В данной заявке испрашивается приоритет предварительной заявки США №62/670,323, поданной 11 мая 2018 г., раскрытие которой включено в данную заявку посредством отсылки, как если бы она была приведена в данной заявке полностью.
Настоящее изобретение относится к новым соединениям и композициям и их применению в качестве фармацевтических препаратов для лечения заболеваний.
Ингибирование фермента KDM1A (также известного как лизинспецифическая деметилаза 1, LSD1, белок, содержащий аминооксидазный домен, содержащий флавин, AOF2, BRAF35-HDAC комплексный белок BHC110, FAD-связывающий белок BRAF35-HDAC комплекс) может изменить экспрессию генов в клетках, достаточную для восстановления их надлежащей физиологической функции или функции ткани, органа или пациента в целом. Это может быть достигнуто либо за счет усиления транскрипции гена или генов, которые патологически подавлены, например, как в случае некоторых раковых клеток и наследственных заболеваний, либо за счет уменьшения транскрипции гена или генов, участвующих в патологическом состоянии. Как таковое, ингибирование KDM1A может быть полезным для лечения таких заболеваний, как рак, и наследственных заболеваний, таких как болезнь Вильсона, кардиомиопатии и гемоглобинопатии.
Экспрессия генов регулируется посредством рекрутирования транскрипционного аппарата РНК-полимеразы II в матрицу ДНК. Вероятность того, что этот большой мультибелковый комплекс появится рядом или в начале транскрипции ДНК и будет проходить через всю кодирующую область гена, частично определяется специфическими последовательностями ДНК, называемыми промоторами и энхансерами, модификациями последовательности ДНК в непосредственной близости от начала транскрипции, белками, связанными с ДНК и топологией самой матрицы ДНК. Факторы, повышающие вероятность синтеза РНК генов, кодирующих белок, известны как факторы транскрипции, некоторые из которых участвуют в транскрипции всех генов, кодирующих белок, а некоторые из которых являются специфичными для транскрипции отдельных генов.
Один из основных механизмов контроля транскрипции состоит в ограничении физической доступности регуляторных областей транскрипции для белков, которые могут активировать или завершать транскрипцию; белки, связанные с промоторными или энхансерными последовательностями ДНК, могут препятствовать связыванию активирующих факторов с этими последовательностями ДНК, что приводит к меньшему количеству инициации транскрипции или расширению активированного прогрессирующего комплекса РНК-полимеразы. Точно так же топологические ограничения, которые не позволяют матричной ДНК раскручиваться в достаточной степени, чтобы обеспечить устойчивое развитие РНК-полимеразы на матрице, также служат для ограничения скорости транскрипции.
Наиболее важными общими факторами, влияющими на синтез РНК с использованием матрицы ДНК in vivo, являются модификации гистоновых белков, которые контролируют среди других факторов топологию матрицы ДНК для транскрипции и ее доступность для комплекса РНК-полимеразы. Небольшое семейство гистоновых белков - Н2А, Н2В, Н3 и Н4 - объединяется для создания каркаса, называемого гистоновым октамером, на котором ДНК пространственно и топологически организована в регулярную повторяющуюся структуру, называемую нуклеосомой, вдоль всей длины ДНК. Конгломерат гистонов, других белков, различных РНК и ДНК называется хроматином. Как ДНК, так и гистоны химически модифицированы таким образом, чтобы привлекать и связывать или отталкивать другие белки с эффектом усиления или репрессии транскрипции.
Модификация ДНК и ассоциированных РНК и белков, которые влияют на регуляцию транскрипции и репликации, которая не предполагает замены канонических оснований ДНК, называется эпигенетической. Эти эпигенетические влияния включают обратимые химические модификации самих четырех оснований ДНК или посттрансляционные химические изменения белков хроматина и RND, которые связаны с ДНК. Эти эпигенетические процессы могут играть ключевую роль в активации или подавлении экспрессии гена; кроме того, эпигенетические модификации могут сохраняться на протяжении всей жизни организма или могут динамически изменяться в ответ на специфические биохимические сигналы, которые исходят либо внутри клетки, либо вне клетки. Эти изменения хроматина могут происходить быстро или могут быть очень стабильными, например, во время гормональной индукции экспрессии гена структура хроматина в определенном локусе может радикально измениться в течение секунд, чтобы обеспечить максимальную транскрипцию, или структура хроматина может быть изменена для полного подавления экспрессии гена, состояния хроматина, которое может стабильно сохраняться во время мнократного деления клеток и даже трансгенерационно.
Метилирование цитозина в 5' положении представляет собой обычную модификацию основания ДНК, которая, в свою очередь, распознается классом белков, наиболее часто ассоциированных с репрессией транскрипции. Точно так же гистоновые белки являются химически модифицироваными, но с более широким спектром химических аддуктов, каждый из которых либо по отдельности, либо в комбинации усиливает или подавляет транскрипцию близлежащих генов. Эти модификации гистонов включают, среди прочего, метилирование, ацетилирование, сумоилирование, фосфорилирование, убиквитилирование и миристоилирование, которые распознаются другими ассоциированными с хроматином белками, которые, в свою очередь, влияют на скорость транскрипции и репликацию ДНК. Динамическое состояние экспрессии гена и ассоциированные состояния хроматина подразумевают, что модификации гистонов не являются постоянными, а вместо этого добавляются и удаляются в соответствии с потребностями клетки в определенных генных продуктах в определенные периоды онтогенеза, взрослой жизни и меняющихся влияний окружающей среды. В самом деле, каждая конкретная химическая модификация гистонов производится классами ферментов, действующих на определенных участках. Эти ферменты, модифицирующие гистоны, в свою очередь, подвергаются жесткой регуляции. Эти ферменты потенциально могут быть нацелены на соединения, которые ингибируют их активность, что приводит к терапевтическому изменению экспрессии генов.
В настоящее время известно, что изменения в состоянии метилирования гистонов играют критическую роль в нормальной регуляции клеточного цикла и роста, реакции на повреждение ДНК и стресс, и внутриутробном развитии, включая дифференцировку. Патологические состояния, такие как рак, связаны с измененными профилями модификаций гистонов и нарушением регуляции белков, модифицирующих гистоны, включая ферменты, модифицирующие хроматин. О необходимости тщательного регулирования модификаций гистонов свидетельствует связь статуса метилирования гистонов с заболеваемостью человека, включая старение.
Метилирование гистона может происходить на любом из трех основных аминокислотных остатков - лизине (K), аргинине (R) и гистидине (H). Метилирование гистона Н3 по лизинам в положениях 4 (H3K4), 9 (H3K9), 27 (H3K27), 36 (H3K36) и 79 (H3K79) являются одними из наиболее изученных модификаций гистонов, влияющих на экспрессию генов. Триметилирование лизина (Kme3) гистона 3 (Н3) в положении 4 (H3K4me3) представляет собой гистоновую метку, обычно связанную с активацией экспрессии гена, тогда как H3K9mel или H3K27me3 связаны с репрессией транскрипции гена. H3K4mel связан с ДНК-энхансерами транскрипции гена, тогда как H3K4me3 связан с активностью промотора гена. Точно так же потеря метальной группы на H3K4 связана с репрессией экспрессии гена. Таким образом, добавление и удаление метальной группы на H3K4 представляет собой переключатель транскрипции гена. Также очевидно, что лизин может быть модифицирован моно-, ди- или три-метильной группой, каждая модификация имеет различный биологический эффект за счет привлечения различных белков, распознающих эти специфические модификации метилирования на этом сайте.
Критическим аспектом регуляции состояния метилирования гистонов является рекрутирование метилтрансфераз и деметилаз к конкретным генетическим локусам. Связывающие белки, специфичные для последовательности ДНК, включая факторы транскрипции, представляют собой один класс белков, ответственных за это рекрутирование посредством сборки белковых комплексов, которые связывают эти ферменты, переносящие метил. Хорошо изученным примером являются элементы ответа (TRE) группы Drosophila melanogaster trithrorax (TrxG), которые рекрутируют H3K4-метилтрансферазу, TRX, к конкретным генам через факторы транскрипции, которые распознают последовательность ДНК TRE.
Метки метилирования гистонов распознаются метил-связывающими доменами в разнообразной группе белков; эти домены включают в себя PHD-''пальцы", WD40 и анкириновые повторы, CW и PWWP домены и суперсемейство белков Royal. Эти белки, в свою очередь, определяют, какие дополнительные активности рекрутированы в сайтах хроматина и, в конечном итоге, состояние транскрипции в данном локусе. Действительно, в зависимости от того, какой белок распознавания метила связывает меченый гистон, одна и та же модификация метил-лизина может иметь противоположные эффекты на транскрипцию. H3K4me2 и H3K4me3 связаны с активацией транскрипции, но при связывании с содержащим PHD-домен корепрессорным белком-ингибитором членом семейства роста 2 (ING2), связанный комплекс гистондеацетилазы стабилизирует экспрессию репрессивного гена. Таким образом, эти эффекторные белки, распознающие модификации гистонов метил-лизина, значительно влияют на уровень транскрипционной активности.
Способность избирательно изменять экспрессию генов путем изменения состояния хроматина позволяет использовать новую терапевтическую стратегию для индукции или отмены репрессии экспрессии генов, которые могут принести пользу, особенно для генов, экспрессия которых подавлена патологическим механизмом, как в случае некоторых видов рака, или подавлена физиологическим механизмом, но они при дерепрессии могут фенотипически подавлять мутации в паралогичных генах с дополнительной функцией.
Многие гены в геноме являются членами генных семейств вследствие дупликации генов. Эти гены называют паралогами друг друга. После дупликации генов профили экспрессии двух генов будут развиваться по-разному, отчасти для контроля эффектов дозы генов. После дупликации гена случайный генетический дрейф, возникающий из-за встречающихся в природе мутаций, и последующий выбор нуклеотидной последовательности обычно сначала наблюдаются в некодирующих областях дублированных генов, часто в регуляторных областях транскрипции. Изменения ДНК в регуляторных последовательностях могут влиять на любой или все аспекты экспрессии гена: величину экспрессии, время ее развития, индукцию стимулами вне клетки, включая гормональные или метаболические сигналы, и тип клетки, в котором экспрессия ограничена. В случаях, когда дупликация произошла недавно в эволюционном периоде или когда естественный отбор сохранил высокую степень сходства последовательностей, кодирующих белок, генный продукт одного паралога, ген А, может дополнять патологическую потерю или сайленсинг другого паралога, гена В, если экспрессия гена А не ограничивается в той же клетке.
Изменение профилей экспрессии генов может иметь значительные терапевтические преимущества при генетических состояниях, при которых усиленная экспрессия паралогичного гена избавляется от фенотипа, возникшего вследствие мутации в паралоге. Это можно назвать комплементацией аутологичных генов. В случае болезни Вильсона, возникший вследствие мутаций в АТР7В, усиленная экспрессия за счет фармакологической индукции ATP1A7 близкородственного белка-переносчика меди, может избавить от мутаций в АТР7В, другом переносчике меди. Основная функция каждого белка-переносчика меди была сохранена, но после дупликации общего предкового гена экспрессия этих двух генов была пространственно разделена, один ограничен кишечными энтероцитами, другой - гепатоцитами. Это один из многих примеров паралогичного гена, в котором один ген может компенсировать потерю второго, если он соответствующим образом экспрессируется в той же самой клетке или ткани.
Ярким примером семейства паралоговых генов является хорошо изученное семейство альфа и бета глобиновых генов, кодирующих альфа и бета субъединицы гемоглобина. Пять бета-подобных генов, каждый из которых возникает в результате дупликации генов, выстраиваются рядом друг с другом на хромосоме 16, причем каждый ген транскрибируется в зависимости от времени в течение 9 месяцев эмбрионального и внутриутробного развития человека. Пять бета-подобных глобиновых белков обладают высокой степенью сходства белковых последовательностей, настолько, что генетические мутации, инактивирующие бета-глобиновый ген взрослого человека, могут быть клинически незаметными, если экспрессия любой из четырех других субъединиц семейства бета-подобных глобинов является достаточной. Активация экспрессии и последующий транскрипционный сайленсинг каждого конкретного эмбрионального и фетального бета-подобного глобинового гена частично регулируется эпигенетическими механизмами. Избавление от мутаций в бета-глобиновом гене, мутаций, которые ответственны за такие заболевания, как большая талассемия или серповидноклеточная анемия, путем транскрипционной индукции одного или нескольких других бета-подобных генов посредством фармакологических манипуляций с эпигенетическим сайленсингом было бы клинически полезным. Аутологичная активация фармакологическим агентом функционально комплементарного паралога мутантного или патологически молчащего гена может быть более успешной терапевтической стратегией, чем замена или восстановление мутированного гена копией дикого типа (нормальной).
Интерес к влиянию на активность модификаций гистонов для терапевтического эффекта возник из наблюдений за тем, что экспрессия конкретных генов под эпигенетическим контролем может быть изменена путем изменения эпигенетических меток, таких как метилирование. В случае рака потеря специфических меток метилирования гистонов, сопровождающаяся сверхэкспрессией гистонде метил аз, связана с рецидивом этих видов рака с сопутствующими худшими исходами. Эти исследования предполагают, что определенные гены-супрессоры опухолей "выключаются" вследствие потери модификаций метилирования, которые, в свою очередь, повышают выживаемость и потенциал к росту неопластических клеток. Это привело к предположению, что ингибирование активности гистондеметилазы может иметь терапевтическое значение.
KDM1A (также известная как лизин-специфическая деметилаза 1 (LSD1) или AOF2 или BHC110) была первым описанным ферментом со специфической активностью лизинде метил азы, однозначно демонстрирующим, что модификации гистонов обратимы, а не постоянны. Среди субстратов деметилазы KDM1A представляет собой лизиндеметилазу гистона Н3, которая катализирует окислительное деметилирование H3K4mel или me2 и H3K9mel или те2, но не субстрата H3K4me3. Фермент также деметилирует негистоновые белки, такие как р53 и Gfi1. KDM1A содержит домен аминоксидазы, который деметилирует субстрат H3Kme зависимым от флавинадениндинуклеотида (FAD) способом, аналогичным другим ингибиторам моноамин-(МАО) и полиаминоксидазы. Действительно, неспецифические ингибиторы ферментов МАО могут подавлять активность деметилазы KDM1A.
KDM1A сверхэкспрессируется при многих раковых заболеваниях человека, включая опухоль Вильма, мелкоклеточный рак легкого, рак мочевого пузыря, рак предстательной железы, рак молочной железы, рак головы и шеи, рак толстой кишки и рак яичников, и ассоциируется с более частыми рецидивами. KDM1A необходима для регуляции транскрипции, опосредованной рецептором андрогена при раке предстательной железы, рецептором эстрогена при карциноме молочной железы и рецептором TLX при нейробластоме. Нокдаун экспрессии KDM1A снижает пролиферацию раковых клеток. KDM1A также сверхэкспрессируется в раковых клетках, которые не зависят от ядерных гормональных рецепторов, включая ER-отрицательную молочную железу. Эффективные селективные низкомолекулярные ингибиторы KDM1A должны быть полезны для лечения этих и других видов рака, при которых активность KDM1A является чрезмерной.
Структура и состояние хроматина также могут влиять на способность патогенного вируса встраиваться в ДНК хозяина, подвергаться транскрипции и реплицироваться. Заражение принадлежащими к альфа-герпесвирусам вирусом простого герпеса (ВПГ) и вирусом ветряной оспы (VSV) влияет на ремоделирование хроматина после заражения клеток-хозяев, чтобы противодействовать быстрому отложению нуклеосом, содержащих гистоны, с репрессивными метками транскрипции за счет использования кодируемых вирусом факторов транскрипции для рекрутирования комплекса коактиватора хозяина HCF-1, который содержит KDM1A и члены семейства Set1 или MLL гистон H3K4 метилтрансферазы. Было продемонстрировано, что ингибирование KDM1A в клетках, инфицированных HSV1, подавляет экспрессию гена IE HSV, подавляет литическую инфекцию и снижает вирусную нагрузку. Аналогичным образом, ингибирование KDM1A вызывает снижение экспрессии немедленно-ранних генов в клетках, инфицированных цитомегаловирусом и аденовирусом человека, что указывает на более широкую роль KDM1A в вирусном патогенезе.
Влияние активности KDM1A на транскрипцию конкретных генов зависит от рекрутирования KDM1A в промоторную область определенного гена через ДНК-связывающие белки. В случае андроген-зависимой экспрессии гена KDM1A связывается с андрогеновым рецептором стероидов, который специфически нацелен на сайты связывания ДНК в промоторах андроген-чувствительных генов. Таким образом, белки, связывающие KDMl А, определяют, где вдоль хромосомы нацелена активность деметилазы. Сообщалось, что многие белки взаимодействуют с KDM1A, включая CoREST, CtBP, NuRD, BRAF35 комплексы, DNMT1, МТА1/2, Mi2beta, RbAp46/48, HDAC1, 2 и 3, TIF1beta, Blimp-1, ZNF217 и ZNF198, подмножество которых образуют более крупные и в некоторых случаях комплексы, взаимно исключающие друг друга. Комплекс KDM1A/CoREST, который может также включать DNMT1 и NuRD среди других факторов, особенно важен для подавления экспрессии определенных генов.
KDM1A рекрутируется в промоторную область генов через сайт-специфические факторы транскрипции. Такие факторы включают, среди прочего, андрогеновый рецептор, рецептор эстрогена альфа, Snail1, Slug, HIV Tat, ZEB1, RBP-J, PIT1, REST, NR2C1, NR2C2 и изоформы Gfi1b. Эти факторы транскрипции могут рекрутировать KDM1A для участия в активации экспрессии генов или подавлении экспрессии генов в зависимости от типа клеток и конкретных факторов транскрипции.
Многие ферментативные активности, регулирующие состояние хроматина, подвергаются аллостерическому влиянию или требуются в качестве кофакторов промежуточных продуктов метаболизма, медиаторов или конечных продуктов клеточного метаболизма. Эти межмолекулярные отношения между экспрессией генов и метаболизмом обеспечивают клетки сигнальными путями, связывающими внешнюю и внутреннюю клеточную среду, включая питательные вещества, с механизмами, модулирующими экспрессию генов. Это клеточное восприятие может изменить как краткосрочные, так и долгосрочные корректировки профилей экспрессии генов, составляющих эпигенетическую память об исторических состояниях метаболизма и условиях окружающей среды. Например, бета-гидроксибутират, продукт метаболизма длинноцепочечных жирных кислот и основной источник энергии для млекопитающих во время голодания или длительной нагрузки, ингибирует гистондеацетилазы класса I (HDAC), но не HDAC класса 2b. Таким образом, эффекты голодания и потери питательных веществ могут быть закодированы эпигенетически и сохранены. Ацетил-кофермент А, никотинамидадениндинуклеотид (НАД) и альфа-кетоглутарат также влияют на состояния метилирования и ацетилирования гистонов.
Флавинадениндинуклеотид (FAD) является необходимым кофактором для KDM1A. FAD в сочетании с NAD и NADP действуют как клеточные окислительно-восстановительные сенсоры. KDM1A временно преобразует FAD в FADH, после чего акцептор электронов, скорее всего О2 и другие, завершает каталитический цикл, регенерируя FAD и Н2О2. Таким образом, окислительно-восстановительное состояние клетки влияет на активность KDM1A как за счет ее способности окислять FADH, так и за счет других акцепторов электронов. В общем смысле состояния хроматина, а следовательно, и экспрессия генов, могут быть изменены различными концентрациями метаболических промежуточных продуктов, а в конкретном случае KDM1A, эта активность полностью зависит от FAD, концентрация которого колеблется в зависимости от энергетической экономии клетки. Кроме того, было показано, что ингибирование KDM1A может привести к снижению уровня глюкозы в сыворотке крови, снижению уровня гликогена в печени и является мощным секретором инсулина. Таким образом, фармацевтические манипуляции с активностью KDM1A могут оказаться полезными для лечения заболеваний, которые представляют собой патологические отклонения энергетического статуса клетки, включая метаболический синдром, дислипидемии, диабет, ожирение, анорексию, задержку развития, кахексию, липодистрофии и стеатогепатит.
Стероидные гормоны эстрадиол, тестостерон и родственное соединение играют ключевую роль как в нормальном развитии, так и в патологических состояниях, таких как рак молочной железы и предстательной железы, при которых рост опухолевых клеток зависит от гормональной передачи сигналов. Биологические эффекты стероидных гормонов опосредуются структурно и функционально различными лиганд-связывающими рецепторами, которые функционируют как фактор транскрипции, рекрутируемый в конкретный сайт связывания ДНК. Связанные с лигандом стероидные рецепторы действуют как главный регулятор транскрипции гормональных эффектов. Транскрипционная активация экспрессии генов всех стероид-зависимых гормонов зависит от структуры хроматина и наличия кофакторов. Рецептор эстрогена использует, например, кофакторы SRC1, SRC2, AIB1, PELP1, СВР, р300, PCAF, CARM1, PRMT1 и корепрессоры, такие как NCoR, SMRT и МТА1. Транскрипционная реакция на гормональную стимуляцию зависит от взаимодействия этих кофакторов и репрессоров, а также от состояния хроматина, особенно от модификации гистонов ферментами, модифицирующими гистоны, связанными с ко-регуляторами. Стимуляция как эстрогенного, так и андрогенного гормона вызывает несколько модификаций гистонов в промоторах генов-мишеней, которые изменяют состояние ацетилирования, фосфорилирования и метилирования локальных гистонов. Чтобы повлиять на максимальную скорость транскрипции гормонального гена, необходима активность KDMl А. Таким образом, KDMA1 должна оказаться полезной в качестве терапевтической мишени фармацевтических препаратов при снижении или устранении гормональной зависимости опухолевых клеток. Та же самая терапевтическая логика применима к другим лиганд-зависимым факторам транскрипции, активация транскрипции которых частично или полностью зависит от активности KDM1A, чтобы изменить состояния хроматина в достаточной степени для облегчения транскрипции - примеры этого могут включать рецепторы, активируемые витамином D, ретиноидами и липидами.
Были идентифицированы многочисленные терапевтические агенты, которые обладают эффектом изменения экспрессии генов, действуя либо непосредственно на белки, как правило, ферменты, которые изменяют состояния хроматина, либо опосредованно. Хотя точные механизмы их действия не все полностью выяснены, этот механизм может быть выведен из нашего понимания белковых комплексов, которые участвуют в активации специфической экспрессии генов. Эти агенты включают 5'-азацитадин и 5'-аза-2'дезоксицитидин (децитабин), которые ингибируют DNMT1 или другие ДНК-метилтрансферазы, которые, как известно, присутствуют и активны в промоторных сайтах молчащих генов, такие как промотор гамма-глобина; вориностат и панобиностат или другие ингибиторы ферментов гистондеацетилазы (HDAC); гидроксимочевина (HU), вальпроат и бутират натрия и его аналоги, каждый из которых может влиять на активность "сиротских" ядерных рецепторов. Все эти агенты имеют некоторое клиническое применение, главным образом при лечении опухолевых заболеваний. Хотя была продемонстрирована некоторая клиническая полезность этих агентов при других болезненных состояниях, они не получили широкого распространения из-за их умеренных терапевтических эффектов и их токсичности.
Применение агентов, которые ингибируют любую ферментативную активность, присущую белковому комплексу, связанному с промотором гена, потенциально может нарушить подавление экспрессии гена гамма-глобина и привести к повышению уровня гемоглобина плода, также известного как гемоглобин F (HbF). К таким мишеням относятся любые интерфейсы конкретных белок-белковых контактов, например, комплекс NuRD и KDM1 А; домены распознавания связывания ДНК, например, NR2C1 и NR2C2; лиганд-связывающие домены, например, NR2C1 и NR2C2; ферментативные активности, такие как лизиндеметилаза, например, KDM1A; гистондеацетилазы (HDAC), например HDAC1, 2 или 3; ДНК метилтрансферазы, например, DNMT1.
Остается потребность в композициях и способах изменения экспрессии генов в клетках и тканях, достаточной для восстановления нормальной физиологической функции клетки или ткани, включая, например, соответствующий апоптоз в случае рака, или для изменения патологического фенотипа клетки, ткани, органа или организма путем индукции экспрессии одного или нескольких генов в достаточной степени для подавления патологического состояния.
Соответственно, в данной заявке авторы изобретения раскрывают новые соединения, композиции и способы лечения заболеваний, связанных с активностью KDM1A
В данной заявке представлен Вариант осуществления 1: соединение, имеющее структурную Формулу I:
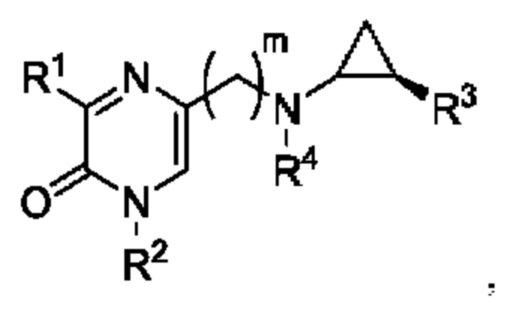
или его соль или сложный эфир, в котором:
m выбран из 0, 1, 2, 3 и 4;
R1 представляет собой азотсодержащий гетероциклоалкил или гетероарил, каждый из которых необязательно замещен 1, 2 или 3 группами R5;
R2 представляет собой H или выбран из алкила, циклоалкила, галогеналкила, гетероциклоалкила, арила, гетероарила, циклоалкилалкила, гетероциклоалкилалкила, арилалкила и гетероарилалкила, любой из которых необязательно замещен 1, 2 или 3 группами R6;
R3 выбран из арила и гетероарила, каждый из которых необязательно замещен 1, 2 или 3 группами R7;
каждый R4 независимо выбран из водорода, алкила, алкенила, алкинила и циклоалкила;
каждый R5 независимо выбран из галогена, алкила, алкенила, алкинила, гидрокси, амино, оксо, циано, COR8, CONR8R9, COOR8, NHCOR8, NHCONR8R9, SOR8, SO2R8, NHSO2R8 и SO2NR8R9;
каждый R6 независимо выбран из водорода, галогена, алкила, алкилсульфониларила, алкенила, алкинила, циклоалкила, галогеналкила, галогеналкокси, галогенарила, алкоксиарила, арила, арилокси, аралкила, гетероциклоалкила, гетероарила, алкилгетероарила, гетероарилалкила, циано, алкокси, алкоксиарила, амино, алкиламино, диалкиламино, оксо, COR8, SO2R8, NHSO2R8, NHSO2NHR8, SO2NR8R9, NHCOR8, NHCONHR8, CONHR8 и CONR8R9;
каждый R7 независимо выбран из алкила, амино, циано, галогена и гидрокси; и
R8 и R9 независимо выбраны из водорода, арила и низшего алкила; или R8 и R9 могут быть взяты вместе с образованием азотсодержащего гетероциклоалкильного или гетероарильного кольца, которое необязательно замещено низшим алкилом.
Определенные соединения, раскрытые в данной заявке, могут обладать полезной ингибирующей KDM1A активностью и могут применяться для лечения или профилактики заболевания или состояния, при котором KDM1A играет активную роль. Таким образом, в широком аспекте определенные варианты осуществления также обеспечивают фармацевтические композиции, содержащие одно или несколько соединений, раскрытых в данной заявке, вместе с фармацевтически приемлемым носителем, а также способы получения и применения соединений и композиций. Некоторые варианты осуществления обеспечивают способы ингибирования KDM1A. Другой вариант осуществления обеспечивает способы лечения расстройства, опосредованного KDM1A, у пациента, нуждающегося в таком лечении, включающие введение указанному пациенту терапевтически эффективного количества соединения или композиции в соответствии с настоящим изобретением. Также обеспечено применение определенных соединений, раскрытых в данной заявке, в производстве лекарственного средства для лечения заболевания или состояния, улучшающегося за счет ингибирования KDM1A.
Также в данной заявке представлен Вариант осуществления 2: соединение, имеющее структурную Формулу Ia:
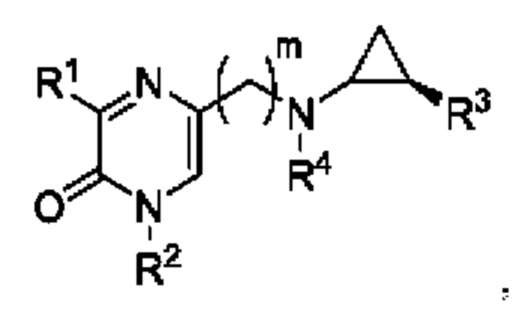
или его соль или сложный эфир, в котором:
m выбран из 0, 1, 2, 3 и 4;
R1 представляет собой азотсодержащий гетероциклоалкил или гетероарил, каждый из которых необязательно замещен 1, 2 или 3 группами R5;
R2 представляет собой H или выбран из алкила, циклоалкила, галогеналкила, гетероциклоалкила, арила и гетероарила, любой из которых необязательно замещен 1, 2 или 3 группами R6;
R3 выбран из арила и гетероарила, каждый из которых необязательно замещен 1, 2 или 3 группами R7;
каждый R4 независимо выбран из водорода, алкила, алкенила, алкинила и циклоалкила;
каждый R5 независимо выбран из галогена, алкила, алкенила, алкинила, гидрокси, амино, оксо, циано, COR8, CONR8R9, COOR8, NHCOR8, NHCONR8R9, SOR8, SO2R8, NHSO2R8 и SO2NR8R9;
каждый R6 независимо выбран из водорода, галогена, алкила, алкилсульфониларила, алкенила, алкинила, циклоалкила, галогеналкила, галогеналкокси, галогенарила, алкоксиарила, арила, арилокси, аралкила, гетероциклоалкила, гетероарила, алкилгетероарила, гетероарилалкила, циано, алкокси, алкоксиарила, амино, алкиламино, диалкиламино, оксо, COR8, SO2R8, NHSO2R8, NHSO2NHR8, SO2NR8R9, NHCOR8, NHCONHR8, CONHR8 и CONR8R9;
каждый R7 независимо выбран из алкила, амино, циано, галогена и гидрокси; и
R8 и R9 независимо выбраны из водорода, арила и низшего алкила; или R8 и R9 могут быть взяты вместе с образованием азотсодержащего гетероциклоалкильного или гетероарильного кольца, которое необязательно замещено низшим алкилом.
Также в данной заявке представлен Вариант осуществления 3: соединение, имеющее структурную Формулу II:
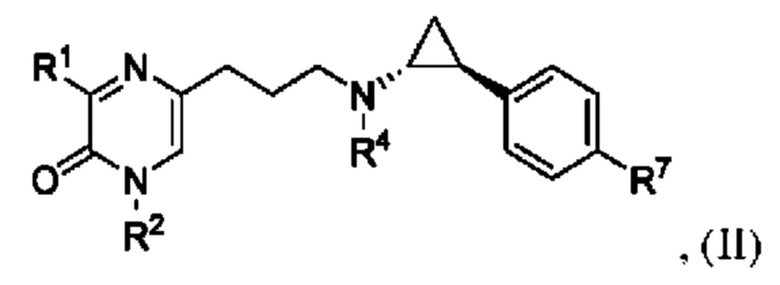
или его соль или сложный эфир, в котором:
R1 представляет собой азотсодержащий гетероциклоалкил или гетероарил, каждый из которых необязательно замещен 1, 2 или 3 группами R5;
R2 представляет собой H или выбран из алкила, циклоалкила, галогеналкила, гетероциклоалкила, арила и гетероарила, любой из которых необязательно замещен 1, 2 или 3 группами R6;
каждый R4 независимо выбран из водорода, алкила, алкенила, алкинила и циклоалкила;
каждый R5 независимо выбран из галогена, алкила, алкенила, алкинила, гидрокси, амино, оксо, циано, COR8, CONR8R9, COOR8, NHCOR8, NHCONR8R9, SOR8, SO2R8, NHSO2R8 и SO2NR8R9;
каждый R6 независимо выбран из водорода, галогена, алкила, алкилсульфониларила, алкенила, алкинила, циклоалкила, галогеналкила, галогеналкокси, галогенарила, алкоксиарила, арила, арилокси, аралкила, гетероциклоалкила, гетероарила, алкилгетероарила, гетероарилалкила, циано, алкокси, алкоксиарила, амино, алкиламино, диалкиламино, оксо, COR8, SO2R8, NHSO2R8, NHSO2NHR8, SO2NR8R9, NHCOR8, NHCONHR8, CONHR8 и CONR8R9;
каждый R7 независимо выбран из водорода, алкила, амино, циано, галогена и гидрокси; и
каждый R8 и R9 независимо выбран из водорода, арила и низшего алкила; или R8 и R9 могут быть взяты вместе с образованием азотсодержащего гетероциклоалкильного или гетероарильного кольца, которое необязательно замещено низшим алкилом.
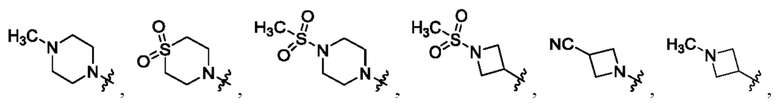
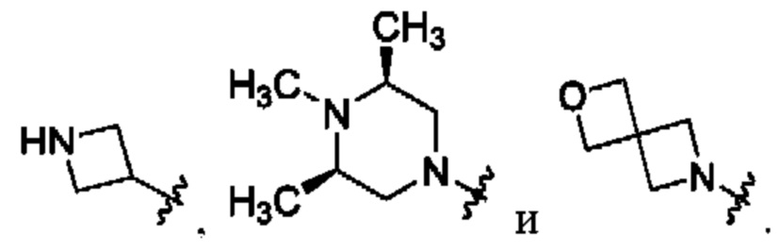
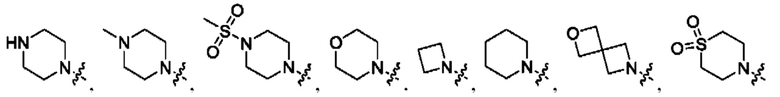
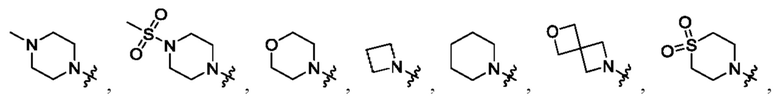
В определенных вариантах осуществления, R1 выбран из пиперидина, морфолина, тиоморфолина, пиперазина, пирролидина, азетидина, 2-азаспиро[3,3]гептана, 2,6-диазаспиро[3,3]гептана и 2-окса-6-азаспиро[3,3]гептана, и необязательно замещен 1, 2 или 3 группами R5.
В определенных вариантах осуществления, R1 выбран из пиперидина, морфолина, тиоморфолина, пиперазина, пирролидина, азетидина, 2-азаспиро[3,3]гептана, 2,6-диазаспиро[3,3]гептана и 2-окса-6-азаспиро[3,3]гептана, и необязательно замещен 1 или 2 группами R5.
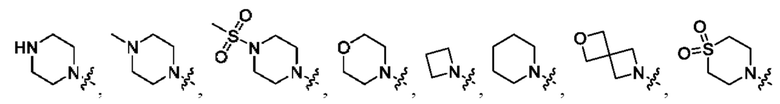
В определенных вариантах осуществления, R1 выбран из 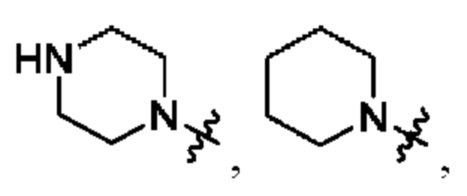
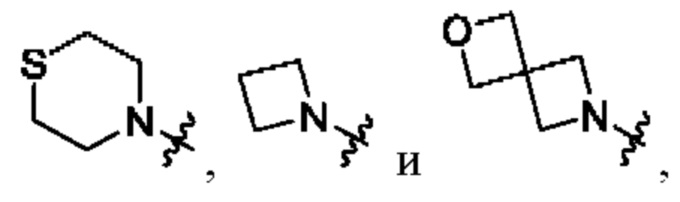 и необязательно замещен 1, 2 или 3 группами R5. В определенных вариантах осуществления, R1 выбран из
и необязательно замещен 1, 2 или 3 группами R5. В определенных вариантах осуществления, R1 выбран из 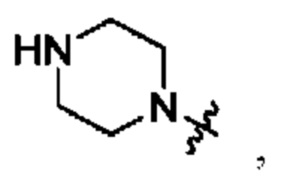
В определенных вариантах осуществления, R2 выбран из арила и гетероарила, каждый из которых необязательно замещен 1 или 2 группами R6.
В определенных вариантах осуществления, R2 выбран из арила и гетероарила, каждый из которых необязательно замещен 1 группой R6.
В определенных вариантах осуществления, R2 выбран из фенила, пирролила, пиразолила, имидазолила, оксазолила, изоксазолила, триазолила, тиазолила, пиридинила, пиразинила, пиридазинила и пиримидинила, любой из которых необязательно замещен 1 или 2 группами R6.
В определенных вариантах осуществления, R2 выбран из фенила, пиридинила и пиримидинила, любой из которых необязательно замещен 1 или 2 группами R6.
В определенных вариантах осуществления, R2 выбран из фенила, пиридинила и пиримидинила, любой из которых необязательно замещен 1 группой R6.
В определенных вариантах осуществления, R2 представляет собой водород.
В определенных вариантах осуществления, R4 представляет собой водород.
В определенных вариантах осуществления, каждый R6 независимо выбран из галогена, алкила, циклоалкила, гетероциклоалкила, гетероарила, алкилгетероарила, гетероарилалкила, циано, COR8, SO2R8, NHSO2R8, NHSO2NHR8, SO2NR8R9, NHCOR8 и NHCONHR8.
В определенных вариантах осуществления, каждый R6 независимо выбран из галогена, гетероарила, алкилгетероарила, SO2R8, NHSO2R8, NHSO2NHR8, SO2NR8R9, NHCOR8 и NHCONHR8.
В определенных вариантах осуществления, каждый R7 независимо выбран из водорода и фтора.
В определенных вариантах осуществления, R7 представляет собой фтор.
Также представлены следующие Варианты осуществления:
Вариант осуществления 4: соединение в соответствии с Вариантом осуществления 1, в котором R3 выбран из фенила, пиридинила, пиридазинила, пиримидинила и пиразинила, любой из которых замещен 1, 2 или 3 группами R7.
Вариант осуществления 5: соединение в соответствии с Вариантом осуществления 4, в котором R3 представляет собой фенил, который необязательно замещен 1, 2 или 3 группами R7.
Вариант осуществления 6: соединение в соответствии с любым из Вариантов осуществления 1, 4 и 5, в котором R3 необязательно замещен 1 или 2 группами R7.
Вариант осуществления 7: соединение в соответствии с Вариантом осуществления 6, в котором R3 замещен 1 или 2 группами R7.
Вариант осуществления 8: соединение в соответствии с Вариантом осуществления 6, в котором R3 необязательно замещен 1 группой R7.
Вариант осуществления 9: соединение в соответствии с Вариантом осуществления 6, в котором R3 выбран из 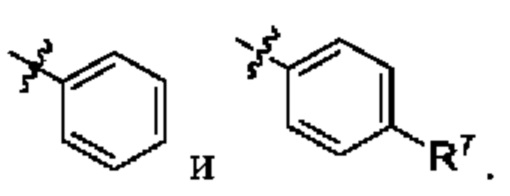
Вариант осуществления 10: соединение в соответствии с любым из Вариантов осуществления 1 и 4-9, в котором каждый R7 независимо выбран из NH2, циано, галогена и гидрокси.
Вариант осуществления 11: соединение в соответствии с Вариантом осуществления 10, в котором каждый R7 независимо выбран из циано и галогена.
Вариант осуществления 12: соединение в соответствии с Вариантом осуществления 11, в котором каждый R7 независимо выбран из брома, хлора и фтора.
Вариант осуществления 13: соединение в соответствии с Вариантом осуществления 12, в котором R7 представляет собой фтор.
Вариант осуществления 14: соединение в соответствии с Вариантом осуществления 6, в котором R3 не замещен группой R7.
Вариант осуществления 15: соединение в соответствии с Вариантом осуществления 3, в котором каждый R7 независимо выбран из NH2, циано, галогена и гидрокси.
Вариант осуществления 16: соединение в соответствии с Вариантом осуществления 15, в котором каждый R7 независимо выбран из циано и галогена.
Вариант осуществления 17: соединение в соответствии с Вариантом осуществления 16, в котором каждый R7 независимо выбран из брома, хлора и фтора.
Вариант осуществления 18: соединение в соответствии с Вариантом осуществления 17, в котором R7 представляет собой фтор.
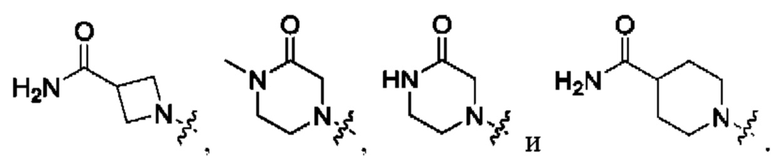
Вариант осуществления 19: соединение в соответствии с Вариантом осуществления 17, в котором R3 представляет собой 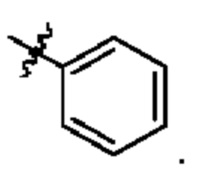
Вариант осуществления 20: соединение в соответствии с Вариантом осуществления 17, в котором R3, с замещением R7, где это приемлемо, представляет собой 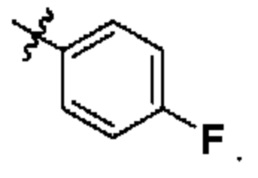
Вариант осуществления 21: соединение в соответствии с любым из Вариантов осуществления 1 и 3-20, в котором R2 выбран из алкила, циклоалкила, галогеналкила, гетероциклоалкила, арила, гетероарила, циклоалкилалкила, гетероциклоалкилалкила, арилалкила и гетероарилалкила, любой из которых необязательно замещен 1, 2 или 3 группами R6.
Вариант осуществления 22: соединение в соответствии с Вариантом осуществления 21, в котором R2 выбран из алкила, циклоалкила, галогеналкила, гетероциклоалкила, арила, гетероарила, циклоалкилметила, гетероциклоалкилметила, арилметила и гетероарилметила, любой из которых необязательно замещен 1, 2 или 3 группами R6.
Вариант осуществления 23: соединение в соответствии с Вариантом осуществления 22, в котором R2 выбран из циклоалкила, арила, гетероарила, арилметила и гетероарилметила, любой из которых необязательно замещен 1, 2 или 3 группами R6.
Вариант осуществления 24: соединение в соответствии с Вариантом осуществления 23, в котором R2 выбран из С3-6циклоалкила, фенила, пиридила, пиридазинила, пиримидинила, пиразинила, фенилметила, пиридилметила, пиридазинилметила, пиримидинилметила и пиразинилметила, любой из которых необязательно замещен 1, 2 или 3 группами R6.
Вариант осуществления 25: соединение в соответствии с любым из Вариантов осуществления 1 24, в котором R2 необязательно замещен 1 или 2 группами R6.
Вариант осуществления 26: соединение в соответствии с Вариантом осуществления 25, в котором R2 замещен 1 или 2 группами R6.
Вариант осуществления 27: соединение в соответствии с Вариантом осуществления 25, в котором R2 необязательно замещен 1 группой R6.
Вариант осуществления 28: соединение в соответствии с Вариантом осуществления 27, в котором R2 замещен 1 группой R6.
Вариант осуществления 29: соединение в соответствии с любым из Вариантов осуществления 1-28, в котором каждый R6 независимо выбран из галогена, алкила, циклоалкила, галогеналкила, галогеналкокси, арила, арилокси, гетероциклоалкила, гетероарила, циано, алкокси, COR8, SO2R8, NHSO2R8, NHSO2NHR8, SO2NR8R9, NHCOR8, NHCONHR8, CONHR8 и CONR8R9.
Вариант осуществления 30: соединение в соответствии с Вариантом осуществления 29, в котором каждый R6 независимо выбран из галогена, алкила, галогеналкокси, арила, гетероарила, циано, алкокси, SO2R8, NHSO2R8, SO2NR8R9, CONHR8 и CONR8R9.
Вариант осуществления 31: соединение в соответствии с Вариантом осуществления 30, в котором каждый R6 независимо выбран из фтора, хлора, фенила, пиридинила, пиридазинила, пиримидинила, пиразинила, пирролила, пиразолила, имидазолила, триазолила, циано, алкокси, SO2R8, SO2NR8R9, CONHR8 и CONR8R9.
Вариант осуществления 32: соединение в соответствии с любым из Вариантов осуществления 1-31, в котором каждый R8 и R9 независимо выбран из водорода и С1-4алкила.
Вариант осуществления 33: соединение в соответствии с Вариантом осуществления 32, в котором каждый R8 и R9 независимо выбран из водорода и метила.
Вариант осуществления 34: соединение в соответствии с Вариантом осуществления 27, в котором R2 не замещен группой R6.
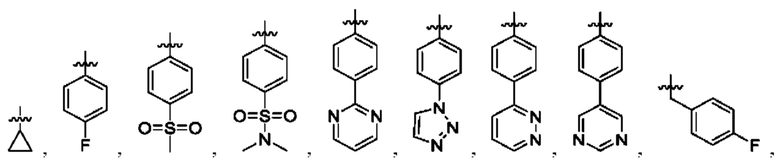
Вариант осуществления 35: соединение в соответствии с любым из Вариантов осуществления 1-18, в котором R2, с замещением R6, где это приемлемо, и дополнительно с замещениями R8 и R9, где это приемлемо, выбран из:
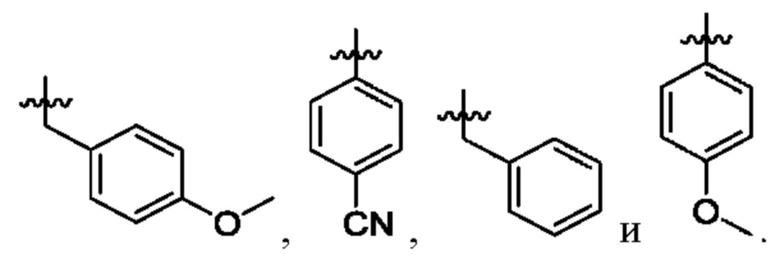
Вариант осуществления 36: соединение в соответствии с любым из Вариантов осуществления 1-18, в котором R2 представляет собой Н.
Вариант осуществления 37: соединение в соответствии с любым из Вариантов осуществления 1 36, в котором R1 представляет собой 5-7-членный азотсодержащий гетероциклоалкил или гетероарил, каждый из которых необязательно замещен 1, 2 или 3 группами R5.
Вариант осуществления 38: соединение в соответствии с Вариантом осуществления 37, в котором R1 представляет собой 5-7-членный гетероарил, который необязательно замещен 1, 2 или 3 группами R5.
Вариант осуществления 39: соединение в соответствии с Вариантом осуществления 38, в котором R1 выбран из пиридила, пиридазинила, пиримидинила, пиразинила, пирролила, пиразолила, имидазолила и триазолила, любой из которых необязательно замещен 1, 2 или 3 группами R5.
Вариант осуществления 40: соединение в соответствии с Вариантом осуществления 39, в котором R1 выбран из пиридила, пиримидинила и пиразолила, любой из которых необязательно замещен 1, 2 или 3 группами R5.
Вариант осуществления 41: соединение в соответствии с Вариантом осуществления 37, в котором R1 представляет собой 5-7-членный азотсодержащий гетероциклоалкил, который необязательно замещен 1, 2 или 3 группами R5.
Вариант осуществления 42: соединение в соответствии с Вариантом осуществления 41, в котором R1 выбран из пиперидина, морфолина, тиоморфолина, пиперазина, пирролидина, азетидина, 2-азаспиро[3,3]гептана, 2,6-диазаспиро[3,3]гептана и 2-окса-6-азаспиро[3,3]гептана, любой из которых необязательно замещен 1, 2 или 3 группами R5.
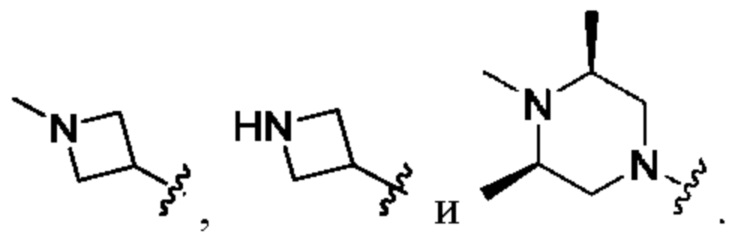
Вариант осуществления 43: соединение в соответствии с Вариантом осуществления 42, в котором R1, с замещением R5, где это приемлемо, и дополнительно с замещениями R8 и R9, где это приемлемо, выбран из 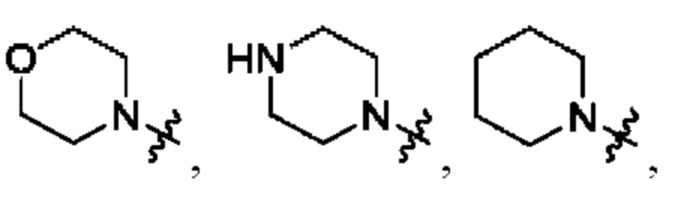
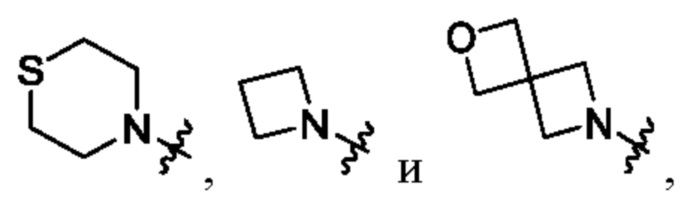 любой из которых необязательно замещен 1, 2 или 3 группами R5.
любой из которых необязательно замещен 1, 2 или 3 группами R5.
Вариант осуществления 44: соединение в соответствии с Вариантом осуществления 43, в котором R1 представляет собой 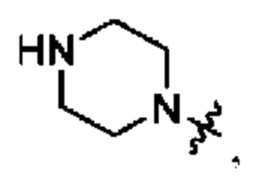 который необязательно замещен 1, 2 или 3 группами R5.
который необязательно замещен 1, 2 или 3 группами R5.
Вариант осуществления 45: соединение в соответствии с любым из Вариантов осуществления 1-44, в котором R1 необязательно замещен 1 или 2 группами R5.
Вариант осуществления 46: соединение в соответствии с Вариантом осуществления 45, в котором R1 замещен 1 или 2 группами R5.
Вариант осуществления 47: соединение в соответствии с Вариантом осуществления 45, в котором R1 необязательно замещен 1 группой R5.
Вариант осуществления 48: соединение в соответствии с любым из Вариантов осуществления 1-47, в котором каждый R5 независимо выбран из галогена, алкила, гидрокси, NH2, оксо, циано, COR8, CONR8R9, COOR8, NHCOR8, NHCONR8R9, SOR8, SO2R8, NHSO2R8 и SO2NR8R9.
Вариант осуществления 49: соединение в соответствии с Вариантом осуществления 48, в котором каждый R5 независимо выбран из С1-6алкила, гидрокси, NH2, оксо, циано, CONR8R9 и SO2R8.
Вариант осуществления 50: соединение в соответствии с Вариантом осуществления 49, в котором каждый R5 независимо выбран из CH3, оксо, CONH2 и SO2CH3.
Вариант осуществления 51: соединение в соответствии с Вариантом осуществления 50, в котором R5 представляет собой SO2CH3.
Вариант осуществления 52: соединение в соответствии с Вариантом осуществления 45, в котором R1 не замещен группой R5.
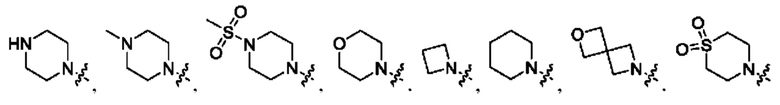
Вариант осуществления 53: соединение в соответствии с Вариантом осуществления 38, в котором R1 выбран из 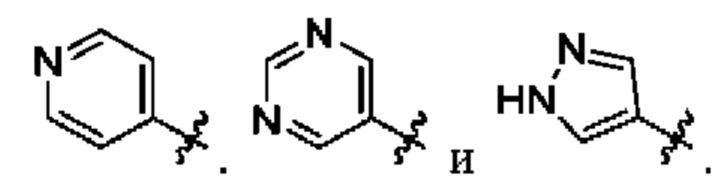
Вариант осуществления 54: соединение в соответствии с Вариантом осуществления 41, в котором R1, с замещением R5, где это приемлемо, и дополнительно с замещениями R8 и R9, где это приемлемо, выбран из:
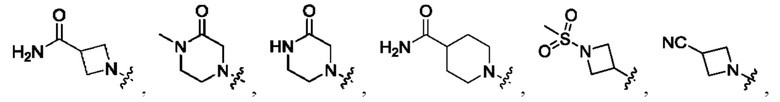
Вариант осуществления 55: соединение в соответствии с Вариантом осуществления 41, в котором R1, с замещением R5, где это приемлемо, и дополнительно с замещениями R8 и R9, где это приемлемо, выбран из:
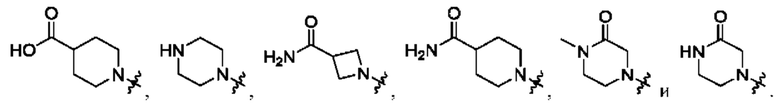
Вариант осуществления 56: соединение в соответствии с Вариантом осуществления 41, в котором R1, с замещением R5, где это приемлемо, и дополнительно с замещениями R8, где это приемлемо, выбран из:
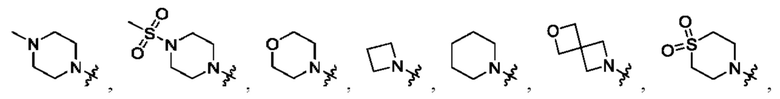
Вариант осуществления 57: соединение в соответствии с Вариантом осуществления 41, в котором R1, с замещением R5, где это приемлемо, и дополнительно с замещениями R8 и R9, где это приемлемо, выбран из:
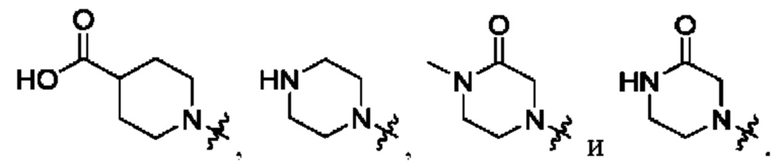
Вариант осуществления 58: соединение в соответствии с Вариантом осуществления 41, в котором R1, с замещением R5, где это приемлемо, и дополнительно с замещениями R8 и R9, где это приемлемо, выбран из:
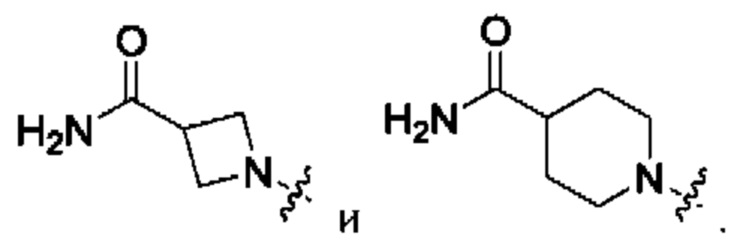
Вариант осуществления 59: соединение в соответствии с любым из Вариантов осуществления 1-58, в котором m выбран из 1, 2, 3 и 4.
Вариант осуществления 60: соединение в соответствии с Вариантом осуществления 59, в котором m выбран из 2 и 3.
Вариант осуществления 61: соединение в соответствии с Вариантом осуществления 60, в котором m означает 2.
Вариант осуществления 62: соединение в соответствии с Вариантом осуществления 60, в котором m означает 3.
Также представлены варианты осуществления, в которых любой вариант осуществления, приведенный выше, может быть объединен с любым одним или несколькими из этих вариантов осуществления, при условии, что комбинация не является взаимоисключающей.
Как используется в данной заявке, два варианта осуществления являются "взаимоисключающими", когда один определяется как нечто отличное от другого. Например, вариант осуществления, в котором две группы объединяются с образованием циклоалкила, является взаимоисключающим с вариантом осуществления, в котором одна группа представляет собой этил, а другая группа представляет собой водород. Аналогичным образом вариант осуществления, в котором одна группа представляет собой CH2, является взаимоисключающим с вариантом осуществления, в котором такая же группа представляет собой NH.
Также обеспечивается соединение, выбранное из Примеров, раскрытых в данной заявке.
Настоящее изобретение также относится к способу ингибирования, по меньшей мере, одной функции KDM1A, включающему стадию приведения KDM1A в контакт с соединением, описанным в данной заявке. Можно отслеживать фенотип клеток, пролиферацию клеток, активность KDM1A, изменение биохимического выхода, продуцируемого активной KDM1A, экспрессию KDM1A или связывание KDM1A с естественным партнером по связыванию. Такие способы могут представлять собой способы лечения заболевания, биологические анализы, клеточные анализы, биохимические анализы и подобные.
В данной заявке также представлен способ лечения заболевания, опосредованного KDM1A, включающий введение терапевтически эффективного количества соединения, раскрытого в данной заявке, или его соли пациенту, нуждающемуся в этом.
В определенных вариантах осуществления, заболеванием является рак.
В определенных вариантах осуществления, рак выбран из саркомы Юинга, множественной миеломы, Т-клеточного лейкоза, опухоли Вильма, мелкоклеточного рака легкого, рака мочевого пузыря, рака предстательной железы, рака молочной железы, рака головы и шеи, рака толстой кишки и рака яичников.
Другие расстройства или состояния, которые преимущественно лечатся описанными в данной заявке соединениями, включают профилактику или лечение гиперпролиферативных заболеваний, особенно рака, либо отдельно, либо в сочетании со стандартами лечения, особенно с теми агентами, которые нацелены на рост опухоли путем восстановления генов-супрессоров опухоли в злокачественных клетках. Гематологические и негематологические злокачественные новообразования, которые можно лечить или предотвращать, включают, но не ограничиваются, множественную миелому, острый и хронический лейкозы и гематопоэтические пролиферативные и неопластические заболевания, включая миелодиспластический синдром (MDS), острый миелогенный лейкоз (AML), острый лимфоцитарный лейкоз (ALL), хронический лимфоцитарный лейкоз (CLL) и хронический миелоидный лейкоз (CML), лимфомы, включая лимфому Ходжкина и неходжкинскую лимфому (низкой, средней и высокой степени), а также солидные опухоли и злокачественные новообразования головного мозга, головы и шеи, молочной железы, легкого (включая не мелкоклеточный рак легкого), половых путей, верхних отделов желудочно-кишечного тракта, поджелудочной железы, печени, почек, мочевого пузыря, предстательной железы и толстой кишки. Настоящие соединения и способы также можно использовать для лечения фиброза, такого как фиброз, возникающий при лучевой терапии. Настоящие соединения и способы можно использовать для лечения субъектов, имеющих или у которых нужно предотвратить прогрессирование аденоматозных полипов, в том числе субъектов с семейным аденоматозным полипозом (FAP) или саркоидозом. К незлокачественным пролиферативным заболеваниям дополнительно относятся псориаз, экзема и дерматит.
В определенных вариантах осуществления, заболеванием является миелоидное заболевание.
В определенных вариантах осуществления, миелоидное заболевание выбрано из миелофиброза, истинной полицитемии, эссенциальной тромбоцитемии, миелодиспластического синдрома (MDS), острого миелогенного лейкоза (AML) и хронического миелоидного лейкоза (CML).
В определенных вариантах осуществления, миелоидное заболевание выбрано из группы, включающей истинную полицитемию (PV), эссенциальную тромбоцитемию (ET), миелофиброз (MF), хронический миелоидный лейкоз (CML), хронический нейтрофильный лейкоз (CNL) и хронический эозинофильный лейкоз (CEL). В определенных вариантах осуществления, миелоидное заболевание выбрано из группы, включающей истинную полицитемию (PV), эссенциальную тромбоцитемию (ET) и миелофиброз (MF). В определенных вариантах осуществления, миелоидным заболеванием является миелофиброз, выбранный из первичного миелофиброза (PMF) и пост-PV/ET миелофиброз. В определенных вариантах осуществления, миелоидным заболеванием является первичный миелофиброз (PMF). В определенных вариантах осуществления, миелоидным заболеванием является пост-PV/ET миелофиброз. В определенных вариантах осуществления, миелоидным заболеванием является эссенциальная тромбоцитемия. В определенных вариантах осуществления, миелоидным заболеванием является истинная полицитемия. В определенных вариантах осуществления, миелоидным заболеванием является хронический миелоидный лейкоз. В определенных вариантах осуществления, миелоидным заболеванием является хронический нейтрофильный лейкоз. В определенных вариантах осуществления, миелоидным заболеванием является хронический эозинофильный лейкоз. В определенных вариантах осуществления, пациентом является человек.
В определенных вариантах осуществления, заболеванием является воспалительное заболевание.
В определенных вариантах осуществления, воспалительное заболевание выбрано из воспалительного заболевания кишечника, ревматоидного артрита или системной красной волчанки.
В настоящем изобретении также предложено соединение, раскрытое в данной заявке, для применения в качестве лекарственного средства.
В настоящем изобретении также предложено соединение, раскрытое в данной заявке, для применения в качестве лекарственного средства для лечения заболевания, опосредованного KDM1A.
Также предложено применение соединения, раскрытого в данной заявке, в качестве лекарственного средства.
Также предложено применение соединения, раскрытого в данной заявке, в качестве лекарственного средства для лечения заболевания, опосредованного KDM1A.
Также предложено соединение, раскрытое в данной заявке, для применения в производстве лекарственного средства для лечения заболевания, опосредованного KDM1A
Также предложено применение соединения, раскрытого в данной заявке, для лечения заболевания, опосредованного KDM1A.
В настоящем изобретении также предложен способ ингибирования KDM1A, включающий приведение KDM1A в контакт с соединением, раскрытым в данной заявке, или его солью.
В настоящем изобретении также предложен способ достижения эффекта у пациента, включающий введение пациенту терапевтически эффективного количества соединения, раскрытого в данной заявке, или его соли, причем эффект выбран из улучшения когнитивных функций.
Также предложен способ модуляции функции, опосредованной KDM1A, у субъекта, включающий введение терапевтически эффективного количества соединения, раскрытого в данной заявке.
Также обеспечена фармацевтическая композиция, содержащая соединение, раскрытое в данной заявке, вместе с фармацевтически приемлемым носителем.
В определенных вариантах осуществления, фармацевтическая композиция разработана для перорального введения.
В определенных вариантах осуществления, фармацевтическая композиция для перорального введения выбрана из таблетки и капсулы.
Термины
Используемые в данной заявке термины имеют указанные значения.
Когда приведены диапазоны значений и используется обозначение "от n1… до n2" или "между n1… и n2", где n1 и n2 означают числа, то, если не указано иное, данное обозначение предназначено для включения самих чисел и диапазон между ними. Этот диапазон может быть целым или непрерывным между конечными значениями и включать конечные значения. В качестве примера предполагается, что диапазон "от 2 до 6 атомов углерода" включает два, три, четыре, пять и шесть атомов углерода, поскольку атомы углерода представлены в целых единицах. Для сравнения, в качестве примера, диапазон "от 1 до 3 мкМ (микромолярный)" должен включать 1 мкМ, 3 мкМ и все, что между ними, с любым количеством значащих цифр (например, 1,255 мкМ, 2,1 мкМ, 2,9999 мкМ и т.д.).
Термин "примерно", как используется в данной заявке, предназначен для определения числовых значений, которые он модифицирует, обозначая такое значение как переменную в пределах погрешности. Когда не указывается какой-либо конкретный предел погрешности, такой как стандартное отклонение от среднего значения, указанного в диаграмме или таблице данных, термин "примерно" следует понимать как обозначающий диапазон, который будет охватывать указанное значение и диапазон, который будет также включен путем округления в большую или меньшую сторону до этой цифры с учетом значащих цифр.
Термин "ацил", как используется в данной заявке, отдельно или в комбинации, относится к карбонилу, присоединенному к алкенилу, алкилу, арилу, циклоалкилу, гетероарилу, гетероциклу или любому другому фрагменту, в котором атом, присоединенный к карбонилу, представляет собой углерод. "Ацетильная" группа относится к группе С(O)СН3. "Алкилкарбонильная" или "алканоильная" группа относится к алкильной группе, присоединенной к фрагменту исходной молекулы через карбонильную группу. Примеры таких групп включают метилкарбонил и этилкарбонил. Примеры ацильных групп включают формил, алканоил и ароил.
Термин "алкенил", как используется в данной заявке, отдельно или в комбинации, относится к углеводородному радикалу с линейной или разветвленной цепью, имеющему одну или несколько двойных связей и содержащему от 2 до 20 атомов углерода. В определенных вариантах осуществления, указанный алкенил будет содержать от 2 до 6 атомов углерода. Термин "алкенилен" относится к системе двойных связей углерод-углерод, присоединенной в двух или более положениях, такой как этенилен [(-СН=СН-), (-С::С-)]. Примеры приемлемых алкенильных радикалов включают этенил, пропенил, 2-метилпропенил, 1,4-бутадиенил и подобные. Если не указано иное, термин "алкенил" может включать "алкениленовые" группы.
Термин "алкокси", как используется в данной заявке, отдельно или в комбинации, относится к алкилэфирному радикалу, в котором термин алкил имеет значения, указанные ниже. Примеры приемлемых алкилэфирных радикалов включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изо-бутокси, втор-бутокси, трет-бутокси и подобные.
Термин "алкил", как используется в данной заявке, отдельно или в комбинации, относится к алкильному радикалу с линейной или разветвленной цепью, содержащему от 1 до 20 атомов углерода. В определенных вариантах осуществления указанный алкил будет содержать от 1 до 10 атомов углерода. В дополнительных вариантах осуществления указанный алкил будет содержать от 1 до 8 атомов углерода. Алкильные группы могут быть необязательно замещены, как определено в данной заявке. Примеры алкильных радикалов включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, гексил, октил, нонил и подобные. Термин "алкилен", как используется в данной заявке, отдельно или в комбинации, относится к насыщенной алифатической группе, полученной из насыщенного углеводорода с линейной или разветвленной цепью, присоединенного в двух или более положениях, такой как метилен (-CH2-). Если не указано иное, термин "алкил" может включать "алкиленовые" группы.
Термин "алкиламино", как используется в данной заявке, отдельно или в комбинации, относится к алкильной группе, присоединенной к фрагменту исходной молекулы через амино группу. Приемлемые алкиламино группы могут быть моно- или диалкилированными, образуя группы, такие как, например, N-метиламино, N-этиламино, N,N-диметиламино, N,N-этилметиламино и подобные.
Термин "алкилиден", как используется в данной заявке, отдельно или в комбинации, относится к алкенильной группе, в которой один атом углерода двойной связи углерод-углерод принадлежит фрагменту, к которому присоединена алкенильная группа.
Термин "алкилтио", как используется в данной заявке, отдельно или в комбинации, относится к алкилтиоэфирному (R-S-) радикалу, в котором термин алкил имеет значения, указанные выше, а сера может быть однократно или двукратно окислена. Примеры приемлемых алкилтиоэфирных радикалов включают метилтио, этилтио, н-пропилтио, изопропилтио, н-бутилтио, изо-бутилтио, втор-бутилтио, трет-бутилтио, метансульфонил, этансульфинил и подобные.
Термин "алкинил", как используется в данной заявке, отдельно или в комбинации, относится к углеводородному радикалу с линейной или разветвленной цепью, имеющему одну или несколько тройных связей и содержащему от 2 до 20 атомов углерода. В определенных вариантах осуществления, указанный алкинил содержит от 2 до 6 атомов углерода. В дополнительных вариантах осуществления, указанный алкинил содержит от 2 до 4 атомов углерода. Термин "алкинилен" относится к тройной связи углерод-углерод, присоединенной в двух положениях, такой как этинилен (-С:::С-, -C=C-). Примеры алкинильных радикалов включают этинил, пропинил, гидроксипропинил, бутин-1-ил, бутин-2-ил, пентин-1-ил, 3-метилбутин-1-ил, гексин-2-ил и подобные. Если не указано иное, термин "алкинил" может включать "алкиниленовые"группы.
Термины "амидо" и "карбамоил", как используются в данной заявке, отдельно или в комбинации, относятся к амино группе, как описано ниже, присоединенной к фрагменту исходной молекулы через карбонильную группу, или наоборот. Термин "С амидо", как используется в данной заявке, отдельно или в комбинации, относится к группе -C(O)N(RR'), в которой R и R' являются такими, как определено в данной заявке, или как определено специально перечисленными группами, обозначенными "R". Термин "N-амидо", как используется в данной заявке, отдельно или в комбинации, относится к группе RC(O)N(R')-, в которой R и R' являются такими, как определено в данной заявке, или как определено специально перечисленными группами, обозначенными "R". Термин "ациламино", как используется в данной заявке, отдельно или в комбинации, включает ацильную группу, присоединенную к исходному фрагменту через амино группу. Примером "ациламино" группы является ацетиламино (CH3C(O)NH-).
Термин "амино", как используется в данной заявке, отдельно или в комбинации, относится к -NRR', где R и R' независимо выбраны из водорода, алкила, ацила, гетероалкила, арила, циклоалкила, гетероарила и гетероциклоалкила, любой из которых сам может быть необязательно замещен. Дополнительно, R и R' могут быть объединены с образованием гетероциклоалкила, каждый из которых может быть необязательно замещен.
Термин "арил", как используется в данной заявке, отдельно или в комбинации, обозначает карбоциклическую ароматическую систему, содержащую одно, два или три кольца, при этом такие полициклические кольцевые системы конденсированы вместе. Термин "арил" охватывает ароматические группы, такие как фенил, нафтил, антраценил и фенантрил.
Термин "арилалкенил" или "аралкенил", как используется в данной заявке, отдельно или в комбинации, относится к арильной группе, присоединенной к фрагменту исходной молекулы через алкенильную группу.
Термин "арилалкокси" или "аралкокси", как используется в данной заявке, отдельно или в комбинации, относится к арильной группе, присоединенной к фрагменту исходной молекулы через алкокси группу.
Термин "арилалкил" или "аралкил", как используется в данной заявке, отдельно или в комбинации, относится к арильной группе, присоединенной к фрагменту исходной молекулы через алкильную группу.
Термин "арилалкинил" или "аралкинил", как используется в данной заявке, отдельно или в комбинации, относится к арильной группе, присоединенной к фрагменту исходной молекулы через алкинильную группу.
Термин "арилалканоил" или "аралканоил" или "ароил", как используется в данной заявке, отдельно или в комбинации, относится к ацильному радикалу, полученному из арил-замещенной алканкарбоновой кислоты, такому как бензоил, нафтоил, фенилацетил, 3-фенилпропионил (гидроциннамоил), 4-фенилбутирил, (2-нафтил)ацетил, 4-хлоргидроциннамоил и подобные.
Термин "арилокси", как используется в данной заявке, отдельно или в комбинации, относится к арильной группе, присоединенной к фрагменту исходной молекулы через окси.
Термины "бензо" и "бенз", как используются в данной заявке, отдельно или в комбинации, относятся к бивалентному радикалу C6H4=, полученному из бензола. Примеры включают бензотиофен и бензимидазол.
Термин "карбамат", как используется в данной заявке, отдельно или в комбинации, относится к сложному эфиру карбаминовой кислоты (-NHCOO-), который может быть присоединен к фрагменту исходной молекулы либо с азотного конца, либо с кислотного конца, и который может быть необязательно замещен, как определено в данной заявке.
Термин "О-карбамил", как используется в данной заявке, отдельно или в комбинации, относится к группе -OC(O)NRR', в которой R и R' являются такими, как определено в данной заявке.
Термин "N-карбамил", как используется в данной заявке, отдельно или в комбинации, относится к группе ROC(O)NR'-, в которой R и R' являются такими, как определено в данной заявке.
Термин "карбонил", как используется в данной заявке, если отдельно, то включает формил [-C(O)H], а в комбинации представляет собой группу -С(О)-.
Термин "карбоксил" или "карбокси", как используется в данной заявке, относится к -C(O)OH или соответствующему "карбоксилатному" аниону, такому как в соли карбоновой кислоты. Группа "О-карбокси" относится к группе RC(O)O-, где R является таким, как определено в данной заявке. Группа "С-карбокси" относится к группе -C(O)OR, где R является таким, как определено в данной заявке.
Термин "циано", как используется в данной заявке, отдельно или в комбинации, относится к -CN.
Термин "циклоалкил" или, альтернативно, "карбоцикл", как используется в данной заявке, отдельно или в комбинации, относится к насыщенной или частично насыщенной моноциклической, бициклической или трициклической алкильной группе, в которой каждый циклический фрагмент содержит от 3 до 12 атомов углерода в кольце, и которая необязательно может быть бензоконденсированной кольцевой системой, которая необязательно замещена, как определено в данной заявке. В определенных вариантах осуществления, указанный циклоалкил будет содержать от 5 до 7 атомов углерода. Примеры таких циклоалкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, тетрагидронафтил, инданил, октагидронафтил, 2,3-дигидро-1H-инденил, адамантил и подобные. "Бициклический" и "трициклический", как используется в данной заявке, предназначены для включения обеих конденсированных кольцевых систем, таких как декагидронафталин, октагидронафталин, а также полициклического (многоцентрового) насыщенного или частично ненасыщенного типа. Последний тип изомера в общем представлен бицикло[1,1,1]пентаном, камфорой, адамантаном и бицикло[3,2,1]октаном.
Термин "сложный эфир", как используется в данной заявке, отдельно или в комбинации, относится к карбокси группе, соединяющей как мостик два фрагмента, связанные с атомами углерода.
Термин "простой эфир", как используется в данной заявке, отдельно или в комбинации, относится к окси группе, соединяющей как мостик два фрагмента, связанные с атомами углерода.
Термин "гало" или "галоген", как используется в данной заявке, отдельно или в комбинации, относится к фтору, хлору, брому или йоду.
Термин "галогеналкокси", как используется в данной заявке, отдельно или в комбинации, относится к галогеналкильной группе, присоединенной к фрагменту исходной молекулы через атом кислорода.
Термин "галогеналкил", как используется в данной заявке, отдельно или в комбинации, относится к алкильному радикалу, имеющему значение, как определено выше, в котором один или несколько атомов водорода заменены галогеном. В частности, включены моногалогеналкильные, дигалогеналкильные и полигалогеналкильные радикалы. Моногалогеналкильный радикал, например, может иметь атом йода, брома, хлора или фтора в радикале. Дигалоген- и полигалогеналкильные радикалы могут содержать два или более одинаковых атома галогена или комбинацию различных галогеновых радикалов. Примеры галогеналкильных радикалов включают фторметил, дифторметил, трифторметил, хлорметил, дихлорметил, трихлорметил, пентафторэтил, гептафторпропил, дифторхлорметил, дихлорфторметил, дифторэтил, дифторпропил, дихлорэтил и дихлорпропил. "Галогеналкилен" относится к галогеналкильной группе, присоединенной в двух или более положениях. Примеры включают фторметилен (-CFH-), дифторметилен (-CF2-), хлорметилен (-CHCl-) и подобные.
Термин "гетероалкил", как используется в данной заявке, отдельно или в комбинации, относится к стабильной линейной или разветвленной цепи или их комбинациям, полностью насыщенным или содержащим от 1 до 3 степеней ненасыщенности, состоящим из указанного числа атомов углерода и от одного до трех гетероатомов, выбранных из N, О и S, и при этом атомы N и S необязательно могут быть окислены, а гетероатом N необязательно может быть кватернизован. Гетероатом(ы) может быть размещен в любом внутреннем положении гетероалкильной группы. До двух гетероатомов могут быть последовательными, как например -CH2-NH-OCH3.
Термин "гетероарил", как используется в данной заявке, отдельно или в комбинации, относится к 3-15-членному ненасыщенному гетеромоноциклическому кольцу или конденсированной моноциклической, бициклической или трициклической кольцевой системе, в которой, по меньшей мере, одно из конденсированных колец является ароматическим, которая содержит, по меньшей мере, один атом, выбранный из N, О и S. В определенных вариантах осуществления, указанный гетероарил будет содержать от 1 до 4 гетероатомов в качестве кольцевых членов. В дополнительных вариантах осуществления, указанный гетероарил будет содержать от 1 до 2 гетероатомов в качестве кольцевых членов. В определенных вариантах осуществления указанный гетероарил будет содержать от 5 до 7 атомов. Термин также включает конденсированные полициклические группы, в которых гетероциклические кольца конденсированы с арильными кольцами, в которых гетероарильные кольца конденсированы с другими гетероарильными кольцами, в которых гетероарильные кольца конденсированы с гетероциклоалкильными кольцами, или в которых гетероарильные кольца конденсированы с циклоалкильными кольцами. Примеры гетероарильных групп включают пирролил, пирролинил, имидазолил, пиразолил, пиридил, пиримидинил, пиразинил, пиридазинил, триазолил, пиранил, фурил, тиенил, оксазолил, изоксазолил, оксадиазолил, тиазолил, тиадиазолил, изотиазолил, индолил, изоиндолил, индолизинил, бензимидазолил, хинолил, изохинолил, хиноксалинил, хиназолинил, индазолил, бензотриазолил, бензодиоксолил, бензопиранил, бензоксазолил, бензоксадиазолил, беизотиазолил, бензотиадиазолил, бензофурил, бензотиенил, хромонил, кумаринил, бензопиранил, тетрагидрохинолинил, тетразолопиридазинил, тетрагидроизохинолинил, тиенопиридинил, фуропиридинил, пирролопиридинил и подобные. Примеры трициклических гетероциклических групп включают карбазолил, бензидолил, фенантролинил, дибензофуранил, акридинил, фенантридинил, ксантенили подобные.
Термины "гетероциклоалкил" и, взаимозаменяемо, "гетероцикл", как используется в данной заявке, отдельно или в комбинации, каждый относится к насыщенной, частично ненасыщенной или полностью ненасыщенной (но не ароматической) моноциклической, бициклической или трициклической гетероциклической группе, содержащей, по меньшей мере, один гетероатом в качестве кольцевого члена, при этом каждый указанный гетероатом может быть независимо выбран из азота, кислорода и серы. В определенных вариантах осуществления, указанный гетероциклоалкил будет содержать от 1 до 4 гетероатомов в качестве кольцевых членов. В дополнительных вариантах осуществления, указанный гетероциклоалкил будет содержать от 1 до 2 гетероатомов в качестве кольцевых членов. В некоторых вариантах осуществления, указанный гетероциклоалкил будет содержать от 3 до 8 кольцевых членов в каждом кольце. В дополнительных вариантах осуществления, указанный гетероциклоалкил будет содержать от 3 до 7 кольцевых членов в каждом кольце. В других вариантах осуществления, указанный гетероциклоалкил будет содержать от 5 до 6 кольцевых членов в каждом кольце. "Гетероциклоалкил" и "гетероцикл" предназначены для включения сульфонов, сульфоксидов, N-оксидов третичных азотных кольцевых членов и карбоциклических конденсированных и бензоконденсированных кольцевых систем; кроме того, оба термина также включают системы, в которых гетероциклическое кольцо конденсировано с арильной группой, как определено в данной заявке, или дополнительной гетероциклической группой. Примеры гетероциклических групп включают азиридинил, азетидинил, 1,3-бензодиоксолил, дигидроизоиндолил, дигидроизохинолинил, дигидроциннолинил, дигидробензодиоксинил, дигидро[1,3]оксазоло[4,5-b]пиридинил, бензотиазолил, дигидроиндолил, дигидропиридинил, 1,3-диоксанил, 1,4-диоксанил, 1,3-диоксоланил, изоиндолинил, морфолинил, пиперазинил, пирролидинил, тетрагидропиридинил, пиперидинил, тиоморфолинил и подобные. Гетероциклические группы могут быть необязательно замещены, если специально не запрещено.
Термин "гидразинил", как используется в данной заявке, отдельно или в комбинации, относится к двум амино группам, соединенным одинарной связью, т.е. -N-N-.
Термин "гидрокси", как используется в данной заявке, отдельно или в комбинации, относится к -ОН.
Термин "гидроксиалкил", как используется в данной заявке, отдельно или в комбинации, относится к гидрокси группе, присоединенной к фрагменту исходной молекулы через алкильную группу.
Термин "имино", как используется в данной заявке, отдельно или в комбинации, относится к=N-.
Термин "иминогидрокси", как используется в данной заявке, отдельно или в комбинации, относится к=N(OH) и=N-O-.
Фраза "в главной цепи" относится к самой длинной непрерывной или смежной цепочке атомов углерода, начинающейся в точке присоединения группы к соединениям любой из формул, раскрытых в данной заявке.
Термин "изоцианато" относится к группе -NCO.
Термин "изотиоцианато" относится к группе -NCS.
Фраза "линейная цепь атомов" относится к самой длинной прямой цепи атомов, независимо выбранных из углерода, азота, кислорода и серы.
Термин "низший", как используется в данной заявке, отдельно или в комбинации, если специально не указано иное, означает содержание от 1 до, и включая, 6 атомов углерода (т.е. C1-C6алкил).
Термин "низший арил", как используется в данной заявке, отдельно или в комбинации, означает фенил или нафтил, каждый из которых может быть необязательно замещен, как предусмотрено.
Термин "низший гетероарил", как используется в данной заявке, отдельно или в комбинации, означает либо 1) моноциклический гетероарил, содержащий пять или шесть кольцевых членов, в котором от одного до четырех указанных членов могут быть гетероатомы, выбранные из N, О и S, либо 2) бициклический гетероарил, в котором каждое из конденсированных колец содержит пять или шесть кольцевых членов, содержащих от одного до четырех гетероатомов, выбранных из N, О и S.
Термин "низший циклоалкил", как используется в данной заявке, отдельно или в комбинации, означает моноциклический циклоалкил, содержащий от трех до шести кольцевых членов (т.е. С3-С6 циклоалкил). Низшие циклоалкилы могут быть ненасыщенными. Примеры низшего циклоалкила включают циклопропил, циклобутил, циклопентил и циклогексил.
Термин "низший гетероциклоалкил", как используется в данной заявке, отдельно или в комбинации, означает моноциклический гетероциклоалкил, содержащий от трех до шести кольцевых членов, из которых от одного до четырех могут быть гетероатомы, выбранные из N, О и S (т.е. С3-C6 гетероциклоалкил). Примеры низших гетероциклоалкилов включают пирролидинил, имидазолидинил, пиразолидинил, пиперидинил, пиперазинил и морфолинил. Низшие гетероциклоалкилы могут быть ненасыщенными.
Термин "низший амино", как используется в данной заявке, отдельно или в комбинации, относится к -NRR', где R и R' независимо выбраны из водорода и низшего алкила, каждый из которых может быть необязательно замещен.
Термин "меркаптил", как используется в данной заявке, отдельно или в комбинации, относится к группе RS-, в которой R является таким, как определено в данной заявке.
Термин "нитро", как используется в данной заявке, отдельно или в комбинации, относится к -NO2.
Термины "окси" или "окса", как используется в данной заявке, отдельно или в комбинации, относится к -О-.
Термин "оксо", как используется в данной заявке, отдельно или в комбинации, относится к=O.
Термин "пергалогеналкокси" относится к алкокси группе, в которой все атомы водорода заменены атомами галогена.
Термин "пергалогеналкил", как используется в данной заявке, отдельно или в комбинации, относится к алкильной группе, в которой все атомы водорода заменены атомами галогена.
Термины "сульфонат", "сульфоновая кислота" и "сульфоновый", как используется в данной заявке, отдельно или в комбинации, относится к группе SO3H и ее анион как сульфоновая кислота используется в образовании соли.
Термин "сульфанил", как используется в данной заявке, отдельно или в комбинации, относится к -S-.
Термин "сульфинил", как используется в данной заявке, отдельно или в комбинации, относится к S(O).
Термин "сульфонил", как используется в данной заявке, отдельно или в комбинации, относится к -S(O)2-.
Термин "N-сульфонамидо" относится к группе RS(=O)2NR'-, в которой R и R' являются такими, как определено в данной заявке.
Термин "S-сульфонамидо" относится к группе -S(=O)2NRR', в которой R и R' являются такими, как определено в данной заявке.
Термины "тиа" и "тио", как используется в данной заявке, отдельно или в комбинации, относятся к группе -S- или простому эфиру, в котором атом кислорода заменен атомом серы. Окисленные производные тио группы, а именно сульфинил и сульфонил, включены в определение тиа и тио.
Термин "тиол", как используется в данной заявке, отдельно или в комбинации, относится к группе -SH.
Термин "тиокарбонил", как используется в данной заявке, если отдельно, включает тиоформил C(S)H, а в комбинации означает группу -C(S)-.
Термин "N-тиокарбамил" относится к группе ROC(S)NR', в которой R и R' являются такими, как определено в данной заявке
Термин "О-тиокарбамил" относится к группе -OC(S)NRR', в которой R и R' являются такими, как определено в данной заявке.
Термин "тиоцианато" относится к группе CNS.
Термин "тригалогенметансульфонамидо" относится к группе X3CS(O)2NR-, в которой X представляет собой галоген, и R является таким, как определено в данной заявке.
Термин "тригалогенметансульфонил" относится к группе X3CS(O)2- в которой X представляет собой галоген.
Термин "тригалогенметокси" относится к группе Х3СО, в которой X представляет собой галоген.
Термин "тризамещенный силил", как используется в данной заявке, отдельно или в комбинации, относится к силоксановой группе, замещенной в трех ее свободных валентностях группами, как указано в данной заявке под определением замещенного амино. Примеры включают триметилсилил, трет-бутилдиметилсилил, трифенилсилил и подобные.
Любое определение в данной заявке может использоваться в сочетании с любым другим определением для описания составной структурной группы. Как принято, завершающим элементом любого такого определения является элемент, который присоединяется к исходному фрагменту. Например, составная группа алкиламидо будет представлять собой алкильную группу, присоединенную к исходной молекуле через амидо группу, а термин алкоксиалкил будет представлять собой алкокси группу, присоединенную к исходной молекуле через алкильную группу.
Когда группа определяется как "нулевая", имеется в виду, что указанная группа отсутствует.
Термин "необязательно замещенный" означает, что предшествующая группа может быть замещенной или незамещенной. В случае замещения заместители "необязательно замещенной" группы могут включать, без ограничения, один или несколько заместителей, независимо выбранных из следующих групп или конкретного обозначенного набора групп, отдельно или в комбинациях: низший алкил, низший алкенил, низший алкинил, низший алканоил, низший гетероалкил, низший гетероциклоалкил, низший галогеналкил, низший галогеналкенил, низший галогеналкинил, низший пергалогеналкил, низший пергалогеналкокси, низший циклоалкил, фенил, арил, арилокси, низший алкокси, низший галогеналкокси, оксо, низший ацилокси, карбонил, карбоксил, низший алкилкарбонил, низший карбоксиэфир, низший карбоксамидо, циано, водород, галоген, гидрокси, амино, низший алкиламино, ариламино, амидо, нитро, тиол, низший алкилтио, низший галогеналкилтио, низший пергалогеналкилтио, арилтио, сульфонат, сульфоновая кислота, тризамещенный силил, N3, SH, SCH3, С(O)СН3, СО2СН3, CO2H, пиридинил, тиофен, фуранил, низший карбамат и низшая мочевина. Если структурно возможно, два заместителя могут быть соединены вместе с образованием конденсированного пяти-, шести- или семичленного карбоциклического или гетероциклического кольца, содержащего от нуля до трех гетероатомов, например с образованием метилендиокси или этилендиокси. Необязательно замещенная группа может быть незамещенной (например, -СН2СН3), полностью замещенной (например, -CF2CF3), монозамещенной (например, -CH2CH2F) или замещенной на любом промежуточном уровне между полностью замещенной и монозамещенной (например, -CH2CF3). Если заместители указаны без уточнения в отношении замещения, охватываются как замещенные, так и незамещенные формы. Если заместитель квалифицируется как "замещенный", то конкретно подразумевается замещенная форма. Кроме того, при необходимости могут быть определены различные наборы необязательных заместителей для конкретного фрагмента; в этих случаях необязательное замещение будет таким, как определено, часто сразу после фразы "необязательно замещен".
Термин R или термин R', появляющийся сам по себе и без обозначения номера, если не указано иное, относится к фрагменту, выбранному из водорода, алкила, циклоалкила, гетероалкила, арила, гетероарила и гетероциклоалкила, любой из которых может быть необязательно замещен. Следует понимать, что такие группы R и R' могут быть необязательно замещенными, как определено в данной заявке. Независимо от того, имеет ли группа R числовое обозначение или нет, каждая группа R, включая R, R' и Rn, где n=(1, 2, 3, …n), каждый заместитель и каждый термин следует понимать как независимые от всех остальных при выборе из группы. Если какая-либо переменная, заместитель или термин (например, арил, гетероцикл, R и т.д.) встречаются более одного раза в формуле или общей структуре, их определение в каждом случае не зависит от определения во всех других случаях. Специалисты в данной области техники также поймут, что определенные группы могут быть присоединены к исходной молекуле или могут занимать положение в цепи элементов с любого конца, как написано. Например, несимметричная группа, такая как -C(O)N(R)-, может быть присоединена к исходному фрагменту либо у углерода, либо у азота.
В соединениях, раскрытых в данной заявке, существуют асимметричные центры. Эти центры обозначаются символами "R" или "S" в зависимости от конфигурации заместителей вокруг хирального атома углерода. Следует понимать, что изобретение охватывает все стереохимические изомерные формы, включая диастереомерные, энантиомерные и эпимерные формы, а также d-изомеры и 1-изомеры и их смеси. Индивидуальные стереоизомеры соединений могут быть получены синтетически из коммерчески доступных исходных веществ, которые содержат хиральные центры, или путем получения смесей энантиомерных продуктов с последующим разделением, таким как преобразование в смесь диастереомеров с последующим разделением или перекристаллизацией, хроматографическими методами, прямым разделением энантиомеров на хиральных хроматографических колонках или любым другим подходящим методом, известным из уровня техники. Исходные соединения определенной стереохимии являются либо коммерчески доступными, либо могут быть получены и разделены методами, известными из уровня техники. Кроме того, соединения, раскрытые в данной заявке, могут существовать в виде геометрических изомеров. Настоящее изобретение включает все цис-, транс-, син-, анти-изомеры, entgegen (E) и zusammen (Z) изомеры, а также их соответствующие смеси. Кроме того, соединения могут существовать в виде таутомеров; все таутомерные изомеры обеспечены настоящим изобретением. Кроме того, соединения, раскрытые в данной заявке, могут существовать в несольватированных, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и подобные. Как правило, сольватированные формы считаются эквивалентными несольватированным формам.
Термин "связь" относится к ковалентной связи между двумя атомами или двумя группами, когда атомы, соединенные связью, считаются частью более крупной подструктуры. Связь может быть одинарной, двойной или тройной, если не указано иное. Пунктирная линия между двумя атомами на чертеже молекулы указывает на то, что дополнительная связь может присутствовать или отсутствовать в этом положении.
Термин "заболевание", как используется в данной заявке, в целом является синонимом и используется взаимозаменяемо с терминами "расстройство", "синдром" и "состояние" (как в случае медицинского состояния), поскольку все они отражают ненормальное состояние тела человека или животного или одной из его частей, которая нарушает нормальное функционирование, и обычно проявляется в виде отличительных признаков и симптомов и приводит к сокращению продолжительности или качества жизни человека или животного.
Термин "миелоидное заболевание", как используется в данной заявке, предназначен для включения заболеваний, которые можно классифицировать под термином миелопролиферативное новообразование.
Термин "миелопролиферативное новообразование" (MPN) относится к раку крови, который возникает, когда в организме вырабатывается слишком много белых или красных кровяных телец или тромбоцитов в результате соматических мутаций, которые активируют пути передачи гормональных сигналов, которые контролируют продуцирование этих типов клеток крови. Они представляют собой "клональные заболевания гемопоэтических стволовых клеток", учитывая, что неопластические клетки возникают из единственного мутантного клона, происходящего из клеток костного мозга (Campregher et al. Rev Bras Hematol Hemoter. 2012; 34 (2): 150-5). MPN включают истинную полицитемию (PV), миелофиброз, включая первичный миелофиброз (PMF, включая, в определенных вариантах осуществления, как префиброзную/раннюю стадию, так и выраженную фиброзную стадию) и пост-PV/ET миелофиброз (PPV-MF и PET-MF), эссенциальную тромбоцитемию (ET), хронический нейтрофильный лейкоз (CNL), хронический эозинофильный лейкоз, если не указано иное (CEL-NOS), и хронический миелоидный лейкоз (CML), а также другие неклассифицируемые MPN. Для более подробного обсуждения MPN и связанных миелоидных новообразований и острого лейкоза, а также диагностических критериев для PV, ET, PMF и других MPN см. Arber et al. "The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia", Blood 2016, 127(20):2391-2405. Для всестороннего обсуждения критериев диагностики и ответа на миелофиброз см. Tefferi A et al., "Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report," Blood, 122(8): 1395-98 (2013).
Термин "комбинированная терапия" означает введение двух или более терапевтических агентов для лечения терапевтического состояния или расстройства, описанного в настоящем изобретении. Такое введение включает совместное введение этих терапевтических агентов, по существу, одновременным образом, например, в одной капсуле с фиксированным соотношением активных ингредиентов или в нескольких отдельных капсулах для каждого активного ингредиента. Кроме того, такое введение также включает применение каждого типа терапевтического агента последовательным образом. В любом случае схема лечения будет обеспечивать благоприятные эффекты комбинации лекарственных средств при лечении состояний или расстройств, описанных в данной заявке.
"Ингибитор KDM1A", как используется в данной заявке, относится к соединению, которое демонстрирует IC50 в отношении активности KDM1A не более, чем примерно 100 мкМ и, как правило, не более, чем примерно 50 мкМ, как измерено в анализе ингибирования KDM1A, описанном в целом в данной заявке. "IC50" представляет собой концентрацию ингибитора, которая снижает активность фермента (например, KDM1A) до полумаксимального уровня. Было обнаружено, что некоторые соединения, раскрытые в данной заявке, демонстрируют ингибирование против KDM1A. В определенных вариантах осуществления, соединения будут демонстрировать IC50 по отношению к KDM1A не более, чем примерно 10 мкМ; в дополнительных вариантах осуществления, соединения будут демонстрировать IC50 по отношению к KDM1A не более, чем примерно 200 нМ; в еще дополнительных вариантах осуществления, соединения будут демонстрировать IC50 по отношению к KDM1A не более, чем примерно 50 нМ; в еще дополнительных вариантах осуществления, соединения будут демонстрировать IC50 по отношению к KDM1A не более, чем примерно 10 нМ; в еще дополнительных вариантах осуществления, соединения будут демонстрировать IC50 по отношению к KDM1A не более, чем примерно 2 нМ, как измерено в анализе KDM1A, описанном в данной заявке.
Фраза "терапевтически эффективный" предназначена для определения количества активных ингредиентов, используемых при лечении заболевания или расстройства или влияющих на клинический результат.
Термин "терапевтически приемлемый" относится к тем соединениям (или солям, пролекарствам, таутомерам, цвиттерионным формам и т.д.), которые подходят для применения в контакте с тканями пациентов без чрезмерной токсичности, раздражения и аллергической реакции, и являются соразмерными разумному соотношению польза/риск и эффективными для предполагаемого применения.
Как используется в данной заявке, ссылка на "лечение" пациента подразумевает профилактику. Лечение также может быть превентивным, т.е. может включать предупреждение заболевания. Предупреждение заболевания может включать полную защиту от заболевания, например, как в случае предотвращения заражения патогеном, или может включать предотвращение прогрессирования заболевания. Например, предотвращение заболевания может не означать полный отказ от любого эффекта, связанного с заболеванием на любом уровне, а вместо этого может означать предотвращение симптомов заболевания до клинически значимого или обнаруживаемого уровня. Предотвращение заболеваний также может означать предотвращение прогрессирования заболевания до более поздней стадии заболевания.
Термин "пациент" обычно является синонимом термина "субъект" и включает всех млекопитающих, включая человека. Примеры пациентов включают людей, домашний скот, такой как коровы, козы, овцы, свиньи и кролики, и домашних животных, таких как собаки, кошки, кролики и лошади. Предпочтительно пациентом является человек.
Термин "пролекарство" относится к соединению, которое становится более активным in vivo. Некоторые соединения, раскрытые в данной заявке, также могут существовать в форме пролекарств, как описано в Hydrolysis in Drug and Prodrug Metabolism: Chemistry, Biochemistry, and Enzymology (Testa, Bernard and Mayer, Joachim M. Wiley-VHCA, Zurich, Switzerland 2003). Пролекарства соединений, описанных в данной заявке, представляют собой структурно модифицированные формы соединения, которые легко претерпевают химические изменения в физиологических условиях с получением соединения. Кроме того, пролекарства можно превратить в соединение химическими или биохимическими методами в среде ex vivo. Например, пролекарства могут медленно превращаться в соединение при помещении в резервуар трансдермального пластыря с приемлемым ферментом или химическим реагентом. Пролекарства часто являются полезными, потому что в некоторых ситуациях их легче вводить, чем соединение или исходное лекарственное средство. Например, они могут быть биодоступными при пероральном введении, тогда как исходное лекарственное средство - нет. Пролекарство также может иметь улучшенную растворимость в фармацевтических композициях по сравнению с исходным лекарственным средством. В данной области техники известно большое количество производных пролекарств, таких как те, которые основаны на гидролитическом расщеплении или окислительной активации пролекарства. Примером пролекарства, без ограничения, могло бы быть соединение, которое вводят в форме сложного эфира ("пролекарство"), а затем метаболически гидролизуют до карбоновой кислоты, активного объекта. Дополнительные примеры включают пептидильные производные соединения.
Соли
Соединения, раскрытые в данной заявке, могут существовать в форме терапевтически приемлемых солей. Настоящее изобретение включает соединения, перечисленные выше, в форме солей, включая кислотно-аддитивные соли. Приемлемые соли включают соли, образованные как с органическими, так и с неорганическими кислотами. Такие кислотно-аддитивные соли обычно являются фармацевтически приемлемыми. Однако, соли неприемлемых с фармацевтической точки зрения кислот могут быть полезными при получении и очистке рассматриваемого соединения. Также могут быть образованы основно-аддитивные соли, которые могут быть фармацевтически приемлемыми. Для более полного обсуждения приготовления и выбора солей см. Pharmaceutical Salts: Properties, Selection, and Use (Stahl, P. Heinrich. Wiley-VCHA, Zurich, Switzerland, 2002).
Термин "терапевтически приемлемая соль", как используется в данной заявке, представляет собой соли или цвиттерионные формы соединений, раскрытых в данной заявке, которые являются водо- или маслорастворимыми или диспергируемыми и являются терапевтически приемлемыми, как определено в данной заявке. Соли могут быть получены во время окончательного выделения и очистки соединений или отдельно путем взаимодействия подходящего соединения в форме свободного основания с подходящей кислотой. Типичные кислотно-аддитивные соли включают ацетат, адипат, альгинат, L-аскорбат, аспартат, бензоат, бензолсульфонат (безилат), бисульфат, бутират, камфорат, камфорсульфонат, цитрат, диглюконат, формиат, фумарат, гентизат, глутарат, глицерофосфат, гликолат, гемисульфат, гептаноат, гексаноат, гиппурат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат (изетионат), лактат, малеат, малонат, DL-манделат, мезитиленсульфонат, метансульфонат, нафтиленсульфонат, никотинат, 2-нафталинсульфонат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфонат, пикрат, пивалат, пропионат, пироглутамат, сукцинат, сульфонат, тартрат, L-тартрат, трихлорацетат, трифторацетат, фосфат, глутамат, бикарбонат, пара-толуолсульфонат (п-тозилат) и ундеканоат.Также основные группы в соединениях, раскрытых в данной заявке, могут быть кватернизованы метил, этил, пропил и бутилхлоридами, бромидами и йодидами; диметил, диэтил, дибутил и диамилсульфатами; децил, лаурил, миристил и стерил хлоридами, бромидами и йодидами; и бензил и фенэтил бромидами. Примеры кислот, которые можно использовать для образования терапевтически приемлемых аддитивных солей, включают неорганические кислоты, такие как соляная, бромистоводородная, серная и фосфорная, и органические кислоты, такие как щавелевая, малеиновая, янтарная и лимонная. Соли также могут быть образованы путем координации соединений с ионом щелочного металла или щелочноземельного металла. Следовательно, в настоящем изобретении рассматриваются натриевые, калиевые, магниевые и кальциевые соли соединений, раскрытых в данной заявке, и т.п.
Основно-аддитивные соли могут быть получены во время окончательного выделения и очистки соединений путем взаимодействия карбокси группы с подходящим основанием, таким как гидроксид, карбонат или бикарбонат катиона металла, или с аммиаком или органическим первичным, вторичным или третичным амином. Катионы терапевтически приемлемых солей включают катионы лития, натрия, калия, кальция, магния и алюминия, а также нетоксичные катионы четвертичных аминов, такие как аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, диэтиламин, этиламин, трибутиламин, пиридин, N,N-диметиланилин, N-метил пиперидин, N-метилморфолин, дициклогексиламин, прокаин, дибензиламин, N,N-дибензилфенэтиламин, 1-эфенамин и N,N'-дибензилэтилендиамин. Другие типичные органические амины, используемые для образования основно-аддитивных солей, включают этилендиамин, этаноламин, диэтаноламин, пиперидин и пиперазин.
Композиции
Хотя соединения в соответствии с настоящим изобретением можно вводить в виде сырьевого химического вещества, их также можно представить в виде фармацевтического состава. Соответственно, в настоящей заявке обеспечены фармацевтические составы, которые содержат одно или несколько определенных соединений, раскрытых в данной заявке, или одну или несколько их фармацевтически приемлемых солей, сложных эфиров, пролекарств, амидов или сольватов вместе с одним или несколькими фармацевтически приемлемыми носителями и необязательно с одним или несколькими другими терапевтическими ингредиентами. Носитель(и) должен быть "приемлемым" в том смысле, что он должен быть совместимым с другими ингредиентами состава и быть безвредным для реципиента. Подходящий состав зависит от выбранного пути введения. Любые из хорошо известных методик, носителей и эксципиентов могут быть использованы в качестве приемлемых и как известно в данной области техники. Фармацевтические композиции, раскрытые в данной заявке, могут быть изготовлены любым способом, известным из уровня техники, например, посредством традиционных процессов смешивания, растворения, гранулирования, изготовления драже, взбалтывания, эмульгирования, инкапсулирования, улавливания или прессования.
Составы включают те, которые подходят для перорального, парентерального (включая подкожное, внутрикожное, внутримышечное, внутривенное, внутрисуставное и внутрикостное), внутрибрюшинного, трансмукозального, трансдермального, ректального и местного (включая кожное, трансбуккальное, сублингвальное и внутриглазное) введения, хотя наиболее подходящий путь может зависеть, например, от состояния и расстройства реципиента. Составы для удобства могут быть представлены в стандартной лекарственной форме и могут быть приготовлены любым из способов, хорошо известных в области фармации. Обычно эти способы включают стадию объединения соединения в соответствии с настоящим изобретением или его фармацевтически приемлемой соли, сложного эфира, амида, пролекарства или сольвата ("активный ингредиент") с носителем, который представляет собой один или несколько дополнительных ингредиентов. Как правило, составы получают путем однородного и тщательного объединения активного ингредиента с жидкими носителями или тонко измельченными твердыми носителями или с обоими и, если необходимо, приданием продукту формы желаемого состава.
Составы соединений, раскрытых в данной заявке, приемлемые для перорального введения могут быть представлены в виде дискретных единиц, таких как капсулы, облатки или таблетки, каждая из которых содержит заранее определенное количество активного ингредиента; в виде порошка или гранул; в виде раствора или суспензии в водной жидкости или неводной жидкости; или в виде жидкой эмульсии масло-в-воде или жидкой эмульсии вода-в-масле. Активный ингредиент также может быть представлен в виде болюса, электуария или пасты.
Фармацевтические препараты, которые можно применять перорально, включают таблетки, твердые капсулы, изготовленные из желатина, а также мягкие герметичные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Таблетки могут быть изготовлены прессованием или формованием, необязательно с одним или несколькими дополнительными ингредиентами. Спрессованные таблетки могут быть получены прессованием в подходящем устройстве активного ингредиента в сыпучей форме, такой как порошок или гранулы, необязательно смешанного со связующими веществами, инертными разбавителями или смазывающими веществами, поверхностно-активны ми веществами или диспергирующими агентами. Формованные таблетки могут быть получены формованием в подходящем устройстве смеси порошкообразного соединения, смоченного инертным жидким разбавителем. Таблетки необязательно могут быть покрыты оболочкой или на них могут быть нанесены насечки, и они могут быть составлены так, чтобы обеспечивать медленное или контролируемое высвобождение активного ингредиента из них. Все составы для перорального введения должны быть в дозировках, подходящих для такого введения. Твердые капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими веществами, такими как крахмалы, и/или смазывающими веществами, такими как тальк или стеарат магния, и, необязательно, стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы. Сердцевины драже имеют подходящие покрытия. Для этой цели могут использоваться концентрированные растворы сахаров, которые могут необязательно содержать гуммиарабик, тальк, поливинилпирролидон, карбополовый гель, полиэтиленгликоль и/или диоксид титана, растворы лаков и подходящие органические растворители или смеси растворителей. К покрытиям таблеток или драже могут быть добавлены красители или пигменты для идентификации или для характеристики различных комбинаций доз активного соединения.
Соединения могут быть составлены для парентерального введения путем инъекции, например, путем болюсной инъекции или непрерывной инфузии. Составы для инъекций могут быть представлены в виде стандартной дозированной формы, например, в ампулах или в многодозовых контейнерах, с добавленным консервантом. Композиции могут быть в таких формах, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать вспомогательные вещества, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Составы могут быть представлены в однодозовых или многодозовых контейнерах, например, в запечатанных ампулах и флаконах, и могут храниться в форме порошка или в сублимированном (лиофилизированном) состоянии, требующих только добавления стерильного жидкого носителя, например, физиологического раствора или стерильной апирогенной воды непосредственно перед использованием. Растворы и суспензии для инъекций для немедленного приема могут быть приготовлены из стерильных порошков, гранул и таблеток, описанных ранее.
Составы для парентерального введения включают водные и неводные (масляные) стерильные инъекционные растворы активных соединений, которые могут содержать антиоксиданты, буферы, бактериостатические вещества и растворенные вещества, которые делают состав изотоничным с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Подходящие липофильные растворители или носители включают жирные масла, такие как кунжутное масло, или синтетические сложные эфиры жирных кислот, такие как этилолеат, или триглицериды, или липосомы. Водные суспензии для инъекций могут содержать вещества, повышающие вязкость суспензии, такие как натрий карбоксиметилцеллюлоза, сорбит или декстран. Необязательно, суспензия также может содержать подходящие стабилизаторы или агенты, которые увеличивают растворимость соединений, что позволяет приготовить высококонцентрированные растворы.
В дополнение к составам, описанным ранее, соединения также могут быть составлены в виде депо-препаратов. Такие препараты длительного действия можно вводить путем имплантации (например, подкожно или внутримышечно) или внутримышечной инъекцией. Таким образом, например, соединения могут быть составлены с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами или в форме умереннорастворимых производных, например, в форме умереннорастворимой соли.
Для трансбуккального или сублингвального введения композиции могут принимать форму таблеток, таблеток для рассасывания, пастилок или гелей, приготовленных обычным способом. Такие композиции могут содержать активный ингредиент в ароматизированной основе, такой как сахароза и гуммиарабик или трагакант.
Соединения также могут быть приготовлены в форме ректальных композиций, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы суппозиториев, такие как масло какао, полиэтиленгликоль или другие глицериды.
Некоторые соединения, раскрытые в данной заявке, можно вводить местно, то есть внесистемно. Такое введение включает нанесение соединения, раскрытого в данной заявке, снаружи на эпидермис или ротовую полость и закапывание такого соединения в ухо, глаз и нос, так что соединение существенно не попадает в кровоток.
Напротив, системное введение относится к пероральному, внутривенному, внутрибрюшинному и внутримышечному введению.
Составы, подходящие для местного применения, включают жидкие или полужидкие препараты, подходящие для проникновения через кожу в место воспаления, такие как гели, линименты, лосьоны, кремы, мази или пасты и капли, подходящие для введения в глаза, уши или нос. Активный ингредиент для местного применения может составлять, например, от 0,001% до 10% масс./масс, (по массе) состава. В определенных вариантах осуществления, активный ингредиент может составлять до 10% масс/масс. В других вариантах осуществления, он может составлять менее, чем 5% масс./масс. В определенных вариантах осуществления, активный ингредиент может составлять от 2% масс/масс, до 5% масс/масс. В других вариантах осуществления, он может составлять от 0,1% до 1% масс./масс. состава.
Для введения путем ингаляции соединения можно удобно доставлять из инсуффлятора, аэрозольных баллонов, находящихся под давлением, или других удобных средств доставки аэрозольной струи. Баллоны под давлением могут содержать подходящий пропеллент, такой как дихлордифтор метан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другой подходящий газ. В случае аэрозоля под давлением стандартная дозировка может быть определена путем обеспечения клапана для доставки отмеренного количества. Альтернативно, для введения путем ингаляции или вдувания соединения в соответствии с изобретением могут быть в форме сухой порошковой композиции, например порошковой смеси соединения и подходящей порошковой основы, такой как лактоза или крахмал. Порошковая композиция может быть представлена в стандартной дозированной форме, например, в капсулах, картриджах, желатиновых или блистерных упаковках, из которых порошок можно вводить с помощью ингалятора или инсуффлятора.
Предпочтительными стандартными дозированными формами являются те, которые содержат эффективную дозу, как изложено в данной заявке ниже, или ее соответствующую часть, активного ингредиента.
Следует понимать, что в дополнение к ингредиентам, конкретно упомянутым выше, составы, описанные выше, могут включать другие агенты, которые являются традиционными в данной области техники, с учетом типа рассматриваемого состава, например, составы, которые подходят для перорального введения, могут включать ароматизаторы.
Соединения можно вводить перорально или путем инъекции в дозе от 0,1 до 500 мг/кг в сутки. Диапазон доз для взрослых людей обычно составляет от 5 мг до 2 г/день. Таблетки или другие формы выпуска, представленные в дискретных единицах, могут для удобства содержать количество одного или нескольких соединений, которые являются эффективными при такой дозировке или в количестве, кратном одному и тому же, например, в единицах, содержащих от 5 мг до 500 мг, обычно примерно от 10 мг до 200 мг.
Количество активного ингредиента, которое может быть объединено с материалами носителя для получения однократной дозированной формы, будет варьироваться в зависимости от пациента, которого лечат, и конкретного способа введения.
Соединения можно вводить различными способами, например, перорально, местно или путем инъекции. За точное количество соединения, вводимого пациенту, отвечает лечащий врач. Конкретный уровень дозы для любого конкретного пациента будет зависеть от множества факторов, включая активность конкретного применяемого соединения, возраст, массу тела, общее состояние здоровья, пол, режим питания, время введения, путь введения, скорость выведения, комбинацию лекарственных средств, конкретное заболевание, которое лечат, и тяжесть показания или состояния, которое лечат. Кроме того, способ введения может варьироваться в зависимости от состояния и его тяжести.
Комбинации и комбинированная терапия
В некоторых случаях может быть подходящим введение, по меньшей мере, одного из описанных в данной заявке соединений (или его фармацевтически приемлемой соли, сложного эфира или пролекарства) в комбинации с другим терапевтическим агентом. Только в качестве примера, если одним из побочных эффектов, испытываемых пациентом при приеме одного из указанных в данной заявке соединений, является воспаление, то может оказаться целесообразным введение противовоспалительного средства в комбинации с исходным терапевтическим агентом. Альтернативно, только в качестве примера, терапевтическая эффективность одного из описанных в данной заявке соединений может быть усилена введением адъюванта (т.е. адъювант сам по себе может иметь только минимальную терапевтическую пользу, но в комбинации с другим терапевтическим агентом общая терапевтическая польза для пациента увеличивается). Существует даже возможность того, что два соединения, одно из соединений, описанных в данной заявке, и второе соединение, вместе могут достичь желаемого терапевтического эффекта, которого невозможно достичь при введении одного из них. Альтернативно, только в качестве примера, польза, которую испытывает пациент, может быть увеличена путем введения одного из описанных в данной заявке соединений с другим терапевтическим агентом (который также включен в терапевтический режим), который также имеет терапевтический эффект. Только в качестве примера, при лечении острого миелогенного лейкоза или серповидноклеточной анемии, включающем введение одного из соединений, описанных в данной заявке, усиление терапевтического эффекта может быть результатом также обеспечения пациента другим терапевтическим агентом для лечения серповидноклеточной анемии или острого миелогенного лейкоза. В любом случае, независимо от заболевания, расстройства или состояния, которые лечат, общая польза, которую испытывает пациент, может быть просто суммарной от двух терапевтических агентов, или два агента могут иметь синергетический терапевтический эффект у пациента.
Эффективная комбинированная терапия может быть достигнута при использовании одной композиции или фармакологического состава, который включает оба агента, или двух разных композиций или составов в одно и то же время, при этом одна композиция включает соединение в соответствии с настоящим изобретением, а другая включает второй агент(ы). Альтернативно, терапия может предшествовать или следовать за лечением другим агентом с интервалами от нескольких минут до месяцев. Введение пациенту соединений в соответствии с настоящим изобретением будет осуществляться в соответствии с общими протоколами введения фармацевтических препаратов, принимая во внимание токсичность лекарственного средства, если таковая имеется. Ожидается, что циклы лечения будут повторяться по мере необходимости.
Конкретные неограничивающие примеры возможных комбинированных терапий включают применение определенных соединений в соответствии с настоящим изобретением со следующими агентами и классами агентов: агенты, которые ингибируют ДНК-метилтрансферазы, такие как децитабин или 5'-аза-цитадин; агенты, которые ингибируют активность гистондеацетилаз, гистондесумоилаз, гистондеубиквитиназ или гистонфосфатаз, такие как гидроксимочевина; антисмысловые РНК, которые могут ингибировать экспрессию других компонентов белкового комплекса, связанных с сайтом DR в промоторе гамма-глобина; агенты, которые ингибируют действие Klf1 или экспрессию KLF1; агенты, которые ингибируют действие Bel11a или экспрессию BCL11A; и агенты, которые ингибируют прогрессию клеточного цикла, такие как гидроксимочевина, ара-С или даунорубицин; агенты, которые индуцируют дифференцирование в лейкозных клетках, такие как полностью транс-ретиноевая кислота (ATRA).
Само по себе ингибирование активности KDM1A (LSD1) может быть достаточной терапией для лечения некоторых заболеваний; для таких, как рак, комбинированные терапии часто являются аддитивными или синергетическими по своим терапевтическим эффектам и даже могут быть необходимы для достижения полного желаемого клинического эффекта. Существуют конкретные научные доказательства, позволяющие рационализировать комбинацию ингибитора KDM1A с полностью транс-ретиноевой кислотой (ATRA), триоксидом мышьяка, ингибиторами ДНК-метилтрансфераз, такими как 5'-азацитидин или 5'-аза 2'-дезоксицитидин, ингибиторами NFκB сигнализации, такими как сулиндак, или стандартными противоопухолевыми агентами, такими как антрациклины, или аналогами нуклеозидов, такими как цитозинарабинозид. Аналогичным образом, агенты, которые вовлекают лейкозные стволовые клетки в клеточный цикл (G-CSF, GM-CSF, фактор стволовых клеток, тромбопоэтин (ТРО)) или агенты, которые сводят на нет роль цитокинов (ТРО, CCL3(MIP-1)), которые участвуют в ремоделирование ниши раковых стволовых клеток, могут быть полезными как часть комбинации, включающей ингибитор LSD1.
Конкретные неограничивающие примеры возможных комбинированных терапий включают применение определенных соединений в соответствии с настоящим изобретением с противораковыми (химиотерапевтическими) лекарственными средствами. Классы противораковых лекарственных средств включают, но не ограничиваются: алкилирующие агенты, антиметаболиты, антимитотики, ингибиторы контрольных точек, растительные алкалоиды и терпеноиды, ингибиторы топоизомеразы, цитотоксические антибиотики, ингибиторы ароматазы, ингибиторы ангиогенеза, антистероиды и антиандрогены, ингибиторы mTOR, ингибиторы тирозинкиназы и другие.
Для применения при раке и неопластических заболеваниях ингибитор СВР/Р300 оптимально может быть использован вместе с одним или несколькими из следующих неограничивающих примеров противораковых агентов:
(1) алкилирующие агенты, включая, но не ограничиваясь, кармустин, хлорамбуцил (LEUKERAN), цисплатин (PLATIN), карбоплатин (PARAPLATIN), оксалиплатин (ELOXATIN), стрептозоцин (ZANOSAR), бусульфан (MYLERAN), дакарбазин, ифосфамид, ломустин (CCNU), мелфалан (ALKERAN), прокарбазин (MATULAN), темозоломид (TEMODAR), тиотепа и циклофосфамид (ENDOXAN);
(2) антиметаболиты, включая, но не ограничиваясь, кладрибин (LEUSTATIN), меркаптопурин (PURINETHOL), тиогуанин, пентостатин (NIPENT), цитозина арабинозид (цитарабин, ARA-C), гемцитабин (GEMZAR), фторурацил (5-FU, CARAC), капецитабин (XELODA), лейковорин (FUSILEV), метотрексат (RHEUMATREX), ралтитрексед;
(3) антимитотики, которые часто являются растительными алкалоидами и терпеноидами, или их производными, включая, но не ограничиваясь, таксаны, такие как доцетаксел (TAXITERE) и паклитаксел (ABRAXANE, TAXOL); алкалоиды барвинка, такие как винкристин (ONCOVIN), винбластин, виндезин и винорелбин (NAVELBINE);
(4) ингибиторы контрольных точек, такие как анти-PD-1 или PD-L1 антитела, пембролизумаб (KEYTRUDA), ниволумаб (OPDIVO), MEDI4736 и MPDL3280A; анти-CTLA-4 антитело ипилимумаб (YERVOY); и те, которые нацелены на LAG3 (белок ген 3 активации лимфоцитов), KIR (иммуноглобулинподобный рецептор киллерных клеток), 4-1ВВ (член суперсемейства рецептора фактора некроза опухолей 9), TIM3 (домен иммуноглобулина Т-клеток и домен 3 муцина) и ОХ40 (член суперсемейства рецептора фактора некроза опухолей 4);
(5) ингибиторы топоизомеразы, включая, но не ограничиваясь, камптотецин (СТР), иринотекан (CAMPTOSAR), топотекан (HYCAMTIN), тенипозид (VUMON) и этопозид (EPOSIN);
(6) цитотоксические антибиотики, включая, но не ограничиваясь, актиномицин D (дактиномицин, COSMEGEN), блеомицин (BLENOXANE), доксорубицин (ADRIAMYCIN), даунорубицин (CERUBIDINE), эпирубицин (ELLENCE), флударабин (FLUDARA), идарубицин, митомицин (MITOSOL), митоксантрон (NOVANTRONE), пликамицин;
(7) ингибиторы ароматазы, включая, но не ограничиваясь, аминоглютетимид, анастрозол (ARIMIDEX), летрозол (FEMARA), ворозол (RIVIZOR), эксеместан (AROMASIN);
(8) ингибиторы ангиогенеза, включая, но не ограничиваясь, генистеин, сунитиниб (SUTENT) и бевацизумаб (AVASTIN);
(9) антистероиды и антиандрогены, такие как аминоглютетимид (CYTADREN), бикалутамид (CASODEX), ципротерон, флутамид (EULEXIN), нилутамид (NILANDRON);
(10) ингибиторы тирозинкиназы, включая, но не ограничиваясь, иматиниб (GLEEVEC), эрлотиниб (TARCEVA), лапатиниб (TYKERB), сорафениб (NEXAVAR) и акситиниб (INLYTA);
(11) mTOR ингибиторы, такие как эверолимус, темсиролимус (TORISEL) и сиролимус;
(12) моноклональные антитела, такие как трастузумаб (HERCEPTIN) и ритуксимаб (RITUXAN);
(13) другие средства, такие как амсакрин; вакцина Бацилла Кальметта-Герена (БЦЖ); бусерелин (ЕТILАМИД); хлорохин (ARALEN); клодронат, памидронат и другие бисфосфонаты; колхицин; деметоксивиридин; дихлорацетат; эстрамустин; филграстим (NEUPOGEN); флудрокортизон (FLORINEF); гозерелин (ZOLADEX); интерферон; лейковорин; лейпролид (LUPRON); левамизол; лонидамин; месна; метформин; митотан (o,p'-DDD, LYSODREN); нокодазол; октреотид (SANDOSTATIN); перифосин; порфимер (особенно в сочетании с фото-и лучевой терапией); сурамин; тамоксифен; титаноцена дихлорид; третиноин; анаболические стероиды, такие как флуоксиместерон (HALOTESTIN); эстрогены, такие как эстрадиол, диэтилстилбестрол (DES) и диенэстрол; прогестины, такие как медроксипрогестерона ацетат (МРА) и мегестрол; и тестостерон.
Таким образом, в другом аспекте, определенные варианты осуществления обеспечивают способы лечения расстройств, опосредованных KDM1A, у человека или животного, нуждающегося в таком лечении, включающие введение указанному субъекту количества соединения, раскрытого в данной заявке, эффективного для уменьшения или предотвращения указанного расстройства у субъекта, в комбинации, по меньшей мере, с одним дополнительным агентом для лечения указанного расстройства, известным в данной области техники. В связанном аспекте, определенные варианты осуществления, обеспечивают терапевтические композиции, содержащие, по меньшей мере, одно соединение, раскрытое в данной заявке, в комбинации с одним или несколькими дополнительными агентами для лечения расстройств, опосредованных KDM1A.
Конкретные заболевания, которые можно лечить с помощью соединений, композиций и способов, раскрытых в данной заявке, включают рак, миелоидные заболевания и воспалительные заболевания.
Конкретные виды рака, которые можно успешно лечить раскрытыми в данной заявке соединениями, включают саркому Юинга, множественную миелому, Т-клеточный лейкоз, опухоль Вильма, мелкоклеточный рак легкого, рак мочевого пузыря, рак предстательной железы, рак молочной железы, рак головы и шеи, рак толстой кишки и рак яичников.
Конкретные миелоидные заболевания, которые можно успешно лечить раскрытыми в данной заявке соединениями, включают миелофиброз, истинную полицитемию, эссенциальную тромбоцитемию, миелодиспластический синдром (MDS), острый миелогенный лейкоз (AML) и хронический миелоидный лейкоз (CML).
Конкретные воспалительные заболевания, которые можно успешно лечить раскрытыми в данной заявке соединениями, включают, без ограничения: артрит, включая подтипы и связанные состояния, такие как ревматоидный артрит, спондилоартропатии, подагрический артрит, остеоартрит, системная красная волчанка, ювенильный артрит, острый ревматоидный артрит, энтеропатический артрит, невропатический артрит, псориатический артрит и пиогенный артрит; остеопороз, тендинит, бурсит и другие связанные заболевания костей и суставов; желудочно-кишечные заболевания, такие как рефлюксный эзофагит, диарея, воспалительное заболевание кишечника, болезнь Крона, гастрит, синдром раздраженного кишечника, язвенный колит, острое и хроническое воспаление поджелудочной железы; воспаление легких, как, например, связанное с вирусными инфекциями и кистозным фиброзом; кожные заболевания, такие как псориаз, экзема, ожоги, солнечный ожег, дерматит (такой как контактный дерматит, атопический дерматит и аллергический дерматит) и крапивница; панкреатит, гепатит, зуд и витилиго. Кроме того, соединения в соответствии с настоящим изобретением также применимы для пациентов с трансплантацией органов либо отдельно, либо в комбинации со стандартными иммуномодуляторами.
Раскрытые в данной заявке соединения могут быть использованы для лечения заболеваний, при которых увеличение транскрипции за счет манипулирования эпигенетическими регуляторными факторами, такими как ингибирование KDM1A, может быть полезным для пациента. Это относится к заболеваниям, включая, но не ограничиваясь, мутации с потерей функции, мутации, приводящие к гаплонедостаточности, делеции и дупликации генетического материала, или эпигенетические регуляторные механизмы, которые изменили нормальный профиль экспрессии гена или генов, которые имеют эффект изменения дозы генного продукта(ов). Такие заболевания могут включать как приобретенные, так и наследственные заболевания, при которых изменяется экспрессия, например, цитокинов, влияющих на иммунную функцию, синдром ломкой Х-хромосомы и другие формы нарушения когнитивной или двигательной функции, такие как болезнь Альцгеймера и Паркинсона, независимо от того, являются ли они приобретенными или наследственными формами, липидные нарушения, такие как повышенный холестерин, липопротеины низкой плотности, липопротеины очень низкой плотности или триглицериды, диабет первого и второго типа и менделевские генетические заболевания.
Другие расстройства или состояния, которые можно успешно лечить с помощью раскрытых в данной заявке соединений, включают воспаление и воспалительные заболевания. Воспалительные заболевания включают, без ограничения: артрит, включая подтипы и связанные состояния, такие как ревматоидный артрит, спондилоартропатии, подагрический артрит, остеоартрит, системная красная волчанка, ювенильный артрит, острый ревматоидный артрит, энтеропатический артрит, невропатический артрит, псориатический артрит, и пиогенный артрит; остеопороз, тендинит, бурсит и другие связанные заболевания костей и суставов; желудочно-кишечные заболевания, такие как рефлюксный эзофагит, диарея, воспалительное заболевание кишечника, болезнь Крона, гастрит, синдром раздраженного кишечника, язвенный колит, острое и хроническое воспаление поджелудочной железы; воспаление легких, как, например, связанное с вирусными инфекциями и кистозным фиброзом; кожные заболевания, такие как псориаз, экзема, ожоги, солнечный ожег, дерматит (такой как контактный дерматит, атопический дерматит и аллергический дерматит) и крапивница; панкреатит, гепатит, зуд и витилиго. Кроме того, соединения в соответствии с настоящим изобретением также применимы для пациентов с трансплантацией органов либо отдельно, либо в комбинации со стандартными иммуномодуляторами.
Аутоиммунные расстройства можно облегчить путем терапии раскрытыми в данной заявке соединениями. Аутоиммунные расстройства включают болезнь Крона, язвенный колит, дерматит, дерматомиозит, сахарный диабет первого типа, синдром Гудпасчера, базедову болезнь, синдром Гийена-Барре (GBS), аутоиммунный энцефаломиелит, тиреоидит Хашимото, идиопатическую тромбоцитопеническую пурпуру, красную волчанку, смешанное заболевание соединительной ткани, рассеянный склероз (MS), миастения гравис, нарколепсию, вульгарный пемфигус, пернициозную анемию, псориаз, псориатический артрит, болезнь Вагнера, первичный билиарный цирроз, ревматоидный артрит, синдром Шегрена, склеродермию, височный артериит (также известный как "гигантоклеточный артериит"), васкулит и гранулематоз Вегенера.
Соединения, раскрытые в данной заявке, также полезны для лечения повреждения органов и тканей, связанных с тяжелыми ожогами, сепсисом, травмами, ранами и гипотонией, вызванной кровотечением или реанимацией, а также при таких заболеваниях, как сосудистые болезни, мигреневые головные боли, узелковый периартериит, тиреоидит, гипопластическая анемия, болезнь Ходжкина, склеродермия, ревматический полиартрит, диабет первого типа, болезнь нервно-мышечного синапса, включая миастения гравис, болезнь белого вещества головного мозга, включая рассеянный склероз, саркоидоз, нефрит, нефротический синдром, синдром Бехчета, болезнь Вагнера, гингивит, пародонтит, отек, возникший после травмы, ишемия, включая ишемию миокарда, сосудисто-сердечную ишемию и ишемию, вторичную по отношению к остановке сердца, и подобные.
Раскрытые в данной заявке соединения также полезны для лечения некоторых заболеваний и расстройств нервной системы. Расстройства центральной нервной системы, при которых полезно ингибирование KDM1A, включают кортикальные деменции, включая болезнь Альцгеймера, повреждение центральной нервной системы в результате инсульта, ишемий, включая ишемию головного мозга (как очаговую ишемию, так и тромботический инсульт, и глобальную ишемию (например, вторичную по отношению к остановке сердца) и травмы. Нейродегенеративные расстройства, при которых полезно ингибирование KDM1A, включают дегенерацию нервных волокон или некроз нервных волокон при расстройствах, таких как гипоксия, гипогликемия, эпилепсия, и при травме центральной нервной системы (CNS) (такие как травма спинного мозга и головы), судороги и токсичность, вызванные гипербарической оксигенацией, деменцию, например, пресенильную деменцию и СПИД-ассоциированную деменцию, кахексию, хорею Сиденгама, болезнь Хантингтона, болезнь Паркинсона, боковой амиотрофический склероз (ALS), корсаковский психоз, когнитивные расстройства, связанные с заболеванием сосудов головного мозга, гиперчувствительность, расстройства сна, шизофрению, депрессию, депрессию или другие симптомы, связанные с предменструальным синдромом (PMS), и тревогу.
Другие расстройства или состояния, которые преимущественно лечатся раскрытыми в данной заявке соединениями, включают профилактику или лечение гиперпролиферативных заболеваний, особенно рака, либо отдельно, либо в комбинации со стандартами лечения, особенно с теми агентами, которые нацелены на рост опухоли путем восстановления генов-супрессоров опухоли в злокачественных клетках. Гематологические и негематологические злокачественные новообразования, которые можно лечить или предотвращать, включают, но не ограничиваются, множественную миелому, острый и хронический лейкоз и гемопоэтические пролиферативные и неопластические заболевания, включая миелодиспластический синдром (MDS), острый миелогенный лейкоз (AML), острый лимфоцитарный лейкоз (ALL), хронический лимфоцитарный лейкоз (CLL) и хронический миелоидный лейкоз (CML), лимфомы, включая лимфому Ходжкина и неходжкинскую лимфому (низкой, средней и высокой степени), а также солидные опухоли и злокачественные новообразования головного мозга, головы и шеи, молочной железы, легкого (включая немелкоклеточный рак легкого), половых путей, верхних отделов желудочно-кишечного тракта, поджелудочной железы, печени, почек, мочевого пузыря, предстательной железы и толстой кишки. Настоящие соединения и способы также можно использовать для лечения фиброза, такого как фиброз, возникающий при лучевой терапии. Настоящие соединения и способы можно использовать для лечения субъектов, имеющих или у которых нужно предотвратить прогрессирование аденоматозных полипов, в том числе субъектов с семейным аденоматозным полипозом (FAP) или саркоидозом. К незлокачественным пролиферативным заболеваниям дополнительно относятся псориаз, экзема и дерматит.
Настоящие соединения могут также использоваться в комбинированной терапии, частично или полностью, вместо других традиционных противовоспалительных методов лечения, как например вместе со стероидами, NSAID, селективными ингибиторами СОХ-2, ингибиторами 5-липоксигеназы, антагонистами LTB4 и ингибиторами гидролазы LTA4. Раскрытые в данной заявке соединения также могут быть использованы для предотвращения повреждения тканей при терапевтическом сочетании с антибактериальными или противовирусными средствами.
Соединения, раскрытые в данной заявке, также полезны для лечения метаболических нарушений. KDM1A, используя флавинаденозиндинуклеотид (FAD) в качестве кофактора, эпигенетически регулирует гены, отвечающие за расход энергии, в адипоцитах в зависимости от доступности клеточного FAD. Кроме того, потеря функции KDM1A индуцирует ряд регуляторов расхода энергии и митохондриальный метаболизм, что приводит к активации митохондриального дыхания. Кроме того, в жировых тканях мышей, получавших диету с высоким содержанием жиров, снижается экспрессия генов-мишеней KDM1A.
Метаболический синдром (также известный как метаболический синдром X) характеризуется наличием, по меньшей мере, трех из следующих симптомов: инсулинорезистентность; абдоминальный жир -у мужчин это определяется как талия 40 дюймов или больше, у женщин 35 дюймов или больше; высокий уровень сахара в крови -по меньшей мере, 110 миллиграммов на децилитр (мг/дл) натощак; высокий уровень триглицеридов по меньшей мере, 150 мг/дл в кровотоке; низкий уровень ЛПВП -менее 40 мг/дл; протромботическое состояние (например, высокий уровень фибриногена или ингибитора активатора плазминогена в крови); или артериальное давление 130/85 мм рт.ст. или выше. Была обнаружена связь между метаболическим синдромом и другими состояниями, такими как ожирение, высокое кровяное давление и высокий уровень холестерина ЛПНП, которые являются факторами риска сердечнососудистых заболеваний. Например, показана повышенная связь между метаболическим синдромом и атеросклерозом. Люди с метаболическим синдромом также более склонны к развитию диабета 2 типа, а также к PCOS (синдрома поликистозных яичников) у женщин и рака предстательной железы у мужчин.
Как описано выше, инсулинорезистентность может проявляться несколькими способами, включая диабет 2 типа. Диабет 2 типа является состоянием, безусловно связанным с инсулинорезистентностью. Компенсирующая гиперинсулинемия часто помогает поддерживать нормальный уровень глюкозы в течение десятилетий, прежде чем разовьется явный диабет. Со временем бета-клетки поджелудочной железы не могут преодолеть инсулинорезистентность за счет гиперсекреции. Повышаются уровни глюкозы и может быть поставлен диагноз диабета. Пациенты с диабетом 2 типа остаются гиперинсулинемичными до тех пор, пока они не достигнут поздней стадии заболевания. Как описано выше, инсулинорезистентность также может коррелировать с гипертонией. Половина пациентов с эссенциальной гипертонией является инсулинорезистентной и гиперинсулинемической, и есть доказательства того, что артериальное давление связано со степенью инсулинорезистентности. Гиперлипидемия тоже связана с инсулинорезистентностью. Липидный профиль пациентов с диабетом 2 типа включает повышенный уровень холестерина липопротеинов очень низкой плотности (ЛПОНП) и триглицеридов в сыворотке крови и, иногда, пониженный уровень холестерина липопротеинов низкой плотности (ЛПНП). Инсулинорезистентность была обнаружена у людей с низким уровнем липопротеидов высокой плотности (ЛПВП). Уровни инсулина также связаны с синтезом ЛПОНП и уровнями триглицеридов в плазме.
Характерные метаболические заболевания и симптомы, которые нужно лечить с помощью соединений, композиций и способов, раскрытых в данной заявке, являются такими, которые опосредованы, по меньшей мере, частично, KDM1A. Соответственно, в данной заявке раскрыты способы: лечения инсулинорезистентности у субъекта; уменьшения накопления гликогена у субъекта; повышения ЛПВП, понижения ЛПНП, сдвига размера частиц ЛПНП от малых плотных частиц до нормальных частиц ЛПНП, понижения ЛПОНП, понижения триглицеридов или ингибирования абсорбции холестерина у субъекта; снижения инсулинорезистентности, повышения потребления глюкозы или снижения артериального давления у субъекта; уменьшения висцерального жира у субъекта; снижения сывороточных трансаминаз у субъекта; индукции митохондриального дыхания у субъекта; или лечения заболеваний; все эти способы включают введение терапевтического количества соединения, описанного в данной заявке, пациенту, нуждающемуся в этом. В дополнительных вариантах осуществления, заболевание, подлежащее лечению, может быть метаболическим заболеванием. В дополнительном варианте осуществления, метаболическое заболевание может быть выбрано из группы, состоящей из: ожирения, сахарного диабета, особенно диабета 2 типа, гиперинсулинемии, непереносимости глюкозы, метаболического синдрома X, дислипидемии, гипертриглицеридемии, гиперхолестеринемии и стеатоза печени. В других вариантах осуществления, заболевание, подлежащее лечению, может быть выбрано из группы, состоящей из сердечно-сосудистых заболеваний, включая сосудистое заболевание, атеросклероз, ишемическую болезнь сердца, цереброваскулярное заболевание, сердечную недостаточность и заболевание периферических сосудов. В предпочтительных вариантах осуществления, указанные выше способы не приводят к индукции или поддержанию гипогликемического состояния.
Помимо того, что они полезны для лечения человека, определенные соединения и составы, раскрытые в данной заявке, также могут быть полезны для ветеринарного лечения домашних животных, экзотических животных и сельскохозяйственных животных, включая млекопитающих, грызунов и т.п. Более предпочтительные животные включают лошадей, собак и кошек.
Список сокращений
ACN = MeCN = CH3CN = ацетонитрил; Вос = трет-бутилоксикарбонил; BPin = 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил; Br2 = бром; Bu = n-бутил; t-Bu = трет-бутил = 2,2-диметилэтил; °С = градусы Цельсия; CBz = карбоксибензил; CDCl3 = дейтерированный хлороформ; CD3CN = дейтерированный ацетонитрил; DBN = 1,5-диазабицикло(4,3,0)нон-5-ен; DBU = 1,8-диазабицикло(5,4,0)ундец-7-ен; DCM = CH2Cl2 = дихлорметан; DDTT = 3-((диметиламинометилиден)амино)-3Н-1,2,4-дитиазол-5-тион; DIPEA = iPr2NEt = диизопропилэтиламин; DMAP = 4-диметиламинопиридин; DMEDA = N,N'-диметилэтилендиамин; ДМФА = диметилформамид; ДМФА-d7 = диметилформамид-d7; ДМСО = диметилсульфоксид; ДМСО-d6 = диметилсульфоксид-d6;, DMTr = диметокситритил = (4-метоксифенил)2(фенил)метил; D2O = дейтерированная вода; dppf = 1,1'-бис(дифенилфосфино) ферроцен; ЕА = EtOAc = этил ацетат; ES+ =ионизация электрораспылением положительно заряженных ионов; ES-=ионизация электрораспылением отрицательно заряженных ионов; Et = этил; EtOH = этанол; ч = час; Н = водород; HCl = хлороводород; HCO2NH4 = формиат аммония; H2O = вода; ВЭЖХ = высокоэффективная жидкостная хроматография, также известная как препаративная высокоэффективная жидкостная хроматография; пром. соед. = промежуточное соединение; iPr = изопропил = 2-пропил; IPA = iPrOH = изопропанол = 2-пропанол; М = молярный; тСРВА = м-хлорпербензойная кислота; МеОН = метанол; МГц = мегагерц; мл = миллилитр; мин = минуты; МС = масс-спектрометрия; MsCl = метансульфонил хлорид; MW = микроволновое излучение; N2 = азот; NH3 = аммиак; NH4OH = гидроксид аммония; NMP = N-Метил-2-пирролидон; 1H-ЯМР = протонный ядерный магнитный резонанс; 31Р-ЯМР = ядерный магнитный резонанс фосфора; PBS = фосфатно-буферный солевой раствор; РЕ = петролейный эфир; Pin = пинакол = 2,3-диметилбутан-2,3-диол; Pin2B2 = 4,4,4', 4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан); Piv = пивалоил = (СН3)3С-С(=O)-; преп-ВЭЖХ = препаративная высокоэффективная жидкостная хроматография, также известная как жидкостная хроматография при высоком давлении; КТ = комнатная температура; NaOH = гидроксид натрия; Pd(dppf)Cl2 = [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) дихлорид; RuPhos = дициклогексил(2',6'-диизопропокси-[1,1'-бифенил]-2-ил)фосфин; ТГФ = тетрагидрофуран; Ру = пиридин; SFC = сверхкритическая жидкостная хроматография; TBSCl = трет-бутилдиметилсилил хлорид; ТЕА = триэтиламин; ТЕАВ = тетраэтил аммония бикарбонат; TfOH = трифторметансульфокислота; TMSCl = триметилсилил хлорид; TFA = трифторуксусная кислота; K2CO3 = карбонат калия; мкл = микролитр.
Общие способы синтеза соединений
Следующие схемы можно использовать для практического осуществления настоящего изобретения.
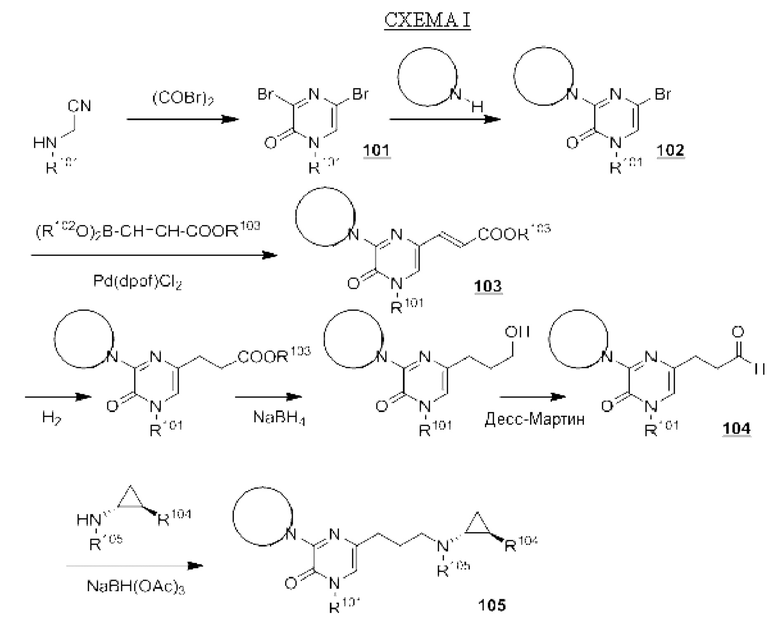
Некоторые примеры, раскрытые в данной заявке, могут быть синтезированы с использованием следующей общей методики синтеза, представленной на Схеме I. Соответствующим образом замещенный амин о ацетонитрил вводят в реакцию с оксалил бромидом с образованием пиразинона 101. Нуклеофильное замещение циклическим амином обеспечивает селективно монозамещенный пиразинон 102. Боковую цепь вводят посредством связывания, опосредованного Pd(II), с фу национализированным винилборонатным реагентом с образованием дважды замещенного пиразинона 103. Серии стадий восстановления и окисления дают альдегид 104, который соединяется с замещенным циклопропиламином в условиях восстановительного аминирования с получением 105.
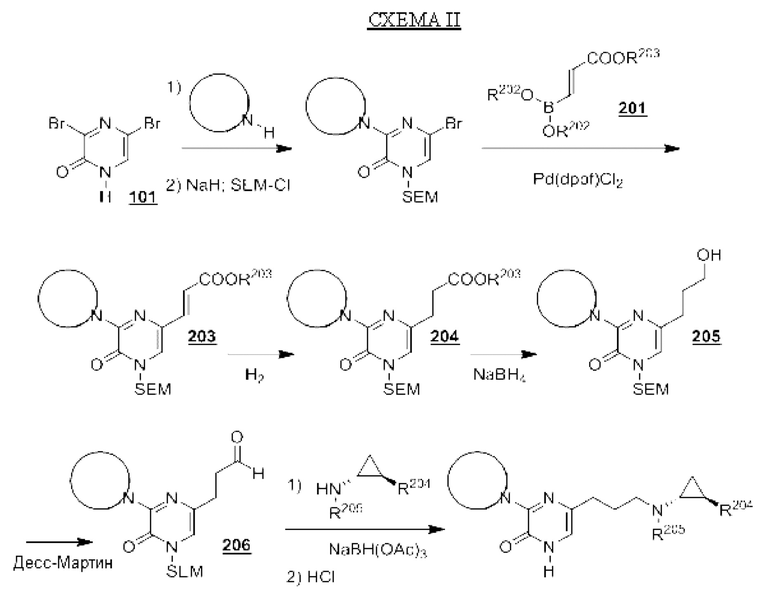
Некоторые примеры, раскрытые в данной заявке, могут быть синтезированы с использованием следующей общей методики синтеза, представленной на Схеме II. Нуклеофнльное замещение дибром пиразинона циклическим амином приводит к появлению первого заместителя. Связывание, опосредованное Pd(II), с винилборонатом 202 вводит боковую цепь. Полученное соединение 203 превращают в альдегид 206 посредством трехстадийной последовательности, состоящей из гидрирования, восстановления гидрида и окисления Десса-Мартина. Альдегид 206 соединяется с замещенным циклопропиламнном в условиях восстановительного аминирования. Наконец, группу SEM удаляют в кислых условиях.
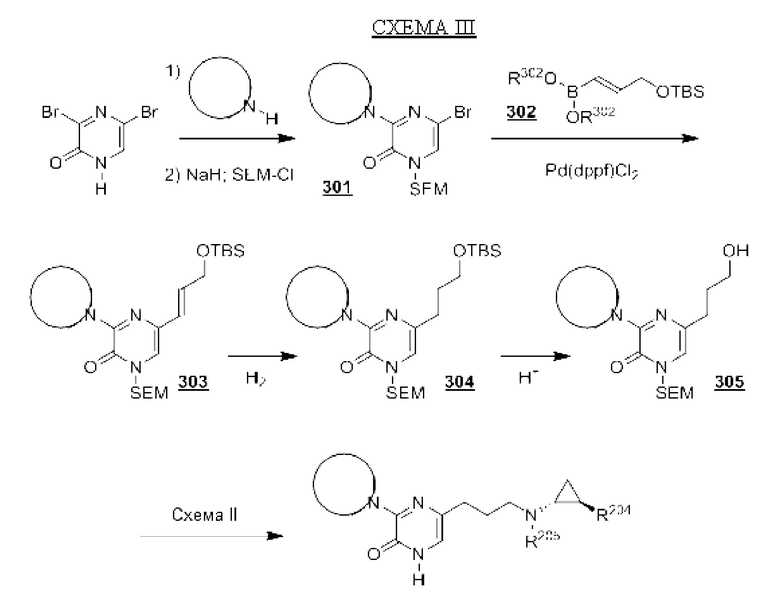
Некоторые примеры, раскрытые в данной заявке, могут быть синтезированы с использованием следующей общей методики синтеза, представленной на Схеме III. Нуклеофильное замещение дибром пиразинона циклическим амином приводит к появлению первого заместителя. Связывание, опосредованное Pd(II), с винилборонатом 302 вводит боковую цепь. Полученное соединение 303 превращают в альдегид 305 посредством двух стад и иной последовательности, состоящей из гидрирования и снятия защиты силилового эфира. Превращение 305 в желаемый продукт осуществляется, как описано для Схемы II.
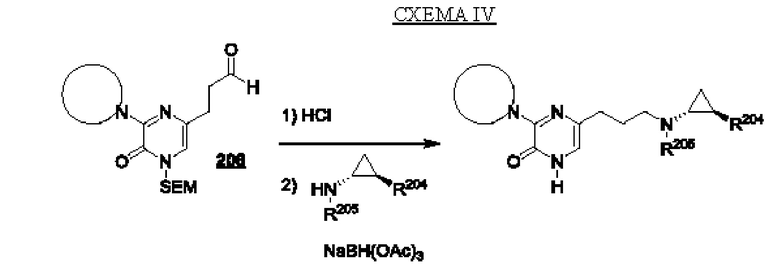
Некоторые примеры, раскрытые в данной заявке, могут быть синтезированы с использованием следующей общей методики синтеза, представленной на Схеме IV.
Альдегид 206, полученный способами, представленными на Схеме II или Схеме III, или другими способами, известными из уровня техники, вводят в реакцию с кислотой для удаления группы SEM. Полученное вещество затем соединяют с замещенным циклопропиламином в условиях восстановительного аминирования.
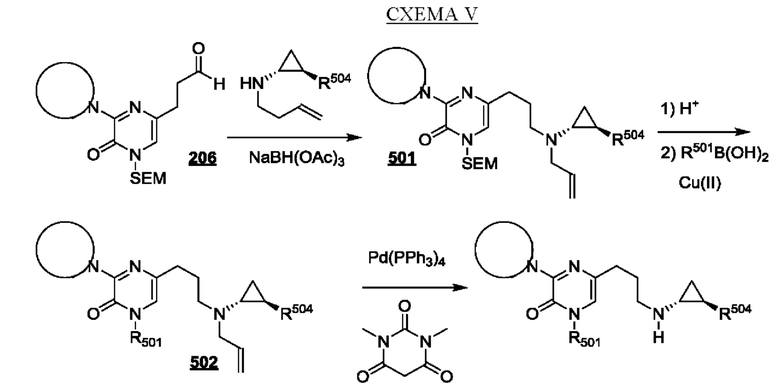
Некоторые примеры, раскрытые в данной заявке, могут быть синтезированы с использованием следующей общей методики синтеза, представленной на Схеме V. Альдегид 206, полученный способами, представленными на Схеме II или Схеме III, или другими способами, известными из уровня техники, затем соединяют с аллил циклопропиламином в условиях восстановительного аминирования. Продукт амин 501 вводят в реакцию с кислотой для удаления группы SEM и затем связывают с подходящим эфиром бороновой кислоты в присутствии Cu(II) с получением 502. Синтез завершают удалением аллильной группы с помощью катализатора Pd(II).
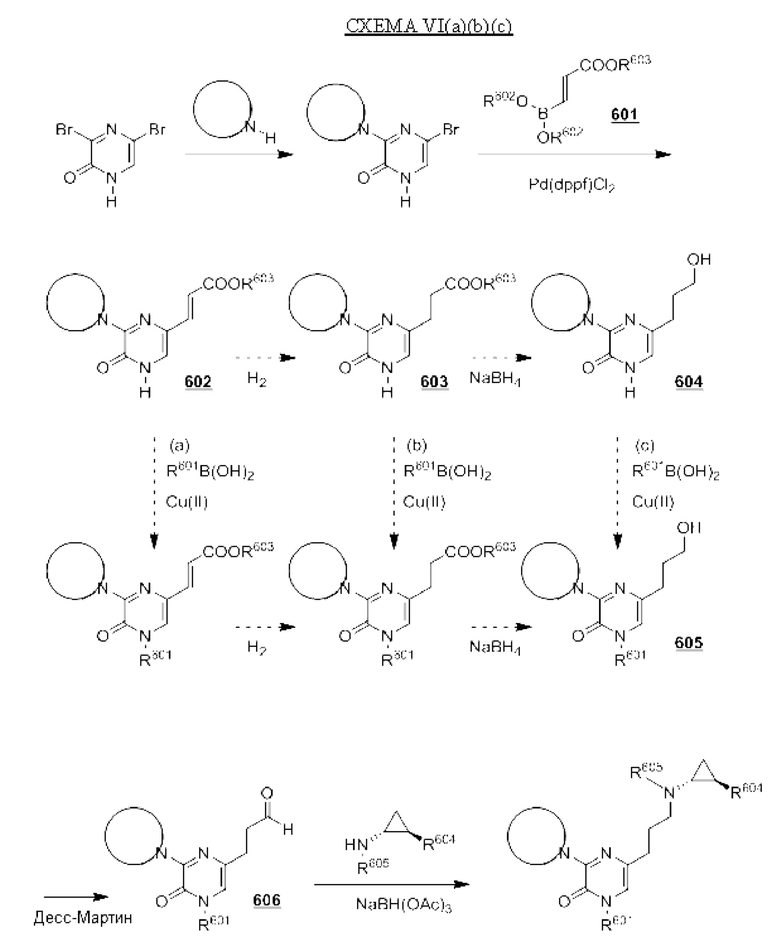
Некоторые примеры, раскрытые в данной заявке, могут быть синтезированы с использованием следующей общей методики синтеза, представленной на Схеме VI. Нуклеофильное замещение дибром пиразинона циклическим амином приводит к появлению первого заместителя. Связывание, опосредованное Pd(H), с винилборонатом 601 вводит боковую цепь. На этой стадии доступны три возможности для опосредованного Cu(II) связывания азота пиразинона с реагентом органобороновой кислоты: (а) до каталитического гидрирования алкена, т.е. связывание с 602, (b) после каталитического гидрирования алкена и до гидридного восстановления сложного эфира, т.е. связывание с 603, или (с) после и каталитического гидрирования алкена, и гидридного восстановления сложного эфира, т.е. связывание с 604. Эти пути показаны на Схеме II. Продукт, полученный в результате любого из трек путей, используют для получения спирта 605, который превращают в желаемый продукт цикл о про пил амина путем окисления Десса-Мартина до альдегида 606 с последующим восстановительным аминированием.
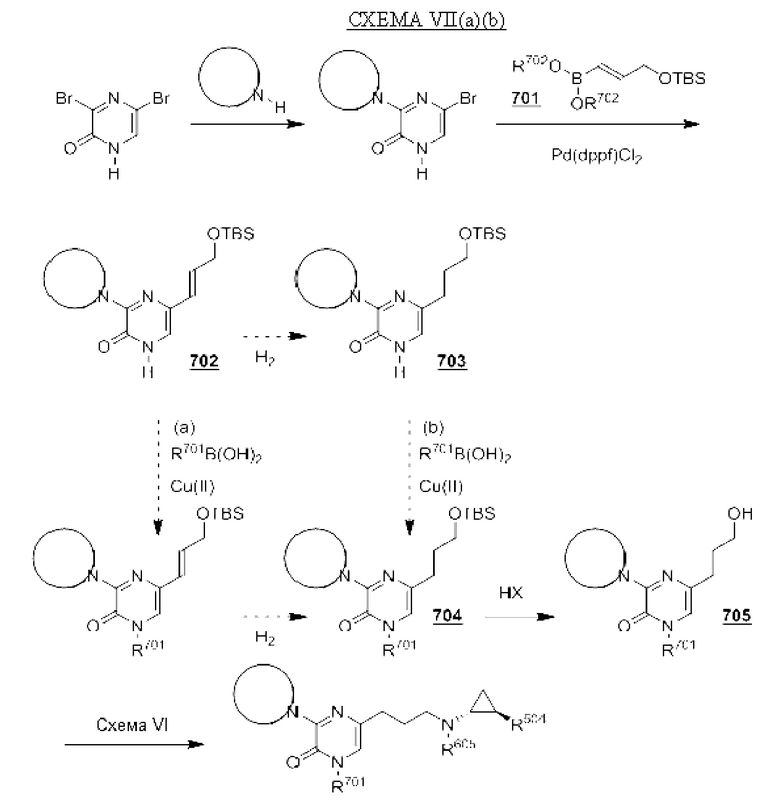
Некоторые примеры, раскрытые в данной заявке, могут быть синтезированы с использованием следующей общей методики синтеза, представленной на Схеме VII. Нуклеофильное замещение дибром пиразинона циклическим амином приводит к появлению первого заместителя. Связывание, опосредованное Pd(II), с винилборонатом 701. вводит боковую цепь. На этой стадии доступны две возможности для опосредованного Cu(II) связывания азота пиразинона с реагентом органобороновой кислоты: (а) до каталитического гидрирования алкена, т.е. связывание с 702, или (b) каталитическое гидрирование алкена, т.е. связывание с 703. Эти два пути показаны на Схеме VII. В обоих случаях с эфира TBS 704 удаляют защитные группы кислотой или другими методами, известными из уровня техники. Полученный спирт 705 превращают в желаемый цикл о про пил а мин с помощью способа, описанного для Схемы VI.
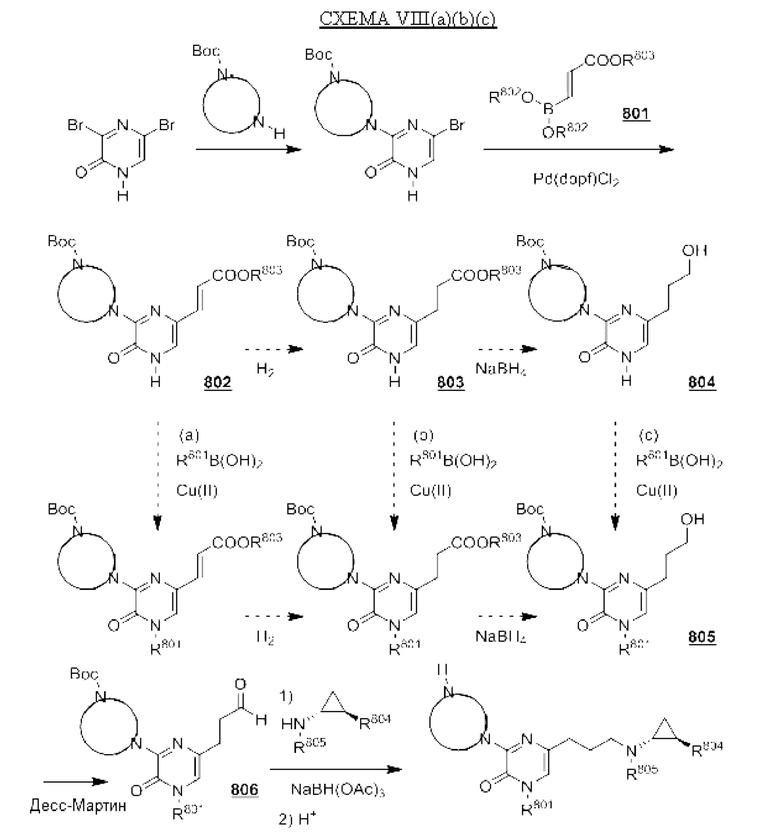
Некоторые примеры, раскрытые в данной заявке, могут быть синтезированы с использованием следующей общей методики синтеза, представленной на Схеме VIII. Нуклеофильным замещением дибром пиразинона циклическим амином, который имеет Вос-защищенную амино-функциональную группу, вводят первый заместитель. Связывание, опосредованное Pd(II), с винилборонатом 301 вводит боковую цепь. Как и на Схеме II, доступны три возможности для опосредованного Cu(II) связывания азота пиразинона с реагентом органобороновой кислоты, и показаны на Схеме III. Полученный спирт 805 превращают в альдегид 306, который, в свою очередь, подвергают условиям восстановительного аминирования. Синтез завершается удалением защитной группы Вое кислотой.
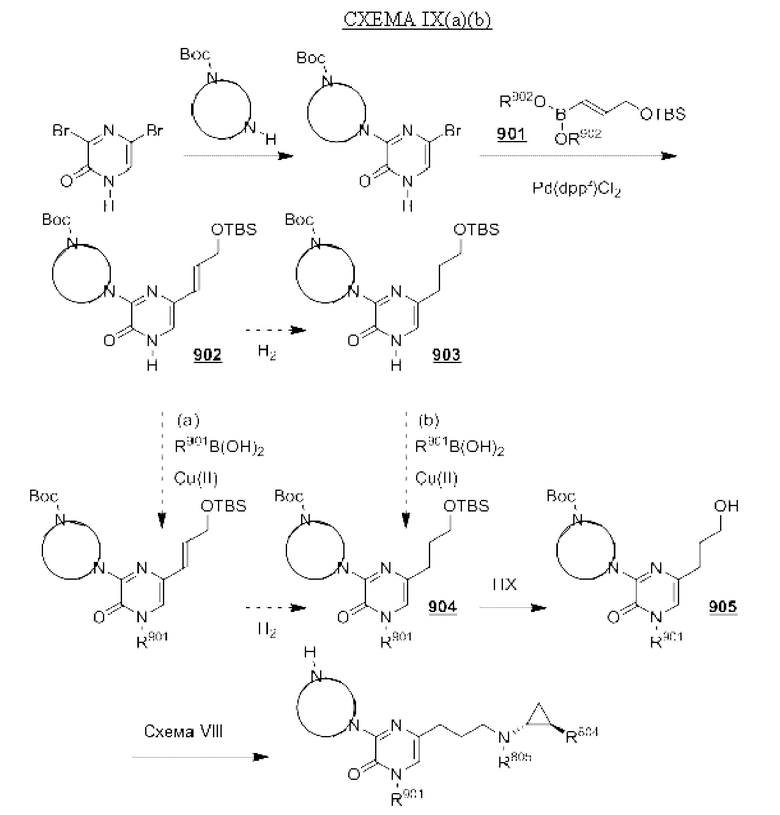
Некоторые примеры, раскрытые в данной заявке, могут быть синтезированы с использованием следующей общей методики синтеза, представленной на Схеме IX. Нуклеофильным замещением дибром пиразинона циклическим амином, который имеет Вос-защищенную амино-функциональную группу, вводят первый заместитель.
Связывание, опосредованное Pd(II), с винил бор онатом 901 вводит боковую цепь. Как и на Схеме VII, доступны две возможности для опосредованного Cu(II) связывания азота пиразинона с реагентом органобороновой кислоты: (а) до каталитического гидрирования алкена, т.е. связывание с 902, или (б) каталитическое гидрирование алкена, т.е. связывание с 903. Эти два пути показаны на Схеме IX. В обоих случаях с эфира TBS 904 удаляют защитные группы кислотой или другими методами, известными из уровня техники. Полученный спирт 905 превращают в желаемый циклопропиламин способами, раскрытыми на Схеме VIII.
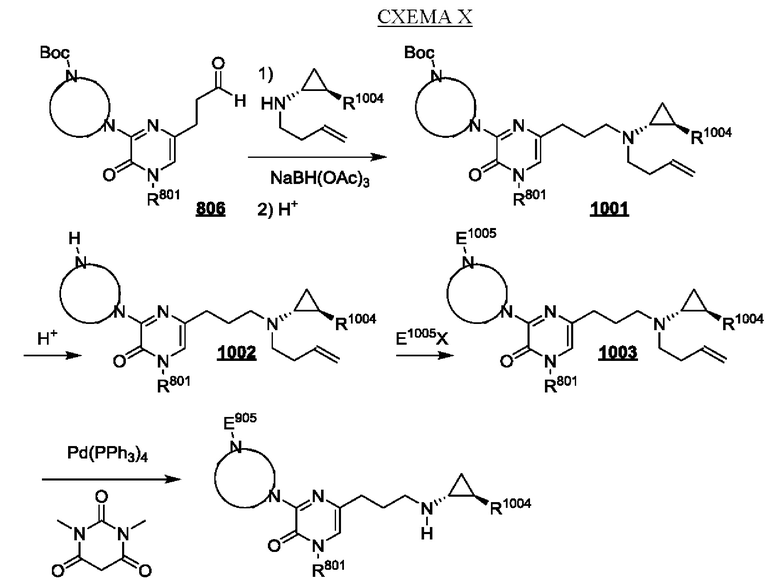
Некоторые примеры, раскрытые в данной заявке, могут быть синтезированы с использованием следующей общей методики синтеза, представленной на Схеме X. Синтез альдегида 806 осуществляют, как приведено на Схеме VIII, или с использованием других методов, известных из уровня техники. Альдегид 806 подвергают условиям восстановительного аминирования с аллиламином, как указано выше. Группу Вое селективно удаляют кислотой с получением амина 1002. и она может быть функционализирована электрофильным компонентом, обозначенным как Е405Х, с получением 1003. Наконец, аллильная группа может быть удалена в присутствии подходящего катализатора Pd(0).
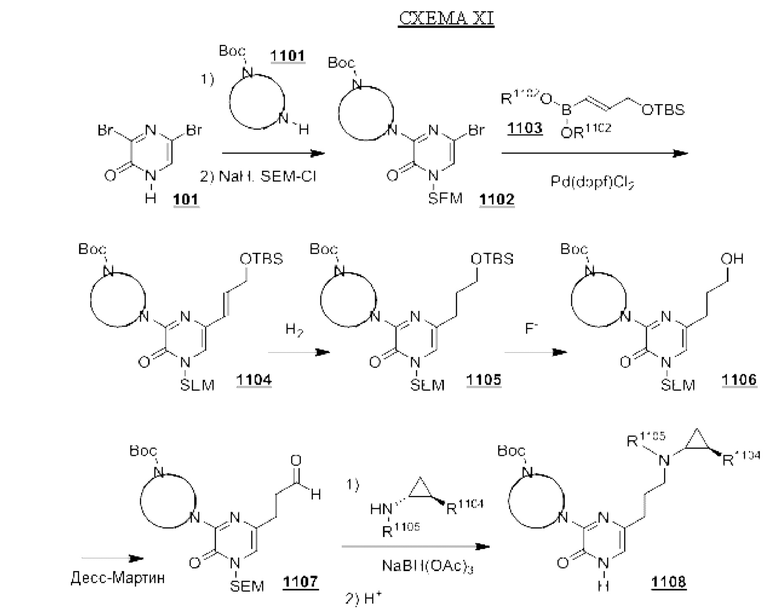
Некоторые примеры, раскрытые в данной заявке, могут быть синтезированы с использованием следующей общей методики синтезы, представленной на Схеме XI. Дибром-предшественник 101 вводят в реакцию с монозащищенным диамином 1101 с получением селективно замещенного пиразинона 1102. Связывание с эфиром бороновой кислоты 1103 дает дизамещенный пиразинон 1104. Манипуляции с функциональными группами осуществляют, как и раньше, с получением альдегида 1107. который затем связывается в условиях восстановительного аминирования с получением 1103.
Изобретение дополнительно проиллюстрировано следующими примерами.
Примеры
Хроматографические методы
Для очистки соединений, раскрытых ниже, могут быть использованы следующие хроматографические методы.
Метод А: Препаративная колонка Sunfire Prep С18 OBD, 10 мкм, 19×250 мм, мобильная фаза Н2О (0,05% TFA)/CH3CN, скорость потока: 20 мл/мин, детектор, УФ 254/210 нм.
Метод В: (2#-Анализ ВЭЖХ-SHIMADZU(ВЭЖХ-10)), препаративная колонка XBridge С18 OBD, размер пор: 100Å, размер частиц: 10 мкм, размер колонки: 19 мм × 250 мм, мобильная фаза: H2O (0,05% TFA)/CH3CN, детектор, УФ 254/220 нм.
Метод С: (2#-Анализ ВЭЖХ-SHIMADZU(ВЭЖХ-10)), препаративная колонка XBridge С18 OBD, размер пор: 100Å, размер частиц: 10 мкм, размер колонки: 19 мм × 250 м, мобильная фаза: H2O (10 мМ NaHCO3)/CH3CN, детектор, УФ 254/220 нм.
Метод D: (2#-Анализ ВЭЖХ-SHIMADZU(ВЭЖХ-10)), колонка XBridge Shield RP18 OBD, размер пор: 130Å, размер частиц 10 мкм, размер колонки: 19×250 мм, мобильная фаза: H2O (0,05% TFA)/CH3CN, скорость потока: 25 мл/мин, детектор, УФ 254/220 нм.
Метод Е: (2#-Анализ ВЭЖХ-SHIMADZU(ВЭЖХ-10)), колонка XBridge Shield RP18 OBD, размер пор: 130Å, размер частиц 10 мкм, размер колонки: 19×250 мм, мобильная фаза: Н2О (10 мМ NaHCO3)/CH3CN, скорость потока: 25 мл/мин, детектор, УФ 254/220 нм.
Метод F: (2#SHIMADZU (ВЭЖХ-01)): колонка Xselect CSH OBD, размер частиц 5 мкм, размер колонки: 30×150 мм; мобильная фаза, Н2О (0,05%TFA) и CH3CN, скорость потока 60 мл/мин, детектор, УФ 220/254 нм.
Метод G: (2#SHIMADZU (ВЭЖХ-01)): колонка: Xselect CSH Fluoro Phenyl OBD, размер частиц 5 мкм, размер колонки: 19×250 мм, 5 мкм; мобильная фаза Н2О (0,05%TFA) и CH3CN, скорость потока: 25 мл/мин, детектор 254/210 нм.
ПРИМЕР 1
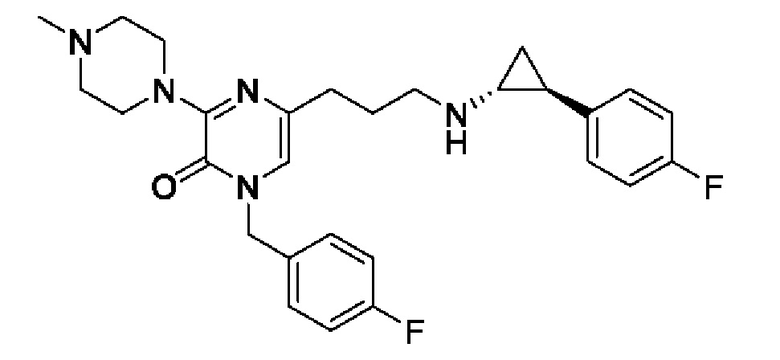
1-[4-Фторбензил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[4-метилпиперазин-1-ил]пиразин-2(1Н)-он
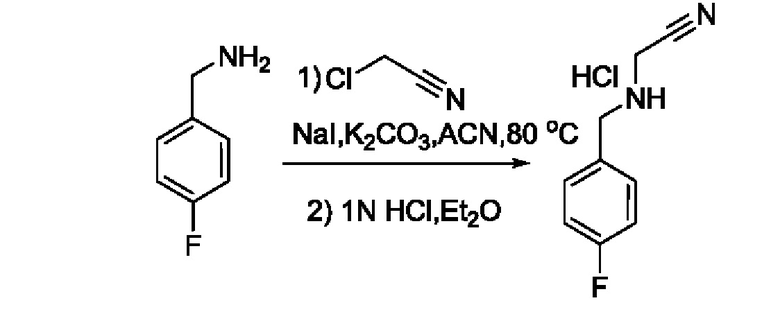
2-[((4-Фторфенил)метил)амино]ацетонитрил
Смесь 2-хлорацетонитрила (6,6 г, 87,90 ммоль, 1,1 эквив.), (4-фторфенил)метанамина (10 г, 79,91 ммоль, 1 эквив.), K2CO3 (33,1 г, 239,72 ммоль, 3 эквив.) и NaI (119,8 мг, 0,80 ммоль, 0,01 эквив.) в CH3CN (200 мл) перемешивали в течение 16 ч при 80°С. Твердые вещества удаляли фильтрованием и фильтрат концентрировали в вакууме. Остаток растворяли в 200 мл Et2O. Полученный в результате раствор разбавляли 50 мл HCl в диоксане. Полученное твердое вещество собирали фильтрованием, получая 10 г (62,37%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
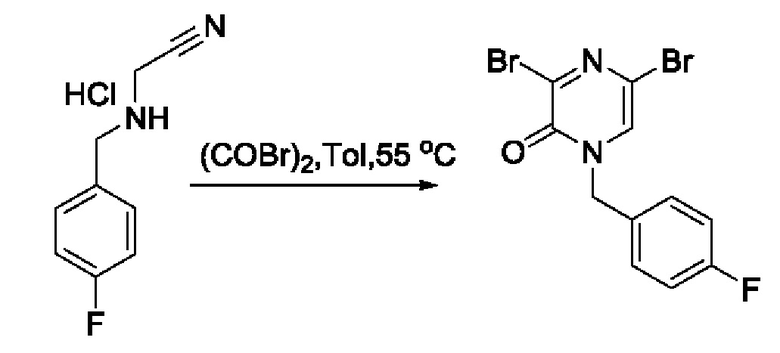
1-((4-Фторфенил)метил)-3,5-дибромпиразин-2(1H)-он
Раствор продукта, полученного на предыдущей стадии, (12 г, 59,81 ммоль, 1 эквив.) и оксалил бромид (64,5 г, 299,04 ммоль, 5 эквив.) в толуоле (200 мл) перемешивали в течение 16 ч при 55°С, затем концентрировали в вакууме. Остаток растворяли в 200 мл CH2Cl2, промывали 2×100 мл водн. Na2CO3 и сушили над безводным Na2SO4. Остаток очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 10 г (46,19%) указанного в заглавии соединения в виде масла желтого цвета.
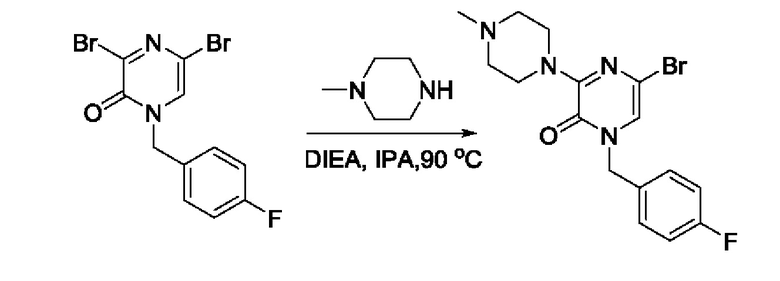
3-(4-Метилпиперазин-1-ил)-5-бром-1-((4-фторфенил)метил)пиразин-2(1Н)-он (Промежуточное соединение 1-3)
Раствор продукта, полученного на предыдущей стадии, (10 г, 27,62 ммоль, 1,00 эквив.) в IPA (500 мл), 1-метилпиперазина (3,31 г, 33,15 ммоль, 1,20 эквив.) и DIEA (7,13 г, 55,25 ммоль, 2,01 эквив.) перемешивали в течение ночи при 90°С. Остаток очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (5:1), получая 10 г (95%) указанного в заглавии соединения в виде масла желтого цвета.
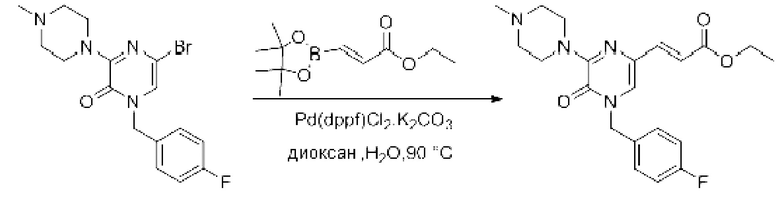
Этил (2E)-3-[6-(4-метилпиперазин-1-ил)-5(4H)-оксо-4-((4-фторфенил)метил)-пиразин-2-ил]пропеноат (Промежуточное соединение 1-4)
Смесь продукта, полученного на предыдущей стадии, (6 г, 15,75 ммоль, 1 эквив.), этил (2Е)-3-(тетраметил-1,3,2-диокса6оролан-2-ил)проп-2-еноата (5,34 г, 23,62 ммоль, 1,5 эквив.), K2CO3 (6,52 г, 47,24 ммоль, 3 эквив.), Pd(dppf)Cl2 (1,15 г, 1,57 ммоль, 0,1 эквив.), диоксана (300 мл) и Н2О (100 мл) перемешивали в течение ночи при 90°С под N2. Остаток очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир, получая 3 г (48%) указанного в заглавии соединения в виде масла желтого цвета.
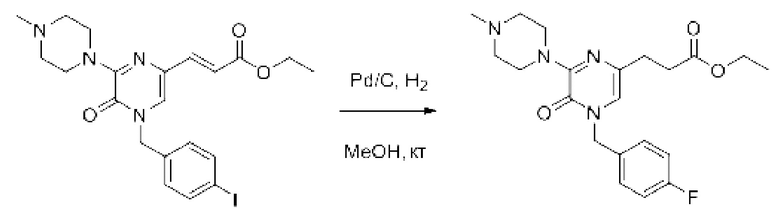
Этил 3-[6-(4-метилпиперазин-1-ил)-5(4Н)-оксо-4-((4-фторфенил)метил)-пиразин-2-ил]пропаноат (Промежуточное соединение 1-5)
Раствор продукта, полученного на предыдущей стадии, (3 г, 7,5 ммоль, 1,00 эквив.) в МеОН (50 мл) перемешивали в течение 1 ч над Pd/С (1,0 г) в атмосфере Н2 при кт. Твердые вещества удаляли фильтрованием и фильтрат концентрировали в вакууме, получая 2,6 г (86%) указанного в заглавии соединения в виде масла желтого цвета.
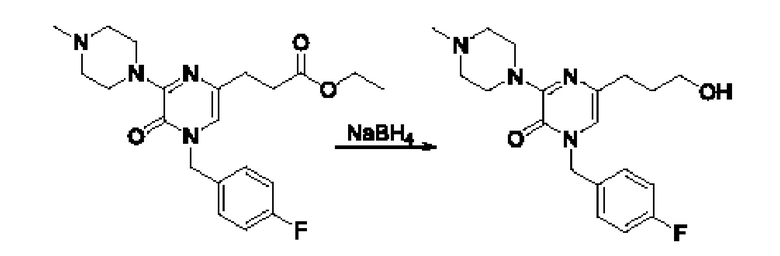
3-[6-(4-Метилпиперазин-1-ил)-4-((4-фторфенил)метил)-5(4Н)-оксопиразин-2-ил1пропан-1-ол (Промежуточное соединение 1-6)
К перемешиваемому раствору продукта, полученного на предыдущей стадии, (1,3 г, 3,23 ммоль, 1 эквив.) в МеОН (50 мл) порциями добавляли NaBH4 (2,46 г, 64,68 ммоль, 20 эквив.). Полученный в результате раствор перемешивали в течение 16 ч при кт. Реакционную смесь затем гасили добавлением 200 мл Н2О. Полученный в результате раствор экстрагировали 3×100 мл CH2Cl2, затем концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (10:1), получая 700 мг (60%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
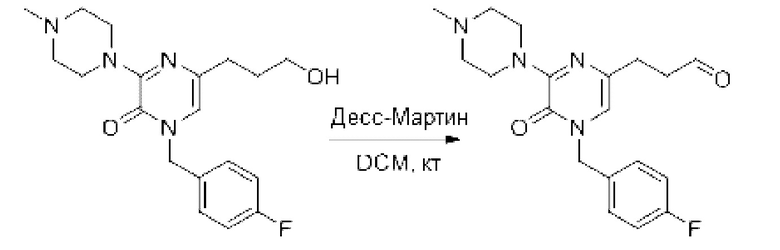
3-[6-(4-Метилпиперазин-1-ил)-4-((4-фторфенил)метил)-5(4Н)-оксопиразин-2-ил]пропанал (Промежуточное соединение 1-7)
Раствор продукта, полученного на предыдущей стадии, (600 мг, 1,67 ммоль, 1,00 эквив.) и реактив Десса-Мартина (848 мг, 2,00 ммоль, 1,20 эквив.) в CH2Cl2 (30 мл) перемешивали в течение 1 ч при кт, затем концентрировали в вакууме и очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (10:1), получая 400 мг (67%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
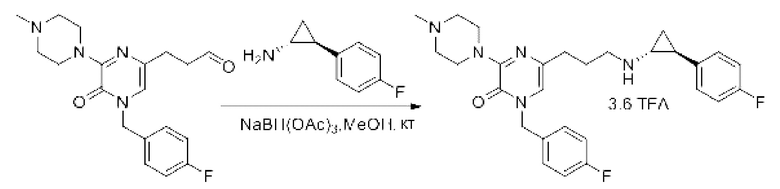
l-[4-Фторбензил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)-пропил]-3-[4-метилпиперазин-1-ил]пиразин-2(1Н)-он (Пример 1)
Раствор продукта, полученного на предыдущей стадии, (400 мг, 1,12 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)-циклопропан-1-амина (202 мг, 1,34 ммоль, 1,2 эквив.) в МеОН (20 мл) перемешивали в течение 30 мин при кт. К раствору затем добавляли NaBH(OAc)3 (568 мг, 2,68 ммоль, 2,4 эквив.) при кт. Полученный в результате раствор перемешивали в течение 30 мин при кт. Реакционную смесь затем гасили добавлением 30 мл Н2О. Полученный в результате раствор экстрагировали 3×30 мл CH2Cl2. Органические слои объединяли, концентрировали при пониженном давлении, и очищали, используя хроматографический Метод А (от 30% до 33% CH3CN за 9 мин), получая 88,2 мг (9%) указанного в заглавии соединения в виде масла желтого цвета.
ЖХ-МС: (ES, m/z): 494 [М+Н]+. 1Н ЯМР (400 МГц, MeOD-d4) δ м.д.: 7,39-7,36 (м, 2Н), 7,18-7,15 (м, 2Н), 7,07-7,01 (м, 5Н), 5,04 (с, 2Н), 4,87481 (м, 2Н), 3,52-3,47 (м, 2Н), 3,21-3,12 (м, 6Н), 2,94-2,90 (м, 4Н), 2,53-2,42 (м, 3Н), 2,06-1,98 (м, 2Н), 1,49-1,32 (м, 2Н).
ПРИМЕР 2
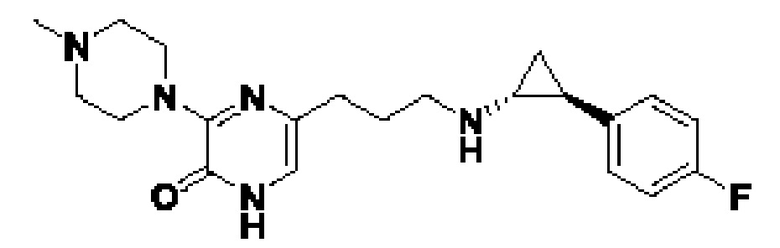
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]3-[4-метилпиперазин-1-ил]-пиразин-2(1Н)-он
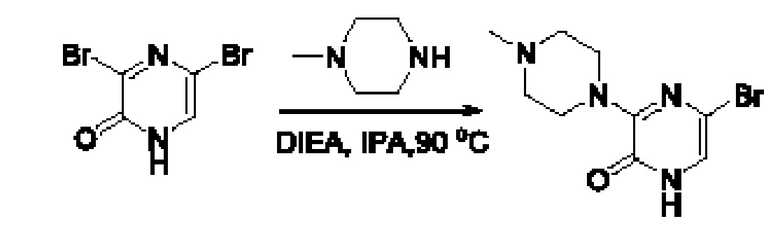
5-Бром-3-(4-метилпиперазин-1-ил)-пиразин-2(1Н)-он (Промежуточное соединение 2-1)
Раствор 3,5-дибром-1,2-дигидропиразин-2-она (10 г, 39,84 ммоль, 1,00 эквив.), 1-метилпиперазина (4,38 г, 1,1 эквив.) и DIEA (15,41 г, 3,0 эквив.) в IPA (50 мл) перемешивали в течение 16 ч при 90°С, затем охлаждали и концентрировали в вакууме, получая 10 г (91%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
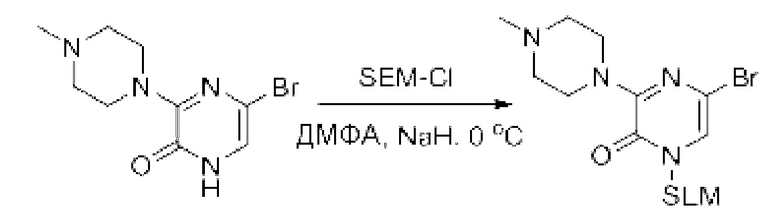
5-Бром-3-(4-метилпиперазин-1-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1Н)-он (Промежуточное соединение 2-2)
К раствору продукта, полученного на предыдущей стадии, (10 г, 36,76 ммоль, 1 эквив.) в ДМФА (500 мл) добавляли NaH (60%) (2,21 г, 55,25 ммоль, 1,5 эквив.). Полученный в результате раствор перемешивали в течение 1 ч при 0°С, затем по каплям добавляли раствор [2-(хлорметокси)этил]триметилсилана (9,15 г, 55,12 ммоль, 1,5 эквив.) в ДМФА (100 мл) при перемешивании в течение 30 мин. Полученный в результате раствор перемешивали еще в течение 4 ч при кт, затем разбавляли 500 мл Н2О и экстрагировали 3×500 мл of EtOAc. Объединенные органические слои концентрировали и очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 10,5 г (55,08%о) указанного в заглавии соединения в виде масла желтого цвета.
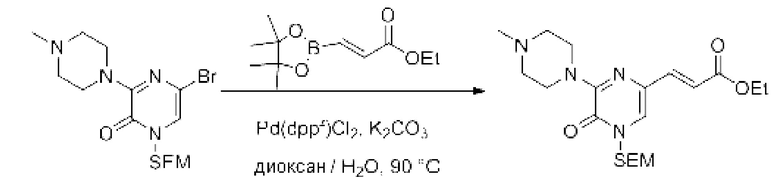
Этил (2E)-3-[6-(4-метилпиперазин-1-ил)-5(4H)-оксо-4-[(2-(триметилсилил)-этокси)метил]-пиразин-2-ил]пропеноат (Промежуточное соединение 2-3)
Методику получения Промежуточного соединения 1-4 использовали с продуктом, полученным на предыдущей стадии, (5 г, 12,39 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 2,5 г (55%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
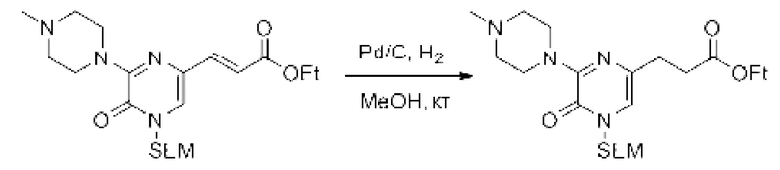
Этил 3-[6-(4-метилпиперазин-1-ил)-5(4H)-оксо-4-[(2-(триметилсилил)-этокси)метил]-пиразин-2-ил]пропаноат (Промежуточное соединение 2-4)
Методику получения Промежуточного соединения 1-5 использовали с Промежуточным соединением 2-3 (2,5 г, 5,92 ммоль, 1,00 эквив.), получая 2,2 г (93%о) указанного в заглавии соединения в виде твердого вещества.
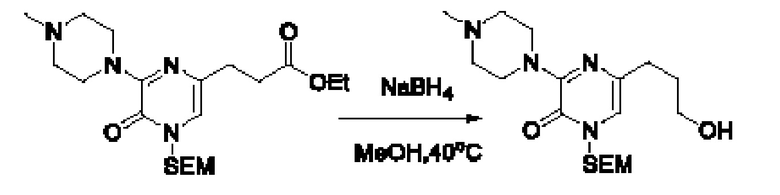
3-[6-(4-Метилпиперазин-1-ил)-5(4Н)-оксо-4-[[2-(триметилсилил)этокси]метил]пиразин-2-ил]пропан-1-ол (Промежуточное соединение 2-5)
Методику получения Промежуточного соединения 1-6 использовали с Промежуточным соединением 2-4 (2,2 г, 5,92 ммоль, 1,00 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (1:10), получая 1,3 г (52%) указанного в заглавии соединения в виде твердого вещества.
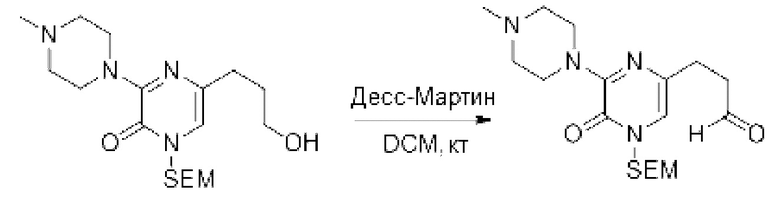
3-[6-(4-Метилпиперазин-1-ил)-5(4Н)-оксо-4-[[2-(триметилсилил)этокси]метил]пиразин-2-ил]пропаналь (Промежуточное соединение 2-6)
Методику получения Промежуточного соединения 1-7 использовали с Промежуточным соединением 2-5 (1,3 г, 3,40 ммоль, 1,00 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 700 мг (74%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
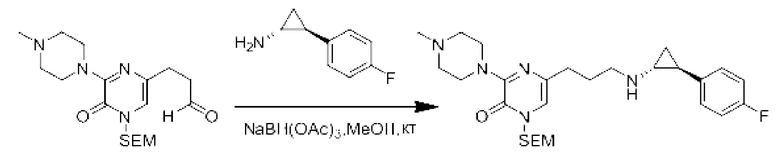
5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил]амино]пропил)-3-(4-метилпиперазин-1-ил)-1-[[2-(триметилсилил)этокси]метил]пиразин-2(1Н)-он (Промежуточное соединение 2-7)
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с Промежуточным соединением 2-6 (700 мг, 1,84 ммоль, 1,00 эквив.). Остаток очищали с использованием хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 400 мг (69%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
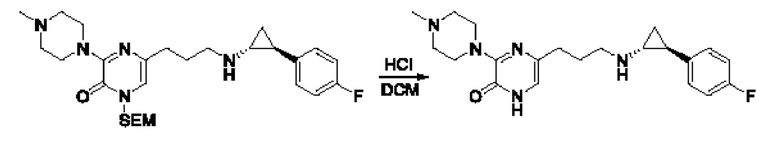
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]3-[4-метилпиперазин-1-ил]-пиразин-2(1Н)-он (Пример 2)
Раствор Промежуточного соединения 2-7 (300 мг, 0,58 ммоль, 1,00 эквив.) в водн. HCl (25 мл)/CH2Cl2 (25 мл) перемешивали в течение 1 ч при кт. Полученную смесь концентрировали в вакууме, затем очищали, используя хроматографический Метод Е (от 28% до 60% CH3CN за 7 мин), Rt: 6,20 мин, получая 44,1 мг (20%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
ЖХ-МС: (ES, m/z): 386 [М+Н]+. 1Н ЯМР (400 МГц, Метанол-d4) δ 7,06-7,03 (м, 2Н), 6,97-6,93 (м, 2Н), 6,65-6,62 (с, 1Н), 3,82-3,72 (с, 4Н), 2,78-2,72 (м, 2Н), 2,58-2,50 (м, 4Н), 2,48-2,42 (м, 2Н), 2,36-2,30 (м, 4Н), 1,90-1,85 (м, 3Н), 1,10-1,02 (м, 1Н), 1,02-0,95 (м, 1Н).
ПРИМЕР 3
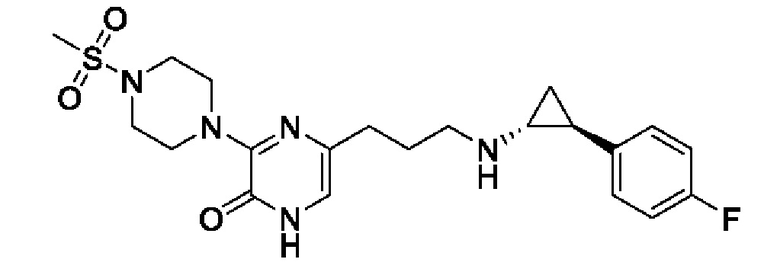
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[4-(метилсульфонил)пиперазин-1-ил]пиразин-2(1H)-он
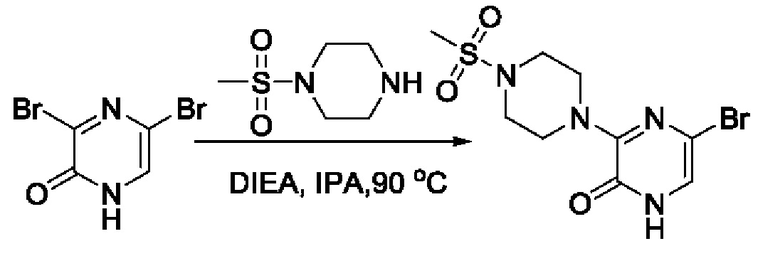
5-Бром-3-(4-(метилсульфонил)пиперазин-1-ил)-пиразин-2(1H)-он (Промежуточное соединение 3-1)
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (10 г, 39,39 ммоль, 1,00 эквив.) и 1-(метилсульфонил)пиперазином (7,80 г, 47,56 ммоль, 1,21 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (4:1), получая 10 г (75%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
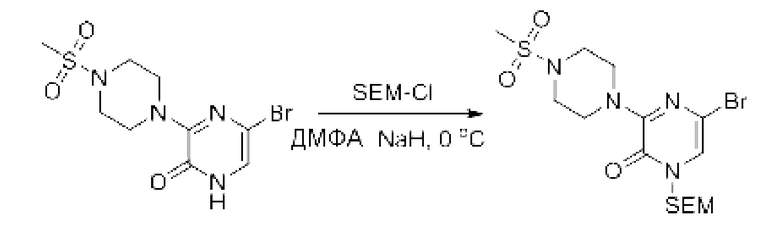
5-Бром-3-(4-(метилсульфонил)пиперазин-1-ил)-1-[(2-(триметилсилил)-этокси)метил]-пиразин-2(1Н)-он (Промежуточное соединение 3-2)
Методику получения Промежуточного соединения 2-2 использовали с Промежуточным соединением 3-1. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 8,2 г (59%) указанного в заглавии соединения в виде масла желтого цвета.
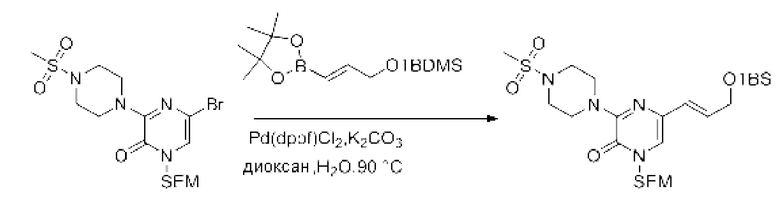
(E)-5-[[3-((трет-бутилдиметил)силил)окси]пропен-1-ил]-3-(4-(метилсульфонил)пиперазин-1-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1Н)-он (Промежуточное соединение 3-3)
Смесь Промежуточного соединения 3-2 (8,2 г, 17,56 ммоль, 1 эквив.), трет-бутилдиметил[([2Е]-3-[тетраметил-1,3,2-диокса6оролан-2-ил]проп-2-ен-1-ил)окси]силана (7,85 г, 26,34 ммоль, 1,5 эквив.), K2CO3 (7,27 г, 52,68 ммоль, 3 эквив.), Pd(dppi)Cl2 (1,28 г, 1,74 ммоль, 0,1 эквив.), диоксана (450 мл), и H2O (150 мл) перемешивали в течение ночи при 90°С, концентрировали в вакууме и очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:2), получая 3,2 г (33%) указанного в заглавии соединения в виде масла желтого цвета.
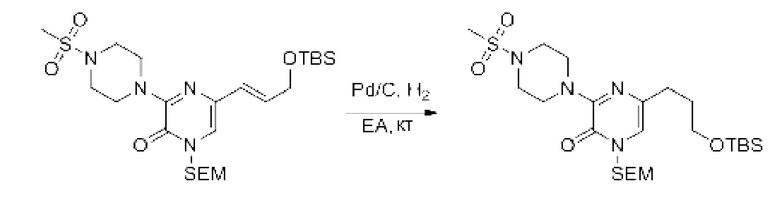
5-[[3-((трет-бутилдиметил)силил)окси]пропил]-3-(4-(метилсульфонил)-пиперазин-1-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1H)-он (Промежуточное соединение 3-4)
Раствор Промежуточного соединения 3-3 (1,5 г, 2,68 ммоль, 1,00 эквив.) в EtOAc (100 мл) перемешивали над Pd/С (0,15 г) в атмосфере Н2 в течение 2 ч при кт. Твердые вещества удаляли фильтрованием и фильтрат концентрировали в вакууме, получая 1,2 г (80%) указанного в заглавии соединения в виде масла желтого цвета.
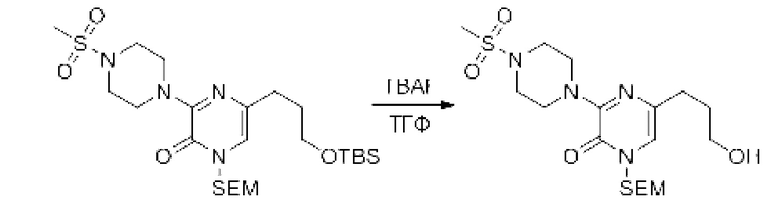
3-[6-(4-(метилсульфонил)пиперазин-1-ил)-5(4Н)-оксо-4-[(2-(триметилсилил)этокси)метил]-пиразин-2ил]пропан-1-ол (Промежуточное соединение 3-5)
Раствор Промежуточного соединения 3-4 (1,2 г, 2,14 ммоль, 1 эквив.) и TBAF (10 мл, ТГФ) в ТГФ (50 мл) перемешивали в течение 2 ч при кт. Остаток очищали с помощью хроматографии на силикагеле, используя CH3CN/H2O (1:3), получая 750 мг (78%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
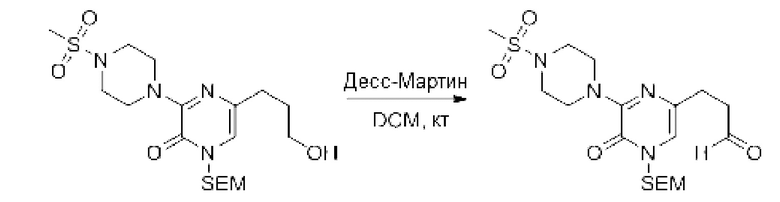
3-[6-(4-(метилсульфонил)пиперазин-1-ил)-5(4Н)-оксо-4-[(2-(триметилсилил)этокси)метил]-пиразин-2ил]пропаналь
Методику получения Промежуточного соединения 1-7 использовали с Промежуточным соединением 3-5 (600 мг, 1,34 ммоль), получая 400 мг (67%) указанного в заглавии соединения в виде масла желтого цвета.
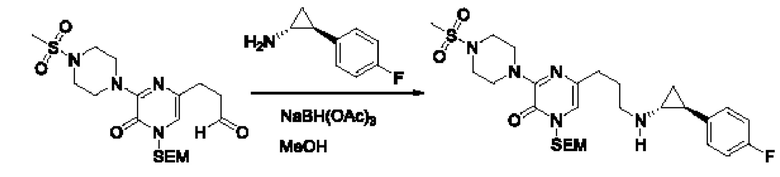
5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил]амино]пропил)-3-(4-(метилсульфонил)пиперазин-1-ил)-1-[[2-(триметилсилил)этокси]метил]пиразин-2(1Н)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (400 мг, 0,899 ммоль, 1 эквив.). Неочищенный продукт реакции очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (2:1), получая 300 мг (58%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
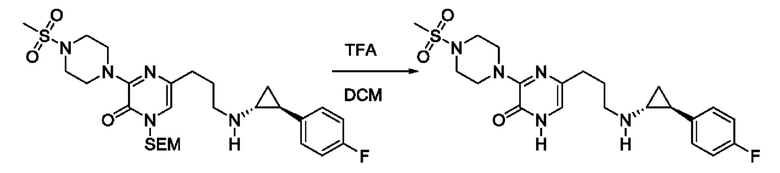
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[4-(метилсульфонил)пиперазин-1-ил]пиразин-2(1H)-он Раствор продукта, полученного на предыдущей стадии, (300 мг, 0,52 ммоль, 1 эквив.) и TFA (2 мл) в CH2Cl2 (10 мл) перемешивали в течение 1 ч при кт. Неочищенный продукт (5 мл) очищали, используя хроматографический Метод В (от 10,0% до 29,0% CH3CN за 9 мин), получая 49,1 мг (21%) указанного в заглавии соединения в виде масла желтого цвета.
ЖХ-МС: (ES, m/z): 450 [М+Н]+. 1Н ЯМР (400 МГц, MeOD-d4) δ м.д.: 7,20-7,17 (м, 2Н), 7,07-7,02 (м, 2Н), 6,69 (с, 1Н), 3,89-3,80 (м, 4Н), 3,30-3,27 (м, 4Н), 3,26-3,22 (м, 2Н), 2,98-2,94 (м, 1Н), 2,85 (с, 3Н), 2,55-2,51 (м, 2Н), 2,47-2,42 (м, 1Н), 2,08-2,01 (м, 2Н), 1,50-1,46 (м, 1Н), 1,40-1,34 (м, 1Н).
ПРИМЕР 4
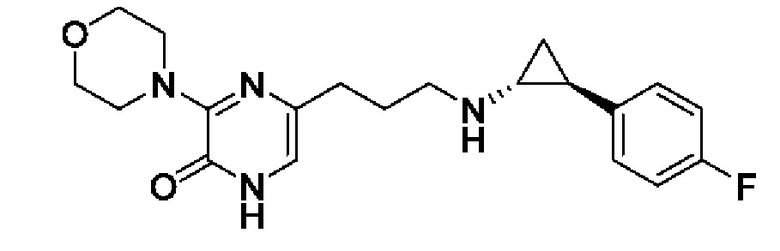
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-морфолинопиразин-2(1Н)-он
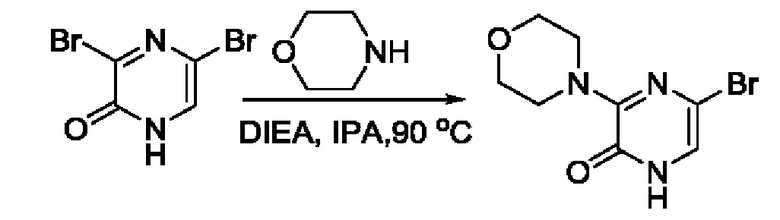
5-Бром-3-(морфолин-4-ил)-пиразин-2(1H)-он (Промежуточное соединение 4-1)
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (10 г, 39,39 ммоль, 1,00 эквив.) и морфолином (5,1 г, 58,54 ммоль, 1,50 эквив.), с временем реакции 2 ч при 90°С, получая 9 г (88%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
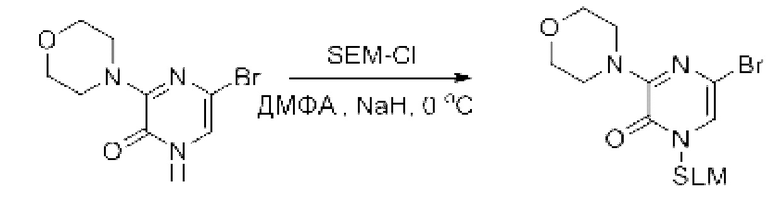
5-Бром-3-(морфолин-4-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1H)-он (Промежуточное соединение 4-2)
Смесь Промежуточного соединения 4-1 (9 г, 34,60 ммоль, 1,00 эквив.), NaH (2,4 г, 100,00 ммоль, 3,00 эквив.) и [2-(хлорметокси)этил]триметилсилана (8,6 г, 51,58 ммоль, 1,50 эквив.) в ДМФА (80 мл) перемешивали в течение 3 ч при 0-10°С. Реакционную смесь гасили и разбавляли 500 мл EtOAc. Полученную смесь промывали 5×200 мл Н2О. Органическую фазу сушили над Na2SO4 и концентрировали в вакууме, получая 7 г (52%) указанного в заглавии соединения в виде масла светло-желтого цвета.
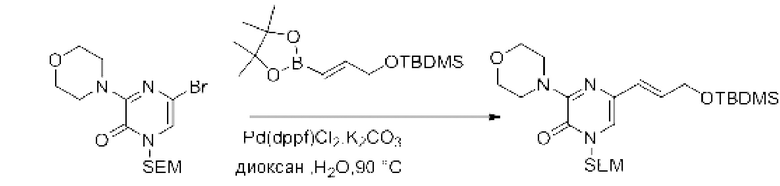
(E)-5-[[3-((трет-бутилдиметил)силил)окси]пропен-1-ил]-3-(морфолин-4-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1Н)-он (Промежуточное соединение 4-3)
Методику получения Промежуточного соединения 3-3 использовали с Промежуточным соединением 4-2 (7 г, 17,93 ммоль), с временем реакции 2 ч при 90°С, получая 3,6 г (42%) указанного в заглавии соединения в виде масла светло-желтого цвета.
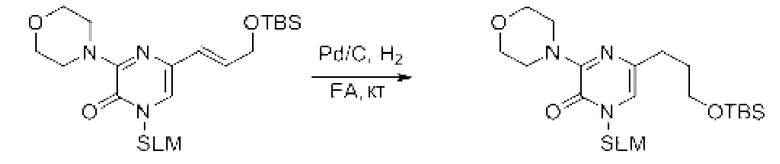
5-[[3-((трет-бутилдиметил)силил)окси]пропил]-3-(морфолин-4-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1Н)-он (Промежуточное соединение 4-4)
Методику получения Промежуточного соединения 3-4 использовали с Промежуточным соединением 4-3 (3,6 г, 7,47 ммоль, 1,00 эквив.), получая 3,2 г (89%) указанного в заглавии соединения в виде масла светло-желтого цвета.
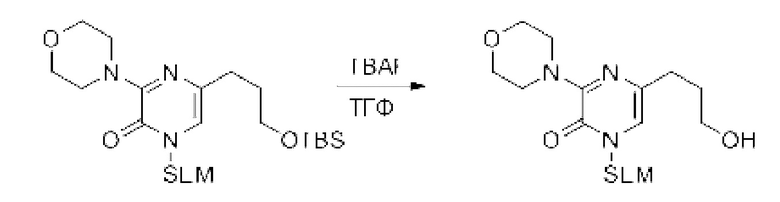
3-[6-(морфолин-4-ил)-5(4Н)-оксо-4-[(2-(триметилсилил)этокси)метил]-пиразин-2-ил]-пропан-1-ол (Промежуточное с о единение 4-5)
Раствор Промежуточного соединения 4-4 (1,4 г, 2,89 ммоль, 1,00 эквив.) и TBAF (5 мл, 1,20 эквив.) в ТГФ (30 мл) перемешивали в течение 2 ч при 25°С. Остаток очищали с помощью колонки с силикагелем, используя H2O/MeCN (2:1), получая 0,73 г (68%) указанного в заглавии соединения в виде масла светло-желтого цвета.
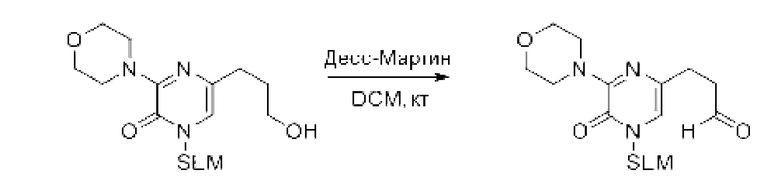
3-[6-(морфолин-4-ил)-5(4H)-оксо-4-[(2-(триметилсилил)этокси)метил]-пиразин-2-ил]-пропаналь (Промежуточное соединение 4-6)
Методику получения Промежуточного соединения 1-7 использовали с Промежуточным соединением 4-5 (700 мг, 1,89 ммоль, 1,00 эквив.), получая 0,35 г (50%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
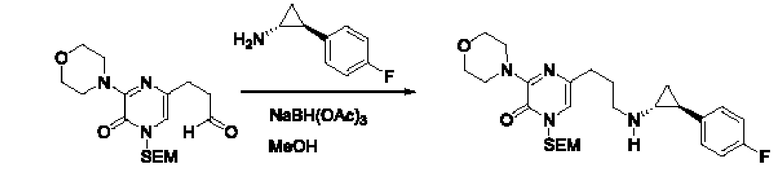
5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил]амино]пропил)-3-(морфолин-4-ил)-1-[[2-(триметилсилил)этокси]метил]пиразин-2(1Н)-он (Промежуточное соединение 4-7)
Смесь Промежуточного соединения 4-6 (350 мг, 0,95 ммоль, 1,00 эквив.), (1R,2S)-2-(4-фторфенил)циклопропан-1-амина (170 мг, 1,12 ммоль, 1,10 эквив.), NaBH(OAc)3 (480 мг, 2,26 ммоль, 2,40 эквив.) и МеОН (20 мл) перемешивали в течение 2 ч при 25°С, затем разбавляли 100 мл CH2Cl2, промывали 3×20 мл Н2О, сушили над Na2SO4 и концентрировали при пониженном давлении, получая 0,32 г (67%) указанного в заглавии соединения в вид е масла светло-желтого цвета.
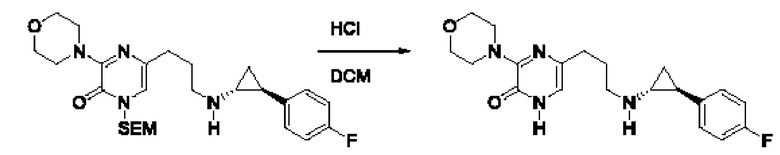
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-морфолиногтразин-2(1H)-он
Стадию удаления защитных групп для получения Примера 2 из Промежуточного соединения 2-7 использовали с продуктом, полученным на предыдущей стадии, (320 мг, 0,64 ммоль). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод В (от 30,0% до 50,0% CH3CN за 8 мин), получая 115,4 мг (49%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-МС: (ES,m/z): 373 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 7,23-7,13 (м, 2Н), 7,13-6,96 (м, 2Н), 6,70-6,60 (с, 1Н), 3,80-3,68 (м, 8Н), 3,22-3,18 (м, 1Н), 3,00-2,82 (м, 1Н), 2,59-2,36 (м, 3Н), 2,07-1,97 (м, 3Н), 1,52-1,30 (м, 2Н).
ПРИМЕР 5
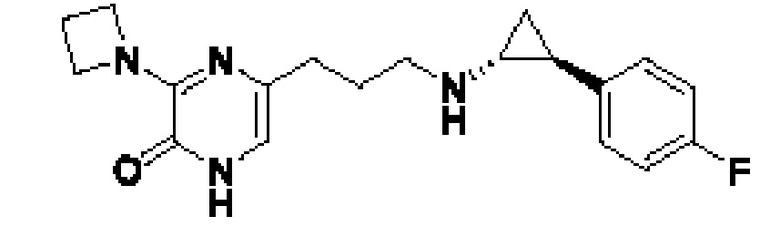
3-[Азетидин-1-ил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-пиразин-2(1Н)-он
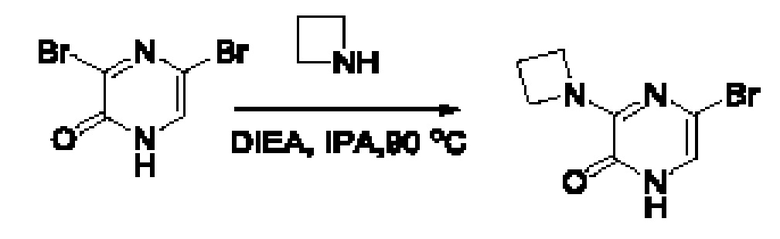
5-Бром-3-(азетидин-1-ил)-ггиразин-2(1Н)-он
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (10 г, 39,39 ммоль, 1,00 эквив.) и азетидином (2,75 г, 47,56 ммоль, 1,21 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (4:1), получая 7,1 г (78,1%) указанного в заглавии соединения в виде твердого вещества белого цвета.
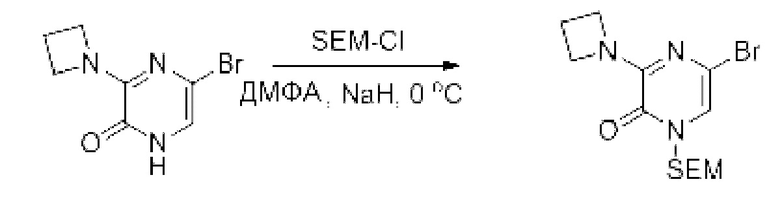
5-Бром-3-(азетидин-1-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1H)-он
Методику получения Промежуточного соединения 2-2 использовали с продуктом, полученным на предыдущей стадии, (7,1 г, 30,87 ммоль, 1 эквив.), получая 7,2 г (64,8%)указанного в заглавии соединения в виде масла желтого цвета.
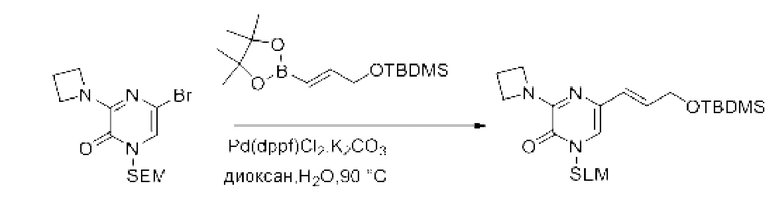
(Е)-5-[[3-((трет-бутилдиметил)силил)окси]пропен-1-ил]-3-(азетидин-1-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1H)-он
Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (7,2 г, 20,0 ммоль), получая 3,30 г (36,4%) указанного в заглавии соединения в виде масла желтого цвета.
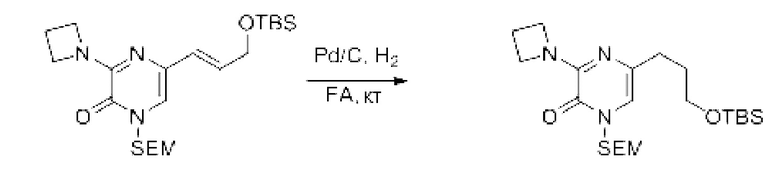
5-[[3-((трет-бутилдиметил)силил)окси]пропил]-3-(азетидин-1-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1H)-он
Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (3,30 г, 7,30 ммоль, 1,00 эквив.), получая 3,10 г (94%) указанного в заглавии соединения в вид е масла оранжевого цвета.
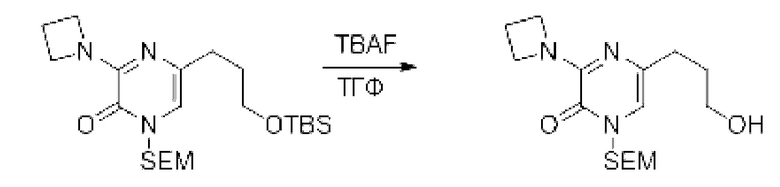
3-[6-(азетидин-1-ил)-5(4H)-оксо-4-[(2-(триметилсилил)этокси)метил]-пиразин-2-ил]-пропан-1-ол
Методику получения Промежуточного соединения 3-5 использовали с продуктом, полученным на предыдущей стадии, (3,0 г, 6,61 ммоль, 1 эквив.), получая 1,5 г (66,76%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
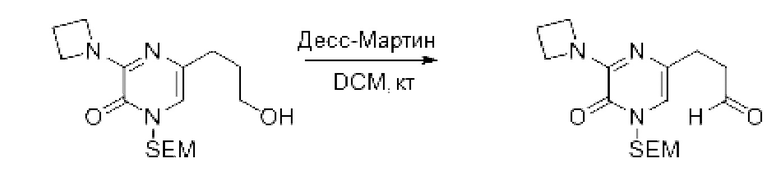
3-[6-(азетидин-1-ил)-5(4H)-оксо-4-[(2-(триметилсилил)этокси)метил]-1шразин-2-ил]-пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (1,00 г, 2,94 ммоль, 1,00 эквив.), получая 0,6 г (60,35%) указанного в заглавии соединения в виде масла желтого цвета.
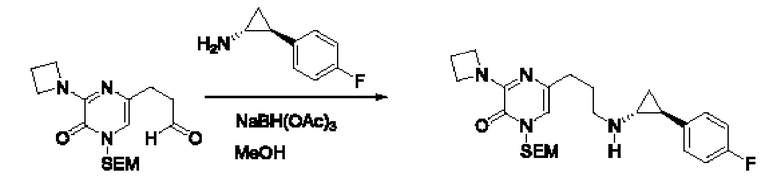
5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил]амино]пропил)-3-(азетидин-1-ил)-1-[[2-(триметилсилил)этокси]метил]пиразин-2(1Н)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (600 мг, 1,78 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (2:1), получая 500 мг (59,6%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
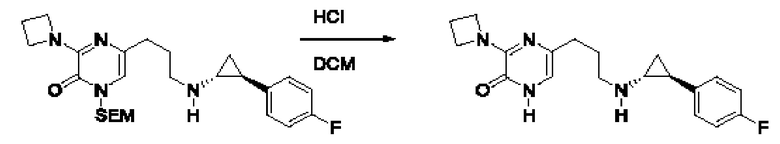
3-[Азетидин-1-ил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-пиразин-2(1H)-он
Стадию удаления защитных групп для получения Примера 2 из Промежуточного соединения 2-7 использовали с продуктом, полученным на предыдущей стадии, (500 мг, 1,06 ммоль, 1 эквив.). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод А (от 10%о до 58%) CH3CN), получая 39,1 мг (10,81%)) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-MC: (ES, m/z): 343 [М+Н]+. 1Н ЯМР (400 МГц, MeOD-d4) δ м.д.: 7,23-7,18 (м, 2Н), 7,05 (т, J=8,6 Гц, 2Н), 6,42 (с, 1Н), 4,61 (с, 4Н), 3,22 (т, J=8,6 Гц, 2Н), 2,98-2,94 (м, 1Н), 2,55-2,42 (м, 5Н), 2,04-1,96 (м, 2Н), 1,54-1,47 (м, 1Н), 1,40-1,33 (м, 1Н).
ПРИМЕР 6
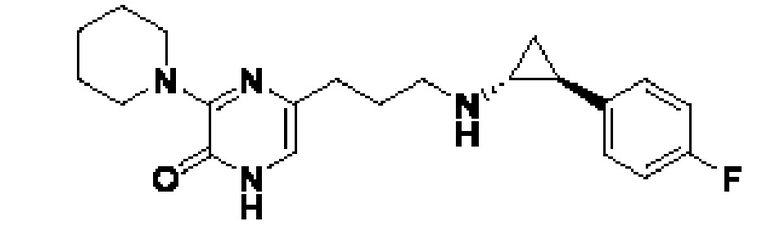
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[пиперидин-1-ил]пиразин-2(1Н)-он
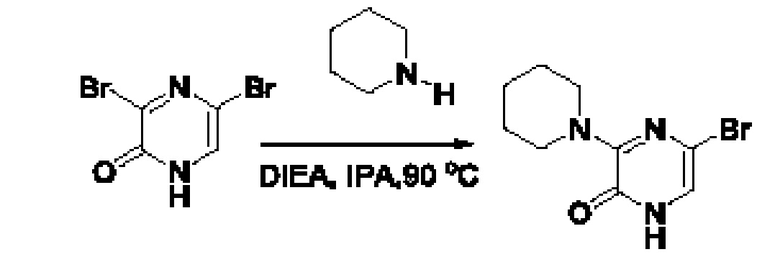
5-Бром-3-(пиперидин-1-ил)-пиразин-2(1Н)-он (Промежуточное соединение 6-1)
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (15 г, 59,08 ммоль, 1 эквив.) и пиперидином (7,5 г, 88,62 ммоль, 1,5 эквив.), получая 12 г (78,69%) указанного в заглавии соединения в виде твердого вещества гряз но-6 ел о го цвета.
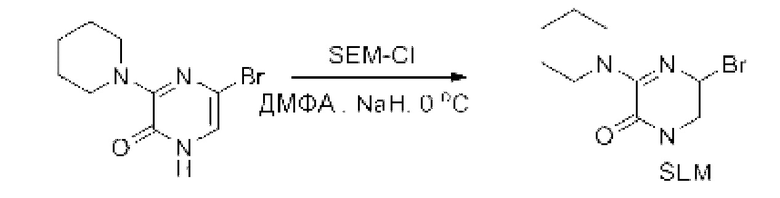
5-Бром-3-(пиперидин-1-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1Н)-он
Методику получения Промежуточного соединения 4-2 использовали с продуктом, полученным на предыдущей стадии, (3,9 г, 15,11 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:30), получая 4,2 г (71,6%) указанного в заглавии соединения в виде масла светло-желтого цвета.
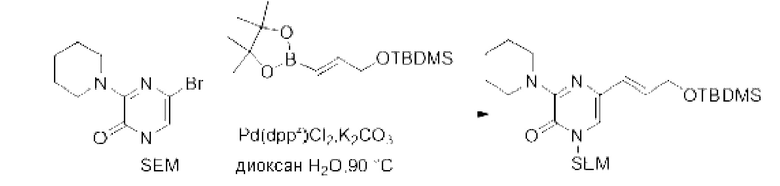
(E)-5-[[3-((трет-бутилдиметил)силил)окси]пропен-1-ил]-3-(пиперидин-1-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1Н)-он
Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (4 г, 10,30 ммоль), с временем реакции 12 ч при 90°С, получая 1,2 г (24,3%) указанного в заглавии соединения в виде масла светло-желтого цвета.
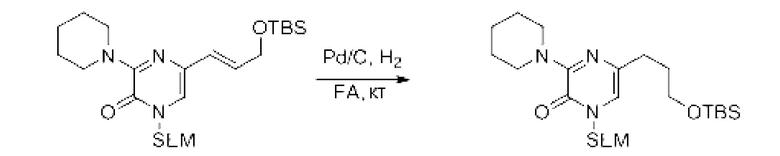
5-[[3-((трет-бутилдиметил)силил)окси]пропил]-3-(пиперидин-1-ил)-1-[(2-(триметилсилил)этокси)метил]-пиразин-2(1Н)-он
Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (1,2 г, 2,50 ммоль, 1 эквив.), получая 1,1 г (91,28%) указанного в заглавии соединения в виде масла светло-желтого цвета.
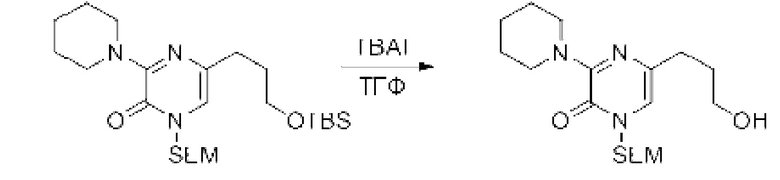
3-[6-(пиперидин-1-ил)-5(4Н)-оксо-4-[(2-(триметилсилил)этокси)метил]-пиразин-2-ил]-пропан-1-ол
Методику получения Промежуточного соединения 3-5 использовали с продуктом, полученным на предыдущей стадии, (1,1 г, 2,28 ммоль, 1 эквив.), получая 0,51 г (60,78%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
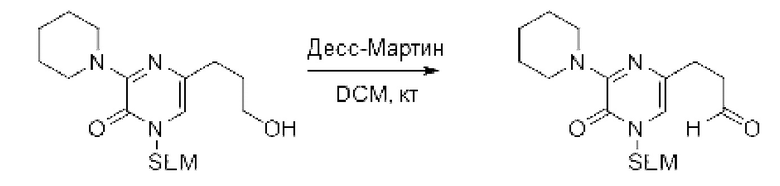
3-[6-(пиперидин-1-ил)-5(4Н)-оксо-4-[(2-(триметилсилил)этокси)метил]-пиразин-2-ил]-пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (510 мг, 1,39 ммоль), с временем перемешивания 2 ч, получая 300 мг (59%) указанного в заглавии соединения в виде масла светло-желтого цвета.
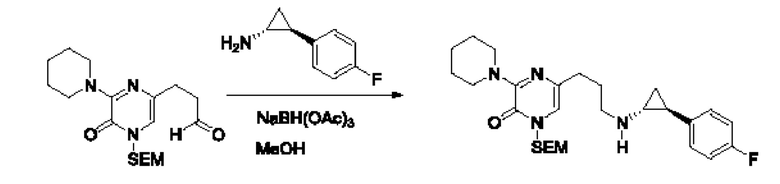
5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил]амино]пропил)-3-(пиперидин-1-ил)-1-[[2-(триметилсилил)этокси]метил]пиразин-2(1Н)-он
Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,82 ммоль, 1 эквив.), получая 210 мг (51,10%) указанного в заглавии соединения в виде масла светло-желтого цвета.
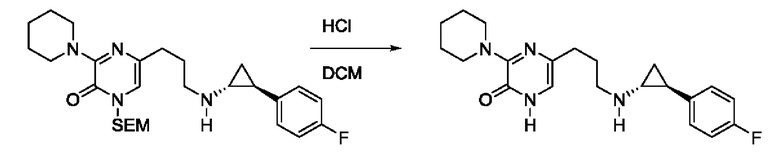
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[пиперидин-1-ил]пиразин-2(1Н)-он
Стадию удаления защитных групп для получения Примера 2 из Промежуточного соединения 2-7 использовали с продуктом, полученным на предыдущей стадии, (210 мг, 0,42 ммоль, 1 эквив.). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод D (от 10% до 51% CH3CN за 7 мин), получая 19,2 мг (9,77%) указанного в заглавии соединения в виде масла светло-желтого цвета.
ЖХ-МС: (ES, m/z): 371 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 7,28-7,15 (м, 2Н), 7,15-6,99 (м, 2Н), 6,70-6,61 (м, 1Н), 3,86-3,65 (м, 4Н), 3,28-3,20 (м, 2Н), 3,02-2,98 (м, 1Н), 2,66-2,36 (м, 3Н), 2,16-1,96 (м, 2Н), 1,80-1,59 (м, 6Н), 1,59-1,31 (м, 2Н).
ПРИМЕР 7
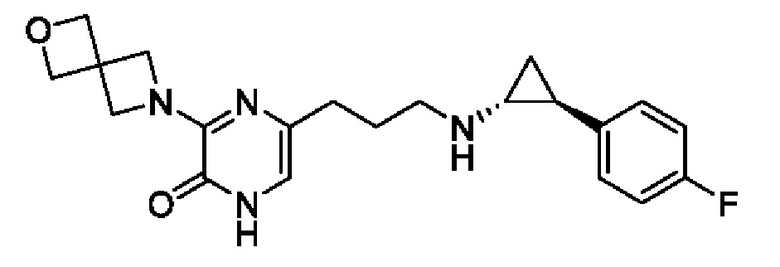
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]пиразин-2(1Н)-он
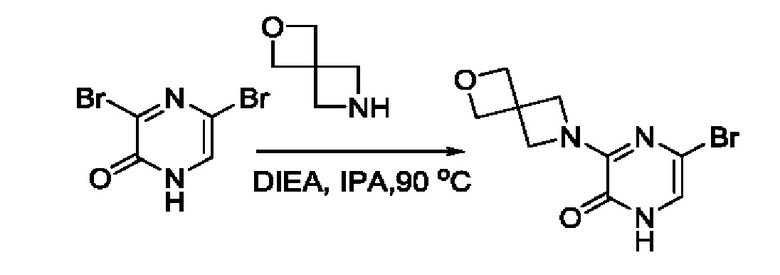
5-Бром-3-(2-Окса-6-азаспиро[3,3]гептан-6-ил)-пиразин-2(1Н)-он (Промежуточное соединение 7-1)
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (10 г, 39,39 ммоль, 1,00 эквив.) и 2-окса-6-азаспиро[3,3]гептаном (5,9 г, 59,52 ммоль, 1,50 эквив.), с временем реакции 6 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (4:1), получая 9 г (34%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
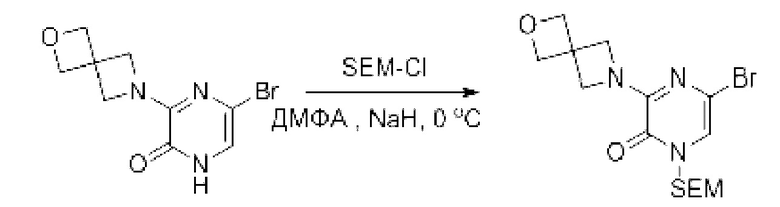
5-Бром-3-(2-окса-6-азаспиро[3,3]гептан-6-ил)-1-[(2-(триметилсилил)этокси)-метил]-пиразин-2(1Н)-он
Методику получения Промежуточного соединения 2-2 использовали с Промежуточным соединением 7-1 (9 г, 33,08 ммоль, 1,00 эквив.), получая 3,6 г (27%) указанного в заглавии соединения в виде масла гряз но-6 ел ого цвета.
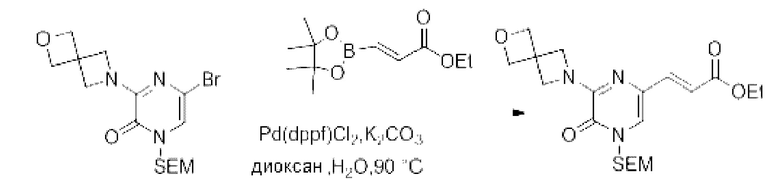
Этил (2Е)-3-[6-(2-окса-6-азаспиро[3,3]гептан-6-ил)-5(4Н)-оксо-4-[(2-(триметилсилил)этокси)метил]-пиразин-2-ил]пропеноат
Методику получения Промежуточного соединения 1-4 использовали с продуктом, полученным на предыдущей стадии, (3,6 г, 8,95 ммоль, 1,00 эквив.), получая 2,8 г (74%) указанного в заглавии соединения в виде масла желтого цвета.
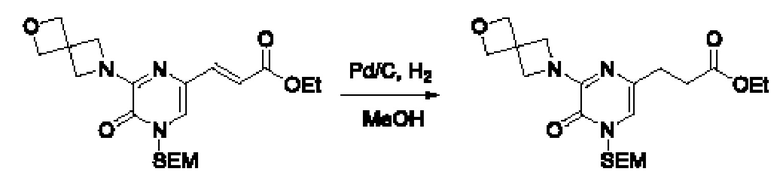
Этил 3-[6-(2-окса-6-азаспиро[3,3]гептан-6-ил)-5(4Н)-оксо-4-[(2-(триметилсилил)этокси)метил]-пиразин-2-ил]пропаноат
Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (2,8 г, 6,64 ммоль, 1,00 эквив.), получая 2,7 г (96%) указанного в заглавии соединения в виде масла желтого цвета.
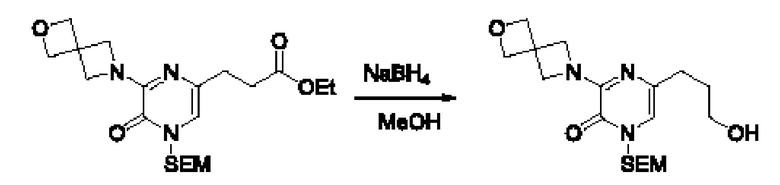
3-[б-(2-Окса-6-азаспиро[3,3]гептан-6-ил)-5(4Н)-оксо-4-[(2-(триметилсилил)-этокси)метил]-пиразин-2-ил]-пропал-1-ол
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии, (2,7 г, 6,37 ммоль, 1,00 эквив.), получая 1,8 г (74%) указанного в заглавии соединения в виде масла желтого цвета.
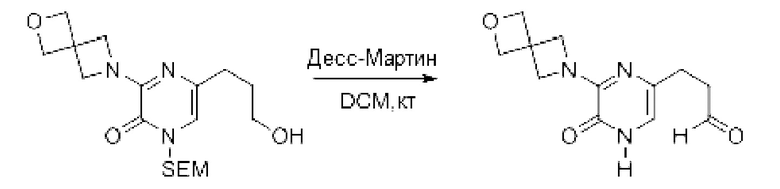
3-[6-(2-Окса-6-азаспиро[3,3]гептан-6-ил)-5(4Н)-оксо-4-[(2-(триметилсилил)-этокси)метил]-пиразин-2-ил]-пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (1,8 г, 4,72 ммоль, 1,00 эквив.), получая 0,5 г (28%о) указанного в заглавии соединения, для которого группа SEM была отщеплена в условиях реакции, в виде твердого вещества белого цвета.
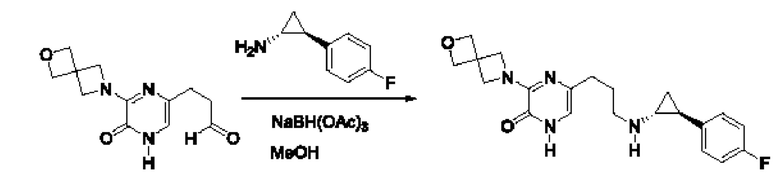
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]пиразин-2(1Н)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (500 мг, 1,32 ммоль, 1,00 эквив.). Неочищенный продукт (3 мл) очищали с помощью препаративной ВЭЖХ (Колонка: XBndge Prep Phenyl OBD, размер частиц: 5 мкм, размер колонки 19×150 мм, H2O (20 мМ NH4HCO3)/CH3CN, скорость потока: 20 мл/мин, градиент: от 25% до 30% CH3CN за 10 мин, Rt: 11,3 мин, детектор, УФ 254 нм), получая 50 мг (10%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХМС: (ES,m/z): 385 [М+Н]+. 1H ЯМР (300 МГц, Метанол-d4) δ м.д.: 7,09-6,97 (м, 2H)1 7,03-6,87 (м, 2Н), 6,37 (с, 1H), 4,83-4,78 (м, 4H), 4,45-4,25 (м, 4Н), 2,74-2,63 (м, 2Н), 2,35 (м, 2Н), 2,3-2,2 (м, 1Н), 1,93-1,63 (м, 3Н), 1,09-0,88 (м, 2S).
ПРИМЕР 8
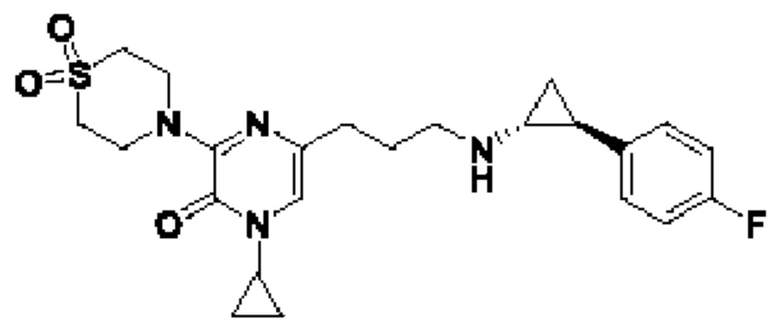
1-Циклопропил-3-[1,1-диоксидотиоморфолино]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]пиразин-2(1H)-он
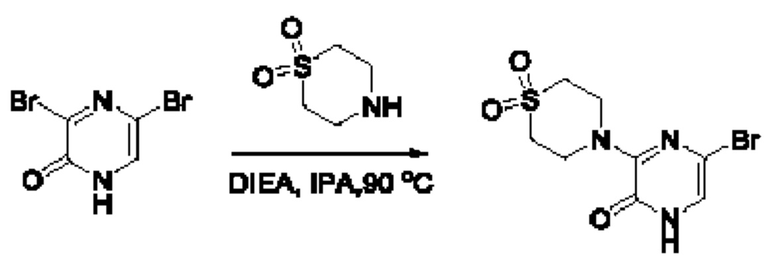
5-Бром-3-(1,1-диоксотиоморфолин-4-ил)-пиразин-2(1H)-он (Промежуточное соединение 8-1)
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (10 г, 39,39 ммоль, 1,00 эквив.) и 4-тиоморфолин-1,1-дионом (6,43 г, 47,56 ммоль, 1,21 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (4:1), получая 4,0 г (33%) указанного в заглавии соединения в виде твердого вещества белого цвета.
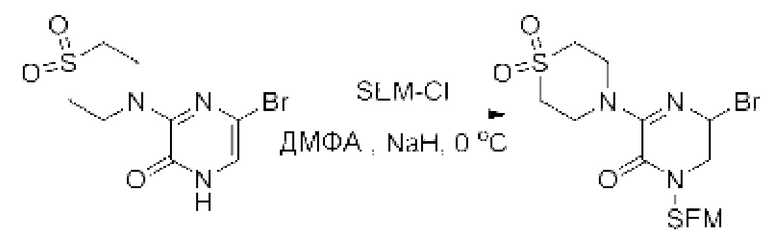
5-Бром-3-(1,1-диоксотиоморфолин-4-ил)-1-[(2-(триметилсилил)этокси)-метил]-пиразин-2(1H)-он (Промежуточное соединение 8-2)
Методику получения Промежуточного соединения 2-2 использовали с Промежуточным соединением 8-1 (3,0 г, 9,74 ммоль, 1 эквив.), получая 2,35 г (55,08%) указанного в заглавии соединения в виде масла желтого цвета.
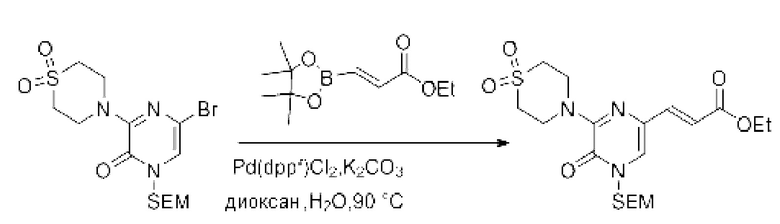
Этил 3-[6-(1,1-диоксотиоморфолин-4-ил)-5(4H)-оксо-4-[(2-(триметилсилил)этокси)метил]-пиразин-2-ил]пропеноат (Промежуточное соединение 8-3)
Методику получения Промежуточного соединения 1-4 использовали с Промежуточным соединением 8-2 (2,35 г, 5,36 ммоль, 1 эквив.), получая 1,80 г (73,25%) указанного в заглавии соединения в виде масла оранжевого цвета.
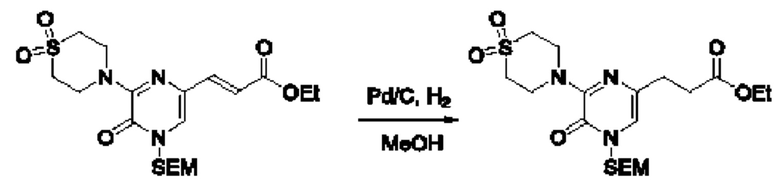
Этил 3-[6-(1,1-диоксотиоптрфолин-4-ил)-5(4H)-оксо-4-[(2-(триметилсилил)-этокси)метил]-пиразин-2-ил]пропаноат (Промежуточное соединение 8-4)
Методику получения Промежуточного соединения 1-5 использовали с Промежуточным соединением 8-3 (1,80 г, 3,93 ммоль, 1,00 эквив.), получая 1,80 г (99%) указанного в заглавии соединения в виде масла оранжевого цвета.
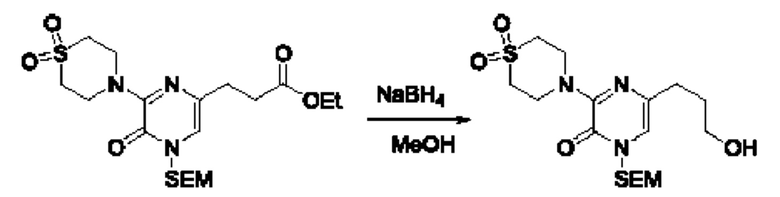
3-[6-(1,1-диоксотиоморфолин-4-ил)-5(4H)-оксо-4-[(2-(триметилсилил)-этокси)метил]-пиразин-2-ил]-пропан-1-ол (Промежуточное соединение 8-5)
Методику получения Промежуточного соединения 1-6 использовали с Промежуточным соединением 8-4 (1,8 г, 3,92 ммоль, 1 эквив.) с временем реакции 2 ч. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (3:2), получая 1,3 г (79,47%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
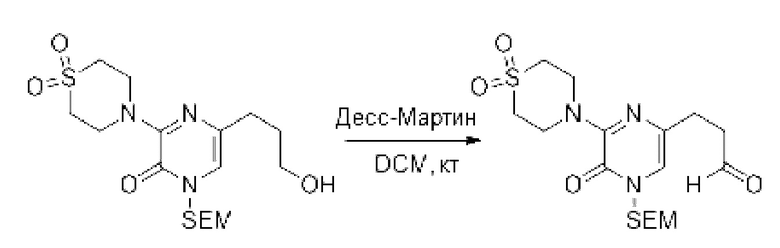
3-[6-(1,1-диоксотиоморфолин-4-ил)-5(4H)-оксо-4-[(2-(триметилсилил)-этокси)метил]-пиразин-2-ил]-пропаналь (Промежуточное соединение 8-6)
Методику получения Промежуточного соединения 1-7 использовали с Промежуточным соединением 8-5 (1,30 г, 3,11 ммоль, 1,00 жвив.), получая 1,0 г (77,29%) указанного в заглавии соединения в виде масла желтого цвета.
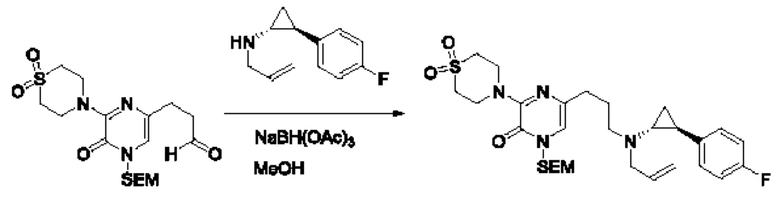
5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил](2-пропен-1-ил)амино]пропил)-3-(1,1-диоксотиоморфолин-4-ил)-1-[[2-(триметилсилил)этокси]метил]пиразин-2(1H)-он (Промежуточное соединение 8-7)
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с Промежуточным соединением 8-6 (1,0 г, 2,40 ммоль, 1 эквив.), получая 900 мг (63,35%) указанного в заглавии соединения в виде масла оранжевого цвета.
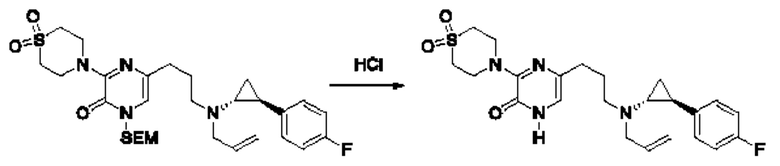
5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил](2-пропен-1-ил)амино]пропил)-3-(1,1-диоксотиоморфолин-4-ил)-пиразин-2(1H)-он (Промежуточное соединение 8-8)
Стадию удаления защитных групп для получения Примера 2 из Промежуточного соединения 2-7 использовали с Промежуточным соединением 8-7 (900 мг, 1,52 ммоль, 1 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (15:1), получая 700 мг (99,71%) указанного в заглавии соединения в виде тверд ого вещества оранжевого цвета.
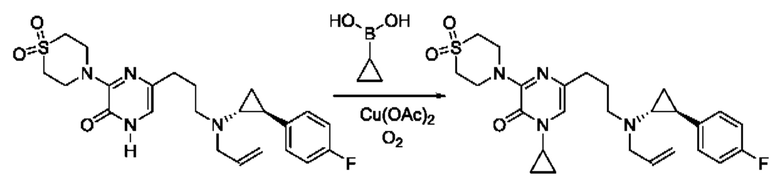
5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил](2-пропен-1-ил)амино]пропил)-3-(1,1-диоксотиоморфолин-4-ил)-1-циклопроиилпиразин-2 (1Н)-он (Промежуточное соединение 8-9)
Промежуточное соединение 8-8 (700 мг, 1,52 ммоль, 1 эквив.) объединяли с циклопропилбороновой кислотой (196 мг, 2,28 ммоль, 1,5 эквив.), Cu(ОАс)2 (276,0 мг, 1,52 ммоль, 1 эквив.) и TEA (461,5 мг, 4,56 ммоль, 3 эквив.) в CH2Cl2 (40 мл). К смеси добавляли кислород и полученный в результате раствор перемешивали в течение 16 ч при кт, затем очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 300 мг (39,43%) указанного в заглавии соединения в виде масла желтого цвета.
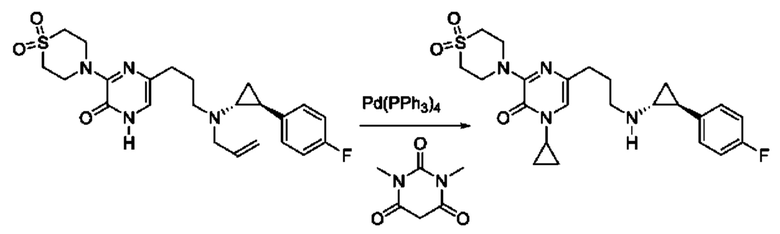
1-Циклопропил-3-[1,1-диоксидотиоморфолино]-5-[3-([(1R,25)-2-(4-фторфенил)циклопропил]амино)пропил]пиразин-2(1H)-он
Раствор продукта, полученного на предыдущей стадии, (300 мг, 0,60 ммоль, 1 эквив.), 1,3-диметил-1,3-диазинан-2,4,6-триона (280,5 мг, 1,80 ммоль, 3 эквив.) и Pd(PPh3)4 (140 мг, 0,12 ммоль, 0,2 эквив.) в ТГФ (30 мл) перемешивали в течение 2 ч под N2 при 50°С. Неочищенный продукт очищали, используя хроматографический Метод А (от 10% до 58% CH3CN за 9 мин), получая 52,1 мг (18,88%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХ-МС: (ES,m/z): 461 [М+Н]+. 1H ЯМР (300 МГц, MeOD-d4) δ м.д.: 7,18-7,14 (м, 2Н), 7,05-6,99 (м, 2Н), 6,87 (с, 1H), 4,28-4,26 (м, 4Н), 3,28-3,19 (м, 3Н), 3,16-3,11 (м, 4Н), 2,96-2,91 (м, 1H), 2,53-2,48 (м, 2Н), 2,45-2,38 (м, 1H), 2,06-1,98 (м, 2Н), 1,46-1,34 (м, 2Н), 1,08-1,03 (м, 2Н), 0,87-0,82 (м, 2Н).
ПРИМЕР 9
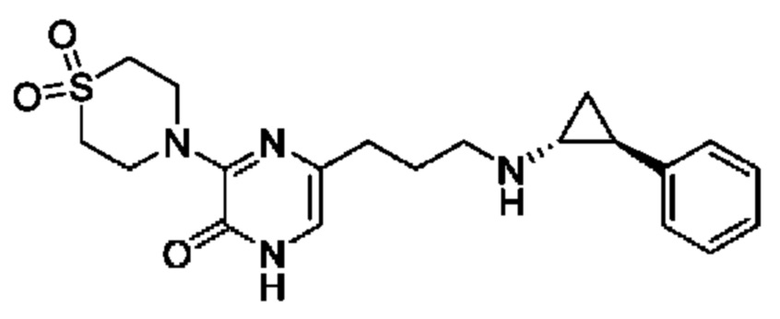
3-(1,1-Диоксидотиоморфолино)-5-(3-(((1R,2S)-2-фенилциклопропил)амино)пропил)пиразин-2(1H)-он
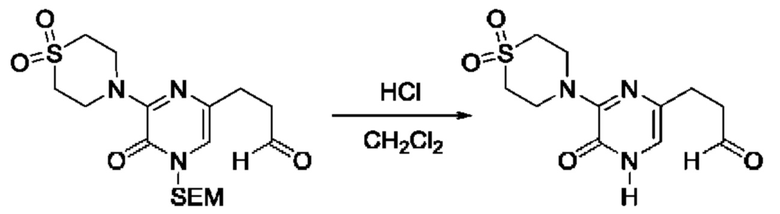
3-[6-(1,1'-диоксотиоморфолин-4-ил)-5(4H)-оксопиразин-2-ил]-пропаналь
Стадию удаления защитных групп для получения Примера 2 из Промежуточного соединения 2-7 использовали с Промежуточным соединением 8-6 (600 мг, 1,44 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (15:1), получая 400 мг (96,97%) указанного в заглавии соединения в виде масла оранжевого цвета.
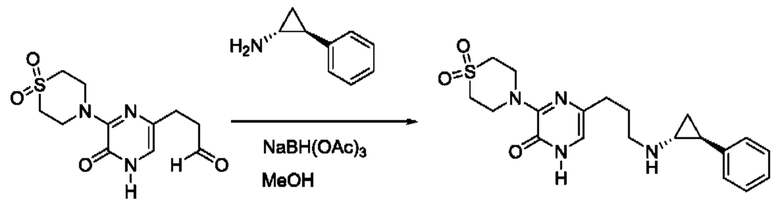
3-(1,1-Диоксидотиоморфолино)-5-(3-(((1R,2S)-2-фенилциклопропил)амино)-пропил)пиразин-2(1H)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (400 мг, 1,40 ммоль, 1 эквив.). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод E (от 28% до 50% CH3CN за 8 мин, Rt: 7,02 мин), получая 85,3 мг (15%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-МС: (ES,m/z): 403 [М+Н]+. 1H ЯМР (300 МГц, MeOD-d4) δ м.д.: 7,27-7,17 (м, 2Н), 7,13-7,06 (м,1Н), 7,02-6,99 (м, 2Н), 6,66 (с, 1H), 4,27-4,24 (м, 4Н), 3,11-3,04 (м, 4Н), 2,73-2,68 (м, 2Н), 2,46-2,40 (м, 2Н), 2,30-2,25 (м, 1H), 1,90-1,80 (м, 3Н), 1,06-0,94 (м, 2Н).
ПРИМЕР 10
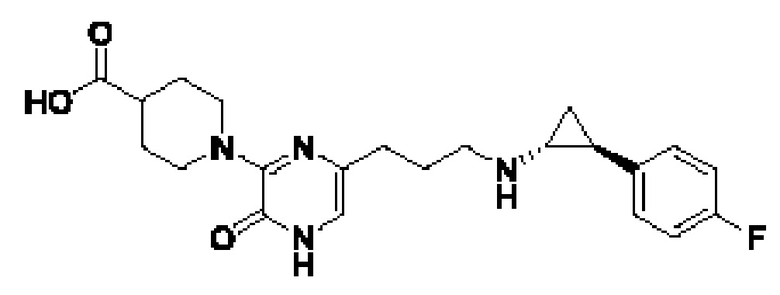
1-[6-(3-[([1R,2S]-2-[4-фторфенил]циклопропил)амино]пропил)-3-оксо-3,4-дигидропиразин-2-ил]пиперидин-4-карбоновая кислота
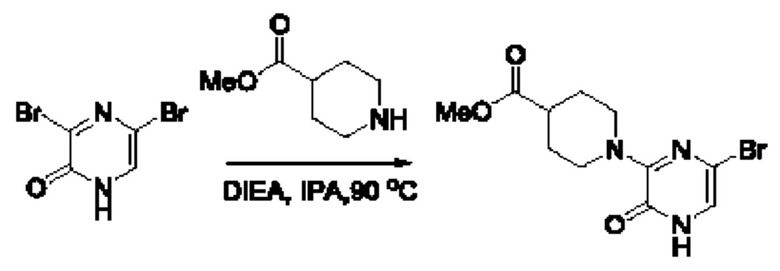
Метил 1-(6-бром-3(4H)-оксопиразин-2-ил)пиперидин-4-карбоксилат (Промежуточное соединение 10-1)
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (10 г, 39,39 ммоль, 1,00 эквив.) и метилпиперидин-4-карбокеилатом (8,4 г, 58,67 ммоль, 1,49 эквив.), с временем реакции 12 ч при 90°C, получая 10 г (80%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
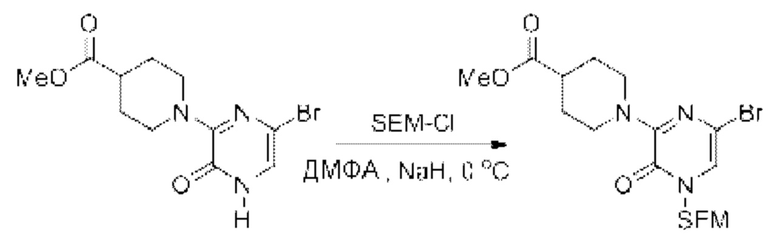
Метил 1-(б-бром-3(4H)-оксо-4-[[2-(триметилсилил)этокси]метил]-пиразин-2-ил)пиперидин-4-карбоксилат
Методику получения Промежуточного соединения 4-2 использовали с Промежуточным соединением 10-1 (5 г, 15,82 ммоль, 1,00 эквив.), получая 4 г (57%) указанного в заглавии соединения в виде масла светло-желтого цвета.
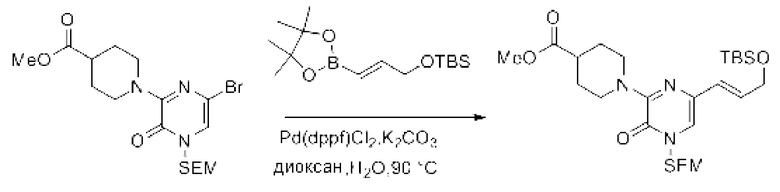
Метил 1-[6-[(1E)-3-[(трет-бутилдиметилсилил)окси]проп-1-ен-1-ил]-3(4H)-оксо-4-[[2-(триметилсилил)этокси]метил]-3,4-дигидропиразин-2-ил]пиперидин-4-карбоксилат
Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (4 г, 8,96 ммоль), с временем реакции 2 ч, получая 1,7 г (35%) указанного в заглавии соединения в виде масла светло-желтого цвета.
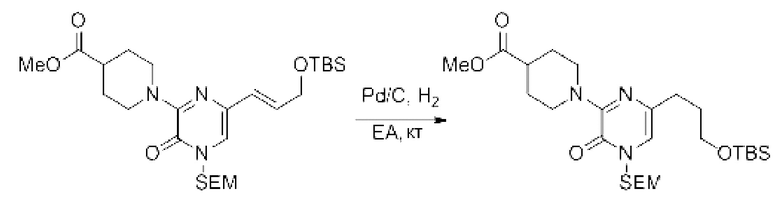
Метил 1-(6-[3-[(трет-бутилдиметилсилил)окси]пропил]-3(4H)-оксо-4-[[2-(триметилсилил)этокси]метил]-пиразин-2-ил)пилеридин-4-карбоксилат
Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (1,8 г, 3,35 ммоль, 1,00 эквив.), получая 1,7 г (94%) указанного в заглавии соединения в виде масла светло-желтого цвета.
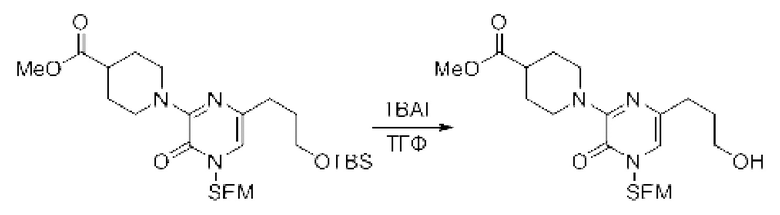
Метил 1-[6-(3-гидроксипропил)-3(4H)-оксо-4-[[2-(триметилсилил)этокси]метил]-пиразин-2-ил]пиперидин-4-карбоксилат
Методику получения Промежуточного соединения 3-5 использовали с продуктом, полученным на предыдущей стадии, (1,7 г, 3,15 ммоль, 1,00 эквив.), получая 0,8 г (60%) указанного в заглавии соединения в виде масла светло-желтого цвета.
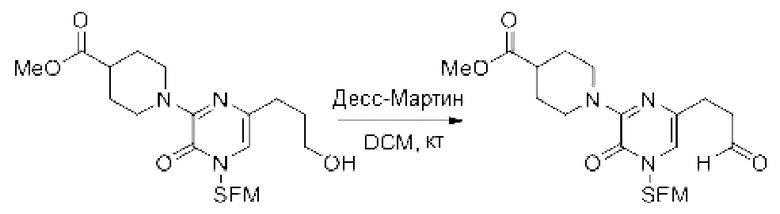
Метил 1-(3(4H)-оксо-6-(3-оксопропил)-пиразин-2-ил)пиперидин-4-карбоксилат
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (800 мг, 1,88 ммоль, 1,00 эквив.), получая 0,45 г (82%) указанного в заглавии соединения в виде масла светло-желтого цвета.
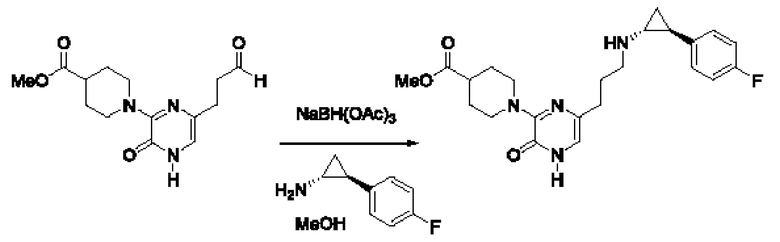
Метил 1-[6-(3-[[(1R,1S)-2-(4-фторфенил)циклопропил]амино]пропил)-3(4H)-оксопиразин-2-ил]пиперидин-4-карбоксилат
Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (450 мг, 1,53 ммоль, 1,00 эквив.), получая 0,3 г (46%) указанного в заглавии соединения в виде масла светло-желтого цвета, который использовали дальше без дополнительной очистки.
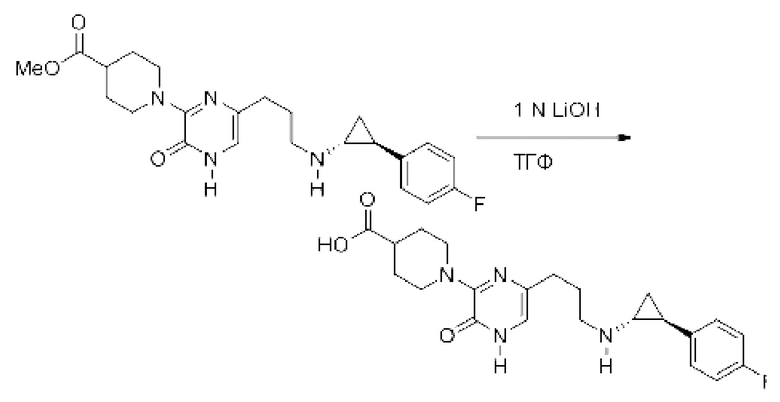
1-[6-(3-[([1R,2S]-2-[4-фторфенил]циклопропил)амино]пропил)-3-оксо-3,4-дигидропиразин-2-ил]пиперидин-4-карбоновая кислота
Раствор продукта, полученного на предыдущей стадии, (300 мг, 0,70 ммоль, 1,00 эквив.) и LiOH (80 мг, 3,34 ммоль, 5,00 эквив.) в ТГФ (20 мл) и Н2О (3 мл) перемешивали в течение 2 ч при 25°C, затем концентрировали в вакууме и очищали, используя хроматографический Метод С (от 38,0% до 50,0% CH3CN за 8,2 мин), получая 82,6 мг (28%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХ-МС: (ES,m/z): 415 [M+H]+, 1H ЯМР (300 МГц, MeOD-d4) δ м.д.: 7,23-7,07 (м, 2Н), 7,07-6,93 (м, 2Н), 6,82-6,69 (с, 1H), 4,784,32 (м, 2H), 3,02-2,78 (м, 4Н), 2,72-2,55 (м, 1H), 2,52-2,28 (м, 3Н), 2,27-2,02 (м, 1H), 2,02-1,81 (м, 4H), 1,81-1,51 (м, 2Н), 1,38-1,01 (м, 2Н).
ПРИМЕР 11
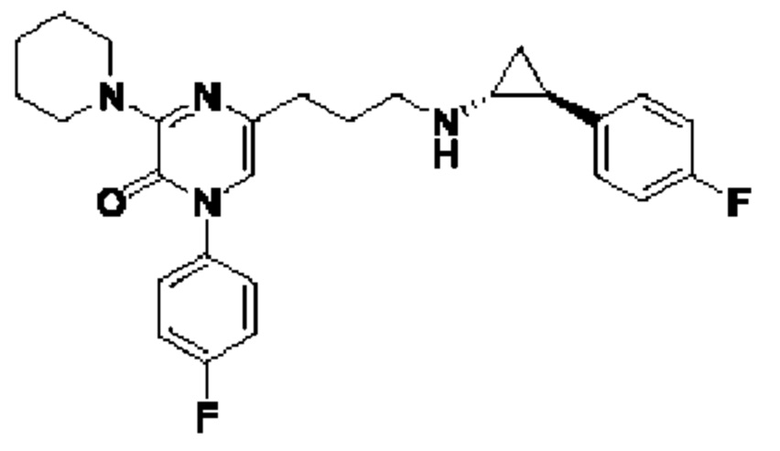
1-[4-Фторфенил]-3-[пиперидин-1-ил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-пиразин-2(1H)-он
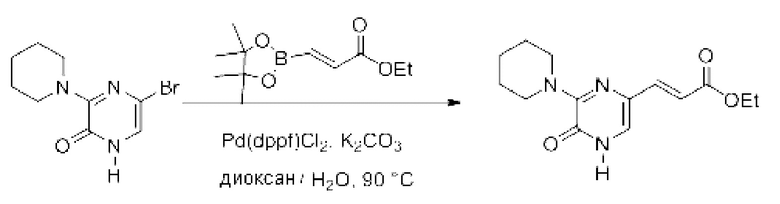
Этил (2Е)-3-[6-(пиперидин-1-ил)-5(4H)-оксопиразин-2-ил]пропеноат
Методику получения Промежуточного соединения 1-4 использовали с Промежуточным соединением 6-1, получая 2 г (47%) указанного в заглавии соединения в виде масла желтого цвета.
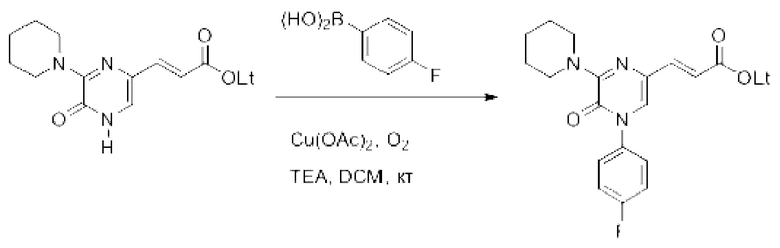
Этил (2E)-3-[4-(4-фторфенил)-6-(пилеридин-1-ил)-5(4H)-оксопиразин-2-ил]-пропеноат
Методику получения Промежуточного соединения 8-9 использовали с продуктом, полученным на предыдущей стадии, (2 г, 7,22 ммоль, 1 эквив.) и 4-фторфенилбороновой кислотой, получая 1 г (37%) указанного в заглавии соединения в виде масла желтого цвета.
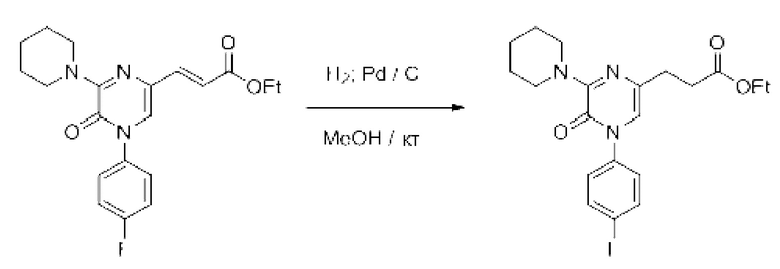
Этил (2E)-3-[4-(4-фторфенил)-6-(пилеридин-1-ил)-5(4H)-оксопиразин-2-ил]-пропаноат
Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (1 г, 2,70 ммоль, 1,00 эквив.), получая 1 г (99%) указанного в заглавии соединения в виде масла желтого цвета.
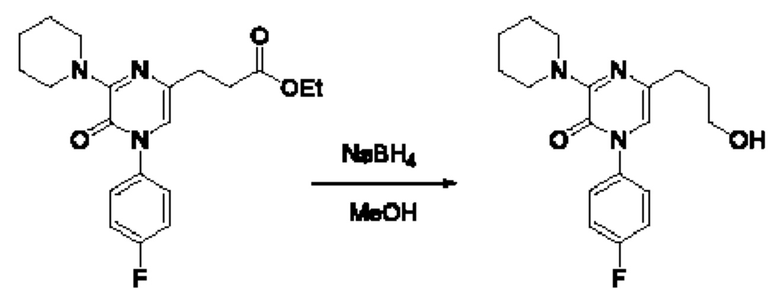
(2E)-3-[4-(4-Фторфенил)-6-(пилеридин-1-ил)-5(4H)-оксопиразин-2-ил]-пропан-1-ол
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии, (1 г, 2,68 ммоль, 1 эквив.), с временем реакции 6 ч, получая 500 мг (56%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
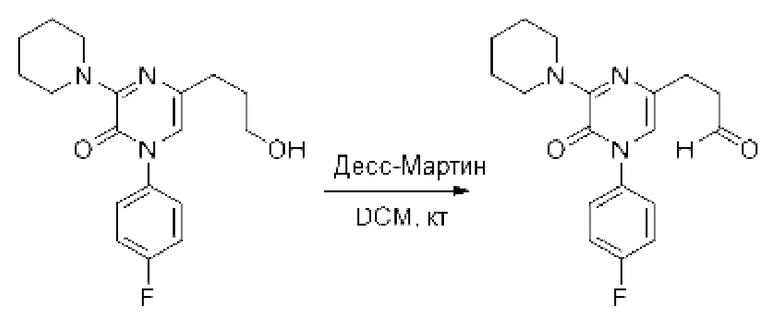
(2E)-3-[4-(4-Фторфенил)-6-(пилеридин-1-ил)-5(4H)-оксопиразин-2-ил]-пропаналь
Методику получения Промежуточного соединения 1-7 применяли к продукту, полученному на предыдущей стадии, (500 мг, 1,51 ммоль, 1,00 эквив.), получая 300 мг (60%) указанного в заглавии соединения в виде масла желтого цвета.
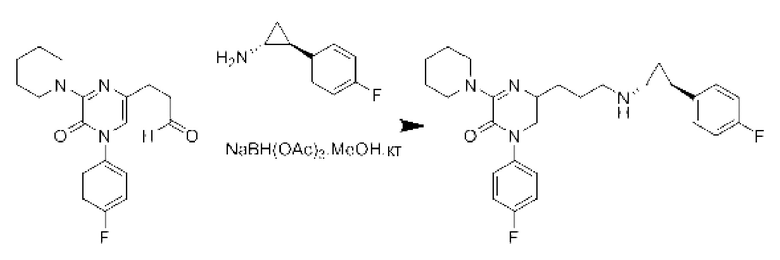
1-[4-Фторфенил]-3-[пиперидин-1-ил]-5-[3-([(1R,1S)-2-(4-фторфенил)-циклопропил]амино)пропил]-пиразин-2(1H)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,912 ммоль, 1 эквив.). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод А (от 35% до 40% CH3CN за 10 мин, Rt: 8,12 мин), получая 68,9 мг (11%) указанного в заглавии соединения в виде масла желтого цвета.
ЖХ-МС: (ES,m/z): 465 [М+Н]+. 1H ЯМР (400 МГц, MeOD-d4) δ м.д.: 7,45-7,42 (м, 2Н), 7,30-7,26 (t, J=17,2 Гц, 2Н), 7,23-7,20 (м, 2Н), 7,09-7,04 (т, J=17,2 Гц, 2Н), 6,81 (с, 1H), 3,77-3,75 (м, 4Н), 3,30-3,28 (м, 2Н), 3,02-2,98 (м, 1H), 2,58-2,54 (м, 2Н), 2,49-2,46 (м, 1H), 2,12-2,08 (м, 2Н), 1,67-1,65 (м, 6Н), 1,53-1,48 (м, 1H), 1,41-1,36 (м, 1H).
ПРИМЕР 12
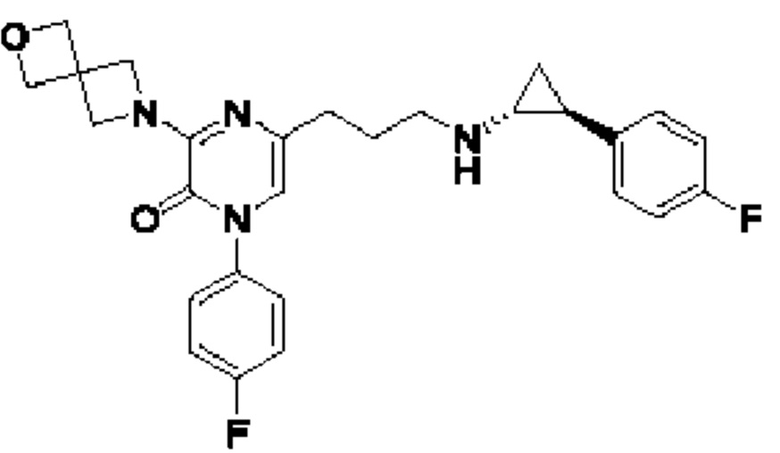
1-[4-Фторфенил]-5-[3-([(1R,2S)-2-(4-фторфенил)пиклопропил]амино)пропил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]пиразин-2(1H)-он
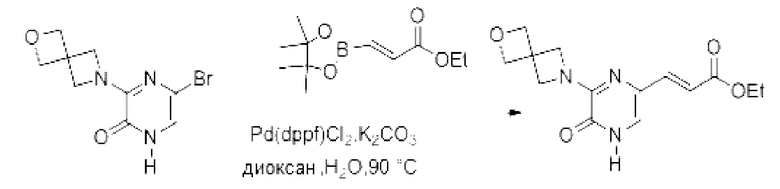
Этил (E)-3-(6-[2-окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксопиразин-2-ил)проп-2-еноат (Промежуточное соединение 12-1)
Методику получения Промежуточного соединения 1-4 использовали с Промежуточным соединением 7-1 (3,6 г, 13,23 ммоль) с временем реакции 2 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 1,7 г (44%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
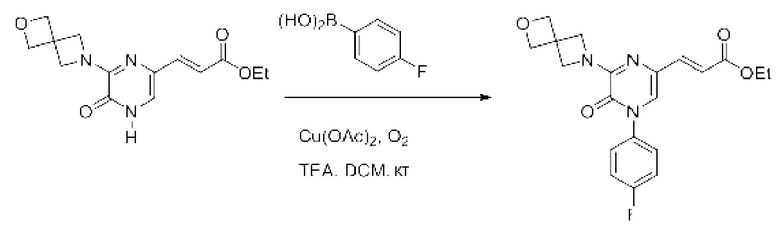
Этил (E)-3-[4-(4-фторфенил)-6-[2-окса-6-азаспиро[3,3]гелтан-6-ил]-5(4H)-оксопиразин-2-ил]проп-2-еноат
Методику получения Промежуточного соединения 8-9 использовали с Промежуточным соединением 12-1 (2 г, 6,87 ммоль, 1,00 эквив.) и 4-фторфенилбороновую кислоту (1,4 г, 10,01 ммоль, 1,50 эквив.). Неочищенный продукт очищали с использованием хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 0,7 г (26%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
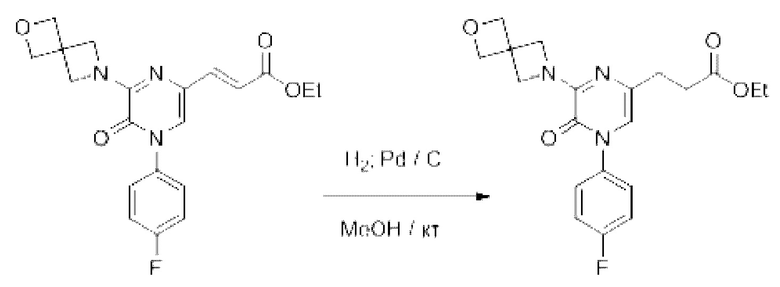
Этил 3-[4-(4-фторфенил)-6-[2-окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксопиразин-2-ил]пропаноат
Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (850 мг, 2,21 ммоль, 1,00 эквив.), получая 0,7 г (82%) указанного в заглавии соединения в виде масла желтого цвета.
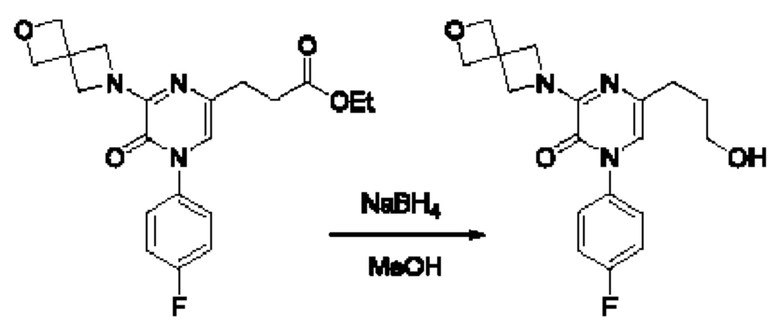
3-[4-(4-Фторфенил)-6-[2-окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксопиразин-2-ил]пропан-1-ол
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии, (700 мг, 1,81 ммоль, 1,00 эквив.), получая 0,4 г (64%) указанного в заглавии соединения в виде бесцветного масла.
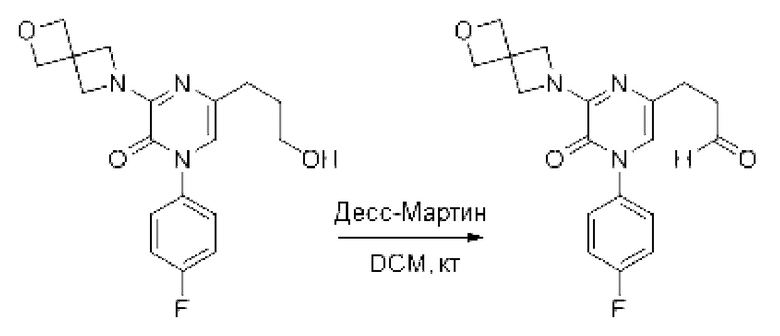
3-[4-(4-Фторфенил)-6-[2-окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксопиразин-2-ил]пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (500 мг, 1,45 ммоль, 1,00 эквив.), получая 0,28 г (56%) указанного в заглавии соединения в виде масла желтого цвета.
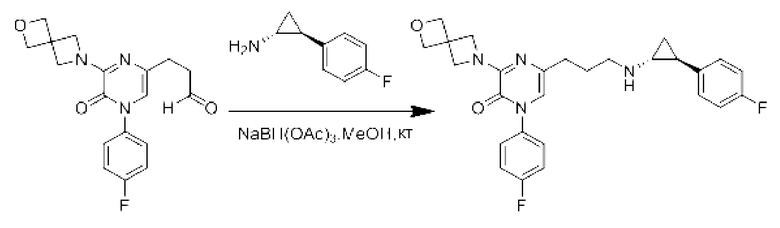
1-[4-Фтор фенил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]пиразин-2(1H)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (280 мг, 0,82 ммоль, 1,00 эквив.). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод С (от 35,0% до 52,0% CH3CN за 8,8 мин), получая 75,9 мг (19%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХМС: (ES,m/z): 479 [М+Н]+. 1H ЯМР (300 МГц, Метанол-d4) δ м.д.: 7,45-7,31 (м, 2H), 7,29-7,14 (м, 2Н), 7,09-6,98 (м, 2Н), 7,00-6,85 (м, 2H), 6,55 (с, 1Н), 4,70 (с, 4Н), 4,40 (с, 4Н), 2,79-2,68 (м, 2Н), 2,38 (т, J=7,2 Гц, 2H), 2,33-2,23 (м, 1H), 1,95-1,74 (м, 3Н), 1,10-0,89 (м, 2Н).
ПРИМЕР 13
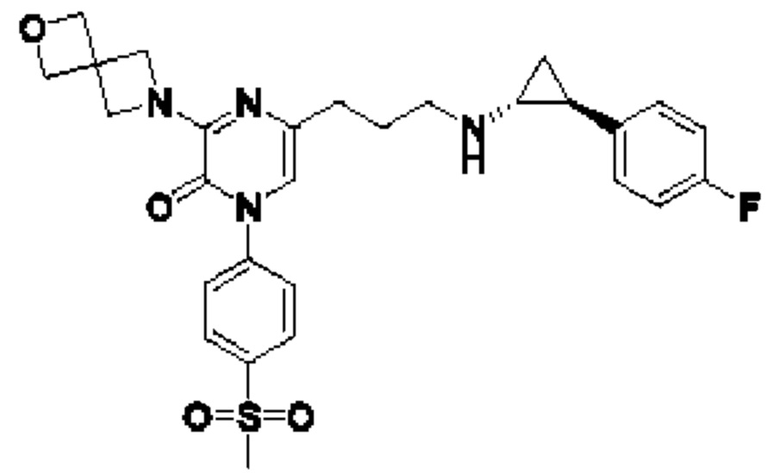
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-1-[4-(метилсульфонил)фенил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]пиразин-2(1H)-он
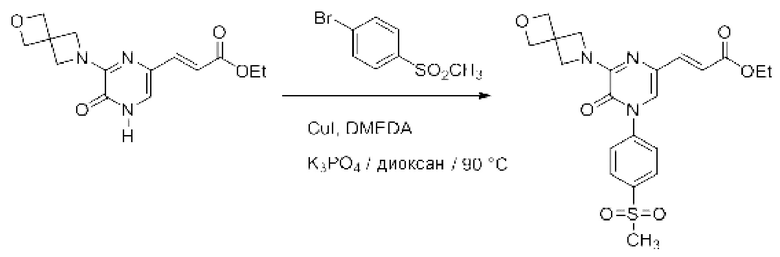
Этил (Е)-3-[4-(4-метилсуль фонилфенил)-6-[2-океа-6-азаспиро[3,3]гептан-6-ил]-5(4H)-океопиразин-2-ил]проп-2-еноат (Промежуточное соединение 13-1)
Раствор Промежуточного соединения 12-1 (2 г, 6,87 ммоль, 1,00 эквив.), 1-бром-метансуль фонил бензол а (2,4 г, 10,21 ммоль, 1,50 эквив.), N,N-диметилэтиле иди амина (480 мг, 5,45 ммоль, 0,80 эквив.), K3PO4 (4,3 г, 20,26 ммоль, 3,00 эквив.), CuI (520 мг, 2,73 ммоль, 0,40 эквив.) в диоксане (100 мл) перемешивали в течение 6 ч при 90°С. Полученную смесь концентрировали в вакууме и очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 1,3 г (43%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
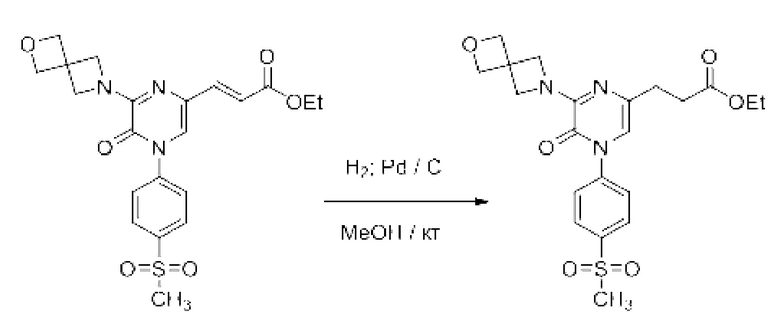
Этил 3-[4-(4-метилсульфонилфенил)-6-[2-окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксопиразин-2-ил]пропаноат
Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (1,3 г, 2,92 ммоль, 1,00 эквив.), получая 1,0 г (77%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
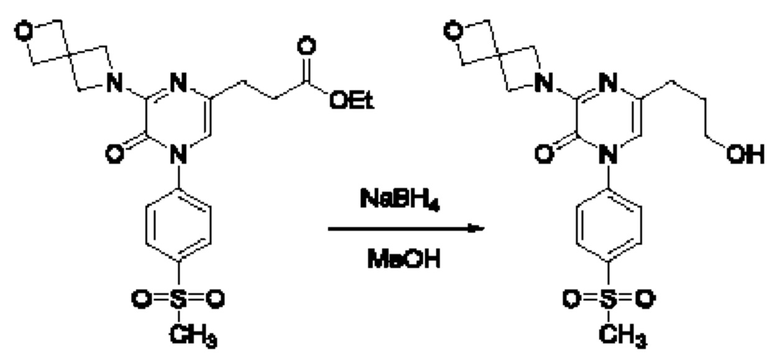
3-[4-(4-Метилсульфонилфенил)-6-[2-окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксопиразин-2-ил]пропан-1-ол
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии (1,0 г, 2,23 ммоль, 1,00 эквив.), с временем реакции 4 ч при кт, получая 0,4 г (44%) указанного в заглавии соединения в виде масла желтого цвета.
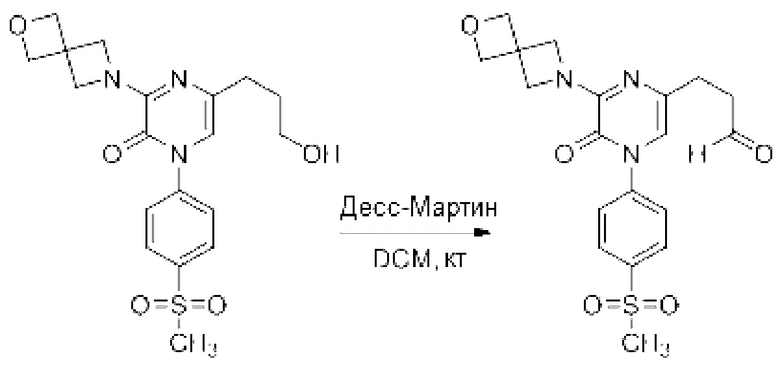
3-[4-(4-Метилсульфонилфенил)-6-[2-окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксопиразин-2-ил]пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (400 мг, 0,99 ммоль, 1,00 эквив.), получая 150 мг (38%) указанного в заглавии соединения в виде бесцветного масла.
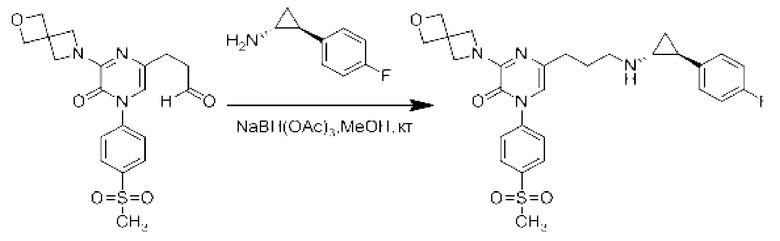
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-1-[4-(метил-сульфонил)фенил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]пиразин-2(1H)-он
Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (150 мг, 0,37 ммоль). Неочищенный продукт очищали, используя хроматографический Метод E (от 42% до 45% CH3CN за 7 мин, Rt: 6,7 мин), получая 11,1 мг (6%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХМС: (ES,m/z): 539 [М+Н]+. 1H ЯМР (300 МГц, Метанол-d4) δ м.д.: 8,12-8,01 (м, 2S), 7,72-7,62 (м, 2H), 7,09-6,86 (м, 4Н), 6,62 (с, 1H), 4,72 (с, 4Н), 4,42 (с, 4Н), 3,14 (с, 3Н), 2,73 (т, J=7,5 Гц, 2Н), 2,39 (т, J=7,2 Гц, 2Н), 2,31-2,22 (м, 1H), 1,89-1,79 (м, 3Н), 1,10-0,88 (м, 2Н).
ПРИМЕР 14
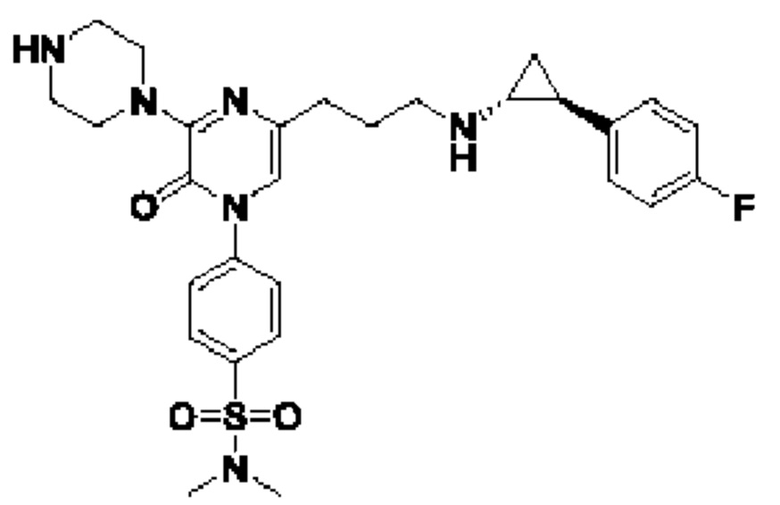
4-[5-(3-[([1R,2S]-2-[4-фторфенил]пиклопропил)амино]пропил)-2-оксо-3-(пиперазин-1-ил)пиразин-1(2H)-ил]-N,N-диметилбензолсульфонамид
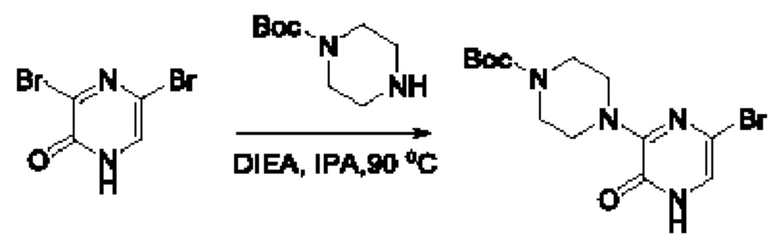
трет-Бутил 4-(6-бром-3(4H)-оксопиразин-2-ил)пиперазин-1-карбоксилат (Промежуточное соединение 14-1)
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (20 г, 78,78 ммоль, 1,00 эквив.) и трет-Бутил пиперазин-1-карбоксилатом (22 г, 118,12 ммоль, 1,50 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:20), получая 25 г (88%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
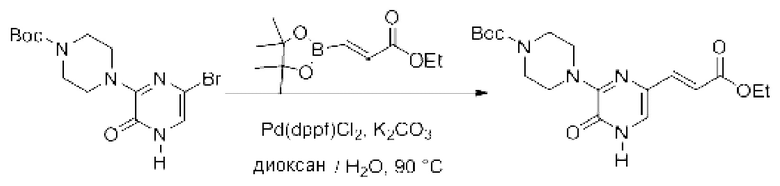
трет-Бутил 4-[6-[(Е)-3-этокси-3-оксопроп-1-ен-1-ил]-3(4H)-оксопиразин-2-ил]пиперазин-1-карбоксилат (Промежуточное соединение 14-2)
Методику получения Промежуточного соединения 1-4 использовали с Промежуточным соединением 14-1 (4 г, 11,14 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:10), получая 1 г (24%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
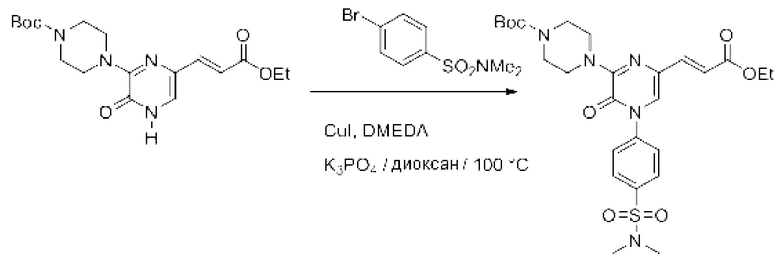
трет-Бутил 4-[4-[4-((диметиламино)сульфонил)фенил]-6-[(Е)-3-этокси-3-оксопроп-1-ен-1-ил]-3(4H)-оксо-пиразин-2-ил]пиперазин-1-карбоксилат (Промежуточное соединение 14-3)
Методику получения Промежуточного соединения 13-1 использовали с Промежуточным соединением 14-2 (2 г, 5,29 ммоль, 1,00 эквив.) и 4-бром-N,N-диметилбензол-1-сульфонамидом (2,1 г, 7,95 ммоль, 1,50 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (10:1), получая 1 г (34%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
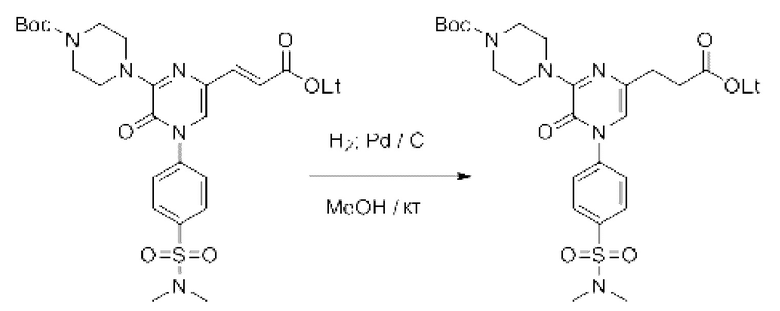
трет-Бутил 4-[4-[4-((диметиламино)сульфонил)фенил]-6-(3-этокси-3-оксопропил)-3(4H)-оксопиразин-2-ил]пиперазин-1-карбоксилат (Промежуточное соединение 14-4)
Методику получения Промежуточного соединения 1-5 использовали с Промежуточным соединением 14-3 (1 г, 1,78 ммоль, 1,00 эквив.), получая 0,8 г (80%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
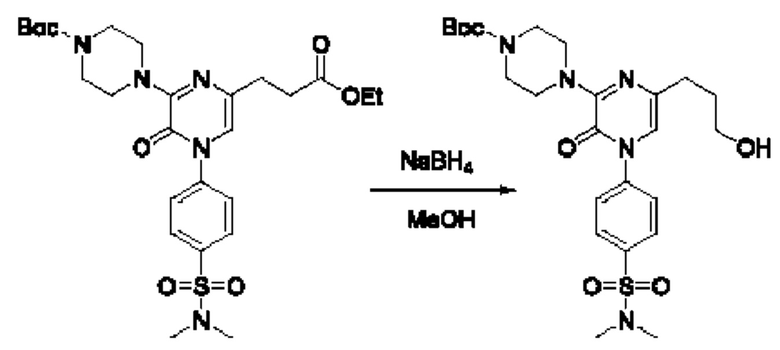
трет-Бутил 4-[4-[4-((диметиламино)сульфонил)фенил]-6-(3-гидроксипропил)-3(4H)-оксопиразин-2-ил]пиперазин-1-карбоксилат (Промежуточное соединение 14-5)
Методику получения Промежуточного соединения 1-6 использовали с Промежуточным соединением 14-4 (800 мг, 1,42 ммоль, 1,00 эквив.), получая 520 мг (70%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
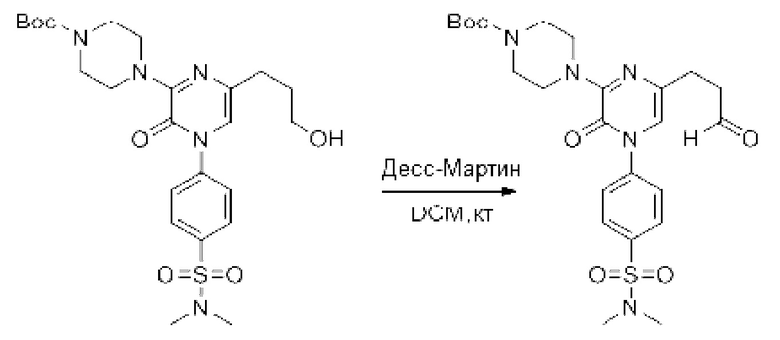
трет-Бутил 4-[4-[4-((диметиламино)сульфонил)фенил]-3(4H)-оксо-6-(3-оксопропил)-пиразин-2-ил]пиперазин-1-карбоксилат (Промежуточное соединение 14-6)
Методику получения Промежуточного соединения 1-7 использовали с Промежуточным соединением 14-5 (520 мг, 1,00 ммоль, 1,00 эквив.), получая 370 мг (71%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
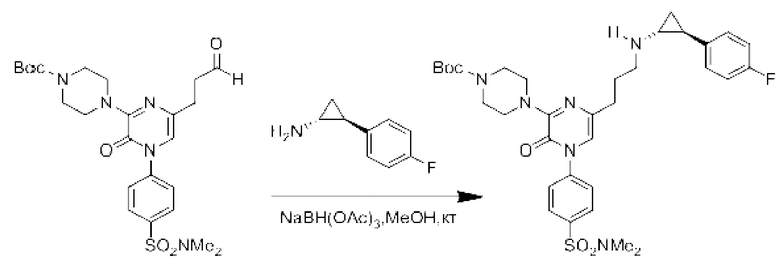
трет-Бутил 4-[4-[4-((диметиламино)сульфонил)фенил]-6-(3-[[(1R,2S)-2-(4-фторфенил)циклопропил]амино]пропил)-3(4H)-оксопиразин-2-ил]пиперазин-1-карбоксилат (Промежуточное соединение 14-7)
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с Промежуточным соединением 14-6 (370 мг, 0,71 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc, получая 300 мг (64%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
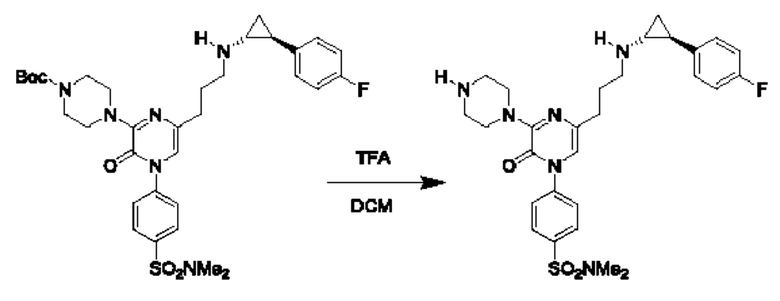
4-[5-(3-[([1R,2S]-2-[4-фторфенил]циклопропил)амино]пропил)-2-оксо-3-(пиперазин-1-ил)пиразин-1(2H)-ил]-N,N-диметилбензолсульфонамид (Пример 14)
Раствор Промежуточного соединения 14-7 (300 мг, 0,46 ммоль, 1,00 эквив.) и TFA (3 мл) в CH2Cl2 (15 мл) перемешивали в течение 1 ч при 25°C рН корректировали до 7 добавлением NaHCO3 (5 мМ). Полученную смесь концентрировали в вакууме.
Неочищенный продукт (2 мл) очищали с помощью препаративной БЭЖХ (2#-Анализ ВЭЖХ-SHIMADZU (ВЭЖХ-10), колонка: Atlantis HILIC OBD, размер частиц: 5 мкм, размер колонки: 19 к 150 мм, мобильная фаза: Н2О (10 мМ NH4HCO3+0,1% NH3)/CH3CN, от 42,0% до 44,0% CH3CN за 8 мин, детектор, УФ 254/210 нм), получая 56,9 мг (22%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
ЖХ-МС: (ES,m/z): 555 [М+Н]+. 1H ЯМР (400 МГц, MeOD-d4) δ м.д.: 7,96-7,89 (м, 2Н), 7,72-7,64 (м, 2Н), 7,06 (м, 2Н), 7,00-6,90 (м, 2Н), 6,85 (с, 1H), 3,77 (м, 4Н), 2,91 (м, 4Н), 2,81-2,71 (м, 8H), 2,47 (м, 2Н), 2,29 (м, 1Н), 1,90 (м, 3Н), 1,10-0,92 (м, 2Н).
ПРИМЕР 15
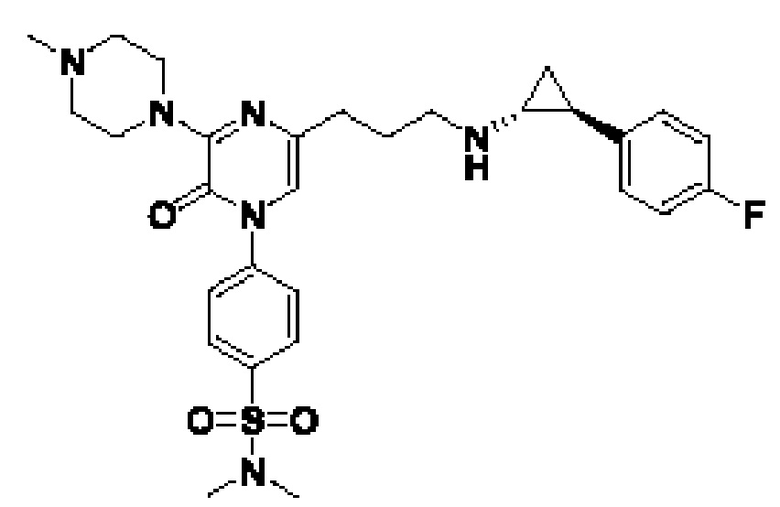
4-[5-(3-[([1R,2S]-2-[4-фторфенил]циклопропил)амино]пропил)-3-(4-метилпиперазин-1-ил)-2-оксопиразин-1(2H)-ил]-N,N-диметилбензолсульфонамид
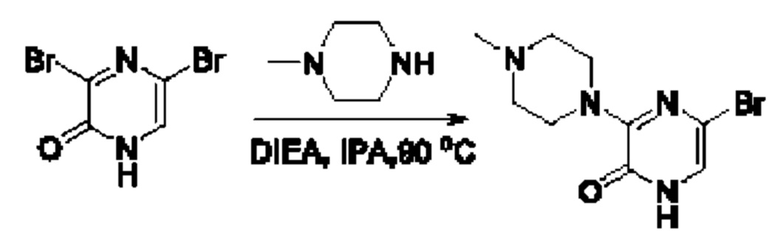
5-Бром-3-(4-метилпиперазин-1-ил)-пиразин-2(1H)-он (Промежуточное соединение 15-1)
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (10 г, 39,39 ммоль, 1,00 эквив.) и 1-мети л пи пер аз ином (4,78 г, 47,72 ммоль, 1,21 эквив.), получая 10,2 г (95%) указанного в заглавии соединения в виде твердого вещества желтовато-коричневого цвета.
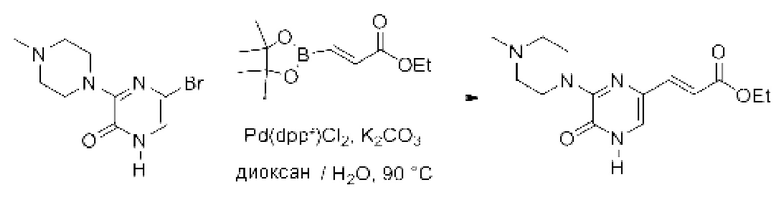
Этил (Е)-3-[6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил]проп-2-еноат (Промежуточное соединение 15-2)
Методику получения Промежуточного соединения 1-4 использовали с Промежуточным соединением 15-1 (5 г, 18,31 ммоль, 1,00 эквив.), получая 2 г (37%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
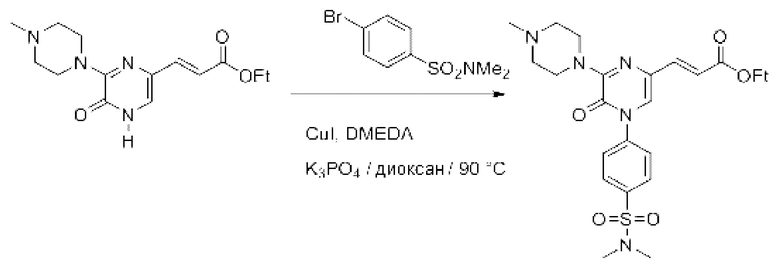
Этил (E)-3-[4-(4-[N,N-диметилсульдрамоил]фенил)-6-(4-метилпиперазин-1-ил)-5-оксо-4,5-дигидропиразин-2-ил)проп-2-еноат (Промежуточное соединение 15-3)
Методику получения Промежуточного соединения 13-1 использовали с Промежуточным соединением 15-2 (2,0 г, 6,84 ммоль, 1,00 эквив.) и 4-бром-N,N-диметилбензол-1-сульфонамидом и 4-бром-7-N,N-диметилбензол-1-сульфонамидом (2,7 г, 10,26 ммоль, 1,50 эквив.), с временем реакции 16 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (10:1), получая 1,25 г (38,4%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
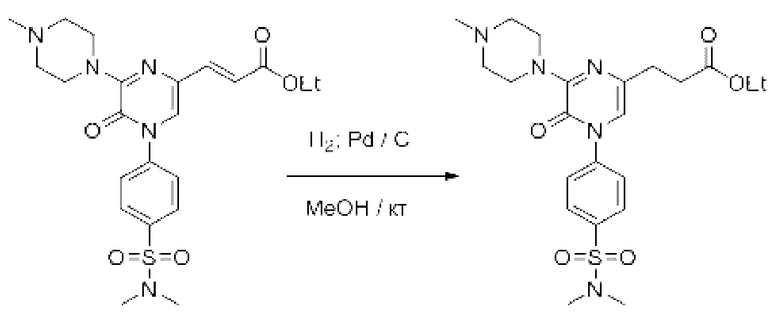
Этил 3-[4-(4-[N,N-диметилсульфамоил]фенил)-6-(4-метилпиперазин-1-ил)-5-оксо-4,5-дигидропиразин-2-ил]пропаноат
Методику получения Промежуточного соединения 1-5 использовали с Промежуточным соединением 15-3 (1,25 г, 2,63 ммоль, 1,00 эквив.) и Pd/C (125 мг), с временем реакции 2 ч, получая 1,2 г (95%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
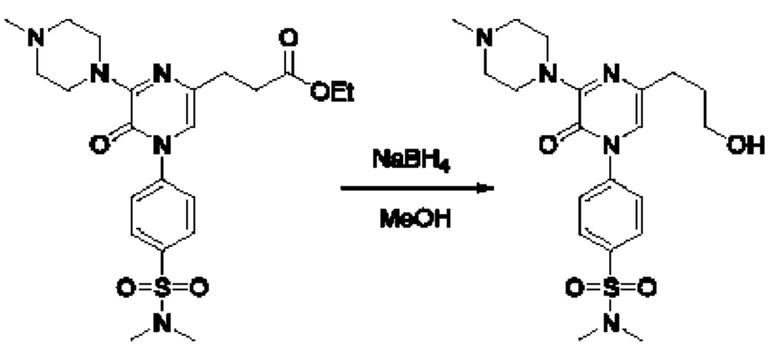
4-[5-(3-Гидроксипропил)-3-(4-метилпиперазин-1-ил]-2-оксопиразин-1(2H)-ил]-N,N-диметилбензолсульфонамид
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии, (1,2 г, 2,52 ммоль, 1,00 эквив.), с временем реакции 5 ч при кт, получая 600 мг (55%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
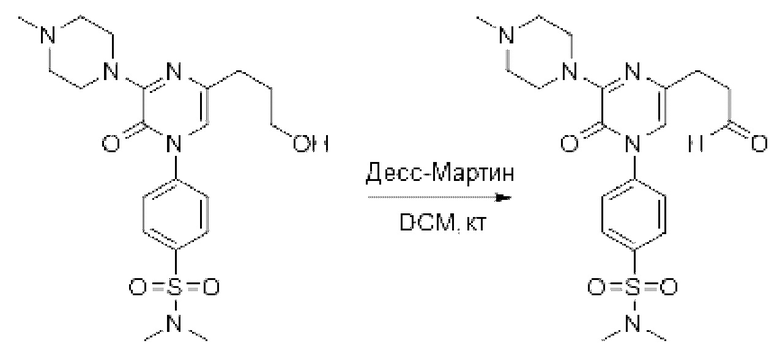
N,N-диметил-4-[3-(4-метилпиперазин-1-ил)-2-оксо-5-(3-оксопропил)пиразин-1(2H)-ил]бензолсульфонамид
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (600 мг, 1,38 ммоль, 1,00 эквив.), получая 300 мг (78%) указанного в заглавии соединения в вид е тверд ого вещества светло-желтого цвета.
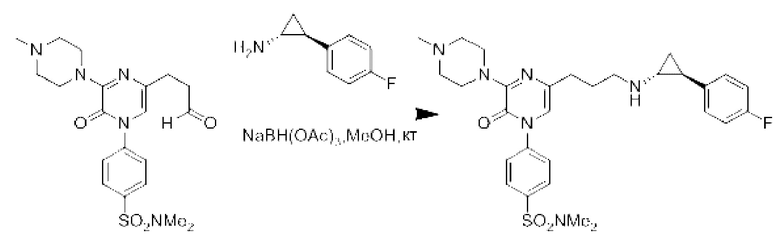
4-[5-(3-[([1R,2S]-2-[4-фторфенил]циклопропил)амино]пропил)-3-(4-метил-пиперазин-1-ил)-2-оксопиразин-1(2Н)-ил]-N,N-диметилбензолсульфонамид
Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,69 ммоль, 1,00 эквив.). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод В (от 10,0% до 29,0%) CH3CN за 8 мин), получая 15,1 мг (4%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
ЖХ-МС: (ES,m/z): 569 [М+Н]+. 1H ЯМР (400 МГц, Метанол-d4) δ 7,95-7,92 (м, 2Н), 7,71-7,67 (м, 2H), 7,23-7,16 (м, 2Н), 7,08-6,99 (м, 2Н), 4,954,80 (м, 2H), 3,65-3,45 (с, 2Н), 3,35-3,15 (м, 6H), 3,00-2,90 (м, 4H), 2,76-2,71 (с, 6H)1 2,62-2,54 (м, 2H), 2,54-2,45 (м, 1H), 2,15-2,05 (м, 2H), 1,55-1,47 (м, 1H), 1,41-1,32 (м, 1H).
ПРИМЕР 16
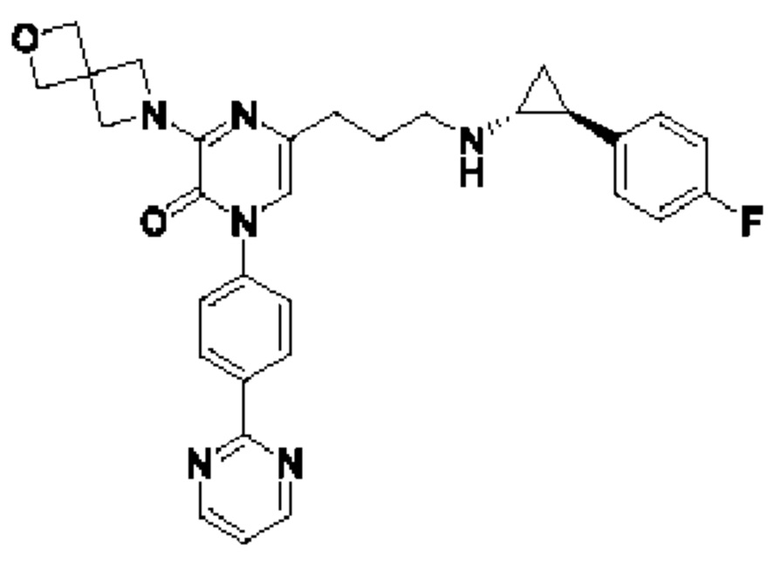
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-1-[4-(пиримидин-2-ил)фенил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]пиразин-2(1H)-он
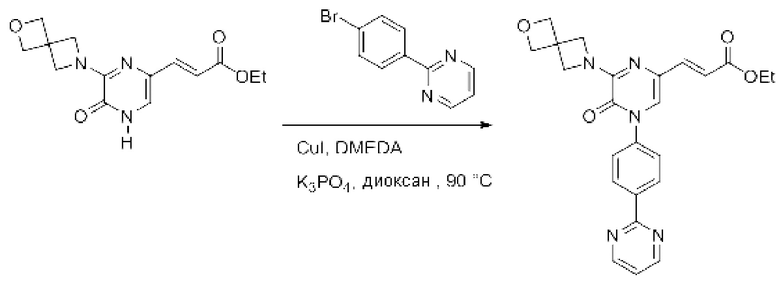
Этил (E)-3-(6-[2-окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксо-4-[4-(пиримидин-2-ил)фенил]-пиразин-2-ил)проп-2-еноат
Методику получения Промежуточного соединения 15-3 использовали с Промежуточным соединением 13-1 и 2-(4-бромфенил)пиримидином с временем реакции 6 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 2,0 г (44%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
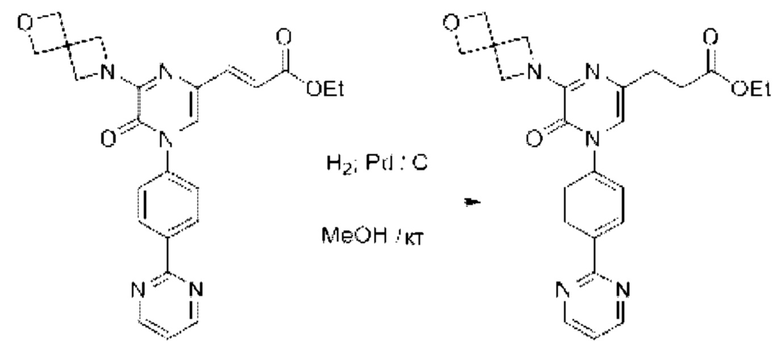
Этил 3-(6-[2-окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксо-4-[4-(пиримидин-2-ил)фенил]-пиразин-2-ил)пропаноат
Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (2 г, 4,49 ммоль, 1,00 эквив.), получая 1,8 г (90%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
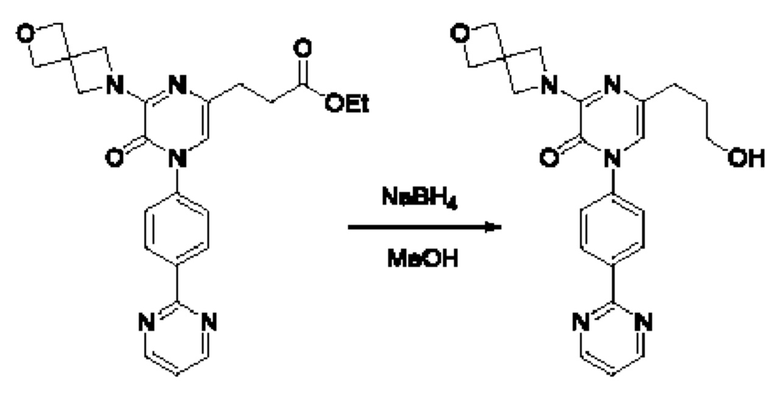
3-[4-(4-(Пиримидин-2-ил)фенил)-6-[2-окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксопиразин-2-ил]пропан-1-ол
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии (1,8 г, 4,02 ммоль, 1,00 эквив.), получая 0,4 г (25%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
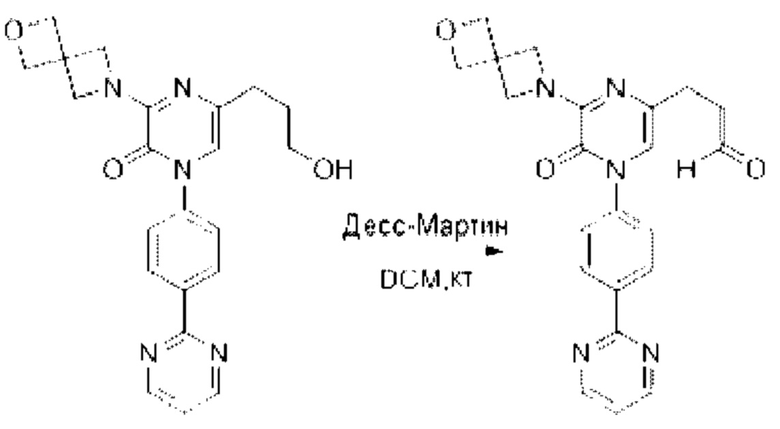
3-[4-(4-(Пиримидин-2-ил)фенил)-6-[2-окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксопиразин-2-ил]пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (400 мг, 0,99 ммоль, 1,00 эквив.), получая 0,12 г (30%) указанного в заглавии соединения в виде масла светло-желтого цвета.
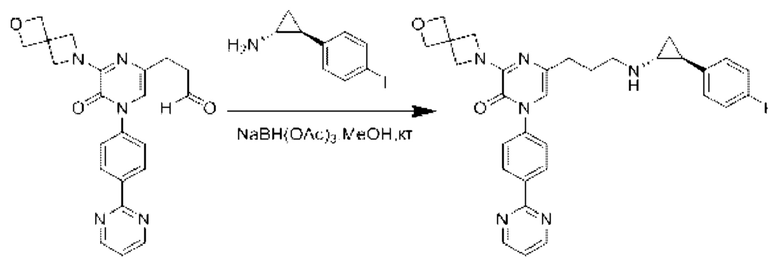
1-(4-(Пиримидин-2-ил)-фенил)-3-(2-окса-6-азаспиро[3,3]гептан-6-ил)-5-(3-[[(1R,2S)-2-(4-фторфенил)циклопропил]амино]пропил)пиразин-2(1H)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (120 мг, 0,30 ммоль). Неочищенный продукт (4 мл) очищали, используя хроматографический Метод E (от 50% до 55% CH3CN за 7 мин, Rt 6,35 мин), получая 13,1 мг (8%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХМС (ES,m/z): 539 [М+Н]+. 1H ЯМР (300 МГц, Метанол-d4) δ м.д.: 8,85 (д, J=4,8 Гц, 2Н), 8,59-8,48 (м, 2H), 7,56-7,46 (м, 2Н), 7,37 (м, 1H), 7,10-6,86 (м, 4Н), 6,65 (с, 1H), 4,83 (с, 4Н), 4,43 (с, 4Н), 2,75 (т, J=7,2 Гц, 2Н), 2,41 (т, J=7,2 Гц, 2Н), 2,33-2,24 (м, 1Н), 1,96-1,76 (м, 3Н), 1,11-0,90 (м, 2H).
ПРИМЕР 17
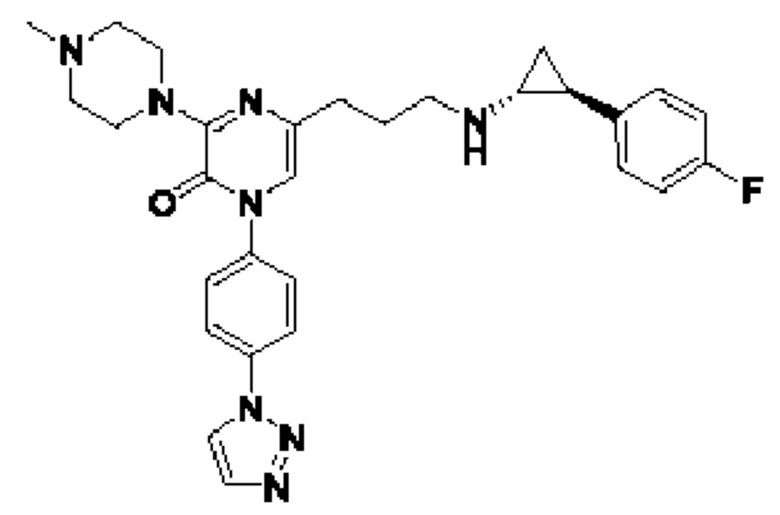
1-[4-(1H-1,2,3-триазол-1-ил)фенил]-5-[3-([(1R,2S)-2-(4-фторфенил)-циклопропил]амино)пропил]-3-[4-метилпиперазин-1-ил]пиразин-2(1H)-он
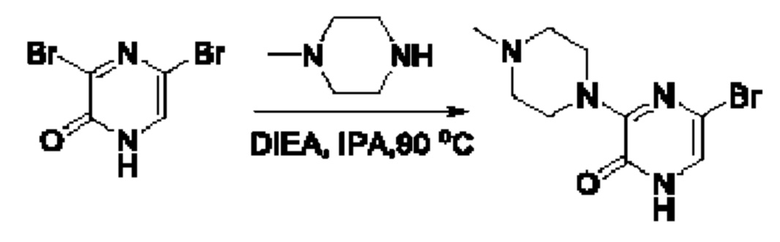
5-Бром-3-(4-метилпиперазин-1-ил)-пиразин-2(1H)-он (Промежуточное соединение 17-1)
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (20 г, 78,78 ммоль, 1,00 эквив.) и 1-метилпиперазином (9,49 г, 94,90 ммоль, 1,20 эквив.). Неочищенный продукт очищали с использованием хроматографии на силикагеле, используя EtOAc/петролейный эфир (8:1), получая 16,0 г (74,1%) указанного в заглавии соединения в виде твердого вещества белого цвета.
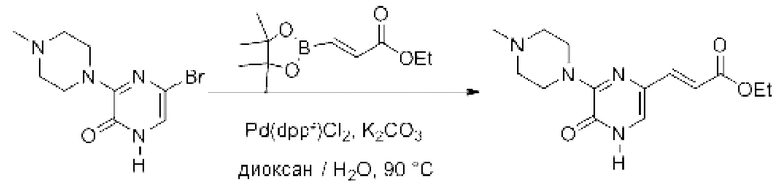
Этил (E)-3-[6-(4-метилпиперазин-1-ил)-5(4Н)-оксопиразин-2-ил]проп-2-еноат (Промежуточное соединение 17-2)
Методику получения Промежуточного соединения 1-4 использовали с Промежуточным соединением 17-1 (16,0 г, 58,61 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (6:1), получая 9,0 г (52,4%) указанного в заглавии соединения в виде масла желтого цвета.
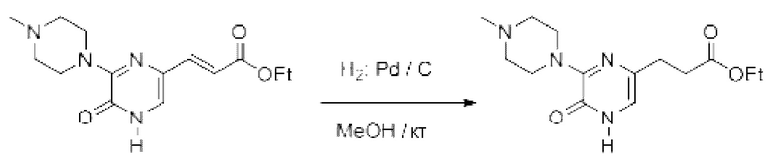
Этил 3-[6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил]пропаноат (Промежуточное соединение 17-3)
Методику получения Промежуточного соединения 1-5 использовали с Промежуточным соединением 17-2 (9,0 г, 30,72 ммоль, 1,00 эквив.), получая 8,9 г (98,2%) указанного в заглавии соединения в виде масла желтого цвета.
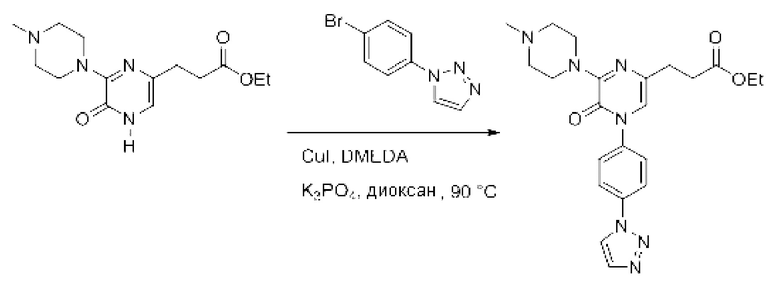
Этил 3-(4-(4-(1H-1,2,3-триазол-1-ил)фенил)-6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил)пропаноат
Методику получения Промежуточного соединения 13-1 использовали с Промежуточным соединением 17-3 (2,9 г, 9,83 ммоль, 1 эквив.) и 1-(4-бромф енил)-1H-1,2,3-триазолом (3,3 г, 14,73 ммоль, 1,5 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 1,2 г (27,9%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
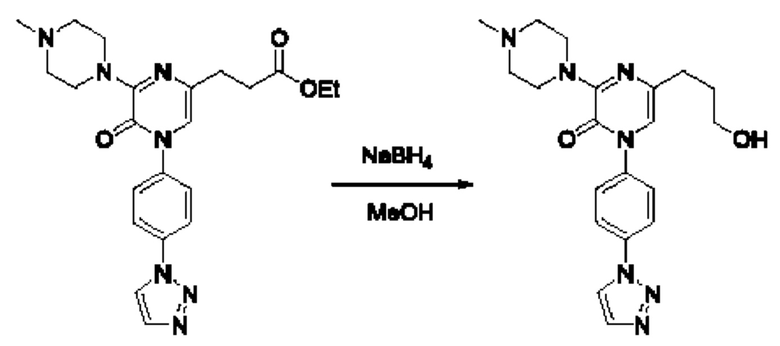
3-(4-(4-(1H-1,2,3-Триазол-1-ил)фенил)-6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил)пропан-1-ол
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии, (1,2 г, 2,74 ммоль, 1 эквив.), получая 400 мг (36,9%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
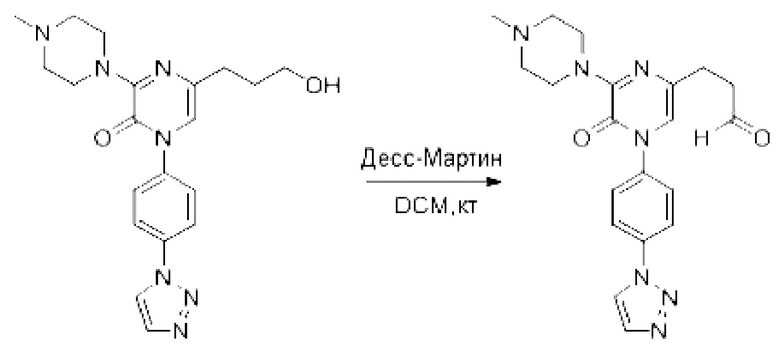
3-(4-(4-(1H-1,2,3-Триазол-1-ил)фенил)-6-(4-метилпиперазин-1-ил)-5(4H)-оксогшразин-2-ил)пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (350 мг, 0,88 ммоль, 1,00 эквив.), получая 160 мг (46. %) указанного в заглавии соединения в виде твердого вещества желтого цвета.
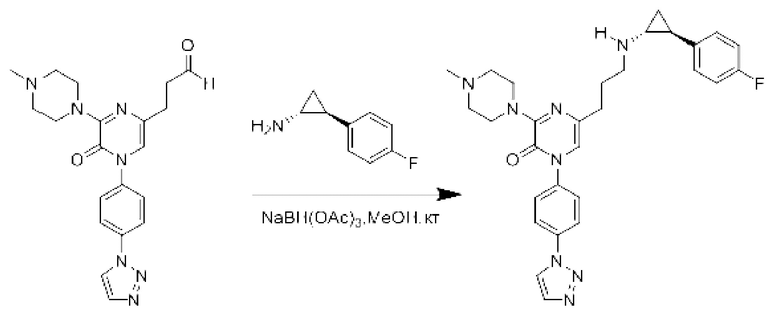
1-[4-(1H-1,2,3-триазол-1-ил)фенил]-5-[3-([(1R,2S)-2-(4-фторфенил)-циклопропил]амино)пропил]-3-[4-метилпиперазин-1-ил]пиразин-2(1H)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (150 мг, 0,41 ммоль). Неочищенный продукт (5 мл) очищали, используя хроматограф ич ее кий Метод E (от 27% до 60% CH3CN, Rt: 10,13 мин), получая 42,8 мг (21,25%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-МС: (ES,m/z): 529 [М+Н]+. 1H ЯМР (300 МГц, MeOD-d4) δ м.д.: 8,60 (с, 1Н), 8,05-7,95 (м, 2H), 7,91 (с, 1H), 7,67-7,57 (м, 2Н), 7,06-7,01 (м, 2H), 6,97-6,88 (м, 2Н), 6,85 (с, 1Н), 3,78 (м, 4H), 2,75 (т, J=7,4 Гц, 2Н), 2,56-2,40 (м, 6Н), 2,29 (с, 3Н), 2,30-2,21 (м, 1Н), 1,95-1,79 (м, 3Н), 1,10-0,88 (м, 2H).
ПРИМЕР 18
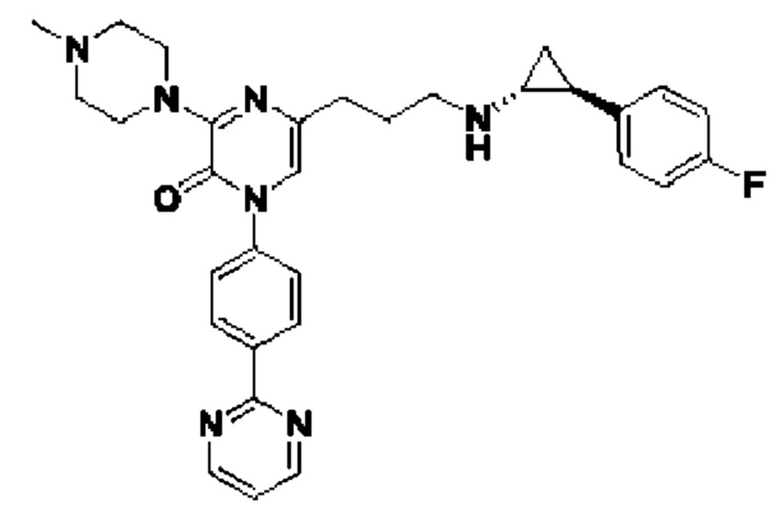
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[4-метилпиперазин-1-ил]-1-[4-(пиримидин-2-ил)фенил]пиразин-2 (1H)-он
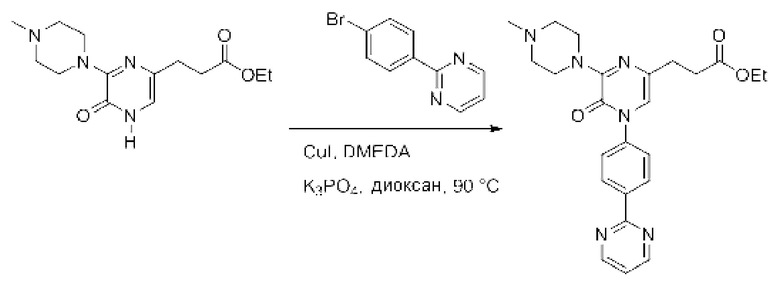
Этил 3-(4-(Пиримидин-2-ил) фенил)-6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил)пропаноат
Методику получения Промежуточного соединения 13-1 использовали с Промежуточным соединением 17-3 (2,2 г, 7,46 ммоль, 1 эквив.) и 2-(4-6ромфенил)пиримидином (2,63 г, 11,12 ммоль, 1,5 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 1,0 г (29,86%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
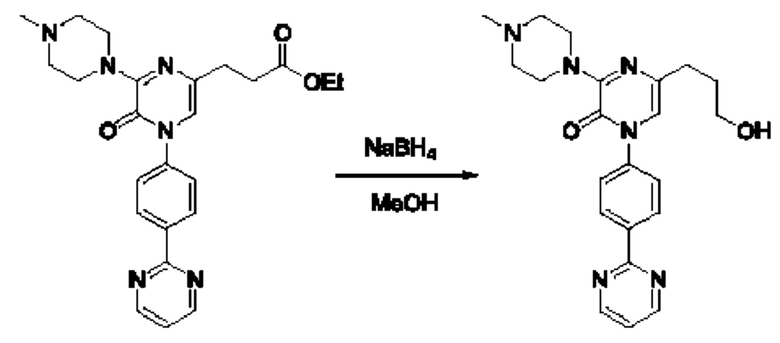
3-(4-(4-(Пиримидин-2-ил)фенил)-6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил)пропан-1-ол
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии, (800 мг, 1,78 ммоль, 1 эквив.), получая 300 мг (41,37%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
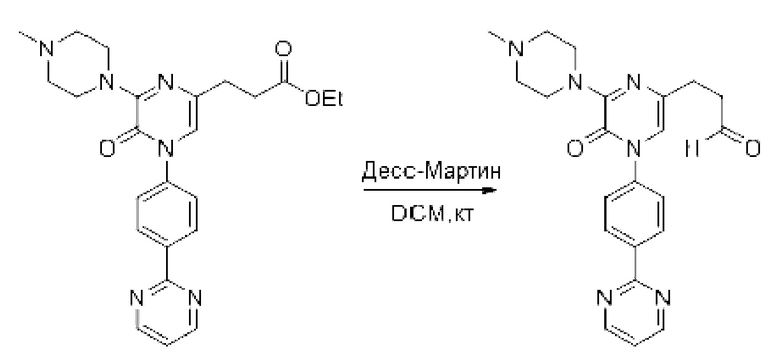
3-(4-(4-(Пиримидин-2-ил)фенил)-6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил)пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,74 ммоль, 1,00 эквив.), получая 150 мг (50,25%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
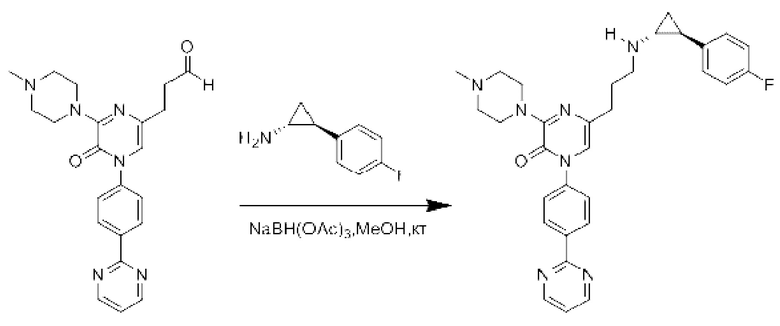
5-[3-([(1R,2S)-2-(4-фторфенил)пиклопропил]амино)пропил]-3-[4-метил-пиперазин-1-ил]-1-[4-(пиримидин-2-ил)фенил]пиразин-2(1H)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (150 мг, 0,37 ммоль). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод D (от 27% до 60% CH3CN, Rt: 10,13 мин), получая 51,5 мг (25,75%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-МС: (ES,m/z): 540 [М+Н]+. 1H ЯМР (300 МГц, MeOD-d4) δ м.д.: 8,86 (д, J=4,9 Гц, 2H), 8,58-8,52 (м, 2Н), 7,57-7,50 (м, 2Н), 7,38 (т, J=4,9 Гц, 1H), 7,21-7,13 (м, 2H), 7,07-6,97 (м, 3H), 4,92-4,85 (м, 2Н), 3,60-3,47 (м, 2Н), 3,25-3,12 (м, 6Н), 3,00-2,85 (м, 4H), 2,57 (т, J=7,3 Гц, 2Н), 2,50-2,40 (м, 1Н), 2,13-2,00 (м, 2H), 1,53-1,42 (м, 1H), 1,40-1,23 (м, 1H).
ПРИМЕР 19
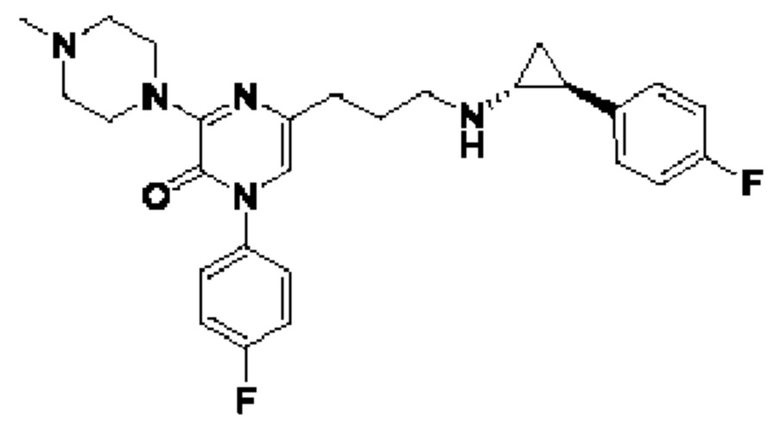
1-[4-Фторфенил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[4-метилпиперазин-1-ил]пиразин-2(1H)-он
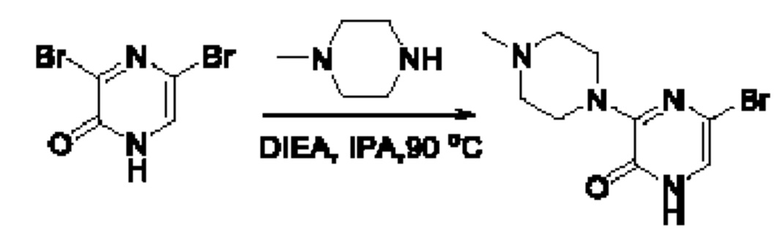
5-Бром-3-(4-метилпиперазин-1-ил)-пиразин-2(1H)-он
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (10 г, 39,37 ммоль, 1,00 эквив.) и 1-метил пи пер аз ином (4,72 г, 47,24 ммоль, 1,20 эквив.). Неочищенный продукт очищали с использованием хроматографии на силикагеле, используя EtOAc/петролейный эфир (8:1), получая 10 г (93%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
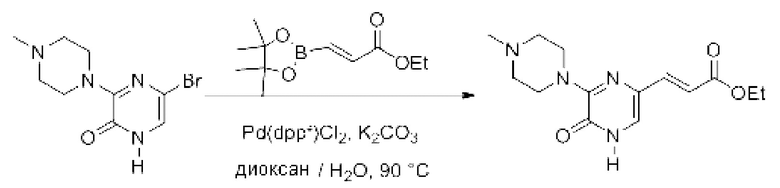
Этил (E)-3-[6-(4-метилпиперазин-1-ил)-5(4Н)-оксопиразин-2-ил]проп-2-еноат
Методику получения Промежуточного соединения 1-4 использовали с продуктом, полученным на предыдущей стадии, (10 г, 36,63 ммоль). Неочищенный продукт очищали с использованием хроматографии на силикагеле, используя EtOAc/петролейный эфир (6:1), получая 5,6 г (52%) указанного в заглавии соединения в виде масла желтого цвета.
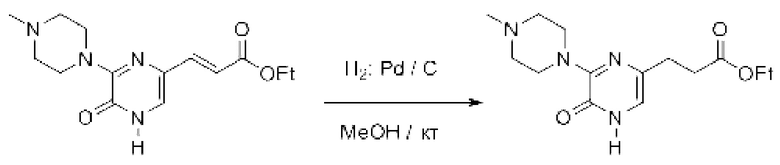
Этил 3-[6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил]пропаноат
Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (5,6 г, 19,18 ммоль, 1,00 эквив.), получая 5,5 г (98%) указанного в заглавии соединения в виде масла желтого цвета.
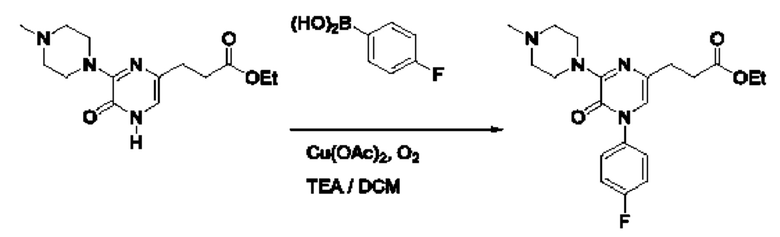
Этил 3-(4-(4-фторфенил)-6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил)пропаноат
Методику получения Промежуточного соединения 8-9 использовали с продуктом, полученным на предыдущей стадии, (1,5 г, 5,10 ммоль, 1 эквив.) и 4-фторфенилбороновой кислотой (1,07 г, 7,65 ммоль, 1,5 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 1 г (51%) указанного в заглавии соединения в виде масла желтого цвета.
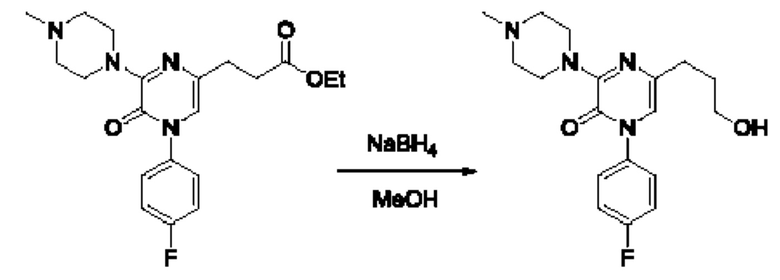
3-(4-(4-(4-Фторфенил)-6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил)пропан-1-ол
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии, (1 г, 2,58 ммоль, 1 эквив.), получая 600 мг (67%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
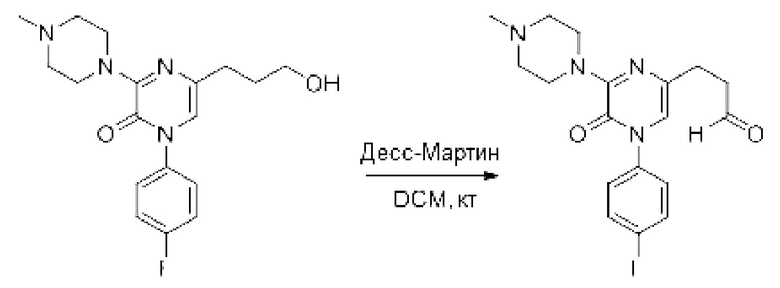
3-(4-(4-Фтор фенил)-6-(4-метилпиперазин-1-ил)-5(4H)-океопиразин-2-ил)пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (600 мг, 1,73 ммоль, 1,00 эквив.), получая 300 мг (50%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
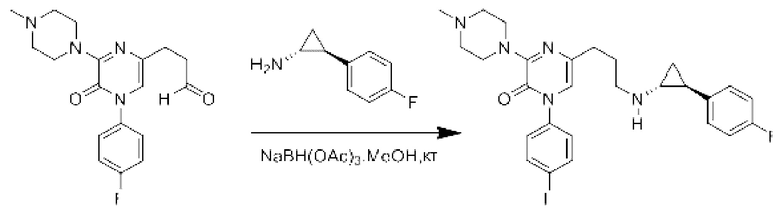
1-[4-Фторфенил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[4-метилпиперазин-1-ил]пиразин-2(1Н)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,87 ммоль). Неочищенный продукт очищали, используя хроматографический Метод В (от 48%) до 70%) CH3CN за 9 мин), получая 34 мг (8%)) указанного в заглавии соединения в виде масла желтого цвета.
ЖХ-МС: (ES,m/z): 480 [М+Н]+. 1H ЯМР (400 МГц, MeOD-d4) δ м.д.: 7,47-7,44 (с, 2Н), 7,31-7,27 (т, J=17,2 Гц, 2Н), 7,23-7,20 (м, 2Н), 7,08-7,04 (т, J=17,6 Гц, 2Н), 6,98 (с, 1Н), 4,934,88 (м, 2Н), 3,59-3,54 (с, 2Н), 3,33-3,24 (м, 6H), 3,00-2,96 (м, 4Н), 2,61-2,57 (м, 2Н), 2,53-2,48 (м, 1H), 2,15-2,07 (м, 2H), 1,52-1,49 (м, 1Н), 1,41-1,37 (м, 1H).
ПРИМЕР 20
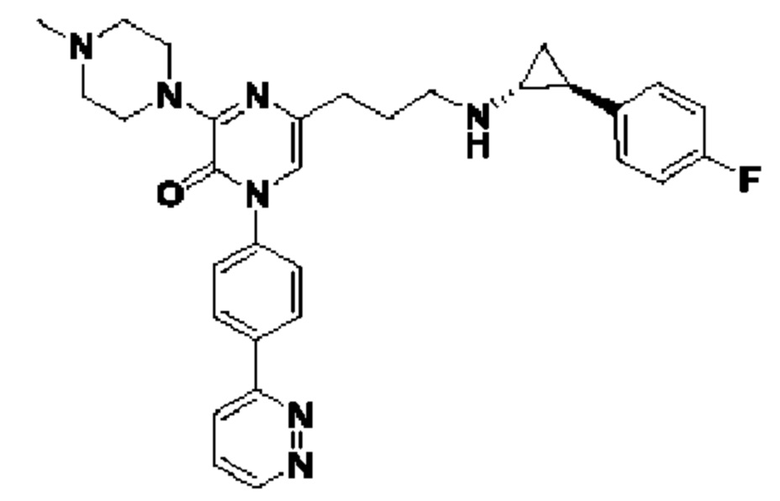
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[4-метилпиперазин-1-ил]-1-[4-(пиридазин-3-ил)фенил]пиразин-2(1H)-он
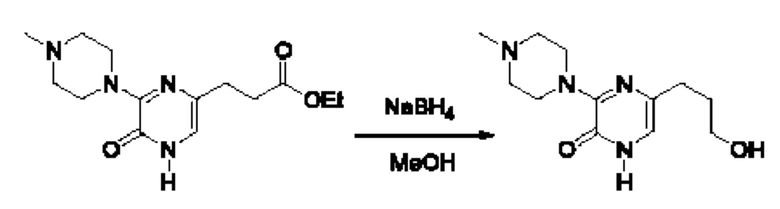
5-(3-Гидроксипропил)-3-(4-метилпиперазий-1-ил)пиразин-2(1H)-он
Методику получения Промежуточного соединения 1-6 использовали с Промежуточным соединением 17-3 (1,0 г, 3,34 ммоль, 1 эквив.) с временем реакции 1 ч, получая 600 мг (69,96%) указанного в заглавии соединения в виде масла желтого цвета.
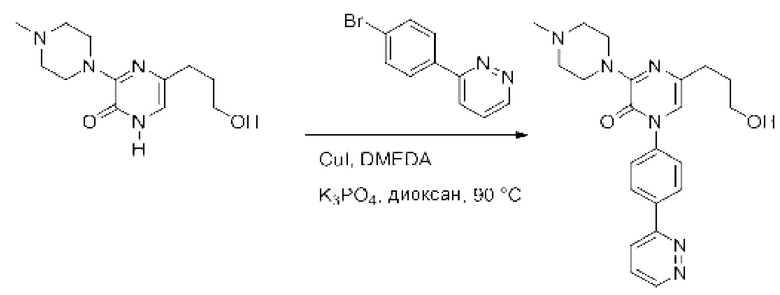
3-(4-(4-(Пиридазин-3-ил)фенил)-6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил)пропан-1-ол
Методику получения Промежуточного соединения 13-1 использовали с продуктом, полученным на предыдущей стадии, (600 мг, 2,37 ммоль, 1 эквив.) и 3-(4-6ромфенил)пиридазином (836 мг, 3,56 ммоль, 1,5 эквив.), в течение ночи при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (10:1), получая 600 мг (61,2%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
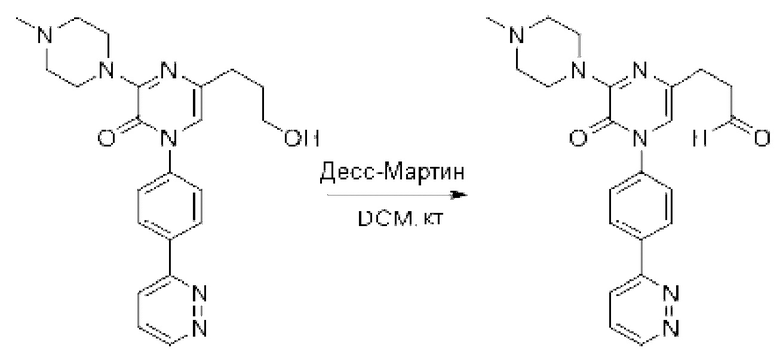
3-(4-(4-(Пиридазин-3-ил)фенил)-6-(4-метилпиперазин-1-ил)-5(4H)-оксопиразин-2-ил)пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,74 ммоль, 1,00 эквив.), получая 150 мг (50,3%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
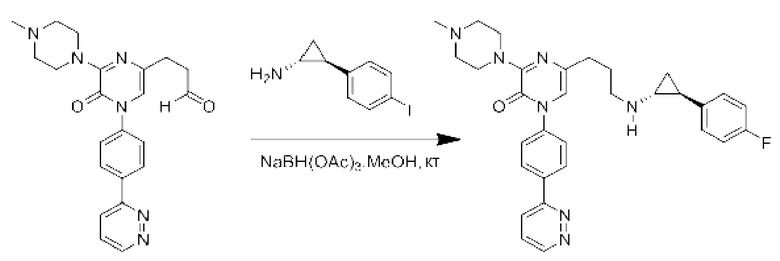
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[4-метил-пиперазин-1-ил]-1-[4-(пиридазин-3-ил)фенил]пиразин-2(1H)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (150 мг, 0,37 ммоль). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод E (от 31% до 68% CH3CN за 8 мин, Rt 7,13 мин), получая 29,7 мг (14,85%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-МС: (ES,m/z): 540 [М+Н]+. 1H ЯМР (300 МГц, MeOD-d4) δ м.д.: 9,16 (dd, J=4,9, 1,5 Гц, 1Н), 8,28-8,18 (м, 3Н), 7,85-7,77 (м, 1Н), 7,62-7,54 (м, 2Н), 7,09-6,98 (м, 2Н), 6,96-6,84 (м, 3Н), 3,85-3,73 (м, 4Н), 2,76 (т, J=7,4 Гц, 2H), 2,57-2,41 (м, 6Н), 2,34-2,24 (м, 4Н), 1,95-1,82 (м, 3Н), 1,10-0,89 (м, 2Н).
ПРИМЕР 21
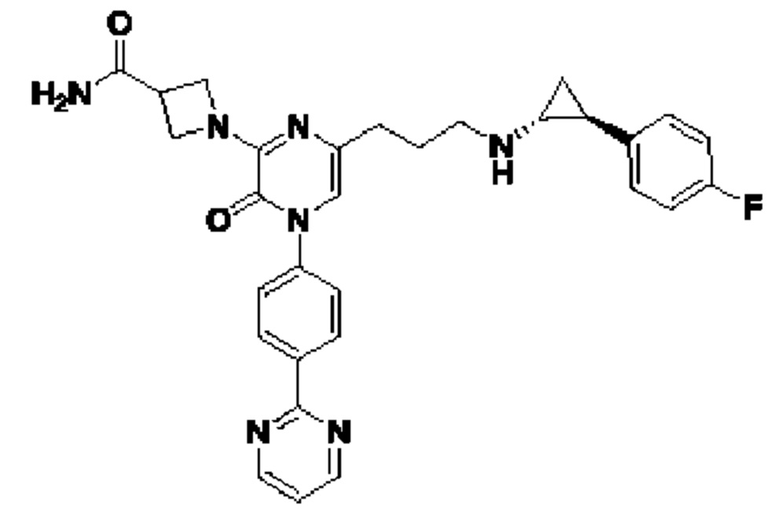
1-[6-(3-[([1R,2S]-1-[4-фторфенил]циклопропил)амино]пропил)-3-оксо-4-(4-[пиримидин-2-ил]фенил)-3,4-дигидропиразин-2-ил]азетидин-3-карбоксамид
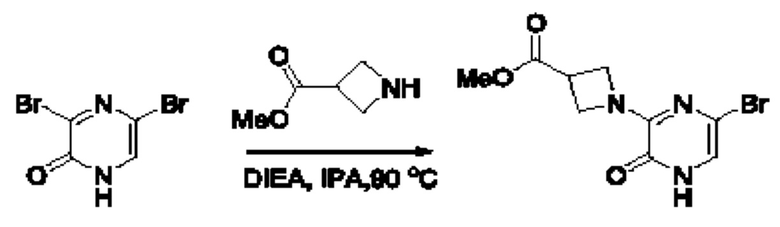
Метил 1-(6-бром-3(4H)-оксопиразин-2-ил)азетидин-3-карбоксилат
Методику получения Промежуточного соединения 2-1 использовали с 3,5-ди6ром-1,2-дигидропиразин-2-оном (10 г, 39,39 ммоль, 1,00 эквив.) и метил азетидин-3-карбоксилатом (6,8 г, 59,36 ммоль, 1,50 эквив.), с временем реакции 2 ч при 90°С, получая 8 г (67%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
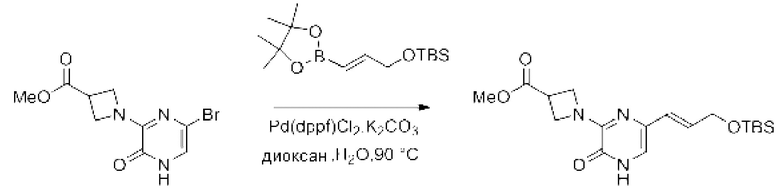
(E)-Метил 1-(6-(3-(трет-бутилдиметилсилилокси)проп-1-енил)-3(4H)-оксопиразин-2-ил)азетидин-3-карбоксилат
Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (8 г, 31,63 ммоль), с временем реакции 2 ч при 90°C Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:20), получая 3 г (28,4%) указанного в заглавии соединения в виде масла светло-желтого цвета.
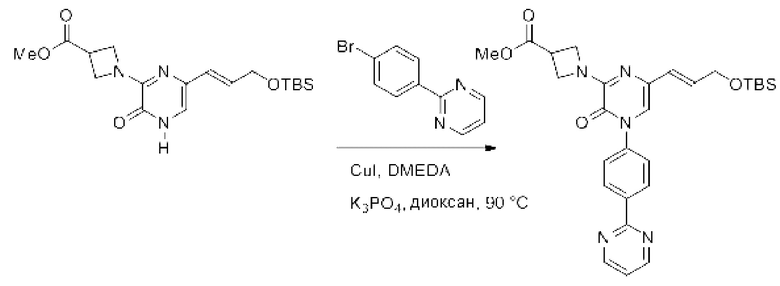
(E)-Метил 1-(6-(3-(трет-бутилдиметилсилилокси)проп-1-енил)-3(4H)-оксо-4-(4-(пиримидин-2-ил)фенил)-пиразин-2-ил)азетидин-3-карбоксилат
Методику получения Промежуточного соединения 13-1 использовали с продуктом, полученным на предыдущей стадии, (3 г, 9,81 ммоль, 1,00 эквив.) и 2-(4-6ромфенил)пиримидином (3,1 г, 14,89 ммоль, 1,50 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир, получая 2,3 г (53%) указанного в заглавии соединения в виде масла желтого цвета.
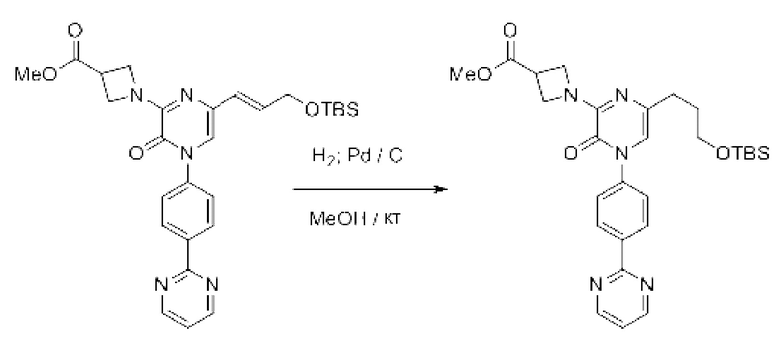
Метил 1-(6-(3-(трет-бутилдиметилсилилокси)пропил)-3(4H)-оксо-4-(4-(пиримидин-2-ил)фенил)-пиразин-2-ил)азетидин-3-карбоксилат
Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (2,3 г, 3,38 ммоль, 1,00 эквив.), получая 2,1 г (89%) указанного в заглавии соединения в виде масла светло-желтого цвета.
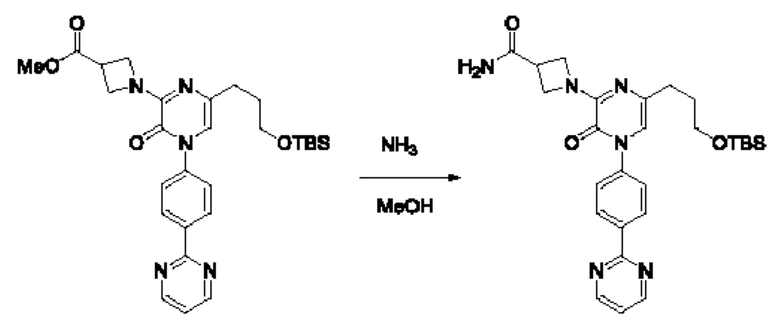
1-(6-(3-(трет-бутилдиметилсилилокси)пропил)-3(4H)-оксо-4-(4-(пиримидин-2-ил)фенил)-пиразин-2-ил)азетидин-3-карбоксамид
Раствор продукта, полученного на предыдущей стадии, (2,1 г, 3,02 ммоль, 1,00 эквив.) в MeOH (10 мл) объединяли с раствором NH3 (4 г, 5,00 эквив.) в MeOH (5 мл). Полученный в результате раствор перемешивали в течение 16 ч при 90°С, затем охлаждали и концентрировали в вакууме, получая 1,5 г (70%) указанного в заглавии соединения в виде масла светло-желтого цвета.
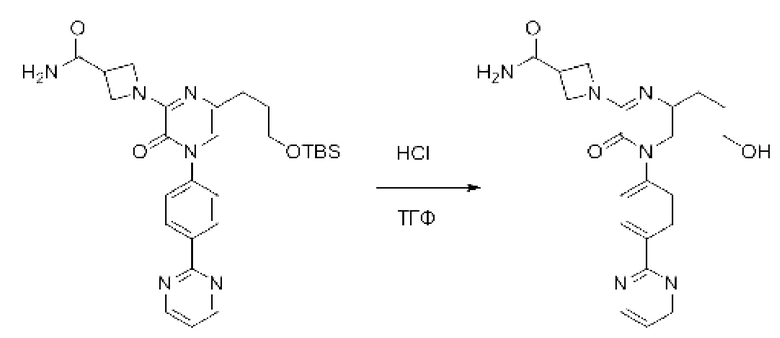
1-(6-(3-Гидроксипропил)-3(4H)-оксо-4-(4-(пиримидин-2-ил)фенил)-пиразин-2-ил)азетидин-3-карбоксамид
Раствор продукта, полученного на предыдущей стадии, (1,5 г, 2,00 ммоль, 1,00 эквив.) в ТГФ (20 мл) объединяли с раствором HCl (3 мл, 2,00 эквив.) в Н2О (1 мл). Полученный в результате раствор перемешивали в течение 1 ч при 25°C. Значение рН раствора корректировали до 7 добавлением 1 M Na2CO3 и остаток затем очищали с помощью хроматографии на силикагеле, используя H2O/CH3CN (5:1), получая 760 мг (64%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
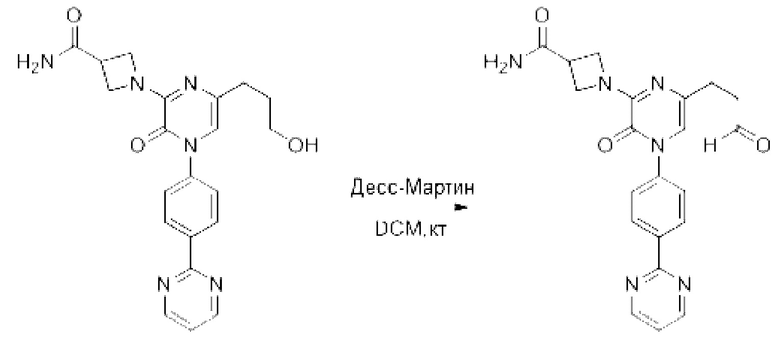
1-(3(4H)-оксо-6-(3-оксопропил)-4-(4-(пиримидин-2-ил)фенил)-пиразин-2-ил)-азетидин-3-карбоксамид
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (760 мг, 1,15 ммоль), получая 360 мг (47%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
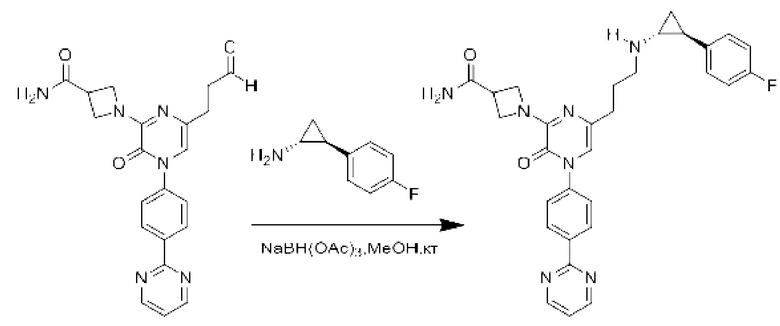
1-[6-(3-[([1R,2S]-2-[4-фторфенил]циклопропил)амино]пропил)-3-оксо-4-(4-[пиримидин-2-ил]фенил)-3,4-дигидропиразин-2-ил]азетидин-3-карбоксамид
Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (126 мг, 0,33 ммоль, 1,20 эквив.). Неочищенный продукт (3 мл) очищали, используя хроматографический Метод С (от 38,0% до 50,0% CH3CN за 8 мин), получая 47,7 мг (10%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХ-МС: (ES,m/z): 540 [М+Н]+. 1H ЯМР (300 МГц, MeOD-d4) δ м.д.: 8,95-8,61 (м, 2Н), 8,65-8,45 (м, 2S), 7,62-7,48 (м, 2S), 7,48-7,31 (м, 1H), 7,11-6,89 (м, 4S), 6,82 (с, 1Н), 4,664,19 (с, 4Н), 3,59-3,40 (м, 1H), 2,85-2,65 (м, 2S), 2,62-2,36 (м, 2S), 2,36-2,17 (м, 1H), 2,00-1,61 (м, 3Н), 1,12-0,89 (м, 2S).
ПРИМЕР 22
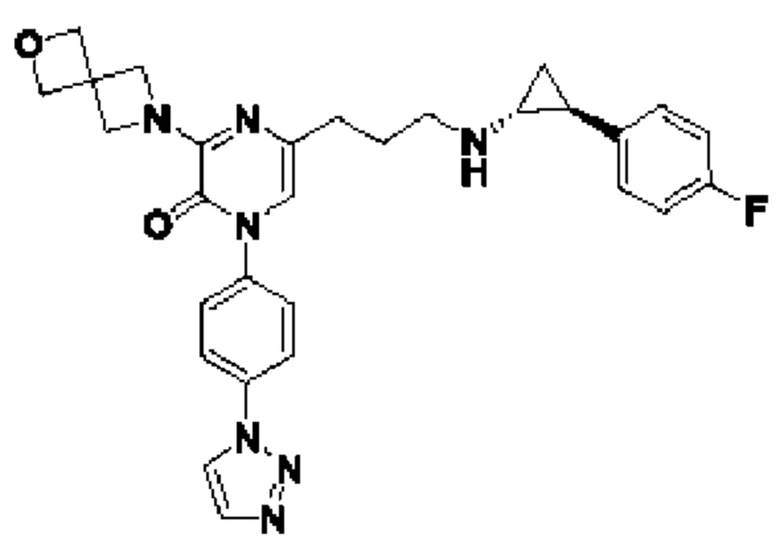
1-[4-(1H-1,2,3-триазол-1-ил)фенил]-5-[3-([(1R,2S)-2-(4-фторфенил)-циклопропил)амино]пропил)-3-(2-окса-6-азаспиро[3,3]гептан-6-ил)пиразин-2(1H)-он
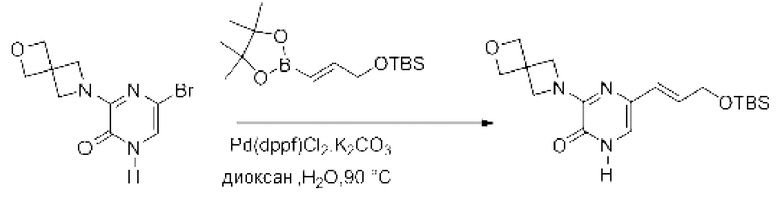
5-(E)-[3-[(трет-Бутилдиметилсилил)окси]пропил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]пиразин-2(1H)он (Промежуточное соединение 22-1)
Методику получения Промежуточного соединения 3-3 использовали с Промежуточным соединением 7-1 (4 г, 14,70 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:15), получая 1 г (19%) указанного в заглавии соединения в виде масла светло-желтого цвета.
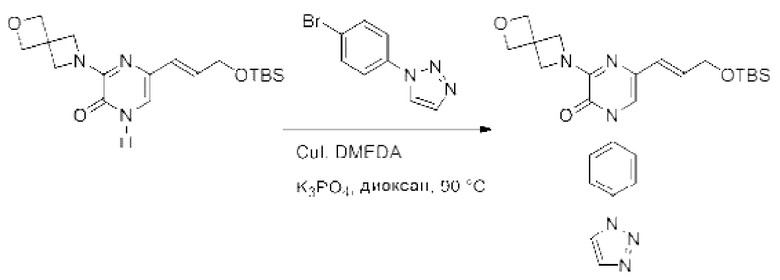
5-(E)[3-[(трет-Бутилдиметилсилил)окси]проп-1-ен-1-ил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]-1-[4-(1H-1,2,3-триазол-1-ил)фенил]-пиразин-2(1H) он (Промежуточное соединение 22-2)
Методику получения Промежуточного соединения 13-1 использовали с Промежуточным соединением 22-1 (1 г, 2,75 ммоль, 1,00 эквив.) и 1-(4-6ромфенил)-1H-1,2,3-триазолом (950 мг, 4,24 ммоль, 1,50 эквив.), с временем реакции 16 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:20), получая 1 г (72%) указанного в заглавии соединения в виде масла светло-желтого цвета.
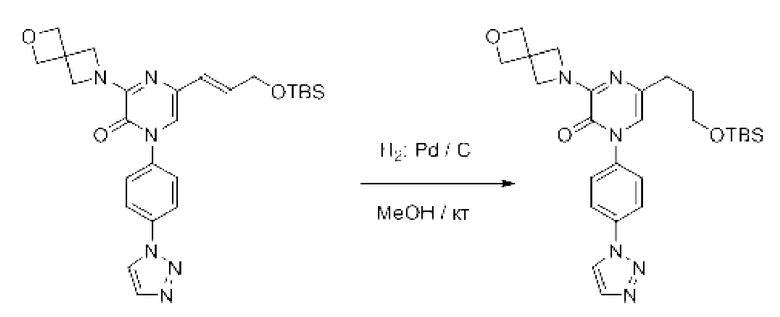
5-[3-[(трет-Бутилдиметилсилил)окси]пропил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]-1-[4-(1H-1,2,3-триазол-1-ил)фенил]-пиразин-2(1H)-он (Промежуточное соединение 22-3)
Методику получения Промежуточного соединения 1-5 использовали с Промежуточным соединением 22-2 (1 г, 1,97 ммоль, 1,00 эквив.), получая 0,9 г (90%) указанного в заглавии соединения в виде масла светло-желтого цвета.
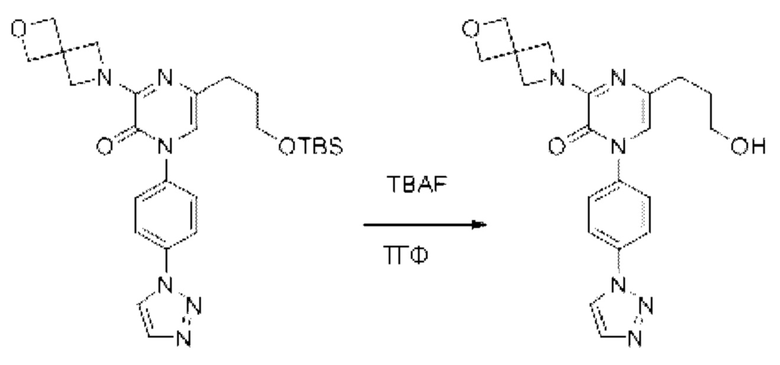
3-(6-[2-Окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксо-4-[4-(1H-1,2,3-триазол-1-ил)фенил]-пир азин- 2-ил) пропан-1-ол (Промежуточное соединение 22-4)
Раствор Промежуточного соединения 22-3 (900 мг, 1,77 ммоль, 1,00 эквив.) и Bu4NF (700 мг, 2,68 ммоль, 1,50 эквив.) в ТГФ (20 мл), перемешивали в течение 1 ч при 25°С, затем очищали с помощью хроматографии на силикагеле, используя H2O/MeCN (5:1), получая 0,52 г (75%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
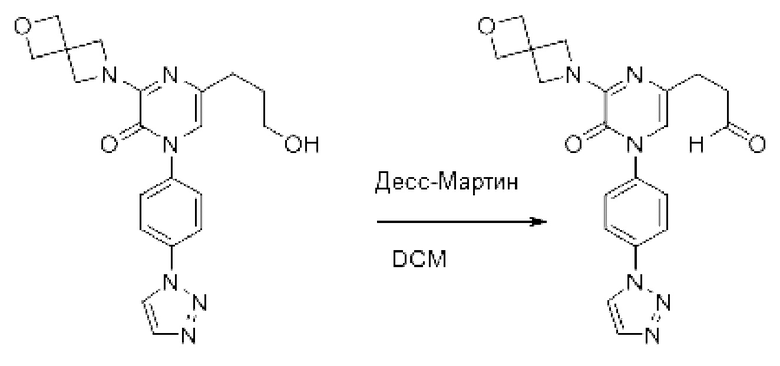
3-(6-[2-Окса-6-азаспиро[3,3]гептан-6-ил]-5(4Н)-оксо-4-[4-(1H-1,2,3-триазол-1-ил)фенил]-пиразин-2-ил)пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (520 мг, 1,32 ммоль, 1,00 эквив.), получая 300 мг (58%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
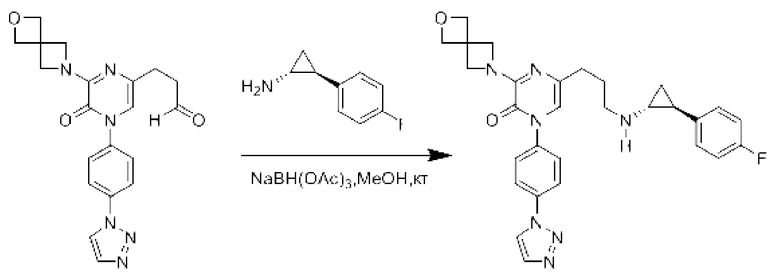
1-[4-(1H-1,2,3-триазол-1-ил)фенил]-5-[3-([(1R,2S)-2-(4-фторфенил)-циклопропил)амино]пропил)-3-(2-окса-6-азаспиро[3,3]гептан-6-ил)пиразин-2(1H)-он
Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,76 ммоль). Неочищенный продукт (2 мл) очищали, используя хроматографический Метод С (от 38,0% до 50,0% CH3CN за 8 мин), получая 11,8 мг (3%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-МС: (ES,m/z): 528 [М+Н]+. 1H ЯМР (300 МГц, MeOD-d4) δ м.д.: 8,70-8,51 (м, 1Н), 8,11 -7,99 (м, 2Н), 7,99-7,80 (м, 1H), 7,79-7,47 (м, 2Н), 7,13-6,80 (м, 4Н), 6,64 (с, 1Н), 4,82 (с, 4Н), 4,43 (с, 4H), 2,88-2,58 (м, 2Н), 2,52-2,35 (м, 2Н), 2,35-2,20 (с, 1Н), 2,09-1,71 (м, 3Н), 1,12-0,82 (м, 2H).
ПРИМЕР 23
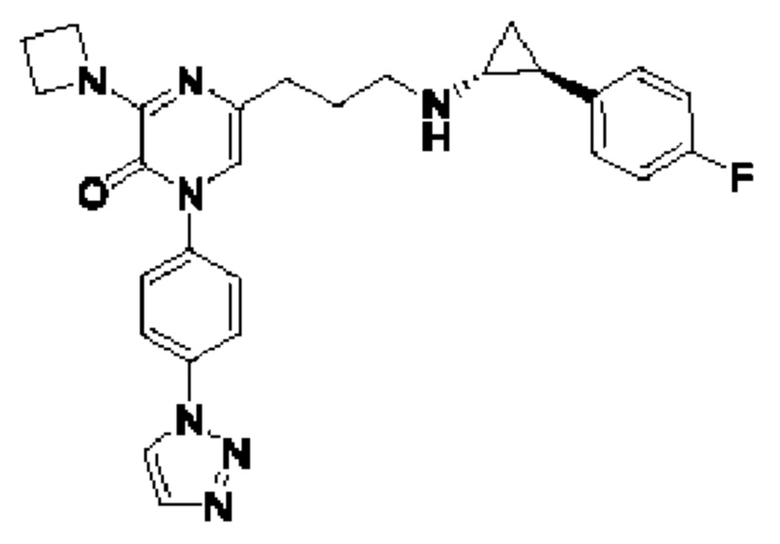
1-[4-(1H-1,2,3-триазол-1-ил)фенил]-3-[азетидин-1-ил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]пиразин-2(1H)-он
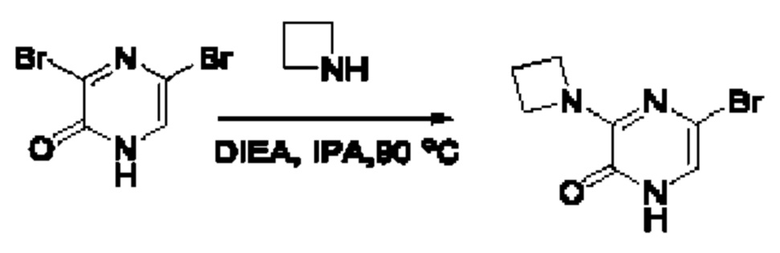
5-Бром-3-(азетидин-1-ил)-пиразин-2(1H)-он
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (5,0 г, 19,76 ммоль, 1 эквив.) и азетидином (1,46 г, 25,61 ммоль, 1,3 эквив.), получая 4,0 г (88%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
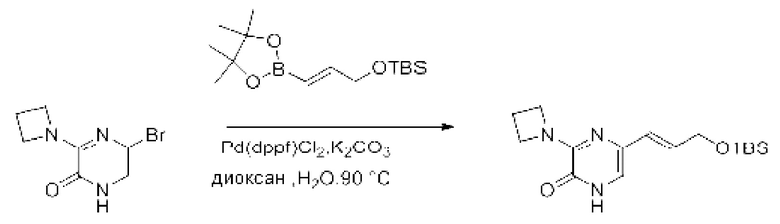
5-(E)-[3-[(трет-Бутилдиметилсилил)окси]пропил]-3-(азетидин-1-ил)-пиразин-2(1H) он
Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (4 г, 17,39 ммоль), с временем реакции 16 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:4), получая 1,4 г (25,04%) указанного в заглавии соединения в виде масла коричневого цвета.
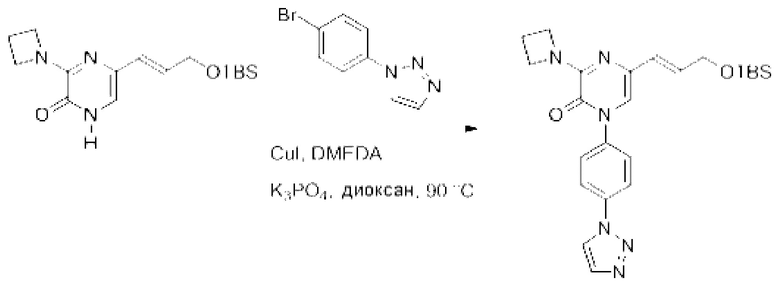
5-(E)[3-[(трет-Бутилдиметилсилил)окси]проп-1-ен-1-ил]-3-(азетидин-1-ил)-1-[4-(1Н-1,2,3-триазол-1-ил)фенил]-пиразин-2(1H)он Методику получения
Промежуточного соединения 13-1 использовали с продуктом, полученным на предыдущей стадии, (1,4 г, 4,35 ммоль, 1 эквив.) и 1-(4-6ромфенил)-1Н-1,2,3-триазолом (1,46 г, 6,53 ммоль, 1,5 эквив.), с временем реакции 16 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир, получая 1,1 г (54,37%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
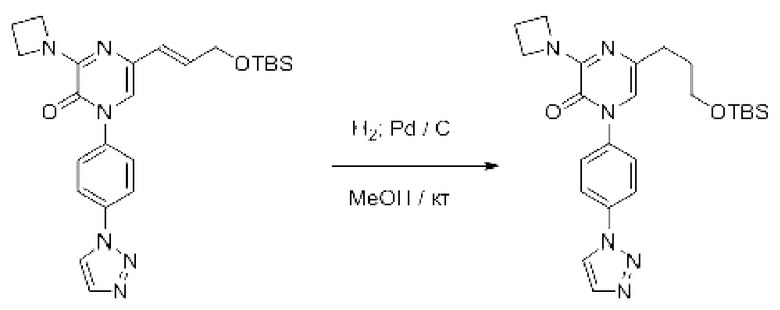
5-[3-[(трет-Бутилдиметилсилил)окси]пропил]-3-(азетидин-1-ил)-2(1H)-оксо-1-[4-(1Н-1,2,3-триазол-1-ил)фенил]-пиразин
Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (1,0 г, 2,15 ммоль, 1 эквив.), получая 1,0 г (99,57%) указанного в заглавии соединения в виде масла желтого цвета.
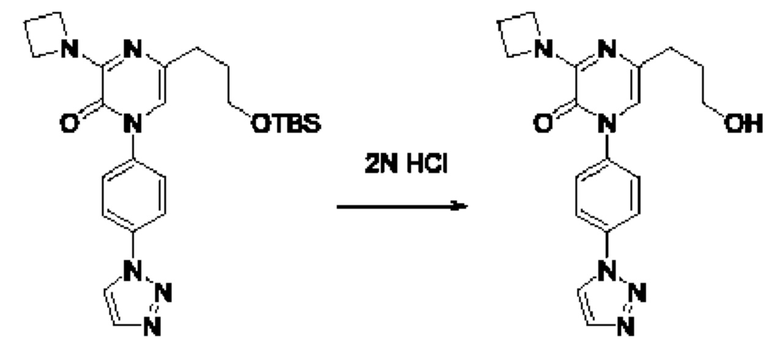
3-(6-(азетидин-1-ил)-5(4H)-оксо-4-[4-(1H-1,2,3-триазол-1-ил)фенил]-пиразин-2-ил)пропан-1-ол
Раствор продукта, полученного на предыдущей стадии, (900 мг, 1,93 ммоль, 1 эквив.) и 2 Nводн. HCl (3 мл) в ТГФ (30 мл) перемешивали в течение 15 мин при кт. Значение рН раствора корректировали до 8 добавлением Na2CO3. Полученный в результате раствор экстрагировали 3×30 мл CH2Cl2 и объединенные органические слои очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (8:1), получая 400 мг (58,84%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
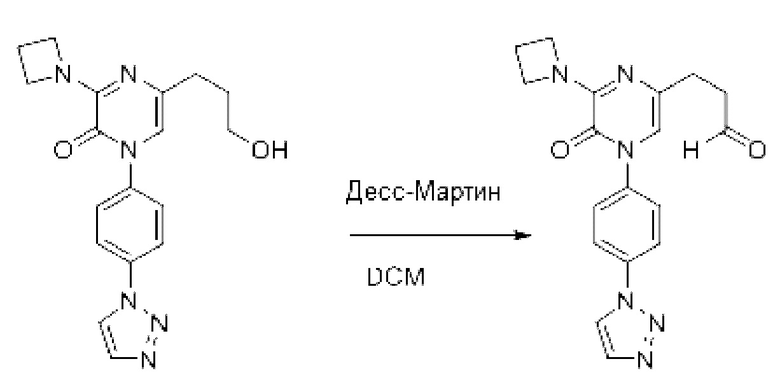
3-(6-(азетидин-1-ил)-5(4H)-оксо-4-[4-(1H-1,2,3-триазол-1-ил)фенил]-пиразин-2-ил)пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (380 мг, 1,08 ммоль, 1,00 эквив.), получая 200 мг (52,9%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
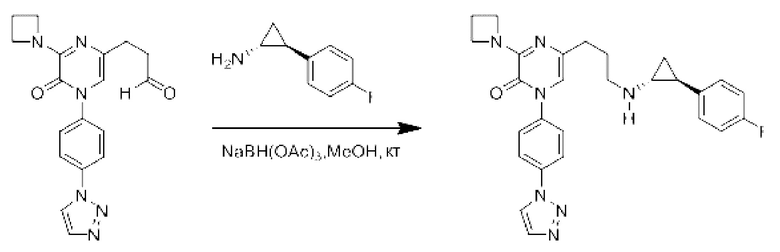
1-[4-(1H-1,2,3-триазол-1-ил)фенил]-3-[азетидин-1-ил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]пиразин-2(1H)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (200 мг, 0,57 ммоль). Неочищенный продукт очищали, используя хроматографии ее кий Метод E (от 30,0% до 65,0% CH3CN за 8,1 мин), получая 7,9 мг (2,85%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-МС: (ES,m/z): 486 [М+Н]+. 1H ЯМР (400 МГц, MeOD-d4) δ м.д.: 8,62 (с, 1Н), 8,03 (д, J=8,8 Гц, 2Н), 7,93 (с, 1H), 7,64 (д, J=8,8 Гц, 2H), 7,15-7,10 (м, 2Н), 6,99 (t, J=8,8 Гц, 2H), 6,65 (с, 1H), 4,424,27 (м, 4Н), 3,04-2,96 (м, 2Н), 2,65-2,56 (м, 1H), 2,47 (т, J=6,8 Гц, 2Н), 2,41-2,30 (м, 2Н), 2,20-2,10 (м, 1Н), 2,00-1,92 (м, 2Н), 1,20-1,10 (м, 2Н).
ПРИМЕР 24
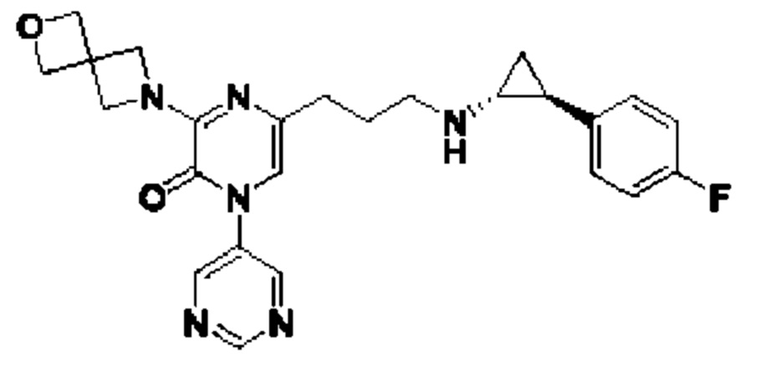
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-1-[пиримидин-5-ил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]пиразин-2(1Н)-он
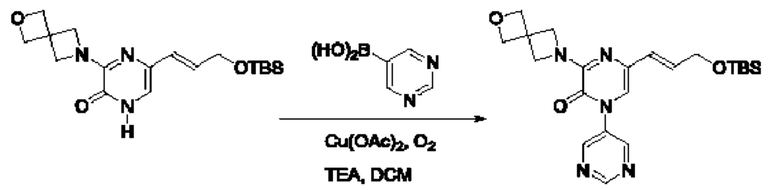
5-(Е)[3-[(трет-Бутилдиметилсилил)окси]проп-1-ен-1-ил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]-1-(пиримидин-5-ил)-пиразин-2(1Н)он
Методику получения Промежуточного соединения 8-9 использовали с Промежуточным соединением 22-1 (2 г, 5,51 ммоль, 1 эквив.) и (пиримидин-5-ил)бороновой кислотой (1,1 г, 8,26 ммоль, 1,5 эквив.). Неочищенный продукт очищали с использованием хроматографии на силикагеле, используя CH2Cl2/MeOH (1:10), получая 1 г (41,1%) указанного в заглавии соединения в виде твердого вещества.
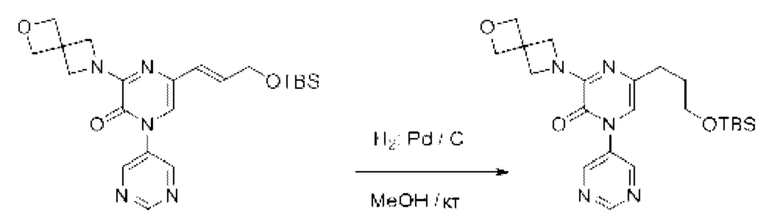
5-[3-[(трет-Бутилдиметилсилил)окси]пропил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]-1-(пиримидин-5-ил)-пиразин-2(1Н)-он
Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (1 г, 2,27 ммоль, 1 эквив.), получая 900 мг (89,6%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
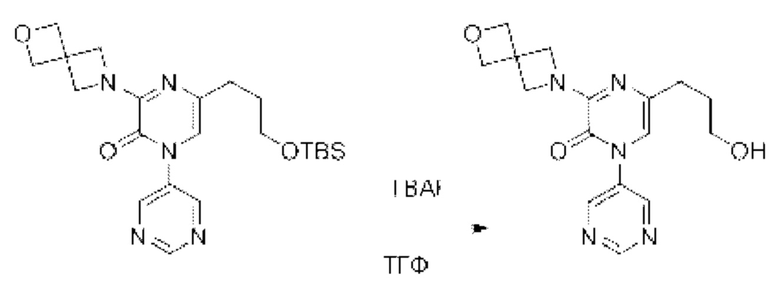
3-(6-[2-Окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксо-4-(пиримидин-5-ил)-пиразин-2-ил)пропан-1-ол
Методику получения Промежуточного соединения 22-4 использовали с продуктом, полученным на предыдущей стадии, (900 мг, 2,03 ммоль). Неочищенный продукт (10 мл)очищали с помощью флеш-препаративной ВЭЖХ (Intel Flash-1: MeCN/H2O=1:10 увеличивая до MeCN/H2O=5:10 за 40 мин; детектор, 220 нм), получая 400 мг (59,86%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
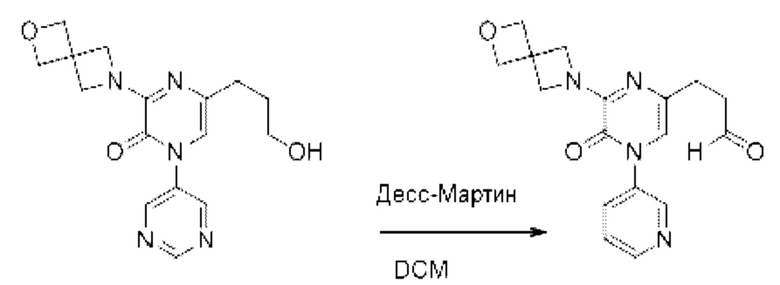
3-(6-[2-Окса-6-азаспиро[3,3]гептан-6-ил]-5(4H)-оксо-4-(пиримидин-5-ил)-пиразин-2-ил)пропаналь
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (400 мг, 1,21 ммоль, 1 зквив.), получая 200 мг (50,31%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
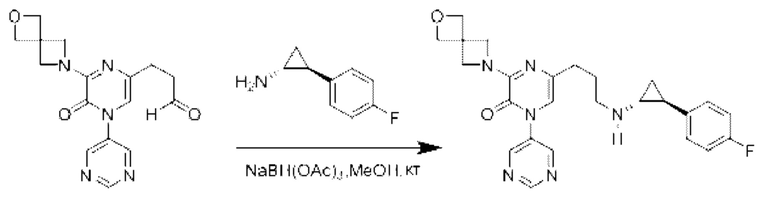
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-1-[пиримидин-5-ил]-3-[2-окса-6-азаспиро[3,3]гептан-6-ил]пиразин-2(1H)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (200 мг, 0,61 ммоль). Неочищенный продукт (5 мл)очищали, используя хроматографический Метод С (от 38,0% до 52,0% CH3CN за 7 мин), получая 24,8 мг (8,78%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХ-МС: (ES, m/z): 463 [М+Н]+. 1H ЯМР (400 МГц, MeOD-d4) δ м.д.: 9,20 (с, 1Н), 8,97 (с, 2Н), 7,11-7,055 (м, 2Н) 7,01-6,94 (м, 2Н), 6,73 (с, 1H), 4,83 (с, 4Н), 4,09 (уш. с, 4Н), 2,82-2,73 (м, 2Н), 2,48-2,39 (м, 2Н), 2,35-2,27 (м, 1Н), 1,98-1,82 (м, 3Н), 1,12-1,04 (м, 1Н), 1,03-0,96 (м, 1H).
ПРИМЕР 25
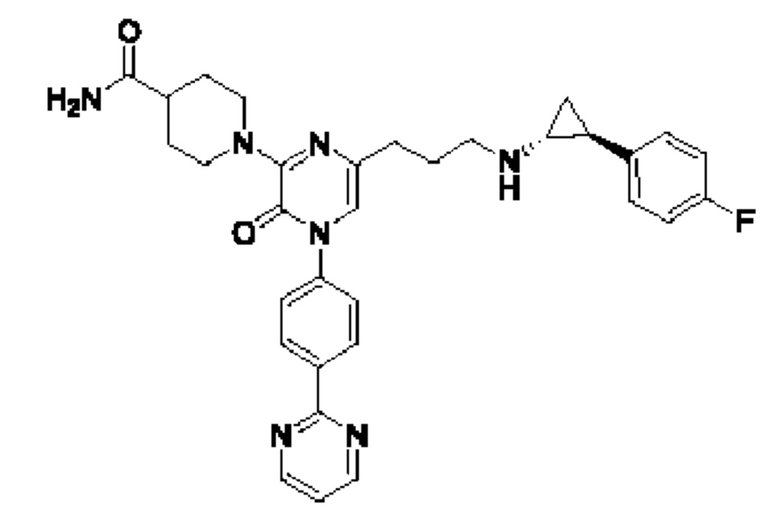
1-[6-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил]амино]пропил)-3-оксо-4-[4-(пиримидин-2-ил)фенил]-3,4-дигидропиразин-2-ил]пиперидин-4-карбоксамид
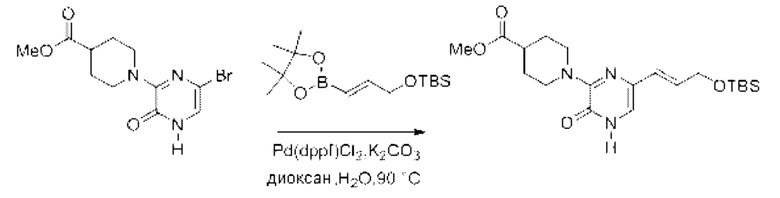
Метил 1-[6-[(Е)-3-[(трет-бутилдиметилсилил)окси]проп-1-ен-1-ил]-3(4Н)-оксопиразин-2-ил]пиперидин]-4-карбоксилат
Методику получения Промежуточного соединения 3-3 использовали с Промежуточным соединением 10-1 (10 г, 31,63 ммоль), с временем реакции 3 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:20), получая 4 г (31%) указанного в заглавии соединения в виде масла светло-желтого цвета.
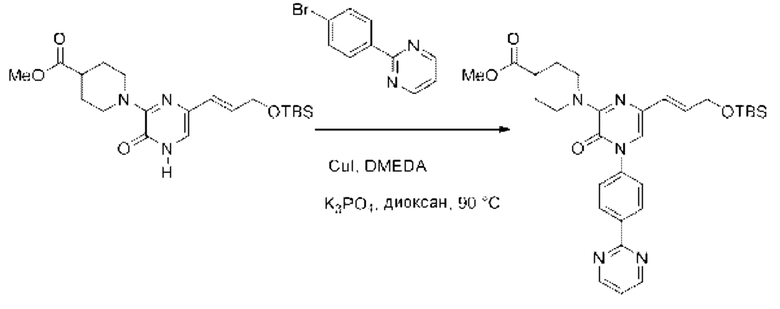
Метил 1-[6-[(Е)-3-[(трет-бутилдиметилсилил)окси]проп-1-ен-1-ил]-3(4H)-оксо-4-[4-(пиримидин-2-ил)фенил]-пиразин-2-ил]пиперидин-4-каобоксилат
Методику получения Промежуточного соединения 13-1 использовали с продуктом, полученным на предыдущей стадии, (4 г, 9,81 ммоль, 1,00 эквив.) и 2-(4-бромфенил)пиримидином (3,5 г, 14,39 ммоль, 1,50 эквив.), с временем реакции 6 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир, получая 1,9 г (34%) указанного в заглавии соединения в виде масла желтого цвета.
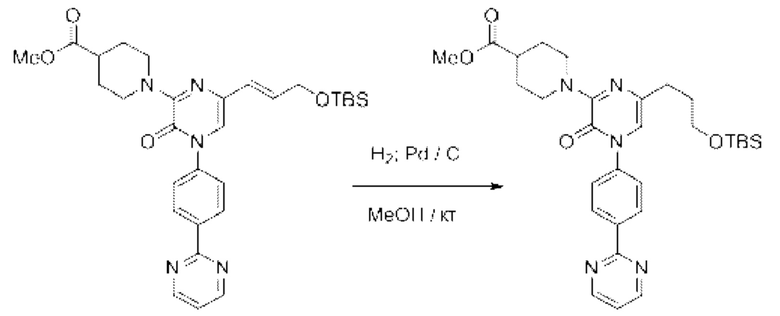
Метил 1-(6-[3-[(трет-бутилдиметилсилил)окси]пропил]-3(4Н)-оксо-4-[4-(пиримидин-2-ил)фенил]-пиразин-2-ил)пиперидин-4-карбоксилат
Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (1,9 г, 3,38 ммоль, 1,00 эквив.), получая 1,7 г (89%) указанного в заглавии соединения в виде масла светло-желтого цвета.
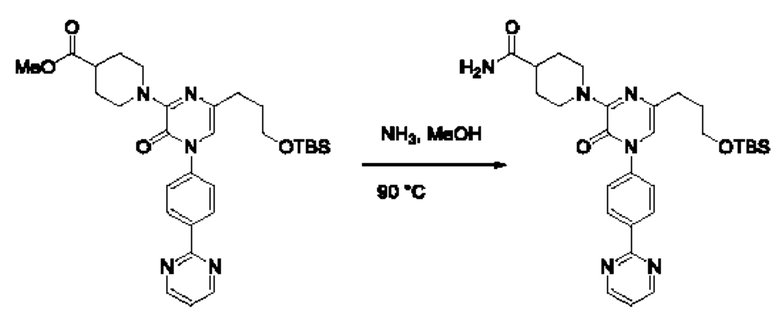
1-(6-[3-[(трет-Бутилдиметилсилил)окси]пропил]-3(4Н)-оксо-4-[4-(пиримидин-2-ил)фенил]-пиразин-2-ил)пиперидин-4-карбоксамид
Раствор продукта, полученного на предыдущей стадии, (1,7 г, 3,02 ммоль, 1,00 эквив.) в МеОН (10 мл)объединяли с раствором NH3 (4 г, 5,00 эквив.) в МеОН (5 мл). Полученный в результате раствор перемешивали в течение 96 ч при 90°С, затем охлаждали и концентрировали в вакууме, получая 1,1 г (66%) указанного в заглавии соединения в виде масла светло-желтого цвета.
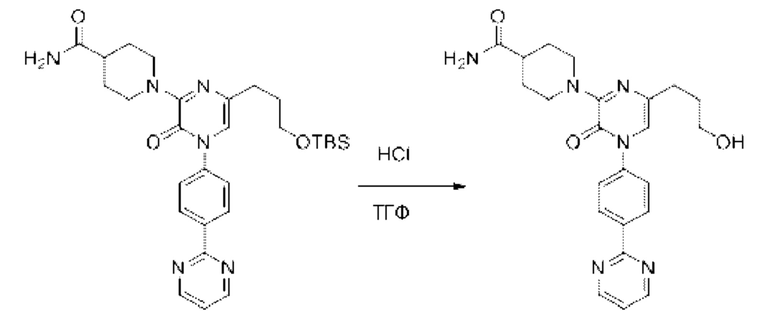
1-[6-(3-Гидроксипропил)-3(4H)-оксо-4-[4-(пиримидин-2-ил)фенил]пиразин-2-ил]пиперидин-4-карбоксамид
Раствор продукта, полученного на предыдущей стадии, (1,1 г, 2,00 ммоль, 1,00 эквив.), HCl (3 мл, 2,00 эквив.) в Н2О (1 мл) и ТГФ (20 мл)перемешивали в течение 1 ч при 25°С. рН корректировали до 7 добавлением Na2CO3 (1 М). Неочищенный продукт очищали с помощью хроматографии на силикагеле с H2O/MeCN (5:1), получая 500 мг (57%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
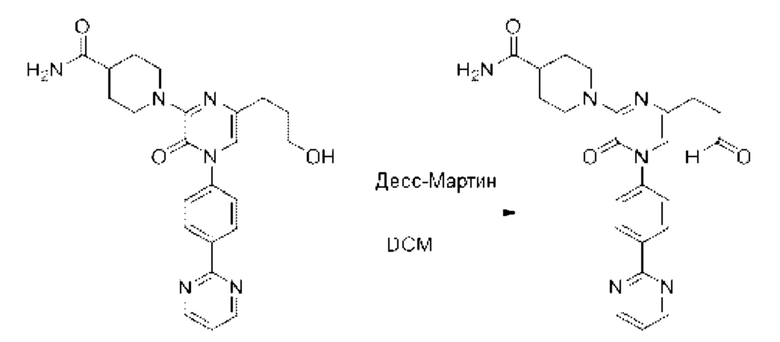
1-[3(4H)-Оксо-6-(3-оксопропил)-4-[4-(пиримидин-2-ил)фенил]-пиразин-2-ил]пиперидин-4-карбоксамид
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (500 мг, 1,15 ммоль, 1,00 эквив.), получая 300 мг (60%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
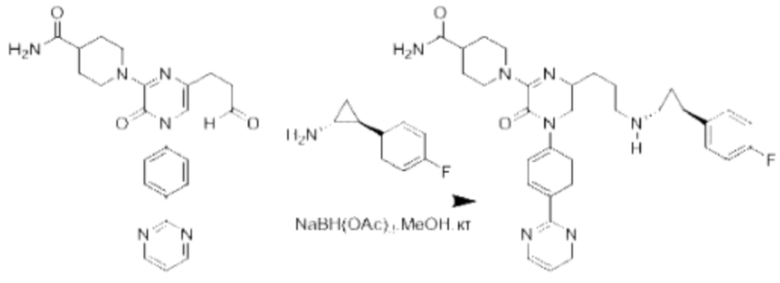
1-[6-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил]амино]пропил)-3-оксо-4-[4-(пиримидин-2-ил)фенил]-3,4-дигидропиразин-2-ил]пиперидин-4-карбоксамид
Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,69 ммоль). Неочищенный продукт (3 мл)очищали, используя хроматографический Метод С (от 38,0% до 50,0% CH3CN за 8 мин), получая 73,9 мг (19%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХ-МС: (ES, m/z): 568 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 8,95-8,61 (д, J=4,8Hz, 2Н), 8,65-8,45 (м, 2Н), 7,62-7,48 (м, 2Н), 7,48-7,31 (м, 1H, 7,11-6,89 (м, 4Н), 6,82 (с, 1Н), 4,82-4,66 (м, 2Н), 2,95-2,85 (м, 2Н), 2,85-2,69 (м, 2Н), 2,62-2,45 (м, 3Н), 2,29 (с, 1Н), 2,01-1,61 (м, 7Н), 1,12-0,92 (м, 2Н).
ПРИМЕР 26
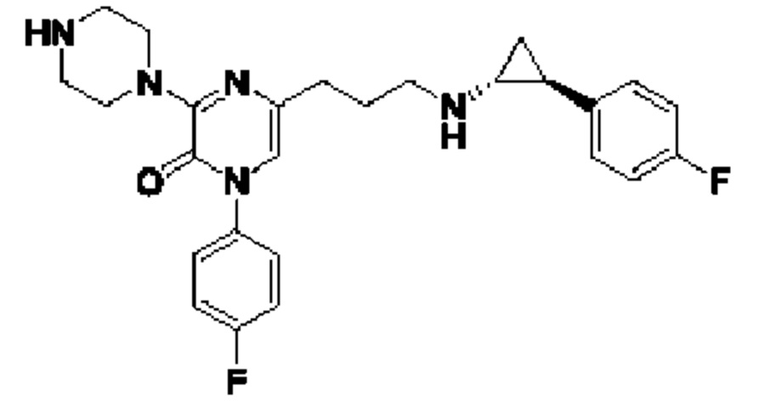
1-[4-Фторфенил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-(гашеразин-1-ил)пиразин-2(1H)-он
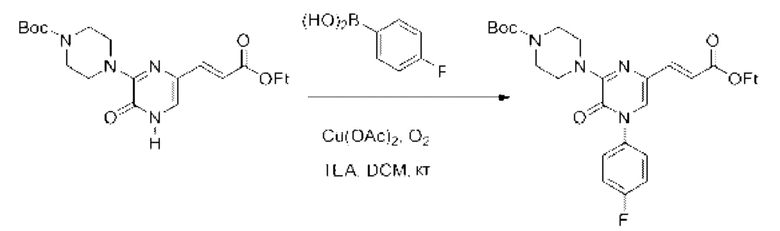
трет-Бутил 4-[6-[(E)-3-этокси-3-оксопроп-1-ен-1-ил]-4-(4-фторфенил)-3(4H)-оксо-пиразин-2-ил]пиперазин-1-карбоксилат
Методику получения Промежуточного соединения 8-9 использовали с Промежуточным соединением 14-2 (2,5 г, 6,61 ммоль, 1,00 эквив.) и (4-фторфенил)6ороновой кислотой (1,65 г, 11,79 ммоль, 1,50 эквив.). Неочищенный продукт очищали с использованием хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 1,6 г (51%) указанного в заглавии соединения в виде масла желтого цвета.
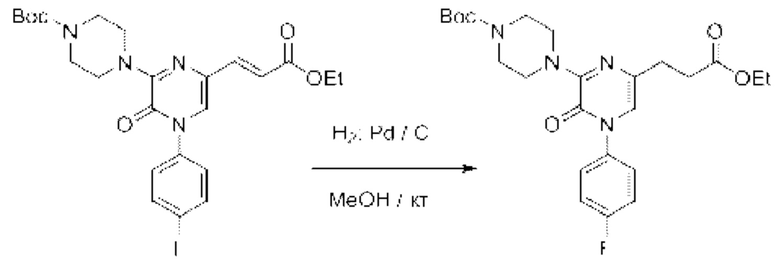
трет-Бутил 4-[6-(3-этокси-3-оксопропил)-4-(4-фторфенил)-3(4Н)-оксопиразин-2-ил]пиперазин-1-карбоксилат (Промежуточное соединение 26-2)
Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (1,6 г, 3,39 ммоль, 1,00 эквив.), получая 1,5 г (93%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
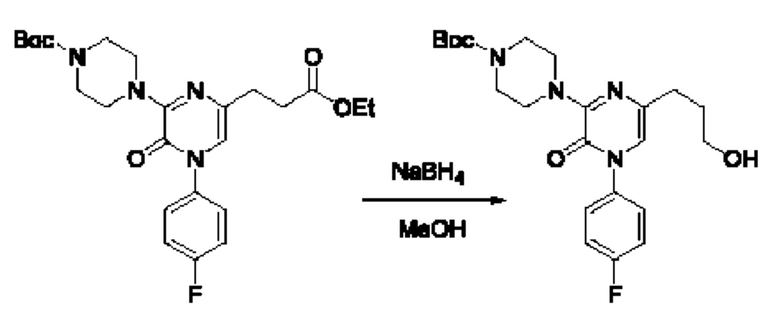
трет-Бутил. 4-[4-(4-фторфенил)-6-(3-гидроксипропил)-3(4H)-оксопиразин-2-ил]пиперазин-1-карбоксилат
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии (1,5 г, 3,16 ммоль, 1,00 эквив.), получая 1,1 г (80%) указанного в заглавии соединения в виде масла желтого цвета.
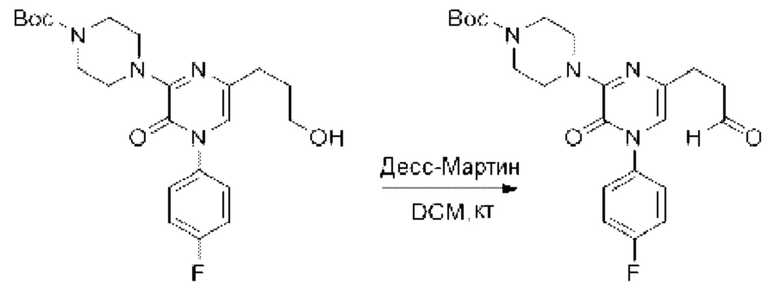
трет-Бутил 4-[4-(4-фторфенил)-3(4Н)-оксо-6-(3-оксопропил)-пиразин-2-ил]-пиперазин-1-карбоксилат
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (600 мг, 1,39 ммоль, 1,00 эквив.), получая 400 мг (66,98%) указанного в заглавии соединения в виде масла желтого цвета.
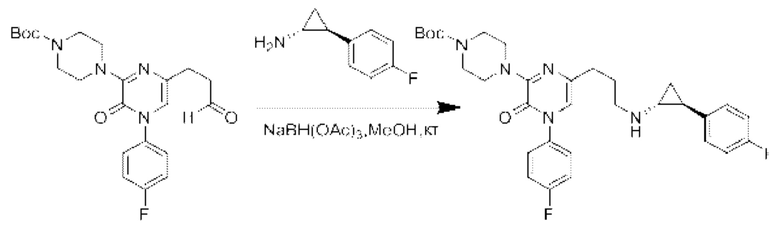
трет-Бутил 4-[4-(4-фторфенил)-6-(3-[[(1R,2S)-2-(4-фторфенил)-циклопропил]амино]пропил)-3(4Н)-оксопиразин-2-ил]пиперазин-1-карбоксилат
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (400 мг, 0,93 ммоль), получая 300 мг (57%) указанного в заглавии соединения в виде масла желтого цвета, которое использовали далее без дополнительной очистки.
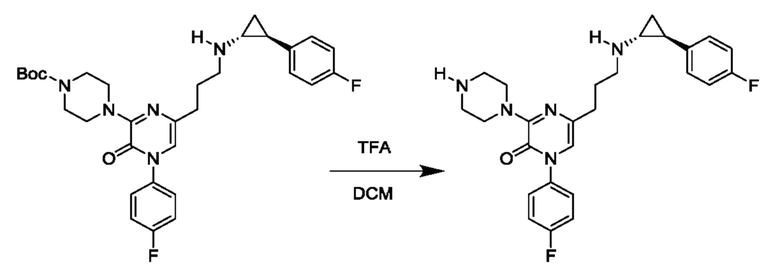
1-[4-Фторфенил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)-пропил]-3-(пиперазин-1-ил)пиразин-2(1H)-он Стадию удаления защитных групп для получения Примера 14 из Промежуточного соединения 14-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,53 ммоль, 1,00 эквив.). Неочищенный продукт (3 мл)очищали, используя хроматографический Метод D (от 15% до 60% CH3CN за 6,5 мин, Rt: 6,88 мин), получая 113,4 мг (46%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХМС: (ES, m/z): 466 [М+Н]+. 1Н ЯМР (300 МГц, Метанол-d4) δ м.д.: 7,51-7,41 (м, 2Н), 7,30-7,27 (м, 2Н), 7,27-7,17 (м, 2Н), 7,07-7,0 (м, 2Н), 6,98 (с, 1H), 4,10-4,0 (м, 4Н), 3,40-3,20 (м, 6Н), 3,04-2,94 (м, 1H), 2,60-2,50 (м, 2Н), 2,56-2,46 (м, 1H), 2,11-2,0 (м, 2Н), 1,58-1,47 (м, 1Н), 1,44-1,29 (м, 1H).
ПРИМЕР 27
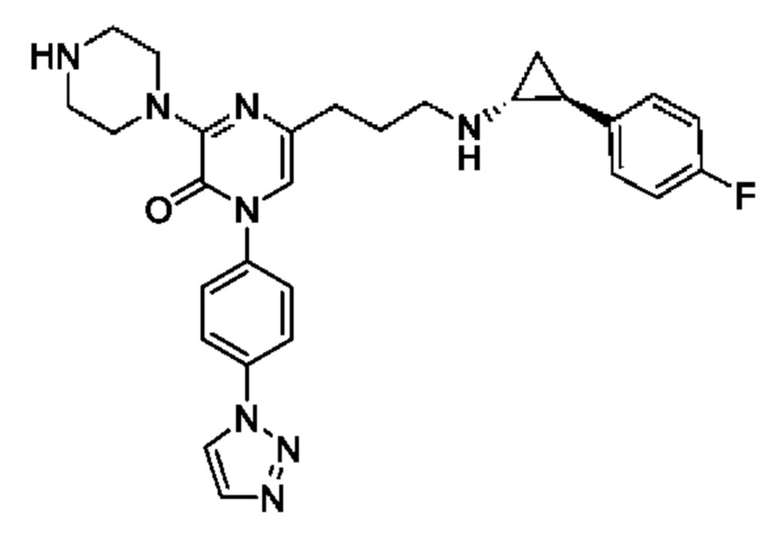
1-[4-(1H-1,2,3-триазол-1-ил)фенил]-5-[3-([(1R,2S)-2-(4-фторфенил)-циклопропил]амино)пропил]-3-[пиперазин-1-ил]пиразин-2(1H)-он
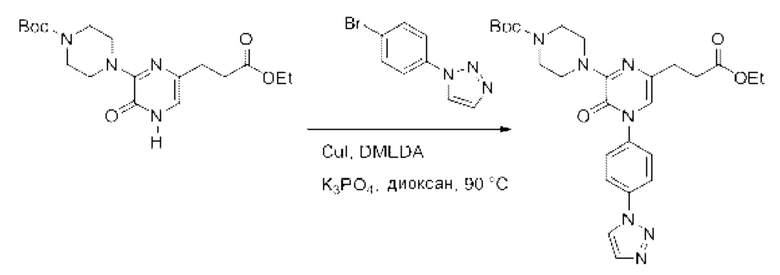
трет-Бутил 4-(4-(4-(1Н-1,2,3-триазол-1-ил)фенил)-6-(3-этокси-3-оксопропил)-3(4Н)-оксопиразин-2-ил)пиперазин-1-каобоксилат (Промежуточное соединение 27-1)
Методику получения Промежуточного соединения 13-1 использовали с Промежуточным соединением 26-2 (1,5 г, 3,95 ммоль, 1 эквив.) и 1-(4-бромфенил)-1H-1,2,3-триазолом (1,34 г, 5,92 ммоль, 1,5 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 0,7 г (33,98%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
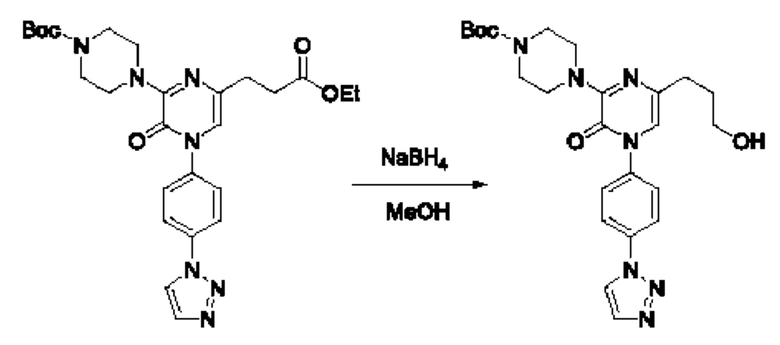
трет-Бутил 4-(4-(4-(1H-1,2,3-триазол-1-ил)фенил)-6-(3-гидроксипропил)-3(4H)-оксопиразин-2-ил)пиперазин-1-карбоксилат
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии, (0,7 г, 1,34 ммоль, 1 эквив.) с временем реакции 1 ч, получая 400 мг (62,21%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
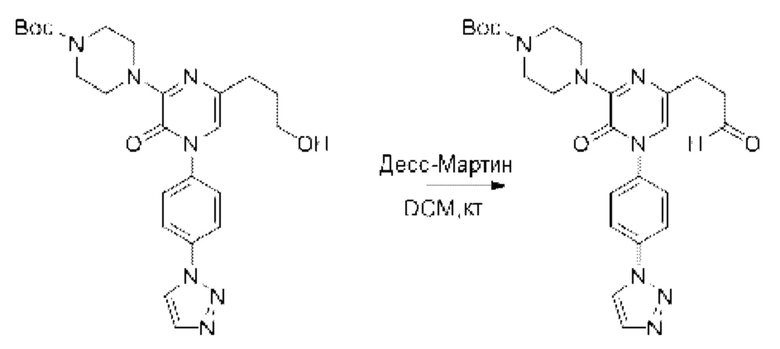
трет-Бутил 4-(4-(4-(1H-1,2,3-триазол-1-ил)фенил)-3(4H)-оксо-6-(3-оксопропил)-пиразин-2-ил)пиперазин-1-карбоксилат
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (400 мг, 0,83 ммоль, 1,00 эквив.), получая 300 мг (75,37%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
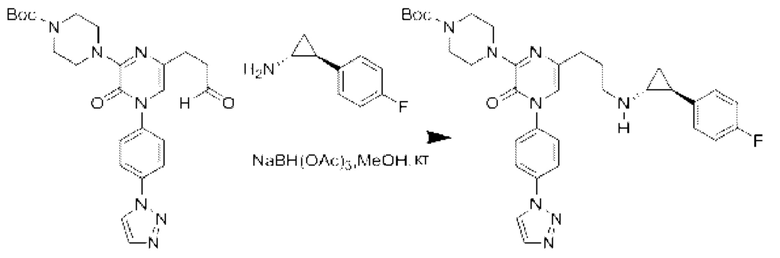
трет-Бутил 4-[4-(4-[1H-1,2,3-триазол-1-ил]фенил)-6-(3-[(1R,2S)-2-(4-фтор-фенил)циклопропиламино]пропил)-3(4H)-оксопиразин-2-ил]пиперазин-1-карбоксилат
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,63 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (10:1), получая 220 мг (57%) указанного в заглавии соединения в виде твердого вещества белого цвета.
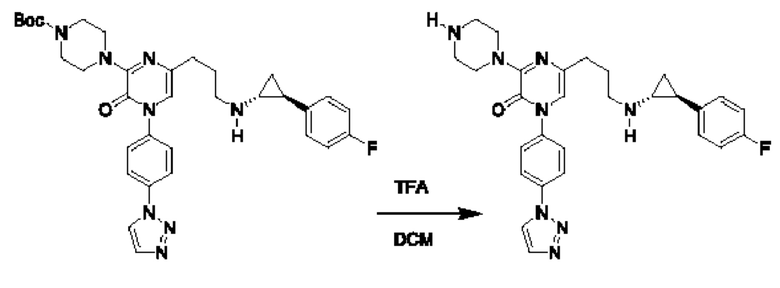
1-[4-(1Н-1,2,3-триазол-1-ил)фенил]-5-[3-([(1R,2S)-2-(4-фторфенил)-циклопропил]амино)пропил]-3-[пиперазин-1-ил]пиразин-2 (1H)-он
Стадию удаления защитных групп для получения Примера 14 из Промежуточного соединения 14-7 использовали с продуктом, полученным на предыдущей стадии, (220 мг, 0,41 ммоль, 1 эквив.). Неочищенный продукт (5 мл)очищали, используя хроматографический Метод Е (от 27% до 60% CH3CN, Rt: 10,13 мин), получая 52 мг (13,44%)) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-МС: (ES, m/z): 515 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 8,59 (с, 1Н), 8,03-8,01 (д, J=8,7 Гц, 2Н),7,90 (с, 1Н)7,63-7,60 (д, J=8,7 Гц, 2Н), 7,08-6,89 (м, 2Н), 6,97-6,87 (м, 2Н), 6,86-6,83 (с, 1Н), 3,78-3,68 (м, 4Н), 2,92-2,84 (м, 4Н), 2,79-2,71 (м, 2Н), 2,50-2,41 (м, 2Н), 2,31-2,23 (м, 1H), 1,95-1,80 (м, 3Н), 1,10-0,95 (м, 2Н).
ПРИМЕР 28
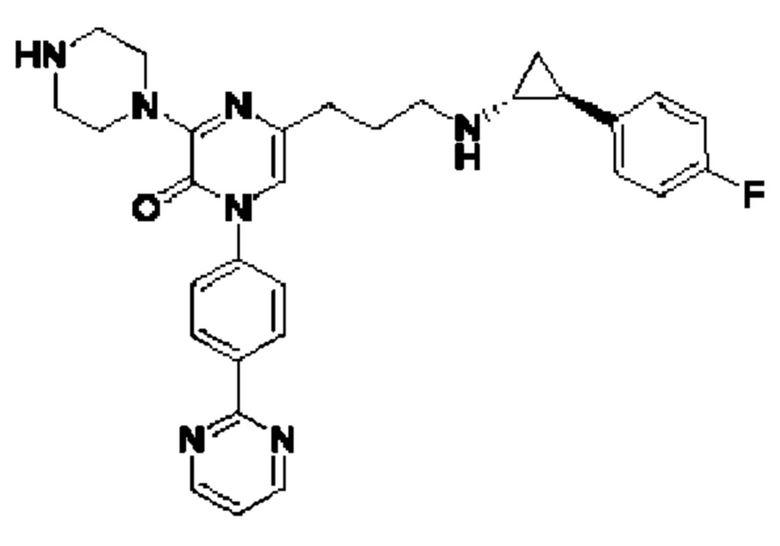
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[пиперазин-1-ил]-1-[4-(пиримидин-2-ил)фенил]пиразин-2(1Н)-он
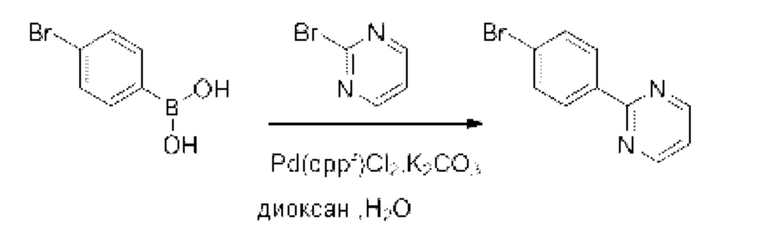
2-(4-Бромфенил)пиримидин
Раствор 4-бромфенилбороновой кислоты (5,0 г, 25,12 ммоль, 1 эквив.), 2-бромпиримидина (5,92 г, 37,68 ммоль, 1,5 эквив.), K2CO3 (10,40 г, 75,37 ммоль, 3 эквив.) и Pd(dppf)Cl2 (1,84 г, 2,51 ммоль, 0,1 эквив.) в диоксане (450 мл)/H2O (100 мл)перемешивали в течение ночи при 90°С под N2. Остаток очищали с использованием хроматографии на силикагеле, используя EtOAc/петролейный эфир (6:1), получая 2,0 г (34%) указанного в заглавии соединения в виде масла желтого цвета.
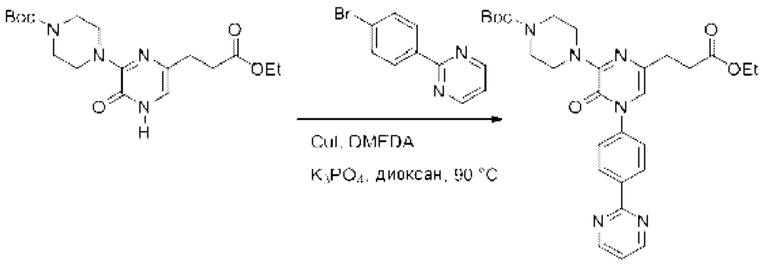
трет-Бутил 4-(6-(3-этокси-3-оксопропил)-3(4H)-оксо-4-(4-(пиримидин-2-ил)фенил)-пиразин-2-ил)пиперазин-1-карбоксилат
Методику получения Промежуточного соединения 13-1 использовали с Промежуточным соединением 27-1 (1,79 г, 4,72 ммоль, 1,1 эквив.) и 2-(4-бромфенил)пиримидином (1,0 г, 4,29 ммоль, 1 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 0,6 г (27,87%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
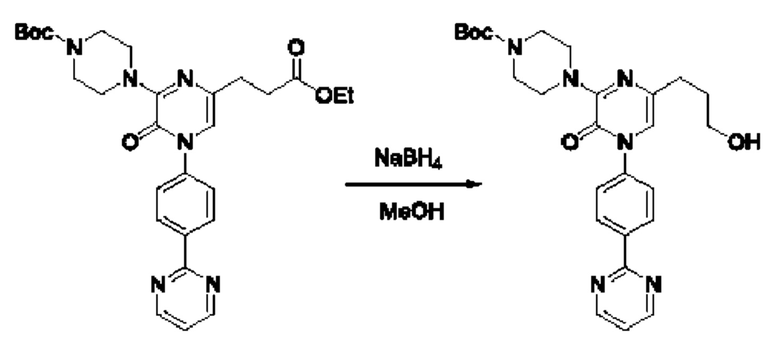
трет-Бутил 4-(6-(3-гидроксипропил)-3(4H)-оксо-4-(4-(пиримидин-2-ил)фенил)-пиразин-2-ил)пиперазин-1-карбоксилат
Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии, (0,6 г, 2,74 ммоль, 1 эквив.), с временем реакции 1 ч, получая 350 мг (39,8%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
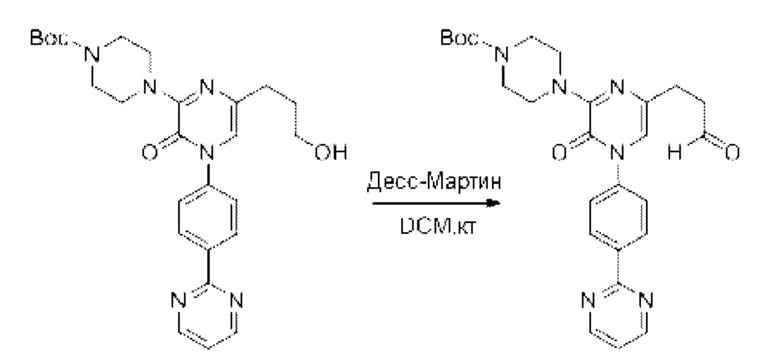
трет-Бутил 4-(3(4H)-оксо-6-(3-оксопропил)-4-(4-(пиримидин-2-ил)фенил)-пиразин-2-ил)пиперазин-1-карбоксилат
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (350 мг, 0,71 ммоль, 1,00 эквив.), получая 210 мг (60%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
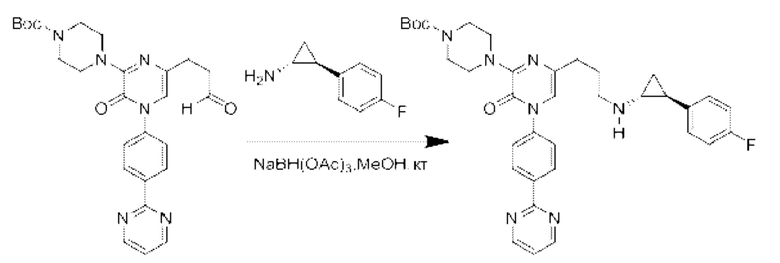
трет-Бутил 4-(6-(3-((1R,2S)-2-(4-фторфенил)циклопропиламино)пропил)-3(4H)-оксо-4-(4-(пиримидин-2-ил)фенил)-пиразин-2-ил)пиперазин-1-карбоксилат
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (210 мг, 0,41 ммоль), получая 190 мг (52%) указанного в заглавии соединения в виде твердого вещества белого цвета, которое использовали далее без дополнительной очистки.
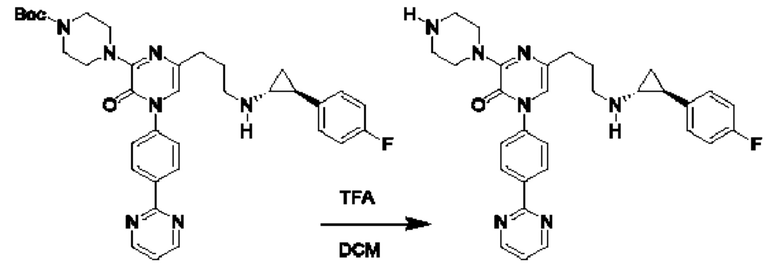
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[пиперазин-1-ил]-1-[4-(пиримидин-2-ил)фенил]пиразин-2(1Н)-он Стадию удаления защитных групп для получения Примера 14 из Промежуточного соединения 14-7 использовали с продуктом, полученным на предыдущей стадии, (190 мг, 0,30 ммоль, 1 эквив.). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод D (от 27% до 60% CH3CN, Rt: 8,3 мин), получая 23,8 мг (14,96%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-МС: (ES, m/z): 526 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 8,95-8,90 (м, 2Н), 8,62-8,56 (м, 2Н), 7,64-7,56 (м, 2Н), 7,49-7,43 (м, 1H, 7,27-7,17 (м, 2Н), 7,10-7,01 (м, 3H), 4,14-4,04 (м, 4Н), 3,43-3,32 (м, 4Н), 3,31-3,23 (м, 2Н), 3,05-2,95 (м, 1H, 2,68-2,52 (м, 3H), 2,23-2,09 (м, 2Н), 1,62-1,52 (м, 1H, 1,42-1,32 (м, Н).
ПРИМЕР 29
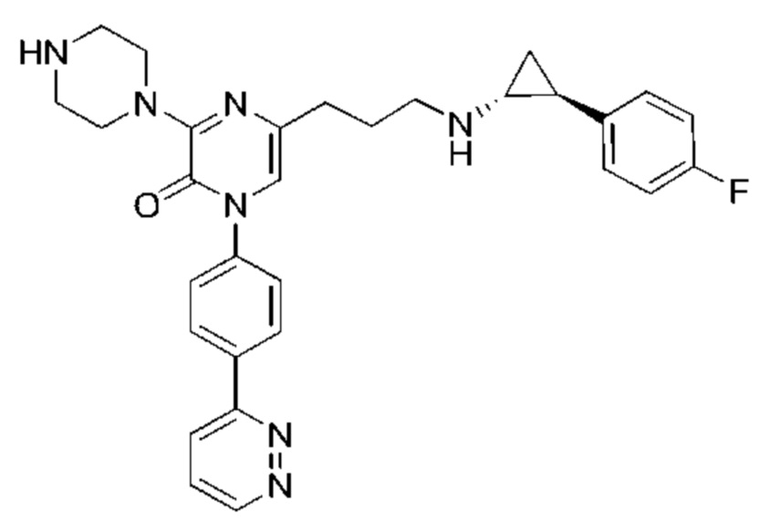
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[пиперазин-1-ил]-1-[4-(пиридазин-3-ил)фенил]пиразин-2(1H)-он
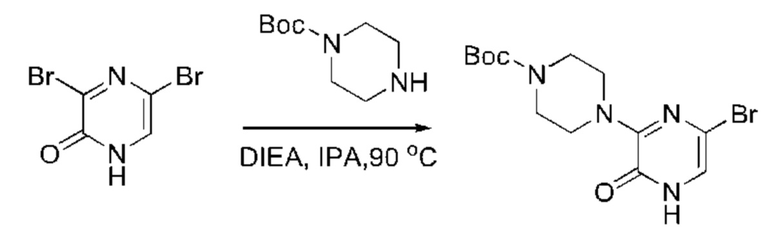
трет-Бутил 4-(6-бром-3(4H)-оксопиразин-2-ил)пиперазин-1-карбоксилат
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (5 г, 19,84 ммоль, 1,00 эквив.) и дареда-бутил пиперазин-1-карбоксилатом (4,06 г, 21,82 ммоль, 1,1 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (4:1), получая 5 г (70%) указанного в заглавии соединения в виде твердого вещества белого цвета.
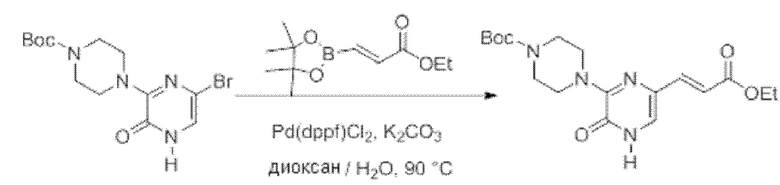
трет-Бутил 4-[6-[(Е)-3-этокси-3-оксопроп-1-ен-1-ил]-3(4H)-оксопиразин-2-ил]пиперазин-1-карбоксилат Методику получения Промежуточного соединения 1-4 использовали с продуктом, полученным на предыдущей стадии, (5,0 г, 13,96 ммоль, 1 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:2), получая 2,20 г (41%) указанного в заглавии соединения в виде масла оранжевого цвета.
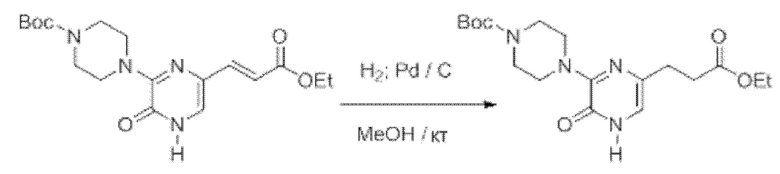
трет-Бутил 4-(6-(3-этокси-3-оксопропил)-3(4H)-оксопиразин-2-ил)пиперазин-1-карбоксилат Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (2,20 г, 5,82 ммоль, 1,00 эквив.), получая 2,1 г (95%) указанного в заглавии соединения в виде масла оранжевого цвета.
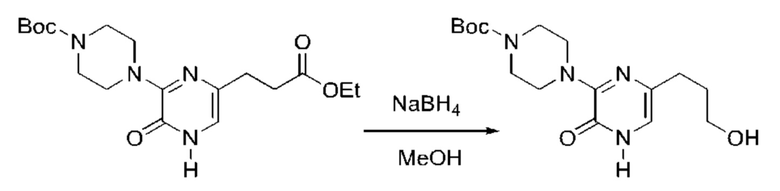
трет-Бутил 4-[6-(3-гидроксипропил)-3(4H)-оксопиразин-2-ил]пиперазин-1-карбоксилат Методику получения Промежуточного соединения 1-6 использовали с продуктом, полученным на предыдущей стадии (1,5 г, 3,94 ммоль, 1,00 эквив.), получая 700 мг (52%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
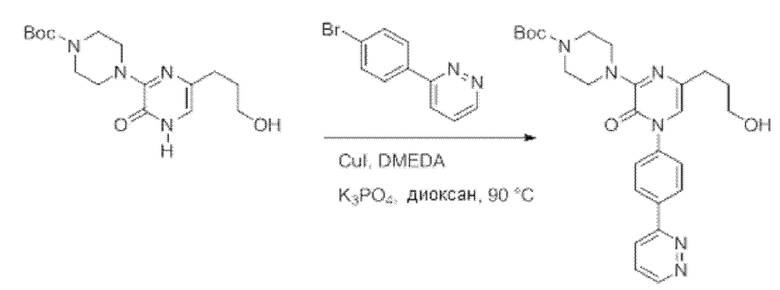
трет-Бутил 4-[6-(3-гидроксипропил)-3(4H)-оксо-4-[4-(пиридазин-3-ил)фенил]-пиразин-2-ил]пиперазин-1-карбоксилат Методику получения Промежуточного соединения 13-1 использовали с продуктом, полученным на предыдущей стадии, (700 мг, 2,07 ммоль, 1,00 эквив.) и 3-(4-бромфенил)пиридазином (723 мг, 3,08 ммоль, 1,49 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 400 мг (39%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
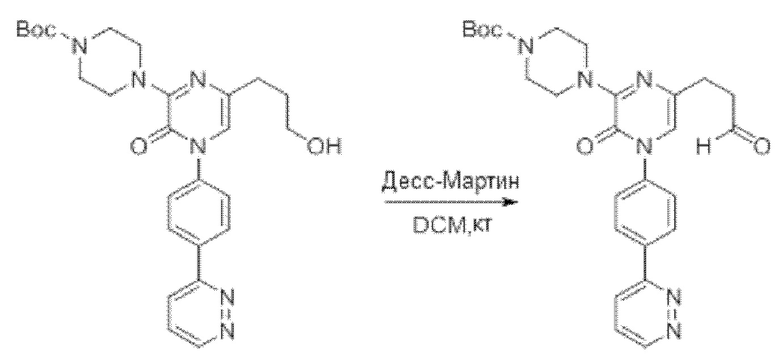
трет-Бутил 4-[3(4H)-оксо-6-(3-оксопропил)-4-[4-(пиридазин-3-ил)фенил]-пиразин-2-ил]пиперазин-1-карбоксилат Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (400 мг, 0,81 ммоль, 1,00 эквив.), получая 350 мг (88%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
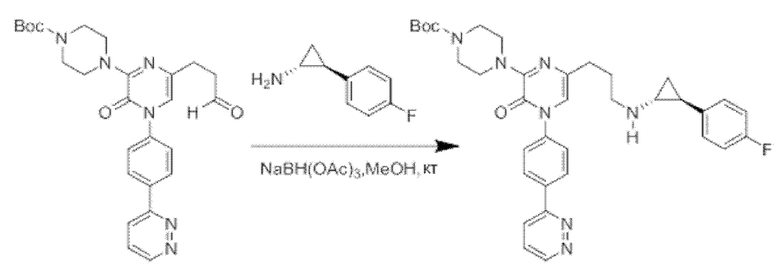
трет-Бутил 4-[6-(3-[[(1R,2S)-2-(4-фторфенил)циклопропил]амино]пропил)-3(4H)-оксо-4-[4-(пиридазин-3-ил)фенил]-пиразин-2-ил]пиперазин-1-карбоксилат
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (350 мг, 0,71 ммоль). Неочищенный продукт очищали с использованием хроматографии на силикагеле, используя CH2Cl2/МеОН (1:10), получая 300 мг (67%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
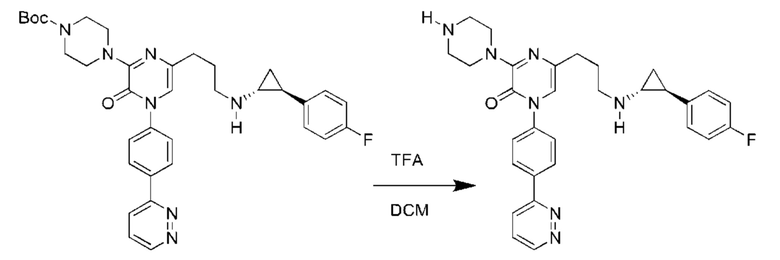
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[пиперазин-1-ил]-1-[4-(пиридазин-3-ил)фенил]пиразин-2(1H)-он Стадию удаления защитных групп для получения Примера 14 из Промежуточного соединения 14-7 использовали с продуктом, полученным на предыдущей стадии, (400 мг, 0,64 ммоль, 1,00 эквив.). Неочищенный продукт (2 мл) очищали с помощью флеш-препаративной ВЭЖХ (IntelFlash-1, колонка: силикагель, детектор, УФ 254 нм), получая 57,4 мг (17%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХ-МС: (ES,m/z): 526 [М+Н]+. 1Н ЯМР (400 МГц, Метанол-d4) δ 9,25-9,22 (с, 1H, 8,34-8,28 (м, 3H), 7,92-7,86 (м, 1H, 7,68-7,64 (м, 2Н), 7,25-7,19 (м, 2Н), 7,11-7,04 (м, 3H), 4,11-4,03 (м, 4Н), 3,41-3,33 (м, 4Н), 3,32-3,27 (м, 2Н), 3,03-2,97 (м, 1H, 2,67-2,59 (м, 2Н), 2,56-2,48 (м, 1H, 2,20-2,09 (м, 2Н), 1,58-1,49 (м, 1H, 1,44-1,36 (м, 1H)
ПРИМЕР 30
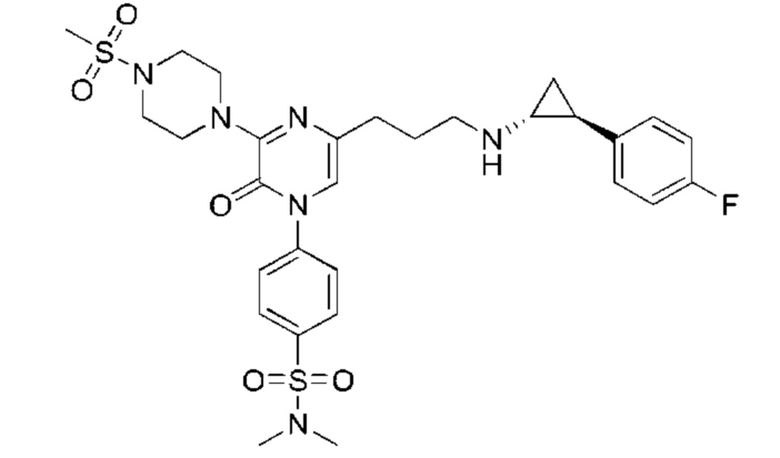
4-[5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил]амино]пропил)-3-(4-метансульфонилпиперазин-1-ил)-2(1H)-оксопиразин-1-ил]-N,N-диметилбензол-1-сульфонамид
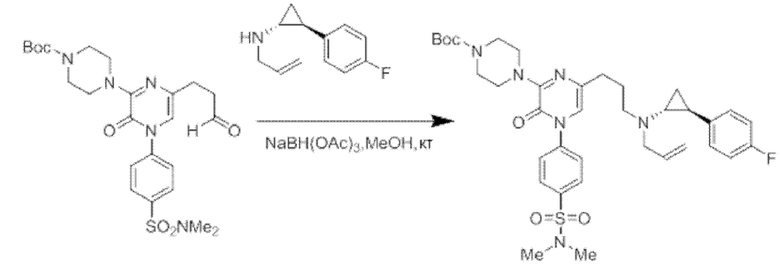
трет-Бутил 4-[4-[4-((диметиламино)сульфонил)фенил]-6-(3-[[(1R,2S)-2-(4-фторфенил)циклопропил](проп-2-ен-1-ил)амино]пропил)-3(4H)-оксопиразин-2-ил]пиперазин-1-карбоксилат Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с Промежуточным соединением 14-6 (760 мг, 1,46 ммоль, 1,00 эквив.) и (1R,2S)-2-(4-фторфенил)-N-(проп-2-ен-1-ил)циклопропан-1-амином (560 мг, 2,93 ммоль, 2,00 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 0,8 г (79%) указанного в заглавии соединения в виде масла светло-желтого цвета.
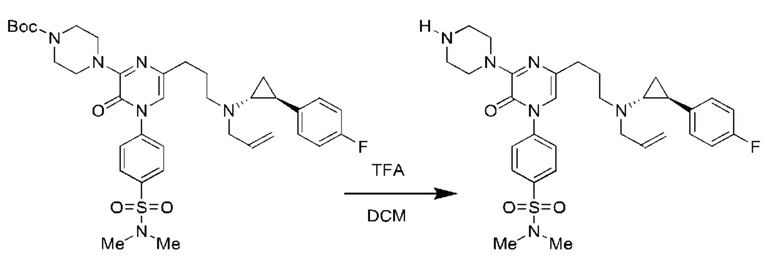
4-[5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил](проп-2-ен-1-ил)амино]-пропил)-2(1H)-оксо-3-(пиперазин-1-ил)-пиразин-1-ил]-N,N-диметилбензол-1-сульфонамид Стадию удаления защитных групп для получения Примера 14 из Промежуточного соединения 14-7 использовали с продуктом, полученным на предыдущей стадии, (800 мг, 1,15 ммоль, 1,00 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2 / МеОН (10:1), получая 560 мг (82%) указанного в заглавии соединения в виде масла коричневого цвета.
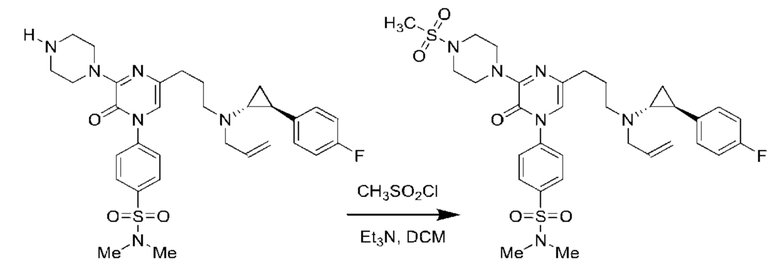
4-[5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил](проп-2-ен-1-ил)амино]пропил)-3-(4-метансульфонилпиперазин-1-ил)-2(1H)-оксопиразин-1-ил]-N,N-диметилбензол-1-сульфонамид Раствор соединения, полученного на предыдущей стадии, (560 мг, 0,94 ммоль, 1,00 эквив.) и Et3N (286 мг, 2,83 ммоль, 3,00 эквив.) в CH2Cl2 (10 мл) перемешивали в течение 30 мин при 0°С, затем добавляли CH3SO2Cl (161 мг, 1,41 ммоль, 1,50 эквив.). Полученный в результате раствор перемешивали в течение 60 мин при кт и экстрагировали 3 х 30 мл CH2Cl2. Объединенные органические слои сушили над Na2SO4 и очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:10), получая 0,5 г (79%) указанного в заглавии соединения в виде масла светло-желтого цвета.
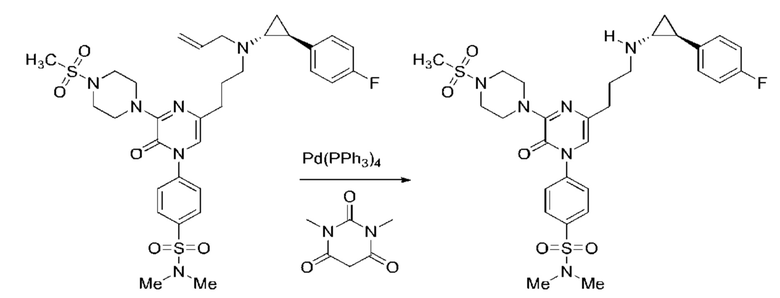
4-[5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил]амино]пропил)-3-(4-метансульфонилпиперазин-1-ил)-2(1H)-оксопиразин-1-ил]-N,N-диметилбензол-1-сульфонамид Раствор продукта, полученного на предыдущей стадии, (500 мг, 0,74 ммоль, 1,00 эквив.), Pd(PPh3)4 (172 мг, 0,15 ммоль, 0,20 эквив.) и 1,3-диметил-1,3-диазинан-2,4,6-триона (348 мг, 2,23 ммоль, 3,00 эквив.) в ТГФ (15 мл) перемешивали в течение 2 ч при 50°С под N2. Неочищенный продукт очищали сначала с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (50:1), а затем с помощью хроматографического Способа Е (от 28% до 80% CH3CN, Rt: 7,75 мин), получая 182 мг (39%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХМС: (ES,m/z): 633 [М+Н]+. 1Н ЯМР (300 МГц, Метанол-d4) δ м.д.: 7,96 - 7,85 (д, J=8,4 Гц, 2Н), 7,71 - 7,60 (д, J=8,4 Гц, 2Н), 7,09 - 6,84 (м, 5Н), 3,85 (м, 4Н), 3,26 (м, 4Н), 2,85-2,65 (м, ПН), 2,45 - 2,31 (м, 2Н), 2,29-2,21 (м, 1Н),1,95 - 1,78 (м, 3H), 1,10 -0,88 (м, 2Н).
ПРИМЕР 31
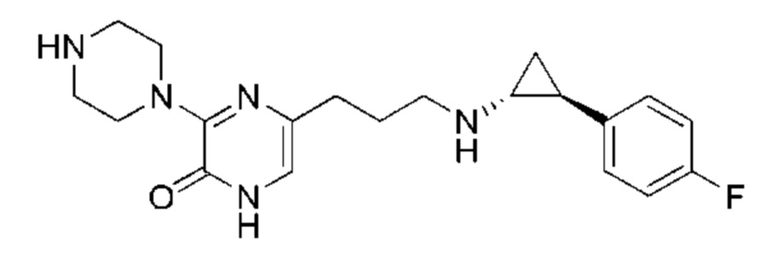
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[пиперазин-1-ил] пиразин-2(1Н)-он
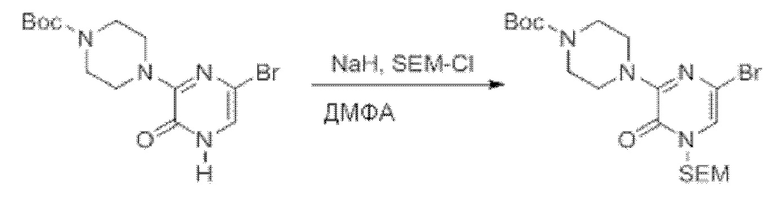
трет-Бутил 4-(6-бром-3(4H)-оксо-4-[[2-(триметилсилил)этокси]метил]-пиразин-2-ил)пиперазин-1-карбоксилат Методику получения Промежуточного соединения 2-2 использовали с Промежуточным соединением 14-1 (10 г, 27,84 ммоль, 1,00 эквив.) и [2-(хлорметокси)этил]триметилсиланом (6,8 г, 40,79 ммоль, 1,50 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:30), получая 10,5 г (77%) указанного в заглавии соединения в виде масла грязно-белого цвета.
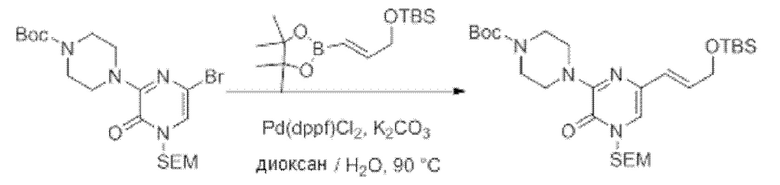
трет-Бутил 4-[6-[(1Е)-3-[(трет-бутилдиметилсилил)окси]проп-1-ен-1-ил]-3(4H)-оксо-4-[[2-(триметилсилил)этокси]метил]-пиразин-2-ил]пиперазин-1-карбоксилат Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (4 г, 8,17 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:50), получая 1,5 г (32%) указанного в заглавии соединения в виде масла светло-желтого цвета.
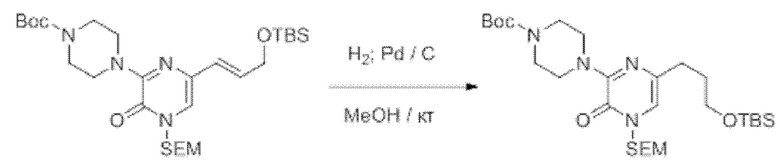
трет-бутил 4-(6-[3-[(трет-бутилдиметилсилил)окси]пропил]-3(4H)-оксо-4-[[2-(триметилсилил)этокси]метил]-пиразин-2-ил)пиперазин-1-карбоксилат
Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (1,5 г, 2,58 ммоль, 1,00 эквив.), получая 1,0 г (66%) указанного в заглавии соединения в виде масла светло-желтого цвета.
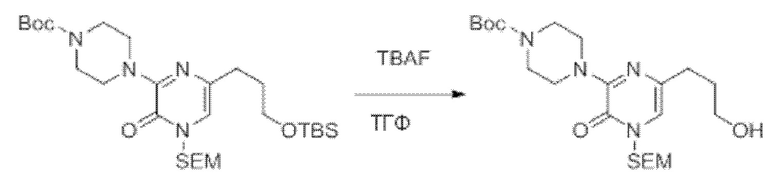
трет-Бутил 4-[6-(3-гидроксипропил)-3(4H)-оксо-4-[[2-(триметилсилил)-этокси]метил]-пиразин-2-ил]пиперазин-1-карбоксилат Методику получения Промежуточного соединения 22-4 использовали с продуктом, полученным на предыдущей стадии, (1 г, 1,72 ммоль, 1,00 эквив.), получая 0,6 г (75%) указанного в заглавии соединения в виде масла светло-желтого цвета.
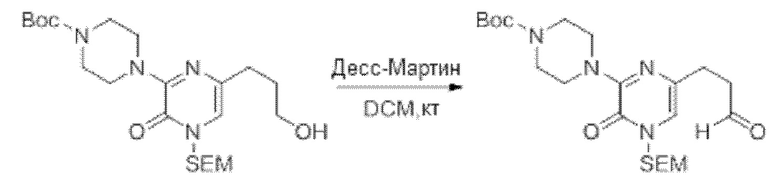
трет-Бутил 4-[3(4H)-оксо-6-(3-оксопропил)-4-[[2-(триметилсилил)этокси]метил]-пиразин-2-ил]пиперазин-1-карбоксилат
Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (600 мг, 1,28 ммоль, 1,00 эквив.), получая 0,3 г (50%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
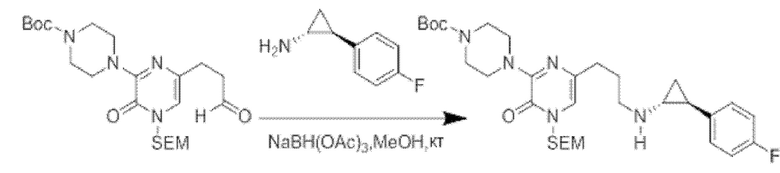
трет-Бутил 4-[6-(3-[[(1R,2S)-2-(4-фторфенил)циклопропил]амино]пропил)-3(4H)-оксо-4-[[2-(триметилсилил)этокси]метил]-пиразин-2-ил]пиперазин-1-карбоксилат Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,64 ммоль), получая 0,2 г (52%) указанного в заглавии соединения в виде масла светло-желтого цвета. Продукт использовали дальше без дополнительной очистки.
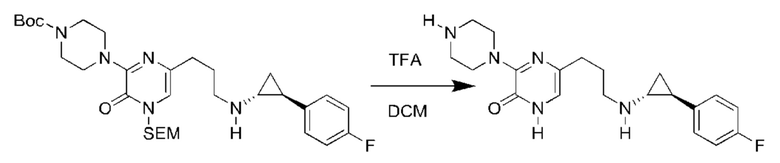
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[пиперазин-1-ил]пиразин-2(1Н)-он Стадию удаления защитных групп для получения Примера 14 из Промежуточного соединения 14-7 использовали с продуктом, полученным на предыдущей стадии, (200 мг, 0,17 ммоль, 1,00 эквив.). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод В (от 30,0% до 50,0% CH3CN за 8 мин), получая 114,6 мг (45%) указанного в заглавии соединения в виде масла светло-желтого цвета.
ЖХ-МС: (ES,m/z): 372 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 7,23 - 7,07 (м, 2Н), 7,07- 6,93 (м, 2Н), 6,82 - 6,69 (с, 1H, 4,02-3,89 (м, 4Н), 3,28-3,14 (м, 6Н), 2,99-2,81 (м, 1H, 2,66 - 2,31 (м, 3H), 2,13-1,89 (м, 2Н), 1,60 - 1,41 (м, 1H, 1,41-1,22 (м, 1H.
ПРИМЕР 32
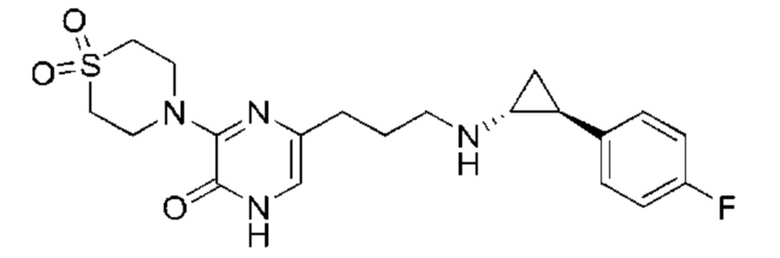
3-[1,1-Диоксидотиоморфолино]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]пиразин-2(1H)-он
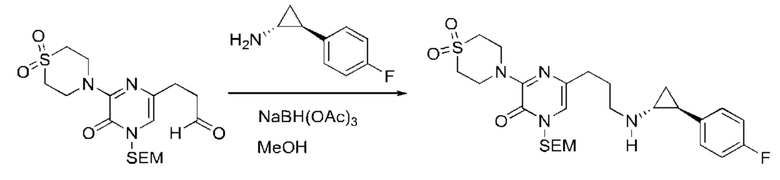
5-(3-[[(1R,2S)-2-(4-Фторфенил)циклопропил]амино]пропил)-3-(1,1-диоксотиоморфолин-4-ил)-1-[[2-(триметилсилил)этокси]метил]-пиразин-2(1H)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с Промежуточным соединением 8-6 (150 мг, 0,36 ммоль, 1 эквив.) и (17R,2S)-2-(4-фторфенил)-циклопропан-1-амином (109 мг, 0,72 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (3:2), получая 140 мг (70,46%) указанного в заглавии соединения в виде масла оранжевого цвета.
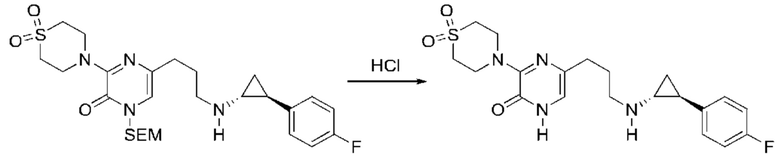
3-[1,1-Диоксидотиоморфолино]-5-[3-([(1R,2S)-2-(4-фторфенил)-циклопропил]амино)пропил]пиразин-2(1H)-он Стадию удаления защитных групп для получения Примера 2 из Промежуточного соединения 2-7 использовали с продуктом, полученным на предыдущей стадии, (140 мг, 0,25 ммоль, 1 эквив.). Неочищенный продукт (5 мл) очищали, используя хроматографический Метод С (от 20,0% до 50,0% CH3CN за 8 мин), получая 61 мг (28%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-МС: (ES,m/z): 421 [М+Н]+. 1НЯМР (300 МГц, MeOD-d4) δ м.д.: 7,04-6,95 (м, 2Н), 6,93-6,89 (м, 2Н), 6,66 (с, 1H, 4,29-4,25 (м, 4Н), 3,13-3,09 (м, 4Н), 2,74-2,68 (м, 2Н), 2,46-2,41 (м, 2Н), 2,29-2,24 (м, 1H, 1,92-1,80 (м, 3H), 1,06-0,92 (м, 2Н).
ПРИМЕР 33
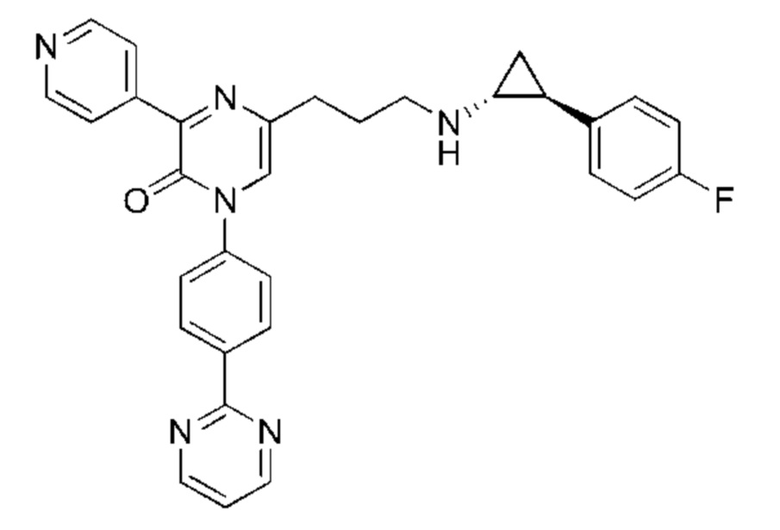
5-[3-([(1R,2S)-2-(4-Фторфенил)циклопропил]амино)пропил]-3-(пиридин-4-ил)-1-[4-(пиримидин-2-ил)фенил] пиразин-2(1H)-он
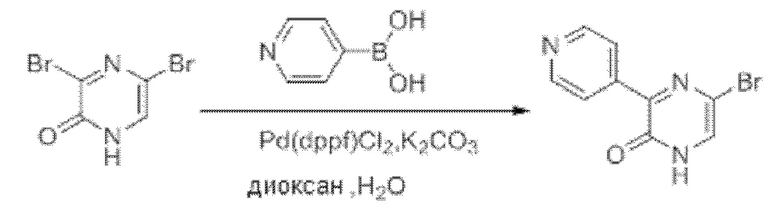
5-бром-3-(пиридин-4-ил)пиразин-2(1H)-он (Промежуточное соединение 33-1)
Раствор 3,5-дибром-1,2-дигидропиразин-2-она (20 г, 0,08 ммоль, 1 эквив.), (пиридин-4-ил)бороновой кислоты (10,7 г, 0,09 ммоль, 1,11 эквив.), К2СО3 (32,7 г, 0,24 ммоль, 3 эквив.) и Pd(dppf)Cl2 (11,5 г, 0,02 ммоль, 0,2 эквив.) в диоксане (200 мл) и Н2О (50 мл) перемешивали в течение 16 ч при 100°С под N2. Полученную смесь концентрировали и очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (10:1), получая 4 г (20%) указанного в заглавии соединения в виде масла желтого цвета.
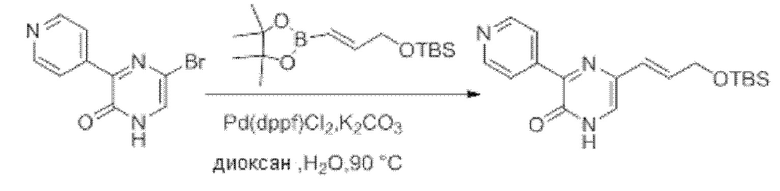
(Е)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-3-(пиридин-4-ил)пиразин-2(1H)-он (Промежуточное соединение 33-2) Методику получения Промежуточного соединения 3-3 использовали с Промежуточным соединением 33-1 (4 г, 15,87 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (10:1), получая 1,5 г (28%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
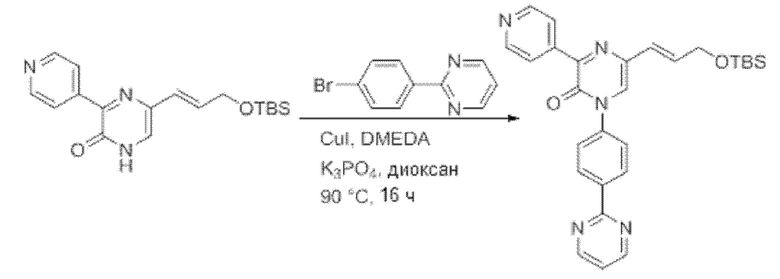
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-3-(пиридин-4-ил)-1-(4-(пиримидин-2-ил)фенил)пиразин-2(1H)-он (Промежуточное соединение 33-3)
Методику получения Промежуточного соединения 13-1 использовали с Промежуточным соединением 33-2 (1,5 г, 4,37 ммоль, 1 эквив.) и 2-(4-бромфенил)пиримидином (1,23 г, 5,24 ммоль, 1,200 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 800 мг (37%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
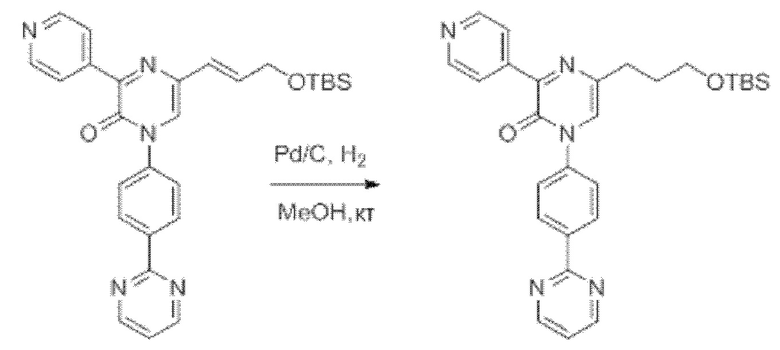
5-(3-((трет-Бутилдиметилсилил)окси)пропил)-3-(пиридин-4-ил)-1-(4-(пиримидин-2-ил)фенил)пиразин-2(1H)-он (Промежуточное соединение 33-4)
Раствор Промежуточного соединения 33-3 (600 мг, 1,15 ммоль, 1 эквив.) в МеОН (20 мл) перемешивали над Pd/C (59,7 мг, 0,56 ммоль, 0,49 эквив.) в атмосфере Н2 в течение 60 мин при кт. Продукт фильтровали и фильтрат концентрировали, получая 400 мг (70%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
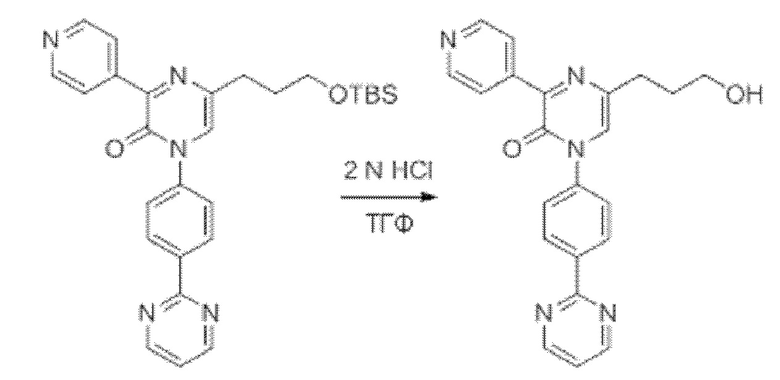
5-(3-Гидроксипропил)-3-(пиридин-4-ил)-1-(4-(пиримидин-2-ил)фенил)пиразин-2(1H)-он (Промежуточное соединение 33-5) Раствор Промежуточного соединения 33-4 (400 мг, 0,18 ммоль) в HCl (2N, 4 мл) и ТГФ (8 мл) перемешивали в течение 60 мин при кт. рН корректировали до 7 добавлением Na2CO3. Полученный в результате раствор экстрагировали 3x30 мл CH2Cl2. Объединенные органические слои концентрировали, получая 210 мг (88%) указанного в заглавии соединения в виде масла желтого цвета.
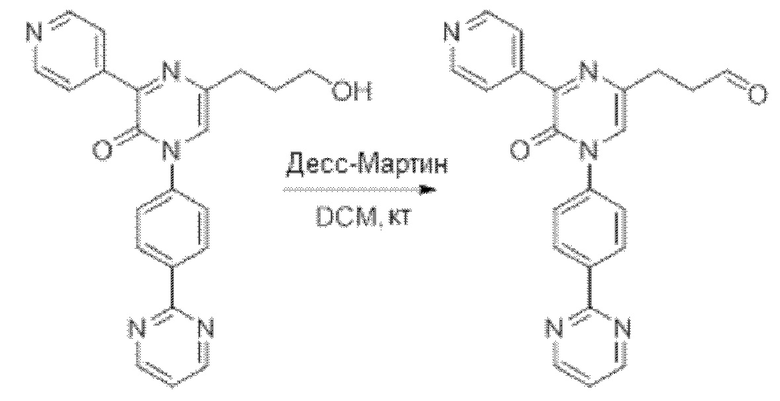
3-(5-Оксо-6-(пиридин-4-ил)-4-(4-(пиримидин-2-ил)фенил)-4,5-дигидропиразин-2-ил)пропаналь Методику получения Промежуточного соединения 1-7 использовали с Промежуточным соединением 33-5 (210 мг, 0,54 ммоль, 1 эквив.) и реактивом Десса-Мартина (346,6 мг, 0,82 ммоль, 1,5 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 140 мг (67%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
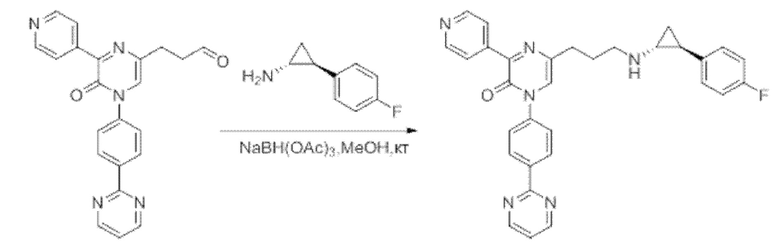
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-(пиридин-4-ил)-1-[4-(пиримидин-2-ил)фенил]пиразин-2(1H)-он Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (140 мг, 0,37 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (99,4 мг, 0,66 ммоль, 1,8 эквив.). Неочищенный продукт очищали, используя хроматографический Метод Е (от 42% до 45% CH3CN за 7 мин), получая 49,2 мг (25,98%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
ЖХМС: (ES,m/z): 519 [М+Н]+. 1Н ЯМР (300 МГц, Метанол-d4) δ м.д.: 8,92-8,90 (д, J=4,8 Гц, 2Н), 8,75-8,64 (м, 5Н), 8,77-8,33 (м, 2Н), 7,74-7,61 (т, J=8,7 Гц, 3H), 7,49-7,34 (т, J=4,8 Гц, 1H, 7,11-6,68 (м, 4Н), 2,90-2,81 (т, J=7,35 Гц, 2Н), 2,81-2,71 (т, J=7,35 Гц, 2Н), 2,39-2,29 (м, 1H, 2,1-1,88 (м, 3H), 1,16-0,94 (м, 2Н).
ПРИМЕР 34
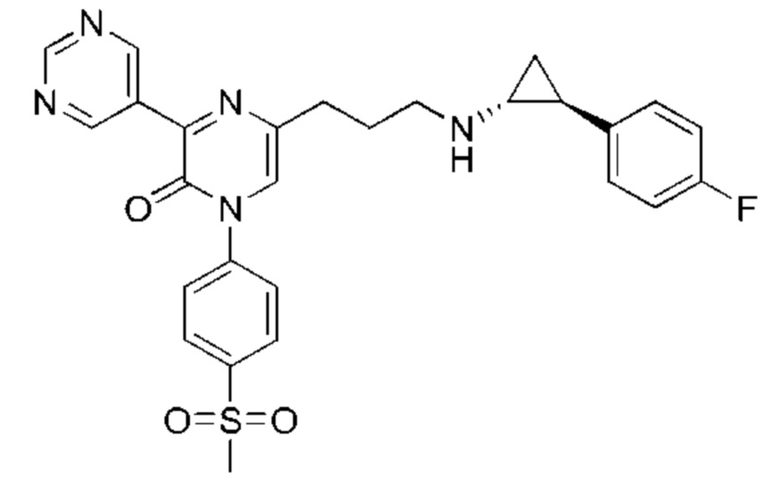
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-1-[4-(метилсульфонил)фенил]-3-(пиримидин-5-ил)пиразин-2(1H)-он
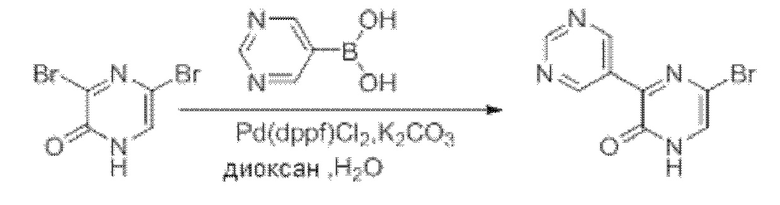
5-бром-3-(пиримидин-5-ил)пиразин-2(1H)-он (Промежуточное соединение 34-1) Раствор 3,5-дибром-1,2-дигидропиразин-2-она (20 г, 78,78 ммоль, 1 эквив.), К2СО3 (32,7 г, 236,60 ммоль, 3,003 эквив.), (пиримидин-5-ил)бороновой кислоты (14,6 г, 117,83 ммоль, 1,496 эквив.) и Pd(dppf)Cl2 (5,8 г, 7,93 ммоль, 0,101 эквив.) в диоксане (200 мл) и Н2О (20 мл) перемешивали в течение 2 ч при 90°С, затем концентрировали, получая 8 г (40%) указанного в заглавии соединения в виде масла светло-желтого цвета.
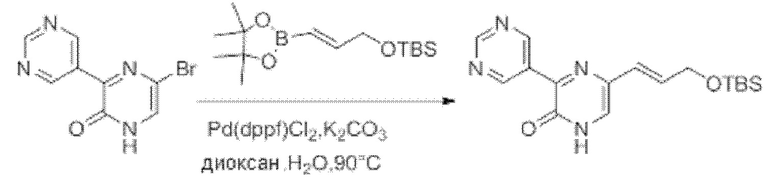
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-3-(пиримидин-5-ил)пиразин-2(1Н)-он (Промежуточное соединение 34-2) Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (4,5 г, 17,78 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (4:1), получая 3,8 г (61,99%) указанного в заглавии соединения в виде масла желтого цвета.
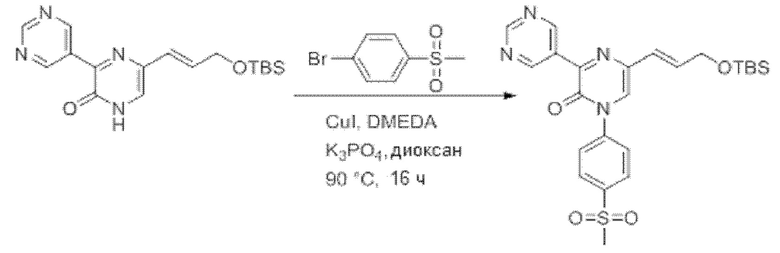
(Е)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-1-(4-(метил-сульфонил)фенил)-3-(пиримидин-5-ил)пиразин-2(1Н)-он Раствор продукта, полученного на предыдущей стадии, (3,8 г, 11,03 ммоль, 1 эквив.), 1-бром-4-метансульфонилбензола (3,9 г, 16,55 ммоль, 1,5 эквив.), CuI (2,1 г, 11,03 ммоль, 1 эквив.), DMEDA (1,9 г, 22,06 ммоль, 2 эквив.) и K3PO4 (7,0 г, 33,09 ммоль, 3 эквив.) в диоксане (50 мл) перемешивали в течение 16 ч при 90°С, затем концентрировали и очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (1:10). Вторую партию подвергали таким же условиям реакции и очистки, получая общий выход 800 мг (7,3%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
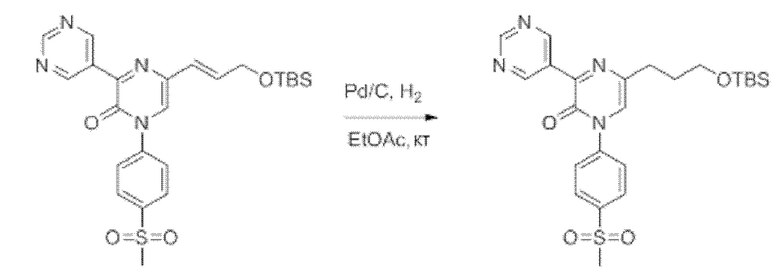
5-(3-((трте-Бутилдиметилсилил)окси)пропил)-1-(4-(метилсульфонил)фенил)-3-(пиримидин-5-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (800 мг, 0,40 ммоль, 1 эквив.), получая 600 мг (79,6%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
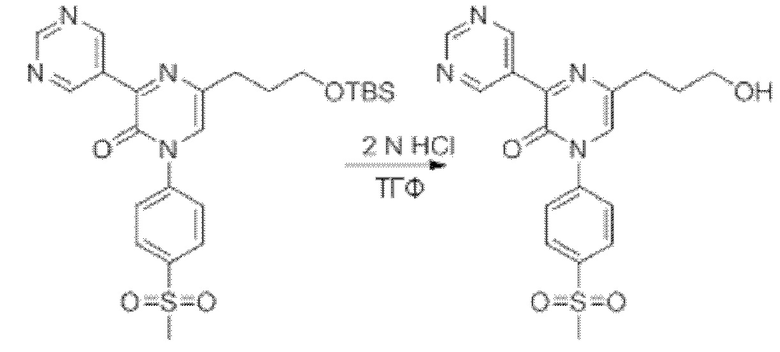
5-(3-Гидроксипропил)-1-(4-(метилсульфонил)фенил)-3-(пиримидин-5-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 33-5 использовали с продуктом, полученным на предыдущей стадии, (600 мг). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2 / МеОН (1:10), получая 300 мг (64,7%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
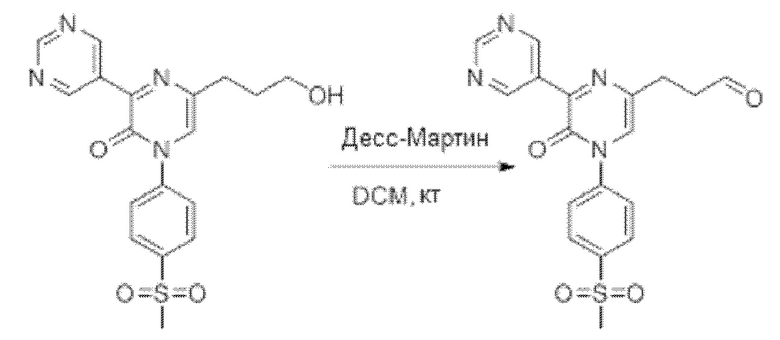
3-(4-(4-(Метилсульфонил)фенил)-5-оксо-6-(пиримидин-5-ил)-4,5-дигидропиразин-2-ил)пропаналь Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,77 ммоль, 1 эквив.) и реактивом Десса-Мартина (394,4 мг, 0,93 ммоль, 1,2 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (1:10), получая 200 мг (67,45%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
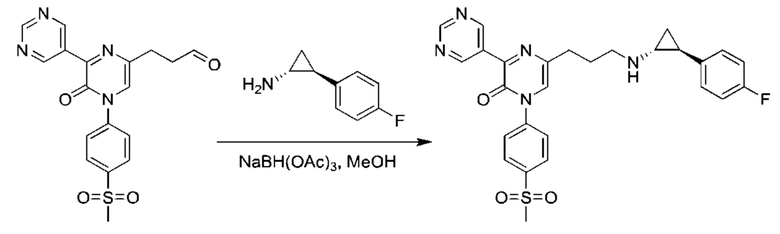
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-1-[4-(метил-сульфонил)фенил]-3-(пиримидин-5-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (200 мг, 0,52 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (118,0 мг, 0,78 ммоль, 1,5 эквив.). Неочищенный продукт очищали, используя хроматографический Метод F (от 18% до 28% CH3CN за 7 мин), получая 20,9 мг (7,7%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХ-МС: (ES,m/z): 520 [М+Н]+
1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 9,67 (с, 2Н), 9,21(с, 1H, 8,19-8,16 (д, J=9,0 Гц, 2Н), 7,84-7,81 (д, J=9,0 Гц, 2Н), 7,63 (с, 1H, 7,20-7,16 (м, 2Н), 7,04-6,99 (м, 2Н), 3,36-3,30 (м, 2Н), 3,20 (с, 3H), 3,17-2,98 (м, 1H, 2,85-2,80 (м, 2Н), 2,49-2,44 (м, 1H, 2,24-2,19 (м, 2Н), 1,50-1,37 (м, 2Н).
ПРИМЕР 35
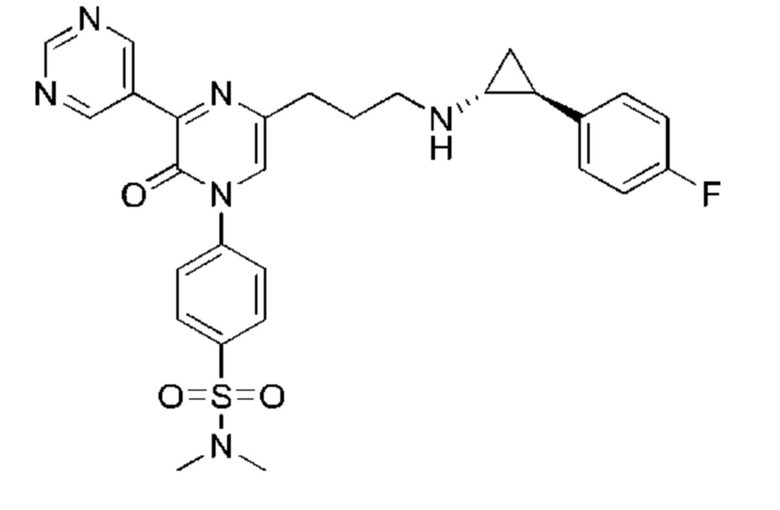
4-[5-(3-[([1R,2S]-2-[4-фторфенил]циклопропил)амино]пропил)-2-оксо-3-(пиримидин-5-ил)пиразин-1(2H)-ил]-N,N-диметилбензолсульфонамид
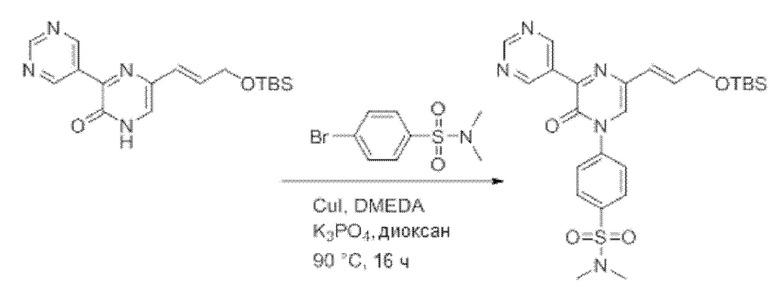
(E)-4-(5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-2-оксо-3-(пиримидин-5-ил)пиразин-1(2H)-ил)-N,N-диметилбензолсульфонамид Методику получения Промежуточного соединения 13-1 использовали с Промежуточным соединением 34-2 (2,6 г, 7,55 ммоль, 1 эквив.) и 4-бром-N,N-диметилбензол-1-сульфонамидом (3,0 г, 11,32 ммоль, 1,5 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (2:1), получая 550 мг (10,13%) указанного в заглавии соединения в виде масла светло-желтого цвета.
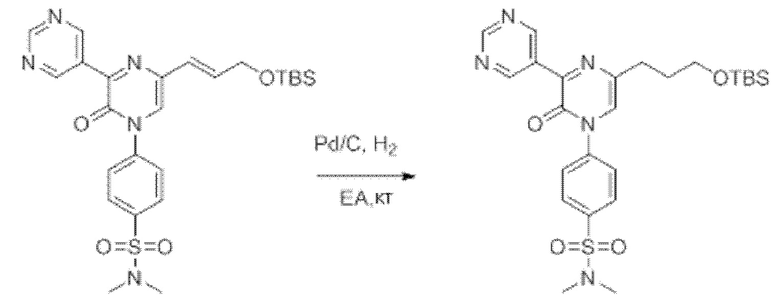
4-(5-(3-((трет-Бутилдиметилсилил)окси)пропил)-2-оксо-3-(пиримидин-5-ил)пиразин-1(2Н)-ил)-N,N-диметилбензолсульфонамид Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (550 мг, 1,04 ммоль, 1 эквив.), получая 420 мг (76,07%) указанного в заглавии соединения в виде масла светло-желтого цвета.
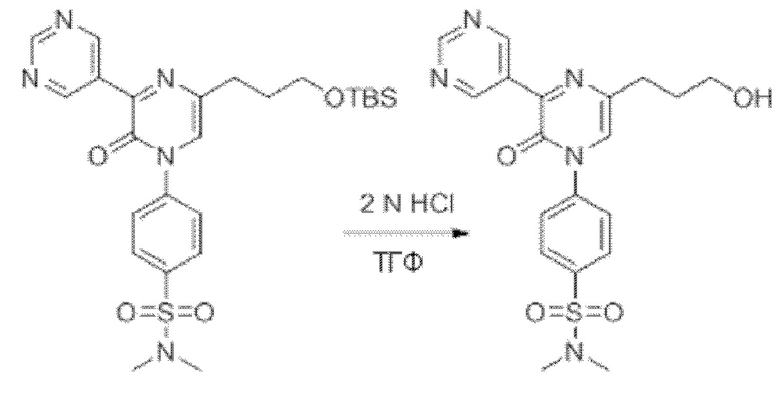
4-(5-(3-Гидроксипропил)-2-оксо-3-(пиримидин-5-ил)пиразин-1(2H)-ил)-N,N-диметилбензолсульфонамид Методику получения Промежуточного соединения 33-5 использовали с продуктом, полученным на предыдущей стадии, (420 мг, 0,79 ммоль, 1 эквив.), получая 180 мг (55%) указанного в заглавии соединения в виде масла светло-желтого цвета.
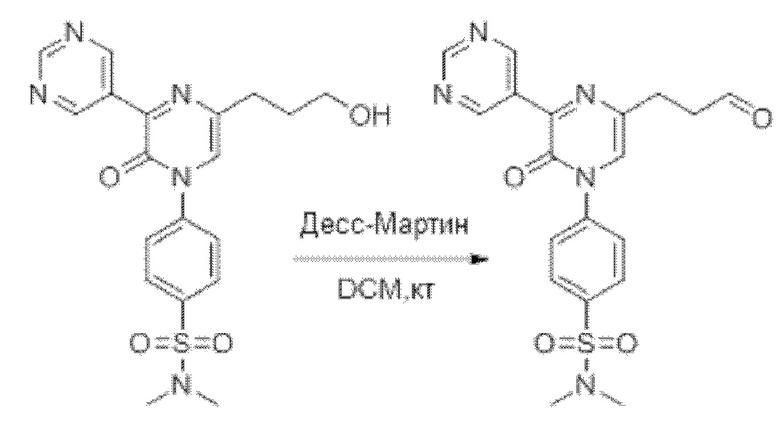
N,N-Диметил-4-(2-оксо-5-(3-оксопропил)-3-(пиримидин-5-ил)пиразин-1(2H)-ил)бензолсульфонамид Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (180 мг, 0,43 ммоль, 1 эквив.) и реактивом Десса-Мартина (220,5 мг, 0,52 ммоль, 1,20 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (40:1), получая 120 мг (67%) указанного в заглавии соединения в виде масла светло-желтого цвета.
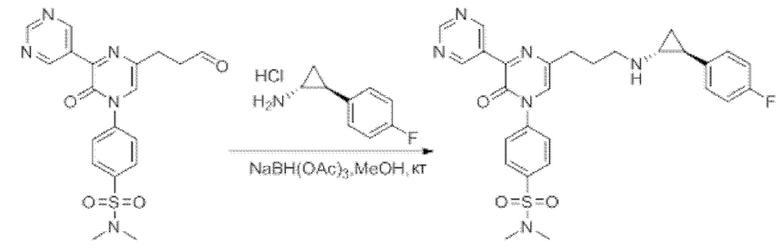
4-(5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-2-оксо-3-(пиримидин-5-ил)пиразин-1(2H)-ил)-N,N-диметилбензолсульфонамид Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (120 мг, 0,29 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)-циклопропан-1 -амином (52,9 мг, 0,35 ммоль, 1,2 эквив.). Неочищенный продукт очищали, используя хроматографический Метод А (от 15% до 45% CH3CN), получая 13,3 мг (5,57%) указанного в заглавии соединения в виде бесцветного твердого вещества.
ЖХ-МС: (ES,m/z): 549 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 9,67 (с, 2Н), 9,21 (с, 1H, 8,19-8,16 (д, J=9,0 Гц, 2Н), 7,84-7,81 (д, J=9,0 Гц, 2Н), 7,63 (с, 1H, 7,20-7,16 (м, 2Н), 7,04-6,99 (м, 2Н), 3,36-3,30 (м, 2Н), 3,29-3,21 (м, 3H), 3,17-2,98 (м, 1H, 2,85-2,80 (м, 2Н), 2,78 (с, 6Н), 2,49-2,44 (м, 1H, 2,24-2,19 (м, 2Н), 1,50-1,37 (м, 2Н).
ПРИМЕР 36
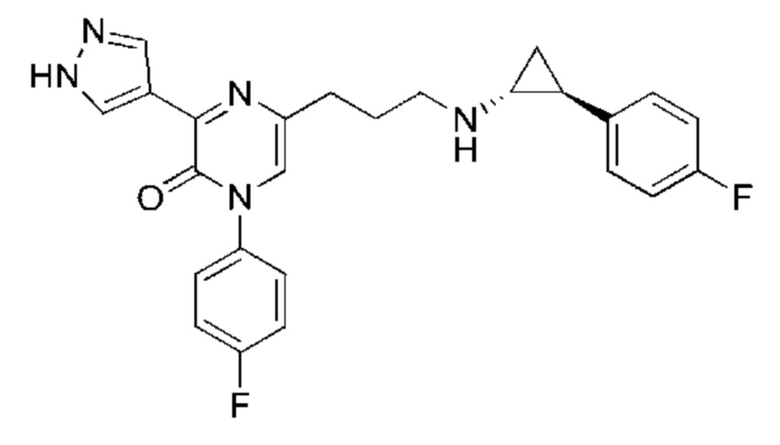
1-[4-фторфенил]-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[1H-пиразол-4-ил]пиразин-2(1H)-он
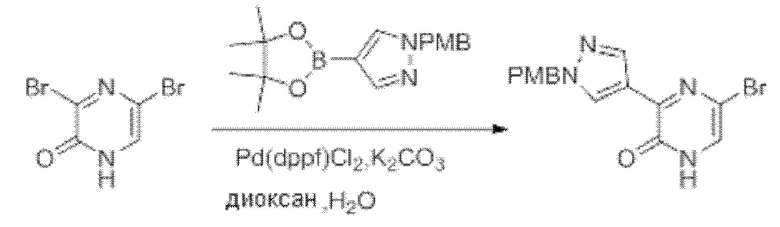
5-Бром-3-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразин-2(1H)-он
Раствор 3,5-дибром-1,2-дигидропиразин-2-она (4,8 г, 19,10 ммоль, 1 эквив.), 1-[(4-метоксифенил)метил]-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразола (6 г, 19,10 ммоль, 1 эквив.), К2СО3 (7,9 г, 57,29 ммоль, 3 эквив.) и Pd(dppf)Cl2 (1,4 г, 1,91 ммоль, 0,1 эквив.) в диоксане (100 мл) и Н2О (10 мл) перемешивали в течение 4 ч под N2 при 90°С. Полученную смесь концентрировали в вакууме и очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (30:1), получая 2,4 г (34,8%) указанного в заглавии соединения в виде масла желтого цвета.
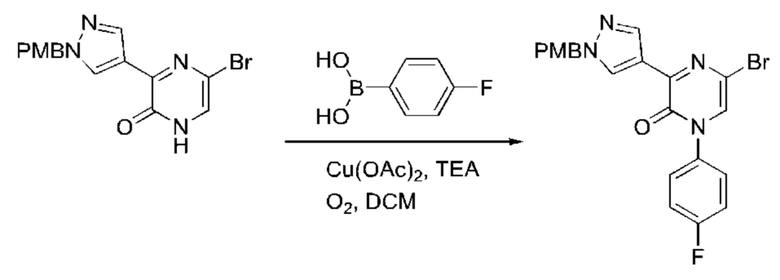
5-Бром-1-(4-фторфенил)-3-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразин-2(1H)-он Раствор продукта, полученного на предыдущей стадии, (2,4 г, 6,64 ммоль, 1 эквив.), (4-фторфенил)бороновой икслоты (1,9 г, 13,29 ммоль, 2,00 эквив.), TEA (1,3 г, 13,29 ммоль, 2 эквив.) и Cu(ОАс)2 (1,8 г, 9,97 ммоль, 1,5 эквив.) в CH2Cl2 (50 мл) перемешивали в течение 16 ч при кт, затем концентрировали в вакууме и очищали с помощью хроматографии на силикагеле, используя CH2Cl2 / МеОН (50:1), получая 1,4 г (46,3%) указанного в заглавии соединения в виде масла желтого цвета.
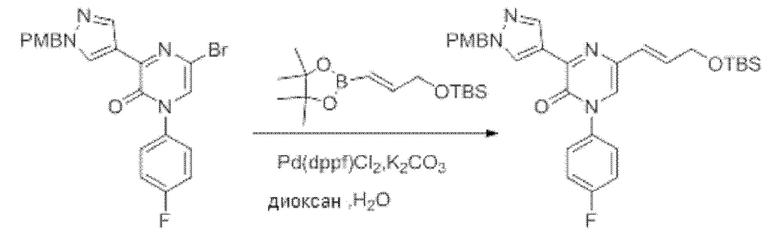
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-1-(4-фторфенил)-3-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (1,3 г, 2,86 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (20:1), получая 1 г (64%) указанного в заглавии соединения в виде масла желтого цвета.
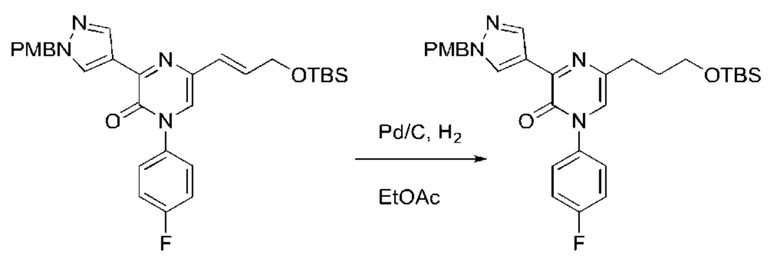
5-(3-((трет-Бутилдиметилсилил)окси)пропил)-1-(4-фторфенил)-3-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (1 г, 1,83 ммоль, 1 эквив.), получая 900 мг (90%) указанного в заглавии соединения в виде масла желтого цвета.
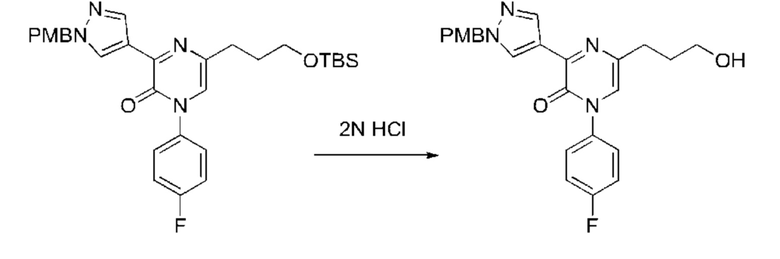
1-(4-Фторфенил)-5-(3-гидроксипропил)-3-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразин-2(1H)-он Раствор продукта, полученного на предыдущей стадии, (800 мг, 1,46 ммоль, 1 эквив.) и HCl (2 мл, 4M в диоксане) в диоксане (2 мл) перемешивали в течение 2 ч при кт, затем концентрировали в вакууме, получая 400 мг (63,2%) указанного в заглавии соединения в виде масла желтого цвета.
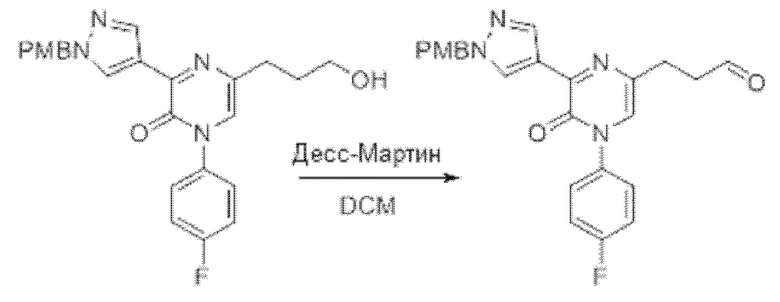
3-(4-(4-Фторфенил)-6-(1-(4-метоксибензил)-1H-пиразол-4-ил)-5-оксо-4,5-дигидропиразин-2-ил)пропаналь Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (350 мг, 0,81 ммоль, 1 эквив.) и реактивом Десса-Мартина (410,0 мг, 0,97 ммоль, 1,2 эквив.), с временем реакции 4 ч. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (10:1), получая 250 мг (71,8%) указанного в заглавии соединения в виде масла желтого цвета.
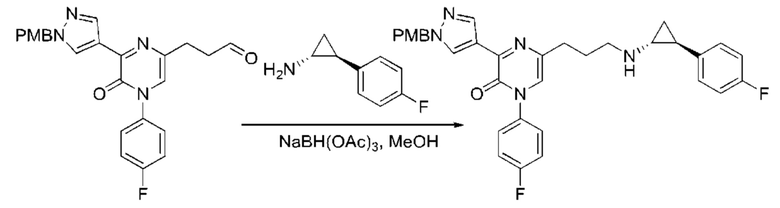
1-(4-Фторфенил)-5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-3-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (250 мг, 0,58 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (104,9 мг, 0,69 ммоль, 1,2 эквив.), с временем реакции 16 ч. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/МеОН (5:1), получая 200 мг (61%) указанного в заглавии соединения в виде масла желтого цвета.
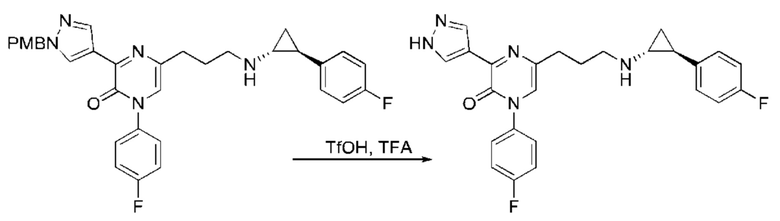
1-(4-Фторфенил)-5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-3-(1H-пиразол-4-ил)пиразин-2(1H)-он
Раствор продукта, полученного на предыдущей стадии, (200 мг, 0,35 ммоль, 1 эквив.) в смеси TFA (1 мл), TfOH (1 мл) и СН2О2 (5 мл) перемешивали в течение 6 ч при кт. Полученную смесь концентрировали в вакууме. Неочищенный продукт (200 мг) очищали, используя хроматографический Метод F (от 25% до 35% CH3CN за 7 мин), получая 18,6 мг (9,4%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
ЖХ-МС: (ES,m/z): 448 [М+Н]+ 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 8,47 (с, 2Н), 7,57-7,52 (м, 2Н), 7,36-7,30 (м, 3H), 7,22-7,17 (м, 2Н), 7,06-7,01 (т, J=8,7 Гц, 2Н), 3,37-3,34 (м, 2Н), 3,03-2,98 (м, 1H, 2,79-2,71 (т, J=7,2 Гц, 2Н), 2,51-2,44 (м, 1H, 2,36-2,18 (м, 2Н), 1,54-1,47 (м, 1H, 1,43-1,31 (м, 1H).
ПРИМЕР 37
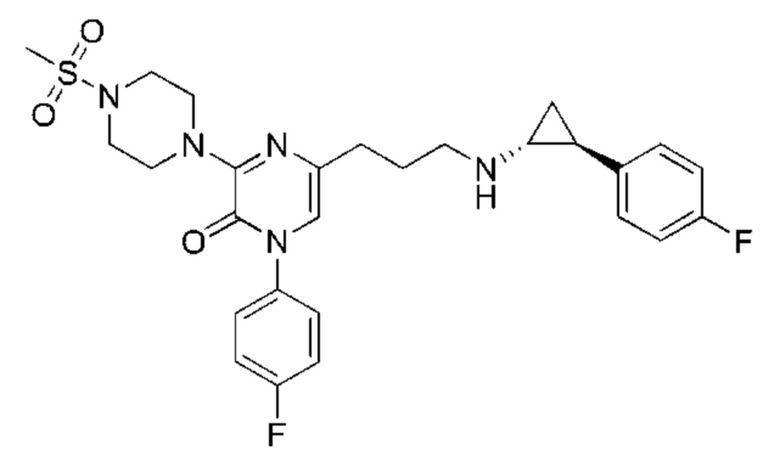
1-(4-Фторфенил)-5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он
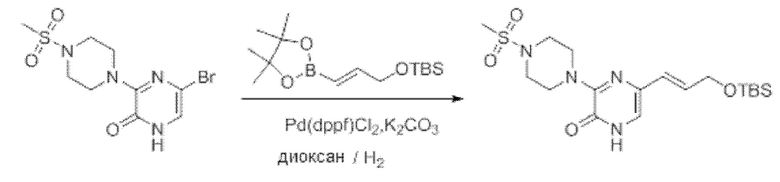
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-3-(4-(метил-сульфонил)пиперазин-1-ил)пиразин-2(1H)-он (Промежуточное соединение 37-1)
Методику получения Промежуточного соединения 3-3 использовали с Промежуточным соединением 3-1 (10 г, 29,66 ммоль) с временем реакции 3 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 2,8 г (22%) указанного в заглавии соединения в виде масла желтого цвета.
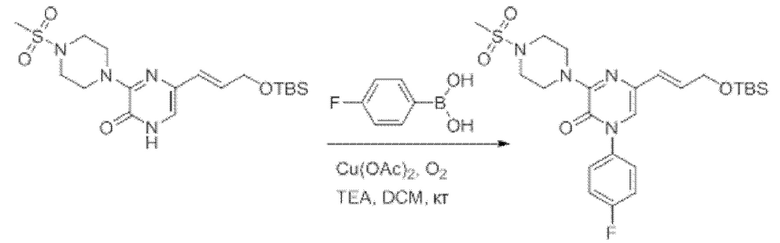
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-1-(4-фторфенил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 8-9 использовали с продуктом, полученным на предыдущей стадии, и 4-фторфенилбороновой икслотой (1,1 г, 1,5 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 1,6 г (58%) указанного в заглавии соединения в виде масла желтого цвета.
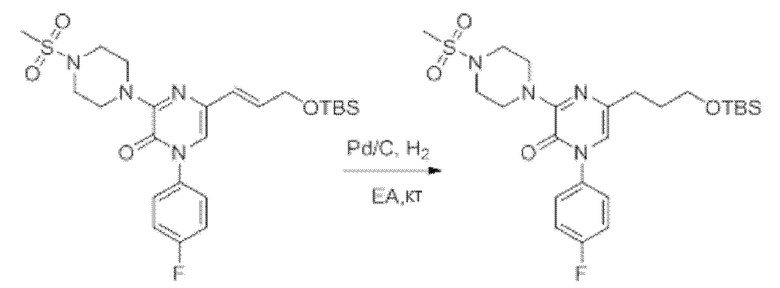
5-(3-((трет-Бутилдиметилсилил)окси)пропил)-1-(4-фторфенил)-3-(4-(метил-сульфонил)пиперазин-1-ил)пиразин-2(1Н)-он Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (1,6 г, 1,0 эквив.), получая 1 г (60%) указанного в заглавии соединения в виде масла желтого цвета.
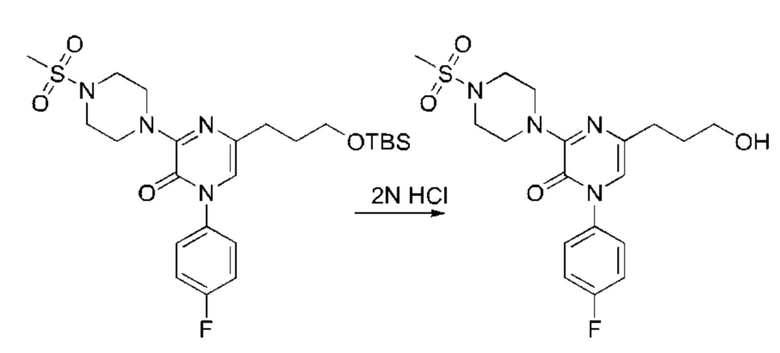
1-(4-Фторфенил)-5-(3-гидроксипропил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 33-5 использовали с продуктом, полученным на предыдущей стадии, (1 г, 1,0 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 390 мг указанного в заглавии соединения в виде твердого вещества желтого цвета.
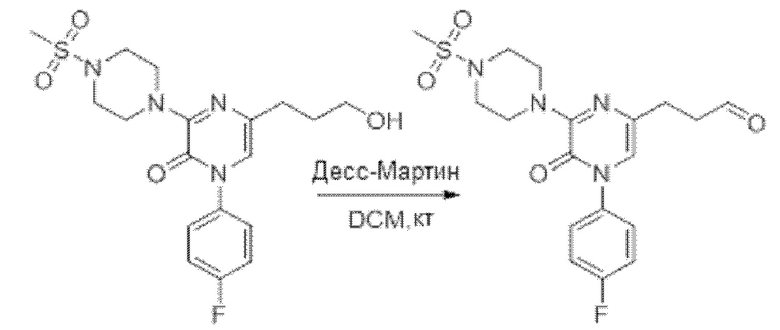
3-(4-(4-Фторфенил)-6-(4-(метилсульфонил)пиперазин-1-ил)-5-оксо-4,5-дигидропиразин-2-ил)пропаналь Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (380 мг, 0,93 ммоль, 1 эквив.) и реактивом Десса-Мартина (589,0 мг, 1,39 ммоль, 1,500 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 340 мг (89,9%) указанного в заглавии соединения в виде масла желтого цвета.
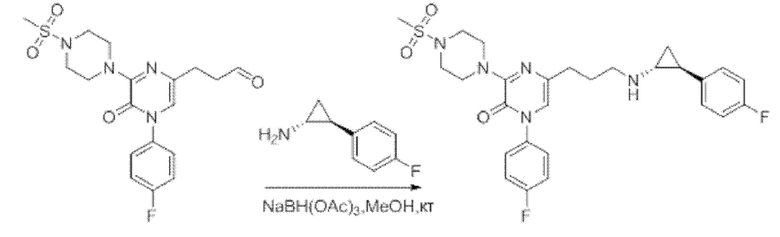
1-(4-Фторфенил)-5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)-пропил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (340 мг, 0,83 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (226,5 мг, 1,50 ммоль, 1,8 эквив.). Неочищенный продукт очищали, используя хроматографический Метод С (от 40% до 56% CH3CN за 16,5 мин), получая 67,1 мг (14,9%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХМС: (ES,m/z): 544 [М+Н]+. 1Н ЯМР (300 МГц, Метанол-d4) δ м.д.: 7,50-7,39 (м, 2Н), 7,35-7,22 (м, 2Н), 7,13-6,91 (м, 4Н), 6,86 (с, 1H, 3,91-3,81 (м, 4Н), 3,32-3,30 (м, 4Н), 2,87 (с, 3H), 2,84-2,73 (т, J=7,5 Гц, 2Н), 2,50 (т, J=7,3 Гц, 2Н), 2,36-2,26 (м, 1H, 1,95-1,85 (м, 3H), 1,14-0,93 (м, 2Н).
ПРИМЕР 38
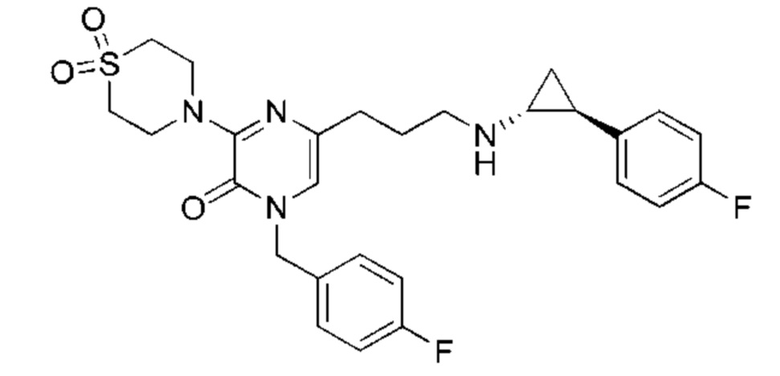
3-(1,1-диоксидотиоморфолино)-1-(4-фторбензил)-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]пиразин-2(1H)-он
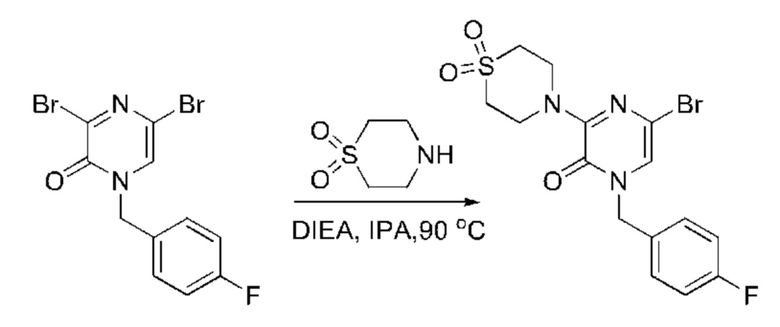
5-бром-3-(1,1-диоксидотиоморфолино)-1-(4-фторбензил)пиразин-2(1H)-он
Раствор тиоморфолин-1,1 -диоксида (2,8 г, 20,72 ммоль, 1,5 эквив.), 3,5-дибром-1-[(4-фторфенил)метил]-1,2-дигидропиразин-2-она (5 г, 13,81 ммоль, 1 эквив.), IPA (30 мл), DIEA (5,4 г, 41,44 ммоль, 3 эквив.). Полученный в результате раствор перемешивали в течение 2 ч при 90°С. Полученную смесь концентрировали. Полученное твердое вещество промывали 100 мл CH2Cl2. Твердые вещества собирали фильтрованием, получая 4 г (70%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
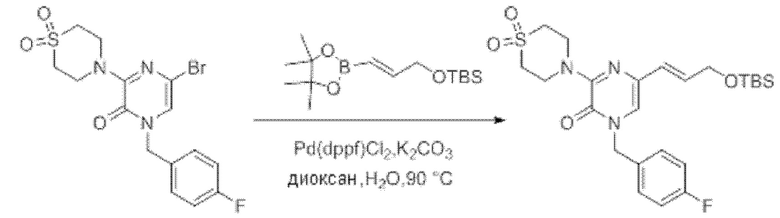
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-3-(1,1-диоксидотиоморфолино)-1-(4-фторбензил)пиразин-2(1H)-он Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (4 г, 9,61 ммоль), с временем реакции 2 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:4), получая 3 г (61%) указанного в заглавии соединения в виде масла светло-желтого цвета.
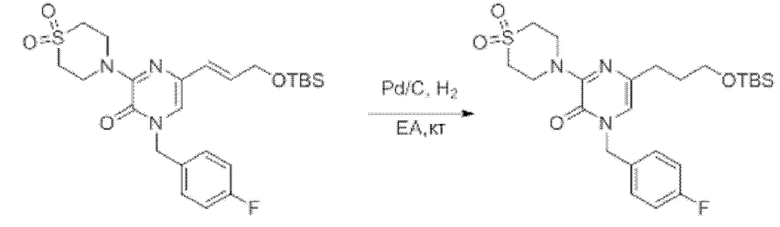
5-(3-((трет-Бутилдиметилсилил)окси)пропил)-3-(1,1-диоксидотиоморфолино)-1-(4-фторбензил)пиразин-2(1H)-он Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (3 г, 5,91 ммоль, 1 эквив.), получая 2,5 г (83%) указанного в заглавии соединения в виде масла светло-желтого цвета.
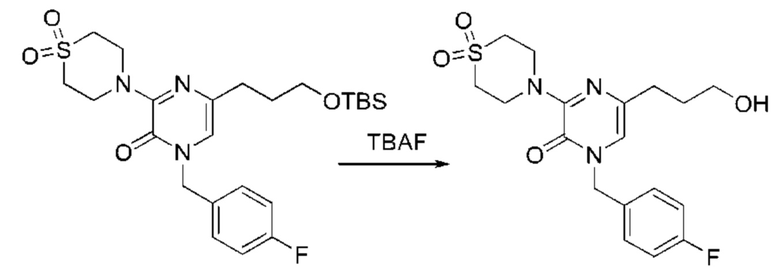
3-(1,1-Диоксидотиоморфолино)-1-(4-фторбензил)-5-(3-гидроксипропил)пиразин-2(1H)-он Методику получения Промежуточного соединения 22-4 использовали с продуктом, полученным на предыдущей стадии, (2,5 г, 4,90 ммоль), с временем реакции 2 ч. Неочищенный продукт очищали с помощью Ci8 обращенно-фазовой хроматографии, используя H2O/MeCN (3:1), получая 1,2 г (61,9%) указанного в заглавии соединения в виде масла светло-желтого цвета.
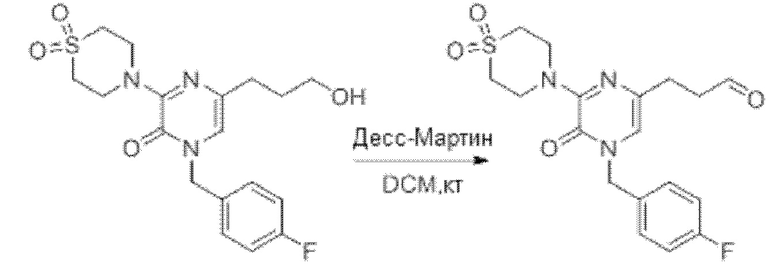
3-(6-(1,1-Диоксидотиоморфолино)-4-(4-фторбензил)-5-оксо-4,5-дигидропиразин-2-ил)пропаналь Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (600 мг, 1,52 ммоль, 1 эквив.) и реактивом Десса-Мартина (772,2 мг, 1,82 ммоль, 1,200 эквив.), с временем реакции 2 ч. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:2), получая 400 мг (67%) указанного в заглавии соединения в виде масла светло-желтого цвета.
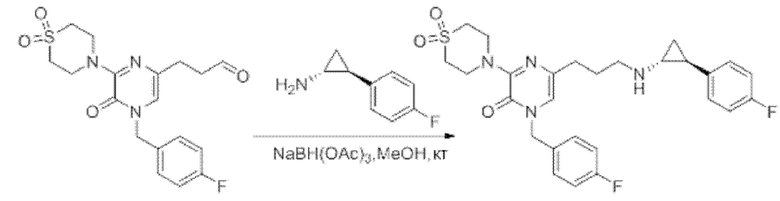
3-(1,1-Диоксидотиоморфолино)-1-(4-фторбензил)-5-(3-(((1R,2S)-2-(4-фтор-фенил)циклопропил)амино)пропил)пиразин-2(1H)-он Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (400 мг, 1,02 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (184,4 мг, 1,22 ммоль, 1,2 эквив.). Неочищенный продукт очищали, используя хроматографический Метод Е (от 44% до 74% CH3CN), получая 69,3 мг (12,9%) указанного в заглавии соединения в виде полутвердого вещества грязно-белого цвета.
ЖХ-МС: (ES,m/z): 529 [М+Н]+. 1НЯМР (300 МГц, MeOD-d4) δ м.д.: 7,41-7,36 (м, 2Н), 7,09-7,02 (м, 4Н), 7,02-6,93 (м, 3H), 5,12-5,00 (с, 2Н), 4,31-4,28 (м, 4Н), 3,20-3,10 (м, 4Н), 2,73-2,66 (т, J=7,2 Гц, 2Н), 2,48-2,43 (т, J=7,2 Гц, 2Н), 2,29-2,24 (м, 1H, 2,09-1,82 (м, 3H), 1,06-0,95 (м, 2Н).
ПРИМЕР 39
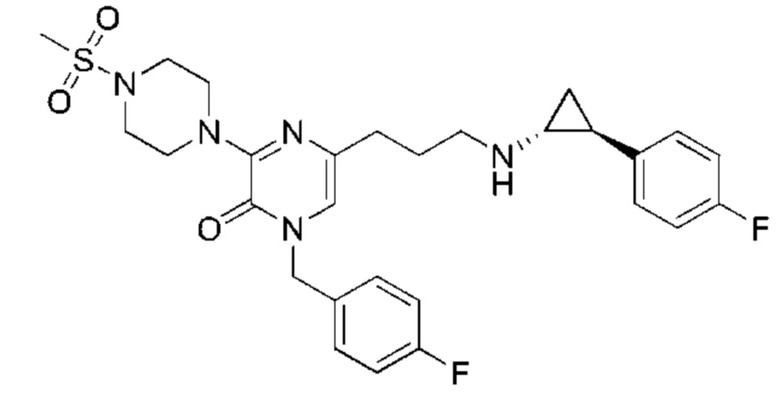
1-(4-фторбензил)-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[4-(метилсульфонил)пиперазин-1-ил] пиразин-2(1H)-он
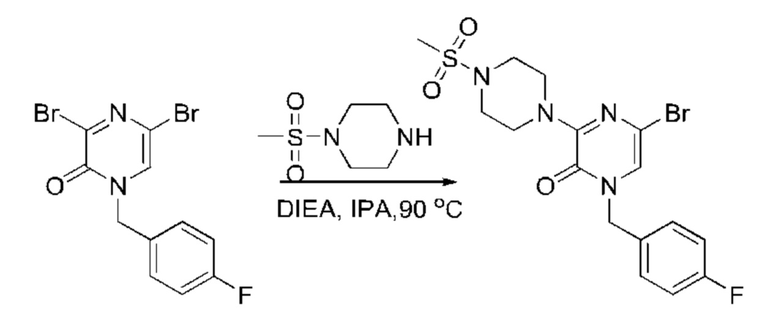
5-Бром-1-(4-фторбензил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1-[(4-фторфенил)метил]-1,2-дигидропиразин-2-оном (5 г, 13,81 ммоль, 1 эквив.) и 1-метансульфонилпиперазином (3,4 г, 0,02 ммоль, 1,5 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 4,8 г (78%) указанного в заглавии соединения в виде масла желтого цвета.
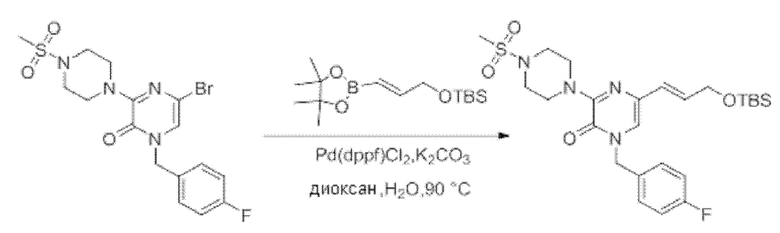
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-1-(4-фторбензил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (4,8 г, 10,78 ммоль), с временем реакции 4 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 3 г (49,78%) указанного в заглавии соединения в виде масла желтого цвета.
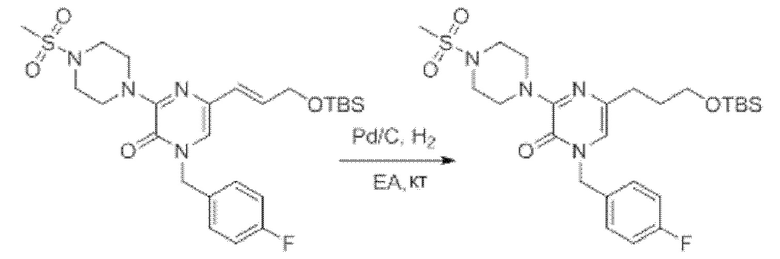
5-(3-((трет-Бутилдиметилсилил)окси)пропил)-1-(4-фторбензил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (3 г, 5,58 ммоль, 1,0 эквив.), получая 2 г (63%) указанного в заглавии соединения в виде масла желтого цвета.
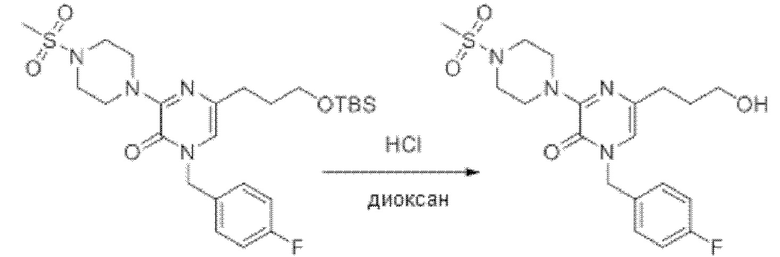
1-(4-Фторбензил)-5-(3-гидроксипропил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 33-5 использовали с продуктом, полученным на предыдущей стадии, (2 г, 3,72 ммоль), получая 360 мг (23%) указанного в заглавии соединения в виде масла желтого цвета.
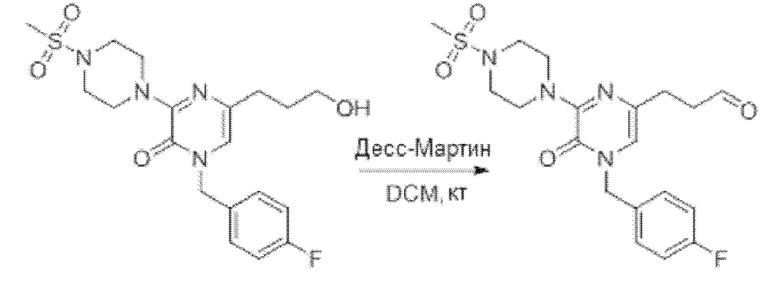
3-(4-(4-Фторбензил)-6-(4-(метилсульфонил)пиперазин-1-ил)-5-оксо-4,5-дигидропиразин-2-ил)пропаналь Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (360 мг, 0,85 ммоль, 1 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 300 мг (83,73%) указанного в заглавии соединения в виде твердого вещества белого цвета.
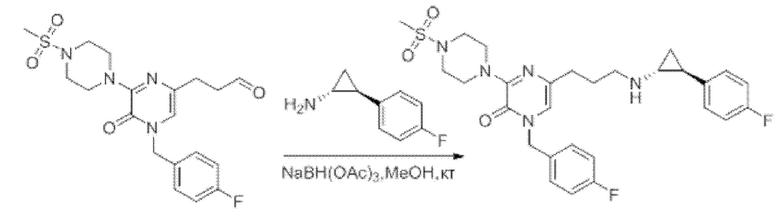
1-(4-Фторбензил)-5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)-пропил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,71 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (128,8 мг, 0,85 ммоль, 1,20 эквив.). Неочищенный продукт очищали, используя хроматографический Способ G (от 25% В до 37% CH3CN за 8 мин), получая 135,7 мг (35,5%) указанного в заглавии соединения в виде масла коричневого цвета.
ЖХМС: (ES,m/z): 558 [М+Н]+. 1Н ЯМР (300 МГц, Метанол-d4) δ м.д.: 7,44-7,34 (м, 2Н), 7,25-7,14 (м, 2Н), 7,14-7,02 (м, 4Н), 7,00 (с, 1H, 5,06 (с, 2Н), 3,87-3,78 (м, 4Н), 3,40-3,18 (м, 6Н), 3,02-2,92 (м, 1H, 2,87 (с, 3H), 2,63-2,31 (м, 3H), 2,11-2,01 (м, 2Н), 1,53-1,43 (м, 1H, 1,42-1,32 (м, 1H.
ПРИМЕР 40
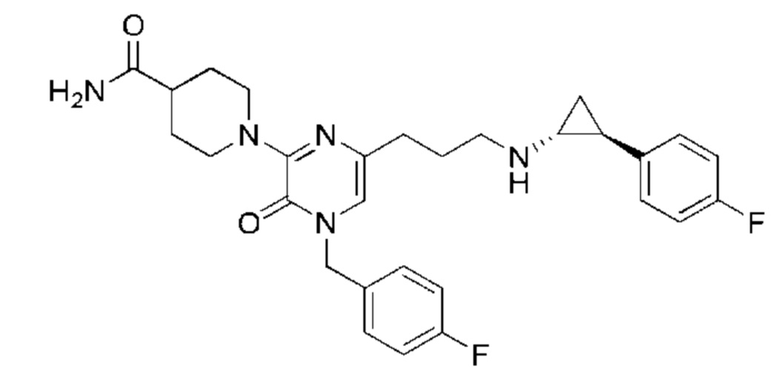
1-[4-(4-фторбензил)-6-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-3-оксо-3,4-дигидропиразин-2-ил)пиперидин-4-карбоксамид
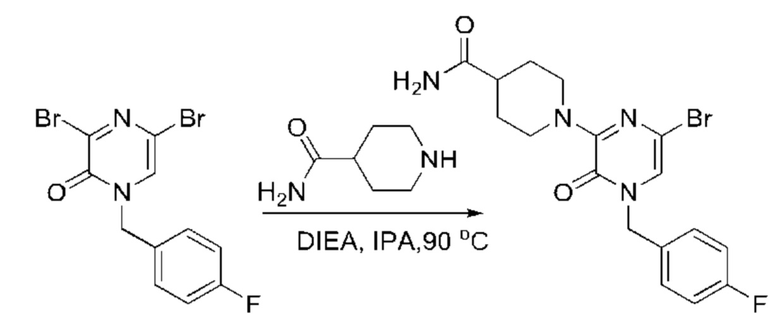
1-(6-Бром-4-(4-фторбензил)-3-оксо-3,4-дигидропиразин-2-ил)пиперидин-4-карбоксамид Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1-(4-фторбензил)пиразин-2(1H)-оном (5 г, 13,81 ммоль, 1,00 эквив.) и пиперидин-4-карбоксамидом (1,94 г, 15,19 ммоль, 1,10 эквив.), с временем реакции 3 ч при 90°С, получая 5 г (88%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
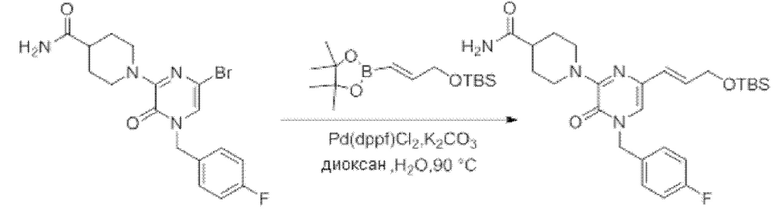
(E)-1-(6-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-4-(4-фторбензил)-3-оксо-3,4-дигидропиразин-2-ил)пиперидин-4-карбоксамид
Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (5 г, 12,2 ммоль, 1,00 эквив.) и трет-бутилдиметил [([2E]-3-[тетраметил-1,3,2-диоксаборолан-2-ил] проп-2-ен-1 -ил)окси]силаном (4,73 г, 15,88 ммоль, 1,30 эквив.), с временем реакции 1 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 2,2 г (24%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
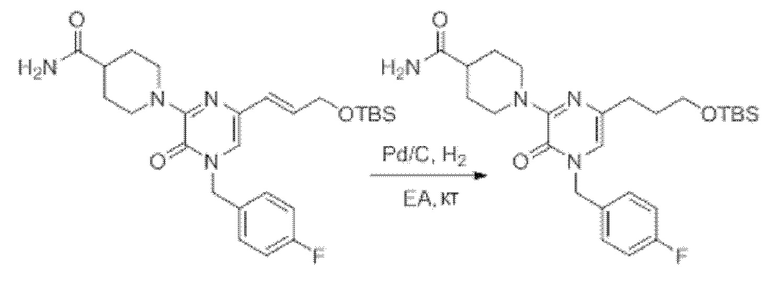
1-(6-(3-((трет-Бутилдиметилсилил)окси)пропил)-4-(4-фторбензил)-3-оксо-3,4-дигидропиразин-2-ил)пиперидин-4-карбоксамид Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (2,2 г, 1,78 ммоль, 1,00 эквив.), получая 2 г (80%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
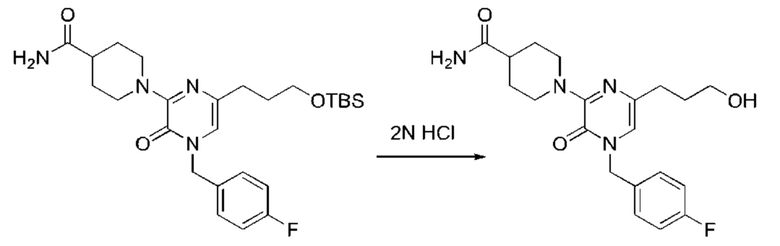
1-(4-(4-Фторбензил)-6-(3-гидроксипропил)-3-оксо-3,4-дигидропиразин-2-ил)-пиперидин-4-карбоксамид Методику получения Промежуточного соединения 33-5 использовали с продуктом, полученным на предыдущей стадии, (2 г, 1,42 ммоль, 1,00 эквив.), с временем реакции 2 ч при 25°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc, получая 1,1 г (70%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
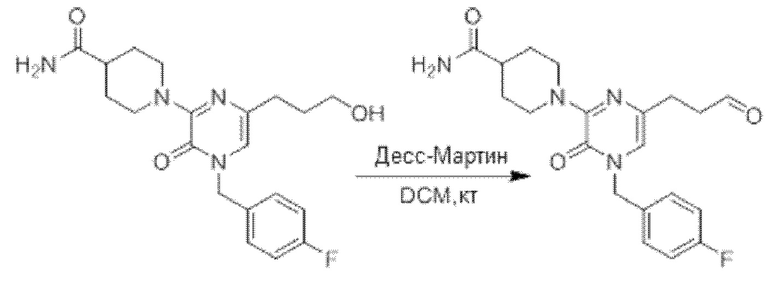
1-(4-(4-Фторбензил)-3-оксо-6-(3-оксопропил)-3,4-дигидропиразин-2-ил)пиперидин-4-карбоксамид Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (1 г, 1,00 ммоль, 1,00 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (2:1), получая 310 мг (71%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
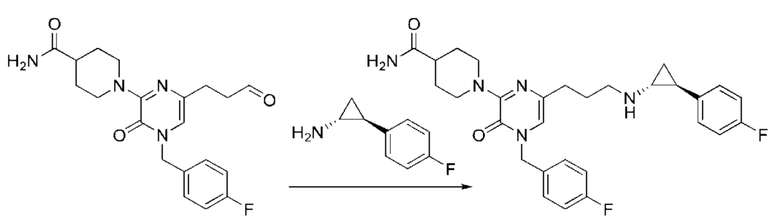
1-(4-(4-Фторбензил)-6-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)-пропил)-3-оксо-3,4-дигидропиразин-2-ил)пиперидин-4-карбоксамид Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (310 мг, 0,71 ммоль, 1,00 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (162 мг, 1,07 ммоль, 1,50 эквив.). Неочищенный продукт (4 мл) очищали, используя хроматографический Метод F (от 22% до 32% CH3CN за 7 мин), получая 40 мг (22%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
ЖХ-МС: (ES,m/z): 522 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 7,38-7,33 (м, 2Н), 7,19-7,14 (м, 2Н), 7,08-7,01 (м, 4Н), 6,87 (с, 1H, 5,01 (с, 2Н), 4,88-4,71 (м, 2Н), 3,31-3,29 (м, 2Н), 2,96-2,88 (м, 3H), 2,51-2,41 (м, 4Н), 2,05-1,99 (м, 2Н), 1,77-1,74 (м, 4Н), 1,45-1,33 (м, 2Н).
ПРИМЕР 41
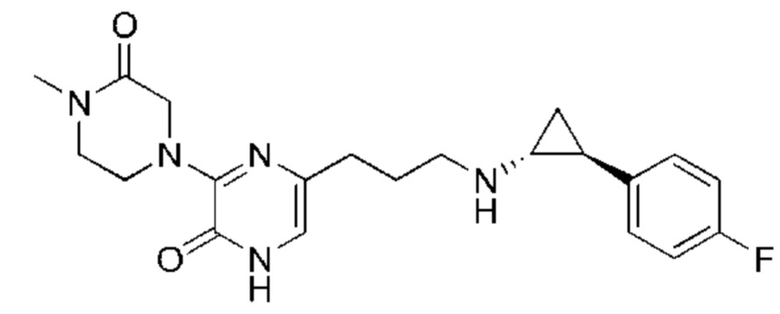
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[4-метил-3-оксопиперазин-1-ил]пиразин-2(1H)-он
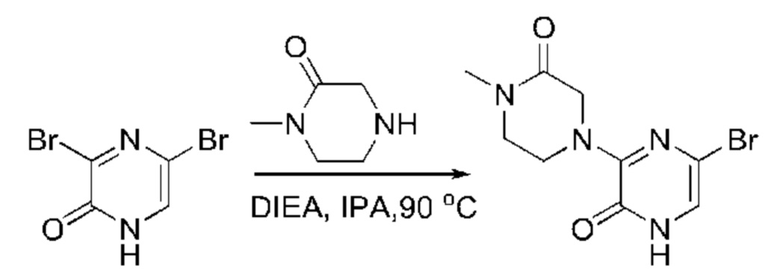
5-Бром-3-(4-метил-3-оксопиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (20 г, 78,78 ммоль, 1 эквив.) и 1-метилпиперазин-2-оном (10,8 г, 94,54 ммоль, 1,20 эквив.), с временем реакции 6 ч при 90°С, получая 18 г (80%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
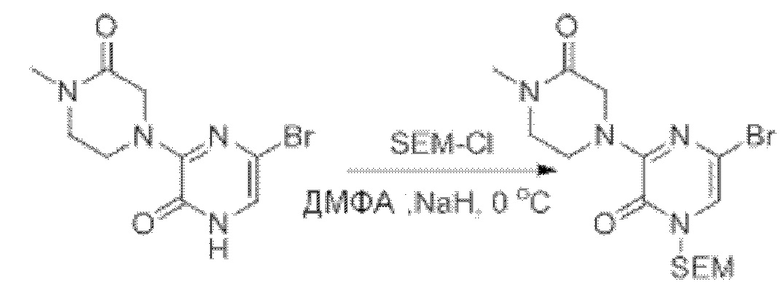
5-Бром-3-(4-метил-3-оксопиперазин-1-ил)-1-((2-(триметилсилил)этокси)-метил)пиразин-2(1H)-он Смесь продукта, полученного на предыдущей стадии, (12 г, 41,80 ммоль, 1 эквив.) и NaH (5,0 г, 125,4 ммоль, 3,0 эквив.) в ДМФА (200 мл) перемешивали в течение 1 ч при 0°С, затем добавляли [2-(хлорметокси)-этил]триметилсилан (10,5 г, 62,7 ммоль, 1,5 эквив.) и смесь перемешивали еще час при кт. Реакционную смесь гасили и экстрагировали 5 х 500 мл EtOAc. Полученную смесь концентрировали, получая 6 г (34%) указанного в заглавии соединения в виде масла желтого цвета.
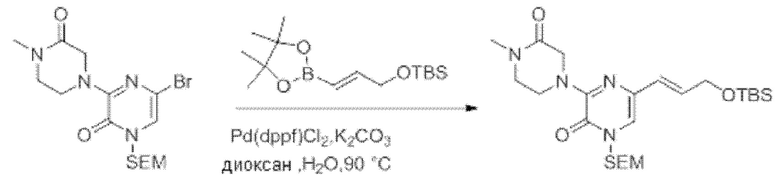
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-3-(4-метил-3-оксопиперазин-1-ил)-1-((2-(триметилсилил)этокси)метил)пиразин-2(1H)-он
Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (6 г, 14,38 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 6 г (82%) указанного в заглавии соединения в виде масла желтого цвета.
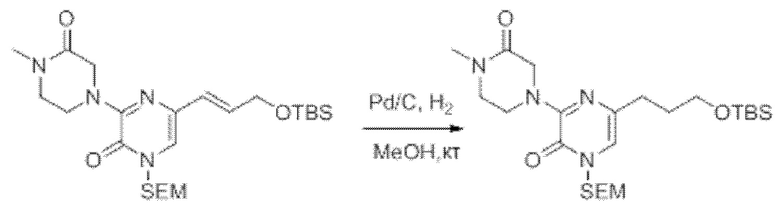
5-(3-((трет-Бутилдиметилсилил)окси)пропил)-3-(4-метил-3-оксопиперазин-1-ил)-1-((2-(триметилсилил)этокси)метил)пиразин-2(1H)-он Методику получения Промежуточного соединения 1-5 использовали с продуктом, полученным на предыдущей стадии, (6 г, 11,81 ммоль, 1,0 эквив.), получая 5,5 г (91%) указанного в заглавии соединения в виде масла желтого цвета.
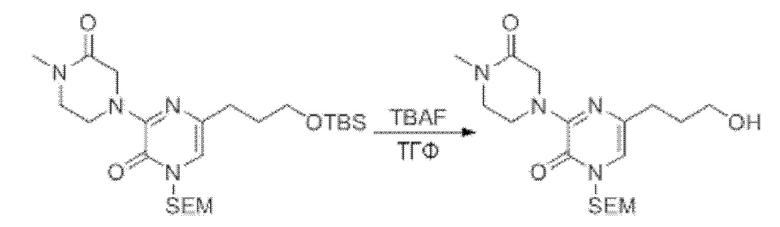
5-(3-Гидроксипропил)-3-(4-метил-3-оксопиперазин-1-ил)-1-((2-(триметилсилил)этокси)метил)пиразин-2(1H)-он Методику получения Промежуточного соединения 22-4 использовали с продуктом, полученным на предыдущей стадии, (5,5 г, 10,78 ммоль), с временем реакции 6 ч. Неочищенный продукт очищали с помощью флеш ВЭЖХ с MeCN/Н2О, получая 4 г (63%) указанного в заглавии соединения в виде масла желтого цвета.
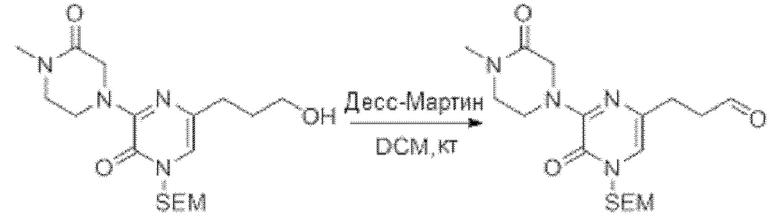
3-(6-(4-Метил-3-оксопиперазин-1-ил)-5-оксо-4-((2-(триметилсилил)этокси)-метил)-4,5-дигидропиразин-2-ил)пропаналь Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (1,5 г, 3,78 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 800 мг (80,03%) указанного в заглавии соединения в виде масла светло-желтого цвета.
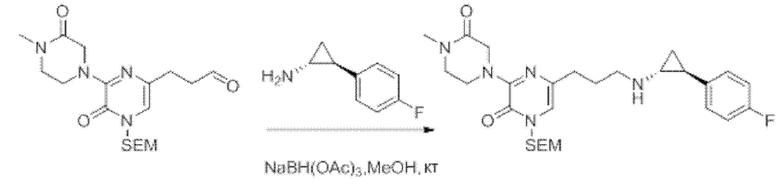
5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-3-(4-метил-3-оксопиперазин-1-ил)-1-((2-(триметилсилил)этокси)метил)пиразин-2(1H)-он
Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (800 мг, 2,03 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (551,8 мг, 3,65 ммоль, 1,8 эквив.). Неочищенный продукт очищали с помощью ТСХ с МеОН/CH2Cl2 (5:1), получая 350 мг (32,62%) указанного в заглавии соединения в виде масла желтого цвета.
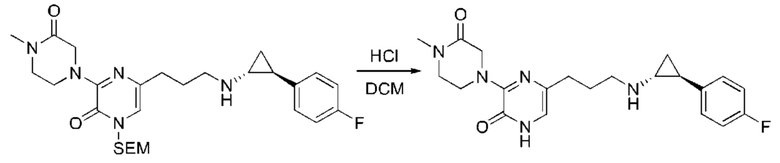
5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-3-(4-метил-3-оксопиперазин-1-ил)пиразин-2(1H)-он Раствор продукта, полученного на предыдущей стадии, (320 мг, 603 ммоль, 1 эквив.) в HCl (4N, 10 мл) и CH2Cl2 (20 мл) перемешивали в течение 2 ч при кт. рН корректировали до 7 добавлением Na2CO3. Полученную смесь концентрировали. Неочищенный продукт очищали, используя хроматографический Метод В (от 15% В до 43% CH3CN за 7 мин), получая 85 мг (10,5%) указанного в заглавии соединения в виде масла желтого цвета.
ЖХМС: (ES,m/z): 400 [М+Н]+. 1Н ЯМР (400 МГц, Метанол-d4) δ м.д.: 7,25-7,17 (м, 2Н), 7,07 (т, J=6,8 Гц, 2Н), 6,72 (с, 1H, 4,25 (с, 2Н), 4,23-4,16 (м, 2Н), 3,51-3,49 (т, J=5,6 Гц, 2Н), 3,30-3,22 (м, 2Н), 3,03-2,99 (м, 4Н), 2,61-2,55 (м, 2Н), 2,51-2,41 (м, 1H, 2,12-2,02 (м, 2Н), 1,54-1,45 (м, 1H, 1,45-1,35 (м, 1H.
ПРИМЕР 42
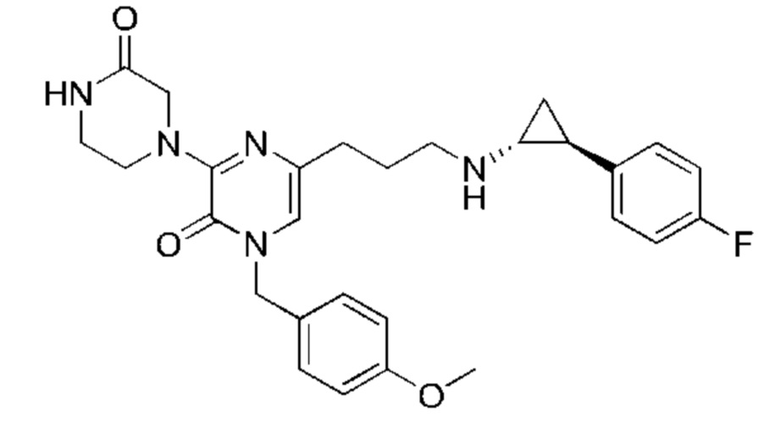
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-1-[4-метоксибензил]-3-[3-оксопиперазин-1-ил]пиразин-2(1H)-он
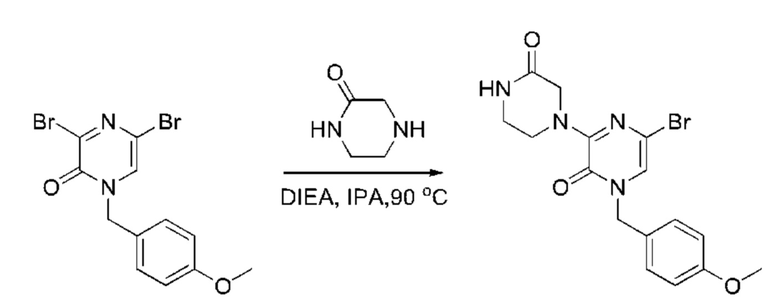
5-бром-1-(4-метоксибензил)-3-(3-оксопиперазин-1-ил)пиразин-2(1H)-он
Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1-(4-метоксибензил)пиразин-2(1H)-оном (6 г, 16,04 ммоль, 1,00 эквив.) и пиперазин-2-оном (1,6 г, 19,28 ммоль, 1,20 эквив.), с временем реакции 3 ч при 90°С, получая 6,0 г (88%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
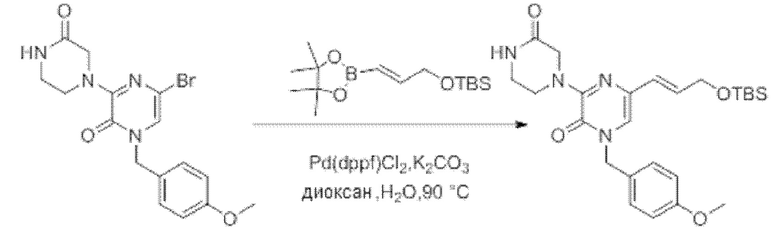
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-1-(4-метоксибензил)-3-(3-оксопиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (4,3 г, 11,14 ммоль, 1,00 эквив.), с временем реакции 1 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 1,7 г (24%) указанного в заглавии соединения в виде твердого вещества оранжевого цвета.
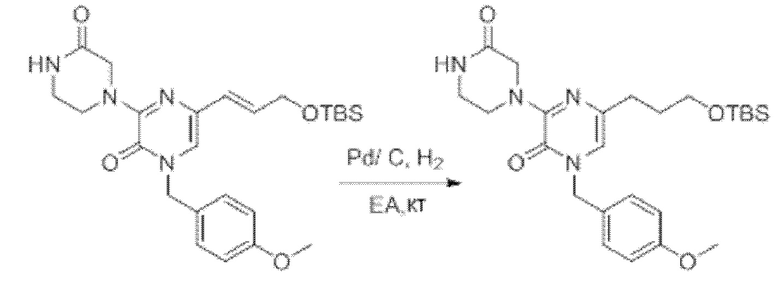
5-(3-((трет-Бутилдиметилсилил)окси)пропил)-1-(4-метоксибензил)-3-(3-оксопиперазин-1-ил)пиразин-2(1Н)-он Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (1,7 г, 1,78 ммоль, 1,00 эквив.), получая 1,3 г (80%) указанного в заглавии соединения в виде масла светло-желтого цвета.
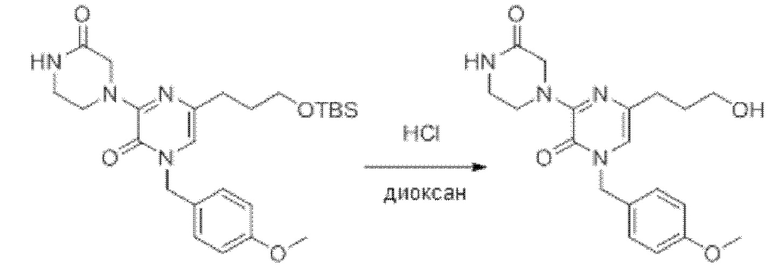
5-(3-Гидроксипропил)-1-(4-метоксибензил)-3-(3-оксопиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 33-5 использовали с продуктом, полученным на предыдущей стадии. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc, получая 500 мг (70%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
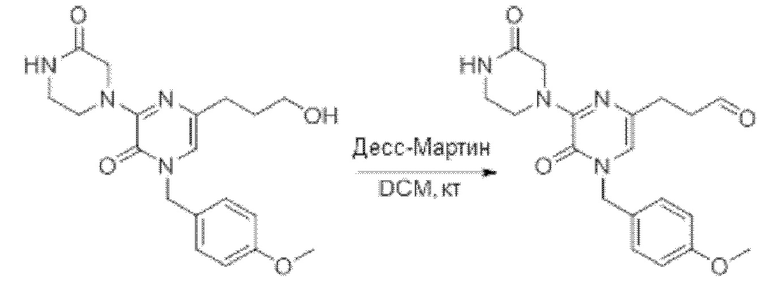
3-(4-(4-Метоксибензил)-5-оксо-6-(3-оксопиперазин-1-ил)-4,5-дигидропиразин-2-ил)пропаналь Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 1,00 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (2:1), получая 220 мг (71%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
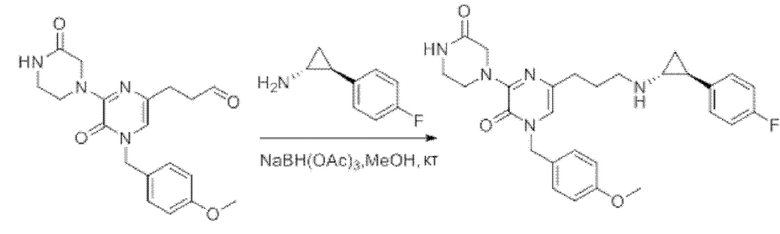
5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-1-(4-метокси-бензил)-3-(3-оксопиперазин-1-ил)пиразин-2(1H)-он Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (220 мг, 0,71 ммоль, 1,00 эквив.) и (1R, 2,5)-2-(4-фторфенил) циклопропан-1-амином (162 мг, 1,07 ммоль, 1,50 эквив.). Неочищенный продукт очищали, используя хроматографический Метод С (от 45% до 60%) CH3CN за 7 мин), получая 16,4 мг (22%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
ЖХ-МС: (ES,m/z): 506 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 7,28-7,25 (д, J=9,0 Гц, 2Н), 7,01-6,84 (м, 7Н), 5,00 (с, 2Н), 4,25 (с, 2Н), 4,04-4,00 (td, J=5,0, 1,7 Гц, 2Н), 3,74 (с, 3H), 3,42-3,38 (т, J=5,4 Гц, 2Н), 2,70-2,65 (т, J=7,5 Гц, 2Н), 2,44-2,39 (т, J=7,2 Гц, 2Н), 2,26-2,21 (м, 1H, 1,88-1,79 (м, 3H), 1,14-0,90 (м, 2Н).
ПРИМЕР 43
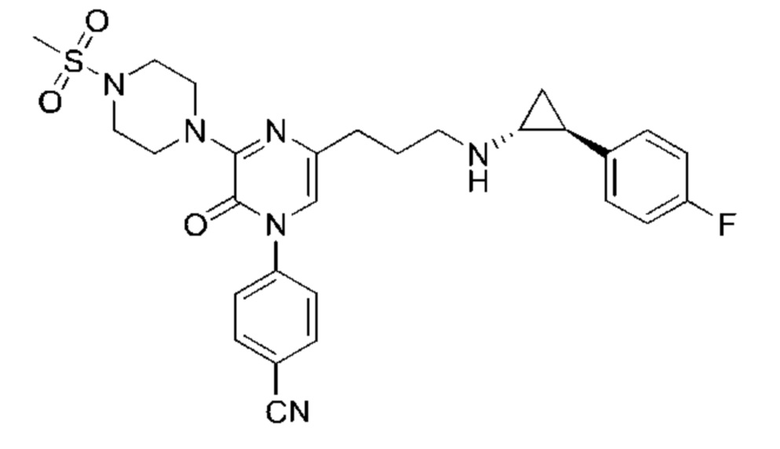
4-[5-(3-[([1R,2S]-2-[4-фторфенил]циклопропил)амино]пропил)-3-(4-[метилсульфонил]пиперазин-1-ил)-2-оксопиразин-1(2Н)-ил]бензонитрил
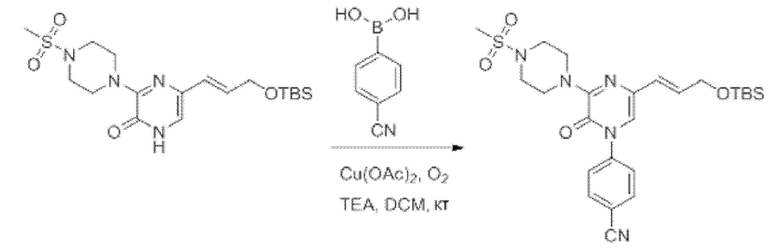
(E)-4-(5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-3-(4-(метилсульфонил)пиперазин-1-ил)-2-оксопиразин-1(2H)-ил)бензонитрил
Методику получения Промежуточного соединения 8-9 использовали с Промежуточным соединением 37-1 (9 г, 21,00 ммоль, 1 эквив.) и (4-цианофенил)бороновой кислотой (3,6 г, 25,2 ммоль, 1,2 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя РЕ/EtOAc (3:1), получая указанное в заглавии соединение (1,2 г, 10,79%) в виде полутвердого вещества темно-коричневого цвета.
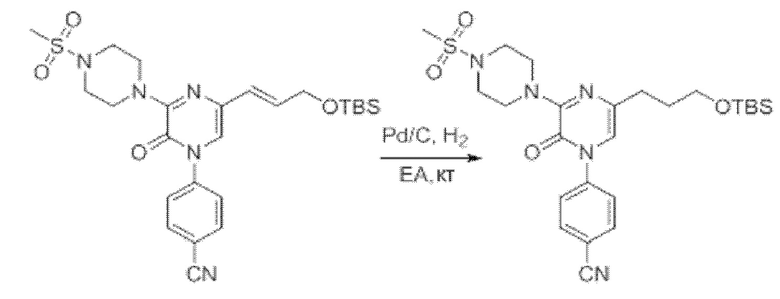
4-(5-(3-((трет-Бутилдиметилсилил)окси)пропил)-3-(4-(метилсульфонил)пиперазин-1-ил)-2-оксопиразин-1(2H)-ил)бензонитрил
Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (900 мг, 1,0 эквив.), получая 600 мг указанного в заглавии соединения в виде масла желтого цвета.
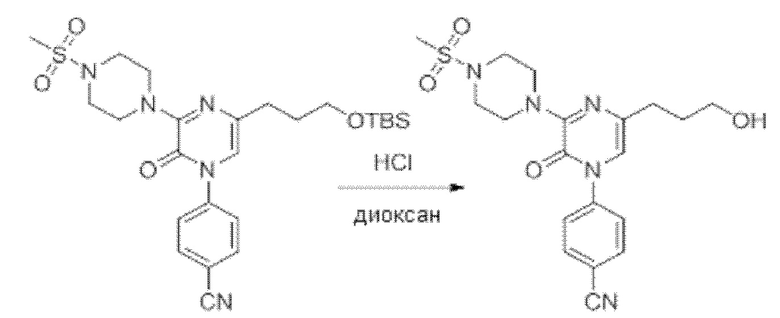
4-(5-(3-Гидроксипропил)-3-(4-(метилсульфонил)пиперазин-1-ил)-2-оксопиразин-1(2H)-ил)бензонитрил Методику получения Промежуточного соединения 33-5 использовали с продуктом, полученным на предыдущей стадии, (600 мг, 1,0 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CHCl2/МеОН (15:1), получая 330 мг указанного в заглавии соединения в виде масла желтого цвета.
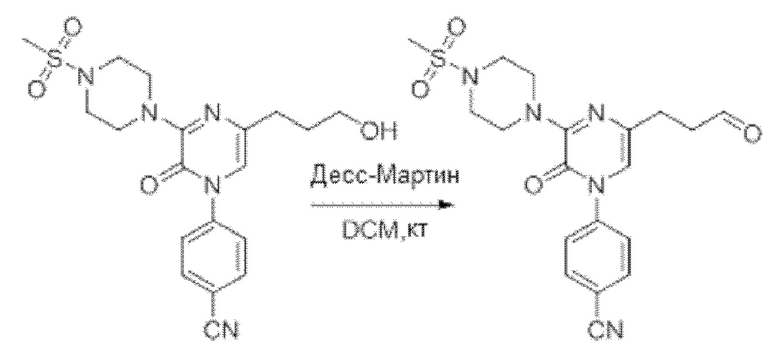
4-(3-(4-(Метилсульфонил)пиперазин-1-ил)-2-оксо-5-(3-оксопропил)пиразин-1(2H)-ил)бензонитрил Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (300 мг, 0,72 ммоль, 1 эквив.) и реактивом Десса-Мартина (396,2 мг, 0,93 ммоль, 1,300 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 230 мг (77,04%) указанного в заглавии соединения в виде масла светло-желтого цвета.
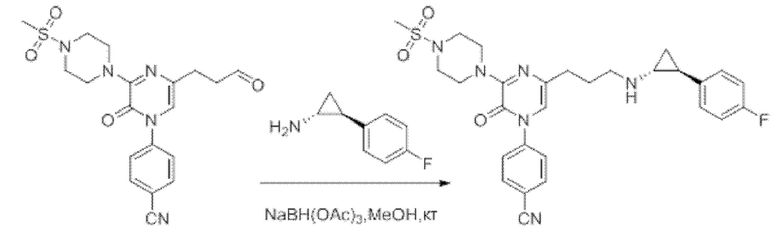
4-(5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-3-(4-(метилсульфонил)пиперазин-1-ил)-2-оксопиразин-1(2H)-ил)бензонитрил Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (230 мг, 0,55 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (125,5 мг, 0,83 ммоль, 1,5 эквив.). Неочищенный продукт очищали, используя хроматографический Метод Е (от 44% до 74% CH3CN за 8 мин), получая 78,5 мг (25,75%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
ЖХ-МС: (ES,m/z): 551 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 8,02-7,82 (д, J=6,0 Гц, 2Н), 7,75-7,52 (д, J=6,0 Гц, 2Н), 7,16-7,03 (м, 2Н), 7,03-6,95 (м, 2Н), 6,88 (с, 1H, 4,00-3,80 (м, 4Н), 3,32-3,28 (м, 4Н), 2,87 (с, 3H), 2,88-2,72 (т, J=7,2 Гц, 2Н), 2,63-2,40 (т, J=7,2 Гц, 2Н), 2,39-2,20 (м, 1H, 2,00-1,80 (м, 3H), 1,15-0,89 (м, 2Н).
ПРИМЕР 44
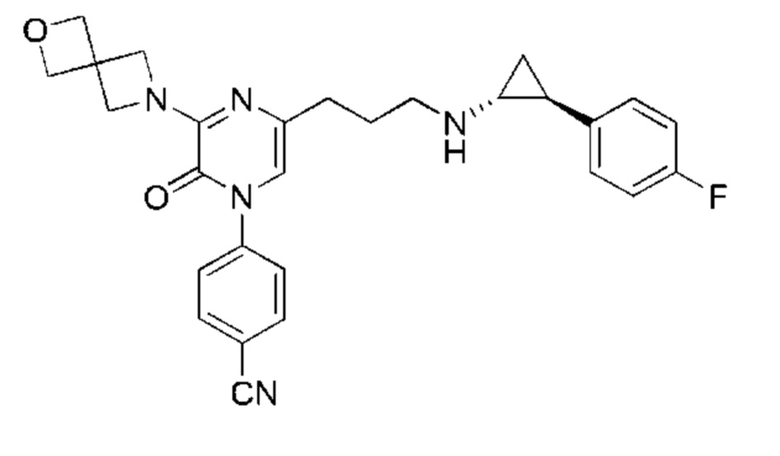
4-[5-(3-[([1R,2S]-2-[4-фторфенил]циклопропил)амино]пропил)-2-оксо-3-(2-окса-6-азаспиро [3,3] гептан-6-ил)пиразин-1 (2Н)-ил] бензонитрил
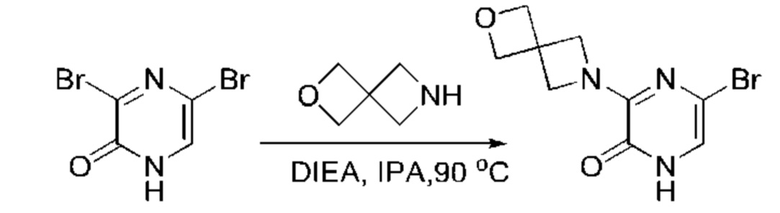
5-Бром-3-(2-окса-6-азаспиро [3,3] гептан-6-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 2-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (20 г, 78,78 ммоль, 1 эквив.) и 2-окса-6-азаспиро[3,3]гептаном (11,7 г, 118,17 ммоль, 1,5 эквив.), с временем реакции 2 ч при 90°С, получая 18 г (83,97%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
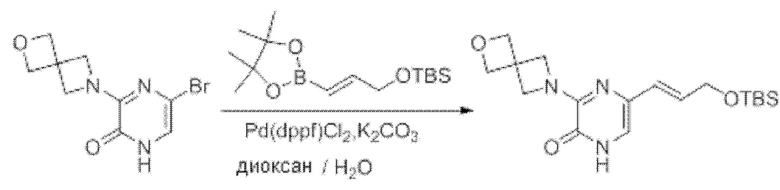
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-3-(2-окса-6-азаспиро[3,3]гептан-6-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (8 г, 29,40 ммоль), с временем реакции 2 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (4:1), получая 2,5 г (23,4%) указанного в заглавии соединения в виде масла светло-желтого цвета.
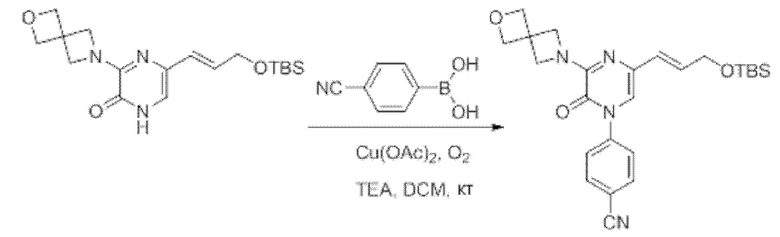
(E)-4-(5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-2-оксо-3-(2-окса-6-азаспиро[3,3]гептан-6-ил)пиразин-1(2H)-ил)бензонитрил Методику получения Промежуточного соединения 8-9 использовали с продуктом, полученным на предыдущей стадии, (2,1 г, 5,78 ммоль, 1 эквив.) и (4-цианофенил)бороновой кислотой (1,3 г, 0,01 ммоль, 1,5 эквив.), с временем реакции 6 ч при кт. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 1,0 г (37,26%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
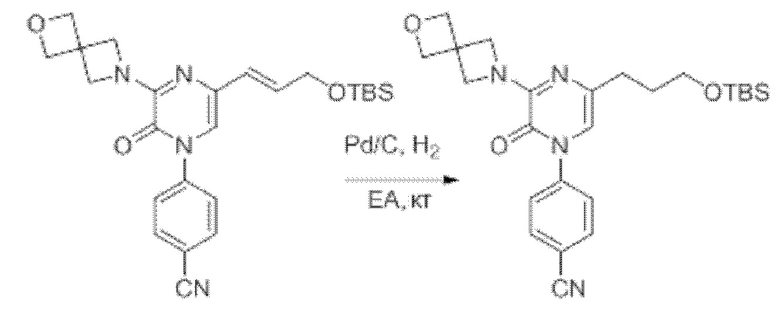
4-(5-(3-((трет-Бутилдиметилсилил)окси)пропил)-2-оксо-3-(2-окса-6-азаспиро[3,3]гептан-6-ил)пиразин-1(2Н)-ил)бензонитрил Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (1 г, 1 эквив.), получая 800 мг (79%) указанного в заглавии соединения в виде масла желтого цвета.

4-(5-(3-Гидроксипропил)-2-оксо-3-(2-окса-6-азаспиро[3,3]гептан-6-ил)пиразин-1(2H)-ил)бензонитрил Методику получения Промежуточного соединения 22-4 использовали с продуктом, полученным на предыдущей стадии, (800 мг). Полученную смесь концентрировали и очищали с помощью флеш ВЭЖХ с MeCN/Н2О, получая 540 мг (53%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
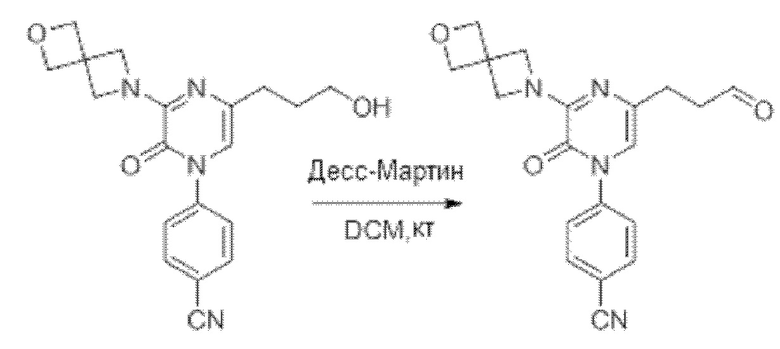
4-(2-Оксо-5-(3-оксопропил)-3-(2-окса-6-азаспиро[3,3]гептан-6-ил)пиразин-1(2H)-ил)бензонитрил Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (470 мг, 1,33 ммоль, 1 эквив.) и реактивом Десса-Мартина (848,6 мг, 2,00 ммоль, 1,5 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:3), получая 280 мг (59,92%) указанного в заглавии соединения в виде масла желтого цвета.
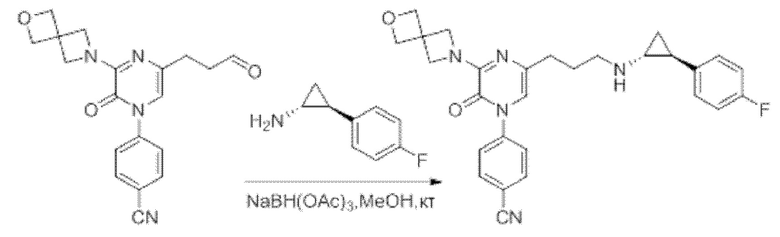
4-(5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-2-оксо-3-(2-окса-6-азаспиро[3,3]гептан-6-ил)пиразин-1(2H)-ил)бензонитрил Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (280 мг, 0,80 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (218,7 мг, 1,45 ммоль, 1,8 эквив.). Неочищенный продукт очищали, используя хроматографический Метод С (от 43% до 60% CH3CN за 7 мин), получая 43,9 мг (11,25%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
ЖХМС: (ES,m/z): 486 [М+Н]+. 1Н ЯМР (400 МГц, Метанол-d4) δ м.д.: 7,94-7,87 (д, J=2,0 Гц, 2Н), 7,68-7,61 (д, J=2,0 Гц, 2Н), 7,12-7,04 (м, 2Н), 7,03-6,92 (м, 2Н), 6,65 (с, 1H, 4,82 (с, 4Н), 4,46 (с, 4Н), 2,77 (т, J=7,5 Гц, 2Н), 2,43 (т, J=7,4 Гц, 2Н), 2,36-2,26 (м, 1H, 1,98-1,81 (м, 3H), 1,12-1,04 (м, 1H, 1,04-0,96 (м, 1H.
ПРИМЕР 45
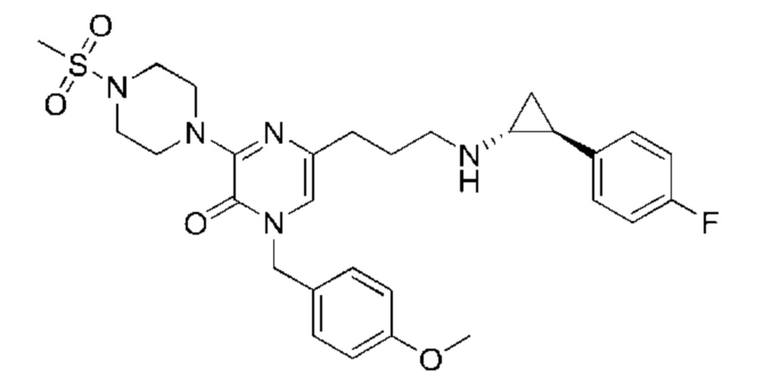
5-(3-(((1R,2S)-2-(4-Фторфенил)циклопропил)амино)пропил)-1-(4-метоксибензил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он
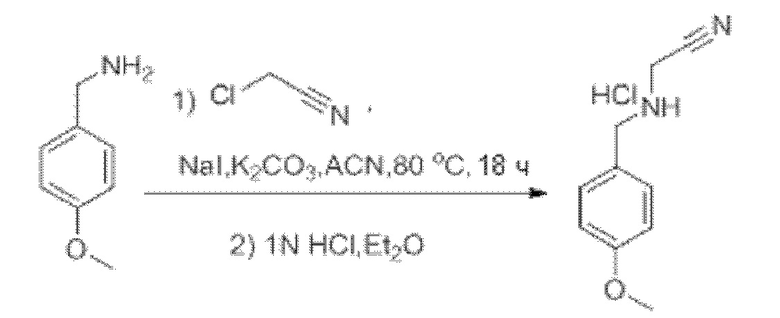
2-((4-Метоксибензил)амино)ацетонитрил гидрохлорид Смесь (4-метоксифенил)метанамина (60 г, 437,37 ммоль, 1 эквив.), 2-хлорацетонитрила (39,6 г, 524,85 ммоль, 1,2 эквив.), Nal (6,57 г, 43,74 ммоль, 0,1 эквив.) и K2CO3 (78,6 г, 568,58 ммоль, 1,3 эквив.) в CH3CN (400 мл) перемешивали в течение 18 ч при 80°С. Реакцию контролировали с помощью ЖХМС. Смесь оставляли охлаждаться до кт. Полученную смесь фильтровали; осадок на фильтре промывали CH3CN (3x350 мл). Фильтрат концентрировали при пониженном давлении. Полученную смесь разбавляли Et2O (400 мл). К раствору добавляли HCl (1N, 300 мл). Выпавшие в осадок твердые вещества собирали фильтрованием и промывали Et2O (3 х 70 мл), получая указанное в заглавии соединение (51 г, 66,17%) в виде твердого вещества серого цвета.
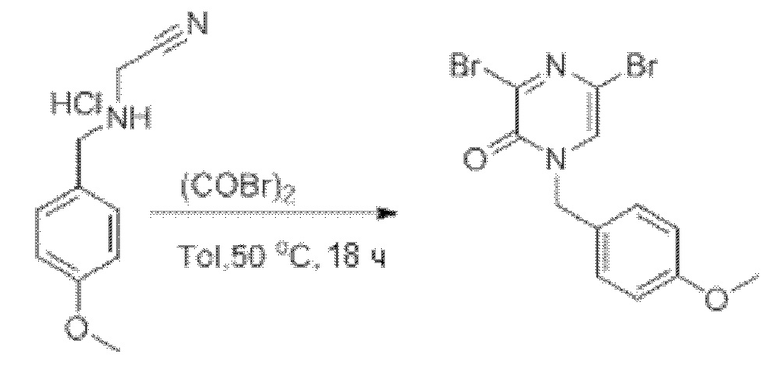
3,5-дибром-1-(4-метоксибензил)пиразин-2(1H)-он В круглодонную колбу на 500 мл, содержащую толуол (200 мл), по каплям добавляли дибромид щавелевой кислоты (150 г, 693,6 ммоль, 3,0 эквив.) в течение 30 мин при кт. Полученную смесь перемешивали еще в течение 15 мин при такой же температуре. К перемешанному раствору порциями добавляли продукт, полученный на предыдущей стадии, (49,2 г, 231,33 ммоль, 1 эквив.) при кт. Полученную смесь перемешивали в течение 18 ч при 50°С. Реакцию контролировали с помощью ЖХМС. Смесь оставляли охлаждаться до кт и затем разбавляли нас. NaH2PO4 (500 мл). Водный слой экстрагировали EtOAc (4 х 400 мл). Объединенные органические слои промывали 500 мл соляного раствора и сушили над Na2SO4. Полученную смесь фильтровали; осадок на фильтре промывали EtOAc (2 х 400 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, используя РЕ/EtOAc (5:1), получая указанное заглавии соединение (9 г, 10,40%) в виде масла темно-желтого цвета.
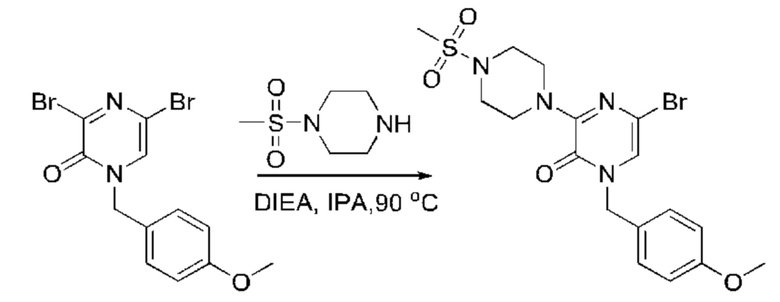
5-Бром-1-(4-метоксибензил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 1-3 использовали с продуктом, полученным на предыдущей стадии, (6,9 г, 18,45 ммоль, 1 эквив.) и 1-метансульфонилпиперазином (3,6 г, 22,14 ммоль, 1,2 эквив.), с временем реакции 2 ч при 90°С, получая указанное в заглавии соединение (6,6 г, неочищенное) в виде твердого вещества желтого цвета.
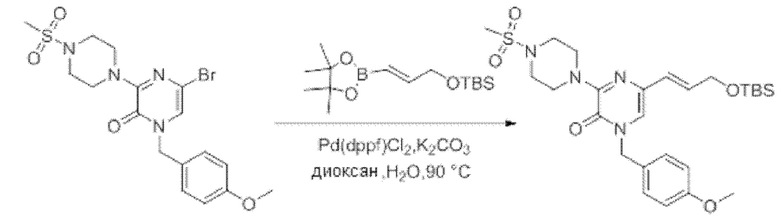
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-1-(4-метоксибензил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он
Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (4,5 г, 9,84 ммоль), с временем реакции 1 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя РЕ/EtOAc (5:1), получая указанное в заглавии соединение (3,9 г, 72,23%) в виде масла желтого цвета.
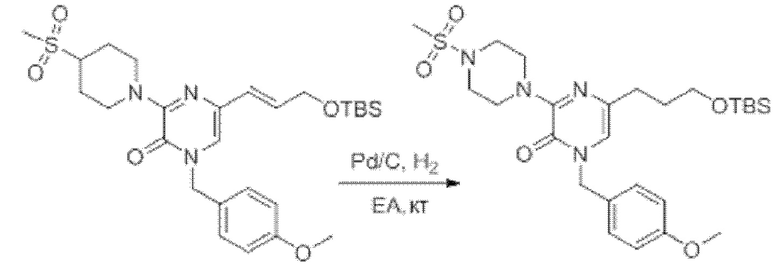
5-(3-((трет-Бутилдиметилсилил)окси)пропил)-1-(4-метоксибензил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (3,5 г, 6,38 ммоль, 1 эквив.), получая указанное в заглавии соединение (3,3 г, 93,94%) в виде масла желтого цвета.
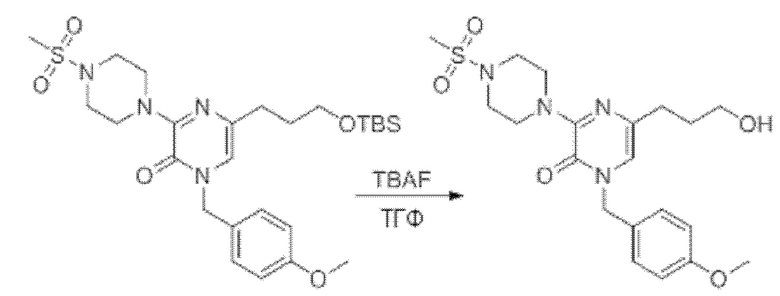
5-(3-Гидроксипропил)-1-(4-метоксибензил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения
Промежуточного соединения 22-4 использовали с продуктом, полученным на предыдущей стадии, (3,3 г, 5,99 ммоль). Неочищенный продукт очищали с помощью ЖХСД в следующих условиях (Мобильная фаза А: вода, Мобильная фаза В: CH3CN; Скорость потока: 100 мл/мин; Градиент: от 0 В до 100% В за 50 мин; 220/254 нм; Rt: 31,26 мин), получая указанное в заглавии соединение (1,1 г, 42,06%) в виде масла желтого цвета.
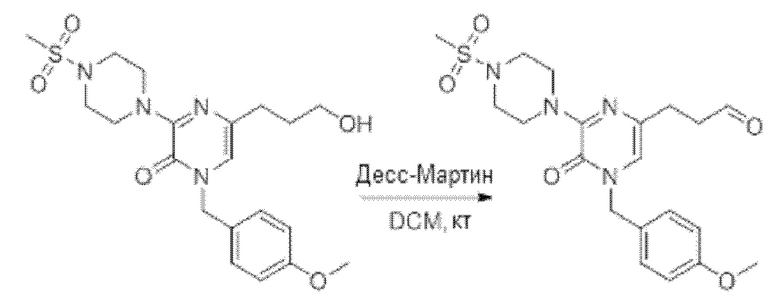
3-(4-(4-Метоксибензил)-6-(4-(метилсульфонил)пиперазин-1-ил)-5-оксо-4,5-дигидропиразин-2-ил)пропаналь Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (500 мг, 1,15 ммоль, 1 эквив.) и реактивом Десса-Мартина (583,0 мг, 1,37 ммоль, 1,2 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя CH2Cl2/EtOAc (1:5), получая указанное в заглавии соединение (380 мг, 76%) в виде твердого вещества грязно-белого цвета.
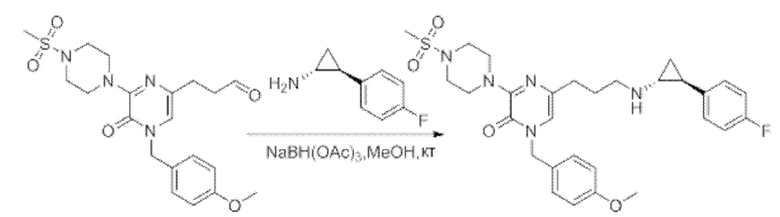
5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-1-(4-метокси-бензил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (380 мг, 0,87 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (246 мг, 1,63 ммоль, 1,861 эквив.). Неочищенный продукт (380 мг) очищали, используя хроматографический Метод С (от 44% до 54% CH3CN за 10 мин), получая указанное в заглавии соединение (69,6 мг, 13,98%) в виде твердого вещества грязно-белого цвета.
ЖХ-МС: (ES,m/z): 570 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 7,30 (д, J=8,7 Гц, 2Н), 7,07-6,88 (м, 7Н), 5,01 (с, 2Н), 3,84-3,80 (м, 4Н), 3,78 (с, 3H), 3,35-3,29 (м, 4Н), 2,87 (с, 3H), 2,71 (т, J=7,2 Гц, 2Н), 2,45 (т, J=7,2 Гц, 2Н), 2,29-2,24 (м, 1H, 1,90-1,82 (м, 3H), 1,06-0,96 (м, 2Н).
ПРИМЕР 46
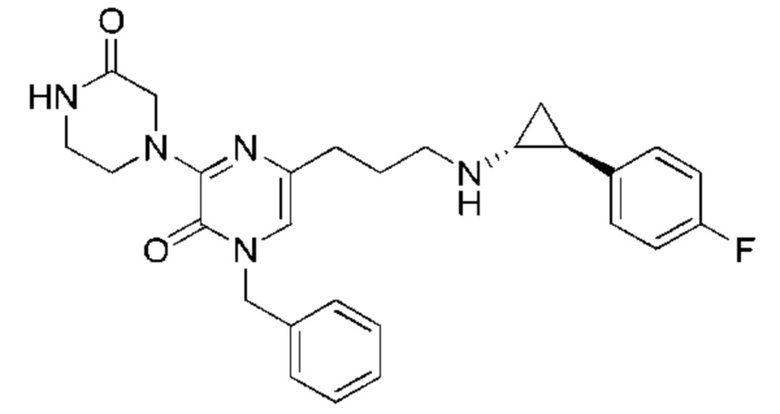
1-бензил-5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[3-оксопиперазин-1-ил]пиразин-2(1H)-он
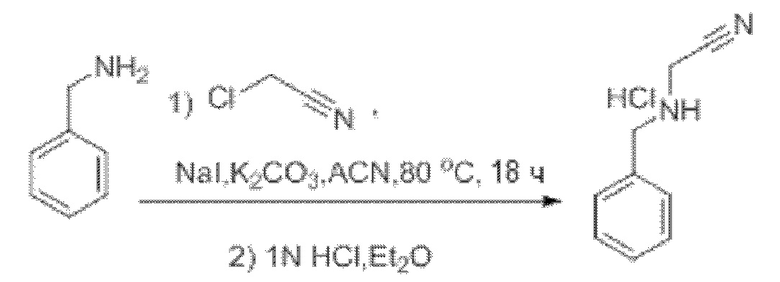
2-(Бензиламино)ацетонитрил гидрохлорид Смесь бензиламина (40 г, 291,58 ммоль, 1 эквив.), 2-хлорацетонитрила (26,4 г, 349,9 ммоль, 1,2 эквив.), NaI (4,37 г, 29,16 ммоль, 0,1 эквив.) и K2CO3 (52,4 г, 379,06 ммоль, 1,3 эквив.) в CH3CN (200 мл) перемешивали в течение 18 ч при 80°С. Реакцию контролировали с помощью ЖХМС. Смесь оставляли охлаждаться до кт. Полученную смесь фильтровали; осадок на фильтре промывали CH3CN (3x150 мл). Фильтрат концентрировали при пониженном давлении. Полученную смесь разбавляли Et2O (200 мл). Затем к раствору добавляли HCl (2 М, 100 мл). Полученное твердое вещество собирали фильтрованием и промывали Et2O (3 х 70 мл), получая указанное в заглавии соединение (34 г, 66,17%) в виде твердого вещества серого цвета.
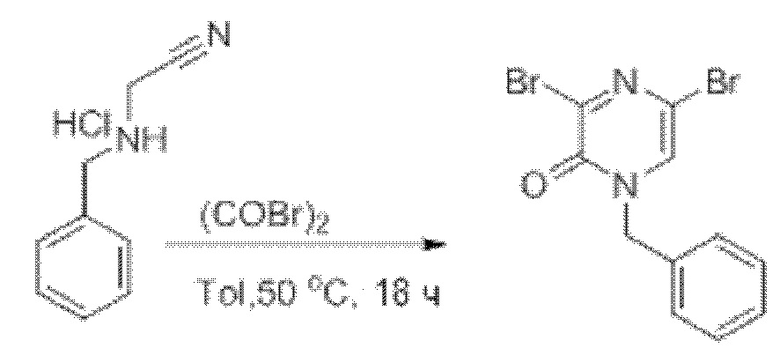
1-бензил-3,5-дибромпиразин-2(1H)-он В круглодонную колбу на 500 мл, содержащую толуол (200 мл), по каплям добавляли дибромид щавелевой кислоты (100 г, 462,40 ммоль, 3,0 эквив.) в течение 30 мин при кт. Полученную смесь перемешивали еще в течение 15 мин при такой же температуре. К перемешанному раствору порциями добавляли продукт, полученный на предыдущей стадии, (32,8 г, 154,22 ммоль, 1 эквив.) при кт. Полученную смесь перемешивали в течение 18 ч при 50°С. Реакцию контролировали с помощью ЖХМС. Смесь оставляли охлаждаться до кт. Полученную смесь разбавляли нас. NaH2PO4 (500 мл). Водный слой экстрагировали EtOAc (4 х 200 мл). Объединенные органические слои промывали 500 мл соляного раствора и сушили над Na2SO4. Полученную смесь фильтровали; осадок на фильтре промывали EtOAc (2 х 200 мл). Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, используя РЕ / EtOAc (5:1), получая указанное в заглавии соединение (6 г, 10%) в виде масла темно-желтого цвета.
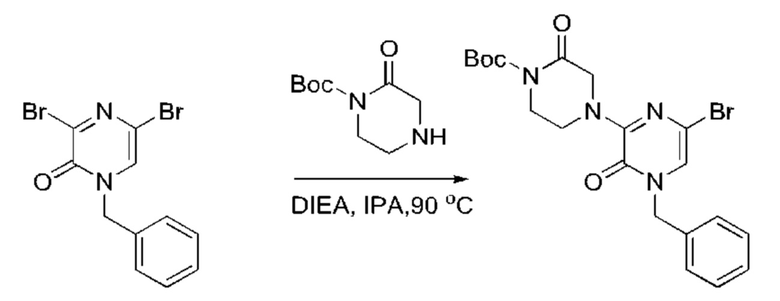
трет-Бутил 4-(4-бензил-6-бром-3-оксо-3,4-дигидропиразин-2-ил)-2-оксопиперазин-1-карбоксилат Методику получения Промежуточного соединения 4-1 использовали с продуктом, полученным на предыдущей стадии, (5 г, 14,5 ммоль, 1,00 эквив.) и дареда-бутил 2-оксопиперазин-1-карбоксилатом (4,36 г, 21,8 ммоль, 1,50 эквив.), получая 3,5 г (88%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
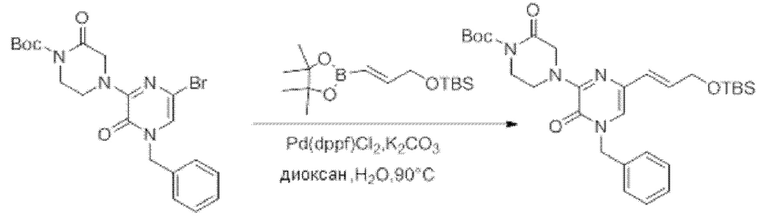
трет-Бутил (Е)-4-(4-бензил-6-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-3-оксо-3,4-дигидропиразин-2-ил)-2-оксопиперазин-1-карбоксилат Методику получения Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (3,5 г, 11,14 ммоль, 1,00 эквив.) и дареда-бутилдиметил[([2Е]-3-[тетраметил-1,3,2-диоксаборолан-2-ил]проп-2-ен-1-ил)окси]силаном (3,3 г, 14,60 ммоль, 1,30 эквив.), с временем реакции 1 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 2,3 г (24%) указанного в заглавии соединения в виде масла оранжевого цвета.
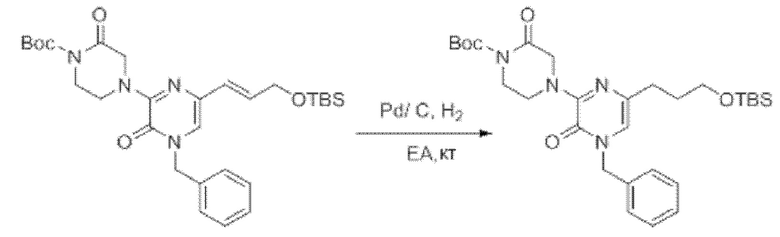
трет-Бутил 4-(4-бензил-6-(3-((трет-бутилдиметилсилил)окси)пропил)-3-оксо-3,4-дигидропиразин-2-ил)-2-оксопиперазин-1-карбоксилат Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (2,3 г, 1,78 ммоль, 1,00 эквив.), получая 2 г (80%) указанного в заглавии соединения в виде масла светло-желтого цвета.
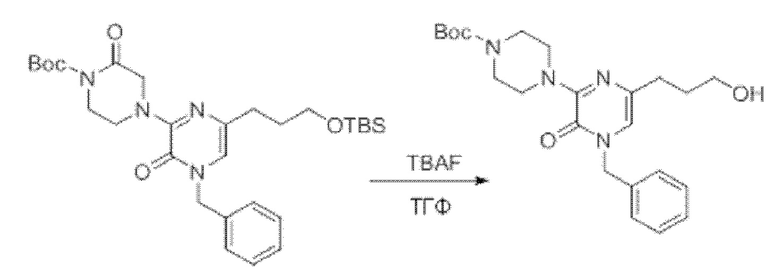
трет-Бутил 4-(4-бензил-6-(3-гидроксипропил)-3-оксо-3,4-дигидропиразин-2-ил)пиперазин-1-карбоксилат Методику получения Промежуточного соединения 22-4 использовали с продуктом, полученным на предыдущей стадии, (2 г, 1,42 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:2), получая 1,1 г (70%) указанного в заглавии соединения в виде масла желтого цвета.
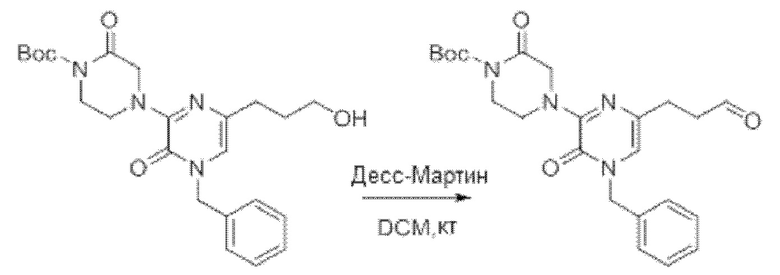
трет-Бутил 4-(4-бензил-3-оксо-6-(3-оксопропил)-3,4-дигидропиразин-2-ил)-2-оксопиперазин-1-карбоксилат Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (800 мг, 1,00 ммоль, 1,00 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (2:1), получая 400 мг (71%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
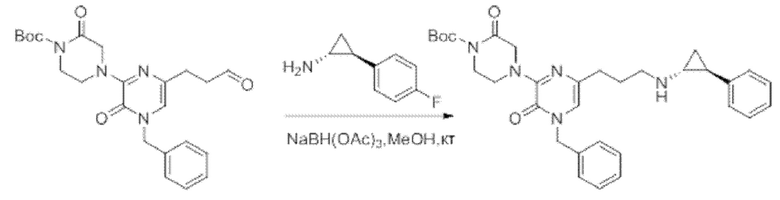
трет-Бутил 4-(4-бензил-3-оксо-6-(3-(((1R,2S)-2-фенилциклопропил)амино)-пропил)-3,4-дигидропиразин-2-ил)-2-оксопиперазин-1-карбоксилат Стадию восстановительного аминирования для получения Примера 1 из Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (400 мг, 0,71 ммоль, 1,00 эквив.) и (1R,2S)-2-(4-фторфенил)циклопропан-1-амином (162 мг, 1,07 ммоль, 1,50 эквив.). Неочищенный продукт очищали с помощью препаративной ТСХ с EtOAc, получая 310 мг (22%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
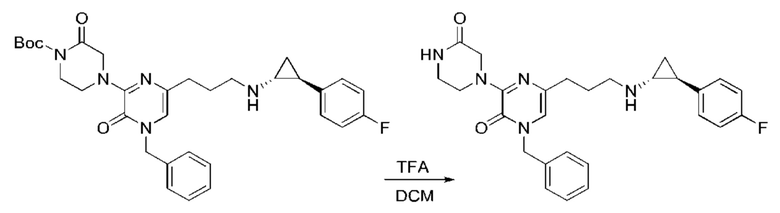
1-Бензил-5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-3-(3-оксопиперазин-1-ил)пиразин-2(1H)-он Стадию удаления защитных групп для получения Примера 14 из Промежуточного соединения 14-7 использовали с продуктом, полученным на предыдущей стадии, (100 мг, 0,71 ммоль, 1,00 эквив.). Неочищенный продукт очищали, используя хроматографический Метод F (от 22% до 32% CH3CN), получая 35,6 мг (22%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
ЖХ-МС: (ES,m/z): 476[М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 7,32-7,30 (м, 5Н), 7,29-6,99 (м, 4Н), 6,94 (с, 1H, 5,10 (с, 2Н), 4,22 (с, 2Н), 4,13-4,09 (т, J=5,4 Гц, 2Н), 3,42-3,40 (т, J=5,4 Гц, 2Н), 3,24-3,18 (м, 2Н), 2,96-2,93 (м, 1H, 2,53-2,50 (т, J=7,2Hz, 2Н), 2,50-2,37 (м, 1H, 2,05-2,0 (м, 2Н), 1,47-1,34 (м, 2Н).
ПРИМЕР 47
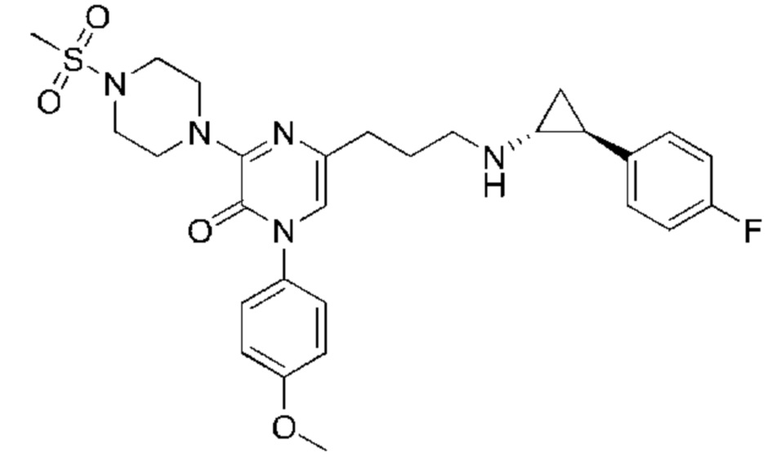
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-1-[4-метоксифенил]-3-[4-(метилсульфонил)пиперазин-1-ил]пиразин-2(1H)-он
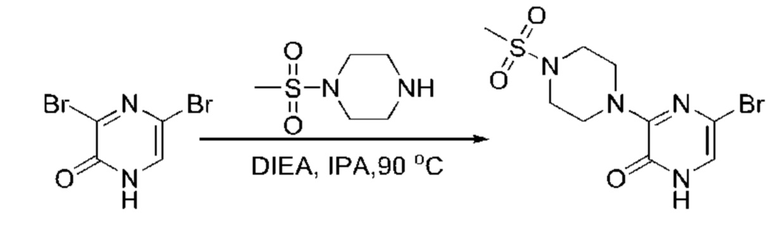
5-Бром-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 4-1 использовали с 3,5-дибром-1,2-дигидропиразин-2-оном (20 г, 78,8 ммоль, 1 эквив.), 1-метансульфонилпиперазином (15,6 г, 94,54 ммоль, 1,2 эквив.), получая указанное в заглавии соединение (16 г, 60%) в виде твердого вещества темно-желтого цвета.
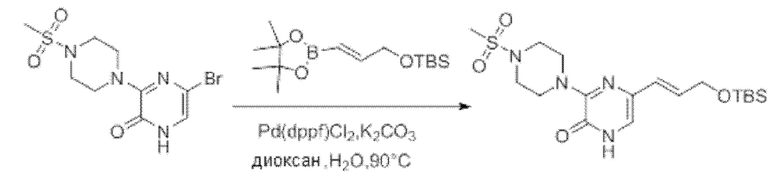
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения
Промежуточного соединения 3-3 использовали с продуктом, полученным на предыдущей стадии, (8 г, 23,73 ммоль), с временем реакции 1 ч при 90°С. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя РЕ / EtOAc (1:1), получая указанное в заглавии соединение (4,26 г, 41,9%) в виде масла желтого цвета.
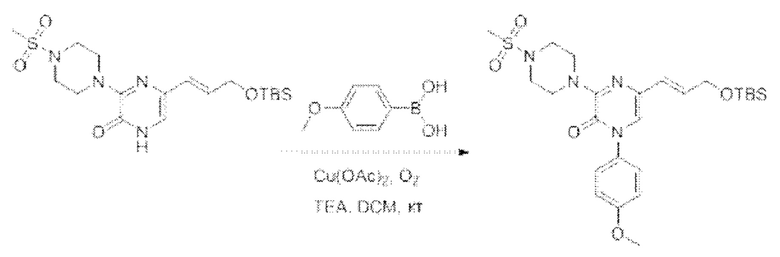
(E)-5-(3-((трет-бутилдиметилсилил)окси)проп-1-ен-1-ил)-1-(4-метоксифенил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он
Методику получения Промежуточного соединения 8-9 использовали с продуктом, полученным на предыдущей стадии, (3,5 г, 8,17 ммоль, 1 эквив.) и (4-метоксифенил)бороновой кислотой (1,5 г, 9,80 ммоль, 1,2 эквив.). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя РЕ/EtOAc (1:1), получая указанное в заглавии соединение (2,1 г, 48%) в виде твердого вещества желтого цвета.
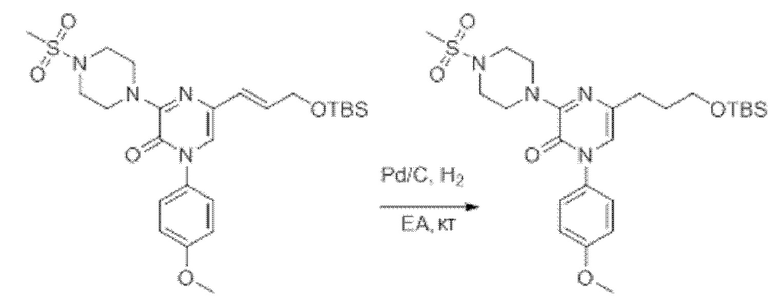
5-(3-((трет-Бутилдиметилсилил)окси)пропил)-1-(4-метоксифенил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1Н)-он Методику получения Промежуточного соединения 3-4 использовали с продуктом, полученным на предыдущей стадии, (2,1 г, 3,92 ммоль, 1,0 эквив.), получая указанное в заглавии соединение (1,9 г, 95%) в виде твердого вещества грязно-белого цвета.
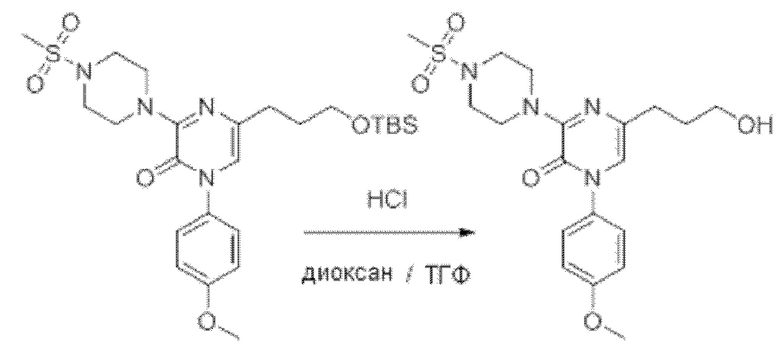
5-(3-гидроксипропил)-1-(4-метоксифенил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 33-5 использовали с продуктом, полученным на предыдущей стадии, (1,9 г, 3,54 ммоль, 1,0 эквив.), получая 1,06 г (71%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
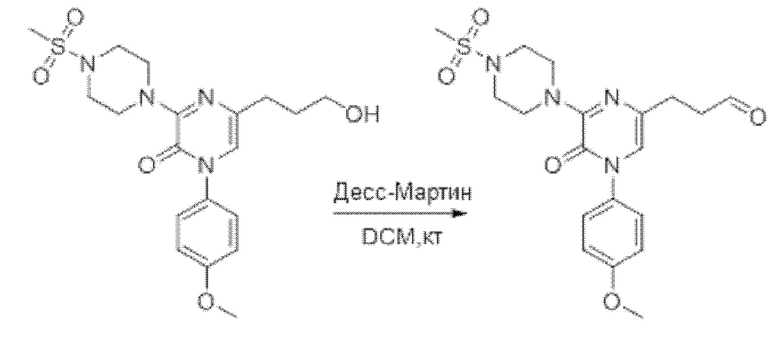
3-(4-(4-Метоксифенил)-6-(4-(метилсульфонил)пиперазин-1-ил)-5-оксо-4,5-дигидропиразин-2-ил)пропаналь Методику получения Промежуточного соединения 1-7 использовали с продуктом, полученным на предыдущей стадии, (500 мг, 1,18 ммоль). Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя EtOAc/петролейный эфир (1:1), получая 380 мг (76%) указанного в заглавии соединения в виде масла желтого цвета.
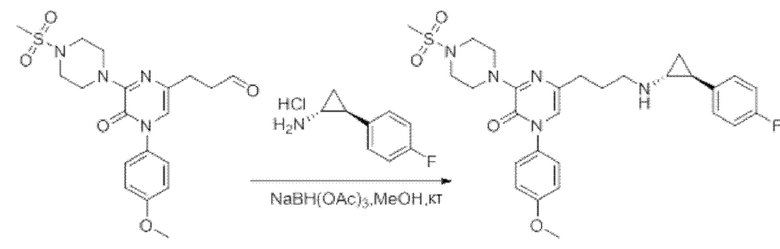
5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-1-(4-метокси-фенил)-3-(4-(метилсульфонил)пиперазин-1-ил)пиразин-2(1H)-он Методику получения Промежуточного соединения 4-7 использовали с продуктом, полученным на предыдущей стадии, (380 мг, 0,90 ммоль, 1 эквив.) и (1R,2S)-2-(4-фторфенил)-циклопропан-1 -амином (204,9 мг, 1,36 ммоль, 1,500 эквив.). Неочищенный продукт очищали, используя хроматографический Метод С (49% В за 8 мин), получая 66,7 мг (13,3%) указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
ЖХМС: (ES,m/z): 556 [М+Н]+. 1Н ЯМР (300 МГц, Метанол-d4) δ м.д.: 7,36-7,26 (д, J=2,1 Гц, 2Н), 7,13-6,91 (м, 6Н), 6,85 (с, 1H, 3,90-3,80 (м, 7Н), 3,30-3,20 (м, 4Н), 2,87 (с, 3H), 2,84-2,73 (м, 2Н), 2,49 (т, J=6,6 Гц, 2Н), 2,36-2,26 (м, 1H, 1,95-1,85 (м, 3H), 1,14-0,93 (м, 2Н).
ПРИМЕР 48
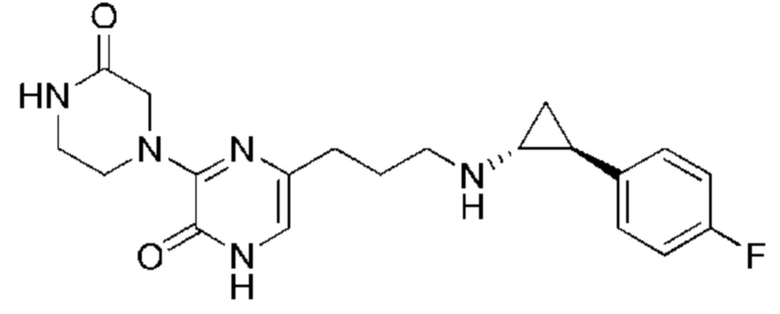
5-[3-([(1R,2S)-2-(4-фторфенил)циклопропил]амино)пропил]-3-[3-оксопиперазин-1-ил]пиразин-2(1H)-он
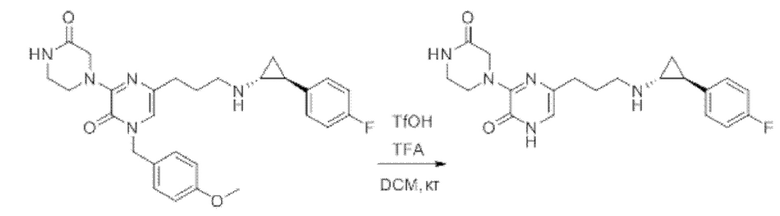
Раствор 5-(3-(((1R,2S)-2-(4-фторфенил)циклопропил)амино)пропил)-1-(4-метоксибензил)-3-(3-оксопиперазин-1-ил)пиразин-2(1H)-она (Пример 42, 400 мг, 0,79 ммоль, 1 эквив.), TFA(10 мл) и TfOH (5 мл) в CH2Cl2 (20 мл) перемешивали в течение 1 ч при 25°С. Полученную смесь концентрировали в вакууме. Остаток разбавляли нас. NaHCO3 (100 мл). Водный слой экстрагировали EtOAc (4x50 мл). Объединенные органические слои промывали 1x100 мл соляного раствора, сушили над Na2SO4 и очищали используя хроматографический Метод С (от 25% до 35% CH3CN за 7 мин), получая 78,5 мг (33%) указанного в заглавии соединения в виде твердого вещества белого цвета.
ЖХ-МС: (ES,m/z): 386 [М+Н]+. 1Н ЯМР (300 МГц, MeOD-d4) δ м.д.: 7,09-6,93 (м, 4Н), 6,69-6,66 (м, 1H, 4,36 (с, 2Н), 4,12-4,05 (м, 2Н), 3,43-3,42 (т, J=5,4 Гц, 2Н), 2,76-2,71 (т, J=7,5 Гц, 2Н), 2,49-2,44 (т, J=7,3 Гц, 2Н), 2,32-2,27 (м, 1H, 1,94-1,82 (м, 3H), 1,09-0,95 (м, 2Н).
Активность соединений из Примеров 1-32 в качестве ингибиторов KDM1A проиллюстрирована в следующем анализе. Предполагается, что другие перечисленные выше соединения, которые еще не были получены и/или протестированы, также обладают активностью в этом анализе.
Анализ биологической активности
Активность приведенных выше Примеров может быть проиллюстрирована в следующих анализах. Предполагается, что перечисленные выше соединения, которые, возможно, еще не были получены и/или протестированы, обладают активностью в этих анализах.
Анализ ингибирования KDM1A можно определить in vitro, в культивируемых клетках и у животных. Существует множество спектрофотометрических методов для обнаружения результатов деметилирования метилированных лизинов, а именно обнаружения продуктов окислительной активности деметилазы KDM1A на пептидном фрагменте, состоящем, по меньшей мере, из 18 аминокислот, представляющих N-конец субстрата гистона Н3, который содержит монометил на четвертом остатке лизина. Пероксид водорода, один из продуктов реакции деметилазы KDM1A, реагирует с пероксидазой хрена и дигидроксифеноксазином (ADHP) с образованием флуоресцентного соединения резоруфина (возбуждение=530-560 нм: излучение=590 нм). Активность фермента деметилазы KDM1A может быть получена из клеток или тканей млекопитающих, экспрессирующих KDM1A из эндогенного или рекомбинантного гена, и очищена или проанализирована из экстракта цельных клеток. Эти методы можно использовать для определения концентрации раскрытых соединений, способной ингибировать пятьдесят процентов активности фермента (IC50). В одном аспекте, раскрытые соединения демонстрируют пятидесятипроцентное ингибирование активности фермента KDM1A при концентрации менее 500 нМ, менее 100 нМ, менее 50 нМ или менее 10 нМ.
Связь KDM1A с другими белками может быть определена множеством способов как in vitro, так и in vivo, известных специалистам в данной области техники. Например, разрушение KDM1A ассоциированными белками можно определить с помощью анализа сдвига электромобильности (EMSA). В различных аспектах нарушение физической ассоциации KDM1A с CoRest описанными соединениями можно наблюдать с помощью EMSА. В другом примере, разрушение KDM1A ассоциированными белками может быть определено иммунопреципитацией с последующим разделением соосажденных белков с помощью масс-спектроскопии или электрофореза. В другом примере нарушение ассоциации KDM1A с CoRest может быть определено по способности KDM1A действовать на нуклеосомный субстрат, содержащий К4 или К9 метилированный гистон Н3, субстрат, который требует присутствия как KDM1 А, так и CoRest. Раскрытые соединения можно использовать для анализа ингибирования ассоциации CoRest с KDM1A с использованием нуклеосомного субстрата; такие соединения могут не ингибировать ферментативную активность KDM1A, как определено с использованием пептидного субстрата, К4 метилированного гистоном НЗ.
Ингибирование KDM1A можно определить с помощью клеточного анализа. Например, KDM1A является существенно важным ферментом, и продолжительное ингибирование KDM1A приведет к гибели клеток, таким образом, можно оценить ингибирование роста клеток, остановку роста клеток или гибель клеток. В другом аспекте, гены, индуцируемые андрогенами и эстрогенами, нуждаются в активности KDM1A; ингибирование описанными соединениями KDM1A подавляет индукцию экспрессии гена в клетках, обработанных андрогенами или эстрогенами. Эти эффекты могут быть измерены, например, с помощью количественной ПЦР мРНК для измерения величины экспрессии генов андроген- и эстроген-зависимых генов. Активность KDM1A необходима для подавления транскрипции определенных генов. Ингибирование KDM1A описанными соединениями может вновь активировать экспрессию таких генов в клетке. Эти гены включают Meisl, VEG-A, АIМ1, НМОХ1, VIM, SKAP1, BMP, EOMES, FOXA2, HNF4, SOX17, GH, PSA, pS2, GREB1, GR-1b, PRL, TSHB, SYN1, HBG, SCN1A, SCN2a и SCN3A, экспрессия которых может быть проанализирована с использованием количественной ПЦР мРНК до и в различное время после обработки клеток описанными соединениями. В другом аспекте, KDM1A является регулятором потенциала лейкозных стволовых клеток и необходима для онкогенной трансформации миелоидных клеток в острый миелоидный лейкоз (AML) с помощью MLL-AF9. Ингибирование KDM1A в клетках, трансформированных MLL-AF9, выращенных в культуре, преодолевает задержку дифференцировки и приводит к более зрелой клетке, экспрессирующей поверхностный антиген CD11b, антиген моноцитарных клеток. Таким образом, ингибирование KDM1A может быть проанализировано с использованием линии клеток AML, такой как ТНР-1, выращенной в культуре, для количественной оценки доли клеток, вновь экспрессирующих антиген CD11b, с использованием активированной флуоресценцией сортировки клеток (FACS). Можно также использовать аналогичный анализ с использованием FACS для подсчета клеток, экспонирующих CD14 или CD86, каждый из которых характерен для более зрелых клеток линии макрофагов/моноцитов. Для этого анализа можно использовать другие линии клеток, полученные от пациентов с острым миелоидным лейкозом, такие как клетки MV4;11 или MOLM-13. Другие маркеры дифференцировки по линии макрофагов/моноцитов могут быть аналогичным образом проанализированы с помощью FACS, такие как CD14 и CD86. Другие линии клеток AML, такие как MPLM-13 или MV4;11, могут быть проанализированы на индукцию либо специфических генов, упомянутых выше, либо маркеров дифференцировки, а также роста клеток или апоптоза путем окрашивания аннексином V и подсчета FACS.
Селективность раскрытых соединений в отношении KDM1A может быть определена путем анализа IC50 раскрытых соединений для других FAD-зависимых аминооксидаз, таких как моноаминоксидаза А (МАО-А), моноаминоксидаза В (МАО-В), IL4I1, KDM1B или SMOX. Таким образом, раскрытое соединение буде ингибировать KDM1A с IC50, которая в 50 или 100, или 250, или 500 раз меньше, чем для МАО-А или МАО-В.
Дополнительные анализы на деметилазу
Анализ гистондеметилазы можно проводить, по существу, как описано в Shi, Y et al. Cell 199, 941-953 (2004). Вкратце, основные гистоны, гистоновые пептиды или нуклеосомы инкубируют с очищенной человеческой рекомбинантной KDM1A, в буфере для анализа 1 активности гистондеметилазы (HDM) (50 мМ Tris рН 8,5, 50 мМ KCl, 5 мМ MgCl, 0,5% BSA и 5% глицерин) от 30 минут до 4 часов при 37°С. Типичную реакцию проводят в 100 микролитрах, в которых в качестве субстратов используют либо 20 микрограммов очищенных основных гистонов, либо 3 микрограмма модифицированных гистоновых пептидов. В реакции используются различные количества KDM1A в диапазоне от 1 до 20 микрограммов вместе с, при необходимости, другими кофакторами, такими как FAD или CoREST, в зависимости от выбранного субстрата. Реакционную смесь анализируют с помощью SDS-PAGE и вестерн-блоттинга с использованием гистон-метил-специфических антител или с помощью анализа образования формальдегида для проверки удаления и превращения метальной группы в формальдегид, или с помощью масс-спектрометрии в случае пептидных субстратов для идентификации деметилированного гистонового пептида.
Основные гистоны (например, 4 мг) инкубируют с указанными количествами рекомбинантных белков или комплексов в буфере для анализа А гистондеметилазы (HDM) (50 мМ Tris рН 8,5, 50 мМ KCl, 5 мМ MgCl, 5% глицерин, 0,2 мМ фенилметилсульфонил фторид и 1 мМ дитиотреитол) в конечном объеме 10 мл в течение 12-16 ч при 37°С. Для нуклеосом (0,3 мг) или мононуклеосом (0,3 мг) можно использовать буфер A HDM, содержащий 0,1% NP40. Реакционная смесь затем может быть проанализирована с помощью SDS-PAGE с последующим вестерн-блоттингом. Антитела против моно- или ди-метил К4 в гистоне Н3 и ацетил-К9/К14 гистона Н3 используют для определения степени метилирования и ацетилирования соответственно. Вестерн-блоттинг затем количественно определяют денситометрией или интенсивностью люминесценции.
В качестве альтернативы можно использовать стандартный флурогенный анализ, в котором гистон-метилированный субстрат привязан ко дну 96-луночного планшета (или к гранулам, находящимся в планшете) с использованием биотина, конъюгированного с гистон-метилированным субстратом и стрепавидином (SA) на гранулах, или SA, прикрепленного к планшету для закрепления биотинилированного субстрата. После инкубации фермента KDM1A в буфере А гистондеметилазы гистон-деметиллированный субстрат может быть обнаружен с использованием антител, специфичных для деметилированного субстрата Н3К4, конъюгированного с фтором или каким-либо другим агентом, который может быть обнаружен. В одном из вариантов этого метода анализа может использоваться антитело, направленное против метилированной версии гистона, в котором количество субстрата определяется количественно до и после инкубации с ферментом. В еще одной версии подобного анализа будет использоваться система обнаружения с резонансным переносом энергии флуоресценции (FRET), в которой антитело, распознающее метилированную версию, конъюгировано или иным образом связано с объектом, например, с гранулой или большой молекулой-носителем, к которой прикреплен флуорофор (донор), а флуорофор (акцептор) связан с объектом, связанным с субстратом.
В качестве альтернативы продуцирование Н2О2 во время реакции KDM1A можно определить флуорометрически. В этой системе продуцирование Н2О2 определяется в буфере для анализа HDM после воздействия субстрата, кофактора и фермента с использованием ADHP (10-ацетил-3,7-дигидроксифеноксазин) в качестве флуорогенного субстрата для пероксидазы хрена (HRP). ADHP (также известный как реактив Amplex Red) является наиболее стабильным и чувствительным флуорогенным субстратом для HRP. Флуоресцентным продуктом является резоруфин. Чувствительность может составлять всего 10-15 М целевого белка. Сигнал считывают с помощью флуоресцентного микропланшет-ридера при длинах волн возбуждения и излучения 530-560 нм и 590 нм, соответственно.
Кроме того, реакция KDM1A может включать другие факторы, которые могут влиять на активность KDM1A. Такие факторы могут включать CoREST, комплексы NuRD, DNMT1, HDAC1, HDAC2 и HDAC3, например, как белки, которые, как известно, связываются с KDM1A или KDM1 А-содержащими комплексами. Могут быть проанализированы взаимодействия, которые влияют на любой аспект активности KDM1A, включая специфичность к матрице, субстрату, Km, Kcat или чувствительность к концентрациям FAD. Например, анализ взаимодействия между KDM1A и CoREST in vitro может быть выполнен с добавлением рекомбинантной KDM1A (например, 10 мг) и CoREST (например, 5 мг), смешанных и инкубированных в течение 1 ч при 4-8°С, фракционированных на гель-фильтрационной колонке Superdex 200 в буфере, содержащем 20 мМ трис-HCl рН 7,9, 500 мМ KCl, 10% глицерин, 0,2 мМ EDTA, 1 мМ дитиотреитол, 0,1% Nonidet Р40 и 0,2 мМ фенилметилсульфонил фторид, и затем анализировали окрашиванием серебром.
Для совместной иммунопреципитации мононуклеосом с KDM1A и CoREST нуклеосомы (1,5 мг) можно расщеплять микрококковой нуклеазой и инкубировать с рекомбинантной KDM1A (например, 1 мг), CoREST (например, 500 нг) или обоими белками в буфере A HDM, содержащем 0,1% NP40, в течение 1 ч при 4-8°С. Добавляют антитела, направленные против KDM1A или CoREST, прикрепленные к аффинной смоле, и после тщательной промывки буфером A HDM, содержащим 0,1% NP40, связанные белки элюируют промывочным буфером. Активность KDM1A можно оценить в элюате или концентрацию KDM1A можно определить с помощью количественного вестерн-блоттинга.
Соединения тестировали в анализе флуоресцентного связывания фермента на 10 доз в режиме IC50 с 3-кратным серийным разведением в двух экземплярах, начиная с 100 мкМ. Продуцирование FAD-зависимой Н2О2 в результате деметилазной активности LSD1 на 10 мкМ пептидном субстрате гистона Н3 (1-21) K4me2 измеряли путем связывания с HRP и Amplex Red с получением резоруфина (флуоресценция измерялась при Ex/Em=535/590 нм на En Vision, Perkin Elmer). Результаты приведены ниже в Таблице 1.
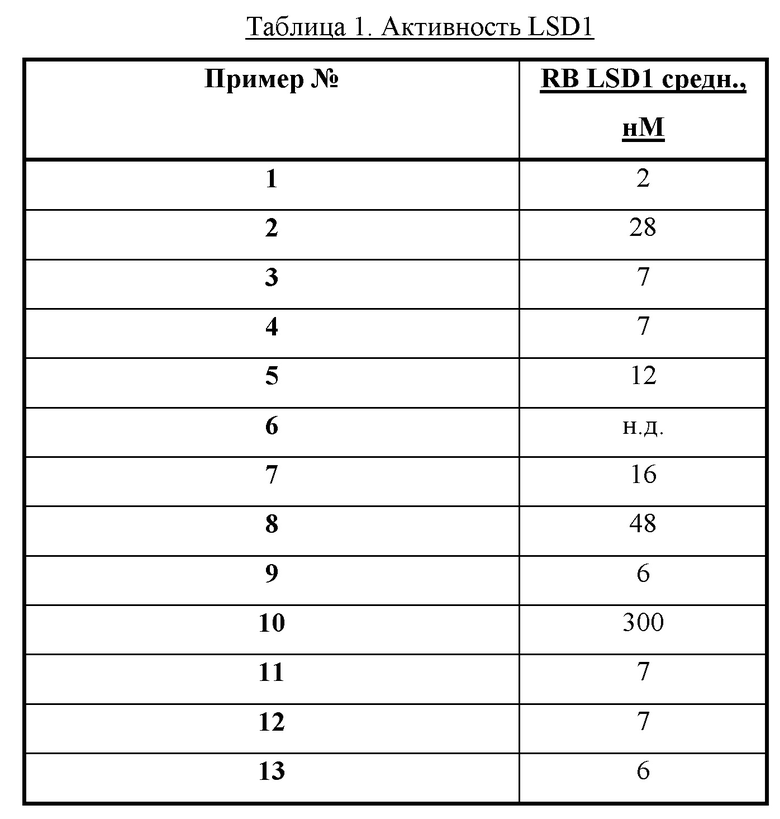
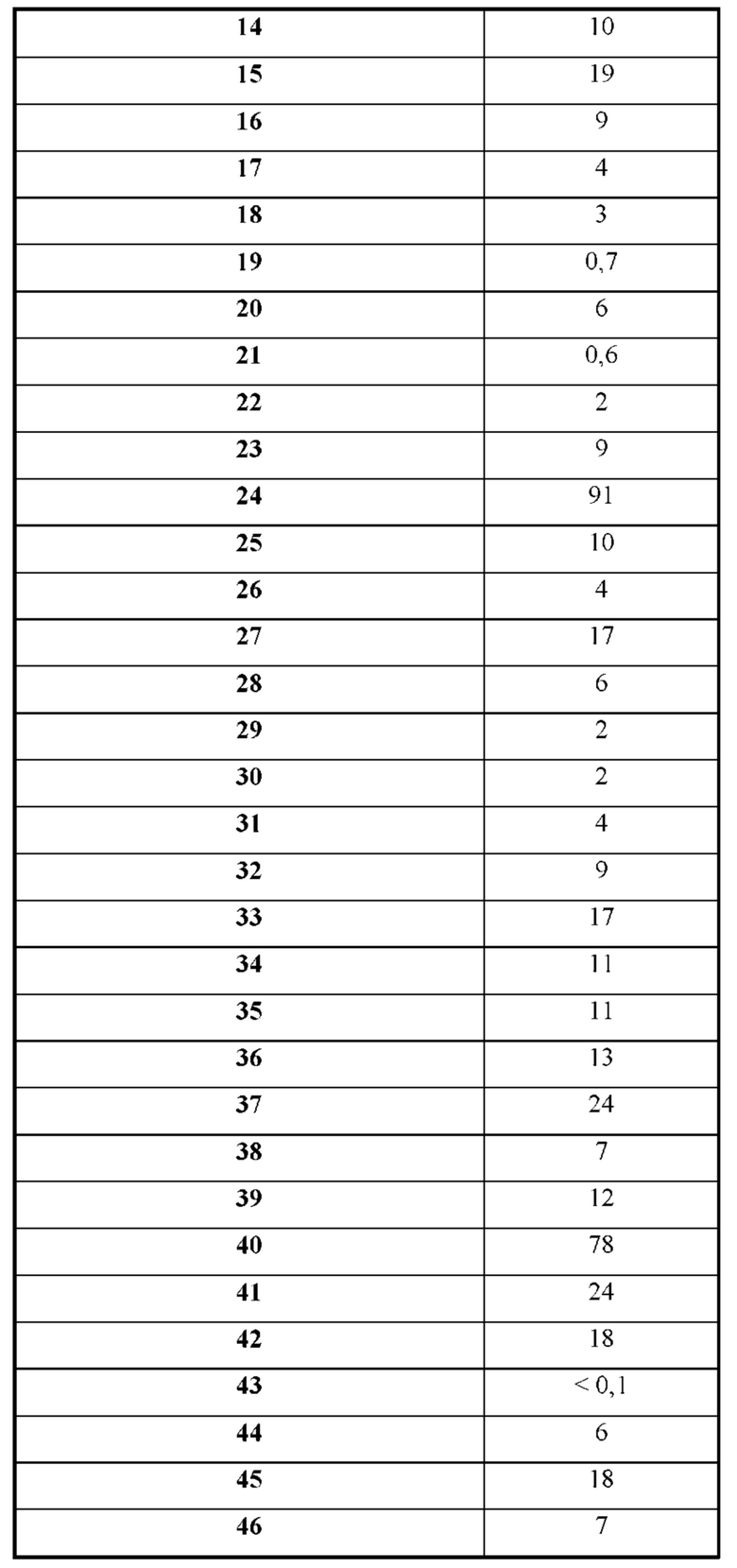

Все ссылки, патенты или заявки, американские или иностранные, процитированные в заявке, включены в нее посредством ссылки, как если бы они были написаны в данной заявке во всей их полноте. При возникновении каких-либо неточностей руководствуются материалом, буквально раскрытым в данной заявке.
Из вышеприведенного описания специалист в данной области техники может легко установить существенные характеристики настоящего изобретения и, не выходя за пределы его сущности и объема, может внести различные изменения и модификации изобретения, чтобы адаптировать его к различным применениям и условиям.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| ПРОИЗВОДНЫЕ 1-(4-ИЗОКСАЗОЛ-5-ИЛ)-1Н-ПИРАЗОЛ-1-ИЛ)-2-МЕТИЛПРОПАН-2-ОЛА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИРОРОВ ИЛ-17 И ИФН-ГАММА ДЛЯ ЛЕЧЕНИЯ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ И ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ | 2018 |
|
RU2785342C2 |
| Соединения циклопентана | 2019 |
|
RU2794997C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ПАРА-АМИНОБЕНЗИЛЬНЫЕ ЛИНКЕРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КОНЪЮГАТАХ | 2021 |
|
RU2839675C1 |
| PROTAC, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ТАУ-БЕЛОК, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2805523C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
Изобретение относится к соединению структурной формулы I или его фармацевтически приемлемой соли, которые могут найти применение для ингибирования активности лизинспецифической гистондеметилазы 1A (KDM1A) и лечения связанных с этой активностью заболеваний. В формуле I m выбран из 1, 2, 3 и 4; R1 представляет собой азотсодержащий гетероциклоалкил или гетероарил, каждый из которых необязательно замещен 1, 2 или 3 группами R5, где гетероциклоалкил относится к насыщенной моноциклической или бициклической 3-8-членной гетероциклической группе, содержащей от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота, кислорода и серы; и где гетероарил относится к ароматическому моноциклическому 5-6-членному гетероциклическому кольцу, которое содержит от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота; R2 представляет собой Н или выбран из C1-С6 алкила, С3-С7 циклоалкила, С6 арила и гетероарила, любой из которых необязательно замещен 1, 2 или 3 группами R6, где гетероарил относится к ароматическому моноциклическому 5-6-членному гетероциклическому кольцу, которое содержит от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота; R3 выбран из С6 арила, который необязательно замещен 1, 2 или 3 группами R7; каждый R4 представляет собой водород; каждый R5 независимо выбран из C1-С6 алкила, оксо, CONR8R9, COOR8 и SO2R8; каждый R6 независимо выбран из галогена, С6 галогенарила, C1-С6 алкокси-С6 арила, С6 арила, гетероарила, циано, C1-С6 алкокси, SO2R8 и SO2NR8R9, где гетероарил относится к ароматическому моноциклическому 5-6-членному гетероциклическому кольцу, которое содержит от 1 до 3 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота; каждый R7 независимо выбран из галогена и каждый R8 и R9 независимо выбран из водорода и C1-С6 алкила. Изобретение относится также к фармацевтической композиции, содержащей указанное соединение. 2 н. и 30 з.п. ф-лы, 1 табл., 48 пр.
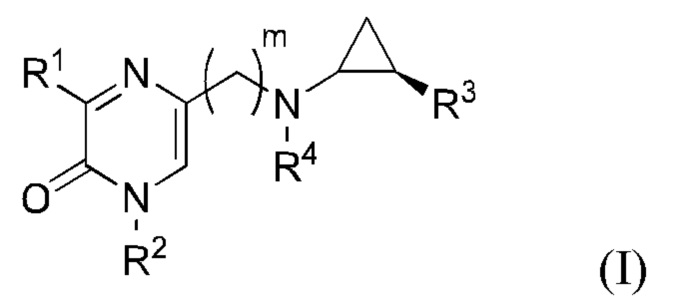
1. Соединение структурной формулы I
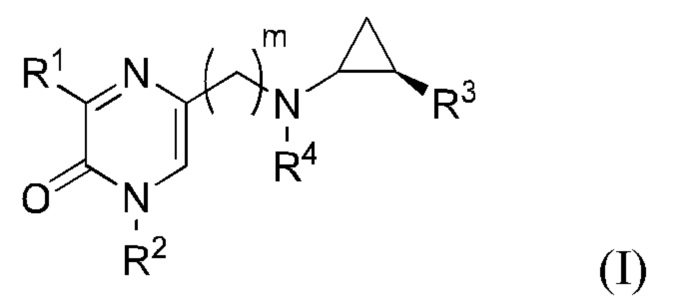
или его фармацевтически приемлемая соль, в котором
m выбран из 1, 2, 3 и 4;
R1 представляет собой азотсодержащий гетероциклоалкил или гетероарил, каждый из которых необязательно замещен 1, 2 или 3 группами R5, где гетероциклоалкил относится к насыщенной моноциклической или бициклической 3-8-членной гетероциклической группе, содержащей от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота, кислорода и серы; и где гетероарил относится к ароматическому моноциклическому 5-6-членному гетероциклическому кольцу, которое содержит от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота;
R2 представляет собой Н или выбран из C1-С6 алкила, С3-С7 циклоалкила, С6 арила и гетероарила, любой из которых необязательно замещен 1, 2 или 3 группами R6, где гетероарил относится к ароматическому моноциклическому 5-6-членному гетероциклическому кольцу, которое содержит от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота;
R3 выбран из С6 арила, который необязательно замещен 1, 2 или 3 группами R7;
каждый R4 представляет собой водород;
каждый R5 независимо выбран из C1-С6 алкила, оксо, CONR8R9, COOR8 и SO2R8;
каждый R6 независимо выбран из галогена, С6 галогенарила, C1-С6 алкокси-С6 арила, С6 арила, гетероарила, циано, C1-С6 алкокси, SO2R8 и SO2NR8R9, где гетероарил относится к ароматическому моноциклическому 5-6-членному гетероциклическому кольцу, которое содержит от 1 до 3 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота;
каждый R7 независимо выбран из галогена и
каждый R8 и R9 независимо выбран из водорода и C1-С6 алкила.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, в котором R3 представляет собой фенил и необязательно замещен 1 или 2 группами R7.
3. Соединение по п. 2 или его фармацевтически приемлемая соль, в котором каждый R7 представляет собой фтор.
4. Соединение по п. 1, имеющее структурную формулу II
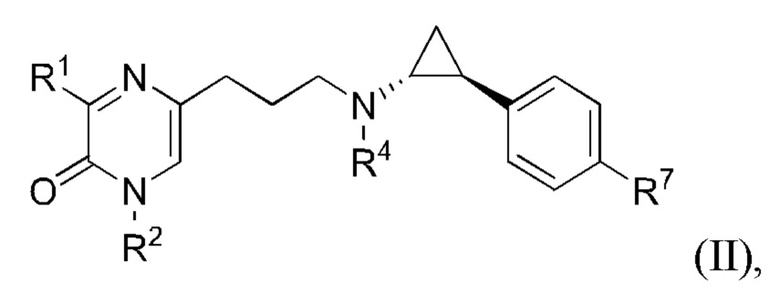
или его фармацевтически приемлемая соль, в котором
R1 представляет собой азотсодержащий гетероциклоалкил или гетероарил, каждый из которых необязательно замещен 1, 2 или 3 группами R5, где гетероциклоалкил относится к насыщенной моноциклической или бициклической 3-8-членной гетероциклической группе, содержащей от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота, кислорода и серы; и где гетероарил относится к ароматическому моноциклическому 5-6-членному гетероциклическому кольцу, которое содержит от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота;
R2 представляет собой Н или выбран из C1-С6 алкила, С3-С7 циклоалкила, С6 арила и гетероарила, любой из которых необязательно замещен 1, 2 или 3 группами R6, где гетероарил относится к ароматическому моноциклическому 5-6-членному гетероциклическому кольцу, которое содержит от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота;
каждый R4 представляет собой водород;
каждый R5 независимо выбран из C1-С6 алкила, оксо, CONR8R9, COOR8 и SO2R8;
каждый R6 независимо выбран из галогена, С6 галогенарила, C1-С6 алкокси-С6 арила, С6 арила, гетероарила, циано, C1-С6 алкокси и SO2R;
каждый R7 независимо выбран из галогена и
каждый R8 и R9 независимо выбран из водорода и C1-С6 алкила.
5. Соединение по п. 4 или его фармацевтически приемлемая соль, в котором R1 представляет собой азотсодержащий гетероциклоалкил, который необязательно замещен 1, 2 или 3 группами R5, где гетероциклоалкил относится к насыщенной моноциклической или бициклической 3-8-членной гетероциклической группе, содержащей от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота, кислорода и серы.
6. Соединение по п. 5 или его фармацевтически приемлемая соль, в котором R1 выбран из
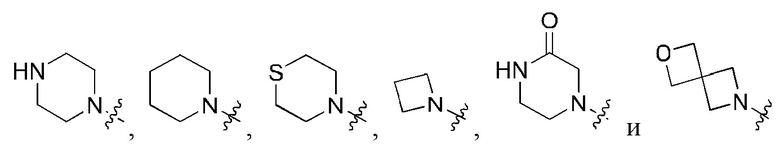
и необязательно замещен 1, 2 или 3 группами R5.
7. Соединение по п. 5 или его фармацевтически приемлемая соль, в котором R1 с заместителем R5, где это приемлемо, и дополнительно с заместителями R8 и R9, где это приемлемо, выбран из
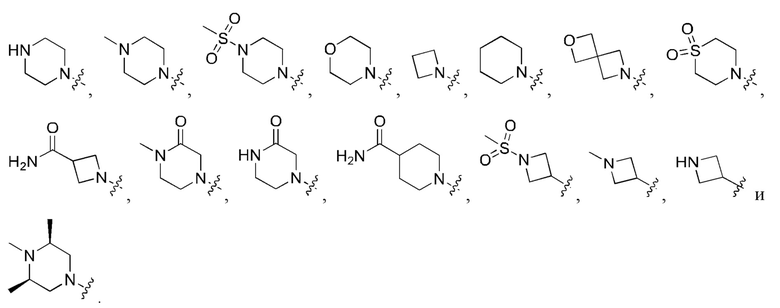
8. Соединение по п. 5 или его фармацевтически приемлемая соль, в котором R1 с заместителем R5, где это приемлемо, и дополнительно с заместителями R8 и R9, где это приемлемо, выбран из
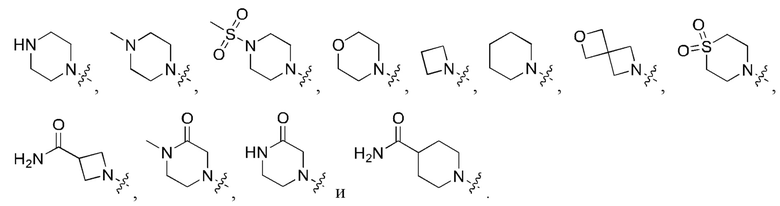
9. Соединение по п. 5 или его фармацевтически приемлемая соль, в котором R1 с заместителем R5, где это приемлемо, и дополнительно с заместителями R8 и R9, где это приемлемо, выбран из
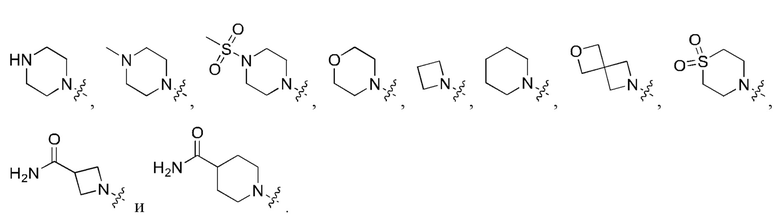
10. Соединение по п. 4 или его фармацевтически приемлемая соль, в котором R1 представляет собой азотсодержащий гетероарил, который необязательно замещен 1, 2 или 3 группами R5, где гетероарил относится к ароматическому моноциклическому 5-6-членному гетероциклическому кольцу, которое содержит от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота.
11. Соединение по п. 10 или его фармацевтически приемлемая соль, в котором R1 выбран из
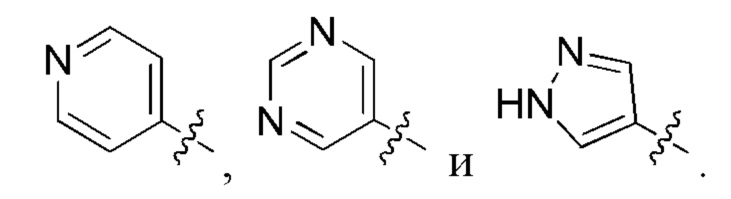
12. Соединение по п. 4 или его фармацевтически приемлемая соль, в котором R2 представляет собой Н.
13. Соединение по п. 4 или его фармацевтически приемлемая соль, в котором R2 выбран из арила и гетероарила, каждый из которых необязательно замещен 1 или 2 группами R6, где гетероарил относится к ароматическому моноциклическому 5-6-членному гетероциклическому кольцу, которое содержит от 1 до 2 гетероатомов в качестве кольцевых членов, при этом каждый указанный гетероатом может быть независимо выбран из азота.
14. Соединение по п. 13 или его фармацевтически приемлемая соль, в котором каждый R6 независимо выбран из галогена, гетероарила, циано и SO2R8.
15. Соединение по п. 14 или его фармацевтически приемлемая соль, в котором каждый R6 независимо выбран из галогена, гетероарила и SO2R8.
16. Соединение по п. 15 или его фармацевтически приемлемая соль, в котором R2 выбран из фенила, пирролила, пиразолила, имидазолила, пиридинила, пиразинила, пиридазинила и пиримидинила, любой из которых необязательно замещен 1 или 2 группами R6.
17. Соединение по п. 16 или его фармацевтически приемлемая соль, в котором R2 выбран из фенила, пиридинила и пиримидинила, любой из которых необязательно замещен 1 или 2 группами R6.
18. Соединение по п. 17 или его фармацевтически приемлемая соль, в котором R2 представляет собой фенил, замещенный одной группой R6, и R6 представляет собой гетероарил.
19. Соединение по п. 13 или его фармацевтически приемлемая соль, в котором R6 представляет собой гетероарил, выбранный из триазолила, пиримидинила и пиридазинила.
20. Соединение по п. 13 или его фармацевтически приемлемая соль, в котором каждый R6 независимо выбран из галогена, циано, C1-С6 алкокси и C1-С6 алкокси-С6 арила.
21. Соединение по п. 13 или его фармацевтически приемлемая соль, в котором R1 выбран из пиперидина, морфолина, тиоморфолина, пиперазина, пирролидина, азетидина, 2-азаспиро[3,3]гептана, 2,6-диазаспиро[3,3]гептана и 2-окса-6-азаспиро[3,3]гептана и необязательно замещен 1, 2 или 3 группами R5.
22. Соединение по п. 21 или его фармацевтически приемлемая соль, в котором R1 выбран из
и необязательно замещен 1, 2 или 3 группами R5.
23. Соединение по п. 13 или его фармацевтически приемлемая соль, в котором R1 с заместителем R5, где это приемлемо, и дополнительно с заместителями R8 и R9, где это приемлемо, выбран из
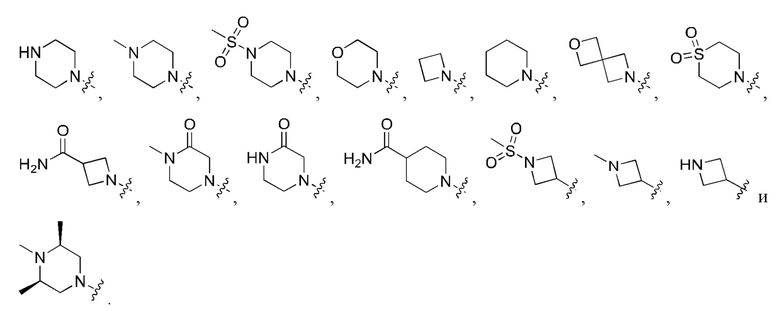
24. Соединение по п. 13 или его фармацевтически приемлемая соль, в котором R1 с заместителем R5, где это приемлемо, и дополнительно с заместителями R8 и R9, где это приемлемо, выбран из
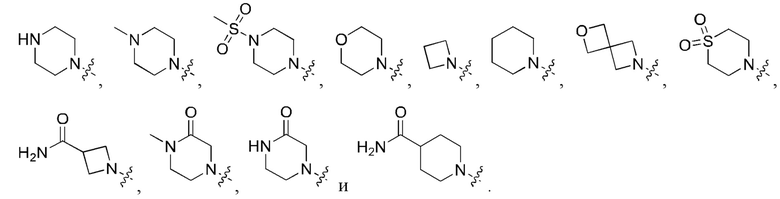
25. Соединение по п. 13 или его фармацевтически приемлемая соль, в котором R1 с заместителем R5, где это приемлемо, и дополнительно с заместителями R8 и R9, где это приемлемо, выбран из
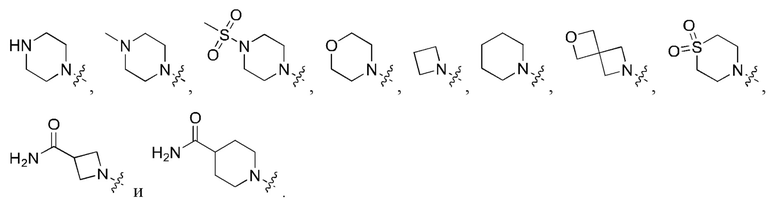
26. Соединение по п. 1 или его фармацевтически приемлемая соль, причем структура выбрана из
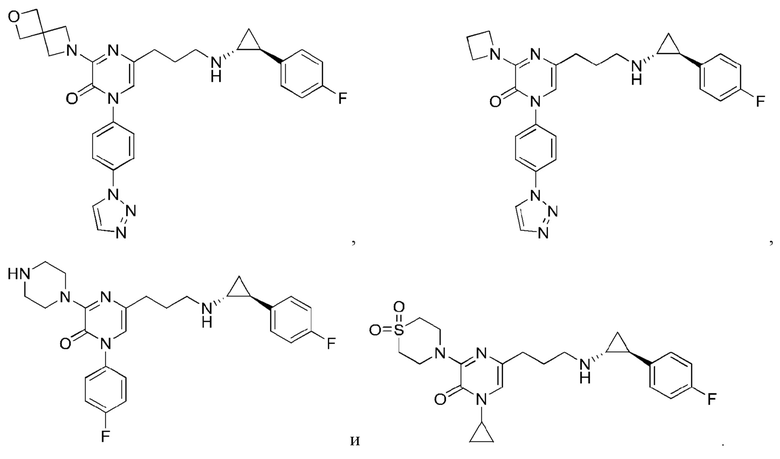
27. Соединение по п. 1 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства.
28. Соединение по п. 1 или его фармацевтически приемлемая соль для применения при лечении заболевания, опосредованного KDM1A.
29. Соединение по п. 1 или его фармацевтически приемлемая соль для применения в производстве лекарственного средства для предотвращения или лечения заболевания или состояния, улучшаемого ингибированием KDM1A.
30. Фармацевтическая композиция для ингибирования активности KDM1A, содержащая терапевтически эффективное количество соединения по п. 1 или его фармацевтически приемлемой соли вместе с фармацевтически приемлемым носителем.
31. Фармацевтическая композиция по п. 30, составленная для перорального введения.
32. Фармацевтическая композиция по п. 30, дополнительно содержащая другой терапевтический агент.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| ИНГИБИТОРЫ ДЕМЕТИЛАЗЫ LSD1 НА ОСНОВЕ АРИЛЦИКЛОПРОПИЛАМИНА И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2011 |
|
RU2611437C2 |

