9 (2а-СН3), 24,5, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2),32,7, 32,8 (СН), 37,3, 37,5, 37,5, 39,4, 40,1 (СН2), 74,9 (2-С), 116,7, 119,0, 122,0, 129,8, 131,2, 132,8, 136,8 (арил/винил С), 150,9 (арил С-O); HRMS (CI, m/z): 440,401602 (М+Н)+, вычисл. для С31Н52O 440,401812.
Раствор промежуточного продукта 6-винилхромана (0,12 г, 0,27 ммоль) в безводном ТГФ (1 мл) охлаждали до 0°С и обрабатывали 9-BBN (0,60 мл, 0,5 М в ТГФ, 0,3 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 8 ч. Реакцию гасили водой (1,5 мл), обрабатывали NaBO3·4Н2O и полученную суспензию перемешивали в течение ночи. Диэтиловый эфир (4 мл) и реакционную смесь экстрагировали СН2Cl2 (2×20 мл). Органические фазы концентрировали до прозрачной маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование этилацетатом (50 об.%) в гексане. В результате получали желаемый промежуточный продукт 6-(2-гидроксиэтил)хроман в виде бесцветной маслянистой жидкости (30 мг, 24%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8'-, 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН2), 1,81 (м, 2Н, 3-СН2), 2,17, 2,24, 2,28 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,68 (т, J=6,8 Гц, 2Н, 4-СН2), 3,01 (т, J=7,5 Гц, 2Н, Ar-СН2), 3,74 (т, J=7,5 Гц, 2Н, ОСН2); 13С-ЯМР (CDCl3, м.д.): 12,0, 15,1, 16,0 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (СН2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (СН2), 62,2 (ОСН2), 72,6 (2-С), 116,8, 122,3, 124,9, 132,4, 133,9 (арил С), 150,1 (арил С-O); HRMS (CI, m/z): 458,412154 (M+H+), вычисл. для C31H54O2 458,412384.
Раствор хлорхромата пиридиния (32 мг, 0,1 ммоль) в безводном СН2Cl2 (0,5 мл) обрабатывали с помощью раствора промежуточного продукта 6-(2-гидроксиэтил)хромана (32 мг, 0,07 ммоль) в СН2Cl2 (0,5 мл). Реакционную смесь перемешивали в течение 2 ч, после этого периода исходное вещество не определялось с помощью тонкослойной хроматографии. Добавляли диэтиловый эфир (2 мл) и полученный раствор фильтровали через тонкий пад из целита. Фильтрат концентрировали с получением желтой маслянистой жидкости (20 мг). Данную маслянистую жидкость растворяли в трет-BuOH (0,5 мл) и обрабатывали фосфатным буфером (0,5 мл, 1 н., рН=4,0), 2-метил-2-бутеном (0,1 мл) и NaClO2 (5,4 мг, 0,05 ммоль). После перемешивания в течение 40 мин реакционную смесь экстрагировали CHCl3 (6×10 мл) и объединенные органические фазы сушили Na2SO4. Раствор CHCl3 концентрировали до желтой маслянистой жидкости, которую очищали с помощью препаративной тонкослойной хроматографии, проводя элюирование этилацетатом (30 об.%) и уксусной кислотой (1%) в гексане. В результате получали 22 в виде бесцветной маслянистой жидкости (20 мг, 63%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,87 (м, 12Н, 4а'-, 8а'-, 12а'-, 13'-СН3), 1,0-1,6 (м, 24Н, 4'-, 8', 12'-СН, 1'-, 2'-, 3'-, 5'-, 6'-, 7'-, 9'-, 10'-, 11'-СН2, 2а-СН3), 1,81 (м, 2Н, 3-СН2), 2,17, 2,24, 2,28 (3 х с, 9Н, 5а-, 7а-, 8а-СН3), 2,66 (т, J=6,8 Гц, 2Н, 4-СН2), 3,71 (с, 2Н, СН2СООН); 13С-ЯМР (CDCl3, м.д.): 12,0, 15,3, 16,2 (5а-, 7а-, 8а-СН3), 19,6, 19,7 (СН3), 20,6, 21,0 (CH2), 22,6, 22,7 (СН3), 23,8 (2а-СН3), 24,4, 24,8 (СН2), 28,0 (СН), 28,9, 31,2 (3-СН2), 32,7, 32,8 (СН), 37,3, 37,4, 37,5, 39,4, 40,0 (CH2), 72,6 (2-С), 117,1, 122,2, 124,9, 132,4, 132,7 (арил С), 150,2 (арил С-O), 179,2 (СООН); HRMS (CI, m/z): 472,391583 (M+H+), вычисл. для С31H52O3 472,391644.
2,5,7,8-Тетраметил-(2R-(гептил)хроман-6-илокси)уксусная кислота (23)
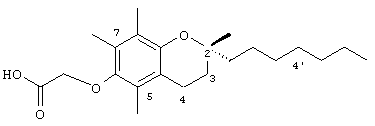
Раствор бромида гексилтрифенилфосфония (0,880 г, 2,05 ммоль) в 11,2 мл безводного DME перемешивали при комнатной температуре, добавляя при этом 0,86 мл (2,06 ммоль) 2,4 М н-бутиллития в гексане. Полученный красный раствор перемешивали в течение 2 ч при комнатной температуре, далее добавляли раствор ((+)S-6-бензилокси-2,5,7,8-тетраметил)хроман-2-карбальдегида (306 мг, 0,944 ммоль) в 3 мл безводного DME и продолжали перемешивать в течение 3 ч при 65-75°С. После охлаждения реакционную смесь вносили в холодную разбавленную H2SO4 и проводили обработку эфира обычным способом. Эфир концентрировали в вакууме с получением маслянистого вещества. Продукт выделяли с помощью колоночной хроматографии и элюирования хлороформом с получением продукта с выходом 46%.
Смесь цис- и транс-изомеров алкена растворяли в 30 мл этилацетата и добавляли 50 мг 5% палладия на углероде, смесь помешивали при 40 фунтах/кв.дюйм Н2 в течение 10 ч и затем фильтровали через целит и хорошо промывали этилацетатом. Фильтрат концентрировали и очищали с помощью хроматографии на силикагеле, проводя элюирование EtOAc в гексане (1:9), с получением (2R)-2,5,1, 8-тетраметил-2-(гептил)-6-хроманола (выход 60%). 1H-ЯМР (CDCl3/TMC, м.д.): 0,89 (с, 3Н), 1,3-1,5 (м, 15Н), 1,89 (м, 2Н), 2,2 (с, 3Н), 2,08 (с, 3Н), 2,23 (с, 3Н) и 2,48 (т, J=6,5 Гц, 2Н); MS (CI, m/z): 305,35 (М+Н+), вычисл. для С20Н32O2 304,4746.
Раствор 2,5,7,8-тетраметил-2-(гептил)хроманола (0,353 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли 5 н. HCl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали Н2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Раствор эфира концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 19 об.% EtOAc и 2% уксусной кислотой в гексане. Полученную жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали и сушили в вакууме в течение 48 ч. В результате получали соединение 23 с выходом 36%. 1H-ЯМР (CDCl3/TMC, м.д.): 0,88 (с, 3Н), 1,2-1,5 (м, 15Н), 1,88 (м, 2Н), 2,1 (с, 3Н), 2,18 (с, 3Н), 2,2 (с, 3Н), 2,55 (т, J=6,5 Гц, 2Н) и 4,78 (с, 2Н); HRMS (CI, m/z): 363,2535 (М+Н+), вычисл. для С22Н35O4 363,2541.
2,5,7,8-Тетраметил-(2R-(тридецил)хроман-6-илокси)уксусная кислота (24)
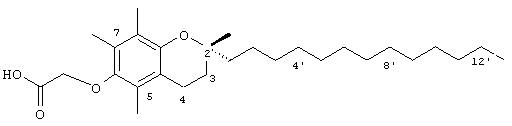
Соединения 24 и 25 синтезировали аналогично синтезу 23, применяя соответствующий бромид фосфония.
1H-ЯМР (CDCl3/TMC, м.д.): 0,83 (с, 3Н), 1,25-1,57 (м, 27Н), 1,88 (м, 2Н), 2,1 (с, 3Н), 2,18 (с, 3Н), 2,20 (с, 3Н), 2,55 (т, J=6,6 Гц, 2Н) и 4,48 (с, 2Н); MS (CI, m/z): 447,14 (М+Н+), вычисл. для С28Н4бO4 446,6732.
2,5,7,8-Тетраметил-(2R-(гептадецил)хроман-6-илокси)уксусная кислота (25)
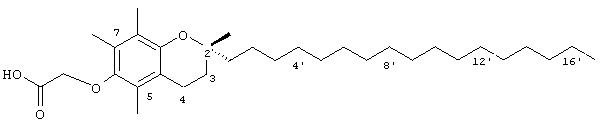
1H-ЯМР (CDCl3/TMC, м.д.): 0,86 (с, 3Н), 1,15-1,67 (м, 35Н), 1,88 (м, 2Н), 2,16 (с, 3Н), 2,20 (с, 3Н), 2,23 (с, 3Н), 2,55 (т, J=6,4 Гц, 2Н) и 4,78 (с, 2Н); MS (CI, m/z): 503,45(М+Н+), вычисл. для С32Н54O4 502,781.
2,5,1,8-Тетраметил-(2R-(4,8-диметил-1,3,7-Е:Z-нонотриен)хроман-6-илокси)уксусная кислота (26)
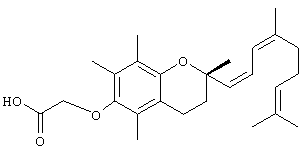
Соединение 26 синтезировали аналогично синтезу соединения 18, применяя нерол взамен псевдоионола. 1H-ЯМР (CDCl3/TMC, м.д.): 1,24 (с, 3Н, 2аСН3), 1,63 (м, 1Н), 1,68 (с, 3Н), 1,74 (с, 6Н), 1,92 (м, 6Н), 2,18 (с, 3Н), 2,29 (с, 6Н), 2,43 (т, J=6,6 Гц, 2Н, 4СН2), 4,68 (с, 2Н, ОСН2), 5,64 (м, 2Н) и 5,27 (м, 1Н); MS (CI, m/z): 413,24 (М+Н+), вычисл. для С26Н36O4 412,0115.
EZ,RS,RS,RS-(фитилтриметилбензолтиол-6-илокси)уксусная кислота (27)
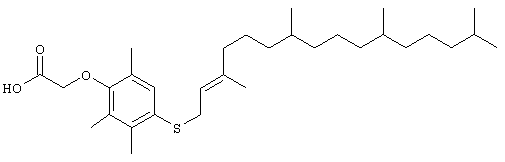
2,3,6-Триметилфенол (1,6 г, 11,8 ммоль) растворяли в 50 мл безводного метанола, который дезоксигенировали барботированием азота. Добавляли тиоцианат аммония (2,2 г, 28,9 ммоль) к данному раствору, который далее охлаждали до 0°С и пропускали газообразный хлор. Первоначально бесцветный гомогенный раствор становился розовым и затем зеленым с образованием белого осадка. Раствор перемешивали в течение 1 ч при 0°С и далее в течение еще одного часа при 20°С. Растворенный хлор удаляли пропусканием азота и осадок удаляли с помощью фильтрации. Проводили упаривание фильтрата при пониженном давлении с последующим высушиванием в глубоком вакууме (0,1 торр) с получением 2,20 г (97%) 2,3,5-триметил-4-гидроксифенилтиоцианата в достаточно чистом виде для следующей стадии синтеза. Аналитический образец перекристаллизовывали из гексанов: белые кристаллы, точка плавления 100,3°С. 1H ЯМР (CDCl3) δ: 7,2 (с, 1Н), 5,0 (с, 1Н) 2,4 (с, 3Н), 2,2 (с, 6Н).
2,3,5-Триметил-4-гидроксифенилтиоцианат (2 г, 10,35 ммоль) растворяли в 100 мл безводного эфира, содержащего 25 мл безводного тетрагидрофурана. Данный раствор добавляли по капле в течение 1 ч к 100 мл безводного эфира, содержащего LiAlH4 (0,9 г, 24 ммоль) при комнатной температуре. После дополнительного часа при 20°С непрореагировавший LiAlH4 разрушали охлаждением гетерогенной смеси до 0°С и добавлением влажного эфира (50 мл), Н2O (50 мл), и 1 н. HCl (50 мл). Добавляли дополнительно 50 мл воды и отделяли органическую фазу и промывали водой (2×50 мл), раствором NaHCO3 (2×50 мл), водой (2×50 мл) и насыщенным NaCl (50 мл). Органическую фазу сушили над безводным MgSO4 и фильтровали, растворитель удаляли при пониженном давлении. Проводили колоночную хроматографию на силикагеле с 5% этилацетатом в гексане с получением 1,8 г (90%) 2,3,5-триметил-4-гидроксибензолтиола в виде белого порошка, точка плавления 86°С (Lit.1 точка плавления 86°С).
Раствор 2,3,5-триметил-4-гидроксибензолтиола (3 г, 17,83 ммоль), изофитола (4,8 г, 16,19 ммоль), безводного хлорида цинка (1,2 г, 8,8 ммоль) и 0,2 мл ледяной уксусной кислоты в 30 мл абсолютного эфира нагревали с обратным холодильником в течение 1 ч. Далее растворитель удаляли в вакууме при 50°С и полученную красную маслянистую жидкость растворяли в смеси 50 мл петролейного эфира и 20 мл 70% водного метанола. Эфирную фазу сушили (Na2SO4) и упаривали в вакууме с получением красной маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование гексан:эфир (9:1) с получением 3 г (38%) E.Z,RS,RS,RS-фитилтриметилгидроксибензолтиола в виде желтой маслянистой жидкости. 1H-ЯМР (CDCl3) δ: 7,11 (с, 1 Н, Ar-H), 5,23 (т, 1 Н, винильный Н), 4,62 (с, 1Н, ОН), 3,34 (д, 2 Н, Ar-S-CH2-), 2,41 (с, 3 Н, Ar-СН3), 2,19 (с, 3 Н, Ar-СН3), 2,18 (с, 3 Н, Ar-СН3), 0,83-1,92 (м, 39 Н, фитоловая цепь).
Раствор фитилтриметилгидроксибензолтиола (3 г, 6,7 ммоль) в N,N-диметилформамиде (80 мл) обрабатывали метилбромацетатом (7,4 г, 48,3 ммоль) и избытком порошкообразного NaOH (7 г, 175 ммоль). Полученную розовую маслянистую жидкость перемешивали при комнатной температуре в течение 24 ч. Реакционную смесь подкисляли 5 н. HCl и экстрагировали эфиром (3×150 мл). Объединенные эфирные фазы промывали H2O (3×150 мл) и солевым раствором (1×150 мл), затем сушили (Na2SO4). Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 20 об.% EtOAc в гексане с получением 3 г (88%) E.Z,RS,RS,RS-(фитилтриметилбензолтиол-6-илокси)уксусной кислоты в виде желтой маслянистой жидкости. 1H-ЯМР (CDCl3) δ: 10,90 (с, 1 Н, СООН), 8,08 (с, 1 Н, Ar-Н), 5,30 (т, 1Н, винильный-Н), 4,35 (с, 2 Н, СН2СООН), 3,42 (д, 2 Н, Ar-S-CH2-), 2,34 (с, 3 Н, Ar-СН3), 2,25 (с, 3 Н, Ar-СН3), 2,22 (с, 3 Н, Ar-СН3), 0,83-1,94 (м, 39 Н, фитильная цепь). HRMS (CI, m/z): 504,362821 (М+Н+),вычисл. для С31Н53О3S 504,363718.
(R)-2((2,5,7,8-Тетраметил-2-(3-пропенметиловый эфир)хроман-6-илокси)уксусная кислота (28)
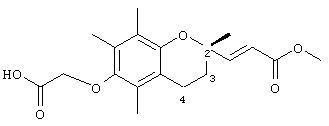
К суспензии бромида (карбометоксиметил)трифенилфосфония (1,8 г, 4,32 ммоль) в 12 мл ТГФ при °С добавляли по капле 1,66 мл н-BuLi (2,5 М в гексане). Полученный раствор переводили до состояния комнатной температуры в течение 2 ч и затем добавляли через канюлю раствор (+)S-6-(диметил-(1,1-диметилэтил)силил)-2,5,7,8-тетраметилхроман-2-карбальдегида (1,31 г, 3,76 ммоль) в 7 мл ТГФ. Раствор перемешивали при комнатной температуре в течение 44 ч и затем добавляли 10 мл 1 н. водн. HCl. Фазы разделяли и далее водную фазу экстрагировали эфиром (3×15 мл). Объединенную органическую фазу промывали солевым раствором, сушили над Na2SO4 и фильтровали. После концентрирования фильтрата неочищенный алкен очищали с помощью тонкослойной хроматографии, проводя элюирование дихлорметаном, с получением смеси цис- и транс-изомеров алкена с выходом 93%. Силилэфирную смесь цис- и транс-изомеров алкена (3,76 ммоль) растворяли в ТГФ и добавляли фторид тетра-н-бутиламмония (0,041 моль). После перемешивания при 23°С в течение 1,5 ч смесь вносили в воду и экстрагировали эфиром. Эфирный экстракт сушили, концентрировали и очищали с помощью хроматографии на силикагеле, проводя элюирование EtOAc в гексане (3:7), и оба цис- и транс-изомера 2,5,7,8-тетраметил-2R-(3'-пропенметиловый эфир)-6-хроманола выделяли и характеризовали (выход 68%).1H-ЯМР (CDCl3/TMC, м.д.): 1,65 (с, 3Н, 2а СН3), 2,12 (м, 2Н, 3СН2), 2,39 (с, 9Н, СН3), 2,48 (м, 2Н, 4 СН2), 3,78 (с, 3Н, ОСН3), 6,11 (д, 1Н, СН=) и 7,13 (д, 1Н, СН=).
Раствор 2,5,7,8-тетраметил-2R-(3'-пропенметиловый эфир)-6-хроманола (0,353 г, 1,16 ммоль) в N,N-диметилформамиде (20 мл) обрабатывали метилбромацетатом (3,4 г, 8,3 ммоль) и избытком порошкообразного NaOH (1,2 г, 30 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли 5 н. HCl и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали H2O (3×30 мл) и солевым раствором (1×30 мл) и затем сушили над Na2SO4. Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование 19 об.% EtOAc и 2% уксусной кислотой в гексане. Полученную жидкость растворяли в диэтиловом эфире (30 мл), промывали Н2O (3×20 мл) и солевым раствором (1×20 мл) и затем сушили над Na2SO4. Полученный раствор концентрировали и сушили в вакууме в течение 48 ч. В результате получали соединение 28 с выходом 40%. 1H-ЯМР (CDCl3/TMC, м.д.): 1,68 (с, 3Н, 2а СН3), 2,11 (м, 2Н, 3СН3), 2,36 (с, 9Н, СН3), 2,56 (м, 2Н, 4 СН2), 3,70 (с, 3Н, ОСН3), 4,78 (с, 2Н, ОСН2), 6,03 (д, 1Н, СН=) и 7,03 (д, 1Н, СН=); MS (CI, m/z): 337,24 (М+Н+), вычисл. для C18H24O6 336,3867.
2,5,7,8-Тетраметил-(2R-(пропионат)хроман-6-илокси)уксусная кислота (29)
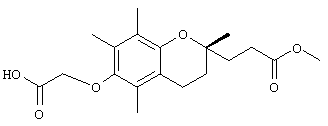
Смесь алкеновых цис- и транс-изомеров 2,5,7,8-тетраметил-2R-(3-пропенметиловый эфир)-6-хроманола растворяли в 30 мл этилацетата и добавляли 50 мг 5% палладия на углероде, смесь помешивали при 40 фунтах/кв.дюйм Н2 в течение 24 ч и затем фильтровали через целит и хорошо промывали этилацетатом. Фильтрат концентрировали и очищали с помощью хроматографии на силикагеле, проводя элюирование EtOAc в гексане (1:9), с получением соединения 29. 1H-ЯМР (CDCl3/TMC, м.д.): 1,62 (с, 3Н, 2а СН3), 2,0-2,3 (м, 6Н, СН2), 2,41 (с, 9Н, СН3), 2,53 (м/ 2Н, 4 СН2), 3,67 (с, 3Н, ОСН3) и 4,88 (с, 2Н, OCH2); MS (CI, m/z): 339,34 (М+Н+), вычисл. для С18Н26О6 338,4025.
ПРИМЕР 3
Синтез всех рацемических аналогов 1-аза-α-токоферола
Фитилтриметилгидрохинон (31)
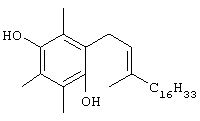
Раствор триметилгидрохинона (соединение 30, 7,5 г, 49,28 ммоль), изофитола (12 г, 40,47 ммоль), безводного хлорида цинка (3 г, 22,01 ммоль) и 0,4 мл ледяной уксусной кислоты в 60 мл абсолютного эфира нагревали с обратным холодильником в течение 1 ч. Далее растворитель удаляли в вакууме при 50°С и полученную суспензию растворяли в смеси 50 мл петролейного эфира и 20 мл 70% водного метанола. Образовавшуюся эмульсию разрушали добавлением 20 мл эфира. Эфирную фазу сушили (Na2SO4) и упаривали в вакууме с получением красной твердой пасты, которую энергично растирали с 70 мл петролейного эфира. После охлаждения до -78°С суспензию центрифугировали и сцеживали супернатантный петролейный эфир. К кристаллической массе добавляли холодный петролейный эфир (10 мл) и процедуру повторяли дважды с получением 10 г (47%) липкого белого вещества, которое применяли на следующей стадии без дополнительной очистки.
Фитилтриметилгидрохинондиацетат (32)
Неочищенный фитилтриметилгидрохинон (31, 9 г, 20,1 ммоль) растворяли в безводном пиридине (90 г) и уксусном ангидриде (90 г). После выдерживания при комнатной температуре в течение 10 ч смесь помещали на лед (200 мл) и экстрагировали эфиром (2×100 мл). Эфирный раствор промывали 3 н. серной кислотой (2×100 мл), 10% раствором бикарбоната натрия (2×100 мл) и опять водой (2×100 мл), сушили (Na2SO4) и упаривали с получением 10,2 г (95%) диацетата (32) в виде светло-желтой маслянистой жидкости. Маслянистую жидкость применяли на следующей стадии без дополнительной очистки.
Триметил-(3,7,11,15-тетраметил-3-бензамидогексадецил)гидрохинондиацетат (33)
К смеси бензонитрила (10 г, 96,97 ммоль), концентрированной серной кислоты (10 г) и ледяной уксусной кислоты (40 мл) добавляли при перемешивании диацетат (32, 10 г, 19,43 ммоль) при 0°С. После выдерживания при комнатной температуре в течение 10 ч раствор помещали на лед (150 мл) и экстрагировали эфиром (3×100 мл). Эфирные экстракты промывали до нейтрального состояния водой (3×100 мл) и насыщенным раствором бикарбоната натрия, сушили (Na2SO4) и упаривали в вакууме. Неочищенный продукт (21 г) подвергали хроматографии через силикагель, проводя элюирование петролейным эфиром:эфиром (9:1). Первым удалялся непрореагировавший бензонитрил. Далее элюция эфиром приводила к получению 11 г (88%) продукта, который перекристаллизовывали из петролейного эфира для получения аналитического образца (33), точка плавления 90-92°С (lit. 1 94-95°C).
2,5,7,8-Тетраметил-2-(4,8,12-триметилтридецил)-3,4-дигидрохинолин-6-(2Н)-он (37)
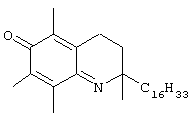
К раствору диацетата (33, 13 г, 20,4 ммоль) в 60 мл метанола добавляли твердый гидроксид калия (11 г) и смесь нагревали с обратным холодильником в атмосфере аргона в течение 45 мин. После охлаждения раствор разбавляли водой со льдом (100 мл) и экстрагировали в атмосфере диоксида углерода с помощью эфира (3×100 мл) с получением 10 г (88%) неочищенного гидрохинона (34).
Раствор данного продукта (34, 10 г, 18,1 ммоль) в абсолютном эфире (110 мл) добавляли по капле при перемешивании в течение 20 мин к суспензии алюмогидрида лития (20 г, 527 ммоль) в абсолютном эфире (110 мл). Смесь нагревали с обратным холодильником в течение 14 ч и затем гидролизовали в атмосфере диоксида углерода с помощью добавления по каплям метанола (10 мл) с последующим добавлением 3 н. соляной кислоты (30 мл). После экстрагирования эфиром (3×200 мл) получали неочищенное твердое бензиламинопроизводное (35, 8,4 г, 86%).
Данное вещество (35, 8,4 г, 15,6 ммоль) растворяли в ледяной уксусной кислоте (120 мл) и гидрировали при 50°С и атмосферном давлении в присутствии 2 г палладия на углероде (5%) при комнатной температуре в течение 22 ч (или до прекращения поглощения водорода). После охлаждения катализатор отфильтровывали и раствор разбавляли водой (50 мл), экстрагировали эфиром (3×80 мл) и сушили (Na2SO4).
К данному безводному и нейтральному эфирному раствору соединения 36 добавляли только что приготовленный оксид серебра (2,1 г, 8,93 ммоль) и суспензию перемешивали в течение 14 ч. После фильтрации в атмосфере аргона и упаривания получали 4 г неочищенного иминохинона (37), который подвергали хроматографии на силикагеле, проводя элюирование гексан:эфир (200:1) с получением 2,4 г (63%) чистой желтой маслянистой жидкости. HMRS (CI, m/z):428,389883 (М+Н+), вычисл. для C29H49NO 428,389241).
1-Аза-α-токоферол (38)

Иминохинон (37, 1,34 г, 3,133 ммоль) в 10 мл эфира гидрировали при комнатной температуре и атмосферном давлении в присутствии 190 мг катализатора Lindlar. Через 20 ч катализатор отфильтровывали в атмосфере аргона и фильтрат упаривали в вакууме с получением 810 мг (60%) неочищенной красной маслянистой жидкости, которую применяли на следующей стадии без дополнительной очистки.
1-Аза-α-токоферол-6-илоксилуксусная кислота (39) и 1-аза-α-токоферол-6-илоксилметилацетат (40)
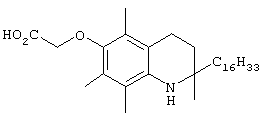
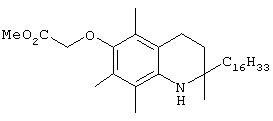
Раствор 1-аза-α-токоферола (38, 800 мг, 1,86 ммоль) в ДМФ (30 мл) обрабатывали метилбромацетатом (2,1 г, 13,4 ммоль) и избытком порошкообразного NaOH (1,9 г, 48,4 ммоль). Полученную желтую суспензию энергично перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь подкисляли 5 н. HCl до рН 6 и экстрагировали диэтиловым эфиром (3×30 мл). Объединенные эфирные фазы промывали H2O (3×40 мл), солевым раствором (1×40 мл) и затем сушили (Na2SO4). Эфирный раствор концентрировали до желтой маслянистой жидкости, которую очищали с помощью хроматографии на силикагеле, проводя элюирование CH2Cl2:MeOH:Hac (97:2:1) с получением 300 мг (33%) липкого желтого твердого вещества (39). (1H-ЯМР (CDCl3, м.д.): 4,30 (с, 2Н, ОСН2), 3,30 (с, 1Н, NH), 2,59 (т, 2Н, Ar-СН2), 2,18 (с, 3Н, Ar-СН3), 2,12 (с, 3Н, Ar-СН3), 2,00 (с, 3Н, Ar-СН3), 1,80-0,84 (м, 38Н, Ar-CH2CH2, NCCH3 и фитильная цепь); 13С-ЯМР (CDCl3, м.д.): 12,01, 12,77, 12,96, 19,60, 19,67, 19,72, 21,09, 22,60, 22,71, 24,42, 24,79, 26,56, 27,95, 32,11, 32,74, 37,26, 37,37, 37,42, 37,55, 37,65 39,34, 41,36, 50,92, 69,50 (алифатический С), 117,69, 118,55, 125,99, 126,39, 138,07, 146,05 (Ar-C), 173,18 (СООН); HRMS (CI, m/z): 488,411055 (М+Н+) вычисл. для С31Н51NO3 488,410370) и 150 мг (17%) желтой маслянистой жидкости (40); 1H-ЯМР (CDCl3/TMC, м.д.) 4,31 (с, 2Н, ОСН2), 3,85 (с, 3Н, CO2СН3), 3,32 (с, 1Н, NH), 2,61 (т, 2Н, Ar-СН2), 2,23 (с, 3Н, Ar-СН3), 2,17 (с, 3Н, Ar-СН3), 2,01 (с, 3Н, Ar-СН3), 1,81-0,86 (м, 38Н, Ar-CH2CH2, NCCH3 и фитильная цепь); 13С-ЯМР (CDCl3, м.д.): 11,96, 12,67, 12,90, 19,57, 19,63, 19,69, 21,06, 21,95, 22,57, 22,68, 24,38, 24,74, 26,49, 27,91, 32,25, 32,67, 32,70, 37,21, 37,33, 37,53, 39,31, 41,68, 50,43, 51,87, 69,97 (алифатический С), 116,98, 117,72, 126,01, 126,36, 138,43,.146,18 (арил С), 169,83 (C=O); HRMS (CI, m/z): 502,425330 (М+Н+), вычисл. для С32Н55NO3 502,426020.
1-Аза-N-метил-α-токоферол-6-илоксилметилацетат (41)
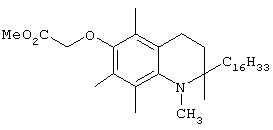
Согласно описанной в литературе схеме 2 смесь порошкообразного КОН (500 мг, 7,77 ммоль) в ДМСО (30 мл) перемешивали при 0°С в течение 5 мин. Добавляли метилацетат (40, 1,3 г, 23,32 ммоль) с последующим немедленным добавлением СН3I (3,3 г, 23,32 ммоль). Перемешивание продолжали в течение 0,5 ч (0°С), после чего смесь вносили в воду. Экстракты (СН2Cl2, 3×40 мл) конечной смеси объединяли, промывали водой (40 мл), солевым раствором (40 мл), сушили (MgSO4) и затем упаривали. Полученную желтую маслянистую жидкость подвергали хроматографии на силикагеле, проводя элюирование 5% EtOAc в гексане с получением 810 мг (60%) светло-желтой маслянистой жидкости. 1H-ЯМР (CDCl3/TMC, м.д.): 4,34 (с, 2Н, ОСН3), 3,84 (с, 3Н, СО2СН3), 2,55 (т, 2Н, Ar-СН2), 2,49 (с, 3Н, NCH3), 2,24 (с, 3Н, Ar-СН3), 2,20 (с, 3Н, Ar-СН3), 2,13 (с, 3Н, Ar-СН3), 1,82-0,82 (м, 38Н, Ar-СН2СН2, NCCH3 и фитильная цепь); 13С-ЯМР (CDCl3, м.д.): 11,72, 13,12, 14,27, 19,55, 19,68, 20,93, 22,57, 22,66, 22,88, 24,35, 24,74, 25,33, 25,37, 27,90, 32,59, 32,71, 37,23, 37,33, 37,51, 37,63, 38,49, 39,06, 39,29, 51,14, 69,57 (алифатический С), 125,34, 126,36, 127,42, 130,32, 143,89, 150,33 (Ar-C), 169,73 (С=O). Соединение 41 использовали на следующей стадии без допонительной очистки.
1-Аза-N-метил-α-токоферол-6-илоксиуксусная кислота (42)
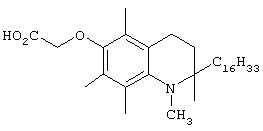
К смеси сложного эфира (41, 740 мг, 14,34 ммоль) в этаноле (34 мл) добавляли по капле 2 н. NaOH (9 мл, 10 экв., 14,34 ммоль). После перемешивания светло-желтого раствора при комнатной температуре в течение 4 ч реакционную смесь подкисляли 2 н. HCl до рН 6, затем экстрагировали EtOAc (3×100 мл) и объединенные органические фазы промывали водой (2×100 мл), солевым раствором (1×100 мл), сушили (MgSO4) и упаривали с получением светло-желтой маслянистой жидкости. Маслянистую жидкость подвергали хроматографии через силикагель, проводя элюирование 1% НОАс, 30% EtOAc в гексане с получением 650 мг (90%) продукта (42) в виде бесцветной маслянистой жидкости. 1H-ЯМР (CDCl3/TMC, м.д.): 10,43 (с, 1Н, CO2Н), 4,37 (с, 1Н, ОСН2), 2,51 (т, 2Н, Ar-СН2), 2,43 (с, 3Н, NCH3), 2,22 (Ar-СН3), 2,12 (с, 3Н, Ar-СН3), 1,81-0,80 (м, 38Н, Ar-СН2СН2, NCCH3 и фитильная цепь); 13С-ЯМР (CDCl3, м.д.): 11,77, 13,18, 14,28, 19,68, 19,73, 20,80, 20,96, 22,62, 22,72, 23,00, 24,00, 24,41, 24,80, 25,37, 27,80, 32,77, 37,28, 37,30, 37,74, 38,00, 39,00, 39,35, 54,69, 69,17 (алифатический С), 125,11, 126,55, 127,20, 130,63, 142,00, 149,60 (Ar-C), 173,62 (COOH); HRMS (CI, m/z): 502,426327 (М+Н)+, вычисл. для С32Н55NO3 502,426020.
6-(2,4-Динитрофенил)азо-(2,5,7,8-тетраметил)-2-(4,8,12-триметилтридецил)-1,2,3,4-тетрагидрохинолин (43)
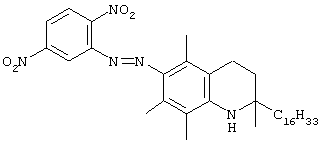
2,4-Динитрофенилгидразин (1,95 г, 9,82 ммоль) растворяли в 34 мл горячего абсолютного этанола и осторожно добавляли 3,4 мл концентрированной серной кислоты. Горячий раствор красного цвета охлаждали до комнатной температуры и добавляли иминохинон (37, 1,00 г, 2,34 ммоль) в 12 мл абсолютного этанола при перемешивании. Смесь оставляли при комнатной температуре на 70 ч, далее разбавляли 120 мл воды и экстрагировали эфиром (3×100 мл). Объединенные эфирные экстракты промывали до нейтрального состояния водой (6×100 мл), сушили (NaSO4) и упаривали в вакууме с получением 2,9 г частично кристаллической темно-фиолетовой массы. Данный продукт подвергали хроматографии через силикагель, проводя элюирование 5% эфиром в петролейном эфире, с получением 1,35 г (95%) фиолетовых кристаллов (43), точка плавления 72-73°С (lit.1 73-75). HRMS (CI, m/z): 608,417199 (M+Н)+, вычисл. для C35H53N5O4 608,417581.
ПРИМЕР 4
Условия клеточного культивирования
Все тестируемые клеточные линии культивировали при 37°С в 5% СО2 в стандартной среде с добавлением эмбриональной сыворотки теленка, применяя установленные стандартные условия. Прилипающие к пластику клетки разделяли трипсином, промывали, подсчитывали и непосредственно применяли в экспериментах. Все клетки проверяли обычным образом на заражение микоплазмами.
ПРИМЕР 5
Растворимость и растворение новых соединений токоферола и токотриенола
Все соединения обрабатывали так, как будто они обладали светочувствительностью (фоторазлагаемые). Все соединения первоначально растворяли в абсолютном этаноле и последовательно разбавляли до конечной концентрации этанола 0,5% соответствующей средой.
ПРИМЕР 6
Определение эффективной концентрации (EC50), индуцирующей апоптоз
Поскольку парентеральные не структурно-модифицированные формы токоферолов не проявляют эффективных апоптотических свойств в отношении ряда малигнизированных клеток, пятнадцать из двадцати девяти соединений RRR-α-токоферола и два из пяти аналогов 1-аза-α-токоферола, структурно модифицированных с помощью присоединенных эфирной связью последовательностей различного состава и размера, являются чрезвычайно эффективными в отношении индуцирования опухолевых клеток к апоптозу, при этом не обладая индуцирующим апоптотическим действием на нормальные клетки. Соединения 1, 2, 3, 7, 8, 9, 12, 15, 17, 19, 20, 21, 22, 25, 26, 27, 39 и 42 проявляют эффективные в отношении ингибирования роста (индукция апоптоза) свойства, специфические для малигнизированных клеток человека большего ряда клеточного происхождения, включающего (i) молочной железы (эстроген-чувствительная клеточная линия злокачественной опухоли молочной железы человека Michigan Cancer Foundation номер 7, MCF-7 McGuire; эстроген-нечувствительная клеточная линия метастазирующей злокачественной опухоли молочной железы человека М. D. Anderson, MDA-MB-435; эстроген-нечувствительная клеточная линия метастазирующей злокачественной опухоли молочной железы человека М. D. Anderson, MDA-MB-231); (ii) предстательной железы (андроген-чувствительная клеточная линия злокачественной опухоли предстательной железы человека LnCaP и андроген-нечувствительная клеточная линия злокачественной опухоли предстательной железы человека РС-3 и DU-145); (iii) промиелоцитные лейкозные клетки (промиелоцитная лейкозная клеточная линия человека HL-60), лимфоидные клеточные линии Jurkat и HL-60; (iv) цервикального происхождения (клеточная линия злокачественной опухоли шейки матки человека ME-180); (v) яичника (клеточная линия злокачественной опухоли яичника человека, клетки С-170); (vi) эндометриального происхождения (клеточная линия злокачественной опухоли эндометрия человека, клетки RL-95-2); (vii) линии клеток толстого кишечника DLD-1 и (viii) линия клеток легочного происхождения А-549. Нормальные исходные клетки молочной железы (нормальные исходные после пересева эпителиальные клетки молочной железы человека, НМЕС) и иммортализованные не опухолевогенные клетки молочной железы (иммортализованные, но не опухолевогенные клетки молочной железы человека Michigan Cancer Foundation номер 10А, MCF-10A) не подвергаются апоптозу при культивировании вместе с вышеуказанными фармакодинамически созданными формами токоферола.
Эффективная терапевтическая доза новых веществ для контроля роста злокачественной опухоли рассматривается как концентрация, ингибирующая рост (IC50) или эффективная концентрация (EC50), которая блокирует 50% роста злокачественной опухоли посредством ингибирования синтеза ДНК, блокирования клеточного цикла и/или гибели клетки. Апоптотическая ЕС50 для ряда тестируемых малигнизированных клеток для двадцати девяти новых соединений RRR-α-токоферола и двух из пяти аналогов 1-аза-α-токоферола согласно данному изобретению представлена в таблицах 1 и 2.
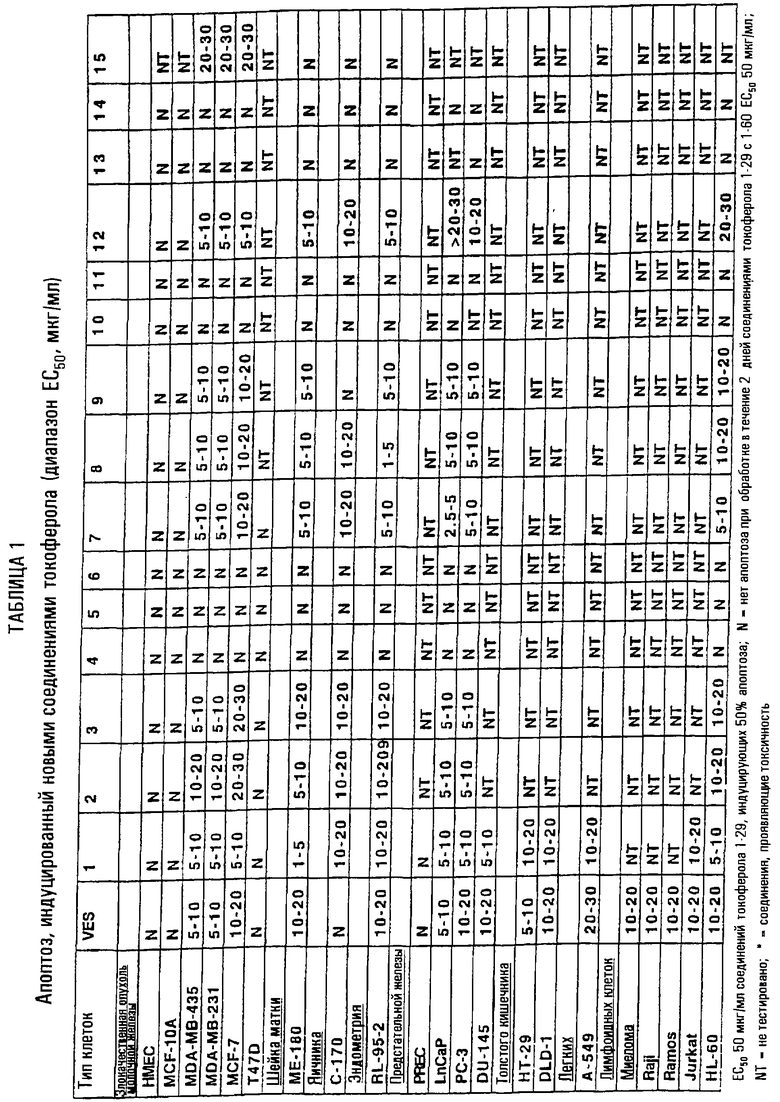
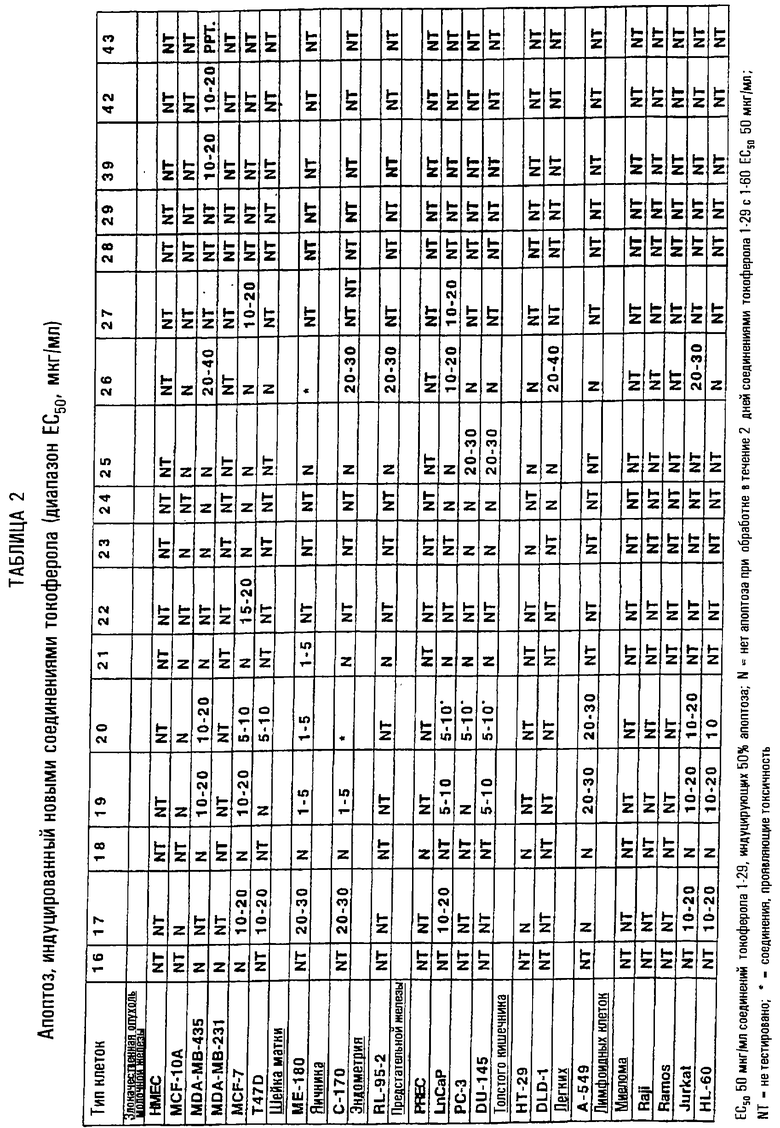
Пример 7
Биоанализ апоптоза
Клетки культивировали в 12-луночной плашке при 1,5×105 клеток/лунку. Клетки оставляли на ночь для прилипания, далее инкубировали с новыми тестируемыми соединениями при 0,01, 0,1, 1, 5, 10 и 20 мкг/мл в течение 1, 2 и 3 дней. После обработки клетки (плавающие+высвобожденные трипсином прилипающие клетки) осаждали центрифугированием, промывали и инкубировали с 2 мкг/мл DAPI (дигидрохлоридом 4',6-диамидин-2'-фенилиндола) в 100% метанола на 15 минут при 37°С и/или инкубировали TUNEL, далее визуализировали с помощью микроскопа Zeiss ICM 405. Клетки, ядро которых содержало явно конденсированный или фрагментированный хроматин, считали за апоптотические. Данные представляли в процентном содержании клеток, прошедших апоптоз.
ПРИМЕР 8
Биоанализ прекращения синтеза ДНК
Для анализа синтеза ДНК все клетки применяли в концентрации 2,5×105/мл. Клетки обрабатывали каждым из соединений 1-29 (таблицы 1 и 2) в концентрациях 0,01, 0,1, 1, 5, 10 и 20 мкг/мл и 200 мкл каждой обработанной группы вносили в четырех экземплярах в 96-луночную культуральную плашку (Corning, Corning NY). Эксперименты проводили в дублях. Одну плашку использовали для оценки жизнеспособности и другую плашку для оценки захвата 3H-TdR для контроля синтеза ДНК. Плашки культивировали в течение 48 часов при 37°С, 5% CO2. За восемь часов до окончания инкубации добавляли 3H-TdR к одной из дублирующих плашек и инкубацию продолжали в течение 8 часов. Клетки потом собирали (для сбора прилипающих клеток требовалась обработка трипсином) и захват изотопа определяли как число импульсов в минуту (cpm). Для изучения жизнеспособности по окончанию инкубации клетки удаляли из лунок и жизнеспособность контролировали по способу включения трипанового синего. Рассчитывали процент жизнеспособности и процент синтеза ДНК по сравнению с необработанными или обработанными носителем клетками каждой обрабатываемой группы.
ПРИМЕР 9
Биоанализ остановки клеточного цикла
Клетки культивировали вместе с новыми тестируемыми соединениями в течение 2-3 дней, фиксировали в 95% этаноле и окрашивали иодистым пропидием в течение ночи. Содержание ДНК определяли с помощью Coulter Epics Elite Flow Cytometer с аргоновым лазером, установленным на 488 нм. Одновременно определяли размер клеток и данные анализировали как процентное содержание клеток в каждой фазе клеточного цикла с помощью Coulter Multicycle Program.
ПРИМЕР 10
Биоанализ клеточной дифференцировки
Для определения индуцирования новыми соединениями дифференцировки клеток клетки культивировали на покровных стеклах, фиксировали в 95% этаноле и инкубировали с краской, специфичной по отношению к липидам, для определения молочных липидов. Дополнительно клетки оценивали иммуногистохимически и с помощью Вестерн-анализа на наличие казеина - белка молока, применяя поликлональные антитела, полученные в лаборатории.
ПРИМЕР 11
Результаты прекращения синтеза ДНК
Клетки культивировали в течение 48 часов, метили в течение 8 часов с помощью меченного тритием тимидина, собирали и подсчитывали. Данные представляли в виде числа импульсов в минуту. Установление прекращения синтеза ДНК определяли по снижению захвата меченного тритием тимидина клетками, обработанными тестируемыми соединениями. Дополнительное подтверждение прекращения синтеза ДНК получали с помощью окрашивания иодистым пропидием и стандартного анализа клеточного цикла.
ПРИМЕР 12
Механизмы индукции апоптоза
Механизмы индукции апоптоза данными соединениями включает три различных пути передачи сигнала к апоптозу, а именно активацию латентного трансформирующего фактора роста-бета (TGF-β), активацию пути передачи сигнала лиганда Fas/Fas и путь передачи сигнала с помощью стресс-киназы (c-Jun, N-концевая киназа).
TGF-β представляют собой эффективные молекулы, ингибирующие рост, которые известны в отношении ингибирования клеточного роста с помощью ингибирования синтеза ДНК и с помощью индукции апоптоза. TGF-β участвуют только в индукции апоптотического пути, т.е. в настоящее время нет подтверждения, что TGF-β осуществляют прекращение синтеза ДНК; однако данная возможность полностью не исключена. TGF-β синтезируются и секретируются клетками в латентной неактивной форме. Для преобразования в эффективную форму ингибиторов опухолевого роста латентные TGF-β должны быть активированы с помощью индукции белков клеточной поверхности, которая обеспечивает характерную структуру для процессинга и активации протеаз, отщепляющих латентный белок и высвобождающих активный TGF-β.
Соединения согласно настоящему изобретению способны активировать протеазы, такие как протеазы семейства катепсина D, и положительно регулировать манноза-6-фосфатный рецептор, который связывает неактивный TGF-β и позволяет активировать посредством протеаз. Активный TGF-β передает сигнал через рецепторы I и II TGF-β на клеточной мембране для активирования нижележащих киназ, относящихся к стресс-киназам или N-концевым киназам c-Jun (JNK), которые фосфорилируют и активируют факторы транскрипции c-Jun, ATF-2 и Elk-1. Длительная активация фактора транскрипции c-Jun приводит к вступлению в апоптоз малигнизированных клеток. Данные факторы транскрипции, действуя как гомодимеры или гетеродимеры вместе с большим числом других факторов транскрипции, активируют проапоптотические гены и/или отрицательно регулируют противоапоптотические гены, приводя к фрагментации ДНК. Соединения согласно настоящему изобретению не вызывают антипролиферативный эффект в ответ на сигнал TGF-β в нормальных немалигнизированных клетках.
Второй механизм индукции апоптоза, называемый путем передачи сигнала к апоптозу лигандом Fas/Fas, активируется новыми соединениями согласно настоящему изобретению. Активация пути передачи сигнала лигандом Fas/Fas может привести к быстрой гибели клетки путем апоптоза. Таким образом, для избежания малигнизированными клетками гибели посредством лиганда Fas/Fas они должны инактивировать данный наиболее важный апоптотический путь. Механизм инактивации пути передачи сигнала лигандом Fas/Fas малигнизированными клетками различен; однако многие малигнизированные клетки отрицательно регулируют экспрессию Fas-рецептора и Fas-лиганда на своих мембранах.
Более важно, показано, что R,R,R-2-(2,5,7,8-тетраметил-2-(4,8,12-триметилтридецил)хроман-6-илокси)уксусная кислота (1) побуждает малигнизированные клетки, не отвечающие на сигнал лиганда Fas/Fas, становиться чувствительными к сигналу лиганда Fas/Fas. Соединение 1 также обладает способностью усиливать экспрессию Fas-лиганда на мембране клеток предстательной железы LNCaP. Исследование показали, что клетки злокачественной опухоли молочной железы человека, не отвечающие на сигнал Fas, задерживают Fas-рецептор в своей цитоплазме, но при культивировании вместе с соединением 1 Fas-рецептор транспортируется из цитоплазмы к мембране, таким образом приводя клетки в состояние чувствительности к сигналу Fas. Более того, данное соединение обладает синергическим действием на запуск апоптоза не Fas-путем, при этом наблюдается больший уровень гибели клеток как для клеток злокачественной опухоли молочной железы человека, так и для клеток злокачественной опухоли предстательной железы при совместной обработке по сравнению с обработкой отдельно. Способность соединения 1 преобразовывать малигнизированные клетки, не отвечающие на сигнал лиганда Fas, в чувствительные к сигналу лиганда Fas малигнизированные клетки и проявлять синергическое действие в отношении гибели клетки представляет крайне важный механизм уничтожения малигнизированных клеток как посредством системы иммунного надзора хозяина, так и с помощью фармацевтического вмешательства. Соединения согласно настоящему изобретению не активируют путь передачи сигнала Fas в нормальных не малигнизированных клетках.
Данные соединения активируют путь передачи сигнала киназы JNK возможно посредством передачи сигнала TGF-β и лигандом Fas/Fas. Продолжительная активация JNK приводит к продолжительной активации факторов транскрипции c-Jun и ATF-2, которые предположительно играют роль в экспрессии или подавлении проапоптотических и противоапоптотических генов соответственно.
ПРИМЕР 13
Механизм индукции прекращения синтеза ДНК, остановки клеточного цикла и клеточной дифференцировки
Механизм ингибирования роста в результате прекращения синтеза ДНК и индукции дифференцировки клетки не описан полностью как механизм ингибирования роста посредством апоптоза. Исследования показали, что соединения согласно настоящему изобретению оказывают сильное влияние на клеточный цикл, индуцируя прекращение синтеза ДНК в приблизительно 95% малигнизированных клеток в течение 24-часовой обработки. Малигнизированные клетки, культивированные вместе с соединениями, описываемыми здесь, прекращают рост на фазе клеточного цикла G1, подвергаются морфологическим изменениям и экспрессируют молочные липиды, являющиеся маркером того, что клетки, блокированные в отношении прохождения клеточного цикла, подвергаются дифференцировке. Ген Р21, известный как ингибитор перехода клеток из фазы G1 клеточного цикла в фазу S клеточного цикла, и мРНК также как и белок гена Р21, подвергаются положительной регуляции при обработке малигнизированных клеток молочной железы человека MDA-MB-435 соединением 1.
ПРИМЕР 14
Возможности в отношении малигнизированных клеток человека in vivo.
Настоящее изобретение обладает возможностью применения в качестве терапевтических средств. Проводили исследования in vivo роста опухоли и метастазирования малигнизированных клеток человека, эктопически или ортотопически трансплантированных иммунокомпромитированным животным, таким как мыши nude, или исследования in vivo с применением широко признанных животных моделей. Ингибирование роста малигнизированных клеток человека, трансплантированных иммунокомпромитированным мышам, представляет доклинические данные для клинических испытаний. Исследования in vivo включают две модели малигнизированных клеток человека, модель метастазирующей эстроген-нечувствительной злокачественной опухоли молочной железы MDA-MB-435 и модель андроген-нечувствительной злокачественной опухоли предстательной железы РС-3.
Модель злокачественной опухоли молочной железы MDA-MB-435
Неинфицированные клетки злокачественной опухоли молочной железы человека MDA-MB-435, устойчиво трансфицированные маркерным белком (флуоресцирующий зеленый белок), выращивали в виде солидной опухоли у иммунокомпромитированных мышей nude или SCID. Опухоли удаляли и срезы толщиной 1 мм одинакового размера ортотопически трансплантировали в липидное окружение молочной железы или эктопически трансплантировали в заднебоковую область самок мышей nude. Исследовали рост опухоли, метастазирование и гибель животных. Рост опухоли измеряли с помощью оценок кавернометром (caliper) размера опухоли. Во время забивания опухоли удаляли, определяли размер и применяли для гистохимической оценки. Такие органы, как селезенка, лимфатические узлы, легкие и костный мозг, оценивали на наличие метастатических клеток MDA-MB-435 с помощью гистохимичекого окрашивания срезов тканей на экспрессию маркерного флуоресцирующего зеленого белка.
Модель злокачественной опухоли предстательной железы РС-3
Неинфицированные клетки злокачественной опухоли предстательной железы человека
РС-3, устойчиво трансфицированные маркерным белком (флуоресцирующий зеленый белок), выращивали в виде солидной опухоли у мышей nude. Опухоли удаляли и срезы толщиной 1 мм одинакового размера эктопически трансплантировали в заднебоковую область самцов мышей nude. Исследовали рост опухоли, метастазирование и гибель животных. Во время забивания опухоли удаляли, определяли размер и применяли для гистохимической оценки. Такие органы, как селезенка, лимфатические узлы, легкие, костный мозг, оценивали на наличие метастатических клеток РС-3 с помощью гистохимического окрашивания тканей на экспрессию маркерного флуоресцирующего зеленого белка.
Модель рака кожи животных
Рак кожи вызывали у безволосых мышей SENCAR и SKH-1 с помощью ультрафиолетового облучения или химической (DMBA) обработки. Кроме того, применяли мышей, специфически экспрессирующих онкоген Her-2/neu в базальных клетках кожи, который приводит к спонтанному развитию рака кожи. Соединения, описываемые здесь, применяли местно на кожу ежедневно, до и после начала развития рака кожи, и оценивали развитие кожного папилломного образования. Контрольных мышей обрабатывали идентично за исключением того, что они получали обработку носителем, применяемым местно на кожу. Контролировали эффективность данных соединений для лечения папиллом, также как и их способность препятствовать переходу в злокачественное состояние при применении до прогрессирования предракового состояния.
ПРИМЕР 15
Дополнение к новым соединениям
Перед началом проведения экспериментов in vivo соединения согласно данному изобретению, обладающие наибольшим эффектом в отношении гибели малигнизированных клеток, вводили мышам nude, SCID, трансгенным и другим мышам в различных концентрациях для определения максимальной концентрации соединения, которая может безопасно вводиться без нежелательных эффектов. Соединения вводили способом, соответствующим модели, например, перорально, инъекционно, включая инъекции непосредственно в орган-мишень, или местно. После определения максимальной концентрации соединения, которая может быть переносима, и эффективных путей введения, новые соединения вводили мышам ежедневно и исследовали рост и прогрессию опухоли, как описано выше.
ПРИМЕР 16
Определение максимально переносимой дозы (MTD) соединения 1
Перед проведением доклинических химиотерапевтических исследований соединение 1 анализировали на безопухолевых самках мышей Balb/c для проведения определения максимально переносимой дозы. Соединение 1 растворяли в 100% этаноле для получения исходного раствора (2 грамма соединения 1 в 5 мл 100% этанола) и растворяли в арахисовом масле (не содержащем витамина Е) с получением 20, 10, и 5 мг/0,1 мл. Сукцинат RRR-α-токоферола (VES; сукцинатная составляющая присоединена к RRR-α-токоферолу через сложноэфирную связь) растворяли и разбавляли, как описано выше, с получением 20 мг/0,1 мл/зондовая порция, служащих дополнительным контролем. Все 30 самок мышей Balb/с разделяли на 6 групп (5 мышей в группе), они получали путем зондового питания соединение 1 ежедневно в течение всех 23 дней. Мыши группы 1 не обрабатывались и служили контролем; мыши группы 2 представляли собой контрольную группу с носителем и получали 0,1 мл носителя (этанол+арахисовое масло эквивалентно обрабатываемым группам)/мышь/сутки в течение всех 23 дней, мыши группы 3 получали 20 мг/мл VES ежедневно в течение всех 23 дней. Обрабатываемые группы 3, 4 и 5 получали путем зондового питания ежедневно в течение 23 дней 5, 10 и 20 мг соединения 1. Соединение 1 в дозе 5, 10 и 20 мг/сутки, вводимое ежедневно путем зондового питания мышам групп 3, 4 и 5 соответственно, в течение 23 дней хорошо переносилось мышами. Каждую неделю определяли массы тела, и не выявилось существенных различий в массах тела мышей, получавших соединение 1 в дозе 5, 10 и 20 мг/сутки, по сравнению с массами тела VES-обработанных, необработанных и обработанных носителем контрольных мышей (фиг.8, данные для 11 дней и 23 дней обработки). Мыши оставались активными и не проявляли симптомов токсичности. После завершения изучения MTD мышей забивали, и при аутопсии не было обнаружено проявлений токсичности.
ПРИМЕР 17
Химиотерапевтическая эффективность соединения 1
Изучение максимально переносимой дозы показало, что соединение 1, вводимое на максимальном уровне (20 мг/сутки/23 дня) не является токсичным; однако вследствие применения нетоксичных концентраций максимальная переносимая доза не установлена. Для доклинических химиотерапевтических исследований в отсутствие максимально переносимой дозы мышам дополнительно вводили 30 и 20 мг/0,1 мл путем зондового питания ежедневно в течение 21 дня. Соединение 1 растворяли в 100% этаноле до 2 грамм соединения 1/5 мл этанола и растворяли в арахисовом масле (не содержащем витамина Е) с получением 30 и 20 мг соединения 1 в 0,1 мл. Контроль, содержащий носитель, включал смесь эквивалентного количества этанола и арахисивого масла. Доклинические химиотерапевтические исследования проводили с соединением 1, применяя клетки злокачественной опухоли молочной железы человека MDA-MB-435, клетки злокачественной опухоли предстательной железы человека DU-145 и клетки опухоли толстого кишечника человека НТ-20, трансплантированные иммунокомпромитированным Balb/c мышам nude.
Все 40 иммунокомпромитированных мышей nude применяли для оценки каждое тестируемое соединение (две обрабатываемые группы по 20 и 30 мг/сутки) для каждого типа малигнизированных клеток. Четыре экспериментальные обрабатываемые группы по 10 мышей каждая применяли для каждого типа малигнизированных клеток (носитель/0,1 мл/зондовой порции ежедневно в течение 21 дня, соединение 1: по 30 мг соединения 1/0,1 мл зондовой порции/ежедневно в течение 21 дня, и соединение 1: по 20 мг соединения 1/0,1 мл зондовой порции ежедневно в течение 21 дня). При исследовании ксенотрансплантатов применяли установленные дозы эффективных химиотерапевтических средств. Таксол в дозе 20 мг/кг, вводимый внутрибрюшинно ежедневно в течение 5 дней, применяли в качестве положительного контроля для ксенотрансплантатов злокачественной опухоли молочной железы человека MDA-MB-435; митоксантрон в дозе 1 мг/кг, вводимый внутривенно ежедневно в течение 5 дней, применяли в качестве положительного контроля для малигнизированных клеток предстательной железы человека DU-145, и 5-фторурацил (5-FU) в дозе 30 мг/кг, вводимый ежедневно в течение 5 дней, применяли в качестве положительного контроля для малигнизированных клеток толстого кишечника человека НТ-29. Малигнизированные клетки трансплантировали подкожно на левый бок и позволяли расти приблизительно до размера 70 мг, начинали вводить тестируемое соединение ежедневно путем зондового питания и продолжали в течение всех дней (21). Мышей контролировали ежедневно в отношении размеров опухоли путем измерения кавернометром. Мышей обрабатывали в течение 21 дня; однако мышей забивали, если опухоли в контроле, содержащем носитель, достигали 500 мг по массе. После завершения протокола опухоли удаляли и размер опухоли определяли по массе. После забивания брали сыворотку, опухолевую ткань и сердечную мышечную ткань у животных двух групп, принимавших тестируемое соединение 1, для определения содержания соединения 1 с помощью ВЭЖХ-анализа (данные не включены). Химиотерапевтическую эффективность соединения 1, вводимого ежедневно в дозе 30 и 20 мг/сутки, определяли путем сравнения размера опухоли двух обрабатываемых групп с группой контроля, содержащего носитель, для каждого из трех типов опухолей и путем сравнения размера опухоли двух обрабатываемых групп с опухолевой группой, обрабатываемой положительным контролем. Обработку проводили в течение 21 дня, эксперименты продолжали до тех пор, когда средняя масса опухоли в контроле, содержащем носитель, достигала 500 мг.
Доклинические химиотерапевтические данные in vivo представлены в таблицах 3 и 4 и на фиг.9А-С и представляют обработку в течение 21-22 дней. Обработка мышей nude с трансплантацией клеток злокачественной опухоли молочной железы человека MDA-MB-435 с введением соединения 1 в течение 21 дня в дозе 20 и 30 мг/сутки путем зондового питания снижало среднюю массу опухоли (мг) трансплантированных малигнизированных клеток на 20,7 и 44,6% соответственно при сравнении со средней массой опухоли группы контроля, содержащего носитель, на 21 день после обработки. Положительной контроль Таксол снижал среднее значение прироста опухоли по сравнению со средним значением прироста опухоли в группе контроля, содержащего носитель, на 90,3% (таблица 3, 4 и фиг.9А). Данные преобразованы в процентное значение среднего прироста опухоли в виде прироста массы от начала лечения (1 сутки) и представлены в таблице 4. Лечение мышей nude с трансплантацией клеток злокачественной опухоли предстательной железы человека DU-145 с введением соединения 1 в течение 21 дней в дозе 20 и 30 мг/сутки путем зондового питания снижало среднюю массу опухоли (мг) трансплантированных малигнизированных клеток на 31,5 и 24,8% соответственно при сравнении со средней массой опухоли группы контроля, содержащего носитель, на 22 день (таблица 3, 4 и фиг.9В). Положительной контроль митоксантрон снижал среднее значение прироста опухоли по сравнению со средним значением прироста опухоли в группе контроля, содержащего носитель, на 27,9%. Лечение иммунокомпромитированных мышей nude с трансплантацией клеток злокачественной опухоли толстого кишечника человека НТ-29 с введением соединения 1 в течение 21 дня в дозе 20 и 30 мг/сутки путем зондового питания снижало среднюю массу опухоли (мг) трансплантированных малигнизированных клеток на 33,3 и 34,5% соответственно при сравнении со средней массой опухоли группы контроля, содержащего носитель, на 22 день (таблица 3, 4 и фиг.9С). Положительный контроль 5-фторурацил снижал среднее значение прироста опухоли по сравнению со средним значением прироста опухоли в группе контроля, содержащего носитель, на 31,4%.
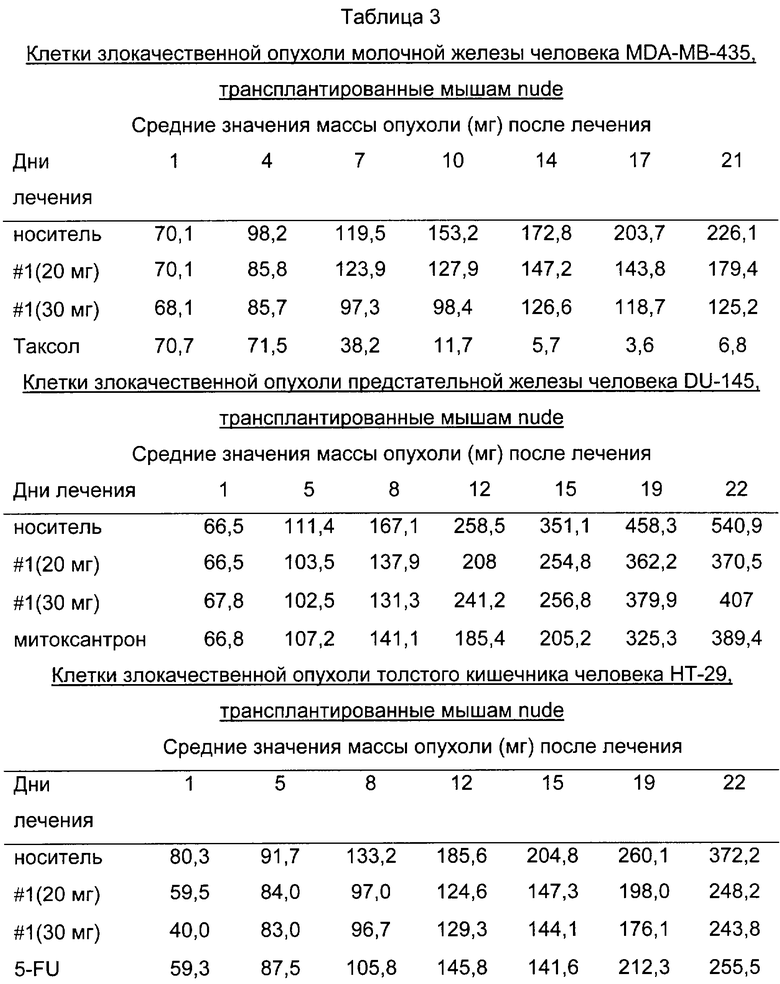
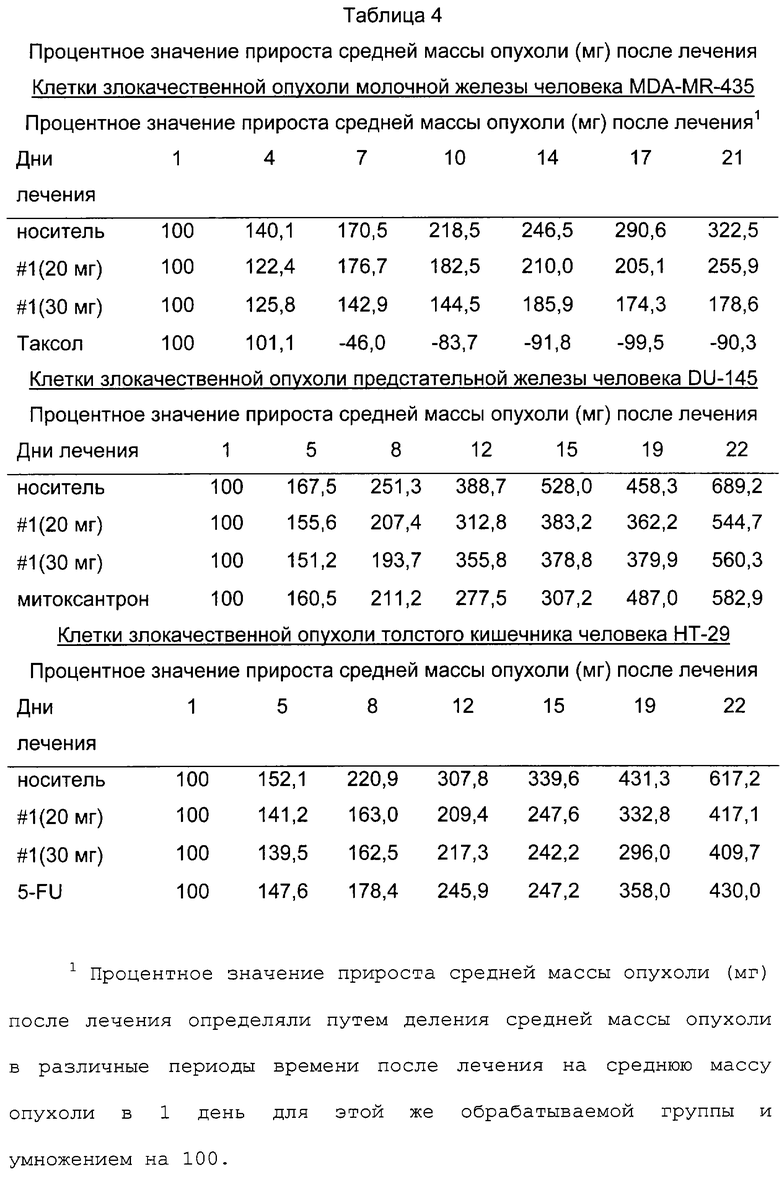
ПРИМЕР 18
Приготовление исходного раствора, разбавление носителя и соединения 1
Соединение 1 получали каждую неделю и хранили при 4°С. Процедура приготовления представляла собой следующую.
Исходный раствор соединения 1
Растворяли 2 грамма соединения 1 в 5 мл 100% этанола (EtOH) и встряхивали при 37°С. Это представляло собой максимальное количество соединения 1, которое может перейти в раствор.
Для последующих разведениий добавляли сухое соединение 1 для получения соответствующей концентрации соединения 1, при этом сохраняя концентрации этанола равными для двух экспериментальных групп и группы контроля, содержащего носитель.
Соединение 1 в дозе 30 мг/0,1 мл зондовой порции/мышь
Объединяли 1 мл исходного раствора соединения 1, 3 мл арахисового масла, не содержащего витамина Е, и 800 мг соединения #1 (сухого) и встряхивали при 37°С.
Соединение 1 в дозе 20 мг/0,1 мл зондовой порции/мышь
Объединяли 1 мл исходного раствора соединения 1, 3 мл арахисового масла, не содержащего витамина Е, и 400 мг соединения #1 (сухого) и встряхивали при 37°С приблизительно 2 ч до получения раствора.
Соединение 1 в дозе 10 мг/0,1 мл зондовой порции/мышь
Объединяли 1 мл исходного раствора соединения 1 и 3 мл арахисового масла, не содержащего витамина Е.
Носитель
Объединяли 1 мл EtOH и 3 мл арахисового масла, не содержащего витамина Е.
ПРИМЕР 19
Химиопрофилактические свойства соединения 1 на модели злокачественной опухоли крыс ACI
Соединение 1 применяли in vivo для воздействия на трансплантированные злокачественные опухоли молочной железы, предстательной железы и толстого кишечника человека, трансплантированные иммунокомпромитированным мышам nude. Химиопрофилактическая эффективность соединения 1 in vivo против злокачественной опухоли молочной железы человека показана на модели злокачественной опухоли молочной железы крыс ACI, вызванной влиянием эстрогена. Приблизительно у 90% крыс с имплантированными пилюлями с эстрогеном развивалася злокачественная опухоль молочной железы в течение 6 месяцев после имплантации эстрогена.
Соединение 1 растворяли в 100% этаноле и разбавляли до соответствующей дозы с помощью арахисового масла, не содержащего витамина Е. Максимально переносимую дозу (MTD, максимальная доза соединения, которая может быть введена без нежелательных эффектов) определяли, как описано в примерах 14 и 15. Соединение 1 вводили в максимально переносимой дозе и 50% максимально переносимой дозы. Крысам ACI 4-недельного возраста имплантировали в подкожную жировую клетчатку лопаточной области пилюли с эстрогеном. Соединение 1 в максимально переносимой дозе и 50% максимально переносимой дозы вводили путем зондового питания. Опухоли молочной железы определяли в контрольной группе в течение приблизительно 100 дней после имплантирования эстрогена. У 90% крыс контрольной группы обнаруживали злокачественную опухоль молочной железы в течение 6 месяцев после имплантирования эстрогена. Животных, имеющих опухоли, из контрольной и обрабатываемой групп забивали в различные интервалы времени после начала обработки и ткань молочной железы оценивали на наличие опухоли и далее оценивали с помощью гистологического анализа.
Здесь упоминались следующие ссылки.
1. Colowisk, Sidney, P. and Kaplan, Nathan, O. Methods in Enzymology, Vol. XVIII, Vitamins and Coenzymes, Part C. Edited by Donald B. McCormick and Lemuel D. Wright. Section XII, pp.335.
2. Dhar, A., Liu, S., Klucik, J., Berlin, K.D., Madler, M.M., Lu, S., Ivey, R.T., Zacheis, D., Brwon, C.W., Nelson, E.C., Birchbichler, P.J., Benbrook, D.M. Synthesis, Structure-Activity Relationships, and RARy-Ligand Interactions of Nitrogen Heteroarotinoids. J. Med. Chem. 42, pp.3602-3614 (1999).
Все патенты или публикации, упомянутые в данном описании, показывают уровень специалистов в данной области, которым принадлежит изобретение. Кроме того, данные патенты и публикации включены здесь в ссылку в том же объеме, как если бы каждая отдельная публикация была специально и отдельно указана для включения в ссылку.
Специалисту в данной области будет легко понятно, что настоящее изобретение хорошо адаптировано для осуществления объектов и достижения целей и указанных преимуществ, также как они представлены здесь. Настоящие примеры вместе со способами, процедурами, обработками, молекулами и конкретными соединениями, описанными здесь, являются сами образцами предпочтительных воплощений, являются иллюстративными и не предназначены для ограничения объема изобретения. Изменения данного и другие применения будут возникать у специалистов в данной области, и они подпадают под суть изобретения, что указано объемом формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТОКОФЕРОЛЫ, ТОКОТРИЕНОЛЫ И ДРУГИЕ ПРОИЗВОДНЫЕ ХРОМАНА И БОКОВЫХ ЦЕПЕЙ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ИСПОЛЬЗОВАНИЕМ | 1999 |
|
RU2232758C2 |
| СПОСОБ ПОЛУЧЕНИЯ (2RS)-2,5,7,8-ТЕТРАМЕТИЛ-2-[(4RS,8RS)-4,8,12-ТРИМЕТИЛТРИДЕЦИЛ]-ХРОМАН-6-ИЛ-N-[3-ОКСОЛУП-20(29)-ЕН-28-ОИЛ]-ГЛИЦИНАТА | 2008 |
|
RU2440366C2 |
| ЛИГАНДЫ, ТРОПНЫЕ К ПРОСТАТИЧЕСКОМУ СПЕЦИФИЧЕСКОМУ МЕМБРАННОМУ АНТИГЕНУ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ДВОЙНЫХ КОНЪЮГАТОВ С ТЕРАПЕВТИЧЕСКИМИ АГЕНТАМИ НА ИХ ОСНОВЕ ДЛЯ КОМБИНИРОВАННОЙ ТЕРАПИИ ПСМА ЭКСПРЕССИРУЮЩИХ ОПУХОЛЕЙ | 2023 |
|
RU2841078C1 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2018 |
|
RU2793315C2 |
| СПОСОБ ПОЛУЧЕНИЯ (S)-(-)-6-БЕНЗИЛОКСИ-3,4-ДИГИДРО-2,5,7,8-ТЕТРАМЕТИЛ-2Н-1-БЕНЗОПИРАН-2-ИЛМЕТАНОЛА | 2010 |
|
RU2443695C1 |
| ПРОИЗВОДНЫЕ 1-(4-ИЗОКСАЗОЛ-5-ИЛ)-1Н-ПИРАЗОЛ-1-ИЛ)-2-МЕТИЛПРОПАН-2-ОЛА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИРОРОВ ИЛ-17 И ИФН-ГАММА ДЛЯ ЛЕЧЕНИЯ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ И ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ | 2018 |
|
RU2785342C2 |
| СОЕДИНЕНИЯ, ОБРАЗУЮЩИЕ ПРОЛЕКАРСТВА | 2014 |
|
RU2667942C2 |
| Способ получения (+)-(3S, 4R)-3,4,7,11-тетраметил-6,10-додекадиеналя | 1987 |
|
SU1533625A3 |
| СОЕДИНЕНИЕ ДЛЯ ДИАГНОСТИКИ ОПУХОЛЕЙ, ЭКСПРЕССИРУЮЩИХ ПСМА, И КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2019 |
|
RU2730507C1 |
| СПОСОБЫ ПРОИЗВОДСТВА И ИСПОЛЬЗОВАНИЯ ЭТОКСИКОМБРЕТАСТАТИНОВ И ПРОЛЕКАРСТВА НА ИХ ОСНОВЕ | 2007 |
|
RU2451664C2 |
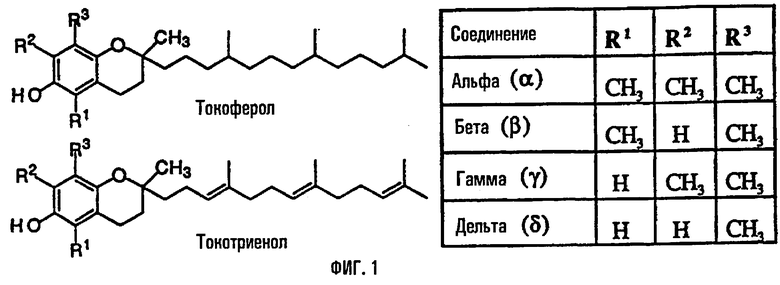
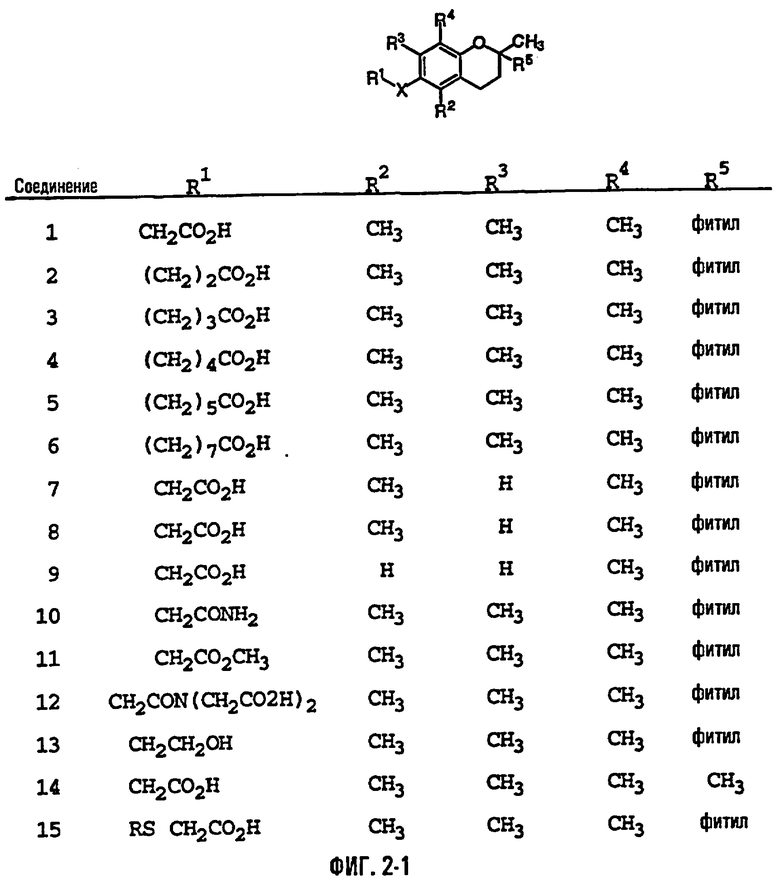
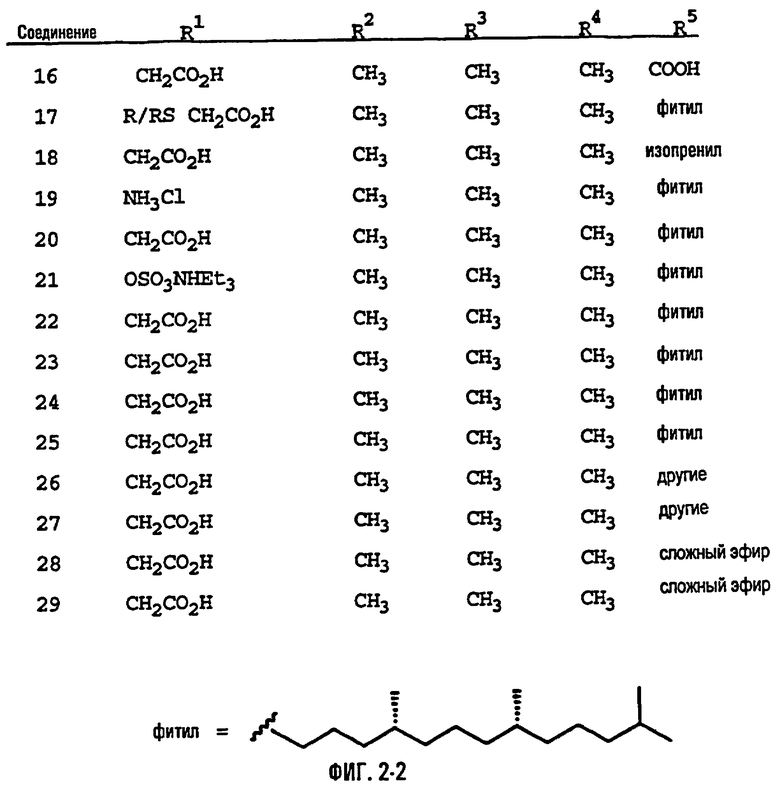
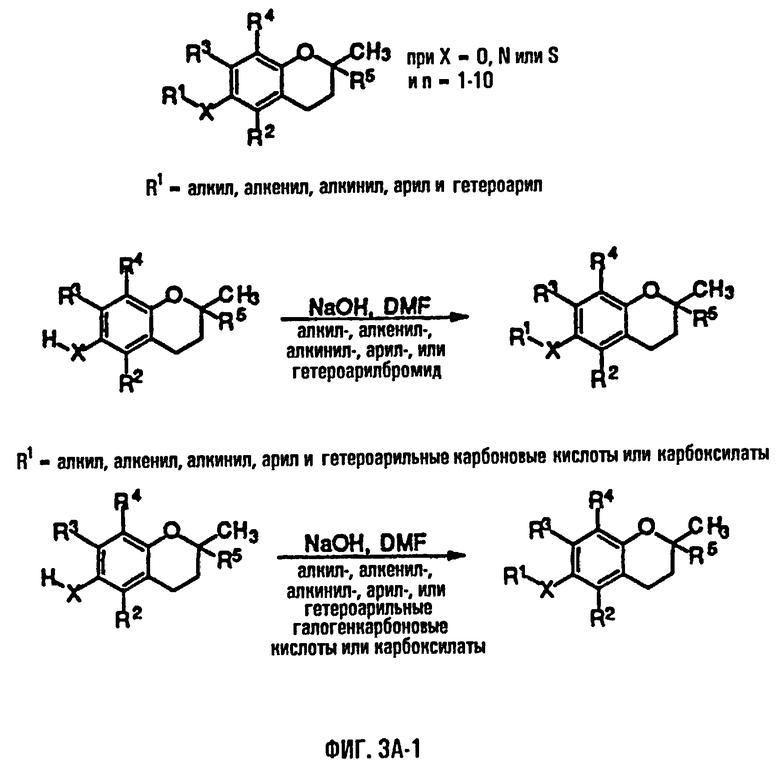
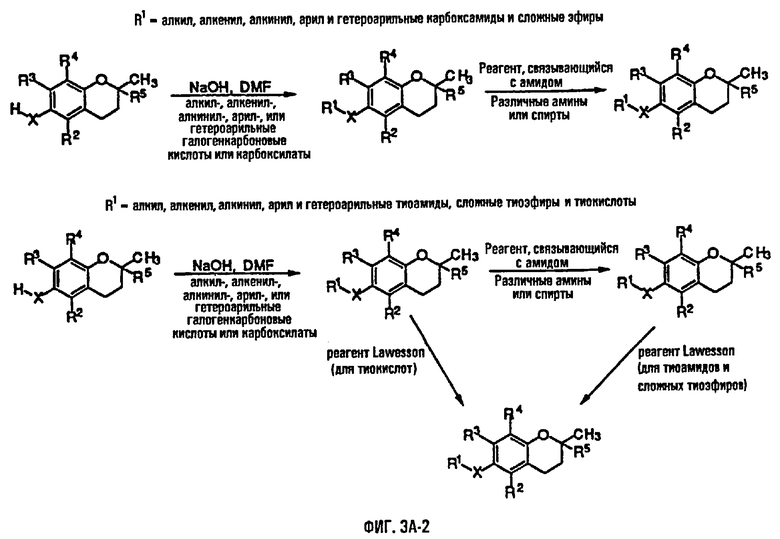
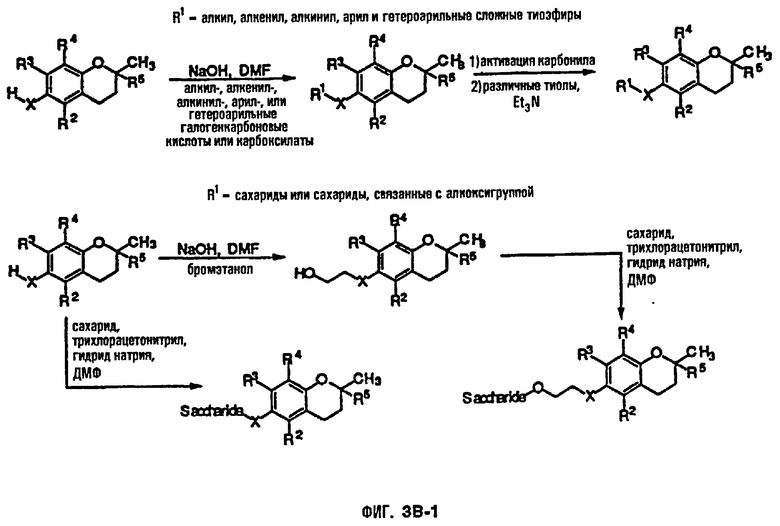
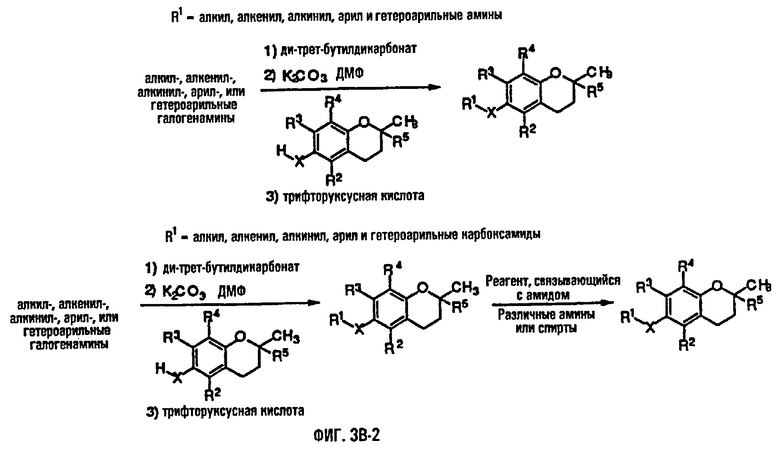
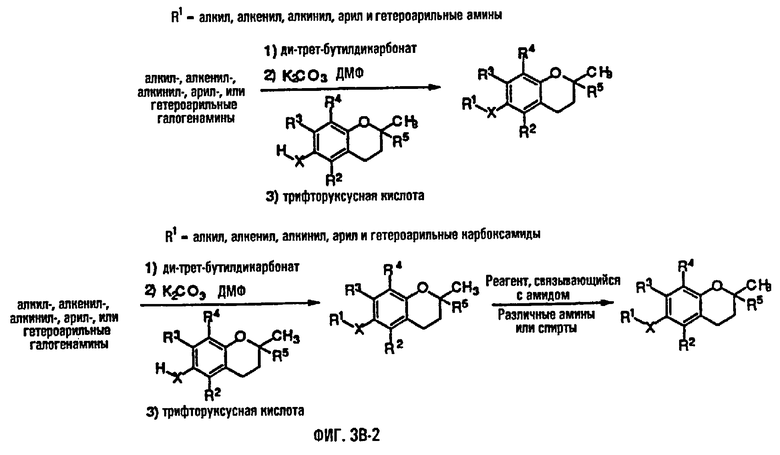
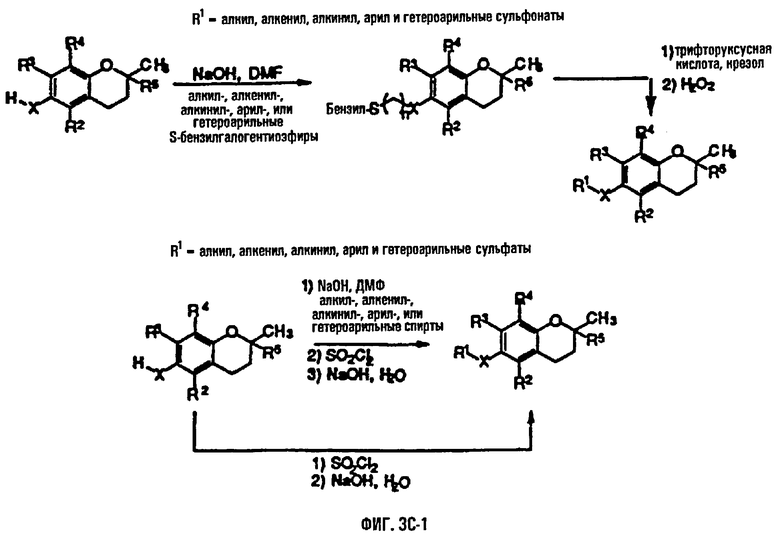
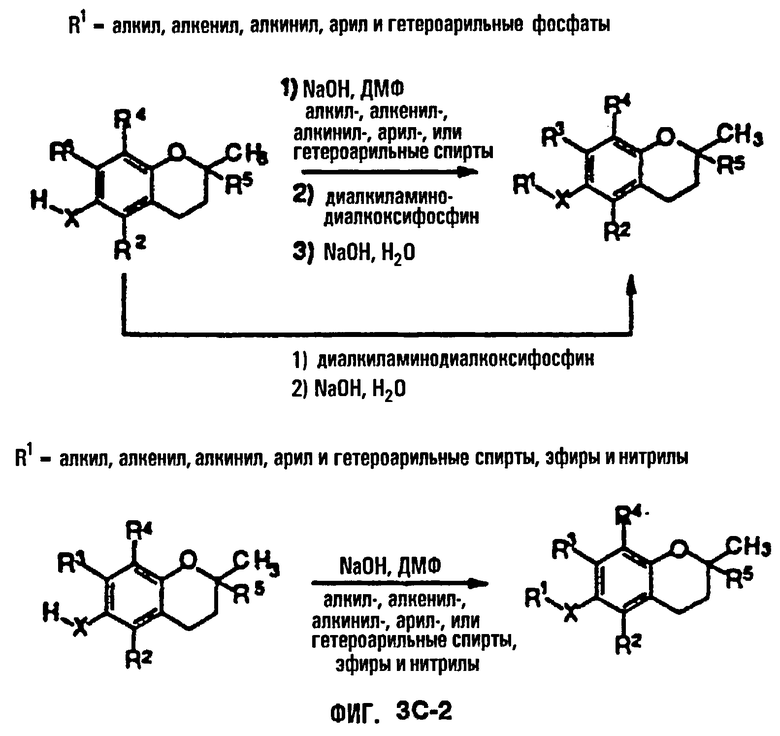

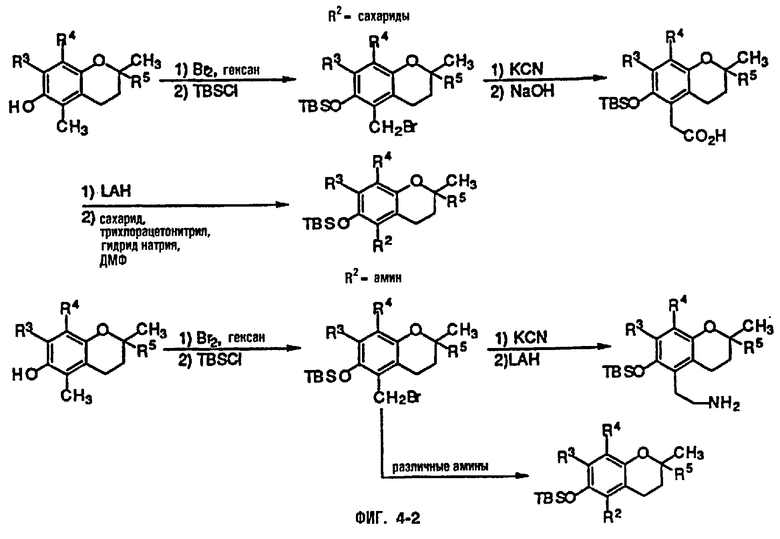
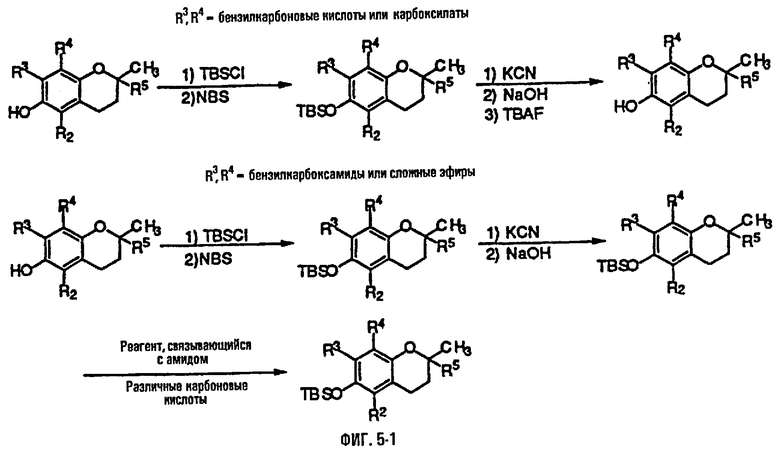
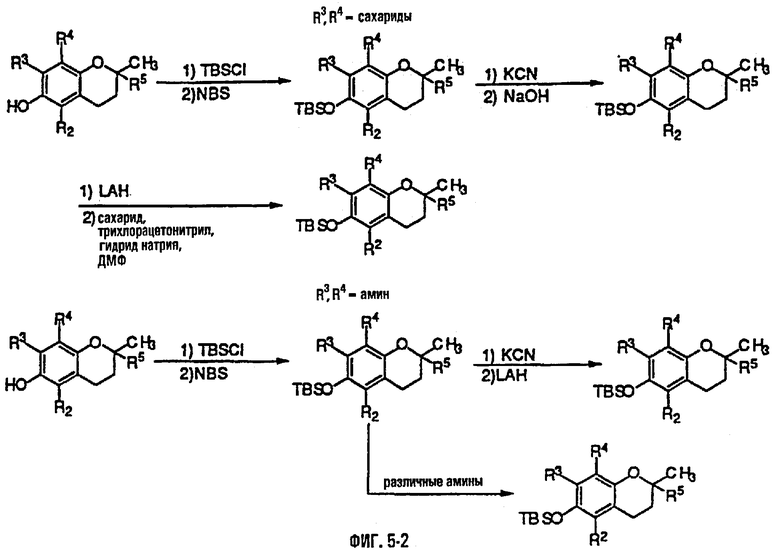
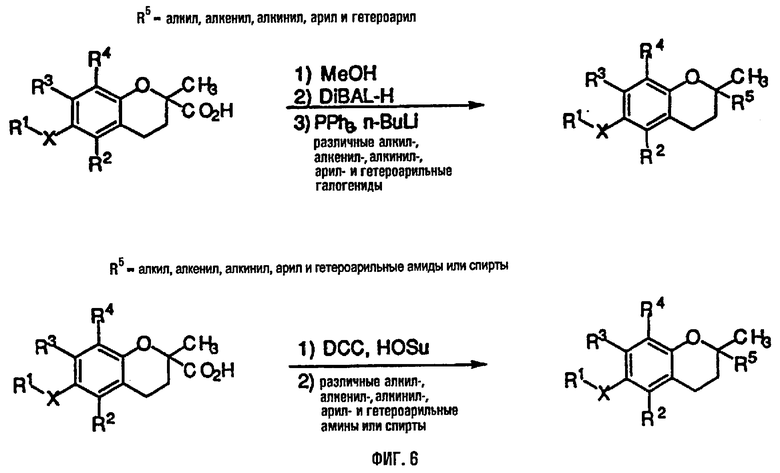
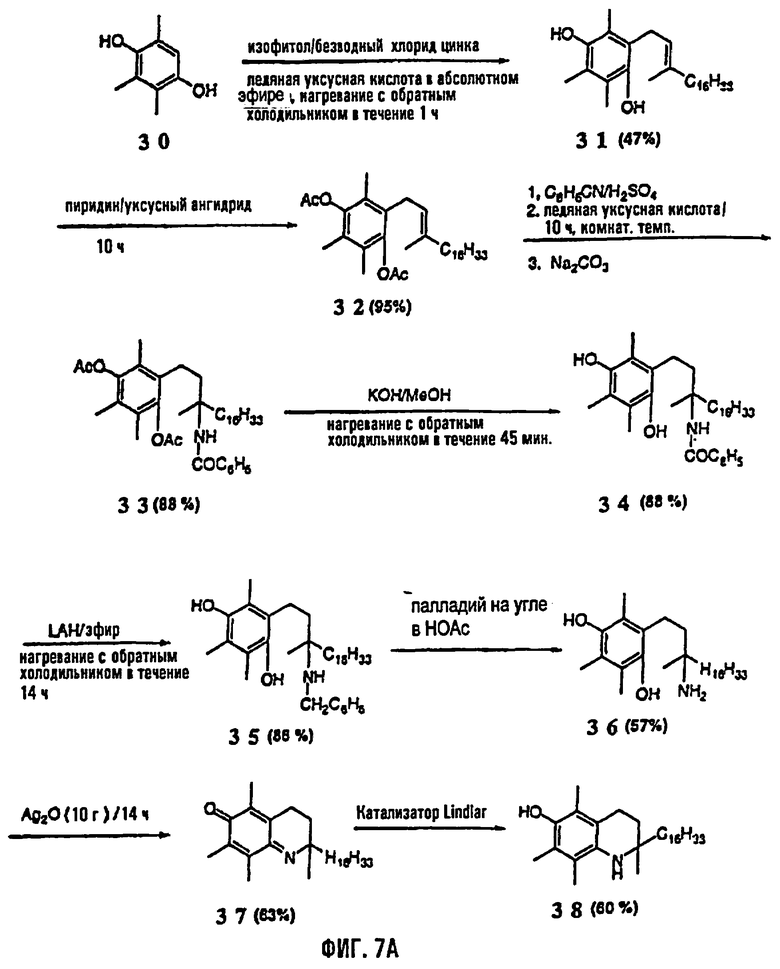
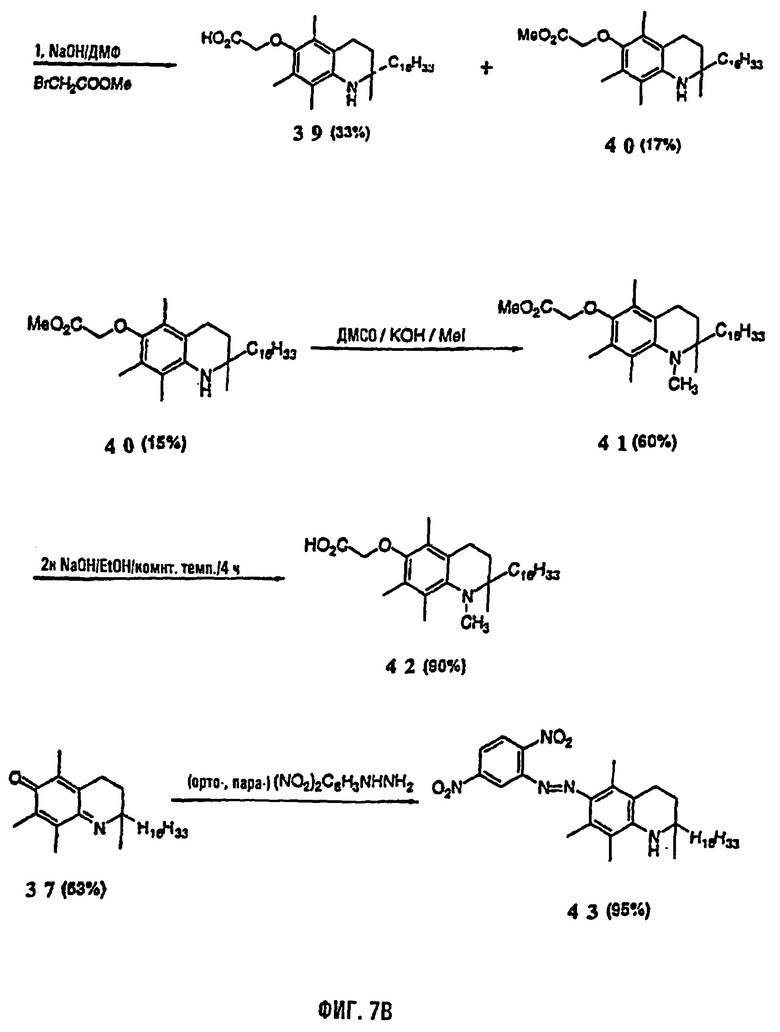
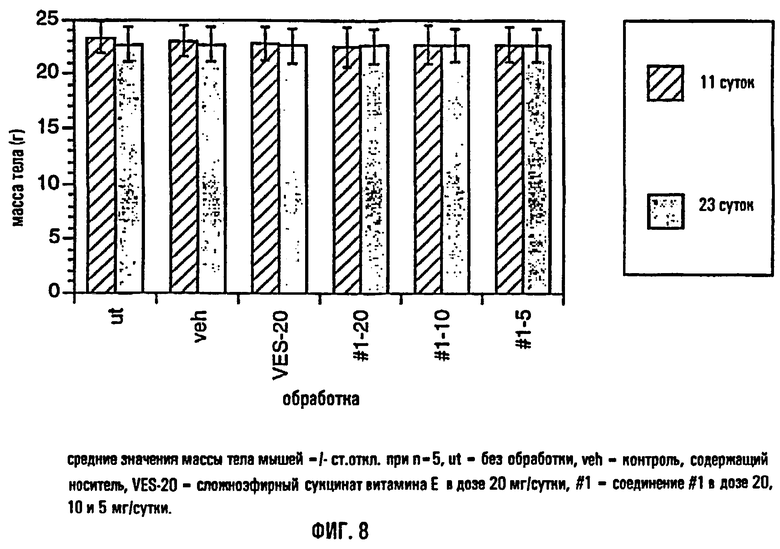
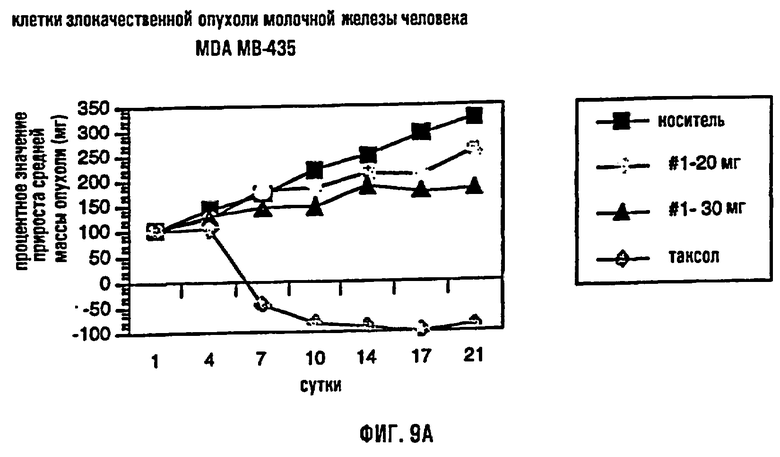
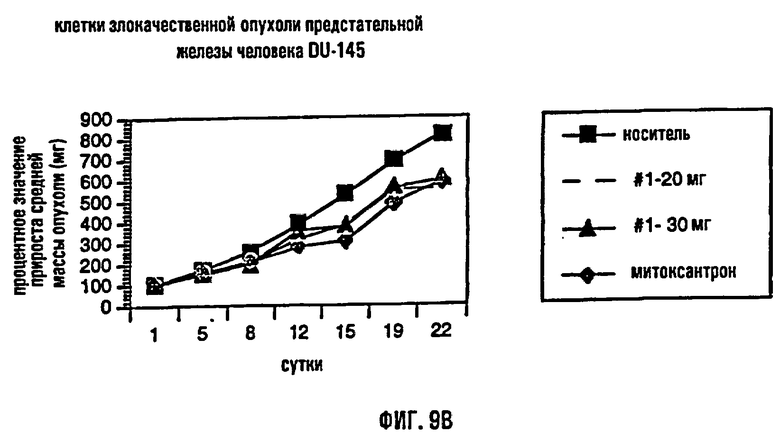
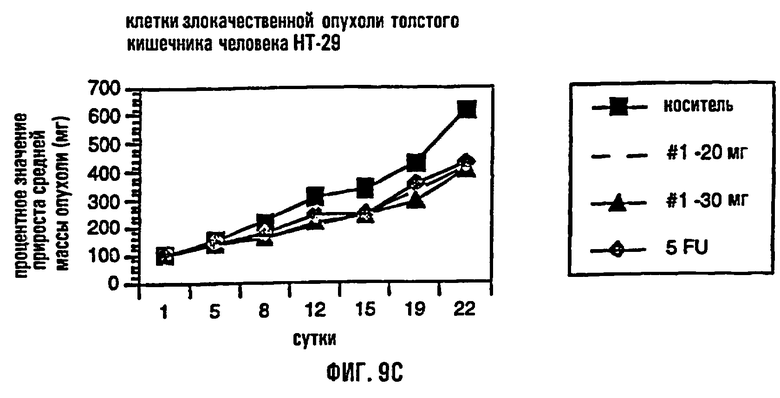
Изобретение относится к новым производным токоферола, токотриенола и другим производным хромана общей формулы I
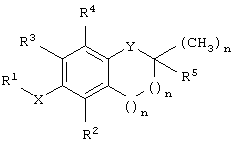
где Х выбран из группы, включающей кислород и азот;
Y выбран из группы, включающей кислород, азот и серу, где, когда Y представляет собой кислород или азот, n равно 1, и, когда Y представляет собой серу, n равно 0;
R1 представляет собой остаток карбоновой кислоты, карбоксамида, сложного эфира, спирта, амина или сульфата; R2 представляет собой метил; R3 представляет собой метил; R4 представляет собой метил и R5 выбран из группы, включающей алкил, алкенил, алкинил, карбоксил и сложноэфирный остаток, где, когда Y представляет собой азот, указанный азот замещен группой R6, где R6 представляет собой водород или метил;
где, когда Х представляет собой кислород, Y представляет собой кислород и R5 представляет собой фитил, тогда R1 не является бутановой кислотой. Изобретение также относится к фармацевтической композиции на основе этих соединений, способу лечения клеточно-пролиферативного заболевания и способу индукции апоптоза клеток.
Технический результат - получения новых соединений, обладающих антипролиферативным действием. 8 н. и 28 з.п. ф-лы., 4 табл. 19 ил.
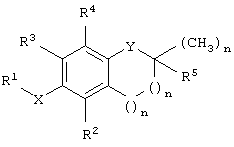
где Х выбран из группы, включающей кислород и азот;
Y выбран из группы, включающей кислород, азот и серу, где, когда Y представляет собой кислород или азот, n равно 1 и, когда Y представляет собой серу, n равно 0;
R1 представляет собой остаток карбоновой кислоты, карбоксамида, сложного эфира, спирта, амина или сульфата;
R2 представляет собой метил;
R3 представляет собой метил;
R4 представляет собой метил и
R5 выбран из группы, включающей алкил, алкенил, алкинил, карбоксил и сложноэфирный остаток;
где, когда Y представляет собой азот, указанный азот замещен группой R6, где R6 представляет собой водород или метил;
где, когда Х представляет собой кислород, Y представляет собой кислород, и R5 представляет собой фитил, тогда R1 не является бутановой кислотой.
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)пропионовую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)валериановую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)гексановую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)октановую кислоту,
2,5,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,7,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,8-диметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетамид,
метил 2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетат,
2-(N,N-(карбоксиметил)-2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси))этан-1-ол,
2-(2,5,7,8-пентаметилхроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2RS-(4RS,8RS,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(карбокси)хроман-6-илокси))уксусную кислоту,
2,5,7,8-тетраметил-(2R-(2RS,6RS,10-триметилундецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(2,6,10-триметил-1,3,5,9-Е:Z-декатетраен)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептадецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(тридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептадецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(4,8-диметил-1,3,7-Е:Z-нонотриен)хроман-6-илокси)уксусную кислоту,
Е,Z,RS,RS-(фитилтриметилбензолтиол-6-илокси)уксусную кислоту,
(R)-2-((2,5,7,8-тетраметил-2-(3-пропенметиловый эфир)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(пропионат)хроман-6-илокси)уксусную кислоту,
1-аза-α-токоферол-6-илоксилуксусную кислоту,
1-аза-α-токоферол-6-илоксилметилацетат,
1-аза-N-метил-α-токоферол-6-илоксил-метилацетат и
1-аза-N-метил-α-токоферол-6-илоксилуксусную кислоту.
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)пропионовую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)валериановую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)гексановую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)октановую кислоту,
2,5,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,7,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,8-диметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетамид,
метил-2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетат,
2-(N,N-(карбоксиметил)-2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси))этан-1-ол,
2-(2,5,7,8-пентаметилхроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2RS-(4RS,8RS,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(карбокси)хроман-6-илокси))уксусную кислоту,
2,5,7,8-тетраметил-(2R-(2RS,6RS,10-триметилундецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(2,6,10-триметил-1,3,5,9-Е:Z-декатетраен)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептадецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(тридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептадецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(4,8-диметил-1,3,7-Е:Z-нонотриен)хроман-6-илокси)уксусную кислоту,
E,Z,RS,RS-(фитилтриметилбензолтиол-6-илокси)уксусную кислоту,
(R)-2-((2,5,7,8-тетраметил-2-(3-пропенметиловый эфир)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(пропионат)хроман-6-илокси)уксусную кислоту,
1-аза-α-токоферол-6-илоксилуксусную кислоту,
1-аза-α-токоферол-6-илоксилметилацетат,
1-аза-N-метил-α-токоферол-6-илоксил-метилацетат и
1-аза-N-метил-α-токоферол-6-илоксилуксусную кислоту.
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)пропионовую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)валериановую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)гексановую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)октановую кислоту,
2,5,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,7,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,8-диметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетамид, метил
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетат,
2-(N,N-(карбоксиметил)-2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси))этан-1-ол,
2-(2,5,7,8-пентаметилхроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2RS-(4RS,8RS,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(карбокси)хроман-6-илокси))уксусную кислоту,
2,5,7,8-тетраметил-(2R-(2RS,6RS,10-триметилундецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(2,6,10-триметил-1,3,5,9-Е:Z-декатетраен)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептадецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(тридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептадецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(4,8-диметил-1,3,7-Е:Z-нонотриен)хроман-6-илокси)уксусную кислоту,
E,Z,RS,RS-(фитилтриметилбензолтиол-6-илокси)уксусную кислоту,
(R)-2-((2,5,7,8-тетраметил-2-(3-пропенметиловый эфир)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(пропионат)хроман-6-илокси)уксусную кислоту,
1-аза-α-токоферол-6-илоксилуксусную кислоту,
1-аза-α-токоферол-6-илоксилметилацетат,
1-аза-N-метил-α-токоферол-6-илоксил-метилацетат и
1-аза-N-метил-α-токоферол-6-илоксилуксусную кислоту.
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)пропионовую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)валериановую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)гексановую кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)октановую кислоту,
2,5,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,7,8-триметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,8-диметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетамид,
метил-2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)ацетат,
2-(N,N-(карбоксиметил)-2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2-(2,5,7,8-тетраметил-(2R-(4R,8R,12-триметилтридецил)хроман-6-илокси))этан-1-ол,
2-(2,5,7,8-пентаметилхроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2RS-(4RS,8RS,12-триметилтридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(карбокси)хроман-6-илокси))уксусную кислоту,
2,5,7,8-тетраметил-(2R-(2RS,6RS,10-триметилундецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(2,6,10-триметил-1,3,5,9-Е:Z-декатетраен)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептадецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(тридецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(гептадецил)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(4,8-диметил-1,3,7-Е:Z-нонотриен)хроман-6-илокси)уксусную кислоту,
E,Z,RS,RS-(фитилтриметилбензолтиол-6-илокси)уксусную кислоту,
(R)-2-((2,5,7,8-тетраметил-2-(3-пропенметиловый эфир)хроман-6-илокси)уксусную кислоту,
2,5,7,8-тетраметил-(2R-(пропионат)хроман-6-илокси)уксусную кислоту,
1-аза-α-токоферол-6-илоксилуксусную кислоту,
1-аза-α-токоферол-6-илоксилметилацетат,
1-аза-N-метил-α-токоферол-6-илоксил-метилацетат и
1-аза-N-метил-α-токоферол-6-илоксилуксусную кислоту.
| RU 96104250 А, 10.06.1998 | |||
| RU 94041687 А, 10.07.1996 | |||
| US 4665204 А, 12.05.1987 | |||
| US 5315017 А, 24.05.1994 | |||
| US 5917060 А, 29.06.1999 | |||
| US 4457918 А, 03.07.1984 | |||
| US 5114957 А, 19.05.1992. |

