В настоящем изобретении описывается способ специфического выделения полного ДНК-содержимого бактериальных возбудителей инфекции в образцах различного происхождения и набор для практического осуществления соответствующего способа.
Количественное и качественное определение бактериальных возбудителей инфекции является важной частью микробиологических исследований при санитарном мониторинге питьевой воды, воды водоемов, пищевых продуктов, рабочих зон, различных поверхностей в общественных местах и в окружающей среде. Для таких исследований - кроме культивирования бактерий in vitro на селективных и неселективных средах и их микроскопического определения, а также биохимических реакций, позволяющих выявлять бактерии на определенных метаболических этапах - в настоящее время доступны методы молекулярной диагностики на основе нуклеиновой кислоты, например, амплификация последовательностей нуклеиновых кислот in vitro с помощью полимеразной цепной реакции (далее ПЦР-амплификации), методы секвенирования, способные выявить последовательности оснований нуклеиновых кислот, а также метод электрофореза в агарозном геле, выявляющий размер полученных фрагментов нуклеиновых кислот [Nucleic Acid Isolation and Purification, 2nd Edition, Roche Diagnostics GmbH, 2003]. В отличие от традиционных методик, эти молекулярные методы диагностики на основе нуклеиновой кислоты делают возможным проведение анализа даже при малом объеме исходных образцов. Матрицей для этих молекулярных методов диагностики на основе нуклеиновой кислоты является бактериальная нуклеиновая кислота с концентрацией и степенью чистоты, требуемых применяемым аналитическим методом. Однако эти два параметра (концентрация нуклеиновой кислоты и степень ее чистоты) варьируются в зависимости от применяемого преаналитического способа, т.е. в зависимости от методов разрушения бактериальных клеток в образце и методов выделения нуклеиновых кислот. Разрушение бактериальных клеток в образце для доступа к матричным нуклеиновым кислотам является сложным процессом, в течение которого подходящее согласованное использование отдельных этапов может проводиться по отдельности или комбинированно друг с другом. Традиционно, разрушение бактериальных клеток начинается с их лизиса, за которым следует селективное связывание-экстракция и очистка целевой дезоксирибонуклеиновой кислоты (ДНК) в ходе последующих этапов; после этого процесс завершается качественным и количественным определением очищенной выделенной нуклеиновой кислоты. Разрушение может происходить, когда обеспечены соответствующие условия: уровень рН, температура и ионное состояние, - механическим путем (например, путем набухания клеток, ультразвуковой дезинтеграции, дробления); химическим путем (например, с помощью буфера, содержащего ферменты, сурфактанты, детергенты, ионные хелаторы), а также комбинированной смесью белкового денатурата и ингибирующих добавок внутриклеточной дезоксирибонуклеазы. Из лизата, полученного при разрушении клеток, целевая нуклеиновая кислота может выделяться и очищаться с помощью экстракции осаждением, центрифугирования, электрофореза или хроматографии. Полуколичественный анализ выхода нуклеиновых кислот, очищенных от клеточных макромолекул и клеточных фрагментов, можно проводить, например, с помощью измерения флуоресцентного излучения полиароматической цепочки, включающей интеркалированную ДНК (например, этидия бромида), индуцированной ультрафиолетовым светом (УФ). Количественный анализ выделенной нуклеиновой кислоты может проводиться на основании измерения оптической плотности (ОП) при длине волны λ260 с помощью УФ-спектрофотометрии. Одна единица ОП соответствует концентрации ДНК 50 мкг/мл. Для определения чистоты выделенной ДНК используется соотношение оптических плотностей, измеренное при двух различных длинах волн УФ-спектра (ОП260/ОП280), это соотношение представляет собой соотношение величин поглощения света, измеренных при длине волны, характерной для водного раствора нуклеиновой кислоты, и при длине волны, характерной для водного раствора белков (ОП260 и ОП280). В предпочтительном случае значение соотношения ОП260/ОП280 варьируется в диапазоне от 1,4 до 2,0. Более низкие значения указывают на загрязнение белками (например, на присутствие остаточных белковых компонентов клеточного лизата), тогда как более высокие значения (>2) указывают на ошибку спектрофотометрических измерений или другие возможные загрязнения. При применении ПЦР-амплификации желательно, чтобы соотношение ОП260/ОП280 составляло 1,4-1,8.
В традиционной практике общественного здравоохранения бактериальная ДНК чаще всего выделяется с помощью смеси фенола и хлороформа [Sambrook J. et al.: Molecular Cloning, A Laboratory Manual, Cold Spring Harbor Laboratory Press, 3rd Edition, 2001]. Этот метод основан на том, что бактериальные клетки разрушаются буфером для лизиса, содержащим детергенты и ферменты, разрушающие белки; затем к лизату добавляется смесь фенол-хлороформа, в результате чего кислая фенольная фаза экстрагирует среди прочих веществ белковые и РНК компоненты лизата [Cohn E.J., Conant J.B.(1926): PNAS 12: 433-438], а ДНК может быть выделена органическим растворителем, смешиваемом с водой, например, изопропанолом [Kirby K.S. (1956): Biochem. J. 64: 405-408].
Использование вышеупомянутых органических растворителей, безусловно, является эффективным в процессе разрушения клеток и выделения ДНК, однако их потенциальная токсичность может угрожать безопасности лабораторного персонала; также не следует забывать об окружающей среде. Достаточно распространенным решением, чтобы избежать использование вышеупомянутых органических растворителей является решение, когда после разрушения биологического образца хаотропный компонент [Chomczynski P. et al. (1997): Biotechniques 22: 550-553] добавляется в буферную систему, служащую для выделения геномного ДНК-содержимого. Благодаря наличию хаотропного компонента, денатурирующего белки (например, NaJ, KJ, перхлората Na, гуанидиния гидрохлорида - ГХ, гуанидиния тиоцианата - GTC), и соответствующему уровню рН, связанные нуклеиновой кислотой белки диссоциируют, при этом функциональное ингибирование нуклеиновой кислоты, разрушающей нуклеазы, облетает эффективное выделение ДНК. В патенте US 4900677 описывается способ, который может применяться для разрушения клеток как грамположительных, так и грамотрицательных бактерий, после которой хромосомное ДНК-содержимое Pseudomonas aeruginosa и Streptococcus faecalis выделяется среди прочих в результате быстрого процесса обработки образца раствором, не содержащим органических растворителей. Чтобы экстрагировать хромосомную ДНК, исходную массу, содержащую 107-108 клеток, разрушают в коктейле на основе Трис-буфера (рН=8,0), содержащем ферменты с различными целевыми спектрами (лизоцим, эндо-N-ацетил-мураминидаза, ахромопептидаза и т.д.), растворяющие вещества (например, диметлсульфоксид, диметилформамид), сурфактанты (например, додецилсульфат натрия. Тритон X-100, CHAPS, CHAPSO) и ионные хелаторы (например, EDTA), путем перемешивания в вихревой мешалке. Содержимое контаминирующей рибонуклеиновой кислоты (РНК) обрабатывается ферментом рибонуклеазы (РНК-азы), а остаточные ферментные или неферментные белки нейтрализуются путем добавления протеинкиназы К. Хаотропный компонент (например, натрия трифторацетат, натрия перхлорат, NaJ), обеспечивающий выделение ДНК, добавляется к системе, при этом ДНК, получаемая примерно через час, очищается, посредством использования диализа через коллодиевую мембрану. Разрушение клеток в образце и нейтрализация остаточных белков также обеспечивается варьированием температуры реакции (от 37°C до 60°C). В соответствии с описанием патента US 5595876, нейтрализация остаточных белков также обеспечивается путем повышения температуры в процессе так называемой in situ экстракции ДНК грамположительных и грамотрицательных бактерий в единственной пробирке.
Прекращение использования органических растворителей в процессе разрушения клеток и выделения ДНК, как описано выше, является успешным, однако, выход экстракции и чистота выделенной ДНК снижается.
Повышение выхода ДНК возможно достигнуть, применяя промежуточное решение, которое обеспечивает выборочное выделение ДНК путем добавления небольшого количества органического растворителя, смешиваемого с водой в щелочной буферной среде (например, при рН=8-9). В описании патента US 5945515 в одном из примеров с эукариотами из лизирующей системы, содержащей также хаотропный компонент (GTC) в щелочном буфере, РНК предварительно осаждается путем добавления небольшого количества органического растворителя; затем преципитат отделяется путем центрифугирования, после чего в растворе оставалась лишь ДНК, которую было необходимо выделить. В дальнейших примерах, приведенных в описании патента, для ускорения описанного выделения ДНК сокращалось число этапов центрифугирования, затем они и вовсе опускались. Во время оценивания чистоты ДНК, полученной таким путем, контаминирующие остатки РНК были также обнаружены при агарозном гель-электрофорезе.
Кроме описанных выше традиционных и промежуточных методик выделения ДНК в течение последних двух десятилетий широкое распространение на практике получили альтернативные методики выделения ДНК, применяющие другие принципы. В одном типе таких альтернативных методик для выделения биологических макромолекул используется принцип хроматографии [например, очистка плазмидной ДНК из лизата E.coli с использованием колоночной хроматографиеской системы в соответствии с описанием патента US 6428703 или отделение нуклеиновых кислот на силанизированной хроматографической колонке в соответствии с описанием заявки WO 9105606]. Эти способы основываются на идее, что после лизиса клеток ДНК-содержимое лизата селективно и обратимо связывается на поверхности носителя. Такими поверхностями носителя могут являться фильтры (нитроцеллюлозный, найлоновый, целлюлозно-ацетатный, металоксидный), пластмассовые (ПВДФ) или стеклянные (силикагель SiO2, SiO2-TiO2) матрицы [Boom R. et al. (1990): J.Clin.Microbiol. 28: 495-503, Mackey К. et al. (1998): Mol.Biotechnol. 9: 1-5, Boom R. et al. (1999): J.Clin.Microbiol. 37: 615-619, Dames S. et al. (2006): J.Mol.Diagn. 8: 16-21, Gushikem Y., Rosatto S.S. (2001): J.Braz.Chem.Soc. 12: 695-705] или микрочастицы и гранулы, полученные путем сочетания различных ионов и полимеров, при использовании которых этапы разрушения клеток и выделения ДНК могут быть автоматизированы [Youngman L.D. et al. (2002): Clin.Chem. 48: 1629-1630, Smith К. et al. (2003): J.Clin.Microbiol. 41: 2440-2443]. В патенте WO 2004033707 описывается использование Na-силикатного носителя; в патенте WO 2004046231 описывается использование кремнезема, силикагеля, связанного с полипропиленовым, полистиреновым и поликарбонатным носителем (в пробирке, лунке микропланшета) в ходе выделения митохондриальной ДНК (мтДНК) эукариотов или полного выделения ДНК-содержимого крови. В одноступенчатом методе, описанном в патенте WO 1998023630, роль гидроксилированного ароматического гидрофобного полимера полигидроксистирена описывается как удержание контаминирующих компонентов клеточного лизата, тогда как нуклеиновая кислота остается в элюенте. В патенте WO 2002000930 описывается выделение ДНК из мазка кала, нанесенного на носитель, импрегнированный найлоном или гваяком, с использованием соли гуанидиния, тогда как в патенте US 20080319182 связывание ДНК обеспечивается специальной катионной поверхностью. Нуклеиновые кислоты, включающие ДНК, связываются на поверхности таких носителей при специфических физико-химических обстоятельствах (при определенной температуре, воздействии определенной центробежной силы, в определенных электролитных условиях и т.д.) с образованием обратимых вторичных связей и при специфических характеристиках селективной способности носителя. После удаления носителя со связанной на его поверхности ДНК из раствора и после соответствующего обессоливания связанная ДНК может быть выделена путем элюирования его из носителя.
Для последующего молекулярного применения (например, проведения ПЦР-амплификации, ферментного расщепления, секвенирования, электрофореза и т.д.) наиболее важными параметрами выделенной ДНК являются чистота (см. ниже), количество (концентрация, мкг/мл) и целостность/степень фрагментации, т.е. физическая сохранность молекулы.
Преимуществами традиционных методов выделения с помощью вышеописанных органических растворителей являются относительно удовлетворительная чистота (показатель ОП260/ОП280 составляет 1,4-1,7) и высокая концентрация (>50 мкг/мл). Недостатки этих методов заключаются в том, что остаточные растворители могут влиять на чувствительность последующего молекулярного применения (например, проведение ПЦР-амплификации образцов гидролиза в режиме реального времени, микроанализ), а также в том, что способ выделения занимает много времени (минимум 1,5-2 часа). Что касается эффективности таких методик, они характеризуются низкой воспроизводимостью; среднее расхождение между двумя независимыми изолятами (CV%) довольно высоко, поэтому такие методики сложно привести к стандартным лабораторным протоколам.
Альтернативные методики и реагенты, основанные на выборочном связывании ДНК из раствора с использованием носителей, описанных выше, имеют более хорошие технические параметры, их показатель воспроизводимости лучше; они выполняются согласно более быстрому протоколу, и, вследствие стандартизованного характера, их легче использовать для инструментального выделения ДНК.
В патенте US 5234809 описывается способ выделения нуклеиновой кислоты, среди прочих, с использованием которой могут быть выделены одноцепочечная и двухцепочечная ДНК. В ходе описанного способа целевая нуклеиновая кислота выделяется без предварительного лизиса непосредственно из цельного биологического образца (например, цельной крови, сыворотки крови, мочи); в этом случае в единственной пробирке с буферной системой создается единое реакционное пространство, в котором находятся ионный хелатор (например, EDTA), связывающий двухвалентные ионы, хаотропное соединение (например, GTC), осаждающее белковые молекулы из их раствора, и поверхность матрицы (например, кремнезем, частицы латекса или нитроцеллюлозный фильтр), связывающая целевые нуклеиновые кислоты. В случае бактериальных образцов на результат применения способа в значительной степени влияет исходная концентрация клеток, поэтому диапазон размеров частиц кремнезема матрицы, связывающей нуклеиновые кислоты, варьируется в зависимости от конкретных условий методики.
В патенте WO 9534569 приводится еще один пример разрушения клеток и выделения ДНК в едином реакционном пространстве, в ходе которого значительно контаминированный маленький образец (например, 5×10 клеток, 0,5 мкл крови) лизируется в той же лунке микропланшета, в которой происходит экстрагирование ДНК на непористую, дисперсную, гомогенную поверхность матрицы, состоящей из частиц SiO2 размером 40 нм. Размер молекул ДНК, выделенных в ходе применения этого способа, варьируется в диапазоне от 50 до 60000 нуклеотидов. Аналогично, в патенте DE 4422044 описывается способ разрушения клеток и выделения ДНК в едином реакционном пространстве, в ходе которой ДНК-содержимое бактериальной плазмиды или клеточных культур HeLa выделяется с похожей эффективностью из такого же небольшого объема исходного образца.
Преимуществом единого реакционного пространства является минимизация технического контаминирования (например, пипетирования). Однако нельзя забывать о том, что кроме нуклеиновых кислот, которые должны быть выделены, на матричных поверхностях могут абсорбироваться или связываться другие компоненты (например, липиды, одноцепочечные нуклеиновые кислоты, пигменты, малые молекулы), которые могут быть потенциальными ингибиторами последующего молекулярного применения (например, проведения ПЦР-амплификации). Удаление таких потенциальных ингибиторов требует дополнительных этапов разделения в процессе выделения нуклеиновых кислот. Поэтому элюированию нуклеиновых кислот, связанных на кремнеземных носителях, перечисленных выше, элюентом, содержащим также ионный хелатор, предшествует промывание несколько раз буфером и органическим растворителем. Как указывалось ранее, фундаментальной проблемой выделения ДНК является сохранение целостности молекулы на протяжении всего процесса. Применяемые механические воздействия, такие как пипетирование, смешивание, центрифугирование, а также контакт с различными детергентами, ионными хелаторами и хаотропными агентами могут привести к искажению структуры или даже разрушению нуклеиновой кислоты. Осуществление лизиса клеток и выделения нуклеиновой кислоты в едином реакционном пространстве, как это было описано выше, минимизирует необходимость центрифугирования и других механических воздействий, что увеличивает сохранение целостности ДНК. В то же время, нельзя забывать о том, что кроме целостности ДНК, для последующего молекулярного применения требуются чистота и определенный выход нуклеиновых кислот. Для достижения этой комплексной цели, в отличие от методов единого пространства, в способе, описываемом в настоящем изобретении, проводится лизис клеток и последующее выборочное выделение ДНК в отдельных местах реакции, в результате чего упрощаются этапы промывания, изолят ДНК получается примерно в течение 30 минут, а его чистота согласно соотношению ОП260/ОП280 (в среднем составляет 1,8) и выход ДНК (20-100 мкг/100 мг исходного образца) являются предпочтительными для проведения ПЦР-амплификации и другого последующего молекулярного применения. В способе, описанном в настоящем изобретении, для выборочного связывания бактериальной двухцепочечной ДНК создается -SiO2-TiO2- матрица с повышенной химический и температурной стабильностью, на поверхности которой находятся химически активированные группы -ОН (силанол, титанол) и додециламиновые группы, при этом матрица в предпочтительном случае предварительно поперечно прошита додециламиновыми линкерами.
В патенте US 6787307 описывается раствор для выделения нуклеиновой кислоты, хромосомной ДНК бактерии, ДНК плазмиды на матричной поверхности, состоящей из намагниченных частиц кремнезема. В этом способе намагниченные частицы кремнезема вместе со связанной на их поверхности нуклеиновой кислотой, которую следует выделить из лизата, могут быть легко отделены с использованием соответствующего магнитного поля. На вышеупомянутом этапе выделения кроме целевых молекул ДНК лизата, полученного при разрушении клеток, на матричной поверхности также задерживаются и другие клеточные компоненты лизата, являющиеся ингибиторами последующего молекулярного применения (например, проведения ПЦР-амплификации). Удаление этих последних компонентов является длительным многоступенчатым процессом (см. выше). В отличие от этого, в способе, описанном в настоящем изобретении, ингибиторы ПЦР-амплификации не прикрепляются к созданной нами поверхности матрицы, связывающей ДНК.
В соответствии с одним из примеров реализации патента US 20100021905, смесь ДНК и РНК из ткани млекопитающего связывается поверхностью матрицы на основе кремнезема в буферных условиях, обеспечивающих связывание обеих нуклеиновых кислот. Связанная ДНК элюируется в щелочной реагент (рН=10) отдельно от белков и РНК. Селективность способа возрастает в зависимости от размера аниона соли гуанидиния, таким образом, что согласно описанию изобретения, вместо ионов хлорида предпочтительно присутствие ионов тиоцианата для более эффективного экстрагирования ДНК. При хаотропном обеспечении выделения ДНК на носителе согласно настоящему изобретению поверхность матрицы из -SiO2-TiO2- и гидрохлорид гуанидиния также оказались предпочтительными.
Трудности известных альтернативных методик, прежде всего, могут быть обусловлены следующими факторами:
a) если они обеспечивают выделение ДНК с высокой выборочностью, то выход чистой ДНК, свободной от компонентов, делающих невозможным последующее молекулярное применение (например, проведение ПЦР-амплификации), остается ниже 20 мкг общего количества выделенных ДНК;
или
b) если возможен более высокий выход ДНК (20-100 мкг/мл), то чистота полученной ДНК довольно сомнительна, т.е. соотношение ОП260/ОП280 является низким (1,3-1,4).
Более того, эти альтернативные методики, в целом, характеризуются следующим:
a) в большинстве случаев параметры выборочное™ и количественные параметры нуклеиновой кислоты, связывающейся с носителем, являются спорными, это значит, что помимо выделяемых двухцепочечных ДНК, они также связывают одноцепочечные ДНК и РНК, и присутствие этих последних может искажать или нарушать ход последующего молекулярного применения и молекулярную специфичность;
b) количество выделенной ДНК в значительно степени зависит от связывающей способности носителей;
c) качество выделенной ДНК и содержание ингибиторов в значительно степени зависит от характера образца;
d) они в большинстве случаев эффективны для селективно обогащенных микробных образцов и не столь эффективны для выделения из сложных и смешанных бактериальных образцов,
e) что важнее всего, почти все методики выделяют ДНК как из жизнеспособных, так и из нежизнеспособных бактериальных клеток, при этом хромосомные ДНК, полученные из нежизнеспособных клеток, приводят к получению ложных положительных сигналов в ходе применения способов определения.
В отношении доступных в настоящее время химических веществ и реагентов следует сказать, что в современной практике не существует способа приготовления ДНК, который бы отвечала одновременно всем следующим требованиям:
a. способ может оперативно выполняться быстро даже в условиях применения лабораторного оборудования общего назначения;
b. способ позволяет выделять ДНК исключительно из жизнеспособных клеток и получать динамичный результат;
c. способ обеспечивает получение положительного результата даже в случае относительно низкого количества микробов;
d. способ специфичен только в отношении двухцепочечных ДНК;
e. способ подходит для получения ДНК с надлежащей высокой степенью химической чистоты (без ингибиторов) и в надлежащей высокой концентрации;
f. способ может характеризоваться высокой воспроизводимостью и низким числом комбинированных вариаций;
g. микробиологические образцы могут отбираться из многих различных источников.
Целью способа, описываемого в настоящем изобретении, является устранение недостатков известных методик и разработка препаративного процесса, в котором специфическое выделение полного ДНК-содержимого бактериальных возбудителей инфекции имеет преимущества, описанные в пунктах a-g, и может быть использовано в качестве надежного достоверного метода на практике в микробиологической диагностике (в общественном здравоохранении, клинической практике, контроле санитарно-гигиенического состояния воды и продуктов питания).
Из описанных выше базовых примеров видно, что ни один из способов разрушения клеток и экстракции нуклеиновой кислоты, применяющихся в преаналитической фазе, которая предшествует молекулярной диагностике на основе бактериальной нуклеиновой кислоты не предполагает разделения в исходном образце жизнеспособных и нежизнеспособных клеточных популяций. Проведение различия между этими двумя состояниями клеток может в значительной степени повлиять на результат выделения полного бактериального ДНК-содержимого, а также на последующее молекулярное применение.
Как и у эукариотов, динамика популяций бактериальных клеток также регулируется пролиферацией и смертью клеток, на которые влияют как внутриклеточные, так и внеклеточные факторы. По этой причине при выделении полного бактериального ДНК-содержимого из образцов, исследуемых в гигиене общественного здоровья, нельзя пренебрегать таким явлением, как смерть клеток.
В биологических системах одним из видов смерти клеток является некроз - повреждение клеток, которое вызывается физическими, химическими и/или биологическими воздействиями. Некроз - это процесс, который можно описать, обычно, как разрушение клетки, например, как повышение пассивной проницаемости клеточной мембраны и клеточной стенки, разрушение компонентов клетки, нарушение клеточной организации и выход содержимого клетки наружу [Robbins and Cotran Pathologic Basis of Disease: 8th edition, Saunders, Elsevier, 2010], в процессе которого продукты распада, получающиеся из клетки в результате некроза, обладают токсичным влиянием на соседние клетки. В биологических системах существует другой вид смерти клеток, называемый апоптозом, который представляет собой запрограммированную смерть клетки; это явление хорошо определяется у эукариотов [Diaz L.F. et al. (2005): Cell Death and Differentiation 12: 1449-1456]. Этот вид смерти клеток является активным процессом, контролируемым на генетическом уровне, на этот процесс влияют стимулы окружающей среды (т.е. морфологические перестройки, сопровождающие изменения в метаболической активности различных периодов онтогенеза), в ходе этого процесса вследствие структурных изменений клеточной мембраны (например, потери фосфатидилсериновой асимметрии, изменений процессов транспорта) снижается способность прилипания и тургор клеток, наблюдается сморщивание клеток, которое сопровождается внутриклеточным ферментативным расщеплением, в ходе которого ДНК-содержимое становится фрагментированным, и в конце этого процесса клетка, подвергающаяся апоптозу, утилизируется соседними клетками. Повышается количество доказательств роли апоптотических механизмов в так называемой адаптации популяций прокариотов к стрессам окружающей среды [Lewis K. (2000): Microbiol.Mol.Biol.Rev. 64: 503-514, Koonin E.V., Aravind L. (2002): Cell Death and Differentiation 9: 394-404]. Примером такой адаптации является множественное деление грамположительных бактерий родов Bacillus и Streptomyces и грамотрицательных бактерий рода Myxobacteria, которое приводит к программируемой смерти клеток посредством аутолитических ферментов, индуцированных в материнской клетке. В качестве примера также можно упомянуть адаптивное появление мутаций, ведущих к невозможности культивирования, у различных грамположительных бактерий [Hochman A. (1997): Critical Rev.Microbiol. 23: 207-214].
Для выделения полного ДНК-содержимого бактериальных возбудителей инфекции в пробных образцах, исследуемых в общественной гигиене, первоначальный этап для разделения жизнеспособных и нежизнеспособных популяций клеток, представляет диагностическую ценность. Соответственно, дальнейшей целью способа, описываемого в настоящем изобретении, является разработка способа разделения клеток на жизнеспособные и нежизнеспособные бактериальные клетки и обеспечение полного выделения ДНК-содержимого исключительно из жизнеспособных клеток.
Описанные выше критерии смерти клеток могут объяснить данные наблюдений, согласно которым в исследованных образцах некротические комплексы появляются на поверхности нежизнеспособных бактериальных клеток, что приводит к агрегации этой клеточной популяции. Нами было также отмечено, что для нежизнеспособных клеток характерно изменение гидратации; их более низкая суспензируемость отлична от суспензируемости жизнеспособных клеток; при этом соотношение клеточной поверхности к клеточному объему тоже изменяется. Мы поняли, что, базируясь на указанных выше физико-химических характеристиках, возможно четко разделить жизнеспособные и нежизнеспособные бактериальные клетки при использовании раствора, содержащего буферную смесь подходящих детергента и электролита, предпочтительно Тритон Х-100, этилендиаминтетрауксусную кислоту, KCl или NaCl, моногидрат лимонной кислоты, т.е. при использовании так называемого буфера, удаляющего нежизнеспособные клетки; в этом случае клетки осаждаются при изменении центробежной силы, которая влияет на осаждение. Мы предположили, что в результате изменения гидратации некротизированных бактериальных клеток происходит изменение клеточной поверхности, объема клеток, т.е их размера и плотности, что, в совокупности с надлежащим образом выбранной центробежной силой, является дифференцирующим фактором при осаждении путем центрифугирования.
При исследовании на примере эукариотов разделение жизнеспособных и нежизнеспособных клеток путем центрифугирования при соответствующей центробежной силе происходит на основании различных размеров клеток, их плотности и гидратации. В результате этого масса нежизнеспособных клеток оказывается в супернатанте, тогда как жизнеспособные клетки попадают в осадок [Fisher D. et al.: Cell Separation: A Practical Approach, Oxford Univ.Press, 1998]. Однако следует учитывать, что даже при соответствующей центробежной силе разделение часто является неполным, и небольшое количество жизнеспособных клеток может быть определено также и в супернатанте. Еще одной возможностью разделения, дополняющей вышеуказанный способ, является центрифугирование в градиенте плотности, используемое для разделения клеток с различными размерами и плотностью;
при необходимости, дополнительным методом разделения может быть in vitro культивирование отдельных осажденных фракций. Серия этапов разделения, описанная выше, безусловно, занимает много времени; в ходе этих этапов повышается риск потери клеточной массы, что уменьшает точность дальнейшего применения. Однако сохранение клеточной массы исходного образца является существенным в разделении жизнеспособных и нежизнеспособных клеток для специфического выделения полного ДНК-содержимого исключительно жизнеспособных бактериальных возбудителей инфекции.
На основании вышеизложенного, в способе, описанном в настоящем изобретении, разделение жизнеспособных и нежизнеспособных клеток основывается на разных характеристиках размеров клеток, характерных для двух состояний клеток, а также на их различной проницаемости и степени гидратации, что обусловлено структурной трансформацией и изменененным аффинитетом ионов клеточной мембраны. Согласно описываемому способу разделение клеток было проверено с помощью проточного цитометра.
Использование проточного цитометра основано на том, что гидродинамически сфокусированная клеточная суспензия проходит через монохромный лазерный пучок таким образом, что клетки, проходящие через окно детекции одна за одной за определенное время, рассеивают свет вперед и в стороны в зависимости от их размера и внутренней организации (рассеяние вперед - размер, рассеяние в стороны - внутренняя организация); при этом свет направляется на детекторы с использованием оптической системы. Сигналы, поступающие на детектор, суммируются блоком электронной обработки и отображаются в форме диаграммы рассеяния или гистограммы (кривой нормального распределения). В нашем случае за основу принимается размер клеток, что позволяет разделить жизнеспособные и нежизнеспособные клетки (см. описание). Диаграмма рассеяния и гистограмма отражают число клеток, которые могут быть сгруппированы по заданному размеру клеток, поэтому квалифицированному специалисту будет понятно, что поточная цитометрия подходит для инструментальной количественной оценки популяций жизнеспособных и нежизнеспособных клеток в исследуемом образце [Shapiro Н.М.: Practical Flow Cytometry, 2nd Edition, John Wiley & 20 Sons, New York, 1988].
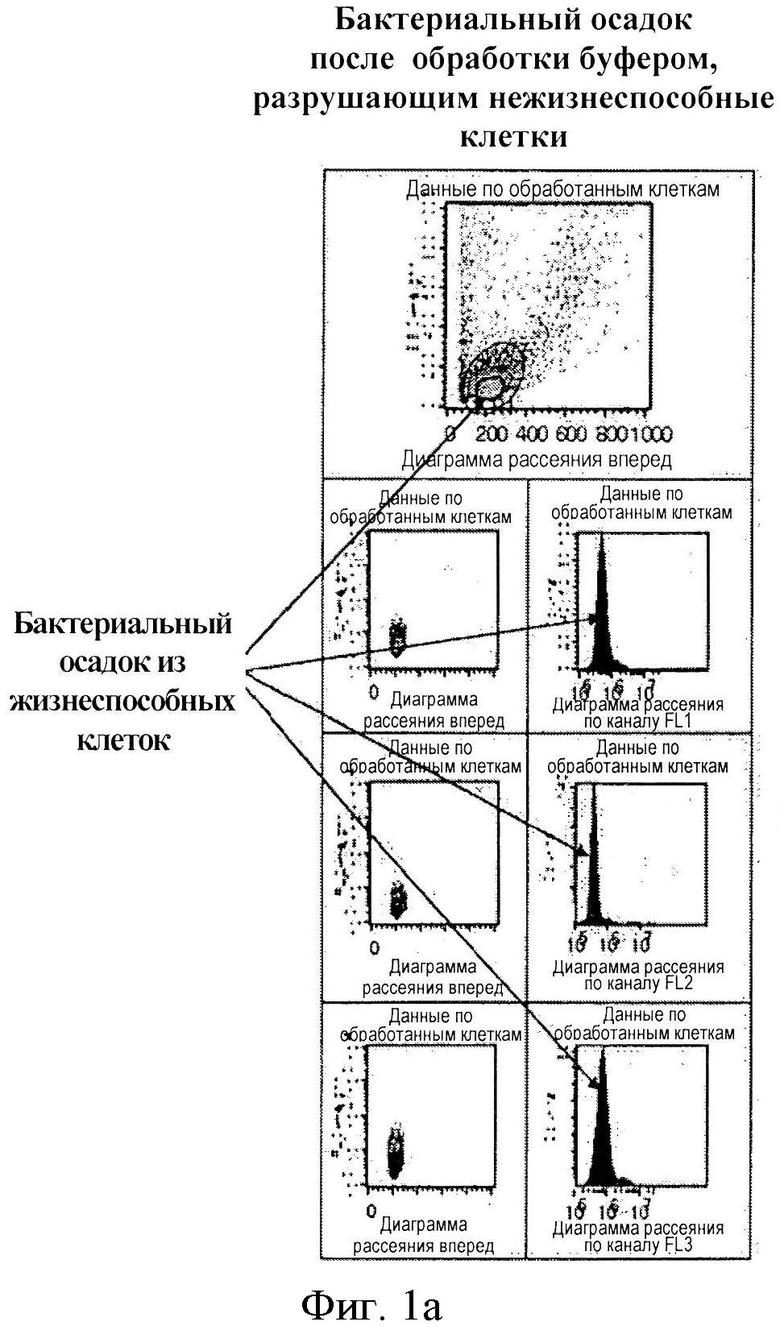
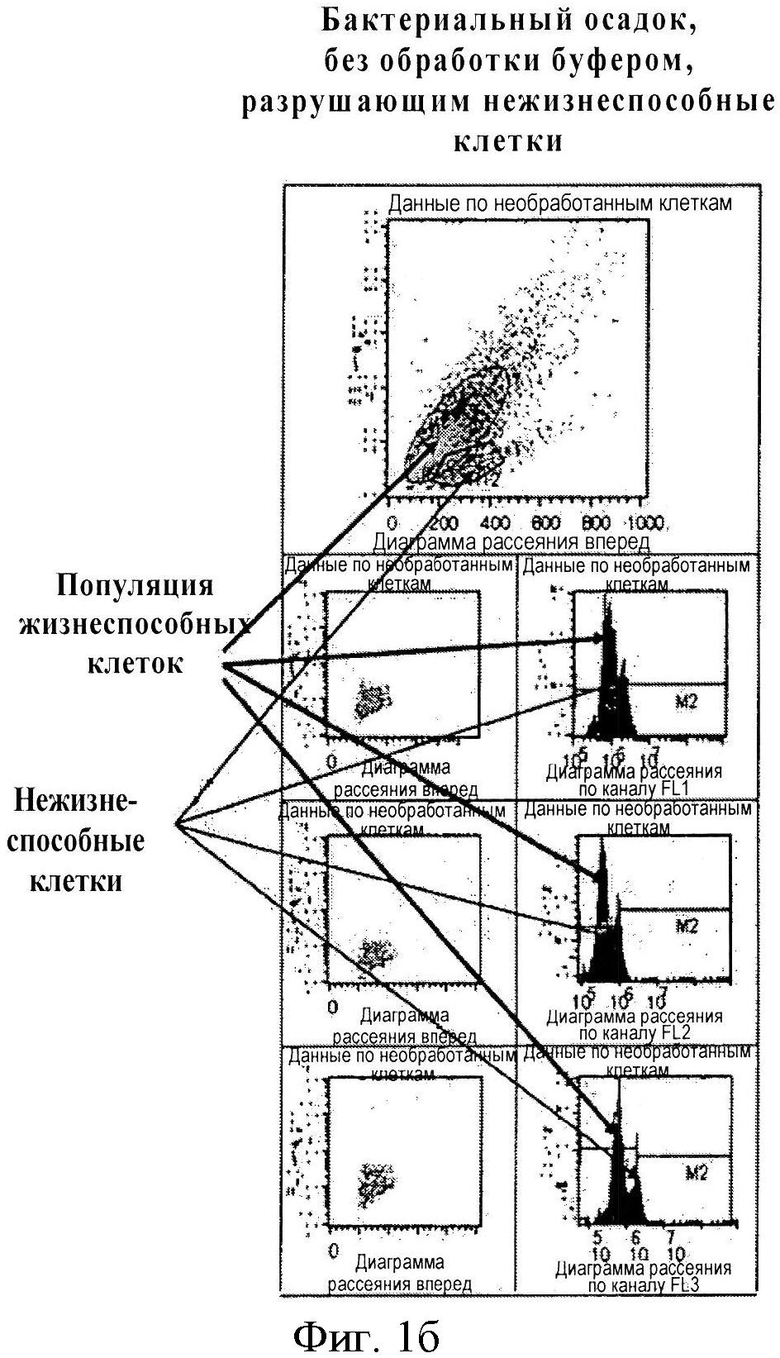
Мы предположили, что после неоднократного промывания образца нашим буфером, удаляющим нежизнеспособные клетки, а также после центрифугирования образца при определенных условиях нежизнеспособные клетки остаются в супернатанте, тогда как жизнеспособные бактериальные клетки могут быть выборочно осаждены и перенаправлены для дальнейшего выделения ДНК. Соответствующее предположение было подтверждено вышеописанным методом проточной цитометрии, в ходе которого проверялось наличие жизнеспособных и нежизнеспособных клеток в осадке и в супернатанте. Полученные результаты представлены на фигуре 1 в виде диаграмм рассеяния и гистограмм 1а и 1b (подробное описание представлено в разделе «Описание фигур»).
На данном этапе, имея осадок, содержащий жизнеспособные клетки, выделенные из исследуемого образца путем обработки разработанным нами буфером, удаляющим нежизнеспособные клетки, можно начинать процесс выделения ДНК (лизис, связывание ДНК, промывание и элюирование).
При проведении экспериментов мы успешно разработали связывающий матричный носитель для двухцепочечной ДНК, который непосредственно и выборочно связывает только двухцепочечные молекулы ДНК, получаемые после лизиса клеток, и не связывает контаминирующие ингибиторы лизированного образца. В нашем матричном носителе, связывающем двухцепочечную ДНК, предпочтительно применение матрицы -SiO2-TiO2-, содержащей химически активированные гидроксильные и додециламиновые группы. Особенно предпочтительно, если додециламиновые группы предварительно поперечно сшиты между собой додециламиновыми линкерами. Посредством этой поперечно-сшитой матрицы создаются условия для специфической реакции целевого связывания, тогда как связывание контаминирующих и остаточных ингибиторов сложного образца (крови, кала, почвы) подавляется.
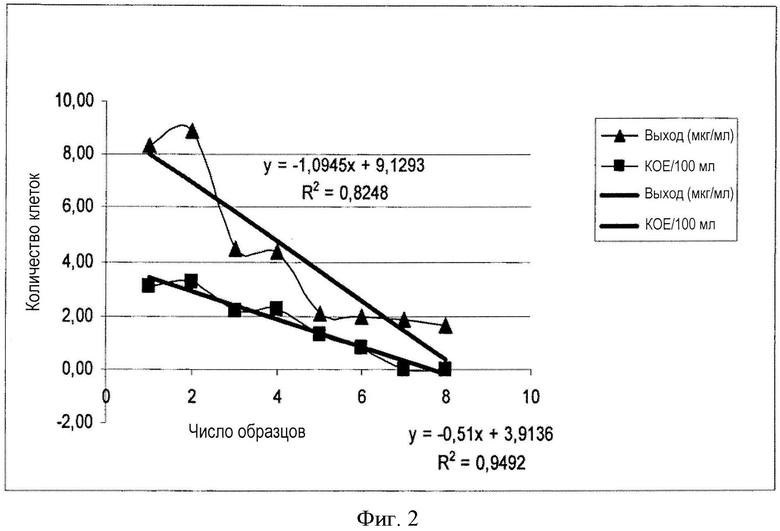
Чувствительность описываемого нами способа продемонстрирована выборочным выделением полного ДНК-содержимого из обедненных исследуемых образцов с низким титром КОЕ (колониеобразующих единиц) (см. фигуру 2 и ее подробное описание).
Задачей настоящего изобретения является способ специфического выделения полного ДНК-содержимого бактериальных возбудителей инфекции в различных образцах, в ходе которой происходит удаление нежизнеспособных клеток; сохраненные жизнеспособные клетки лизируются, после чего из лизата выборочно связывается полное ДНК-содержимое; связанное полное ДНК-содержимое промывается и обессоливается; затем обессоленная нуклеиновая кислота элюируется из связывающей поверхности водным раствором. Под образцом понимаются материалы, исследуемые в общественной гигиене, такие как вода и пищевые продукты, клинические, твердые и жидкие материалы окружающей среды, а также пузырьки воздуха в жидкости, популяция бактериальных клеток, которые обрабатываются разработанным нами буферным раствором, удаляющим нежизнеспособные клетки сразу же после отбора или перед этапом обогащения, который предшествует литическому разрушению клеток. Сущность использования разработанного нами буфера, удаляющего нежизнеспособные клетки, заключается в том, что нежизнеспособные и жизнеспособные клетки разделяются на основе различий в физико-химических характеристиках поверхности этих клеток, в результате чего на ДНК-связывающем матричном носителе из лизата связывается двухцепочечная ДНК, полученная исключительно из жизнеспособных клеток; в предпочтительном случае матричный носитель представляет собой матрицу -SiO2-TiO2-, содержащую химически активированные группы -ОН и додециламиновые группы, предпочтительно предварительно поперечно-сшитые с додециламиновыми линкерами. Неспецифичные вторичные связи на ДНК-связывающем матричном носителе блокируются путем добавления трегалозы или декстрана и БСА (бычьего сывороточного альбумина).
В предпочтительном варианте осуществления способа жизнеспособные и нежизнеспособные клетки распознаются на основании различной проницаемости клеточной поверхности и аффинитета ионов, как это указано выше.
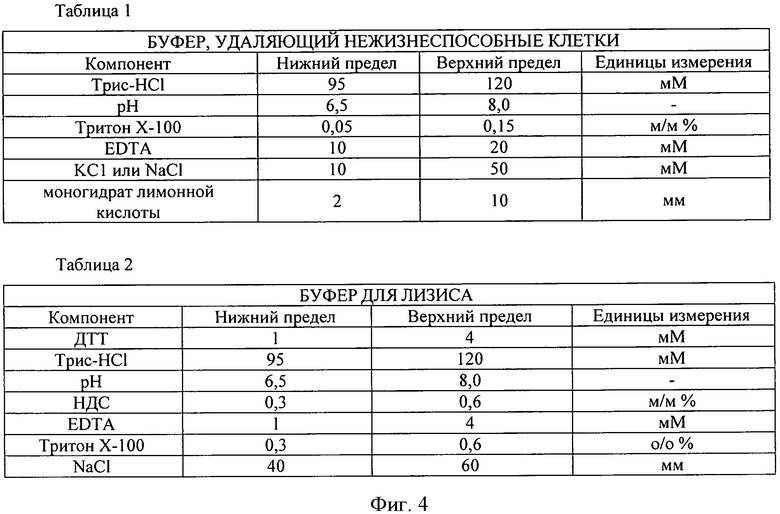
В случае еще одного предпочтительного варианта реализации изобретения для распознавания жизнеспособных и нежизнеспособных бактериальных клеток используется промывочный раствор с различной проникающей способностью. Например, может использоваться промывочный раствор, содержащий детергент, электролит, предпочтительно Тритон Х-100, этилендиаминтетрауксусную кислоту, KCl, NaCl, моногидрат лимонной кислоты в буферной смеси (состав представлен на фигуре 4 в таблице 1); при этом изменяется центробежная сила, влияющая на осаждение клеток.
В предлагаемом способе лизис жизнеспособных клеток проводится путем их механического разрушения или путем совместного механического и ферментативного разрушения, наиболее предпочтительно путем расщепления лизоцимом в специальных пробирках для разрушения клеток. Механическое разрушение бактериальных клеток обеспечивается использованием настольного оборудования.
В способе, описываемом в настоящем изобретении, из бактериального образца, содержащего по крайней мере 104 КОЕ/мл, двухцепочечная ДНК изолируется с помощью буфера для лизиса, содержащего окислительно-восстановительный компонент, хелатирующий агент и детергенты, предпочтительно дитиотреитол (ДТТ), этилендиаминтетрауксусную кислоту (EDTA), Тритон Х-100 и натрия додецилсульфат (НДС). Контаминирующие красящие вещества, получаемые из жизнеспособных бактериальных клеток, и другие ингибиторы ПЦР-амплификации (порфирины, гемовые соединения, полиароматические вещества) отделяются во время осаждения бактериального лизата, поскольку буфер для лизиса также является благоприятной физико-химической средой для осаждения этих веществ.
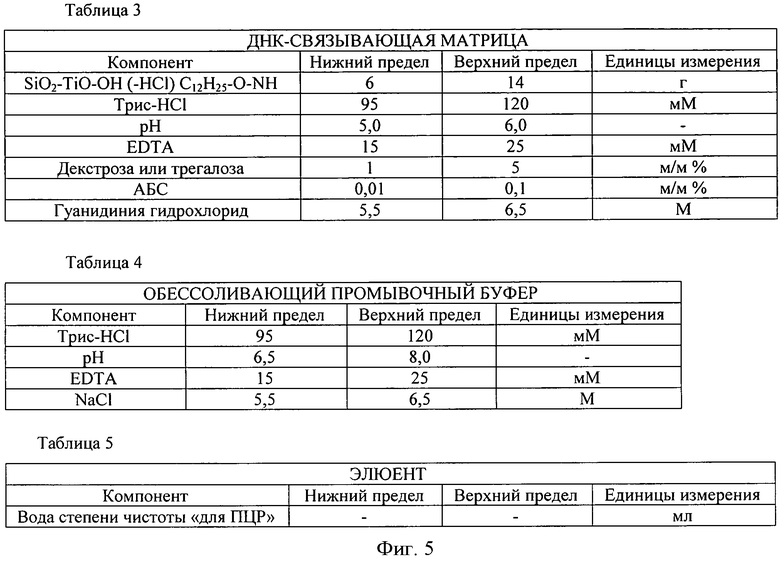
В варианте осуществления способа, в соответствии с настоящим изобретением, выборочное связывание двухцепочечной ДНК из лизата жизнеспособных бактериальных клеток без ее ухудшения ее качественных характеристик проводится на матрице -SiO2-TiO2 (состав представлен на фигуре 5 в таблице 3), содержащей химически активированные группы -ОН и додециламиновые группы и предварительно поперечно-сшитой с помощью додециламиновых линкеров. Образование неспецифических вторичных связей с матрицей (РНК, белки, другие макромолекулы) блокируется добавлением трегалозы или декстрана и БСА (бычьего сывороточного альбумина).
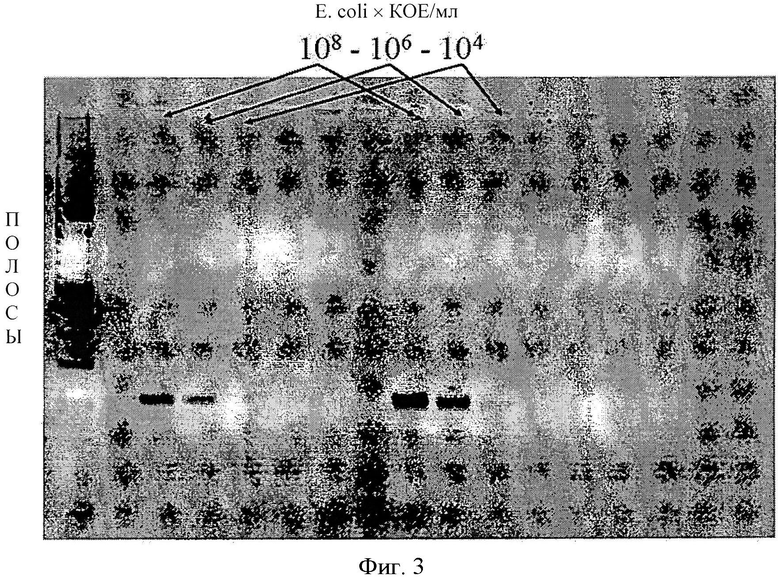
Количество и качество (чистота) выделенной двухцепочечной ДНК проверяется с помощью УФ-спектрофотометра с длиной волны 260 нм и 280 нм, и сравниваются коэффициенты ослабления ОП260 и ОП280. В качестве полуколичественного теста применяется традиционный электрофорез в агарозном геле, следующий за ПЦР-амплификацией выделенной ДНК in vitro (на фигуре 3 приводится пример и его подробное описание).
В изобретении также описывается НАБОР, подходящий для экстрагирования полного ДНК-содержимого бактериальных возбудителей инфекции путем описанного выше способа, который, прежде всего, предназначен для анализа воды, пищевых продуктов, различных поверхностей мест общественного пользования и образцов, исследуемых в общественном здравоохранении (в санитарной медицине). Структурное изображение состава НАБОРА представлено на фигуре 6 (подробное объяснение приводится в описании фигуры).
Ниже приводится подробное описание фигур.
Фигура 1. С помощью поточной цитометрии показана эффективность удаления нежизнеспособных клеток перед этапом выделения полного ДНК-содержимого бактериальных возбудителей инфекции. По оси Х откладывается размер клеток, по оси Y - количество клеток определенного размера. На фигуре 1а показаны поточно-цитометрическая диаграмма рассеяния (точки) и гистограмма (кривая нормального распределения) осадочной фракции бактериальных клеток после центрифугирования, обработанных разработанным нами буфером, удаляющим нежизнеспособные клетки (состав буфера представлен на фигуре 4 в таблице 1). На фигуре 1b представлены поточно-цитометрическая диаграмма рассеяния (точки) и гистограмма (кривая нормального распределения) осадочной фракции бактериальных клеток после центрифугирования, не обработанных разработанным нами буфером, удаляющим нежизнеспособные клетки. При сравнении двух фигур можно отметить, что, в соответствии с фигурой 1а, жизнеспособные гидратированные клетки одинакового размера осаждаются в виде гомогенной фракции (которая на фигуре называется «осадком»), тогда как малогидратированные или негидратированные некротические клетки остаются в супернатанте, что является результатом обработки буфером, удаляющим нежизнеспособные клетки. В соответствии с фигурой 1b, если образец клеток не обрабатывается буфером, удаляющим нежизнеспособные клетки, то состав осадка в пробирке гетерогенен; кроме того, в нем, помимо жизнеспособных клеток, появляется дополнительная фракция; гистограмма оказывается ложно расширенной.
Фигура 2. Выделение полного ДНК-содержимого бактериальных возбудителей инфекции по описанному в настоящем изобретении способу из водного раствора с низким титром КОЕ (КОЕ/100 мл), сравнение логарифмических значений количества клеток (ось Y, нижняя кривая с квадратами), которое можно соотнести с числом образцов (ось X) и выходом выделенной ДНК (ось Y, верхняя кривая с треугольниками). Полное ДНК-содержимое бактериальных возбудителей инфекции было выделено из водных образцов с различными разведениями, т.е. из образцов с различными титрами КОЕ Legionella pneumophila (ось Y, нижняя кривая с квадратами). Характер линии регрессии выхода бактериальной ДНК (мкг/мл), полученной по описанному нами способу, подобен характеру линии регрессии исходного числа клеток, но в случае выхода ДНК наклон более выражен. На основании подобного характера линий регрессии можно сделать заключение, что выделение полного ДНК-содержимого бактериальных возбудителей инфекции по описанному способу следует из числа клеток в исходном образце. Тот факт, что ДНК может быть успешно выделена (значения мкг/мл на верхней кривой по оси Y соотносились с числом образцов 5-8 по оси X) даже в случае большего наклона линии регрессии выхода ДНК, а также в случае значительного разведения (значения КОЕ на нижней кривой по оси Y соотносились с числом образцов 5-8 по оси X), позволяет сделать заключение, что выделение двухцепочечной ДНК в соответствии с описанным способом, надежно осуществимо в случае низкого титра клеток, т.е. при выраженном разведении образца. Следовательно, выделение ДНК по описанному способу доказывает его достаточную чувствительность.
Фигура 3. Полуколичественный анализ бактериальной полной ДНК, выделенной по описанному в настоящем изобретении способу, на примере Е.coli, с помощью электрофореза в агарозном геле. В ходе полу количественного анализа сначала ДНК, не содержащая ингибиторов, элюированная с матричной поверхности, подвергается ПЦР-амплификации, затем увеличенное количество полинуклеотидов анализируется методом электрофореза в агарозном геле с использованием ДНК-интеркалирующего красителя этидиума бромида для флуоресцентного излучения, индуцированного УФ-освещением. На основе полос плотности, видимых на фигуре, порог чувствительности описанного способа выделения ДНК составляет 104 КОЕ/мл. В крайнем левом столбике агарозного геля можно увидеть электрофореграмму полинуклеотида с исходной молекулярной массой, в других столбиках показана электрофоретическая картина амплифицированной с использованием ПЦР-амплификации полной ДНК, выделенной из Е.coli водного образца. Выделения ДНК из образцов с бактериальных титром 108 КОЕ/мл; 106 КОЕ/мл; 104 × КОЕ/мл, показанные в верхней части столбиков, проводились с 10 независимыми повторами.
Фигуры 4 и 5. Таблицы 1-5 содержат подробные сведения о возможных условиях осуществления способа, описанного в настоящем изображении, такие как компоненты и диапазоны изменения рН буфера, удаляющего нежизнеспособные клетки, буфера для лизиса, матрицы связывания, обессоливающего промывочного буфера и элюента.
Фигура 6. Состав НАБОРА для точного выделения полного ДНК-содержимого бактериальных возбудителей инфекции. Показан набор для обработки 42 образцов. На фигуре показана комплексная интегральная система для выделения микробной ДНК. Описываемый НАБОР содержит растворы, которые используются на этапах способа, следующих за удалением нежизнеспособных клеток, таких как буфер для лизиса, матрицу связывания, обессоливающий промывочный буфер и элюент, а также пробирки для разрушения клеток, описанные в примере использования и установленные в соответствии с текущими потребностями.
Ниже приводится краткое резюме преимуществ и возможностей реализации настоящего изобретения. В ходе реализации предпочтительно следовать рекомендациям таблиц на фигурах 4 и 5, приведенных ниже.
1) В таблице 1 показан предпочтительный состав буферного промывочного раствора, подходящего для отделения жизнеспособных и нежизнеспособных клеток согласно нашему изобретению.
В результате отделения жизнеспособных и нежизнеспособных клеток получена возможность выделить полное ДНК-содержимое исключительно жизнеспособных клеток, присутствующих в образцах, исследуемых в санитарной медицине.
2) В отличие от обычных буферов для лизиса целостность ДНК в разрушенных бактериальных клетках весьма успешно сохраняется в буфере для лизиса, соответствующем таблице 2, содержащем восстанавливающий дитиотреитол (ДТТ), анионный натрия додецилсульфат (НДС) и неионный детергент (Тритон Х-100), в щелочной среде буфера Трис-HCl, в присутствии двухвалентного ионного хелатора (EDTA).
Если применение буфера для лизиса, соответствующего настоящему изобретению, сочетается с применением изготовленной нами пробирки для разрушения клеток, содержащей стеклянные опилки и стальной шарик, то получается метод разрушения клеток, который можно адаптировать к автоматизированной подготовке ДНК и системе обработки образца на основе механического разрушения.
3) Буфер для лизиса, описанный в пункте 2, также может эффективно сочетаться с традиционным методом разрушения, проводящимся с помощью лизоцима.
4) Из смеси компонентов, показанных в таблице 3, нами разработан матричный носитель, посредством которого ДНК связывается напрямую после лизиса бактериальных клеток в достаточно высоком объеме; при порядке возрастания величины связывающей способности выше 20 мкг/100 мг.
ДНК-связывающая поверхность в соответствии с описанным способом является -SiO2-TiO2- матрицей, содержащей химически активированные группы -ОН и додециламиновые группы, предпочтительно поперечно-сшитые между собой с помощью додециламиновых линкеров. Эта матрица связывает двухцепочечные ДНК выборочно и исключительно в среде, соответствующей таблице 3, и определенно не связывает наиболее распространенные ингибиторы ПЦР-амплификации, присутствующие в биологических образцах (например, порфирины, билирубин). Неспецифические вторичные связи на матрице (РНК, белки, другие макромолекулы) блокируются добавлением трегалозы или декстрана и БСА (бычьего сывороточного альбумина).
Согласно нашему опыту предпочтителен денатурирующий хаотропный компонент гидрохлорид гуанидиния.
5) С использованием описанного выше обессоливающего промывочного буфера в соответствии с таблицей 4 примесь из ДНК, связанной на поверхности ДНК-связывающего матричного носителя, легко удаляется в присутствии двухвалентного ионного хелатора; для этого проводится два этапа промывания с короткими периодами центрифугирования.
6) ДНК, связанная на поверхности ДНК-связывающего матричного носителя, соответствующего таблице 3 и обработанного описанным выше обессоливающим промывочным буфером, соответствующим таблице 4, элюируется из связывающей поверхности водой, имеющей степень чистоты «для ПЦР-амплификации», в соответствии с таблицей 5.
7) Среднее значение чистоты ДНК, элюируемой согласно пункту 6, составляет ОП260/ОП280=1,8.
Значение комбинированного коэффициента вариации (KB), характеризующее чистоту двух независимых изолятов, полученное с помощью описанного способа, составляет 9%. Средняя концентрация ДНК, которая элюируется согласно пункту 6, составляет 60 мкг/мл, значение типичного комбинированного KB составляет 7,5%. Средняя фрагментация ДНК, которая элюируется согласно пункту 6, составляет 5-12 тысяч пар нуклеотидов, значение типичного комбинированного KB составляет 15%. Наименьшее исходное число бактериальных возбудителей инфекции для выделения ДНК по описанному в настоящем исследовании способу, составляет 104 КОЕ/мл, значение типичного комбинированного KB составляет 10,5%.
8) Количество полной ДНК, выделенной из образцов с различным титром возбудителей инфекции с помощью ДНК-связывающего матричного носителя, может быть соответствующим образом проанализировано с помощью полуколичественного определения (см. фигуру 3 и объяснение к ней). В процессе полуколичественного определения сначала ДНК, не содержащая ингибиторов, элюированная с матричной поверхности, подвергается ПЦР-амплификации, затем увеличенное количество полинуклеотидов анализируется посредством метода электрофореза в агарозном геле с использованием флуоресцентной плотности, излучаемой ДНК-интеркалирующим красителем этидиумом бромида при УФ-освещении (см. описание).
9) Еще одной специфической особенностью описанного способа является то, что размер бактериальной ДНК, которая выделяется таким способом, составляет не менее 500-1500 пар оснований линейной полимерной нуклеиновой кислоты, при этом использование описанного способа для получения ДНК размеров меньших, чем этот, не желательно.
Еще одним преимуществом описанного способа является то, что физико-химическая сохранность экстрагированной ДНК, ее чистота и концентрация делают возможным ее последующее использование для проведения ПЦР-амплификации.
Еще одним преимуществом описанного изобретения является то, что методика может осуществляться быстро и может сочетаться с традиционными и альтернативными методами выделения ДНК с помощью приготовленного для этих целей буфера для лизиса.
Несколько возможных вариантов реализации настоящего изобретения описано ниже; данное описание не ограничивает объем запатентованного изобретения соответствующими примерами.
Примеры реализации
Идеальные возможности выделения полного ДНК-содержимого бактериальных возбудителей инфекции в образцах воды, пищевых продуктов, клинических образцах, твердых и жидких образцах, полученных из окружающей среды, а также пузырьках воздуха в жидкости и т.д.
В ходе реализации предпочтительно руководствоваться таблицами, представлеными на фигурах 4 и 5, приведенных ниже.
А/ Разрушение клеток образца механическим и ферментативным способом
Реакция оптимизирована для лабораторных инструментов, которые применяются в молекулярной биологии, и выделяющих устройств (например, FastPrep™).
Первая фаза способа / удаление нежизнеспособных бактериальных клеток из образца
1. Жидкий культивируемый бактериальный образец осаждается при комнатной температуре (КТ) в течение 10 минут.
2. Жидкая фаза образца переносится в чистую стерильную пробирку для центрифугирования.
3. Перенесенный образец центрифугируется со скоростью 3000 об/мин в течение 1 мин при температуре 25°C.
4. Супернатант удаляется, затем осадок тщательно суспендируется в 5 мл БУФЕРА, УДАЛЯЮЩЕГО НЕЖИЗНЕСПОСОБНЫЕ КЛЕТКИ (состав буфера и соответствующее значение рН приводится в таблице 1).
5. Суспензия тщательно перемешивается в вихревой мешалке.
6. Суспензия центрифугируется со скоростью 3000 об/мин в течение 1 мин при температуре 25°C.
7. Супернатант удаляется, затем преципитат тщательно суспендируется в 5 мл БУФЕРА, УДАЛЯЮЩЕГО НЕЖИЗНЕСПОСОБНЫЕ КЛЕТКИ (состав буфера и соответствующее значение рН приводится в таблице 1).
8. Суспензия тщательно перемешивается в вихревой мешалке.
9. Суспензия центрифугируется со скоростью 2000 об/мин в течение 1 мин при температуре 25°C.
10. Супернатант удаляется, затем осадок тщательно суспендируется в 1 мл БУФЕРА, УДАЛЯЮЩЕГО НЕЖИЗНЕСПОСОБНЫЕ КЛЕТКИ (состав буфера и соответствующее значение рН приводится в таблице 1).
11. Суспензия тщательно перемешивается в вихревой мешалке.
12. Суспензия центрифугируется со скоростью 1000 об/мин в течение 3 мин. при температуре 4°C.
13. Осадок растворяется в 0,2-0,5 мл буфера с 0,1 М Трис-HCl (рН=7,5).
14. Образец готов для проведения лизиса клеток.
Ниже приводятся обязательные отличительные особенности первой фазы способа:
a) Маточные растворы хранятся при температуре +4°C.
b) При приготовлении маточных растворов необходимо строго следовать стандартным лабораторным протоколам, относящимся к способам проведения ПЦР-амплификации (EN ISO 20838: 2006, GLP (GLP - Надлежащая лабораторная практика)).
c) Работа должна проводиться строго в чистом помещении (ISO 209, FS 209, BS5295, ISO 14644-1: 1999) при минимальном уровне безопасности BSL2 (уровень биологической безопасности 2).
Вторая фаза способа / лизис клеток и связывание двухцепочечной ДНК на поверхности матричного носителя
1. Перенести образец, обработанный в соответствии с этапами 1-14 первой фазы способа, в пробирку для разрушения клеток емкостью 2 мл, содержащую 0,1 г стеклянных опилок и шарик из стали Cr-Va размером 4,5" (4,5×25,4 мм).
2. Поместить пробирку для разрушения, содержащую образец, в гомогенизирующий асимметрический дезинтегратор (например, FastPrep™) и затем начать процедуру циклического разрушения. Через 30 секунд образец можно использовать для дальнейших операций.
3. Добавить фермент лизоцим к образцу до конечной концентрации 100 мкг/мл и инкубировать образец в течение 15 минут при температуре 37°C. После этого образец можно использовать для дальнейших операций.
4. Добавить 1 мл БУФЕРА ДЛЯ ЛИЗИСА (состав буфера и соответствующее значение рН приводится в таблице 2) к механически и ферментативно обработанному образцу. Затем оставить его при комнатной температуре на 1-2 минуты.
5. Центрифугировать суспензию со скоростью 13000 об/мин в течение 5 мин при температуре 4°C.
6. Перенести 800 мкл супернатанта в чистую стерильную пробирку для центрифугирования емкостью 2,5 мл.
7. Добавить 800 мкл ДНК-СВЯЗЫВАЮЩЕЙ МАТРИЦЫ (состав матрицы и соответствующее значение рН приводится в таблице 3). Укупорить пробирку для центрифугирования.
8. Осторожно повернуть пробирку с суспензией 10-15 раз. На этом этапе запрещается использовать вихревую мешалку при любых обстоятельствах.
9. Инкубировать пробирку в течение 2 минут при комнатной температуре, медленно поворачивая ее с широкими интервалами.
10. Центрифугировать суспензию со скоростью 12000 об/мин в течение 1 мин. Удалить супернатант.
11. Суспендировать преципитат в 700 мкл ОБЕССОЛИВАЮЩЕГО ПРОМЫВОЧНОГО БУФЕРА (состав буфера и соответствующее значение рН приводится в таблице 4).
12. Центрифугировать суспензию со скоростью 12000 об/мин в течение 1 мин. Удалить супернатант.
13. Суспендировать преципитат в 500 мкл ОБЕССОЛИВАЮЩЕГО ПРОМЫВОЧНОГО БУФЕРА (состав буфера и соответствующее значение рН приводится в таблице 4).
14. Центрифугировать суспензию со скоростью 12000 об/мин в течение 30 сек. Удалить супернатант.
15. Тщательно удалить остаточный супернатант над преципитатом, не прикасаясь к матрице и не перемешивая ее.
16. Дать возможность матрице высохнуть в течение 2 мин при температуре 37°C.
17. Добавить 100-120 мкл ЭЛЮЕНТА (см. таблицу 5) к сухому матричному преципитату. Суспендировать преципитат.
18. Оставить суспензию выстояться при комнатной температуре на 1 мин.
19. Центрифугировать суспензию со скоростью 12000 об/мин в течение 1 мин.
20. Перенести полученный супернатант в чистую стерильную пробирку для центрифугирования без ДНК-азы.
21. Выделенная ДНК готова для последующего использования.
Ниже приводятся обязательные отличительные особенности второй фазы способа:
a) Растворы и компоненты хранятся при температуре от +4 до +8°C, за исключением ДНК-СВЯЗЫВАЮЩЕЙ МАТРИЦЫ, которую необходимо хранить при комнатной температуре.
b) При приготовлении маточных растворов необходимо строго следовать стандартным лабораторным протоколам, относящимся к способам проведения ПЦР-амплификации (EN ISO 20838: 2006, GLP (GLP - Надлежащая лабораторная практика)).
c) Работа должна проводиться строго в чистом помещении (ISO 209, FS 209, BS5295, ISO 14644-1: 1999) при минимальном уровне безопасности BSL2 (уровень биологической безопасности 2). Третья фаза способа / количественный и качественный контроль Концентрация выделенной ДНК определяется по традиционной лабораторной методике с помощью УФ-спектрофотометрии при длине волны λ=260 нм. Чистота выделенной ДНК характеризуется соотношением значений оптической плотности, измеренной при длинах волн λ=260 нм и λ=280 нм.
Ниже приводятся обязательные отличительные особенности третьей фазы способа:
a) При выполнении спектрофотомерии необходимо строго следовать стандартным лабораторным протоколам, относящимся к способам проведения ПЦР-амплификации (EN ISO 20838: 2006, GLP (GLP - Надлежащая лабораторная практика)).
b) Работа должна проводиться строго в чистом помещении (ISO 209, FS 209, BS5295, ISO 14644-1: 1999) при минимальном уровне безопасности BSL2 (уровень биологической безопасности 2).
В/ Разрушение клеток образца посредством механической обработки
Реакция оптимизирована для обычных лабораторных инструментов, которые применяются в молекулярной биологии, и выделяющих устройств (например, FastPrep™).
Первая фаза способа / удаление нежизнеспособных клеток из образца.
Этапы и обязательные отличительные особенности способа аналогичны соответствующим этапам и обязательным характеристикам первой фазы разрушения образца (А).
Вторая фаза способа / лизис клеток и связывание двухцепочечной ДНК на поверхности матричного носителя
1) Перенести образец, обработанный в соответствии с этапами 1-14 первой фазы способа, в пробирку для разрушения клеток емкостью 2 мл, содержащую 0,1 г стеклянных опилок и шарик из стали Cr-Va размером 4,5" (4,5×25,4 мм).
2) Поместить пробирку для разрушения с образцом в гомогенизирующий асимметрический дезинтегратор (например, FastPrep™) и затем начать процедуру циклического разрушения.
Через 30 секунд образец можно использовать для дальнейших операций.
3) Добавить 1 мл БУФЕРА ДЛЯ ЛИЗИСА (состав буфера и соответствующее значение рН приводится в таблице 2) к механически обработанному образцу. Оставить образец при комнатной температуре на 1-2 минуты.
4) Суспензия центрифугируется со скоростью 13000 об/мин в течение 5 мин при температуре 4°C.
5) Перенести 800 мкл супернатанта в чистую стерильную пробирку для центрифугирования емкостью 2,5 мл.
6) Добавить 800 мкл ДНК-СВЯЗЫВАЮЩЕЙ МАТРИЦЫ (состав матрицы и соответствующее значение рН приводится в таблице 3). Укупорить пробирку для центрифугирования.
7) Осторожно повернуть пробирку с суспензией 10-15 раз. На этом этапе запрещается использовать вихревую мешалку при любых обстоятельствах.
8) Инкубировать пробирку в течение 2 минут при комнатной температуре, медленно поворачивая ее с широкими интервалами.
9) Центрифугировать суспензию со скоростью 12000 об/мин в течение 1 мин. Удалить супернатант.
10) Суспендировать преципитат в 700 мкг ОБЕССОЛИВАЮЩЕГО ПРОМЫВОЧНОГО БУФЕРА (состав буфера и соответствующее значение рН приводится в таблице 4).
11) Центрифугировать суспензию со скоростью 12000 об/мин в течение 1 мин. Удалить супернатант.
12) Суспендировать преципитат в 500 мкл ОБЕССОЛИВАЮЩЕГО ПРОМЫВОЧНОГО БУФЕРА (состав буфера и соответствующее значение рН приводится в таблице 4).
13) Центрифугировать суспензию со скоростью 12000 об/мин в течение 30 сек. Удалить супернатант.
14) Тщательно удалить остаточный супернатант над преципитатом, не прикасаясь к матрице и не перемешивая ее.
15) Дать возможность матрице высохнуть в течение 2 мин при температуре 37°C.
16) Добавить 100-120 мкл ЭЛЮЕНТА (см. таблицу 5) к сухому матричному преципитату. Суспендировать преципитат.
17) Оставить суспензию выстояться при комнатной температуре на 1 мин.
18) Центрифугировать суспензию со скоростью 12000 об/мин в течение 1 мин.
19) Перенести полученный супернатант в чистую стерильную пробирку для центрифугирования без ДНК-азы.
20) Выделенная ДНК готова для последующего использования.
Ниже приводятся обязательные отличительные особенности второй фазы этого способа:
a) Растворы и компоненты хранятся при температуре от +4 до +8°C, за исключением ДНК-СВЯЗЫВАЮЩЕЙ МАТРИЦЫ, которую необходимо хранить при комнатной температуре.
b) При приготовлении маточных растворов необходимо строго следовать стандартным лабораторным протоколам, относящимся к способам проведения ПЦР-амплификации (EN ISO 20838: 2006, GLP (GLP - Надлежащая лабораторная практика)).
c) Работа должна проводиться строго в чистом помещении (ISO 209, FS 209, BS5295, ISO 14644-1: 1999) при минимальном уровне безопасности BSL2 (уровень биологической безопасности 2).
Третья фаза способа / количественный и качественный контроль Концентрация выделенной ДНК определяется по традиционной лабораторной методике с помощью УФ-спектрофотометрии при длине волны λ=260 нм. Чистота выделенной ДНК характеризуется соотношением значений оптической плотности, измеренной при длинах волн λ=260 нм и λ=280 нм.
Ниже приводятся обязательные отличительные особенности третьей фазы способа:
a) При выполнении спектрофотомерии необходимо строго следовать стандартным лабораторным протоколам, относящимся к способам проведения ПЦР-амплификации (EN ISO 20838: 2006, GLP (GLP - Надлежащая лабораторная практика)).
b) Работа должна проводиться строго в чистом помещении (ISO 209, FS 209, BS5295, ISO 14644-1: 1999) при минимальном уровне безопасности BSL2 (уровень биологической безопасности 2).
С/ Разрушение образца с помощью ферментативного лизиса
Реакция оптимизирована для обычных лабораторных инструментов, которые применяются в молекулярной биологии.
Первая фаза способа / удаление нежизнеспособных клеток из образца.
Этапы и обязательные отличительные особенности способа аналогичны соответствующим этапам и обязательным отличительным особенностям первой фазы разрушения образца (А).
Вторая фаза способа / лизис клеток и связывание двухцепочечной ДНК на поверхности матричного носителя
1. Добавить фермент лизоцим к образцу, обработанному в соответствии с этапами 1-14 первой фазы способа, до конечной концентрации 100 мкг/мл, и инкубировать образец в течение 15 минут при температуре 37°C. После этого образец можно использовать для дальнейших операций.
2. Добавить 1 мл БУФЕРА ДЛЯ ЛИЗИСА к ферментативно обработанному образцу (состав буфера и соответствующее значение рН приводится в таблице 2). Оставить образец при комнатной температуре на 1-2 минуты.
3. Центрифугировать суспензию со скоростью 13000 об/мин в течение 5 мин. при температуре 4°C.
4. Перенести 800 мкл супернатанта в чистую стерильную пробирку для центрифугирования емкостью 2,5 мл.
5. Добавить 800 мкл ДНК-СВЯЗЫВАЮЩЕЙ МАТРИЦЫ (состав матрицы и соответствующее значение рН приводится в таблице 3). Укупорить пробирку для центрифугирования.
6. Осторожно повернуть пробирку с суспензией 10-15 раз. На этом этапе запрещается использовать вихревую мешалку при любых обстоятельствах.
7. Инкубировать пробирку в течение 2 минут при комнатной температуре, медленно поворачивая ее с широкими интервалами.
8. Центрифугировать суспензию со скоростью 12000 об/мин в течение 1 мин. Супернатант удаляется.
9. Суспендировать преципитат в 700 мкг ОБЕССОЛИВАЮЩЕГО ПРОМЫВОЧНОГО БУФЕРА (состав буфера и соответствующее значение рН приводится в таблице 4).
10. Центрифугировать суспензию со скоростью 12000 об/мин в течение 1 мин. Супернатант удаляется.
11. Суспендировать преципитат в 500 мкл ОБЕССОЛИВАЮЩЕГО ПРОМЫВОЧНОГО БУФЕРА (состав буфера и соответствующее значение рН приводится в таблице 4).
12. Центрифугировать суспензию со скоростью 12000 об/мин в течение 30 сек. Супернатант удаляется.
13. Тщательно удалить остаточный супернатант над преципитатом, не прикасаясь к матрице и не перемешивая ее.
14. Дать возможность матрице высохнуть в течение 2 мин при температуре 37°C.
15. К сухому матричному преципитату добавляется 100-120 мкл элюента (см. таблицу 5). Суспендировать преципитат.
16. Оставить суспензию выстояться при комнатной температуре на 1 мин.
17. Центрифугировать суспензию со скоростью 12000 об/мин в течение 1 мин.
18. Перенести полученный супернатант в чистую стерильную пробирку для центрифугирования без ДНК-азы.
19. Выделенная ДНК готова для последующего использования.
Ниже приводятся обязательные отличительные особенности второй фазы способа:
a) Растворы и компоненты следует хранятся при температуре от +4 до +8°C, за исключением ДНК-СВЯЗЫВАЮЩЕЙ МАТРИЦЫ, которую необходимо хранить при комнатной температуре.
b) При приготовлении маточных растворов необходимо строго следовать стандартным лабораторным протоколам, относящимся к способам проведения ПЦР-амплификации (EN ISO 20838: 2006, GLP (GLP - Надлежащая лабораторная практика)).
c) Работа должна проводиться строго в чистом помещении (ISO 209, FS 209, BS5295, ISO 14644-1: 1999) при минимальном уровне безопасности BSL2 (уровень биологической безопасности 2).
Третья фаза способа / количественный и качественный контроль
Концентрация выделенной ДНК определяется по традиционной лабораторной методике с помощью УФ-спектрофотометрии при длине волны λ=260 нм. Чистота выделенной ДНК характеризуется соотношением значений оптической плотности, измеренной при длинах волн λ=260 нм и λ=280 нм.
Ниже приводятся обязательные характеристики третьей фазы способа:
a) При выполнении спектрофотомерии необходимо строго следовать стандартным лабораторным протоколам, относящимся к способам проведения ПЦР-амплификации (EN ISO 20838: 2006, GLP (GLP - Надлежащая лабораторная практика)).
b) Работа должна проводиться строго в чистом помещении (ISO 209, FS 209, BS5295, ISO 14644-1: 1999) при минимальном уровне безопасности BSL2 (уровень биологической безопасности 2).
Для осуществления этапов способа, описанных в примерах реализации настоящего изобретения, предпочтительно использование следующих устройств:
ламинарный кабинет (BSL2, класс II, тип А2), вихревая мешалка, охлаждаемая лабораторная центрифуга и охлаждаемая микроцентрифуга, инкубатор для пробирок емкостью 2 мл, возможно, водяная баня. Кроме того, опционально возможно использование механического дезинтегратора, обеспечивающего автоматизированное разрушение клеток (например, FastPrep™, TeenPrep™, BigPrep™ и т.д.).
Одно из технических преимуществ настоящего изобретения представлено хорошими показателями измерительной технологии. В соответствии с этим, между двумя независимыми изолятами значение комбинированного коэффициента вариации (КВ%), характеризующее чистоту, является низким и составляет 7,5-8,5% для бактериальной колонии с численностью популяции 106 КОЕ/мл. Специфичность выделения составляет 95%, надежность воспроизведения составляет 85%. Динамический диапазон нашего способа выделения двухцепочечной ДНК составляет 104-108 КОЕ/мл (образец 100 мг) бактериальной колонии.
Еще одним техническим преимуществом настоящего изобретения является чувствительность методики: минимальное требование к количеству клеток, необходимому для выделения ДНК, т.е. абсолютное значение нижнего предела применимости описанного метода составляет 104 КОЕ/мл ±15%.
Третьим преимуществом настоящего изобретения является быстрота выполнения способа, что значит, что следующее за быстрым удалением нежизнеспособных клеток выделение полного ДНК-содержимого бактериальных возбудителей инфекции может быть выполнено в течение получаса.
Среди экономических преимуществ настоящего изобретения, в первую очередь следует повторно упомянуть быстроту выполнения соответствующего способа. После быстрого удаления нежизнеспособных клеток выделение ДНК, длящееся полчаса, обеспечивает быстрый результат в последующем применении при санитарно-гигиеническим мониторинге, например, при проведении анализа с помощью ПЦР-амплификации, что при необходимости обеспечивает своевременность вмешательства в деятельность компаний в соответствии с системой контроля качества (предприятия в сфере водоснабжения и пищевого производства). Это позволяет избежать потерь времени при необходимости оперативного проведения контрольных тестов или оперативного изъятия готовой продукции.
Предполагаемые пользователи и области применения описанного способа выделения полного ДНК-содержимого бактериальных возбудителей инфекции включают исследование питьевой воды, исследование сточных вод, санитарные бактериологические исследовательские лаборатории и лаборатории исследования водопроводной воды, станции исследования продуктов питания, исследовательские лаборатории пищевой промышленности, общие бактериологические лаборатории, промышленные лаборатории и лаборатории общественной гигиены и гигиены рабочего места.
Возможная версия коммерческого НАБОРА для практической реализации способа выделения ДНК, описанной в настоящем изобретении, представлена на фигуре 6. На фигуре показана комплексная интегральная система выделения микробиологической ДНК. НАБОР включает растворы, используемые в ходе выполнения различных этапов способа, следующих за удалением нежизнеспособных клеток (буфер для лизиса, матрица связывания, обессоливающий промывочный буфер, элюент), а также пробирки для разрушения клеток, описанные в примере реализации настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ И НАБОР ДЛЯ ДЕТЕКЦИИ МИКРООРГАНИЗМОВ | 2010 |
|
RU2527897C2 |
| СПОСОБ ВЫДЕЛЕНИЯ ДНК COCCIDIOIDES IMMITIS ДЛЯ ПРОВЕДЕНИЯ ПОЛИМЕРАЗНОЙ ЦЕПНОЙ РЕАКЦИИ | 2005 |
|
RU2295569C1 |
| Универсальный способ выделения ДНК и лизирующая смесь для его осуществления | 2022 |
|
RU2807254C1 |
| СИСТЕМА АНАЛИЗА ДЛЯ ОРТОГОНАЛЬНОГО ДОСТУПА К БИОМОЛЕКУЛАМ И ИХ МЕЧЕНИЯ В КЛЕТОЧНЫХ КОМПАРТМЕНТАХ | 2017 |
|
RU2771892C2 |
| СМЕННЫЙ МИКРОФЛЮИДНЫЙ МОДУЛЬ ДЛЯ АВТОМАТИЗИРОВАННОГО ВЫДЕЛЕНИЯ И ОЧИСТКИ НУКЛЕИНОВЫХ КИСЛОТ ИЗ БИОЛОГИЧЕСКИХ ОБРАЗЦОВ И СПОСОБ ВЫДЕЛЕНИЯ И ОЧИСТКИ НУКЛЕИНОВЫХ КИСЛОТ С ЕГО ИСПОЛЬЗОВАНИЕМ | 2008 |
|
RU2380418C1 |
| СПОСОБЫ ДИСКРЕТНОЙ АМПЛИФИКАЦИИ ПОЛНОГО ГЕНОМА | 2016 |
|
RU2736351C2 |
| АВТОМАТИЗИРОВАННЫЙ ПРИБОР ДЛЯ ВЫДЕЛЕНИЯ, ОЧИСТКИ И АНАЛИЗА НУКЛЕИНОВЫХ КИСЛОТ МЕТОДОМ ПЦР-РВ | 2020 |
|
RU2784821C2 |
| ПРОБИРКА ДЛЯ УДАРНОЙ ОБРАБОТКИ ШАРИКАМИ И СПОСОБ ЭКСТРАКЦИИ ДЕЗОРИБОНУКЛЕИНОВОЙ КИСЛОТЫ И/ИЛИ РИБОНУКЛЕИНОВОЙ КИСЛОТЫ ИЗ МИКРООРГАНИЗМОВ | 2017 |
|
RU2743140C2 |
| СПОСОБЫ АМПЛИФИКАЦИИ ДНК ДЛЯ СОХРАНЕНИЯ СТАТУСА МЕТИЛИРОВАНИЯ | 2018 |
|
RU2754038C2 |
| ПРОБИРКА ДЛЯ УДАРНОЙ ОБРАБОТКИ ШАРИКАМИ И СПОСОБ ЭКСТРАКЦИИ ДЕЗОРИБОНУКЛЕИНОВОЙ КИСЛОТЫ И/ИЛИ РИБОНУКЛЕИНОВОЙ КИСЛОТЫ ИЗ МИКРООРГАНИЗМОВ | 2018 |
|
RU2762314C2 |

Изобретение относится к области биотехнологии. Предложен способ специфического выделения полного ДНК-содержимого бактериальных возбудителей инфекции. Нежизнеспособные клетки отделяют от жизнеспособных путем центрифугирования. Используют промывочный раствор, содержащий детергент и электролит. Проводят лизис жизнеспособных клеток. ДНК лизата связывают с матрицей, промывают и элюируют. Преимуществом изобретения являются высокие показатели измерительной технологии, высокий выход ДНК бактерий, чувствительность методики, быстрота выполнения способа. 3 з.п. ф-лы, 6 ил., 3 пр.
1. Способ специфического выделения полного ДНК-содержимого бактериальных возбудителей инфекции из образцов, в ходе которого клетки лизируют, ДНК-содержимое лизата выборочно связывают, промывают и затем обессоленную линейную полимерную нуклеиновую кислоту элюируют со связывающей поверхности водой, имеющей степень чистоты для ПЦР-амплификации, отличающийся тем, что на предварительном этапе нежизнеспособные клетки отделяют от жизнеспособных путем центрифугирования, используя промывочный раствор, содержащий детергент и электролит, предпочтительно буферную смесь из Тритона X-100 и Трис-HCl, этилендиаминтетрауксусной кислоты, KCl или NaCl, моногидрата лимонной кислоты; при этом изменяют центробежную силу, влияющую на осаждение клеток, затем проводят лизис жизнеспособных клеток, после чего двухцепочечные ДНК лизата связывают с матрицей -SiO2-TiO2-, содержащей химически активированные группы -OH и додециламиновые группы, и на которой образование неспецифических вторичных связей, в частности связей РНК, белков и других макромолекул, блокируют путем добавления трегалозы или декстрана и бычьего сывороточного альбумина (БСА).
2. Способ по п. 1, отличающийся тем, что лизис жизнеспособных бактериальных клеток проводят:
а) путем механического разрушения, производимого с использованием настольного оборудования; или
б) путем ферментативного разрушения, предпочтительно расщепления лизоцимом, в специально изготовленных пробирках для разрушения клеток; или
с) путем комбинированного ферментативного и механического разрушения.
3. Способ по п. 2, отличающийся тем, что двухцепочечное ДНК-содержимое бактериального образца, содержащего не менее 104 КОЕ/мл, выделяют с использованием буфера для лизиса, содержащего окислительно-восстановительный компонент, хелатирующий агент и детергенты, предпочтительно дитиотреитол (ДТТ), этилендиаминтетрауксусную кислоту (EDTA), Тритон Х-100 и натрия додецилсульфат (НДС); при этом возможные окрашивающие агенты, получающиеся из жизнеспособных бактериальных клеток, и ингибиторы ПЦР-амплификации: порфирины, гемовые соединения, полиароматические материалы, отделяют при осаждении бактериального лизата, и свободное от деградации выборочное связывание двухцепочечных ДНК, полученных из лизата жизнеспособных бактериальных клеток, проводят на матрице -SiO2-TiO2-, содержащей химически-активированные группы -OH и додециламиновые группы и предварительно поперечно-сшитой додециламиновыми линкерами.
4. Способ по п. 3, отличающийся тем, что количество и чистоту выделенной двухцепочечной ДНК проверяют с использованием УФ-спектрофотометра при длине волны 260 нм, и количество и чистоту выделенной двухцепочечной ДНК также проверяют с использованием УФ-спектрофотометра при длине волны 280 нм, после чего проводят сравнение значений оптической плотности ОП260 и оптической плотности ОП280.
| NOCKER ANDREAS et al | |||
| Selective Removal of DNA from Dead Cells of Mixed Bacterial Communities by Use of Ethidium Monoazide // Applied and Environmental Microbiology, vol | |||
| Термосно-паровая кухня | 1921 |
|
SU72A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Электрическое сопротивление для нагревательных приборов и нагревательный элемент для этих приборов | 1922 |
|
SU1997A1 |
| Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs | |||
| dead bacteria by selective removal | |||

