Предпосылки изобретения
Описание федерального финансирования
Настоящее изобретение было осуществлено отчасти с использованием средств, полученных по гранту HL59652 от National Institutes of Health. Следовательно, федеральное правительство обладает некоторыми правами на настоящее изобретение.
Область изобретения
Настоящее изобретение в целом относится к области физиологии и молекулярной биологии. Более конкретно настоящее изобретение касается повреждений ДНК и влияния повреждений ДНК на развитие атеросклероза.
Описание данной области техники
Было установлено, что активные формы кислорода играют ключевую роль в патогенезе атеросклеротических повреждений [1-6], но лежащие в основе этого механизмы выяснены не были. Например, механизмы, опосредованные активными формами кислорода, по-видимому, являются важным фактором в окислении LDL (ox-LDL), что является ключевым событием атерогенеза [3, 7, 8]. Исследования показали, что и супероксид (O
Митохондрия является основным источником активных форм кислорода (О
Атеросклеротическая ишемическая болезнь сердца является ведущей причиной смертности в западных странах. Хотя существует существенное противоречие в определении точной последовательности событий, приводящих к атеросклерозу коронарных сосудов, продолжают накапливаться доказательства того, что атеросклеротические повреждения обусловлены факторами, опосредуемыми активными формами кислорода. Макрофаги распознают и интернализуют ox-LDL с участием рецепторов-“скэвенджеров”, становясь ксантомными клетками. Накопление таких ксантомных клеток связано с долговременными изменениями в физиологии сосудов, включая миграцию и пролиферацию клеток гладкой мускулатуры, синтез белков внеклеточного матрикса и далее дисфункцию клеток эндотелия, причем все это является центральными компонентами атеросклеротических бляшек. Сходным образом многие факторы риска развития ишемической болезни сердца связаны с повышением выработки активных форм кислорода (например, курение и гиперхолестеринемия). В артерии выработка активных форм кислорода может индуцироваться метаболическими процессами (окислительное фосфорилирование в митохондриях), активацией цитокинов или факторов роста, стимуляцией макрофагов или нейтрофилов (воспалительный ответ) и взаимодействием оксида азота с супероксидом с образованием пероксинитрита, который, в свою очередь, образует синглетный кислород и гидроксильный радикал. Следовательно, хотя имеется ряд процессов, важных с точки зрения атерогенеза, механизмы, опосредованные активными формами кислорода, и их проявления входят в группу наиболее важных из них.
Многочисленные исследования были нацелены на митохондрии, как уязвимую мишень для активных форм кислорода. Связь митохондриальной ДНК с матричной зоной внутренней мембраны делает ее чувствительной к повреждениям мембраны и потенциальной мишенью для электрофильных факторов, образованных на мембране. Помимо тесной связи митохондриальной ДНК с внутренней мембраной и ОК-ФОС, дополнительными факторами, делающими ее чувствительной к повреждению, являются отсутствие защитных гистонов и негистоновых белков и ее ограниченная репарационная способность. Проведенные ранее исследования показали, что митохондрии восприимчивы к повреждениям, опосредуемым активными формами кислорода, что проявляется в интенсификации перекисного окисления липидов и повреждений митохондриальной ДНК. В частности, было установлено, что воздействие активных форм кислорода на эндотелиальные клетки приводит к предпочтительному повреждению митохондриальной ДНК, снижению содержания транскриптов с мтДНК и нарушению митохондриального ОК-ФОС.
В известном уровне техники имеется дефицит способов измерения оксидативного стресса, который влияет на атерогенез. Настоящее изобретение восполняет этот давно существующий пробел и потребность данной области техники.
Краткое содержание изобретения
Настоящее изобретение показывает, что активные формы кислорода опосредуют повреждения митохондрий и дисфункцию клеток эндотелия пупочной вены человека (HUVEC) и клеток гладкой мускулатуры аорты человека (HASMC) in vitro. Повреждения ДНК, экспрессию генов и синтез митохондриальных белков оценивали в клетках, обработанных Н2O2, пероксинитритом и О
Принимая во внимание то, что митохондриальная ДНК более чувствительна к повреждению, опосредуемому активными формами кислорода, а также учитывая, что усиленный оксидативный стресс, как считается, участвует в начальных событиях атерогенеза, настоящее изобретение демонстрирует, что митохондриальная ДНК в тканях аорты, готовых к тому, чтобы стать атеросклеротическими, характеризуется повышенной поврежденностью. Для этого уровни повреждений митохондриальной ДНК, имеющихся в тканях аорты в гиперхолестеринемической мышиной модели атеросклероза (нулевые по аполипопротеину-Е мыши), сравнивали с показателями здоровых контрольных мышей аналогичного возраста. Оценка повреждений ДНК показала, что в тканях аорты у мышей ароЕ имеет место существенное повышение уровня повреждений митохондриальной ДНК до и после развития патологически выявляемых повреждений (по сравнению со здоровым контролем).
Кроме того, уровень повреждений митохондриальной ДНК увеличивается с возрастом у всех мышей, однако только у мышей ароЕ имеется достоверная (Р<0,05) связь увеличения уровня повреждений с возрастом. Напротив, рацион коррелировал с уровнем повреждений митохондриальной ДНК только у мышей C57BL в возрасте 10 недель, причем "западный" рацион приводил к увеличению повреждений. Наконец, было обнаружено, что снижение пищевого белка достоверно (Р<0,05) коррелировало со снижением поврежденности митохондриальной ДНК в аорте и у ароЕ-, и у контрольных мышей. Не было выявлено отчетливых связей повреждений с локусом β-глобина, являющимся маркером повреждений ядерной ДНК. Гистохимический анализ аорт в каждой группе показал присутствие атеросклеротических повреждений только у старых мышей ароЕ, выкармливавшихся на смешанном (4% жиров) или "западном" (21% жиров) рационах. Как ожидалось, уровни пероксидов липидов и холестерина достоверно возрастали у мышей ароЕ по сравнению с контролем C57BL при тех же возрастах (р<0,05). Однако, уровни пероксидов липидов не увеличивались существенно в группе ароЕ при высокожирном "западном" рационе по сравнению со смешанным рационом. Таким образом, полученные данные подтверждают, что: (1) в мышиной модели атеросклероза in vivo повреждения митохондриальной ДНК происходят до развития атеросклеротических повреждений или одновременно с ним; (2) повреждения митохондриальной ДНК в аорте усиливаются с возрастом in vivo; (3) генотип ароЕ в большей степени влияет на уровень повреждений митохондриальной ДНК по сравнению с составом рациона; и (4) влияние рациона на повреждения митохондриальной ДНК, по-видимому, более выражены у молодых мышей C57BL. Следовательно, повреждения митохондриальной ДНК происходят на ранних этапах развития атеросклероза и могут являться инициирующим событием в атерогенезе.
Одним объектом настоящего изобретения является представление способов прогнозирования атеросклеротической ишемической болезни сердца и любого иного заболевания, вызываемого оксидативным стрессом, основываясь на степени повреждения митохондриальной ДНК или на сходных измерениях дисфункции митохондрий, которые обусловливаются повреждениями митохондриальной ДНК, включая изменения в выработке митохондриальных белков, изменения в митохондриальном окислительном фосфорилировании или изменения в выработке АТФ в митохондриях.
Осуществлением настоящего изобретения является способ прогнозирования атеросклероза у субъекта с повышенным риском, включающий следующие стадии: (а) отбор у субъекта образцов интересующих тканей; (b) определение количественного уровня повреждений митохондриальной ДНК (мтДНК) в интересующей ткани; и (с) сравнение количественного уровня повреждений митохондриальной ДНК в интересующей ткани субъекта с повышенным риском с количественным уровнем повреждений митохондриальной ДНК в интересующей ткани контрольного субъекта, у которого атеросклероз отсутствует; где более высокий количественный уровень повреждений митохондриальной ДНК у субъекта с повышенным риском, чем у контрольного субъекта, является прогностическим признаком атеросклероза для субъекта из группы риска.
Другое осуществление настоящего изобретения представляет собой способ измерения количественного уровня оксидативного стресса у субъекта, включающий следующие стадии: (а) отбор образца интересующей ткани у субъекта; (b) определение количественного уровня повреждений митохондриальной ДНК (мтДНК) в интересующей ткани; (с) определение количественного уровня повреждений ДНК в ядерном гене в интересующей ткани; и (d) сравнение количественного, в расчете на длину ДНК, уровня повреждений ДНК между митохондриальной ДНК и ядерным геном, где более высокий количественный уровень повреждений митохондриальной ДНК в расчете на длину ДНК, чем уровень повреждений ядерной ДНК в расчете на длину ДНК, является индикатором повышенного уровня оксидативного стресса у субъекта.
Еще одно осуществление настоящего изобретения представляет собой способ определения эффективности лечения, призванного снизить риск развития ишемической болезни сердца, у субъекта, включающий следующие стадии: (а) отбор лейкоцитов периферической крови у субъекта до лечения; (b) отбор образца интересующей ткани у субъекта после лечения; и (с) определение количественного уровня повреждений митохондриальной ДНК (мтДНК) в интересующей ткани, взятой до лечения и после лечения, где снижение уровня повреждений митохондриальной ДНК в результате лечения является индикатором того, что лечение снизило риск развития ишемической болезни сердца.
Другие и дополнительные аспекты, свойства и преимущества настоящего изобретения будут ясны из нижеследующего описания представляемых предпочтительных вариантов изобретения. Данные варианты даны для целей описания.
Краткое описание фигур
Для того, чтобы материал, в котором указанные выше свойства, преимущества и объекты настоящего изобретения, равно как и другие, которые станут понятными, был понятен в деталях, более конкретные описания изобретения, кратко описанного выше, могут быть выполнены путем отсылки к некоторым его осуществлениям, которые проиллюстрированы на прилагаемых фигурах. Данные фигуры образуют часть настоящей заявки. Необходимо отметить, однако, что прилагаемые фируры лишь иллюстрируют предпочтительные осуществления изобретения и, следовательно, не должны рассматриваться как ограничивающие его объем.
На фигуре 1А показан пример повреждений ДНК, связанных с воздействием на HUVEC пероксида водорода. Клетки обрабатывали в течение 60 минут 0,2 мМ H2O2, собирали и проводили КолПЦР. Контрольные культуры инкубировали только в бессывороточной среде. Меньшее количество продукта указывает на повышение поврежденности матрицы. На фигуре 1В показан анализ обработанных H2O2 клеток HASMC и HUVEC в зависимости от времени. Клетки обрабатывали 0,2 мМ H2O2 в течение 0-60 минут, экстрагировали геномную ДНК, и повреждения в расчете на 10 тысяч пар нуклеотидов (т.п.н.) оценивали путем сравнения с необработанным контролем (нулевой класс). В каждом отдельном эксперименте проводили по крайней мере по две ПЦР в расчете на образец. Звездочки (*) указывают на статистически значимое (Р<0,05) повышение поврежденности по сравнению с необработанным контролем. Величины являются средними значениями (± s.e.).
На фигуре 2А показан образец повреждений ДНК в связи с воздействием пероксинитрита на клетки HUVEC и HASMC. Клетки обрабатывали в течение 60 минут указанными дозами пероксинитрита, собирали и проводили КолПЦР. Контрольные культуры инкубировали только в бессывороточной среде. Меньшее количество продукта указывает на повышенную поврежденность матрицы. На фигуре 2В показано воздействие 1 мМ SIN-1 на HUVEC и HASMC. SIN-1 образует эквимолярные количества оксида азота и О
На фигуре 3 отражены уровни транскриптов митохондриальной ДНК в клетках, обработанных пероксинитритом. Диаграмма представляет относительные уровни транскриптов (среднее ± s.e.; нормализованы по β-актину) 16S-pPHK, ND2 и Cyt-b после 60-минутой обработки (0,1 мМ и 0,5 мМ; культуральная среда без сыворотки) по отношению к необработанному контролю (бессывороточная среда). Уровни транскриптов определяли методом Нозерн-блоттинга и гибридизации с соответствующим радиоактивно помеченным зондом. Звездочки (*) указывают на уровни транскриптов, которые статистически значимо (Р<0,05) отличаются от необработанного контроля.
На фигуре 4 отображено включение 35S-метионина в синтезируемые митохондриальные белки. На фигуре 4А показан пример мечения белка после воздействия пероксинитрита на клетки HASMC (левая часть). Клетки обрабатывали 0,1 мМ или 0/5 мМ пероксинитрита в течение 60 минут и затем помечали 35S-метионином в течение примерно 2 часов с последующим электрофорезом в ДСН-ПААГ. В правой части показан тот же гель, окрашенный кумасси голубым. На фигуре 4В изображена таблица, обобщающая проценты включения 35S-метионина в клетки и HUVEC, и HASMC, обработанные активными формами кислорода, по сравнению с контролем (“пустые” обработки в бессывороточной среде). Сокращения: н/д - нет данных.
На фигуре 5 показана диаграмма, отображающая митохондриальное дыхание (оцененное по восстановлению МТТ) и общее клеточное содержание АТФ. Клетки HASMC и HUVEC обрабатывали соответствующей концентрацией пероксинитрита в течение 1 часа при 37°С. На фигуре 5А показана диаграмма, отражающая относительные уровни восстановления МТТ. После обработки клетки промывали в ФСБ и инкубировали в течение 1 часа в кондиционной среде, содержащей 2,0 мкг/мл МТТ. После этого среду удаляли и клетки лизировали, измеряя поглощение при 570 нм. Восстановление МТТ (среднее ± s.e.) приводится в виде доли от уровня восстановления в необработанном контроле. На фигуре 5В показано определение общего содержания АТФ в HASMC и HUVEC, обработанных пероксинитритом. После такой обработки клетки обрабатывали АТФ-высвобождающим реагентом (Labsystems), и содержание АТФ определяли с помощью люминесценции (Molecular Probes). Значения приведены относительно необработанных клеток (100%).
На фигуре 6 показано, что мыши ароЕ характеризуются существенно большей поврежденностью митохондриальной ДНК в аорте и тканях сердца по сравнению с контролем того же возраста. На фигуре 6А показаны повреждения митохондриальной ДНК, оцененные методом КолПЦР, в аорте у ароЕ- и контрольных мышей, на основании чего определяли уровень амплификации по отношению к контрольной группе возраста 10 недель. Меньшее количество амплифицированного продукта соответствует повышенной поврежденности митохондриальной ДНК. Звездочки (*) указывают на статистически значимое различие между аортами в контроле и группе ароЕ. На фигуре 6В показаны повреждения митохондриальной ДНК, оцененные методом КолПЦР, в левом желудочке мышей ароЕ и из контроля, на основании чего определяли уровень амплификации относительно контрольной группы возраста 10 недель. Меньшее количество амплифицированного продукта соответствует повышенной поврежденности митохондриальной ДНК. Звездочки (*) указывают на статистически значимое различие между аортами ароЕ и контроля.
На фигуре 7 показано то, что снижение усвоения пищевого белка коррелирует с меньшим уровнем повреждений митохондриальной ДНК у контрольных и ароЕ-мышей. Контрольных и ароЕ-мышей выкармливали рационом с 16% или 24% белка в течение 4 недель, начиная с 6-недельного возраста, и повреждения митохондриальной ДНК оценивали методом КолПЦР. На фигуре 7А показан уровень амплификации митохондриальной ДНК в аортах контрольных и ароЕ-мышей по отношению к контрольной группе, выкармливаемой рационом с 24% белка. Меньшее количество амплифицированного продукта означает повышение уровня поврежденности митохондриальной ДНК. Звездочки (*) указывают на статистически значимое различие между мышами, выкармливавшимися при 16% и 24% белка. Р-величины, определенные с помощью критерия Стьюдента, даны в скобках. На фигуре 7В показан относительный уровень амплификации митохондриальной ДНК контроля и ароЕ в левом желудочке по сравнению с контрольной группой, выкармливавшейся при 24% белка. Меньшее количество амплифицированного продукта означает повышение уровня повреждений митохондриальной ДНК. Звездочки (*) указывают на статистически значимое различие между мышами, выкармливавшимися при 16% и 24% белка. Величины Р, определенные с помощью критерия Стьюдента, даны в скобках.
На фигуре 8 показано возрастание повреждений мтДНК в аортах у 3-недельных мышей ароЕ-/- по сравнению с контролем. Продукты 16,0-т.п.н. и 0,08-т.п.н. представляют длинный и короткий КолПЦР-фрагменты мтДНК соответственно. В таблице ниже приведены частоты относительной поврежденности (число повреждений на 10 т.п.н.) в аортах у 3-недельных контрольных (нулевой класс: А0) и ароЕ-/- мышей. Величины являются средними ± s.е.
На фигуре 9 показано, что перекисное окисление липидов в существенной степени повышено у мышей ароЕ по сравнению с контрольными мышами, хотя перекисное окисление липидов значительно возрастает у контрольных мышей, которым давали "западный" рацион (21% жиров). Диаграмма иллюстрирует уровень перекисного окисления липидов у ароЕ- и контрольных мышей при выкармливании на смешанном (4% жиров) и "западном" (21%) рационах. Значения выражены относительно контрольных мышей, выкармливаемых на смешанном рационе. Хотя мыши ароЕ характеризовались существенно более высокими уровнями перекисного окисления липидов по сравнению с контрольными мышами независимо от рациона, достоверные различия в уровне перекисного окисления липидов между мышами ароЕ, которым давали смешанный или "западный" рационы, отсутствовали. Напротив, у контрольных мышей, получавших "западный" рацион, наблюдались существенно более высокие уровни перекисного окисления липидов (отмечено “*”) по сравнению с контрольными мышами, получавшими смешанный рацион.
На фигурах 10А, 10В и 10С показано, что у гибридных мышей ароЕ-/-, SOD2-/+ имелись более интенсивные атеросклеротические повреждения и повреждения мтДНК. На фигуре 10А цельные аорты у родственных гибридов ароЕ-/-, SOD2-/+ и ароЕ-/- окрашивали масляным красным О. В верхней левой части показана окрашенная масляным красным О аорта 17-недельного самца ароЕ-/-, SOD2-/+ (4% жиров в рационе), а в верхней правой части показаны данные для родственных самцов ароЕ-/- (4% жиров в рационе). На нижних левой и правой частях показаны окрашенные аорты 34-недельных родственных особей, соответственно ароЕ-/-, SOD2-/+ и ароЕ-/-. Черные стрелки указывают на присутствие крупных атеросклеротических повреждений у ароЕ-/-, SOD2-/+ (пока отсутствующих у 17-недельного родственника ароЕ-/- того же помета), а красная стрелка в каждой части указывает на пример мелких повреждений, образованных на артериальных ответвлениях, которые являются более частыми и характерными для мышей ароЕ-/-, SOD2-/+. На фигуре 10В приведена диаграмма, отражающая частоту атеросклеротических повреждений в цельных аортах мышей ароЕ-/-, SOD2-/+ и ароЕ-/-, визуализованных окрашиванием масляным красным О (n=4). На фигуре 10С показана относительная поврежденность мтДНК в аорте у родственников ароЕ-/-, SOD2-/+ и ароЕ-/- (n=12). Родственные особи ароЕ-/- в этой серии экспериментов являлись контролем, поэтому в частотах повреждений их обозначили как “нулевой класс” (Ао). Частоты повреждений приведены как средние ± s.е.
На фигуре 11 показано увеличение встречаемости повреждений митохондриальной ДНК у больных, характеризующихся наличием факторов риска инфаркта миокарда. Образцы крови были отобраны во время проведения коронарной ангиографии с целью определения уровня повреждений митохондриальной ДНК методом КолПЦР. На данной фигуре уровни повреждений митохондриальной ДНК в существенной степени повышались по отношению к средней для “нормальной” популяции. Диаграмма отражает долю больных в каждой категории, характеризующихся повышенной поврежденностью митохондриальной ДНК. Наличие повышенной поврежденности митохондриальной ДНК оказывается более частым у субъектов, страдающих артериальной гипертонией, курящих, страдающих сахарным диабетом и сочетанием этих факторов, определяющих риск инфаркта миокарда.
На фигуре 12 показана поврежденность митохондриальной ДНК до и после 20-мильного “тренировочного забега”. Были взяты пробы крови с получением образца лейкоцитарной пленки, на материале которой уровень повреждений митохондриальной ДНК определяли с помощью КолПЦР. Хотя повреждения митохондриальной ДНК появляются сразу после тренировочного забега, как здесь показано, их уровень быстро возвращается к нормальной величине.
На фигуре 13 показано, что у спортсменов-сверхмарафонцев имеется существенно меньший уровень повреждений митохондриальной ДНК после включения в рацион высокожирной пищи по сравнению с контрольными субъектами. Во всех случаях повреждения митохондриальной ДНК были оценены до, сразу после употребления жирной пищи и через 4-6 часов. Выявленные уровни повреждений митохондриальной ДНК в контроле отражают мгновенный эффект пищевого усвоения АФК в форме жиров и холестерина. Относительная “защищенность”, обнаруженная у сверхмарафонцев, может определяться позитивной регуляцией антиокислительных защитных механизмов у этих людей.
На фигуре 14 показано, что у сверхмарафонцев имеется существенно меньший уровень повреждаемости белков после употребления высокожирной пищи по сравнению с контрольными субъектами.
На фигурах 15А и 15В показаны результаты КолПЦР мтДНК из нормальной и атеросклеротизованной аорты человека. На фигуре 15А показаны результаты КолПЦР-сравнения “нормальной” и атеросклеротизованной аорты при одном и том же возрасте. Продукты 16,2 т.п.н. (верхний ряд) и 0,22 т.п.н. (нижний ряд) представляют собой длинный и короткий КолПЦР-амплификоны человека соответственно. Методы КолПЦР мтДНК человека были описаны ранее (Ballinger, 1996, 1999; Yakes, 1997). На фигуре 15В показаны оценки повреждений мтДНК в расчете на 10 т.п.н. из здоровой и атеросклеротизованной аорты человека. Средняя частота повреждений (± s.e.) для атеросклеротизованных аорт выражена относительно нормальной группы (обозначенной как “нулевой класс” - 0,0±0,06 повреждений на 10 т.п.н.). Статистический анализ (независимый критерий Стьюдента) показал наличие достоверных различий между нормальными и атеросклеротизованными аортами.
Подробное описание изобретения
Было установлено, что активные формы кислорода играют ключевую роль в патогенезе атеросклеротических повреждений, однако лежащие в основе этого механизмы выяснены не были. В среде кровеносных сосудов супероксидный ион (О2
Повреждения ДНК также можно оценивать в ткани аорты у 10- и 34-недельных мышей с атеросклерозом (ароЕ) и контрольных особей. Мыши ароЕ лишены аполипопротеина-Е, являющегося высокоаффинным лигандом для рецепторов липопротеина, которые важны в связи с поглощением LDL из кровяного русла. Следовательно, такие мыши характеризуются существенно увеличенным уровнем сывороточных холестерина и триглицеридов, и у них атеросклеротические бляшки начинают развиваться в возрасте 20 недель. Для определения уровня повреждений ядерной и митохондриальной ДНК в аорте в каждой из групп геномную ДНК экстрагировали из проксимальной и дистальной областей аорты и подвергали количественной ПЦР (КолПЦР). Мыши ароЕ характеризовались повышенной поврежденностью митохондриальной ДНК в аортах по сравнению с контрольными мышами C57BL во всех группах, за исключением возраста 10 недель в группе, выкармливаемой на "западном" рационе. При том, что уровень повреждений митохондриальной ДНК увеличивался с возрастом у всех мышей, только у мышей ароЕ имелся статистически значимый (Р <0,05) уровень повреждений митохондриальной ДНК, связанный с возрастом, "западный" рацион коррелировал с повышенной поврежденностью митохондриальной ДНК (по сравнению со смешанным рационом) только у 10-недельных мышей линии C57BL. Напротив, отсутствовали достоверные различия по ядерной ДНК (β-глобин). Кроме того, уровни пероксидов липидов и холестерина достоверно возрастали у мышей ароЕ по сравнению с контрольными мышами C57BL того же возраста (р<0,05), а гистохимический анализ аорт показал присутствие атеросклеротических повреждений в возрастных группах ароЕ при "западном" и смешанном рационах. Полученные данные подтверждают, что: (1) повреждения митохондриальной ДНК связаны с атерогенезом в мышиной модели атеросклероза in vivo; (2) повреждения митохондриальной ДНК аорты увеличиваются с возрастом in vivo; (3) генотип ароЕ оказывает более существенное влияние на уровень повреждений митохондриальной ДНК по сравнению с фактором рациона; и (4) влияние рациона на повреждения митохондриальной ДНК, по-видимому, сильнее сказывается у молодых мышей C57BL. Следовательно, повреждения митохондриальной ДНК рассматриваются как инициирующее атерогенез событие.
Настоящее изобретение относится к способам прогнозирования развития атеросклеротической ишемической болезни сердца на основании уровней повреждений митохондриальной ДНК. Более конкретно, настоящее изобретение относится к способу оценки “атеросклеротического состояния” субъекта, включающему следующие стадии: (а) отбор образца интересующей ткани указанного субъекта; (b) определение количественного уровня повреждений митохондриальной ДНК в указанной интересующей ткани; и (с) сравнение количественного уровня повреждений митохондриальной ДНК в интересующей ткани указанного субъекта с количественным уровнем повреждений митохондриальной ДНК в интересующей ткани контрольного субъекта, у которого нет атеросклероза, где более высокий количественный уровень повреждений митохондриальной ДНК у указанного субъекта с повышенным риском, чем у указанного контрольного субъекта, является индикатором атеросклероза у указанного субъекта. Хотя повреждение митохондриальной ДНК может быть определено с использованием любого метода, известного специалистам в данной области техники, одним из их примеров является количественная ПЦР. Данный метод может быть использован для идентификации и количественного определения уровня повреждений митохондриальной ДНК у любого субъекта. Предпочтительно обследуемый субъект связан по крайней мере с одним фактором риска, ассоциированным с атеросклерозом. Такие факторы риска хорошо известны в данной области техники и включают, например, курение или жевание табака, артериальную гипертонию, сахарный диабет, ожирение, гиперхолестеринемию и гиперлипидемию.
Также настоящее изобретение относится к способу определения количественного уровня оксидативного стресса у субъекта, включающему стадии: (а) отбор образца интересующей ткани указанного субъекта; (b) определение количественного уровня повреждений митохондриальной ДНК в указанной интересующей ткани; (с) определение количественного уровня повреждений ДНК в ядерном гене в указанной интересующей ткани; и (d) сравнение количественного уровня повреждений в расчете на длину ДНК у указанной митохондриальной ДНК и указанного ядерного гена, где более высокий количественный уровень повреждений митохондриальной ДНК в расчете на длину ДНК по сравнению таковым у ядерной ДНК в расчете на длину ДНК является индикатором повышенного уровня оксидативного стресса у указанного субъекта. Ядерный ген выбирают из группы, которая включает β-глобиновый локус, транскрипционно активные или неактивные гены, что зависит от того, нужно ли определять уровень повреждений ядерной ДНК в активно транскрибирующихся генах или в группе ядерных генов. Повреждения митохондриальной ДНК и повреждения ДНК в указанных ядерных генах могут быть определены с помощью количественной ПЦР. В целом, повышенные количественные уровни оксидативных стрессов являются прогностическими признаками атерогенеза, артериальной гипертонии, сахарного диабета, гиперхолестеринемии, табакокурения, дегенеративных заболеваний, связанных со старением, и раковых опухолей.
Также настоящее изобретение направлено на способ определения эффективности лекарственного средства по снижению риска развития атеросклероза у субъекта, включающий стадии: (а) взятие пробы интересующей ткани от указанного субъекта до и после введения указанного лекарства указанному субъекту; (b) определение количественного уровня повреждений митохондриальной ДНК в указанной интересующей ткани, взятой в качестве пробы, где снижение поврежденности митохондриальной ДНК после указанного лечения является индикатором того, что лечение снижает риск атеросклероза.
По использованию в данном тексте термин “атерогенез” или “атеросклероз” обозначает биологический процесс, который приводит к образованию бляшек, стенозу и закупорке периферических, коронарных или мозговых артерий, что обусловливает ишемию, инфаркт миокарда, инсульт и последующие болезненные состояния и смертность.
По использованию в данном тексте термин “интересующая ткань” обозначает любую кроветворную клетку или образец ткани.
По использованию в данном тексте термин “оксидативный стресс” обозначает патофизиологические воздействия активных форм кислорода, таких как H2O2, супероксид, пероксинитрит, все их производные и другие активные формы кислорода, на нормальную функцию клеток. Мишенями оксидативных стрессов могут быть белки, антигены, липиды, РНК, ДНК или любой другой клеточный компонент.
По использованию в данном тексте термин “введение антиоксиданта” указывает на любые органические или неорганические вещества, которые взаимодействуют с активной формой кислорода, обнуляя его патофизиологические проявления.
По использованию в данном тексте термин “низкобелковый рацион” в целом обозначает рацион, содержащий менее 16% белка, но который может быть откорректирован в соответствии с количеством белка, содержащегося в анализируемом рационе.
По использованию в данном тексте термин “повреждение мтДНК” в целом указывает на любой тип повреждения (например, замены нуклеотидов, апуриновые сайты, разрывы цепи, образование аддуктов и т.п.) или мутацию размера мтДНК (делеции, вставки и дупликации), которые потенциально могут быть выявлены либо непосредственно с помощью КолПЦР (за счет блокирования полимеразы или образования КолПЦР-продукта, размер которого отличается от ожидаемого вследствие мутаций размера мтДНК) или в сочетании с ферментной обработкой (например, ДНК перед проведением КолПЦР можно обработать FAPY-гликозилазой с целью выявления 8-оксогуанина).
Для специалиста в данной области техники будет понятно, что определение уровня повреждений митохондриальной ДНК является лишь одним возможным способом идентификации оксидативного стресса. Любой “далее расположенный” или результирующий эффект повреждений митохондриальной ДНК будет отражать тот же самый процесс развития заболевания. Например, измерение выработки митохондриальных белков, изменений митохондриального окислительного фосфорилирования или изменений выработки АТФ в митохондриях будет решать такую же задачу.
Нижеследующие примеры приведены с целью проиллюстрировать различные варианты настоящего изобретения и не призваны как бы то ни было ограничить настоящее изобретение.
Пример 1
Клетки in vitro и мыши
Клетки эндотелия пупочной вены человека (HUVEC) и клетки гладкой мускулатуры аорты человека (HASMC) поддерживали при 37°С и 5% CO2 + 95% воздуха в модифицированной по Дальбекко культуральной среде Игла (HASMC: Cell Gro) или среде M199 (HUVEC: Cell Gro), дополненных 10% (HASMC) или 20% (HUVEC) инактивированной нагреванием плодной телячьей сыворотки (Gibco BRL), буфером HEPES (10 мМ), глутамином, пенициллином и стрептомицином. Колбы стандартным образом разделяли каждые 3-4 дня и дисассоциировали для экспериментов с использованием трипсина-ЭДТА (Gibco BRL). Клетки обрабатывали различными активными формами кислорода (“пустая” контрольная обработка - бессывороточной средой) при степени слияния 70-80% между 5-7-ми пересевами.
“Выключенные” по аполипопротеину-Е мыши (ароЕ) (-/-), как было подтверждено, являются эффективной моделью развития атеросклероза. Мыши ароЕ(-/-) лишены аполипопротеина-Е, являющегося высокоаффинным лигандом для рецепторов липопротеинов, которые играют важную роль в связывании LDL из кровяного русла. Следовательно, такие мыши характеризуются существенно повышенными уровнями сывороточных холестерина и триглицеридов, и атеросклеротические бляшки начинают у них развиваться в возрасте 20 недель при выкармливании на "западном" высокожирном рационе. Среди мышиных моделей атеросклероза мыши линии ароЕ(-/-) наиболее точно воспроизводят параметры атеросклероза человека.
Контрольные С57 и мыши ароЕ (на основе линии C57BL) были закуплены (Jackson Laboratories, Bar Harbor) в возрасте 5 недель, после чего их акклиматизировали в виварии UTMB Animal Research Facilities в течение недели и затем выкармливали либо на смешанном (4% жиров: рацион Harlan Teklad 7001), либо на "западном" (21% жиров: Harlan Teklad # 88137) рационах, начиная с 6-недельного возраста в течение 4 (молодая, 10-недельная группа) или 28 (старая, 34-недельная группа) недель до взятия проб ткани. Для эксперимента с белковыми рационами 6-недельных мышей выкармливали рационами либо с 16% (16% белка, 4% жира: NIH31 # 101034), либо 24% (24% белка, 4% жира: рацион Harlan Teklad # 7001) белка в течение 4 недель (при умерщвлении возраст составил 10 недель). В каждую группу входили 4 мыши ароЕ и 4 мыши контрольной линии C57BL. Образцы тканей были взяты после внутрибрюшинной инъекции кетасет-ксилазином (1 мг и 20 мг). По одной аорте из группы использовали для гистохимического анализа, в то время как образцы остальных проксимальных и дистальных частей аорты, сердца, печени, легких и головного мозга вырезали, немедленно замораживали на жидком азоте и хранили при -80°С до использования. Образцы плазмы были взяты и использованы для определения общего содержания холестерина (Boehringer Mannheim, IN) и содержания пероксидов липидов (Calbiochem-Novabiochem, La Jolla, CA).
Пример 2
Обработка активными формами кислорода
Концентрированный маточный раствор H2O2 (30%, Fisher) разбавляли фосфатно-солевым буфером (ФСБ), и концентрацию определяли по поглощению при 230 нм [30]. Пероксинитрит синтезировали из нитрита натрия и подкисленной H2O2 и анализировали его количественно [31]. В качестве доноров низкой дозы О
Пример 3
Тест с количественной ПЦР (КолПЦР)
В тесте с КолПЦР измеряют средний уровень повреждений ДНК в расчете на цепь для двух матричных цепей представляющего интерес геномного сегмента. Выявление повреждений ДНК с помощью КолПЦР основывается на следующем. (1) На предпосылке, согласно которой любая ДНК-матрица, включающая повреждение, будет останавливать термостабильную полимеразу либо напрямую [25,32], либо в сочетании с воздействием фермента перед КолПЦР (т.е. обработка образцов FAPY-гликозилазой позволит выявить с использованием КолПЦР повреждения типа 8-осогуанина), и (2) на том, что размерные мутации (делеции и вставки) будут изменять размер ожидаемого КолПЦР-продукта, что приведет к сниженному выходу ожидаемого КолПЦР-продукта (т.к. делеции в мтДНК будут приводить к образованию КолПЦР-продуктов, размеры которых будут меньше размера ожидаемого продукта). Следовательно повреждения ДНК (т.е. разрывы цепей, модификации нуклеотидов, аддукты ДНК и апуриновые сайты) и размерные мутации (т.е. делеции мтДНК) будут либо блокировать работу полимеразы (например, повреждения), либо давать ПЦР-продукты измененного размера (например, делеции мтДНК), что приведет к снижению амплификации последовательности-мишени (ожидаемого размера). Следовательно, только те ДНК-матрицы, которые не включают размерных мутаций ДНК и/или выявляемых повреждений ДНК, будут давать ожидаемый амплификационный продукт. Повреждения анализируют в митохондриальном геноме по амплификации продукта длиной 16,2 т.п.н. на матрице митохондриальной ДНК, а в ядерном геноме - по продукту длиной 17,7 т.п.н. из кластера генов β-глобинов. Повышенная поврежденность ДНК связана со снижением выходов амплификационных продуктов. Поскольку различия в КолПЦР-амплификации могут иногда быть связаны с различиями в числе копий матрицы или просто с качеством ДНК вне связи с повреждениями, вызванными in vivo и in vitro, то с целью контроля качества проводят количественную амплификацию небольшого сегмента [32]. Маловероятно, что небольшие сегменты-мишени в составе ДНК подвержены каким-либо аномалиям, и, следовательно, они могут служить в качестве индикаторов относительного числа копий и качества ПЦР геномного экстракта.
Также можно использовать альтернативные средства для количественного анализа продуктов КолПЦР: ими могут являться флуоресцентные, люминесцентные, радиоизотопные и иммунологические средства (антитела). Примерами этого являются использование флуоригенных зондов, помеченных гасителем и/или репортерными красителями, помеченных антителами или олигонуклеотидными зондами, способы связывания (например, биотинилированные зонды) и т.п. Такой тип количественной ПЦР снижает необходимость в электрофорезе и получении фосфорных отпечатков. Наконец, одноцепочечную КолПЦР с использованием перечисленных выше способов количественного анализа можно также применить в качестве способа количественного анализа повреждений в отдельных цепях ДНК.
Пример 4
Выделение ДНК и КолПЦР
Общую клеточную ДНК выделяли с помощью набора Qiagen genomic tip 20G kit в соответствии с описанным у изготовителя. В результате выделения ДНК данным способом получают геномные препараты, пригодные для протяженной КолПЦР. Концентрации общей клеточной ДНК определяли по флуоресценции в бромистом этидии с использованием флуориметра с фильтром А4, у которого фильтр пропускания полосы возбуждения настроен на 360 нм, а отсекающий эмиссионный фильтр - на 600 нм (Optical Technology Devices, Elmsford, NY), с использованием в качестве стандарта λ-HindIII-ДНК. Вначале, перед КолПЦР, количество ДНК оценивали с помощью электрофореза в пульсирующем поле. Качество образца также анализировали с помощью КолПЦР 222-нуклеотидного фрагмента митохондриальной ДНК и 84-нуклеотидного фрагмента генов β-глобинов (праймеры для митохондриальной ДНК 14619FOR, 1484REV; праймеры для β-глобинов - 48440FOR, 48634REV), ожидая, что равные концентрации матриц дадут сходные концентрации (коротких) КолПЦР-продуктов. В экспериментах на мышах качество образцов анализировали с помощью КолПЦР 80-нуклеотидного фрагмента митохондриальной ДНК и 143-нуклеотидного фрагмента генов β-глобинов (праймеры для митохондриальной ДНК - прямой 13281-13306, обратный 13335-13361; праймеры для β-глобинов - прямой 21582-21605, обратный 21704-21725).
КолПЦР проводили в системе GeneAmp PCR 2400 с набором реактивов GeneAmp XLPCR (Perkin Elmer). Реакционные смеси содержали 15 нг геномной ДНК в качестве матрицы. Параметры реагентов для КолПЦР описаны [25,32] для продукта митохондриальной ДНК длиной 16,2 т.п.н. (координаты прямого праймера - 15149-15174; координаты обратного праймера - 14841-14816) и продукта β-глобина длиной 17,7 т.п.н. (координаты прямого праймера - 44330-44351; координаты обратного праймера - 61989-61968). Каждая реакция содержала по 1х XL-буфера II (Perkin Elmer Cetus), 1,1 мМ Мg(ОАс)2, 0,1 мг/мл БСА, 0, 6 мМ праймеров, 2 мКи α-32Р-дАТФ (Dupon-NEN) и 1 единицу полимеразы rTth (Perkin Elmer Cetus). Каждую КолПЦР инициировали горячим стартом при 75°С с добавлением ДНК-полимеразы rTth. В экспериментах на мышах количественный контроль с половиной (7,5 нг) контрольной геномной матрицы включали в каждую ПЦР-серию для обеспечения количественных условий. По завершении КолПЦР 15 мкл каждого КолПЦР-продукта разгоняли (методом вертикального электрофореза) в 1%-ном агарозном геле (ТВЕ) при напряжении 80-90 вольт в течение 4 часов. Высушенные гели экспонировали на фосфорные экраны в течение 12-14 часов и оценивали количественно с помощью программы IMAGEQUANT (Molecular Dynamics PhosphoImager 425). Хотя было описано использование для КолПЦР праймеров с конкретной локализацией и взаиморасположением, специалист в данной области техники сможет сконструировать много других праймерных последовательностей и положений, что будет диктоваться основным подходом, описанным в настоящей заявке.
Подсчитывают частоту повреждений ДНК [33]. Вкратце, амплификацию поврежденных образцов (Аd) нормализовали к амплификации неповрежденных контролей (Ао), в результате чего получали относительную степень амплификации (Ао для групп возрастов 10 и 34 недель являлась контрольная группа в возрасте 10 недель; Ао для 3-недельных мышей являлись мыши С57 в возрасте 3 недель; Ао для мышей ароЕ-/-, SOD2-/+ и ароЕ-/- являлись мыши ароЕ-/-). Приняв случайный характер распределения повреждений и используя уравнение Пуассона [f(x)=eλλx/x!, где λ равна средней частоте повреждений], для неповрежденных матриц (т.е. для “нулевого класса”: x=0) была определена средняя частота повреждений в расчете на цепь ДНК: λ=-lnAd/Ao. Статистический анализ был проведен с использованием независимого критерия Стьюдента. Поскольку при использовании КолПЦР не было выявлено существенных различий в поврежденности ДНК в проксимальной и дистальной отделах аорты в составе каждой соответствующей группы мышей ароЕ или C57BL, то при проведении межгрупповых сравнений аорт результаты для каждой части (проксимальной и дистальной) объединяли для каждой группы.
Пример 5
Анализ транскриптов методом Нозерн-блоттинга
Контрольные и обработанные пероксинитритом культуры собирали с помощью 4 М гуанидинизотиоцианата, и тотальную клеточную РНК выделяли центрифугированием через 5,7 М хлорида цезия [34]. Для анализа стабильности транскриптов 2,5 мг/мл актиномицина-D добавляли перед добавлением пероксинитрита. Ткани сердца солюбилизовали с использованием тканевого солюбилизатора Polytron в 4 М гуанидинизотиоцианата. Затем гомогенаты центрифугировали в течение 15 минут (3000 g), и надосадочные фракции собирали с целью выделения тотальной клеточной РНК центрифугированием через 5,7 М хлорида цезия [34]. Тотальную РНК разгоняли с помощью электрофореза в агарозном геле, переносили на нейлоновые мембраны и прегибридизовали и гибридизовали [35] с соответствующим зондом. Зонды для транскриптов митохондриальной ДНК получали на материале очищенной митохондриальной ДНК [36] с помощью ПЦР (16S-рРНК: прямой праймер - 2005-2022, обратный - 2982-3001; ND2: прямой праймер - 4831-4847, обратный - 5464-5481; Cyt-b: прямой праймер - 14730-14749, обратный - 15845-15863) и очищали в геле (Qiagen) с последующим случайным мечением с 32Р-дЦТФ (Stratagene).
В экспериментах, в которых РНК экстрагировали из ткани мыши, очищенные в геле ПЦР-продукты, охватывающие участки генов 16S-рРНК (прямой праймер - нуклеотиды 1330-1354, обратный праймер - нуклеотиды 2072-2097), ND2 (прямой праймер - нуклеотиды 4234-4259, обратный праймер - нуклеотиды 4916-4941) и Cyt-B (прямой праймер - нуклеотиды 14196-14220, обратный праймер - нуклеотиды 14967-14992), служили в качестве матриц для случайно помеченных 32Р-дЦТФ зондов (Stratagene). Фильтры экспонировали на кодаковскую пленку XAR при -80°С. Уровни РНК в каждом образце нормализовали гибридизацией с зондом β-актина, имеющимся в продаже (Clontech). Авторадиографические отпечатки сканировали на денситометре (денситометр SI от Molecular Dynamics) и оценивали количественно с использованием программы IMAGEQUANT (Molecular Dynamics). В связи с количественной оценкой уровней митохондриальных транскриптов может быть использован любой метод количественного анализа уровней мРНК-транскриптов. Статистический анализ проводили с использованием независимого критерия Стьюдента.
Пример 6
Синтез митохондриальных белков
Методы анализа синтеза митохондриальных белков описаны [37]. Вкратце, контрольные и обработанные клетки промывали не содержащей метионина культуральной средой и инкубировали в течение 2 часов с 400 мКи/мл 35S-метионина в присутствие 100 мг/мл эметина (ингибитор синтеза ядерных белков) с последующим 20-30-минутным чейзингом в 0,1 мМ холодного L-метионина.
Клетки обрабатывали трипсином и промывали в ФСБ. Собирали клеточные центрифугаты, ресуспендировали их в солюбилизирующем буфере (4% ДСН), обрабатывали ультразвуком (6 пульсов при 30%-ной нагрузке, выход 5) и определяли общий белок. Равные количества (общего белка) помеченных продуктов синтеза разгоняли электрофоретически в 10-20%-ном градиентном ДСН-ПААГ. Гели высушивали с помощью фильтровальной ватманской бумаги и экспонировали на пленку Kodak XAR при -80°С. Процент мечения продуктов трансляции определяли денситометрически (Molecular Dynamics Densitometer SI) для всех полос для обработанных и необработанных образцов. Сумму мечения для полос каждого образца использовали для подсчета относительных уровней включения. С точки зрения количественного определения уровней митохондриальных белков пригодным является любой метод, применяемый для количественного анализа синтеза митохондриальных белков. Статистический анализ был проведен с использованием независимого критерия Стьюдента.
Пример 7
Тесты на МТТ и АТФ
Восстановление МТТ комплексом II использовали для оценки митохондриального дыхания [38-42]. Клетки высевали в 96-луночные планшеты при плотности 8000 клеток на лунку и инкубировали при 37°С. После культивирования в течение 48 часов среду заменяли на бессывороточную среду, содержащую 0,2 мМ H2O2, 0,1 мМ, 0,5 мМ или 1,0 мМ пероксинитрита, на 1 час, и реакцию восстановления проводили в кондиционной среде в течение 1 часа с МТТ в конечной концентрации 2,0 мг/мл, лизировали и измеряли поглощение при 570 нм [25]. Показатели поглощения преобразовывали в восстановление МТТ с использованием калибровочной кривой, выстроенной для известных количеств живых клеток. Восстановление МТТ для обработанных образцов затем нормализовали по необработанным контрольным образцам и выражали в виде доли от контроля. Общее содержание АТФ определяли с использованием набора для определения содержания АТФ (Molecular Probes, A-6608) и микроинъекторного люминометра MicroLumat Plus LB (EG&G Berthold). Вкратце, данный тест основывается на люциферин-люциферазной биолюминесценции (≈560 нм) в присутствии АТФ. Данный тест является исключительно чувствительным: большинство люминометров могут выявлять по меньшей мере 0,1 пикомоль уже существующего АТФ или АТФ после выработки в кинетических системах. Статистический анализ был проведен с использованием независимого критерия Стьюдента.
Пример 8
Гистологический анализ, перекисное окисление липидов и уровни холестерина
По одной аорте из каждой группы фиксировали в 4% параформальдегиде, заливали в парафин, приготавливали срезы толщиной 5 мкм, депарафинизировали, регидрировали и окрашивали гематоксилином и эозином, после чего анализировали развитие атеросклеротических повреждений. Уровни перекисного окисления липидов определяли по образцам плазмы, используя колориметрический тест (586 нм: Calbiochem-Novabiochem, La Jolla, CA), специфичный для малонового альдегида (MDA) и 4-гидрокси-2(Е)-ноненаля (4-HNE), которые являются конечными продуктами, образующимися в результате перекисного окисления полиненасыщенных жирных кислот и родственных сложных эфиров. Измерение таких продуктов дает индекс перекисного окисления липидов. Образцы сравнивали с калибровочными кривыми для 4-HNE и MDA. Уровни общего холестерина определяли на образцах плазмы мышей по калибровочной кривой с использованием набора реактивов для определения холестерина (Boehringer Mannheim, Indianapolis, IN) на основании инструкций, предоставляемых изготовителем.
Пример 9
Результаты экспериментов in vitro
В среде сосудистой системы супероксид (О
Воздействие пероксида водорода обусловливало повышение поврежденности митохондриальной ДНК в обеих клеточных линиях, в то время как повреждения ядерной ДНК также происходили в эндотелиальных клетках. И HUVEC, и HASMC обрабатывали 0,2 мМ пероксида водорода в течение 1 часа и оценивали на повреждения митохондриальной ДНК и ядерной ДНК (β-глобин) по сравнению с необработанным контролем (табл. 1, фиг.1). Уровень поврежденности митохондриальной ДНК и яДНК (генный кластер β-глобина) был статистически достоверным (по сравнению с необработанным контролем - Р<0,005) в клетках HUVEC, в то время как в HASMC существенные повреждения имелись только в митохондриальной ДНК (Р<0,005). Напротив, в HASMC не отмечено статистически достоверного повышения повреждений яДНК по отношению к необработанному контролю (Р=0,695).
Анализ временной динамики воздействия пероксида водорода показал, что повреждения митохондриальной ДНК происходят быстро в обеих клеточных линиях. Клетки обрабатывали 0,2 мМ пероксида водорода в течение 0-60 минут, и повреждения ДНК оценивали в каждый отрезок времени (фиг.1). Достоверная поврежденность митохондриальной ДНК имела место в течение 10 минут в клетках HUVEC (Р=0,037) и 15 минут в HASMC (Р=0,047) по сравнению с необработанным контролем. Напротив, локус β-глобина не проявил быстрого накопления повреждений яДНК (фиг.1), т.к. требуется 60-минутная обработка для того, чтобы достичь проявления достоверного уровня повреждений в клетках HUVEC (Р=0,005). Следовательно, повреждения митохондриальной ДНК происходят быстро и в HUVEC, и в HASMC, подвергнутых действию активных форм кислорода in vitro.
Обработка пероксинитритом приводила к преимущественным повреждениям митохондриальной ДНК в HUVEC и HASMC. Для анализа влияния пероксинитрита клетки обрабатывали 0,1 мМ и 0,5 мМ пероксинитрита в течение 1 часа (табл.1, фиг.2). Обработка HUVEC 0,1 мМ и 0,5 мМ пероксинитрита обусловливала существенное повышение поврежденности митохондриальной ДНК (Р<0,005), в то время как воздействие 0,5 мМ пероксинитрита во всех случаях приводило к существенным повреждениям в HASMC (Р<0,005) по сравнению с необработанными образцами (табл.1).
В попытке оценить действие постоянных низких доз пероксинитрита in vitro клетки HUVEC и HASMC обрабатывали донорами О
Обработка HUVEC и HASMC пероксинитритом обусловливала существенное снижение уровней транскриптов кодируемых митохондриальной ДНК генов - НАДФ-дегидрогеназы-2 (ND2) и цитохрома-b (Cyt-b), - но не 16S-pPHK. Обработка клеток HASMC 0,5 мМ пероксинитрита приводила к снижению на 55% уровней транскриптов ND2 и Cyt-b, в то время как снижение уровней 16S-pPHK составило 14% (фиг.3). Сходным образом обработанные образцы HUVEC характеризовались снижением уровней транскриптов ND2 и Cyt-b на 45-50% при обработке 0,5 мМ пероксинитрита, в то время как уровни 16S-pPHK снижались на 26% по отношению к необработанным контролям (фиг.3). При использовании меньшей дозы пероксинитрита (0,1 мМ) уровни транскриптов 16S-pPHK, ND2 и Cyt-b снижались на 25-30%, а снижение в HASMC составило 5-15% (фиг.3).
Предварительная обработка клеток актиномицином-D, являющимся транскрипционным ингибитором, показала, что воздействие пероксинитрита обусловливает и подавление транскрипции (ND2 и Cyt-b), и разрушение отдельных транскриптов (Cyt-b). Напротив, уровни транскриптов ядерного гена β-актина от обработки пероксинитритом не зависели. Следовательно, пероксинитрит способен дифференцированно подавлять и транскрипцию, и стабильность транскриптов в отношении митохондриальных генов.
Обработка активными формами кислорода также приводила к ослаблению синтеза митохондриальных белков в обеих клеточных линиях. Обработка HUVEC и HASMC 0,2 мМ H2O2 обусловливала снижение общего уровня синтеза митохондриальных белков на 23-33%, в то время как обработка 0,5 мМ пероксинитрита проявлялась в 55-70%-ной потере синтеза митохондриальных белков по сравнению с необработанными клетками (фиг.4). Меньшие дозы пероксинитрита (0,1 мМ) в HASMC проявили небольшое снижение - на 12% - включения 35S-метионина. Следовательно, воздействие активных форм кислорода также может быть связано с ослаблением синтеза митохондриальных белков.
Также воздействие пероксинитрита приводило к снижению общих уровней АТФ и ослаблению митохондриального дыхания (комплекс II) в HUVEC и HASMC (фиг.5). Оценка общих уровней АТФ показала, что, в то время как доза 0,1 мМ пероксинитрита не приводит к существенному снижению общего АТФ в любой клеточной линии, доза 0,5 мМ обусловливала существенное снижение количества АТФ (HUVEC-Р=0,02; HASMC-Р=0,04). Сходным образом, воздействие 0,1 мМ пероксинитрита не приводило к существенному восстановлению МТТ в комплексе II (митохондриальное дыхание), в то время как доза 0,5 мМ обусловливала существенное снижение (HUVEC-Р=0,02; HASMC-Р=0,008).
Восстановление МТТ сукцинатдегидрогеназой, являющейся компонентом комплекса II ОК-ФОС, является индикатором функции митохондрий и часто используется в качестве средства для оценки дыхательных и окислительно-восстановительных функций клеток. Следовательно, восстановление МТТ будет отражать окислительно-восстановительные способности митохондрий. В параллельных экспериментах клетки окрашивали трипановым синим с целью определения степени клеточной гибели, связанной с каждым воздействием активных форм кислорода. Обе клеточные линии виртуально не характеризовались гибелью клеток (<5%) в те моменты времени, когда оценивали МТТ и АТФ. Следовательно, предполагается, что обработка активными формами кислорода обладает влиянием на общее содержание АТФ и дыхание в HUVEC и HASMC.
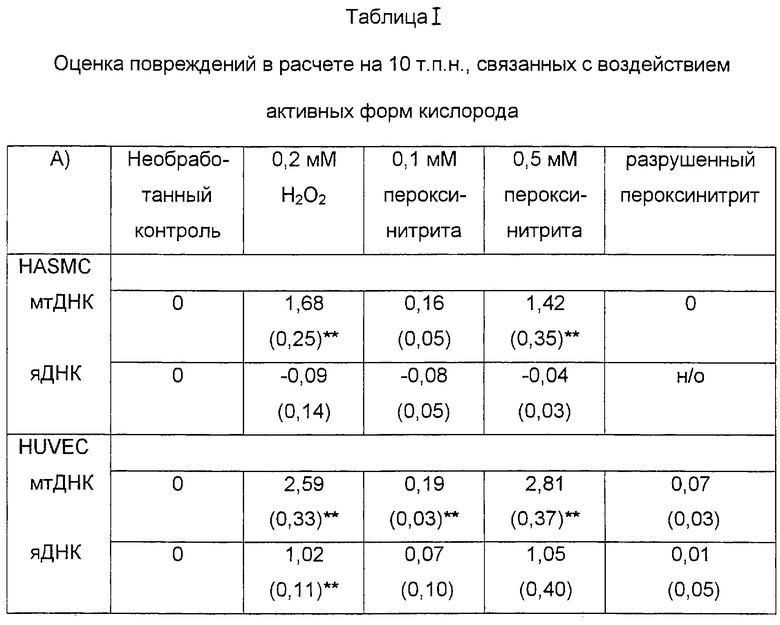
В таблице I показаны значения частоты повреждений (на 10 т.п.н.), оцененную в митохондриальной ДНК и яДНК (кластер генов β-глобина), по сравнению с “нулевым классом” необработанного контроля в случае воздействий перокисида водорода (H2O2) и пероксинитрита. Разрушенный пероксинитрит указывает на пероксинитрит, проинкубированный в культуральной среде в течение 1 часа при комнатной температуре перед добавлением (0,5 мМ) к культивируемым клеткам. Значения даны в виде средних (± s.e.).
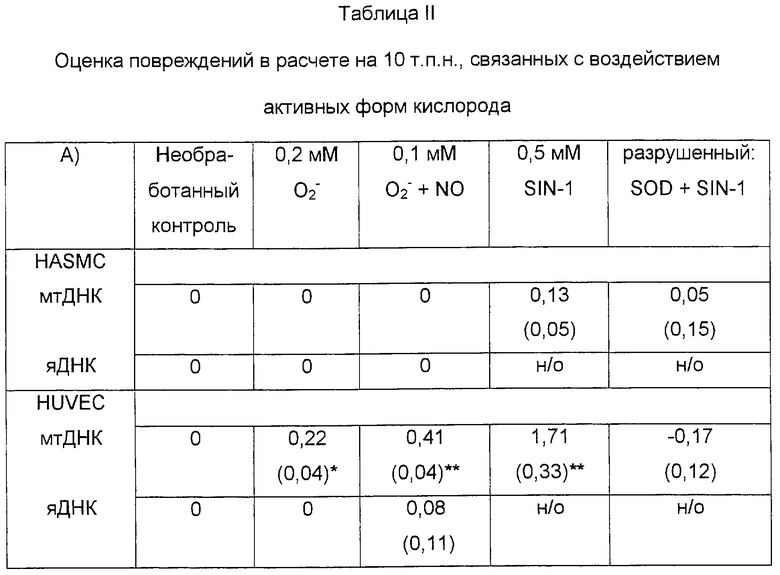
Таблица II показывает оцененную частоту повреждений (на 10 т.п.н.) в митохондриальной ДНК и яДНК (кластер генов β-глобина), связанных с донорами супероксида (О
Пример 10
Повреждения ДНК у мышей
Мышей с гиперхолестеринемией ароЕ и здоровых контрольных мышей разделяли на две возрастные группы (возраст в момент умерщвления - 10 недель или 34 недели) и выкармливали либо на смешанном (4% жиров), либо на "западном" (21% жиров) рационе, начиная с возраста 6 недель. Ткани аорты и сердца (левый желудочек) оценивали на повреждения ДНК с использованием КолПЦР, в то время как кровяную плазму использовали для определения уровней холестерина и перекисного окисления липидов (4-HNE и MDA). По одной аорте из каждой группы фиксировали для гистологического исследования.
Ткани аорты мышей ароЕ с гиперхолестеринемией, выкормленных на смешанном рационе, характеризовались существенно повышенными уровнями повреждений митохондриальной ДНК по сравнению с контролем (Р<0,05; фиг.6А). Данные различия были выявлены у мышей в возрасте и 10, и 34 недель: это отчетливо показало, что аорты у ароЕ сохраняют более высокий уровень поврежденности ДНК in vivo по сравнению со здоровыми мышами. В аорте 10-недельных мышей ароЕ выявлено снижение амплификации на 61% (сниженная амплификация коррелирует с повышенной поврежденностью ДНК) по отношению к нулевому классу контроля, или оцененный уровень повреждений митохондриальной
ДНК на 10 тысяч пар нуклеотидов (т.п.н.) составил 0,582±0,123 по сравнению с 0,0±0,198 повреждений на 10 т.п.н. у 10-недельных контрольных мышей (Р=0,018). Сходным образом, 34-недельные особи группы ароЕ характеризовались 3-кратным увеличением оцененной поврежденности митохондриальной ДНК по сравнению с контрольной группой того же возраста (1/325±0,257 повреждений на 10 т.п.н. против 0,453±0,162 повреждений на 10 т.п.н.: Р=0,007). Такие же параметры также были выявлены в ткани сердца (левый желудочек; фиг.6В). Например, 10-недельные мыши ароЕ характеризовались 67%-ным снижением амплификации по сравнению с нулевым классом контроля (0,685±0,093 повреждений на 10 т.п.н. против 0,0±0,048 повреждений на 10 т.п.н.: Р<0,001), в то время как у 34-недельных мышей ароЕ сохранялось приблизительно 4-кратное повышение оцененного уровня повреждений митохондриальной ДНК по сравнению с контролями того же возраста (0,819±0,151 повреждений на 10 т.п.н. против 0,213±0,295 повреждений на 10 т.п.н.: Р=0,056).
Влияние "западного" рациона также оценивали у ароЕ и контрольных мышей (фиг.6В). У контрольных мышей "западный" рацион был ассоциирован с более высокими уровнями повреждений митохондриальной ДНК в аортах 10-недельных контрольных мышей по сравнению с контрольными мышами, выкормленными на смешанном рационе (0,31±0,17 повреждений на 10 т.п.н. против 0,0±0,20 повреждений на 10 т.п.н. соответственно при "западном" и смешанном рационе), однако, это различие было менее достоверным (Р=0,25). Эти различия были менее очевидными у контрольных мышей в возрасте 34 недель (а именно 0,45±0,16 повреждений на 10 т.п.н. и 0,49±0,25 повреждений на 10 т.п.н. при смешанном и "западном" рационе соответственно: Р=0,90). Сходным образом, 10-недельные контрольные мыши характеризовались повышенным выявленным уровнем повреждений митохондриальной ДНК сердца при "западном" рационе (фиг.6В; снижение амплификации на 58%: 0,54±0,23 повреждений на 10 т.п.н. и 0,0±0,05 повреждений на 10 т.п.н. при "западном" и смешанном рационе соответственно), и что похоже на данные, полученные для аорты, не было выявлено различий у контрольных мышей в возрасте 34 недель (0,21±0,29 повреждений на 10 т.п.н. и 0,25±0,21 повреждений на 10 т.п.н. при смешанном и "западном" рационе, соответственно). Напротив, не было выявлено увеличения поврежденности митохондриальной ДНК в связи с "западным" рационом ни в аорте, ни в ткани сердца у мышей ароЕ. Следовательно, повышение жирности пищи, по-видимому, связано с повышенной поврежденностью митохондриальной ДНК только у контрольных мышей в возрасте 10 недель.
Возраст также был ассоциирован с увеличением повреждений митохондриальной ДНК в аортах и у ароЕ, и у контрольных мышей (табл. 2). Оцененное 2,3-5,8-кратное увеличение числа повреждений наблюдалось в аортах мышей ароЕ в возрасте 10 недель и 34 недель (смешанный рацион - Р=0,013; "западный" рацион - Р=0,011), в то время как контрольные мыши не проявляли достоверного возрастания уровней поврежденности, хотя увеличение и было значительным (табл. 2). Более старший возраст, по-видимому, влияет на контрольных мышей, выкармливаемых на смешанном рационе (0,0±0,20 повреждений на 10 т.п.н. против 0,45±0,17 повреждений на 10 т.п.н. в возрасте 10 и 34 недель соответственно - Р=0,09), в то время как меньшие различия были выявлены в связи с возрастом при выкармливании мышей на "западном" рационе (0,31±0,17 повреждений на 10 т.п.н. против 0,49±0,25 повреждений на 10 т.п.н. в возрасте 10 и 34 недель соответственно - Р=0,556). Напротив, в тканях сердца статистически достоверных связей выявлено не было. Следовательно, возраст достоверно коррелирует с повышенной поврежденностью митохондриальной ДНК в аорте у мышей ароЕ, в то время как тенденция считается подтвержденной при выкармливании мышей смешанным рационом.
Поскольку не было выявлено существенных различий по повреждениям митохондриальной ДНК между смешанным и "западным" рационами при одинаковых возрасте и генотипе, исследовали влияние белка на мышей в возрасте 10 недель. И мышей ароЕ, и контрольных мышей C57BL выкармливали на рационах, содержащих 16% или 24% белка, в течение 4 недель, начиная с возраста 6 недель. Хотя оба рациона были связаны с более высокими повреждениями митохондриальной ДНК у мышей ароЕ по сравнению с контролем (аорта - Р=0,015; сердце - Р=0,005), рацион с меньшим количеством белка обусловливал достоверное снижение поврежденности в аорте и в контроле (Р=0,007), и у ароЕ (Р<0,001) (фиг.7). Аналогичная тенденция была также выявлена в сердце у ароЕ (Р=0,002). Следовательно, рацион с меньшим содержанием белка связан со сниженной поврежденностью митохондриальной ДНК у обоих типов мышей.
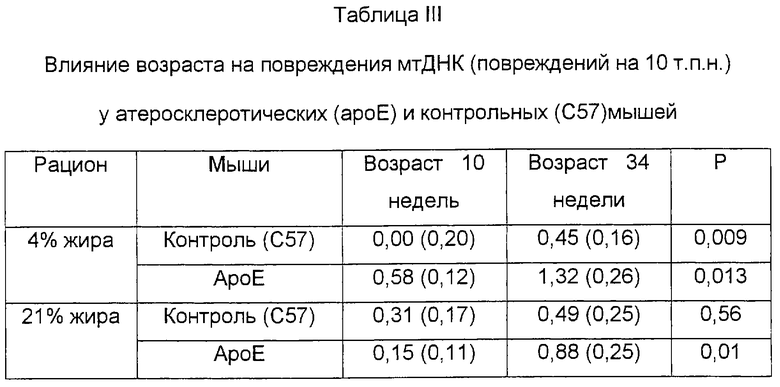
В таблице III приведены оцененные [среднее (s.e.)] величины повреждений мтДНК в расчете на 10 т.п.н. по отношению к контрольной группе (С57) при рационе с 4% жиров. Все рационы давали мышам, начиная с возраста 6 недель, и неизменно вплоть до достижения возраста 10 или 34 недель. Для статистического анализа использовали независимый критерий Стьюдента.
Оцененные частоты повреждений митохондриальной ДНК в расчете на 10 т.п.н. соотносили с 10-недельными контрольными мышами C57BL, выкормленными на смешанном рационе (повреждения нулевого класса). Данные приведены в виде средних значений (±s.e.). Значения Р критерия Стьюдента даны для сравнений между группами в возрасте 10 против 34 недель для каждого генотипа (ароЕ или С57) и рациона. Звездочки (*) указывают на статистически значимые отличия от 10-недельного референсного варианта.
Пример 11
Повреждения мтДНК в аорте у 3-недельных мышей ароЕ-/- и контрольных мышей
Повреждения мтДНК аорты также оценивали у 3-недельных мышей ароЕ-/- и контрольных мышей. Аорты 3-недельных мышей ароЕ-/- характеризовались существенно более высокими уровнями повреждений мтДНК по сравнению с контрольными животными того же возраста (фиг.8; Р=0,001): это подтверждает, что повреждения мтДНК происходят в раннем возрасте в аортах, предрасположенных к развитию атеросклеротических повреждений.
Пример 12
Содержание холестерина у мышей
Как ожидалось, уровни холестерина были существенно повышены во всех группах мышей ароЕ по сравнению с контрольными мышами (выше примерно в 4-5 раз: Р<0,001). Хотя "западный" рацион был связан с наивысшими уровнями холестерина у всех мышей, такое увеличение (по отношению к смешанному рациону) было статистически достоверным только у мышей ароЕ (выше примерно в 2,2 раз; смешанный - 225,8±33,4 мг/дл; "западный" - 490,2±56,7 мг/дл; Р=0,0019). Напротив, контрольные мыши C57BL проявили не намного более высокие уровни холестерина (выше примерно в 1,6 раза; смешанный - 53,4±15,1 мг/дл; "западный" - 85,2±10,8 мг/дл) при использовании "западного" рациона, хотя различия не были статистически достоверными (Р=0,17). Следовательно, мыши ароЕ характеризовались существенно более высокими уровнями холестерина по сравнению с контролем C57BL того же возраста, а наивысшие уровни холестерина были связаны с западным рационом.
Пример 13
Перекисное окисление липидов у мышей
Уровни перекисного окисления липидов измеряли путем определения уровней MDA и 4-HNE. Мыши ароЕ с гиперхолестеринемией характеризовались существенно повышенными уровнями пероксидов липидов по сравнению с контрольными мышами (Р<0,05). Однако в противоположность результатам, полученным для общих уровней холестерина, степень перекисного окисления липидов не возрастала существенно у мышей ароЕ при сравнении "западного" со смешанным рационом (фиг.9). Напротив, у контрольных мышей, выкармливаемых на "западном" рационе, сохранялось повышенное перекисное окисление липидов по сравнению со смешанным рационом (Р<0,05). Следовательно, хотя мыши ароЕ часто имеют наивысшие уровни пероксидов липидов по сравнению с контрольными мышами, степень перекисного окисления липидов у ароЕ не увеличивается в случае использования "западного" рациона по сравнению со смешанным рационом.
Пример 14
Гистология мышей
Аорту от каждой группы заливали в парафин и приготавливали поперечные срезы толщиной 5 мкм, которые окрашивали гематоксилином и эозином. Хотя атеросклеротические повреждения отсутствовали во всех группах 10-недельных мышей (контрольных и ароЕ), причем вне зависимости от рациона (смешанный либо "западный"), атеросклеротические повреждения имелись в обеих группах 34-недельных мышей ароЕ, но отсутствовали у 34-недельных контрольных мышей. На количественном уровне и частота, и размер повреждений очевидно возрастали у мышей ароЕ, выкармливаемых на "западном" рационе, по сравнению с мышами ароЕ, выкармливаемыми на смешанном рационе.
Пример 15
Анализ атерогенеза и повреждений мтДНК у мышей ароЕ-/-, SOD2-/+
Для определения того, влияет ли нарушение функции митохондрий и антиоксидантной активности (MnSOD, SOD2) на атерогенез, сформировали линию мышей ароЕ-/-, SOD2-/+. Ранее проведенные исследования показали, что гетерозиготные по SOD2 мыши (-/+) характеризуются сниженными функцией митохондрий и активностью SOD2 по сравнению с мышами дикого типа. Гибридные мыши ароЕ-/-, SOD2-/+ имели 40% активности SOD2 от той, которая выявлена у мышей ароЕ-/-. Развитие атеросклеротических повреждений у этих мышей было отчетливым по сравнению с их родственниками ароЕ-/- того же возраста (фиг.10А), при том, что у мышей ароЕ-/-, SOD2-/+ отмечено 2,5-кратное увеличение (Р=0,02) числа атеросклеротических повреждений по сравнению с родственниками ароЕ-/- (фиг.10В). Следовательно, поврежденность мтДНК аорты возрастала у мышей ароЕ-/-, SOD2-/+ по сравнению с родственными им мышами генотипа ароЕ-/- (фиг.10С; Р=0,006).
Пример 16
Обсуждение экспериментов in vitro
Настоящее исследование было проведено с целью продемонстрировать то, что пероксид водорода и пероксинитрит опосредуют повреждения и дисфункцию митохондрий в клетках HUVEC и HASMC in vitro. Повреждения митохондриальной ДНК были достоверными в обоих типах клеток под воздействием пероксида водорода, а у HUVEC были существенными и повреждения ядерной ДНК. Сходным образом, митохондриальная ДНК оказывается достоверно повреждена при действии острых доз пероксинитрита на HUVEC (0,1 мМ и 0,5 мМ) и HASMC (0,5 мМ). Более того, при действии низких постоянных концентраций пероксинитрита достоверные повреждения митохондриальной ДНК по сравнению с контролями были выявлены в клетках HUVEC, в то время как влияние на яДНК отсутствовало. Также обработка SIN-1 приводила к достоверному возрастанию поврежденности митохондриальной ДНК в HUVEC. Напротив, обработка SIN-1 клеток HASMC не обусловливала существенных уровней повреждений, что, по-видимому, противоречит влиянию обработки пероксинитритом (табл.1). Причиной таких различий может быть относительная способность HASMC восстанавливать О
Уровень транскриптов ND2 и Cyt-b снижался после воздействия пероксинитрита в обоих типах клеток. Хотя уровни и ND2, и Cyt-b снижались значительно, только снижение уровней транскрипта Cyt-b было статистически достоверным (HASMC-Р=0,033; HUVEC-Р=0,001). Снижение содержания транскриптов ND2 и Cyt-b, по-видимому, является временным, поскольку большинство культур восстанавливали содержания, характерные для необработанного контроля, после удаления среды, обработанной активными формами кислорода, и 2-часового нахождения в кондиционной среде. Следовательно, снижение уровня транскриптов непосредственно связано с острой обработкой активными формами кислорода. Поскольку транскрипт Cyt-b наиболее удален от сайта инициации транскрипции полицистронной мтРНК [43-46], то гены, которые расположены ближе в сайту инициации, могут быть в большей степени (на транскрипционном уровне) подавлены случайным повреждением митохондриальной ДНК. С другой стороны, стабильность транскриптов в присутствии конкретных активных форм кислорода может влиять на уровни РНК. Результаты экспериментов с актиномицином-D подтверждают, что и подавление транскрипции, и нестабильность транскриптов имеют место в ответ на воздействие пероксинитрита в случае с Cyt-b, в то время как транскрипт ND2, по-видимому, менее восприимчив к нестабильности, индуцируемой пероксинитритом. Наконец, митохондриальные рРНК являются субъектом предпочтительной экспрессии [47, 48], что потенциально делает их менее чувствительными к влиянию обработки пероксинитритом, проведенной в настоящем исследовании.
Согласуясь со снижением уровней транскрипции, аналогичное ослабление синтеза митохондриальных белков было выявлено и в HUVEC, и в HASMC после обработки острыми дозами активных форм кислорода. Синтез белков снижался на 23-33% в обеих линиях клеток под воздействием 0,2 мМ H2O2, в то время как обработка 0,5 мМ пероксинитрита обусловливала более существенные потери в мечении белка (HASMC - 30% от контроля; HUVEC - 45% от контроля). Также с этими данными согласуется снижение выработки клеточного АТФ и интенсивности дыхания, что происходит после воздействия активных форм кислорода. Следовательно, эти полученные in vitro данные подтверждают, что активные формы кислорода могут опосредовать ряд родственных событий, связанных с функционированием митохондрий в клетках HUVEC и HASMC, что в конечном счете приводит к дисфункции клеток.
Хотя митохондрии являются основными продуцентами клеточного АТФ, они также являются и постоянными генераторами активных форм кислорода [12-16], образуя О
Хотя известно, что оксид азота в первую очередь выполняет антиатерогенную роль [50-52], такие эффекты ослабляются в атеросклеротизованных сосудах из-за взаимодействия оксида азота с О
Роль оксида азота также была исследована путем удаления окислителей, происходящих от митохондрий клеток-трофобластов плаценты человека (НРСТ) [61]. Когда выработка оксида азота снижена в плацентарных клетках-трофобластах человека действием L-NAME, являющимся ингибитором синтазы оксида азота, как и ожидается, образование клеточных окислителей усиливается. Обработка клеток-трофобластов плаценты человека CuZnSOD и каталазой не ослабляет вызванный окислительный ответ вследствие подавления NOS. Использование дополнительных ингибиторов ксантиноксидазы, циклооксигеназ и митохондриального ОК-ФОС показывает, что только ингибиторы митохондриального ОК-ФОС усиливают окислительный ответ, индуцированный подавлением синтазы оксида азота, в то время как ингибиторы ХО и циклооксигеназ такого эффекта не проявляют. Это означает, что активные формы кислорода, образовавшиеся в митохондриях, являются основными повреждающими агентами в клетке, и что регуляция синтазы оксида азота может играть важную роль с точки зрения активных форм кислорода, образованных в митохондриях.
В оксидативной среде артерии клетки эндотелия и гладкой мускулатуры сосудов подвержены хроническому стрессу, вызываемому активными формами кислорода. Следовательно, клетки, которые больше не способны адекватно реагировать на оксидативный стресс, характеризуются более высоким риском неблагоприятного проявления, вызванного повреждением, опосредованным активными формами кислорода. Здесь было показано, что HASMC и HUVEC (особенно HUVEC) чувствительны к такому повреждению in vitro и, следовательно, что хроническая выработка in vitro активных форм кислорода с наибольшей вероятностью вызывает дисфункцию клеток сосудов за счет инициации ослабления митохондрий, что обусловливает потерю многих важных клеточных функций.
Пример 17
Обсуждение экспериментов на мышах
Исходной целью данного исследования являлось изучение степени повреждения ДНК в тканях аорты и сердца у мышей с гиперхолестеринемией ароЕ и контрольных мышей одинакового возраста. У мышей ароЕ развивались атеросклеротические повреждения по сходному с человеком типу. В целом, атерогенез ускоряется тогда, когда мышей ароЕ выкармливают на "западном" рационе с высоким содержанием жира по сравнению со смешанным рационом. Например, присоединение моноцитов к эндотелиальным клеткам происходит в возрасте не более 6 ("западный" рацион) или 8 (смешанный рацион) недель, после чего ксантомные клетки образуются не позднее возраста 8 ("западный" рацион) или 10 (смешанный рацион) недель, а повреждения развиваются не позднее возраста 15 ("западный" рацион) или 20 (смешанный рацион) недель. Степень и прогрессия атерогенеза у мышей ароЕ, использованных в данной серии экспериментов, согласовывались с ранее полученными наблюдениями.
Поврежденность митохондриальной ДНК статистически значимо возрастала в аортах и у 10- и у 34-недельных мышей ароЕ с гиперхолестеринемией, выкармливаемых на смешанном рационе, по сравнению с контролями C57BL того же возраста. Возраст также был ассоциирован с непосредственным увеличением выявленной частоты повреждений митохондриальной ДНК у мышей ароЕ (Р<0,05), причем независимо от рациона. Поскольку дополнительные исследования, в которых сравнивали относительные уровни повреждений митохондриальной ДНК между проксимальной и дистальной концами аорты у мышей ароЕ, показали отсутствие достоверных различий в поврежденности митохондриальной ДНК в пределах каждой из групп, то маловероятным является то, что повреждения митохондриальной ДНК, выявленные в аортах ароЕ, являются артефактом атерогенных факторов, специфичных для проксимальной области аорты. Напротив, повышенная поврежденность митохондриальной ДНК имела место по всей длине аорты у ароЕ: следовательно, она не является продуктом развития атеросклеротических повреждений, но является потенциальным фактором, участвующим в атерогенезе.
У мышей ароЕ, выкормленных на "западном" рационе, не было существенного повышения поврежденности митохондриальной ДНК по сравнению с вариантом ароЕ, выкормленным на смешанном рационе. В связи с этим в каждой группе оценивали уровни общего холестерина и перекисного окисления липидов. Хотя "западный" рацион у мышей ароЕ также был ассоциирован с существенно более высокими уровнями холестерина по сравнению со смешанным рационом (Р<0,05), это не приводило к достоверному возрастанию перекисного окисления липидов у мышей ароЕ по сравнению со смешанным рационом. Следовательно, полученные данные подтверждают, что уровни перекисного окисления липидов уже достигли максимума у мышей ароЕ на смешанном рационе (возможно, вследствие их генотипической предрасположенности к гиперхолестеринемии). Соответственно, "западный" рацион с более высоким содержанием жиров не способен усиливать перекисное окисление липидов у мышей ароЕ по сравнению со смешанным рационом, что согласуется с данными по повреждениям митохондриальной ДНК, которые не позволили выявить различий между западным и смешанным рационами у мышей ароЕ. Следовательно, хотя "западный" рацион может обусловливать повышение общего холестерина по сравнению со смешанным рационом, тем не менее "западный" рацион в существенной степени не увеличивает общих уровней перекисных окислений липидов у мышей ароЕ, что, следовательно, может отражать то, что поврежденность митохондриальной ДНК у мышей ароЕ более тесно коррелирует с генотипом и возрастом, чем с рационом. Как ожидалось, мыши ароЕ с гиперхолестеринемией характеризуются существенно повышенными уровнями холестерина (в 4,2-5,7 раз выше: Р<0,001) и перекисного окисления липидов (в 1,9-2,7 раз выше: Р<0,05) по сравнению со “здоровым” контролем того же возраста. Хотя было показано, что по сравнению с контрольными мышами одинакового возраста имеет место связанное с возрастом усиление перекисного окисления липидов у мышей ароЕ, не удалось найти сообщений, в которых бы сравнивались уровни перекисного окисления липидов при смешанном и "западном" рационах. Следовательно, изложенные здесь результаты указывают на то, что, хотя "западный" рацион приводит у мышей ароЕ к существенно повышенным уровням холестерина по сравнению со смешанным рационом, он не изменяет общий уровень продуктов перекисного окисления липидов.
Хотя возраст и высокая жирность рациона достоверно не связаны с существенным повышением поврежденности митохондриальной ДНК у мышей C57BL, значительное повышение поврежденности митохондриальной ДНК было выявлено у контроля на смешанном рационе в связи с возрастом, а тенденция к повышенной поврежденности при использовании "западного" рациона была отмечена у мышей в возрасте 10 недель. По-видимому, повышение возраста оказывает влияние на контрольных мышей на смешанном рационе, в то время как у мышей, выкармливаемых "западным" рационом, были найдены меньшие различия. Контрольные мыши возраста 10 недель, выкармливаемые на "западном" рационе, также характеризовались более высоко оцененной частотой повреждений митохондриальной ДНК по сравнению с выкармливанием на смешанном рационе, в то время как не удалось обнаружить связи между рационом и поврежденностью митохондриальной ДНК у 34-недельных контрольных мышей: это подтверждает, что влияние рациона на повреждения митохондриальной ДНК имеет наибольший эффект у молодых контрольных мышей. В противоположность данным, полученным для мышей ароЕ, использование "западного" рациона было связано с достоверным (Р<0,05) повышением уровня перекисного окисления липидов у контрольных мышей по сравнению со смешанным рационом: это говорит о том, что перекисное окисление липидов не “вышло” на максимальный уровень у “здоровых” мышей, выкормленных на смешанном рационе, и в результате повышения содержания субстрата (т.е. "западного" рациона) количество продуктов перекисного окисления липидов возрастает у контрольных мышей, что может согласовываться с увеличенной поврежденностью митохондриальной ДНК, выявленной у контрольных мышей на "западном" рационе.
Ограничение калорийности и снижение содержания пищевого белка, как известно, имеет положительное значение для различных организмов, увеличивая продолжительность жизни. Следовательно, тот факт, что обе линии мышей характеризовались меньшим уровнем повреждений митохондриальной ДНК при выкармливании на рационе с меньшим количеством белка по сравнении с высокобелковым рационом, подтверждает эту точку зрения. Для более полного понимания этого факта необходимо провести долговременные исследования на мышах ароЕ с использованием низкокалорийных рационов. Поскольку низкокалорийные рационы связаны со снижением уровней активных форм кислорода, данные наблюдения согласуются с точкой зрения, в соответствии с которой снижение поврежденности, обусловливаемой активными формами кислорода, подавляет атерогенез.
За последнее десятилетие были накоплены доказательства роли митохондрий (их повреждения и дисфункции) в различных хронических связанных с возрастом заболеваниях. Основной идеей является то, что повреждения митохондрий накапливаются со временем в тканях, вызывая снижение потенциала клеточного ОК-ФОС (энергетической производительности), в то время как выработка активных форм кислорода, опосредуемая ОК-ФОС, возрастает, приводя к дисфункции клеток. Хотя было показано, что митохондрии эндотелиальных клеток чувствительны к повреждениям in vitro, опосредуемым активными формами кислорода, имеется информация о повышении числа патогенных мутаций митохондриальной ДНК в сердечно-сосудистых тканях у людей с атеросклерозом, равно как и информация о патогенных мутациях митохондриальной ДНК, ассоциированных с факторами повышенного риска болезней сердца (возраст, курение, сахарный диабет и др.). Интересно, что такие факторы риска, как курение, повышенное артериальное давление, гиперхолестеринемия и т.п., опосредуют повышенное образование активных форм кислорода, и в недавних исследованиях было показано, что митохондрий являются восприимчивой мишенью для повреждений. Хотя атерогенез на самом деле является сложным процессом, вовлекающим различные обязательные стадии, в данной заявке предполагается, что основной механизм развития атеросклеротических повреждений начинается с накопления повреждений митохондрии в тканях сосудов, что в конечном счете приводит к дисфункции ОК-ФОС и снижению энергетической производительности. Действуя вместе, эти процессы приводят к повышению образования активных форм кислорода (свойство, уже отмеченное в атеросклеротических тканях) и дисфункции сосудистых клеток, создавая тем самым атерогенную среду в артерии.
Представлено первое сообщение об исследованиях уровня повреждений митохондриальной ДНК в тканях аорты в мышиной модели атеросклероза при сравнении с контролями аналогичного возраста. Полученные результаты показывают, что отчетливое повышение поврежденности митохондриальной ДНК ассоциировано с атерогенезом и возрастом. На основе присутствия существенного уровня повреждений митохондриальной ДНК у мышей ароЕ в возрасте 10 недель можно предположить, что в данной модели атеросклероза in vivo повреждение происходит до развития атеросклеротического повреждения или одновременно с ним. Кроме того, поврежденность митохондриальной ДНК в аорте увеличивается с возрастом in vivo. Более того, генотип ароЕ, по-видимому, обладает более сильным влиянием на уровень повреждений митохондриальной ДНК по сравнению с рационом, хотя влияние рациона на повреждения митохондриальной ДНК может иметь место у молодых “здоровых” мышей C57BL. Следовательно, повреждения митохондриальной ДНК, выявленные у мышей ароЕ, могут в конечном счете приводить к нарушению процессов ОК-ФОС, тем самым инициируя дисфункцию клеток эндотелия, что является ключевым событием атерогенеза.
Пример 18
Измерения повреждений митохондриальной ДНК in vivo
Больные, которым проводили коронарную ангиографию, были отобраны для исследования in vivo повреждений митохондриальной ДНК. Все отобранные больные дали информированное согласие на взятие дополнительной пробы крови во время коронарной ангиографии. Всего группа состояла из 75 больных. Лейкоциты выделяли с помощью стандартных методов (препарат “лейкоцитарной пленки”) с выделением из них ДНК (и ядерной, и митохондриальной). Повреждения митохондриальной ДНК определяли методом количественной ПЦР с использованием и “коротких фрагментов митохондриальной ДНК”, и повреждений в пределах гена β-глобина в качестве контролей. На фиг.11 отражена повышенная встречаемость повреждений митохондриальной ДНК у больных, сгруппированных в соответствии с факторами риска, связанными с инфарктом миокарда.
Основываясь на исходных результатах, некоторый уровень повреждений митохондриальной ДНК был определен как норма. Превышение этой нормы рассматривалось как высокий уровень повреждений. Затем была определена доля больных, у которых повреждения митохондриальной ДНК являлись функцией факторов повышенного риска заболеваний сердца. Например, среди курящих приблизительно половина имела повышенную поврежденность митохондриальной ДНК, в то время как среди некурящих менее чем у 20% имело место повышение поврежденности митохондриальной ДНК. В некоторых случаях влияние носило аддитивный характер: например, у 100% курящих диабетиков отмечена повышенная поврежденность митохондриальной ДНК. В проведенном анализе выявлено достоверное различие между теми, у кого есть факторы риска, и теми, у кого их нет (например, курение - Р<0,05; курение плюс диабет - Р<0,01; курение плюс артериальная гипертония - Р<0,05). Также было установлено наличие тенденции, направленной на существование ишемической болезни сердца в зависимости от степени поврежденности митохондриальной ДНК. Однако для определения степени существенности данной тенденции необходимо обследование большей группы больных.
Для дальнейшего изучения степени поврежденности митохондриальной ДНК при различных состояниях in vivo были исследованы 5 “сверхмарафонцев” и группа из 6 субъектов аналогичного возраста, здоровых и ведущих активный образ жизни, но не бегающих сверхмарафоны. Сначала повреждения митохондриальной ДНК определяли у сверхмарафонцев до и после 20-мильного “тренировочного забега”. Были взяты пробы крови с получением лейкоцитарной пленки и оценкой повреждений митохондриальной ДНК.
На фиг.12 показано, что имеет место небольшое увеличение поврежденности митохондриальной ДНК сразу после забега, уровень которой возвращается к норме в течение 4 часов. В другой день сверхмарафонцы и контрольные субъекты получали высокожирную пищу. Высокожирная пища была представлена блюдом "El Grande Platter" из местного ресторана с мексиканской кухней, калорийность которого составляла 3000 калорий, 60-70% которой приходилось на калорийность жиров. Пробы крови были взяты непосредственно перед едой и через 2 и 6 часов после нее. Получали лейкоцитарные пленки и выделяли ДНК. Также количественно оценивали повреждения митохондриальных белков с использованием в качестве источника лейкоцитарного митохондриального белка. На фиг.13 и 14 показано, что имеется постепенное небольшое увеличение повреждений митохондриальной ДНК в контроле, а также повышение повреждений митохондриальных белков через 2 часа со снижением через 4 часа. У сверхмарафонцев повреждения митохондриальной ДНК и повреждения белков на самом деле снижаются ниже фоновых уровней на 2-м часу и начинают возвращаться к фоновому уровню через 6 часов. Предположительно, поскольку сверхмарафонцы подвержены повышенному оксидативному стрессу во время бега, их антиоксидантная защитная система более эффективна. Таким образом, при воздействии стимула к повреждениям ДНК у сверхмарафонцев происходит даже меньше повреждений, чем в норме. Следовательно, у них активируются антиокислительные системы и уровни окислителей становятся ниже уровней, характерных для состояния покоя.
Пример 19
Анализ повреждений мтДНК в атеросклеротизованных и здоровых аортах
Для определения того, имеется ли увеличение повреждений мтДНК в атеросклеротизованных тканях человека, анализ повреждений ДНК был проведен на тканях аорт в группах здоровых и с атеросклерозом людей, аналогичных по возрастному составу. Аорты от каждой группы фиксировали в 4%-ном параформальдегиде, заливали в парафин, готовили срезы толщиной 5 мкм, депа-рафинизировали, регидратировали, окрашивали гематоксилином и эозином и оценивали развитие атеросклеротических повреждений.
Анализ методом КолПЦР показал достоверное повышение поврежденности мтДНК в атеросклеротизованных тканях по сравнению с аортами здоровой контрольной группы (фиг.15А и 15В). Напротив, маркер повреждений ядерной ДНК - кластер генов β-глобина - не проявил существенной поврежденности в атеросклеротизованной аорте по сравнению с контролем (Р=0,15). Следовательно, мтДНК проявляла существенную и предпочтительную повреждаемость в атеросклеротизованных аортах человека.
Пример 20
Влияние повреждений мтДНК на атерогенез
Сравнение уровней повреждений мтДНК в тканях атеросклеротизованной аорты с контролем аналогичного возраста показывает, что повышенная поврежденность мтДНК тесно связана с атерогенезом. Из-за присутствия существенного уровня повреждений мтДНК у мышей ароЕ-/- возраста 3 и 10 недель считается, что повреждение происходит in vivo до развития атеросклеротического повреждения или одновременно с ним. Более того, поскольку и частота атеросклеротических повреждений, и повреждения мтДНК в аорте повышены у гибридов ароЕ-/-, SOD2-/+ по сравнению с родственными особями ароЕ-/-, то понятно, что функция митохондрий важна с точки зрения атерогенеза. Следовательно, полученные данные подтверждают, что: (1) повреждения мтДНК достоверно повышены в атеросклеротизованной аорте и у мышей, и у человека; (2) поврежденность мтДНК в аорте увеличивается с возрастом in vivo; (3) повреждения мтДНК происходят in vivo до развития атеросклеротических повреждений или согласованно с ними; (4) повреждения митохондрий являются важным фактором атерогенеза.
Следующие ссылки были цитированы в данном тексте:
1. Carpenter et al. (1995) Athersclerosis 118, 169-172.
2. Halliwell, В. (1989) British J. Exper. Path. 70, 737-757.
3. Berliner, et al. (1996) Free Rad Biol Med 20, 707-727.
4. Massaeli, et al. (1995) Cardiovascular Res 29, 597-603.
5. Freeman et al. (1995) Adv Pharmacol 34, 45-69.
6. Corral-Debrinski et al. (1992) Mut Res 275, 169-180.
7. Parthasarathy, et al. (1992) Prog Lipid Res 31, 127-143.
8. Berliner et al, (1995) Circulation 91, 2499-2496.
9. Kimura et al. (1995) Atherosclerosis 118, 1-8.
10. Darley-Usmar et al. (1992) Free Radical Res Commun 17, 9-20.
11. Graham et al. (1993) FEBS Lett 330, 181-185.
12. Forman, H.J., (1982) In Free radicals in biology, Vol V, ed. Prior, W.A. (Academic Press, Orlando), pp.65-90.
13. Turrens, J., (1980) Biochem J 191, 421-427.
14. Boveris, A., Turrens, J.F. (1980) in Chemical and biochemical aspects of superoxide and superoxide dismutase, Vol I, eds. Bannister, J.V., (Elsevier, Amsterdam), pp.84-91.
15. Chance et al. (1979) Physiol Rev 59, 527-604.
16. Breen et al. (1995) Free Rad Biol Med 18, 1033-1077.
17. Chen, et al. (1994) Proc Natl Acad Sci 91, 4130-4134.
18. Zhang et al. (1990) J Biol Chem 265, 16330-16336.
19. Bandy, et al. (1990) Free Rad Biol Med 8, 523-539.
20. Ferrari et al. (1985) J Mol Cell Cardiol 17, 937-45.
21. McCord. J.M. (1988) Free Rad Biol Med 4, 9-14.
22. Giulivi et al. (1995) Arch Biochem Biophys. 316, 909-916.
23. Bindoli, A. (1988) Free Rad Biol Med 5, 247-261.
24. Hruszkewyca, A.M. (1992) Mut Res 275, 243-248.
25. Yakes, et al. (1997) Proc Natl Acad Sci 94, 514-519.
26. Adachi et al. (1993) Biochem Biophys Res Commun 195, 945-951.
27. Trounce et al. (1989) Lancet 1, 637-639.
28. Cortopassi, et al. (1990) Nucleic Acids Res 18, 6927-6933.
29. Sastre et al. (1996) Hepatology 24, 1199-1205.
30. Iyer et al. (1961) Nature 192, 535-541.
31. Beckman et al. (1990) Proc. Natl. Acad. Sci. USA 87, 1620-1624.
32. Ballinger et al. (1996) Cancer Res 56, 5692-5697.
33. Yakes et al. (1996) In Technolgies for detection of DNA damage and mutations, ed. Pfeifer, G.P. (Plenum, NY), pp.169-182.
34. Chirgwin et al, (1979) Biochemistry 18, 5294-5299.
35. Torroni et al. (1990) J. Biol. Chem. 265, 20589-20593.
36. Bogenhagen, et al. (1978) J. Mol. Biol. 119, 69-81.
37. Ballinger et al. (1992) Nature Genet 1, 11-15.
38. Slater et al. (1963) Biochim Biophys Acta 77, 383-393.
39. Berg et al. (1990) APMIS 98. 152-162.
40. Lippold HJ. (1982) Histochemistry 76, 381-405.
41. Schanenstein et al. (1972) Monatshefte fur Chemie 103, 1271-1275.
42. Huet et al. (1992) Cytometry 13, 532-539.
43. Ojala et al. (1980) Cell 22,393-403.
44. Ojala et al. (1981) Nature 290, 470-474.
45. Attardi et al. (1985) In Achievements and Perspectives of Mitochondrial Research, eds. Qkuagliarello, et al. (Elsevier Science, NY), pp.145-163.
46. Doerson et al. (1985) J. Biol. Chem. 260, 5942-5949.
47. Christianson, et al. (1986) Proc. Natl. Acad. Sci. USA 83, 6277-6281.
48. Christianson, et al. (1988) Mol. Cell. Biol. 8,4502-4509.
49. Huie, et al. (1993) Free Radical Res. Commun. 18, 195-199.
50. Palmer et al. (1988) Nature 333, 664-666.
51. Yao et al. (1992) Circulation 86, 1302-1309.
52. Garg, U.C, Hassid, A. (1989) J Clin Invest 83, 1774-1777.
53. Fossel et al. (1994) Cancer Res 54, 1240-1248.
54. Holland et al. (1996) J Cell Phys 166, 144-151.
55. Leeuwenburgh et al. (1997) J Biol Chem 272, 1433-1436.
56. Beckmann et al. (1994) Biol Chem 375, 81-88.
57. MacMillan-Crow et al. (1996) Proc, Natl. Acad. Sci. USA 93, 11853-11858.
58. MacMillan-Crow et al. (1998) Biochemistry 37, 1613-1622.
59. Lloyd-Jones, et al. (1996) Annu Rev Med 47, 365-375.
60. Darley-Usmar et al. (1995) FEBS Letters 369, 131-135.
61. Goda et al. (1996) Am J Physiol 271, H1893-H1899.
Любые патенты или публикации, упоминавшиеся в данной заявке, определены в соответствии с уровнем кругозора специалистов в данной области техники, к которым обращено изобретение. Кроме того, данные патенты и публикации включены здесь для сведения в виде библиографических ссылок в той же степени, в какой каждая отдельная публикация по отдельности и в частности определена, как включенная для сведения в виде ссылки.
Для специалиста в данной области техники будет понятно, что настоящее изобретение полностью приспособлено для получения его объектов и достижения конечных целей и преимуществ, упоминавшихся выше, а также тех объектов, конечных целей и преимуществ, вытекающих из него. Представленные здесь примеры наряду с методами, процедурами, обработками, молекулами и конкретными соединениями, описанными в данном тексте, представляют предпочтительные варианты, служат примерами и не призваны ограничить объем настоящего изобретения. Изменения в них и другие пути применения будут ясны специалистам в данной области техники, и они соответствуют сущности настоящего изобретения, что определяется объемом формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПРОГНОЗИРОВАНИЯ ИСХОДА ОСТРОЙ ИШЕМИИ МИОКАРДА | 2016 |
|
RU2649774C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ УСИЛЕНИЯ ИЛИ ПОДДЕРЖАНИЯ МЫШЕЧНОЙ ДЕЯТЕЛЬНОСТИ | 2011 |
|
RU2606763C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ УЛУЧШЕНИЯ ФУНКЦИИ МИТОХОНДРИЙ И ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ И КОГНИТИВНЫХ РАССТРОЙСТВ | 2011 |
|
RU2745439C2 |
| КОМПОЗИЦИЯ ДЛЯ ПОВЫШЕНИЯ ЭКСПРЕССИИ PGC-1α | 2017 |
|
RU2814560C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ГЕНОТОКСИЧНОСТИ КСЕНОБИОТИКОВ НА ОСНОВЕ АНАЛИЗА ПОВРЕЖДЕНИЙ МИТОХОНДРИАЛЬНОЙ ДНК ЗЕМЛЯНОГО ШМЕЛЯ (BOMBUS TERRESTRIS) | 2017 |
|
RU2663719C1 |
| ЛЕЧЕНИЕ МИТОХОНДРИАЛЬНЫХ ЗАБОЛЕВАНИЙ | 2016 |
|
RU2745611C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ АМИНОКИСЛОТЫ, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, АССОЦИИРОВАННЫХ С МИТОХОНДРИАЛЬНЫМИ ДИСФУНКЦИЯМИ | 2018 |
|
RU2770660C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ АМИНОКИСЛОТЫ, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, АССОЦИИРОВАННЫХ С МИТОХОНДРИАЛЬНЫМИ ДИСФУНКЦИЯМИ | 2018 |
|
RU2778296C2 |
| ЛЕЧЕНИЕ МИТОХОНДРИАЛЬНЫХ ЗАБОЛЕВАНИЙ | 2016 |
|
RU2834339C1 |
| Применение мембранных везикул мультипотентных стромальных клеток, индуцированных цитохалазином В, для восстановления и повышения митохондриальной функции | 2019 |
|
RU2727540C1 |
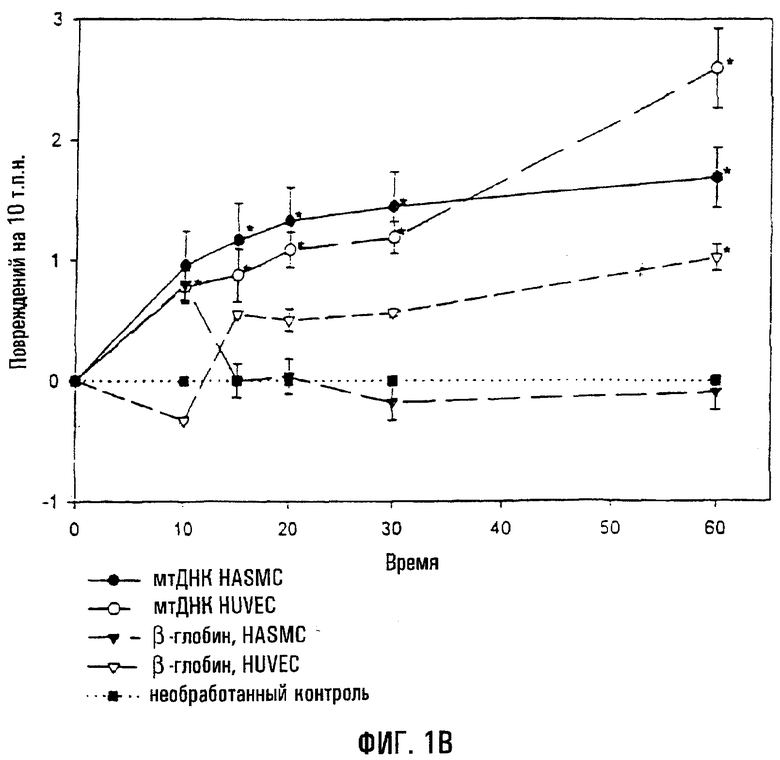
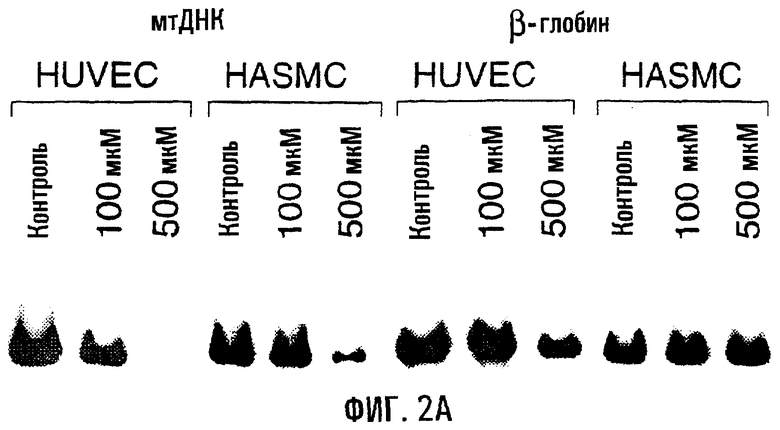
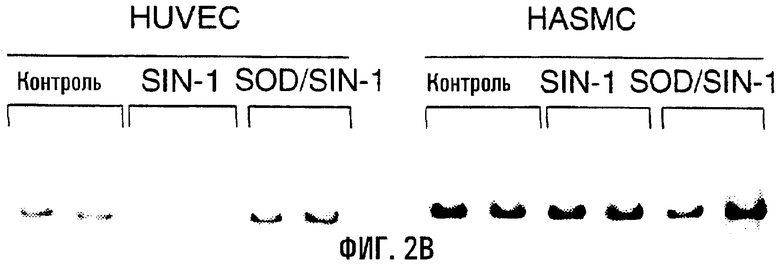
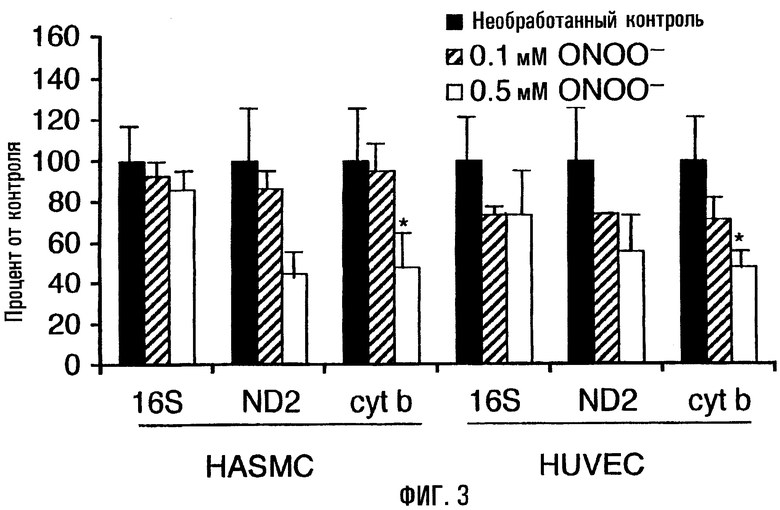
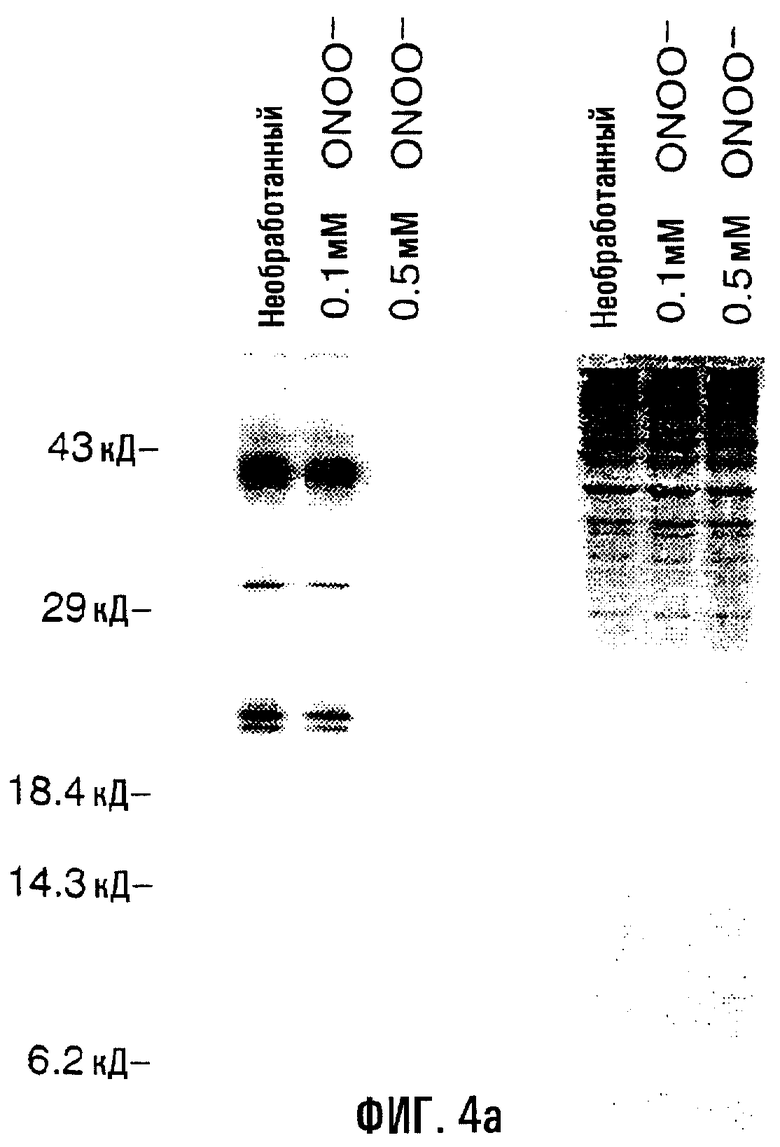
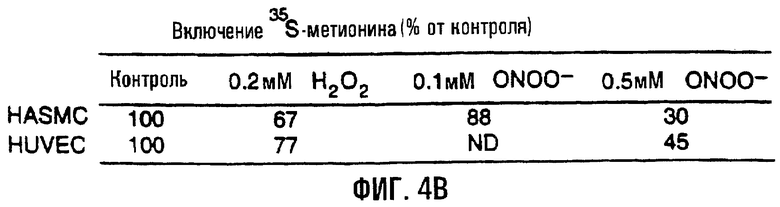
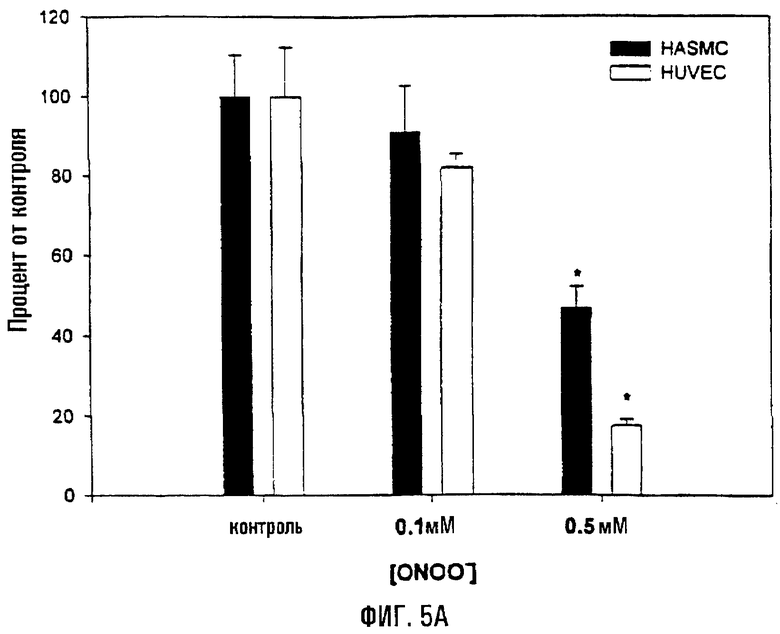
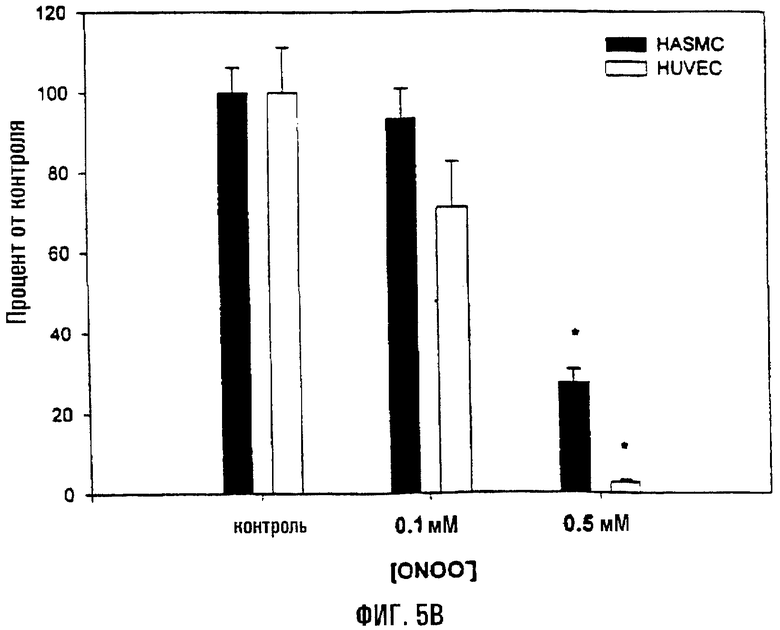
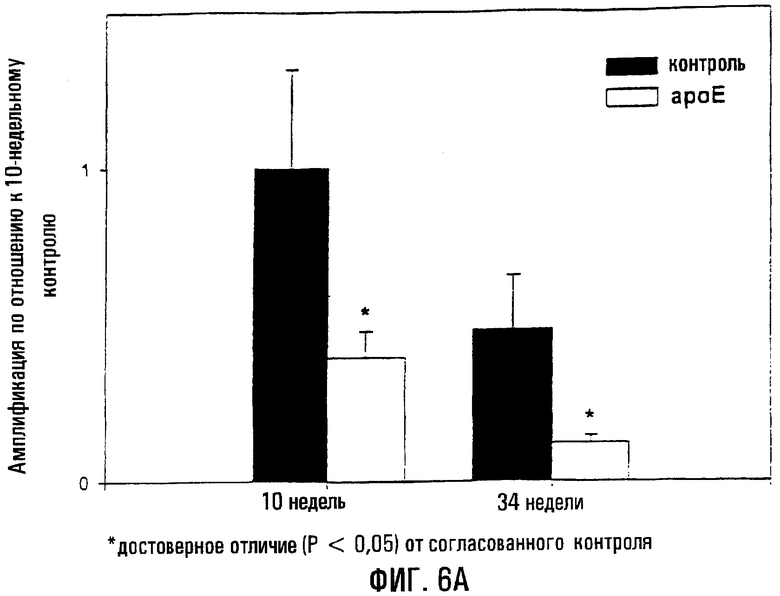
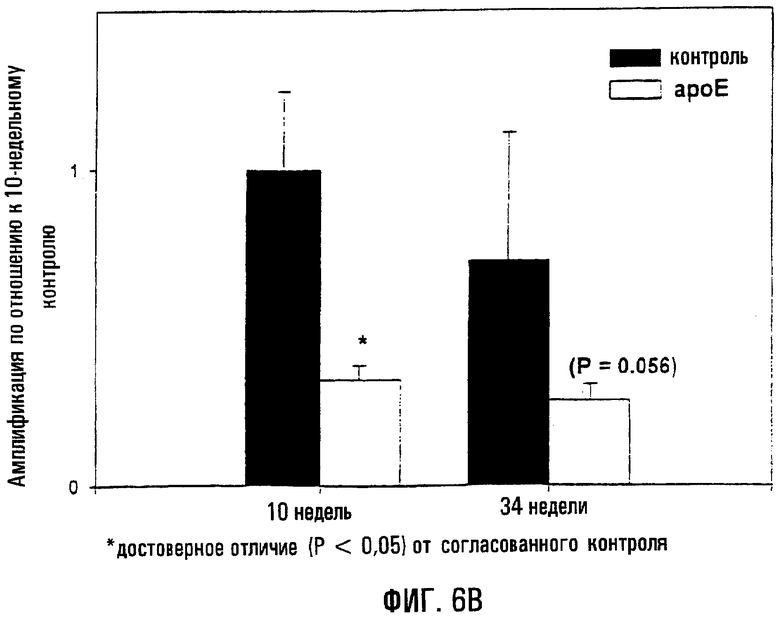
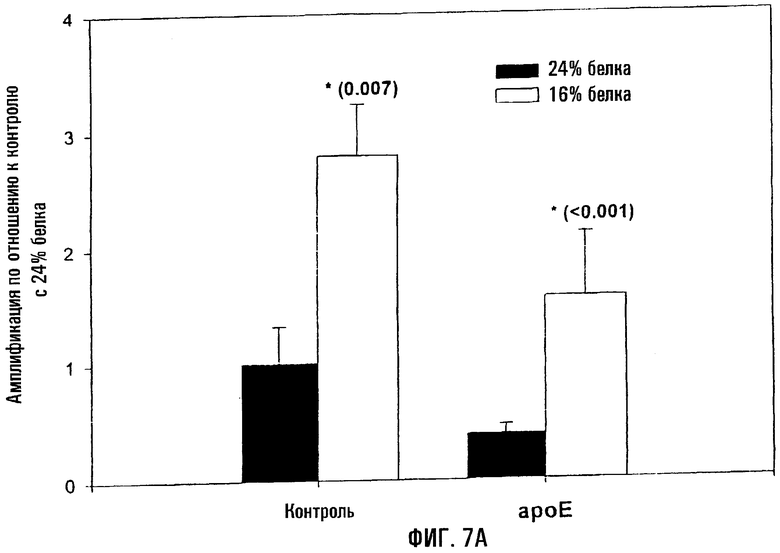
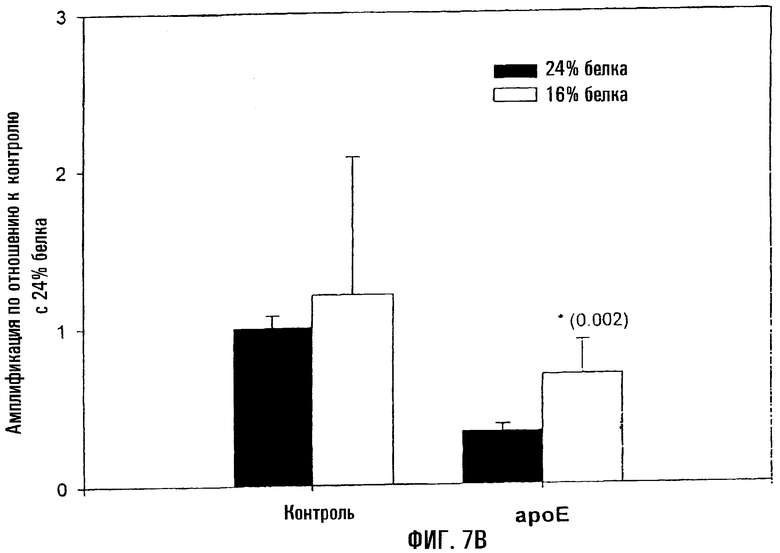
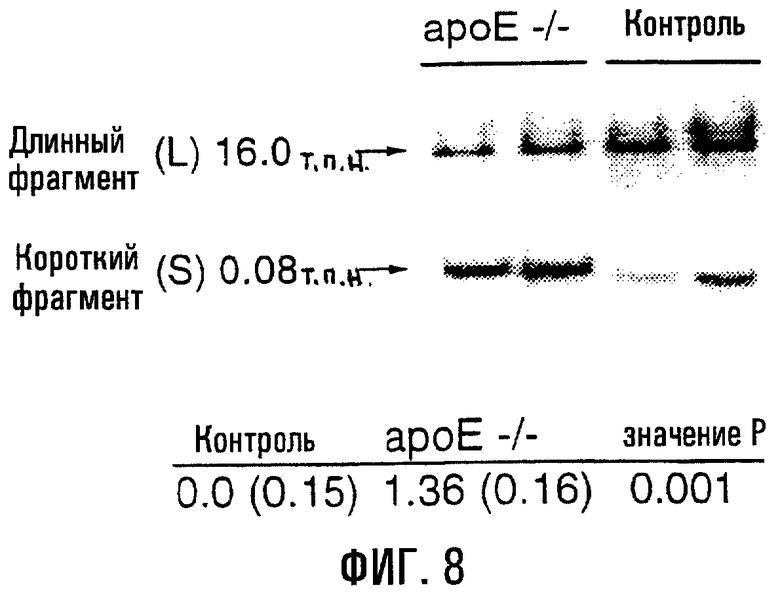
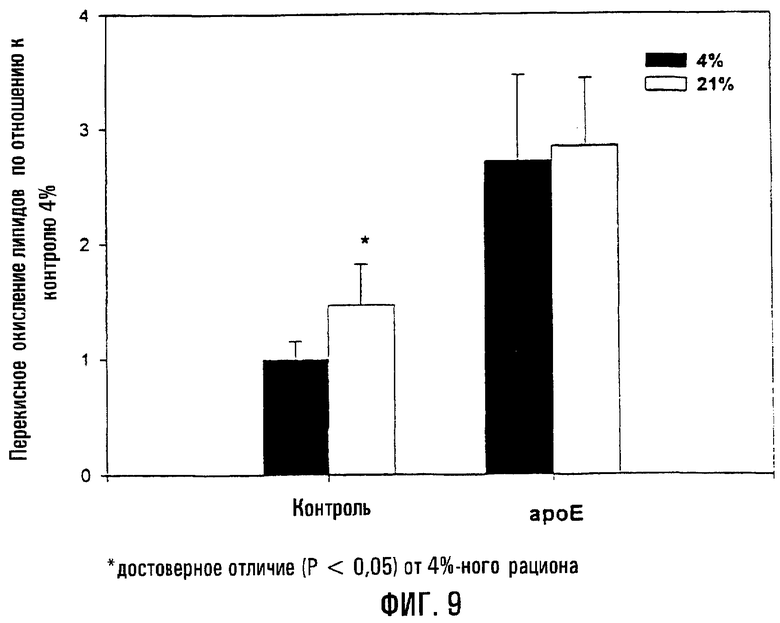
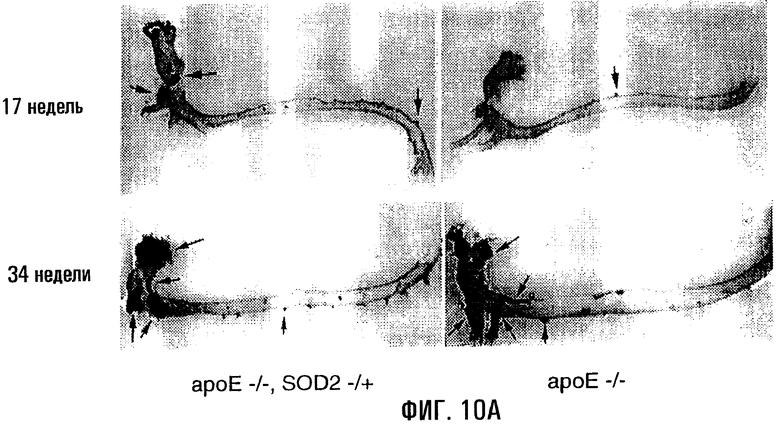
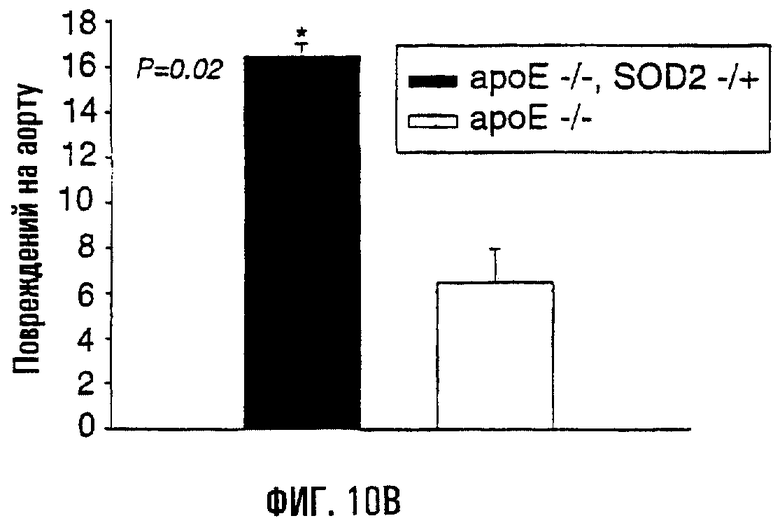
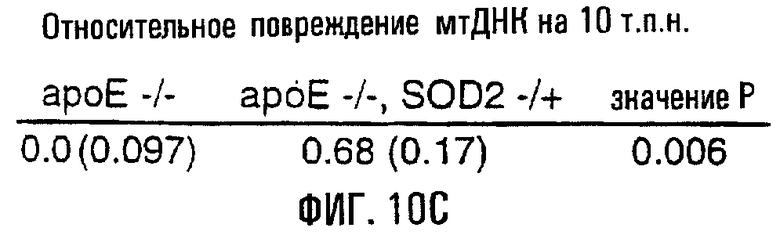
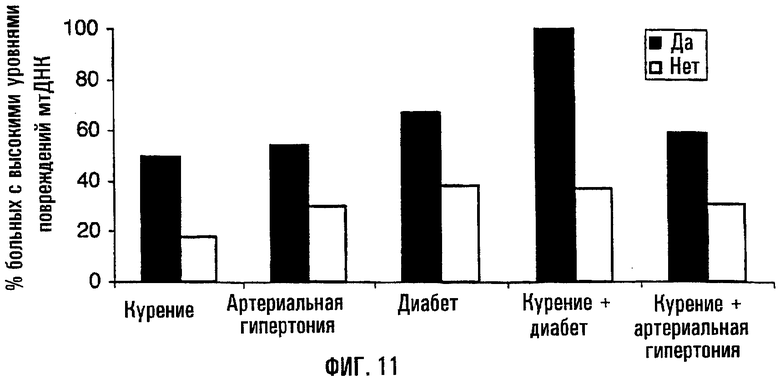
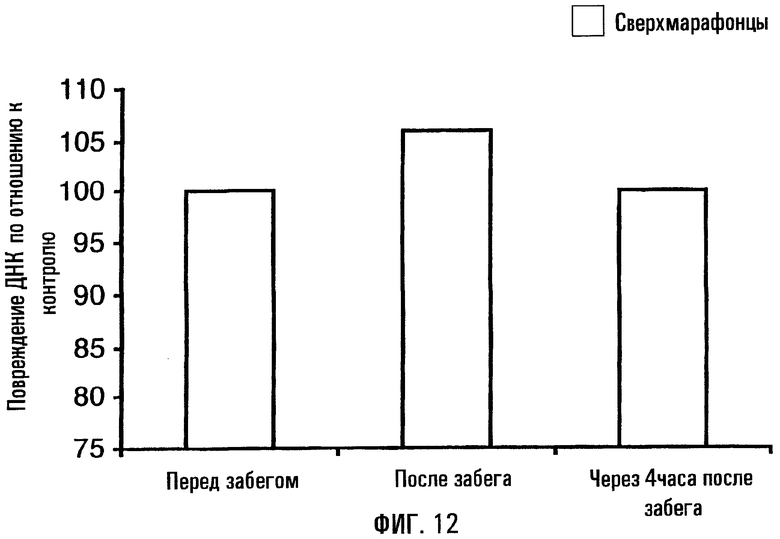
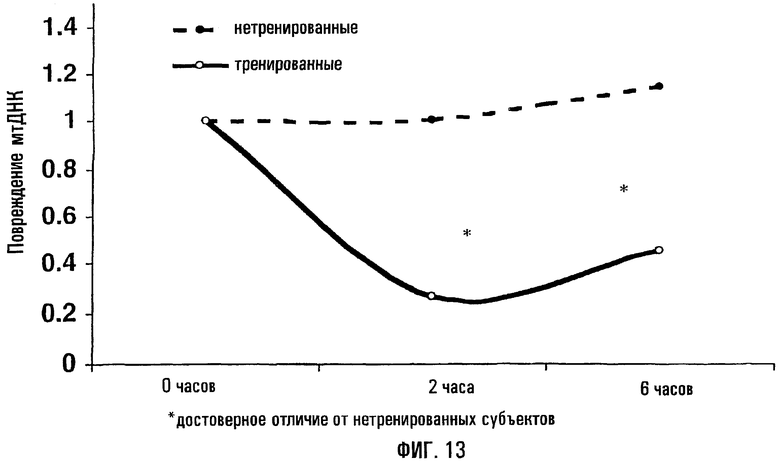
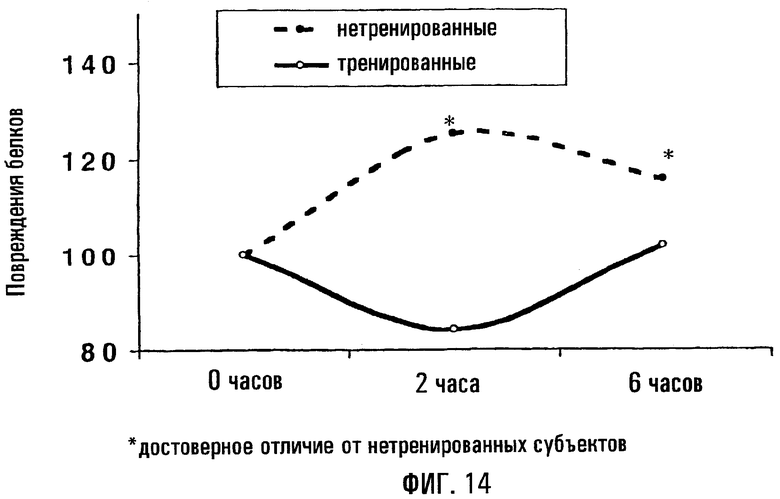
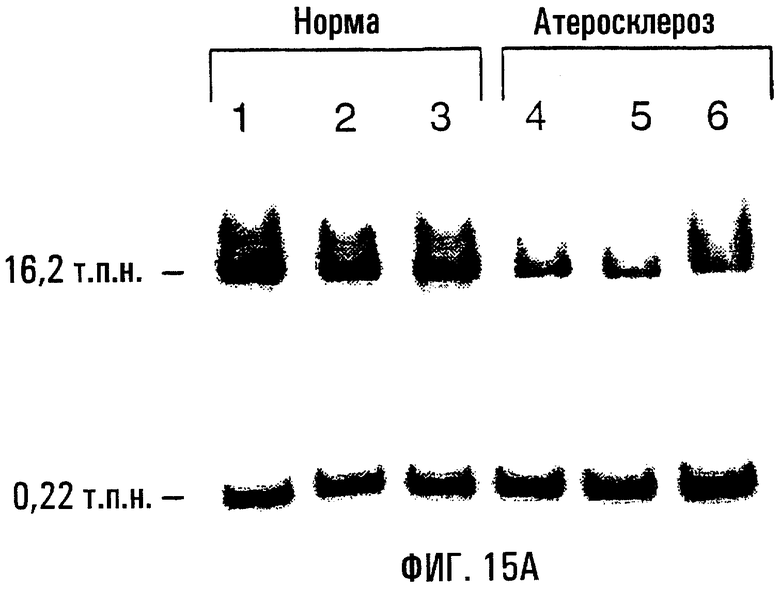

Изобретение относится к медицине и касается повреждений митохондриальной ДНК как прогностический признак атеросклеротической ишемической болезни сердца. Изобретение показывает, что повреждения митохондриальной ДНК происходят до развития атеросклеротических повреждений или одновременно с ними, что поврежденность митохондриальной ДНК в аорте увеличивается с возрастом и что и генотип, и рацион влияют на уровень повреждений митохондриальной ДНК. Следовательно, настоящее изобретение показывает, что повреждения митохондриальной ДНК происходят на ранней стадии атерогенеза и могут являться инициирующим событием в атерогенезе, а также представляет способы прогнозирования атеросклеротической ишемической болезни сердца, основываясь на количественном уровне повреждений митохондриальной ДНК, преимущество изобретения заключается в повышении чувствительности способа прогнозирования атеросклеротической ишемической болезни сердца. 3 с. и 6 з.п. ф-лы, 3 табл., 15 ил.
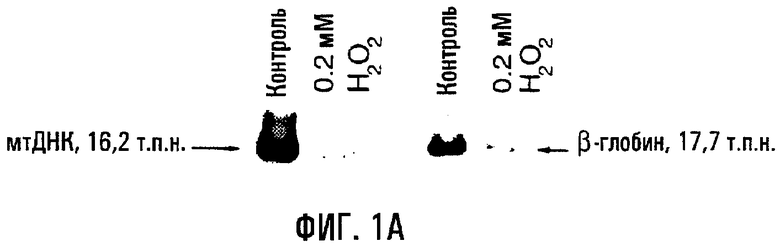
| CORRAL-DEBRINSKY M | |||
| et al | |||
| Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease | |||
| Mutation Res | |||
| Пуговица для прикрепления ее к материи без пришивки | 1921 |
|
SU1992A1 |
| CORRAL-DEBRINSKY M | |||
| et al | |||
| Hypoxemia is associated with mitochondrial DNA damage and gene induction: Implications for cardiac disease | |||
| JAMA, 1991, v.266, p.1812-1816. | |||

