Область техники
Настоящее изобретение относится к фармацевтической композиции для предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания, в частности, к фармацевтической композиции, содержащей в качестве активного ингредиента соединение, которое ингибирует Pin1 (пептидил-пролилцис-транс-изомераза, взаимодействующая с 1) для предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания.
Предпосылки изобретения
Pin1 (пептидил-пролилцис-транс-изомераза, взаимодействующая с 1), пролил-изомераза, представляет собой фермент, который катализирует цис/транс-изомеризацию амидного остатка пролина путем связывания с фосфорилированным сайтом Ser/Thr-Pro (Lu, et al. Science 1999, 283(5406):1325-8). Во время внутриклеточного сигнального процесса сериновые (Ser), треониновые (Thr) и тирозиновые (Tyr) остатки белков фосфорилируются. После этого фосфорилирования Pin1 изменяет структуру различных белков посредством цис/транс-изомеризации амида остатка пролина, позволяя этим белкам начать выполнять свои функции в клетке.
Согласно исследованиям, проведенным на сегодняшний день, известно, что фермент Pin1 участвует в возникновении и прогрессировании воспалительных заболеваний, нарушений обмена веществ и злокачественного новообразования. В частности, сообщалось, что фермент Pin1 активирует 56 онкогенов, участвующих в онкогенезе, таких как бета-катенин, AKT, AR, CyclinD1, Plk, NF-kB, Stat3, Myc, c-Jun, c-Fos, c-Myb, Raf-1, HIF-1, Nanog, Notch1, Oct4 и т. д., и ингибирует активность 26 генов-супрессоров опухолей, таких как ATR, Bax, Btk, FADD, Fbw7, PML и т.д., тем самым индуцируя метастазирование рака и ангиогенез рака в общем механизме развития рака. Поэтому проводится ряд исследований белка Pin1 в качестве мишени для разработки реагентов для диагностики рака, прогностических факторов и полезных новых противораковых средств (Nature reviews Cancer 2016, 16:463-478; Cell Death and Disease 2018, 9:883).
Как сообщалось при исследовании образцов, удаленных хирургическим путем, является известным, что Pin1 сильно экспрессируется при различных типах рака, таких как рак толстой кишки, рак шейки матки, рак молочной железы, рак легких, рак поджелудочной железы, рак желудка, рак печени и т.д. Фактически, сообщалось, что трансгенные мыши со сверхэкспрессией Pin1 проявляют канцерогенный фенотип. Кроме того, известно, что митотическая остановка и апоптоз могут быть индуцированы, а свойства стволовых клеток рака могут быть подавлены в раковых клетках, нокаутированных по Pin1 с использованием специфического ингибитора или siRNA. В частности, фермент Pin1 выполняет функцию индукции самообновления, метастазирования и образования опухолей стволовых клеток рака молочной железы.
Когда Pin1 избыточно экспрессируется, эпителиальные клетки молочной железы человека дифференцируются в псевдостволовые клетки, и происходит переход от эпителия к мезенхиме. Другими словами, когда раковые клетки инвазивно растут и метастазируют, чтобы разорвать адгезию с окружающими клетками и двигаться по кровеносным или лимфатическим сосудам, связь между клетками ослабевает, скелет клеток изменяется и приобретается подвижность. Фермент Pin1 известен как важный фактор самообновления стволовых клеток, поддерживающий стабильность белков Nanog, Oct4 и Myc.
Хотя Pin1 участвует во многих механизмах в живом организме, как описано выше, известно, что мыши с нокаутом Pin1 хорошо растут без серьезных проблем. Поэтому ожидается, что ингибирование Pin1 не окажет существенного влияния на организм человека. Однако хотя имеются сообщения о том, что Pin1 может оказать помощь в головном мозге при лечении болезни Альцгеймера, есть сообщения о том, что он действует обратным образом при болезни Паркинсона, и есть много частей, которые еще не идентифицированы. В заключение, Pin1 играет очень важную роль в возникновении злокачественного новообразования в целом, и его ингибирование в областях, отличных от головного мозга, очень полезно для эффективного и безопасного лечения рака.
Кроме того, сообщалось, что при ингибировании Pin1 или подавлении экспрессии гена Pin1 продукция простагландина E2 и оксида азота, стимулируемая липополисахаридом (LPS) и никотином, ослабевает, а экспрессия циклооксигеназы-2 (COX-2) и индуцибельная синтазы оксида азота (iNOS) также ослабляются, что приводит к противовоспалительному эффекту (Journal of Dental Research, 2015, 94(2):371-380). Хорошо известно, что, когда Pin1 присоединен к CRTC2, продукция комплекса CBP-CRTC2-CREB, который способствует глюконеогенезу, ингибируется, так что CRTC2 может регулироваться в соответствии с уровнем экспрессии Pin1 и, таким образом, может участвовать в регуляции метаболизма глюкозы (J. Biol. Chem., 2010, 285(43):33018-27). Следовательно, препараты, влияющие на экспрессию или активность Pin1, можно использовать в качестве терапевтических средств при воспалительном заболевании или метаболическом заболевании, таком как диабет.
Таким образом, авторы настоящего изобретения провели фармацевтический химический синтез и биологическую и фармакологическую оценку синтезированных соединений, чтобы обнаружить соединения, которые ингибируют активность Pin1. Благодаря этому авторы настоящего изобретения обнаружили соединения, проявляющие превосходный противораковый эффект за счет сильного ингибирования Pin1, и разработали настоящее изобретение. Кроме того, считается, что соединения по настоящему изобретению можно использовать в качестве терапевтических средств для лечения заболеваний, основанных на роли Pin1 в воспалительных и метаболических заболеваниях.
Описание изобретения
Техническая задача
Целью настоящего изобретения является разработка нового соединения, его изомера или его фармацевтически приемлемой соли, которое ингибирует Pin1 (пептидил-пролил-цис/транс-изомераза, взаимодействующая с NIMA 1).
Другой целью настоящего изобретения является способ получения соединения.
Другой целью настоящего изобретения является фармацевтическая композиция, содержащая соединение, его изомер или его фармацевтически приемлемую соль в качестве активного ингредиента, для предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания.
Другой целью настоящего изобретения является композиция функциональной оздоровительной продукции, содержащая соединение, его изомер или его фармацевтически приемлемую соль в качестве активного ингредиента, для предупреждения или облегчения злокачественного новообразования, воспалительного заболевания или метаболического заболевания.
Техническое решение
Для достижения вышеуказанных целей в одном аспекте настоящего изобретения настоящее изобретение относится к соединению, представленному формулой 1, его изомеру или его фармацевтически приемлемой соли:
[Формула 1]

(В формуле 1
Ar представляет собой 6-10-членный арил или 5-10-членный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O;
R1 и R2, независимо, представляют собой водород, галоген или 5-8-членный гетероциклоалкил C1-5 алкокси, содержащий по меньшей мере один N;
R3 представляет собой водород, галоген, незамещенный или замещенный прямой или разветвленный C1-8 алкокси, незамещенный или замещенный прямой или разветвленный C1-8 алкил, NRa1Ra2 или ORa3, где Ra1, Ra2 и Ra3, независимо, представляют собой водород, незамещенный или замещенный прямой или разветвленный C1-8 алкил, незамещенный или замещенный фенил или 5-8-членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O,
при этом замещенный алкокси, замещенный алкил, замещенный фенил и замещенный гетероарил, независимо, замещены одним или несколькими заместителями, выбранными из группы, включающей 4-8-членный незамещенный или замещенный гетероциклоалкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 4-8-членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, NRb1Rb2, 3-6-членный циклоалкил, галоген, гидрокси и сульфонил, или замещенный алкокси и замещенный алкил могут быть дополнительно замещены с образованием 3-6-членного циклоалкила с каждым независимо замещенным атомом углерода,
замещенный гетероциклоалкил и замещенный гетероарил замещены одним или несколькими заместителями, выбранными из группы, включающей прямой или разветвленный C1-5 алкил незамещенный или замещенный одним или несколькими галогенами, прямой или разветвленный C1-5 алкилкарбонил, NRb1Rb2, галоген, гидрокси и оксо,
Rb1 и Rb2, независимо, представляют собой водород или прямой или разветвленный C1-6 алкил;
L1 представляет собой 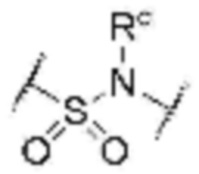 или
или  , где Rc представляет собой водород или L2 и образует 5-8-членный гетероциклоалкилен, содержащий N, вместе с атомом азота, к которому они присоединены;
, где Rc представляет собой водород или L2 и образует 5-8-членный гетероциклоалкилен, содержащий N, вместе с атомом азота, к которому они присоединены;
L2 представляет собой одинарную связь, прямой или разветвленный C1-8 алкилен, C3-8 циклоалкилен или фенилен, незамещенный или замещенный одним гидрокси и оксо;
Z представляет собой незамещенный или замещенный C6-10 арил, C3-8, циклоалкил, незамещенный или конденсированный с 5-9-членным незамещенным или замещенным гетероарилом, содержащим один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 4-8-членный незамещенный или замещенный гетероциклоалкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 5-9-членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, или 5-9-членный незамещенный или замещенный гетероарил C1-5 алкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O,
замещенный арил замещен галогеном, фенилом, карбокси, 4-8-членным незамещенным или замещенным гетероциклоалкилом, содержащим один или несколько гетероатомов, выбранных из группы, включающей N, S и O, или прямым или разветвленным C1-8 алкоксикарбонилом,
при этом замещенный гетероциклоалкил, замещенный гетероарил и замещенный гетероарилалкил, независимо, замещены одним или несколькими заместителями, выбранными из группы, включающей прямой или разветвленный C1-5 алкил, прямой или разветвленный C1-8 алкокси, фенил, бензил, галоген, гидрокси или оксо.)
В другом аспекте настоящего изобретения способ получения соединения, представленного формулой 1, включает стадию получения соединения, представленного формулой 1, путем взаимодействия соединения, представленного формулой 2, с соединением, представленным формулой 3, как показано на схеме реакции 1 ниже:
[Схема реакции 1]
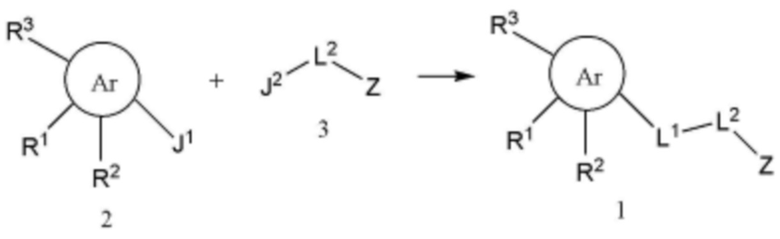
(На схеме реакции 1 Ar, R1, R2, R3, L1, L2 и Z имеют значения, определенные в формуле 1 пункта 1 формулы изобретения,
J1 представляет собой хлорсульфон или карбокси; и
J2 представляет собой амин или пиперидин.)
В другом аспекте настоящего изобретения настоящее изобретение относится к фармацевтической композиции, содержащей в качестве активного ингредиента соединение, представленное формулой 1, его изомер или его фармацевтически приемлемую соль, для предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания.
В другом аспекте настоящего изобретения настоящее изобретение относится к композиции функциональной оздоровительной продукции, содержащей в качестве активного ингредиента соединение, представленное формулой 1, его изомер или его фармацевтически приемлемую соль, для предупреждения или облегчения рака, воспалительного заболевания или метаболического заболевания.
В другом аспекте настоящего изобретения настоящее изобретение относится к способу предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания, включающему стадию введения субъекту, нуждающемуся в этом, фармацевтической композиции или композиции функциональной оздоровительной продукции, содержащей в качестве активного ингредиента соединение, представленное формулой 1, или его фармацевтически приемлемую соль.
В другом аспекте настоящего изобретения настоящее изобретение относится к применению фармацевтической композиции или композиции функциональной оздоровительной продукции, содержащей в качестве активного ингредиента соединение, представленное формулой 1, или его фармацевтически приемлемую соль, для предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания.
Положительные эффекты изобретения
Поскольку новое соединение по настоящему изобретению проявляет превосходную ингибирующую активность в отношении Pin1, фармацевтическая композиция, содержащая соединение в качестве активного ингредиента, может быть эффективно использована для предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания.
Лучший способ осуществления изобретения
Далее настоящее изобретение описано подробно.
В одном аспекте настоящего изобретения настоящее изобретение относится к соединению, представленному формулой 1, его изомеру или его фармацевтически приемлемой соли:
[Формула 1]
(В формуле 1
Ar представляет собой 6-10-членный арил или 5-10-членный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O;
R1 и R2, независимо, представляют собой водород, галоген или 5-8-членный гетероциклоалкил C1-5 алкокси, содержащий по меньшей мере один N;
R3 представляет собой водород, галоген, незамещенный или замещенный прямой или разветвленный C1-8 алкокси, незамещенный или замещенный прямой или разветвленный C1-8 алкил, NRa1Ra2 или ORa3, где Ra1, Ra2 и Ra3, независимо, представляют собой водород, незамещенный или замещенный прямой или разветвленный C1-8 алкил, незамещенный или замещенный фенил или 5-8 -членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O,
при этом замещенный алкокси, замещенный алкил, замещенный фенил и замещенный гетероарил, независимо, замещены одним или несколькими заместителями, выбранными из группы, включающей 4-8 -членный незамещенный или замещенный гетероциклоалкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 4-8 -членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, NRb1Rb2, 3-6 -членный циклоалкил, галоген, гидрокси и сульфонил, или замещенный алкокси и замещенный алкил могут быть далее замещены с образованием 3-6 -членный циклоалкил с каждым независимо замещенным атомом углерода,
замещенный гетероциклоалкил и замещенный гетероарил замещены одним или несколькими заместителями, выбранными из группы, включающей прямой или разветвленный C1-5 алкил незамещенный или замещенный одним или несколькими галогенами, прямой или разветвленный C1-5 алкилкарбонил, NRb1Rb2, галоген, гидрокси и оксо,
Rb1 и Rb2, независимо, представляют собой водород или прямой или разветвленный C1-6 алкил;
L1 представляет собой или , где Rc представляет собой водород или L2 и образует 5-8-членный гетероциклоалкилен, содержащий N вместе с атомом азота, к которому они присоединены;
L2 представляет собой одинарную связь, прямой или разветвленный C1-8 алкилен, C3-8 циклоалкилен или фенилен, незамещенный или замещенный одним гидрокси и оксо;
Z представляет собой незамещенный или замещенный C6-10 арил, C3-8, циклоалкил, незамещенный или конденсированный с 5-9-членным незамещенным или замещенным гетероарилом, содержащим один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 4-8-членный незамещенный или замещенный гетероциклоалкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 5-9-членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, или 5-9-членный незамещенный или замещенный гетероарил C1-5 алкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O,
замещенный арил замещен галогеном, фенилом, карбокси, 4-8-членным незамещенным или замещенным гетероциклоалкилом, содержащим один или несколько гетероатомов, выбранных из группы, включающей N, S и O, или прямым или разветвленным C1-8 алкоксикарбонилом,
при этом замещенный гетероциклоалкил, замещенный гетероарил и замещенный гетероарилалкил, независимо, замещены одним или несколькими заместителями, выбранными из группы, включающей прямой или разветвленный C1-5 алкил, прямой или разветвленный C1-8 алкокси, фенил, бензил, галоген, гидрокси или оксо.)
Соединение, его изомер или его фармацевтически приемлемая соль в соответствии с п.1, где:
Ar представляет собой 6-10-членный арил или 5-6-членный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O;
R1 и R2, независимо, представляют собой водород или фтор;
R3 представляет собой водород, бром, незамещенный или замещенный прямой или разветвленный C1-6 алкокси, незамещенный или замещенный прямой или разветвленный C1-6 алкил, NRa1Ra2 или ORa3, где Ra1, Ra2 и Ra3, независимо, представляют собой водород, незамещенный или замещенный прямой или разветвленный C1-6 алкил, незамещенный или замещенный фенил или 5-7-членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O,
при этом замещенный алкокси, замещенный алкил, замещенный фенил и замещенный гетероарил, независимо, замещены одним или несколькими заместителями, выбранными из группы, включающей 4-7-членный незамещенный или замещенный гетероциклоалкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 4-7-членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, NRb1Rb2, 3-5-членный циклоалкил, фтор, бром, гидрокси и сульфонил, или замещенный алкокси и замещенный алкил могут быть далее замещены с образованием 3-5-членный циклоалкил с каждым независимо замещенным атомом углерода,
замещенный гетероциклоалкил и замещенный гетероарил замещены одним или несколькими заместителями, выбранными из группы, включающей прямой или разветвленный C1-4 алкил незамещенный или замещенный одним или несколькими галогенами, прямой или разветвленный C1-4 алкилкарбонил, NRb1Rb2, хлор, фтор, бром, гидрокси и оксо,
Rb1 и Rb2, независимо, представляют собой водород или прямой или разветвленный C1-3 алкил;
L1 представляет собой или , где Rc представляет собой водород или L2 и образует пиперидинилен вместе с атомом азота, к которому они присоединены;
L2 представляет собой одинарную связь, прямой или разветвленный C1-6 алкилен, C4-6 циклоалкилен или фенилен, незамещенный или замещенный одним гидрокси и оксо;
Z представляет собой незамещенный или замещенный C6-9 арил, C4-7, циклоалкил, незамещенный или конденсированный с 5-9-членным незамещенным или замещенным гетероарилом, содержащим один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 5-6-членный незамещенный или замещенный гетероциклоалкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 6-9-членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, или 6-9-членный незамещенный или замещенный гетероарил C1-4 алкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O,
замещенный арил замещен хлором, фенил, карбокси, морфолинил или прямой или разветвленный C1-3 алкоксикарбонил,
при этом замещенный гетероциклоалкил, замещенный гетероарил и замещенный гетероарилалкил, независимо, замещены одним или несколькими заместителями, выбранными из группы, включающей прямой или разветвленный C1-4 алкил, прямой или разветвленный C1-4 алкокси, бензил, фенил, фтор, хлор, бром, гидрокси или оксо.
Соединение, его изомер или его фармацевтически приемлемая соль в соответствии с п.1, где:
Ar представляет собой фенил, нафтил, пиридин или триазол;
R1 и R2, независимо, представляют собой водород или фтор;
R3 представляет собой водород, бром, незамещенный или замещенный прямой или разветвленный C1-5 алкокси, незамещенный или замещенный прямой или разветвленный C1-5 алкил, NRa1Ra2 или ORa3, где Ra1, Ra2 и Ra3, независимо, представляют собой водород, незамещенный или замещенный прямой или разветвленный C1-5 алкил, незамещенный или замещенный фенил или 5-6-членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O,
при этом замещенный алкокси, замещенный алкил, замещенный фенил и замещенный гетероарил, независимо, замещены одним или несколькими заместителями, выбранными из группы, включающей 4-7-членный незамещенный или замещенный гетероциклоалкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 5-7-членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, NRb1Rb2, 3-4-членный циклоалкил, фтор, бром, гидрокси и сульфонил, или замещенный алкокси и замещенный алкил могут быть далее замещены с образованием циклопропил с каждым независимо замещенным атомом углерода,
замещенный гетероциклоалкил и замещенный гетероарил замещены одним или несколькими заместителями, выбранными из группы, включающей прямой или разветвленный C1-3 алкил незамещенный или замещенный одним или несколькими атомами фтора, прямой или разветвленный C1-4 алкилкарбонил, NRb1Rb2, хлор, фтор, бром, гидрокси и оксо,
Rb1 и Rb2, независимо, представляют собой водород, метил или этил;
L1 представляет собой или , где Rc представляет собой водород или L2 и образует пиперидинилен вместе с атомом азота, к которому они присоединены;
L2 представляет собой одинарную связь, прямой или разветвленный C1-4 алкилен, циклогексилен или фенилен, незамещенный или замещенный одним гидрокси и оксо;
Z представляет собой незамещенный или замещенный фенил, циклогексил, нафтил, C5-6 циклоалкил, конденсированный с 5-9-членным незамещенным или замещенным гетероарилом, содержащим один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 5-6-членный незамещенный или замещенный гетероциклоалкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, 6-9-членный незамещенный или замещенный гетероарил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O, или 6-9 -членный незамещенный или замещенный гетероарил C1-3 алкил, содержащий один или несколько гетероатомов, выбранных из группы, включающей N, S и O,
замещенный фенил замещен хлором, фенилом, карбокси, морфолинилом или метоксикарбонилом,
при этом замещенный гетероциклоалкил, замещенный гетероарил и замещенный гетероарилалкил, независимо, замещены одним или несколькими заместителями, выбранными из группы, включающей прямой или разветвленный C1-3 алкил, метокси, бензил, фенил, фтор, хлор, бром, гидрокси или оксо.
Соединение, его изомер или его фармацевтически приемлемая соль в соответствии с п.1, где:
Ar представляет собой фенил, нафтил, пиридин или триазол;
R1 и R2, независимо, представляют собой водород или фтор;
R3 представляет собой водород, бром,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 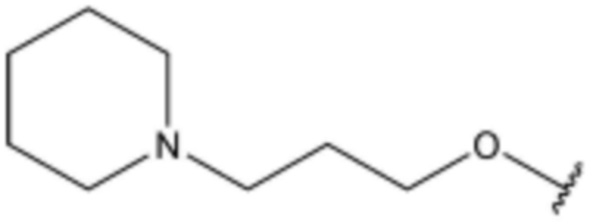 ,
,  ,
, 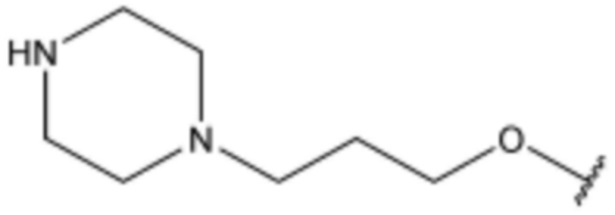 ,
,  ,
, 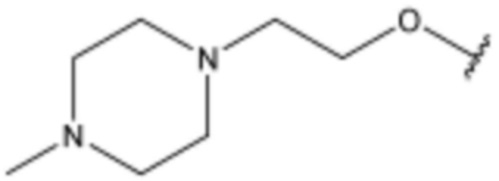 ,
,  ,
,  ,
,  ,
, 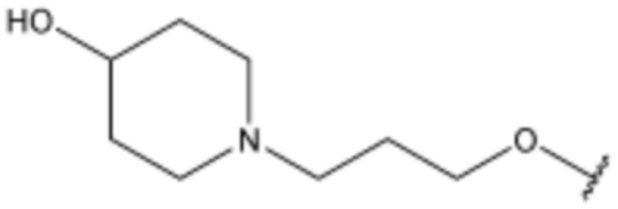 ,
,  ,
,  ,
,  ,
, 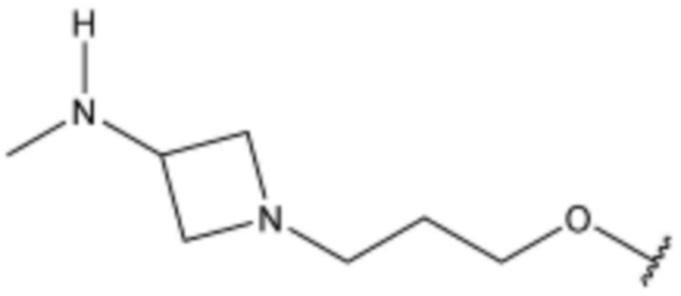 ,
, 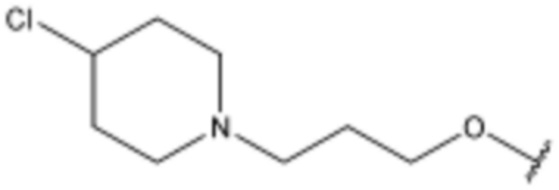 ,
, 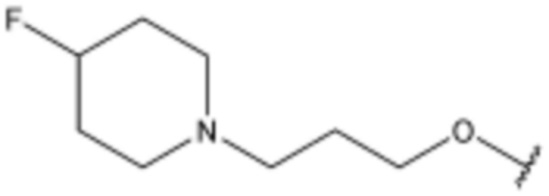 ,
,  ,
,  ,
,  ,
, 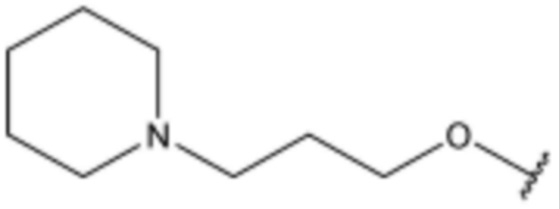 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 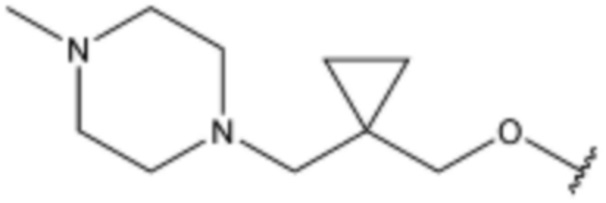 ,
,  ,
,  ,
, 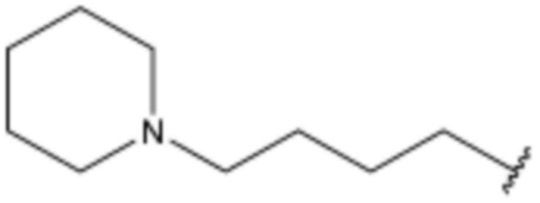 ,
, 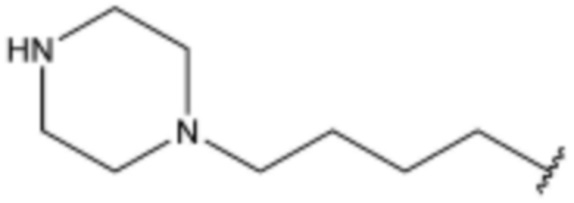 ,
,  ,
,  ,
, 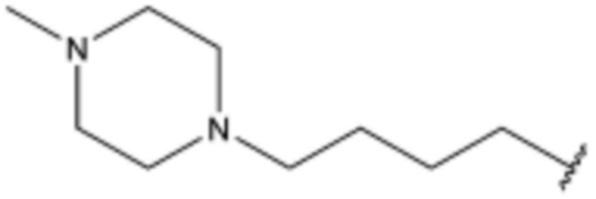 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 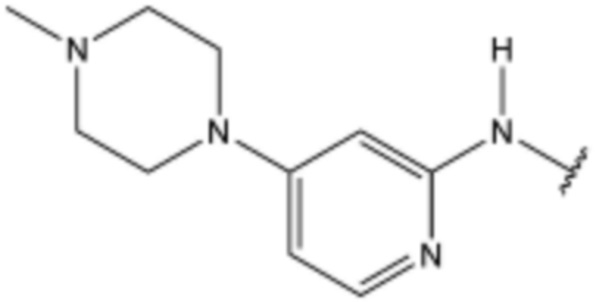 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 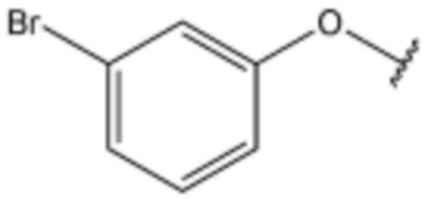 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 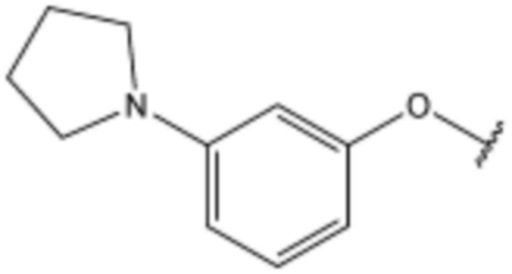 ,
,  ,
,  ,
, 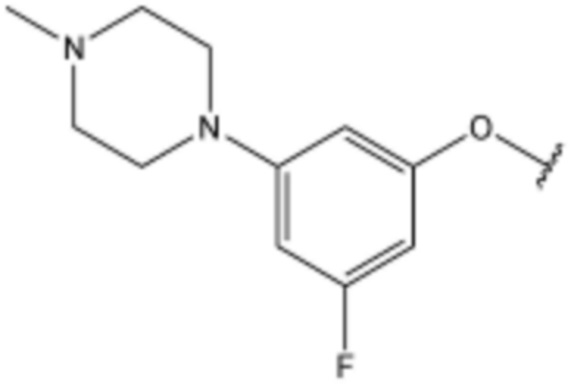 ,
,  ,
, 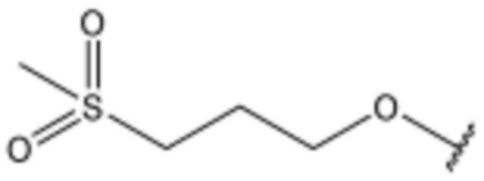 ,
, 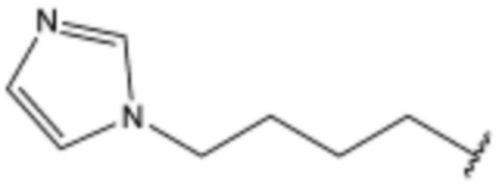 ,
,  ,
,  ,
,  ,
, 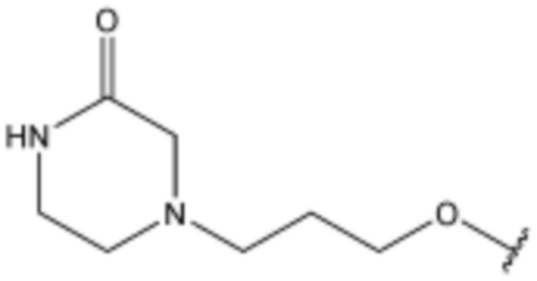 ,
,  ,
,  ,
,  ,
, 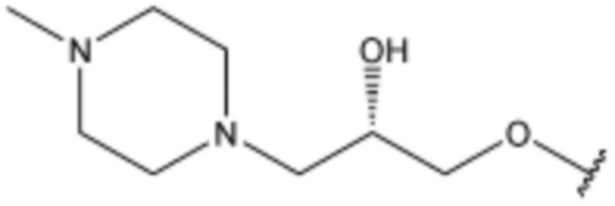 ,
,  ,
, 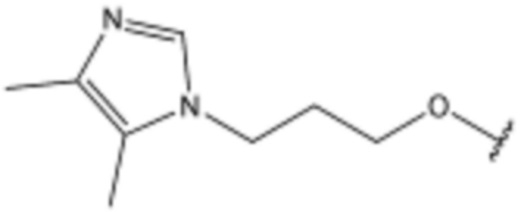 ,
,  ,
,  или
или 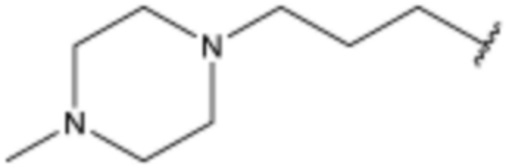 ;
;
L1 представляет собой 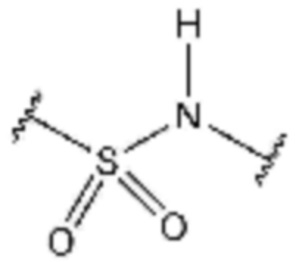 ,
,  или
или  ;
;
L2 представляет собой одинарную связь, 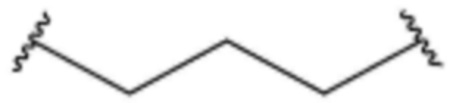 ,
, 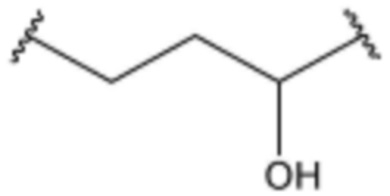 ,
,  ,
,  ,
, 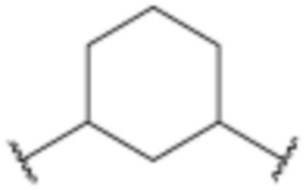 ,
,  ,
,  или
или  ;
;
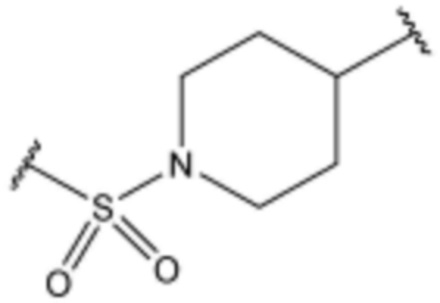
Z представляет собой  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 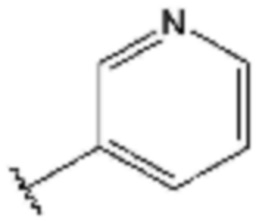 ,
,  ,
, 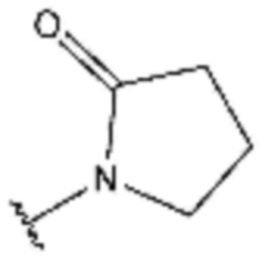 ,
, 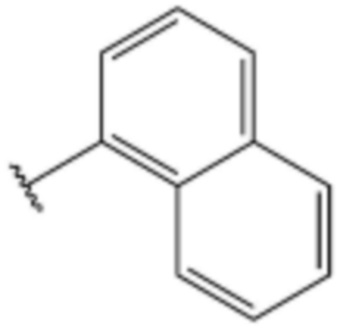 ,
, 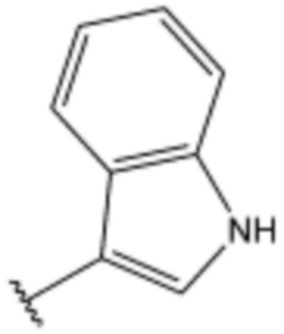 ,
,  ,
,  ,
,  ,
,  ,
, 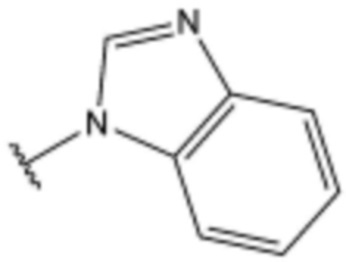 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 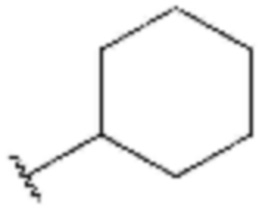 ,
,  ,
,  ,
,  ,
,  ,
, 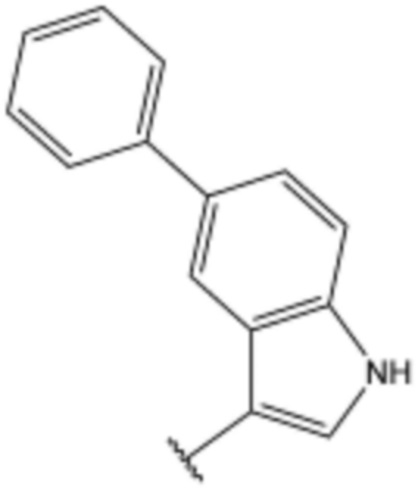 ,
, 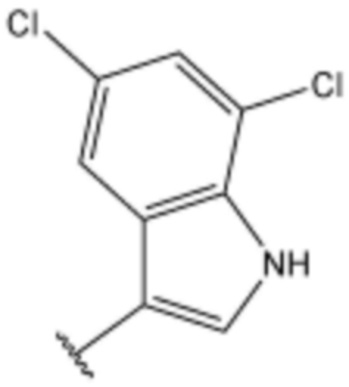 ,
,  ,
,  ,
,  ,
,  ,
, 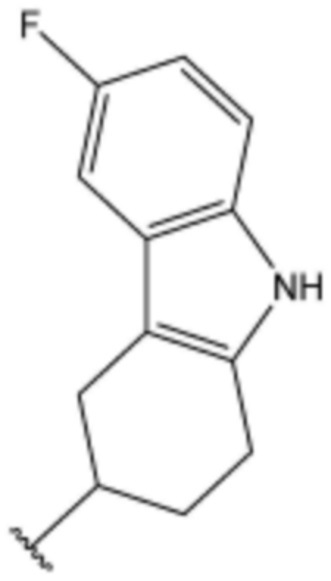 ,
,  или
или  .
.
Соединение, его изомер или его фармацевтически приемлемая соль в соответствии с п.1, где соединение, представленное формулой 1, выбрано из группы, включающей следующие соединения:
<1> N-(3-([1,1'-бифенил]-4-ил)пропил)-4-бутоксибензолсульфонамид;
<2> 4-бутокси-N-(3-(4-изопропилпиперазин-1-ил)пропил)бензолсульфонамид;
<3> 4-бутокси-N-(3-(4-хлорфенил)пропил)бензолсульфонамид;
<4> 4-бутокси-N-(3-циклогексилпропил)бензолсульфонамид;
<5> 4-бутокси-N-(3-(пиридин-3-ил)пропил)бензолсульфонамид;
<6> 4-бутокси-N-(3-морфолинопропил)бензолсульфонамид;
<7> 4-бутокси-N-(4-морфолинофенетил)бензолсульфонамид;
<8> 4-(2-(4-бутоксифенилсульфонамидо)этил)бензойная кислота;
<9> метил 4-(2-(4-бутоксифенилсульфонамидо)этил)бензоат;
<10> N-(2-(4-бензилпиперидин-1-ил)этил)-4-бутоксибензолсульфонамид;
<11> 4-бутокси-N-(3-гидрокси-3-фенилпропил)бензолсульфонамид;
<12> 4-бутокси-N-(3-оксо-3-фенилпропил)бензолсульфонамид;
<13> 4-бутокси-N-(3-(2-оксопирролидин-1-ил)пропил)бензолсульфонамид;
<14> 4-(3-(диметиламино)пропокси)-N-фенетилбензолсульфонамид;
<15> 4-(3-(диметиламино)пропокси)-N-(3-фенилпропил)бензолсульфонамид;
<16> метил 4-(2-(4-(3-(диметиламино)пропокси)фенилсульфонамидо)этил)бензоат;
<17> 4-(2-(4-(3-(диметиламино)пропокси)фенилсульфонамидо)этил)бензойная кислота;
<18> 4-(3-(диметиламино)пропокси)-N-(3-(2-оксопирролидин-1-ил)пропил)бензолсульфонамид;
<19> 4-бутокси-N-(3-(нафталин-1-ил)пропил)бензолсульфонамид;
<20> N-(3-(1H-индол-3-ил)пропил)-4-бутоксибензолсульфонамид;
<21> N-(3-(1H-индол-3-ил)пропил)-4-(2-(диметиламино)этокси)бензолсульфонамид;
<22> N-(3-(1H-индол-3-ил)пропил)-4-(3-(диметиламино)пропокси)бензолсульфонамид;
<23> N-(2-(1H-бензо[d]имидазол-2-ил)этил)-4-бутоксибензолсульфонамид;
<24> N-(2-(1H-индол-3-ил)этил)-4-бутоксибензолсульфонамид;
<25> N-(2-(1H-индол-2-ил)этил)-4-бутоксибензолсульфонамид;
<26> N-(3-(1H-индол-3-ил)пропил)-4-(изопентилокси)бензолсульфонамид;
<27> N-(3-(1H-индол-3-ил)пропил)-4-(пентилокси)бензолсульфонамид;
<28> N-(3-(1H-бензо[d]имидазол-2-ил)пропил)-4-бутоксибензолсульфонамид;
<29> 4-бутокси-N-(2-(5-гидрокси-1H-индол-3-ил)этил)бензолсульфонамид;
<30> N-(3-(1H-индол-3-ил)пропил)-4-(3-морфолинопропокси)бензолсульфонамид;
<31> N-(3-(1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<32> N-(3-(1H-индол-1-ил)пропил)-4-бутоксибензолсульфонамид;
<33> N-(3-(1H-бензо[d]имидазол-1-ил)пропил)-4-бутоксибензолсульфонамид;
<34> N-(3-(1H-бензо[d]имидазол-1-ил)пропил)-4-бутоксибензолсульфонамид;
<35> N-(3-(1H-индол-3-ил)пропил)-4-(3-(диэтиламино)пропокси)бензолсульфонамид;
<36> N-(3-(1H-индол-3-ил)пропил)-4-(3-(4-этилпиперазин-1-ил)пропокси)бензолсульфонамид;
<37> N-(2-(1H-бензо[d]имидазол-2-ил)этил)-4-(3-(диметиламино)пропокси)бензолсульфонамид;
<38> N-(2-(1H-индол-2-ил)этил)-4-(3-(диметиламино)пропокси)бензолсульфонамид;
<39> N-(3-(1H-индол-1-ил)пропил)-4-(3-(диметиламино)пропокси)бензолсульфонамид;
<40> N-(3-(1H-бензо[d]имидазол-1-ил)пропил)-4-(3-(диметиламино)пропокси)бензолсульфонамид;
<41> N-(3-(1H-индол-1-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<42> N-(3-(1H-бензо[d]имидазол-1-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<43> N-(3-(1H-индол-3-ил)пропил)-4-(3-(пиперидин-1-ил)пропокси)бензолсульфонамид;
<44> N-(3-(1H-индол-3-ил)пропил)-4-(3-(2-оксопирролидин-1-ил)пропокси)бензолсульфонамид;
<45> N-(3-(1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<46> 4-(3-(диметиламино)пропокси)-N-(3-(1-метил-1H-индол-3-ил)пропил)бензолсульфонамид;
<47> N-(3-(1H-индол-3-ил)пропил)-4-бромбензолсульфонамид;
<48> N-(3-(1H-индол-3-ил)пропил)-4-(3-(4-изопропилпиперазин-1-ил)пропокси)бензолсульфонамид;
<49> N-(2-(5-метокси-1H-индол-3-ил)этил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<50> N-(2-(1H-индол-3-ил)этил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<51> N-(2-(1H-индол-3-ил)этил)-4-(3-(4-изопропилпиперазин-1-ил)пропокси)бензолсульфонамид;
<52> N-(3-(1H-индол-3-ил)пропил)-4-(2-(4-метилпиперазин-1-ил)этокси)бензолсульфонамид;
<53> N-(3-(1H-индол-3-ил)пропил)-4-(2-(4-изопропилпиперазин-1-ил)этокси)бензолсульфонамид;
<54> N-(2-(2-метил-1H-индол-3-ил)этил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<55> N-(3-(1H-индол-3-ил)пропил)-4-(2-(1-метилпиперидин-4-ил)этокси)бензолсульфонамид;
<56> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<57> 3-(1-((4-(3-(4-метилпиперазин-1-ил)пропокси)фенил)сульфонил)пиперидин-4-ил)-1H-индол;
<58> N-(3-(2-метил-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<59> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(1-метилпиперидин-4-ил)пропокси)бензолсульфонамид;
<60> N-(3-(1H-индол-3-ил)пропил)-4-(3-(1-метилпиперидин-4-ил)пропокси)бензолсульфонамид;
<61> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<62> N-(3-(1H-индол-3-ил)пропил)-3-фтор-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<63> 4-(3-(4-этилпиперазин-1-ил)пропокси)-N-(3-(5-фтор-1H-индол-3-ил)пропил)бензолсульфонамид;
<64> 5-метокси-3-(1-((4-(3-(4-метилпиперазин-1-ил)пропокси)фенил)сульфонил)пиперидин-4-ил)-1H-индол;
<65> 5-метил-3-(1-((4-(3-(4-метилпиперазин-1-ил)пропокси)фенил)сульфонил)пиперидин-4-ил)-1H-индол;
<66> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<67> N-(3-(5-метил-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<68> N-(3-(5-метокси-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<69> N-(3-(1H-индол-3-ил)пропил)-4-(3-(4-гидроксипиперидин-1-ил)пропокси)бензолсульфонамид;
<70> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<71> 5-фтор-3-(1-((4-(3-(4-метилпиперазин-1-ил)пропокси)фенил)сульфонил)пиперидин-4-ил)-1H-индол;
<72> 4-(3-((3S,5R)-3,5-диметилпиперазин-1-ил)пропокси)-N-(3-(5-фтор-1H-индол-3-ил)пропил)бензолсульфонамид;
<73> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(4-изобутирилпиперазин-1-ил)пропокси)бензолсульфонамид;
<74> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(4-(2,2,2-трифторэтил)пиперазин-1-ил)пропокси)бензолсульфонамид;
<75> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-гидроксипиперидин-1-ил)пропокси)бензолсульфонамид;
<76> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-(2,2,2-трифторэтил)пиперазин-1-ил)пропокси)бензолсульфонамид;
<77> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-(метиламино)азетидин-1-ил)пропокси)бензолсульфонамид;
<78> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-хлорпиперидин-1-ил)пропокси)бензолсульфонамид;
<79> N-(3-(5-хлор-1H-индол-3-ил)пропил)-6-(3-(пиперазин-1-ил)пропокси)пиридин-3-сульфонамид;
<80> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-фторпиперидин-1-ил)пропокси)бензолсульфонамид;
<81> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-(трифторметил)пиперидин-1-ил)пропокси)бензолсульфонамид;
<82> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4,4-дифторпиперидин-1-ил)пропокси)бензолсульфонамид;
<83> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метил-1,4-диазепан-1-ил)пропокси)бензолсульфонамид;
<84> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(пиперидин-1-ил)пропокси)бензолсульфонамид;
<85> N-(3-(1H-индол-3-ил)циклогексил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<86> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)нафталин-1-сульфонамид;
<87> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперидин-1-ил)пропокси)бензолсульфонамид;
<88> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(пиперидин-4-ил)пропил)амино)бензолсульфонамид;
<89> N-(3-(1H-индол-3-ил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<90> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(пиперидин-1-ил)бутил)бензолсульфонамид;
<91> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(пиперазин-1-ил)бутил)бензолсульфонамид;
<92> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(1-метилпиперидин-4-ил)пропокси)бензолсульфонамид;
<93> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперидин-4-ил)пропокси)бензолсульфонамид;
<94> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-(3-(пиперидин-4-ил)пропокси)бензолсульфонамид;
<95> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-(3-(пиперидин-1-ил)пропокси)бензолсульфонамид;
<96> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<97> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(пиперидин-4-ил)пропокси)бензолсульфонамид;
<98> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(4-метилпиперазин-1-ил)пропил)амино)бензолсульфонамид;
<99> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(пиперидин-4-ил)бутил)бензолсульфонамид;
<100> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пирролидин-1-ил)пропокси)бензолсульфонамид;
<101> 4-(3-(азепан-1-ил)пропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<102> 4-(3-(1,4-диазепан-1-ил)пропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<103> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-фтор-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<104> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-фтор-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<105> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-фтор-4-(3-(4-гидроксипиперидин-1-ил)пропокси)бензолсульфонамид;
<106> 4-(3-(1,4-диазепан-1-ил)пропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-фторбензолсульфонамид;
<107> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<108> (R)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<109> (S)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<110> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-((3S,5R)-3,5-диметилпиперазин-1-ил)пропокси)бензолсульфонамид;
<111> (S)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3,4-диметилпиперазин-1-ил)пропокси)бензолсульфонамид;
<112> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(4-метилпиперазин-1-ил)бутил)бензолсульфонамид;
<113> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(пиперазин-1-ил)пропил)амино)бензолсульфонамид;
<114> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(пиперидин-1-ил)пропил)амино)бензолсульфонамид;
<115> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(1-метилпиперидин-4-ил)бутил)бензолсульфонамид;
<116> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((1-((4-метилпиперазин-1-ил)метил)циклопропил)метокси)бензолсульфонамид;
<117> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((1-(пиперазин-1-илметил)циклопропил)метокси)бензолсульфонамид;
<118> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)фенокси)бензолсульфонамид;
<119> 4-(3-бромфенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<120> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперидин-1-ил)фенокси)бензолсульфонамид;
<121> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)фенокси)бензолсульфонамид;
<122> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метил-1,4-диазепан-1-ил)фенокси)бензолсульфонамид;
<123> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(4-метилпиперазин-1-ил)фенил)амино)бензолсульфонамид;
<124> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(пиперазин-1-ил)фенил)амино)бензолсульфонамид;
<125> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((4-(4-метилпиперазин-1-ил)пиримидин-2-ил)амино)бензолсульфонамид;
<126> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-морфолинофенил)амино)бензолсульфонамид;
<127> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((4-(пиперазин-1-ил)пиримидин-2-ил)амино)бензолсульфонамид;
<128> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-морфолинофенокси)бензолсульфонамид;
<129> 4-(3-(1,4-диазепан-1-ил)фенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<130> 4-(3,5-бис(4-метилпиперазин-1-ил)фенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<131> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((6-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)бензолсульфонамид;
<132> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((6-(пиперазин-1-ил)пиридин-2-ил)амино)бензолсульфонамид;
<133> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((4-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)бензолсульфонамид;
<134> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((4-(пиперазин-1-ил)пиридин-2-ил)амино)бензолсульфонамид;
<135> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пирролидин-1-ил)фенокси)бензолсульфонамид;
<136> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-гидроксипиперидин-1-ил)фенокси)бензолсульфонамид;
<137> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-фтор-5-(4-метилпиперазин-1-ил)фенокси)бензолсульфонамид;
<138> 4-(3-бром-5-(4-метилпиперазин-1-ил)фенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<139> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(4-гидроксипиперидин-1-ил)фенил)амино)бензолсульфонамид;
<140> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((6-хлор-4-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)бензолсульфонамид;
<141> 4-((4,6-бис(4-метилпиперазин-1-ил)пиридин-2-ил)амино)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<142> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((4-(4-метилпиперазин-1-ил)фенил)амино)бензолсульфонамид;
<143> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензамид;
<144> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензамид;
<145> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(метилсульфонил)пропокси)бензолсульфонамид;
<146> N-(3-(5-хлор-1H-индол-3-ил)пропил)-2-((3-(пиперазин-1-ил)пропил)амино)триазол-5-сульфонамид;
<147> N-(3-(5-хлор-2-метил-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<148> 4-(4-(1H-имидазол-1-ил)бутил)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<149> N-(2-((5-хлор-1H-индол-3-ил)метил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<150> N-(2-((5-хлор-1H-индол-3-ил)метил)фенил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<151> N-(4-(5-хлор-1H-индол-3-ил)бутан-2-ил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<152> N-(4-(5-хлор-1H-индол-3-ил)бутан-2-ил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<153> N-(3-(5-бром-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<154> N-(3-(5-бром-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<155> N-(3-(5-фенил-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<156> 4-(3-(4-метилпиперазин-1-ил)пропокси)-N-(3-(5-фенил-1H-индол-3-ил)пропил)бензолсульфонамид;
<157> N-(3-(5-хлор-2-метил-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<158> N-(2-(5-хлор-1H-индол-3-ил)этил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<159> N-(2-(5-хлор-1H-индол-3-ил)этил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<160> 4-(3-(1H-1,2,4-triazol-1-ил)пропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<161> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(2-метил-1H-имидазол-1-ил)пропокси)бензолсульфонамид;
<162> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метил-3-оксопиперазин-1-ил)пропокси)бензолсульфонамид;
<163> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-оксопиперазин-1-ил)пропокси)бензолсульфонамид;
<164> N-(3-(5,7-дихлор-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<165> N-(3-(5,7-дихлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<166> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4,5-дихлор-1H-имидазол-1-ил)пропокси)бензолсульфонамид;
<167> N-(3-(5-хлор-1H-индазол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<168> N-(3-(5-хлор-1H-индазол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<169> N-(3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<170> N-(3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<171> (R)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(2-гидрокси-3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<172> (R)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(2-гидрокси-3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<173> (S)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(2-гидрокси-3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<174> (S)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(2-гидрокси-3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<175> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4,5-диметил-1H-имидазол-1-ил)пропокси)бензолсульфонамид;
<176> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(2,4,5-триметил-1H-имидазол-1-ил)пропокси)бензолсульфонамид;
<177> N-((6-хлор-2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<178> N-((6-хлор-2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<179> N-(3-(5-хлор-1H-индол-3-ил)циклогексил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<180> N-(3-(5-хлор-1H-индол-3-ил)циклогексил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<181> N-(2-(2-(5-хлор-1H-индол-3-ил)пропан-2-ил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<182> N-(2-(2-(5-хлор-1H-индол-3-ил)пропан-2-ил)фенил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<183> N-(3-(5,6-дихлор-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<184> N-(3-(5,6-дихлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<185> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиридин-4-илокси)пропил)бензолсульфонамид;
<186> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропил)бензолсульфонамид;
<187> N-((6-фтор-2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<188> N-((6-фтор-2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<189> 4-(3-(4-метилпиперазин-1-ил)пропокси)-N-((2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)бензолсульфонамид;
<190> 4-(3-(пиперазин-1-ил)пропокси)-N-((2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)бензолсульфонамид.
Соединение, представленное формулой 1 по настоящему изобретению, может быть использовано в виде фармацевтически приемлемой соли, в которой соль, предпочтительно, представляет собой кислотно-аддитивную соль, образованную фармацевтически приемлемыми свободными кислотами. Кислотно-аддитивная соль по настоящему изобретению может быть получена из неорганических кислот, таких как хлористоводородная кислота, азотная кислота, фосфорная кислота, серная кислота, бромистоводородная кислота, йодистоводородная кислота, азотистая кислота и фосфористая кислота; нетоксичных органических кислот, таких как алифатические моно/дикарбоксилаты, фенилзамещенные алканоаты, гидроксиалканоаты, алкандиоаты, ароматические кислоты и алифатические/ароматические сульфокислоты; или органических кислот, таких как уксусная кислота, бензойная кислота, лимонная кислота, молочная кислота, малеиновая кислота, глюконовая кислота, метансульфокислота, 4-толуолсульфокислота, винная кислота и фумаровая кислота. Примерами фармацевтически нетоксичных солей являются сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, йодид, фторид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутилат, капрат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, кабакат, фумарат, малиат, бутин-1,4-диоат, гексан-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, терефталат, бензолсульфонат, толуолсульфонат, хлорбензолсульфонат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутилат, цитрат, лактат, гидроксибутилат, гликолят, малат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат и манделат.
Кислотно-аддитивная соль по настоящему изобретению может быть получена обычным способом, известным специалистам в данной области. Например, производное, представленное формулой 1, растворяют в органическом растворителе, таком как метанол, этанол, ацетон, дихлорметан и ацетонитрил, к которому добавляют органическую кислоту или неорганическую кислоту, чтобы вызвать осаждение. Затем осадок фильтруют и сушат, получая соль. Или растворитель и избыточную кислоту перегоняют при пониженном давлении и сушат, получая соль. Или осадок кристаллизуют в органическом растворителе, чтобы получить то же самое.
Фармацевтически приемлемая соль металла может быть получена с использованием основания. Соль щелочного металла или щелочноземельного металла получают следующими способами: растворением соединения в избыточном растворе гидроксида щелочного металла или гидроксида щелочноземельного металла; фильтрованием нерастворимого соединения соли; выпариванием оставшегося раствора и его сушкой. В это время соль металла, предпочтительно, получают в фармацевтически подходящем виде соли натрия, калия или кальция. И соответствующую соль серебра получают взаимодействием соли щелочного металла или щелочноземельного металла с соответствующей солью серебра (например, нитратом серебра).
Кроме того, настоящее изобретение включает не только соединение, представленное формулой 1, и его фармацевтически приемлемую соль, но также сольваты, изомеры, гидраты и т.д., которые из них могут быть получены.
Термин «изомер» относится к соединению по настоящему изобретению или его соли, имеющей ту же самую химическую формулу или молекулярную формулу, но отличающуюся структурно или стерически. Такой изомер включает структурные изомеры, такие как таутомеры, R- или S-изомеры, имеющие асимметричный углеродный центр, стереоизомеры, такие как геометрические изомеры (транс-, цис-) и оптические изомеры. Все эти изомеры и их смеси также входят в объем настоящего изобретения.
Соединение, представленное формулой 1 по настоящему изобретению, может быть получено в соответствии со способом получения, продемонстрированном в следующем примере, но это только пример и им не ограничивается, и каждая стадия получения может быть осуществлена с использованием способа, хорошо известного специалистам в данной области.
В другом аспекте настоящего изобретения настоящее изобретение относится к способу получения соединения, представленного формулой 1, включающий стадию получения соединения, представленного формулой 1, путем взаимодействия соединения, представленного формулой 2, с соединением, представленным формулой 3, как показано на схеме реакции 1 ниже.
[Схема реакции 1]
На схеме реакции 1 Ar, R1, R2, R3, L1, L2 и Z имеют значения, определенные в формуле 1 выше,
J1 представляет собой хлорсульфон или карбокси; и
J2 представляет собой амин или пиперидин.
Далее подробно описан способ получения, показанный на схеме реакции 1.
В способе получения соединения, представленного формулой 1 по настоящему изобретению, стадия схеме реакции 1 представляет собой стадию получения соединения, представленного формулой 1, путем взаимодействия соединения, представленного формулой 2, с соединением, представленным формулой 3. В частности, это стадия, на которой соединение, представленное формулой 1, образуется путем взаимодействия хлорсульфона или карбоксигруппы соединения, представленного формулой 2, с амином соединения, представленного формулой 3.
В то же время эта стадия конкретно не ограничивается, если она является способом получения соединения, представленного формулой 1, и входит в объем настоящего изобретения. Соединение, представленное формулой 2, можно понимать как соединение, имеющее группу, которая легко принимает электроны, такую как сульфон, и удаляемую группу, которая легко вступает в реакцию, такую как хлор, или соединение, содержащее карбоксильную группу, которая может взаимодействовать с амином с образованием амида, и соединение, представленное формулой 3, можно понимать как амин, способный к реакции нуклеофильного замещения или образования амида, но не всегда этим ограничивается. Соединение, имеющее группу, которая легко принимает электроны, и удаляемую группу и амин, обладающий достаточной нуклеофильностью для взаимодействия с ним, подвергаются нуклеофильному замещению, или карбоксигруппа и амин подвергаются реакции образования амида, в результате чего получается конечное соединение.
Более подробно это можно понять применительно к способу получения примера соединения по настоящему изобретению, но может быть изменено каждое условие реакции (условие реакции, которое может быть рассмотрено специалистом в области органического синтеза, такое как температура реакции, время, атмосферные условия, условия давления и т. д.). Можно понять, что изобретение не ограничивается этим, и соединения и их производные, используемые на каждой стадии, включают модифицированные производные, которые можно модифицировать простой модификацией, изменением или удалением заместителя, в дополнение к описанным соединениям, которые включены в настоящее изобретение.
В качестве предпочтительных вариантов осуществления способа получения можно привести способы получения, описанные в примерах с 1 по 190 ниже, но настоящее изобретение ими не ограничивается.
В другом аспекте настоящего изобретения настоящее изобретение относится к фармацевтической композиции, содержащей в качестве активного ингредиента соединение, представленное формулой 1, его изомер или его фармацевтически приемлемую соль, для предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания.
Соединение, представленное формулой 1 по настоящему изобретению, его изомер или его фармацевтически приемлемая соль могут ингибировать Pin1 (пептидил-пролилцис-транс-изомераза, взаимодействующая с 1).
Pin1, пролил-изомераза, представляет собой фермент, который катализирует цис/транс-изомеризацию амидного остатка пролина путем связывания с фосфорилированным сайтом Ser/Thr-Pro. Согласно исследованиям, проведенным на сегодняшний день, известно, что Pin1 участвует в возникновении и прогрессировании воспалительного заболевания, метаболического заболевания и злокачественного новообразования.
В частности, сообщалось, что Pin1 активирует 56 онкогенов, участвующих в онкогенезе, и ингибирует активность 26 генов-супрессоров опухолей, тем самым индуцируя метастазирование рака и ангиогенез рака в общем механизме развития рака.
Кроме того, сообщалось, что при ингибировании Pin1 или подавлении экспрессии гена Pin1 продукция простагландина E2 и оксида азота, стимулируемая липополисахаридом (LPS) и никотином, ослабевает, и экспрессия циклооксигеназы-2 (COX-2) и индуцибельная синтазы оксида азота (iNOS) также ослабляются, что приводит к противовоспалительному эффекту.
Хорошо известно, что, когда Pin1 присоединен к CRTC2, продукция комплекса CBP-CRTC2-CREB, который способствует глюконеогенезу, ингибируется, так что CRTC2 может регулироваться в соответствии с уровнем экспрессии Pin1 и, таким образом, может участвовать в регуляции глюкозы. метаболизм.
Таким образом, препараты, влияющие на экспрессию или активность Pin1, можно использовать в качестве терапевтических средств при злокачественном новообразовании, воспалительном заболевании или метаболическом заболевании, таком как диабет.
Таким образом, соединение, представленное формулой 1 по настоящему изобретению, его изомер или его фармацевтически приемлемая соль могут быть эффективно использованы в качестве фармацевтической композиции для предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания, или функциональной оздоровительной продукции, содержащей то же самое в качестве активного ингредиента, для предупреждения или улучшения состояния при злокачественном новообразовании, воспалительном заболевании или метаболическом заболевании.
Злокачественное новообразование может представлять собой по меньшей мере одно, выбранное из группы, включающей псевдомиксому, внутрипеченочную холангиокарциному, гепатобластому, рак печени, рак щитовидной железы, рак толстой кишки, рак яичек, миелодиспластический синдром, глиобластому, рак ротовой полости, рак губы, грибовидный микоз, острый миелогенный лейкоз, острый лимфоцитарный лейкоз, базальноклеточный рак, эпителиальную карциному яичников, герминогенную карциному яичников, рак молочной железы у мужчин, рак головного мозга, аденому гипофиза, множественную миелому, рак желчного пузыря, рак желчевыводящих путей, колоректальный рак, хронический миелогенный лейкоз, хронический лимфолейкоз, ретинобластому, меланому хориоидеи, ампулярный рак фатера, рак мочевого пузыря, рак брюшины, рак паращитовидной железы, рак надпочечников, рак полости носа, немелкоклеточный рак легкого, рак языка, астроцитому, мелкоклеточный рак легкого, педиатрический рак мозга, педиатрическую лимфому, детский лейкоз, рак тонкой кишки, менингиому, рак пищевода, глиому, рак почки, рак почки, рак сердца, рак двенадцатиперстной кишки, злокачественный рак мягких тканей, злокачественный рак костей, злокачественную лимфому, злокачественную мезотелиому, злокачественную меланому, рак глаза, рак вульвы, рак уретры, рак неизвестной первичной локализации, лимфому желудка, рак желудка, рак желудка, интерстициальный рак желудочно-кишечного тракта, Рак Вильмса, рак молочной железы, саркому, рак полового члена, рак глотки, болезнь ворсинок беременных, рак шейки матки, рак эндометрия, саркому матки, рак предстательной железы, метастатический рак костей, метастатический рак головного мозга, рак средостения, рак прямой кишки, карциноидную опухоль прямой кишки, рак влагалища, рак спинного мозга, вестибулярную шванному, рак поджелудочной железы, рак слюнных желез, саркому Капоши, болезнь Педжета, рак миндалин, плоскоклеточный рак, аденокарциному легкого, рак легкого, плоскоклеточный рак легкого, рак кожи, анальный рак, рабдомиосаркому, рак гортани, рак плевры, рак крови и рак тимуса.
Воспалительное заболевание может представлять собой по меньшей мере одно, выбранное из группы, включающей артрит, энцефаломиелит, менингит, перитонит, остеомиелит, энцефалит, анкилозирующий спондилоартрит, васкулит, увеит, илеит, атеросклероз, миозит, поражение лейкоцитов, воспалительное заболевание кишечника, язвенный колит, отслойку сетчатки, пигментный ретинит, дегенерацию желтого пятна, панкреатит, атопический дерматит, подагру, васкулит, неалкогольный стеатогепатит, первичный склерозирующий холангит, нефрит, внутрибрюшинное заболевание, сепсис, синдром системной воспалительной реакции, инфаркт миокарда, аллергические заболевания, астму, атопический дерматит, гранулематоз Вегенера, легочный саркоидоз, болезнь Бехчета, хроническую обструктивную болезнь легких и пародонтит.
Метаболическое заболевание может представлять собой по меньшей мере одно, выбранное из группы, включающей ожирение, диабет, гипертонию, гиперлипидемию, гиперхолестеринемию, атеросклероз, ожирение печени, подагру, инсульт и болезни сердца.
В другом аспекте настоящего изобретения настоящее изобретение относится к функциональной оздоровительной продукции, содержащей в качестве активного ингредиента соединение, представленное формулой 1, его изомер или его фармацевтически приемлемую соль, для предупреждения или облегчения злокачественного новообразования, воспалительного заболевания или метаболического заболевания.
Термин «предупреждение», используемый в настоящем изобретении, относится к любому действию, которое подавляет или отсрочивает начало неврологического расстройства путем введения субъекту фармацевтической композиции по настоящему изобретению.
Термин «лечение», используемый в настоящем изобретении, относится к любому действию по улучшению или облегчению симптомов неврологического расстройства путем введения субъекту фармацевтической композиции по настоящему изобретению.
Когда композицию по настоящему изобретению используют в качестве лекарственного препарата, фармацевтическую композицию, содержащую в качестве активного ингредиента соединение, представленное формулой 1, его изомер или его фармацевтически приемлемую соль, можно составить и вводить в виде различных пероральных или парентеральных форм как следует согласно клиническому введению, но не всегда ограничивается этим.
Примерами составов для перорального введения являются таблетки, пилюли, твердые/мягкие капсулы, растворы, суспензии, эмульсии, сиропы, гранулы, эликсиры, пастилки и т. д. Эти составы в дополнение к активному ингредиенту могут включать разбавители (например, лактозу, декстрозу, сахарозу, маннит, сорбит, целлюлозу и/или глицин) и смазывающие вещества (например, диоксид кремния, тальк, стеарат и его соли магния или кальция и/или полиэтиленгликоль). Таблетки могут включать связующие агенты, такие как алюмосиликат магния, крахмальная паста, желатин, метилцеллюлоза, натрийкарбоксиметилцеллюлоза и/или поливинилпирролидон, и при необходимости разрыхляющие агенты, такие как крахмал, агароза, альгиновая кислота или ее натриевая соль или азеотропные смеси и/или абсорбенты, дополнительно могут быть включены красители, ароматизаторы и подсластители.
Когда соединение, представленное формулой 1, его изомер или его фармацевтически приемлемая соль применяют в качестве фармацевтической композиции для предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболическогох заболевания, его можно вводить как индивидуальное терапевтическое средство или его можно использовать в комбинации с другими применяемыми терапевтическими средствами.
Фармацевтическую композицию, содержащую в качестве активного ингредиента соединение, представленное формулой 1 или его фармацевтически приемлемую соль, можно вводить парентерально, и парентеральное введение включает подкожную инъекцию, внутривенную инъекцию, внутримышечную инъекцию или внутригрудную инъекцию.
Для получения соединения, представленного формулой 1, его изомера или его фармацевтически приемлемой соли в виде лекарственной формы для парентерального введения соединение, представленное формулой 1, его изомер или его фармацевтически приемлемую соль смешивают со стабилизатором или буферным агентом в воде с получением раствора или суспензии, которые затем готовят в виде ампул или флаконов. Композиция в настоящем документе может быть стерилизована и дополнительно содержит консерванты, стабилизаторы, смачивающиеся порошки или эмульгаторы, соли и/или буферы для регулирования осмотического давления и другие терапевтически полезные материалы, и композиция может быть составлена в соответствии с обычными способами, такими как диспергирование и гелеобразование.
Дозировка фармацевтической композиции, содержащей соединение, представленное формулой 1, в качестве активного ингредиента, для организма человека может быть определена в соответствии со способом приготовления, возрастом, массой тела, полом, способом введения, состоянием здоровья и тяжестью заболевания. Предпочтительная доза композиции по настоящему изобретению составляет 0,001~1000 мкг/мл в день, которую можно вводить перорально или парентерально несколько раз в день или, предпочтительно, 1~3 раза в день по решению врача или фармацевта.
Фармацевтическую композицию по настоящему изобретению можно использовать в виде отдельного препарата. Фармацевтическая композиция по настоящему изобретению также может быть приготовлена и использована в виде комбинированного препарата, дополнительно включающего одно или несколько других терапевтических агентов.
В другом аспекте настоящего изобретения настоящее изобретение относится к способу лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания, включающему стадию введения фармацевтической композиции нуждающемуся в этом субъекту. Фармацевтическая композиция относится к фармацевтической композиции для предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания, содержащая в качестве активного ингредиента соединение, представленное формулой 1, его изомер или его фармацевтически приемлемую соль.
В другом аспекте настоящего изобретения настоящее изобретение относится к применению фармацевтической композиции или композиции функциональной оздоровительной продукции, содержащей в качестве активного ингредиента соединение, представленное формулой 1, его изомер или его фармацевтически приемлемую соль, для предупреждения или лечения злокачественного новообразования, воспалительного заболевания или метаболического заболевания.
Метод осуществления изобретения
Далее настоящее изобретение подробно описано с помощью следующих примеров и экспериментальных примеров.
Однако следующие примеры и экспериментальные примеры предназначены только для иллюстрации настоящего изобретения, и содержание настоящего изобретения ими не ограничивается.
<Пример 1> Получение N-(3-([1,1'-бифенил]-4-ил)пропил)-4-бутоксибензолсульфонамида
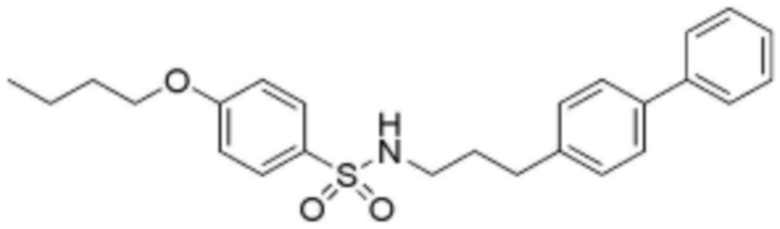
Стадия 1: Получение (E)-4-(2-изоциановинил)-1,1'-бифенил

Гидрид натрия (60% мас, 0,263 г, 6,59 ммоль) растворяли в безводном тетрагидрофуране (13 мл), при температуре 0°C добавляли по каплям диэтил(цианометил)фосфонат (0,977 мл, 6,04 ммоль). В реакционную смесь добавляли по каплям [1,1'-бифенил]-4-карбальдегид (1 г, 5,49 ммоль), затем перемешивали при комнатной температуре в течение 30 минут. После завершения реакции реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. В результате получали (E)-4-(2-изоциановинил)-1,1'-бифенил в виде твердого вещества белого цвета без дополнительной очистки.
Стадия 2: Получение 3-([1,1'-бифенил]-4-ил)пропаннитрила

Соединение (1,126 г, 5,49 ммоль), полученное на стадии 1 выше, растворяли в смеси этилацетат:метанол (1:1) (24 мл), при температуре 0°C добавляли по каплям Pd/C (10% мас), затем перемешивали при комнатной температуре в течение 12 часов в атмосфере водорода. После завершения реакции реакционную смесь фильтровали через целит и растворитель удаляли при пониженном давлении. В результате получали 3-([1,1'-бифенил]-4-ил)пропаннитрил в виде твердого вещества белого цвета без дополнительной очистки.
Стадия 3: Получение 3-([1,1'-бифенил]-4-ил)пропан-1-амина

Соединение 3-([1,1'-бифенил]-4-ил)пропаннитрил (1,137 г, 5,49 ммоль), синтезированное на стадии 2 выше, растворяли в безводном тетрагидрофуране (10 мл), при температуре 0°C добавляли по каплям 1 M литийалюминийгидрид (10,97 мл, 10,97 ммоль), затем перемешивали при комнатной температуре в течение 2 часов. Реакцию завершали добавлением по каплям воды и реакционную смесь фильтровали через фильтр и растворитель удаляли при пониженном давлении. В результате получали 3-([1,1'-бифенил]-4-ил)пропан-1-амин в виде липкого соединения желтого цвета без дополнительной очистки.
Стадия 4: Получение N-(3-([1,1'-бифенил]-4-ил)пропил)-4-бутоксибензолсульфонамида

4-Бутокси бензолсульфонилхлорид (40 мкл, 0,200 ммоль) растворяли в дихлорметане (1 мл), добавляли 3-([1,1'-бифенил]-4-ил)пропан-1-амин (127 мг, 0,601 ммоль), синтезированный на стадии 3 выше, затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (14,4 мг, 16%, твердое вещество белого цвета).
Соединения по примерам 2-7 получали аналогично способу, описанному на стадии 4 примера 1.
<Пример 8> Получение 4-(2-(4-бутоксифенилсульфонамидо)этил)бензойной кислоты

Стадия 1: Получение метил 4-(2-((4-бутоксифенил)сульфонамидо)этил)бензоата

Целевое соединение (6,7 мг, 28%, твердое вещество белого цвета) получали аналогично способу, описанному на стадии 4 примера 1.
Стадия 2: Получение 4-(2-(4-бутоксифенилсульфонамидо)этил)-бензойной кислоты

Соединение метил 4-(2-((4-бутоксифенил)сульфонамидо)этил)бензоат (23 мг, 0,059 ммоль), полученное на стадии 1 выше, растворяли в метаноле (0,5 мл), добавляли по каплям 4 M водный раствор гидроксида калия (0,147 мл, 0,588 ммоль), затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционную смесь нейтрализовали, используя 1 M водный раствор HCl. Реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (3,4 г, 15%, твердое вещество белого цвета).
Соединение примера 9 получали таким же способом, как описано на стадии 1 примера 8.
<Пример 10> Получение N-(2-(4-бензилпиперидин-1-ил)этил)-4-бутоксибензолсульфонамида

Стадия 1: Получение трет-бутил (2-(4-бензилпиперидин-1-ил)этил)карбамата

4-Бензилпиперидин (8 г, 45,6 ммоль) и карбонат калия (K2CO3) (9,46 г, 68,5 ммоль) растворяли в ДМФ (228 мл), затем перемешивали при комнатной температуре в течение 5 минут. В реакционную смесь добавляли по каплям трет-бутил(2-бромэтил)карбамат (12,27 г, 54,8 ммоль), затем перемешивали при температуре 60°C в течение 12 часов. К продукту реакции опять добавляли по каплям трет-бутил(2-бромэтил)карбамат (12,27 г, 54,8 ммоль), затем в течение 12 часов перемешивали при температуре 60°C. После завершения реакции добавляли по каплям 1 M водный раствор HCl, затем экстрагировали дихлорметаном. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением трет-бутил(2-(4-бензилпиперидин-1-ил)этил)карбамата (12,9 г, 89%, твердое вещество белого цвета).
Стадия 2: Получение 2⋅HCl соли 2-(4-бензилпиперидин-1-ил)этанамина

Трет-бутил (2-(4-бензилпиперидин-1-ил)этил)карбамат (12,9 г, 40,5 ммоль), полученный на стадии 1 выше, напрямую обрабатывали 4 M раствором HCl в диоксане (10,13 мл, 40,5 ммоль), затем перемешивали при комнатной температуре в течение 2 часов. После завершения реакции избыток HCl удаляли при пониженном давлении. В результате получали 2⋅HCl соль 2-(4-бензилпиперидин-1-ил)этанамина (8,9 г, 75%, твердое вещество белого цвета).
Стадия 3: Получение N-(2-(4-бензилпиперидин-1-ил)этил)-4-бутоксибензолсульфонамида

Целевое соединение (7,6 мг, 14%, прозрачное масло) получали аналогично способу, описанному на стадии 4 примера 1, с использованием соединения, полученного на стадии 2 выше.
Соединение примера 11 получали аналогично способу, описанному на стадии 4 примера 1.
<Пример 12> Получение 4-бутокси-N-(3-оксо-3-фенилпропил)бензолсульфонамида

Стадия 1: Получение (4-бутокси-N-(3-гидрокси-3-фенилпропил)бензолсульфонамида
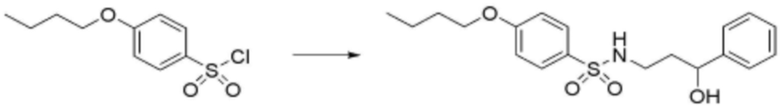
Целевое соединение получали аналогично способу, описанному на стадии 4 примера 1.
Стадия 2: Получение 4-бутокси-N-(3-оксо-3-фенилпропил)бензолсульфонамида

Соединение (30 мг, 0,121 ммоль), полученное на стадии 1 выше, растворяли в дихлорметане (0,5 мл), добавляли по каплям оксид марганца (IV) (31,1 мг, 0,358 ммоль), затем перемешивали при комнатной температуре в течение 12 часов. После завершения реакции реакционную смесь фильтровали через целит и растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (9,2 мг, 71%, твердое вещество белого цвета).
Соединение примера 13 получали аналогично способу, описанному на стадии 4 примера 1.
<Пример 14> Получение 4-(3-(диметиламино)пропокси)-N-фенетилбензолсульфонамида

Стадия 1: Получение N,N-диметил-3-феноксипропан-1-амина

Фенол (600 мг, 6,38 ммоль), 3-(диметиламино)пропан-1-ол (0,88 мл, 7,44 ммоль) и трифенилфосфин (2,508 г, 9,56 ммоль) растворяли в тетрагидрофуране (12 мл) при температуре 0°C, добавляли по каплям раствор диэтил азодикарбоксилата (DEAD) (4,34 мл, 9,56 ммоль), затем перемешивали при комнатной температуре в течение 12 часов в атмосфере азота. После завершения реакции растворитель удаляли при пониженном давлении. Реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением N,N-диметил-3-феноксипропан-1-амина (922 мг, 81%, масло желтого цвета).
Стадия 2: Получение 4-(3-(диметиламино)пропокси)бензол-1-сульфонилхлорида
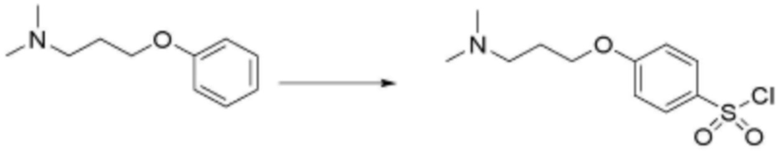
N,N-диметил-3-феноксипропан-1-амин (922 мг, 5,14 ммоль), полученный на стадии 1 выше, растворяли в дихлорметане (6 мл), добавляли по каплям хлорсульфоновую кислоту (1,37 мл, 20,57 ммоль), затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции добавляли лед. Реакционную смесь экстрагировали дихлорметаном, и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. В результате получали 4-(3-(диметиламино)пропокси)бензол-1-сульфонил хлорид (451 мг, 32%, твердое вещество желтого цвета) без дополнительной очистки.
Стадия 3: Получение 4-(3-(диметиламино)пропокси)-N-фенетилбензолсульфонамида
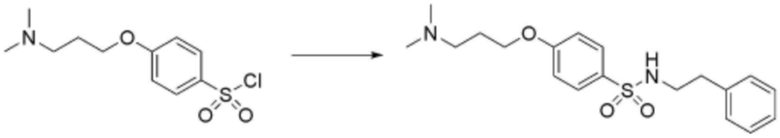
4-(3-(Диметиламино)пропокси)бензол-1-сульфонил хлорид (100 мг, 0,318 ммоль), полученный на стадии 2 выше растворяли в дихлорметане (2 мл), добавляли по каплям 2-фенилэтанамин (116 мг, 0,955 ммоль) и карбонат калия (K2CO3) (48,4 мг, 0,35 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (88 мг, 75%, прозрачное масло).
Соединения по примерам 15 и 16 получали аналогично способу, описанному на стадии 3 примера 14.
<Пример 17> Получение 4-(2-(4-(3-(диметиламино)пропокси)фенилсульфонамидо)этил)бензойной кислоты

Стадия 1: Получение метил 4-(2-(4-(3-(диметиламино)пропокси)фенилсульфонамидо)этил)бензоата

Целевое соединение получали аналогично способу, описанному на стадии 3 примера 14.
Стадия 2: Получение 4-(2-(4-(3-(диметиламино)пропоксифенилсульфонамидо)этил)бензойной кислоты
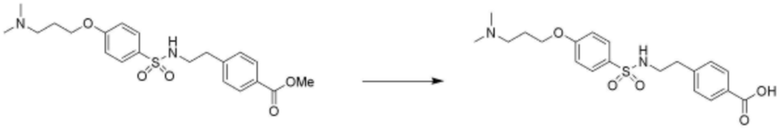
Соединение (58 мг, 0,138 ммоль), полученное на стадии 1 выше, растворяли в метаноле (1 мл), добавляли по каплям 4 M водный раствор гидроксида калия (0,69 мл, 2,76 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь нейтрализовали 1 M водным раствором HCl, экстрагировали дихлорметаном, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ТСХ с получением целевого соединения (1,7 мг, 3%, твердое вещество белого цвета).
Соединение примера 18 получали аналогично способу, описанному на стадии 3 примера 14.
<Пример 19> Получение 4-бутокси-N-(3-(нафталин-1-ил)пропил)бензолсульфонамида
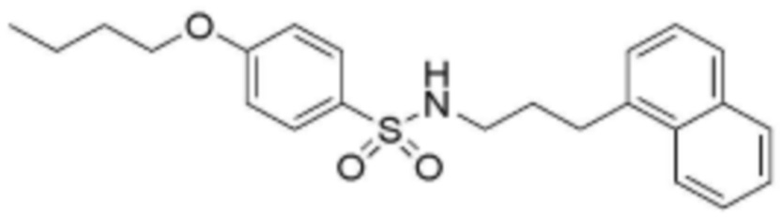
Стадия 1: Получение трет-бутил(3-(нафталин-1-ил)пропил)карбамата

Трет-бутилаллилкарбамат (1 г, 6,36 ммоль) растворяли в тетрагидрофуране (6 мл), добавляли по каплям 9-BBN (10 ммоль), растворенный в 0,5 M тетрагидрофуране, затем перемешивали при комнатной температуре в течение 2,5 часов. В реакционную смесь добавляли по каплям 1-йоднафталин (0,925 мл, 6,36 ммоль), PdCl2(dppf)-CH2Cl2 (308 мг, 0,377 ммоль) и 1 M водный раствор гидроксида натрия (10 мл), затем перемешивали при комнатной температуре в течение 3 часов. Добавляли по каплям PdCl2(dppf)-CH2Cl2 (55 мг, 0,07 ммоль), затем перемешивали при комнатной температуре в течение 14 часов. После завершения реакции добавляли по каплям насыщенный водный раствор хлорида аммония, и реакционную смесь экстрагировали этилацетатом, сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Затем к смеси тетрагидрофурана (20 мл), 15% водного раствора гидроксида натрия (5 мл) и 30% перекиси водорода (10 мл) при температуре 0°C добавляли по каплям соединение (1,6 г), полученное выделением и очисткой реакционной смеси с помощью ЖХСД, затем перемешивали при той же температуре в течение 2 часов. После завершения реакции добавляли по каплям этилацетат, и реакционную смесь промывали перенасыщенным водным раствором гидрокарбоната натрия и насыщенным солевым раствором, сушили над безводным сульфатом магния, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением трет-бутил(3-(нафталин-1-ил)пропил)карбамата (1,4 г, 77%).
Стадия 2: Получение 3-(нафталин-1-ил)пропан-1-амина

Трет-бутил(3-(нафталин-1-ил)пропил)карбамат (1,4 г, 4,9 ммоль), полученный на стадии 1 выше, растворяли в дихлорметане (20 мл), добавляли по каплям трифторуксусную кислоту (10 мл, 130 ммоль), затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционную смесь нейтрализовали путем добавления по каплям 10% водного раствора гидроксида натрия. Реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении с получением 3-(нафталин-1-ил)пропан-1-амина (925 мг, 100%).
Стадия 3: Получение 4-бутокси-N-(3-(нафталин-1-ил)пропил)бензолсульфонамида

Целевое соединение (66 мг, 82%, прозрачное твердое вещество) получали аналогично способу, описанному на стадии 4 примера 1 с использованием соединения, полученного на стадии 2 выше.
Соединение примера 20 получали аналогично способу, описанному на стадии 4 примера 1.
<Пример 21> Получение N-(3-(1H-индол-3-ил)пропил)-4-(2-(диметиламино)этокси)бензолсульфонамида

Стадия 1: Получение N,N-диметил-2-феноксиэтан-1-амина

Фенол (500 мг, 5,31 ммоль), 2-(диметиламино)этан-1-ол (521 мг, 5,84 ммоль) и трифенилфосфин (2,089 г, 797 ммоль) при температуре 0°C растворяли в тетрагидрофуране (20 мл), добавляли по каплям раствор диэтил азодикарбоксилата (DEAD) (3,61 мл, 7,97 ммоль), затем перемешивали при комнатной температуре в течение 12 часов в атмосфере азота. После завершения реакции растворитель удаляли при пониженном давлении. Реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением N,N-диметил-2-феноксиэтан-1-амина (870 мг, 99%, твердое вещество желтого цвета).
Стадия 2: Получение 4-(2-(диметиламино)этокси)бензолсульфонилхлорида

N,N-диметил-3-феноксиэтан-1-амин (533 мг, 3,23 ммоль), полученный на стадии 1 выше, растворяли в дихлорметане (3 мл), при температуре 0°C добавляли по каплям хлорсульфоновую кислоту (0,858 мл, 12,90 ммоль), затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции добавляли лед. Реакционную смесь экстрагировали дихлорметаном и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. В результате получали 4-(2-(диметиламино)этокси)бензолсульфонилхлорид (622 мг, 73%, масло желтого цвета) получали без дополнительной очистки.
Стадия 3: Получение N-(3-(1H-индол-3-ил)пропил)-4-(2-(диметиламино)этокси)бензолсульфонамида

4-(2-(Диметиламино)этокси)бензолсульфонил хлорид (45 мг, 0,171 ммоль), полученное на стадии 2 выше, растворяли в дихлорметане, добавляли по каплям 3-(1H-индол-3-ил)пропан-1-амин (131 мг, 0,751 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (11 мг, 17%, твердое вещество желтого цвета).
Соединение примера 22 получали аналогично способу, описанному в примере 14.
Соединения по примерам 23-25 получали аналогично способу, описанному на стадии 4 примера 1.
<Пример 26> Получение N-(3-(1H-индол-3-ил)пропил)-4-(изопентилокси)бензолсульфонамида


4-(Изопентилокси)бензолсульфонилхлорид (20 мг, 0,076 ммоль) растворяли в дихлорметане (1,5 мл), добавляли по каплям 3-(1H-индол-3-ил)пропан-1-амин (30 мг, 0,172 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (26 мг, 85%, твердое вещество желтого цвета).
<Пример 27> Получение N-(3-(1H-индол-3-ил)пропил)-4-(пентилокси)бензолсульфонамида


4-(Пентилокси)бензолсульфонил хлорид (20 мг, 0,076 ммоль) растворяли в дихлорметане, добавляли по каплям 3-(1H-индол-3-ил)пропан-1-амин (30 мг, 0,172 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (25 мг, 81%, твердое вещество белого цвета).
Соединение примера 28 получали аналогично способу, описанному на стадии 4 примера 1.
<Пример 29> Получение 4-бутокси-N-(2-(5-гидрокси-1H-индол-3-ил)этил)бензолсульфонамида


4-Бутокси бензолсульфонилхлорид (50 мг, 0,201 ммоль) растворяли в дихлорметане (1 мл), добавляли по каплям 3-(2-аминоэтил)-1H-индол-5-олгидрохлорид (128 мг, 0,603 ммоль) и триэтиламин (0,084 мл, 0,603 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (10,2 мг, 13%, твердое вещество белого цвета).
<Пример 30> Получение N-(3-(1H-индол-3-ил)пропил)-4-(3-морфолинопропокси)бензолсульфонамида

Стадия 1: Получение 4-(3-феноксипропил)морфолина
Фенол (600 мг, 6,38 ммоль), 4-(3-гидроксипропил) морфолин (1,11 г, 7,65 ммоль) и трифенилфосфин (2,5 г, 9,56 ммоль) при температуре 0°C растворяли в тетрагидрофуране (16 мл), добавляли по каплям раствор диэтил азодикарбоксилата (DEAD) (4,34 мл, 9,56 ммоль), затем перемешивали при комнатной температуре в течение 12 часов в атмосфере азота. После завершения реакции растворитель удаляли при пониженном давлении. Реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением 4-(3-феноксипропил)морфолина (1,41 г, 100%, прозрачное масло).
Стадия 2: Получение 4-(3-морфолинопропокси)бензол-1-сульфонилхлорида

4-(3-Феноксипропил)морфолин (1,41 г, 6,38 ммоль), полученный на стадии 1 выше, растворяли в дихлорметане (12 мл), при температуре 0°C добавляли хлорсульфоновую кислоту (1,69 мл, 25,5 ммоль), затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции добавляли лед. Реакционную смесь экстрагировали дихлорметаном и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и затем растворитель удаляли при пониженном давлении. В результате получали 4-(3-морфолинопропокси)бензол-1-сульфонилхлорид (2,22 г, 67%, твердое вещество желтого цвета) без дополнительной очистки.
Стадия 3: Получение N-(3-(1H-индол-3-ил)пропил)-4-(3-морфолинопропокси)бензолсульфонамида

4-(3-Морфолинопропокси)бензол-1-сульфонилхлорид (150 мг, 0,469 ммоль), полученный на стадии 2 выше, растворяли в дихлорметане (1,5 мл), добавляли по каплям 3-(1H-индол-3-ил)пропан-1-амин (245 мг, 1,407 ммоль) и карбонат калия (194 мг, 1,407 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь экстрагировали дихлорметаном и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (10,6 мг, 5%, твердое вещество желтого цвета).
<Пример 31> Получение N-(3-(1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида
Стадия 1: Получение 4-(3-бромпропокси)бензол-1-сульфонил хлорида

(3-Бромпропокси)бензол (1 г, 4,65 ммоль) растворяли в дихлорметане (15 мл), при температуре 0°C добавляли по каплям хлорсульфоновую кислоту (1,236 мл, 18,60 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции добавляли лед. Реакционную смесь экстрагировали дихлорметаном и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и затем растворитель удаляли при пониженном давлении. В результате получали 4-(3-бромпропокси)бензол-1-сульфонилхлорид (1,1 г, 38%, твердое вещество желтого цвета) получали без дополнительной очистки.
Стадия 2: Получение N-(3-(1H-индол-3-ил)пропил)-4-(3-бромпропокси)бензолсульфонамида

4-(3-Бромпропокси)бензол-1-сульфонил хлорид (538 мг, 1,716 ммоль), полученный на стадии 1 выше, растворяли в дихлорметане (5 мл), добавляли по каплям 3-(1H-индол-3-ил)пропан-1-амин (897 мг, 5,15 ммоль) и карбонат калия (K2CO3) (711 мг, 5,15 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь экстрагировали дихлорметаном и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением N-(3-(1H-индол-3-ил)пропил)-4-(3-бромпропокси)бензолсульфонамида (230 мг, 29%, твердое вещество белого цвета).
Стадия 3: Получение N-(3-(1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

N-(3-(1H-индол-3-ил)пропил)-4-(3-бромпропокси)бензолсульфонамид (175 мг, 0,388 ммоль), полученный на стадии 2 выше, растворяли в этаноле (1,5 мл), добавляли по каплям 1-метилпиперазин (0,172 мл, 1,551 ммоль) и карбонат калия (K2CO3) (80 мг, 0,582 ммоль), затем перемешивали при температуре 70°C в течение 12 часов. После завершения реакции реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (66 мг, 36%, твердое вещество белого цвета).
Соединения по примерам 23-25 получали аналогично способу, описанному на стадии 4 примера 1.
Соединения по примерам 34 и 35 получали аналогично способу, описанному в примере 30.
Соединение примера 36 получали аналогично способу, описанному в примере 31.
Соединения по примерам 37-40 получали аналогично способу, описанному в примере 14.
Соединения по примерам 41 и 42 получали аналогично способу, описанному в примере 30.
Соединения по примерам 43-45 получали аналогично способу, описанному в примере 31.
<Пример 46> Получение 4-(3-(диметиламино)пропокси)-N-(3-(1-метил-1H-индол-3-ил)пропил)бензолсульфонамида
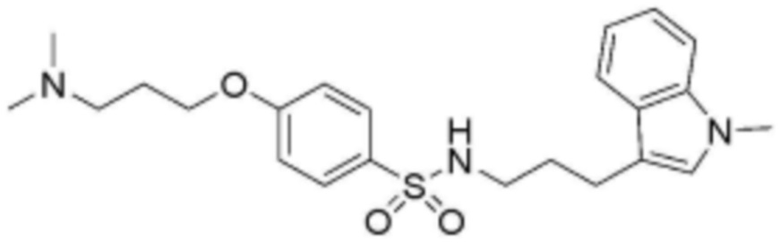
Стадия 1: Получение N-(3-(1H-индол-3-ил)пропил)-4-(3-(диметиламино)пропокси)бензолсульфонамид
N-(3-(1H-индол-3-ил)пропил)-4-(3-(диметиламино)пропокси)бензолсульфонамид получали аналогично способу, описанному в примере 14.
Стадия 2: Получение 4-(3-(диметиламино)пропокси)-N-(3-(1-метил-1H-индол-3-ил)пропил)бензолсульфонамида

N-(3-(1H-индол-3-ил)пропил)-4-(3-(диметиламино)пропокси)бензолсульфонамид (150 мг, 0,981 ммоль), полученный на стадии 1 выше, растворяли в ДМФ (1 мл), добавляли по каплям гидрид натрия (60%) (51,5 мг, 1,287 ммоль) при температуре -20°C, затем перемешивали в течение 30 минут. При той же температуре медленно по каплям добавляли йодметан (23 мкл, 0,368 ммоль), растворенный в ДМФ (0,1 мл), затем перемешивали при комнатной температуре в течение 12 часов. После завершения реакции реакционную смесь экстрагировали этилацетатом, и нейтрализовали с использованием перенасыщенного водного раствора хлорида аммония. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (13,7 мг, 8%, прозрачное масло).
<Пример 47> Получение N-(3-(1H-индол-3-ил)пропил)-4-бромбензолсульфонамида

Стадия 1: Получение 4-бромбензол-1-сульфонилхлорида

Бромбензол (0,067 мл, 0,637 ммоль) растворяли в дихлорметане (2 мл), при температуре 0°C добавляли по каплям хлорсульфоновую кислоту (0,169 мл, 2,55 ммоль), затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции добавляли лед. Реакционную смесь экстрагировали дихлорметаном и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и затем растворитель удаляли при пониженном давлении. В результате получали 4-бромбензол-1-сульфонилхлорид (74 мг, 45%, твердое вещество белого цвета) получали без дополнительной очистки.
Стадия 2: Получение N-(3-(1H-индол-3-ил)пропил)-4-бромбензолсульфонамида

4-Бромбензол-1-сульфонил хлорид (74 мг, 0,29 ммоль), полученный на стадии 1 выше, в дихлорметане (1 мл), добавляли по каплям 3-(1H-индол-3-ил)пропан-1-амин (151 мг, 0,869 ммоль) и карбонат калия (K2CO3) (120 мг, 0,869 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь экстрагировали дихлорметаном, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (71 мг, 62%, твердое вещество белого цвета).
Соединения по примерам 48-54 получали аналогично способу, описанному в примере 31.
Соединение примера 55 получали аналогично способу, описанному в примере 30.
Соединения по примерам 56-58 получали аналогично способу, описанному в примере 31.
Соединения по примерам 59 и 60 получали аналогично способу, описанному в примере 30.
Соединение примера 61 получали аналогично способу, описанному в примере 31.
Соединение примера 62 получали аналогично способу, описанному в примере 30.
Соединения по примерам 63-71 получали аналогично способу, описанному в примере 31.
<Пример 72> Получение 4-(3-((3S,5R)-3,5-диметилпиперазин-1-ил)пропокси)-N-(3-(5-фтор-1H-индол-3-ил)пропил)бензолсульфонамида
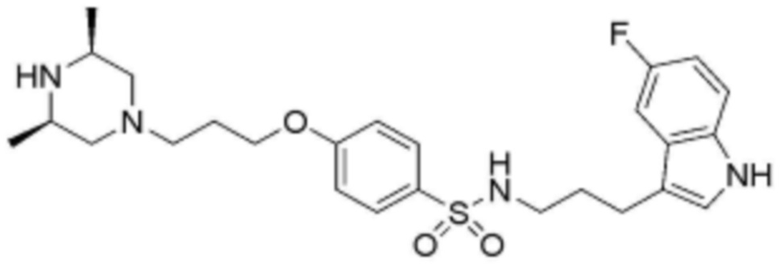
Стадия 1: Получение (2S,6R)-трет-бутил 4-(3-(4-(N-(3-(5-фтор-1H-индол-3-ил)пропил)сульфамоил)фенокси)пропил)-2,6-диметилпиперазин-1-карбоксилата
(2S,6R)-трет-бутил 4-(3-(4-(N-(3-(5-фтор-1H-индол-3-ил)пропил)сульфамоил)фенокси)пропил)-2,6-диметилпиперазин-1-карбоксилат получали аналогично способу, описанному в примере 31.
Стадия 2: Получение 4-(3-((3S,5R)-3,5-диметилпиперазин-1-ил)пропокси)-N-(3-(5-фтор-1H-индол-3-ил)пропил) бензолсульфонамида

(2S,6R)-трет-бутил 4-(3-(4-(N-(3-(5-фтор-1H-индол-3-ил)пропил)сульфамоил)фенокси)пропил)-2,6-диметилпиперазин-1-карбоксилат (80 мг, 0,133 ммоль), полученный на стадии 1 выше, растворяли в дихлорметане (1 мл), при комнатной температуре добавляли трифторуксусную кислоту (0,2 мл, 2,65 ммоль), затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции добавляли лед. Реакционную смесь экстрагировали этилацетатом и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (5,2 мг, 7%, твердое вещество белого цвета).
Соединения по примерам 73-76 получали аналогично способу, описанному в примере 31.
<Пример 77> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-(метиламино)азетидин-1-ил)пропокси)бензолсульфонамида
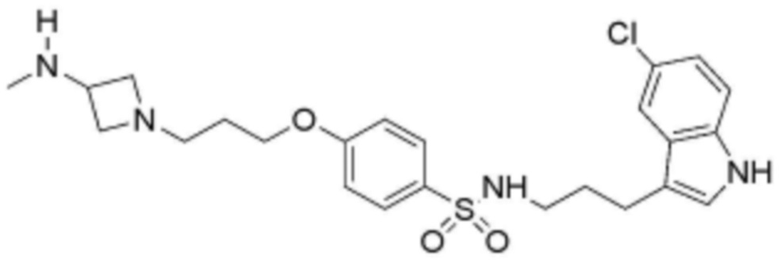
Стадия 1: Получение трет-бутил(1-(3-(4-(N-(3-(5-хлор-1H-индол-3-ил)пропил)сульфамоил)фенокси)пропил)азетидин-3-ил)(метил)карбамата
Трет-бутил(1-(3-(4-(N-(3-(5-хлор-1H-индол-3-ил) пропил)сульфамоил)фенокси)пропил)азетидин-3-ил(метил)карбамат получали аналогично способу, описанному в примере 31.
Стадия 2: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-(метиламино)азетидин-1-ил)пропокси)бензолсульфонамида

Трет-бутил(1-(3-(4-(N-(3-(5-хлор-1H-индол-3-ил)пропил)сульфамоил)фенокси)пропил)азетидин-3-ил)(метил)карбамат (38 мг, 0,064 ммоль) растворяли в дихлорметане (0,5 мл), используя трет-бутилазетидин-3-ил(метил)карбамат, полученный на стадии 1 выше, при комнатной температуре добавляли по каплям трифторуксусную кислоту (0,2 мл, 2,61 ммоль), затем перемешивали при комнатной температуре в течение 2 часов. После завершения реакции добавляли лед. Реакционную смесь экстрагировали этилацетатом и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (7 мг, 22%, твердое вещество желтого цвета).
Соединение примера 78 получали аналогично способу, описанному в примере 31.
<Пример 79> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-6-(3-(пиперазин-1-ил)пропокси)пиридин-3-сульфонамида

Стадия 1: Получение трет-бутил-4-(3-((5-бромпиридин-2-ил)окси)пропилпиперазин-1-карбоксилата

5-Бром-2-фторпиридин (0,326 мл, 2,84 ммоль) растворяли в ДМФ (12 мл), при температуре 0°C добавляли по каплям гидрид натрия 60% мас (0,159 г, 3,98 ммоль), затем перемешивали в течение 5 минут. Добавляли по каплям трет-бутил-4-(3-гидроксипропил)пиперазин-1-карбоксилат, затем перемешивали при температуре 70°C в течение 6 часов. После завершения реакции добавляли лед. Реакционную смесь экстрагировали дихлорметаном и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. В результате получали трет-бутил-4-(3-((5-бромпиридин-2-ил)окси)пропилпиперазин-1-карбоксилат (1,35 г, 110%) без дополнительной очистки.
Стадия 2: Получение трет-бутил 4-(3-((5-((4-метоксибензил)тио)пиридин-2-ил)окси)пропил)пиперазин-1-карбоксилата
Трет-бутил-4-(3-((5-бромпиридин-2-ил)окси)пропил пиперазин-1-карбоксилат (0,52 г, 1,3 ммоль), полученный на стадии 1 выше, растворяли в 1,4-диоксане (12 мл), добавляли по каплям (4-метоксифенил)метантиол (0,199 мл, 1,23 ммоль) и N-этил-N-изопропилпропан-2-амин (0,681 мл, 3,90 ммоль), затем давали взаимодействовать в течение 5 минут в атмосфере азота. Добавляли по каплям Pd2(dba)3 (9,91 мг, 0,032 ммоль), Xantphos (0,038 г, 0,065 ммоль) и трет-бутил-4-(3-гидроксипропил)пиперазин-1-карбоксилат, затем давали взаимодействовать при температуре 150 °C в течение 30 минут в микроволновой печи Biotage. После завершения реакции реакционную смесь экстрагировали дихлорметаном и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. В результате получали трет-бутил 4-(3-((5-((4-метоксибензил)тио)пиридин-2-ил)окси)пропил)пиперазин-1-карбоксилат (0,51 г, 51,8%) без дополнительной очистки.
Стадия 3: Получение трет-бутил 4-(3-((5-(хлорсульфонил)пиридин-2-ил)окси)пропил)пиперазин-1-карбоксилата

Трет-бутил 4-(3-((5-((4-метоксибензил)тио)пиридин-2-ил)окси)пропил)пиперазин-1-карбоксилат (0,469 г, 0,99 ммоль), полученный на стадии 2 выше, растворяли в уксусной кислоте (8 мл) и воде (2 мл), добавляли по каплям N-хлорсукцинимид (0,529 г, 3,96 ммоль), затем давали взаимодействовать при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь экстрагировали дихлорметаном и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. В результате получали трет-бутил 4-(3-((5-(хлорсульфонил)пиридин-2-ил)окси)пропил)пиперазин-1-карбоксилат (0,25 г, 60%) без дополнительной очистки.
Стадия 4: Получение трет-бутил 4-(3-((5-(N-(3-(5-хлор-1H-индол-3-ил)пропил)сульфамоил)пиридин-2-ил)окси)пропил)пиперазин-1-карбоксилата

Трет-бутил 4-(3-((5-(хлорсульфонил)пиридин-2-ил)окси)пропил)пиперазин-1-карбоксилат (0,1 г, 0,238 ммоль), полученный на стадии 3 выше, растворяли в ДМФ (2 мл), добавляли по каплям 3-(5-хлор-1H-индол-3-ил)пропан-1-амин (0,055 г, 0,262 ммоль) и карбонат калия (0,066 г, 0,476 ммоль), затем перемешивали при комнатной температуре в течение 2 часов. После завершения реакции реакционную смесь экстрагировали дихлорметаном и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ТСХ с получением трет-бутил 4-(3-((5-(N-(3-(5-хлор-1H-индол-3-ил)пропил)сульфамоил)пиридин-2-ил)окси)пропил)пиперазин-1-карбоксилата (18 мг, 12,8%).
Стадия 5: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-6-(3-(пиперазин-1-ил)пропокси)пиридин-3-сульфонамида

4-(3-((5-(N-(3-(5-хлор-1H-индол-3-ил)пропил) сульфамоил)пиридин-2-ил)окси)пропил)пиперазин-1-карбоксилат (18 мг, 0,034 ммоль), полученный на стадии 4 выше, растворяли в тетрагидрофуране (1 мл), при комнатной температуре добавляли по каплям трифторуксусную кислоту (0,5 мл, 6,49 ммоль), затем перемешивали при комнатной температуре в течение 2 часов. После завершения реакции добавляли лед. Реакционную смесь экстрагировали этилацетатом и нейтрализовали с использованием перенасыщенного раствора карбоната натрия. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и затем растворитель удаляли при пониженном давлении. Затем реакционную смесь разделяли и очищали с помощью ТСХ с получением целевого соединения (4,5 мг, 30%).
Соединения по примерам 80-84 получали аналогично способу, описанному в примере 31.
<Пример 85> Получение N-(3-(1H-индол-3-ил)циклогексил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Стадия 1: Получение 3-(1H-индол-3-ил)циклогексан-1-она

1H-индол (1 г, 8,54 ммоль) и циклогекс-2-енон (1,64 г, 17,1 ммоль) растворяли в CH3CN (30 мл), добавляли Sc(OTf)3 (0,42 г, 0,854 ммоль), затем перемешивали при температуре 30°C в течение 3 часов в атмосфере N2. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали с помощью преп. HPLC с получением целевого соединения (805 мг, 44%).
Стадия 2: Получение 3-(1H-индол-3-ил)циклогексан-1-амина

3-(1H-индол-3-ил)циклогексан-1-он (800 мг, 3,75 ммоль), полученное на стадии 1 выше, растворяли в дихлорэтане (10 мл) и уксусной кислоте (0,5 мл), при комнатной температуре добавляли ацетат аммония (8,67 г, 113 ммоль), затем перемешивали в течение 2 часов. Затем при комнатной температуре добавляли триацетоксиборгидрид натрия (3,18 г, 15,0 ммоль), потом перемешивали в течение 5 часов. После завершения реакции реакционную смесь, полученную концентрированием смеси при пониженном давлении, разделяли и очищали с помощью ЖХСД с получением целевого соединения (250 мг, 31%, твердое вещество желтого цвета).
Стадия 3: Получение N-(3-(1H-индол-3-ил)циклогексил)-4-(3-бромпропокси)бензолсульфонамида

Целевое соединение (100 мг, 46%, твердое вещество белого цвета) получали аналогично способу, описанному на стадии 2 примера 31 с использованием 3-(1H-индол-3-ил)циклогексан-1-амина (105 мг, 0,491 ммоль), полученного на стадии 2 выше, и 4-(3-бромпропокси)бензолсульфонилхлорида (140 мг, 0,446 ммоль), полученного аналогично способу, описанному на стадии 2 примера 31.
Стадия 4: Получение N-(3-(1H-индол-3-ил)циклогексил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Целевое соединение (21,6 мг, выход 69%) в виде твердого вещества белого цвета аналогично способу, описанному на стадии 3 примера 31 с использованием N-(3-(1H-индол-3-ил)циклогексил)-4-(3-бромпропокси)бензолсульфонамида (30 мг, 0,061 ммоль), полученного на стадии 3 выше, и 1-метилпиперазина (24,5 мг, 0,244 ммоль).
<Пример 86> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)нафталин-1-сульфонамида
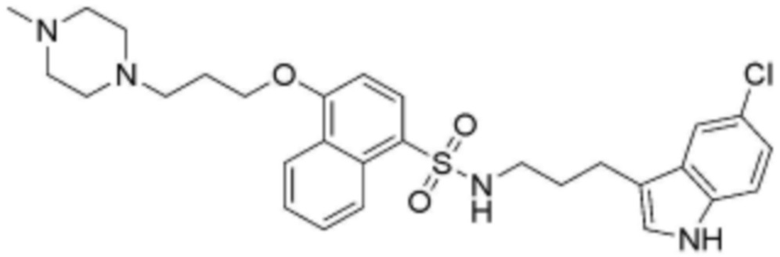
Стадия 1: Получение натриевой (I) соли 4-(3-бромпропокси)нафталин-1-сульфоната

4-Гидроксинафталин-1-сульфонат, натриевую (I) соль (0,3 г, 1,218 ммоль) растворяли в этаноле (2,5 мл), добавляли 1,3-дибромпропан (0,211 мл, 2,071 ммоль), тетрабутиламмоний сульфат (0,033 г, 0,097 ммоль) и водный раствор гидроксида калия (1,56 мл, 1,218 ммоль), затем кипятили с обратным холодильником в течение 3 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении, опять растворяли в этанол и вновь концентрировали. После добавления в реакционный раствор горячего метанола нерастворимое вещество фильтровали и фильтрат концентрировали. Полученный твердый продукт промывали дихлорметаном и фильтровали с получением целевого соединения (0,4 г, 99%, твердое вещество белого цвета).
Стадия 2: Получение 4-(3-бромпропокси)нафталин-1-сульфонилхлорида

К соединению (0,2 г, 0,545 ммоль), полученному на стадии 1 выше, добавляли тионилхлорид (0,286 мл, 3,92 ммоль) и диметилформамид (1,91 мкл), затем кипятили с обратным холодильником в течение 4 часов. После завершения реакции температуру понижали до комнатной температуры, и реакционную смесь разбавляли этилацетатом и промывали водой и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали, и использовали на следующей стадии без дополнительной очистки.
Стадия 3: Получение 4-(3-бромпропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)нафталин-1-сульфонамида

Целевое соединение получали аналогично способу, описанному на стадии 2 примера 31.
Стадия 4: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)нафталин-1-сульфонамида
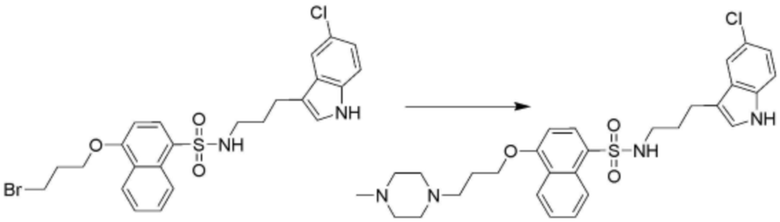
Целевое соединение получали аналогично способу, описанному на стадии 3 примера 31.
Соединение примера 87 получали аналогично способу, описанному в примере 31.
<Пример 88> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(пиперидин-4-ил)пропил)амино)бензолсульфонамида
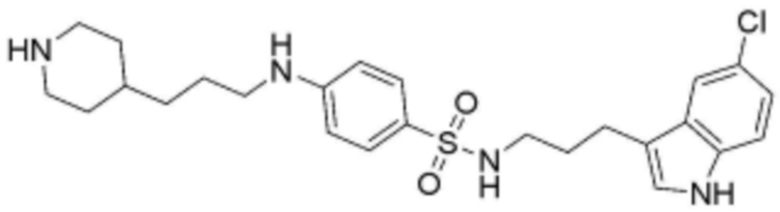
Стадия 1: Получение трет-бутил-4-(3-оксопропил)пиперидин-1-карбоксилата

Трет-бутил-4-(3-гидроксипропил)пиперидин-1-карбоксилат (0,3 г, 1,233 ммоль) растворяли в дихлорметане (6 мл), при температуре 0°C добавляли периодинан Десса-Мартина (0,680 г, 1,603 ммоль), затем перемешивали при комнатной температуре в течение 4 часов. После завершения реакции реакционный раствор фильтровали и фильтрат промывали водным раствором гидрокарбоната натрия и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Концентрат очищали с помощью ЖХСД с получением целевого соединения (0,24 г, 79%, прозрачное масло).
Стадия 2: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-нитробензолсульфонамида

Целевое соединение получали аналогично способу, описанному на стадии 2 примера 31.
Стадия 3: Получение 4-амино-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамида
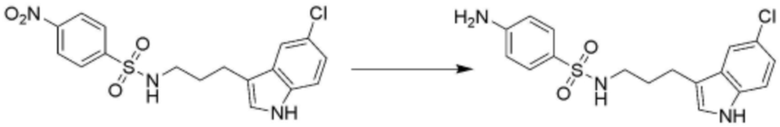
Соединение (0,72 г, 1,840 ммоль), полученное на стадии 2 выше, растворяли в этилацетате (12 мл), добавляли дигидрат хлорида олова (II) (4,15 г, 18,40 ммоль), затем нагревали при температуре 60°C в течение 5 часов. После завершения реакции в реакционный раствор добавляли аммиачную воду для установления pH 5, и добавляли карбонат натрия для установления pH 7. Реакционный раствор фильтровали через целит, и фильтрат концентрировали при пониженном давлении, который использовали на следующей стадии без дополнительной очистки.
Стадия 4: Получение трет-бутил 4-(3-((4-(N-(3-(5-хлор-1H-индол-3-ил)пропил)сульфамоил)фенил)амино)пропил)пиперидин-1-карбоксилата
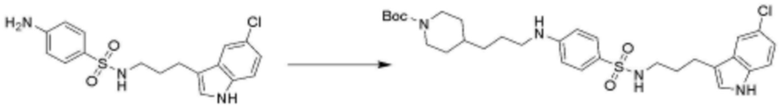
Соединение (0,05 г, 0,137 ммоль), полученное на стадии 3 выше, растворяли в дихлорэтане (1 мл), добавляли соединение (0,046 г, 0,192 ммоль), полученное на стадии 1 выше, и уксусную кислоту (0,024 мл, 0,412 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. Добавляли триацетоксиборгидрид натрия (0,087 г, 0,412 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь разбавляли дихлорметаном и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали, очищали с помощью преп. HPLC и нейтрализовали путем добавления водного раствора гидрокарбоната натрия с получением целевого соединения (0,017 г, 5%, твердое вещество желтого цвета).
Стадия 5: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(пиперидин-4-ил)пропил)амино)бензолсульфонамида

Целевое соединение получали аналогично способу, описанному на стадии 5 примера 79.
<Пример 89> Получение N-(3-(1H-индол-3-ил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида
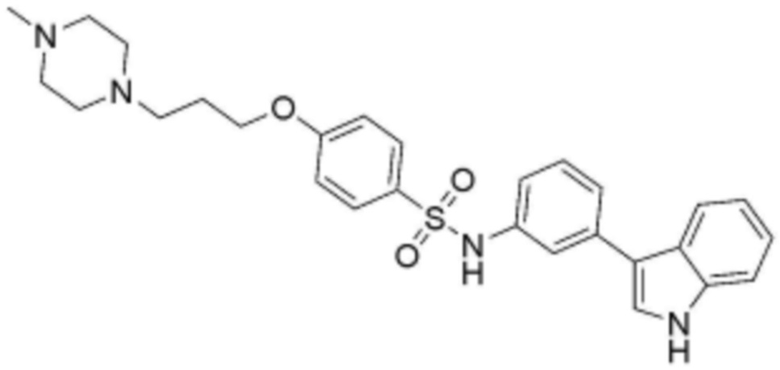
Стадия 1: Получение 3-(3-нитрофенил)-1H-индола
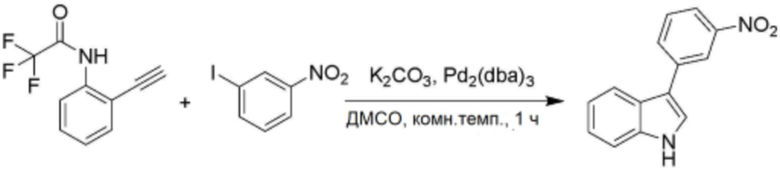
N-(2-этинилфенил)-2,2,2-трифторацетамид и 1-йод-3-нитробензол растворяли в ДМСО, обрабатывали избыточным количеством K2CO3 в присутствии каталитического количества Pd2(dba)3 с получением целевого соединения 3-(3-нитрофенил-1H-индол).
Стадия 2: Получение 3-(1H-индол-3-ил)анилина

Соединение (0,047 г, 0,197 ммоль), полученное на стадии 1 выше, растворяли в метаноле (2 мл), добавляли Pd/C (0,004 г, 3,76 мкмоль), затем перемешивали при комнатной температуре в течение 3 часов в атмосфере водорода. После завершения реакции реакционный раствор фильтровали через целит и промывали метанолом. Фильтрат концентрировали при пониженном давлении и использовали на следующей стадии без дополнительной очистки.
Стадия 3: Получение N-(3-(1H-индол-3-ил)фенил)-4-(3-бромпропокси)бензолсульфонамида

Соединение (0,041 г, 0,197 ммоль), полученное на стадии 2 выше, растворяли в пиридине (1 мл), при температуре добавляли 0°C соединение (0,068 г, 0,217 ммоль), полученное на стадии 1 примера 31, затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционную смесь разбавляли этилацетатом и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали и очищали с помощью ЖХСД с получением целевого соединения (0,047 г, 49%, твердое вещество желтого цвета).
Стадия 4: Получение N-(3-(1H-индол-3-ил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Целевое соединение получали аналогично способу, описанному на стадии 3 примера 31.
<Пример 90> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(пиперидин-1-ил)бутил)бензолсульфонамида

Стадия 1: Получение 4-(4-бромбутил)бензол-1-сульфонилхлорида

Целевое соединение (1,4 г, 96%, твердое вещество белого цвета) получали аналогично способу, описанному на стадии 1 примера 31 с использованием (4-бромбутил)бензола (1,0 г, 4,69 ммоль) и хлорсульфоновой кислоты (0,936 мл, 14,1 ммоль).
Стадия 2: Получение 4-(4-бромбутил)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфониламида

4-(4-Бромбутил)бензол-1-сульфонилхлорид (300 мг, 0,963 ммоль), полученный на стадии 1 выше, растворяли в дихлорметане (5 мл), при комнатной температуре добавляли 3-(5-хлор-1H-индол-3-ил)пропан-1-амин (211 мг, 1,01 ммоль) и триэтиламин (0,268 мл, 1,93 ммоль), затем перемешивали в течение 3 часов. После завершения реакции смесь концентрировали при пониженном давлении и очищали с помощью ЖХСД с получением целевого соединения (330 мг, 71%, твердое вещество белого цвета).
Стадия 3: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(пиперидин-1-ил)бутил)бензолсульфонамида

Целевое соединение (16 мг, 53%, твердое вещество белого цвета) получали аналогично способу, описанному на стадии 3 примера 31, с использованием 4-(4-бромбутил)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфониламида (30 мг, 0,062 ммоль), полученного на стадии 2 выше, пиперидина (15,9 мг, 0,186 ммоль) и карбоната калия (12,9 мг, 0,093 ммоль).
Соединение примера 91 получали аналогично способу, описанному в примере 90.
<Пример 92> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(1-метилпиперидин-4-ил)пропокси)бензолсульфонамида
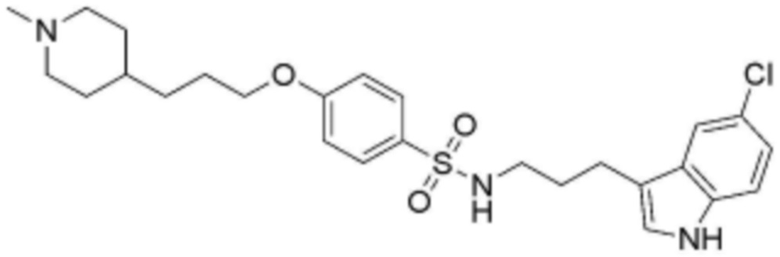
Стадия 1: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-гидроксибензолсульфонамида

4-Гидроксибензол-1-сульфонилхлорид (0,5 г, 2,60 ммоль) растворяли в дихлорметане (8,7 мл), добавляли 3-(5-хлор-1H-индол-3-ил)пропан-1-амин (0,596 г, 2,86 ммоль) и триэтиламин (0,724 мл, 5,19 ммоль), затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционную смесь разбавляли этилацетатом и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали, и затем очищали с помощью ЖХСД с получением целевого соединения (0,19 г, 21%, твердое вещество белого цвета).
Стадия 2: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(1-метилпиперидин-4-ил)пропокси)бензолсульфонамида

Соединение (0,05 г, 0,137 ммоль), полученное на стадии 1 выше, растворяли в тетрагидрофуране (1,5 мл) в атмосфере азота при температуре 0°C, добавляли 3-(1-метилпиперидин-4-ил)пропан-1-ол (0,026 г, 0,164 ммоль), PPh3 (0,054 г, 0,206 ммоль) и DEAD (0,093 мл, 0,206 ммоль), затем перемешивали в течение 1 часа. После завершения реакции реакционную смесь разбавляли дихлорметаном и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали, очищали с помощью преп. HPLC и нейтрализовали путем добавления водного раствора гидрокарбоната натрия с получением целевого соединения (0,004 г, 7%, твердое вещество белого цвета).
<Пример 93> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперидин-4-ил)пропокси)бензолсульфонамида


Соединение (0,035 г, 0,061 ммоль), полученное аналогично способу, описанному в примере 92, растворяли в дихлорметане (2 мл), добавляли трифторуксусную кислоту (1 мл, 12,98 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении, очищали с помощью преп. HPLC и нейтрализовали путем добавления водного раствора гидрокарбоната натрия с получением целевого соединения (0,01 г, 37%, твердое вещество белого цвета).
Соединение примера 94 получали аналогично способу, описанному в примере 93.
Соединения по примерам 95 и 96 получали аналогично способу, описанному в примере 94.
Соединение примера 97 получали аналогично способу, описанному в примере 31.
<Пример 98> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(4-метилпиперазин-1-ил)пропил)амино)бензолсульфонамида

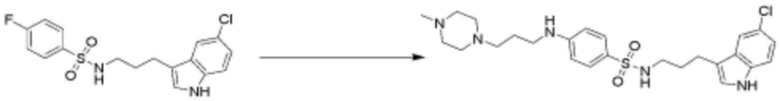
Соединение (0,05 г, 0,136 ммоль), полученное аналогично способу, описанному на стадии 2 примера 47, растворяли в диметилформамиде (1 мл), добавляли 3-(4-метилпиперазин-1-ил)пропан-1-амин (0,043 г, 0,273 ммоль) и карбонат калия (0,021 г, 0,150 ммоль), затем перемешивали при температуре 100°C в течение 3 дней. После завершения реакции реакционный раствор концентрировали при пониженном давлении, очищали с помощью преп. HPLC и нейтрализовали путем добавления водного раствора гидрокарбоната натрия с получением целевого соединения (0,01 г, 16%, твердое вещество белого цвета).
<Пример 99> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(пиперидин-4-ил)бутил)бензолсульфонамида

Стадия 1: Получение трет-бутил 4-(бут-3-ен-1-ил)пиперидин-1-карбоксилата

Метилтрифенилфосфоний бромид (1,184 г, 3,32 ммоль) растворяли в безводном тетрагидрофуране (2 мл), при температуре 0°C медленно добавляли бистриметилсилиламид лития (3,32 мл, 3,32 ммоль). После растворения при температуре 0°C медленно по каплям добавляли трет-бутил 4-(3-оксопропил)пиперидин-1-карбоксилат (0,4 г, 1,66 ммоль), полученный на стадии 1 примера 88, в безводном тетрагидрофуране (2 мл). Смесь перемешивали при температуре 0°C в течение 30 минут и затем перемешивали при комнатной температуре в течение 20 часов. После завершения реакции реакционную смесь концентрировали при пониженном давлении, промывали насыщенным солевым раствором и экстрагировали этилацетатом. Экстрагированный органический слой сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали с помощью ЖХСД с получением целевого соединения (0,2 г, 50%, прозрачное масло).
Стадия 2: Получение трет-бутил 4-(4-(4-(N-(3-(5-хлор-1H-индол-3-ил)пропил)сульфамоил)фенил)бутил)пиперидин-1-карбоксилата

Трет-бутил 4-(бут-3-ен-1-ил)пиперидин-1-карбоксилат (50 мг, 0,209 ммоль), полученный на стадии 2 выше, растворяли в безводном тетрагидрофуране (1,5 мл), при комнатной температуре добавляли 9-борабицикло[3,3,1]нонан (0,796 мл, 0,398 ммоль). Реакционную смесь постепенно нагревали и перемешивали при кипячении с обратным холодильником в течение 1 часа. После завершения реакции реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток растворяли в 1,4-диоксане (1,5 мл), добавляли 4-бром-N-(3-(5-хлор-1H-индол-3)-ил)пропил)бензолсульфонамид (85 мг, 0,199 ммоль), полученный аналогично способу, описанному на стадии 2 примера 90, и 1 M водный раствор карбоната натрия (0,597 мл, 0,597 ммоль). В реакционную смесь добавляли Pd(PPh3)4 (23 мг, 0,02 ммоль) и смесь постепенно нагревали и перемешивали при температуре 90 °C в течение 3 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, фильтровали через целит, концентрировали при пониженном давлении и очищали с помощью преп. HPLC с получением целевого соединения (15 мг, 13%, твердое вещество желтого цвета).
Стадия 3: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(пиперидин-4-ил)бутил)бензолсульфонамида
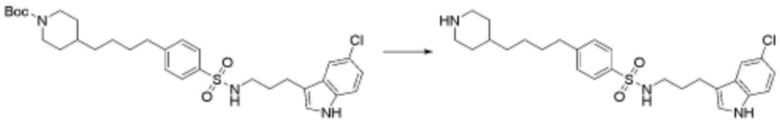
Трет-бутил 4-(4-(4-(N-(3-(5-хлор-1H-индол-3-ил)пропил)сульфамоил)фенил)бутил)пиперидин-1-карбоксилат (15 мг, 0,013 ммоль), полученный на стадии 2 выше, растворяли в дихлорметане (2 мл), при комнатной температуре добавляли трифторуксусную кислоту (1 мл, 13,1 ммоль), затем перемешивали в течение 1 часа. После завершения реакции остаток, полученный концентрированием смеси, нейтрализовали перенасыщенным водным раствором гидрокарбоната натрия и затем экстрагировали этилацетатом. Экстрагированный органический слой промывали дистиллированной водой, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением целевого соединения (5 мг, 56%, твердое вещество белого цвета).
Соединения по примерам 100-102 получали аналогично способу, описанному в примере 31.
<Пример 103> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-фтор-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Стадия 1: Получение 1-(3-бромпропокси)-2-фторбензола
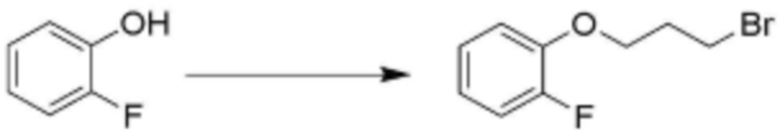
2-Фторфенол (5 г, 44,6 ммоль) растворяли в ацетонитриле (74 мл), добавляли 1,3-дибромпропан (90 г, 446 ммоль) и карбонат калия (6,16 г, 44,6 ммоль), затем перемешивали при температуре 80°C в течение 16 часов. После завершения реакции реакционный раствор фильтровали, концентрировали при пониженном давлении и использовали на следующей стадии без дополнительной очистки.
Стадия 2: Получение 4-(3-бромпропокси)-3-фторбензолсульфонилхлорида

Соединение (5 г, 19,31 ммоль), полученное на стадии 1 выше, растворяли в дихлорметане (38 мл), добавляли хлорсульфоновую кислоту (5,13 мл, 77 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь разбавляли дихлорметаном и нейтрализовали водным раствором карбоната натрия при температуре 0°C. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали и использовали на следующей стадии без дополнительной очистки.
Стадия 3: Получение 4-(3-бромпропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-фторбензолсульфонамида

Соединение (1 г, 3,02 ммоль), полученное на стадии 2 выше, растворяли в дихлорметане (6 мл), добавляли 3-(5-хлор-1H-индол-3-ил)пропан-1-амин (0,818 г, 3,92 ммоль) и карбонат калия (0,542 г, 3,92 ммоль), затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционную смесь разбавляли дихлорметаном, и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали и затем очищали с помощью ЖХСД с получением целевого соединения (1,1 г, 77%, твердое вещество белого цвета).
Стадия 4: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-фтор-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида
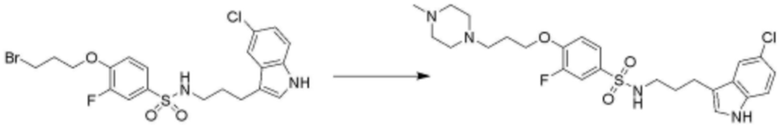
Соединение (0,05 г, 0,099 ммоль), полученное на стадии 3 выше, растворяли в этаноле (1 мл), добавляли карбонат калия (0,021 г, 0,149 ммоль) и 1-метилпиперазин (0,020 г, 0,198 ммоль), затем перемешивали при температуре 70°C в течение 16 часов. После завершения реакции реакционную смесь разбавляли дихлорметаном и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали, очищали с помощью преп. HPLC и затем нейтрализовали путем добавления водного раствора гидрокарбоната натрия с получением целевого соединения (0,011 г, 22%, твердое вещество белого цвета).
Соединения по примерам 104-106 получали аналогично способу, описанному в примере 103.
<Пример 107> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-метилпиперазин-1-ил)пропокси)бензолсульфонамида


Целевое соединение (15 мг, 72%) получали в виде твердого вещества бледно-желтого цвета аналогично способу, описанному на стадии 4 примера 99, с использованием трет-бутил 4-(3-(4-(N-(3-(5-хлор-1H-индол-3-ил)пропил)сульфамоил)фенокси)пропил)-2-метилпиперазин-1-карбоксилата (25 мг, 0,041 ммоль), полученного аналогично способу, описанному в примере 31, и трифторуксусной кислоты (1 мл, 13,1 ммоль).
Соединения по примерам 108-111 получали аналогично способу, описанному в примере 107.
Соединение примера 112 получали аналогично способу, описанному в примере 90.
<Пример 113> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(пиперазин-1-ил)пропил)амино)бензолсульфонамида


Соединение (0,0142 г, 0,024 ммоль), полученное аналогично способу, описанному в примере 98, растворяли в дихлорметане (2 мл), добавляли трифторуксусную кислоту (1 мл, 12,98 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении, очищали с помощью преп. HPLC и нейтрализовали путем добавления водного раствора гидрокарбоната натрия с получением целевого соединения (0,005 г, 50%, твердое вещество белого цвета).
Соединение примера 114 получали аналогично способу, описанному в примере 98.
Соединение примера 115 получали аналогично способу, описанному на стадиях 1-3 примера 99.
<Пример 116> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((1-((4-метилпиперазин-1-ил)метил)циклопропил)метокси)-бензолсульфонамида
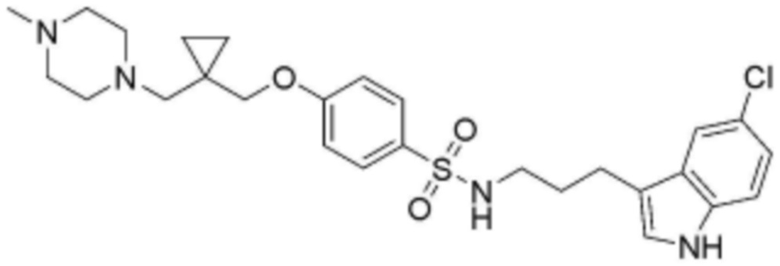
Стадия 1: Получение (1-(((трет-бутилдиметилсилил)окси)-метил)циклопропил)метанола

Циклопропан-1,1-диилдиметанол (5 г, 49,0 ммоль) растворяли в дихлорметане (130mL), добавляли имидазол (5 г, 73,4 ммоль) и трет-бутилдиметилсилил хлорид (7,75 г, 51,4 ммоль). Реакционную смесь перемешивали при температуре 0°C в течение 30 минут и затем перемешивали при комнатной температуре в течение 16 часов. После завершения реакции реакционную смесь нейтрализовали перенасыщенным водным раствором гидрокарбоната натрия и экстрагировали этилацетатом. Экстрагированный органический слой промывали дистиллированной водой, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении. Реакционную смесь очищали с помощью ЖХСД с получением целевого соединения (4,7 г, 44%, прозрачное масло).
Стадия 2: Получение ((1-(бромметил)циклопропил)метокси)-(трет-бутил)диметилсилана

(1-(((Трет-бутилдиметилсилил)окси)метил)циклопропил)метанол (1,1 г, 5,08 ммоль), полученный на стадии 1 выше, растворяли в дихлорметане (10 мл), добавляли тетрабромид углерода (1,85 г, 5,59 ммоль) и трифенилфосфин (1,47 г, 5,59 ммоль). Реакционную смесь перемешивали при температуре 0°C в течение 30 минут и затем перемешивали при комнатной температуре в течение 16 часов. После завершения реакции реакционную смесь концентрировали при пониженном давлении и очищали с помощью ЖХСД с получением целевого соединения (1 г, 70%, прозрачное масло).
Стадия 3: Получение (1-(бромметил)циклопропил)метанола

((1-(Бромметил)циклопропил)метокси)(трет-бутил)диметилсилан (1 г, 3,58 ммоль), полученный на стадии 2 выше, растворяли в уксусной кислоте/тетрагидрофуран/дистиллированная вода (12 мл/4 мл/4 мл), затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционную смесь нейтрализовали перенасыщенным водным раствором гидрокарбоната натрия и экстрагировали этилацетатом. Экстрагированный органический слой промывали дистиллированной водой, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении. Полученную реакционную смесь очищали с помощью ЖХСД с получением целевого соединения (0,3 г, 51%, масло желтого цвета).
Стадия 4: Получение 4-((1-(бромметил)циклопропил)метокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамида

Целевое соединение (45 мг, 48%, твердое вещество желтого цвета) получали аналогично способу, описанному на стадии 2 примера 92, с использованием (1-(бромметил)циклопропил)метанола (30 мг, 0,182 ммоль), полученного на стадии 3 выше, N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-гидроксибензолсульфонамида (66 мг, 0,182 ммоль), трифенилфосфина (72 мг, 0,273 ммоль) и диэтил азодикарбоксилата (0,124 мл, 0,273 ммоль).
Стадия 5: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((1-((4-метилпиперазин-1-ил)метил)циклопропил)метокси)-бензолсульфонамид
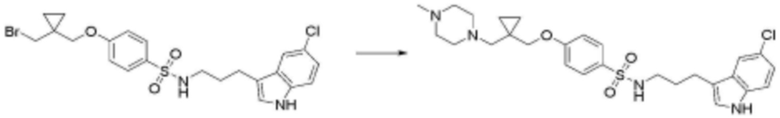
Целевое соединение (12,3 мг, 59%, твердое вещество желтого цвета) получали аналогично способу, описанному на стадии 3 примера 31, с использованием 4-((1-(бромметил)циклопропил)метокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамида (20 мг, 0,039 ммоль), полученного на стадии 4 выше, 1-метилпиперазина (0,013 мл, 0,117 ммоль) и карбоната калия (8,1 мг, 0,059 ммоль).
Соединение примера 117 получали аналогично способу, описанному в примере 116.
<Пример 118> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)фенокси)бензолсульфонамида

Стадия 1: Получение 4-(3-бромфенокси)бензолсульфоновой кислоты
1-Бром-3-феноксибензол (300 мг, 1,20 ммоль) растворяли в дихлорметане, медленно при температуре 0°C добавляли хлорсульфоновую кислоту (0,088 мл, 1,33 ммоль). Реакционную смесь перемешивали при температуре 0°C в течение 30 минут и затем перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционную смесь концентрировали при пониженном давлении с получением целевого соединения (395 мг, 100%, масло желтого цвета).
Стадия 2: Получение 4-(3-бромфенокси)бензолсульфонилхлорида

4-(3-Бромфенокси)бензолсульфоновую кислоту (395 мг, 1,20 ммоль), полученную на стадии 1 выше, растворяли в SnCl2⋅H2O (3 мл, 41,4 ммоль) при температуре 0°C, затем перемешивали. Затем при температуре 0°C добавляли две капли диформамида и реакционную смесь постепенно нагревали и перемешивали при кипячении с обратным холодильником в течение 2 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, концентрировали при пониженном давлении и очищали с помощью ЖХСД с получением целевого соединения (380 мг, 91%, масло желтого цвета).
Стадия 3: Получение 4-(3-бромфенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамида

Целевое соединение (170 мг, 76%, твердое вещество желтого цвета) получали аналогично способу, описанному на стадии 2 примера 90, с использованием 4-(3-бромфенокси)бензолсульфонил-хлорида (150 мг, 0,432 ммоль), полученного на стадии 2 выше, 3-(5-хлор-1H-индол-3-ил)пропан-1-амина (90 мг, 0,432 ммоль) и триэтиламина (0,090 мл, 0,647 ммоль).
Стадия 4: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)фенокси)бензолсульфонамида

4-(3-Бромфенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид (90 мг, 0,173 ммоль), полученный на стадии 3 выше, растворяли в 1,4-диоксане (1,5 мл), при комнатной температуре добавляли трет-BuOK (29,1 мг, 0,260 ммоль) и 1-метилпиперазин (0,03 мл, 0,260 ммоль). Реакционную смесь нагревали при температуре 70°C и перемешивании и дегазировали в течение 5 минут с использованием азота из баллона, добавляли Pd(t-Bu3P)2 (8 мг, 0,016 ммоль), затем перемешивали при кипячении с обратным холодильником в течение 3 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, фильтровали и концентрировали через целит и очищали с помощью преп. HPLC. Полученное трифторацетатное соединение нейтрализовали путем добавления перенасыщенного водного раствора гидрокарбоната натрия и экстрагировали 10%-ным раствором метанол/дихлорметан. Органический слой сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением целевого соединения (6,5 мг, 53%, твердое вещество белого цвета).
Соединение примера 119 получали аналогично способу, описанному на стадиях 1-3 примера 118.
Соединения по примерам 120-122 получали аналогично способу, описанному в примере 118.
<Пример 123> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(4-метилпиперазин-1-ил)фенил)амино)бензолсульфонамида


Соединение (0,05 г, 0,117 ммоль), полученное аналогично способу, описанному на стадии 2 примера 47, растворяли в 2-бутаноле (1,5 мл), добавляли 3-(4-метилпиперазин-1-ил)анилин (0,02 г, 0,117 ммоль) и карбонат калия (0,08 г, 0,584 ммоль), затем дегазировали в течение 1 минуты. Затем при температуре 80°C добавляли Pd2(dba)3 (0,01 г, 0,012 ммоль) и Xphos (0,005 г, 0,012 ммоль), далее перемешивали при температуре 100°C в течение 1 часа. После завершения реакции реакционный раствор фильтровали через целит и промывали этилацетатом и дихлорметаном. Фильтрат концентрировали при пониженном давлении, очищали с помощью преп. HPLC и нейтрализовали путем добавления водного раствора гидрокарбоната натрия с получением целевого соединения (0,023 г, 37%, твердое вещество желтого цвета).
Соединения по примерам 124-127 получали аналогично способу, описанному в примере 123.
Соединения по примерам 128 и 129 получали аналогично способу, описанному в примере 119.
<Пример 130> Получение 4-(3,5-бис(4-метилпиперазин-1-ил)фенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамида

Стадия 1: Получение 1,3-дибром-5-феноксибензола

Фенол (100 мг, 1,06 ммоль) растворяли в N-метил-2-пирролидиноне (2 мл), при комнатной температуре добавляли карбонат цезия (173 мг, 0,531 ммоль), 1,3-дибром-5-фторбензол (0,201 мл, 1,59 ммоль), TMHDA (0,057 мл, 0,266 ммоль) и CuCl (52,6 мг, 0,531 ммоль). Реакционную смесь дегазировали при комнатной температуре с использованием азота из баллона, затем перемешивали в течение 20 минут. Реакционную смесь постепенно нагревали и перемешивали при температуре 120°C в течение 16 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, фильтровали через целит, экстрагировали этилацетатом и концентрировали при пониженном давлении. Полученную реакционную смесь очищали с помощью ЖХСД с получением целевого соединения (220 мг, 63%, твердое вещество желтого цвета).
Стадия 2: Получение 4-(3,5-дибромфенокси)бензолсульфоновой кислоты
Целевое соединение (274 мг, 100%, твердое вещество желтого цвета) получали аналогично способу, описанному на стадии 1 примера 118, с использованием 1,3-дибром-5-феноксибензола (220 мг, 0,671 ммоль), полученного на стадии 1 выше, и хлорсульфоновой кислоты (0,049 мл, 0,738 ммоль).
Стадия 3: Получение 4-(3,5-дибромфенокси)бензолсульфонилхлорида

Целевое соединение (230 мг, 80%, твердое вещество желтого цвета) получали аналогично способу, описанному на стадии 2 примера 118, с использованием 4-(3,5-дибромфенокси)бензолсульфоновой кислоты (274 мг, 0,671 ммоль), полученной на стадии 2 выше, и тионилхлорида (4 мл, 55,1 ммоль).
Стадия 4: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3,5-дибромфенокси)бензолсульфонамида
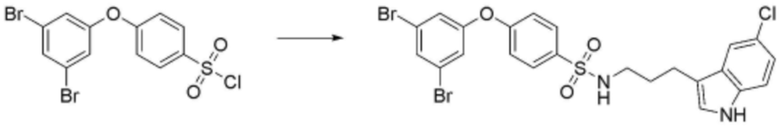
Целевое соединение (300 мг, 93%, твердое вещество белого цвета) получали аналогично способу, описанному на стадии 2 примера 90, с использованием 4-(3,5-дибромфенокси)-бензолсульфонилхлорида (230 мг, 0,539 ммоль), полученного на стадии 3 выше, 3-(5-хлор-1H-индол-3-ил)пропан-1-амина (118 мг, 0,566 ммоль) и триэтиламина (0,113 мл, 0,809 ммоль).
Стадия 5: Получение 4-(3,5-бис(4-метилпиперазин-1-ил)фенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамида

Целевое соединение (5 мг, 9%, твердое вещество желтого цвета) получали аналогично способу, описанному на стадии 4 примера 118, с использованием N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3,5-дибромфенокси)бензолсульфонамид (50 мг, 0,084 ммоль), полученного на стадии 4 выше, 1-метилпиперазина (0,037 мл, 0,334 ммоль), трет-бутоксида калия (37,5 мг, 0,334 ммоль) и Pd(t-Bu3P)2 (8,54 мг, 0,017 ммоль).
Соединения по примерам 131-134 получали аналогично способу, описанному в примере 123.
Соединения по примерам 135 и 136 получали аналогично способу, описанному в примере 118.
Соединение примера 137 получали аналогично способу, описанному в примере 138.
<Пример 138> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-бром-5-(4-метилпиперазин-1-ил)фенокси)бензолсульфонамида

Стадия 1: Получение 1-бром-3-фтор-5-феноксибензола

Целевое соединение (0,45 г, 32%, прозрачное масло) получали аналогично способу, описанному на стадии 1 примера 130, с использованием фенола (0,5 г, 5,31 ммоль), карбоната цезия (0,866 г, 2,66 ммоль), 1-бром-3,5-дифторбензола (1,54 г, 7,97 ммоль), TMHDA (0,284 мл, 1,33 ммоль) и CuCl (0,263 мг, 2,66 ммоль).
Стадия 2: Получение 4-(3-бром-5-фторфенокси)бензол-1-сульфонилхлорида

Целевое соединение (0,44 г, 71%, масло желтого цвета) получали аналогично способу, описанному на стадии 1 примера 31, с использованием 1-бром-3-фтор-5-феноксибензола (0,45 г, 1,685 ммоль), полученного на стадии 1 выше, и хлорсульфоновой кислоты (0,123 мл, 1,853 ммоль).
Стадия 3: Получение 4-(3-бром5-фторфенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамида
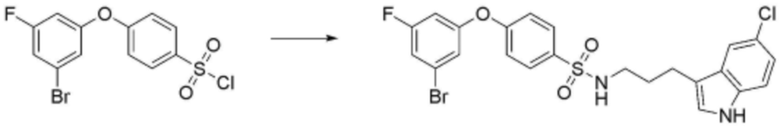
Целевое соединение (300 мг, 93%, твердое вещество желтого цвета) получали аналогично способу, описанному на стадии 2 примера 90, с использованием 4-(3-бром-5-фторфенокси)бензол-1-сульфонилхлорида (200 мг, 0,547 ммоль), полученного на стадии 2 выше, 3-(5-хлор-1H-индол-3-ил))пропан-1-амина (115 мг, 0,553 ммоль) и триэтиламина (0,114 мл, 0,821 ммоль).
Стадия 4: Получение 4-(3-бром-5-(4-метилпиперазин-1-ил)фенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамида

4-(3-Бром-5-фторфенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид (30 мг, 0,056 ммоль), полученный на стадии 3 выше, растворяли в диметилсульфоксиде (1 мл), при комнатной температуре добавляли 1-метилпиперазин (0,025 мл, 0,223 ммоль) и карбонат калия (30,8 мг, 0,223 ммоль). Затем реакционную смесь нагревали при температуре 120°C и перемешивали в течение 16 часов. После завершения реакции смесь охлаждали до комнатной температуры, промывали дистиллированной водой и насыщенным солевым раствором и экстрагировали этилацетатом. Экстрагированный органический слой сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали с помощью преп. HPLC. Полученное трифторацетатное соединение нейтрализовали путем добавления перенасыщенного водного раствора гидрокарбоната натрия, затем экстрагировали 10%-ным раствором метанол/дихлорметан. Экстрагированный органический слой сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении с получением целевого соединения (6,2 мг, 22%, твердое вещество цвета слоновой кости).
Соединения по примерам 139-142 получали аналогично способу, описанному в примере 123.
<Пример 143> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензамида

Стадия 1: Получение метил 4-(3-бромпропокси)бензоата

Метил 4-гидроксибензоат (0,5 г, 3,29 ммоль) растворяли в ацетонитриле (10 мл), добавляли 1,3-дибромпропан (0,995 г, 4,93 ммоль) и карбонат калия (0,681 г, 4,93 ммоль). Реакционную смесь нагревали при температуре 80°C и перемешивали в течение 5 часов. После завершения реакции смесь охлаждали до комнатной температуры, фильтровали через целит, и концентрировали при пониженном давлении. Полученный остаток очищали с помощью ЖХСД с получением целевого соединения (0,7 г, 78%, прозрачное масло).
Стадия 2: Получение 4-(3-бромпропокси)бензойной кислоты

Метил 4-(3-бромпропокси)бензоат (0,7 г, 2,56 ммоль), полученный на стадии 1 выше, растворяли в смеси тетрагидрофуране/метанол/дистиллированная вода (6,0 мл/3 мл/3 мл), при комнатной температуре добавляли LiOH·H2O (0,323 г, 7,69 ммоль). Затем реакционную смесь нагревали при температуре 50°C и перемешивали в течение 2 часов. После завершения реакции смесь охлаждали до комнатной температуры и по каплям медленно добавляли 1н водный раствор соляной кислоты до установления pH 2. Полученный твердый продукт промывали дистиллированной водой и затем сушили в вакууме с получением целевого соединения (0,35 г, 53%, твердое вещество белого цвета).
Стадия 3: Получение 4-(3-бромпропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензамида

4-(3-Бромпропокси)бензойную кислоту (0,1 г, 0,386 ммоль), полученную на стадии 2 выше, растворяли в дихлорметане (2 мл), при комнатной температуре добавляли 3-(5-хлор-1H-индол-3-ил)пропан-1-амин (0,081 г, 0,390 ммоль), HATU (0,294 г, 0,772 ммоль) и N,N-диизопропилэтиламин (0,15 г, 1,158 ммоль), затем перемешивали при комнатной температуре в течение 16 часов. После завершения реакции смесь промывали дистиллированной водой и экстрагировали дихлорметаном. Экстрагированный органический слой сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали с помощью ЖХСД с получением целевого соединения (0,1 г, 58%, твердое вещество цвета слоновой кости).
Стадия 4: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензамида
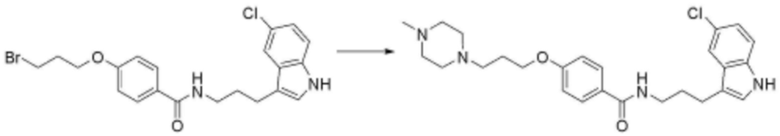
Целевое соединение (8 мг, 38%, твердое вещество цвета слоновой кости) получали аналогично способу, описанному на стадии 3 примера 31, с использованием 4-(3-бромпропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензамида (20 мг, 0,044 ммоль), полученного на стадии 3 выше, 1-метилпиперазина (0,02 мл, 0,178 ммоль) и карбоната калия (12,3 мг, 0,089 ммоль).
Соединение примера 144 получали аналогично способу, описанному в примере 143.
Соединение примера 145 получали аналогично способу, описанному в примере 92.
<Пример 146> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-2-((3-(пиперазин-1-ил)пропил)амино)триазол-5-сульфонамида

Стадия 1: Получение 2-хлор-N-(3-(5-хлор-1H-индол-3-ил)пропил)триазол-5-сульфонамида
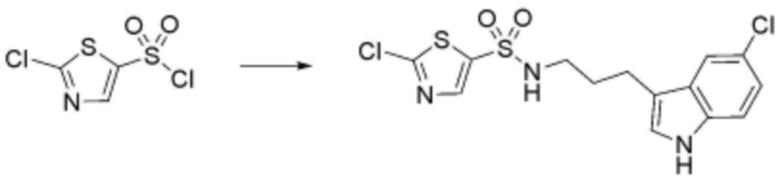
Целевое соединение получали аналогично способу, описанному на стадии 2 примера 31.
Стадия 2: Получение трет-бутил 4-(3-((5-(N-(3-(5-хлор-1H-индол-3-ил)пропил)сульфамоил)тиазол-2-ил)амино)пропил)пиперазин-1-карбоксилата
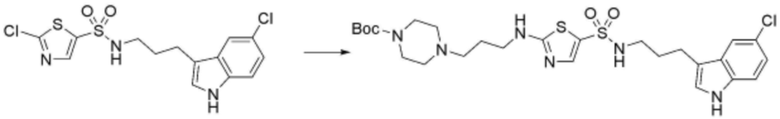
2-Хлор-N-(3-(5-хлор-1H-индол-3-ил)пропил)триазол-5-сульфонамид (30 мг, 0,077 ммоль), полученный на стадии 1 выше, растворяли в диметилформамиде, добавляли трет-бутил 4-(3-аминопропил)пиперазин-1-карбоксилат (28 мг, 0,115 ммоль) и DIPEA (0,022 мл, 0,154 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения реакции растворитель удаляли при пониженном давлении и затем реакционную смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (37 мг, 81%, твердое вещество желтого цвета).
Стадия 3: Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-2-((3-(пиперазин-1-ил)пропил)амино)триазол-5-сульфонамида

Целевое соединение (30 мг, 97%, твердое вещество бледно-желтого цвета) получали аналогично способу, описанному на стадии 3 примера 99.
<Пример 147> Получение N-(3-(5-хлор-2-метил-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Стадия 1: Получение гидрохлорида 3-(5-хлор-2-метил-1H-индол-3-ил)пропан-1-амина

Хлорфенил гидразин (1,40 г, 9,81 ммоль) и 6-хлоргексан-2-он (1,10 г, 8,17 ммоль) растворяли в этаноле (30 мл), затем перемешивали при кипячении с обратным холодильником в течение 20 часов. После завершения реакции смесь охлаждали до комнатной температуры и растворитель удаляли при пониженном давлении с получением гидрохлорида 3-(5-хлор-2-метил-1H-индол-3-ил)пропан-1-амина (2,114 г, количественный, твердое вещество бледно-желтого цвета).
Стадия 2: Получение 4-(3-бромпропокси)-N-(3-(5-хлор-2-метил-1H-индол-3-ил)пропил)бензолсульфонамида

3-(5-Хлор-2-метил-1H-индол-3-ил)пропан-1-амин гидрохлорид (0,413 г, 1,59 ммоль), полученный на стадии 1 выше, растворяли в этиленхлориде (6 мл), добавляли по каплям триэтиламин (0,444 мл, 3,19 ммоль), затем перемешивали в течение 5 минут. Добавляли по каплям 4-(3-бромпропокси)бензол-1-сульфонил хлорид (0,200 г, 0,638 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. Для завершения реакции добавляли по каплям воду, затем экстрагировали дихлорметаном. Экстрагированный органический слой сушили над безводным сульфатом натрия и фильтровали и растворитель удаляли при пониженном давлении. Затем смесь разделяли и очищали с помощью ЖХСД с получением целевого соединения (177 мг, 56%, твердое вещество коричневого цвета).
Стадия 3: Получение N-(3-(5-хлор-2-метил-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида
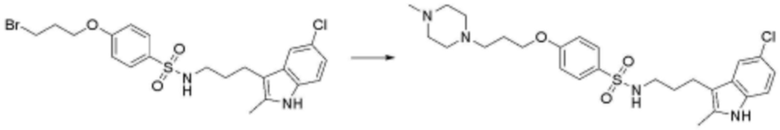
Целевое соединение (19,8 мг, 64%, твердое вещество бледно-желтого цвета) получали аналогично способу, описанному на стадии 3 примера 31.
<Пример 148> Получение N-(3-(5-хлор-2-метил-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида
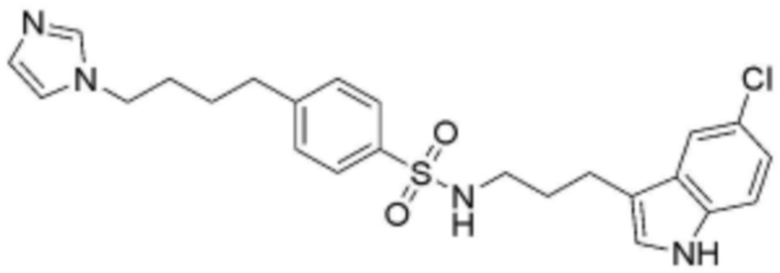

4-(4-Бромбутил)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфониламид (30 мг, 0,062 ммоль), полученный на стадии 2 примера 90, растворяли в ацетонитриле, при комнатной температуре добавляли карбонат калия (25,7 мг, 0,186 ммоль) и 1H-имидазол (8,44 мг, 0,124 ммоль), затем перемешивали при температуре 60°C в течение 4 часов. После завершения реакции смесь фильтровали через целит. Фильтрат концентрировали при пониженном давлении и затем разделяли и очищали с помощью ЖХСД с получением целевого соединения (13 мг, 45%, твердое вещество белого цвета).
<Пример 149> Получение N-(2-((5-хлор-1H-индол-3-ил)метил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида
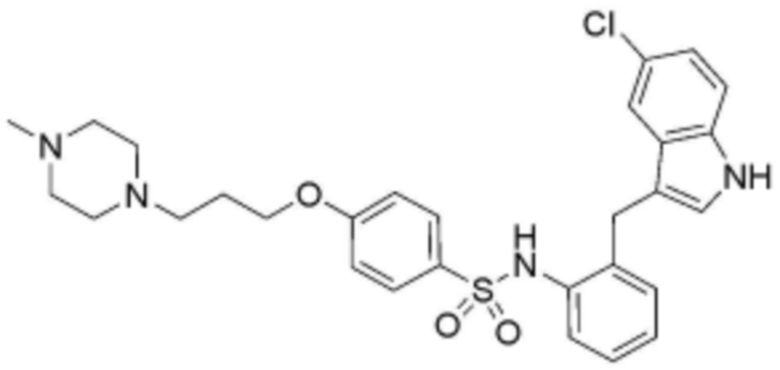
Стадия 1: Получение 2-((5-хлор-1H-индол-3-ил)метил)анилина
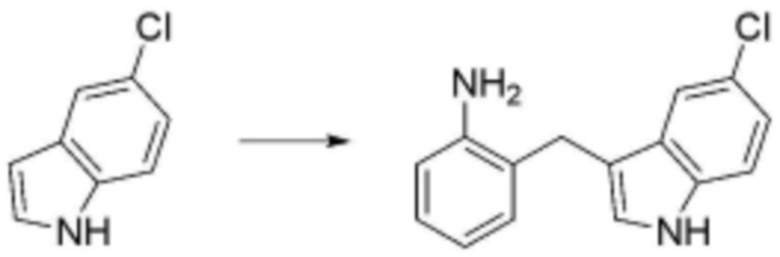
5-Хлор-1H-индол (1 г, 6,60 ммоль) и (2-аминофенил)метанол (0,812 г, 6,60 ммоль) растворяли в 1,2-дихлорэтане (20 мл), при комнатной температуре добавляли трифторуксусную кислоту (0,151 мл, 1,979 ммоль), затем перемешивали при температуре 50°C в течение 16 часов. После охлаждения смеси до комнатной температуры для завершения реакции добавляли по каплям перенасыщенным водным раствором карбоната натрия, затем экстрагировали этилацетатом. Экстрагированный органический слой сушили над безводным сульфатом натрия, фильтровали и затем разделяли и очищали с помощью ЖХСД с получением целевого соединения (0,55 г, 33%, твердое вещество желтого цвета).
Стадия 2: Получение 4-(3-бромпропокси)-N-(2-((5-хлор-1H-индол-3-ил)метил)фенил)бензолсульфонамида

Целевое соединение получали аналогично способу, описанному на стадии 2 примера 31.
Стадия 3: Получение N-(2-((5-хлор-1H-индол-3-ил)метил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси) бензолсульфонамида

Целевое соединение (13,2 мг, 64%, твердое вещество бледно-желтого цвета) получали аналогично способу, описанному в примере 148.
Соединение примера 150 получали аналогично способу, описанному в примере 149.
<Пример 151> Получение N-(2-((5-хлор-1H-индол-3-ил)метил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Стадия 1: Получение 4-((5-хлор-1H-индол-3-ил)бутан-2-она

5-Хлор-1H-индол (1 г, 6,60 ммоль) растворяли в дистиллированной воде (20 мл), при комнатной температуре добавляли 3-бутен-2-он (1,387 г, 19,79 ммоль) и трифторметансульфоновую кислоту (10 мг, 0,066 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После завершения реакции смесь промывали перенасыщенным водным раствором карбоната натрия и затем экстрагировали этилацетатом. Экстрагированный органический слой сушили над безводным сульфатом натрия, фильтровали и затем разделяли и очищали с помощью ЖХСД с получением целевого соединения (0,64 г, 44%, твердое вещество бледно-желтого цвета).
Стадия 2: Получение 4-(5-хлор-1H-индол-3-ил)бутан-2-амина

4-((5-Хлор-1H-индол-3-ил)бутан-2-он (200 мг, 0,902 ммоль), полученный на стадии 1 выше, растворяли в метаноле (8 мл), при комнатной температуре добавляли ацетат аммония (695 мг, 9,02 ммоль) и цианоборгидрид натрия (283 мг, 4,51 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После завершения реакции смесь промывали перенасыщенным водным раствором карбоната натрия и затем экстрагировали этилацетатом. Экстрагированный органический слой сушили над безводным сульфатом натрия, фильтровали и разделяли и очищали с помощью ЖХСД с получением целевого соединения (0,2 г, 100%, масло бледно-желтого цвета).
Стадия 3: Получение 4-(3-бромпропокси)-N-(4-(5-хлор-1H-индол-3-ил)бутан-2-ил)бензолсульфонамида

Целевое соединение получали аналогично способу, описанному на стадии 2 примера 31.
Стадия 4: Получение N-(2-((5-хлор-1H-индол-3-ил)метил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Целевое соединение (16 мг, 77%, твердое вещество белого цвета) получали аналогично способу, описанному в примере 149.
Соединение примера 152 получали аналогично способу, описанному в примере 151.
<Пример 153> Получение N-(3-(5-бром-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамида

Стадия 1: Получение 3-(5-бром-1H-индол-3-ил)пропан-1-ола

(4-Бромфенил)гидразин гидрохлорид (1 г, 4,47 ммоль) растворяли в ацетонитриле (10 мл), добавляли 4% водный раствор серной кислоты (10 мл). Медленно при температуре 100°C добавляли 3,4-дигидро-2H-пиран (0,40 мл, 4,47 ммоль) в течение 2 минут, затем перемешивали при температуре 100°C в течение 2 часов. После завершения реакции реакционную смесь разбавляли этилацетатом и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали и затем очищали с помощью ЖХСД с получением целевого соединения (0,61 г, 54%).
Стадия 2: Получение 5-бром-3-(3-бромпропил)-1H-индола

Соединение (0,31 г, 1,231 ммоль), полученное на стадии 1 выше, растворяли в дихлорметане (2 мл), при температуре 0°C добавляли тетрабромид углерода (0,49 г, 1,477 ммоль) и трифенилфосфин (0,38 г, 1,477 ммоль). Реакционную смесь перемешивали в течение 30 минут и затем перемешивали при комнатной температуре в течение 4 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и затем очищали с помощью ЖХСД с получением целевого соединения (0,29 г, 77%).
Стадия 3: Получение 3-(3-азидопропил)-5-бром-1H-индола

Соединение (0,99 г, 3,25 ммоль), полученное на стадии 2 выше, растворяли в диметилформамиде (11 мл), добавляли азид натрия (1,05 г, 16,24 ммоль), затем перемешивали при комнатной температуре в течение 4 часов. После завершения реакции реакционную смесь разбавляли этилацетатом и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали и затем очищали с помощью ЖХСД с получением целевого соединения (0,54 г, 62%).
Стадия 4: Получение 3-(5-бром-1H-индол-3-ил)пропан-1-амина

Соединение (0,1 г, 0,358 ммоль), полученное на стадии 3 выше, растворяли в тетрагидрофуране (3 мл) и воде (0,2 мл), добавляли трифенилфосфин (0,47 г, 1,791 ммоль), затем перемешивали при комнатной температуре в течение 16 часов. После завершения реакции реакционную смесь разбавляли дихлорметаном и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали и затем очищали с помощью ЖХСД с получением целевого соединения (0,075 г, 83%).
Стадия 5: Получение N-(3-(5-бром-1H-индол-3-ил)пропил)-4-(3-бромпропокси)бензолсульфонамида

Соединение (0,094 г, 0,299 ммоль), полученное на стадии 1 примера 31, растворяли в дихлорметане (1 мл), добавляли соединение (0,075 г, 0,299 ммоль), полученное на стадии 4 выше, и карбонат калия (0,124 г, 0,896 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь разбавляли дихлорметаном и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали и затем очищали с помощью ЖХСД с получением целевого соединения (0,12 г, 80%).
Стадия 6: Получение N-(3-(5-бром-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамида

Соединение (0,06 г, 0,113 ммоль), полученное на стадии 5 выше, растворяли в этаноле (1,5 мл), добавляли карбонат калия (0,031 г, 0,226 ммоль) и пиперазин (0,02 г, 0,339 ммоль), затем перемешивали при температуре 70°C в течение 16 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении, разбавляли дихлорметаном и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали, очищали с помощью преп. HPLC и нейтрализовали путем добавления водного раствора гидрокарбоната натрия с получением целевого соединения (0,032 г, 53%, твердое вещество желтого цвета).
Соединения по примерам 154-156 получали аналогично способу, описанному в примере 153.
Соединение примера 147 получали аналогично способу, описанному в примере 147.
Соединения по примерам 158 и 159 получали аналогично способу, описанному в примере 31.
Соединения по примерам 160 и 161 получали аналогично способу, описанному в примере 148.
Соединения по примерам 162 и 163 получали аналогично способу, описанному в примере 31.
Соединения по примерам 164 и 165 получали аналогично способу, описанному в примере 153.
Соединение примера 148 получали аналогично способу, описанному в примере 148.
<Пример 167> Получение N-(3-(5-хлор-1H-индазол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Стадия 1: Получение (цианометил) трифенилфосфония

2-Бромацетонитрил (1,328 мл, 19,06 ммоль) растворяли в дихлорметане (80 мл), при комнатной температуре добавляли трифенилфосфин (5 г, 19,06 ммоль), затем перемешивали в течение 6 часов. После завершения реакции смесь промывали диэтиловым эфиром без дополнительной очистки с получением целевого соединения (7 г, 96%, твердое вещество белого цвета).
Стадия 2: Получение (E)-3-(5-хлор-1H-индазол-3-ил)акрилонитрила
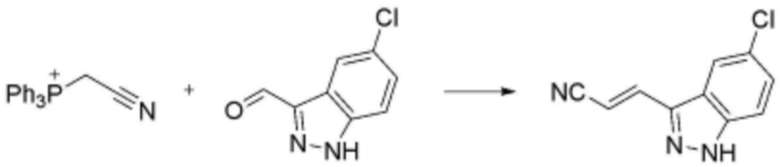
(Цианометил)трифенилфосфоний (3,17 г, 8,31 ммоль), полученный на стадии 1 выше, растворяли в безводном тетрагидрофуране (10 мл), при температуре -78°C добавляли трет-бутоксид натрия (0,8 г, 8,31 ммоль), затем перемешивали в течение 30 минут. При температуре -78°C добавляли 5-хлор-1H-индазол-3-карбальдегид (1,0 г, 5,44 ммоль), растворенный в безводном тетрагидрофуране. Температуру реакционной смеси медленно повышали до комнатной температуры, затем перемешивали в течение 1 часа. После завершения реакции смесь промывали перенасыщенным водным раствором хлорида аммония и экстрагировали этилацетатом. Экстрагированный органический слой сушили над безводным сульфатом натрия, фильтровали и затем разделяли и очищали с помощью ЖХСД с получением целевого соединения (0,3 г, 27%, твердое вещество бледно-желтого цвета).
Стадия 3: Получение 3-(5-хлор-1H-индазол-3-ил)пропан-1-амина

(E)-3-(5-хлор-1H-индазол-3-ил)акрилонитрил (300 мг, 1,473 ммоль), полученный на стадии 2 выше, растворяли в эфире (5 мл), при температуре -78°C добавляли LAH (168 мг, 4,42 ммоль). Температуру реакционной смеси медленно повышали до комнатной температуры, затем перемешивали в течение 2 часов. Реакцию завершали путем добавления дополнительного количества LAH (112 мг, 2,95 ммоль). После завершения реакции смесь фильтровали через целит. Фильтрат концентрировали при пониженном давлении и затем разделяли и очищали с помощью ЖХСД с получением целевого соединения (200 мг, 65%, масло желтого цвета).
Стадия 4: Получение 4-(-бромпропокси)-N-(3-(5-хлор-1H-индазол-3-ил)пропил)бензолсульфонамида

Целевое соединение (75 мг, 18%, твердое вещество белого цвета) получали аналогично способу, описанному на стадии 2 примера 31.
Стадия 5: Получение N-(3-(5-хлор-1H-индазол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида
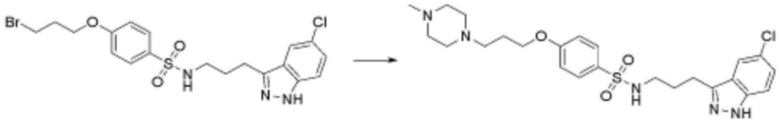
Целевое соединение (13,3 мг, 85%, твердое вещество бледно-желтого цвета) получали аналогично способу, описанному в примере 148.
Соединения по примерам 168-170 получали аналогично способу, описанному в примере 167.
<Пример 171> Получение (R)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(2-гидрокси-3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Стадия 1: Получение (R)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(оксиран-2-илметокси)бензолсульфонамида

Соединение (0,41 г, 1,026 ммоль), полученное на стадии 1 примера 92, растворяли в диметилформамиде (3,4 мл), добавляли (R)-2-(хлорметил)оксиран (0,095 г, 1,026 ммоль) и карбонат калия (0,21 г, 1,539 ммоль), затем перемешивали при температуре 60°C в течение 16 часов. После завершения реакции реакционную смесь разбавляли дихлорметаном и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали и затем очищали с помощью ЖХСД с получением целевого соединения (0,16 г, 37%).
Стадия 2: Получение (R)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(2-гидрокси-3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Соединение (0,083 г, 0,198 ммоль), полученное на стадии 1 выше, растворяли в ацетоне (1 мл), добавляли 1-метилпиперазин (0,19 г, 1,981 ммоль) и карбонат калия (0,274 г, 1,981 ммоль), затем перемешивали при температуре 80°C в течение 16 часов. После завершения реакции реакционную смесь разбавляли дихлорметаном и промывали водой и насыщенным солевым раствором. Остаток органического слоя сушили над безводным сульфатом натрия, фильтровали и концентрировали, очищали с помощью преп. HPLC и нейтрализовали путем добавления водного раствора гидрокарбоната натрия с получением целевого соединения (0,030 г, 30%, твердое вещество желтого цвета).
Соединения по примерам 172-174 получали аналогично способу, описанному в примере 171.
Соединения по примерам 175 и 176 получали аналогично способу, описанному в примере 148.
<Пример 177> Получение N-((6-хлор-2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил) 4-(3-(4-метилпиперазин-1-ил)пропокси)-бензолсульфонамида

Стадия 1: Получение 4-(3-бромпропокси)-N-(3-(5-хлор-1H-индазол-3-ил)пропил)бензолсульфонамида

Целевое соединение получали аналогично способу, описанному на стадии 2 примера 31 с использованием (6-хлор-2,3,4,9-тетрагидро-1H-карбазол-3-ил)метанамина, полученного аналогично способу, описанному в ссылочной литературе [WO2006/89053].
Стадия 2: Получение N-((6-хлор-2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Целевое соединение (6,3 мг, 20%, твердое вещество бледно-желтого цвета) получали аналогично способу, описанному в примере 148.
Соединение примера 178 получали аналогично способу, описанному в примере 177.
Соединения по примерам 179 и 180 получали аналогично способу, описанному в примере 85.
<Пример 181> Получение N-(2-(2-(5-хлор-1H-индол-3-ил)пропан-2-ил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Стадия 1: Получение N-(2-(2-(5-хлор-1H-индол-3-ил)пропан-2-ил)фенил)-4-метилбензолсульфонамида

5-Хлор-1H-индол (0,400 г, 5,28 ммоль) и N-(2-(2-гидроксипропан-2-ил)фенил)-4-метилбензолсульфонамид (0,967 г, 3,17 ммоль) растворяли в 1,2-дихлорэтане (40 мл), добавляли PdCl2·(CH3CN)2 (0,0340 г, 0,132 ммоль), затем перемешивали в течение 3 часов в атмосфере азота. После завершения реакции реакционную смесь охлаждали до комнатной температуры, растворитель удаляли при пониженном давлении и разделяли и очищали с помощью ЖХСД с получением целевого соединения (1,02 г, 88%, твердое вещество белого цвета).
Стадия 2: Получение 2-(2-(5-хлор-1H-индол-3-ил)пропан-2-ил)анилина

N-(2-(2-(5-хлор-1H-индол-3-ил)пропан-2-ил)фенил)-4-метилбензолсульфонамид (0,540 г, 1,23 ммоль), полученное на стадии 1 выше, растворяли в серной кислоте (6 мл), затем перемешивали при комнатной температуре в течение 40 минут. После завершения реакцию путем добавления льда к продукту реакции для нейтрализации добавляли по каплям 2н водный раствор гидроксида натрия. Реакционную смесь экстрагировали этилацетатом, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении и затем разделяли и очищали с помощью ЖХСД с получением целевого соединения (0,175 г, 50%, твердое вещество бледно-желтого цвета).
Стадия 3: Получение 4-(3-бромпропокси)-N-(2-(2-(5-хлор-1H-индол-3-ил)пропан-2-ил)фенил)бензолсульфонамида

Целевое соединение получали аналогично способу, описанному на стадии 2 примера 31.
Стадия 4: Получение N-(2-(2-(5-хлор-1H-индол-3-ил)пропан-2-ил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамида

Целевое соединение (17,2 мг, 33%, твердое вещество бледно-желтого цвета) получали аналогично способу, описанному в примере 148.
Соединение примера 182 получали аналогично способу, описанному в примере 181.
Соединения по примерам 183 и 184 получали аналогично способу, описанному в примере 153.
<Пример 185> Получение N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиридин-4-илокси)пропил)бензолсульфонамида

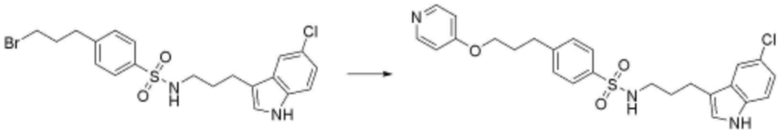
Целевое соединение (18,5 мг, 60%, твердое вещество белого цвета) получали аналогично способу, описанному в примере 148, с использованием 4-(3-бромпропил)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамида, полученного аналогично способу, описанному на стадиях 1 и 2 примера 90.
Соединение примера 186 получали аналогично способу, описанному в примере 185.
Соединения по примерам 187-190 получали аналогично способу, описанному в примере 177.
Названия соединений, структурные формулы и результаты анализа 1H ЯМР и МС соединений по примерам 1-190 суммированы в таблице 1 ниже.

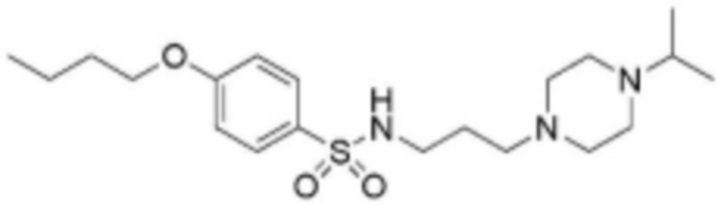
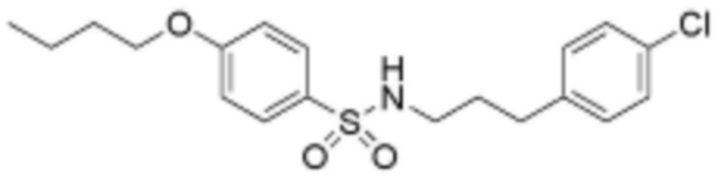
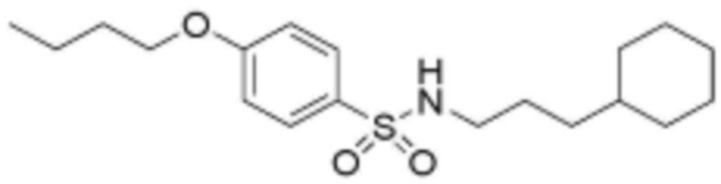



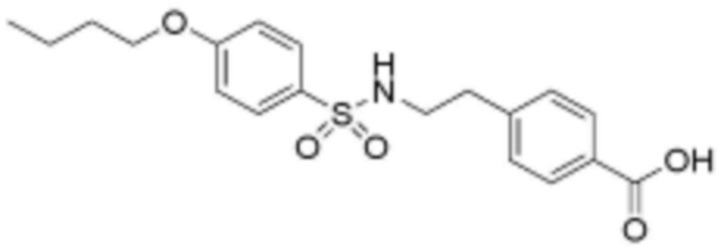





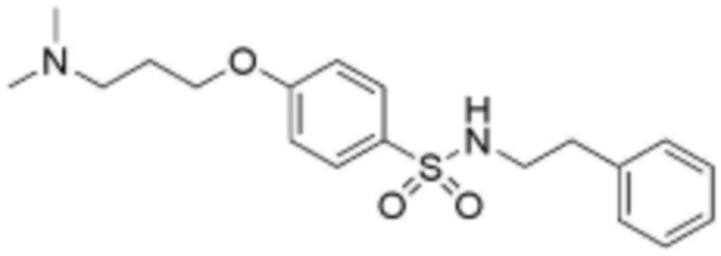




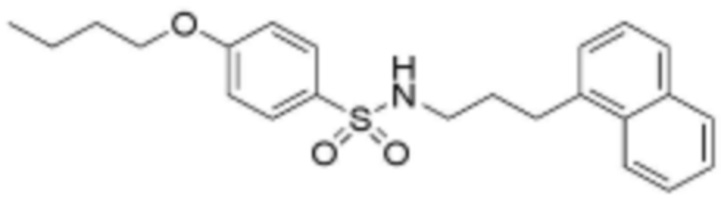


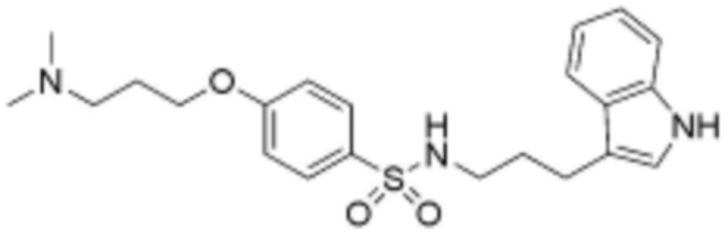
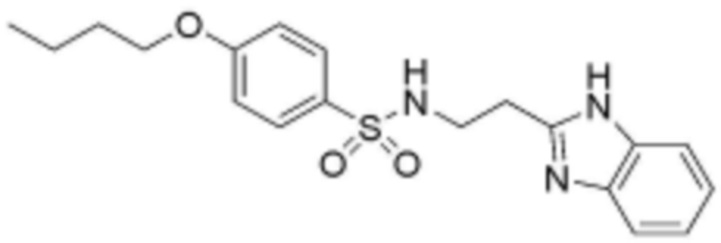

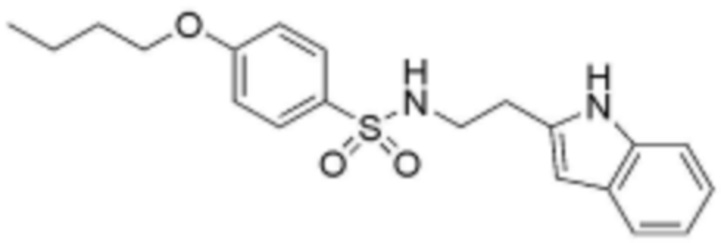





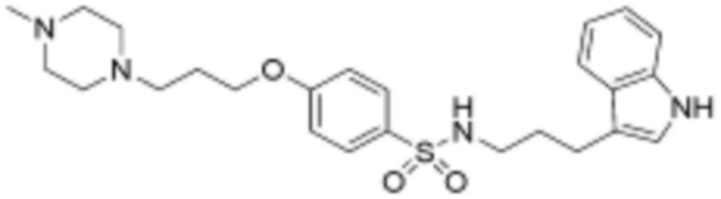

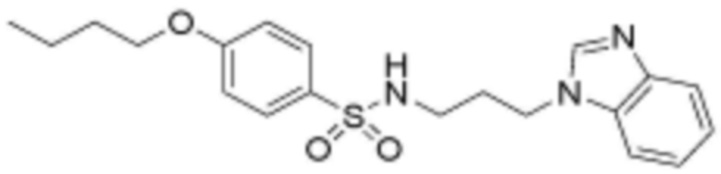


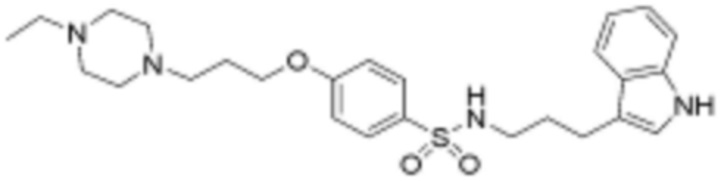

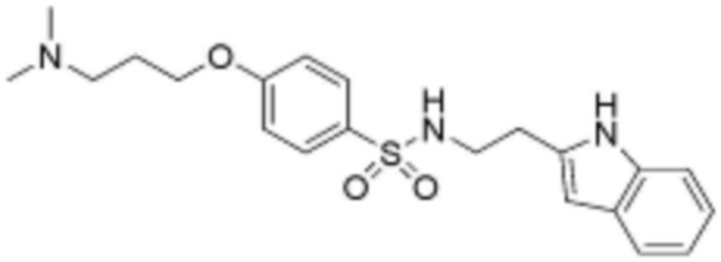





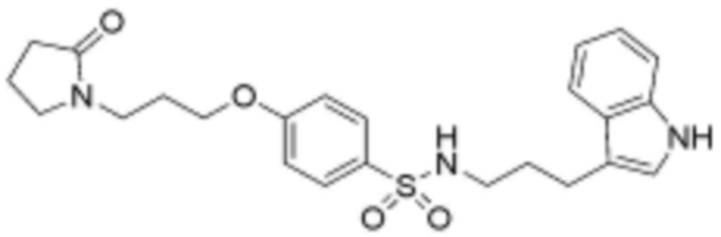








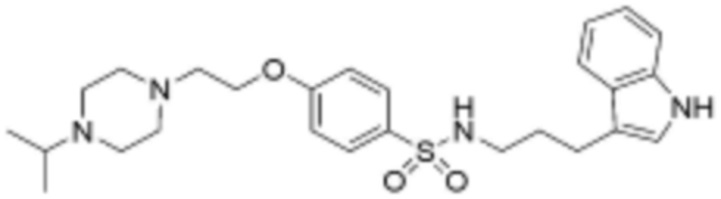


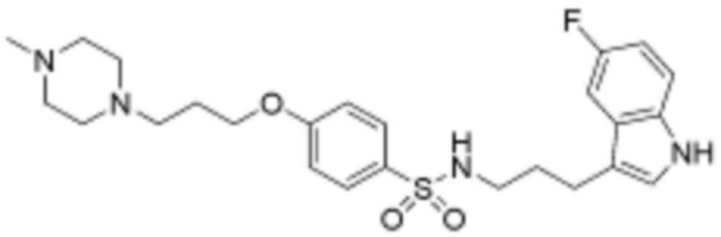
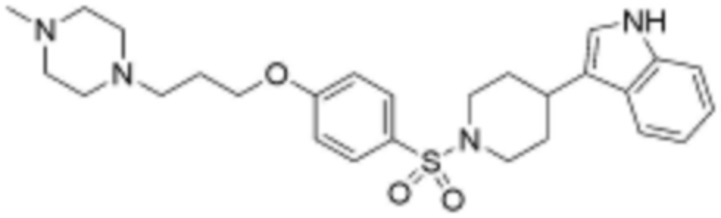
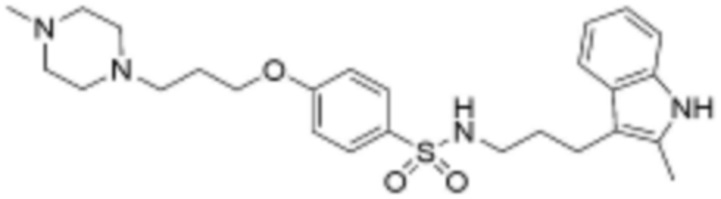
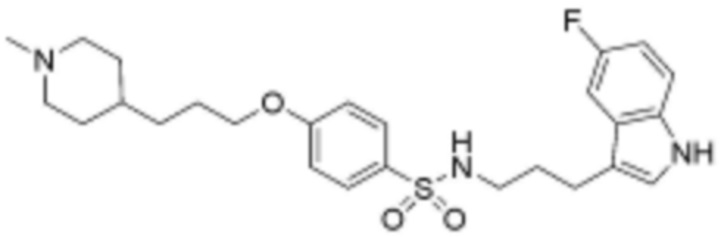


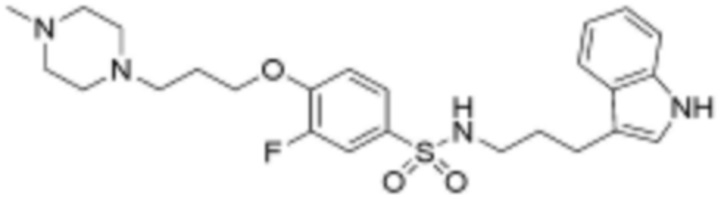
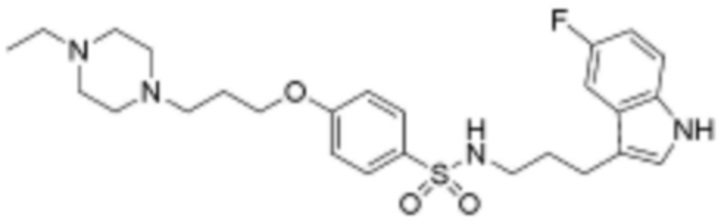


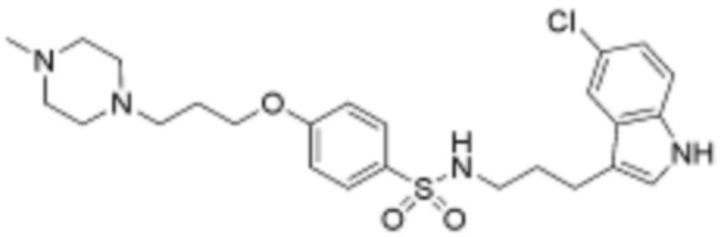




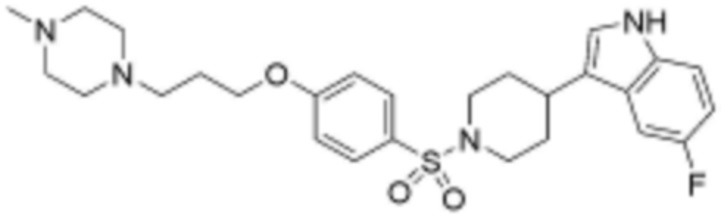




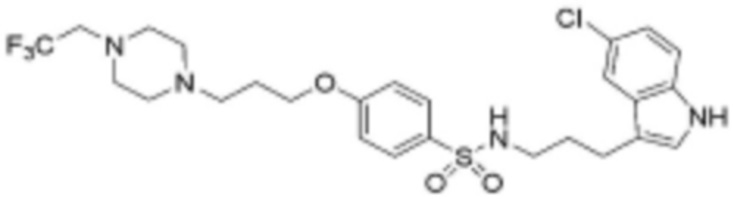
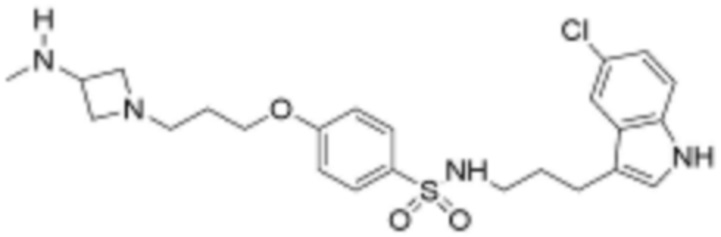



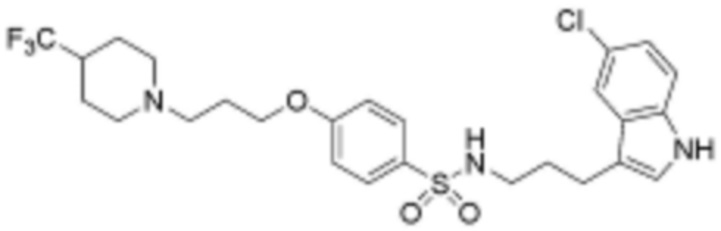

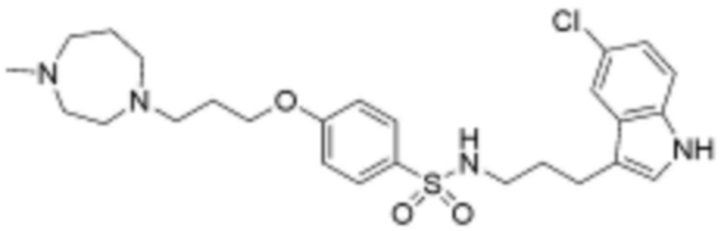



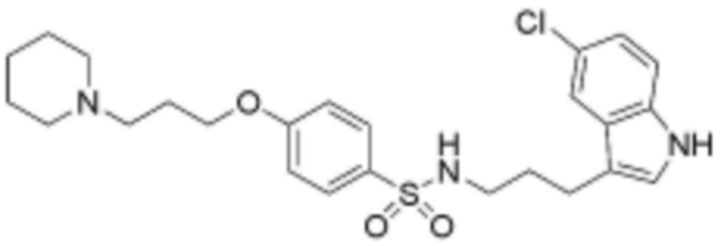







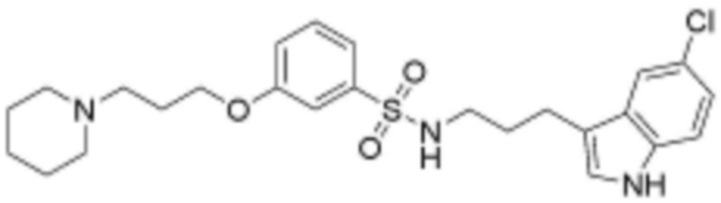




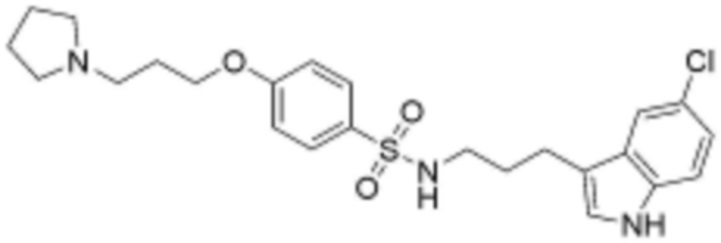

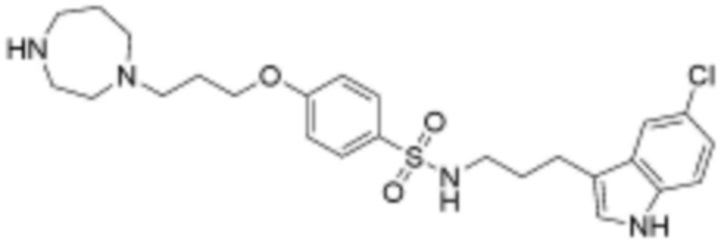



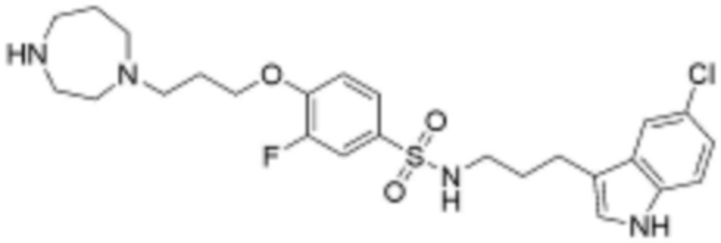
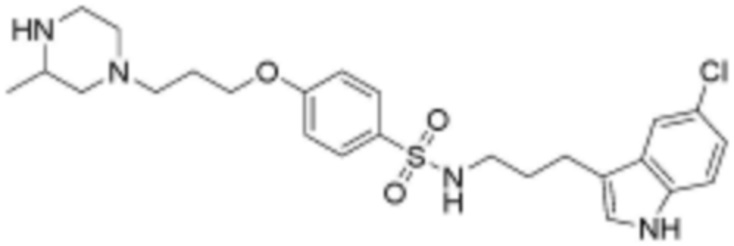


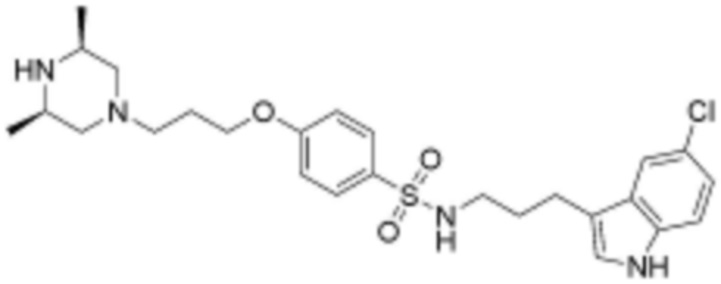



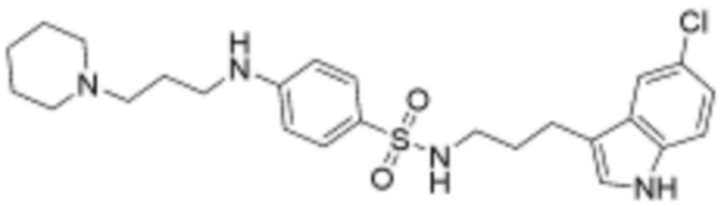
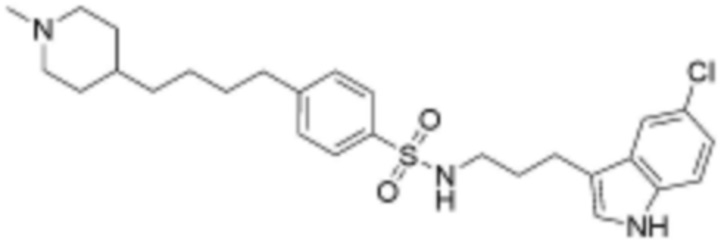








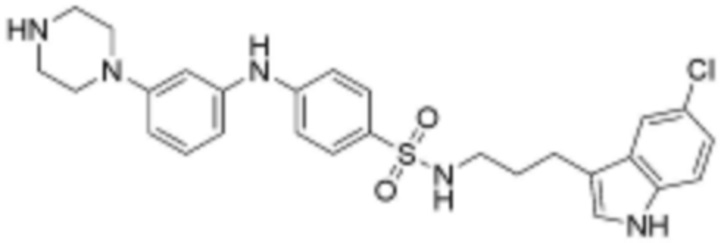






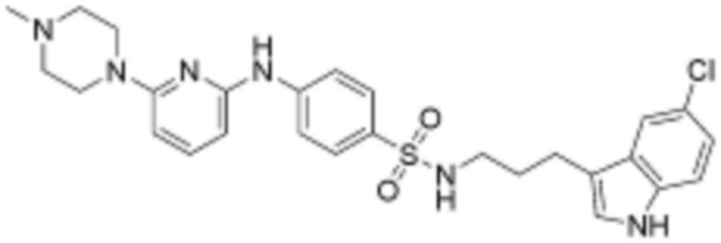
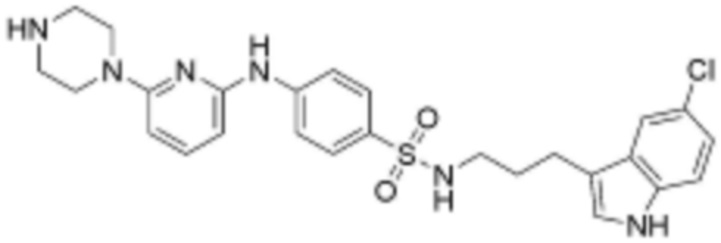

















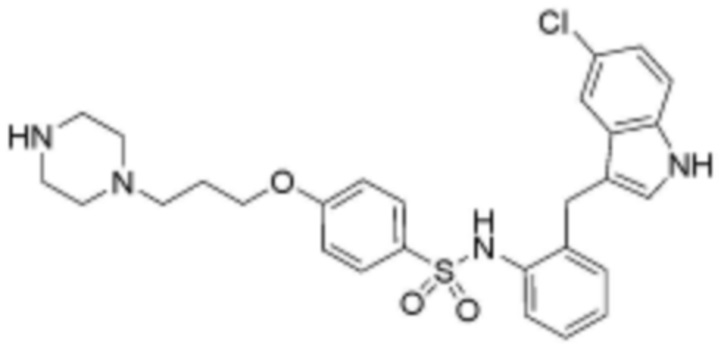
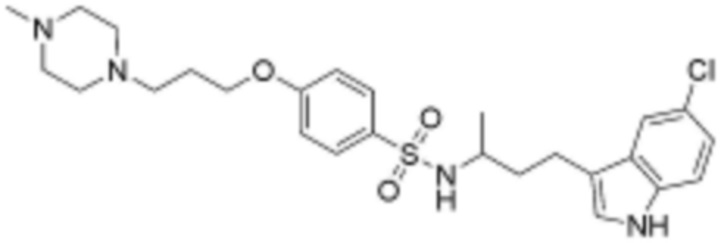
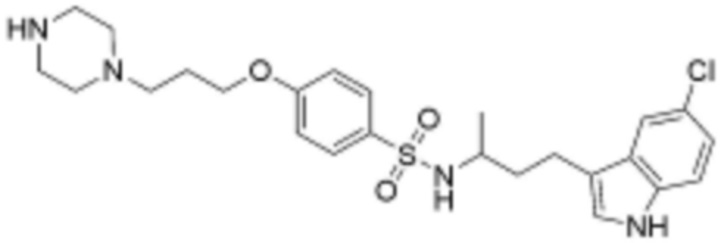

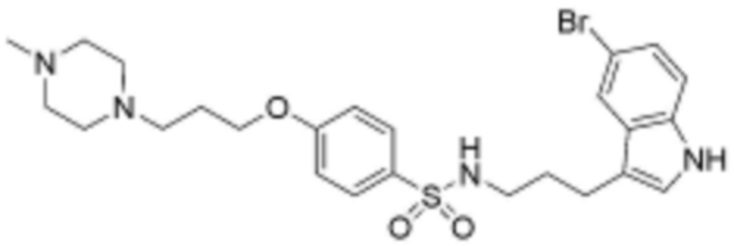

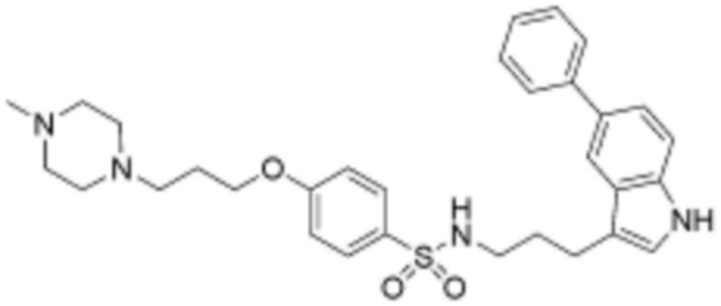





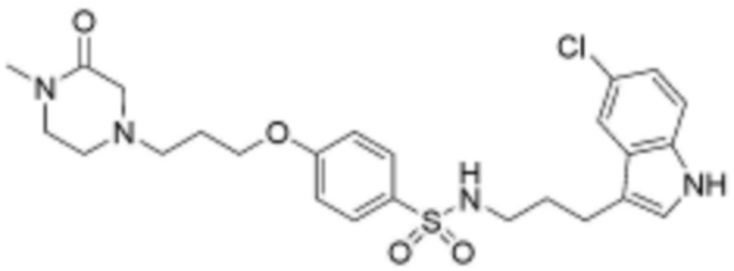

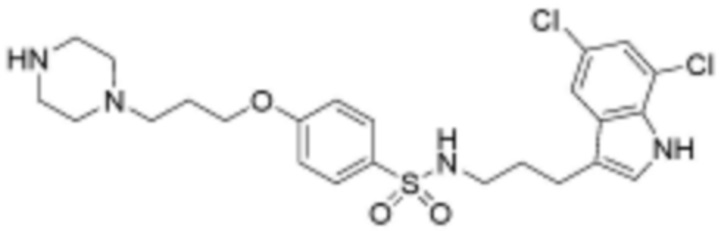

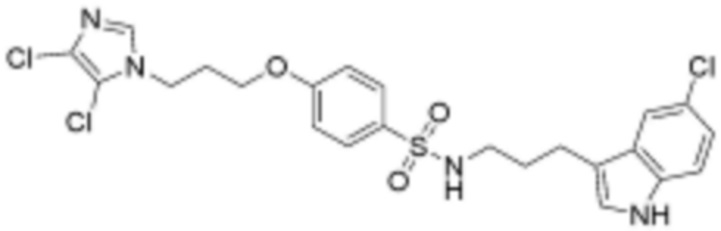
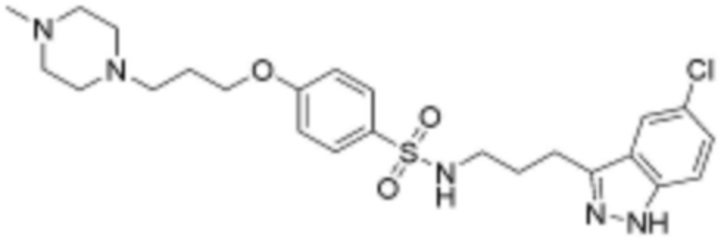



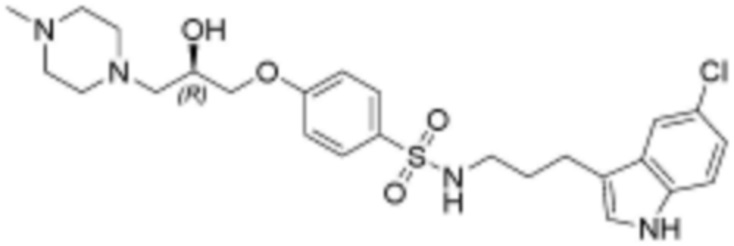
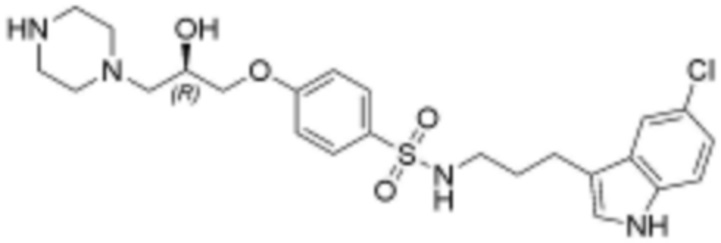




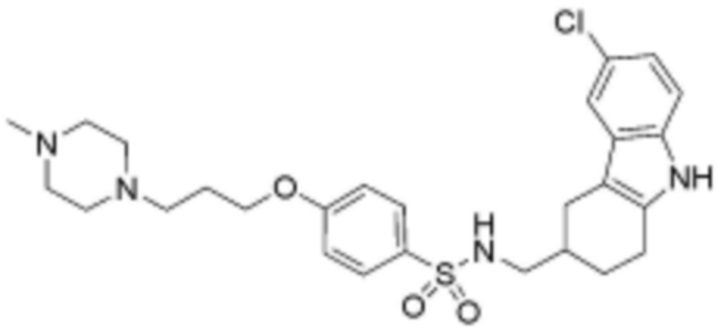








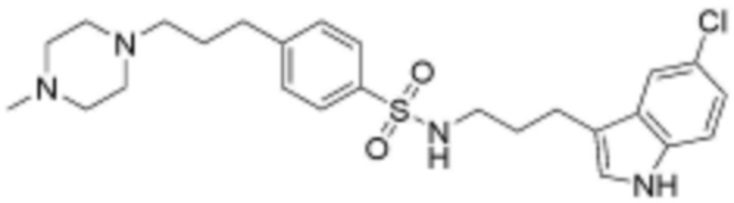
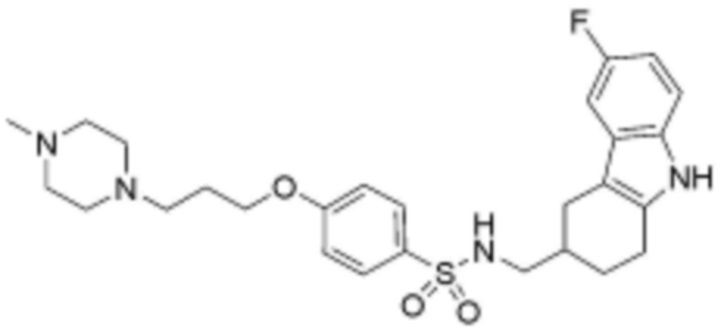


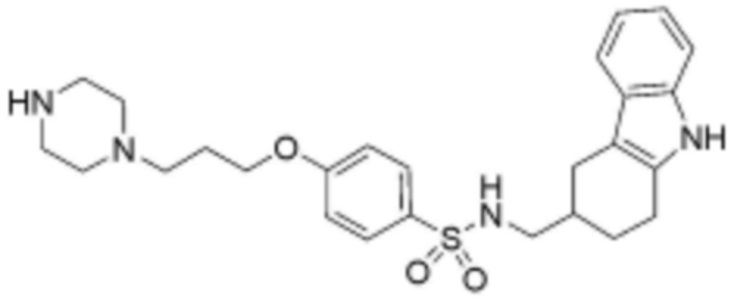
<Условия анализа и очистки>
[Условия анализа ЖХ-МС]
Название устройства: Shimadzu LCMS-2020
Колонка: ACE Excel2 C18, 75×2,1 мм
Подвижная фаза: ацетонитрил/H2O+0,1% ТФУ
Скорость потока: 0,5 мл/мин
УФ-детектор: 254 нм
[Условия очистки ЖХСД]
Название устройства: CombiFlash®Rf+
УФ-детектор: 254 нм
[Условия очистки препаративной ВЭЖХ]
Название устройства: Gilson GX-281, насос 321, UV/VIS-155
Колонка: Luna® 10 νM C18 (2) 100 Å, 250×21,2 мм
Подвижная фаза: ацетонитрил/0,1% ТФУ H2O
Скорость потока: 18 мл/мин
УФ-детектор: 254 нм
[1H ЯМР]
Название устройства: Bruker Avance (400 МГц)
<Экспериментальный пример 1> Оценка ингибирующей активности фермента Pin1
Для оценки ингибирующей активности соединения по настоящему изобретению, представленного формулой 1, ингибирующего фермент Pin1, был проведен следующий эксперимент, и результаты показаны в таблице 2 ниже.
Ферментативную активность белка Pin1 измеряли с использованием набора для анализа sensolyte Green Pin1 компании Anaspec. Субстрат разводили в буфере в соотношении 1:100 для получения концентрации 1 мкМ, а белок разводили в буфере в соотношении 1:20 для приготовления концентрации 50 мкг/мл. Кроме того, готовили проявляющий раствор, разбавляя его буфером в соотношении 1:100.
Соединение, полученное в каждом примере, разбавляли ДМСО и готовили в концентрации 100 мкМ, 10 мкМ, 1 мкМ или 0,1 мкМ с использованием буфера. После последовательного добавления в каждую лунку черного 96-луночного планшета по 10 мкл соединения, полученного в каждом примере, разведенного до четырех концентраций по вышеуказанной методике, 10 мкл белка Pin1 и 30 мкл проявляющего раствора, наконец добавляли 50 мкл субстрата. Наконец, в каждую лунку планшета добавляли по 50 мкл субстрата и проводили ферментативную реакцию после встряхивания планшета в течение 10 секунд.
Затем планшет инкубировали в течение 2 часов при комнатной температуре в среде без света и измеряли значения флуоресценции при длине волны 490 нм/520 нм с помощью устройства Synergy Neo (Tecan). Результаты показаны в таблице 2 ниже путем деления степеней от A до D для каждой концентрации (мкМ), при которой ингибируется активность фермента Pin1.
* A<1 мкМ, 1 мкМ<B<10 мкМ, 10 мкМ<C<100 мкМ, 100 мкМ<D.
Как показано в таблице 2, было подтверждено, что, когда обрабатывали соединением по примеру 56, 61, 66, 70, 75, 77, 79, 83, 86, 87, 88, 90, 91, 92, 93, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 112, 113, 114, 115, 116, 117, 118, 121, 123, 124, 125, 127, 130, 133, 134, 137, 138, 140, 141, 143, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 158, 159, 160, 161, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 174, 175, 176, 177, 178, 183, 184, 185, 186, 187, 188, 189 или 190, ингибирующая активность Pin1 проявлялась при концентрации менее 1 мкМ.
Приведенные выше результаты показывают, что соединения примеров по настоящему изобретению превосходно ингибируют фермент Pin1, и, следовательно, можно видеть, что производное бензолсульфонамида по настоящему изобретению может быть эффективно использовано в качестве фармацевтической композиции для предупреждения или лечения злокачественного новообразования.
<Экспериментальный пример 2> Оценка ингибирующей способности экспрессии белка MDA-MB 231 тройной негативной линии клеток рака молочной железы
Чтобы подтвердить способность соединения, представленного формулой 1, по настоящему изобретению ингибировать экспрессию белка в клеточной линии тройного негативного рака молочной железы MDA-MB 231, был проведен следующий эксперимент, и результаты показаны в таблице 3 ниже.
Клетки MDA-MB-231 культивировали в инкубаторе с 5% CO2 при 37°C c использованием среды RPMI с добавлением 10% FBS и 1% пенициллина/стрептомицина.
Клетки промывали PBS один раз в день, а затем заменяли новой средой. Клетки, выращенные при плотности не менее 90%, за 1 день до эксперимента открепляли с использованием трипсина/ЭДТА, а затем высевали при плотности 100000 на лунку в 6-луночный планшет.
На следующий день культивирования соединение, приготовленное в каждом примере, обрабатывали в концентрации 10 мкМ, а ДМСО обрабатывали в концентрации 0,1% от общего количества в качестве контроля с последующей инкубацией в течение 72 часов. Через 72 часа клетки дважды промывали PBS, собирали соскобом и центрифугировали при 13000 об/мин в течение 3 минут, чтобы удалить весь супернатант, и брали только клетки.
К подготовленным клеткам добавляли RIPA-буфер (содержащий 1Х ингибитор протеазы и ингибитор фосфатазы) (80 мкл/лунку) и встряхивали планшет каждые 5 минут для разрушения клеток в течение 15 минут. Разрушенные клетки центрифугировали при 13000 об/мин в течение 15 минут, чтобы собрать только супернатант белка. Полученный белок количественно определяли с использованием набора для количественного определения белка (Pierece, MA, USA), и для вестерн-блоттинга использовали всего 10 мкг белка.
Образец белка выделяли с помощью 10% SDS-PAGE и переносили на мембрану PVDF. Антитело против циклина D1 человека (Cell signal) и антитело против винкулина связывали с образцом белка, и экспрессию белка измеряли с использованием реагента ECL и оборудования LAS4000. Результаты измерений были количественно определены и обобщены с использованием ImageJ. Результаты показаны в таблице 3 ниже путем деления оценок от A до D в соответствии с количеством белка по сравнению с контрольной группой, обработанной ДМСО.
* 0<A<35%, 35%<B<50%, 50%<C<70%, 70%<D<100%.
Как показано в таблице 3, было обнаружено, что при обработке соединением по примеру 35, 61, 66, 70, 75, 77, 78, 79, 81, 83, 91, 92, 93, 98, 99 или 121 уровень экспрессии белка MDA-MB 231 тройной негативной клеточной линии рака молочной железы составляет менее 35% по сравнению с контрольной группой, обработанной ДМСО.
Приведенные выше результаты показывают, что соединения примеров по настоящему изобретению обладают превосходной способностью ингибировать экспрессию белка MDA-MB 231 тройной негативной клеточной линии рака молочной железы, и поэтому можно видеть, что производное бензолсульфонамида по настоящему изобретению можно эффективно использовать в качестве фармацевтической композиции для профилактики или лечения рака.
<Экспериментальный пример 3> Оценка жизнеспособности клеток MCF7, MCF7-SP, A2780 или A2780-SP
Для подтверждения ингибирующего действия соединения по настоящему изобретению, представленного формулой 1, на рост клеток MCF7, MCF7-SP, A2780 или A2780-SP был проведен следующий эксперимент, результаты которого представлены в таблицах 4 и 5 ниже.
Клетки MCF7 (клетки рака молочной железы) высевали в 96-луночный планшет с плотностью 3000 жизнеспособных клеток на лунку и культивировали в среде DMEM/с высоким содержанием глюкозы с добавлением 10% FBS и 1% пенициллина/стрептомицина.
На следующий день культивирования клетки обрабатывали соединением примеров 71, 76, 80, 84, 92, 99, 100 или 119 с последующим культивированием в течение 3 дней. Жизнеспособность клеток оценивали с помощью Cell-titer Glo (Promega, WC, USA), и активность люциферазы определяли с помощью планшетного ридера (Biocompare, USA).
С другой стороны, клетки MCF7-SP (стволовые клетки рака молочной железы) высевали в круглодонный 96-луночный планшет Ultra-Low Attachment (посев) с плотностью 1500 жизнеспособных клеток на лунку и культивировали в среде раковых стволовых клеток. Среда для раковых стволовых клеток состоит из нейробазальной среды с добавлением 20 нг/мл bFGF, 10 нг/мл EGF, 2,5 нг/мл амфотерицина B, HEPES, Glutamax и B27.
На следующий день культивирования клетки обрабатывали соединением по примеру 71, 76, 80, 84, 92, 99, 100 или 119 с последующим культивированием в течение 8 дней. Среду, содержащую соединение, снова добавляли на 4-й день. Жизнеспособность клеток сферы оценивали с помощью Cell-titer Glo (Promega, WC, USA), и активность люциферазы определяли с помощью планшет-ридера Tecan (Biocompare, USA).
Кроме того, клетки A2780 (клетки рака яичников) высевали в 96-луночный планшет с плотностью 3000 жизнеспособных клеток на лунку и культивировали в среде RPMI-1640 с добавлением 10% FBS и 1% пенициллина/стрептомицина.
На следующий день культивирования клетки обрабатывали соединением по примерам 57 и 69-144 с последующим культивированием в течение 3 дней. Жизнеспособность клеток оценивали с помощью Cell-titer Glo (Promega, WC, USA), и активность люциферазы определяли с помощью планшет-ридера Tecan (Biocompare, USA).
С другой стороны, клетки A2780-SP (стволовые клетки рака яичников) высевали в круглодонный 96-луночный планшет Ultra-Low Attachment (посев) с плотностью 1500 жизнеспособных клеток на лунку и культивировали в среде раковых стволовых клеток. Среда для раковых стволовых клеток состоит из нейробазальной среды с добавлением 20 нг/мл bFGF, 10 нг/мл EGF, 2,5 нг/мл амфотерицина B, HEPES, Glutamax и B27.
На следующий день культивирования клетки обрабатывали соединением по примерам 57 и 69-144 с последующим культивированием в течение 8 дней. Среду, содержащую соединение, снова добавляли туда на 4-й день. Жизнеспособность клеток сферы оценивали с помощью Cell-titer Glo (Promega, WC, USA), и активность люциферазы определяли с помощью планшет-ридера Tecan (Biocompare, USA).
* A<1 мкМ, 1 мкМ<B<10 мкМ, 10 мкМ<C.
* A<1 мкМ, 1 мкМ<B<10 мкМ, 10 мкМ<C, -: не исследовали.
Как показано в таблице 4, концентрация клеток MCF7 составляла менее 10 мкМ при обработке соединением по примеру 70, 75, 83, 91, 99 или 118, а концентрация клеток MCF7-SP была менее 10 мкМ, когда обрабатывали соединением по примерам 70, 83, 91, 99 или 118.
Как показано в таблице 5, концентрация клеток A2780 составляла менее 10 мкМ, когда обрабатывали соединением по примеру 56, 59, 61, 66, 70, 75, 83, 93, 98, 99, 100, 106, 108, 109, 121, 123, 124, 130, 153, 154, 164, 165, 167, 169, 170, 173, 175, 176, 177 или 178, и концентрация была менее 1 мкМ, когда обрабатывали соединением по примеру 56, 59, 61, 66, 70, 79, 83, 88, 90, 91, 92, 93, 99, 100, 103, 104, 105, 106, 108, 109, 110, 111, 112, 115, 118, 121, 134, 150, 151, 152, 153, 154, 157, 158, 159, 161, 164, 165, 167, 168, 169, 170, 171, 172, 173, 174, 175, 176, 177, 178, 183, 184, 186, 187, 188, 189 или 190.
Приведенные выше результаты показывают, что соединения по примерам по настоящему изобретению обладают превосходным ингибирующим действием на рост клеток MCF7, MCF7-SP, A2780 или A2780-SP, и поэтому можно видеть, что производное бензолсульфонамида в соответствии с настоящим изобретением может быть эффективно использовано в качестве фармацевтической композиции для предупреждения или лечения злокачественного новообразования.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ДИАМИНОПИРИМИДИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2012 |
|
RU2587981C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПУРИНИЛПИРИДИНИЛАМИНО-2,4-ДИФТОРФЕНИЛСУЛЬФОНАМИДА, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ RAF-КИНАЗЫ, СОДЕРЖАЩАЯ ДАННОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2011 |
|
RU2550038C2 |
| СОЕДИНЕНИЕ, ИНГИБИРУЮЩЕЕ ОБРАЗОВАНИЕ КОМПЛЕКСА С-MYC/MAX/ДНК | 2017 |
|
RU2727177C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2021 |
|
RU2826628C1 |
| НОВОЕ ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ, ОБЛАДАЮЩЕЕ ЭФФЕКТОМ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2744168C1 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2833354C1 |
| ПРОИЗВОДНЫЕ ДИАМИНОПИРИМИДИНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2012 |
|
RU2587493C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| СПОСОБ СИНТЕЗА БИЛИРУБИНА | 2022 |
|
RU2838556C2 |
| ПРОТИВОРАКОВАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2017 |
|
RU2724340C1 |
Изобретение относится к соединению, ингибирующему Pin1 (пептидил-пролил-цис/транс-изомеразу, взаимодействующую с NIMA 1), или к его фармацевтически приемлемой соли, способу получения указанного соединения, фармацевтической композиции, содержащей в качестве активного ингредиента соединение, которое ингибирует Pin1, и его применению для предупреждения или лечения злокачественного новообразования. 9 н. и 9 з.п. ф-лы, 5 табл., 185 пр.
1. Соединение, представленное формулой 1, или его фармацевтически приемлемая соль:
[Формула 1]
 ,
,
где
Ar представляет собой 6-10-членный арил или 5-6-членный гетероарил, содержащий один или два гетероатома, выбранных из группы, состоящей из N, S и O;
R1 и R2 независимо представляют собой водород или галоген;
R3 представляет собой замещенный прямой или разветвленный C1-8алкил, NRa1Ra2, ORa3,  или
или  , где Ra1 представляет собой водород, Ra2 и Ra3 независимо представляют собой замещенный прямой или разветвленный C1-8алкил, незамещенный или замещенный фенил или незамещенный или замещенный 5-6-членный гетероарил, содержащий один-три гетероатома, выбранных из группы, состоящей из N, S и O,
, где Ra1 представляет собой водород, Ra2 и Ra3 независимо представляют собой замещенный прямой или разветвленный C1-8алкил, незамещенный или замещенный фенил или незамещенный или замещенный 5-6-членный гетероарил, содержащий один-три гетероатома, выбранных из группы, состоящей из N, S и O,
где замещенный алкил замещен одним заместителем, выбранным из группы, состоящей из незамещенного или замещенного 4-8-членного гетероциклоалкила, содержащего один или два гетероатома, выбранных из группы, состоящей из N, S и O, и незамещенного или замещенного 4-6-членного гетероарила, содержащего один-три гетероатомов, выбранных из группы, состоящей из N, S и O, и необязательно дополнительно замещен гидрокси, или образует 3-6-членный циклоалкил с замещенным атомом углерода,
где замещенный фенил и замещенный 5-6-членный гетероарил независимо замещены одним-тремя заместителями, выбранными из группы, состоящей из 4-8-членного незамещенного или замещенного гетероциклоалкила, содержащего один или два гетероатома, выбранных из группы, состоящей из N, S и O, и галогена, и
замещенный 4-8-членный гетероциклоалкил замещен одним-тремя заместителями, выбранными из группы, состоящей из прямого или разветвленного С1-5алкила, незамещенного или замещенного одним или более атомами галогена, прямого или разветвленного С1-5алкилкарбонила, NRb1Rb2, галогена, гидрокси и оксо, и
замещенный 4-6-членный гетероарил замещен одним-тремя заместителями, выбранными из группы, состоящей из прямого или разветвленного C1-5алкила и галогена,
Rb1 и Rb2 независимо представляют собой водород или прямой или разветвленный C1-6алкил;
L1 представляет собой 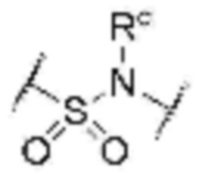 , где Rc представляет собой водород;
, где Rc представляет собой водород;
L2 представляет собой прямой или разветвленный C1-8алкилен, незамещенный или замещенный одним гидрокси, или фенилен;
Z представляет собой C3-8циклоалкил, конденсированный с незамещенным или замещенным 9-членным гетероарилом, содержащим один или два гетероатома, представляющих собой N, замещенный 9-членный гетероарил, содержащий один или два гетероатома, представляющих собой N, или замещенный 9-членный гетероарилC1-5алкил, содержащий один или два гетероатома, представляющих собой N,
где Z представляет собой замещенный 9-членный гетероарилC1-5алкил, содержащий один или два гетероатома, представляющих собой N, когда L2 представляет собой фенилен,
где замещенный 9-членный гетероарил в замещенном 9-членном гетероариле и замещенном гетероарилС1-5алкиле независимо замещен галогеном, и необязательно дополнительно замещен прямым или разветвленным С1-5алкилом.
2. Соединение, или его фармацевтически приемлемая соль по п.1, где:
Ar представляет собой 6-10-членный арил или 5-6-членный гетероарил, содержащий один или два гетероатома, выбранных из группы, состоящей из N, S и O;
R1 и R2 независимо представляют собой водород или фтор;
R3 представляет собой замещенный прямой или разветвленный C1-6алкил, NRa1Ra2, ORa3, или ,
где Ra1 представляет собой водород, Ra2 и Ra3 независимо представляют собой замещенный прямой или разветвленный C1-6алкил, незамещенный или замещенный фенил или незамещенный или замещенный 5-6-членный гетероарил, содержащий один или два гетероатома, выбранных из группы, состоящей из N, S и O,
где замещенный алкил замещен одним заместителем, выбранным из группы, состоящей из незамещенного или замещенного 4-7-членного гетероциклоалкила, содержащего один или два гетероатома, выбранных из группы, состоящей из N, S и O, и незамещенного или замещенного 4-6-членного гетероарила, содержащего один-три гетероатома, выбранных из группы, состоящей из N, S и O, и дополнительно замещен гидрокси, или образует 3-5-членный циклоалкил с замещенным атомом углерода,
где замещенный фенил и замещенный 5-6-членный гетероарил независимо замещены одним-тремя заместителями, выбранными из группы, состоящей из 4-7-членного незамещенного или замещенного гетероциклоалкила, содержащего один или два гетероатома, выбранных из группы, состоящей из N, S и O, и галогена, и
замещенный 4-7-членный гетероциклоалкил независимо замещен одним-тремя заместителями, выбранными из группы, состоящей из прямого или разветвленного C1-4алкила, незамещенного или замещенного одним или несколькими галогенами, прямого или разветвленного C1-4алкилкарбонила, NRb1Rb2, хлора, фтора, брома, гидрокси и оксо, и
замещенный 4-6-членный гетероарил замещен одним-тремя заместителями, выбранными из группы, состоящей из прямого или разветвленного C1-4 алкила, хлора, фтора и брома,
Rb1 и Rb2 независимо представляют собой водород или прямой или разветвленный C1-3 алкил;
L1 представляет собой , где Rc представляет собой водород;
L2 представляет собой прямой или разветвленный C1-6алкилен, незамещенный или замещенный одним заместителем, выбранным из гидрокси и фенилена;
Z представляет собой C4-7циклоалкил, конденсированный с незамещенным или замещенным 9-членным гетероарилом, содержащим один или два гетероатома, представляющих собой N, замещенный 9-членный гетероарил, содержащий один или два гетероатома, представляющих собой N, или замещенный 9-членный гетероарилC1-4алкил, содержащий один или два гетероатома, представляющих собой N,
где Z представляет собой замещенный 9-членный гетероарилC1-4алкил, содержащий один или два гетероатома, представляющих собой N, когда L2 представляет собой фенилен,
где замещенный 9-членный гетероарил в замещенном 9-членном гетероариле и замещенном гетероарилС1-4алкиле независимо замещен одним или несколькими заместителями, выбранными из группы, состоящей из фтора, хлора, и брома, и необязательно дополнительно замещен прямым или разветвленным С1-4алкилом.
3. Соединение, или его фармацевтически приемлемая соль по п.1, где:
Ar представляет собой фенил, нафтил, пиридил или тиазолил;
R1 и R2 независимо представляют собой водород или фтор;
R3 представляет собой замещенный прямой или разветвленный C1-5алкил, NRa1Ra2, ORa3, или ,
где Ra1 представляет собой гидрокси, Ra2 и Ra3 независимо представляют собой замещенный прямой или разветвленный C1-5алкил, незамещенный или замещенный фенил или незамещенный или замещенный 5-6-членный гетероарил, содержащий один-три гетероатома, выбранных из группы, состоящей из N, S и O,
где замещенный алкил замещен одним или несколькими заместителями, выбранными из группы, состоящей из незамещенного или замещенного 4-7-членного гетероциклоалкила, содержащего один или два гетероатома, выбранных из группы, состоящей из N, S и O, и незамещенного или замещенного 5-6-членного гетероарила, содержащего один-три гетероатома, выбранных из группы, состоящей из N, S и O, или необязательно дополнительно замещен гидрокси, или образует циклопропил с замещенным атомом углерода,
где замещенный фенил и замещенный 5-6-членный гетероарил независимо замещены одним или несколькими заместителями, выбранными из группы, состоящей из незамещенного или замещенного 4-7-членного гетероциклоалкила, содержащего один или два гетероатома, выбранных из группы, состоящей из N, S и O, незамещенного или замещенного 5-6-членного гетероарила, содержащего один-три гетероатома, выбранных из группы, состоящей из N, S и O, NRb1Rb2, 3-4-членного циклоалкила, галогена, гидрокси и сульфонила, и
замещенный 4-7-членный гетероциклоалкил замещен одним-тремя заместителями, выбранными из группы, состоящей из прямого или разветвленного C1-3алкила, незамещенного или замещенного одним или несколькими атомами фтора, прямого или разветвленного C1-4 алкилкарбонила, NRb1Rb2, хлора, фтора, брома, гидрокси и оксо, и
замещенный 5-6-членный гетероарил замещен одним-тремя заместителями, выбранными из группы, состоящей из прямого или разветвленного C1-3 алкила, хлора, фтора и брома,
Rb1 и Rb2 независимо представляют собой водород, метил или этил;
L1 представляет собой , где Rc представляет собой водород;
L2 представляет собой прямой или разветвленный C1-4алкилен, незамещенный или замещенный гидрокси, или фенилен;
Z представляет собой C5-6циклоалкил, конденсированный с незамещенным 9-членным гетероарилом, содержащим один или два гетероатома, представляющих собой N, замещенный 9-членный гетероарил, содержащий один или два гетероатома, представляющих собой N, или 9-членный гетероарилC1-3алкил, содержащий один или два гетероатома, представляющих собой N,
где Z представляет собой 9-членный гетероарилC1-4алкил, содержащий один или два гетероатома, представляющих собой N, когда L2 представляет собой фенилен,
где замещенный 9-членный гетероарил в замещенном 9-членном гетероариле и замещенном 9-членном гетероарилС1-3алкиле независимо замещены одним-тремя заместителями, выбранными из группы, состоящей из фтора, хлора и брома, и необязательно дополнительно замещен прямым или разветвленным С1-3алкилом.
4. Соединение, или его фармацевтически приемлемая соль по п.1, где:
Ar представляет собой фенил, нафтил, пиридин или тиазол;
R1 и R2 независимо представляют собой водород или фтор;
R3 представляет собой  ,
,  ,
,  ,
,  ,
,  ,
, 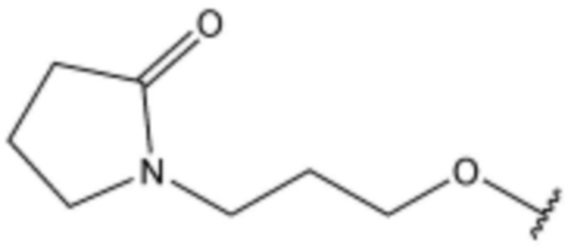 ,
,  ,
,  ,
, 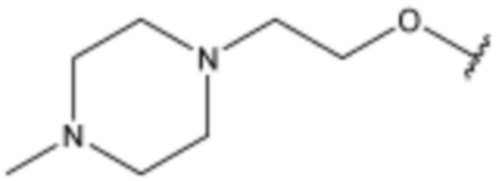 ,
, 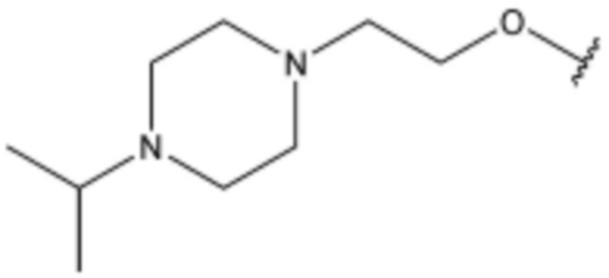 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 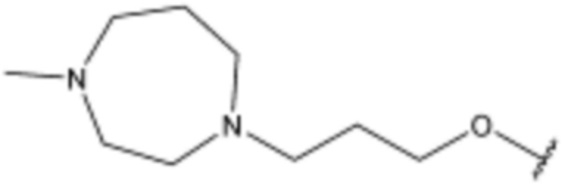 ,
, 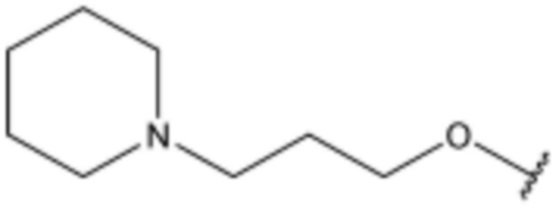 ,
,  ,
,  ,
,  ,
,  ,
, 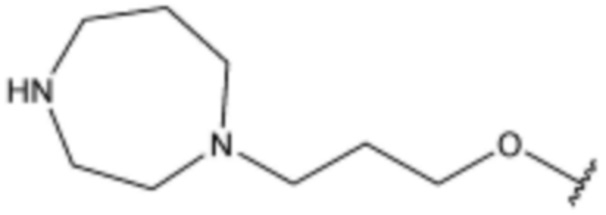 ,
,  ,
,  ,
,  ,
, 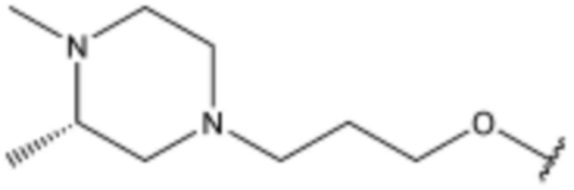 ,
, 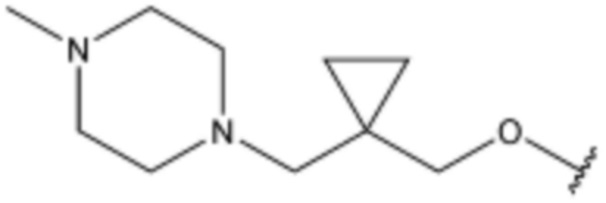 ,
, 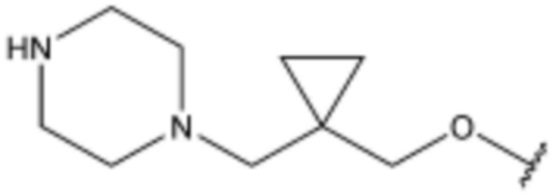 ,
, 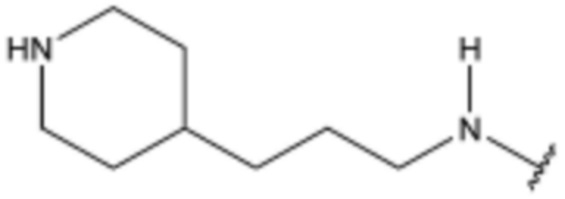 ,
, 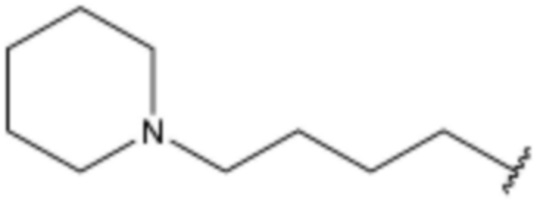 ,
, 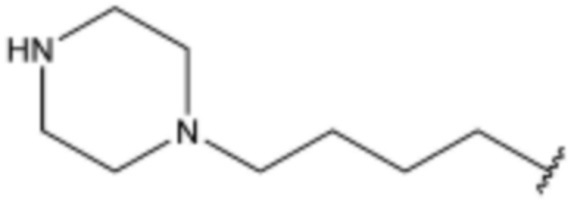 ,
,  ,
,  ,
,  ,
, 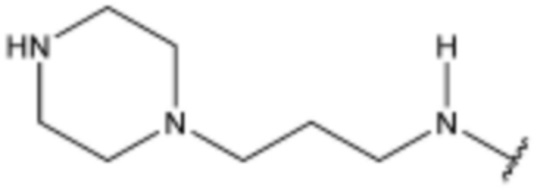 ,
,  ,
, 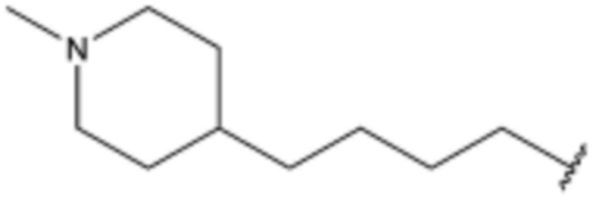 ,
, 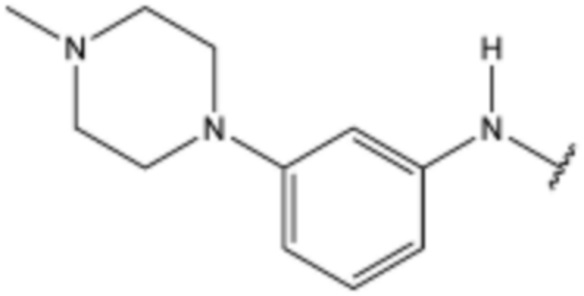 ,
,  ,
,  ,
, 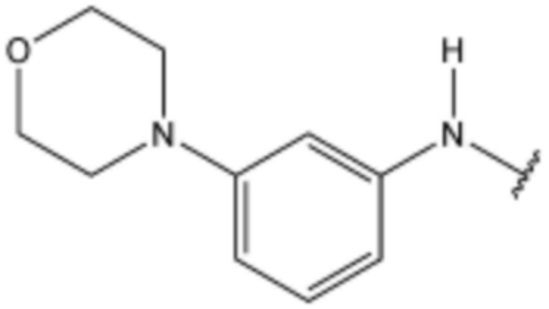 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 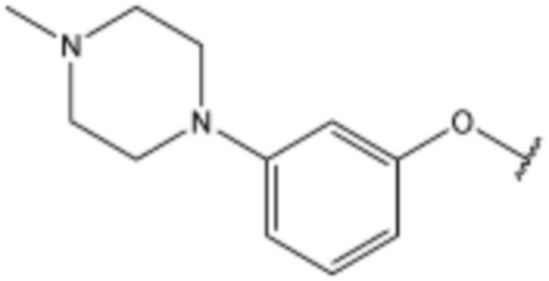 ,
,  ,
,  ,
, 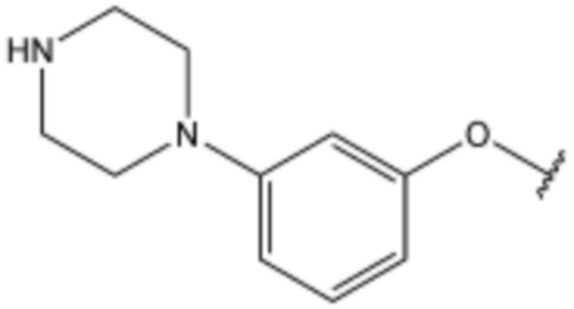 ,
, 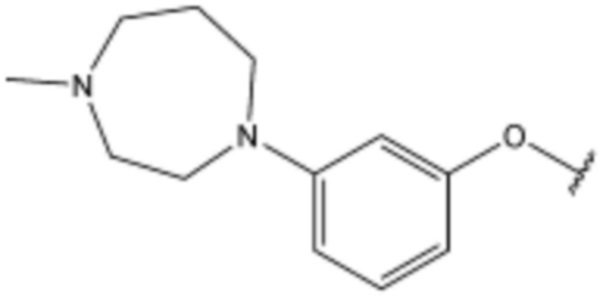 ,
,  ,
, 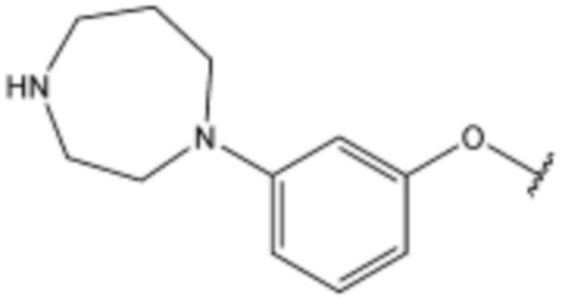 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 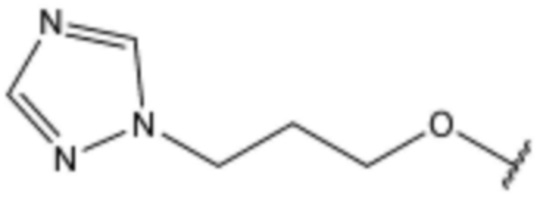 ,
,  ,
,  ,
, 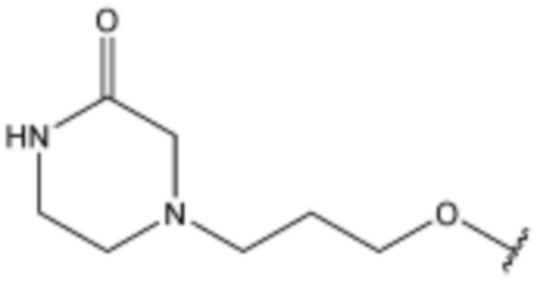 ,
, 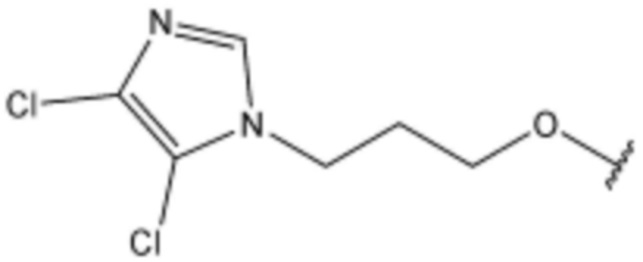 ,
,  ,
,  ,
,  ,
, 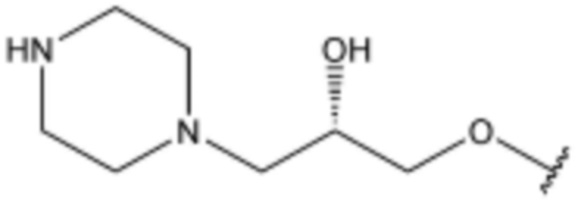 ,
,  ,
,  ,
,  или
или 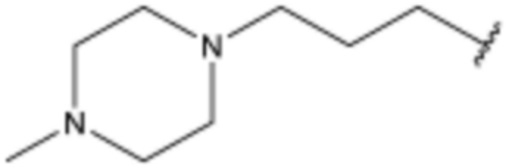 ;
;
L1 представляет собой  ;
;
L2 представляет собой  ,
,  ,
,  ,
,  ,
,  или
или  ;
;
Z представляет собой  ,
, 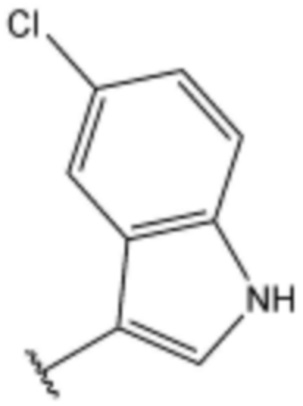 ,
,  ,
,  ,
, 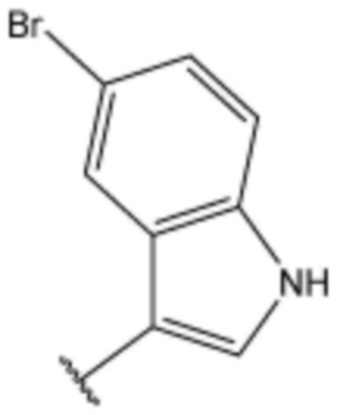 ,
,  ,
, 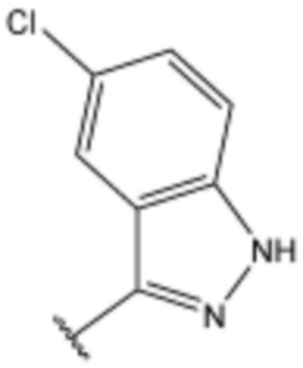 ,
,  ,
,  ,
, 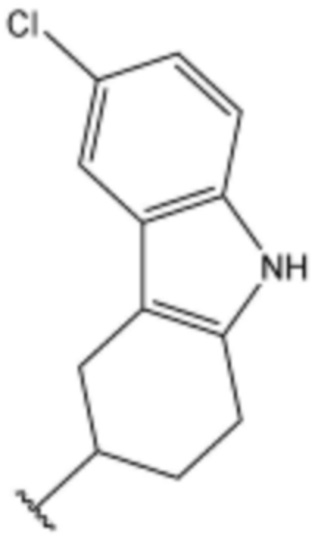 ,
,  ,
, 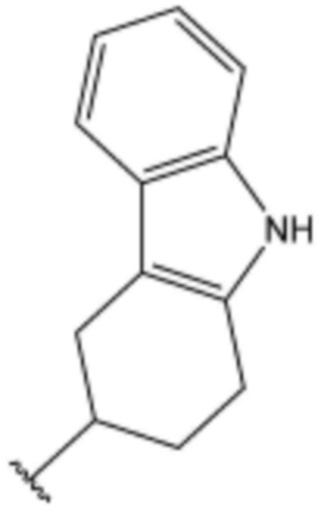 или
или  .
.
5. Соединение, его стереоизомер или его фармацевтически приемлемая соль где соединение выбрано из группы, состоящей из следующих соединений:
<56> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<59> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(1-метилпиперидин-4-ил)пропокси)бензолсульфонамид;
<61> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<63> 4-(3-(4-этилпиперазин-1-ил)пропокси)-N-(3-(5-фтор-1H-индол-3-ил)пропил)бензолсульфонамид;
<66> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<70> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<71> 5-фтор-3-(1-((4-(3-(4-метилпиперазин-1-ил)пропокси)фенил)сульфонил)пиперидин-4-ил)-1H-индол;
<72> 4-(3-((3S,5R)-3,5-диметилпиперазин-1-ил)пропокси)-N-(3-(5-фтор-1H-индол-3-ил)пропил)бензолсульфонамид;
<73> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(4-изобутирилпиперазин-1-ил)пропокси)бензолсульфонамид;
<74> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(4-(2,2,2-трифторэтил)пиперазин-1-ил)пропокси)бензолсульфонамид;
<75> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-гидроксипиперидин-1-ил)пропокси)бензолсульфонамид;
<76> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-(2,2,2-трифторэтил)пиперазин-1-ил)пропокси)бензолсульфонамид;
<77> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-(метиламино)азетидин-1-ил)пропокси)бензолсульфонамид;
<78> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-хлорпиперидин-1-ил)пропокси)бензолсульфонамид;
<79> N-(3-(5-хлор-1H-индол-3-ил)пропил)-6-(3-(пиперазин-1-ил)пропокси)пиридин-3-сульфонамид;
<80> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-фторпиперидин-1-ил)пропокси)бензолсульфонамид;
<81> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-(трифторметил)пиперидин-1-ил)пропокси)бензолсульфонамид;
<82> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4,4-дифторпиперидин-1-ил)пропокси)бензолсульфонамид;
<83> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метил-1,4-диазепан-1-ил)пропокси)бензолсульфонамид;
<84> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(пиперидин-1-ил)пропокси)бензолсульфонамид;
<86> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)нафталин-1-сульфонамид;
<87> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперидин-1-ил)пропокси)бензолсульфонамид;
<88> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(пиперидин-4-ил)пропил)амино)бензолсульфонамид;
<90> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(пиперидин-1-ил)бутил)бензолсульфонамид;
<91> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(пиперазин-1-ил)бутил)бензолсульфонамид;
<92> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(1-метилпиперидин-4-ил)пропокси)бензолсульфонамид;
<93> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперидин-4-ил)пропокси)бензолсульфонамид;
<94> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-(3-(пиперидин-4-ил)пропокси)бензолсульфонамид;
<95> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-(3-(пиперидин-1-ил)пропокси)бензолсульфонамид;
<96> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<97> N-(3-(5-фтор-1H-индол-3-ил)пропил)-4-(3-(пиперидин-4-ил)пропокси)бензолсульфонамид;
<98> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(4-метилпиперазин-1-ил)пропил)амино)бензолсульфонамид;
<99> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(пиперидин-4-ил)бутил)бензолсульфонамид;
<100> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пирролидин-1-ил)пропокси)бензолсульфонамид;
<101> 4-(3-(азепан-1-ил)пропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<102> 4-(3-(1,4-диазепан-1-ил)пропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<103> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-фтор-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<104> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-фтор-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<105> N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-фтор-4-(3-(4-гидроксипиперидин-1-ил)пропокси)бензолсульфонамид;
<106> 4-(3-(1,4-диазепан-1-ил)пропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-3-фторбензолсульфонамид;
<107> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<108> (R)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<109> (S)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<110> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-((3S,5R)-3,5-диметилпиперазин-1-ил)пропокси)бензолсульфонамид;
<111> (S)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3,4-диметилпиперазин-1-ил)пропокси)бензолсульфонамид;
<112> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(4-метилпиперазин-1-ил)бутил)бензолсульфонамид;
<113> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(пиперазин-1-ил)пропил)амино)бензолсульфонамид;
<114> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(пиперидин-1-ил)пропил)амино)бензолсульфонамид;
<115> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(4-(1-метилпиперидин-4-ил)бутил)бензолсульфонамид;
<116> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((1-((4-метилпиперазин-1-ил)метил)циклопропил)метокси)бензолсульфонамид;
<117> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((1-(пиперазин-1-илметил)циклопропил)метокси)бензолсульфонамид;
<118> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)фенокси)бензолсульфонамид;
<119> 4-(3-бромфенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<120> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперидин-1-ил)фенокси)бензолсульфонамид;
<121> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)фенокси)бензолсульфонамид;
<122> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метил-1,4-диазепан-1-ил)фенокси)бензолсульфонамид;
<123> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(4-метилпиперазин-1-ил)фенил)амино)бензолсульфонамид;
<124> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(пиперазин-1-ил)фенил)амино)бензолсульфонамид;
<125> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((4-(4-метилпиперазин-1-ил)пиримидин-2-ил)амино)бензолсульфонамид;
<126> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-морфолинофенил)амино)бензолсульфонамид;
<127> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((4-(пиперазин-1-ил)пиримидин-2-ил)амино)бензолсульфонамид;
<128> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-морфолинофенокси)бензолсульфонамид;
<129> 4-(3-(1,4-диазепан-1-ил)фенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<130> 4-(3,5-бис(4-метилпиперазин-1-ил)фенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<131> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((6-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)бензолсульфонамид;
<132> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((6-(пиперазин-1-ил)пиридин-2-ил)амино)бензолсульфонамид;
<133> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((4-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)бензолсульфонамид;
<134> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((4-(пиперазин-1-ил)пиридин-2-ил)амино)бензолсульфонамид;
<135> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пирролидин-1-ил)фенокси)бензолсульфонамид;
<136> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-гидроксипиперидин-1-ил)фенокси)бензолсульфонамид;
<137> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-фтор-5-(4-метилпиперазин-1-ил)фенокси)бензолсульфонамид;
<138> 4-(3-бром-5-(4-метилпиперазин-1-ил)фенокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<139> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((3-(4-гидроксипиперидин-1-ил)фенил)амино)бензолсульфонамид;
<140> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((6-хлор-4-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)бензолсульфонамид;
<141> 4-((4,6-бис(4-метилпиперазин-1-ил)пиридин-2-ил)амино)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<142> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-((4-(4-метилпиперазин-1-ил)фенил)амино)бензолсульфонамид;
<145> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(метилсульфонил)пропокси)бензолсульфонамид;
<146> N-(3-(5-хлор-1H-индол-3-ил)пропил)-2-((3-(пиперазин-1-ил)пропил)амино)тиазол-5-сульфонамид;
<147> N-(3-(5-хлор-2-метил-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<148> 4-(4-(1H-имидазол-1-ил)бутил)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<149> N-(2-((5-хлор-1H-индол-3-ил)метил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<150> N-(2-((5-хлор-1H-индол-3-ил)метил)фенил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<151> N-(4-(5-хлор-1H-индол-3-ил)бутан-2-ил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<152> N-(4-(5-хлор-1H-индол-3-ил)бутан-2-ил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<153> N-(3-(5-бром-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<154> N-(3-(5-бром-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<157> N-(3-(5-хлор-2-метил-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<158> N-(2-(5-хлор-1H-индол-3-ил)этил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<159> N-(2-(5-хлор-1H-индол-3-ил)этил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<160> 4-(3-(1H-1,2,4-triazol-1-ил)пропокси)-N-(3-(5-хлор-1H-индол-3-ил)пропил)бензолсульфонамид;
<161> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(2-метил-1H-имидазол-1-ил)пропокси)бензолсульфонамид;
<162> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метил-3-оксопиперазин-1-ил)пропокси)бензолсульфонамид;
<163> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(3-оксопиперазин-1-ил)пропокси)бензолсульфонамид;
<164> N-(3-(5,7-дихлор-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<165> N-(3-(5,7-дихлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<166> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4,5-дихлор-1H-имидазол-1-ил)пропокси)бензолсульфонамид;
<167> N-(3-(5-хлор-1H-индазол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<168> N-(3-(5-хлор-1H-индазол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<169> N-(3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<170> N-(3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<171> (R)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(2-гидрокси-3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<172> (R)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(2-гидрокси-3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<173> (S)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(2-гидрокси-3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<174> (S)-N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(2-гидрокси-3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<175> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4,5-диметил-1H-имидазол-1-ил)пропокси)бензолсульфонамид;
<176> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(2,4,5-триметил-1H-имидазол-1-ил)пропокси)бензолсульфонамид;
<177> N-((6-хлор-2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<178> N-((6-хлор-2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<179> N-(3-(5-хлор-1H-индол-3-ил)циклогексил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<180> N-(3-(5-хлор-1H-индол-3-ил)циклогексил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<181> N-(2-(2-(5-хлор-1H-индол-3-ил)пропан-2-ил)фенил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<182> N-(2-(2-(5-хлор-1H-индол-3-ил)пропан-2-ил)фенил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<183> N-(3-(5,6-дихлор-1H-индол-3-ил)пропил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<184> N-(3-(5,6-дихлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<185> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(пиридин-4-илокси)пропил)бензолсульфонамид;
<186> N-(3-(5-хлор-1H-индол-3-ил)пропил)-4-(3-(4-метилпиперазин-1-ил)пропил)бензолсульфонамид;
<187> N-((6-фтор-2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)-4-(3-(4-метилпиперазин-1-ил)пропокси)бензолсульфонамид;
<188> N-((6-фтор-2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)-4-(3-(пиперазин-1-ил)пропокси)бензолсульфонамид;
<189> 4-(3-(4-метилпиперазин-1-ил)пропокси)-N-((2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)бензолсульфонамид;
<190> 4-(3-(пиперазин-1-ил)пропокси)-N-((2,3,4,9-тетрагидро-1H-карбазол-3-ил)метил)бензолсульфонамид.
6. Способ получения соединения, представленного формулой 1, включающий стадию получения соединения, представленного формулой 1, путем взаимодействия соединения, представленного формулой 2, с соединением, представленным формулой 3, как показано на схеме реакции 1 ниже:
[Схема реакции 1]

где в реакции 1 Ar, R1, R2, R3, L1, L2 и Z имеют значения, определенные в формуле 1 по п.1,
J1 представляет собой хлорсульфон; и
J2 представляет собой амин.
7. Фармацевтическая композиция, содержащая соединение, или его фармацевтически приемлемую соль по п.1 в качестве активного ингредиента, для предупреждения или лечения злокачественного новообразования.
8. Фармацевтическая композиция в соответствии с п.7, где соединение ингибирует Pin1 (пептидил-пролилцис-транс-изомеразу, взаимодействующую с 1).
9. Фармацевтическая композиция в соответствии с п.7, где злокачественное новообразование представляет собой по меньшей мере одно, выбранное из группы, включающей внутрипеченочную холангиокарциному, гепатобластому, рак печени, рак щитовидной железы, рак толстой кишки, рак яичек, миелодиспластический синдром, глиобластому, рак ротовой полости, рак губы, грибовидный микоз, острый миелогенный лейкоз, острый лимфоцитарный лейкоз, базальноклеточный рак, эпителиальную карциному яичников, герминогенную карциному яичников, рак молочной железы у мужчин, рак головного мозга, аденому гипофиза, множественную миелому, рак желчного пузыря, рак желчевыводящих путей, колоректальный рак, хронический миелогенный лейкоз, хронический лимфолейкоз, ретинобластому, меланому хориоидеи, ампулярный рак фатера, рак мочевого пузыря, рак брюшины, рак паращитовидной железы, рак надпочечников, рак полости носа, немелкоклеточный рак легкого, рак языка, астроцитому, мелкоклеточный рак легкого, педиатрический рак мозга, педиатрическую лимфому, детский лейкоз, рак тонкой кишки, менингиому, рак пищевода, глиому, рак почки, рак почки, рак сердца, рак двенадцатиперстной кишки, злокачественный рак мягких тканей, злокачественный рак костей, злокачественную лимфому, злокачественную мезотелиому, злокачественную меланому, рак глаза, рак вульвы, рак уретры, рак неизвестной первичной локализации, лимфому желудка, рак желудка, рак желудка, интерстициальный рак желудочно-кишечного тракта, Рак Вильмса, рак молочной железы, саркому, рак полового члена, рак глотки, болезнь ворсинок беременных, рак шейки матки, рак эндометрия, саркому матки, рак предстательной железы, метастатический рак костей, метастатический рак головного мозга, рак средостения, рак прямой кишки, карциноидную опухоль прямой кишки, рак влагалища, рак спинного мозга, вестибулярную шванному, рак поджелудочной железы, рак слюнных желез, саркому Капоши, болезнь Педжета, рак миндалин, плоскоклеточный рак, аденокарциному легкого, рак легкого, плоскоклеточный рак легкого, рак кожи, анальный рак, рабдомиосаркому, рак гортани, рак плевры, рак крови и рак тимуса.
10. Фармацевтическая композиция, содержащая соединение, или его фармацевтически приемлемую соль по п.1 в качестве активного ингредиента, для предупреждения или лечения состояний, связанных с ингибированием Pin1 (пептидил-пролилцис-транс-изомеразы, взаимодействующей с 1).
11. Способ предупреждения или лечения рака, включающий введение нуждающемуся в этом субъекту соединения по п.1 или его фармацевтически приемлемой соли.
12. Способ по п.11, где соединение ингибирует Pin1 (пептидил-пролилцис-транс-изомеразу, взаимодействующую с 1).
13. Способ по п.11, где злокачественное новообразование представляет собой по меньшей мере одно, выбранное из группы, включающей псевдомиксому, внутрипеченочную холангиокарциному, гепатобластому, рак печени, рак щитовидной железы, рак толстой кишки, рак яичек, миелодиспластический синдром, глиобластому, рак ротовой полости, рак губы, грибовидный микоз, острый миелогенный лейкоз, острый лимфоцитарный лейкоз, базальноклеточный рак, эпителиальную карциному яичников, герминогенную карциному яичников, рак молочной железы у мужчин, рак головного мозга, аденому гипофиза, множественную миелому, рак желчного пузыря, рак желчевыводящих путей, колоректальный рак, хронический миелогенный лейкоз, хронический лимфолейкоз, ретинобластому, меланому хориоидеи, ампулярный рак фатера, рак мочевого пузыря, рак брюшины, рак паращитовидной железы, рак надпочечников, рак полости носа, немелкоклеточный рак легкого, рак языка, астроцитому, мелкоклеточный рак легкого, педиатрический рак мозга, педиатрическую лимфому, детский лейкоз, рак тонкой кишки, менингиому, рак пищевода, глиому, рак почки, рак почки, рак сердца, рак двенадцатиперстной кишки, злокачественный рак мягких тканей, злокачественный рак костей, злокачественную лимфому, злокачественную мезотелиому, злокачественную меланому, рак глаза, рак вульвы, рак уретры, рак неизвестной первичной локализации, лимфому желудка, рак желудка, рак желудка, интерстициальный рак желудочно-кишечного тракта, Рак Вильмса, рак молочной железы, саркому, рак полового члена, рак глотки, болезнь ворсинок беременных, рак шейки матки, рак эндометрия, саркому матки, рак предстательной железы, метастатический рак костей, метастатический рак головного мозга, рак средостения, рак прямой кишки, карциноидную опухоль прямой кишки, рак влагалища, рак спинного мозга, вестибулярную шванному, рак поджелудочной железы, рак слюнных желез, саркому Капоши, болезнь Педжета, рак миндалин, плоскоклеточный рак, аденокарциному легкого, рак легкого, плоскоклеточный рак легкого, рак кожи, анальный рак, рабдомиосаркому, рак гортани, рак плевры, рак крови и рак тимуса.
14. Способ предупреждения или лечения состояний, связанных с ингибированием Pin1 (пептидил-пролилцис-транс-изомеразы, взаимодействующей с 1), включающий введение нуждающемуся в этом субъекту соединения по п.1 или его фармацевтически приемлемой соли.
15. Применение соединения по по п.1 или его фармацевтически приемлемой соли для производства лекарственного средства для предупреждения или лечения злокачественного новообразования.
16. Применение соединения в соответствии с п.15, где соединение ингибирует Pin1 (пептидил-пролилцис-транс-изомераза, взаимодействующая с 1).
17. Применение соединения в соответствии с п.15, где злокачественное новообразование представляет собой по меньшей мере одно, выбранное из группы, включающей псевдомиксому, внутрипеченочную холангиокарциному, гепатобластому, рак печени, рак щитовидной железы, рак толстой кишки, рак яичек, миелодиспластический синдром, глиобластому, рак ротовой полости, рак губы, грибовидный микоз, острый миелогенный лейкоз, острый лимфоцитарный лейкоз, базальноклеточный рак, эпителиальную карциному яичников, герминогенную карциному яичников, рак молочной железы у мужчин, рак головного мозга, аденому гипофиза, множественную миелому, рак желчного пузыря, рак желчевыводящих путей, колоректальный рак, хронический миелогенный лейкоз, хронический лимфолейкоз, ретинобластому, меланому хориоидеи, ампулярный рак фатера, рак мочевого пузыря, рак брюшины, рак паращитовидной железы, рак надпочечников, рак полости носа, немелкоклеточный рак легкого, рак языка, астроцитому, мелкоклеточный рак легкого, педиатрический рак мозга, педиатрическую лимфому, детский лейкоз, рак тонкой кишки, менингиому, рак пищевода, глиому, рак почки, рак почки, рак сердца, рак двенадцатиперстной кишки, злокачественный рак мягких тканей, злокачественный рак костей, злокачественную лимфому, злокачественную мезотелиому, злокачественную меланому, рак глаза, рак вульвы, рак уретры, рак неизвестной первичной локализации, лимфому желудка, рак желудка, рак желудка, интерстициальный рак желудочно-кишечного тракта, Рак Вильмса, рак молочной железы, саркому, рак полового члена, рак глотки, болезнь ворсинок беременных, рак шейки матки, рак эндометрия, саркому матки, рак предстательной железы, метастатический рак костей, метастатический рак головного мозга, рак средостения, рак прямой кишки, карциноидную опухоль прямой кишки, рак влагалища, рак спинного мозга, вестибулярную шванному, рак поджелудочной железы, рак слюнных желез, саркому Капоши, болезнь Педжета, рак миндалин, плоскоклеточный рак, аденокарциному легкого, рак легкого, плоскоклеточный рак легкого, рак кожи, анальный рак, рабдомиосаркому, рак гортани, рак плевры, рак крови и рак тимуса.
18. Применение соединения по п.1 или его фармацевтически приемлемой соли для производства лекарственного средства для предупреждения или лечения состояний, связанных с ингибированием Pin1 (пептидил-пролилцис-транс-изомеразы, взаимодействующей с 1).
| WO 2018178384 A1, 04.10.2018 | |||
| KR 1020170110017 A, 10.10.2017 | |||
| US 20110130387 A1, 02.06.2011 | |||
| WO 2012015723 A1, 02.02.2012 | |||
| WO 2004037251 A1, 06.05.2004 | |||
| Фильтр для очистки воды | 1978 |
|
SU1022721A1 |

