Изобретение касается замещенных N-(арилоксиалкил)-гетероарилпиперидинов и -гетероарилпиперазинов, обладающих антипсихотической и/или анальгетической активностью, их использования для получения лекарственного средства с указанной активностью, их использования в качестве антипсихотических лекарственных средств. Изобретение относится также к новым промежуточным соединениям для получения производных N-(арилоксиалкил)-гетероарилпиперидина и -гетероарилпиперазина и к способу лечения психозов с использованием указанных производных гетероарилпиперидина и -пиперазина.
Широко распространено терапевтическое лечение больных шизофренией введением нейролептических лекарственных средств, таких как хлорпромазин, галоперидол, сульфирид и химически близкородственных соединений. Если регулирование симптомов шизофрении ведется успешно, то лечение этими препаратами не ведет к выздоровлению психотического больного, который почти определенно рецидивирует, если прекращает прием лекарств. Постоянно существует потребность в области нейролептических лекарственных средств для лечения психозов.
Более того, некоторые известные антипсихотические средства вызывают нежелательные побочные действия. Например, побочные действие многих антипсихотических средств включают экстрапирамидальные симптомы, такие как ригидность и дрожание, непрерывное возбужденное хождение, лицевое гримасничание и непроизвольные лицевые движения и подергивание конечностей. Обычной также является гипотензия. Таким образом, имеет также место потребность в области антипсихотических лекарственных средств, которые вызывают менее тяжелые проявления этих обычных побочных эффектов.
Кроме того, существует потребность в лекарственных средствах, которые могут вызывать другие биологические действия, например, снятие боли при аспирации в пожилом возрасте, которое привело к открытию природных и синтетических аналгетиков. И, тем не менее, потребность в надежных и эффективных аналгетиках остается и в настоящее время.
Сущность изобретения
Настоящее изобретение преследует цель - восполнить эти потребности в данной области предложением производного N-(арилоксиалкил)-гетероарилпиперидина и гетероарилпиперазина формулы (I):
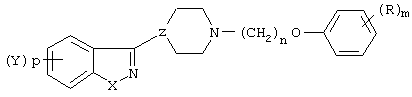
в которой Х представляет собой -О-, -S-, -NH- или -N(R2)-;
р - целое число, равное единице;
Y является водородом, Сl, Вr или F;
R2 представляет бензоил;
Z представляет собой 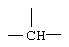 или
или 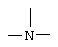 ;
;
n равно 2, 3, 4 или 5;
R представляет собой водовод, гидрокси, C1-6-алкил, C1-6-алкокси, Вr, алкиламино, алкилтио, трифторацетил, аминокарбонил, -СО-алкил, -СОО-алкил, -CO-Ph, -СН(ОR3)алкил, где алкил является низшим алкилом;
R3 представляет собой водород или низший алкил;
m равно 1, 2 или 3 при условии, что Z не является группой 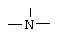 , когда Х означает -S- и R означает водород, или их фармацевтически приемлемые соли.
, когда Х означает -S- и R означает водород, или их фармацевтически приемлемые соли.
Производные формулы (I) используются для производства лекарственного средства, обладающего антипсихотической и/или анальгетической активностью.
Это изобретение предусматривает также новые промежуточные соединения, используемые для получения производных формулы (I).
Эти новые промежуточные соединения представляют собой: [3-(3-бромпропокси)-4-метоксифенил]фенилметанон;
1-[4-(3-бромпропокси)-3-бромфенил]этанон;
1-[4-(3-бромпропокси)-3,5-дибромфенил]этанон;
1-[4-(3-бромпропокси)-3-(метилмеркапто)фенил]этанон;
1-[4-(3-бромпропокси)-3-метилфенил]этанон;
1-[4-(3-бромпропокси)-3-метоксифенил]фенилметанон;
6-хлор-3-(1-пиперазинил)-lH-индазол и
4-(3-бромпропокси)-3-метоксибензонитрил.
Изобретение касается также способа лечения психозов, включающего введение млекопитающему эффективного для лечения психозов количества соединения формулы (I).
Детальное описание изобретения
Соединения формулы (I) согласно изобретению являются полезными в качестве антипсихотических средств и в качестве аналгезирующих средств и используются для получения таких средств в качестве активного начала. Соединения согласно изобретению могут содержать разнообразие различных заместителей и химических групп. Использованный в описании термин "низший", упомянутый в связи с описанием конкретной группы, означает, что описываемая группа содержит от 1 до 6 углеродных атомов.
Термин алкил, использованный в описании, касается углеводородной группы с прямой или разветвленной цепью, не содержащей ненасыщенных связей, например метил, этил, изопропил, 2-бутил, неопентил или н-гексил.
Термин алкокси, использованный в описании, касается одновалентного заместителя, содержащего алкильную группу, связанную через эфирный кислород, имеющую свою свободную валентную связь от эфирного кислорода, т.е. метокси, этокси, пропокси, бутокси или пентокси.
Термин низший алкилтио касается одновалентного заместителя, имеющего формулу: низший алкил -S-.
На протяжении описания и в прилагаемой формуле изобретения данная химическая формула или название будет включать все геометрические и стереоизомеры соединения, если таковые существуют.
А. Соединения согласно изобретению
Соединения согласно изобретению могут быть представлены следующей формулой:
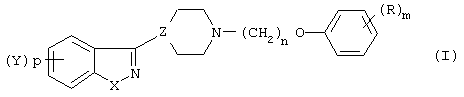
в которой Х представляет собой -О-, -S-, -NH- или -N(R2)-;
р - целое число, равное единице;
Y является водородом, Сl, Вr или F;
R2 представляет бензоил;
Z представляет собой  или
или 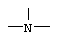 ;
;
n равно 2, 3, 4 или 5;
R представляет собой водовод, гидрокси, C1-6-алкил, C1-6-алкокси, Вr, алкиламино, алкилтио, трифторацетил, аминокарбонил, -СО-алкил, -СОО-алкил, -CO-Ph, -СН(ОR3) алкил, где алкил является низшим алкилом и R3 представляет собой водород или низший алкил;
m равно 1, 2 или 3, при условии, что Z не является группой 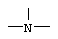 , когда Х означает -S- и R означает водород, или их фармацевтически приемлемые соли.
, когда Х означает -S- и R означает водород, или их фармацевтически приемлемые соли.
Заместитель Y предпочтительно находится в 5- или 6-положении кольца. Кроме того, в предпочтительных осуществлениях изобретения заместитель Y является водородом, хлором или фтором и в особенно предпочтительных соединениях согласно изобретению Y является фтором, в особенности в 6-положении кольца.
Значение n в формуле (I) может быть два, три, четыре или пять и предпочтительно 2, 3 или 4. В особенно предпочтительных соединениях согласно изобретению n равно трем.
Если X в соединениях согласно изобретению представляет собой -N(R2)-, то заместитель R2 представляет бензоил.
Заместитель Z в формуле (I) может быть  , и в этом случае соединения согласно изобретению являются производными гетероарилпиперидина, или
, и в этом случае соединения согласно изобретению являются производными гетероарилпиперидина, или  , и в этом случае соединения являются производными гетероарилпиперазина. Предпочтительными соединениями согласно изобретению являются гетероарилпиперидины.
, и в этом случае соединения являются производными гетероарилпиперазина. Предпочтительными соединениями согласно изобретению являются гетероарилпиперидины.
Соединения согласно изобретению могут содержать один, два или три R-заместителя. Заместитель R может быть водородом, C1-6-алкилом, C1-6-алкокси, гидроксилом, атомом брома, низшим алкилтио, алкиламино, трифторацетилом (т.е.  ), аминокарбонилом (т.е.
), аминокарбонилом (т.е. 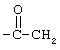 ),
), 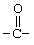 алкилом,
алкилом, 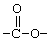 алкилом,
алкилом,  фенилом,
фенилом, 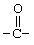 гетероарилом или
гетероарилом или  алкилом, алкил представляет собой низший алкил,
алкилом, алкил представляет собой низший алкил,
R3 представляет собой водород или низший алкил,
m равное единице, двум или трем.
Если соединения согласно изобретению содержат два или три R-заместителя, то каждый заместитель R может быть выбран независимо друг от друга среди перечисленных выше заместителей. Предпочтительно каждый из R-заместитель выбран из группы, включающей водород, алкил с 1-3 углеродными атомами, алкокси с 1-3 углеродными атомами, окси, атом бора, C1-С3-алкиламино,  алкил и
алкил и 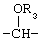 алкил.
алкил.
Соединения согласно настоящему изобретению получают следующим ниже образом. Заместители R, R1, R2, R3, X, Y и Z и целые числа m, n и р имеют указанные выше значения, если не оговорено специально.
В. Получение соединении согласно изобретению
Соединения согласно изобретению могут быть получены взаимодействием пиперидина или пиперазина формулы:

в алкилирующих условиях с соединением формулы:
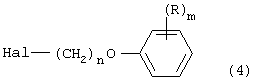
в которой Hal означает атом хлора, брома или йода. Процесс, который может быть использован для получения пиперидинов, пиперазинов и алкилирующих агентов указанных выше формул, будет далее описан подробно.
1. Получение 3-(1-незамещенных 4-пиперазинил)-lH-индазолов
Соединения формул
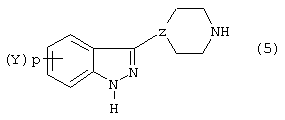
и
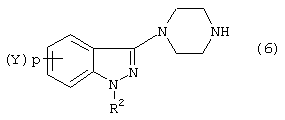
используемые в синтезе индазоилзамещенных пиперазинов согласно изобретению, могут быть получены следующим образом.
Выбирали замещенный ариловый сложный эфир формулы (7):

в которой R3 представляет собой низший алкил и Hal выбран из группы галогенов, включающий атом хлора, брома и йода. Сложный эфир формулы (7) подвергали взаимодействию с гидразином формулы H2NNH2 в стандартных условиях образования гидразида. Типично реакцию проводят в нереакционноспособном растворителе, т.е. в этаноле, метаноле или толуоле при температуре от комнатной до температуры образования флегмы растворителя в течение от 4 до 16 часов с образованием гидразида формулы (8):

Гидразид формулы (8) подвергали реакции с галоидом фенилсульфонилом формулы:
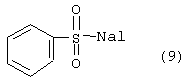
в которой Hal является галогеном, выбранным из группы, включающей атом хлора и брома, с образованием соединения формулы:
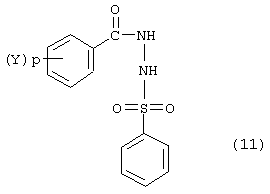
Соединение формулы (11) взаимодействует с соединением формулы (12):

в которой R6 является низшим алкилом в традиционных условиях нуклеофильных реакций, например, в инертном растворителе, таком как тетрагидрофуран, толуол или диэтиловый эфир, при температуре от 5 до 50° С в течение от 1 до 16 часов с образованием соединения, имеющего формулу:
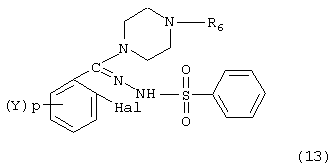
Соединение формулы (13) подвергают затем взаимодействию с конденсирующим агентом, таким как медь, сплав меди и бронзы или окись меди, в растворителе, таком как диметилформамид, диметилацетамид или тетраметилмочевина, в интервале температур от 120 до 177° С в течение от 1 до 16 часов с образованием пиперазинзамещенного фенилсульфонилиндазола формулы:

Цианозамещенный пиперазинфенилсульфонилиндазол образуется затем при взаимодействии соединения формулы (14) с традиционным источником цианирования, таким как галоидный цианид, т.е. BrCN или ClCN, в обычных условиях цианирования, типично в инертном растворителе, например в диметилсульфоксиде или хлороформе, при температуре окружающей среды в течение от 2 до 16 часов с образованием соединения формулы (15):
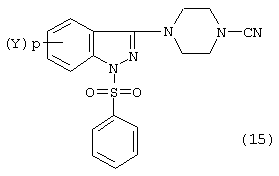
Соединение формулы (15) затем подвергают восстановлению с помощью гидрида металла, т.е. алюмогидрида лития (LiA1H4). Обычно восстановление проводят в стандартных восстановительных условиях в растворителе, таком как тетрагидрофуран или диэтиловый эфир, в интервале температур от 35 до 67° С в течение от 6 до 16 часов с образованием соединения формулы (16):
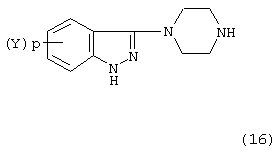
Соединение формулы (16), где (Y)p - атом хлора в 6 положении индазольного кольца, представляет собой новое промежуточное соединение - 6-хлор-3-(1-пиперазинил)-1H-индазол.
Соединение формулы (16) может быть получено альтернативным способом путем взаимодействия соединения формулы (14) с сильным основанием, таким как алкоголят металла, т.е. метилат натрия, этилат натрия или бутилат натрия, или гидроокисью калия в тетрагидрофуране с образованием соединения формулы (17):

Эту реакцию обычно проводят в полярном растворителе, таком как, например, метанол или этанол, в интервале температур от комнатной до 50° С в течение от 1 до 16 часов.
Альтернативно соединение формулы (17) может быть получено восстановлением соединения формулы (14) алюмогидридом лития (LiAlH4) в ранее описанных условиях.
Соединение формулы (17), в свою очередь, можно подвергнуть взаимодействию с реагентом цианирования, как описано выше, с образованием цианозамещенного пиперазининдазола формулы:
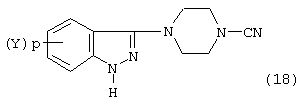
которое в свою очередь может быть восстановлено гидридом металла, как описано ранее, с образованием соединения формулы 16.
В альтернативном осуществлении соединение формулы (18) может быть подвергнуто взаимодействию с водной минеральной кислотой, например серной или хлористоводородной кислотой, в интервале температур от 50 до 120° С в течение 2-16 часов с образованием соединения формулы (16).
2. Получение 3(-1-незамещенных 4-пиперазинил)-1,2-бензизоксазолов
Соединение формулы (19):

может быть получено традиционными методами. Подходящие методы описаны в J. Меd. Сhem., 1986, 29: 359. Соединения формулы (19) полезны для синтеза бензизоксазол-замещенных пиперазинов согласно изобретению.
3. Получение 3-(1-незамещенных 4-пиперазинил)-1,2-бензизотиазолов
Соединение формулы:
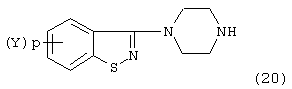
используемое в синтезе бензизотиазол-замещенных пиперазинов согласно изобретению, может быть получено методом, описанным в J. Med. Chem., 1986, 29: 359 и в патенте Великобритании №2163432 А.
4. Получение 3(1-незамещенных 4-пиперидинил)-1Н-индазолов
Соединение формул (21) и (22):

или
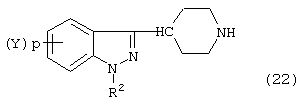
используемое в синтезе индазол-замещенных пиперидинов согласно изобретению, может быть получено с использованием известных методов. Например, подходящие методы описаны в существенных деталях в патенте СШA №4710573.
5. Получение 3-(1-незамещенных 4-пиперидинил)-1,2-бензизоксазолов
Соединение формулы:

может быть получено известными из нескольких источников методами. Например, патент США №4355037 содержит подробное описание соединений формулы (23) и способы получения соединений. Дополнительное раскрытие методов получения соединений формулы (23) можно найти в патенте США №4327103 и у Strupzewski et al., J. Med. Chem., 28: 761-769 (1985). Соединения формулы (23) могут быть использованы в синтезе бензизоксазол-замещенных пиперидинов согласно изобретению.
6. Получение 3-(1-незамещенных 4-пиперидинил)-1,2-бензизотиазолов
Некоторые 3-(4-пиперидинил)-1,2-бензизотиазолы можно использовать в синтезе N-(арилоксиалкил)-гетероарилпиперидинов согласно изобретению. Специфически бензизотиазол формулы:
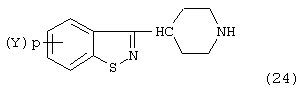
может взаимодействовать с алкилирующим агентом, описанным ранее, с образованием N-(арилоксиалкил)-гетероарилпиперидинов согласно изобретению. Соединения формулы (24) и способы их получения подробно описаны в патенте США №4458076.
7. Получение алкилирующих средств
Соединения, описанные в разделах 1-6, могут взаимодействовать с алкилирующими средствами формулы:
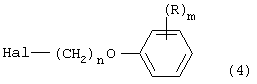
с образованием N-(арилоксиалкил)-гетероарилпиперидинов и -пиперазинов согласно изобретению. Алкилирующие средства и способы их получения описаны в патенте США №4366162. Дополнительные сведения можно найти в публикации Южной Африки ZA 8614522.
Ряд из алкилирующих средств формулы (4) представляет один из аспектов настоящего изобретения - являются новыми промежуточными соединениями для получения производных формулы (I). Эти новые алкилирующие средства следующие:
[3-(3-бромпропокси)-4-метоксифенил]фенилметанон;
1-[4-(3-бромпропокси)-3-бромфенил]этанон;
1-[4-(3-бромпропокси)-3,5-дибромфенил]этанон;
1-[4-(3-бромпропокси)-3-(метилмеркапто)фенил]этанон;
1-[4-(3-бромпропокси)-3-метилфенил]этанон;
1-[4-(3-бромпропокси)-3-метоксифенил]фенилметанон и
4-(3-бромпропокси)-3-метоксибензонитрил.
8. Алкилирование гетероарилпиперидинов и -пиперазинов для получения соединений согласно изобретению
Гетероарилпиперидины и -пиперазины, описанные в разделах 1-6 выше, могут взаимодействовать в алкилирующих условиях с алкилирующими агентами, описанными в разделе 7, с образованием соединений согласно изобретению. Реакция может быть проведена растворением реагентов в инертном растворителе, таком как диметилформамид, ацетонитрил или бутанол, и взаимодействием реагентов в интервале температур от 50° С до температуры образования флегмы растворителя в присутствии кислотного рецептора, такого как основание. Примерами подходящих оснований являются карбонаты щелочных металлов, таких как карбонат кадия, карбонат натрия или бикарбонат натрия. Реакция может быть проведена с каталитическим количеством щелочного йодида или без него, например йодистого калия или йодистого натрия, в течение времени, достаточного для образования соединения формулы (I) согласно изобретению. Вообще реакцию алкилирования проводят в течение 4-16 часов в зависимости от реакционной способности реагентов. Температура реакции может варьировать от 50 до 120° С. Продукт реакции может быть выделен обработкой водой, экстрагированием продукта в органический растворитель, не смешивающийся с водой, промывкой, сушкой и концентрированием органического растворителя для получения свободного основания и затем, если показано, обращением полученного соединения в кислотно-аддитивную соль традиционным образом.
Следующие примеры являются типичными для соединений согласно изобретению, которые могут быть получены описанным выше способом:
1-[4-[3-[4-(1H-индазол-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанон
1-[4-[3-[4-(1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
1-[4-[3-[4-(6-фтор-1,2-бенэизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-мотоксифенил]этанон
1-[4-[4-[4-(1,2-бензизоксаэол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанон
1-[4-[4-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанон
фумарат 1-[4-[2-[4-(1,2-бензизоксазол-3-ил)-1-пиперидинил]-этокси]-3-метоксифенил]этанона
фумарат 1-[4-[4-[4-(1Н-индазол-3-ил)-1-пиперазинил]-бутокси]-3-метоксифенил]этанона
1-[4-[2-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидил]-этокси]-3-метоксифенил]этанон
4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метокси-α -метилбензолметанол
1-[4-[3-[4-(1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-оксифенил]этанон
1-[4-[3-[4-(6-фтор-1Н-индазол-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанон
1-[4-[4-[4-(6-фтор-1Н-индазол-3-ил)-1-пиперазинил]-бутокси]-3-метоксифенил]этанон
1-[4-[3-[4-(1H-индазол-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанон
1-[4-[3-[4-(6-хлор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
фумарат 1-[4-[4-[4-(6-хлор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанона
1-[4-[3-[4-(5-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
фумарат 6-фтор-3-[1-[3-(2-метоксифенокси)-пропил]-4-пиперидинил]-1,2-бензизоксазола
[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]фенилметанон
1-[4-[4-[4-(1H-индазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанон
1-[4-[2-[4-(6-хлор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-этокси]-3-метоксифенил]этанон
фумарат 1-[3-[3-[4-(6-фтор-1,2-бензиоксазол-3-ил)-1-пиперидинил]-пропокси]-фенил]этанона
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-2-метилфенил]этанон
1-[2-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-5-метилфенил]этанон
полуфумарат N-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]ацетамида
6-хлор-3-(1-пиперазинил)-1H-индазол
1-[4-[3-[4-(6-фтор-1Н-индазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
полуфумарат 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанона
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-фенил]этанон
1-[4-[3-[4-(6-хлор-1Н-индазол-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанон
1-[4-[4-[4-(1,2-бензизотиазол-3-ил)-1-пиперазинил]-бутокси]-3-метоксифенил]этанон
4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксибензонитрил
1-[4-[4-[4-(6-фтор-1Н-индазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанон
сесквифумарат 1-[4-[3-[4-(1-бензоил-6-фтор-1Н-индазол-3-ил)-1-пидеразинил]-пропокси]-3-метоксифенил]этанона
1-[4-[4-[4-(6-хлор-1Н-индазол-3-ил)-1-пиперазинил]-бутокси]-3-метоксифенил]этанон
полуфумарат 1-[4-[3-[4-(1,2-бензизотиазол-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанона
1-[3,5-дибром-4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-фенил]этанон
1-[4-[2-[4-(1,2-бензизотиазол-3-ил)-1-пиперазинил]-этокси]-3-метоксифенил]этанон
6-фтор-3-[1-(3-феноксипропил)-4-пиперидинил]-1,2-бензизоксазол
1-[4-[2-[4-(6-хлор-1Н-индазол-3-ил)-1-пиперазинил]-этокси]-3-метоксифенил]этанон
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метилмеркаптофенил]этанон
1-[4-[4-[4-(1,2-бензизотиазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанон
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]фенилметанон
1-[3-бром-4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-фенил]этанон
гидрохлорид 3-[1-[3-[4-(1-этоксиэтил)-2-метоксифенокси]-пропил]-4-пиперидинил]-6-фтор-1,2-бензизоксазола
фумарат 3-[1-[3-[4-(1-ацетоксиэтил)-2-метоксифенокси)-пропил]-4-пиперидинил]-6-фтор-1,2-бензизоксазола
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-метоксифенил]пентанон
полуфумарат 2-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-N-метилбензоламина
3-[1-[3-(4-бром-2-метоксифенокси)-пропил]-4-пиперидинил]-6-фтор-1,2-бензизоксазол
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]пропанон
4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксибензамид
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-(метиламино)-фенил]этанон и
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-этоксифенил]этанон.
Соединения согласно настоящему изобретению полезны для лечения психозов благодаря их способности вызывать антипсихотическую реакцию у млекопитающих, и способ лечения психозов является следующим аспектом настоящего изобретения.
Антипсихотическую активность определяли тестом карабкания мышей на стенки клеток по методу, аналогичному описанному P. Protais et al., Psychopharmacol. 50:1, (1976) and В. Costall, Eur. J. Pharmacol., 50:39, (1978).
Подопытными животными служили самцы мышей линии СК-1 весом 23-27 г, которых содержали группами в стандартных лабораторных условиях. Мышей размещали отдельно в проволочные клетки (размером 10× 25 см) и оставляла на один час для адаптации и осмотра новой окружающей среды. Затем подкожно инъецировали 1,5 мг/кг апоморфина, дозу, вызывающую вскарабкивание на стенку у всех подопытных животных в течение 30 минут. Соединения, подлежащие испытаниям на антипсихотическую активность, инъецировали внутрибрюшинно или вводили пероральные дозы с различными временными интервалами, т.е. 30 минут, 60 минут и так далее до введения апоморфина в провоцирующей дозе 10-60 мг/кг.
Для оценки карабкания снимали три показания через 10, 20 и 30 минут после введения апоморфина по шкале, приведенной в табл. А.

Мыши, последовательно карабкавшиеся на стенку до инъекции апоморфина, не учитывались.
При полном развитии вызванного апоморфином поднимания вверх животные висели на стенках клетки, часто без движения, в течение длительного периода времени, в противоположность этому карабкание вследствие двигательного стимулирования обычно длится лишь несколько секунд.
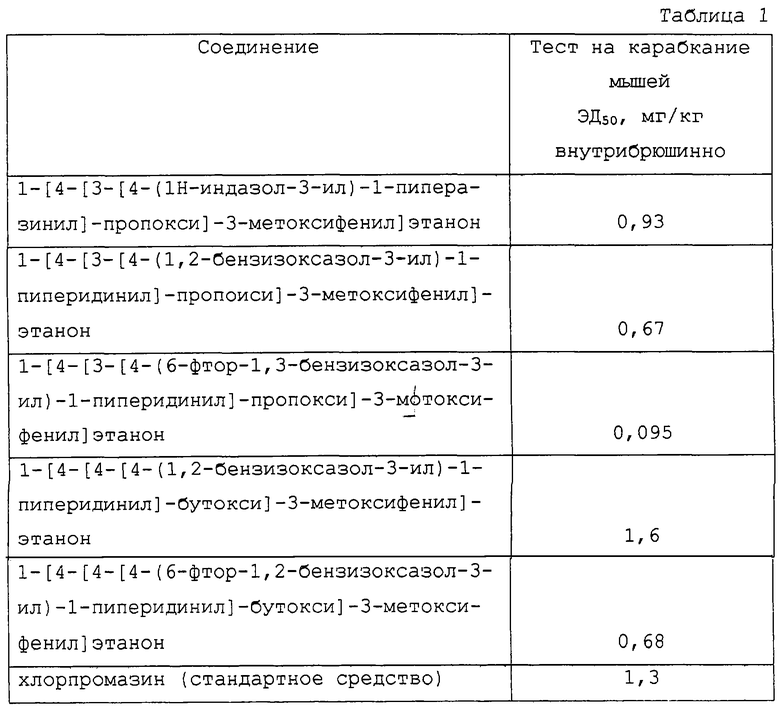
Показатели карабканий на высоту суммировались индивидуально (максимальный балл 6 от трех показаний для мыши) и общий показатель контрольной группы (получавшей носитель внутрибрюшинно и апоморфин подкожно) принимали за 100%. Значения эффективной дозы (ЭД50) с 95% пределом достоверности, рассчитанные методом линейной регрессии для некоторых соединений согласно настоящему изобретению, а также для стандартного антипсихотического средства, представлены в таблице I.

Антипсихотическая реакция достигается, если соединения согласно настоящему изобретению вводятся субъекту, требующему такое лечение, в виде эффективной пероральной, парентеральной или внутривенной дозы от 0,01 до 50 мг/кг веса тела в день. Следует иметь, однако, в виду, что для каждого конкретного больного должны быть установлены специфические дозные режимы, согласующиеся с индивидуальной потребностью и профессиональной подготовкой лиц, вводящих или контролирующих введение упомянутых соединений. Следует также понимать, что определенные здесь дозы являются только примерными и они не должны никоим образом ограничивать объем или практику осуществления этого изобретения.
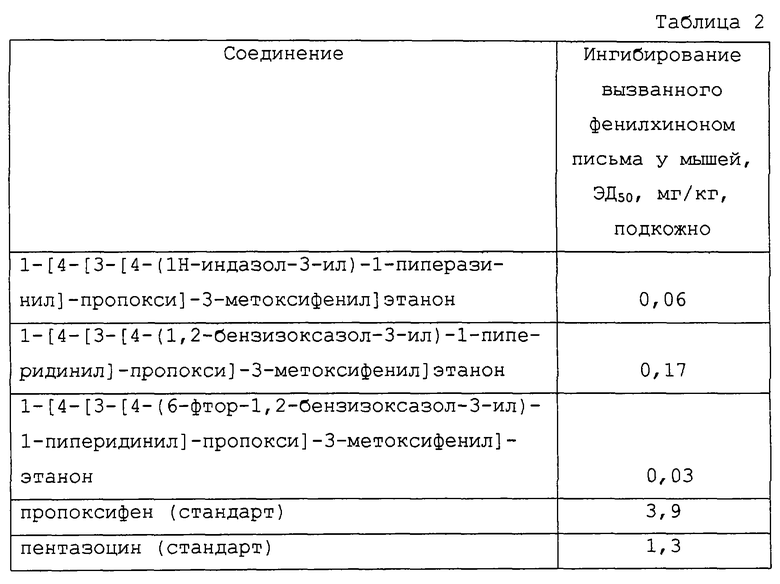
Некоторые соединения согласно настоящему изобретению полезны также в качестве аналгезирущих средств благодаря их способности смягчать боль у млекопитающих. Аналгетическую полезность демонстрировали в тесте письма (царапание лапками) у мышей, вызванного введением фенил-пара-хинона, стандартного теста на обезболивание, описанного в Proc. Soc. Exptl. Biol. Med. 95:729 (1957). Так, например, подкожная доза, вызывающая приблизительно 50%-ное ингибирование симптома письма у мышей (ЭД50), полученная в этом эксперименте, показана в таблице 2.
Аналгезия достигается, если больному, нуждающемуся в таком лечении, вводят эффективную пероральную, парентеральную или внутрибрюшинную дозу от 0,01 до 100 мг/кг веса тела в день. Следует понимать, однако, что схема применения лекарственного средства приводится в соответствие с персональной потребностью больного и профессиональной подготовкой лица, вводящего или контролирующего введение упомянутого соединения. Далее следует понимать, что установленные выше дозы являются только примерными и они ни в коей мере не ограничивают объема или осуществления настоящего изобретения.
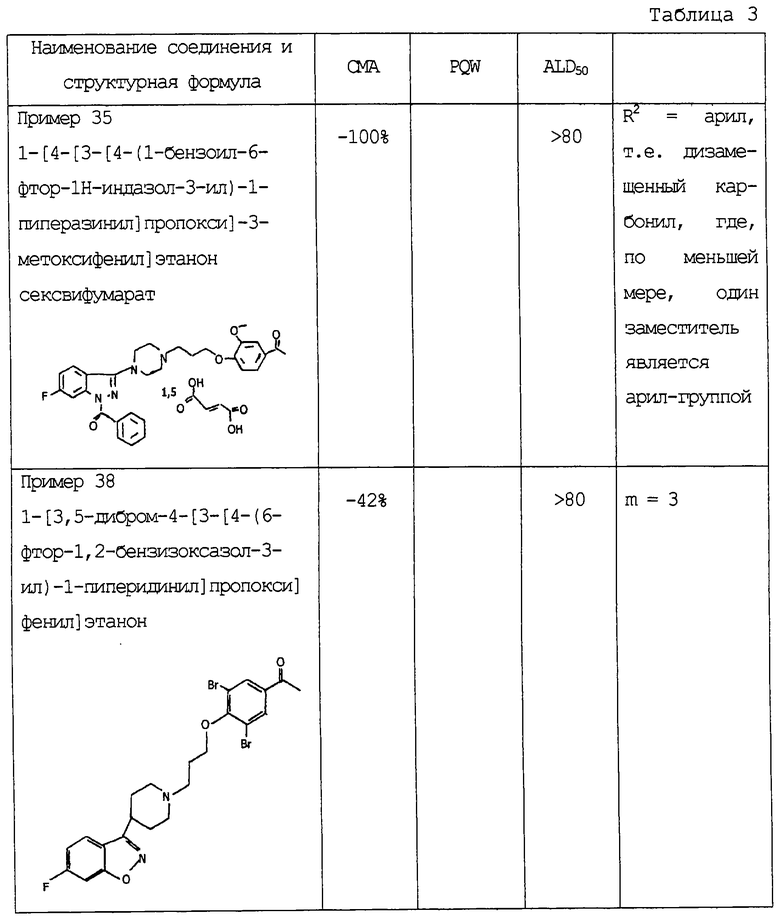
Дополнительно проводились следующие эксперименты для определения антипсихотической активности (СМА), аналгетической активности (PQW) и токсичности (ALD50) некоторых соединений изобретения.
Антипсихотическая активность определялась посредством ингибирования апоморфин-индуцированного подъема вверх (карабкания) мыши при 20 мг/кг внутрибрюшинно 30 минут. Аналгетическая активность определялась на мышах ингибированием вызванных фенилхиноном болевых судорог мыши при 20 мг/кг подкожно 30 минут. Токсичность была измерена при острой летальной дозе (ALD50 мг/кг, внутрибрюшинно). Полученные результаты представлены в таблице 3.

Для подтверждения преимуществ изобретения, связанных с меньшей токсичностью, приводятся результаты испытания представителя соединений изобретения 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]пропокси]-3-метоксифенил]-этанона (в дальнейшем называемого для краткости "соединение") в сравнении с хлорпромазином. Проводились испытания: (а) на стереотипию у крыс, вызванную апоморфином (APO-S) и (b) на индуцирование каталепсии у крыс (методика испытаний приведена выше).
Испытание APO-S было проведено с соединением и хлорпромазином, соединениями, которые предотвращают антагонизм APO-S допаминовыми рецепторами в нигростриатальной системе головного мозга, и антагонизм этого поведения является предсказателем склонности к развитию экстрапирамидальных побочных действий (EPS) и запоздалых (поздних) дискинезий (расстройств произвольных движений) (TD); результаты, полученные в этом стандартном испытании на животных, следующие:
хлорпромазин: ЭД50=5,75 мг/кг, i.p.;
соединение: ЭД50=34,8 мг/кг, i.p.
Следовательно, соединение с меньшей вероятностью (при соотношении 1/6) по сравнению с хлорпромазином, будет проявлять побочные действия EPS и TD (т.е. токсичность).
Из таблицы 1 видно, что соединение в 13,6 раз является более сильнодействующим, чем хлорпромазин в Испытании на карабкание мышей (СМА), что является показателем нейролептической эффективности; из результатов СМА и результатов APO-S, приведенных выше, был вычислен терапевтический индекс следующим образом:

хлорпромазин: TR=5,75
соединение: TR=366
и соответственно соединение значительно превосходит хлорпромазин.
Испытание на каталепсию у крыс было проведено с соединением и хлорпромазином (нейролептики, которые оказывают ингибирующее действие на нигростриатальную допаминную систему, индуцирующую каталепсию), каталептические симптомы у грызунов сравнивались с Паркинсоно-образными экстрапирамидальными побочными действиями, наблюдаемыми клинически при назначении нейролептических средств; результаты, полученные в этом стандартном испытании на животных, следующие:
хлорпромазин: 83% индуцирование каталепсии при 1,00 мг/кг, i.р.,
соединение: 10% индуцирование каталепсии при 10,00 мг/кг, i.p.;
следовательно, соединение превосходит хлорпромазин с точки зрения меньших побочных действий (токсичности).
1. Ингибирование апоморфиновой стереотипии у крыс
Цель:
Отбор нейролептических соединений, которые действуют непосредственно на допаминергическую систему путем блокирования действия аломорфина на постсинаптические допаминовые рецепторы (Andenetal, 1967; Ernst, 1967).
Методика:
Использовали группы крыс Wistar мужского пола (125-200 граммов), пища и вода были доступны без ограничений (досыта). Лекарства приготавливались с использованием дистиллированной воды и, если они были нерастворимы, добавляли подходящее поверхностно-активное вещество. Способ назначения мог меняться, объем дозы 10 мл/кг.
Для первичного скрининга использовали группу из шести крыс. Средство назначалось за один час до оценки показателей, и животных помещали в индивидуальные чистые пластиковые клетки (24× 14× 13 см). Контрольная группа получала наполнитель. Апоморфин· НСl (Merck и Со.) приготавливали в концентрации 15 мг/10 мл в 0,03% исходном растворе аскорбиновой кислоты, приготовленном с использованием 30 мг аскорбиновой кислоты в 100 мл 1 солевого раствора, чтобы увеличить стабильность апоморфина· НСl в растворе. Апоморфин· НСl назначался в дозе 1,5 мг/кг s.с. (подкожно) с объемом дозы 1 мл/кг, через 50 минут после введения дозы средства наблюдали стереотипическое поведение. Стереотипическая активность определяется как принюхивание, облизывание или жевательное поведение, которое подразделяется следующим образом: постоянное принюхивание, облизывание или жевание без прерывания; животное считается защищенным, если это поведение прерывается.
Процентная эффективность средства определяется по количеству защищенных животных в каждой группе.
Определение ответной реакции на дозу проводится таким же образом, как и первичный скрининг, за исключением того, что использовалась группа из 10 крыс и животным давали дозу препарата случайным образом. Одна группа получала наполнитель. Показатель ЭД50 для стереотипии вычислялся с помощью пробит анализа.
Ссылки:
Anden, N.E., Rubenson, A., Fuxe, K., Hokfelt, Т.: Evidenee for dopancine receptor stimulation by apomorphine. J. Pharm. Pharmacol., 19: 627-629, 1967. Ernst, A.M.: Mode of action of apomorphine and dexamphetamine on gnawing compulsion in rats. Psychopharmacological (Berl.) 10: 316-323, 1967.
2. Каталепсия
Методика:
Использовали группы крыс Wistar мужского пола (150-300 граммов). Животные содержались в колонии с контролируемыми климатическими условиями. Пища и вода были доступны без ограничений. В день испытания животных приносили в лабораторию и делили на группы по шесть.
Лекарства приготавливались в дистиллированной воде и назначались i.p. (внутрибрюшинно) в объеме дозы 10 мл/кг.
Испытание выполнялось через 1, 2, 3, 4, 5 и 6 часов после введения дозы и проводилось в присутствии "белого шума". Испытание на каталепсию состоит в помещении отдельного животного в белый полупрозрачный пластиковый ящик (26× 20× 15 см) с деревянным штифтом, установленным горизонтально на 10 см от пола и 4 см от одного края ящика. Пол был покрыт приблизительно 2 см слоистого материала. Животным позволялось адаптироваться к ящику в течение двух минут. После двух минут каждое животное мягко охватывалось вокруг плеч и за передние лапы и аккуратно помещалось на перекладину (барьер). Определялось количество времени, которое животное проводит с, по крайней мере, одной передней лапой на перекладине. Когда животное убирало свои лапы, регистрировалось время и животное вновь помещалось на перекладину. Проводилось три опыта для каждого животного при каждом времени испытания, и время, проведенное на перекладине, регистрировалось для каждого опыта при каждом времени проведения испытания, в то время когда животное остается на перекладине в течение 60 секунд, никаких дополнительных испытаний не требуется.
Животное считалось каталептиком, если оно оставалось на перекладине в течение 60 секунд.
Процент каталепсии вычисляется следующим образом:

Соединения согласно изобретению, будучи эффективными сами, могут быть приготовлены как лекарственные препараты (что является одним из аспектов данного изобретения) и введены в форме своих фармацевтически приемлемых аддитивных солей по причине их стабильности, удобства кристаллизации, повышенной растворимости и тому подобного. Предпочтительные фармацевтически приемлемые аддитивные соли включают соли минеральных кислот, например хлористоводородной кислоты, серной кислоты, азотной кислоты и тому подобных; соли одноосновных карбоновых кислот, например уксусной кислоты, пропионовой кислоты и тому подобных; соли двухосновных карбоновых кислот, например малеиновой кислоты, фумаровой кислоты и тому подобных, и соли трехосновных карбоновых кислот, таких как карбоксиянтарная кислота, лимонная кислота и тому подобные.
Эффективные количества соединений согласно изобретению могут быть введены перорально, например, с инертным разбавителем или со съедобным носителем. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. Для целей перорального терапевтического введения соединения согласно изобретению могут быть внедрены в носитель и использованы в форме таблеток, гранул, капсул, эликсиров, суспензий, сиропов, облаток, жевательной резины и тому подобного. Эти препараты могут содержать, по меньшей мере, 0,5% активного соединения согласно изобретению, но это количество может варьироваться в зависимости от конкретной формы и может обычно составлять от 4 до 70 процентов от веса лекарственной формы. Количество активного соединения в такой композиции таково, что обеспечивает достижение подходящей дозы. Предпочтительные композиции и препараты согласно настоящему изобретению готовят так, чтобы форма дозированной единицы для перорального введения содержала от 1,0 до 300 миллиграммов активного соединения согласно изобретению.
Таблетки, пилюли и капсулы, облатки и тому подобное могут также содержать следующие ингредиенты: связующее, такое как микрокристаллическая целлюлоза, трагакант, или желатин; наполнитель, такой как крахмал или лактоза; диспергирующее средство, такое как альгиновая кислота, примогель, кукурузный крахмал и тому подобные; смазка, такая как стеарат магния или стеротес, скользящее средство, такое как коллоидная двуокись кремния, и сладости, такие как сахароза или сахарин, или ароматическое средство, такое как мята, метилсилацилат или апельсиновый аромат. Если дозированной единичной формой является капсула, то она может содержать в дополнение к материалам перечисленного выше типа жидкий носитель, такой как жировое масло. Другие формы дозированных единиц могут содержать различные материалы, чтобы модифицировать физическую форму дозированной единицы, например покрытия. Так, например, таблетки или пилюли могут быть покрыты сахаром, шеллаком или другими растворяющимися в кишечнике покровными средствами. Сироп может содержать в дополнение к активным соединениям сахарозу в качестве сладости и некоторые консерванты, красители и ароматические средства. Материалы, используемые для приготовления этих различных композиций, должны быть фармацевтически чистыми и нетоксичными в используемых количествах.
Для целей парентерального терапевтического введения активные соединения согласно изобретению могут быть введены в раствор или суспензию.
Эти препараты должны содержать по меньшей мере 0,1% активного соединения, но это количество может варьироваться от 0,5 до 50 процентов от веса препарата. Количество активного соединения в таких композициях таково, что обеспечивается нужная доза. Предпочтительные композиции и препараты согласно настоящему изобретению приготовлены так, чтобы парентеральная дозированная единица содержала от 0,5 до 100 миллиграммов активного соединения.
Растворы или суспензии могут также включать следующие компоненты: стерильный разбавитель, такой как вода для инъекции, физиологический раствор, нелетучее масло, полиэтиленгликоли, глицерин, пролиленгликоль или другие синтетические растворители; противомикробные средства, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия, хелатообразующие средства, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетатные, нитратные или фосфатные буферы, и средства для поддержания тонуса, такие как хлористый натрий или декстроза. Препараты для парентерального введения могут быть заключены в ампулы, свободные шприцы или сосуды дли многократных доз, сделанные из стекла или пластика.
Следующие ниже примеры имеют лишь иллюстративные цели и не могут рассматриваться как ограничивающие объем изобретения. Все температуры даны в градусах Цельсия (° С), если не указано особо.
Пример 1
Получение 1-[4-[3-[4-(1Н-индазод-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанона
(А) Синтез 2-фенилсульфонилгидразида 2-бромбензойной кислоты
К раствору гидразида 2-бромбензойной кислоты (132 г) в пиридине (~500 мл) (1,2^ Р), охлажденному до 10° С на ледяной бане, добавляли хлористый бензолсульфонил (78,3 мл). После окончания добавки реакционную смесь перемешивали при комнатной температуре 4 часа, затем выливали в смесь льда и хлористоводородной кислоты для осаждения желтого твердого вещества, 135 г. Продукт перекристаллизовывали из изопропанола и получали 125 г 2-фенилсульфонилгидразида 3-бромбензойной кислоты, точка плавления 154-156° С.
(B) Синтез α -хлор-2-бромбензальдегида фенилсульфонилгидразона
Смесь фенилсульфонилгидразида 2-бромбензойной кислоты (125 г, 0,35 моль) и хлористого тионила (265 мл) перемешивали и нагревали с обратным холодильником два часа. После 15 минут кипячения с обратным холодильником твердое вещество переходило в раствор. Реакционную смесь охлаждали и затем выливали в гексан. Полученное белое твердое вещество собирали и получали 124 г α -хлор-2-бромбензальдегида фонилсульфонилгидразона, точка плавления 120-122° С.
(C) Синтез 1-[[фенилсульфонил)-гидразоно]-(2-бромфенил)метил]-4-метилпиперазина
К перемешиваемому раствору α -хлор-2-бромбензальдегида фенилсульфонилгидразона (271,1 г, 0,72 моль) в тетрагидрофуране (тетрагидрофурана два литра) в атмосфере азота по каплям добавляли N-метилпиперазин (159 г, 1,6 моль). Реакционную смесь перемешивали при температуре окружающей среды три часа и затем выдерживали 16 часов при той же температуре. Реакционную смесь охлаждали на ледяной бане и затем фильтровали для удаления образовавшегося гидрохлорида пиперазина. Фильтрат концентрировали до получения коричневой смолы. Смолу растирали с горячим ацетонитрилом, смесь охлаждали на ледяной бане и охлажденную смесь фильтровали для удаления нежелательного побочного продукта. Затем фильтрат концентрировали для получения 392,9 г коричневой смолы неочищенного 1-[[(фенилсульфонил)-гидразоно]-(2-бромфенил)метил]-4-метилпиперазина.
(D) Синтез 3-(4-метил-1-пиперазинил)-1-фенилсульфонил-1Н-индазола
Смесь 1-[[(фенилсульфонил)-гидразоно]-(2-бромфенил)метил]-4-метилпиперазина (31,0 г, 0,08 моль) бронзовой меди (3,1 г), углекислого калия (11,5 г) и диметилформамида (500 мл) перемешивали и нагревали с обратным холодильником 1,5 часа. Реакционную смесь выливали в воду и водную суспензию энергично перемешивали с этилацетатом. Содержащую две фазы смесь фильтровали через целит и последовательно разделяли слои. Водную часть экстрагировали другой частью этилацетата и объединенные экстракты промывали водой и сушили (сульфат магния). Концентрацией экстракта получали твердое вещество, которое при растирании с простым эфиром давало 19,7 г твердого вещества. Твердое вещество перекристаллизовывали из изопропанола и получали 17,7 г (60%) продукта, точка плавления 158-161° С. Чистые для анализа образцы получали второй перекристаллизацией из изопропанола (с обработкой древесным углем) в форме бесцветных кристаллов индазола, 3-(4-метил-1-пиперазинил)-1-фенилсульфонил-1Н-индазола, точка плавления 160-161° С.
Элементный анализ: для C18H20N4O2S (%):
вычислено: С 60,66%; Н 5,66%; N 15,72%,
найдено: С 60,45%; Н 5,62%; N 15,61%.
(Е) Синтез 4-[1-(фенилсульфонил)-lH-индазол-3-ил]-1-пиперазинкарбонитрила
К перемешиваемой смеси 3-(4-метил-1-пиперазинил)-1-фенилсульфонил-1Н-индазола (237 г, 0,67 моль), углекислого калия (102 г, 0,74 моль) и диметилсульфоксида (2000 мл) в атмосфере азота добавляли бромистый циан (72 г, 0,68 моль), растворенный в диметилсульфоксиде (525 мл). Реакционную смесь перемешивали при температуре окружающей среды 5,5 часа и затем переливали в воду (7,0 л). Твердое вещество, выпавшее в осадок из раствора, собирали фильтрацией, хорошо промывали водой и получали 168 г (68%) продукта. Образец весом 5,2 г два раза перекристаллизовывали из смеси этанол - вода и получали 4,0 г 4-[1-(фенилсульфонил)-1H-индазол-3-ил]-1-пиперазинкарбонитрила, точка плавления 178-180° С.
Элементный анализ для С18Н17N3О2S (%):
вычислено: С 59,01; Н 4,63; N 19,06,
найдено: С 59,01; Н 4,63; N 19,09.
(F) Синтез 3-(1-пиперазинил)-1Н-индазола
К перемешиваемой смеси 4-[1-(фенилсульфонил)-1H-индазол-3-ил]-пиперазинкарбонитрила (163 г, 0,44 моль) в тетрагидрофуране (2,0 л) по каплям добавляли алюмогидрид лития (880 мл, 0,88 моль 1 М раствора алюмогидрида лития в тетрагидрофуране). После окончания добавки реакционную смесь нагревали с обратным холодильником и перемешивали в течение 6 часов, перемешивали при комнатной температуре один час и оставляли на ночь при комнатной температуре. Реакцию гасили осторожным добавлением воды по каплям. После превращения выделения водорода реакционную смесь фильтровали и осадок на фильтре литиевой соли тщательно промывали тетрагидрофураном, фильтрат объединяли с фильтратом второй серии (исходный материал от двух процессов вместе составлял 300 г, т.е. 0,82 моль) и объединенные экстракты концентрировали до получения 372 г желтого твердого вещества, суспендированного в воде. Была предпринята попытка распределить продукт в воде и в дихлорметане, но продукт оказался лишь слабо растворимым в дихлорметане. Поэтому двухфазную суспензию продукта фильтровали через воронку с горизонтальным расположением слоев и собранный белый продукт сушили и получали 121 г. Две фазы фильтрата разделяли и водную фазу снова экстрагировали дихлорметаном. Все фазы дихлорметана объединяли, два раза промывали водой, сушили сульфатом магния и концентрировали до получения 41 г коричневого остатка. Остаток растирали с диэтиловым эфиром. Фильтровали и получали 10 г твердого вещества цвета беж. Точка плавления 139-150° С. Спектр ЯМР и масс-спектр находились в согласии со структурой. Перекристаллизацией 10 г продукта из толуола получали 7,5 г 3-(1-пиперазинил)-lH-индазола, точка плавления 153-156° С.
(G) 3-(4-мeтил-1-пиперазинил)-1Н-индазол
Перемешиваемую смесь 3-(4-метил-1-пиперазинил)-1-фенил-сульфонил-1Н-индазола (13,5 г, 0,038 моль), метанола (150 мл) и 25% метилата натрия в метаноле (15,3 мл) нагревали с обратным холодильником 2,5 часа. Реакционную смесь концентрировали примерно до одной десятой объема и к смеси добавляли воду, получали красного цвета раствор. Раствор экстрагировали дихлорметаном, экстракт промывали водой, сушили сульфатом магния, растворитель концентрировали и получали 6,6 г окрашенного в розовый цвет твердого вещества. Перекристаллизацией из смеси толуол-гексан получали 4,3 г (52%) 3-(4-метил-1-пиперазинил)-1H-индазола в виде твердого вещества не совсем белого цвета, точка плавления 111-113° С.
Элементный анализ для C12H16N4:
вычислено (%): С 66,64; Н 7,46; N 25,91,
найдено (%): С 66,83; Н 7,4; N 25,69.
(Н) 4-(1Н-индазол-3-ил-)-1-пиперазинкарбонитрил
К перемешиваемой смеси бромистого циана (5,3 г, 0,05 моль), углекислого калия (К2СО3, 7,1 г) и диметилсульфоксида (40 мл) добавляли по каплям 3-(4-метил-1-пиперазинил)-1Н-индазол (11,0 г, 0,051 моль), растворенный в диметилсульфоксиде (60 мл). Реакционную смесь перемешивали при температуре окружающей среды один час и затем выливали в воду. Водную суспензию экстрагировали этилацетатом, этилацетат промывали водой, сушили сульфатом магния, концентрировали и получали 7,8 г (67%) твердого вещества, окрашенного в желтый цвет.
Этот образец объединяли с другим и два раза перекристаллизовывали из толуола, получали чистый для анализа 4-(1H-индазол-3-ил)-1-пиперазинкарбонитрил в виде белого твердого вещества, точка плавления 120-122° C.
Элементный анализ для C12H13N5:
вычислено (%): С 63,42; Н 5,75,
найдено (%): С 63,04; Н 5,84.
(I) Синтез 3-(1-пиперазинил)-1Н-индазола
Смесь 4-(1H-индазол-3-ил)-1-пиперазинкарбонитрила (8,0 г, 0,04 моль) и 25% серной кислоты (100 мл) перемешивали при нагревании с обратным холодильником в течение 4,5 часа. Реакционную смесь охлаждали на ледяной бане и подщелачивали добавлением по каплям 50% гидроокиси натрия. Основный раствор экстрагировали этилацетатом. Этилацетат промывали водой, сушили сульфатом магния и получали 5,2 г (73%) желаемого соединения в виде твердого вещества. Твердое вещество два раза кристаллизовали из толуола и получали 3,0 г 3-(1-пиперазинил)-1Н-индазола, точка плавления 153-155° С.
Элементный анализ для C11H14N4:
вычислено (%): С 65,32; Н 6,98; N 27,70,
найдено (%): С 65,21; Н 6,99; N 27,80.
(J) Синтез 1-[4-[3-[4-(1Н-индазол-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанона
Смесь 3-(1-пиперазинил)-1H-индазола (4,0 г, 0,02 моль), карбоната калия (К2СО3, 3,0 г, 0,022 моль), 1-[4-(3-хлор-пропокси)-3-метоксифенил]этанона (5,3 г, 0,002 моль), несколько кристаллов йодистого калия и диметилформами да (60 мл) перемешивали при 90° С пять часов. Реакционную смесь выливали в воду и водную смесь экстрагировали этилацетатом. Экстракт промывали рассолом, сушили сульфатом магния, растворитель концентрировали и получали белое твердое вещество, которое растирали с диэтиловым эфиром, собирали и получали 7,0 г продукта. После двух перекристаллизаций из абсолютного этилового спирта получали 5,3 г (64%) чистого для анализа 1-[3-[4-(1Н-индазол-3-ил)-1-пиперазинил]-пропокси]-3-мотоксифенил]этанона, точка плавления 155-157° С.
Элементный анализ для С23Н28Н4O3:
вычислено (%): С 67,62; Н 6,91; N 13,72,
найдено (%): С 67,45; Н 6,74; N 13,56.
Пример 2
1-[4-[3-[4-(1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
Смесь гидрохлорида 3-(4-пиперидинил)-1,2-бензизоксазола (4,8 г, 0,02 моль), карбоната калия (5,2 г, 0,04 моль), 1-[4-(3-хлорпропокси)-3-метоксифенил]этанона (5,3 г, 0,022 моль), несколько кристаллов йодистого калия и диметилформамида (60 мл) перемешивали при 90° С в течение 16 часов. Реакционную смесь выливали в воду и водную смесь экстрагировали этилацетатом. Экстракт промывали водой, сушили сульфатом магния, концентрировали и получали коричневое масло. Масло хроматографировали на Waters Prep 500, используя колонки силикагеля и смесь этилацетат - диэтиламин (2%) в качестве элюента. Концентрированием соответствующих фракций получали 3,9 г продукта в виде твердого вещества не совсем белого цвета. Перекристаллизацией из абсолютного этилового спирта получали 2,6 г (33%) 1-[4-[3-[4-(1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил] этанона, точка плавления 102-104° С, в виде бесцветных иголок.
Элементный анализ для C24H28N2O4:
вычислено (%): С 70,56; Н 6,91; N 6,66,
найдено (%): С 70,73; Н 6,93; N 6,85.
Пример 3
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
Перемешиваемую смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола гидрохлорида (5,1 г, 0,02 моль), карбоната калия (5,2 г, 0,04 моль), 1-[4-(3-хлорпропокси)-3-метоксифенил]этанона (5,3 г, 0,022 моль) и диметилформамида (60 мл) нагревали при 90° С в течение 16 часов. Реакционную смесь выливали в воду и водную смесь экстрагировали этилацетатом. Этилацетат промывали водой, сушили сульфатом магния, концентрировали до получения влажного твердого вещества. Перекристаллизацией (два раза) из этилового спирта получали 5,0 г (58%) 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанона в виде твердого вещества цвета беж, точка плавления 118-120° С.
Элементный анализ для C24H27FN2O4:
вычислено (%): С 67,60; Н 6,38; N 6,57,
найдено (%): С 67,47; Н 6,40; N 6,53.
Пример 4
1-[4-[4-[4-(1,2-бензизоксазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанон
Смесь гидрохлорида 3-(4-пиперидинил)-1,2-бензизоксазола (4,3 г, 0,018 моль), карбоната калия (5,5 г, 0,04 моль) и 1-[4-(4-бромбутокси)-3-метоксифенил]этанона (5,5 г, 0,018 моль) в диметилформамиде (60 мл) перемешивали и нагревали при 75° С в течение 16 часов. Реакционную смесь выливали в воду и экстрагировали этилацетатом. Этилацетат промывали водой, сушили сульфатом магния, растворитель концентрировали и получали 7,2 г твердого вещества цвета беж. Перекристаллизацией (двойной) из этилового спирта получали 3,3 г (43%) 1-[4-[4-[4-(1,2-бензизоксазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанона, точка плавления 99-101° С.
Элементный анализ для С25Н30Н2O4:
вычислено (%): С 71,11; Н 7,16; N 6,63,
найдено (%): С 70,76; Н 7,24; N 6,58.
Пример 5
1-[4-[4-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанон
Перемешиваемую смесь гидрохлорида 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (5,1 г, 0,02 моль), карбоната калия (5,2 г, 0,04 моль), 1-[4-(4-бромбутокси)-3-метоксифенил]этанона (6,6 г, 0,022 моль) и диметилформамида (60 мл) нагревали при 75° С пять часов. Реакционную смесь выливали в воду и водную смесь экстрагировали этилацетатом. Этилацетат промывали водой, сушили сульфатом магния и растворитель концентрировали первоначально до масла, которое отверждалось при выдерживании. Твердое вещество растирали с гексаном, собирали и получали 7,7 г продукта в виде воскового твердого вещества. Соединение хроматографировали на Waters Prep 500, используя колонки силикагеля и элюируя смесью дихлорметан - метанол (5%). Концентрированном соответствующих фракций получали 5,1 г не совсем белого твердого вещества 1-[4-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанона, который перекристаллизовывали из этилового спирта с выходом 3,2 г (36%) в виде пушисто-белых иголок, точка плавления 88-90° С. Элементный анализ для C25H29FN2O4:
вычислено (%): С 68,16; Н 6,64; N 6,36,
найдено (%): С 67,96 Н; 6,49; N 6,29.
Пример 6
Фумарат 1-[4-[2-[4-(1,2-бензизоксазол-3-ил)-1-пиперидинил]-этокси]-3-метоксифенид]этанон
Смесь гидрохлорида 3-(4-пиперидинил)-1,2-бензизоксазола (4,8 г, 0,02 моль), карбоната калия (5,2 г, 0,04 моль), 1-[4-(2-хлорэтокси)-3-метоксифенил]этанона (5,0 г, 0,022 моль), и диметилформамида (90 мл) нагревали при 90° С в течение 16 часов. Реакционную смесь переливали в воду и водную смесь экстрагировали этилацетатом. Этилацетат промывали водой, сушили сульфатом магния и растворитель концентрировали до получения масла. При выдерживании масло отверждалось до вещества бежевого цвета. Неочищенное твердое вещество дважды перекристаллизовывали из этилового спирта и получали 5,9 г не совсем белого твердого вещества. Твердое вещество растворяли в этилацетате и добавляли фумаровую кислоту (1,2 г, 1,1 эквивалента). Совсем нагревали недолго на паровой бане и затем перемешивали при температуре окружающей среды два часа. Первоначальное зеленое масло выпадало в осадок и надосадочный слой сливали. Добавляли эфир к надосадочному слою декантированному и собирали 4,0 г белой фумаратной соли. Соль два раза перекристаллизовывали из смеси этанол - эфир и получали 1,7 г (17%) фумарата 1-[4-[2-[4-(1,2-бензизоксазол-3-ил)-1-пиперидинил]этокси]-3-метоксифенил]этанона, точка плавления 127-129° С.
Элементный анализ для С23Н26N2О4:
вычислено (%): С 63,52; Н 5,92; N 5,49,
найдено (%): С 63,00; Н 5,87; N 5,42.
Пример 7
Фумарат 1-[4-[4-[4-(1Н-индазод-3-ил)-1-пиперазинил]-бутокси]-3- метоксифенил]этанона
Перемешиваемую смесь 3-(1-пиперазинил)-1Н-индазола (4,0 г, 0,02 моль), карбоната калия (3,0 г, 0,023 моль), 1-[4-(4-бромбутокси)-3-метоксифенил]этанона (5,3 г) и диметилформамида (60 мл) нагревали при 75° С шесть часов.
Реакционную смесь выливали в воду, и белое твердое вещество выпадало в осадок из раствора. Твердое вещество собирали, сушили и получали 7,2 г неочищенного продукта. Неочищенное твердое вещество дважды перекристаллизовывали из этилового спирта и получали 4,1 г свободного основания, которое обращали в его фумаратную соль добавлением фумаровой кислоты (1,1 г) к соединению, растворенному в нагретом с обратным холодильником ацетоне. Полученную фумаратную соль перекристаллизовывали из этилового спирта и получали 3,8 г (35%) фумарата 1-[4-[4-[4-(1H-индазол-3-ил)-1-пиперазинил]-бутокси]-3-метоксифенил]этанона в виде белого твердого вещества, точка плавления 163-165° С.
Элементный анализ С24Н20N4О3·С4Н4О4:
вычислено (%): С 62,44; Н 6,36; N 10,40,
найдено (%): С 62,28; Н 6,62; N 10,34.
Пример 8
1-[4-[2-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-этокси]-3-метоксифенил]этанон
Перемешиваемую смесь гидрохлорида 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (5,1 г, 0,02 моль), карбоната калия (5,2 г), 1-[4-(2-хлорэтокси)-3-метоксифенил]этанона (5,0 г, 1,022 моль) и диметилформамида (90 мл) нагревали при 90° С 16 часов. Реакционную смесь выливали в воду и водную смесь экстрагировали этилацетатом. Этилацетат промывали водой, сушили сульфатом магния и концентрировали до получения 7,4 г твердого вещества желтого цвета. Твердое вещество перекристаллизовывали из этилового спирта и получали 3,1 г (38%) 1-[4-[2-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-этокси]-3-метоксифенил]этанона в виде слегка желтых хлопьев, точка плавления 133-134° С.
Элементный анализ для С23Н25FN2O4:
вычислено (%): С 66,98; Н 6,13; N 6,79,
найдено (%): С 66,90; Н 6,20; N 6,74.
Пример 9
4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метокси-α -метилбензолметанол
К смеси 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси-3-метоксифенил]этанона (4,0 г, 0,009 моль) в смеси метанол - тетрагидрофуран (60 мл в отношении 1:1) при перемешивании добавляли боргидрид натрия (0,4 г, 0,01 моль). После начального выделения газа все нерастворенные вещества переходили в раствор. Реакционную смесь перемешивали при температуре окружающей среды три часа и тонкослойная хроматография в это время показывала присутствие очень малого количества исходного кетона. Поэтому добавляли еще 0,1 г боргидрида натрия и перемешивание продолжали дополнительные полчаса. Теперь тонкослойная хроматография показывала полное исчезновение исходного материала. Реакционную смесь концентрировали до получения не совсем белого остатка, который разбавляли водой, собирали и получали 3,4 г спирта. Его перекристаллизовывали из толуола (дважды, с обработкой древесным углем) и получали 2,7 г (67%) 4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-3-метокси-α -метилбензолметанола в виде белого твердого вещества, точка плавления 136-138° С.
Элементный анализ для C24H29FN2О4:
вычислено (%): С 67,27; Н 6,82; N 6,54,
найдено (%): С 67,39; Н 6,89; N 6,47.
Пример 10
1-[4-[3-[4-(1,2-бензизотиазол-3-ил]-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
Смесь 3-(4-пиперидинил)-1,2-бензизотиазола (3,0 г, 0,0137 моль), карбоната калия (2,3 г, 0,0155 моль), 1-[4-(3-хлорпропокси)-3-метоксифенил]этанола (4,0 г, 0,0165 моль), йодистого калия (200 мг) и ацетонитрила (100 мл) перемешивали при нагревании с обратным холодильником в атмосфере азота в точение 24 часов. Охлажденную реакционную смесь фильтровали и остаток на фильтре промывали хорошо ацетонитрилом. Фильтрат концентрировали до маслянистого остатка, который распределяли между водой и этилацетатом. Экстракт этилацетата хорошо промывали водой, сушили сульфатом магния, концентрировали до получения масла цвета беж, которое отверждалось при выдерживании. Продукт растирали с диэтиловым эфиром, фильтровали и получали 4,2 г твердого вещества бежевого цвета. Соединение перекристаллизовывали из этилового спирта и получали 35 г, а вторая перекристаллизация из этилового спирта (с использованием обесцвечивающего угля) давала 2,4 г (419) 1-[4-[3-[4-(1,2-бензизотиазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанона, точка плавления 93-95° С.
Элементный анализ для C24H28N2O3S:
вычислено (%): С 67,90; Н 6,65; N 6,60,
найдено (%): С 67,89; Н 6,61; N 6,39.
Пример 11
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-оксифенил]этанон
(А) Синтез 1-[-4-(3-хлорпропокси)-3-оксифенил]этанона
К перемешиваемому раствору 1-[4-(3-хлорпропокси)-3-метоксифенил]этанона (10,0 г, 0,041 моль) в хлористом метилене (120 мл), охлажденному до -50° С (смесью сухой лед-метанол), по каплям добавляли 1 М трибромид бора в хлористом метилене (123 г, 0,12 моль). Температуру сохраняли между -40 и -50° С. После окончания добавки реакционной смеси позволяли повысить температуру до -30° С и контролировали тонкослойной хроматографией (примерно через 15 минут после окончания добавки трифторида бора). Добавляли по каплям насыщенный кислый углекислый натрий, не позволяя температуре повышаться до 0° С в большей части процесса добавки. Когда было добавлено достаточное количество кислого углекислого натрия для подщелачивания раствора, органический слой собирали. Слой промывали рассолом, сушили сульфатом магния, концентрировали до получения 8,1 г темно-коричневого масла, которое отверждалось при выдерживании. Продукт хроматографировали на хроматографе Waters Prep 500 (две колонки силикагеля, смесь 2% метанола - хлористого метилена в качестве элюента). После концентрирования соответствующих фракций получали 5,8 г липкого коричневого твердого вещества. Его перекристаллизовывали из изопропилового эфира (со сливом желтого надосадочного слоя изопропилового эфира и темно-коричневого масляного остатка) и получали первоначально 2,5 г желтого твердого вещества. Концентрированном маточного раствора получали дополнительно 0,5 г продукта, точка плавления 110-113° С.
(В) Синтез 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-оксифенил]этанона
Перемешиваемую смесь 6-фтор-(4-пиперидинил)-1,2-бензизоксазола (2,8 г, 0,013 моль), двууглекислого натрия (1,1 г), нескольких кристаллов йодистого калия, 1-[4-(3-хлорпропокси)-3-оксифенил]этанона и ацетонитрила (100 мл) нагревали с обратным холодильником 16 часов. Реакционную смесь выливали в воду и водную смесь экстрагировали этилацетатом. Органический экстракт промывали водой, сушили сульфатом магния, растворитель концентрировали и получали 5,7 г вязкого желтого масла. Масло хроматографировали на жидком хроматографе Waters Prep 500 на силикагеле, элюируя смесью 7% метанол - хлористый метилен. Концентрированием соответствующих фракций получали желтое масло, которое после выдерживания давало 3,5 г соединения в виде бледно-желтого твердого вещества. Твердое вещество перекристаллизовывали из этилового спирта и получали 2,7 г (50%) 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-оксифенил]этанона в виде бледно-желтого твердого вещества, точка плавления 122-124° С.
Элементный анализ дли С23Н25FN2O4:
вычислено (%): С 66,98; Н 6,11; N 6,79,
найдено (%): С 66,97; Н 6,20; N 6,69.
Пример 12
Перемешиваемую смесь 6-фтор-3-(1-пиперазинил)-lH-индазола (2,3 г, 0,01 моль), карбоната калия (1,5 г), 1-[4-(3-хлорпропокси)-3-метоксифенил]этанона (2,8 г, 0,011 моль), нескольких кристаллов йодистого калия и диметилформамида (50 мл) нагревали при 90° С в течение 16 часов. Реакционную смесь выливали в воду и водную суспензию экстрагировали этилацетатом. Этилацетат промывали водой, сушили сульфатом магния, концентрировали и получали 5,0 г желтого масла. Масло хроматографировали на Waters Prep 500, используя колонки силикагеля и элюируя смесью хлористый метилен - метанол (7%). Концентрированном желаемых фракций получали не совсем белое твердое вещество (2,0 г, 46%). Этот образец объединяли с 1,0 г предыдущего образца и перекристаллизовывали из толуола. Получали 2,6 г 1-[4-[3-[4-(6-фтор-1Н-индазол-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанона в виде белого твердого вещества, точка плавления 135-137° С.
Элементный анализ для С23Н27FN4О3:
вычислено (%): С 64,77; Н 6,38; N 13,14,
найдено (%): С 64,66; Н 6,21; N 13,02.
Пример 13
1-[4-[4-[4-(6-Фтор-1-1Н-индазол-3-ил)-1-пиперазинил]бутокси]-3-метоксифенил]этанон
Перемешиваемую смесь гидрохлорида 6-фтор-3-(1-пиперазинил)-1H-индазола (5 г, 0,019 моль), карбоната калия (5,8 г), 1-[4-(4-бромбутокси)-3-метоксифенил]этанона (6,3 г, 0,021 моль) и диметилформамида (80 мл) нагревали при 75° С шесть часов. Реакционную смесь выливали в воду, и из раствора выпадало не очень белое твердое вещество. Твердое вещество собирали, сушили и получали 4,5 г неочищенного продукта. Соединение перекристаллизовывали три раза из этанола и получали 3,0 г не совсем белого твердого вещества. Твердое вещество хроматографировали на Waters Prep 500, используя колонки из силикагеля и элюируя смесью хлористый метилен - метанол (7%). Концентрированием соответствующих фракций получали 2,3 г не совсем белого твердого вещества, которое затем перекристаллизовывали из этанола и получали 1,9 г (26%) чистого для анализа 1-[4-[4-[4-(6-фтор-1-1Н-индазол-3-ил)-1-пиперазинил]бутокси]-3-метоксифенил]этанона, точка плавления 156-158° С.
Элементный анализ для С24Н29FN4O3:
вычислено (%): С 65,44; Н 6,54; N 12,72,
найдено (%): С 65,38; Н 6,49; N 12,60.
Пример 14
1-[4-[3-[4-(1H-индазол-3-ил)-1-пиперидинил]пропокси]-3-метоксифенид]этанон
Смесь 3-(4-пиперидинил)-1H-индазола (3,0 г, 0,015 моль), карбоната калия (1,6 г), 1-[4-(3-хлорпропокси)-3-метоксифенил]этанона (5,3 г, 0,022 моль), несколько кристаллов йодистого калия и ацетонитрила (100 мл) перемешивали и нагревали с обратным холодильником в течение 16 часов. Реакционную смесь выливали в воду и из раствора выделялся осадок белого твердого вещества. Твердое вещество собирали, сушили и получали 5,1 г продукта. Перекристаллизацией из этанола получали 3,6 г соединения, которое подвергали высоко производительной препаративной жидкостной хроматографии на силикагеле, элюируя смесью хлористый метилен - метанол в отношении 9:1 и получали 3,0 г (49%) не совсем белого твердого вещества. Перекристаллизацией из этанола получали чистый для анализа 1-[4-[3-[4-(1H-индазол-3-ил)-1-пиперидинил]-пропокси)-3-метоксифенил]-этанон в виде белого твердого вещества, точка плавления 171-173° С.
Элементный анализ для С24Н29N3О3:
вычислено (%): С 70,74; Н 7,17; N 10,31,
найдено (%): С 70,52; Н 7,27; N 10,42.
Пример 15
1-[4-[3-[4-(6-хлор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
Перемешиваемую смесь 6-хлор-3-(4-пиперидинил)-1,2-бензоксазола (4,7 г, 0,02 моль), 1-[4-(3-хлорпропокси)-3-метоксифенил]этанона (4,8 г, 0,02 моль), карбоната калия (2,8 г) нескольких кристаллов йодистого калия и ацетонитрила (120 мл) нагревали с обратном холодильником 16 часов. Реакционную смесь фильтровали, фильтрат концентрировали и получали желтую смесь твердого вещества с маслом. Остаток хроматографировали на Waters Prep 500, используя колонки силикагеля и элюируя смесью хлористый метилен - метанол (5%). Концентрированием желаемых фракций получали 3,2 г твердого вещества цвета беж, которое после перекристаллизации из этанола давало 2,7 г (31%) 1-[4-[3-[4-(6-хлор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанона в виде твердого вещества бежевого цвета, точка плавления 116-118° С.
Элементный анализ для СН24H27СlN2O4:
вычислено (%): С 65,08; Н 6,14; N 6,32,
найдено (%): С 65,35; Н 6,22; N 6,28.
Пример 16
Фумарат 1-[4-[4-[4-(6-хлор-1,2-бензизоксазол-3-ил)-пиперидинил]-бутокси]-3-метоксифенил]этанона
Перемешиваемую смесь 6-хлор-3-(4-пиперидинил)-1,2-бензизоксазола (4,7 г, 0,02 моль), 1-[4-(4-бромбутокси)-3-метоксифенил]этанона (6,0 г, 0,02 моль), карбоната калия (2,8 г) и ацетонитрила (120 мл) нагревали с обратным холодильником 16 часов. Реакционную смесь охлаждали, фильтровали и фильтрат концентрировали до 9,9 г коричневого масла. Масло хроматографировали на Waters Prep 500, используя колонки силикагеля и элюируя смесью хлористый метилен - метанол (5%). Концентрирование соответствующих фракций давало 2,3 г не совсем белого твердого вещества. Твердое вещество растворяли в этаноле, этанол испаряли и полученное коричневое твердое вещество растворяли в нагреваемом с обратным холодильником ацетоне. Посла охлаждения белое твердое вещество кристаллизовалось из раствора с выходом 2,2 (19%) фумарата 1-[4-[4-[4-(6-хлор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]-этанона в виде белого твердого вещества, точка плавления 139-141° С.
Элементный анализ для C25H29ClN2O4·C4H4O4:
вычислено (%): С 60,78; Н 5,80; N 4,89,
найдено (%): С 60,69; Н 5,74; N 4,85.
Пример 17
1-[4-[3-[4-(5-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
Смесь 5-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (2,2 г 0,01 моль), 1-[4-(3-хлорфенокси)-3-метоксифенил]этанона (2,4 г, 0,01 моль), карбоната калия (1,4 г), нескольких кристаллов йодистого калия и ацетонитрила (100 мл) перемешивали и нагревали с обратным холодильником восемь часов. Реакционную смесь выливали в воду и водную смесь экстрагировали этилацетатом. Этилацетатный экстракт промывали рассолом, сушили сульфатом магния, концентрировали и получали 4,0 г белого твердого вещества. Твердое вещество хроматографировали на высокоэффективном жидкостном хроматографе Waters Prop 500 используя колонки силикагеля и элюируя смесью хлористый метилен - метанол (3%). Концентрированием соответствующих фракций получали 2,0 г (47%) 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанона в виде целого кристаллического твердого вещества, точка плавления 103-105° С.
Элементный анализ для C24H27FN2О4:
вычислено (%): С 67,59; Н 6,38; N 6,57,
найдено (%): С 67,50; Н 6,74; N 6,53.
Пример 18
Фумарат 6-фтор-3-[1-[3-(2-метоксифенокси)-пропил]-4-пиперидинил]-1,2-бензизоксазода
Перемешиваемую смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (2,45 г, 11,1 ммоль), карбоната калия (2,0 г) и 3-(2-метоксифенокси)-пропилхлорида (3,5 г, 17,4 ммоль) в ацетонитриле (40 мл) нагревали при 90° С четыре часа. По окончании реакции растворитель удаляли и твердые вещества растворяли в дихлорметане (100 мл). Раствор промывали водой и рассолом, затем сушили над сульфатом магния. Неочищенный материал объединяли с 1,2 г неочищенного материала, полученного таким же образом (используя 0,5 г исходного материала). Объединенный материал очищали быстрой хроматографией на колонке силикагеля (49 г, со смесью 0,5% диэтиламина - 1% метанола и 98,5% дихлорметана в качестве элюента, 568 мл). Фракции, содержащие чистый продукт, собирали и концентрировали до светлого масла (3,68 г). Это масло обрабатывали фумаровой кислотой (1,14 г, 9,8 ммоль) в этаноле (13 мл). Получены кристаллы фумарата 6-фтор-3-[1-[3-(3-метоксифенокси)-пропил]-4-пиперидинил]-1,2-бензизоксазола, весившие 4,01 г (60%), точка плавления 169-170° С.
Элементный анализ C22H25FN2O3·C4H4O4:
вычислено (%): С 62,39; Н 5,84; N 5,60,
найдено (%): С 62,37; Н 5,88; N 5,60.
Пример 19
1-[3-[4-[6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-4-метоксифенил]фенилметанон
Перемешиваемую смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (2,01 г, 9,13 моль), карбоната калия (2,0 г), 1-[3-(3-хлорпропокси)-4-метоксифенил]-фенилметанона (3,93 г, 11,3 ммоль) и ацетонитрила (50 мл) нагревали с обратным холодильником четыре часа. В конце реакции растворитель выпаривали и остаток распределяли между водой (150 мл) и дихлорметаном (400 мл). Дихлорметановый раствор промывали водой и рассолом (100 мл), сушили над сульфатом магния, затем концентрировали до масла. Очистку проводили быстрой хроматографией на колонке силикагеля (двуокись кремния 40 г, элюированная дихлорметаном, 300 мл: смесью 1% метанол - дихлорметан, 850 мл). Полученный таким образом материал в виде бесцветного масла отверждался при выдерживании. Перекристаллизацией из этанола (150 мл) получали 1-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-4-метоксифенил]фенилметанона в форме белых кристаллов, 0,07 г (63%), точка плавления 140-141° С.
Элементный анализ для C29H29FN2О4:
вычислено (%): С 73,30; Н 5,98; N 5,73,
найдено (%): С 71,09; Н 5,98; N 5,73.
Пример 20
1-[4-[4-[4-(1Н-индазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
Смесь 3-(4-пиперидинил)-lH-индазола (3,4 г, 0,016 моль), 1-[4-[4-бромбутокси)-3-метоксифенил]этанона (5,0 г, 0,016 моль), карбоната калия (2,2 г) и ацетонитрила (100 мл) перемешивали и нагревали с обратным холодильником шесть часов. Реакционную смесь выливали в воду и образовавшееся желтое твердое вещество собирали и получали 5,3 г продукта. Соединение перекристаллизовывали из ацетонитрила и затем из этилацетата и получали 3,0 г (45%) слегка желтого твердого вещества 1-[4-[4-[4-(1Н-индазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанона, точка плавления 133-135° С.
Элементный анализ для С25Н31N3О3:
вычислено (%): С 71,23; Н 7,41; N 9,97,
найдено (%): С 70,85; Н 7,61; N 9,81.
Пример 21
1-[4-[2-[4-(6-хлор-1,2-бензизоксазол-3-ил)-1-пиперидинид]-этокси]-3-метоксифенил]этанон
Перемешиваемую смесь 6-хлор-3-(4-пиперидинил)-1,2-бензизоксазола (4,6 г, 0,019 моль), 1-[4-(2-хлорэтокси)-3-метоксифенил]этанона (4,3 г, 0,019 моль), карбоната калия (2,8 г), нескольких кристаллов йодистого калия и ацетонитрила (120 мл) нагревали с обратным холодильником в течение 16 часов.
Реакционную смесь фильтровали, фильтрат концентрировали до получения 8,0 г желтого твердого вещества. Твердое вещество хроматографировали на жидкостном хроматографе Waters Prep 500 (колонки кремнезема, с элюентом из смеси хлористый метилен - метанол, 5%). Концентрированней соответствующих фракций получали 3,2 г светло-желтого твердого вещества, из которого после перекристаллизации из этилацетата получали 2,3 г (28%) 1-[4-[2-[4-(6-хлор-1,2-бензизоксазол-3-ил)-1-пиперидинил]этокси]-3-метоксифенил]этанона (28% выход) в виде бледно-желтого твердого вещества, точка плавления 133-135° С.
Элементный анализ для С23Н25СlN2О4:
вычислено (%): С 64,41; Н 5,88; N 6,53,
найдено (%): С 64,35; Н 5,87; N 6,41.
Пример 22
3-(3-бромпропокси-4-метоксифенил)фенилметанон
Раствор 3-окси-4-метоксибензофенона (4,6 г, 20 ммоль) в диметилформамиде (35 мл) обрабатывали гидридом натрия (600 мг, 25 ммоль) при 0° С в течение 20 минут, затем одной порцией добавляли 1,3-дибромпропан (5 г, 24,7 ммоль). Смесь нагревали при 90° С один час и затем перемешивали при комнатной температуре два часа. В конце реакции смесь выливали в воду (500 мл) и экстрагировали этилацетатом (400 мл). Этилацетатный раствор промывали водой, рассолом и сушили над безводным сульфатом магния. Растворитель удаляли и неочищенное масло очищали быстрой хроматографией на колонке силикагеля (кремнезем, 85 г, элюент - смесь, смесь гексан - дихлорметан в отношении 3:1, 1,6 л, смесь гексан - дихлорметан в отношении 3:7 1,4 л). Полученный таким образом чистый продукт в виде масла весил 4,67 г (66%). Перекристаллизация два раза из изопропилового эфира (500 мл) давала 2,42 г чистого для анализа 5-(3-бромпропокси-4-метоксифенил)фенилметанона, точка плавления 81-83° С.
Элементный анализ для С17Н17ВrО3:
вычислено (%): С 58,47; Н 4,91,
найдено (%): С 58,63; Н 4,82.
Пример 23
Фумарат 1-[3-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-фенил]этанона
Смесь гидрохлорида 6-фтор-(4-пиперидинил)-1,2-бензизоксазола (4,53 г, 20,5 ммоль), карбоната калия (4,5 г), 1-[3-(3-хлорпропокси)-фенил]этанона (6,4 г, 29 ммоль) в ацетонитриле (60 мл) нагревали с обратным холодильником пять часов. По окончании реакции растворитель удаляли и остаток экстрагировали в дихлорметан (300 мл). Неорганические нерастворимые вещества отфильтровывали. Дихлорметановый раствор концентрировали до небольшого объема (10 мл) и очищали хроматографией на колонке (кремнезем, 75 г, элюентом служили дихлорметан, 900 мл и смесь 2% метанол - дихлорметан, 800 мл). Фракции, содержащие чистый продукт, объединяли и концентрировали до масла (2,87 г, 35%). Масло растворяли в этаноле и обрабатывали раствором фумаровой кислоты (841 мг). Двойной перекристаллизацией из этанола получали 2,53 г фумарата 1-[3-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-фенил]-этанона в виде белых кристаллов, т. пл. 172-174° С.
Элементный анализ для C22H25FN2O3·C4H4O4:
вычислено (%): С 63,27; Н 5,70; N 5,47,
найдено (%): С 63,04; Н 5,63; N 5,43.
Пример 24
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-2-метилфенил]этанон
Перемешиваемую смесь гидрохлорида 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (5,5 г, 21,6 ммоль), карбоната калия (3,5 г), 1-[4-(3-бромпропокси)-2-метилфенил]этанона (4,83 г, 17,8 ммоль) в диметилформамиде (25 мл) нагревали при 120° С пять часов. По окончании реакции растворитель удаляли, остаток экстрагировали в дихлорметан (300 мл) и раствор промывали водой и рассолом. Органический раствор сушили и выпаривали до неочищенного масла. Очистку проводили колоночной хроматографией на силикагеле (80 г, элюировали дихлорметаном, 1,0 л, смесью 1% метанол - дихлорметан, 1,2 л: смесью 2% этанол - дихдорметан, 1,2 л). Самые чистые фракции объединяли и получали 2,91 г твердого вещества. Перекристаллизацией из дихлорметана и этанола получали 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-2-метилфенил]-этанон в виде не совсем белых кристаллов, точка плавления 113-114° С.
Элементный анализ для С24Н27FN2О3:
вычислено (%): С 70,22; Н 6,63; N 6,82,
найдено (%): С 70,13; Н 6,63; N 6,77.
Пример 25
1-[3-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]пропокси]-5-метилфенил]этанон
Смесь гидрохлорида 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (2,87 г, 11,23 ммоль), карбоната калия (2,50 г), 1-[2-(3-бромпропокси)-5-метилфенил]этанона (3,74 г, 13,8 ммоль) в диметилформамиде (10 мл) и ацетонитриле (50 мл) нагревали при 95° С шесть часов. По окончании реакции растворитель концентрировали и смесь экстрагировали в дихлорметан (300 мл). Органический раствор промывали водой и рассолом, сушили над сульфатом магния, затем концентрировали до неочищенного масла. Очистку проводили быстрой хроматографией на колонке силикагеля (двуокись кремния, 60 г, элюировали смесями 1% метанол - дихлорметан, 1,2 л и 3% метанол - дихлорметан, 600 мл). Полученный таким образам материал кристаллизовали из небольшого объема эфира и гексана и получали 2,13 г (46%) не совсем белого 1-[3-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-5-метилфенил]этанона, точка плавления 92-93° С.
Элементный анализ для С24H27FN2О3:
вычислено (%): С 70,22; Н 6,63; N 6,82,
найдено(%): С 70,21; Н 6,96; N 6,81.
Пример 26
Полуфумарат N-[3-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-4-метоксифенил]ацетамида
Смесь гидрохлорида 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (3,94 г, 15,4 ммоль), карбоната калия (3,67 г, 26,6 ммоль), N-[3-(3-бромпропокси)-4-метоксифенил]-ацетамида (5,56 г, 18,6 ммоль) в диметилформамиде (75 мл) и ацетонитриле (100 мл) нагревали при 100° С три часа. По окончании реакции растворитель концентрировали и смесь экстрагировали в дихлорметан (500 мл). Органический раствор промывали водой (500 мл) и рассолом (400 мл), сушили, затем концентрировали до неочищенного масла. Очистку осуществляли быстрой хроматографией через колонку силикагеля (двуокись кремния 65 г, элюировали смесью 1% метанол - дихлорметан, 1,2 л и смесью 3% метанол - дихлорметан, 500 мл). Полученный таким образом материал в виде масла весил 2,33 г (34,3%). Материал растворяли в этаноле и обрабатывали раствором фумаровой кислоты (661 мг) в этаноле. Получали N-[3-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-4-метоксифенил]ацетамид, полуфумарат в виде не совсем белых кристаллов, весивших 2,17 г, точка плавления 205-206° С.
Элементный анализ для С24Н28FN3O4·0,5 С4Н4O4:
вычислено (%): С 62,50; Н 6,05; N 8,41,
найдено (%): С 62,30; Н 6,05; N 8,32.
Пример 27
6-хлор-3-(1-пиперазинил)-1H-индазол
К перемешиваемой суспензии 4-(6-хлор-1-фенилсульфонил-1H-издазол-3-ил)-1-пиперазинкарбонитрила (192,5 г, 0,479 моль) в сухом тетрагидрофуране (3,5 л) в атмосфере азота добавляли по каплям алюмогидрид лития (958 мл 1,0 М раствора алюмогидрида лития в тетрагидрофуране, 0,958 моль). После завершения добавки реакционную смесь нагревали с обратным холодильником и перемешиванием под азотом в течение 4 часов. Реакционную смесь охлаждали до 4° С на ледяной бане с солью и избыток алюмогидрида лития разлагали осторожным, по каплям, добавлением воды. Смесь энергично перемешивали дополнительно 30 минут и затем фильтровали через стеклянную воронку с горизонтальными слоями. Остаток на фильтре хорошо промывали тетрагидрофураном (три раза по 500 мл) и затем метанолом (два раза по 600 мл), фильтрат концентрировали до получения 151,0 г бежевой смолы. Растиранием с диэтиловым эфиром получали твердое вещество, которое собирали, сушили и получали 75,0 г (66%) желаемого индазола. Образец весом 4,0 г перекристаллизовывали из толуола и получали 3,2 г, из которого после второй перекристаллизации из толуола (используя обесцвечивающий уголь) получали 2,1 г (35%) твердого вещества бежевого цвета: 6-хлор-3-(1-пиперазинил)-lH-индазол, точка плавления 135-137° С.
Элементный анализ для C11H13ClN4:
вычислено (%): С 55,82; Н 5,54; N 23,67,
найдено (%): С 55,91; Н 5,54; N 23,41.
Пример 28
1-[4-[3-[4-(6-фтор-1Н-индазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
Перемешиваемую смесь 6-фтор-3-(4-пиперидинил)-lH-индазола (3,5 г, 0,016 моль), карбоната калия (2,2 г), 1-[4-(3-хлорпропокси)-3-метоксифенил]этанона (3,8 г, 0,016 моль) и ацетонитрила (90 мл) нагревали с обратным холодильником 16 часов. Реакционную смесь выливали в воду и образовавшееся белое твердое вещество, которое выпало из раствора, собирали и получали 5,5 г желаемого продукта. Соединение перекристаллизовывали из метилформамида (дважды) и получали 3,0 г (44%) 1-[4-[3-[4-(6-фтор-1Н-индазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанона в виде белого твердого вещества, точка плавления 202-204° С.
Элементный анализ для С24Н28FN3О3:
вычислено (%): С 67,57; Н 6,63; N 9,88,
найдено (%): С 67,59; Н 6,61; N 9,96.
Пример 29
Полуфумарат 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метилфенил]этанона
Перемешиваемую смесь гидрохлорида 6-фтор-3-(4-пиперидинил)-1,2-бензизоксаэола (3,0 г, 11,7 ммоль), карбоната калия (3,0 г) и 1-[4-(3-бромпропокси)-3-метилфенил]этанона (3,19 г) в диметилформамиде (20 мл) и ацетонитриле (50 мл) нагревали при 95° С четыре часа. По окончании реакции растворитель концентрировали до 30 мл, затем распределяли между водой (200 мл) и дихлорметаном (300 мл). Дихлорметановый раствор отделяли, промывали водой и рассолом, затем сушили над сульфатом магния. Неочищенный продукт после выпаривания растворителя очищали быстрой хроматографией на колонке силикагеля (кремнезем, 60 г, элюировали 1% метанолом в дихлорметане, 600 мл 2% метанолом в дихлорметане, 600 мл). Полученный таким образом материал представлял собой светло-желтое масло, вес 2,07 г (43%). Это масло растворяли в этаноле и обрабатывали раствором фумаровой кислоты (585 мг) в метаноле. После охлаждения до 0° С образовались кристаллы полуфумарата 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метилфенил]этанона. Его собирали и получали 1,5 г, точка плавления 185-187° С.
Элементный анализ для С24Н27FN2O3·0,5 С4Н4O4:
вычислено (%): С 66,65; Н 6,24; N 5,98,
найдено (%): С 66,69; Н 6,23; N 5,95.
Пример 30
1-[4-[3-[4-(6-фтор-1,2-бeнзизoкcaзoл-3-ил)-1-пиперимидин]-пропокси]-фенил]этанон
Смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (3,27 г, 14,8 ммоль), карбоната калия (3,0 г), 1-[4-(3-бромпропокси)-фенил]этанона (4,5 г, 17,5 ммоль) в ацетонитриле (60 мл) нагревали с обратным холодильником четыре часа. Растворитель удаляли. Остаток растворяли в дихлорметане (300 мл) и промывали водой и рассолом, затем сушили над сульфатом магния. Неочищенный продукт после выпаривания раствора очищали быстрой хроматографией (кремнезем, 60 г, элюировали 1% метанолом в дихлорметане, 1,0 литр). Наиболее чистые фракции объединяли и получали 2,8 г (48%) 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-пиперидинил]-пропокси]-фенил]этанона, точка плавления 111-112° С.
Элементный анализ для С23Н25FN2O3:
вычислено (%): С 69,69; Н 6,36; N 7,07,
найдено (%): С 69,60; Н 6,38; N 7,07.
Пример 31
1-[4-[3-[4-(6-хлор-1Н-индазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]этанон
Смесь 6-хлор-[3-(1-пиперазинил)]-1Н-индазола (3,4 г, 0,0 моль), карбоната калия (2,5 г, 0,018 моль), 1-[4-(3-хлорпропокси)-3-метоксифенил]этанона (3,8 г, 0,016 моль), йодистого калия (200 мг) и ацетонитрила (125 мл) перемешивали в атмосфере азота при нагревании с обратным холодильником в течение 30 часов. После выдерживания при комнатной температуре в течение 40 часов реакционную смесь фильтровали и остаток на фильтре хорошо промывали ацетонитрилом. Фильтрат концентрировали до маслянистого твердого вещества, которое распределяли между водой и этилацетатом. Этилацетатный экстракт промывали водой, сушили над сульфатом магния и концентрировали до получения 6,9 г темного масла, которое отверждалось после двух дней выдержки в вакууме. Продукт очищали препаративной высокоэффективной жидкостной хроматографией (Waters Associates Prep LC/System 500, с использованием двух колонок силикагеля и смеси 6% метанола с хлористым метиленом в качестве элюента) и получали 4,2 г продукта. Материал перекристаллизовывали из этанола и получали 3,4 г блестящего бежевого цвета 1-[4-[3-[4-(6-хлор-1Н-индазол-3-ил)-3-пиперазинил]-пропокси]-3-метоксифенил]этанона в форме кристаллов, точка плавления 132-134° С.
Элементный анализ для С23Н27СlN4O3S:
вычислено (%): С 62,37; Н 6,14; N 12,65,
найдено (%): С 62,49; Н 6,16; N 12,60.
Пример 32
1-[4-[4-[4-(1,2-бензизотиазол-3-ил)-1-пиперизинил]-бутокси]-3-метоксифенил]этанон
Смесь 3-(1-пиперазинил)-1,2-бензизотиазола (4,0 г, 0,0182 моль), 1-[4-(4-бромбутокси)-3-метоксифенил]этанона (6,0 г, 0,0200 моль), карбоната калия (3,0 г, 0,0218 моль), йодистого калия (200 мг) и ацетонитрила (125 мл) перемешивали при нагревании с обратным холодильником в атмосфере азота пять часов. Большую часть растворителя удаляли в вакууме и полученный смолистый остаток распределяли между этилацетатом и водой. Органический экстракт промывали водой, сушили сульфатом магния, концентрировали и получали 7,8 г. Очисткой препаративной высокопроизводительной жидкостной хроматографией (Waters Associates Prep LC/System 500 с использованием двух колонок силикагеля и смеси 4% метанола и хлористого метилена в качестве элюента) получали 6,5 г влажного не совсем белого твердого вещества. Продукт два раза перекристаллизовывали из толуола и получали 3,1 г (39%) 1-[4-[4-(1,2-бензизотиазол-3-ил)-1-пиперазинил]-бутокси]-3-метоксифенил]этанона в виде твердого вещества белого цвета, точка плавления 114-116° С.
Элементный анализ для С24Н29N3О3S:
вычислено (%): С 65,58; Н 6,65; N 9,56,
найдено (%) С 65,74; Н 6,66; N 9,54.
Пример 33
4-[3-[4-(6-фтор-1,2-бензизотиазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксибензонитрил
Смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизотиазола (3,0 г 13,6 ммоль), карбоната калия (2,8 г), 4-(3-бромпропокси)-3-метоксибензонитрила (4,0 г, 14,8 ммоль) в ацетонитриле (70 мл) нагревали с обратным холодильником три часа. К окончанию реакции растворитель удаляли на роторном испарителе. Органический материал экстрагировали в дихлорметан (250 мл), а неорганические вещества отфильтровывали. Дихлорметановый раствор концентрировали до неочищенного масла. Очистку проводили хроматографией на колонке силикагеля (двуокись кремния, 55 г, элюировали дихлорметаном, 600 мл). Полученный таким образом материал кристаллизовали из малого количества дихлорметана. Перекристаллизацией из этанола (25 мл) получали 3,8 мг (68%) 4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксибензонитрила в виде белых кристаллов, точка плавления 107-108° С.
Элементный анализ дли С23Н24FN3О3:
вычислено (%): С 67,47; Н 5,91; N 10,26,
найдено (%): С 67,32; Н 5,90; N 10,24.
Пример 34
1-[4-[4-[4-(6-фтор-1Н-индазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанон
Перемешиваемую смесь 6-фтор-3-(4-пиперидинил)-1Н-индола (1,9 г, 0,0086 моль), 1-[4-(4-бромбутокси)-3-метоксифенил]-этанона (2,6 г, 0,0086 моль), карбоната калия (1,2 г) и ацетонитрила (75 мл) нагревали с обратным холодильником шесть часов. Реакционную смесь выливали в воду, и белое твердое вещество осаждалось из раствора. Его собирали, сушили и получали 3,2 г продукта. Продукт перекристаллизовывали из этанола и получали 2,7 г (71%) 1-[4-[4-[4-(6-фтор-1Н-индазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанона в виде блестящих белых хлопьев, точка плавления 158-160° С.
Элементный анализ для С25Н30FN3О:
вычислено (%): С 68,32; Н 6,88; N 9,56,
найдено (%): С 68,00; Н 6,93; N 9,51.
Пример 35
Сесквифумарат 1-[4-[3-[4-(1-бензоил-6-фтор-1Н-индазол-3-ил) -1-пиперазинил]-пропокси]-3-метоксифенил]этанона
Смесь 1-[4-[3-[4-(6-фтор-1Н-индазол-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанона (3,2 г, 0,0075 моль) и хлористого бензоила (15 мл) нагревали на паровой бане 15 минут. Реакционную смесь охлаждали и добавляли эфир. Нерастворимое не совсем белое вещество собирали и получали 4,4 г продукта в форме хлористоводородной соли. Соль обращали в свободное основание водной гидроокисью аммония и после экстрактивной обработки хлористым метиленом выделяли 3,0 г свободного основания в виде твердого белого вещества. Свободное основание растворяли в этилацетате и добавляли фумаровую кислоту (0,72 г, 1,1 эквивалента) и смесь нагревали на паровой бане 15 минут. После выдерживания при температуре окружающей среды в течение 4 дней собирали 2,0 г не совсем белой фумаратной соли, в то время как концентрирование фильтрата давало дополнительно 1,0 г соли. Перекристаллизацией сначала из этилацетата и затем из этанола получали 1,4 г (26%) сесквифумарата 1-[4-[3-[4-(1-бензоил-6-фтор-1Н-индазол-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанона, точка плавления 138-140° С.
Элементный анализ для С30Н31FN4O4·1,5 C4H4O4:
вычислено (%): С 61,35; Н 5,29; N 7,95,
найдено (%): С 61,68; Н 5,31; N 8,25.
Пример 36
1-[4-[4-[4-(6-хлор-1Н-индазол-3-ил)-1-пиперазинил]-бутокси]-3-метоксифенил]этанон
Смесь 6-хлор-[3-(1-пиперазинил)]-1Н-индазола (4,0 г, 0,017 моль), карбоната калия (2,8 г, 0,020 моль), 1-[4-(4-бромбутокси)-3-метоксифенил]этанона (5,7 г, 0,019 моль), йодистого калия (100 мг) и ацетонитрила (125 мл) перемешивали при нагревании с обратным холодильником в атмосфере азота 18 часов. Охлажденную реакционную смесь выливали в воду, образовавшееся не совсем белое твердое вещество собирали фильтрацией, сушили и получали 7,0 г продукта. Соединение два раза перекристаллизовывали из толуола и получали 6,2 г. Дальнейшую очистку проводили высокоэффективной жидкостной хроматографией (Waters Associates Prep LC/System 500, используя 5%-ный метанол - хлористый метилен в качестве элюента и две колонки силикагеля) и получали 5,3 г блестящих кристаллов цвета беж, которые перекристаллизовывали четыре раза из толуола и получали 3,1 г белого твердого вещества. Чистый для анализа материал получали последующей перекристаллизацией из диметилформамида и получали 2,5 г (32%) 1-[4-[4-[4-(6-хлор-1Н-индазол-3-ил)-1-пиперазинил]-бутокси]-3-метоксифенил]этанона в форме не совсем белого порошка, точка плавления 189-191° С.
Элементный анализ для C24Н29ClN4O3:
вычислено (%): С 63,08; Н 6,40; N 12,26,
найдено (%): С 62,86; Н 6,57; N 12,49.
Пример 37
Полуфумарат 1-[4-[3-[4-(1,2-бензизотиазол-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанона
Смесь 3-(1-пиперазинил)-1,2-бензизотиазола (4,0 г, 0,0182 моль), карбоната калия (3,0 г, 0,0218 моль), йодистого калия (200 мг), 1-[4-(3-хлорпропокси)-3-метоксифенил]этанона (5,3 г, 0,0200 моль) и ацетонитрила (125 мл) нагревали с обратным холодильником при перемешивании в атмосфере азота в течение 36 часов. Охлажденную реакционную смесь фильтровали и остаток на фильтре хорошо промывали ацетонитрилом.
Фильтрат концентрировали до получения 10,7 г маслянистого остатка, который экстрагировали этилацетатом. Этилацетатный экстракт промывали водой, сушили сульфатом магния и концентрировали до получения 8,0 г темного масла. Масло очищали препаративной высокопроизводительной жидкостной хроматографией (Waters Associates Prep LC/System 500, используя две колонки силикагеля и смесь 3% метанола с хлористым метиленом в качестве элюента). Концентрированием соответствующих фракций получали 4,6 г красного масла, которое отвергалось при выдерживании. Образец 3,4 г растворяли в этилацетате (100 мл) и добавляли фумаровую кислоту (0,95 г). Смесь перемешивали при нагревании при умеренной температуре обратного холодильника один час и затем при температуре окружающей среды полтора часа. Полученное бежевое твердое вещество собирали фильтрацией, сушили и получали 4,0 г продукта. Продукт два раза кристаллизовали из этанола и получали 2,7 г (27%) полуфумарата 1-[4-[3-[4-(1,2-бензизотиазол-3-ил)-1-пиперазинил]-пропокси]-3-метоксифенил]этанона в виде порошка цвета беж, точка плавления 186-188° С.
Элементный анализ для С23Н27N3О3S· 0,5 C4H4O4:
вычислено (%): С 62,09; Н 6,06; N 8,69,
найдено (%): С 62,01; Н 6,06; N 8,68.
Пример 38
Перемешиваемую смесь 6-фтор-3-(4-пиперидил-1,2-бензизоксазола (2,0 г, 9,0 ммоль), карбоната калия (1,3 г), 1-[4-(3-бромпропокси)-3,5-дибромфенил]этанона (2,65 г, 9,0 ммоль) и ацетонитрила (50 мл) нагревали с обратным холодильником три часа. В конце реакции растворитель выпаривали и остаток экстрагировали в дихлорметан (150 мл). Нерастворимые вещества отфильтровывали. Дихлорметановый раствор концентрировали до масла. Очистки проводили быстрой хроматографией на колонке силикагеля (двуокись кремния 47 г, в качестве элюента служили дихлорметан, 300 мл и 1% метанол в дихлорметане, 600 мл). Очищенный таким образом продукт до бесцветного масла отверждался при выдерживании. Перекристаллизацией из этанола получали 1-[3,5-дибром-4-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидил]-пропокси]-фенил]этанон в виде белых кристаллов (2,93 г,57%), точка плавления 102-103° С.
Элементный анализ для С23Н23Вr2FN2O3:
вычислено (%): С 49,84; Н 4,18; N 5,05,
найдено (%): С 49,91; Н 4,11; N 4,98.
Пример 39
1-[4-[2-[4-(1,2-бензизотиазол-3-ил)-1-пиперазинил]-этокси]-3-метоксифенил]этанон
Смесь 3-(1-пиперазинил)-1,2-бензизотиазола (4,0 г, 0,182 моль), 1-[4-(2-хлорэтокси)-3-метоксифенил]этанона (4,3 г, 0,0200 моль), карбоната калия (3,0 г, 0,0218 моль), ацетонитрила (125 мл) и каталитического количества йодистого калия кипятили с обратным холодильником и перемешивали в атмосфере азота 24 часа. В этот момент в реакционную смесь добавляли дополнительное количество карбоната калия (1,0 г, 0,0072 моль) и алкилирующий агент (0,4 г, 0,0017 моль) и нагревание с обратным холодильником возобновляли в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Осадок на фильтре промывали ацетонитрилом и фильтрат концентрировали до получения темного масла. Масло экстрагировали хлористым метиленом и органический экстракт промывали водой, сушили сульфатом магния, концентрировали и получали 9,2 г масла. Очистка препаративной высокопроизводительной жидкостной хроматографией (Waters Associates Prep LC/System 500, используя две колонки силикагеля и смесь 3% метанол - хлористый метилен в качестве элюента) давало 3,8 г сочной бежевого цвета смолы, которая быстро отверждалась. Соединение два раза перекристаллизовывали из этанола и получали 2,1 г (28%) 1-[4-[2-[4-(1,2-бенэизотиазол-3-ил)-1-пиперазинил]-этокси]-3-метоксифенил]этанона в виде твердого вещества цвета беж, точка плавления 98-100° С.
Элементный анализ для С22Н25N3О3:
вычислено (%): С 64,21; Н 6,12; N 10,2,
найдено (%): С 64,05; Н 6,09; N 10,12.
Пример 40
6-фтор-3-[1-(3-феноксипропил)-4-пиперидинил]-1,2-бензизоксазол
Смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (4,0 г 0,0182 моль), карбоната калия (3,0 г, 0,0218 моль), йодистого калия (100 мг), 3-хлорпропоксибензола (3,4 г, 0,0200 моль) и ацетонитрила перемешивали при нагревании с обратным холодильником в атмосфере азота 30 часов. Реакционную смесь выливали в воду и водную смесь экстрагировали этилацетатом. Этилацетатный экстракт промывали рассолом, сушили сульфатом магния, концентрировали и получали 6,2 г вязкого твердого вещества бежевого цвета. Соединение два раза перекристаллизовывали из этанола и получали 6-фтор-3-[1-(3-феноксипропил)-4-пиперидинил]-1,2-бензизоксазол в виде твердого вещества светло-бежевого цвета с выходом 47%, точка плавления 78-80° С.
Элементный анализ для C21H33FN2O2:
вычислено (%): С 71,17; Н 6,54; N 7,90,
найдено (%): С 71,00; Н 6,52; N 7,81.
Пример 41
1-[4-[2-[4-(6-хдор-1Н-индазол-3-ил)-пиперазинил]-этокси]-3-метоксифенил]этанон
Смесь 6-хлор-[3-(1-пиперазинил)]-lH-индазола (2,1 г, 0,0089 моль), карбоната калия (1,5 г, 0,0107 моль), йодистого калия (100 мг), 1-[4-(2-хлорэтокси)-3-метоксифенил]этанона (2,2 г, 0,0098 моль) и ацетонитрила (70 мл) перемешивали при нагревании с обратным холодильником 48 часов в атмосфере азота. Охлажденную реакционную смесь выливали в воду и водную смесь экстрагировали этилацетатом. Органический экстракт промывали водой, сушили сульфатом магния, концентрировали и получали 6,0 г светло-желтого масла. Масло очищали препаративной высокопроизводительной жидкостной хроматографией (Waters Associates Prep LC/System 500, используя две колонки силикагеля и смесь 5,3% метанола и хлористого метилена в качестве элюента). Концентрированием последних фракций получали 1,6 г не совсем белого твердого вещества. Его объединяли с дополнительным образцом (всего 3,4 г) и после двух перекристаллизации из этанола получали 2,1 г (23%) 1-[4-[2-[4-(6-хлор-1Н-индазол-3-ил)-1-пиперазинил]-этокси]-3-метоксифенил]этанона в виде не совсем белого твердого вещества, точка плавления 154-156° С.
Элементный анализ для С22Н25СlN4O3:
вычислено (%): С 61,61; Н 5,88; N 13,06,
найдено (%): С 61,66; Н 5,87; N 13,06.
Пример 42
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]-2,2,2-трифторэтанон
Смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (1,5 г, 0,0067 моль), карбоната калия (0,88 г), йодистого калия (0,1 г) и ацетонитрила (50 мл) перемешивали и нагревали с обратным холодильником 16 часов. После охлаждения реакционную смесь выливали в воду и водную смесь экстрагировали этилацетатом. Экстракт промывали водой, сушили сульфатом магния и растворитель концентрировали до масла, которое при откачке в высоком вакууме давало 3,2 г воскообразного твердого вещества. Твердое вещество хроматографировали на препаративном жидкостном хроматографе Waters (колонки кремнезема, элюируемые смесью 3% метанол - дихлорметан). Концентрирование соответствующих фракций давало 1,8 г (56%) 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]-2,2,2-трифторметилэтанона в виде твердого вещества с точкой плавления 94-96° С.
Элементный анализ для C24Н24F4N2O4:
вычислено (%): С 60,00; Н 5,03; N 5,83,
найдено (%): С 60,01; Н 5,06; N 5,68.
Пример 43
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинид]-пропокси]-3-метилмеркаптофенил]этанон
Перемешиваемую смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (1,88 г, 8,5 ммоль), карбоната калия (1,8 г) и 1-[4-(3-бромпропокси)-3-метилмеркаптофенил]этанона (2,3 г, 7,6 ммоль) в ацетонитриле (100 мл) нагревали с обратным холодильником четыре часа. К окончанию реакции растворитель концентрировали, затем разбавляли дихлорметаном (250 мл). Нерастворимые вещества отфильтровывали. Дихлорметановый раствор концентрировали досуха и получали масло. Масло очищали быстрой хроматографией на колонке силикагеля (двуокись кремния, 54 г, элюируемая дихлорметаном, 500 мл, смесью 1% метанол - дихлорметан, 1,1 литра). Наиболее чистые фракции объединяли и получали бесцветное масло, которое отверждалось до не совсем белого твердого вещества (2,4 г). Перекристаллизацией из этанола (100 мл) получали 2,15 г 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метилмеркаптофенил]этанона в форме игольчатых кристаллов, точка плавления 150-152° С.
Элементный анализ для С24Н27FN2O3S:
вычислено (%): С 65,14; Н 6,15; N 6,33,
найдено (%): С 65,09; Н 6,10; N 6,25.
Пример 44
1-[4-(3-бромпропокси)-3-бромфенил]этанон
Перемешиваемую смесь 3-бром-4-оксиацетофенона (4,5 г, 21,2 ммоль), карбоната калия (4,0 г) и 1,3-дибромпропана (7,6 г) в ацетонитриле (200 мл) нагревали с обратным холодильником два часа. В конце реакции растворитель удаляли и остаток растворяли в дихлорметане (400 мл) и фильтровали. Дихлорметановый раствор концентрировали до масла. Масло добавляли к изопропиловому эфиру и перемешивали, чтобы вызвать кристаллизацию (4,1 г, 58%). Твердое вещество перекристаллизовывали из изопропилового эфира и получали 3,5 г 1-[4-(3-бромпропокси)-3-бромфенил]этанона в форме блестящих кристаллов, точка плавления 83-84° С.
Элементный анализ для С11Н12Вr2О2:
вычислено (%): С 39,31; Н 3,60,
найдено (%): С 39,80; Н 3,55.
Пример 45
1-[4-(3-бромпропокси)-3,5-дибромфенил]этанон
Перемешиваемую смесь 3,5-дибром-4-оксиацетофенона (3,0 г, 10,1 ммоль), карбоната калия (2,6 г, 20,3 ммоль), 1,3-дибромпропана (4,0 г, 19,8 ммоль) в ацетонитриле (100 мл) нагревали с обратным холодильником пять часов. Растворитель удаляли. Неочищенный продукт экстрагировали в дихлорметан (150 мл) и нерастворимые неорганические вещества отфильтровывали.
Раствор снова концентрировали досуха. Очистку проводили быстрой хроматографией на силикагеле (45 г, двуокись кремния, элюировали смесью гексан - дихлорметан в отношении 1:1). Таким образом, полученный материал (2,8 г) дважды перекристаллизовывали из изопропилового эфира и получали чистый для анализа 1-[4-(3-бромпропокси)-3,5-дибромфенил]этанон, точка плавления 87-88° С.
Элементный анализ для С11H11Br3O2:
вычислено (%): С 31,84; Н 2,67,
найдено (%): С 31,97; Н 2,63.
Пример 46
1-[4-[4-[4-(1,2-бензизотиазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанон
Перемешиваемую смесь 3-(4-пиперидинил)-1,2-бензизотиазола (2,6 г, 0,0119 моль), 1-[4-(4-бромбутокси)-3-метоксифенил] этанола (3,9 г, 0,0131 моль), карбоната калия (2,0 г, 0,0143 моль), йодистого калия (200 мг) и ацетонитрила (125 ми) нагревали с обратным холодильником в атмосфере азота 18 часов. Реакционную смесь охлаждали до температуры окружающей среды и фильтровали. Остаток на фильтре хорошо промывали свежим ацетонитрилом и фильтрат концентрировали до получения влажного коричневого твердого вещества. Остаток разбавляли водой и водную суспензию экстрагировали хлористым метиленом. Органический экстракт промывали водой, сушили сульфатом магния и концентрировали до получения 6,5 г темного масла. Масло очищали препаративной высокопроизводительной жидкостной хроматографией (Waters Associates Prep LC/System 500, используя 2 колонки силикагеля и смесь 5% метанол - хлористый метилен) и получали 4,5 г твердого вещества цвета беж. Образец 3,1 г (0,0071 моль) растворяли в абсолютном этиловом спирте (80 мл) и добавляли щавелевую кислоту (0,067 г, 0,0074 моль). Раствор нагревали с обратным холодильником при средней температуре на паровой бане в течение 45 минут и затем перемешивали при комнатной температуре один час. Полученную суспензию разбавляли безводным эфиром (150 мл) и перемешивали пять минут. Твердое вещество собирали, сушили и получали 3,1 г твердого вещества светло-бежевого цвета. Соль перекристаллизовывали из этанола и получали 2,8 г продукта. Это соединение снова обращали в свободное основание 50% гидроокисью натрия и получали 2,4 г, которые немедленно кристаллизовались из этанола и давали 1,5 г (28%) 1-[4-[4-[4-(1,2-бензизотиазол-3-ил)-1-пиперидинил]-бутокси]-3-метоксифенил]этанона в форме порошка цвета беж, точка плавления 78-80° С.
Элементный анализ для С25Н30N2О3S:
вычислено (%): С 68,46; Н 6,91; N 6,39,
найдено (%): С 68,34; Н 6,85; N 6,33.
Пример 47
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]фенилметанон
Смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (2,2 г, 10 ммоль), карбоната калия (2,3 г), 1-[4-(3-бромпропокси)-3-метоксифенил]фенилметанона (3,47 г, 10 ммоль) в ацетонитриле (100 мл) нагревали с обратным холодильником три часа. В конце реакции ацетонитрил концентрировали и смесь экстрагировали в дихлорметан (200 мл). Нерастворимые вещества отфильтровывали и растворитель выпаривали до масла. Очистку проводили быстрой хроматографией на колонке силикагеля двуокись кремния, 50 г, элюировали дихлорметаном, 600 мл: смесью 2% метанола 98% дихлорметан, 600 мл). Фракции, содержащие чистый продукт, объединяли, концентрировали и получали 4,24 г (87%) не совсем белого твердого вещества. Перекристаллизация из этанола (75 мл) давала 3,9 г [4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]фенилметанона в форме не совсем белых кристаллов, точка плавления 128-130° С.
Элементный анализ для C29H29FN2O4:
вычислено (%): С 71,30; Н 5,98; N 5,73,
найдено (%): С 71,31; Н 5,99; N 5,75.
Пример 48
1-[3-бром-4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-фенил]этанон
Смесь 6-фтор-3-(4-пиперидинил)-1,3-бензизоксазола (2,1 г, 9,5 ммоль), карбоната калия (2,0 г), 1-[3-бром-4-(3-бромпропокси)-фенил]этанона (3,1 г, 9,2 ммоль) в ацетонитриле (100 мл) нагревали с обратным холодильникам три часа. В конце реакции растворитель упаривали и смесь экстрагировали в дихлорметан (200 мл). Нерастворимые фракции отфильтровывали. Дихлорметан снова концентрировали. Неочищенный остаток очищали хроматографией на силикагеле (одна колонка двуокиси кремния, 49 г, элюентом служили: дихлорметан, 500 мл: смесь 3% метанол - 97% дихлорметан, 600 мл). Полученный таким образом продукт (3,26 г, 72%) перекристаллизовывали из этанола (40 мл) и получали 3,0 г 1-[3-бром-4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-фенил]этанона в форме светло-желтых кристаллов, точка плавления 126-128° С.
Элементный анализ для С23Н24ВrFN2О3:
вычислено (%): С 58,12; Н 5,09; N 5,89,
найдено (%): С 57,64; Н 5,35; N 5,55.
Пример 49
Гидрохлорид 3-[1-[3-[4-(1-этоксиэтил)-2-метоксифенокси]-пропил]-4-пиперидинил]-6-фтор-1,2-бензизоксазола
К смеси 4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метокси-α -метилбензолметанола (3,8 г, 0,089 моль) в пиридине (25 мл) добавляли уксусный ангидрид (5 мл). Смесь недолго нагревали на паровой бане, чтобы ускорить растворение, и затем смесь оставляли при комнатной температуре на 16 часов. Большую часть пиридина выпаривали при пониженном давлении и полученное масло разбавляли водой. Водный раствор подщелачивали разбавленной гидроокисью натрия и затем экстрагировали этилацетатом. Органический экстракт промывали водой, сушили сульфатом магния, растворитель концентрировали и получали 3,7 г O-ацетил-производного в виде бесцветного масла. Соединение растворяли в диэтиловом эфире и эфирную хлористоводородную кислоту добавляли к выпавшей в осадок смолоподобной хлористоводородной соли, которая при обработке флегмой этилацетата давала 3,4 г кристаллической соли, точка плавления 143-145° С. Попытка кристаллизации соли из смеси этанол - диэтиловый эфир приводила к замещению ацетата в пользу этилового эфира. Соль этого продукта (2,8 г) кристаллизовали из смеси этанол - диэтиловый эфир и получали 2,1 г (48%) гидрохлорида 3-[1-[3-[4-(1-этоксиэтил)-2-метоксифенокси)-пропил]4-пиперидинил]-6-фтор-1,2-бензизоксазола, точка плавления 139-141° С.
Элементный анализ для С26Н33FN2O4·НСl:
вычислено (%): С 63,34; Н 6,95; N 5,68,
найдено (%): С 63,06; Н 6,80; N 5,63.
Пример 50
Фумарат 3-[1-[3-[4-(1-ацетоксиэтил)-2-метоксифенокси]-пропил]-4-пиперидинил]-6-фтор-1,2-бензизоксазола
Смесь 4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-3-мотокси-α -метилбензолметанола (4,8 г, 0,011 моль) в пиридине (45 мл) недолго нагревали для ускорения образования раствора и затем добавляли уксусный ангидрид (6,3 мл). Реакционную смесь выдерживали при температуре окружающей среды 16 часов, концентрировали в вакууме и оставшееся бесцветное масло растворяли в воде. Водный раствор подщелачивали насыщенным раствором карбоната калия и смесь экстрагировали диэтиловым эфиром. Экстракт промывали водой, сушили сульфатом магния, концентрировали и получали 5,2 г вязкого бесцветного масла. Масло (4,8 г) растворяли в безводном диэтиловом эфире и добавляли фумаровую кислоту (1,2 г, 0,01 моль). Смесь перемешивали при температуре окружающей среды четыре часа и затем выдерживали при той же температуре 16 часов. Полученный белого цвета фумарат 3-[1-[3-[4-(1-ацетоксиэтил)-2-метоксифенокси]-пропил]-4-пиперидинил]-6-фтор-1,2-бензизоксазола собирали и получали 3,0 г материала. Фильтрат обрабатывали дополнительным количеством фумаровой кислоты (0,3 г) и собирали еще 0,9 г фумарата 3-[1-[3-[4-(1-ацетоксиэтил)-2-метоксифенокси]-пропил]-4-пиперидинил]-6-фтор-1,2-бензизоксазола. Две загрузки объединяли и кристаллизовали из ацетонитрила (два раза) и получали 2,3 г (43%) ацетата, точка плавления 150-152° С.
Элементный анализ для C26H31FN2O3·C4H4О4:
вычислено (%): С 61,43; Н 6,01; N 4,78,
найдено (%): С 61,06; Н 5,87; N 4,73.
Пример 51
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]пентанон
Смесь 6-фтор-3-(4-липеридинил)-1,2-бензизоксазола (2,9 г, 0,01 моль), карбоната калия (3,0 г), 1-[4-(3-бромпропокси)-3-метоксифенил]пентанона (3,7 г, 0,0113 моль) в ацетонитриле (140 мл) нагревали с обратным холодильником четыре часа. По окончании реакции смесь охлаждали и фильтровали. Фильтрат концентрировали до масла. Очистку проводили быстрой хроматографией на колонке силикагеля (кремнезем, 55 г, элюировали смесью 1% метанола в дихлорметане, 600 мл, 3% метанола - 97% дихлорметана, 400 мл). Фракции, содержавшие чистый продукт, собирали, концентрировали и получали 4,3 г (91%). Перекристаллизацией из этанола (10 мл) получали порошок 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]пентанона (3,22 г), точка плавления 79-80° С.
Элементный анализ для С27Н33FN2O4:
вычислено (%): С 69,21; Н 7,10; N 5,98,
найдено (%): С 69,00; Н 6,94; N 6,39.
Пример 52
Полуфумарат 2-[3-[4-(6-фтор-1,2-бендизоксазол-3-ил)-1-пиперидинил]-пропокси]-N-метилбензоламина
Смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (2,5 г, 0,0114 моль), карбоната калия (1,8 г, 0,0130 моль), 4-(3-хлорпропокси)-2-метиламинобензола (2,4 г, 0,120 моль) и ацетонитрила (100 мл) нагревали с обратным холодильником 18 часов. Реакционную смесь охлаждали до температуры окружающей среды и выливали в воду. Водную смесь экстрагировали этилацетатом и этилацетатный экстракт промывали водой, сушили сульфатом магния, концентрировали и получали 4,1 г коричневого масла. Масло очищали препаративной высокопроизводительной жидкостной хроматографией (Waters Associates Prep LC/System 500, используя две колонки с силикагелем и элюируя смесью 4% метанол - хлористый метилен). Концентрированном соответствующих фракций получали 2,45 г масла цвета беж. Продукт растворяли в этилацетате (50 мл) и добавляли фумаровую кислоту (0,78 г). Смесь перемешивали при умеренном кипячении с обратным холодильником в течение 45 минут и затем при комнатной температуре 1,5 часа. Продукт выделяли вакуумной фильтрацией и получали 2,5 г твердого вещества бледно-желтого цвета. Перекристаллизацией из этанола получали 2,0 г (40%) 2-[3-[4-(6-фтор-1,3-бензизоксазол-3-ил)-1-ппиперидинил]-пропокси]-N-метилбензоламина, полуфумарата в форме кристаллов цвета беж, точка плавления 180-182° С.
Элементный анализ для C22H26FN3O2·0,5 C4Н4О4:
вычислено (%): С 65,28; Н 6,40; N 9,52,
найдено (%): С 65,08; Н 6,35; N 9,45.
Пример 53
3-[1-[3-(4-бром-2-метоксифенокси)-пропил]-4-пиперидил]-6-фтор-1,2-бензизоксазол
Смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (2,6 г, 0,0117 моль), карбоната калия (2,0 г, 0,0144 моль), 4-(3-хлорпропокси)-3-метоксибромбензола (3,6 г, 0,0129 моль), ацетонитрила (100 мл) и нескольких кристаллов йодистого калия перемешивали при нагревании с обратным холодильником в атмосфере азота 18 часов. Охлажденную реакционную смесь выливали в воду и водную смесь экстрагировали этилацетатом. Этилацетатный экстракт промывали водой, сушили сульфатом магния, концентрировали и получали 5,0 г зеленого масла. Образец очищали препаративной высокопроизводительной жидкостной хроматографией (Waters Associates Prep LC/System 500, используя две колонки с силикагелем и смесь 4% метанол - хлористый метилен в качестве элюента). Концентрированием соответствующих фракций получали 3,15 г твердого вещества рыжевато-коричневого цвета. Соединение два раза перекристаллизовывали из этанола и получали 2,0 г (37% 3-[1-[3-[4-бром-2-метоксифенокси)-пропил]-4-пиперидинил]-6-фтор-1,2-бензизоксазола в виде твердого вещества бежевого цвета, точка плавления 88-90° С.
Элементный анализ для C22Н24BrFN2O3:
вычислено (%) С 57,03; Н 5,22; N 6,05,
найдено (%) С 57,04; Н 5,19; N 6,06.
Пример 54
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]пропанон
Смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (2,8 г 15,2 ммоль), карбоната калия (3,0 г), 1-[4-(3-бромпропокси)-3-метоксифенил]пропанона (4,6 г, 18,2 ммоль) в ацетонитриле (100 мл) нагревали с обратным холодильником два часа. По окончании реакции смесь фильтровали, растворитель концентрировали и остаток экстрагировали в дихлорметан (300 мл). Дихлорметан снова фильтровали и концентрировали. Неочищенный материал (6,4 г) очищали быстрой хроматографией на колонке силикагеля (двуокись кремния, 50 г, элюировали дихлорметаном, 700 мл, смесью 1% метанол - дихлорметан, 1,4 литра). Очищенный таким образом материал (2,87 г, 51%) перекристаллизовывали из этанола (25 мл) и получали 2,13 г 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксифенил]пропанона в форме кристаллов, окрашенных в бежевый цвет, точка плавления 118-119° С.
Элементный анализ для C25H29FN2О4:
вычислено (%): С 68,16; Н 6,64; N 6,36,
найдено (%): С 68,32; Н 6,63; N 6,29.
Пример 55
4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксибензамид
Смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (2,2 г, 10,0 ммоль), карбоната калия (2,0 г) и 4-(3-бромпропокси)-3-метоксибензамида (2,32 г, 8,0 ммоль) в ацетонитриле (80 мл) нагревали с обратным холодильником пять часов. По окончании реакции растворитель выпаривали. Остаток экстрагировали в дихлорметан. Неорганические нерастворимые фракции отфильтровывали. Дихлорметан снова концентрировали. Неочищенный остаток очищали быстрой хроматографией через колонку с силикагелем (55 г двуокиси кремния, элюировали смесью 1% метанола в дихлорметане, 1,0 литр, 2% метанола в дихлорметане, 1,0 литр). Полученный материал весил 2,93 г (84%) и представлял собой белые кристаллы. Перекристаллизация из горячего этанола (60 мл) давала 2,2 г 4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-метоксибеизамида в форме белых кристаллов, точка плавления 163-164° С.
Элементный анализ для С23Н26FN3O4:
вычислено (%): С 64,62; Н 6,13; N 9,83,
найдено (%): С 64,20; Н 6,06; N 9,71.
Пример 56
1-[4-[3-[4-(6-фтор-1,2-бензизоксазод-3-ил)-1-пиперидинил]-пропокси]-3-(метиламино)-фенил]этанон
Смесь 6-фтор-3-(4-пиперидинил)-1,2-бензизоксазола (2,3 г, 0,0103 моль), карбоната калия (1,4 г, 0,0103 моль), 1[4-(3-хлорпропокси)-3-(метиламино)-фенил]этанона (2,5 г, 0,010 моль), йодистого калия (0,10 г) и ацетонитрила (100 мл) перемешивали при нагревании с обратным холодильником в атмосфере азота 23 часа. Реакционную смесь охлаждали до температуры окружающей среды, выливали в воду и водную смесь экстрагировали этилацетатом. Этилацетатный экстракт дважды промывали водой, сушили над сульфатом магния, концентрировали и получали 4,8 г влажного коричневого твердого вещества. Соединение выделяли препаративной высокопроизводительной жидкостной хроматографией (Waters Associates Prep LC/System 500, используя две колонки с силикагелем и смесь 4% метанола с хлористым метиленом в качестве элюента.) Концентрацией соответствующих фракций получали 2,4 г продукта. Перекристаллизацией из этанола получали 2,1 г 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-(метиламино)-фенил]этанона в виде твердого вещества бежевого цвета, точка плавления 151-153° С.
Элементный анализ для С24Н28FN3О3:
вычислено (%): С 67,76; Н 6,63; N 9,88,
найдено (%): С 67,83; Н 6,76; N 9,90.
Пример 57
1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-этоксифенил]этанон
Суспензию гидрида натрия (0,28 г 50% масляной дисперсии, 0,0059 моль) в диметилформамиде (20 мл) охлаждали до 4° С на ледяной бане. К суспензии по каплям добавляли 1-[4-[3-[4-(6-фтор-1,2-бензоксазол-3-ил)-1-пиперидинил]-пропокси]-3-оксифенил]этанон (2,3 г, 0,0056 моль), растворенный в диметилформамиде (40 мл). После добавления всего количества смесь перемешивали в атмосфере азота один час, сохраняя температуру ниже 10° С. Затем добавляли к реакционной смеси по каплям раствор бромэтана (1,3 г, 0,0018 моль). Перемешивание в атмосфере азота продолжали три часа, позволяя температуре смеси подняться медленно до температуры окружающей среды. Реакционную смесь охлаждали на ледяной бане, добавляли воду и водную смесь экстрагировали этилацетатом. Этилацетатный экстракт промывали водой, сушили сульфатом магния, концентрировали до получения 3,9 г влажного твердого вещества цвета беж. Твердое вещество растирали с диэтиловым эфиром, фильтровали и получали 1,5 г. Их объединяли с дополнительным образцом (всего 3,5 г) и перекристаллизацией из этанола получали 3,0 г (57%) блестящих кристаллов бежевого цвета соединения 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]-пропокси]-3-этоксифенил]этанона, точка плавления 113-114° С.
Элементный анализ для C25H29FN2О4:
вычислено (%): С 68,16 Н 6,64 N 6,36
найдено (%): С 68,10 Н 7,03 N 6,35.
Пример 58
1-[4-(3-бромпропокси)-3-(метилмеркапто)фенил]этанон
Смесь 1-[4-окси-3-(метилмеркапто)фенил]этанона (5,4 г, 0,03 моль), карбоната калия (4,2 г), 1,3-дибромпропана (8,0 г, 0,039 моль) в ацетонитриле (150 мл) нагревали с обратным холодильником три часа и оставляли на ночь при перемешивании при комнатной температуре. Ацетонитрил удаляли при пониженном давлении и остаток экстрагировали в дихлорметан (250 мл). Нерастворимые фракции отфильтровывали. Дихлорметановый раствор концентрировали. Неочищенный продукт очищали на колонке силикагеля (двуокись кремния 100 г, элюировали смесью гексан - дихлорметан в отношении 1:1, 1,6 литра). Соединение кристаллизовалось при концентрировании и продукт (3,5 г, 33%) перекристаллизовывали из этанола (40 мл) и получали 2,0 г 1-[4-(3-бромпропокси)-3-(метилмеркапто)фенил]этанона в форме белых иголок, точка плавлении 120-122° С.
Элементный анализ для C12H15BrO2S:
вычислено (%): С 47,53; Н 4,99,
найдено (%): С 47,74; Н 4,91.
Пример 59
4-(3-бромпропокси)-3-метоксибензонитрил
Смесь 4-оксо-3-метоксибензонитрила (7,5 г, 50 ммоль), карбоната калия (12,5 г) и 1,3-дибромпропана (15 г, 75 ммоль) в ацетонитриле (100 мл) нагревали с обратным холодильником три часа и оставляли на ночь при комнатной температуре. Растворитель реакционной смеси удаляли на вращающемся испарителе и неочищенное твердое вещество экстрагировали в хлористый метилен (500 мл). Нерастворимые вещества отфильтровывали. Дихлорметановый раствор концентрировали и материал очищали колоночной хроматографией (двуокись кремния, 105 г, элюировали смесью дихлорметан - гексан в отношении 2:3 и затем дихлорметаном). Желаемый продукт после такого рода очистки весил 7,74 г (52%). Перекристаллизацией (дважды) из этанола получали чистый для анализа 4-(3-бромпропокси)-3-метоксибензонитрил, точка плавления 99-101° С.
Элементный анализ для C4H12BrNO2:
вычислено (%): С 48,91; Н 4,48; N 5,19,
найдено (%): С 49,49; Н 4,47; N 5,21.
Пример 60
1-[4-(3-бромпропокси)-3-метоксифенил]этанон
Смесь 4-окси-3-метилацетофенона (14,5 г, 96 ммоль), карбоната калия (17,5 г, 144 ммоль) и 1,3-дибромпропана (30 г, 144 ммоль) в эцетонитриле (400 мл) нагревали с обратным холодильником шесть часов. По окончании реакции растворитель удаляли на роторном испарителе и неочищенное твердое вещество экстрагировали в дихлорметан (750 мл). Нерастворимые неорганические вещества отфильтровывали. Дихлорметановый раствор снова концентрировали до неочищенного масла (34,5 г). Очистку проводили быстрой хроматографией на колонке силикагеля (двуокись кремния, 150 г, элюируя смесью гексан - дихлорметан в отношении 7:3, 2 литра и дихлорметаном, 2 литра). Очищенный таким образом материал весил 14,6, (56%) и был перекристаллизован из этанола. Перекристаллизация повторно из этанола давала чистый для анализа 1-[4-(3-бромпропокси)-3-метилфенил]этанон, точка плавления 59-61° С.
Элементный анализ для C12H15BrO2:
вычислено (%): С 53,15; Н 5,58,
найдено (%): С 53,35; Н 5,52.
Пример 61
1-[4-(3-бромпропокси)-3-метоксифенил]фенилметанон
Смесь 1-(4-окси-3-метоксифенил)фенилметанона (14 г, 61,4 ммоль), карбоната калия (13,0 г, 92,1 ммоль) и 1,3-дибромпропана (28,0 г, 86 ммоль) в ацетонитриле (400 мл) нагревали с обратным холодильником четыре часа. Течение реакции контролировали тонкослойной хроматографией. По окончании реакции неорганические вещества отфильтровывали и растворитель удаляли на вращающемся испарителе. Остаток очищали быстрой хроматографией на колонке (двуокись кремния, 140 г, элюировали смесью гексан - дихлорметан в отношении 4:1, 1,2 литра) и получали частично отвержденный материал, 15,44 г (729). Двойной перекристаллизацией из этанола получали 2,84 г 1-[4-(3-бромпропокси)-3-метоксифенил]фенилметанона в форме белых кристаллов, точка плавления 88-89° С.
Элементный анализ для С17Н17ВrО3:
вычислено (%): С 58,47; Н 4,91,
найдено (%): С 59,03; Н 4,87.
Настоящее изобретение, таким образом, предусматривает соединения формулы (I), способные вызывать антипсихотическое действие, и могут быть способны влиять на отрицательные симптомы шизофрении положительным образом. В дополнение многие соединения могут также понижать тенденцию вызывать побочные экстрапирамидальные явления у млекопитающих. Соединения формулы (I) будучи эффективными сами, могут использоваться для получения лекарственных препаратов и введены в форме своих фармацевтически приемлемых солей.
Пример лекарственного средства 1
Таблетка, содержащая 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил)-1-пиперидинил]пропокси]-3-метоксифенил]этанон, мг:
1-[4-[3-[4-(6-фтор-1,2-
бензизоксазол-3-ил)-1-
пиперидинил]пропокси]-3-
метоксифенил]этанон 2
Крахмал 40
Тальк 10
Стеарат магния 10
Пример лекарственного средства 2
Капсула, содержащая 1-[4-[3-[4-(6-фтор-1,2-бензизоксазол-3-ил) 1-пиперидинил]пропокси]-3-метоксифенил]этанон, мг:
1-[4-[3-[4-(6-фтор-1,2-
бензизоксазол-3-ил)-1-
пиперидинил]пропокси]-3-
метоксифенил]этанон 2
Тальк 40
Натрийкарбоксиметилцеллюлоза 40
Крахмал 120
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ N-(АРИЛОКСИАЛКИЛ)-ГЕТЕРОАРИЛПИПЕРИДИНА ИЛИ ГЕТЕРОАРИЛПИПЕРАЗИНА | 1990 |
|
RU2062776C1 |
| ПРОИЗВОДНЫЕ N-(АРИЛОКСИАЛКИЛ)-ГЕТЕРОАРИЛПИПЕРИДИНА И ГЕТЕРОАРИЛПИПЕРАЗИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1990 |
|
RU2147583C1 |
| ГЕТЕРОАРИЛПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2127731C1 |
| БЕНЗИЗОТИАЗОЛ- ИЛИ БЕНЗИЗОКСАЗОЛ-3-КАРБОКСАМИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИПСИХОТИЧЕСКОЙ АКТИВНОСТЬЮ | 1992 |
|
RU2039057C1 |
| ХИНОЛОНОВОЕ СОЕДИНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2544530C2 |
| 2-ФЕНИЛИМИДАЗО[1,2-А]ПИРИМИДИНЫ В КАЧЕСТВЕ ВИЗУАЛИЗИРУЮЩИХ СРЕДСТВ | 2014 |
|
RU2665580C2 |
| ПРОИЗВОДНОЕ ХИНОЛОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2008 |
|
RU2490259C2 |
| ИНДАЗОЛЫ, БЕНЗИЗОКСАЗОЛЫ И БЕНЗИЗОТИАЗОЛЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЭСТРОГЕННЫХ СРЕДСТВ | 2005 |
|
RU2402536C2 |
| НОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2814103C1 |
| Способ получения производных пиперидина или их солей с кислотами | 1990 |
|
SU1838299A3 |
Изобретение относится к химико-фармацевтической промышленности и касется производных N-(арилоксиалкил)-гетероарилпиперидина и -гетероарилпиперазина общей формулы (I), используемых для получения лекарственного средства, обладающего антипсихотической или анальгетической активностью, и способа лечения психозов с использованием указанных производных. Изобретение обеспечивает высокую эффективность лечения. 2 с. и 4 з.п. ф-лы, 4 табл.


в которой Х представляет собой -O-, -S-, -NН- или –N(R2)-;
р - целое число, равное единице;
Y является водородом, Сl, Вr или F;
R2 представляет бензоил;
Z представляет собой  или
или  ;
;
n = 2, 3, 4 или 5;
R представляет собой водород, гидрокси, С1-6-алкил, С1-6-алкокси, Вr, алкиламино, алкилтио, трифторацетил, аминокарбонил, -СО-алкил, -СОО-алкил, -СО-Рh, -СН(OR3)алкил, где алкил является низшим алкилом, где R3 представляет собой водород или низший алкил;
m = 1, 2 или 3, при условии, что 2 не является группой
 , когда Х означает -S- и R означает водород,
, когда Х означает -S- и R означает водород,
или его фармацевтически приемлемые соли, используемые для получения лекарственного средства, обладающего антипсихотической и/или анальгетической активностью.
Приоритет по пунктам:
| ЕР 315316, А2,10.05.1989 | |||
| Способ получения производных пирролидина | 1983 |
|
SU1240359A3 |
| DE 3530089, А1, 15.08.1986 | |||
| Машина для отделения шкурки от кусков свинины | 1960 |
|
SU135781A1 |
| US 4366162, А, 28.12.1982. | |||

