Изобретение относится к области аналитической химии, в частности к вольтамперометрическому способу определения гормона инсулина, и может быть использовано в дифференциальной диагностике сахарного диабета.
Инсулин - гормон, вырабатывающийся в β-клетках островков Лангерганса поджелудочной железы, по химической структуре представляющий соединение белковой природы, содержащее 51 аминокислотный остаток. Молекула инсулина состоит из двух цепей: из цепи А (21 аминокислотный остаток) и цепи В (30 остатков), соединенных дисульфидным мостиком. Биологический предшественник - проинсулин представляет собой одну цепь, состоящую из субъединиц А и В, соединенных между собой так называемым С-пептидом (31 аминокислотный остаток у человека) [1].
На сегодняшний день в клинической практике для количественной оценки инсулина используются радиоиммунологический и иммуноферментный методы.
Радиоиммунный метод заключается в проведении реакции связывания свободных лигандов: инсулина пробы и меченого изотопа 125I инсулиноспецифичными антителами. Радиоактивность меченого лиганда, связанного с антителами, обратно пропорциональна концентрации инсулина в исследуемой жидкости [2; 3].
Иммуноферментный метод заключается в количественной оценке антител способом встречного электроиммуноосмофореза. Для осуществления данного метода проводят анализ антигена методом встречного иммуноэлектрофореза и после окрашивания пластинок вырезают участки геля с окрашенным преципитатом, элюируют их 0,1% раствором натрия додецилсульфата в 0,1 моль/л ацетатном буфере, рН 5,0, с последующим фотометрированием элюата [4].
Наиболее близким способом является вольтамперометрический метод определения инсулина на классическом ртутном капающем электроде в растворе Брдички (0,1 моль/л раствор амммония гидроксида в 0,1 моль/л растворе аммония хлорида в присутствии 0,0016 моль/л раствора кобальта хлорида) [5]. Данный способ выбран в качестве прототипа.
Недостатками указанного метода является то, что ртутный капающий электрод является очень токсичным и громоздким, а содержащийся в фоновом растворе аммония гидроксид быстро улетучивается из ячейки, поэтому использование условий, приведенных в прототипе, делают невозможным применение данного способа.
Целью изобретения является увеличение чувствительности и экспрессности способа.
Поставленная цель достигается техническим решением, представляющим собой вольтамперометрический способ определения инсулина на приборе ТА-2 путем регистрации катодных кривых без предварительного электрохимического концентрирования вещества на поверхности электрода. Раствор инсулина предварительно перемешивают и деаэрируют. Для этого через раствор пропускают азот с содержанием кислорода менее 0,001%. Пропускание азота проводят в течение 150 с при потенциале - 1,45 В. В качестве рабочего используют ртутно-пленочный игольчатый электрод. Затем регистрацию поляризационных кривых проводят при линейной скорости развертки потенциала 50 мВ/с. Концентрацию инсулина определяют по высоте пика в интервале потенциалов от -0,8 до -0,9 В относительно хлор-серебряного электрода. Определение проводят на фоне 0,01 моль/л раствора калия хлорида, в который добавляют 5 моль/л раствор винной кислоты до рН 3,0.
Новым в способе является то, что проводят предварительное перемешивание и деаэрацию раствора путем пропускания азота с содержанием кислорода менее 0,001% в течение 150 с при потенциале - 1,45 В на приборе ТА-2. В качестве рабочего электрода используют ртутно-пленочный (игольчатый). Регистрацию поляризационных кривых проводят после перемешивания при линейной скорости развертки потенциала 50 мВ/с, а концентрацию инсулина определяют по высоте пика в интервале потенциалов от -0,8 до -0,9 В относительно хлор-серебряного электрода на фоне 0,01 моль/л раствора калия хлорида с добавлением 5 моль/л раствора винной кислоты до рН 3,0.
Отличительные признаки проявили в заявляемой совокупности новые свойства: увеличение чувствительности (10-4-10-5 мг/л) и экспрессности анализа.
С учетом изложенного следует считать заявляемое решение соответствующим критерию “существенные отличия”.
Все условия определения инсулина подобраны экспериментально. Приготовление фоновых и стандартных растворов органического вещества в воде являются общепринятыми.
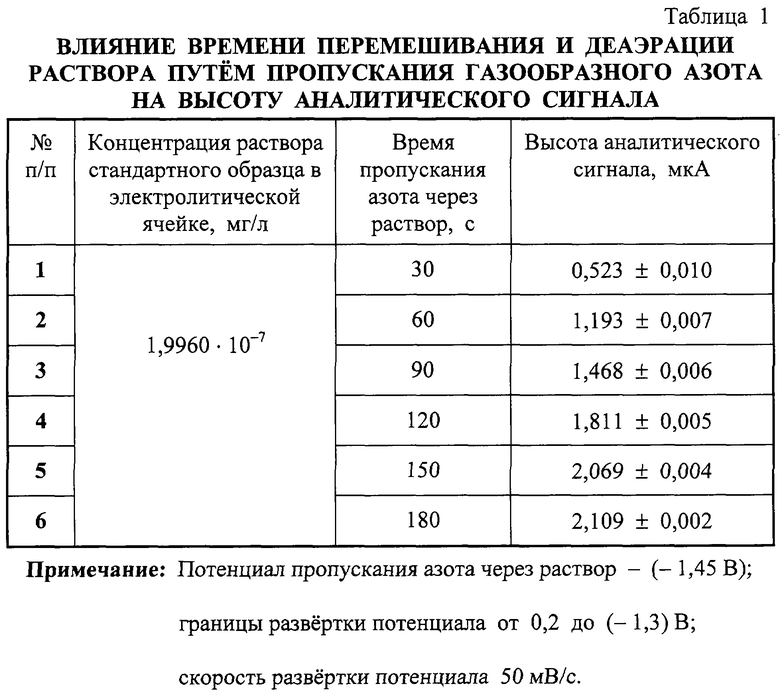
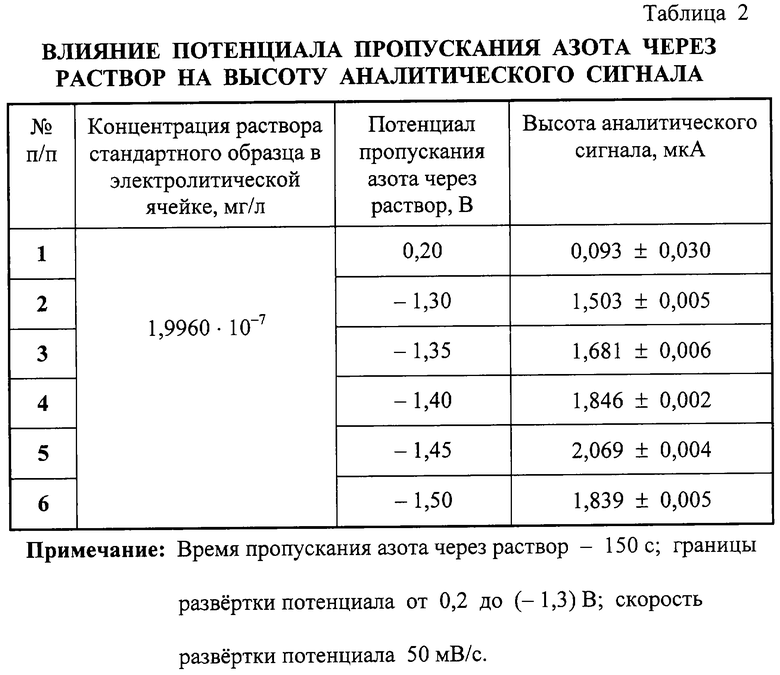
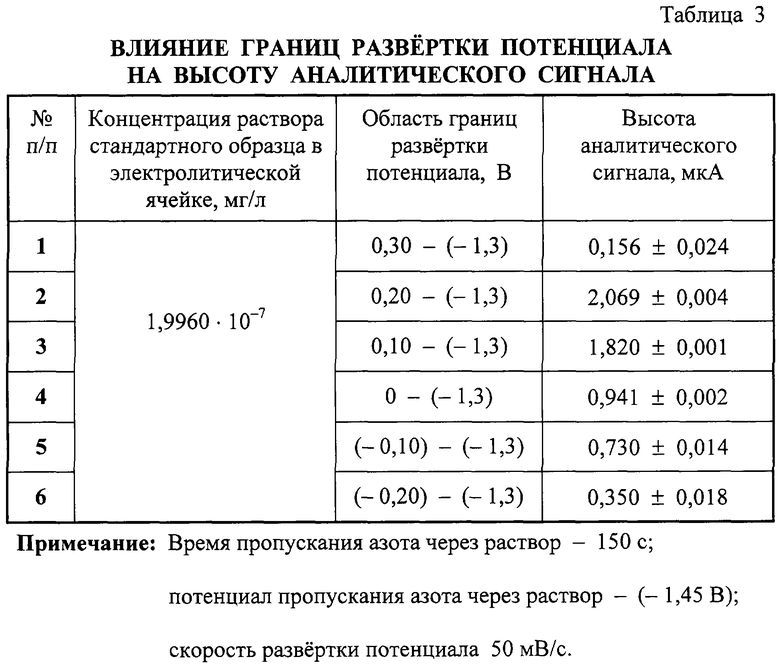
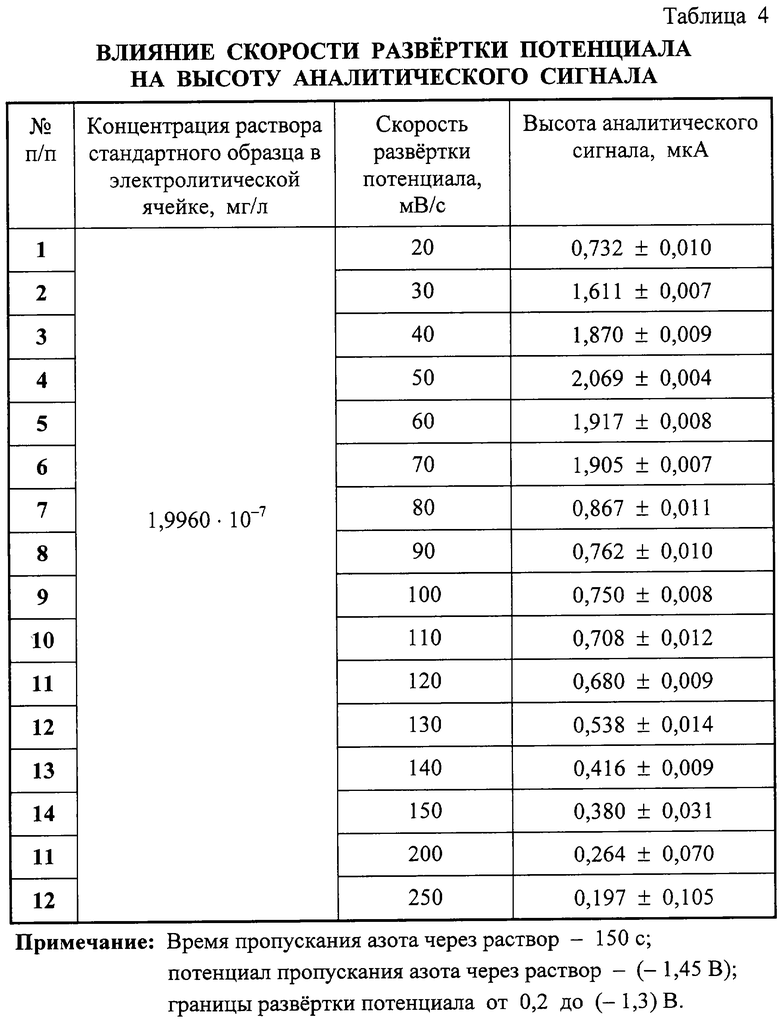
В процессе поиска оптимальных условий вольтамперометрического определения инсулина было изучено влияние ряда факторов (индикаторный электрод, фоновый электролит, его концентрация и рН, время и потенциал перемешивания путем пропускания азота, границы и скорость развертки потенциала) на высоту аналитического сигнала (табл.1-5).
Учитывая, что прототип для определения инсулина в качестве фонового электролита предлагает раствор Брдички, были использованы предложенные условия. Однако при работе на приборе ТА-2 нами не было получено четкого пика при использовании раствора Брдички, причем находящийся в растворе аммония гидроксид быстро улетучивался, вследствие чего дальнейший анализ был невозможен. Также были исследованы растворы аммония нитрата, натрия гидрофосфата, калия хлорида, лития хлорида, смесь лития хлорида и разведенной серной кислоты различной концентрации. Исходя из полученных результатов, в качестве фонового электролита был выбран раствор калия хлорида, так как на нем наблюдалась четкая волна восстановления инсулина, кроме того, данный раствор обеспечивал хорошую электропроводность, широкую рабочую область и необходимую площадь для обработки сигнала, был прост в приготовлении, к преимуществам также можно отнести продолжительный срок годности.
Оптимальная концентрация раствора калия хлорида составила 0,01 моль/л. В более концентрированных растворах мы не наблюдали прироста от добавки при наличии большого остаточного тока, тогда как более разбавленный раствор был неустойчив во времени.
В предлагаемом способе в качестве индикаторного электрода использовали пленку ртути на серебряной проволоке, упакованной во фторопластовый пакет (игольчатый электрод). Преимуществом такого электрода является возможность получения более узких и высоких пиков, служащих аналитической характеристикой определяемого вещества, что повышает разрешающую способность метода. Органические вещества способны образовывать с ртутью устойчивые комплексы или труднорастворимые соли.
Оптимальное значение рН фонового электролита составило 3,0. В щелочной среде при подобранных условиях сигнал инсулина отсутствовал, тогда как в нейтральной был слабо выражен, при изменении рН в более кислую сторону (менее 3,0) сужалась рабочая область электрода, при этом невозможно было зафиксировать пик инсулина. Оптимальным подкисляющим агентом была выбрана винная кислота, так как серная загрязняла фоновую линию, а на хлороводородной мы не наблюдали воспроизводимости результатов.
Оптимальное время перемешивания и деаэрации раствора путем пропускания азота составило 150 с, так как до данного значения линия фона была загрязнена кислородом и высота сигнала возрастала пропорционально времени, а после выбранного значения была получена чистая фоновая линия и высота сигнала почти не изменялась, поэтому именно указанное время мы определили как наиболее приемлемое (табл.1).
Оптимальный потенциал перемешивания путем пропускания азота через раствор составил - 1,45 В. При потенциале 0,2 В высота сигнала была минимальной, а в области от -1,5 до -1,3 В имела колоколообразную форму с максимумом при - 1,45 В (табл.2).
Оптимальный параметр границ развертки потенциала был установлен при постоянстве конечного потенциала соответствующего -1,3 В и переменной величине начального значения. Сдвиг нижней границы потенциала в более отрицательную область был нецелесообразным, так как наблюдали процесс восстановления водорода, кроме того, при данном значении рабочая область электрода достигала предела, тогда как при начальном потенциале 0,3 В сигнал инсулина резко уменьшался, что связано с десорбцией молекул ртути из электрода в раствор, тогда как при начальном потенциале 0,2 В мы наблюдали появление выраженного пика, но при сужении области развертки сигнал заметно уменьшался (табл.3).
Важным для определения инсулина вольтамперометрическим методом является выбор скорости развертки потенциала. Оптимально экспериментально установленной является 50 мВ/с. Изменение скорости развертки потенциала в сторону увеличения или уменьшения заметно понижало высоту аналитического сигнала, при этом уменьшалась и разрешающая способность метода, что затрудняло обработку полярограмм, увеличивало время анализа и не позволяло определять очень низкие концентрации инсулина (табл.4).
Пример I. Определение инсулина вольтамперометрическим методом в растворе
В кварцевый стаканчик, емкостью 20 мл, наливают 10 мл 0,01 моль/л раствора калия хлорида, добавляют 5 моль/л раствор винной кислоты до рН 3,0. При потенциале - 1,45 В раствор перемешивают и деаэрируют путем пропускания азота с содержанием кислорода менее 0,001% в течение 150 с. Затем отключают газ и фиксируют вольтамперограмму при скорости развертки потенциала 50 мВ/с, начиная от потенциала 0,2 В. Отсутствие пиков свидетельствует о чистоте фона.
Затем добавляют N капель объемом 0,01 мл стандартного раствора инсулина 1·10-4 мг/л и при потенциале - 1,45 В раствор перемешивают и деаэрируют путем пропускания азота в течение 150 с. Затем отключают газ и фиксируют вольтамперограмму при скорости развертки потенциала 50 мВ/с, начиная от потенциала 0,2 В. Аналитический сигнал для указанной концентрации инсулина регистрируют в диапазоне потенциалов от -0,8 до -0,9 В.
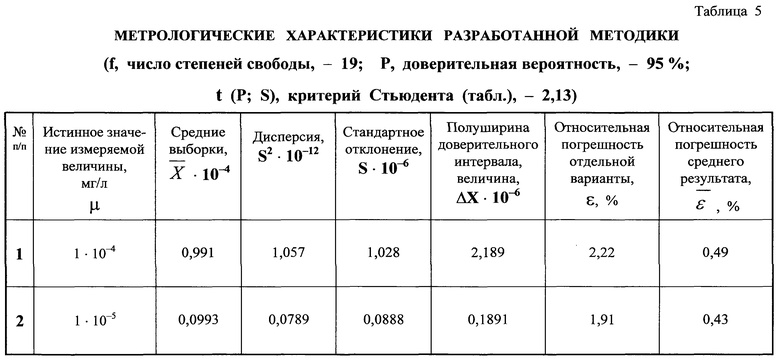
Установленные условия впервые позволили количественно определить инсулин путем регистрации вольтамперных кривых при потенциалах от -0,8 до -0,9 В на фоне 0,01 моль/л раствора калия гидроксида при рН 3,0. Нижняя граница определяемых содержаний инсулина составляет 1·10-5 мг/л. Относительное стандартное отклонение для диапазона концентраций 1·10-4-1·10-5 мг/л, соответствующих содержанию данного гормона в крови, не превышает 0,5 (табл.5).
Время единичного анализа не превышает 10 мин.
Предложенный авторами способ позволяет увеличить чувствительность и экспрессность определения инсулина в водной и биологических средах, а также позволяют разработать методику определения содержания микроколичеств инсулина в плазме, сыворотке крови.
Источники информации
1. Руководство по клинической лабораторной диагностике / Под ред. В.В. Меньшикова. - М.: Медицина, 1982. - 576 с.
2. Кауфман А.С., Северин О.В., Ли Д.Х. Способ радиоиммуннохимического анализа // Патент РФ №2149405.
3. Кауфман А.С., Северин О.В., Ли Д.Х. Способ радиоиммуннохимического анализа // Патент РФ №2454831.
4. Чигрин В.В., Соколов И.И., Каневчева И.С. Способ иммунноэлектроосмофореза // Патент РФ №4655809114.
5. Мискиджьян С.П. Полярография лекарственных препаратов / Мискиджьян С.П., Кравченюк Л.П. - Киев: Вища школа, 1976. – 232 с.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ АНГИОТЕНЗИНА II МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2003 |
|
RU2260797C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КАРВЕДИЛОЛА МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2007 |
|
RU2334510C1 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЛИДОКАИНА ГИДРОХЛОРИДА | 2007 |
|
RU2348925C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ФОЗИНОПРИЛА НАТРИЯ МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2005 |
|
RU2288469C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ АМИОДАРОНА (КОРДАРОНА) МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2003 |
|
RU2246722C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СПИРАПРИЛА ГИДРОХЛОРИДА МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2004 |
|
RU2280860C2 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ L-ТИРОКСИНА | 2010 |
|
RU2428690C1 |
| СПОСОБ ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ ИОНОВ КОБАЛЬТА (II) В РАСТВОРАХ СУЛЬФАТА ЦИНКА | 2001 |
|
RU2216014C2 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ГЛИЦИРРИЗИНОВОЙ КИСЛОТЫ В ФАРМАЦЕВТИЧЕСКИХ СУБСТАНЦИЯХ | 2015 |
|
RU2603363C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ БЕНАЗЕПРИЛА ГИДРОХЛОРИДА (ЛОТЕНЗИНА) МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2004 |
|
RU2280861C2 |
Изобретение относится к области медицины, а именно к методам исследования, и предназначено для дифференциальной диагностике сахарного диабета. Регистрируют катодные кривые без предварительного электрохимического концентрирования вещества на поверхности электрода. Проводят предварительное перемешивание и деаэрацию раствора путем пропускания азота с содержанием кислорода менее 0,001% в течение 150 с при потенциале -1,45 В. В качестве рабочего электрода используют ртутно-пленочный игольчатый. Регистрируют поляризационные кривые при линейной скорости развертки потенциала 50 мВ/с. Концентрацию инсулина определяют по высоте пика в интервале потенциалов от -0,8 до -0,9 В относительно хлор-серебряного электрода на фоне 0,01 моль/л раствора калия хлорида с добавлением 5 моль/л раствора винной кислоты до рН 3,0. Способ позволяет увеличить чувствительность и повысить экспрессность диагностики. 5 табл.
Вольтамперометрический способ определения инсулина, заключающийся в регистрации катодных кривых без предварительного электрохимического концентрирования вещества на поверхности электрода, отличающийся тем, что проводят предварительное перемешивание и деаэрацию раствора путем пропускания азота с содержанием кислорода менее 0,001% в течение 150 с при потенциале -1,45 В, в качестве рабочего электрода используют ртутно-пленочный игольчатый, затем регистрируют поляризационные кривые при линейной скорости развертки потенциала 50 мВ/с, а концентрацию инсулина определяют по высоте пика в интервале потенциалов от (-0,8) до (-0,9) В относительно хлорсеребряного электрода на фоне 0,01 моль/л раствора калия хлорида с добавлением 5 моль/л раствора винной кислоты до рН, равной 3,0.
| Способ определения инсулина у новорожденных детей | 1990 |
|
SU1805404A1 |
| СПОСОБ ДИАГНОСТИКИ САХАРНОГО ДИАБЕТА | 1994 |
|
RU2104537C1 |
| СПОСОБ ДИАГНОСТИКИ СИНДРОМА ИНСУЛИНОРЕЗИСТЕНТНОСТИ | 1999 |
|
RU2153170C1 |
| СПОСОБ РАДИОИММУНОХИМИЧЕСКОГО АНАЛИЗА | 1999 |
|
RU2149405C1 |

