Область техники, к которой относится изобретение
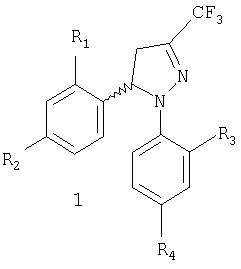
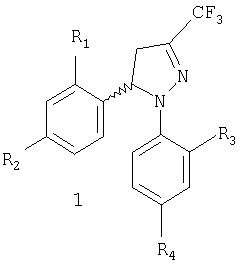
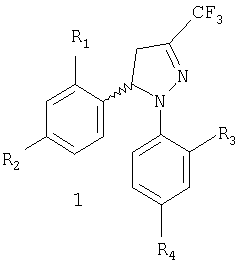
Настоящее изобретение относится к новому коммерчески пригодному способу получения соединений, имеющих общую формулу 1, которая включает рацемическую смесь (±)-1 и энантиомерно чистые соединения (-)-1 и (+)-1.
Уровень техники
Патент WO 9962884 описывает новые производные Δ2-пиразолинов, известные также как 4,5-дигидро-1H-пиразолы, которые ингибируют фермент циклооксигеназу-2 и являются пригодными для лечения человека и/или животных в качестве противовоспалительных агентов, а также других заболеваний, в протекание которых включена циклооксигеназа-2 и для которых характерна низкая или нулевая желудочная и почечная токсичность, так что они являются противовоспалительными агентами с более широким профилем безопасности. Из приведенного выше патента известен ряд рацемических смесей (±)-1 и энантиомерно чистых стереоизомеров (-)-1 и (+)-1, которые в настоящее время находятся на стадии клинического испытания. Вышеупомянутый патент описывает получение (±)-1 смеси при взаимодействии (E)-1,1,1-трифтор-4-арил-3-бутен-2-она с 4-(аминосульфонил)фенилгидразином или 4-(метилсульфонил)фенил-гидразином или при взаимодействии 4-(арил)фенилгидразина с (E)-1,1,1-трифтор-4-(4-аминосульфонилфенил)-3-бутен-2-оном или (Е)-1,1,1 -трифтор-4-(4-метилсульфонил-фенил)-3-бутен-2-оном. Он также описывает получение (-)-1 и (+)-1 стереоизомеров путем разделения рацемической смеси (±)-1 с помощью жидкостной хроматографии высокого разрешения с использованием колонки CHIRALPAK AS с размером частиц до 10 мкм и размерами 25×2 см (Daicel), подвижной фазой 0,1% диэтиламина в метаноле и скоростью потока 8 мл/минуту.
Кроме того, из уровня техники известны многочисленные способы разделения рацемических смесей, которые имеют широкое применение, смотри [а) монографию о свойствах рацематов и их разделении, Jacques, Collet, Wilen "Enantiomers Racemates и Resolutions", Wiley: New York, 1981; обзоры: b) Wilen, Top. Stereochem., 1971, 6, 107; с) Boyle, Q. Rev. Chem. Soc., 1971, 25; d) Buss, Vermeulen, Ind. Eng. Chem., 1968, 60, 12]. Однако в уровне техники имеется мало примеров, касающихся разделения Δ2-пиразолинов [Toda, J. Chem. Soc., Chem. Commun., 1995, 1453]. Эти работы раскрывают разделение Δ2-пиразолина путем образования комплекса включения. Работа [Mukai, Can. J. Chem., 1979, 57, 360-366] раскрывает выделение оптически активного Δ2-пиразолинов-бензолсульфата натрия из соответствующих рацематов путем использования в качестве агентов разделения цинконидина, (-)-α-метилбензиламина и бруцина, в зависимости от субстрата. Недостаток указанного способа заключается в необходимости проведения последующих перекристаллизации как в процессе образовании сульфоната натрия (от 3 до 7 перекристаллизации), так и в процессе образования и отделения смеси диастереоизомерных солей (от 4 до 7 перекристаллизации), что приводит к значительному снижению выхода продукта.
Раскрытие изобретения
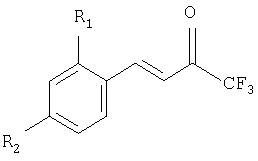
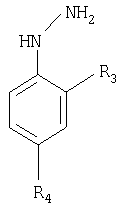
Авторы настоящего изобретения нашли путь, по которому можно получить соединения общей формулы 1, включающий использование производных бензальдегида, которые являются более дешевыми, чем 4-(аминосульфонил)бензальдегид или 4-(метилсульфонил)бензальдегид для получения (E)-1,1,1-трифтор-4-арил-3-бутен-2-она и производных фенилгидразина, которые являются более дешевыми, чем 4-(аминосульфонил)фенилгидразин или 4-(метилсульфонил)фенилгидразин. Эти енон и гидразин используют, чтобы получить кольцо Δ2-пиразолина, причем этот процесс, когда последовательно объединяют с процессом сульфирования и оптического разрешения, чтобы получить энантиомерно чистые соединения рацемической сульфокислоты, используя оптически активное основание или смесь оснований, в которой, по крайней мере, одно основание является оптически активным, приводит к образованию диастереоизомерных солей. Продолжение процесса заключается в отделении этих солей, преобразования их в соль натрия, образования хлорида кислоты и получения энантиомерно чистого сульфонамида или сульфона 1.
Осуществление изобретения
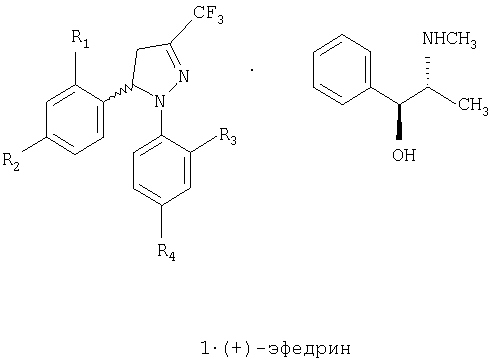
Задачей настоящего изобретения является обеспечение коммерчески пригодного способа получения соединений общей формулы 1, которая включает рацемическую смесь (±)-1 и энантиомерно чистые соединения (-)-1 и (+)-1, где
R1 и R3, одинаковые или различные, представляют собой атом водорода, хлора, фтора, метил, трифторметил или метоксигруппу,
R2 представляет собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси, метилсульфонил или аминосульфонильную группу,
R4 представляет собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси, метилсульфонил или аминосульфонильную группу,
при условии что один из заместителей R2 или R4 является метилсульфонильной или аминосульфонильной группой.
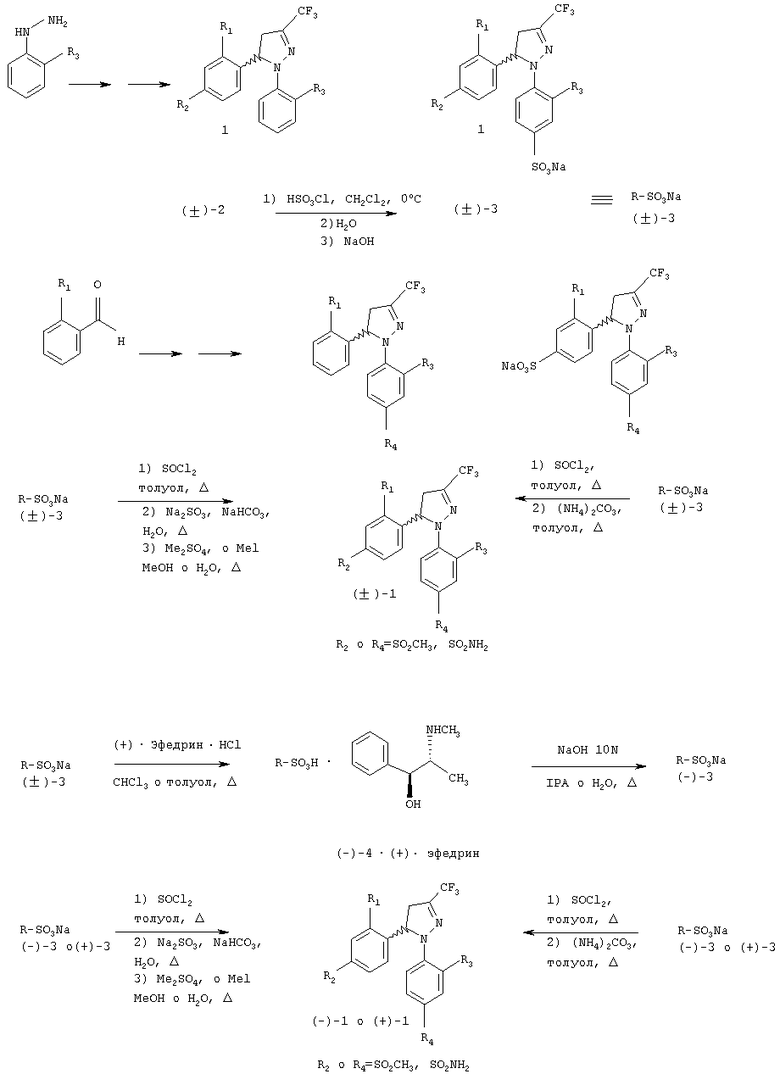
Настоящее изобретение раскрывает способ получения рацемической смеси (±)-1, которая является менее дорогой, чем одна из смесей ранее описанных в WO 9962884 и использования в нем фенилгидразина вместо 4-(аминосульфонил)фенилгидразина или 4-(метилсульфонил)фенилгидразина или бензальдегида вместо 4-(аминосульфонил)бенза-льдегида или 4-(метилсульфонила)бензальдегида, чтобы образовать кольцо Δ2-пиразолина в формуле 1 как соединение (±)-2. С помощью сульфирования получают хлорид кислоты, который подвергают взаимодействию с аммиаком или карбонатом аммония, чтобы получить сульфонамид (R2 или R4=SO2NH2) или с сульфитом натрия и сульфинатом натрия, полученным с метилсульфатом или метилиодидом, чтобы получить метилсульфон (R2 или R4=SO3СН3), (±)-1. Также можно выделить соответствующую соль натрия: с помощью сульфирования и обработки гидроксидом натрия с получением соли (±)-3, которая взаимодействует с тионилхлоридом и полученный хлорид кислоты взаимодействует с аммиаком или карбонатом аммония, с образованием сульфонамида (R2 или R4=SO2NH2) или вместо этого с сульфитом натрия и полученный сульфинат натрия с метилсульфатом или метилиодидом, чтобы получить метилсульфон (R2 или R4=SO2СН3), (±)-1.
Кроме того, обеспечивается промышленное применение способа получения энантиомерно чистых стереоизомеров (+)-1 и (-)-1. Одна пара энантиомеров может быть разделена с помощью различных способов, а именно, с превращением их в диастереоизомерные соли, с последующим разделением путем фракционной кристаллизации, которая обычно используется. Когда диастереоизомерные соли получены и отделены, энантиомеры (кислоты или основания) могут быть легко выделены и хиральная кислота или основание восстановлены, так что этот простой и недорогой способ широко используют для промышленного применения. Если рацемическое соединение содержит в своей структуре аминогруппу, то возможно образовывать диастереоизомерные соли с оптически активной кислотой, а если рацемическое соединение содержит кислотную группу, то возможно образовывать диастереоизомерные соли с оптически активным основанием. Поскольку соединение 1 лишено кислотной и основной группы, достаточно сильных для образования диастереоизомерных солей, настоящее изобретение предлагает способ, приведенный ниже на схеме 1, для получения рацемической смеси (±)-1 и энантиомерно чистых соединений (-)-1 и (+)-1.
Схема 1
Способ, предлагаемый в настоящем изобретении, описан в виде схем, приведенных ниже для двух конкретных примеров: первый (Схема 2) для получения энантиомерно чистого соединения (-)-8.
Схема 2
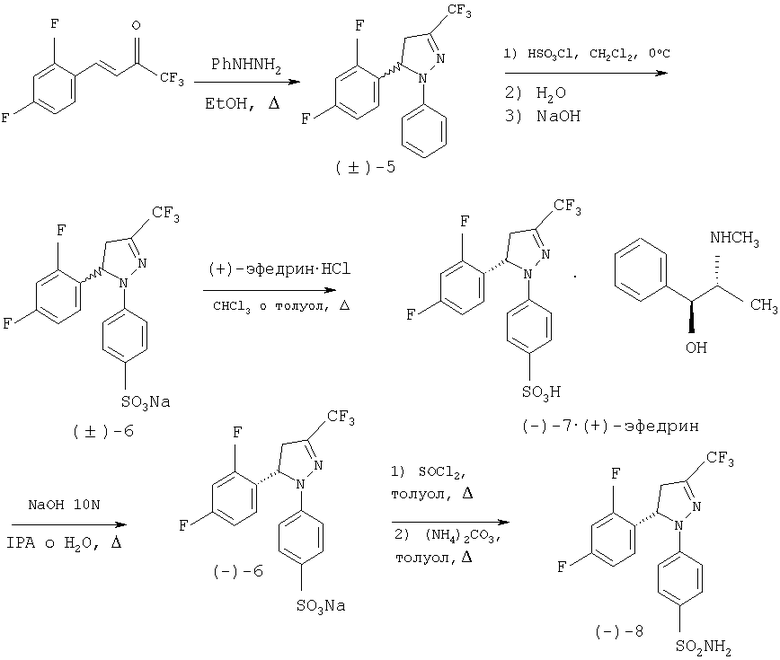 Соединение (-)-8 синтезируют, в соответствии с настоящем изобретением, с помощью способа, описанного ниже со схематическим определением ряда предпочтительных условий. Третья стадия представляет собой разделение рацемической смеси (±)-6 на два энантиомера, с использованием в качестве агента разрешения (+)-эфедрина. Эфедрин является отличным разделяющим агентом, поскольку оба энантиомера, полученных при разделении, являются доступными с высокой энантиомерной концентрацией, имеют коммерческую ценность и легко превращаются и кристаллизуются. Соединение (+)-8 получают с помощью тех же самых стадий синтеза, как в предыдущем случае, лишь заменяя на стадии разделения (стадия 3) эфедриновый энатиомер. Синтез рацемического соединения (±)-8 осуществляют тем же самьм способом, исключая стадии, связанные с разделением, то есть получают при непосредственном взаимодействии хлорида кислоты с аммиаком или карбонатом аммония.
Соединение (-)-8 синтезируют, в соответствии с настоящем изобретением, с помощью способа, описанного ниже со схематическим определением ряда предпочтительных условий. Третья стадия представляет собой разделение рацемической смеси (±)-6 на два энантиомера, с использованием в качестве агента разрешения (+)-эфедрина. Эфедрин является отличным разделяющим агентом, поскольку оба энантиомера, полученных при разделении, являются доступными с высокой энантиомерной концентрацией, имеют коммерческую ценность и легко превращаются и кристаллизуются. Соединение (+)-8 получают с помощью тех же самых стадий синтеза, как в предыдущем случае, лишь заменяя на стадии разделения (стадия 3) эфедриновый энатиомер. Синтез рацемического соединения (±)-8 осуществляют тем же самьм способом, исключая стадии, связанные с разделением, то есть получают при непосредственном взаимодействии хлорида кислоты с аммиаком или карбонатом аммония.
Первая стадия включает получение пиразолина (±)-5 из (Е)-1,1,1-трифтор-4-(2,4-дифторфенил)-3-бутен-2-она и фенилгидразина в подходящем растворителе, например в спиртах, таких как метанол, этанол или изопропанол или в отсутствии растворителя. Реакция протекает в кислой среде, которая может быть органической, такой как уксусная кислота или п-толуолсульфоновая кислота, или неорганической, такой как соляная кислота, или смесью обеих кислот или вместо этого в щелочной среде, такой как в пиперидине, пиперазине, гидроксиде натрия, гидроксиде калия, метилате натрия или этилате натрия, или в их смеси. Та же самая кислая или щелочная среда может действовать как растворитель. Наиболее подходящим температурным интервалом является интервал от температуры окружающей среды до 150°С и время реакции находится между 2 и 48 часами. Очистку пиразолина (±)-5 проводят с помощью кристаллизации.
На второй стадии проводят сульфирование пиразолина (±)-5 с помощью хлорсульфоновой кислоты без растворителя или в растворителе, используя хлорированный растворитель, такой как дихлорметан при температурах, находящихся в интервале между 0°С и температурой кипения растворителя, обеспечивая образование соответствующей сульфокислоты после водной обработки. Добавление гидроксида натрия приводит к осаждению сульфоната натрия (±)-6.
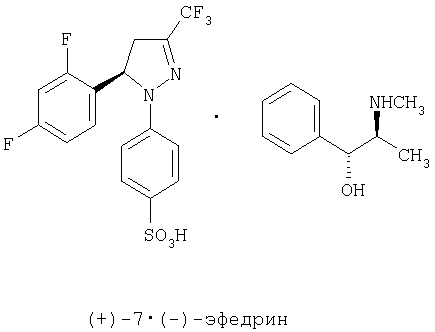
На третьей стадии рацемическую смесь (±)-6 разделяют на два энантиомера путем образования смеси двух диастереоизомерных солей и последующего отделения одного из этих осадков в той же самой среде взаимодействия. Способ, который является объектом по настоящему изобретению, не имеет вышеупомянутых недостатков при разделении аналогичного продукта, как описано у Mukai et al. [Can. J. Chem. 1979 57, 360-366], при этом разделение двух диастереоизомерных солей проводят в той же самой среде взаимодействия во время процесса образования смеси диастереоизомерных солей, благодаря чему требуется единственная кристаллизация. Применяемый разделяющий агент является (+)-эфедрином, который при взаимодействии рацемической смеси (±)-6 с хлоргидратом (+)-эфедрина в хлорированном растворителе, таком как хлороформ, и при температурах, колеблющихся между температурой окружающей среды и температурой кипения, обеспечивает получение смеси диастереоизомерных солей и в процессе охлаждения приводит к осаждению только энатиомера (-)-7 в форме соли (+)-эфедрина с энантиомерной чистотой выше 98%. Возможно получить из маточного раствора диастероизомерную соль (+)-7 и (+)-эфедрина путем упаривания растворителя и последующей перекристаллизации из спирта, такого как изопропиловый спирт, или смеси спирта и воды. Кроме того, с помощью того же самого способа на стадии 3 схемы, но используя хлоргидрат (-)-эфедрина, диастереоизомерную соль, образованную из (+)-7 и (-)-эфедрина, получают путем осаждения и из маточного раствора может быть получена диатероизомерная соль (-)-7 и (-)-эфедрина путем упаривания растворителя и последующей перекристаллизации из спирта, такого как изопропиловый спирт, или смеси спирта и воды.
На четвертой стадии, показанной в схеме, сульфонат натрия (-)-6 выделяют в энантиомерно чистом виде путем основного гидролиза соли (-)-7-(+)-эфедрина с водным раствором гидроксида натрия и используя спирт, такой как изопропанол, как растворитель. Из маточного раствора легко выделить эфедрин путем удаления растворителя и подкисления остатка, растворенного в этаноле соляной кислотой в спирте. Энатиомер (+)-6 получают теми же самыми способами из соли (+)-7-(+)-эфедрина или (+)-7-(-)-эфедрина.

Пятая и последняя стадия, показанная на схеме, отражает получение стереоизомера (-)-8 с энантиомерной чистотой выше 98% при взаимодействии оптически активного натрий сульфоната(-)-6 с тионилхлоридом в отсутствии растворителя или в подходящем растворителе, таком как толуол, при температурах между температурой окружающей среды и температурой кипения, с последующим образованием сульфонамида при добавлении аммиака или карбоната аммония к реакционной среде. Тем же самым способом энатиомер (+)-8 может быть получен из (+)-6. Исключая стадии, связанные с разделением, возможно получить рацемическое соединение (±)-8.
Схема 3 показывает другой конкретный пример получения соединений, являющихся объектом изобретения: получение (-)-13.
Схема 3
 Соединение (-)-13 синтезируют, в соответствии с настоящем изобретением, с помощью способа, описанного ниже, предпочтительно в условиях, показанных на схеме. Третья стадия включает разделение рацемической смеси (±)-11 на два энантиомера путем образования смеси диастереоизомерных солей, используя в качестве агента разделения (+)-эфедрина, чтобы получить энатиомер (-)-13. Соединение (+)-13 получают с помощью того же синтеза, как и в предыдущем примере, заменяя лишь энатиомер эфедрина на стадии разделения (стадия 3). Синтез рацемического соединения (±)-13 осуществляют тем же способом, исключая процесс образования соли эфедрина и ее последующего гидролиза.
Соединение (-)-13 синтезируют, в соответствии с настоящем изобретением, с помощью способа, описанного ниже, предпочтительно в условиях, показанных на схеме. Третья стадия включает разделение рацемической смеси (±)-11 на два энантиомера путем образования смеси диастереоизомерных солей, используя в качестве агента разделения (+)-эфедрина, чтобы получить энатиомер (-)-13. Соединение (+)-13 получают с помощью того же синтеза, как и в предыдущем примере, заменяя лишь энатиомер эфедрина на стадии разделения (стадия 3). Синтез рацемического соединения (±)-13 осуществляют тем же способом, исключая процесс образования соли эфедрина и ее последующего гидролиза.
Первая стадия состоит из получения пиразолина (±)-10 из (Е)-1,1,1-трифтор-5-фенил-3-бутен-2-она и хлоргидрата 2,4-дифторфенилгидразина в подходящем растворителе, таком как спирты, такие как этанол, или в отсутствии растворителя. Реакция проходит в кислой среде, такой как уксусная кислота или п-толуолсульфокислота. Наиболее подходящие температуры лежат в интервале между температурой окружающей среды и 110°С и время взаимодействия - между 2 и 24 часами. Очистку пиразолин (±)-10 выполняют путем кристаллизации.
На второй стадии сульфирование пиразолина (±)-10 хлорсульфоновой кислотой проводят без растворителя или с хлорированным растворителем, таким как дихлорметан, при температурах между 0°С и точкой кипения растворителя, с получением соответствующей сульфоновой кислоты после водной обработки. Добавление гидроксида натрия высаживает сульфонат натрия (±)-11.
На третьей стадии рацемическую смесь (±)-11 разделяют на два энантиомера путем образования смеси двух диастереоизомерных солей и последующего отделения одной из них путем осаждения из той же реакционной среды, после чего требуется одна кристаллизация. Смесь диастереоизомерных солей получают путем взаимодействия рацемической смеси (±)-11 с хлоргидратом (+)-эфедрина в подходящем растворителе, таком как толуол, при температурах, лежащих в интервале между температурой окружающей среды и температурой кипения. В процессе охлаждения осаждается только энатиомер (-)-12 в виде соли с (+)-эфедрином с энантиомерной чистотой 84%. Из маточного раствора может быть получена диастереоизомерная соль (+)-12 и (+)-эфедрина. Кроме того, посредством той же самой стадии 3 схемы, но используя хлоргидрат (-)-эфедрина, получают путем осаждения диастереоизомерную соль, образованную из (+)-12 и (-)-эфедрина и из маточного раствора получают диастереоизомерную соль (-)-12 и (-)-эфедрина.
На четвертой стадии, приведенной на схеме, сульфонат натрия (-)-11 получают энантиомерно чистым путем щелочного гидролиза соли (-)-12•(+) эфедрина с водным раствором гидроксида натрия, используя воду как растворитель. Из маточного раствора легко получить эфедрин, как описано выше, подкисляя остаток, растворенный в этаноле с помощью соляной кислоты в этаноле. Получение энатиомера (+)-11 осуществляют тем же самом способом из соли (+)-12•(+)-эфедрина или (+)-12•(-)-эфедрина.
На пятой и последней стадии, показанной на схеме, стереоизомер (-)-13 получают путем взаимодействия оптически активного сульфоната натрия (-)-11 с тионилхлоридом в отсутствии растворителя или в подходящем растворителе, таком как толуол, при температурах, лежащих в интервале между температурой окружающей среды и температурой кипения, с последующим образованием сульфината натрия при взаимодействии хлорида кислоты с сульфитом натрия в основной водной среде и, наконец, при взаимодействии сульфината натрия, полученного с метилиодидом или метилсульфатом в спиртовой или водной среде. Аналогично получают энатиомер (+)-13 из (+)-11. Удаляя стадии, связанные с разделением, получают рацемическое соединение (±)-13.
Способ разделения, являющийся объектом по настоящему изобретению, может быть использован или для рацемических смесей (такие, в которых два энантиомера присутствует в соотношении 1:1) или для не рацемических смесей, в которых один из энантиомеров является доминирующим, полученным с помощью любого физического или химического способа.
Ниже приведены основные примеры, в соответствии с которыми проводят способ получения некоторых из соединений, к которым относится настоящее изобретение. Эти примеры показаны только с целью иллюстрации и не должны рассматриваются с точки зрения ограничения рамок изобретения.
Пример 1: Получение (-)-4-[5-(2,4-дифторфенил)-4,5-дигидро-3-(трифторметил)-1H-пиразол-1-ил]-бензолсульфонамида, (-)-8
Получение (±)-1-фенил-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразола, (±)-5
В стакан, объемом 50 мл помещают (Е)-1,1,1 -трифтор-4-(2,4-дифторфенил)-3-бутен-2-он (2,66 г, 11,2 ммоль), моногидрат п-толуолсульфокислоты (2,1 г, 11,2 ммоль) и хлоргидрат фенилгидразина (1,33 г, 12,3 ммоль) и нагревают до температуры 110°С. Небольшое количество этилового спирта может быть использовано для растворения исходной смеси. Приблизительно через 2 часа (контроль с помощью CCF) смеси дают возможность остыть и ее разбавляют этилацетатом. Затем ее промывают насыщенным раствором NaHCO3, сушат MgSO4, фильтруют и растворитель упаривают при низком давлении. Полученный затем сырой продукт (3,9 г) перекристаллизовывают из метанола (2 мл), чтобы осадить 3,67 г (65%) пиразолина (±)-5: т.пл.=83-84°С; ИК (KBr) μ max (см-1) 1600, 1505, 1326; 1Н-ЯМР (CDCl3) δ (частей на млн.): 7,28-6,72 (м, 8Н), 5,64 (дд, J-13 Гц, J=7,5 Гц, 1Н), 3,8-3,6 (м., 1Н), 2,94 (дд, J=17,2 Гц, J=7 Гц, 1H).
Получение (±)-4-[5-(2,4-дифторфенил)-4,5-дигидро-3-(трифторметил)-1H-пиразол-1-ил]-натрий бензолсульфоната, (±)-6
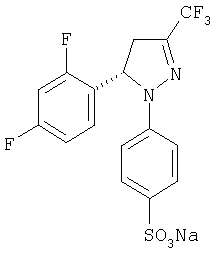
В стакане объемом 100 мл пиразолин (±)-5 (4,0 г, 12,27 ммоль) растворяют в дихлорметане (12 мл). Смесь охлаждают при температуре 0°С, после чего добавляют по капле хлорсульфоновую кислоту (0,82 мл, 12,27 ммоль). Перемешивание и температуру поддерживают в течение 20 минут, после чего к реакционной смеси медленно добавляют воду (20 мл) при температуре 4°С, перемешивают состав в течение 14 часов при температуре окружающей среды. Две фазы отделяют и водную фазу промывают дихлорметаном (5 мл). Водную фазу концентрируют до двух третей от исходного объема, после чего к ней добавляют при перемешивании водный раствор гидроксида натрия 1М (12,27 мл, 12,27 ммоль). Полученный осадок белого цвета, который соответствует натрий сульфонату(±)-6, фильтруют, промывают большим количеством воды и сушат (3,93 г, 75% выход): т.пл.=292-295°С; ИК (KBr) μ max (см-1) 3430, 1600, 1570, 1425; 1Н-ЯМР (CDCl3/CD3OD:10/1) δ (частей на млн.): 7,6 (д, J=8,8 Гц, 2Н), 7,1-6,7 (м, 3Н), 6,9 (д, J=8,8 Гц, 2Н), 5,69 (дд, J=12,6 Гц, J= 6,3 Гц, 1Н).
Получение (-)-4-[5-(2,4-дифторфенил)-4,5-дигидро-3-(трифторметил)-1Н-пиразол-1-ил]-бензолсульфоната и (+)-эфедрина, (-)-7-(+)-эфедрин.
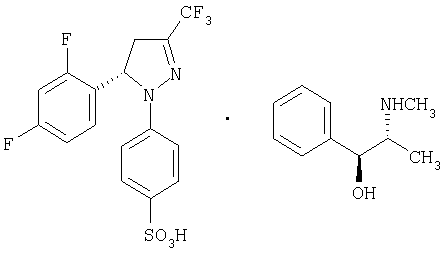
В стакан объемом 2 л помещают сульфонат натрия (±)-6 (3,95 г, 9,23 ммоль), (+)-хлоргидрат эфедрина (1,86 г, 9,23 ммоль) и хлороформ (79 мл). Смесь встряхивают и нагревают до температуры кипения в течение 10 минут. Смеси дают возможность медленно остыть до температуры окружающей среды, при этом осаждается твердое вещество (2,49 г) в виде смеси соли (-)-7•(+)-эфедрина (энантиомерный избыток выше 98%) и хлорида натрия, который образуется в ходе процесса. Этот образец используют непосредственно в следующей реакции. Если образец растворяют с небольшим количеством AcOEt, его промывают водой, сушат над MgSO4 и растворитель упаривают, то получают чистую фракцию соли (-)-7-(+)-эфедрина: [α]20D=-94,6 (с=2, МеОН); ИК (KBr) μ max (см-1): 3410, 3040, 2860, 2780, 1595, 1570, 1500, 1420; 1Н-ЯМР (CDCl3/CD3OD:10/1) 5 (частей на млн.): 7,7 (д, J=9 Гц, 2Н), 7,4-7,2 (м., 5Н), 7,1-6,7 (м., 3Н), 6,95 (д, J=9 Гц, 2Н), 5,65 (дд, J=12,5 Гц, J=6,5 Гц, 1Н), 5,3 (д, J=2,2 Гц, 1Н), 3,9-3,6 (м., 1Н), 3,4-3,1 (м., 1Н), 3,0 (дд, J=18,4 Гц, J=5,8 Гц, 1Н), 2,76 (с, 3Н), 1,9 (широкая полоса частот, 1H), 1,0 (д, J=6 Гц, 3Н).
Получение (-)-4-[5-(2,4-дифторфенил)-4,5-дигидро-3-(трифторметил)-1 Н-пиразол-1-ил]-натрия бензолсульфоната, (-)-6
В стакан объемом 50 мл помещают изопропиловый спирт (50 мл) и смесь соли (-)-7•(+)-эфедрина (энантиомерный избыток выше 98%) и хлорид натрия (2,49 г). Полученную суспензию встряхивают и к ней добавляют гидроксид натрия 10М (0,4 мл). Раствор нагревают до температуры кипения и 10 минут спустя ей дают возможность медленно остыть до температуры окружающей среды. Полученный осадок, который еще раз фильтруют, промывают изопропиловьм спиртом и сушат, соответствует смеси натрий сульфоната(-)-6 и хлорида натрия (1,86 г), который непосредственно используют для получения (-)-8. Для того, чтобы определить оптическое вращение соединения (-)-6 возможно очистить часть образца с помощью промывания водой: [α]20D=-170,1 (с=1, МеОН).
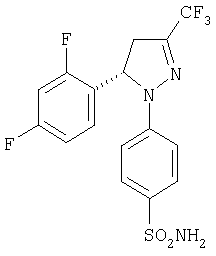
Получение (-)-4-[5-(2,4-дифторфенил)-4,5-дигидро-3-(трифторметил)-1Н-пиразол-1-ил]-бензолсульфонамида, (-)-8.
В стакан, объемом 1 л, помещают образец (72 г) смеси сульфоната натрия (-)-6 (48,3 г, 112,8 ммоль) и NaCl (23,7 г) в толуоле (250 мл). Суспензию нагревают до температуры 60°С, добавляют тионилхлорид (18 мл, 247,5 ммоль) и нагревают до температуры кипения, после чего оставляют при этой температуре, по крайней мере, на 2 часа. После образования хлорида кислоты реакция заканчивается, избыток тионилхлорида отделяют с помощью азеотропной отгонки с толуолом (190 мл; 76°С при 60 мм рт.ст.). Затем добавляют еще толуол (190 мл) и снова перегоняют в тех же самых условиях.
Для получения сульфонамида предыдущий образец разбавляют толуолом (190 мл), смесь охлаждают до температуры 70°С, добавляют твердый карбонат аммония (22,6 г, 235 ммоль), нагревают до температуры 90°С и встряхивают при той же самой температуре в течение 2 часов. Когда реакции заканчивается (если необходимо, то вводят дополнительное количество карбоната аммония), добавляют воду (300 мл) и смесь выдерживают в течение 30 минут при температуре 90°С. Затем смесь охлаждают до температуры окружающей среды и добавляют водный раствор 17,5% HCl до тех пор, пока значение рН не станет равным 6-7 и оставляют перемешиваться в течение еще 10 минут. Осажденное твердое вещество фильтруют, промывают толуолом и сушат, чтобы получить сульфонамид (-)-8 (38,4 г, 84% выход). Продукт может быть перекристаллизован из смеси изопропилового спирта и воды (60:40), получая, как указано выше 99%: т.пл.=173-174°С; [α]20D=-192,8 (с=1, МеОН); ИК (KBr) μ max (см-1): 3310, 3230, 1600, 1500, 1430; 1Н-ЯМР (CDCl3) 6 (частей на млн.): 7,76 (д, J=9 Гц, 2Н), 7,04 (д, J=9 Гц, 2Н), 7,1-6,75 (м., 3Н), 5,71 (дц, J=12,4 Гц, J=6,2 Гц, 1H), 4,74 (с, 2Н), 3,9-3,7 (м., 1Н), 3,03 (дд, J=19,8 Гц, J=6,2 Гц, 1H).
Пример 2: Получение (-)-1-(2,4-дифторфенил)-4,5-дигидро-5-(4-метилсульфонилфенил)-3-(трифторметил)-1H-пиразола, (-)-13
Получение (±)-1-(2,4-дифторфенил)-4,5-дигидро-5-фенил-3-(трифторметил)-1Н-пиразола, (±)-10
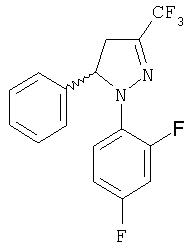
В стакан объемом 50 мл помещают (Е)-1,1,1-трифтор-5-фенил-3-бутен-2-она (3,04 г, 15,2 ммоль), моногидрат п-толуолсульфокислоты (2,9 г, 15,2 ммоль) и хлоргидрат 2,4-дифторфенилгидразина (3,01 г, 16,7 ммоль), после чего нагревают до температуры 110°С. Может быть использовано небольшое количество этилового спирта для растворения исходной смеси. После приблизительно 2 часов (контролирование с помощью CCF, хроматография на силуфоле), смеси дают возможность остыть и ее разбавляют этилацетатом. Затем ее промывают насыщенным раствором NaHCO3, сушат над MgSO4, фильтруют и растворитель упаривают при низком давлении. Полученный сырой продукт повторно кристаллизуют из изопропилового спирта (1 г/1 мл), осаждая 3,95 г (80%) пиразолина (±)-10: т.пл.=52-54°С; ИК (KBr) μ max (см-1) 1598, 1511, 1414, 1324; 1Н-ЯМР (CDCl3) δ (частей на млн.): 7,4-6,6 (м., 8Н), 5,7-5,4 (м., 1Н), 3,8-3,5 (м., 1Н), 3,3-3.0 (м., 1Н).
Получение (±)-4-[1-(2,4-дифторфенил)-4,5-дигидро-3-(трифторметил)-1Н-пиразол-5-ил]-натрий бензолсульфоната, (±)-11
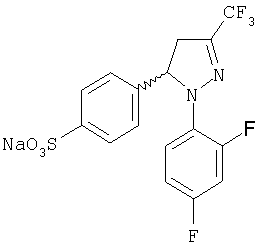
В стакане объемом 100 мл растворяют пиразолин (±)-10 (3,0 г, 9,2 ммоль) в дихлорметане (1,5 мл). Смесь охлаждают до температуры 0°С и в нее по каплям добавляют хлорсульфоновую кислоту (6,1 мл, 92 ммоль). Подсоединяют хладагент, при этом температура повышается до 50°С. Встряхивание и температуру поддерживают в течение 5 часов (контролирование с помощью CCF), затем смеси дают возможность остыть и ее разбавляют дихлорметаном (90 мл), в течение этого времени к реакционной массе медленно добавляют воду (90 мл) при температуре 4°С. Две фазы отделяют и проводят две экстракции водной фазы дихлорметаном (25 мл). Органическую фазу сушат над MgSO4, фильтруют и растворитель упаривают при низком давлении. Полученный таким образом сырой продукт (3,6 г) помещают в стакан объемом 25 мл, подсоединяют к хладагенту и к нему добавляют воду (13,4 мл). Суспензию нагревают до температуры 70°С и в нее медленно добавляют, перемешивая, водный раствор гидроксида натрия 10М (1,7 мл, 17,04 ммоль). Смесь нагревают до температуры кипения и оставляют при этой температуре на 10 минут. Смеси дают возможность медленно остыть до температуры окружающей среды, при этом осаждается твердое вещество белого цвета, которое соответствует натрий сульфонату (±)-11, который фильтруют, промывают большим количеством воды и сушат (3,0 г, 82% выход): т.пл.=271-273°С; ИК (KBr) μ max (см-1) 3477, 1617, 1513, 1416; 1Н-ЯМР (CDCl3) δ (частей на млн.): 7,59 (д, J=8,5 Гц, 2Н), 7,3-7,0 (м., 3Н), 6,95 (д, J=8,5 Гц, 2Н), 5,4 (м., 1Н), 3,5 (м, 1Н), 2,9 (м, 1Н).
Получение (-)-4-[1-(2,4-дифторфенил)-4,5-дигидро-3-(трифторметил)-1Н-пиразол-5-ил]-бензолсульфонат (+)-эфедрина, (-)-12•(+)-эфедрин.
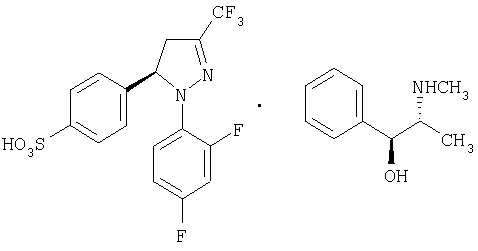
В стакан объемом 50 мл помещают сульфонат натрия (±)-11 (2,45 г, 5,72 ммоль), хлоргидрат (+)-эфедрина (1,15 г, 5,72 ммоль) и толуол (24,5 мл). Смесь встряхивают и нагревают до температуры кипения в течение 10 минут. Затем ей дают возможность медленно остыть до температуры окружающей среды, при этом осаждается твердое вещество, которое фильтруют и промывают дополнительным количеством толуола. Это обеспечивает получение 1,18 г, образуемой в процессе, смеси соли (-)-12•(+)-эфедрина (энантиомерный избыток 84%) и хлорида натрия. Полученный образец непосредственно используют на следующей реакции; ИК (KBr) μ max (см-1) 3377, 3031, 1603, 1515, 1399; 1Н-ЯМР (CDCl3/CD3OD:10/1) 5 (частей на млн.): 7,76 (д, J=8 Гц, 2Н), 7,4-7,2 (м., 6Н), 7,19 (д, J=8 Гц, 2Н), 6,75 (м., 2Н), 5.6 (м., 1Н), 5,35 (с, 1Н), 3,65 (м., 1Н), 3,3 (м., 1Н), 3,15-3,0 (м., 1H), 2,76 (с, 3Н), 2,65 (м., 2Н), 1,07 (д, J=7 Гц, 3Н).
Получение (-)-4-[1-(2,4-дифторфенил)-4,5-дигидро-3-(трифторметил)-1H-пиразол-5-ил]-натрий бензолсульфоната, (-)-11
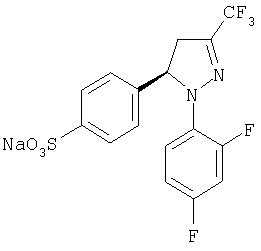
В стакан объемом 10 мл помещают воду (2,8 мл) и смесь (1 г) соли (-)-7•(+)-эфедрина и хлорида натрия (28% от общего веса). Полученную суспензию встряхивают и добавляют к ней гидроксид натрия 10М (0,3 мл). Раствор нагревают до температуры кипения и 10 минут спустя ему дают возможность медленно остыть до температуры окружающей среды. Полученный остаток отфильтровывают, промывают водой и сушат, он соответствует натрий сульфонату(-)-11 (0,34 г), который используют непосредственно для получения соединения (-)-13: [μ]20D=-104,3 (с=1, МеОН).
Получение (-)-1-(2,4-дифторфенил)-4,5-дигидро-5-(4-метилсульфонилфенил)-3-(трифторметил)-1H-пиразол, (-)-13.
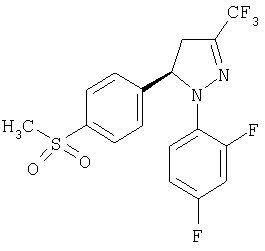
В стакане объемом 10 мл растворяют соединение (-)-11 (230 мг, 0,54 ммоль) в толуоле (1,1 мл). Суспензию нагревают до температуры 60°С, добавляют тионилхлорид (88 мкл, 1,18 ммоль) и смесь оставляют при указанной температуре в течение, по крайней мере, 2 часов. В конце реакции образования хлорида кислоты избыток тионилхлорида отделяют с помощью азеотропной перегонки с толуолом (76°С при 60 мм рт.ст.). Вводят дополнительное количество толуола (1 мл) и реакционную массу снова перегоняют при тех же самых условиях. В сырой продукт, полученный таким образом, добавляют воду (1,15 мл), NaHCO3 (95 мг, 1,13 ммоль) и Na2SO3 (124 мг, 0,97 ммоль), нагревая до температуры 75°С. Реакционную массу оставляют при этой температуре в течение 2 часов и затем ей дают возможность остыть до комнатной температуры. Растворитель упаривают при низком давлении и в сырой продукт добавляют метиловый спирт (14 мл). После часа кипения с обратным холодильником горячую реакционную массу фильтруют и растворитель упаривают при пониженном давлении. Твердое вещество, полученное таким образом (297 мг), растворяют в метиловом спирте (2,8 мл) и к нему добавляют метилиодид (44 мкл, 0,7 ммоль). Полученное вещество нагревают до температуры 55°С и оставляют при этой температуре в течение 16 часов. Растворитель выпаривают при низком давлении, получая на выходе 168 мг (77%) сырого продукта. Этот продукт может быть перекристаллизован из смеси толуола и циклогексана: т.пл.=86-9°; [α]20D=-86,1 (c=1, СН3ОН); ИК (KBr) μ max (см-1): 1598, 1513, 1416; 1Н-ЯМР (CDCl3) δ (частей на млн.): 7,87 (д, J=8,4 Гц, 2Н), 7,5-7,2 (м., 3Н), 6,9-6,6 (м., 2Н), 5,7 (дд, J=6,5 Гц, J=2,6 Гц, 1Н), 3,8-3,6 (м., 1Н), 3,2-2,9 (м., 1Н), 3,02 (с, 3Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРАЗОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1999 |
|
RU2233272C2 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ ПИРАЗОЛИНА ПРИ ПОЛУЧЕНИИ ЛЕЧЕБНОГО СРЕДСТВА ДЛЯ ПРЕДУПРЕЖДЕНИЯ И/ИЛИ ЛЕЧЕНИЯ БОЛЕЗНЕЙ, СВЯЗАННЫХ С ПРОЛИФЕРАЦИЕЙ КЛЕТОК | 2002 |
|
RU2305545C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ АЛЬФА-АМИНОАЦЕТАЛЕЙ | 2009 |
|
RU2507194C2 |
| СТЕРЕОИЗОМЕРНО ОБОГАЩЕННЫЕ 3-АМИНОКАРБОНИЛЬНЫЕ БИЦИКЛОГЕПТЕНОВЫЕ ПИРИМИДИНДИАМИНЫ И ИХ ПРИМЕНЕНИЯ | 2005 |
|
RU2416604C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА | 2020 |
|
RU2836442C2 |
| ЗАМЕЩЕННЫЕ АРИЛПИРАЗОЛЫ В КАЧЕСТВЕ АГЕНТОВ ДЛЯ УНИЧТОЖЕНИЯ ПАРАЗИТОВ | 2006 |
|
RU2381218C2 |
| БЕНЗОЛСУЛЬФАНОМИДНЫЕ CОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, КОТОРЫЕ ВОСПРИИМЧИВЫ К МОДУЛЯЦИИ ДОФАМИНОВОГО РЕЦЕПТОРА D | 2008 |
|
RU2485103C2 |
| ПИРРОЛОПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ | 2001 |
|
RU2254335C2 |
| КОНФОРМАЦИОННО ОГРАНИЧЕННЫЕ ИНГИБИТОРЫ PI3K И mTOR | 2014 |
|
RU2669696C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2315758C2 |
Предлагается способ получения соединений общей формулы 1, который включает рацемические смеси (±)-1, и энантиомерно чистые соединения (-)-1 и (+)-1, где R1 и R3, одинаковые или различные, представляют собой атом водорода, хлора, фтора, метил, трифторметил или метоксигруппу; R2 представляет собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси, метилсульфонил или аминосульфонильную группу; R4 представляет собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси, метилсульфонил или аминосульфонильную группу, при условии, что один из заместителей R2 или R4 является метилсульфонилом или аминосульфонильной группой; который включает получение рацемической смеси соединений общей формулы (±)-1 при взаимодействии (Е)-1,1,1-трифтор-4-арил-3-бутен-2-она с фенилгидразином, с последующей обработкой хлорсульфокислотой или при взаимодействии с хлорсульфокислотой, с последующей реакцией с гидроксидом натрия и, наконец, с тионилхлоридом. Продукт, полученный с помощью любого из этих способов, подвергают взаимодействию с карбонатом аммония или аммиаком или с сульфитом натрия и метилиодидом или метилсульфонатом. Кроме того, чтобы получить энатиомерно чистые соединения общей формулы 1 при разделении рацемической смеси общей формулы (±)-1, реакцию осуществляют с оптически активным эфедрином, что приводит к образованию натриевой соли каждого из энатиомеров, реакции с тионилхлоридом и карбонатом аммония или аммиаком или вместо тионилхлорида с сульфитом натрия и метилиодидом или метилсульфонатом, чтобы, таким образом, получить отдельно энантиомерно чистые соединения общей формулы (-)-1 и (+)-1. Технический результат - разработка альтернативного коммерчески пригодного способа получения соединений формулы 1 за счет использования более дешевых и доступных исходных реагентов. 5 з.п. ф-лы.
где R1 и R2, одинаковые или различные, представляют собой атом водорода, хлора, фтора, метил, трифторметил или метоксигруппу,
R2 представляет собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси, метилсульфонил или аминосульфонильную группу,
R4 представляет собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси, метилсульфонил или аминосульфонильную группу,
при условии, что один из заместителей R2 или R4 является метилсульфонильной или аминосульфонильной группой;
включающий получение рацемической смеси общей формулы (±)-1 путем взаимодействия (Е)-1,1,1-трифтор-4-арил-3-бутен-2-она с фенилгидразином, где R1 и R3, одинаковые или различные, представляют собой атом водорода, хлора, фтора, метил, трифторметил или метоксигруппу и R3 и R4 представляют собой атом водорода, хлора, фтора, метил, трифторметил, метокси или трифторметоксигруппу, при условии, что по крайней мере, один из заместителей R2 или R4 представляет собой атом водорода
с образованием пиразолина общей формулы (±)-1, где R1 и R3, одинаковые или различные, представляют собой атом водорода, хлора, фтора, метил, трифторметил или метоксигруппу, и R2 и R4 представляют собой атом водорода, хлора, фтора, метил, трифторметил, метокси или трифторметоксигруппу, при условии, что, по крайней мере, один из заместителей R2 или R4 представляет собой атом водорода;
который также включает взаимодействие полученного пиразолина с хлорсульфоновой кислотой с образованием пиразолина общей формулы (±)-1, где R1 и R3, одинаковые или различные, представляют собой атом водорода, хлора, фтора, метила, трифторметил или метоксигруппу и R2 и R4 представляют собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси или сульфонилхлоридную группу, при условии, что один из заместителей R2 или R4 представляет собой сульфонилхлоридную группу (SO2Cl); или с помощью реакции с хлорсульфоновой кислотой и последующей реакции с гидроксидом натрия с образованием пиразолина общей формулы (±)-1, где R1 и R3, одинаковые или различные, представляют собой атом водорода, хлора, фтора, метил, трифторметил или метоксигруппу, и R2 и R4 представляют собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси или натрийсульфонатную группу, при условии, что один из заместителей R2 или R4 представляет натрийсульфонатную группу (SO3Na);
который также включает взаимодействие последнего пиразолина с тионилхлоридом с образованием пиразолина общей формулы (±)-1, где R1 и R3, одинаковые или различные, представляют собой атом водорода, хлора, фтора, метил, трифторметил или метоксигруппу и R2 и R4 представляют собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси или сульфонилхлорид, при условии, что один из заместителей R2 или R4 представляет собой сульфонилхлоридную группу (SO2Cl);
который также включает взаимодействие последнего пиразолина с карбонатом аммония или аммиаком или с сульфитом натрия и метилиодидом или метилсульфатом с образованием рацемической смеси общей формулы (±)-1 где R1 и R3, одинаковые или различные, представляют собой атом водорода, хлора, фтора, метил, трифторметил или метоксигруппу и R2 и R4 представляют собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси, метилсульфонил или аминосульфонильную группу, при условии, что один из заместителей R2 или R4 представляет собой метилсульфонильную группу (SO2CH3) или аминосульфонильную группу (SO2NH2);
и который, наконец, включает получение энантиомерно чистых соединений общей формулы 1 путем разделения рацемической смеси общей формулы (±)-1, где R1 и R3, одинаковые или различные, представляют собой атом водорода, хлора, фтора, метил, трифторметил или метоксигруппу, и R2 и R4 представляют собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси или натрийсульфонатную группу, при условии, что один из заместителей R2 или R4 представляет собой натрийсульфонатную группу (SO3Na), на его энантиомеры с помощью реакции с оптически активным эфедрином, с последующим образованием натриевой соли каждого из энатиомеров, реакции с тионилхлоридом и карбонатом аммония или аммиаком или с тионилхлоридом, а затем с сульфитом натрия и метилиодидом или метилсульфатом, с образованием отдельных энантиомерно чистых соединений общей формулы (-)-1 и (+)-1, где R1 и R3, одинаковые или различные, представляют собой атом водорода, хлора, фтора, метил, трифторметил или метоксигруппу и R2 и R4 представляют собой атом водорода, хлора, фтора, метил, трифторметил, метокси, трифторметокси, метилсульфонил или аминосульфонильную группу, при условии, что один из заместителей R2 или R4 представляет собой метилсульфонильную группу (SO2СН3) или аминосульфонильную группу (SO2NH2).
раздельное образование солей натрия (-)-1 и (+)-1 в водной среде или спирте, включая изопропиловый спирт, где один из заместителей R2 или R4 представляет собой натрийсульфонатную группу (SO3Na) и заместители R1, R2, R3 и R4 являются теми, как определено в п.1, взаимодействие каждого из энатиомеров с тионилхлоридом, а затем с карбонатом аммония или аммиаком, или вместо этого, с тионилхлоридом, а затем с сульфитом натрия и метилиодидом или метилсульфатом, по п.3, для образования отдельных энантиомерно чистых пиразолинов (-)-1 и (+)-1, где один из заместителей R2 или R4 представляет собой метилсульфонильную группу (SO2СН3) или аминосульфонильную группу (SO2NH2) и заместители R1, R2, R3 и R4 являются теми, как определено в п.1.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| WO 00/76503 A1, 21.12.2000 | |||
| Способ получения 1-(4 -сульфамоилфенил)-3-(4"-хлорфенил)-пиразолина | 1975 |
|
SU654616A1 |
| MUKAI et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Canadian journal chemistry | |||
| Дверной замок, автоматически запирающийся на ригель, удерживаемый в крайних своих положениях помощью серии парных, симметрично расположенных цугальт | 1914 |
|
SU1979A1 |
| Способ получения на волокне оливково-зеленой окраски путем образования никелевого лака азокрасителя | 1920 |
|
SU57A1 |

