Изобретение относится к области органической химии и медицины, конкретно к синтетическим производным пиримидина -2,8-дитиоксо-1Н-пирано[2,3-(1; 6,5-d']дипиримидина и их 10-аза-аналогам, а также их комплексам и солям, обладающим противовирусной и противобактериальной активностью. Изобретение предназначено в основном для использования в медицине и ветеринарии для лечения вирусных заболеваний и заболеваний, вызываемых бактериями, а также в косметологии в качестве добавки для профилактики и лечения инфекций.
Как известно, многие производные пиримидина обладают выраженной биологической активностью и участвуют в процессах жизнедеятельности организмов [1 - Досон Р. и др. Справочник биохимика. М., Мир, 1991, 544]. Большое значение имеют и синтетические производные пиримидина, из которых широкое использование в медицине приобрели замещенные барбитуровые кислоты, урацилы и др. [2 - Машковский М.Д. Лекарственные средства, 14-е изд., М., "Новая Волна", 2000]. Данные о биологической активности 5-илиденпроизводных барбитуровых кислот суммированы в обзоре [3 - Sans R.G., Chosas M.G. // Pharmazie, 1988, Bd 43, N12, S.827-829], где отмечено антиконвульсантное, антимикробное, спазмолитическое, жаропонижающее, противоопухолевое действие этих веществ. Получены данные о биологической активности некоторых 5-арилиденбарбитуровых кислот Singh А. и др. // Pharmacol. Res., 1989, Vol.21, N1, P.59-64 (Chemical Abstracts, Vol.11, 49906z); Патент Японии No 05213755, опубл. 24.08.1993 (Chemical Abstracts, 1993, Vol.119, 262520 г); Kumar А., и др. // Indian J., Chem. Sect. 1988, Vol.27, N5, P.443-447], 5-аминометиленбарбитуровых кислот [9 -.Kreutzberger А. и др. // Arch. Pharm., 1983, Bd 316, Н. 1, S.6-9], 5-арилкарбамоилбарбитуровых кислот [10, 11 - Minatelli J.A. и др. заявка Германии №3446371, опубл. 27.06.1985; Brewer A.D. Патент США №4920126, опубл. 24.04.1990]. Высокая активность обнаружена также у аннелированных производных пиримидина, например у пиразоло[3,4-d]пиримидинов, [12 -Naka Т. и др. заявка ЕПВ №. 237289 (1987)], 5-деазафлавинов [13, 14-.Kimachi Т. и др. // J. Heterocycl. Chem., 1992, Vol.29, N4, P.763-765; 5-диалкиламинометилуридинов [19 -Motawia M.S. и др. // Monatsh. Chem., 1993, Bd 124, Н. 1, S.55-64] и пиримидо[4,5-с]пиридазинов [20 - Billings B.K. и др. // J. Heterocycl. Chem., 1975, Vol.12, N6, P.1221-1224]. Перечисленные соединения обладают пестицидным, противоопухолевым, антимикробным, иммуносупрессивным, ноотропным, антигипертензионным и антиаллергическим действием.
В то же время многие группы производных пиримидина остаются практически неисследованными, с одной стороны, из-за их синтетической труднодоступности, а с другой стороны, из-за отсутствия объективных критериев, позволяющих заранее ожидать, что они проявят желаемую биологическую активность, и не будут при этом обладать высокой токсичностью и другими побочными эффектами. Поэтому, несмотря на теоретические и практические сложности, синтез новых групп производных пиримидина и изучение их биологической активности, остается актуальным направлением поиска эффективных средств лечения основных болезней человечества. В связи с этим особенно интересен синтез соединений редкой и малоисследованной группы - производных пирано[2,3-d: 6,5-d']дипиримидина. К настоящему времени известно лишь несколько единичных примеров образования пиранодипиримидиновой системы, в частности, при взаимодействии барбитуровых кислот с 3-ацилхромонами [21, 22 - Eiden F. и др. // Chemische Berichte, 1968, Bd 101, N8, S.2894-2898; [21, 22 - Eiden F. и др. // Chemische Berichte, 1968, Bd 101, N 8, S.2894-2898; Eiden F. и др..// Arch. Pharm., 1972, Bd 305, N 3, S. 187-193]. Сведения об их биологической активности отсутствуют.
Наиболее близкими к заявляемым соединениям по химическому строению и назначению являются производные 5Н-пирано[2,3-d:6,5-d']дипиримидина, обладающие антибактериальным, антивирусным и иммуномодулирующим действием [23 - Ашкинази Р.И. PCT/RU97/00371 от 19.11.1997, патент RU 2188201]. Это соединения выбраны нами в качестве прототипа. При этом вещества прототипа обладают ограниченным спектром противовирусного и антибактериального действия: они не активны против вирусов гриппа не или мало активны против ретровирусов.
Задачей изобретения является получение новых, более эффективных химических соединений с противовирусным и антибактериальным действием.
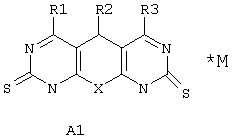
Поставленная задача решается синтезом новых веществ общей формулы А1*М:
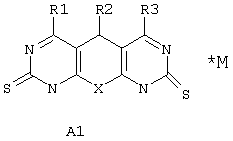
где Х выбран из группы: О, NH, N-Alkyl;
R1 выбран из группы: Н, ОН, Cl, O-Alkyl, NH2, NH-Alkyl, NH-Ar, N(Alkyl)2, SH, S-Alkyl;
R2 выбран из группы: фенилнезамещенный или замещенный, нафтил, тиенил;
R3 выбран из группы: Н, Cl, O-Alkyl, NH2, NH-Alkyl, S-дигидроксипиримидинил;
М либо отсутствует, либо выбран из группы: катион Na, К, Li, аммония, или любой другой фармакологически приемлемый катион; либо комплекс фармакологически приемлемого катиона (см. выше) с анионом одного из производных А1 (варианты R1-R3 заданы выше).
В данной серии экспериментов наилучшую активность среди заявленных веществ А1*М (где М отсутствует) проявили следующие производные:
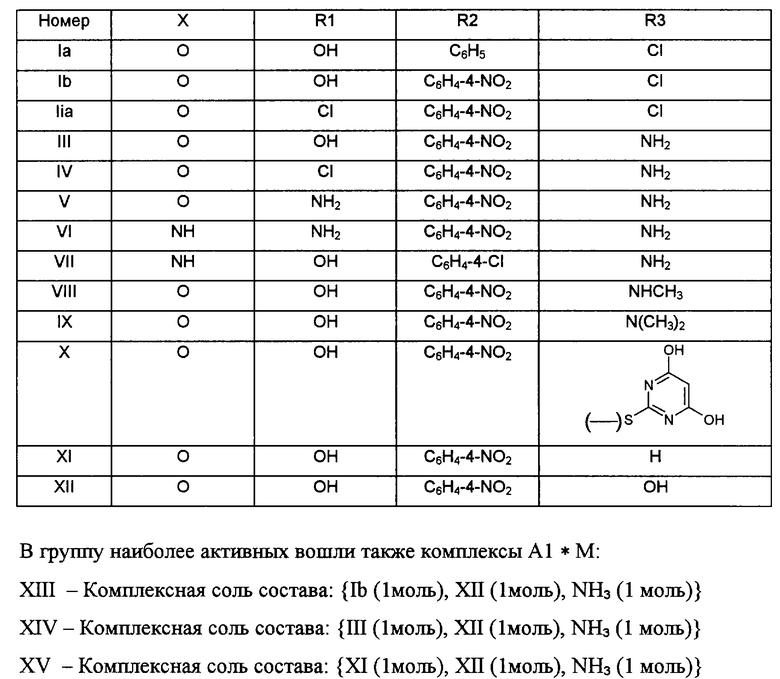
Как видно из вышеприведенных материалов, заявляемые нами производные пирано[2,3-d: 6,5-d']дипиримидина А1*М отличаются от прототипа наличием других функциональных групп в пиримидиновых фрагментах и не могут быть получены по методам, указанным в прототипе. Необходимо отметить, что наличие высокой биологической активности у заявляемых веществ не вытекает из существующего уровня техники, так как пирано[2,3-d;6,5-d']дипиримидины и их 10-аза-аналоги являются сложной и на настоящий момент малоисследованной группой, для новых представителей которой спектр и уровень активности невозможно предсказать заранее. Заявляемые вещества являются новыми, так как они отсутствуют в известных источниках информации. Таким образом, изобретение является новым, неочевидным и обладает преимуществами перед известными на настоящий момент образцами.
Следует отметить, что изобретение распространяется не только на наиболее активные соединения (Ia, Ib, IIа, III-XII) и их комплексные соли (XIII-XV), но также и на все производные А1*М, предусмотренные формулой изобретения. Проведенные нами исследования показали, что все вещества, полученные на основе предложенного нами общего способа синтеза, обладают заявленными видами активности в той или иной степени. Это позволяет заключить, что как для процесса синтеза, так и для биологических свойств заявляемых соединений наиболее существенное значение имеют не частные особенности строения радикалов R1, R2, R3, а их принадлежность к указанным в общей формуле химическим группам.
Сущность изобретения поясняется приведенными далее сведениями:
А) Синтез и анализ заявляемых соединений:
Общий способ синтеза - два этапа.
Примеры синтеза заявленных веществ, где:
Пример 1 - вариант выполнения первого этапа - синтеза промежуточного (и одновременно целевого) продукта, а именно -4-хлор-6-гидрокси-5-арил-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (Iа).
Пример 2 - Вариант выполнения первого этапа -синтеза промежуточного (и одновременно целевого) продукта, а именно-4,6-дихлор-5-арил-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (IIа).
Пример 3 - Вариант выполнения второго этапа - синтеза целевого продукта, а именно -4-амино-6-гидрокси-5-арил-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (III).
Пример 4 - Вариант выполнения второго этапа - синтеза целевого продукта, а именно -4-хлор-6-амино-5-(4-нитрофенил)-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (IV).
Пример 5 - Вариант выполнения второго этапа - синтеза целевого продукта, а именно -4,6-диамино-5-арил-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (V).
Пример 6 - Вариант выполнения второго этапа - синтеза целевого продукта, а именно -4,5-диамино-10-арил-9,10-дигидро-1Н, 8Н-1, 3, 6, 8, 9-пентаазаантрацен-2,7-дитиона (VI).
Пример 7. - Вариант выполнения второго этапа - синтеза целевого продукта, а именно -4-(4,6-дигидроксипиримидин-2-сульфанил)-6-гидрокси-5-арил-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (X).
Пример 8 - Вариант выполнения второго этапа - синтеза целевого продукта, а именно -6-гидрокси-5-(4-нитрофенил)-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (XI).
Пример 9 - Вариант выполнения второго этапа - синтеза целевого продукта, а именно -комплексной соли XIII, в состав которой входят соединения Ib, XII и аммиак {1b (1 моль), XII (1 моль), NH3 (1 моль)}.
Пример 10 - Вариант выполнения второго этапа - синтеза целевого продукта, а именно -комплексной соли XIV, в состав которой входят соединения III, XII и аммиак {III (1 моль), XII (1 моль), NН3 (1 моль)}.
Физико-химические характеристики - с двумя сводными таблицами:
Таблица 1 - спектры ПМР заявляемых соединений
Таблица 2 - температуры разложения и результаты элементного анализа.
Б) Экспериментальное определение биологических свойств заявленных соединений:
Пример 11 - определение действия на вирус герпеса (с Таблицей 3).
Пример 12 - определение действия на Chlamydia trachomatis (с Таблицей 4).
Пример 13 - определение действия на вирусы гриппа А и Б (с Таблицей 5)
Пример 14 - определение активности против вируса иммунодефицита человека (с Таблицей 6).
Пример 15 - Использование заявляемых соединений совместно с препаратами, применяемыми для лечения СПИДа (с Таблицей 7).
Пример 16 - определение острой токсичности заявляемых веществ.
А. СИНТЕЗ И АНАЛИЗ ЗАЯВЛЯЕМЫХ СОЕДИНЕНИЙ
Общий способ синтеза заявленных соединений (два этапа).
Предлагаемый нами синтез включает в себя два основных этапа:
Первый этап: Синтезируют 5-арилпроизводные 4-хлор-6-гидрокси-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (Ia, b) или 4,6-дихлор-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (IIа) из соответствующих ароматических альдегидов и 2-тиобарбитуровой кислоты с последующей обработкой РОСl3 или другими хлорирующими и водоотнимающими агентами. Полученные при этом промежуточные соединения (Ia, b IIа и их аналоги) являются одновременно и целевыми, так как обладают достаточно высоким уровнем заявляемой биологической активности.
Второй этап.Синтезируют целевые соединения (III-V, VIII-XII и их аналоги, где Х=О, R1=ОН, незамещенная или замещенная аминогруппа, R2=незамещенное или замещенное бензольное кольцо, R3 -незамещенная или замещенная аминогруппа, или алкоксигруппа, незамещенная или замещенная меркаптогруппа) из полученных на первом этапе промежуточных веществ (I и II) путем замещения одного или двух атомов хлора в трициклической 1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитионовой системе на NH2-, алкиламино-или диалкиламиногруппу, алкоксигруппу, SH-, алкилтио-, арилтио-или гетарилтиогруппу с помощью обработки соответствующими нуклеофильными реагентами (аминами, алкоголятами или тиолятами);
- Синтезируют целевые соединения (VI, VII и их аналоги, где X=NH или N-Alkyl, R1=ОН, незамещенная или замещенная аминогруппа, R2 -незамещенное или замещенное бензольное кольцо, R3 - незамещенная или замещенная аминогруппа) из полученных на первом этапе промежуточных веществ (Ia, b и IIа) путем замещения одного или двух атомов хлора в трициклической 1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитионовой системе на NH2 или алкиламиногруппу, с одновременным обменом пиранового атома кислорода (O10) на аминогруппу;
- Синтезируют целевые соединения (XI и его аналоги, где R2 - незамещенное или замещенное бензольное кольцо) из полученного на первом этапе промежуточного вещества (Iа) путем его восстановительного дегалогенирования;
- Синтезируют целевые соединения (комплексные соли XIII-XV) путем растворения эквимольных количеств соединений III и ХII или XI и XII, или Ib и XII в избытке аммиака и с дальнейшим подкислением раствора.
Пример 1. Вариант выполнения первого этапа - синтеза промежуточного (и одновременно целевого) продукта, а именно 4-хлор-6-гидрокси-5-фенил-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (Iа).
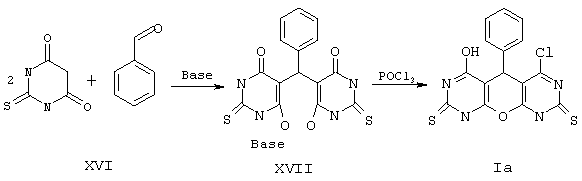
Растворяют 0.1 моль 2-тиобарбитуровой кислоты (XVI) в 50 мл диметилацетамида. К этому раствору при перемешивании прибавляют смесь 0.05 моль бензальдегида * и 0.05 моль основания (триэтиламина) в растворе диметилацетамида. Через несколько часов разбавляют эфиром, осадок промывают эфиром и сушат триэтиламмониевую соль XVII.
К полученной соли XVII (27 г) приливают 0.3 моль РОСl3, 100 мл хлороформа и кипятят 3 ч. Затем растворитель отгоняют, к остатку прибавляют воду и отделяют твердое вещество, промывают его водой и сушат. Получают продукт Iа с выходом 81%.
*Примечание. 4-Хлор-6-гидрокси-5-(4-нитрофенил)-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитион (Ib) и 4-хлор-6-гидрокси-5-(4-хлорфенил)-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитион (Iс) получают по аналогичной методике (выход 84 и 77%) при использовании п-нитробензальдегида или п-хлорбензальдегида вместо бензальдегида соответственно.
Аналогично при использовании других ароматических альдегидов получаются соответствующие 5-арилпроизводные 4-хлор-6-гидрокси-5,9-дигидро-1Н-пирано [2,3-d; 6,5-d']дипиpимидин-2,8-дитиoнa (см. общую формулу, Х=О, R1=ОН, R2=Aryl, R3=Cl, M - нет). Соответственно получены производные Id-Li (расшифровка радикалов - см. общую формулу):
n-метилбензальдегида - Id (R1=OH, R2=n-толил, R3=С1);
из n-фторбензальдегида - Ie (R1=OH, R2=C6H4-F(p), R3=C1);
из n-этоксибензальдегида - If(R1=OН, R2=C6H4-OEt(p), R3=C1);
из 3,4-диметоксибензальдегида - Ig (R1=OH, R2=C6H3-3,4-(OMe)2, R3=C1);
из 1-нафтальдегида- Ih (R1=OH, R2=1-Нафтил, R3=C1);
из тиофен-2-альдегида - Ii (R1=OH, R2=2-Tиeнил, R3=C1).
Мы не перечисляем все эти варианты, ограничиваясь наиболее активными представителями (Iа, Ib), приведенными в таблице 1.
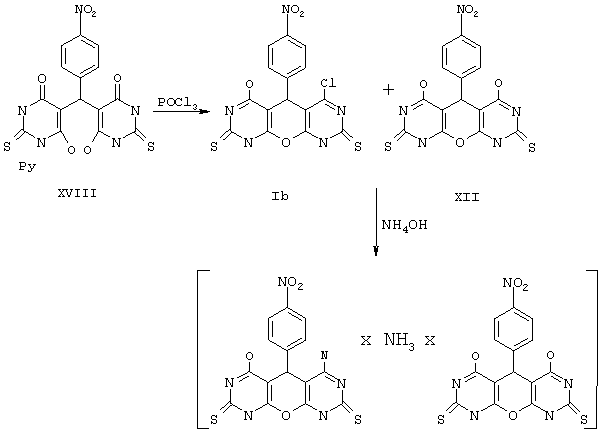
Пример 2. Вариант выполнения первого этапа - синтеза промежуточного (и одновременно целевого) продукта, а именно 4,6-дихлор-5-(4-нитрофенил)-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (IIа).
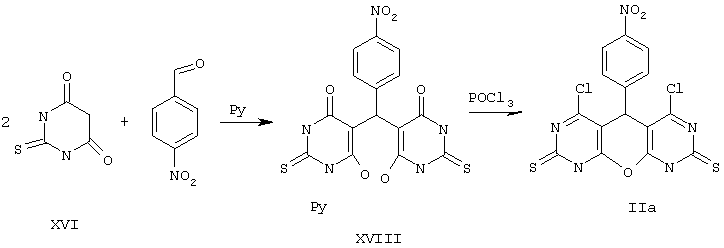
К 0.1 моль 2-тиобарбитуровой кислоты (XII) в 50 мл пиридина прибавляют 0.05 моль п-нитробензальдегида и нагревают смесь до полного растворения. Через несколько часов разбавляют эфиром, осадок промывают эфиром и сушат пиридиниевую соль (XVIII).
К полученной соли XVIII (26,5 г) приливают 0,5 моль POCl3* и нагревают с обратным холодильником около 1 ч до растворения осадка. Затем избыток POCl3 отгоняют, к остатку прибавляют воду и отделяют твердое вещество, промывают его водой и сушат. К этому веществу вновь прибавляют 0,5 моль РОСl3 и повторяют вышеописанную процедуру. После промывки и сушки получают продукт IIа с выходом 77%.
*Примечание. По аналогичной методике, при использовании вместо РОСl3 трифторуксусного ангидрида, получают 4,6-дигидрокси-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитион XII и его аналоги (см. общую формулу, Х=О, R1=R3=ОН, R2=Aryl, M отсутствует)
Пример 3. Вариант выполнения второго этапа -синтеза целевого продукта, а именно 4-амино-6-гидрокси-5-(4-нитрофенил)-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (III).
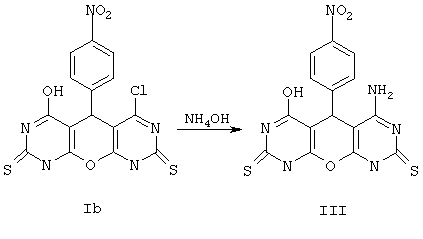
0.01 моль соединения Iб при перемешивании порциями растворяют в 30 мл 25%-ного аммиака *. Нерастворенное вещество отделяют, а раствор выдерживают сутки при комнатной температуре, затем разбавляют водой и подкисляют до рН 5-6. Выделившийся осадок отделяют, промывают водой, спиртом и сушат. Получают продукт III с выходом 71%.
*Примечание. По аналогичной методике, при использовании метиламина вместо аммиака, получают 4-метиламино-6-гидрокси-5-(4-нитрофенил)-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитион (VIII), выход 78% и 4-диметиламино-6-гидрокси-5-(4-нитрофенил)-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитион (IX), выход 73%. Методика, приведенная в примере 3, является общей и позволяет аналогично, исходя из других 5-арилпроизводных 4-хлор-6-гидрокси-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона, замещением атом хлора в реакции с аммиаком (или алкиламинами) на аминогруппу, получать соответствующие 5-арил-4-аминопроизводные 6-гидрокси-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (см. общую формулу, где Х=О, R1=ОН, R2=незамещенный или замещенный фенил или др. арил, R3=NH2 или NHAlk, или NAlk2, M отсутствует). Мы не перечисляем все эти варианты, ограничиваясь наиболее активными представителями (III, VIII, IX), приведенными в таблице 1.
Пример 4. Вариант выполнения второго этапа - синтеза целевого продукта, а именно 4-хлор-6-амино-5-(4-нитрофенил)-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (IV).
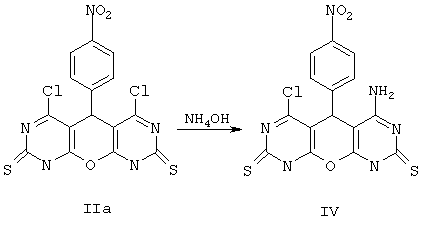
К 0.01 моль соединения IIa приливают 10 мл спирта, содержащего 0.02 моль NH4OH, и перемешивают. Смесь выдерживают сутки при комнатной температуре, затем разбавляют водой и подкисляют до рН 5-6. Выделившийся осадок отделяют, промывают водой и сушат. Получают продукт IV с выходом 54%.
Пример 5. Вариант выполнения второго этапа синтеза и получения целевого продукта, а именно 4,6-диамино-5-(4-нитрофенил)-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (V).*
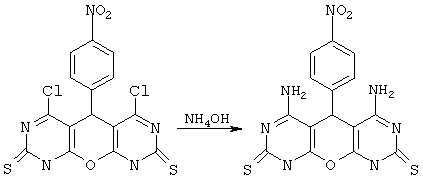
0.01 моль соединения IIa при перемешивании порциями растворяют в 30 мл 25%-ного аммиака. Нерастворенное вещество отделяют, а раствор выдерживают сутки при комнатной температуре, затем разбавляют водой и подкисляют до рН 5-6. Выделившийся осадок отделяют, промывают водой, спиртом и сушат. Получают продукт V с выходом 66%.
*Примечание. Методика, приведенная в примере 5, является общей и позволяет аналогично, исходя из других 5-арилпроизводных 4,6-дихлор-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона, замещать атомы хлора в реакции с аммиаком (или алкиламинами) на аминогруппы и получать соответствующие 5-арил-4,6-диаминопроизводные 5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (см. общую формулу, где Х=О, R1, R3=NH2 или NHAlk, или NAlk2, R2=незамещенный или замещенный фенил или др. арил, М отсутствует). Мы не перечисляем все эти варианты, ограничиваясь наиболее активными представителями (III, VIII, IX), приведенными в таблице 1.
Пример 6. Вариант выполнения второго этапа синтеза и получения целевого продукта, а именно 4,5-диамино-10-(4-нитрофенил)-9,10-дигидро-1Н, 8Н-1, 3, 6, 8, 9-пентаазаантрацен-2,7-дитиона(VI).
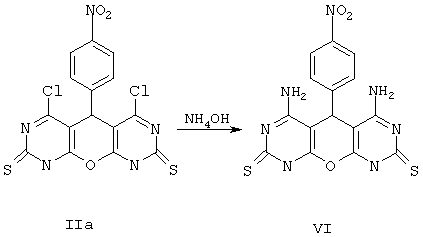
0.01 моль соединения IIа растворяют в 80 мл 25%-ного аммиака. Раствор фильтруют от нерастворенных частиц и нагревают сутки с обратным холодильником. Затем упаривают до 40 мл, охлаждают, выпавший осадок промывают водой, спиртом и сушат. Получают продукт VI с выходом 31%.
Примечание. По аналогичной методике, из соединения Iс (см. Пример 1) получают 4-гидрокси-5-амино-10-(4-хлорфенил)-9,10-дигидро-1Н, 8Н-1, 3, 6, 8, 9 -пентаазаантрацен-2,7-дитион(VII).
Пример 7. Вариант выполнения второго этапа -синтеза целевого продукта, а именно 4-(4,6-дигидроксипиримидин-2-сульфанил)-6-гидрокси-5-(4-нитрофенил)-5,9-дигидро 1Н-пирано[2,3-d; 6,5-d']дипиримидин 2,8-дитиона (X).
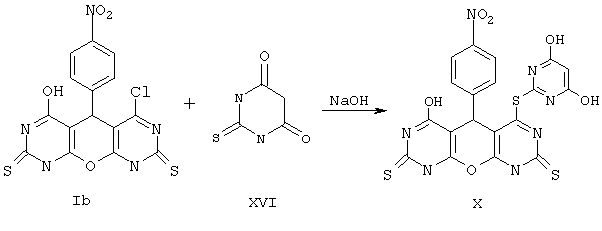
0.015 моль 2-тиобарбитуровой кислоты (XVI) при перемешивании растворяют в 20 мл воды, содержащей 0.03 моль NaOH. К полученному раствору приливают 20 мл диметилсульфоксида, затем добавляют раствор 0.01 моль соединения Ib в диметилсульфоксиде и перемешивают несколько часов при комнатной температуре. Раствор разбавляют водой, подкисляют до рН 5-6, выпавший осадок отделяют, промывают его водой, спиртом и сушат. Получают продукт Х с выходом 42%.
Пример 8. Вариант выполнения второго этапа -синтеза целевого продукта, а именно -6-гидрокси-5-(4-нитрофенил)-5,9-дигидро-1Н-пирано[2,3-d; 6,5-d']дипиримидин-2,8-дитиона (XI).
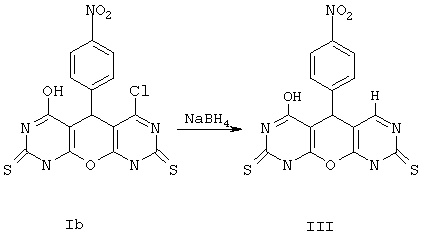
0.01 моль соединения Ib растворяют в 25 мл ледяной уксусной кислоты. К этому раствору в течение 0.5 ч при перемешивании прибавляют небольшими порциями 0.02 моль NaBH4, поддерживая температуру не выше 30°С. Перемешивают еще 4 ч и выливают смесь в воду, выпавший осадок отделяют, промывают водой, спиртом и перекристаллизовывают из диметилформамида. Получают продукт XI с выходом 36%.
Пример 9 - Вариант выполнения второго этапа -синтеза целевого продукта, а именно комплексной соли XIII, в состав которой входят следующие вещества: соединение Ib (1 моль), соединение XII (1 моль) и NH3 (1 моль).
0.01 моль соединения Ib * и 0.01 моль соединения XII растворяют при перемешивании без нагревания в 200 мл 0.5% аммиака. Нерастворившийся остаток отделяют и обрабатывают новой порцией 50 мл 0.5% аммиака. Объединенный прозрачный раствор подкисляют уксусной кислотой и выдерживают несколько часов при комнатной температуре. Выпавший осадок отделяют, промывают его водой, спиртом и сушат. Получают 6.9 г комплексной соли XIII, выход 80%.
*Примечание 1. По аналогичной методике, при использовании вместо Ib других соединений (III или XI) получают соответственно комплексные соли XIV и XV.
Пример 10 - Вариант выполнения второго этапа синтеза и получения целевого продукта, а именно комплексной соли XIV, в состав которой входят следующие вещества: III (1 моль), XII (1 моль) и NН3 (1 моль).
К 0.01 моль пиридиниевой соли XVIII (см. Пример 2) приливают 12 мл РОСl3 и нагревают с обратным холодильником около 40-50 мин до тех пор, пока подавляющая часть осадка не перейдет в раствор. Декантируют раствор от осадка и отгоняют 5 мл РОСl3, выливают в лед и после гидролиза отделяют твердое вещество и промывают его водой. К полученному продукту прибавляют 40 мл воды и 10 мл конц. аммиака и перемешивают 4 часа без нагревания, при этом подавляющая часть осадка растворяется. Раствор фильтруют от нерастворенных частиц и подкисляют уксусной кислотой. Через несколько часов фильтруют выпавший осадок, промывают водой и сушат. Получают продукт XIV с выходом 71%.
ФИЗИКО-ХИМИЧЕСКИЕ ХАРАКТЕРИСТИКИ ЗАЯВЛЯЕМЫХ ВЕЩЕСТВ
Спектры ПМР заявляемых веществ (ДМСО-d6, δ, м.д., J, Гц)
Температуры разложения и данные элементного анализа заявляемых веществ
Б. ЭКСПЕРИМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ БИОЛОГИЧЕСКИХ СВОЙСТВ ЗАЯВЛЯЕМЫХ СОЕДИНЕНИЙ.
Пример 11. Определение действия на вирус простого герпеса.
Антивирусную активность изучали по отношению к вирусу герпеса I типа (ВПГ - I/ Ленинград/248/88) по общепринятому методу [24 - Gentry G.A. и др. J. of Clinical Microbiology, 1985, 22, 2, P.I 99-204]. Вирусы выращивали на перевиваемой культуре клеток Vero, полученной из банка клеточных культур Института цитологии Российской Академии Наук.
Действие заявляемых соединений на вирус простого герпеса.
** - препарат в данной концентрации не исследовали.
*** - число клеток в 100 полях зрения,
**** процент защиты клеток от инфекции
Полученные результаты, приведенные в таблице 3, показывают, что заявляемые соединения обладают активностью против вируса герпеса.
Пример 12. Определение действия на Chlamydia trachomatis
Антимикробную активность заявляемых соединений изучали по отношению к С.trachomatis D323 -стандартному штамму из коллекции кафедры микробиологии Санкт-Петербургского Государственного Медицинского университета им. ак. И.П.Павлова. Данный штамм, выделенный от больного с хламидийным уретритом, имеет морфологию и физиологическую активность характерную для представителей данного вида, чувствителен к действию препаратов, используемых для лечения хламидийной инфекции.
Эффективность действия препаратов оценивали по защите клеток МсСоу и L929, при инфицировании хламидиями в концентрации 1·106 кл/мл [25, 26 - Judson В.А. и др. J. of Clinical Microbiology, 1988, vol.26, N12, р.2657-2658; Fenelon L.E. и др. J, of Antiimcrobial Chemotherapy, 1990, 26, р.763-767]. Оценку результатов проводили путем выявления хламидийных цитоплазматических включений методом иммунофлюоресценции (MicroTrac Chlamydia trachomatis Direct Specimen Test) и хламидийных антигенов методом CylaMonoScreen (Russian-British Joint Venture 66 Regent's Pare Road London NW1 7SX) [26 - Fenelon L.E. и др. J. of Antimicrobial Chemotherapy, 1990, 26, р.763-767].
Эффект действия препарата определяли, анализируя состояние монослоя и число клеток с ЦПВ по сравнению с контролем (культура клеток, зараженная C.trachomatis D323), при этом учитывали число неизмененных клеток в 100 полях зрения, полученных при использовании специальной сетки окуляра микроскопа.
Действие заявляемых соединений на C.trachomatis.
Полученные данные свидетельствуют, что исследованные заявляемые соединения могут быть применены для лечения заболеваний, вызванных хламидиями.
Пример 13. Активность против вирусов гриппа.
Определение противовирусной активности соединений в отношении вируса гриппа проводили на модели хорион аллантоисной оболочки (ХАО).
Соединения в исследуемых концентрациях растворяли в среде для ХАО и вносили в лунки панелей с фрагментами ХАО, куда затем добавляли вирус, и инкубировали при температуре 33-34°С в течение 48 (для гриппа типа А) и 72 (для гриппа типа В) часов. Вирусингибирующее действие исследуемых соединений оценивали по реакции гемагглютинации (РГА) при добавлении 1% куриных эритроцитов в культуральную жидкость.
Эффективность соединений оценивали по снижению инфекционной активности вируса в опыте по сравнению с контролем - индекс нейтрализации (ИН). При значении ИН до 1,0 препарат считали неактивным, при ИН от 1,0 до 2,0, при ИН выше 2,0 - активным.
Титр вируса рассчитывали по методу Рида и Мэнча.
Активность заявляемых препаратов против вирусов гриппа.
Таким образом, заявляемые соединения обладают активностью против вирусов гриппа А и В.
Пример 14. Активность против вируса иммунодефицита человека. Активность по отношению к вирусу иммунодефицита человека определяли по защите Т лимфобластоидных клеток МТ4 при инфицировании вируссодержащей жидкостью культуры HTHIV27. Клетки, инфицированные вирусом, анализировали методами 1) непрямой иммунофлюоресценции (ИФА) с поликлональной пулированной антисывороткой от ВИЧ-инфицированных и больных СПИД (титр антител в ИФА равен 1:1000000). В тестах использовали разведение 1:40. 2) В конкурентном ИФА с моноклональными антителами (МоnАb) к р24 ВИЧ и поликлональной подложкой. В качестве контроля использован азидотимидин (AZT) (Sigma).
Анти-ВИЧ активность заявленных соединений.
Таким образом, испытанные образцы угнетают репродукцию вируса иммунодефицита 1.
Пример 15. Использование заявляемых соединений совместно с препаратами, применяемыми для лечения СПИДа.
Совместное действие оценивали по защите клеток при одновременном добавлении испытуемых веществ и азидотимидина. Активность определяли по защите Т лимфобластоидных клеток МТ4 при инфицировании вируссодержащей жидкостью культуры HTHIV27. Клетки, инфицированные вирусом, анализировали методами 1) непрямой иммунофлюоресценции (ИФА) с поликлональной пулированной антисывороткой от ВИЧ-инфицированных и больных СПИД (титр антител
в ИФА равен 1:1000000). В тестах использовали разведение 1:40. 2) В конкурентном ИФА с моноклональными антителами (МоnАb) к р24 ВИЧ и поликлональной подложкой.
Угнетение репродукции вируса иммунодефицита
Полученные результаты показывают, что заявляемые соединения могут быть использованы совместно со стандартным препаратом - азидотимидином, применяемым для лечения СПИДа.
Пример 16. Определение острой токсичности заявляемых соединений Испытуемые соединения вводили через рот с помощью желудочного зонда (1000 мг/кг) или внутрибрюшинно (200 мг/кг) белым нелинейным мышам массой 20-25 г (по 5 самцов и 5 самок в каждой из испытуемых групп), после чего наблюдали за их состоянием на протяжении 14 дней. Отсутствие симптоматики, свойственной токсическим эффектам, и отсутствие гибели животных в течение указанного времени позволяет сделать вывод, что в пределах исследованных доз вещества не проявляют острой токсичности в использованной модели.
Промышленная применимость
Примеры 1-8 практического синтеза и химико-физический анализ заявляемых соединений, приведенный в таблицах 1 и 2, подтверждают возможность лабораторного и промышленного синтеза всех заявляемых соединений средствами, освоенными современной фармацевтической промышленностью, а также их четкую идентификацию общепринятыми методами контроля.
Серия приведенных экспериментов по определению биологических свойств показала, что заявляемые соединения обладают биологической активностью по отношению к различным микроорганизмам и прежде всего вирусам иммунодефицита, герпеса, гриппа А и В), что указывает на возможность их использования при лечении различных вирусных инфекций, вызванных ДНК содержащими вирусами (вирус простого герпеса) и РНК содержащими (вирусы гриппа и иммунодефицита), а также и некоторых бактериальных заболеваний, вызванных хламидиями. На основе полученных веществ могут быть созданы фармацевтически приемлемые композиции и модификации с совместимыми лекарственными препаратами, а также системами доставки лекарств, обеспечивающими максимальную системную циркуляцию действующего компонента в плазме крови. Основной путь введения - внутривенный. Однако могут быть использованы другие методы введения, обеспечивающие поступление действующего компонента в системную циркуляцию - подкожный, внутримышечный, ингаляционный, внутрибрюшинный и др. Дозы и режимы введения при этом определяются природой используемого активного ингредиента, зависят от пути введения.
Полученные результаты свидетельствуют о достижении задач, поставленных изобретением - синтезированы производные 2,8-дитиоксо-1Н-пирано[2,3-d; 6,5-d']дипиримидина и их 10-аза-аналоги. Для синтеза заявляемых соединений использованы новые технологии, описанные в заявке. Таким образом, по нашему мнению, заявляемые средства (новые вещества) удовлетворяют всем требованиям, предъявляемым к изобретению: они новы, неочевидны и промышленно применимы.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ВОЗДЕЙСТВИЯ НА ВИРУСЫ ПУТЕМ ИСПОЛЬЗОВАНИЯ ВЕЩЕСТВА НА ОСНОВЕ 2,8-ДИТИОКСО-1H-ПИРАНО[2,3-d; 6,5-d`] ДИПИРИМИДИНА И ИХ 10-АЗА-АНАЛОГОВ (ВАРИАНТЫ) | 2005 |
|
RU2294367C2 |
| ПРОИЗВОДНЫЕ 5H-ПИРАНО[2,3-D:6,5-D']ДИПИРИМИДИНА, ОБЛАДАЮЩИЕ АНТИМИКРОБНЫМ, ПРОТИВОВИРУСНЫМ И ИММУНОМОДУЛИРУЮЩИМ ДЕЙСТВИЕМ | 1997 |
|
RU2188201C2 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО С ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ (ВАРИАНТЫ) | 2015 |
|
RU2595038C1 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ АКТИВНОСТИ ИНТЕРЛЕЙКИНА-1 | 2018 |
|
RU2792143C2 |
| 4-АМИНО-6,6-ДИМЕТИЛ-5,6-ДИГИДРО-8Н-ПИРАНО /4′,3′:4,5/ ПИРРОЛО [2,3-D]ПИРИМИДИН | 1982 |
|
SU1088331A1 |
| ПРОИЗВОДНЫЕ 2,2-ДИМЕТИЛ-1,2-ДИГИДРО-4Н-ПИРАНО[4,3-D]ФУРО[2,3-B]ПИРИДИНА, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1990 |
|
SU1786804A1 |
| СПОСОБ ПОЛУЧЕНИЯ ИМИДАЗОЛОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ СОЛИ | 1989 |
|
RU2028293C1 |
| ЗАМЕЩЕННЫЕ ПИРИДО[4',3':5,6]ПИРАНО[2,3-d]ПИРИМИДИНЫ И КОМБИНАТОРНАЯ БИБЛИОТЕКА | 2004 |
|
RU2269538C1 |
| СОЕДИНЕНИЯ ДИОКСИН- И ОКСАЗИН[2,3-D]ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОИНОЗИТИД-3-КИНАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2612251C2 |
| СПИРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА | 2008 |
|
RU2470933C2 |
Изобретение относится к новому веществу, обладающему антивирусной и антибактериальной активностью на основе производных 2,8-дитиоксо-1Н-пирано[2,3-d; 6,5-d']дипиримидина и их 10-аза-аналогов, отличающееся тем, что включает производное указанной группы общей формулы А1 * М:
где Х выбран из группы: О, NH, N-Alkyl;
R1 выбран из группы: Н, ОН, Cl, O-Alkyl, NH2, NH-Alkyl, NH-Ar, N(Alkyl)2, SH, S-Alkyl;
R2 выбран из группы: фенилнезамещенный или замещенный, нафтил,тиенил;
R3 выбран из группы: Н, Cl, O-Alkyl, NH2, NH-Alkyl, S-
дигидроксипиримидинил;
М либо отсутствует, либо выбран из группы: катион Na, К, Li,
аммония, или любой другой фармакологически приемлемый катион; либо комплекс фармакологически приемлемого катиона (см. выше) с анионом одного из производных А1 (варианты R1-R3 заданы выше).
Технический результат - получение новых соединений, обладающих противовирусной и антибактериальной активностью. 1 н. и 16 з.п. ф-лы, 7 табл.
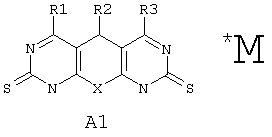
где Х выбран из группы: О, NH, N-Alkyl;
R1 выбран из группы: Н, ОН, Cl, O-Alkyl, NH2, NH-Alkyl, NH-Ar. N(Alkyl)2, SH, S-Alkyl, S-Ar, S-Hetaryl;
R2 выбран из группы: C6H5, Aryl;
R3 выбран из группы: Н, Cl, O-Alkyl, NH2, NH-Alkyl, NH-Ar, S-Hetaryl;
М либо отсутствует, либо выбран из группы: катион Na, К, Li, аммония, или любой другой фармакологически приемлемый катион; либо комплекс фармакологически приемлемого катиона (см. выше) с анионом одного из производных А1 (варианты R1-R3 заданы выше).

| ПРОИЗВОДНЫЕ 5H-ПИРАНО[2,3-D:6,5-D']ДИПИРИМИДИНА, ОБЛАДАЮЩИЕ АНТИМИКРОБНЫМ, ПРОТИВОВИРУСНЫМ И ИММУНОМОДУЛИРУЮЩИМ ДЕЙСТВИЕМ | 1997 |
|
RU2188201C2 |
| УЗЕЛ КРЕПЛЕНИЯ ОПТИЧЕСКИХ ДЕТАЛЕЙ В ОПРАВЕ | 0 |
|
SU203971A1 |
| WO 9925718 A1 27.05.1999. | |||

