Настоящее изобретение касается циклических производных, содержащих псевдосимметрические имидазольной цис-амидной связи, которые селективно связываются подтипами рецепторов соматостатина. Данное изобретение касается также способов получения соединений данного изобретения.
Соматостатин (SRIF) представляет собой циклический тетрадекапептидный гормон, содержащий дисульфидный мостик между положениями 3 и 14 и обладающий свойствами ингибирования выделения гормона роста (GH) и тиреотропного гормона (TSH), ингибирования выделения инсулина и глюкагона и уменьшения секреции желудка. Метаболизм соматостатина посредством аминопептидаз и карбоксипептидаз приводит к малой продолжительности действия.
Соматостатин связывается пятью определенными подтипами рецепторов (SSTR) при относительно высоком сродстве к каждому подтипу. Меньшие, более жесткие аналоги настоящего изобретения демонстрируют высокую селективность относительно некоторых подтипов рецепторов. Присоединение к различным типам рецепторов соматостатина связано с лечением нижеследующих состояний и/или заболеваний. Активация типа 2 и типа 5 связана с подавлением гормона роста, более конкретно, с аденомами, выделяющими GH (акромегалии), и аденомами, выделяющими TSH. Активация типа 2, но не типа 5, связана с лечением аденом, выделяющих пролактин. Другими показаниями, связанными с активацией подтипов рецепторов соматостатина, являются рестеноз, ингибирование инсулина и/или глюкагона и, более конкретно, сахарный диабет, гиперлипидемия, невосприимчивость к инсулину, синдром X, ангиопатия, пролиферативная ретинопатия, феномен "утренней зари" и нефропатия; ингибирование желудочного выделения кислоты и, более конкретно, пептическая язва, наружные свищи кишечника и поджелудочной железы, слизистый колит, синдром Дампинга (Dumping), синдром водянистого стула, диарея вследствие СПИДа, диарея, вызванная химиотерапией, острый или хронический панкреатит и желудочно-кишечные опухоли, выделяющие гормон; лечение рака, такого как гепатома; ингибирование ангиогенеза, лечение воспалительных заболеваний, таких как артрит; хроническое отторжение аллотрансплантатов; ангиопластика; профилактика кровотечения трансплантатов сосудов и желудочно-кишечного кровотечения. Агонисты соматостатина можно также применять для снижения веса пациента.
Агонисты соматостатина раскрыты также как препараты, полезные для ингибирования пролиферации helicobacter pylori.
Краткое содержание изобретения
Один аспект данного изобретения обеспечивает соединение формулы (I)
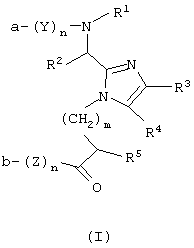
или его фармацевтически приемлемую соль, где
Y и Z каждый для каждого случая независимо представляет собой D- или L-природную или неприродную α-аминокислоту;
n для каждого случая независимо равно от 0 до 50, при условии, что оба n одновременно не могут быть равными 0;
m равно 0 или целому числу от 1 до 10;
а представляет собой Н или R1;
b представляет собой ОН, -OR1 или -NR9R9;
или а вместе с b образуют амидную связь;
R1 независимо представляет собой Н, (С1-С4) алкил или арил-(C1-C4) алкил;
R2 представляет собой Н или необязательно замещенный фрагмент, выбранный из группы, состоящей из (C1-C4) алкила, фенила, фенил-(C1-C4) алкила и гетероциклил-(C1-C4) алкила, причем этот необязательно замещенный фрагмент необязательно замещен одним заместителем или более, каждый из которых независимо выбирают из группы, состоящей из (C1-C4) алкила, (С3-С8)циклоалкила, -O-R6, -S(O)q-R7, -N(R9R9), -NHCO-R6, -NHSO2R9, -CO2R9, -CONR9R9 и -SO2NR9R9, где q равно 0, 1, 2 или 3;
R3 и R4 каждый независимо представляет собой Н, галоген или необязательно замещенный фрагмент, выбранный из группы, состоящей из (C1-C4) алкила, (С3-C8) циклоалкила, арила и арил-(C1-C4)алкила; причем этот необязательно замещенный фрагмент необязательно замещен одним заместителем или более, выбранными из группы, состоящей из ОН, (C1-C4) алкила, (C1-C4)алкокси, арилокси, арил-(C1-C4) алкокси, -NR9R9, -COOH, -CONR9R9 и галогена; или
R3 и R4, взятые вместе с углеродами, к которым они присоединены, образуют необязательно замещенный арил, причем этот арил необязательно замещен одним заместителем или более, каждый из которых независимо выбран из группы, состоящей из ОН, (C1-C4) алкила, (C1-C4) алкокси, арилокси, арил-(C1-C4) алкокси, -NR9R9, -COOR5, -CONR9R9 и галогена;
R5 для каждого случая независимо представляет собой Н или необязательно замещенный фрагмент, выбранный из группы, состоящей из (C1-C4) алкила и арил-(C1-C4) алкила, причем этот необязательно замещенный фрагмент необязательно замещен одним заместителем или более, каждый из которых независимо выбран из группы, состоящей из (C1-С4) алкила, ОН, (C1-C4) алкокси, арилокси, NO2, арил-(C1-C4) алкокси, -NR9R9, -COOH, -CONR9R9 и галогена;
R6 для каждого случая независимо выбирают из группы, состоящей из Н, (C1-C4) алкила, (C1-C4) алкокси, арил-(C1-C4) алкила и арил-(C1-C4) алкокси;
R7 представляет собой Н, если q равно 3, или R7 для каждого случая независимо выбирают из группы, состоящей из (C1-C4) алкила, арила или арил-(C1-C4) алкила, если q равно 0, 1 или 2; и
R9 для каждого случая независимо выбирают из группы, состоящей из Н, NO2, (C1-C4) алкила, арила и арил-(C1-C4) алкила.
Предпочтительным соединением формулы (I) является Н-Trp-D-Trp-Lys-Abu-Phe ψ (4-(3-метоксифенил)имидазол)-Gly-OH.
Другой аспект настоящего изобретения обеспечивает соединение формулы (II)
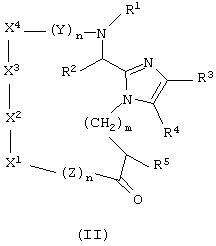
или его фармацевтически приемлемую соль, где
Y и Z каждый для каждого случая независимо представляет собой D- или L-природную или неприродную α-аминокислоту;
m равно 0 или целому числу от 1 до 10;
n для каждого случая независимо равно от 0 до 6;
R1 для каждого случая независимо представляет собой Н, (C1-C4)алкил или арил-(C1-C4) алкил;
R2 представляет собой Н или необязательно замещенный фрагмент, выбранный из группы, состоящей из (C1-C4) алкила, фенила, фенил-(C1-C4) алкила и гетероциклил-(C1-C4) алкила, причем этот необязательно замещенный фрагмент необязательно замещен одним заместителем или более, каждый из которых независимо выбирают из группы, состоящей из (C1-C4) алкила, циклоалкила, -O-R6, -S(O)q-R7, -N(R9R9), -NHCO-R6, -NHSO2R9, -CO2R9, -CONR9R9 и -SO2NR9R9, где q равно 0, 1, 2 или 3;
R3 и R4 каждый независимо представляет собой Н, галоген или необязательно замещенный фрагмент, выбранный из группы, состоящей из (C1-C4) алкила, циклоалкила, арила и арил-(С1-С4)алкила; причем этот необязательно замещенный фрагмент необязательно замещен одним заместителем или более, выбранными из группы, состоящей из ОН, (C1-C4) алкила, (C1-C4) алкокси, арилокси, арил-(C1-C4) алкокси, -NR9R9, -COOH, -CONR9R9 и; галогена; или
R3 и R4 вместе с углеродами, к которым они присоединены, образуют необязательно замещенный арил, причем этот арил необязательно замещен одним заместителем или более, каждый из которых независимо выбирают из группы, состоящей из ОН, (C1-C4) алкила, (C1-C4) алкокси, арилокси, арил-(C1-C4) алкокси, -NR9R9, -COOR5, -CONR9R9 и галогена;
R5 для каждого случая независимо представляет собой Н или необязательно замещенный фрагмент, выбранный из группы, состоящей из (C1-C4) алкила и арил-(C1-C4) алкила, причем этот необязательно замещенный фрагмент необязательно замещен одним заместителем или более, независимо выбранными из группы, состоящей из (C1-C4) алкила, ОН, (C1-C4) алкокси, арилокси, NO2, арил-(C1-C4) алкокси, -NR9R9, -COOH, -CONR9R9 и галогена;
R6 для каждого случая независимо выбирают из группы, состоящей из Н, (C1-C4) алкила, (C1-C4) алкокси, арил-(C1-C4) алкила и арил-(C1-C4) алкокси;
R7 представляет собой Н, если q равно 3, или R7 для каждого случая независимо выбирают из группы, состоящей из (C1-C4) алкила, арила или арил-(C1-C4) алкила, если q равно 0, 1 или 2; и
R9 для каждого случая независимо выбирают из группы, состоящей из Н, NO2, (C1-C4) алкила, арила и арил-(C1-C4) алкила.
X1 представляет собой природную или неприродную D- или L-α-аминокислоту, причем, если X1 является Phe, Nal, Trp, Туr, Pal или His, его ароматическое кольцо необязательно замещено по углероду или азоту радикалом R6, или, если X1 является Ser или Thr, кислород боковой цепи необязательно замещен одним или более R1;
X2 представляет собой D- или L-Trp, N-метил-D-Trp или N-метил-L-Тrр;
X3 представляет собой Lys, α-N-метил-Lуs, или ε-N-(C1-C4)алкил-Lys, или ε-N-[арил-(C1-C4)алкил]-Lys;
X4 представляет собой природную или неприродную D- или L-α-аминокислоту, причем, если X4 является Phe, Nal, Trp, Туr или His, его ароматическое кольцо необязательно замещено по углероду или азоту радикалом R8, или, если X4 является Ser, Туr или Thr, кислород боковой цепи необязательно замещен одним или более R1.
Связи между Х1, X2, X3 и X4 являются амидными связями как связь между X1 и Z и связь между X4 и Y.
Предпочтительную группу соединений формулы (II), обозначенную как группа А, составляют соединения, в которых:
каждое n равно 2;
m равно 0 или от 1 до 5;
R1 для каждого случая независимо представляет собой Н, метил или арил-(C1-C4) алкил;
R2 представляет собой необязательно замещенный фрагмент, выбранный из группы, состоящей из фенил-(C1-C4) алкила и гетероциклил-(C1-C4) алкила, причем этот необязательно замещенный фрагмент необязательно замещен заместителем, выбранным из группы, состоящей из (C1-C4)алкила и -O-R6; и
R3 и R4 каждый независимо представляет собой Н, галоген или необязательно замещенный фрагмент, выбранный из группы, состоящей из (C1-C4) алкила и арила; причем этот необязательно замещенный фрагмент необязательно замещен заместителем, выбранным из группы, состоящей из ОН, (C1-C4)алкокси, арилокси и галогена.
Предпочтительную группу соединений группы А, обозначенную как группа В, составляют соединения, в которых Х1 представляет собой Phe, Nal, Trp, Туr, Pal или His, в которых ароматическое кольцо необязательно замещено по углероду или азоту радикалом R6; и
X4 представляет собой Val, Abu, Ser, Thr, Nal, Trp, Tyr или His, где ароматическое кольцо Nal, Trp, Туr и His необязательно замещено по углероду и/или азоту радикалом R8, или, если X4 является Ser, Туr или Thr, кислород боковой цепи необязательно замещен R1.
Предпочтительную группу соединений группы В, обозначенную как группа С, составляют соединения, в которых X1 представляет собой Phe, Trp или Туr, ароматическое кольцо которых необязательно замещено по углероду или азоту радикалом R6;
X2 представляет собой D-Trp или N-метил-D-Тrр;
X3 представляет собой Lys или α-N-метил-Lуs;
X4 представляет собой Val, Thr, Abu, Nal или Туr, где кислород гидроксильной группы Thr и Туr боковой цепи необязательно замещен R1;
R1 для каждого случая независимо представляет собой Н, метил или бензил;
R2 представляет собой необязательно замещенный фрагмент, выбранный из группы, состоящей из фенилметила и гетероциклилметила, причем этот необязательно замещенный фрагмент необязательно замещен заместителем, выбранным из группы, состоящей из (C1-C4)алкила и -O-R6;
R3 представляет собой (C1-C4)алкил или необязательно замещенный арил; где необязательно замещенный арил замещен заместителем, выбранным из группы, состоящей из ОН, (C1-C4) алкокси, арилокси и галогена;
R4 представляет собой Н; и
R6 для каждого случая независимо выбирают из группы, состоящей из Н и арил-(C1-C4) алкокси.
Предпочтительную группу соединений группы С, обозначенную как группа D, составляют соединения, в которых X1 представляет собой Phe, Trp, Туr или Tyr (Bzl);
X4 представляет собой Val, Thr, Abu, Nal или Туr, где гидроксильная группа Thr и Туr необязательно замещена бензилом;
m равно 0, 2 или 4;
R2 представляет собой необязательно замещенный фрагмент, выбранный из группы, состоящей из фенилметила и 3-индолилметила, причем этот необязательно замещенный фрагмент необязательно замещен радикалом -O-R6; и
R3 представляет собой 1,1-диметилэтил или необязательно замещенный арил; где необязательно замещенный арил необязательно замещен заместителем, выбранным из группы, состоящей из ОН, (C1-C4) алкокси и галогена.
Предпочтительную группу соединений группы D, обозначенную как группа Е, составляют соединения, в которых R2 является фенилметилом;
R3 является 1,1-диметилэтилом или необязательно замещенным фенилом, где необязательно замещенный фенил необязательно замещен ОН или ОСН3; и
R6 для каждого случая независимо выбирают из группы, состоящей из Н и бензилметокси.
Предпочтительную группу соединений группы Е, обозначенную как группа F, составляют:
цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Туr(Bzl)-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr(Bzl)-Pheψ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Abu-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-Рhеψ(4-(1,1-диметилэтил)-имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Рhеψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Tyr-D-Trp-Lys-Val-Pheψ(-4(3-гидроксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Nal-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Nal-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)имидазол)-(γ)Abu],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Рhеψ(4-(4-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Рhеψ(4-(фенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)-имидазол)-(ε)Ahx] и
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-гидроксифенил)-имидазол)-(γ)Abu].
Предпочтительную группу соединений группы F, обозначенную как группа G, составляют:
цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Туr(Bzl)-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr(Bzl)-Рhеψ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
циклo[Trp-D-Trp-Lys-Abu-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(1,1-диметилэтил)-имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr-Pheψ(4-(3-гидроксифенил)имидазол)-Gly] и
цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly].
Предпочтительную группу соединений группы G, обозначенную как группа Н, составляют:
цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Abu-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly] и
цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly].
Предпочтительную группу соединений группы Н, обозначенную как группа I, составляют:
цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr-Pheψ(4-(3-гидроксифенил)имидазол)-Gly] и
цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly].
Еще одну предпочтительную группу соединений группы F, обозначенную как группа J, составляют:
цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr(Bzl)-Рhеψ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло [Trp-D-Trp-Lys-Abu-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(1,1-диметилэтил)-имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Nal-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Nal-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)-имидазол-(γ)Abu],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(4-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(фенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)-имидазол)-(ε)Ahx] и
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-гидроксифенил)-имидазол)-(γ)Abu].
Предпочтительную группу соединений группы J, обозначенную как группа К, составляют:
цикло[Trp-D-Trp-Lys-Thr-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Nal-Pheψ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Рhеψ(4-(3-метоксифенил)имидазол-(γ)Abu],
цикло[Тrр-D-Тrр-Lуs-Туr(Вzl)-Рhеψ(4-(4-метоксифенил)имидазол)-Glу] и
цикло[Тrр-D-Тrр-Lуs-Туr(Вzl)-Pheψ(4-(фенил)имидазол)-Gly].
Предпочтительную группу соединений группы К, обозначенную как группа L, составляют:
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Рhеψ(4-(3-метоксифенил)имидазол-(γ)Abu],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(4-метоксифенил)имидазол)-Gly] и
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(фенил)имидазол)-Gly].
Еще один аспект настоящего изобретения обеспечивает фармацевтическую композицию, содержащую эффективное количество соединения формулы (I) или формулы (II) или их фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
Еще один аспект настоящего изобретения обеспечивает способ выявления агонистического эффекта рецепторов соматостатина у нуждающегося в этом млекопитающего, который включает введение указанному млекопитающему эффективного количества соединения формулы (I) или формулы (II) или их фармацевтически приемлемой соли.
Еще один аспект настоящего изобретения обеспечивает способ выявления антагонистического эффекта рецепторов соматостатина у нуждающегося в этом млекопитающего, который включает введение указанному млекопитающему эффективного количества соединения формулы (I) или формулы (II) или их фармацевтически приемлемой соли.
Другой аспект настоящего изобретения обеспечивает способ лечения аденом, выделяющих пролактин, рестеноза, сахарного диабета, гиперлипидемии, невосприимчивости к инсулину, синдрома X, ангиопатии, пролиферативной ретинопатии, феномена “утренней зари”, нефропатии, выделения кислоты в желудке, пептических язв, наружного свища кишечника и наружного свища поджелудочной железы, слизистого колита, синдрома Дампинга (Dumping), синдрома водянистого стула, диареи вследствие СПИДа, диареи, вызванной химиотерапией, острого или хронического панкреатита, желудочно-кишечных опухолей, выделяющих гормоны, рака, гепатомы, ангиогенеза, воспалительных заболеваний, артрита, хронического отторжения аллотрансплантатов, ангиопластики, кровотечения трансплантатов сосудов или желудочно-кишечного кровотечения у нуждающегося в таком лечении млекопитающего, который включает введение указанному млекопитающему соединения формулы (I) или формулы (II) или их фармацевтически приемлемой соли.
Еще один аспект данного изобретения обеспечивает способ ингибирования пролиферации helicobacter pylori у нуждающегося в этом млекопитающего, который включает введение указанному млекопитающему соединения формулы (I) или формулы (II) или их фармацевтически приемлемой соли.
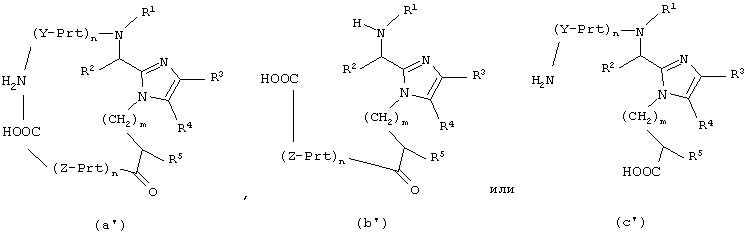
Еще один аспект данного изобретения обеспечивает способ получения соединения формулы
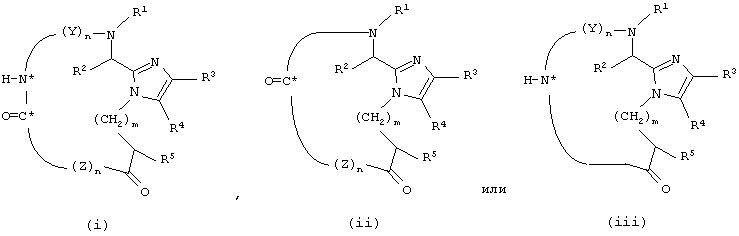
который включает снятие защиты с соединения формулы
посредством отщепления Prt-группы;
где
Prt обозначает защитную группу аминокислоты боковой цепи;
Y и Z каждый независимо представляет собой D- или L-природную или неприродную α-аминокислоту, необязательно имеющую защищенную боковую цепь, где H-N* является аминогруппой N-концевой аминокислоты, обозначенной Y, и O=С* является карбоксильной группой С-концевой аминокислоты, обозначенной Z;
n для каждого случая независимо равно от 1 до 50;
и все остальные радикалы такие, как определено для приведенной здесь выше формулы (I).
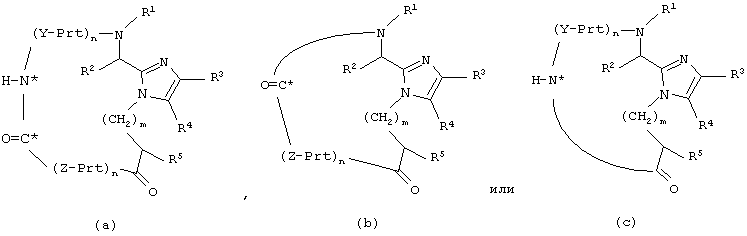
Другой аспект настоящего изобретения обеспечивает способ получения соединения формулы
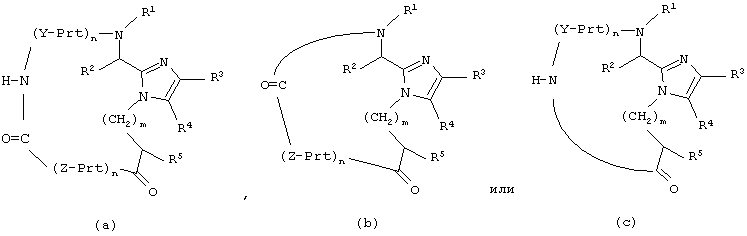
который включает:
для соединения формулы (а) образование амидной связи между концевой аминогруппой последней аминокислоты, обозначенной Y, и концевой карбоксильной группой последней аминокислоты, обозначенной Z, при взаимодействии соединения формулы (а’) с реагентом конденсации пептидов и добавкой; или
для соединения формулы (b) образование амидной связи между концевой аминогруппой и концевой карбоксильной группой последней аминокислоты, обозначенной Z, при взаимодействии соединения формулы (b’) с реагентом конденсации пептидов и добавкой; или
для соединения формулы (с) образование амидной связи между концевой аминогруппой последней аминокислоты, обозначенной Y, и концевой карбоксильной группой, при взаимодействии соединения формулы (с’) с реагентом конденсации пептидов и добавкой;
где Prt обозначает защитную группу аминокислоты боковой цепи;
Y и Z каждый независимо представляет собой D- или L-природную или неприродную α-аминокислоту, необязательно имеющую защищенную боковую цепь, где H-N* является аминогруппой N-концевой аминокислоты, обозначенной Y, и O=С* является карбоксильной группой С-концевой аминокислоты, обозначенной Z;
n для каждого случая независимо равно от 1 до 50;
и все остальные переменные такие, как определено для приведенной здесь выше формулы (I).
Еще один аспект настоящего изобретения обеспечивает способ получения соединения формулы
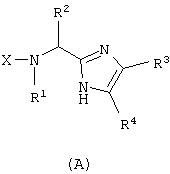
который включает реакцию соединения формулы
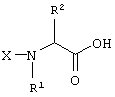
с α-галогенкетоном формулы X’-СН(R3)CO(R4) в присутствии основания и полярного апротонного растворителя по существу до завершения реакции; выпаривание полярного апротонного растворителя с получением твердого вещества; растворение этого твердого вещества в апротонном органическом растворителе и избыточном количестве водного NH4OAc с образованием раствора; кипячение этого раствора с обратным холодильником и последующее удаление полярного слоя с получением соединения формулы (А);
в котором Х представляет собой защитную группу для аминогруппы;
X’ представляет собой галоген;
и все остальные радикалы такие, как определено для приведенной здесь выше формулы (I).
Еще один аспект данного изобретения обеспечивает способ получения соединения формулы (I)
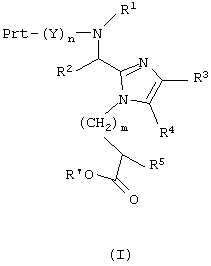
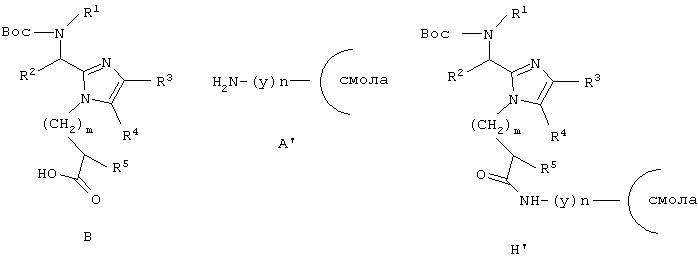
который включает сочетание соединения формулы (В)
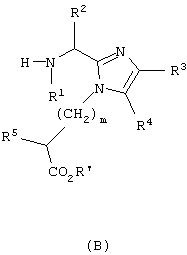
с Nα-защищенной аминокислотой (Prt)-Y, где Nα-защищенная аминокислота присутствует в виде своего активированного сложного эфира, ангидрида или галогенангидрида, в присутствии основания по существу до завершения реакции с получением соединения формулы (С),
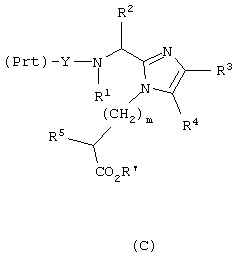
необязательное снятие защиты с аминогруппы Nα-защищенной аминокислоты (Prt)-Y, применяя обычные реакции снятия защиты, и повторение реакции сочетания с другой Nα-защищенной аминокислотой повторно до получения требуемого соединения формулы (I);
Y для каждого случая независимо представляет собой D- или L-природную или неприродную α-аминокислоту, необязательно имеющую боковую цепь с защитной группой;
Prt обозначает защитную группу для аминогруппы;
R’ представляет собой сложный алкиловый эфир или сложный бензиловый эфир;
n равно от 1 до 100;
и все остальные радикалы такие, как определено для приведенной здесь выше формулы (I).
Другой аспект данного изобретения обеспечивает способ получения соединения формулы (I), определенного здесь выше, который включает связывание соединения формулы (В), активированного в виде его активного сложного эфира, ангидрида или галогенангидрида кислоты, с пептид-смолой (А’) со снятой N-защитной группой, полученной способами, хорошо известными тем, кто знаком с синтезом пептидов, удаление N-концевой Fmoc-защитной группы, с использованием пиперидина в DMF, ТАЕА [трис(2-аминоэтил)амин] или аналогичного основания, снятие защиты и отщепление полученного промежуточного продукта (В’) от смолы с использованием сильной кислоты. Все радикалы такие, как определено для приведенной здесь выше формулы (I).
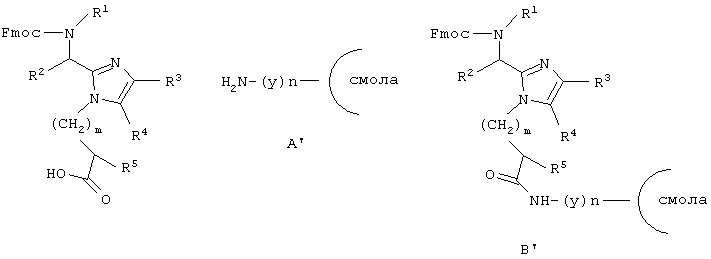
Другой аспект данного изобретения обеспечивает способ получения соединения формулы (I), который включает связывание соединения формулы (В), активированного в виде его активного сложного эфира, ангидрида или галогенангидрида кислоты, с пептид-смолой (Н), со снятой N-защитной группой, полученной способами, хорошо известными тем, кто знаком с синтезом пептидов, удаление N-концевой Fmoc-защитной группы с использованием пиперидина в DMF, ТАЕА или аналогичного основания, ацилирование освобожденной N-концевой аминогруппы посредством Nα-Fmoc-защищенной аминокислоты (х), используя хорошо известные специалистам в данной области реакции сочетания пептидов, повторение снятия защиты при помощи основания и стадий сочетания, которые требуются для включения дополнительных аминокислот (х), снятие защиты и отщепление полученного промежуточного продукта (С’) от смолы с использованием сильной кислоты. Все радикалы такие, как определено для приведенной здесь выше формулы (I).
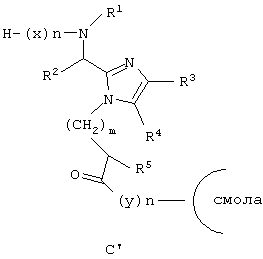
Другой аспект данного изобретения обеспечивает способ получения соединения формулы (I), который включает связывание соединения формулы (В), активированного в виде его активного сложного эфира, ангидрида или галогенангидрида кислоты, с амино-замещенной смолой, такой как трис-(алкокси)-бензиламиновая смола (PAL Resin), 4-(2’,4’-диметоксифенил-аминометил)фенокси-смола (смола Rinka со снятой N-защитной группой), или бензгидриламино-смола, удаление N-концевой Fmoc-защитной группы с использованием пиперидина в DMF, ТАЕА или аналогичного основания, ацилирование освобожденной N-концевой аминогруппы посредством Nα-Fmос-защищенной аминокислоты (х), используя хорошо известные специалистам в данной области реакции сочетания пептидов, повторение снятия защиты при помощи основания и стадий сочетания, которые требуются для включения дополнительных аминокислот (х), снятие защиты и отщепление полученного промежуточного продукта (D’) от смолы с использованием сильной кислоты. Все радикалы такие, как определено для приведенной здесь выше формулы (I).
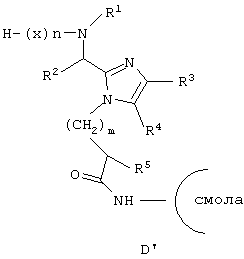
Еще один аспект данного изобретения обеспечивает способ получения соединения формулы (I), который включает взаимодействие соединения формулы (В) с основанием, таким как Сs2СО3, связывание полученной фенольной соли цезия (Е’) с галогенметилированной полистирольной смолой, такой как пептидная смола Merrified, удаление Fmoс-защитной группы посредством пиперидина или аналогичного органического основания, ацилирование освобожденной N-концевой аминогруппы посредством Nα-Fmoc-защищенной аминокислоты (х), используя хорошо известные специалистам в данной области реакции сочетания пептидов, повторение снятия защиты при помощи основания и стадий сочетания, которые требуются для включения дополнительных аминокислот (х), удаление защиты с конечной защищенной последовательности пептидов по N-концу при помощи пиперидина или аналогичного органического основания и по С-концу при помощи трифторуксусной кислоты (Tfa), циклизацию полученного промежуточного продукта (F’), применяя хорошо известные специалистам в данной области реакции сочетания пептидов, и отщепление полученного промежуточного продукта (G’) от смолы с использованием сильной кислоты. Все радикалы такие, как определено для приведенной здесь выше формулы (I).
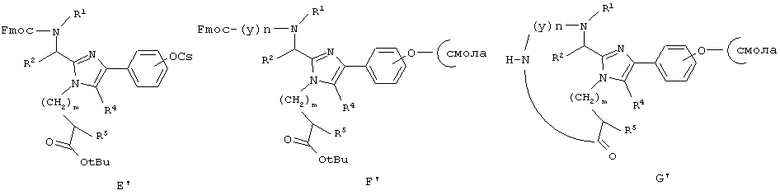
Еще один аспект данного изобретения обеспечивает способ получения определенного выше соединения формулы (I), который включает связывание соединения формулы (В), активированного в виде его активного сложного эфира, ангидрида или галогенангидрида кислоты, с пептид-смолой (А’) со снятой N-защитной группой, полученной хорошо известными специалистам в данной области способами, удаление N-концевой Вос-защитной группы с использованием Tfa, удаление защитных групп боковой цепи и отщепление полученного промежуточного продукта (Н’) от смолы с использованием сильной кислоты. Все радикалы такие, как определено для приведенной здесь выше формулы (I).
Еще один аспект данного изобретения обеспечивает способ получения соединения формулы (I), который включает связывание соединения формулы (В), активированного в виде его активного сложного эфира, ангидрида или галогенангидрида кислоты, с пептид-смолой (Н) со снятой N-защитной группой, полученной хорошо известными специалистам в данной области способами, удаление концевой Вос-защитной группы с использованием Tfa, ацилирование освобожденной N-концевой аминогруппы посредством Nα-Fmoc-защищенной аминокислоты (х), используя хорошо известные специалистам в данной области реакции сочетания пептидов, повторение снятия защиты при помощи Tfa и стадий сочетания, которые требуются для включения дополнительных аминокислот (х), снятие защиты и отщепление полученного промежуточного продукта (I’) от смолы с использованием сильной кислоты. Все радикалы такие, как определено для приведенной здесь выше формулы (I).
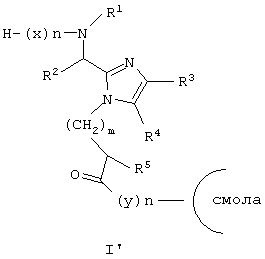
Еще один аспект данного изобретения обеспечивает способ получения соединения формулы (I), который включает реакцию соединения формулы (В) с основанием, таким как Сs2СО3, связывание полученной фенольной соли цезия (J’) с галогенметилированной полистирольной смолой, такой как пептидная смола Merrified, удаление Вос-защитной группы посредством Tfa, ацилирование освобожденной N-концевой аминогруппы посредством Nα-Вос-защищенной аминокислоты (х), используя хорошо известные специалистам в данной области реакции сочетания пептидов, повторение снятия защиты при помощи Tfa и стадий сочетания, которые требуются для включения дополнительных аминокислот (х), снятие защиты с конечной защищенной последовательности пептидов по N-концу при помощи Tfa и по С-концу при помощи неорганического основания, такого как LiOH, в водном DMF, циклизацию полученного промежуточного продукта (К’), применяя хорошо известные специалистам в данной области реакции сочетания пептидов, и отщепление полученного промежуточного продукта (L’) от смолы с использованием сильной кислоты. Все радикалы такие, как определено для приведенной здесь выше формулы (I).
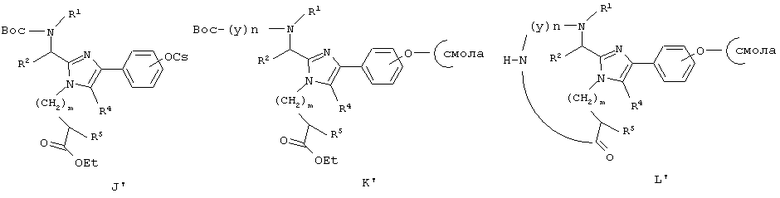
Еще один аспект данного изобретения обеспечивает способ получения соединения формулы (I), который включает связывание соединения формулы (В), активированного в виде его активного сложного эфира, ангидрида или галогенангидрида кислоты с 4-нитробензофеноноксим пептид-смолой (М’) со снятой N-защитной группой, полученной хорошо известными специалистам в данной области способами, удаление N-концевой Вос-защитной группы с использованием Tfa, ацилирование освобожденной N-концевой аминогруппы посредством Nα-Boc-защищенной аминокислоты (х), используя хорошо известные специалистам в данной области реакции сочетания пептидов, повторение снятия защиты при помощи Tfa и стадий сочетания, которые требуются для включения дополнительных аминокислот (х), удаление N-концевой Вос-защитной группы при помощи Tfa, циклизацию и отщепление полученного промежуточного продукта (N’) со снятой N-защитной группой посредством нейтрализации подходящим органическим основанием и удаление защитных групп из боковой цепи посредством сильной кислоты, такой как HF. Все радикалы такие, как определено для приведенной здесь выше формулы (I).
Подробное описание изобретения
Использованный здесь термин "гетероцикл" обозначает гетероцикл, который может появиться в боковой цепи аминокислоты.
Примеры включают (но не ограничены этим) такие гетероциклы, как бензотиенил, кумарил, имидазолил, индолил, пуринил, пиридил, пиримидинил, хинолинил, тиазолил, тиенил и триазолил.
Подразумевается, что использованный здесь термин "арил" обозначает любое стабильное моноциклическое или бициклическое углеродное кольцо, содержащее до 7 членов в каждом кольце, в котором, по меньшей мере, одно кольцо является ароматическим. Примеры арильных групп включают бифенил, инданил, нафтил, фенил и 1,2,3,4-тетрагидронафталин.
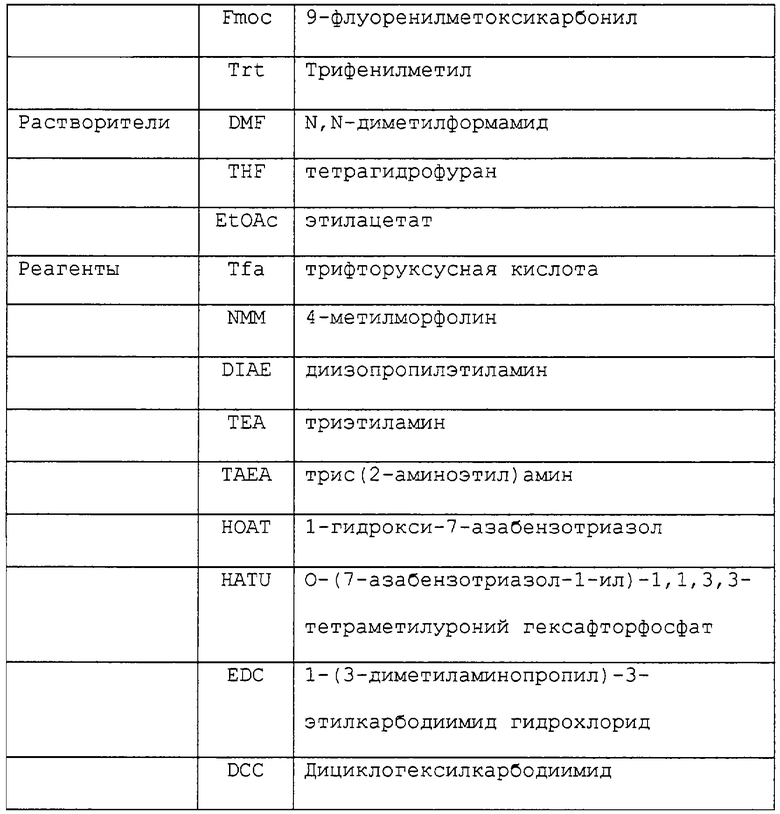
В данной заявке использованы некоторые сокращенные обозначения компонентов аминокислот, некоторых предпочтительных защитных групп, реагентов и растворителей. Значения таких сокращенных обозначений приведены в таблице 1.
Исследование in vitro
Посредством измерения ингибирования связывания [125I-Tyr11]SRIF-14 с СНО-К1 трансфицированными клетками определяют сродство соединения к рецепторам подтипов 1-5 соматостатина человека (sst1, sst2, set3, sst4 и sst5, соответственно).
sst1 рецепторный ген человека клонируют как геномный фрагмент. 1,5 т.п.н. PstI-XmnI сегмент, содержащий 100 пар нуклеотидов 5’-нетранслируемой области, 1,17 т.п.н. полной кодирующей области и 230 пар нуклеотидов 3’-нетранслируемой области, модифицируют путем добавления BglII линкера. Полученный фрагмент ДНК субклонируют в ВаmHI сайт pCMV-81, продуцируя экспрессионную плазмиду млекопитающего (предоставлена Dr. Graeme Bell, Univ. Chicago). Получают клонированную клеточную линию, стабильно экспрессирующую sstt1 рецептор, путем трансфекции в клетки СНО-К1 (АТСС), применяя метод соосаждения фосфатом кальция (1). В качестве селектируемого маркера включают плазмиду pRSV-neo (АТСС). Клонированные клеточные линии отбирают в среде RPMI 1640, содержащей 0,5 мг/мл G418 (Gibco), клонируют в кольце и размножают в культуре.
sst2 соматостатин-рецепторный ген человека, выделенный как 1,7 т.п.н. фрагмент BamHI-HindIII геномной ДНК и субклонированный в плазмидный вектор pGEM3Z (Promega), любезно предоставлен Dr. G. Bell (Univ. Chicago). Конструируют вектор клеточной экспрессии млекопитающего, вставляя 1,7 т.п.н. фрагмент BamHI-HindII в совместимые сайты рестрикционной эндонуклеазы в плазмиде pCMV5. Получают клонированную клеточную линию путем трансфекции в клетки СНО-К1, применяя метод соосаждения фосфатом кальция. В качестве селектируемого маркера включают плазмиду pRSV-neo.
sst3 человека выделяют как геномный фрагмент, и полная кодирующая последовательность содержится внутри 2,4 т.п.н фрагмента BamHI/HindIII. Конструируют экспрессионную плазмиду млекопитающего pCMV-h3, вставляя 2,0 т.п.н. фрагмент NcoI-HindIII в сайт EcoR1 вектора pCMV после модификации концов и добавления линкеров EcoR1. Получают клонированную клеточную линию, стабильно экспрессирующую рецептор sst3, путем трансфекции в клетки СНО-К1 (АТСС), применяя метод соосаждения фосфатом кальция. В качестве селектируемого маркера включают плазмиду pRSV-neo (АТСС). Клонированные клеточные линии отбирают в среде RPMI 1640, содержащей 0,5 мг/мл G418 (Gibco), клонируют в кольце и размножают в культуре.
Экспрессионная плазмида рецептора sst4 человека, pCMV-НХ, предоставлена Dr. Graeme Bell (Univ. Chicago). Вектор содержит 1,4 т.п.н NheI-NheI геномный фрагмент, кодирующий sst4 человека, 456 пар нуклеотидов 5’-нетранслируемой области и 200 пар нуклеотидов 3’-нетранслируемой области, клон вставляют в XbaI-EcoRl сайты pCMV-HX. Получают клонированную клеточную линию, стабильно экспрессирующую sst4, путем трансфекции в клетки СНО-К1 (АТСС), применяя метод соосаждения фосфатом кальция. В качестве селектируемого маркера включают плазмиду pRSV-neo (АТСС). Клонированные клеточные линии отбирают в среде RPMI 1640, содержащей 0,5 мг/мл G418 (Gibco), клонируют в кольце и размножают в культуре.
Ген sst5 человека получен посредством PCR (полимеразной реакции синтеза цепи), используя геномный клон λ в качестве матрицы, и любезно предоставлен Dr. Graeme Bell (Univ. Chicago). Полученный 1,2 т.п.н PCR фрагмент содержит 21 пару нуклеотидов 5’-нетранслируемой области, полную кодирующую область и 55 пар нуклеотидов 3’-нетранслируемой области. Клон вставляют в EcoRl сайт плазмиды pBSSK(+). Вставку извлекают как 1,2 т.п.н фрагмент HindIII-XbaI для субклонирования в вектор экспрессии pCVM5 млекопитающего. Получают клонированную клеточную линию, стабильно экспрессирующую рецептор sst5, путем трансфекции в клетки СНО-К1 (АТСС), применяя метод соосаждения фосфатом кальция. В качестве селектируемого маркера включают плазмиду pRSV-neo (АТСС). Клонированные клеточные линии отбирают в среде RPMI 1640, содержащей 0,5 мг/мл G418 (Gibco), клонируют в кольце и размножают в культуре.
Клетки СНО-К1 (АТСС), стабильно экспрессирующие один из рецепторов sst человека, культивируют в RPMI 1640, содержащей 10% фетальной сыворотки теленка и 0,4 мг/мл генетицина. Клетки собирают при помощи 0,5 мМ EDTA и центрифугируют в течение 5 минут при 500 g и примерно 4°С. Осадок повторно суспендируют в 50 мМ Трис, рН 7,4 и дважды центрифугируют в течение 5 минут при 500 g и примерно 4°С. Клетки лизируют ультразвуком и центрифугируют около 10 минут при 39000 g и примерно 4°С. Осадок ресуспендируют в том же буфере и центрифугируют при 50000 g около 10 мин при 4°С и мембраны в полученном осадке хранят при -80°С.
Эксперименты по конкурентному ингибированию связывания [125I-Tyr11] SRIF-14 проводят в планшетах на 96 лунок, воспроизводя их дважды. Клеточные мембраны (10 мкг белка/лунка) инкубируют с [125I-Tyr11] SRIF-14 (0,05 нМ) в течение примерно 60 минут и при температуре около 37°С в 50 мМ HEPES (рН 7,4), 0,2% BSA, 5 мМ MgCl2, 200 KIU/мл Trasylol, 0,02 мг/мл бацитрацина и 0,02 мг/мл фенилметилсульфонилфторида.
Связанный [125I-Tyr11] SRIF-14 отделяют от свободного путем немедленной фильтрации через предварительно пропитанную 0,1% полиэтиленимином (PEI) стекловолоконную фильтровальную пластину GF/C (Unifilter, Packard), используя коллектор клеток Filtermate 196 (Packard). Фильтры в течение 4 секунд промывают 50 мМ HEPES при температуре примерно 0-4°С и исследуют на радиоактивность, применяя счетчик Packard Top Count.
Специфическое связывание получают путем вычитания неспецифического связывания (определенного в присутствии 0,1 мкМ SRIF-14) из общего связывания. Данные по связыванию анализируют методом компьютерного нелинейного регрессионного анализа (MDL) и определяют величины констант ингибирования (Ki).
Является ли соединение настоящего изобретения агонистом или антагонистом, определяют в следующем исследовании.
Функциональное исследование: ингибирование внутриклеточного производства сАМР:
Клетки СНО-К1, экспрессирующие рецепторы подтипа соматостатина (SRIF-14) человека, высевают в чашки на 24-ячейки с культурой ткани в среду RPMI 1640 с 10% фетальной сывороткой теленка (FSC) и 0,4 мг/мл генетицина. Среду меняют за день до эксперимента.
Клетки (105 клеток/ячейка) дважды промывают 0,5 мл свежей RPMI с 0,2% BSA с добавлением 0,5 мМ (1) 3-изобутил-1-метилксантина (IBMX) и инкубируют примерно 5 минут при температуре около 37°С.
- Производство циклической АМР стимулируют, добавляя 1 мМ форсколина (FSK) в течение 15-30 минут при температуре около 37°С.
- Агонистическое действие соединения измеряют, одновременно добавляя FSK (1 мкМ), SRIF-14 (10-12 -10-6 М) и исследуемое соединение (10-10 -10-5 М).
- Антагонистическое действие соединения измеряют, одновременно добавляя FSK (1 мкМ), SRIF-14 (1-10 нМ) и исследуемое соединение (10-10 -10-5 M).
Реакционную среду удаляют и добавляют 200 мл 0,1N HCl. CAMP определяют, применяя радиоиммунологический метод (Kit FlashPlate SMP001A, New England Nuclear).
Исследование связывания радиолиганда
Мембраны для in vitro исследований связывания рецепторов получают путем гомогенизации (Polytron, набор 6, 15 секунд) клеток СНО-К1, экспрессирующих подтипы рецепторов hsst, в охлажденном льдом 50 мМ Трис-НСl и центрифугирования два раза при 39000 g (10 мин) с промежуточным ресуспендированием в свежем буфере. Конечные осадки ресуспендируют в 10 мМ Трис-НСl для исследования. Для исследования hsst1, hsst3, hsst4, hsst5 инкубируют аликвоты мембранных препаратов (примерно 30 мин при температуре около 37°С) с 0,05 нМ [125I-Tyr11]SRIF-14 в 50 мМ HEPES (рН 7,4), содержащем BSA (10 мг/мл), MgCl2 (5 мМ), Trasylol (200 KIU/мл), бацитрацин (0,02 мг/мл) и фенилметилсульфонилфторид (0,02 мг/мл). Конечный объем для исследования составлял 0,3 мл.
Для исследования hsst2 используют в качестве радиолиганда [125I]MK-678 (0,05 нМ), и время инкубации составляет 90 мин при температуре около 25°С. Инкубацию заканчивают быстрым фильтрованием через GF/C фильтры (предварительно пропитанные 0,3% полиэтиленимином), используя фильтрационный распределитель Brandel filtration manifold. Затем каждую трубку и фильтр трижды промывают аликвотами по 5 мл охлажденного льдом буфера.
Специфическое связывание определяют как общее связывание радиолиганда минус связывание в присутствии 1000 нМ SRIF-14 (hsstl,3,4,5) или 1000 нМ МК-678 для hsst2.
Соединения настоящего изобретения можно исследовать in vivo для применений, ассоциированных со связыванием с рецептором соматостатина, включая специфическое связывание с подтипами рецептора (рецепторов) соматостатина, согласно хорошо известным специалистам в данной области способам, примеры которых приведены в следующих ссылках: I. Shimon и др. "Somatoctatin receptor subtype specificity in human fetal pituitary ciltures", J.Clin. Invest., т.99, №4, 789-798, 1997; С.Gilon и др. "A backbone-cyclic, receptor 5-selective somatostatin analogue: Synthesis, bioactivity, and nuclear magnetic resonance conformational analysis", J. Med. Chem., 1998, 41, 919-929.
Как хорошо известно в данной области, известные и потенциальные применения агонистов и/или антагонистов соматостатина разнообразны и многочисленны. Эти различные применения соматостатина можно суммировать следующим образом:
Агонисты соматостатина можно применять для подавления гормона роста и, более конкретно, аденом, выделяющих GH (акромегалии), и аденом, выделяющих TSH; лечения аденом, выделяющих пролактин; ингибирования инсулина и/или глюкагона и, более конкретно, сахарного диабета, ангиопатии, пролиферативной ретинопатии, феномена "утренней зари" и нефропатии; ингибирования желудочного выделения кислоты и, более конкретно, пептических язв; наружного свища кишечника и наружного свища поджелудочной железы; слизистого колита; синдрома Дампинга (Dumping); синдрома водянистого стула; диареи вследствие СПИДа; диареи, вызванной химиотерапией; острого или хронического панкреатита и желудочно-кишечных опухолей, выделяющих гормоны; лечения рака, такого как гепатома; ингибирования ангиогенеза; лечения воспалительных заболеваний, таких как артрит; ретинопатии; хронического отторжения аллотрансплантатов; лечения при ангиопластике; профилактики кровотечения трансплантатов сосудов или желудочно-кишечного кровотечения.
Соответственно, объем настоящего изобретения включает фармацевтические композиции, содержащие в качестве активного ингредиента, по меньшей мере, одно из описанных здесь соединений данного изобретения в соeдинении с фармацевтически приемлемым носителем.
Соединение данного изобретения можно вводить пероральным, парентеральным (например, в виде внутримышечной, внутрибрюшинной, внутривенной или подкожной инъекции или имплантата), назальным, вагинальным, ректальным, подъязычным или локальным путями и можно готовить в виде препаратов с фармацевтически приемлемыми носителями, обеспечивая дозированные формы, подходящие для каждого способа введения.
Твердые дозированные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких дозированных твердых формах активный компонент смешан, по меньшей мере, с одним инертным фармацевтически приемлемым носителем, таким как сахароза, лактоза или крахмал. Такие дозированные формы могут также включать, что является обычной практикой, отличные от инертных разбавителей дополнительные вещества, например, смазывающие агенты, такие как стеарат магния. В случае капсул, таблеток и пилюль дозированные формы могут также включать буферные агенты. Кроме того, таблетки и пилюли могут иметь энтеросолюбильные оболочки.
Жидкие дозированные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры, содержащие обычно используемые на практике инертные разбавители, такие как вода. Кроме этих инертных разбавителей композиции могут включать вспомогательные средства, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, агенты для подслащивания, вкуса и аромата.
Препараты данного изобретения для парентерального введения включают стерильные водные или неводные растворы, суспензии или эмульсии. Примерами неводных растворителей или наполнителей являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и кукурузное масло, желатин и органические сложные эфиры для инъекций, такие как этилолеат. Такие дозированные формы могут также содержать вспомогательные средства, такие как консерванты, смачивающие агенты, эмульгаторы и диспергирующие агенты. Их можно стерилизовать, например, фильтрованием через фильтр, задерживающий бактерии, или включением в композиции стерилизующих агентов, облучением композиции, или нагреванием композиции. Их можно также производить в виде стерильных твердых композиций, которые можно растворять непосредственно перед использованием в стерильной воде или других стерильных средах для инъекций.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые в дополнение к активному веществу могут содержать эксципиенты, такие как масло какао или свечной воск.
Композиции для назального или подъязычного введения также готовят со стандартными эксципиентами, хорошо известными в данной области.
Кроме того, соединение данного изобретения можно вводить в виде композиции с замедленным высвобождением активного компонента, такой, как описана в нижеследующих патентах. Патент США №5672659 описывает композиции с замедленным высвобождением, включающие биологически активный агент и полиэфир. Патент США №5595760 описывает композиции с замедленным высвобождением, включающие биологически активный агент в желеобразном виде. Патент США №5821221 описывает полимерные композиции с замедленным высвобождением, включающие биологически активный агент и хитозан. Патент США №5916883 описывает композиции с замедленным высвобождением, включающие биологически активный агент и циклодекстрин. Заявка США №09/015394, поданная 29 января 1998 (в настоящее время отозванная), описывает абсорбируемые композиции биологически активного агента с замедленным высвобождением. Описания перечисленных выше патентов и заявок включены здесь в виде ссылок.
Дозировку активного ингредиента в композициях данного изобретения можно варьировать; однако, необходимо, чтобы количество активного ингредиента обеспечивало получение подходящей дозированной формы. Выбранная дозировка зависит от требуемого терапевтического эффекта, способа приема и продолжительности лечения. Для получения терапевтического эффекта людям и другим животным, например, млекопитающим, обычно вводят дозы от 0,0001 до 100 мг/кг веса тела в день.
Предпочтительны дозы в диапазоне от 0,01 до 5,0 мг/кг веса тела в день, которые можно вводить как разовую дозу или поделить на несколько доз.
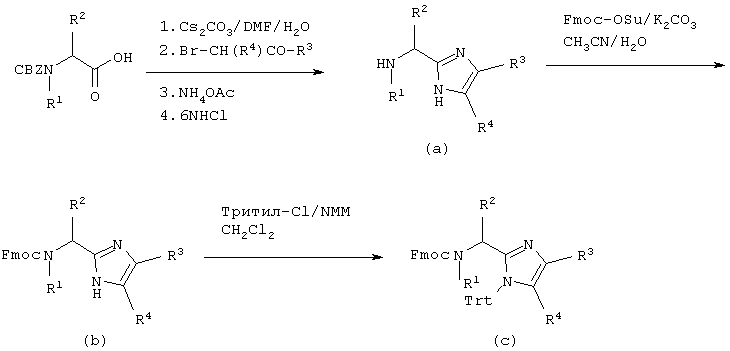
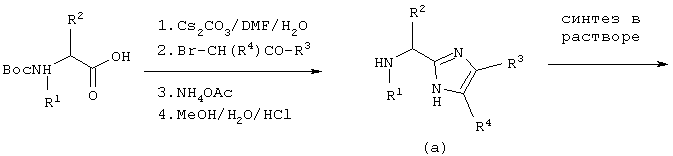
Соединение данного изобретения можно синтезировать согласно нижеследующему описанию и Схеме I. На первой стадии аминокислоту, защищенную по α-аминогруппе Воc, Cbz или другой подходящей группой, превращают в карбоксилатную соль при помощи неорганического основания, например, NaOH, КОН, К2СО3 или, наиболее предпочтительно, Сs2СО3 в полярном растворителе, таком как H2O, DMF, THF или подобные. Растворитель удаляют в вакууме, а оставшуюся соль повторно растворяют в полярном апротонном растворителе, таком как DMF, и добавляют подходящий α-галогенкетон при перемешивании и температуре от -20°С до 100°С, предпочтительно при комнатной температуре. Перемешивание продолжают от 10 минут до 24 часов или до тех пор, пока не завершится образование сложного эфира (по данным тонкослойной хроматографии), тогда раствор концентрируют в вакууме при температуре от 0°С до 100°С, наиболее предпочтительно, от 40°С до 70°С. Промежуточный продукт повторно растворяют в апротонном органическом растворителе, таком как бензол, толуол или, наиболее предпочтительно, ксилолы и добавляют NH4OAc, примерно (5-100)-кратный молярный избыток, наиболее предпочтительно, 15-20 молярный избыток. Двухфазную смесь нагревают при кипячении с обратным холодильником и через 1-4 часа полностью удаляют полярный слой при помощи ловушки Дина-Старка, получая сырой промежуточный продукт (А), который можно использовать сырым или очистить кристаллизацией или колоночной хроматографией.
СХЕМА I
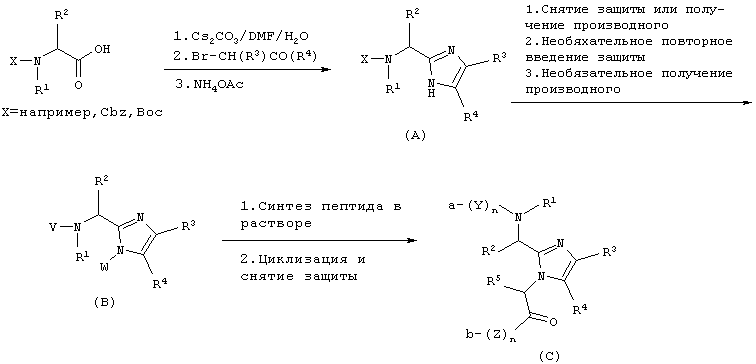
На второй стадии снимают защиту с промежуточного продукта (А), применяя каталитическое гидрирование или сильные кислоты, такие как HF, HCl, НВr или Tfa. Далее можно ввести защиту на α-азот при помощи чувствительной к основаниям защитной группы, такой как Fmoc, используя коммерчески доступный N-(9-флуоренилметоксикарбонилокси)сукцинимид и К2СО3, например, в ацетонитриле и воде. Альтернативно, азот Nα-Cbz-защищенного имидазола можно алкилировать защищенным галогенидом сложного эфира карбоновой кислоты и снять защиту с α-аминогруппы, применяя каталитическое гидрирование и получая В’ (V=H, W=(CH2)mCR5CO2R’, где R’ обозначает алкиловый или бензиловый эфир). Азот имидазола можно защитить, используя коммерчески доступный трифенилметилхлорид и третичное аминное основание, такое как 4-метилморфолин, диизопропилэтиламин или триэтиламин, и получая Fmoc-защищенный промежуточный продукт, в котором затем последовательно снимают защиту с α-аминогруппы, используя такое основание как, например, ТАЕА, получая промежуточный продукт (В) (V=H, W=Trt). Альтернативно, имидазол В’’ (V=H, W=H) со снятой N-защитной группы можно использовать без дополнительной модификации.
На третьей стадии промежуточный продукт В, В’ или В’’ используют как анкерную группу для непрерывного синтеза в растворе целевого пептида. Таким образом, анкерную группу растворяют в этилацетате при концентрации примерно 50-200 ммоль на литр и добавляют примерно 1-5 молярных эквивалентов (или, более предпочтительно, 1,1-1,5 молярных эквивалентов) Nα-Fmoc-защищенной аминокислоты в виде ее активированного сложного эфира, ангидрида или галогенангидрида кислоты. Смесь перемешивают над вторым слоем слабого основания, такого как водный раствор Na2CO3 или, более предпочтительно, водный раствор NаНСО3, до завершения реакции. Водный слой удаляют, добавляют примерно 1-10 мл/ммоль или, более предпочтительно, примерно 2-4 мл/ммоль ТАЕА или пиридина и перемешивают смесь в течение примерно 30 минут. Затем раствор промывают насыщенным раствором NaCl (2 раза по 30 мл/ммоль) и затем 10% фосфатным буферным раствором доводят рН до 5,5 (3 раза примерно по 10 мл/ммоль). Следующие циклы проводят аналогично первому циклу. Конечную аминокислоту можно защитить по Nα группой Воc или Fmoc.
На четвертой стадии N-концевые и С-концевые защитные группы удаляют при помощи водного основания или сильной кислоты и полученный пептидный промежуточный продукт можно циклизовать, применяя классические способы сочетания пептидов, как описано в "The Practice of Peptide Synthesis", Bodanszky and Bodanszky, Springer-Varlag, 1984. Согласно этому, пептидный промежуточный продукт растворяют в апротонном растворителе, таком как DMF, и этот раствор делают основным, добавляя третичное аминное основание, такое как 4-метилморфолин. Карбоксилатную часть промежуточного продукта активируют, добавляя (1-6)-кратный молярный избыток карбодиимида, такого как DCC или EDC, и добавки, такие как, например, 1-гидроксибензотриазол. Смесь перемешивают при температуре от 0 до 100°С, наиболее предпочтительно, при комнатной температуре до завершения реакции.
На конечной стадии защищенный пептид освобождают от защитных групп, применяя каталитическое гидрирование или сильные кислоты, такие как HF, HCl, НВr или Tfa, получая конечный продукт (С), где радикалы с R1 по R5, a, b, Y, Z и n определены выше для формулы (I).
Данные масс-спектроскопии с инфузией получены на спектрометре Finnigan SSQ 7000, оборудованном источником ESI (ионизация при электрораспылении). Данные ЯМР получены на спектрометре Varian Unity 300 МГц на образцах с концентрациями около 10-20 мг/мл в соответствующих растворителях.
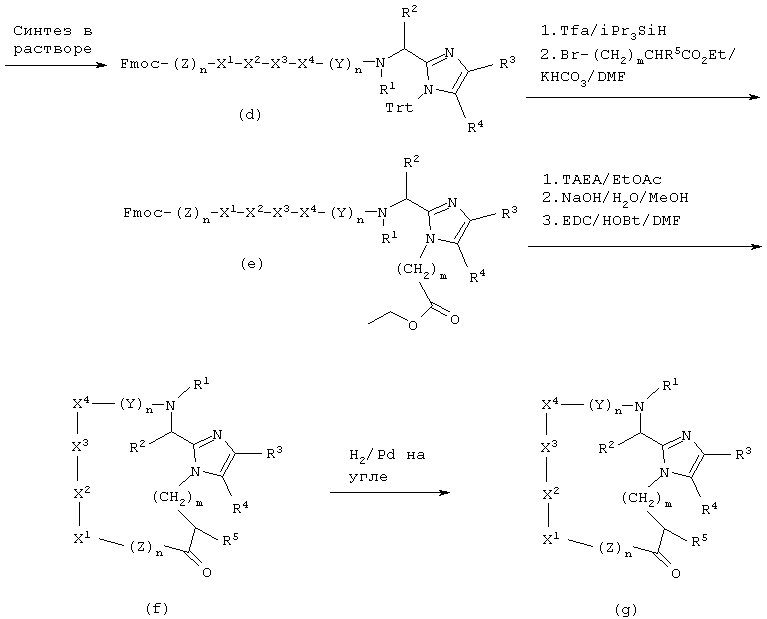
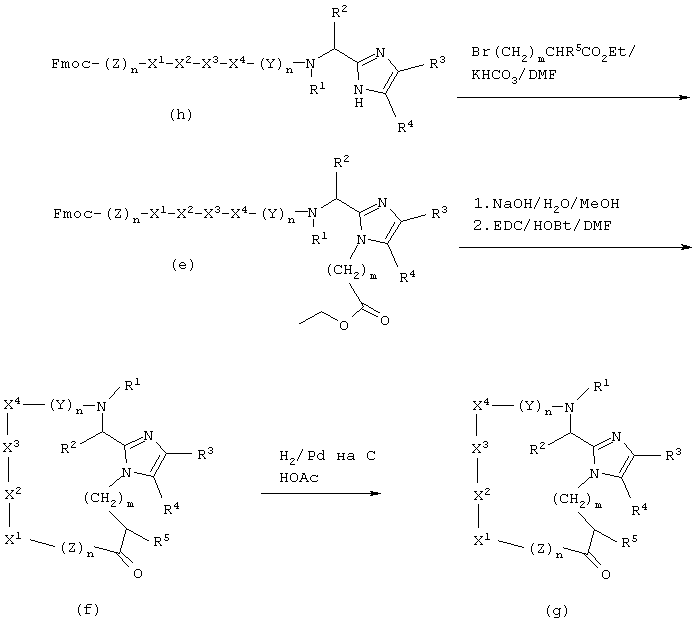
Альтернативно, соединения настоящего изобретения можно получать, применяя способы твердофазного синтеза пептидов. Таким образом, промежуточный продукт А(Х=Вос) алкилируют, например, этилбромацетатом и в присутствии подходящего основания, например, К2СО3 в апротонном растворителе, например, DMF, и полученный промежуточный продукт - этиловый сложный эфир - гидролизуют, используя водное основание, например, NaOH, получая промежуточный продукт В (V=Boc, W=-CH2CO2H). Промежуточный продукт В (V=Boc, W=-CH2CO2H) можно активировать, применяя известные способы активации, как описано в "The Practice of Peptide Synthesis", Bodanszky and Bodanszky, Springer-Varlag, 1984, и использовать непосредственно для сочетания с растущим пептидом на твердой подложке или, начиная твердофазный синтез, можно присоединить промежуточный продукт В (V=Boc, W=-CH2CO2H) непосредственно к твердой подложке. Удаление N-концевой защитной Вос-группы, например, при помощи Tfa позволяет продолжать синтез пептида в условиях, известных специалистам в данной области.
Промежуточный продукт В (например, V=Fmoc, W=-CH2CO2T-Вu) можно обработать кислотой, например, Tfa, для удаления защищающего карбоксилат трет-бутилового эфира и полученный промежуточный продукт В (например, V=Fmoc, W=-CH2CO2H) можно использовать для твердофазного синтеза пептидов, применяя стратегию Fmoc. Таким образом, промежуточный продукт В (V=Fmoc, W=-CH2CO2H) можно активировать известными приемами активации, как описано в "The Practice of Peptide Synthesis", Bodanszky and Bodanszky, Springer-Varlag, 1984, и использовать непосредственно для сочетания с растущим пептидом на твердой подложке. Удаление N-концевой защитной Fmoc-группы, например, пиридином позволяет продолжать синтез пептида в условиях, известных специалистам в данной области.
Твердофазный синтез циклических аналогов можно также проводить согласно приведенной ниже Схеме II.
СХЕМА II
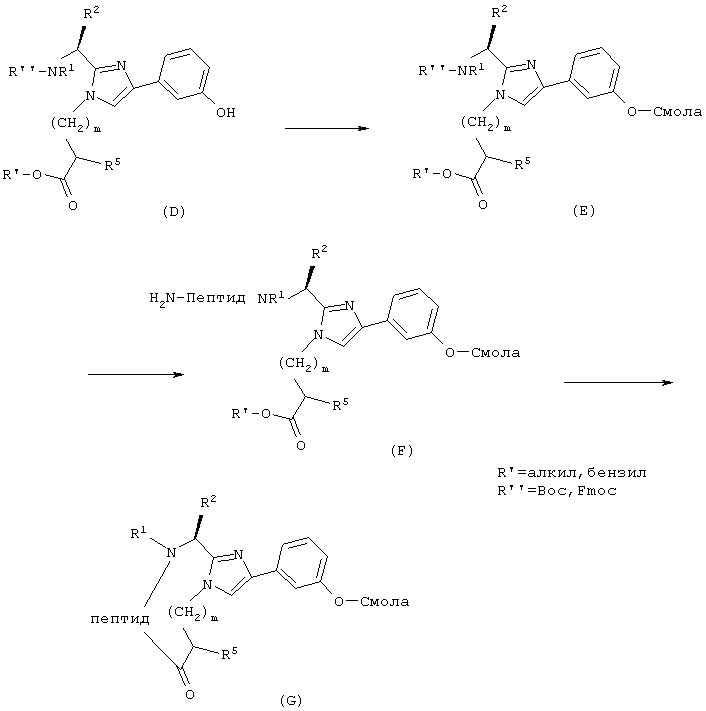
Для получения свободного фенола А (Х=Н, R3=2-гидpoкcифенил, 3-гидроксифенил или 4-гидроксифенил) промежуточный продукт А (Х=Вос или Cbz, R3=2-мeтoкcифeнил, 3-метоксифенил или 4-метоксифенил) может быть обработан 1М ВВr3 в CH2Cl2 в течение примерно 1/2 часа. Затем можно ввести на α-азот защитную группу, чувствительную к кислоте, такую как группа Вос, используя ди-трет-бутилдикарбонат и основание, например, NaOH в смеси смешивающегося с водой органического растворителя, например, диоксана, и воды. Промежуточный продукт А (Х=Вос, R3=2-гидpoкcифeнил, 3-гидроксифенил или 4-гидроксифенил) алкилируют, например, этилбромацетатом и подходящим основанием, например, К2СО3, в апротонном растворителе, например, DMF, получая в результате промежуточный продукт D в виде сложного этилового эфира. Затем промежуточный продукт D превращают в его цезиевую соль взаимодействием с карбонатом цезия. Избыток этой соли цезия подвергают реакции со смолой Меррифилда (Merrifield resin), давая промежуточный продукт Е. Промежуточный продукт Е перерабатывают, применяя стандартный Вос-твердофазный синтез пептидов или стандартный Fmoc-твердофазный синтез пептидов, описанный выше для получения промежуточного продукта F. Когда сконструирована полная аминокислотная последовательность, снимают защиту с С-концевого этилового эфира, используя подходящее основание, например, LiOH в водном DMF, и пептид циклизуют, применяя стандартный способ активации, например, карбодиимид с, например, гидроксибензотриазолом и третичным аминным основанием, например, диизопропилэтиламином, получая промежуточный продукт G. Заключительное снятие защиты с боковой цепи и отщепление от смолы при добавлении очень сильной кислоты, например, HF, дает соединения данного изобретения.
Данное изобретение проиллюстрировано следующими примерами, но не ограничено их подробностями.
Пример 1
цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly]
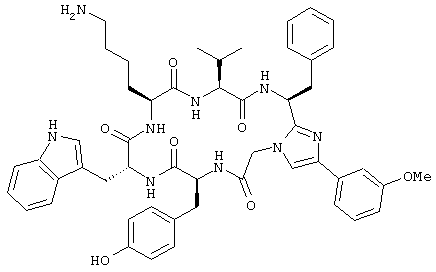
Соединение примера 1 синтезируют согласно приведенной ниже Схеме синтеза 1:
Схема 1
Стадия а: 2-(1-(S)-амино-2-фенилэтил)-4-(3-метоксифенил)имидазол.
Cbz-(L)-Фенилаланин (10,0 г, 33,4 ммоль) и Сs2СО3 (5,44 г, 16,7 ммоль) объединяют в смеси 2:1=DMF:H2O (75 мл) и смесь перемешивают до гомогенности. Растворители удаляют при пониженном давлении, остаток растворяют в DMF (70 мл) и добавляют 2-бром-3’-метоксиацетофенон (7,65 г, 33,4 ммоль) в DMF (30 мл). Эту смесь перемешивают при комнатной температуре примерно 30 минут, а затем концентрируют при пониженном давлении. Полученный кетоэфир растворяют в ксилоле (150 мл) и отфильтровывают CsBr. Добавляют ацетат аммония (40,0 г, 0,52 моль) и смесь нагревают при кипении с обратным холодильником в течение примерно 2 часов, удаляя избыток NН4OАс и выделяющуюся H2O при помощи ловушки Дина-Старка. Реакционную смесь охлаждают и промывают насыщенным раствором NaHCO3 (50 мл) и насыщенным раствором NaCl (50 мл). Ксилольный слой сушат над Na2SO4, фильтруют и концентрируют в вакууме.
Остаток растворяют в диоксане (30 мл), добавляют 6N НСl (115 мл) и нагревают смесь при кипении с обратным холодильником примерно 3 часа. Раствор концентрируют в вакууме и обрабатывают этиловым эфиром (4×100 мл). Остаток сушат в вакууме до постоянного веса, получая 12,15 г (99%) промежуточного продукта 1а. Масс-спектр 294,2 МН+.
Стадия b: 2-(1-(S)-((флуоренилметокси)карбонил)амино-2-фенилэтил)-4-(3-метоксифенил)имидазол.
Промежуточный продукт 1а (11,8 г, 32,2 ммоль) растворяют в смеси 1:1=ацетонитрил: Н2О (200 мл) и осторожно частями добавляют К2СО3 (5,38 г, 39 ммоль). Добавляют 9-флуоренил-метилсукцинимидилкарбонат и полученную смесь энергично перемешивают в течение примерно 20 минут. Продукт экстрагируют EtOAc (100 мл) и EtOAc слой промывают H2O (2×50 мл). EtOAc слой сушат над Na2SO4, фильтруют и концентрируют в вакууме. Продукт очищают хроматографически на силикагеле (150 г), элюируя смесью 2:2:1=СН2Сl2:гексан: EtOAc, а затем смесью 1:1=гексан:EtOAc. Фракции продукта объединяют и концентрируют в вакууме, получая промежуточный продукт 1b в виде светло-желтой пены, 14,77 г (85%). Масс-спектр 516,3 МН+; ЯМР (300 МГц, DMSO-d6): 11,8-12,0 (1Н, с); 7,8-8,0 (3Н, д); 7,6-7,8 (2Н, д); 7,5 (1H, с); 7,1-7,5 (12Н, м); 6,7-6,9 (1H, д); 4,8-5,0 (1H, м); 4,1-4,3 (3Н, м); 3,7-3,9 (3Н, с); 3,0-3,4 (2Н, м).
Стадия с: 2-(1-(S)-((флуоренилметокси)карбонил)амино-2-фенилэтил)-4-(3-метоксифенил)-1-трифенилметилимидазол.
Промежуточный продукт 1b (13,9 г, 26,9 ммоль) растворяют в СН2Сl2 (50 мл) в атмосфере N2, добавляют 4-метилморфолин (2,96 мл, 26,9 ммоль) и хлортрифенилметан (7,51 г, 26,9 ммоль) и раствор перемешивают при комнатной примерно 45 минут. Твердые вещества удаляют фильтрованием, а фильтрат очищают флэш-хроматографией на силикагеле (300 г), используя в качестве элюента смесь 70:30=гексан:ЕtОАс. Фракции продукта объединяют и концентрируют в вакууме, получая промежуточный продукт 1с в виде пены, 18,0 г (88%). ЯМР (300 МГц, DMSO-d6): 7,84-7,95 (2Н, д); 7,7-7,8 (1Н, д); 7,6-7,7 (1Н, д); 6,7-7,5 (29Н, м); 4,3-4,5 (1Н, м); 3,75-3,95 (2Н, м); 3,75-3,85 (3Н, с); 3,6-3,7 (1Н, м); 2,65-2,85 (1Н, д, д); 2,05-2,2 (1Н, м).
Стадия d: 2-(1-(S)-((Fmoc-Tyr(Bzl)-D-Trp-Lys(Cbz)-Val)амино-2-фенилэтил)-4-(3-метоксифенил)-1-трифенилметил)имидазол.
Промежуточный продукт 1с (1,89 г, 2,50 ммоль) растворяют в EtOAc (40 мл), добавляют Трис(2-аминоэтил)амин (9 мл) и смесь энергично перемешивают примерно 1/2 часа. EtOAc-слой промывают насыщенным раствором NaCl (2×120 мл) и затем доводят рН примерно до 5,5 при помощи 10% фосфатного буферного раствора (3×40 мл). EtOAc-слой перемешивают над насыщенным раствором NaHCO3 (40 мл) и добавляют Fmoc-Val-F (1,02 г, 3,00 ммоль). Реакционную смесь перемешивают примерно 1 час и водный слой удаляют.
Затем удаляют защиту с промежуточного продукта и присоединяют к нему Fmoc-Lys(Cbz)-Osu, Fmoc-D-Trp-Osu и Fmoc-Tyr(Bzl)-Osu аналогичным образом, как в описанном выше цикле для Fmoc-Val-F. EtOAc-слой разбавляют 1,5 объемами гексана и наносят на колонку с силикагелем для очистки флэш-хроматографией, используя в качестве элюентов сначала смесь 50:30:20=СН2Сl2:ЕtOАс:гексан, а затем смесь 4:1=ЕtOАс:гексан. Фракции продукта объединяют и концентрируют в вакууме, получая промежуточный продукт 1d в виде белой пены, 1,90 г (46%). Масс-спектр: 1581,2 MNa+, 1559,5 МН+.
Стадия е: 1-((2-этокси-2-оксо)этил)-2-(1-(S)-((Fmoc-Tyr-(Bzl)-D-Trp-Lys(Cbz)-Val)амино-2-фенилэтил)-4-(3-метоксифенил)имидазол.
Промежуточный продукт 1d (519 мг, 0,33 ммоль) растворяют в Tfa (10 мл), содержащей и - Рr3SiН (205 мкл, 1,0 ммоль) и смесь перемешивают примерно 15 минут. Промежуточный продукт осаждают, добавляя простой этиловый эфир (60 мл), и отфильтровывают. Масс-спектр: 1316 МН+. Этот промежуточный продукт растворяют в DMF (3 мл), добавляют КНСО3 (198 мг, 2,0 ммоль) и этилбромацетат (721 мкл, 6,5 ммоль) и смесь перемешивают в течение ночи при комнатной температуре. Смесь концентрируют в вакууме, растворяют в CH2Cl2 (10 мл) и промывают Н2О (10 мл). СН2Сl2-слой сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая сырой промежуточный продукт 1е (540 мг), который используют без дополнительной очистки.
Стадия f: цикло[Туr(Bzl)-D-Trp-Lys(Cbz)-Val-Pheψ4-(3-метоксифенил)имидазол-Gly].
Промежуточный продукт 1e (540 мг, 0,33 ммоль) суспендируют в EtOAc (10 мл), добавляют трис(аминоэтил)амин (1 мл) и смесь энергично перемешивают примерно 1/2 часа. Добавляют EtOAc (10 мл) и промывают раствор насыщенным раствором NaCl (2×25 мл), а затем 10% фосфатным буферным раствором (рН 5,5, 3×10 мл). Промежуточный продукт осаждают, добавляя гексан (40 мл), и растворители декантируют. Остаток растворяют в метаноле (10 мл) и перемешивают в течение ночи при комнатной температуре с 2,5N NaOH (0,5 мл). Смесь разбавляют до помутнения Н2О и доводят рН до 6,7. Промежуточный продукт с удаленной защитой отфильтровывают и сушат в вакууме. Твердое вещество переносят в DMF (25 мл) и добавляют DCC (340 мг, 1,65 ммоль) и HOBt (252 мг, 1,65 ммоль). Смесь перемешивают при комнатной температуре в течение примерно 2 часов и концентрируют при пониженном давлении. Сырой продукт очищают флэш-хроматографией на силикагеле, используя в качестве элюента EtOAc. Фракции продукта объединяют и концентрируют в вакууме, получая промежуточный продукт 1f в виде стекла (180 мг, 48% от промежуточного продукта 1d). Масс-спектр 1134,5 МН+.
Стадия g: цикло[Tyr-D-Trp-Lys-Val-Pheψ4-(3-метоксифенил)имидазол-Gly].
Промежуточный продукт If (180 мг, 0,16 ммоль) растворяют в уксусной кислоте (10 мл), содержащей 10% Pd на угле (24 мг), и смесь встряхивают в атмосфере H2 (1,758 кг/м2=25 psi) при комнатной температуре в течение 8 часов. Катализатор отфильтровывают, а остаток концентрируют в вакууме. Сырая смесь состоит из материала с полностью удаленной защитой (удалены Cbz и бензиловый эфир) и материала с частично удаленной защитой (Cbz удалена, а бензиловый эфир не тронут). Смесь очищают способом препаративной высокоэффективной жидкостной хроматографии на колонке VYDAC® Protein & Peptide С18 (The Nest Group Inc., Southborough, MA), используя градиент от 20% до 70% CH3CN/0,1% Tfa за 55 минут. Чистые фракции более полярного пика объединяют, концентрируют и лиофилизуют (2×10 мл 0,5% НСl, затем 1×10 мл Н2O), получая указанное в заголовке примера 1 соединение, 45 мг (29%). Масс-спектр 910,4 МН+.
Пример 2
цикло[Туr(Bzl)-D-Тrр-Lуs-Vаl-Рhеψ(4-(3-метоксифенил)имидазол)-Gly]
Соединение примера 2 получают в основном согласно Схеме синтеза 1, пример 1, но используя подходящие аминокислоты. Объединяют чистые фракции менее полярного пика от очистки 1 г, концентрируют их и лиофилизуют (2×10 мл 0,5% НСl, затем 1×10 мл Н2O), получая указанный в заголовке Примера 2 продукт, 33 мг (21%). Масс-спектр 1000,4 МН+.
Пример 3
цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)Gly]
Соединение примера 3 получают согласно Схеме синтеза 1, способом, по существу аналогичным примеру 1, но со следующими различиями:
Стадия d: 2-(1-(S)-((Fmoc-Trp-D-Trp-Lys(Cbz)-Val)амино-2-фенилэтил)-4-(3-метоксифенил)-1-(трифенилметил)имидазол.
Промежуточный продукт 1с (757 мг, 1,0 ммоль) растворяют в EtOAc (20 мл), добавляют трис(2-аминоэтил)амин (3 мл) и смесь энергично перемешивают примерно 1/2 часа. EtOAc-слой промывают насыщенным раствором NaCl (2×60 мл) и затем 10% фосфатным буферным раствором доводят рН примерно до 5,5 (3×20 мл). EtOAc-слой перемешивают над насыщенным раствором NаНСО3 (20 мл) и добавляют Fmoc-Val-F (825 мг, 2,33 ммоль). Реакцию перемешивают примерно 1 час и удаляют водный слой.
Затем снимают защиту с промежуточного продукта и присоединяют к нему Fmoc-Lys(Cbz)-OSu, Fmoc-D-Trp-OSu и Fmoc-Trp-OSu аналогичным образом, как в описанном выше цикле для Fmoc-Val-F. EtOAc-слой разбавляют 1,5 объемами гексана и наносят на колонку с силикагелем для очистки флэш-хроматографией, используя в качестве элюентов сначала смесь 50:30:20=CH2Cl2:EtOAc:гeкcaн, а затем смесь 4:1=ЕtOАс:гексан. Фракции продукта объединяют и концентрируют в вакууме, получая промежуточный продукт 3d в виде белой пены, 1,02 г (68%). Масс-спектр: 11492,0 MNa+, 1514,2 МН+.
Стадия е: 1-((2-этокси-2-оксо)этил)-2-(1-(S)-((Fmoc-Trp-D-Trp-Lys (Cbz)-Vаl)амино-2-фенилэтил)-4-(3-метоксифенил)имидазол.
Промежуточный продукт 3d (1,00 г, 0,67 ммоль) растворяют в смеси CH2Cl2 (10 мл), Tfa (1 мл) и - Рr3SiН (205 мкл, 1,0 ммоль) и смесь перемешивают примерно 20 минут. Добавляют смесь 1:1=Еt2O:гексан (100 мл) и промежуточный продукт отфильтровывают и сушат (0,88 г). Этот промежуточный продукт растворяют в DMF (10 мл), добавляют КНСО3 (200 мг, 2,00 ммоль) и этилбромацетат и реакционную смесь перемешивают в течение ночи при комнатной температуре. Смесь концентрируют при пониженном давлении, получая сырой промежуточный продукт 3е, который используют без дополнительной очистки. Масс-спектр 1335,7 МН+.
Стадия f: цикло[Тrр-D-Тrр-Lуs(Cbz)-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly].
Промежуточный продукт 3е (сырой, 0,67 ммоль) растворяют в метаноле (10 мл) и перемешивают при комнатной температуре с 2,5N NaOH (1,0 мл) примерно 45 минут. Смесь разбавляют до помутнения Н2О и доводят рН до 6,9. Растворители декантируют, а остаток растирают с Н2О, получая светло-желтый порошок (650 мг. Масс-спектр 1085,5 МН+). Этот порошок (629 мг) растворяют в DMF (20 мл) и затем добавляют NMM (220 мкл, 2,0 ммоль), EDC (192 мг, 1,0 ммоль) и HOBt (153 мг, 1,0 ммоль). Смесь перемешивают при комнатной температуре в течение примерно 2 часов и концентрируют в вакууме. Сырой продукт растворяют в CH2Cl2 (15 мл) и промывают 10% фосфатным буферным раствором (доводят рН до 5,5). СН2Сl2-слой сушат над Na2SO4, отфильтровывают и концентрируют до 2 мл. Добавляют этиловый эфир для осаждения продукта, который отфильтровывают и сушат, получая промежуточный продукт 3f (440 мг, 71%). Масс-спектр 1067,4 МН+.
Стадия g: цикло [Тrр-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly]
Промежуточный продукт 3f (200 мг, 0,19 ммоль) растворяют в уксусной кислоте (15 мл), содержащей 10% Pd на угле (40 мг), и смесь встряхивают в атмосфере H2 (1,758 кг/м2=25 psi) при комнатной температуре в течение 2 дней. Катализатор отфильтровывают и остаток концентрируют в вакууме. Сырую смесь очищают препаративной высокоэффективной жидкостной хроматографией на колонке C18 (Rainin Microsorb™ 80-22-C5), используя градиент от 20% до 70% СН3СN/0,1% Tfa примерно за 55 минут. Для получения хорошего разделения необходимо второе пропускание с градиентом от 30% до 50% СН3СN/0,1% Tfa примерно за 55 минут. Чистые фракции объединяют, концентрируют и лиофилизуют (2×10 мл 0,5% НСl, затем 1×10 мл Н2О), получая указанное в заголовке Примера 3 соединение, 26 мг (14%). Масс-спектр 933,5 МН+.
Пример 4
цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly]
Соединение примера 4 получают согласно Схеме синтеза 1, способом, по существу аналогичным Примеру 1, со следующими различиями:
Стадия g: цикло[Trp-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly].
Промежуточный продукт 3f (150 мг, 0,14 ммоль) растворяют в CH2Cl2 (12 мл) и добавляют 1М ВВr3 в гексане в атмосфере N2. Полученную суспензию перемешивают примерно 1/2 часа. Добавляют метанол (10 мл) и концентрируют смесь в вакууме. Сырую смесь очищают препаративной высокоэффективной жидкостной хроматографией на колонке C18, используя градиент от 24% до 48% СН3СN/0,2% NH4OAc примерно за 50 минут. Чистые фракции объединяют, концентрируют и лиофилизуют (2×10 мл Н2O), получая указанное в заголовке Примера 4 соединение, 40 мг (29%). Масс-спектр 919,4 МН+.
Пример 5
цикло[Trp-D-Trp-Lys-Thr(Bzl)-Pheψ(4-(3-метоксифенил)-имидазол)-Gly]
Соединение примера 5 получают согласно Схеме 1, способом, по существу аналогичным Примеру 3, за исключением того, что на стадии d вместо Fmoc-Val-F используют Fmoc-Thr(OBzl)-F. Масс-спектр 1025,5 МН+.
Пример 6
цикло[Trp-D-Trp-Lys-Thr-Pheψ(4-(3-гидроксифенил)имидазол)-Gly]
Соединение примера 6 получают согласно Схеме 1, способом, по существу аналогичным Примеру 4, за исключением того, что на стадии g вместо промежуточного продукта 3f используют промежуточный продукт 5f, цикло[Trp-D-Trp-Lys(Cbz)-Thr(Bzl)-Pheψ(4-(3-гидроксифенил)имидазол)-Gly]. Масс-спектр 1025,5 МН+.
Пример 7
H-Trp-D-Trp-Lys-Abu-Pheψ(4-(3-метоксифенил)имидазол)-Gly-OH
Соединение примера 7 получают согласно Схеме 1, способом, по существу аналогичным примеру 3, за исключением того, что на стадии d вместо Fmoc-Val-F используют Fmoc-Abu-F и на стадии f не проводят циклизации с карбодиимидом и HOBt. Масс-спектр 937,3 МН+.
Пример 8
цикло[Тrр-D-Trp-Lys-Abu-Pheψ(4-(3-метоксифенил)имидазол)-Gly]
Соединение примера 8 получают согласно Схеме 1, способом, по существу аналогичным примеру 3, за исключением того, что на стадии d вместо Fmoc-Val-F используют Fmoc-Abu-F. Масс-спектр 919,5 МН+.
Пример 9
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(1,1-диметилэтил)имидазол)-Gly]
Соединение примера 9 получают согласно Схеме 2.
СХЕМА 2
Стадия a: 2-(1-(S)-амино-2-фенилэтил)-4-(1,1-диметилэтил)имидазол.
Boc-(L)-Фенилаланин (5,31 г, 20,0 ммоль) и Сs2СО3 (3,26 г, 10,0 ммоль) объединяют в смеси 1:1=DMF:H2O (50 мл) и перемешивают до получения гомогенной смеси. Растворители удаляют при пониженном давлении, остаток растворяют в DMF (50 мл) и добавляют 1-хлорпинаколон (2,63 мл, 20,0 ммоль). Эту смесь перемешивают при комнатной температуре в течение ночи, а затем концентрируют при пониженном давлении. Полученный кетоэфир растворяют в ксилоле (100 мл) и отфильтровывают CsBr. Добавляют ацетат аммония (25,0 г, 0,33 моль) и смесь нагревают при кипении с обратным холодильником в течение примерно 2 часов, удаляя избыток NH4OAc и выделяющуюся Н2О при помощи ловушки Дина-Старка. Реакционную смесь охлаждают, промывают насыщенным раствором NаНСО3 (50 мл), сушат над Na2SO4, фильтруют и концентрируют в вакууме. Очистка защищенного промежуточного продукта флэш-хроматографией на силикагеле с использованием в качестве элюента смеси 80:20=гексан: ЕtOАс дает 3,45 г (50%) кристаллического промежуточного продукта (Масс-спектр 344,3 МН+). Этот промежуточный продукт растворяют в метаноле (30 мл) и добавляют концентрированную НСl (5,0 мл), смесь перемешивают в течение 3 часов. Раствор концентрируют в вакууме и осаждают остаток из THF и этилового эфира. Твердое вещество сушат в вакууме, получая 1,89 г (95%) промежуточного продукта 9а. ЯМР (300 МГц, DMSO-d6): 8,5-10,5 (3Н, шир.с); 7,3-7,4 (1Н, с); 7,15-7,35 (3Н, м); 7,0-7,1 (2Н, м); 4,9-5,1 (1Н, т); 3,5-3,65 (2Н, д); 1,2-1,3 (9Н, с).
Стадия h: 2-(1-(S)-(Fmoc-Phe-D-Trp-Lys(Воc)-Туr(Bzl))-амино-2-фенилэтил)-4-(1,1-диметилэтил)-1H-имидазол.
Промежуточный продукт 9а (790 мг, 2,50 ммоль) растворяют в EtOAc (40 мл), добавляют трис(2-аминоэтил)амин и смесь энергично перемешивают примерно 1/2 часа. EtOAc-слой промывают насыщенным раствором NaCl (2×120 мл) и затем доводят рН примерно до 5,5 при помощи 10% фосфатного буферного раствора (3×40 мл). EtOAc-слой перемешивают над насыщенным раствором NaHCO3 (40 мл) и добавляют Fmoc-Tyr(Bzl)-OSu (1,02 г, 3,00 ммоль). Реакционную смесь перемешивают примерно 1,5 часа и удаляют водный слой.
Затем снимают защиту с промежуточного продукта и присоединяют к нему Fmoc-Lys(Cbz)-Osu, Fmoc-D-Trp-OSu и Fmoc-Phe-OSu аналогичным образом, как в описанном выше цикле для Fmoc-Val-F. EtOAc-слой наносят на колонку с силикагелем для очистки флэш-хроматографией, используя в качестве элюента 1% уксусную кислоту в EtOAc. Фракции продукта объединяют и концентрируют в вакууме. Сырой продукт повторно растворяют в EtOAc, осаждают, добавляя гексан, и отфильтровывают. Твердое вещество сушат в вакууме, получая промежуточный продукт 9h, 1,67 г (52%). Масс-спектр: 1280,7 МН+.
Стадия е: 4-(1,1-диметилэтил)-2-(1-(S)-((Fmoc-Phe-D-Trp-Lys(Boc)-Tyr(Bzl))амино-2-фенилэтил)-1-(2-этокси-2-оксоэтил)имидазол.
Промежуточный продукт 9h (128 мг, 0,10 ммоль) растворяют в DMF (2 мл), добавляют К2СО3 (35 мг, 0,25 ммоль) и этилбромацетат (28 мкл, 0,25 ммоль) и смесь перемешивают в течение ночи при комнатной температуре. Смесь концентрируют в вакууме, растворяют в EtOAc (10 мл) и промывают H2O (10 мл). EtOAc-слой сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая сырой промежуточный продукт 9е (126 мг, 92%), который используют без дополнительной очистки.
Стадия f: цикло[Рhе-D-Тrр-Lуs(Воc)-Туr(Bzl)-Pheψ4-(3-метоксифенил)имидазол-Glу].
Промежуточный продукт 9е (116 мг, 0,085 ммоль) суспендируют в EtOAc (2 мл), добавляют трис(аминоэтил)амин (0,5 мл) и смесь энергично перемешивают примерно 1/2 часа. Добавляют EtOAc (10 мл) и промывают раствор насыщенным раствором NaCl (2×5 мл), а затем 10% фосфатным буферным раствором (рН 5,5, 3×5 мл). Промежуточный продукт осаждают, добавляя гексан (40 мл), и отфильтровывают (76 мг). Остаток растворяют в метаноле (2 мл) и перемешивают в течение ночи при комнатной температуре с 2,5N NaOH (0,1 мл). Смесь разбавляют до помутнения Н2O и доводят рН до 6,0. Промежуточный продукт с удаленной защитой отфильтровывают и сушат в вакууме. Твердое вещество переносят в DMF (20 мл) и добавляют DCC (126 мг, 0,60 ммоль) и HOBt (90 мг, 0,60 ммоль). Смесь перемешивают при комнатной температуре в течение примерно 6 часов, концентрируют в вакууме, растворяют в EtOAc (5 мл) и промывают насыщенным раствором NaHCO3 (1×5 мл) и насыщенным раствором NaCl (5 мл), сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая промежуточный продукт 9f. Масс-спектр 1098,5 МН+.
Стадия g: цикло[Рhе-D-Тrр-Lуs-Туr-Рhеψ(4-(1,1-диметилэтил)имидазол-Glу].
Промежуточный продукт 9f (сырой, 0,085 ммоль) растворяют в Tfa (9,4 мл), содержащей и - Рr3SiН и Н2О (0,5 мл), перемешивают в течение примерно 20 минут и концентрируют в вакууме. Сырую смесь чистят препаративной высокоэффективной жидкостной хроматографией на колонке С18, используя градиент от 30% до 60% CH3CN/0,1% Tfa примерно за 50 минут. Для получения хорошего разделения требуется второе пропускание с использованием градиента от 32% до 80% СН3СN/0,2% и лиофилизуют (2×10 мл 0,5% HCl, затем 1×10 мл H2O), получая указанное в заголовке Примера 9 соединение, 9 мг (10%). Масс-спектр 998,4 МН+.
Пример 10
цикло[Phe-D-Trp-Lys-Val-Pheψ(4-(3-метоксифенил)имидазол)-Gly]
Соединение примера 10 получают согласно Схеме 1, способом, аналогичным примеру 3, за исключением того, что на стадии d вместо Fmoc-Trp-F используют Fmoc-Phe-OSu. Масс-спектр 894,4 МН+.
Пример 11
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)имидазол)-Gly]
Соединение примера 11 получают согласно Схеме 1, способом, аналогичным примеру 3, за исключением того, что на стадии d вместо Fmoc-Trp-F используют Fmoc-Phe-OH и вместо Fmoc-Val-F используют Fmoc-Tyr(OBzl)-ОН. Fmoc-Tyr(OBzl)-ОН активируют посредством DCC и коммерчески доступного HOAt. Сырая смесь на стадии g состоит из материала с полностью удаленной защитой (удалены и Cbz, и бензиловый эфир) и материала с частично удаленной защитой (Cbz удалена, а бензиловый эфир не затронут). Менее полярный пик, полученный в результате частичного удаления защиты, дает соединение, указанное в заголовке Примера 11. Масс-спектр 1048,5 МН+.
Пример 12
цикло[Phe-D-Trp-Lys-Tyr-Pheψ(4-(3-метоксифенил)имидазол)-Gly]
Соединение примера 12 получают согласно Схеме 1, способом, аналогичным примеру 3, за исключением того, что вместо Fmoc-Trp-F используют Fmoc-Phe-OH и вместо Fmoc-Val-F используют Fmoc-Tyr(Bzl)-ОН на стадии d. Fmoc-Tyr(Bzl)-ОН активируют посредством DCC и коммерчески доступного HOAt. Сырая смесь на стадии g состоит из материала с полностью удаленной защитой (удалены и Cbz, и бензиловый эфир) и материала с частично удаленной защитой (Cbz удалена, а бензиловый эфир не затронут). Более полярный пик, полученный в результате полного удаления защиты, дает соединение, указанное в заголовке Примера 12. Масс-спектр 958,4 МН+.
Пример 13
цикло[Phe-D-Trp-Lys-Tyr-Pheψ(4-(3-гидроксифенил)имидазол)-Gly]
Соединение примера 13 получают согласно Схеме 1, способом, аналогичным примеру 4, за исключением того, что на стадии g вместо промежуточного продукта 3f используют промежуточный продукт 11f, цикло[Phe-D-Trp-Lys(Cbz)-Туr(Bzl)-Рhеψ(4-(3-метоксифенил)имидазол)-Gly]. Масс-спектр 944,6 МН+.
Пример 14
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)имидазол)-Gly]
Соединение примера 14 получают согласно Схеме 2, способом, по существу аналогичным примеру 9, со следующими различиями:
Стадия h: 2-(1-(S)-(Fmoc-Trp-D-Trp-Lys(Cbz)-Tyr(Bzl))-амино)-2-фенилэтил)-4-(3-метоксифенил)-1-(трифенилметил)имидазол.
Синтез пептида проводят способом, аналогичным стадии 9h, за исключением того, что используют Fmoc-Trp-Osu вместо Fmoc-Phe-Osu, используют Fmoc-Lys(Воc)-Osu вместо Fmoc-Lys(Cbz)-Osu и используют Fmoc-Tyr(Bzl)-Osu вместо Fmoc-Val-OSu. Выход 783 мг (57%). Масс-спектр: 1370,6 МН+.
Стадия е: 1-((2-этокси-2-оксо)этил)-2-(1-(S)-((Fmoc-Trp-D-Trp-Lys (Воc)-Туг(Bzl))амино)-2-фенилэтил)-4-(3-метоксифенил) имидазол.
Алкилирование промежуточного продукта 14h выполняют способом, аналогичным реакции 9h, получая 640 мг (80%). Масс-спектр 1456,3 МН+.
Стадия f: цикло [Trp-D-Trp-Lys(Boc)-Tyr(Bzl)-Pheψ4-(3-метоксифенил)имидазол)-Gly].
Промежуточный продукт 14е (640 мг, 0,44 ммоль) растворяют в 15 мл метанола и добавляют 2,5N NaOH (1 мл, 2,5 ммоль). Смесь перемешивают примерно 1/2 часа и затем доводят рН примерно до 7,0, добавляя 5% раствор НСl. Удаляют метанол при пониженном давлении и декантируют водный слой. Остаток тщательно сушат при пониженном давлении, затем растворяют в 15 мл DMF. Добавляют DCC (206 мг, 1,0 ммоль) и HOBt (153 мг, 1,0 ммоль) и оставляют реакцию перемешиваться в течение ночи. Реакционную смесь концентрируют в вакууме, растворяют в EtOAc (10 мл) и дважды промывают 10% фосфатным буфером, рН 5,5. Затем EtAc-слой наносят на колонку с силикагелем и элюируют продукт дополнительным количеством EtOAc. Фракции продукта объединяют и концентрируют, получая 240 мг (46%) промежуточного продукта 14f.
Стадия g: цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Рhеψ(4-(3-метоксифенил)имидазол-Glу].
Промежуточный продукт 14f (240 мг, 0,20 ммоль) растворяют в СН2Сl2 (10 мл) и добавляют Tfa (10 мл), содержащую и - Pr3SiH (205 мкл, 1,0 ммоль). Смесь перемешивают примерно 15 минут при комнатной температуре. CH2Cl2 выпаривают при пониженном давлении и осаждают сырой продукт, добавляя эфир. Растворители декантируют, а остаток очищают препаративной высокоэффективной жидкостной хроматографией на колонке C18, используя градиент от 30% до 50% СН3СN/0,1% Tfa примерно за 55 минут. Чистые фракции объединяют, концентрируют и лиофилизуют (2×10 мл 0,5% НСl, затем 1×10 мл H2O), получая соединение, указанное в заголовке Примера 14, 25 мг (11%). Масс-спектр 1087,4 МН+.
Пример 15
цикло[Tyr-D-Trp-Lys-Val-Pheψ4-(3-гидроксифенил)имидазол)-Gly]
Соединение примера 15 получают согласно Схеме 1, способом, по существу аналогичным примеру 4, со следующими различиями:
Стадия g: цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly].
Промежуточный продукт 1f (130 мг, 0,115 ммоль) растворяют в СН2Сl2 (5 мл) и добавляют раствор 1М ВВr3 в гексане (5 мл) в атмосфере N2. Полученную суспензию перемешивают примерно 1/2 часа, затем охлаждают до температуры примерно 0°С. Добавляют метанол (10 мл) и смесь концентрируют в вакууме. Сырую смесь наносят на колонку C18, промывают 1% раствором NH4OAC, промывают раствором 0,1% Tfa и затем элюируют, используя градиент от 20% до 35% СН3СN/0,1% Tfa примерно за 50 минут. Чистые фракции объединяют, концентрируют и лиофилизуют (2×10 мл 0,5% НСl), получая соединение, указанное в заголовке Примера 15, 60 мг (54%). Масс-спектр 896 МН+.
Пример 16
цикло[Phe-D-Trp-Lys-Nal-Pheψ(4-(3-гидроксифенил)имидазол)-Gly]
Соединение примера 16 получают согласно Схеме 1, способом, по существу аналогичным примеру 3, со следующими различиями:
Стадия d: 2-(1-(S)-((Fmoc-Phe-D-Trp-Lys(Сbz)-Nаl)амино-2-фенилэтил)-4-(3-метоксифенил)-1-(трифенилметил)имидазол.
Промежуточный продукт 16d получают способом, по существу аналогичным получению промежуточного продукта Id, за исключением того, что используют Fmoc-Nal-OAt вместо Fmoc-Val-F и используют Fmoc-Phe-OH вместо Fmoc-Tyr(Bzl)-ОН.
Стадия g: цикло[Tyr-D-Trp-Lys-Val-Pheψ(4-(3-гидроксифенил)имидазол)-Gly].
Стадию 16g проводят по существу аналогично стадии 4g, получая соединение, указанное в заголовке Примера 16. Масс-спектр.
Пример 17
цикло[Phe-D-Trp-Lys-Nal-Pheψ(4-(3-метоксифенил)имидазол)-Gly]
Соединение примера 17 получают согласно Схеме 1, способом, по существу аналогичным примеру 16, за исключением следующего:
Стадия g: цикло[Туr-D-Тrр-Lуs-Vаl-Рhеψ(4-(3-метоксифенил)имидазол-Glу].
Промежуточный продукт 17f (310 мг, 0,27 ммоль) суспендируют в анизоле (3 мл) и обрабатывают эту суспензию примерно 12 мл безводной HF. Смесь перемешивают примерно 1 час при температуре 0°С. HF отгоняют, а продукт осаждают, добавляя эфир. Сырой продукт отфильтровывают и дополнительно очищают препаративной высокоэффективной жидкостной хроматографией, используя градиент от 20% до 80% СН3СN/0,1% Tfa примерно за 40 минут. Чистые фракции объединяют, концентрируют и дважды лиофилизуют из разбавленного раствора НСl. Выход 56 мг (19%). Масс-спектр 992,4 МН+.
Пример 18
цикло[Тrр-D-Тrр-Lуs-Туr(Вzl)-Рhеψ(4-(3-метоксифенил)имидазол)-(γ)Abu]
Соединение примера 18 получают согласно Схеме синтеза 3.
СХЕМА 3
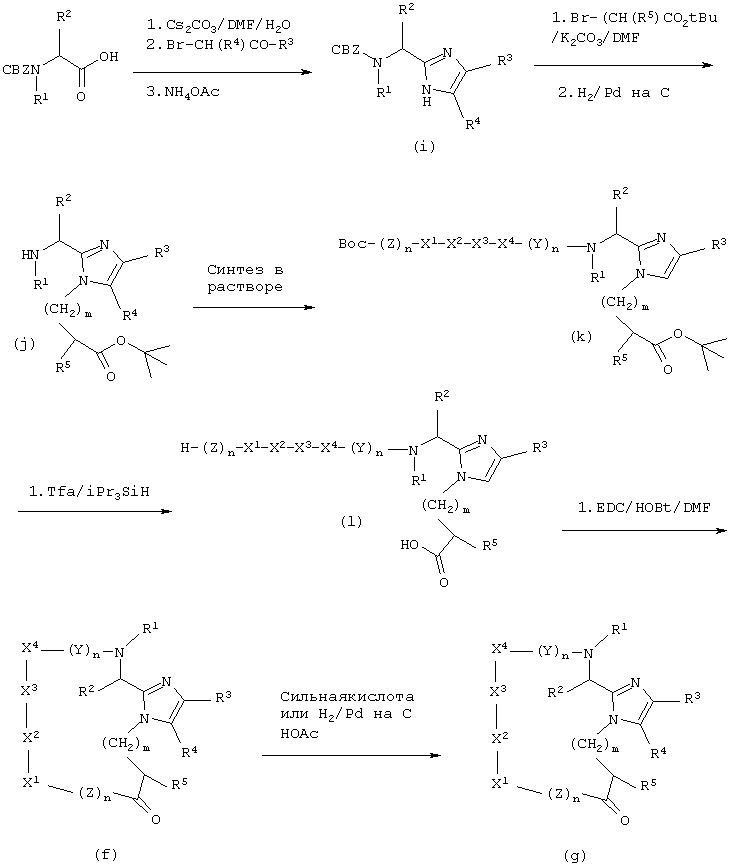
Стадия i: 2-(1-(S)-((фенилметокси)карбонил)амино-2-фенилэтил)-4-(4-метоксифенил)имидазол.
Cbz-(L)-Фенилаланин (10,0 г, 33,4 ммоль) и Сs2СО3 (5,44 г, 16,7 ммоль) объединяют в смеси 2:1=DMF:H2O (75 мл) и смесь перемешивают до гомогенности. Растворители удаляют при пониженном давлении, остаток растворяют в DMF (70 мл) и добавляют 2-бром-3’-метоксиацетофенон (7,65 г, 33,4 ммоль) в DMF (30 мл). Эту смесь перемешивают при комнатной температуре примерно 1/2 часа, затем концентрируют при пониженном давлении. Полученный кетоэфир растворяют в ксилоле (150 мл) и отфильтровывают CsBr. Добавляют ацетат аммония (40,0 г, 0,52 моль) и смесь нагревают при кипении с обратным холодильником в течение примерно 2 часов, удаляя избыток NH4OAc и выделяющуюся H2O при помощи ловушки Дина-Старка. Реакционную смесь охлаждают и промывают насыщенным раствором NаНСО3 (50 мл) и насыщенным раствором NaCl (50 мл). Ксилольный слой сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая промежуточный продукт 18i в виде рыжевато-коричневого твердого вещества (13,8 г, 96%). Масс-спектр 428,2 МН+.
Стадия j: 2-(1-(S)-амино-2-фенилэтил)-1-(4-(1,1-диметилэтокси)-4-оксобутил)-4-(4-метоксифенил)имидазол.
Промежуточный продукт 18i (2,14 г, 5,0 ммоль) растворяют в DMF (11,5 мл) и обрабатывают КНСО3 (1,50 г, 15,0 ммоль) 4-бром-трет-бутилбутиратом (6,69 г, 30 ммоль) в три порции, перемешивая смесь при температуре примерно 50°С в течение 18 часов. Смесь разбавляют эфиром и промывают один раз насыщенным раствором NaHCO3 и один раз насыщенным раствором NaCl. Эфирный слой сушат над Na2SO4, фильтруют и концентрируют до масла. Колоночная хроматография на силикагеле с использованием CH2Cl2 в качестве элюента дает продукт в виде масла.
В сыром алкилированном продукте удаляют защиту посредством гидрирования в уксусной кислоте с использованием 10% Pd на угле в качестве катализатора. Катализатор отфильтровывают, а растворители выпаривают при пониженном давлении. Остаток растворяют в EtOAc, промывают насыщенным раствором NаНСО3 и насыщенным раствором NaCl, сушат над Na2SO4, фильтруют и концентрируют, получая промежуточный продукт 18j в виде масла (450 мг), которое используют на следующей стадии без дополнительной очистки.
Стадия k: 2-(1-(S)-((Boc-Trp-D-Trp-Lys(Cbz)-Vаl)амино-2-фенилэтил)-1-((4-(1,1-диметилэтокси)-4-оксо)бутил)-4-(3-метоксифенил)имидазол.
Синтез в растворе проводят способом, аналогичным синтезу, подробно описанному на стадии Id, за исключением того, что используют Boc-Trp-Osu вместо Fmoc-Tyr(OBzl)-Osu и используют Fmoc-Tyr(OBzl)-OAt вместо Fmoc-Val-F, получая промежуточный продукт 18k. Выход 1,03 г (81%).
Стадия 1: 2-(1-(S)-((H-Trp-D-Trp-Lys(Сbz)-Vаl)амино-2-фенилэтил)-1-((4-гидрокси-4-оксо)бутил)-4-(3-метоксифенил)имидазол.
Промежуточный продукт 18k обрабатывают раствором, содержащим и - Рr3SiН (593 мкл, 2,90 ммоль) в Tfa (10 мл) и смесь перемешивают примерно 1 час. Реакционную смесь концентрируют и растирают с раствором 1:1=эфир:гексан, получая промежуточный продукт 18l в виде рыжевато-коричневого твердого вещества, которое используют на следующей стадии без дополнительной очистки. Масс-спектр 1267,7 МН+.
Стадия f: цикло[Trp-D-Trp-Lys(Cbz)-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)имидазол)-(γ)Abu].
Промежуточный продукт 18l растворяют в DMF (20 мл) и добавляют 4-метилморфолин (159 мкл, 1,45 ммоль), HOBt (196 мг, 1,45 ммоль) и EDC (278 мг, 1,45 ммоль) и реакционную смесь перемешивают в течение ночи. Реакционную смесь концентрируют в вакууме и очищают флэш-хроматографией на силикагеле, используя в качестве элюента смесь CH2Cl2:MeOH=9:1 и получая промежуточный продукт 18f. Выход 220 мг (24%). Масс-спектр 1249,7 МН+.
Стадия g: циклo[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-мe-токсифенил)имидазол)-(γ)Abu].
Промежуточный продукт 18f (220 мг, 176 мкмоль) частично гидрируют в уксусной кислоте при давлении Н2 (2,11 кг/м2=30 psi), используя 10% Pd на угле в качестве катализатора, при комнатной температуре в течение примерно 14 часов. Катализатор отфильтровывают, а фильтрат концентрируют в вакууме. Сырую смесь очищают препаративной высокоэффективной жидкостной хроматографией на колонке C18, используя градиент от 0% до 75% СН3СN/0,1% Tfa за 40 минут. Чистые фракции пика продукта объединяют, концентрируют и лиофилизуют (2×10 мл 0,5% НСl, затем 1×10 мл Н2О), получая соединение, указанное в заголовке примера 18, 72 мг (37%). Масс-спектр 1115,6 МН+.
Пример 19
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(4-метоксифенил)имидазол)-Gly]
Соединение примера 19 получают согласно Схеме 3, способом, по существу аналогичным примеру 18, со следующими различиями:
Стадия j: 2-(1-(S)-амино-2-фенилэтил)-1-(2-(1,1-диметилэтокси)-2-оксоэтил)-4-(4-метоксифенил)имидазол.
Промежуточный продукт 18i (854 мг, 2,0 ммоль) растворяют в DMF (10 мл) и добавляют трет-бутилбромацетат (646 мкл, 4,0 ммоль) и К2СО3 (552 мг, 4,0 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Удаляют растворители при пониженном давлении, а остаток растворяют в EtOAc и промывают насыщенным раствором NaCl. EtOAc-слой сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая 1,02 г пены. Масс-спектр 428,2 МН+. ЯМР (300 МГц, DMSO-d6): 7,85-8,0 (1Н, д); 7,6-7,75 (2Н, д); 7,85-7,95 (1Н, с); 7,1-7,35 (10Н, м); 6,9-7,0 (2Н, д); 4,75-5,05 (5Н, м); 3,7-3,9 (3Н, с); 3,1-3,4 (2Н, м); 1,3-1,5 (9Н, с).
Белую промежуточную пену (1,02 г, 1,88 ммоль) растворяют в НОАс (50 мл), содержащей 10% Pd на угле (50 мг), и гидрируют при давлении H2 2,11 кг/м2 (30 psi) при комнатной температуре в течение 10 часов. Катализатор отфильтровывают и добавляют 2N НСl (940 мкл, 1,88 ммоль). Смесь лиофилизуют один раз и затем лиофилизуют повторно из 20% СН3СN/Н2О, получая промежуточный продукт 19j в виде светло-коричневого твердого вещества (862 мг), который используют без дополнительной очистки. Масс-спектр 408,2 МН+.
Стадия g: Каталитическое гидрирование и обработку проводят способом, по существу аналогичным стадии 18g, получая указанный в заголовке продукт (22 мг, 4%) в виде белого твердого вещества. Масс-спектр 1088,2 МН+.
Пример 20
цикло[Тrр-D-Тrр-Lуs-Туr(Вzl)-Pheψ(4-(фенил)имидазол)-Gly]
Соединение примера 20 получают согласно Схеме 3, способом, по существу аналогичным примеру 18, за исключением того, что вместо 2-бром-3’-метоксиацетофенона на стадии i используют 2-бромацетофенон. Масс-спектр 1057,4 МН+.
Пример 21
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-Pheψ(4-(3-метоксифенил)имидазол)-(ε)Ahx]
Соединение примера 21 получают согласно Схеме 3, способом, по существу аналогичным примеру 18, со следующими различиями:
Стадия j: 2-(1-(S)-амино-2-фенилэтил)-1-(6-(этокси)-6-оксогексил)-4-(3-метоксифенил)имидазол
Промежуточный продукт 21i (855 мг, 2,0 ммоль) растворяют в DMF (8,0 мл) и обрабатывают КНСО3 (200 мг, 2,0 ммоль) и этил 6-бромгексаноатом (3,56 мл, 20 ммоль) при температуре около 120°С в течение 8 часов. Смесь концентрируют, а остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь 2:1=гексан:ЕtOАс и получая чистый продукт в виде смолы (0,94 г).
С сырого алкилированного продукта удаляют защиту посредством гидрирования в уксусной кислоте с использованием 10% Pd на угле в качестве катализатора. Катализатор отфильтровывают и выпаривают растворители при пониженном давлении. Остаток растворяют в разбавленной НСl, замораживают и лиофилизуют, получая промежуточный продукт 21j в виде бледно-желтого твердого вещества (720 мг, 91%), которое используют на следующей стадии без дополнительной очистки. Масс-спектр 436,2 МН+.
Стадия k и стадия 1: 2-(1-(S)-((H-Trp-D-Trp-Lys(Воc)-Туr(Bzl)-2-(1-(S)-амино-2-фенилэтил)-1-(6-(этокси)-6-оксогексил)-4-(3-метоксифенил)имидазол.
Fmoc-Tyr(Bzl)-OAt получают, перемешивая Fmoc-Tyr(Bzl) -ОН (493 мг, 1,00 ммоль), HOAt (136 мг, 1,0 ммоль) и DCC (206 мг, 1,0 ммоль) в 8 мл EtOAc в течение примерно 1/2 часа и отфильтровывая затем дициклогексилмочевину. Полученный раствор добавляют к раствору промежуточного продукта 21j (457 мг, 0,9 ммоль) в EtOAc (4 мл) и смесь перемешивают над насыщенным раствором NаНСО3 (10 мл) до завершения реакции по данным масс-спектрального анализа. Водный слой удаляют, a EtOAc-слой сушат над Na2SO4, фильтруют и обрабатывают трис(2-аминоэтил)амином (2,7 мл). Смесь энергично перемешивают примерно в течение 1/2 часа. EtOAc-слой промывают насыщенным раствором NaCl (2×60 мл), а затем доводят рН раствора примерно до 5,5 при помощи 10% фосфатного буфера (3×15 мл).
Затем с промежуточного продукта последовательно снимают защиту и сочетают с Fmoc-Lys(Cbz)-OSu, Fmoc-D-Trp-OSu и Fmoc-Trp-Osu способом, по существу аналогичным описанному выше для цикла Fmoc-Tyr(Bzl)-OAt. Финальное удаление защитной группы Fmoc дает промежуточный этиловый эфир со снятой защитой с N-конца. EtOAc-слой сушат над Na2SO4, фильтруют и разбавляют 4 объемами гексана. Растворители декантируют, а остаток растирают с гексаном, получая продукт в виде твердого вещества (0,67 г, 58%), который используют без дополнительной очистки. Масс-спектр 1289,6 МН+.
Этиловый эфир удаляют путем обработки промежуточного продукта в МеОН (5,0 мл) 1,7М раствором NaOH (1,7 мл) в течение ночи. рН смеси доводят примерно до 8,2 при помощи 5% раствора НСl, растворители удаляют при пониженном давлении. Остаток переносят в DMF, удаляют NaCl фильтрованием, а раствор в DMF используют на следующей стадии без дальнейшей очистки.
Циклизацию и удаление защиты проводят способом, аналогичным примеру 18, получая соединение, указанное в заголовке примера 21, в виде белого порошка (62 мг). Масс спектр 1143,9 МН+.
Пример 22
цикло[Тrр-D-Тrр-Lуs-Туr(Вzl)-Рhеψ(4-(3-гидроксифенил)имидазол)-(γ)Abu]
Соединение примера 22 получают согласно Схеме 3, способом, по существу аналогичным примеру 18, со следующими различиями:
Стадия j: 2-(1-(S)-амино-2-фенилэтил)-1-(4-(этокси)-4-оксобутил)-4-(4-гидроксифенил)имидазол.
Промежуточный продукт 22i (2,0 г, 4,68 ммоль) растворяют в DMF (7,0 мл) и обрабатывают КНСО3 (468 мг, 4,68 ммоль) и этил 4-бромбутиратом (6,70 мл, 46,8 ммоль) и перемешивают при температуре около 100°С в течение примерно 30 часов. Смесь разбавляют эфиром и промывают один раз насыщенным раствором NаНСО3 и один раз насыщенным раствором NaCl. Эфирный слой сушат над Na2SO4, фильтруют и концентрируют до масла. Колоночная хроматография на силикагеле с применением CH2Cl2 в качестве элюента дает продукт в виде масла (2,53 г, 94%). Масс-спектр 542,3 МН+.
Сырой алкилированный продукт (2,53 г, 4,67 ммоль) в CH2Cl2 (50 мл) добавляют по капле в течение 15 минут при температуре примерно -10°С к 1М раствору ВВr3/гексан (23,4 мл) в CH2Cl2 (250 мл). Смеси дают нагреться до комнатной температуры и перемешивают примерно 2 часа. Добавляют этанол (40 мл) и концентрируют смесь примерно до 50 мл. Этот раствор разбавляют этанолом (100 мл) и оставляют на ночь перемешиваться при комнатной температуре. Смесь концентрируют при пониженном давлении, а остаток распределяют между EtOAc и насыщенным раствором NаНСО3. EtOAc-слой сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении до масла (1,41 г, 76%), которое используют на следующих стадиях без защиты фенола. Масс-спектр 394,3 МН+.
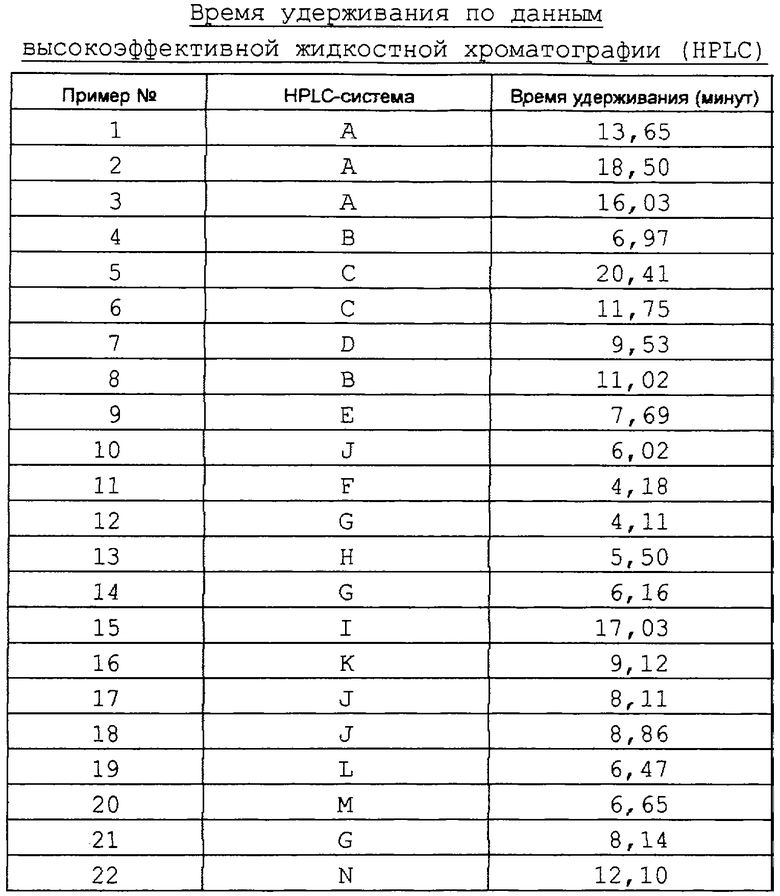
HPLC-системы:
А Градиент: 20-80% СН3СN/0,1% Tfa, 24 мин
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: VYDAC® Protein and Peptide C18
В Градиент: 35-50% СН3СN/0,1% Tfa, 24 мин
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: VYDAC® Protein and Peptide C18
С Градиент: 32-64% CH3CN/0,1 NH4OAc, 24 мин
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: VYDAC® Protein and Peptide C18
D Градиент: 20-60% СН3СN/0,1% Tfa, 24 мин
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: VYDAC® Protein and Peptide C18
E Градиент: 55-75% CH3CN/0,1% Tfa, 24 мин
Скорость потока: 1,0 мл/мин
Детектирование: 220 нм
Колонка: Phenomenex LICHROSPHERE® 5RP18 (Phenomenex, 2320 205th St., Torrance, CA)
F Градиент: 60% СН3СN/0,1% Tfa, 24 мин
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: VYDAC® Protein and Peptide C18
G Градиент: 50% СН3СN/0,1% Tfa, 24 мин изократический
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: VYDAC® Protein and Peptide C18
H Градиент: 38% СН3СN/0,1% Tfa, изократический
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: VYDAC® Protein and Peptide C18
I Градиент: 20-40% СН3СN/0,1% Tfa, 24 мин
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: VYDAC® Protein and Peptide C18
J Градиент: 50% СН3СN/0,1% Tfa, изократический
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: NUCLEOSIL™ C18, 5 мкм (Alltech Associates, 2051 Waukegan Rd., Deerfield, Illinois)
K Градиент: 40% CH3CN/0,1% Tfa, изократический
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: NUCLEOSIL™ С18, 5 мкм
L Градиент: 52% CH3CN/0,1% Tfa, изократический
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: NUCLEOSIL™ С18, 5 мкм
М Градиент: 50% CH3CN/0,1% Tfa, изократический
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: NUCLEOSIL™ С18, 5 мкм
N Градиент: 48% CH3CN/0,1% Tfa, изократический
Скорость потока: 1,0 мл/мин
Детектирование: 254 нм
Колонка: NUCLEOSIL™ С18, 5 мкм
Данные биологических испытаний соединений по примерам 1-22 представлены в таблице 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| АГОНИСТЫ РЕЦЕПТОРОВ НЕЙРОМЕДИНА В И СОМАТОСТАТИНА | 2000 |
|
RU2263680C2 |
| ПЕПТИДНЫЕ ВЕКТОРЫ | 2004 |
|
RU2361876C2 |
| ХИМЕРНЫЕ АНАЛОГИ ЛИГАНДОВ СОМАТОСТАТИНОВЫХ И ДОПАМИНОВЫХ РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБЫ ВОЗДЕЙСТВИЯ НА РЕЦЕПТОРЫ СОМАТОСТАТИНА И/ИЛИ ДОПАМИНА | 2004 |
|
RU2329273C2 |
| АНАЛОГИ GLP-1 | 1999 |
|
RU2288232C9 |
| АНТАГОНИСТЫ РЕЦЕПТОРОВ СОМАТОСТАТИНА | 1997 |
|
RU2179172C2 |
| АНАЛОГИ ПЕПТИДА ЛГ-РФ, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1998 |
|
RU2212247C2 |
| ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, РАДИОФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ, ЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ИМЕЮЩИЕ РАДИОАКТИВНУЮ МЕТКУ | 1994 |
|
RU2145608C1 |
| ХИМЕРНЫЕ АНАЛОГИ СОМАТОСТАТИНА-ДОФАМИНА | 2002 |
|
RU2277539C2 |
| ПРОИЗВОДНЫЕ МЕТАСТИНА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2454425C2 |
| Ингибиторы фактора VIIa | 1999 |
|
RU2223967C2 |
Изобретение относится к соединениям формулы (I) или (II)
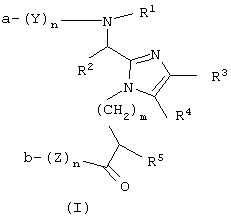
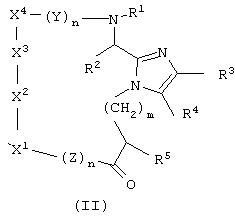
или их фармацевтически приемлемым солям, где Y и Z каждый для каждого случая независимо представляет собой D- или L-природную или неприродную α-аминокислоту; n для каждого случая независимо равно 0 или 4, (I) при условии, что оба n одновременно не могут быть равными 0; и 0 или 4 (II),
причем указанные аминокислоты (в I) составляют цепь: Х1-X2-X3-X4, где Х1 представляет собой Tyr или Trp, который может быть защищен Вос-группой; X2 представляет собой D-Trp; X3 представляет собой Lys, который может быть защищен Вос-группой; X4 представляет собой Nal, Tyr или Thr; m равно 0; а означает Н или R1; b означает -ОН или -OR1; для (II) X1 представляет собой природный или неприродный D- или L-изомер Phe, Trp или Tyr, где в том случае, когда Х1 является Tyr, ароматическое кольцо в его боковой цепи необязательно замещено R6; X2 представляет собой D- или L-изомер Trp; X3 представляет собой Lys; X4 представляет собой природную или неприродную D- или L-аминокислоту Nal, Thr, Tyr или Ser, где в том случае, когда X4 является Tyr, ароматическое кольцо, расположенное в его боковой цепи, может быть необязательно замещено R6, или в том случае, когда X4 является либо Ser, либо Thr, атом кислорода, расположенный в его боковой цепи, необязательно может быть замещен одним или несколькими R1. R1 независимо представляет собой Н или (C1-С4)алкил (I); для (II) R1=Н, (C1-С4)алкил или арил-(C1-С4)алкил; R2 представляет собой Н или замещенный фенил-(C1-С4)алкил для (I); для (II): R2 – замещенный фенил –(С1-С4)алкил; R3 и R4 каждый независимо представляет собой Н или замещенный арил, который может быть замещен одним или несколькими заместителями, выбранными из группы, состоящей из ОН, (C1-C4) алкила и (C1-C4) алкоксигруппы; и R5 для каждого случая представляет собой Н. R3 и R4 каждый независимо представляет собой Н или необязательно замещенный фрагмент, выбранный из группы, состоящей из (C1-C4)алкила и арила, причем необязательно замещенный фрагмент замещен одним или несколькими заместителями, выбранными из группы, состоящей из -ОН и (C1-C4) алкоксигруппы; R5 для каждого случая независимо представляет собой Н; R6 для каждого случая независимо представляет собой арил-(C1-C4)алкил. К двум фармацевтическим композициям, используемым для избирательного связывания, по меньшей мере, с одним из подтипов рецептора соматостатина человека, содержащим в эффективном количестве соединение формулы I или соединение формулы II. 4 н. и 13 з.п. ф-лы, 2 табл.
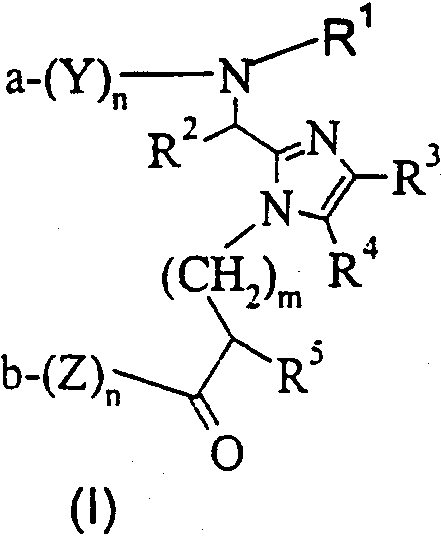
или его фармацевтически приемлемая соль,
где Y и Z каждый для каждого случая независимо представляет собой D- или L- природную или неприродную α-аминокислоту;
n для каждого случая независимо равно 0 или 4 при условии, что оба n одновременно не могут быть равными 0;
причем указанные аминокислоты составляют цепь X1-X2-X3-X4, где
X1 представляет собой Tyr или Trp, который может быть защищен Boc-группой;
X2 представляет собой D-Trp;
X3 представляет собой Lys, который может быть защищен Boc-группой;
X4 представляет собой Nal, Tyr или Thr;
m равно 0;
a означает H или R1;
b означает –OH или-OR1;
R1 независимо представляет собой H или (C1-C4)алкил;
R2 представляет собой H или замещенный фенил-(C1-C4)алкил;
R3 и R4 каждый независимо представляет собой H или замещенный арил, который может быть замещен одним или несколькими заместителями, выбранными из группы, состоящей из OH, (C1-C4)алкила и (C1-C4)алкоксигруппы;
R5 для каждого случая представляет собой H.
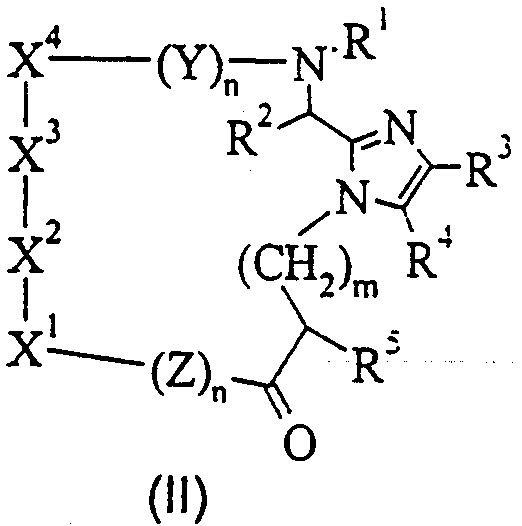
или его фармацевтически приемлемая соль,
где каждый Y и Z для каждого случая независимо представляет собой D- или L- природную или неприродную α-аминокислоту;
m равно 0;
n для каждого случая независимо равно 0 или 1;
R1 для каждого случая независимо представляет собой H, (C1-C4)алкил или арил-(C1-C4)алкил;
R2 представляет собой замещенный фенил-(C1-C4)алкил;
R3 и R4 каждый независимо представляет собой H или необязательно замещенный фрагмент, выбранный из группы, состоящей из (C1-C4)алкила и арила, причем необязательно замещенный фрагмент замещен одним или несколькими заместителями, выбранными из группы, состоящей из –OH и (C1-C4)алкоксигруппы;
R5 для каждого случая независимо представляет собой H;
R6 для каждого случая независимо представляет собой арил-(C1-C4)алкил.
X1 представляет собой природный или неприродный D- или L-изомер Phe, Trp или Tyr, где в том случае, когда X1 является Tyr, ароматическое кольцо в его боковой цепи необязательно замещено R6;
X2 представляет собой D- или L-изомер Trp;
X3 представляет собой Lys;
X4 представляет собой природную или неприродную D- или L-аминокислоту Nal, Thr, Tyr или Ser, где в том случае, когда X4 является Tyr, ароматическое кольцо, расположенное в его боковой цепи, может быть необязательно замещено R6, или в том случае, когда X4 является либо Ser, либо Thr, атом кислорода, расположенный в его боковой цепи, необязательно может быть замещен одним или несколькими R1.
R4 представляет собой Н.
цикло[Tyr-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Tyr(Bzl)-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr(Bzl)-PheΨ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Abu-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(1,1-диметилэтил)-имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Tyr-D-Trp-Lys-Val-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Nal-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Nal-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)-имидазол-( γ)Abu],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(4-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(фенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)-имидазол)-(ε)Ahx] или
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-гидроксифенил)-имидазол)-(γ)Abu].
цикло[Tyr-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Tyr(Bzl)-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr(Bzl)-PheΨ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Abu-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(1,1-диметилэтил)-имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr-PheΨ(4-(3-гидроксифенил)имидазол)-Gly] или
цикло[Tyr-D-Trp-Lys-Val-PheΨ(4-(3-гидроксифенил)-имидазол)-Gly].
цикло[Tyr-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Abu-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)имидазол)-Gly] или
цикло[Tyr-D-Trp-Lys-Val-PheΨ(4-(3-гидроксифенил)-имидазол)-Gly].
цикло[Tyr-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr-PheΨ(4-(3-гидроксифенил)имидазол)-Gly] или
цикло[Tyr-D-Trp-Lys-Val-PheΨ(4-(3-гидроксифенил)-имидазол)-Gly].
цикло[Tyr-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Val-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr(Bzl)-PheΨ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Thr-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Abu-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(1,1-диметилэтил)-имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)-имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Tyr-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Tyr-D-Trp-Lys-Val-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Nal-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Nal-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)-имидазол-( γ)Abu],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(4-метоксифенил)-имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(фенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)-имидазол)-( ∈)Ahx] или
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-гидроксифенил)-имидазол)-( γ)Abu].
цикло[Trp-D-Trp-Lys-Thr-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Phe-D-Trp-Lys-Nal-PheΨ(4-(3-гидроксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)-имидазол-( γ)Abu],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(4-метоксифенил)-имидазол)-Gly] или
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(фенил)имидазол)-Gly].
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)имидазол)-Gly],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(3-метоксифенил)имидазол-( γ)Abu],
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(4-метоксифенил)имидазол)-Gly] или
цикло[Trp-D-Trp-Lys-Tyr(Bzl)-PheΨ(4-(фенил)имидазол)-Gly].
| Способ получения производных соматостатина | 1974 |
|
SU586837A3 |
| Чугунный экономайзер с вертикально-расположенными трубами с поперечными ребрами | 1911 |
|
SU1978A1 |
| Gilon E.A | |||
| J.Med | |||
| Chemistry,1998, v.41, N5, p.919-929.Shimon L | |||
| et.al | |||
| Journal of clinical investigation, 1997, v | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

