Настоящее изобретение относится к соединению формулы
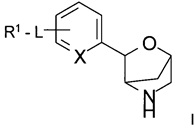 ,
,
где
L представляет собой связь, -C(O)NH-, -NHC(O)-, -CH2NHC(O)-, -CH2C(O)NH-, -CH2NH-, -NH- или -NHC(O)NH-;
R1 представляет собой атом водорода, низший алкил, атом галогена, низший алкокси-алкил, низший алкокси, замещенный атомом галогена, низший алкил, замещенный атомом галогена, или
представляет собой фенил или гетероарил, выбранный из группы, состоящей из пиридинила, пиримидинила, пиразинила или пиразолила, и при этом фенил и гетероарил возможно замещены одним, двумя или тремя заместителями, выбранными из группы, состоящей из атома галогена, низшего алкила, низшего алкокси, низшего алкила, замещенного атомом галогена, низшего алкокси, замещенного атомом галогена, циклоалкила или -O-СН2-циклоалкила;
X представляет собой СН или N;
или к его фармацевтически приемлемой соли присоединения кислоты, ко всем рацемическим смесям, всем соответствующим энантиомерам и/или оптическим изомерам.
К настоящему времени установлено, что соединения формул I обладают высокой аффинностью к рецепторам, ассоциированным со следовыми аминами (trace amine associated receptors; TAAR), особенно к TAAR1.
Данные соединения можно применять при лечении депрессии, тревожных расстройств, биполярного расстройства, синдрома дефицита внимания и гиперактивности (attention deficit hyperactivity disorder; ADHD), расстройств, вызванных стрессом, психотических расстройств, таких как шизофрения, неврологических заболеваний, таких как болезнь Паркинсона, нейродегенеративных расстройств, таких как болезнь Альцгеймера, эпилепсии, мигрени, гипертензии, злоупотребления алкоголем или наркотиками, и метаболических расстройств, таких как расстройства приема пищи, диабет, осложнения диабета, ожирение, дислипидемия, расстройств потребления и ассимиляции энергии, расстройств и нарушений теплового гомеостаза, расстройств сна и циркадного ритма и сердечно-сосудистых заболеваний.
Некоторые физиологические эффекты (т.е. воздействия на сердечнососудистую систему, гипотензия, вызывание седативного эффекта), описанные для соединений, которые могут связываться с адренергическими рецепторами, (WO 02/076950, WO 97/12874 или ЕР 0717037) могут рассматриваться как нежелательные побочные эффекты в случае лекарственных средств, предназначенных для лечения заболеваний центральной нервной системы, которые описаны выше. Поэтому желательно получить лекарственные средства, обладающие селективностью в отношении рецептора TAAR1 в противоположность адренергическим рецепторам. Объекты настоящего изобретения демонстрируют селективность в отношении рецептора TAAR1 по сравнению с адренергическими рецепторами, в частности высокую селективность, в противоположность адренергическим рецепторам альфа1 и альфа2 человека и крысы.
Классические биогенные амины (серотонин, норэпинефрин, эпинефрин, допамин, гистамин) как нейромедиаторы играют важную роль в центральной и периферической нервной системе [1]. Их синтез и хранение, а также их деградация и обратный захват после высвобождения строго регулируются. Известно, что дисбаланс уровней биогенных аминов ответственен за изменение функции головного мозга при многих патологических состояниях [2-5]. Соединения, образующие второй класс эндогенных аминов, так называемые следовые амины (trace amine; ТА), очень схожи с классическими биогенными аминами по своей структуре, метаболизму и субклеточной локализации. ТА включают пара-тирамин, β-фенилэтиламин, триптамин и октопамин, и их уровень в нервной системе млекопитающих существенно ниже уровней классических биогенных аминов [6].
Нарушение их регуляции связано с различными психическими заболеваниями, такими как шизофрения и депрессия [7], и другими состояниями, такими как синдром дефицита внимания и гиперактивности, головная боль типа мигрени, болезнь Паркинсона, злоупотребление алкоголем или наркотиками и расстройства приема пищи [8, 9].
В течение долгого времени существование ТА-специфичных рецепторов являлось всего лишь гипотезой, основанной на присутствии в центральной нервной системе (ЦНС) человека и других млекопитающих анатомически дискретных сайтов связывания, обладающих высокой аффинностью к ТА [10, 11]. Соответственно, считалось, что фармакологическое действие ТА опосредовано хорошо известными механизмами, как и действие классических биогенных аминов, то есть либо стимуляцией их высвобождения, либо ингибированием их обратного захвата, либо "перекрестной реактивностью" с их рецепторными системами [9, 12, 13]. За последнее время данная точка зрения претерпела значительные изменения в связи с идентификацией нескольких членов нового семейства G-белок-связанных рецепторов (G protein-coupled receptor; GPCR), рецепторов, ассоциированных со следовыми аминами (TAAR) [7, 14]. У человека имеются 9 кодирующих TAAR генов (в том числе 3 псевдогена), а у мыши 16 генов (в том числе 1 псевдоген). Гены, кодирующие TAAR, не содержат интронов (за одним исключением, TAAR2 содержит 1 интрон) и расположены друг за другом на одном и том же хромосомном сегменте. Филогенетическое родство генов этих рецепторов, выявленное при углубленном сравнении с GPCR фармакофором (GPCR, от англ. G-protein coupled receptor - рецептор, сопряженный с G-белком) на предмет их сходства, и фармакологические данные подтверждают, что эти рецепторы образуют три различных подсемейства [7, 14]. TAAR1 относится к первому подклассу, состоящему из четырех генов (TAAR1-4), являющихся высококонсервативными в геномах человека и грызунов. ТА активируют TAAR1 через субъединицы Gα. Было показано, что нарушение регуляции ТА связано с этиологией различных заболеваний, таких как депрессия, психоз, синдром дефицита внимания и гиперактивности, злоупотребление алкоголем или наркотиками, болезнь Паркинсона, головная боль типа мигрени, расстройства приема пищи, метаболические расстройства, и поэтому применение лигандов TAAR1 при лечении этих заболеваний является весьма перспективным.
Таким образом, получение новых знаний о рецепторах, ассоциированных со следовыми аминами, весьма актуально.
Использованные ссылки
1. Deutch, A.Y. and Roth, R.H. (1999) Neurotransmitters. В: Fundamental Neuroscience (2oe издание) (Zigmond, M.J., Bloom, F.E., Landis, S.C., Roberts, J.L and Squire, L.R., eds.), pp. 193-234, Academic Press.
2. Wong, M.L. and Licinio, J. (2001) Research and treatment approaches to depression. Nat. Rev. Neurosci., 2, 343-351.
3. Carlsson, A. et al. (2001) Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu. Rev. Pharmacol. Toxicol., 41, 237-260.
4. Tuite, P. and Riss, J. (2003) Recent developments in the pharmacological treatment of Parkinson's disease. Expert Opin. Investig. Drugs, 12, 1335-1352.
5. Castellanos, F.X. and Tannock, R. (2002) Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nat. Rev. Neurosci., 3, 617-628.
6. Usdin, Earl; Sandler, Merton; Editors. Psychopharmacology Series, Vol. 1: Trace Amines and the Brain [Proceedings of a Study Group at the 14th Annual Meeting of the American College of Neuropsychoparmacology, San Juan, Puerto Rico] (1976).
7. Lindemann, L. and Hoener, M. (2005) A renaissance in trace amines inspired by a novel GPCR family. Trends in Pharmacol. Sci., 26, 274-281.
8. Branchek, T.A. and Blackburn, T.P. (2003) Trace amine receptors as targets for novel therapeutics: legend, myth and fact. Curr. Opin. Pharmacol., 3, 90-97.
9. Premont, R.T. et al. (2001) Following the trace of elusive amines. Proc. Natl. Acad. Sci. USA, 98, 9474-9475.
10. Mousseau, D.D. and Butterworth, R.F. (1995) A high-affinity [3H] tryptamine binding site in human brain. Prog. Brain Res., 106, 285-291.
11. McCormack, J.K. et al. (1986) Autoradiographic localization of tryptamine binding sites in the rat and dog central nervous system. J. Neurosci., 6, 94-101.
12. Dyck, L.E. (1989) Release of some endogenous trace amines from rat striatal slices in the presence and absence of a monoamine oxidase inhibitor. Life Sci., 44, 1149-1156.
13. Parker, E.M. and Cubeddu, L.X. (1988) Comparative effects of amphetamine, phenylethylamine and related drugs on dopamine efflux, dopamine uptake and mazindol binding. J. Pharmacol. Exp. Ther., 245, 199-210.
14. Lindemann, L. et al. (2005) Trace amine associated receptors form structurally and functionally distinct subfamilies of novel G protein-coupled receptors. Genomics, 85, 372-385.
Объектами настоящего изобретения являются новые соединения формулы I и их фармацевтически приемлемые соли, их применение для изготовления лекарственных средств для лечения заболеваний, связанных с биологической функцией рецепторов, ассоциированных со следовыми аминами, их получение и лекарственные средства на основе соединения по изобретению для регулирования и предупреждения таких заболеваний, как депрессия, тревожные расстройства, биполярное расстройство, синдром дефицита внимания и гиперактивности, расстройства, вызванные стрессом, психотические расстройства, такие как шизофрения, неврологические заболевания, такие как болезнь Паркинсона, нейродегенеративные расстройства, такие как болезнь Альцгеймера, эпилепсия, мигрень, злоупотребление алкоголем или наркотиками и метаболические расстройства, такие как расстройства приема пищи, диабет, осложнения диабета, ожирение, дислипидемия, расстройства потребления и ассимиляции энергии, расстройства и нарушения теплового гомеостаза, расстройства сна и циркадного ритма и сердечно-сосудистые заболевания.
Предпочтительными показаниями к применению соединений по настоящему изобретению являются депрессия, психоз, болезнь Паркинсона, тревога, синдром дефицита внимания и гиперактивности (ADHD) и диабет.
Использованный в данном описании термин "низший алкил" означает группу с насыщенной прямой или разветвленной цепью, содержащую от 1 до 7 атомов углерода, например, метил, этил, пропил, изопропил, н-бутил, изобутил, 2-бутил, трет-бутил и тому подобное. Предпочтительными алкильными группами являются группы, содержащие 1-4 атома углерода.
Использованный в данном описании термин "низший алкокси" означает группу, где алкильный остаток является таким, как определено выше, и присоединен через атом кислорода.
Термин "галоген" означает хлор, йод, фтор и бром. Предпочтительной группой галогена является атом фтора.
Использованный в данном описании термин "низший алкил, замещенный атомом галогена" означает группу с насыщенной прямой или разветвленной цепью, содержащую от 1 до 7 атомов углерода, как определено для термина "низший алкил", где по меньшей мере один атом водорода заменен на атом галогена. Предпочтительным атомом галогена является атом фтора. Примерами таких групп являются CF3, CHF2, CH2F, CH2CF3 или CH2CHF2.
Использованный в данном описании термин "низший алкокси, замещенный атомом галогена" означает низшую алкоксигруппу, которая определена выше, где по меньшей мере один атом водорода заменен на атом галогена. Примерами таких групп являются OCF3, OCHF2, OCH2F, OCH2CF3 или OCH2CHF2.
Термин "циклоалкил" означает насыщенное углеродное кольцо, содержащее от 3 до 6 атомов углерода, например, циклопропил, циклобутил, циклопентил или циклогексил.
Термин "фармацевтически приемлемые соли присоединения кислоты" включает в себя соли с неорганическими и органическими кислотами, такими как соляная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфоновая кислота, пара-толуолсульфоновая кислота и тому подобное.
Одним из воплощений данного изобретения являются соединения формулы I, где R1 представляет собой атом водорода, низший алкил, атом галогена, низший алкокси-алкил, низший алкокси, замещенный атомом галогена или низший алкил, замещенный атомом галогена, и L является таким, как описано выше, например, приведенные ниже соединения:
(1R,3S,4R)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан;
(1S,3R,4S)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан;
N-бутил-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин;
(1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан;
(1R,3S,4R)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан;
N-(3-метоксипропил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)ацетамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(3,3,3-трифторпропокси)ацетамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)ацетамид;
4,4,4-трифтор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бутанамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(3,3,3-трифторпропокси)ацетамид;
4,4,4-трифтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бутанамид;
(1R,3R,4R)-3-(2-пиридил)-2-окса-5-азабицикло[2.2.1]гептан;
(1S,3S,4S)-3-(2-пиридил)-2-окса-5-азабицикло[2.2.1]гептан или
(1R,3S,4R)-3-(2-фторфенил)-2-окса-5-азабицикло[2.2.1]гептан.
Одним из воплощений данного изобретения являются другие соединения формулы I, где R1 представляет собой фенил, который возможно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из атома галогена, низшего алкила, низшего алкокси, низшего алкила, замещенного атомом галогена, низшего алкокси, замещенного атомом галогена, циклоалкила или -O-СН2-циклоалкилфенила, и L является таким, как описано выше, например, приведенные ниже соединения:
3-хлор-N-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-хлор-N-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
1-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-[4-(трифторметил)фенил]мочевина;
1-(4-хлорфенил)-3-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]мочевина;
1-(3-хлорфенил)-3-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]мочевина;
4-хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
3-хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
3-хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-(циклопропилметокси)-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-этокси-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-этокси-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-(циклопропилметокси)-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
1-(4-хлорфенил)-3-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]мочевина;
N-[(4-хлорфенил)метил]-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин;
4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметил)фенил]метил]анилин;
N-[(4-фторфенил)метил]-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин;
4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметокси)фенил]метил]анилин;
N-(4-хлорфенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(4-бромфенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(4-фторфенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(4-этоксифенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[4-(трифторметил)фенил]бензамид;
4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметил)фенил]метил]бензамид;
N-[(4-хлорфенил)метил]-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
4,4,4-трифтор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бутанамид;
N-(4-бромфенил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(4-фторфенил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(4-этоксифенил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметил)фенил]метил]бензамид или
N-[(4-хлорфенил)метил]-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид.
Одним из воплощений данного изобретения являются соединения формулы I, где R1 представляет собой пиридинил, пиримидинил, пиразинил или пиразолил, которые возможно замещены одним, двумя или тремя заместителями, выбранными из группы, состоящей из атома галогена, низшего алкила, низшего алкокси, низшего алкила, замещенного атомом галогена, низшего алкокси, замещенного атомом галогена, циклоалкила или -O-СН2-циклоалкилфенила, и L является таким, как описано выше, например, приведенные ниже соединения:
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-амин;
6-этокси-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-3-карбоксамид;
6-этокси-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-3-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(2,2,2-трифторэтокси)пиридин-3-карбоксамид;
2-циклопропил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-амин;
5-хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-2-амин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиримидин-4-амин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиразин-2-амин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиримидин-2-амин;
5-хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-2-амин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиридин-4-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиридин-4-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(2,2,2-трифторэтокси)пиридин-3-карбоксамид;
2-циклопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиразин-2-амин;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиримидин-2-амин;
2-этил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиридин-4-амин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-карбоксамид;
4-хлор-3-циклопропил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиримидин-4-карбоксамид;
3-изопропил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(трифторметил)пиридин-3-карбоксамид;
4-хлор-3-этокси-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-хлор-3-метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-хлор-1-метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-пропил-пиразол-3-карбоксамид;
4-хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-пропил-1Н-пиразол-5-карбоксамид;
3-этил-4-метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(2,2,2-трифторэтокси)пиридин-2-карбоксамид;
N-(6-хлор-3-пиридил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(трифторметил)пиридин-3-амин;
N-(6-этокси-3-пиридил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
3-этил-4-метил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-пропил-1Н-пиразол-5-карбоксамид;
3-циклопропил-4-метил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(трифторметил)пиридин-3-карбоксамид;
2-этил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид;
3-изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-хлор-3-этил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
3-циклопропил-4-фтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-фтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-пропил-1Н-пиразол-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-4-(2,2,2-трифторэтокси)пиримидин-2-амин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)пиримидин-4-амин;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиримидин-4-карбоксамид;
4-хлор-3-циклопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)пиримидин-4-амин;
2-изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(2,2,2-трифторэтокси)пиразол-3-карбоксамид;
3-бутил-4-фтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
3-бутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-(6-хлор-3-пиридил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(6-этокси-3-пиридил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
4-хлор-3-этокси-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-бром-3-этил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-фтор-3-изобутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
3-изобутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-хлор-3-изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид или
4-фтор-3-изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид.
Получение соединений формулы I по настоящему изобретению может быть осуществлено путем последовательного или конвергентного синтеза. Синтез соединений по изобретению показан на приведенных далее схемах 1-8 и в описании 106 конкретных примеров. Навыки, необходимые для выполнения данной реакции и очистки полученных продуктов, известны специалистам в данной области техники. Заместители и индексы, использованные в следующем далее описании способов, имеют значения, приведенные в данном описании ранее, если не указано иное.
Более подробно, соединения формулы I могут быть получены способами, приведенными ниже, способами, приведенными в разделе Примеры, или аналогичными способами. Условия реакции, подходящие для отдельных стадий реакции, известны специалисту в данной области техники. Последовательность реакций не ограничена той, которая показана на схемах 1-8, однако в зависимости от исходных веществ и соответствующей им реакционной способности последовательность реакционных стадий можно менять по своему усмотрению. Исходные вещества либо имеются в продаже, либо могут быть получены способами, аналогичными способам, приведенным ниже, способами, описанными в ссылках, цитированных в данном описании или в разделе Примеры, или способами, известными в данной области техники.
Соединения по настоящему изобретению формулы I и их фармацевтически приемлемые соли могут быть получены способами, известными в данной области техники, например, способами, описанными ниже, при этом способ включает:
а) отщепление N-защитной группы (PG) от соединений формулы
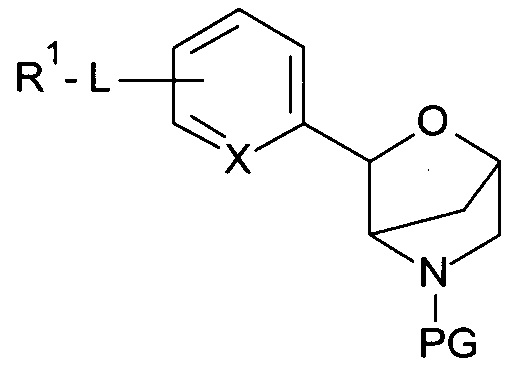
с получением соединения формулы
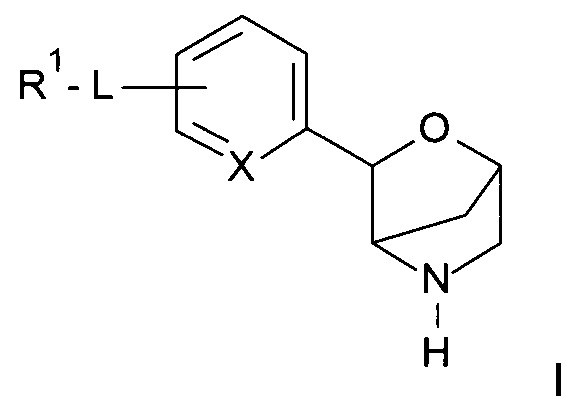 ,
,
где PG представляет собой N-защитную группу, выбранную из -С(O)O-трет-бутила (ВОС), и другие определения являются такими, как описано выше, и
при желании превращение полученных соединений в фармацевтически приемлемые соли присоединения кислоты.
ОБЩАЯ МЕТОДИКА
Схема 1
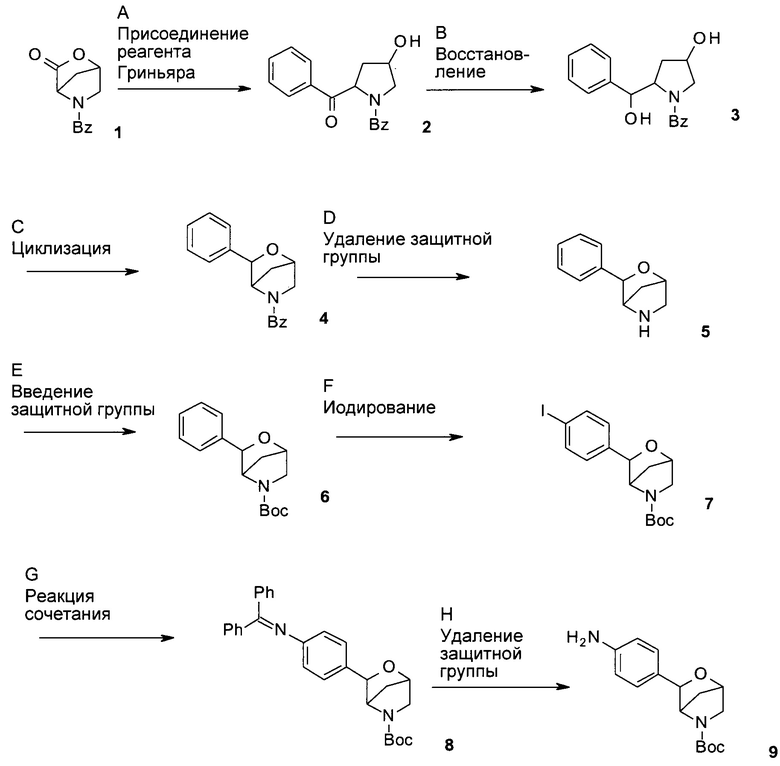
Стадия А: превращение лактона 1 в кетон 2 можно осуществить посредством присоединения фенильного реагента Гриньяра к лактону 1 и Me(MeO)NH⋅HCl в безводных апротонных органических растворителях, таких как тетрагидрофуран (ТГФ), диэтиловый эфир, 1,2-диметоксиэтан (DME, от англ. - dimethoxyethane), трет-бутил-метиловый эфир (ТВМЕ, от англ. - tert-butyl methyl ether), при температуре от -78°С до 0°С в атмосфере инертного газа.
Предпочтительные условия относятся к использованию фенилмагнийбромида в ТГФ при -70°С в течение 10 часов.
Стадия В: восстановление кетона 2 до соответствующего диола 3 можно осуществить посредством обработки восстанавливающим реагентом, таким как NaBH4, LiBH4, ZnBH4, 9-борабицикло[3.3.1]нонан (9-BBN), комплекс боран-ТГФ, LiAlH4 или диизобутилалюминийгидрид (DIBAL-H), в растворителях, таких как ТГФ, диэтиловый эфир, DME, 1,4-диоксан и ТВМЕ, метанол или этанол.
Предпочтительные условия относятся к использованию NaBH4 в качестве восстанавливающего реагента в МеОН при 0°С в течение 2 часов.
Стадия С: циклизацию диола 3 можно осуществить путем взаимодействия по типу реакции Мицунобу, путем опосредуемой кислотой катионной циклизации, или путем постадийного способа с участием промежуточных сложных сульфонатных эфиров.
В случае взаимодействия по типу реакции Мицунобу такое превращение можно осуществить посредством обработки трифенилфосфином и азодикарбоксилатом, таким как диэтилазодикарбоксилат (DEAD) или диизопропилазодикарбоксилат (DIAD), в эфирных растворителях, таких как диэтиловый эфир, диоксан, ТГФ или ТВМЕ, или других апротонных органических растворителях, таких как толуол и бензол.
В случае опосредуемой кислотой катионной циклизации такое превращение можно осуществить посредством обработки неорганическими кислотами, такими как H2SO4, H3PO4, при повышенных температурах, или посредством обработки органическими кислотами, такими как трифторуксусная кислота, BF3⋅Et2O, возможно с использованием вспомогательного вещества, такого как Et3SiH, в растворителях, таких как дихлорметан, 1,2-дихлорэтан или толуол, при температуре от 0°С до комнатной температуры.
В случае постадийного способа такое превращение можно осуществить посредством обработки диола 3 одним эквивалентом сульфонилхлорида, такого как метансульфонилхлорид или толуолсульфонилхлорид, в присутствии органического основания, такого как пиридин, триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, в эфирных растворителях, таких как диэтиловый эфир, диоксан, ТГФ или ТВМЕ, или с использованием органического основания в качестве растворителя, при температуре от 0°С до 50°С. Полученный сложный сульфонатный эфир можно превратить в защищенный имеющий мостиковую связь морфолин 4 посредством обработки ненуклеофильным основанием, таким как гидрид натрия, трет-бутилат калия или 2-метил-2-бутилат калия, в эфирных растворителях, таких как диэтиловый эфир, диоксан, ТГФ или ТВМЕ.
Предпочтительные условия относятся к способу по типу реакции Мицунобу: к обработке диола 3 DIAD и трифенилфосфином в толуоле при 0°С и продолжению реакции при комнатной температуре в течение 16 часов.
Стадия D: удаление защитной группы можно осуществить, используя или индуцированную основанием реакцию, или постадийный способ с участием бензил-защищенного промежуточного соединения.
В случае индуцированной основанием реакции, удаление защитной группы может быть выполнено посредством обработки основанием, таким как гидразин, KOH, NaOH или Cs2CO3, в растворителях, таких как метанол, этанол, при повышенных температурах, как например, от 90°С до 150°С.
В случае постадийного способа бензоильную защитную группу можно превратить в бензильную защитную группу посредством обработки восстанавливающими реагентами, такие как LiAlH4, ВН3⋅ТГФ и BH3⋅Me2S, в эфирных растворителях, таких как диэтиловый эфир, диоксан, ТГФ или ТВМЕ, при температуре от 0°С до 60°С. Полученную бензильную группу можно удалить, используя или реакцию гидрирования, катализируемую Pd катализатором, или обработку хлорформиатами, такими как ClCOOCH2CH2Cl, ClCOOCH(Cl)Me, ClCOOCH2Ph и ClCOOCH2CCl3, и возможно в присутствии основания, такого как триэтиламин, диизопропилэтиламин и гидроксид натрия, в растворителях, таких как толуол, ТГФ, диэтиловый эфир, диоксан или ТВМЕ, при температурах от комнатной до повышенной температуры.
Предпочтительные условия относятся к постадийному способу с использованием LiAlH4 в ТГФ при температуре от 0°С до комнатной температуры в течение 2 часов для первой стадии с последующей обработкой ClCOOCH2CH2Cl в толуоле при 110°С в течение 16 часов.
Стадия Е: введение защитной группы в имеющий мостиковую связь морфолин 5 можно осуществить посредством обработки ди-трет-бутилкарбонатом, возможно в присутствии органического или неорганического основания, такого как триэтиламин, N,N-диизопропилэтиламин, N-метилморфолин, карбонат калия, карбонат натрия или карбонат цезия, в галогенированных растворителях, таких как дихлорметан или 1,2-дихлорэтан, или эфирных растворителях, таких как диэтиловый эфир, диоксан, ТГФ или ТВМЕ.
Предпочтительные условия относятся к использованию ТГФ в присутствии карбоната калия в качестве основания при комнатной температуре в течение 10 часов.
Стадия F: иодирование имеющего мостиковую связь морфолина 6 можно осуществить посредством обработки галогенирующими реагентами, такими как йод и иодсукцинимид, или комплексы соединений поливалентного йода с йодом, такие как [бис(трифторацетокси)иод]бензол/йод и бис(ацетокси)фенилиод/йод, в галогенированных растворителях, таких как дихлорметан, хлороформ или тетрахлорметан, при температуре от комнатной до 80°С.
Предпочтительные условия относятся к использованию комплекса [бис(трифторацетокси)иод]бензол/йод в тетрахлорметане при комнатной температуре.
Стадия G: сочетание иодида 7 с бензофенонимином можно осуществить в присутствии катализатора на основе палладия или меди, лиганда и основания в растворителях, таких как диоксан, DME, ТГФ, толуол и диметилсульфоксид (DMSO), при повышенных температурах, например, с использованием катализируемой палладием реакции Бухвальда-Хартвига.
Предпочтительные условия относятся к использованию каталитических количеств трис(дибензилиденацетон)дипалладия(0), каталитических количеств 4,5-бис(дифенилфосфино)-9,9-диметилксантена (Xantphos) и Cs2CO3 в толуоле при 100°С в течение 5 часов.
Стадия Н: удаление N-дифенилметиленовой группы в соединении 8 можно осуществить посредством гидрирования водородом при нормальном или повышенном давлении либо посредством гидрирования с переносом водорода с использованием формиата аммония или циклогексадиена в качестве источника водорода в присутствии катализатора, такого как PtO2, Pd-C или никель Ренея, в растворителях, таких как МеОН, EtOH, H2O, диоксан, ТГФ, EtOAc, дихлорметан, хлороформ, диметилформамид (DMF) или их смеси.
Превращение также может быть выполнено посредством обработки гидроксиламина гидрохлоридом в присутствии основания, такого как ацетат натрия, ацетат калия, карбонат натрия, карбонат калия, карбонат цезия, в растворителях, таких как МеОН, EtOH, диоксан, ТГФ, DMF или их смеси.
Предпочтительные условия относятся к использованию гидроксиламина гидрохлорида в присутствии ацетата натрия в МеОН при комнатной температуре в течение 2 часов.
Альтернативно, N-дифенилметилен-защищенный анилин 8 может быть получен согласно последовательности реакций, приведенной на схеме 2.
Схема 2
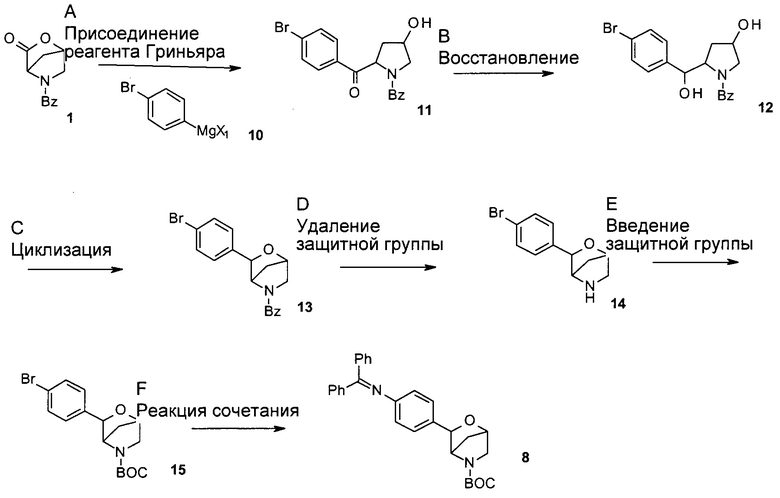
Стадия А: реакцию присоединения реагента Гриньяра можно осуществить посредством присоединения фенильного реагента Гриньяра (соединения 10, где Х1 представляет собой Cl или Br, образованного in situ в результате обработки пара-бромфенил-бромида или -иодида) к лактону 1 в безводных апротонных органических растворителях, таких как ТГФ и диэтиловый эфир, при температуре от -78°С до 0°С в атмосфере инертного газа.
Предпочтительные условия относятся к использованию пара-бромфенилмагнийбромида (соединения 10, где X1 представляет собой Br) в безводном ТГФ при -78°С в течение 30 минут.
Стадия В: восстановление кетона 11 до соответствующего диола 12 может быть осуществлено путем обработки восстанавливающим реагентом, таким как NaBH4, LiBH4, ZnBH4, 9-BBN, комплекс боран-ТГФ, LiAlH4 или DIBAL-H, в растворителях, таких как ТГФ, диэтиловый эфир, DME, 1,4-диоксан и ТВМЕ, метанол или этанол.
Предпочтительные условия относятся к использованию NaBH4 в качестве восстанавливающего реагента в МеОН при 0°С в течение 2 часов.
Стадия С: циклизацию диола 12 можно осуществить путем взаимодействия по типу реакции Мицунобу, путем опосредуемой кислотой катионной циклизации или путем постадийного способа с участием промежуточных сложных сульфонатных эфиров.
В случае взаимодействия по типу реакции Мицунобу такое превращение можно осуществить посредством обработки трифенилфосфином и азодикарбоксилатом, таким как диэтилазодикарбоксилат (DEAD) или диизопропилазодикарбоксилат (DIAD), в эфирных растворителях, таких как диэтиловый эфир, диоксан, ТГФ или ТВМЕ, или других апротонных органических растворителях, таких как толуол и бензол.
В случае опосредуемой кислотой катионной циклизации такое превращение можно осуществить посредством обработки неорганическими кислотами, такими как H2SO4, H3PO4, при повышенных температурах, или посредством обработки органическими кислотами, такими как трифторуксусная кислота, BF3⋅Et2O, возможно с использованием вспомогательного вещества, такого как Et3SiH, в растворителях, таких как дихлорметан, 1,2-дихлорэтан или толуол, при температуре от 0°С до комнатной температуры.
В случае постадийного способа такое превращение можно осуществить посредством обработки диола 12 одним эквивалентом сульфонилхлорида, такого как метансульфонилхлорид или толуолсульфонилхлорид, в присутствии органического основания, такого как пиридин, триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, в эфирных растворителях, таких как диэтиловый эфир, диоксан, ТГФ или ТВМЕ, или с использованием органического основания в качестве растворителя, при температуре от 0°С до 50°С. Полученный сложный сульфонатный эфир можно превратить в защищенный имеющий мостиковую связь морфолин 13 посредством обработки ненуклеофильным основанием, таким как гидрид натрия, трет-бутилат калия или 2-метил-2-бутилат калия, в эфирных растворителях, таких как диэтиловый эфир, диоксан, ТГФ или ТВМЕ.
Предпочтительные условия относятся к способу по типу реакции Мицунобу: к обработке диола 12 DIAD и трифенилфосфином в толуоле при 0°С и продолжению реакции при комнатной температуре в течение 12 часов.
Стадия D: удаление защитной группы можно осуществить, используя или индуцированную основанием реакцию, или постадийный способ с участием бензил-защищенного промежуточного соединения.
В случае индуцированной основанием реакции, удаление защитной группы может быть выполнено посредством обработки основанием, таким как гидразин, KOH, NaOH или Cs2CO3, в растворителях, таких как метанол, этанол, при повышенных температурах, как например, от 90°С до 150°С.
В случае постадийного способа бензоильную защитную группу можно превратить в бензильную защитную группу посредством обработки восстанавливающими реагентами, такие как LiAlH4, ВН3⋅ТГФ и BH3⋅Me2S, в эфирных растворителях, таких как диэтиловый эфир, диоксан, ТГФ или ТВМЕ, при температуре от 0°С до 60°С. Полученную бензильную группу можно удалить, используя или реакцию гидрирования, катализируемую Pd катализатором, или обработку хлорформиатами, такими как ClCOOCH2CH2Cl, ClCOOCH(Cl)Me, ClCOOCH2Ph и ClCOOCH2CCl3, и возможно в присутствии основания, такого как триэтиламин, диизопропилэтиламин и гидроксид натрия, в растворителях, таких как толуол, ТГФ, диэтиловый эфир, диоксан или ТВМЕ, при температурах от комнатной до повышенной температуры.
Предпочтительные условия относятся к индуцированной основанием реакции с использованием KOH в качестве основания и МеОН в качестве растворителя в герметично закрытой пробирке при 110°С в течение 30 минут.
Стадия Е: введение защитной группы в имеющий мостиковую связь морфолин 14 можно осуществить посредством обработки ди-трет-бутилкарбонатом, возможно в присутствии органического или неорганического основания, такого как триэтиламин, N,N-диизопропилэтиламин, N-метилморфолин, карбонат калия, карбонат натрия или карбонат цезия, в галогенированных растворителях, таких как дихлорметан или 1,2-дихлорэтан, или эфирных растворителях, таких как диэтиловый эфир, диоксан, ТГФ или ТВМЕ.
Предпочтительные условия относятся к использованию ТГФ в присутствии карбоната калия в качестве основания при комнатной температуре в течение 10 часов.
Стадия F: сочетание иодида 15 с бензофенонимином можно осуществить в присутствии катализатора на основе палладия или меди, лиганда и основания в растворителях, таких как диоксан, DME, ТГФ, толуол и DMSO, при повышенных температурах, например, с использованием катализируемой палладием реакции Бухвальда-Хартвига.
Предпочтительные условия относятся к использованию каталитических количеств трис(дибензилиденацетон)дипалладия(0), каталитических количеств 2,2'-бис(дифенилфосфино)-1,1'-бинафтила (BINAP) и KOtBu в толуоле при 90°С в течение 30 минут при нагревании микроволнами.
Схема 3
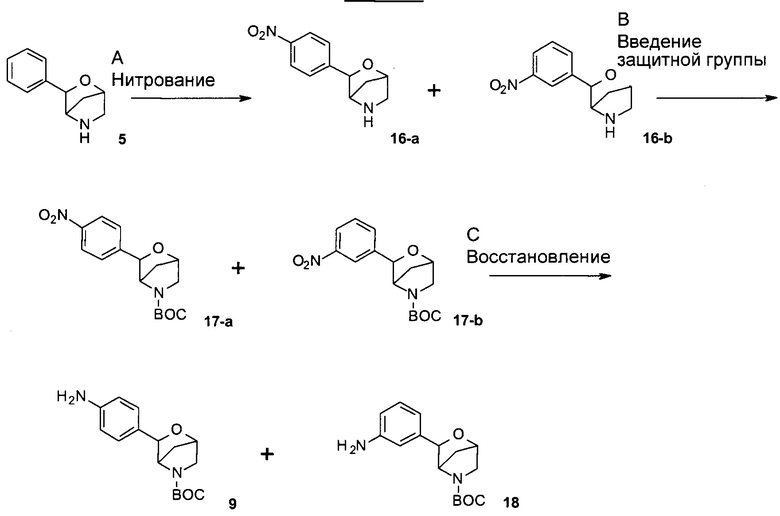
Стадия А: нитрование фенилморфолина 5 можно осуществить посредством обработки дымящей азотной кислотой или азотной кислотой в присутсвии других органических и неорганических кислот, таких как трифторуксусная кислота и серная кислота, при температуре от -40°С до комнатной температуры, возможно в углеводородном или галогенированном углеводородном растворителе, таком как гексаны, дихлорметан или 1,2-дихлорэтан. Альтернативно, данная реакция может быть осуществлена в результате обработки фенилморфолина 5 солями азотной кислоты, такими как нитрат калия, нитрат натрия или нитрат цезия, в других органических и неорганических кислотах, таких как трифторуксусная кислота и серная кислота, при температуре от -40°С до комнатной температуры. Соединения 16-a и 16-b могут быть либо разделены хроматографией, либо перенесены на следующую стадию в виде такой смеси.
Предпочтительные условия относятся к обработке дымящей азотной кислотой при 0-5°С.
Стадия В: введение защитной группы в имеющие мостиковую связь морфолины 16-а, 16-b или их смесь со стадии А можно осуществить посредством обработки ди-трет-бутилкарбонатом, возможно в присутствии органического или неорганического основания, такого как триэтиламин, N,N-диизопропилэтиламин, N-метилморфолин, карбонат калия, карбонат натрия или карбонат цезия, в галогенированных растворителях, таких как дихлорметан или 1,2-дихлорэтан, или эфирных растворителях, таких как диэтиловый эфир, диоксан, ТГФ или ТВМЕ.
Предпочтительные условия относятся к использованию ТГФ в присутствии карбоната калия в качестве основания при комнатной температуре в течение 10 часов. Соединения 17-а и 17-b могут быть либо разделены хроматографией, либо перенесены на следующую стадию в виде такой смеси.
Стадия С: восстановление нитрогруппы в соединениях 17-а, 17-b или их смеси со стадии В можно осуществить посредством обработки восстанавливающим реагентом, таким как SnCl2, Na2S2O4 или порошок Zn, возможно в присутствии уксусной кислоты или трифторуксусной кислоты в качестве вспомогательного вещества, в МеОН или EtOH в качестве растворителей при повышенных температурах. Альтернативно, превращение может быть выполнено посредством гидрирования водородом при нормальном или повышенном давлении либо посредством гидрирования с переносом водорода с использованием формиата аммония или циклогексадиена в качестве источника водорода в присутствии катализатора, такого как PtO2, Pd-C или никель Ренея, в растворителях, таких как МеОН, EtOH, H2O, диоксан, ТГФ, НОАс, EtOAc, CH2Cl2, DMF или их смеси. На этой стадии анилины 9 и 18 могут быть разделены хроматографией на диоксиде кремния.
Предпочтительные условия относятся к использованию SnCl2 в качестве восстанавливающего реагента, в присутствии уксусной кислоты в качестве вспомогательного вещества, в EtOH при температуре дефлегмации.
Схема 4

где X1 представляет собой галогена, L1 представляет собой связь, -С(О)-, -СН2С(O)-, -СН2- или -NHC(O)-; и R1 представляет собой фенил или гетероарил, выбранный из группы, состоящей из пиридинила, пиримидинила, пиразинила или пиразолила, и при этом фенил и гетероарил возможно замещены одним, двумя или тремя заместителями, выбранными из группы, состоящей из атома галогена, низшего алкила, низшего алкокси, низшего алкила, замещенного атомом галогена, низшего алкокси, замещенного атомом галогена, циклоалкила или -O-СН2-циклоалкила.
Стадия А: сочетание арилгалогенида 19 (включая соединение 7, где Х1 представляет собой I, и соединение 15, где X1 представляет собой Br) с ариламином (20-а), ариламидом (20-b), арилмочевиной (20-с) или арилметанамином (20-d) можно осуществить посредством обработки в присутствии катализатора на основе палладия или меди, лиганда и основания в растворителях, таких как диоксан, DME, ТГФ, толуол и DMSO, при повышенных температурах, например, с использованием катализируемой палладием реакции Бухвальда-Хартвига.
Предпочтительные условия относятся к использованию каталитических количеств трис(дибензилиденацетон)дипалладия(0), каталитических количеств 4,5-бис(дифенилфосфино)-9,9-диметилксантена (Xantphos) и Cs2CO3 в диоксане при 90°С в течение 16 часов.
Стадия В: удаление N-защитной группы ВОС может быть выполнено с использованием минеральных кислот, таких как HCl, H2SO4 или H3PO4, или органических кислот, таких как CF3COOH, CHCl2COOH, НОАс или пара-толуолсульфоновая кислота, в растворителях, таких как CH2Cl2, CHCl3, ТГФ, МеОН, EtOH или H2O, при 0-80°С.
Предпочтительные условия относятся к использованию CF3COOH в качестве кислоты в CH2Cl2 при комнатной температуре в течение 2 часов.
Схема 5
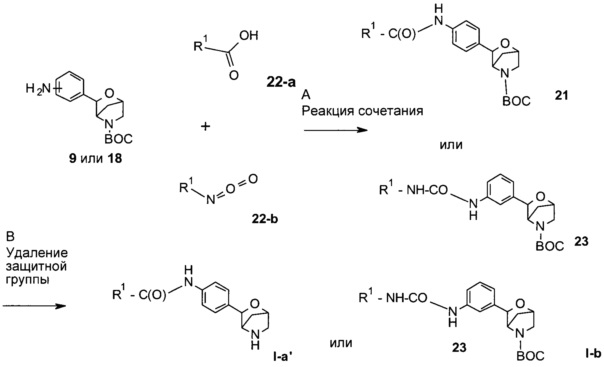
Стадия А. Образование амида может быть осуществлено в результате проведения реакции между анилином 9 или 18 и карбоновой кислотой 22-а в присутствии реагента сочетания, такого как DCC (дициклогексилкарбодиимид), EDC (1-этил-3-(3-диметиламинопропил)карбодиимид), TBTU (тетрафторборат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония), HBTU (гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония) или HATU (гексафторфосфат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония), в присутствии органического основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, в растворителях, таких как дихлорметан или 1,2-дихлорэтан, DMF, DMSO или эфирные растворители, включая диэтиловый эфир, диоксан, ТГФ, DME или ТВМЕ.
Предпочтительные условия относятся к использованию HATU в присутствии N,N-диизопропилэтиламина в DMF при комнатной температуре в течение 12 часов.
Образование мочевины может быть осуществлено в результате проведения реакции между анилином 9 или 18 и изоцианатом 22-b в присутствии органического основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, в галогенированных растворителях, таких как дихлорметан, 1,2-дихлорэтан, хлорбензол.
Предпочтительные условия относятся к использованию триэтиламина в качестве основания в дихлорметане при комнатной температуре в течение 16 часов.
Стадия В: удаление N-защитной группы ВОС может быть выполнено с использованием минеральных кислот, таких как HCl, H2SO4 или H3PO4, или органических кислот, таких как CF3COOH, CHCl2COOH, НОАс или пара-толуолсульфоновая кислота, в растворителях, таких как CH2Cl2, CHCl3, ТГФ, МеОН, EtOH или H2O, при 0-80°С.
Предпочтительные условия относятся к использованию CF3COOH в качестве кислоты в CH2Cl2 при комнатной температуре в течение 2 часов.
Схема 6
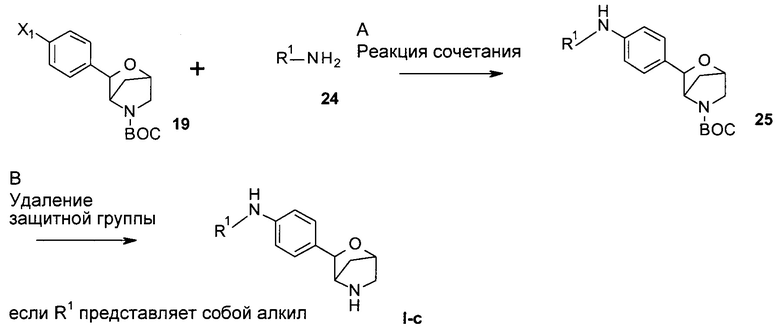
Стадия А: сочетание арилгалогенида 19 (включая соединение 7, где Х1 представляет собой I, и соединение 15, где Х1 представляет собой Br) с алкиламином (24) можно осуществить посредством обработки в присутствии катализатора на основе палладия или меди, лиганда и основания в растворителях, таких как диоксан, DME, ТГФ, толуол и DMSO, при повышенных температурах, например, с использованием катализируемой палладием реакции Бухвальда-Хартвига.
Предпочтительные условия относятся к использованию каталитических количеств трис(дибензилиденацетон)дипалладия(0), каталитических количеств 4,5-бис(дифенилфосфино)-9,9-диметилксантена (Xantphos) и Cs2CO3 в диоксане при 90°С в течение 16 часов.
Стадия В: удаление N-защитной группы ВОС может быть выполнено с использованием минеральных кислот, таких как HCl, H2SO4 или H3PO4, или органических кислот, таких как CF3COOH, CHCl2COOH, НОАс или пара-толуолсульфоновая кислота, в растворителях, таких как CH2Cl2, CHCl3, ТГФ, МеОН, EtOH или H2O, при 0-80°С.
Предпочтительные условия относятся к использованию CF3COOH в качестве кислоты в CH2Cl2 при комнатной температуре в течение 2 часов.
Схема 7
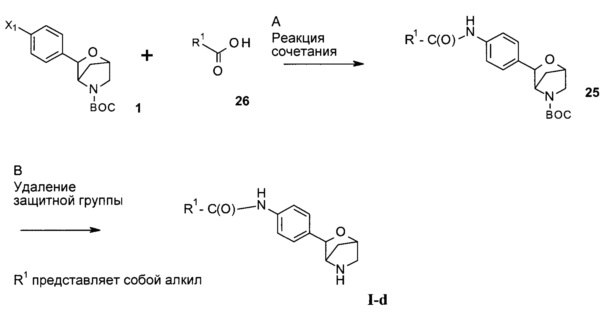
Стадия А: сочетание арилгалогенида 19 (включая соединение 7, где Х1 представляет собой I, и соединение 15, где Х1 представляет собой Br) с алкилкарбоновой кислотой (26) может быть осуществлено в результате проведения реакции в присутствии реагента сочетания, такого как DCC, EDC, TBTU, HBTU или HATU, в присутствии органического основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, в растворителях, таких как дихлорметан или 1,2-дихлорэтан, DMF, DMSO или эфирные растворители, включая диэтиловый эфир, диоксан, ТГФ, DME или ТВМЕ.
Предпочтительные условия относятся к использованию HATU в присутствии N,N-диизопропилэтиламина в DMF при комнатной температуре в течение 12 часов.
Стадия В: удаление N-защитной группы ВОС может быть выполнено с использованием минеральных кислот, таких как HCl, H2SO4 или H3PO4, или органических кислот, таких как CF3COOH, CHCl2COOH, НОАс или пара-толуолсульфоновая кислота, в растворителях, таких как CH2Cl2, CHCl3, ТГФ, МеОН, EtOH или H2O, при 0-80°С.
Предпочтительные условия относятся к использованию CF3COOH в качестве кислоты в CH2Cl2 при комнатной температуре в течение 2 часов.
Схема 8
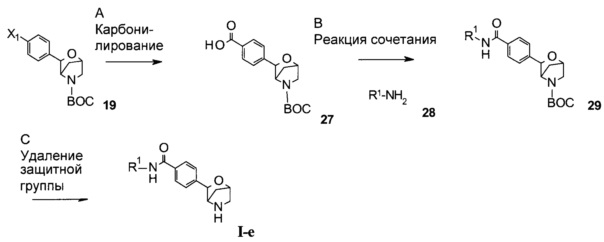
Стадия А: осуществления карбонилирования галогенида 19 можно достичь в результате литирования с использованием алкиллитиевых реагентов в безводном растворителе, таком как диэтиловый эфир, диоксан, ТГФ, DME или ТВМЕ, с последующим добавлением CO2. Альтернативно, такого превращения можно достичь в результате сочетания с СО в присутствии катализаторов на основе переходных металлов, таких как катализаторы на основе Pd, Mo, Co, Cu, в присутствии лиганда и вспомогательных веществ. В качестве растворителей могут быть выбраны DMF, ТГФ, диоксан, DMSO, этанол и вода.
Предпочтительные условия относятся к литированию с использованием н-BuLi при -78°С в безводном ТГФ с последующим барботированием реакционного раствора обезвоженным CO2.
Стадия В: сочетания кислоты 27 с амином 28 можно достичь в результате осуществления реакции в присутствии реагента сочетания, такого как DCC, EDC, TBTU, HBTU или HATU, в присутствии органического основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, в растворителях, таких как дихлорметан или 1,2-дихлорэтан, DMF, DMSO или эфирные растворители, включая диэтиловый эфир, диоксан, ТГФ, DME или ТВМЕ.
Предпочтительные условия относятся к использованию HATU в присутствии N,N-диизопропилэтиламина в DMF при комнатной температуре в течение 2 часов.
Стадия С: удаление N-защитной группы ВОС может быть выполнено с использованием минеральных кислот, таких как HCl, H2SO4 или H3PO4, или органических кислот, таких как CF3COOH, CHCl2COOH, НОАс или пара-толуолсульфоновая кислота, в растворителях, таких как CH2Cl2, CHCl3, ТГФ, МеОН, EtOH или H2O, при 0-80°С.
Предпочтительные условия относятся к использованию CF3COOH в качестве кислоты в CH2Cl2 при комнатной температуре в течение 2 часов.
Выделение и очистка соединений
Выделение и очистка соединений и промежуточных соединений, описанных в данной заявке, при желании могут быть выполнены посредством любой подходящей методики разделения или очистки, такой как, например, фильтрация, экстракция, кристаллизация, колоночная хроматография, тонкослойная хроматография, толстослойная хроматография, препаративная жидкостная хроматография низкого или высокого давления либо комбинация этих методик. Конкретные иллюстрации подходящих методик разделения и выделения могут быть приведены посредством ссылки на подготовительные примеры и примеры, представленные в данном описании ниже. Однако, несомненно, также могут быть использованы и другие эквивалентные методики разделения и выделения. Рацемические смеси хиральных соединений формулы I могут быть разделены с использованием хиральной ВЭЖХ (высокоэффективная жидкостная хроматография). Рацемические смеси хиральных синтезированных промежуточных соединений также могут быть разделены с использованием хиральной ВЭЖХ.
Соли соединений формулы I
Соединения формулы I являются основными и могут быть превращены в соответствующую соль присоединения кислоты. Такое превращение осуществляется путем обработки по меньшей мере стехиометрическим количеством соответствующей кислоты, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное, и органических кислот, таких как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, пара-толуолсульфоновая кислота, салициловая кислота и тому подобное. Обычно, свободное основание растворяют в инертном органическом растворителе, таком как диэтиловый эфир, этилацетат, хлороформ, этанол или метанол и тому подобное, и добавляют кислоту в том же растворителе. Температуру поддерживают в диапазоне от 0°С до 50°С. Полученная соль выпадает в осадок самопроизвольно или может быть выделена из раствора с использованием менее полярного растворителя.
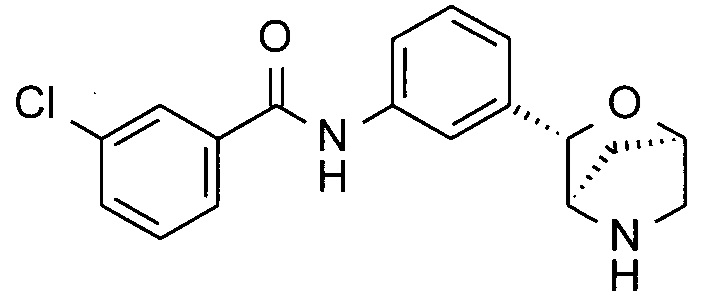
Пример 1
3-Хлор-N-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
a) [(2R,4R)-1-Бензоил-4-гидрокси-пирролидин-2-ил]-фенил-метанон
К раствору (1R,4R)-5-бензоил-2-окса-5-азабицикло[2.2.1]гептан-3-она (21,7 г; CAS (Chemical abstracts service - химическая реферативная служба): 444313-68-2, полученого согласно методике, приведенной в Tetrahedron, 2007, 63(32), 7523-7531) и N,O-диметилгидроксиламина гидрохлорида (11,6 г; CAS: 6638-79-5) в безводном тетрагидрофуране (1,5 л) добавляли фенилмагнийбромид (133 мл; 3М в диэтиловом эфире; CAS: 100-58-3) при -78°С в атмосфере N2. Реакционную смесь перемешивали при -70°С в течение 10 часов. LCMS (liquid chromatography/mass spectrometry - жидкостная хроматография в сочетании с масс-спектрометрией) показывала завершение реакции. Реакцию гасили насыщенным водным раствором NH4Cl (100 мл). Смесь экстрагировали этилацетатом (2×500 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. После флэш-хроматографии (силикагель; петролейный эфир : этилацетат составляет 1:1 по объему) получали [(2R,4R)-1-бензоил-4-гидрокси-пирролидин-2-ил]-фенил-метанон (6,67 г; выход 23%) в виде белого твердого вещества.
MS (ESI) (mass spectrometry - масс-спектрометрия/electrospray ionization - электрораспылительная ионизация): 296,1 ([M+H]+).
b) [(2R,4R)-4-Гидрокси-2-[(S)-гидрокси(фенил)метил]пирролидин-1-ил]-фенил-метанон
К раствору [(2R,4R)-1-бензоил-4-гидрокси-пирролидин-2-ил]-фенил-метанона (6 г) в МеОН (200 мл) при 0°С порциями добавляли NaBH4 (3 г). Реакционную смесь перемешивали в течение 1,5 ч до тех пор, пока анализ посредством TLC (thin layer chromatography - тонкослойная хроматография) не показывал завершение реакции. Для погашения избытка NaBH4 добавляли ацетон (10 мл). Смесь концентрировали при пониженном давлении. Добавляли насыщенный водный раствор NH4Cl (100 мл). Смесь экстрагировали этилацетатом (2×100 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир/EtOAc составляет 1:2 по объему), получая [(2R,4R)-4-гидрокси-2-[(S)-гидрокси(фенил)метил]пирролидин-1-ил]-фенил-метанон (5,5 г; 92%) в виде белого твердого вещества. MS (ESI): 298,1 ([М+Н]+).
c) Фенил-[(1R,3S,4R)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-ил]метанон
К смеси [(2R,4R)-4-гидрокси-2-[(S)-гидрокси(фенил)метил]пирролидин-1-ил]-фенил-метанона (5,5 г) и PPh3 (5,82 г) в толуоле (100 мл) добавляли диизопропилазодикарбоксилат (DIAD; 4,49 г; CAS: 2446-83-5) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении и разбавляли трет-бутил-метиловым эфиром. Суспензию перемешивали и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир/EtOAc составляет 1:1 по объему), получая фенил-[(1R,3S,4R)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-ил]метанон (4,1 г; выход 79%) в виде белого твердого вещества.
MS (ESI): 280,1 ([М+Н]+).
1Н-ЯМР (ядерный магнитный резонанс) (CDCl3, 400 МГц): δ 7.625-7.285 (10Н), 5.25 (1Н), 4.96 (1Н), 4.56 (1Н), 3.65 (2Н), 1.93 (1Н), 1.51 (1Н).
d) (1R,3S,4R)-5-Бензил-3-фенил-2-окса-5-азабицикло[2.2.1]гептан
К смеси фенил-[(1R,3S,4R)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-ил]метанона (10 г) в ТГФ (200 мл) добавляли LiAlH4 (5,3 г) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов до тех пор, пока LCMS не показывала полного расходования исходного вещества. Для гашения избытка LiAlH4 добавляли Na2SO4⋅10H2O (10 г). Смесь фильтровали. Фильтрат концентрировали при пониженном давлении и далее сушили под высоким вакуумом, получая неочищенный (1R,3S,4R)-5-бензил-3-фенил-2-окса-5-азабицикло[2.2.1]гептан (10 г; количественный выход), который использовали на следующей стадии непосредственно без очистки. MS (ESI): 266,1 ([М+Н]+).
1Н-ЯМР (CDCl3, 400 МГц): δ 7.46-7.24 (10Н), 5.29 (1Н), 4.63 (1Н), 4.01-3.92 (2Н), 3.40 (1Н), 3.02 (1Н), 3.00 (1Н), 1.72 (1Н), 1.67 (1Н).
e) (1R,3S,4R)-3-Фенил-2-окса-5-азабицикло[2.2.1]гептан
В атмосфере азота к раствору (1R,3S,4R)-5-бензил-3-фенил-2-окса-5-азабицикло[2.2.1]гептана (16 г) в толуоле (250 мл) по каплям добавляли ClCOOCH2CH2Cl (17 г). Реакционную смесь нагревали в условиях кипячения с обратным холодильником в течение 16 часов до тех пор, пока TLC не показывала полного расходования исходного вещества. Затем реакционную смесь охлаждали до комнатной температуры. Добавляли МеОН (5 мл) и реакционную смесь перемешивали в течение часа. Летучие вещества удаляли при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; CH2Cl2:МеОН от 10:0 до 5:1 по объему), получая (1R,3S,4R)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан (4,5 г; выход 45%) в виде желтого масла.
1Н-ЯМР (CDCl3, 400 МГц): δ 7.37-7.24 (5Н), 4.91 (1Н), 4.70 (1Н), 3.62 (1Н), 3.16 (1Н), 3.02 (1Н), 1.83 (1Н), 1.51 (1Н).
f) (1R,3S,4R)-3-(3-Нитрофенил)-2-окса-5-азабицикло[2.2.1]гептан и (1R,3S,4R)-3-(4-нитрофенил)-2-окса-5-азабицикло[2.2.1]гептан
К дымящей азотной кислоте (15 мл) при -20°С добавляли раствор (1R,3S,4R)-3-фенил-2-окса-5-азабицикло[2.2.1]гептана (1,4 г) в CH2Cl2 (1 мл). Реакционную смесь перемешивали в течение 0,5 ч. Смесь выливали в ледяную воду. Для подведения pH до значения примерно 10 добавляли NaOH. Смесь концентрировали при пониженном давлении и разбавляли МеОН (50 мл). Суспензию фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; CH2Cl2:MeOH от 10:0 до 5:1 по объему), получая смесь (1R,3S,4R)-3-(3-нитрофенил)-2-окса-5-азабицикло[2.2.1]гептана и (1R,3S,4R)-3-(4-нитрофенил)-2-окса-5-азабицикло[2.2.1]гептана (1,25 г; общий выход 71,0%) в виде желтого масла.
1Н-ЯМР (CDCl3, 400 МГц): δ 8.23-8.11 (2Н), 7.76-7.47 (2Н), 4.98 (1Н), 4.78-4.77 (1Н), 3.78-3.70 (1Н), 3.18-3.08 (2Н), 1.76-1.56 (2Н).
g) трет-Бутил-(1R,3S,4R)-3-(3-нитрофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат и трет-бутил-(1R,3S,4R)-3-(4-нитрофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К смеси (1R,3S,4R)-3-(3-нитрофенил)-2-окса-5-азабицикло[2.2.1]гептана и (1R,3S,4R)-3-(4-нитрофенил)-2-окса-5-азабицикло[2.2.1]гептана (1,1 г) в ТГФ (20 мл) добавляли K2CO3 (2,1 г) и Boc2O (1,3 г) при комнатной температуре. Реакционную смесь перемешивали в течение 16 часов до тех пор, пока TLC-анализ не показывал полного расходования исходных веществ. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Остаток разбавляли рассолом (20 мл). Смесь экстрагировали CH2Cl2 (3×20 мл). Объединенные органические слои сушили над Na2SO4 и далее концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир : этилацетат от 10:0 до 5:1 по объему), получая смесь трет-бутил-(1R,3S,4R)-3-(3-нитрофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата и трет-бутил-(1R,3S,4R)-3-(4-нитрофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (1,45 г; общий выход 91%) в виде белого твердого вещества.
1Н-ЯМР (CDCl3, 400 МГц): δ 8.24-8.14 (2Н), 7.68-7.45 (2Н), 5.10 (1Н), 4.83 (1Н), 4.51-4.37 (1Н), 3.59-3.37 (2Н), 1.75-1.69 (2Н), 1.57 (9Н).
h) трет-Бутил-(1R,3S,4R)-3-(3-аминофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К смеси трет-бутил-(1R,3S,4R)-3-(3-нитрофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата и трет-бутил-(1R,3S,4R)-3-(4-нитрофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (1,2 г) в этаноле (50 мл) добавляли хлорид олова(II) дигидрат (4,51 г; CAS: 10025-69-1) и уксусную кислоту (2,4 г). Реакционную смесь перемешивали при 50°С в течение 4 часов в атмосфере N2 до тех пор, пока TLC-анализ не показывал полного расходования исходных веществ. Смесь разбавляли насыщенным водным раствором NaHCO3, экстрагировали CH2Cl2, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали препаративной TLC (петролейный эфир/EtOAc составляет 1:1 по объему), получая трет-бутил-(1R,3S,4R)-3-(3-аминофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (240 мг; выход 20,6%) в виде желтого твердого вещества.
MS (ESI): 291,0 ([М+Н]+); 235,0 ([М-С4Н8+Н]+); 191,0 ([М-С4Н8-CO2+Н]+).
i) трет-Бутил-(1R,3S,4R)-3-[3-[(3-хлорбензоил)амино]фенилъ-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
Раствор 3-хлорбензойной кислоты (38 мг; CAS: 535-80-8), HATU (114 мг; CAS: 148893-10-1) и DIPEA (0,17 мл; CAS: 7087-68-5) в CH2Cl2 (2 мл) перемешивали при комнатной температуре в течение 30 мин. К реакционной смеси добавляли трет-бутил-(1R,3S,4R)-3-(3-аминофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (58 мг). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов до тех пор, пока TLC-анализ не показывал полного расходования исходного вещества. Реакционную смесь разбавляли CH2Cl2 (50 мл). Раствор промывали водным раствором NH4Cl (50 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. После дальнейшей сушки под высоким вакуумом получали неочищенный трет-бутил-(1R,3S,4R)-3-[3-[(3-хлорбензоил)амино]фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат в виде коричневого твердого вещества.
j) 3-Хлор-N-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
Неочищенный трет-бутил-(1R,3S,4R)-3-[3-[(3-хлорбензоил)амино]фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат в виде коричневого твердого вещества со стадии (i) растворяли в смеси CH2Cl2 (2 мл) и трифторуксусной кислоты (TFA; 0,5 мл; CAS: 76-05-1). Раствор перемешивали при комнатной температуре в течение часа. Летучие вещества удаляли при пониженном давлении. Остаток очищали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,1% TFA; колонка С18), получая 3-хлор-N-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид (18 мг; выход 27% за две стадии) в виде белого твердого вещества.
MS: 331,1 ({37Cl}M+H)+; 329,1 ({35Cl}M+H)+.
1Н-ЯМР (400 МГц, метанол-d4): δ 7.98 (1Н), 7.89 (1Н), 7.68 (1Н), 7.60 (2Н), 7.54 (1Н), 7.36 (1Н), 7.12 (1Н), 4.95 (1Н), 4.73 (1Н), 3.65 (1Н), 3.05 (1Н), 2.96 (1Н), 1.88 (1Н), 1.54 (1Н).
Пример 2
4-Хлор-N-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
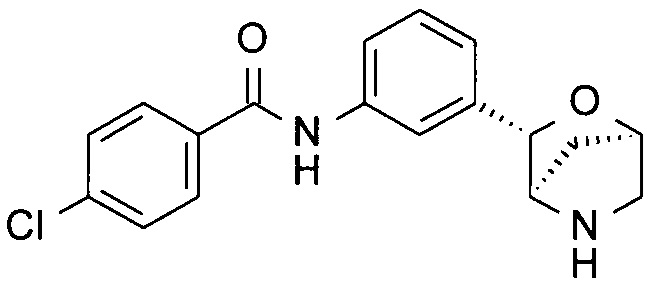
Указанное в заголовке соединение получали по аналогии с соединением примера 1, используя 4-хлорбензойную кислоту (CAS: 74-11-3) вместо 3-хлорбензойной кислоты на стадии (i). Беловатое твердое вещество. MS (ESI): 331,0 ([{37Cl}M+H]+); 329,1 ([{35Cl}M+H]+).
Пример 3
1-[3-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-[4-(трифторметил)фенил]мочевина
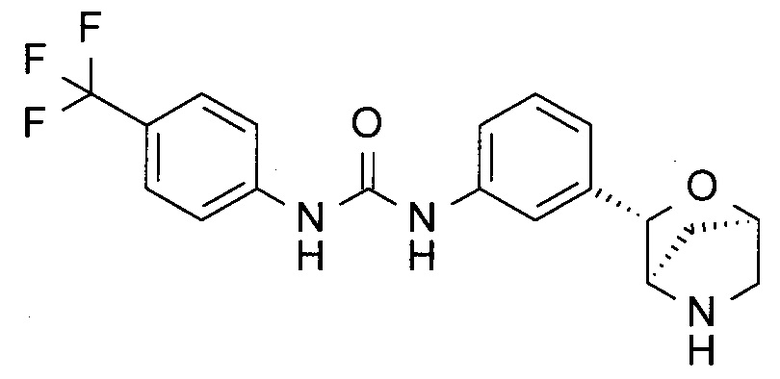
а) трет-Бутил-(1R,3S,4R)-3-[3-[[4-(трифторметил)фенил]карбамоиламино]фенилъ-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К раствору трет-бутил-(1R,3S,4R)-3-(3-аминофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (44 мг) и Et3N (23 мг; CAS: 121-44-8) в CH2Cl2 (1 мл) добавляли 4-(трифторметил)фенилизоцианат (34 мг; CAS: 1548-13-6) при комнатной температуре. Реакционную смесь перемешивали в течение ночи. Реакционную смесь разбавляли CH2Cl2 (20 мл), промывали водным раствором NaHCO3 (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. После дальнейшей сушки под высоким вакуумом получали трет-бутил-(1R,3S,4R)-3-[3-[[4-(трифторметил)фенил]карбамоиламино]фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (50 мг; выход 70%) в виде коричневого твердого вещества.
b) 1-[3-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-[4-(трифторметил)фенил]мочевина
К раствору трет-бутил-(1R,3S,4R)-3-[3-[[4-(трифторметил)фенил]-карбамоиламино]фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (50 мг) со стадии (а) в CH2Cl2 (2 мл) добавляли TFA (0,5 мл; CAS: 76-05-1). Реакционную смесь перемешивали при комнатной температуре в течение часа. Летучие вещества удаляли при пониженном давлении. Неочищенную смесь очищали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,1% TFA; колонка С18), получая 1-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-[4-(трифторметил)фенил]мочевину (10 мг; выход 26%) в виде белого твердого вещества.
MS(ESI): 378,1 ([М+Н]+).
1Н-ЯМР: (метанол-d4, 400 МГц) δ 7.663-7.583 (4Н), 7.44 (1Н), 7.33-7.28 (2Н), 7.00 (1Н), 4.92 (1Н), 4.72 (1Н), 3.63 (1Н), 3.06-2.94 (2Н), 1.86 (1Н), 1.53 (1Н).
Пример 4
1-(4-Хлорфенил)-3-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]мочевина
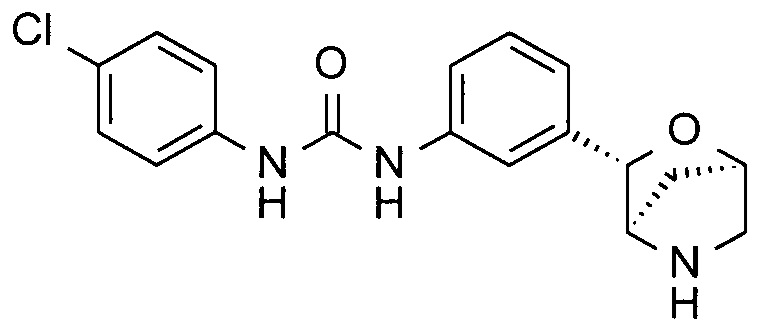
Указанное в заголовке соединение получали по аналогии с соединением примера 3, используя 4-хлорфенилизоцианат (CAS: 104-12-1) вместо 4-(трифторметил)фенилизоцианата на стадии (а). Белое твердое вещество. MS (ESI): 346,1 ([{37Cl}M+H]+); 344,1 ([{35Cl}M+H]+).
Пример 5
1-(3-Хлорфенил)-3-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]мочевина
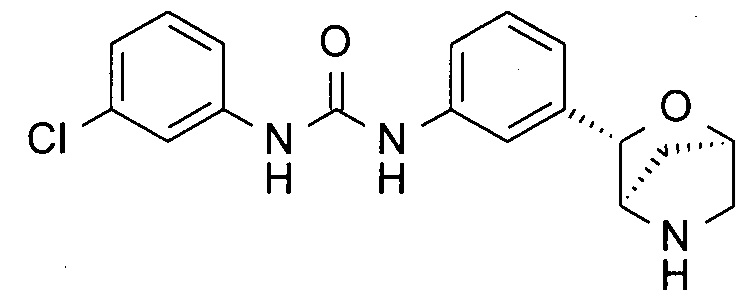
Указанное в заголовке соединение получали по аналогии с соединением примера 3, используя 3-хлорфенилизоцианат (CAS: 2909-38-8) вместо 4-(трифторметил)фенилизоцианата на стадии (а). Белое твердое вещество. MS (ESI): 346,1 ([{37Cl}M+H]+); 344,1 ([{35Cl}M+H]+).
Пример 6
(1R,3S,4R)-3-Фенил-2-окса-5-азабицикло[2.2.1]гептан
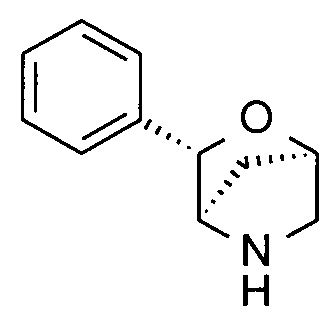
Указанное в заголовке соединение получали на стадии (е) примера 1.
MS(ESI): 176,1 ([М+Н]+).
Пример 7
4-Хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
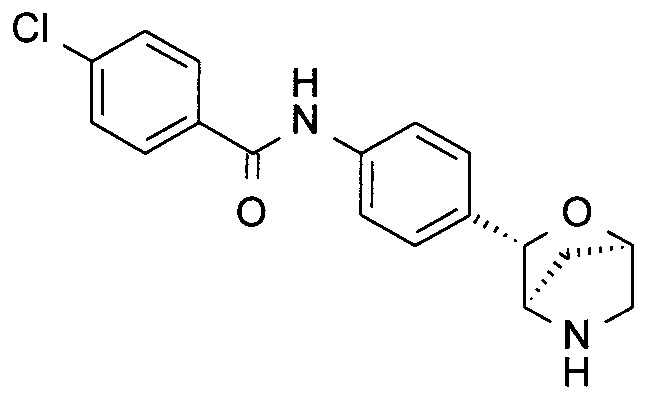
a) трет-Бутил-(1R,3S,4R)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К раствору (1R,3S,4R)-3-фенил-2-окса-5-азабицикло[2.2.1]гептана (3,85 г) в ТГФ (100 мл) добавляли K2CO3 (5,88 г; CAS: 584-08-7) и Boc2O (5,88 г; CAS: 24424-99-5) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение ночи. TLC-анализ показывал завершение реакции. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир : этилацетат от 10:0 до 5:1 по объему), получая трет-бутил-(1R,3S,4R)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (5,9 г; выход 98%) в виде белого твердого вещества.
1Н-ЯМР (CDCl3, 400 МГц) δ 7.36-7.285 (m, 5Н), 5.056 (1Н), 4.78 (1Н), 4.41 (1Н), 3.51 (1Н), 3.37 (1Н), 1.86 (1Н), 1.67-1.52 (10Н).
b) трет-Бутил-(1R,3S,4R)-3-(4-иодфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
В атмосфере N2 раствор трет-бутил-(1R,3S,4R)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (1,1 г), [бис(трифторацетокси)иод]-бензола (2,1 г; CAS: 2712-78-9) и йода (1,1 г; CAS: 7553-56-2) в CCl4 (10 мл) перемешивали при комнатной температуре в течение ночи. LC-MS анализ показывал превращение более чем на 90%. Реакционную смесь разбавляли водным раствором NaHSO3, экстрагировали CH2Cl2 и промывали рассолом. Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (С-18, MeCN/0,1% NH3 в воде), получая трет-бутил-(1R,3S,4R)-3-(4-иодфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (560 мг; выход 35%) в виде коричневого твердого вещества.
MS (ESI): 424,0 (M+Na)+; 346,0 (М-С4Н8+Н)+; 302,0 (М-С4Н8-CO2+Н)+.
c) 4-Хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
В атмосфере N2 раствор трет-бутил-(1R,3S,4R)-3-(4-иодфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (40 мг), 4-хлорбензамида (23 мг; CAS: 619-56-7), трис(дибензилиденацетон)дипалладия(0) (18 мг; CAS: 51364-51-3), бис(дифенилфосфино)-9,9-диметилксантена (Xantphos, 10 мг; CAS: 161265-03-8) и Cs2CO3 (162 мг; CAS: 534-17-8) в диоксане (1 мл) перемешивали при 90°С в течение ночи. TLC-анализ показывал завершение реакции. Смесь фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (колонка С-18; 0,1% NH3 в смеси H2O/MeCN), получая неочищенное промежуточное соединение в виде коричневого масла. Неочищенное промежуточное соединение растворяли в CH2Cl2 (2 мл). Добавляли TFA (0,5 мл; CAS: 76-05-1). Раствор перемешивали при комнатной температуре в течение часа. Летучие вещества удаляли при пониженном давлении. Остаток очищали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,1% TFA; колонка С-18), получая 4-хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид (10 мг; выход 30% за 2 стадии) в виде белого твердого вещества. MS (ESI): 331,0 ([{37Cl}M+H]+); 329,0 ([{35Cl}M+H]+).
1Н-ЯМР (метанол-d4, 400 МГц): δ 7.95 (2Н), 7.76 (2Н), 7.56 (2Н), 7.38 (2Н), 5.18 (1Н), 4.96 (1Н), 4.42 (1Н), 3.47 (1Н), 3.37 (1Н), 2.15 (1Н), 1.83 (1Н).
Пример 8
3-Хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
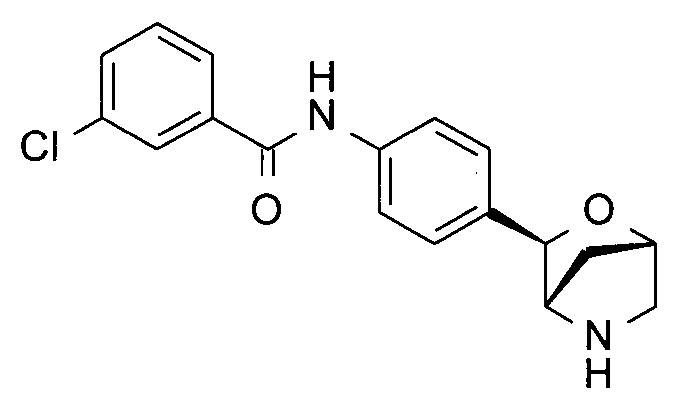
a) [(2S,4S)-1-Бензоил-4-гидрокси-пирролидин-2-ил]-фенил-метанон
(1S,4S)-5-Бензоил-2-окса-5-азабицикло[2.2.1]гептан-3-он (CAS: 31560-25-5) может быть получен согласно опубликованной методике (Tetrahedron, 1971, 27(5), 961-967).
В атмосфере азота к раствору (1S,4S)-5-бензоил-2-окса-5-азабицикло[2.2.1]гептан-3-она (32 г) и N,O-диметилгидроксиламина гидрохлорида (17 г; CAS: 6638-79-5) в безводном ТГФ (2,0 л) добавляли фенилмагнийбромид (133 мл; 3 М в диэтиловом эфире; CAS: 100-58-3) при -70°С в течение 10 часов. TLC-анализ показывал завершение реакции. Затем реакцию гасили насыщенным водным раствором NH4Cl и экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир/EtOAc составляет 1:1 по объему), получая [(2S,4S)-1-бензоил-4-гидрокси-пирролидин-2-ил]-фенил-метанон (50 г; выход 38%) в виде белого твердого вещества. MS (ESI): 296,2 ([М+Н]+).
b) [(2S,4S)-4-Гидрокси-2-[(R)-гидрокси(фенил)метил]пирролидин-1-ил]-фенил-метанон
К раствору [(2S,4S)-1-бензоил-4-гидрокси-пирролидин-2-ил]-фенил-метанона (50 г) в МеОН (500 мл) при 0°С порциями добавляли NaBH4 (25 г; CAS: 16940-66-2). Реакционную смесь перемешивали при 0°С в течение 1,5 часа до тех пор, пока LCMS-анализ не показывал завершения реакции. Для погашения избытка NaBH4 добавляли ацетон. Летучие вещества удаляли при пониженном давлении. Добавляли насыщенный водный раствор NH4Cl. Смесь экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир/EtOAc составляет 1:2 по объему), получая [(2S,4S)-4-гидрокси-2-[(R)-гидрокси(фенил)метил]пирролидин-1-ил]-фенил-метанон (33 г; выход: 65%) в виде белого твердого вещества. MS (ESI): 298,2 ([М+Н]+).
c) Фенил-[(1S,3R,4S)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-ил]метанон
К раствору [(2S,4S)-4-гидрокси-2-[(R)-гидрокси(фенил)метил]пирролидин-1-ил]-фенил-метанона (16 г) и PPh3 (17 г; CAS: 603-35-0) в безводном толуоле (200 мл) добавляли диизопропилазодикарбоксилат (DIAD; 14 г; CAS: 2446-83-5) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в трет-бутил-метиловом эфире (МТВЕ; 200 мл; CAS: 1634-04-4). Суспензию фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир/EtOAc составляет 1:1 по объему), получая фенил-[(1S,3R,4S)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-ил]метанон (11,5 г; выход: 77%) в виде белого твердого вещества. MS (ESI): 280,2 ([М+Н]+).
1Н-ЯМР (CDCl3, 400 МГц) δ 7.61-7.13 (10Н), 5.20 (1Н), 4.96 (1Н), 4.82 (1Н), 3.70-3.51 (2Н), 1.90 (1Н), 1.67 (1Н).
d) (1S,3R,4S)-5-Бензил-3-фенил-2-окса-5-азабицикло[2.2.1]гептан
К раствору фенил-[(1S,3R,4S)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-ил]метанона (3 г) в безводном ТГФ (30 мл) добавляли LiAlH4 (1,7 г; CAS: 16853-85-3) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Для гашения избытка LiAlH4 добавляли твердый Na2SO4⋅10H2O (10 г; CAS: 7727-73-3). Смесь фильтровали. Фильтрат концентрировали при пониженном давлении и сушили под высоким вакуумом, получая неочищенный (1S,3R,4S)-5-бензил-3-фенил-2-окса-5-азабицикло[2.2.1]гептан в виде желтого масла (3 г; количественный выход), который использовали на следующей стадии без очистки. MS (ESI): 266,2 ([М+Н]+).
e) (1S,3R,4S)-3-Фенил-2-окса-5-азабицикло[2.2.1]гептан
К раствору неочищенного (1S,3R,4S)-5-бензил-3-фенил-2-окса-5-азабицикло[2.2.1]гептана (3 г; 11 ммоль) со стадии (d) в толуоле (40 мл) по каплям добавляли 2-хлорэтилхлорформиат (3,2 г; CAS: 627-11-2). Реакционную смесь перемешивали при 110°С в атмосфере N2 в течение ночи. Добавляли МеОН (20 мл) и реакционную смесь перемешивали при комнатной температуре в течение часа. Летучие вещества удаляли при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; CH2Cl2:MeOH составляет 10:1 по объему, в подвижную фазу добавляли 1% NH3⋅H2O), получая (1S,3R,4S)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан (350 мг; выход: 17%) в виде желтого твердого вещества.
MS (ESI): 176,1 ([М+Н]+).
1Н-ЯМР (CDCl3, 400 МГц): δ 7.38-7.29 (5Н), 5.55 (1Н), 4.88 (1Н), 4.36 (1Н), 3.57 (1Н), 3.42 (1Н), 2.10 (1Н), 1.98 (1Н).
f) трет-Бутил-(1S,3R,4S)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К раствору (1S,3R,4S)-3-фенил-2-окса-5-азабицикло[2.2.1]гептана (10 г) в безводном ТГФ (150 мл) добавляли K2CO3 (24 г) и Boc2O (14 г; CAS: 24424-99-5). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Летучие вещества удаляли при пониженном давлении. Добавляли насыщенный раствор NaCl (100 мл). Смесь экстрагировали EtOAc (3×50 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир/EtOAc составляет 20:1 по объему), получая трет-бутил-(1S,3R,4S)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (4,2 г; выход: 25%) в виде желтого твердого вещества.
1Н-ЯМР (CDCl3, 400 МГц): δ 7.38-7.26 (5Н), 5.04 (1Н), 4.76 (1Н), 4.39 (1Н), 3.50 (1Н), 3.35 (1Н), 1.84 (1Н), 1.63 (1Н), 1.51 (9Н).
g) трет-Бутил-(1S,3R,4S)-3-(4-иодфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К раствору трет-бутил-(1S,3R,4S)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (1,0 г) в CCl4 (12 мл) добавляли [бис(трифторацетокси)иод]бензол (1,88 г; CAS: 2712-78-9) и йод (1,0 г; CAS: 7553-56-2). Реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение ночи. Смесь разбавляли хлороформом (200 мл). Раствор промывали 5%-ным водным раствором NaHSO3 (2×50 мл) и 10%-ным водным раствором NaCl (5×50 мл). Летучие вещества удаляли при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир/EtOAc составляет 20:1 по объему), получая трет-бутил-(1S,3R,4S)-3-(4-иодфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (600 мг) в виде коричневого масла.
MS (ESI): 345,8 (М-С4Н8+Н)+, 301,9 (М-С4Н8-CO2+Н)+.
h) 3-Хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
В атмосфере N2 раствор трет-бутил-(1S,3R,4S)-3-(4-иодфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (40 мг), 3-хлорбензамида (22 мг; CAS: 618-48-4), трис(дибензилиденацетон)дипалладия(0) (18 мг; CAS: 51364-51-3), бис(дифенилфосфино)-9,9-диметилксантена (Xantphos, 19 мг; CAS: 161265-03-8) и Cs2CO3 (163 мг; CAS: 534-17-8) в диоксане (1 мл) перемешивали при 90°С в течение ночи. TLC-анализ показывал завершение реакции. Смесь фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (колонка С-18; 0,1% NH3 в смеси H2O/MeCN), получая неочищенное промежуточное соединение в виде коричневого масла. Неочищенное промежуточное соединение растворяли в CH2Cl2 (2 мл). Добавляли TFA (0,5 мл; CAS: 76-05-1). Раствор перемешивали при комнатной температуре в течение часа. Летучие вещества удаляли при пониженном давлении. Остаток подвергали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,1% TFA; колонка С-18), получая 3-хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид (10 мг; выход 30% за 2 стадии) в виде белого твердого вещества. MS (ESI): 331,0 ([{37Cl}M+H]+); 329,0 ([{35Cl}M+H]+).
1Н-ЯМР (метанол-d4, 400 МГц): δ 7.95 (1Н), 7.87 (1Н), 7.75 (2Н), 7.60 (1Н), 7.53 (1Н), 7.37 (2Н), 5.15 (1Н), 4.95 (1Н), 4.39 (1Н), 3.46 (1Н), 3.36 (1Н), 2.13 (1Н), 1.82 (1Н).
Пример 9
(1S,3R,4S)-3-Фенил-2-окса-5-азабицикло[2.2.1]гептан
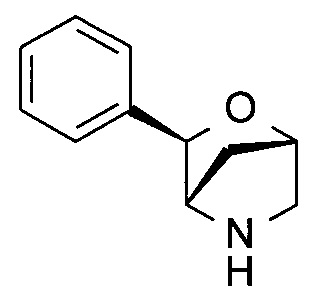
Указанное в заголовке соединение получали на стадии (е) примера 8.
MS(ESI): 176,1 ([М+Н]+).
Пример 10
3-Хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
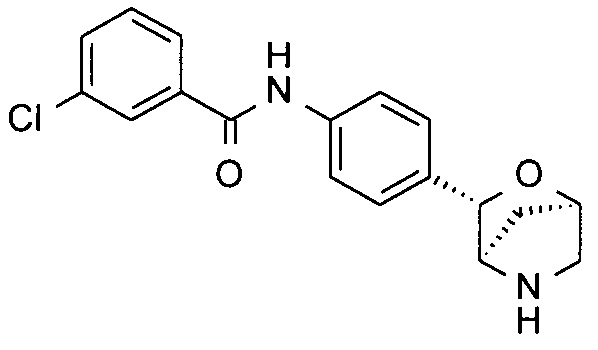
Указанное в заголовке соединение получали по аналогии с соединением примера 7, используя 3-хлорбензамид (CAS: 618-48-4) вместо 4-хлорбензамида на стадии (с). Белое твердое вещество. MS (ESI): 331,0 ([{37Cl}M+H]+); 329,0 ([{35Cl}M+H]+).
Пример 11
N-[4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-амин
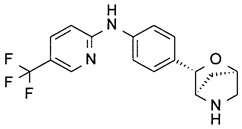
В атмосфере N2 раствор трет-бутил-(1R,3S,4R)-3-(4-иодфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (40 мг), 2-амино-5-(трифторметил)пиридина (24 мг; CAS: 74784-70-6), трис(дибензилиденацетон)дипалладия(0) (18 мг; CAS: 51364-51-3), бис(дифенилфосфино)-9,9-диметилксантена (Xantphos; 10 мг; CAS: 161265-03-8) и Cs2CO3 (162 мг; CAS: 534-17-8) в диоксане (1 мл) перемешивали при 90°С в течение ночи. TLC-анализ показывал завершение реакции. Смесь фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (колонка С-18; 0,1% NH3 в смеси H2O/MeCN), получая неочищенное промежуточное соединение в виде коричневого масла. Неочищенное промежуточное соединение растворяли в CH2Cl2 (2 мл). Добавляли TFA (0,5 мл; CAS: 76-05-1). Раствор перемешивали при комнатной температуре в течение часа. Летучие вещества удаляли при пониженном давлении. Остаток подвергали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,1% TFA; колонка С-18), получая указанное в заголовке соединение (12 мг; выход 36% за 2 стадии) в виде желтого твердого вещества.
MS (ESI): 336,2 ([М+Н]+).
1Н-ЯМР (метанол-d4, 400 МГц): δ 8.40 (1Н), 7.78 (1Н), 7.76 (2Н), 7.33 (2Н), 6.93 (1Н), 5.17 (1Н), 4.91 (1Н), 4.38 (1Н), 3.46 (1Н), 3.37 (1Н), 2.16 (1Н), 1.84 (1Н).
Пример 12
4-(Циклопропилметокси)-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
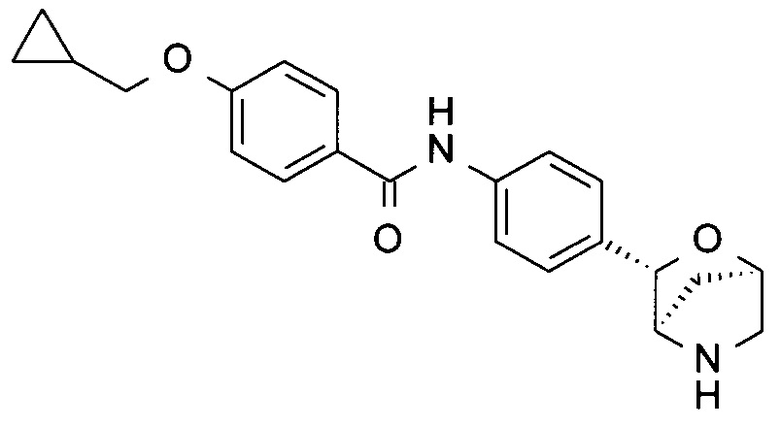
а) 4-(Циклопропилметокси)бензамид
К раствору 4-(циклопропилметокси)бензойной кислоты (384 мг; CAS: 355391-05-8), HATU (836 мг; CAS: 148893-10-1) и Et3N (606 мг; С AS: 121-44-8) в DMF (2,0 мл) добавляли NH3 в воде (25%-28%; 1,0 мл) при комнатной температуре. Реакционную смесь перемешивали в течение ночи. Летучие вещества удаляли при пониженном давлении. Смесь очищали посредством обращенно-фазовой хроматографии (колонка С-18, подвижная фаза: А, H2O; В, CH3CN с 0,5% NH3⋅H2O), получая 4-(циклопропилметокси)бензамид в виде белого твердого вещества (275 мг; выход 72%). MS(ESI): 192,1 (М+Н)+.
b) 4-(Циклопропилметокси)-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
Указанное в заголовке соединение получали по аналогии с соединением примера 7, используя 4-(циклопропилметокси)бензамид вместо 4-хлорбензамида на стадии (с). Белое твердое вещество. MS (ESI): 365,1 ([М+Н]+).
Пример 13
6-Этокси-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-3-карбоксамид
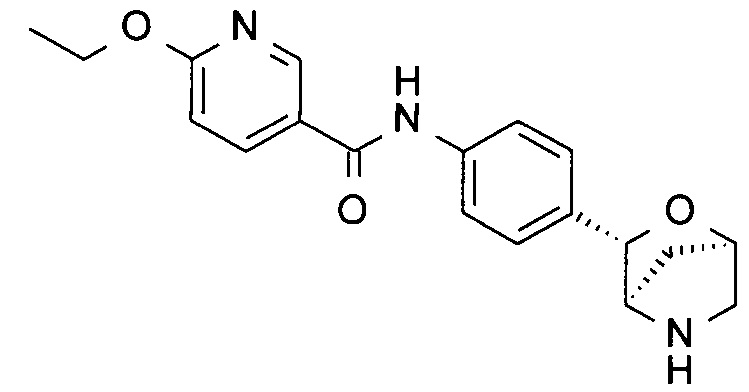
Указанное в заголовке соединение получали по аналогии с соединением примера 7, используя 6-этоксипиридин-3-карбоксамид (CAS: 473693-84-4) вместо 4-хлорбензамида на стадии (с). Белое твердое вещество. MS (ESI): 340,2 ([М+Н]+).
Пример 14
4-Хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
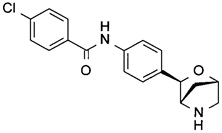
Указанное в заголовке соединение получали по аналогии с соединением примера 8, используя 4-хлорбензамид (CAS: 619-56-7) вместо 3-хлорбензамида на стадии (h). Белое твердое вещество. MS (ESI): 331,1 ([{37Cl}M+H]+); 329,1 ([{35Cl}M+H]+).
Пример 15
6-Этокси-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-3-карбоксамид
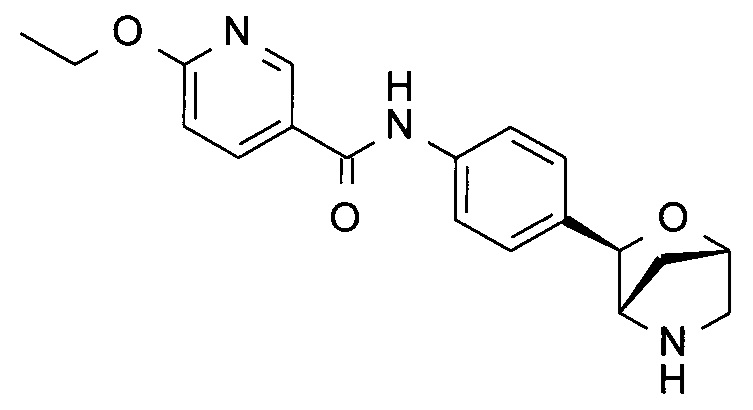
Указанное в заголовке соединение получали по аналогии с соединением примера 8, используя 6-этоксипиридин-3-карбоксамид (CAS: 473693-84-4) вместо 3-хлорбензамида на стадии (h). Белое твердое вещество. MS (ESI): 340,2 ([M+H]+).
Пример 16
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(2,2,2-трифторэтокси)пиридин-3-карбоксамид
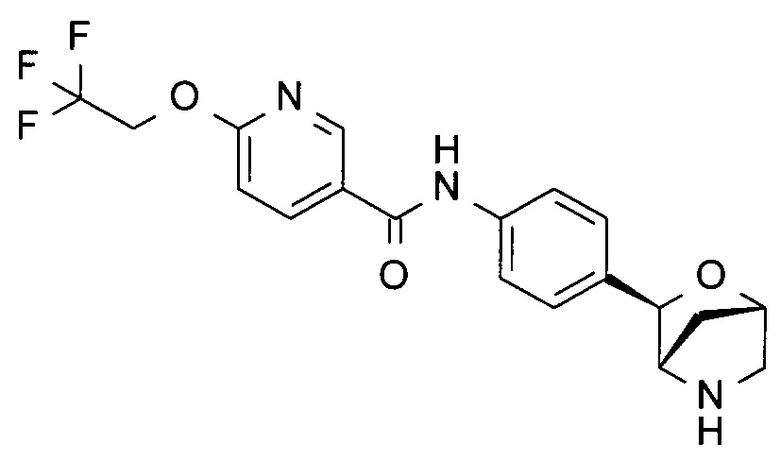
Указанное в заголовке соединение получали по аналогии с соединением примера 8, используя 6-(2,2,2-трифторэтокси)пиридин-3-карбоксамид (CAS: 676533-51-0) вместо 3-хлорбензамида на стадии (h). Белое твердое вещество. MS (ESI): 394,1 ([М+Н]+).
Пример 17
4-Этокси-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид

Указанное в заголовке соединение получали по аналогии с соединением примера 8, используя 4-этоксибензамид (CAS: 55836-71-0) вместо 3-хлорбензамида на стадии (h). Белое твердое вещество. MS (ESI): 339,2 ([М+Н]+).
Пример 18
2-Циклопропил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид
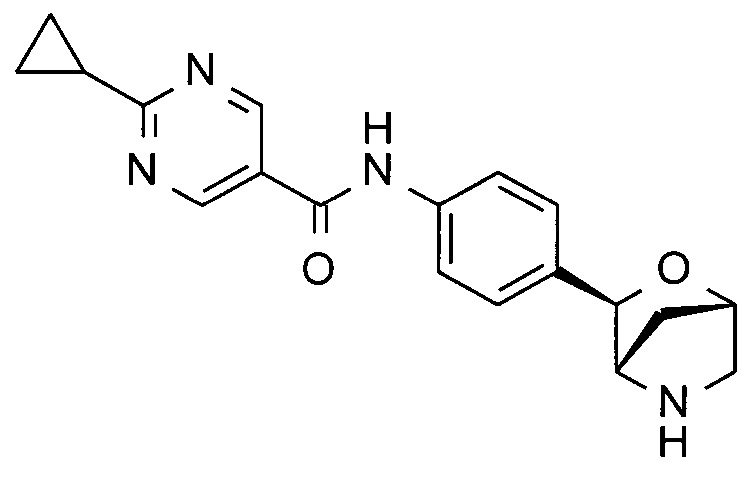
a) 2-Циклопропилпиримидин-5-карбоксамид
К раствору 2-циклопропилпиримидин-5-карбоновой кислоты (328 мг; СAS: 648423-79-4), HATU (836 мг; CAS: 148893-10-1) и Et3N (606 мг; CAS: 121-44-8) в DMF (2,0 мл) добавляли NH3 в воде (25%-28%; 1,0 мл) при комнатной температуре. Реакционную смесь перемешивали в течение ночи. Летучие вещества удаляли при пониженном давлении. Смесь очищали посредством обращенно-фазовой хроматографии (колонка С-18, подвижная фаза: А, H2O; В, CH3CN с 0,5% NH3⋅H2O), получая 2-циклопропилпиримидин-5-карбоксамид в виде белого твердого вещества (241 мг; выход 74%). MS (ESI): 164,1 (М+Н)+.
b) 2-Циклопропил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид
Указанное в заголовке соединение получали по аналогии с соединением примера 8, используя 2-циклопропилпиримидин-5-карбоксамид (CAS: 1447607-18-2) вместо 3-хлорбензамида на стадии (h). Белое твердое вещество. MS (ESI): 337,2 ([М+Н]+).
Пример 19
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-амин
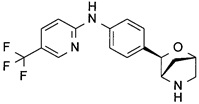
В атмосфере N2 раствор трет-бутил-(1S,3R,4S)-3-(4-иодфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (50 мг), 2-амино-5-(трифторметил)-пиридина (80 мг; CAS: 74784-70-6), трис(дибензилиденацетон)дилалладия(0) (20 мг; CAS: 51364-51-3), бис(дифенилфосфино)-9,9-диметилксантена (Xantphos; 20 мг; CAS: 161265-03-8) и Cs2CO3 (120 мг; CAS: 534-17-8) в диоксане (3 мл) перемешивали при 90°С в течение ночи. TLC-анализ показывал завершение реакции. Смесь фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (колонка С-18; 0,1% NH3 в смеси H2O/MeCN), получая неочищенное промежуточное соединение в виде коричневого масла. Неочищенное промежуточное соединение растворяли в CH2Cl2 (2 мл). Добавляли TFA (0,5 мл; CAS: 76-05-1). Раствор перемешивали при комнатной температуре в течение часа. Летучие вещества удаляли при пониженном давлении. Остаток подвергали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,1% TFA; колонка С-18), получая указанное в заголовке соединение (16 мг; выход 32% за 2 стадии) в виде желтого твердого вещества.
MS(ESI): 336,1 ([М+Н]+).
Пример 20
5-Хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-2-амин

Указанное в заголовке соединение получали по аналогии с соединением примера 19, используя 2-амино-5-хлорпиридин (CAS: 1072-98-6) вместо 2-амино-5-(трифторметил)пиридина. Белое твердое вещество. MS (ESI): 304,0 ([{37Cl}M+H]+); 302,0 ([{35Cl}М+Н]+).
Пример 21
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиримидин-4-амин
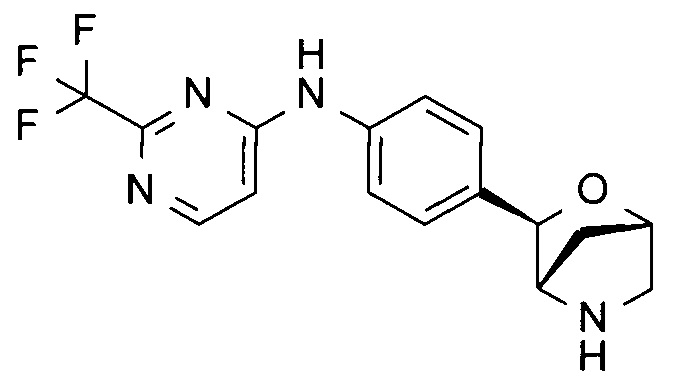
Указанное в заголовке соединение получали по аналогии с соединением примера 19, используя 2-(трифторметил)пиримидин-4-амин (CAS: 672-42-4) вместо 2-амино-5-(трифторметил)пиридина. Белое твердое вещество. MS (ESI): 337,1 ([М+Н]+).
Пример 22
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиразин-2-амин
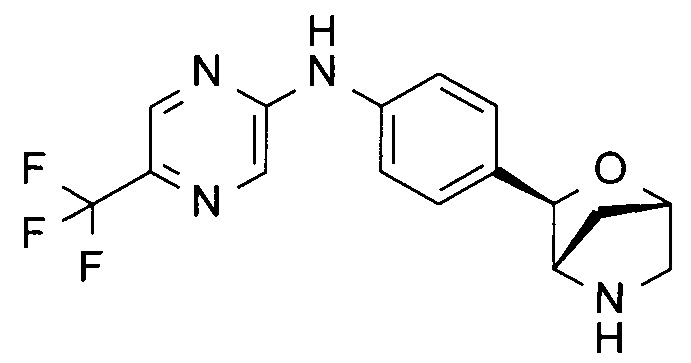
Указанное в заголовке соединение получали по аналогии с соединением примера 19, используя 5-трифторметил-2-аминопиразин (CAS: 69816-38-2) вместо 2-амино-5-(трифторметил)пиридина. Белое твердое вещество. MS (ESI): 337,0 ([М+Н]+).
Пример 23
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиримидин-2-амин
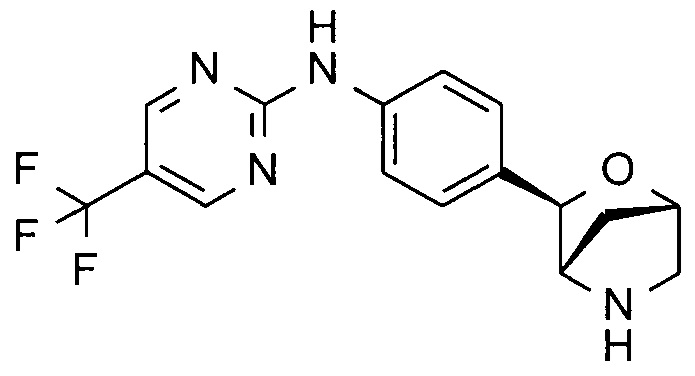
Указанное в заголовке соединение получали по аналогии с соединением примера 19, используя 5-(трифторметил)пиримидин-2-амин (CAS: 69034-08-8) вместо 2-амино-5-(трифторметил)пиридина. Белое твердое вещество. MS (ESI): 337,0 ([М+Н]+).
Пример 24
5-Хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-2-амин
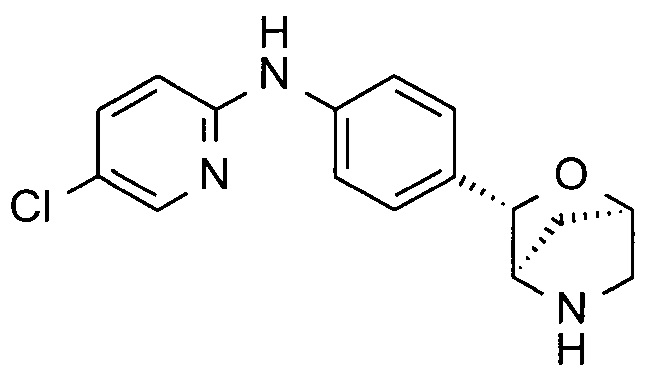
Указанное в заголовке соединение получали по аналогии с соединением примера 11, используя 2-амино-5-хлорпиридин (CAS: 1072-98-6) вместо 2-амино-5-(трифторметил)пиридина. Белое твердое вещество. MS (ESI): 304,1 ([{37Cl}M+H]+); 302,1 ([{35Cl}M+H]+).
Пример 25
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиридин-4-карбоксамид

Указанное в заголовке соединение получали по аналогии с соединением примера 8, используя 4-амино-2-(трифторметил)пиридин (CAS: 147149-98-2) вместо 3-хлорбензамида на стадии (h). Белое твердое вещество. MS (ESI): 364,1 ([М+Н]+).
Пример 26
N-[4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиридин-4-карбоксамид
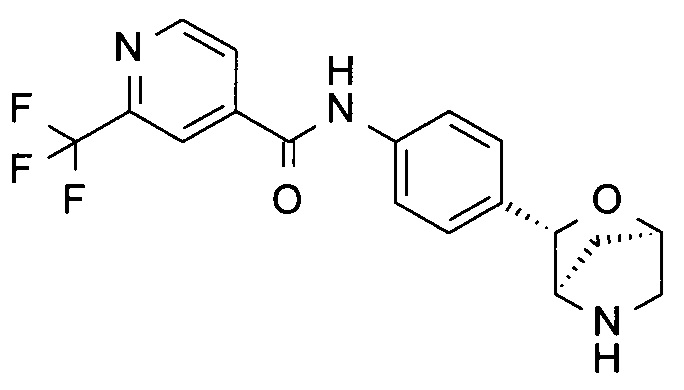
Указанное в заголовке соединение получали по аналогии с соединением примера 11, используя 4-амино-2-(трифторметил)пиридин (CAS: 147149-98-2) вместо 2-амино-5-(трифторметил)пиридина. Белое твердое вещество. MS (ESI): 364,1 ([М+Н]+).
Пример 27
N-[4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(2,2,2-трифторэтокси)пиридин-3-карбоксамид
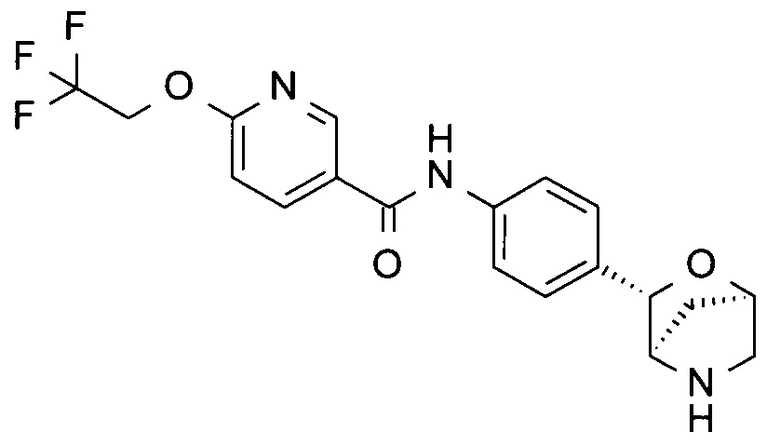
Указанное в заголовке соединение получали по аналогии с соединением примера 11, используя 6-(2,2,2-трифторэтокси)пиридин-3-карбоксамид (CAS: 676533-51-0) вместо 2-амино-5-(трифторметил)пиридина. Белое твердое вещество. MS (ESI): 394,2 ([М+Н]+).
Пример 28
4-Этокси-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
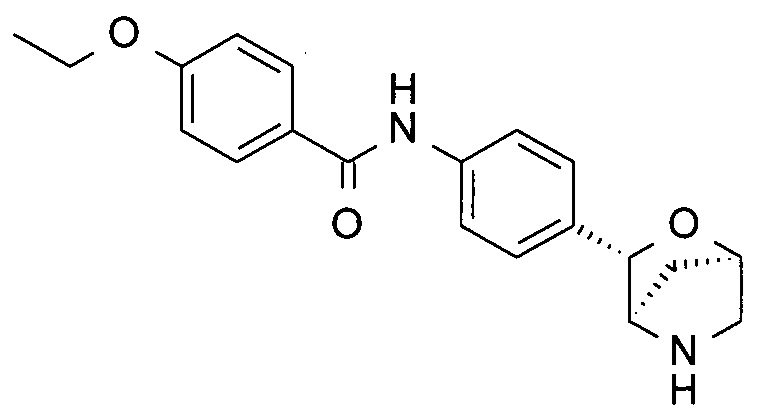
Указанное в заголовке соединение получали по аналогии с соединением примера 11, используя 4-этоксибензамид (CAS: 55836-71-0) вместо 2-амино-5-(трифторметил)пиридина. Белое твердое вещество. MS (ESI): 339,2 ([М+Н]+).
Пример 29
2-Циклопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид
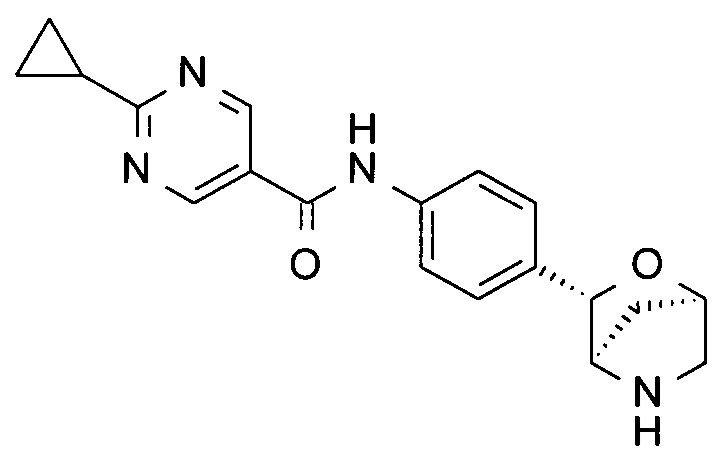
Указанное в заголовке соединение получали по аналогии с соединением примера 11, используя 2-циклопропилпиримидин-5-карбоксамид (CAS: 1447607-18-2) вместо 2-амино-5-(трифторметил)пиридина. Белое твердое вещество. MS (ESI): 337,2 ([М+Н]+).
Пример 30
N-[4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиразин-2-амин
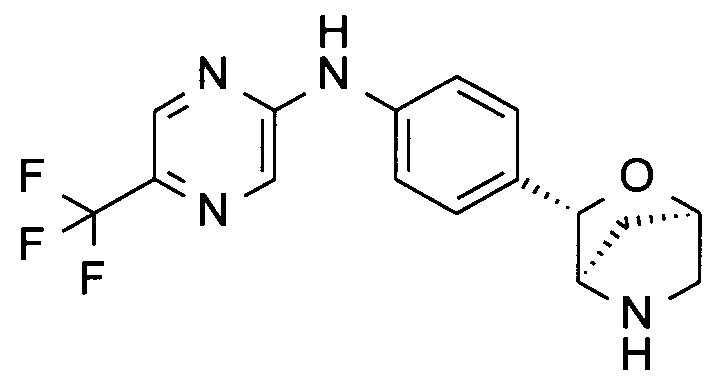
Указанное в заголовке соединение получали по аналогии с соединением примера 11, используя 5-трифторметил-2-аминопиразин (CAS: 69816-38-2) вместо 2-амино-5-(трифторметил)пиридина. Белое твердое вещество. MS (ESI): 337,1 ([М+Н]+).
Пример 31
N-[4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиримидин-2-амин
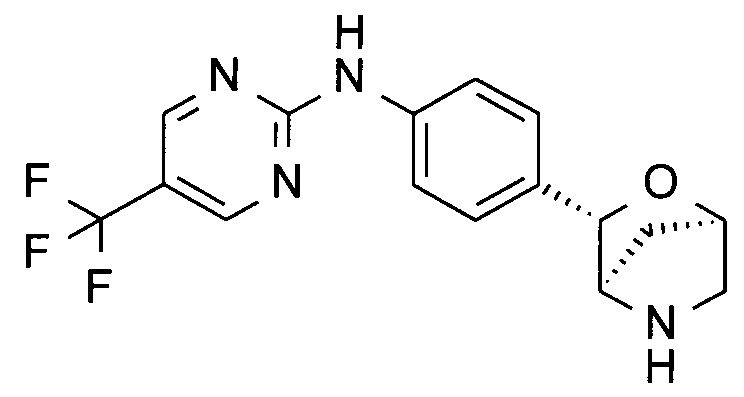
Указанное в заголовке соединение получали по аналогии с соединением примера 11, используя 5-(трифторметил)пиримидин-2-амин (CAS: 69034-08-8) вместо 2-амино-5-(трифторметил)пиридина. Белое твердое вещество. MS (ESI): 337,2 ([М+Н]+).
Пример 32
4-(Циклопропилметокси)-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид
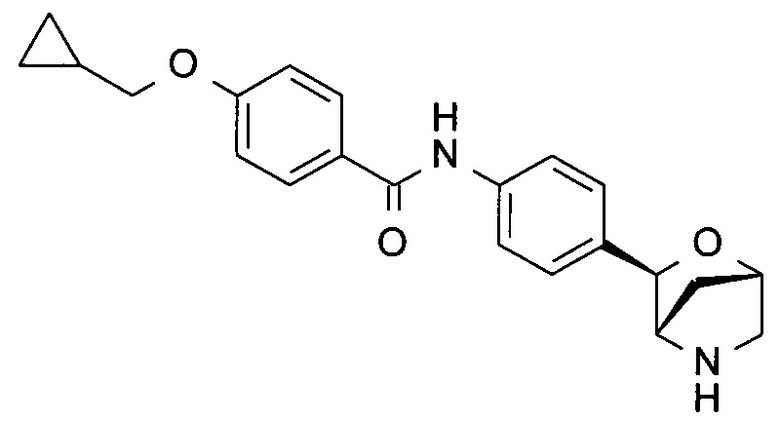
Указанное в заголовке соединение получали по аналогии с соединением примера 8, используя 4-(циклопропилметокси)бензамид вместо 3-хлорбензамида на стадии (h). Белое твердое вещество. MS (ESI): 365,1 ([М+Н]+).
Пример 33
1-(4-Хлорфенил)-3-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]мочевина
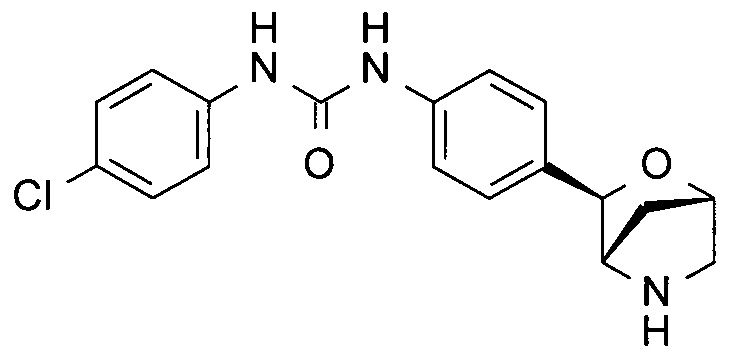
В атмосфере N2 раствор трет-бутил-(1S,3R,4S)-3-(4-иодфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (40 мг), 4-хлорфенилмочевины (22 мг; CAS: 140-38-5), трис(дибензилиденацетон)дипалладия(0) (18 мг; CAS: 51364-51-3), бис(дифенилфосфино)-9,9-диметилксантена (Xantphos; 19 мг; CAS: 161265-03-8) и Cs2CO3 (163 мг; CAS: 534-17-8) в диоксане (1 мл) перемешивали при 90°С в течение ночи. TLC-анализ показывал завершение реакции. Смесь фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (колонка С-18; 0,1% NH3 в H2O/MeCN), получая неочищенное промежуточное соединение в виде коричневого масла. Неочищенное промежуточное соединение растворяли в CH2Cl2 (2 мл). Добавляли TFA (0,5 мл; CAS: 76-05-1). Раствор перемешивали при комнатной температуре в течение часа. Летучие вещества удаляли при пониженном давлении. Остаток подвергали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,1% TFA; колонка С-18), получая указанное в заголовке соединение (4 мг; выход 12% за 2 стадии) в виде белого твердого вещества.
MS (ESI): 346,1 ([{37Cl}M+H]+); 344,1 ([{35Cl}M+H]+).
Пример 34
2-Этил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид
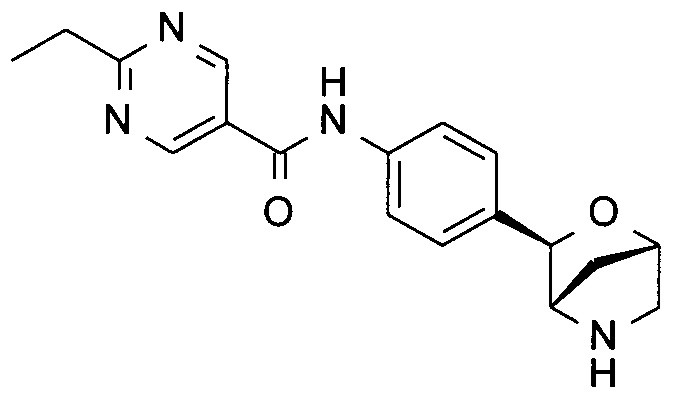
а) 2-Этилпиримидин-5-карбоксамид
К раствору 2-этилпиримидин-5-карбоновой кислоты (304 мг; CAS: 72790-16-0), HATU (836 мг; СAS: 148893-10-1) и Et3N (606 мг; CAS: 121-44-8) в DMF (2,0 мл) добавляли NH3 в воде (25%-28%; 1,0 мл) при комнатной температуре. Реакционную смесь перемешивали в течение ночи. Летучие вещества удаляли при пониженном давлении. Смесь очищали посредством обращенно-фазовой хроматографии (колонка С-18, подвижная фаза: А, H2O; В, CH3CN с 0,5% NH3⋅H20), получая 2-этилпиримидин-5-карбоксамид в виде белого твердого вещества (120 мг; выход 40%). MS (ESI): 152,2 (М+Н)+.
b) 2-Этил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид
Указанное в заголовке соединение получали по аналогии с соединением примера 8, используя 2-этилпиримидин-5-карбоксамид вместо 3-хлорбензамида на стадии (h). Воскообразное твердое вещество. MS (ESI): 325,0 ([М+Н]+).
Пример 35
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиридин-4-амин
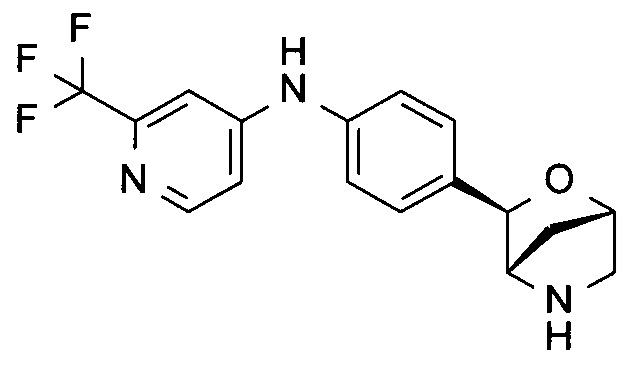
а) [(2S,4S)-1-Бензоил-4-гидрокси-пирролидин-2-ил]-(4-бромфенил)метанон
К раствору 1,4-дибромбензола (30 г; CAS: 106-37-6) в безводном ТГФ (500 мл) по каплям добавляли раствор н-бутиллития (2,5 М в гексанах; 52 мл; CAS: 109-72-8) при -78°С. Перемешивание продолжали в течение 30 минут. Затем этот раствор медленно добавляли к раствору (1S,4S)-5-бензоил-2-окса-5-азабицикло[2.2.1]гептан-3-она (25 г) в безводном ТГФ (1,0 л) при -78°С. Реакционную смесь перемешивали в течение 30 минут. Для гашения реакции добавляли насыщенный водный раствор NH4Cl (300 мл). Летучие вещества удаляли при пониженном давлении. Остаток растворяли в CH2Cl2 (1,0 л). Органический слой собирали. Водный слой экстрагировали CH2Cl2 (3×500 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир: этил ацетат составляет 1:3 по объему), получая [(2S,4S)-1-бензоил-4-гидрокси-пирролидин-2-ил]-(4-бромфенил)метанон (20 г; 53 ммоль; выход: 41%) в виде белого твердого вещества.
MS (ESI): 375,9 ([{81Br}M+H]+); 373,9 ([{79Br}M+H]+).
b) [(2S,4S)-2-[(R)-(4-Бромфенил)-гидрокси-метил]-4-гидрокси-пирролидин-1-ил]-фенил-метанон
К раствору [(2S,4S)-1-бензоил-4-гидрокси-пирролидин-2-ил]-(4-бромфенил)метанона (20 г; 53 ммоль; со стадии а) в МеОН (100 мл) при 0°С добавляли NaBH4 (8,1 г; 213 ммоль). Реакционную смесь перемешивали при 0°С в течение 1,5 часа. Для погашения избытка NaBH4 добавляли ацетон. Летучие вещества удаляли при пониженном давлении. Добавляли насыщенный водный раствор NH4Cl. Смесь экстрагировали EtOAc, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир : этилацетат составляет 1:1 по объему), получая [(2S,4S)-2-[(R)-(4-бромфенил)-гидрокси-метил]-4-гидрокси-пирролидин-1-ил]-фенил-метанон (15 г; выход: 41%) в виде белого твердого вещества.
MS (ESI): 378,0 ([{81Br}M+H]+); 376,0 ([{79Br}M+H]+).
c) [(1S,3R,4S)-3-(4-Бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-фенил-метанон
К раствору полученного [(2S,4S)-2-[(R)-(4-бромфенил)-гидрокси-метил]-4-гидрокси-пирролидин-1-ил]-фенил-метанона (9,5 г) и PPh3 (8,5 г; CAS: 603-35-0) в безводном толуоле (100 мл) добавляли DIAD (6,6 г; CAS: 2446-83-5) при 0°С. Смесь перемешивали при комнатной температуре в течение ночи. Летучие вещества удаляли при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир : этилацетат составляет 5:1 по объему), получая [(1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-фенил-метанон (4 г; выход: 45%) в виде белого твердого вещества. MS (ESI): 360,1 ([{81Br}M+H]+); 358,1 ([{79Br}M+H]+).
d) (1S,3R,4S)-3-(4-Бромфенил)-2-окса-5-азабицикло[2.2.1]гептан
К раствору [(1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-фенил-метанона (10 г) в МеОН (28 мл) добавляли KOH (31 г). Реакционную смесь перемешивали при температуре дефлегмации в течение 30 минут до тех пор, пока TLC-анализ не показывал завершения реакции. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в CH2Cl2 (200 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; MeOH:CH2Cl2 составляет 1:20 по объему), получая (1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан (6,3 г; выход: 89%) в виде прозрачного масла.
MS (ESI): 256,0 ([{81Br}M+H]+); 254,0 ([{79Br}M+H]+).
1Н-ЯМР (CDCl3, 400 МГц): δ 7.44 (2Н), 7.15 (2Н), 4.81 (1Н), 4.67 (1Н), 3.56 (1Н), 3.11 (1Н), 2.99 (1Н), 1.73 (1Н), 1.49 (1Н).
e) трет-Бутил-(1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К раствору (1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептана (6,3 г) в ТГФ (100 мл) добавляли K2CO3 (10,3 г) и Boc2O (6,5 г). Смесь перемешивали при комнатной температуре в течение ночи до тех пор, пока TLC-анализ не показывал завершения реакции. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в CH2Cl2 (200 мл) и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир : этилацетат составляет 20:1 по объему), получая трет-бутил-(1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (4,0 г; выход: 45%) в виде белого твердого вещества.
1Н-ЯМР (CDCl3, 400 МГц): δ 7.48 (2Н), 7.22 (1Н), 7.14 (1Н), 4.97 (1Н), 4.75 (1Н), 4.35 (1Н), 3.48 (1Н), 3.34 (1Н), 1.77 (1Н), 1.63 (1Н), 1.554-1.499 (9Н).
f) N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиридин-4-амин
К раствору 4-амино-2-(трифторметил)пиридина (33 мг; CAS: 147149-98-2) и трет-бутил-(1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (50 мг) в диоксане (3 мл) добавляли Cs2CO3 (136 мг; CAS: 534-17-8), трис(дибензилиденацетон)дипалладий(0) (20 мг; CAS: 51364-51-3) и бис(дифенилфосфино)-9,9-диметилксантен (Xantphos; 20 мг; CAS: 161265-03-8). Реакционную смесь перемешивали при 90°С в атмосфере N2 в течение ночи. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в CH2Cl2 (10 мл) и фильтровали через набивку диоксида кремния в виде тонкого слоя. Фильтрат концентрировали и сушили под высоким вакуумом, получая неочищенный трет-бутил-(1S,3R,4S)-3-[4-[[2-(трифторметил)-4-пиридил]амино]фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (40 мг) в виде коричневого масла, которое использовали на следующей стадии непосредственно.
К раствору трет-бутил-(1S,3R,4S)-3-[4-[[2-(трифторметил)-4-пиридил]амино]фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (40 мг) в безводном CH2Cl2 (2 мл) добавляли TFA (0,5 мл; CAS: 76-05-1). Смесь перемешивали при комнатной температуре в течение 30 минут. Затем летучие вещества удаляли при пониженном давлении. Остаток очищали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,1% TFA, колонка С-18), получая N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиридин-4-амин (24 мг; выход 34% за 2 стадии) в виде белого твердого вещества. MS (ESI): 336,1 ([М+Н]+).
1Н-ЯМР (метанол-d4, 400 МГц): δ 8.27 (1Н), 7.47 (2Н), 7.36 (2Н), 7.31 (1Н), 7.18 (1Н), 5.20 (1Н), 4.95 (1Н), 4.43 (1Н), 3.45 (1Н), 3.36 (1Н), 2.11 (1Н), 1.84 (1Н).
Пример 36
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-карбоксамид
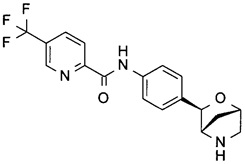
a) трет-Бутил-(1S,3R,4S)-3-[4-(бензгидрилиденамино)фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К раствору трет-бутил-(1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (1,0 г), 2,2'-бис(дифенилфосфино)-1,1'-бинафталина (BINAP; 200 мг; CAS: 98327-87-8), трис(дибензилиденацетон)дипалладия(0) (200 мг; CAS: 51364-51-3) и трет-бутилата натрия (806 мг; CAS: 865-48-5) в безводном толуоле (10 мл) добавляли бензофенонимин (610 мг; CAS: 1013-88-3). Реакционную смесь перемешивали в атмосфере N2 в течение 30 минут при 90°С в микроволновом реакторе. LCMS показывала завершение реакции. Смесь охлаждали до комнатной температуры, разбавляли водой (20 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические слои промывали рассолом (3×20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир : этилацетат составляет 5:1 по объему), получая трет-бутил-(1S,3R,4S)-3-[4-(бензгидрилиденамино)фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (1,15 г; выход 89%) в виде желтого твердого вещества. MS (ESI): 455,2 ([М+Н]+).
b) трет-Бутил-(1S,3R,4S)-3-(4-аминофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К раствору трет-бутил-(1S,3R,4S)-3-[4-(бензгидрилиденамино)фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (1,15 г) в безводном МеОН (10 мл) добавляли ацетат натрия (802 мг; CAS: 127-09-3) и гидроксиламина гидрохлорид (348 мг; CAS: 5470-11-1) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 минут до тех пор, пока TLC-анализ не показывал завершения реакции. Смесь разбавляли CH2Cl2 (100 мл) и промывали насыщенным водным раствором Na2CO3 (50 мл). Органический слой сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали хроматографией (силикагель; петролейный эфир : этилацетат составляет 1:1 по объему), получая трет-бутил-(1S,3R,4S)-3-(4-аминофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (500 мг; выход 68%) в виде желтого твердого вещества.
MS(ESI): 291,2 ([М+Н]+).
1Н-ЯМР (CDCl3, 400 МГц): δ 7.11 (1Н), 7.05 (1Н), 6.68 (2Н), 4.94 (1Н), 4.72 (1Н), 4.31 (1Н), 3.66 (2Н), 3.47 (1Н), 3.32 (1Н), 1.86 (1Н), 1.61 (1Н), 1.54-1.50 (9Н).
с) N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-карбоксамид
К раствору трет-бутил-(1S,3R,4S)-3-(4-аминофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (40 мг) в безводном DMF (1 мл) добавляли HATU (67 мг; CAS: 148893-10-1) и DIPEA (104 мг; CAS: 7087-68-5). Смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли 5-(трифторметил)пиридин-2-карбоновую кислоту (50 мг; CAS: 80194-69-0). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов до тех пор, пока LCMS-анализ не показывал завершения реакции. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в CH2Cl2 (20 мл) и промывали водой и рассолом. Органический слой концентрировали при пониженном давлении и сушили под высоким вакуумом, получая неочищенный трет-бутил-(1S,3R,4S)-3-[4-[[5-(трифторметил)пиридин-2-карбонил]амино]фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (40 мг) в виде желтого твердого вещества, которое использовали на следующей стадии непосредственно.
К раствору неочищенного трет-бутил-(1S,3R,4S)-3-[4-[[5-(трифторметил)пиридин-2-карбонил]амино]фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (40 мг) в безводном CH2Cl2 (2 мл) добавляли TFA (0,5 мл; CAS: 76-05-1). Смесь перемешивали при комнатной температуре в течение 30 минут. Летучие вещества удаляли при пониженном давлении. Остаток очищали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,1% TFA, колонка С-18), получая N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-карбоксамид (6 мг) в виде белого твердого вещества. MS (ESI): 455,2 ([М+Н]+).
1Н-ЯМР (метанол-d4, 400 МГц): δ 9.04 (1Н), 8.39 (2Н), 7.87 (2Н), 7.40 (2Н), 5.16 (1Н), 4.95 (1Н), 4.40 (1Н), 3.46 (1Н), 3.36 (1Н), 2.14 (1Н), 1.82 (1Н).
Пример 37
4-Хлор-3-циклопропил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
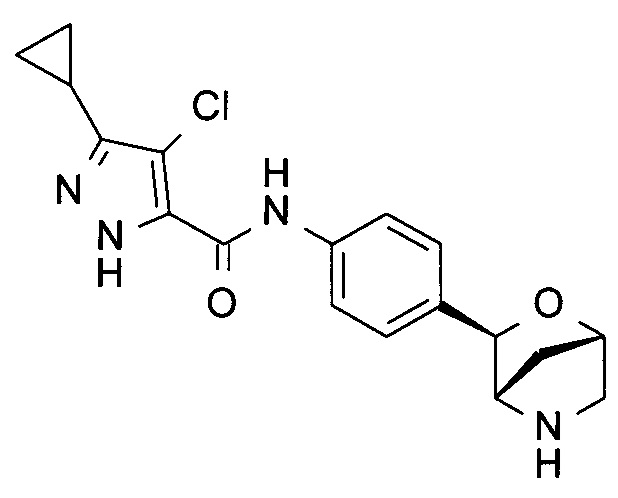
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 4-хлор-5-циклопропил-2Н-пиразол-3-карбоновую кислоту (CAS: 1291275-83-6) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 361,2 ([{37Cl}M+H]+); 359,2 ([{35Cl}M+H]+).
Пример 38
N-[(4-Хлорфенил)метил]-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин

Указанное в заголовке соединение получали по аналогии с соединением примера 35, используя 4-хлорбензиламин (CAS: 104-86-9) вместо 4-амино-2-(трифторметил)пиридина на стадии (f). Белое твердое вещество. MS (ESI): 317,1 ([{37Cl}M+H]+); 315,2 ([{35Cl}М+Н]+).
Пример 39
4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметил)фенил]метил]анилин
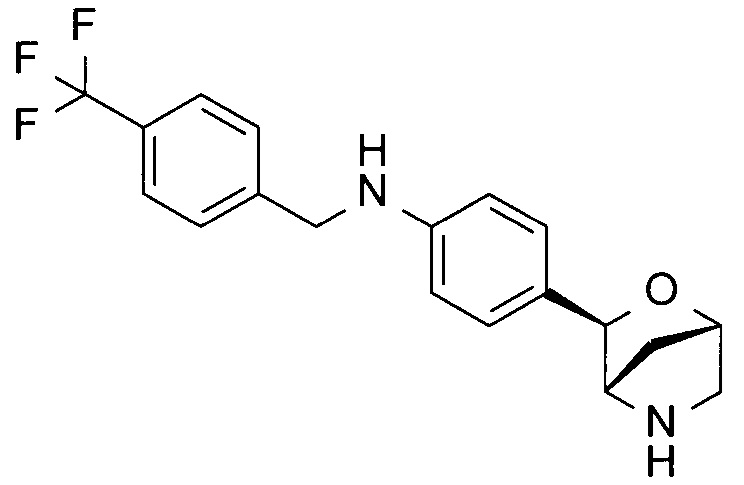
Указанное в заголовке соединение получали по аналогии с соединением примера 35, используя 4-(трифторметил)бензиламин (CAS: 3300-51-4) вместо 4-амино-2-(трифторметил)пиридина на стадии (f). Белое твердое вещество. MS (ESI): 349,2 ([М+Н]+).
Пример 40
N-[(4-Фторфенил)метил]-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин
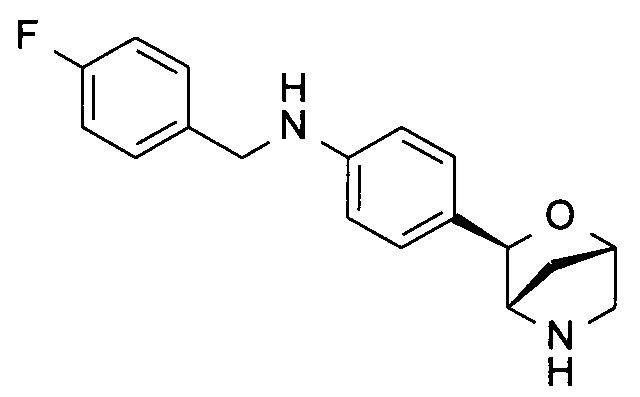
Указанное в заголовке соединение получали по аналогии с соединением примера 35, используя 4-фторбензиламин (CAS: 140-75-0) вместо 4-амино-2-(трифторметил)пиридина на стадии (f). Белое твердое вещество. MS (ESI): 299,2 ([М+Н]+).
Пример 41
N-Бутил-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин
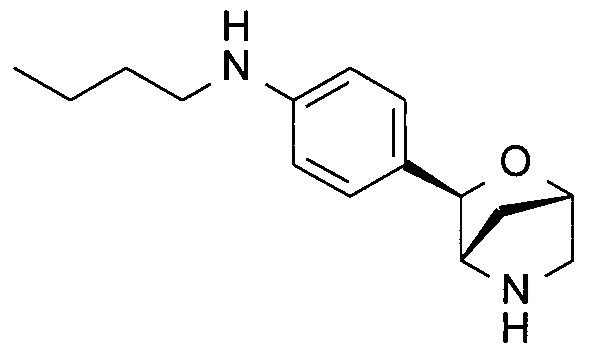
Указанное в заголовке соединение получали по аналогии с соединением примера 35, используя н-бутиламин (CAS: 109-73-9) вместо 4-амино-2-(трифторметил)пиридина на стадии (f). Белое твердое вещество.
MS (ESI): 247,2 ([М+Н]+).
Пример 42
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиримидин-4-карбоксамид
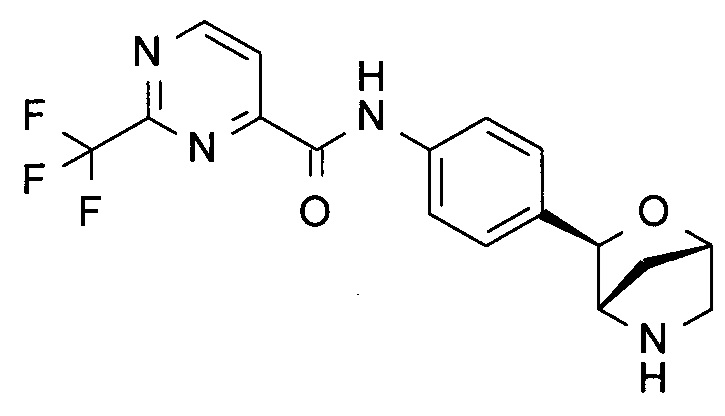
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 2-(трифторметил)пиримидин-4-карбоновую кислоту (CAS: 878742-59-7) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 365,2 ([М+Н]+).
Пример 43
3-Изопропил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
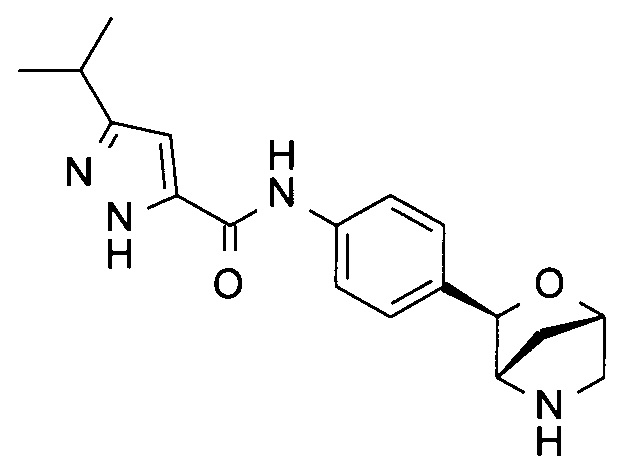
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 3-изопропилпиразол-5-карбоновую кислоту (CAS: 92933-47-6) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 327,3 ([М+Н]+).
Пример 44
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(трифторметил)пиридин-3-карбоксамид
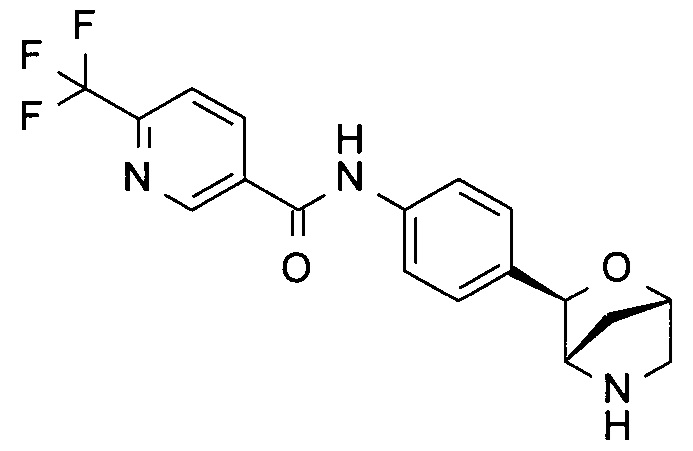
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 6-трифторметилникотиновую кислоту (CAS: 231291-22-8) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Воскообразное твердое вещество. MS (ESI): 364,2 ([М+Н]+).
Пример 45
4-Хлор-3-этокси-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид

а) Метил-3-гидрокси-1Н-пиразол-5-карбоксилат
К раствору гидразина моногидрата (3,85 г; CAS: 7803-57-8) в толуоле (30 мл) добавляли уксусную кислоту (15 мл) и диметилацетилендикарбоксилат (10 г; CAS: 762-42-5). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Смесь выливали в ледяную воду. Собирали осадок посредством фильтрования, промывали водой и сушили под высоким вакуумом, получая метил-3-гидрокси-1Н-пиразол-5-карбоксилат (7,5 г; выход 75%) в виде белого твердого вещества.
1Н-ЯМР (DMSO-d6, 400 МГц): δ 12.81 (1Н), 10.03 (1Н), 5.96 (1Н), 3.77 (3Н).
b) Метил-3-этокси-1Н-пиразол-5-карбоксилат
К раствору метил-3-гидрокси-1Н-пиразол-5-карбоксилата (4 г) в DMF (25 мл) добавляли K2CO3 (5,83 г) и иодэтан (4,8 г; CAS: 75-03-6). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Смесь выливали в воду и экстрагировали этилацетатом (100 мл). Органический слой промывали рассолом (30 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали перекристаллизацией из CH2Cl2 (10 мл), получая метил-3-этокси-1Н-пиразол-5-карбоксилат (2,2 г; выход 46%) в виде белого твердого вещества.
1Н-ЯМР (DMSO-d6, 400 МГц): δ 13.12 (1Н), 6.22 (1Н), 4.13-4.08 (2Н), 3.81-3.75 (3Н), 1.32-1.25 (3Н).
c) Метил-4-хлор-3-этокси-1Н-пиразол-5-карбоксилат
К раствору метил-3-этокси-1Н-пиразол-5-карбоксилата (2,2 г) в DMF (40 мл) добавляли N-хлорсукцинимид (2,06 г; CAS: 128-09-6) при 0°С. Реакционную смесь нагревали до 50°С и перемешивание продолжали в течение 15 часов. Летучие вещества удаляли при пониженном давлении. Смесь выливали в воду. Собирали осадок посредством фильтрования, промывали водой и сушили под высоким вакуумом, получая метил-4-хлор-3-этокси-1Н-пиразол-5-карбоксилат (1,65 г; выход 63%) в виде белого твердого вещества.
1Н-ЯМР (DMSO-d6, 400 МГц): δ 13.44 (1Н), 4.26-4.20 (2Н), 3.85 (3Н), 1.33-1.30 (3Н).
d) 4-Хлор-3-этокси-1Н-пиразол-5-карбоновая кислота
К раствору метил-4-хлор-3-этокси-1Н-пиразол-5-карбоксилата (1,65 г) в смеси ТГФ/МеОН (об./об. составляет 1:1, 30 мл) добавляли 1 М водный раствор NaOH (16 мл) с охлаждением в ледяной бане. Реакционную смесь перемешивали при нагревании с обратным холодильником в течение 3 часов. Реакционный раствор выливали в воду и pH подводили до 1, используя концентрированный раствор HCl. Осадок собирали фильтрованием, промывали водой и сушили под высоким вакуумом, получая 4-хлор-3-этокси-1Н-пиразол-5-карбоновую кислоту (1,4 г; выход 92%) в виде белого твердого вещества.
1Н-ЯМР (DMSO-d6, 400 МГц): δ 13.25 (1Н), 4.25-4.20 (2Н), 1.33-1.30 (3Н).
e) 4-Хлор-3-этокси-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 4-хлор-3-этокси-1Н-пиразол-5-карбоновую кислоту вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 365,1 ([{37Cl}M+H]+); 363,2 ([{35Cl}M+H]+).
Пример 46
4-Хлор-3-метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
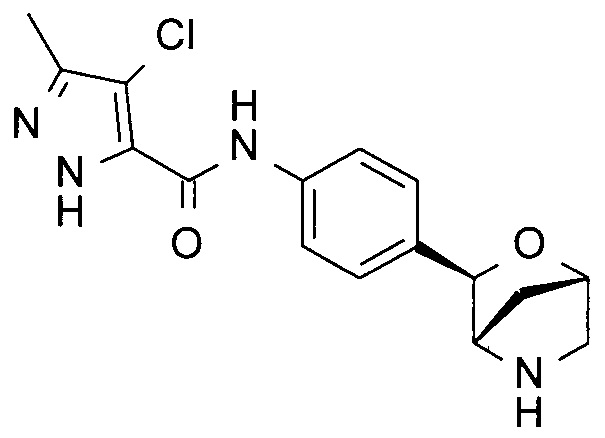
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 4-хлор-3-метил-1Н-пиразол-5-карбоновую кислоту (CAS: 29400-84-8) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 335,2 ([{37Cl}M+H]+); 333,2 ([{35Cl}M+H]+).
Пример 47
4-Метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
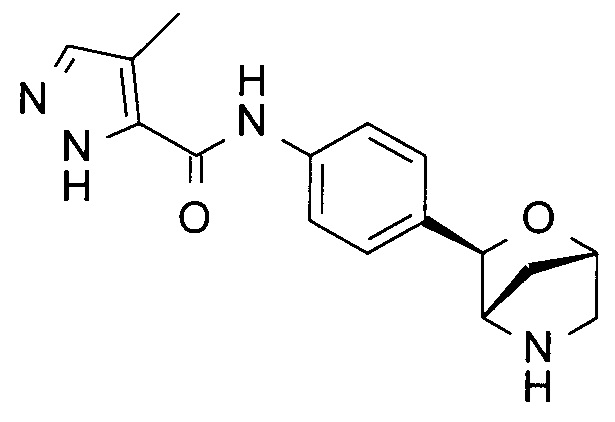
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 4-метилпиразол-3-карбоновую кислоту (CAS: 82231-51-4) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 299,0 ([М+Н]+).
Пример 48
4-Хлор-1-метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-пропил-пиразол-3-карбоксамид
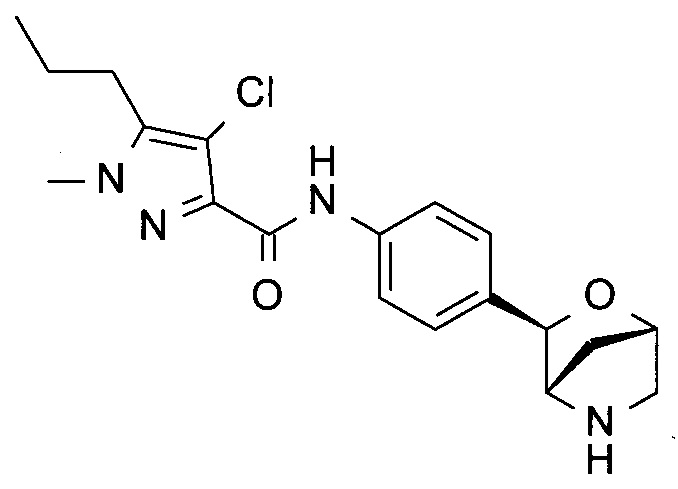
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 4-хлор-1-метил-5-пропил-пиразол-3-карбоновую кислоту (CAS: 1248078-41-2) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 377,2 ([{37Cl}M+H]+); 375,2 ([{35Cl}M+H]+).
Пример 49
4-Хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-пропил-1Н-пиразол-5-карбоксамид
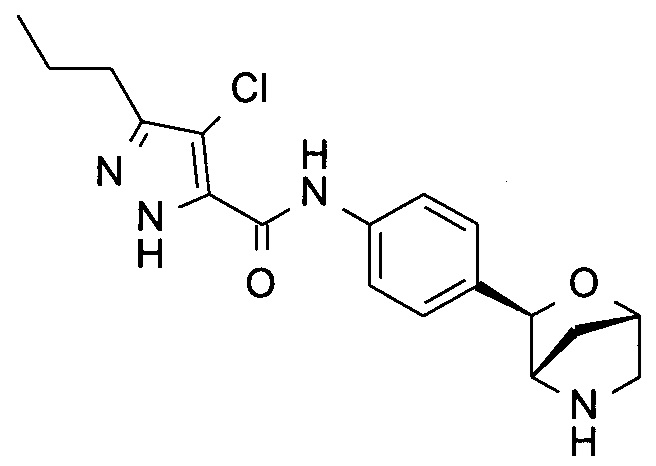
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 4-хлор-3-пропил-1Н-пиразол-5-карбоновую кислоту (CAS: 1340578-20-2) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 363,2 ([{37Cl}M+H]+); 361,2 ([{35Cl}M+H]+).
Пример 50
3-Этил-4-метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
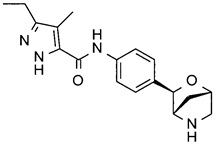
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 3-этил-4-метил-1Н-пиразол-5-карбоновую кислоту (CAS: 957129-38-3) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 327,2 ([М+Н]+).
Пример 51
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(2,2,2-трифторэтокси)пиридин-2-карбоксамид

Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 5-(2,2,2-трифторэтокси)пиридин-2-карбоновую кислоту (CAS: 881409-53-6) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 394,3 ([М+Н]+).
Пример 52
4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметокси)фенил]метил]анилин
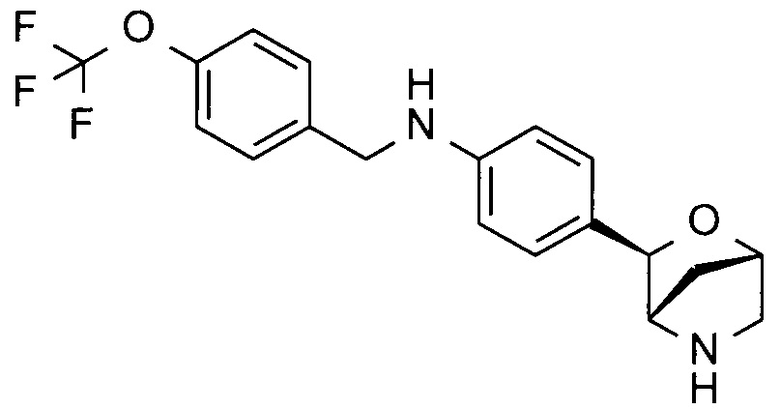
Указанное в заголовке соединение получали по аналогии с соединением примера 35, используя 4-(трифторметокси)бензиламин (CAS: 93919-56-3) вместо 4-амино-2-(трифторметил)пиридина на стадии (f). Белое твердое вещество. MS (ESI): 365,1 ([М+Н]+).
Пример 53
(1S,3R,4S)-3-(4-Бромфенил)-2-окса-5-азабицикло[2.2.1]гептан

Указанное в заголовке соединение получали на стадии (d) в процессе получения соединения примера 35.
MS (ESI): 256,0 ([{81Br}M+H]+); 254,0 ([{79Br}M+H]+).
Пример 54
(1R,3S,4R)-3-(4-Бромфенил)-2-окса-5-азабицикло[2.2.1]гептан
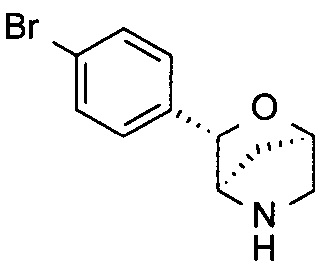
а) [(2R,4R)-1-Бензоил-4-гидрокси-пирролидин-2-ил]-(4-бромфенил)метанон
К раствору 1,4-дибромбензола (54,5 г; CAS: 106-37-6) в безводном ТГФ (1,0 л) по каплям добавляли раствор н-бутиллития (2,5 М в гексанах; 91 мл; CAS: 109-72-8) при -78°С. Перемешивание продолжали в течение 30 минут. Затем этот раствор медленно добавляли к раствору (1R,4R)-5-бензоил-2-окса-5-азабицикло[2.2.1]гептан-3-она (50 г) в безводном ТГФ (1,0 л) при -78°С. Реакционную смесь перемешивали в течение 30 минут. Для гашения реакции добавляли насыщенный водный раствор NH4Cl (300 мл). Летучие вещества удаляли при пониженном давлении. Остаток растворяли в CH2Cl2 (1,0 л). Органический слой собирали. Водный слой экстрагировали CH2Cl2 (3×500 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир : этилацетат от 5:1 до 1:3 по объему), получая [(2R,4R)-1-бензоил-4-гидрокси-пирролидин-2-ил]-(4-бромфенил)метанон (30 г; выход: 35%) в виде белого твердого вещества.
MS (ESI): 376,0 ([{81Br}М+Н]+); 374,0 ([{79Br}М+Н]+).
b) [(2R,4R)-2-[(S)-(4-Бромфенил)-гидрокси-метил]-4-гидрокси-пирролидин-1-ил]-фенил-метанон
К раствору [(2R,4R)-1-бензоил-4-гидрокси-пирролидин-2-ил]-(4-бромфенил)метанона (33 г) в МеОН (300 мл) при 0°С добавляли NaBH4 (13 г). Реакционную смесь перемешивали при 0°С в течение 1,5 часа. Для погашения избытка NaBH4 добавляли ацетон. Летучие вещества удаляли при пониженном давлении. Добавляли насыщенный водный раствор NH4Cl (200 мл). Смесь экстрагировали EtOAc (3×500 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир : этилацетат составляет 1:2 по объему), получая [(2R,4R)-2-[(S)-(4-бромфенил)-гидрокси-метил]-4-гидрокси-пирролидин-1-ил]-фенил-метанон (25 г; выход: 76%) в виде белого твердого вещества.
MS (ESI): 378,0 ([{81Br}M+H]+); 376,0 ([{79Br}M+H]+).
c) [(1R,3S,4R)-3-(4-Бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-фенил-метанон
К раствору полученного [(2R,4R)-2-[(S)-(4-бромфенил)-гидрокси-метил]-4-гидрокси-пирролидин-1-ил]-фенил-метанона (22 г) и PPh3 (18 г) в безводном толуоле (300 мл) добавляли DIAD (14 г; CAS: 2446-83-5) при 0°С. Смесь перемешивали при комнатной температуре в течение ночи. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в трет-бутил-метиловом эфире (600 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир : этилацетат от 5:1 до 1:1 по объему), получая [(1R,3S,4R)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-фенил-метанон (12 г; выход: 57%) в виде белого твердого вещества. MS (ESI): 360,0 ([{81Br}M+H]+); 358,0 ([{79Br}M+H]+).
d) (1R,3S,4R)-3-(4-Бромфенил)-2-окса-5-азабицикло[2.2.1]гептан
К раствору [(1R,3S,4R)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-фенил-метанона (12,5 г) в МеОН (35 мл) добавляли KOH (39 г). Реакционную смесь перемешивали при температуре дефлегмации в течение часа до тех пор, пока LCMS-анализ не показывал завершения реакции. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в CH2Cl2 (200 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель, MeOH:CH2Cl2 от 1:20 до 1:5 по объему), получая (1R,3S,4R)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан (7 г; выход: 75%) в виде прозрачного масла.
MS (ESI): 256,0 ([{81Br}M+H]+); 254,0 ([{79Br}M+H]+).
1Н-ЯМР (метанол-d4, 400 МГц): δ 7.51 (d, 2Н), 7.24 (d, 2Н), 4.90 (s, 1Н), 4.73 (s, 1Н), 3.66 (s, 1Н), 3.07 (d, 1Н), 2.98 (d, 1Н), 1.79 (d, 1Н), 1.54 (d, 1Н).
Пример 55
N-(4-Хлорфенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
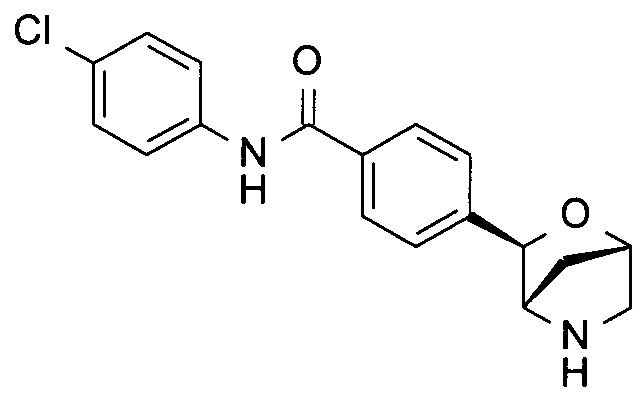
а) 4-[(1S,3R,4S)-5-трет-Бутоксикарбонил-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензойная кислота
К раствору трет-бутил-(1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (800 мг; 2,25 ммоль) в ТГФ (20 мл) добавляли раствор н-бутиллития (2,5 М в гексанах; 1,2 мл; 2,9 ммоль, CAS: 109-72-8) при -78°С. Перемешивание продолжали при -78°С в течение 30 минут. Через раствор в течение 10 минут барботировали обезвоженный CO2. Раствор нагревали до комнатной температуры. В реакционный раствор добавляли 1 М водный раствор HCl для подведения pH до 4-5. Смесь экстрагировали CH2Cl2 (3×30 мл). Объединенные органические слои сушили над Na2SO4. Летучие вещества удаляли при пониженном давлении. Остаток очищали посредством флэш-хроматографии (силикагель; CH2Cl2/MeOH от 100/1 до 30/1 по объему), получая 4-[(1S,3R,4S)-5-трет-бутоксикарбонил-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензойную кислоту (450 мг; выход 62%) в виде желтого твердого вещества.
1Н-ЯМР (400 МГц, CDCl3): δ 8.10 (m, 2Н), 7.42 (dd, 2Н), 5.09 (d, 1Н), 4.80 (s, 1Н), 4.43 (d, 1Н), 3.52 (m, 1Н), 3.37 (t, 1Н), 1.78 (d, 1Н), 1.67 (m, 1Н), 1.57-1.55 (m, 9Н).
b) N-(4-Хлорфенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
К раствору 4-[(1S,3R,4S)-5-трет-бутоксикарбонил-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензойной кислоты (40 мг; 0,125 ммоль) в DMF (1 мл) добавляли HATU (52 мг; 0,14 ммоль; CAS: 148893-10-1), DIPEA (49 мг; 0,38 ммоль; CAS: 7087-68-5) и 4-хлоранилин (16 мг; 0,125 ммоль; CAS: 106-47-8). Раствор перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь разбавляли CH2Cl2 (10 мл). Раствор промывали рассолом (20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток растворяли в смеси CH2Cl2 (1 мл) и трифторуксусной кислоты (1 мл). Раствор перемешивали при комнатной температуре в течение 2 часов. Летучие вещества удаляли при пониженном давлении. Остаток очищали посредством препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,1% TFA, колонка С-18), получая N-(4-хлорфенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид (25 мг; выход 61%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, метанол-d4): δ 7.98 (d, 2Н), 7.71 (d, 2Н), 7.52 (d, 2Н), 7.36 (d, 2Н), 5.22 (s, 1Н), 4.98 (s, 1Н), 4.47 (s, 1Н), 3.48 (d, 1Н), 3.36 (d, 1Н), 2.09 (d, 1Н), 1.84 (d, 1Н).
MS (ESI): 330,9 ([{37Cl}M+H]+); 329,0 ([{35Cl}M+H]+).
Пример 56
N-(4-Бромфенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
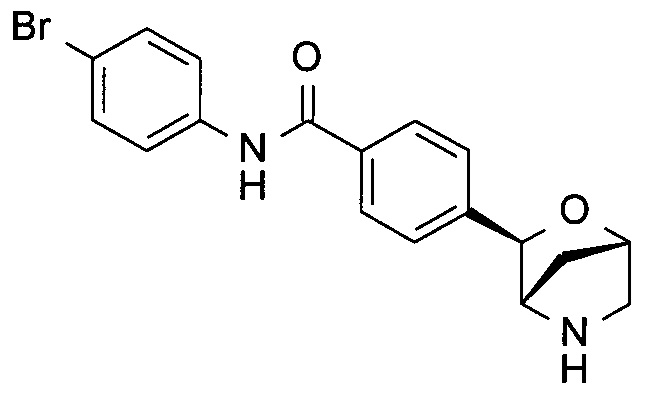
Указанное в заголовке соединение получали по аналогии с соединением примера 55, используя 4-броманилин (CAS: 106-40-1) вместо 4-хлоранилина на стадии (b). Белое твердое вещество.
MS (ESI): 374,9 ([{81Br}M+H]+); 372,9 ([{79Br}M+H]+).
Пример 57
N-(4-Фторфенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
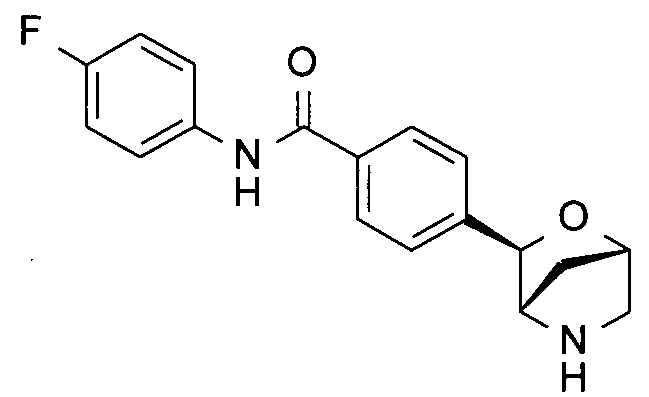
Указанное в заголовке соединение получали по аналогии с соединением примера 55, используя 4-фторанилин (CAS: 371-40-4) вместо 4-хлоранилина на стадии (b). Белое твердое вещество. MS (ESI): 313,0 ([М+Н]+).
Пример 58
N-(4-Этоксифенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид

Указанное в заголовке соединение получали по аналогии с соединением примера 55, используя 4-этоксианилин (CAS: 156-43-4) вместо 4-хлоранилина на стадии (b). Белое твердое вещество. MS (ESI): 339,0 ([М+Н]+).
Пример 59
4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[4-(трифторметил)фенил]бензамид
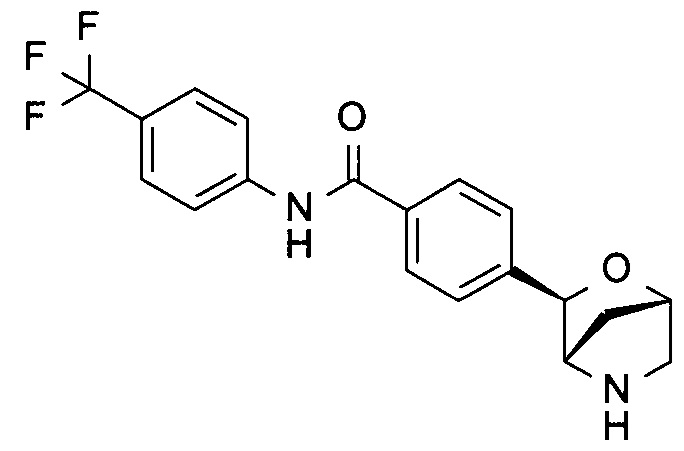
Указанное в заголовке соединение получали по аналогии с соединением примера 55, используя 4-(трифторметил)анилин (CAS: 455-14-1) вместо 4-хлоранилина на стадии (b). Белое твердое вещество. MS (ESI): 363,0 ([М+Н]+).
Пример 60
N-(6-Хлор-3-пиридил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
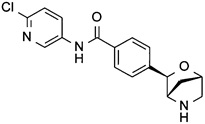
Указанное в заголовке соединение получали по аналогии с соединением примера 55, используя 3-амино-6-хлорпиридин (CAS: 5350-93-6) вместо 4-хлоранилина на стадии (b). Белое твердое вещество.
MS (ESI): 332,1 ([{37Cl}M+H]+); 330,1 ([{35Cl}M+H]+).
Пример 61
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(трифторметил)пиридин-3-амин
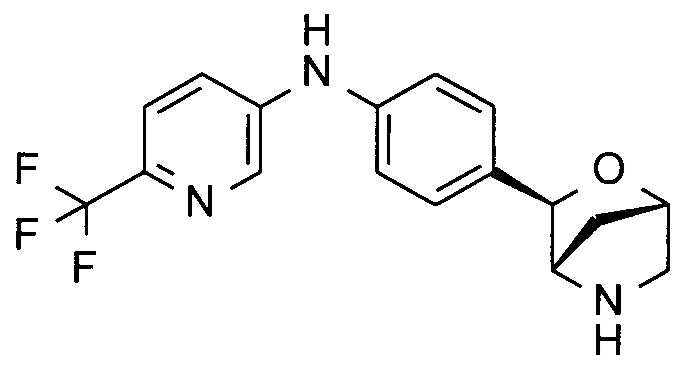
Указанное в заголовке соединение получали по аналогии с соединением примера 35, используя 5-амино-2-трифторметилпиридин (CAS: 106877-33-2) вместо 4-амино-2-(трифторметил)пиридина на стадии (f). Белое твердое вещество. MS (ESI): 336,2 ([М+Н]+).
Пример 62
N-(6-Этокси-3-пиридил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
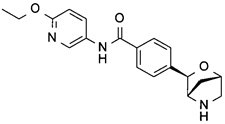
Указанное в заголовке соединение получали по аналогии с соединением примера 55, используя 6-этокси-3-пиридинамин (CAS: 52025-34-0) вместо 4-хлоранилина на стадии (b). Белое твердое вещество. MS (ESI): 340,2 ([М+Н]+).
Пример 63
4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметил)фенил]метил]бензамид

Указанное в заголовке соединение получали по аналогии с соединением примера 55, используя 4-(трифторметил)бензиламин (CAS: 3300-51-4) вместо 4-хлоранилина на стадии (b). Белое твердое вещество. MS (ESI): 377,1 ([М+Н]+).
Пример 64
N-[(4-Хлорфенил)метил]-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
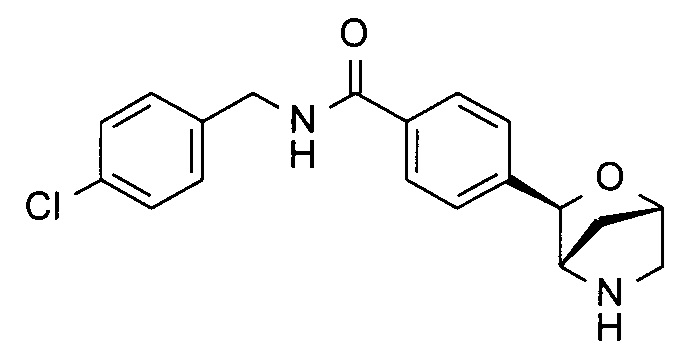
Указанное в заголовке соединение получали по аналогии с соединением примера 55, используя 4-хлорбензиламин (CAS: 104-86-9) вместо 4-хлоранилина на стадии (b). Белое твердое вещество.
MS(ESI): 345,1 ([{37Cl}M+H]+); 343,1 ([{35Cl}M+H]+).
Пример 65
N-(3-Метоксипропил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин
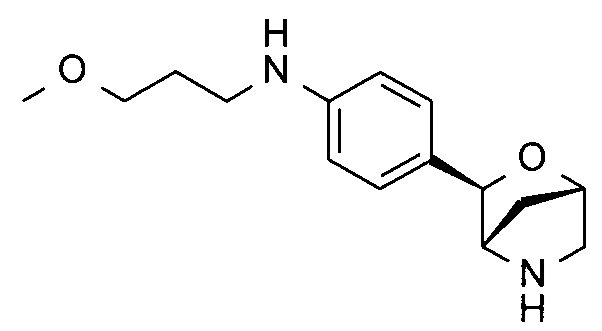
Указанное в заголовке соединение получали по аналогии с соединением примера 35, используя 3-метоксипропиламин (CAS: 5332-73-0) вместо 4-амино-2-(трифторметил)пиридина на стадии (f). Белое твердое вещество. MS (ESI): 263,1 ([М+Н]+).
Пример 66
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)ацетамид
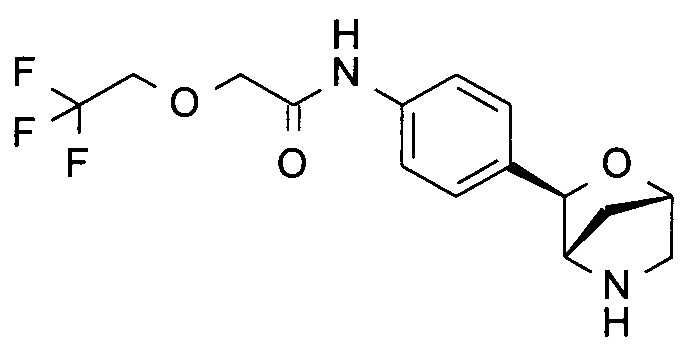
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 2-(2,2,2-трифторэтокси)уксусную кислоту (CAS: 675-67-2) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 331,1 ([М+Н]+).
Пример 67
3-Этил-4-метил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
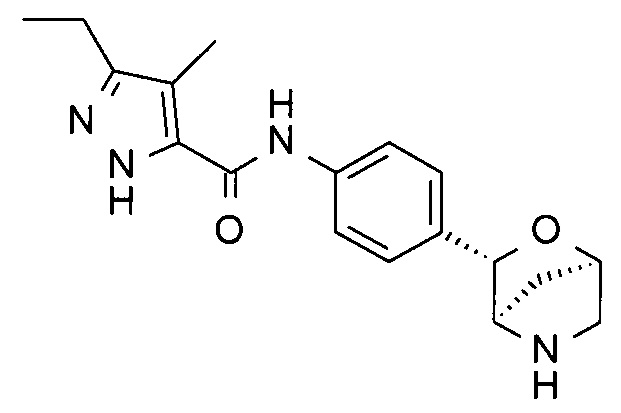
а) трет-Бутил-(1R,3S,4R)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К раствору (1R,3S,4R)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептана (6,8 г; 27 ммоль) в ТГФ (150 мл) добавляли K2CO3 (11 г; 81 ммоль) и Boc2O (7 г; 32 ммоль; CAS: 24424-99-5). Раствор перемешивали при комнатной температуре в течение ночи. Смесь фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир : этилацетат составляет 20:1 по объему), получая трет-бутил-(1R,3S,4R)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (4,0 г; выход: 45%) в виде белого твердого вещества.
1Н-ЯМР (CDCl3, 400 МГц): δ 7.49 (m, 2Н), 7.23 (d, 1Н), 7.16 (d, 1Н), 4.98 (d, 1Н), 4.77 (s, 1Н), 4.36 (d, 1Н), 3.50 (dd, 1Н), 3.36 (t, 1Н), 1.78 (d, 1Н), 1.65-1.51 (m, 10Н).
b) трет-Бутил-(1R,3S,4R)-3-[4-(бензгидрилиденамино)фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К раствору трет-бутил-(1R,3S,4R)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (1,0 г; 3 ммоль), 2,2'-бис(дифенилфосфино)-1,1'-бинафталина (BINAP; 373 мг; 0,6 ммоль; CAS: 98327-87-8), трис(дибензилиденацетон)дипалладия(0) (275 мг; 0,3 ммоль; CAS: 51364-51-3) и трет-бутилата натрия (873 мг; 9 ммоль; CAS: 865-48-5) в безводном толуоле (10 мл) добавляли бензофенонимин (652 мг; 3,6 ммоль; CAS: 1013-88-3). Реакционную смесь перемешивали в атмосфере N2 в течение 30 минут при 90°С в микроволновом реакторе. LCMS показывала завершение реакции. Смесь охлаждали до комнатной температуры, разбавляли водой (10 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические слои промывали рассолом (3×20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; петролейный эфир : этил ацетат составляет 10:1 по объему), получая трет-бутил-(1R,3S,4R)-3-[4-(бензгидрилиденамино)фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (1,1 г; выход 81%) в виде желтого твердого вещества.
MS (ESI): 455,2 ([М+Н]+).
c) трет-Бутил-(1R,3S,4R)-3-(4-аминофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат
К раствору трет-бутил-(1R,3S,4R)-3-[4-(бензгидрилиденамино)фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (454 мг; 1 ммоль) в безводном МеОН (5 мл) добавляли ацетат натрия (328 мг; 4 ммоль; CAS: 127-09-3) и гидроксиламина гидрохлорид (138 мг; 2,0 ммоль; CAS: 5470-11-1) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 минут до тех пор, пока TLC-анализ не показывал завершения реакции. Смесь разбавляли CH2Cl2 (100 мл) и промывали водным раствором NaOH (50 мл). Органический слой сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали хроматографией (силикагель; петролейный эфир : этилацетат от 1:0 до 1:1; с 0,1% триэтиламина), получая трет-бутил-(1R,3S,4R)-3-(4-аминофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (200 мг; выход 72%) в виде желтого твердого вещества.
1Н-ЯМР (CDCl3, 400 МГц): δ 7.13 (d, 1Н), 7.06 (d, 1Н), 6.69 (2Н), 4.96 (d, 1Н), 4.73 (s, 1Н), 4.33 (1Н), 3.48 (dd, 1Н), 3.34 (t, 1Н), 1.87 (d, 1Н), 1.65-1.51 (m, 10Н).
d) 3-Этил-4-метил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
К раствору 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты (20 мг; 0,129 ммоль; CAS: 957129-38-3) в безводном DMF (1 мл) добавляли HATU (52 мг; 0,129 ммоль; CAS: 148893-10-1) и DIPEA (26 мг; 0,258 ммоль; CAS: 7087-68-5). Раствор перемешивали при комнатной температуре в течение 30 минут. Добавляли трет-бутил-(1R,3S,4R)-3-(4-аминофенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат (25 мг; 0,086 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. TLC-анализ показывал завершение реакции. Летучие вещества удаляли при пониженном давлении. Остаток очищали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,3% NH3⋅H2O; колонка С-18), получая трет-бутил-(1R,3S,4R)-3-[4-[(3-этил-4-метил-1Н-пиразол-5-карбонил)амино]фенил]-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат в виде белого твердого вещества, которое затем растворяли в смеси безводного CH2Cl2 (2 мл) и TFA (1 мл; CAS: 76-05-1). Смесь перемешивали при комнатной температуре в течение 2 часов. Летучие вещества удаляли при пониженном давлении. Остаток очищали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,5% NH3⋅H2O; колонка С-18), получая 3-этил-4-метил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид (6 мг; 21% за 2 стадии) в виде белого твердого вещества. MS (ESI): 327,2 ([М+Н]+).
1Н-ЯМР (метанол-d4, 400 МГц): δ 7.70 (d, 2Н), 7.31 (d, 2Н), 5.04 (s, 1Н), 4.84 (s, 1Н), 4.05 (s, 1Н), 3.27 (d, 1Н), 3.17 (d, 1Н), 2.68 (m, 2Н), 2.25 (s, 3Н), 2.01 (d, 1Н), 1.69 (d, 1Н), 1.24 (t, 3Н).
Пример 68
4-Хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-пропил-1Н-пиразол-5-карбоксамид
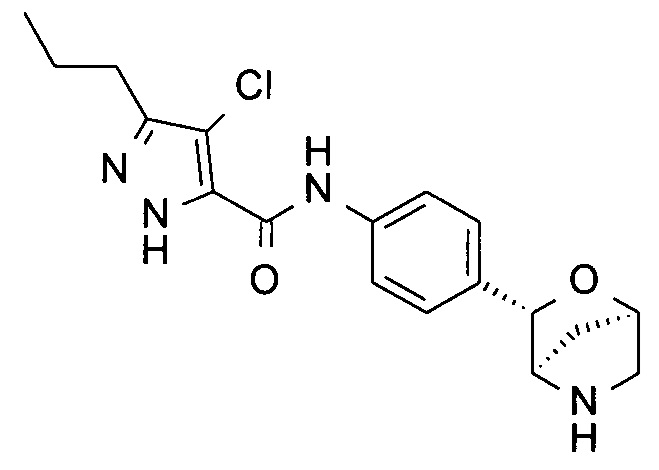
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 4-хлор-3-пропил-1Н-пиразол-5-карбоновую кислоту (CAS: 1340578-20-2) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 363,0 ([{37Cl}M+H]+); 361,0 ([{35Cl}M+H]+).
Пример 69
3-Циклопропил-4-метил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
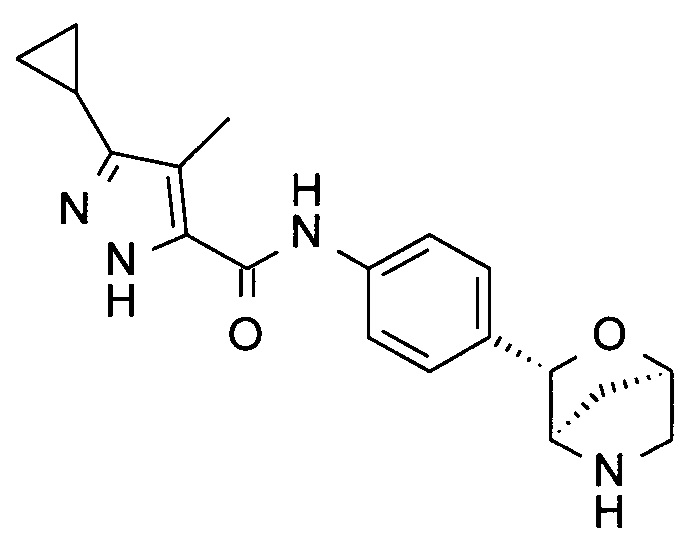
а) Этил-3-циклопропил-4-метил-1Н-пиразол-5-карбоксилат
К раствору этилата натрия (7,6 г; 0,112 моль; CAS: 141-52-6) в безводном этаноле (150 мл) порциями добавляли диэтилоксалат (16,4 г; 0,112 моль; CAS: 95-92-1) при комнатной температуре в атмосфере N2. После этого добавляли этилциклопропилкетон. Смесь перемешивали при 50°С в течение 20 часов. Раствор охлаждали до комнатной температуры. Добавляли уксусную кислоту (9 г; 0,15 ммоль) и гидразина моногидрат (98%-ная чистота; 8,1 г; 0,15 моль; CAS: 7803-57-8). Реакционную смесь перемешивали при 50°С в течение 12 часов до тех пор, пока LCMS-анализ не показывал завершения реакции. Реакционный раствор охлаждали до комнатной температуры. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в дихлорметане (400 мл) и промывали рассолом (40 мл). Раствор сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. После очистки флэш-хроматографией (силикагель; петролейный эфир : этилацетат составляет 20:1 по объему) получали продукт, этил-3-циклопропил-4-метил-1Н-пиразол-5-карбоксилат, (1 г; выход 4,6%) в виде белого твердого вещества. MS (ESI): 195,1 ([М+Н]+).
b) 3-Циклопропил-4-метил-1Н-пиразол-5-карбоновая кислота
К раствору этил-3-циклопропил-4-метил-1Н-пиразол-5-карбоксилата (1 г; 5 ммоль) в смеси этанол/H2O (об./об. составляет 5:1, 12 мл) добавляли NaOH (0,6 г; 15 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Смесь концентрировали при пониженном давлении и подкисляли до значения pH, приблизительно равного 2, используя 2 н. раствор HCl (20 мл). Смесь экстрагировали CH2Cl2 (2×50 мл). Объединенные органические экстракты промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. После дальнейшей сушки под высоким вакуумом получали 3-циклопропил-4-метил-1Н-пиразол-5-карбоновую кислоту (250 мг; выход 30%) в виде белого твердого вещества.
MS(ESI): 167,1 ([М+Н]+).
1Н-ЯМР (метанол-d4, 400 МГц) δ 2.28 (s, 3Н), 1.82 (m, 1Н), 0.93 (m, 2Н), 0.76 (m, 2Н).
c) 3-Циклопропил-4-метил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 3-циклопропил-4-метил-1Н-пиразол-5-карбоновую кислоту вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 339,0 ([М+Н]+).
Пример 70
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(3,3,3-трифторпропокси)ацетамид
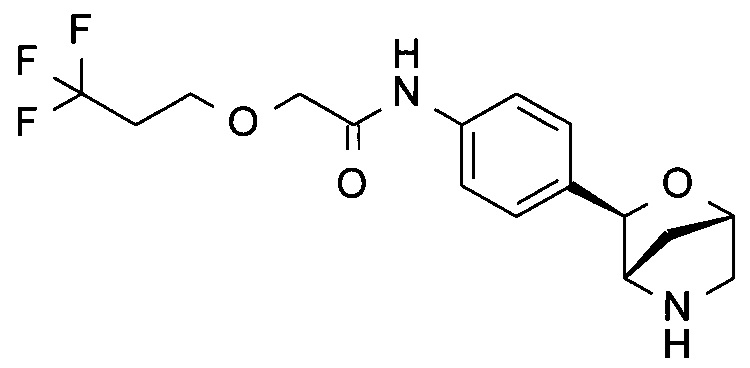
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 2-(3,3,3-трифторпропокси)уксусную кислоту (CAS: 840489-14-7) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 345,2 ([М+Н]+).
Пример 71
N-[4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-карбоксамид
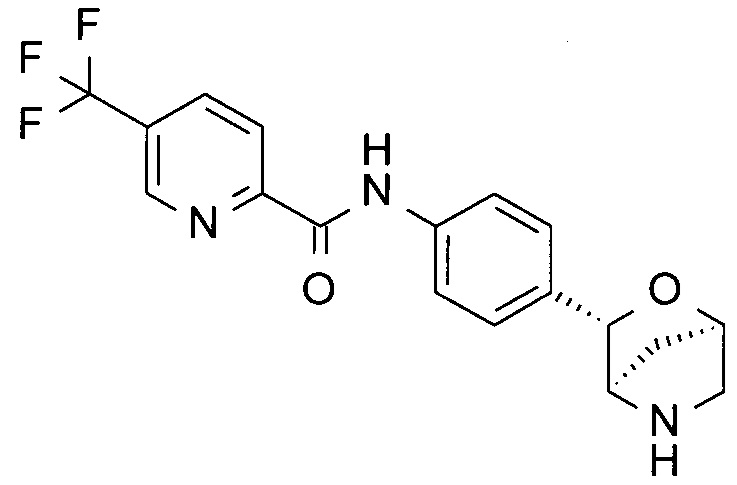
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 5-(трифторметил)пиридин-2-карбоновую кислоту (CAS: 80194-69-0) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 364,1 ([М+Н]+).
Пример 72
N-[4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(трифторметил)пиридин-3-карбоксамид
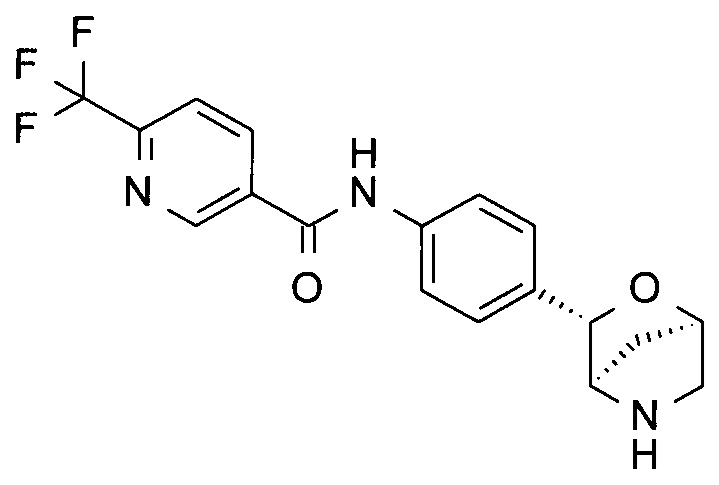
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 6-(трифторметил)пиридин-3-карбоновую кислоту (CAS: 231291-22-8) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 364,1 ([М+Н]+).
Пример 73
N-[4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)ацетамид
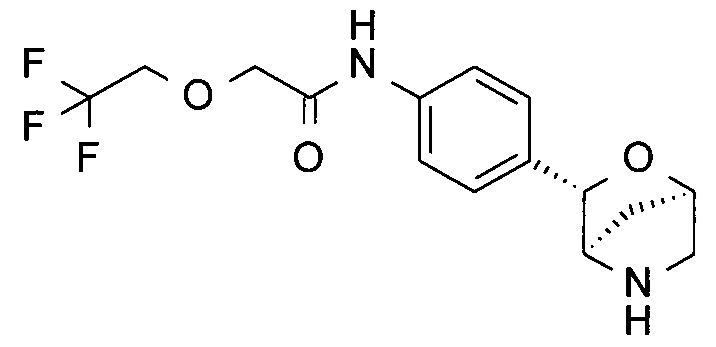
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 2-(2,2,2-трифторэтокси)уксусную кислоту (CAS: 675-67-2) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 331,1 ([М+Н]+).
Пример 74
2-Этил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид
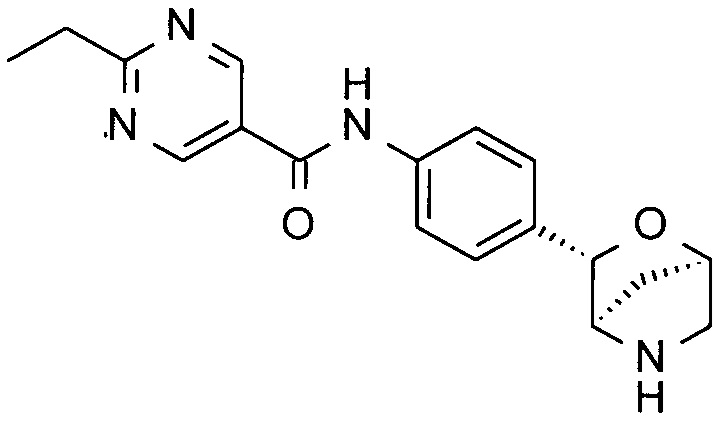
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 2-этилпиримидин-5-карбоновую кислоту (CAS: 72790-16-0) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 325,1 ([М+Н]+).
Пример 75
3-Изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
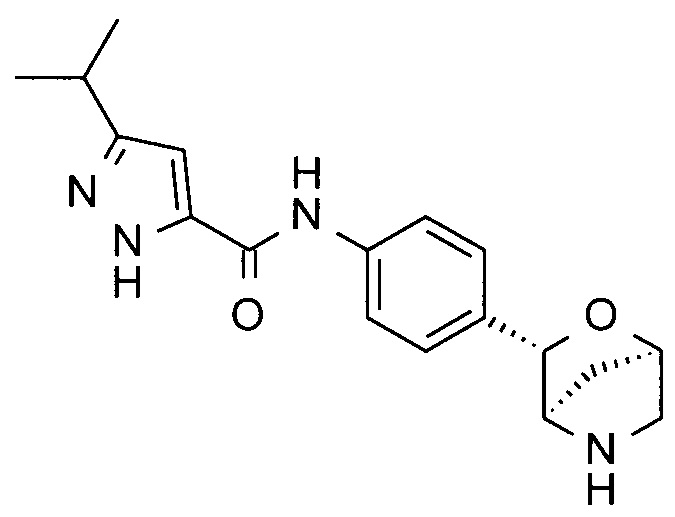
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 3-изопропилпиразол-5-карбоновую кислоту (CAS: 92933-47-6) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 327,2 ([М+Н]+).
Пример 76
4-Хлор-3-этил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
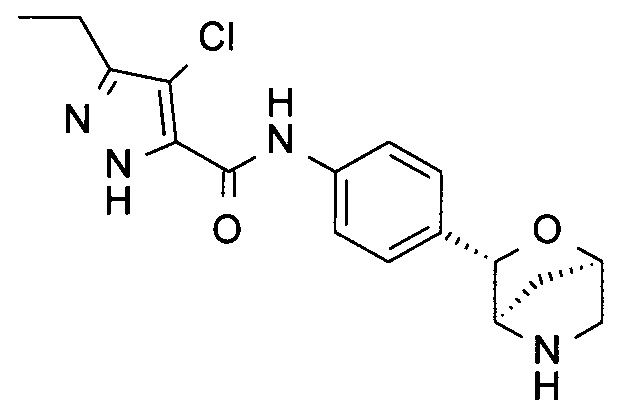
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 4-хлор-3-этил-1Н-пиразол-5-карбоновую кислоту (CAS: 158668-22-5) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 349,1 ([{37Cl}M+H]+); 347,1 ([{35Cl}M+H]+).
Пример 77
3-Циклопропил-4-фтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
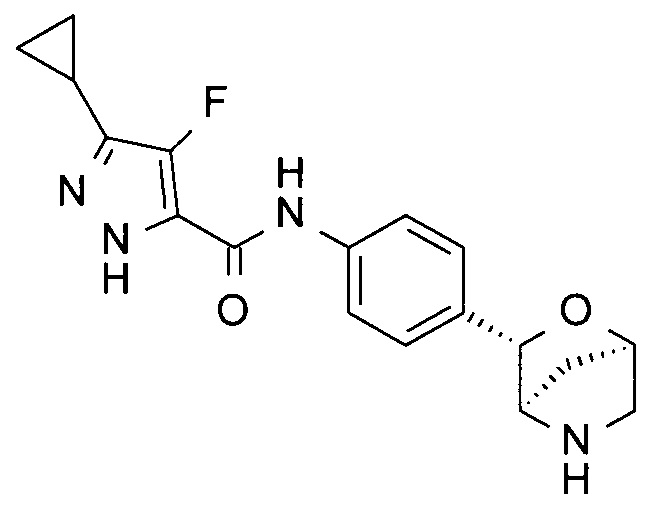
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 5-циклопропил-4-фтор-1Н-пиразол-3-карбоновую кислоту (CAS: 681034-74-2) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 343,2 ([М+Н]+).
Пример 78
4-Фтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-пропил-1Н-пиразол-5-карбоксамид
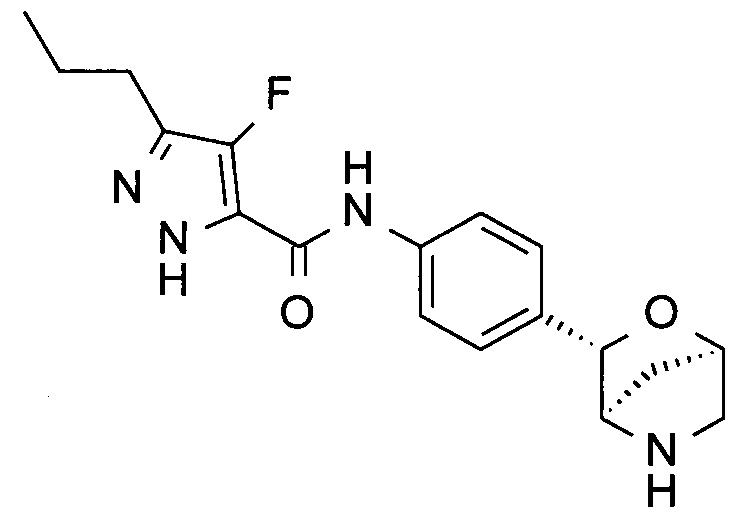
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 4-фтор-5-пропил-1Н-пиразол-3-карбоновую кислоту (CAS: 681034-64-0) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 345,2 ([М+Н]+).
Пример 79
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-4-(2,2,2-трифторэтокси)пиримидин-2-амин
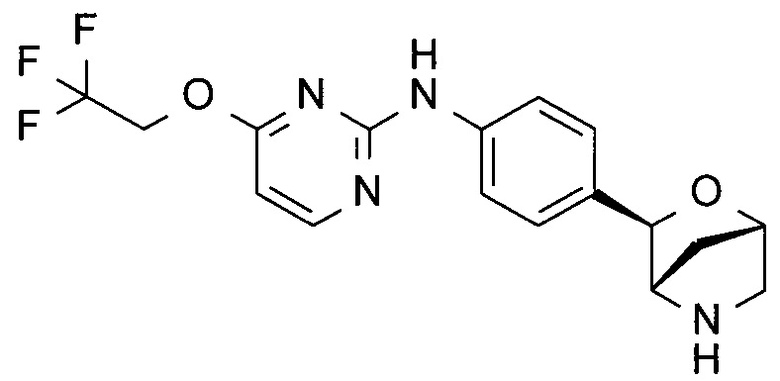
Указанное в заголовке соединение получали по аналогии с соединением примера 35, используя 2-амино-4-(2,2,2-трифторэтокси)пиримидин (CAS: 852921-89-2) вместо 4-амино-2-(трифторметил)пиридина на стадии (f). Белое твердое вещество. MS (ESI): 367,1 ([М+Н]+).
Пример 80
N-[4-[(1S,3R,4S)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)пиримидин-4-амин
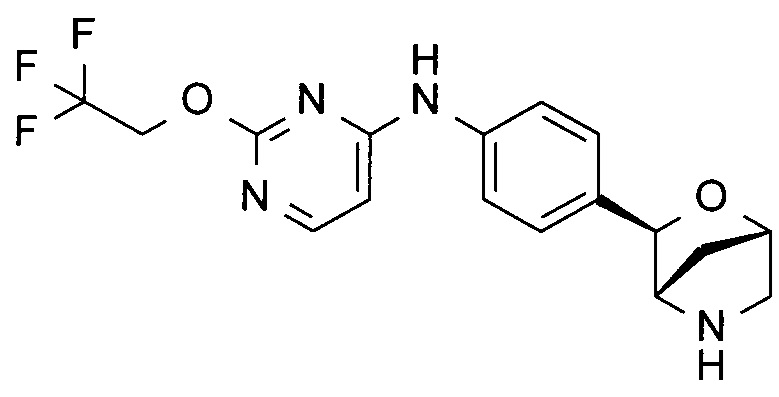
Указанное в заголовке соединение получали по аналогии с соединением примера 35, используя 2-(2,2,2-трифторэтокси)пиримидин-4-амин (CAS: 1431654-73-7) вместо 4-амино-2-(трифторметил)пиридина на стадии (f). Белое твердое вещество. MS (ESI): 367,1 ([М+Н]+).
Пример 81
N-[4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиримидин-4-карбоксамид
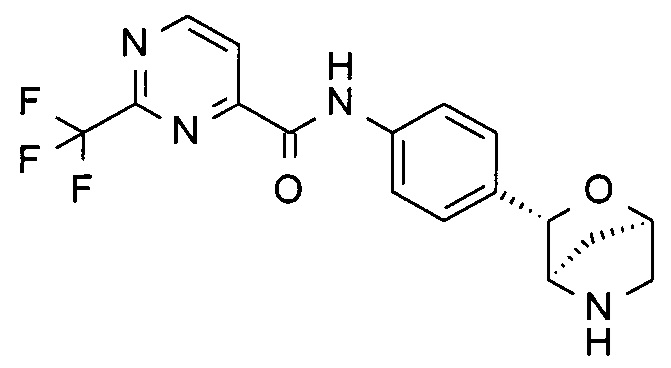
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 2-(трифторметил)пиримидин-4-карбоновую кислоту (CAS: 878742-59-7) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 365,1 ([М+Н]+).
Пример 82
4-Хлор-3-циклопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
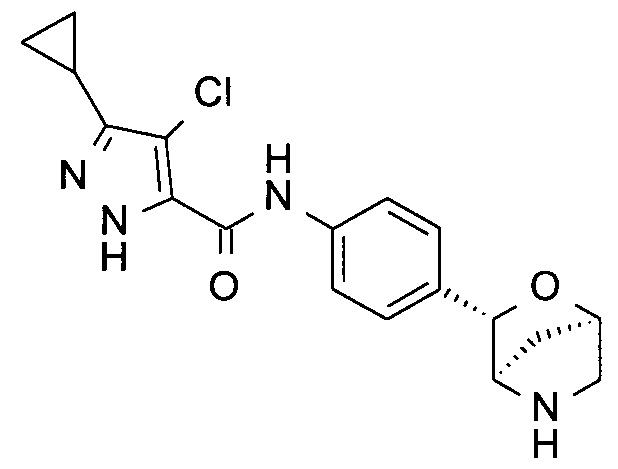
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 4-хлор-3-циклопропил-1Н-пиразол-5-карбоновую кислоту (CAS: 1291275-83-6) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 361,1 ([{37Cl}M+H]+); 359,1 ([{35Cl}M+H]+).
Пример 83
N-[4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)пиримидин-4-амин
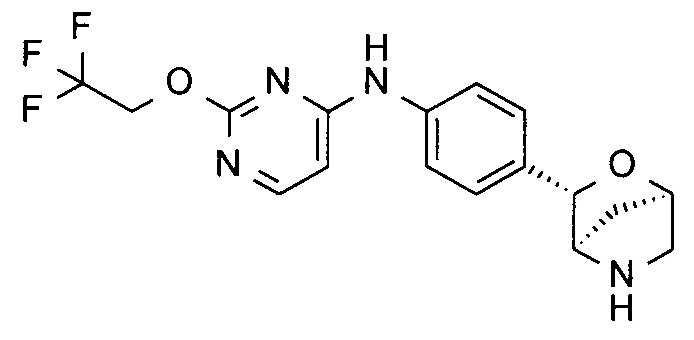
Указанное в заголовке соединение получали по аналогии с соединением примера 80, используя трет-бутил-(1R,3S,4R)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилат вместо трет-бутил-(1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата на катализируемой палладием стадии сочетания. Белое твердое вещество. MS (ESI): 367,2 ([М+Н]+).
Пример 84
4,4,4-Трифтор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бутанамид
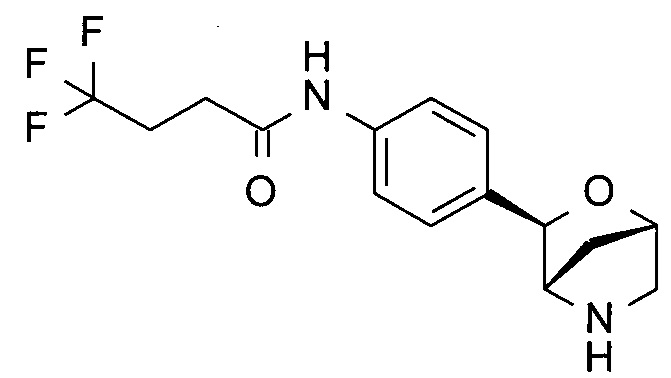
Указанное в заголовке соединение получали по аналогии с соединением примера 36, используя 4,4,4-трифтормасляную кислоту (CAS: 406-93-9) вместо 5-(трифторметил)пиридин-2-карбоновой кислоты на стадии (с). Белое твердое вещество. MS (ESI): 315,1 ([М+Н]+).
Пример 85
4,4,4-Трифтор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бутанамид
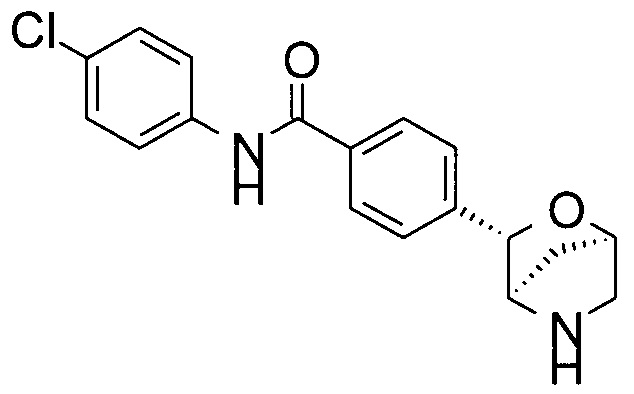
а) 4-[(1R,3S,4R)-5-трет-Бутоксикарбонил-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензойная кислота
К раствору трет-бутил-(1R,3S,4R)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан-5-карбоксилата (800 мг; 2,25 ммоль; полученного на стадии а, пример 67) в ТГФ (20 мл) добавляли раствор н-бутиллития (2,5 М в гексане; 1,17 мл; 2,93 ммоль) при -78°С. Перемешивание продолжали в течение 30 минут. Через раствор в течение 10 минут барботировали обезвоженный CO2. Раствор нагревали до комнатной температуры. TLC-анализ показывал полное расходование исходного вещества. В реакционный раствор добавляли 1 М водный раствор HCl для подведения pH до 4-5. Смесь экстрагировали CH2Cl2. Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток разбавляли петролейным эфиром (100 мл) и перемешивали в течение 30 минут. Осадок собирали фильтрованием и сушили под высоким вакуумом, получая 4-[(1R,3S,4R)-5-трет-бутоксикарбонил-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензойную кислоту (300 мг; выход 42%) в виде желтого твердого вещества.
1Н-ЯМР (400 МГц, CDCl3): δ 8.10 (t, 2Н), 7.46 (d, 1Н), 7.39 (d, 1Н), 5.09 (d, 1Н), 4.80 (s, 1Н), 4.43 (d, 1Н), 3.52 (m, 1Н), 3.37 (t, 1Н), 1.78 (d, 1Н), 1.65 (m, 1Н), 1.55 (d, 9Н).
b) 4,4,4-Трифтор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бутанамид
К раствору 4-[(1R,3S,4R)-5-трет-бутоксикарбонил-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензойной кислоты (40 мг; 0,125 ммоль) в безводном DMF (1 мл) добавляли HATU (52,4 мг; 0,138 ммоль; CAS: 148893-10-1), диизопропилэтиламин (48,6 мг; 0,376 ммоль) и 4-хлоранилин (16 мг; 0,125 ммоль; CAS: 106-47-8). Раствор перемешивали при комнатной температуре в течение ночи. Реакционный раствор разбавляли CH2Cl2 (10 мл). Смесь промывали рассолом (20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении.
Остаток растворяли в смеси CH2Cl2 (1 мл) и трифторуксусной кислоты (1 мл). Раствор перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (подвижная фаза А: H2O, В: CH3CN с 0,1% TFA, колонка С-18), получая 4,4,4-трифтор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бутанамид (20 мг; 0,061 ммоль; выход 48%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, метанол-d4): δ 7.97 (d, 2Н), 7.71 (dd, 2Н), 7.52 (d, 2Н), 7.36 (dd, 2Н), 5.22 (s, 1Н), 4.98 (s, 1Н), 4.47 (s, 1Н), 3.4 (d, 1Н), 3.37 (d, 1Н), 2.09 (d, 1Н), 1.84 (dd, 1Н).
MS (ESI): 331,0 ([{37Cl}M+Н]+); 329,0 ([{35Cl}M+H]+).
Пример 86
N-(4-Бромфенил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
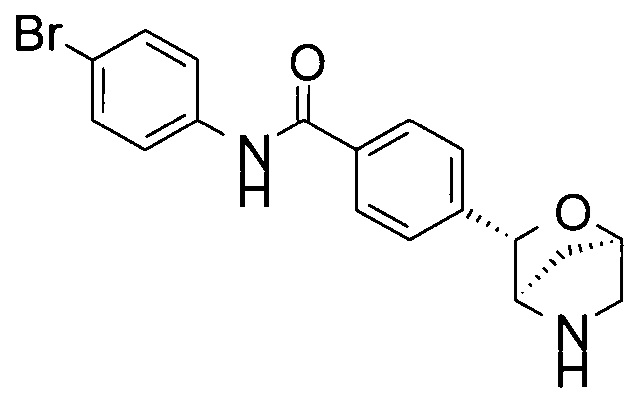
Указанное в заголовке соединение получали по аналогии с соединением примера 85, используя 4-броманилин (CAS: 106-40-1) вместо 4-хлоранилина на стадии (b). Белое твердое вещество.
MS (ESI): 374,9 ([{81Br}M+H]+); 372,9 ([{79Br}M+H]+).
Пример 87
N-(4-Фторфенил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
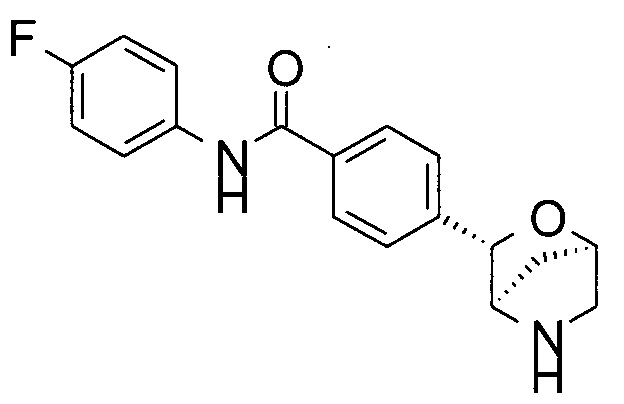
Указанное в заголовке соединение получали по аналогии с соединением примера 85, используя 4-фторанилин (CAS: 371-40-4) вместо 4-хлоранилина на стадии (b). Белое твердое вещество.
MS(ESI): 313,0 ([М+Н]+).
Пример 88
N-(4-Этоксифенил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
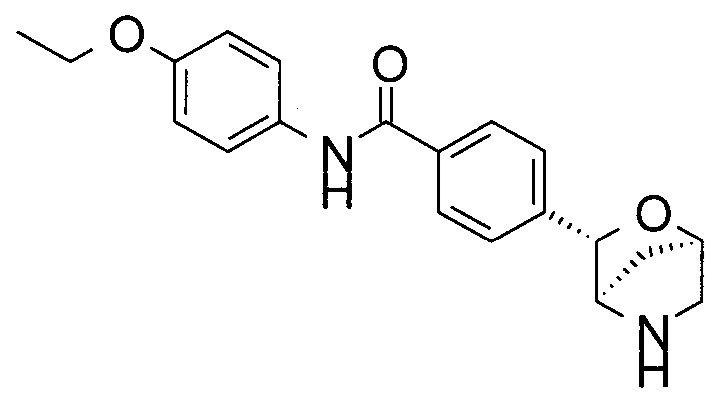
Указанное в заголовке соединение получали по аналогии с соединением примера 85, используя 4-этоксианилин (CAS: 156-43-4) вместо 4-хлоранилина на стадии (b). Белое твердое вещество.
MS (ESI): 339,0 ([М+Н]+).
Пример 89
2-Изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(2,2,2-трифторэтокси)пиразол-3-карбоксамид

а) Метил-5-гидрокси-1Н-пиразол-3-карбоксилат
К раствору гидразина моногидрата (44,8 г; 0,894 моль; CAS: 7803-57-8) в смеси толуола (300 мл) и уксусной кислоты (180 мл) добавляли диметилацетилендикарбоксилат (100 мл; 0,813 моль; CAS: 762-42-5). Раствор перемешивали при комнатной температуре в течение 3 часов. Смесь выливали в ледяную воду. Осадок собирали фильтрованием, промывали водой, сушили под высоким вакуумом, получая метил-5-гидрокси-1Н-пиразол-3-карбоксилат (67,5 г; выход 59%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, DMSO-d6): δ 12.81 (s, 1Н), 10.03 (br, 1Н), 5.91 (br, 1Н), 3.78 (s, 3Н).
b) Метил-5-(2,2,2-трифторэтокси)-1Н-пиразол-3-карбоксилат
К раствору метил-5-гидрокси-1Н-пиразол-3-карбоксилата (10 г; 70,4 ммоль) в DMF (100 мл) добавляли Cs2CO3 (25 г; 77.5 ммоль) и 2,2,2-трифторэтил-трифторметансульфонат (16,3 г; 70,4 ммоль; CAS: 6226-25-1). Раствор перемешивали при комнатной температуре в течение ночи. Затем смесь осторожно выливали в 500 мл ледяной воды. Осадок собирали посредством фильтрования, промывали холодной водой и сушили под высоким вакуумом, получая метил-5-(2,2,2-трифторэтокси)-1Н-пиразол-3-карбоксилат (12 г; выход 76%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, DMSO-d6): δ 13.41 (s, 1Н), 6.73 (s, 1Н), 4.86 (m, 2Н), 3.84 (s, 3Н).
c) Метил-2-изопропил-5-(2,2,2-триФторэтокси)пиразол-3-карбоксилат
К раствору метил-5-(2,2,2-трифторэтокси)-1Н-пиразол-3-карбоксилата (12,0 г; 53,4 ммоль), Cs2CO3 (52,0 г; 161 ммоль) в DMF (100,0 мл) порциями добавляли 2-бромпропан (7,2 г; 56,0 ммоль, CAS: 75-26-3). Раствор перемешивали при комнатной температуре в течение ночи. Затем раствор концентрировали при пониженном давлении и выливали в воду. Осадок собирали посредством фильтрования, промывали холодной водой и сушили под высоким вакуумом, получая метил-2-изопропил-5-(2,2,2-трифторэтокси)пиразол-3-карбоксилат (10,3 г; выход 74%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, DMSO-d6): δ 6.45 (s, 1Н), 5.35 (m, 1Н), 4.82 (m, 2Н), 3.83 (s, 3Н), 1.36 (d, 6Н).
d) 2-Изопропил-5-(2,2,2-трифторэтокси)пиразол-3-карбоновая кислота
Раствор метил-2-изопропил-5-(2,2,2-трифторэтокси)пиразол-3-карбоксилата (4,3 г; 16,2 ммоль), NaOH (1,9 г; 48,5 ммоль) в смеси МеОН/вода (об./об. составляет 3:1; 50,0 мл) перемешивали при комнатной температуре в течение ночи. Затем pH раствора подводили до 4-5 с использованием концентрированной HCl (5 мл) при 0°С. Раствор осторожно выливали в 500 мл ледяной воды. Осадок собирали фильтрованием, промывали охлажденной водой и сушили под высоким вакуумом, получая 2-изопропил-5-(2,2,2-трифторэтокси)пиразол-3-карбоновую кислоту (3,86 г; выход 95%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, DMSO-d6): δ 6.37 (s, 1Н), 5.38 (m, 1Н), 4.79 (m, 2Н), 1.34 (d, 6Н).
e) 2-Изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(2,2,2-трифторэтокси)пиразол-3-карбоксамид
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 2-изопропил-5-(2,2,2-трифторэтокси)пиразол-3-карбоновую кислоту вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 425,2 ([М+Н]+).
Пример 90
N-[4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(3,3,3-трифторпропокси)ацетамид
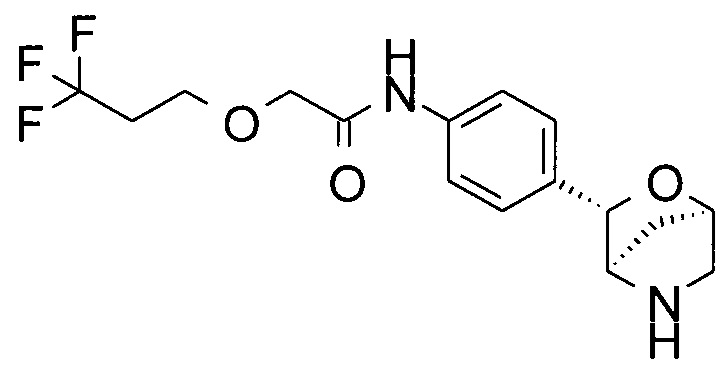
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 2-(3,3,3-трифторпропокси)уксусную кислоту (CAS: 840489-14-7) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 345,1 ([М+Н]+).
Пример 91
4,4,4-Трифтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бутанамид
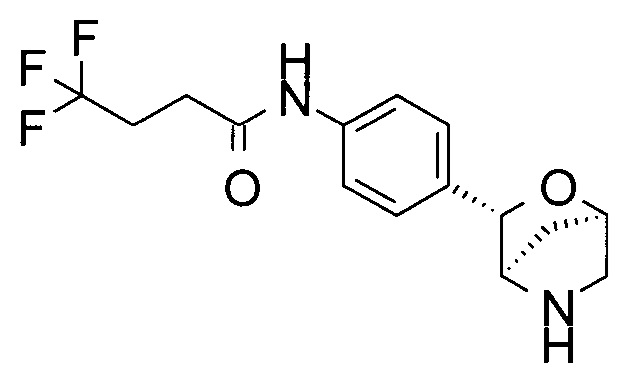
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 4,4,4-трифтормасляную кислоту (CAS: 406-93-9) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 315,1 ([М+Н]+).
Пример 92
3-Бутил-4-фтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
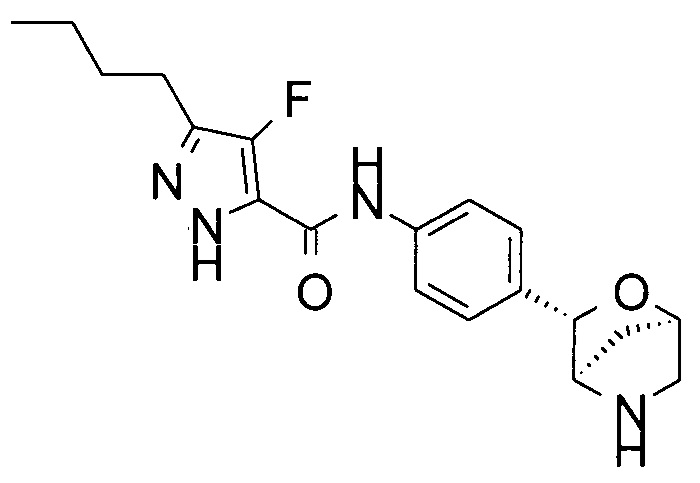
a) Этил-3-бутил-1Н-пиразол-5-карбоксилат
К раствору EtONa (13,2 г; 0,2 моль; CAS: 141-52-6) в безводном этаноле (400 мл) добавляли диэтилоксалат (29,2 г; 0,2 моль; CAS: 95-92-1) и 2-гексанон (20 г; 0,2 моль; CAS: 591-78-6) при 0-5°С. Раствор нагревали до 50°С и перемешивание продолжали в течение ночи. Смесь охлаждали до 0-5°С. Добавляли уксусную кислоту (12 г; 0,2 моль), затем гидразина моногидрат (10 г; 0,2 моль; CAS: 7803-57-8). Смесь перемешивали при 30°С в течение 12 часов. Летучие вещества удаляли при пониженном давлении. Остаток разбавляли насыщенным водным раствором NaHCO3 (500 мл). Смесь экстрагировали этилацетатом (1 л). Органический слой промывали рассолом и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; CH2Cl2/MeOH от 200/1 до 80/1 по объему), получая этил-3-бутил-1Н-пиразол-5-карбоксилат (13 г; выход 33%) в виде желтого твердого вещества.
b) Этил-3-бутил-4-фтор-1Н-пиразол-5-карбоксилат
К раствору этил-3-бутил-1Н-пиразол-5-карбоксилата (8,0 г; 40,8 ммоль) в CH3CN (500 мл) добавляли Selectfluor (17,3 г; 48,9 ммоль; CAS: 140681-55-6) при 0°С. Затем раствор перемешивали при 70°С в течение 15 часов. Летучие вещества удаляли при пониженном давлении. Остаток разбавляли водным раствором HCl (3 н., 200 мл) и экстрагировали CH2Cl2 (500 мл). Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (силикагель; CH2Cl2/MeOH от 200/1 до 100/1 по объему), получая этил-3-бутил-4-фтор-1Н-пиразол-5-карбоксилат (1,4 г; выход 16%) в виде желтого масла.
c) 3-Бутил-4-фтор-1Н-пиразол-5-карбоновая кислота
К раствору этил-3-бутил-4-фтор-1Н-пиразол-5-карбоксилата (1,4 г; 6,53 ммоль) в ТГФ/МеОН (20/20 мл) добавляли 1 М водный раствор NaOH (13,1 мл; 13,1 ммоль) при 0°С. Затем раствор кипятили с обратным холодильником в течение 3 часов. Реакционную смесь выливали в воду и подкисляли до pH приблизительно 1, используя концентрированную HCl. Смесь экстрагировали этилацетатом (100 мл). Органический слой промывали рассолом и концентрировали при пониженном давлении. Остаток перекристаллизовывали из этилацетата (10 мл), получая 3-бутил-4-фтор-1Н-пиразол-5-карбоновую кислоту (0,4 г; выход 33%) в виде желтого твердого вещества.
1Н-ЯМР(400 МГц, DMSO-d6): δ 13.30 (br, 1Н), 2.55 (t, 2Н), 1.55 (m, 2Н), 1.29 (m, 2Н), 0.88 (t, 3Н).
MS(ESI): 187,0 ([М+Н]+); 209,0 ([M+Na]+).
d) 3-Бутил-4-фтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенилъ-1Н-пиразол-5-карбоксамид
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 3-бутил-4-фтор-1Н-пиразол-5-карбоновую кислоту вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество.
1Н-ЯМР (400 МГц, метанол-d4): δ 7.73 (d, 2Н), 7.34 (d, 2Н), 5.15 (s, 1Н), 4.93 (s, 1Н), 4.37 (s, 1Н), 3.46 (m, 1Н), 3.35 (m, 1Н), 2.70 (t, 2Н), 2.12 (d, 1Н), 1.82 (d, 1Н), 1.67 (m, 2Н), 1.39 (m, 2Н), 0.96 (t, 3Н).
MS (ESI): 359,2 ([М+Н]+).
Пример 93
3-Бутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
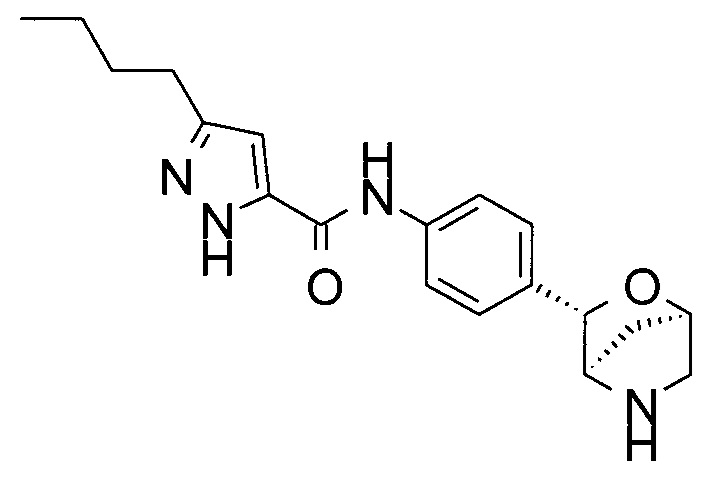
а) 3-Бутил-1Н-пиразол-5-карбоновая кислота
К раствору этил-3-бутил-1Н-пиразол-5-карбоксилата (5,0 г; 25,5 ммоль) в ТГФ/МеОН (30/30 мл) добавляли 1 М водный раствор NaOH (51 мл; 51 ммоль) при 0°С. Затем раствор кипятили с обратным холодильником в течение 3 часов. Реакционную смесь выливали в воду и подкисляли до pH приблизительно 1, используя концентрированную HCl. Смесь экстрагировали этилацетатом (100 мл). Органический слой промывали рассолом и концентрировали при пониженном давлении. Остаток перекристаллизовывали из этилацетата (50 мл), получая 3-бутил-1Н-пиразол-5-карбоновую кислоту (2,0 г; выход 47%) в виде желтого твердого вещества.
1Н-ЯМР (400 МГц, DMSO-d6): δ 12.9 (br, 2Н), 6.47 (s, 1Н), 2.59 (t, 2Н), 1.55 (m, 2Н), 1.29 (m, 2Н), 0.89 (t, 3Н).
b) 3-Бутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 3-бутил-1Н-пиразол-5-карбоновую кислоту вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 341,2 ([М+Н]+).
Пример 94
N-(6-Хлор-3-пиридил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
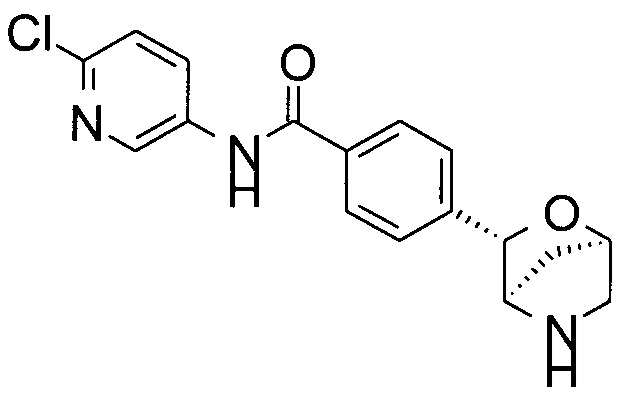
Указанное в заголовке соединение получали по аналогии с соединением примера 85, используя 3-амино-6-хлорпиридин (CAS: 5350-93-6) вместо 4-хлоранилина на стадии (b). Белое твердое вещество.
MS (ESI): 332,1 ([{37Cl}M+H]+); 330,1 ([{35Cl}M+H]+).
Пример 95
N-(6-Этокси-3-пиридил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
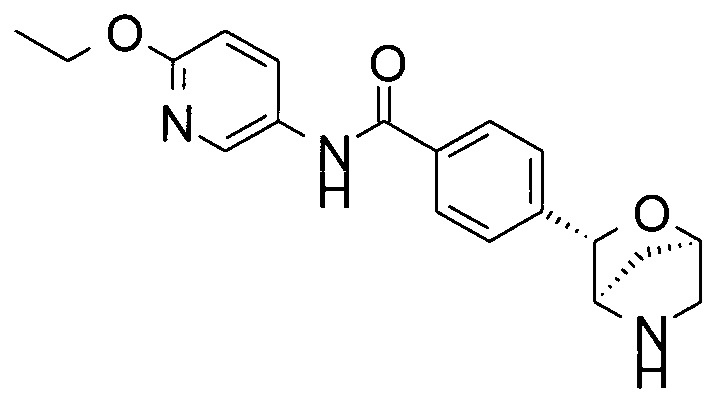
Указанное в заголовке соединение получали по аналогии с соединением примера 85, используя 6-этокси-3-пиридинамин (CAS: 52025-34-0) вместо 4-хлоранилина на стадии (b). Белое твердое вещество.
MS (ESI): 340,2 ([M+H]+).
Пример 96
4-[(1R,3S,4R)-2-Окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметил)фенил]метил]бензамид
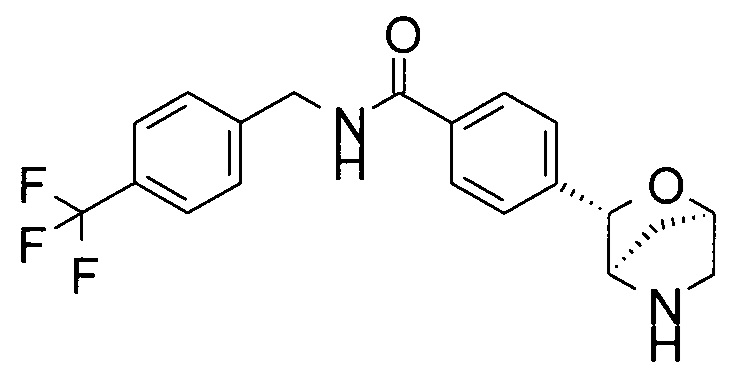
Указанное в заголовке соединение получали по аналогии с соединением примера 85, используя 4-(трифторметил)бензиламин (CAS: 3300-51-4) вместо 4-хлоранилина на стадии (b). Белое твердое вещество. MS (ESI): 377,2 ([М+Н]+).
Пример 97
N-[(4-Хлорфенил)метил]-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид
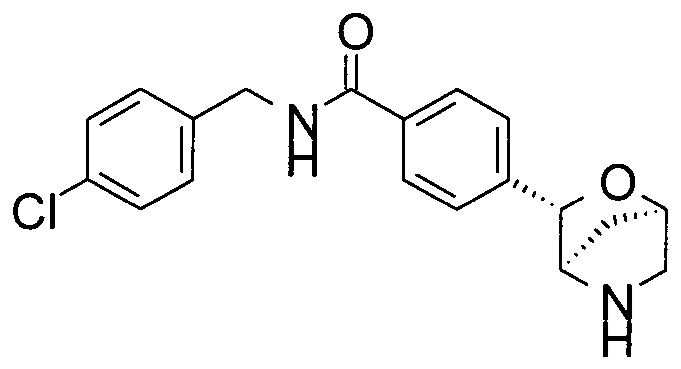
Указанное в заголовке соединение получали по аналогии с соединением примера 85, используя 4-хлорбензиламин (CAS: 104-86-9) вместо 4-хлоранилина на стадии (b).
MS (ESI): 345,1 ([{37Cl}M+H]+); 343,1 ([{35Cl}M+H]+).
Пример 98
4-Хлор-3-этокси-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
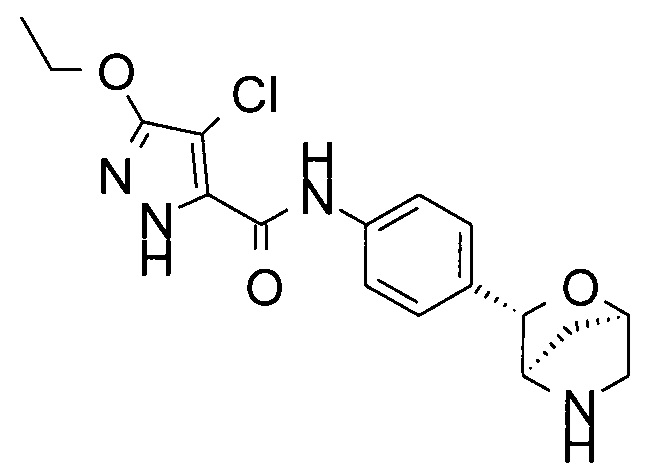
а) Метил-5-гидрокси-1Н-пиразол-3-карбоксилат
К раствору гидразина моногидрата (3,85 г; 0,077 моль; CAS: 7803-57-8) в толуоле (30 мл) добавляли уксусную кислоту (15 мл) и диметил-ацетилендикарбоксилат (10 г; 0,07 моль; CAS: 762-42-5). Раствор перемешивали при комнатной температуре в течение 3 часов. Смесь выливали в ледяную воду. Осадок собирали фильтрованием, промывали водой и сушили под высоким вакуумом, получая метил-5-гидрокси-1Н-пиразол-3-карбоксилат (7,5 г; выход 75%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, DMSO-d6): δ 12.81 (s, 1Н), 10.04 (br, 1Н), 5.96 (br, 1Н), 3.77 (s, 3Н).
b) Метил-5-этокси-1Н-пиразол-3-карбоксилат
К раствору метил-5-гидрокси-1Н-пиразол-3-карбоксилата (4 г; 28,17 ммоль) в DMF (25 мл) добавляли K2CO3 (5,83 г; 42,2 ммоль) и CH3CH2I (4,8 г; 31 ммоль, CAS: 75-03-6). Раствор перемешивали при комнатной температуре в течение 15 часов. Затем смесь выливали в воду и экстрагировали этилацетатом (100 мл). Органический слой промывали рассолом (30 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали перекристаллизацией из CH2Cl2 (10 мл), получая метил-5-этокси-1Н-пиразол-3-карбоксилат (2,2 г; выход 46%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, DMSO-d6): δ 13.13 (s, 1Н), 6.23 (s, 1Н), 4.11 (m, 2Н), 3.81 (s, 3Н), 1.28 (m, 3Н).
с) Метил-4-хлор-5-этокси-1Н-пиразол-3-карбоксилат
К раствору метил-5-этокси-1Н-пиразол-3-карбоксилата (2,2 г; 12,94 ммоль) в DMF (40 мл) добавляли N-хлорсукцинимид (2,06 г; 15,5 ммоль; CAS: 128-09-6) при 0°С. Реакционную смесь нагревали до 50°С и перемешивание продолжали в течение 15 часов. Большую часть летучих веществ удаляли при пониженном давлении. Остаток выливали в воду. Осадок собирали фильтрованием, промывали водой и сушили под высоким вакуумом, получая метил-4-хлор-5-этокси-1Н-пиразол-3-карбоксилат (1,65 г; выход 63%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, DMSO-d6): δ 13.45 (br, 1Н), 4.23 (m, 2Н), 1.32 (t, 3Н).
d) 4-Хлор-5-этокси-1Н-пиразол-3-карбоновая кислота
К раствору метил-4-хлор-5-этокси-1Н-пиразол-3-карбоксилата (1,65 г; 8,06 ммоль) в смеси ТГФ/МеОН (об./об. составляет 1:1; 30 мл) добавляли 1М водный раствор NaOH (16,1 мл; 16,1 ммоль) при 0°С. Затем раствор кипятили с обратным холодильником в течение 3 часов. Реакционную смесь выливали в воду и подкисляли до значения pH, приблизительно равного 1, используя концентрированную HCl. Осадок собирали фильтрованием, промывали водой и сушили под высоким вакуумом, получая 4-хлор-5-этокси-1Н-пиразол-3-карбоновую кислоту (1,4 г; выход 92%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, DMSO-d6): δ 13.25 (s, 1Н), 4.23 (m, 2Н), 1.32 (t, 3Н).
e) 4-Хлор-3-этокси-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 4-хлор-5-этокси-1Н-пиразол-3-карбоновую кислоту вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество.
1Н-ЯМР (400 МГц, метанол-d4): δ 7.65 (d, 2Н), 7.29 (d, 2Н), 4.91 (s, 1Н), 4.69 (s, 1Н), 4.30 (m, 2Н), 3.57 (s, 1Н), 3.00 (m, 2Н), 1.80 (d, 1Н), 1.50 (d, 1Н), 1.40 (t, 3Н).
MS (ESI): 365,1 ([{37Cl}M+H]+); 363,1 ([{35Cl}M+H]+).
Пример 99
4-Бром-3-этил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
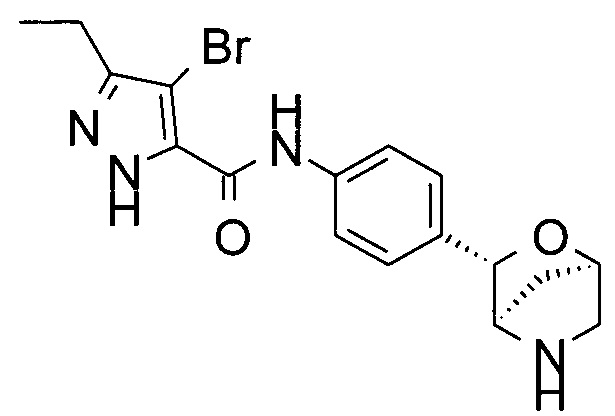
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 4-бром-3-этил-1Н-пиразол-5-карбоновую кислоту (CAS: 1291177-22-4) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество.
MS(ESI): 393,1 ([{81Br}М+Н]+); 391,1 ([{79Br}М+Н]+).
Пример 100
4-Фтор-3-изобутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
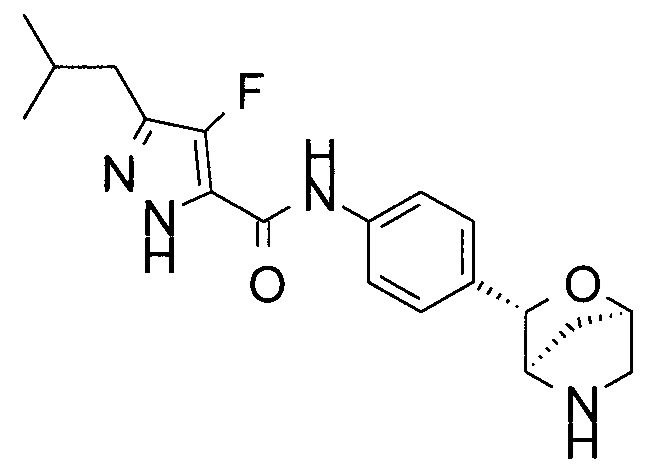
a) Этил-5-изобутил-1Н-пиразол-3-карбоксилат
К раствору CH3CH2ONa (7 г; 0,1 моль) в безводном этаноле (150 мл) добавляли диэтилоксалат (15 г; 0,1 моль; CAS: 95-92-1) и 4-метил-2-пентанон (10 г; 0,1 моль; CAS: 108-10-1). Смесь перемешивали при 50°С в течение 20 часов. Реакционный раствор, содержащий этил-6-метил-2,4-диоксо-гептаноат, использовали на следующей стадии непосредственно.
К полученному выше раствору этил-6-метил-2,4-диоксо-гептаноата добавляли уксусную кислоту (9 г; 0,15 моль) и гидразина моногидрат (8,1 г; 0,15 моль; CAS: 7803-57-8). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Летучие вещества удаляли при пониженном давлении. Остаток растворяли в этилацетате и промывали водой и рассолом. Органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали флэш-хроматографией (силикагель; петролейный эфир : этилацетат от 20:1 до 2:1 по объему), получая этил-5-изобутил-1Н-пиразол-3-карбоксилат в виде белого твердого вещества (13 г; выход 68%).
MS(ESI): 197,2 ([М+Н]+).
b) Этил-4-фтор-5-изобутил-1Н-пиразол-3-карбоксилат
К раствору этил-5-изобутил-1Н-пиразол-3-карбоксилата (5,0 г; 25,5 ммоль) в CH3CN (300 мл) добавляли Selectfluor (18,0 г; 51,0 ммоль, CAS: 140681-55-6) при 0°С. Раствор перемешивали при 70°С в течение 15 часов. Затем реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разбавляли водным раствором HCl (3 н., 200 мл) и экстрагировали CH2Cl2 (100 мл). Органический слой промывали рассолом (20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. После очистки флэш-хроматографией (силикагель; CH2Cl2/MeOH от 200/1 до 100/1 по объему) получали этил-4-фтор-5-изобутил-1Н-пиразол-3-карбоксилат (1,4 г; выход 26%) в виде желтого масла.
1Н-ЯМР (400 МГц, CDCl3): δ 4.42 (m, 2Н), 2.55 (d, 2Н), 2.00 (m, 1Н), 1.40 (t, 3Н), 0.95 (d, 6Н).
c) 4-Фтор-5-изобутил-1Н-пиразол-3-карбоновая кислота
К раствору этил-4-фтор-5-изобутил-1Н-пиразол-3-карбоксилата (1,4 г; 6,54 ммоль) в смеси ТГФ/МеОН (об./об. составляет 1:1; 20 мл) добавляли 1 М водный раствор NaOH (13,1 мл; 13,1 ммоль) при 0°С. Затем раствор кипятили с обратным холодильником в течение 3 часов. Реакционный раствор выливали в воду и подкисляли до pH приблизительно 1, используя концентрированную HCl. Смесь экстрагировали этилацетатом (100 мл). Органический слой промывали рассолом (20 мл) и концентрировали при пониженном давлении. Остаток перекристаллизовывали из этилацетата (30 мл), получая 4-фтор-5-изобутил-1Н-пиразол-3-карбоновую кислоту (1,2 г; выход 99%) в виде желтого твердого вещества.
1Н-ЯМР (400 МГц, DMSO-d6): δ 3.37 (br, 1Н), 2.44 (d, 2Н), 1.89 (m, 1Н), 0.87 (d, 6Н).
MS (ESI): 187,1 ([М+Н]+).
d) 4-Фтор-3-изобутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 4-фтор-5-изобутил-1Н-пиразол-3-карбоновую кислоту вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 359,2 ([М+Н]+).
Пример 101
3-Изобутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
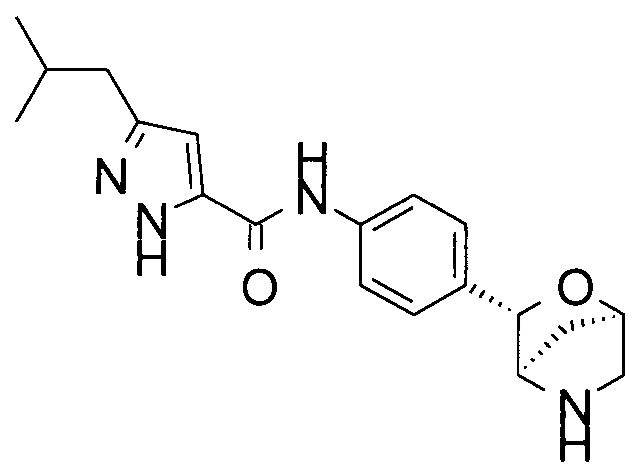
a) 5-Изобутил-1Н-пиразол-3-карбоновая кислота
К раствору этил-5-изобутил-1Н-пиразол-3-карбоксилата (3 г; 15,3 ммоль) в смеси этанол/вода (об./об. составляет 5:1, 60 мл) добавляли NaOH (1,8 г; 45,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении. Остаток дважды экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали рассолом (40 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. После дальнейшей сушки под высоким вакуумом получали 5-изобутил-1Н-пиразол-3-карбоновую кислоту (2 г; выход 80%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, метанол-d4): δ 6.58 (s, 1Н), 2.57 (d, 2Н), 1.95 (m, 1Н), 0.96 (d, 6Н).
MS(ESI): 169,2 ([M+H]+).
b) 3-Изобутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 5-изобутил-1Н-пиразол-3-карбоновую кислоту вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество. MS (ESI): 341,2 ([М+Н]+).
Пример 102
4-Хлор-3-изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
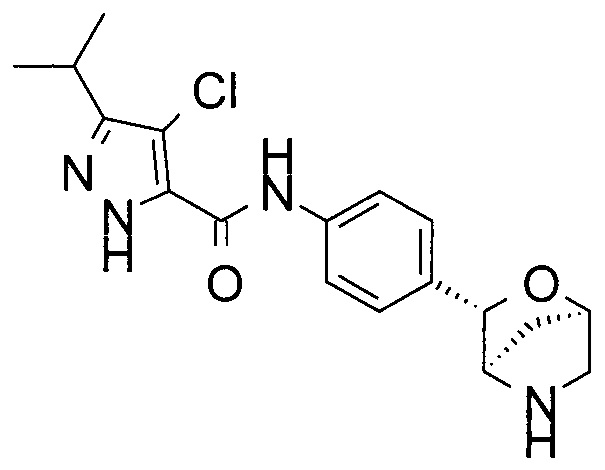
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 4-хлор-3-изопропил-1Н-пиразол-5-карбоновую кислоту (CAS: 1291271-55-0) вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество.
MS (ESI): 363,1 ([{37Cl}M+H]+); 361,1 ([{35Cl}M+H]+).
Пример 103
4-Фтор-3-изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
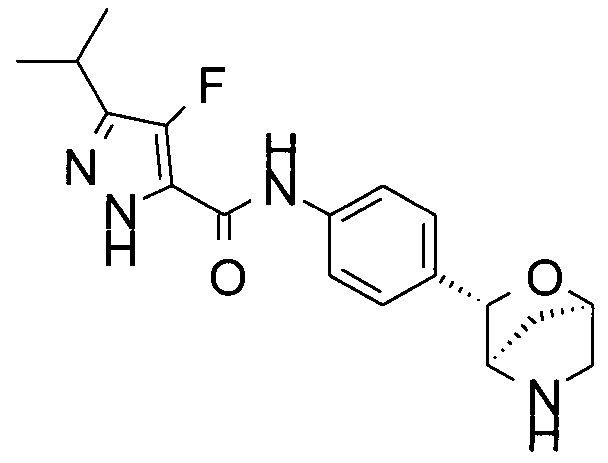
а) Этил-5-изопропил-1Н-пиразол-3-карбоксилат
К раствору CH3CH2ONa (23 г; 0,34 моль) в безводном EtOH (500 мл) добавляли диэтилоксалат (50 г; 0,34 моль; CAS: 95-92-1) и 3-метил-2-бутанон (29 г; 0,34 моль; CAS: 563-80-4) при 0°С. Раствор перемешивали при 50°С в течение ночи. Смесь охлаждали до 0-5°С и добавляли уксусную кислоту (20,4 г; 0,34 моль), затем гидразина моногидрат (17,2 г; 0,34 моль; CAS: 7803-57-8). Смесь перемешивали при 30°С в течение ночи и после этого охлаждали до комнатной температуры. Летучие вещества удаляли при пониженном давлении. Остаток разбавляли насыщенным водным раствором NaHCO3 (500 мл) и экстрагировали этилацетатом (1 л). Органический слой промывали рассолом и концентрировали при пониженном давлении, получая желаемый неочищенный продукт. После очистки флэш-хроматографией (силикагель; CH2Cl2/MeOH от 200/1 до 80/1 по объему) получали неочищенный этил-5-изопропил-1Н-пиразол-3-карбоксилат (30 г; выход 48%) в виде желтого твердого вещества.
MS(ESI): 183,1 ([М+Н]+).
b) Этил-4-фтор-5-изопропил-1Н-пиразол-3-карбоксилат
К раствору этил-5-изопропил-1Н-пиразол-3-карбоксилата (5,0 г; 0,027 ммоль) в CH3CN (300 мл) добавляли Selectfluor (12,65 г; 35,67 ммоль, CAS: 140681-55-6) при 0°С. Раствор перемешивали при 70°С в течение 15 часов и после этого охлаждали до комнатной температуры. Летучие вещества удаляли при пониженном давлении. Остаток разбавляли водным раствором HCl (3 н., 200 мл) и экстрагировали CH2Cl2 (100 мл × 2). Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали при пониженном давлении. После очистки флэш-хроматографией (силикагель; CH2Cl2/MeOH от 200/1 до 100/1 по объему) получали этил-4-фтор-5-изопропил-1Н-пиразол-3-карбоксилат (900 мг; выход 17%) в виде желтого масла.
MS (ESI): 201,1 ([М+Н]+).
c) 4-Фтор-5-изопропил-1Н-пиразол-3-карбоновая кислота
К раствору этил-4-фтор-5-изопропил-1Н-пиразол-3-карбоксилата (900 мг; 4,49 ммоль) в смеси ТГФ/МеОН (10/10 мл) добавляли 1 М водный раствор NaOH (9 мл; 8,98 ммоль) при 0°С. Затем раствор кипятили с обратным холодильником в течение 3 часов. Реакционный раствор выливали в воду и подкисляли с использованием концентрированной HCl до pH приблизительно 1. Смесь экстрагировали EtOAc (100 мл × 2). Объединенные органические слои промывали рассолом, концентрировали при пониженном давлении. Остаток перекристаллизовывали из этилацетата (10 мл), получая 4-фтор-5-изопропил-1Н-пиразол-3-карбоновую кислоту (450 мг; выход 58%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, метанол-d4): δ 3.08 (m, 1Н), 1.32 (d, 6Н).
MS (ESI): 173,1 ([М+Н]+).
d) 4-Фтор-3-изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид
Указанное в заголовке соединение получали по аналогии с соединением примера 67, используя 4-фтор-5-изопропил-1Н-пиразол-3-карбоновую кислоту вместо 5-этил-4-метил-2Н-пиразол-3-карбоновой кислоты на стадии (d). Белое твердое вещество.
MS (ESI): 345,2 ([М+Н]+).
Пример 104
(1R,3R,4R)-3-(2-Пиридил)-2-окса-5-азабицикло[2.2.1]гептан
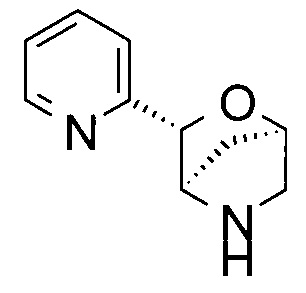
a) [(2R,4R)-4-Гидрокси-2-(пиридин-2-карбонил)пирролидин-1-ил]-фенил-метанон
К раствору 2-бромпиридина (3,64 г; 0,023 моль; CAS: 109-04-6) в ТГФ (60 мл) добавляли раствор н-BuLi (2,5 М; 9,2 мл; 0,023 моль) при -70°С. Смесь перемешивали в течение 30 минут. Затем полученный выше раствор по каплям добавляли к раствору (1R,4R)-5-бензоил-2-окса-5-азабицикло[2.2.1]гептан-3-она (5 г; 0,023 моль; CAS: 444313-68-2) в ТГФ (100 мл) при -70°С. Реакционную смесь перемешивали в течение 30 минут. Затем реакционный раствор гасили добавлением водного раствора NH4Cl (100 мл). Смесь экстрагировали CH2Cl2 (100 мл × 2), промывали рассолом (50 мл × 2), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали посредством флэш-хроматографии (силикагель; CH2Cl2/MeOH от 200/1 до 50/1 по объему), получая [(2R,4R)-4-гидрокси-2-(пиридин-2-карбонил)пирролидин-1-ил]-фенил-метанон (1,0 г; выход 15%) в виде желтого масла.
MS (ESI): 297,0 ([М+Н]+).
b) [(2R,4R)-4-Гидрокси-2-[(R)-гидрокси(2-пиридил)метил]пирролидин-1-ил]-фенил-метанон
К раствору [(2R,4R)-4-гидрокси-2-(пиридин-2-карбонил)пирролидин-1-ил]-фенил-метанона (1,0 г; 3,35 ммоль) в МеОН (20 мл) добавляли NaBH4 (255 мг; 6,7 моль) при 0°С. Раствор перемешивали при комнатной температуре в течение 2 часов. Затем реакционный раствор выливали в воду (50 мл). Смесь экстрагировали CH2Cl2 (50 мл × 2). Объединенные органические слои промывали рассолом (50 мл × 2), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (CH2Cl2/MeOH от 200/1 до 50/1 по объему), получая [(2R,4R)-4-гидрокси-2-[(R)-гидрокси(2-пиридил)метил]пирролидин-1-ил]-фенил-метанон (720 мг; выход 72%) в виде белого твердого вещества.
MS (ESI): 299,0 ([М+Н]+).
с) Фенил-[(1R,3R,4R)-3-(2-пиридил)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]метанон
К раствору [(2R,4R)-4-гидрокси-2-[(R)-гидрокси(2-пиридил)метил]-пирролидин-1-ил]-фенил-метанона (0,72 г; 2,41 ммоль) в толуоле (20 мл) добавляли PPh3 (758 мг; 2,89 моль) и диизопропилазодикарбоксилат (584 мг; 2,89 ммоль; CAS: 2446-83-5) при 0°С. Раствор перемешивали при комнатной температуре в течение ночи. Летучие вещества удаляли при пониженном давлении. Остаток очищали посредством флэш-хроматографии (силикагель; CH2Cl2/этилацетат от 10/1 до 1/1 по объему), получая фенил-[(1R,3R,4R)-3-(2-пиридил)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]метанон (350 мг; выход 52%) в виде желтого твердого вещества.
MS (ESI): 281,1 ([М+Н]+).
d) (1R,3R,4R)-3-(2-Пиридил)-2-окса-5-азабицикло[2.2.1]гептан
К раствору фенил-[(1R,3R,4R)-3-(2-пиридил)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]метанона (0,75 г; 2,67 ммоль) в МеОН (3 мл) добавляли KOH (3 г; 53,5 ммоль). Смесь перемешивали при температуре дефлегмации в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли МеОН (50 мл). Добавляли концентрированную HCl для подведения pH до приблизительно 7. Осадок удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (подвижная фаза А: H2O с 0,5% NH3⋅H2O, В: CH3CN; колонка С18), получая (1R,3R,4R)-3-(2-пиридил)-2-окса-5-азабицикло[2.2.1]гептан (10 мг; выход 2,1%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, метанол-d4): δ 8.51 (d, 1Н), 7.84 (m, 1Н), 7.48 (d, 1Н), 7.31 (m, 1Н), 4.94 (s, 1Н), 4.77 (s, 1Н), 3.94 (s, 1Н), 3.14 (d, 1Н), 3.01 (d, 1Н), 1.79 (d, 1Н), 1.59 (d, 1Н).
MS (ESI): 177,1 ([М+Н]+).
Пример 105
(1S,3S,4S)-3-(2-Пиридил)-2-окса-5-азабицикло[2.2.1]гептан
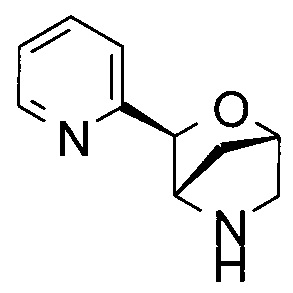
Указанное в заголовке соединение получали по аналогии с соединением примера 104, используя (1S,4S)-5-бензоил-2-окса-5-азабицикло[2.2.1]гептан-3-он (CAS: 31560-25-5) вместо (1R,4R)-5-бензоил-2-окса-5-азабицикло[2.2.1]гептан-3-она на стадии (a). MS (ESI): 177,1 ([М+Н]+).
Пример 106
(1R,3S,4R)-3-(2-Фторфенил)-2-окса-5-азабицикло[2.2.1]гептан
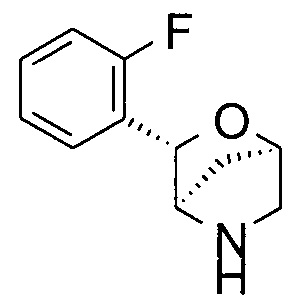
Указанное в заголовке соединение получали по аналогии с соединением примера 104, используя 1-фтор-2-иодбензол (CAS: 348-52-7) вместо 2-бромпиридина на стадии (а).
MS (ESI): 194,0 ([М+Н]+).
1Н-ЯМР (400 МГц, метанол-d4): δ 7.38 (m, 1Н), 7.28 (m, 1Н), 7.17 (m, 1Н), 7.05 (m, 1Н), 5.07 (s, 1Н), 4.70 (s, 1Н), 3.62 (s, 1Н), 3.00 (d, 1Н), 2.92 (d, 1Н), 1.74 (d, 1Н), 1.51 (d, 1Н).
Материалы и методы
Конструирование TAAR-экспрессирующих плазмид и стабильно трансфицированных клеточных линий
Для конструирования экспрессирующих плазмид амплифицировали кодирующие TAAR1 последовательности из геномной ДНК человека, крысы и мыши по существу так, как описано Lindemann и др. [14]. Использовали систему полимеразной цепной реакции (ПЦР) с высокоточным наращиванием цепи (Roche Diagnostics) с 1,5 мМ Mg2+ и очищенные ПЦР-продукты клонировали в клонирующий вектор pCR2.1-TOPO (Invitrogen), следуя инструкциям производителя. ПЦР-продукты субклонировали в вектор pIRESneo2 (BD Clontech, Palo Alto, California) и последовательности экспрессирующих векторов подтверждали путем секвенирования перед введением их в клеточные линии.
Клетки HEK293 (линия 293 клеток почки эмбриона человека) (АТСС (American type culture collection - Американская коллекция типовых культур) № CRL-1573) культивировали по существу так, как описано Lindemann и др. (2005). Для получения стабильно трансфицированных клеточных линий клетки HEK293 трансфицировали экспрессирующими плазмидами pIRESneo2, содержащими TAAR-кодирующие последовательности (описанные выше), используя липофектамин 2000 (Invitrogen) в соответствии с инструкциями производителя, и через 24 часа после трансфекции в культуральную среду добавляли G418 (Sigma, Buchs, Switzerland) в концентрации 1 мг/мл. После культивирования в течение приблизительно 10 суток клоны выделяли, рассевали и тестировали в отношении их восприимчивости к следовым аминам (все соединения были приобретены в Sigma) с использованием системы для иммуноферментного анализа (enzyme immunoassay; EIA) "cAMP Biotrak" (Amersham), следуя предоставленной производителем методике EIA без ацетилирования. Для всех последующих исследований использовали моноклональные клеточные линии, которые показывали стабильную ЕС50 (эффективную концентрацию, вызывающую эффект величиной 50%) в течение периода культивирования, составляющего 15 пассажей.
Анализ связывания радиоактивного лиганда с использованием TAAR1 крысы
Приготовление мембран и связывание радиоактивного лиганда
Клетки HEK-293, стабильно экспрессирующие TAAR1 крысы, поддерживали при 37°С и 5% CO2 в среде DMEM (Dulbecco modified Eagle's medium - модифицированная Дульбекко среда Игла) с высоким содержанием глюкозы, в состав которой входит фетальная телячья сыворотка (10%, инактивированная нагреванием в течение 30 мин при 56°С), пенициллин/стрептомицин (1%) и генетицин в концентрации 375 мкг/мл (Gibco). Клетки высвобождали из культуральных флаконов, используя трипсин/EDTA (этилендиаминтетрауксусная кислота), собирали, дважды промывали охлажденным во льду забуференным фосфатом физиологическим раствором (PBS) (без Ca2+ и Mg2+), собирали центрифугированием при 1000 об./мин в течение 5 мин при 4°С, замораживали и хранили при -80°С. Замороженные осадки после центрифугирования суспендировали в 20 мл буфера на основе 2-гидроксиэтил-пиперазин-2-этансульфоновой кислоты (HEPES) и NaOH (20 мМ; pH 7,4), содержащего 10 мМ EDTA, и гомогенизировали с использованием политрона (РТ 6000, Kinematica) при 14000 об./мин в течение 20 с. Гомогенат центрифугировали при 48000×g в течение 30 мин при 4°С. После этого супернатант извлекали и отбрасывали и осадок после центрифугирования ресуспендировали в 20 мл буфера HEPES-NaOH (20 мМ; pH 7,4), содержащего 0,1 мМ EDTA, используя политрон (20 с при 14000 об./мин). Эту процедуру повторяли и конечный осадок ресуспендировали в буфере HEPES-NaOH, содержащем 0,1 мМ EDTA, и гомогенизировали, используя политрон. Обычно порции по 2 мл аликвот мембран хранили при -80°С. Для каждой новой партии мембран определяли константу диссоциации (Kd) на основании кривой насыщения. Радиоактивный лиганд для TAAR1 3[Н]-(S)-4-[(этил-фенил-амино)-метил]-4,5-дигидро-оксазол-2-иламин (описанный в WO 2008/098857) использовали в концентрации, равной рассчитанной величине Kd, обычно составляющей примерно 2,3 нМ, при которой связывается приблизительно 0,2% от всего добавленного радиоактивного лиганда, и специфическое связывание составляет приблизительно 85% от общего связывания. Неспецифическое связывание определяли как количество [3Н]-(S)-4-[(этил-фенил-амино)-метил]-4,5-дигидро-оксазол-2-иламина, связавшегося в присутствии 10 мкМ немеченого лиганда. Все соединения тестировали в широком диапазоне концентраций (от 10 пМ до 10 мкМ) в двух повторах. Тестируемые соединения (20 мкл/лунка) переносили в 96-луночный планшет с глубокими лунками (TreffLab) и добавляли по 180 мкл буфера HEPES-NaOH (20 мМ; pH 7,4), содержащего MgCl2 (10 мМ) и CaCl2 (2 мМ) (буфер для связывания), по 300 мкл раствора радиоактивного лиганда 3[Н]-(S)-4-[(этил-фенил-амино)-метил]-4,5-дигидро-оксазол-2-иламина в концентрации 3,3х Kd в нМ и по 500 мкл препарата мембран (ресуспендированных из расчета 50 мкг белка в одном мл). 96-луночные планшеты с глубокими лунками инкубировали в течение 1 ч при 4°С. Инкубирование останавливали путем быстрого фильтрования через 96-луночные планшеты UniFilter (Packard Instrument Company) и стеклянные фильтры GF/C (Perkin Elmer), которые предварительно вымачивали в течение 1 ч в полиэтиленимине (0,3%-ном), и промывали 3 раза, используя по 1 мл холодного буфера для связывания. После добавления 45 мкл сцинтилляционной жидкости микросцинт 40 (PerkinElmer) 96-луночный планшет UniFilter герметично закрывали и через 1 ч измеряли радиоактивность, используя сцинтилляционный счетчик для микропланшетов TopCount (Packard Instrument Company).
Анализ связывания радиоактивного лиганда с использованием TAAR1 мыши
Приготовление мембран и связывание радиоактивного лиганда
Клетки HEK-293, стабильно экспрессирующие TAAR1 мыши, поддерживали при 37°С и 5% CO2 в среде DMEM с высоким содержанием глюкозы, в состав которой входит фетальная телячья сыворотка (10%, инактивированная нагреванием в течение 30 мин при 56°С), пенициллин/стрептомицин (1%) и генетицин в концентрации 375 мкг/мл (Gibco). Клетки высвобождали из культуральных флаконов, используя трипсин/EDTA, собирали, дважды промывали охлажденным во льду PBS (без Са2+ и Mg2+), собирали центрифугированием при 1000 об./мин в течение 5 мин при 4°С, замораживали и хранили при -80°С. Замороженные осадки после центрифугирования суспендировали в 20 мл буфера HEPES-NaOH (20 мМ; pH 7,4), содержащего 10 мМ EDTA, и гомогенизировали с использованием политрона (РТ 6000, Kinematica) при 14000 об./мин в течение 20 с. Гомогенат центрифугировали при 48000×g в течение 30 мин при 4°С. После этого супернатант извлекали и отбрасывали и осадок после центрифугирования ресуспендировали в 20 мл буфера HEPES-NaOH (20 мМ; pH 7,4), содержащего 0,1 мМ EDTA, используя политрон (20 с при 14000 об./мин). Эту процедуру повторяли и конечный осадок ресуспендировали в буфере HEPES-NaOH, содержащем 0,1 мМ EDTA, и гомогенизировали, используя политрон. Обычно порции по 2 мл аликвот мембран хранили при -80°С. Для каждой новой партии мембран определяли константу диссоциации (Kd) на основании кривой насыщения. Радиоактивный лиганд для TAAR1 3[Н]-(S)-4-[(этил-фенил-амино)-метил]-4,5-дигидро-оксазол-2-иламин (описанный в WO 2008/098857) использовали в концентрации, равной рассчитанной величине Kd, обычно составляющей примерно 0,7 нМ, при которой связывается приблизительно 0,5% от всего добавленного радиоактивного лиганда, и специфическое связывание составляет приблизительно 70% от общего связывания. Неспецифическое связывание определяли как количество [3Н]-(S)-4-[(этил-фенил-амино)-метил]-4,5-дигидро-оксазол-2-иламина, связавшегося в присутствии 10 мкМ немеченого лиганда. Все соединения тестировали в широком диапазоне концентраций (от 10 пМ до 10 мкМ) в двух повторах. Тестируемые соединения (20 мкл/лунка) переносили в 96-луночный планшет с глубокими лунками (TreffLab) и добавляли по 180 мкл буфера HEPES-NaOH (20 мМ; pH 7,4), содержащего MgCl2 (10 мМ) и CaCl2 (2 мМ) (буфер для связывания), по 300 мкл раствора радиоактивного лиганда 3[Н]-(S)-4-[(этил-фенил-амино)-метил]-4,5-дигидро-оксазол-2-иламина в концентрации 3,3х Kd в нМ и по 500 мкл препарата мембран (ресуспендированных из расчета 60 мкг белка в одном мл). 96-луночные планшеты с глубокими лунками инкубировали в течение 1 ч при 4°С. Инкубирование останавливали путем быстрого фильтрования через 96-луночные планшеты UniFilter (Packard Instrument Company) и стеклянные фильтры GF/C (Perkin Elmer), которые предварительно вымачивали в течение 1 ч в полиэтиленимине (0,3%-ном), и промывали 3 раза, используя по 1 мл холодного буфера для связывания. После добавления 45 мкл сцинтилляционной жидкости микросцинт 40 (PerkinElmer) 96-луночный планшет UniFilter герметично закрывали и через 1 ч измеряли радиоактивность, используя сцинтилляционный счетчик для микропланшетов TopCount (Packard Instrument Company).
В анализе с использованием TAAR1 мыши или крысы соединения демонстрируют значение Ki (в мкМ), показанное в приведенной ниже таблице.
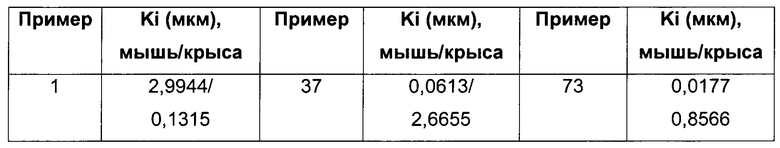
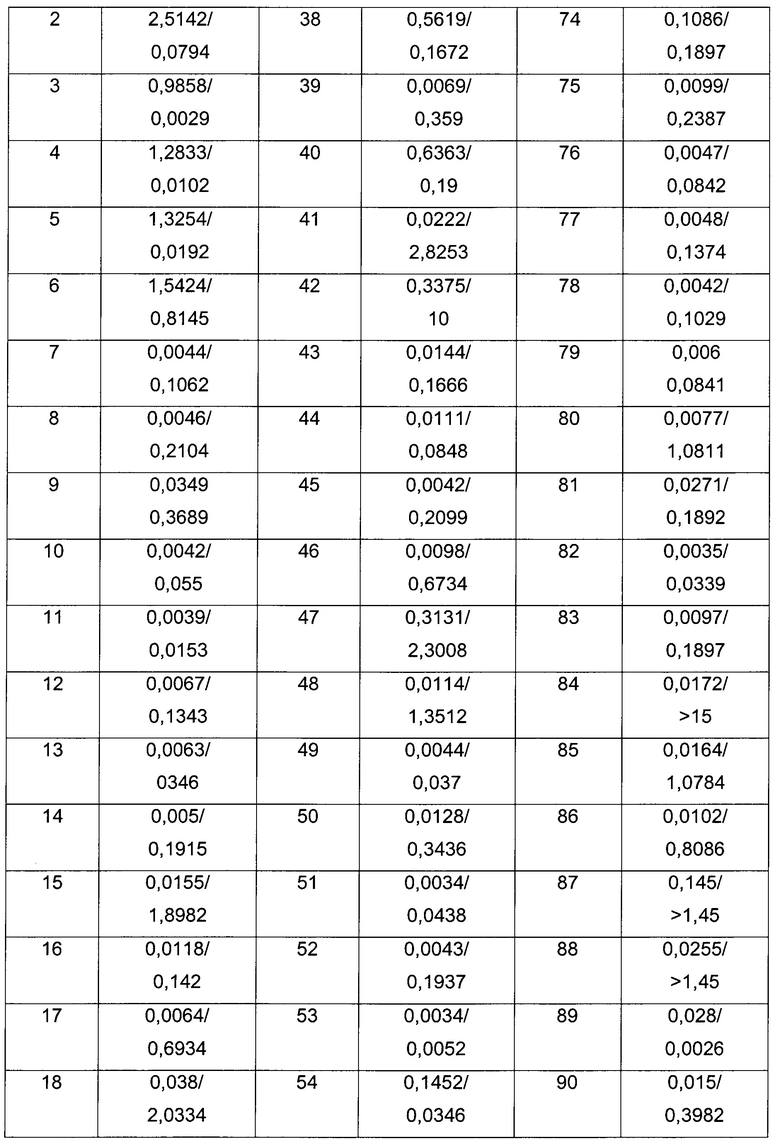
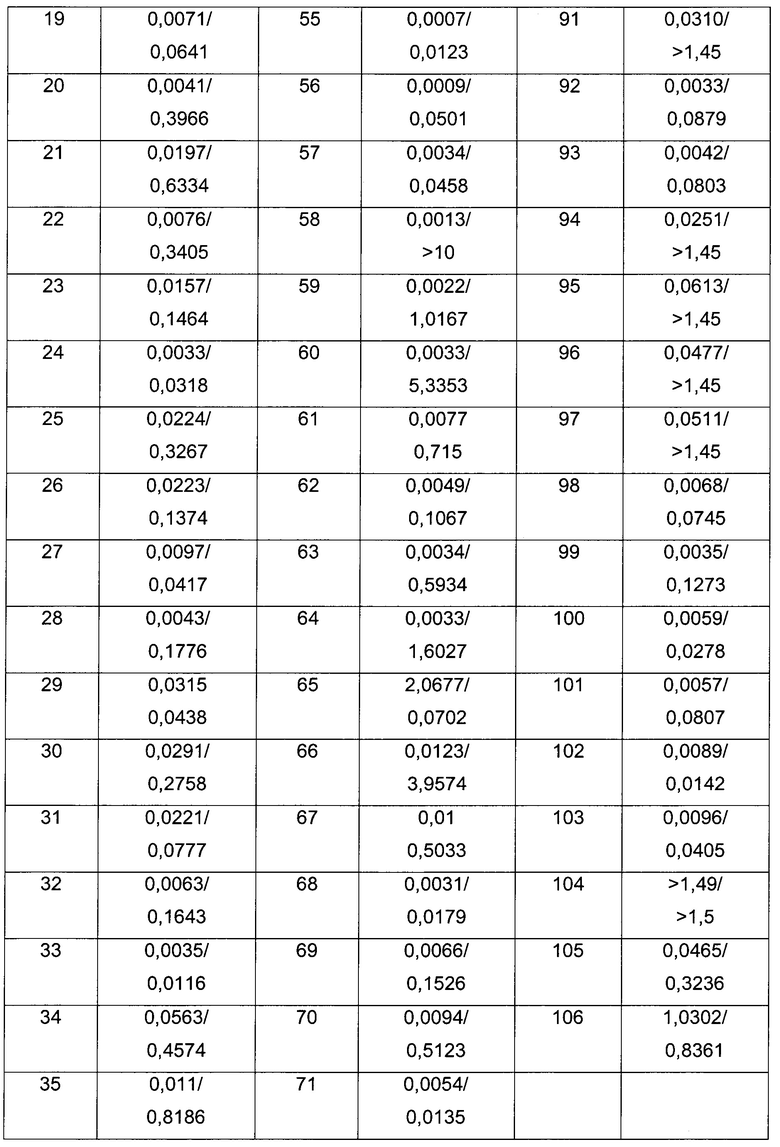

Соединения формулы I и фармацевтически приемлемые соли соединений формулы I можно использовать в качестве лекарственных средств, например в форме фармацевтических препаратов. Эти фармацевтические препараты можно вводить перорально, например в форме таблеток, таблеток, покрытых оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако их также можно вводить ректально, например в форме суппозиториев, или парентерально, например в форме растворов для инъекций.
В способе изготовления фармацевтических препаратов на основе соединений формулы I можно использовать фармацевтически инертные неорганические или органические носители. Например, в качестве таких носителей для таблеток, таблеток, покрытых оболочкой, драже и твердых желатиновых капсул можно использовать лактозу, кукурузный крахмал или его производные, тальк, стеариновые кислоты или их соли и тому подобное. Подходящими носителями для мягких желатиновых капсул являются, например растительные масла, воски, жиры, полутвердые и жидкие полиолы и тому подобное. В случае мягких желатиновых капсул никаких носителей обычно не требуется, однако это зависит от природы активного вещества. Подходящими носителями для изготовления растворов и сиропов являются, например вода, полиолы, глицерин, растительное масло и тому подобное. Подходящими носителями для суппозиториев являются, например природные или отвержденные жиры, воски, жиры, полужидкие или жидкие полиолы и тому подобное.
Помимо этого фармацевтические препараты могут содержать консерванты, солюбилизаторы, стабилизаторы, увлажняющие агенты, эмульгаторы, подсластители, красители, корригенты, соли для изменения осмотического давления, буферы, маскирующие агенты или антиоксиданты. Они также могут содержать другие дополнительные терапевтически полезные вещества.
Лекарственные средства, содержащие соединение формулы I или его фармацевтически приемлемую соль и терапевтически инертный носитель, также представляют собой объект настоящего изобретения, как и способ их изготовления, который включает внесение одного или более чем одного соединения формулы I и/или одной или более чем одной фармацевтически приемлемой соли присоединения кислоты и, при желании, одного или более чем одного другого терапевтически полезного вещества в форму галенова препарата для введения вместе с одним или более чем одним терапевтически инертным носителем.
Наиболее предпочтительными показаниями согласно настоящему изобретению являются те, которые включают расстройства центральной нервной системы, например, лечение или предупреждение депрессии, психоза, болезни Паркинсона, тревоги, синдрома дефицита внимания и гиперактивности (ADHD) и диабета.
Дозировку можно варьировать в широких пределах, и, несомненно, в каждом конкретном случае она должна быть подобрана с учетом индивидуальных потребностей. В случае перорального введения дозировка для взрослых может изменяться от примерно 0,01 мг до примерно 1000 мг в сутки для соединения общей формулы I или соответствующего количества его фармацевтически приемлемой соли. Суточную дозу можно вводить в виде разовой дозы или в виде разделенных доз, и помимо этого также может быть превышен верхний предел, если на то имеется показание.

Методика приготовления
1. Смешать ингредиенты по позициям 1, 2, 3 и 4 и провести гранулирование с использованием очищенной воды.
2. Высушить гранулы при 50°С.
3. Пропустить гранулы через подходящее для размола оборудование.
4. Добавить ингредиент по позиции 5 и перемешать в течение трех минут; выполнить прессование на подходящем прессе.

Методика приготовления
1. Смешать ингредиенты по позициям 1, 2 и 3 в подходящем смесителе в течение 30 минут.
2. Добавить ингредиенты по позициям 4 и 5 и перемешать в течение 3 минут.
3. Поместить в подходящую капсулу.
| название | год | авторы | номер документа |
|---|---|---|---|
| СРЕДСТВА, ИНДУЦИРУЮЩИЕ АПОПТОЗ, ДЛЯ ЛЕЧЕНИЯ РАКА, ИММУННЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2011 |
|
RU2568611C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОПЕНТАНА | 2010 |
|
RU2572555C2 |
| СШИТАЯ ИСКУССТВЕННАЯ НУКЛЕИНОВАЯ КИСЛОТА ALNA | 2019 |
|
RU2820226C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2625309C2 |
| ИНДАЗОЛЫ, БЕНЗОТИАЗОЛЫ, БЕНЗОИЗОТИАЗОЛЫ, БЕНЗОИЗОКСАЗОЛЫ, ПИРАЗОЛОПИРИДИНЫ, ИЗОТИАЗОЛОПИРИДИНЫ, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ | 2006 |
|
RU2450003C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2697712C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| АМИДНОЕ ПРОИЗВОДНОЕ ПИРАЗОЛА | 2015 |
|
RU2658827C2 |
| МОДУЛЯТОРЫ MAS-СВЯЗАННОГО РЕЦЕПТОРА G-БЕЛКА X2 И СВЯЗАННЫЕ С НИМИ ПРОДУКТЫ И СПОСОБЫ | 2021 |
|
RU2841536C1 |
Изобретение относится к соединениям формулы (I), а также к фармацевтическим композициям и применению. Технический результат: получены новые соединения, которые обладают активностью в отношении TAAR1. 6 н. и 5 з.п. ф-лы, 1 табл., 106 пр.
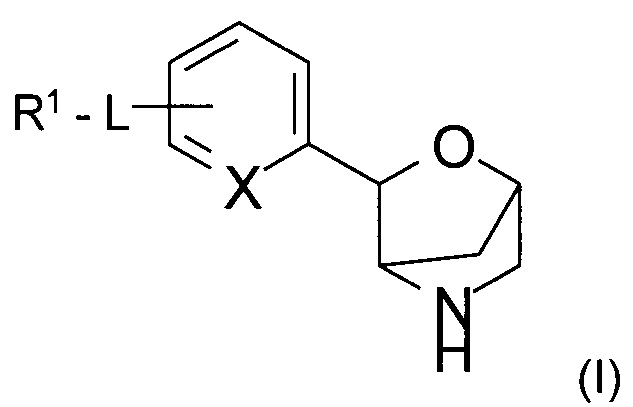
1. Соединение формулы
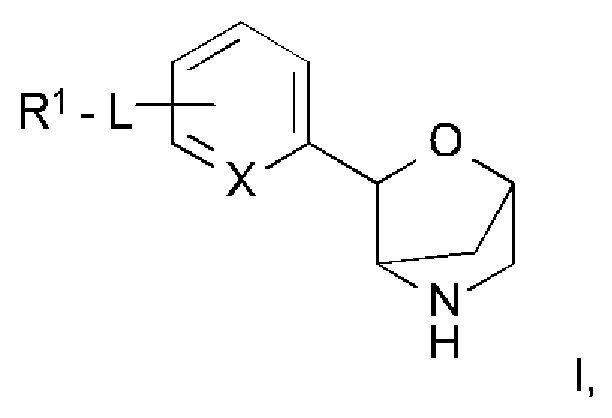
где
L представляет собой связь, -C(O)NH-, -NHC(O)-, -CH2NHC(O)-, -CH2C(O)NH-, -CH2NH-, -NH- или -NHC(O)NH-;
R1 представляет собой атом водорода, С1-7алкил, атом галогена, С1-7алкокси-алкил, С1-7алкокси, замещенный атомом галогена, С1-7алкил, замещенный атомом галогена, или
представляет собой фенил или гетероарил, выбранный из группы, состоящей из пиридинила, пиримидинила, пиразинила или пиразолила, и при этом фенил и гетероарил возможно замещены одним или двумя заместителями, выбранными из группы, состоящей из атома галогена, С1-7алкила, C1-7алкокси, С1-7алкила, замещенного атомом галогена, С1-7алкокси, замещенного атомом галогена, С3-6циклоалкила или -O-СН2-С3-6циклоалкила;
X представляет собой СН или N;
или его фармацевтически приемлемая соль присоединения кислоты.
2. Соединение формулы I по п. 1, где R1 представляет собой атом водорода, С1-7алкил, атом галогена, С1-7алкокси-алкил, С1-7алкокси, замещенный атомом галогена, или С1-7алкил, замещенный атомом галогена, и L является таким, как описано в п. 1.
3. Соединение формулы I, выбранное из:
(1R,3S,4R)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан;
(1S,3R,4S)-3-фенил-2-окса-5-азабицикло[2.2.1]гептан;
N-бутил-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин;
(1S,3R,4S)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан;
(1R,3S,4R)-3-(4-бромфенил)-2-окса-5-азабицикло[2.2.1]гептан;
N-(3-метоксипропил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)ацетамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(3,3,3-трифторпропокси)ацетамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)ацетамид;
4,4,4-трифтор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бутанамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(3,3,3-трифторпропокси)ацетамид;
4,4,4-трифтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бутанамид;
(1R,3R,4R)-3-(2-пиридил)-2-окса-5-азабицикло[2.2.1]гептан;
(1S,3S,4S)-3-(2-пиридил)-2-окса-5-азабицикло[2.2.1]гептан или
(1R,3S,4R)-3-(2-фторфенил)-2-окса-5-азабицикло[2.2.1]гептан.
4. Соединение формулы I по п. 1, где R1 представляет собой фенил, который возможно замещен одним или двумя заместителями, выбранными из группы, состоящей из атома галогена, С1-7алкила, С1-7алкокси, С1-7алкила, замещенного атомом галогена, С1-7алкокси, замещенного атомом галогена, С3-6циклоалкила или -O-СН2-С3-6циклоалкила, и L является таким, как описано в п. 1.
5. Соединение формулы I, выбранное из:
3-хлор-N-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-хлор-N-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
1-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-[4-(трифторметил)фенил]мочевина;
1-(4-хлорфенил)-3-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]мочевина;
1-(3-хлорфенил)-3-[3-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-ил]фенил]мочевина;
4-хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
3-хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
3-хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-(циклопропилметокси)-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-этокси-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-этокси-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
4-(циклопропилметокси)-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бензамид;
1-(4-хлорфенил)-3-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]мочевина;
N-[(4-хлорфенил)метил]-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин;
4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметил)фенил]метил]анилин;
N-[(4-фторфенил)метил]-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]анилин;
4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметокси)фенил]метил]анилин;
N-(4-хлорфенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(4-бромфенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(4-фторфенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(4-этоксифенил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[4-(трифторметил)фенил]бензамид;
4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметил)фенил]метил]бензамид;
N-[(4-хлорфенил)метил]-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
4,4,4-трифтор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]бутанамид;
N-(4-бромфенил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(4-фторфенил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(4-этоксифенил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]-N-[[4-(трифторметил)фенил]метил]бензамид или
N-[(4-хлорфенил)метил]-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид.
6. Соединение формулы I по п. 1, где R1 представляет собой пиридинил, пиримидинил, пиразинил или пиразолил, которые возможно замещены одним или двумя заместителями, выбранными из группы, состоящей из атома галогена, С1-7алкила, С1-7алкокси, С1-7алкила, замещенного атомом галогена, С1-7алкокси, замещенного атомом галогена, С3-6циклоалкила или -O-СН2-С3-6циклоалкила, и L является таким, как описано в п. 1.
7. Соединение формулы I, выбранное из:
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-амин;
6-этокси-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-3-карбоксамид;
6-этокси-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-3-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(2,2,2-трифторэтокси)пиридин-3-карбоксамид;
2-циклопропил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-амин;
5-хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-2-амин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиримидин-4-амин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиразин-2-амин;
N-[4-[(1S,3R,4S)2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиримидин-2-амин;
5-хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиридин-2-амин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиридин-4-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиридин-4-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(2,2,2-трифторэтокси)пиридин-3-карбоксамид;
2-циклопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиразин-2-амин;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиримидин-2-амин;
2-этил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиридин-4-амин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-карбоксамид;
4-хлор-3-циклопропил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиримидин-4-карбоксамид;
3-изопропил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(трифторметил)пиридин-3-карбоксамид;
4-хлор-3-этокси-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-хлор-3-метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-хлор-1-метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-пропил-пиразол-3-карбоксамид;
4-хлор-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-пропил-1Н-пиразол-5-карбоксамид;
3-этил-4-метил-N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(2,2,2-трифторэтокси)пиридин-2-карбоксамид;
N-(6-хлор-3-пиридил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(трифторметил)пиридин-3-амин;
N-(6-этокси-3-пиридил)-4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
3-этил-4-метил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-хлор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-пропил-1Н-пиразол-5-карбоксамид;
3-циклопропил-4-метил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(трифторметил)пиридин-2-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-6-(трифторметил)пиридин-3-карбоксамид;
2-этил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]пиримидин-5-карбоксамид;
3-изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-хлор-3-этил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
3-циклопропил-4-фтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-фтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-3-пропил-1Н-пиразол-5-карбоксамид;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-4-(2,2,2-трифторэтокси)пиримидин-2-амин;
N-[4-[(1S,3R,4S)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)пиримидин-4-амин;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(трифторметил)пиримидин-4-карбоксамид;
4-хлор-3-циклопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-2-(2,2,2-трифторэтокси)пиримидин-4-амин;
2-изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-5-(2,2,2-трифторэтокси)пиразол-3-карбоксамид;
3-бутил-4-фтор-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
3-бутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
N-(6-хлор-3-пиридил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
N-(6-этокси-3-пиридил)-4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]бензамид;
4-хлор-3-этокси-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-бром-3-этил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-фтор-3-изобутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
3-изобутил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид;
4-хлор-3-изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид или
4-фтор-3-изопропил-N-[4-[(1R,3S,4R)-2-окса-5-азабицикло[2.2.1]гептан-3-ил]фенил]-1Н-пиразол-5-карбоксамид.
8. Фармацевтическая композиция для лечения заболевания, связанного с биологической функцией TAAR1 (рецептора, ассоциированного со следовыми аминами), содержащая эффективное количество соединения по любому из пп. 1-7 и фармацевтический приемлемый носитель и/или адъювант.
9. Соединения по любому из пп. 1-7 для применения в качестве лиганда TAAR1.
10. Соединения по любому из пп. 1-7 для применения в качестве терапевтически активных веществ при лечении заболевания, связанного с биологической функцией TAAR1.
11. Применение соединения по любому из пп. 1-7 для изготовления лекарственных средств для терапевтического и/или профилактического лечения заболевания, связанного с биологической функцией TAAR1.
| WO2014072257 A1, 15.05.2014 | |||
| RU2016149984 A, 02.07.2018 | |||
| ТЕРМОМЕТР ДЛЯ ОПРЕДЕЛЕНИЯ ТЕМПЕРАТУРЫ ПОВЕРХНОСТИ ТЕЛА ЧЕЛОВЕКА И ЖИВОТНЫХ | 1929 |
|
SU17802A1 |

