Изобретение относится к новым соединениям, левовращающему и правовращающему, оптически чистым энантиомерам 1-[(4-хлорфенил)фенилметил] -4-[(метилфенил)сульфонил]-пиперазина формулы I: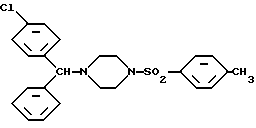
к способу получения этих соединений, также к их использованию для получения левовращающего и правовращающего, оптически чистых энантиомеров 1-[(4-хлорфенил)фенилметил] -пиперизина. Эти последние соединения представляют собой ценные промежуточные продукты для получения терапевтически активных соединений в лево- и правовращающей формах, оптически чистых.
Эти терапевтически активные соединения могут быть использованы для лечения астмы, аллергий, воспаления, беспокойства и в качестве седативных или транквиллизирующих агентов. Эти соединения проявляют высокую периферическую и/или центральную антигистаминную активность, которая лежит в основе их использования в качестве медикаментов.
Хорошо известно, что на биологические свойства многочисленных соединений, например лечебных, гормонов, гербицидов, инсектицидов или подслащивающих веществ, влияют стереохимические факторы. На фактор зависимости между оптической активностью и биологическими свойствами было обращено внимание в работе /A.R. GUSHNY, Biological Relations of Optically Isоmeric Substances, Williams and Williams Co., Baltimore, 1926/. С того времени многочисленные примеры подтверждают тот факт, что рацемическое соединение и его лево- и правовращающие энантиомеры проявляют себя фармакологически совершенно различно. Оптическая активность, которая является отражением асимметрической структуры органического соединения, представляет собой один из важных факторов, которые модулируют фармакологическую активность этого соединения и его биологический ответ. В самом деле, в зависимости от использования лево- или правовращающей формы соединения с фармакологической активностью могут появляться глубокие модификации свойств соединения, такие, как его транспортировка, его распределение в организме или его удаление. Эти свойства являются решающими для концентрации медикамента в организме или времени его выдержки на месте воздействия. Более того, фармакологическая активность обоих изомеров может быть значительно различной. Например, один из оптически активных изомеров может быть отчетливо более активным, чем другой, и, в конечном счете, этот изомер может обладать один всей фармакологической активностью; причем другой изомер является полностью неактивным и тогда играет роль простого разбавителя. Также может случиться, что фармакологические активности обоих изомеров различны и тогда два соединения обладают терапевтически различными свойствами. Кроме того, метаболизм и токсичность изомеров могут сильно различаться один от другого в такой степени, что один из оптически активных изомеров может быть более токсичен, чем другой. Одним из наиболее удивительных примеров в этой области является пример талидомида, оба энантиомера которого обладают подобными гипнотическими эффектами, но из которых один лишь энантиомер S проявляет тератогенные эффекты.
Наконец, оптические изомеры представляют собой особо ценные зонды для изучения химических взаимодействий с физиологическими механизмами (например, селективность фиксации на рецепторе).
На этом основании многие фармацевтические исследования направлены на выделение и синтез энантиомеров фармакологически активного соединения и для изучения их терапевтических свойств.
В патенте Великобритании 2 225 321 описывается способ получения энантиомеров дихлоргидрата 2-[2-{4[(4-хлорфенил)фенилметил]-1- пиперазинил}этокси] -уксусной кислоты, известного в качестве антигистаминного, не седативного медикамента под общим названием Цетиризин. Этот способ основан на использовании лево- или правовращающего 1-[(4-хлорфенил)фенилметил]пиперазина в качестве исходного продукта. В этом патенте энантиомеры 1-[(4-хлорфенил)фенилметил]пиперазина получают путем химического расщепления рацемической формы известными способами, в особенности путем образования соли с надлежащим образом выбранным оптическим изомером винной кислоты.
Основными недостатками этого способа являются, во-первых, крайне незначительный (только 12,7%) выход стадии расщепления рацемического 1-[(4-хлорфенил) фенилметил] пиперазина и, с другой стороны, оптическая чистота таким образом полученных лево- и правовращающего анантиомеров недостаточна и не позволяет получать целевой продукт с оптической чистотой выше 95%.
В соответствии с этим необходимо располагать новыми путями получения энантиомеров 1-[(4-хлорфенил)фенилметил] пиперазина с улучшенной оптической чистотой и с лучшими выходами, чтобы располагать отличными исходными продуктами для получения оптически активных изомеров для лечебных целей, обладающих также очень большой оптической чистотой.
Для достижения этой цели необходимо найти предшественники, которые бы обладали точной стереохимической конфигурацией и которые, с одной стороны, могли бы быть получены легко и экономично с достаточной оптической чистотой и, с другой стороны, легко бы превращались с высоким выходом в по существу оптически чистые энантиомеры 1-[(4-хлорфенил)фенилметил]пиперазина.
Эта цель достигается с помощью новых соединений, в частности 1-[(4-хлорфенил)фенилметил] -4-[(4-метилфенил)сульфонил] -пиперазина, в форме лево- и правовращающего энантиомеров.
Изобретение, следовательно, относится к лево- и правовращающему энантиомеру 1-[(4-хлорфенил)фенилметил]-4-[(4-метилфенил)сульфонил]пиперазина формулы I: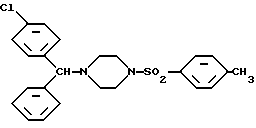
Согласно настоящему изобретению энантиомеры соединения формулы I предпочтительно находятся по существу в оптически чистой форме.
В настоящем описании под выражением "по существу оптически чистый" понимают оптическую чистоту выше 98% и эта оптическая чистота соответствует избытку, выражаемому в процентах, оптически активного изомера, имеющегося в большем количестве по отношению к оптически активному изомеру, имеющемуся в меньшем количестве, и определяется с помощью высокоэффективной жидкостной хроматографии /ВЭЖХ/ на стационарной хиральной фазе; она может быть определена по уравнению, описанному на с. 107 работы J. MARCH" Advanced Organic Chemistry", John Wiley and Sons, 1 пс, Нью-Йорк, 3-е издание, 1985:
Оптическая чистота, %
[(+)]-](-)]/[(+)]+[(-)]•100,
где
[(+)] = концентрация правовращающего энантиомера;
[(-)] = концентрация левовращающего энантиомера.
Предметом настоящего изобретения является также способ получения лево- и правовращающего энантиомеров 1-[(4-хлорфенил/фенилметил] -4-[(4-метилфенил)сульфонил] пиперазина формулы I, отличающийся тем, что энантиомер /4-хлорфенил/фенилметиламина формулы II: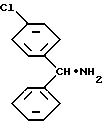
вводят во взаимодействие с N,N-диэтил-4-метил-бензолсульфонамидом формулы III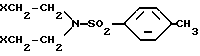
в которой X обозначает атом хлора, брома или иода или (4-метилфенил)-сульфонилокси- или метилсульфонилокси-группу, в присутствии 2,2 - 4,4 эквивалентов органического или неорганического основания на эквивалент энантиомера (4-хлорфенил)фенилметиламина и при температуре кипения реакционной смеси.
Основаниями, которые пригодны для получения соединений формулы I, являются либо органические основания, такие, как этилдиизопропиламин, N-этилморфолин, 2,4,6-триметилпиридин или триэтиламин, предпочтительно этилдиизопропиламин; либо неорганические основания, как карбонат натрия.
Лево- и правовращающий энантиомеры (4-хлорфенил)фенилметиламина формулы II, используемые в качестве исходных продуктов, представляют собой известные соединения; их можно получать путем химического расщепления рацемического (4-хлорфенил)фенилметиламина с помощью винной кислоты само по себе известными способами. Они имеют оптическую чистоту по крайней мере 98%.
Исходные соединения формулы III также являются известными продуктами; они легко получаются исходя из бис/2-гидроксиэтил/амина само по себе известными способами.
Предметом настоящего изобретения, кроме того, является использование новых лево- и правовращающего энантиомеров 1-[(4-хлорфенил)-фенилметил]-4-[(4-метилфенил)сульфонил] пиперазина формулы I для получения по существу оптически чистых энантиомеров 1-[(4-хлорфенил)-фенилметил]-пиперазина формулы IV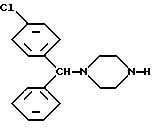
Согласно настоящему изобретению получают лево- и правовращающий энантиомеры соединения формулы IV способом, который отличается тем, что с помощью бромоводородной кислоты в среде уксусной кислоты и в присутствии фенольного соединения, предпочтительно 4-гидроксибензойной кислоты, гидролизуют энантиомер 1-[(4-хлорфенил)фенилметил]-4-[(метилфенид)сульфонил]пиперазина формулы I.
Этот гидролиз обычно осуществляют при температуре 18 - 100oC, предпочтительно при температуре, близкой к 25oC.
Преимущества, связанные с получением 1-[(4-хлорфенил)фенилметил]-4-[(4-метилфенил)сульфонил] пиперазина формулы I в виде его лево- и правовращающего энантиомеров и их использованием многочисленны.
Во-первых, оказалось, что только энантиомеры соединения формулы I, тозилированные по аминной функции, практически могут быть синтезированы с достаточно большим выходом. В самом деле, если для получения этих соединений заменить N, N-диэтил-4-метилбензолсульфонамид формулы III соответствующим соединением, в котором 4-метилфенилсульфонильная группа заменена водородом или другой защитной для аминофункции группой, например карбонильной или алкильной группой, в частности трифторметильным радикалом, то в процессе образования энантиомера соединения формулы I наблюдают значительную рацемизацию исходного амина формулы II и/или соединения формулы I, а также образование многочисленных продуктов разложения.
Кроме того, исходные соединения формулы III, в которых 4-метилфенилсульфонильная группа заменена водородом, известны как крайне токсичные из-за присутствия свободного амина.
Напротив, используя в качестве исходного продукта N,N-диэтил-4-метилбензолсульфонамид формулы III, указанные недостатки отсутствуют. В самом деле, предлагаемый способ получения энантиомеров формулы I не вызывает рацемизацию продуктов, обеспечивает высокий выход энантиомеров, который может достигать 89%, а их оптическая чистота выше 98%, а часто близка к 100%. Кроме того, используемые сульфоновые вещества относительно мало токсичны и намного менее вредны при обращении с ними. Этот последний момент создает также немалое преимущество для промышленной реализации способа изобретения.
Во-вторых, использование энантиомеров соединения формулы I для получения энантиомеров 1-[/4-хлорфенил/фенилметил] пиперазина формулы IV имеет следующие преимущества:
- более высокий выход соединений формулы IV - выше 80%. Этот выход отчетливо выше выхода, указанного в патенте Великобритании 2 225 321;
- реакция гидролиза, приводящая к образованию энантиомеров, протекает без рацемизации и обеспечивает очень высокую оптическую чистоту получаемых продуктов, равную 98%, даже близкую к 100%.
Таким образом, энантиомеры 1-[/4-хлорфенил/фенилметил]-4- [(4-метилфенил)сульфонил] -пиперазина формулы I, согласно изобретению предлагают в высшей степени эффективный путь получения энантиомеров 1-[/4-хлорфенил/фенилметил] пиперазина формулы IV.
Лево- и правовращающие практически оптически чистые энантиомеры формулы IV представляют основной интерес в качестве предшественников для получения ценных терапевтически активных соединений, а именно лево- и правовращающих форм практически оптически чистых 1-[(4-хлорфенил)фенилметил]пиперазинов формулы V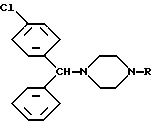
в которой R обозначает метильный, (3-метилфенил)метильный, (4-трет.-бутилфенил)метильный, 2-(2-гидроксиэтокси)этильный, 2-[2-(2-гидркосиэтокси)этокси]этильный, 2-(карбамоилметокси)этильный, 2[(метоксикарбонилметокси)этильный и 2-(карбоксиметокси)этильный радикал.
Эти соединения, уже известные в рацемической форме, обладают интересными фармакологическими свойствами и могут быть использованы для лечения астмы, аллергий, воспаления или в качестве седативных, транквиллизирующих или анксиолитических агентов.
Предпочтительными соединениями формулы V являются лево- и правовращающий энантиомеры 1-[(4-хлорфенил)фенилметил] -4-метилпиперазина; 1-[(4-хлорфенил)фенилметил] -4-[/3-метилфенил/метил] пиперазина; 1-[/4-трет.-бутилфенил/метил] -4-[/4-хлорфенил/фенилметил] пиперазина; 2-(2-[4-{ /4-хлорфенил/фенилметил] -2-пиперазинил] этокси} этанола; 2-[2-{ 2-/4-[(4-хлорфенил)фенилметил] -1-пиперазинил/этокси} этокси] -этанола; 2-[2-{4-(4-хлорфенил)фенилиметил] -1-пиперазинил} этокси] -ацетамида, метил-2-[2-{4-/(4-хлорфенил)фенилметил] -1-пиперазинил} этокси] -ацетата и 2-[2-{ 4-[(4-хлорфенил)фенилметил] -1-пиперазинил}этокси]-уксусной кислоты, также, как фармацевтически приемлемые соли этих энантиомеров.
Получение этих оптически чистых энантиомеров осуществляют известными способами и оно заключается во введении во взаимодействие при нагревании энантиомера соединения формулы V с галогенидом формулы RX, в которой R имеет вышеуказанное значение и X обозначает атом галогена. Энантиомеры формулы V представляют собой новые соединения, за исключением соединений, где R обозначает 2-(карбоксиметокси)этильный радикал, и обладают интересными антигистаминными свойствами, в особенности они очень четко отличаются друг от друга в том, что касается ингибирования рецептора H1 гистамина, т.е. один из энантиомеров является активным ингибитором, а другой неактивным.
Нижеописанные фармакологические испытания подчеркивают эти свойства.
Нижеследующие примеры иллюстрируют изобретение, однако не ограничивая его объема охраны. В этих примерах температуры плавления определяются путем дифференциальной колориметрии с продувкой /D.S.C./ с градиентом температуры 20oC/мин. Оптическая чистота определяется путем высокоэффективной жидкостной хроматографии на стационарной хиральной фазе /колонка CHIRAL PAK AD, 250x4,6 мм; элюирующее средство: смесь 50:50:0,1 /по объему/ гексанэтанол-диэтиламин; давление 104 бара; температура 25oC, дебит 1 мл/мин/.
Пример 1. Получение лево- и правовращающего энантиомеров /4-хлорфенил/метиламина формулы II.
1. (-)-/4-Хлорфенил/-фенилметиламин (левовращающий).
Это соединение получают путем расщепления рацемического (4-хлорфенил)фенилметиламина с помощью /+/-винной кислоты согласно методу, описанному R.CLEMO и др. /J.Chem. Soc. (1939), с. 1958 - 1960/.
2. (+)-/4-Хлорфенил/фенилметиламин (правовращающий).
Это соединение получают путем расщепления рацемического /4-хлорфенил/фенилметиламина с помощью /-/-винной кислоты согласно методу, описанному R.CLEMO и др. /цитировано выше/.
3. Рекуперация неиспользованного энантиомера /4-хлорфенил/фенилметиламина.
С целью рекуперации и рециркуляции неиспользованного энантиомера /4-хлорфенил/фенилметиламина это соединение подвергают реакции рацемизации и затем таким образом полученный рацемический /4-хлорфенил/-фенилметиламин используют в новой стадии расщепления с помощью изомера винной кислоты согласно способу, описанному в п.п. 1 или 2, указанных выше.
4,35 г /0,02 моль/ правовращающего /+/-/4-хлорфенил/фенилметиламина, 244 мг /0,002 моль/ 2-гидркосибензальдегида и 1,1 г /0,02 моль/ метилата натрия суспендируют в 21,8 мл метанола. Смесь кипятят с обратным холодильником в течение 5,5 часов, затем оставляют стоять для снижения температуры до комнатной и прикапывают 6,7 мл концентрированной соляной кислоты. Метанол выпаривают, остаток обрабатывают с помощью 50 мл воды и добавляют еще 25 мл концентрированной соляной кислоты. Спустя 1 час, отфильтровывают и промывают водой осадок белого цвета, который образовался, и сушат его в вакууме при 40oC. Получают 3,7 г рацемического /4-хлорфенил/фенилметиламина.
Выход = 73%, (α)
Пример 2. Получение N,N-диэтил-4-метилбензолсульфонамидов формулы III.
1. 4-Метил-N,N-бис[2-{/4-метилфенил/сульфонилокси}этил] бензолсульфонамид /формула III, X = /4-метилфенил/сульфонилокси/.
Это соединение получают из N,N-бис-/2-гидркосиэтил/-4-метилбезолсульфонамида согласно способу, описанному D.H.PEACOCK и U.C.DUTTA /J.Chem. Soc. /1934/, с. 1303 - 1305/. Т. пл. 75,9oC. Выход = 79,7%.
2. 4-Метил-N,N-бис[2-/метилсульфонилокси/этил]бензолсульфонамид /формула III, X = метилсульфонилокси/.
Раствор 11,4 г /0,1 моль/ метансульфонилхлорида в 17,1 мл дихлорметана охлаждают до 5oC. Затем при перемешивании прикапывают растор 13 г /0,05 моль/ N, N-бис/2-гидроксиэтил/-4- метилбензолсульфонамида и 10,1 г /0,1 моль/ триэтиламина в 52 мл дихлорметана. Оставляют реакционную смесь стоять до повышения температуры до комнатной и продолжают перемешивание в течение 3 часов. Реакционную смесь экстрагируют три раза по 40 мл воды. Органическую фазу сушат над сульфатом натрия, отфильтровывают и концентрируют на ротационном испарителе. Полученное масло кристаллизуют из этанола. Получают 17,8 г 4-метил-N, N- бис/2-/метилсульфонилокси/этил/ бензолсульфонамида. Т. пл. 64,6oC. Выход: 85,7%.
3. N,N-Бис(2-хлорэтил)-4-метилбензолсульфонамид (формула III, X = Cl).
Это соединение получают согласно способу, описанному K.A.AL-RASHOOD и др. /Arzneim - Forch./Drug. Res. с. 1242 - 1245/. Т. пл. = 45,8oC. Выход = 69,0%.
4. N,N-Бис(2-иодэтил)-4-метил-бензолсульфонамид /формула III, X = иод/
5,7 г (0,01 моль) 4-Метил-N,N-бис[2-{(4-метилфенил)сульфонилокси}-этил]- бензолсульфонамида /получен как указано в п. 1 выше/ растворяют в 57 мл ацетона и добавляют туда 4,5 г (0,03 моль) иодида натрия. Смесь кипятят с обратным холодильником в течение 22 часов. Оставляют охлаждаться и ацетон выпаривают. Твердый остаток обрабатывают смесью 10 мл воды и 25 мл дихлорметана и разделяют две фазы. Водную фазу экстрагируют с помощью 25 мл дихлорметана и органические фазы объединяют. Эту органическую фазу промывают последовательно с помощью 10 мл 10%-ного водного раствора тиосульфата натрия, затем с помощью 10 мл воды. Органическую фазу сушат над сульфатом натрия, отфильтровывают и выпаривают. Полученное твердое вещество белого цвета сушат в вакууме при 25oC. Получают 4,7 г N,N-бис/2-иодэтил/-4-метилбензолсульфонамида. Т. пл. 93,8oC. Выход = 98%.
5. N,N-Бис(2-бромэтил)-4-метилбензолсульфонамид (формула III, X = бром).
Это соединение получают согласно способу, описанному в п. 4 выше, но заменяя иодид натрия на бромид натрия. Реакционную смесь кипятят с обратным холодильником в ацетоне в течение 16 дней. Т. пл. 69,2oC. Выход = 98,7%.
Пример 3. Получение энантиомеров 1-[(4-хлорфенил)фенилметил-4-[(4-метилфенил)сульфонил]пиперазина формулы I.
A1. Левовращающий /-/-1-[/4-хлорфенил/фенилметил] -4-[(4-метилфенил)-сульфонил]пиперазин.
В колбе емкостью 25 мл смешивают 3,4 г /0,0156 моль/ левовращающего /-/-(4-хлорфенил)фенилметиламина [получен в примере 1.1] и 5,1 г /0,0172 моль/ N,N-бис,2-хлорэтил/-4-метилбензолсульфонамида [получен в примере 2.3] в 6 мл /4,4 г или 0,0343 моль/ этилдиизопропиламина. Смесь кипятят с обратным холодильником /127oC/ в течение 4 часов. При перемешивании охлаждают до 86oC и добавляют за один раз 13,8 мл метанола. После этого смесь охлаждают на ледяной бане и выдерживают при перемешивании в течение 1 часа. Образовавшийся осадок отфильтровывают, промывают его с помощью 10 мл метанола и сушат его в вакууме при 40oC. Продукт перекристаллизуют из смеси 3:1 /по объему/ метанола с ацетоном. Получают 6 г левовращающего /-/-1-[(4-хлорфенил)фенилметил] -4-[(4-метилфенил)сульфонил]пиперазина.
Т. пл. 171,1oC. Выход: 87,2% (α)
Оптическая чистота составляет 100%.
Анализ для C24H25ClN2O2S, %:
Рассчитано,%: C 65,37; H 5,71; N 6,35; Cl 8,04; S 7,27.
Найдено, %: C 65,95; H 5,80; N 6,60; Cl 8,12; S 7,33.
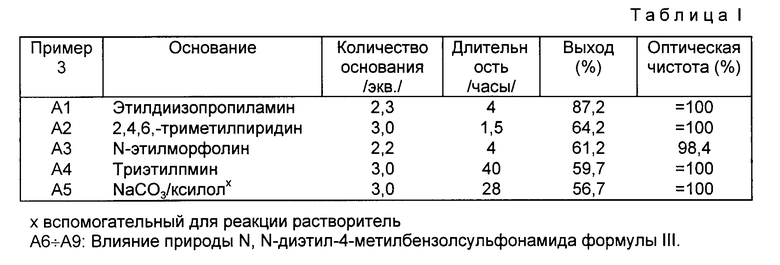
A2 - A5: Исследование влияния природы основания.
Получают левовращающий /-/-1-[(4-4-хлорфенил)фенилметил]-4- [(4-метилфенил)сульфонил] пиперазин из N,N-бис/2-хлорэтил-4-метилбензолсульфонамида, используя способ, описанный выше в п. A1, но заменяя этилдиизопропиламин различными другими основаниями.
В табл. I указано: в первой колонке: номер примера; во второй колонке: используемое основание; в третьей колонке: количество используемого основания, выраженное в эквивалентах на эквивалент /-/-(4-хлорфенил)-фенилметиламина; в четвертой колонке: время(часы), в течение которого реакционную смесь кипятят с обратным холодильником; в пятой колонке: полученный выход левовращающего /-/-1-[(4-хлорфенил)фенилметил] -4-[(4-метилфенил)сульфонил] пиперазина и в шестой колонке: оптическая чистота полученного продукта, выраженная в %.
При рассмотрении табл. I видно, что природа основания оказывает очень небольшое влияние на оптическую чистоту получаемого продукта. Однако видно, что этилдиизопропиламин более предпочтителен с точки зрения выхода реакции.
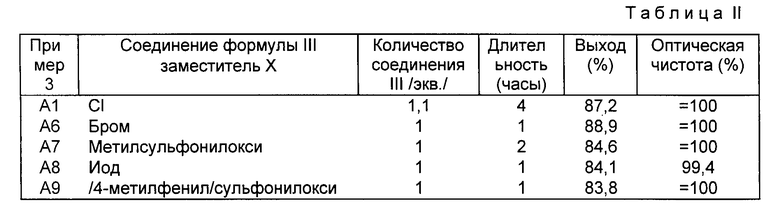
Получают левовращающий /-/-1-[(4-хлорфенил)фенилметил]-4- [(4-метилфенил)сульфонил] пиперазин, используя способ, описанный в п. A1 выше, но заменяя N, N-бис-(2-хлорэтил)-4-метилбензолсульфонамид формулы III (X = Cl), используемый в качестве исходного продукта, на бромсодержащее (X = бром), иодсодержащее (X = иод), тозилированное [X = (4-метилфенил)сульфонилокси] и мезилированное [X = метилсульфонилокси] производное, получаемые соответственно в примерах 2.5, 2.4, 2.1 и 2.2.
В табл. II указано: в первой колонке: номера примера; во второй колонке: природа заместителя X в исходном соединении формулы III; в третьей колонке: количество используемого соединения формулы III, выраженное в эквивалентах на эквивалент /-/-(4-хлорфенил)фенилметиламина; в четвертой колонке: время, выраженное в часах, в течение которого реакционную смесь кипятят с обратным холодильником; в пятой колонке: полученный выход левовращающего /-/-1-[/4-хлорфенил] -фенилметил(-4-[/4-метилфенил)сульфонил] пиперазина и в шестой колонке: оптическая чистота продукта, выраженная в %.
При рассмотрении табл. II видно, что природа соединения формулы III оказывает очень мало влияния на оптическую чистоту получаемого продукта. Более того, видно, что она очень незначительно влияет на выход реакции, однако наилучший выход получен с бромсодержащим производным.
Б. Правовращающий /+/-1-[(-4-хлорфенил)фенилметил]-4-[(4-метилфенил)-сульфонил]-пиперазин.
В трехгорлую колбу емкостью 500 мл вводят 57 г (0,2618 моль) правовращающего /+/-(4-хлорфенил)фенилметиламина (получен в в примере 1,2) и 86,4 г (0,2917 моль) N,N-бис(2-хлорэтил)-4- метилбензолсульфонамида (получен в примере 2,3) в 200 мл (1,15 моль) этилдиизопропиламина. Смесь кипятят с обратным холодильником в течение 3 ч, затем выливают ее в 400 мл метанола и охлаждаемую на ледяной бане смесь перемешивают в течение 1 ч. Образовавшийся осадок отфильтровывают, промывают его метанолом и сушат в вакууме при 50oC. Получают 88,6 г правовращаюшего /+/-1-[/4-хлорфенил/фенилметил]-4-[/4-метилфенил/сульфонил]пиперазина. Т.пл. 173,3oC. Выход = 76,7%.
(α)
Анализ для C24H25ClN2O2S, %:
Рассчитано,%: C 65,38; H 5,71; N 6,35; Cl 8,04; S 7,27.
Найдено, %: C 64,98; H 5,70; N 6,40; Cl 7,96; S 7,35.
Пример 4. Получение лево- и правовращающего энантиомера 1-[(4-хлорфенил)фенилметил]пиперазина формулы IV.
1. Левовращающий /-/-1-[(4-хлорфенил)фенилметил]пиперазин.
В 1 л 30%-ного раствора бромоводородной кислоты в уксусной кислоте вводят 370 г (0,839 моль) левовращающего /-/-1-[(4-хлорфенил)-фенилметил]-4- [(4-метилфенил)сульфонил] пиперазина /получен в примере 3.А1/ и 405 г 4-гидроксибензойной кислоты. Суспензию перемешивают в течение 17 ч. при 25oC. Затем добавляют 2 л воды и охлаждают на ледяной бане. Образуется осадок, который отфильтровывают и промывают с помощью 750 мл воды. К фильтрату добавляют 2 л толуола и 0,9 л водного 50%-ного раствора гидроксида натрия. Декантируют и промывают органическую фазу с помощью 100 мл воды, затем еще один раз с помощью 1 л водного насыщенного раствора хлорида натрия. Органическую фазу сушат над сульфатом натрия, отфильтровывают и растворитель выпаривают при пониженном давлении. Остаток перекристаллизуют при кипении из 600 мл гексана. Отфильтровывают в горячем состоянии для удаления небольшого количества нерастворимой части и фильтрат оставляют кристаллизоваться сначала при комнатной температуре, затем путем охлаждения на ледяной бане в течение 24 часов. Кристаллы отфильтровывают, промывают гексаном и сушат в вакууме при 40oC. Получают 204,15 г левовращающего /-/-1-[(4-хлорфенил)фенилметил] пиперазина.
Т.пл. = 90,5oC. Выход = 84,8%. (α)
Оптическая чистота = ≥ 99,8%.
Анализ для C17H19ClN2, %:
Рассчитано,%: C 71,19; H 6,68; N 9,97; Cl 12,36.
Найдено, %: C 71,19; H 6,84; N 9,55; Cl 11,48.
2. Правовращающий (+) 1-[(4-хлорфенил)фенилметил]пиперазин.
Работают согласно способу, описанному выше в п. 4.1. но заменяя исходный левовращающий энантиомер 1-[(4-хлорфенил)фенилметил] -4-[(4- метилфенил)сульфонил] пиперазина на правовращающий энантиомер (получен в примере 3. Б)
Т.пл. = 91,5oC. Выход = 97,9%. (α)
Оптическая чистота: 100%.
Анализ для C17H19ClN2, %:
Рассчитано,%: C 71,19; H 6,68; N 9,77; Cl 12,36.
Найдено, %: C 70,90; H 6,74; N 9,72; Cl 12,23.
Пример 5. Использование энантиомеров 1-[(4-хлорфенил)фенилметил] -пиперазина для получения терапевтически активных соединений формулы V.
1. Левовращающий дихлоргидрат 1-[(4-хлорфенил)фенилметил]-4-[(3-метилфенил)метил]пиперазина.
При 50oC нагревают раствор, содержащий 10 г /0,0348 моль/ правовращающего /+/-1-[(4-хлорфенил)фенилметил] пиперазина /получен в примере 4.2/ в 100 мл н-бутанола. Туда добавляют 5,5 мл (0,0417 моль) 1-хлорметил-3-метилбензола, 8,9 г (0,0836 моль) карбоната натрия и 0,5 г /0,0030 моль/ иодида калия. Смесь кипятят с обратным холодильником в течение 3 часов. Охлаждают, твердые остатки удаляют путем фильтрации и споласкивают их с помощью 200 мл толуола. Органические фазы объединяют и растворители выпаривают до получения остаточного масла. Масло снова растворяют в 500 мл этанола, к которым добавляют 15 мл концентрированной соляной кислоты, растворенной в 35 мл этанола. Охлаждают на ледяной бане, осадок отфильтровывают и фильтрат выпаривают. Полученный после выпаривания остаток и осадок объединяют и суспендируют в 100 мл изопропилового спирта. Суспензию отфильтровывают, твердое вещество промывают небольшим количеством изопропилового спирта и сушат в вакууме при 50oC. Получают 12,7 г левовращающего дихлоргидрата 1-[(4-хлорфенил)фенилметил] -4-[(3- метилфенил)метил]пиперазина. Т. пл. 252,3oC. Выход = 78,6%.
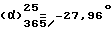 (c = 1, метанол). Оптическая чистота = 100%.
(c = 1, метанол). Оптическая чистота = 100%.
Анализ для C25H27ClN2•2HCl, %:
Рассчитано,%: C 64,73; H 6,30; N 6,04; Cl- 15,29.
Найдено, %: C 64,45; H 6,42; N 5,93; Cl- 15,18.
2. Правовращающий дихлоргидрат 1-[(4-хлорфенил)фенилметил]-4- [(3-метилфенил)метил]пиперазина.
Работают как описано в предыдущем способе 5.1, но исходный правовращающий /+/-1-[/4-хлорфенил/фенилметил] пиперазин заменяют на левовращающий энантиомер /получен в примере 4.1/, используя те же количества реагентов. Получают 13 г правовращающего дихлоргидрата 1-[/4-хлорфенил/фенилметил]-4-[/3-метилфенил/метил]-пиперазина. Т.пл. 252,9oC. Выход = 80,4%.
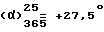 (c = 1, метанол).
(c = 1, метанол).
Оптическая чистота: 100%.
Анализ для C25H27ClN2•2HCl, %:
Рассчитано,%: C 64,73; H 6,30; N 6,04; Cl- 15,29.
Найдено, %: C 64,47; H 6,32; N 5,88; Cl- 15,18.
3. Левовращающий дихлоргидрат 1-[(4-трет.-бутилфенил)метил]-4- [(4-хлорфенил)фенилметил]пиперазина.
Нагревают при 50oC раствор, содержащий 10 г (0,0348 моль) правовращающего /+/-1-[(4-хлорфенил)фенилметил] пиперазина (получен в примере 4.2) в 100 мл н-бутанола. Туда добавляют 7,6 мл (0,0418 моль) 1-хлорметил-4-трет.-бутилбензола, 8,9 г (0,0836 моль) карбоната натрия и 0,5 г (0,0030 моль) иодида калия. Смесь кипятят с обратным холодильником в течение 1 часа. Затем охлаждают, удаляют твердые продукты путем фильтрования и споласкивают их с помощью 200 мл толуола. Органические фазы объединяют и растворители выпаривают до получения остаточного масла. Это масло снова растворяют в 300 мл ацетона и добавляют 15 мл концентрированной соляной кислоты, растворенной в 35 мл ацетона, затем добавляют еще 200 мл ацетона. Охлаждают на ледяной бане, затем образовавшийся осадок отфильтровывают и сушат его в вакууме при 50oC. Получают 14,68 г левовращающего дихлоргидрата 1-[(4-трет.-бутилфенил)метил] -4- [/4-хлорфенил/фенилметил] пиперазина. Т. пл. 257,7oC. Выход = 83,3% 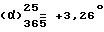 (c = 0,2, метанол). Оптическая чистота = 100%.
(c = 0,2, метанол). Оптическая чистота = 100%.
Анализ для C28H33ClN2•2HCl, %:
Рассчитано,%: C 66,47; H 6,97; N 5,54; Cl- 14,01.
Найдено, %: C 66,35; H 7,39; N 5,45; Cl- 13,85.
4. Правовращающий дихлоргидрат 1-[(4-трет.-бутилфенил)метил]-4- [(4-хлорфенил)фенилметил]пиперазина.
Это соединение получают при использовании способа, описанного в п. 3 выше для получения левовращающего энантиомера, однако используя 4 г левовращающего /-/-1-[(4-хлорфенил)фенилметил]пиперазина [получен в примере 4.1]. Получают 4,75 г правовращающего дихлоргидрата 1-[(4-трет.-бутилфенил)метил] -4-[(4-хлорфенил)фенилметил]пиперазина.
Т.пл. 273,9 oC. Выход = 67,4%. 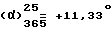 (c = 0,2, метанол).
(c = 0,2, метанол).
Оптическая чистота = 100%.
Анализ для C28H33ClN2•2HCl, %:
Рассчитано,%: C 66,47; H 6,97; N 5,54; Cl- 14,01.
Найдено, %: C 66,37; H 7,16; N 5,27; Cl- 13,85.
5. Левовращающий дихлоргидрат 2-/2-{4-[(4-хлорфенил)фенилметил] -1-пиперазинил (этокси)этанола.
Нагревают при 50oC раствор, содержащий 10 г /0,0348 моль/ правовращающего /+/-1-[(4-хлорфенил)фенилметил] пиперазина [получен в примере 4.2] в 100 мл н-бутанола. Туда добавляют 5 мл (0,0464 моль) 2-(2-хлорэтокси)этанола, 8,9 г (0,0836 моль) карбоната натрия и 0,5 г (0,0030 моль) иодида калия. Смесь кипятят с обратным холодильником в течение 16 ч. Затем добавляют еще 2 мл 2-(2-хлорэтокси)этанола и кипятят с обратным холодильником дополнительно 4 ч. Охлаждают, отфильтровывают и промывают осадок с помощью 200 мл толуола. Органические фазы выпаривают до получения масла, которое растворяют в 100 мл этанола. Добавляют 12 мл концентрированной соляной кислоты, растворенных в 38 мл этанола. Растворитель выпаривают и остаток перекристаллизуют из этанола. Отфильтровывают и промывают осадок с помощью небольшого количества изопропилового спирта (1-я порция). Фильтрат выпаривают и твердый остаток промывают небольшим количеством изопропилового спирта (2-я порция). Обе порции перекристаллизуют вместе из смеси 30:1 (по объему) изопропилового спирта с метанолом. Получают 10,57 г левовращающего дихлоргидрата 2-[-{ 4-[(4-хлорфенил)фенилметил]-1-пиперазинил}этокси]этанола. Т.пл. 229,8oC.
Выход = 67,8%. 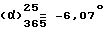 (c = 1, вода). Оптическая чистота = 100%.
(c = 1, вода). Оптическая чистота = 100%.
Анализ для C21H27ClN2О2•2HCl, %:
Рассчитано,%: C 56,32; H 6,53; N 6,26; Cl- 15,83.
Найдено, %: C 56,32; H 6,79; N 6,08; Cl- 15,63.
6. Правовращающий дихлоргидрат 2-[2-{4-[(4-хлорфенил)фенилметил]-1-пиперазинил}этокси]этанола.
Используя такие же количества реагентов, используемые в способе, описанном в п. 5 выше, получают таким же образом правовращающий энантиомер, исходя из левовращающего /-/-1-[(4-хлорфенил)фенилметил]пиперазина (получен в примере 4.1). Получают 11,7 г правовращающего дихлоргидрата 2-[-{4-/(4-хлорфенил) фенилметил/-1-пиперазинил}этокси]этанола.
Т.пл. = 231,3oC. Выход 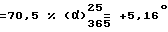 (c = 1, вода).
(c = 1, вода).
Оптическая чистота = 100%.
Анализ для C21H27ClN2О2•2HCl, %:
Рассчитано,%: C 56,32; H 6,52; N 6,25; Cl- 15,83.
Найдено, %: C 55,75; H 6,54; N 6,10; Cl- 15,81.
7. Левовращающий дихлоргидрат 2-[2-{2-[(4-хлорфенил)фенилметил]-1-пиперазинил/этокси}этокси]этанола.
При 40oC нагревают раствор, содержащий 10 г /0,0348 моль/ правовращающего /+/-1-[(4-хлорфенил)фенилметил] пиперазина /получен в примере 4,2/ в 100 мл н-бутанола. Туда добавляют 6,1 мл /0,0419 моль/ 2-[2-(2-хлорэтокси)этокси] этанола, 8,9 г (0,0836 моль) карбоната натрия и 0,5 г (0,0030 моль) иодида калия. Смесь кипятят с обратным холодильником в течение 6 часов. Охлаждают, удаляют твердые продукты путем фильтрования и промывают их небольшим количеством толуола. Объединяют фильтрат и растворитель от промывки и растворители выпаривают. Полученный остаток обрабатывают с помощью 50 мл толуола, который затем выпаривают. Остаток снова обрабатывают с помощью 100 мл толуола, промывают с помощью 100 мл воды и органическую фазу выпаривают. Полученное после выпаривания масло растворяют в 100 мл изопропилового спирта. Туда добавляют раствор, содержащий 12 мл концентрированной соляной кислоты в 38 мл изопропилового спирта. Растворитель выпаривают. Твердый остаток обрабатывают при нагревании с помощью 150 мл изопропилового спирта, добавляют туда 100 мл гексана и раствор кипятят с обратным холодильником. Затем охлаждают на ледяной бане. Осадок отфильтровывают и промывают с помощью 50 мл смеси 1:1 (по объему) изопропилового спирта с гексаном, затем с помощью 50 мл гексана. Таким образом полученный твердый продукт сушат в вакууме при 50oC. Получают 12,2 г левовращающего дихлоргидрата 2-[2-{ 2-[4-(4-хлорфенил)фенилметил] -1-пиперазинил]этокси}этокси]-этанола. Т.пл. 198oC. Выход 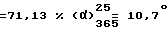 (с = 1, метанол).
(с = 1, метанол).
Оптическая чистота: 100%.
Анализ для C23H31ClN2О2•2HCl, %:
Рассчитано,%: C 56,16; H 6,76; N 5,69; Clобщ. 21,62.
Найдено, %: C 56,34; H 7,00; N 7,00; Clобщ. 21,76.
8. Правовращающий дихлоргидрат 2-[2-{2-[4-/(4-хлорфенил)фенилметил]-1-пиперазинил/этокси}этокси]этанола.
С помощью того же самого способа, который описан в п. 7, получают правовращающий энантиомер из левовращающего /-/-1-[/4-хлорфенил]-фенилметил] пиперазина (получен в примере 4.1). Т.пл. 196,1oC.
Выход 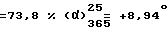 (с = 1, метанол). Оптическая чистота = 100%.
(с = 1, метанол). Оптическая чистота = 100%.
Анализ для C23H31ClN2О3•2HCl, %:
Рассчитано,%: C 56,16; H 6,76; N 5,69; Clобщ. 21,62.
Найдено, %: C 56,48; H 6,96; N 5,65; Clобщ. 21,1.
9. Левовращающий (-)2-[2-{4-[(4-хлорфенил)фенилметил]-1- пиперазинил}-этокси]ацетамид.
77 г (0,2685 моль) Левовращающего (-)-1-[(4-хлорфенил)фенилметил)-пиперазина [получен в примере 4.1] 40,5 г (0,2932 моль) 2-(2-хлорэтокси)ацетамида, 62,8 г (0,591 моль) карбоната натрия и 2 г (0,0120 моль) иодида калия вводят в 700 мл толуола. Смесь кипятят с обратным холодильником в течение 24 часов. Затем добавляют 10 г Норита и фильтруют в горячем состоянии через Dicalite. Фильтрат промывают с помощью 500 мл воды, затем с помощью 500 мл водного насыщенного раствора хлорида натрия. Органическую фазу отделяют и сушат над 250 г сульфата натрия. Отфильтровывают и растворитель выпаривают. Остаточное масло при нагревании обрабатывают с помощью 1500 мл диизопропилового эфира. Раствор кипятят с обратным холодильником и оставляют кристаллизоваться путем охлаждения на ледяной бане. Кристаллы отфильтровывают, промывают их небольшим количеством диизопропилового эфира и сушат в вакууме при 40oC. Получают 82,91 г левовращающего (-)-2-[2-{4-[(4-хлорфенил)фенилметил] -1-пиперазина} этокси] ацетамида. Т. пл. 94,3oC. Выход 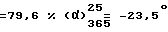 (с = 1, метанол).
(с = 1, метанол).
Оптическая чистота = 100%.
Анализ для C21H26ClN3О2, %:
Рассчитано,%: C 65,02; H 6,76; N 70,83; Cl 8,14.
Найдено, %: C 65,19; H 6,70; N 10,99; Cl 9,23.
10. Правовращающий (+)-2-[2-{4-[(4-хлорфенил)фенилметил]-1- пиперазинил} -этокси]ацетамид.
15 г (0,0523 моль) правовращающего (+)-1-[(4-хлорфенил)фенилметил]пиперазина [получен в примере 4.2] , 8,3 г (0,0601 моль) 2-(2-хлорэтокси)ацетамида, 12,8 г (0,1203 моль) карбоната натрия и 0,5 г (0,0030 моль) иодида калия вводят в смесь 100 мл п-ксилола и 150 мл толуола. Реагенты смешивают все одновременно при комнатной температуре, затем смесь нагревают до 140oC и выдерживают при этой температуре в течение 17 часов. Добавляют немного Норита и фильтруют при нагревании через Dicalite. Остаток на фильтре промывают небольшим количеством толуола и фильтрат объединяют с промывным раствором. Растворители выпаривают и остаток обрабатывают с помощью 100 мл толуола. Органическую фазу промывают последовательно с помощью 100 мл воды и два раза с помощью 100 мл водного насыщенного раствора хлорида натрия. Органическую фазу отделяют и растворитель выпаривают. На этой стадии полученный неочищенный остаток может быть очищен способом, аналогичным таковому, описанному в п. 9 выше, для получения правовращающего /+/-2-[2-{4-[(4-хлорфенил) фенилметил]-1-пиперазинил} этокси]ацетамида в виде свободного основания. Однако, если желательно, неочищенный остаток может быть превращен в соответствующий дихлоргидрат следующим образом: полученный неочищенный остаток обрабатывают 100 мл ацетона, охлаждают на ледяной бане и прикапывают 15 мл концентрированной соляной кислоты. Затем добавляют еще 200 мл ацетона и охлаждаемую на ледяной бане смесь перемешивают в течение 1 часа. Осадок отфильтровывают и сушат в вакууме при 50oC. Получают 19 г левовращающего дихлоргидрата 2-[2-{4-[(4-хлорфенил)фенилметил]-1- пиперазинил}этокси]ацетамида.
Т.пл. 237,4oC. Выход 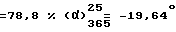 (с = 1, метанол).
(с = 1, метанол).
Оптическая чистота = 100%.
Анализ для C21H26ClN3О2•2HCl, %:
Рассчитано,%: C 54,73; H 6,12; N 9,12; Clобщ. 23,08; Cl- 15,38.
Найдено, %: C 65,19; H 6,70; N 10,99; Clобщ. 23,08; Cl- 15,61.
11. Левовращающий дималеатметил-2-[2-{4-[(4-хлорфенил) фенилметил]-1-пиперазинил}этокси]ацетата.
46 г /0,16 моль/ левовращающего /-/-1-[/4-хлорфенил/фенилметил]- пиперазина [получен в примере 4.1] , 36,6 г /0,24 моль/ метил-(2-хлорэтокси)ацетата, 37,3 г /0,35 моль/ безводного карбоната натрия и 1,05 г /0,0064 моль/ иодида калия суспендируют в 46 мл толуола. Суспензию кипятят с обратным холодильником при перемешивании в течение 18 часов. Затем охлаждают до комнатной температуры и суспензию отфильтровывают. Твердые продукты промывают с помощью 100 мл толуола и объединяют фильтрат с промывным раствором. Толуол выпаривают на ротационном испарителе при 50oC при пониженном давлении. Получают 76 г коричневого масла, которое обрабатывают 80 мл дихлорметана. Раствор очищают путем хроматографии {колонка с диоксидом кремния [15 -40 мкм, 1 кг]; элюирующее средство: чистый дихлорметан, к которому постепенно добавляют метанол вплоть до максимально 2% метанола /по объему/}. Таким образом получают 43,5 г метил-2-[2-{4-[(4-хлорфенил)фенилметил]-1-пиперазинил}этокси]ацетата в виде масла. Выход = 67,5%.
Это соединение может быть превращено в соответствующий дималеат следующим образом: 15 г /0,037 моль/ метил-2-[2-{4-[(4-хлорфенил)-фенилметил]-1-пиперазинил} этокси]ацетата, полученного выше, растворяют в 45 мл метанола при температуре кипения с обратным холодильником и добавляют туда за один раз 9,1 г (0,078 моль) малеиновой кислоты. Смесь кипятят с обратным холодильником до тех пор, пока полностью не растворится малеиновая кислота, затем оставляют раствор возвращаться к комнатной температуре, все время перемешивая. Образовавшиеся кристаллы отфильтровывают и суспендируют их в 15 мл метанола. Суспензию перемешивают в течение 1,5 часов при комнатной температуре, затем еще раз в течение 1,5 часов при 0oC. Кристаллы отфильтровывают, промывают их с помощью 15 мл метанола при 0oC и высушивают их до постоянного веса. Получают 19,5 г левовращающего дималеата метил-2-[2-{4-[(4-хлорфенил)фенилметил] -1-пиперазинил} этокси] ацетата. Т. пл. 143,5oC. Выход 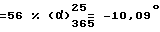 (с = 1, метанол). Оптическая чистота = 100%.
(с = 1, метанол). Оптическая чистота = 100%.
Анализ для C22H27ClN2О3 • 2C4H4O4, %:
Рассчитано,%: C 56,79; H 5,56; N 4,41.
Найдено, %: C 56,81; H 5,68; N 4,12.
12. Правовращающий дималеат метил-2-[2-{4-[(4-хлорфенил)фенилметил]-1-пиперазинил}этокси]ацетата.
14,3 г (0,05 моль) правовращающего /+/-1-[/4-хлорфенил/фенилметил]пиперазина [получен в примере 4.2] , 8,4 г /0,055 моль/ метил-(2-хлорэтокси)ацетата, 11,7 г (0,11 моль) безводного карбоната натрия и 0,332 г (0,002 моль) иодида калия суспендируют в 14,3 мл толуола. Суспензию при перемешивании кипятят с обратным холодильником в течение 17 часов. Добавляют еще 1,52 г (0,01 моль) иодида калия суспендируют в 14,3 мл толуола. Суспензию при перемешивании кипятят с обратным холодильником в течение 17 часов. Добавляют еще 1,52 г (0,01 моль) метил-(2-хлорэтокси)ацетата и суспензию снова кипятят с обратным холодильником в течение 3 часов при перемешивании. Затем охлаждают до комнатной температуры и суспензию отфильтровывают. Твердые продукты промывают с помощью 50 мл толуола и фильтрат объединяют с промывным раствором. Толуол выпаривают на ротационном испарителе при 50oC и при пониженном давлении. Получают 22,8 г коричневого масла, которое обрабатывают 45 мл дихлорметана. Раствор очищают путем хроматографии /колонка с диоксидом кремния /15 - 40 мкм, 1 кг/; элюирующее средство: чистый дихлорметан, к которому постепенно добавляют метанол вплоть до максимально 2% метанола /по объему//. Получают 11,1 г метил-2[2-{4- [(4-хлорфенил)фенилметил]-1-пиперазинил} этокси]-ацетата в виде масла. Выход = 55,1%.
Это соединение может быть превращено в соответствующий дималеат следующим образом: 8 г /0,0198 моль/ метил-2-[2-{4-[(4-хлорфенил)фенилметил]-1-пиперазинил} этокси] ацетата, полученного выше, растворяют в 16 мл метанола при температуре кипячения с обратным холодильником и добавляют туда за один раз 4,85 г /0,0417 моль/ малеиновой кислоты. Смесь кипятят с обратным холодильником до тех пор, пока малеиновая кислота полностью не растворится, затем раствор оставляют до возвращения температуры к комнатной, все время перемешивая. Образовавшиеся кристаллы отфильтровывают и суспендируют их в 16 мл метанола. Суспензию перемешивают в течение 2 часов при комнатной температуре. Кристаллы отфильтровывают, промывают их 10 мл метанола и сушат их до постоянного веса. Получают 7,3 г правовращающего дималеата метил-2-[2-{4-[(4-хлорфенил)фенилметил]-1-пиперазинил}этокси]ацетата. Т.пл. 143,2oC.
Выход 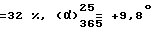 (с = 1, метанол). Оптическая чистота = 100%.
(с = 1, метанол). Оптическая чистота = 100%.
Анализ для C22H27ClN2О3 • 2C4H4O4, %:
Рассчитано,%: C 56,79; H 5,56; N 4,41.
Найдено, %: C 56,71; H 5,58; N 4,17.
13. Левовращающий дихлогидрат 2-[2-{4-[(4-хлорфенил)фенилметил] -1-пиперазинил}этокси]уксусной кислоты.
К суспензии 25,2 г /0,065 моль/ правовращающего /+/-2-[2-{4-[(4-хлорфенил)фенилметил]-1-пиперазинил}этокси]ацетамида [получен в п. 10 выше] в 70 мл воды прикапывают 26 мл концентрированной соляной кислоты. Температура смеси повышается до 38oC. Затем смесь в течение 17 часов нагревают при 50oC. После этого реакционную смесь охлаждают на ледяной бане и доводят до pH-значения = 4-5 путем добавления 4 н. водного раствора гидроксида натрия. Раствор экстрагируют последовательно с помощью 100 мл, затем два раза по 50 мл, дихлорметаном. Органические фазы объединяют и сушат над сульфатом магния. Отфильтровывают и растворитель выпаривают. Остаточное масло растворяют в 243 мл ацетона, раствор обрабатывают с помощью 3,5 г Норита и фильтруют через целит. Промывают с помощью 35 мл ацетона. Раствор кипятят с обратным холодильником и добавляют туда по каплям 198 мл /0,13 моль/ концентрированной соляной кислоты. Охлаждают на ледяной бане и смесь оставляют стоять в течение часа. Образовавшийся осадок отфильтровывают, промывают его 100 мл ацетона и сушат его в вакууме при 50oC. Получают 24,1 г левовращающего дихлоргидрата 2-[2-{ 4-[(4-хлорфенил)фенилметил]-1-пиперазинил}этокси]уксусной кислоты. Т.пл. 229,3oC. Выход = 80,3%,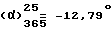 (с = 1, вода). Оптическая чистота = 100%.
(с = 1, вода). Оптическая чистота = 100%.
Анализ для C21H25ClN2О3•2HCl, %:
Рассчитано,%: C 54,61; H 5,90; N 6,07; Cl- 15,35; Clобщ. 23,03.
Найдено, %: C 54,67; H 5,91; N 6,03; Cl- 15,35; Clобщ. 23,28.
14. Правовращающий дихлоргидрат 2-[2-{4-[(4-хлорфенил)фенилметил]-1-пиперазинил}этокси]уксусной кислоты.
Правовращающий дихлоргидрат 2-[2-{4-[(4-хлорфенил)фенилметил]-1-пиперазинил} этокси] уксусной кислоты получают согласно способу, описанному выше в п. 13, используя 25,2 г /0,065 моль/ левовращающего /-/-2-[2-{4-[(4-хлорфенил)фенилметил] -1-пиперазинил} этокси] ацетамида /получен в п. 9, выше/. Таким образом получают 25,6 г целевого продукта. Т. пл. 227,9oC. Выход 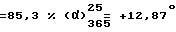 (с = 1, вода).
(с = 1, вода).
Оптическая чистота: 99,87%.
Анализ для C21H25ClN2О3•2HCl, %:
Рассчитано,%: C 54,61; H 5,90; N 6,07; Cl- 15,35; Clобщ. 23,03.
Найдено, %: C 54,71; H 5,92; N 6,04; Cl- 15,34; Clобщ. 23,19.
15. Правовращающий дихлоргидрат 2-[2-{4-[(4-хлорфенил)фенилметил]-1-пиперазинил}этокси]уксусной кислоты.
При перемешивании и при комнатной температуре 13,75 г /0,00216 моль/ левовращающего дималеата метил-2-[2-{4-[(4-хлорфенил)фенилметил]-1-пиперазинил}этокси]ацетата [получен в п. 11 выше] вводят в 54 мл водного 2 н. раствора гидроксида натрия. Реакционную смесь экстрагируют последовательно с помощью 100 мл и 75 мл диэтилового эфира и органические фазы объединяют. Эту органическую фазу сушат над безводным сульфатом натрия, отфильтровывают и остаток от фильтрации промывают с помощью 50 мл диэтилового эфира. Органические фазы объединяют и диэтиловый эфир выпаривают. Таким образом полученное масло /8,4 г/ обрабатывают 50 мл этанола и добавляют 1,3 г /0,0229 моль/ твердого гидроксида калия. Смесь кипятят с обратным холодильником в течение 1 часа, затем оставляют для возвращения температуры к комнатной. Отфильтровывают и фильтрат выпаривают. Остаток обрабатывают с помощью 50 мл воды и концентрируют на ротационном испарителе для удаления остаточного этанола. К частично концентрированному раствору добавляют 10 мл воды и pH раствора доводят до величины 4-5 путем добавления водного 10%-ного раствора соляной кислоты. Раствор экстрагируют с помощью 50 мл дихлорметана, снова доводят до pH значения раствора 4-5 путем добавления водного 10%-ного раствора соляной кислоты и экстрагируют еще раз с помощью 50 мл дихлорметана. Органические фазы объединяют и сушат над безводным сульфатом магния. Отфильтровывают и дихлорметан выпаривают. Таким образом полученное вязкое масло /9,8 г/ растворяют в 68,6 мл ацетона, слегка мутный раствор обрабатывают с помощью 1 г активного угля и отфильтровывают в нагретом состоянии через диатомовую землю. К таким образом полученному горячему, прозрачному, желтого цвета раствору добавляют 3,6 мл /0,043 моль/ концентрированной соляной кислоты. Оставляют суспензию принимать комнатную температуру при перемешивании, затем перемешивают еще в течение 1 часа при 0oC. Образовавшийся осадок отфильтровывают, промывают его с помощью 50 мл ацетона и сушат его в вакууме при 40oC. Таким образом получают 6,8 г правовращающего дихлоргидрата 2-[2-{4-[(4-хлорфенил)фенилметил] -1-пиперазинил} этокси] уксусной кислоты. Т. пл. 227,8oC. Выход = 70,8%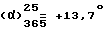 (с = 1, вода). Оптическая чистота = 100%.
(с = 1, вода). Оптическая чистота = 100%.
Анализ для C21H25ClN2О3•2HCl, %:
Рассчитано,%: C 54,61; H 5,90; N 6,07.
Найдено, %: C 54,18; H 6,02; N 5,68.
Следующие соединения подвергают фармакологическим испытаниям, результаты которых приводятся ниже:
-/-/-1-[(4-хлорфенил/фенилметил] пиперазин [соединение A, получено в примере 4.1];
- /+/-1-[/4-хлорфенил/фенилметил] пиперазин [соединение B, получено в примере 4.2];
- левовращающий дихлоргидрит 1-[/4-хлорфенил/фенилметил]-4-[/3 метилфенил/метил]пиперазина [соединение C, получено в примере 5.1];
- правовращающий дихлоргидрат 1-[/4-хлорфенил/фенилметил]-4- [/3-метилфенил/метил]пиперазина [соединение D, получено в примере 5.2];
- левовращающий дихлоргидрат 1-[/4-трет.-бутилфенил/метил]-4- [/4-хлорфенил/фенилметил]пиперазина [соединение E, получено в примере 5.3];
- правовращающий дихлоргидрат 1-[/4-трет. -бутилфенил/метил]-4- [/4-хлорфенил/фенилметил]пиперазина [соединение F, получено в примере 5.4];
- левовращающий дихлоргидрат 2-[2-[4-{/4-хлорфенил/фенилметил}-1-пиперазинил/этокси]этанола [соединение G, получено в примере 5.5];
- правовращающий дихлоргидрат 2-[2-/4-{/4-хлорфенил/фенилметил}- 1-пиперазинил/этокси]этанола [соединение H, получено в примере 5.6];
- левовращающий дихлоргидрат 2-[2-/2-{4-/(4-хлорфенил)фенилметил/-1-пиперазинил}этокси]этокси] этанола /соединение I, получено в примере 5.7/;
- правовращающий дихлоргидрат-2-{2-[2-/4-{/4-хлорфенил/ фенилметил}-1-пиперазинил/этокси]этокси}этанола /соединение F, получено в примере 5.8/;
- /-/-2-[2-/4-{ /хлорфенил/фенилметил} -1-пиперазинил/ этокси]-ацетамид /соединение K, получено в примере 5.9/;
/+/-2-[2-/4-{ /4-хлорфенил/фенилметил} -1-пиперазинил/ этокси]-ацетамид /соединение L, получено в примере 5.10/;
- левовращающий дималеат метил-2-[2-/4-{/4-хлорфенил/ фенилметил}-1-пиперазинил/этокси]ацетата /соединение M, получено в примере 5.11/;
- правовращающий дималеат метил 2-[2-/4-{/4-хлорфенил/фенилметил}-1-пиперазинил/этокси]ацетата [соединение N, получено в примере 5.12];
- левовращающий дихлоргидрат 2-[2-/4-{/4-хлорфенил/фенилметил}-1-пиперазинил-/этокси]-уксусной кислоты /соединение O, получено в примере 5.1,; и
- праворащающий дихлоргидрат 2-[2-/4-{/4-хлорфенил/фенилметил}-1-пиперазинил/этокси]уксусной кислоты /соединение P, получено в примере 5.14/.
1. Средство по отношению к рецептору H1 гистамина
Средство соединений к рецептору H1 гистамина коры головного мозга крысы определяют с помощью метода, описанного M.M. BIL L AH и сотр., J. Pharmacol. Exp. Ther. 252, /3/, /1990/, 1090 - 1096.
Эти классические испытания позволяют выявить способность в фиксации на рецепторе H1 гистамина, с одной стороны, изучаемого соединения, и, с другой стороны, радиолиганда, который в частном случае рецептора H1 гистамина представляет собой /3H/ мепирамин и является известным селективным антагонистом этого рецептора.
Кривые сдвига фиксации /3H/ мепирамина строят при использовании различных концентраций изучаемых соединений, включая величины от 10-10 до 10-4 моль/л и при использовании 4,5•10-9 моль/л /3H/ мепирамина [24,8 Ci/ммоль; выпускается New England Nuclear, Бельгия].
Кору головного мозга самцов крыс Sprague-Dawley гомогенизируют в 2 мл буфера Трис-HCl, 20 мМ /pH = 7,4/, содержащего 250 мМоль сахарозы.
Гомогенизаты центрифугируют в течение 30 минут при 30000 g, при 4oC. Осадки после центрифугирования снова суспендируют в том же самом свежем буфере и хранят в жидком азоте.
Для определения фиксации на рецепторе H1 образцы, содержащие 0,5 мг мембранного протеина коры, в 0,5 мл 50 мМ Трис-HCl-буфера /pH = 7,4/, содержащего 2 ммоль хлорида магния, инкубируют при 25oC в течение 60 минут в присутствии /3H/ мепирамина и изучаемого соединения. Фиксированный /3H/ мепирамин отделяют от свободного радиолиганда путем быстрой фильтрации через фильтр Whatman GF/C, предварительно пропитанный в течение по крайней мере 2 часов 0,1%-ным раствором полиэтиленимина, чтобы уменьшить возможность неспецифической фиксации радиолиганда с другими протеинами. Затем остаток от фильтрации промывают 4 раза по 2 мл 50 мМ Трис-HCl-буфера /pH = 7,4/, охлажденного на ледяной бане. Радиоактивность измеряют с помощью спинтилляционного счетчика частиц бета Tri-corb 1090 /Camberra-Pakcard, Бельгия/. Неспецифическую фиксацию оценивают в присутствии водного 10 мкмоль раствора цетиризина и она составляет 30% от общей фиксации. Величины IC50 изучаемых соединений [концентрация в моль /л, необходимые для ингибирования 30% фиксации радиолиганда на рецепторе Hi] определяют путем анализа кривых, отражающих способность фиксации /A.DEL EAN и сотр., Mol. Pharmacol., 21, /1982/, 5 - 16/ и значения констант ингибирования /Ki/ рассчитывают с помощью уравнения CHENG и FRUSOFF /Y.C.CHENG и W.H.PRUSOFF, Biochem, Pharmacl., 22, /1973/, 3099 - 3108/.
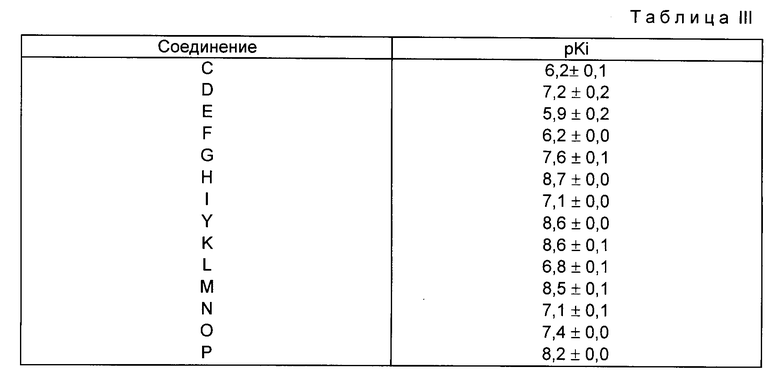
В нижеприведенной таблице III приводятся значения pKi [кологарифм Ki/, рассчитанные на Ki] среднее значение + отклонение от среднего /n = 2/.
Из этой таблицы следует, что соединения формулы V обладают хорошей антигистаминной активностью. Эти результаты также показывают, что между энантиомерами одного и того же соединения существует разница в pKi, которая соответствует разнице в относительном сродстве /следовательно, в Ki/ фактора от 2 до 64 к рецептору H1 коры головного мозга крысы. Такое различие указывает на то, что можно специфически использовать энантиомер, который обладает наибольшим сродством к этому типу рецептора [например, соединение F по сравнению с его энантиомером I] в качестве анксиолотического или транквилизирующего агента для лечения патологий, которые увеличивают возбуждение центральной нервной системы.
2. Антигистаминные периферические свойства
Периферические антигистаминные свойства соединений демонстрируют путем измерения ингибирования сокращения изолированной трахеи морской свинки, вызываемого гистамином, согласно методу, описанному M.H. AMIRI и G.CABELLA /Anal. Embryol., 178 /1988/, 389 - 397/.
Извлекают трахеи у морских свинок Dunkin-Hartley обоего пола (вес: 250 - 500 г) и разрезают их на 4 фрагмента по 3 сегмента хряща в каждом. Эти фрагменты погружают в раствор Krebs-Heinseleit при 37oC, содержащий 10-7 мол/л атропина и 10-5 моль/л индометацина, и натягивают с помощью веса 1 г. Раствор аэрируют путем пропускания тока кислорода, содержащего 5% диоксида углерода. Каждую модификацию натяжения регистрируют с помощью индикатора изометрической силы K 30 /фирмы Hugo Sachs Elektronik/, связанного с амплификатором и регистратором Sanborn 7700 /фирмы Хьюлетт Паккард/. Таким образом полученный препарат оставляют стабилизироваться в течение 1 часа, в течение которого, если необходимо, снова выверяют базовую лини натяжения.
Каждый препарат предварительно сокращают путем добавления в среды 10-4 моль/л гистамина; наблюдаемое сжатие принимают за стандарт /100%/. После промывки и стабилизации в качестве контроля чертят кумулятивную кривую воздействий гистамина в зависимости от концентрации /10-6, 10-5 и 10-4 моль/л/.
На том же самом препарате регистрируют еще 4 других кумулятивных кривых воздействий гистамина в зависимости от его концентрации для четырех возрастающих концентраций испытуемых соединений.
Испытуемые соединения вводят в препарат за 5 минут до гистамина. Между каждым измерением препараты промывают по крайней мере 4 раза с интервалом 5 минут между каждой промывкой. Каждое соединение испытывают по крайней мере на 6-и фрагментах трахеи. Когда вычерчена последняя кривая, то в среду добавляют дополнительные количества гистамина 3.2•10-4 и 10-3 моль/л для определения, имеет ли антагонизм конкурентноспособную природу или нет.
Когда наблюдают неконкурентноспособное ингибирование, то рассчитывают pD2, т. е. кологарифм концентрации испытуемого соединения, которая вызывает ингибирование 50% максимально зарегистрированного сокращения /J.M.VAN ROSSUM, Arch. Int. Pharmacodyn., 143 /1963/, 299 - 230/. Когда наблюдают конкурентноспособное ингибирование, то рассчитывают pA2, т.е. кологарифм концентрации испытуемого соединения, которая вызывает удваивание дозы гистамина, необходимой для достижения того же самого эффекта сжатия.
В нижеприведенной таблице IV даются pA или pD, рассчитанные /среднее значение ± стандартное отклонение/ для испытуемых соединений.
Этот тест демонстрирует удивительную характеристику пар лево- и правовращающего испытуемых энантиомеров. За исключением пары энантиомеров A и B, установлено, что для всех других пар один энантиомер является конкурентноспособным ингибитором, тогда как другой представляет собой неконкурентноспособный ингибитор, вследствие чего представляет интерес получение оптически чистых производных 1-[/4-хлорфенил/фенилметил]-пиперазина.
Конкурентноспособные ингибиторы представляют интерес вследствие того, что они обычно обладают худшим сродством к рецептору H1 гистамина коры головного мозга крысы, которое позволяет предсказать, что антиаллергические свойства этих соединений мало ассоциированы или вообще неассоциированы с нежелательными воздействиями на центральную нервную систему, такими, как, например, успокоение или сонливость.
Неконкурентноспособные ингибиторы обладают преимуществом точно также ингибировать воздействие гистамина, даже когда он присутствует в высоких локальных концентрациях. Следовательно, они лучше применимы при топическом лечении патологий кожи или слизистой оболочки.
3. Ингибирование кожной реакции, вызванной гистамином у собаки.
Среди видов животных собаку рассматривают как вид, который имеет чувствительность к гистамину, относительно близкую к таковой человека. Следовательно, рассматривают, что антигистаминная активность соединения, наблюдаемая в случае собаки, предсказывает таковую, которая будет наблюдаться у человека.
В этом тесте, используют 9 собак Beagle со средним весом 12,6 кг и в возрасте около 2-х лет, брюхо которых локально выбрито. В таким образом выбритую зону подкожно инъекцируют 50 мкл водного 0,9%-ного раствора хлорида натрия, содержащего 10 мкг/мл гистамина. Одновременно внутрикожно в дозе 0,1 мл/кг инъекцируют раствор синьки Эванса [60 мг/мл в водном 0,9%-ном растворе хлорида натрия] . Аллергическая реакция проявляется по месту подкожной инъекции и видно появление папулы, поверхность которой измеряют точно спустя 30 минут после обеих инъекций. Эту поверхность принимают за стандарт /100%/.
Затем вводят перорально испытуемое соединение, в дозе 0,15 мг/кг /0,32•10-6 моль/кг/.
Спустя 0,5; 1,5; 6; 9; 12; 24 и 32 часа, считая от введения испытуемого соединения, индуктируют новые папулы в разных местах брюха за счет инъекции гистамина. Каждый раз измеряют поверхность индуктированной папулы спустя 30 минут после инъекции гистамина.
Антигистаминное воздействие соединения на кожную аллергическую реакцию определяется путем измерения уменьшения поверхности папул, индуктированных после введения соединения, по отношению к поверхности принятой за стандарт папулы, и выражают в процентах.
В нижеприведенной таблице V представлена антигистаминная активность, полученная для соединения P. В табл. V:
первая колонка указывает время, выраженное в часах, прошедшее с момента введения испытуемого соединения;
вторая колонка: поверхность, выраженная в мм2, папул, индуктированных гистамином [среднее значение, наблюдаемое для 9 собак, + стандартное отклонение]
третья колонка: уменьшение в процентах поверхности папул, наблюдаемое с течением времени, по сравнению со стандартной поверхностью, и
четвертая колонка: статистическое значение эффекта, наблюдаемого с течением времени, оцениваемое с помощью теста Wilcoxon.
Констатируют, что уменьшение поверхности папул, наблюдаемое спустя 30 минут после введения соединения P, составляет 15%. Максимальное ингибирование наблюдается спустя 3 часа и достигает 57%. Спустя 32 часа еще наблюдают статистически заметное ингибирование 33%.
4. Токсичность.
Соединения формулы V обладают слабой токсичностью. Летальная доза [вызывающая гибель 2-х мышей из 3-х во время интраперитонеальной инъекции соединений] отчетливо выше дозы, которая ингибирует кожную реакцию, вызываемую гистамином у собаки. В таблице VI даны значения летальных доз для нескольких соединений формулы V.
5. Дозировка и введение.
Соединения формулы V в особенности обладают антиаллергической, антигистаминной активностями, с одной стороны, транквилизирующей и анксиолитической активностями, с другой стороны. Фармацевтические композиции, включающие эти соединения, могут вводиться орально, парентерально или ректально; их можно также вводить в виде ингаляций или аэрозолей для введения в нос или в форме кремов или мази.
Для орального введения используют жидкие или твердые формы, такие, как таблетки, желатиновые капсулы с лекарством, драже, гранулы, растворы, сиропы и т.д.
Для парентерального введения используют, например, водные или масляные растворы, суспензии или эмульсии.
Для ректального/кишечного/введения используют свечи,
Вышеуказанные фармацевтические формы готовят способами, обычно используемыми фармацевтами, и они могут содержать обычные добавки в фармацевтически нетоксичных дозах, такие, как диспергаторы, стабилизаторы, консерваторы, подслащивающие вещества, красители, и т.д.
Процентное количество действующего начала может изменяться, в зависимости от фармацевтических форм, в широких пределах, зависящих от способа введения и в особенности от частоты введения. Что касается суточной дозировки, то она может изменяться в широкий гамме, от 0,5 до 100 мг действующего начала, предпочтительно 2 - 20 мг.


Изобретение может быть использовано в химии гетероциклических соединений и медицине. Описываются лево- и правовращающие энантиомеры 1-[(4-хлорфенил)фенилметил]-4-[(4-метилфенил)сульфонил]пиперазина формулы I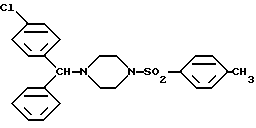
способ их получения и их использования для получения оптически чистых энантиомеров 1-[(4-хлорфенил)фенилметил]пиперазина, которые являются ценными промежуточными продуктами для получения терапевтических оптически активных антигистаминных соединений общей формулы V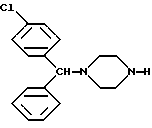
где R - метил, (3-метилфенил)метил, (4-трет. бутилфенил)-метил, 2-(2-гидроксиэтокси)этил, 2-(2-)-2-гидроксиэтокси-(этокси)этил, 2-(карбамоилметокси)этил, 2-(метоксикарбонилметокси)этил, 2-(карбометокси)этил, очень высокой оптической чистоты. 5 с. и 3 з.п. ф-лы, 6 табл.
\\\1 1. Энантиомеры 1-[(4-хлорфенил)фенилметил]-4-[(4-метилфенил)сульфонил]пиперазина лево- и правовращающих форм формулы I \\\6 $$$ \\\2 2. Энантиомеры формулы I по п.1 в качестве исходных соединений для получения энантиомеров 1-[(4-хлорфенил)фенилметил]пиперазина формулы IV \\\6 $$$ \\\1 предшественников антигистаминных соединений. \\\2 3. Способ получения лево- и правовращающего энантиомеров 1-[(4-хлорфенил)фенилметил]-4-[(4-метилфенил)сульфонил] пиперазина формулы I, отличающийся тем, что энантиомер (4-хлорфенил)фенилметиламина формулы II \\\6 $$$ \\\1 подвергают взаимодействию с N, N-диэтил-4-метилбензолсульфонамидом общей формулы III \\\6 $$$ \\\1 где X - хлор, бром или йод, или (4-метилфенил)сульфонилокси- или метилсульфонилоксигруппа, \ \ \ 1 в присутствии 2,2 - 4,4 эквивалентов органического или неорганического основания на эквивалент энантиомера (4-хлорфенил) фенилметиламина и при температуре кипения реакционной смеси. \\\2 4. Способ по п.3, отличающийся тем, что основание выбирают из группы, включающей этилдиизопропиламин, N-этилморфолин, 2,4,6-триметилпиридин, триэтиламин и карбонат щелочного металла. \\\2 5. Способ по п.4, отличающийся тем, что основанием является этилдиизопропиламин. \\\2 6. Способ получения лево- и правовращающего энантиомеров 1-[(4-хлорфенил)фениламетил]пиперазина формулы IV по п.2, отличающийся тем, что энантиомер 1-[(4-хлорфенил)фенилметил)-4-[(4-метилфенил)сульфонил] пиперазина формулы I по п.1 гидролизуют с помощью бромоводородной кислоты в среде уксусной кислоты в присутствии фенольного соединения, предпочтительно 4-гидроксибензойной кислоты, и при 18 - 100<198>C. \\\2 7. Лево- и правовращающие энантиомеры 1-[(4-хлорфенил)-фенилметил]пиперазина формулы IV в качестве полупродукта для получения лево- и правовращающей формы терапевтически активных производных 1-[(4-хлорфенил)фенилметил]пиперазинов общей формулы V \\\6 $$$ \\\1 где R - метильный, (3-метилфенил)метильный, (4-трет-бутилфенил)метильный, 2-(2-гидроксиэтокси)этильный, 2-(2-(2-гидроксиэтокси)этокси)этильный, 2-(карбамоилметокси)этильный, 2-(метоксикарбонилметокси)этильный или 2-(карбоксиметокси)этильный радикал, \\ \ 1 путем взаимодействия при нагревании с галогенидом общей формулы \\\6 RX, \ \ \ 1 где R имеет указанное значение; \\\4 X - галоген. \\\2 8. Соединения общей формулы V, представляющие собой левовращающий дихлоргидрат 1-[(4-хлорфенил)фенилметил]-4-[(3-метилфенил)метил]пиперазина, правовращающий дихлоргидрат 1-[(4-хлорфенил)фенилметил] -4-[(3-метилфенил)метил] пиперазина, левовращающий дихлоргидрат 1-[(4-третбутилфенил)метил]-4-[(4-хлорфенил)фенилметил] пиперазина, правовращающий дихлоргидрат 1-[(4-трет-бутилфенил)метил]-4-[(4-хлорфенил)фенилметил]пиперазина, левовращающий дихлоргидрат 2-[2-/4-{(4-хлорфенил)фенилметил}-1-пиперазинил/этокси]этанола, правовращающий дихлоргидрат 2-[2-/4-{(4-хлорфенил)фенилметил}-1-пиперазинил/этокси]этанола, левовращающий дихлоргидрат 2-[2-/2-{4-/(4-хлорфенил)-фенилметил/-1-пиперазинил} этокси/этокси]этанола, правовращающий дихлоргидрат 2-[2-/2-{4-/(4-хлорфенил)-фенилметил/-1-пиперазинил}этокси/этокси]этанола, левовращающий 2-[2-/4-{ /4-хлорфенил/фенилметил} -1-пиперазинил/этокси] ацетамид и его правовращающий дихлоргидрат, правовращающий 2-[2-/4-{4-хлорфенил/фенилметил} -1-пиперазинил/-этокси] ацетамид и его левовращающий дихлоргидрат, левовращающий дималеат метил-2-[2-{/4-хлорфенил/фенилметил}-1-пиперазинил/этокси]ацетат, правовращающий дималеат метил-2-[2-/4-{/4-хлорфенил/-фенилметил}-1-пиперазинил/этокси]ацетат.
| GB, патент, 2225321, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, 0598123 A, кл | |||
| УСТРОЙСТВО ПАРОПЕРЕГРЕВАТЕЛЯ | 1920 |
|
SU295A1 |

