Предметом изобретения являются композиции без хаотропных компонентов для выделения нуклеиновых кислот при связывании с твердой фазой, в особенности ДНК, из любых комплексных исходных материалов и количеств, содержащие буферную систему для лизиса/связывания, которая включает по меньшей мере один антихаотропный солевой компонент, твердую фазу и само по себе известные буферы для промывки и элюирования. Буферная система для лизиса/связывания может находиться в виде водного раствора или в виде твердой композиции в готовых для использования реакционных сосудах. В качестве твердой фазы могут служить все носители, которые используют для выделения с помощью хаотропных реагентов, предпочтительно стекловолокнистые носители, стеклянные мембраны, кремнийсодержащие носители, керамические изделия, цеолиты или материалы, обладающие отрицательно функционализированными поверхностями или химически модифицированными поверхностями, которые можно переводить в отрицательный потенциал заряда.
Предметом изобретения, далее, является способ выделения нуклеиновых кислот, в особенности ДНК, из любых комплексных исходных материалов при использовании предлагаемых согласно изобретению композиций, отличающийся тем, что он включает лизис исходного материала, связывание нуклеиновых кислот с носителем, промывку связанных с носителем нуклеиновых кислот и элюирование нуклеиновых кислот, причем в случае необходимости можно осуществлять последующую амплификацию выбранных участков последовательности и в случае необходимости последующий анализ амплифицированных участков генов в одной и той же реакционной лунке. Областями применения способа являются все, занимающиеся выделениями ДНК лаборатории, как судебная медицина, экспертиза пищевых продуктов, медицинская диагностика, молекулярная биология, биохимия, генная инженерия и все другие смежные области.
В классических условиях выделение ДНК из клеток и тканей осуществляют тем, что содержащие нуклеиновые кислоты исходные материалы в сильно денатурирующих и восстанавливающих условиях, отчасти также при использовании расщепляющих протеины ферментов, подвергают рестрикции, образующиеся фракции нуклеиновых кислот очищают путем осуществления стадии экстракции фенолом/хлороформом и нуклеиновые кислоты выделяют из водной фазы путем диализа или преципитации этанолом (Sambrook J., Fritsch E.F. и Maniatis Т., "Молекулярное клонирование", 1989, CSH).
Эти "классические способы" выделения нуклеиновых кислот из клеток и особенно из тканей требуют очень больших затрат времени (частично продолжительнее 48 часов) и значительных расходов на аппаратуру и, сверх того, также нереализуемы в полевых условиях. Кроме того, такие способы из-за используемых химикалиев, как фенол и хлороформ, в немалой степени опасны для здоровья.
Различные альтернативные способы выделения нуклеиновых кислот из различных биологических исходных материалов позволяют избегать дорогостоящей и вредной для здоровья экстракции нуклеиновых кислот с помощью фенола и/или хлороформа, а также снижать затраты времени.
Все эти способы базируются на разработанном и впервые описанном Vogelstein и Gillespie (Proc. Natl. Acad. Sci. USA, 76, 615-619 (1979)) методе препаративной и аналитической очистки фрагментов ДНК из агарозных гелей. Согласно этому методу комбинируют растворение агарозы, содержащей выделяемые зоны ДНК, в насыщенном растворе хаотропной соли (NaJ) со связыванием ДНК со стеклянной частицей. Фиксированную на стеклянной частице ДНК затем промывают с помощью раствора для промывки (20 мМ трис-НСl [рН 7,2]; 200 мМ NaCl; 2 мМ ЭДТУ (этилендиаминтетрауксусная кислота); 50% объем/объем этанола) и потом отделяют от частиц носителя обратным растворением.
Этот метод до настоящего времени претерпел ряд модификаций и его применяют в настоящее время для различных способов экстракции и очистки нуклеиновых кислот из различного сырья (Marko M.A., Chipperfield R. и Birnboim H.G., Anal. Biochem., 121, 382-387 (1982)).
Сверх того, в настоящее время во всем мире существует множество систем реагентов, прежде всего для очистки фрагментов ДНК из агарозных гелей и для выделения плазмидной ДНК из бактериальных лизатов, однако также для выделения нуклеиновых кислот с длинными цепями (геномная ДНК, клеточная общая РНК) из крови, тканей или также клеточных культур.
Все эти коммерчески доступные наборы базируются на достаточно известном принципе связывания нуклеиновых кислот с минеральными носителями в присутствии растворов различных хаотропных солей и в качестве носителей в них используют суспензии тонкоизмельченных стеклянных порошков (например, Glasmilk ("стеклянное молоко"), BIO 101, La Jolla, Канада), диатомовые земли (фирма Sigma) или также силикагели (Diagen, заявка на патент ФРГ 4139664-А1).
Используемый для множества различных применений способ выделения нуклеиновых кислот представлен в патенте США 5234809 (Boom). Там описан способ выделения нуклеиновых кислот из содержащих нуклеиновые кислоты исходных материалов путем инкубации исходного материала с хаотропным буфером и связывающей ДНК твердой фазой. Хаотропные буферы осуществляют как лизис исходного материала, так и также связывание нуклеиновых кислот с твердой фазой. Способ хорошо пригоден для выделения нуклеиновых кислот из маленьких количеств образцов и свое практическое применение находит особенно в области выделения вирусных нуклеиновых кислот.
Специфические модификации этих способов касаются использования нового рода носителей, которые обладают применимыми для определенных целей преимуществами (Invitek GmbH, Международная заявка А-95/34569).
Основные недостатки способов выделения нуклеиновых кислот из комплексных исходных материалов на основе инкубации исходного материала с хаотропным буфером и твердой фазой, однако, состоят, в частности, в том, что осуществляемое за счет хаотропных буферов расщепление клеток применимо не для всех материалов, соответственно также для больших количеств исходных материалов крайне неэффективно и происходит при больших затратах времени. Сверх того, необходимы механические способы гомогенизации, когда, например, нужно выделять ДНК из образцов тканей. Далее, для различных целей также всегда нужно использовать высокие концентрации различных хаотропных буферов. Способ тем самым никоим образом не является универсальным.
Проблемы, возникающие за счет затруднительного при известных условиях лизиса исходного материала, правда, можно решить благодаря ряду коммерчески доступных продуктов для выделения нуклеиновых кислот (особенно для выделения геномной ДНК из комплексных исходных материалов), однако имеется большой недостаток в том, что речь больше не идет о классическом, осуществляемом "в одной пробирке" способе, что характеризует способ согласно патенту США, так как лизис исходного материала протекает в обычном буфере при включении протеолитического фермента. Необходимые для последующего связывания нуклеиновых кислот, например, с мембранами для центрифугирования хаотропные ионы нужно дополнительно добавлять к смеси для лизиса после осуществленного лизиса. Они, однако, не могут быть компонентом буфера для лизиса, так как расщепляющая протеин функция хаотропных солей известна и, естественно, точно также тотчас разрушался бы необходимый для эффективного лизиса протеолитический фермент.
Несмотря на ряд недостатков, методы выделения нуклеиновых кислот с применением хаотропных солей осуществляют во всем мире и миллионы раз используют при применении коммерчески доступных продуктов. Эти системы крайне просты в своем осуществлении и действуют всегда по принципу лизиса исходного материала, последующего связывания нуклеиновой кислоты с твердой фазой, стеклянной мембраной или мембраной из диоксида кремния, которая находится в колоночках для центрифугирования с суспензией носителя, промывки связанных нуклеиновых кислот и последующего элюирования нуклеиновых кислот с помощью буфера меньшей ионной силы.
Все эти системы основаны на связывании нуклеиновых кислот с соответствующими поверхностями носителя в присутствии хаотропных солей, то есть по меньшей мере один буферный раствор в качестве основного компонента содержит хаотропную соль. Это относится, смотря по обстоятельствам, уже к буферу для лизиса или, в случае систем при включении протеолитических ферментов, к необходимому буферу для связывания, который добавляют после осуществленного лизиса исходного материала.
Базисом хаотропных солей являются ряды Гофмейстера для высаливания из отрицательно заряженных, нейтральных или основных растворов протеинов. Хаотропные соли характеризуются тем, что денатурируют протеины, повышают растворимость в воде неполярных веществ, а также разрушают гидрофобные взаимодействия. Именно эти свойства согласно уровню техники также вместе с буферными системами хаотропных солей вызывают разрушение переупорядоченной структуры водной среды, чтобы таким образом способствовать связыванию нуклеиновых кислот с выбранными твердыми фазами. Самыми известными представителями хаотропных солей для выделения нуклеиновых кислот являются перхлорат натрия, иодид натрия, йодид калия, гуанидинтиоцианат и гуанидингидрохлорид. Они, однако, во-первых, дорогостоящие и, во-вторых, являются отчасти токсичными или оказывающими разъедающее действие (едкими).
В этом уровне техники вплоть до сегодняшнего дня существует очень большое число заявок на патенты, а также выданных патентов, причем при этом речь идет всегда о вариантах способа, как, например, применение новых носителей или более эффективные буферы для промывки, и т.д., причем основным принципом всегда является применение хаотропных солей для связывания с твердой фазой из материалов на основе диоксида кремния.
Физико-химический принцип связывания нуклеиновых кислот с минеральными носителями в присутствии хаотропных солей считается признанным специалистами во всем мире. Связывание нуклеиновых кислот с поверхностями минеральных носителей заключается в нарушении переупорядоченных структур водной среды, благодаря которому нуклеиновые кислоты адсорбируются на поверхности минеральных материалов, в особенности частиц стекла, или соответственно диоксида кремния. Для нарушения переупорядоченных структур водной среды всегда требуется присутствие хаотропных ионов. В случае высоких концентраций хаотропных солей реакция протекает почти количественно. На основании этих описанных физико-химических данных поэтому специалисты исходят из того, что все коммерчески доступные системы для выделения нуклеиновых кислот должны содержать буферные композиции с высокими концентрациями хаотропных солей для связывания нуклеиновых кислот со связывающей нуклеиновые кислоты твердой фазой.
Тем неожиданнее оказались данные согласно изобретению, что композиции, содержащие антихаотропные соли в буферной системе для лизиса/связывания, точно также и лучше пригодны для выделения нуклеиновых кислот из любых, в особенности комплексных, исходных материалов.
Предметом изобретения являются способ и композиции без хаотропных компонентов для выделения нуклеиновых кислот при связывании с твердой фазой, в особенности ДНК, из любых комплексных исходных материалов, которые содержат буферную систему для лизиса/связывания, включающую по меньшей мере один антихаотропный солевой компонент, твердую фазу и само по себе известные буферы для промывки и элюирования.
Антихаотропными компонентами согласно изобретению являются соли аммония, цезия, натрия и/или калия, предпочтительно хлорид аммония.
Буферная система для лизиса/связывания, кроме того, включает само по себе известные детергенты и в случае необходимости добавки, как, например, трис-HCl, этилендиаминтетрауксусная кислота (ЭДТУ), поливинилпирролидон, цетилтриметил-аммонийбромид (СТАВ), тритон Х-100, N-лаурилсаркозин, цитрат натрия, трео-1,4-димеркаптобутан-2,3-диол (DTT), додецилсульфат натрия и/или твин. В предпочтительном варианте осуществления буферная система для лизиса/связывания с твердой фазой содержит спирт, как, например, этанол и изопропанол и в случае необходимости ферменты, предпочтительно расщепляющие протеины ферменты, как, например, протеиназа.
С помощью изобретения можно использовать соответствующий уровню техники принцип для решения специфической проблемы выделения нуклеиновых кислот, соответственно существующий вариант в отношении оптимизации и повышения эффективности определенных существенных параметров. Так, этот принцип пригоден для осуществления полностью автоматизированного, высокопроизводительного способа.
В отличие от известного уровня техники согласно настоящему изобретению совершенно неожиданно с помощью буферных систем для лизиса/связывания без хаотропных солей в качестве компонента можно связывать нуклеиновые кислоты, в особенности геномную ДНК, с минеральным носителем и также элюировать в обычных реакционных условиях.
Далее показано, что множество совершенно различных солей пригодно в качестве компонентов в случае необходимости само по себе обычных буферных систем для лизиса/связывания для связывания нуклеиновых кислот с классическими носителями на основе стекла или диоксида кремния.
Особенно неожиданно наилучшие результаты достигнуты с помощью солей, которые по своим физико-химическим характеристикам обладают абсолютно противоположными действиями в отношении хаотропных солей, до сих пор используемых для связывания нуклеиновых кислот. Эти соли, таким образом, можно назвать антихаотропными.
Так, с помощью буферов для лизиса/связывания, основным компонентом которых являются, например, соли аммония вместо хаотропных солей (коммерчески доступные наборы для экстракции), при экстракциях геномной ДНК из различных комплексных исходных материалов (как, например, кровь, ткань, растения) при постоянстве до сих пор обычных других реакционных компонентов, носителей, а также при совершенно одинаковом протекании реакции, можно достигать по меньшей мере таких же количественных, а также качественных результатов.
В особенности при этом ион аммония с физико-химической точки зрения в ряду Гофмейстера представляет собой ион, который обладает абсолютно противоположными признаками по отношению к известным хаотропным ионам этого ряда.
Только путем обмена до сих пор используемого хаотропного солевого компонента на антихаотропный солевой компонент в буфере для лизиса/связывания при существующем постоянстве всех других параметров на само по себе известных твердых поверхностях носителей возможно по меньшей мере адекватное количественное выделение нуклеиновых кислот.
Это означает, что с помощью соли, которая не денатурирует, а стабилизирует протеины, которая не повышает, а снижает растворимость неполярных веществ в воде, а также которая не разрушает, а усиливает гидрофобные взаимодействия, нуклеиновые кислоты точно также можно выделять из комплексных исходных материалов, очищать и использовать для само по себе обычных применений.
С помощью настоящего изобретения предлагается нового рода альтернативный механизм связывания нуклеиновых кислот с твердыми, предпочтительно минеральными, носителями и на этом основании универсально применимый нового рода способ выделения нуклеиновых кислот из комплексных исходных материалов.
Таким образом, согласно изобретению за счет применения нового рода составов буферов для лизиса/связывания на основе антихаотропных солей для выделения нуклеиновых кислот, особенно выделения геномной ДНК, базирующегося на связывании нуклеиновых кислот с само по себе обычными различными твердыми фазами из диоксида кремния или соответственно стекломатериалов, возможно использование альтернативного химизма в качестве существенной составной части соответствующих тест-наборов (композиций).
Предлагаемый согласно изобретению способ при использовании антихаотропных солей при этом следует из известных из лабораторной практики осуществлений способов выделения нуклеиновых кислот и отличается тем, что осуществляют:
1. лизис исходного материала;
2. связывание нуклеиновых кислот с твердой фазой (колонка для центрифугирования или суспензия);
3. промывка связанных нуклеиновых кислот;
4. элюирование нуклеиновых кислот с помощью само по себе известного буфера с низким содержанием соли.
Согласно изобретению можно осуществлять высокоэффективное и быстрое выделение нуклеиновых кислот, особенно геномной ДНК, из любого и в случае необходимости комплексного исходного материала. Необходимые для связывания антихаотропные ионы могут быть компонентом буфера для лизиса/связывания даже при включении протеолитических ферментов. Предлагаемый согласно изобретению способ таким образом является простым в осуществлении и универсально применимым.
Выделение нуклеиновых кислот, в особенности ДНК, из любых исходных материалов осуществляют путем инкубации содержащего нуклеиновую кислоту исходного материала без использования хаотропных веществ в контакте
- с буферной системой для лизиса/связывания, которая включает водный раствор, один антихаотропный солевой компонент, по меньшей мере один детергент, в случае необходимости добавки и в случае необходимости фермент;
- и с любой твердой фазой, предпочтительно со стекловолокнистыми прочесами, стеклянными мембранами, стеклами, цеолитами, керамическими изделиями, а также другими кремнийсодержащими носителями, благодаря чему происходит лизис исходного материала и последующее связывание ДНК с твердой фазой. Затем связанную нуклеиновую кислоту промывают само по себе известными способами и ДНК отделяют от твердой фазы путем растворения.
Согласно определенным протоколам экстракции в смесь для лизиса в случае необходимости можно примешивать дополнительный детергент, спирт или смесь детергента со спиртом.
Предпочтительными исходными материалами являются компактные растительные материалы, как, например, плоды, семена, листья, иглы и т.д.; клинически релевантные образцы, как, например, цельная кровь, ткань, микробиоптаты, парафинированные материалы, пробы эндоскопической холангиопанкреатографии, тупферный материал мазков; пищевые продукты, как, например, рыба, колбаса, консервы, молоко; образцы для судебно-медицинской экспертизы, как, например, корни волос, окурки, следы крови, и другие образцы, которые содержат ДНК.
Предпочтительными ионами согласно изобретению являются представленные в ряду Гофмейстера антихаотропные ионы аммония, ионы цезия, а также ионы калия и натрия, или комбинации этих ионов, предпочтительно хлорид аммония. Для лизиса/связывания их используют в концентрациях от 0,1 М до 8 М.
Для связывания нуклеиновых кислот, особенно ДНК, с твердыми носителями при этом достаточны уже незначительные концентрации этих солей, составляющие предпочтительно ≤1 М, при определенных применениях предпочтительно даже ≤0,5 М, причем для количественного выделения нуклеиновых кислот из больших количеств исходных материалов достаточны более высокие концентрации ионов.
Благодаря использованию антихаотропных солей, оказывающих стабилизирующее действие на протеины, в качестве существенных компонентов буфера для лизиса согласно предпочтительному варианту осуществления изобретения могут быть также протеолитические ферменты, как, например, протеиназа К, для обеспечения и повышения эффективности процесса лизиса, вследствие чего для необходимого расщепления клеток также добавляют высокие концентрации антихаотропных солей, например, 5 М, так что становится возможным количественное выделение нуклеиновых кислот.
Буферные системы согласно уровню техники с известными хаотропными солями в необходимых высоких концентрациях, которые вообще требуются для количественного выделения нуклеиновых кислот, не содержат никаких протеолитических ферментов. Таким образом, их всегда нужно дополнительно добавлять для связывания нуклеиновых кислот с твердой фазой.
В качестве детергентов в предлагаемых согласно изобретению буферах для лизиса/связывания предпочтительно используют анионные, катионные или нейтральные детергенты, как, например, додецилсульфат натрия, тритон Х-101, твин или цетилтриметиламмонийбромид.
После осуществленного лизиса исходного материала суспензию в случае необходимости за счет кратковременной стадии центрифугирования отделяют от еще неполностью лизированных составных частей и инкубируют непосредственно вместе со связывающим ДНК материалом, соответственно, как уже описано, инкубируют с твердой фазой после добавления дополнительного детергента, спирта или смеси детергента со спиртом. В случае необходимости в буферной системе для лизиса дополнительно находятся в незначительных концентрациях (<50 мМ) ЭДТУ и/или трис-HCl. Для выделения ДНК из очень сильно загрязненных исходных материалов предпочтительно к буферной системе также добавляют 2-4% поливинилпирролидона или других известных веществ для селективного связывания ингибирующих компонентов.
В качестве связующих материалов для выделяемой ДНК оказываются исключительно пригодными, например, коммерчески доступные стекловолокнистые прочесы в колонках для центрифугирования, кремнийсодержащие соединения, как SiO2 с различным размером частиц. Вместе с тем можно применять все материалы, которые используют для выделения нуклеиновых кислот с помощью хаотропных буферов.
После инкубации со связывающим ДНК материалом осуществляют отделение лизата от связующего материала путем кратковременного центрифугирования. Затем само по себе известным образом промывают с помощью буфера для промывки, который состоит, например, по меньшей мере из 50% этанола и в случае необходимости незначительной концентрации соли, например NaCl, носитель высушивают и связанную ДНК элюируют с помощью само по себе известного буфера с низким содержанием соли (трис-HCl; ТЕ; вода) и при предпочтительной температуре 50-70°С.
Другой вариант осуществления изобретения состоит в том, что для лизиса трудно растворяющихся исходных материалов, например компактных образцов ткани, корней волос, для оптимизации эффективности лизиса и для сокращения необходимых затрат времени для лизиса осуществляют добавление протеолитических ферментов, предпочтительно протеиназ, как, например, протеиназа К.
Таким образом, согласно изобретению на основе нового рода комбинаций антихаотропных солей в качестве существенных компонентов буферных смесей для лизиса предлагается универсально применимый способ выделения нуклеиновых кислот, в особенности ДНК, из всех содержащих ДНК исходных материалов, а также из любых количеств самых различных исходных материалов, причем точно также эффективно можно использовать все до сих пор применяемые носители и их конструкции, а также идентично используемыми являются до сих пор применяемые на практике методики выделения нуклеиновых кислот.
В самом общем варианте осуществления изобретения с помощью предлагаемого согласно изобретению способа из всех соответствующих уровню техники выбранных для экстракции ДНК комплексных исходных материалов можно экстрагировать нуклеиновые кислоты, то есть с помощью новой универсальной буферной системы успешно, крайне просто и очень быстро можно осуществлять высокоэффективный лизис и последующее связывание нуклеиновых кислот с минеральным носителем, исходя из компактных растительных материалов (как, например, плоды, семена, листья, иглы и т.д.), из клинически релевантных проб (как, например, цельная кровь, ткань, микробиоптаты, парафинированные материалы, пробы эндоскопической холангиопанкреатографии, тупферный материал мазков), из пищевых продуктов (как, например, рыба, колбаса, консервы, молоко), из образцов для судебно-медицинской экспертизы (как, например, корни волос, окурки, следы крови), как также из других исходных материалов.
Другое преимущество способа также состоит в том, что при этом с высокой эффективностью можно осуществлять выделение ДНК как из крайне незначительных количеств исходных материалов (например, выделение ДНК из 1 мкл цельной крови; корня волоса, микробиоптатов из количества <1 мг), так и также из очень больших количеств исходных материалов, как, например, из 50 мл цельной крови; 1 г ткани; <1 г растительного материала.
Дальнейшие преимущества замены хаотропных солей антихаотропными солями состоят в том, что используемые буферы благодаря отсутствию хаотропных химических веществ также более не являются токсичными или не обладают разъедающим действием.
Наряду с самым общим вариантом осуществления оптимизация способа экстракции в отношении специфических применений позволяет осуществлять даже почти количественное выделение содержащихся в исходном образце количеств ДНК. Удивительным образом с помощью предлагаемого согласно изобретению способа без используемых согласно уровню техники хаотропных ионов в высокой концентрации для связывания ДНК, можно достигать более высоких выходов ДНК, чем это до сих пор было возможно с помощью коммерчески доступных и высокооптимизированных наборов для экстракции.
Соответствующие сравнительные результаты при использовании коммерчески доступных наборов для экстракции представлены в примерах осуществления. Эти результаты ясно демонстрируют потенциальные возможности изобретения.
Наряду с выделением ДНК из любых, содержащих ДНК комплексных исходных материалов согласно следующему варианту осуществления предлагаемого в изобретении способа также возможно выделение плазмидной ДНК из бактериальных лизатов с высокой эффективностью и без использования хаотропных солей, требующихся согласно уровню техники для связывания плазмидной ДНК с минеральными носителями. Так, после известных специалисту стадий способа выделения плазмидной ДНК путем щелочного лизиса осуществляют необходимую, так называемую реакцию нейтрализации с помощью классического раствора III (Maniatis и Sambroek), и этот раствор III в соответствии с существующей вместе с тем двойственной функцией также одновременно реализует связывание плазмидной ДНК с само по себе обычными твердыми носителями. Для связывания плазмидной ДНК, таким образом, не требуется обычная добавка хаотропного гуанидинийгидрохлорида.
Связанную плазмидную ДНК также само по себе обычным образом промывают и отделяют путем элюирования от носителя. Способ пригоден для выделения плазмидной ДНК из любых используемых исходных количеств (от мини до гига). Достигаемые выходы плазмидной ДНК при этом идентичны по отношению к выходам, получаемым с помощью обычных, коммерчески доступных способов. Предлагаемый согласно изобретению способ, однако, при его осуществлении намного дешевле, чем все другие известные системы, так как хаотропные соли являются очень дорогостоящими.
Способ при применении антихаотропных солей, таким образом, также превосходно пригоден для разработки автоматизируемых систем для выделения плазмид, в случае которых, как известно, решающим критерием выбора является стоимость/приготовление образцов.
Композиции согласно изобретению неожиданно делают возможным доступ к другим, представляющим большой интерес и нового рода применениям в области выделения нуклеиновых кислот и диагностики.
Согласно другому варианту осуществления изобретения имеющиеся новые буферные системы для лизиса/связывания, содержащие по меньшей мере один хаотропный солевой компонент, могут связывать нуклеиновые кислоты с твердыми фазами, которые обладают отрицательно заряженными поверхностями или поверхностями, которые обладают отрицательным потенциалом заряда.
Из уровня техники известны способы и средства для очистки нуклеиновых кислот, причем осуществляют связывание нуклеиновой кислоты с химически модифицированными твердыми фазами (патент США 5523392: Очистка ДНК на силикатах алюминия и фосфосиликатах; патент США 5503816: Силикаты для очистки ДНК; патент США 5674997: Очистка ДНК на модифицированных силикатах; патент США 5438127: Очистка ДНК путем твердофазной экстракции при использовании модифицированной с помощью РСl3 мембраны из стекловолокна; патент США 5606046: Очистка ДНК путем твердофазной экстракции при использовании промытой трифторуксусной кислотой мембраной из стекловолокна. Очистка ДНК путем твердофазной экстракции при использовании мембраны из стекловолокна, предварительно обработанной с помощью трифторуксусной кислоты и затем с помощью фтор-иона, гидроксид-иона или ВСl3; патент США 5610291: Мембраны из стекловолокна, модифицированные путем обработки с помощью SiCl3, АlСl3 или ВСl3 и промытые с помощью раствора NaOH для придания способности адсорбировать ДНК; патент США 5616701: Очистка ДНК путем твердофазной экстракции при использовании промытой гидроксидом мембраны из стекловолокна; патент США 5650506: Модифицированные мембраны из стекловолокна, пригодные для очистки ДНК путем твердофазной экстракции).
Условием для этого связывания нуклеиновой кислоты при этом всегда является то, что в используемые для связывания мембраны за счет химических модифицирующих реакций вводят положительные ионные заряды. Таким образом, очевидно, что между положительно заряженной поверхностью используемых мембран и отрицательным ионным зарядом фосфатной основы нуклеиновых кислот за счет кулоновских взаимодействий происходит связывание. В этом отношении используют достаточно известный в кругу специалистов принцип связывания нуклеиновых кислот с положительно заряженными твердыми фазами, который, как известно, уже многие годы стандартно применяют, например, для методов блотирования ДНК/РНК на положительно заряженных нейлоновых фильтрах.
Довольно существенный недостаток этих описанных способов состоит, однако, в том, что они непригодны для выделения нуклеиновых кислот, то есть совершенно невозможно выделить нуклеиновые кислоты из комплексных исходных материалов. Исходные материалы всегда уже содержат изолированные нуклеиновые кислоты, которые, как представлено в цитированных патентах США, само по себе известным образом нужно выделять. В особенности один аспект при этом специалисту кажется неясным. Описанные условия связывания (связывание в физиологических условиях буферирования) и условия элюирования являются идентичными. Непонятно, как при одних и тех же условиях буферирования для связывания нуклеиновых кислот с положительно заряженной мембраной также снова можно отделять нуклеиновые кислоты от мембран обратным растворением.
В конечном счете, представленные средства и относящийся к этому способ имеют только очень узкое практическое применение. Известны также синтетически получаемые олигонуклеотиды, связывающиеся с положительно заряженными поверхностями. Это происходит при этом снова при использовании кулоновских взаимодействий, то есть благодаря соединению положительных и отрицательных зарядов, например, за счет модифицированных олигонуклеотидов (соединение с аминолинкерами или фосфатными линкерами). Также эти варианты не позволяют выделять нуклеиновые кислоты из комплексных исходных материалов.
Как подробно указано, существуют альтернативные формы связывания нуклеиновых кислот для очистки на мембранах с достаточным положительным зарядом, которые не представляют собой никаких способов выделения нуклеиновых кислот. Связывание нуклеиновых кислот происходит за счет кулоновских сил, базирующихся на взаимодействиях между положительными ионными зарядами мембран и отрицательными ионными зарядами участков нуклеиновых кислот. Этот принцип является вместе с тем логически объяснимым.
Обнаружен неожиданно феномен, базирующийся на предлагаемом согласно изобретению выделении нуклеиновых кислот из комплексных исходных материалов с помощью антихаотропных солей. Так, оказалось, что также отрицательно заряженные поверхности или поверхности, которые можно переводить в отрицательный потенциал заряда, пригодны для связывания нуклеиновых кислот за счет использования предлагаемых согласно изобретению буферных систем для лизиса/связывания. В общем, такой возможности нельзя было ожидать, так как не должно было бы происходить никакого связывания, а должно было бы быть отталкивание из-за одинаковых потенциалов заряда.
Используемые согласно изобретению отрицательно функционализированные поверхности или поверхности с потенциально отрицательными модификациями получают само по себе известными способами. В качестве пригодного показано, например, фотохимическое связывание ацетильной группы, карбоксильной группы или гидроксильной группы с поверхностью реакционного сосуда.
С помощью данного варианта способа открываются совершенно новые перспективы для комплексного анализа нуклеиновых кислот, а именно показано, что для связывания нуклеиновой кислоты с отрицательно заряженными или потенциально отрицательно заряжаемыми поверхностями нуклеиновую кислоту, как в случае всех до сих пор описанных вариантов, уже не нужно выделять. Связывание происходит из реакционной смеси для лизиса, то есть содержащий нуклеиновую кислоту исходный образец лизируется, и высвобождающиеся нуклеиновые кислоты связываются с отрицательно заряженной поверхностью (например, поверхностью лунки микробиологического планшета или реакционного сосуда Эппендорфа).
Благодаря предлагаемому согласно изобретению варианту способа теперь можно реализовать совершенно нового рода осуществляемые "в одной пробирке" или одностадийные способы выделения нуклеиновых кислот из комплексных исходных материалов. Такие способы согласно их спектру применения дают пользователю колоссальные преимущества (простота, дешевизна, уменьшение количества отходов, быстрота, пригодность для рутинного использования, автоматизируемость и прочее).
Следующее применение этого варианта способа, кроме того, состоит не только в реализации экстракции нуклеиновых кислот в реакционной лунке, но и также в возможности осуществления следующей целевой амплификации и в случае необходимости последующего анализа в одном и том же реакционном сосуде, в случае необходимости в возможности проведения реакций гибридизации или осуществления секвенирований на твердых фазах.
На этом основании, например, реакционный сосуд Эппендорфа для полимеразной цепной реакции емкостью 0,5 мл модифицируют известными специалисту способами с помощью отрицательно заряженной или потенциально отрицательной функциональной группы. Для этого пригодно, например, фотохимическое связывание ацетильной группы, карбоксильной группы или гидроксильной группы с поверхностью реакционного сосуда. В реакционный сосуд затем вводят выбранную для выделения нуклеиновой кислоты пробу (например, цельную кровь), смешивают с буфером для лизиса, содержащим антихаотропную солевую фракцию, например хлорид аммония, детергент и протеолитический фермент, и инкубируют в течение 5 минут при температуре 70°С.
Для сведения к максимуму связывания нуклеиновых кислот после лизиса исходного материала можно еще пипетировать смесь детергента со спиртом. Полученную смесь затем кратковременно инкубируют и затем удаляют путем декантации из реакционного сосуда. Нуклеиновая кислота теперь связана с функционализированной поверхностью реакционного сосуда, затем ее кратковременно промывают с помощью спиртового буфера для промывки и спирт удаляют путем инкубации, например, при температуре 70°С. Далее, квалифицированно осуществляют элюирование связанных нуклеиновых кислот путем добавления в реакционный сосуд буфера с низким содержанием соли (например, 10 мМ трис-НСl) и кратковременной инкубации (например, в течение двух минут), например, при температуре 70°С. Таким образом имеют в распоряжении нуклеиновую кислоту для последующих применений.
Как показано, все реакции при выделении нуклеиновой кислоты из комплексного исходного материала протекают в одном реакционном сосуде; то есть лизис исходного материала, связывание нуклеиновых кислот, промывку связанных нуклеиновых кислот и элюирование нуклеиновых кислот реализуют в одном реакционном сосуде.
Чаще всего используемые в настоящее время во всем мире наборы для экстракции фирмы Qiagen для последовательности лизиса, связывания, промывки и элюирования соответственно необходимо включают фильтровальный картуш и по меньшей мере 4 отдельных реакционных сосуда, далее, необходимы многочисленные стадии центрифугирования.
В противоположность этому предлагаемый согласно изобретению вариант способа позволяет осуществлять экстракцию нуклеиновой кислоты без единой стадии центрифугирования. Отсюда также можно сделать вывод об огромном преимуществе в отношении затрат времени. Эти преимущества касаются также описанных способов экстракции нуклеиновых кислот цитированного патента США 5234809 на имя Boom.
Наряду с возможной экстракцией нуклеиновой кислоты, однако, связанную нуклеиновую кислоту также можно оставлять на поверхности описанного реакционного сосуда емкостью 0,5 мл и затем, например, путем добавления полной реакционной смеси для полимеразной цепной реакции (праймер, нуклеотиды, полимеразный буфер, Taq-полимераза, магний) использовать равным образом для осуществления полимеразной цепной реакции, то есть экстракция и амплификация протекают тогда в одном и том же реакционном сосуде.
Эти примеры иллюстрируют огромные преимущества и широкую применимость, которые следуют из изобретения. Изобретение позволяет в одном варианте осуществления реализовать полный процесс от подготовки образца до амплификации и в случае необходимости также анализа, например, в одной реакционной лунке. Вместе с этим за счет приготовления модифицированных реакционных сосудов (или также других твердых поверхностей) и пригодных буферов для лизиса/связывания получают новые стандарты в лабораториях молекулярной биологии и прежде всего по разработке диагностики на основе нуклеиновых кислот, причем благодаря новым, потенциально применимым решениям прежде всего также резко уменьшаются достаточно известные проблемы в отношении загрязнения образцов.
Следующее преимущество и также дальнейшее применение состоит в том, что фиксированные на поверхности нуклеиновые кислоты также по меньшей мере более продолжительное время остаются стабильно фиксированными на поверхности и таким образом имеются в распоряжении для более поздней обработки, то есть полимеразную цепную реакцию можно проводить не вынужденно тотчас после экстракции. Дальнейшей областью применения является полностью автоматизированная экстракция нуклеиновых кислот и в случае необходимости анализ при использовании описанных здесь, имеющих отрицательные или потенциально отрицательные заряды поверхностей, предпочтительно пластиковых поверхностей пригодных реакционных лунок, например, микробиологических планшетов.
Предлагаемые согласно изобретению буферные системы для лизиса/связывания с антихаотропными солями в качестве основных компонентов при включении в случае необходимости протеолитического фермента могут быть приготовлены также в виде твердой композиции. Для этого в обычных реакционных сосудах "аликвотируют" смеси из солей и детергентов, добавок и в случае необходимости ферментов и инкубируют в течение нескольких часов при температуре 95°С или лиофилизируют известными способами и таким образом переводят в твердую композицию.
Эти твердые композиции в готовых комплексных реакционных смесях для выделения нуклеиновых кислот устойчивы при длительном хранении, то есть биологическая активность протеолитического фермента в качестве компонента сохраняется при хранении в течение длительного времени (см. пример осуществления). Приготовление буферных смесей для лизиса в виде твердой композиции при этом осуществляют без добавления само по себе известных защитных добавок, просто путем низкотемпературной лиофилизации.
Все имеющиеся в продаже тест-наборы для экстракции нуклеиновых кислот содержат отдельные необходимые компоненты по отдельности, причем только определенные растворы должны быть приготовлены пользователем, и, сверх того, стойкость растворов ограничена. Другой недостаток состоит в том, что пользователь во время выделения нуклеиновых кислот при применении, например, обычных тест-наборов должен соблюдать многочисленные стадии пипетирования для различных отдельных растворов. Это прежде всего в области медицинской диагностики увеличивает опасность загрязнения. Недостатком, далее, также является то, что за счет существующих, например, пределов загрузок широко используемых обычных колонок для центрифугирования, которые применяют главным образом для выделения нуклеиновых кислот, также сильно ограничено количество исходного материала. Это обосновано тем, что к исходному материалу еще нужно добавлять необходимый для экстракции буфер для лизиса и связывания.
Благодаря приготовлению твердой композиции в виде устойчивой при хранении смеси для лизиса на основе антихаотропных солей совершенно просто решаются все существующие проблемы. Эта композиция имеет следующие преимущества:
1. хранение в течение длительного времени "готовых для использования" буферных смесей для лизиса;
2. стабилизация протеолитических ферментов в готовых смесях для лизиса и их хранение в течение длительного времени;
3. использование гораздо больших количеств исходных материалов при одинаковом размере существующих колонок для центрифугирования (например, утроение исходного количества);
4. уменьшение опасностей загрязнения за счет сокращения стадий пипетирования и растворений;
5. получение образцов в готовых смесях для лизиса также вне лаборатории и в случае необходимости их хранение в течение длительного времени;
6. стабильная отправка образцов и охлаждение.
Готовые твердые стабильные буферные смеси для лизиса состоят из множества отдельных компонентов, при включении в случае необходимости протеолитических ферментов просты в обращении (также для неспециалистов), так как реакцию инициируют просто путем добавления образца, который содержит выделяемую нуклеиновую кислоту. Сверх того, можно исходить из того, что смеси в соответствии со своими ингредиентами обладают устойчивостями по меньшей мере в течение 6 месяцев, благодаря чему также транспортировка образца при температуре окружающей среды больше не представляет собой никакой проблемы.
Преимущество твердых композиций основано на том, что для лизиса содержащих нуклеиновые кислоты (NAs) образцов содержащий эти нуклеиновые кислоты образец лишь вводят в реакционный сосуд с содержащимся, устойчивым при хранении буфером для лизиса и в случае необходимости за счет добавления воды образец подвергают лизису в соответствующем реакционном сосуде. Полностью отпадают многочисленные дорогостоящие и вызывающие загрязнение стадии пипетирования. Прежде всего благодаря предлагаемой согласно изобретению композиции решены известные проблемы в отношении сбора и обработки клинических и для судебно-медицинской экспертизы образцов в полевых условиях, и в распоряжении имеется простая в обращении композиция.
Затем также при осуществлении на практике неожиданно оказалось, что после добавления лизируемого исходного материала в случае необходимости при добавлении твердого образца, после добавления воды твердая композиция в стандартных реакционных условиях без проблем снова переводится в жидкую фазу.
Таким образом предметом изобретения является применение антихаотропных солей в композициях без хаотропных компонентов для выделения нуклеиновых кислот при связывании с твердой фазой, в особенности ДНК, из любых комплексных исходных материалов. Композиции содержат буферные системы для лизиса/связывания, включающие по меньшей мере один антихаотропный солевой компонент, твердую фазу и само по себе известные буферы для промывки и элюирования.
Буферная система для лизиса/связывания может находиться в виде водного раствора или в виде твердой композиции в готовых для использования реакционных сосудах.
В качестве твердой фазы могут служить все носители, которые используют для выделения с помощью хаотропных реагентов, предпочтительно стекловолокнистые прочесы, стеклянные мембраны, кремнийсодержащие носители и аэросилы, или носители, которые имеют отрицательно заряженную поверхность или химически модифицированные поверхности, обладающие отрицательным потенциалом заряда.
Предметом изобретения, далее, является способ выделения нуклеиновых кислот, в особенности ДНК, из любых комплексных исходных материалов при использовании указанных композиций, отличающийся тем, что он включает лизис исходного материала, связывание нуклеиновых кислот с носителем, промывку связанных с носителем нуклеиновых кислот и элюирование нуклеиновых кислот.
Благодаря достигаемому качеству ДНК он также хорошо пригоден для препаративного выделения и очистки ДНК с целью использования в генной терапии.
Предметом изобретения также являются устойчивые при хранении и готовые для использования твердые композиции буферных систем для лизиса для выделения нуклеиновых кислот на основе антихаотропных солей, которые находятся в виде "готовых для использования" смесей в обычных реакционных сосудах. Твердые композиции буферных смесей для лизиса активируют путем добавления только образца в случае жидких образцов (как, например, цельная кровь, слюна, клеточные суспензии, сыворотка, плазма, спинно-мозговая жидкость), в случае же твердых исходных материалов, как ткань, корни волос, следы крови на твердых поверхностях, окурки, депарафинированная ткань и прочее, активируют дополнительно путем добавления воды и осуществляют лизис исходного материала. После осуществленного лизиса исходного материала лизированную смесь само по себе известным образом в случае необходимости после добавления этанольного раствора, соответственно смеси спирта с детергентом, инкубируют вместе с используемыми, связывающими нуклеиновую кислоту твердыми фазами любой формы (суспензия, колонки для центрифугирования). Последующее связывание нуклеиновых кислот с соответствующими твердыми фазами, промывку связанных нуклеиновых кислот и конечное элюирование осуществляют, как уже описано согласно уровню техники.
С помощью этих твердых композиций получены нового рода решения прежде всего для областей применения любого вида диагностики нуклеиновых кислот.
Нужно еще раз подчеркнуть, что вариант согласно изобретению одностадийного способа и осуществляемого "в одной пробирке" способа позволяет осуществлять выделение нуклеиновых кислот из комплексных исходных материалов, в случае необходимости целевые амплификации и в случае необходимости последующий анализ амплифицированного участка нуклеиновой кислоты. Исходный материал при этом не должен быть уже выделенной нуклеиновой кислотой, а представляет собой комплексный, содержащий нуклеиновую кислоту исходный материал. Необходимая для связывания нуклеиновой кислоты поверхность содержит отрицательные или потенциально отрицательные функциональные группы. Связывание нуклеиновой кислоты происходит в буфере для лизиса/связывания, причем ионы, необходимые для связывания отрицательно заряженной нуклеиновой кислоты с отрицательно функционализированной поверхностью, происходят из антихаотропных солей.
При этом реализуют:
1. осуществляемый "в одной пробирке" способ выделения нуклеиновых кислот из комплексных исходных материалов;
2. осуществляемый "в одной пробирке" способ выделения нуклеиновых кислот из комплексных исходных материалов и последующей целевой амплификации;
3. осуществляемый "в одной пробирке" способ выделения нуклеиновых кислот из комплексных исходных материалов, последующей целевой амплификации и последующего анализа амплифицированного участка нуклеиновой кислоты.
Это означает, что и выделение нуклеиновой кислоты из самых различных, содержащих ДНК исходных материалов, и в случае необходимости целевую амплификацию, и в случае необходимости анализ осуществляют в одной и той же реакционной лунке или в случае необходимости на одной и той же реакционной поверхности.
Предлагаемые согласно изобретению композиции и универсальный способ для связывания нуклеиновых кислот с твердыми фазами с целью выделения, очистки и последующего комплексного молекулярного анализа нуклеиновых кислот из любых исходных материалов и количеств, включающих нуклеиновые кислоты, представляют собой нового рода базовую технологию для разработки интеграционных, полностью автоматизированных ген-аналитических систем, которые позволяют реализовать подготовку образцов, целевую амплификацию и целевой анализ в одной реакционной лунке.
Изобретение ниже подробнее поясняется в примерах осуществления.
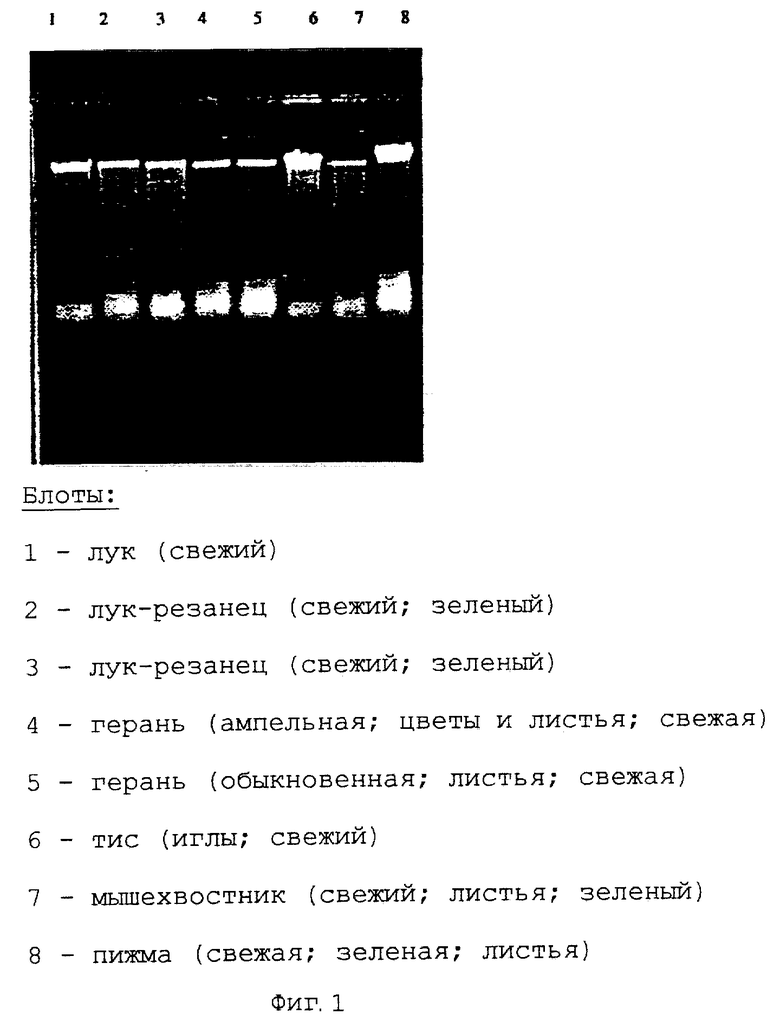
Пример 1. Выделение геномной ДНК из различных растительных материалов
В каждом случае, 50-100 мг исходного растительного материала размельчали в ступке в жидком азоте и затем переносили в реакционный сосуд Эппендорфа емкостью 1,5 мл. Добавляли 500 мкл буфера для лизиса (2% СТАВ; 2% поливинилпирролидона; 10 мМ трис-HCl; 20 мМ ЭДТУ и 1,3 М хлорида аммония) и осуществляли инкубацию по меньшей мере в течение 30 минут при температуре 65°С. Отделяли путем центрифугирования не подвергнутые лизису компоненты и супернатант смешивали с 200 мкл изопропанола. Раствор переносили в колонку для центрифугирования с мембраной из стекловолокна (колонка Micro Spin; фирма LIDA). Центрифугировали в течение двух минут при скорости 12000 оборотов в минуту. Фильтрат отбрасывали и мембрану промывали два раза с помощью буфера для промывки (50 мМ NaCl; 10 мМ трис-HCl; 1 мМ ЭДТУ; 70% объем/объем этанола). После удаления этанола путем кратковременной стадии центрифугирования (2 минуты при скорости 12000 оборотов в минуту) добавляли 200 мкл буфера для элюирования (10 мМ трис-НСl; рН 8,7) и осуществляли элюирование ДНК путем центрифугирования в течение 1 минуты при скорости 10000 оборотов в минуту.
В каждом случае, 20 мкл элюированной ДНК помещали на агарозный гель и выделяли после окрашивания этидиумбромидом (фиг.1).
Пример 2. Одновременное выделение геномной ДНК из различных исходных материалов с помощью универсальной буферной системы
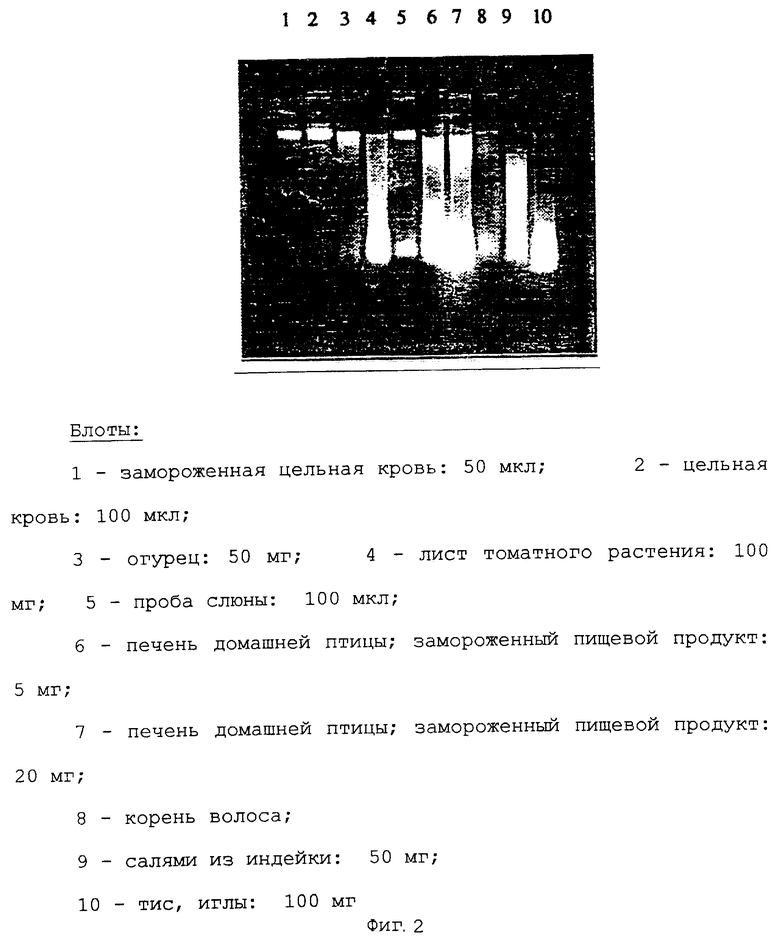
Для выделения использовали следующие образцы:
1 - замороженная цельная кровь: 50 мкл; 2 - цельная кровь: 100 мкл; 3 - огурец: 50 мг; 4 - лист томатного растения: 100 мг; 5 - проба слюны: 100 мкл; 6 - печень домашней птицы, замороженный пищевой продукт: 5 мг; 7 - печень домашней птицы, замороженный пищевой продукт: 20 мг; 8 - корень волоса; 9 - салями из индейки: 50 мг; 10 - тис, иглы: 100 мг.
Все образцы инкубировали при температуре 65°С в 500 мкл буфера для лизиса (2% СТАВ; 2% поливинилпирролидона; 10 мМ трис-HCl; 20 мМ ЭДТУ и 1,5 М хлорида аммония) и за исключением всех растительных образцов при добавлении 20 мкл протеиназы К (20 мг/мл).
Лизаты затем смешивали с 200 мкл изопропанола и переносили в колонку для центрифугирования с мембраной из стекловолокна (колонка Micro Spin; фирма LIDA). Центрифугировали в течение двух минут при скорости 12000 оборотов в минуту. Фильтрат отбрасывали и мембрану промывали два раза с помощью буфера для промывки (50 мМ NaCl; 10 мМ трис-HCl; 1 мМ ЭДТУ; 70% объем/объем этанола). После удаления этанола путем кратковременной стадии центрифугирования (2 минуты при скорости 12000 оборотов в минуту) добавляли 50-200 мкл буфера для элюирования (10 мМ трис-HCl; рН 8,7) и осуществляли элюирование ДНК путем центрифугирования в течение 1 минуты при скорости 10000 оборотов в минуту.
В каждом случае, 1/5 элюированной ДНК помещали на агарозный гель и выделяли после окрашивания этидиумбромидом (фиг.2).
Примеp 3. Выделение геномной ДНК из тупферных образцов мазков слизистой оболочки рта
Выделение ДНК из тупферных образцов мазков слизистой оболочки рта описано ниже.
В каждом случае 400 мкл буфера для лизиса (СТАВ, поливинилпирролидон, хлорид аммония, трис, ЭДТУ) вносили в реакционный сосуд Эппендорфа емкостью 1,5 мл. В этот буфер для лизиса вводили тупфер с материалом мазка и добавляли 20 мкл протеиназы К (20 мг/мл). Затем полученную смесь инкубировали при температуре 70°С в течение 10 минут. После лизиса добавляли 200 мкл смеси детергента с изопропанолом, образец кратковременно встряхивали, после чего переносили на коммерчески доступную колонку для центрифугирования (фирма LIDA; мембрана из стекловолокна) и центрифугировали в течение 1 минуты при скорости 12000 оборотов в минуту. Колонку затем дважды промывали с помощью содержащего этанол буфера для промывки (NaCl, трис-HCl, ЭДТУ, этанол) (центрифугирование в течение 1 минуты при скорости 12000 оборотов в минуту) и мембрану высушивали путем кратковременной стадии центрифугирования. Путем добавления 200 мкл буфера для элюирования (10 мМ трис-НСl) связанную ДНК удаляли из фильтровальной мембраны путем элюирования за счет кратковременной стадии центрифугирования (1 минута; 10000 оборотов в минуту).
В каждом случае 20 мкл выделенной ДНК из обоих способов экстракции для анализа помещали на агарозный гель с 0,7% ТАЕ и анализировали после окрашивания этидиумбромидом (фиг.3).
Пример 4. Сравнение экстракции ДНК согласно изобретению из образцов цельной крови (200 мкл) с коммерчески доступным набором на основе связывания нуклеиновых кислот в присутствии хаотропных солей
Сравнивали выделение геномной ДНК с помощью предлагаемого согласно изобретению способа с коммерчески доступным и обычно используемым способом выделения геномной ДНК при использовании хаотропных солей для связывания нуклеиновых кислот. Экстракцию геномной ДНК с помощью сравнительного способа осуществляли согласно инструкции для применения.
Выделение ДНК с помощью предлагаемого согласно изобретению способа описано ниже.
В каждом случае 200 мкл образца цельной крови (обработана ЭДТУ, свежая) помещали в реакционный сосуд Эппендорфа емкостью 1,5 мл. После добавления 350 мкл буфера для лизиса (СТАВ, поливинилпирролидон, хлорид аммония, трис, ЭДТУ) и 20 мкл протеиназы К (20 мг/мл) осуществляли инкубацию в течение 10 минут при температуре 70°С для лизиса исходного материала.
После лизиса добавляли 180 мкл смеси детергента с изопропанолом, образцы кратковременно встряхивали, затем переносили в коммерчески доступные колонки для центрифугирования (фирма LIDA; мембрана из стекловолокна) и центрифугировали в течение двух минут при скорости 12000 оборотов в минуту. Колонку затем дважды промывали с помощью содержащего этанол буфера для промывки (NaCl, трис-HCl, ЭДТУ, этанол) (цетрифугирование в течение 1 минуты при скорости 12000 оборотов в минуту) и мембрану высушивали путем кратковременной стадии центрифугирования. Путем добавления 200 мкл буфера для элюирования (10 мМ трис-НСl) элюировали связанную ДНК из фильтровальной мембраны путем кратковременного центрифугирования (1 минута, 10000 оборотов в минуту).
В каждом случае 10 мкл выделенной ДНК из обоих способов экстракции для анализа помещали на агарозный гель с 0,7% ТАЕ и после окрашивания этидиумбромидом анализировали.
Проведено сравнение выходов геномной ДНК, ее целостности (чистая отдельная полоса без низкомолекулярных размытых полос) и воспроизводимости способов экстракции. Как видно, с помощью предлагаемого согласно изобретению способа можно достигать лучших результатов, чем с помощью сравнительного способа (фиг.4).
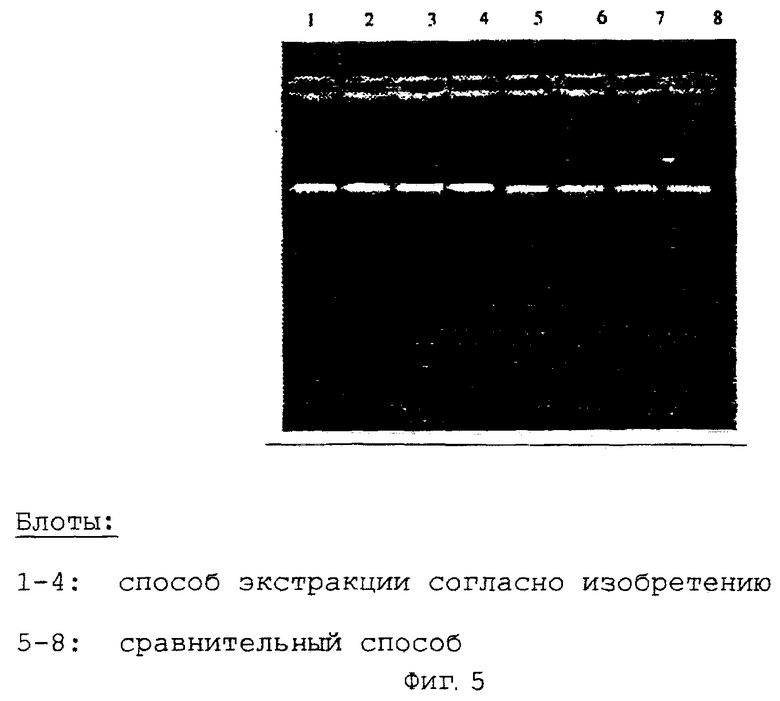
Пример 5. Сравнение предлагаемой согласно изобретению экстракции ДНК из образцов цельной крови (5 мкл) с экстракцией с помощью коммерчески доступного набора на основе связывания нуклеиновых кислот в присутствии хаотропных солей.
Сравнивали выделение геномной ДНК с помощью предлагаемого согласно изобретению способа с коммерчески доступным и обычно используемым способом выделения геномной ДНК при использовании хаотропных солей для связывания нуклеиновых кислот. Экстракцию геномных ДНК с помощью сравнительного способа осуществляли согласно инструкции для применения.
Выделение ДНК с помощью предлагаемого согласно изобретению способа описано ниже.
В каждом случае 5 мкл образца цельной крови (обработана ЭДТУ; свежая) вносили в реакционный сосуд Эппендорфа емкостью 1,5 мл. Образец доводили до объема 200 мкл путем добавления 195 мкл 1 х PBS (забуференный фосфатом физиологический раствор) буфера и после добавления 350 мкл буфера для лизиса (СТАВ, поливинилпирролидон, хлорид аммония, трис, ЭДТУ) и 20 мкл протеинаэы К (20 мг/мл) осуществляли инкубацию при температуре 70°С в течение 10 минут для лизиса исходного материала.
После лизиса добавляли 180 мкл смеси детергента с изопропанолом, образец кратковременно встряхивали, затем переносили на коммерчески доступную колонку для центрифугирования (фирма LIDA; мембрана из стекловолокна) и центрифугировали в течение двух минут при скорости 12000 оборотов в минуту. Колонку затем дважды промывали с помощью содержащего этанол буфера для промывки (NaCl, трис-HCl, ЭДТУ, этанол) (цетрифугирование в течение 1 минуты при скорости 12000 оборотов в минуту) и мембрану высушивали путем кратковременной стадии центрифугирования. Путем добавления 200 мкл буфера для элюирования (10 мМ трис-НСl) элюировали связанную ДНК из фильтровальной мембраны путем кратковременной стадии центрифугирования (1 минута, 10000 оборотов в минуту).
В каждом случае 20 мкл выделенной ДНК из обоих способов экстракции с целью анализа помещали на агарозный гель с 0,7% ТАЕ и после окрашивания этидиумбромидом анализировали.
Доказано и проведено сравнение возможности выделения геномной ДНК из очень незначительных количеств исходного материала и воспроизводимости способов экстракции. Как видно, с помощью предлагаемого согласно изобретению способа можно достигать лучших результатов, чем с помощью сравнительного способа (фиг.5).
Примеp 6. Сравнение предлагаемой согласно изобретению экстракции ДНК из различных видов образцов ткани животных и различных количеств исходного материала с коммерчески доступным набором на основе связывания нуклеиновых кислот в присутствии хаотропных солей.
Сравнивали выделение геномной ДНК с помощью предлагаемого согласно изобретению способа с осуществляемым с помощью коммерчески доступного набора и обычно используемым способом выделения геномной ДНК при использовании хаотропных солей для связывания нуклеиновых кислот. Экстракцию геномной ДНК с помощью сравнительного способа осуществляли согласно инструкции для применения.
Выделение ДНК с помощью предлагаемого согласно изобретению способа описано ниже.
В каждом случае 5 мг или 20 мг образцов ткани из почки, сердца и печени свиньи помещали в реакционный сосуд Эппендорфа емкостью 1,5 мл. К образцу добавляли 400 мкл буфера для лизиса (СТАВ, поливинилпирролидон, хлорид аммония, трис, ЭДТУ) и 40 мкл протеиназы К (20 мг/мл). Лизис исходного материала осуществляли путем инкубации при температуре 52°С.
После лизиса за счет кратковременного центрифугирования (1 минута; 14000 оборотов в минуту) отделяли возможно не подвергнутые лизису компоненты и к супернатанту в новом реакционном сосуде добавляли 200 мкл смеси детергента с изопропанолом, образец кратковременно встряхивали, затем переносили в коммерчески доступную колонку для центрифугирования (фирма LIDA; мембрана из стекловолокна) и центрифугировали в течение двух минут при скорости 12000 оборотов в минуту. Колонку затем дважды промывали с помощью содержащего этанол буфера для промывки (NaCl, трис-HCl, ЭДТУ, этанол) (центрифугирование в течение 1 минуты при скорости 12000 оборотов в минуту) и мембрану высушивали путем кратковременной стадии центрифугирования. Путем добавления 200 мкл буфера для элюирования (10 мМ трис-НСl) элюировали связанную ДНК из фильтровальной мембраны путем кратковременной стадии центрифугирования (1 минута, 10000 оборотов в минуту).
В каждом случае 10 мкл выделенной ДНК из обоих способов экстракции для анализа помещали на агарозный гель с 0,7% ТАЕ и после окрашивания этидиумбромидом анализировали.
Доказано и проведено сравнение возможности выделения геномной ДНК из различных образцов ткани, а также различных количеств исходного материала в отношении выходов геномной ДНК, ее целостности (более чистая отдельная полоса без низкомолекулярных "размытых" полос) и воспроизводимости экстракций. Как видно, с помощью предлагаемого согласно изобретению способа можно достигать лучших результатов, чем с помощью сравнительного способа (фиг.6).
Пример 7. Экстракция ДНК из образцов цельной крови (200 мкл) с помощью предлагаемого согласно изобретению способа и связывание нуклеиновых кислот с различными носителями, используемыми для связывания при посредничестве хаотропных солей
Представлено выделение геномной ДНК с помощью предлагаемого согласно изобретению способа из 200 мкл цельной крови и связывание нуклеиновых кислот с различными, используемыми для выделения нуклеиновых кислот с помощью хаотропных агентов носителями (колоночные мембраны и суспензии).
Экстракцию ДНК осуществляли, как описано в примере осуществления 4, причем вместо мембраны из стекловолокна фирмы LIDA использовали различные другие носители.
В каждом случае 20 мкл выделенной ДНК для анализа помещали на агарозный гель с 0,7% ТАЕ и после окрашивания этидиумбромидом анализировали.
Как видно из фиг.7, согласно предлагаемому в изобретении способу реализуют связывание нуклеиновых кислот с различными носителями, используемыми для известных до сих пор хаотропных способов.
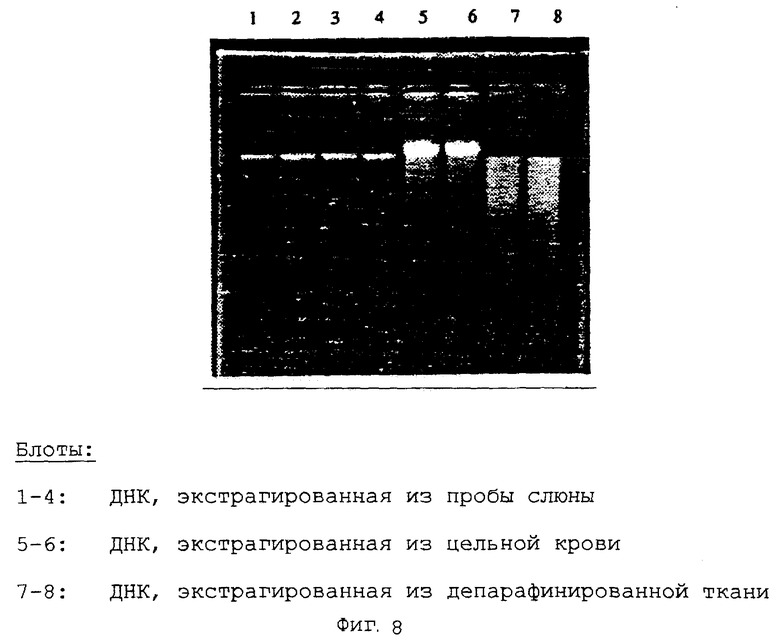
Пример 8. Приготовление устойчивой при хранении буферной системы для лизиса при включении протеолитического фермента (буферная смесь 1) и применение буферной системы для лизиса для выделения геномной ДНК из различных исходных материалов
Приготовление исходного буферного раствора для лизиса, содержащего 3М хлорида калия, 2% СТАВ, 18,2 мМ трис-НСl (рН 8,3), 12,5 мМ ЭДТУ, 2,8% поливинилпирролидона. Введение аликвот в каждом случае по 400 мкл исходного раствора в реакционные сосуды Эппендорфа емкостью 1,5 мл и добавление по 40 мкл протеиназы К (20 мг/мл). Лиофилизация буферных смесей для лизиса в установке для лиофилизации (Alpha 2; фирма Christ). Последующее хранение буферных смесей для лизиса в закрытых реакционных сосудах при комнатной температуре в течение 6 месяцев.
Экстракцию геномной ДНК осуществляли из:
А: 500 мкл цельной крови;
В: 400 мкл пробы слюны;
С: депарафинированной ткани.
а. Экстракция ДНК из цельной крови
Добавление 500 мкл цельной крови к твердой композиции буфера для лизиса и инкубация при температуре 70°С в течение 10 минут. Добавление 200 мкл изопропанола и перенесение суспензии в колонку для центрифугирования (стекловолокнистый прочес). Центрифугирование в течение двух минут при максимальной скорости и отбрасывание центрифугата. Добавление 600 мкл буфера для промывки (70% этанола, NaCl, трис, ЭДТУ), центрифугирование в течение 1 минуты при максимальной скорости и отбрасывание центрифугата. Повторение стадии промывки. Последующее высушивание мембраны путем центрифугирования в течение двух минут при максимальной скорости. Элюирование ДНК из мембраны путем добавления 200 мкл буфера для элюирования (70°С) и центрифугирование в течение 1 минуты при максимальной скорости.
б. Экстракция ДНК из проб слюны
Добавление 500 мкл пробы слюны к твердой композиции буфера для лизиса и инкубация при температуре 70°С в течение 10 минут. Добавление 200 мкл изопропанола и перенесение суспензии в колонку для центрифугирования (стекловолокнистый прочес). Центрифугирование в течение двух минут при максимальной скорости и отбрасывание центрифугата. Добавление 600 мкл буфера для промывки (70% этанола, NaCl, трис, ЭДТУ), центрифугирование в течение 1 минуты при максимальной скорости и отбрасывание центрифугата. Повторение стадии промывки. Последующее высушивание мембраны путем центрифугирования в течение двух минут при максимальной скорости. Элюирование ДНК из мембраны путем добавления 200 мкл буфера для элюирования (70°С) и центрифугирование в течение 1 минуты при максимальной скорости.
в. Экстракция ДНК из депарафинированной ткани
Добавление депарафинированного кусочка ткани к твердой композиции буфера для лизиса, добавление 500 мкл полностью дейтерированной воды и инкубация при температуре 52°С в течение 30 минут. Добавление 200 мкл изопропанола и перенесение суспензии в колонку для центрифугирования (стекловолокнистый прочес). Центрифугирование в течение двух минут при максимальной скорости и отбрасывание центрифугата. Добавление 600 мкл буфера для промывки (70% этанола, NaCl, трис, ЭДТУ), центрифугирование в течение 1 минуты при максимальной скорости и отбрасывание центрифугата. Повторение стадии промывки. Последующее высушивание мембраны путем центрифугирования в течение двух минут при максимальной скорости. Элюирование ДНК из мембраны путем добавления 200 мкл буфера для элюирования (70°С) и центрифугирование в течение 1 минуты при максимальной скорости.
Экстрагированную ДНК затем анализировали путем гель-электрофореза. Для этого наносили в каждом случае 1/10 всего элюата ДНК (фиг.8).
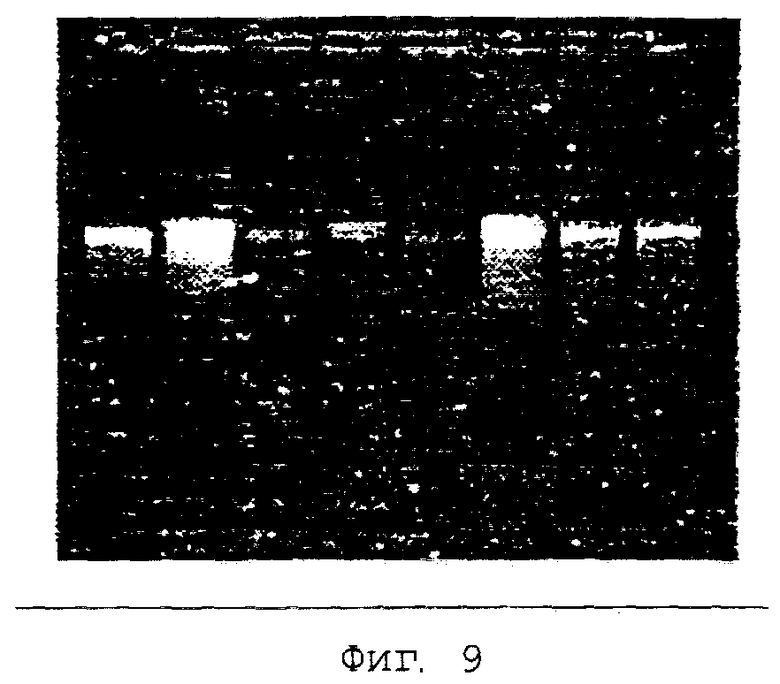
Пример 9. Приготовление устойчивой при хранении буферной системы для лизиса при включении протеиназы К (буферная смесь 2) и применение буферной системы для лизиса для выделения геномной ДНК из 8 индивидуальных по 100 мкл образцов цельной крови
Приготовление исходного буферного раствора для лизиса, содержащего 3 М хлорида аммония, 2% поливинилпирролидона, 16,7 мМ ЭДТУ, 60 мМ трис-HCl, 1,6% СТАВ, 20 мкл протеиназы К (20 мг/мл).
Внесение аликвот в каждом случае по 400 мкл исходного раствора в реакционные сосуды Эппендорфа емкостью 1,5 мл и инкубация открытых реакционных сосудов Эппендорфа в термосмесителе при температуре 95°С вплоть до полного высыхания. Последующее закрывание реакционных сосудов и хранение в течение 12 месяцев при комнатной температуре.
Добавление 100 мкл цельной крови к твердой композиции буфера для лизиса и инкубация при температуре 70°С в течение 10 минут. Добавление 20 мкл суспензии минерального носителя на основе диоксида кремния и кратковременное смешение. Инкубация смеси в течение 1 минуты. Получение осадка носителя путем кратковременного центрифугирования. Промывка осадка после центрифугирования носителя с помощью 800 мкл буфера для промывки (70% этанола, NaCl, трис, ЭДТУ) и последующее удаление остаточного этанола путем инкубации при температуре 70°С. Элюирование ДНК из носителя путем добавления 200 мкл нагретого до температуры 70°С буфера для элюирования (10 мМ трис-HCl, рН 8,69) и отделение нуклеиновой кислоты от носителя путем центрифугирования в течение 1 минуты при максимальной скорости, а также перенесение нуклеиновой кислоты в новый реакционный сосуд.
Экстрагированную ДНК затем анализировали путем гель-электрофореза. Для этого наносили в каждом случае 1/10 всего элюата ДНК (фиг.9).
Пример 10. Выделение геномной ДНК из периферических лимфоцитов крови путем прямого связывания с функционализированными поверхностями микробиологического планшета
В качестве микробиологического планшета использовали коммерчески доступный планшет, “нагруженный” группами СОО-.
В каждом случае для выделения использовали одну полосу планшета (8 лунок) с функциональными группами и одну полосу без групп СОО- в качестве отрицательного контроля.
Во все лунки помещали по 30 мкл периферических лимфоцитов крови в 1 × PDS буфере и смешивали со 180 мкл буфера для лизиса (хлорид аммония, СТАВ, поливинилпирролидон, трис-HCl, ЭДТУ, протеиназа К) и инкубировали при температуре 70°С в течение 5 минут. После этого добавляли 80 мкл смеси детергента с изопропанолом. Смеси кратковременно встряхивали и инкубировали в течение 5 минут. Затем растворы выливали из лунок. Каждую лунку затем промывали дважды с помощью содержащего этанол буфера для промывки и остаточный этанол удаляли путем кратковременной инкубации при температуре 70°С. Элюирование нуклеиновых кислот осуществляли путем добавления 25 мкл 10 мМ трис-НСl и инкубации в течение двух минут.
Элюаты затем анализировали на агарозном геле с 0,7% ТАЕ (фиг.10).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОБНАРУЖЕНИЯ КЛИНИЧЕСКИ РЕЛЕВАНТНЫХ ИЗМЕНЕНИЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДЕЗОКСИРИБОНУКЛЕИНОВОЙ КИСЛОТЫ ОНКОГЕНА KI-RAS, ЕГО ПРИМЕНЕНИЕ И НАБОР ДЛЯ ТЕСТА РАННЕГО ВЫЯВЛЕНИЯ ОПУХОЛЕЙ | 1997 |
|
RU2207378C2 |
| СМЕННЫЙ МИКРОФЛЮИДНЫЙ МОДУЛЬ ДЛЯ АВТОМАТИЗИРОВАННОГО ВЫДЕЛЕНИЯ И ОЧИСТКИ НУКЛЕИНОВЫХ КИСЛОТ ИЗ БИОЛОГИЧЕСКИХ ОБРАЗЦОВ И СПОСОБ ВЫДЕЛЕНИЯ И ОЧИСТКИ НУКЛЕИНОВЫХ КИСЛОТ С ЕГО ИСПОЛЬЗОВАНИЕМ | 2008 |
|
RU2380418C1 |
| СПОСОБ СПЕЦИФИЧЕСКОГО ВЫДЕЛЕНИЯ ПОЛНОГО ДНК-СОДЕРЖИМОГО БАКТЕРИАЛЬНЫХ ВОЗБУДИТЕЛЕЙ ИНФЕКЦИИ | 2010 |
|
RU2567809C2 |
| Универсальный способ выделения ДНК и лизирующая смесь для его осуществления | 2022 |
|
RU2807254C1 |
| СПОСОБ ВЫДЕЛЕНИЯ НУКЛЕИНОВЫХ КИСЛОТ | 2004 |
|
RU2272072C1 |
| СПОСОБ ВЫДЕЛЕНИЯ ДНК ИЗ КОСТНОГО МАТЕРИАЛА | 2019 |
|
RU2724506C1 |
| СПОСОБ АВТОМАТИЗИРОВАННОГО ВЫДЕЛЕНИЯ С ОДНОВРЕМЕННОЙ ОЧИСТКОЙ НУКЛЕИНОВЫХ КИСЛОТ ИЗ НЕСКОЛЬКИХ БИОЛОГИЧЕСКИХ ОБРАЗЦОВ | 2014 |
|
RU2595374C2 |
| СПОСОБ ВЫДЕЛЕНИЯ ГЕНОМНОЙ ДНК ИЗ КЛЕТОК МИКРООРГАНИЗМОВ | 2000 |
|
RU2177035C2 |
| Способ выделения целевых фрагментов ДНК из многокомпонентной смеси | 2024 |
|
RU2832884C1 |
| МАГНИТНЫЙ СОРБЕНТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ВЫДЕЛЕНИЯ МОЛЕКУЛ НУКЛЕИНОВЫХ КИСЛОТ | 2017 |
|
RU2653130C1 |





Изобретение относится к области биохимии и может быть использовано при проведении любых анализов, требующих выделения нуклеиновых кислот из комплексных образцов, в частности в медицинской диагностике, судебно-медицинской экспертизе и экспертизе пищевых продуктов. Предложена композиция для выделения нуклеиновых кислот (НК) из различных комплексных образцов, предусматривающего лизис исходного материала и связывание НК с твердой фазой. Композиция включает буферную систему для лизиса/связывания, содержащую, по меньшей мере, один антихаотропный солевой компонент; твердую фазу и обычные буферы для промывки и элюирования НК. При этом буферная система может находиться как в виде водного раствора, так и в твердом состоянии, в готовых для использования реакционных сосудах. В качестве твердой фазы могут быть использованы стекловолокнистые прочесы, стеклянные мембраны, керамические изделия, цеолиты, а также материалы с отрицательно заряженной поверхностью или химически модифицированной поверхностью, которая может быть переведена в отрицательно заряженную. Предметом изобретения является также основанный на использовании предложенной композиции способ выделения НК, отличающийся простотой исполнения и высокой эффективностью. 4 с. и 21 з.п.ф-лы, 10 ил.
Конвенционный приоритет установлен от 04.12.1998 в соответствии с заявкой 198 56 064.8, поданной в Патентное ведомство Германии.
| US 5234809, 10.08.1993 | |||
| Механизм прерывистого движения барабана-питателя лесопосадочной машины | 1975 |
|
SU648776A1 |
| US 5693785, 02.12.1997. | |||

