Настоящее изобретение относится к соединениям общей формулы
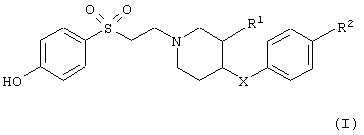
в которой
R1 обозначает водородный атом или гидроксил;
R2 обозначает водородный атом или метил; а
Х обозначает -О- или -СН2-;
и к их фармацевтически приемлемым кислотно-аддитивным солям.
Понятие "фармацевтически приемлемые кислотно-аддитивные соли" охватывает соли неорганических и органических кислот, таких, как соляная кислота, азотная кислота, серная кислота, молочная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфоновая кислота, п-толуолсульфоновая кислота и т.п.
К соединениям по изобретению относятся цисизомеры.
К соединениям по настоящему изобретению относятся селективные блокаторы подтипа NMDA (N-метил-D-аспартат) рецепторов, которые выполняют ключевую функцию в модуляции нейронной активности и пластичности, что делает их ключевыми плеерами в процессах медиации, лежащих в основе развития центральной нервной системы, включая формирование и функционирование процессов усвоения и памяти.
При патологических состояниях, характерных для активных и хронических форм нейродегенерации, чрезмерная активация NMDA рецепторов служит даже ключом для запуска мехенизма гибели нервных клеток. NMDA рецепторы состоят из субъединиц членов двух семейств, а именно, NR-1 (8 различных сплайс-вариантов) и NR-2 (от А до D), кодируемых различными генами. Члены 15 этих двух субъединичных семейств по-разному распределены в различных областях мозга. Результатом образования гетеромерных комбинаций членов NR-1 с различными субъединицами NR-2 является возникновение NMDA рецепторов, обладающих различными фармакологическими свойствами. Возможные терапевтические показания для использования специфических блокаторов подтипа NMDA рецептора включают активные формы нейродегенерации, вызванные, например, параличом или травмой головного мозга; хронические формы нейродегенерации, такие, как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона и АБС (амиотрофический боковой склероз); нейродегенерацию, связанную с бактериальными или вирусными инфекциями; и такие болезни, как шизофрения, страх, депрессия и острая/хроническая боль.
Объектами настоящего изобретения являются новые соединения формулы I, применение при лечении или профилактике заболеваний, вызванных повышенной активацией соответствующих подтипов NMDA рецепторов, которые включают острые формы нейродегенерации, вызванной, например, параличом или травмой мозга; хронические формы нейродегенерации, такие, как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона или АБС (амиотрофический боковой склероз); нейродегенерацию, связанную с бактериальными или вирусными инфекциями; и такие болезни, как шизофрения, страх, депрессия и острая/хроническая боль, применение этих соединений при приготовлении соответствующих лекарственных препаратов, способ получения этих новых соединений и их содержащих лекарственных препаратов.
В общем смысле, но не конкретно, соединения формулы 1 и их соли являются известными соединениями, описанными в WO 95/25721. Сказано, что они проявляют активность в отношении глутаматного рецептора или АМРА рецептора, их используют для лечения заболеваний, связанных с этими рецепторами. К тому же, подобные соединения, в которых пиперидиновое кольцо замещено гидроксильной группой в 4-м положении, описаны в ЕР 824098. Указано, что эти соединения обладают активностью по отношению к NMDA рецептору и эффективны при лечении активных форм нейродегенерации, вызванной, например, параличом или травмой головного мозга, и хронических форм нейродегенерации, таких, как болезнь Альцгеймера, болезнь Паркинсона, АБС (амиотрофический боковой склероз); нейродегенерации, связанной с бактериальными и вирусными инфекциями; острой/хронической боли.
Из ЕР 824098 известно, что эти соединения являются хорошими специфическими блокаторами подтипа NMDA рецептора с высоким сродством к содержащейся в рецепторах NR2B субъединице и с низким сродством к содержащейся в рецепторах NR2A субъединице.
Активность в отношении α-адренергических рецепторов также низка, и при аудиогенных приступах у мышей эти соединения проявляют активность in vivo при низких соотношениях мг/кг. Важно отметить, что эти соединения проявляли нейропротектную активность на модели животного, пораженного параличом, а именно, при перманентной окклюзии средней артерии мозга. Однако изучение кардиотоксичности in vitro и in vivo показало, что эти соединения склонны пролонгировать потенциал действия сердца in vitro и, следовательно, "Q-T" интервал in vivo, так что они возможно несут ответственность за проявление сердечной аритмии. Способность таких соединений пролонгировать потенциал действия сердца объясняли тем, что они влияют на калиевый канал hERG типа, который очень важен для реполяризации потенциала действия у человека и существ других видов; большинство известных соединений, пролонгирующих "Q-T" интервал у человека, активны за счет блокирования этого канала. Таким образом, известные ранее соединения блокируют рекомбинантые ERG калиевые каналы человека гетерологически.
Установлено, что следующие соединения формулы I:
4-[2-(4-бензилпиперидин-1-ил)этансульфонил]фенол (1),
4-[2-(4-п-толилоксипиперидин-1-ил)этансульфонил]фенол (2),
(-)-(3R,4R)- или (3S,4S)-4-бензил-1-[2-(4-гидроксибензолсульфонил)этил]пиперидин-3-ол (3),
(+)-(3S,4S)- или (3R,4R)-4-бензил-1-[2-(4-гидроксибензолсульфонил)этил]пиперидин-3-ол (4),
(3RS,4RS)-4-бензил-l-[2-(4-гидроксибензолсульфонил)этил]пиперидин -3-ол (5),
(-)-(3R,4R)- или (3S,4S)-1-[2-(4-гидроксибензолсульфонил)этил]-4-(4-метилбензил)пиперидин-3-ол (6),
(+)-(3R,4R)- или (3S,4S)-1-[2-(4-гидроксибензолсульфонил)этил]-4-(4метилбензил)пиперидин-3-ол (7) и
(3RS,4RS)-1-[2-(4-гидроксибензолсульфонил)этил]-4-(4-метилбензил)-пиперидин-3-ол (8)
представляют собой селективные антагонисты подтипа NR2B NMDA рецепторов, и несмотря на то, что они являются частью известных в данной области техники соединений, обладающих свойствами селективного блокирования этого высоко специфического подтипа, например, 1-[2-(4-гидроксифенокси)этил]-4-(4-метилбензил)пиперидин-4-ол (9), и проявляют in vivo функции нейропротекторов, они менее активны как блокаторы hERG калиевых каналов, и поэтому с меньшей вероятностью демонстрируют проаритмическую активность у человека.
В таблице проиллюстрирована высокая селективность соединений по настоящему изобретению.
Новые соединения формулы I и их фармацевтически приемлемые соли могут быть получены по методам, которые в данной области техники известны, например, по описанным ниже методам, которые включают:
а) реакцию соединения формулы
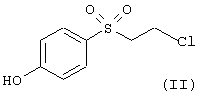
с соединением формулы
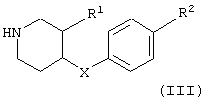
с получением соединения формулы
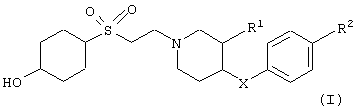
в которых заместители имеют значения, указанные выше, и, если необходимо,
б) превращение полученного соединения формулы I в фармацевтически приемлемые кислотно-аддитивные соли и, если необходимо,
в) превращение рацемической смеси в ее энантиомерный компонент с получением таким образом оптически чистых соединений.
В соответствии с вариантом а) способа 4-(2-хлорэтансульфонил)фенол растворяют в метиленхлориде и добавляют соединение формулы III, например 4-п-толилоксипиперидин, 4-бензилпиперидин, (3R,4R)- или (3S,4S)-4-бензил-пиперидин-3-ол, (3R,4R)- или (3S,4S)-4-(4-метилбензил)пиперидин-3-ол, и в присутствии триэтиламина или избытка пиперидина раствор перемешивают в течение нескольких часов при комнатной температуре. Реакционную смесь очищают хроматографией на силикагеле.
Для применения с фармацевтическими целями особенно хорошо подходят кислотно-аддитивные соли соединений формулы I.
Получение соединений формулы I и соединений формул XIII, XIV и VIII, которые являются промежуточными продуктами, иллюстрируют следующие схемы с 1 по 3. Исходные материалы формул V и XV либо представляют собой известные соединения, либо соединения, которые могут быть получены по известным методам.
Схема 1
Синтез сульфоновых производных и их солей
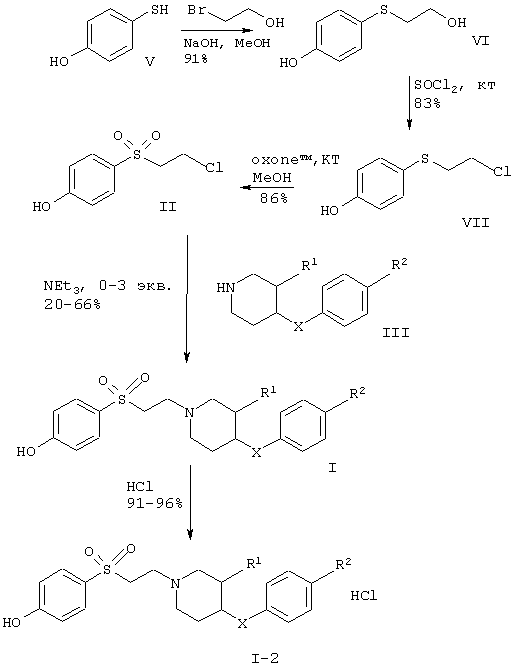
Схема 2
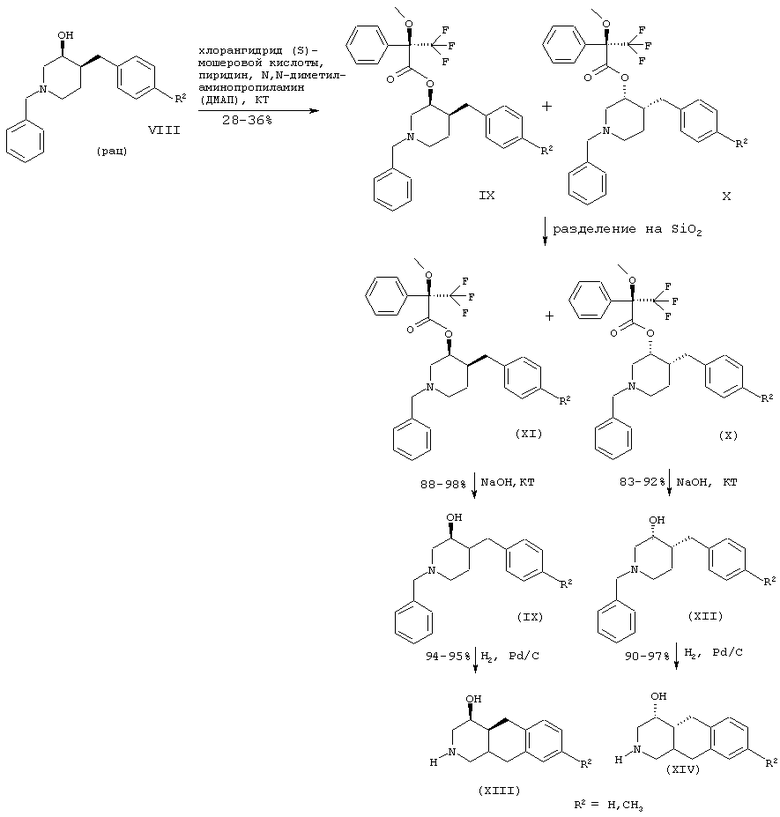
Синтез энантиомерно чистых 3-гидроксибензилпиперидинов
Схема 3
Синтез гидроксибензилпиперидиновых производных
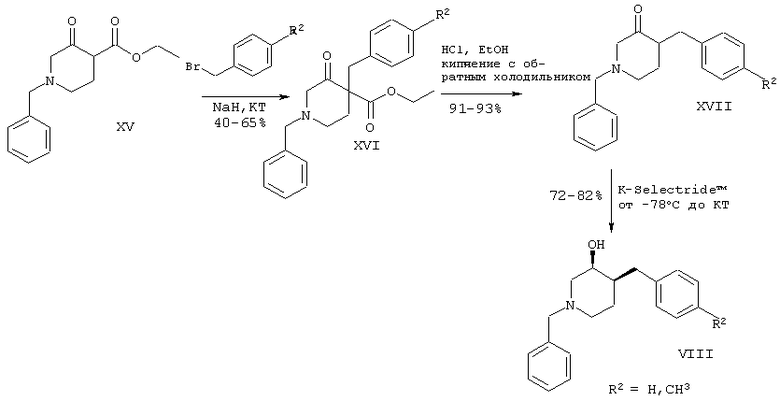
Подробное описание вышеупомянутых способов представлено в примерах с 1 по 31.
Как сказано выше, соединения формулы I и их фармацевтически приемлемые кислотно-аддитивные соли обладают ценными фармакодинамическими свойствами. Они являются селективными блокаторами подтипа NMDA-рецептора, который играет ключевую роль в модуляции нейронной активности и пластичности, что делает их ключевыми плеерами в процессах медиации, лежащих в основе развития ЦНС, а также в формировании и процессах усвоения и памяти.
Эти соединения исследовали в соответствии с приведенным ниже тестом.
Метод 1
Связывание [3H]-Ro 25-6981 (Ro 25-6981 представляет собой [R-(R*,S*)]-α-(4-гидроксифенил)-β-метил-4-(фенилметил)-1-пиперидинпропанол)
Использовали самцов белых крыс линии Fullinsdorf массой от 150 до 200 г каждый. Мембраны готовили гомогенизацией всего головного мозга (кроме мозжечка и мозгового вещества продолговатого мозга) с помощью гомогенизатора Polytron (10000 об/мин, 30 с) в 25 объемах холодного 50 мМ Трис-НСl буфера, содержавшего 10 мМ EDTA, с рН 7,1. Гомогенат центрифугировали при 48000 г в течение 10 мин и при 4°С. Осадок повторно суспендировали, используя Polytron, в том же объеме буфера и гомогенат инкубировали при 37°С в течение 10 мин. После повторного центрифугирования осадок гомогенизировали в том же буфере, замораживали при -80°С и, хранили в течение по крайней мере 16 ч, но не более 10 дней. Для опытов по изучению связывания гомогенат размораживали при 37°С, центрифугировали и осадок промывали 3 раза, как описано выше, охлажденным 5 мМ Трис-НСl буфером с рН 7,4. Полученный осадок вновь суспендировали в том же самом буфере и использовали при конечной концентрации 200 мкг белка/мл.
Опыты по связыванию [3H]-Ro 25-6981 проводили, применяя 50 мМ Трис-НСl буфер с рН 7,4. В опытах по замещению использовали 5 нМ [3H]-Ro 25-6981, а неспецифическое связывание определяли, используя 10 мкМ тетрагидроизохинолин; обычно его количество составляло 10% от суммарного. Время инкубации было равным 2 ч при 40°С, опыт останавливали фильтрацией на стеклянном волокнистом фильтре Whatmann CF/B (Unifilter-96, фирма Packard, Цюрих, Швейцария). Фильтры 5 раз промывали холодным буфером. После добавления 40 мл микросцина 40 (фирма Canberra Packard S.A., Цюрих, Швейцария) с помощью сцинцилляционного счетчика Packard Top-count microplate измеряли радиоактивность фильтра.
Действие соединений определяли, используя минимум 8 концентраций (в каждом случае по крайней мере с одним повтором). Нормализованные значения пула анализировали, используя программу расчета нелинейной регрессии, которая позволяла определять значения ИК50 с верхней и нижней границами доверительного интервала 95% (RS1, BBN, США).
Метод 2
Связывание 3Н-празозина
Использовали самцов белых крыс линии Fullinsdorf массой от 150 до 200 г каждый. Мембраны готовили гомогенизацией всего головного мозга (кроме мозжечка и мозгового вешества продолговатого мозга) с помощью гомогенизатора Polytron (10000 об/мин, 30 с) в 25 объемах холодного 50 мМ Трис-HCl буфера, содержавшего 10 мМ EDTA с рН 7,1. Гомогенат центрифугировали при 48000 г в течение 10 мин и при 4°С. Осадок повторно суспендировали, используя Polytron, в том же объеме буфера и гомогенат инкубировали при 37°С в течение 10 мин. После повторного центрифугирования осадок гомогенизировали в том же буфере, замораживали при -80°С и хранили в течение по крайней мере 16 ч, но не более 10 дней. Для опытов по связыванию гомогенат размораживали при 37°С, центрифугировали и осадок промывали 3 раза, как описано выше, холодным 5 мМ Трис-НСl буфером с рН 7,4. Полученный осадок вновь суспендировали в том же самом буфере и использовали при конечной концентрации 200 мкг белка/мл.
Опыты по связыванию 3Н-празозина проводили, применяя 50 мМ Трис-НСl буфер с рН 7,4. В опытах по замещению использовали 0,2 нМ 3Н-празозин, а неспецифическое связывание определяли, используя 100 мкМ хлорпромазин. Время инкубации составляло 30 мин при комнатной температуре; опыт останавливали фильтрацией на стеклянном волокнистом фильтре Whatmann CF/B (Unifilter-96, фирма Canberra Packard S.A., Цюрих, Швейцария). Фильтры 5 раз промывали холодным буфером. После добавления 40 мл микросцина 40 (фирма Canberra Packard S.A., Цюрих, Швейцария) с помощью сцинцилляционного счетчика Packard Top-count microplate измеряли радиоактивность фильтра. Действие исследуемых соединений определяли, используя минимум 8 концентраций (в каждом случае по крайней мере с одним повтором). Нормализованные значения пула анализировали, используя программу расчета нелинейной регрессии, которая позволяла определить значение ИК50 с верхней и нижней границами доверительного интервала 95% (RS1, BBN, США).
Определенная таким образом активность соединений в соответствии с изобретением примеров с 1 по 3 и 6, представленная в приведенной выше таблице, находится в интервале от 0,011 до 0,024 (в мкМ).
Метод 3
Изучение ингибирования hERG каналов
СНО клетки подвергали трансфекции экспрессирующим вектором pcDNA3-hERG, который содержал для селекции SV40-нео кассету. Клетки высевали тонким слоем на 35-миллиметровые чашки и использовали для электрофизиологических опытов по прошествии от 1/2 до 3 дней.
Во время опытов клетки постоянно обрабатывали экстрацеллюлярным физиологическим раствором, содержавшим (в мМ): 150 NaCl, 10 KCl, 1 MgCl2, 3 CaCl2, 10 HEPES (рН до 7,3 доводили добавлением NaOH). 10 мМ запасный раствор тестируемого соединения готовили в чистом ДМСО. Тестируемый раствор готовили по крайней мере 1000-кратным разбавлением запасного раствора в экстрацеллюлярном физиологическом растворе. Стеклянные микропипетки для регистрации бляшка-зажим целых клеток были наполнены cледующим содержимым (в мМ): 110 KCl, 10 BARTA, 10 HEPES, 4,5 MgCl2, 4 Na2ATP, 20 Nа2-фосфокреатин, а также 200 мкг/мл креатинкиназы (рН доводили до 7,3 прибавлением КОН).
В экспериментах использовали конфигурацию целой клетки техники бляшка-зажим. Клетки прикрепляли к устройству, поддерживавшему потенциал -80 мV, и многократно стимулировали (0,1 Гц) импульсом напряжения, состоявшим из 1-s деполимеризации кондиционирования до 20 мV, за которой немедленно следовала гиперполяризация в течение 50 мс до -120 мV. Ток мембраны записывали в течение по крайней мере 3 мин (18 стимуляций) перед введением соединения (контрольный опыт) и затем в течение двух других интервалов по 3 мин в присутствии исследумого соединения в двух различных концентрациях. Для расчета эффективности действия соединения, выраженной в процентах, амплитуду тока (Iтест) полученную для каждого соединения к концу применяемого интервала, делили на среднее значение амплитуды тока (Iконтрольный), полученной в первоначальный контрольный период:
эффективность (%) (1-Iтест/Iконтрольный)·100.
Концентрацию соединений подбирали ее десятикратными изменениями (обычно 1 или 10 мкМ) вблизи ожидаемой 50%-ной ингибирующей концентрации (ИК50). Если после первого опыта оказывалось, что ИК50 не находилась в интервале между двумя выбранными концентрациями, концентрацию изменяли так, чтобы в следующих опытах она соответствовала ИК50. Соединения испытывали по крайней мере на трех клетках. Затем по совокупности всех значений процент-эффекта с помощью нелинейной регрессии, используя функцию эффект = 100/(1-ИК50/концентрацияHill), устанавливали значение их ИК50.
Концентрации выше 10 мкМ не тестировали. Если эффект соединения в концентрации 10 мкМ оказывался меньше 50%, значение ИК50 выражали как ">10 мкМ" и соединение характеризовали средним эффектом, который наблюдали при 10 мкМ.
Соединения формулы I и их соли, как они представлены в настоящем описании, вместе с фармацевтически инертными наполнителями можно вводить в стандартные фармацевтические дозированные препаративные формы, например для перорального или парентерального применения, с обычными фармацевтическими адъювантами, в частности с органическими или неорганическими инертными носителями, такими, как вода, желатина, лактоза, крахмал, стеарат магния, тальк, растительные масла, камеди, полиалкиленгликоли и т.п. Примерами фармацевтических препаратов в твердой форме служат таблетки, суппозитории, капсулы, а в жидкой форме - растворы, суспензии и эмульсии. К фармацевтическим адъювантам относятся консерванты, стабилизаторы. смачивающие агенты и эмульгаторы, соли для изменения осмотического давления или для выполнения функций буферов. Фармацевтические препараты могут также содержать другие терапевтически активные вещества.
Ежедневную дозу предназначенных для введения в организм соединений формулы I варьируют в зависимости от конкретно применяемого соединения, выбранного пути введения и пациента. Обычным методом введения в организм соединений формулы I является путь перорального и парентерального типов введения. В предпочтительном варианте композицию на основе соединения формулы 1 взрослым пациентам перорально вводят в дозе, находящейся в интервале от 1 до 1000 мг/день. В предпочтительном варианте композицию на основе соединения формулы I взрослым пациентам парентерально вводят в дозе, находящейся в интервале от 5 до 500 мг/день.
Сущность изобретения далее иллюстрируют следующие примеры.
Пример 1
4-[2-(4-бензилпиперидин-1-ил)этансульфонил]фенол
В раствор 40,0 г (181 ммоль) 4-(2-хлорэтансульфонил)фенола в 600 мл СН2Сl2 вводили 69,9 г (399 ммолей) 4-бензилпиперидина. После перемешивания в течение 16 ч при КТ реакционную смесь концентрировали до объема 100 мл и непосредственно очищали хроматографией на силикагеле (CH2Cl2/MеОН/NН3 в соотношении 19/1/0,1). Перекристаллизацией из этилацетата/гексана (2:1) получали 25 г (70 ммолей) продукта (выход: 38%). MS: m/e = 360,2 (М+Н+).
Гидрохлорид 4-[2-(4-бензилпиперидин-1-ил)этансульфонил] фенола (1:1)
В раствор 1,15 г (3,2 ммоля) 4-[2-(4-бензилпиперидин-1-ил)этансульфонил] фенола в 5 мл EtOH вводили 2,6 мл 1,46 М (3,8 ммоля) НСl. Реакционную смесь охлаждали до температуры от 0 до 5°С и перемешивали в течение 10 мин. Далее диэтиловый эфир добавляли до осаждения продукта. После фильтрования в виде белого твердого вещества получали 1,14 г (2,9 ммоля) продукта (выход: 91%).
MS: m/e = 360,2 (М+Н+).
В общем аналогично примеру 1 получали соединения примеров со 2 по 8.
Пример 2
4-[2-(4-п-толилоксипиперидин-1-ил)этансульфонил]фенол
Указанное в заглавии соединение в виде белого твердого вещества получали из 4-(2-хлорэтансульфонил)фенола и 4-п-толилоксипиперидина (полученного в соответствии с J.Med.Chem., 1978, 309) с 59%-ным выходом продукта. MS: m/e = 376,4 (М+Н+).
Гидрохдорид 4-[2-(4-п-толилоксипиперидин-1-ил)этансульфонил]фенола
Указанное в заглавии соединение в виде белого твердого вещества получали из 4-[2-(4-п-толилоксипиперидин-1-ил)этансульфонил]-фенола с 96%-ным выходом продукта.
MS: m/e = 376,4 (M+H+).
Пример 3
(-)-(3R,4R)- или -(3S,4S)-4-бензил-1-[2-(4-гидроксибензолсульфонил)этил]пиперидин-3-ол
Указанное в заглавии соединение в виде белого твердого вещества получали из 4-(2-хлорэтансульфонил)фенола и (3R,4R)- или (3S,4S)-4-бензилпиперидин-3-ола с 66%-ным выходом продукта.
MS: m/e = 376,4 (М+Н+), [α]
Пример 4
(+)-(3S,4S)- или -(3R,4R)-4-бензил-1-[2-(4-гидроксибензолсульфонил)этил] пиперидин-3-ол
Указанное в заглавии соединение получали из 4-(2-хлорэтансульфонил)фенола и (3S,4S)- или (3R,4R)-4-бензилпиперидин-3-ола с 50%-ным выходом в виде белого твердого вещества.
MS: m/e = 376,4 (М+Н+), [α]
Пример 5
(3SR,4SR)-4-бензил-1-[2-(4-гидроксибензолсульфонил)этил]пиперидин-3-ол
Указанное в заглавии соединение получали из 4-(2-хлорэтансульфонил)фенола и (3SR,4SR)-4-бeнзилпипepидин-3-oлa с 20%-ным выходом в виде белой пены.
MS: m/e = 376,4(М+Н+).
Пример 6
(-)-(3R,4R)- или -(3S,4S)-1-[2-(4-гидроксибензолсульфонил)этил] -4-(4-метилбензил)пиперидин-3-ол
Указанное в заглавии соединение получали из 4-(2-хлорэтансульфонил)фенола и (3R,4R)- или (3S,4S)-4-(4-мeтилбeнзил)пипepидин-3-oлa с 51%-ным выходом в виде белой пены.
MS: m/e = 390,2 (М+Н+), [α]
Пример 7
(+)-(3S,4S)- или -(3R,4R)-1-[2-(4-гидроксибензолсульфонил)этил]-4-(4-метилбензил) пиперидин-3-ол
Указанное в заглавии соединение получали из 4-(2-хлорэтансульфонил)фенола и (3S,4S)- или (3R,4R)-4-(4-метилбензил)пиперидин-3-ола с 31%-ным выходом в виде белой пены.
MS: m/e = 390,3 (М+Н+), [α]
Пример 8
(3SR,4SR)-1-[2-(4-гидроксибензолсульфонил)этил]-4-(4-метилбензил)пиперидин-3-ол
Указанное в заглавии соединение получали из 4-(2-хлорэтансульфонил)фенола и (3SR,4SR )-4-(4-мeтилбeнзил)пипepидин-3-oлa с 30%-ным выходом в виде белого твердого вещества.
MS: m/e = 390,3 (М+Н+).
Получение промежуточных продуктов
Пример 9
(3S,4S)- или (3R,4R)-4-бензилпиперидин-3-ол
320 мг (1,1 ммоля) (3S,4S)- или (3R,4R)-1,4-дибензилпиперидин-3-ола растворяли в 10 мл этанола и гидрировали под атмосферным давлением при 50°С в течение 2 ч в присутствии 70 мг 10%-ного Pd на С. Реакционную смесь фильтровали и промывали этанолом с получением 205 мг (1,1 ммоля) продукта в виде белого твердого вещества (выход: 94%).
MS: m/e = 191 (М+Н+), [α]
В общем аналогично примеру 9 получали соединения примеров с 10 по 14.
Пример 10
(3R,4R) или (3S,4S)-4-бензилпиперидин-3-ол
Указанное в заглавии соединение с 97%-ным выходом в виде бесцветного масла получали из (3R,4R)- или (3S,4S)-1,4-дибeнзилпипepидин-3-oлa.
MS: m/e = 191 (М), [α]
Пример 11
(3SR,4SR)-4-бензилпиперидин-3-ол
Указанное в заглавии соединение с 88%-ным выходом в виде бесцветного масла получали из (3SR,4SR)-1,4-дибензилпиперидин-3-ола.
MS: m/e = 191 (М).
Пример 12
(3S,4S)- или (3R,4R)-4-(4-метилбензил)пиперидин-3-ол
Указанное в заглавии соединение с 95%-ным выходом в виде бесцветного масла получали из (3S,4S)- или (3R,4R)-1-бензил-4-(4-метилбензил)пиперидин-3-ола.
MS: m/e = 206,2 (М+Н+), [α]
Пример 13
(3R,4R)- или (3S,4S)-4-(4-мeтилбeнзил)пипepидин-3-oл
Указанное в заглавии соединение с 90%-ным выходом в виде бесцветного масла получали из (3R,4R)- или (3S,4S)-1-бензил-4-(4-метилбензил)пиперидин-3-ола.
MS: m/e = 206,2 (М+Н+), [α]
Пример 14
(3SR,4SR)-4-(4-мeтилбeнзил)пипepидин-3-oл
Указанное в заглавии соединение с количественным выходом в виде бесцветного масла получали из (3SR,4SR)-l-бензил-4-(4-метилбензил)пипеpи-дин-3-ола.
MS: m/e = 206,2 (M+H+).
Пример 15
(3S,4S)- или (3R,4R)-1,4-дибензилпиперидин-3-ол
В раствор 700 мг (1,4 ммоля) (3S,4S)-1,4-дибeнзилпипepидин-3-илoвoro эфира (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты или (3R,4R)-1,4-дибензилпиперидин-3-илового эфира (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты в 15 мл этанола при КТ вводили 7 мл 4н. (28 ммолей) NaOH. По прошествии 16 ч реакционную смесь выливали в смесь воды с CH2Cl2 в соотношении 1:1 и органический слой отделяли. Водную фазу дважды экстрагировали CH2Cl2 и объединенные органические слои промывали водой, сушили над MgSO4 и под пониженным давлением удаляли растворитель с получением в виде желтого твердого вещества 350 мг (12,4 ммоля) продукта (выход: 88%).
MS: m/e = 281 (М), [α]
В общем аналогично примеру 15 получали соединения примеров с 16 по 18.
Пример 16
(3R,4R)- или (3S,4S)-1,4-дибензилпиперидин-3-ол
Указанное в заглавии соединение в виде желтого твердого вещества получали с 83%-ным выходом из (3R,4R)-1,4-дибензилпиперидин-3-илового эфира (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты или (3S,4S)-1,4-дибензилпиперидин-3-илового эфира (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты.
MS: m/e = 281 (М), [α]
Пример 17
(3S,4S)- или (3R,4R)-1-бензил-4-(4-метилбензил)пиперидин-3-ол
Указанное в заглавии соединение в виде желтого масла получали с 98%-ным выходом из (3S,4S)-1-бензил-4-(4-метилбензил)пиперидин-3-илового эфира (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты или (3R,4R)-1-бензил-4-(4-метилбензил)пиперидин-3-илового эфира (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты.
MS: m/e = 296,4 (М+Н+), [α]
Пример 18
(3R,4R)- или (3S,4S)-1-бензил-4-(4-метилбензил)пиперидин-3-ол
Указанное в заглавии соединение в виде бесцветного масла получали с 92%-ным выходом из (3R,4R)-1-бензил-4-(4-метилбензил)пиперидин-3-илового эфира (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты или (3S,4S)-1-бензил-4-(4-метилбензил)пиперидин-3-илового эфира (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты.
MS: m/e = 296,4 (М+Н+), [α]
Пример 19
(3S,4S)-1,4-дибензилпиперидин-3-иловый эфир (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты или (3R,4R)-1,4-дибензилпиперидин-3-иловый эфир (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты
В раствор 1,50 г (53 ммоля) (3SR,4SR)-1,4-дибензилпиперидин-3-ола в 50 мл СН2Сl2 при 0°С вводили 0,515 мл (506 мг, 64 ммоля) пиридина, 912 мг (74,6 ммоля) диметиламинопиридина и 1,19 мл (1,62 г, 64 ммоля) (S)-(+)-альфа-метокси-альфа-трифторметилфенилацетилхлорида. Реакционную смесь перемешивали при КТ в течение 5 ч, реакцию гасили добавлением 50 мл воды и смесь перемешивали в течение 30 мин. Органическую фазу отделяли и дважды промывали 50 мл насыщенного раствора NаНСО3. Объединенные водные фазы экстрагировали СН2Сl2 и объединенные органические фазы сушили над MgSO4. Под пониженным давлением удаляли растворитель и сырой продукт очищали хроматографией на силикагеле (СН2Сl2/гексан/NН3 в соотношении 50/50/1) с получением в виде желтого масла 750 мг (15,1 ммоля) продукта (28%-ный выход).
MS: m/e = 498,2 (M+H+), [α]
В общем аналогично примеру 19 получали соединения примеров с 20 по 22.
Пример 20
(3R,4R)-1,4-дибензилпиперидин-3-иловый эфир (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты или (3S,4S)-1,4-дибензилпиперидин-3-иловый эфир (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты
Указанное в заглавии соединение получали в виде желтого масла с 29%-ным выходом из (3SR,4SR)-1,4-дибензилпиперидин-3-ола и (S)-(+)-альфа-метокси-альфа-трифторметилфенилацетилхлорида.
MS: m/e = 498,3 (М+Н+), [α]
Пример 21
(3S,4S)-1-бензил-4-(4-метилбензил)пиперидин-3-иловый эфир (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты иди (3R,4R)-1-бензил-4-(4-метилбензид)пиперидин-3-иловый эфир (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты
Указанное в заглавии соединение получали в виде желтого масла с 33%-ным выходом из (3SR,4SR)-4-(4-метилбензил)пиперидин-3-ола и (S)-(+)-альфа-метокси-альфа-трифторметилфенилацетилхлорида.
MS: m/e = 512,3 (М+Н+), [α]
Пример 22
(3R,4R)-1-бензил-4-(4-метилбензил)пиперидин-3-иловый эфир (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты или (3S,4S)-1-бензил-4-(4-метилбензил)пиперидин-3-иловый эфир (R)-3,3,3-трифтор-2-метокси-2-фенилпропионовой кислоты
Указанное в заглавии соединение получали в виде желтого масла с 36%-ным выходом из (3SR,4SR)-4-(4-метилбензил)пиперидин-3-ола и (S)-(+)-альфа-метокси-альфа-трифторметилфенилацетилхлорида.
MS: m/e = 512,4 (М+Н+), [α]
Пример 23
(3SR,4SR)-1,4-дибензилпиперидин-3-ол
В раствор 9,0 г (32 ммоля) (SR)-1,4-дибензилпиперидин-3-она в 200 мл сухого ТГФ при -78°С по каплям вводили 48 мл (48 ммолей) продукта К-selectride® (1н. в ТГФ). Реакционную смесь перемешивали в течение 1 ч при -70°С, а затем нагревали до КТ. Реакцию гасили добавлением 100 мл раствора NаНСО3 и водную фазу дважды экстрагировали 200 мл этилацетата. Объединенные органические фазы промывали 100 мл воды и 100 мл рассола. Органическую фазу сушили над MgSO4, фильтровали и под пониженным давлением удаляли растворитель с получением сырого продукта. Очисткой хроматографией (этилацетат/гексан в соотношениях от 1/2 до 2/1) в виде желтого масла получали 6,5 г (23 ммоля) продукта (72%-ный выход).
MS: m/e = 281 (М).
В общем аналогично примеру 23 получали соединение примера 24.
Пример 24
(3SR,4SR)-1-бензил-4-(4-метилбензил)пиперидин-3-ол
Указанное в заглавии соединение в виде оранжевого масла получали с 82%-ным выходом из (SR)-1-бензил-4-(4-метилбензил)пиперидин-3-она.
MS: m/e = 296,4 (М+Н+).
Пример 25
(RS)-1,4-дибензилпиперидин-3-он
В раствор 13,5 г (38,4 ммоля) этилового эфира (SR)-1,4-дибензил-3-оксопиперидин-4-карбоновой кислоты в 20 мл этанола вводили 47,5 мл 37%-ного НСl и желтый раствор кипятили с обратным холодильником в течение 48 ч. Реакционную смесь охлаждали до 0°С и добавляли NaOH до достижения рН 8. Водную фазу три раза экстрагировали 200 мл этилацетата и объединенные органические фазы промывали двумя порциями по 100 мл воды и двумя порциями по 100 мл рассола. Органическую фазу сушили над MgSO4, фильтровали и под пониженным давлением удаляли растворитель с получением в виде коричневого масла 9,8 г (35 ммолей) продукта (91%-ный выход).
MS: m/e = 279 (М).
В общем аналогично примеру 25 получали соединение примера 26.
Пример 26
(SR)-1-бензил-4-(4-метилбензил)пиперидин-3-он
Указанное в заглавии соединение в виде коричневого масла получали с 77%-ным выходом из этилового эфира (SR)-1-бензил-4-(4-метилбензил)-3-оксопиперидин-4-карбоновой кислоты.
MS: m/e = 294 (М+Н+).
Пример 27
Этиловый эфир (SR)-1,4-дибензил-3-оксопиперидин-4-карбоновой кислоты
В 55%-ную суспензию 30,9 г NaH (772 ммоля) в 1000 мл ДМФ в аргоновой атмосфере при температуре от 0 до 5°С порциями вводили 115 г (386 ммолей) технически доступного гидрохлорида этил-(SR)-N-бензил-3-оксо-4-пиперидинкарбоксилата. При КТ реакционную смесь перемешивали в течение 1 ч и при 0°С добавляли раствор 45,9 мл (66,0 г, 386 ммолей) бензилбромида в 200 мл ДМФ. При КТ реакционную смесь перемешивали в течение 1,5 ч и при температуре от 0 до 10°С добавляли 200 мл насыщенного раствора NаНСО3. Объем реакционной смеси уменьшали до 500 мл и добавляли 1000 мл воды. Водную фазу три раза экстрагировали 1000 мл этилацетата и объединенные органические фазы промывали 3 порциями по 200 мл воды и 3 порциями по 200 мл рассола. Органическую фазу сушили над MgSO4, фильтровали и под пониженным давлением удаляли растворитель. Сырой продукт очищали хроматографией на силикагеле (этилацетат/гексан в соотношении 1/8, затем 1/4) с получением в виде коричневого масла 101 г (290 ммолей) продукта (75%-ный выход).
MS: m/e = 352,4 (М+Н+).
В общем аналогично примеру 27 получали соединение примера 28.
Пример 28
Этиловый эфир (SR)-1-бензил-4-(4-метилбензил)-3-оксопиперидин-4-карбоновой кислоты
Указанное в заглавии соединение в виде коричневого масла получали с 73%-ным выходом из (SR)-N-бензил-3-оксо-4-пиперидинкарбоксилата.
MS: m/e = 366,4 (М+Н+).
Пример 29
4-(2-хдорэтансульфонил)фенол
В раствор 4,6 г (24,4 ммоля) 4-(2-хлорэтилсульфанил)фенола в 100 мл МеОН при КТ вводили 22,5 г (36,6 ммоля) продукта oxone®. При КТ реакционную смесь перемешивали в течение 16 ч, фильтровали и твердое вещество промывали МеОН. Фильтрат концентрировали под пониженным давлением, растворяли в этилацетате и дважды промывали водой. Объединенные водные фазы дважды экстрагировали этилацетатом. Объединенные органические слои сушили над MgSO4 и под пониженным давлением удаляли растворитель. Сырой продукт очищали хроматографией на силикагеле (этилацетат/гексан в соотношении 1/3) с получением в виде белого твердого вещества 4,6 г (20,9 30 ммоля) продукта (86%-ный выход).
MS: m/e = 220 (М).
Пример 30
4-(2-хлорэтилсульфанил)фенол
В раствор 5,0 г (29 ммолей) 4-(2-гидроксиэтилсульфанил)фенола в 100 мл СН2Сl2 при 0°С вводили 2,6 мл (32,3 ммоля) пиридина и 2,34 мл (32,3 ммоля) SOCl2, растворенного в 10 мл CH2Cl2. Реакционную смесь перемешивали при КТ в течение 1 ч, а затем реакцию гасили добавлением воды. Органическую фазу отделяли и дважды промывали насыщенным раствором NaHCO3. Объединенные водные фазы дважды экстрагировали CH2Cl2, объединенные органические слои сушили над MgSO4 и растворитель удаляли под пониженным давлением с получением в виде желтого масла 4,6 г (24,3 ммоля) продукта (83%-ный выход).
MS: m/e = 188 (М).
Пример 31
4-(2-гидроксиэтидсульфанил)фенол
В раствор 10,9 г (87 ммолей) 4-гидрокситиофенола в 200 мл МеОН при температуре от 0 до 5°С вводили 87 мл (87 ммолей) 1н. NaOH. После перемешивания реакционной смеси в течение 10 мин добавляли 6,1 мл (86 ммолей) бромэтанола, растворенного в 100 мл МеОН. При КТ реакционную смесь перемешивали в течение 3 ч и под пониженным давлением частично удаляли метанол. Остаток выливали в смесь этилацетата с насыщенным раствором NаНСО3 в соотношении 1:1 и органическую фазу выделяли, сушили над MgSO4, фильтровали и под пониженным давлением удаляли растворитель. Остаток очищали хроматографией на силикагеле (этилацетат/гексан в соотношении от 3/2 до 2/1) с получением в виде белого твердого вещества 13,4 г (78,7 ммоля) продукта (91%-ный выход). MS: m/e = 170 (М).
Пример А
Приготовление таблеток (мокрое гранулирование)

Метод приготовления
1. Смешать компоненты 1, 2, 3 и 4 и гранулировать с использованием очищенной воды.
2. Высушить гранулят при 50°С.
3. Пропустить гранулят через подходящее измельчительное устройство.
4. Добавить компонент 5 и перемешивать в течение трех минут; прессовать в соответствующем прессе.
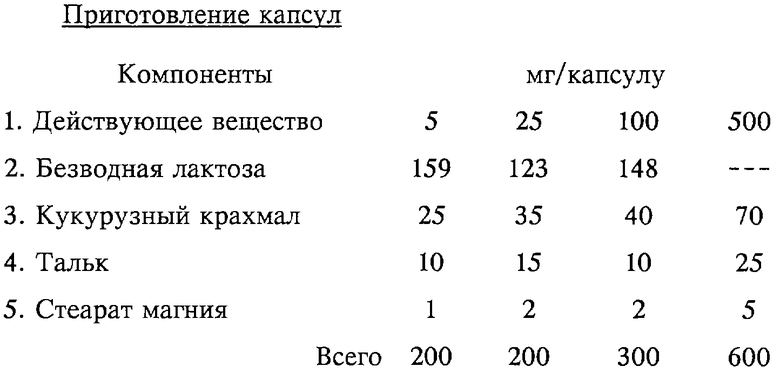
Метод приготовления
1. Перемешивать компоненты 1, 2 и 3 в подходящем смесителе в течение 30 мин.
2. Добавить компоненты 4 и 5 и перемешивать в течение 3 мин.
3. Заполнить соответствующую капсулу.

Метод приготовления
1. Смешать компоненты 1, 2, 3 и 4 и гранулировать с использованием очищенной воды.
2. Высушить гранулят при 50°С.
3. Пропустить гранулят через подходящее измельчительное устройство.
4. Добавить компонент 5 и перемешивать в течение трех минут; прессовать в соответствующем прессе.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2010 |
|
RU2562605C2 |
| ФТОР- И ТРИФТОРАЛКИЛСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ ИНГИБИТОРЫ ОБРАЗОВАНИЯ БЕТА-АМИЛОИДА И ИХ ПРОИЗВОДНЫЕ | 2004 |
|
RU2342374C2 |
| ИМИДАЗОПИРРОЛОПИРАЗИНОВЫЕ ПРОИЗВОДНЫЕ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ АНОМАЛЬНОЙ АКТИВНОСТЬЮ ПРОТЕИНКИНАЗ Jak1, Jak3 ИЛИ Syk | 2010 |
|
RU2570416C2 |
| Имидазопирролопиразиновые производные, полезные для лечения заболеваний, вызванных аномальной активностью протеинкиназ Jak1, Jak3 или Syk | 2010 |
|
RU2711869C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА NK-3 | 2010 |
|
RU2561271C2 |
| СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ РВОТЫ У МЛЕКОПИТАЮЩИХ И ЧЕЛОВЕКА С ИСПОЛЬЗОВАНИЕМ НЕКОТОРЫХ ХИНУКЛИДИНОВЫХ ПИПЕРИДИНОВЫХ, АЗАНОРБОРНАНОВЫХ, ЭТИЛЕНДИАМИНОВЫХ ПРОИЗВОДНЫХ И РОДСТВЕННЫХ ИМ СОЕДИНЕНИЙ | 1994 |
|
RU2135179C1 |
| ТРИАЗОЛОПИРИДИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ КИНАЗЫ PIM | 2012 |
|
RU2598846C2 |
| 2-ОКСА-5-АЗАБИЦИКЛО[2.2.1]ГЕПТАН-3-ИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2015 |
|
RU2697651C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
| НОВЫЕ АМИДНЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНКАРБОНОВОЙ КИСЛОТЫ | 2006 |
|
RU2410374C2 |
Изобретение относится к производным этансульфонилпиперидина формулы (I)
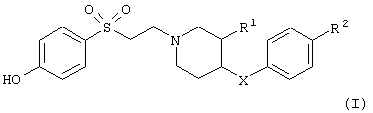
или их фармацевтически приемлемым кислотно-аддитивным солям, где R1 означает водородный атом или гидроксил; R2 означает водородный атом или метил, Х означает –О- или -СН2-. Указанные соединения обладают хорошим сродством к NMDA рецептору и могут быть использованы для лечения заболеваний, вызванных повышенной активностью NR2B подтипов NMDA рецепторов. 2 н. и 8 з.п. ф-лы, 1 табл.
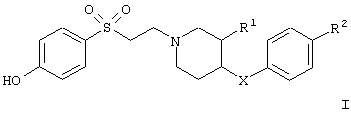
в которой R1 обозначает водородный атом или гидроксил;
R2 обозначает водородный атом или метил;
Х обозначает -О- или -СН2-;
и их фармацевтически приемлемые кислотно-аддитивные соли.
4-[2-(4-бензилпиперидин-1-ил)этансульфонил]фенол.
4-[2-(4-п-толилоксипиперидин-1-ил)этансульфонил]фенол.
(-)-(3R,4R)- или -(3S,4S)-4-бензил-1-[2-(4-гидроксибензолсульфонил)этил]пиперидин-3-ол.
(+)-(3S,4S)- или -(3R,4R)-4-бензил-1-[2-(4-гидроксибензолсульфонил)этил]пиперидин-3-ол.
(3RS,4RS)-4-бензил-1-[2-(4-гидроксибензолсульфонил)этил]пиперидин-3-ол.
(-)-(3R,4R)- или -(3S,4S)-1-[2-(4-гидpoкcибeнзoлcyльфoнил)этил]-4(4-метилбензил)пиперидин-3-ол.
(+)-(3R,4R)- или -(3S,4S)-1-[2-(4-гидроксибензолсульфонил)этил]-4-(4-этилбензил)пиперидин-3-ол.
(3RS,4RS)-1-[2-(4-гидроксибензолсульфонил)этил]-4-(4-метилбензил)пиперидин-3-ол.
| ПРОИЗВОДНЫЕ 2-(4-ГИДРОКСИПИПЕРИДИНО)-1-АЛКАНОЛА И ПРОИЗВОДНЫЕ 2-(4-ГИДРОКСИПИПЕРИДИНО)-1-АЛКАНОНА | 1992 |
|
RU2065859C1 |

