В соответствии с 35 USС §119(е) данная заявка испрашивает более ранний приоритет, основанный на предварительной заявке США сер. номер 60/708438, зарегистрированной 15 августа 2005 г. Полное описание этой заявки включено в описание настоящей заявки в виде ссылки в полном объеме.
Область техники, к которой относится изобретение
В изобретении предлагается новый класс соединений, фармацевтические композиции, включающие такие соединения, и способы применения таких соединений для лечения или профилактики заболеваний или нарушений, ассоциированных с аномальной или разрегулированной активностью ТРО, прежде всего заболеваний или нарушений, которые сопровождаются тромбоцитопенией.
Предпосылки создания изобретения
Мегакариоциты представляют собой клетки костного мозга, которые отвечают за продуцирование тромбоцитов, циркулирующих в кровотоке. Тромбопоэтин (ТРО), кроветворный цитокин, поддерживает процесс клеточной пролиферации и дифференциации кроветворных стволовых клеток и необходим для регуляции мегакариоцитов.
Новые соединения по настоящему изобретению можно использовать в качестве миметиков ТРО для лечения заболеваний или состояний, которые предшествуют и/или приводят к снижению содержания в крови ТРО или тромбоцитов, которые включают, без ограничения перечисленным, лучевую терапию, химиотерапию, иммунотерапию, рак, вирусные инфекции и трансплантацию, такую как трансплантация костного мозга или стволовых клеток.
Краткое изложение сущности изобретения
Одним объектом настоящего изобретения являются соединения формулы I
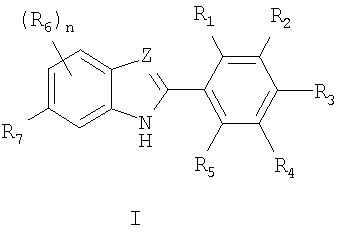
в которых
n выбирают из 0, 1, 2 и 3,
Z выбирают из N и CR8, где R8 выбирают из группы, включающей водород, галоген, C1-С6алкил, галоген(C1-С6)алкил, причем любой алкил в составе R8 необязательно содержит группу метилен, которая заменена на атом или группу, выбранную из группы, включающей -S(O)0-2-, -C(O)-, -NR9- и -O-, причем R9 выбирают из группы, включающей водород и С1-6алкил,
R1, R2, R4 и R5 независимо выбирают из группы, включающей водород, галоген, гидрокси, циано, нитро, -XNR9R10, C1-С6алкил, галоген(C1-С6)алкил, C1-С6алкокси и галоген(C1-С6)алкокси, причем Х выбирают из группы, включающей химическую связь и C1-С6алкилен, а R9 и R10 независимо выбирают из группы, включающей водород и C1-С6алкил,
R3 выбирают из группы, включающей водород, C1-С6алкил, С3-С8гетероциклоалкил, С5-С10гетероарил, -OS(O)2R11, -NR11S(O)2R12, -NR11C(O)R12, -NR11C(O)NR11R12, -NR11C(O)C(O)OR12, -NR11C(O)OR12, -OC(O)NR11R12, -C(O)OR11, -C(O)R13, -NR11R12, -NR11R13, -S(O)2NR11R12 и -C(O)NR11R12, где R11 и R12 независисимо выбирают из группы, включающей водород, C1-С6алкил, галоген(C1-С6)алкил, C1-С6алкокси, циано(C1-С6)алкил, гидрокси(C1-С6)алкил и C1-С6алкил, замещенный группой -NR9R10, R13 означает С3-С8гетероциклоалкил, необязательно замещенный 1-3 группами C1-С6алкил, причем любой гетероциклоалкил или гетероарил R3 необязательно замещен 1-3 группами, независимо выбранными из группы, включающей галоген, C1-С6алкил, C1-С6алкокси, циано(C1-С6)алкил, гидрокси(C1-С6)алкил, галоген(C1-С6)алкил и галоген(C1-С6)алкокси,
R6 выбирают из группы, включающей галоген и C1-С6алкил, а
R7 выбирают из группы, включающей галоген, циано, C1-С6алкил, С2-С6алкенил, С2-С6алкинил, галоген(C1-С6)алкил, С6-С10арил, С5-С10гетероарил, С3-С8гетероциклоалкил и С3-С12циклоалкил, причем любой алкил, алкенил, алкинил, арил, гетероарил, циклоалкил или гетероциклоалкил необязательно замещен 1-5 группами, независимо выбранными из группы, включающей галоген, C1-С6алкил, C1-С6алкокси, циано(C1-С6)алкил, гидрокси(C1-С6)алкил, галоген(C1-С6)алкил и галоген(C1-С6)алкокси, -NR14R15, -XOR14, -S(O)2R14, С3-С12циклоалкил, С3-С8гетероциклоалкил, С6-С10арил и С3-С8гетероарил, где Х означает химическую связь или C1-С6алкилен, a R14 и R15 независимо выбирают из группы, включающей C1-С6алкил, циано(C1-С6)алкил, гидрокси(C1-С6)алкил, галоген(C1-С6)алкил и галоген(C1-С6)алкокси, причем любой арил, гетероарил, циклоалкил и гетероциклоалкил в составе R7 необязательно замещен 1-3 группами, независимо выбранными из группы, включающей галоген, C1-С6алкил, C1-С6алкокси, циано(C1-С6)алкил, гидрокси(C1-С6)алкил, галоген(C1-С6)алкил и галоген(C1-С6)алкокси, и N-оксиды, пролекарства, защищенные производные, индивидуальные изомеры и смеси изомеров, и фармацевтически приемлемые соли и сольваты (например, гидраты) указанных соединений.
Вторым объектом настоящего изобретения является фармацевтическая композиция, которая содержит соединение формулы I или его N-оксид, индивидуальные изомеры и смеси изомеров или фармацевтически приемлемую соль, в смеси с одним или более пригодными эксципиентами.
Третьим объектом настоящего изобретения является способ лечения заболевания или состояния у животного, при котором за счет повышения уровня тромбоцитов в крови можно снизить или уменьшить интенсивность патологического состояния и/или симптомов заболевания или состояния, заключающийся в том, что животному вводят терапевтически эффективное количество соединения формулы I или его N-оксида, индивидуальных изомеров и смеси изомеров или его фармацевтически приемлемой соли.
Четвертым объектом настоящего изобретения является применение соединения формулы I для получения лекарственного средства, предназначенного для лечения заболевания или состояния у животного, при котором пониженный уровень тромбоцитов способствует развитию патологического состояния и/или симптомов заболевания или состояния.
Пятым объектом настоящего изобретения является способ получения соединений формулы I и их N-оксидов, пролекарств, защищенных производных, индивидуальных изомеров и смеси изомеров или фармацевтически приемлемых солей указанных соединений.
Подробное описание изобретения
Определение терминов
"Алкил" в виде отдельной группы или структурного фрагмента в составе других групп, например галогеналкила и галогеналкокси, может содержать прямую или разветвленную цепь. С1-С4алкокси включает метокси, этокси и т.п. Галогеналкил включает трифторметил, пентафторэтил и т.п.
"Арил" означает моноциклическую или конденсированную бициклическую ароматическую систему, содержащую в цикле 6-10 атомов углерода. Например, арил может означать фенил или нафтил, предпочтительно фенил. "Арилен" означает двухвалентный радикал, образованный арильной группой.
"Гетероарил" имеет значения, указанные для арила, в котором один или более атомов в цикле являются гетероатомами. Например, гетероарил включает пиридил, индолил, индазолил, хиноксалинил, хинолинил, бензофуранил, бензопиранил, бензотиопиранил, бензо[1,3]диоксол, имидазолил, бензимидазолил, пиримидинил, фуранил, оксазолил, изоксазолил, триазолил, тетразолил, пиразолил, тиенил и т.п.
"Циклоалкил" означает насыщенную или частично ненасыщенную, моноциклическую, конденсированную бициклическую или мостиковую полициклическую систему, содержащую в цикле указанное число атомов. Например, С3-С10циклоалкил включает циклопропил, циклобутил, циклопентил, циклогексил и т.п.
"Гетероциклоалкил" означает циклоалкил, имеющий значения, указанные в описании заявки, при условии, что один или более атомов углерода в цикле заменен на фрагмент, выбранный из группы, включающей -O-, -N=, -NR-, -С(O)-, -S-, -S(O) - или -S(O)2-, где R означает водород, C1-С4алкил или N-защитную группу. Например, С3-С8гетероциклоалкил, используемый при описании соединений по изобретению, включает морфолино, пирролидинил, пирролидинил-2-он, пиперазинил, пиперидинил, пиперидинилон, 1,4-диокса-8-азаспиро[4.5]дец-8-ил и т.п.
"Галоген" предпочтительно означает хлор или фтор, а также бром или иод.
"Тромбопоэтин (ТРО)" известен также в области техники как лиганд с-Mpl, лиганд mpl, мегапоэтин и фактор роста и развития мегакариоцитов.
"Лечение" означает способ ослабления или подавления заболевания и/или сопутствующих симптомов.
Описание предпочтительных вариантов осуществления изобретения
В настоящем изобретении предлагаются соединения, композиции и способы лечения тромбоцитопении. В общих чертах тромбоцитопению можно интерпретировать как любое снижение числа тромбоцитов в крови ниже нормального уровня, характерного для здорового индивидуума.
Одним вариантом соединений формулы I являются соединения формулы Ia
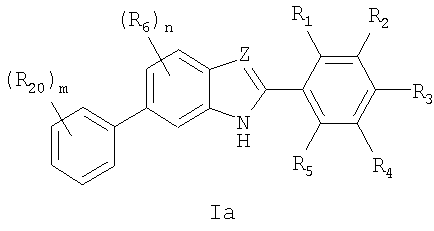
в которых
n выбирают из 0, 1, 2 и 3,
m выбирают из 0, 1, 2, 3, 4 и 5,
Z выбирают из N и CR8, где R8 выбирают из группы, включающей водород, галоген, C1-С6алкил, галоген(C1-С6)алкил, причем любой алкил в составе R8 необязательно содержит метиленовую группу, замененную на атом или группу, выбранную из группы, включающей -S(O)0-2-, -С(O)-, -NR9- и -O-, где R9 выбирают из группы, включающей водород и C1-С6алкил,
R1, R2, R4 и R5 независимо выбирают из группы, включающей водород, галоген, гидрокси, нитро, -XNR9R10, C1-С6алкил, галоген(C1-С6)алкил, где Х выбирают из группы, включающей химическую связь и C1-С6алкилен, а R9 и R10 независимо выбирают из группы, включающей водород и C1-С6алкил,
R3 выбирают из группы, включающей -XCOOR9, -XCONR9R10, -NR11S(O)2R12, -S(O)2NR11R12, -NR11C(O)R12, -NR11C(O)NR11R12, -NR11C(O)C(O)OR12 и -NR11C(O)OR12, где Х выбирают из группы, включающей химическую связь и C1-С6алкилен, а R9 и R10 независимо выбирают из группы, включающей водород и C1-С6алкил,
R6 выбирают из группы, включающей водород и C1-С6алкил, и
R20 выбирают из группы, включающей водород и C1-С6алкил.
В другом варинте R6 означает фтор, а R20 выбирают из группы, включающей фтор, метил и бутил.
В еще одном варианте R3 выбирают из группы, включающей карбоксил, аминокарбонил, аминосульфонил, метилсульфониламино и амино, а R4 выбирают из группы, включающей водород, гидроксил, нитро и амино.
Предпочтительные соединения по изобретению выбирают из группы, включающей 4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]бензойную кислоту, 4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]бензамид, 4-[7-фтор-6-(4-фтор-3-метилфенил)-1Н-индол-2-ил]бензойную кислоту, 4-[7-фтор-6-(4-фтор-3-метилфенил)-1Н-индол-2-ил]бензолсульфонамид, 4-[6-(4-бутилфенил)-3-этил-7-фтор-1Н-индол-2-ил]-2-гидроксибензойную кислоту, 4-[6-(4-бутилфенил)-7-фтор-3-изопропил-1Н-индол-2-ил]-2-гидроксибензойную кислоту, 4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]-2-гидроксибензойную кислоту, 4-[6-(4-бутилфенил)-7-фтор-3-метил-1Н-индол-2-у1]-2-гидроксибензойную кислоту, 4-[6-(4-бутилфенил)-1Н-бензимидазол-2-ил]бензойную кислоту, 4-[6-(4-бутилфенил)-1Н-бензимидазол-2-ил]-2-гидроксибензойную кислоту, N-{4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]фенил}метансульфонамид, N-{4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]фенил}ацетамид, N-{4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]-2-хлорфенил}ацетамид, 4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]-2-хлорфениламин и 2-амино-4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]бензойную кислоту.
Другие предпочтительные соединения по изобретению подробно описаны в разделе Примеры и в таблицах.
Фармакология и промышленная применимость
В широком смысле тромбоцитопению можно интерпретировать как любое снижение числа тромбоцитов в крови ниже нормального уровня, характерного для здорового индивидуума. Установлено, что тромбоцитопения развивается под воздействием множества факторов, включающих, без ограничения перечисленным, лучевую терапию, химиотерапию, иммунотерапию, иммунную тромбоцитопеническую пурпуру, миелодиспластический синдром (MDS), апластическую анемию, острый миелоидный лейкоз (AML), хронический миелолейкоз (CML), вирусное инфекционное заболевание печени (включающее, без ограничения перечисленным, ВИЧ, гепатит С, парвовирус), удаление костного мозга, трансплантацию костного мозга, трансплантацию стволовых клеток, трансплантацию стволовых клеток периферической крови, дефект клеток-предшественников, полиморфизм стволовых клеток и клеток-предшественников, дефекты ТРО, нейтропению, мобилизацию, пролиферацию, активацию или дифференциацию дендритных клеток.
ТРО имеет большое терапевтическое значение при лечении пациентов с пониженным уровнем тромбоцитов. Например, от тромбоцитопении страдают пациенты, больные различными видами рака, поскольку миелодепрессантная химиотерапия или лучевая терапия, которые увеличивают риск кровотечения, часто ограничивают дозу химиотерапевтических агентов, которые необходимы для интенсивной химиотерапии, или при трансплантации костного мозга.
Соединения по настоящему изобретению можно использовать при лечении тромбоцитопении независимо от фактора или факторов, вызывающих заболевание. Соединения по настоящему изобретению можно также использовать при лечении тромбоцитопении, если фактор или факторы, вызывающие заболевание, неизвестны или еще неидентифицированы. Соединения по настоящему изобретению можно использовать в любых случаях, когда возникает опасность кровопотери или снижения числа тромбоцитов в крови, включающих, без ограничения перечисленным, хирургическую трансплантацию, хирургическое вмешательство, анестезию перед родами и операцией на кишечнике.
Поскольку тромбоциты необходимы для свертывания крови и пациент с низким уровнем тромбоцитов рискует погибнуть от внезапного кровотечения, миметики ТРО по изобретению могут найти применение при лечении различных гематологичеких нарушений, например заболеваний, главным образом, вызванных дефектами тромбоцитов.
В соответствии с вышеизложенным, настоящее изобретение также относится к способу профилактики или лечения любых заболеваний или нарушений, описанных выше, у субъекта, который нуждается в таком лечении, который заключается в том, что указанному субъекту вводят терапевтически эффективное количество (см. раздел "Способы введения и фармацевтические композиции") соединения формулы I или его фармацевтически приемлемой соли. При любом из указанных способов применения требуемая доза изменяется в зависимости от способа введения, конкретного состояния, подлежащего лечению, и требуемого результата.
Способы введения и фармацевтические композиции
В общем случае, соединения по изобретению вводят в терапевтически эффективных количествах любыми обычными и приемлемыми способами, отдельно или в комбинации с одним или более терапевтическими агентами. Терапевтически эффективное количество может изменяться в широком интервале в зависимости от тяжести заболевания, возраста и относительного состояния здоровья субъекта, активности используемого соединения и других факторов. В общем случае удовлетворительные результаты достигаются при системном введении суточных доз от приблизительно 0,03 до 2,5 мг/кг массы тела. Для более крупного млекопитающего, например человека, назначаемая суточная доза составляет от приблизительно 0,5 мг до приблизительно 100 мг, которую можно вводить стандартными способами, например раздельными дозами до четырех раз в сутки или в виде формы с замедленным высвобождением. Пригодные стандартные лекарственные формы для перорального введения включают от приблизительно 1 до 50 мг активного ингредиента.
Соединения по изобретению можно вводить в виде фармацевтических композиций любым приемлемым способом, прежде всего энтеральным, например пероральным способом, например, в форме таблеток или капсул, или парентеральным способом, например, в форме инъекционных растворов или суспензий, местным способом, например, в форме лосьонов, гелей, мазей или кремов, или назальным способом, или в форме суппозиториев. Фармацевтические композиции, включающие соединение по настоящему изобретению в свободной форме или в форме фармацевтически приемлемой соли в смеси по меньшей мере с одним фармацевтически приемлемым носителем или разбавителем, получают обычным способом с использованием процессов смешивания, гранулирования или нанесения покрытия. Например, пероральные композиции могут представлять собой таблетки или желатиновые капсулы, включающие активный ингредиент в смеси с а) разбавителями, например лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином, б) замасливателями, например диоксидом кремния, тальком, стеариновой кислотой, ее кальциевой или магниевой солью и/или полиэтиленгликолем, а таблетки могут также включать в) связующие агенты, например силикат магния/алюминия, крахмальную пасту, желатин, трагакант, метилцеллюлозу, Na-соль карбоксиметилцеллюлозы и/или поливинилпирролидон, и при необходимости г) дезинтегрирующие агенты, например крахмалы, агар, альгиновую кислоту или ее натриевую соль, или шипучие смеси, и/или д) абсорбенты, красители, ароматизаторы и подсластители. Инъекционные композиции могут представлять собой водные изотонические растворы или суспензии, а суппозитории получают из жировых эмульсий или суспензий. Композиции можно стерилизовать, и/или они могут содержать адъюванты, такие как консервирующие, стабилизирующие, смачивающие или эмульгирующие агенты, способствующие растворению агенты, соли для регуляции осмотического давления и/или буферные вещества. Дополнительно они могут также содержать другие терапевтически ценные соединения. Пригодные составы для чрескожного применения включают эффективное количество соединения по настоящему изобретению в смеси с носителем. Носитель может включать абсорбируемые фармакологически приемлемые растворители для обеспечения проницаемости через кожу пациента. Например, системы для чрескожного введения представляют собой повязку, включающую основу, резервуар, содержащий соединение необязательно в смеси с носителем, необязательно мембрану, регулирующую доставку соединения к поверхности кожи с заданной скоростью в течение продолжительного времени, и средство, обеспечивающее удерживание устройства на поверхности кожи. Можно также использовать матричные чрескожные составы. Пригодные составы для местного нанесения, например на кожу и в глаза, предпочтительно представляют собой водные растворы, мази, кремы или гели, известные в данной области. Такие составы могут содержать солюбилизирующие, стабилизирующие, тонизирующие агенты, буферные вещества и консерванты.
Соединения по изобретению можно вводить в терапевтически эффективных количествах в комбинации с одним или более терапевтическими агентами (фармацевтические комбинации). Миметики ТРО по настоящему изобретению можно также использовать для действия на клетки с целью повышения жизнеспособности или пролиферации клеток в сочетании с другими агентами, оказывающими на клетки аналогичное действие. Такие агенты включают, без ограничения перечисленным, G-CSF, GM-CSF, ТРО, M-CSF, ЕРО, Gro-beta, IL-11, SCF, лиганд FLT3, LIF, IL-3, IL-6, IL-1, прогенипоэтин, NESP, SD-01, или IL-5, или биологически активное производное любого из вышеуказанных агентов.
Установлено, что дендритные клетки человека экспрессируют рецептор ТРО и ТРО является эффективным стимулятором дендритных клеток. Миметики ТРО по настоящему изобретению можно также использовать в качестве адъювантов в составе вакцин благодаря повышению активности и подвижности дендритных клеток. Фармацевтически активные соединения по настоящему изобретению можно использовать в качестве иммунологического адъюванта в комбинации с вакциной и/или иммуномодулятором при пероральной, чрескожной или подкожной доставке благодаря повышению активности и подвижности дендритных клеток.
Установлено, что ТРО обладает широким спектром действия, который включает антиапоптотическое/повышающее жизнеспособность действие на мегакариоциты, тромбоциты и стволовые клетки и пролиферативное действие на стволовые клетки и мегакариотические клетки. Следовательно, ТРО и/или миметики ТРО по изобретению эффективно повышают уровень стволовых клеток и клеток-предшественников и при использовании ТРО в сочетании с другими цитокинами, индуцирующими дифференциацию, оказывают синергетическое действие.
Если соединения по изобретения вводят в сочетании с другими способами лечения, дозы совместно вводимых соединений изменяются в зависимости от типа совместно вводимого лекарственного средства, конкретного лекарственного средства, состояния, подлежащего лечению, и т.п.
Изобретение также относится к фармацевтическим комбинациям, например набору, включающему а) первый агент, который представляет собой соединение по изобретению, указанное выше, в свободной форме или в форме фармацевтически приемлемой соли, и б) по меньшей мере один второй агент. Набор включает инструкции по введению лекарственных средств.
Термины "совместное введение" или "комбинированное введение" или аналогичные термины, используемые в описании, означают введение выбранных терапевтических агентов одному пациенту, а также курс лечения, согласно которому агенты необязательно вводятся одновременно или одним и тем же способом.
Термин "фармацевтическая комбинация", используемый в описании заявки, означает продукт, который образуется при смешивании или комбинировании более одного активного ингредиента и включает фиксированные и нефиксированные комбинации активных ингредиентов. Термин "фиксированная комбинация" означает, что активные ингредиенты, например соединение формулы I и сопутствующий агент, вводятся пациенту одновременно в форме одного продукта или дозы. Термин "нефиксированная комбинация" означает, что активные ингредиенты, например соединение формулы I и сопутствующий агент, вводятся пациенту раздельно, одновременно, совместно или последовательно, без ограничений по времени, причем такое введение обеспечивает достижение терапевтически эффективных уровней двух соединений в организме пациента. Последнее относится также к комбинированному лечению, например к введению трех или более активных ингредиентов.
Способы получения соединений по изобретению
Настоящее изобретение включает также способы получения соединений по изобретению. В описанных реакциях необходимо защищать реакционно-способные функциональные группы, например гидрокси, амино, имино, тио или карбоксигруппы, если они должны присутствовать в конечном продукте. Введение защитных групп позволяет исключить участие таких функциональных групп в проводимых реакциях. Стандартные защитные группы используются в соответствии с принятой практикой, например см. T.W.Greene и Р.G.M.Wuts, "Protective Groups in Organic Chemistry", John Wiley and Sons (1991).
Соединения формулы I, в которых Z означает N, получают способом, показанным на схеме I.
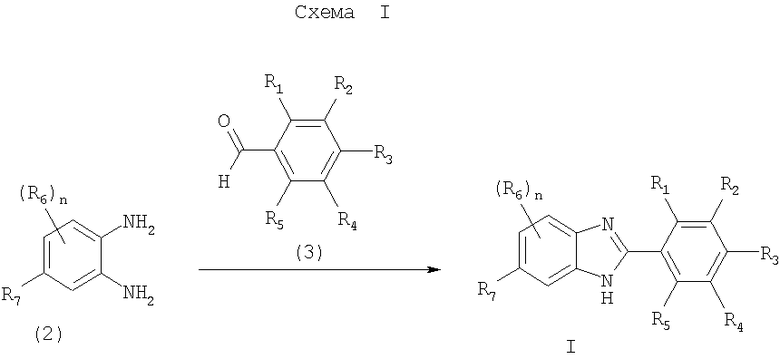
На схеме I n и R1-R7 имеют значения, указанные в кратком описании изобретения. Соединение формулы I синтезируют по реакции соединения формулы 2 с соединением формулы 3 в присутствии гидросульфита натрия в пригодном растворителе (например, DMA и т.п.). Реакцию проводят при температуре от приблизительно 100°С до приблизительно 180°С в течение от приблизительно 24 ч до завершения реакции.
Примеры синтеза соединения формулы I, в которых Z означает CR8, получают по методике, показанной на схеме II.
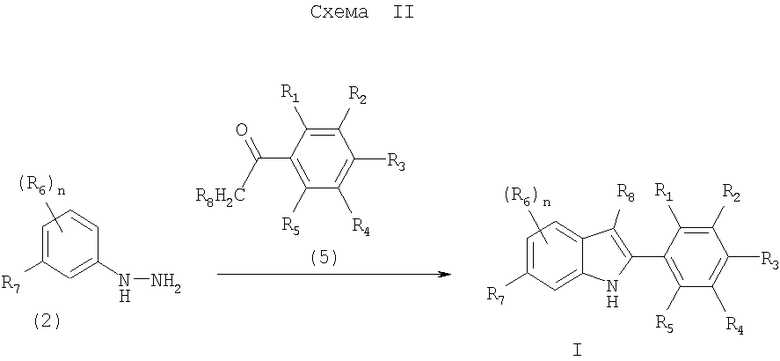
На схеме II n и R1-R8 имеют значения, указанные в кратком описании изобретения. Соединение формулы I синтезируют по реакции соединения формулы 2 с соединением формулы 5 в присутствии пригодной кислоты Льюиса (например, хлорида цинка и т.п.) или протонной кислоты (например, HCl и т.п.) в пригодном растворителе (например, уксусной кислоте, этаноле и т.п.). Реакцию проводят при температуре от приблизительно 80°С до приблизительно 120°С в течение до приблизительно 72 ч, т.е. до завершения реакции.
Примеры синтеза соединения формулы I приводятся в разделе Примеры.
Дополнительные способы получения соединений по изобретению
Соединение по изобретению можно получить в виде фармацевтически приемлемой кислотно-аддитивной соли по реакции свободного основания соединения с фармацевтически приемлемой неорганической или органической кислотой. В другом варианте фармацевтически приемлемую основно-аддитивную соль соединения по изобретению можно получить по реакции свободной кислоты соединения с фармацевтически приемлемым неорганическим или органическим основанием.
В другом варианте соли соединений по изобретению можно получить с использованием солей исходных материалов или промежуточных соединений.
Свободные кислоты или свободные основания соединений по изобретению получают из соответствующих основно-аддитивной соли или кислотно-аддитивной соли соответственно. Например, соединение по изобретению в форме кислотно-аддитивной соли можно превратить в соответствующее свободное основание при обработке соответствующим основанием (например, раствором гидроксида аммония, гидроксида натрия и т.п.). Соединение по изобретению в форме основно-аддитивной соли можно превратить в соответствующую свободную кислоту при обработке соответствующей кислотой (например, соляной кислотой и т.п.).
Соединения по изобретению в неокисленной форме можно получить из N-оксидов соединений по изобретению при обработке восстанавливающим агентом (например, серой, диоксидом серы, трифенилфосфином, боргидридом лития, боргидридом натрия, трихлоридом фосфора, трибромидом фосфора или т.п.) в пригодном инертном органическом растворителе (например, ацетонитриле, этаноле, водном диоксане или т.п.) при температуре от 0°С до 80°С.
Пролекарства соединений по изобретению получают известными методами (например, см. Saulnier и др., Bioorganic and Medicinal Chemistry Letters, 4, с.1985 (1994)). Например, соответствующие пролекарства можно получить при взаимодействии немодифицированного соединения по изобретению с пригодным карбамилирующим агентом (например, 1,1-ацилоксиалкилкарбанохлоридом, пара-нитрофенилкарбонатом или т.п.).
Соединения по изобретению, содержащие защитные группы, можно получить известными методами. Подробное описание методик введения защитных групп и их удаления можно найти в монографии T.W. Greene, "Protecting Groups in Organic Chemistry", 3 изд., John Wiley и Sons, Inc., (1999).
Соединения по изобретению можно получить стандартным способом или способом по изобретению, в виде сольватов (например, гидратов). Гидраты соединений по настоящему изобретению получают стандартным способом при перекристаллизации из смеси вода/органический растворитель, такой как диоксан, тетрагидрофуран или метанол.
Соединения по изобретению можно получить в виде индивидуальных стереоизомеров при взаимодействии рацемической смеси соединения с оптически активным разделяющим агентом с образованием пары диастереоизомерных соединений, разделении диастереомеров и выделении оптически чистых энантиомеров. В то время как разделение энантиомеров проводят с использованием ковалентных диастереомерных производных соединений по изобретению, предпочтительными являются диссоциирующие комплексы (например, кристаллические диастереомерные соли). Диастереомеры обладают разными физическими свойствами (например, температурой плавления, температурой кипения, растворимостью, реакционной способностью и т.п.), и благодаря этому их можно разделить простыми методами. Диастереомеры разделяют хроматографией или предпочтительно по методике, основанной на различии в растворимости. Затем оптически чистый энантиомер выделяют с использованием разделяющего агента любым способом, исключающим рацемизацию. Более подробное описание методик, используемых для выделения стереоизомеров соединений из рацемических смесей, можно найти в монографии Jean Jacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions", John Wiley And Sons, Inc. (1981).
В итоге, соединения формулы I можно получить способом, который заключается в том, что:
(а) проводят реакции, как показано на схемах I или II, и
(б) необязательно превращают соединение по изобретению в фармацевтически приемлемую соль,
(в) необязательно превращают соль соединения по изобретению в несолевую (свободную) форму,
(г) необязательно превращают неокисленную форму соединения по изобретению в фармацевтически приемлемый N-оксид,
д) необязательно превращают N-оксид соединения по изобретению в его неокисленную форму,
(е) необязательно выделяют индивидуальный изомер соединения по изобретению из смеси изомеров,
(ж) необязательно превращают немодифицированное соединение по изобретению в фармацевтически приемлемое пролекарство, и
(з) необязательно превращают пролекарство соединения по изобретению в немодифицированную форму.
Получение исходных материалов подробно не описано, поскольку соединения известны, или их можно получить известными методами, или, как описано ниже в разделе Примеры.
Для специалиста в данной области представляется очевидным, что вышеуказанные синтезы и модификации приводятся для иллюстрации методов получения соединений по настоящему изобретению и что их можно получить с использованием других известных методов.
Примеры
Настоящее изобретение иллюстрируется следующими примерами получения соединений формулы I, не ограничивающими его объем.
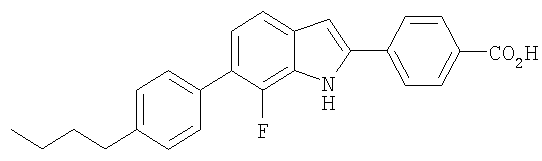
Пример 1
4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]бензойная кислота
Стадия 1
Метиловый эфир 4-(6-хлор-7-фтор-1Н-индол-2-ил)бензойной кислоты
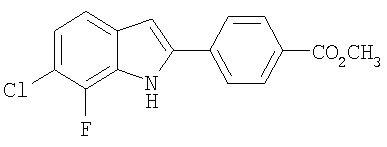
К гидрохлориду (3-хлор-2-фторфенил)гидразина (фирма Apollo Scientific, Ltd., 221 мг, 1,12 ммоль) и метиловому эфиру 4-ацетилбензойной кислоты (200 мг, 1,12 ммоль) добавляли безводный ZnCl2 (382 мг, 2,81 ммоль) и уксусную кислоту (10 мл), реакционную смесь нагревали при 105°С в течение 48 ч. Затем смесь охлаждали до комнатной температуры, разбавляли этилацетатом, последовательно промывали Н2О (5×) и насыщенным раствором NaCl, органический слой сушили над Na2SO4, фильтровали и фильтрат концентрировалии. Неочищенный продукт очищали препаративной ОФ-ЖХ/МС, при этом получали метиловый эфир 4-(6-хлор-7-фтор-1Н-индол-2-ил)бензойной кислоты в виде твердого вещества грязно-белого цвета. МС (ES): m/z 304,0 (М+Н+).
Стадии 2 и 3
В смесь метилового эфира 4-(6-хлор-7-фтор-1Н-индол-2-ил)бензойной кислоты (37 мг, 0,122 ммоль, стадия 1), 4-н-бутилфенилбороновой кислоты (43 мг, 0,244 ммоль) и карбоната цезия (159 мг, 0,487 ммоль) в диоксане (4 мл) добавляли катализатор на основе палладия (CombiPhos-Pd6, фирма Combiphos Catalysts Inc., 3 мг), смесь продували азотом в течение 5 мин и нагревали в закрытой пробирке при 120°С в течение 7 ч. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и последовательно промывали 1н. HCl, Н2О и насыщенным раствором NaCl. Органический слой сушили над Na2SO4, фильтровали и концентрировали. В полученный остаток добавляли этанол/Н2О (2 мл/1 мл), LiOH (26 мг, 0,609 ммоль) и смесь нагревали при 50°С в течение 1 ч. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и промывали 1н. HCl. Органический слой сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали препаративной ОФ-ЖХ/МС, при этом получали 4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]бензойную кислоту в виде твердого вещества белого цвета. МС (ES): m/z 388,2 (М+Н+).
1Н-ЯМР (400 МГц, ацетон-d6): δ 11,13 (ушир. s, 1H), 8,10-8,05 (m, 4H), 7,59 (d, 2H), 7,52 (d, 1H), 7,32 (d, 2H), 7,18-7,12 (m, 2H), 2,71 (t, 2H), 1,70-1,63 (m, 2H), 1,49-1,40 (m, 2H), 0,98 (t, 3H).
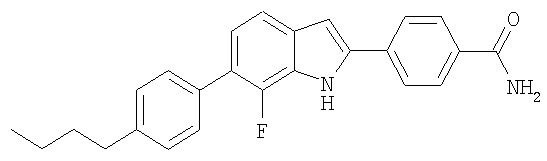
Пример 2
4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]бензамид
Стадия 1
Гидрохлорид (4'-бутил-2-фторбифенил-3 -ил)гидразина
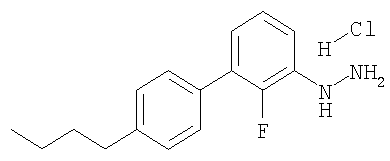
Смесь диоксана (16 мл) и N-метилпирролидинона (8 мл) продували азотом в течение 15 мин для удаления следов кислорода. 3-Хлор-2-фторанилин (3 г, 20,6 ммоль) помещали в круглодонную колбу объемом 250 мл, снабженную насадкой с резьбой, позволяющей закрывать колбу через трехходовой кран, а затем добавляли 4-бутилфенилбороновую кислоту (5,5 г, 30,9 ммоль), фторид цезия (7,82 г, 51,5 ммоль) и бис(три-трет-бутилфосфино)палладий (527 мг, 1,03 ммоль). Колбу вакуумировали, заполняли азотом и закрывали. Затем колбу помещали на масляную баню, нагретую до 130°С, и перемешивали в течение 4 ч. Реакционную смесь охлаждали, фильтровали через слой целита, который промывали EtOAc. Растворители удаляли на роторном испарителе, неочищенную реакционную смесь разбавляли водой и экстрагировали EtOAc. Фазы разделяли, органический слой сушили над MgSO4, фильтровали и концентрировали. Полученное масло разбавляли диэтиловым эфиром и добавляли избыток 4 М раствора HCl в диоксане. Полученное твердое вещество отделяли, промывали диэтиловым эфиром и высушивали, при этом получали гидрохлорид 4'-бутил-2-фторбифенил-3-иламина в виде твердого вещества белого цвета. ЖХ/МС: рассч. для C16H19FN 244,3, найд. 244,2 [М+Н]+. В гидрохлорид 4'-бутил-2-фторбифенил-3-иламина (5,4 г, 0,019 моль) в конц. HCl (50 мл) при 0°С в течение 10 мин добавляли по каплям раствор нитрита натрия (1,3 г, 0,019 моль) в Н2О (20 мл). Полученный раствор перемешивали при 0°С в течение 1 ч, а затем при 0°С добавляли по каплям раствор хлорида олова (II) (13,0 г, 0,058 моль) в конц. HCl (14 мл). Смесь перемешивали в течение 15 мин, а затем фильтровали. Полученное твердое вещество промывали холодным насыщенным раствором NaCl, а затем суспендировали в 50% растворе NaOH. Полученную суспензию разбавляли Н2О и экстрагировали Et2O. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Полученный продукт растворяли в 100 мл Et2O и охлаждали до 0°С. В указанный раствор добавляли по каплям 4,0 М раствор HCl в диоксане (15 мл) и полученный осадок отделяли, при этом получали 4,58 г (81%) требуемого гидрохлорида гидразина. ЖХ/МС: рассч. для C16H20FN2 259,3, найд. 259,2.
Стадия 2
В гидрохлорид (4'-бутил-2-фторбифенил-3-ил)гидразина (90 мг, 0,306 ммоль, стадия 1) и 4-ацетилбензамид (Sigman М.Е. и др., J. Am. Chem. Soc., 110, 4297 (1958), 50 мг, 0,306 ммоль) добавляли безводный ZnCl2 (418 мг, 3,06 ммоля) и уксусную кислоту (4 мл), реакционную смесь нагревали при 105°С в течение 48 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и последовательно промывали Н2О (5×), 1н. HCl и насыщенным раствором NaCl. Органический слой сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали препаративной ОФ-ЖХ/МС, при этом получали 4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]бензамид в виде твердого вещества бежевого цвета. МС (ES): m/z 387,2 (М+Н+).
1Н-ЯМР (400 МГц, ацетон-d6): δ 11,11 (ушир. s, 1Н), 8,08-8,01 (m, 4H), 7,59-7,43 (m, 4H), 7,38-7,29 (m, 2H), 7,18-7,12 (m, 2H), 6,65 (ушир., 1Н), 2,74-2,63 (m, 2H), 1,76-1,64 (m, 2H), 1,50-1,38 (m, 2H), 1,04-0,93 (m, 3H).
Пример 3
4-[7-Фтор-6-(4-фтор-3-метилфенил)-1Н-индол-2-ил]бензойная кислота
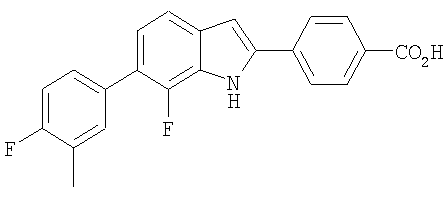
4-[7-Фтор-6-(4-фтор-3-метилфенил)-1Н-индол-2-ил]бензойную кислоту получали в виде твердого вещества белого цвета из соответствующих исходных материалов по методикам, описанным в примере 2. МС (ES): m/z 364,1 (М+Н+).
1Н ЯМР (400 МГц, CD3OD): δ 8,09 (d, 2H), 7,98 (d, 2H), 7,52-7,42 (m, 3H), 7,18-7,05 (m, 3H), 2,32 (s, 3H).
Пример 4
4-[7-Фтор-6-(4-фтор-3-метилфенил)-1Н-индол-2-ил]бензолсульфонамид
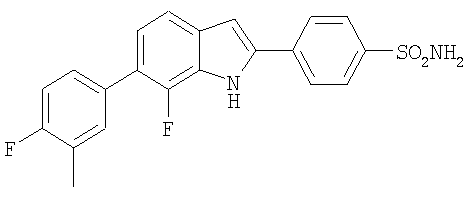
4-[7-Фтор-6-(4-фтор-3-метилфенил)-1Н-индол-2-ил]бензолсульфонамид получали в виде твердого вещества бежевого цвета из соответствующих исходных материалов по методикам, описанным в примере 2. МС (ES): m/z 399,1 (М+Н+).
1Н-ЯМР (400 МГц, ацетон-d6): δ 11,12 (ушир. s, 1H), 8,18-8,13 (m, 2H), 8,06-7,98 (m, 2H), 7,59-7,48 (m, 3H), 7,22-7,12 (m, 3H), 6,68 (ушир., 2H), 2,33 (s, 3H).
Пример 5
4-[6-(4-Бутилфенил)-3-этил-7-фтор-1Н-индол-2-ил]-2-гидроксибензойная кислота
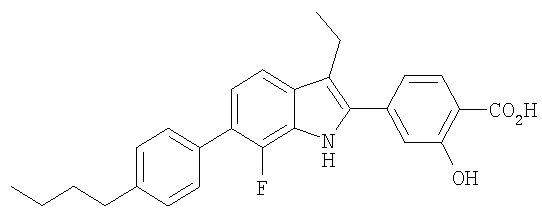
Стадия 1
2-Гидрокси-4-иодбензойная кислота
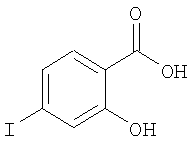
В колбу Эрленмейера объемом 2 л, снабженную большим стержнем для перемешивания, помещали 4-амино-2-гидроксисалициловую кислоту (10 г, 65,3 ммоль), охлаждали на ледяной бане и добавляли конц. серную кислоту (20 мл) и немного воды для образования суспензии (~50 мл). Реакционную смесь перемешивали в течение 20 мин, а затем в течение 10 мин добавляли водный раствор нитрита натрия (4,55 г, 66,0 ммоль, 20 мл). Реакционную смесь перемешивали в течение еще 3 мин, а затем в течение 15 мин добавляли водный раствор иодида калия (16,9 г, 101 ммоль, 30 мл). Баню удаляли, реакционную смесь обрабатывали и перемешивали осторожно, поскольку при этом выделялось большое количество азота. После завершения реакции смесь кратковременно нагревали до 70°С, затем сразу охлаждали до комнатной температуры и выдерживали в течение ночи. Полученное твердое вещество отделяли фильтрованием, промывали водой и высушивали, при этом получали неочищенную 2-гидрокси-4-иодбензойную кислоту, которую использовали на следующей стадии без дополнительной очистки. МС (ES): m/z 265,0 (М+Н+).
Стадия 2
Метиловый эфир 2-гидрокси-4-иодбензойной кислоты
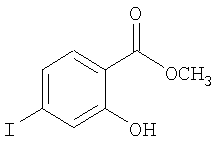
В раствор 2-гидрокси-4-иодбензойной кислоты (4,0 г, 0,015 моль) в ТГФ (25 мл) и МеОН (25 мл) добавляли по каплям (триметилсилил)диазометан (2,0 М раствор в Et2O, 15 мл) и смесь перемешивали при комнатной температуре в течение 2,5 ч. Летучие компоненты удаляли в вакууме и неочищенный остаток разбавляли EtOAc. Органическую фазу последовательно промывали насыщенным раствором NaHCO3 (3×), насыщенным раствором NaCl (1×) и Н2О (1×), сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: градиент этилацетат/гексан, от 0% до 80%), при этом получали метиловый эфир 2-гидрокси-4-иодбензойной кислоты. МС (ES): m/z 279,0 (М+Н+).
Стадия 3
Метиловый эфир 2-гидрокси-4-винилбензойной кислоты
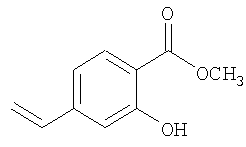
В раствор метилового эфира 2-гидрокси-4-иодбензойной кислоты (8,01 г, 0,0288 моль) в ТГФ (184 мл) и Н2О (46 мл) добавляли дибутиловый эфир винилбороновой кислоты (9,53 мл, 0,0432 моль, 1,5 экв.), Na2CO3 (21,37 г, 0,201 моля, 7 экв.) и дихлор-бис(трифенилфосфин)палладий (1,01 г, 1,44 ммоль, 5 мол.%). Раствор продували N2 в течение 5 мин, а затем кипятили с обратным холодильником в течение 2 ч. Реакционную смесь концентрировали в вакууме, остаток разбавляли EtOAc и последовательно промывали Н2О и насыщенным раствором NaCl. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: 5% этилацетат/гексан), при этом получали метиловый эфир 2-гидрокси-4-винилбензойной кислоты. МС (ES): m/z 179,1 (М+Н+).
Стадия 4
Метиловый эфир 4-формил-2-гидроксибензойной кислоты
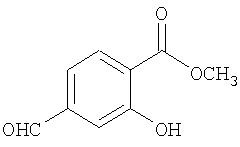
Через раствор метилового эфира 2-гидрокси-4-винилбензойной кислоты (3,28 г, 0,0184 моль) в CH2Cl2 (50 мл) при -78°С в течение 5 мин пропускали непрерывный поток О2, а затем через раствор пропускали поток О3 до приобретения раствором синего/серого цвета. Затем раствор в течение 5 мин продували O2, добавляли DMS (4,05 мл, 0,0552 моль) и нагревали до комнатной температуры в течение ночи. Все летучие компоненты удаляли в вакууме и неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: градиент этилацетат/гексан, от 0% до 100%), при этом получали метиловый эфир 4-формил-2-гидроксибензойной кислоты. МС (ES): m/z 181,0 (М+Н+).
Стадия 5
Метиловый эфир 2-гидрокси-4-(1-гидроксибутил)бензойной кислоты
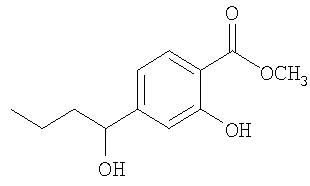
Метиловый эфир 4-формил-2-гидроксибензойной кислоты (360 мг, 2 ммоль) растворяли в безводном ТГФ (10 мл), раствор охлаждали до -78°С и перемешивали в атмосфере азота. Затем в раствор шприцом добавляли по каплям н-пропилмагнийхлорид (2 М раствор в ТГФ, 2 мл, 2 ммоль). Реакционную смесь перемешивали при -78°С в течение 1 ч, затем в течение 1 ч нагревали до комнатной температуры и реакцию останавливали при добавлении насыщенного раствора хлорида аммония. Полученную смесь экстрагировали EtOAc (3×15 мл), объединенную органическую фазу промывали насыщенным раствором NaCl, сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: градиент этилацетат/гексан, от 10% до 20%), при этом получали метиловый эфир 2-гидрокси-4-(1-гидроксибутил)бензойной кислоты в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3): δ 10,74 (s, 1H), 7,79 (d, 1H, J 8,4 Гц), 6,95 (d, 1H, J 1,2 Гц), 6,86 (dd, 1H, J 1,6 Гц), 4,67 (t, 1H, J 6,8 Гц), 3,94 (s, 3H), 1,82 (ушир., 1H), 1,80-1,62 (m, 1H), 1,46-1,28 (m, 2H), 0,93 (t, 3H, J 7,2 Гц).
Стадия 6
Метиловый эфир 4-бутирил-2-гидроксибензойной кислоты
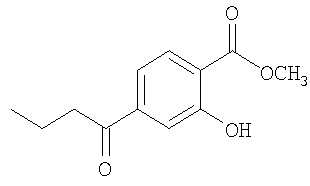
В раствор метилового эфира 2-гидрокси-4-(1-гидроксибутил)бензойной кислоты (132 мг, 0,59 ммоль) в ДХМ (6 мл) одной порцией добавляли PDC (245 мг, 1,1 ммоль) и реакционную смесь перемешивали в течение ночи. Затем реакционную смесь фильтровали через слой силикагеля при элюировании ДХМ и фильтрат концентрировали, при этом получали метиловый эфир 4-бутирил-2-гидроксибензойной кислоты в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, CDCl3): δ 10,77 (s, 1H), 7,92 (d, 1H, J 8,4 Гц), 7,52 (d, 1H, J 1,6 Гц), 7,44 (dd, 1H, J 1,6, 8 Гц), 3,99 (s, 3H), 2,93 (t, 2H, J 7,2 Гц), 1,76 (qt, 2H, J 7,6 Гц), 1,00 (t, 3Н, J 7,6 Гц).
Стадия 7
Метиловый эфир 4-(6-хлор-3-этил-7-фтор-1Н-индол-2-ил)-2-гидроксибензойной кислоты
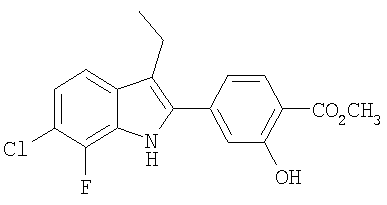
Смесь метилового эфира 4-бутирил-2-гидроксибензойной кислоты (60 мг, 0,27 ммоль), (3-хлор-2-фторфенил)гидразина (фирма Apollo Scientific, Ltd., 54 мг, 0,27 ммоль) и хлорида цинка (110 мг, 0,8 ммоль) в уксусной кислоте (2 мл) продували азтом в течение 5 мин, а затем нагревали в закрытой пробирке при 120°С в течение 2 ч. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом и последовательно промывали насыщенным раствором Na2CO3 и насыщенным раствором NaCl, сушили над сульфатом натрия и концентрировали. Неочищенный продукт очищали экспресс-хроматографией на колонке с силикагелем (элюент: 10% этилацетат/гексан), при этом получали метиловый эфир 4-(6-хлор-3-этил-7-фтор-1Н-индол-2-ил)-2-гидроксибензойной кислоты в виде твердого вещества светло-желтого цвета. МС (ES): m/z 348,1 (М+Н+).
Стадии 8 и 9
В смесь метилового эфира 4-(6-хлор-3-этил-7-фтор-1Н-индол-2-ил)-2-гидроксибензойной кислоты (50 мг, 0,14 ммоль), 4-н-бутил-фенилбороновой кислоты (50 мг, 0,28 ммоль) и фторида цезия (78 мг, 0,52 ммоль) в безводном диоксане (3 мл) добавляли бис(три-трет-бутилфосфин)палладий (6 мг, 10 мол.%). Полученную смесь продували N2 в течение 3 мин, а затем нагревали в закрытой пробирке при 120°С в течение 4 ч. Смесь охлаждали до комнатной температуры, фильтровали и фильтрат концентрировали. Остаток растворяли в этаноле/Н2О (1 мл/0,1 мл) и переносили в пробирку для микроволнового реактора. В смесь добавляли LiOH (12 мг, 0,54 ммоля) и нагревали при 120°С в течение 6 мин в микроволновом реакторе. Смесь фильтровали, фильтрат концентрировали и неочищенный продукт очищали препаративной ОФ-ЖХ/МС, при этом получали 4-[6-(4-бутилфенил)-3-этил-7-фтор-1Н-индол-2-ил]-2-гидроксибензойную кислоту в виде твердого вещества желтого цвета. МС (ES): m/z 432,2 (М+Н+).
1Н-ЯМР (400 МГц, CD3OD): δ 7,96 (d, 1H, J 8,8 Гц), 7,50 (d, 2H, J 8 Гц), 7,42 (d, 1H, J 8,4 Гц), 7,24 (d, 2H, J 8 Гц), 7,21-7,20 (m, 2H), 7,10 (dd, 1H, J 8,8 Гц), 2,95 (q, 2H, J 7,6 Гц), 2,66 (t, 2H, J 8 Гц), 1,65 (m, 2H), 1,40 (m, 2H), 1,34 (t, 3H, J 7,6 Гц), 0,97 (t, 3H, J 7,6 Гц).
Пример 6
4-[6-(4-Бутилфенил)-7-фтор-3-изопропил-1Н-индол-2-ил]-2-гидроксибензойная кислота
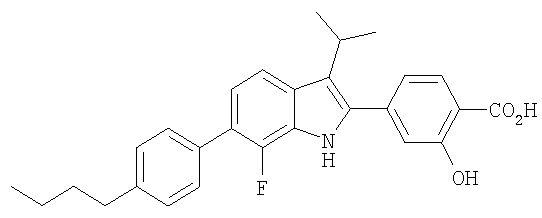
4-[6-(4-Бутилфенил)-7-фтор-3-изопропил-1Н-индол-2-ил]-2-гидроксибензойную кислоту получали в виде твердого вещества желтого цвета из соответствующих исходных материалов по методикам, описанным в примере 5. МС (ES): m/z 446,2 (М+Н+).
1Н-ЯМР (400 МГц, ацетон-d6): δ 10,64 (s, 1H), 8,01 (d, 1H, J 8,4 Гц), 7,68 (d, 1H, J 8 Гц), 7,55 (d, 2H, J 7,6 Гц), 7,31 (d, 2H, J 8 Гц), 7,23-7,13 (m, 3H), 3,46 (m, 1H), 2,68 (t, 2H, J 7,6 Гц), 1,66 (m, 2H), 1,51 (d, 6H, J 7,2 Гц), 1,40 (m, 2H), 0,96 (t, 3H, J 7,6 Гц).
Пример 7
4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]-2-гидроксибензойная кислота
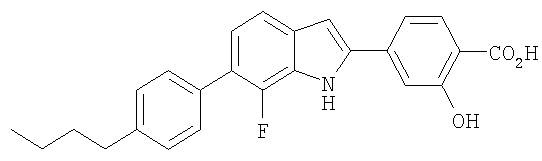
4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]-2-гидроксибензойную кислоту получали в виде твердого вещества желтого цвета из соответствующих исходных материалов по методикам, описанным в примере 5. МС (ES): m/z 404,2 (М+Н+).
1Н-ЯМР (400 МГц, ацетон-d6): δ 11,12 (ушир. s, 1Н), 7,99 (d, 1H), 7,60-7,53 (m, 4H), 7,48 (d, 1H), 7,32 (d, 2H), 7,18-7,10 (m, 2H), 2,67 (t, 2H), 1,72-1,61 (m, 2H), 1,49-1,37 (m, 2H), 0,98 (t, 3H).
Пример 8
4-[6-(4-Бутилфенил)-7-фтор-3-метил-1Н-индол-2-ил]-2-гидроксибензойная кислота
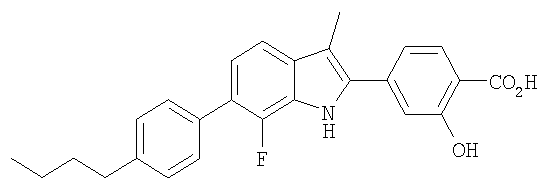
4-[6-(4-Бутилфенил)-7-фтор-3-метил-1Н-индол-2-ил]-2-гидроксибензойную кислоту получали в виде твердого вещества светло-желтого цвета из соответствующих исходных материалов по методикам, описанным в примере 5. МС (ES): m/z 418,2 (М+Н+).
1Н-ЯМР (400 МГц, ацетон-d6): δ 10,75 (ушир. s, 1H), 8,02 (d, 1H), 7,57-7,15 (m, 8H), 2,74-2,62 (m, 2H), 2,58 (s, 3H), 1,71-1,61 (m, 2H), 1,43-1,35 (m, 2H), 0,98 (t, 3H).
Пример 9
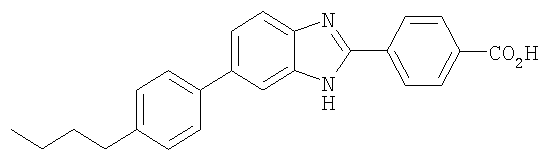
4-[6-(4-Бутилфенил)-1Н-бензимидазол-2-ил]бензойная кислота
Стадия 1
4'-Бутил-3-нитробифенил-4-иламин
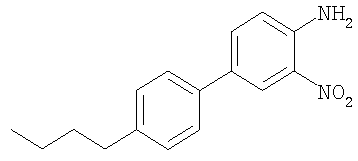
Пробирку для проведения реакции Смита, содержащую 4-бром-2-нитроанилин (205 мг, 0,945 ммоль), 4-бутилфенилбороновую кислоту (336 мг, 1,89 ммоль), фторид цезия (430 мг, 2,83 ммоль), бис(три-трет-бутилфосфин)палладий (24,1 мг, 0,0472 ммоль) и диоксан (2 мл), продували аргоном в течение 5 мин, а затем нагревали в микроволновом реакторе при 120°С в течение 15 мин. Неочищенную реакционную смесь фильтровали через целит при элюировании EtOAc. Фильтрат концентрировали в вакууме и неочищенный продукт очищали экспресс-хроматографией на силикагеле (элюент: градиент этилацетат/гексан, от 0% до 100%), при этом получали 4'-бутил-3-нитробифенил-4-иламин. МС (ES): m/z 271,2 (М+Н+).
Стадия 2
4'-Бутилбифенил-3,4-диамин
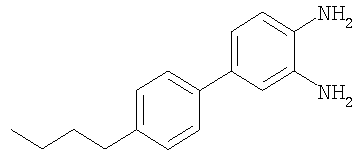
Смесь 4'-бутил-3-нитробифенил-4-иламина (117 мг, 3,70 ммоль) и 10% Pd/C (10 мг) в MeOH/EtOAc (4 мл, 2:1, об./об.) продували H2 в течение 15 мин, а затем перемешивали в атмосфере Н2 (1 атм) в течение 1 ч. Реакционную смесь фильтровали через целит и фильтрат концентрировали в вакууме, при этом получали 4'-бутилбифенил-3,4-диамин. МС (ES): m/z 241,2 (М+Н+).
Стадия 3
В нагретую (140°С) смесь метилового эфира 4-формилбензойной кислоты (21,0 мг, 0,129 ммоль) и NaHSO3 (20,1 мг, 0,193 ммоль, 1,5 экв.) в DMA (250 мкл) в течение 10 мин добавляли по каплям раствор 4'-бутилбифенил-3,4-диамина (31 мг, 0,129 ммоль) в DMA (100 мкл). Полученную смесь перемешивали при 140°С в течение 1 ч, а затем добавляли Н2О (1 мл) и перемешивали в течение еще 1 ч. Реакционную смесь разбавляли EtOAc и фазы разделяли. Органическую фазу сушили над MgSO4, фильтровали и концентрировали. Неочищенный бензимидазол разбавляли 95% этанолом (0,5 мл) и ТГФ (1 мл), переносили в пробирку для проведения реакции Смита, содержащую LiOH (15,4 мг, 0,645 ммоль, 5 экв.) и нагревали в микроволновом реакторе при 165°С в течение 5 мин. Смесь концентрировали и неочищенный продукт очищали препаративной ОФ-ЖХ/МС, при этом получали 4-[6-(4-бутилфенил)-1Н-бензимидазол-2-ил]бензойную кислоту. МС (ES): m/z 371,2 (М+H+).
1Н-ЯМР (400 МГц, CD3OD): δ 9,00 (d, J 8,2 Гц, 1Н), 8,91 (d, J 8,1 Гц, 1Н), 8,63 (s, 1H), 8,53 (s, 1Н), 8,31 (d, J 7,6 Гц, 2Н), 8,23 (d, J 8,3 Гц, 1Н), 8,02 (d, J 7,7 Гц, 1Н), 7,94 (s, 1Н), 7,84 (s, 1Н), 7,62 (d, J 7,7 Гц, 1Н), 7,33 (d, J 7,6 Гц, 1Н), 3,38 (dd, J 7,8, 7,7 Гц, 1Н), 2,69 (dd, J 7,8, 7,6 Гц, 1Н), 2,35 (m, 1H), 2,09 (m, 1H), 1,66 (dd, J 7,4, 7,3 Гц, 3H), 1,41 (m, 1H), 0,97 (dd, J 7,3, 6,9 Гц, 2Н).
Пример 10
4-[6-(4-Бутилфенил)-1Н-бензимидазол-2-ил]-2-гидроксибензойная кислота
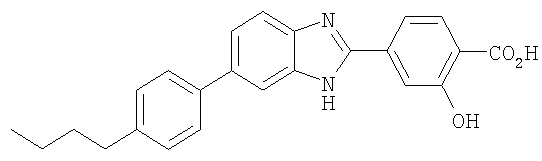
4-[6-(4-Бутилфенил)-1Н-бензимидазол-2-ил]-2-гидроксибензойную кислоту получали из соответствующих исходных материалов по методикам, описанным в примерах 5 и 9. МС (ES): m/z 387,1 (М+Н+).
Пример 11
N-{4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]фенил}метансульфонамид
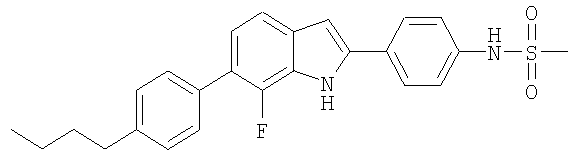
N-{4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]фенил}метансульфонамид получали в виде твердого вещества бежевого цвета из соответствующих исходных материалов по методикам, описанным в примере 2. МС (ES): m/z 437,2 (М+Н+).
1Н-ЯМР (400 МГц, ацетон-d6): δ 11,00 (ушир. s, 1H), 8,79 (s, 1H), 7,99 (d, 2Н), 7,55 (d, 2Н), 7,52-7,44 (m, 3H), 7,32 (d, 2Н), 7,15 (dd, 1H), 7,01 (s, 1H), 3,09 (s, 3H), 2,72 (t, 2Н), 1,75-1,64 (m, 2Н), 1,47-1,36 (m, 2Н), 0,99 (t, 3H).
Пример 12
N-{4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]фенил}ацетамид
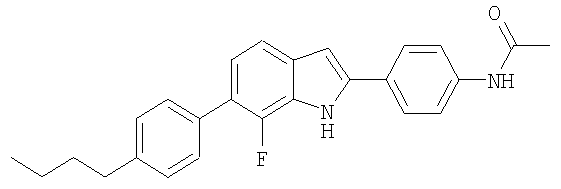
N-{4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]фенил}ацетамид получали в виде твердого вещества бежевого цвета из соответствующих исходных материалов по методикам, описанным в примере 2. МС (ES): m/z 401,1 (М+Н+).
1Н-ЯМР (400 МГц, ацетон-d6): δ 10,95 (ушир. s, 1H), 9,32 (ушир. s, 1H), 7,92 (d, 2H), 7,81 (d, 2H), 7,57 (d, 2H), 7,43 (d, 1H), 7,32 (d, 2H), 7,16 (dd, 1H), 6,97 (s, 1H), 2,70 (t, 2H), 1,69-1,62 (m, 2H), 1,42-1,33 (m, 2H), 0,96 (t, 3H).
Пример 13
N-{4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]-2-хлорфенил}ацетамид
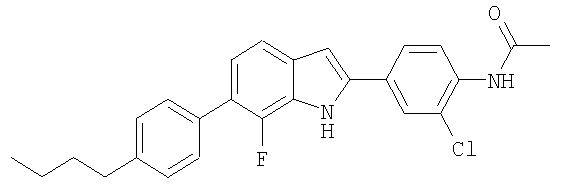
N-{4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]-2-хлорфенил}ацетамид получали в виде твердого вещества из соответствующих исходных материалов по методикам, описанным в примере 2. МС (ES): m/z 435,2 (М+Н+).
1Н-ЯМР (400 МГц, ДМСО-d6): δ 10,1 (s, 1H), 8,62 (s, 1H), 8,19 (d, J 8,6 Гц, 1H), 7,92 (d, J 2,1 Гц, 1H), 7,77 (dd, J 8,6, 2,1 Гц, 1H), 7,42 (dd, J 8,1, 1,6 Гц, 2H), 7,32 (d, J 8,2 Гц, 1H), 7,19 (d, J 8,2 Гц, 1H), 7,03 (dd, J 8,1, 6,9 Гц, 1H), 6,91 (dd, J 3,3, 2,3 Гц, 1H), 2,56 (t, J 7,7 Гц, 2H), 2,11 (s, 3H), 1,54 (m, 2H), 1,28 (m, 2H), 0,83 (t, J 7,3 Гц, 3H).
Пример 14
4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]-2-хлорфениламин
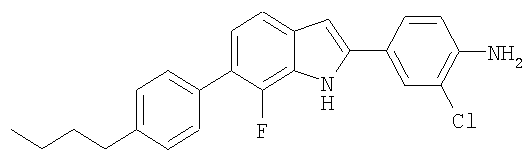
4-[6-(4-Бутилфенил)-7-фтор-1Н-индол-2-ил]-2-хлорфениламин получали в виде твердого вещества из соответствующих исходных материалов по методикам, описанным в примере 2. МС (ES): m/z 393,3 (М+Н+).
Пример 15
2-Амино-4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]бензойная кислота
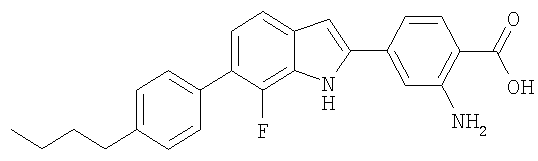
Стадия 1
1-(3-Нитро-4-винилфенил)этанон
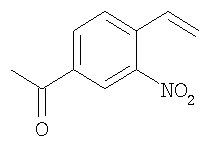
В смесь 4'-бром-3'-нитроацетофенона (600 мг, 2,46 ммоль), дибутилового эфира винилбороновой кислоты (680 мг, 3,69 ммоль) и карбоната натрия (1,83 г, 17,22 ммоль) в ТГФ/Н2О (12 мл/4 мл) добавляли дихлорбис(трифенилфосфин)палладий (II) (86 мг, 5 ммол.%). Пробирку закрывали, продували N2 в течение 3 мин и нагревали при 70°С в течение 1,5 ч. Затем смесь охлаждали до комнатной температуры и выливали в насыщенный раствор хлорида аммония. Смесь экстрагировали этилацетатом (3×20 мл), органические экстракты объединяли, промывали солевым раствором и концентрировали. Неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: 20% этилацетат/гексан), при этом получали 1-(3-нитро-4-винилфенил)этанон в виде твердого вещества желтого цвета (выход 87%). МС (ES): m/z 192,0 (М+Н+).
Стадия 2
Метиловый эфир 4-ацетил-2-нитробензойной кислоты
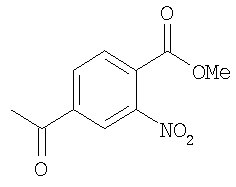
1-(3-Нитро-4-винилфенил)этанон (410 мг, 2,14 ммоль, стадия 1) растеряли в ДХМ (20 мл), охлаждали до -78°С, добавляли NaOH (429 мг, 10,7 ммоль) в МеОН (5 мл) и через раствор пропускали поток О3 до приобретения раствором синего/серого цвета. Затем раствор продували N2 до обесцвечивания, нагревали до комнатной температуры и концентрировали, при этом получали метиловый эфир 4-ацетил-2-нитробензойной кислоты в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3): δ 8,41 (d, 1H, J 1,6 Гц), 8,19 (dd, 1H, J 7,6, 1,6 Гц), 7,79 (d, 1H, J 8 Гц), 3,91 (s, 3H), 2,66 (s, 3H).
Стадии 3-5
Метиловый эфир 4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]-2-нитробензойной кислоты получали при взаимодействии метилового эфира 4-ацетил-2-нитробензойной кислоты (полученного, как описано на стадии 2) и гидрохлорида (4'-бутил-2-фторбифенил-3-ил)гидразина (полученного, как описано в примере 2, стадия 1) по методике, описанной в примере 2. Затем проводили гидрирование нитрогруппы (1 атм Н2, Pd/C) с образованием соответствующего амина и гидролиз метилового эфира (NaOH в EtOH/Н2О) и после очистки продукта методом препаративной ОФ-ЖХ/МС получали 2-амино-4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]бензойную кислоту в виде твердого вещества. МС (ES): m/z 403,1 (М+Н+).
1Н-ЯМР (400 МГц, ацетон-d6): δ 11,07 (s, 1Н), 7,92 (d, 1H), 7,53 (dd, 2H), 7,45 (d, 1H), 7,36 (d, 1H), 7,31 (d, 2H), 7,19-7,13 (m, 2H), 7,02-7,00 (m, 1H), 2,67 (t, 2H), 1,67-1,62 (m, 2H), 1,42-1,37 (m, 2H), 0,94 (t, 3H).
Методы анализа
Соединения по настоящему изобретению оценивали по их эффективности в качестве миметиков ТРО при анализе пролиферации in vitro с использованием клеток линии BaF3 мыши, трансфектированных рецептором ТРО человека (ТРО-R).
Анализы с использованием репортерного гена люциферазы
Клетки Ba/F3-TpoR промывали и ресуспендировали в среде RPMI-1640, содержащей 1% или 20% ЭТС, MS, HS или (смесь сывороточного альбумина человека/кислого α1-гликопротеина человека), 1% Pen-Strep-Glu и 1 мМ или 25 мкМ ZnSO4, при концентрации 8×104 клеток/мл, переносили в 384-луночные планшеты (50 мкл/лунку) и инкубировали в течение ночи (18-20 ч) на минимальной среде. В день 2 истощенные клетки обрабатывали 0,5 мл ДМСО, соединением или rhTpo (30 нг/мл) при 37°С в атмосфере 5% СО2 в течение 7 ч. В каждую лунку добавляли реагент Britelite (25 мкл, фирма Perkin Elmer), разбавленный водой до концентрации 60%, и через несколько мин в планшетах регистрировали интенсивность люминесценции на люминометре CLIPR.
Анализ пролиферации
Клетки Ba/F3-TpoR промывали и ресуспендировали в среде RPMI-1640, содержащей 1% ЭТС, 1% Pen-Strep-Glu и 1 мМ или 25 мкМ ZnSO4, при концентрации 8×104 клеток/мл, переносили в 384-луночные планшеты (50 мкл/лунку) и инкубировали в течение ночи (18-20 ч) на минимальной среде. В день 2 истощенные клетки обрабатывали 0,5 мл ДМСО, соединением или rhTpo (30 нг/мл) при 37°С в атмосфере 5% СО2 в течение 48 ч. В каждую лунку добавляли реагент Alamar Blue (3,5 мкл при ~7% конечной концентрации), планшеты инкубировали в течение 4 ч и регистрировали сигнал флуоресценции в системе Analyst GT.
Анализ CFU-Meg
При проведении анализа использовали клетки CD34+и набор MegaCult-C (фирма StemCell Technologies, Inc., Ванкувер, Канада). Клетки CD34+ смешивали с раствором коллагена MegaCult-C по методике фирмы-производителя в количестве 104 клеток на одно предметное стекло. Затем добавляли ТРО или соединение по изобретению в различных концентрациях, предметные стекла инкубировали при 37°С в атмосфере 5% СО2 в течение 12 сут, фиксировали, проявляли CFU-Meg человека и подсчитывали число колоний с использованием инвертированного микроскопа.
Соединения формулы I в свободной форме или в форме фармацевтически приемлемой соли проявляют ценные фармакологические свойства, например, по данным испытаний in vitro, описанных в настоящей заявке. Соединения по изобретению предпочтительно обладают активностью миметиков ТРО при значении IC50 в интервале от 1×10-9 до 1×10-5 М, предпочтительно менее 500 нМ, более предпочтительно менее 250 нМ. Соединения формулы I проявляют эффективность в интервале от 25% до 150% относительно ТРО.
Подразумевается, что изобретение иллюстрируется примерами и вариантами его осуществления, и для специалиста в данной области техники представляется очевидным, что в пределах сущности и объема изобретения возможны различные модификации и изменения, которые включены в объем заявки и пунктов прилагаемой формулы изобретения. Все публикации, патенты и патентные заявки, цитированные в описании, включены в текст заявки в качестве ссылки в полном объеме.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ АНТАГОНИСТОВ CRTH2 В ТЕРАПИИ | 2004 |
|
RU2412934C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2528046C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2382779C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2556984C2 |
| СПОСОБ МОДУЛЯЦИИ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2525115C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА | 2002 |
|
RU2320658C2 |
| Бензотиофены и родственные соединения в качестве агонистов STING | 2019 |
|
RU2806274C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| ПРОИЗВОДНЫЕ СПИРООКСИНДОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ТЕРАПЕВТИЧЕСКИХ АГЕНТОВ | 2006 |
|
RU2415143C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛА КАК ЛЕКАРСТВЕННЫЕ ВЕЩЕСТВА | 2005 |
|
RU2470916C2 |
1. Изобретение относится к новым соединениям формулы I
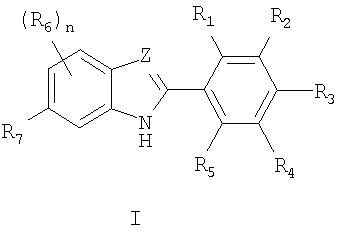
где: n означает 0 или 1; Z означает N, CR8, где R8 означает Н, С1-С6алкил; R1, R2, R4 и R5 независимо означают Н, галоген, ОН, -XNR9R10, где X означает химическую связь, a R9 и R10 означают Н; R3 означает -NR11S(O)2R12, -NR11C(O)R12, -C(O)OR11, -NR11R12, S(O)2NR11R12 и -C(O)NR11R12, где R11 и R12 независимо означают H, С1-С6алкил; R6 означает галоген, С1-С6алкил; R7 означает фенил, замещенный 1 или 2 группами, независимо выбранными из фтора и С1-С6алкила, и его фармацевтически приемлемым солям. Соединения проявляют активность миметика ТРО, что позволяет получать из них лекарственные средства для повышения уровня тромбоцитов в крови животного. 4 н. и 4 з.п. ф-лы.
1. Соединение формулы I
в котором n выбирают из 0 и 1,
Z выбирают из N и CR8, где R8 выбирают из группы, включающей водород, С1-С6алкил,
R1, R2, R4 и R5 независимо выбирают из группы, включающей водород, галоген, гидроксил и -XNR9R10, где X обозначает химическую связь, a R9 и R10 обозначают водород,
R3 выбирают из группы, включающей -NR11S(O)2R12, -NR11C(O)R12, -C(O)OR11, -NR11R12, -S(O)2NR11R12 и -C(O)NR11R12, где R11 и R12 независимо выбирают из группы, включающей водород и С1-С6алкил,
R6 выбирают из группы, включающей галоген и C1-С6алкил, а
R7 обозначает фенил, замещенный 1 или 2 группами, независимо выбранными из фтора и С1-С6алкила, и его фармацевтически приемлемые соли.
2. Соединение по п.1 формулы Ia
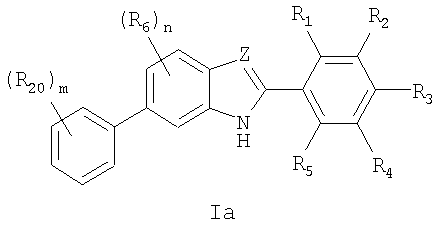
в котором n выбирают из 0 и 1, m выбирают из 1 и 2,
Z выбирают из N и CR8, где R8 выбирают из водорода и С1-С6алкила,
R1, R2, R4 и R5 независимо выбирают из группы, включающей водород, галоген, гидроксил и -XNR9R10, где X обозначает химическую связь, a R9 и R10 обозначают водород,
R3 выбирают из группы, включающей -NR11S(O)2R12, -NR11C(O)R12, -C(O)OR11, -NR11R12, -S(O)2NR11R12 и -C(O)NR11R12, где R11 и R12 независимо выбирают из группы, включающей водород и С1-С6алкил,
R6 выбирают из группы, включающей фтор и С1-С6алкил, а
R20 выбирают из группы, включающей фтор и С1-С6алкил.
3. Соединение по п.2, в котором R20 выбирают из группы, включающей фтор, метил и бутил.
4. Соединение по п.3, в котором R3 выбирают из группы, включающей карбоксил, аминокарбонил, аминосульфонил, метилсульфониламино и амино, a R4 выбирают из группы, включающей водород, гидроксил, хлор и амино.
5. Соединение по п.4, выбранное из группы, включающей 4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]бензойную кислоту, 4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]бензамид, 4-[7-фтор-6-(4-фтор-3-метилфенил)-1Н-индол-2-ил]бензойную кислоту, 4-[7-фтор-6-(4-фтор-3-метилфенил)-1Н-индол-2-ил]бензолсульфонамид, 4-[6-(4-бутилфенил)-3-этил-7-фтор-1Н-индол-2-ил]-2-гидроксибензойную кислоту, 4-[6-(4-бутилфенил)-7-фтор-3-изопропил-1Н-индол-2-ил]-2-гидроксибензойную кислоту, 4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]-2-гидроксибензойную кислоту, 4-[6-(4-бутилфенил)-7-фтор-3-метил-1Н-индол-2-ил]-2-гидроксибензойную кислоту, 4-[6-(4-бутилфенил)-1Н-бензимидазол-2-ил]бензойную кислоту, 4-[6-(4-бутилфенил)-1Н-бензимидазол-2-ил]-2-гидроксибензойную кислоту, N-{4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]фенил}метансульфонамид, N-{4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]фенил}ацетамид, N-{4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]-2-хлорфенил}ацетамид, 4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]-2-хлорфениламин и 2-амино-4-[6-(4-бутилфенил)-7-фтор-1Н-индол-2-ил]бензойную кислоту.
6. Фармацевтическая композиция, проявляющая активность миметика ТРО, включающая терапевтически эффективное количество соединения по п.1 в комбинации с фармацевтически приемлемым эксципиентом.
7. Способ повышения уровня тромбоцитов в крови животного, заключающийся в том, что животному вводят терапевтически эффективное количество соединения по п.1.
8. Применение соединения формулы I по п.1 для получения лекарственного средства, предназначенного для повышения уровня тромбоцитов в крови животного.
| US 6787550 В1, 07.09.2004 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| RU 2004137491 А1, 10.06.2005. | |||

