Область, к которой относится изобретение
Изобретение относится к 1,4-замещенным пиперазинам, 1,4-замещенным пиперидинам и 1-замещенным 4-алкилиденилпиперидинам.
Уровень техники
Лейкотриены являются эффективными местными медиаторами, играющими основную роль в воспалительных и аллергических реакциях, включая артрит, астму, псориаз и тромбоз. Лейкотриены представляют собой линейные эйкозаноиды, получающиеся окислением арахидоновой кислоты с помощью липоксигеназ. Арахидоновая кислота окисляется с помощью 5-липоксигеназы и в результате превращается в лейкотриены А4, В4, С4, D4 или Е4. 15-Липоксигеназа ответственна за превращение арахидоновой кислоты в различные биологически активные метаболиты, включая 15-гидрокси-5,8,11,13-эйкозатетраеновую кислоту (15-НЕТЕ). Оба эти медиатора связаны с патогенезом заболеваний дыхательных путей и аллергических заболеваний, таких как астма, участвуя в бронхостенозе, секреции слизи и миграции эозинофилов. Известно, что смесь с одним или более таких лейкотриенов является сильным бронхосуживающим средством. Так, было показано, что лейкотриены играют важную роль в патологии астмы. Неопровержимое доказательство роли лейкотриенов при астме было получено с помощью нескольких основополагающих клинических испытаний, в ходе которых перорально вводимые ингибиторы 5-липоксигеназы (5-LO) (или антагонисты LTD4-рецептора) оказывали безусловное благотворное терапевтическое воздействие на больных астмой. Это воздействие включает улучшение при применении классической терапии астмы, такой как бета-агонисты и кортикостероиды. Из уровня техники хорошо известно, что некоторые гидроксимочевино- и гидроксиамидозамещенные ароматические соединения могут функционировать как 5-LO-ингибиторы. Например, в международных заявках WO 92/09567 и WO 92/09566 приводится большой ряд соединений N-гидроксимочевины и гидроксамовой кислоты в качестве ингибиторов фермента липоксигеназы.
Было установлено, что гистамин участвует в процессе воспаления в целом. Хорошо известно, что антигистаминные препараты наиболее действенны для контроля за аллергией. Кроме того, полагают, что гистамин связан с астмой. Например, известно, что и гистамин и цистеиниллейкотриены (cLT) являются ключевыми медиаторами для тонуса дыхательных путей. Клинические исследования показали, что комбинированное лечение с помощью антагониста cLT-рецептора и антигистаминного средства, вводимых больным астмой, ослабляют ранние астматические ответы (EAR) и поздние астматические ответы (LAR) в значительно большей степени, чем любой агент, действующий в одиночку (A. Roquet, et al. Am. J. Respir Crit. Care Med., 155, 1856 (1997)). Это указывает на участие гистамина в астматическом заболевании.
Хорошо известно, что некоторые [бис(замещенный и/или незамещенный арил)метил- и метилен-]-1-пиперидинильные соединения обладают антигистаминергической активностью, и многочисленные публикации показывают это. Например, Yanni et al., (патенты США 4 810 713 и 4 950 674) описывают [[бис(арил)метил- или метилен-]-1-пиперидинил]алкокси-арильные и гетероарильные соединения, применяемые для лечения аллергических явлений, включая астму и ринит. Teng et al. (патент США 5 070 087) описывают [бис(арил) метил- или метилен-]-N-[(фенокси и фенилтио)алкил]пиперидины для количественной оценки действия гистамина при аллергии.
В других работах показано применение [бис(арил)метил]пиперазин-1-ильных соединений в качестве противоастматических и противоаллергических средств, которые ингибируют выделение лейкотриенов (например, японский патент 97077754). В патенте США 4 525 358 показано применение 2-[4-(дифенилметил)-1-пиперазинил]-уксусной кислоты и ее амидов в качестве противоаллергических, спазматических и антигистаминных агентов. Японский патент 7138230 описывает применение производных 4-аралкил-1-пиперазинил-ненасыщенных карбоновых кислот в качестве противоаллергических агентов для лечения, например, астмы и ринита. В международной заявке WO 97/23466 описано получение N-диарилметилпиперазинов, применяемых в качестве анальгетиков.
Однако нигде в уровне техники не говорится, не предполагается и не рассматривается сочетание 5-LO- или 15-LO-ингибирующих функциональных свойств остатков гидрокимочевины с антигистаминергическими свойствами [бис(замещенный или незамещенный арил)метил- и метилен]-1-пиперидинильных или -1-пиперазинильных фрагментов в одном целом, дающее соединение, обладающее двойственной функцией как противогистамической, так и в качестве ингибитора 5-LO/15-LO.
Сущность изобретения
Данное изобретение охватывает новые соединения с двойным действием, обладающие как ингибирующими липоксигеназу, так и противогистаминергическими свойствами. В предпочтительном варианте изобретения каждое новое соединение по изобретению действует как ингибитор 5-LO- и/или 15-LO и как антагонист рецептора гистамина H1.
Соединения по изобретению применимы для лечения состояний, в которых участвует гистаминный или лейкотриеновый компонент. Эти состояния включают предпочтительно астму, сезонный или хронический аллергический ринит, синусит, конъюнктивит, пищевую аллергию, скомброидное отравление, псориаз, крапивницу, зуд, экзему, ревматоидный артрит, воспалительные кишечные заболевания, хроническую обструкцию легких, тромбоз и отит. Соответственно изобретение также включает фармацевтические композиции, содержащие соединения по изобретению, и способы лечения астмы и ринита с помощью фармацевтических композиций.
Соединения, входящие в данное описание, также можно использовать для изучения биологических путей, включающих как лейкотриены, так и гистамин, и, в частности, для дальнейшего более глубокого исследования роли гистамина в бронхостенозе.
Краткое описание фигур:
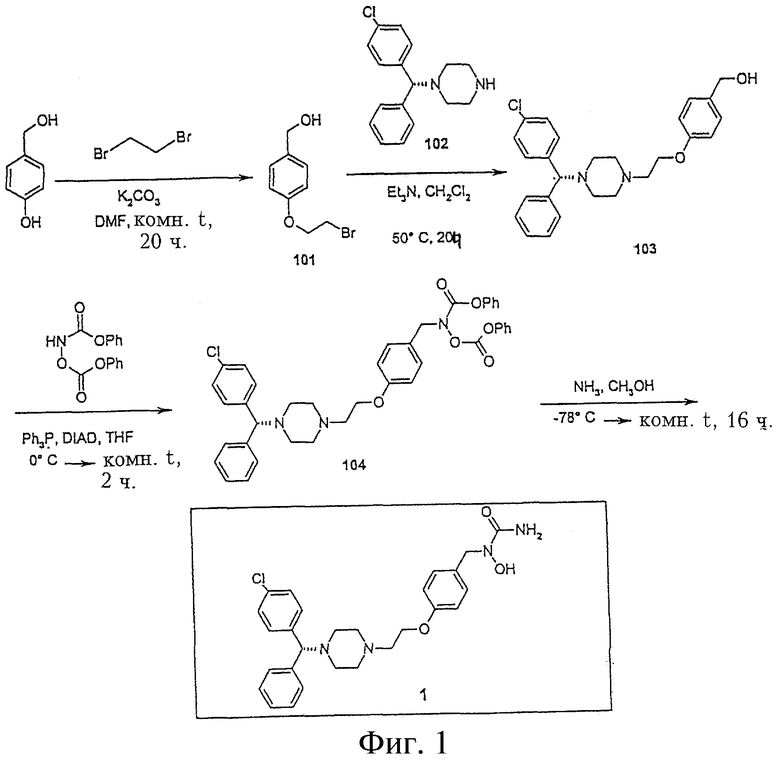
На Фигуре 1 представлен синтез соединения 1.
На Фигуре 2 представлен синтез соединения 12.
На Фигуре 3 представлен синтез соединения 17.
На Фигуре 4 представлен синтез соединений 35 и 36.
На Фигуре 5 представлен синтез соединения 37.
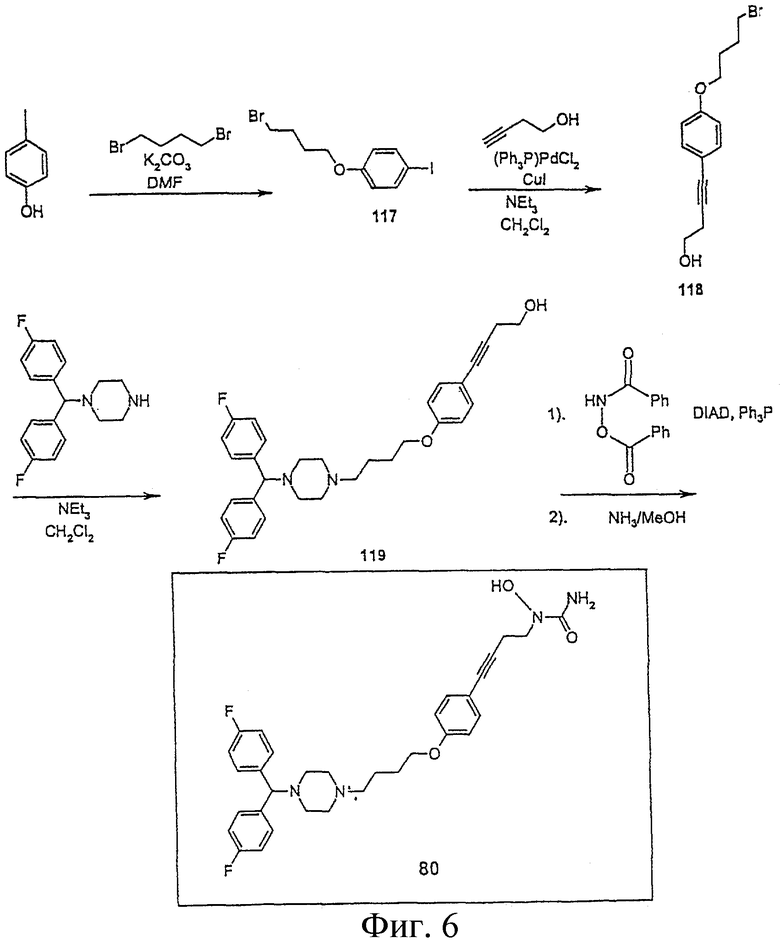
На Фигуре 6 представлен синтез соединения 80.
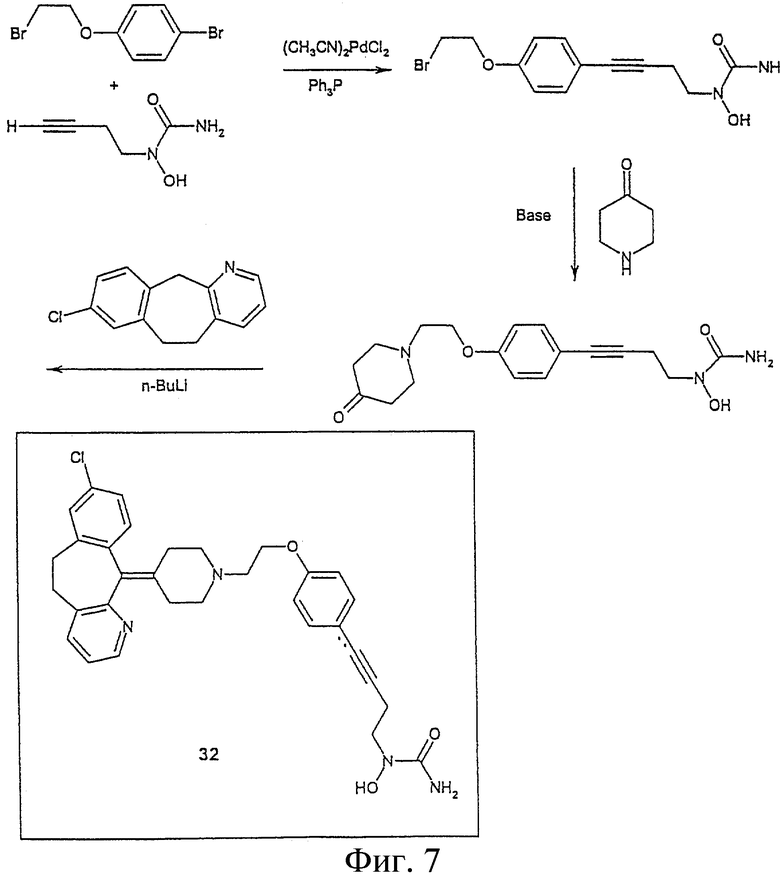
На Фигуре 7 представлен синтез соединения 32.
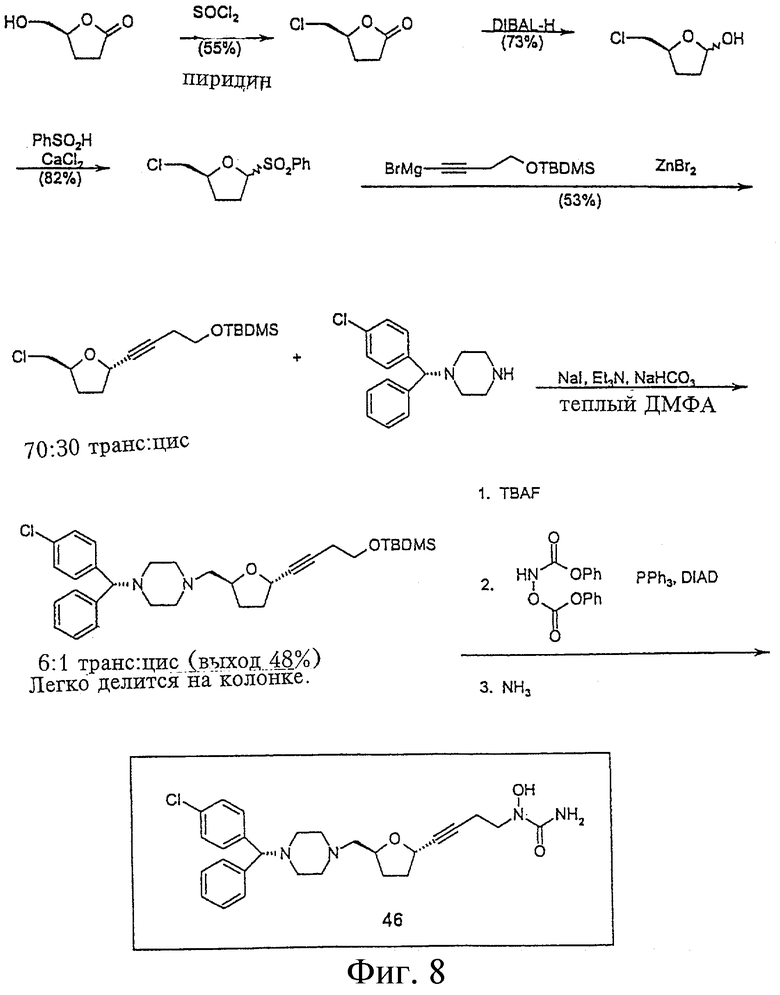
На Фигуре 8 представлен синтез соединения 46.
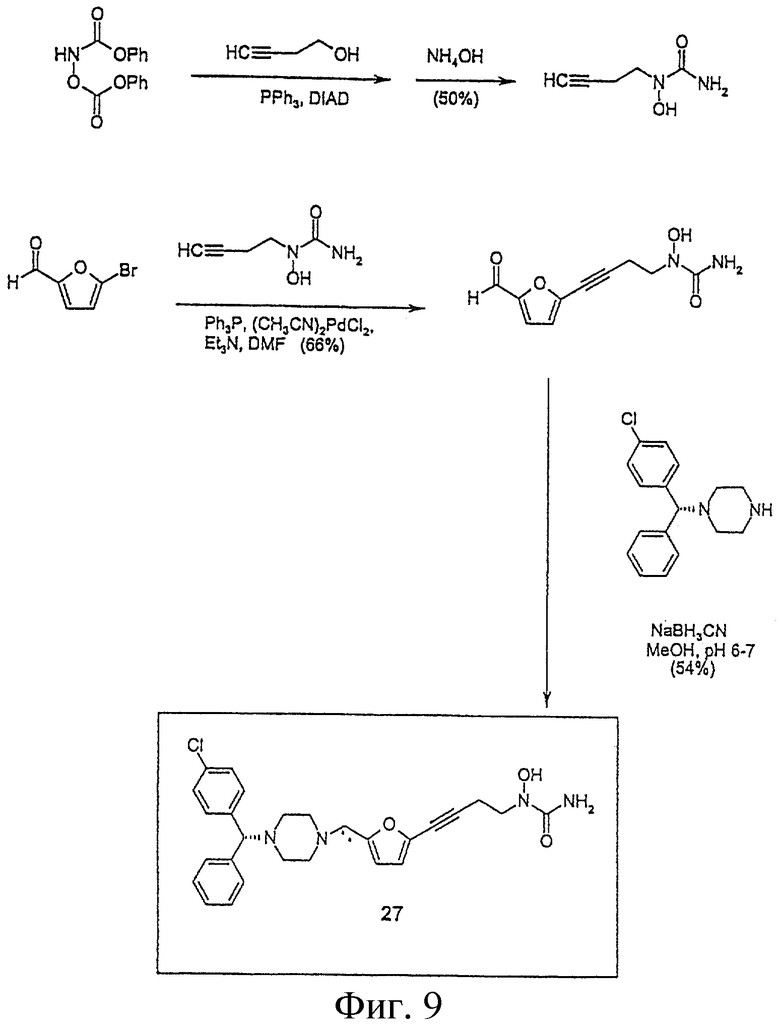
На Фигуре 9 представлен синтез соединения 27.
Подробное описание изобретения
Соединения
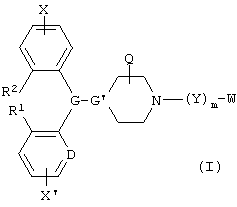
В одном аспекте данное изобретение охватывает соединения формулы I, включая геометрические изомеры, энантиомеры, диастереомеры, рацематы и их фармацевтически приемлемые соли:
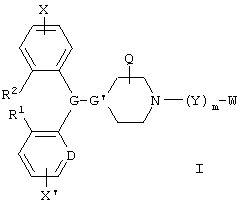
где Х и X' независимо обозначают водород, галоген, алкил, алкенил, алкинил, алкокси, трифторметил или -(Y')m'-W;
G и G' вместе образуют 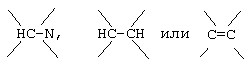
D обозначает -СН = или = N-;
R1 и R2 независимо обозначают водород или вместе обозначают -(CH2)n-, где n равен 0, 1, 2 или 3;
m и m' обозначают независимо 0 или 1;
Y и Y' обозначают -L1- или -L2-V(Z)t-L3, где t обозначает 0 или 1;
L1 обозначает алкилен, алкенилен или один из вышеназванных, где одна или более метиленовых групп замещены на -О-, -S-, -S(O)-, -S(O)2-, -N(Q)- или -N(R3)-;
L2 обозначает (а) алкилен, алкенилен, алкинилен или один из вышеназванных, где одна или более метиленовых групп замещены на -О-, -S-, -S(O)-, -S(O)2-, -N(Q')- или -N(R4)-, или (б) -L4-С(O)-N(Q')- или -L4(Q')-, или (в) простую связь;
L3 обозначает (а) алкилен, алкенилен, алкинилен или один из вышеназванных, где одна или более метиленовых групп замещены на -О-, -S-, -S(O)-, -S(O)2-, -N(Q")- или -N(R5)-, или (б) простую связь;
L4 обозначает (а) алкилен, алкенилен или алкинилен или один из вышеназванных, в котором одна или более метиленовых групп замещены на -О-, -S-, -S(O)-, -S(O)2-, -N(Q")- или -N(R5)-, или (б) простую связь;
V обозначает (а) двухвалентный арен, двухвалентный гетероарен или двухвалентный насыщенный гетероцикл, когда t обозначает 0, или (б) трехвалентный арен или трехвалентный гетероарен, когда t обозначает 1;
Q, Q' и Q" независимо обозначают водород, AC(O)OR6 или AC(O)NR6R7;
W и W' независимо обозначают -N(OM)C(O)N(R8)R9, N(R8)C(O)N(OM)R9, -N(OM)C(O)R8, -C(O)NR8R9 или -C(O)OR8, при условии, что, по меньшей мере, один из W и W' обозначает N(OM)C(O)N(R8)R9, -N(R8)C(O)N(OM)R9 или -N(OM)C(O)R8;
Z обозначает -A"N(OM')C(O)N(R10)R11, -А"N(R10)C(O)N(OM')R11, -A"N(OM')C(O)R11, -A'C(O)N(OM')R11, -A'C(O)NR10R11, -A'C(O)OR10, галоид, СН3, NR3R4, NR3C(O)R4, NO2, CN, CF3, S(O)2NR3R4, S(O)2R3, SR3 или S(O)R3;
А, А' и А" обозначают независимо простую связь, алкилен, алкенилен, алкинилен, илоалкиларил, илоарилалкил или диилоалкиларен или один из вышеназванных, в котором одна или более метиленовых групп замещены на -О-, -NH-, -S-, -S(O)- или -S(O)2- и/или один или более метилиденовых остатков замещены на =N-;
М и М' независимо обозначают водород, фармацевтически приемлемый катион или расщепляемую в процессе метаболизма группу; и
R3, R4, R5, R6, R7, R8, R9, R10 и R11 независимо обозначают водород, алкил, алкенил, алкинил, арил, арилалкил, алкиларил, алкиларилалкил или один из вышеназванных, в котором одна или более метиленовых групп замещены на -О-, -NH-, -S-, -S(O)- или -S(O)2- и/или один или более метилиденовых остатков замещен на =N-;
при условии, что в отличие от атомов кислорода, связанных с серой в -S(O)- и -S(O)2-, когда одна или более метиленовых групп замещены на -О-, -NH-, -S-, -S(O)- или -S(O)2-и когда один или более метилиденовых остатков замещены на =N-, такое замещение не происходит в двух гетероатомах, ковалентно связанных друг с другом;
и кроме того, при условии, что когда m обозначает О, W не обозначает -C(O)NR8R9 или -C(O)OR8,
и кроме того, при условии, что в заместителе -AC(O)OR6 R6 не может обозначать водород, когда А обозначает простую связь.
Предпочтительными соединениями по данному изобретению являются соединения формулы I':
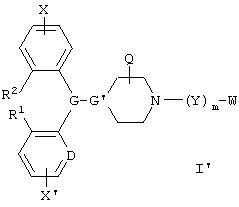
и геометрические изомеры, энантиомеры, диастереомеры и их фармацевтически приемлемые соли, где каждое из переменных имеет описанное выше обозначение, за исключением того, что: Х и X' независимо обозначают водород, галоген, алкил, алкенил, алкинил, алкокси или трифторметил; и
W обозначает -N(ON)C(O)N(R8)R9, N(R8)C(O)N(OM)R9 или -N(OM)C(O)R8.
В другом предпочтительном варианте изобретения соединения по данному изобретению представлены формулой I":
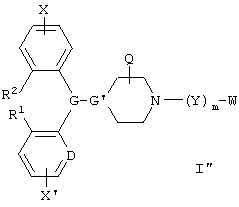
и их геометрические изомеры, энантиомеры, диастереомеры и их фармацевтически приемлемые соли, где каждое из переменных имеет вышеуказанное значение.
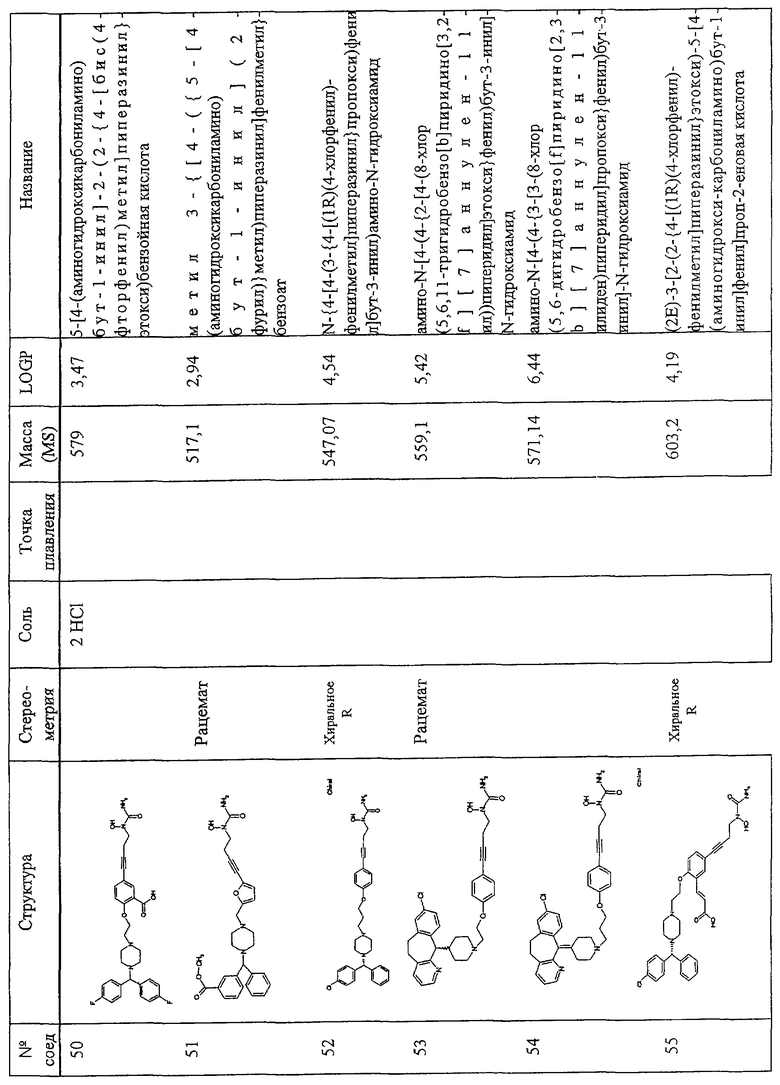
В других предпочтительных вариантах изобретения соединения формулы I представлены формулами II и III:
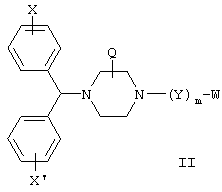
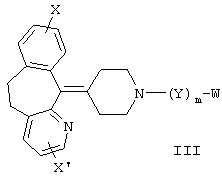
и их геометрические изомеры, энантиомеры, диастереомеры и их фармацевтически приемлемые соли, где каждое из переменных имеет приведенное выше значение.
Более предпочтительные варианты соединений формулы II и формулы III и их геометрические изомеры, энантиомеры, диастереомеры и их фармацевтически приемлемые соли представляют собой такие, в которых каждое из переменных имеет приведенное выше значение, за исключением тех, где:
1. Х обозначает -Cl, X' обозначает водород, m 1 и W обозначает -N(OH)C(O)NH2;
2. X обозначает -Cl, X' обозначает водород, m обозначает 1, Y обозначает -L1-, где L' обозначает алкинилен, илоалкокси или илоалкоксиалкил;
3. Х обозначает -Cl, X' обозначает водород, m обозначает 1, Y обозначает -L2-V(Z)t-L3-, t обозначает 0, V обозначает 1,4-фенилен или 1,3-фенилен, L2 обозначает илоалкокси и L3 обозначает алкилен, алкенилен или алкинилен;
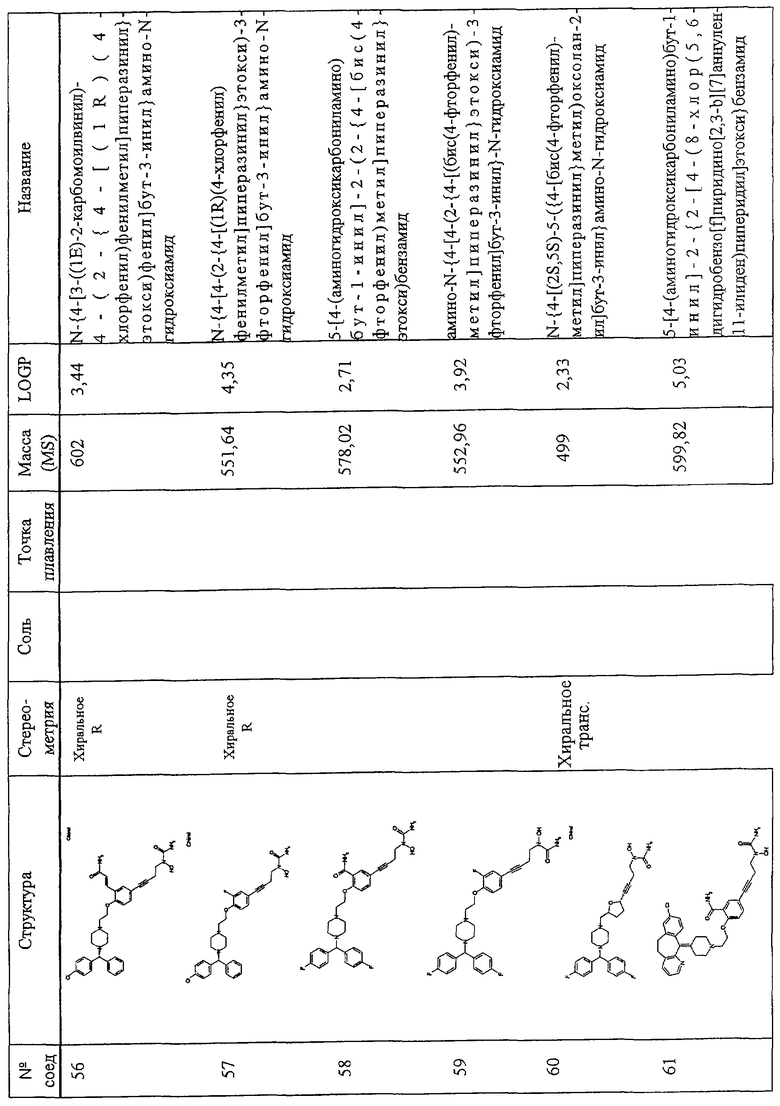
4. Х обозначает -Cl, X' обозначает водород, m обозначает 1, Y обозначает -L2-V(Z)t-L3-t обозначает 0, V обозначает 2,5-фурилен, L2 обозначает алкилен и L3 обозначает алкилен, алкенилен или алкинилен; или
5. Х обозначает -Cl, X' обозначает водород, m обозначает 1, Y обозначает -L2-V(Z)t-L3-, t обозначает 1, L2 обозначает илоалкокси, V обозначает трехвалентный гетероарен, Z обозначает -A'C(O)NR10R11 или -A'C(O)OR10 и W обозначает -N(OH)C(O)NH2;
6. X и X' обозначают F, m обозначает 1, Y обозначает -L2-V(Z)t-L3-, t обозначает 0, V обозначает 1,4-фенилен или 1,3-фенилен, L2 обозначает илоалкокси и L3 обозначает алкилен, алкенилен или алкинилен.
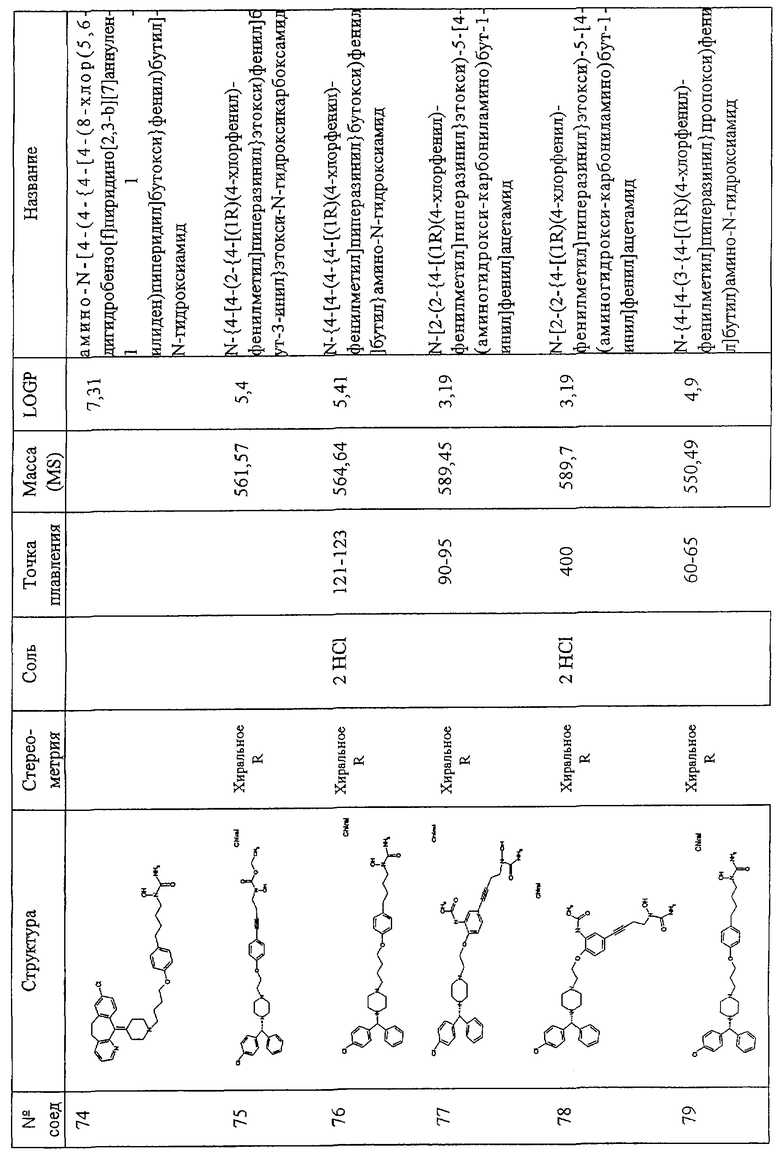
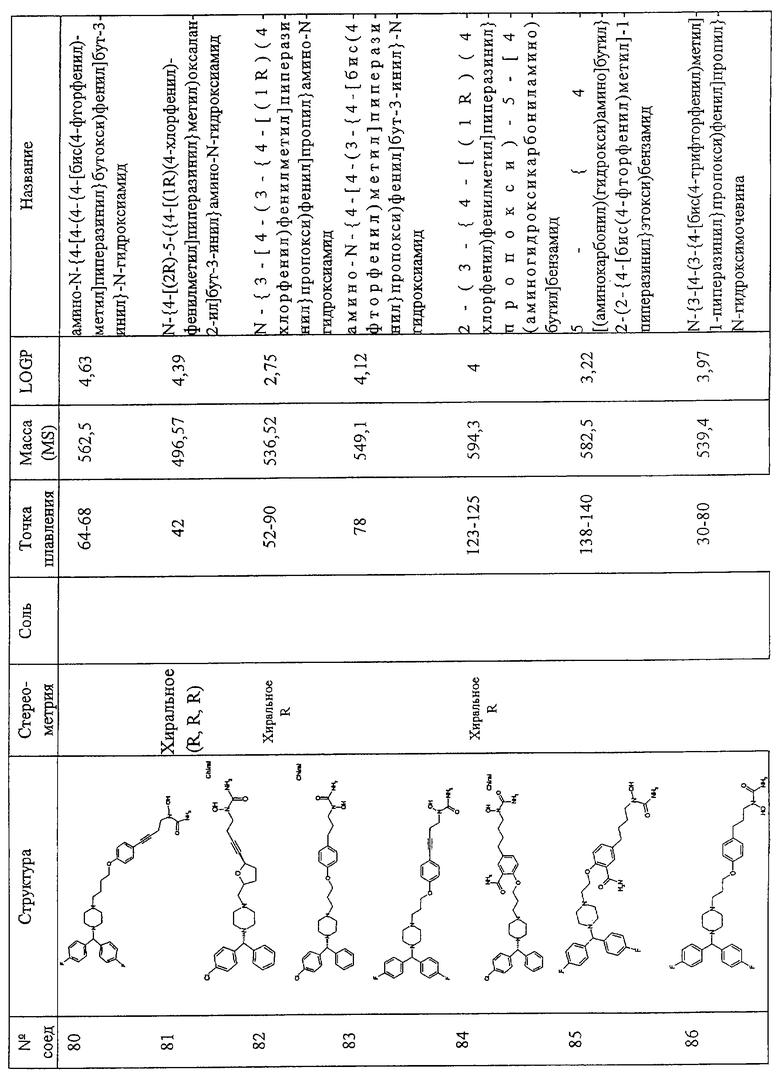
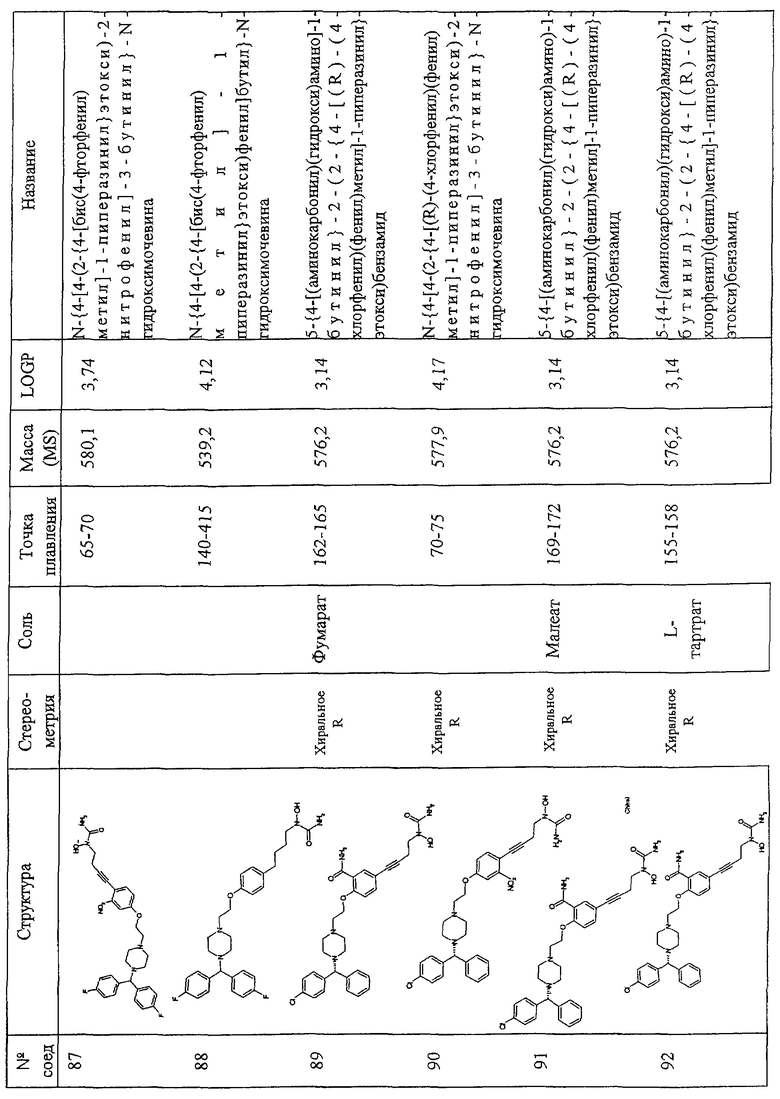
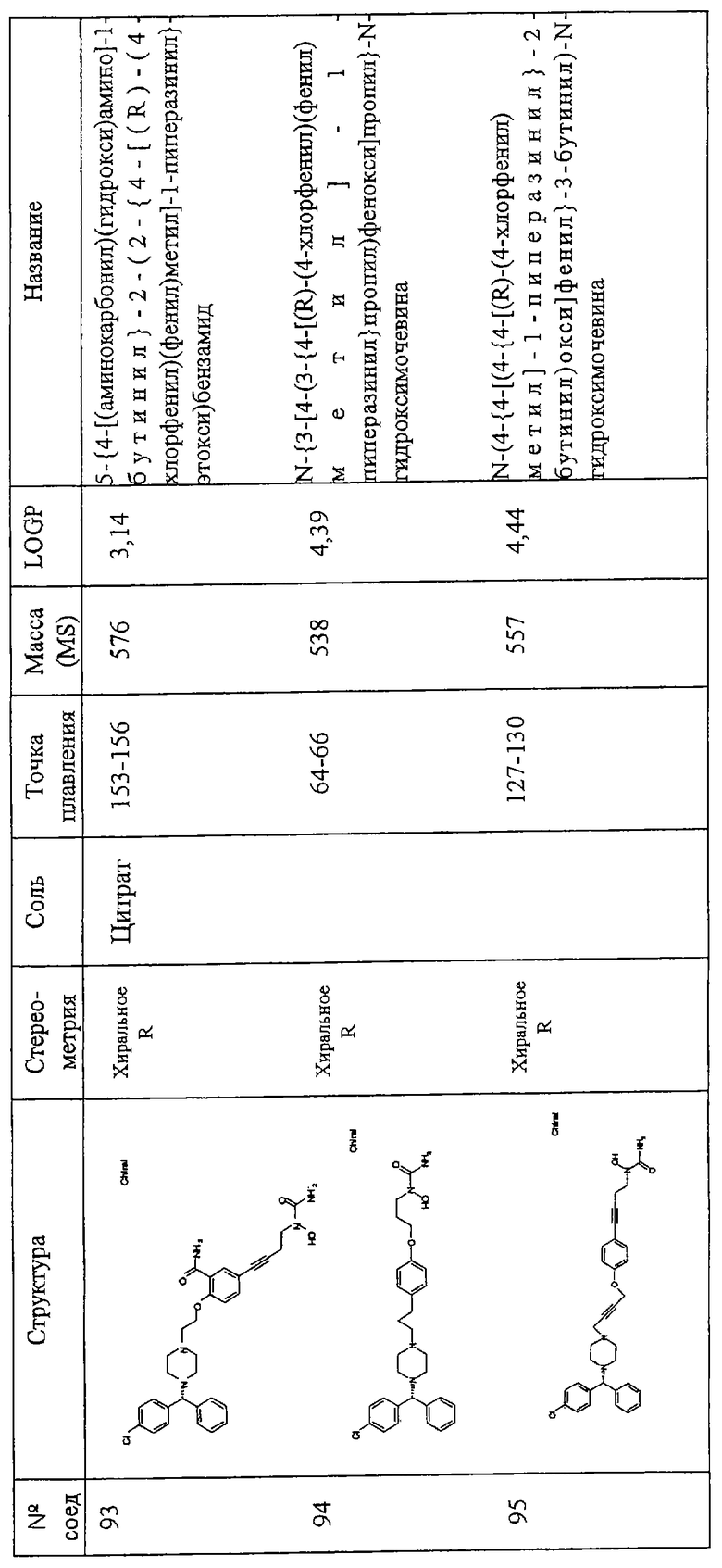
Соединения по изобретению включают соединения, представленные в Таблице 1.
Особенно предпочтительными соединениями являются соединения, перечисленные в Таблице 1
Более предпочтительны соединения N-{[4-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)фенил]метил}амино-N-гидроксиамид, N-{[3-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)фенил]метил}амино-N-гидроксиамид, амино-N-{[4-(2-{4-[бис(4-фторфенил)-метил]пиперазинил}этокси)фенил]этил}-N-гидроксиамид, N-{4-[4-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)фенил] бут-3-инил}амино-N-гидроксиамид N-{[4-(3-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}пропил)фенил]метил}амино-N-гидроксиамид, N-{4-[3-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)фенил]бут-3-инил}амино-N-гидроксиамид, N-{[3-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)фенил]метил}(метил(гидроксиамино)) карбоксиамид, N-{4-[3-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)фенил]бутил}амино-N-гидроксиамид, амино-N-[4-(4-{2-[4-(8-хлор(5,6-дигидробензо[f]пиридино[2,3-b][7]аннулен-11-лиден)пиперидил]этокси}фенил)бут-3-инил]-N-гидроксиамид, N-{3-[5-({4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}метил)(2-фурил)]-1-метилпроп-2-инил}амино-N-гидроксиамид, амино-N-{4-[5-({4-[(бис(4-фторфенил)-метил]пиперазинил}метил)(2-фурил)]бут-3-инил}-N-гидроксиамид, 2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]бензамид, метил 2-[2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси]-5-[4-(аминогидрокси-карбониламино)бут-1-инил]бензоат, 2-[2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси]-5-[4-(аминогидрокси-карбониламино)бут-1-инил]бензойная кислота, метил 2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[4-(аминогидрокси-карбониламино)бутил]бензоат, 2-(2-{4-[(1R)(4-хлорфенил)-фенилметил] пиперазинил}этокси)-5-[4-(аминогидрокси-карбониламино)бутил] бензойная кислота, 2-(2-{4-[(1R)(4-хлорфенил)-фенилметил] пиперазинил}этокси)-5-[4-(аминогидроксикарбониламино)бутил]бензамид, 2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[4-(аминогидрокси-карбониламино)бутил]бензойная кислота, 2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[4-(аминогидрокси-карбониламино)бут-1-инил]бензойная кислота, метил 5-[4-(аминогидроксикарбониламино)бут-1-инил]-2-(2-{4-[бис(4-фторфенил) метил]пиперазинил}-этокси]бензоат, N-{4-[5-({4-[(1R)(4-хлорфенил)-фенилметил] пиперазинил}метил)(2S,5S)оксолан-2-ил]бут-3-инил}амино-N-гидроксиамид, метил (2Е)-3-[2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[4-(аминогидроксикарбониламино)бутил]фенил]проп-2-еноат, метил (2Е)-3-[2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]фенил]проп-2-еноат, 5-[4-(аминогидроксикарбониламино)бут-1-инил]-2-(2-{4-[бис(4-фторфенил)метил]пиперазинил}-этокси) бензойная кислота, N-{4-[4-(3-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}пропокси)фенил]бут-3-инил)амино-N-гидроксиамид, амино-N-[4-(4-{2-[4-(8-хлор(5,6,11-тригидробензо[b]пиридино[3,2-f][7]аннулен-11-ил))пиперидил]этокси}фенил)бут-3-инил]-N-гидроксиамид, амино-N-[4-(4-{3-[3-(8-хлор(5,6-дигидробензо[f]пиридино[2,3-b][7]аннулен-11-илиден)пиперидил]пропокси}фенил)бут-3-инил]-N-гидроксиамид, (2Е)-3-[2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]фенил]проп-2-еновая кислота, N-{4-[3-((1Е)-2-карбомоилвинил)-4-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}-этокси)фенил]бут-3-инил}амино-N-гидроксиамид, N-{4-[4-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}этокси)-3-фторфенил]бут-3-инил}амино-N-гидроксиамид, 5-[4(аминогидрокси-карбониламино)бут-1-инил]-2-(2-{4-[бис(4-фторфенил)метил]пиперазинил}-этокси)бензамид, амино-N-{4-[4-(2-{4-[(бис(4-фторфенил)метил]пиперазинилэтокси)-3-фторфенил]бут-3-инил}-N-гидроксиамид, 5-[4-(аминогидрокси-карбониламино)бут-1-инил]-2-{2-[4-(8-хлор(5,6-дигидробензо[f]пиридино[2,3-b][7]аннулен-11-илиден)пиперидил]этокси}бензамид, 2-(3-{4-[(1R)(4-хлорфенил)-нилметил]пиперазинил}пропокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]бензамид, 2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил} этокси)-5-[5-(аминогидрокси-карбониламино)пент-1-инил]бензамид, 2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[4-(аминогидрокси-карбониламино)бут-1-инил]бензамид, N-{4-[4-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-3-(трифторметил) фенил]бут-3-инил}амино-N-гидроксиамид, N-{4-[4-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-3-(трифторметил)фенил]бут-3-инил}амино-N-гидроксиамид, N-{4-[4-(2-хлорфенил)-фенилметил]пиперазинил}этокси)-3-цианофенил]бут-3-инил}амино-N-гидроксиамид, N-{4-[4-(4-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}бутокси)фенил] бут-3-инил}амино-N-гидроксиамид, 2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[3-(аминогидроксикарбониламино) проп-1-инил]бензамид, N-{4-[4-(4-{4-[(1R)(4-хлорфенил)-фенилметил] пиперазинил}бутокси)фенил]бутил}амино-N-гидроксиамид, N-{4-[4-(2-{4-[(1S)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-3-(трифторметил)фенил]бутил}амино-N-гидроксиамид, N-{4-[4-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-3-(трифторметил) фенил]бутил}амино-N-гидроксиамид, aминo-N-[4-(4-{4-(8-xлop(5,6-дигидpoбeнзo,[f]пиpидинo[2,3-b][7]аннулен-11-илиден))пиперидил] бутокси}фенил)бут-3-инил]-N-гидроксиамид, амино-N-[4-(4-{4-[4-(8-хлор(5,6-дигидробензо[f]пиридино[2,3-b][7]аннулен-11-илиден)пиперидил]бутокси}фенил)бутил]-N-гидроксиамид, N-{4-[4-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)фенил]бут-3-инил} этокси-N-гидроксикарбоксамид, N-{4-[4-(4-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}бутокси)фенил]бутил}амино-N-гидроксиамид, N-[2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]фенил]ацетамид, N-[2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]фенил]ацетамид, N-{4-[4-(3-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}пропокси)фенил]бутил) амино-N-гидроксиамид, амино-N-{4-[4-(4-{4-[бис(4-фторфенил)-метил]пиперазинил}бутокси)фенил]бут-3-инил}-N-гидроксиамид, N-{3-[4-(3-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}пропокси)фенил] пропил}амино-N-гидроксиамид, амино-N-{4-[4-(3-{4-[бис(4-фторфенил) метил]пиперазинил}пропокси)фенил]бут-3-инил}-N-гидроксиамид, 2-(3-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}-пропокси)-5-[4-(аминогидроксикарбониламино)-бутил]бензамид, 5-{4-[(аминокарбонил) (гидрокси)амино]бутил}-2-(2-{4-[бис(4-фторфенил)метил]-1-пиперазинил}этокси)бензамид, N-{3-[4-(3-{4-[бис(4-трифторфенил) метил]-1-пиперазинил}пропокси)фенил]пропил}-N-гидроксимочевина, N-{4-[4-(2-{4-[бис(4-фторфенил)метил]-1-пиперазинил}этокси)-2-нитрофенил]-3-бутинил}-N-гидроксимочевина, -{4-[4-(2-{4-[бис(4-фторфенил)метил]-1-пиперазинил}этокси)фенил]бутил}-N-гидроксимочевина, 5-{4-[(аминокарбонил)(гидрокси)амино]-1-бутинил}-2-(2-{4-[(R)-(4-хлорфенил)(фенил)метил]-1-пиперазинил}-этокси)бензамид, N-{4-[4-(2-{4-[(R)-(4-хлорфенил)(фенил)метил]-1-пиперазинил}этокси)-2-нитрофенил]-3-бутинил}-N-гидроксимочевина, 5-{4-[(аминокарбонил)(гидрокси)амино)-1-бутинил}-2-(2-{4-[(R)-(4-хлорфенил)(фенил)метил]-1-пиперазинил}-этокси)бензамид, 5-{4-[(аминокарбонил)(гидрокси)амино)-1-бутинил}-2-(2-{4-[(R)-(4-хлорфенил)(фенил)метил]-1-пиперазинил}-этокси)бензамид, 5-{4-[(аминокарбонил)(гидрокси)амино]-1-бутинил}-2-(2-{4-[(R)-(4-хлорфенил)(фенил)метил]-1-пиперазинил}-этокси) бензамид, N-{3-[4-(3-{4-[(R)-(4-хлорфенил)(фенил)метил]-1-пиперазинил}пропил) фенокси]пропил}-N-гидроксимочевина.
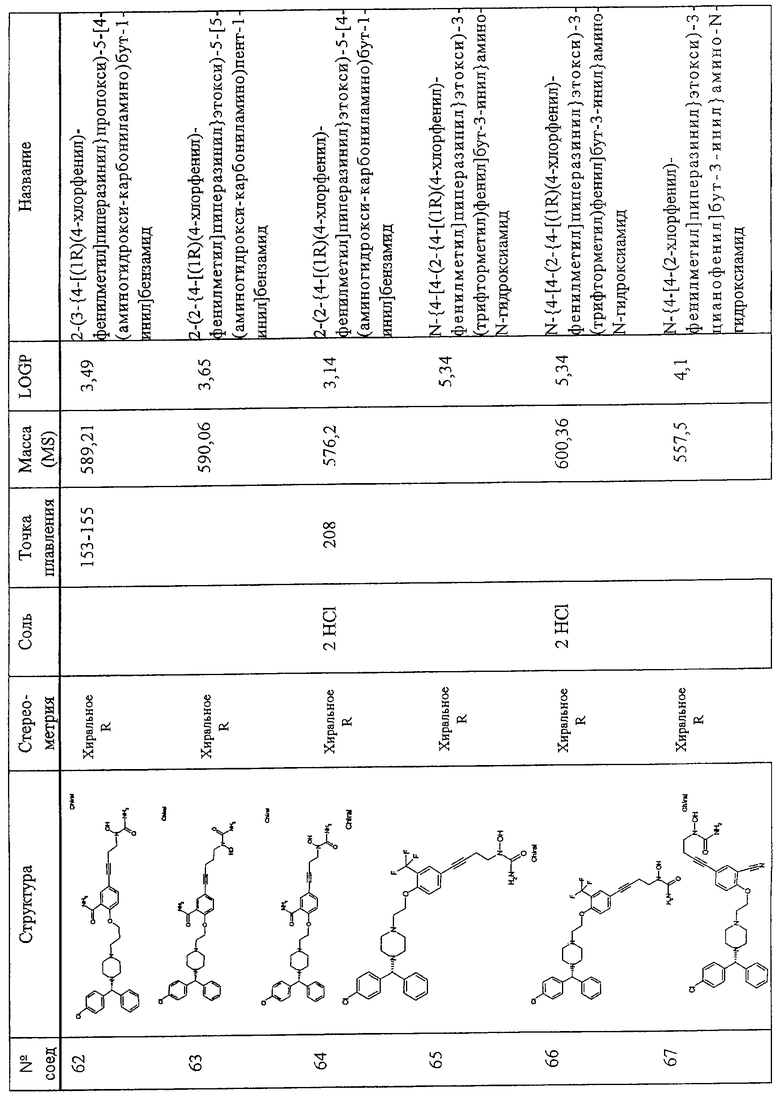
Самыми предпочтительными являются соединения N-{4-[4-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)фенил]бутил}амино-N-гидроксиамид, амино-N-[4-(4-{2-[4-(8-хлор(5,6-дигидробензо[f] пиридино[2,3-b][7]аннулен-11-илиден)пиперидил]этокси}фенил)бут-3-инил]-N-гидроксиамид, амино-N-{4-[5-({4-[(бис(4-фторфенил)-метил] пиперазинил}метил)(2-фурил)]бут-3-инил}-N-гидроксиамид, 2-(2-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}этокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]бензамид, N-{4-[5-({4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил}метил)(2S,5S)оксолан-2-ил]бут-3-инил}амино-N-гидроксиамид, N-{4-[4-(3-{4-[(1R)(4-хлорфенил)-фенилметил]пиперазинил} пропокси)фенил]бут-3-инил)амино-N-гидроксиамид и амино-N-{4-[4-(4-{4-[бис(4-фторфенил)-метил] пиперазинил}бутокси)фенил]бут-3-инил}-N-гидроксиами.
Определения
Ниже даны определения различных химических фрагментов (остатков), которые входят в состав соединений по изобретению, и они остаются неизменными по всему описанию и в Формуле изобретения, если не указано иначе.
Термин “алкил” относится к одновалентному фрагменту насыщенного линейного разветвленного циклического С1-С6-алкана и конкретно охватывает метил, этил, пропил, изопропил, бутил, изобутил, трет. бутил, пентил, циклопентил, изопентил, неопентил, гексил, изогексил, циклогексил, 3-метилпентил, 2,2-диметилбутил и 2,3-диметилбутил. Алкильная группа может при необходимости иметь заместитель, любую подходящую группу, включая без ограничения R3 или один или более фрагментов, выбираемых из группы, состоящей из галоида, гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфорной кислоты, фосфата или фосфоната, либо незащищенных, либо защищенных, если необходимо, как известно специалистам в данной области техники или как описано, например, в Greene, et al., “Protective Groups in Organic Synthesis”, John Wiley and Sons, Third Edition, 1999.
Термин “алкокси” относится к алкильному фрагменту с -O-концом со свободной валентностью, например СН3СН2-О-.
Термин “илакокси” представляет собой алкокси (определенный выше), в котором из алкильного фрагмента удален атом водорода с образованием двухвалентного радикала, например -СН2СH2-О- или -СН(СН3)-O-.
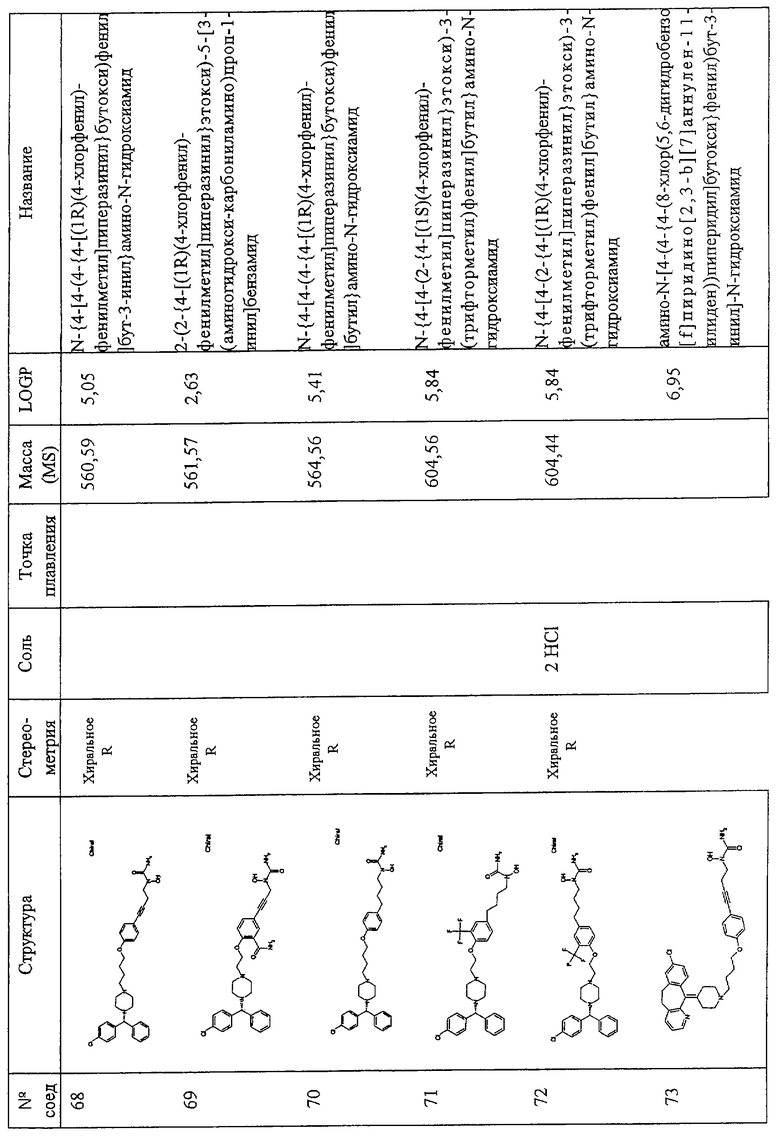
Термин “илалкоксиалкил” относится к двухвалентному фрагменту диалкилового эфира, имеющему одну свободную валентность в каждом из алкильных фрагментов, причем алкильные фрагменты являются одинаковыми или различными, например - СН2СН2СН2-О-СН2-.
Термин “алкилен” относится к алкилъному фрагменту (см. определение выше), в котором удален атом водорода с образованием двухвалентного радикала, например -СН2СН(СН3)СН2СН2-.
Термин “алкенил” относится к одновалентному фрагменту C1-C5 линейного, разветвленного или в случае С5-6 циклического углеводорода с, по меньшей мере, одной двойной связью, при необходимости замещенного, как описано выше.
Термин “алкенилен” относится к алкенильному фрагменту (см. определение выше), в котором удален атом водорода с образованием двухвалентного радикала, например -СН2СН=СНСН2-.
Термин “алкинил” относится к одновалентному фрагменту С2-С6 линейного или разветвленного углеводорода с, по меньшей мере, одной тройной связью (при необходимости замещенному, как описано выше), и конкретно включает ацетиленил, пропинил и -С≡СН2(алкил), включая -С≡С-СН2(СН3).
Термин “алкинилен” относится к алкинильному фрагменту (см. определение выше), в котором удален атом водорода, давая двухвалентный радикал, например, -С≡C-СН(СН3)-.
Термин “арил” относится к одновалентному фенилу (преимущественно), бифенилу или нафтилу. Арильная группа может при необходимости иметь заместитель, любую подходящую группу, включая без ограничения один или более фрагментов, выбираемых из группы, состоящей из галоида, гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, либо незащищенных, либо защищенных, как известно специалистам в данной области техники, например, как описано в Greene, et al., “Protective Groups in Organic Synthesis”, John Wiley and Sons, Third Edition, 1999, и, предпочтительно, галоген (включая без ограничения, фтор), алкокси (включая метокси), арилокси (включая фенокси), W, циано или R3.
Термин “арилен” или “двухвалентный арен” относится к арильному фрагменту (определение дано выше), в котором удален атом водорода с образованием двухвалентного радикала, например -С6Н4-.
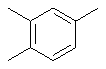
Термин “трехвалентный арен” относится к ариленовому фрагменту (определение см. выше), в котором удален атом водорода с образованием трехвалентного радикала, например
Термин “илалкиларил” относится к двухвалентному алкилзамещенному арильному фрагменту, в котором одна несвязанная валентность относится к алкильной части и одна относится к арильной части, например -СН2 -СН2-С6H4-.
Термин “иларилалкил” относится к двухвалентному арилзамещенному арильному фрагменту, в котором одну несвязанную валентность имеет алкильная часть и одну имеет арильная часть, например -C6H4-CH2-CH2-.
Термин “диилдиалкиларен” относится к двухвалентному диалкилзамещенному арену, в котором по одной несвязанной валентности имеется на каждой из серы, кислорода или азота в ароматическом кольце, которое может быть замещенным, как описано выше для арильных групп. Неограничивающими примерами являются фурилен, пиридилен, 1,2,4-тиадиазолилен, пиримидилен, тиенилен, изотиазолилен, имидазолилен, тетразолилен, пиразинилен, пиримидилен, хинолилен, изохинолилен, бензотиенилен, изобензофурилен, пиразолилен, индолилен, пуринилен, карбазолилен, бензимидазолилен и изоксазолилен.
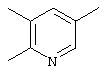
Термин “трехвалентный гетероарен” относится к гетероариленовому фрагменту (см. определение выше), в котором удален один атом водорода с образованием трехвалентного радикала, например
Термин галоид (галоген) относится к атомам хлора, фтора, йода или брома.
Когда метиленовую группу в алкильном, алкенильном или алкинильном (или их двухвалентных радикалах-аналогах) радикале замещают на О, -NH-, -S-, -S(O)-или -S(O)2-, это можно осуществлять в любом подходящем положении во фрагменте, либо в концевом положении, либо в неконцевом, например СН3СН2-О-, СН3-O-СН2-, СН3СН2NН- и СН3NНСН2-.
Несвязанные (“открытые”) валентности радикалов, представленных в данном описании, могут быть при любом одном (или более для двухвалентных радикалов) из атомов фрагмента. Например, одновалентный С3-алкильный фрагмент включает как пропил, так и изопропил. Другой пример, двухвалентный С4-алкиленовый фрагмент включает как тетраметилен (-СН2(СН2)2СН2-), так и этилэтилен (-СН(СН2СН3)СН2-).
Термин “органический” или “неорганический анион” относится к органическому или неорганическому фрагменту, который несет отрицательный заряд и может быть использован в качестве отрицательной части соли.
Термин “фармацевтически приемлемый катион” относится к органическому или неорганическому фрагменту, который несет положительный заряд и который может вводиться связанным с фармацевтическим агентом, например, как противокатион в соли. Фармацевтически приемлемые катионы известны специалистам в данной области техники и включают, не ограничиваясь, натрий, калий и (четвертичный) аммоний.
Термин “метаболически (в процессе обмена) расщепляемая группа” относится к фрагменту, который может отщепляться in vivo от молекулы, с которой она соединена, и включает, не ограничиваясь, органический анион, фармацевтически приемлемый катион, ацил (например, (алкил)С(О), включая ацетил, пропионил и бутирил), алкил, фосфат, сульфат и сульфонат, NH2С(О)- или(алкил)С(O)-.
Термин “ингибитор 5-липоксигеназы” относится к соединению, которое ингибирует фермент при 30 мкМ или ниже. Термин “ингибитор 15-липоксигеназы” относится к соединению, которое ингибирует фермент при 30 мкМ или ниже.
Применяемый в данном описании термин “фармацевтически приемлемые соли или комплексы” относится к солям или комплексам, которые сохраняют заданную биологическую активность вышеприведенных соединений и проявляют минимальный нежелательный токсикологический эффект или совсем его не проявляют. Примеры таких солей включают, но не ограничиваются этим, соли присоединения неорганических кислот (например, хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, азотной кислоты и т.п.) и соли, образованные с органическими кислотами, такими как фумаровая кислота, малеиновая кислота, уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, памовая кислота, альгиновая кислота, полиглутаминовая кислота, нафталинсульфоновая кислота и полигалактуроновая кислота. Соединения также можно вводить в виде фармацевтически приемлемых четвертичных солей, известных специалистам в данной области техники, которые конкретно включают четвертичную аммониевую соль формулы -NR+Z-, где R обозначает водород, алкил или бензил и Z обозначает противоион, включая хлорид, бромид, иодид, -O-алкил, толуолсульфонат, метилсульфонат, сульфонат, фосфат или карбоксилат (такой как фумарат, бензоат, сукцинат, ацетат, гликолят, малеат, малат, цитрат, тартрат, аскорбат, бензоат, циннамат, манделоат, бензилоат и дифенилацетат).
Термин “фармацевтически активное производное” (дериват) относится к любому соединению, которое после введения реципиенту способно превращаться прямо или опосредованно в представленные в данном описании соединения.
Схемы синтеза
Схемы синтеза, представленные на Фигурах 1-9 и в Примерах 1-7, иллюстрируют, как можно получать соединения по изобретению. Специалисты в данной области техники смогут легко модифицировать и/или приспособить эти схемы и описания для синтеза любого соединения по изобретению.
Фармацевтические композиции, методы лечения и введения
Соединения по изобретению применимы для лечения состояний, при которых предположительно существует гистаминовый и/или лейкотриеновый компонент. Эти состояния включают предпочтительно астму, сезонный или хронический аллергический ринит, синусит, конъюнктивит, пищевую аллергию, скомброидное отравление, псориаз, крапивницу, зуд, экзему, ревматоидный артрит, воспалительное заболевание кишечника, хроническую легочную обструкцию, тромбоз и отит. Соединения проявляют эту биологическую активность, действуя как антагонисты рецептора гистамина H1, ингибируя липоксигеназы, такие как 5-липоксигеназа, или проявляя двойственную (двойную) активность, то есть действуя и как антагонист рецептора гистамина H1 и как ингибитор липоксигеназы, такой как 5-липоксигеназа. Субъектов, нуждающихся в лечении опосредуемого лейкотриеном и/или гистамином состояния (предпочтительно астмы, сезонного или хронического аллергического ринита, синусита, конъюнктивита, пищевой аллергии, скомброидного отравления, псориаза, крапивницы, зуда, экземы, ревматоидного артрита, воспалительного заболевания кишечника, хронической легочной обструкции, тромбоза и отита, можно лечить, вводя больному эффективное количество одного или более вышеприведенных соединений, или их фармацевтически приемлемого производного или соли в фармацевтически приемлемом носителе, или разбавителе с целью уменьшить образование кислородных радикалов. Активные вещества можно вводить любым подходящим способом, например перорально, парентерально, внутривенно, внутрикожно, подкожно, внутримышечно или местно, в виде жидкости, крема, геля или твердом виде, в виде спрея для введения через рот или через нос или в виде аэрозоля.
Кроме того, изобретение относится к применению соединений формулы I для получения медицинского препарата для использования в терапии. В частности, изобретение относится к применению соединений формулы I для получения медицинского препарата для лечения состояний, в которых предположительно имеется гистаминный и/или лейкотриеновый компонент. Изобретение относится к применению соединения формулы I для получения медицинского препарата, пригодного для лечения астмы, сезонного или хронического аллергического ринита, синусита, конъюнктивита, пищевой аллергии, скомброидного отравления, псориаза, крапивницы, зуда, экземы, ревматоидного артрита, воспалительного заболевания кишечника, хронической легочной обструкции, тромбоза и отита и предпочтительно астмы, сезонного и хронического аллергического ринита.
Изобретение, кроме того, относится к применению соединений формулы I в качестве лекарственных средств. Изобретение относится к применению соединения формулы I в качестве лекарственного средства для лечения астмы, сезонного или хронического аллергического ринита, синусита, конъюнктивита, пищевой аллергии, скомброидного отравления, псориаза, крапивницы, зуда, экземы, ревматоидного артрита, воспалительного заболевания кишечника, хронической легочной обструкции, тромбоза и отита и предпочтительно астмы, сезонного и хронического аллергического ринита.
Активное соединение вводят в фармацевтически приемлемый носитель или разбавитель в количестве, достаточном для доставки больному терапевтически эффективного количества, не оказывая серьезного токсического действия на проходящего лечение больного. Предпочтительно доза активного соединения для всех вышеуказанных состояний составляет около 0,01-300 мг/кг, предпочтительно 0,1-100 мг/кг в день, более обычно 0,5 - около 25 мг/кг веса тела реципиента в день. Типичная доза для местного применения составляет 0,01-3 вес.% в соответствующем носителе. Интервал эффективных доз фармацевтически приемлемых производных можно рассчитать, исходя из требуемого весового количества исходного соединения. Если производное само по себе проявляет активность, эффективную дозу оценивают как указано выше, используя вес производного, или другим, известным специалистам в данной области техники способом.
Способы по изобретению заключаются во введении млекопитающему (предпочтительно человеку) в опосредуемом лейкотриеном и/или гистамином состоянии (предпочтительно больному астмой или ринитом) фармацевтической композиции по изобретению в количестве, достаточном для того, чтобы облегчить состояние. Соединение вводят соответствующим образом в виде любой подходящей стандартной лекарственной формы, включая без ограничения стандартную лекарственную форму, содержащую 1-3000 мг, предпочтительно 5-500 мг активного ингредиента на унифицированную (стандартную) лекарственную форму. Обычно подходящей является доза для перорального введения 1-500, предпочтительно 10-250, более предпочтительно 25-250 мг. Активный ингредиент следует вводить так, чтобы достичь пика концентрации активного соединения в плазме, составляющего около 0,001-30 мкМ, предпочтительно около 0,01-10 мкМ. Этого можно достичь, например, внутривенной инъекцией раствора или препарата активного ингредиента при необходимости в физиологическом растворе или в водной среде, или вводя в виде болюса активного ингредиента.
Концентрация активного соединения в лекарственном препарате зависит от скорости всасывания, распространения, инактивирования и выделения лекарственного средства, а также от других факторов, известных специалистам в данной области техники. Следует отметить, что величины доз также меняются в зависимости от тяжести состояния, которое нужно облегчить. Кроме того, следует понимать, что для каждого конкретного больного схему приема лекарственного средства нужно со временем корректировать в соответствии с индивидуальной необходимостью и профессиональной оценкой того, кто назначает или наблюдает за введением препаратов, следует также понимать, что представленные в данном описании концентрации даны только в качестве примера, но не претендуют на то, чтобы ограничить объем или практическое применение заявляемой композиции. Активный ингредиент можно вводить сразу или можно разделить на несколько меньших доз и вводить с различными интервалами.
Препараты для перорального введения включают обычно инертный разбавитель или “съедобный” носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. Для перорального введения активное соединение можно объединять с эксципиентами и применять в виде таблеток, пастилок и капсул. Фармацевтически совместимые связующие и/или адъюванты можно включать как часть композиции.
Таблетки, пилюли, капсулы, драже и т.п. могут содержать любые из следующих ингредиентов или соединений, им подобных: связующее, такое как микрокристаллическая целлюлоза, смола трагаканта или желатин; экспипиент, такой как крахмал или лактоза; диспергатор, такой как альгиновая кислота, Primogel (примогель) или крахмал; смазку, такую как стеарат магния или Sterores; вещество, способствующее проглатыванию (глидант), такое как коллоидная двуокись кремния; подсластитель, такой как сахароза или сахарин; или вкусовую добавку, такую как перечная мята, метилсалицилат или добавка со вкусом апельсина. Когда стандартная лекарственная форма представляет собой капсулу, она может содержать, помимо вышеуказанных веществ, жидкий носитель, такой как жирное масло. Кроме того, лекарственные формы могут содержать различные другие материалы, которые могут модифицировать физическую форму стандартной дозы, например покрытие из сахара, шеллака или энтеросолюбильных агентов.
Активное соединение или его фармацевтически приемлемые соль или производное можно вводить в качестве компонента эликсира, суспензии, сиропа, облатки, жевательной резинки и т.п. Сироп может содержать, помимо активных соединений, сахарозу в качестве подсластителя и некоторые консерванты, красители и корригенты.
Активное соединение или его фармацевтически приемлемые производные или соли можно также смешивать с другими активными веществами, которые не ослабляют заданного эффекта, или с веществами, которые дополняют заданный эффект, такими как адренергические агонисты, подобные псевдоэфедрину, антибиотики, противогрибковые вещества, другие противовоспалительные или противовирусные соединения.
Растворы или суспензии для парентерального, интрадермального, подкожного, внутривенного, внутримышечного или местного применения, могут содержать следующие компоненты: стерильный разбавитель (растворитель), такой как вода для инъекций, физиологический раствор, жирные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты; и агенты для корректировки тонуса, такие как хлористый натрий или декстроза. Нужный препарат можно заключать в ампулы, одноразовые шприцы или флаконы с многократной дозой из стекла или пластика.
При внутривенном введении предпочтительными носителями являются физиологический солевой раствор или забуференный фосфатом физиологический раствор (PBS).
В одном варианте изобретения активные соединения готовят с носителями, которые защищают соединения от быстрого удаления из организма, в препарате пролонгированного действия, включая имплантаты и микроинкапсулированные системы доставки. Можно использовать биоразрушаемые, биосовместимые полимеры, такие как сополимер этилена и винилацетата, полиангидриды, полигликоливая кислота, коллаген, полиортоэфиры и полимолочная кислота. Методы получения таких препаратов будут очевидны для специалистов в данной области техники. Материалы можно получать, например, от Alza Corporation (CA) и Guilford Pharmaceuticals (Baltimore, Md). Липосомные суспензии также могут быть фармацевтически приемлемыми носителями. Их можно готовить по способам, известным специалистам в данной области техники, например, как описано в патенте США 4 522 811. Например, липосомные рецептуры можно готовить, растворяя соответствующий(-ие) липид(-ы) (такие как стеароилфосфатидилэтаноламин, стеароилфосфатидилхолин, арахадоилфосфатидилхолин и холестерин) в неорганическом растворителе, который затем упаривают и при этом остается пленка высушенного липида на поверхности контейнера. Водный раствор активного соединения или его монофосфатные, дифосфатные и/или трифосфатные производные вводят затем в контейнер. Затем контейнер перемешивают (встряхивают) рукой, чтобы “оторвать” липид от боковых стенок контейнера и диспергировать липидные агенты, тем самым образуя липосомную суспензию.
Следующие примеры даны только с целью иллюстрации и не претендуют и не должны никоим образом истолковываться как ограничивающие. Специалисты в данной области техники поймут, что простые вариации и модификации следующих примеров можно делать, не отходя от сущности и объема изобретения. Примеры
Пример 1. Получение N-{[4-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил} этокси)-фенил]метил}амино-N-гидроксиамида (соединение 1, Фигура 1).
4-(2-Бромэтокси)бензиловый спирт (соединение 101).
К раствору 4-гидроксибензилового спирта (2,0 г, 16,11 ммол) в ДМФА (10 мл) добавляют карбонат калия (2,67 г, 19,3 ммол). Реакционную смесь перемешивают при комнатной температуре в течение 30 минут и затем добавляют 1,2-дибромэтан (3,03 г, 16,13 ммол). Реакционную смесь перемешивают при комнатной температуре еще в течение 20 часов и затем добавляют воду и экстрагируют этилацетатом. Органический слой промывают водой и рассолом, упаривают, получая масло, которое очищают флеш-хроматографией на колонке (силикагель, 3:1 гексан/этилацетат), получая соединение 101 (1,7 г, 45,7%); 1H ЯМР (CDCl3) d 3,64 (т, 2Н), 4,29 (т, 2Н), 4,62 (с, 2Н), 6,91 (д, 2Н), 7,30 (д, 2Н).
4-{2-[4-((1R)(4-хлорфенил)фенилметил)пиперазинил]этокси} бензиловый спирт (соединение 103).
К раствору соединения 101 (205 мг, 0,89 ммол), [(1R)(4-хлорфенил)фенилметил]-пиперазина (102) (203 мг, 0,80 ммол) в хлористом метилене (2,5 мл) добавляют триэтиламин (122,0 мг, 1,21 ммол). Реакционную смесь перемешивают при 50°С в течение 20 часов. Растворитель упаривают и остаток очищают флеш-хроматографией на колонке (силикагель, 3:1 гексан/этилацетат), получая соединение 103 (330 мг, 94,1%); 1H ЯМР (CDCl3) d 2,45 (м, 4Н), 2,62 (м, 4Н), 2,81 (т, 2Н), 4,08 (т, 2Н), 4,22 (с, 1Н), 4,51(с, 2Н), 6,87 (д, 2Н), 7,28 (м, 6Н), 7,39 (м, 5Н).
N-{[4-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил} этокси)фенил]метил} феноксикарбониламинофеноксиформиат (соединение 104)
К перемешиваемому раствору соединения 103 (330 мг, 0,76 ммол), феноксикарбониламинофеноксиформиата (251,6 мг, 0,92 ммол) и трифенилфосфина (225,2 мг, 0,86 ммол) в ТГФ (8 мл) при 0°С добавляют диизопропилазодикарбоксилат (171,1 мг, 0,86 ммол). После добавления реакционную смесь нагревают до комнатной температуры и перемешивают при комнатной температуре в течение 2 часов. Растворитель упаривают и остаток очищают флеш-хроматографией на колонке (силикагель, гексан/этилацетат 2:1), получая соединение 104 (410 мг, 78,4%): 1H ЯМР (CDCl3) d 2,47 (м, 4Н), 2,65 (м, 4Н), 2,84 (т, 2Н), 4,12 (т, 2Н), 4,23 (с, 2Н), 4,95(с, 2Н), 6,92 (д, 2Н), 7,20 (м, 5Н), 7,26 (м, 6Н), 7,40 (м, 10Н).
N-{[4-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}этокси) фенил]метил}-амино-N-гидроксиамид (соединение 1)
В сосуд с завинчивающейся крышкой помещают раствор соединения 104 (410 мг, 0,59 ммол) в метаноле (15 мл) и охлаждают до -78°С в бане сухой лед-ацетон. В этот сосуд добавляют жидкий аммиак (2-3 мл) и закрывают. Баню “ацетон-сухой лед” затем удаляют и реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь снова охлаждают в ацетоне с сухим льдом и спускают давление. Емкость открывают и растворитель упаривают. Соединение 1 отделяют на колонке флеш-хроматографией (силикагель, СН2Сl2/СН3ОН 19:1) (215 мг, 73,2%): 1H ЯМР (CDCl3) d 2,42 (м, 4Н), 2,59 (м, 4Н), 2,74 (т, 2Н), 3,98 (т, 2Н), 4,20 (с, 1Н), 4,57(с, 2Н), 5,22 (уш.с, 2Н), 6,77 (д, 2Н), 7,25 (м, 6Н), 7,36 (м, 5Н).
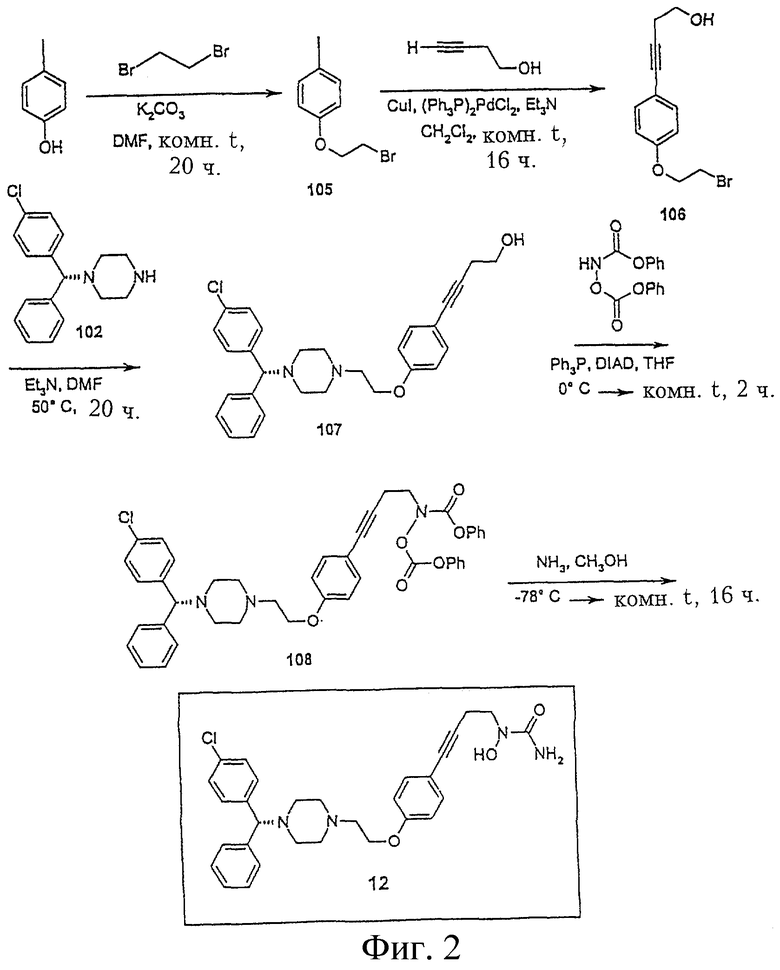
Пример 2. Получение N-{4-[4-(2-{4-[(1R)(4-хлорфенил)-фенилметил] пиперазинил}этокси)фенил]бут-3-инил}амино-N-гидроксиамида (соединение 12, Фигура 2).
4-(2-Бромэтокси)-1-иодбензол (соединение 105)
К раствору 4-иодфенола (10,0 г, 45,45 ммол) в ДМФА (50 мл) добавляют карбонат калия (12,6 г, 91,17 ммол). Реакционную смесь перемешивают при комнатной температуре 30 минут и затем добавляют 1,2-дибромэтан (17,07 г, 90,91 ммол). Реакционную смесь перемешивают при комнатной температуре еще в течение 16 часов и затем “гасят” водой и экстрагируют хлористым метиленом. Органический слой промывают водой и рассолом, упаривают, получая масло, которое очищают флеш-хроматографией на колонке (силикагель, гексан), получая соединение 105 (2,7 г, 18,2%): 1H ЯМР (CDCl3) d 3,63 (т, 2Н), 4,26 (т, 2Н), 6,70 (д, 2Н), 7,58 (д, 2Н).
4-[4-(2-Бромэтокси)фенил]бут-3-ин-1-ол (соединение 106)
К смеси соединения 105 (2,7 г, 8,26 ммол), 3-бутин-1-ола (696,3 мг, 9,94 ммол), дихлорбис(трифенилфосфин)палладия (II) (1,15 г,1,64 ммол) и иодида меди (317,1 мг, 1,67 ммол) добавляют триэтиламин (45 мл). Реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Растворитель упаривают и остаток очищают на колонке флеш-хроматографией (силикагель, гексан/этилацетат, 3:1), получая соединение 106 (1,3 г, 58,6%): 1H ЯМР (CDCl3) d 2,70 (м, 4Н), 3,65 (т, 2Н), 3,82 (м, 2Н), 4,30 (т, 2Н), 6,83 (д, 2Н), 7,37 (д, 2Н).
4-{4-[2-(4-((1R)(4-хлорфенил)фенилметил)пиперазинил)этокси] фенил}бут-3-ин-1-ол (соединение 107)
К раствору соединения 106 (1,5 г, 5,58 ммол), [(1R)(4-хлорфенил)фенилметил]-пиперазина (102) (1,6 г, 5,59 ммол) в ДМФА (15 мл) добавляют триэтиламин (871,2 мг, 8,63 ммол). Реакционную смесь перемешивают при 50°С в течение 20 часов, добавляют воду и реакционную смесь экстрагируют этилацетатом. Органический слой промывают водой и рассолом, сушат сульфатом магния, фильтруют и упаривают, полученное масло очищают на колонке флеш-хроматографией (силикагель, гексан/этилацетат, 1:1), получая соединение 107 (2,6 г, 98,1%): 1H ЯМР (CDCl3) d 2,42 (м, 4Н), 2,61 (м, 4Н), 2,68 (т, 2Н), 2,82 (т, 2Н), 3,80 (т, 2Н), 4,10 (т, 2Н), 4,21 (с, 1Н), 6,80 (д, 2Н), 7,26 (м, 5Н), 7,35 (м, 6Н).
N-{4-[4-(2-(4-((1R)(4-хлорфенил)фенилметил)пиперазинил)этокси) фенил]бут-3-инил}феноксикарбониламинофеноксиформиат (соединение 108)
К перемешиваемому раствору соединения 107 (1,5 г, 3,16 ммол), феноксикарбониламинофеноксиформиата (1,05 г, 3,85 ммол) и трифенилфосфина (937,1 мг, 3,57 ммол) в ТГФ (35 мл) при 0°С прибавляют диизопропилазодикарбоксилат (721,4 мг, 3,57 ммол). После прибавления реакционную смесь нагревают до комнатной температуры и перемешивают при комнатной температуре в течение 2 часов. Растворитель упаривают и остаток чистят флеш-хроматографией на колонке (силикагель, гексан/этилацетат, 2:1), получая соединение 108 (1,4 г, 60,6%): 1H ЯМР (CDCl3) d 2,44 (м, 4Н), 2,62 (м, 4Н), 2,82 (м, 2Н), 2,91 (т, 2Н), 4,10 (м, 4Н), 4,21 (с, 1Н), 6,80 (д, 2Н), 7,18 (м, 5Н), 7,30 (м, 8Н), 7,37 (м, 8Н).
N-{4-[4-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}этокси)фенил]бут-3-инил}амино-N-гидроксиамид (соединение 12)
В сосуд с завинчивающейся крышкой помещают раствор соединения 108 (1,4 г, 1,92 ммол) в метаноле (50 мл) и охлаждают до -78°С в бане “ацетон-сухой лед”. В этот реактор добавляют жидкий NH3 (6 мл) и закрывают. Затем оставляют баню с сухим льдом в ацетоне и реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь снова охлаждают в ацетоне с сухим льдом и спускают давление. Реактор открывают и растворитель упаривают. Соединение 12 выделяют флеш-хроматографией на колонке (силикагель, СН2Cl2/СН3ОН 19:1) (580 мг, 56,9%): 1H ЯМР (CDCl3) d 2,45 (м, 4Н), 2,65 (м, 4Н), 2,72 (т, 2Н), 2,84 (т, 2Н), 3,80 (т, 2Н), 4,10 (т, 2Н), 4,22 (с, 1Н), 5,25 (уш.с., 2Н), 6,80 (д, 2Н), 7,25 (м, 5Н), 7,36 (м, 6Н).
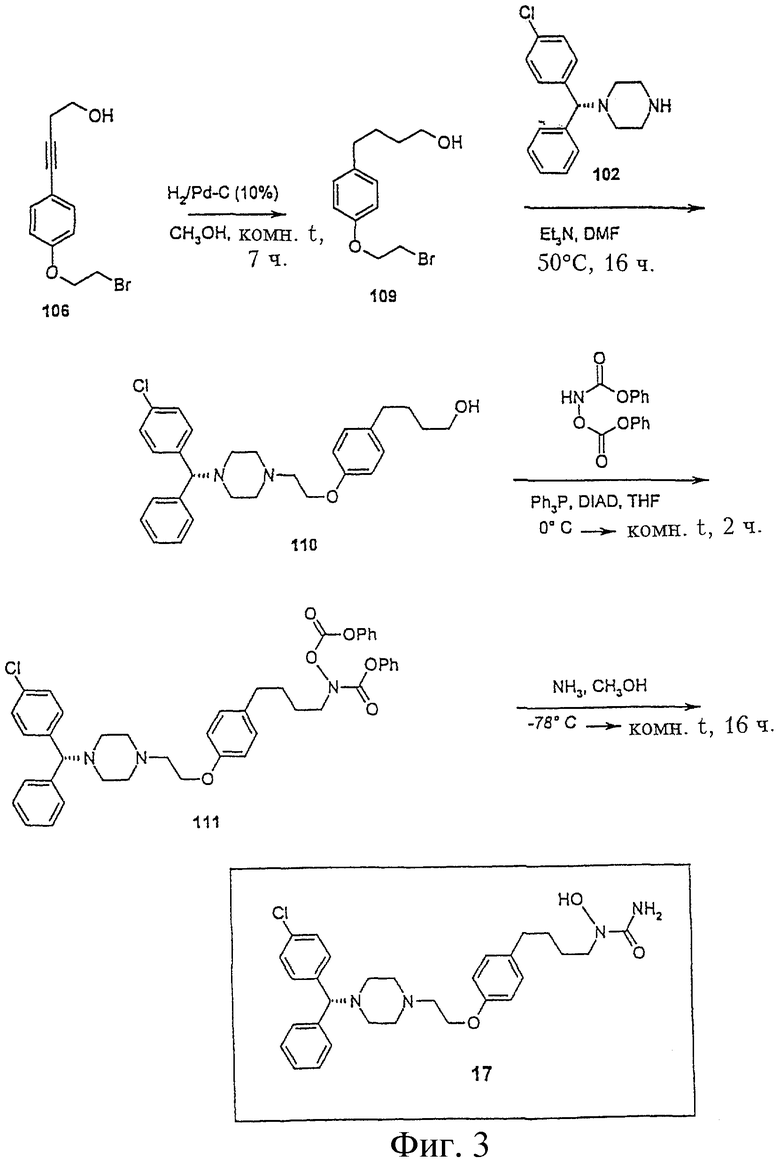
Пример 3. Получение N-{4-[4-(2-{4-[(1R)(4-хлорфенил)-фенилметил] пиперазинил}этокси)фенил]бутил}амино-N-гидроксиамида (соединение 17, Фигура 3)
4-[4-(2-Бромэтокси)фенил]бутан-1-ол (соединение 109)
Раствор соединения 106 (1,3 г, 4,83 ммол) в метаноле (15 мл) гидрируют на 10% палладии на угле (130 мг) в течение 7 часов под давлением из баллона. Катализатор отфильтровывают и фильтрат упаривают, получая соединение 109 (1,31 г, 99,2%): 1H ЯМР (CDCl3) d 1,65 (м, 4Н), 2,60 (т, 2Н), 3,66 (м, 4Н), 4,28 (м, 2Н), 6,83 (д, 2Н), 7,10 (д, 2Н).
4-{4-[2-(4-((1R)(4-хлорфенил)фенилметил)пиперазинил)этокси] фенил}бутан-1-ол (соединение 110)
К раствору соединения 109 (1,3 г, 4,76 ммол), [(1R)(4-хлорфенил)фенилметил]-пиперазина (102) (1,39 г, 4,86 ммол) в ДМФА (12 мл) добавляют триэтиламин (762,3 мг, 7,55 ммол). Реакционную смесь перемешивают при 50°С в течение 15 часов, добавляют воду и реакционную смесь экстрагируют хлористым метиленом. Органический слой промывают водой и рассолом, сушат сульфатом магния, фильтруют и упаривают, получая масло, которое очищают флеш-хроматографией на колонке (силикагель, гексан/этилацетат, 1:1), получая соединение 110 (2,42 г, 104%): 1H ЯМР (CDCl3) d 1,65 (м, 4Н), 2,45 (м, 4Н), 2,62 (м, 6Н), 2,81 (т, 2Н), 3,66 (т, 2Н), 4,08 (т, 2Н), 4,21 (с, 1Н), 6,81 (д, 2Н), 7,08 (д, 2Н), 7,25 (м, 4Н), 7,36 (м, 5Н), 8,02 (уш.с., 1Н).
N-{4-[4-(2-(4-((1R)(4-хлорфенил)фенилметил)пиперазинил)этокси) фенил]бутан-1-ол}феноксикарбониламинофеноксиформиат (соединение 111)
К перемешиваемому раствору соединения 110 (1,5 г, 3,14 ммол), феноксикарбониламинофеноксиформиата (1,05 г, 3,85 ммол) и трифенилфосфина (938,0 мг, 3,58 ммол) в ТГФ (35 мл) при 0°С прибавляют диизопропилазодикарбоксилата (724,0 мг, 3,58 ммол). После прибавления реакционную смесь нагревают до комнатной температуры и перемешивают при комнатной температуре 2 часа. Растворитель упаривают и остаток очищают флеш-хроматографией на колонке (силикагель, гексан/этилацетат, 2:1), получая соединение 111 (1,58 г, 68,7%).
N-{4-[4-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}этокси) фенил]бутил}-амино-N-гидроксиамид (соединение 17)
В реактор (сосуд) с завинчивающейся крышкой помещают раствор соединения 111 (1,58 г, 2,16 ммол) в метаноле (50 мл) и охлаждают в ацетоне с сухим льдом. В этот реактор добавляют жидкий аммиак (6 мл) и закрывают. Отставляют баню с сухим льдом в ацетоне и реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь затем снова охлаждают сухим льдом в ацетоне и убирают давление. Реактор открывают и растворитель упаривают. Соединение 17 отделяют флеш-хроматографией на колонке (силикагель, 19:1 СН2Сl2/СН3ОН) и дополнительно очищают перекристаллизацией из смеси этилацетат-гексан (550 мг, 47,4%): 1H ЯМР (CDCl3) d 1,60 (м, 4Н), 2,44 (м, 4Н), 2,52 (т, 2Н), 2,67 (м, 4Н), 2,83 (т, 2Н), 3,48 (т, 2Н), 4,08 (т, 2Н), 4,21 (с, 1Н), 6,78 (д, 2Н), 7,04 (д, 2Н), 7,25 (м, 4Н), 7,35 (м, 5Н).
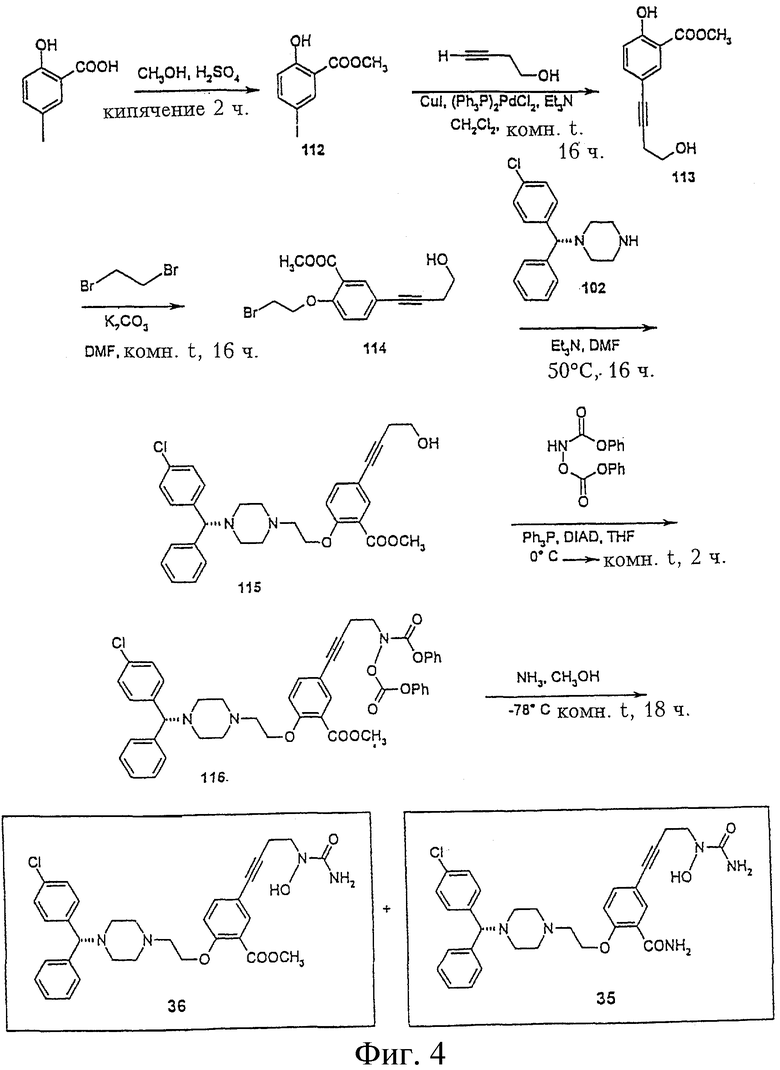
Пример 4. Получение метил-2-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}этокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]бензоата (соединение 36, Фигура 4), 2-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}этокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]бензамида (соединение 35, Фигура 4) и 2-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил} этокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]бензойной кислоты (соединение 37, Фигура 5)
4-иодфенол, метиладетат (соединение 112).
К раствору 5-иодсалициловой кислоты (5,0 г, 18,94 ммол) в метаноле (100 мл) прибавляют несколько капель серной кислоты. Реакцию перемешивают при кипении в течение 24 часов. Растворитель (метанол) упаривают до малого объема, добавляют воду и экстрагируют хлористым метиленом. Органический слой промывают 10% NaHCO2-раствором, водой и рассолом, сушат сульфатом магния, фильтруют и упаривают, получая заданное соединение (3,5 г, 66,5%): 1H ЯМР (CDCl3) d 3,96 (с, 3Н), 6,78 (д, 1Н), 7,70 (дд, 1Н), 8,12 (д, 1Н).
Метил 2-гидрокси-5-(4-гидроксибут-1-инил)бензоат (соединение 113)
К смеси соединения 112 (2,0 г, 7,19 ммол), 3-бутин-1-ола (655,2 мг, 9,35 ммол), дихлорбис(трифенилфосфин)палладия (II) (1,0 г, 1,42 ммол) и иодида меди (276,3 мг, 1,45 ммол) добавляют триэтиламин (40 мл). Реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Растворитель упаривают и остаток очищают флеш-хроматографией на колонке (силикагель, гексан/этилацетат, 2:1), получая соединение 113 (1,6 г, 101,3%): 1H ЯМР (CDCl3) d 2,68 (т, 2Н), 3,81 (м, 2Н), 3,96 (с, 3Н), 6,92 (д, 1Н), 7,50 (дд, 1Н), 7,93 (д, 1Н).
Метил 2-(2-бромэтокси)-5-(4-гидроксибут-1-инил)бензоат (соединение 114)
К раствору соединения 113 (1,6 г, 7,27 ммол) в ДМФА (8 мл) добавляют карбонат калия (1,51 г, 10,91 ммол). Реакционную смесь перемешивают при комнатной температуре 30 минут и затем добавляют 1,2-дибромэтан (5,47 г, 29,09 ммол). Реакционную смесь перемешивают при комнатной температуре еще в течение 16 часов и затем добавляют воду и экстрагируют хлористым метиленом. Органический слой промывают водой и рассолом, упаривают, получая масло, которое очищают флеш-хроматографией на колонке (силикагель, гексан/этилацетат 2:1), получая соединение 114 (710 мг, 29,8%): 1H ЯМР (CDCl3) d 2,70 (т, 2Н), 3,68 (т, 2Н), 3,82 (т, 2Н), 3,90 (с, 3Н), 4,35 (т, 2Н), 6,90 (д, 1Н), 7,50 (дд, 1Н), 7,88 (д, 1Н).
Метил 2-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}этокси)-5-(4-гидроксибут-1-инил)бензоат (соединение 115)
К раствору соединения 114 (300,0 мг, 0,92 ммол), [(1R)(4-хлорфенил)-фенилметил]пиперазина (102) (262,4 мг, 0,92 ммол) в ДМФА (2 мл) добавляют триэтиламин (139,0 мг, 1,38 ммол). Реакционную смесь перемешивают при 50°С в течение 20 часов и экстрагируют хлористым метиленом. Органический слой промывают водой и рассолом, сушат сульфатом магния, фильтруют и упаривают, получая масло, которое очищают флеш-хроматографией на колонке (силикагель, этилацетат), получают соединение 115 (510 мг, 102,4%): 1H ЯМР (CDCl3) d 2,44 (м, 4Н), 2,68 (м, 6Н), 2,90 (м, 2Н), 3,81 (т, 2Н), 3,84 (с, 3Н), 4,08 (м, 2Н), 4,21 (с, 1Н), 6,90 (д, 1Н), 7,25 (м, 4Н), 7,38 (м, 5Н), 7,49 (дд, 1Н), 7,85 (д, 1Н).
N-{4-[4-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}этокси)-3-(метоксикарбонил)фенил]бут-3-инил}фенилкарбониламинофеноксиформиат (соединение 116)
К перемешиваемому раствору соединения 115 (320,0 мг, 0,60 ммол) феноксикарбониламинофеноксиформиата (198,4 мг, 0,73 ммол) и трифенилфосфина (55,7 мг, 0,21 ммол) в ТГФ (2 мл) при 0°С добавляют диизопропилазодикарбоксилат (78,2 мг, 0,68 ммол). После добавления реакционную смесь нагревают до комнатной температуры и перемешивают при этой температуре в течение 2 часов. Растворитель упаривают и остаток очищают флеш-хроматографией на колонке (силикагель, гексан/этилацетат, 1:1), получая соединение 116 (350 мг, 73,9%): 1H ЯМР (CDCl3) d 2,42 (м, 4Н), 2,65 (м, 6Н), 2,90 (м, 2Н), 3,82 (с, 3Н), 4,15 (м, 4Н), 4,21 (с, 1Н), 6,85 (д, 1H), 7,25 (м, 8Н), 7,40 (м, 12Н), 7,82 (с, 1Н).
Метил-2-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}этокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]бензоат (соединение 36) и 2-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}этокси)-5-[4-(аминогидроксикарбонил-амино)бут-1-инил]бензамид (соединение 35).
В сосуд (реактор) с завинчивающейся крышкой помещают раствор соединения 116 (350 мг, 0,44 ммол) в метаноле (20 мл) и охлаждают до -78°С сухим льдом в ацетоне. В этот реактор наливают жидкий аммиак (3 мл) и закрывают. Охлаждающую баню затем отставляют и реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь снова охлаждают сухим льдом в ацетоне и давление снимают. Реактор открывают и растворитель упаривают. Соединение 36 выделяют флеш-хроматографией на колонке (силикагель, СН2Cl2/СН3ОН 9:1) в виде белого твердого вещества. Смесь соединений 35 и 36 дополнительно очищают флеш-хроматографией на колонке (силикагель, СН2Сl2/СН3ОН 9:1), получая дополнительно соединение 36 (всего 31 мг) и соединение 35 (содержащее около 5% соединения 36). Соединение 35 дополнительно отделяют от соединения 36 перекристаллизацией, используя в качестве растворителя смесь этилацетат-гексан (35 мг).
Соединение 36: 1H ЯМР (CDCl3) d 2,45 (м, 4Н), 2,70 (м, 6Н), 2,90 (т, 2Н), 3,75 (т, 2Н), 3,83 (с, 3Н), 4,18 (т, 2Н), 4,21 (с, 1Н), 5,34 (уш.с, 2Н), 6,85 (д, 1Н), 7,25 (м, 4Н), 7,37 (м, 5Н), 7,43 (дд, 1Н), 7,80 (с, 1Н). Соединение 35: 1H ЯМР (CDCl3) d 2,40 (м, 4Н), 2,54 (м, 4Н), 2,75 (т, 2Н), 2,80 (т, 2Н), 3,80 (т, 2Н), 4,20 (м, 3Н), 5,42 (уш.с, 2Н), 5,80 (уш.с, 1Н), 6,87 (д, 1Н), 7,25 (м, 4Н), 7,36 (м, 5Н), 7,45 (дд, 1Н), 8,14 (д, 1Н), 8,75 (уш.с, 1Н).
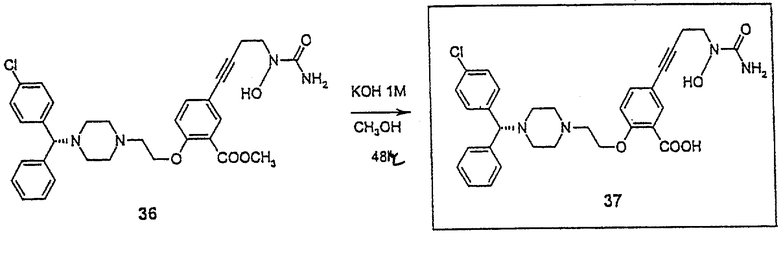
2-(2-{4-[(1R)(4-хлорфенил)фенилметил)пидеразинил}этокси)-5-[4-(аминогидроксикарбониламино)бут-1-инил]бензойная кислота (соединение 37)
В небольшую круглодонную колбу помещают соединение 36 (30 мг, 0,05 ммол). В эту колбу добавляют 1М КОН/СН3ОН (0,30 мл, 0,30 ммол). Реакционную смесь перемешивают при комнатной температуре в течение 48 часов и затем охлаждают в бане со льдом. Прибавляют 1М HCl/эфир (0,30 мл, 0,30 ммол) и смесь очищают флеш-хроматографией на колонке (силикагель, 9:1 СН2Сl2/СН3ОН), получая соединение 37 в виде белого твердого вещества (9 мг, 31,4%) 1H ЯМР (CD3OD) d 2,56 (м, 4Н), 2,66 (т, 2Н), 2,96 (м, 4Н), 3,10 (т, 2Н), 3,68 (т, 2Н), 4,32 (т, 2Н), 4,34 (с, 1H), 6,98 (д, 2Н), 7,20 (д, 1Н), 7,30 (м, 4Н), 7,44 (м, 6Н).
Пример 5. Получение амино N-{4-[4-(2-{4-(8-хлор(5,6-дигидробензо[f] пиридино[2,3-b][7]-аннулен-11-илиден))пиперидил}этокси)фенил]бут-3-инил}-N-гидроксиамида (соединение 32, Фигура 7)
4-(2-Бромэтокси)-1-иодбензол
К перемешиваемому раствору 4-иодфенола (25 г, 110 ммол) и K2CO3 (31 г, 220 ммол) в ДМФА (250 мл) в течение 1 часа добавляют 1,2-дибромэтан (5 мл, 55 ммол). Раствор греют при 50°С при перемешивании под Ar в течение ночи. Для завершения реакции прибавляют дополнительно 1,2-дибромэтан (20 мл, 220 ммол) и K2CO3 (6 г, 43 ммол) и смесь нагревают в атмосфере Ar при 50°С еще в течение 12 часов. Добавляют воду и реакционную смесь экстрагируют хлористым метиленом, сушат Na2SO4, фильтруют и растворитель упаривают в вакууме. Сырую смесь очищают хроматографией на силикагеле, элюируя 10% этилацетатом в гексане, получая искомое соединение в виде белого твердого вещества (5,5 г, 17 ммол).
4-[4-(2-Бромэтокси)фенол]бут-3-ин-1-ол
К смеси 4-(2-бромэтокси)-1-иодбензола (5,5 г, 17 ммол), 3-бутин-1-ола (1,9 мл, 25 ммол), CuJ (952 мг, 5 ммол) и дихлорбис(трифенилфосфин)палладия (II) (3,5 г, 5 ммол) в хлористом метилене (100 мл) прибавляют по каплям Et3N (3,5 мл, 25 ммол). Реакционную смесь перемешивают в течение ночи при комнатной температуре под аргоном. Растворитель упаривают в вакууме и добавляют этилацетат, чтобы растворить реакционную смесь, отфильтровывают через целит для удаления большей части Pd. Сырой продукт очищают хроматографией на силикагеле, элюируя смесью гексан/этилацетат (2:1). 4 г Заглавного соединения получают в виде светло-коричневого твердого вещества.
4-[4-(2-{4-(8-хлор-5,6-дигидробензо[f]пиридино[2,3-b][7]аннулен-11-илиден)-пиперидил}этокси) бут-3-ин-1-ол
8-хлор-11-(4-пиперидилиден)-5,6-дигидробензо[а]пиридино[2,3-d][7]аннулен (2,5 г, 7,75 ммол) и 4-[4-(2-бромэтокси)фенол]бут-3-ин-1-ол (2,5 г, 9,2 ммол) растворяют в хлористом метилене. К этому раствору добавляют Et3N (2,6 мл, 18,5 ммол) и реакционную смесь кипятят под аргоном в течение ночи. Хлористый метилен упаривают в вакууме. Непрореагировавшие исходные вещества регенерируют после очистки хроматографией, элюируя 10% МеОН в хлористом метилене. Заглавное соединение получают в виде белого твердого вещества (1,9 г, 3,76 ммол).
Фенил-{N-{4-[4-(2-{4-(8-хлор(5,6-дигидробензо[f] пиридино[2,3-b][7]аннулен-11-илиден)пиперидил}этокси)фенил]бут-3-инил}феноксикарбониламиноокси}формиат
Раствор 4-[4-(2-{4-(8-хлор-5,6-дигидробензо[f]пиридино[2,3-b][7]аннулен-11-илиден)пиперидил}этокси)бут-3-ин-1-ола (1,9 г, 3,76 ммол), трифенилфосфина (1,2 г, 4,7 ммол) и N,O-бис(феноксикарбонил) гидроксиламина (1,3 г, 4,7 ммол) в ТГФ (20 мл) охлаждают при 0°С в бане со льдом. К раствору при перемешивании по каплям добавляют диизопропилазодикарбоксилат (950 мг, 4,7 ммол). Реакционная смесь нагревается до комнатной температуры и перемешивается один час. По завершении реакции растворитель упаривают в вакууме. Продукт очищают хроматографией на силикагеле с 10% МеОН в хлористом метилене в качестве элюента. Получают 4,5 г заглавного соединения (с небольшим количеством примесей).
Амино-N-{4-[4-(2-{4-(8-хлор(5,6-дигидробензо[f]пиридино[2,3-b][7]-аннулен-11-илиден)пиперидил}этокси)фенил]бут-3-инил}-N-гидроксиамид
Фенил-{N-{4-[4-(2-{4-(8-хлор(5,6-дигидробензо[f]пиридино[2,3-b][7]аннулен-11-илиден)пиперидил}этокси)фенил]бут-3-инил}феноксикарбониламиноокси}-формиат
(4,5 г) Растворяют в МеОН, насыщенном NH3 (100 мл). Систему закрывают резиновой мембраной и перемешивают при комнатной температуре в течение ночи. Растворитель упаривают в вакууме и сырой продукт очищают хроматографией на силикагеле с 10% МеОН, насыщенном NH3, в хлористом метилене в качестве элюента, получают заглавное соединение 32 (800 мг) (или же реакцию можно проводить в ампуле под давлением).
Пример 6. Получение N-{4-[4-(3-{4-[(1R)(4-хлорфенил)-фенилметил] пиперазинил}пропокси)фенил]бут-3-инил)амино-N-гидроксиамида (соединение 52)
4-(2-Бромэтокси)-1-иодбензол
К раствору 4-иодфенола (15 г, 70 ммол) и K2CO3 (12,4 г, 90 ммол) в ДМФА (30 мл) при перемешивании добавляют в течение 1 часа 1,2-дибромпропан (7,8 мл, 90 ммол). Раствор нагревают при 50°С при перемешивании в течение ночи в атмосфере аргона. Добавляют воду (500 мл) и экстрагируют хлористым метиленом, сушат Na2SO4, фильтруют и растворитель упаривают в вакууме. Очищают хроматографией на силикагеле, элюируют 10% этилацетатом в гексане, получая заглавное соединение в виде белого твердого вещества (10 г, 29 ммол).
4-[4-(2-Бромпропокси)фенил]бут-3-ин-1-ол
К раствору 4-(2-бромпропокси)-1-иодбензола (10 г, 29 ммол), 3-бутин-1-ола (2,6 мл, 37 ммол), CuJ (980 мг, 5,2 ммол) и дихлорбис(трифенилфосфин)палладия(II) (3,6 г, 5,2 ммол) в хлористом метилене (40 мл) добавляют Et3N (6,0 мл, 44 ммол) по каплям. Реакционную смесь перемешивают в течение ночи при комнатной температуре в атмосфере аргона. Растворитель упаривают в вакууме и добавляют этилацетат для растворения соединения, фильтруют через целит для удаления большей части Pd. Сырой продукт очищают хроматографией на силикагеле, элюируют смесью гексан/этилацетат (2:1). Получают 2,6 г заглавного соединения в виде светло-коричневого твердого соединения.
4-{4-[3-(4-((1R)(4-хлорфенил)фенилметил)пиперазинил)пропокси] фенил}бут-3-ин-1-ол
[(1R)(4-хлорфенил)фенилметил]пиперазин (1,6 г, 5,6 ммол) и 4-[4-(2-бромпропокси)фенил]бут-3-ин-1-ол (2,0 г, 7,04 ммол) растворяют в хлористом метилене (10 мл). Добавляют по каплям Et3N (1 мл, 7,04 ммол), раствор кипятят в атмосфере аргона в течение ночи. Растворитель упаривают и соединение очищают хроматографией на силикагеле, элюируют этилацетатом. Получают 2,0 г заглавного соединения в виде белого вещества.
N-{4-[4-(3-(4-((1R)(4-хлорфенил)фенилметил)пиперазинил)пропокси)фенил]бут-3-инил}феноксикарбониламинофеноксиформиат
Раствор 4-{4-[3-(4-((1R)(4-хлорфенил)фенилметил)пиперазинил) пропокси]фенил}-бут-3-ин-1-ола (1,6 г, 5,6 ммол), трифенилфосфина (1,3 г, 5,1 ммол) и N,О-бис(феноксикарбонил)гидроксиламина (1,4 г, 5,1 ммол) в ТГФ (20 мл) охлаждают до 0°С в бане со льдом. К раствору при перемешивании по каплям добавляют диизопропилазодикарбоксилат (1 г, 5,1 ммол). Затем реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают один час. По завершении реакции растворитель упаривают в вакууме. Вещество дополнительно не чистят.
N-{4-[4-(2-{4-[(1R)(4-хлорфенил)фенилметил]пиперазинил}пропокси)фенил]бут-3-инил}амино-N-гидроксиамид (соединение 52)
N-{4-[4-(3-(4-((1R)(4-хлорфенил)фенилметил)пиперазинил) пропокси)фенил]бут-3-инил}феноксикарбониламинофеноксиформиат растворяют в МеОН и добавляют к 20 мл жидкого (сухой лед/ацетон) NH3 в пробирке для работы под давлением. Пробирку закрывают, оставляют нагреваться до комнатной температуры. Перемешивают в течение ночи, медленно снимают давление, снимают крышку (пробку), открывая систему на воздух, затем растворитель упаривают в вакууме. Очистка хроматографией на силикагеле с 10% МеОН, насыщенным NH3, в хлористом метилене в качестве элюента дает заглавное соединение 52 (1,05 г).
Пример 7. Получение амино-N-{4-[4-(4-{4-[бис(4-фторфенил)-метил] пиперазинил}бутокси)фенил]бут-3-инил}-N-гидроксиамида (соединение 80, Фигура 6)
1-(4-Бромбутокси)-4-иодбензол (117)
К раствору 4-иодфенола (100 г, 0,5 мол) и K2CO3 (70 г, 0,5 мол) в ДМФА (400 мл) при перемешивании добавляют 1,4-дибромэтан (100 мл, 0,84 мол) в течение 1 часа. Раствор перемешивают в течение ночи при комнатной температуре под аргоном. Добавляют Н2О (1000 мл) и экстрагируют СН2Сl2. Затем органический слой промывают 1000 мл рассола, сушат MgSO4, упаривают, получая белое твердое вещество (100 г); 1H ЯМР (CD3Cl) d 2,15-1,87 (м, 6Н), 3,50-3,20 (м, 4Н), 3,94 (т, 2Н), 6,85 (д, 2Н), 7,55 (д, 2Н).
4-[4-(4-Бромбутокси)фенол]бут-3-ин-1-ол (118)
Раствор соединения 117 (100 г, 0,3 мол), 3-бутин-1-ола (45 мл, 0,6 мол), CuJ (800 мг, 4,2 ммол) и дихлорбис(трифенилфосфин) палладия(II) (2,9 г, 4,2 ммол) в хлористом метилене (400 мл) охлаждают при 0°С (баня со льдом). При низкой температуре по каплям добавляют Et3N (84 мл, 0,6 мол). Затем смесь нагревают в до комнатной температуры и перемешивают под аргоном в течение ночи. Хлористый метилен упаривают в вакууме. Полученное полужидкое соединение растворяют в минимальном количестве СН2Сl2 и пропускают через большой слой силикагеля, элюируя 10% EtOAc в гексане, затем 50% EtOAc:50% гексана. Получают 75 г светло-коричневого твердого вещества: 1H ЯМР (CDCl3) d 2,10-1,80 (м, 4Н), 2,66 (т, 2Н), 3,25 (т, 1Н), 3,50 (т, 2Н), 3,80 (т, 2Н), 3,94 (т, 2Н), 6,85 (д, 2Н), 7,55 (д, 2Н).
Соединение (119). 4-Бис(4-фторфенил)метилпиперазин (58 г, 0,2 мол) и 118 (74 г, 0,25 мол) растворяют в СН2Сl2 (500 мл). К этому раствору добавляют NEt3 (43 мл, 0,31 мол). Смесь перемешивают в течение 48 часов при комнатной температуре под аргоном. После упаривания растворителя в вакууме полученное полужидкое вещество растворяют в минимальном количестве СН2Сl2, пропускают через большой слой силикагеля и элюируют 50% EtOAc:50% гексана, а затем EtOAc, выделяя искомое соединение. Упариванием раствора получают пену грязно-белого цвета (70 г) 90% чистоты; 1H ЯМР (CDCl3) d 1,78-1,75 (м, 6Н), 2,72-2,45 (м, 12Н), 3,78 (т, 2Н), 3,94 (т, 2Н), 4,23 (с, 1Н), 6,76 (д, 2Н), 6,97 (т, 4Н), 7,37-7,25 (м, 6Н).
Соединение 80: Раствор 119 (70 г, 0,14 мол) трифенилфосфина (45 г, 0,17 мол) и N,O-бис(феноксикарбонил)гидроксиламина (46 г, 0,17 мол) в ТГФ (500 мл) охлаждают до 0°С льдом. К раствору при перемешивании добавляют по каплям диизопропилазодикарбоксилат (34 мл, 0,17 мол). Отставляют баню со льдом, реакционная смесь нагревается до комнатной температуры, ее перемешивают один час. Окончание реакции контролируют ТСХ. Растворитель упаривают в вакууме, сырой продукт растворяют в 700 мл МеОН, насыщенном аммиаком. Смесь перемешивают в течение ночи в круглодонной колбе, закрытой резиновыми мембранами. Реакционную смесь обрабатывают, экстрагируя кислотой/основанием, и пропускают через высокий слой силикагеля (45 г), элюируя 10% МеОН в хлористом метилене. Продукт перекристаллизовывают из 500 мл кипящего EtOAc и охлаждают при комнатной температуре в течение ночи, получают 20 г чистого соединения; 1H ЯМР (CDCl3) d 1,78-1,75 (м, 6Н), 2,57- 2,45 (м, 10Н), 2,72 (т, 2Н), 3,78 (т, 2Н), 3,94(т, 2Н), 4,23 (с, 1Н), 5,34 (уш.с, 2Н), 6,76 (д, 2Н), 6,97 (т, 4Н), 7,37-7,25 (м, 6Н).
В Таблице 2 представлены данные ЯМР для предпочтительных соединений.
Пример 8. Протокол СНО-К1 HIR-связывания
Этот тест обычно проводят для того, чтобы определить способность соединения вести себя как лиганд, связывающий рецептор гистамина H1. Так как в этом тесте используют клонированные рецепторы H1 человека, он может дать хорошее приближение к тому, чего следует ожидать, когда соединение вводят человеку.
Подробности методики теста даны ниже. Клетки СНО-K1, экспрессирующие человеческий клонированный H1 рецептор, выращивают до монослоя в плашках для культур тканей. Клетки собирают, используя D-PBS-буфер (JRH Biosciences), хранят при 4°С, центрифугируют (4°С, 500 г, 10 мин). Конечный клеточный осадок гомогенизируют и снова суспендируют с помощью Tris/сахарозного буфера (20 нМ Tris, 250 мМ сахарозы, рН 7,4 при 4°С). Аликвоты препарата мембраны хранят при -70°С.
В день проведения анализа препарат мембраны размораживают и центрифугируют (ротор TLA 100.3, 4°С, 15 мин, 23 000 об/мин). Осадок снова суспендируют сначала в Tris/сахарозном буфере, а затем дополнительно разбавляют, если необходимо, аналитическим буфером А (50 мМ Na/KPO4, 2 мМ MgCl2, 0,5% (вес/объем) BSA, рН 7,5).
Для анализа связывания препарат мембраны, испытуемое соединение и 3Н-пириламин (2 нМ в конце) в буфере А с конечным 1 вес.% ДМСО инкубируют в 96-луночном планшете из полипропилена в течение 3 часов при 37°С. Неспецифическое связывание определяют в присутствии 10 мкМ пириламина. Для переноса с 96-луночного планшета на предварительно обработанный 0,1 об.% PEI фильтр-планшет gF/B используют 96-луночный харвестер (Packard). Планшет “считают” на Packard Topcounter после добавления сцинтилляционной жидкости Microscint 20 (Packard). Из этих данных рассчитывают Ki для каждого соединения, связанного с рецепторами гистамина H1. Результаты даны ниже в Таблице 3.
Пример 9. Ингибирование продуцирования LTB4 в цельной человеческой крови
В этом тесте исследуется способность соединения ингибировать продуцирование лейкотриена В4 в человеческой крови, стимулированное кальций-ионофором. Так как это продуцирование лейкотриена В4 опосредуется активацией ферментом 5-липоксигеназой, то этот тест позволяет предсказывать способность соединения ингибировать человеческий фермент 5-липоксигеназу.
Анализ проводят следующим образом. Кровь нормальных людей-добровольцев отбирают в пробирки с гепарином. 1 мл Гепаринизированной крови переносят пипеткой в полипропиленовую пробирку на 1,5 мл. К этому образцу добавляют либо различные концентрации испытуемого соединения (5 мкл), растворенного в ДМСО, либо 5 мкл ДМСО в качестве контрольного носителя. Эти образцы инкубируют на водяной бане при 37°С в течение 15 минут. 5 мкл Кальций-ионофора А 23187 (при конечной концентрации 50 мкМ) добавляют затем в каждый образец, образец встряхивают и ставят обратно в водяную баню на 30 минут. Затем образцы центрифугируют при 2500 об/мин 10 минут при 4°С. 50 мкл Супернатанта переносят в предварительно охлажденные пробирки Эппендорфа, содержащие по 950 мкл буфера для ферментного иммуноанализа (EIA). Выпускаемый промышленностью набор EIA (Cayman Chemical Co., Ann Arbor, MI, USA), применяемый для последующего измерения продуцирования LTB4 в образцах. Уровни LTB4, продуцированного в контрольном образце носителя, сравнивают затем с уровнями при добавлении испытуемого соединения. Исходя из этого рассчитывают ингибирование (в процентах) продуцирования LTB4 при каждой концентрации испытуемого соединения и определяют IC50 для ингибирования продуцирования LTB4 для каждого испытуемого соединения. Результаты даны в Таблице 3.
Пример 10. Антигистаминергическая активность in vivo
Самцы морских свинок Hartley весом 350-400 грамм получены из Charles River Labs. Ингибирование гистаминной активности определяют по методу Konzett и Rossler (Концетт и Ресслер) (Naonyn-Schmiedebergs Arch. Exp. Path. Pharmacol. 195, 71-74 (1940)). Усыпленных морских свинок подвергают искусственной вентиляции. Определяют эндотрахеальное давление. Бронхостеноз индуцируется последовательными внутривенными инъекциями гистамина. Испытуемые соединения вводят перорально в виде 1% метоцеллюлозной суспензии в несколько (в группу) временных точек перед введением гистамина.
Результаты (Таблица 4) показывают - в процентах - ингибирование гистамининдуцированного бронхостеноза выбранными соединениями в несколько временных точек после перорального приема. Считается значительным 50%-ное ингибирование или более.
Из таблицы 4 видно, что соединения по данному изобретению обладают высокой активностью в том, что касается их способности ингибировать гистамининдуцированный бронхостеноз. Кроме того, некоторые из соединений, вводимых в виде однократной дозы, обладают антигистаминергической активностью пролонгированного действия. Например, соединение 27 при дозе 2 мг/кг все еще ингибирует гистамининдуцированный бронхостеноз на 91% через 6 часов после перорального приема.
Эти эксперименты также указывают, что испытуемые соединения являются биодоступными при пероральном приеме.
Пример 11. 5-Липоксигеназная ингибирующая активность in vivo
Самцы морских свинок Hartley, весом 350-400 грамм, получены из Charles River Labs. Соединения готовят в объеме [1-2 мг/мл] в 1% метилцеллюлозе для перорального дозирования. Животных делят на группы по 5 свинок (5). Каждый тест включает контрольную группу, которой дают в виде дозы носитель. Каждой группе животных дают либо дозу носителя, либо соединения перорально через зонд. Животным дают отдыхать один, два, три или шесть часов после введения дозы. Через соответствующие промежутки времени животных подвергают анестезии с помощью Urethane, 1,5 г/кг, вб. Кровь берут “гепаринизированным” шприцом с помощью сердечной пункции.
Аликвоты крови (5 мл) помещают в различным образом помеченные пробирки Эппендорфа. Каждый образец нагружают 5 мкл [15 мМ] арахидоновой кислоты и помещают на 5 минут в водяную баню с температурой 37°С. Через 5 минут кровь стимулируют с помощью 5 мкл [5 мМ] А23187 (кальций-ионофора) и оставляют в водяной бане еще на 30 минут. Через тридцать минут образцы крови убирают из водяной бани и центрифугируют при 14 000 об/мин в течение 2 минут. Плазму разбавляют EIA-буфером и осуществляют EIA согласно инструкциям производителя (Cayman Chemical Co., Ann Arbor, MI, USA).
Результаты (Таблица 5) показывают ингибирование - в процентах - 5-липоксигеназы выбранными соединениями в различные временные точки после перорального введения доз. Ингибирование 50% или более считается значительным.
Из таблицы 5 видно, что соединения по данному изобретению обладают высокой активностью в том, что касается их способности ингибировать фермент 5-липоксигеназу. Кроме того, некоторые из этих соединений, вводимые в виде однократной дозы, обладают 5-липоксигеназной ингибирующей активностью пролонгированного действия. Например, соединение 87 при дозе 2 мг/кг все еще ингибирует 5-липоксигеназую активность на 94% через 6 часов после перорального введения дозы.
Пример 12. Ингибирование 15-липоксигеназы
С помощью данного теста исследуется способность соединения ингибировать продуцирование 15-гидрокси-5,8,11,13-эйкозатетраеновой кислоты (15-НЕТЕ) за счет действия 15-липоксигеназы на арахидоновую кислоту. 15-Липоксигеназу очищают от кроличьих перитонеальных полиморфно-ядерных лейкоцитов. Фермент отвечает за превращение арахидоновой кислоты (путем окисления при атоме углерода 15 арахидоновой кислоты) в 15-гидроперокси-5,8,11,13-эйкозатетраеновую кислоту (15-НЕТЕ), которая затем восстанавливается в 15-гидрокси-5,8,11,13-эйкозатетраеновую кислоту (15-НЕТЕ).
Ниже подробно описана методика. Арахидоновую кислоту совместно инкубируют с 15-НЕТЕ в течение 5 минут при 37°С в присутствии или в отсутствие различных концентраций испытуемого соединения (10-8-10-5 М). Продуцирование 15-НЕТЕ в каждом образце затем определяют радиоиммуноанализом. Уровни 15-НЕТЕ, продуцированной в контрольном образце с носителем, сравнивают затем с уровнями в образцах с добавленным испытуемым соединением. Исходя из этого определяют ингибирование продуцирования 15-НЕТЕ для каждого испытуемого соединения. IC50 (нМ) равны 1300, 170, 46, 61 и 110 для соединений 1, 32, 35, 52 и 80 соответственно.
Пример 13. Антигистаминергическая и 5-липоксигеназная ингибирующая активность in vivo
UCB 62045: Фармакология нового противопоспалительного агента с двойной функцией с активностью актнагониста рецептора гистамина-1 и с активностью ингибитора 5-липоксигеназы
Полагают, что как гистамин, так и лейкотриены являются основными медиаторами астмы, а также других воспалительных или аллергических патофизиологических нарушений. Имеющиеся данные наводят на мысль, что комбинация антагонистов лейкотриена (LT) и антигистаминных препаратов (зафирлукаст + лоратидин или монтелукаст + лоратидин) может ослаблять как экспериментальные, так и клинические случаи астмы или ринита. Было синтезировано соединение, фигурирующее в Таблице 3 под номером 80, которое противостоит действию рецептора гистамина-1 (H1) (26 нМ К, в клетках СНО, трансфецированных человеческим рецептором H1), и ингибирует также 5-липоксигеназу (5-LO; IC50 193 нМ и 88 нМ против человеческой рекомбинантной 5-LO и ингибирование индуцированного с помощью А23187 образования LTB4 в цельной человеческой крови соответственно). Перорально вводимое соединение (2 мг/кг в 1% метилцеллюлозе) в зависимости от дозы ингибировало как индуцированный гистамином бронхостеноз (0, 98 и 97% через 1, 3 и 6 ч после введения дозы), так и ех vivo индуцированное с помощью А23187 образование LTB4 у морских свинок (66, 73 и 88% через 1, 3 и 6 ч после введения дозы). Кроме того, соединение (2 мг/кг) также снижает содержание гистамин- и лейкотриензависимых компонентов аллергического бронхостеноза (овальбумин, 0.1 мг/мл в.в.) в модели, находящейся под наркозом, страдающей аллергией морской свинки (37, 86 и 91% через 1, 3 и 6 ч после введения дозы) Эти результаты показывают, что заявленные соединения могут оказаться клинически полезными для лечения заболеваний дыхательных путей благодаря противовоспалительной активности за счет уменьшения содержания всех лейкотриенов в сочетании с антигистаминэргическим действием.
| название | год | авторы | номер документа |
|---|---|---|---|
| Ингибиторы лизинспецифической гистондеметилазы 1A (KDM1A) для терапии заболеваний | 2019 |
|
RU2813145C2 |
| ЛИГАНДЫ ЦЕРЕБЛОНА И БИФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ | 2018 |
|
RU2795146C2 |
| 2-ОКСА-5-АЗАБИЦИКЛО[2.2.1]ГЕПТАН-3-ИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2015 |
|
RU2697651C2 |
| ЗАМЕЩЕННЫЙ 2-АМИНОПИРИДИН В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2014 |
|
RU2671212C2 |
| ПРОИЗВОДНЫЕ 1-(4-ИЗОКСАЗОЛ-5-ИЛ)-1Н-ПИРАЗОЛ-1-ИЛ)-2-МЕТИЛПРОПАН-2-ОЛА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИРОРОВ ИЛ-17 И ИФН-ГАММА ДЛЯ ЛЕЧЕНИЯ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ И ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ | 2018 |
|
RU2785342C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АЦЕТАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И ИНГИБИТОРЫ ПРОТЕАЗ НА ИХ ОСНОВЕ | 1997 |
|
RU2181360C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АММОНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2743098C2 |
| ПРОИЗВОДНЫЕ ПИРАЗИНОНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ЛЕГОЧНЫХ ЗАБОЛЕВАНИЙ | 2008 |
|
RU2479580C2 |
| N-(1-(1-БЕНЗИЛ-4-ФЕНИЛ-1Н-ИМИДАЗОЛ-2-ИЛ)-2,2-ДИМЕТИЛПРОПИЛ)БЕНЗАМИДНЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНЕЗИНОВОГО БЕЛКА ВЕРЕТЕНА (KSP) ДЛЯ ЛЕЧЕНИЯ РАКА | 2005 |
|
RU2427572C2 |
| СЕЛЕКТИВНЫЕ К BCL-2 АГЕНТЫ, ВЫЗЫВАЮЩИЕ АПОПТОЗ, ДЛЯ ЛЕЧЕНИЯ РАКА И ИММУННЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2621052C2 |
Настоящее изобретение относится к 1,4-замещенным пиперизанам или 4-алкилиденнилпиперидинам общей формулы I
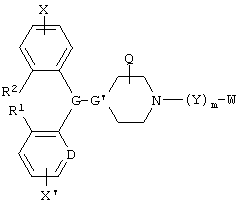
и его геометрические изомеры, энантиомеры, диастереомеры и фармацевтически приемлемые соли,
где G и G' - вместе образуют
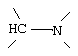 ,
, 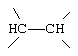 или
или 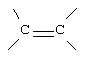
D - обозначает -CH= или =N—;
Y обозначает –L2-V(Z)t-L3-, где t обозначает 0 или 1;
W обозначает –N(ОМ)C(О)N(R8)R9.
Соединения обладают как свойствами ингибировать лейкотриен, так и антигистаминергическими свойствами, и применимы для лечения астмы, сезонного и хронического аллергического ринита. Описаны также фармацевтическая композиция на основе соединений формулы I и способ лечения 4 н. и 18 з.п. ф-лы, 9 ил., 5 табл.
и его геометрические изомеры, энантиомеры, диастереомеры и фармацевтически приемлемые соли,
где Х и X' независимо обозначают водород, галоген, или -(Y')m'-W';
G и G' вместе образуют
 или
или 
D обозначает -СН= или =N-;
R1 и R2 независимо обозначают водород или вместе обозначают -(CH2)n-, где n = 2;
m и m' обозначают независимо 0 или 1;
Y обозначает -L1- или -L2-V(Z)t-L3, где t = 0 или 1;
Y' обозначает -L1-;
L1 обозначает алкилен, алкенилен или один из вышеназванных, где одна или более метиленовых групп замещены на -О-;
L2 обозначает (а) алкилен, алкинилен или один из вышеназванных, где одна или более метиленовых групп замещены на -О-, или (б) -L4-C(O)-N(Q')-, или -L4(Q')-;
L3 обозначает (а) алкилен и алкинилен или один из вышеназванных, где одна или более метиленовых групп замещены на -О-;
L4 обозначает алкилен, у которого одна или более метиленовых групп замещены на -N(R5)-;
V обозначает (а) двухвалентный арен, двухвалентный гетероарен или двухвалентный насыщенный гетероцикл, когда t = 0, или (б) трехвалентный арен или трехвалентный гетероарен, когда t = 1;
Q обозначает водород, AC(O)OR6 или AC(O)NR6R7;
W обозначает -N(OM)C(O)N(R8)R9, -N(R8)C(O)N(OM)R9, -N(OM)C(O)R8, или -C(O)OR8;
W' обозначает -N(OM)C(O)N(R8)R9 или -C(O)OR8, при условии, что хотя бы один из W и W′ представляет собой N(OM)C(O)N(R8)R9;
Z обозначает -A'C(O)NR10R11, -A'C(O)OR10, галоид, NR3C(O)R4, NO2, CN, или СF3;
А и А' обозначают независимо простую связь, алкилен, алкинилен;
М и М' независимо обозначают водород, фармацевтически приемлемый катион или расщепляемую в процессе метаболизма группу;
R3-R11 независимо обозначают водород, алкил, арилалкил или один из вышеназванных, в котором одна или более метиленовых групп замещены на -О-,
при условии, что в отличие от атомов кислорода, связанных с серой в -S(O)-и -S(O)2-, когда одна или более метиленовых групп замещены на -О-, -NH-, -S-, -S(O)- или -S(O)2- и когда один или более метилиденовых остатков замещены на =N-, такое замещение не происходит в двух гетероатомах, ковалентно связанных друг с другом; и, кроме того, при условии, что, когда m = 0, W не обозначает -C(O)OR8; и, кроме того, при условии, что в заместителе -AC(O)OR6 R6 не может обозначать водород, когда А обозначает простую связь.
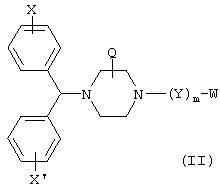
в которой значение заместителей определено в п.1,
и его геометрические изомеры, энантиомеры, диастереомеры и фармацевтически приемлемые соли.
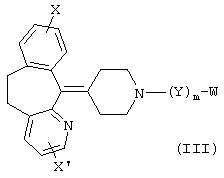
в которой значение заместителей определено в п.1,
и его геометрические изомеры, энантиомеры, диастереомеры и фармацевтически приемлемые соли.
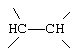 или
или 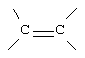
D обозначает -СН= или =N-; R1 и R2 независимо обозначают водород или вместе обозначают -(СН2)2-; m и m' независимо обозначают 0 или 1; Y обозначает -L1- или -L2-V(Z)t-L3, где t = 0 или 1; Y' обозначает -L1-; L1 обозначает алкилен, алкинилен или один из вышеперечисленных, в котором одна или более метиленовых групп замещены на -О-; L2 обозначает (а) алкилен, алкинилен или один из вышеперечисленных, в котором одна или более метиленовых групп замещены на -О- или (б) -L4-C(O)-N(Q')-; L3 обозначает (а) алкилен, алкинилен или один из вышеперечисленных, в котором одна или более метиленовых групп замещены на -О-; L4 обозначает алкилен; V обозначает (а) двухвалентный арен, двухвалентный гетероарен или двухвалентный насыщенный гетероцикл, когда t = 0, или (б) трехвалентный арен или трехвалентный гетероарен, когда t = 1; Q обозначает водород; Q' обозначает -AC(O)OR6 или -AC(O)NR6R7; W обозначает -N(OM)C(O)N(R8)R9, -N(R8)C(O)N(OM)R9, N(OM)C(O)R8 или -C(O)OR8; W обозначает -N(OM)C(O)N(R8)R9 или -C(O)OR8, при условии, что хотя бы один из W и W' представляет собой N(OM)C(O)N(R8)R9; Z обозначает -A'C(O)NR10R11, -A'C(O)OR10, галоид, NR3C(O)R4, NO2, CN или СF3; А и А' независимо обозначают простую связь, алкилен или алкенилен; М и М' независимо обозначают водород, фармацевтически приемлемый катион или метаболически расщепляемую группу; R3-R11, если присутствуют, независимо обозначают водород или алкил, в котором одна или несколько метиленовых групп заменены на -О-, при условии, что в отличие от атомов кислорода, связанных с атомами серы в -S(O)- и -S(O)2-, когда одна или более метиленовых групп замещены на -О- или -NH- и когда одна или более метилиденовых групп замещены на =N-, при таком замещении не образуется двух ковалентно связанных друг с другом гетероатомов и при условии, что, когда m = 0, W не обозначает -C(O)OR8, и еще при условии, что в заместителе -AC(O)OR6R6 не может обозначать водород, когда А обозначает простую связь.
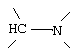 или
или 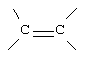
Y обозначает -L2-V(Z)t-L3-, где t = 0 или 1; L2 обозначает алкилен C1-C6, в котором одна или более метиленовых групп могут быть заменены на -О-; V(Z)t обозначает фенилен, при необходимости замещенный группами -A'C(O)NR10R11, -A'C(O)OR10, галоид, NR3C(O)R4, NO2, CN или СF3 или фурилен или оксоланилен; L3 обозначает алкилен C1-C6, в котором одна или более метиленовых групп могут быть заменены на -О-, или С2-С6-алкинилен; W обозначает -N(OM)C(O)N(R8)R9, -N(R8)C(O)N(OM)R9 или -N(OM)C(O)R8; А' обозначает метилен, винилен или простую связь; R3-R11, если присутствуют, независимо обозначают водород или С1-С6, -алкил, в котором одна или более метиленовых групп может быть заменена на -О-.

