Изобретение относится к аналитической химии и может быть использовано для автоматического или экспресс-анализа технологических растворов, оборотных и сточных вод цветной металлургии.
Известен способ определения кобальта в присутствии больших концентраций цинка методом дифференциальной импульсной полярографии (см. статью: Geibler M. , Maia R.D. Determination of cobalt in the presence of hight concentrations of zinc by differential pulse polarography // Fresenius Z. Anal. Chem. 1988. 330. 7. Р.624-626).
Сущность известного способа состоит в полярографическом определении ионов Со(II) на фоновом электролите состава 0,1М Na3С6Н5O7+0,1М NH4Cl+0,08%C4H8O2N2 по высоте катодного пика при потенциале -1,02В (отн. н. к. э. ). Способ осуществляют следующим образом. Смешивают в 100 мл мерной колбе 50 мл основного раствора (2 моль/л лимоннокислого натрия + 0,2 моль/л хлорида аммония), 8 мл 1% спиртового раствора диметилглиоксима, 1 мл пробы и доводят объем раствора в колбе до метки бидистиллятом. Часть подготовленного к анализу раствора деаэрируют в электрохимической ячейке потоком азота в течение 5 мин. Регистрируют дифференциальную импульсную полярограмму (ДИП) на ртутно-капающем электроде от -0,75В (по Н.К.Э.) до потенциала восстановления цинка при амплитуде импульса 50 мВ и скорости развертки потенциала 2 мВ/с. Концентрацию кобальта в пробе рассчитывают по методу стандартных добавок. Нижний предел обнаружения кобальта (II) составляет 2,5 мг/л (4,2•10-5 моль/литр). Калибровочная кривая линейна в пределах от 1•10-7 до 5•10-6 моль/л кобальта без цинка и в присутствии 5•10-3 моль/л Zn. Предельно допустимое соотношение концентрации ионов цинка и кобальта в анализируемом растворе составляет 50000:1.
К недостаткам известного способа, ограничивающим возможности его применения для контроля ионов Со(II) в технологических растворах цинкового производства, относятся низкая чувствительность и недостаточно высокая селективность. Кобальт входит в число примесей, оказывающих наиболее вредное воздействие на процесс электролиза цинка, резко снижая выход по току и вызывая коррозию цинкового листа. В этой связи для достижения высоких технологических показателей электролиза необходимо поддерживать концентрацию ионов Со(II) на уровне ≤0,5-1 мг/л (см., например, книгу Снурников А.П. Гидрометаллургия цинка. М.: Металлургия, 1981. С.238). На современных зарубежных цинковых заводах концентрация ионов кобальта (II) в очищенном растворе доводится до следовых количеств.
Важнейшим условием качественной очистки цинковых растворов от примесей является высокоизбирательный автоматический контроль низких концентраций ионов тяжелых металлов. В настоящее время для этих целей преимущественно используются вольтамперометрические методы анализа (см., например, книги Раннев Г.Г., Салин А.А. Методы и приборы автоматического контроля качества растворов. Ташкент: Фан, 1968. 158 с.; Зарецкий Л.С. Импульсный полярографический концентратомер. М.: Энергия, 1970. 80 с.; Раннев Г.Г. Хронопотециографы М.: Энергия, 1979 г., статью Bond A.M., Knight R.W., Newman O.M.G. // Analytical Chemistry. 1988. Vol.60. 21. P.2445-2448, статью Боровков Г.А., Зарецкий Л. С. , Бабицкий Л.Б. Внедрение анализатора АЖЭ-11 на предприятиях свинцово-цинковой отрасли // Цветная металлургия. Бюл. ЦНИИЭИЦМ. 1987. 1. С.49-51). С помощью вольтамперометрических анализаторов осуществляется непрерывный автоматический контроль ионов Cu2+, Sb3+, Cd2+. Однако для определения кобальта (II) с нижним пределом обнаружения 0,5 мг/л в растворах, содержащих до 150 г/л цинка (т.е. при соотношении ионных концентраций [Zn]:[Co]=300000:l), вольтамперометрические методы до настоящего времени не применялись. Это объясняется близостью потенциалов электрохимического восстановления катионов Со24+ и Zn2+, в большинстве использующихся в аналитической практике фоновых электролитов. Применение же для повышения селективности анализа различных комплексообразователей не всегда позволяет достичь необходимого результата. В известном способе в качестве такого комплексообразователя использован диметилглиоксим, позволяющий существенно повысить селективность полярографического определения кобальта (II) в присутствии избыточных концентраций цинка. Вместе с тем разрешающая способность известного способа не удовлетворяет современным требованиям оперативного контроля ионного состава технологических растворов цинкового производства. Помимо цинка мешающее влияние на полярографическое определение кобальта (II) на фоне 0,1М Na3C6H5O7+0,1M NH4Cl+0,08% C4H8O2N2 оказывают также марганец (VII) и никель (II), потенциалы ДИП пиков которых, по нашим данным, соответственно составляют -0,98 и -0,97 В. Причем, если концентрации ионов Ni2+ и Со2+ в очищенном цинковом растворе, как правило, соизмеримы, то содержание Mn(VII) более чем в 1000 раз превышает концентрацию ионов двухвалентного кобальта (см., например, книгу Лакерник М.М., Пахомова Г.Н. Металлургия цинка и кадмия М.: Металлургия, 1969. 488 с.). Катодные вольтамперные кривые Mn(VII) и Ni(II) маскируют ДИП пик Со(II) и уже при соотношениях ионных концентраций [Ni]:[Co]=4:l и [Mn]:[Со]=5:1 полярографическое определение кобальта на фоне 0,1М Na3С6Н5О7+0,1М NH4Cl+0,08%C4H8O2N2 становится невозможным. Ионы Mn(II), концентрация которых в поступающем на электролиз растворе достигает 10-13 г/л, восстанавливаются при более электроотрицательных потенциалах, чем двухвалентный кобальт и не мешают его определению.
Существенным недостатком известного способа является также изменение наклона калибровочной кривой Со(II) в зависимости от содержания цинка в анализируемом растворе, что неизбежно приводит к снижению точности результатов измерений. Кроме того, необходимость продувки контролируемого раствора потоком азота для обескислороживания значительно затрудняет автоматизацию полярографического анализа в производственных условиях.
Известен также способ полярографического определения кобальта в присутствии меди, никеля и цинка (Авторское свидетельство СССР 440593 МКИ5 G 01 27/48 авторов Л.Н. Васильевой и Н.Б. Когана. Б.И. 31. 1974 г.). Способ осуществляется следующим образом. К 2-3 мл синтетического раствора, содержащего кобальт (II) медь, цинк и никель, с целью окисления кобальта (II) до кобальта (III) добавляют 50 мл аммиака (25%), 5 г хлористого аммония и 0,22 г феррицианида калия. Раствор перемешивают и вводят 2 мл пиридина. Затем полученный раствор доводят водой до 100 мл и снова перемешивают. Из аликвотной части раствора - 20 мл удаляют кислород путем продувания азота. Обработанный таким образом кобальтсодержащий раствор полярографируют в интервале потенциалов от -0,3 до -0,9 В (донная ртуть). Измерение тока пиридинового комплекса кобальта (II) осуществляют при потенциале -0,7 В на вектор-полярографе или ППТ-1. Известный способ позволяет определять содержание кобальта от 0,25 мг/л и выше, при этом допускаются следующие соотношения кобальта и мешающих элементов: [Co]:[Cu]=1:20; [Co]:[Ni]=l:100 и [Co]:[Zn]=l:100.
К недостаткам известного способа относятся низкая селективность измерения ионов кобальта в присутствии цинка, сложная, практически не поддающаяся ватоматизации пробоподготовка, включающая применение такого высокотоксичного реагента, как пиридин.
Известен способ полярографического определения меди, никеля и кобальта (Авторское свидетельство СССР 502303 МКИ5 G 01 N 27/48 авторов Л.Н. Васильевой и З.Л. Юстус. Б.И. 5. 1976 г.). Способ осуществляется следующим образом. Навеску пробы 0,5-1,0 г разлагают в 30 мл соляной кислоты с 2 г фторида аммония, приливают 20 мл азотной кислоты; раствор выпаривают, дважды обрабатывают соляной кислотой по 5-10 мл, выпаривая раствор. Приливают 2 мл соляной кислоты и 15 мл воды, кипятят, добавляют 20% раствор едкого кали до рН 1,0-1,5 и прибавляют 2 г фторида натрия; раствор с осадком переносят в мерную колбу 50 мл; доводят водой до метки, перемешивают (раствор А). Через 20 мин отфильтровывают 10 мл раствора А в мерную колбу 50 мл, приливают 30 мл воды и 5 мл 1н. хлорида аммония + 1н. аммиака, водой уровень раствора доводят до метки, перемешивают, пропускают азот и фиксируют полярограммы меди и никеля. К 25 мл раствора А прибавляют 2 г хлорида аммония, 5 г однозамещенного фосфата натрия и аммиака до рН 6-7, осадок оставляют на 20-30 мин, переносят раствор с осадком в мерную колбу 50 мл, прибавляют 1 мл 1% спиртового раствора диметилглиоксима, уровень раствора водой повышают до метки, перемешивают, отфильтровывают 35 мл раствора в сосуд для пропускания азота, добавляют 5-6 капель аммиака до рН 8-9, пропускают азот и полярографируют. Расчет проводят методом добавок.
Таким образом, сущность известного способа состоит в том, что в электролит при рН 4 вводят однозамещенный фосфат натрия, повышают рН до 6-7, выдерживают раствор с осадком 20-30 мин и вводят диметилглиоксим, а затем повышают рН до 8-9 и полярографируют. Способ дает возможность определения меди, никеля и кобальта при соотношениях цинк:кобальт=0000:1; никель:кобальт=10000:1; медь:кобальт=10000:1.
Существенным недостатком известного способа является его сложность и трудоемкость, а его разрешающая способность не позволяет производить измерение концентрации ионов кобальта в технологических растворах цинкового производства.
Наиболее близким по технической сущности к заявляемому является способ полярографического определения кобальта на фоне 1М раствора хлорида аммония с добавлением аммиачного раствора диметилглиоксима (см. книгу: Манита М.Д., Салихджанова Р. М. -Ф., Яворская С.Ф. Современные методы определения атмосферных загрязнений населенных мест. М.: Медицина, 1980. С.198-199). Способ основан на каталитическом восстановлении водорода на ртутном капающем электроде в присутствии диметилглиоксимата кобальта.
Анализ производится в режиме переменнотоковой полярографии. Концентрация кобальта в контролируемом растворе определяется по высоте катодного пика при потенциале -1,09 В. Нижний предел обнаружения ионов кобальта составляет 0,01 мкг/мл. Способ обеспечивает возможность одновременного определения кобальта и никеля, потенциал катодного пика которого равен -0,82 В. Известный способ предназначен для определения кобальта в атмосферном воздухе и предусматривает аспирационный отбор проб с последующим переводом сконцентрированного на фильтре вещества в водный раствор. Способ осуществляется следующим образом. Воздух протягивают со скоростью 150 л/мин через фильтр, помещенный в патрон. Для анализа отбирают 2000-4500 л воздуха. Фильтр с пробой переносят в фарфоровый тигель и озоляют в муфельной печи. После охлаждения растворяют в 5 мл концентрированной соляной кислоты. Затем переносят по 2,5 мл в две мерные колбы на 10 мл, в каждую добавляют по 5 мл дистиллированной воды.
Раствор одной из колб оттитровывают по каплям раствором аммиака до рН 9,0; приливают 0,25 мл аммиачного раствора сульфита натрия и 0,25 мл 4•10-2 аммиачного раствора диметилглиоксима. Затем доводят раствор в колбе до метки 5М раствором хлорида аммония и полярографируют. По результатам полярографического анализа определяют концентрацию в растворе кобальта. Расчет производят по калибровочному графику. Рабочие растворы для построения калибровочного графика готовят с содержанием 0,02; 0,05; 0,1; 0,5; 1,0; 1,5 мкг/мл разбавлением раствором фона, содержащим 1М хлорида аммония и 10-3М диметилглиоксима. Раствор, находящийся во второй мерной колбе, после соответствующей пробоподготовки анализируют на содержание никеля.
К достоинствам известного способа относятся высокая чувствительность и простота пробоподготовки, позволяющие использовать его для экспресс-анализа кобальта в различных водных растворах, включая промышленные сточные воды.
Существенным недостатком данного способа является низкая селективность. Известный способ обеспечивает полярографическое измерение кобальта (II) в присутствии не более чем 20-кратных избытков марганца (VII) и цинка (II), что полностью исключает возможность его применения для контроля технологических растворов цинкового производства. Недостатком известного способа является необходимость строгого поддержания величины рН анализируемого раствора на уровне 9 при полярографическом определении кобальта (см. статью Астфьева В. В. , Прохорова Г.В. и Р.М.-Ф. Салихджанова. Изучение восстановления диметилглиоксиматов меди, никеля и кобальта с помощью вектор-полярографии // ЖАХ. 1976. Т. 21. Вып. 2. С.260-264). Изменение величины рН как в сторону увеличения, так и в сторону уменьшения приводит к существенному снижению чувствительности полярографического анализа. Кроме того, недостатком известного способа является изменение наклона калибровочного графика кобальта в зависимости от концентрации цинка в контролируемом растворе, что приводит к снижению точности результатов измерений.
Целью предлагаемого изобретения является повышение избирательности и чувствительности анализа.
Поставленная цель достигается тем, что согласно способу вольтамперометрического определения кобальта, включающему наложение линейно-изменяющегося потенциала на ртутный индикаторный электрод и регистрацию вольтамперной кривой на фоне хлоридноаммиачного буферного электролита, содержащего диметилглиоксим, в анализируемый раствор дополнительно вводят солянокислый гидроксиламин, полярографирование ведут на фоне 2M NH4Cl+2M NH4OH+2•10-3M C4H8O2N2+0,2M NH2OH•HCl в диапазоне напряжений от -1,05 до -1,30 В, а концентрацию кобальта измеряют по высоте катодного пика при потенциале -1,15±0,01 В.
В результате проведения патентных исследований не было выявлено каких-либо известных способов вольтамперометрического определения кобальта, предусматривающих полярографирование на фоне 2М NH4Cl+2M NH4OH+2•10-3M C4H8O2N2+0,2M NH2OH•HCl в диапазоне напряжений от -1,05 до-1,30 В, с измерением концентрации кобальта по высоте катодного пика при потенциале -1,15±0,01 В, т. е. имеющих признаки, сходные с признаками, отличающими заявленный способ от прототипа.
В среде хлоридно-аммиачного буферного электролита, содержащего диметилглиоксим, происходят сложные взаимодействия ионов кобальта с компонентами фона, приводящие в конечном итоге к образованию нового электрохимически активного комплекса, восстанавливающегося на ртутном электроде в более положительной области потенциалов.
Установлено (см., например, Стромберг А.Г., Землянская А.И.// Ж.общ. химии, 1945. Т. 15. С. 303; Burger К., Syrek G., Farsang Gy., // Acta Chim. Acad. Sci. Hung. 1966.V.49. P.113), что в аммиачном растворе диметилглиоксима в присутствии кобальта возникает каталитическая волна, которая может быть использована в аналитических целях. В работе Виноградова Е.Н. и Прохорова Г.В. О природе каталитических волн водорода в аммиачном буферном растворе в присутствии диметилглиоксиматов никеля и кобальта // ЖАХ. 1968. Т. 23. Вып.11. С.1666-1668 предложена схема возникновения каталитического тока водорода в аммиачном буферном растворе в присутствии диметилглиоксиматов кобальта и никеля (МеА2): катализатор присоединяет протон от донора протонов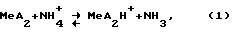
далее при присоединении электрона образуется незаряженная частица с характером свободного радикала
которой энергетически выгодно вступить в бимолекулярное взаимодействие
2MeA2H_→2MeA2+H2. (3)
В результате реакции (3) регенерируется катализатор в основной форме и выделяется молекула водорода. Регенерированная основная форма катализатора присоединяет протон по уравнению (1), и весь цикл повторяется.
Как отмечается в работе К. Бургер. Органические реагенты в неорганическом анализе. М.: Мир, 1975. С.104, ни один из комплексов диметилглиоксима не дает полярографической волны, подобной каталитической волне комплекса кобальта (II). Причина этого явления заключается, по-видимому, в восстановлении того атома водорода в комплексе, который участвует в водородной связи, и этот процесс ответственен за каталитическое действие молекулы комплекса. Согласно результатам ИК-спектроскопического анализа, проведенного автором, в комплексах кобальта (II) с диметилглиоксимом обнаружены самые слабые водородные связи по сравнению с комплексами диметилглиоксима с двухвалентными переходными металлами. Экспериментальные данные работы Астафьева В.В., Прохорова Г.В. и Салихджанова Р.М.-Ф. Изучение восстановления диметилглиоксиматов меди, никеля и кобальта с помощью вектор-полярографии // ЖАХ. 1976. Т.31. Вып. 2. С. 260-264 подтверждают, что по силе каталитического эффекта диметилглиоксиматы Со(II) существенно превосходят диметилглиоксиматы Cu(II) и Ni(II). В работе Geibler М., Maia R.D. Determination of cobalt in the presence of hight concentrations of zinc by differetial pulse polarography // Fresenius Z. Anal. Chem. 1988. 330. 7. Р.624-626 отмечено, что вольтамперные кривые кобальта на фоне 0,1М Na3C6H5O7+0,1M NH4Cl+0,08%C4H8O2N2, записанные методом циклической вольтамперометрии с треугольной волной, не выявляют анодного пика. Это показывает, что восстановление кобальт-диметилглиоксимового хелата полностью необратимо. Аналогичный результат был получен нами при регистрации катодных и анодных вольтамперных кривых кобальта (II) на хлоридно-аммиачном фоновом электролите, содержащем диметилглиоксим, методом ДИП на стационарном ртутно-капельном электроде (СРКЭ).
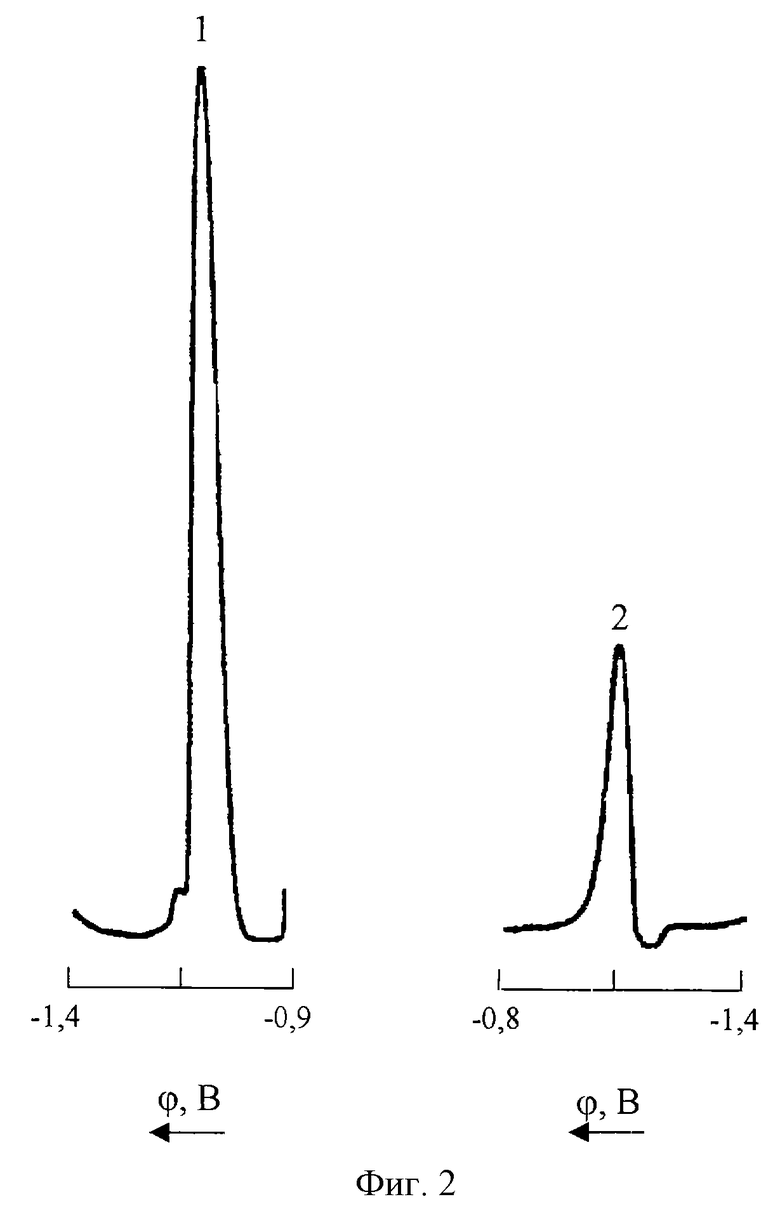
На фиг. 1 представлены катодная (1) и анодная (2) ДИП кривые Со(II) 0,5 мг/л на фоне
1М NH4Cl+1•10-3М C4H8O2N2 (pH 9 NH4OH).
На фиг. 2 представлены катодная (1) и анодная (2) ДИП кривые Со(II) 0,1 мг/л на фоне
1М NH4Cl+1•10-3M C4H8O2N2+0,2M NH2OH•HCl (pH 9 NH4OH).
На фиг.3 показано влияние гидроксиламина на чувствительность полярографического определения кобальта в среде хлоридно-аммиачного фонового электролита
1. CCo(II)=0,05 мг/л; фон 1М NH4Cl+1•10-3M C4H8O2N2 (pH 9 NH4OH).
2. CCo(II)=0,005 мг/л; фон 1М NH4Cl+1•10-3М C4H8O2N2+0,2M NH2OH•HCl (pH 9 NH4OH).
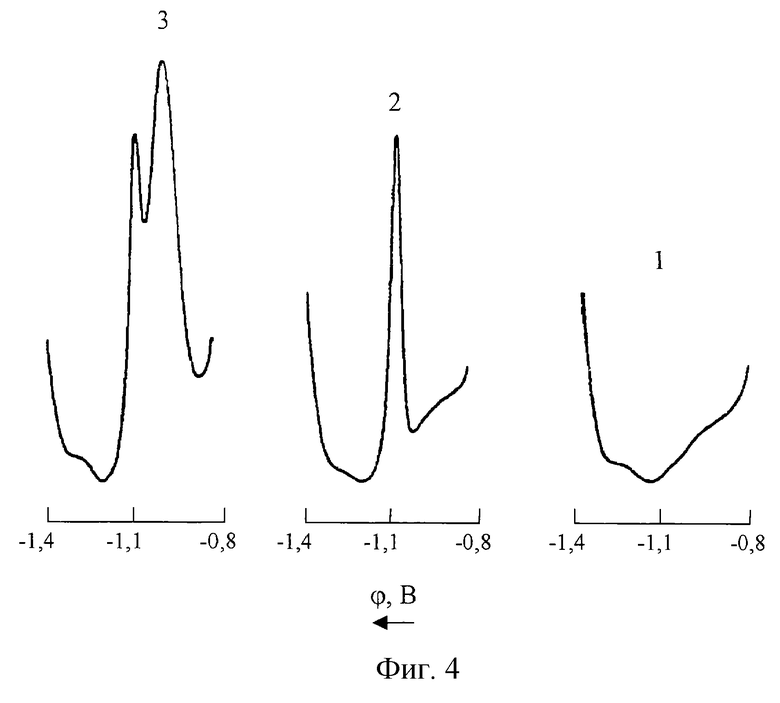
На фиг. 4 представлены ДИП кривые кобальта на фоне 1М NH4CI+1•10-3M C4H8O2N2 (pH 9 Н4OН) до и после введения в анализируемый раствор марганца
1 - фон;
2 - СCo(II)=0,2 мг/л; СMn(VII)=0;
3 - СCо(II)=0,2 мг/л; CMn(VII)=20 мг/л.
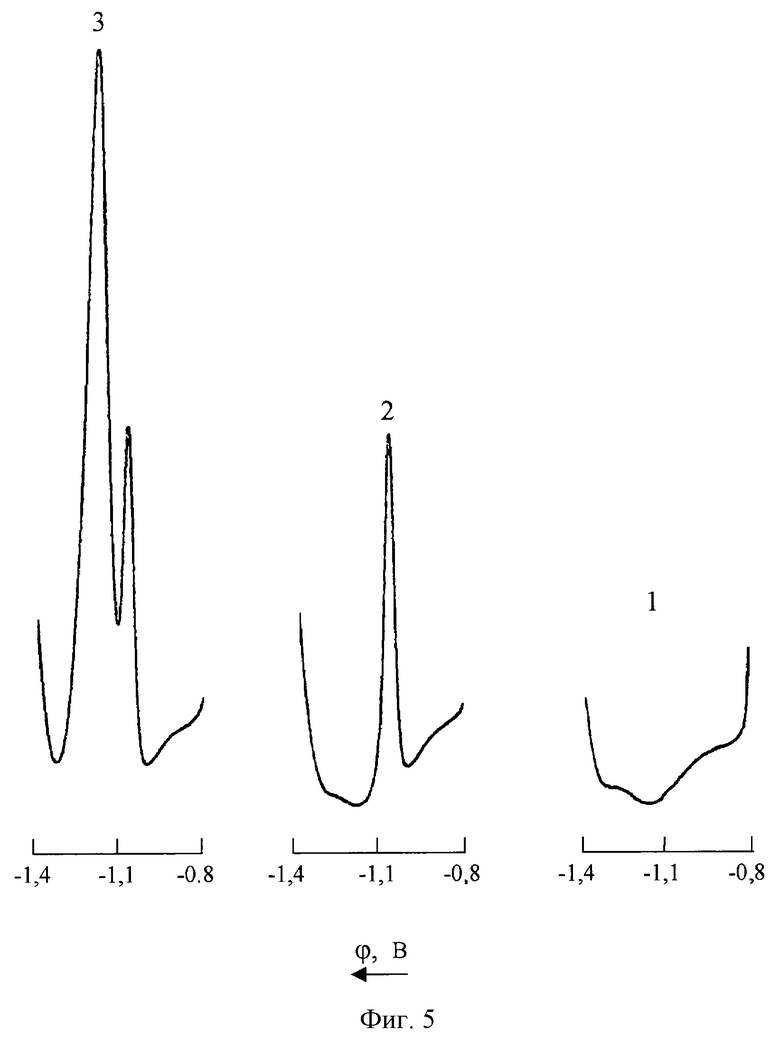
На фиг. 5 представлены ДИП кривые кобальта на фоне 1М NH4CI+1•10-3M C4H8O2N2 (pH 9 NH4OH) до и после введения в анализируемый раствор цинка
1 - фон;
2 - СCo(II)=0,2 мг/л; CZn(II)=0;
3 - СCo(II)=0,2 мг/л; СZn(II)=20 мг/л.
На фиг. 6 представлены ДИП кривые кобальта на фоне 1М NH4Cl+1•10-3M C4H8O2N2 (pH 9 NH4OH) до и после введения в анализируемый раствор цинка и марганца
1 - фон;
2 - СCo(II)=0,2 мг/л; CZn(II)=0; СMn(VII)=0;
3 - СCo(II)=0,2 мг/л; СZn(II)=20 мг/л; CMn(VII)=20 мг/л.
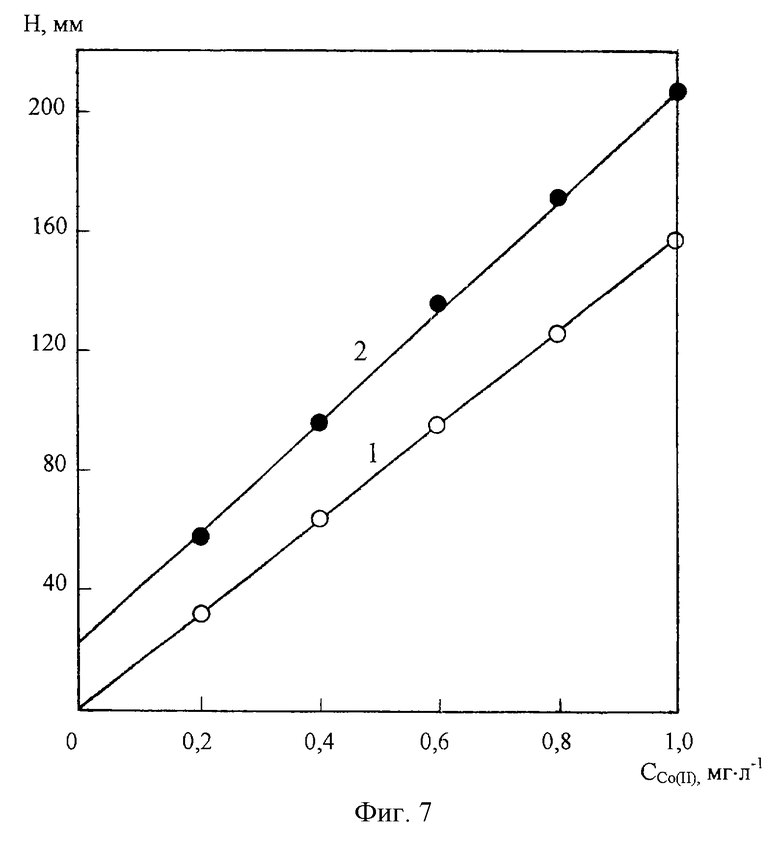
На фиг. 7 представлены градуировочные графики для определения Со(II) в диапазоне концентраций 0-1 мг/л в отсутствие Zn (1) и в присутствии 25 мг/л Zn (2) на фоне 1М NH4Cl+1•10-3М C4H8O2N2 (pH 9 NH4OH).
На фиг. 8 представлены ДИП пики Zn(II) 20 мг/л до и после введения в анализируемый раствор солянокислого гидроксиламина
1 - фон 1М NH4Cl+1•10-3М C4H8O2N2 (pH 9 NH4OH)
2 - фон 1М NH4Cl+1•10-3М C4H8O2N2+0,2M NH2OH•HCl (pH 9 NH4OH).
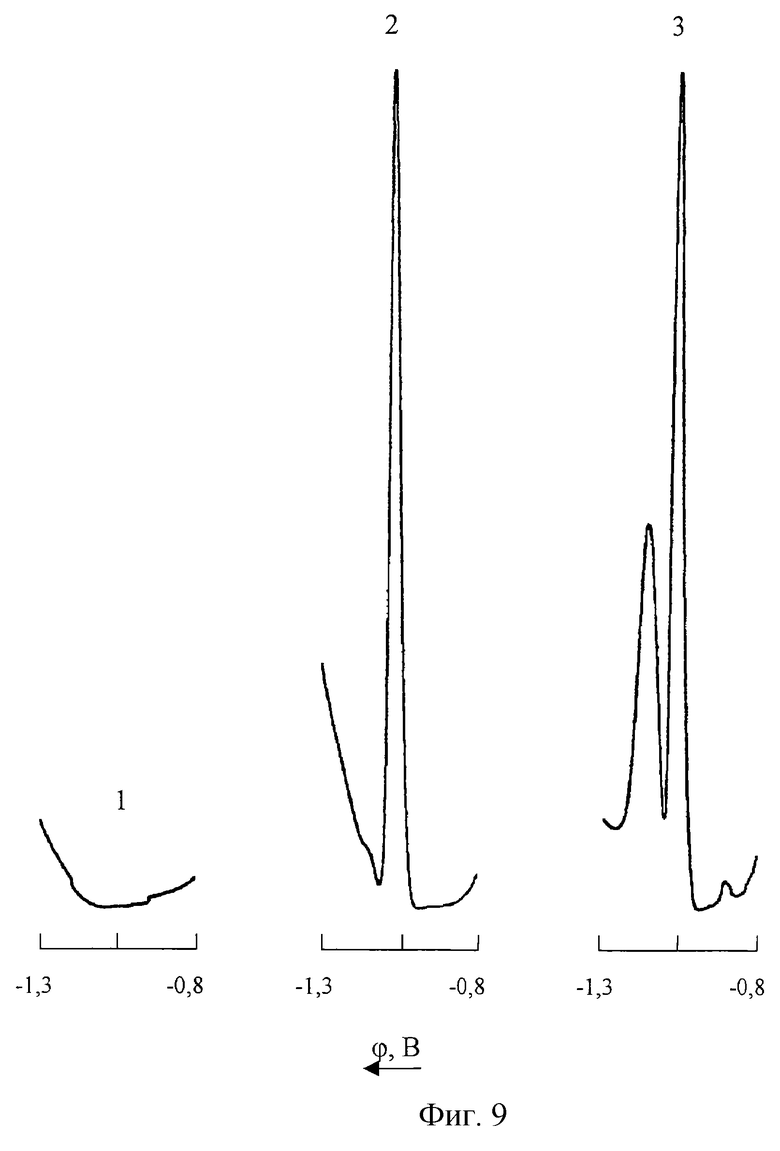
На фиг. 9 представлены ДИП пики кобальта на фоне 1М NH4Cl+1•10-3М C4H8O2N2+0,2М NH2OH•HCl (pH 9 NH4OH) до (2) и после (3) введения в анализируемый раствор цинка и марганца
1 - фон;
2 - СCo(II)=0,005 мг/л; CMn(VII)=0; CZn(II)=0;
3 - СCo(II)=0,005 мг/л. СMn(VII)=100 мг/л; СZn(II)=20 мг/л.
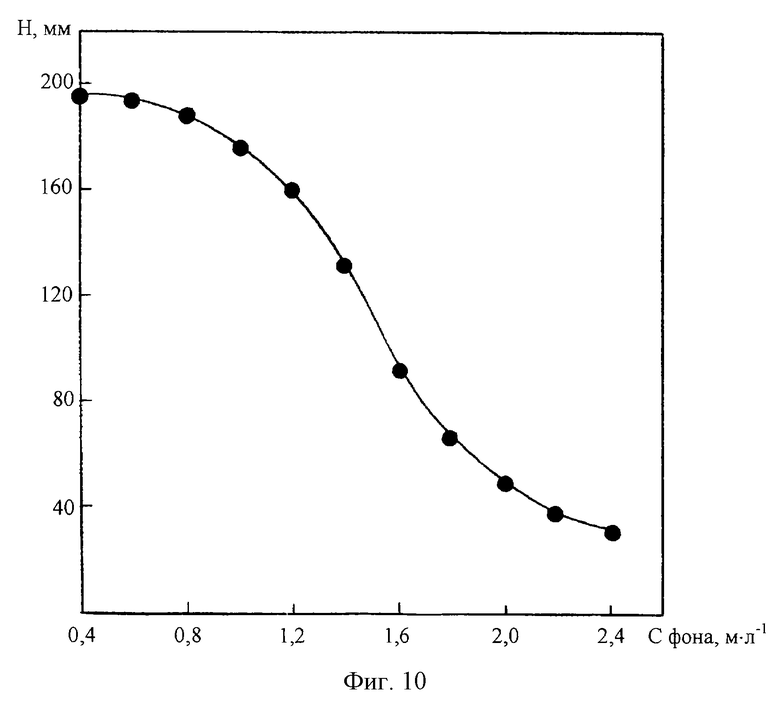
На фиг.10 представлена зависимость высоты ДИП пика Со(II) 0,005 мг/л от концентрации хлоридно-аммиачного фонового электролита CC4H8O2N2=2•10-3 м/л; СNH2OH•HCI=0,2 м/л.
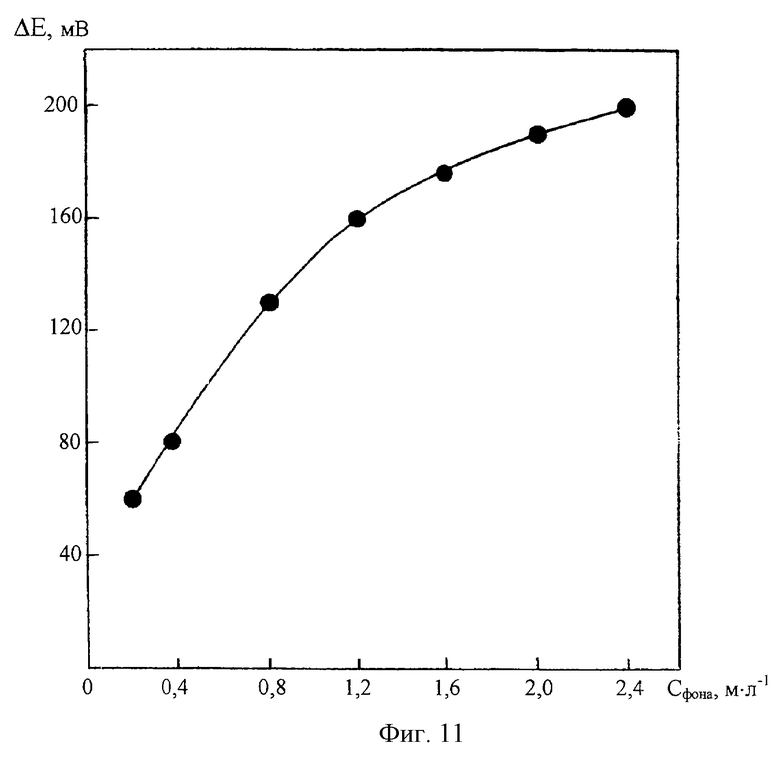
На фиг.11 представлена зависимость разности потенциалов ΔЕ, мВ ДИП пиков Со(II) 0,025 мг/л и Zn(II) 6 мг/л от концентрации хлоридно-аммиачного фонового электролита СC4Н8O2N2=2•10-3 м/л; CNH2OH•HCI=0,2 м/л.
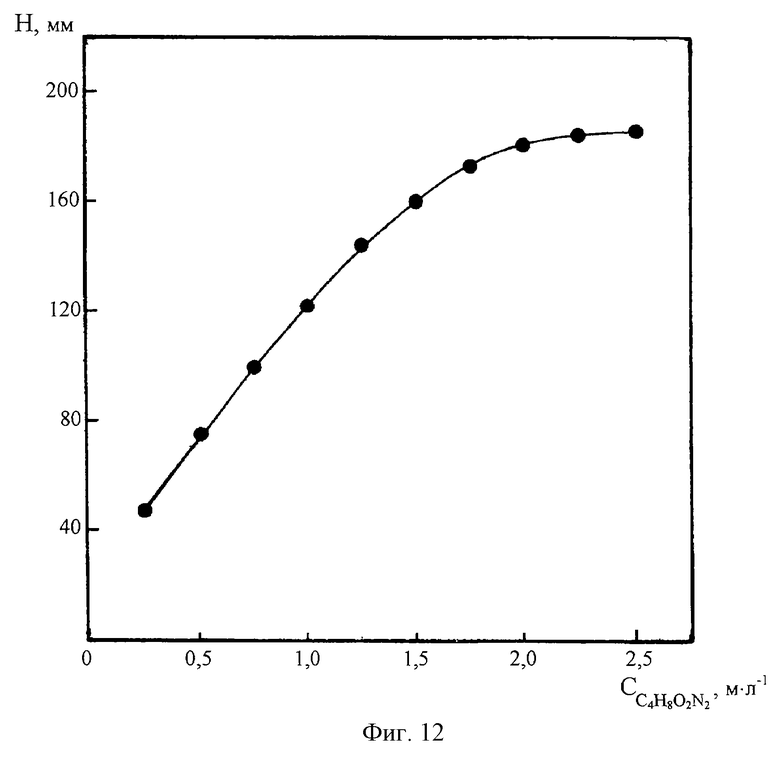
На фиг. 12 представлена зависимость высоты ДИП пика Со(II) 0,01 мг/л на фоне 2М NH4Cl+2M NH4OH+0,2M NH2OH•HCl от концентрации диметилглиоксима.
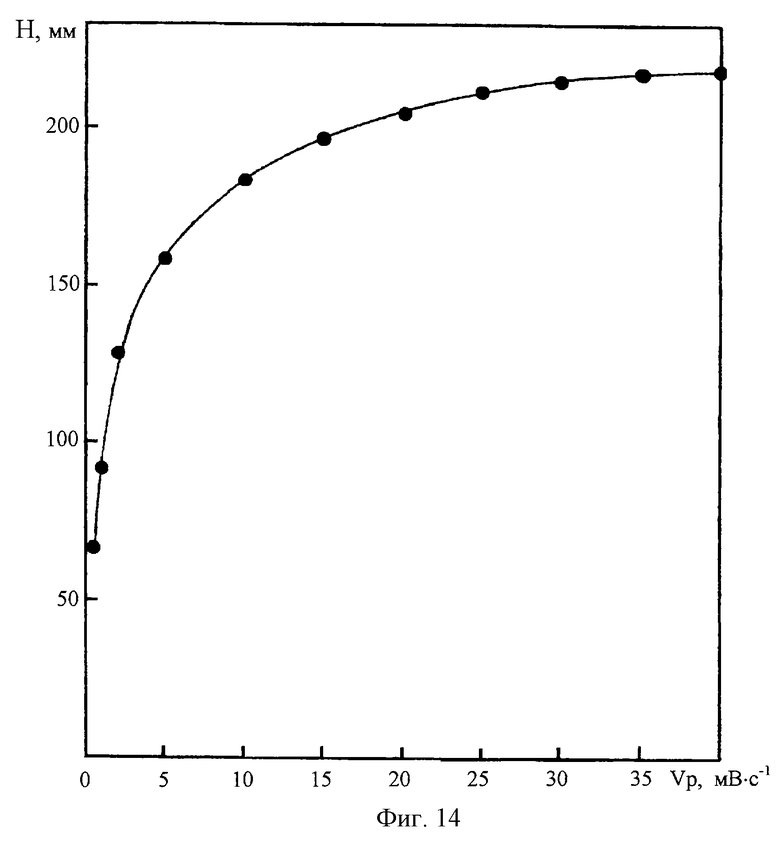
На фиг. 13 представлена зависимость высоты ДИП пика Со(II) 0,01 мг/л на фоне 2М NH4Cl+2M NH4OH+2•10-3M C4H8O2N2 от концентрации гидроксиламина. На фиг.14 представлена зависимость высоты ДИП пика Со(II) 0,01 мг/л на фоне 2М NH4Cl+2M NH4OH+2•10-3M C4H8O2N2+0,2M NH2OH•HCl от скорости развертки потенциала.
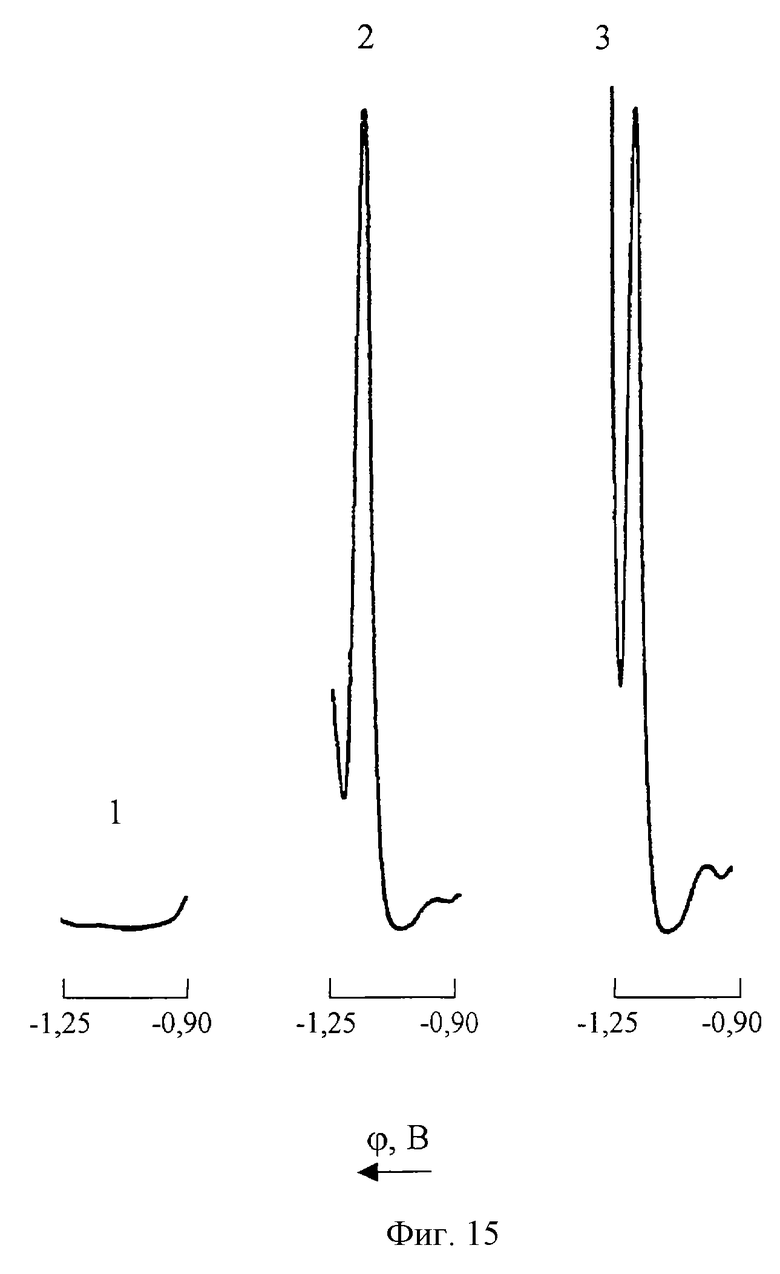
На фиг. 15 представлены ДИП пики кобальта (II) на фоне 2М NH4Cl+2M NH4OH+2•10-3M C4H8O2N2+0,2M NH2OH•HCl; скорость катодной развертки потенциала 10 мВ/с
1 - фон;
2 - СCo(II)=0,005 мг/л; CZn(II)=0; СMn(VII)=0;
3 - CCo(II)=0,005 мг/л; СZn(II)=1500 мг/л; СMn(VII)=200 мг/л.
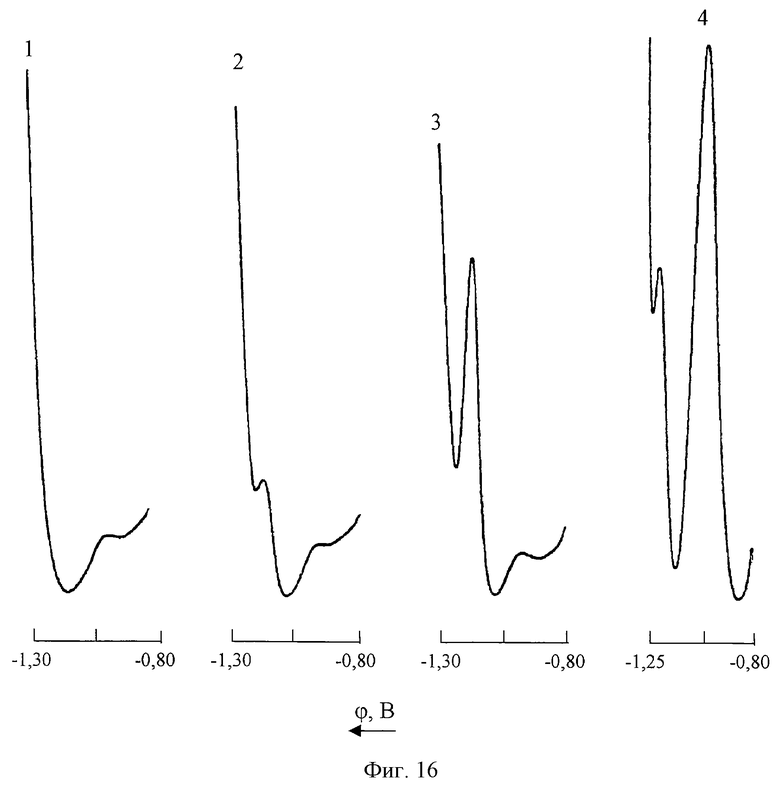
На фиг. 16 представлены вольтамперные кривые кобальта (II) на фоне 2М NH4Cl+2М NH4OH+2•10-3M C4H8O2N2; скорость катодной развертки потенциала 10 мВ/с
1 - фон;
2 - СCo(II)=0,04 мг/л;
3 - СCo(II)=0,1 мг/л; CZn(II)=0; СMn(VII)=0;
4 - СCo(II)=0,1 мг/л; СZn(II)=1000 мг/л; CMn(VII)=20 мг/л.
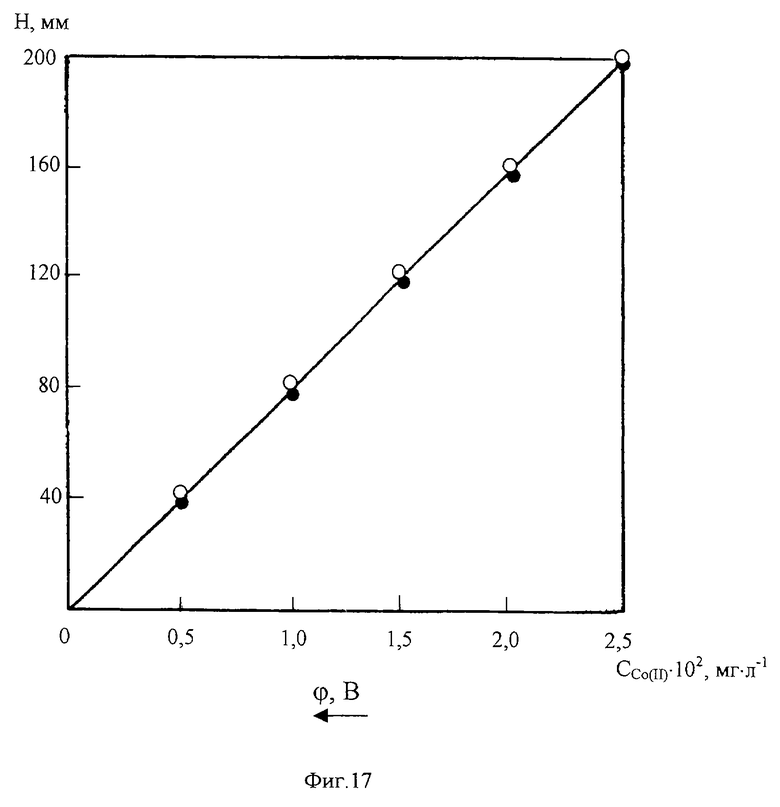
На фиг. 17 представлен градуировочный график для определения Со(II) на фоне 2М NH4Cl+2M NH4OH+2•10-3M C4H8O2N2+0,2M NH2OH•HCl в отсутствии цинка (•) и в присутствии 1500 мг/л Zn2+ (о).
На фиг. 18 представлена структурная схема автоматического вольтамперометрического анализатора ионов Со(II) в очищенных цинковых растворах.
Как видно из фиг. 1, Со(II) восстанавливается на СРКЭ в среде 1М NH4Cl+1•10-3M С4Н8O2N2 (pH 9 NH4OH) необратимо при потенциале ДИП пика -1,04 В (кривая 1); анодная запись в этой области потенциалов не регистрирует ДИП пика (кривая 2).
Измерения проводились на вольтамперометрическом анализаторе АЖЭ-11, изготовленном НПК "Югцветметавтоматика" и предназначенном для работы в режимах прямой и инверсионной дифференциальной импульсной полярографии на датчике со стационарным ртутным электродом (см. статью Боровков Г.А., Зарецкий Л. С., Бабицкий Л.Б. Внедрение анализатора АЖЭ-11 на предприятиях свинцово-цинковой подотрасли // Цветная металлургия. 1987. 1. С.49-51). Схема электрохимической ячейки 3-х электродная (рабочий электрод типа "висящая" ртутная капля, электрод сравнения хлорсеребряный, вспомогательный электрод стеклоуглеродный). Длительность наложения прямоугольных импульсов 40 мс с периодом 80 мс. Остальные режимные параметры (амплитуда импульсного поляризующего напряжения, скорость развертки потенциала, начальное напряжение, амплитуда развертки) выбирались в ходе экспериментальных исследований. Анализатор АЖЭ-11 обеспечивает полную автоматизацию всех стадий измерительного процесса, включая пробоподготовку и подачу анализируемого раствора в электрохимическую ячейку; формирование "висящей" ртутной капли; регистрацию вольтамперной кривой и обсчет ее показателей с выдачей выходного сигнала в единицах концентрации контролируемого вещества; сброс и складирование отработанных ртутных капель и удаление проанализированного раствора в дренаж.
Из сопоставления приведенных на фиг.1 и 2 кривых 1 видно, что введение в анализируемый раствор 0,2М солянокислого гидроксиламина приводит к смещению потенциала катодного ДИП пика кобальта (II) на 60 мВ в отрицательную область напряжений (ϕn = -1,10 B), что свидетельствует об образовании в растворе и электровосстановлении на СРКЭ нового комплексного соединения. При этом высота катодного пика увеличивается более чем в 50 раз, а на анодной вольтамперограмме (фиг.2 кривая 2) регистрируется пик при потенциале -1,09 В.
Основываясь на данных работы Харитонов Ю.А., Саруханов М.А. Химия комплексов металлов с гидроксиламином. М.: Наука, 1977, 296 с., можно предположить, что дополнительное введение в анализируемый раствор солянокислого гидроксиламина приводит к образованию смешанного комплексного соединения, например, типа [Co(Hx)2(DH)2] Cl, где Нх - гидроксиламин, а DH - остаток диметилглиоксима.
Полярографическую волну восстановления комплекса кобальта с диметилглиоксимом связывают с каталитическим восстановлением водорода (см. статью Виноградова Е. Н. , Прохорова Г.В. О природе каталитических волн водорода в аммиачном буферном растворе в присутствии диметилглиоксиматов никеля и кобальта // ЖАХ. 1968. Т.23. Вып.11. С.1666-1668), что косвенно согласуется с электрохимической необратимостью этой волны, проявляющейся при записи анодной кривой в нашем эксперименте (фиг.1). Для комплекса кобальта с двумя лигандами (гидроксиламином и диметилглиоксимом) катодный ДИП пик характеризуется возникновением каталитического тока, существенно превышающего величину каталитического тока комплекса кобальта с диметилглиоксимом (фиг.3), а тем более величину предельного диффузионного тока иона кобальта (см. статью Астафьева В.В., Прохорова Г.В., Салихджанова Р.М.-Ф. Изучение восстановления диметилглиоксиматов меди, никеля и кобальта с помощью вектор-полярографии // ЖАХ. 1976. Т.31. Вып.2. С.260-263). При этом волна становится частично обратимой (фиг. 2). Такой эффект в полярографии каталитических токов координационных соединений отвечает катализу комплексом "ион металла - лиганд", способным образовываться как в объеме раствора, так и в адсорбционном слое (см. работы: Рувинский О. Е. Кинетические, каталитические и адсорбционные эффекты в полярографии комплексных соединений // Полярография. Проблемы и перспективы. Рига: Зинатне, 1977. С.189-215 и Турьян Я.И. Полярографические каталитические токи комплексов металлов при катализе лигандом // Успехи химии. 1973. Т.42. Вып.6. С.969-986). Можно предположить, что в присутствии гидроксиламина образование нового полярографически активного комплекса происходит с участием адсорбированного на поверхности ртутного электрода диметилглиоксимового лиганда (предшествующая редокс-процессу химическая стадия носит поверхностный гетерогенный характер). Система электрод-диметилглиоксим-кобальт-гидроксиламин является электропроводящей и при восстановлении смешанного комплекса осуществляется "мостиковый" механизм переноса электрона. Предполагается также, что электрохимически активные комплексы с двумя или более лигандами (в данном случае с учетом аммиачного фона), адсорбирующиеся на поверхности электрода, имеют более искаженную симметрию, чем аналогичные комплексы в объеме раствора, что, в свою очередь, должно привести к повышению скорости переноса заряда (см. статью Рувинский О.Е. Кинетические, каталитические и адсорбционные эффекты в полярографии комплексных соединений // Полярография. Проблемы и перспективы. Рига: Зинатне, 1977. С.189-215) и соответствующему увеличению каталитического тока, т.е. чувствительности полярографического анализа.
Возможности полярографического определения ионов Со(II) в среде хлоридно-аммиачного фонового электролита, содержащего диметилглиоксим, существенно ограничиваются мешающим влиянием различного рода сопутствующих веществ и в первую очередь ионов Mn(VII) и Zn(II). Как видно из фиг.4 и фиг.5, в присутствии в анализируемом растворе ионов Mn(VII) и Zn(II) искажаются соответственно правая и левая ветви ДИП пика кобальта. При одновременном присутствии в контролируемом растворе марганца (VII) и цинка (II) полярoграфическое определение кобальта на фоне 1М NH4Cl+1•10-3M C4H8O2N2 (pH 9 NH4OH) становится практически невозможным (фиг.6). Кроме того, как следует из фиг. 7, при наличии в анализируемом растворе цинка изменяется наклон калибровочного графика кобальта, что неизбежно приводит к снижению точности результатов измерений.
Проведенные нами экспериментальные исследования показали, что добавление в контролируемый раствор солянокислого гидроксиламина позволяет достичь высокой селективности полярографического определения кобальта в присутствии ионов Mn(VII) и Zn(II).
В щелочной среде гидроксиламин является сильным восстановителем. При взаимодействии с перманганат-ионами гидроксиламин восстанавливает Mn(VII) до Mn(II). При этом сам гидроксиламин окисляется перманганатом до азоксигидроксила. Скорость реакции очень высока, т.к. ионы Mn2+ катализируют процесс окисления гидроксиламина. Таким образом, в растворе одновременно протекают две реакции: нормальная и автокаталитическая (см. книгу Брикун И.К., Козловский М.Т., Никитина Л.В. Гидразин и гидроксиламин и их применение в аналитической химии. Алма-Ата: Наука, 1967, 176 с.). Визуальным признаком протекания реакции является обесцвечивание раствора, содержащего перманганат-ионы, при введении в него гидроксиламина.
В среде аммиачно-хлоридного фонового электролита Mn(II) электрохимически восстанавливается в более отрицательной области потенциалов чем Со(II) и не мешает его полярографическому определению. Например, на фоне 1М NH4Cl+1M NH4OH потенциалы полуволн восстановления ионов Mn(II) и Со(II) соответственно равны -1,68 В и -1,32 В (н.к.э.) (см. книгу Крюкова Т.А., Синякова С.И., Арефьева Т.В. Полярографический анализ. М.: Госхимиздат, 1959, 772 с.).
Повышение избирательности полярографического определения кобальта в присутствии цинка при введении в контролируемый раствор гидроксиламина связано с резким увеличением высоты ДИП пика восстановления ионов Со(II) при неизменности катодного пика Zn(II). Как видно из фиг.8, добавление в анализируемый раствор солянокислого гидроксиламина не влияет на высоту и форму вольтамперной кривой цинка, зарегистрированной на фоне 1М NH4Cl+1•10-3M C4H8O2N2 (pH 9 NH4OH). Таким образом, снижение мешающего влияния цинка в заявляемом способе достигается за счет существенного и селективного повышения чувствительности полярографического анализа кобальта при введении в состав фонового электролита солянокислого гидроксиламина. Из представленных на фиг. 9 вольтамперных кривых видно, что мешающее влияние избыточных концентраций Mn(VII) и Zn(II) значительно снижается при дополнительном введении 0,2М NH2OH•HCl в состав фонового электролита, приготовленного согласно известному способу.
Проведенные нами исследования показали, что при полярографическом определении Со(II) на хлоридно-аммиачном фоне, содержащем диметилглиоксим и гидроксиламин, отпадает необходимость стабилизации величины рН на строго заданном оптимальном уровне, как это предусмотрено в известном способе. При этом экспериментально установлено, что для нейтрализации кислого раствора гидроксиламина и создания щелочной среды, необходимой для полярографического определения Со(II) в присутствии диметилглиоксима, целесообразно в качестве фонового электролита использовать достаточно концентрированный хлоридно-аммиачный буферный раствор, содержащий равные молярные доли хлорида и гидроксида аммония.
Чувствительность и селективность полярографического определения ионов Со(II) в сложных по составу технологических растворах цинкового производства существенно зависит от концентрации фона, диметилглиоксима и гидроксиламина.
Как видно из фиг.10, с уменьшением концентрации хлоридно-аммиачного фонового электролита высота ДИП пика Со(II) заметно возрастает. Однако, как нами установлено, при этом существенно ухудшается селективность определения Со(II) в присутствии избыточных содержаний цинка. Из фиг.11 видно, что разность потенциалов катодных ДИП пиков Со(II) и Zn(II) закономерно увеличивается с повышением концентрации фона.
Таким образом, высокая разрешающая способность полярографического определения низких содержаний кобальта в растворах сульфата цинка может быть достигнута только при использовании достаточно концентрированных фоновых электролитов. Нами установлено, что оптимальным является электролит состава 2М NH4Cl+2M NH4OH, на котором при соответствующем подборе концентраций диметилглиоксима и гидроксиламина обеспечивается возможность прямого полярографического определения Со(II) с нижним пределом обнаружения 0,0025 мг/л при ионном соотношении Zn(II):Co(II)≥300000:1.
Из фиг.12 и 13 видно, что чувствительность определения кобальта на фоне 2М NH4Cl+2M NH4OH существенно возрастает с увеличением концентрации диметилглиоксима и гидроксиламина. Увеличение концентрации NH2OH•HCl и C4H8O2N2 одновременно приводит к повышению избирательности вольтамперометрического измерения кобальта за счет селективного роста высоты катодного (каталитического) пика Со(II) при неизменности высот ДИП пиков сопутствующих ионов металлов, например цинка. В условиях полярографического определения микроколичеств кобальта в цинковых электролитах высокая чувствительность и разрешающая способность заявляемого способа достигается на фоне 2М NH4Cl+2M NH4OH, содержащем 2•10-3÷4•10-3M C4H8O2N2 и 0,15-0,25М NH2OH•HCl.
Дальнейшее увеличение концентрации комплексообразователей не дает заметных преимуществ и поэтому нецелесообразно по экономическим соображениям. Уменьшение концентрации гидроксиламина и диметилглиоксима приводит к некоторому снижению селективности и чувствительности полярографического анализа.
Изучение влияния скорости катодной развертки потенциала (Vp) на чувствительность полярографического определения Со(II) на фоне 2М NH4Cl+2M NH4OH+2•10-3M C4H8O2N2+0,2М NH2OH•HCl показало, что при изменении Vp от 0,5 до 10 мВ/с высота ДИП пика заметно возрастает; дальнейшее увеличение скорости развертки сказывается на чувствительности полярографического анализа кобальта в значительно меньшей степени. Регистрация вольтамперных кривых со скоростью катодной развертки 10 мВ/С позволяет достичь высокой оперативности измерения при хорошей воспроизводимости и точности результатов анализа.
В результате проведенных экспериментальных исследований выбраны следующие оптимальные условия полярографического определения кобальта в растворах сульфата цинка: состав фонового электролита 2М NH4Cl+2М NH4OH+2•10-3M C4H8O2N2+0,2M NH2OH•HCl; начальное напряжение -1,05 В; скорость катодной развертки потенциала 10 мВ/с; амплитуда развертки 0,25В.
Из представленных на фиг. 15 ДИП кривых видно, что при таком режиме вольтамперометрического анализа обеспечиваются высокая чувствительность и селективность полярографического определения Со(II) в растворах сульфата цинка, содержащих избыточные концентрации Mn(VII). Для сравнения из фиг.16 следует, что в отсутствие гидроксиламина нижний предел обнаружения ионов Со(II) на фоне 2М NH4Cl+2M NH4OH+2•10-3M C4H8O2N2 составляет 0,4 мг/л, а допустимые соотношения [Zn] : [Co] = 10000: 1; [Mn]:[Co]=200:l, что свидетельствует о существенном снижении чувствительности и избирательности анализа.
Градуировочные кривые Со(II) на фоне избранного нами 2М NH4Cl+2M NH4OH+2•10-3 М C4H8O2N2+0,2M NH2OH•HCl линейны в диапазонах концентраций 0-0,025 мг/л и 0-2,5 мг/л. Введение в анализируемый раствор ионов Zn(II) не меняет наклона градуировочного графика (фиг.17), что обеспечивает высокую точность измерений Со(II) независимо от изменения концентрации сульфата цинка.
Изучено влияние на полярографическое определение кобальта наиболее характерных для технологических растворов цинкового производства ионных компонентов, электрохимически восстанавливающихся на ртутном электроде в среде 2М NH4Cl+2M NH4OH+2•10-3M C4H8O2N2+0,2M NH2OH•HCl, в том числе Cu(II), Cd(II), Ni(II), Fe(II), Mn(II), Ge(IV).
Из приведенных в таблице данных видно, что заявляемый способ обеспечивает высокую селективность определения Со(II) в присутствии всех сопутствующих ему в очищенных цинковых растворах веществ.
Присутствующие в цинковом электролите ионы Sb(III) в аммиачной среде гидролизуются и выпадают в осадок (см., например, книгу Пац Р.Г., Васильева Л. Н. Методы анализа с использованием полярографии переменного тока М.: Металлургия, 1967, 116 с.). Ионы Fe(III) химически восстанавливаются до Fe(II) входящим в состав фонового электролита гидроксиламином (см., например, книгу Брикун И. К. , Козловский М.Т., Никитина Л.В. Гидразин и гидроксиламин и их применение в аналитической химии. Алма-Ата: Наука, 176 с.).
Мышьяк (III) в хлоридно-аммиачных фоновых электролитах восстанавливается на ртутном электроде при потенциале -1,7 В и полярографическому определению кобальта не мешает (см., например, книгу Немодрук А.А. Аналитическая химия мышьяка. М.: Наука, 1976. С.79).
Способ может быть реализован на различных отечественных и зарубежных полярографических анализаторах, обеспечивающих проведение измерений в режиме дифференциальной импульсной полярографии на стационарных ртутных электродах, в частности, на приборах ПУ-1, ПЛС, PAR 174, PAR 384, PA-4, АЖЭ-11, АЖЭ-12 и др.
Способ осуществляется следующим образом. В мерную колбу на 50 мл вводят пипеткой 0,5 мл анализируемого раствора, например очищенного цинкового раствора, добавляют 25 мл фонового электролита 4М NH4Cl+4M NH4OH+4•10-3M C4H8O2N2+0,4M NH2OH•HCl и доводят объем раствора в колбе до метки дистиллированной водой. Полученную смесь заливают в электрохимическую ячейку и снимают катодную кривую при линейно-изменяющемся во времени потенциале в интервале (-1,05)-(-1,30) В. Развертку потенциала производят со скоростью 10 мВ/с. Регистрируют серию из 3-5 кривых и, измеряя высоту катодных пиков при потенциале -1,15 В, определяют содержание Со(II) в растворе по градуировочному графику. Общая длительность контроля одной пробы на содержание кобальта при ручной пробоподготовке не превышает 3 мин.
Фоновый электролит состава 4М NH4Cl+4M NH4OH+4•10-3M C4H8O2N2+0,4M NH2OH•HCl готовится из исходных растворов: 5М NH4Cl+5M NН4OH; 0,1М C4H8O2N2 (спиртовый раствор); 2М NH2OH•HCl. Как показали специально проведенные экспериментальные исследования, фоновый электролит сохраняет свои свойства в течение 2-3 месяцев.
При проведении анализа в автоматическом режиме смешение пробы с фоновым электролитом и подачу раствора в ячейку вольтамперометрического датчика производят с помощью трехкамерных дозирующих устройств мембранного типа ДЗЖ-4, выполненных с использованием коррозийностойких материалов (см. статью Боровков Г. А. , Зарецкий Л.С. Автоматическое вольтамперометрическое определение индия в технологических растворах завода "Электроцинк" // Заводская лаборатория. 1986. Т.52. 11. С.7-10).
Пример. Количественное определение кобальта (II) в очищенном растворе сульфата цинка выщелачивательного цеха завода "Электроцинк", содержащем 0,5-2,5 мг/л кобальта; 130-140 г/л цинка; 0,5-1,5 г/л марганца (VII); 10-12 г/л марганца (II); 30-50 мг/л железа; 0,5-1,0 мг/л кадмия; 0,2-1,0 мг/л никеля; 0,1-0,2 мг/л меди; 0,2-0,3 мг/л сурьмы; 0,01-0,05 мг/л мышьяка.
Отбирают пипеткой 0,5 мл анализируемого цинкового раствора и помещают в мерную колбу на 50 мл, добавляют 25 мл фонового электролита 4М NH4Cl+4M NH4OH+4•10-3 М C4H8O2N2+0,4М NH2OH•HCl и доливают колбу до метки дистиллированной водой. Полученную смесь переносят в электрохимическую ячейку датчика со стационарным ртутным электродом и регистрируют катодную ДИП кривую кобальта в интервале напряжений (-1,05)-(-1,30) В со скоростью развертки 10 мВ/с. Измеряют высоту пика при потенциале -1,15 В и определяют содержание Со(II) в анализируемом растворе по градуировочному графику. Всего снимают не менее трех ДИП пиков, при этом концентрацию кобальта в пробе цинкового раствора рассчитывают по среднему арифметическому всех результатов анализа.
Заявляемый способ вольтамперометрического определения кобальта может быть реализован с помощью имеющихся технических средств, например анализаторов АЖЭ-11 и АЖЭ-12 (см. статьи Г.А. Боровков, О.В. Щербич. Автоматизация контроля ионного состава сточных вод завода "Рязцветмет" // Заводская лаборатория. 1991. Т.57. 8. С.9-12 и Г.А. Боровков, О.Г. Виноградов, В.В. Зеленский и др. Опыт разработки и внедрения автоматических электрохимических анализаторов ионного состава флотационных пульп.// Цветные металлы. 1990. 9. С. 108-112).
Структурная схема автоматического вольтамперометрического анализатора АЖЭ-11, предназначенного для контроля ионов кобальта (II) в очищенных цинковых растворах, показана на фиг.18. Анализатор состоит из полярографа ПУ-1 (1), блока управления НВ-83 (2), электрохимического преобразователя (вольтамперометрического датчика) ДТЩ-17 (3) со стационарным ртутно-капельным электродом, вторичного регистрирующего прибора "Диск-250" (4), системы пробоподготовки (5), выполненной на базе мембранных дозаторов жидкости и гидравлически связанной с приемной емкостью для пробы цинкового раствора (6), емкостью с фоновым электролитом (7), емкостью с водой (8) и проточной ячейкой электрохимического преобразователя (3). В приемной емкости (6) установлена электродная система (9) контактного датчика уровня (10), подключенного к блоку управления (2). Электрохимический преобразователь (3) гидравлически связан с ртутным резервуаром (11) и ловушкой для отработанной ртути (12).
Отбор и доставка контролируемых растворов к месту установки анализатора осуществляется с помощью центробежного насоса, работающего в сочетании с рукавным динамическим фильтром (на схеме не показаны). Отфильтрованная проба поступает самотеком в приемную емкость (6). Включение центробежного насоса производится автоматически по команде с блока управления (2) либо вручную технологическим персоналом выщелачивательного цеха.
Важнейшим узлом анализатора является электрохимический преобразователь с рабочим стационарным ртутно-капельным электродом, представляющим собой капиллярный дозатор ртути с клапаном игольчатого типа. Размеры формируемой ртутной капли определяются геометрическими параметрами капилляра и длительностью включения электромагнитного привода клапана, в момент открытия которого обеспечивается свободный исток ртути из связанного с атмосферой резервуара. Сброс капли осуществляется принудительно с помощью механической лопатки с электромагнитным приводом. Наличие в электрохимическом преобразователе электрического двигателя обеспечивает также использование лопатки в качестве мешалки анализируемого раствора.
С целью предотвращения капиллярных шумов, связанных с проникновением анализируемого раствора внутрь рабочего электрода, а также во избежание отрыва ртутной капли при механических воздействиях, например при сильных вибрациях, торец (исток) стеклянного капилляра защищен вваренной в него платиновой втулкой. Необходимые свойства капилляру такой конструкции придают предварительной электрохимической амальгамацией платиновой втулки. При этом капля удерживается на торце капилляра не только за счет сил поверхностного натяжения, но и вследствие химического взаимодействия ртути с покрывающим платину слоем амальгамы.
Схема электрохимической ячейки трехэлектродная (электрод сравнения хлорсеребряный, электрод вспомогательный - стеклоуглеродный). При работе анализатора в режиме дифференциальной импульсной полярографии весь измерительный процесс производится на одной ртутной капле.
Основу автоматической системы пробоподготовки составляют дозирующие устройства ДЗЖ-4, обеспечивающие по заданной программе отбор пробы из приемной емкости, разбавление ее водой, смешение с фоновым электролитом, подачу подготовленного к анализу раствора в проточную ячейку вольтамперометрического датчика с последующим сбросом в дренаж через ловушку отработанной ртути. Дозаторы выполнены из химически стойких материалов и представляют собой мембранные насосы с регулируемой производительностью, снабженные электрическими приводами.
Блок управления программирует работу дозаторов системы пробоподготовки, электромагнитных приводов роста и сброса ртутной капли, а также мешалки электрохимического преобразователя, включение развертки потенциала полярографа и привода диаграммной ленты регистрирующего прибора. Входящее в состав блока управления вычислительное устройство обсчитывает показатели вольтамперной кривой (измеряет высоту ДИП пика кобальта) и выдает электрический сигнал на вторичный прибор, отградуированный в единицах концентрации контролируемого вещества.
Анализатор может работать в непрерывном или дискретном режимах измерения. Дискретность анализа определяется режимом работы системы пробоотбора и прободоставки.
Контроль проб очищенного цинкового раствора на содержание кобальта заявляемым способом осуществляется следующим образом. После заполнения фильтратом цинкового раствора приемной емкости (6) до уровня, установленного электродной системой (9), с выхода контактного датчика (10) поступает сигнал на включение блока управления (2). Программа блока управления начинается с команды на включение дозаторов жидкости системы пробоподготовки (5), осуществляющих отбор пробы из приемной емкости (6), разбавление ее в 50 раз водой, дозируемой из емкости (8), и смешение разбавленной пробы в соотношении 1:1 с фоновым электролитом состава 4М NH4Cl+4M NH4OH+4•10-3M C4H8О2N2+0,4M NH2OH•HCl, хранящимся в емкости (7). Подготовленная к анализу проба закачивается в проточную ячейку электрохимического преобразователя (3). При этом анализируемый раствор обновляется за счет вытеснения предыдущей пробы в дренаж через пороговое отверстие в боковой стенке ячейки. Отработанная ртуть удаляется в ловушку (12) через встроенный в днище ячейки затвор, препятствующий свободному истечению контролируемого раствора в дренажную линию.
Таким образом, в результате всех стадий пробоподготовки на анализ поступает разбавленная водой и обработанная фоновым электролитом проба цинкового раствора. После обновления раствора в ячейке электрохимического преобразователя (3) дозаторы жидкости системы пробоподготовки (5) выключаются и производится полярографическое определение кобальта (II). При этом в соответствии с программой блока управления (2) осуществляются сброс отработанной и формирование свежей ртутной капли, включение развертки потенциала на полярографе (1) и регистрация катодного ДИП пика. Полярографический анализ проводится в среде 2М NH4Cl+2M NH4OH+2•10-3М C4H8O2N2, выходные сигналы с полярографа поступают на вычислительное устройство блока управления, обрабатывающее (обсчитывающее) вольтамперную кривую и выдающее выходной сигнал на вторичный регистрирующий прибор (4), отградуированный в единицах концентрации ионов Со2+.
Автоматическая регистрация вольтамперной кривой производится в интервале потенциалов от (-1,05) до (-1,30) В со скоростью катодной развертки потенциала 10 мВ/с. Измерение концентрации кобальта (II) в растворе осуществляется по амплитуде (высоте) ДИП пика при потенциале -1,15 В (относительно хлорсеребряного электрода сравнения).
По окончании измерения блок управления (2) выключает измерительные приборы анализатора. После поступления в приемную емкость (6) свежего фильтрата цинкового раствора по сигналу с контактного датчика уровня (10) цикл анализа полностью повторяется.
Таким образом, заявляемый способ позволяет максимально упростить пробоподготовку контролируемого раствора, исключив из нее все сложные для автоматизации операции и может быть реализован на освоенных промышленностью анализаторах.
Испытания предлагаемого способа проводились на промышленных пробах очищенного цинкового раствора завода "Электроцинк". Перерабатываемое в настоящее время на этом предприятии сырье (цинковые концентраты) содержит значительные количества кобальта, оказывающего вредное влияние на процесс получения катодного цинка. Технология очистки цинковых растворов выщелачивательного цеха предусматривает окисление Со(II) до Co(III) перманганатом калия с последующим осаждением трехвалентного кобальта в виде гидроксида Со(ОН)3. Для оптимизации процесса очистки растворов сульфата цинка от водорастворимых солей кобальта необходим оперативный контроль остаточной концентрации ионов Со(II) в готовом электролите. В настоящее время для этих целей применяется методика фотоколориметрического анализа, основанная на использовании в качестве селективного комплексообразователя нитрозо-R-соли, образующей в водных растворах красную комплексную соль с ионами Со(II) при рН 5,5 (см., например, книгу Новиков Ю.В., Ласточкина К.О., Болдина З.Н. Методы исследования качества воды водоемов. М. : Медицина, 1990. С.185-186). Фотометрическое определение кобальта предусматривает сложную пробоподготовку цинковых растворов, включающую обработку их перекисью водорода, ацетатом натрия, охлаждение, что существенно увеличивает длительность анализа и практически полностью исключает его автоматизацию. Фотометрический контроль кобальта производится в экспресс-лаборатории выщелачивательного цеха не чаще 3-4 раз в смену, что не позволяет достичь качественной очистки цинкового электролита и приводит к перерасходу дорогостоящего перманганата калия.
В период проведения испытаний пробы отбирались из сборников очищенного раствора сульфата цинка. По данным ОТК завода "Электроцинк" концентрация ионных компонентов в растворах, поступающих на электролиз, в среднем изменяется в следующих пределах, мг/л: Zn(130-140)(103; Mn(VII) 500-1500; Ni(II) 0,2-1,0; Cu(II) 0,1-0,2; Cd(II) 0,5-1,0; Fe(II) 30-50; Co(II) 0,5-2,5; Sb 0,2-0,3; As 0,01-0,05.
Во время испытаний в качестве контрольного использовался метод стандартных добавок. Всего было проанализировано 35 проб очищенного цинкового раствора, концентрация ионов Со(II) в которых изменялась в пределах от 0,48 до 2,48 мг/л. При этом приведенная среднеквадратичная погрешность анализа кобальта не превысила 2,11%.
Заявляемый способ может быть использован для автоматического или экспресс-анализа кобальта в технологических растворах цинкового производства, а также в сточных и оборотных водах предприятий цветной металлургии.
Внедрение предлагаемого способа в аналитическую практику цинкового производства позволит значительно повысить оперативность контроля ионов Со(II), оптимизировать технологический процесс очистки растворов сульфата цинка от примесей кобальта и в конечном итоге улучшить технико-экономические показатели производства катодного цинка. Использование заявляемого способа для экомониторинга сложных по составу сточных вод обеспечит снижение выброса ионов Со(II) в открытые водоемы в зоне действия предприятий цветной металлургии.






Изобретение относится к аналитической химии и может быть использовано для автоматического или экспресс-анализа технологических растворов цинкового производства, а также сточных или оборотных вод предприятий цветной металлургии. Сущность: способ основан на вольтамперометрическом определении кобальта на стационарном ртутном электроде в хлоридно-аммиачной среде в присутствии диметилглиоксима и гидроксиламина. Измерение производят на фоне 2М NH4Cl+2M NH4OH+2•10-3M C4H8O2N2+0,2M NH2OH•HCl методом дифференциальной импульсной полярографии. Вольтамперную кривую регистрируют в диапазоне напряжений (-1,05) - (-1,30)В со скоростью развертки потенциала 10 мВ/с. Содержание Со (II) определяют по высоте катодного пика при потенциале -1,5±0,01В (отн. х. с. э). Технический результат изобретения заключается в повышении избирательности и чувствительности анализа. 1 табл., 18 ил.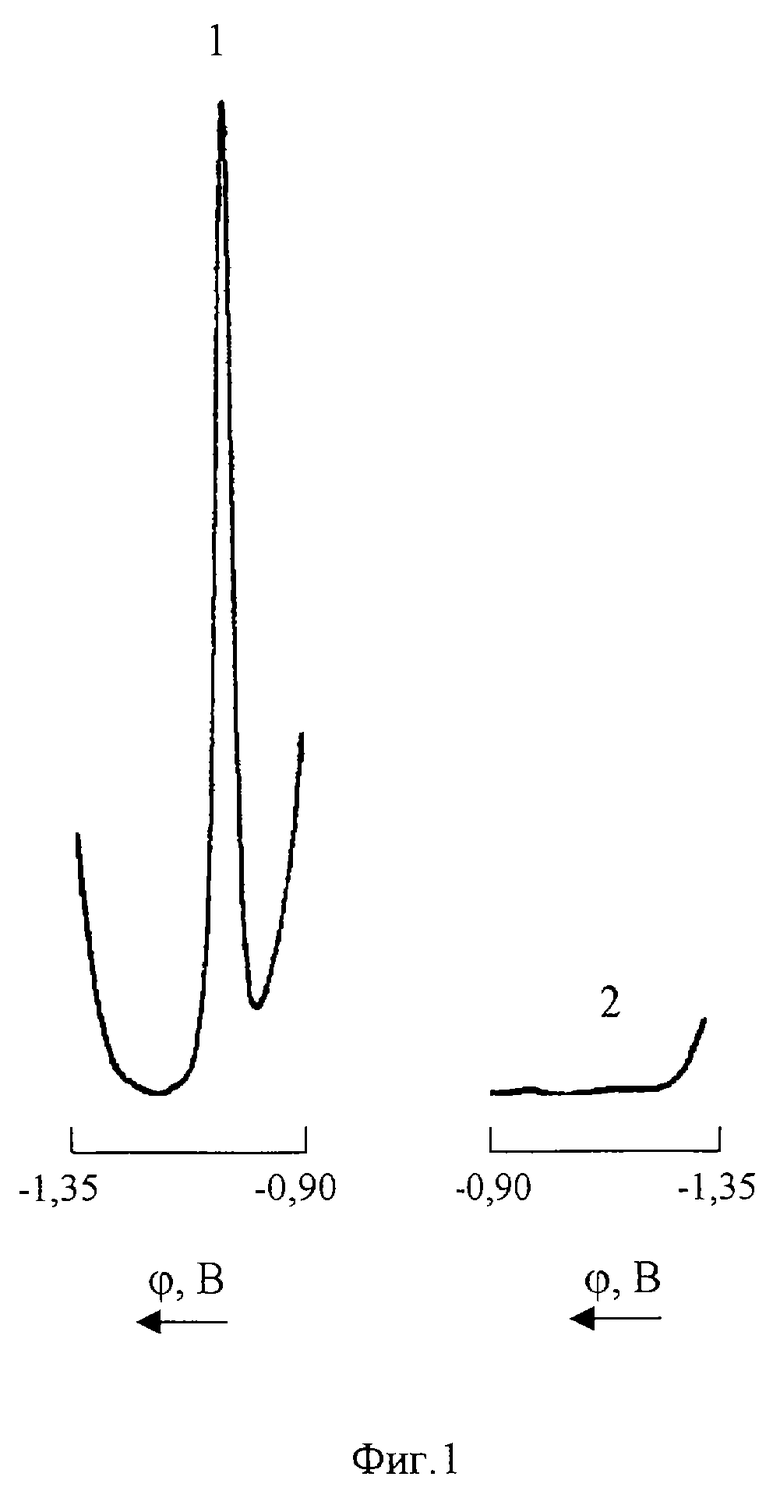
Способ вольтамперометрического определения концентрации ионов кобальта (II) в растворах сульфата цинка, включающий наложение линейно изменяющегося потенциала на индикаторный электрод и регистрацию вольтамперной кривой на фоне хлоридно-аммиачного буферного электролита, содержащего диметилглиоксим, отличающийся тем, что в анализируемый раствор дополнительно вводят соляно-кислый гидроксиламин, а полярографирование ведут на фоне 2М NH4Cl + 2M NH4OH + 2 • 10-3M C4H8O2N2 + 0,2M NH2OH • HCl в диапазоне напряжений (-1,05) - (-1,30) В, а концентрацию кобальта измеряют по высоте катодного пика при потенциале - 1,15±0,01 В.
| МАНИТА М.Д | |||
| и др | |||
| Современные методы определения атмосферных загрязнений населенных мест | |||
| - М.: Медицина, 1980, с.198-199 | |||
| Способ вольтамперометрии | 1987 |
|
SU1518767A1 |
| Способ определения микроколичеств кобальта методом переменнотоковой полярографии | 1982 |
|
SU1022037A1 |
| Способ полягрофического определения меди, никеля и кобальта | 1974 |
|
SU502303A1 |

