Изобретение относится к электрохимическим методам анализа, а именно к катодному инверсионно-вольтамперометрическому способу определения никеля в пищевых продуктах, продовольственном сырье, кормах и кормовых добавках и может быть использовано в пищевой промышленности, сельском хозяйстве и экологическом мониторинге.
В работе [Rocha M., Neto M., Torres M., Varennes. Square wave adsorptive stripping voltammetry of Nickel (II) in flowing systems at a wall-jet mercury film electrode plates in situ // Electroanalysis, 1997, №9, P.145-149] авторами изучено вольтамперометрическое поведение никеля на стеклянно-углеродном электроде, модифицированном ртутной пленкой и диметилглиоксимом. Электролиз никеля проводили после удаления растворенного кислорода из фонового электролита (аэрирование азотом) при потенциале накопления минус 0,8 В в течение 60 с на фоне 8 моль/дм3 аммиачного буферного раствора (pH ~9).
Концентрацию никеля в модельных растворах и почве определяли по высоте пика при потенциале минус 1,1 В в диапазоне до 1,3·10-10 до 8·10-10 моль/дм3 с относительным стандартным отклонением 12, 2%.
Известен способ [Дерябина В.И., Слепченко Г.Б., Фам Кам Ньунг, Хо Ши Линь, Лычева Т.В., Кириллова М.Е. Определение йода в кормах и кормовых добавках методом вольтамперометрии. //Достижения науки и техники АПК - 2011, №11, с.42-44] определения йода в кормах и кормовых добавках методом вольтамперометрии на ОМЭ в пределах 0,002…200 мг/кг с погрешностью не более 20%.
Авторами изучено ВА - поведение йодид-ионов на серебряных электродах, модифицированных солями арилдиазоний тозилата с различными группами заместителей: карбокси - ( MAgЭ -СООН) , амино -(MAgЭ - NH2) и нитро - (MAgЭ - NO2). При сравнении градуировочных зависимостей аналитических сигналов, максимальной чувствительностью обладал электрод, модифицированный арилдиазоний тозилатом с аминогруппой в качестве заместителя. Накопление йодид-ионов в перемешиваемом растворе проводили в течение 10-20 с при потенциале электролиза (0,00±0,05)В на фоне 0,1 моль/дм3 раствора сернокислого гидразина с последующей регистрацией катодных пиков в дифференциально - импульсном режиме при скорости развертки потенциала 20 мВ/с. Концентрацию йодид-ионов определяли по высоте пика в диапазоне потенциалов от минус (0,40±0,05) В методом добавок аттестованных смесей.
Известен способ (прототип) определения массовой концентрации никеля в зерне и продуктах его переработки, силосе из зеленых растений, корме, комбикормах, комбикормовом сырье и кормовых добавках методом адсорбционной инверсионной вольтамперометрии (ИВ) [МУ 08-47/247 Зерно и продукты его переработки, силос из зеленых растений, корма, комбикорма, комбикормовое сырье и кормовые добавки. Инверсионно-вольтамперометрический метод определения содержания элементов (железа, йода, кобальта, марганца, мышьяка, никеля, ртути и селена)].
Принцип метода основан на предварительной адсорбции диметилглиоксимата никеля (II) на ртутно-пленочном электроде при потенциале электролиза минус 0,7 B в течение 30 с последующим катодным восстановлением данного комплекса.
Процесс восстановления комплекса и регистрация аналитического сигнала (катодного пика) на вольтамперограмме проводили при дифференциально-импульсной развертке потенциала от минус 0,70 В до минус 1,30 В относительно хлоридсеребряных электродов при заданной чувствительности прибора. Потенциал максимума регистрируемого пика (аналитического сигнала) для никеля равен минус (1,05±0,05)B. В качестве фонового электролита использовали хлоридно-аммиачный буферный раствор (pH 9,2) с добавкой 0,03 см3 0,1 моль/дм3 диметилглиоксима.
Мешающее влияние растворенного кислорода устраняли пропусканием через электролит инертного газа. Массовую концентрацию никеля в пробе определяли по методу добавок аттестованных смесей (АС) никеля. Диапазон измерения массовых концентраций никеля в исследуемых объектах от 0,1 до 30 мг/кг.
Предложенный в прототипе ртутно-пленочный электрод при обновлении требует нанесение пленки ртути, что небезопасно при проведении серийных анализов. Токсичность ртути, особенно трудности с ее утилизацией, ставят перед аналитиками задачи поиска электродов из нетоксичных материалов.
Задачей заявленного изобретения является использование нетоксичных органо-модифицированных электродов (ОМЭ) для определения никеля методом катодной инверсионной вольтамперометрии в присутствии растворенного кислорода.
Поставленная задача достигается выбором условий вольтамперометрического определения Ni2+ на серебряной подложке, модифицированной арилдиазоний тозилатом с аминогруппой в качестве заместителя. Измерения проводили в течение 30 с при потенциале электролиза равном минус (0,7±0,05) В относительно хлоридсеребряных электродов на фоне хлоридно-аммиачного буферного раствора с добавкой 0,03 см3 0,1 моль/дм3 диметилглиоксима, без удаления из электролита растворенного кислорода, с последующей регистрацией катодного пика в дифференциально-импульсном режиме съемки вольтамперограмм. Концентрацию никеля определяют по высоте пика в диапазоне потенциалов минус (1,00±0,05) В методом добавок аттестованных смесей.
Абсолютной новизной является экспериментально установленная способность Ni2+ адсорбироваться на поверхности индикаторного ОМЭ электрода, в виде диметилглиоксимата никеля (II) при потенциале электролиза минус 0,7 В в течение 30 с последующим катодным восстановлением данного комплекса без удаления кислорода, растворенного в электролите. Измерения проводили на вольтамперометрическом анализаторе СТА (OOO «ИТМ», г. Томск). В качестве индикаторного ОМЭ электрода использовали серебряный электрод модифицированный арилдиазоний тозилатом с амино - группой в качестве заместителя (MAgЭ -NH2) (в прототипе ртутно-пленочный).
Изучено влияние концентрации модификатора и времени контакта серебряного электрода на аналитический сигнал Ni2+ Рабочей концентрацией исходной диазониевой соли для приготовления модифицированного электрода является концентрация 0,1 ммоль/дм3 арилдиазония тозилата с аминогруппой в качестве заместителя. Время экспозиции серебряной подложки с модификатором составляет 5-7 с. Модификацию электрода осуществляли следующим способом.
В чистый кварцевый стаканчик помещали 10 см3 0,1 ммоль/дм3 раствора модификатора (соль арилдиазония с аминогруппой в качестве заместителя). Серебряный электрод подключали к соответствующей клемме прибора СТА и погружали в раствор модификатора. Далее в течение (5-7) с проводили электронакопление при потенциале Ен=(0,0±0,05)B.
При модифицировании вспомогательный электрод и электрод сравнения не использовались.
Предлагаемый вольтамперометрический способ позволил исключить использование ртути, повысить чувствительность определения, а также проводить измерения в присутствии растворенного кислорода. Диапазон измерения массовых концентраций никеля в различных типах вод от 0,003 до 0,8 мг/дм3, в продуктах более сложного состава от 0,3 до 40 мг/кг. Относительное стандартное отклонение (Sr) не более 28%.
Определению не мешают вещества, присутствие которых возможно в водах, пищевых продуктах, кормах, кормовых добавках, фармпрепаратах и биологических объектах: Zn, Co, Pb, Cu, Hg, Fe в соизмеримых количествах.
Пример 1. Определение содержания Ni2+ на уровне 0,01 мг/дм3.
В кварцевый стаканчик вместимостью 25 см3 с помощью пипетки или дозатора вносят 10 см3 раствора фонового электролита, которым является хлоридно-аммиачный буферный раствор (рН=2) с добавкой 0,03 см3 диметилглиоксима концентрации 0,1 моль/дм3. Стаканчик с раствором помещают в электролитическую ячейку. Опускают в раствор электроды (индикаторный - серебряный, модифицированный арилдиазоний тозилатом с аминогруппой в качестве заместителя; вспомогательный и сравнения -хлоридсеребряные). Проводят электронакопление при потенциале минус 0,7 В в течение 30 с при перемешивании раствора. По окончании электролиза начинают регистрацию вольтамперограммы в диапазоне потенциалов от минус 0,7 до минус 1,3 В (Фиг.1, график 1). Отсутствие пиков свидетельствует о чистоте фона. Затем добавляют 0,01 см3 стандартного раствора Ni2+ концентрации 10 мг/дм3 и проводят электронакопление при потенциале минус 0,7 В в течение 30 с последующей регистрацией вольтамперограммы в диапазоне потенциалов от минус 0,7 до минус 1,3 В. Потенциал катодного пика Ni24+ находится в диапазоне минус (1,0±0,05) В (Фиг.1, график 2). Далее в стаканчик с анализируемым раствором с помощью дозатора вносят добавку аттестованной смеси Ni2+ в объеме 0,01 см концентрацией 10 мг/дм3 . Электронакопление и регистрацию аналитического сигнала проводят в тех же условиях (Фиг.1, график 3).
Расчет содержания никеля в анализируемой пробе проводится по формуле:
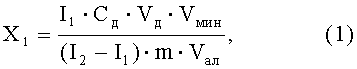
где:
X1 - массовая концентрация элемента в анализируемой пробе, мг/кг (мг/дм3);
Сд - концентрация АС, из которой делается добавка к анализируемой пробе, мг/дм3;
Vд - объем добавки АС элемента, см3;
I1 - величина максимального катодного тока элемента в анализируемой пробе, А;
I2 - величина максимального катодного тока никеля в пробе с добавкой АС, А;
m - масса (объем) пробы, взятая для анализа, г (см3);
Vал - объем аликвоты, взятой для анализа из минерализата, см3;
Vмин - объем минерализата, полученного растворением золы в известном объеме растворителя, см3.
Если для анализа берется вся проба, то Vмин/Vал равно 1 .
Пример 2. Определение содержание никеля в кормах и кормовых добавках
Навеску (0,250±0,050) г тщательно размолотой пробы помещают в кварцевый стаканчик объемом 25 см3, добавляют (2-3) см3 концентрированной перегнанной азотной кислоты. Пробу в стаканчике хорошо перемешивают стеклянной палочкой и помещают в комплекс пробоподготовки «Темос-экспресс» (OOO «ИТМ», г. Томск) с температурой (100-120)°C до полного растворения пробы. После этого температуру увеличивают до 140°C и упаривают раствор досуха. Стаканчики охлаждают до комнатной температуры, осадок растворяют, приливая 4 см3 концентрированной азотной кислоты. Затем добавляют 0,5 см3 перекиси водорода и 1,0 см3 концентрированной азотной кислоты, раствор упаривают при температуре (135-140)°С до влажных солей - данную процедуру повторяют четыре раза. После этого температуру поднимают до 580°С и выдерживают пробу 30 мин. Стаканчики с осадком вынимают, охлаждают, добавляют 10 см3 хлоридно-аммиачный буферного раствора с добавкой 0,03 см3 диметилглиоксима концентрации 0,1 моль/дм3.
Электронакопление и регистрацию аналитического сигнала проводят в тех же условиях. Содержание никеля в анализируемой пробе оценивают методом добавок аттестованных смесей, измеряя высоту катодных пиков по формуле (1). В таблице представлены результаты сравнения способов измерения количества никеля в кормах и кормовых добавках. Как видно из таблицы совпадение результатов показывает, что предлагаемый способ измерения не уступает по точности измерения способу, изложенному в прототипе.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЙОДА МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2011 |
|
RU2459199C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ МОЛОЧНОЙ КИСЛОТЫ МЕТОДОМ ВОЛЬТАМПЕРОМЕТРИИ НА СТЕКЛОУГЛЕРОДНОМ ЭЛЕКТРОДЕ | 2013 |
|
RU2526821C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ВОДОРАСТВОРИМЫХ ВИТАМИНОВ В И В МЕТОДОМ ВОЛЬТАМПЕРОМЕТРИИ НА ОРГАНО-МОДИФИЦИРОВАННЫХ ЭЛЕКТРОДАХ | 2011 |
|
RU2477465C1 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ ОДНОВРЕМЕННОГО ОПРЕДЕЛЕНИЯ СЕЛЕНА И ЙОДА | 2009 |
|
RU2415411C1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ МОДИФИЦИРОВАННОГО ЭЛЕКТРОДА ДЛЯ ОДНОВРЕМЕННОГО ИНВЕРСИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ СЛЕДОВ ТЯЖЕЛЫХ МЕТАЛЛОВ И ИОДИД-ИОНОВ | 2003 |
|
RU2237888C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ АФЛАТОКСИНА В1 МЕТОДОМ ДИФФЕРЕНЦИАЛЬНОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2013 |
|
RU2534732C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ СМЕСИ АФЛАТОКСИНОВ B1, B2, G1, G2 МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2015 |
|
RU2592049C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СУРЬМЫ, ВИСМУТА, МЕДИ В ВОДНЫХ РАСТВОРАХ МЕТОДОМ АНОДНО-КАТОДНОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2010 |
|
RU2419786C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЛЕВОМИЦЕТИНА В ПИЩЕВЫХ ПРОДУКТАХ И ФАРМПРЕПАРАТАХ | 2000 |
|
RU2180748C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ Т-2 ТОКСИНА МЕТОДОМ ДИФФЕРЕНЦИАЛЬНОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2015 |
|
RU2580412C1 |
Использование: для разработки методик анализа никеля в различных типах вод, эко- и биологических объектах, пищевых продуктах, продовольственном сырье, кормах и кормовых добавках. Сущность: заключается в сочетании кислотной минерализации образца на этапе подготовки проб с последующим вольтамперометрическим определением Ni2+ в трехэлектродной ячейке: индикаторный электрод - серебряная подложка, модифицированная арилдиазоний тозилатом с аминогруппой в качестве заместителя, вспомогательный и сравнения - хлоридсеребряные электроды. При этом накопление Ni2+ в перемешиваемом растворе проводят в течение 30 с при потенциале электролиза минус 0,7±0,05 В на фоне хлоридно-аммиачного буферного раствора с добавкой 0,03 см3 0,1 моль/дм диметилглиоксима, без удаления из электролита растворенного кислорода, с последующей регистрацией катодных пиков в дифференциально-импульсном режиме при скорости развертки потенциала 20 мВ/с. Концентрацию никеля определяют по высоте пика в диапазоне потенциалов от минус (1,00±0,05) В методом добавок аттестованных смесей. Технический результат: использование нетоксичных органо-модифицированных электродов (ОМЭ) для определения никеля методом катодной инверсионной вольтамперометрии в присутствии растворенного кислорода. 1 ил., 1 табл.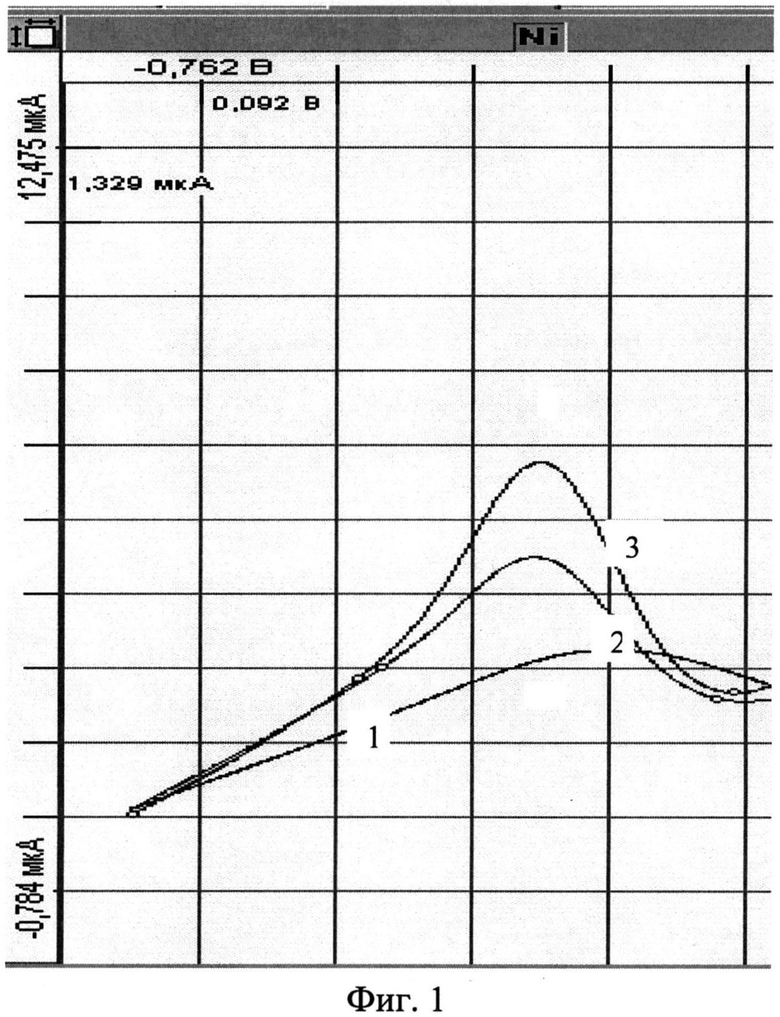
Способ количественного определения никеля методом инверсионной вольтамперометрии на органо-модифицированном электроде, включающий сочетание условий вольтамперометрического определения Ni2+ на серебряной подложке, модифицированной арилдиазоний тозилатом с аминогруппой в качестве заместителя в течение 30 с при потенциале электролиза, равном минус (0,7±0,05) В, относительно хлоридсеребряных электродов на фоне хлоридно-аммиачного буферного раствора с добавкой 0,03 см3 0,1 моль/дм3 диметилглиоксима, без удаления из электролита растворенного кислорода, с последующей регистрацией катодного пика в дифференциально-импульсном режиме съемки вольтамперограмм, концентрацию никеля определяют по высоте пика в диапазоне потенциалов минус (1,00±0,05) В методом добавок аттестованных смесей.
| Rocha M., Neto M., Torres M., Varennes, Square wave adsorptive stripping voltammetry of Nickel (II) in flowing system at a wall-jet mercury film electrode plates in situ, Electroanalysis, 1997, N9, p.145-149 | |||
| Дерябин В.И., Слепченко Г.Б., Фам Кам Ньунг, Хо Ши Линь, Лычева Т.В., Кириллова М.Е | |||
| Определение йода в кормах и кормовых добавках методом |

