Область техники, к которой относится изобретение
Настоящее изобретение относится к соединениям, композициям, способам получения таких соединений и к способам лечения инфекции, вызываемой вирусом гепатита С (HCV). Настоящее изобретение относится, в частности, к новым пептидным аналогам, фармацевтическим композициям, содержащим такие аналоги, и к способам применения этих аналогов для лечения HCV-инфекции.
Предпосылки создания изобретения
Вирус гепатита С (HCV) является основным возбудителем гепатита, не относящегося к гепатитам А и В, который возникает после переливания крови и который поражает людей во всем мире. Установлено, что свыше 170 миллионов людей в мире заражено вирусом. Большой процент носителей становится хронически инфицированным и у многих пациентов это приводит к хроническому заболеванию печени, так называемому хроническому гепатиту С. В свою очередь, эта группа больных имеет высокий риск заболевания такой серьезной болезнью печени, как цирроз печени, печеночно-клеточный рак и последняя стадия болезни печени, приводящая к смерти.
Механизм, с помощью которого происходит сохранение вируса HCV в организме и обеспечивается высокий коэффициент заболеваемости хронической болезнью печени, пока недостаточно изучен. Неизвестно, как HCV взаимодействует с иммунной системой хозяина и преодолевает ее действие. Кроме того, также еще не выявлены роли клеточных и гуморальных иммунных ответов в защите от HCV-инфекции и при заболевании гепатитом. Имеются данные о том, что иммуноглобулины могут применяться для профилактики связанного с переливанием крови вирусного гепатита, однако Центр по контролю заболеваемости в настоящее время не рекомендует лечение с использованием иммуноглобулинов для этой цели. Отсутствие эффективного защитного иммунного ответа затрудняет разработку вакцины или адекватных профилактических мер после экспозиции, поэтому в ближайшее время надежды в основном возлагаются на антивирусные средства.
С целью выявления фармацевтических агентов, обладающих эффективностью в отношении лечения HCV-инфекции у пациентов, страдающих хроническим гепатитом С, были проведены различные клинические испытания. В этих испытаниях применяли интерферон-альфа индивидуально или в сочетании с другими антивирусными агентами. Эти исследования позволили установить, что у основного большинства участников эксперимента не было обнаружено реакции на такие схемы лечения, а из тех участников, которые оказались чувствительными к лечению, у большей части после окончания лечения наблюдали рецидив.
Таким образом, до последнего времени терапия с использованием интерферона (IFN) оставалась единственным доступным методом лечения пациентов, страдающих хроническим гепатитом С, обладающей доказанной в клинических условиях эффективностью. Однако длительность действия такого лечения является небольшой, и кроме того, лечение интерфероном вызывает серьезные побочные действия (т.е. ретинопатию, тиреоидит, острый панкреатит, депрессию), что снижает качество жизни пациентов, подвергающихся лечению. В настоящее время интерферон в сочетании с рибавирином предложен для лечения пациентов, не чувствительных к IFN, применяемому индивидуально. Такой подход в настоящее время рекомендован для лечения не подвергавшихся ранее лечению пациентов и является наилучшим в терапии HCV. Однако побочные действия, вызванные IFN, не уменьшаются при такой совместной терапии.
Таким образом, существует необходимость в разработке эффективных антивирусных агентов для лечения HCV-инфекции, которые были бы лишены недостатков существующих методов лечения, основанных на применении фармацевтических средств.
HCV представляет собой заключенную в оболочку положительную цепь РНК-ового вируса семейства Flaviviridae. Геном HCV, представленный одноцепочечной РНК, состоит приблизительно из 9500 нуклеотидов и имеет одну открытую рамку считывания (ОРС), которая кодирует один большой полипротеин, состоящий примерно из 3000 аминокислот. В зараженных клетках этот полипротеин расщепляется во многих сайтах клеточными и вирусными протеазами с образованием структурных и неструктурных (NS) протеинов. В случае HCV под действием двух вирусных протеаз образуются зрелые неструктурные протеины (NS2, NS3, NS4A, NS4B, NS5A и NS5B). Первая из них, которая пока еще недостаточно охарактеризована, расщепляет связь NS2-NS3; вторая представляет собой серинпротеазу, содержащуюся в N-концевой области NS3 (далее обозначена как NS3-протеаза), и она опосредует все последующие расщепления, происходящие по ходу транскрипции относительно NS3, как в цис-ориентации, в сайте расщепления NS3-NS4A, так и в транс-ориентации в остальных сайтах NS4A-NS4B, NS4B-NS5A, NS5A-NS5B. Протеин NS4A, вероятно, обладает множественными функциями, действуя в качестве кофактора для NS3-прoтеазы и возможно способствуя локализации на мембране NS3 и других вирусных репликаз. Образование комплекса протеина NS3 с NS4A, вероятно, необходимо для процессирования, усиления протеолитической эффективности во всех сайтах. Протеин NS3 также обладает нуклеозидтрифосфатазной активностью и РНК-геликазной активностью. NS5B представляет собой РНК-зависимую РНК-полимеразу, принимающую участие в репликации HCV.
В заявке на патент WO 97/06804 описан (-)-энантиомер нуклеозидного аналога цитозин-1,3-оксатиолана (также известный как 3ТС), обладающий активностью в отношении HCV. Хотя в проведенных ранее клинических испытаниях для этого соединения обнаружена активность в отношении ВИЧ и HBV, пока нет клинического доказательства его активности в отношении HCV и не выявлен его механизм действия в отношении этого вируса.
Общая стратегия разработки антивирусных агентов состоит в инактивации кодируемых вирусом ферментов, которые важны для репликации вируса.
Значительные усилия, предпринятые в этой связи для выявления соединений, которые ингибируют NS3-протеазу или РНК-геликазу HCV привели к следующим результатам:
В патенте US 5633388 описаны замещенные гетероциклом карбоксамиды и их аналоги, обладающие активностью в отношении HCV. Мишенью для этих соединений является геликазная активность протеина NS3 вируса, однако пока нет данных о их клинических испытаниях.
Chu и др. (Tet. Lett., (1996), 7229-7232) описали фенантренхинон, обладающий активностью в отношении NS3-протеазы HCV in vitro. Никакие дополнительные данные о этом соединении не опубликованы.
В научном докладе, представленном на Девятой международной конференции по антивирусным исследованиям (Ninth International Conference on Antiviral Research, Urabandai, Fukyshima, Japan (1996) (Antiviral Research, (1996), 30, 1, A23 (реферат 19)), сообщалось о триазолидиновых производных, обладающих ингибирующей активностью в отношении HCV-протеазы.
В некоторых иследованиях описаны соединения, обладающие ингибирущим действием в отношении других серинпротеаз, таких как человеческая эластаза лейкоцитов. Одна группа таких соединений описана в WO 95/33764 (Hoechst Marion Roussel, 1995). Описанные в данной заявке пептиды представляют собой морфолинилкарбонилбензоилпептидные аналоги, которые структурно отличаются от пептидов по настоящему изобретению.
В WO 98/17679 (на имя VeKTex Pharmaceuticals Inc.) описаны ингибиторы серин-протеазы, в частности NS3-протеазы вируса гепатита С.
Фирмой Hoffman LaRoche (WO 98/22496, US 5866684 и US 6018020) также описаны гексапептиды, являющиеся ингибиторами протеиназы, в качестве антивирусных агентов для лечения HCV-инфекции.
 и др. и Ingallinella и др. опубликовали данные о продукте ингибирования NS4A-4B (Biochemistry (1998), 37, 8899-8905 и 8906-8914).
и др. и Ingallinella и др. опубликовали данные о продукте ингибирования NS4A-4B (Biochemistry (1998), 37, 8899-8905 и 8906-8914).
В WO 97/43310 на имя Schering Corporation описаны состоящие из 20 и 21 аминокислот последовательности пептидов, обладающих активностью в отношении NS3-протеазы HCV.
В WO 98/46597 на имя университета Эмори описаны пептиды и миметики пептидов, обладающие активностью in vitro в отношении серинпротеаз.
В WO 98/46630 на имя Peptide Therapeutics Limited описан депсипептидный субстрат, обладающий способностью ингибировать NS3-протеазу HCV.
Наконец, в патенте US 5869253 описаны ферментативные молекулы РНК, которые обладают способностью ингибировать NS3-протеазу HCV.
Ни в одной из указанных выше заявок на патент не описаны циклические пептиды, обладающие активностью в отношении NS3-протеазы HCV.
В WO 99/07733, WO 99/07734, WO 00/09543 и WO 00/09558 описаны гексатетрапептиды и трипептидные аналоги, обладающие способностью ингибировать NS3-протеазу. Однако в этих описаниях не предложены макроциклические аналоги по настоящему изобретению и представленная информация не может привести к их созданию.
В заявке WO 99/38888 на имя Institute de Richerche di Biologia Moleculare (IRBM), опубликованной 5 августа 1999 г., описаны малые пептидные ингибиторы NS3-npoтeaз HCV.
В этом описании не предложено и не указано циклическое строение пептидов по настоящему изобретению. Кроме того, это описание опубликовано после даты приоритета настоящего описания.
В заявке WO 99/64442 на имя IRBM, которая также опубликована после даты приоритета настоящего описания, описаны олигопептиды, включающие кетокислоты в положении Р1.
Заявка WO 99/50230 на имя VeKTex Pharmaceuticals (опубликованная 7 октября 1999 г.) также опубликована после даты приоритета настоящей заявки. И в этом случае в публикации даже отдаленно не подразумеваются какие-либо циклические пептиды по настоящему изобретению.
Одним из преимуществ настоящего изобретения является то, что в нем заявлены макроциклические пептиды, которые обладают ингибирующей активностью в отношении NS3-протеазы вируса гепатита С.
Еще одним преимуществом объекта настоящего изобретения является тот факт, что эти пептиды специфически ингибируют NS3-протеазу и не проявляют заметной ингибирующей активности в отношении других серинпротеаз, таких как эластаза лейкоцитов человека (HLE), эластаза панкреатической железы свиньи (РРЕ) или бычий химотрипсин поджелудочной железы, или в отношении цистеинпротеаз, таких как катепсин В печени человека (Cat В).
И еще одним преимуществом настоящего изобретения является то, что в нем представлены небольшие пептиды с низкой молекулярной массой, которые обладают способностью проникать через клеточные мембраны и ингибировать NS3-протеазную активность в культуре клеток.
Еще одно преимущество соединений по настоящему изобретению заключается в том, что они обладают активностью в отношении обоих основных генотипов, обнаруженных в клинических изолятах (1а и 1б), что убедительно подтверждает, что эти соединения обладают активностью в отношении всех известных в настоящее время генотипов HCV.
Краткое изложение сущности изобретения
Объектом настоящего изобретения являются соединения формулы (I):

где
W обозначает СН или N,
R21 обозначает Н, галоген, C1-С6алкил, C3-С6циклоалкил, C1-С6галоалкил, C1-С6алкокси, C3-С6циклоалкокси, гидрокси или N(R23)2, где каждый R23 независимо друг от друга обозначает Н, C1-С6алкил или C3-С6циклоалкил; и
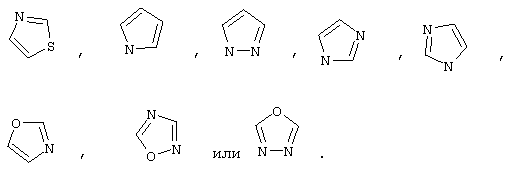
R22 обозначает Н, галоген, C1-С6алкил, C3-С6циклоалкил, C1-С6галоалкил, C1-С6тиоалкил, C1-С6алкокси, C3-С6циклоалкокси, C2-С7алкоксиалкил, C3-С6циклоалкил, С6- или С10арил или Het, где Het обозначает пяти-, шести-, или семичленный насыщенный или ненасыщенный гетероцикл, содержащий один-четыре гетероатома, выбранных из азота, кислорода и серы,
при этом указанный циклоалкил, арил или Het замещен радикалом R24, где
R24 обозначает Н, галоген, C1-С6алкил, C3-С6циклоалкил, C1-С6алкокси, C3-С6циклоалкокси, NO2, N(R25)2, NH-C(O)-R25 или NH-C(O)-NH-R25,
где каждый R25 независимо друг от друга обозначает Н, C1-С6алкил или C3-С6циклоалкил,
или
R24 обозначает NH-C(О)-OR26, где R26 обозначает C1-С6алкил или C3-С6циклоалкил,
R3 обозначает гидрокси, NH2 или группу формулы -NH-R31, где R31 обозначает С6-или С10арил, гетероарил, -C(О)-R32, -C(О)-OR32 или -C(О)-NHR32, где
R32 обозначает C1-С6алкил или C3-С6циклоалкил,
D обозначает состоящую из 5-10 атомов насыщенную или ненасыщенную алкиленовую цепь, необязательно включающую один-три гетероатома, независимо друг от друга выбранных из О, S, или N-R41, где
R41 обозначает Н, C1-С6алкил, C3-С6циклоалкил или -C(О)-R42, где R42 обозначает C1-С6алкил, C3-С6циклоалкил или С6- или С10арил;
R4 обозначает Н или один-три заместителя на любом атоме углерода цепи D, где заместители независимо друг от друга выбирают из группы, включающей C1-С6алкил, C1-С6галоалкил, C1-С6алкокси, гидрокси, галоген, амино, окси, тиогруппу или C1-С6тиоалкил, и
А обозначает амид формулы -C(О)-NH-R5, где
R5 выбирают из группы, включающей C1-С8алкил, C3-С6циклоалкил, С6- или С10арил или С7-С16аралкил; или
А обозначает карбоновую кислоту, или его фармацевтически приемлемая соль или сложный эфир.
Под объем настоящего изобретения также подпадает фармацевтическая композиция, включающая эффективное в отношении вируса гепатита С количество соединения формулы I или его фармацевтически приемлемой соли или его сложного эфира в смеси с фармацевтически приемлемым носителем или вспомогательным веществом.
Важным объектом изобретения является способ лечения инфекции, вызываемой вирусом гепатита С у млекопитающего, предусматривающий введение млекопитающему эффективного в отношении вируса гепатита С количества соединения формулы I, или его терапевтически приемлемой соли, или сложного эфира, или описанной выше композиции.
Другим важным объектом изобретения является способ ингибирования репликации вируса гепатита С путем обработки вируса ингибирующим NS3-протеазу вируса гепатита С количеством соединения формулы I, или его терапевтически приемлемой соли, или сложного эфира, или описанной выше композиции.
Еще одним объектом изобретения является способ лечения инфекции, вызываемой вирусом гепатита С у млекопитающего, предусматривающий введение млекопитающему эффективного в отношении вируса гепатита С количества композиции, включающей соединения формулы I, или его терапевтически приемлемую соль, или сложный эфир. Согласно одному из вариантов осуществления фармацевтические композиции по изобретению включают дополнительный иммуномодулятор. Примеры дополнительных иммуномодуляторов включают (но не ограничиваясь ими) α-, β- или γ-интерфероны.
В альтернативном варианте осуществления фармацевтические композиции по настоящему изобретению могут дополнительно включать антивирусный агент. Примерами антивирусных агентов являются рибавирин и амантадин.
Еще в одном альтернативном варианте осуществления фармацевтические композиции по настоящему изобретению могут дополнительно включать другие ингибиторы HCV-протеазы.
И еще в одном варианте осуществления фармацевтические композиции по изобретению могут дополнительно включать ингибитор других мишеней жизненного цикла HCV, таких как геликаза, полимераза, металлопротеаза или IRES.
Подробное описание предпочтительных вариантов осуществления
Определения
Если не указано иное, то в контексте настоящего описания используемые понятия имеют следующие значения:
В тех случаях, когда для обозначения конфигурации заместителя, например, R4 соединения формулы I, используют символы (R) или (S), то обозначение относится ко всему соединению, а не только к заместителю.
Обозначение "P1, P2 и Р3" в контексте настоящего описания относится к положению аминокислотных остатков, начиная с С-конца пептидных аналогов и простираясь к N-концу [т.е. Р1 обозначает положение 1 относительно С-конца, P2 обозначает положение 2 относительно С-конца и т.д.) (см. Berger А. и Schechter I., Transactions of the Royal Society London series (1970), B257. 249-264].
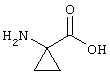
В контексте настоящего описания понятие "1-аминоциклопропилкарбоновая кислота" (АЦКК) относится к соединению формулы:
В контексте настоящего описания понятие "винил-АЦКК" относится к соединению формулы:
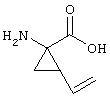
В контексте настоящего описания понятие "гомоаллил-АЦКК" относится к соединению формулы:

Понятие "гало" в контексте настоящего описания обозначает галогеновый заместитель, выбранный из группы, включающей бром, хлор, фтор или йод.
Понятие "C1-С6галоалкил" индивидуально или в сочетании с другим заместителем в контексте настоящего описания обозначает ациклические алкильные заместители с прямой или разветвленной цепью, содержащие 1-6 атомов углерода и имеющие один или несколько заместителей атома водорода, представляющих собой галоген, выбранный из ряда, включающего бром, хлор, фтор или йод.
Понятие "C1-С6тиоалкил" индивидуально или в сочетании с другим заместителем в контексте настоящего описания обозначает ациклические алкильные заместители с прямой или разветвленной цепью, содержащие тиольную группу, такие как тиопропил.
Понятие "C1-С6алкил" или "(низш.)алкил" в контексте настоящего описания, одно или в сочетании с другим заместителем, обозначает ациклические алкильные заместители с прямой или разветвленной цепью, содержащие от 1 до 6 атомов углерода, и включает, например, метил, этил, пропил, бутил, гексил, 1-метилэтил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил.
Понятие "C3-С6циклоалкил" в контексте настоящего описания, одно или в сочетании с другим заместителем, обозначает циклоалкильный заместитель, содержащий от 3 до 6 атомов углерода, и включает циклопропил, циклобутил, циклопентил и циклогексил.
Понятие "ненасыщенный циклоалкил" включает, например, замещенный цикло-гексенил:

Понятие "насыщенный или ненасыщенный алкилен" в контексте настоящего описания обозначает двухвалентный алкильный заместитель, полученный путем удаления одного атома водорода с обоих концов насыщенного или ненасыщенного алифатического углеводорода с прямой или разветвленной цепью и включает, например, -СН2СН2С(СН3)2СН2СН2-, -СН2СН2СН=СНСН2СН2- или -СН2С≡ССН2СН2-. Такая алкильная цепь необязательно может включать гетероатом, такой как кислород (например -СН3-СН2-О-СН2-).
Понятие "С1-С6алкокси" индивидуально или в сочетании с другим заместителем обозначает заместитель -O-С1-С6алкокси, где алкил имеет указанное вше значение и содержит до шести атомов углерода. Понятие алкокси включает метокси, этокси, пропокси, 1-метилэтокси, бутокси и 1,1-диметилэтокси. Последний заместитель обычно обозначают как трет-бутокси.
Понятие "С3-С6циклоалкил" индивидуально или в сочетании с другим заместителем в контексте настоящего описания обозначает заместитель -О-С3-С6циклоалкил, содержащий от 3 до 6 атомов углерода.
Понятие "C1-С6алкоксиалкил" в контексте настоящего описания обозначает заместитель C1-С6алкил-О-C1-С6алкил, где алкил имеет указанные выше значения и содержит до шести атомов углерода. Например, метоксиметил обозначает -СН2-О-СН3.
Понятие "С2-С7ацил" индивидуально или в сочетании с другим заместителем обозначает C1-С6алкильную группу, связанную с карбонильной группой, такую как -С(О)-C1-С6алкил.
Понятие "С6- или С10-арил" в контексте настоящего описания, индивидуально или в сочетании с другим заместителем, обозначает либо ароматическую моноциклическую систему, содержащую 6 атомов углерода, либо ароматическую бициклическую систему, содержащую 10 атомов углерода. Например, арил включает фенильную или нафтильную кольцевую систему.
Понятие "С7-С16аралкил" в контексте настоящего описания, индивидуально или в сочетании с другим заместителем, обозначает арил, как он определен выше, связанный с алкильной группой, где алкил имеет указанные выше значения, и содержит от 1 до 6 атомов углерода. Аралкил включает, например, бензил и бутилфенил.
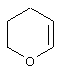
Понятие "Het" в контексте настоящего описания, индивидуально или в сочетании с другим заместителем, обозначает одновалентный заместитель, полученный путем удаления водорода из 5-, 6- или 7-членного насыщенного или ненасыщенного (в том числе ароматического) гетероцикла, содержащего от 1 до 4 гетероатомов, выбранных из ряда, включающего азот, кислород и серу. Примеры пригодных гетероциклов включают тетраги дрофуран, тиофен, диазепин, изоксазол, пиперидин, диоксан, морфолин, пиримидин или
Понятие "Het" также включает гетероцикл как он определен выше, слитый с одним или несколькими другими циклами, которые могут представлять собой гетероциклы или любой другой цикл. Одним из примеров является тиазол[4,5-b]пиридин.
Хотя его обычно обозначают "Het", понятие “гетероарил” в контексте настоящего описания строго обозначает ненасыщенный гетероцикл, двойные связи которого образуют ароматическую систему. Соответствующими примерами гетероароматических систем являются хинолин, индол, пиридин,
Понятие "фармацевтически приемлемый сложный эфир" в контексте настоящего описания индивидуально или в сочетании с другим заместителем, обозначает сложные эфиры соединения формулы I, в которых любая из карбоксильных функций в молекуле, но предпочтительно С-концевая функция, замещена алкоксикарбонильной функцией:
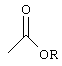
где фрагмент R сложного эфира выбирают из алкила (например, метила, этила, н-пропила, трет-бутила, н-бутила); алкоксиалкила (например, метоксиметила); алкоксиацила (например, ацетоксиметила); аралкила (например, бензила); арилоксиалкила (например, феноксиметила); арила (например, фенила), необязательно замещенного галогеном, C1-С4алкилом или C1-С4алкоксигруппой. Другие пригодные в качестве пролекарства сложные эфиры можно найти в "Design ofprodrugs", Bundgaard H., изд. Elsevier (1985), публикация включена в настоящее описание в качестве ссылки. Такие фармацевтически приемлемые сложные эфиры обычно гидролизуются in vivo при введении млекопитающему и превращаются в кислотную форму соединения формулы I.
Что касается описанных выше сложных эфиров, то, если не указано иное, то любой присутствующий алкильный фрагмент предпочтительно содержит 1-16 атомов углерода, особенно предпочтительно 1-6 атомов углерода. Любой присутствующий в таких сложных эфирах арильный фрагмент предпочтительно представляет собой фенильную группу.
В частности, сложные эфиры могут представлять собой сложный C1-С16алкиловый эфир, незамещенный бензиловый эфир или бензиловый эфир, замещенный по меньшей мере одним атомом галогена, C1-С6алкилом, C1-С6алкокси-, нитрогруппой или трифторметилом.
Понятие "фармацевтически приемлемая соль" в контексте настоящего описания включает соли, полученные из фармацевтически приемлемых оснований. Примеры приемлемых оснований включают холин, этаноламин и этилендиамин. Под объем настоящего изобретения также подпадают соли Na+, K+ и Са++ (см. также "Pharmaceutical salts", Birge S.M. и др., J. Pharm. Sci., (1977), 66, 1-19, публикация включена в настоящее описание в качестве ссылки).
Предпочтительные варианты осуществления
R1:
Предпочтительные варианты осуществления настоящего изобретения включают описанные выше соединения формулы I, в которых радикал R1 выбирают из двух различных диастереоизомеров, где углеродный центр в положении 1 имеет R - конфигурацию, что представлено структурами (i) и (ii):
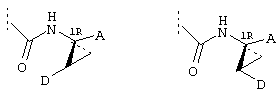
D находится в син-ориентации по отношению к амиду (i), или находится в син-ориентации по отношению к А (ii)
Более предпочтительно линкер D связан с R1 в син -ориентации по отношению к А, как это представлено структурой (ii).
R2:
Предпочтительные варианты осуществления настоящего изобретения включают указанные выше соединения формулы I, где фрагмент R2 обозначает

Предпочтительно R21 обозначает Н, C1-С6алкил, C1-С6алкокси, гидрокси, хлор или N(R23)2, где R23 предпочтительно обозначает Н или C1-С6алкил. Более предпочтительно R21 обозначает Н или C1-С6алкокси. Наиболее предпочтительно R21 обозначает метокси.
Предпочтительно R22 обозначает Н, C1-С6тиоалкил, C1-С6алкокси, фенил или Het, выбранный из группы, включающей:
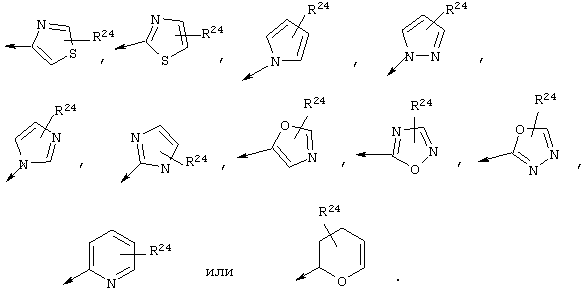
Предпочтительно R24 обозначает Н, C1-С6алкил, NH-R25, NH-C(O)-R25 или NH-C(O)-NH-R25 или NH-C(O)-OR26
Более предпочтительно R2 обозначает C1-С4алкокси, фенил или Het, выбранный из группы, включающей

Более предпочтительно R24 обозначает Н, C1-С6алкил, NH-R25, NH-C(O)-R25; или NH-C(O)-OR26.
Наиболее предпочтительно R22 обозначает этокси, или Het выбирают из группы, включающей:
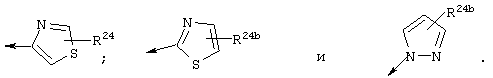
Наиболее предпочтительно R24a обозначает NH-R25, NH-C(O)-R25, или NH-C(O)-OR26. Наиболее предпочтительно R24b обозначает Н или C1-С6алкил.
Предпочтительно каждый R25 независимо друг от друга обозначает Н, C1-С6алкил или C3-С6циклоалкил. Более предпочтительно R25 обозначает C1-С6алкил или C3-С6циклоалкил.
Более предпочтительно R25 обозначает C1-С6алкил. Предпочтительно R26 обозначает C1-С6алкил.
R3:
Предпочтительные варианты осуществления настоящего изобретения включают описанные выше соединения формулы I, где фрагмент R3 предпочтительно обозначает амид формулы NH-C(О)-R32, мочевину формулы NH-C(О)-NH-R32 или карбамат формулы NH-C(О)-OR32. Более предпочтительно R3 обозначает карбамат или мочевину. Наиболее предпочтительно R3 обозначает карбамат.
Предпочтительно R32 обозначает C1-С6алкил или C3-С6циклоалкил. Более предпочтительно R32 обозначает C1-С6алкил или C4-С6циклоалкил. Наиболее предпочтительно R32 обозначает трет-бутил, циклобутил или циклопентил.
D:
Предпочтительные варианты осуществления настоящего изобретения включают соединения формулы I, где линкер D представляет собой насыщенную или ненасыщенную алкиленовую цепь, состоящую из 6 - 8 атомов. Более предпочтительно линкер D представляет собой цепь, состоящую из 7 атомов.
Предпочтительно цепь D содержит один или два гетероатома, выбранные из группы, включающей О, S, NH, N-C1-С6алкил или Т-С2-С7ацил. Более предпочтительно цепь D необязательно содержит один гетероатом, выбранный из группы, включающей NH или Т-C2-С7ацил, наиболее предпочтительно N(Ac), и он находится на атоме в положении 10 цепи. Наиболее предпочтительно цепь, содержащая атом азота, является насыщенной.
В альтернативном варианте D содержит один гетероатом, выбранный из О или S. Предпочтительно, если цепь D содержит 7 атомов, атом О или S находится в положении 9 цепи. Предпочтительно эта цепь имеет заместитель R4, где R4 обозначает Н или C1-С6алкил. Более предпочтительно R4 обозначает Н или метил. Наиболее предпочтительно R4 обозначает Н или 8-(S)-Ме. Еще более предпочтительно цепь D является насыщенной. В альтернативном варианте D содержит одну двойную связь между положениями 11 и 12. Предпочтительно эта двойная связь находится в транс-ориентации.
В альтернативном варианте D представляет собой насыщенную или ненасыщенную алкиленовую цепь, содержащую только атомы углерода. В этом случае цепь D предпочтительно является насыщенной и состоит из 7 атомов. Более предпочтительно цепь D имеет заместитель R4, где R4 обозначает Н, оксо, тио, гидрокси, тиоалкил, алкокси или алкил. Более предпочтительно R4 обозначает Н или C1-С6алкил. Наиболее предпочтительно R4 обозначает Н или метил. Наиболее предпочтительно R4 обозначает Н или 10-(S)-Ме.
В альтернативном варианте D представляет собой алкиленовую цепь, состоящую только из атомов углерода, которая предпочтительно содержит одну двойную связь и включает 7 атомов. Более предпочтительно указанная двойная связь находится между положениями 13 и 14 цепи. Наиболее предпочтительно эта двойная связь находится в цис-ориентации. Предпочтительно эта цепь D замещена R4, где R4 обозначает Н, оксо, гидрокси, алкокси или алкил. Более предпочтительно R4 обозначает Н или C1-С6алкил. Еще более предпочтительно R4 обозначает Н или метил. Наиболее предпочтительно R4 обозначает Н или 10-(S)-Ме.
А:
Предпочтительные варианты осуществления настоящего изобретения включают описанные выше соединения формулы I, где А обозначает карбоновую кислоту.
Конкретные варианты осуществления:
Предпочтительные варианты осуществления настоящего изобретения включают описанные выше соединения формулы I, где R2 обозначает заместитель, представляющий собой хинолин (т.е. W обозначает N);
R3 обозначает группу формулы -NH-C(O)-NHR32 или -NH-C(O)-OR32, где R32 обозначает C1-С4алкил или C4-С6циклолкил;
D обозначает состоящую из 6-8 атомов насыщенную или ненасыщенную алкиленовую цепь, присоединенную к R1 в син-ориентации по отношению к А, необязательно включающую один или два гетероатома, независимо друг от друга выбранных из О, S или N-R41, где R41 обозначает C2-С7ацил;
R4 обозначает Н или один-три заместителя независимо друг от друга выбранных из гидрокси или C1-С6алкила; и
А обозначает карбоновую кислоту, или их фармацевтически приемлемые соли или сложные эфиры.
Более предпочтительными являются соединения формулы I, где R’ имеет указанные выше значения; R21 обозначает Н или метокси;
R22 обозначает C1-С6алкокси, или Het выбирают из группы, включающей:
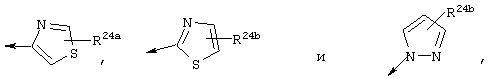
где R24a обозначает Н, C1-С6алкил, NH-R25, NH-C(О)-R25, NH-C(О)-NH-R25,
где R25 обозначает: Н, C1-С6алкил или C3-С6циклоалкил;
или R24а обозначает NH-C(О)-OR26, где R26 обозначает C1-С6алкил или C3-С6циклоалкил;
и R246 обозначает Н или C1-С6алкил;
R3 обозначает мочевину формулы NH-C(О)-NHR32 или карбамат формулы NH-C(O)-
OR32, где R32 обозначает C1-С6алкил или C3-С6циклоалкил;
D обозначает содержащую 7 атомов углерода насыщенную или ненасыщенную алкиленовую цепь, необязательно включающую одну двойную связь между положениями 11, 12 или 13, 14.
При этом цепь D необязательно включает один гетероатом независимо выбранный из О, S, NH, N(Me) или N(Ac); и R4 обозначает Н или C1-С6алкил.
Наиболее предпочтительными являются соединения формулы I, где R21 обозначает метокси и R22 обозначает этокси или:
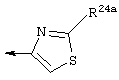
где R24а обозначает NH-(C1-С4алкил), NН-С(О)-(C1-С4алкил); или NH-C(O)-O-(C1-С4алкил); и
D представляет собой насыщенную цепь или содержит одну двойную связь, находящуюся в цис-ориентации между положениями 13, 14.
Наконец, под объем настоящего изобретения подпадают все соединения формулы I, представленные в таблицах 1-9.
Фармацевтические композиции по настоящему изобретению могут вводиться перорально, парентерально или с помощью устройства для имплантации. Предпочтительным является пероральное введение или введение с помощью инъекции. Фармацевтические композиции по настоящему изобретению могут содержать любые общепринятые нетоксичные фармацевтически приемлемые носители, адъюванты или наполнители. В некоторых случаях значение рН композиции может регулироваться с помощью фармацевтически приемлемых кислот, оснований или буферов с целью повышения стабильности соединения, включенного в композицию или в форму для его введения. В контексте настоящего описания понятие "парентеральный" включает подкожный, внутрикожный, внутривенный, внутримышечный, внутрисуставной, интрасиновиальный, интрастернальный, интратекальный путь введения с помощью инъекции или инфузии, а также введение в область повреждения.
Фармацевтические композиции могут иметь форму стерильного препарата для инъекции, например, стерильной инъецируемой водной или жирорастворимой суспензии. Эта суспензия может быть получена с помощью хорошо известных методов с использованием диспергирующих или смачивающих агентов (таких как, например, Твин 80) и суспендирующих агентов.
Фармацевтические композиции по настоящему изобретению могут вводиться перорально в виде любой приемлемой дозируемой формы для перорального введения, включая (но не ограничиваясь ими) капсулы, таблетки и водные суспензии и растворы. В случае таблеток для перорального введения обычно применяемые носители включают лактозу и кукурузный крахмал. Как правило, также добавляют замасливатели, такие как стеарат магния. Для перорального введения в форме капсул приемлемые разбавители включают лактозу и безводный кукурузный крахмал. Когда пероральным путем вводят водные суспензии, действующее вещество объединяют с эмульгирующими и суспендирующими агентами. При необходимости могут быть добавлены определенные подслащивающие вещества и/или корригенты, и/или красители.
Другие пригодные наполнители или носители для указанных выше препаративных форм и композиций можно обнаружить в обычных фармацевтических справочниках, например, в "Remington’s Pharmaceutical Sciences", The Science and Practice of Pharmacy, 19-е изд. Mack Publishing Company, Easton, Penn., (1995).
Дозы в диапазоне от примерно 0,01 до примерно 100 мг/кг веса тела в день, предпочтительно от примерно 0,5 до примерно 75 мг/кг веса тела в день соединения по изобретению, являющегося ингибитором протеазы, могут применяться для монотерапии с целью предупреждения и лечения опосредованной HCV болезни. Как правило, фармацевтические композиции по изобретению могут вводиться от примерно 1 до примерно 5 раз в день или в другом варианте путем непрерывной инфузии. Такое введение может применяться как для экстренного, так и для длительного лечения. Количество действующего вещества, которое может быть объединено с носителями для получения стандартной дозируемой формы, должно варьироваться в зависимости от хозяина, подлежащего лечению, и конкретного пути введения. Обычная композиция может содержать от примерно 5% до примерно 95% действующего вещества (мас.%). Предпочтительно такие композиции содержат от примерно 20% до примерно 80% действующего вещества.
Как должно быть очевидно специалисту в данной области, могут применяться и более низкие или более высокие дозы, чем указанные выше. Специфические схемы дозирования и лечения для любого конкретного пациента могут зависеть от различных факторов, включая активность конкретного применяемого соединения, возраст, вес тела, общее состояние здоровья, пол, рацион, время введения, скорость выведения, сочетание лекарственных средств, серьезность и особенности развития болезни, предрасположенность пациента к инфекции, и они определяются лечащим врачом. Как правило, лечение начинают с небольших доз, существенно более низких, чем оптимальная доза пептида. Затем дозу повышают небольшими порциями до достижения оптимального действия в конкретных условиях. Обычно наиболее предпочтительно вводить соединение в таком диапазоне концентраций, которые обеспечивают антивирусную активность, но не обладают какими-либо вредными или опасными побочными действиями.
Когда композиции по настоящему изобретению включают комбинацию соединения формулы I и одного или нескольких дополнительных терапевтических или профилактических агентов, то соединение и дополнительный агент должны присутствовать в диапазоне доз от примерно 10% до 100% и более предпочтительно от примерно 10 до 80% от дозы, обычно применяемой в режиме монотерапии.
Когда эти соединения или их фармацевтически приемлемые соли объединяют в препаративной форме с фармацевтически приемлемым носителем, то полученная композиция может вводиться in vivo млекопитающему, такому как человек, для ингибирования NS3-протеазы HCV или для лечения или предупреждения инфекции, вызываемой вирусом HCV. Такое лечение также может быть достигнуто при использовании соединения по изобретению в сочетании с агентами, включающими иммуномодуляторы, такие как α-, β- или γ-интерфероны; другие антивирусные агенты, такие как рибавирин, амантадин; другие ингибиторы NS3-протеазы HCV; ингибиторы других мишеней в жизненном цикле HCV, которые включают (но не ограничиваясь ими) геликазу, полимеразу, металлопротеазу или внутренний сайт входа в рибосому (IRES); или их комбинацию. Дополнительные агенты могут быть объединены с соединениями по изобретению для создания однократной дозируемой формы. В альтернативном варианте эти дополнительные агенты могут вводиться млекопитающему по отдельности в виде части многократной дозируемой формы.
Согласно другому варианту осуществления настоящее изобретение относится к способам ингибирования активности NS3-протеазы HCV у млекопитающих путем введения соединения формулы I, в котором заместители имеют указанные выше значения.
Согласно предпочтительному варианту осуществления эти способы могут применяться для понижения активности NS3-протеазы HCV у млекопитающего. Если фармацевтическая композиция включает в качестве действующего вещества только соединение по изобретению, то такие способы могут дополнительно включать стадию введения млекопитающему агента, выбранного из иммуномодулятора, антивирусного агента, ингибитора HCV-протеазы или ингибитора других мишеней в жизненном цикле HCV, таких как геликаза, полимераза или металлопротеаза. Такой дополнительный агент может вводиться млекопитающему до, одновременно или после введения композиций по изобретению.
Согласно еще одному предпочтительному варианту осуществления эти способы могут применяться для ингибирования репликации вируса в организме млекопитающего. Такие способы могут применяться для лечения или предупреждения вызываемой HCV болезни. Если фармацевтическая композиция включает в качестве действующего вещества только соединение по изобретению, то такие способы могут дополнительно включать стадию введения млекопитающему агента, выбранного из иммуномодулятора, антивирусного агента, ингибитора HCV-протеазы или ингибитора других мишеней в жизненном цикле HCV. Такой дополнительный агент может вводиться млекопитающему до, одновременно или после введения композиции по изобретению.
Предложенные в настоящем изобретении композиции также могут применяться в качестве лабораторных реагентов. Заявителями впервые предложены низкомолекулярные соединения, которые обладают высокой активностью и специфичностью в отношении NS3-протеазы HCV. Некоторые из соединений по настоящему изобретению могут представлять собой инструмент для разработки систем анализа репликации вируса, создания систем анализа в организме животных и изучения зависимости между строением и биологической активностью с целью расширения понимания механизмов, связанных с HCV заболеваний.
Соединения по настоящему изобретению также могут применяться для искоренения или предупреждения вирусного загрязнения материалов, и тем самым они понижают риск заражения вирусом персонала лабораторий или медицинских учреждений, которые имеют контакт с такими материалами (например, кровью, тканями, хирургическими инструментами и предметами одежды, лабораторными инструментами и предметами одежды, приборами и материалами для сбора крови).
Методика
Некоторые методы синтеза ациклических промежуточных продуктов для соединений формулы I описаны в WO 00/09543 и WO 00/09558.
Соединения по настоящему изобретению синтезируют согласно общему процессу, проиллюстрированному на схемах I, II и III (где PG обозначает соответствующие защитные группы). [На всех представленных ниже схемах D’ имеет одно и то же значение, что и D, но короче на 2-5 атомов].
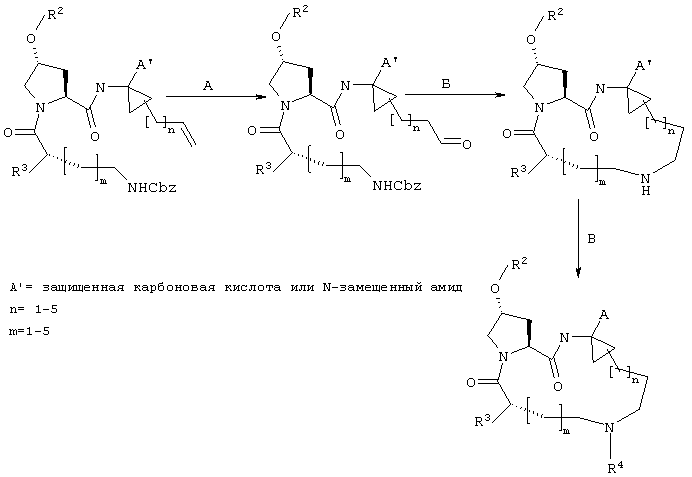
Когда рассматриваются соединения формулы I, где А обозначает N-замещенный амид, то винил-АЦКК или гомоаллил АЦКК (R1) подвергают сочетанию с соответствующим амином перед сочетанием с Р2. Методы такого сочетания хорошо известны специалистам в данной области. Как известно специалистам в данной области, такой амид (А) является незащищенным, но может иметь любой из указанных выше пригодных заместителей R5.
Реакцию замыкания кольца (макроциклизацию) осуществляют либо путем олефинового метатезиса (Схема I), либо, когда линкер содержит атом азота, путем восстановительного аминирования (Схема II), либо путем образования пептидной связи согласно Схеме III.
Эти процессы подробно описаны ниже:
А. Макроциклизация путем олефинового метатезиса
Схема I
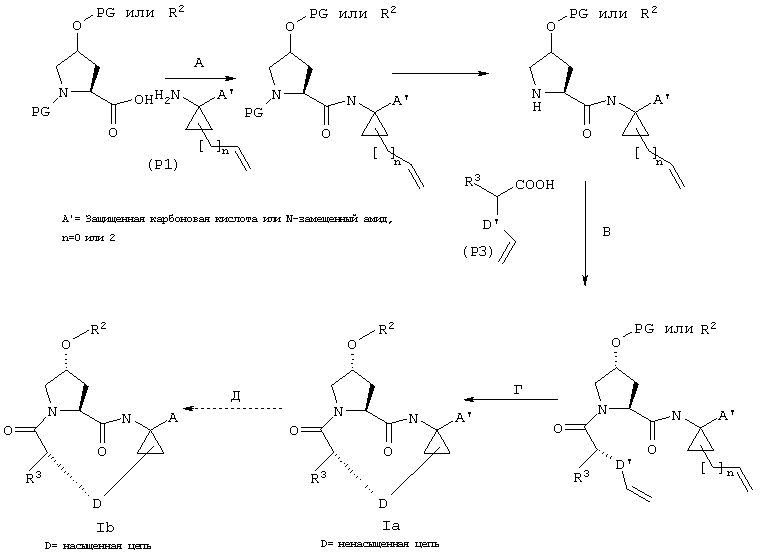
Схема I:
Существует несколько путей осуществления последовательности реакций сочетания, которые хорошо известны специалистам в данной области. Используя в качестве исходного продукта 4-(S)-гидроксипролин, заместитель в 4-гидроксигруппу может быть введен с помощью реакции Мицунобу (согласно методу, описанному у Mitsunobu,
Cunthesis 1981, January, 1-28; Rano и др., Tet. Lett. 1994, 36, 3779-3792; Krchnak и др., Tel. Lett. 1995, 366, 6193-6196) перед осуществлением макроциклизации или после нее. В альтернативном варианте сборка может быть осуществлена с использованием соответствующего 4-(R)-гидроксизамещенного пролина согласно общим процессам, описанным в WO 00/09543 и WO 00/09558 (ниже представлены конкретные примеры этих фрагментов).
Стадии А, Б, В: В целом фрагменты P1, P2 и Р3 могут быть связаны с помощью хорошо известных методов пептидного сочетания и они в целом описаны в WO 00/09543 и WO 00/09558.
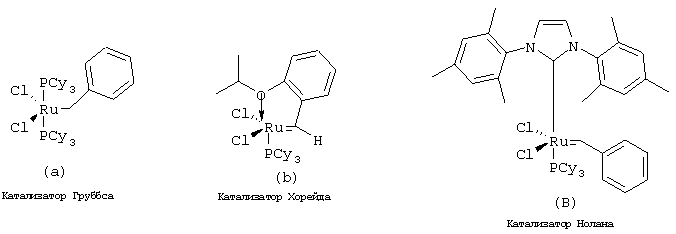
Стадия Г: Получение макроцикла может быть осуществлено путем олефинового метатезиса с использованием содержащего Ru катализатора, как это описано у Miller, S.J.; Blackwell, H.E.; Grubbs, R.H. J. Am. Chem. Soc. 1996, 118, 9606-9614 (a); Kingsbury, J.S.; Harrity, J.P.A.; Bonitatebus, P.J.; Hoveyda, A.H. J. Am. Chem. Soc. 1999, 121, 791-799 (б) и у Huang, J.; Stevens, E.D.; Nolan, S.P.; Petersen, J.L.; J. Am. Chem. Soc. 1999, 121, 2674-2678 (в). Следует также отметить, что для этой реакции могут быть использованы катализаторы, содержащие другие переходные металлы, такие как Мо.
Стадия Е: Двойная связь необязательно может быть восстановлена с помощью стандартных методов гидрирования, хорошо известных в данной области. Если А’ представляет собой защищенную карбоновую кислоту, то также соответствующим образом удаляют защитные группы.
Б. Макроциклизацня путем восстановительного аминирования (для линкеров, содержащих N)
Если линкер содержит атом азота, то макроциклизация может быть осуществлена путем восстановительного аминирования как проиллюстрировано на Схеме II с получением ингибиторов, имеющих общую структуру II.
Схема II
Стадия А: Гидроборирование двойной связи согласно методу Брауна (Н.С. Brown и B.C. Subba Rao, J. Am. Che. Soc. 1959, 81,6434-6437) с последующим окислением образовавшегося спирта (например, с помощью периодината Десс-Мартина, J. Am. Chem. Soc. 1991, 113, 7277-7287) приводит к получению соответстувующего альдегида.
Стадия Б: Гидрирование в присутствии кислоты приводит к удалению аминозащитной группы, после чего проводят макроциклизацию путем восстановительного аминирования.
Фрагмент Р3, используемый в этом синтезе, может быть легко получен из различных аминокислот, таких как лизин, орнитин, глутамин (с помощью реакции Гофмана: Веr. 1881, 14, 2725) и другие; модификации этого метода синтеза хорошо известны в данной области.
Стадия В: Вторичный амин в линкере D (полученный на стадии Г) необязательно алкилируют с помощью алкилгалогенида или ацетилируют с помощью алкил- или арилхлорангидрида с использованием методик, хорошо известных в данной области, с получением ингибиторов, имеющих общую структуру II. Если А’ представляет собой защищенную карбоновую кислоту, то соответствующим образом также удаляют защитные группы.
В. Макроциклизация путем образования лактама
С другой стороны, следует понимать, что указанные макроциклические соединения, имеющие общие структуры I и II могут быть синтезированы другими методами.
Например, Р1 и Р3 могут быть сначала соединены с линкером D, затем подвергнуты сочетанию с Р2 и реакция макроциклизации может привести к образованию лактама, которое может быть осуществлено двумя возможными путями, как это известно специалистам в данной области, что проиллюстрировано на схеме III.
Схема III
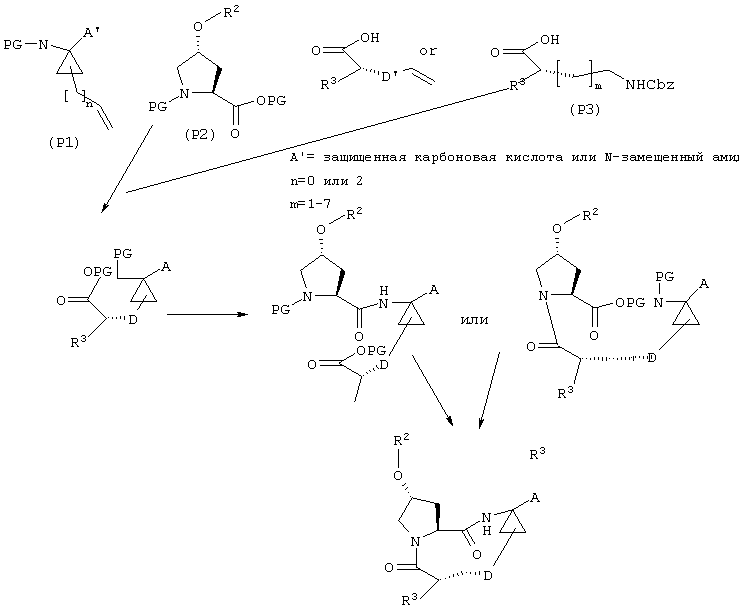 Синтез Р1
Синтез Р1
Для синтеза ингибиторов, имеющих общие структуры I и II, требуются одинаковые фрагменты Р1:
а) винил-АЦКК, синтез и выделение которого описаны в WO 00/09543 и WO 00/09558, или
б) гомоаллил-АЦКК (пример 1, соединение 1f).
Синтез Р2
Некоторые из фрагментов Р2, используемые при синтезе соединений формулы I, описаны в WO 00/09543 и WO 00/09558.
Другие фрагменты Р2 синтезируют следующим образом:
Синтез 2-"Het"-4-гидрокси-7-метоксихинолинового производного
(i) Подход, основанный на использовании в качестве исходного продукта соответствующей "Неt"-карбоновой кислоты IVb
Схема IV
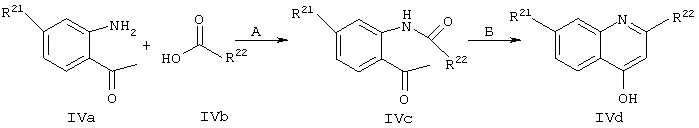
Синтез осуществляют согласно модифицированному методу, описанному у Li и др. J. Med. Chem. 1994, 34, 3400-3407. Промежуточный продукт IVa, где R21 обозначает ОМе (пример 7, соединение 7b) получают согласно методу, описанному у Brown и др., J. Med. Chem. 1989, 32, 807-826.
Стадия А: Промежуточный продукт IVa подвергают сочетанию с гетероциклическими карбоновыми кислотами IVb в основных условиях в присутствии РОСl3 для активации карбоксилатной группы. Для получения ингибиторов используют различные карбоновые кислоты, имеющие общую структуру IVb, которые либо имеются в продаже, либо могут быть синтезированы как показано на схемах V, VI и VII, либо могут быть синтезированы индивидуально с помощью методов, описанных в конкретных примерах.
Стадия Б: Замыкание кольца с последующей дегидратацией осуществляют в основных условиях, получая хинолины, имеющие общую структуру IVd.
(i.a). Синтез "Het-карбоновых кислот общей формулы IVb
Синтез 2-(замещенный)амино-4-карбоксиаминотиазоловых производных (Vс)
Применяют модифицированный процесс, описанный у Berdikhina и др.. Chem. Heterocycl, Compd. (в переводе на английский язык) 1991, 4,427-433.
Схема V
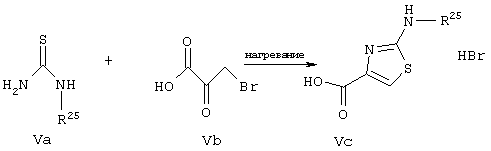
Согласно общей методологии синтеза, представленной на схеме V, с использованием тиомочевин (Va), имеющих различные алкильные заместители (R25 обозначает алкильную группу), и 3-бромпировиноградной кислоты получают различные 2-алкиламинотиазолил-4-карбоновые кислоты, представляющие собой соединения,
имеющие общую структуру Vc. Этот тип реакции конденсации широко известен в данной области.
В альтернативном варианте фрагмент Р2, включающий производные 2-аминозамещенного тиазола, синтезируют из соответствующего 2-карбоксильного производного, как показано на схеме VI, согласно методу, описанному у Unangst, P.С.; Connor, D.T. J. Heterocyc. Chem. 29, 5,1992, 1097-1100.
Схема VI
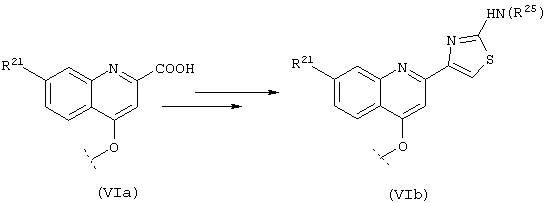
Примеры такого процесса описаны в WO 00/09543 и WO 00/09558.
Синтез 2-карбокси-4-замещенного аминотиазольных производных формулы VIId Согласно общей методологии синтеза, представленной на схеме VII, получают различные 4-алкилтиазолил-2-карбоновые кислоты, представляющие собой соединения, имеющие общую структуру VIId.
Схема VII
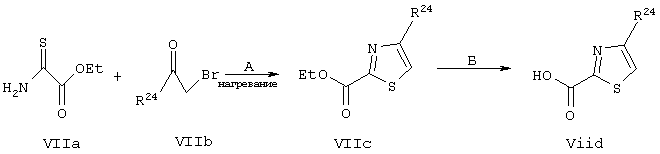
Используют процесс, описанный у Janusz и др., J. Med. Chem. 1998, 41, 3515-3529 со следующими модификациями: этилтиооксамат (Vila) подвергают взаимодействию с различными β-бромкетонами, имеющими общую структуру VIIb (R24 обозначает алкильную группу), получая тиазолилкарбоновые кислоты, имеющие общую структуру VIId. Такой тип реакции конденсации широко известен в данной области техники. Синтез 2-карбокси(замещенный)имидазольных производных (VIIIb). Согласно общей методологии синтеза, представленной на схеме VIII, получают различные алкилимидазолил-2-карбоновые кислоты, представляющие собой соединения, имеющие общую структуру VIIIb.
Схема VIII
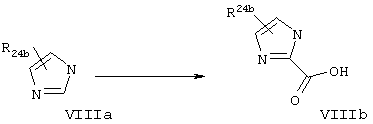
Используют процесс, описанный у Baird и др., J. Amer. Chem. Soc. 1996, 118, 6141-6146: алкилимидазол депротонируют с помощью сильного основания (например, н-BuLi) и затем подвергают взаимодействию с СО2, получая карбоновую кислоту формулы VIIIb. Такой тип реакции конденсации широко известен в данной области.
б. Синтез 4-гидрокси-7-метокси-2-(имидазолил или пиразолил)хинолинов
4-Гидpoкcи-7-R21-xинoлины, имеющие в положении С2 имидазолильный или хи-нолильный фрагмент, получают в целом согласно методологии, представленной на схеме IX.
Схема IX
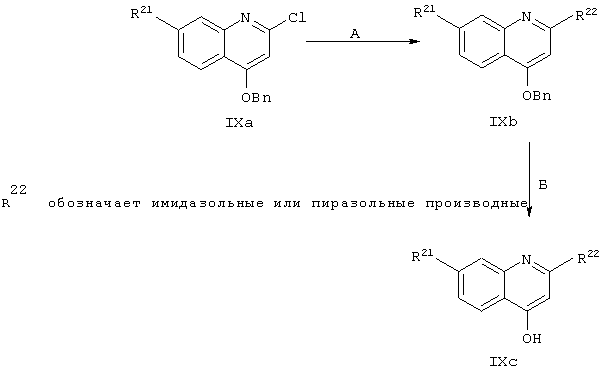
Синтез основного промежуточного продукта, (где R21 обозначает ОМе) 4-бензилокси-2-хлор-7-метоксихинолина формулы IXa подробно описан в примере 6 (соединение 6е).
Стадия А: При высоких температурах для замещения 2-хлор-фрагмента в соединении IXa могут использоваться различные имидазолы, алкилзамещенные имидазолы, пиразолы или алкилзамещенные пиразолы, в результате чего получают соединения, имеющие общую структуру IXb.
Стадия Б: После удаления бензильной защитной группы из 4-гидрокси-фрагмента хинолина с помощью стандартных методов гидрирования получают хинолиновые производные, имеющие общую структуру IХс.
Синтез Р3
Для макроциклизации путем олефинового метатезиса синтезируют различные фрагменты Р3, содержащие соответствующее D-линкерное удлинение. В целом содержащие концевой олефин фрагменты Р3 синтезируют для метатезиса согласно приведенным ниже общим схемам (Схемы X, XI и XII).
Синтез линкеров из класса А
Данный общий метод синтеза используют для получения линкеров, содержащих только атомы углерода (не имеющие гетероатомов) (Схема X).
Схема Х
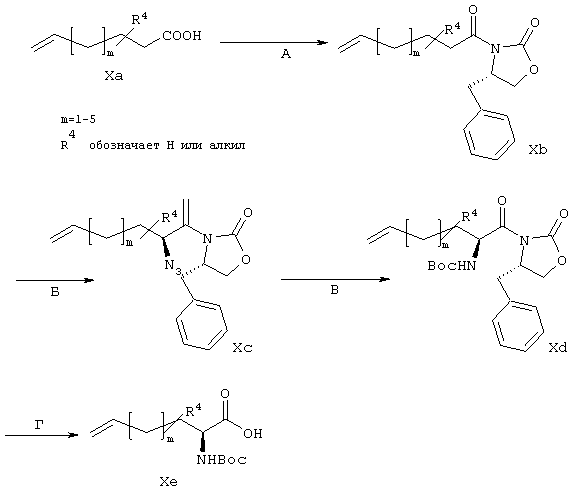
Синтез осуществляют согласно методу, описанному у Evans и др., J. Am. Chem. Soc. 1990, 112,4011-4030.
Исходные карбоновые кислоты (Ха) имеются в продаже или могут быть получены согласно описанным в литературе методам, известным специалистам в данной области.
Стадия А: Карбоновую кислоту Ха активируют с помощью пивалоилхлорида и затем подвергают взаимодействию с анионом хирального вспомогательного 4(S)-4-(фенилметил)-2-оксазолидинона Эванса согласно широко известному методу (см. обзор: D.J.Ager и др., Aldrichimica Acta 1997, 30, 3-11 и приведенные в нем ссылки), получая соединения, имеющие общую структуру Хb.
Стадия Б: Стереоселективное α-азидирование хирального имиденолата, такого, который может быть получен из соединений, имеющих общую структуру Хb, в присутствии основания, такого как KHMDS, с помощью тризилазида также широко известно в данной области (см. обзор: D.J. Ager и др., Aldrichimica Acta 1997, 30, 3-11 и приведенные в нем ссылки).
Стадия В: После восстановления α-азида, катализируемого с помощью SnCl2, осуществляют защиту образовавшегося амина в виде его трет-бутилкарбамата, получая промежуточные продукты, имеющие общую структуру Хс. Такие реакции широко известны в данной области.
Стадия Г: В завершение хиральный вспомогательный продукт гидролизуют в основных условиях, например, в смеси H2O2 с LiOH, получая линкеры аминокислотного типа, имеющие общую структуру Хе.
В альтернативном варианте фрагменты Р3, имеющие общую структуру Хе, синтезируют согласно методу, описанному у M.J. Burk и др., J. Am. Chem. Soc 1998, 120, 657-663, проиллюстрированному на схеме XI. Эти соединения различаются количеством метиленовых звеньев (-СН2-) в линкере (m=1-5) и замещением алкильных групп в R4, но они не содержат гетероатомов.
Схема XI
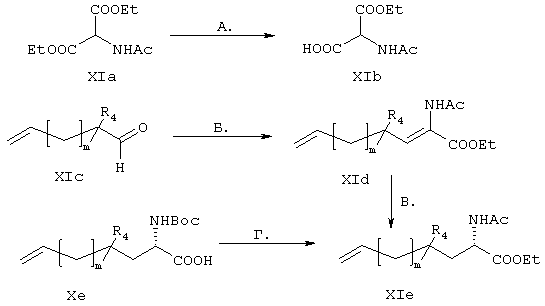
Стадия А: Производное одноосновной кислоты формулы XIb получают из имеющегося в продаже диэтил-2-ацетамидомалоната путем стандартного гидролиза сложного эфира в основных условиях.
Стадия Б: Реакция конденсации типа Кневенагеля между альдегидом, имеющим общую структуру XIc и соединением формулы XIb в присутствии основания, такого как пиридин, и уксусного ангидрида приводит к образованию енамида формулы XId, являющегося промежуточным продуктом, характеризующегося Z-стереохимической конфигурацией относительно вновь образовавшейся двойной связи, как показано на схеме.
Стадия В: Региоселективное и энантиоселективное каталитическое гидрирование промежуточного продукта, представляющего собой енамид формулы XId с получением промежуточного продукта, представляющего собой аминокислоту формулы Xie, осуществляют согласно методу Бурка.
Стадия Г: Затем азот ацетамидового производного формулы XIe дважды защищают путем присоединения заместителя, представляющего собой трет-бутилкарбамат, перед ацетатной группой, а также в виде этилового эфира, продукт гидролизуют в стандартных основных условиях, получая фрагменты Р3, имеющие общую структуру XIf.
Синтез линкеров из класса Б
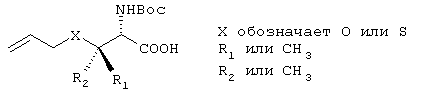
Эту общую схему синтеза применяют для получения линкеров, содержащих кислород или серу.
Схема XII
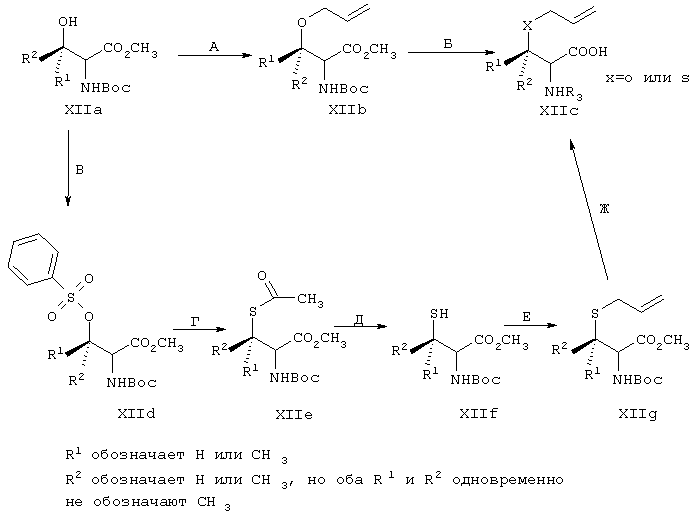
Стадия А: Соответствующим образом защищенные аминокислоты, такие как метиловый эфир Вос-(L)-серина, метиловый эфир Вос-(L)-треонина или метиловый эфир Вос-(L)-аллотреонина, алкилируют с помощью алкилйодида в присутствии Ag2O, получая метиловый эфир формулы XIIb.
Стадия Б: С помощью гидролиза сложного метилового эфира в стандартных основных условиях получают линкеры в виде простого эфира, имеющие общую структуру XIIc (X обозначает О).
Стадия В: Аналог, содержащий серу, получают из той же самой исходной аминокислоты формулы ХIIа (защищенной соответствующим образом, как указано выше) и ее гидроксильную группу превращают в легко удаляемую группу (такую как промежуточный продукт, представляющий собой тозилат формулы XIId), используя стандартные методы, широко известные в данной области.
Стадия Г: После этого тозильный фрагмент замещают анионом тиоацетата, получая путем инверсии хирального центра на β-атоме углерода промежуточный продукт, представляющий собой сложный тиоэфир ХIIе.
Стадия Д: Путем гидролиза тиоэфирного фрагмента в слабых основных условиях получают свободный тиол формулы XIIf.
Стадия Е: Алкилирование тиольного фрагмента легко может быть осуществлено в основных условиях с помощью аллилйодида.
Стадия Ж: В завершение путем гидролиза сложного метилового эфира с помощью стандартных методов получают сульфидный аналог формулы ХIIс (X обозначает S).
Синтез фрагмента R3:
Примеры синтеза фрагментов, где R3 обозначает NH-R31, описаны в WO 00/09543.
ПРИМЕРЫ
Ниже изобретение проиллюстрировано на примерах, не ограничивающих его объем. Другие конкретные методы синтеза или разделения описаны, например, в WO 00/09543 и WO 00/09558.
Температуры даны в градусах Цельсия. Проценты для растворов представляют собой % мас./об., а соотношения в растворах представляют собой соотношения объемов, если не указано иное. Спектры ядерного магнитного резонанса (ЯМР) регистрировали с помощью спектрометра фирмы Brucker при частоте 400 МГц, химические сдвиги (S) выражены в част./млн и отнесены к внутреннему дейтериевому растворителю, если не указано иное. ЯМР-спектры всех конечных соединений (ингибиторов) регистрировали в растворе их солей с трифторуксусной кислотой (ТФК) в ДМСО-d6, если не указано иное. Экспресс-хроматографию на колонке проводили на силикагеле (SiO2) согласно методу Стилла (W.C.Still и др., J. Org. Chem., (1978), 43, 2923). В примерах используются следующие сокращения: Вn: бензил; Воc: трет-бутилоксикарбонил {Ме3СОС(О)}; БСА: бычий сывороточный альбумин; КБЗ: бензилоксикарбонил; ХАПС: 3-[(3-холамидопропил)диметиламмоний]-1-пропансульфонат; ДБУ: 1,8-диазабицикло[5.4.0]ундец-7-ен; СН2Сl2=ДХМ: метиленхлорид; ДЭАД: диэтилазодикарбоксилат; ДИАД: диизопропилазодикарбоксилат; ДИЭА: диизопропилэтиламин; ДИПЭА: диизопропилэтиламин; ДМАП: диметиламинопиридин; ДЦК: 1,3-дициклогексилкарбодиимид; ДМЭ: 1,2-димстилоксиэтан; ДМФ: диметилформамид; ДМСО: диметилсульфоксид; ДТТ: дитиотреитол или трео-1,4-димеркапто-2,3-бутандиол; ДФФА: дифенилфосфорилазид; ЭДТК: этилендиаминтетрауксусная кислота; Et: этил; EtOH: этанол; EtOAc: этилацетат; Et2O: диэтиловый эфир; ЭСМС: масс-спектрометрия с ионизацией электронным пучком; ГАТУ: [гексафторфосфат O-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония]; ЖХВР: жидкостная хроматография высокого разрешения; МС: масс-спектрометрия; MALDI-TOF: времяпролетная масс-спектрометрия с матричной лазерной десорбцией образца; FAB: бомбардировка быстрыми атомами; ЛАГ: алюмогидрид лития; Me: метил; МеОН: метанол; МЭС: (2-{N-морфолино}этансульфоновая кислота); NaHMDS: бис(триметилсилил)амид натрия; NMM: N-метилморфолин; NMMO: оксид N-метилморфолина; NМП: N-метилпирролидин; Рr: пропил; Succ: 3-карбоксипропаноил; pNA: 4-нитрофениламино или пара-нитроанилид; ТБАФ: фторид тетра-н-бутиламмония; ТБТУ: тетрафторборат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония; ТКЭФ: гидрохлорид трис(2-карбоксиэтил)фосфина; ТФК: трифторуксусная кислота; ТГФ: тетрагидрофуран; ТИС: триизопропилсилан; ТСХ: тонкослойная хроматография; ТМСЭ: триметилсилилэтил; Трис/HCl: гидрохлорид трис(гидроксиметил)аминометана.
ФРАГМЕНТЫ Р1
Пример 1
Синтез трет-бутил-(1R,2R)/(1S,2S)-1-амино-2-гомоаллилциклопропилкарбоксилата (1f):
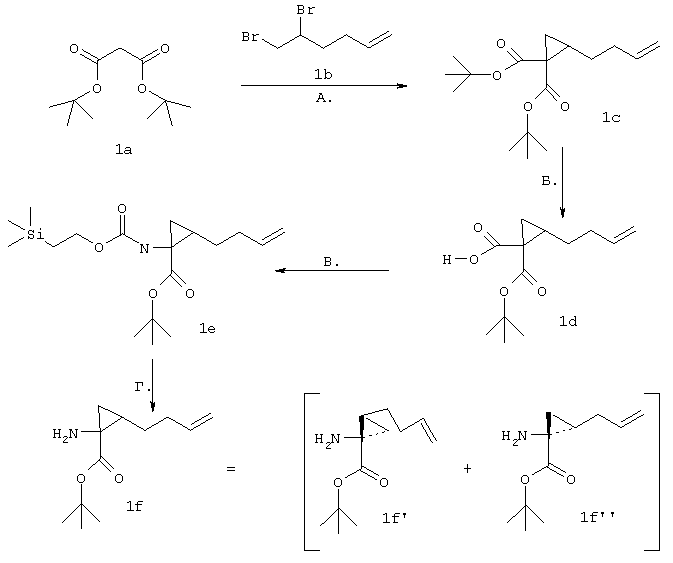
А. К суспензии хлорида бензилтриэтиламмоний (5,08 г, 22,3 ммоля) в 50%-ном водном растворе NaOH (50 мл) последовательно добавляли 1,2-дибром-5-гексен (1b, 8,10 г, 33,46 ммоля) и ди-трет-бутилмалонат (1а, 4,82 г, 22,30 ммоля). Смесь интенсивно перемешивали при КТ в течение 16 ч, затем разбавляли Н2О и экстрагировали CH2Cl2 (3×50 мл). Затем органический слой промывали Н2О (2×50 мл), соляным раствором/Н2О (2/1,2×50 мл), сушили над мгSO4 и упаривали. Неочищенный остаток очищали с помощью экспресс-хроматографии на колонках на силикагеле, используя в качестве элюента градиент от 3 до 5% ЕtOАс в гексане, получая соединение 1с с выходом 38% (2,48 г).
1Н ЯМР (СDСl3, 400 МГц): δ 1,19 (bd, J=7,9 Гц, 2Н), 1,25-1,33 (m, 1H), 1,46 (s, 9H), 1,48 (s, 9H), 1,47-1,60 (m, 1H), 1,75-1,82 (m, 1H), 2,14-2,22 (m, 2H), 4,93-5,50 (m, 2Н), 4,96 (dm, J=10,2 Гц, 1H), 5,18 (dm, J=17,2 Гц, 1H). ES(+) MC m/z 297 (М+Н)+
Б. К суспензии трет-бутоксида калия (5,75 г, 51,25 ммоля) в безводном диэтиловом эфире (150 мл) при 0°, добавляли Н2О (203 мкл, 11,27 ммоля) и реакционную смесь перемешивали при 0° в течение 10 мин. Добавляли раствор соединения 1с в диэтиловом эфире (2,48 г в 10 мл диэтилового эфира, 10,25 ммоля) и смесь перемешивали при КТ в течение 5 ч. Смесь разбавляли охлажденной на льду Н2О и экстрагировали диэтиловым эфиром (3×200 мл). Водный слой подкисляли до рН 3,5-4 с помощью охлажденного на льду 10%-ного водного раствора лимонной кислоты и повторно экстрагировали с помощью ЕtOАс (3×200 мл). ЕtOАс-слой промывали Н2О (2×100 мл), соляным раствором (100 мл), сушили над мгSO4 и упаривали, получая соединение 1d с выходом 85% в пересчете на количество восстановленного исходного продукта.
1Н ЯМР (CDCl3, 400 МГц): δ 1,51 (s, 9H), 1,64-1,68 (m, 1H), 1,68-1,75 (m, 1H). 1,77-1,88 (m, 1H), 1,96-2,01 (m, 1H), 2,03-2,22 (m, 3Н), 5,01 (dm, J=6,4 Гц, 1H), 5,03 (dm, J=14,9Гц, 1H), 5,72-5,83 (m, 1H).
ЭС(+)МS:m/z 241 (M+H)+
В. К раствору кислоты 1d в безводном бензоле (1,14 г в 25 мл бензола, 4,74 ммоля), добавляли Et3N (800 мкл, 5,68 ммоля), а затем добавляли дифенилфосфорилазид (1,13 мл, 5,21 ммоля) и смесь нагревали до температуры дефлегмации в течение 3,5 ч. Затем добавляли триметилсилилэтанол (1,36 мл, 9,48 ммоля) и продолжали перемешивание при температуре дефлегмации еще в течение 4 ч. Затем смесь охлаждали до КТ, упаривали до половины ее первоначального объема, разбавляли диэтиловым эфиром (30 мл) и промывали 5%-ным водным раствором NaHCO3 (2×30 мл), соляным раствором (50 мл), сушили над мгSO4 и упаривали. Остаток в виде масла хроматографировали на силикагеле, используя в качестве элюента 10%-ный ЕtOАс в гексане, в результате чего получали чистое соединение 1е с выходом 88% (1,49 г).
1Н ЯМР (CDCl3, 400 МГц) δ 0,03 (s, 9H), 0,91-0,99 (m, 2H), 1,18-1,29 (m, 2H), 1,45 (bs, 11H), 1,56-1,72 (m, 2H), 2,02-2,18 (m, 2H), 4,12 (t, J=8,3 Гц, 2Н), 4,93 (dm, J=10,2 Гц, 1Н), 4,98 (dm, J=17,2 Гц, 1H), 5,07 (bs, 1H), 5,71-5,83 (m, 1H).
Г. К раствору циклопропильного производного 1е (1,19 г, 3,35 ммоля, в 30 мл ТГФ), добавляли трет-Вu4NF (6,7 мл в виде 1М раствора в ТГФ, 6,7 ммоля) и смесь сначала перемешивали при КТ в течение 16 ч, а затем нагревали до температуры дефлегмации в течение 15 мин. Растворитель осторожно выпаривали при пониженном давлении (вследствие высокой летучести свободного амина 1f необходимо принять меры предосторожности при выпаривании растворителя). Неочищенный остаток повторно растворяли в ЕtOАс (100 мл) и промывали Н2O (2×50 мл), соляным раствором (50 мл), сушили над мг SO4 и затем снова осторожно выпаривали растворитель. Неочищенный продукт 1f (представляющий собой смесь двух энантиомеров 1f’ и 1f’’) использовали для сочетания с производным пролина Р2 без дополнительной очистки. На этой стадии с помощью экспресс хроматографии легко мог быть выделен фрагмент Р1Р2, имеющий требуемое стереохимическое строение Р1 (пример 21, фрагмент 21b).
ФРАГМЕНТЫ Р2
Пример 2
Синтез Вос-4(R)-[(7-метокси-4-хинолинил)окси]пролина (2с):
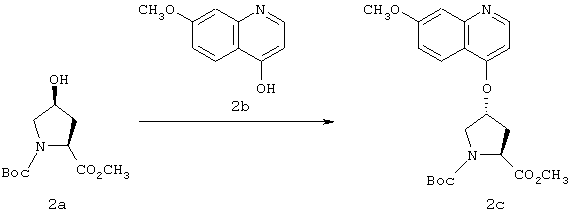
4-Гидрокси-7-метоксихинолин (2b) получали согласно методу, описанному у M.W.Chun, K.K.Olmstead, Y.S.Choi, C.O.Lee, C.-K.Lee, J.H.Kirn, Lee,.J. Bioorg. Med. Chem. Lett. 1997, 7, 789. Раствор соединения 2b (1,88 г, 10,73 ммоля) и ДЭАД (3,4 мл, 21,46 ммоля) в безводном ТГФ добавляли при перемешивании к раствору защищенного цис-гидроксипролина 2а (2,63 г, 10,73 ммоля) и трифенилфосфина (5,63 г, 21,46 ммоля) в безводном ТГФ (160 мл) при 0° в атмосфере N2. Реакционной смеси давали нагреться до КТ и ее перемешивали в течение 14 ч. Затем ТГФ выпаривали и выделяли чистый продукт 2с с помощью экспресс-хроматографии на колонке, используя в качестве элюента 5%-ный МеОН в ЕtOАс; выход 35% (1,5 г).
1Н ЯМР (CDCl3, 400 МГц): δ 1,44 (s, 9H), 1,65 (bs, 1H), 2,34-2,43 (m, 1H), 2,63-2,76 (m, 1H), 3,78 (s, 3Н), 3,75-3,85 и 3,89-3,99 (2m, 1H, 2 ротамера), 3,95 (s. 3Н), 4,51 и 4.60 (2t, J=8 Гц, 1H, 2 ротамера), 5,15 (bs, 1H), 6,53-6,59 (m, 1H), 7,12-7,18 (dd, J=8,9 и 2.2 Гц, 1H), 7,36 (d, J=2,6 Гц, 1H), 8,03 (bd, J=9,2 Гц, 1H), 8,65 (bd, J=5,1 Гц, 1H).
Пример 3
Синтез 2-этокси-4-гидрокси-7-метоксихинолина (3с)
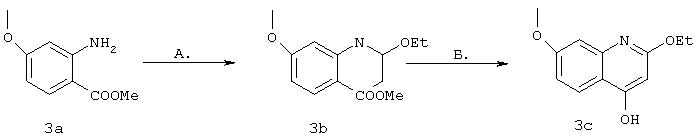
Синтез метил-пара-метоксиантранилата 3а осуществляли согласно методу, описанному у Katzu др., J. Org. Chew., 1953, 18, 1380-1400.
Общая процедура синтеза производного хинолина 3 с представляет собой модификацию метода, описанного у ВАЦККг и др., Indian Journal of Chemistry, 1995, Sat. В, 330-332.
А. Метил-пара-метоксиантранилат 3а (3,069 г, 16,96 ммоля) растворяли в триэтилортоацетате (4,7 мл, 25,4 ммоля), затем добавляли раствор безводной НСl (4 н. раствор в диоксане, 50 мкл, 0,6 ммоля). Образовавшуюся смесь выдерживали при температуре дефлегмации в течение 19 ч. Затем летучие компоненты выпаривали в вакууме, получая продукт 3b (4,92 г, масло янтарного цвета, количественный выход), который использовали на следующей стадии без дальнейшей обработки.
Б. К раствору субстрата 3b (согласно теоретической оценке 16,96 ммоля) в ТГФ (34 мл) при -78°С в атмосфере азота добавляли LiHMDS (1 M раствор в ТГФ, 22 мл, 1,3 экв.). Вскоре после добавления охлаждающую баню удаляли и смесь перемешивали при температуре окружающей среды в течение 1 ч, после чего добавляли еще одну порцию LiHMDS (16 мл). Образовавшуюся смесь перемешивали до полного исчезновения исходного продукта (1 ч) по данным ТСХ (100% ЕtOАс, время удерживания имидата Rf=0,7, время удерживания продукта Rf=0,2). После этого добавляли НСl (4н. раствор в диоксане, 10 мл) и смесь концентрировали в вакууме. Образовавшийся пастообразный продукт растирали со смесью ЕtOАс (10 мл) и водного раствора NaH2PO4 (1M, 10 мл) и облучали ультразвуком. В результате образовывался обильный осадок, который собирали фильтрацией, промывали водой и сушили, получая требуемый продукт 3с в виде твердого вещества бежевого цвета (3,117 г, выход 84% для двух стадий, чистота >99% по данным анализа с помощью ЖХВР).
1Н ЯМР (400 МГц, ДМСО-d) δ (част./млн): 7,88 (d, J=8,9 Гц, 1Н), 6,98 (br. s, 1H), 6,89 (br. d, J=8,6 Гц, 1H), 5,94 (br. s, 1H), 4,30 (br. s, 2H), 3,84 (s, 3Н), 1,34 (t, J=7,0 Гц, 3Н).
Пример 4
Синтез 4-гидрокси-7-метокси-2-(3-метил-1,2,4-оксадиазол-5-ил)хинолина(4d)
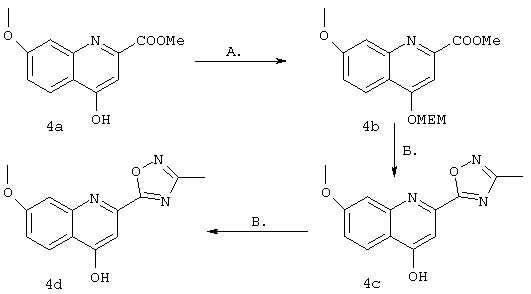
А. К раствору 2-карбометокси-4-гидрокси-7-метоксихинолина 4а (его получение описано в WO 00/09543 и WO 00/09558) (1 г, 4,29 ммоля) в ДМФ (10 мл) в атмосфере азота добавляли NaH (60%-ный в минеральном масле, 190 мг, 4,98 ммоля). Образовавшуюся смесь перемешивали при температуре окружающей среды в течение 1 ч, затем по каплям добавляли хлорид MEM (455 мкл, 4,98 ммоля) и образовавшуюся смесь перемешивали при температуре окружающей среды еще в течение 19,5 ч. Реакционную смесь разбавляли EtOAc (100 мл), промывали Н2О (50 мл), соляньм раствором (50 мл), сушили над MgSO4, концентрировали в вакууме, получая неочищенный продукт реакции (1,37 г). Его очищали с помощью экспресс-хроматографии на колонке, получая продукт 4b (1,04 г, выход 75%) в виде бесцветного масла.
Б. К смеси, содержащей только что активированные молекулярные сита с размером 4 Е (500 мг) и ацетамидоксим (248 мг, 3,35 ммоля) добавляли ТГФ (3 мл). Образовавшуюся смесь перемешивали в течение 15 мин в атмосфере азота при температуре окружающей среды, после чего порциями добавляли NaH (60%-ный в минеральном масле, 124 мг, 3,24 ммоля). Образовавшуюся суспензию перемешивали при температуре окружающей среды в течение 1 ч, после чего добавляли сложный эфир 4b (500 мг, 1,56 ммоля) в виде раствора в ТГФ (5 мл). Образовавшуюся смесь выдерживали при температуре дефлегмации в течение 1 ч, затем фильтровали через целит, промывали EtOAc (3 порции по 20 мл) и концентрировали в вакууме. Образовавшуюся неочищенную смесь очищали с помощью экспресс-хроматографии (100%-ный EtOAc), получая продукт 4 с (352 мг, выход 65%) в виде твердого вещества белого цвета.
В. К простому МЕМ-эфиру 4 с (170 мг, 0,493 ммоля) в ТГФ (4 мл) добавляли водный раствор НСl (1 н., 1 мл). Образовавшуюся смесь перемешивали при температуре окружающей среды в течение 1 ч, затем разбавляли водным раствором NаН2РO4 (1М, 50 мл). Образовавшийся твердый продукт фильтровали, растирали с EtOAc, фильтровали и сушили, получая требуемый продукт (4d) (90 мг, выход 71%) в виде твердого вещества белого цвета. МС (ES+) 258 (М+1), (ES-) 256 (М-1).
1Н ЯМР (400 МГц, ДМСО-d) δ (част./млн): 8,03 (d, J=9,2 Гц, 1Н), 7,38 (d, J=2,2 Гц, 1H), 7,06 (d, J=8,6 Гц, 1H), 6,85 (br. s, 1H), 3,88 (s, 3H), 2,64 (s, 3H).
Пример 5
Синтез 4-гидрокси-7-метокси-2(5-метил-1,3,4-оксадиазол-2-ил)хинолина (5е)
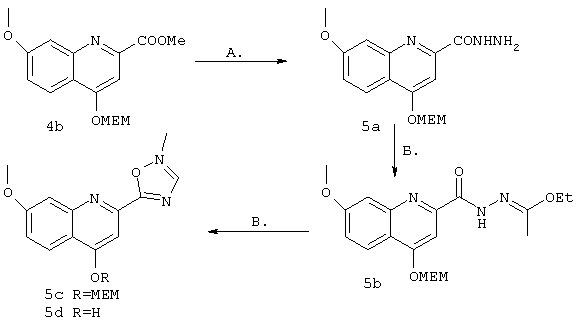
А. К субстрату 4b (465 мг, 1,45 ммоля) в этаноле (5 мл) добавляли безводный гидразин (57 мкл, 1,8 ммоля). Образовавшийся раствор выдерживали при температуре дефлегмации в течение 4 ч, затем концентрировали в вакууме, получая продукт 5а (704 мг, количественный выход неочищенного продукта) в виде твердого вещества желтого цвета, который использовали без очистки на следующей стадии.
Б. Соединение 5а (согласно теоретической оценке 1,45 ммоля) в триэтилортоацетате (5 мл) выдерживали при 100-110°С в атмосфере азота. Затем образовавшуюся смесь разбавляли EtOAc (100 мл), промывали водным насыщенным раствором NaHCO3 (50 мл), соляным раствором (50 мл), сушили над MgSO4, концентрировали в вакууме и очищали с помощью экспресс-хроматографии на колонке (100%-ный EtOAc). Соединение 5b (359 мг, выход 61% для двух стадий) получали в виде масла желтого цвета. МС (ES+) 392 (m+1), (ES-) 390 (m-1).
В. Соединение 5b (333 мг, 0,852 ммоля) выдерживали при 140°С в глубоком вакууме в течение 8,5 ч и очищали с помощью экспресс-хроматографии на колонке (100%-ный ЕtOАс), получая смесь соединения 5b (116 мг, выход 35%, Rf=0,5) и соединения 5с (138 мг, скорректированный выход 72%, rf=0,3). К раствору соединения 5с (138 мг, 0,4 ммоля) в ТГФ (4 мл) добавляли водный раствор НСl (1 н., 1 мл) и образовавшуюся смесь перемешивали до завершения реакции (30 мин). ТГФ выпаривали в вакууме и добавляли водный раствор NaH2PO4 (1 М, 2 мл). Образовавшуюся суспензию облучали ультразвуком, фильтровали и твердый продукт сушили в глубоком вакууме, получая требуемый продукт 5d, (75 мг, выход 73%) в виде твердого вещества бежевого цвета. МС (ES+) 258 (m+1), (ES-) 256 (m-1). 1Н ЯМР (400 МГц, ДМСО-d): δ 8,03 (d. J=9,2 Гц, 1 Н), 7,39 (d, J=2,2 Гц, 1 Н), 7,06 (br. d, J=8,6 Гц, 1 Н), 6,85 (br. s, I H), 3,88 (s, 3Н), 2,64 (s, 3H).
Пример 6
Синтез 4-бензилокси-2-(хлор)-7-метоксихинолина (6е)
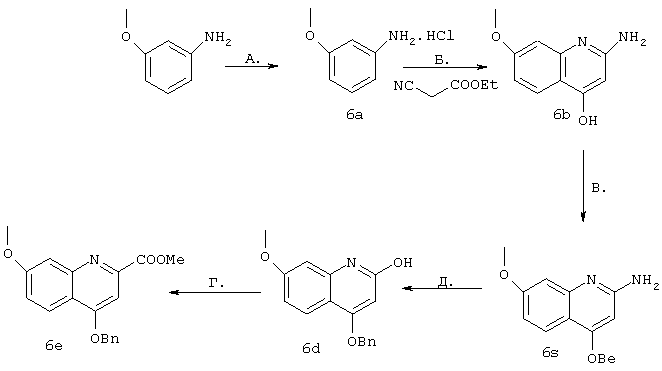
А. Имеющийся в продаже мета-анизидин (25 г, 0,20 моля) в диоксане (80 мл) охлаждали до 0°С и добавляли безводную НСl (4н. раствор в диоксане, 75 мл, 0,30 моля). Затем добавляли Et2O (500 мл) и продолжали перемешивание в течение 1 ч. После этого фильтрацией получали твердый продукт бежевого цвета и сушили в вакууме, получая соль 6a (31,88 г, выход 98%).
Б. К этой соли добавляли этилцианоацетат (21,3 мл, 0,20 моля) и смесь, помещенную в колбу, снабженную дистилляционной насадкой и колбой для сбора продукта, нагревали до 280-300°С. Для наблюдения за ходом реакции производили сбор этанола. После того как накапливалось 9 мл этанола (теоретически рассчитанное количество составляет 11,7 мл), нагревание прекращали, реакционную смесь охлаждали до КТ, разбавляли смесью вода (200 мл) - ЕtOАс (200 мл), затем перемешивали и добавляли водный раствор NаН2РO4 (300 мл). После перемешивания еще в течение 1 ч, фильтрации и сушки получали продукт 6b (19,06 г, чистота 84,5%, выход ~50%) в виде твердого вещества желтого цвета, который использовали при проведении следующей реакции без дополнительной очистки.
В. Соединение 6b (11,0 г, 57,8 ммоля) в ДМФ (100 мл) при 0°С добавляли к NaH (60%-ный раствор в минеральном масле, 2,78 г, 115,6 ммоля). После этого ледяную баню удаляли и смесь перемешивали при температуре окружающей среды в течение 1 ч, затем добавляли бензилбромид (7,6 мл, 63,6 ммоля) и реакционную смесь перемешивали в течение 16 ч. После этого раствор разбавляли смесью ЕtOАс (220 мл) - гексан (220 мл) и фильтровали образовавшийся твердый продукт, растирали с водным насыщенным раствором NаНСО3 (110 мл), промывали водой, смесью гексан-ЕtOАс (соотношение 1:1, 100 мл) и сушили в глубоком вакууме. В результате получали продукт 6с (5,6 г, чистота 91%, выход 35%) в виде твердого вещества желтого цвета.
К соединению 6с (2,67 г, 9,52 ммоля) в уксусной кислоте (21 мл) добавляли изо-амилнитрит (3,8 мл, 28,6 ммоля), образовавшуюся смесь перемешивали при температуре окружающей среды и наблюдали с помощью ЖХВР. Через 2 ч добавляли еще одну порцию изо-амилнитрита (1,3 мл, 9,52 ммоля) и смесь оставляли перемешиваться в течение 90 ч (по данным ЖХВР содержание продукта 81%, субстрата 3%). К образовавшейся суспензии добавляли воду (100 мл) и затем фильтровали. Собранное твердое вещество сушили в глубоком вакууме, получая продукт 6d (2,35 г, чистота 92%, выход 72%).
Г. К соединению 6d (1,5 г, 4,39 ммоля) добавляли оксихлорид фосфора (13 мл, 141 ммоля) и образовавшуюся смесь выдерживали при температуре дефлегмации в течение 1 ч, после чего разбавляли ЕtOАс (150 мл) и реакцию прекращали путем медленного добавления при 0°С водного раствора NaOH (1 н., 150 мл), доводя значение рН до 9. Разделяли два слоя и органический слой сушили над MgSO4 и концентрировали в вакууме, получая твердое вещество коричневого цвета, которое очищали с помощью экспресс-хроматографии на колонке (15%-ный ЕtOАс/гексан). Получали продукт 6е (819 мг, чистота >99%, выход 62%) в виде твердого вещества желтого цвета.
1Н ЯМР (400 МГц, CDCl3): δ 8,07 (d, J=9,2 Гц, 1 Н). 7,50-7,40 (m. 5H), 7,29 (d, J=2,5 Гц. 1 Н), 7,12 (dd, J=9,2, 2,5 Гц, 1 Н), 6,73 (s, 1H), 5,26 (s, 2H), 3,92 (s. 3H).
Пример 7
Синтез 4-гидрокси-2-(1-имидазолил)-7-метоксихинолина (7b); 4-гидрокси-2-(4-метил-1-имидазолил)-7-метоксихинолина(7d); 4-гидрокси-7-метокси-2-(1-пиразолил)хинолина (7f); и 4-гидрокси-2-(3-мeтил-1-пиразолил)-7-метоксихинолина (7h).
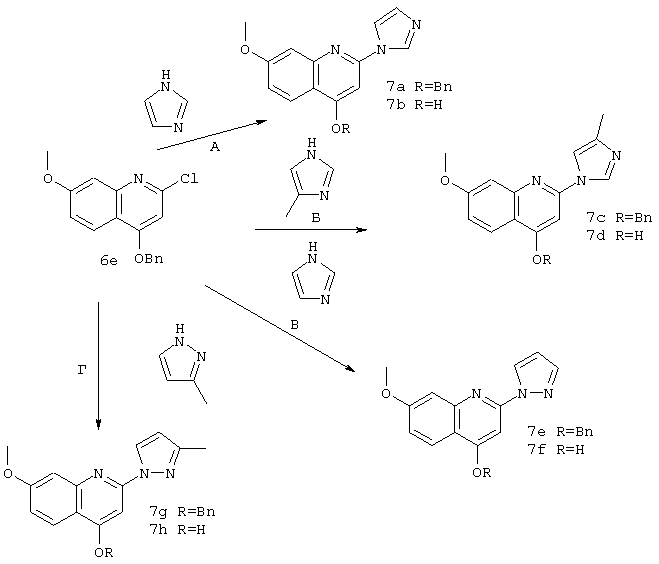
А. Соединение 6e (423 мг, 1,41 ммоля) и имидазол (400 мг, 5,88 ммоля) выдерживали при 110°С в течение 20 ч. Затем смесь разбавляли EtOAc и промывали водой и соляным раствором, сушили над MgSO4, концентрировали при пониженном давлении, получая соединение 7a (422 мг, чистота 96%, выход 90%) в виде твердого вещества желтого цвета. Соединение 7a (319 мг, 0,963 ммоля) в присутствии Pd (5%/C, 64 мг) в смеси этанола (5 мл) и ТГФ (5 мл) продували водородом и оставляли в атмосфере водорода при давлении 1 ат. После перемешивания в течение 7,5 ч при температуре окружающей среды реакционную смесь фильтровали, промывали смесью хлороформ-метанол и концентрировали, получая продукт 7b (130 мг, чистота 97,7%, выход 56%) в виде твердого вещества желтого цвета. МС (ES+) 242 (m+1), (ES-) 240 (m-1).
1Н ЯМР (400 МГц, ДМСО-d): δ 8,51 (s, 1H), 8,03 (d, J=8,9 Гц, 1H), 7,93 (s, 1H), 7,23 (d, J=1,9 Гц, 1H), 7,15 (s, 1H), 7,12 (dd, J=9,2, 2,2 Гц, 1H), 6,92 (br. s, 1H), 3,91 (s, 3H).
Б. Соединение 6е (251 мг, 0,837 ммоля) и 4-метилимидазол (344 мг, 4,19 ммоля) выдерживали при 110°С в течение 20 ч. Затем смесь разбавляли ЕtOАс, промывали водой и соляным раствором, сушили над MgSO4, и концентрировали при пониженном давлении, получая неочищенный продукт, содержащий смесь 10:1 изомеров 4-метил- и 5-метилимидазолила соответственно. Основной требуемый изомер 11с, представляющий собой твердое вещество белого цвета (166 мг, чистота 99%, выход 57%), отделяли от второй более полярной фракции (76 мг, выход 23%). содержащей смесь изомеров 4- и 5-метилимидазолила, с помощью экспресс-хроматографии на колонке (100%-ный ЕtOАс). Соединение 7с (163 мг, 0,472 ммоля) в присутствии Pd (5%/C, 33 мг) в смеси этанола (2,4 мл) и ТГФ (5 мл) продували водородом и оставляли в атмосфере водорода при давлении 1 ат. После перемешивания в течение 18 ч при температуре окружающей среды реакционную смесь фильтровали, промывали смесью хлороформ-метанол и концентрировали, получая соединение 7d (118 мг, чистота 99%, выход 98%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, ДМСО-d): δ 8,42 (br. s, 1H), 8,01 (d, J=9,2 Гц, 1H), 7,64 (br. s, 1H), 7,21 (br.s, 1H),7,10(d, J=8,9 Гц, 1H), 6,89 (br. s, 1H), 3,90 (s, 3H), 2,20 (s, 3H).
В. Соединение 6e (184 мг, 0,614 ммоля) и пиразол (209 мг, 3,07 ммоля) выдерживали при 110°С в течение 17 ч. Затем смесь разбавляли ЕtOАс и промывали водным раствором NaOH (1 н.) и соляным раствором, сушили над MgSO4, концентрировали при пониженном давлении, получая неочищенный продукт, который очищали с помощью экспресс-хроматографии на колонке (смесь 2:1 гексан-ЕtOАс), получая соединение 7е (103 мг, выход 50%) в виде твердого вещества светло-желтого цвета. Соединение 7е (103 мг, 0,311 ммоля) в присутствии Pd (5%/C, 20 мг) в смеси этанола (2 мл) и ТГФ (2 мл) продували водородом и оставляли в атмосфере водорода при давлении 1 ат. После перемешивания в течение 5,5 ч при температуре окружающей среды реакционную смесь фильтровали, промывали смесью хлороформ-метанол и концентрировали, получая соединение 7f(77 мг, чистота 99%, выход 99%) в виде твердого вещества желтого цвета. МС (ES+) 242 (m+1), (ES-) 240 (m-1).
1Н ЯМР (400 МГц, ДМСО-d): δ 8,72 (d, J=2,5 Гц, 1H), 8,31 (s, 1H), 8,00 (d, J=8,9 Гц, 1 Н), 7,83 (br. s, 1 Н), 7,43 (br. s, 1 Н), 7,24 (br. s, 1 Н), 7,10 (d, J=8,6 Гц, 1 Н), 6,59 (br. s, 1H), 3,90 (s,3H).
Г. Соединение 6е (217 мг, 0,724 ммоля) и 4-метилпиразол (594 мг, 7,24 ммоля) выдерживали при 110°С в течение 23 ч. Затем смесь, представляющую собой смесь 1:1 дебензилированного соединения 7h и бензилированного продукта 7g разбавляли EtOAc (2-3 мл) и фильтровали, получая чистый дебензилированный продукт 7h (111 мг, чистота 95%, выход 54%) в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, ДМСО-d): δ 8,58 (d, J=2,6 Гц, 1Н), 7,98 (d, J=9,2 Гц, 1Н), 7.25 (br. s, 1H), 7,20 (s, 1H), 7,04 (br. d, J=9,2 Гц, 1H), 6,38 (s, 1H), 3,89 (s, 3H), 2,30 (s, 3H).
Пример 8
Синтез 4-гидрокси-7-метокси-2[4-(2-изопропиламинотиазолил)]хинолина (8f)
Примечание: [С использованием одной и той же схемы синтеза путем замены соединения 8b другими алкилтиомочевинами получали различные 2-алкиламинотиазолильные заместители].
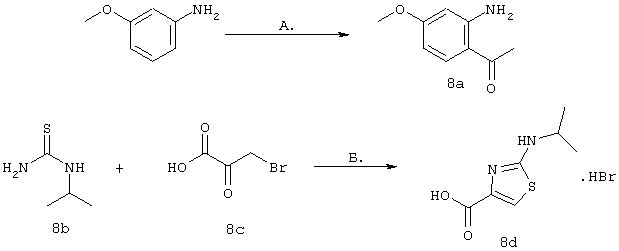
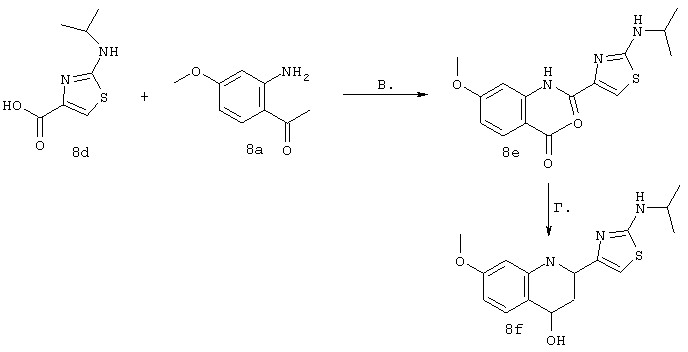
А. Для превращения мета-анизидина в соединение 8а использовали протокол, идентичный описанному в литературе: F.J.Brown и др., J. Med. Chem. 1989, 32, 807-826. Однако процедура очистки была модифицирована для того, чтобы избежать очистки с помощью хроматографии. ЕtOАс-фазу, содержащую требуемый продукт, обрабатывали смесью, содержащей MgSO4, уголь и 5 мас.% (в пересчете на теоретически рассчитанную массу) силикагеля. После фильтрации через целит продукт растирали с простым эфиром. Соединение 8а получали в виде твердого вещества светло-коричневого цвета с чистотой >99% (по данным анализа с помощью ЖХВР).
Б. Суспензию изопропилтиомочевины (8b, 3,55 г, 30 ммолей) и 3-бромпировиноградной кислоты (8с, 5 г, 1 экв.) в диоксане (300 мл, 0,1 М) нагревали до 80°С. После того, как температура достигала 80°С, раствор становился прозрачным и вскоре после этого происходило осаждение продукта в виде твердого вещества белого цвета. После нагревания в течение 2 ч раствор охлаждали до КТ и фильтровали осадок белого цвета, получая соединение 8d с высокой чистотой (чистота >98% по данным ЯМР-анализа) и с выходом 94% (7,51 г).
В. Смесь, содержащую карбоновую кислоты 8d (4,85 г, 18,2 ммоля) и производное анилина 8а (3 г, 1 экв.) в пиридине (150 мл, 0,12 М) охлаждали до -30°С (после охлаждения прозрачный раствор превращался практически в суспензию). Затем медленно в течение периода времени, составляющего 5 мин, добавляли оксихлорид фосфора (3,56 мл, 2,1 экв.). Реакционную смесь перемешивали при -30°С в течение 1 ч, баню удаляли и реакционной смеси давали нагреться до КТ. Через 1,5 ч реакционную смесь сливали на лед, значение рН доводили до 11 с помощью 3н. водного раствора NaOH, экстрагировали с помощью CH2Cl2, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Затем твердое вещество бежевого цвета очищали с помощью экспресс-хроматографии (45%-ный EtOAc в гексане), получая соединение 8е в виде твердого вещества светло-желтого цвета с выходом 73% (6,07 г).
Г. Раствор трет-ВuОК (2,42 г, 21,6 ммоля) в безводном трет-ВuОН (40 мл, 0,14М, очищенный дистилляцией от металлического Mg) нагревали до температуры дефлегмации. В течение 5 мин добавляли порциями соединение 8е (1,8g, 5,4 ммоля) и образовавшийся раствор темно-красного цвета перемешивали при температуре дефлегмации еще в течение 20 мин (завершение реакции оценивали с помощью ЖХВР). Смесь охлаждали до КТ и добавляли НСl (4 н. в диоксане, 1,5 экв.). Затем смесь концентрировали в вакууме для того, чтобы гарантировать, что удалено все количество НСl и диоксана, продукт дважды повторно растворяли в СН2Сl2 и сушили в вакууме, получая в результате гидрохлорид соединения 8f в виде твердого вещества бежевого цвета (1,62 г, чистота 93% по данным ЖХВР). Затем продукт сливали в фосфатный буфер (1н. раствор NaH2PO4, pH=~4,5) и облучали ультразвуком. Твердый продукт бежевого цвета фильтровали и сушили в вакууме, получая соединение 8f (1,38 г, выход 81%) в виде твердого вещества бежевого цвета (чистота 91% по данным ЖХВР).
1Н ЯМР (400 МГц, ДМСО) δ 8,27 (s, 1Н), 8,12 (d, 1H, J=9,2 Гц), 7,97 (br.s, 1H), 7,94 (s, 1H), 7,43 (s, 1H), 7,24 (dd, 1H, J=9,2, 2,2 Гц), 3,97 (m, 1H), 3,94 (s, 3H), 1,24 (d, 2Н, J=6,4 Гц).
Пример 9
Синтез 4-гидрокси-7-метокси-2-[2-(4-изопропилтиазолил)]хинолина (9f).
Примечание: С использованием одной и той же схемы синтеза, в которой соединение 9b замещали другими α-бромкетонами, получали различные 2-(4-алкил)тиазолильные заместители.
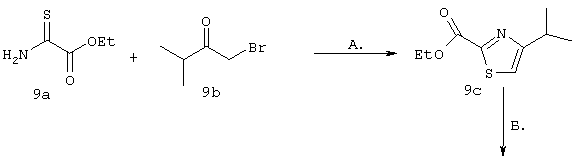
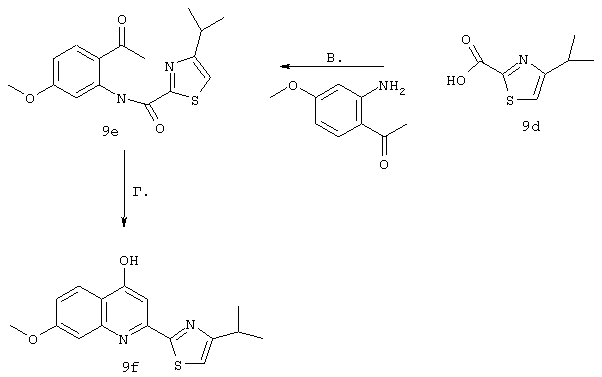
A. К раствору 3-метилбутан-2-она (8 г, 93 ммоля) в МеОН (100 мл) при -30°С, добавляли по каплям в течение 45 мин Вr2 (4,79 мл, 93 ммоля, 1 экв.). Затем образовавшуюся смесь перемешивали при КТ в течение 90 мин. Добавляли пентан и раствор промывали 5%-ным водным раствором NаНСО3, органический слой сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Полученное соединение 9b, представляющее собой неочищенное масло желтого цвета, использовали без дополнительной очистки. Раствор, содержащий этилтиооксамат (9а, 1,8 г, 13,5 ммоля) и производное бромкетона 9b (13,5 ммоля) перемешивали при 70°С в течение 15 ч. После этого смесь концентрировали в вакууме и затем очищали с помощью экспресс-хроматографии на колонке, используя в качестве элюента 15%-ный EtOAc в гексане, получая соединение 9с (740 мг, выход 28%).
Б. Раствор соединения 9с (700 мг, 3,5 ммоля) в смеси ТГФ/МеОН/Н2О (соотношение 3:1:1, 13 мл) обрабатывали при КТ в течение 5h LiOHН2О (148 мг, 3,5 ммоля, 1 экв.). Затем значение рН доводили до 6 с помощью 0,1 н. раствора НСl и смесь концентрировали досуха в вакууме, получая кислоту 13d, которую непосредственно использовали на следующей стадии без дополнительной очистки.
В. Раствор, содержащий 4-метокси-2-аминоацетофенон (промежуточное соединение 8а, 570 мг, 3,45 ммоля) и производное карбоновой кислоты 9d (590 мг, 3,45 ммоля, 1 экв.) в пиридине (30 мл) охлаждали до -20°С. Затем по каплям в течение 5 мин добавляли РОСl3 (0,35 мл, 3,79 ммоля, 1,1 экв.). Образовавшийся раствор перемешивали при -10°С в течение 2 ч. Реакцию прекращали путем добавления Н2О и смесь концентрировали в вакууме. Остаток сливали на насыщенный водный раствор NаНСО3 и экстрагировали EtOAc. Органический слой сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью экспресс-хроматографии на колонке, используя в качестве элюента 25%-ный ЕtOАс в гексане, в результате чего получали соединение 9е в виде твердого вещества белого цвета (740 мг, выход 67%).
Г. Трет-ВuОК (518 мг, 2,1 экв.) добавляли к суспензии соединения 9е (700 мг, 2,2 ммоля) в безводном трет-ВuОН (11 мл). Образовавшуюся смесь нагревали до 75°С в течение 7,5 ч, затем раствор охлаждали до КТ и подкисляли путем добавления НСl (4 н. раствор НСl в диоксане, 2,5 мл). Смесь концентрировали в вакууме и полученный остаток сливали на 1 н. раствор NaH2PO4 и фильтровали. Затем твердый продукт растирали с небольшим количеством ЕtOАс, фильтровали и сушили в вакууме, получая соединение 9f в виде твердого вещества светло-бежевого цвета (270 мг, выход 41%).
1Н ЯМР (400 МГц, ДМСО-d6) δ 8,00 (br. s, Ih), 7,60 (br. s, 1H), 7,51 (br. s, 1H), 7,43 (br. s, 1 H), 7,29 (br. s, 1 H), 7,14 (br. s. 1 Н), 6,95 (br. а, 1 Н), 3,90 (s, 3H), 3,15 (m, 1 Н), 1,33 (d, J-5,4 Гц, 6Н).
Пример 10
Синтез 4-гидрокси-2-(1-метил-2-имидазолил)-7-метоксихинолина (10d)
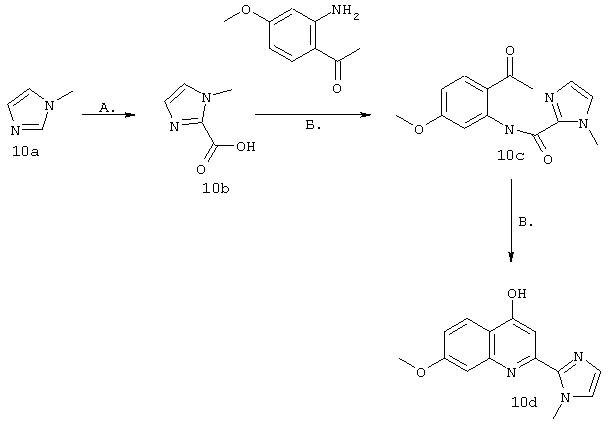
А. Раствор N-метилимидазола 10а (5 г, 61 ммоль) в 100 мл ТГФ охлаждали до -78°C. По каплям в течение 15 мин добавляли н-BuLi (24,4 мл 2,5 М раствора в Et2O, 1 экв.). Образовавшуюся смесь перемешивали в течение 90 мин при -78°С, после чего порциями сливали на избыток твердого СO2. Гетерогенную смесь перемешивали в течение 2 ч и затем давали ей нагреться до КТ. Добавляли 1 н. раствор НСl до достижения значения рН 5, отделяли водный слой и лиофилизировали. Полученный таким образом остаток экстрагировали ЕtOАс (для удаления солей), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении. В результате получали 6,2 г (выход 80%) твердого продукта 10b белого цвета.
Б. Раствор, содержащий 4-метокси-2-аминоацетофенон 8а (394 мг, 2,39 ммоля) и производное карбоновой кислоты 10b (301 мг, 1 экв.) в пиридине (10 мл), охлаждали до -20°С. Затем по каплям в течение 5 мин добавляли РОСl3 (244 мкл, 1,1 экв.). Образовавшийся раствор перемешивали при -10°С а течение 2,5 ч. Затем добавляли воду и смесь концентрировали при пониженном давлении. Остаток сливали на насыщенный раствор NаНСО3 и экстрагировали EtOAc. Органическую фазу сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Продукт очищали с помощью хроматографии на силикагеле (25%-ный ЕtOАс/Нех), получая 530 мг (выход 81%) в виде твердого вещества 10с светло-желтого цвета.
В. К суспензии субстрата 10с (500 мг, 1,8 ммоля) в 8 мл трет-ВuОН добавляли трет-ВuОК (431 мг, 2,1 экв.). Затем образовавшуюся смесь нагревали до 75°С в течение 7 ч. Раствору давали остыть в течение ночи до комнатной температуры и добавляли 2,5 мл НСl (4 н. раствор в диоксане). Смесь концентрировали при пониженном давлении и полученный остаток разбавляли EtOAc. Добавляли 1н. раствор NaOH до достижения значения рН 7. Отделяли органическую фазу и сушили (MgSO4), фильтровали и концентрировали при пониженном давлении, получая 145 мг соединения 10d (выход 31%) в виде твердого вещества светло-бежевого цвета. 1Н ЯМР (400 МГц, ДМСО-d): δ 7,99 (d, J=8,9 Гц, 1Н), 7,49 (s, 1H), 7,37 (s, 1H), 7,18 (s, 1H), 6,92 (d, J=8,9 Гц, 1Н), 6,31 (s, 1H), 3,87 (s,3H), 3,84 (s,3H).
Пример 11
Синтез 4-гидрокси-2-(1-пирролил)-7-метоксихинолина (11b)
А. Раствор, содержащий субстрат На (полученный из соединения 6с путем гидрогенолиза бензильной группы в присутствии 5%-ного Pd/C в растворе этанол-ТГФ) (1 г, 5,25 ммоля) и 2,5-диметокситетрагидрофуран (0,68 мл, 1 экв.) в ледяной уксусной кислоте, кипятили с обратным холодильником в течение 4,5 ч и затем давали охладиться до КТ. Затем смесь концентрировали при пониженном давлении. Остаток разбавляли метанолом и добавляли 1 н. NaOH (водный раствор) до достижения значения рН 7. Продукт очищали с помощью хроматографии на силикагеле (3%-ный МеОН/СH2Сl2, остаток предварительно адборбировали на силикагеле). В результате получали 140 мг (выход 13%) соединения 11b в виде твердого вещества белого цвета.
1Н ЯМР (400 МГц, ДМСО-d): δ 7,98 (d, J=9,2 Гц, 1H), 7,64 (s, 2H), 7,18 (d, J=2,5 Гц, 1H), 7,05 (br. d, J=7,9 Гц, 1H), 6,88 (br. s, 1H), 6,32 (s, 2H), 3,90 (s, 3Н).
Пример 12
Синтез 4-гидрокси-7-метокси-2-(6-метил-2-пиридил)хинолина (12d)

А. 6-Метилпиколиновую кислоту 12а (411 мг, 3,0 ммоля) и SOCl2 (0,520 мл, 7,2 ммоля, 2,4 экв.) кипятили с обратным холодильником в течение 2 ч в бензоле (5 мл). Растворитель и избыток SOCl2 удаляли из реакционной смеси в вакууме и остаток растирали с пентаном. Образовавшийся твердый продукт отфильтровывали и фильтрат концентрировали, получая хлорангидрид 12b (500 мг, 2,6 ммоля).
Б. К раствору неочищенного хлорангидрида 12b в СН2Сl2 (5 мл) при 0°С, добавляли раствор анилина 8a (344 мг, 2,08 ммоля), ДИПЭА (1,45 мл, 8,35 ммоля) и ДМАП (61 мг, 0,5 ммоля) в СН2Сl2 (10 мл). Реакционную смесь перемешивали при КТ в течение 16 ч. Летучие компоненты удаляли в вакууме, остаток растворяли в EtOAc и раствор промывали 5%-ным NаНСО3 (2х), Н2О и соляным раствором. Затем органический слой сушили над MgSO4 и концентрировали в вакууме. Смесь очищали с помощью экспресс-хроматографии на колонке, используя в качестве элюента смесь EtOAc/ гексан (1:2), получая амид 12с (490 мг, выход 82%).
В. К суспензии амида 12с (490 мг, 1,71 ммоля) в трет-ВuОН (10 мл), добавляли трет-ВuОК (410 мг, 3,43 ммоля) и смесь перемешивали при 75°С в течение 6 ч, а затем при КТ в течение 16 ч. Затем смесь сливали на фосфатный буфер (175 мл, рН 7) и перемешивали в течение 30 мин. Твердый продукт дважды растирали с этилацетатом. Органическую фазу промывали соляным раствором, сушили над MgSO4 и концентрировали в вакууме. Полученный твердый продукт растирали с EtOAc, получая производное хинолина 12d (263 мг, выход 58%). 1Н ЯМР: (CDCl3, 400 МГц): 5 2,68 (s, 3 Н), 3,94 (s, 3 Н), 6,85-6,88 (2d, J=8,68 и 9,5 Гц, 2 Н), 6,94 (dd, J=8,9 и 2,2 Гц, 1 Н), 7,27 (dd, J=6,7 и 1,9 Гц, 1 Н), 7,73-7,79 (m, 2 Н). 8,28 (d, J=8,9 Гц, 1 Н), 10,3 (br s, 1 Н).
Пример 13
Синтез 4-гидрокси-7-метокси-2-(5-метокси-2-пиридил)хинолина (13d)

А. К раствору соединения 13а (623 мг, 3,73 ммоля) в МеОН, добавляли NaOH (2М, 4,70 мл) и реакционную смесь перемешивали при КТ в течение 2 ч. Затем раствор подкисляли с помощью НСl (6 н., 2,2 мл) и концентрировали, получая соединение 13b, которое использовали на следующей стадии без очистки.
Б. К раствору неочищенного соединения 13b (~3,73 ммоля) в пиридине (25 мл), добавляли 8а (500 мг, 3,03 ммоля) и раствор охлаждали до -25°С, после чего добавляли РОСl3 (0,35 мл, 3,73 ммоля). Реакционную смесь перемешивали при -10°С в течение 1 ч, а затем при 0°С в течение 2 ч. Затем смесь сливали на Н2О и экстрагировали EtOAc (2-3х). Объединенные органические слои промывали 5%-ным NаНСО3 и соляным раствором, сушили над MgSO4 и концентрировали в вакууме. Неочищенный продукт очищали с помощью экспресс-хроматографии на колонке, используя в качестве элюента смесь ЕtOАс/гексан (1:2), в результате чего получали амид 13с (617 мг, выход 55%).
В. К суспензии амида 13с (617 мг, 2,05 ммоля) в безводном трет-ВuОН (10 мл) добавляли трет-ВuОК (490 мг, 4,11 ммоля) и смесь перемешивали при 75°С в течение 6 ч, а затем при КТ в течение 16 ч. Реакционную смесь сливали на фосфатный буфер (175 мл, рН 7) и перемешивали в течение 30 мин. Образовавшийся твердый продукт фильтровали и растирали с EtOAc, получая производное хинолина 13d (250 мг, выход 43%). 1Н ЯМР: (ДМСО, 400 МГц): δ 3,86 (s, 3 Н), 3,94 (s, 3 Н), 6,72 (bs, 1 Н), 6,91 (dd, J=8,9 и 1,9 Гц, 1 Н), 7,54 (d, J=1,9 Гц, 1 Н), 7,60 (dd, J=8,9 и 2,9 Гц, 1 Н), 7.97 (d, J=8,9 Гц, 1 Н), 8,21 (d, J=8,6 Гц, 1 Н), 8,48 (d, J=1,9 Гц, 1 Н).
Пример 14
Синтез 4-гидрокси-7-метокси-2-(оксазол-5-ил)хинолина (14с):
А. Защищенное производное хинолина 4b из примера 4 (3,8 г, 11,8 ммоля) растворяли в СН2Сl2 (60 мл) и охлаждали до -78°С, после чего очень медленно в течение 15 мин добавляли гидрид диизобутилалюминия (ДИБАЛ) (7,9 мл, 1 экв., 1,5 М раствор в толуоле). После перемешивания в течение 80 мин добавляли дополнительную порцию ДИБАЛ (5,5 мл, 0,7 экв., 1,5 М раствор в толуоле). После перемешивания при -78°С еще в течение 2 ч, реакцию осторожно прекращали с помощью метанола (4 мл) при -78°С и затем сливали на водный раствор сегнетовой соли (1 н. K-Na тартрат). Густую пасту перемешивали с СН2Сl2 (300 мл) в течение 2 ч до тех пор, пока смесь не становилась прозрачной. Фазы разделяли и органическую фазу сушили (MgSO4), фильтровали и концентрировали, получая твердое вещество белого цвета. Путем очистки с помощью экспресс-хроматографии (SiO2, 230-400 меш) с использованием смеси 50%-ный EtOAc/гексан получали альдегид 14а в виде твердого вещества белого цвета (2,5 г, выход 73%).
Б. К суспензии К2СО3 (48 мг, 0,34 ммоля) в МеОН (7 мл) при перемешивании добавляли толуолсульфонилметилизоцианид (66 мг, 0,34 ммоля). Реакционную смесь нагревали до 45°С и добавляли альдегид 14а (0,10 г, 0,34 ммоля). Реакционную смесь нагревали до 80°С в течение 16 ч и затем концентрировали досуха в вакууме. После осуществления очистки с помощью экспресс-хроматографии (SiO2, 230-400 меш) получали требуемый оксазол 14b (0,089 г, выход 80%). МС: 331,0 (М+H)+.
В. МЕМ-защищенный гидроксихинолин 14b растворяли в ТГФ (3 мл) и обрабатывали водным раствором НСl (1 н., 1 мл). Реакционную смесь перемешивали в течение 30 мин при КТ, после чего концентрировали досуха в вакууме. Остаток обрабатывали фосфатным буфером (3 мл, 1 н. раствор, рН 4,5) и перемешивали, после чего продукт отфильтровывали, промывали дистиллированной водой и сушили в течение ночи в глубоком вакууме (60°С, 16 ч). Получали требуемый гидроксихинолин 14с в виде твердого вещества рыжевато-коричневого цвета (0,065 г, выход 100%). МС: 242.9 (М+H)+.
1Н ЯМР (ДМСО-d6): δ 8,65 (s, 1H), 8,02 (bs, 1H), 7,97 (d, J=8,9 Гц, 1Н), 7.19 (s, 1H), 6,93 (d, J=7,9 Гц, 1H), 6,42 (bs, 1H), 3,87 (s, 3H). ES (+) МС: m/z 242,9 (М+Н)+.
ФРАГМЕНТЫ ПЕПТИДНОГО ЛИНКЕРА (Р3)
Пример 15
Синтез (2S)-N-Вос-аминонон-8-еноевой кислоты (15g)
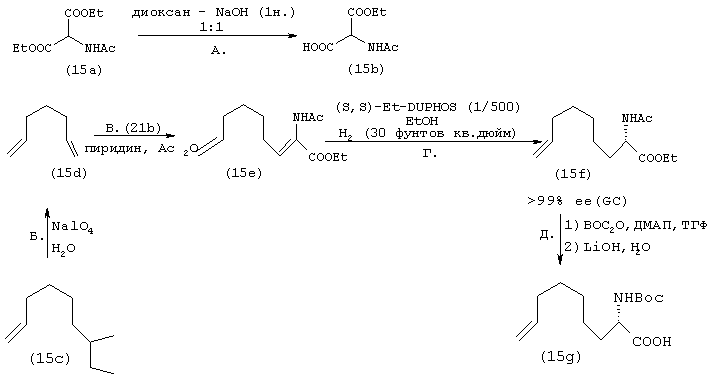
А. К раствору имеющегося в продаже диэтил-2-ацетамидомалоната 15а (100 г, 0,46 моля) в диоксане (500 мл) добавляли по каплям в течение 30-45 мин водный раствор гидроксида натрия (1 М, 1 экв., 460 мл). Образовавшейся смеси давали выстояться в течение 16,5 ч, затем диоксан выпаривали в вакууме, водный раствор экстрагировали тремя порциями по 300 мл этилацетата и подкисляли до рН 1 с помощью концентрированной НСl. Раствор оставляли кристаллизоваться в бане со смесью лед-вода. После появления небольшого количества кристаллов смесь облучали ультразвуком, в результате чего образовывался обильный осадок. После фильтрации и сушки в вакууме получали соединение 15b, (62,52 г, выход 72%) в виде твердого вещества белого цвета.
Б. К перемешиваемой с помощью магнитной мешалки эмульсии, содержащей имеющийся в продаже 7-октен-1,2-диол 15с (25 г, 0,173 моля) и H2O (100 мл), находящейся в круглодонной колбе объемом 1 л, добавляли в течение 20 мин (слабая экзотермическая реакция) водный раствор периодата натрия (40,7 г, 0,190 моля, 1,1 экв., в 475 мл Н2О). Образовавшуюся смесь перемешивали при комнатной температуре еще в течение 1 ч (завершение реакции подтверждали с помощью ТСХ). Затем смесь сливали в делительную воронку и водный слой отделяли от органического слоя. Водный раствор насыщали с помощью NaCl, сливали и еще раз отделяли от органической фракции. Две органические фракции объединяли, сушили над сульфатом натрия и фильтровали через хлопковую пробку (в пипетке Пастера), получая соединение 15d (15,135 г, бесцветное масло, выход 78%). Водный раствор экстрагировали СН2Сl2, сушили над безводным MgSO4, и концентрировали в вакууме (без нагрева, при температуре кипения гептанала 153°С), получая дополнительное количество соединения 15d (1,957 г, бесцветное масло, выход 10%). Общий выход составлял 88%.
В. К твердому этил-2-ацетамидомалонату 15b (7,57 г, 40 ммолей) в течение 1 мин добавляли 6-гептенал 15d (4,48 г, 40 ммолей) в растворе пиридина (32 мл, 10 экв.). Образовавшийся раствор охлаждали в бане при 10°С и в течение 4 мин добавляли уксусный ангидрид (12 мл, 3,2 экв.). Образовавшийся раствор оранжевого цвета перемешивали в течение 3 ч при КТ и добавляли еще одну порцию этил-2-ацетамидомалоната 15b (2,27 г). Образовавшуюся смесь перемешивали при комнатной температуре еще в течение 11 ч. Затем добавляли лед (60 мл) и раствор перемешивали в течение 1,5 ч, после чего смесь разбавляли 250 мл воды и экстрагировали двумя порциями простого эфира. Эфирный раствор промывали 1 н. раствором НСl, насыщенным раствором Na-НСО3, сушили над Nа2SO4, концентрировали и очищали с помощью экспресс-хроматографии (40%-ный ЕtOАс/гексан), получая соединение 15е (4,8 г, выход 50%) в виде масла светло-желтого цвета.
Г. К дегазированному (путем барботации аргоном в течение 30 мин) раствору Z-этил-2-ацетамидо-2,8-нонадиеноата 15е (8,38 г, 35 ммолей) в безводном этаноле (70 мл) добавляли (S,S)-Et-DUPHOS Rh(COD)OTf (51 мг, S/C=496). Смесь помещали в атмосферу водорода при давлении 30 фунтов./кв. дюйм (после 4 циклов вакуум-Н2) и перемешивали с помощью шейкера Парра в течение 2 ч. Образовавшуюся смесь упаривали досуха, получая неочищенное соединение 15f, которое использовали на следующей стадии без очистки.
Д. К раствору неочищенного (S)-этил 2-ацетамидо-8-ноненоата 15f (7,3 г, 30,3 ммоля) в ТГФ (100 мл) добавляли Вос2О (13,2 г, 2 экв.) и ДМАП (740 мг, 0,2 экв.) и реакционную смесь выдерживали при температуре дефлегмации в течение 2,5 ч. Затем большую часть растворителя, представляющего собой ТГФ, выпаривали, неочищенную смесь разбавляли СН2Сl2 и промывали 1 н. НСl для удаления ДМАП. Затем органический слой экстрагировали насыщенным водным раствором NаНСО3, сушили над безводным Na2SO4 и концентрировали в вакууме. После этого неочищенный продукт разбавляли ТГФ (50 мл) и водой (30 мл), добавляли LiOH·Н2О (2,54 г, 2 экв.) и образовавшуюся смесь перемешивали при КТ в течение 25 ч (завершение гидролиза подтверждали с помощью ТСХ). Реакционную смесь концентрировали в вакууме для удаления большей части растворителя, представляющего собой ТГФ, и разбавляли с помощью СН2Сl2. Образовавшийся раствор промывали 1н. раствором НСl, сушили над безводным Na2SO4 и концентрировали в вакууме. Для удаления небольшого количества примесей и избытка Boc2O неочищенный продукт очищали с помощью экспресс-хроматографии (используя в качестве элюента градиент растворителя 100%-ный гексан - 100%-ный EtOAc). В результате получали указанное в заголовке соединение 15g с высокой чистотой в виде масла светло-желтого цвета (5,82 г, выход 71%). 1Н ЯМР (ДМСО, 400 МГц): δ 7,01 (d, J=8 Гц, 1Н), 5,79 (tdd, Jt=6,7 Гц, Jd=17,0, 10,2 Гц, 1Н), 5,00 (md, Jd=17,0 Гц, 1Н), 4,93 (md, Jd=10,2 Гц, 1Н), 3,83 (m, 1H), 2,00 (q, J=6,9 Гц, 2Н), 1,65-1,5 (m, 2Н), 1,38 (s, 9H), 1,35-1,21 (m, 6H).
Пример15а
Альтернативный метод синтеза (2S)-N-Вос-аминонон-8-еновой кислоты (15g):
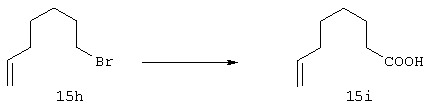
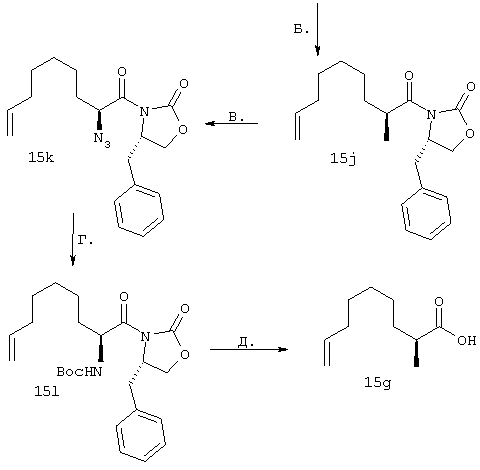
А. К суспензии тонко нарезанной магниевой стружки (0,55 г, 22,5 ммоля) в безводном ТГФ (30 мл), содержащей дибромэтан (0,1 мл), при перемешивании добавляли по каплям в течение 15 мин (реакция является слабо экзотермической) 8-бром-1-октен (15h, 2,52 мл, 15 ммолей). Через 30 мин, смесь нагревали до 38°С в течение 1 ч и затем охлаждали до -78°С, после чего ее добавляли с помощью канюли к избыточному количеству твердого СO2. Смесь разбавляли диэтиловым эфиром (100 мл) и раствор промывали соляным раствором (2×50 мл), сушили над MgSO4 и упаривали. Получали неочищенный продукт в виде масла, который очищали с помощью хроматографии на силикагеле, используя в качестве элюента 15%-ный EtOAc в гексане, в результате чего получали соединение 15i с выходом 62% (1,44 г).
1Н ЯМР (CDCl3, 400 МГц) δ 1,31-1,42 (m, 6H), 1,60-1,69 (m, 2H), 2,02-2,09 (m, 2H), 2,35 (t, J=8,3 Гц, 2H), 4,99 (dm, J=10,0 Гц, 1Н), 5,04 (dm, J=17,0 Гц, 1Н), 5,75-5,86 (m, 1H).
Б. К интенсивно перемешиваемому раствору карбоновой кислоты 15i (1,36 г, 8,7 ммоля) в безводном ТГФ (70 мл) при -78°С добавляли с помощью шприца в безводных условиях продистиллированный непосредственно перед проведением реакции Еt3N (1,6 мл; 11,3 ммоля) и пивалоилхлорид (1,18 мл, 9.58 ммоля). Смесь перемешивали при -78°С в течение 15 мин, а затем при 0°С в течение 45 мин. Смесь снова охлаждали до -78°С и затем переносили с помощью канюли в безводный раствор литиевой соли 4(S)-4-(фенилметил)-2-оксазолидинона в ТГФ при -78°С; для этого предварительно получали реактив, представляющий собой литиевую соль оксазолидинона, путем медленного добавления н-BuLi (2,00 М раствор в гексане, 7,85 мл, 15,7 ммоля) в раствор оксазолидинона (2,78 г, 15,7 ммоля) в ТГФ (20 мл) при -78°С.
Реакционную смесь перемешивали при -78°С в течение 15 мин, а затем при КТ в течение 1,5 ч. В завершение реакцию прекращали с помощью водного раствора бисульфата натрия (100 мл 1 М раствора) и выпаривали ТГФ до достижения ѕ его первоначального объема. Остаток экстрагировали с помощью EtOAc (2×150 мл) и объединенные органические слои промывали 5%-ным раствором NаНСО3 (3×50 мл), соляным раствором (2×50 мл), сушили над MgSO4 и упаривали. Образовавшийся неочищенный продукт в виде масла хроматографировали на силикагеле, используя в качестве элюента 15%-ный EtOAc в гексане, в результате чего получали соединение 15j с выходом 68% (1,88 г).
1Н ЯМР (CDCl3, 400 МГц) δ 1,35-1,47 (m, 6Н), 1,67-1,74 (m, 2H), 2,02-2,09 (m, 2H), 2,65 (dd, J=13,4 и 9,9 Гц, 1Н), 2,84-3,02 (m, 2H), 3,31 (dd, J=13,4 и 3,2 Гц, 1Н), 4.13-4,22 (m, 2H), 4,62-4,71 (m, 1H), 4,93 (d, J=10,2 Гц, 1H), 5,00 (dd, J=17,2 и 1,6 Гц, 1Н), 5,75-5,84 (m, 1H), 7,18-7,38 (m, 5H).
В. К раствору KHMDS (0,8 М раствор в ТГФ, 22 мл, 17,5 ммоля) в безводном ТГФ (50 мл) при -78°С при перемешивании добавляли с помощью канюли раствор производного кислоты 15j (3,25 г, 10,30 ммоля) в безводном ТГФ (40 мл) при -78°С. Смесь перемешивали при -78°С в течение 45 мин. К этой смеси добавляли раствор трисилазида (3,67 г, 11,85 ммоля) в безводном ТГФ (40 мл) при -78°С. Смесь перемешивали при -78°С в течение 3 мин, затем реакцию прекращали с помощью уксусной кислоты (5 мл). После этого смесь перемешивали при КТ в течение 1 ч 45 мин и, наконец, при 40°С в течение 15 мин. Большую часть ТГФ выпаривали. Остаток растворяли в EtOAc (100 мл) и органический раствор промывали Н2O (50 мл), 5%-ным раствором NaHCO3 (3×50 мл) и соляным раствором (50 мл), (MgSO4) и выпаривали. Полученный продукт в виде масла хроматографировали на силикагеле, используя в качестве элюента смесь гексан/СН2Сl2 (1/1), в результате чего получали соединение 15k (2,47 г, выход 67%).
1Н ЯМР (CDCl3, 400 МГц) δ 1,32-1,45 (m, 6H), 1,45-1,6 (m, 1H), 1,75-1,88 (2, 2H, ротамеры), 2,01-2,11 (m, 2H), 2,82-2.87 (m, 1H), 3,33 (dd, J=13,4 и 3,2 Гц, 1Н), 4,10-4,28 (m. 2H), 4,62-4,72 (m, 1 H), 4,90-5,05 (m, 3H), 5,73-5,88 (m, 1 H), 7,17-7,38 (m, 5H).
Г. К раствору безводного SnCl2 (2,61 г, 13.8 ммоля) в безводном МеОН (80 мл) при перемешивании добавляли при 0°С с помощью канюли раствор азида 15k (2,45 г, 6,9 ммоля) в безводном МеОН (20 мл). Смесь перемешивали при КТ в течение 4 ч. Выпаривали МеОН и полученный пенообразный продукт растворяли в смеси диоксан/Н2О (100 мкл/20 мкл) и обрабатывали Вос2О (3,0 г, 13,8 ммоля) и NаНСО3 (2,89 г, 34,5 ммоля) (при необходимости значение рН доводили до 8 с помощью дополнительного количества NаНСО3) и смесь перемешивали при КТ в течение 16 ч. Часть диоксана выпаривали (~50%) и остаток дважды экстрагировали EtOAc. Органический раствор промывали соляным раствором (2×50 мл), сушили и упаривали. Полученный остаток хроматографировали на силикагеле, используя в качестве элюента 20-25%-ный EtOAc в гексане, получая соединение 151 (1,75 г, выход 60%).
1Н ЯМР (CDCl3, 400 МГц) δ 1,27-1,53 (m, 6H), 1,46 (s, 9H), 1,80 (m, 1H), 2,00-2,08 (m, 1H), 2,80 (t, J=12,1 Гц, 1H), 3,34 (d, 14,3 Гц, 1H), 4,17-4,23 (m, 2H), 4,60-4,66 (m, 1 Н), 4,93 (d, J=10,2 Гц, 1 Н), 5,05 (dd, J=17,2 и 1,9 Гц, 1 Н), 5,13 (bs, I H), 5,38-5,43 (m. 1H), 5,74-5,84 (m, 1H), 7,22-7,36 (m, 5H).
Д. К раствору N-Boc-защищенного производного 151 (1,74 г, 4,04 ммоля) в смеси ТГФ/Н2O (75 мл/15 мл) добавляли при перемешивании при 90°С Н2О2 (30% об./мас., 2,05 мл, 16,2 ммоля) и LiOH·H2O (0,34 г, 8,1 ммоля) и раствор перемешивали при 0°С в течение 1 ч. Реакцию прекращали путем добавления Na2SO3 (2,24 г в Н2О, 15 мл, 17,8 ммоля). Значение рН доводили до 4-5 с помощью 10%-ного водного раствора лимонной кислоты и смесь разбавляли EtOAc. Водную фракцию экстрагировали еще один раз EtOAc и органический раствор дважды промывали соляным раствором, сушили и упаривали. Остаток хроматографировали на силикагеле, используя в качестве элюента 20%-ный гексан в EtOAc, в результате чего получали свободную карбоновую кислоту 15g (0,76 г, выход 70%). Это соединение во всех отношениях было идентично соединению из примера 15.
Пример 16
Синтез метилового эфира (2S)-N-Вос-амино-5-оксонон-8-еноевой кислоты (16d):
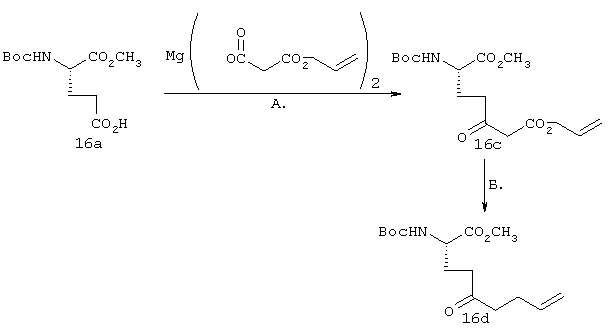
Этот процесс синтеза основан на методе, описанном у Т. Tsuda и др., J. Am. Chem. Soс.,1980, 102,6381-6384.
А. К раствору моноаллилового эфира малоновой кислоты (1,50 г, 10,4 ммоля) в безводном ТГФ (20 мл) в атмосфере N; при -78°С при интенсивном перемешивании в течение 5 мин по каплям добавляли н-Bu2Mg (0,9 М раствор в гексане, 5,8 мл, 5,2 ммоля). Затем густую суспензию перемешивали при КТ в течение 1 ч и упаривали досуха (вакуумный отсос в атмосфере N2). Твердую магниевую соль 16b сушили в вакууме в течение 1 ч.
Производное глутаминовой кислоты 16а сначала смешивали с 1,1’-карбонилидиимидазолом (1,65 г, 10,21 ммоля) в безводном ТГФ и смесь перемешивали при КТ в течение 1 ч для того, чтобы активировать фрагмент свободной кислоты. Затем активированное производное глутаминовой кислоты вносили с помощью канюли в раствор магниевой соли 16b и полученную реакционную смесь перемешивали при КТ в течение 16 ч. Затем ее разбавляли ЕtOАс и органический раствор промывали 0,5 н. охлажденной на льду НСl, промывали соляным раствором, сушили и упаривали. Полученный остаток хроматографировали на силикагеле, используя в качестве элюента 35-40%-ный ЕtOАс в гексане, в результате чего получали соединение 16с (1,85 г, выход 53%).
1Н ЯМР (CDCl3, 400 МГц) δ 1,44 (s, 9H), 1,85-1,95 (m, 1 Н), 2,12-2,22 (m, 1H), 2,58-2,74 (m, 2H), 3,48 (s, 2H), 3,74 (s, 3H), 4,24-4,34 (m, 1H), 4,52 (dm, J=5,7 Гц, 2Н), 5,09 (m, 1H), 5,25 (dm, J=10,2 Гц, 1H), 5,34 (dm, J=17,2 Гц, 1H), 5,91 (m, 1H).
Б. К раствору тeтpaкиc(тpифeнилфocфин)Pd (0) (0,116 г, 5 мол., 0,1 ммоля) в безводном ДМФ (7 мл) при перемешивании добавляли (с помощью шприца в атмосфере N2) сложный диэфир 16с (0,687 г, 2 ммоля) в безводном ДМФ (3 мл). Смесь перемешивали при КТ в течение 3,5 ч. ДМФ выпаривали при пониженном давлении и остаток разбавляли EtOAc (20 мл). ЕtOАс-раствор промывали 0,5 н. охлажденной на льду НСl (5 мл), соляным раствором (10 мл), сушили и упаривали. Остаток хроматографировали на силикагеле, используя в качестве элюента 15-20%-ный EtOAc в гексане, в результате чего получали соединение 16d (0,253 г, выход 42%).
1Н ЯМР (CDCl3, 400 МГц) δ 1,44 (s, 9Н), 1,84-1,94 (m, 1H), 2,08-2,22 (m, 1H), 2,33 (dd, J=14,0 и 7,3 Гц, 2Н), 2,45-2,55 (m, 4H), 3,74 (s, 3Н), 4,28 (bm, 1H), 4,98 (dm, J=10,2 Гц, 1 Н), 5,03 (dm, J=17,2 Гц, 1 Н), 5,00-5,10 (m, 1H), 5,74-5,85 (m, 1H).
Пример 17
Синтез (2S,5R)-N-Boc-2-aмино-5-метилион-8-eнoeвой кислоты (17f)
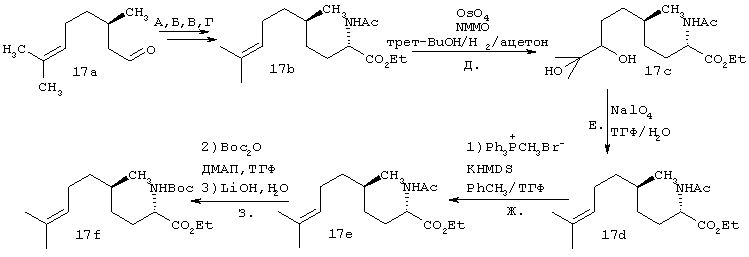
А, Б, В, Г. Имеющийся в продаже (R)-(+)- цитронеллаль 17а сначала превращали в производное аминокислоты 17b, используя те же самые стадии синтеза, которые были ранее описаны в примере 15 для превращения альдегида 15d в промежуточный продукт, представляющий собой аминокислоту 15f.
Д. Соединение 17b (0,675 г, 5,6 ммоля) растворяли в смеси трет-ВuОН/ацетон/Н2О (1:1:1, 18 мл) и помещали в ледяную баню (0°С). Последовательно добавляли NMMO (0,789 г, 6,74 ммоля, 1,2 экв.) и OsO4 (2,5 мас.% в трет-ВuОН, 0,7 мл, 0,067 ммоля, 0,012 экв.) и реакционную смесь перемешивали при КТ в течение 4 ч. Большую часть ацетона удаляли путем выпаривания в вакууме и затем смесь экстрагировали EtOAc. После этого органический слой промывали Н2O и соляным раствором, сушили над безводным MgSO4 и упаривали досуха. С помощью экспресс-хроматографии на колонке с использованием в качестве элюента 1%-ного EtOH в EtOAc получали диол 17с с высокой чистотой с выходом 77% (0,575 г).
Е. К раствору 17с (0,575 г, 1,73 ммоля) в ТГФ/Н2О (1:1, 20 мл) при 0°С добавляли NaIO4 (0,48 г, 2,25 ммоля, 1,3 экв.) и реакционную смесь перемешивали при КТ в течение 3,5 ч. Затем большую часть растворителя, представляющего собой ТГФ, удаляли путем выпаривания в вакууме и оставшуюся смесь экстрагировали EtOAc (2×100 мл). После этого объединенные органические слои промывали 5%-ным водным раствором лимонной кислоты (2×20 мл), 5%-ным водным раствором NаНСО3 (20 мл) и соляным раствором (2×50 мл), затем EtOAc-раствор сушили над безводным MgSO4 и упаривали досуха в вакууме. Промежуточный продукт, представляющий собой альдегид 17d (0,47 г неочищенного продукта) использовали на следующей стадии без дополнительной очистки.
Ж. К раствору Рh3РСН3Вr (925 мг, 2,6 ммоля) в безводном толуоле (15 мл) добавляли KHMDS (0,5 М раствор в толуоле, 5,2 мл, 2,6 ммоля) и образовавшуюся суспензию желтого цвета перемешивали при КТ в течение 30 мин в атмосфере N2. По истечении этого времени суспензию сначала охлаждали до 0°С, добавляли с помощью шприца раствор альдегида 17d (0,47 г, 1,73 ммоля, растворенный в 15 мл безводного ТГФ) и смеси давали нагреться до КТ. После перемешивания при КТ в течение 1 ч большую часть ТГФ удаляли выпариванием в вакууме, к смеси добавляли EtOAc (100 мл) и органический слой промывали Н2О (30 мл), 5%-ным водным раствором NaHCO3 (30 мл) и соляным раствором (30 мл). Затем ЕtOАс-раствор сушили над безводным MgSO4 и упаривали досуха в вакууме. После очистки с помощью экспресс-хроматографии на колонке на силикагеле с использованием в качестве элюента смеси гексан:ЕtOАс (3:2) выделяли чистое соединение 17е с выходом 63% (0,29 г) для двух последних стадий.
Для получения соединения 17f осуществляли гидролиз сложного этилового эфира и одновременную замену N-ацетильной защитной группы на группу Воc в промежуточном продукте 17е, используя такую же процедуру, которая описана для превращения соединения 15f в соединение 15g (17f, 310 мг, количественный выход). 1Н ЯМР (СDСl3, 400 МГц): δ 0,88 (d, J=6,4 Гц, 3Н), 1,18-1,28 (m, 2Н), 1,35-1,48 (m, 3H), 1,45 (s, 9Н), 1,64-1,74 (m. 1H). 1,81-1,89 (m, 1H), 1,94-2,12 (m, 2H), 4,28 (bd, J=~3,2 Гц, 1Н),4,93 (dm, J=11,1 Гц, 1Н), 5,00 (dm, J=16,8 Гц, 1 Н), 5,74-5,84 (m, 1H).
Пример 18
Синтез N-Вос-О-аллил-(L)-треонина (18d)
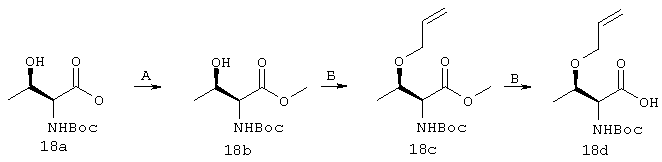
А. Вос-(L)-треонин 18a (500 мг, 2,28 ммоля) частично растворяли в смеси CH2Cl2/MeOH (8 мл/0,5 мл соответственно) при 0°С. Медленно добавляли раствор диазометана в диэтиловом эфире до тех пор, пока не получали выраженную желтую окраску, указывающую на присутствие избытка диазометана. После выпаривания растворителей получали сложный метиловый эфир 18b в виде мутного масла белого цвета (0,534 г).
Б. Затем промежуточный продукт 18b (311 мг, 1,33 ммоля) растворяли в безводном диэтиловом эфире (8 мл), добавляли Ag2O (341 мг, 1,47 ммоля) и активированные непосредственно перед этим 4А молекулярные сита (1 г). В завершение в реакционную колбу добавляли аллилйодид (134 мкл, 1,47 ммоля) и смесь перемешивали при температуре дефлегмации. Через 20 ч и 30 мин добавляли еще две порции аллилйодида (каждый раз по 45 мкл, 0,50 ммоля) и продолжали перемешивание в общей сложности в течение 36 ч. Затем смесь фильтровали через целит и очищали с помощью экспресс-хроматографии на колонке на силикагеле, используя в качестве элюента смесь ЕtOАс/гексан (1:4), в результате чего получали 73 мг (выход 27%) соединения 18с в виде прозрачного масла.
1Н ЯМР (CDCl3, 400 МГц): δ 1,21 (d, J=6,0 Гц, 3Н), 1,45 (s, 9H), 3,75 (s, 3H), 3,82-3,87 (m, 1H), 3,99-4,07 (m, 2H), 4,29 (dd, J=9,5 и 2,5 Гц, 1H), 5,14 (dm, J=10,5 Гц, 1Н), 5,21 (dm, J=17,2 Гц, 1H), 5,75-5,84 (m, 1H).
В. Соединение, представляющее собой сложный эфир 18с (99 мг, 0,362 ммоля) растворяли в смеси ТГФ/МеОН/Н2О (2:1:1,4 мл) и добавляли LiOH·H2O (61 мг, 1,45 ммоля). Раствор перемешивали при КТ в течение 2 ч и затем подкисляли с помощью 1 н. раствора НСl до достижения значения рН ~3, после чего растворители удаляли в вакууме. Образовавшееся соединение 18d в виде масла использовали без дополнительной обработки для синтеза макроциклических ингибиторов.
Пример 19
Синтез (2S,38)-N-Вос-2-амино-3-(меркаптоаллил)бутановой кислоты (19е)
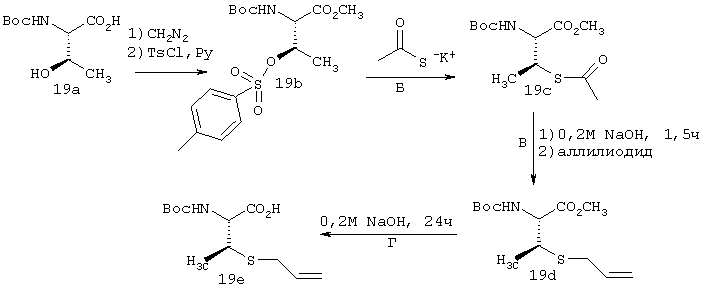
А. Соединение 19а (9,1 ммоля) растворяли в пиридине (5 мл) и раствор охлаждали до 0°С в ледяной бане, добавляли небольшими порциями тозилхлорид (2,3 г, 11,8 ммоля, 1,3 экв.) и рекационную смесь перемешивали при КТ в течение 24 ч. После истечения этого периода времени реакционную смесь распределяли между диэтиловым эфиром (300 мл) и Н2О (100 мл). Затем эфирный слой промывали 0,2 н. раствором НСl (6×100 мл) и соляным раствором (100 мл), сушили над безводным MgSO4, фильтровали и концентрировали досуха в вакууме. После очистки неочищенного продукта с помощью экспресс-хроматографии на колонке с использованием в качестве элюента смеси гексан/ЕtOАс (градиент от 8:2 до 7:3) выделяли производное тозила 19b с выходом 85% (3,05 г).
Б. К раствору промежуточного продукта 19b (775 мг, 2 ммоля) в безводном ДМФ (2,5 мл) добавляли тиоацетат калия (365 мг, 3,2 ммоля, 1,6 экв.) и реакционную смесь перемешивали при КТ в течение 24 ч. Затем большую часть ДМФ выпаривали в вакууме и оставшуюся смесь распределяли между EtOAc и Н2О. Водный слой повторно экстрагировали EtOAc, объединенные органические слои промывали соляным раствором, сушили над безводным MgSO4 и упаривали досуха. Путем очистки неочищенного продукта с помощью экспресс-хроматографии на колонке с использованием в качестве элюента смеси гексан/ЕtOАс (в соотношении 4:1) выделяли соединение 19с с выходом 80% (465 мг).
В. К раствору сложного тиоэфира 19с (465 мг) в смеси Н2О/ЕtOН (соотношение 3:5, 8 мл) добавляли 0,2 М водный раствор NaOH (2,4 мл) и смесь перемешивали при КТ в течение 1,5 ч. Затем добавляли аллилиодид (0,292 мл, 3,2 ммоля, 2 экв.) и продолжали перемешивание при КТ еще в течение 30 мин. Реакционную смесь концентрировали до половины ее первоначального объема и затем экстрагировали EtOAc. Водный слой подкисляли до значения рН~3 с помощью водного 0,5 н. раствора НСl и повторно экстрагировали EtOAc. Объединенные органические слои промывали соляным раствором, сушили над безводным MgSO4 и упаривали досуха в вакууме. Реакционная смесь содержала по крайней мере четыре продукта; все эти продукты выделяли с помощью экспресс-хроматографии на колонке на силикагеле, используя в качестве элюента смесь гексан/ЕtOАс (градиент от 9:1 до 3:1). Структура наименее полярного соединения (ТСХ Rf=0,68 в смеси гексан/ЕtOАс, 4:1) соответствовала требуемому продукту 19d (83 мг, выход 18%).
1Н ЯМР (CDCl3, 400 МГц): δ 1,24 (d, J=7,0 Гц, 3Н), 1,46 (s, 9H), 3,13-3,19 (m, 2Н), 3,24-3,29 (m, 1H), 3,77 (s, 3Н), 4,50 (dd, J=8,6 и 3,8 Гц, 1Н), 5,12 (d, J=12,4 Гц, 1Н), 5,15 (dd, J=18,4 и 1,3 Гц, 1Н), 5,22 (bd, J=7,6 Гц, 1Н), 5,75-5,85 (m, 1Н).
Г. Раствор сложного метилового эфира 19d (83 мг, 0,287 ммоля) в смеси МеОН/Н2O (3:1, 4 мл) смешивали с водным раствором NaOH (0,2 н., 1,3 мл, 0,26 ммоля) в течение 24 ч при КТ и в течение 1 ч при 40°С. Реакционную смесь подкисляли холодным водным раствором НСl (0,5 н. НСl, при 0°С, рН 4-5), МеОН удаляли в вакууме и оставшуюся водную смесь экстрагировали EtOAc. Органический раствор сушили над MgSO4 и упаривали досуха, получая соединение 19е. Соединение 19е использовали для заключительного синтеза ингибиторов без какой-либо дополнительной очистки.
Пример 20
Синтез (S)-N-Вос-2-амино-3-метил-3-(1-меркапто-4-бутенил)бутановой кислоты (20с)
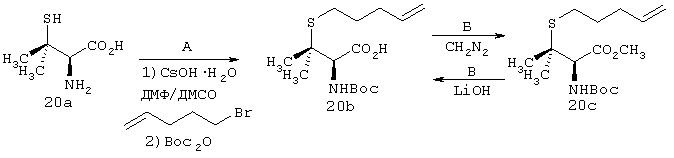
А. L-пеницилламин 20а (448 мг, 3 ммоля) растворяли в смеси ДМФ/ДМСО (соотношение 5:1,6 мл), добавляли 4-бромпентен (0,46 мл, 4,5 ммоля, 1,5 экв.) и CsOH•H2O (1,0 г, 6 мл, 2 экв.) и реакционную смесь перемешивали при КТ. Через 24 ч к реакционной смеси добавляли Вос2О (820 мг, 3,75 ммоля, 1,25 экв.) и продолжали перемешивание еще в течение 12 ч. После этого ДМФ удаляли в вакууме, оставшуюся смесь разбавляли холодным 0,5 н. водным раствором НСl, доводя до рН~4-5 и затем экстрагировали ЕtOАс (2×50 мл). Органический слой промывали соляным раствором (2х), сушили над безводным MgSO4 и упаривали досуха, получая неочищенную карбоновую кислоту 20b.
Б. Очистка соединения 20b оказалась трудной, поэтому неочищенный продукт сначала обрабатывали диазометаном для получения соответствующего сложного метилового эфира 20с, а потом очищали с помощью экспресс-хроматографии на колонке, используя в качестве элюента смесь гексан/ЕtOАс (9:1), в результате чего получали 190 мг (выход 20%) чистого сложного метилового эфира 20с.
1Н ЯМР (400 МГц, CDCl3): δ 1,35 (s, 3H), 1,37 (s, 3Н), 1,44 (s, 9H), 1,59-1,67 (m, 2Н), 2,11-2,17 (m, 2H), 2,51-2,60 (m, 2H), 3,74 (s, 3Н), 4,29 (d, J=8,6 Гц, 1Н), 4,98 (dm, J=10,5 Гц, 1H), 5,03 (dm, J=19 Гц, 1H), 5,35 (bd, J=7 Гц, 1H), 5,72-5,83 (m, 1H).
В. После этого сложный эфир растворяли в смеси ТГФ/МеОН/Н2O (2:2:1, 5 мл), добавляли LiOH•H2O (50 мг, 2,0 ммоля, 2 экв.) и реакционную смесь перемешивали при 40°С в течение 4 ч для того, чтобы гидролизовать сложный эфир 20с до кислоты 20b. Реакционную смесь подкисляли 0,5 н. НСl до рН 4-5, ТГФ и МеОН выпаривали полностью и оставшийся водный раствор экстрагировали ЕtOАс. ЕtOАс-слой сушили над безводным MgSO4 и упаривали досуха, получая соединение 20b, которое использовали в последующем синтезе макроциклических ингибиторов без дополнтельной очистки.
Промежуточные ациклические дипептиды и трипептиды
Общий метод проведения реакций сочетания в растворе и конкретные примеры их осуществления описаны в WO 00/09543 и WO 00/09558.
Эти методы были использованы для синтеза промежуточных дипептидов 26с, 30a и трипептидов 23а, 24а, 31а, 32а и 33а.
Пример 21
Синтез ациклического трипептида 21е
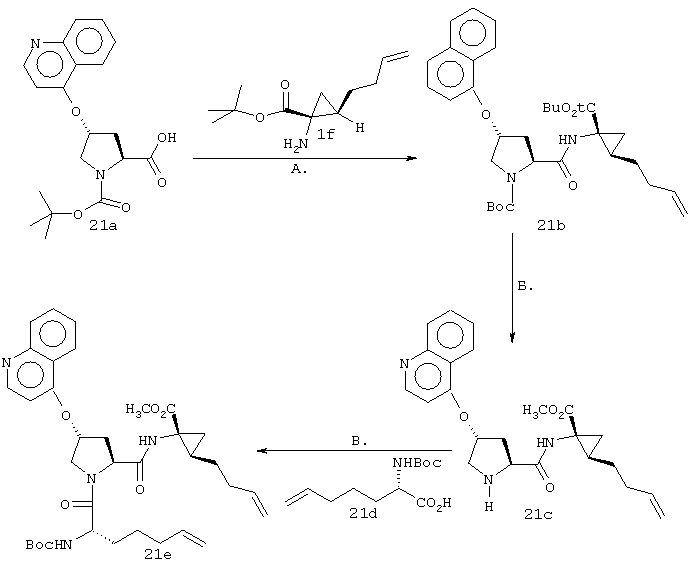
А. К раствору, содержащему производное пролина 21 а (полученное из имеющегося в продаже Вос-4(R)-гидроксипролина и 4-хлорхинолина согласно методу, описанному в WO 00/05543 и WO 00/09558) (1,32 г, 3,68 ммоля) и неочищенный гомоаллил АЦКК 1f (-3,35 ммоля) в СН2Сl2 (10 мл), последовательно добавляли NMM (1,21 мл, 10,05 ммоля) и ГАТУ (1,53, 4,02 ммоля) и суспензию перемешивали при КТ в течение 18 ч. По истечение этого периода времени растворитель выпаривали и неочищенную реакционную смесь повторно растворяли в EtOAc (30 мл). Раствор промывали 5%-ным водным раствором NaHCO3 (2×10 мл), соляным раствором (10 мл), сушили над MgSO4 и упаривали. Неочищенный продукт очищали с помощью хроматографии на силикагеле, используя в качестве элюента 8%-ный диэтиловый эфир в EtOAc, в результате чего получали требуемый диастереомер соединения 21b с выходом 20% (абсолютное стерехимическое строение не определяли).
1Н ЯМР (CDCl3, 400 МГц): δ 0,93 и 1,01 (t, J=8,3 Гц, 1Н, ротамеры в соотношении 3:7), 1,14-1,35 (m, 2H), 1,44 (s, 9H), 1,45 (s, 9H), 1,50-1,82 (m, 4H), 2,08-2,24 (m, 2H), 2,32 (bs, 0,7Н), 2,63 (bs, 0.75H), 2,93 (bs, 0.75H), 3,16 (m, 0.25H), 3,77 (bs, 1,5H), 3,88 (bs, 0,5Н), 4,4-4,55 (m, 1H), 4,98 (d, J=10,2 Гц, 1H), 5,03 (dd, J=17,2 и 1,6 Гц, 1Н), 5,24 (bs, 1H), 5,75-5,88 (m, 1H), 6,57 и 6,78 (2bs, 1H, 2 ротамера). 7,42-7,58 (m, 3H), 7,63-7,73 (m, 2Н), 8,04 (d, J=8,3 Гц, 1H), 8,11 (d, J=8,3 Гц, 1H), 8,74 (d, J=5,1 Гц, 1H).
Б. К раствору дипептида 21b (137 мг, 0,248 ммоля) в безводном СН2Сl2 добавляли раствор НСl в диоксане (4М, 4 мл) и смесь перемешивали при КТ в течение 1,5 ч. Затем растворитель выпаривали и остаток сушили в глубоком вакууме, получая свободную аминокислоту. Смесь растворяли в смеси диэтиловый эфир /МеОН (3 мкл/2 мкл) и обрабатывали небольшим избытком диазометана, растворенного в диэтиловом эфире. Через 30 мин избыток диазометана удаляли путем добавления НСl (4М раствор в диоксане) и смесь упаривали досуха, получая гидрохлорид соединения 21с, который использовали на следующей стадии без какой-либо очистки.
В. К суспензии неочищенного дипептида 21с (0,23 г, 0,48 ммоля) в СН2Сl2 (25 мл) последовательно добавляли при перемешивании (2S)-N-Вос-аминогепт-6-еновую кислоту 21d (0,151 г, 0,62 ммоля), NMM (210 мкл, 1,91 ммоля) и ГАТУ (0,236 г, 0,62 ммоля) и смесь перемешивали при КТ в течение 16ч (при необходимости через 1 ч значение рН доводили до ~8 с помощью NNM). СН2Сl2 выпаривали, остаток растворяли в EtOAc (50 мл) и органический раствор промывали 5% -ным NаНСО3 (2×20 мл), соляным раствором (2×20 мл), сушили и упаривали. Полученное неочищенное соединение хроматографировали на силикагеле (50 мл, 2% EtOH/EtOAc), получая соединение 21 с (0,139 г, выход 46%).
1Н ЯМР (CDCl3, 400 МГц, соотношение ротамеров 6:1) химические сдвиги основного ротамера δ 1,21-1,27 (m, 1H), 1,36 (s, 9Н), 1,45-1,81 (4m, 7H), 2,20-2,22 (m, 4H), 2,28-2,37 (m, 1H), 2,90-2,99 (m, 1H), 3,66 (s, 3H), 3,94-3,98 (m, 1H), 4,29 (bd, J=9,9 Гц, 1H), 4,46-4,50 (m, 1H), 4,81 (dd, J=8,3 и 5,4 Гц, 1H), 4,92-5,06 (m, 4H), 5,16 (d, J=8,3 Гц 1 Н), 5,37 (m, 1 Н), 5,70-5,84 (m, 2H), 6,82 (d, J=5,1 Гц, 1 Н), 7,47-7,55 (m, 2H), 7.71 (dt, J=7,0 и 1,3 Гц, 1H), 8,03 (d, J=8,6 Гц, 1H), 8,17 (d, J=8,0 Гц, 1H), 8,78 (d. J=5,1 Гц, 1H).
Макроциклические пептиды
Пример 22
Общая процедура макроциклизации с помощью олефинового метатезиса
Во всех случаях трипептидный диен растворяли в СН2Сl2 в концентрации 0,01 М и раствор деоксигенировали путем барботирования аргоном (~1 ч для объема 500 мл). Добавляли раствор катализатора (5-30 мол. %, растворенный в небольшом количестве дегазированного СH2Сl2) и реакционную смесь выдерживали при температуре дефлегмации до тех пор, пока весь исходный продукт не превратится в конечный продукт(ы) по данным анализа с помощью ТСХ и ЖХВР. Затем неочищенные реакционные смеси концентрировали практически досуха и фильтровали через короткую подушку из силикагеля, элюируя сначала с помощью СН2Сl2 для удаления большей части катализатора, а затем с помощью EtOAc для элюирования всех макроциклических продуктов(а) (в основном в виде отдельного стереомера). Неочищенные продукты каждой реакции анализировали с помощью хиральной ЖХВР на CHIRALCEL OJ-R-колонке (полученной от фирмы Chiral Technologies Inc, ⊘ 0,46×15 см), используя изократическую смесь растворителей 70% Н2О+0,06% ТФК - 30% СН3СN+0,06% ТФК при 205 нм. Основные макроциклические продукты были полностью охарактеризованы с помощью данных, полученных с использованием 1Н, COSY, TOCSY и ROESY ЯМР-анализов, для подтверждения их структуры и стереохимического строения.
Пример 23
Синтез макроциклического промежуточного продукта (23b)
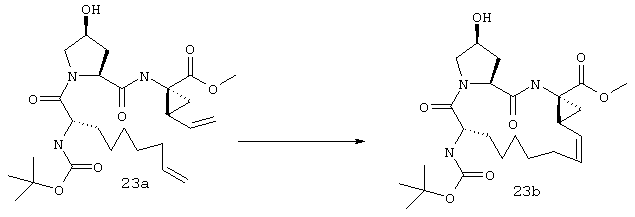
Раствор диена 23а (4,0 г, 7,88 ммоля) в безводном СН2Сl2 (800 мл, фирма Aldrich. безводный) деоксигенировали путем барботирования Аr в течение 2 ч. Затем добавляли катализатор Ховейда (262 мг, 0,434 ммоля, 5,5 мол.%) в виде твердого вещества и реакционную смесь кипятили с обратным холодильником в атмосфере подаваемого из баллона Аr. Через 28 ч выпаривали раствор красно-оранжевого цвета до получения аморфного твердого вещества и затем очищали с помощью экспресс-хроматографии на колонке на силикагеле. В качестве начальной системы растворителей использовали 10%-ный EtOAc в СН2Сl2. Как только заканчивали элюирование катализатора из колонки, растворитель заменяли на чистый EtOAc. Процесс элюирования катализатора из колонки был виден благодаря его цвету. Макроциклический продукт 23b выделяли в виде бесцветной пены и его повторно растворяли в смеси СН2Сl2/гексан (~1:2). После выпаривания растворителя получали продукт в виде порошка белого цвета (3,362 г, выход 89%).
1Н ЯМР (CDCl3, 400 МГц): δ 1,20-1,50 (m, 6H), 1,43 (s, 9H), 1,53 (dd, J=9,5 и 5,4, 1Н), 1,61-1,70 (т, 1Н), 1,76-1,90 (m,2H), 2,05-2,26 (m, 4H), 2,45 (d, J=14,3, 1H), 3,67 (s, 3H), 3,71 (d, J=11,1, 1Н), 3,90 (dd, J=11,1 и 4,3, 1Н), 4,43-4,53 (m, 2H), 4,76 (d, J=8,6, 1Н), 4,86 (bd, J=9,8, 1H), 5,20-5,23 (m, 2H), 5,57 (dt, J=7,0 и 9,8, 1Н), 7,32 (bs, 1H).
Пример 24
Синтез макроциклического промежуточного продукта (24b):

Раствор диена 24а (2,76 г, 3,82 ммоля) в безводном CH2Cl2 (600 мл, безводный) деоксигенировали путем барботирования Аr в течение 1,5 ч. С помощью канюли добавляли раствор катализатора Ховейда (117 мг, 0,19 ммоля, 0,05 экв.) в безводном и дегазированном CH2Cl2 (8 мл) и реакционную смесь перемешивали при температуре дефлегмации в атмосфере Аr, подаваемого из баллона. Через 20 ч реакционная смесь прореагировала приблизительно на 50%, в это время добавляли вторую порцию катализатора (117 мг) и продолжали перемешивание еще в течение 16 ч. Затем раствор концентрировали до объема ~100 мл, наносили на верхнюю поверхность подушки из силикагеля (6×10 см) и прежде всего выделяли катализатор путем элюирования с помощью СН2Сl2. Соединение 24b вымывали из подушки из силикагеля с помощью 3%-ного МеОН в EtOAc и повторно очищали с помощью экспресс-хроматографии на колонке, используя в качестве элюента смесь EtOAc/гексан (2:1), в результате чего получали твердое вещество белого цвета со слабым оливковым оттенком (выход 70%) (1,85 г, чистота 94% по данным анализа с помощью ЖХВР).
1Н ЯМР (400 МГц, ДМСО-d6) δ 8,69 (s, 1H), 8,13 (d, J=9,2 Гц, 1H). 7,50-7,44 (m. 2H), 7,17 (dd, J=9,2, 2,2 Гц, 1H), 7,04 (d, J=6,4 Гц, 1H), 5,60-5,56 (m, 1H), 5,52 (dd, J=9,2 Гц, 1H), 5,25 (dd, J=9,2 Гц, 1H), 4,59 (d. J=11 Гц, 1H), 4,44 (dd, J=9,2 Гц. 1H). 4,05-3,98 (m, 1H), 3,94 (s, 3Н), 3,92 (s, 3H), 3,89-3,82 (m, 1H), 3,55 (s, 3Н), 2,64-2,53 (m, 1 Н), 2,46 (d, J=7,3 Гц, 1Н), 2,40-2,31 (m, 1Н), 2,21 (dd, J=8,9 Гц, 1Н), 1,78-1,65 (m, 2H), 1,55 (dd, J=4,8 Гц, 1Н), 1,485 (dd, J=4,8 Гц, 1Н), 1,41-1,30 (m, 7H), 1,16 (s, 9H). МС; es+: 795,4 (M+Н)+.
Пример 25
Синтез соединений 202 и 203 (таблица 2)
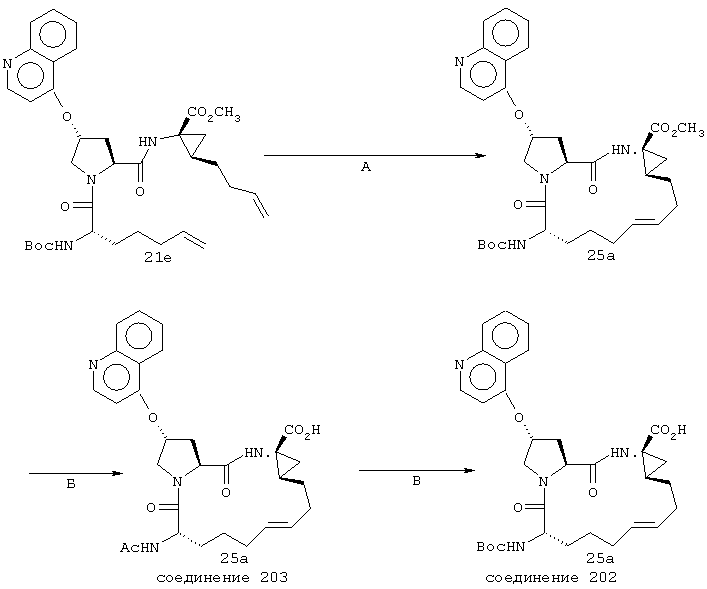
А. Диеновое производное 21e (0,130 г, 0,205 ммоля) подвергали циклизации с использованием каталитических количеств дихлорида бис(трициклогексилфосфин)бензилиденрутения IV (катализатор Грубба, выше) (52 мг, 0,064 ммоля) в СН2Сl2 (60 мл) при температуре дефлегмации в течение 2 ч и после хроматографии на силикагеле (50 мл, 3%-ный EtOH/EtOAc) получали соединение 25а (60,1 мг, выход 48%).
1Н ЯМР (CDCl3, 400 МГц) δ 1,22-1,30 (m, 2H), 1,35 (s, 9H), 1,44-2,35 (m, 13H), 3,07-3,14 и 3,16-3,24 (2m, 1H, соотношение ротамеров 1:3), 3,69 (s, 3Н), 3,96-4,04 (m, 1H0, 4,42-4,50 (m, 1H), 4,95-5,04 (m, 1H), 5,05-5,15 (m, 1H), 5,20-5,30 (m, 1H), 5,55-5,65 (m, 1H), 6,75-6,79 (2d, J=5,4 Гц, 1H, соотношение ротамеров 1:3), 7,36 (s, 1H). 7,46-7,50 (m, 1H), 8,03 (d, J=8,3 Гц, 1H), 8,13 и 8,17 (2d, J=8,0 Гц, 1H, соотношение ротамеров 1:3), 8,77 (d, J=5,1 Гц, 1H).
В. Сложноэфирный фрагмент макроциклического соединения 25а (0,0156 г, 0,026 ммоля) гидролизовали с помощью LiOH•H2O (8,7 мг, 0,206 ммоля) в смеси ТГФ/МеОН/Н2O (4 мл /2 мл /2 мл ). Неочищенный продукт очищали с помощью ЖХВР с обращенной фазой на С 18-колонке типа Whatman (PaKTisil 10,ODS3) 50/2.4 см, используя градиент растворителя от 5%-ного водного раствора СН3СМ до 100%-ного СН3СN, в результате чего получали чистое соединение 202 в виде аморфного твердого вещества белого цвета (11,8 мг).
1Н ЯМР (ДМСО, 400 МГц): δ 1,12 (s, 9H), 1,20-1,24 (m, 2H), 1,32-1,40 (m, 3H), 1,58-1,62 (m, 2H), 1,68-1,78 (m, 3H), 1,95-2,02 (m, 1H), 2,08-2,18 (m, 2H), 2,42-2,59 (m, 2H), 3,97-4,00 (bd, J=9,8 Гц, 2H), 4,47 (t, J=8,6 Гц, 1H), 4,58 (d, J=11,8 Гц, 1H), 5,22-5,29 (m, 1H), 5,46-5,54 (m, 1H), 5,66 (s, 1H), 7,12 (d, J=6,0 Гц, 1H), 7,49 (d, J=3,5 Гц, 1H), 7,68 (dd, J=7,3 Гц, 1Н), 7,98 (dd, J=7,0 Гц, 1H), 8,08 (d, J=8,3 Гц, 1Н), 8,21 (s, 1Н), 8,35 (d, J=8,3 Гц, 1Н), 9,08 (d, J=5 Гц, 1Н).
С. Макроциклическое соединение 25а (20 мг, 0,033 ммоля) в безводном СН2Сl2 (1 мл) перемешивали в присутствии 4 М НСl в диоксане (5 мл) в течение 1 ч. Смесь упаривали и осторожно сушили. Остаток повторно растворяли в СН2Сl2/ДМФ (3 мл /1 мл) и обрабатывали NMM (14,5 мкл, 0,132 ммоля) и уксусным ангидридом (7,0 мкл, 0,073 ммоля) и перемешивали при КТ в течение 14 ч. Смесь упаривали и сушили в глубоком вакууме. Затем остаток растворяли в смеси ТГФ/МеОН/Н2O (4 мл/2 мл/2 мл) и перемешивали в течение ночи с LiOH·2H2O (11 мг, 0,264 ммоля). Остаток, выделенный после подкисления до значения рН 3 с помощью 1н. охлажденной на льду НСl, очищали с помощью ЖХВР с обращенной фазой на колонке С 18, используя градиент растворителя 0-40% водного раствора СН3СN (0,06% ТФК), в результате чего выделяли чистое соединение 203 в виде аморфного твердого вещества белого цвета (12 мг).
1Н ЯМР (50 мМ Nа2РO4-буфер, рН=6,0, 600 МГц): δ 1,22-1,27 (m, 2H), 1,38-1,43 (m, 2H), 1,58-1,64 (m,2H), 1,67-1,76 (m, 2H), 1,77-1,84 (m, 1H), 1,92-1,99 (m, 1H), 2,22-2,08 (m, 1H), 2,12-2,27 (m, 1H), 2,22-2,27 (m, 1H), 2,60-2,67 (m, 1H, Pro-β’), 2,83-2,89 (m, 1H, Pro-β), 4,32 (dd, J=12,1 и 3,5 Гц, 1H, Pro-δ’), 4,41 (dd, J=12,1 и 7,3 Гц, 1H), 4,56 (bd, J=8,0 Гц, 1H, Pro-δ), 4,62 (dd, J=8,9 Гц, 1H, Pro-α), 5,40-5,46 (m, 1H), 5,55-5,61 (m, 1H), 5,73 (bs, 1H, Pro-γ), 7,41 (d, J=6,3 Гц. 1H), 7,64 (bs, 1H, АЦКК-NH), 7,80 (dd, J=7,9 Гц, 1 Н), 8,03 (dd, J=8,0 Гц, 1Н), 8,07 (d, J=9,5 Гц, 1H), 8,16 (d, J=7 Гц, 1H, AcNH). 8,36 (d, J=8,3 Гц, 1H), 8,90 (d, J=6,0 Гц, 1H).
Пример 26
Синтез соединения 508 (таблица 5)
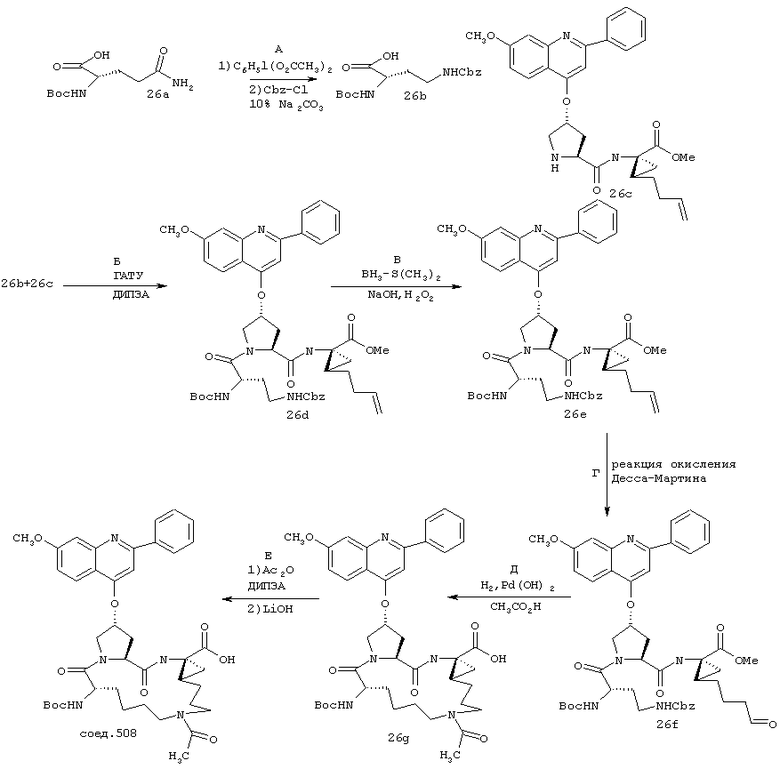
А. Раствор Вос-защищенного L-глутамина 26а (4,93 г, 20 ммолей) и диацетата йодбензола (7,73 г, 24 ммоля, 1,2 экв.) в смеси ЕtOАс/СН3СN/Н2О (2:2:1, 60 мл) перемешивали при 16°С в течение 1 ч и при 20°С в течение 3 ч. Затем реакционную смесь разбавляли Н2О (20 мл), растворители ЕtOАс и CH3CN удаляли в вакууме и оставшуюся водную смесь экстрагировали диэтиловым эфиром (3×50 мл) и ЕtOАс (50 мл) для удаления большей части примесей. Затем водный слой (содержащий промежуточный продукт в виде амина) концентрировали досуха, оставшийся продукт повторно растворяли в 10%-ном Nа2СО3 (30 мл), охлаждали до 0°С в ледяной бане и медленно добавляли (~10 мин) раствор бензилхлорформиата (3,3 мл, 20,4 ммоля, 1,02 экв.) в диоксане (40 мл). Реакционную смесь перемешивали при 0°С в течение 1 ч и при КТ в течение 2 ч.
Затем смесь разбавляли Н2O (50 мл), экстрагировали холодным (~5°С) диэтиловым эфиром (3×50 мл), подкисляли с помощью 4 М НСl до значения рН 3-4 и экстрагировали EtOAc (3×50 мл). Объединенные органические слои сушили над безводным MgSО4 и упаривали досуха в вакууме. Неочищенный продукт очищали с помощью экспресс-хроматографии на колонке, используя в качестве элюента смесь ЕtOАс/гексан/АсОН (7:2,9:0,1), в результате чего получали соединение 26b с общим выходом 43% (3,04 г).
Б. Промежуточный продукт, представляющий собой дипептид 26с (250 мг, 0,41 ммоля), соединение 26b (171 мг, 0,49 ммоля, 1,2 экв.) и ГА ТУ (185 мг, 0,49 ммоля, 1,2 экв.) растворяли в CH2Cl2 (6 мл) и добавляли ДИПЭА (0,29 мл, 1,62 ммоля, 4 экв.). Реакционную смесь перемешивали при КТ в течение 14 ч, после чего СН2Сl2 выпаривали в вакууме и неочищенный продукт повторно растворяли в EtOAc. ЕtOАс-раствор промывали водным 5%-ным раствором NаНСО3 и соляным раствором, сушили над безводным MgSO4 и упаривали досуха. После очистки неочищенного продукта с помощью экспресс-хроматографии на колонке с использованием в качестве элюента смесь EtOAc/гексан (4:1) получали соединение 26d с выходом 98% (338 мг).
В. Раствор соединения 26d (335 мг, 0,394 ммоля) в ТГФ (5 мл) охлаждали до 0°С и добавляли раствор ВН3 в диметилсульфиде (0,12 мл 10 М раствора, 1,2 ммоля, 3 экв.). Реакционной смеси давали нагреться до КТ и ее перемешивали в течение 1 ч. Затем ее снова охлаждали до 0°С, после чего медленно в течение 15 мин добавляли водный раствор NaOH (0,8 мл 2,5 М раствора, 1,97 ммоля, 5 экв.), а затем медленно добавляли (~15 мин) водный раствор H2O; (0,8 мл 8,8 М раствора, 6,9 ммоля, 17,5 экв.). Реакционной смеси давали нагреться до КТ и ее перемешивали в течение 1 ч. После этого реакционную смесь подкисляли до рН ~4 для того, чтобы нейтрализовать избыток ВН3, затем добавляли водный раствор NаНСО3, доводя значение рН до ~9-10, удаляли в вакууме ТГФ и неочищенный продукт распределяли между H2O и EtOAc. Водный слой повторно экстрагировали EtOAc, объединенные органические слои промывали соляным раствором, сушили над безводным MgSO4 и упаривали досуха в вакууме. Неочищенный продукт очищали с помощью экспресс-хроматографии на колонке, используя в качестве элюента смесь ЕtOАс/гексан/NН4OН (8:2:0.5), в результате чего получали чистое соединение 26е с выходом 57% (192 мг).
Г. К раствору соединения 26е в СН2Cl2(8 мл) добавляли перйодинат Десса-Мартина (195 мг, 97%, 0,33 ммоля, 1,5 экв.) и реакционную смесь перемешивали при КТ в течение 1,5 ч. Реакцию прекращали путем добавления водного раствора Nа2S2O3 (3 мл 5%-ного раствора), затем добавляли насыщенный водный раствор NaHCO3(5 мл) и смесь перемешивали при КТ в течение 15 мин. В завершение неочищенную реакционную смесь экстрагировали ЕtOАс, органический слой промывали водным 5%-ным раствором NaHCO3 и соляным раствором, сушили над безводным MgSO4 и упаривали в вакууме, получая 188 мг альдегида 28f, который использовали на следующей стадии без дальнейшей очистки.
Д. Раствор, содержащий соединение 26f(188 мг, 0,22 ммоля), СН3СО2Н (38 мкл) и Рd(ОН)2 (25 мг) в этаноле (5 мл), перемешивали при КТ в атмосфере Н2 при атмосферном давлении в течение 16 ч. По истечении этого периода времени в сосуд добавляли дополнительное количество газообразного H2, Pd(OH)2 (180 мг) и СН3СO2Н (154 мкл) и продолжали перемешивание еще в течение 24 ч. Затем смесь фильтровали и растворитель выпаривали досуха, неочищенный макроциклический продукт очищали с помощью экспресс-хроматографии на колонке, используя в качестве элюента смесь СНСl3/МеОН/АсОН (10:2:1), в результате чего получали соединение 26g c выходом ~30% (48 мг).
Е. Смесь, содержащую соединение 26g (22 мг, 0,031 ммоля), ДИПЭА (27 мкл, 0,155 ммоля, 5 экв.) и уксусный ангидрид (8,7 мкл, 0,093 ммоля, 3 экв.) в СН2Сl2 (5 мл), перемешивали при КТ в течение 16 ч. Затем СH2Сl2 удаляли в вакууме, добавляли смесь ТГФ/МеОН/Н2О (2:2:1, 5 мл) и LiOH·2H2O (13 мг, 0,31 ммоля, 10 экв.) и осуществляли реакцию гидролиза в течение 68 ч при КТ и в течение 2 ч при 50°С. Затем реакционную смесь подкисляли (рН ~4) и очищали с помощью ЖХВР с обращенной фазой, получая конечное соединение 508 (~6 мг, выход ~26% для последних 2 стадий).
1Н ЯМР (ДМСО, 400 МГц) для соединения 508 (смесь ротамеров, выявленная на основе данных COSY, TOCSY и ROESY ЯМР-анализа): δ 1,18 (s, 9H), 1,09-1,85 (перекрывающийся m, 11Н), 1,95 (s, 3Н), 2,30 (m, 1H), 2,63 (m, 1H), 3,18-4,14 (перекрывающийся m, 6H), 3,96 (s, 3Н), 4,44 (m, 1H), 4,62 и 4,69 (2d, J=11,8 Гц, 1H, ротамеры), 5,82 (bs, 1H), 7,20 (m, 2Н), 7,53 (bs, 1H). 7,67 (bs, 4H), 8,19 (bs, 3Н), 8,61 (s, 1H).
Пример 27
Синтез насыщенного макроциклического производного (27а)
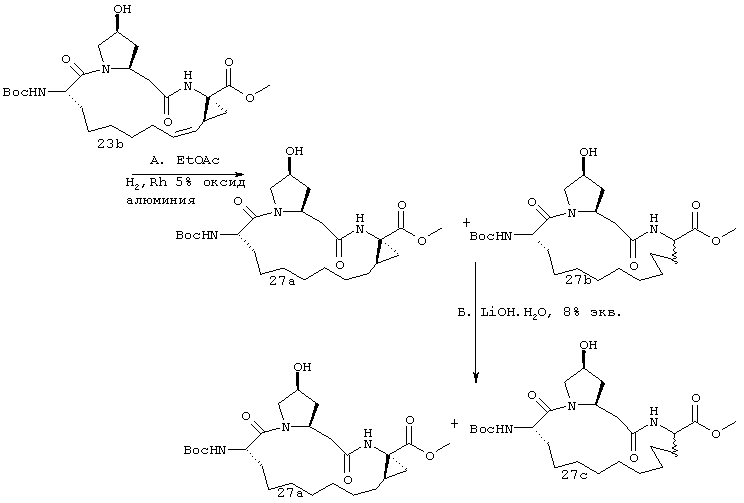
А. Ненасыщенное макроциклическое производное 23b (3,50 г, 7,30 ммоля) растворяли в ЕtOАс (30 мл) и добавляли 700 мг (20 мас.%) 5%-ного Rh на оксиде алюминия. Смесь перемешивали в атмосфере газообразного Н2 при атмосферном давлении и при КТ в течение 1,5 ч. По истечение этого периода времени анализ с помощью ЖХВР подтвердил, что исходный продукт полностью превратился в два продукта, в требуемый продукт 27а и минорный продукт (8% от общей массы), который позднее был идентифицирован как соединение 27b, образующийся в результате размыкания циклопропанового кольца. Реакционную смесь фильтровали и концентрировали, получая твердое вещество светло-зеленого цвета (3,47 г). Твердое вещество подвергали совместному упариванию с ЕtOН для полного удаления ЕtOАс (присутствие ЕtOАс оказывает влияние на ход реакции на следующей стадии). Оказалось, что разделение соединений 27а и 27b с помощью хроматографии является затруднительным, поэтому был выбран альтернативный метод, основанный на разнице относительных скоростей гидролиза их соответствующих фрагментов сложного метилового эфира.
Б. Неочищенную смесь соединений 27а и 27b (3,47 г) растворяли в смесь ТГФ:МеОН (1:1, 20 мл), добавляли водный раствор LiOH·H2O (24 мг в 5 мл H2O, 8% зкв.) и реакционную смесь перемешивали при КТ в течение 16 ч (завершение гидролиза побочного продукта 27b с образованием соответствующей кислоты 27с устанавливали путем анализа с помощью ЖХВР). Реакционную смесь концентрировали в вакууме для удаления большей части ТГФ и МеОН и распределяли между H2O(100 мл) и EtOAc (300 мл). Органический слой промывали 0,5 н. NaOH (3×100 мл), соляным раствором (100 мл), 10%-ным водным раствором лимонной кислоты (2×100 мл), соляным раствором (100 мл), сушили над безводным MgSO4, фильтровали и концентрировали досуха. В результате этого получали требуемый продукт 27а с высокой чистотой (чистота >90% по данным анализа с помощью ЖХВР) в виде пены светло-зеленого цвета с общим выходом 93% (3,28 г) для двух стадий.
1Н ЯМР: (400 МГц, CDCl3): δ 1,1-1,38 (m, 13Н), 1,42 (s, 9Н), 1,51-1,57 (m, 1Н), 1,63-1,67 (dd, J=8,0 и 5,1 Гц, 1Н), 1,81-1,87 (m, 1Н), 1,92-1,99 (m, 1Н), 2,02-2,08 (m, 1Н), 2,62 (d, J=14 Гц, 1Н), 3,4 (d, J=8,3, 1H), 3,65 (s, 3Н), 4,01 (dd, J=10,8 и 4,1 Гц, 1Н), 4,42-4,48 (m, 1Н), 4,51-4,55 (m, 1Н), 4,87 (d, J=8,6 Гц, 1Н), 5,14 (d, J=8.6 Гц, 1Н), 7,97 (br s, 1Н).
Пример 28
Синтез соединения №741 (таблица 7)

Производное хинолина 8f присоединяли к полученному перед этим макроциклическому соединению 23b с помощью реакции Мицунобу. Производное хинолина 8f(30 мг, 0,095 ммоля) растворяли в ТГФ, затем добавляли макроциклическое соединение 23b (45,6 мг, 1 экв.) и РРh3(49,8 мг, 2 экв.). Образовавшуюся смесь охлаждали до 0°С. Затем по каплям добавляли ДИАД (37,4 мкл, 2 экв.). Раствор перемешивали в течение 1 ч при 0°С, а затем перемешивали при комнатной температуре в течение ночи. После этого смесь разбавляли EtOAc (15 мл), промывали насыщенным раствором NaHCO3 (15 мл), а затем соляным раствором. Раствор сушили над MgSO4, фильтровали и концентрировали в вакууме. Получали 202 мг продукта в виде масла желтого цвета. Продукт очищали с помощью экспресс-хроматографии на силикагеле (100%-ный EtOAc). После очистки продукт еще содержал побочный продукт ДИАД. Полученный продукт содержал 55 мас.% требуемого продукта, поэтому выход должен составлять 62%.
Промежуточный продукт в виде сложного эфира (46 мг, 0,06 ммоля) растворяли в смеси ТГФ/МеОН/Н2О (соотношение 2:1:1, 2 мл), добавляли LiOH·H2O (20 мг, 0,48 ммоля) и раствор перемешивали при КТ. Через 16 ч анализ реакционной смеси с помощью ЖХВР показал, что гидролиз был завершен. Органические растворители удаляли в вакууме и оставшийся неочищенный продукт, растворенный в ДМСО, очищали с помощью ЖХВР с обращенной фазой на колонке типа С18, получая чистый ингибитор 741.
1Н ЯМР (400 МГц, ДМСО-d6) δ (част./млн): 8,67 (s, 1H), 8,29-8,14 (m, 2H), 8,08-7,97 (m, 1H), 7,91-7,78 (m, 1H), 7,74 (s, 1H), 7,31-7,20 (m, 1H), 7,10 (d, J=5.7 Гц, 1H), 5,82-5,71 (m, 1H), 5,58-5,47 (m, 1H), 5,32-5,23 (m, 1H), 4,74-4,64 (m, 1H), 4,55-4,47 (m, 1Н), 4,23-4,06 (m, 1H), 4,04-3,94 (m, 1H), 3,97 (s, 3H), 3,92-3,85 (m, 1H), 2,70-2,55 (m, 2H), 2,53-2,36 (m, 2H), 2,20-2,09 (m, 1H), 1,80-1,62 (m, 2H), 1,56-1,43 (m, 2H), 1,42-1.29 (m, 6H), 1,27 (d, J=3,2 Гц, 3H), 1,25 (d, J=2,9 Гц, 3H), 1,12 (s, 9H).
МС: 763,1(М+1), 761,1 (М-1).
Пример 29
Синтез соединения 205 (таблица 2)

К раствору макроциклического соединения 25а (21 мг, 0,035 ммоля) в смеси трет-бутанол/Н2O) (1,5 мл/1,5 мл) при 0°С, добавляли раствор OsO4 в трет-бутаноле (0,36 мл, 35% мас./об., 0,035 ммоля) и смесь перемешивали при КТ в течение 1 ч. Смесь разбавляли EtOAc (20 мл) и органический раствор промывали 5%-ным NaHCO3 (2×10 мл). соляным раствором (2×10 мл), сушили и упаривали досуха. Неочищенное соединение растворяли в смеси ТГФ/МеОН/Н2О (3 мл / 1,5 мл / 1,5 мл) и перемешивали в присутствии LiOH·H2O (13 мг, 0,28 ммоля) в течение 16 ч. Смесь подкисляли до рН 4 с помощью 0,5 н. охлажденной на льду НСl, упаривали и очищали с помощью ЖХВР с обращенной фазой на колонке типа С18, используя градиент растворителя от Н2О (0,06% ТФК) до 40%-ного водного раствора CH3CN (0,06% ТФК). Выделяли диол 205, имеющий син-конфигурацию, в виде аморфного твердого вещества белого цвета с высокой степенью чистоты.
Соединение №205: 1Н ЯМР (ДМСО, 400 МГц): δ 1,01 (s, 9H), 1,06-1,30 (m, 9H), 1,48-1,68 (m,3H), 1,78-1,88 (m, 1H), ≈2,2-2,5 (2m, 2H), 3,78-3,82 (m, 1H), 3,86-3,90 (m, 1Н), 4,39 (t, J=8,9 Гц, 1H), 4,61 (d, J=11,4 Гц, 14), 5,60 (bs, 1H, Pro-γ), 7,03 (d, J=6,0 Гц, 1H), 7,40 (bs, 1H), 7,58-7,62 (m, 1H), 7,87-7,91 (m, 1H), 8,00 (d, J=8,3 Гц, 1H), 8,24 (d, J=8,6 Гц, 1H), 8,60 (s, 1H), 8,99 (bs, 1H).
ЭМС (форма отрицательной ионизации): m/z 625 (М-Н)-.
Пример 30
Синтез соединений 214 и 218 (таблица 2)
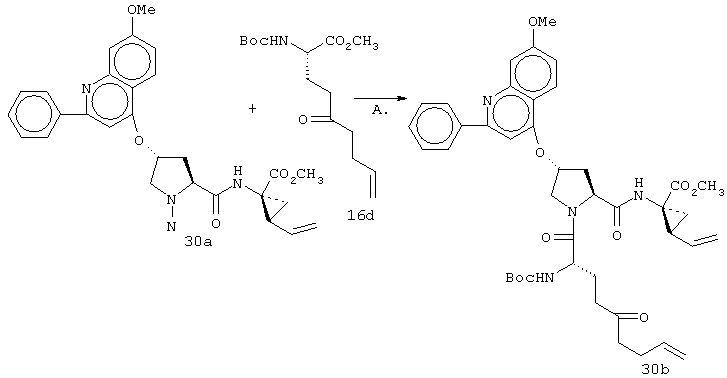
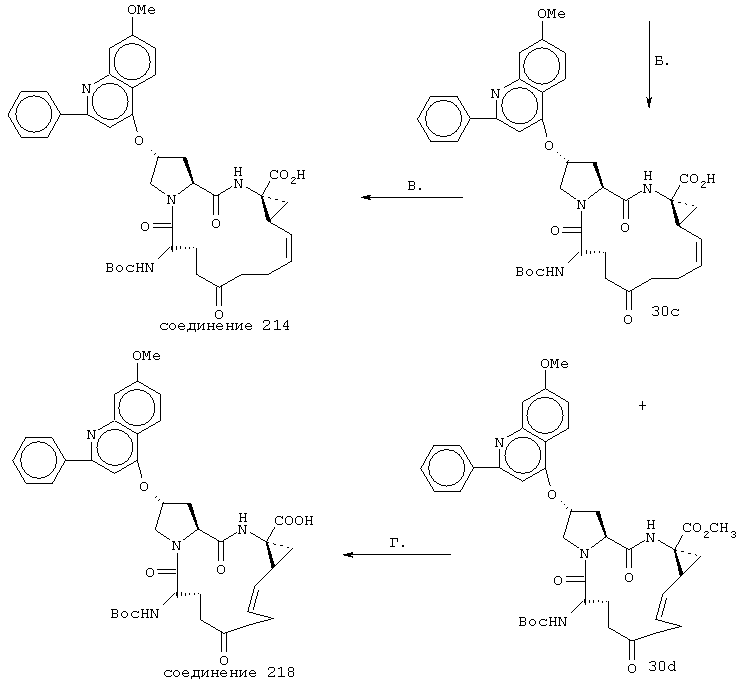
А. Раствор сложного кетононеноатного эфира 16d (0,180 г, 0,6 ммоля) в смеси МеОН/Н2О (5 мл/2 мл) перемешивали при КТ в присутствии LiOH·H2O (50 мг, 1,2 ммоля) в течение 1 ч. Раствор подкисляли до рН 6 с помощью 0,5 н. охлажденной на льду НСl и выпаривали большую часть МеОН. Затем остаток растворяли в ЕtOАс (30 мл) и раствор промывали 0,5 н. охлажденной на льду НСl (10 мл), соляным раствором (10 мл), сушили и упаривали. Затем неочищенный остаток повторно растворяли в CH2Cl2 (10 мл) и подвергали взаимодействию с фрагментом Р1-Р2 30а (0,337 г, 0,6 ммоля) в присутствии ГАТУ (233 мг, 0,612 ммоля) и ДИПЭА (420 мкл, 2,4 ммоля) в течение 16 ч при КТ. Реакционную смесь хроматографировали на силикагеле, используя в качестве элюента смесь ЕtOАс/гексан (1/1), в результате чего выделяли чистое соединение 30b (0,370 г, выход 83%, чистота >95% по данным анализа с помощью ЖХВР).
1Н ЯМР (CDCl3, 400 МГц) δ 1,41 (s, 9H), 1,45-1,54 (m, 1H), 1,58-1,62 (m, 1H), 1,73-1,77 (m, 1Н), 1,86-1,91 (m, 1H), 2,16 (dd, J=17,8 и 8,6 Гц, 1Н), 2,26-2,43 (2m, 2H), 2,46-2,58 (m, 2H), 2,64-2,81 (m, 1H), 2,85-2,92 и 2,95-3,03 (2m, 1H, смесь ротамеров в соотношении 1:3), 3,67 (s, 3Н), 3,95 (s, 3Н), 4,10-4,18 (m, 1H), 4,20-4,30 (m, 1H), 4,40-4,55 (m, 1H), 4,80-4,88 (m, 1H), 4,92-5,10 (m, 2H), 5,14 (dd, J=10,2 и 1,6Гц, 1H), 5,24-5,38 (m, 4Н), 5,42-5,54 (m, 1H), 5,68-5,86 (m, 2H), 7,04-7,14 (m, 2H), 7,42-7,64 (m, 5H), 7,92-8,12 (m, 3H).
Б. Диен 30b (0,370 г, 0,49 ммоля) подвергали циклизации в присутствии катализатора, представляющего собой дихлорид бис(трициклогексилфосфин)бензилиденрутения(IV) (0,125 мг, 0,15 ммоля) в CH2Cl2 (освобожденного путем дистилляции от СаН2 и дегазированного с помощью аргона в течение 30 мин) при температуре дефлегмации в течение 2 ч. После экспресс-хроматографии на колонке на силикагеле с использованием в качестве элюента смеси EtOAc/гексан (3/1) получали соединение в виде смеси стереоизомеров (30с и 30d, соотношение 1:1) с выходом 35% (0,124 г).
1Н ЯМР для смеси соединений 30с и 30d (CDCl3, 400 МГц) δ 1,44 (s, 4H) и 1,37 (s, 4H), 1,60 (m, 2H), 1,83 (m, 0,5H), 2,01 (m, 1H), 2,09 (m, 1H), 2,42 (m, 5H), 2,73 (m, 2H), 3,26 (m, 0,5H), 3,69 (s, 1,5H), 3,76 (s, 1,5H), 3,96 (s, 3H), 4,10 (m, 1H), 4,24 (m, 0,5H), 4,10 (m, 0,5H), 4,58 (m, 1H), 4,73 (m, 1H), 4,89 (m, 0,5H), 4,97 (m, 0,5H), 5,30 (m, 0,5H), 5,44 (m, 2H), 5,64 (m, 1H), 7,1-7,0 (m, 3H), 7,47 (m, 4H), 8,08-7,98 (m, 3H).
В, Г. Гидролиз сложных метиловых эфиров 30с и 30d (24 мг, 0,033 ммоля) осуществляли в смеси ТГФ/МеОН/Н2О (1 мл/0,5 мл/0,5 мл) с помощью LiO•H2O (11 мг, 0,246 ммоля) в течение 16 ч при КТ. По истечении этого периода времени смесь подкисляли до значения рН 4-5 и хроматографировали с помощью ЖХВР с обращенной фазой на колонке типа С 18, используя градиент растворителя от H2O (0,06% ТФК) до 50%-ного водного раствора CH3CN (0,06% ТФК). Из смеси двух соединений выделяли требуемые соединения 214 и 218, имеющие высокую степень чистоты (чистота 94% по данным анализа с помощью ЖХВР), с выходом 15% (3 мг).
Соединение 214: 1Н ЯМР (ДМСО, 400 МГц) δ 1,15 (s,9H), 1,48-1,54 (m, 2H), 1,65-1,74 (m, 1H), 1,77-1,85 (m, 1H), 2,12-2,25 (m, 4H), 2,27-2,34 (m, 1H), 2,61-2,68 (m, 1H), 2,87 (bt, J=11,5 Гц, 1H), 3,92 (dd, J=9,2 и 1,5 Гц, 1H, Pro-5), 3,97 (s, 3H, -ОСН3). 4,14-4,20 (m, 1H), 4,52 (t, J=7,8 Гц, 1H, Pro-α), 4,66 (d, J=11,8 Гц, 1H, Pro-α), 5,45 (t, J=9,9 Гц, 1H), 5,51-5,58 (m, 1H), 5,82 (bs, 1H, Рго-γ), 7,09 (d, J=6,0 Гц, 1Н, BocNH), 7,26 (bs, 1H), 7,53 (s, 1H), 7,67 (bs, 3H), 8,16 (d, J=2 Гц, 1H), 8,18 (s, 1H), 8,83 (s, 1H, АЦКК-NН).
Соединение 218: 1Н ЯМР(ДМСO.400 МГц): δ 1,06-1,10 (m, 1H), l,18(s,9H), 1,52-1,55 (m, 1H), 1,62-1,80 (m, 1H), 2,10-2,68 (перекрываются, 9Н), 3,90 (bd, J=8,3 Гц. 1Н), 3,96 (s, 3H, ОСН3), 4,20-4,27 (m, 1H), 4,58-4,63 (m, 1H, Pro-δ), 4,66 (dd, J=8,3 Гц, 1Н, Pro-α) 4,88 (dd, J=10,2 Гц, 1H), 5,18-5,26 (m, 1H), 5,73-5,79 (m, 1H, Pro-γ), 7,01 (d, J=6,4 Гц, 1H), 7,23 (bs, 1H), 7,50 (bs, 1H), 7,66 (bs, 3Н), 8,20 (bs, 2H), 8,53 (s, 1H).
Пример 31
Синтез соединения 209 (таблица 2)
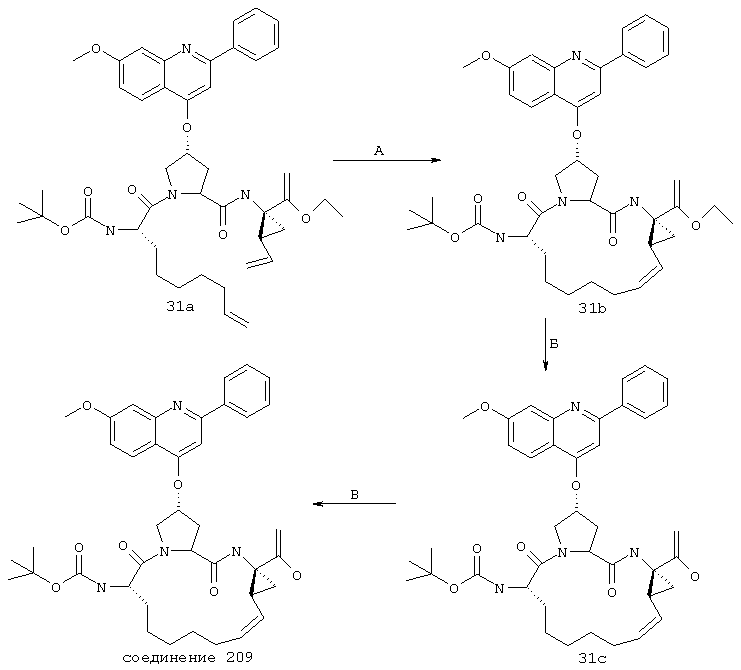
А. Диен 31а (249 мг, 0,330 ммоля) растворяли в 30 мл безводного CH2Cl2 и раствор дегазировали с помощью аргона в течение 15 мин. Катализатор, представляющий собой дихлорид бис(трициклогексилфосфин)бензилиденрутения (IV) (82 мг, 0,100 ммоля) растворяли в 3 мл безводного и дегазированного CH2Cl2 и добавляли к раствору диена. Реакционную смесь выдерживали при температуре дефлегмации в течение 2 ч в атмосфере N2. Раствор концентрировали и очищали с помощью экспресс-хроматографии на колонке, получая соединение 31b в виде твердого вещества коричневого цвета с выходом 71% (171 мг).
1Н ЯМР (CDCl3, 400 МГц): δ 1,22-1,44 (m, 10Н), 1,42 (s, 9H), 1.66-1,74 (m, 1H), 1,87-1,97 (m,2H), 2,13-2,28 (m,3H), 2,32-2,39 (m, 1H), 3,08-3,16 (m, 1H),3,41 (s, 3H), 4,07-4,22 (m, 3H), 4,28-4,34 (m, 1H), 4,58-4,64 (m, 1H), 4,95-4,99 (m, 1H), 5,22-5,29 (m, 2H), 5,38-5,43 (m, 1H), 5,48-5,56 (m, 1H), 7,00-7,12 (m, 3H), 7,43-7,55 (m, 4H), 7,97-8,11 (m, 3H).
ES(+)MC: m/z 727,4 (М+Н)+.
Б. Соединение 31b (0,117 ммоля) перемешивали в растворе НСl (1 мл 4н. раствора в диоксане) в течение 30 мин и концентрировали досуха. Твердый продукт растворяли в СН2Сl2 (2 мл) и последовательно добавляли Еt3N (82 мкл, 0,585 ммоля) и трет-бутилизоцианат (35 мг, 0,351 ммоля). После перемешивания при КТ в течение 20 ч смесь концентрировали досуха и неочищенное соединение 31с использовали на конечной стадии гидролиза без дополнительной очистки.
В. Соединение 31с (85 мг, 0,117 ммоля) растворяли в смеси ТГФ/МеОН/Н2О (2 мл /1 мл/ 1 мл), добавляли LiOH·Н2О (39 мг, 0,936 ммоля) и раствор перемешивали в течение 20 ч при КТ. По истечении этого периода времени добавляли уксусную кислоту (1 мл) и раствор концентрировали для удаления МеОН и ТГФ. После очистки неочищенного продукта с помощью ЖХВР с обращенной фазой на колонке типа С 18 выделяли чистое соединение 209 (25 мг, выход ~31%).
1Н ЯМР (ДМСО, 400 МГц): δ 1,04 (s, 9H), 1,15-1,24 (m, 2H), 1,30-1,40 (m, 5H), 1,44-1,51 (m, 2H), 1,54-1,68 (m, 1 Н), 1,75-1,88 (m, 1H), 2,18 (dd, J=17,2 и 8,5 Гц, 1Н), 2,32-2,45 (m, 1H, Pro-β), 2,54-2,62 (m, 1H), 2,65-2,68 (m, 1H, Pro-β), 3,91 (dd, J=11,1 и 3,5 Гц, 1H, Pro-δ), 3,96 (s, 3Н, -ОСН3), 4,17-4,23 (m, 1H), 4,47 (dd, J=8,6, 1H, Pro-α), 4,67 (bd, J=7,9 Гц, 1H, Pro-δ), 5,30 (dd, J=9,5 Гц, 1Н), 5,52 (bdd, J=19 и 8,3, 1H), 5,68 (s, 1H), 5,78 (bs, 1H, Pro-γ), 5,94 (bs, 1H), 7,21 (bs, 1H), 7,51 (bs, 1H), 7,66 (bs, 4H), 8,19 (s, 2H), 8,40 (d, J=7 Гц, 1H), 8,61 (s, 1H, АЦКК-NH).
ES(+)MC: m/z 698,3 (M+H)+.
Пример 32
Синтез соединений 404 и 407 (таблица 4)
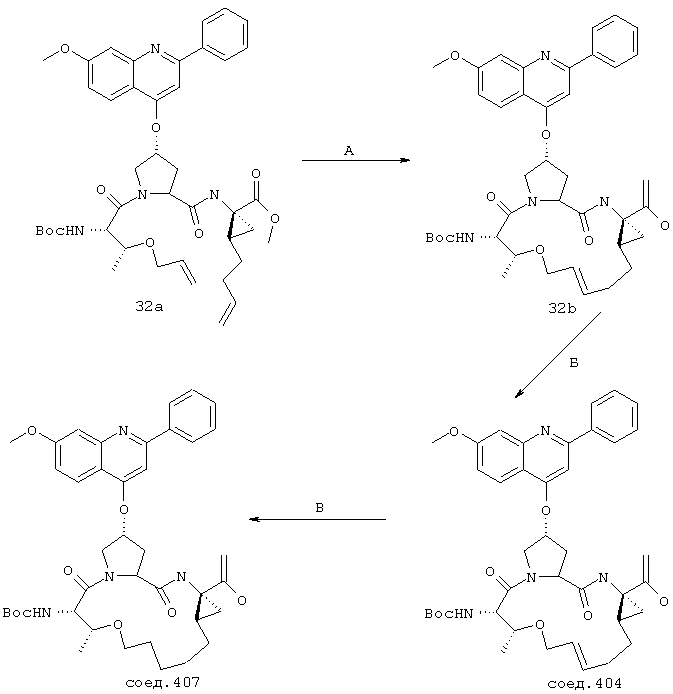
А. Диен 32а (84 мг, 0,11 ммоля) растворяли в безводном CH2Cl2 (11 мл) и раствор дегазировали в течение 15 мин с помощью струи аргона. Катализатор, представляющий собой дихлорид бис(трициклогексилфосфин)бензилиденрутения(IV) (19 мг, 0,023 ммоля) сначала растворяли в 1 мл дегазированного СН2Сl2, а затем его переносили с помощью канюли в реакционную колбу. Реакционную смесь перемешивали а течение 2 ч при температуре дефлегмации. Затем растворитель удаляли в вакууме и реакционную смесь очищали с помощью экспресс-хроматографии на колонке, используя в качестве элюента смесь ЕtOАс/ гексан (1:1), в результате чего получали макроциклическое соединение 32b в виде масла желтого цвета (33 мг, выход 41%).
Б. Промежуточный продукт, представляющий собой сложный эфир 32b (33 мг, 0,045 ммоля), растворяли в смеси ТГФ/MeOH/H2O (соотношение 2:1:1,2 мл), добавляли LiOH·H2O (8 мг, 0,18 ммоля) и раствор перемешивали при КТ. Через 16 ч анализ реакционной смеси с помощью ЖХВР показал, что гидролиз еще не завершился. Поэтому добавляли дополнительное количество LiOH·H2O (4 мг, 0,09 ммоля) и раствор перемешивали при КТ в общей сложности в течение 36 ч. В завершение раствор подкисляли небольшим аликвотным количеством уксусной кислоты, органические растворители удаляли в вакууме и оставшийся неочищенный продукт очищали с помощью ЖХВР с обращенной фазой на колонке типа С 18, получая чистый ингибитор 404.
1Н ЯМР(ДМСО, 400 МГц): δ 1.21 (d, J=6,0 Гц, 3Н, Me), 1,36(s, 9H, Воc), 1,1-1,4 (3m, 3Н), 1,66 (m, 1 Н), 1,80 (m, 1 Н), 2,10 (m, 2Н), 2,57 (m, 2H), 3,90 (m, 4H), 4,47 (bd, J=12,7 Гц, 1H), 4,58 (bd, J=7,3, 1H), 4,66 (dd, J=8,0 Гц, 1H), 5,57 (m, 1H), 5,66 (m, 1H), 5,83 (bs, 1H), 6,18 (bd, J=6,9 Гц, 1H), 7,25 (bd, J=7,3 Гц, 1H), 7,56 (bs, 1H), 7,70 (m, 4H), 8,22 (bd, J=2,9 Гц, 2H), 8,29 (bs, J=9,2 Гц, 1Н).
В. Ингибитор 404 (15 мг, 0,021 ммоля) растворяли в этаноле (2 мл) и добавляли 10%-ный Pd/C (2 мг). Смесь перемешивали в атмосфере водорода при КТ в течение 16 ч. После фильтрации смесь очищали с помощью ЖХВР с обращенной фазой на колонке типа С 18, получая ингибитор 407 в виде твердого вещества белого цвета (10 мг, выход 66%)
1Н ЯМР (ДМСО, 400 МГц): δ 1,04 (m, 1H), 1,17 (d, J=6,0 Гц, 3Н), 1,35 (s, 9H), 1,25-1,75 (m, 12 Н), 2,32-2,45 (m, 1 Н), 3,40-3,50 (m, 2 Н), 3,74-3,83 (m, 1H), 3,85-3,93 (m, 1H), 3,97 (s, 3Н), 4,27-4,36 (dd, J=21,1 и 8,6 Гц, 1H), 4,54 (dd, J=7,95 и 7,95 Гц, 1H), 5,64 (d, J=8,3 Гц, 1H), 5,82 (brs, 1H), 7,27-7,33 (m, 1H), 7,53-7,57 (bs, 1 Н), 7,60-7,74 (m, 4 Н), 8,13-8,27 (m, 3 Н), 8,30-8,35 (br s, 1H).
Пример 33
Синтез соединения 824 (таблица 8)
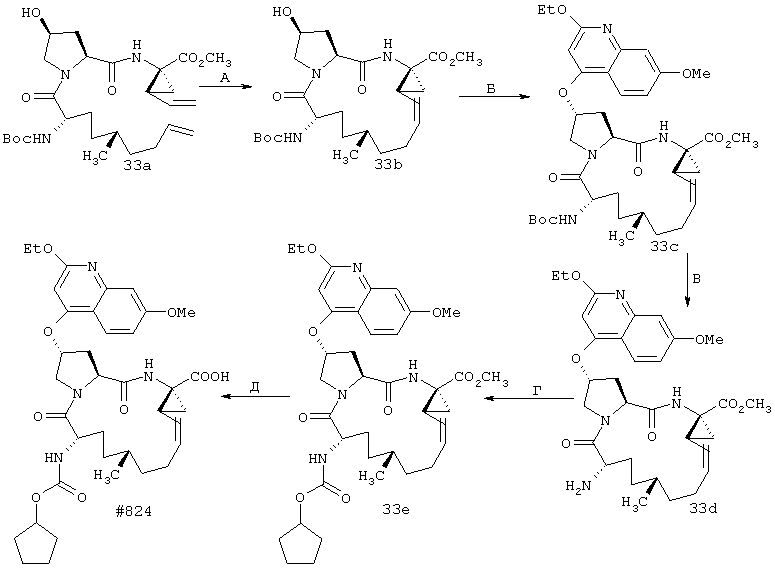
А. Соединение 33а (~0,55 ммоля) растворяли в СН2Сl2 (100 мл) и раствор осторожно дегазировали, после чего добавляли порцию катализатора Ховейда (17 мг, 0,028 ммоля, 0,05 экв.). Затем раствор перемешивали при температуре дефлегмации в течение 5 ч. Реакционную смесь концентрировали и очищали с помощью экспресс-хроматографии на колонке, используя градиент растворителя СН2Сl2/ЕtOАс (от 3:2 до 2:3 ratio), в результате чего получали соединение 33b с выходом 72% (194 мг).
Б. К раствору, содержащему соединение 33b (70 мг, 0,142 ммоля), 2-этокси-4-гидрокси-7-метоксихинолин 3с (63 мг, 0,284 ммоля, 2 экв.) и Рh3Р (186 мг, 0,71 ммоля, 5 экв.) в безводном ТГФ (15 мл), медленно в течение 20 мин добавляли при 0°С ДИАД (140 мкл, 0,71 ммоля, 5 экв.). Реакционной смеси давали нагреться до КТ и ее перемешивали при КТ в течение 2,5 ч. Затем ТГФ выпаривали в вакууме и неощищенный продукт очищали с помощью экспресс-хроматографии на колонке, используя градиент растворителя гексан/ЕtOАс (от 7:3 до 1:1). Выделяли чистое соединение 33с с выходом 73% (72 мг).
В. Соединение 33с (72 мг, 0,104 ммоля) смешивали с СН2Сl2 (5 мл) и 4 М раствором НСl в диоксане (5 мл) и смесь перемешивали при КТ в течение 1,5 ч для удаления защитной группы Воc и получения гидрохлорида промежуточного продукта 33d. Неочищенную реакционную смесь упаривали досуха в вакууме, сушили в вакууме для того, чтобы гарантировать полное удаление НСl, и продукт использовали на следующей стадии без очистки.
Г. К раствору циклопентанола (29 мкл, 0,32 ммоля) в ТГФ (10 мл) по каплям добавляли раствор фосгена в толуоле (1,93М, 274 мкл, 0,528 ммоля) и смесь перемешивали при КТ в течение 2 ч для получения циклопентилхлорформиатного реагента. По истечении этого времени приблизительно половину растворителя удаляли путем выпаривания в вакууме, оставшийся раствор светло-желтого цвета разбавляли путем добавления CH2Cl2 (5 мл) и повторно концентрировали до половины первоначального объема для того, чтобы гарантировать полное удаление всего избытка фосгена. Затем указанный выше раствор циклопентилхлорформиатного реагента разбавляли ТГФ (10 мл), охлаждали до 0°С и добавляли к твердому соединению 33d (0,104 ммоля) при 0°С. К реакционной смеси добавляли Еt2N (75 мкл, 0,534 ммоля, 5,2 экв.) и продолжали перемешивание при 0°С в течение 1,5 ч. Растворители удаляли в вакууме и неочищенный продукт очищали с помощью экспресс-хроматографии на колонке, используя в качестве элюента смесь EtOAc/гексан (1:1), в результате чего получали соединение 33е с почти количественным выходом (75 мг).
Д. Гидролиз сложного метилового эфира проводили путем взаимодействия соединения 33е (75 мг, 0,11 ммоля) с LiOH·H2O (35 мг, 0,84 ммоля, 8 экв.) в смеси растворителей ТГФ/МеОН/Н2О (соотношение 2:2:1, 7,5 мл) при 50°С в течение 2,5 ч. После завершения гидролиза смесь подкисляли до значения рН 4,5 и растворители выпаривали досуха в вакууме. Неочищенный продукт очищали с помощью препаративной ЖХВР с обращенной фазой на колонке типа С 18, используя градиент растворителя от H2O до 58%-ного водного раствора CH3CN (содержащего 0,06% ТФК), в результате чего получали ингибитор №824 в виде аморфного твердого вещества белого цвета (45 мг, выход 65%). 1Н ЯМР для Na+-coли соединения №824 (ДМСО, 400 МГц): δ 0,88 (d, J=6,7 Гц, 3Н), 0,95-1,70 (перекрывающиеся резонансные пики, 17Н), 1,37 (t, J=7 Гц, 3Н), 2,00-2,10 (m, 1H), 2,10-2,33 (m, 3Н), 2,38-2,44 (m, 1H), 3,80-3,85 (m, 1H), 3,85 (s, 3H), 4,02-4,08 (m, 1H), 4,42 (q, J-7 Гц, 2H), 4,35-4,44 (m, 1H), 4,50 (d, J=10,8 Гц, 1H), 4,63 (bs, 1H), 5,28 (dd, J=9,5 Гц, 1H), 5,38 (bs, 1H), 5,42-5,49 (m, 1H), 6,37 (s, 1H), 6,87 (dd, J=8,9 и 2,2 Гц, 1H), 7,07 (d, J-2,2 Гц, 1H), 7,28 (d, J=7,0 Гц, 1H). 7,90 (d, J=8,9 Гц, 1H), 8,57 (s, 1H).
Пример 34
Синтез соединения 812 (таблица 8)
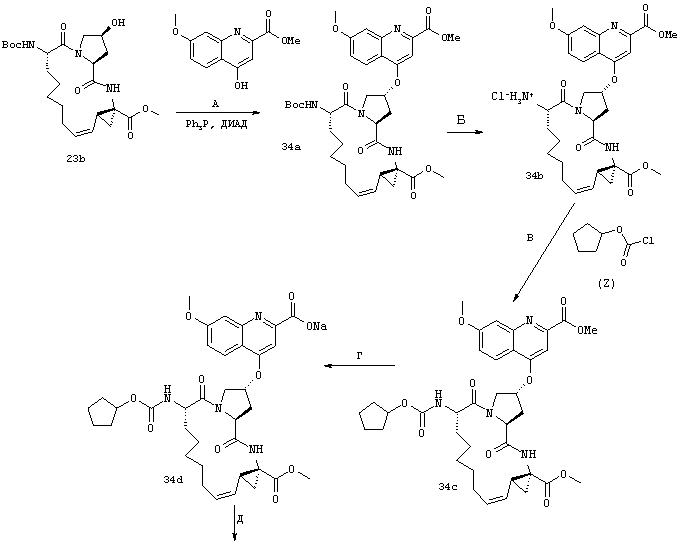
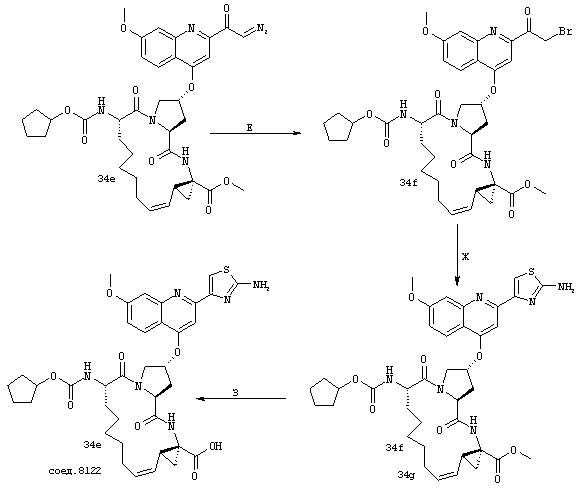
А. К раствору, содержащему макроциклический промежуточный продукт 23b (13,05 г, 27,2 ммоля, 1.0 экв.), Рh3Р (14,28 г, 54,4 ммоля, 2,0 экв.) и 2-карбоксиметокси-4-гидрокси-7-метоксихинолин (WO 00/09543 и WO 00/09558) (6,67 г, 28,6 ммоля, 1,05 экв.) в ТГФ (450 мл), добавляли по каплям в течение 15 мин при 0°С ДИАД (10,75 мл, 54,6 ммоля, 2,0 экв.). Затем ледяную баню удаляли и реакционную смесь перемешивали при КТ в течение 3 ч. После завершения превращения исходного продукта в конечные продукты растворитель выпаривали в вакууме, оставшуюся смесь разбавляли EtOAc, промывали насыщенным раствором NаНСО3 (2х) и соляным раствором (1х), органический слой сушили над безводным MgSO4, фильтровали и упаривали досуха. После экспресс-хроматографии на колонке получали чистое соединение 34а; колонку элюировали сначали смесью гексан/ЕtOАс (50:50), а затем СНСl3/ЕtOАс (95:5) для удаления побочных продуктов Рh3РО и ДИАД, за процессом элюирования примесей наблюдали с помощью ТСХ. В завершение требуемый продукт 34а элюировали из колонки с использованием смеси СНСl3/ЕtOАс (70:30). Как правило, стадию хроматографии необходимо повторять 2-3 раза для того, чтобы соединение 34а могло быть выделено с высокой степенью чистоты в виде твердого вещества белого цвета с общим выходом 68% (12,8 г, чистота 99,5% по данным анализа с помощью ЖХВР).
Б. К раствору Вос-защищенного промежуточного продукта 34а (1,567 г) в СН2Сl2 (15 мл) добавляли 4 н. НСl в диоксане (12 мл) и реакционную смесь перемешивали при КТ в течение 1 ч. [в том случае, когда до окончания реакции начинал образовываться густой гель, добавляли еще 10 мл CH2Cl2]. После завершения удаления защитной группы растворители выпаривали досуха, получая твердое вещество желтого цвета и пастообразный продукт. Смесь повторно растворяли приблизительно в 5%-ном растворе МеОН в CH2Cl2 и повторно упаривали досуха в вакууме, получая соединение 34b в виде твердого вещества желтого цвета, которое использовали на следующей стадии без какой-либо очистки.
В. К раствору циклопентанола (614 мкл, 6,76 ммоля) в ТГФ (15 мл) по каплям добавляли раствор фосгена в толуоле (1,93 М, 5,96 мл, 11,502 ммоля) и смесь перемешивали при КТ в течение 2 ч, получая реагент (z), представляющий собой циклопентил-хлорформиат. По истечении этого периода времени приблизительно половину растворителя удаляли путем выпаривания в вакууме, оставшийся раствор светло-желтого цвета разбавляли путем добавления СН2Сl2 (5 мл) и концентрировали до половины его первоначального объема для того, чтобы гарантировать удаление всего избытка фосгена. Затем полученный выше циклопентилхлорформиатный реагент разбавляли ТГФ (15 мл) и добавляли к аминдигидрохлориду 34b. Смесь охлаждали до 0°С в ледяной бане, значение рН доводили до ~8,5-9 путем добавления Et3N (добавляя по каплям) и реакционную смесь перемешивали при 0°С в течение 1 ч. По истечении этого периода времени смесь разбавляли EtOAc, промывали водой (1х), насыщенным раствором NaHCO3 (2x), Н2О (2x) и соляным раствором (1х). Органический слой сушили над безводным MgSO4, фильтровали и упаривали в вакууме, получая пену светло-янтарного цвета. После очистки с помощью экспресс-хроматографии на колонке (используя в качестве элюента градиент растворителя от 30%-ного гексана до 20%-ного гексана в EtOAc) получали соединение 34с в виде пены белого цвета с выходом 80% (1,27 г) и чистотой >93%.
Г. Сложный диметиловый эфир 34с (1,17 г) растворяли в смеси ТГФ/МеОН/Н2О (20 мл, соотношение 2:1:1) и добавляли водный раствор NaOH (1,8 мл, 1 н., 1 экв.). Реакционную смесь перемешивали при КТ в течение 1 ч, после чего ее упаривали досуха, получая натриевую соль 34d в виде твердого вещества белого цвета (~1,66 ммоля). Соединение 34d использовали на следующей стадии без очистки.
Д. Неочищенную натриевую соль 34d (1,66 ммоля) растворяли в ТГФ (17 мл), добавляли Et3N и смесь охлаждали до 0°С в ледяной бане. По каплям добавляли изобу-тилхлорформиат (322 мкл, 2,5 ммоля) и смесь перемешивали при 0°С в течение 75 мин. По истечении этого периода времени добавляли диазометан (15 мл) и продолжали перемешивание при 0°С в течение 30 мин, а затем еще в течение 1 ч при КТ. Большую часть растворителя удаляли путем выпаривали досуха в вакууме, оставшуюся смесь разбавляли EtOAc, промывали насыщенным раствором NаНСО3 (2х), Н2O (2х) и соляным раствором (1х), сушили над безводным MgSO4, фильтровали упаривали досуха, получая соединение 34е в виде пены светло-желтого цвета (1,2 г, ~1,66 ммоля). Промежуточный продукт, представляющий собой диазокетон 34е использовали на следующей стадии без очистки.
Е. Диазокетон 34е (1,2g, 1,66 ммоля), растворенный в ТГФ (17 мл) охлаждали до 0°С в ледяной бане. По каплям добавляли водный раствор НВr (48%, 1,24 мл) и реакционную смесь перемешивали при 0°С в течение 1 ч. Затем смесь разбавляли EtOAc, промывали насыщенным раствором NaHCO3 (2x), H2O (2х) и соляным раствором (1 х), органический слой сушили над безводным MgSO4, фильтровали и упаривали досуха, получая промежуточный продукт, представляющий собой β-бромкетон 34f, в виде пены светло-желтого цвета (~1,657 ммоля).
Ж. К раствору бромкетона 34f (600 мг, 0,779 ммоля) в изопропаноле (5 мл) добавляли тиомочевину (118 мг, 1,55 ммоля) и реакционную смесь помещали в масляную баню, предварительно нагретую до 75°С, где ее перемешивали в течение 1 ч. Затем изопропанол удаляли в вакууме и продукт растворяли в EtOAc (100 мл). Раствор промывали насыщенным раствором NаНСО3 и соляным раствором, органический слой сушили над безводным Na2SO4, фильтровали и упаривали, получая неочищенный продукт 34g (522 мг) в виде твердого вещества красно-коричневого цвета. Этот продукт использовали на конечной стадии без какой-либо дополнительной очистки.
З. Неочищенный сложный метиловый эфир 34g (122 мг, 0,163 ммоля) растворяли в смеси ТГФ/МеОН/Н2О (соотношение 2:1:1,4 мл) и подвергали омылению с использованием LiOH•H2O (89 мг, 2,14 ммоля). Реакцию гидролиза проводили в течение 12-15 ч при КТ. Затем растворитель удаляли в вакууме и неочищенный продукт очищали с помощью ЖХВР с обращенной фазой на колонке типа С 18, используя градиент растворителя от 10%-ного СН3СN в Н2O до 100%-ного СН3СN, в результате чего получали соединение 812, представляющее собой ингибитор HCV-протеазы, в виде твердого вещества желтого цвета (24 мг, общий выход при превращении промежуточного продукта 34f в ингибитор 812 составлял 20%).
1Н ЯМР (400 МГц, ДМСО-d6) δ 8,63 (s, 1H), 8,26-8,15 (m, 2H), 7,79 (bs, 1H), 7.72 (bs, 1H), 7,50 (bs, 2H), 7,33-7,25 (m, 2H), 5,77 (bs, 1H), 5,52 (dd, J=8,3 Гц, 1H), 5,27 (dd, J=9,2 Гц, 1Н), 4,64 (d, J=10,8 Гц, 1Н), 4,50 (dd, J=8,3 Гц, 1Н), 4,39-4,31 (m, 1Н), 4,08-3,99 (m, 2H), 3,94 (s, 3H), 3,87 (d, J=9,5 Гц, 2H), 2,65-2,53 (m, 2H), 2,46-2.36 (m, 2H), 2,20-2,12 (dd, J=8,6 Гц, 1Н), 1,80-1,64 (m, 2Н), 1,63-1,06 (m, 14H). MC; es+:733,2 (М+H)+, es-: 731,2 (М-Н)-.
Пример 34а
Используя такую же процедуру, которая описана в примере 34, но подвергая взаимодействию бромкетон 34f c имеющейся в продаже N-метилтиомочевиной, получали соединение №811 (таблица 8)
1Н ЯМР (400 МГц, ДМСО-d6): δ 8,63 (s, 1Н), 8,20 (s, 1H), 8,18 (s, 1H), 8,12-7,93 (m, 1H), 7,88-7,69 (m, 2H), 7,32-7,24 (m, 2H), 5,82-5,75 (m, 1H), 5,52 (ddd, J=8,1 Гц, 1Н), 5,28 (dd, J=9,9 Гц, 1H), 4,67-4,61 (m, 1H), 4,51 (dd, J=8,8 Гц, 1H), 4,44-4,37 (m, 1H), 4,08-4,00 (m, 1H), 3,96 (s, 3Н), 3,89 (m, 1H), 3,04 (d, J=4,1 Гц, 3Н), 2,65-2,37 (m, 3Н), 2,16 (m, 1H), 1,77.1,65 (m, 2H), 1,63-1,11 (m, 17H). MC; es+: 747,2 (M+H)+, es-:745,3 (M-H)-.
Пример 34б
Используя такую же процедуру, которая описана в примере 34, но подвергая взаимодействию бромкетон 34f c имеющейся в продаже N-этилтиомочевиной, получали соединение №810 (таблица 8)
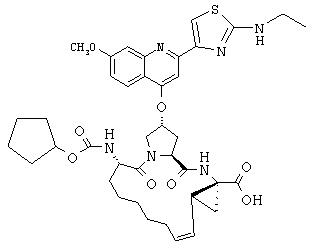
1Н ЯМР (400 МГц, ДМСО-d6): δ 8,63 (s, 1H), 8,27 (bs, 1H), 8,20 (d, J=9,0 Гц, 1H), 8,13-8,07 (m, 1H), 7,86 (bs, 1H), 7,78 (s, 1H), 7,33-7,25 (m, 2H), 5,81 (bs, 1H), 5,54 (dd, J=8,8 Гц, 1H), 5,28 (dd, J=9,7 Гц, 1H), 4,65 (d, J=12,4 Гц, 1H), 4,51 (dd, J=8,8 Гц, 1H), 4,38 (bs, 1H), 4,03 (m. 1H), 3,97 (s, 3H), 3,92-3,87 (m, 1H), 3,54-3,46 (m, 2H), 2,68-2,65 (m, 2H), 2,47-2,38 (m, 1H), 2,15 (dd, J=8,6 Гц, 1H), 1,78-1,65 (m, 2H), 1,60-1,12 (m, 17H), 1,25 (t, J=7,3 Гц, 3H). MC; es+: 783,2 (M+Na)+, es-:761,2 (M+H)-.
Пример 34в
Используя такую же процедуру, которая описана в примере 34, но подвергая взаимодействию бромкетон 34f c имеющейся в продаже N-изопропилтиомочевиной, получали соединение 822
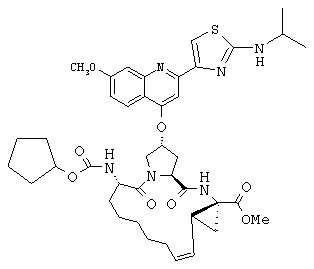
1Н ЯМР (400 МГц,ДМСО-d6) δ 8,63 (s, 1H), 8,33-8,23 (bs, 1H), 8,21 (d, J=9,2 Гц, 1H), 8,04 (d, J=8,3 Гц, 1H), 7,86 (bs, 1H), 7,77 (s, 1H), 7,35-7,23 (m, 2H), 5,81 (bs, 1H), 5,52 (dd, J=8,5 Гц, 1H), 5,27 (dd, J=9,2 Гц, 1H), 4,65 (d, J=11,8 Гц, 1H), 4,51 (dd, J=7,6 Гц, 1H), 4,37 (bs, 1H), 4,15 (bs, 1H), 4.07-3,98 (m, 2H), 3,97 (s, 3H), 3,88 (d, J=8,9 Гц, 1H), 2,60-2,53 (m, 2H), 2,47-2,37 (m, 2H), 2,19-2,10 (dd, J=9,2 Гц, 1H), 1,80-1,64 (m, 2H), 1,63-1,29 (m, 13H), 1,27 и 1,25 (2 x d, J=6,5 Гц, 6Н), 1,23-1,09 (m, 2H). MC; es+:775,0 (M+H)+, es-:772,9 (M-Н)-.
Пример 34г
Используя такую же процедуру, которая описана в примере 34, но подвергая взаимодействию бромкетон 34f c имеющейся в продаже N-ацетилтиомочевиной, получали соединение №809
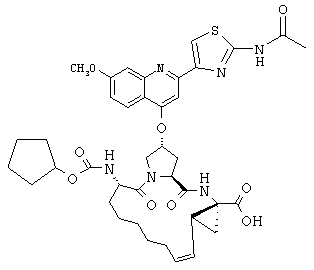
1Н ЯМР (400 МГц, ДМСО-d6): δ 8,62 (s, 1Н), 8,30 (bs, 1H), 8,17 (d, J=8,9 Гц, 1Н), 7,62 (bs, 1H), 7,52 (bs, 1H), 7,28 (d, J=6,4 Гц, 1H), 7,21 (bs, 1H), 5,63 (bs, 1H), 5,54 (dd, J=8,1 Гц, 1H), 5,28 (dd, J=9,5 Гц, 1H), 4,62 (d, J=12,1 Гц, 1H), 4,56-4,46 (m, 2Н), 4,11-4,04 (m, 1H), 3,95 (s, 3H), 3,93-3,88 (m, 1H), 2,62-2,54 (m, 1H), 2,45-2,36 (m, 1H), 2,22 (s, 3Н), 2,21-2,13 (m, 1H), 1,79-1,69 (m, 2H), 1,65-1,30 (m, 16H), 1,26-1,12 (m, 2H). MC; es+: 775,3 (M+H)+, es-: 773,3 (M-Н)-.
Пример 34д
К раствору промежуточного продукта, представляющего собой 2-амино-4-тиазолил 34g (0,24 г, 0,32 ммоля), в СН2Сl2(5 мл) при КТ добавляли ДИПЭА (0,55 мл, 3,18 ммоля, 10 экв.) и метилхлорформиат (0,13 мл, 1,6 ммоля, 5 экв.). Реакционную смесь перемешивали в течение 6,5 ч, после чего ее концентрировали в вакууме. После этого неочищенный выделенный продукт подвергали гидролизу как описано в примере 34, с образованием требуемой карбоновой кислоты, получая соединение №818
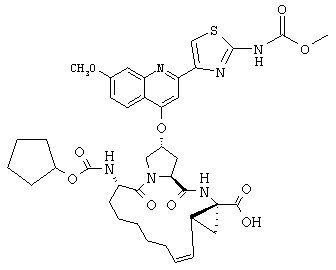
1Н ЯМР (400 МГц, ДMCO-d6): δ 8,61 (s, 1H), 8,21-8,07 (m, 2H), 7,61-7,38 (m, 2H), 7,26 (d, J=6,4 Гц, 1H), 7,19-7,10 (m, 1H), 5,60-5,47 (m, 2H), 5,27 (dd, J=9,2 Гц, 1H), 4,63-4,53 (m, 1H), 4,47 (d, J=7,9 Гц, 1H), 4,13-4,04 (m, 1H), 3,93 (s, 3Н), 3,92-3,87 (m, 2H), 3,79 (s, 3Н), 2,42-2,30 (m, 2H), 2,17 (dd, J=9,2 Гц, 1H), 1,81-1,68 (m, 2H). 1,63-1,29 (m, 16H), 1,23-1,10 (m, 2H). MC; es+: 791,1 (M+H)+, es-: 789,1 (M-H)-.
Пример 34е
Используя условия, описанные в примере 34д, но применяя изобутилхлорформиат, получали промежуточный продукт, представляющий собой соответствующий замещенный изобутилхлорформиат. Затем неочищенный выделенный продукт подвергали гидролизу, получая требуемое №819
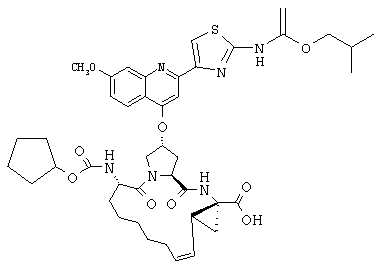
1Н ЯМР (400 МГц, ДМСО-d6): δ 8,62 (s, 1H), 8,47-8,27 (bs, 1H), 8,18 (d, J=8,6 Гц, 1H), 7,69-7,60 (m, 1H), 7,60-7,51 (m, 1H), 7,28 (d, J=6,7 Гц, 1H), 7,28-7,19 (m, 1H), 5,70-5,60 (m, 1H), 5,52 (dd, J=8,3 Гц, 1Н), 5,27 (dd, J=9,8 Гц, 1H), 4,63 (d, J=11,8 Гц, 1Н), 4,53-4,44 (m, 2H), 4,10-3,99 (m, 1H), 4,04 (d, J=6,7 Гц, 2Н), 3,95 (s, 3H), 3,94-3,87 (m, 1H), 2,65-2,53 (m, 1H), 2,46-2.34 (m, 1H), 2,16 (dd,J=8,1 Гц, 1H), 2,03-1,91 (m, 1H), 1,79-1,09 (m, 20H), 0,95 (d, J=6,7 Гц, 6Н). MC; es+: 833,2 (M+H)+, es-: 831,2 (M-Н)-.
Пример 35
Синтез соединения №908
Используя в качестве исходного продукта производное 27а и такую же процедуру синтеза, которая описана в примере 34, получали указанный ниже макроцикл, соединение №908 (таблица 9).
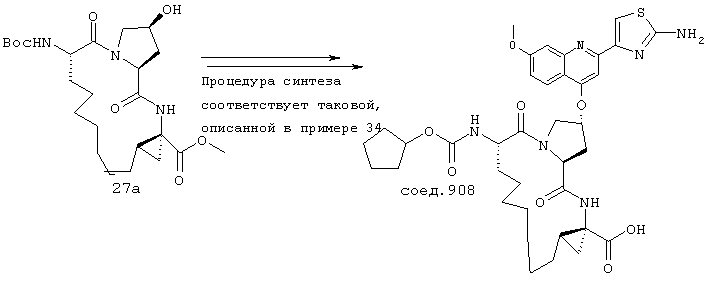
1Н ЯМР (400 МГц, ДМСО-d6): δ 8,47 (s, 1H), 8,16 (d, J=10 Гц, 1H), 8,15-8,07 (m, 1H), 7,82-7,63 (m, 2H), 7,53-7.43 (m, 2H), 7,33-7,22 (m, 1H), 7,13 (d, J=7 Гц, 1H), 5,77-5,65 (m, 1H), 4,62-4,52 (m, 2H), 4,50-4,4 (m, 1H), 4,20-4,10 (m, 1H), 3,94 (s, 3H), 3,89-3,83 (m, 1H), 2,59-2,53 (m, 1H), 2,48-2,40 (m, 1H), 1,79-1,0 (m, 25H). MC; es+: 735,2 (M+H)+, es-: 733,2 (M-H)-.
Пример 35а
Синтез соединения №909
Используя такую же процедуру синтеза, которая описана в примере 35, но применяя соответствующую N-ацетилтиомочевину, получали соединение №909 (таблица 9).

1Н ЯМР (400 МГц, ДМСО-d6): δ 8,53-8,41 (m, 2H), 8,20 (d, J=9,2 Гц, 1Н), 7,68 (bs, 1H), 7,68 (bs, 1H), 7,27 (dd, J=9,2 Гц, 1H), 7,15 (d, J=6,4 Гц,1Н), 5,67 (bs, 1H), 4,65-4,50 (m, ЗН), 4,44-4,37 (m, 1H), 4,21-4,13 (m, 1H), 3,96 (s, 3H), 3,99-3,86 (m, 1H), 2,62-2,39 (m, 2H), 2,24 (s, 3H), 1,78-1,67 (m, 3H), 1,67-1,01 (m, 22H).
MC; es+: 798,0 (M+Na)+, es-: 777,0 (M+Н)+.
Пример 35б
Синтез соединения №910
Используя такую же процедуру синтеза, которая описана в примере 35, но применяя соответствующую N-этилтиомочевину, получали соединение №910 (таблица 9).
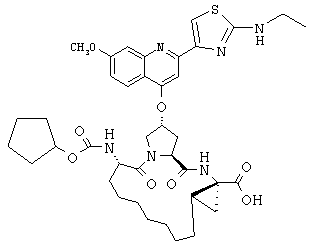
1Н ЯМР (400 МГц, ДМСО-d6): δ 8,47 (s, 1H), 8,29 (bs, 1H), 8,20 (d, J=9,2 Гц, 1H), 8,09 (bs, 1H), 7,87 (s, 1H), 7,77 (s, 1H), 7,32 (dd, J=9,2 Гц, 1H), 7,14 (dd, J=6,7 Гц, 1H), 5,78 (bs, 1H), 4,58 (dd, J=8,1 Гц, 2H), 4,43 (bs, 1H), 4,18-4,12 (m, 1H), 3,97 (s, 3Н), 3,87 (d, J=8,9 Гц, 1H), 3,55-3,46 (m, 2H), 2,63-2,53 (m, 1H), 2,47-2,41 (m, 1H), 1,78-1.00 (m. 25Н), 1,25 (t, J=7,3 Гц, 3Н).). MC; es+: 763,1 (M+H)+, es-: 761,1 (M-H)-.
Пример 35в
Синтез соединения №911
Используя такую же процедуру синтеза, которая описана в примере 35, но применяя соответствующую N-изопропилтиомочевину, получали соединение №911 (таблица 9).
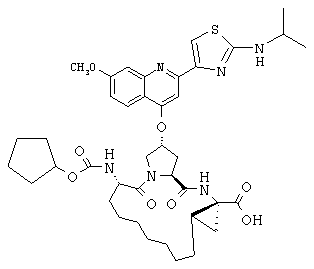
1Н ЯМР (400 МГц, ДМСO-d6): δ 8,47 (s, 1H), 8,29-8,19 (m, 1H), 8,19 (d, J=9,2 Гц, 1H), 8,09-8,0 (m, 1H), 7,83 (bs, 1H), 7,74 (bs, 1H), 7,31 (d, J=8 Гц, 1H), 7,14 (d, J=6,4 Гц, 1H), 5,76 (bs, 1H), 4,64-4,53 (m, 2H), 4,44 (bs, 1H), 4,22-4,09 (m, 3H), 3,97 (s, 3H), 3,87 (d, J=8,6 Гц, 1H), 2,63-2,58 (m, 1H), 2,46-2,41 (m, 1H), 1,79-1,10 (m. 24H), 1,27 и 1,26 (2 x d, J=6,5 Гц, 6Н). MC; es+: 777,0 (M+H)+, es-: 775,0 (M-Н)-.
Пример 36
Синтез соединения №716
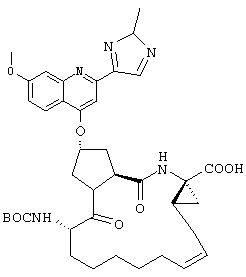
1Н ЯМР (400 МГц, ДМСО-d6): δ (част./млн): 8,62 (s, 1H), 8,13 (d, J=9,2 Гц, 1H), 7,64-7,54 (m, 2H), 7,47 (d, J=2,6 Гц. 1H), 7,16 (dd, J=9,2, 2,2 Гц, 1H), 7,03 (d, J=6,0 Гц, 1H), 5,63 (s, 1H), 5,52 (q, J=9,9 Гц, 1H), 5,26 (t, J=8,9 Гц, 1H),4.62 (d, J=11,45, 1H), 4,45 (dd, J=9,2, 8,27 Гц, 1H), 4,02 (m, 1H) 3,93 (s, 3H), 3,7 (dd, J=7,6, 1,0 Гц, 1H), 2,66 (s, 3H), 2,55-2,65 (m, 1H), 2,35-2,45 (m, 1H), 2,17 (q, J=8,6 Гц, 1H), 1,65-1,75 (m, 2H), 1,5-1,35 (m, 7H), 1,15(s, 9H).
MC:705(M+1),703(M-1).
Пример 37
Синтез соединения №717
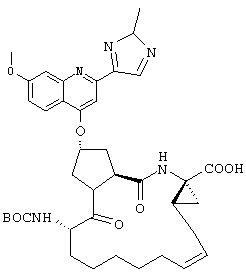
1H ЯМР (400 МГц, ДМСО-d6): δ (част./млн): 8,62 (s, 1H), 8,15 (d, J=8,9 Гц, 1Н), 7,62 (s, 1H), 7,49 (s, 1H), 7,19 (dd, J=9,2, 2,2 Гц, 1H), 7,02 (d, J=5,4 Гц, 1H), 5,64 (s, 1H), 5,52 (q, J=9,9 Гц, 1H), 5,26 (t, J=9,2 Гц, 1H), 4,63 (d, J=11,44, 1H), 4,45 (t, J=9,2 Гц, 1Н), 3,94 (s, ЗН), 3,9-3,8 (m, 1H), 2,7-2,55 (m, 1H), 2,4-2,3 (m, 1H), 2,18 (q, J=8,9 Гц, 1H), 1,75-1,65 (m, 2Н), 1,5-1,2 (m, 7H), 1,14 (s, 9H).
MS:705(M+1),703(M-1).
Пример 38
Синтез соединения №718

1Н ЯМР (400 МГц, ДMCO-d6): δ (част./млн): 9,55 (s, 1H), 8,63 (s, 1H), 8,43 (s, 1H), 8,13 (d, J=9,2 Гц, 1 H), 7,66 (s, 1 H), 7,46 (s, 1 H), 7,32 (d, J=2,6 Гц, 1 H), 7,10-7,07 (m, 2Н), 5,64-5,54 (m, 1H), 5,59-5,48 (m, 1H), 5,33-5,23 (m, 1H), 4,73-4,61 (m, 1H), 4,45 (dd, J=7,5, 9,1 Гц, 1H), 4,09-4,00 (m, 1H), 3,92 (s, 3H), 3,93-3,83 (m, 1H), 2,67-2,55 (m, 2H), 2,53-2,43 (m, 1H), 2,42-2,31 (m, 1H), 2,23-2,12 (m, 1Н), 1,81-1,66 (m, 2H), 1,52-1,42 (m, 2H), 1,42-1,25 (m, 6H), 1,21 (s, 9H).
МС: 689,3 (М+1), 687,3 (М-1).
Пример 39
Синтез соединения №722
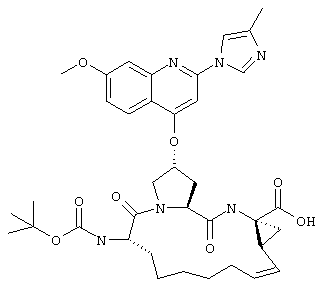
1Н ЯМР (400 МГц, ДМСО-d6): δ (част./млн): 9,70 (s, 1Н), 8,64 (s, 1H), 8,26 (s, 1H), 8,14 (d, J=9,2 Гц, 1H), 7,45 (s, 1H), 7,30 (d, J=2,5 Гц, 1H), 7,14-7,06 (m, 2H), 5,60-5,54 (m, 1H), 5,58-5,48 (m, 1H), 5,31-5,23 (m, 1H), 4,71-4,62 (m, 1H), 4,49-4,40 (m, 1H), 4,08-3,99 (m, 1H), 3,92 (s, 3H), 3,92-3,84 (m, 1H), 2,69-2,54 (m, 2H), 2,53-2,46 (m, 1H), 2,42-2,31 (m, 1H), 2,37 (s, 3H), 2,22-2,13 (m, 1H), 1,81-1,64 (m, 2H), 1,54-1,42 (m, 2H), 1,42-1,27(m, 6H), 1,22 (s,9H).
МС: 703,3 (М+1), 701,3 (М-1)
Пример 40
Синтез соединения №733
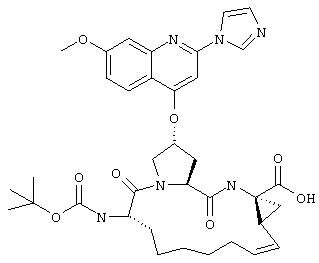
1Н ЯМР (400 МГц, ДМСО-d6): δ (част./млн): 8,75 (m, 1H), 8,62 (s, 1H), 8,06 (d, J=9,2 Гц, 1H), 7,88-7,87 (m, 1H), 7,48 (s, 1H), 7,28 (d, J=2,6 Гц, 1H), 7,05-7,00 (m, 2H), 6,64-6,63 (m, 1H), 5,62-5,58 (m, 1H), 5,55-5,49 (m, 1H), 5,28-5,24 (m, 1H), 4,64-4,61 (m, 1H), 4,48-4,44 (m, 1H), 4,07-4,03 (m, 1H), 3,91 (s, 3H), 3,92-3,85 (m, 1H), 2,67-2,54 (m, 2H), 2,53-2,45 (m, 1H), 2,41-2,34 (m, 1H), 2,20-2,14 (m, 1H), 1,75-1,69 (m, 2H), 1,50-1,43 (m, 2H), 1,41 -1,32 (m, 6H), 1,17 (s, 9H).
МС: 689,3 (М+1), 687,2 (М-1).
Пример 41
Синтез соединения №703
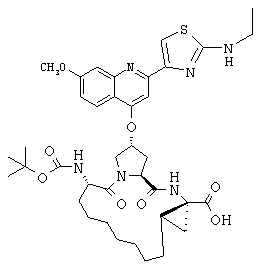
1Н ЯМР (400 МГц, ДМСО-d6): δ 8,50 (s, 1H), 8,19 (s, 1Н), 8,17 (s, 1H), 8,11-8,00 (m, 1H), 7,88-7,77 (m, 1H), 7,73 (s, 1H), 7,25 (d, J=8,6 Гц, 1 H), 6,93 (d, J=6 Гц, 1H), 5,89-5,68 (m, 1H), 4,62 (d, J=11 Гц, 1Н), 4,53 (dd, J=8,3 Гц, 1Н), 4,16-4,07 (m, 1H), 3,96 (s, ЗН), 3,88 (bd, J=9,5 Гц, 1H), 3,53-3,43 (m, 2H), 2,63-2,51 (m, 1H), 2,46-2,36 (m, 1H), 1,81-1,62(m, 2H), 1,60-1,01 (m, 15Н), 1,24 (t, J=7,4 Гц, ЗН), 1,17 (s, 9H). МС: es+:751,1 (М+Н)+, es-: 749,1-(М-Н)-.
Пример 42
Синтез соединения №734
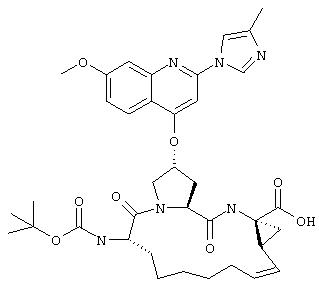
1H ЯМР (400 МГц, ДМСО-d6): δ (част./млн): 8,62 (s, 1H), 8,54 (s, 1H), 8,04 (d, J=9,2 Гц, 1H), 7,70 (s, 1H), 7,43 (s, 1H), 7,24 (d, J=2,6 Гц, 1H), 7,05-6,98 (m, 2H), 5,57-5,54 (m, 1H), 5,55-5,48 (m, 1H), 5,28-5,24 (m, 1H), 4,63-4,59 (m, 1H), 4,47-4,43 (m, 1H), 4,13-3,99 (m, 1H), 3,90 (s, 3Н), 3,92-3,83 (m, 1H), 2,67-2,55 (m, 2H), 2,53-2,46 (m, 1H), 2,43-2,31 (m, 1H), 2,22-2,15 (m, 1H), 2,15 (3Н), 1,75-1,70 (m, 2H), 1,51-1,42 (m, 2H), 1,41-1,28 (m, 6H), l,17(s,9H).
MC: 703,2 (М+1), 701,3 (М-1).
Пример 43
Синтез соединения №738
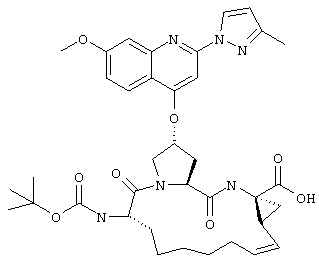
1Н ЯМР (400 МГц, ДМСО-d6): δ (част./млн): 8,64 (d, J=2,5 Гц, 1H), 8,62 (s, 1 Н), 8,04 (d, J=9,2 Гц, 1H), 7,39 (s, 1H), 7,24 (d, J=2,5 Гц, 1H), 7,04 (d, J=6,0 Гц, 1Н), 6,99 (dd, J=2,2, 9,2 Гц, 1H), 6,43 (d, J=2,2 Гц, 1H), 5.62-5,57 (m, 1H), 5,56-5,47 (m, 1H), 5,31-5,22 (m, 1H), 4,65-4,56 (m, 1H), 4,45 (dd, J=7,6, 8,9 Гц, 1H), 4,07-4,00 (m, 1H), 3,90 (s, 3Н), 3,88-3,84 (m, 1H), 2,68-2,56 (m, 2H), 2,54-2,43 (m, 1H), 2,42-2,31 (m, 1H), 2,34 (s, ЗН), 2,24-2,14 (m, 1H), 1,80-1,64 (m, 2H), 1,52-1,43 (m, 2H), 1,43-1,27 (m, 6H), 1,18(s, 9Н).
МС: 703,2 (М+1), 701,2 (М-1)
Пример 44
Синтез соединения №725
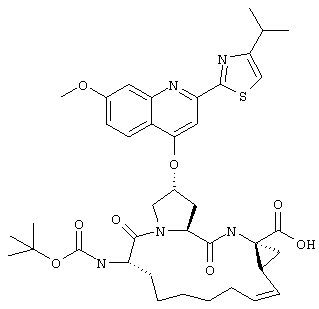
1Н ЯМР (400 МГц, ДМСО-d6): δ (част./млн): 8,62 (s, 1H), 8,10 (d, J=9,2 Гц, 1Н), 7,57 (s, 1H), 7,49 (s, 1H), 7,35 (d, J=2,2 Гц, 1H), 7,09-7,03 (m, 2H), 5,65-5,61 (m, 1H), 5,55-5,49 (m, 1H), 5,28-5,24 (m, 1H), 4,62-4,57 (m, 1H), 4,49-4,45 (m, 1H), 4,08-4,01 (m. 1H), 3,93 (s, 3Н), 3,92-3,86 (m, 1H), 3,20-3,14 (m, 1H), 2,65-2,56 (s, 1H), 2,53-2,47 (m, 1H), 2,42-2,35 (m, 1H), 2,22-2,15 (m, 1H), 1,79-1,68 (m, 2H), 1,50-1,43 (m, 2H), 1,41-1,28 (m, 12H), 1,18(s,9H).
MC: 748,2 (M+1), 746,2 (M-1).
Пример 45
Синтез соединения №726
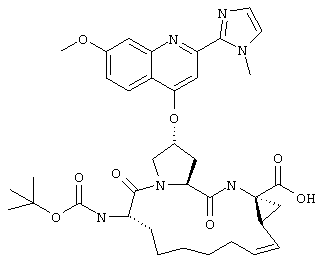
1H ЯМР (400 МГц, ДМСО-d6): δ (част./млн): 8,64 (s, 1H), 8,10 (d, J=9,5 Гц, 1Н), 7,83-7,76 (m, 2H), 7,60 (s, 1H), 7,44-7,42 (m, 1H), 7,18-7,01 (m, 2H), 5,56-5,49 (m, 2H), 5,29-5,24 (m, 1H), 4,66-4,63 (m, 1H), 4,47-4,42 (m, 1H), 4,28 (s, ЗН), 4,06-4,02 (m, 2H), 3,94 (s, ЗН), 3,93-3,86 (m, 1H), 2,66-2,55 (m, 2H), 2,42-2,31 (m, 2H), 2,22-2,14 (m, 1H), 1,79-1,65 (m, 2H), 1,52-1,27 (m, 7H), 1,22 (s, 9H).
МС:703,2(М+1),701,3(М-1).
Пример 46
Синтез соединения №906
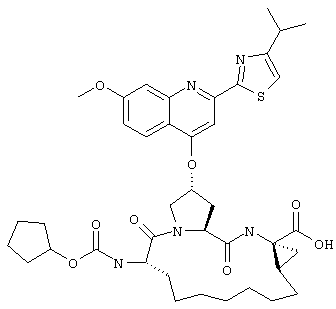
1H ЯМР (400 МГц, ДМСО-d6): δ (част./млн): 8,46 (s, 1H), 8,06 (d, J=9,2 Гц, 1H), 7,57 (s, 1H), 7,49 (s, 1H), 7,34 (m, 1H), 7,14-7,05 (m, 2H), 5,63-5,58 (m, 1H), 4,66-4,61 (m, 1H), 4,54-4,44 (m, 2H), 4,23-4,18 (m, 1H), 3,93 (s, 3Н), 3,92-3,88 (m, 1H), 3,21-3,14 (m, I H), 2,44-2,33 (m, 1H), 1,35 (d, J=7 Гц, 6Н), 1,73-1,01 (m, 26H)
MC: 762,0 (M+1), 759,9 (M-1).
Пример 47
Синтез соединения №907
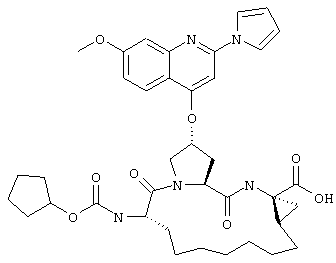
1Н ЯМР (400 МГц, ДМСО-d6): δ (част./млн): 8,46 (s, 1Н), 7,98 (d, J-8,9 Гц, 1Н), 7,91-7,89 (m, 2H), 7,23-7,21 (m, 2Н), 7,07-7,00 (m, 2H), 6,35-6,32 (m, 2H), 5,64-5,58 (m, 1H), 4,65-4,61 (m, 1H), 4,53-4,47 (m, 2H), 4,24-4,19 (m, 1H), 3,90 (s, 3H), 3,86-3,84 (m, 1H), 2,40-2,33 (m, 1H), 1,73-1,01 (m, 26H).
MC: 702,0 (M+1), 699,9 (M-1).
Пример 48
Флуорогенный анализ полноразмерного гетеродимерного протеина NS3-NS4A
3’-Некодирующую область NS2-NS5B клонировали с помощью ОТ-ПЦР в векторе pCR®3 (фирма Invitrogen), используя РНК, экстрагированную из сыворотки человека, зараженного генотипом HCV 1b (предоставленной Др. Bernard Willems, Hopital St-Luc, Монреаль, Квебек, Канада). Затем NS3-NS4A-o6nacTb (NS3-NS4AFL) субклонировали с помощью ПЦР в экспрессионном бакусоловирусном векторе pFastBac™HTa (фирма Gibco/BRL). Последовательность вектора включает область, кодирующую состоящую из 28 остатков N-концевую последовательность, которая содержит метку из шести остатков гистидина. Для получения рекомбинантного бакуловируса использовали бакуловирусную систему экспрессии Вас-to-Вас™ (фирма Gibco/BRL). Протеин His-NS3-NS4AFL экспрессировали, заражая при 27°С 106 клеток линии Sf21/MA рекомбинантным бакуловирусом с фактором заражения 0,1-0,2. При экспрессии происходил аутентичный автопротеолиз, в результате чего происходило образование нековалентного и стабильного протеинового комплекса NS3-NS4A (обозначенный как "FL"). Зараженную культуру собирали через 48-64 ч центрифугированием при 4°С. Клеточный дебрис гомогенизировали в смеси, содержащей 50мМ NaPO4, pH 7,5, 40% глицерина (маc./об.), 2мМ / β-меркаптоэтанол, в присутствии смеси ингибиторов протеаз. Затем His-NS3-NS4AFL экстрагировали из клеточного лизата с помощью 1,5% NP-40, 0,5% Тритон X-100, 0,5 М NaCl и обработки ДНКазой. После ультрацентрифугирования растворимый экстракт разбавляли в 4 раза и связывали с Ni-хелатирующей колонкой типа Pharmacia Hi-Trap. His-NS3-NS4AFL элюировали в форме, имеющей чистоту >90% (что подтверждено с помощью ДСН-ПААГ), используя градиент имидазола от 50 до 400 мМ. His-NS3-NS4AFL хранили при -80°С в смеси, содержащей 50 мМ фосфат натрия, рН 7,5, 10% (мас./об.) глицерина, 0,5 М NaCl, 0,25 M имидазол, 0,1% NP-40. Перед применением его подвергали оттаиванию на льду и разбавляли.
Протеазную активность His-NS3-NS4AFL оценивали в 50 мМ Tris-HCl, рН 8,0, 0,25М цитрате натрия, 0,01% (мас./об.) н-додецил-β-D-мальтозида, 1 мМ ТКЭФ. Инкубировали 5мкМ субстрата aнтpaнилил-DDIVPAbu[C(O)-O]-AMY(3-NO2)TW-OH, котором была прекращена внутренняя реакция, в присутствии различных концентраций ингибитора с 1,5М His-NS3-NS4AFL в течение 45 мин при 23°С. Конечная концентрация ДМСО не превышала 5,25%. Реакцию прекращали, добавляя 1 М МЭС, рН 5,8. Флуоресценцию N-концевого продукта определяли с помощью флуориметра типа Perkin-Elmer LS-50B, снабженного 96-луночным планшет-ридером (длина волны возбуждения: 325 нм; длина волны испускания: 423 нм).
Ингибирование (в %) определяли из следующего уравнения:
100-[(количество импульсовинг.-количество импульсовчист.контр.)/(количество импульсовконтр.-количество импульсовчист.конт. 100]
Данные о зависимости ингибирования от концентрации аппроксимировали нелинейной кривой с использованием модели Хилла и величину эффективной концентрации, при которой происходит 50%-ное ингибирование (IС50), рассчитывали с помощью программного обеспечения SAS (Statistical Software System; SAS Institute, Inc. Cary, N.C.).
Пример 49
Радиометрический анализ NS3-npoтеазы HCV
Субстрат, который используется для радиометрического анализа NS3-npoтеазы HCV, т.е. DDIVPC-SMSYTW, расщепляется с помощью фермента между остатками цистеина и серина. Последовательность DIVPC-SMSYTW соответствует естественному сайту расщепления NS5A/NS5B, в котором остаток цистеина в Р2 заменен на пролин. Пептидный субстрат DDIVPC-SMSYTW и метку биoтин-DDIVPC-SMS[125I-Y]TW инкубировали с рекомбинантой NS3-пpoтeaзoй в присутствии ингибиторов или без них. Отделение субстрата от продуктов проводили, добавляя покрытые авидином агарозные гранулы к смеси для анализа, с последующей фильтрацией. Количество продукта SMS[125I-Y]TW, обнаруженное в фильтрате (с ингибитором и без него), позволяет рассчитать процент превращения субстрата и процент ингибирования.
А. Реагенты
Трис и Трис-НСl (высокой степени чистоты (UltraPure)) получали от фирмы Life Technologies. Глицерин (UltraPure), МЭС и БСА получали от фирмы Sigma®. ТКЭФ получали от фирмы Pierce, ДМСО получали от фирмы Aldrich® и NaOH от фирмы Anachemia®.
Буфер для анализа: 50 мМ Трис-HCl, рН 7,5, 30% (мас./об.) глицерина, 2% (мас./об.) CHAPS, 1 мг/мл БСА, 1 мМ ТКЭФ (ТКЭФ добавляли непосредственно перед применением из 1 М маточного раствора в воде). Субстрат: DDIVPC-SMSYTW, конечная концентрация 25 мкМ (из 2 мМ маточного раствора в ДМСО, хранившегося -20°С для того, чтобы избежать окисления).
Метка: восстановленный монойодированный субстрат (биотин-DDIVPC-SMS[125I-Y]TW (конечная концентрация ~1 нМ).
NS3-протеаза типа 1b HCV, конечная концентрация 25 нМ (из маточного раствора, содержащего 50 мМ фосфат натрия, рН 7,5, 10% глицерина, 300 мМ NaCl, 5 мМ ДТТ, 0,01%NP-40).
Б. Протокол
Анализ проводили в 96-луночном полипропиленовом планшете. Каждая лунка содержала:
20 мкл субстрата/метки в буфере для анализа;
10 мкл ± ингибитора в 20% ДМСО/буфере для анализа;
10 мкл NS3-протеазы 1b.
На этом же планшете для анализа также готовили чистый контроль (без ингибитора и без фермента) и контроль (без ингибитора).
Ферментативную реакцию инициировали, добавляя раствор фермента, и анализируемую смесь инкубировали в течение 60 мин при 23°С при осторожном перемешивании. Добавляли 20 мкл 0,025н. NaOH для прекращения ферментативной реакции.
В фильтрационный планшет типа Millipore® MADP N65 добавляли 20 мкл покрытых авидином агарозных гранул (полученных от фирмы Pierce®). Смесь для анализа, в которой прекращена ферментативная реакция, переносили в фильтрационный планшет и инкубировали в течение 60 мин при 23°С при осторожном перемешивании.
Планшеты фильтровали с помощью устройства для вакуумной фильтрации типа Millipore® MultiScreen Vacuum Manifold Filtration и 40 мкл фильтрата переносили в непрозрачный 96-луночный планшет, содержащий по 60 мкл сцинтилляционной жидкости на лунку.
Радиоактивность фильтратов подсчитывали с помощью счетчика Packard® Top-Count, используя протокол, основанный на применении 125I-жидкости в течение 1 мин.
Ингибирование (в %) рассчитывали из следующего уравнения:
100-[(количество импульсовинг.-количество импульсовчист.котр.)/(количество импульсовинг.-количество импульсовчист.котр.)×100]
Данные о зависимости ингибирования от концентрации аппроксимировали нелинейной кривой с использованием модели Хилла и величину эффективной концентрации, при которой происходит 50%-ное ингибирование (IС50), рассчитывали с помощью программного обеспечения SAS (Statistical Software System; SAS Institute, Inc. Cary, N.C.).
Пример 50
Определение специфической активности
Специфическую активность соединений определяли в отношении различных серинпротеаз: эластазы лейкоцитов человека, эластазы поджелудочной железы свиньи и бычьего α-химотрипсина поджелудочной железы и одной цистеинпротеазы: катепсина В печени человека. Во всех случаях использовали протокол, основанный на применении 96-луночных планшетов и специфичного для каждого фермента хромогенного субстрата. Каждый анализ включал предварительную инкубацию в течение 1 ч фермента-ингибитора при комнатной температуре с последующим добавлением субстрата и гидролизом до ≈30% превращения, которое оценивали с помощью планшет-ридера для титрационннных микропланшетов типа UV Thermomax® или флуоресцентного планшет-ридера типа Perkin-Elmer® LS50B. Концентрации субстрата поддерживали на возможно более низком уровне по сравнению с КМ с целью уменьшения конкуренции субстрата. Концентрации соединений варьировали от 300 до 0,06 мкМ в зависимости от их активности. Конечные условия каждого анализа были следующими:
50 мМ Трис-НСl рН 8, 0,5М Nа2SO4 50 мМ NaCl, 0,1мМ ЭДТК, 3% ДМСО, 0,01% Твин-20 с
[100 мкМ Succ-AAPF-pNA и 250пМ α-химотрипсин], [133 мкМ Succ-AAA-pNA и 8 нМ эластаза свиньи], [133 мкМ Succ-AAV-pNA и 8 нМ эластаза лейкоцитов], или
[100 мМ NaHPO4 pH 6, 0,1 мМ ЭДТК, 3% ДМСО, 1 мМ ТСЭФ, 0,01% Твин-20, 4 мкМ Z-FR-AMC (7-амино-4-метилкумарин) и 0,5 нМ катепсин В (исходный фермент активировали перед применением в буфере, содержащем 20 мМ ТСЭФ)].
Ниже в качестве характерного примера обобщены условия для эластазы поджелудочной железы свиньи:
В плоскодонный полистироловый 96-луночный планшет (типа Cellwells, фирма Corning) добавляли, используя ручной заборник для жидкостей типа Biomek (фирма Beckman):
40 мкл буфера для анализа (50 мМ Трис-HCl, pH 8, 1 М Na2SO4 50 мМ NaCl, 0,1мМ ЭДТК);
20 мкл раствора фермента (50 мМ Трис-HCl, pH 8, 50 мМ NaCl, 0,1 мМ ЭДТК, 0,02% Твин-20, 40 нМ эластаза поджелудочной железы свиньи); и
20 мкл раствора ингибитора (50 мМ Трис-HCl, pH 8, 50 мМ NaCl, 0,1 мМ ЭДТК, 0,02% Твин-20, 1,5 мМ-0,3 мкМ ингибитор, 15 об.% ДМСО).
После предварительной инкубации в течение 60 мин при комнатной температуре в каждую лунку добавляли по 20 мкл раствора субстрата (50 мМ Трис-HCl, pH 8,0,5 М Na2SO4 50 мМ NaCl, 0,1 мМ ЭДТК, 665 мкМ Succ-AAA-pNA) и реакционную смесь дополнительно инкубировали в течение 60 мин при комнатной температуре, после чего определяли абсорбцию с помощью планшет-ридера типа UV Thermomax®. Ряды лунок оставляли для контрольного варианта (без ингибитора) и для чистого контроля (без ингибитора и без фермента).
Проводили последовательные двукратные разбавления раствора ингибитора на отдельном планшете с помощью ручного заборника жидкости, используя 50 мМ Трис-HCl, pH 8, 50 мМ NaCl, 0,1 мМ ЭДТК, 0,02% Твин-20, 15 об. % ДМСО.
Все опыты по определению специфической активности проводили аналогичным образом.
Ингибирование (в %) определяли из следующего уравнения:
[1-((УФинг.-УФчист.контр.)/(УФконтр.-Уфчист.контр.))]×100
Данные о зависимости ингибирования от концентрации аппроксимировали нелинейной кривой с использованием модели Хилла и величину эффективной концентрации, при которой происходит 50%-ное ингибирование (IС50), рассчитывали с помощью программного обеспечения SAS (Statistical Software System; SAS Institute, Inc. Cary, N.C.).
Пример 51
Анализ NSS-протеазы на клеточном уровне
Этот анализ проводили с использованием клеток линии Huh-7, представляющих собой линию клеток человека, полученную из гепатомы, которую совместно трансфектировали двумя конструкциями ДНК:
первая (обозначенная как NS3), экспрессирующая часть неструктурного полипротеина HCV, слитую с протеином tTA через сайт расщепления NS5A-NS5B в следующей последовательности: NS3-NS4A-NS4B-NS5A-(NS5B)tTA, где (NS5B) обозначает первые 6 аминоксилот NS5B.
Этот полипротеин экспрессируют под контролем промотора CMV, - вторая (обозначена как SEAP), экспрессирующая репортерный протеин, секретирующий щелочную фосфатазу (SEAP) под контролем чувствительного к tTA промотора.
Первая конструкция позволяет осуществить экспрессию полипротеина, из которого после расщепления NS3-пpoтeaзoй высвобождаются различные зрелые протеины. Вероятно, зрелые вирусные протеины формируют комплекс на мембране эндоплазматического ретикулума. tTA представляет собой слитый протеин, описанный Gossen и Bujard (Proc. Natl. Acad. Sci. USA, 89 (1992): 5547-5551), который содержит ДНК-связывающий домен и активатор транскрипции. Высвобождение протеина tTA требует зависящего от NS3 расщепления в сайте расщепления NS5A-NS5B между NS5A и им самим. Это последнее расщепление дает tTA возможность мигрировать в ядро и тран-сактивировать ген SEAP. Таким образом, восстановление протеолитической активности NS3 должно привести к ограничению области нахождения tTA цитоплазмой и способствовать понижению активности SEAP.
Для оценки на клеточном уровне других видов активности соединения, отличных от ингибирования NS3-npoтеазы, осуществляли параллельную совместную трансфекцию конструкцией, экспрессирующей только tTA, и такой же репортерной конструкцией, в которой SEAP-активность не зависела от протеазы NS3.
Протокол анализа: клетки линии Huh-7, выращенные в CHO-SFMII (фирма Life Technologies) + 10% ФТС (фетальная телячья сыворотка), совместно трансфектировали двумя конструкциями ДНК, взятыми в следующих соотношениях:
7 мкг NS3 + 500 нг SEAP + 800 мкл FuGenel (фирма Boehringer Mannheim) на 4×106 клеток Huh-7. После инкубации в течение 5 ч при 37° клетки промывали, обрабатывали трипсином и высевали (80000 клеток/лунку) в 96-луночные планшеты, содержащие различные концентрации тестируемых соединений. После 24-часового периода инкубации активность SEAP в среде определяли с помощью набора Phospha-Light (фирма Tropix).
Оценивали процент ингибирования активности SEAP в зависимости от концентрации соединения с помощью программного обеспечения SAS, получая значение IC50.
Таблицы соединений
В следующих таблицах перечислены репрезентативные соединения по изобретению. Обнаружено, что все соединения, представленные в таблицах 1-9, обладают ферментативной активностью, выявленной с помощью анализа, описанного в примере 48. Число, помеченное звездочкой (*) обозначает ферментативную активность, которая была определена с помощью радиометрического анализа, описанного в примере 49, для которой IС50 ниже примерно 50 мкМ. В этих ферментативных анализах используют следующие градации: А≥1 мкм; 1 мкм>В>0,1 мкМ и С≤0,1 мкМ.
Некоторые соединения тестировали в отношении их специфической активности с помощью анализа, описанного в примере 50, и было обнаружено, что они обладают специфичностью в отношении NS3-протеазы. В целом, результаты анализов по определению специфичности позволяют установить, что изученные соединения обладают следующей активностью: HLE>300 мкМ; РРЕ>300 мкМ; α-Chym.>300 мкМ; Cat.В>300 мкМ, что свидетельствует о том, что они обладают высокой специфичностью в отношении NS3-протеазы и следует ожидать отсутствие у них серьезных побочных воздействий.
Кроме того, некоторые из этих соединений тестировали на клеточном уровне в опыте, описанном в примере 51, и было установлено, что они обладают активностью, причем значение ЕС50 составляло менее 10 мкМ, что с большой вероятностью позволяет предположить, что эти соединения могут пересекать клеточную мембрану. В частности, соединения из таблиц 7, 8 и 9 были протестированы на клеточном уровне и данные об их активности приведены в последней колонке. При описании результатов, полученных на клеточном уровне, используют следующие градации: А>1 мкМ; В≤1 мкМ. В приведенных далее таблицах использовали следующие сокращения:
МС: данные масс-спектрометрии с использованием электроспрея; значение m/z представляет собой МН4, за исключением обозначенных звездочкой (*) случаев, когда m/z представляет собой МН-; Ас: ацетил; Вn: бензил; Воc: трет-бутоксикарбонил Ph: фенил: Рr: пропил.
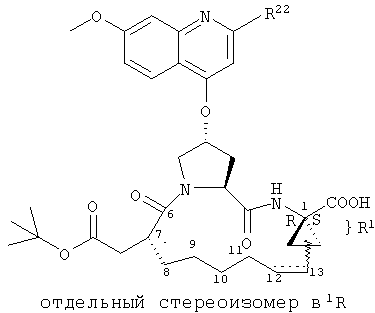

12-OH цис
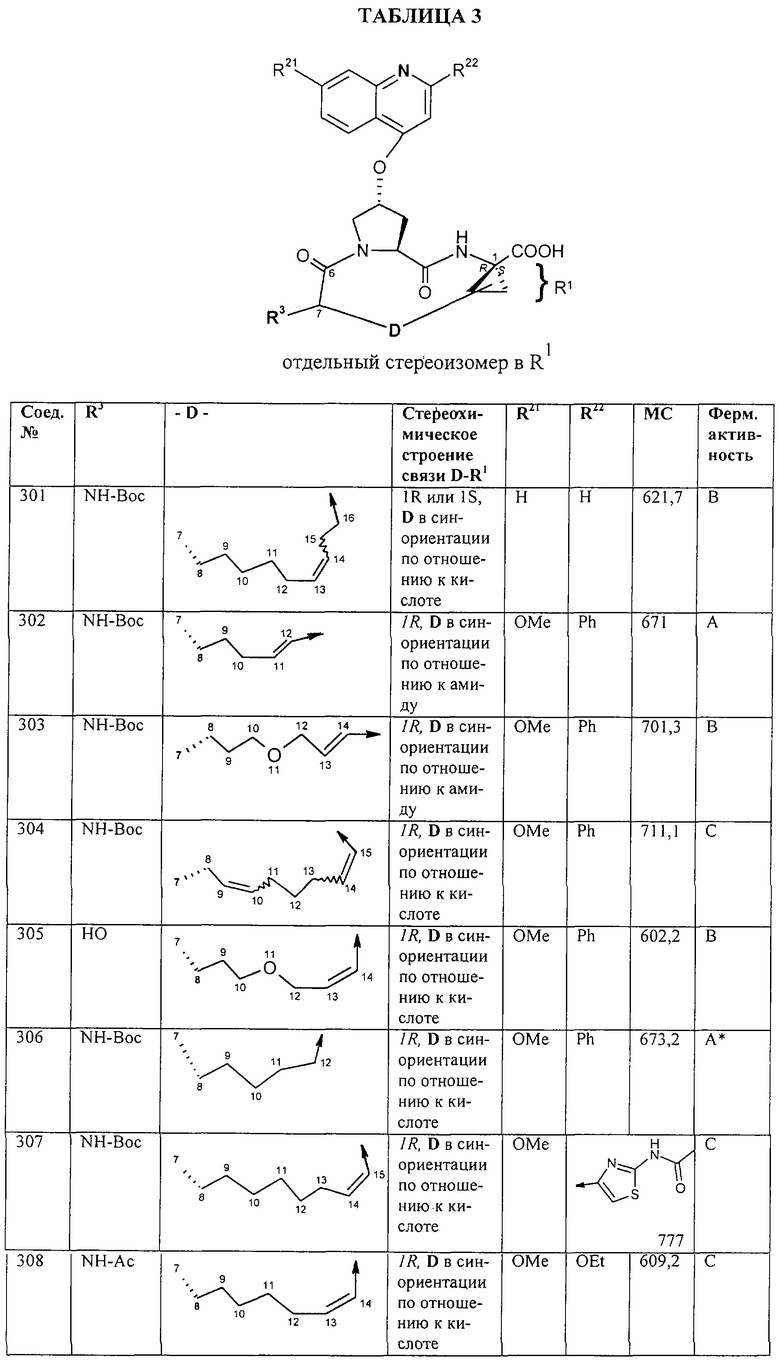
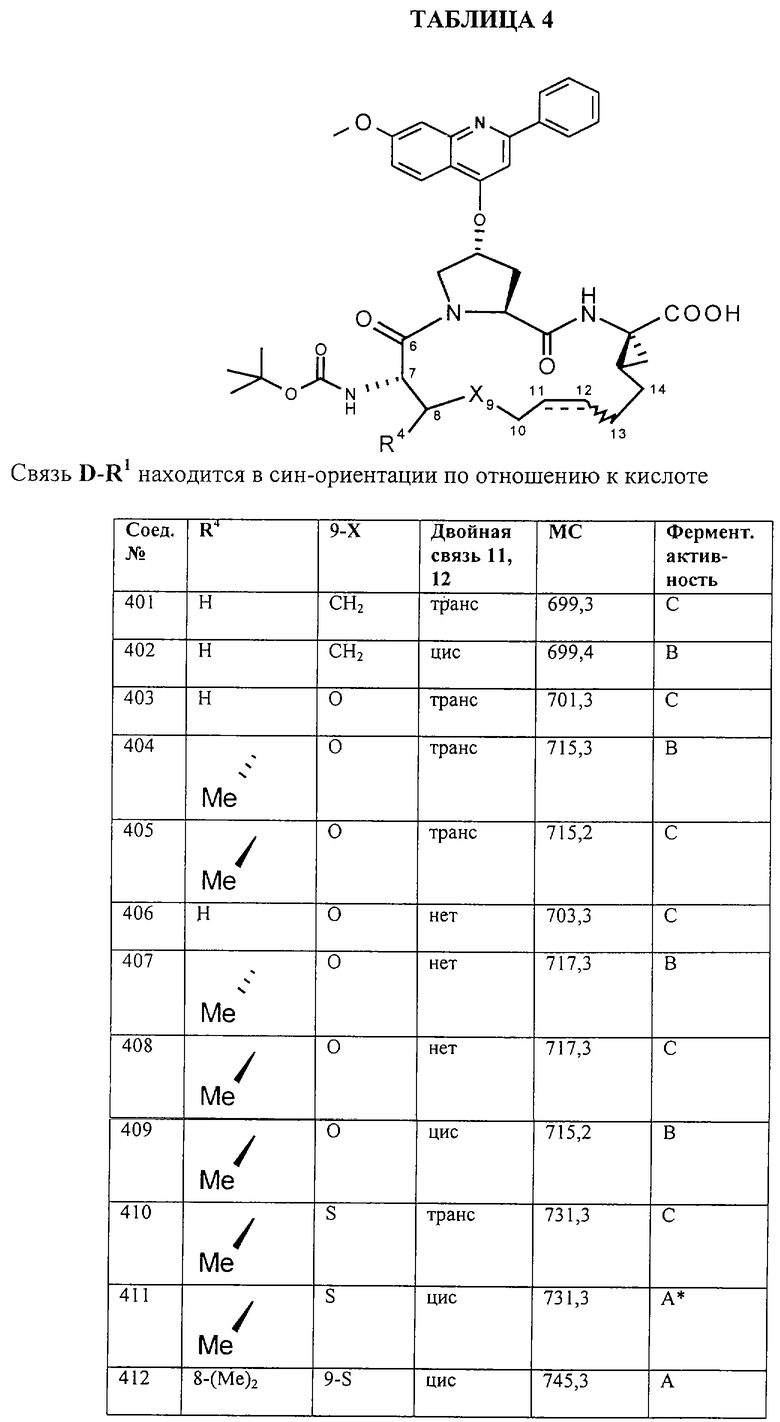
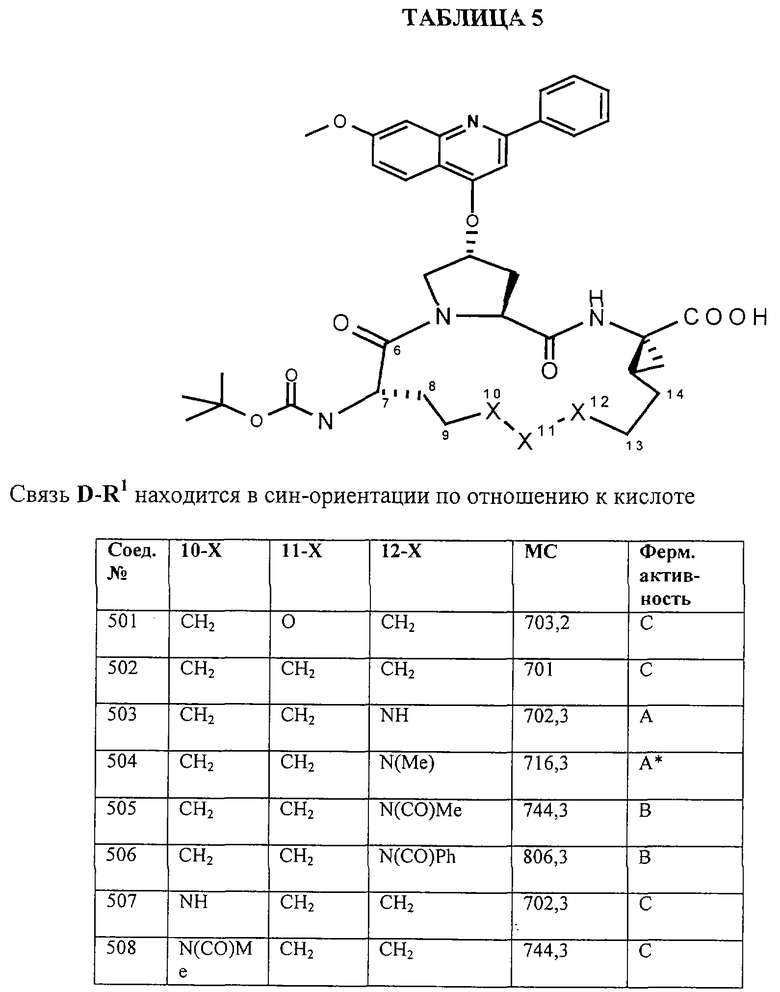
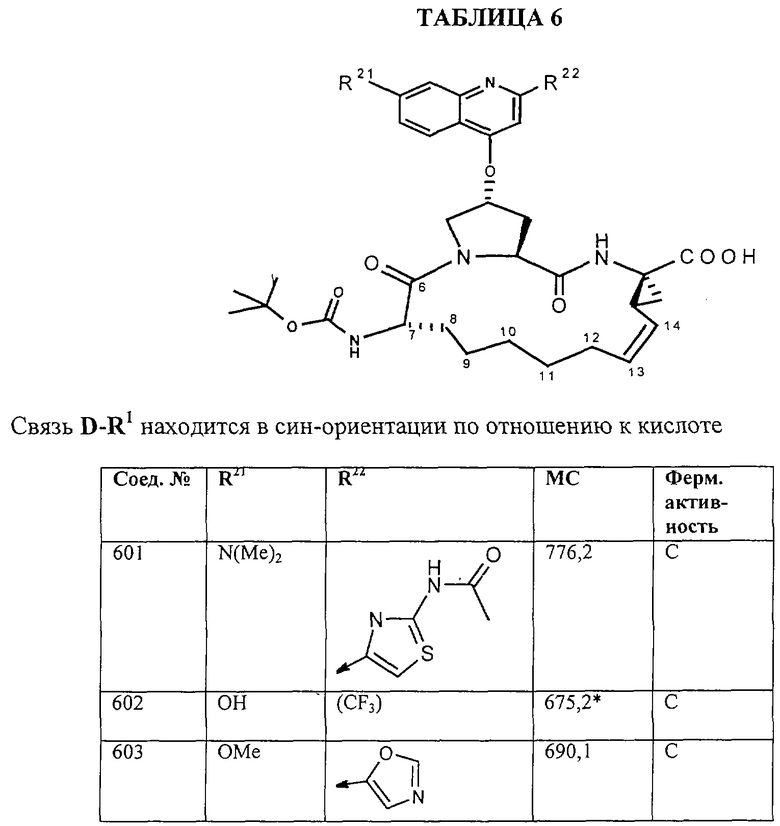

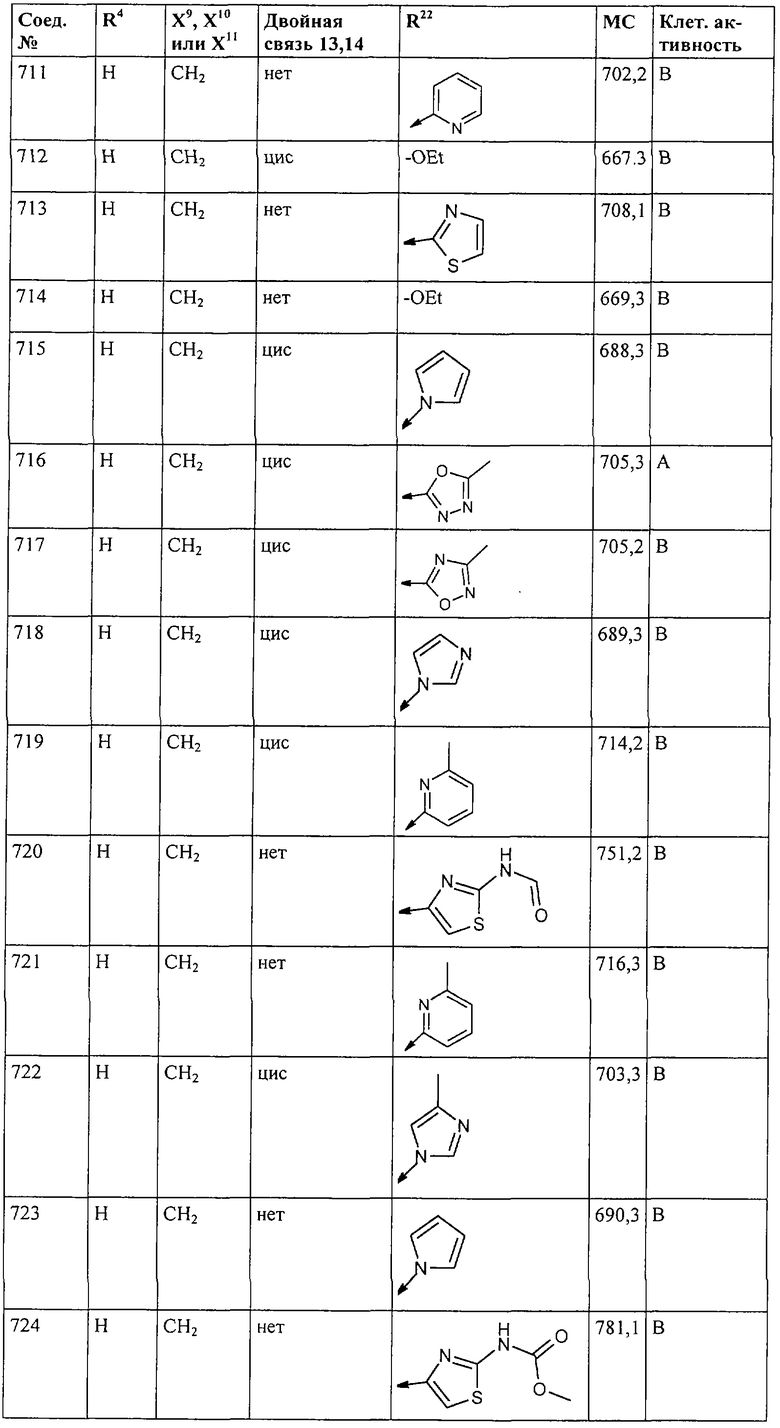
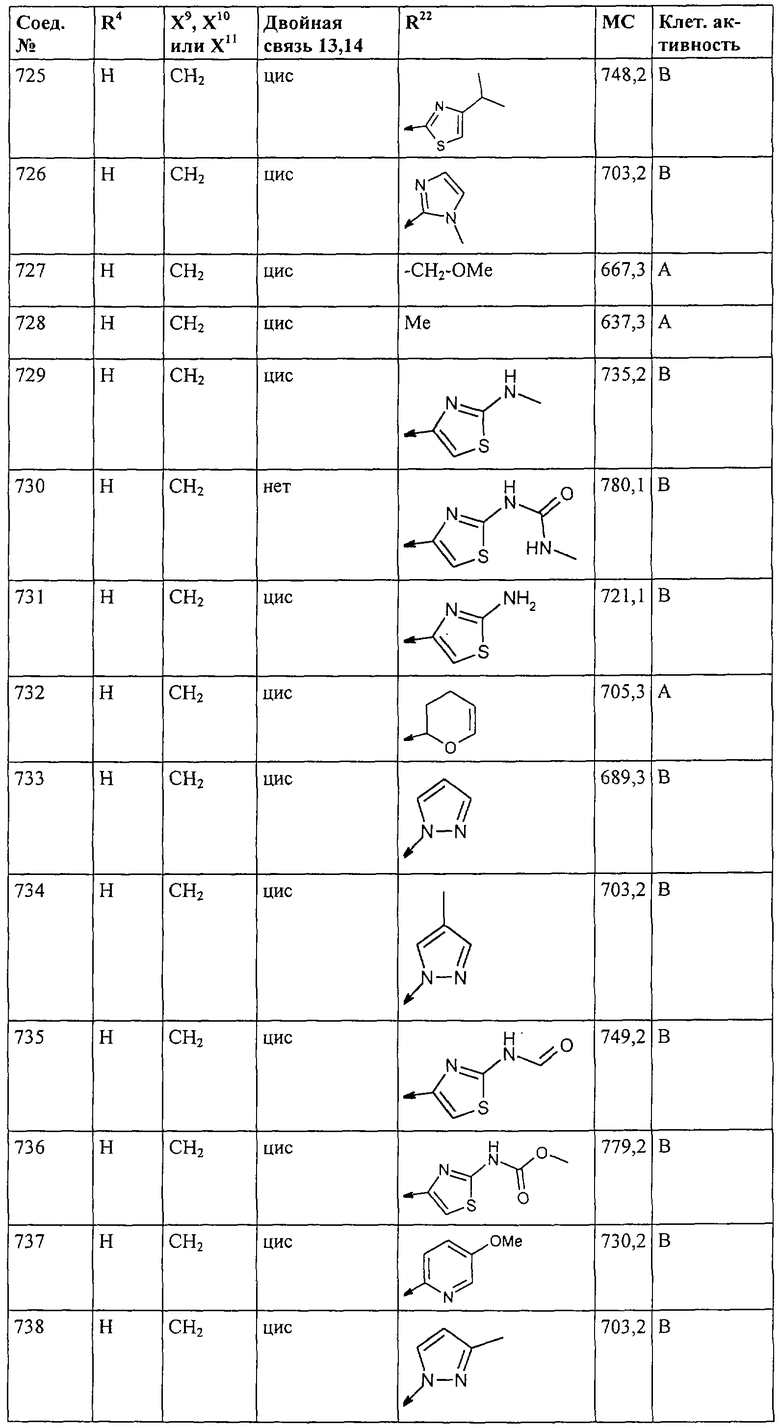
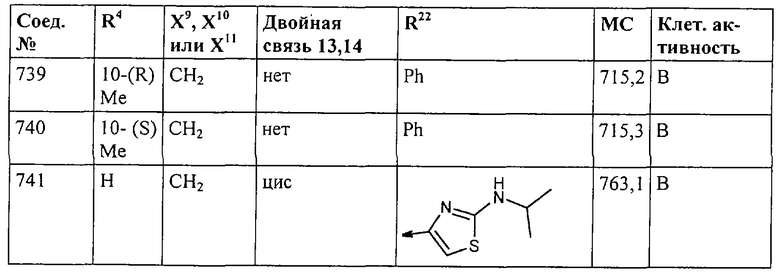
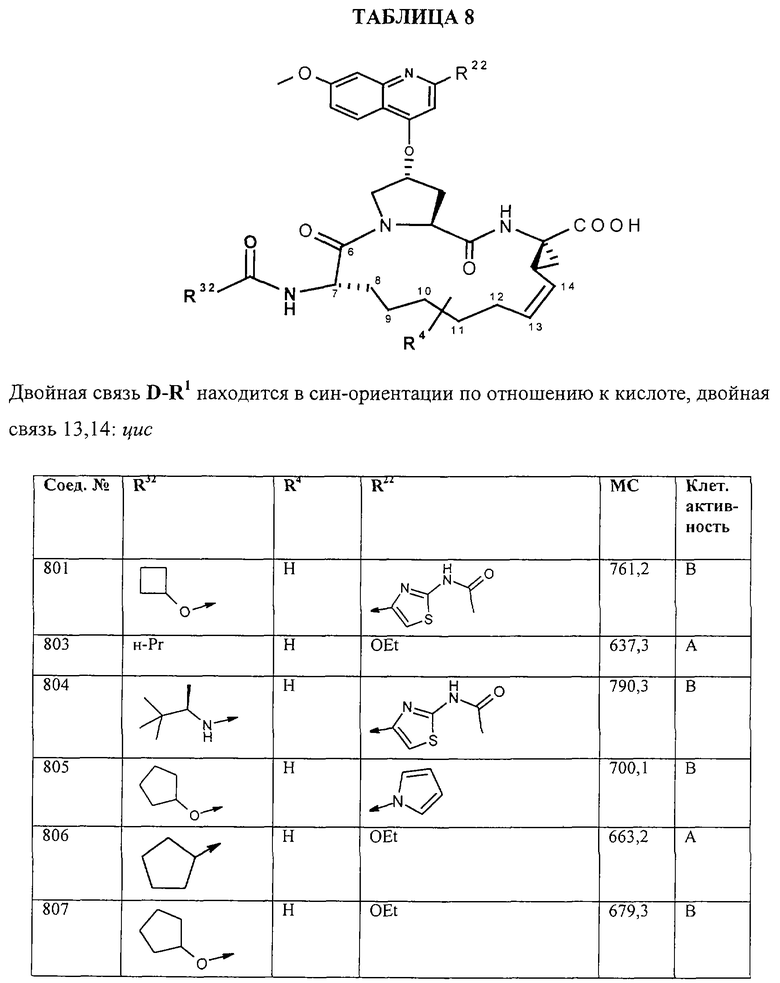
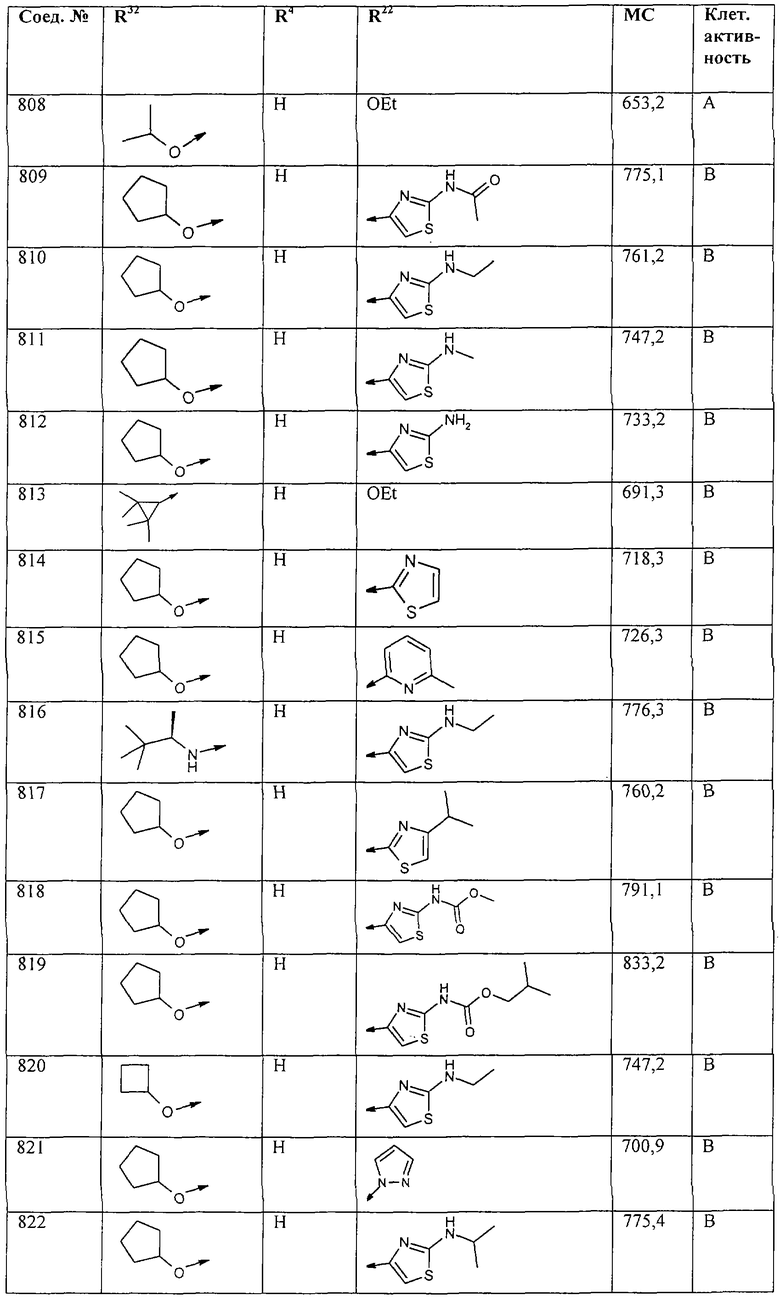
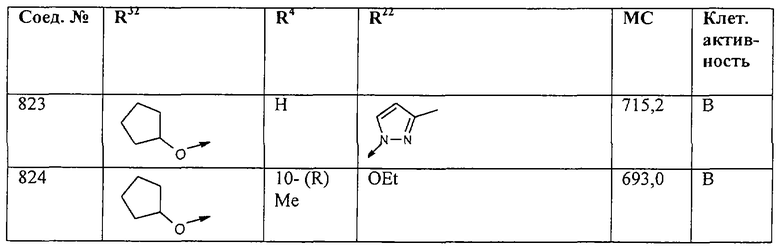
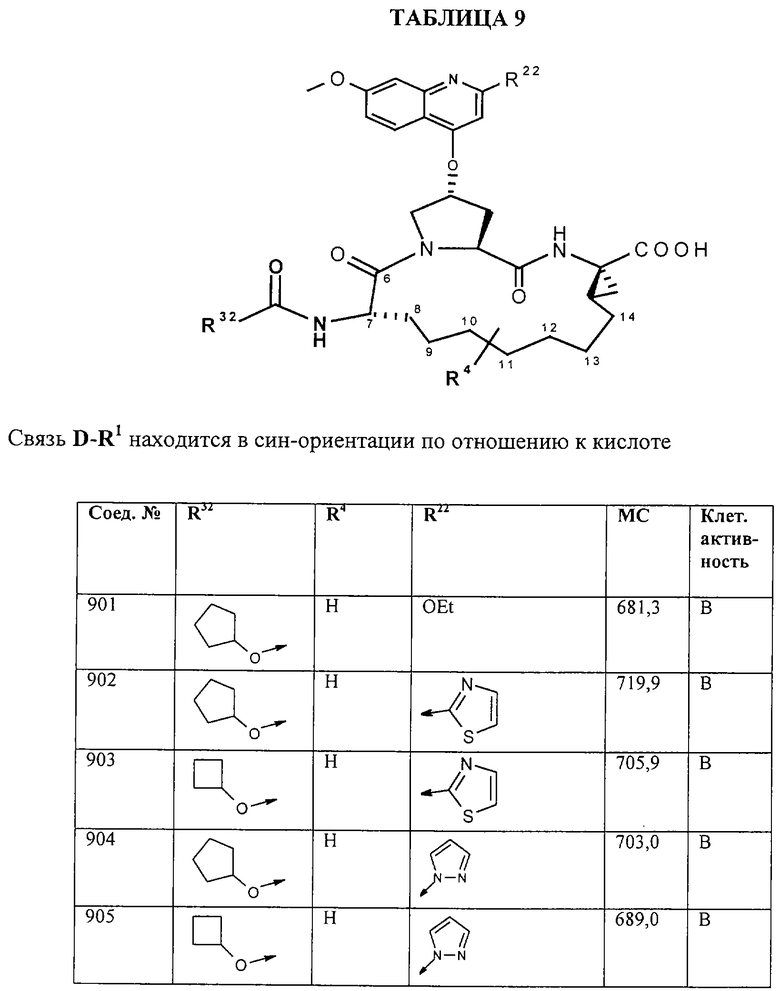

| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛЗАМЕЩЕННЫХ 2-ДЕЗОКСИ-2-ФТОР-D-РИБОФУРАНОЗИЛ-ПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2407747C2 |
| ИНГИБИТОРЫ АКТИВНОСТИ ПРОТЕИНТИРОЗИНКИНАЗЫ | 2009 |
|
RU2533827C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| ИНДОЛИЛМАЛЕИМИДНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ НАРУШЕНИЙ ИЛИ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ Т-ЛИМФОЦИТАМИ И/ИЛИ ПКС | 2003 |
|
RU2340610C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ МИМЕТИКОВ ТРО | 2006 |
|
RU2385865C2 |
| СИММЕТРИЧНЫЕ И НЕСИММЕТРИЧНЫЕ ПРОИЗВОДНЫЕ ДИФЕНИЛМОЧЕВИНЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОДАВЛЕНИЯ РОСТА ОПУХОЛЕВЫХ КЛЕТОК, ОПОСРЕДОВАННОГО КИНАЗОЙ RAF | 1998 |
|
RU2247109C9 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| ПРОИЗВОДНЫЕ 3-ЗАМЕЩЕННОГО 4-АРИЛХИНОЛИН-2-ОНА В КАЧЕСТВЕ МОДУЛЯТОРОВ КАЛИЕВЫХ КАНАЛОВ | 1999 |
|
RU2240998C2 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2804468C2 |
Изобретение относится к макроциклическим пептидам общей формулы I
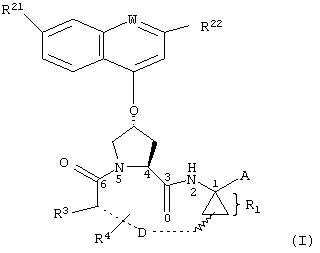
где W обозначает N, R21 обозначает Н, C1-С6алкокси, гидрокси или N(C1-С6алкил)2; R22 обозначает Н, C1-С6алкил, CF3, C1-С6алкокси, С2-С7алкоксиалкил, С6-арил или Het, где Het обозначает пяти- или шестичленный насыщенный или ненасыщенный гетероцикл, содержащий два гетероатома, выбранных из азота, кислорода и серы, при этом указанный Het замещен радикалом R24, где R24 обозначает Н, -NH-C(O)-R26, OR26, -NHR26, -NHC(O)-NH-R26, -NHC(O)-OR26, R26, где R26 обозначает водород, C1-С6алкил; R3 обозначает гидрокси или группу формулы -NH-R31, где R31 обозначает -C(O)-R32, -C(O)-NHR32 или -C(O)-OR32, где R32 обозначает C1-С6алкил или C3-С6циклоалкил, D обозначает состоящую из 5-10 атомов насыщенную или ненасыщенную алкиленовую цепь, необязательно включающую один-три гетероатома, независимо друг от друга выбранных из О, S, или N-R41, где R41 обозначает Н, -C(O)-R42, где R42 обозначает C1-С6алкил, С6-арил; R4 обозначает Н или один-три заместителя на любом атоме углерода цепи D, где заместители независимо друг от друга выбирают из группы, включающей C1-С6алкил, гидроксил; А обозначает карбоновую кислоту или ее сложные алкиловые эфиры, или их производным; фармацевтическим композициям, содержащим указанные соединения и обладающим активностью в отношении вируса гепатита С, при этом эти пептиды специфически ингибируют NS3-протеазу и не проявляют заметной ингибирующей активности в отношении других серинпротеаз. 12 с. и 94 з.п. ф-лы, 9 табл.
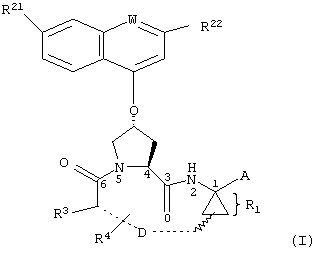
где
W обозначает N,
R21 обозначает Н, С1-С6алкокси, гидрокси или N(С1-С6алкил)2;
R22 обозначает Н, С1-С6алкил, СF3, С1-С6алкокси, С2-С7алкоксиалкил, С6-арил или Het,
где Het обозначает пяти- или шестичленный насыщенный или ненасыщенный гетероцикл, содержащий два или три гетероатома, выбранных из азота, кислорода и серы, при этом указанный Het замещен радикалом R24, где
R24 обозначает Н, C1-C6алкил, С1-С6алкокси-NH-C(O)-R26, -OR26, -NHR26, -NHC(O)-NH-R26, -NHC(O)-OR26 и R26,
где R26 обозначает водород, С1-С6алкил;
R3 обозначает гидрокси, NH2 или группу формулы -NH-R31, где
R31 обозначает -C(O)-R32, -C(O)-NHR32 или -С(O)-OR32, где
R32 обозначает С1-С6алкил или С3-С6циклоалкил,
D обозначает состоящую из 5-10 атомов насыщенную или ненасыщенную алкиленовую цепь, необязательно включающую один-три гетероатома, независимо друг от друга выбранных из О, S, или N-R41, где
R41 обозначает Н, C1-C6алкил, -С(О)-R42, где R42 обозначает С1-С6алкил, С6-арил;
R4 обозначает Н или один-три заместителя на любом атоме углерода цепи D, где заместители независимо друг от друга выбирают из группы, включающей С1-С6алкил, гидроксил или оксо;
А обозначает карбоновую кислоту или ее сложные алкиловые эфиры.
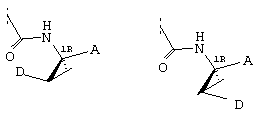
при этом D находится в син-ориентации по отношению к амиду (i) или D находится в син-ориентации по отношению к А (ii).
где
W обозначает N;
R21 обозначает Н, С1-С6алкокси, гидрокси, или N(С1-С6алкил)2;
R22 обозначает Н, С1-С6алкил, С1-С6алкокси, фенил или Het, выбранный из группы, включающей
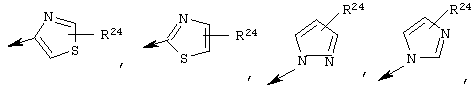
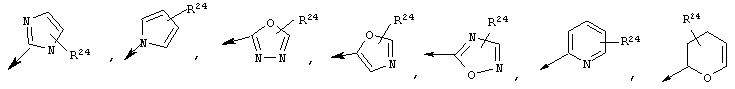
где R24 обозначает Н или группу NH-C(O)-OR26, где R26 обозначает
С1-С6алкил.
R21 обозначает Н или С1-С6алкокси.

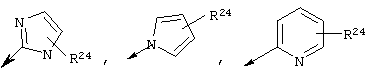 ,
,
где R24 обозначает Н, С1-С6алкил, NH-R26, NH-C(O)-R26 или
NH-C(O)-OR26, где R26 имеет значения, указанные в п.4.
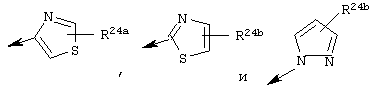 ,
,
или R24a обозначает NH-C(O)-OR26, где R26 С1-С6алкил, и R24b обозначает Н или С1-С6алкил.
W обозначает N,
R3 обозначает группу формулы -NH-C(O)-NHR32 или -NH-C(O)-OR32, где
R32 обозначает С1-С4алкил или С4-С6циклоалкил,
D обозначает состоящую из 6-8 атомов насыщенную или ненасыщенную алкиленовую цепь, присоединенную к R1 в син-ориентации по отношению к А, необязательно включающую один или два гетероатома, независимо друг от друга выбранных из О, S или N-R41, где R41 обозначает Н или С2-С7ацил,
R4 обозначает Н или один-три заместителя, выбранных независимо друг от друга из гидрокси или С1-С6алкила, и
А обозначает карбоновую кислоту,
или ее сложный эфир.
R21 обозначает Н или метокси,
R22 обозначает С1-С6алкокси или Het, выбранный из группы, включающей
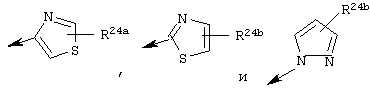
R24a обозначает NH-C(O)-OR26, где
R26 обозначает С1-С6алкил; и
R24b обозначает Н или С1-С6алкил;
R3 обозначает мочевину формулы N-C(O)-NHR32 или карбамат формулы N-C(O)-OR32,
где R32 обозначает С1-С6алкил или С3-С6циклоалкил;
D обозначает содержащую 7 атомов алкиленовую цепь, необязательно включающую одну двойную связь между положениями 11 и 12 или 13 и 14,
при этом цепь D необязательно включает один гетероатом, независимо выбранный из O, S, NH, N(Me) или N(Ac), и
R4 обозначает Н или С1-С6алкил.
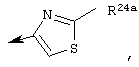
где R24a обозначает NH(C1-C4)алкил, NH-CO-(C1-C4алкил), NH-C(O)-O-(C1-C4алкил), NH-C(O)-NH-(С1-С4алкил), и
D обозначает С7- цепь, все атомы которой представляют собой углерод и которая является насыщенной или содержит одну двойную цис-связь между положениями 13 и 14.
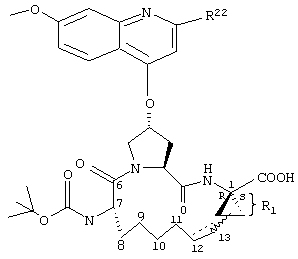
включающее один стереоизомер на R1, где двойная связь, стереохимическое строение связи D-R1 и R22 имеют следующие значения
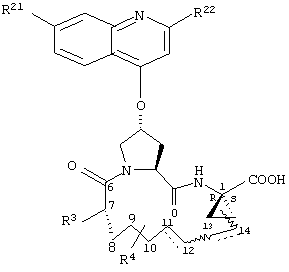
включающее один стереоизомер на R1, где R3, R4, положение двойной связи, стереохимическое строение связи D-R1, R21 и R22 имеют следующие значения:
кислоте
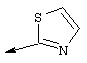
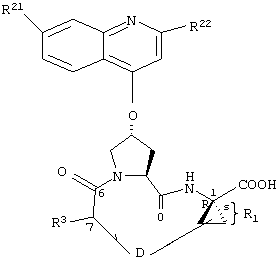
включающее один стереоизомер на R1, где R3, D, стереохимическое строение связи D- R1, R21 и R22 имеют следующие значения:
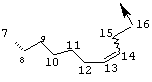

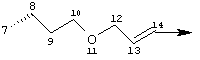
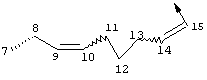
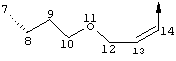
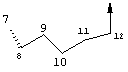
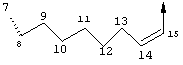
ориентации по отношению к
кислоте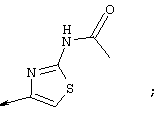
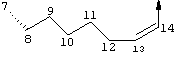
ориентации по отношению к
кислоте
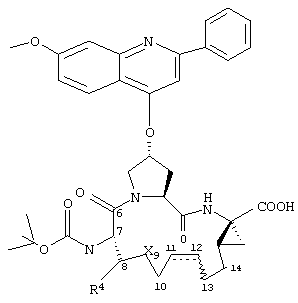
где связь D-R1 находится в син-ориентации по отношению к кислоте, а R4, Х9, и двойная связь 11, 12 имеют следующие значения:
11, 12:

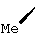
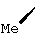
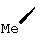
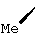
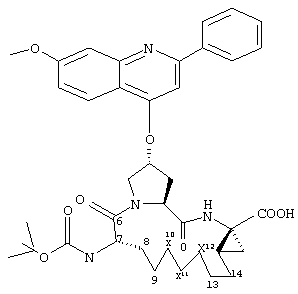
где связь D-R1 находится в син-ориентации по отношению к кислоте, а Х10, Х11 и Х12 имеют следующие значения:
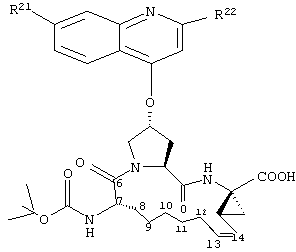
где связь D-R1 находится в син-ориентации по отношению к кислоте, а R21 и R22 имеют следующие значения:
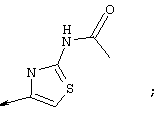
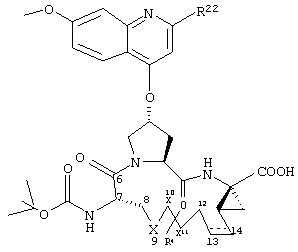
где связь D-R1 находится в син-ориентации по отношению к кислоте, R4, X9, X10, X11, а двойная связь между положениями 13 и 14 и R22 имеют следующие значения:

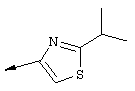
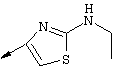
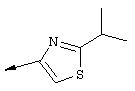
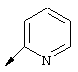
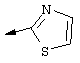
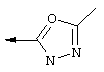
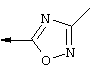
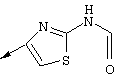

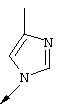
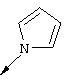
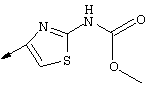
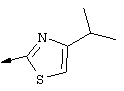
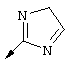
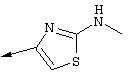
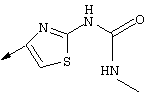
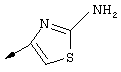
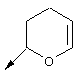

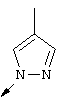
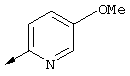

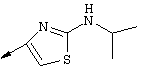
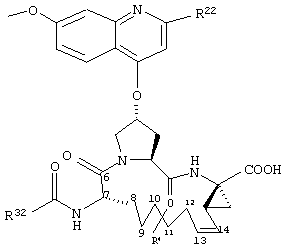
где связь D-R1 находится в син-ориентации по отношению к кислоте, двойная связь между положениями 13 и 14 находится в цис-ориентации, а R32, R4 и R22 имеют следующие значения:
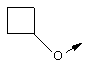
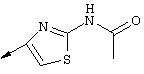

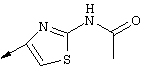
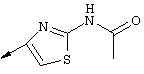

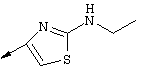


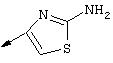

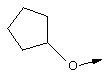
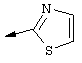
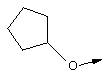
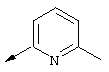
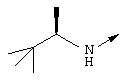
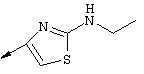
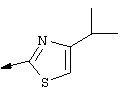

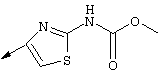
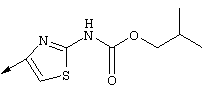
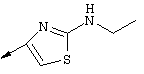
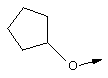
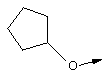
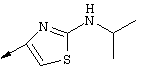
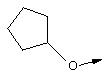
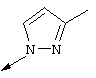
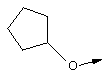
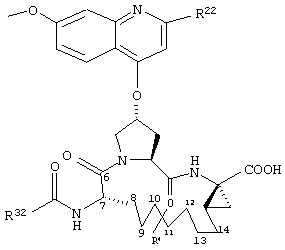
где связь D-R1 находится в син-ориентации по отношению к кислоте, а R32, R4 и R22 имеют следующие значения:
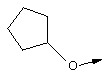
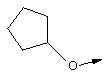
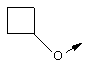
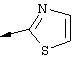
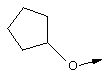
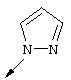



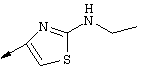
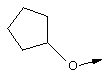


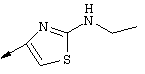
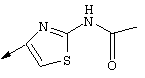
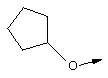
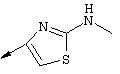
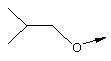
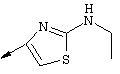
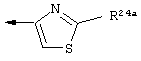
где R24а обозначает NH-(С1-С4алкил); NH-C(O)- (С1-С4алкил); NH-C(O)-O-(С1-С4алкил); или NH-C(O)-NH-(С1-С4алкил).
| ЦИКЛИЧЕСКИЕ ПЕПТИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2096415C1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

