Изобретение относится к способу получения эргостерола и его промежуточных продуктов с использованием рекомбинантных дрожжей и плазмид для трансформации дрожжей.
Эргостерол представляет собой конечный продукт синтеза стерола в дрожжах и грибах. Экономическое значение этого соединения состоит, с одной стороны, в том, что из эргостерола с помощью УФ-облучения получают витамин D2, а с другой стороны, в том, что из эргостерола посредством биотрансформации получают стероидные гормоны. Для синтеза терпенов в качестве компонента используют сквален. Он находит применение в гидрированной форме под названием сквалан в дерматологии и косметике, а также в виде различных производных в качестве ингредиента в средствах по уходу за кожей и волосами. Экономическое значение также имеют промежуточные продукты пути метаболизма эргостерола. В качестве наиболее важных продуктов в данном случае следует отметить фарнезол, гераниол и сквален. Кроме того, практическое значение имеют такие стеролы, как, например, зимостерол и ланостерол, причем ланостерол является основным исходным продуктом и имеет решающее значение при химическом синтезе сапонинов и стероидных гормонов. Благодаря своей хорошей способности проникать через кожу и хорошим характеристикам растворимости ланостерол применяется в качестве вспомогательного и действующего вещества при приготовлении эмульсий, используемых в кремах для кожи.
Широко известны гены пути метаболизма эргостерола в дрожжах и способы их клонирования, например, такие как ген HMG-CoA-редуктазы (HMGl) (Basson и др. (1988)), ген скваленсинтетазы (ERG9) (Fegueur и др. (1991)), ген ацил-СоА:стеролацилтрансферазы (SAT1) (Yu и др. (1996)) и ген скваленэпоксидазы (ERG1) (Jandrositz и др. (1991)). Скваленсинтетаза катализирует реакцию превращения фарнезилпирофосфата в сквален через получение промежуточного продукта прескваленпирофосфата. Механизмы действия стеролацилтрансферазы не полностью изучены. Ранее проводились опыты с использованием сверхэкспрессии генов указанных выше ферментов, однако это не привело к какому-либо заметному повышению количества эргостерола. Имеются данные о том, что сверхэкспрессия HMG1 приводит к сверхпродуцированию сквалена, при этом вводили дополнительные мутации для прерывания пути метаболизма сквалена (ЕР 0486290). Также описано сверхпродуцирование гераниола и фарнезола, однако при этом осуществляли не сверхэкспрессию генов пути метаболизма эргостерола, а прерывали путь реакции в направлении образования гераниола и фарнезола (ЕР 0313465).
Определенные ингибиторы биосинтеза эргостерола также могут приводить к накоплению повышенных количеств некоторых промежуточных продуктов, например аллиламина, которые препятствуют превращению сквалена в эпоксид сквалена. В результате этого накапливаются большие количества сквалена (до 600 раз превышающие нормальный уровень) (Jandrositz и др. (1991)).
Хотя с использованием ингибиторов может быть достигнуто значительное накопление продукта, например сквалена, тем не менее дальнейшее добавление этих веществ может оказаться неэффективным, так как даже незначительные их количества проявляют в организме такое же действие, как и их повышенные количества, поэтому предпочтительно получение продуктов биосинтеза эргостерола за счет сверхпродуцирования.
Задачей настоящего изобретения является разработка микробиологического способа получения эргостерола и его промежуточных продуктов, создание необходимых для этого микроорганизмов, таких как штаммы дрожжей, которые способны синтезировать увеличенные количества эргостерола, соответственно необходимых для его синтеза промежуточных продуктов и плазмид, необходимых для трансформации штаммов дрожжей.
При создании изобретения было установлено, что количество эргостерола и его промежуточных продуктов может быть увеличено, если в микроорганизмы, такие как, например, дрожжи, встраивают в измененной форме гены HMG1 (Basson и др. (1988)), ERG9 (Fegueur и др. (1991), Current Genetics 20:365-372), SAT1 (Yu и др. (1996)) и ERG1 (Jandrositz и др. (1991)), при этом гены могут быть локализованы либо по отдельности на одной плазмиде, либо в виде комбинации друг с другом на одной или нескольких плазмидах, и они могут быть встроены в хозяина одновременно или последовательно.
Таким образом, объектом настоящего изобретения является способ, отличающийся тем, что
а) сначала конструируют плазмиду, в которую встраивают несколько соответствующих генов пути метаболизма эргостерола в измененной форме или
б) сначала конструируют плазмиды, в каждую из которых встраивают один из генов путем метаболизма эргостерола в измененной форме,
в) созданными таким путем плазмидами трансформируют микроорганизмы, причем микроорганизмы трансформируют одной плазмидой, указанной в пункте а) или трансформируют одновременно или последовательно несколькими плазмидами, указанными в пункте б),
г) с использованием созданных таким путем микроорганизмов осуществляют ферментацию с получением эргостерола,
д) после ферментации эргостерол и его промежуточные продукты экстрагируют из клеток и анализируют и в завершение
е) полученный в результате эргостерол и его промежуточные продукты очищают и выделяют с помощью хроматографии на колонках.
Объектом настоящего изобретения является прежде всего способ, отличающийся тем, что
а-I) сначала конструируют плазмиду, в которую встраивают следующие гены:
I) ген HMG-CoA-редуктазы (t-HMG),
II) ген скваленсинтетазы (ERG9),
(III) ген ацил-СоА:стеролацилтрансферазы (SAT1) и
(IV) ген скваленэпоксидазы (ERG1), или
а-II) сначала конструируют плазмиду, в которую встраивают следующие гены:
I) ген HMG-CoA-редуктазы (t-HMG) и
II) ген скваленсинтетазы (ERG9), или
a-III) сначала конструируют плазмиду, в которую встраивают следующие гены:
I) ген HMG-CoA-редуктазы (t-HMG) и
(III) ген ацил-СоА:стеролацилтрансферазы (SAT1), или
a-IV) сначала конструируют плазмиду, в которую встраивают следующие гены:
I) ген HMG-CoA-редуктазы (t-HMG) и
(IV) ген скваленэпоксидазы (ERG1), или
a-V) сначала конструируют плазмиду, в которую встраивают следующие гены:
II) ген скваленсинтетазы (ERG9) и
(III) ген ацил-СоА:стеролацилтрансферазы (SAT1), или
а-VI) сначала конструируют плазмиду, в которую встраивают следующие гены:
II) ген скваленсинтетазы (ERG9) и
(IV) ген скваленэпоксидазы (ERG1), или
а-VII) сначала конструируют плазмиду, в которую встраивают следующие гены:
(III) ген ацил-СоА:стеролацилтрансферазы (SAT1) и
(IV) ген скваленэпоксидазы (ERG1), или
б) сначала конструируют плазмиды, в каждую из которых встраивают один из генов, указанных в пункте а-1), и
в) созданными таким путем плазмидами трансформируют микроорганизмы, причем микроорганизмы трансформируют одной плазмидой, указанной в пунктах с a-I) no a-VII), или трансформируют одновременно или последовательно несколькими плазмидами, указанными в пункте б),
г) с использованием созданных таким образом микроорганизмов осуществляют ферментацию с получением эргостерола,
д) после ферментации эргостерол и его промежуточные продукты экстрагируют из клеток и анализируют и в завершение
е) полученный в результате эргостерол и его промежуточные продукты очищают и выделяют с помощью хроматографии на колонках.
В плазмиды, перечисленные в пунктах а-II), a-III) и a-V), может быть дополнительно встроен ген скваленэпоксидазы (ERG1), а в плазмиду, указанную в пункте а-II), может быть дополнительно встроен ген ацил-СоА:стеролацилтрансферазы (SAT1). Эти плазмиды также являются объектом настоящего изобретения.
Под промежуточными продуктами в контексте настоящего описания следует понимать сквален, фарнезол, гераниол, ланостерол, зимостерол, 4,4-диметилзимостерол, 4-метилзимостерол, эргост-7-енол и эргоста-5,7-диенол, прежде всего стерол с 5,7-диеновой структурой.
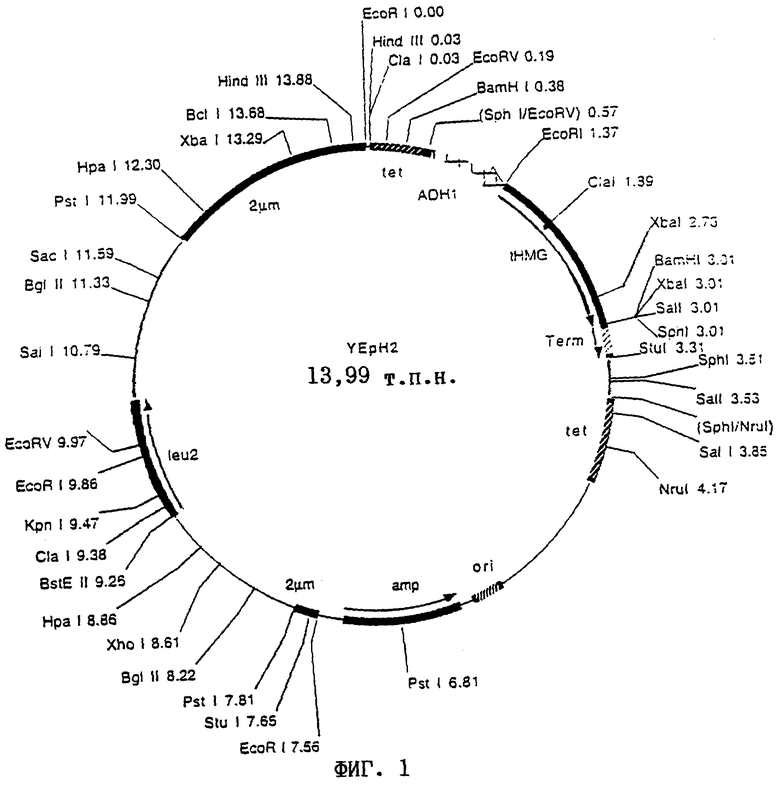
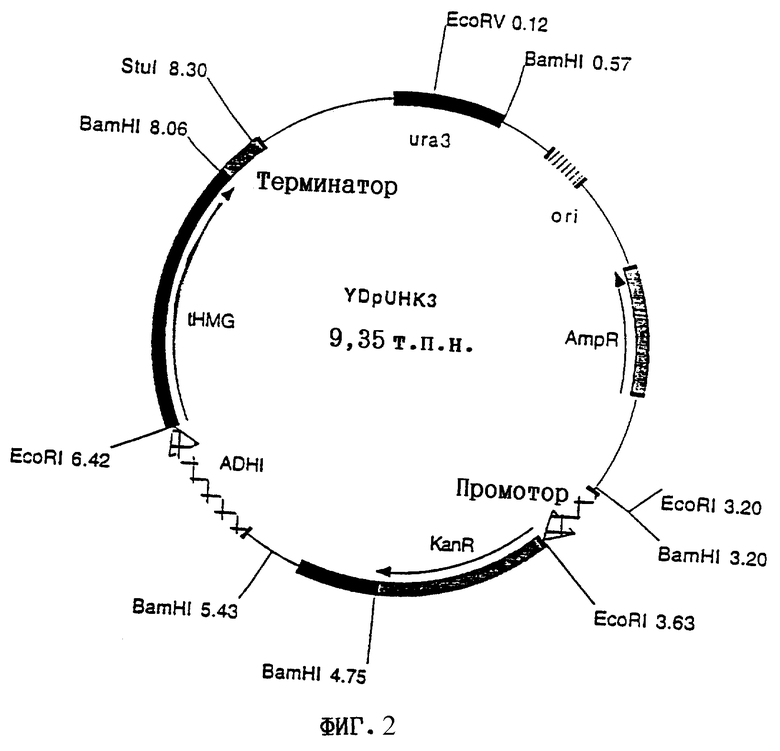
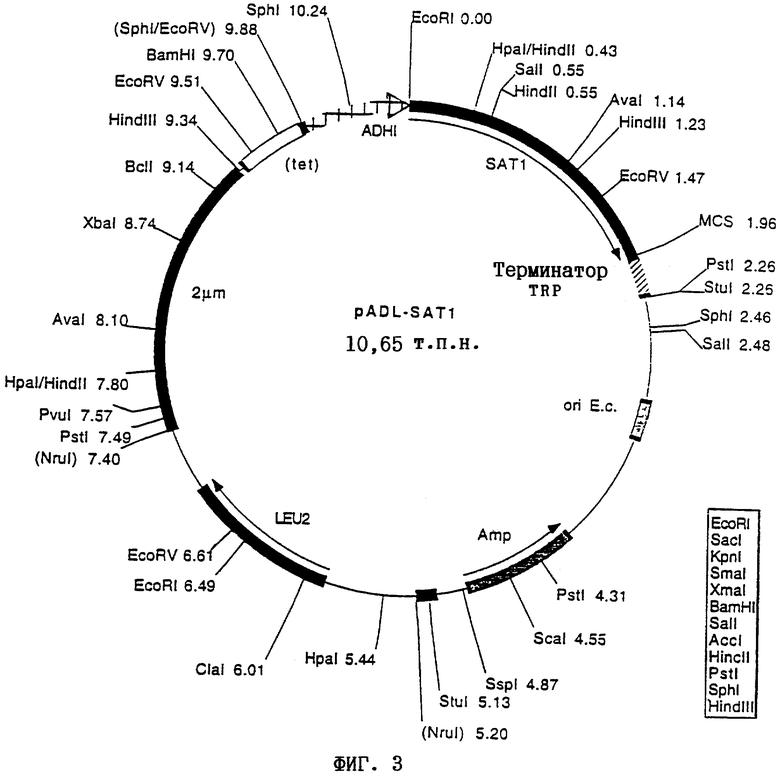
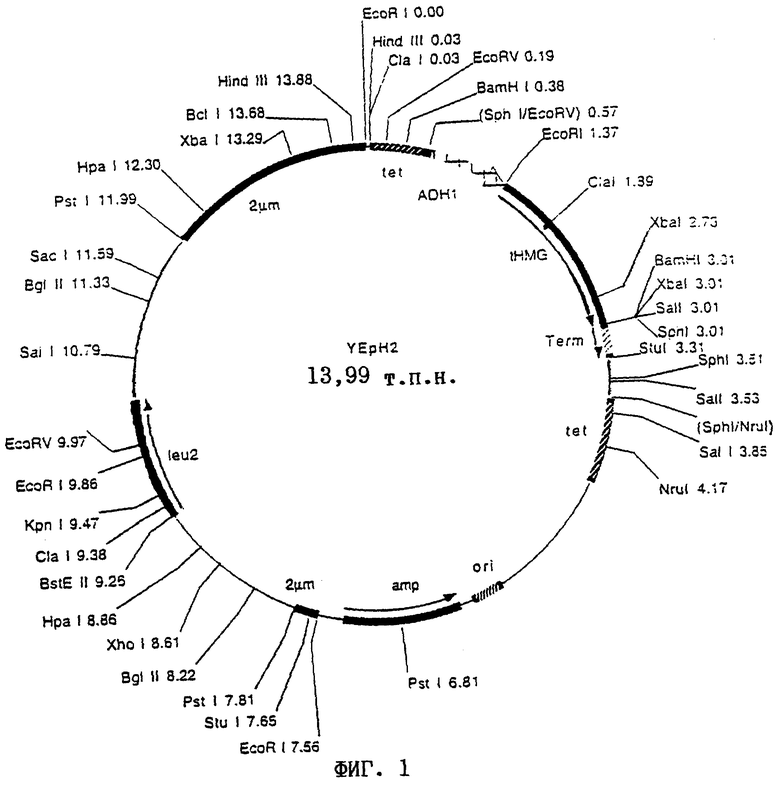
В качестве плазмид предпочтительно используют плазмиду YEpH2, которая содержит средний промотор ADH, t-HMG (модифицированный вариант HMG1) и терминатор TRP (см. фиг.1), плазмиду YDpUHKS, которая содержит средний промотор ADH, t-HMG (модифицированный вариант HMG1) и терминатор TRP, ген, придающий устойчивость к канамицину, и ген ura3 (см. фиг.2) и плазмиду pADL-SAT1, которая содержит ген SAT1 и ген LEU2 из YEpl3.
Эти плазмиды и их применение для получения эргостерола и его промежуточных продуктов, таких как сквален, фарнезол, гераниол, ланостерол, зимостерол, 4,4-диметилзимостерол, 4-метилзимостерол, эргост-7-енол и эргоста-5,7-диенол, прежде всего стерол с 5,7-диеновой структурой, также являются объектом настоящего изобретения.
В качестве хозяина для встраивания плазмид по изобретению в принципе пригодны любые микроорганизмы, прежде всего дрожжи.
Предпочтительным является вид S. cerevisiae, прежде всего штамм S. cerevisiae AH22.
Объектом настоящего изобретения также является штамм дрожжей S. cerevisiae AH22, несущий один или несколько генов, указанных в пункте а-I) способа.
Также объектом настоящего изобретения является штамм дрожжей S. cerevisiae AH22, несущий плазмиду pADL-SAT1.
Кроме того, предпочтительной является комбинированная трансформация микроорганизмов, прежде всего дрожжей, таких как штамм S. cerevisiae AH22, плазмидами pADL-SAT1 и YDpUHKS.
В целом на интенсивность пути метаболизма эргостерола воздействуют следующим образом:
Интенсивность синтеза эргостерола максимально повышают за счет того, что одновременно усиливают активность нескольких наиболее важных ферментов. При этом важную роль играют различные ферменты, причем решающее значение для повышения выхода эргостерола имеет определенное сочетание дерегуляции, соответственно сверхэкспрессии. Для получения такого действия ферменты, соответственно их гены HMG1 (Basson и др. (1988)), ERG9 (Fegueur и др. (1991)), ацил-СоА:стеролацилтрансферазы (SAT1) (Yu и др. (1996)) и/или скваленэпоксидазы (ERG1) (Jandrositz и др. (1991)) встраивают в штамм дрожжей в измененной форме, причем встраивание генов осуществляют с помощью одной или нескольких плазмид, при этом последовательности ДНК в плазмиде(ах) содержатся по отдельности или в комбинации друг с другом. В случае гена HMG1 "измененный" означает, что у соответствующего гена экспрессируется только каталитическая область без связывающегося с мембраной домена. Такие изменения уже были описаны ранее (ЕР-0486290). Цель изменения гена HMG1 заключается в том, чтобы воспрепятствовать регуляции по типу обратной связи с помощью промежуточных продуктов биосинтеза эргостерола. Как у гена HMG1, так и у обоих других названных генов одинаковым образом нарушают регуляцию транскрипции. Для этого промотор генов заменяют "средним" промотором ADH1. Этот промоторный фрагмент промотора ADH1 обладает почти конститутивной экспрессией (Ruohonen и др. (1995)), в результате чего регуляция транскрипции больше не распространяется на промежуточные продукты биосинтеза эргостерола.
Образующиеся в результате сверхэкспрессии продукты могут быть использованы для биотрансформаций, соответственно других химических и терапевтических целей, например для получения витамина D2 из эргостерола под действием УФ-облучения и для получения стероидных гормонов путем биотрансформации с использованием в качестве исходного продукта эргостерола.
Объектом настоящего изобретения также являются микроорганизмы, прежде всего штаммы дрожжей, которые в результате сверхэкспрессии генов, указанных в пункте а-I) способа, могут продуцировать увеличенное количество эргостерола и эргостерола в сочетании с увеличенными количествами сквалена.
Предпочтительным является измененный вариант гена HMGl, в котором происходит экспрессия только каталитической области без связанного с мембраной домена. Эта модификация описана в ЕР 0486290.
Объектом настоящего изобретения также является способ получения эргостерола и его промежуточных продуктов, отличающийся тем, что гены (комбинации из двух, трех и четырех генов), указанные в пункте а) способа, прежде всего в пунктах с a-I) no a-VII) способа, сначала встраивают с помощью плазмид независимо друг от друга в микроорганизмы одного и того же вида и с их помощью проводят общую ферментацию с получением эргостерола, полученный таким образом эргостерол экстрагируют из клеток, анализируют и затем очищают и выделяют с помощью хроматографии на колонках.
Объектами настоящего изобретения также являются кассеты экспрессии, включающие средний промотор ADH, ген t-HMG, терминатор TRP и ген SAT1 со средним промотором ADH и терминатором TRP, и кассеты экспрессии, включающие средний промотор ADH, ген t-HMG, терминатор TRP, ген SAT1 со средним промотором ADH и терминатором TRP, и ген ERG9 со средним промотором ADH и терминатором TRP.
Объектом настоящего изобретения также является такая комбинация кассет экспрессии, которая состоит из
а) первой кассеты экспрессии, в которой локализованы промотор ADH, ген t-HMG и терминатор TRP,
б) второй кассеты экспрессии, в которой локализованы промотор ADH, ген SAT1 и терминатор TRP, и
в) третьей кассеты экспрессии, в которой локализованы промотор ADH, ген ERG9 и терминатор TRP.
Кроме того, объектом настоящего изобретения является применение этих кассет экспрессии для трансформации микроорганизмов, применяемых для ферментативного получения эргостерола, причем микроорганизмы предпочтительно представляют собой дрожжи.
Объектом настоящего изобретения являются далее микроорганизмы, такие как дрожжи, содержащие эти кассеты экспрессии, а также их применение для ферментативного получения эргостерола и промежуточных продуктов эргостерола.
Ниже подробно рассмотрены методы, необходимые для осуществления изобретения, поясняемого на примерах.
1. Рестрикция
Рестрикцию (расщепление с помощью рестриктаз) плазмид (от 1 до 10 мкг) проводили в смесях объемом по 30 мкл. Для этого ДНК разводили в 24 мкл Н2О и добавляли 3 мкл соответствующего буфера, 1 мкл БСА (бычий сывороточный альбумин) и 2 мкл фермента. Концентрация фермента составляла 1 ед./мкл или 5 ед./мкл в зависимости от количества ДНК. В отдельных случаях к смеси добавляли еще 1 мкл РНКазы для разложения тРНК. Смесь для рестрикции инкубировали в течение 2 ч при 37°С. Рестрикцию контролировали с помощью мини-геля.
2. Гель-электрофорез
Гель-электрофорез проводили с использованием устройств для мини-геля или широкого мини-геля. Мини-гели (приблизительно 20 мл, 8 карманов) и широкие мини-гели (50 мл, 15 или 30 карманов) состояли из 1%-ной агарозы в ТАЕ. В качестве буфера для разделения использовали однократный буфер ТАЕ (1×TAE). Образцы (10 мкл) дополняли 3 мкл стоп-раствора и загружали. В качестве стандарта использовали 1-ДНК (индикаторную ДНК), расщепленную с помощью HindIII (полосы соответствуют 23,1 т.п.н., 9,4 т.п.н., 6,6 т.п.н., 4,4 т.п.н., 2,3 т.п.н., 2,0 т.п.н., 0,6 т.п.н.). Для разделения применяли напряжение 80 В в течение 45-60 мин. После этого гель окрашивали раствором бромида этидия и регистрировали при УФ-свете с помощью видеозаписывающей системы INTAS или фотографировали с использованием оранжевого фильтра.
3. Элюирование из геля
Элюированием из геля выделяли нужные фрагменты. Смесь для рестрикции вносили в несколько карманов с мини-гелем и разделяли. Раствором бромида этидия окрашивали только λ-HindIII и продукты, полученные в результате расщепления ("след жертвы"), анализировали при УФ-свете и маркировали нужный фрагмент. При этом принимали меры для того, чтобы бромид этидия и УФ-свет не повреждали ДНК в остальных карманах. После наложения окрашенных и неокрашенных кусочков геля с использованием маркировки можно было выделить из неокрашенного кусочка геля требуемый фрагмент. Кусочек агарозы с выделенным фрагментом переносили в камеру для диализа, покрывали небольшим количеством буфера ТАЕ, не содержащего пузырьков воздуха, и вносили в устройство для анализа мини-геля фирмы BioRad. В качестве буфера для разделения использовали 1×TAE при напряжении в 100 В, приложенном в течение 40 мин. После этого на 2 мин изменяли полярность тока с целью снова отделить ДНК, приклеившуюся к камере для диализа. Содержащий фрагменты ДНК буфер из камеры для диализа переносили в реакционный сосуд и осаждали этанолом. Для этого к раствору, содержащему ДНК, добавляли 1/10 объема 3М ацетата натрия, тРНК (1 мкл на 50 мкл раствора) и 2,5-кратный объем 96%-ного ледяного этанола. Смесь инкубировали в течение 30 мин при -20°С, после чего центрифугировали при 12000 об/мин в течение 30 мин при 4°С. Содержащий ДНК дебрис сушили и растворяли в 10-50 мкл Н2О (в зависимости от количества ДНК).
4. Обработка по методу Кленова
Путем обработки по методу Кленова дефосфорилировали выступающие концы фрагментов ДНК таким образом, чтобы получить "затупленные концы". Пипеткой вносили следующую смесь (на 1 мкг ДНК):

При этом необходимо использовать ДНК, полученную осаждением из этанола во избежание ингибирования полимеразы Кленова примесями. Инкубацию проводили в течение 30 мин при 37°С, затем реакцию прекращали, выдерживая еще в течение 5 мин при 70°С. ДНК выделяли из смеси осаждением этанолом и растворяли в 10 мкл Н2О.
5. Лигирование
Очищали предназначенные для лигирования фрагменты ДНК. Конечный объем, составляющий 13,1 мкл, содержал приблизительно 0,5 мкг ДНК с соотношением вектор-вставка, равным 1:5. Образец инкубировали в течение 45 с при 70°С, охлаждали до комнатной температуры (приблизительно в течение 3 мин) и затем инкубировали в течение 10 мин на льду. После этого добавляли буфер для лигирования, содержащий 2,6 мкл 500 мМ Трис-HCl, рН 7,5 и 1,3 мкл 100 мМ MgCl2, и инкубировали на льду еще в течение 10 мин. После добавления 1 мкл 500 мМ ДТТ и 1 мкл 10 мМ АТФ и выдержки на льду еще в течение 10 мин добавляли 1 мкл лигазы (1 ед./мл). Всю процедуру осуществляли по возможности без встряхивания во избежание повторного разъединения соприкасающихся концов ДНК. Лигирование осуществляли в течение ночи при 14°С.
6. Трансформация Е. coli
Компетентные клетки штамма Escherichia coli (E. coli) NM522 трансформировали с помощью продуктов лигирования ДНК. В качестве позитивного контроля применяли продукт, содержащий 50 нг плазмиды pScL3, а в качестве нулевого контроля - продукт, не содержащий ДНК. Для получения каждого продукта трансформации в центрифужные пробирки горизонтальной центрифуги вносили пипеткой по 100 мкл 8%-ного раствора ПЭГ, 10 мкл ДНК и 200 мкл компетентных клеток (штамм Е. coli NM522). Продукты выдерживали в течение 30 мин на льду и периодически встряхивали. После этого воздействовали тепловым шоком в течение 1 мин при 42°С. Для регенерации клетки переносили в 1 мл LB-среды и инкубировали в течение 90 мин при 37°С на шейкере. На пластины с LB-средой, дополненной ампициллином, высевали каждый раз по 100 мкл неразведенного продукта, продукта, полученного в результате разведения 1:10, и продукта, полученного в результате разведения 1:100, и инкубировали в течение ночи при 37°С.
7. Выделение плазмиды из Е. coli (мини-препарат)
Колонии Е. coli выращивали в течение ночи в центрифужных пробирках горизонтальной центрифуги, содержащих 1,5 мл LB-среды, дополненной ампициллином, при 37°С и 120 об/мин. На следующий день клетки центрифугировали в течение 5 мин при 4°С и 5000 об/мин и клеточный дебрис переносили в 50 мкл ТЕ-буфера. В каждую порцию смеси добавляли 100 мкл 0,2 н. раствора NaOH, 1%-ный раствор додецилсульфата натрия (ДСН), перемешивали и выдерживали на льду в течение 5 мин (лизис клеток). После этого добавляли 400 мкл раствора ацетата натрия/NaCl (230 мкл Н2О, 130 мкл 3М ацетата натрия, 40 мкл 5М NaCO, смесь перемешивали и выдерживали на льду еще в течение 15 мин (осаждение протеина). После центрифугирования в течение 15 мин при 11000 об/мин надосадочную жидкость, содержащую плазмидную ДНК, переносили в сосуд Эппендорфа. Если надосадочная жидкость была недостаточно прозрачной, ее еще раз подвергали центрифугированию. В надосадочную жидкость вносили 360 мкл охлажденного на льду изопропанола и инкубировали в течение 30 мин при -20°С (осаждение ДНК). ДНК центрифугировали (15 мин, 12000 об/мин, 4°С), отбрасывали надосадочную жидкость, клеточный дебрис промывали в 100 мкл охлажденного на льду 96%-ного этанола, инкубировали в течение 15 мин при -20°С и снова центрифугировали (15 мин, 12000 об/мин, 4°С). Клеточный дебрис сушили в устройстве типа Speed Vac и переносили в 100 мкл Н2О. Характеристики плазмидной ДНК определяли с помощью рестрикционного анализа. Для этого подвергали рестрикции по 10 мкл каждого образца и разделяли с помощью гель-электрофореза в широком мини-геле (см. выше).
8. Выделение плазмиды из Е. coil (макси-препарат)
Для выделения больших количеств плазмидной ДНК применяли метод макси-препаратов. В две колбы, содержащие 100 мл LB-среды, дополненной ампициллином, вносили одну колонию, соответственно 100 мкл замороженной культуры, содержащей выделенную плазмиду, и инкубировали в течение ночи при 37°С и 120 об/мин. Выращенный продукт (200 мл) переносили на следующий день в GSA-стакан и центрифугировали в течение 10 мин при 4000 об/мин (2600×g). Клеточный дебрис переносили в 6 мл ТЕ-буфера. Для разложения клеточной стенки добавляли 1,2 мл раствора лизозима (20 мг/мл ТЕ-буфера) и инкубировали в течение 10 мин при комнатной температуре. После этого проводили лизис клеток с помощью 12 мл 0,2 н. раствора NaOH, 1%-ного раствора ДСН и инкубировали еще в течение 5 мин при комнатной температуре. Протеин осаждали путем добавления 9 мл охлажденного 3М раствора ацетата натрия (рН 4,8) и 15-минутной инкубации на льду. После центрифугирования (GSA: 13000 об/мин (27500×g), 20 мин, 4°С) надосадочную жидкость, содержащую ДНК, переносили в новый GSA-стакан и осаждали ДНК с помощью 15 мл ледяного изопропанола и инкубации в течение 30 мин при -20°С. Содержащий ДНК дебрис промывали 5 мл охлажденного на льду этанола и сушили на воздухе (приблизительно 30-60 мин). После этого его переносили в 1 мл Н2О. С помощью рестрикционного анализа исследовали плазмиду. Концентрацию определяли путем нанесения разведений на мини-гель. Для уменьшения содержания соли в течение 30-60 мин проводили микродиализ (размер пор 0,025 мкм).
9. Трансформация дрожжей
Для трансформации дрожжей применяли предварительно выращенную культуру штамма Saccharomyces cerevisiae AH22. В колбу, содержащую 20 мл YE-среды, вносили 100 мкл замороженной культуры и инкубировали в течение ночи при 28°С и 120 об/мин. Основную культуру выращивали в таких же условиях в колбах со 100 мл YE-среды, в которые вносили 10 мкл, 20 мкл или 50 мкл предварительной культуры.
9.1 Получение компетентных клеток
На следующий день с помощью камеры Тома определяли концентрацию клеток в колбах и колбы, в которых концентрация составляла 3-5×107 клеток/мл, подвергали дальнейшей обработке. Клетки собирали центрифугированием (GSA: 5000 об/мин (4000×g), 10 мин). Клеточный дебрис переносили в 10 мл ТЕ-буфера и распределяли по двум центрифужным пробиркам горизонтальной центрифуги (в каждую вносили по 5 мл). Клетки центрифугировали в течение 3 мин при 6000 об/мин и еще дважды промывали, используя каждый раз по 5 мл ТЕ-буфера. Затем клеточный дебрис вносили в литийацетатный буфер (330 мкл на 109 клеток), переносили в стерильные колбы Эрленмейера объемом 50 мл и встряхивали в течение 1 ч при 28°С. Таким образом получали компетентные для трансформации клетки.
9.2 Трансформация
Для получения каждой порции смеси для трансформации в центрифужные пробирки горизонтальной центрифуги вносили пипеткой 15 мкл ДНК спермы сельди (10 мг/мл), 10 мкл предназначенной для трансформации ДНК (приблизительно 0,5 мкг) и 330 мкл компетентных клеток и инкубировали в течение 30 мин при 28°С (без встряхивания!). После этого добавляли 700 мкл 50%-ного ПЭГ 6000 и инкубировали еще в течение 1 ч при 28°С без встряхивания. Затем смесь подвергали действию теплового шока в течение 5 мин при 42°С.
100 мкл суспензии высевали на среду для селекции (YNB, фирма Difco) с целью проведения отбора по признаку прототрофии в отношении лейцина. В случае отбора по признаку устойчивости к G418 после теплового шока проводили регенерацию клеток (см. ниже п.9.3 "Фаза регенерации").
9.3 Фаза регенерации
Поскольку селектируемый маркер придает устойчивость к G418, клеткам требуется некоторое время для экспрессии гена, обусловливающего устойчивость. Продукты трансформации переносили в 4 мл YE-среды и инкубировали в течение ночи при 28°С на шейкере (120 об/мин). На следующий день отделенные центрифугированием клетки (6000 об/мин, 3 мин) вносили в 1 мл YE-среды и высевали 100 мкл, соответственно 200 мкл этого продукта на пластины, содержащие среду YE+G418. Пластины инкубировали в течение нескольких дней при 28°С.
10. Реакционные условия для ПЦР
Реакционные условия для полимеразной цепной реакции необходимо оптимизировать в каждом конкретном случае и нельзя использовать одинаковые условия для всех продуктов. Так, можно среди прочего варьировать используемые количества ДНК, концентрации соли и температуры плавления. Для рассматриваемых целей оказалось предпочтительным в колпачке Эппендорфа, предназначенном для загрузки в термоячейку, объединять следующим образом перечисленные ниже вещества: к 2 мкл (-0,1 ед.) полимеразы Super Taq добавлять 5 мкл "супер"-буфера, по 8 мкл каждого из дНТФ (в каждом случае в концентрации 0,625 мкМ), 5'-праймер, 3'-праймер и 0,2 мкг матричной ДНК, растворенных в таком количестве воды, чтобы получить суммарный объем смеси для ПЦР, равный 50 мкл. Смесь центрифугировали в течение небольшого промежутка времени и покрывали каплей масла. Амплификацию проводили с использованием от 37 до 40 циклов.
В приведенных ниже примерах описано создание плазмид и штаммов дрожжей по изобретению, а также их применение, однако эти примеры не ограничивают объем изобретения.
Пример 1
Экспрессия tHMG в S. cerevisiae AH22
Последовательность ДНК tHMG (Basson и др. (1988)) амплифицировали с помощью ПЦР из геномной ДНК штамма Saccharomyces cerevisiae S288C (Mortimer и Johnston (1986)) с использованием стандартных методов. Используемые для этого праймеры представляли собой ДНК-олигомеры tHMG-5' и tHMG-3' (см. SEQ ID N0:l и SEQ ID N0:2). Полученный фрагмент ДНК после обработки по методу Кленова встраивали в клонирующий вектор pUC19 (Yanisch-Perron и др. (1985)), получая вектор pUC19-tHMG. После выделения плазмиды и расщепления pUC19-tHMG эндонуклеазами (рестриктазами) EcoRI и BamHI полученный фрагмент встраивали в экспрессионный вектор рРТ2b дрожжей (Lang и Looman (1995)), также расщепленный EcoRI и BamHI. Образовавшаяся плазмида pPT2b-tHMG содержала промотор ADH1 (Bennetzen и Hall (1982)) и терминатор TRP1 (Tschumper и Carbon (1980)), между которыми находился фрагмент ДНК tHMG. Из вектора pPT2b-tHMG с помощью эндонуклеаз EcoRV и NruI выделяли участок ДНК, который содержал так называемый средний промотор ADH1, tHMG и терминатор TRP1. Этот участок ДНК встраивали в вектор YEpl3 дрожжей (Fischhoff и др. (1984)), расщепленный эндонуклеазой SphI и ДНК-полимеразой. Полученный таким образом вектор YEpH2 (фиг.1) обрабатывали эндонуклеазами EcoRV и NruI. В результате получали фрагмент ДНК, содержащий следующие области: активирующую транскрипцию область из гена, придающего устойчивость к тетрациклину (Sidhu и Bollon (1990)), средний промотор ADH1, tHMG и терминатор TRP1 (кассета экспрессии). Этот фрагмент ДНК встраивали в вектор YDpU (Berben и др. (1991)), обработанный StuI. Полученный в результате вектор YDpUH2/12 обрабатывали эндонуклеазой SmaI и лидировали с последовательностью ДНК, кодирующей ген устойчивости к канамицину (Webster и Dickson (1983)). Полученную конструкцию (YDpUHKS, фиг.2) обрабатывали EcoRV. Этой конструкцией трансформировали штамм дрожжей Saccharomyces cerevisiae AH22. Трансформация дрожжей линеаризованным вектором, как он описан в данном примере, приводила к хромосомной интеграции всего вектора в локусе гена URA3. Для исключения из интегрированного вектора областей, не относящихся к кассете экспрессии (точка начала транскрипции в Е. coli, ген Е. coli, придающий устойчивость к ампициллину, промотор ТЕF и ген, придающий устойчивость к канамицину), трансформированные дрожжи подвергали давлению отбора с помощью ФОК-отбора (отбора с помощью 5-фтороротовой кислоты) (Boecke и др. (1987)), что позволяло отобрать дрожжи, ауксотрофные в отношении урацила. Полученный в результате отбора штамм, ауксотрофный в отношении урацила, обозначили как AH22/tH3ura8, и он несет кассету экспрессии tHMGl, интегрированную в хромосому гена URA3.
Штамм дрожжей AH22/tH3ura8 и исходный штамм АН22 культивировали в YE-среде в колбах с дефлекторами в течение 48 ч при 28°С и 160 об/мин.
Условия культивирования: предварительную культуру WMVIII готовили следующим образом: в 20 мл среды WMVIII + гистидин (20 мкг/мл) + урацил (20 мкг/мл) вносили 100 мкл замороженной культуры и инкубировали в течение 2 дней при 28°С и 120 об/мин (на вибрационном шюттль-аппарате). 20 мл предварительной культуры вносили в 100 мл среды WMVIII + гистидин (20 мкг/мл) + урацил (20 мкг/мл). Для получения основной культуры в 50 мл YE-среды (в колбах с дефлекторами объемом 250 мл) вносили 1х109 клеток. Колбы инкубировали в течение 48 ч при 28°С на круговом шейкере при 160 об/мин. Оценивали активность HMG-CoA-редуктазы (по методу Qureshi и др. (1981)) и получали следующие значения (см. таблицу 1).

Экстрагировали стерол (Parks и др. (1985)) и анализировали с помощью газовой хроматографии. Были получены следующие значения (см. таблицу 2).

Пример 2
Экспрессия SAT1 в штамме S. cerevisiae AH22
Последовательность ацил-СоА:стеролтрансферазы (SAT1; Yang и др. (1996)) получали по описанной выше методике с помощью ПЦР из геномной ДНК штамма Saccharomyces cerevisiae S288C. Используемые для этого праймеры представляли собой ДНК-олигомеры SAT1-5' и SAT1-3' (см. SEQ ID N0:3 и SEQ ID N0:4). Полученный фрагмент ДНК клонировали в клонирующем векторе pGEM-T (Mezei и Storts (1994)), в результате чего получали вектор pGEM-SATl. После обработки pGEM-SAT1 с помощью EcoRI получали фрагмент, который клонировали в экспрессионном векторе дрожжей pADH1001, также обработанном EcoRI. Полученный таким образом вектор pADH-SAT1 обрабатывали эндонуклеазой NruI и лигировали с фрагментом из YEpl3, содержащим ген LEU2.
В результате получали экспрессионный вектор дрожжей pADL-SAT1 (фиг.3), который встраивали в штамм дрожжей AH22. Полученный таким образом штамм AH22/pADL-SATl инкубировали в течение 7 дней в минимальной среде WMVIII (Lang и Looman (1995)). Условия культивирования: (для предварительной культуры см. выше) для основной культуры: в 50 мл среды WMVIII + гистидин (20 мкг/мл) + урацил (20 мкг/мл) (в колбах с дефлекторами объемом 250 мл) вносили 1×109 клеток культуры. Колбы инкубировали в течение 7 дней при 28°С на круговом шейкере при 160 об/мин. Образовавшийся стерол анализировали с помощью газовой хроматографии (см. таблицу 3).

Пример 3
Комбинированная экспрессия укороченной 3-гидрокси-3-метилглутарил-СоА-редуктазы (tHMG) и ацил-СоА:стероладилтрансферазы (SAT1)
Пример 3.1
Штамм дрожжей AH22/tH3ura8 трансформировали экспрессионным вектором pADL-SAT1, экспрессирующим SAT1, и получали штамм AH22/tH3ura8/pADL-SATl. Этот комбинированный штамм культивировали в течение 7 дней в среде WMVIII. Экстрагировали стерол (см. выше) и анализировали с помощью газовой хроматографии. Были получены следующие значения (см. таблицу 4).

Пример 3.2
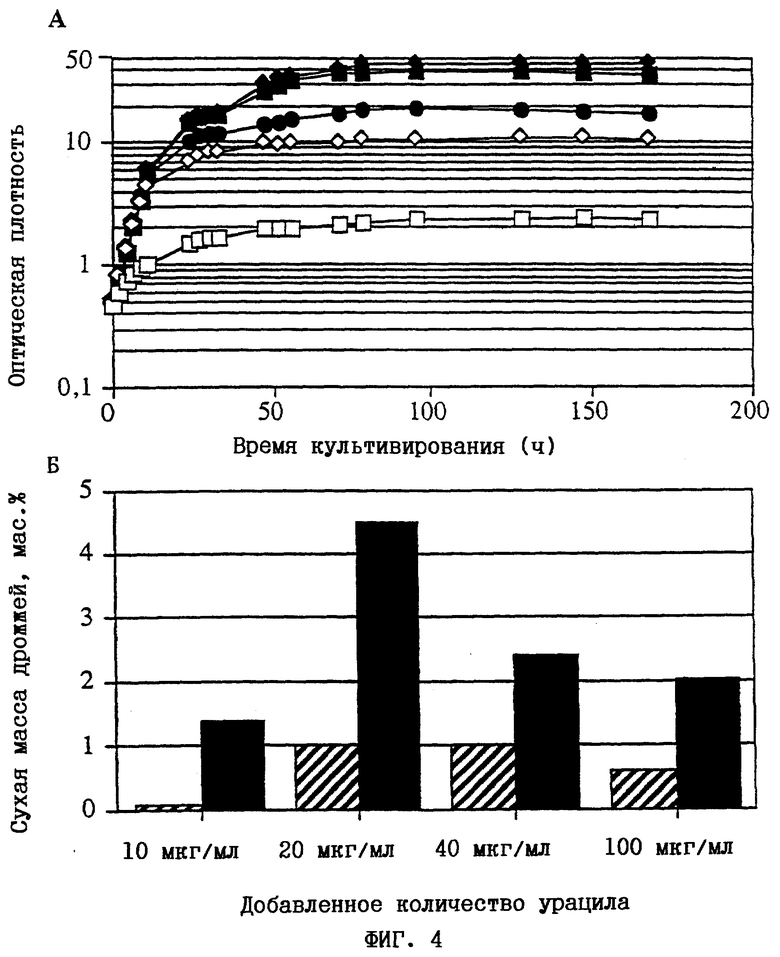
Дрожжевые культуры выращивали в течение 7 дней в среде WMVIII, при этом в культуры вносили различные количества урацила. Концентрации урацила в среде доводили до 10, 20, 40 и 100 мкг/мл. При добавлении 20 мкг/мл урацила получали максимальные количества эргостерола и сквалена. Данные представлены на фиг.4.
Очевидно, что продупирование эргостерола и сквалена штаммом AH22/tH3ura8/pADL-SATl в значительной степени зависит от количества урацила, добавленного в среду для культивирования WMVIII.
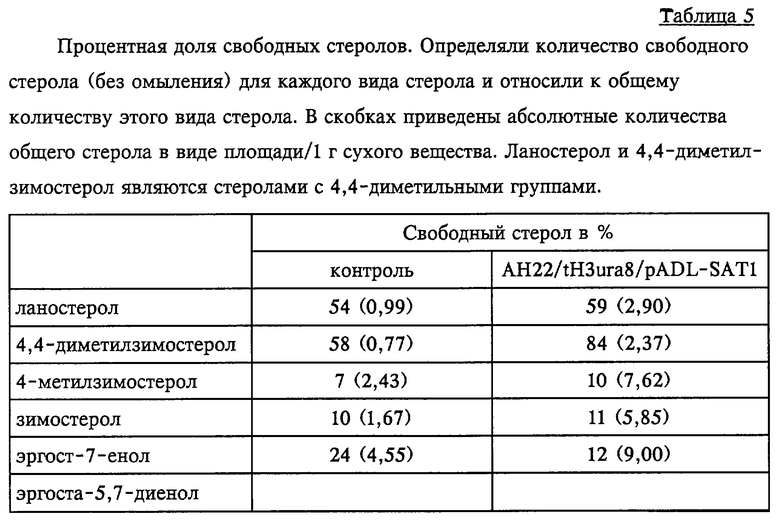
Пример 3.3
Дрожжевые культуры культвировали в течение 7 дней в среде WMVIII. После этого определяли общее количество стерола согласно описанному выше методу. Количество свободного стерола из дрожжей, собранных с помощью стеклянных бусинок и экстрагированных н-гексаном, определяли с помощью газовой хроматографии.
Данные представлены в таблице 5.
Из представленных данных видно, что фермент стеролацилтрансфераза (SAT1) наиболее эффективно этерифицирует стеролы, в которых отсутствуют 4,4-диметильные группы. Это можно рассматривать как практическое применение в данной области техники для разделения 4,4-диметилстеролов и соответствующих деметилированных форм.
Описание чертежей
На фиг.1 показана плазмида YEpH2 с соответствующими сайтами рестрикции.
На фиг.2 показана плазмида YDpUHK3 с соответствующими сайтами рестрикции.
На фиг.3 показана плазмида pADL-SAT1 с соответствующими сайтами рестрикции.
На фиг.4 представлены данные о росте и содержании эргостерола и сквалена при различных добавленных количествах урацила.
Источники информации
1. Basson M.E., Thorsness M., Finer-Moore J., Stroud R.M., Rine J., Structural and functional conservation between yeast and human 3-hydroxy-3-methylgluataryl coenzyme A reductases, the rate-limiting enzyme of sterol biosynthesis, Mol. Cell. Biol. 8: 3793-3808 (1988).
2. Bennetzen J.L., Hall B.D. The primary structure of the Saccharomyces cerevisiae gene for alcohol dehydrogenase, J. Biol. Chem. 257: 3018-3025 (1982).
3. Berben G., Dumont J., Gilliquet V., Bolle P.A., Hilger F. The YDp plasmids: a uniform set of vectors bearing versatile gene disruption cassetes for Saccharomyces cerevisiae, Yeast 7: 475-477 (1991).
4. Boeke J.D., Trueheart J., Natsoulis G., Fink G., 5-Fluorootic acid as a selective agent in yeast molecular genetics, Methods in Enzymology 154: 164-175 (1987).
5. Fegueur M., Richard L., Charles A.D., Karst F. Isolation and primary structure of the ERG9 gene of Saccharomyces cerevisiae encoding squalene synthetase, Current Genetics 20: 365-372 (1991).
6. Fischhoff D.A., Waterston R.H., Olson M.V., The yeast cloning vector YEpl3 contains a tRNALeu3 gene that can mutate to an amber suppressor, Gene 27:239-251 (1984).
7. Jandrositz A. Turnowsky F.,  G. The gene encoding squalen epoxidase from Saccharomyces cerevisiae: cloning and characterization, Gene 107: 155-160 (1991).
G. The gene encoding squalen epoxidase from Saccharomyces cerevisiae: cloning and characterization, Gene 107: 155-160 (1991).
8. Mezei L.M., Storts D.R. PCR technology: current innovations, под ред. Griffin H.G и Griffin A.M. CRC Press, Boca Raton, 21 (1994).
9. Mortimer R.K., Johnston J.R. Genealogy of principal strains of the yeast genetic stock center. Genetics 113: 35-43 (1986).
10. Lang С., Looman A.C. Efficient expression and secretion of Aspergillus niger RH5344 polygalacturonase in Saccharomyces cerevisiae, Appl. Microbiol. Biotechnol. 44: 147-156 (1995).
11. Parks L.W., Bottema C.D.K., Rodriguez R.J., Lewis T.A. Yeast sterols: yeast mutants as tools for the study of sterol metabolism, Meth. Enzymol. 1ll: 333-346 (1985).
12. Qureshi N., Nimmannit S., Porter J.W. 3-Hydroxy-3-methylglutaryl-CoA reductase from Yeast, Meth. Enzymol. 71: 455-461 (1981).
13. Ruohonen L., Aalto M.K. Keranen S. Modification to the ADH1 promotor of Saccharomyces cerevisiae for efficient production of heterologous proteins, Journal of Biotechnology 39: 193-203 (1995).
14. Siduh R.S., Bollon A.P. Bacterial plasmid pBR322 sequences serve as upstream activating sequences in Saccharomyces cerevisiae, Yeast 6: 221-229 (1990).
15. Tschumper G., Carbon J. Sequence of yeast DNA fragment containing a chromosomal replicator and the TRP1 gene, Gene 10: 157-166 (1980).
16. Webster T.D., Dickson R.C. Direct selection of Saccharomyces cerevisiae resistant to the antibiotic G418 following transformation with a DNA vector carring the canamycin-resistant gene of Tn903, Gene 26: 243-252 (1983).
17. Yang H., Bard M., Bruner D.A., Gleeson A., Deckelbaum R.J., Aljinovic G., Pohl T.M., Rothstein R., Sturley S.L. Sterol esterification in yeast: a two-gene process, Science 272: 1353-13561.
18. Yanisch-Perron С., Vieira J., Messing J. Gene 33: 103-119 (1985).
19. Yu С., Rothblatt J.A. Cloning and characterization of the Saccharomyces cerevisiae acyl-CoA:sterol acyltransferase. The Journal of Biological Chemistry, 271: 24157-24163 (1996).
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕННО-МОДИФИЦИРОВАННЫЕ ОРГАНИЗМЫ ДЛЯ ПОЛУЧЕНИЯ ЛИПИДОВ | 2010 |
|
RU2617963C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЛОГИЧНОГО ПОЛИПЕПТИДА В ЭУКАРИОТИЧЕСКИХ МИКРООРГАНИЗМОВ | 1986 |
|
RU2091490C1 |
| ИНТЕГРАТИВНЫЙ ПЛАЗМИДНЫЙ ВЕКТОР ДЛЯ ЭКСПРЕССИИ ГЕНОВ В ДРОЖЖАХ | 2008 |
|
RU2388823C1 |
| Способ получения человеческой М @ О @ -дисмутазы | 1988 |
|
SU1741610A3 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДА PFS 19, ОПРЕДЕЛЯЮЩАЯ СИНТЕЗ ПОВЕРХНОСТНОГО АНТИГЕНА S ВИРУСА ГЕПАТИТА B ЧЕЛОВЕКА | 1992 |
|
RU2115730C1 |
| ШТАММ ДРОЖЖЕЙ SACCHAROMYCES CEREVISIAL, СОДЕРЖАЩИЙ РЕКОМБИНАНТНУЮ ПЛАЗМИДУ YEP 63/AB, - ПРОДУЦЕНТ ПРОИЗВОДНОГО М-БЕЛКА ВИРУСА ГЕПАТИТА В ЧЕЛОВЕКА | 1994 |
|
RU2082759C1 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК pZEN16 ДЛЯ ПЕРЕНОСА И ЭКСПРЕССИИ ГЕНОВ В МИЦЕЛИАЛЬНОМ ГРИБЕ ACREMONIUM CHRYSOGENUM | 2009 |
|
RU2434944C2 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДА ДЛЯ ЭКСПРЕССИИ В ДРОЖЖАХ PICHIA PASTORIS ГЕНА ФОСФОЛИПАЗЫ, ШТАММ ДРОЖЖЕЙ PICHIA PASTORIS - ПРОДУЦЕНТ ФОСФОЛИПАЗЫ | 2009 |
|
RU2409671C1 |
| Трансформант дрожжей Yarrowia lipolytica, продуцирующий кантаксантин | 2023 |
|
RU2827562C1 |
| ДВУГИБРИДНАЯ СИСТЕМА, ОСНОВАННАЯ НА ОБЕСПЕЧЕНИИ УМОЛЧАНИЯ ГЕНОВ ПУТЕМ ТРАНСКРИПЦИОННОЙ ИНТЕРФЕРЕНЦИИ | 2005 |
|
RU2403287C2 |

Изобретение относится к биотехнологии. Способ получения эргостерола и его промежуточных продуктов основан на использовании рекомбинантных дрожжей, способных синтезировать увеличенные количества эргостерола, и плазмид для трансформации дрожжей. Предпочтительным видом дрожжей является вид S. cerevisiae. В качестве плазмид используются вектора экспрессии YEpH2, pADL-SAT1 и YDpUHK3, в каждый из которых встроена кассета экспрессии, содержащая средний ADH1-промотор и терминатор TRP. Образующиеся в результате сверхэкспрессии продукты могут быть использованы для различных биотрансформаций. 9 н. и 7 з.п. ф-лы, 4 ил., 5 табл.
II) ген скваленсинтетазы (ERG9), (III) ген ацил-СоА: cтеролацилтрансферазы (SAT1) и (IV) ген скваленэпоксидазы (ERG1);
или а-II) сначала конструируют вектор-экспрессии, в который встроены следующие гены: I) ген HMG-CoA-редуктазы (t-HMG) и II) ген скваленсинтетазы (ERG9);
или a-III) сначала конструируют вектор-экспрессии, в который встроены следующие гены:
I) ген HMG-Co-A-редуктазы (t-HMG) и
(III) ген ацил-СоА: стеролацилтрансферазы (SAT1);
или a-IV) сначала конструируют вектор-экспрессии, в который встроены следующие гены: I) ген HMG-Co-A-редуктазы (t-HMG) и (IV) ген скваленэпоксидазы (ERG1);
или a-V) сначала конструируют вектор-экспрессии, в который встроены следующие гены: II) ген скваленсинтетазы (ERG9) и (III) ген ацил-СоА:стеролацилтрансферазы (SAT1);
или а-VI) сначала конструируют вектор-экспрессии, в который встроены следующие гены: II) ген скваленсинтетазы (ERG9) и (IV) ген скваленэпоксидазы (ERG1);
или а-VII) сначала конструируют вектор-экспрессии, в который встроены следующие гены: (III) ген ацил-СоА:стеролацилтрансферазы (SAT1) и (IV) ген скваленэпоксидазы (ERG1);
или б) сначала конструируют векторы-экспрессии, в каждый из которых встроен один из генов, перечисленных в подпункте а-1), и в) созданными таким путем векторами-экспрессии трансформируют дрожжи, причем дрожжи трансформируют одним из векторов-экспрессии, указанных в подпунктах с а-I) по а-VII), или трансформируют одновременно или последовательно несколькими векторами-экспрессии, указанными в подпункте б), г) с использованием созданных таким путем дрожжей осуществляют ферментацию с получением эргостерола, д) после ферментации эргостерол и его промежуточные продукты экстрагируют из клеток и анализируют и в завершение е) полученный в результате эргостерол и его промежуточные продукты очищают и выделяют с помощью хроматографии на колонках.
| US 5460949 А, 24.10.1995 | |||
| УСТРОЙСТВО для КОНТРОЛЯ СОПРОТИВЛЕНИЯ изоляции | 0 |
|
SU313465A1 |
| КАРТЕЛЬ Н.А | |||
| и др | |||
| Генетика, Энциклопедический словарь | |||
| - Минск | |||
| Тэхналогiя, 1999, с.172. | |||

