Область техники, к которой относится изобретение
Настоящее изобретение относится к ингибиторам протеиназ (протеаз) и, в частности, к применению производных ароматической сульфонгидроксамовой кислоты, которые, помимо прочего, являются избирательными ингибиторами матриксных металлопротеиназ, предназначенными для лечения состояний, связанных с патологической активностью матриксных металлопротеиназ, к самим избирательным ингибиторам, к композициям ингибиторов протеиназ, к промежуточным продуктам, предназначенным для синтеза ингибиторов протеиназ, и к способу получения ингибиторов протеиназ.
Предпосылки создания изобретения
Соединительная ткань, составляющие внеклеточного матрикса и базальные мембраны, являются необходимыми компонентами организма всех млекопитающих. Эти компоненты представляют собой биологический материал, который обеспечивает жесткость, дифференциацию, присоединение, а в некоторых случаях и эластичность биологических систем, включая организм человека и других млекопитающих. Компоненты соединительной ткани включают, например, коллаген, эластин, протеогликаны, фибронектин и ламинин. Они представляют собой биохимические составляющие или являются компонентами таких структур, как кожа, кость, зуб, сухожилие, хрящ, базальная мембрана, кровеносные сосуды, роговица и стекловидное тело.
В нормальных состояниях процессы превращения и/или репарации соединительной ткани контролируются и находятся в состоянии равновесия. Нарушение по какой-либо причине этого баланса приводит к многочисленным болезненным состояниям. Ингибирование ферментов, ответственных за нарушение равновесия, обеспечивает механизм контроля указанного разложения ткани и, следовательно, является средством лечения этих заболеваний.
Разложение соединительной ткани или компонентов соединительной ткани происходит под воздействием ферментов протеиназ, которые выделяются из расположенных в ткани клеток и/или из внедрившихся воспалительных или опухолевых клеток. Основным классом ферментов, осуществляющих эту функцию, являются зависимые от цинка металлопротеиназы (металлопротеазы).
Ферменты металлопротеазы подразделяют на классы, некоторые представители которых имеют несколько различных принятых названий. Их примерами являются коллагеназа I (матриксная металлопротеаза-1 (ММП-1), коллагеназа фибробластов, КФ 3.4.24.3), коллагеназа II (ММП-8, коллагеназа нейтрофилов, КФ 3.4.24.34), коллагеназа III (ММП-13), стромелизин 1 (ММП-3, КФ 3.4.24.17), стромелизин 2 (ММП-10, КФ 3.4.24.22), протеогликаназа, матрилизин (ММП-7), желатиназа А (ММП-2, желатиназа с молекулярной массой 72 кДа, коллагеназа базальной мембраны, КФ 3.4.24.24), желатиназа В (ММП-9, желатиназа с молекулярной массой 92 кДа, КФ 3.4.24.35), стромелизин 3 (МММ-11), металлоэластаза (ММП-12, ЧМЭ, эластаза макрофага человека) и мембранная ММП (ММП-14). ММП является аббревиатурой или акронимом от понятия "матриксная металлопротеаза", а присоединенные к этому акрониму цифры позволяют отличать друг от друга различных конкретных представитей групп ММП.
Неконтролируемое разложение соединительной ткани металлопротеазами является причиной многих патологических состояний. Их примерами являются ревматоидный артрит, остеоартрит, септический артрит, изъязвление роговицы, эпидермиса или желудка, периодонтит, протеинурия, болезнь Альцгеймера, коронарный тромбоз и заболевание кости. Также могут иметь место нарушения процессов репарации повреждения. Это может вызывать нарушение процесса заживления ран, приводящее к слабой репарации, сращению и рубцеванию. Последние из указанных нарушений могут привести к физическому недостатку и/или к временной потере трудоспособности, например к появлению спаек после хирургического вмешательства.
Металлопротеазы также участвуют в биосинтезе фактора некроза опухоли (TNF) и ингибируют продуцирование или активность TNF и родственных ему соединений, что является важным клиническим методом лечения болезней. Например, TNF-α представляет собой цитокин, который, как предполагается в настоящее время, первоначально продуцируется в виде связанной с клеткой молекулы с молекулярной массой 28 кДа. Затем происходит выделение активной формы с молекулярной массой 17 кДа, которая может обусловливать многочисленные вредные воздействия in vitro и in vivo. Например, TNF может вызывать и/или оказывать воздействия на воспалительные процессы, ревматоидный артрит, аутоиммунное заболевание, рассеянный склероз, отторжение трансплантата, фиброзные заболевания, рак, инфекционные болезни, малярию, инфекции, вызываемые микобактериями, менингит, лихорадку, псориаз, сердечно-сосудистые/легочные явления, такие как постишемическое перфузионное повреждение, застойная сердечная недостаточность, кровотечение, коагуляция, альвеолярное повреждение вследствие гипероксии, радиационное повреждение и острые фазы реакций типа тех, которые обнаруживаются при инфекциях и сепсисе и во время шока, например септического шока и гемодинамического шока. Хроническое выделение активного TNF может вызвать кахексию и анорексию. TNF может привести к смерти и одноврменно с этим TNF может помогать контролировать рост опухолевых клеток.
TNF-α -конвертаза представляет собой металлопротеазу, участвующую в образовании растворимого TNF-α . Ингибирование TNF-α -конвертазы (ТАСЕ) ингибирует продуцирование активного TNF-α . Соединения, которые ингибируют как активность ММП, так и производство TNF-α , описаны в WO 94/24140, WO 94/02466 и WO 97/20824. Установлено, что соединения, которые ингибируют такие ММП, как коллагеназа, стромелизин и желатиназа, также обладают способностью ингибировать выделение TNF (Gearing и др., Nature 376. 555-557 (1994), McGeehan и др.. Nature 376. 558-561 (1994)). Существует необходимость в эффективных ингибиторах ММП. Кроме того, сохраняется необходимость в агентах, являющихся эффективными ингибиторами TNF-α -конвертазы.
ММП также принимают участие в других биохимических процессах у млекопитающих. Они участвуют в регуляции овуляции, послеродовой инволюции матки, возможно, в имплантации, в расщеплении АПП (β -амилоидный предшественник протеина) с образованием амилоидной бляшки и в инактивации ингибитора α 1-протеазы (α 1-ПИ). Ингибирование этих металлопротеаз позволяет регулировать фертильность и лечить или предупреждать болезнь Альцгеймера. Кроме того, увеличение и сохранение уровней эндогенного или введенного лекарства, являющегося ингибитором серинпротеазы, или биохимического агента, такого как α 1-ПИ, способствует лечению или предупреждению таких болезней, как эмфизема, заболевания легких, воспалительные болезни и болезни, связанные со старением, такие как потеря упругости и эластичности кожи или органа.
Ингибирование определенных ММП также может оказаться целесообразным в других случаях. Лечение рака и/или ингибирование метастазов и/или ингибирование ангиогенеза являются примерами таких подходов к лечению болезней, когда избирательное ингибирование такого фермента или ферментов, как стромелизин, желатиназа А или В или коллагеназа III, вероятно, существенно более важно, чем ингибирование коллагеназы I (ММП-1). Лекарственное средство, которое не ингибирует коллагеназу I, может обладать наиболее ценным терапевтическим профилем. Еще одной важной болезнью, при которой разложение хряща в воспаленных суставах, вероятно, по меньшей мере частично обусловлено ММП-13, выделяющейся из таких клеток, как стимулированные хондроциты, является остеопороз, наибольший эффект при лечении которого может быть достигнут за счет применения лекарственных средств, одним из механизмов действия которых является ингибирование ММП-13 (см., например, Mitchell и др., J. Clin. Invest., 97: 761-768 (1996) и Reboul и др., J. Clin. Invest., 97: 2011-2019 (1996)).
Известен ряд ингибиторов металлопротеаз. Их примеры включают природные биохимические агенты, такие как тканевые ингибиторы металлопротеиназ (ТИМП), α 2-макроглобулин и их аналоги или производные. Эти эндогенные ингибиторы представляют собой молекулы протеинов с большой молекулярной массой, которые образуют неактивные комплексы с металлопротеазами. Описан целый ряд пептидоподобных соединений меньшего размера, которые ингибируют металлопротеазы. Установлено, что меркаптоамидпептидильные производные ингибируют АСЕ in vitro и in vivo. Ангиотензинпревращающий фермент (АСЕ) участвует в продуцировании ангиотензина II, соединения, являющегося эффективным сосудосуживающим агентом у млекопитающих, и ингибирование этого фермента приводит к снижению кровяного давления.
Ингибиторы металлопротеаз (ММП), представляющие собой амид- или пептидиламидсодержащие соединения, несущие тиольную группу, известны например, из WO 95/12389, WO 96/11209 и патента US 4595700. Ингибиторы ММП, несущие гидроксаматную группу, известны из многочисленных публикаций, таких как WO 95/29892, WO 97/24117, WO 97/49679 и ЕР 0780386, в которых описаны соединения, имеющие углеродный скелет, и в WO 90/05719, WO 93/20047, WO 95/09841 и WO 96/06074, в которых описаны гидроксаматы, имеющие пептидильные скелеты или пептидоподобные скелеты, и такие соединения также описаны в статьях Schwartz и др., Progr. Med. Chem., 29: 271-334 (1992), а также Rasmussen и др., Pharmacol. Ther., 75(1): 69-75 (1997) и Denis и др., Invest. New Drugs, 15(3): 175-185 (1997).
Одной из возможных проблем, связанных с известными ингибиторами ММП, является то, что такие соединения часто проявляют одинаковое или близкое ингибирующее действие в отношении всех ферментов ММП. Так, например, для соединения из класса пептидоподобных гидроксаматов, известного под названием батимастат, установлено, что значения IС50 составляют от примерно 1 до примерно 20 нмоль (нМ) в отношении каждой из таких ММП, как ММП-1, ММП-2, ММП-3, ММП-7 и ММП-9. Для маримастата, другого пептидоподобного гидроксамата, который является еще одним ингибитором широкого спектра ММП, установлено, что спектр его ингибирующей активности очень близок с таковым батимастата за исключением того, что значение IC50 маримастата в отношении ММП-3 составляет 230 нМ (Rasmussen и др., Pharmacol. Ther., 75(1): 69-75 (1997)).
Предварительный анализ данных, полученных на фазе I/II при исследовании пациентов, у которых в результате применения маримастата достигнуто успешное быстро прогрессирующее лечение плотных злокачественных опухолей (прямой кишки, поджелудочной железы, яичника, предстательной железы), показал зависящее от дозы уменьшение уровня карциномных антигенов, которые использовались в качестве косвенных маркеров биологической активности. Хотя маримастат и проявил определенный уровень активности в отношении этих маркеров, тем не менее при этом обнаружены побочные токсичные действия. Наиболее часто встречающимся связанным с лекарством токсичным действием маримастата, выявленным при клинических испытаниях, оказалась боль скелетных мышц и ригидность, часто затрагивающая небольшие суставы кистей рук, распространяясь далее на руку и плечо. Короткий перерыв в 1-3 недели в лечении с последующим уменьшением доз позволяет продолжить лечение (Rasmussen и др., Pharmacol. Ther., 75(1): 69-75 (1997)). Возможно, причиной этого является отсутствие специфического ингибирующего действия в отношении ММП.
В WO 98/38163, опубликованной 3 сентября 1998 г., описана большая группа ингибиторов ММП и ТАСЕ из класса гидроксаматов. Соединения, описанные в WO 98/38163, содержат один или два заместителя, соседних с функционально активным гидроксаматом, и заместитель, который может представлять собой ароматическую сульфонильную группу в соседнем положении по отношению к одному или к обоим этим заместителям.
В WO 98/37877, опубликованной 3 сентября 1998 г., описаны соединения, которые содержат 5-7-членное гетероциклическое кольцо в соседнем положении относительно функционально активного гидроксамата и могут содержать ароматическую сульфонильную группу по соседству с гетероциклическим кольцом.
Хотя многие из известных ингибиторов ММП, такие как батимастат, маримастат и гидроксаматы, описанные в WO 98/37877 и WO 98/38163, и обладают широким спектром активности в отношении ММП, однако эти соединения не обладают выраженным избирательным действием с точки зрения их ингибирующей активности. Это отсутствие избирательности может быть причиной боли скелетных мышц и ригидности, которые обнаружены при их применении. Кроме того, может оказаться терапевтически целесообразным применять лекарственное средство, которое является избирательным с точки зрения активности по сравнению с обычно применяемыми активными соединениями, чтобы лечение могло быть более точно направлено на патологическое состояние, имеющееся у хозяина-млекопитающего. Ниже описан способ лечения хозяина-млекопитающего, имеющего состояние, связанное с патологической активностью матриксных металлопротеаз, с использованием соединения, которое избирательно ингибирует одну или несколько ММП, но при этом является менее активным в отношении по крайней мере ММП-1.
Краткое изложение сущности изобретения
Настоящее изобретение относится к способу лечения, включающему введение хозяину-млекопитающему, который имеет состояние, связанное с патологической активностью матриксных металлопротеаз, эффективного количества рассматриваемой ароматической сульфонгидроксамовой кислоты, которая является ингибитором металлопротеаз. Рассматриваемая молекула проявляет, помимо прочего, очень высокую ингибирующую активность в отношении одного или большего количества ферментов металлопротеаз (ММП), таких как ММП-2, ММП-9 и ММП-13, но обладает существенно меньшей ингибирующей активностью по крайней мере в отношении ММП-1. Под понятием "существенно меньшая" подразумевают, что для рассматриваемого соединения в приведенном ниже в данном описании опыте по оценке ингибирования in vitro соотношение значений IС50 в отношении одной или нескольких ММП, таких как ММП-2, ММП-9 или ММП-13, и его значения IС50 в отношении ММП-1, например IС50 ММП-2, IС50 ММП-1, составляет менее примерно 1:10, предпочтительно менее примерно 1:100 и наиболее предпочтительно менее примерно 1:1000. Изобретение также относится к определенным соединениям, которые избирательно ингибируют активность одной или нескольких таких ММП, как ММП-2, ММП-9 и ММП-13, но обладают существенно меньшей ингибирующей активностью в отношении по крайней мере ММП-1, а также к композиции, содержащей такой ингибитор ММП в качестве действующего вещества. Изобретение также относится к промежуточным продуктам, предназначенным для получения рассматриваемой молекулы ароматической сульфонгидроксамовой кислоты, и к способу получения молекулы ароматической сульфонгидроксамовой кислоты.
В целом, одним из объектов настоящего изобретения является способ лечения, включающий введение эффективного количества рассматриваемой ароматической сульфонгидроксамовой кислоты, являющейся ингибитором металлопротеаз, которая избирательно ингибирует активность матриксных металлопротеаз, хозяину-млекопитающему, имеющему состояние, связанное с патологической активностью металлопротеаз. Применяемый ингибитор фермента имеет строение, представленное приведенной ниже формулой (I), или является его фармацевтически приемлемой солью:

где R1 и R2 оба обозначают гидрид или R1 и R2 вместе с атомами, к которым они присоединены, образуют 5-8-членное кольцо, содержащее в кольце один, два или три гетероатома, представляющих собой кислород, серу или азот.
R3 в формуле I обозначает необязательно замещенный арильный или необязательно замещенный гетероарильный радикал. Когда R3 обозначает замещенный арильный или гетероарильный радикал, то указанный заместитель выбирают из группы, включающей арил, гетероарил, аралкил, гетероаралкил, арилокси-, арилтио-, аралкокси-, гетероаралкоксигруппу, аралкоксиалкил, арилоксиалкил, аралканоилалкил, арилкарбонилалкил, аралкиларил, арилоксиалкиларил. аралкоксиарил, арилазоарил, арилгидразиноарил, алкилтиоарил, арилтиоалкил, алкилтиоаралкил, аралкилтиоалкил, аралкилтиоарил, сульфоксид или сульфон любого из тиозаместителей, и сконденсированную кольцевую структуру, включающую два или большее количество 5-6-членных колец, выбранных из группы, включающей арильный, гетероарильный, карбоциклический и гетероциклический радикалы.
Заместитель, связанный с арильным или гетероарильным радикалом, который включает радикал R3, сам может быть замещен одним или большим количеством заместителей; т.е. замещающий заместитель необязательно является замещенным. Когда арильный или гетероарильный радикал является замещенным и замещающий фрагмент (группа, заместитель или радикал) сам является замещенным, последний заместитель независимо друг от друга выбирают из группы, включающей цианогруппу, перфторалкил, трифторметокси-, трифторметилтиогруппу, галоалкил, трифторметилалкил, аралкоксикарбонил, арилоксикарбонил, гидроксигруппу, галоген, алкил, алкокси-, нитрогруппу, тиол, гидроксикарбонил, арилокси-, арилтиогруппу, аралкил, арил, арилкарбониламино-, гетероарилокси-, гетероарилтиогруппу, гетероаралкил, циклоалкил, гетероциклоокси-, гетероциклотио-, гетероциклоамино-, циклоалкилокси-, циклоалкилтио-, гетероаралкокси-, гетероаралкилтио-, аралкокси-, аралкилтио-, аралкиламино-, гетероциклогруппу, гетероарил, арилазо-, гидроксикарбонилалкокси-, алкоксикарбонилалкоксигруппу, алканоил, арилкарбонил, аралканоил, алканоилокси-, аралканоилоксигруппу, гидроксиалкил, гидроксиалкокси-, алкилтио-, алкоксиалкилтиогруппу, алкоксикарбонил, арилоксиалкоксиарил, арилтиоалкилтиоарил, арилоксиалкилтиоарил, арилтиоалкоксиарил, гидроксикарбонилалкокси-, гидроксикарбонилалкилтио-, алкоксикарбонилалкокси-, алкоксикарбонилалкилтио-, аминогруппу, где азот аминогруппы является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, гетероарил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил, арилкарбонил, аралканоил, гетероарилкарбонил, гетероаралканоил и алканоил, или (III) где азот аминогруппы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо, содержащее 0-2 дополнительных гетероатома, представляющих собой азот, кислород или серу, и само этот кольцо является (а) незамещенным или (б) замещенным одной или двумя группами, независимо друг от друга выбранными из группы, включающей арил, алкил, гетероарил, аралкил, гетероаралкил, гидрокси-, алкоксигруппу, алканоил, циклоалкил, гетероциклоалкил, алкоксикарбонил, гидроксиалкил, трифторметил, сконденсированный с бензольным кольцом гетероциклоалкил, гидроксиалкоксиалкил, аралкоксикарбонил, гидроксикарбонил, арилоксикарбонил, сконденсированную с бензольным кольцом гетероциклоалкоксигруппу, сконденсированный с бензольным кольцом циклоалкилкарбонил, гетероциклоалкилкарбонил и циклоалкилкарбонил, карбониламиногруппу,
где азот карбониламиногруппы является (I) незамещенным или (II) представляет собой реакционноспособный амин аминокислоты, или (III) является замещенным одним или двумя радикалами, выбранными из группы, включающей алкил, гидроксиалкил, гидроксигетероаралкил, циклоалкил, аралкил, трифторметилалкил, гетероциклоалкил, сконденсированный с бензольным кольцом гетероциклоалкил, сконденсированный с бензольным кольцом циклоалкил и N,N-диалкилзамещенную алкиламиноалкильную группу, или (IV) азот карбоксамидогруппы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло-, гетероарильное или сконденсированное с бензольным кольцом гетероциклоалкильное кольцо, которое само может быть незамещенным или может быть замещено одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей алкил, алкоксикарбонил, нитрогруппу, гетероциклоалкил, гидроксигруппу, гидроксикарбонил, арил, аралкил, гетероаралкил
и аминогруппу, где азот аминогруппы является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил и гетероарил. или (III) где азот аминогруппы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо,
и аминоалкильную группу где азот аминоалкила является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил и алканоил, или (III) где азот аминоалкила и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо.
В предпочтительном варианте осуществления R и R вместе с атомами, к которым они присоединены, образуют 6-членное кольцо.
Радикал R3 предпочтительно имеет длину, превышающую длину пентильной группы[-(СН2)4СН3-цепь] и более предпочтительно превышающую длину примерно гексильной группы[-(СН2)5СН3-цепь]. Радикал R3 предпочтительно имеет длину, меньшую чем длина икозильной группы[-(СН2)19CH3-цепь], и более предпочтительно длину, меньшую чем длина стеарильной группы[-(СН2)17СН3-цепь]. Предпочтительно группа R3 содержит 2 или большее количество 5-6-членных колец. Рассматриваемая группа R3 при вращении вокруг оси, проходящей через связанное с SO2 1-положение и связанное с заместителем 4-положение 6-членного кольца или через связанное с SO2 1-положение и связанное с заместителем 3-или 4-положение 5-членного кольца, образует трехмерное пространство, наибольший размер которого в направлении, перпендикулярном указанной оси вращения, равен длине от примерно одного фуранильного кольца до примерно двух фенильных колец.
Предпочтительно также, чтобы радикал R3 представлял собой арильную или гетероарильную группу с одним кольцом, которая является 5- или 6-членной и сама замещена в ее 4 положении, когда она представляет собой 6-членное кольцо, или в ее 3 или 4 положении, когда она представляет собой 5-членное кольцо, необязательно замещенным заместителем, выбранным из группы, включающей другую арильную или гетероарильную группу с одним кольцом, С3-С14алкильную группу, N-пиперидильную группу, N-пиперазильную группу, феноксигруппу, тиофеноксигруппу, 4-тиопиридильную группу, фенилазогруппу и бензамидогруппу. Заместитель 5- или 6-членной арильной или гетероарильной группы сам может быть замещен, как указано выше.
Предпочтительное соединение, предназначенное для применения в рассматриваемом способе, имеет строение, соответствующее приведенной ниже формуле II, или является его фармацевтически приемлемой солью:
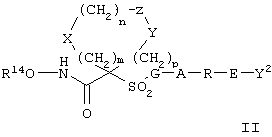
где
R14 обозначает гидрид, фармацевтически приемлемый катион или C(W)R5, где W обозначает О или S, а R15 выбирают из группы, включающей С1-С6алкил, арил, С1-С6алкоксигруппу, гетероарилС1-С6алкил, С3-С8 циклоалкил-С1-С6алкил, арилокси-, арС1-С6алкоксигруппу, арС1-С6алкил, гетероарил и аминоС1-С6алкил, где азот аминоалкила является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арил, арС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, арС1-С6алкоксикарбонил, С1-С6алкоксикарбонил и С1-С6алканоил, или (III) азот аминоС1-С6алкильной группы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо,
m равно 0, 1 или 2,
n равно 0, 1 или 2
р равно 0, 1 или 2,
сумма m+n+p равна 1, 2, 3 или 4,
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя из группы, включающей X, Y и Z, обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей X, Y и Z, обозначает CR8R9, или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей
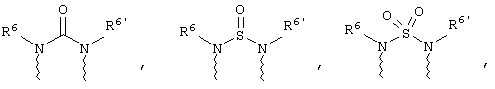
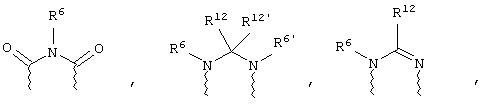
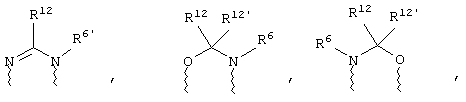

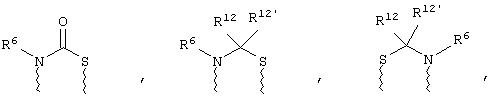

где
волнистые линии обозначают связи с атомами изображенного кольца, R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6лерфторалкил, С1-С6трифторметилалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетеропиклоалкил, С3-С8гетероциклоалкилкарбонил, С6арил, С5-С6гетероцикло, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкоксиС1-С6алкил, гетероарилтиоС1-С6алкил, С6арилсульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4 алкоксикарбонил-С1-С6алкил, аминокарбонил, С1-С6алкилиминокарбонил, С1-С6арилиминокарбонил, С1-С6гетероциклоиминокарбонил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С4алкокси-С1-С4алкил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5алкилкарбонил, гидроксиС1-С5алкил, аминокарбонил, где азот в аминокарбониле является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, гидроксиаминокарбонил, аминосульфонил, где азот в аминосульфонильной группе является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, аминоС1-С6алкилсульфонил, где азот аминоС1-С6алкилсульфонильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиал,
R8 и R9, и R10, и R11 независимо друг от друга выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6 алкил, гетероарилтиоС1-С6алкил, сульфоксид или сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил, или где R8 и R9 или R10 и R11 вместе с атомами, к которым они присоединены, образуют карбонильную группу, или R8 и R9 или R10 и R11, или R8 и R10 вместе с атомами, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, которые представляют собой азот, кислород или серу, при условии, что только один из радикалов R8 и R9 или R10 и R11 обозначает гидроксигруппу,
R12 и R12' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероаралкил, С1-С6алкинил, С1-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонил С1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид или сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С1-С6алкинил, С2-С6алкенил и С1-С6гидроксиалкил, и
G-A-R-E-Y представляет собой заместитель, который предпочтительно имеет длину, превышающую длину пентильной группы, и более предпрочтительно имеет длину, превышающую длину гексильной группы. Заместитель G-A-R-E-Y предпочтительно имеет длину, меньшую чем длина икозильной группы, и более предпочтительно меньшую чем длина стеарильной группы.
В этом заместителе G обозначает арильную или гетероарильную группу;
А выбирают из группы, включающей
(1) -О-,
(2) -S-,
(3) -NR17-,
(4) -CO-NR17-или-NR17-CO-, где R17 обозначает водород, С1-С4алкил или фенил,
(5) -СО-O- или -O-СО-,
(6) -O-СО-О-,
(7) -НС=СН-,
(8) -NH-CO-NH-,
(9) -С=С-,
(10) -NH-CO-O-или-O-CO-NH-,
(11) -N=N-.
(12) -NH-NH-и
(13) -CS-N(R18)-или-N(R18)-CS-, где R18 обозначает водород, С1-С4алкил или фенил, или
(14) А отсутствует, a G непосредственно присоединен к R,
R обозначает фрагмент, выбранный из группы, включающей алкил, алкоксиалкил, арил, гетероарил, циклоалкил, гетероциклоалкил, аралкил, гетероаралкил, гетероциклоалкилалкил, циклоалкилалкил, циклоалкоксиалкил, гетероциклоалкоксиалкил, арилоксиалкил, гетероарилоксиалкил, арилтиоарил, гетероарилтиоарил, циклоалкилтиоалкил и гетероциклоалкилтиоалкил, где арильный или гетероарильный, или циклоалкильный, или гетероциклоалкильный заместитель является (I) незамещенным или (II) замещенным одним или двумя радикалами, выбранными из группы, включающей галоген, алкил, перфторалкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, аминогруппу, алкоксикарбонилалкил, алкокси-, С1-С2алкилендиоксигруппу, гидроксикарбонилалкил, гидроксикарбонилалкиламино-, нитро-, гидроксигруппу, гидроксиалкил, алканоиламиногруппу и алкоксикарбонил, и R не обозначает алкил или алкоксиалкил, когда А обозначает -О- или -S-,
Е выбирают из группы, включающей
(1) -CO(R19)- или -(R19)СО-, где R19 обозначает гетероциклоалкил или циклоалкил,
(2) -CONH-или-HNCO-,
(3) -СО-,
(4) -SO2-R19- или -R19-SO2-,
(5) -SO2- и
(6) -NH-SO2- или -SO2-NH, или
(7) Е отсутствует и R непосредственно присоединен к Y, и Y отсутствует или его выбирают из группы, включающей гидрид, алкил, алкоксигруппу, галоалкил, арил, аралкил, циклоалкил, гетероарил, гидрокси-, арилокси-, аралкокси-, гетероарилоксигруппу, гетероаралкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, алкенил, гетероциклоалкил, циклоалкил, трифторметил, алкоксикарбонил и аминоалкил, где арильная или гетероарильная, или гетероциклоалкильная группа является (I) незамещенной или (II) замещенной одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей алканоил, галоген, нитрогруппу, аралкил, арил, алкокси- и аминогруппу, где азот в аминогруппе является (I) незамещенным или (II) замещенным одним или двумя группами, независимо друг от друга выбранными из группы, включающей гидрид, алкил и аралкил. Особенно предпочтительное соединение, предназначенное для применения в предлагаемом способе, имеет строение, соответствующее приведенной ниже формуле III, или представляет собой его фармацевтически приемлемую соль:
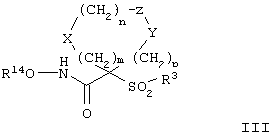
где m, n, р, X, Z, Y и R14 имеют значения, указанные выше для формулы II, а значения радикала R, которые приведены ниже, представляют собой подгруппу значений, приведенных ранее для заместителей G-A-R-E-Y.
Таким образом, R3 обозначает радикал, включающий арильную или гетероарильную группу с одним кольцом, которая является 5- или 6-членной и сама замещена в 4-положении, когда она представляет собой 6-членное кольцо, и в 3- или в 4-положении, когда она представляет собой 5-членное кольцо, заместителем, выбранным из группы, включающей тиофенокси-, 4-хлорфенокси-, 3-хлорфенокси-, 4-метоксифенокси-, 3-бензодиоксол-5-илокси-, 3,4-диметилфенокси-, 4-фторфенокси-, 4-фтортиофенокси-, фенокси-, 4-трифторметоксифенокси-, 4-трифторметилфенокси-, 4-(трифторметилтио)фенокси-, 4-(трифторметилтио) тиофенокси-, 4-хлор-3-фторфенокси-, 4-изопропоксифенокси-, 4-изопропилфенокси-, (2-метил-1,3-бензотиазол-5-ил)окси-, 4-(1Н-имидазол-1-ил)фенокси-, 4-хлор-3-метилфенокси-, 3-метилфенокси-, 4-этоксифенокси-, 3,4-дифторфенокси-, 4-хлор-3-метилфенокси-, 4-фтор-3-хлорфенокси-, 4-(1Н-1,2,4-триазол-1-ил)фенокси-, 3,5-дифторфенокси-, 3,4-дихлорфенокси-, 4-циклопентилфенокси-, 4-бром-3-метилфенокси-, 4-бромфенокси-, 4-метилтиофенокси-, 4-фенилфенокси-, 4-бензилфенокси-, 6-хинолинилокси-, 4-амино-3-метилфенокси-, 3-метоксифенокси-, 5,6,7,8-тетрагидро-2-нафталинилокси-, 3-гидроксиметилфеноксигруппу, N-пиперидил, N-пиперазинил и 4-бензилоксифеноксигруппу.
Более предпочтительное соединение, предназначенное для применения в предлагаемом способе, имеет строение, соответствующее приведенной ниже формуле IV, или представляет собой его фармацевтически приемлемую соль:
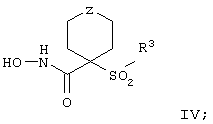
где R3 имеет значения, указанные выше для формулы I, более предпочтительно указанные для формулы II (где эта группа R3 представляет собой заместитель G-A-R-E-Y), и более предпочтительно также значения, указанные для формулы III, и Z выбирают из группы, включающей О, S, NR6, SO, SO2 и NSO2R7, где R6 выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алканоил, бензил, бензоил, С3-С5алкинил, С3-С5алкенил, С1-С3алкокси-С1-С4алкил, С3-С6циклоалкил, гетероарилС1-С6алкил, С1-С5гидроксиалкил, С1-С5карбоксиалкил, С1-С5алкокси-С1-С5алкилкарбонил и NR8R9-С1-С6алкилкарбонил или NR8R9-С1-С6алкил, где R8 и R9 независимо друг от друга обозначают гидрид, С1-С5алкил, С1-С5алкоксикарбонил или арилС1-С5алкоксикарбонил, или NR8R9 вместе образуют гетероциклическое кольцо, содержащее 5-8 атомов в кольце, а R выбирают из группы, включающй арилалкил, арил, гетероарил, гетероцикло-, С1-С6алкил, С3-С6алкинил, С1-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил.
Еще более предпочтительная группа соединений, предназначенных для применения согласно предлагаемому способу, имеет строение, соответствующее приведенной ниже формуле V, или представляет собой фармацевтически приемлемую соль этой группы соединений:
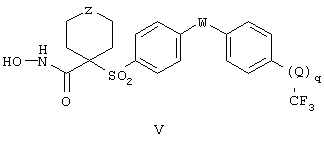
где Z имеет значения, указанные ранее для формулы IV, W и Q независимо друг от друга обозначают кислород (О), NR6 или серу (S), где R6 имеет значения, указанные для формулы IV, и q равно 0 или 1, причем когда q равно 0, то трифторметильная группа присоединена непосредственно к указаному фенильному концу.
Применение соединения формул I-V или фармацевтически приемлемой соли одного из этих соединений рассмотрено в описанном ранее способе. Кроме того, соединения формул II, III, IV и V и их фармацевтически приемлемые соли являются соединениями по настоящему изобретению.
Настоящее изобретение также относится к предшественнику или промежуточному соединению, пригодному для получения соединения формул I-V. Такое промежуточное соединение имеет строение, соответствующее приведенной ниже формуле VI:
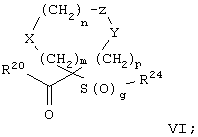
где m, n, p, X, Z и Y имеют значения, указанные выше для формулы II, g равно 0, 1 или 2 и R24 обозначает R3, как этот радикал определен для формул I, III или IV, заместитель G-A-R-E-Y формулы II (формула VIA) или обозначает R3', в котором арильная или гетероарильная группа замещена связывающим заместителем, реакционноспособным в отношении связывания с другим фрагментом (формула VIB), таким как замещаемая нуклеофилом уходящая группа D.
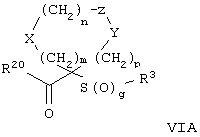
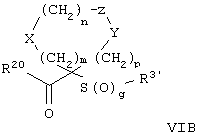
Примерами замещаемой нуклеофилом уходящей группы D являются галоген (фтор, хлор, бром или йод), нитро-, азидо-, фенилсульфоксидо-, арилокси-, С1-С6алкоксигруппа, С1-С6алкилсульфонатная или арилсульфонатная группа и тризамещенная аммонийная группа, в которой три заместителя независимо друг от друга выбирают из группы, включающей арил, арС1-С6алкил или С1-С6алкил.
R20 обозначает (a) -O-R, где R21 выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил и фармацевтически приемлемый катион, или (б) -NH-O-R22, где R22 обозначает избирательно удаляемую защитную группу, такую, как 2-тетрагидропиранил, С1-С6ацил, ароил, бензил, пара-метоксибензилоксикарбонил (MOZ), бензилоксикарбонил, С1-С6алкоксикарбонил, С1-С6алкокси-СН2-, С1-С6алкокси-С1-С6алкокси-СН2-, тризамещенная силильная группа или орто-нитрофенильная группа, смола для синтеза пептидов и т.п. Тризамещенная силильная группа может быть замещена С1-С6алкилом, арилом или apС1-С6алкилом.
Особенно предпочтительным предшественником или промежуточным соединением формулы VI является промежуточное соединение формулы VII
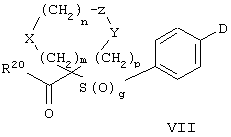
где m, n, p, g, X, Z, Y, D и R20 имеют значения, указанные выше для формулы VI.
Настоящее изобретение позволяет достичь целого ряда преимуществ благодаря предложенным в нем соединениям и композициям эффективных в качестве ингибиторов активности матриксных металлопротеиназ, т.е. благодаря таким соединениям и композициям, которые обладают эффективностью в отношении ингибирования металлопротеиназ, обусловливающих болезни и нарушения, связанные с неконтролируемым разрушением соединительной ткани.
Настоящее изобретение позволяет достичь целого ряда преимуществ, в частности благодаря предложенным в нем соединению и композиции, эффективным в отношении избирательного ингибирования определенных металлопротеиназ, таких как одна или несколько ММП, выбранных из группы, включающей ММП-2, ММП-9 или ММП-13, с которым связаны такие патологические состояния, как, например, ревматоидный артрит, остеоартрит, септический артрит, изъязвление роговицы, эпидермиса или желудка, периодонтит, протеинурия, болезнь Альцгеймера, коронарный тромбоз и заболевание кости.
Еще одно преимущество достигается согласно изобретению благодаря предлагаемым в нем соединениям, композициям и способам, эффективным в отношении лечения таких патологических состояний путем избирательного ингибирования металлопротеиназ, таких как ММП-2, ММП-9 или ММП-13, с которыми связаны указанные состояния, с минимальными побочными воздействиями, возникающими в результате ингибирования других металлопротеиназ, таких как ММП-1, активность которых необходима или желательна для нормальной функции организма.
Еще одно преимущество достигается согласно изобретению благодаря предлагаемому в нем способу получения таких соединений.
Следующее преимущество достигается согласно изобретению благодаря предлагаемому в нем способу лечения патологического состояния, связанного с аномальной активностью матриксных металлопротеиназ.
Еще одно преимущество достигается согласно изобретению благодаря предлагаемому в нем способу получения указанных композиций.
Другие преимущества и задачи изобретения очевидны для специалиста в данной области техники из приведенного ниже описания.
Подробное описание изобретения
Согласно настоящему изобретению было установлено, что определенные ароматические сульфонгидроксамовые кислоты (гидроксаматы) обладают эффективностью в отношении ингибирования матриксных металлопротеиназ ("ММП"), с которыми, вероятно, связано неконтролируемое или какое-либо иное патологическое разрушение соединительной ткани. В частности, было установлено, что такие определенные ароматические сульфонгидроксаматы обладают эффективностью в отношении ингибирования одного или нескольких ферментов, таких как ММП-2, ММП-9 и ММП-13, которые могут вызывать особенно значительные разрушения ткани, если присутствуют или образуются в аномальных количествах или концентрациях и, таким образом, проявляют патологическую активность. Это понятие патологической активности включает способность содействовать процессу проникновения опухолей или опухолевых клеток через базальную мембрану и развитию нового или усиленного кровоснабжения, т.е. ангиогенезу.
Кроме того, было установлено, что эти ароматические сульфонгидроксаматы обладают избирательностью в отношении ингибирования одной или нескольких из ММП-2, ММП-9 и ММП-13, не обладая при этом выраженным ингибирующим действием в отношении других коллагеназ, важных для нормальной функции организма, такой как процессы превращения и репарации ткани. В частности, было установлено, что указанный ароматический сульфонгидроксамат по изобретению или его фармацевтически приемлемая соль особенно активны в отношении ингибирования одной или нескольких из М МП-2, ММП-9 и ММП-13 в опытах in vitro, что позволяет предположить наличие активности in vivo. Кроме того, обладая избирательным действием в отношении одной или нескольких из ММП-2, ММП-9 и ММП-13, указанный ароматический сульфонгидроксамат или его соль обладают ограниченным или минимальным ингибирующим действием в отношении ММП-1 in vivo.
Таким образом, существует выраженное отличие в активности соединения, которое применяется в рассматриваемом способе, в отношении одной или нескольких из ММП-2, ММП-9 и ММП-13 и в отношении ММП-1. Это выраженное различие выявляют с помощью анализа ингибирования in vitro, который описан в примерах. Выраженное различие в активности заключается в том, что значение IC50 соединения в отношении одной или нескольких из ММП-2, ММП-9 и ММП-13 составляет примерно 0,1 от значения IC50 соединения в отношении ММП-1 и более предпочтительно 0,01 от значения в отношении ММП-1 и наиболее предпочтительно 0,001 или более от значения в отношении ММП-1. Так, для некоторых соединений коэффициент избирательного действия при оценке по величине значений IC50 превысил значения, полученные в аналогичном опыте, в 100000 раз. Эта избирательность действия проиллюстрирована ниже в таблицах, в которых приведены данные по ингибированию.
Рассматриваемое соединение также может по-разному ингибировать активность ММП-2 по сравнению с ММП-9 или ММП-13 и ММП-1. Аналогично этому рассматриваемое соединение может ингибировать активность ММП-13 и ММП-2 и при этом обладать меньшей способностью ингибировать активность ММП-1 и ММП-9. Кроме того, рассматриваемое соединение может ингибировать активность фермента ММП, но при этом проявлять меньшее воздействие на выделение фактора некроза опухоли.
Не вдаваясь в теоретические подробности, преимущества избирательного действия рассматриваемого соединения можно оценить на основе терапевтической ценности соединений. Например, предполагается, что ингибирование ММП-1 является нежелательным вследствие ее роли в качестве конститутивных ферментов (ферментов "домашнего хозяйства"), способствующих поддержанию нормальных процессов превращения и репарации соединительной ткани. Ингибирование ММП-1 может привести к токсичности или побочным действиям, таким как разрушение сустава или соединительной ткани и боль. С другой стороны, предполагается, что ММП-13 непосредственно участвует в деструкции компонентов сустава при таких заболеваниях, как остеопороз. Так, весьма желательным является эффективное и избирательное ингибирование ММП-13 по сравнению с ингибированием ММП-1, поскольку ингибитор ММП-13 в дополнение к его противовоспалительному действию может оказывать положительное действие в отношении прогрессивного развития болезни у пациента.
Ингибирование ММП-2 и ММП-9 может оказаться желательным для ингибирования роста опухоли, метастазов, инвазии и/или ангиогенеза. Профиль избирательного ингибирования ММП-2 и ММП-9 по сравнению с ММП-1 может обеспечить терапевтическое преимущество.
Еще одним преимуществом рассматриваемого соединения является избирательность действия в отношении выделения фактора некроза опухоли и/или выделения рецептора фактора некроза опухоли, что является для лечащего врача еще одной возможностью выбора наиболее эффективного лекарственного средства для конкретного пациента. Не основываясь на какой-либо теории, можно предположить, что существует несколько аспектов этого типа избирательного действия, которые следует учитывать.
Во-первых, присутствие фактора некроза опухоли может быть желательным для контроля развития злокачественной опухоли в организме, если TNF не присутствует в избыточном количестве, вызывающем токсичное действие. Так, неконтролируемое ингибирование выделения TNF может иметь отрицательные последствия и фактически может рассматриваться как вредное побочное действие даже у страдающих раком пациентов. Кроме того, также может оказаться желательной избирательность в отношении ингибирования выделения рецептора фактора некроза опухоли. Присутствие этого рецептора может оказаться желательным для поддержания контролируемого уровня некроза опухоли у млекопитающего путем связывания избытка TNF.
Рассматриваемое соединение, являющееся избирательным ингибитором ММП, согласно рассматриваемому процессу может быть введено различными путями и обеспечивать требуемые терапевтические уровни в крови активного в отношении фермента ингибитора. Соединения может вводиться, например, оральным (в желудок (IG), перорально (РО)) или внутривенным (IV) путем. Оральный путь введения является предпочтительным, если пациент находится в амбулаторных условиях, не госпитализирован, физически способен и достаточно ответственен для того, чтобы самостоятельно принимать лекарство через определенные промежутки времени. Это относится даже к случаю, когда курс лечения предусматривает прием более одного лекарства при лечени более чем одной болезни. С другой стороны, внутривенное введение лекарства является предпочтительным в больничных условиях, при этом доза и связанный с ней уровень в крови хорошо поддаются контролю. Рассматриваемый ингибитор также может быть при необходимости введен в композицию для внутримышечного (IM) введения. Этот способ применения может оказаться желательным для введения пролекарств или для регулярного введения лекарства пациенту, который либо является физически слабым, либо плохо соблюдает режим и схему лечения, либо когда требуются постоянные уровни лекарства в крови.
Таким образом, в одном из вариантов осуществления настоящего изобретения предлагаемый способ лечения предусматривает введение эффективного количества рассматриваемой ароматической сульфонгидроксамовой кислоты, являющейся ингибитором металлопротеаз, или ее фармацевтически приемлемой соли хозяину-млекопитающему, который имеет состояние, связанное с патологической активностью матриксных металлопротеаз.
Рассматриваемый ароматический сульфонгидроксамат, являющийся соединением-ингибитором, которое применяется согласно этому варианту осуществления способа, ингибирует активность одной или нескольких из ММП-2, ММП-9 и ММП-13 и обладает существенно меньшей ингибирующей активностью в отношении по крайней мере ММП-1 в указанном ранее опыте in vitro, который описан подробно ниже. Ароматический сульфонгидроксамат, являющийся соединением-ингибитором, которое применяется согласно рассматриваемому варианту способа, имеет строение, соответствующее приведенной ниже формуле I:
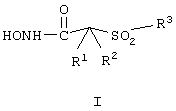
в которой заместители имеют следующие значения.
В одном из вариантов осуществления R1 и R2 оба обозначают гидрид. В другом варианте осуществления R1 и R2 вместе с атомами, к которым они присоединены, образуют 5-8-членное кольцо, содержащее в кольце один, два или три гетероатома, представляющих собой кислород, серу или азот.
Предпочтительно, чтобы R1 и R2 вместе с атомами, к которым они присоединены, образовывали 5-8-членное кольцо, содержащее в кольце один или два гетероатома, хотя R1 и R2 вместе с атомами, к которым они присоединены, могут образовывать 5-8-членное кольцо, содержащее в кольце один, два или три гетероатома. Гетероциклическое кольцо само может иметь в качестве заместителей до шести С1-С6алкильных групп или групп, включающих другое 5-8-членное карбоциклическое или гетероциклическое кольцо, аминогруппу, или может содержать одну или две оксо-(карбонил) группы. R3 в формуле I обозначает необязательно замещенный арильный или необязательно замещенный гетероарильный радикал. Этот радикал R3 выбирают из группы, включающей арил, гетероарил, аралкил, гетероаралкил, аралкокси-, гетероаралкоксигруппу, аралкоксиалкил, арилоксиалкил, аралканоилалкил, арилкарбонилалкил, аралкиларил, арилоксиалкиларил, аралкоксиарил,
арилазоарил, арилгидразиноарил, алкилтиоарил, арилтиоалкил, алкилтиоаралкил, аралкилтиоалкил, аралкилтиоарил, сульфоксид или сульфон любого из тиозаместителей и сконденсированную кольцевую структуру, включающую два или большее количество 5-6-членных колец, выбранных из группы, включающей арил, гетероарил, карбоциклический и гетероциклический радикал.
Заместитель, входящий в радикал R3, сам может быть незамещенным или может быть замещен одним или несколькими заместителями, независимо друг от друга выбранными из группы, включающей цианогруппу, перфторалкил, трифторметилалкил, гидроксигруппу, галоген, алкил, алкокси-, нитрогруппу, тиол, гидроксикарбонил, арилокси-, арилтиогруппу, аралкил, арил, гетероарилокси-, гетероарилтиогруппу, гетероаралкил, циклоалкил, гетероциклоокси-, гетероциклотио-, гетероциклоамино-, циклоалкилокси-, циклоалкилтио-, гетероаралкокси-, гетероаралкилтио-, аралкокси-, аралкилтио-, аралкиламино-, гетероциклогруппу, гетероарил, арилазо-, гидроксикарбонилалкокси-, алкоксикарбонилалкоксигруппу, алканоил, арилкарбонил, аралканоил, алканоилокси-, аралканоилоксигруппу, гидроксиалкил, гидроксиалкокси-, алкилтио-, алкоксиалкилтиогруппу, алкоксикарбонил, арилоксиалкоксиарил, арилтиоалкилтиоарил, арилоксиалкилтиоарил, арилтиоалкоксиарил, гидроксикарбонилалкокси-, гидроксикарбонилалкилтио-, алкоксикарбонилалкокси-, алкоксикарбонилалкилтио-, аминогруппу, где азот аминогруппы является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, гетероарил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил, арилкарбонил, аралканоил, гетероарилкарбонил, гетероаралканоил и алканоил, или (III) где азот аминогруппы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо, содержащее 0-2 дополнительных гетероатома, представляющих собой азот, кислород или серу, и само этот кольцо является (а) незамещенным или (б) замещенным одной или двумя группами, независимо друг от друга выбранными из группы, включающей арил, алкил, гетероарил, аралкил, гетероаралкил, гидрокси-, алкоксигруппу, алканоил, циклоалкил, гетероциклоалкил, алкоксикарбонил, гидроксиалкил, трифторметил, сконденсированный с бензольным кольцом гетероциклоалкил, гидроксиалкоксиалкил, аралкоксикарбонил, гидроксикарбонил, арилоксикарбонил, сконденсированную с бензольным кольцом гетероциклоалкоксигруппу, сконденсированный с бензольным кольцом циклоалкилкарбонил, гетероциклоалкилкарбонил и циклоалкилкарбонильную группу,
карбониламиногруппу, где азот карбоксамидогруппы является (I) незамещенным или (II) представляет собой реакционноспособный амин аминокислоты, или (III) является замещенным одним или двумя радикалами, выбранными из группы, включающей алкил, гидроксиалкил, гидроксигетероаралкил, циклоалкил, аралкил, трифторметилалкил, гетероциклоалкил, сконденсированный с бензольным кольцом гетероциклоалкил, сконденсированный с бензольным кольцом циклоалкил и N,N-диалкилзамещенную алкиламиноалкильную группу, или (IV) азот карбоксамидогруппы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло-, гетероарильное или сконденсированное с бензольным кольцом циклоалкильное кольцо, которое само может быть незамещенным или может быть замещено одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей алкил, алкоксикарбонил, нитрогруппу, гетероциклоалкил, гидроксигруппу, гидроксикарбонил, арил, аралкил, гетероаралкил
и аминогруппу,
где азот аминогруппы является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил и гетероарил, или (III) где азот аминогруппы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо,
и аминоалкильную группу где азот аминоалкила является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил и алканоильную группу, или (III) где азот аминоалкила и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо. Соединение формулы I также может применяться в форме фармацевтически приемлемой соли.
Радикал R3 имеет длину, превышающую длину пентильной группы [а-(СН2)4СН3-цепь] и более предпочтительно превышающую длину примерно гексильной группы [-(СН2)5СН3-цепь]. Радикал R3 имеет длину, меньшую чем длина икозильной группы [эйкозил-(СН2)19СН3-цепь], и более предпочтительно длину, меньшую чем длина стеарильной группы [а-(СН2)17СН3-цепь]. Рассматриваемая группа R при вращении вокруг оси, проходящей через связанное с SO2 1-положение и связанное с заместителем 4-положение 6-членного кольца, или через связанное с SO2 1-положение и связанное с заместителем 3-или 4-положение 5-членного кольца, образует трехмерное пространство, наибольший размер которого в направлении, перпендикулярном указанной оси вращения, равен длине от примерно одного фуранильного кольца до примерно двух фенильных колец.
Если, например, связанный с SО2-группой радикал R обозначает 4-феноксифенил, то данное соединение может рассматриваться как феноксифенилсульфоновое производное, т.е. требуемый 5-8-членный N-гидроксикарбоксамид. Примерами таких соединений, которые следует упомянуть являются следующие:
N-гидрокси-1-метил[4-(феноксифенилсульфонил)]-4-пиперидинкарбоксамид,
N-гидрокси[4-(феноксифенилсульфонил)]тетрагидро-2Н-пиран-4-карбоксамид,
N-гидрокси-1-метил[2,6-диоксо-4-(феноксифенилсульфонил)]-4-пиперидинкарбоксамид,
N-гидрокси-2,2-диметил[5-(феноксифенилсульфонил)]-1,3-диоксан-5-карбоксамид,
N-гидрокси-1,2-диметил-6-оксо[4-(феноксифенилсульфонил)]-4-пиперидинкарбоксамид,
N-гидрокси-2,2,6,6-тетраметил[4-(феноксифенилсульфонил)]-4-пиперидинкарбоксамид,
N-гидрокси-1,3-диметил[5-(феноксифенилсульфонил)]гексагидро-5-пиримидинкарбоксамид,
2-амино-N-гидрокси[5-(феноксифенилсульфонил)]-1,4,5,6-тетрагидро-5-пиримидинкарбоксамид,
N-гидрокси-1,1-диоксо[4-(феноксифенилсульфонил)]-1(λ 6),2,6-тиадизинан-4-карбоксамид,
N-гидрокси-2-оксо[5-(феноксифенилсульфонил)]гексагидро-5-пиримидинкарбоксамид,
N-гидрокси[2-(феноксифенилсульфонил)]тетрагидро-2-фуранкарбоксамид,
N-гидрокси-1-метил[2-(феноксифенилсульфонил)]-2-пирролидинкарбоксамид,
N-гидрокси-2-метил[4-(феноксифенилсульфонил)]-4-пиперидинкарбоксамид,
N-гидрокси[3-(феноксифенилсульфонил)]-8-азабицикло[3.2.1]октан-3-карбоксамид,
N-гидрокси-1,1-диоксо[4-(феноксифенилсульфонил)]гексагидро-1 (λ 6)-тиопиран-4-карбоксамид,
N-гидрокси[3-(феноксифенилсульфонил)]тетрагидро-3-фуранкарбоксамид,
N-гидрокси[3-(феноксифенилсульфонил)]-3-пирролидинкарбоксамид,
моногидрохлорид N-гидpoкcи-4-[[4-(фенилтио)фенил]сульфонил)]-1-(2-пропинил)-4-пиперидинкарбоксамида,
монометансульфонат N-гидpoкcи-4-[[4-(фенилтито)фенил]сульфонил)]-1-(2-пропинил)-4-пиперидинкарбоксамида,
тетрагидро-N-гидрокси-4-[[4-[4-[(трифторметил]фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамид,
гидрохлорид N-гидрокси-1-(4-пиридинилметил)-4-[[4-[4-(трифторметил]фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида,
дигидрохлорид N-гидрокси-1-(3-пиридинилметил)-4-[[4-[4-(трифторметил]фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида,
дигидрохлорид N-гидрокси-1-(2-пиридинилметил)-4-[[4-[4-(трифторметил]фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида,
дигидрохлорид гидрокси-1-(3-пиридинилметил)-4-[[4-[4-(трифторметокси)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида,
моногидрохлорид N-гидрокси-1-(2-метоксиэтил)-4-[[4-[4-(трифторметокси)фенокси]фенил]сульфонил)]-4-пиперидинкарбоксамида,
моногидрохлорид N-гидрокси-1-(2-метоксиэтил)-4-[[4-[4-(трифторметил)фенокси]фенил]сульфонил)]-4-пиперидинкарбоксамида,
моногидрохлорид N-гидрокси-1-(2-метоксиэтил)-4-[[4-[4-[(трифторметил)тио]фенокси]фенил]сульфонил)]-4-пиперидинкарбоксамида,
моногидрохлорид 1-циклопропил-N-гидрокси-4-[[4-[4-(трифторметил)фенокси]фенил]сульфонил)]-4-пиперидинкарбоксамида и т.п.
Несколько примеров групп R1 и R2, которые вместе образуют указанное гетероциклическое кольцо, представлено в приведенных ниже таблицах, а также в описаниях этих 5-8-членных колец и в конкретных примерах, в качестве нескольких указанных производных ароматической сульфонгидроксамовой кислоты.
В более предпочтительном варианте в формуле I R1 и R2 вместе с атомами, к которым они присоединены, образуют 5-8-членное кольцо, содержащее в кольце один, два или три гетероатома. Более предпочтительно это кольцо представляет собой 6-членное кольцо, которое содержит один гетероатом, расположенный в положении 4 относительно положения, в котором присоединена группа SO2. Другие предпочтительные соединения, предназначенные для применения согласно указанному способу, имеют строение, соответствующее одной или нескольким приведенным ниже формулам II, III, IV или V.
В одном из вариантов осуществления соединение, предназначенное для применения согласно указанному способу, имеет строение, соответствующее приведенной ниже формуле II:
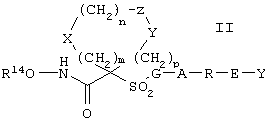
где
R14 обозначает гидрид, фармацевтически приемлемый катион или C(W)R15, где
W обозначает О или S, а
R15 выбирают из группы, включающей С1-С6алкил, арил, С1-С6алкоксигруппу, гетероарилС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, арилокси-, арС1-С6алкоксигруппу, арС1-С6алкил, гетероарил и аминоС1-С6алкил, где азот аминоалкила является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арил, арС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, арС1-С6алкоксикарбонил, С1-С6алкоксикарбонил и С1-С6алканоил, или (III) азот аминоС1-С6алкильной группы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо,
m равно 0, 1 или 2,
n равно 0, 1 или 2,
р равно 0, 1 или 2,
сумма m+n+p равна 1, 2, 3 или 4,
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя X, Y и Z обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей X, Y и Z, обозначает CR8R9, или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей
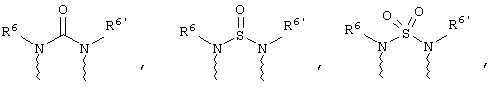
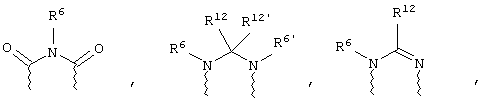
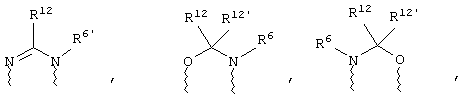
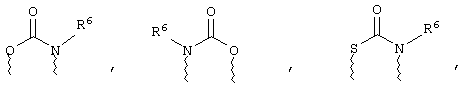
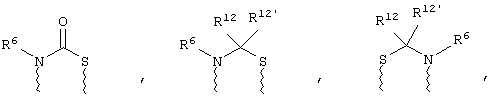
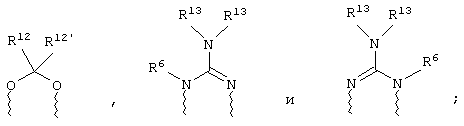
где волнистые линии обозначают связи с атомами изображенного кольца, R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6перфторалкил, С1-С6трифторметилалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкил-карбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкоксиС1-С6алкил, гетероарилтиоС1-С6алкил, C6арилсульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С6алкоксикарбонил-С1-С6алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С5-С6гетероциклоиминокарбо-нил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С4алкокси-С1-С4алкил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5алкилкарбонил, гидроксиС1-С5алкил, аминокарбонил, где азот в аминокарбониле является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, гидроксиаминокарбонил, аминосульфонил, где азот в аминосульфонильной группе является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, аминоС1-С6алкилсульфонил, где азот аминоС1-С6алкилсульфонильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещен одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещен одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей бензил, фенил, С1-С6алкил, С3-С6алкинил, С3-С6алкенил и С1-С6гидроксиалкил,
R8 и R9, и R10, и R11 независимо друг от друга выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С1-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид или сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил, или где R8 и R9 или R10 и R11 вместе с атомами, к которым они присоединены, образуют карбонильную группу, или R8 и R9 или R10 и R11, или R8 и R10 вместе с атомами, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, которые представляют собой азот, кислород или серу, при условии, что только один из радикалов R8 и R9 или R10 и R11 обозначает гидроксигруппу,
R12 и R12’ независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероаралкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, арилоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид или сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С2-С6алкинил, С2-С6алкенил и С1-С6гидроксиал, и
G-A-R-E-Y представляет собой заместитель, который предпочтительно имеет длину, превышающую длину пентильной группы, и более предпочтительно имеет длину, превышающую длину гексильной группы. Заместитель G-A-R-E-Y предпочтительно имеет длину, меньшую, чем длина икозильной группы и более предпочтительно, меньшую, чем длина стеарильной группы.
В этом заместителе:
G обозначает арильную или гетероарильную группу,
А выбирают из группы, включающей
(1) -О-,
(2) -S-,
(3) -NR17-,
(4) -CO-NR17- или -NR17-CO-, где R17 обозначает атом водорода, С1-С4алкил или фенил,
(5) -СО-O- или -O-СО-,
(6) -O-СО-О-,
(7) -НС=СН-,
(8) -NH-CO-NH-,
(9) -С=С-,
(10) -NH-CO-O-или-O-CO-NH-,
(11) -N=N-,
(12) -NH-NH и
(13) -CS-N(R18)- или -N(R18)-CS-, где R18 обозначает атом водорода, С1-С4алкил или фенил, или
(14) А отсутствует и G непосредственно присоединен к R, R обозначает фрагмент, выбранный из группы, включающей алкил, алкоксиалкил, арил, гетероарил, циклоалкил, гетероциклоалкил, аралкил, гетероаралкил, гетероциклоалкилалкил, циклоалкилалкил, циклоалкоксиалкил, гетероциклоалкоксиалкил, арилоксиалкил, гетероарилоксиалкил, арилтиоалкил, гетероарилтиоалкил, циклоалкилтиоалкил и гетероциклоалкилтиоалкил, где арильный или гетероарильный или циклоалкильный или гетероциклоалкильный заместитель является (I) незамещенным или (II) замещенным одним или двумя радикалами, выбранными из группы, включающей галоген, алкил, перфторалкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, аминогруппу, алкоксикарбонилалкил, алкокси-, С1-С2алкилендиоксигруппу, гидроксикарбонилалкил, гидроксикарбонилалкиламино-, нитро-, гидроксигруппу, гидроксиалкил, алканоиламиногруппу и алкоксикарбонил, и R не обозначает алкил или алкоксиалкил, когда А обозначает -О- или -S-, Е выбирают из группы, включающей
(1) -CO(R19)- или -(R19)CO-, где R19 обозначает гетероциклоалкильную или циклоалкильную группу,
(2) -CONH- или -HNCO-,
(3) -СО-,
(4) -SO2-R19- или -R19-SO2-,
(5) -SO2-,
(6) -NH-SO2- или -SO2-NH, или
(7) Е отсутствует и R непосредственно присоединен к Y, и Y отсутствует или его выбирают из группы, включающей гидрид, алкил, алкоксигруппу, галоалкил, арил, аралкил, циклоалкил, гетероарил, гидрокси-, арилокси-, аралкокси-, гетероарилоксигруппу, гетероаралкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, алкенил, гетероциклоалкил, циклоалкил, трифторметил, алкоксикарбонил и аминоалкил, где арильная или гетероарильная, или гетероциклоалкильная группа является (I) незамещенной или (II) замещеной одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей алканоил, галоген, нитрогруппу, аралкил, арил, алкокси- и аминогруппу, где азот в аминогруппе является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей гидрид, алкил и аралкил.
Заместитель -G-A-R-E-Y предпочтительно содержит 2-4 карбоциклических или гетероциклических кольца, включая арильную или гетероарильную группу G. Более предпочтительно каждое из этих колец является 6-членным. В дополнительных конкретных предпочтительных вариантах заместители в соединении формулы II имеют следующие значения: (а) А обозначает -О- или -S-, (б) R обозначает арил, гетероарил, циклоалкил или гетероциклоалкил, (в) Е отсутствует и (г) Y выбирают из группы, включающей гидрид, алкил, алкокси-, перфторалкокси- и перфторалкилтиогруппу.
Более предпочтительное соединение, предназначенное для применения согласно указанному способу, имеет строение, соответствующее приведенной ниже формуле III:
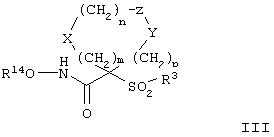
где R3 обозначает радикал, включающий 5-или 6-членную арильную группу или гетероарильную группу с одним кольцом, которая сама замещена в 4-положении, когда она представляет собой 6-членное кольцо, и в 3- или в 4-положении, когда она представляет собой 5-членное кольцо, заместителем, выбранным из группы, включающей тиофенокси-, 4-хлорфенокси-, 3-хлорфенокси-, 4-метоксифенокси-, 3-бензодиоксол-5-илокси-, 3,4-диметилфенокси-, 4-фторфенокси-, 4-фтортиофенокси-, фенокси-, 4-трифторметоксифенокси-, 4-трифторметилфенокси-, 4-(трифторметилтио)фенокси-, 4-(трифторметилтио) тиофенокси-, 4-хлор-3-фторфенокси-, 4-изопропоксифенокси-, 4-изопропилфенокси-, (2-метил-1,3-бензотиазол-5-ил)окси-, 4-(1H-имидазол-1-ил) фенокси-, 4-хлор-3-метилфенокси-, 3-метилфенокси-, 4-этоксифенокси-, 3,4-дифторфенокси-, 4-хлор-3-метилфенокси-, 4-фтор-3-хлорфенокси-, 4-(1Н-1,2,4-триазол-1-ил)фенокси-, 3,5-дифторфенокси-, 3,4-дихлорфенокси-, 4-циклопентилфенокси-, 4-бром-3-метилфенокси-, 4-бромфенокси-, 4-метилтиофенокси-, 4-фенилфенокси-, 4-бензилфенокси-, 6-хинолинилокси-, 4-амино-3-метилфенокси-, 3-метоксифенокси-, 5,6,7,8-тетрагидро-2-нафталинилокси-, 3-гидроксиметилфенокси- и 4-бензилоксифеноксигруппу,
R14 обозначает гидрид, фармацевтически приемлемый катион или C(W)R15, где W обозначает О или S, а R выбирают из группы, включающей С1-С6алкил, арил, С1-С6алкоксигруппу, гетероарилС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, арилокси-, apС1-С6алкоксигруппу, арС1-С6алкил, гетероарил и аминоС1-С6алкил, где азот аминоалкила является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арил, apС1-С6алкил, С1-С6циклоалкил-С1-С6алкил, арС1-С6алкоксикарбонил, С1-С6алкоксикарбонил и С1-С6алканоил, или (III) азот аминоС1-С6алкильной группы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо, m равно 0, 1 или 2, n равно 0, 1 или 2, р равно 0, 1 или 2, сумма m+n+p равна 1, 2, 3 или 4,
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя X, Y и Z обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O), NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей X, Y и Z, обозначает CR8R9 или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей

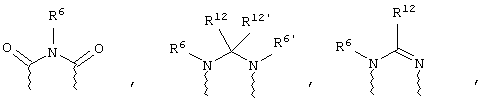
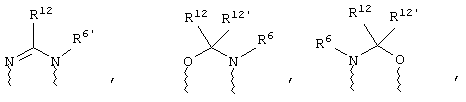
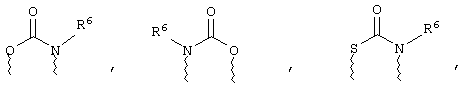
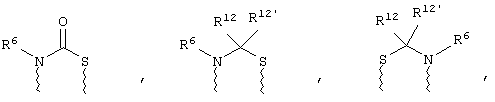
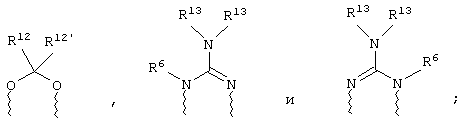
где волнистые линии обозначают связи с атомами изображенного кольца, R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6перфторалкил, С1-С6трифторметилалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкил-карбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкоксиС1-С6алкил, гетероарилтиоС1-С6алкил, С6арил сульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4алкоксикарбонил-С1-С6алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С5-С6гетероциклоиминокарбонил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С4алкокси-С1-С4алкил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5алкилкарбонил, гидроксиС1-С5алкил, аминокарбонил, где азот в аминокарбониле является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, гидроксиаминокарбонил, аминосульфонил, где азот в аминосульфонильной группе является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, аминоС1-С6алкилсульфонил, где азот аминоС1-С6алкилсульфонильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей бензил, фенил, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил, R8 и R9, и R10, и R11 независимо друг от друга выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбо-нилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтио С1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид или сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или где R8 и R9 или R10 и R11 вместе с атомами, к которым они присоединены, образуют карбонильную группу, или R8 и R9 или R10 и R11, или R8 и R10 вместе с атомами, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, которые представляют собой азот, кислород или серу, при условии, что только один из радикалов R8 и R9 или R10 и R11 обозначает гидроксигруппу,
R12 и R12' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероаралкил, С1-С6алкинил, С1-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, арилоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид или сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С1-С6алкинил, С2-С6алкенил и С1-С6гидроксиалкил. И в этом случае подразумевается, что соединение формулы III можно применять в виде фармацевтически приемлемой соли.
Предпочтительными вариантами соединения формулы III являются такие же варианты, как и в случае соединения формулы II, независимо друг от друга включающие следующие условия: (а) сумма m+n+p равна 1 или 2 и более предпочтительно равна 2, (б) Z обозначает О, S или NR6, (в) R6 выбирают из группы, включающей С3-С6циклоалкил, С1-С6алкил, С3-С6алкенил, С3-С6алкинил, С1-С6алкокси-С1-С6алкил, аминоС1-С6алкил, аминосульфонил, гетероарилС1-С6алкил, арилоксикарбонил и С1-С6алкоксикарбонил, и (г) m=n=0, р=1 и Y обозначает NR6. Другим предпочтительным вариантом как соединения формулы II, так и соединения формулы III являются варианты, когда R14 обозначает гидрид или когда W группы C(W)R15 пролекарства обозначает О, где R15 обозначает С1-С6алкил, арил, С1-С6алкоксигруппу, гетероарилС1-С6алкил, С3-С6циклоалкил-С1-С6алкил или арилоксигруппу.
Еще более предпочтительное соединение, предназначенное для применения согласно указанному способу, имеет строение, соответствующее приведенной ниже формуле IV:
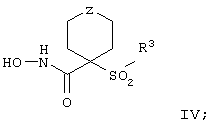
где
R3 имеет значения, указанные выше для формул I, III, более предпочтительно имеет значения, указанные для формулы II (где радикал R3 представляет собой заместитель G-A-R-E-Y), наиболее предпочтительно R3 имеет значения, указанные для формулы III,
Z выбирают из группы, включающей О, S, NR6, SO, SO2 и NSO2R7,
где R6 выбирают из группы, включающей гидрид, С1-С6алкил, С1-С6алканоил, бензил, бензоил, С3-С5алкинил, С3-С5алкенил, С1-С3алкокси-С1-С4алкил, С3-С6циклоалкил, гетероарилС1-С6алкил, С1-С5гидроксиалкил, С1-С5карбоксиалкил, С1-С5алкокси-С1-С5алкилкарбонил и NR8R9-С1-С5алкилкарбонил или NR8R9-С1-С5алкил, где R8и R9независимо друг от друга обозначают гидрид, С1-С5алкил, С1-С5алкоксикарбонил или арилС1-С5алкоксикарбонил, или NR8R9 вместе образуют гетероциклическое кольцо, содержащее 5-8 атомов в кольце, а
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил. Наиболее предпочтительно Z обозначает О или NR6. И в этом случае подразумевается, что соединение формулы III можно применять в виде фармацевтически приемлемой соли.
Более предпочтительная группа рассматриваемых соединений, предназначенных для применения согласно указанному способу, имеет строение, соответствующее приведенной ниже формуле V:
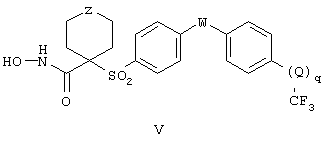
где Z имеет значения указанные ранее для формулы IV, W и Q независимо друг от друга обозначают кислород (О), NR или серу (S), где R6 имеет значения, указанные ранее для формулы IV, и q равно 0 или 1, причем когда q равно 0, то Q отсутствует, а трифторметильная группа непосредственно присоединена к указанному фенильному кольцу. И в этом случае подразумевается, что соединение формулы IV можно применять в виде фармацевтически приемлемой соли.
Особенно предпочтительные соединения в группе, представленной формулой V, имеют строение, соответствующее приведенной ниже формуле:
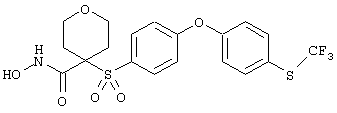
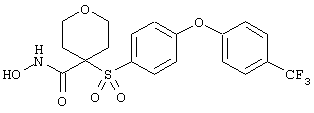
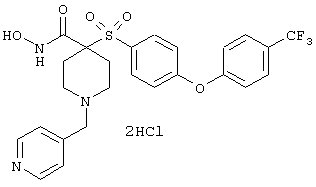
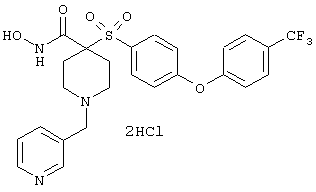
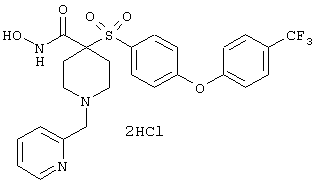
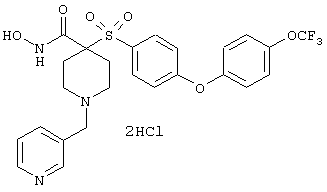
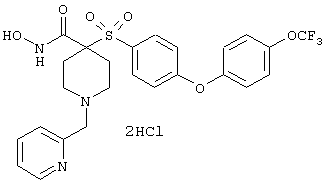
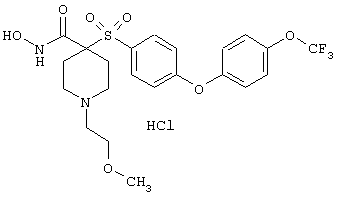
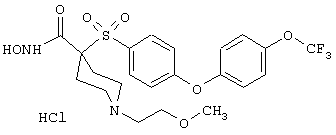
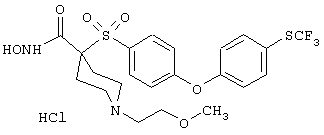
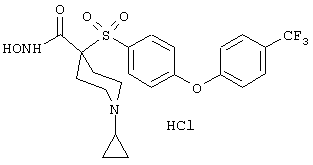
Также особенно предпочтительными являются следующие соединения:
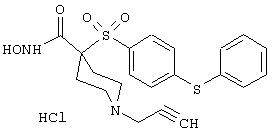
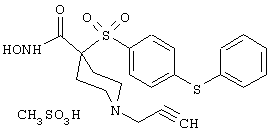
Несколько особенно предпочтительных соединений, имеющих строение, соответствующее формулам I-V, приведены ниже в таблицах и в примерах.
Как уже было отмечено выше, соединения формул II, III, IV и V и их фармацевтически приемлемые соли являются соединениями, подпадающими под объем изобретения.
В предпочтительном варианте осуществления соединенный с SO2-группой радикал R3 представляет собой арильную или гетероарильную группу, имеющую одно 5-6-членное кольцо, которое замещено одной другой арильной группой или гетероарильной группой с одним кольцом или алкильной или алкоксигруппой с длиной цепи от 3 до примерно 16 атомов углерода (более предпочтительно с длиной примерно до 14 атомов углерода), феноксигруппой, тиофеноксигруппой [С6Н5-S-], фенилазогруппой [С6Н5-N2-], N-пиперидилом [C5H10N-], N-пиперазилом [NC4H9N-] или бензамидогруппой [NHC(O)C6H5]. В этом случае соединенная с SО2-группой арильная или гетероарильная группа R3 с одним кольцом замещена в ее 4-положении, если она представляет собой 6-членное кольцо, и замещена в ее 3-или 4-положении, если она представляет собой 5-членное кольцо.
Соединенная с SО2-группой арильная или гетероарильная группа радикала R3 предпочтительно замещена в ее 4-положении, если она представляет собой 6-членное кольцо, и замещена в ее 3-или 4-положении, если она представляет собой 5-членное кольцо. Особенно предпочтительным заместителем является арильная или гетероарильная группа с одним кольцом, фенокси-, тиофенокси-, фенилазагруппа, N-пиперидил, N-пиперазил или бензамидогруппа, которая может быть незамещенной или сама может быть замещена.
Указанные 4- и 3-положения в кольцах пронумерованы начиная с мест присоединения заместителя в соответствии с официально действующей нумерацией положений, принятой в номенклатуре гетероарильных производных, как приведено ниже в настоящем описании. Так, предпочтительными являются отдельные атомы, такие как атомы галогена (фтор, хлор, бром или йод), или заместители, которые содержат в цепи от одного до примерно пяти атомов, отличных от водорода, такие как фенил, С1-С4алкил, трифторметил, трифторметоксигруппа, трифтортиометил или карбоксиэтил, хотя могут применяться и заместители, имеющие большую длину, такие, у которых общая длина не превышает длину икозильной группы.
Примеры особенно предпочтительных замещенных связанных с SO2-группой радикалов R включают 4-(фенил)фенил[бифенил], 4-(4’-метоксифенил)фенил, 4-(фенокси)фенил, 4-(тиофенил)фенил[4-(фенилтио)фенил], 4-(азофенил)фенил, 4-[(4’-трифторметилтио)фенокси]фенил, 4-[(4’-трифторметилтио)тиофенил]фенил, 4-[(4’-трифторметил) фенокси]фенил, 4-[(4’-трифторметил)тиофенил]фенил, 4-[(4’-трифторметокси)фенокси]фенил, 4-[(4’-трифторметокси)тиофенил]фенил, 4-[(4’-фенил)-N-пиперидил]фенил, 4-[(4’-ацетил-N-пиперазил]фенил и 4-(бензамидо)фенил.
Поскольку рассматриваемый связанный с SО2-группой арильный или гетероарильный радикал R3 сам предпочтительно замещен 6-членным кольцом, с целью облегчить понимание расположений заместителей в настоящем описании одновременно используются две номенклатурных системы. В первой системе используются номера положений кольца, непосредственно связанного с SO2-группой, а во второй системе используется обозначения орто-, мета- или пара-положения одного из заместителей 6-членного кольца, соединенного со связанным с SО2-группой арильным или гетероарильным радикалом. Хотя, как правило, для алифатических кольцевых систем не используется номенклатура в виде орто-, мета- или пара-положения, вероятно, существенно легче понять строение соединений, приведенных в настоящем описании, в сочетании с системой нумерации для первого кольца, связанного с SО2-группой. Если радикал R3 не обозначает 6-членное кольцо, то положение заместителей нумеруется от положения связи с ароматическим или гетероароматическим кольцом. Для названия конкретных соединений используется официально действующая номенклатура.
Так, 1-положение описанных выше связанных с SО2-группой арильных или гетероарильных радикалов обозначает положение, в котором SO2-группа присоединена к кольцу. Указанные 3- и 4-положения колец пронумерованы начиная с мест присоединения заместителя относительно места присоединения SO2-групп в соответствии с официально действующей нумерацией положений, принятой в номенклатуре гетероарильных производных.
С учетом самой длинной цепи атомов радикала R3, включающей его собственные заместители, его общая длина превышает длину насыщенной цепи, состоящей из 5 атомов углерода (пентильная группа), предпочтительно радикал имеет длину, превышающую длину насыщенной цепи, состоящей из 6 атомов углерода (гексильная группа), т.е. длина радикала примерно соответствует гептильной цепи или превышает ее. Радикал R3 также имеет длину, меньшую, чем длина насыщенной цепи, состоящей примерно из 20 атомов углерода[изокильная группа (икозил ранее назывался эйкозил)], более предпочтительно примерно из 18 атомов углерода (стеарильная группа). Наиболее предпочтительно длина цепи R3 составляет от примерно 8 до примерно 12 атомов углерода, хотя в кольцевых структурах и в заместителях может присутствовать существенно большее число атомов. Требования к их длине еще будут рассмотрены ниже.
В целом, без учета конкретных фрагментов, из которых он конструируется, радикал R3 (группа или фрагмент) имеет длину, превышающую длину пентильной группы. Кроме того, радикал R3 также имеет длину, меньшую чем длина икозильной (дидецильной) группы. Это означает, что R3 представляет собой радикал, имеющий минимальную длину, превышающую длину насыщенной цепи, состоящей из 5 атомов углерода, и предпочтительно, превышающую длину гексильной группы, но более короткую, чем длина насыщенной цепи, состоящей из 20 атомов углерода, и предпочтительно цепи, состоящей из 18 атомов углерода. Наиболее предпочтительно R3 имеет длину, превышающую длину октильной группы, но меньшую, чем длина лаурильной группы.
В частности, группа R3 имеет минимальную длину, равную длине гексильной группы только тогда, когда заместитель включает два кольца, которые могут быть сконденсированы или просто ковалентно связаны друг с другом экзоциклической связью. Если R3 не содержит два связанных или сконденсированных кольца, например если радикал R3 включает алкил или второй, третий или четвертый кольцевой заместитель, R3 имеет длину, превышающую длину гексильной группы. Примерами таких двух кольцевых систем групп R3 являются 2-нафтильная группа или 2-хинолинильная группа (каждая с 6 атомами углерода в цепи) и 8-пуринильная группа (с 5 атомами углерода в цепи). Не основываясь на какой-либо теории, можно предположить, что присутствие нескольких колец в радикале R3 повышает избирательность профиля ферментативной активности ингибитора.
Длины цепей радикалов измеряют по наиболее длинной линейной цепи атомов в радикале, при необходимости следуя вдоль атомов скелета кольца. Для облегчения расчетов каждый атом в цепи, например углерод, кислород, сера и азот, считается атомом углерода.
Длины цепей могут быть легко определены с помощью опубликованных данных об углах связей, длинах связей и радиусах атомов, которые при необходимости применяются для изображения и измерения требуемой, обычно зигзагообразной, цепи или с помощью построения моделей с использованием имеющихся в продаже наборов, в которых углы, длины связей и радиусы атомов приведены в соответствие с опубликованным данными. Длины радикалов (заместителей) также могут быть определены с несколько меньшей точностью, если принять, что все атомы имеют длины связей, соответствующие насыщенному атому углерода, что ненасыщенные связи имеют такие же длины, как и насыщенные связи, и что все углы для ненасыщенных связей такие же, как и углы для насыщенных связей, хотя приведенные выше методы измерения являются предпочтительными. Например, при использовании такого метода измерения фенильная и пиридильная группа имеют длину, соответствующую длине цепи, состоящей из 4 атомов углерода, равно как и пропоксигруппа, в то время как длина бифенильной группы соответствует длине цепи, состоящей примерно из 8 атомов углерода.
Кроме того, группа R3 при вращении вокруг оси, проходящей через связанное с SO2 1-положение и связанное с заместителем 4-положение 6-членного кольца или через связанное с SO2 1-положение и связанное с заместителем 3-или 4-положение 5-членного кольца, образует трехмерное пространство, наибольший размер которого в направлении, перпендикулярном указанной оси вращения, равен длине от примерно одного фуранильного кольца до примерно двух фенильных колец.
Так, группа R3 имеет размер, соответствующий 2-нафтильному заместителю или 8-пуринильному заместителю, при оценке с помощью вышеописанного критерия, основанного на оценке радиуса вращения, а также с помощью описанных выше методов. С другой стороны, 1-нафтильная группа или 7-или 9-пуринильная группа имеет слишком большой радиус вращения и не подпадает под обозначение группы R3.
Как следствие таких требований, предъявляемых к длине и радиусу вращения, особенно предпочтительными в качестве радикалов R3 являются такие радикалы R3, как 4-(фенил)фенил[бифенил], 4-(4’-метоксифенил)фенил, 4-(фенокси)фенил, 4-(тиофенил)фенил[4-(фенилтио)фенил], 4-(азофенил)фенил, 4-[(4’-трифторметилтио)фенокси]фенил, 4-[(4’-трифторметилтио)тиофенил]фенил, 4-[(4’-трифторметил)фенокси]фенил, 4-[(4’-трифторметил)тиофенил]фенил, 4-[(4’-трифторметокси)фенокси]фенил, 4-[(4’-трифторметокси)тиофенил]фенил, 4-[(4’-фенил)-N-пиперидил]фенил, 4-[(4’-ацетил-N-пиперазил]фенил и 4-(бензамидо)фенил. Эти заместители сами могут быть замещены во втором кольце относительно SО2-группы в мета- или пара-положении или в обоих положениях одним атомом или заместителем, самая длинная цепь которого может иметь длину до 5 атомов, исключая атом водорода.
Не основываясь на какой-либо теории, полагают, что длина связанного с SO2-группой заместителя радикала R3, вероятно, играет определенную роль в общей активности рассматриваемого соединения, обладающего ингибирующей активностью в отношении ферментов ММП. Длина радикала R3, вероятно, также играет некоторую роль в избирательной активности соединения, обладающего ингибирующей активностью в отношении определенных ферментов ММП.
В особенно предпочтительном варианте осуществления R3 обозначает группу PhR23, где Ph обозначает фенил. Фенильное кольцо (Ph) группы PhR23 замещено в его пара-положении (4-положение) группой R23, которая может представлять собой другую арильную или гетероарильную группу с одним кольцом, пиперидильную группу, пиперазинильную группу, феноксигруппу, тиофеноксигруппу [C6Н5-S-], фенилазогруппу [C6H5-N2-]или бензамидогруппу [-NHC(O)C6H5].
В одном из вариантов осуществления в особенно предпочтительном ароматическом сульфонгидроксамовом соединении, обладающем ингибирующей активностью, заместитель R23 обозначает феноксигруппу и сам замещен в его пара-положении фрагментом, который выбирают из группы, включающей галоген, С1-С4алкоксигруппу, С1-С4алкильную группу, диметиламиогруппу, карбоксилС1-С3алкиленовую группу, С1-С4алкоксикарбонил-С1-С3алкиленовую группу, трифторметилтиогруппу, трифторметоксигруппу, трифторметильную группу и карбоксамидоС1-С3алкиленовую группу, или замещен в мета- и в пара-положениях метилендиоксигруппой. Очевидно, что любой заместитель R23 может быть замещен фрагментом из числа вышеперечисленных. Предпочтительным является такое замещение в пара-положении.
Настоящее изобретение также относится к промежуточному продукту, пригодному для получения соединения формул I-V. Такое промежуточное соединение имеет строение, соответствующее приведенной ниже формуле VI:
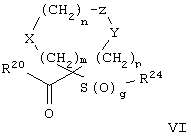
где q равно 0, 1 или 2,
R20 обозначает (a) -O-R21, где R21 выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил и фармацевтически приемлемый катион, или (б) -NH-O-R22 где R22 обозначает избирательно удаляемую защитную группу, такую как 2-тетрагидропиранил, С1-С6ацил, ароил, бензил, пара-метоксибензил (MOZ), карбонилС1-С6алкоксигруппа, тризамещенная силильная группа или орто-нитрофенильная группа, смола для синтеза пептидов и т.п., причем тризамещенная силильная группа замещена С1-С6алкилом, арилом или арС1-С6алкилом,
m равно 0, 1 или 2,
n равно 0, 1 или 2,
р равно 0, 1 или 2,
сумма m+n+p равна 1, 2, 3 или 4,
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя из группу, включающей X, Y и Z, обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей X, Y и Z, обозначает CR8R9 или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей
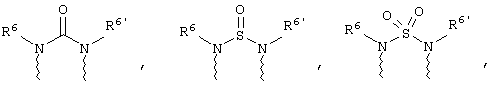
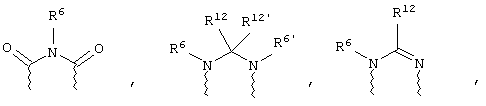
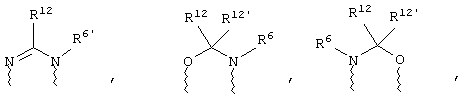
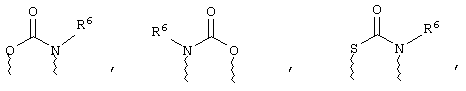
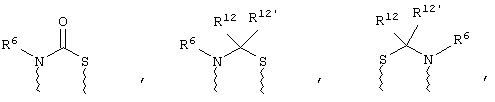
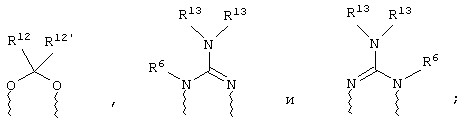
где волнистые линии обозначают связи с атомами изображенного кольца, R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бисС1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6перфторалкил, С1-С6трифторметилалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкилкарбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкоксиС1-С6алкил, гетероарилтиоС1-С6алкил, арилсульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4алкоксикарбонил-С1-С6алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С1-С6гетероциклоиминокарбонил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С4алкокси-С1-С4алкил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5алкилкарбонил, гидроксиС1-С5алкил, аминокарбонил, где азот в аминокарбониле является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, гидроксиаминокарбонил, аминосульфонил, где азот в аминосульфонильной группе является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, аминоС1-С6алкилсульфонил, где азот аминоС1-С6алкилсульфонильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей бензил, фенил, С1-С6алкил, С3-С6алкинил, С3-С6алкенил и С1-С6гидроксиалкил,
R8 и R9, и R10, и R11 независимо друг от друга выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид или сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или где R8 и R9 или R10 и R11 и атом углерода, к которому они присоединены, образуют карбонильную группу, или где R8 и R9 или R10 и R11, или R8 и R10 вместе с атомами, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, которые представляют собой азот, кислород или серу, при условии, что только один из радикалов R8 и R9 или R10 и R11 обозначает гидроксигруппу,
R12 и R12' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероаралкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, арилоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид или сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкильной группы является (I) незамещенным или (II) замещенным одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С2-С6алкинил, С2-С6алкенил и С1-С6гидроксиалкил, и
R24 обозначает R3, как он определен в формулах I, III, IV, или обозначает заместитель G-A-R-E-Y формулы II (формула VIA).
В альтернативном варианте R24 обозначает R3', арильную или гетероарильную группу, которая замещена связывающим заместителем, реакционноспособным в отношении связывания с другим фрагментом (формула VIВ), таким как замещаемая нуклеофилом уходящая группа D.
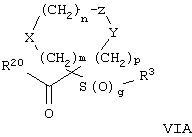
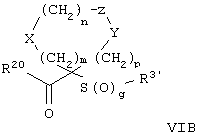
Примерами замещаемых нуклеофилом уходящих групп D являются галоген (фтор, хлор, бром или йод), нитро-, азидо-, фенилсульфоксидо-, арилокси-, С1-С6алкоксигруппа, С1-С6алкилсульфонатная или арилсульфонатная группа и тризамещенная аммонийная группа, в которой три заместителя независимо друг от друга выбирают из группы, включающей арил, арС1-С6алкил или С1-С6алкил. Дополнительные связывающие заместители включают гидроксильную группу и аминогруппу, которые могут быть связаны с содержащими карбонил фрагментами с образованием сложных эфиров, уретанов, карбонатов, амидов и мочевин, но не ограничены этими заместителями. Аналогично этому карбоксильный связывающий заместитель может использоваться для получения сложного эфира, тиоэфира или амида. Так связывающий заместитель пригоден для прекращения связывающей содержащей заместитель арильной или гетероарильной группы в такой заместитель, как описанный выше заместитель G-A-R-E-Y, путем образования ковалентной связи.
Соединение формулы VI может быть связано с другим фрагментом по связывающему заместителю R3 с образованием соединения, в котором вновь образованная группа R3 соответствует таковой в формулах I, III, IV или -G-A-R-E-Y-. Примером таких реакций сочетания является нуклеофильное замещение с образованием простых эфиров и тиоэфиров, а также с образованием сложного эфира, амида, мочевины, карбоната, уретана и тому подобных связей.
Особенно предпочтительным предшественником для промежуточного соединения формулы VI является промежуточное соединение приведенной ниже формулы VII:

где m, n, р, g, X, Z, Y, D и R имеют значения, указанные выше для формулы VI.
При этом R20 предпочтительно обозначает -NH-O-R, где R22 обозначает избирательно удаляемую защитную группу, такую как 2-тетрагидропиранил, С1-С6ацил, ароил, бензил, пара-метоксибензил (MOZ), карбонилС1-С6алкоксигруппа, орто-нитрофенильная группа, смола для синтеза пептидов, так называемая Merrifield’s Peptide Resin фирмы Sigma Chemical Co., и т.п., при этом предпочтительным является 2-тетрагидропиранил. Таким образом, очевидно, что группа -NH-O-R (R20) в формулах VI и VII является продуктом реакции гидроксиламина, кислород которого связан с избирательно удаляемой защитной группой и карбоксильной группой.
В соединении каждой из формул VI и VII букву "g" в виде нижнего индекса применяют для того, чтобы указать состояние окисления атома серы. Когда g равно 0, атом серы находится в неокисленном состоянии и указанное соединение, как правило, является сульфидным продуктом реакции содержащего серу синтона, как это проиллюстрировано ниже в примерах. Когда g равно 1, сера окислена до сульфоксида, а когда g равно 2, сера окислена до сульфона, что также проиллюстрировано ниже. Соединения формул VI или VII, в которых g равно 0 или 1, как правило, сами являются промежуточными продуктами для получения аналогичного соединения, в котором g равно 2, и промежуточный продукт предпочтительно является сульфоном.
Таким образом, предпочтительный промежуточный продукт имеет строение, соответствующее приведенной ниже формуле VIIA
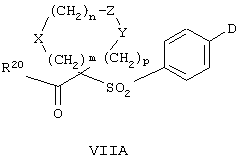
В настоящем описании в обозначении молекул или групп молекулярные дескрипторы могут быть объединены с целью написания слов или фраз, которые обозначают структурные группы, или объединены с целью обозначения структурных групп. Такие дескрипторы применяются в настоящем описании. Примерами таких обычно применяемых понятий являются аралкил (или арилалкил), гетероаралкил, гетероциклоалкил, циклоалкилалкил, аралкоксиалкоксикарбонил и т.п. Конкретным примером соединения, подпадающего под последнее понятие аралкоксиалкоксикарбонил, является С6Н5-СН2-СН2-O-СН2-O-(С=O), где С6H5-обозначает фенил. Также следует отметить, что в данной области структурная группа может обозначаться более чем одним словом или фразой, например гетероарилоксиалкилкарбонил также может быть назван гетероарилоксиалканоилом. Такие комбинации применяются в настоящем описании при описании процессов, соединений и композиций по настоящему изобретению, а ниже приведены другие такие примеры. Следующий список не следует рассматривать как исчерпывающий или ограничивающий, поскольку он дан в качестве иллюстративных примеров слов или фраз (понятий), которые применяются в контексте настоящего описания.
В контексте настоящего описания понятие "алкил" отдельно или в сочетании обозначает алкильный радикал с прямой или разветвленной цепью, содержащий от 1 до примерно 12 атомов углерода, предпочтительно от 1 до примерно 10 атомов углерода и более предпочтительно от 1 до примерно 6 атомов углерода. Примеры таких радикалов включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, гексил, октил и т.п.
Понятие "алкенил" отдельно или в сочетании обозначает углеводородный радикал с прямой или разветвленной цепью, содержащий одну или более двойных связей и от 2 до примерно 12 атомов углерода, предпочтительно от 2 до примерно 10 атомов углерода и более предпочтительно от 2 до примерно 6 атомов углерода. Примеры приемлемых алкенильных радикалов включают этенил (винил), 2-пропенил, 3-пропенил, 1,4-пентадиенил, 1,4-бутадиенил, 1-бутенил, 2-бутенил, 3-бутенил, деценил и т.п.
Понятие "алкинил" отдельно или в сочетании обозначает углеводородный радикал с прямой или разветвленной цепью, содержащий одну или более тройных связей и от 2 до примерно 12 атомов углерода, предпочтительно от 2 до примерно 10 атомов углерода и более предпочтительно от 2 до примерно 6 атомов углерода. Примеры приемлемых алкинильных радикалов включают этинил, 2-пропинил, 3-пропинил, децинил, 1-бутинил, 2-бутинил, 3-бутинил, и т.п.
Понятие "карбонил" или "оксогруппа" отдельно или в сочетании обозначает -С(=O)-группу, в которой две оставшихся связи (валентности) могут быть замещены независимо друг от друга. Подразумевается, что понятие карбонил также включает гидратированную карбонильную группу -С(ОН)2-.
Понятие "тиол" или "сульфгидрил" отдельно или в сочетании обозначает -SH-группу. Понятие "тиогруппа" или "тиагруппа" отдельно или в сочетании обозначает простую тиаэфирную группу, т.е. простую эфирную группу, в которой кислород простого эфира заменен на атом серы.
Понятие "аминогруппа" отдельно или в сочетании обозначает амин или -NH2-группу, в которой понятие "монозамещенная аминогруппа" отдельно или в сочетании обозначает замещенный амин, т.е. группу-N(H) (заместитель), в которой один атом водорода заменен заместителем, а понятие "дизамещенный амин" обозначает группу-N(заместитель)2, в которой два атома водорода аминогруппы заменены заместителями, независимо друг от друга выбранными из определенной группы заместителей.
Амины, аминогруппы и амиды представляют собой соединения, которые могут быть обозначены как первичные (I° ), вторичные (II° ) или третичные (III° ), или незамещенные, монозамещенные или N,N-дизамещенные в зависимости от степени замещения азота аминогруппы. Четвертичный амин (аммоний) (IV° ) обозначает азот с 4 заместителями [-N+(заместитель)4], который является положительно заряженным и которому сопутствует противоион, а понятие "N-оксид" обозначает, что один заместитель представляет собой кислород, и эта группа может быть обозначена как [-N+(заместитель)3-О-], т.е. заряды внутренне уравновешены.
Понятие "цианогруппа" отдельно или в сочетании обозначает -С-тройная связь-N-группу, т.е. (-C≡ N). Понятие "азидогруппа" отдельно или в сочетании обозначает -N-тройная связь-N-группу, т.е. (-N≡ N). Понятие "гидроксил" отдельно или в сочетании обозначает -ОН-группу. Понятие "нитрогруппа" отдельно или в сочетании обозначает -NO2-группу. Понятие "азогруппа" отдельно или в сочетании обозначает -N=N-группу, в которой связи в концевых положениях могут быть независимо друг от друга замещены.
Понятие "гидразиногруппа" отдельно или в сочетании обозначает -NH-NH-группу, в которой указанные оставшиеся две связи (валентности) могут быть независимо друг от друга замещены. Атомы водорода гидразиногруппы могут быть независимо друг от друга замещены заместителями, а атомы азота могут образовывать кислотно-аддитивные соли или могут образовывать четвертичные основания.
Понятие "сульфонил" отдельно или в сочетании обозначает -SO2-группу, в которой указанные оставшиеся две связи (валентности) могут быть независимо друг от друга замещены. Понятие "сульфоксидогруппа" отдельно или в сочетании обозначает -SO-группу, в которой указанные оставшиеся две связи (валентности) могут быть независимо друг от друга замещены.
Понятие "сульфон" отдельно или в сочетании обозначает -SO2-группу, в которой указанные оставшиеся две связи (валентности) могут быть независимо друг от друга замещены. Понятие "сульфенамид" отдельно или в сочетании обозначает группу -SON=, в которой указанные оставшиеся три связи (валентности) могут быть независимо друг от друга замещены. Понятие "сульфид" отдельно или в сочетании обозначает -S-группу, в которой указанные оставшиеся две связи (валентности) могут быть независимо друг от друга замещены.
Понятие "алкоксигруппа" отдельно или в сочетании обозначает радикал простого алкильного эфира, где понятие алкил имеет указанные выше значения. Примеры приемлемых радикалов простых алкильных эфиров включают метокси-, этокси-, н-пропокси-, изопропокси-, н-бутокси-, изобутокси-, втор-бутокси-, трет-бутоксигруппу и т.п.
Понятие "циклоалкил" отдельно или в сочетании обозначает циклический алкилъный радикал, содержащий от 3 до примерно 8 атомов углерода. Понятие "циклоалкилалкил" обозначает алкильный радикал, как он определен выше, замещенный циклоалкильным радикалом, содержащим от 3 до примерно 8, предпочтительно от 3 до примерно 6 атомов углерода. Примеры таких циклоалкильных радикалов включают циклопропил, циклобутил, циклопентил, циклогексил и т.п.
Гетероциклический (гетероцикло-) или гетероциклический фрагмент гетероциклокарбонильной, гетероциклооксикарбонильной, гетероциклоалкоксикарбонильной или гетероциклоалкильной группы или т.п. представляет собой насыщенный или частично ненасыщенный моноциклический, бициклический или трициклический гетероцикл, который содержит один или несколько гетероатомов, выбранных из группы, включающей азот, кислород и серу. Такой фрагмент необязательно может быть замещен по одному или нескольким кольцевым атомам углерода галогеном, алкилом, алкокси-, оксогруппой и т.п. и/или по атому вторичного азота (т.е. -NH-) кольца - алкилом, аралкоксикарбонилом, алканоилом, арилом или арилалкилом и по атому третичного азота (т.е. =N-) - оксидогруппой, присоединенной через атом углерода. Также может быть присоединен атом третичного азота с 3 заместителями с образованием N-оксида [=N(О)-группа].
Понятие "арил" отдельно или в сочетании обозначает 5- или 6-членный карбоциклический ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле только атомы углерода, т.е. карбоциклический арильный радикал. Примеры карбоциклических арильных радикалов включают фенил, инденил и нафтил.
Понятие "гетероарил" отдельно или в сочетании обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему (радикал), содержащую два или три кольца, которые содержат в цикле (ах) атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот. Примерами таких гетероциклических или гетероарильных групп являются пирролидинил, пиперидил, пиперазинил, морфолинил, тиаморфолинил, пирролил, имидазолил (например имидазол-4-ил, 1-бензилоксикарбонилимидазол-4-ил и т.п.), пиразолил, пиридил, пиразинил, пиримидинил, фурил, тетрагидрофурил, тиенил, триазолил, оксазолил, оксадиазоил, тиазолил, тиадиазоил, индолил (например, 2-индолил и т.п.), хинолинил (например, 2-хинолинил, 3-хинолинил, 1-оксидо-2-хинолинил и т.п.), изохинолинил (например, 1-изохинолинил, 3-изохинолинил и т.п.), тетрагидрохинолинил (например, 1,2,3,4-тетрагидро-2-хинолил и т.п.), 1,2,3,4-тетрагидроизохинолинил (например, 1,2,3,4-тетрагидро-1-оксоизохинолинял и т.п.), хиноксалинил, β -карболинил, 2-бензофуранкарбонил, бензотиофенил, 1-, 2-, 4- или 5-бензимидазолил и аналогичные радикалы.
Если арильный или гетероарильный радикал представляет собой замещающий фрагмент (группу, заместитель или радикал), то он сам может быть замещен, при этом последний из указанных заместитель представляет собой заместитель, независимо друг от друга выбранный из группы, включающей цианогруппу, перфторалкил, трифторметокси-, трифторметилтиогруппу, галоалкил, трифторметилалкил, аралкоксикарбонил, арилоксикарбонил, гидроксигруппу, галоген, алкил, алкокси-, нитрогруппу, тиол, гидроксикарбонил, арилокси-, арилтиогруппу, аралкил, арил, арилкарбониламино-, гетероарил окси-, гетероарилтиогруппу, гетероаралкил, циклоалкил, гетероциклоокси-, гетероциклотио-, гетероциклоамино-, циклоалкил окси-, циклоалкилтио-, гетероаралкокси-, гетероаралкилтио-, аралкокси-, аралкилтио-, аралкиламино-, гетероциклогруппу, гетероарил, арилазо-, гидроксикарбонилалкокси-, алкоксикарбонилалкоксигруппу, алканоил, арилкарбонил, аралканоил, алканоилокси-, аралканоилоксигруппу, гидроксиалкил, гидроксиалкокси-, алкилтио-, алкоксиалкилтиогруппу, алкоксикарбонил, арилоксиалкоксиарил, арилтиоалкилтиоарил, арилоксиалкилтиоарил, арилтиоалкоксиарил, гидроксикарбонилалкокси-, гидроксикарбонилалкилтио-, алкоксикарбонилалкокси-, алкоксикарбонилалкилтио-,
аминогруппу,
где азот аминогруппы является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, гетероарил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил, арилкарбонил, аралканоил, гетероарилкарбонил, гетероаралканоил и алканоил, или (III) где азот аминогруппы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо, содержащее 0-2 дополнительных гетероатома, представляющих собой азот, кислород или серу, и само этот кольцо является (а) незамещенным или (б) замещенным одной или двумя группами, независимо друг от друга выбранными из группы, включающей арил, алкил, гетероарил, аралкил, гетероаралкил, гидрокси-, алкоксигрулпу, алканоил, циклоалкил, гетероциклоалкил, алкоксикарбонил, гидроксиалкил, трифторметил, сконденсированный с бензольным кольцом гетероциклоалкил, гидроксиалкоксиалкил, аралкоксикарбонил, гидроксикарбонил, арилоксикарбонил, сконденсированную с бензольным кольцом гетероциклоалкоксигруппу, сконденсированный с бензольным кольцом циклоалкилкарбонил, гетероциклоалкилкарбонил и циклоалкилкарбонил, карбониламиногруппу,
где азот карбониламиногруппы является (I) незамещенным или (II) представляет собой реакционноспособный амин аминокислоты, или является (III) замещенным одним или двумя радикалами, выбранными из группы, включающей алкил, гидроксиалкил, гидроксигетероаралкил, циклоалкил, аралкил, трифторметилалкил, гетероциклоалкил, сконденсированный с бензольным кольцом гетероциклоалкил, сконденсированный с бензольным кольцом циклоалкил и N,N-диалкилзамещенную алкиламиноалкильную группу, или (IV) азот карбоксамидогруппы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло-, гетероарильное или сконденсированное с бензольным кольцом гетероциклоалкильное кольцо, которое само может быть незамещенным или может быть замещено одним или двумя радикалами, независимо друг от друга выбранными из группы, включающей алкил, алкоксикарбонил, нитрогруппу, гетероциклоалкил, гидроксигруппу, гидроксикарбонил, арил, аралкил, гетероаралкил
и аминогруппу, где азот аминогруппы является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил и гетероарил, или (III) где азот аминогруппы и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо,
и аминоалкильную группу где азот аминоалкила является (I) незамещенным или (II) замещенным одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил и алканоил, или (III) где азот аминоалкила и два присоединенных к нему заместителя образуют 5-8-членное гетероцикло- или гетероарильное кольцо. Понятие "аралкил" отдельно или в сочетании обозначает алкильный радикал, как он определен выше, в котором один атом водорода заменен арильным радикалом, как он определен выше, такой как бензил, 2-фенилэтил и т.п.
Понятие "аралкоксикарбонил" отдельно или в сочетании обозначает радикал формулы аралкил -О-С(О)-, в котором понятие "аралкил" имеет указанные выше значение. Примером аралкоксикарбонильного радикала является бензилоксикарбонил.
Понятие "арилоксигруппа" обозначает радикал формулы арил-О-, в котором понятие "арил" имеет указанные выше значения. Примером арилоксигрупы является феноксигруппа.
Понятия "гетероаралкил" и "гетероарилоксигруппа" обозначают аналогичные по строению аралкилу и арилоксигруппе радикалы, которые образованы из гетероарильных радикалов. Примеры таких радикалов включают 4-пиколинил и 2-пиримидиноксигруппу соответственно.
Понятия "алканоил" или "алкилкарбонил" отдельно или в сочетании обозначают ацильный радикал, образованный из алканкарбоновой кислоты. Примерами такого радикала являются формил, ацетил, пропионил, бутирил, валерил, 4-метилвалерил и т.п.
Понятие "циклоалкилкарбонил" обозначает ацильную группу, полученную из моноциклической или содержащей мостик циклоалканкарбоновой кислоты, такую как циклопропанкарбонил, циклогексанкарбонил, адамантанкарбонил и т.п., или полученную из сконденсированной с бензолом моноциклической циклоалканкарбоновой кислоты, которая является необязательно замещенной, например, алканоиламиногруппой, такую как 1,2,3,4-тетрагидро-2-нафтоил, 2-ацетамидо-1,2,3,4-тетрагидро-2-нафтоил.
Понятия "аралканоил" или "аралкилкарбонил" обозначают ацильный радикал, полученный из замещенной арилом алканкарбоновой кислоты, такой как фенилацетил, 3-фенилпропионил (гидроксициннамоил), 4-фенилбутирил, (2-нафтил)ацетил, 4-хлоргидроксициннамоил, 4-аминогидроксициннамоил, 4-метоксигидроциннамоил и т.п.
Понятия "ароил" или "арилкарбонил" обозначают ацильный радикал, полученный из ароматической карбоновой кислоты. Примеры таких радикалов включают радикалы ароматических карбоновых кислот, необязательно замещенных бензойной или нафтойной кислоты, такие как бензоил, 4-хлорбензоил, 4-карбоксибензоил, 4-(бензилоксикарбонил) бензоил, 1-нафтоил, 2-нафтоил, 6-карбокси-2-нафтоил, 6-(бензилоксикарбонил)-2-нафтоил, 3-бензилокси-2-нафтоил, 3-гидрокси-2-нафтоил, 3-(бензилоксиформамидо)-2-нафтоил и т.п.
Понятие "циклоалкилалкоксикарбонил" обозначает ацильную группу формулы циклоалкилалкил -O-СО-, где "циклоалкилалкил" имеет указанные выше значения. Понятие "арилоксикарбонил" обозначает ацильный радикал формулы арил-O-алканоил, где "арил" и "алканоил" имеют указанные выше значения. Понятие "гетероциклооксикарбонил" обозначает ацильную группу, имеющую формулу гетероцикло-O-СО-, где "гетероциклогруппа" имеет указанные выше значения.
Понятие "гетероциклоалканоил" обозначает ацильный радикал формулы "замещенная гетероциклогруппой алканкарбоновая кислота", где "гетероциклогруппа" имеет указанные выше значения. Понятие "гетероарилоксикарбонил" обозначает ацильный радикал, представленный формулой гетероарил-O-СО-, где "гетероарил" имеет указанные выше значения.
Понятие "аминокарбонил" (карбоксамид) отдельно или в сочетании обозначает замещенную аминогруппой карбонильную (карбамоильную) группу, полученную из амина в результате взаимодействия с карбоновой кислотой, где аминогруппа (азот амидогруппы) является незамещенной (-NH2) или может быть замещена первичной или вторичной аминогруппой, содержащей один или два заместителя, выбранных из группы, включающей гидрид, алкил, арил, аралкил, циклоалкил, циклоалкилалкил и т.п., как указано выше. Гидроксамат представляет собой N-гидроксикарбоксамид.
Понятие "аминоалканоил" обозначает ацильную группу, полученную из алканкарбоновой кислоты, замещенной аминогруппой, где аминогруппа может представлять собой первичную или вторичную аминогруппу, содержащую заместители, независимо друг от друга выбранные из группы, включающей гидрид, алкил, арил, аралкил, циклоалкил, циклоалкилалкил и т.п.
Понятие "галоген" обозначает фтор, хлор, бром или йод. Понятие "галоалкил" обозначает алкильный радикал, имеющий указанные выше значения, в котором один или несколько атомов водорода заменены галогеном. Примеры таких галоалкильных радикалов включают хлорметил, 1-бромэтил, фторметил, дифторметил, трифторметил, 1,1,1-трифторэтил и т.п.
Понятие "перфторалкил" обозначает алкильную группу, в которой каждый атом водорода заменен атомом фтора. Примерами таких перфторалкильных групп помимо указанного выше трифторметила являются перфторбутил, перфторизопропил, перфтордодецил и перфтордецил.
Понятие "перфторалкоксигруппа" отдельно или в сочетании обозначает радикал простого перфторалкильного эфира, где "перфторалкил" имеет указанные выше значения. Примерами таких перфторалкоксигрупп помимо указанной трифтометоксигруппы (F3C-O-) являются перфторбутокси-, перфторизопропокси-, перфтордодекокси- и перфтордекоксигруппа.
Понятие "перфторалкилтиогруппа" отдельно или в сочетании обозначает тиоэфирный перфторалкильный радикал, в котором "перфторалкил" имеет указанные выше значения. Примерами таких перфторалкилтиогрупп помимо указанной трифтометилтиогруппы (F3C-S-) являются перфторбутилтио-, перфторизопропилтио-, перфтордодецилтио- и перфтордецилтиогруппа.
Понятие "ароматическое кольцо" в сочетании, например, с ароматическим кольцом, замещенным сульфоном, или с ароматическим кольцом, замещенным сульфоксидом, обозначает арил или гетероарил, как они определены выше.
Понятие "фармацевтически приемлемый" при его использовании в качестве имени прилагательного в контексте настоящего описания обозначает, что изменяемое понятие, использованное в качестве имени существительного, пригодно для применения в фармацевтическом продукте. Фармацевтически приемлемые катионы включают ионы металлов и органические ионы. Более предпочтительные ионы металлов включают ионы солей щелочных металлов (группа Iа), солей щелочно-земельных металлов (группа IIа) и ионы других физиологически приемлемых металлов, но не ограничены ими. Примеры ионов включают ионы алюминия, кальция, лития, магния, калия, натрия и цинка с обычными для них валентностями. Предпочтительные органические ионы включают протонированные третичные амины и катионы четвертичного аммония, включая, в частности, триметиламин, диэтиламин, N,N’-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, меглумин (N-метилглукамин) и прокаин. Примеры фармацевтически приемлемых кислот включают соляную кислоту, бромистоводородную кислоту, фосфорную кислоту, серную кислоту, метансульфоновую кислоту, уксусную кислоту, муравьиную кислоту, винную кислоту, малеиновую кислоту, яблочную кислоту, лимонную кислоту, изолимонную кислоту, янтарную кислоту, молочную кислоту, глюконовую кислоту, глюкуроновую кислоту, пировинограцную кислоту, щавелевоуксусную кислоту, фумаровую кислоту, пропионовую кислоту, аспарагиновую кислоту, глутаминовую кислоту, бензойную кислоту и т.п., но не ограничены ими.
Получение требуемых соединений
В приведенных ниже реакционных схемах "М" обозначает уходящую группу, такую как галоген, сложный фосфатный эфир или сложный сульфатный эфир.
На приведенных ниже схемах А-В и на схемах 1-19 проиллюстрированы химические процессы и превращения, которые могут использоваться для получения соединений, пригодных для применения согласно изобретению, т.е. соединений формул I, II, III, IV и V и аналогичных циклических соединений, обладающих ингибирующей активностью. Кроме того, проиллюстрировано получение соединений формулы VI и формулы VII. Соединения формулы VI и формулы VII могут применяться в качестве промежуточных продуктов для получения соединений формул I, II, III, IV и V или пролекарств или ингибиторов ММП.
На схемах А-В символы J независимо друг от друга обозначают R20 или другие пригодные для синтеза группы, такие как амиды, хлорангидриды, смешанные ангидриды и т.п. Символ n обозначает 0, 1 или 2, а на схеме В предпочтительно обозначает 1 или 2. На указанных схемах n соответствует g в формулах VI и VII и обозначает 0, 1 или 2. Символ m обозначает 1 или 2. Символы r независимо друг от друга обозначают 1, 2 или 3. Символ Р обозначает защитную группу, которая также может быть представителем группы R. На схеме А для упрощения позиционные изомеры изображены стандартным образом со связью, проходящей через кольцо. На последующих схемах, как правило, показан только один из позиционных изомеров, однако подразумевается, что эти структуры и реакционные схемы включают позиционные изомеры, характерные для приведенных выше формул I, II, III, IV, V, VI и VII. Аналогично этому символ В обозначает О, S, SO, SO2 и NR6. Символы С и С’ независимо друг от друга обозначают электрофильные группы или группы, способные участвовать в реакции конденсации. В связи с этим также следует отметить, что 6-членное кольцо приведено исключительно с целью иллюстрации, а процессы и/или реагенты применимы в отношении и представляют собой комбинации, позволяющие получать 5-8-членные кольцевые системы.
Структуры, приведенные на схемах 1-19, также соответствуют соединениям, представляющим собой другую группу соединений по настоящему изобретению. Ароматическое кольцо на схеме В обозначает арил и гетероарил. Фрагменты-A-R-E-Y имеют указанные выше значение. Приведенные в качестве иллюстрации реакции, в которых участвует спирогетероциклический атом азота, могут быть не пригодны для соединений, включающих серу или кислород.
Схема А
На стадии 1 схемы А показано восстановление гетероарильного соединения с образованием карбоксильного производного. Как правило, первый продукт представляет собой гетероциклический содержащий водород амин, если в качестве исходного продукта используется ароматический радикал, или представляет собой содержащий R6 гетероцикл, если в качестве исходного продукта используется частично ненасыщенный гетероцикл.
Соединение формулы 2 может быть подвергнуто обработке несколькими способами в зависимости от требований, предъявляемых химиком. На стадии 2 азот может быть защищен путем получения, например, карбобензокси- (Z) или трет-бутоксикарбонильного производного. Такие реакции ацилирования можно проводить по методам, хорошо известным в данной области, прежде всего в области синтеза аминокислот и пептидов. Процесс ацилирования с помощью реагентов, содержащих активированную карбоксильную группу или активированную сульфонильную группу, для получения указанных соединений осуществляют аналогичным образом. Примерами таких ацилирующих групп являются карбонилазиды, галогениды, ангидриды, смешанные ангидриды, производные карбодиимида или другие менее традиционные активированные сложноэфирные группы, такие как производное гидроксибензотриазола (ГОБТ). Эти реакции ацилирования при необходимости могут протекать в присутствии основания, включая слабые основания, такие как триэтиламин или N-этилморфолин. Получение некоторых активированных сложноэфирных реагентов и их применение для получения других соединений по изобретению описано ниже. Следует еще раз подчеркнуть, что обозначенные символом Р группы, которые служат в качестве избирательно удаляемой защитной группы, также могут представлять собой часть группы R6.
На стадии 4 схемы А показан процесс алкилирования или ацилирования соединения формулы 2 с получением соединения формулы 5. Процесс алкилирования или ацилирования приведен в настоящем описании. На стадии 5 группа J при необходимости может быть заменена. Примером такой замены является обмен сложного эфира на ТГП-защищенный гидроксамат при превращении ТГП-защищенного гидроксамата в гидроксамат или превращении кислоты в защищенный гидроксамат или т.п.
На стадиях 3, 7 и 8 показано получение содержащих серу производных указанных соединений или промежуточных продуктов этих соединений. На указанных выше стадиях исходный продукт (например соединения формул 2, 5 и 6) может быть обработан основанием для депротонирования альфа-углерода с целью получения функционально активного карбонила. Такой анион может быть подвергнут взаимодействию с содержащим серу электрофилом с получением сульфона, сульфоксида или сульфида. Такие электрофилы могут буть представлены, например, в таких формах, как R24S-SR24, R24SO2Cl, R24SCl, R24SOCl, R24S(O)-SR24 и т.п., где R24 имеет указанные выше значения, или могут обозначать арильный или гетероарильный содержащий серу продукт, включающий связывающий заместитель, т.е. R3, который может использоваться для получения одной из содержащих R24 групп. Для получения аниона необходимо основание, причем может требоваться сильное основание, такое как один из указанных в настоящем описании амидов, гидридов или алкилов металлов. Применяются непротонные растворители, а предпочтительными являются биполярные апротонные растворители наряду с применением атмосферы инертного газа. На следующих схемах для упрощения, как правило, предполагается, что группа R24 обозначает R3.
Следует отметить, что с помощью указанных процессов в зависимости от исходного продукта получают сульфиды (простые тиоэфиры), сульфоксиды или сульфоны. Кроме того, сульфиды могут быть окислены с образованием сульфоксидов или сульфонов, а сульфоксиды могут быть окислены с образованием соответствующих сульфоновых производных. Выбор в процессе синтеза стадии, на которой следует изменять степень окисления серы, а также принятие решения об изменении степени окисления серы осуществляется специалистом в области химии. Способы окисления серы описаны ниже.
На стадиях 6, 9, 10 и 12 схемы А независимо друг от друга проиллюстрирована взаимная конверсия групп, обозначенных символом J. Примеры таких взаимных конверсий включают замену сложного эфира на гидроксамовую кислоту или на производное гидроксамовой кислоты, превращение карбоновой кислоты в производное с активированной карбонильной функцией или в гидроксамовую кислоту или в производное гидроксамовой кислоты (пролекарство или защищенное производное) или удаление защитной группы гидроксаматного производного. Получение соединений с активированной карбонильной функцией, их взаимодействие с нуклеофилами, такими как гидроксамовая кислота, защищенные гидроксаматы или пролекарства на основе гидроксамовой кислоты, описано ниже на примере превращения защищенных производных гидроксамовой кислоты в гидроксамовые кислоты. Получение, например, продуктов, являющихся производными гидроксибензотриазола/карбодиимида, описано ниже. Получение или гидролиз сложных эфиров, амидов, амидных производных, хлорангидридов, ангидридов кислот, смешанных ангидридов и т.п. являются методами синтеза, хорошо известными в данной области, и поэтому они не рассматриваются подробно в настоящем описании. На стадии 6 проиллюстрировано превращение соединения формулы 4 в соединение формулы 9 без осуществления предварительного превращения в соединение формулы 7.
Схема Б

На схеме Б представлен альтернативный путь получения указанных соединений. Реагент, показанный стрелкой на приведенной выше стадии 1, представляет собой реагент с двумя активными группами в дополнение к гетероатомам (В), указанным ранее. И в этом случае конкретный приведенный в качестве иллюстрации реагент выбирают для упрощения описания реакции, при этом подразумевается, что могут использоваться реагенты, которые определяют положение гетероатома и позволяют получать соединения с размером кольца, состоящим из 5, 7 и 8 членов. Эти реагенты могут быть легко выбраны специалистами в данной области.
Символы С и С’ в реагенте, приведенном на стадии 1, независимо друг от друга обозначают электрофильную группу или группу, которая может превращаться в электрофил. Такие группы включают галогениды, эфиры сульфоновой кислоты, эпоксиды, тиоэпоксиды, гидроксильные группы и т.п. Этот реагент вступает во взаимодействие с нуклеофильным анионом содержащего серу карбонильного соединения, такого как соединение формулы 1. Анион образуется в результате депротонирования соединения формулы 1, а примеры оснований, пригодных для такого депротонирования, приведены ниже. Обработку указанным выше электрофильным реагентом проводят в условиях алкилирования, хорошо известных в данной области и рассмотренных в настоящем описании. Продуктом этой реакции может быть либо соединение формулы 2, либо соединение формулы 3, т.е. реакция при необходимости может быть проведена как реакция "в одной пробирке", либо как двухстадийный процесс.
На стадии 3 проиллюстрирована взаимная конверсия обозначенных символом J групп, которую при необходимости проводят аналогично тому, как это описано выше для схемы А. На стадии 4 используют реагент, в котором С, например, обозначает нуклеофил, как указано выше, а С’ обозначает электрофил или нуклеофил, такой как гидроксил, тиол или R6-аминогруппа. Следует отметить, что символы С’ могут обозначать независимо друг от друга нуклеофил или электрофил, когда m равно 2, т.е. когда m равно 2, не существует обязательного условия, чтобы группы С’ имели одинаковое значение. Когда m равно 2, обработка вторым молем основания является для специалиста-химика альтернативным методом получения соединения формулы 5. Когда С’ обозначает гидроксил, тиол или R6-аминогруппу, a m равно 2, специалист в данной области может конденсировать соединение формулы 4, например, с альдегидом или кетоном, в восстановительных условиях или с последующим восстановлением с получением рассматриваемого соединения. Как указано выше, соединение, в котором m равно 2, может быть получено в одну стадию (процесс в одной пробирке) или в две стадии, что дает возможность специалисту в области химии выбрать, должен (-ны) ли быть реагент(-ы) одинаковыми (одна пробирка) или различными (двухстадийный процесс).
На схеме Б также проиллюстрированы взаимные конверсии групп, обозначенных символом J, степени окисления серы и группы у атома азота, т.е. R-группы, с помощью которых можно получить рассматриваемые соединения. Эти методы и процессы рассмотрены выше при описании реакций по схеме А.
Схема В
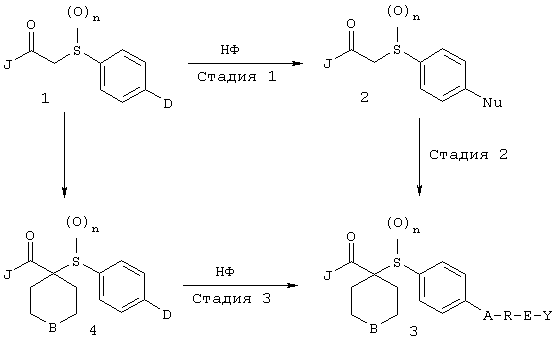
На схеме В проиллюстрировано нуклеофильное замещение группы D, имеющей указанные выше значения. Эту реакцию проводят способом, аналогичным способу проведения реакций замещения, который рассмотрен в настоящем описании. Степень окисления серы выбирается специалистом в данной области, однако сульфоксидные или сульфоновые группы являются предпочтительными, а сульфон является наиболее предпочтительным. Замещение может быть осуществлено либо до, либо после взаимодействия метилена, расположенного за карбонильной группой, с получением спирогетероциклической группы.
На стадиях 1, 2 и 3 также показано, что хотя нуклеофильное замещение может быть осуществлено с использованием одного нуклеофила (НФ), продукт этой реакции может быть модифицирован с помощью способов, хорошо известных в данной области, с получением согласно настоящему описанию группы -A-R-E-Y-, как она определена выше.
Пример такого способа, когда D обозначает фторид, приведен с целью иллюстрации и не ограничивает объем изобретения. Фторидная уходящая группа может быть непосредственно замещена анионом 4-трифтометилфенола, 4-трифтометоксифенола, 4-трифтометилтиофенола и т.п. с получением рассматриваемого соединения, что является примером проводимого в одной пробирке способа получения с использованием в качестве исходного продукта соединения формулы 4. Другие соединения, включающие группу -A-R-E-Y-, могут быть получены замещением фторидной уходящей группы на аммиак с получением амина, который затем может быть подвергнут ацилированию описанными методами, например с помощью 4-трифторметилбензоилхлорида, с получением в качестве продукта реакции другого рассматриваемого соединения.
Если это является целесообразным или необходимым, функционально активная группа R6 может быть заменена и/или дополнительно модифицирована специалистом в данной области в соединениях или на приведенных на схемах стадиях с целью получения рассматриваемых соединений. Следует упомянуть взаимную конверсию бифункциональных групп, представляющих собой защитные группы, предназначенные для кратковременной или продолжительной защиты, в другие группы R6. В данной области хорошо известно множество других общепринятых и/или приемлемых конверсии, включая получение синтетических промежуточных продуктов. Некоторые не ограничивающие объем изобретения примеры таких превращений или реакций включают реакции восстановления, реакции нуклеофильного замещения/замены, реакции обмена или получения карбоновых или сульфоновых кислот, амидов, сложных эфиров, галогенангидридов, смешанных ангидридов и т.п., реакции электрофильного замещения/замены, реакции окисления, реакции превращения кольца/цепи, реакции размыкания кольца, реакции конденсации, включая реакции, в которых используются сульфонильные или карбонильные группы и/или углерод-водородные связи, на которые оказывают влияние любая или обе эти группы. Специалист в данной области техники может самостоятельно выбрать приемлемый метод получения или превращения рассматриваемых соединений и порядок проведения реакции(й). Следует отметить, что если определенная последовательность или метод окажутся нежелательными, то следует выбрать и использовать альтернативный метод. Также можно осуществлять выбор в пользу получения/добавления групп в одну стадию с использованием стратегии конвергентного ингибирования или получения конечной группы R6 с использованием постадийного подхода.
Таким образом, в целом выбор исходных продуктов и условий реакции может изменяться, что хорошо известно специалистам в данной области. Как правило, соответствующий метод не ограничен одним набором условий, поскольку при необходимости могут применяться их вариации. Условия также при необходимости выбирают в зависимости от конкретной цели, такой как получение небольших количеств продукта или крупномасштабное получение. В любом случае применение более опасных для человека или более опасных для окружающей среды продуктов или реагентов, как правило, необходимо свести к минимуму. Примерами таких продуктов являются диазометан, диэтиловый эфир, соли тяжелых металлов, диметилсульфид, хлороформ, бензол и т.п.
Эти реакции при необходимости можно проводить в атмосфере сухого инертного газа, такого как азот или аргон. Определенные реакции, известные специалистам в данной области, можно проводить в сухой атмосфере, такой как атмосфера сухого воздуха, в то время как другие стадии синтеза, например гидролиз сложного эфира или амида с помощью водной кислоты или основания, можно проводить в атмосфере лабораторного воздуха. Кроме того, некоторые стадии этого синтеза можно проводить в сосудах, предназначенных для проведения реакций под давлением, при давлении выше, равном или ниже атмосферного давления. Применение таких сосудов позволяет контролировать газообразные реагенты, такие как водород, аммиак, триметиламин, метиламин, кислород и т.п., а также позволяет защитить протекающую реакцию от действия воздуха или влажности. Сказанное выше не является исчерпывающим, и представляется очевидным, что специалистом-химиком могут быть разработаны и могут применяться дополнительные или альтернативные методы, условия, реакции или системы.
Приведенные в качестве иллюстрации реакции, как правило, проводят в диапазоне температур от -25° С до температуры дефлегмации растворителя в атмосфере инертного газа, такого как азот или аргон. Растворитель или смесь растворителей может широко варьироваться в зависимости от реагентов и других условий, и они могут включать указанные полярные или биполярные апротонные растворители или смеси этих растворителей. Реакции, например, такие как реакции металлирования или образования аниона с помощью сильных оснований, при необходимости можно проводить при пониженных температурах, таких как температура сухого льда/ацетона или жидкого азота.
В некоторых случаях амины, такие как триэтиламин, пиридин или другие нереакционноспособные основания, могут служить реагентами и/или растворителями и/или сорастворителями. В некоторых случаях в этих реакциях и в других реакциях, приведенных на указанных схемах, для поддержания или сохранения групп в других частях молекулы (молекул) в положениях, в которых не требуется наличие реакционноспособного(ых) центра (ов), применяются защитные группы. Примеры таких групп, которые специалист может поддерживать или сохранять, включают амины, другие гидроксильные группы, тиолы, кислоты и т.п. Такие защитные группы могут включать ацильные группы, арилалкильные группы, карбамоильные группы, простые эфиры, простые алкоксиалкильные эфиры, простые циклоалкилоксиэфиры, арилалкильные группы, силильные группы, включая тризамещенные силильные группы, сложноэфирные группы и т.п. Примеры таких защитных групп включают ацетил, трифторацетил, тетрагидропиран (ТГП), бензил, трет-бутоксикарбонил (ВОС или т-ВОС), бензилоксикарбонил (Z или КБЗ), трет-бутилдиметилсилил (ТБДМС) или метоксиэтоксиметилен (МЭМ). Получение таких защищенных соединений, а также метод удаления защитных групп хорошо известны в данной области. Защитные группы также могут применяться в качестве заместителей в рассматриваемых соединениях, которые скорее используются в качестве лекарства, а не синтетического промежуточного продукта.
Во многих реакциях и процессах принимают участие основания, которые могут выступать в качестве реактантов, реагентов, депротонирующих агентов, акцепторов кислоты, солеобразующих реагентов, растворителей, сорастворителей и т.п. Основания, которые могут применяться, включают, например, гидроксиды металлов, такие как гидроксид натрия, калия, лития, цезия или магния, оксиды, например оксиды натрия, калия, лития, кальция или магния, карбонаты металлов, такие как карбонаты натрия, калия, лития, цезия, кальция или магния, бикарбонаты металлов, такие как бикарбонат натрия или бикарбонат калия, первичные (I° ), вторичные (II° ) или третичные (III° ) органические амины, такие как алкиламины, арилалкиламины, алкиларилалкиламины, гетероциклические амины или гетероарильные амины, гидроксиды аммония или гидроксиды четвертичного аммония. Не ограничивающие объем изобретения примеры таких аминов включают триэтиламин, триметиламин, диизопропиламин, метилдиизопропиламин, диазабициклононан, трибензиламин, диметилбензиламин, морфолин, N-метилморфолин (N-MM), N,N’-диметилпиперазин, N-этилпиперидин, 1,1,5,5-тетраметилпиперидин, диметиламинопиридин, пиридин, хинолин, тетраметилэтилендиамин и т.п. Не ограничивающие объем изобретения примеры гидроксидов аммония, обычно полученных из аминов и воды, могут включать гидроксид аммония, гидроксид триэтиламмония, гидроксид триметиламмония, гидроксид метилдиизопропиламмония, гидроксид трибензиламмония, гидроксид диметилбензиламмония, гидроксид морфолиния, гидроксид N-метилморфолиния, гидроксид N,N’-диметилпиперазиния, гидроксид N-этилпиперидиния и т.п. В качестве не ограничивающих объем изобретения примеров гидроксидов четвертичного аммония могут служить гидроксид тетраэтиламмония, гидроксид тетраметиламмония, гидроксил диметилдиизопропиламмония, гидроксид бензилметилдиизопропиламмония, гидроксид метилдиазабициклонониламмония, гидроксид метилтрибензиламмония, гидроксид N,N-диметилморфолиния, гидроксид N,N,N’,N’-тетраметилпиперазиния и гидроксид N-этил-N’-гeкcилпипepидиния и т.п.
Также пригодными реагентами являются гидриды, амиды и алкоголяты металлов, такие как гидрид кальция, гидрид натрия, гидрид калия, гидрид лития, гидрид алюминия, гидрид диизобутилалюминия (ДИБАЛ), метоксид натрия, трет-бутоксид калия, этоксид кальция, этоксид магния, амид натрия, диизопропиламид калия и т.п. Металлорганические депротонирующие агенты, такие как реагенты на основе алкила или арила лития, такие как метиллитий, фениллитий, трет-бутиллитий, ацетилидлитий или бутиллитий, реагенты Гриньяра, такие как бромметилмагний или хлорметилмагний, кадмийорганические реагенты, такие как диметилкадмий и т.п., также могут служить в качестве оснований, способствующих образованию солей, или катализаторов реакций. Гидроксиды четвертичного аммония или смешанные соли также могут применяться для облегчения сочетаний фазового переноса или служат в качестве агентов фазового переноса. Фармацевтически приемлемые основания могут подвергаться взаимодействию с кислотами с получением рассматриваемых фармацевтически приемлемых солей. Также следует отметить, что оптически активные основания могут применяться для получения оптически активных солей, которые могут использоваться для оптических разделений.
Как правило, реакционная среда может включать один растворитель, смешанные растворители из одного или из различных классов или служить в качестве реагента в системе, состоящей из одного или из смешанных растворителей. Растворители могут быть протонными, непротоннными или биполярными апротонными. Не ограничивающие объем изобретения примеры протонных растворителей включают воду, метанол (МеОН), денатурованный или чистый 95%-ный или абсолютный этанол, изопропанол и т.п. Типичные непротонные растворители включают ацетон, тетрагидрофуран (ТГФ), диоксан, диэтиловый эфир, метил-трет-бутиловый эфир (ТБМЭ), ароматические соединения, такие как ксилол, толуол или бензол, этилацетат (ЭА), метилацетат, бутилацетат, трихлорэтан, метиленхлорид, этилендихлорид (ЭДХ), гексан, гептан, изооктан, циклогексан и т.п. Биполярные апротонные растворители включают такие соединения, как диметилформамид (ДМФ), диметилацетамид (ДМАц), ацетонитрил, ДМСО, триамид гексаметилфосфора (ГМФА), нитрометан, тетраметилмочевину, N-метилпирролидон и т.п. Не ограничивающими объем изобретения примерами реагентов, которые могут использоваться в качестве растворителей или части системы смешанных растворителей, являются органические или неорганические моно- или мультипротонные кислоты или основания, такие как соляная кислота, фосфорная кислота, серная кислота, уксусная кислота, муравьиная кислота, лимонная кислота, янтарная кислота, триэтиламин, морфолин, N-метилморфолин, пиперидин, пиразин, пиперазин, пиридин, гидроксид калия, гидроксид натрия, спирты или амины, предназначенные для получения сложных эфиров или амидов, или тиолы, предназначенные для получения рассматриваемых продуктов, и т.п.
Получение рассматриваемых в настоящем описании соединений может требовать окисления азота или серы с получением производных в виде N-оксидов либо сульфоксидов или сульфонов. Реагенты для этого процесса могут включать в виде не ограничивающих объем изобретения примеров пероксимоносульфат (оксон, OXONE®), перекись водорода, мета-хлорпербензойную кислоту (м-ХПБК), пербензойную кислоту, перуксусную кислоту, пермолочную кислоту, перекись трет-бутила, гипохлорит трет-бутила, гипохлорит натрия, хлорноватистую кислоту, мета-перйодат натрия, перйодную кислоту и т.п., причем для получения сульфонов и сульфоксидов наиболее пригодны более слабые агенты. Могут быть выбраны либо чистые, либо смешанные протонные, непротоннные, биполярные апротоннные растворители, например смесь метанол/вода.
Окисление можно проводить при температуре от примерно -78° С до примерно 50° С, как правило, в диапазоне от -10° С до примерно 40° С. Сульфоксиды наиболее целесообразно получать, используя один эквивалент окислителя. При применении более активных окислителей может оказаться целесообразным, но не необходимым, чтобы реакции протекали в атмосфере инертного газа с дегазацией растворителей или без нее. Следует отметить, что окисление сульфидов с получением сульфонов можно проводить в одну или в две стадии через сульфоксид в зависимости от требований, предъявляемых специалистом.
Восстановление является хорошо известным процессом в данной области, причем наиболее распространенным методом является гидрирование. В таких случаях (каталитическое восстановление) могут применяться металлические катализаторы, такие как Rh, Pd, Pt, Ni или т.п. с дополнительным носителем, таким как уголь, карбонат бария и т.п., или без него. Растворители при необходимости могут представлять собой протонные или непротонные чистые растворители или смешанные растворители. Реакции восстановления можно проводить при давлении от атмосферного до давления в несколько атмосфер, причем предпочтительным является давление от атмосферного до примерно 40 фунтов на квадратный дюйм (фунтов/кв.дюйм) или очень высокое давление, достигаемое с помощью устройства для гидрирования, хорошо известного в данной области.
Восстановительное алкилирование аминов или активных метиленовых соединений также является приемлемым методом получения соединений. Такие реакции алкилирования можно проводить в описанных выше условиях восстановительного гидрирования с использованием, например, альдегидов или кетонов. Реагенты, предназначенные для переноса гидрида, такие как цианборогидрид натрия, гидрид алюминия, алюмогидрид лития, боран, борогидрид натрия, гидрид диизобутилалюминия и т.п., также являются пригодными реагентами для восстановительного алкилирования. Аналогичным путем могут быть восстановлены ацильные группы с получением замещенных аминов.
Альтернативным методом алкилирования углерода или азота является прямое алкилирование. Такое алкилирование, как это хорошо известно в данной области, может заключаться в обработке активированного углерода, содержащего по меньшей мере один атом водорода, основанием с получением соответствующего аниона, в добавлении электрофильного реагента и в осуществлении SN2-взаимодействия. Подлежащий алкилированию амин обрабатывают аналогичным образом за исключением того, что при этом может не потребоваться депротонирования. Электрофилы включают производные галогенов, сложные эфиры сульфонатов, эпоксиды и т.п.
Основания и растворители для реакций алкилирования являются такими же, которые описаны ранее. Предпочтительными являются основания, при использовании которых конкуренция с электрофилом является минимальной. Дополнительными предпочтительными основаниями являются гидриды металлов, анионы амидов или металлорганические основания, такие как н-бутиллитий. Приемлемыми являются описанные растворители, смеси растворителей или смеси растворителей/реагентов, при этом непротонные или биполярные апротонные растворители, такие как ацетон, ацетонитрил, ДМФ и т.п, являются предпочтительными примерами.
Кислоты используются во многих реакциях во время различных процессов синтеза. Например, для удаления защитной группы ТГП с получением гидроксамовой кислоты. Кислота может быть моно-, ди- или трипротонной органической или неорганической кислотой. Примеры кислот включают соляную кислоту, фосфорную кислоту, серную кислоту, уксусную кислоту, муравьиную кислоту, лимонную кислоту, янтарную кислоту, бромистоводородную кислоту, фтористоводородную кислоту, угольную кислоту, фосфористую кислоту, пара-толуолсульфоновую кислоту, трифторметансульфоновую кислоту, трифторуксусную кислоту (ТФК), дифторуксусную кислоту, бензойную кислоту, метансульфоновую кислоту, бензолсульфоновую кислоту, 2,6-диметилбензолсульфоновую кислоту, трихлоруксусную кислоту, нитробензойную кислоту, динитробензойную кислоту, тринитробензойную кислоту и т.п. Они также могут представлять собой кислоты Льюиса, такие как хлорид алюминия, боронтрифторид, пентафторид сурьмы и т.п. Кислоты в форме протонов также могут использоваться для гидролиза сложных эфиров, амидов и т.п., а также в качестве катализаторов в реакциях обмена.
Защищенную карбоновую кислоту в виде эфира или амида можно превращать также в гидроксамовую кислоту или в производное гидроксамовой кислоты, такое как простой O-арилалкиловый эфир или простой O-циклоалкоксиалкиловый эфир. В том случае когда используют гидроксиламин, гидроксамовая кислота может быть получена непосредственно после обработки эфира или амида одним или несколькими эквивалентами гидрохлорида гидроксиламина при комнатной температуре или при повышенной температуре в растворителе или в растворителях, как правило, в протонном или в частично протонном растворителе, таком как указанные выше. Это процесс обмена может, кроме того, происходить при добавлении другой кислоты в качестве катализатора. В альтернативном варианте для получения in situ гидроксиламина из его гидрохлорида может применяться основание, такое как спиртовой раствор соли, используемое в качестве растворителя, например метоксид натрия в метаноле, которое может вступать в реакции обмена с эфиром или амидом. Как указывалось выше, реакция обмена может происходить с участием защищенного гидроксиламина, такого как тетрагидропиранилгидроксиамин (ТГПОНН2), бензилгидроксиламина (BnONH2) и т.п., в том случае когда продуктами реакций, которые приведены на схемах А, Б и В, являются производные гидроксамовой кислоты, защищенные тетрагидропиранилом (ТГП) или бензилом (Вn). Удаление защитных групп, если это требуется, например, после дополнительных трансформаций в другой части молекулы или после хранения, осуществляют стандартными методами, хорошо известными в данной области, такими как кислотный гидролиз ТГП-группы, как это описано выше, или восстановительное удаление бензильной группы водородом в присутствии металлического катализатора, такого как палладий, платина, палладий на угле или никель.
В том случае когда R20 обозначает гидроксил, т.е. когда промежуточным продуктом является карбоновая кислота, могут использоваться стандартные реакции сочетания. Например, кислота может быть превращена в хлорангидрид, смешанный ангидрид или активированный эфир, такой как гидроксибензотриазол, и подвергнута обработке гидроксиламином или защищенным гидроксиламином в присутствии неконкурирующего основания с получением ацилированного азотом соединения, которое является тем же самым продуктом, который описан выше. Сочетания такой природы хорошо известны в данной области и прежде всего в области, связанной с химией пептидов и аминокислот.
Рассматриваемые в данном описании соединения могут нести один или несколько асимметричных атомов углерода и поэтому могут существовать в форме оптических изомеров, энантиомеров, диастереоизомеров, а также в форме рацемических или нерацемических смесей. Соединение также может существовать в других изомерных формах, таких как орто-, мета- и пара-изомеры, цис- и транс-изомеры, син- и антиизомеры, Е- и Z-изомеры, таутомерные изомеры, альфа- и бэта-изомеры, осевые и экваториальные изомеры и изомеры, полученные в результате заторможенного вращения. Изомер может существовать в состоянии равновесия с другим изомером в организме млекопитающих или в другой опытной системе. Такое соединение также может существовать в виде системы с изомерным равновесием с растворителем или водой, например в виде гидратированното кетона или альдегида, что хорошо известно в данной области. Все такие изомеры подпадают под понятие "соединение по изобретению".
Описанные выше химические реакции, как правило, рассмотрены в понятиях их наиболее широкого возможного применения для получения соединений по изобретению. Иногда может оказаться, что реакции не применимы в том виде, как они описаны, для каждого соединения, подпадающего под объем изобретения. Соединения, к которым это относится, могут быть легко определены специалистами в данной области. Во всех таких случаях либо реакции можно успешно осуществлять с помощью обычных модификаций, известных специалистам в данной области, например соответствующей защитой взаимодействующих групп, заменой на альтернативные обычные реагенты, общепринятой модификацией условий реакции и т.п., либо для получения соответствующих рассматриваемых соединений могут использоваться другие реакции, указанные в данном описании или являющиеся общепринятыми.
Схема 1
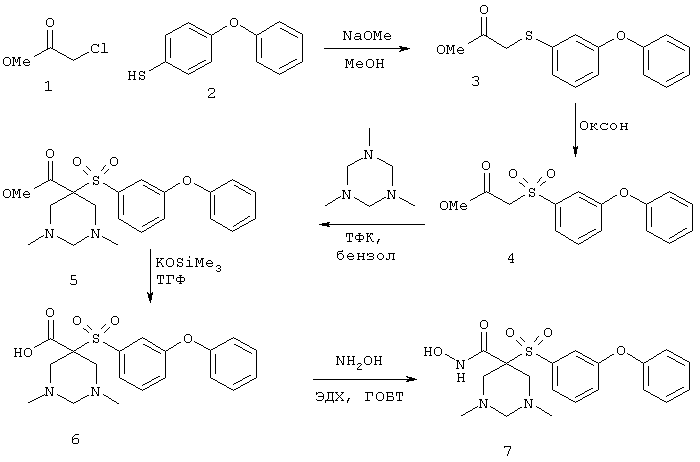 Схема 2
Схема 2
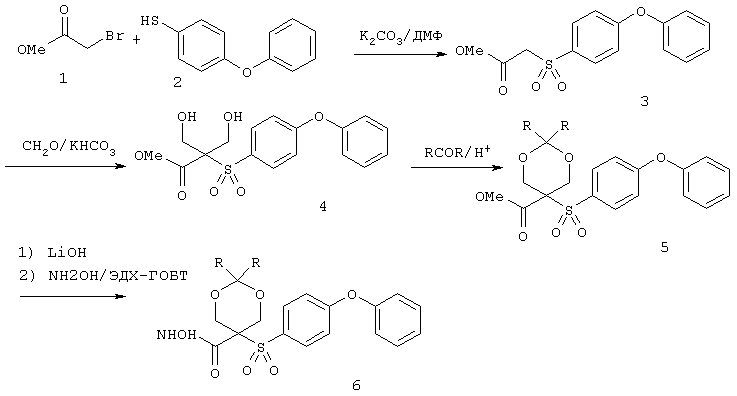
Схема 3
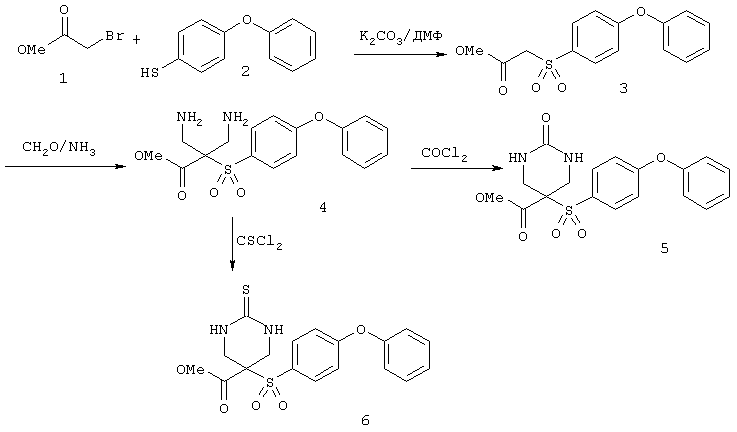 Схема 4
Схема 4
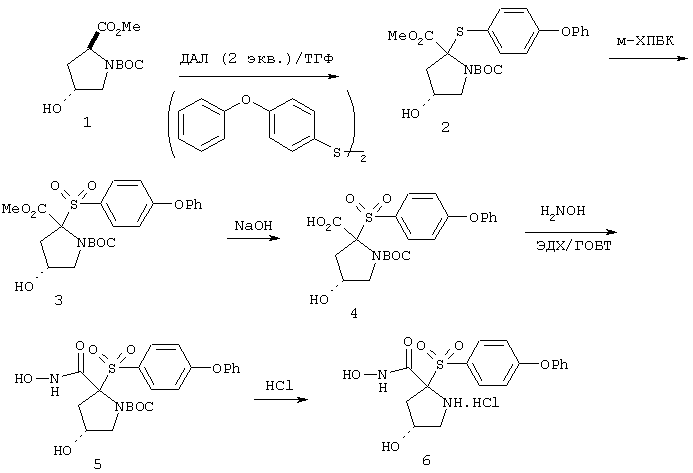
Схема 5
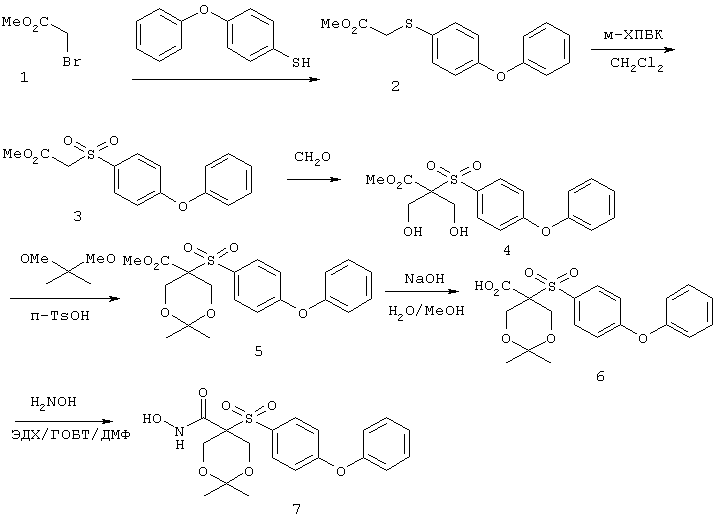
Схема 6
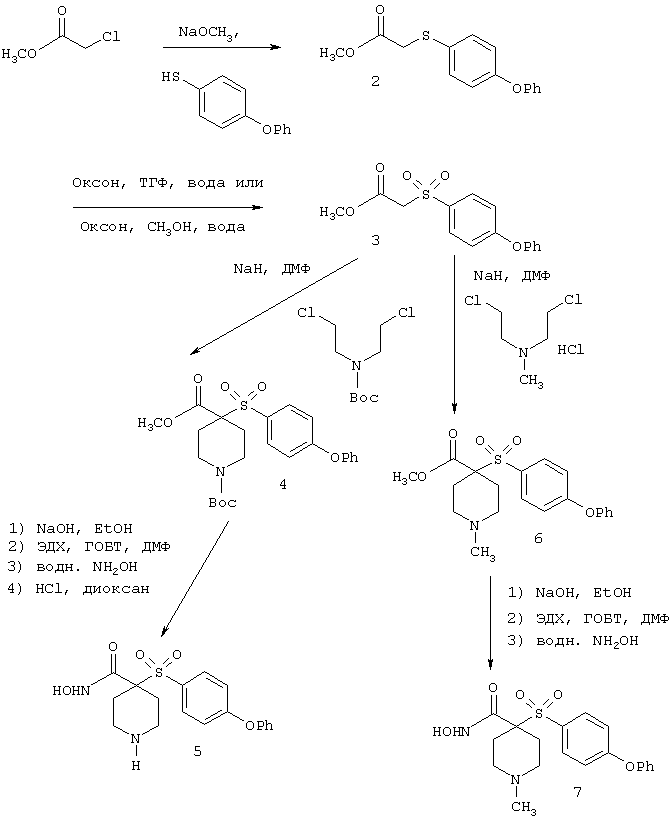
Схема 7
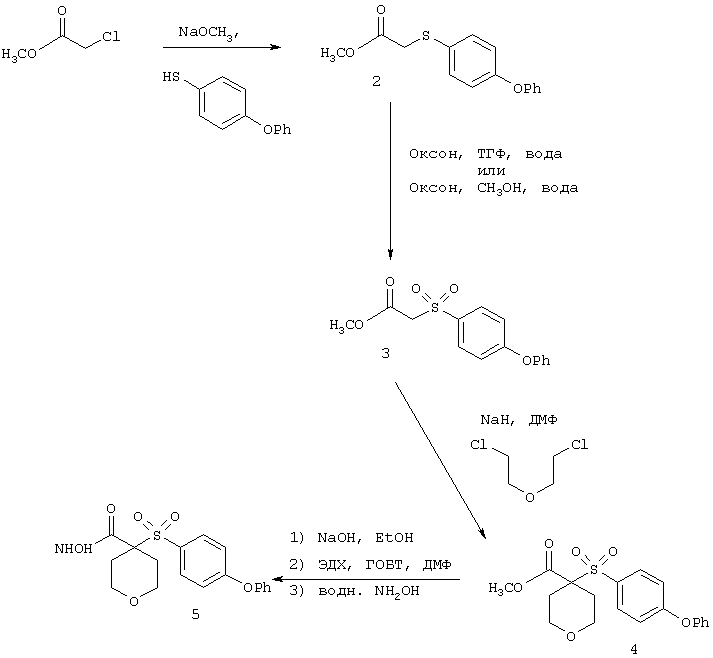
Схема 8
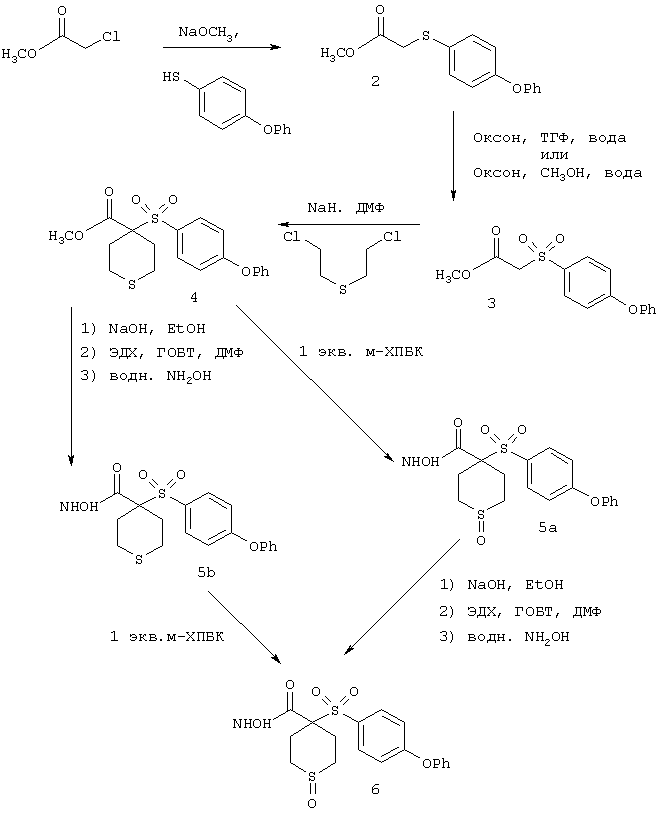
Схема 9
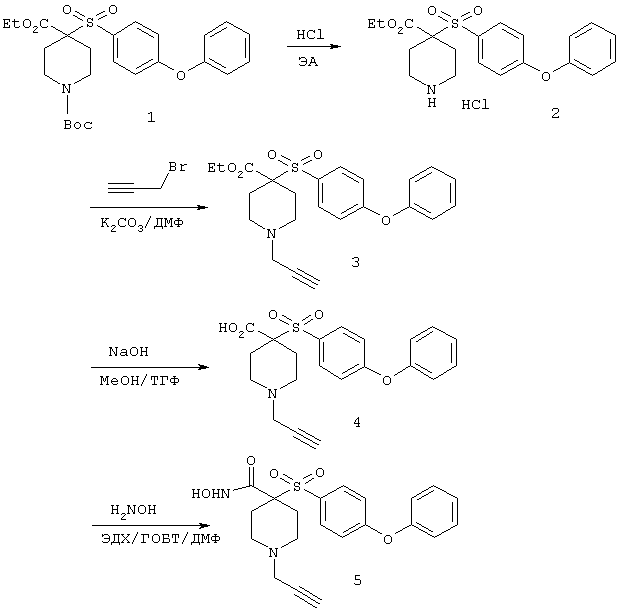
Схема 10
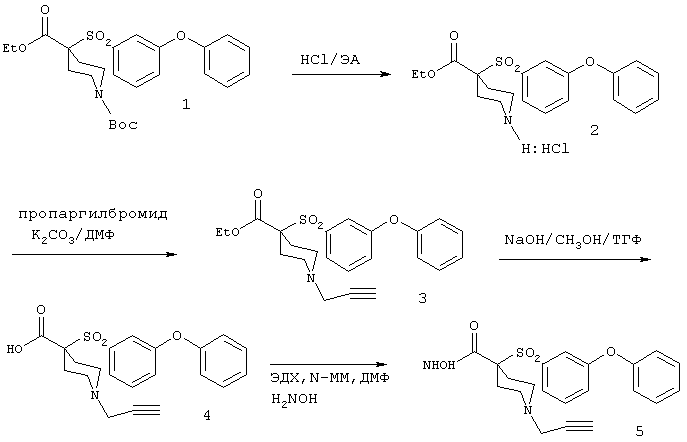 Схема 11
Схема 11

Схема 12
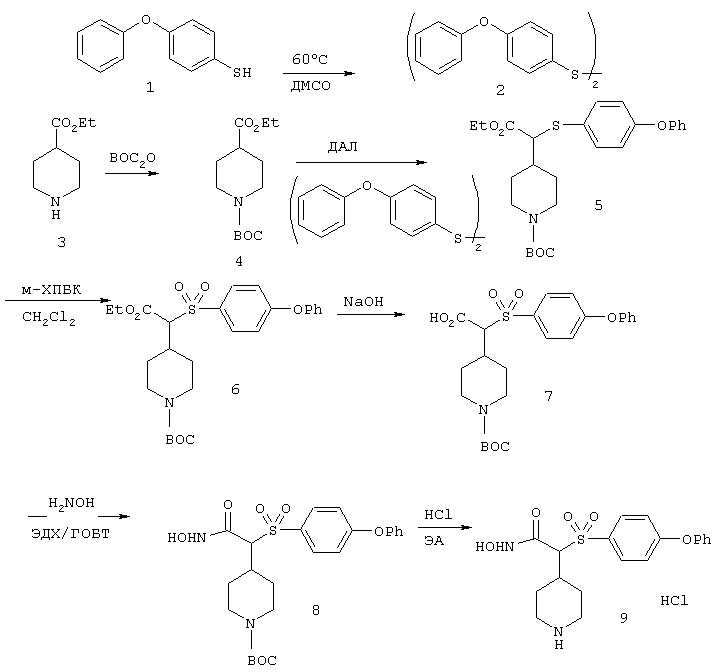
Схема 13
Аналогичным путем могут быть получены следующие аналоги

Схема 14
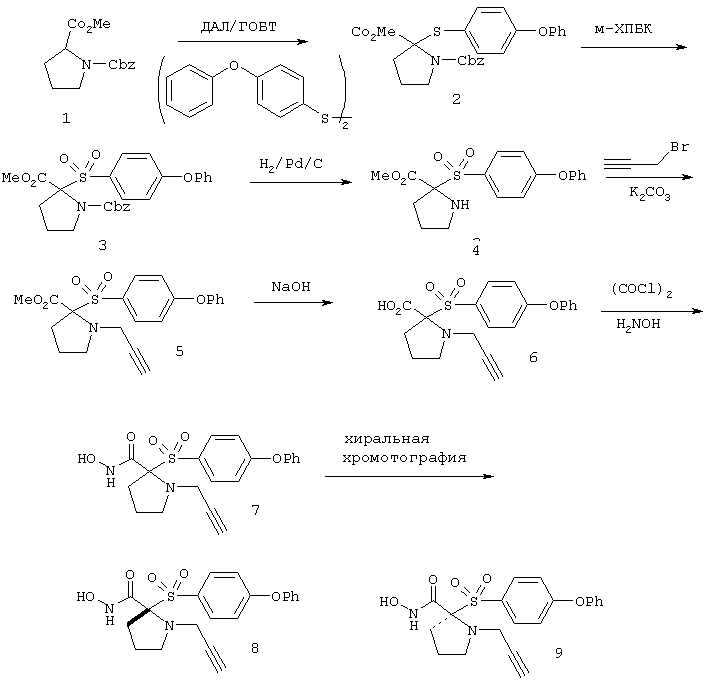
Схема 15
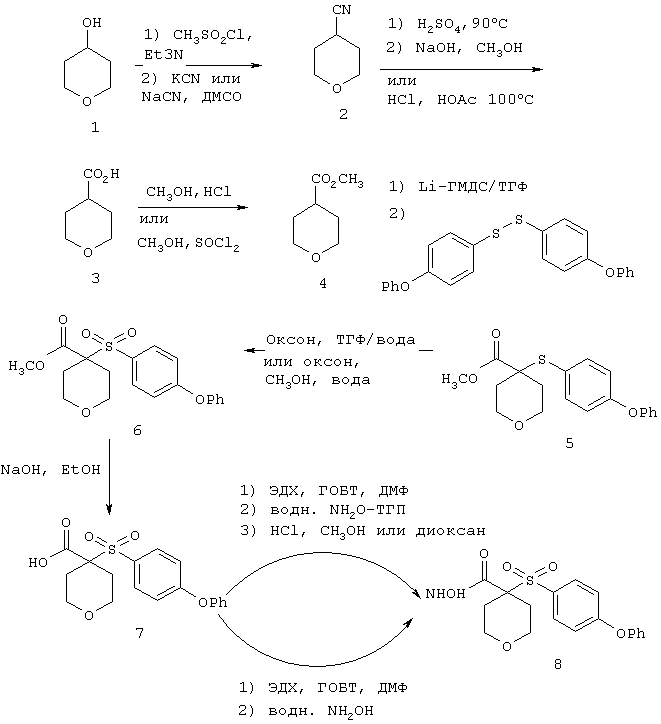
Схема 16
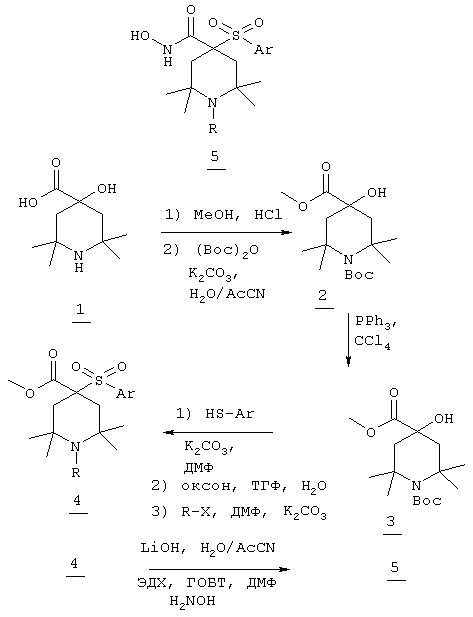
Схема 17
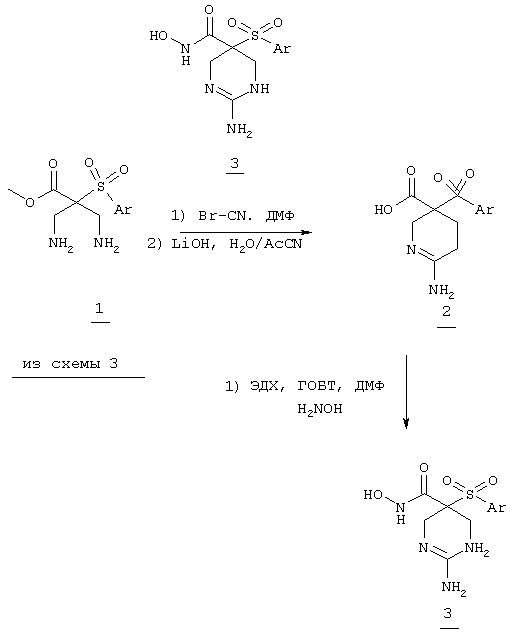
Схема 18
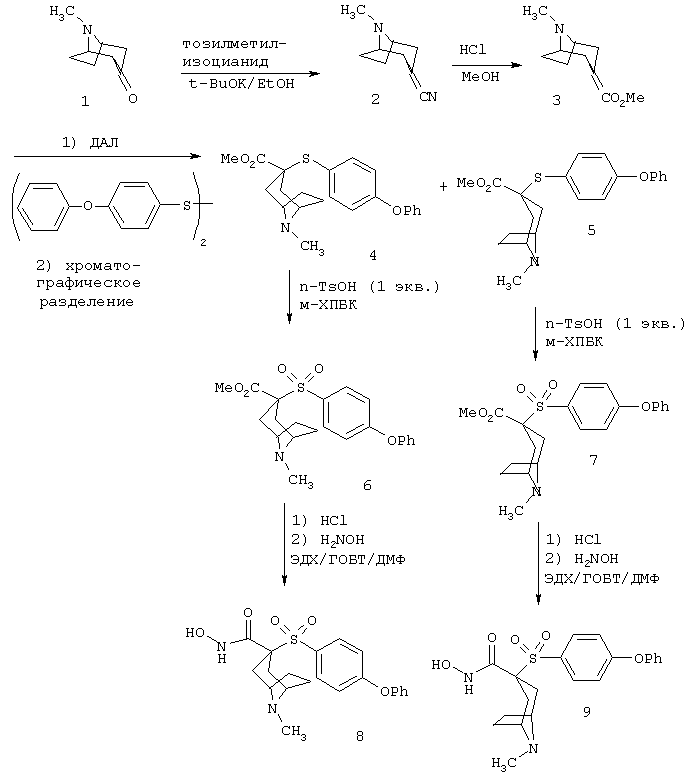
Схема 19
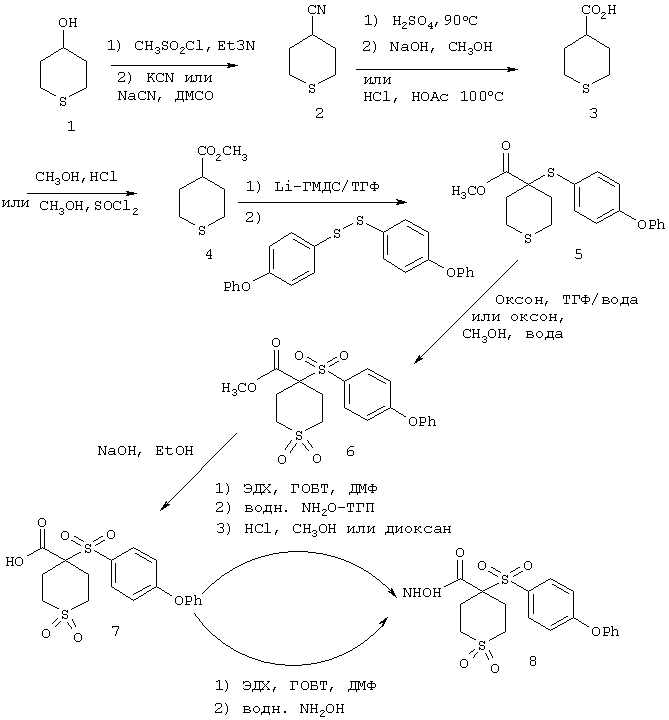
В приведенных ниже таблицах 1-150 представлены несколько рассматриваемых производных ароматических сульфонгидроксамовых кислот, обладающих ингибирующей активностью, или структурные формулы, иллюстрирующие группы заместителей. Каждая группа соединений проиллюстрирована в виде общей формулы или формул, за которой(ыми) следуют серии предпочтительных фрагментов или групп, представляющих собой различные заместители, которые могут быть присоединены в положении, точно указанном на общей формуле. Соответствующие заместителям символы, например R1, R2 и R3, имеют значения, приведенные в каждой таблице и, как правило, не имеют указанных ранее значений. Одна или две связи (волнистые линии) показаны для тех заместителей, для которых следует указать соответствующие положения присоединения в проиллюстрированном соединении. Эта система является хорошо известной для передачи информации в области химии и широко используется в научных статьях и других документах. Например, в таблице 2 R1 и R2 вместе с атомами, к которым они присоединены, обозначают вариабельную группу с приведенными в этой таблице структурными элементами, которые могут замещать одновременно R1 и R2.
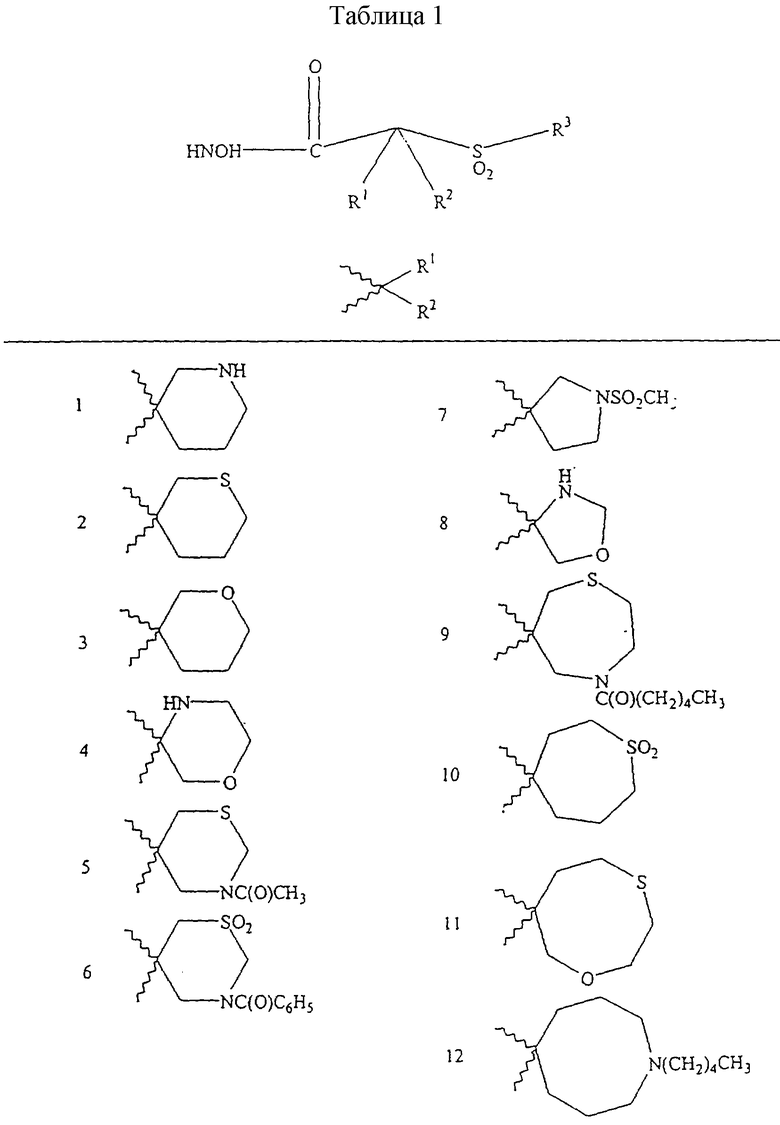
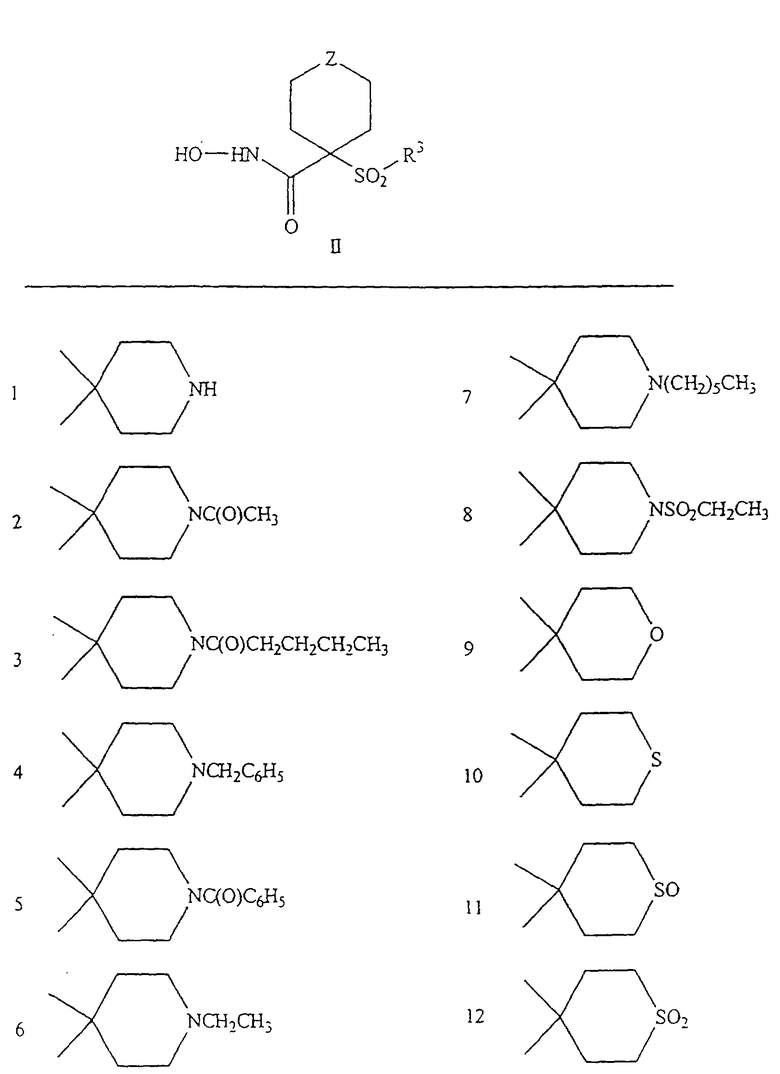
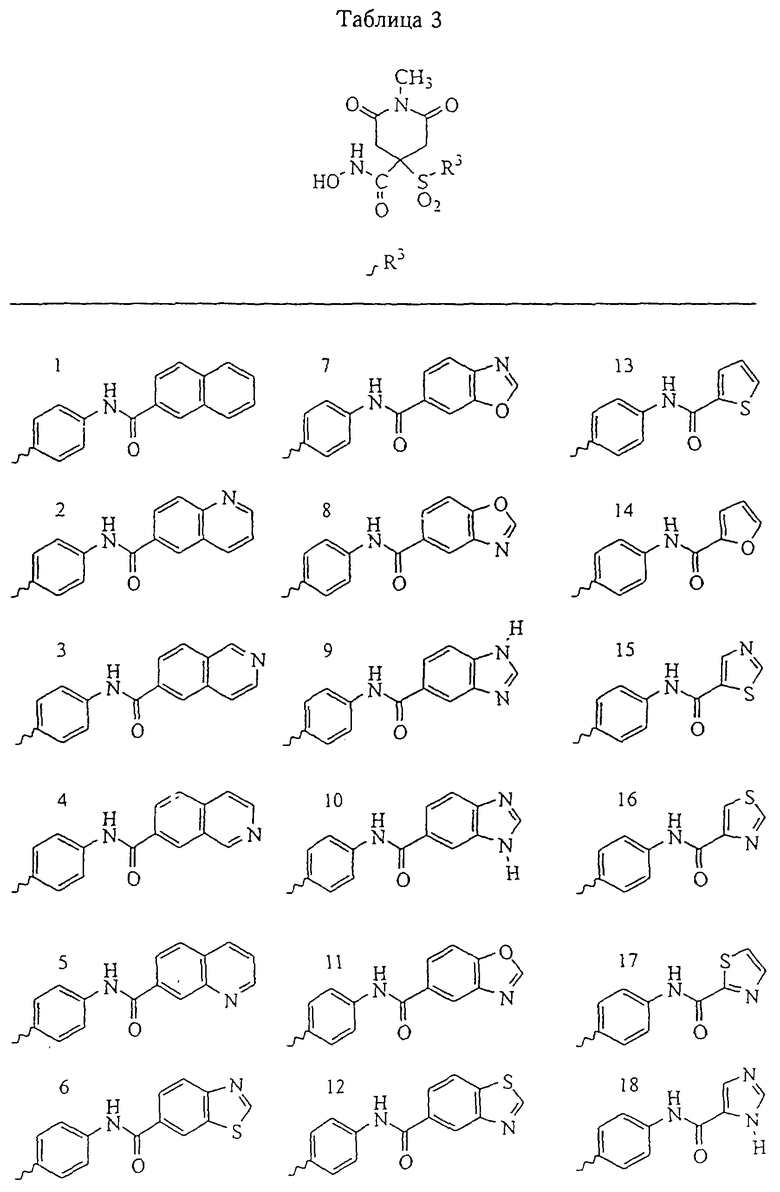
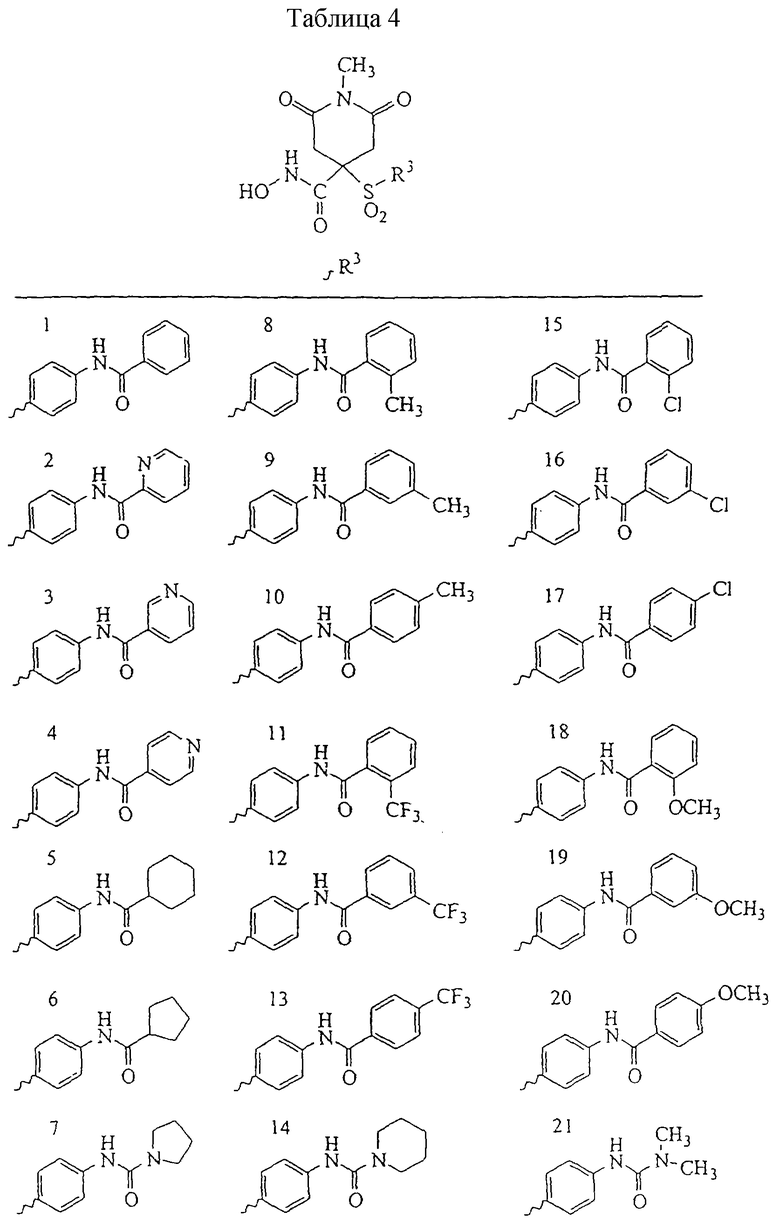
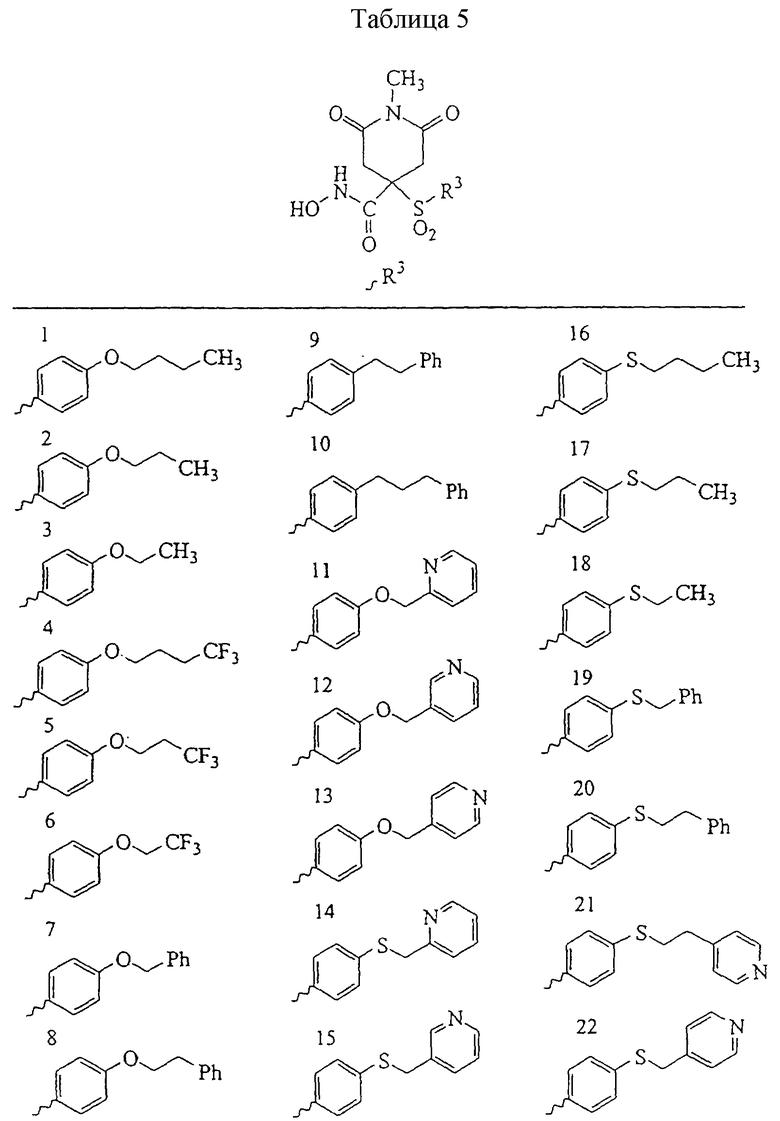
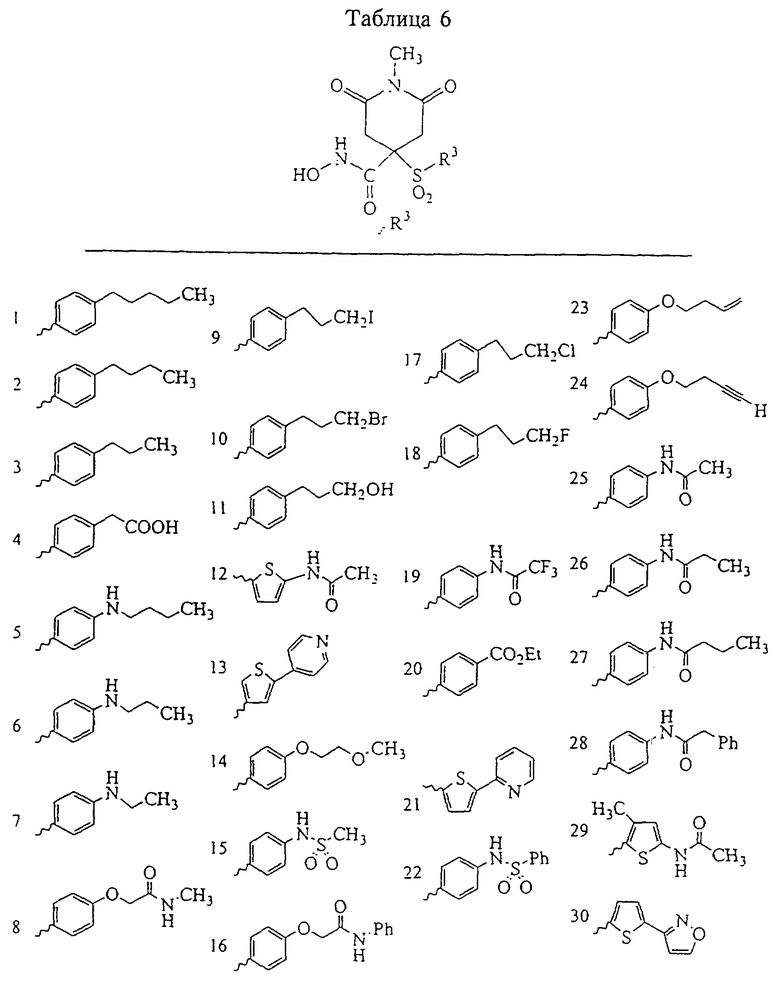
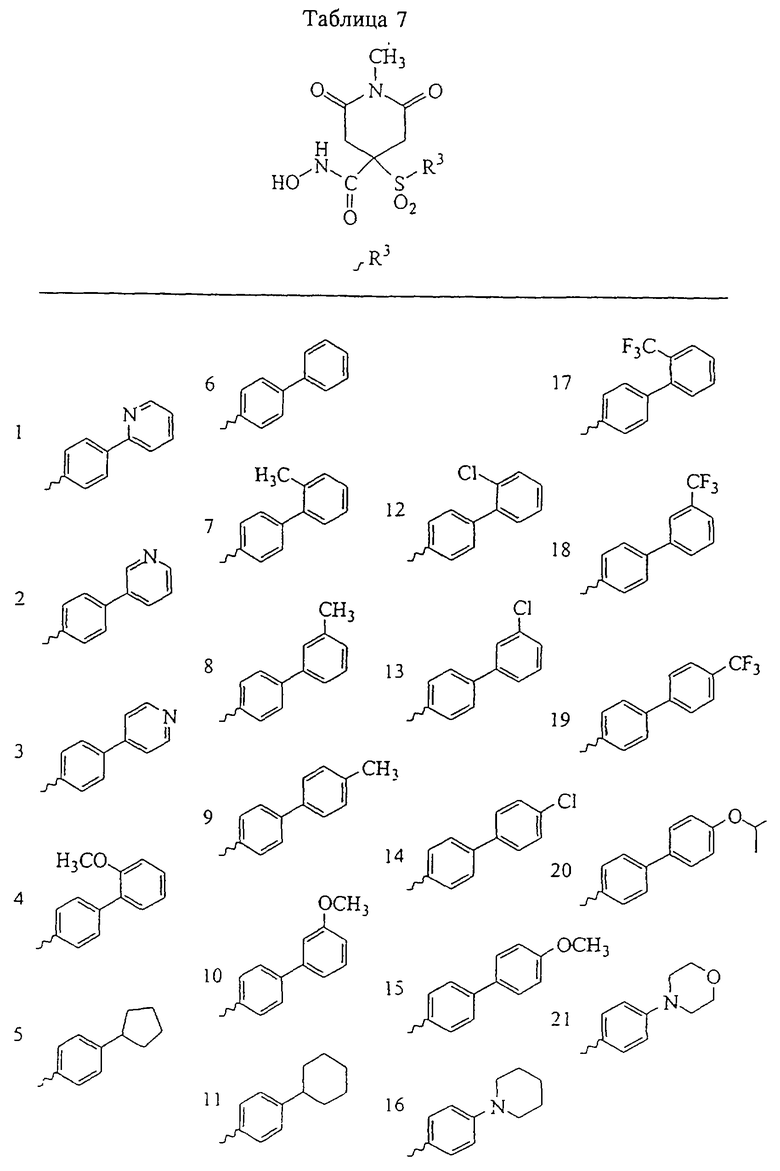
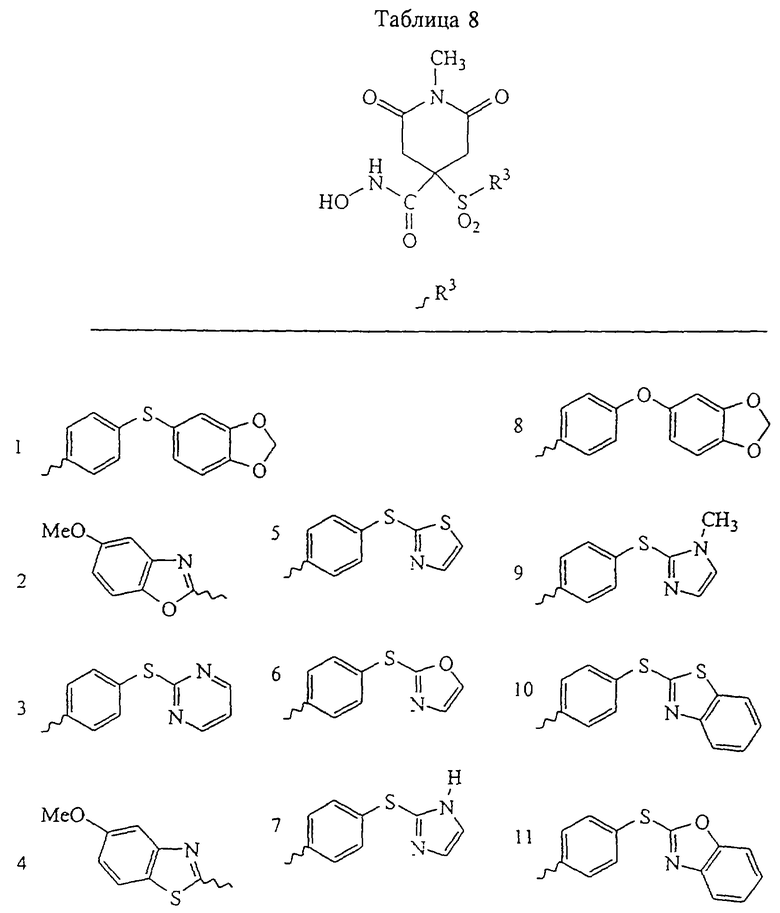
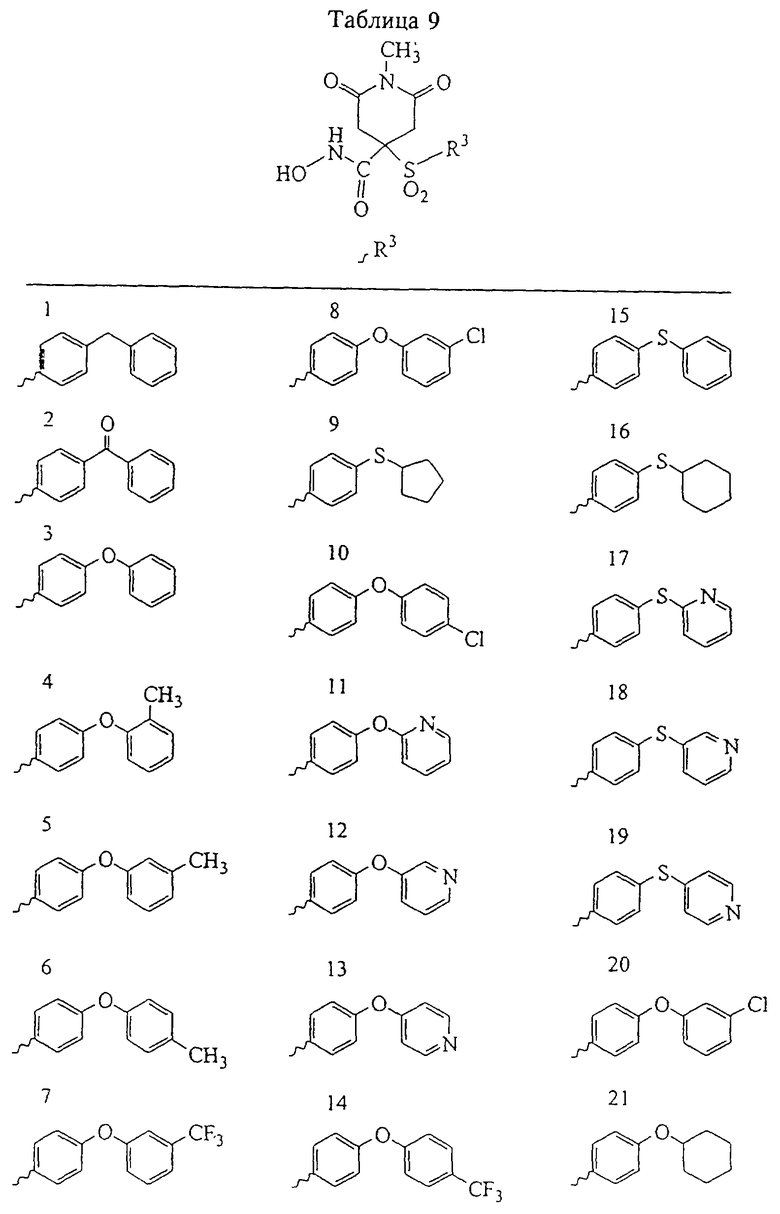
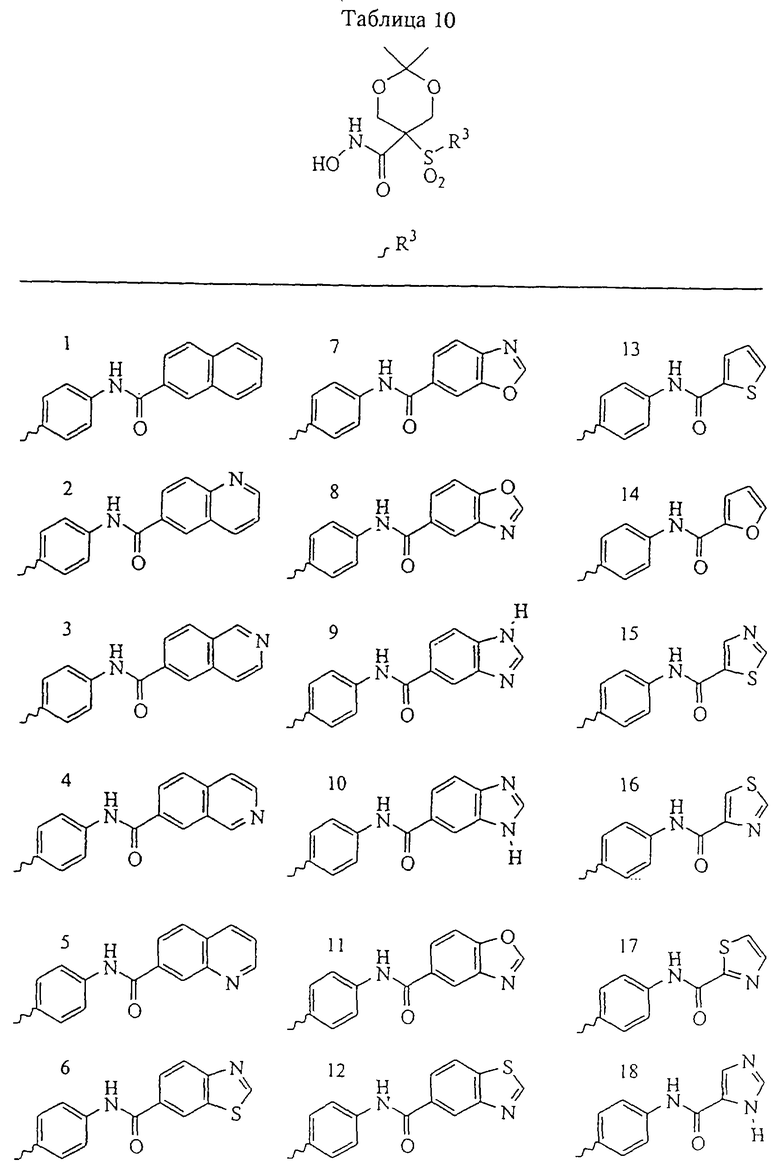
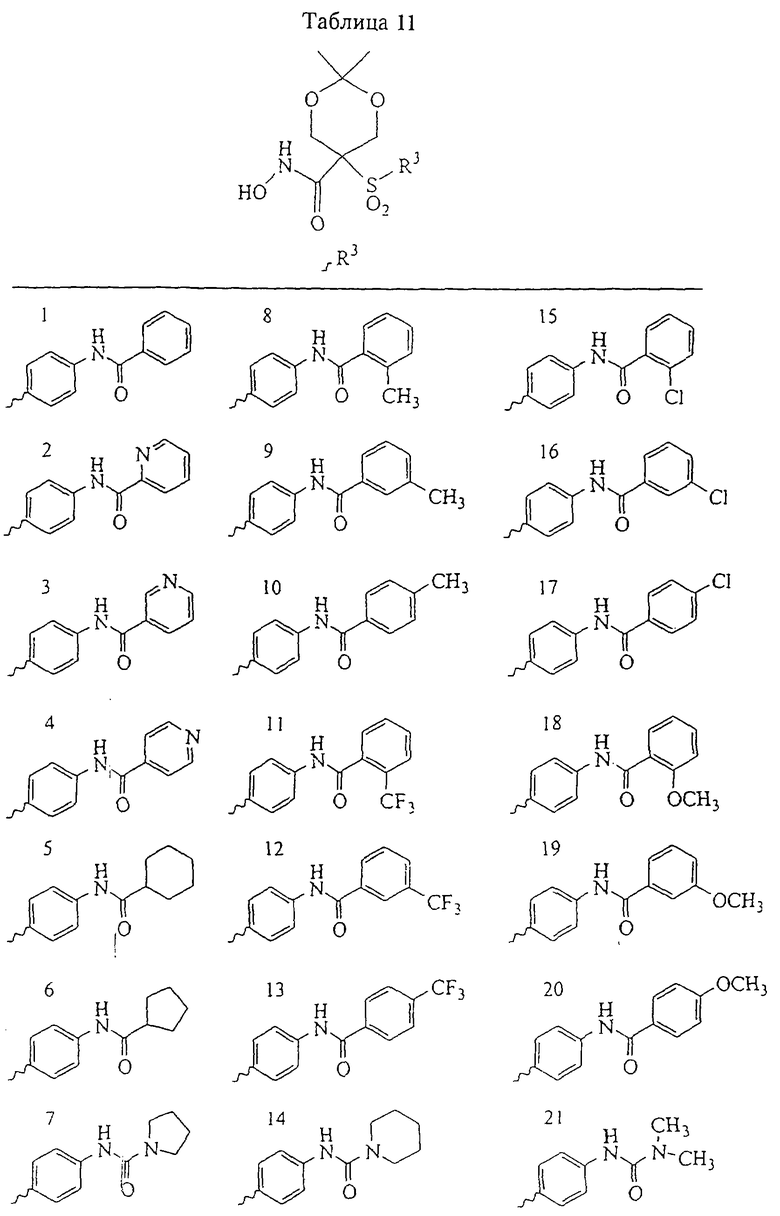
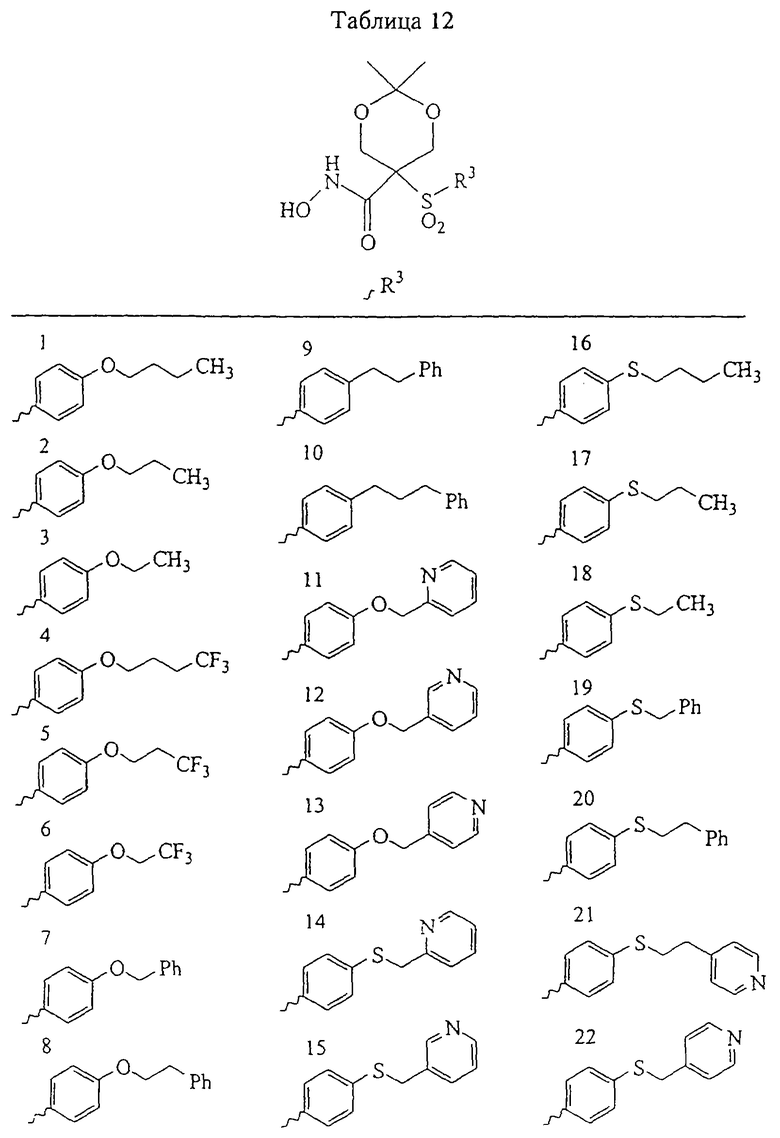
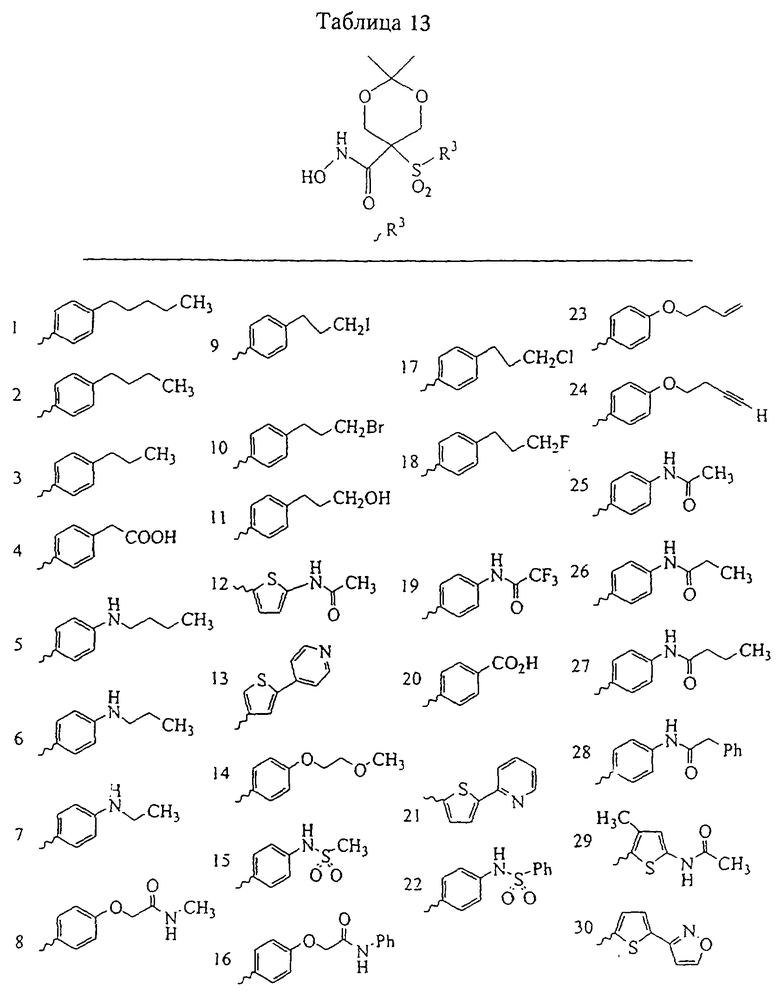
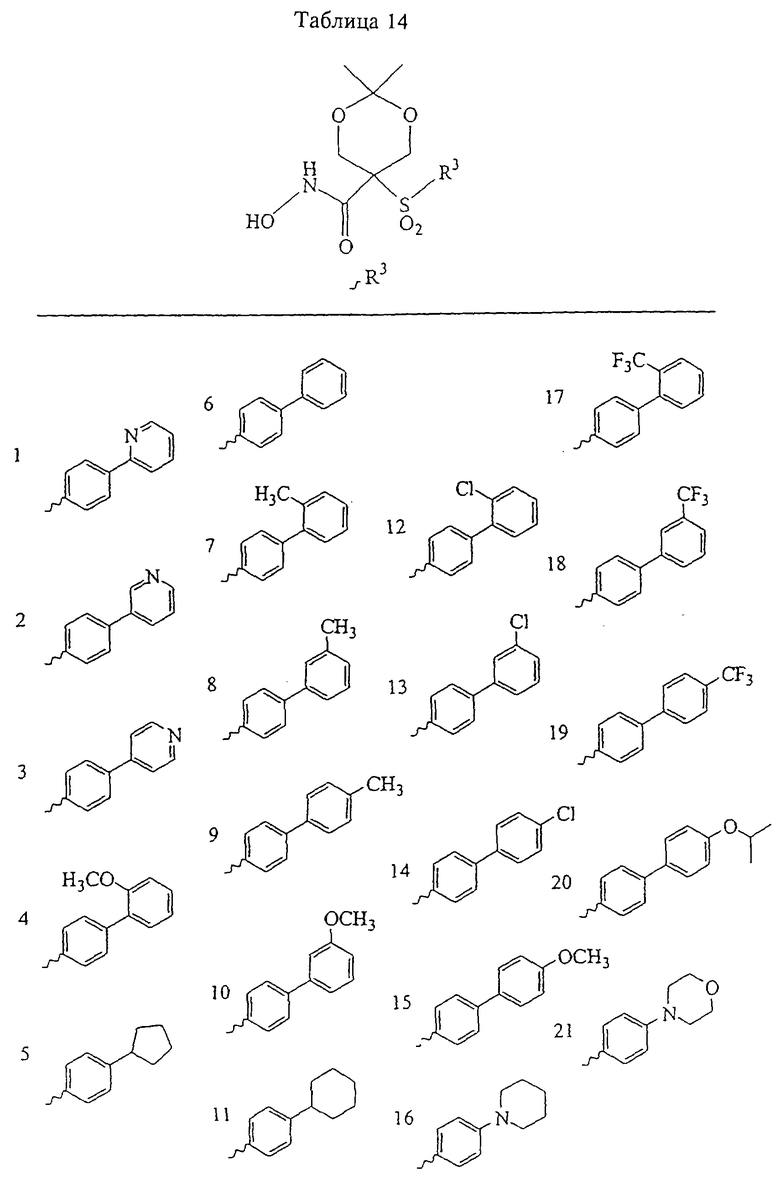
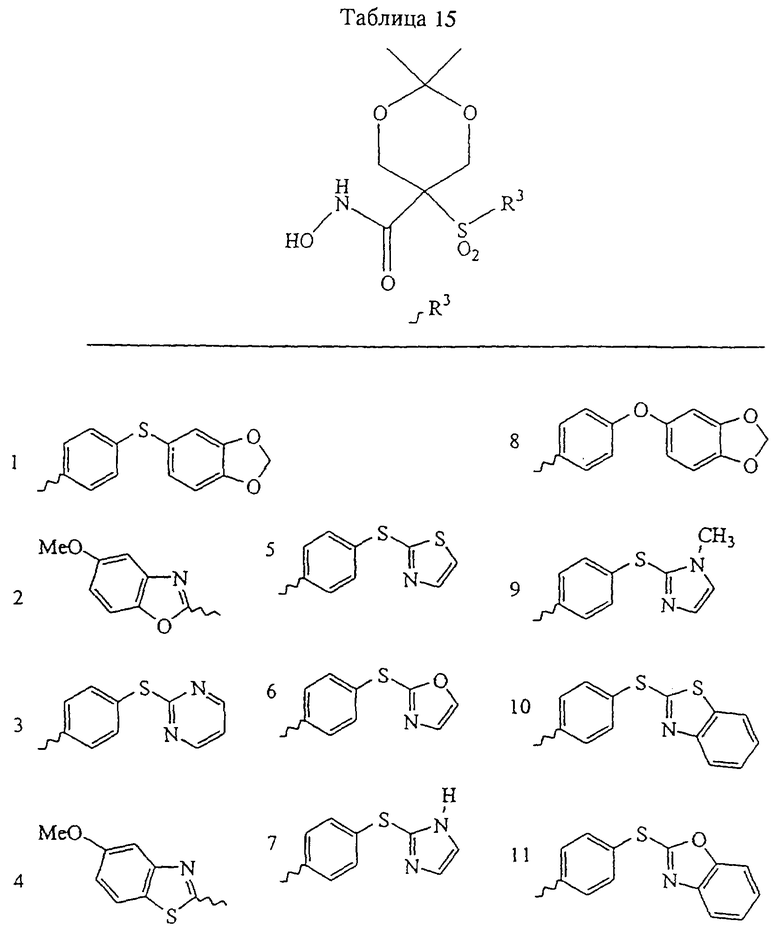
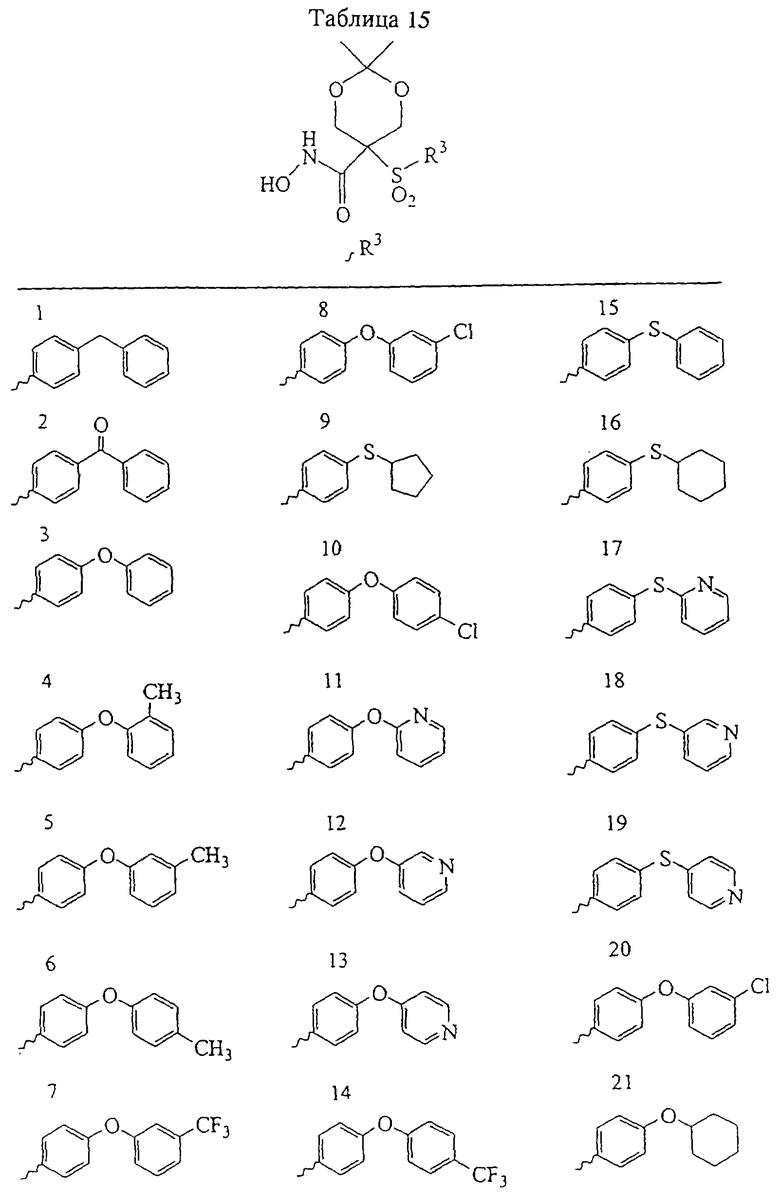

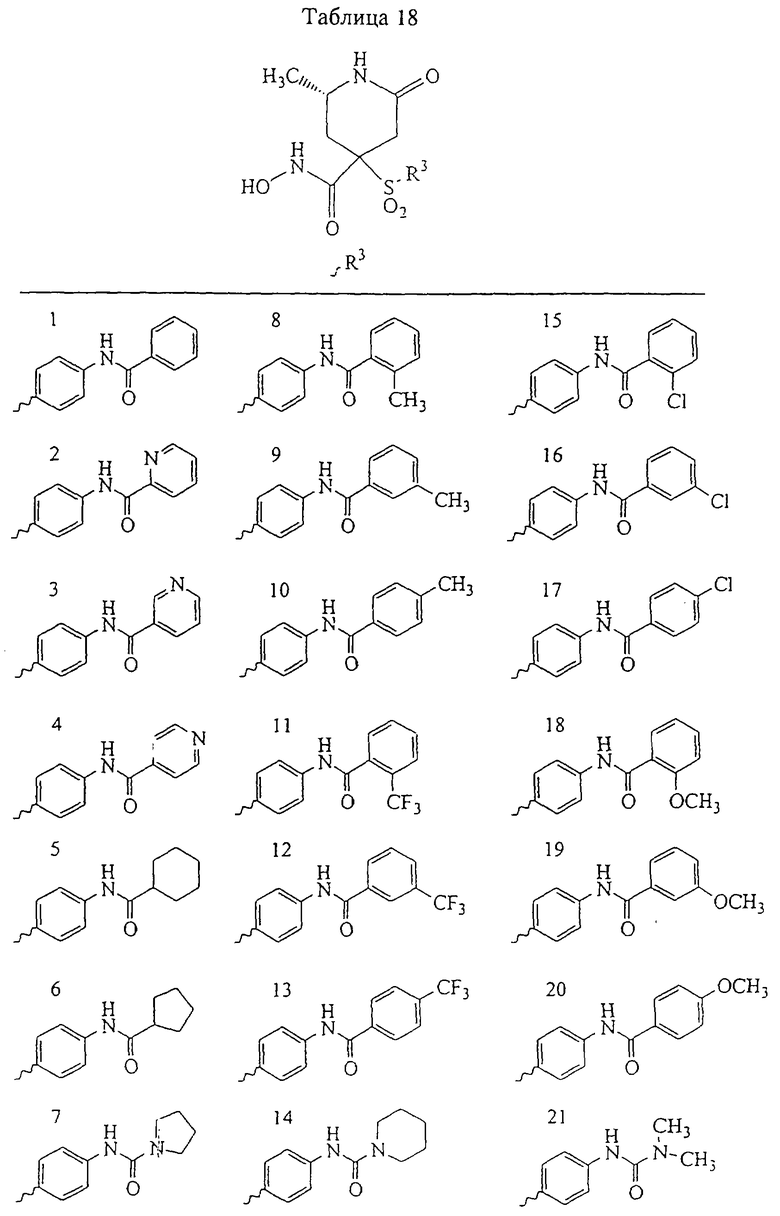
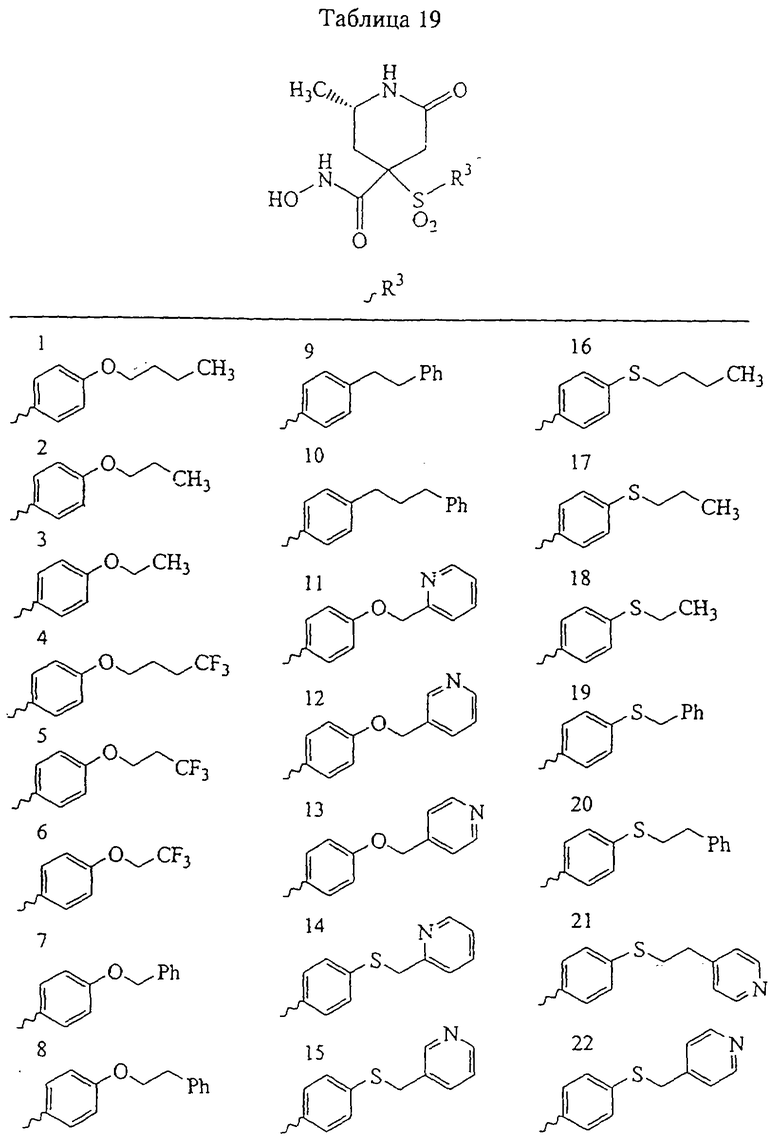
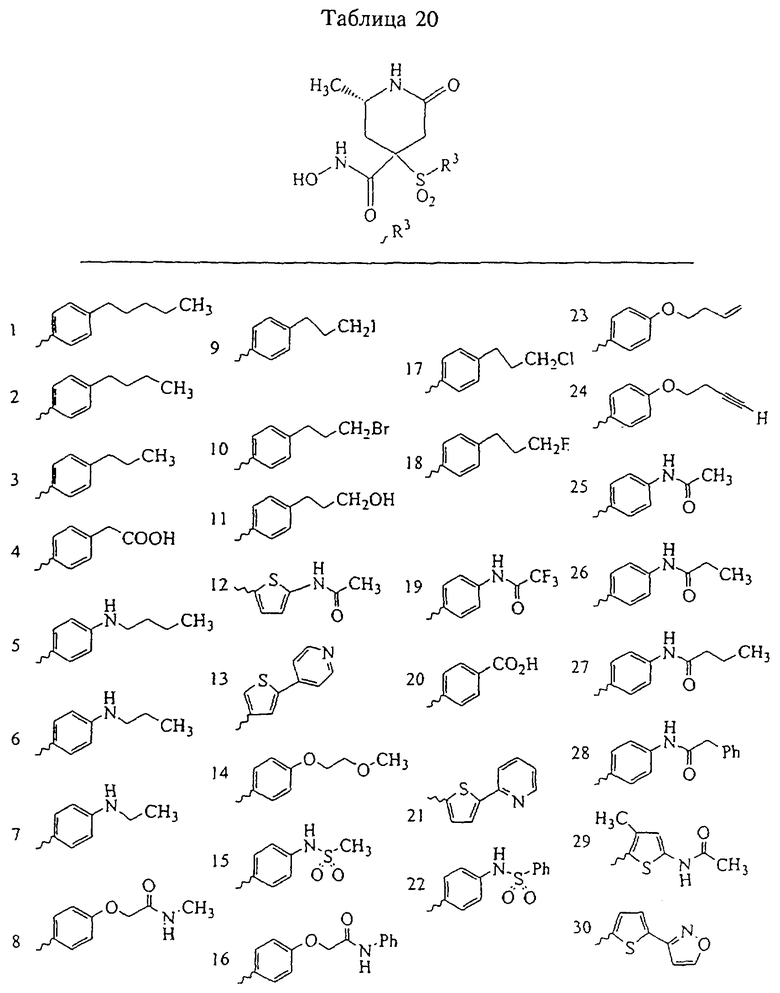
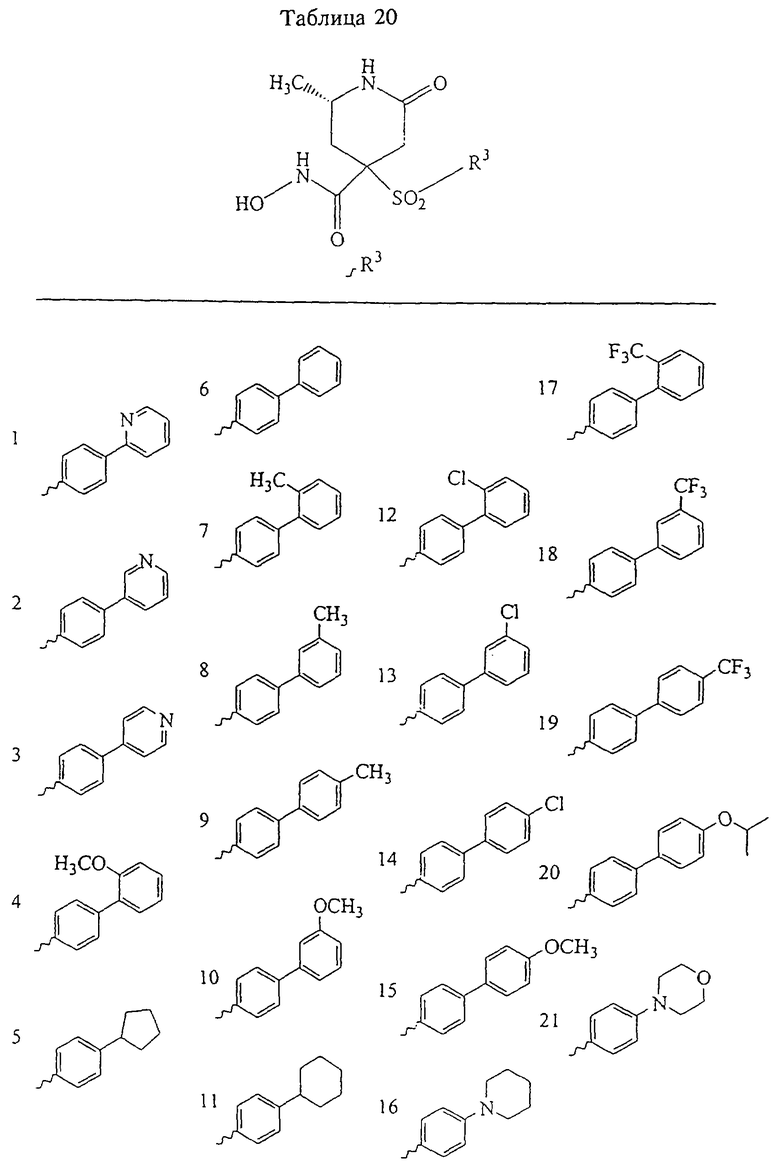
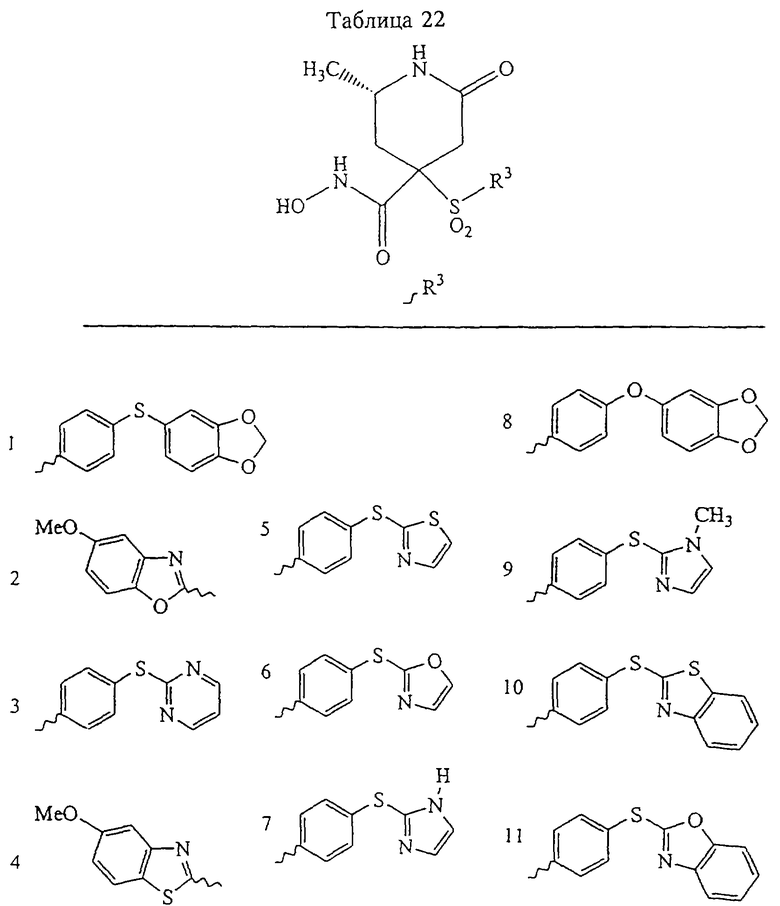
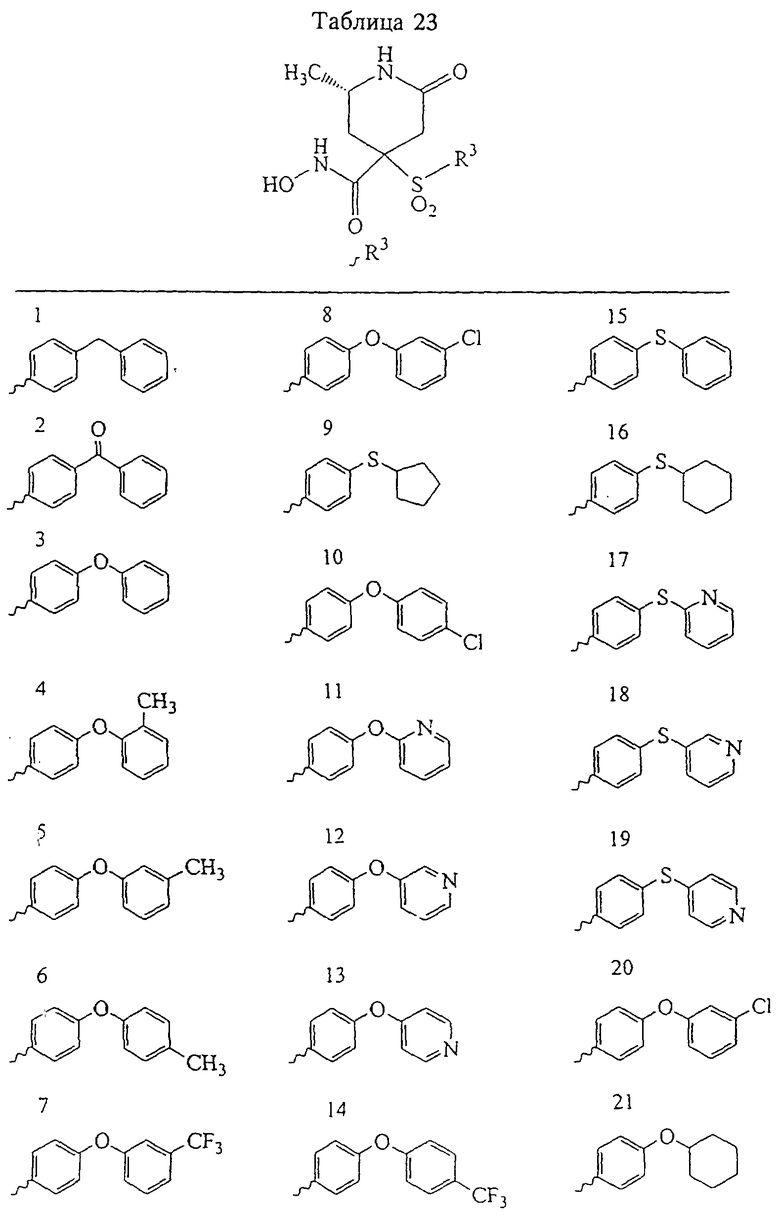
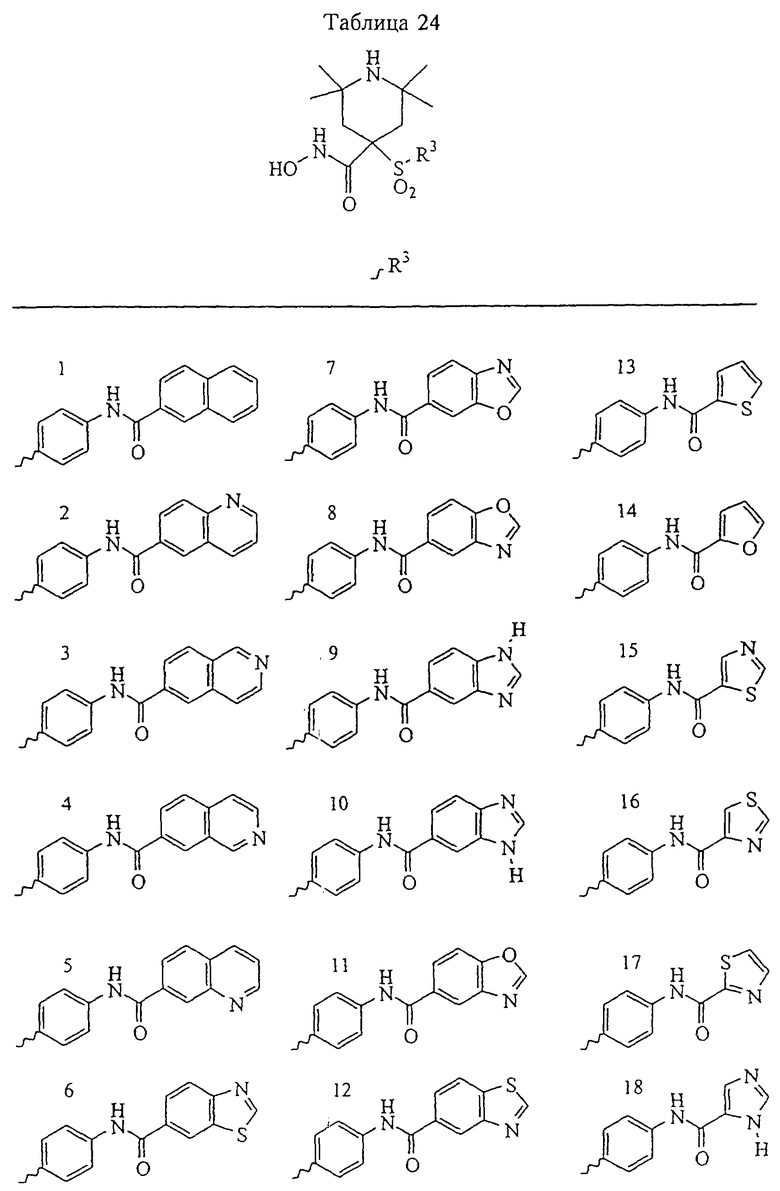
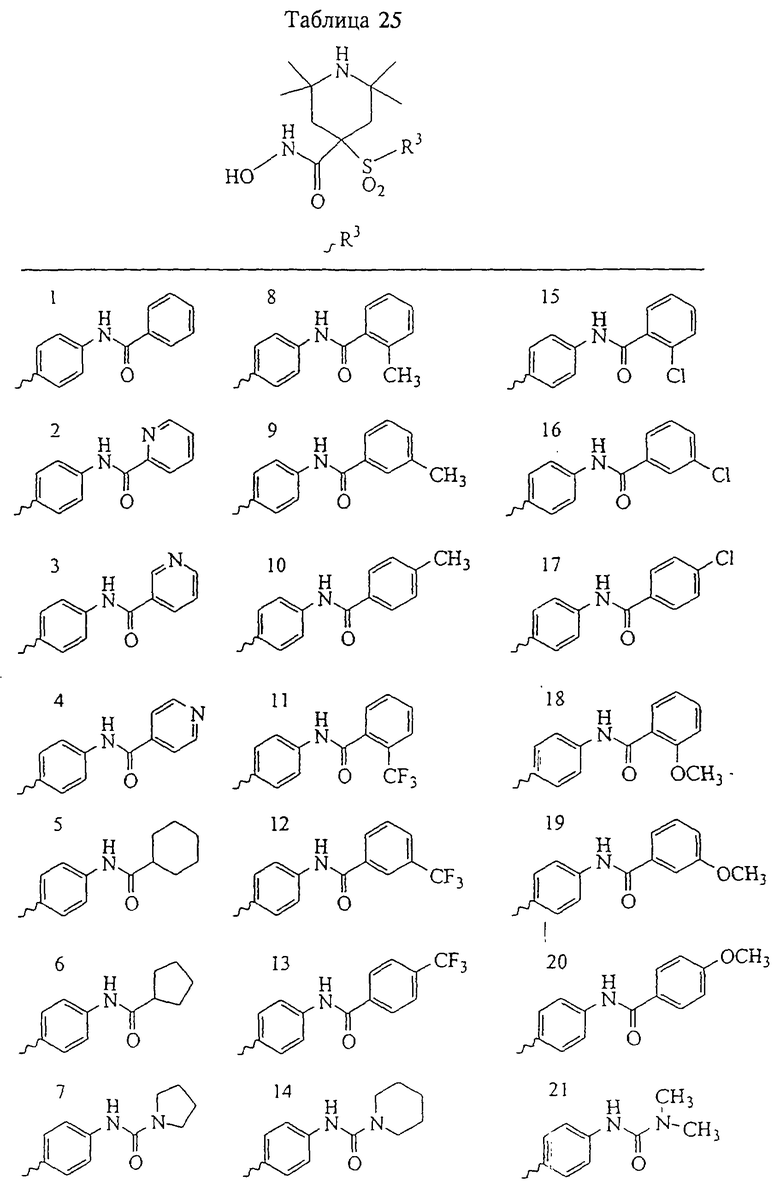
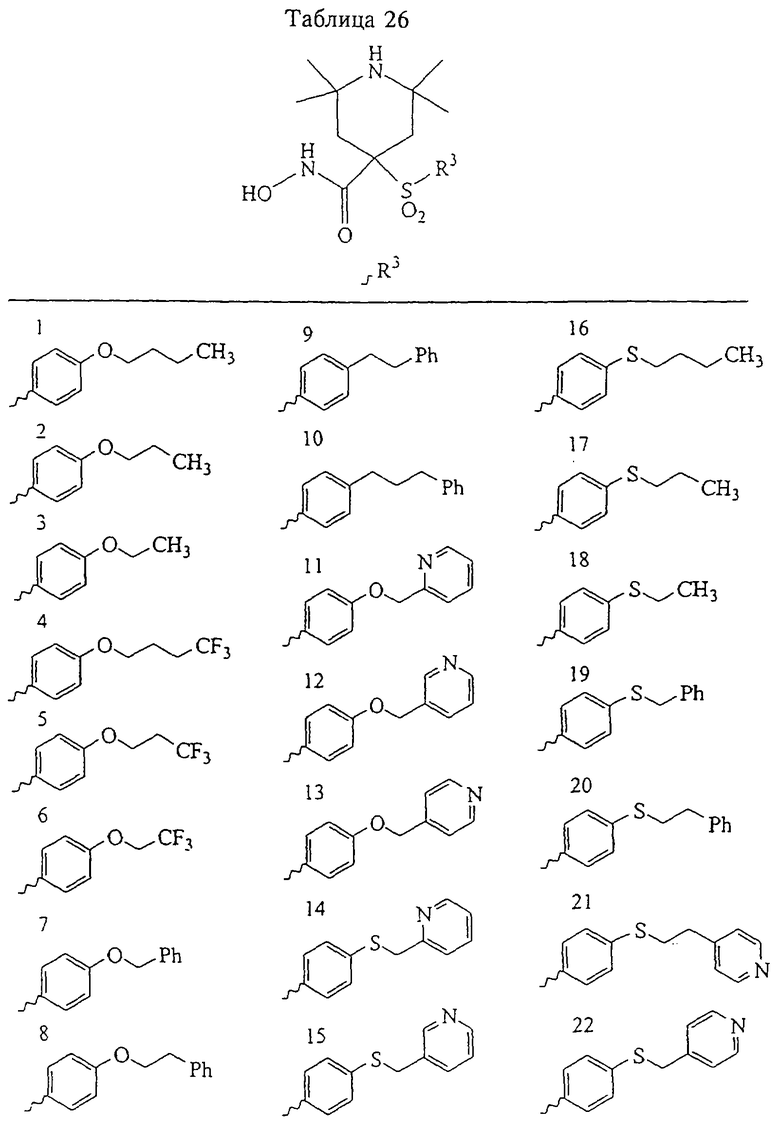
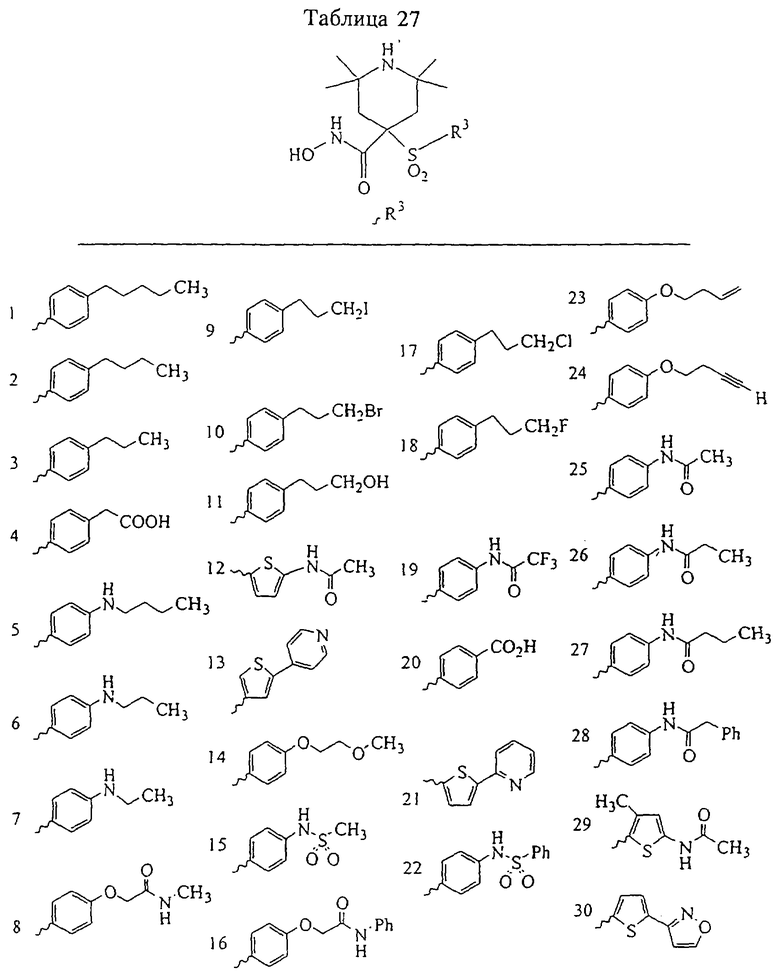
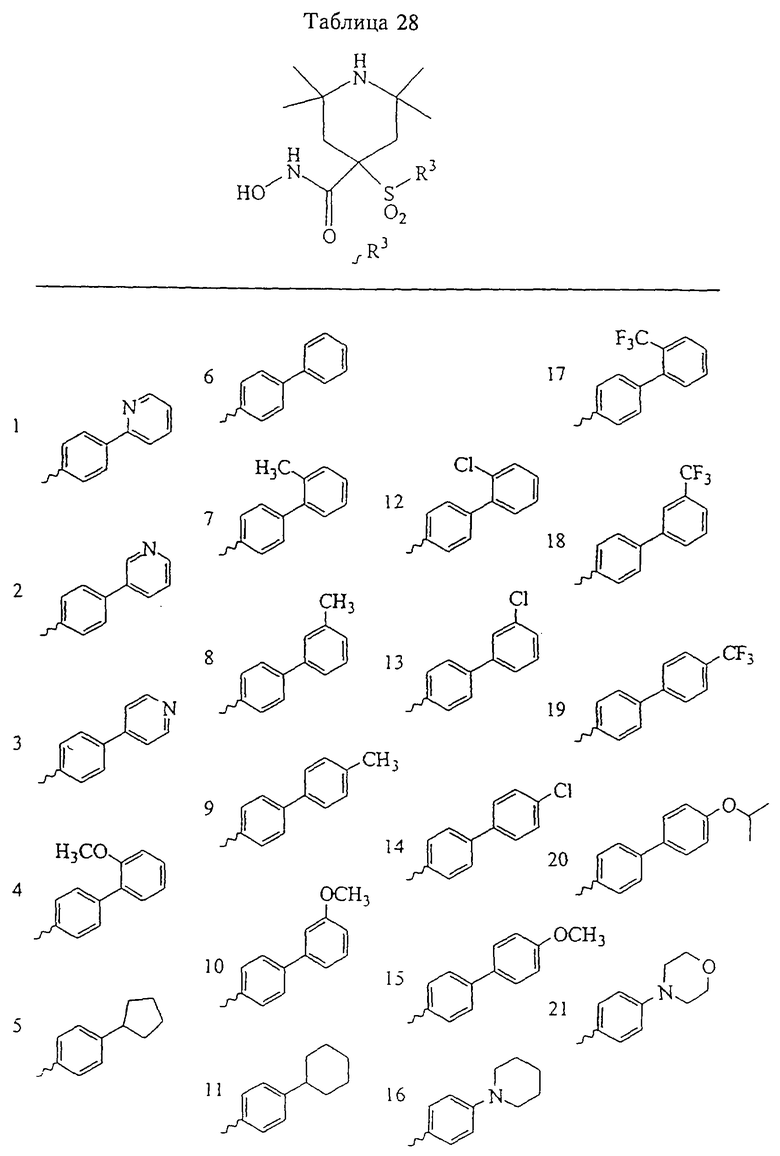
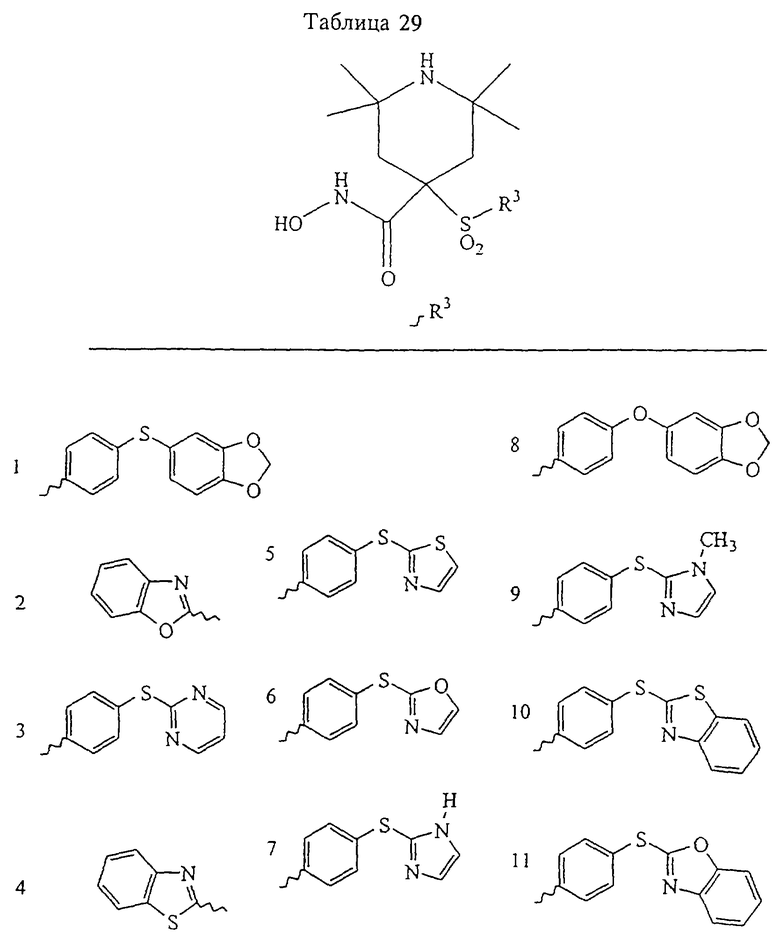
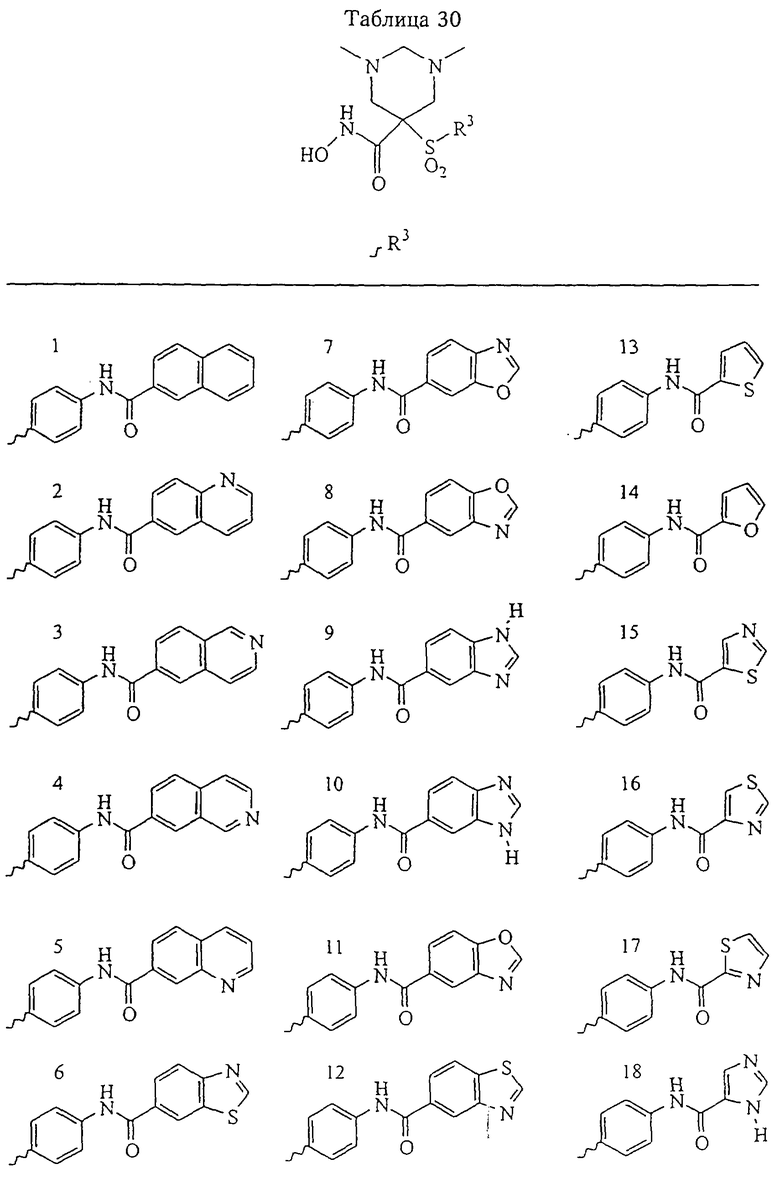
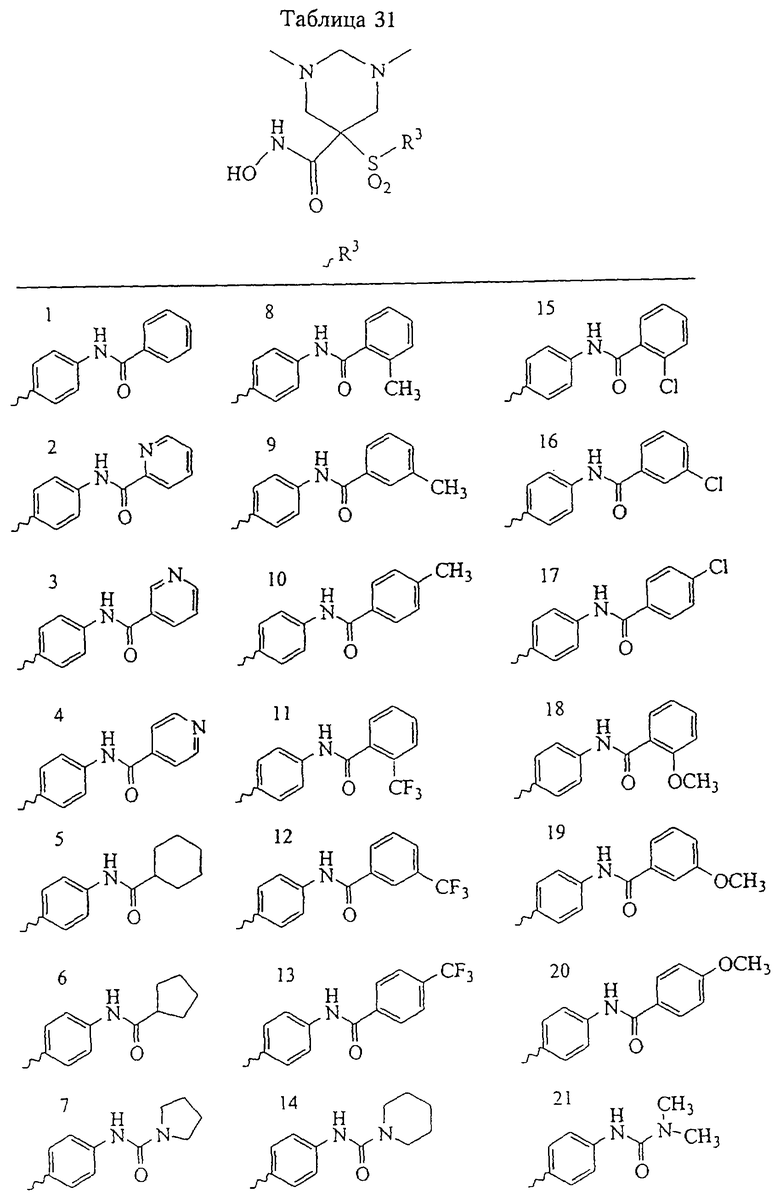
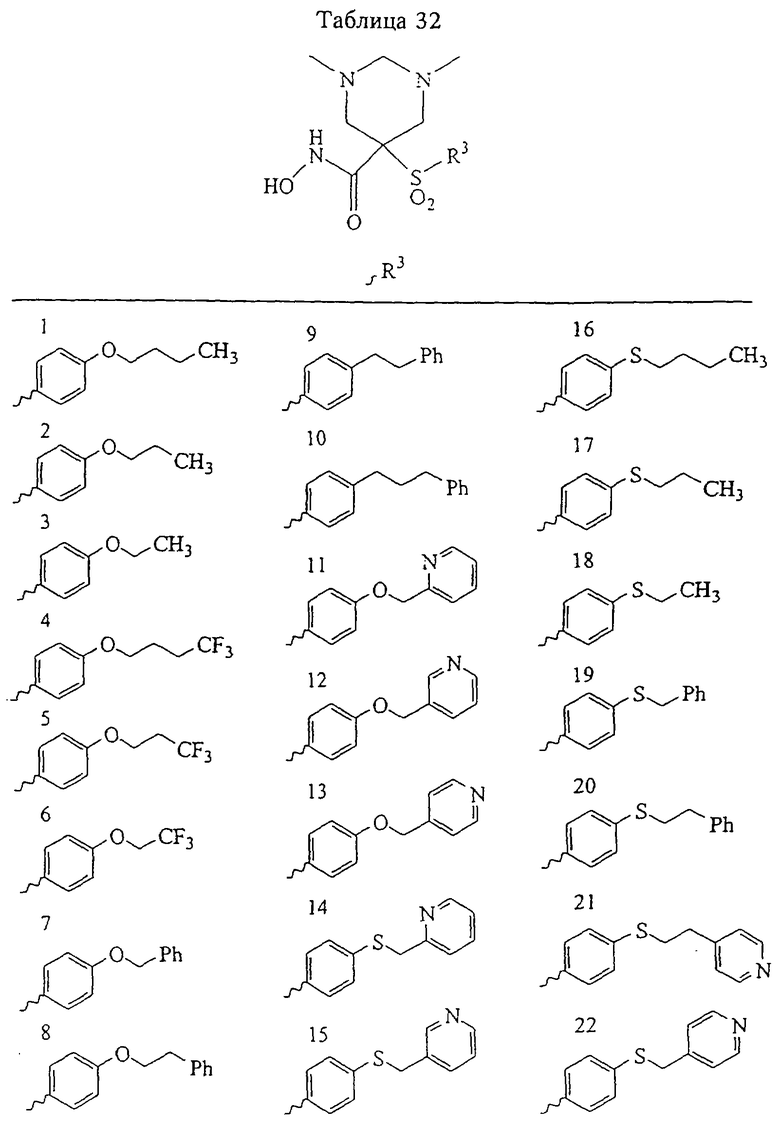
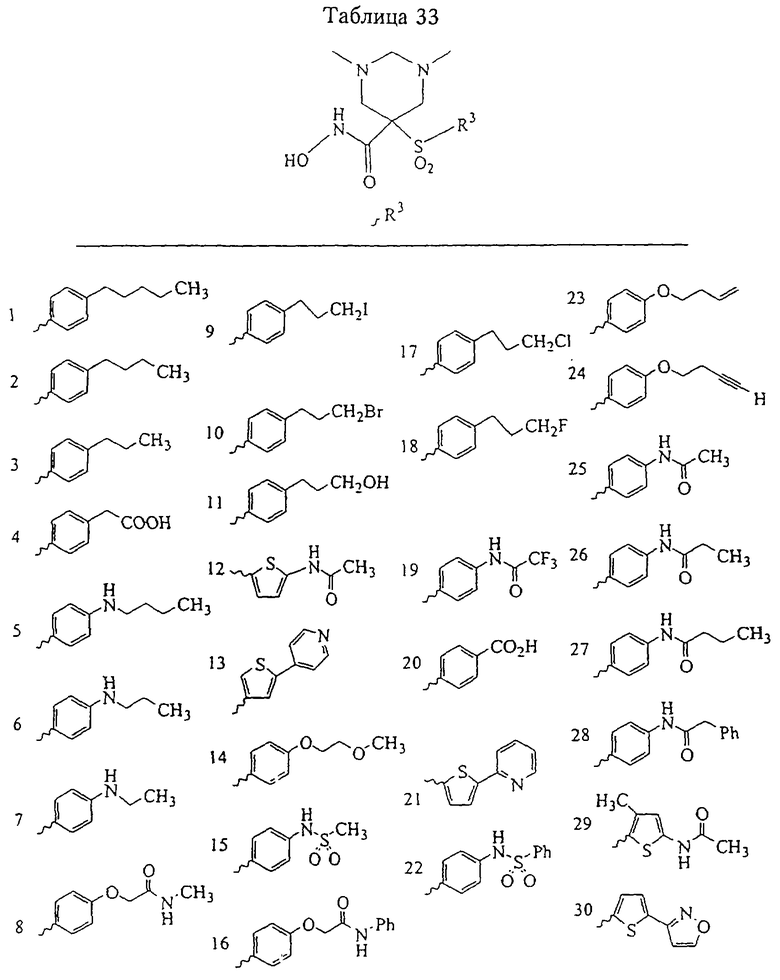
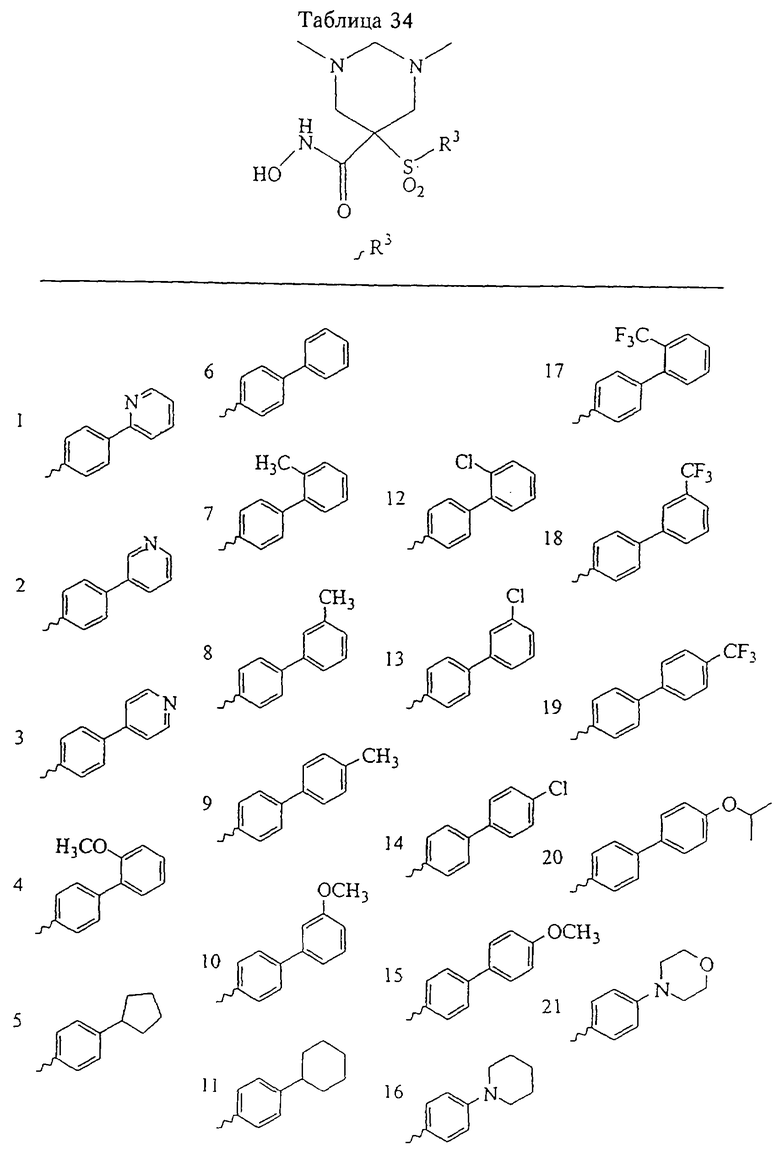
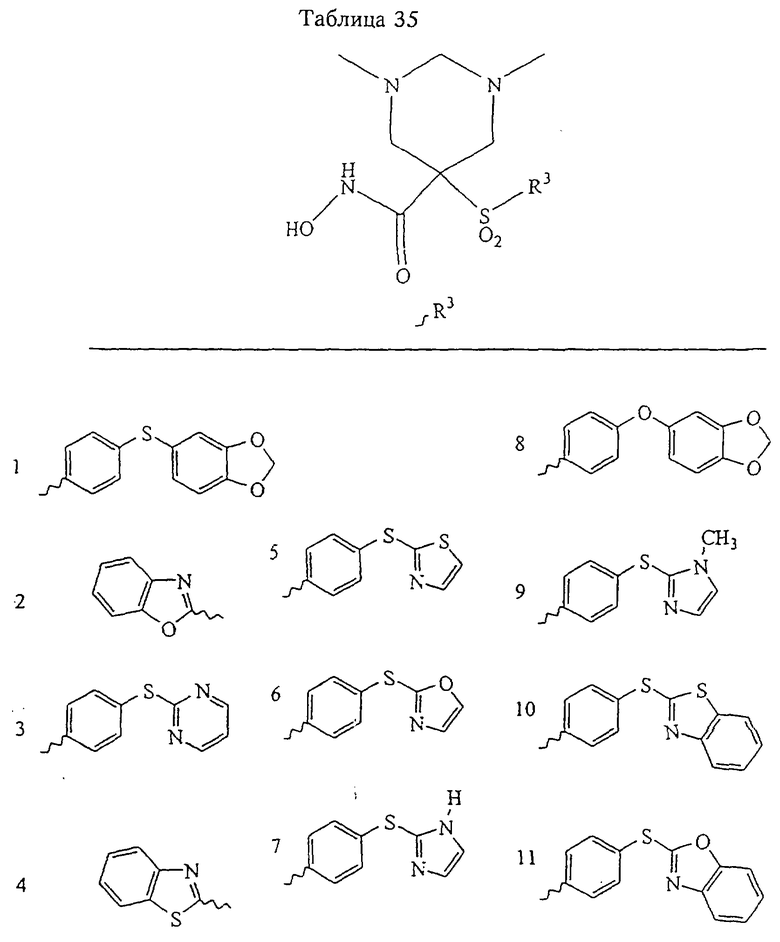
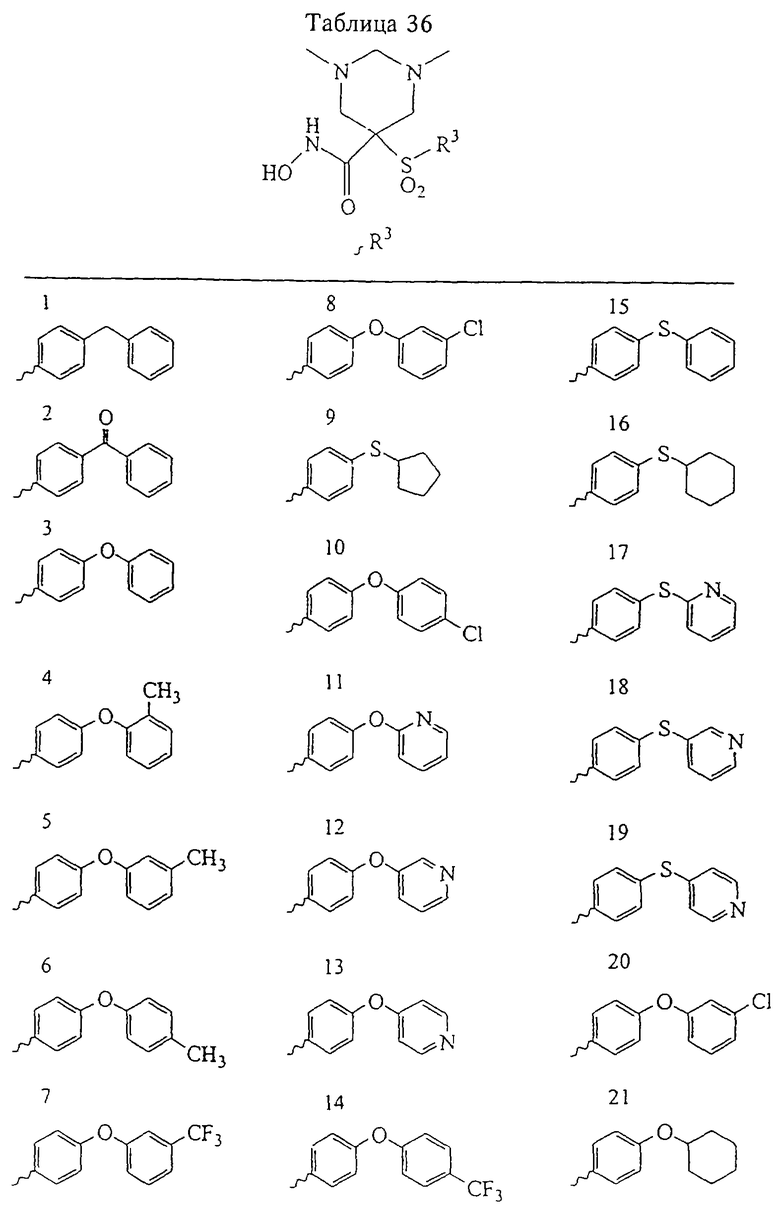
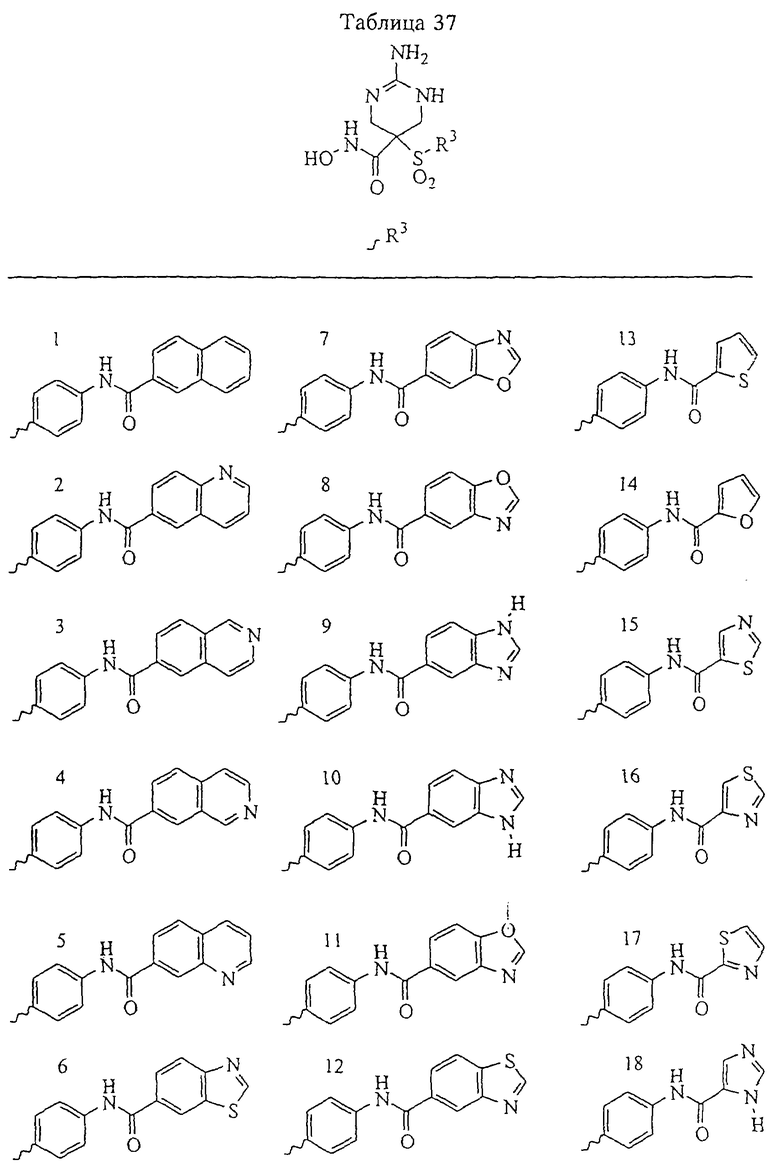
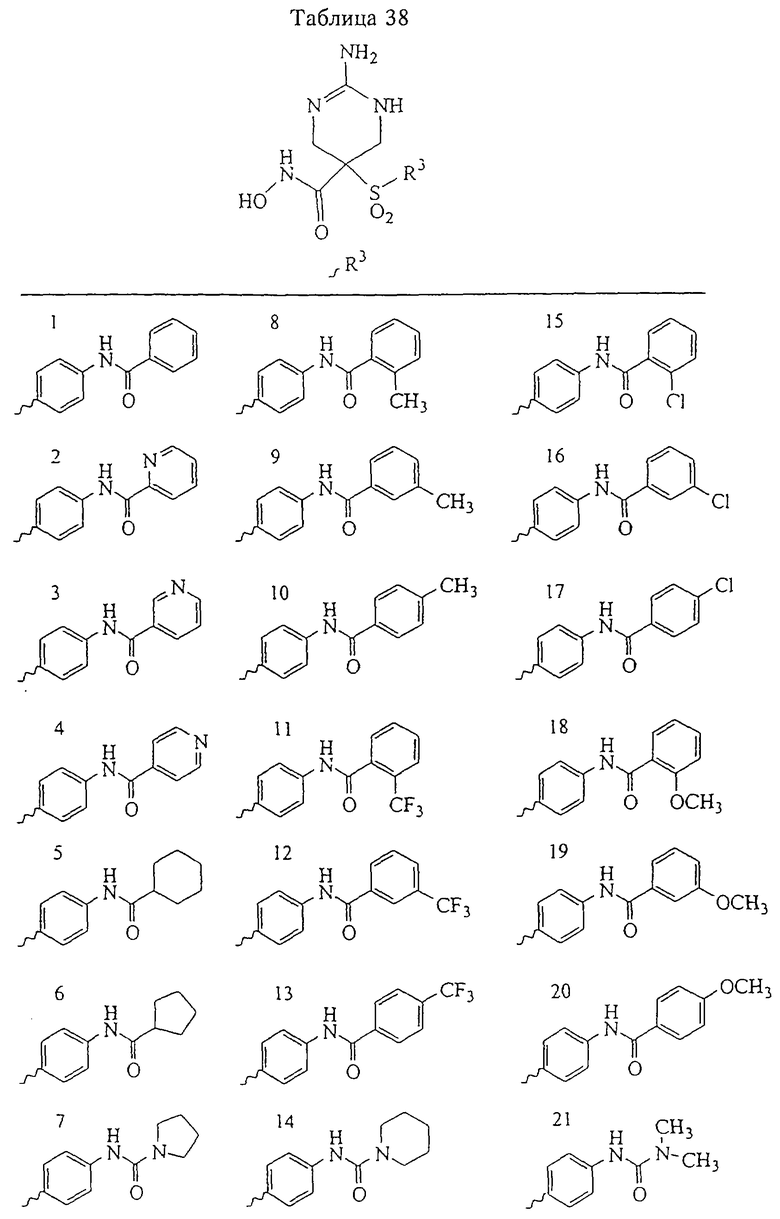
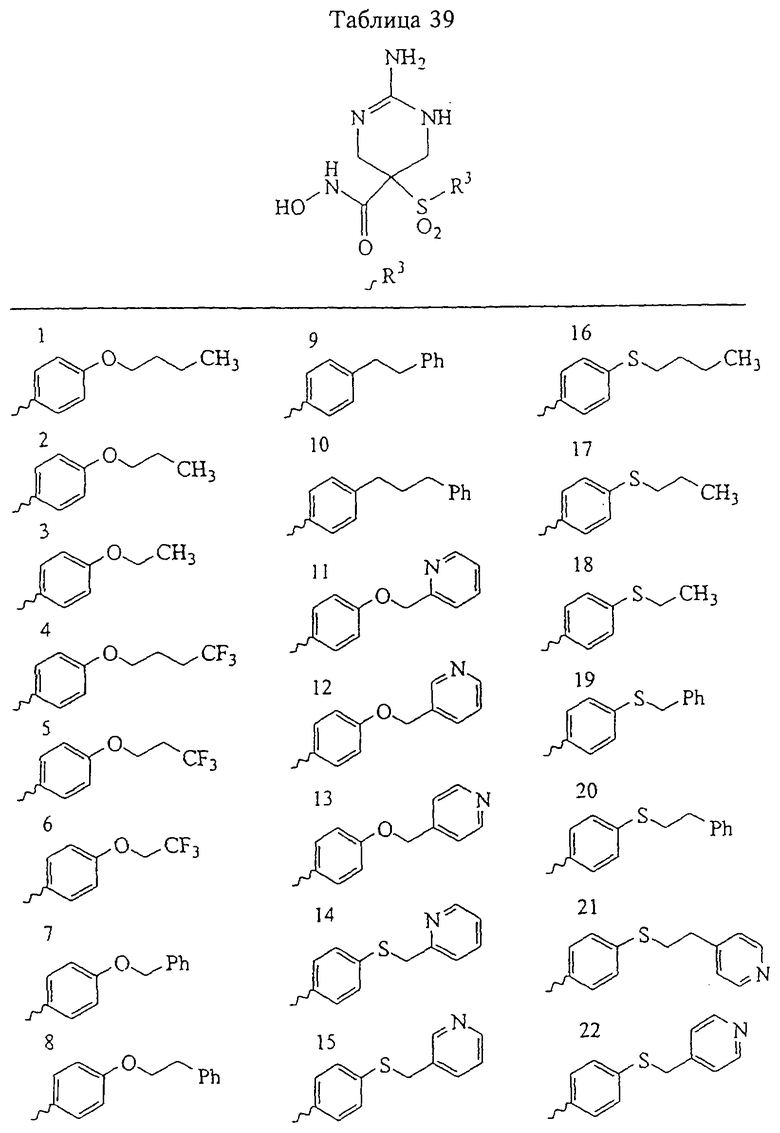
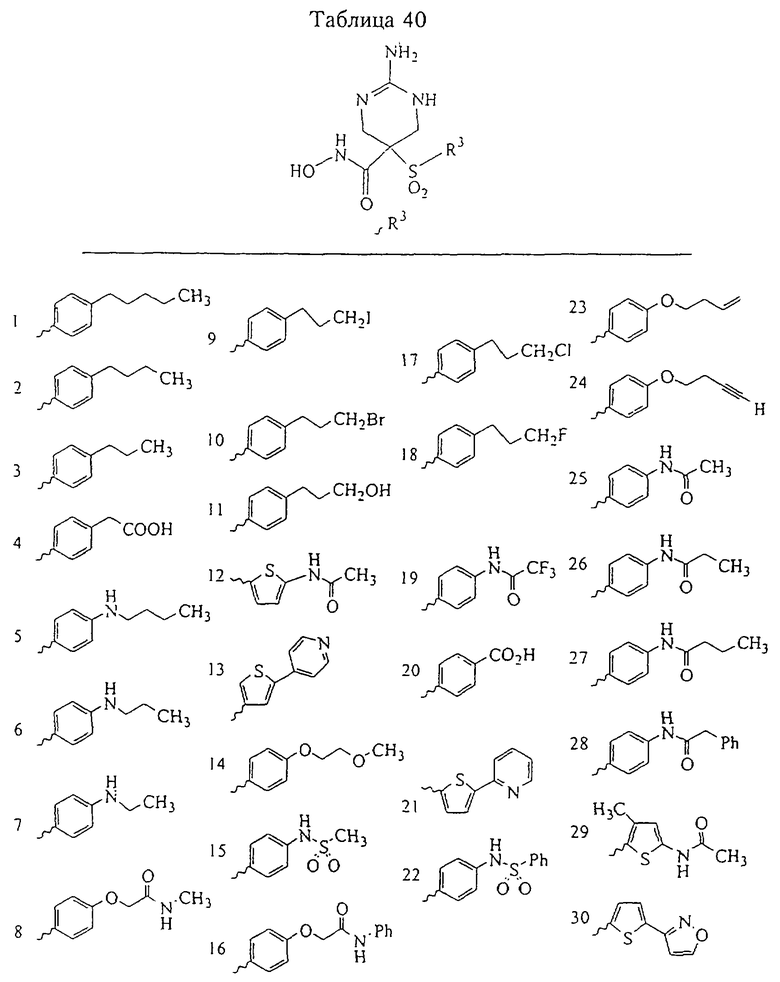
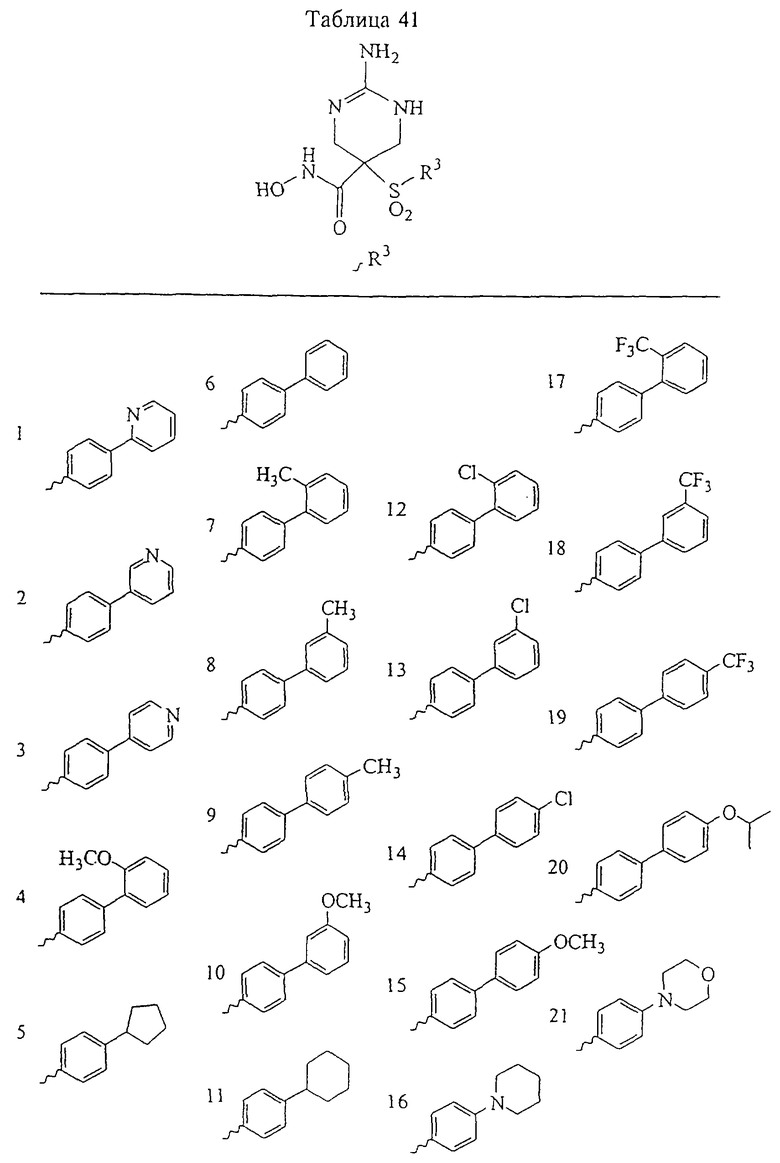
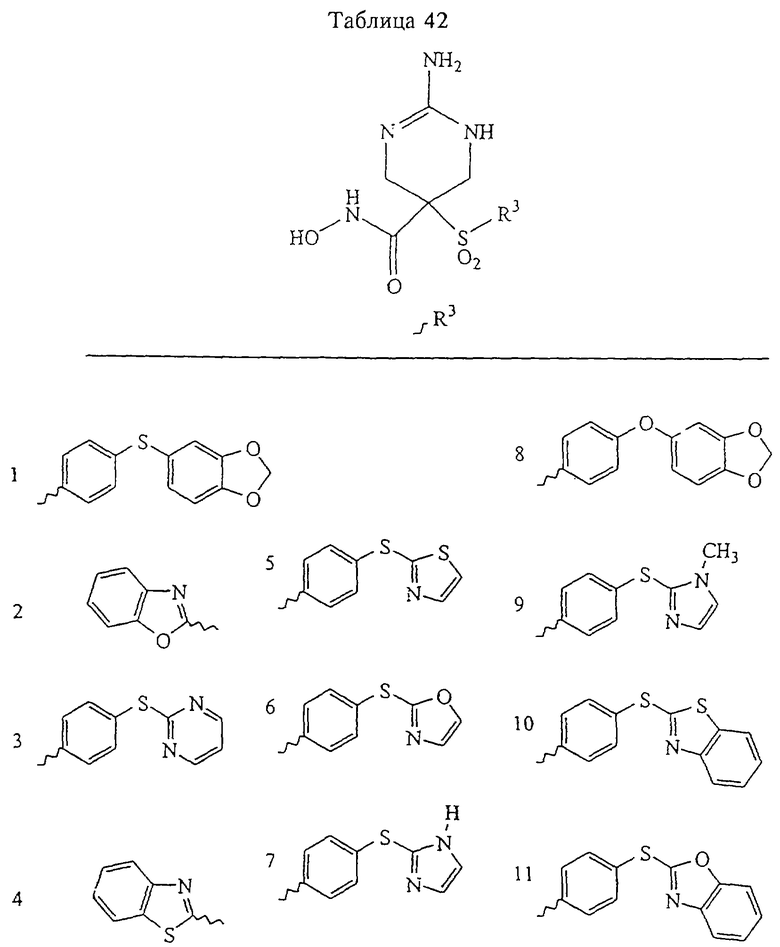
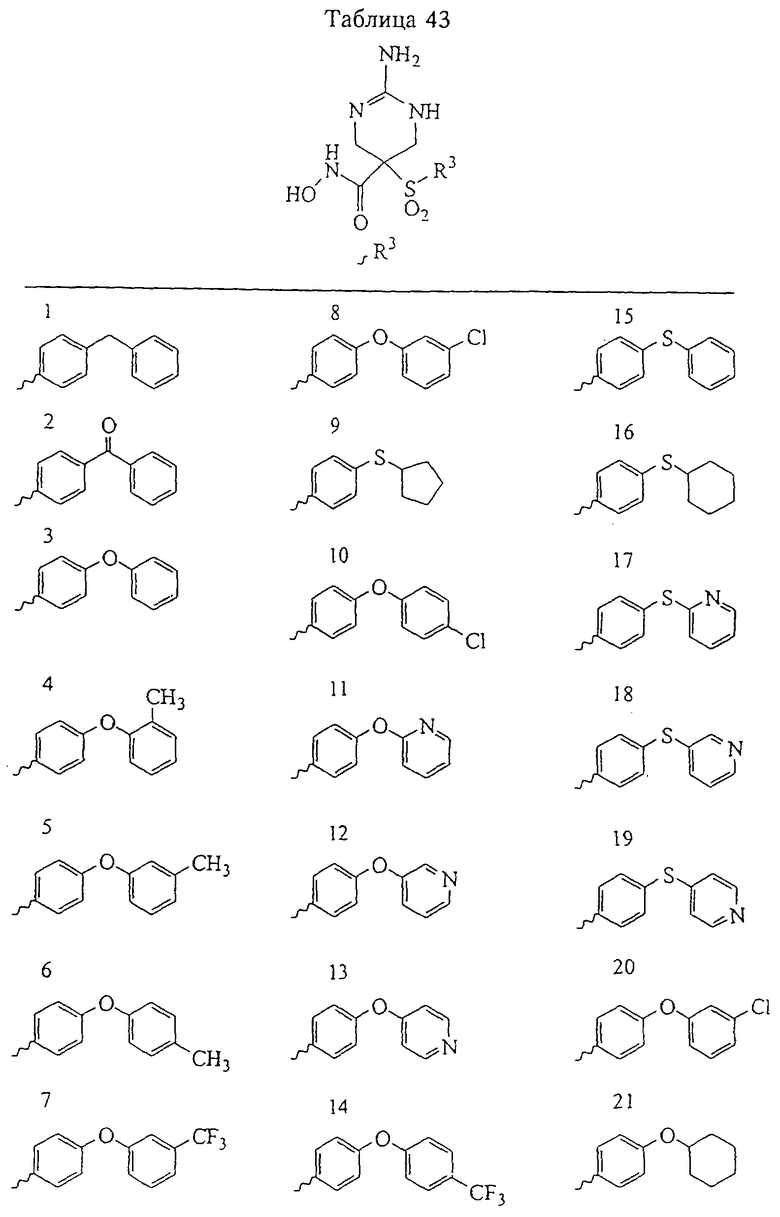
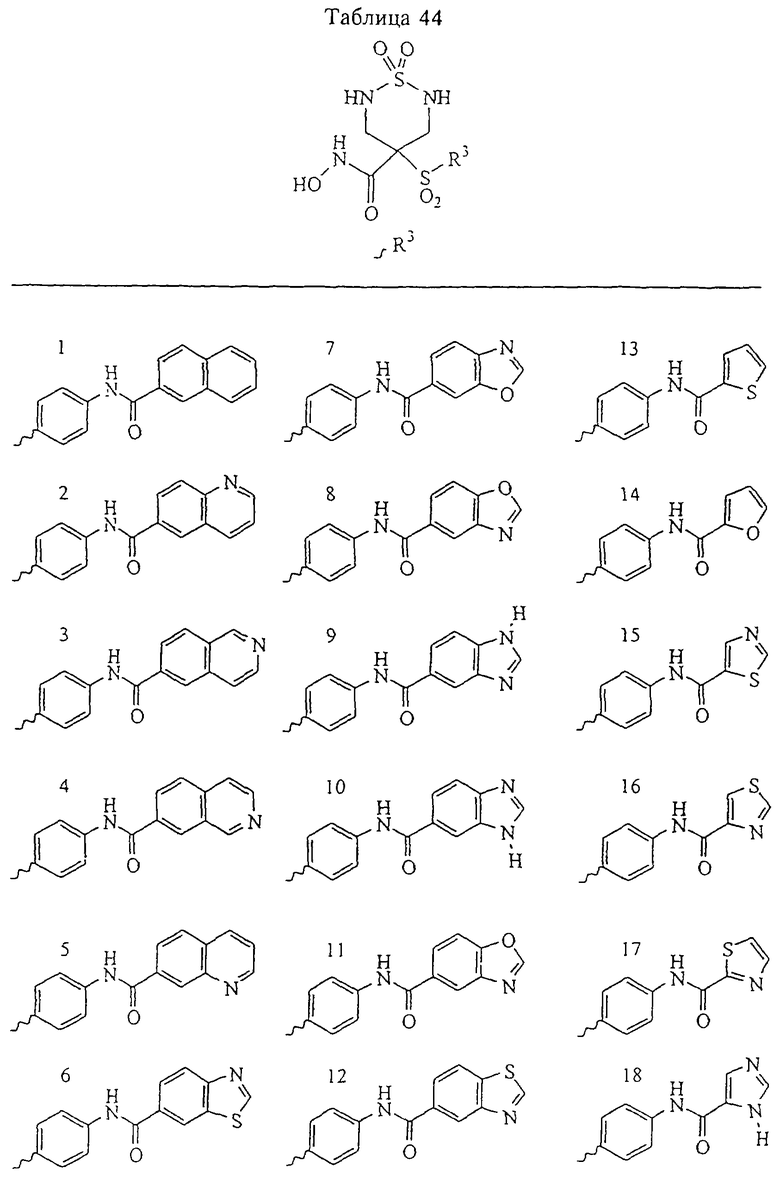

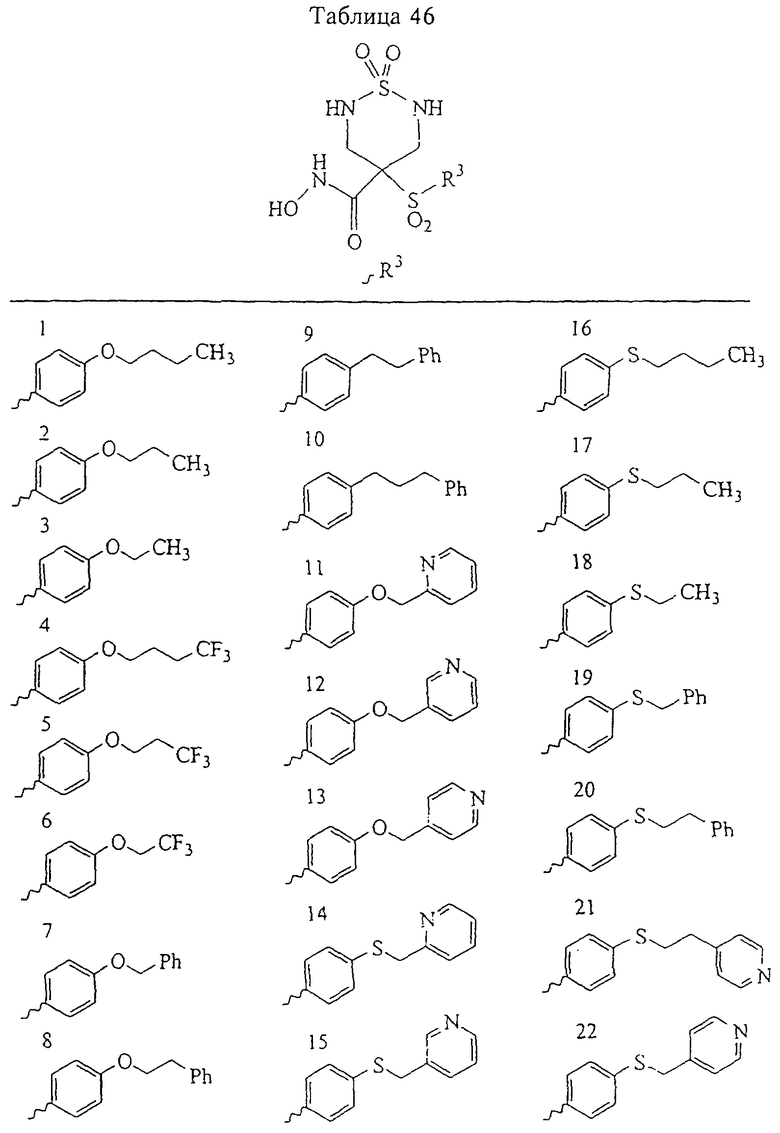
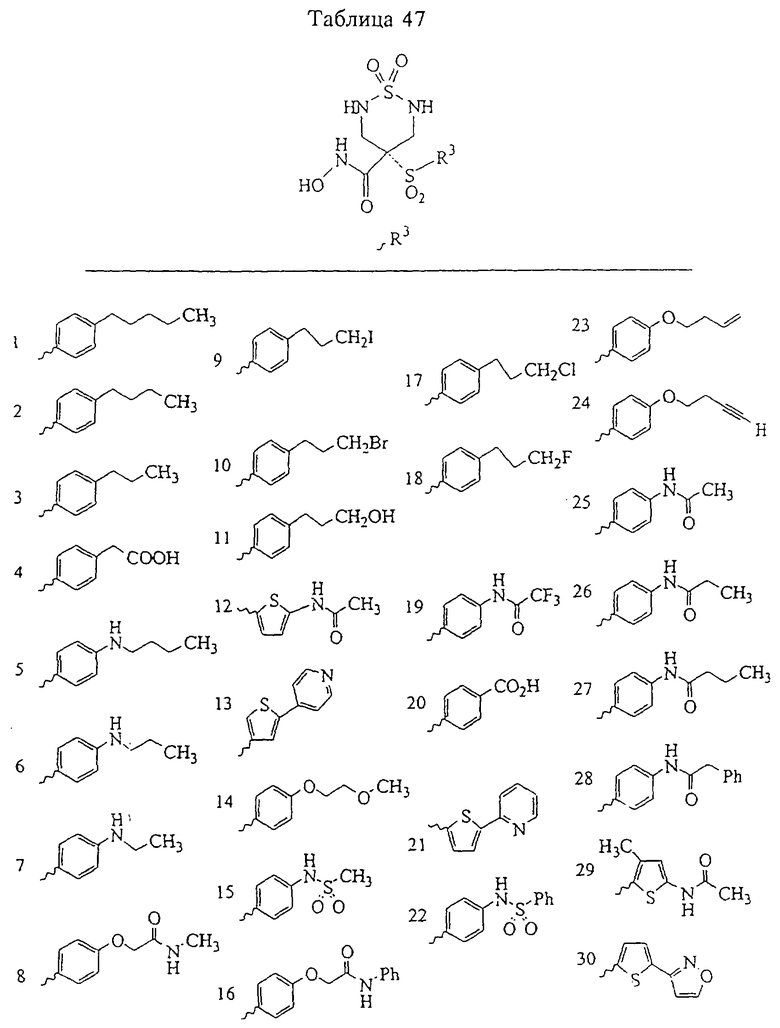
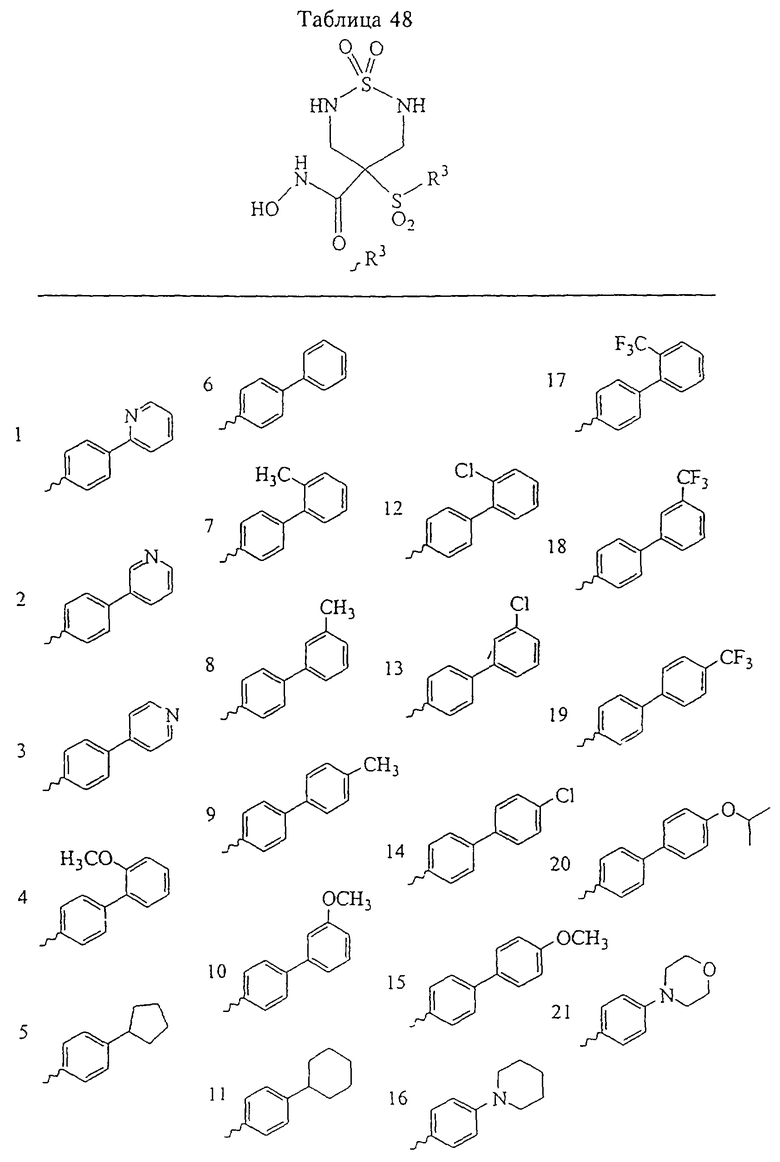
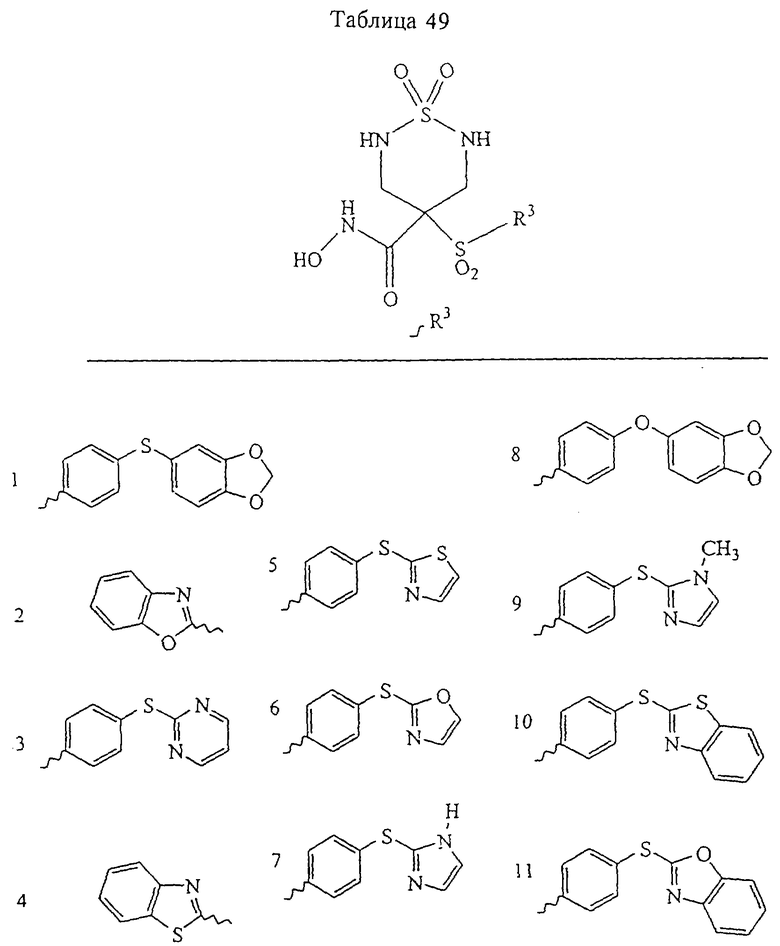

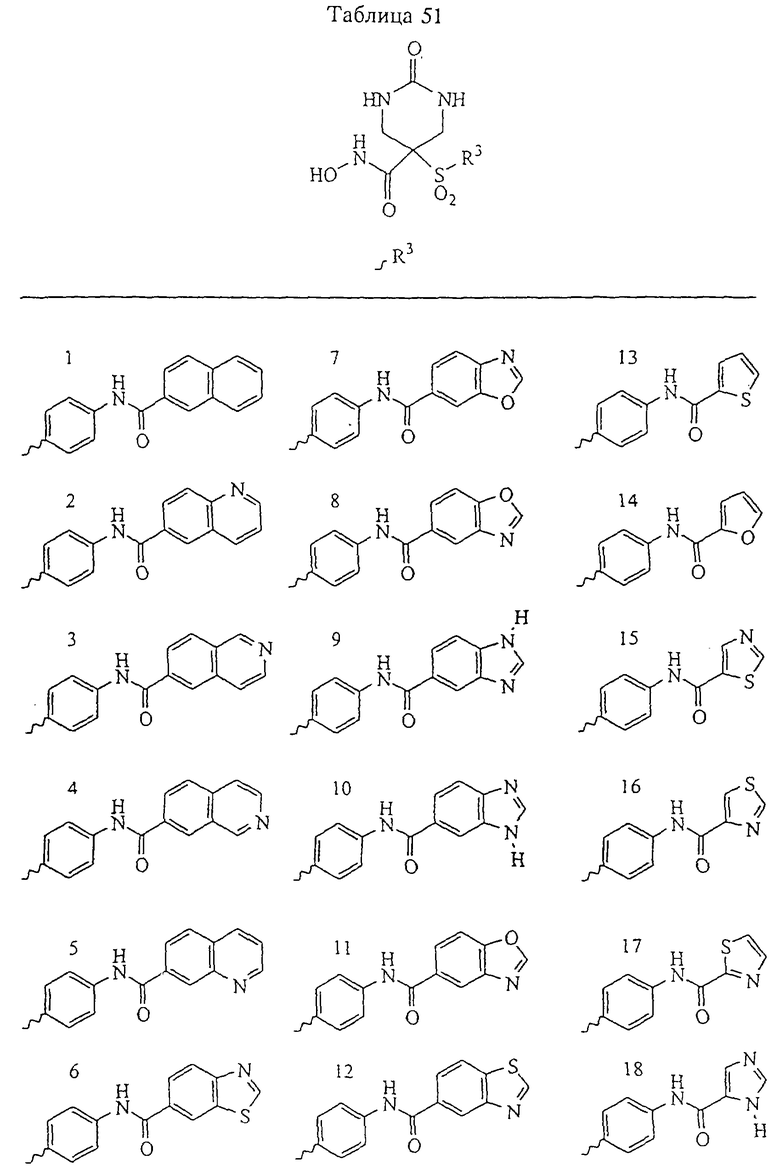
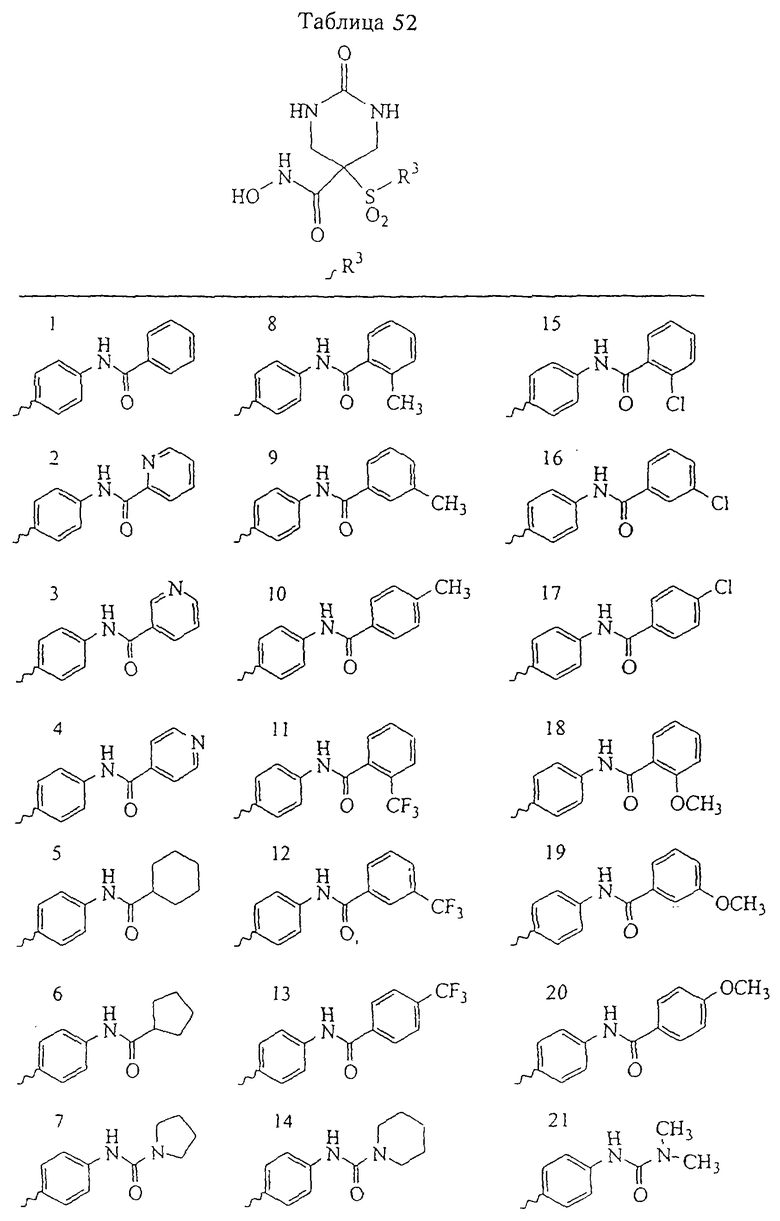
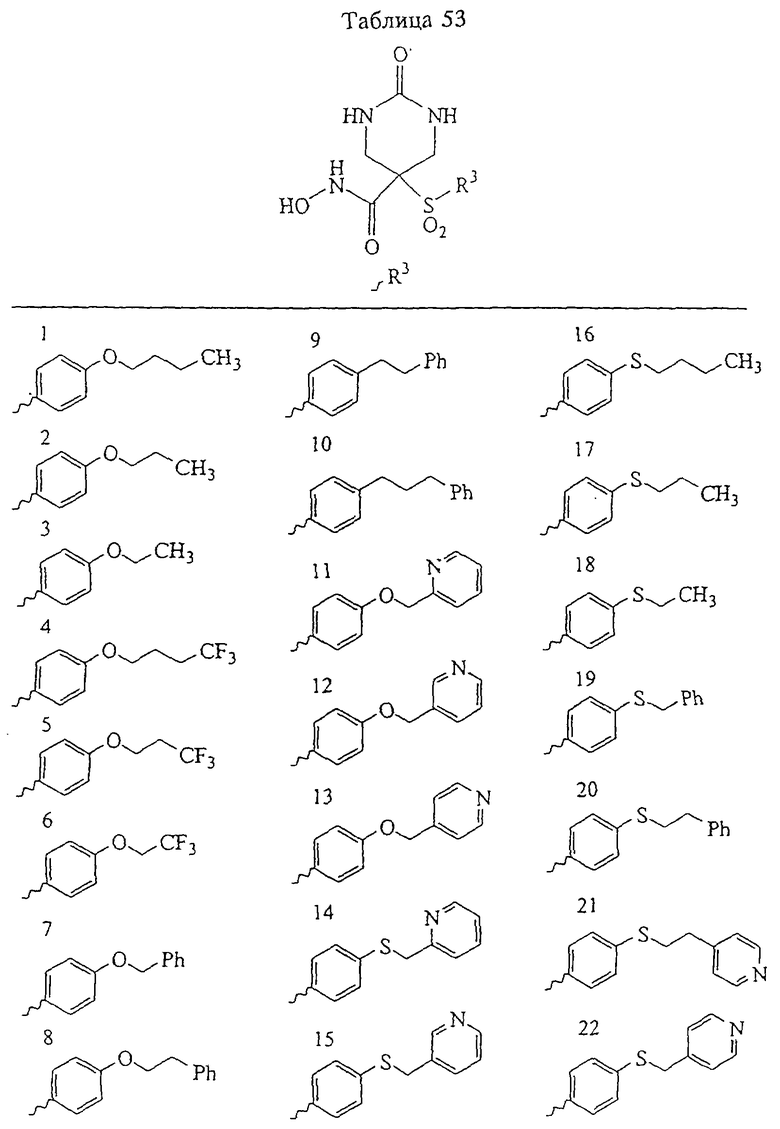
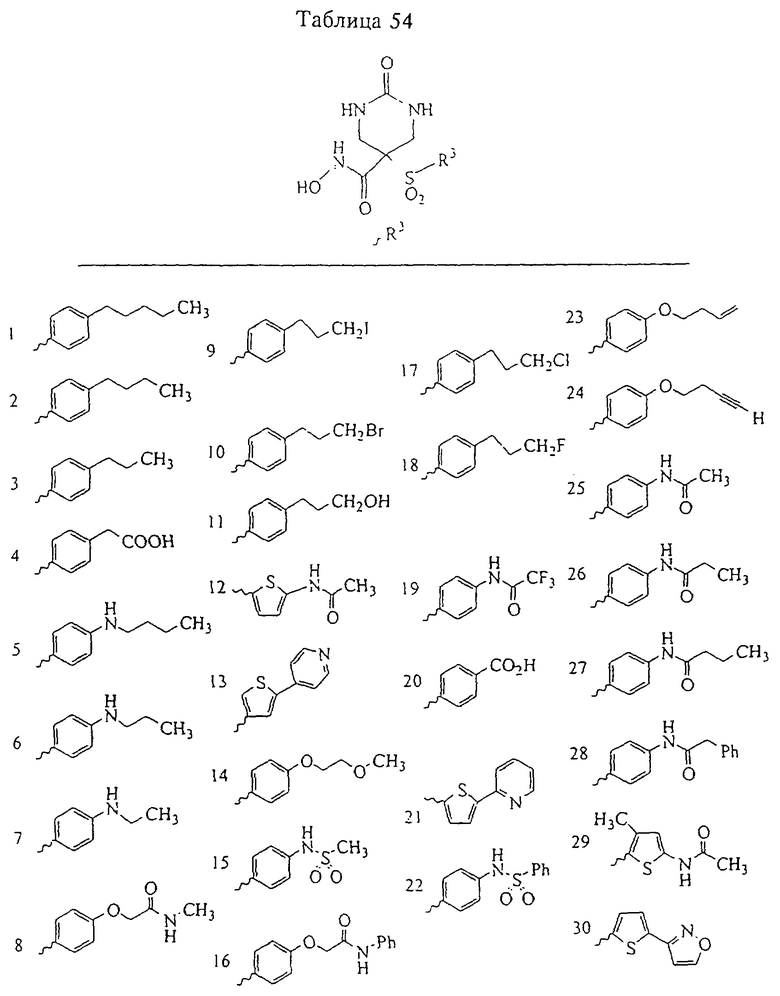
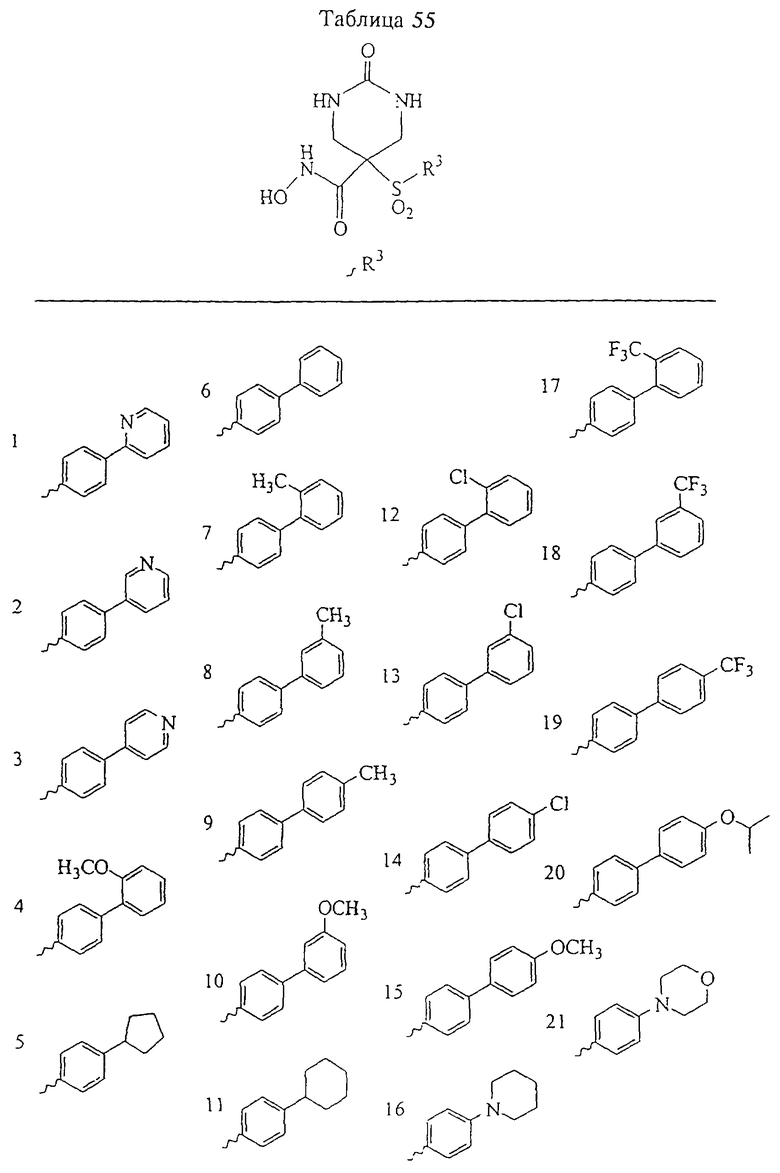
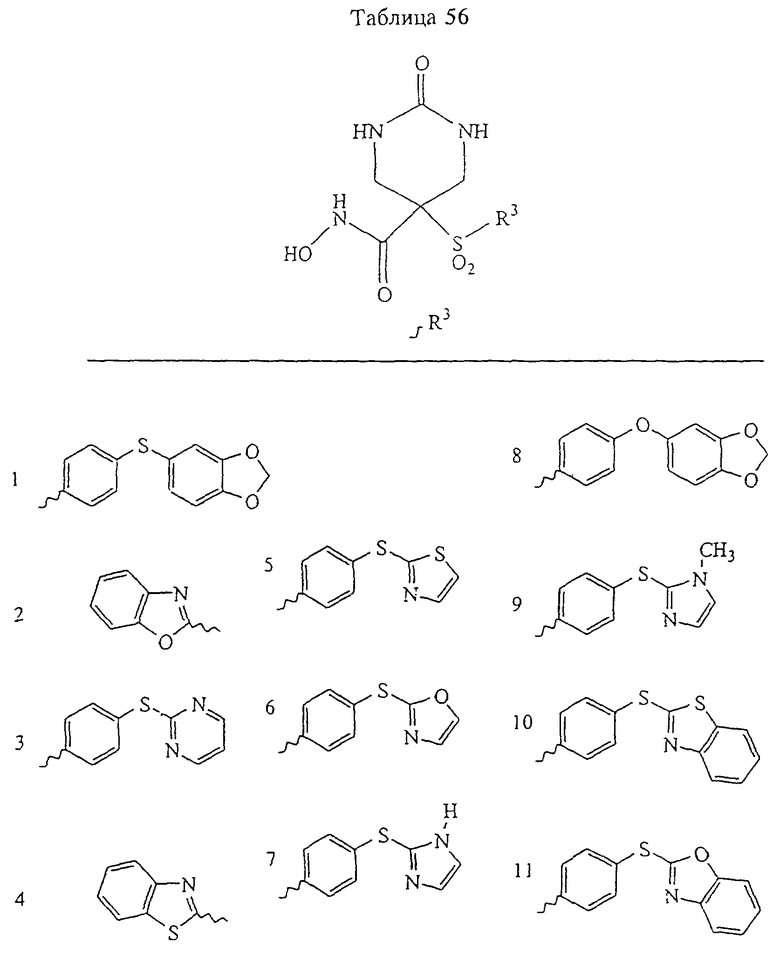
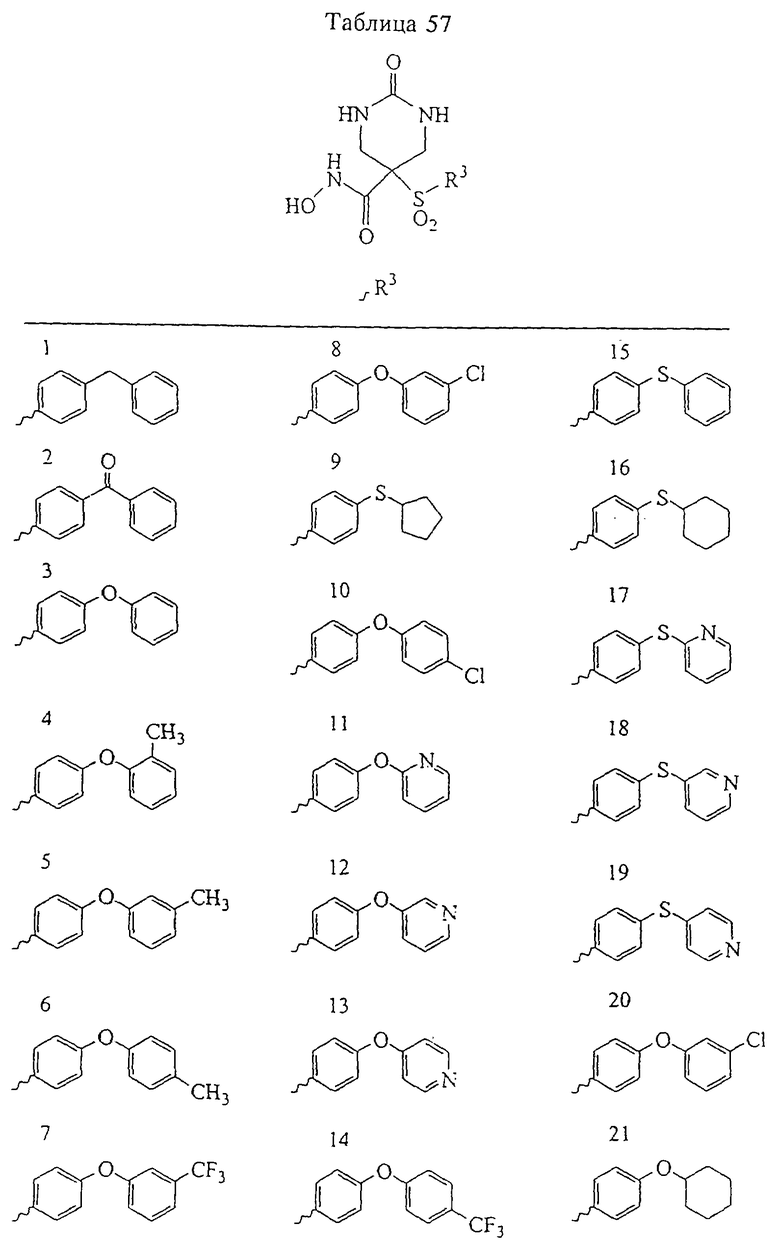
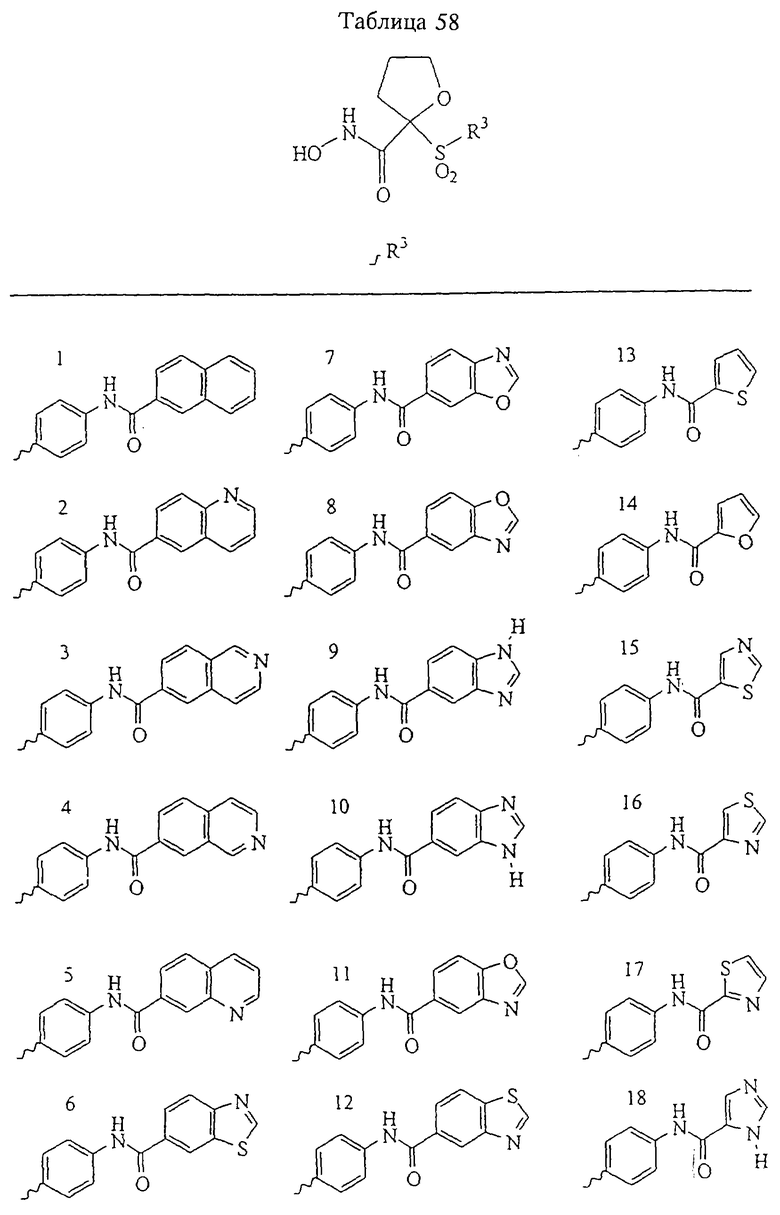
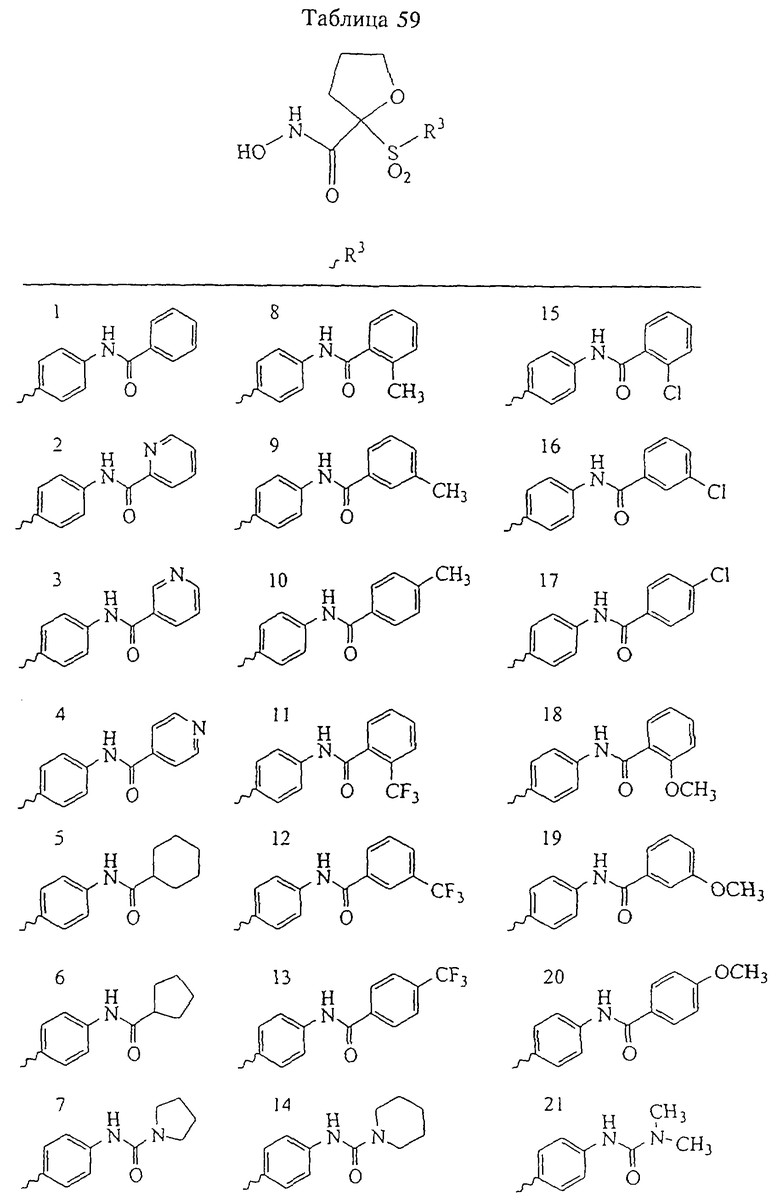
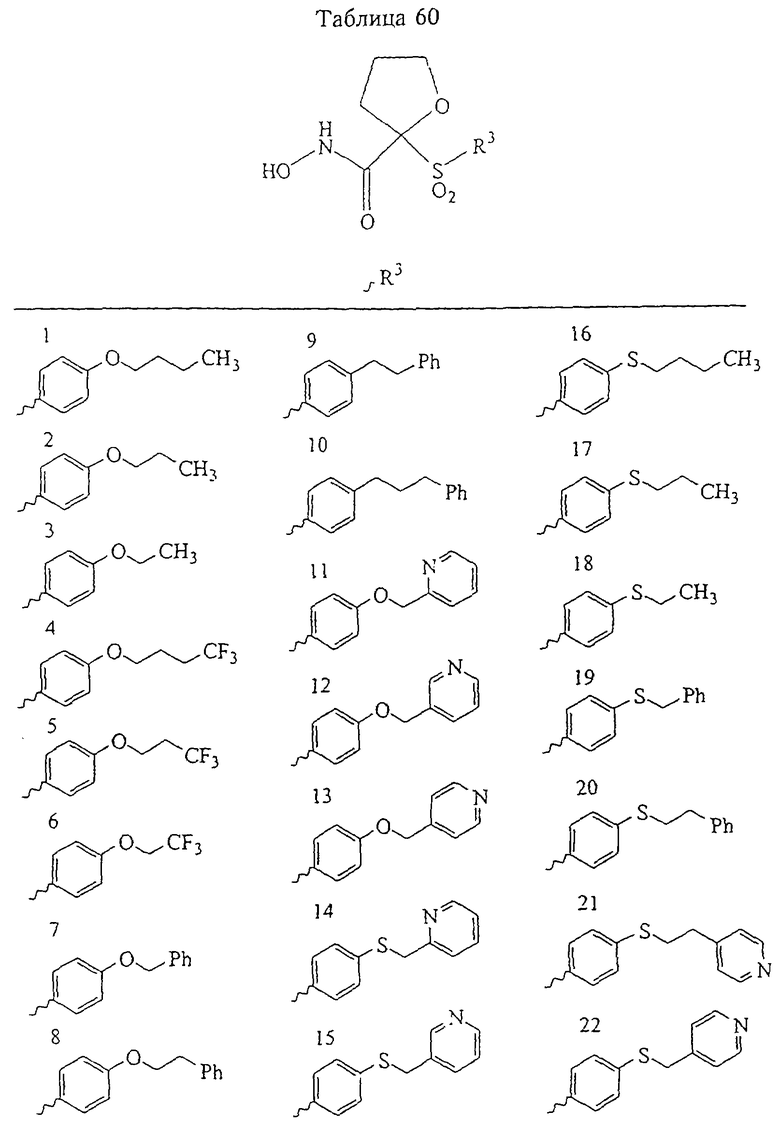

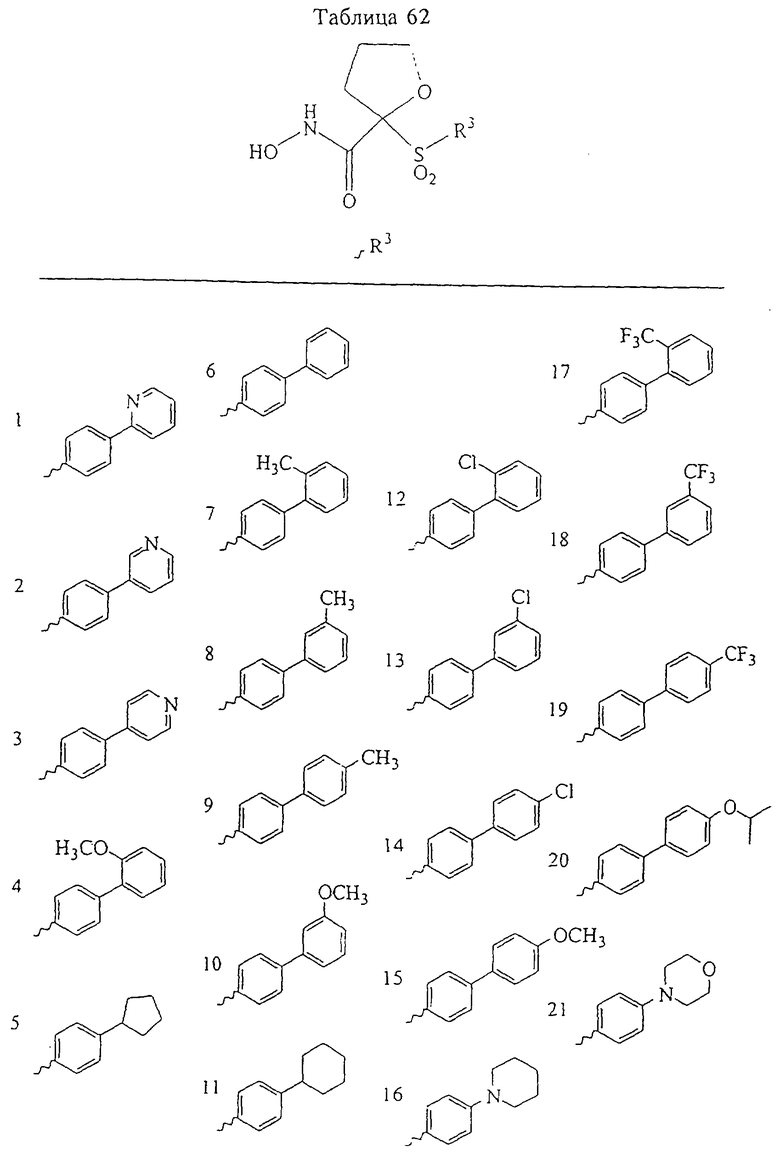
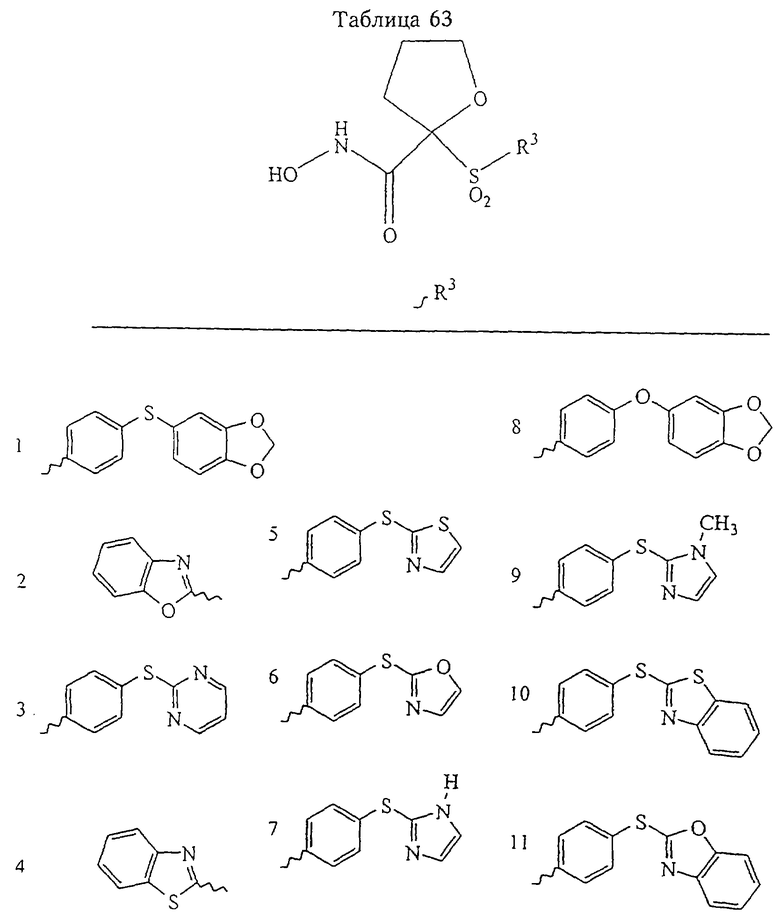
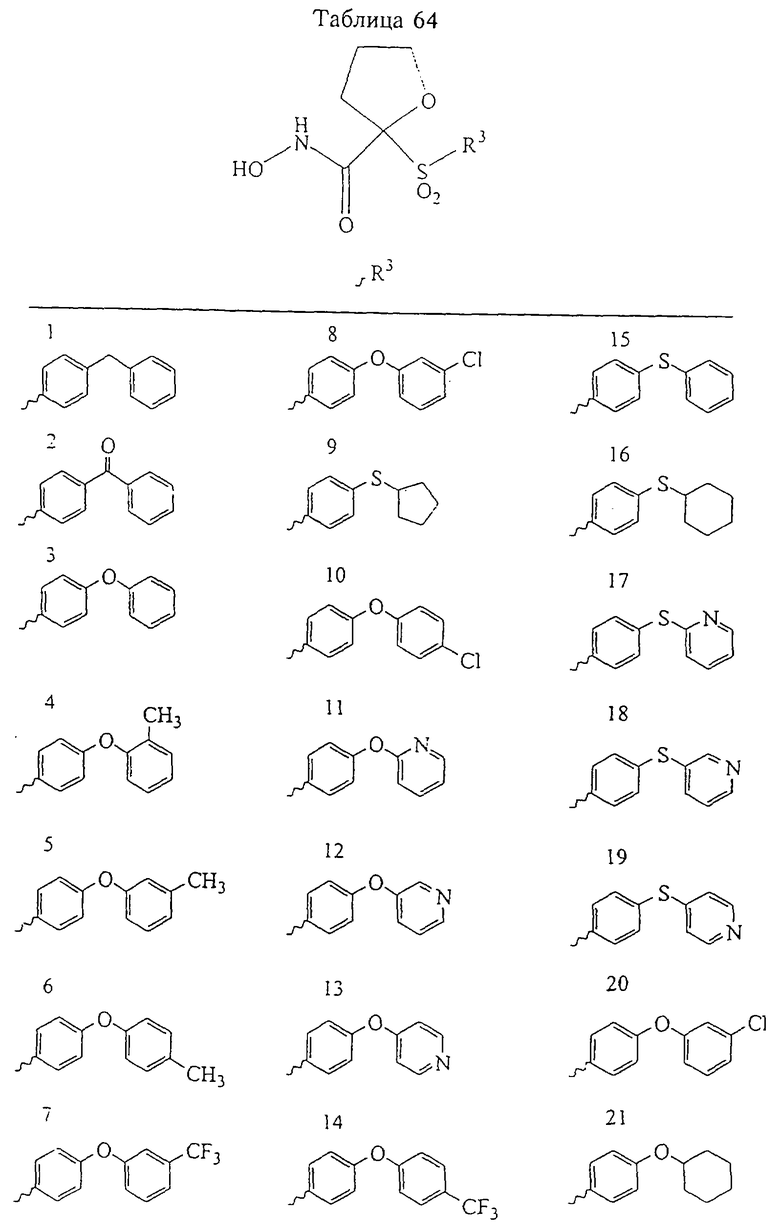
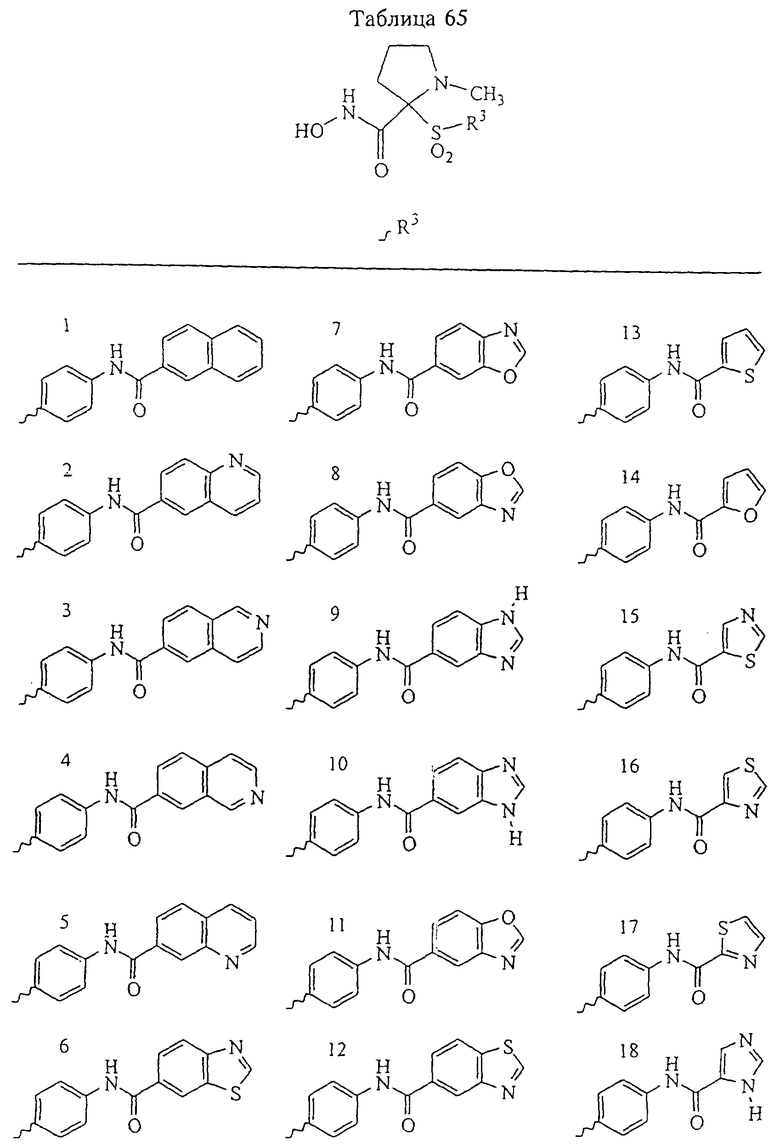
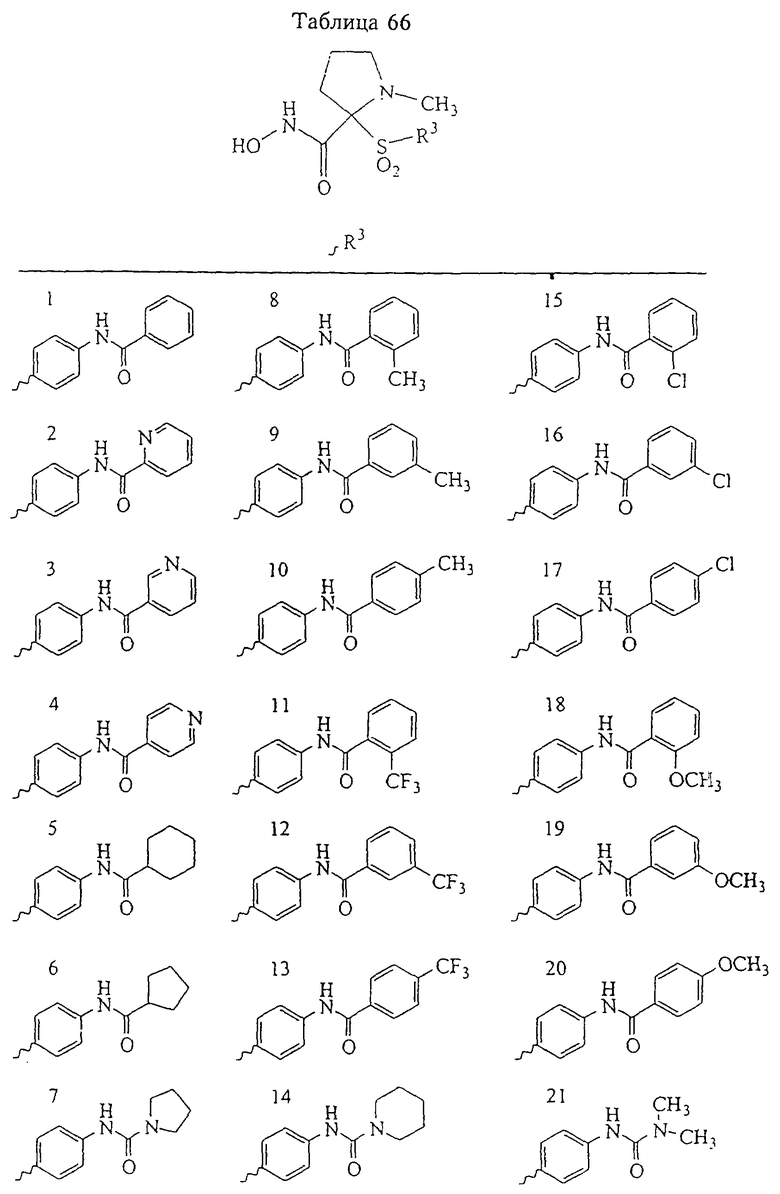
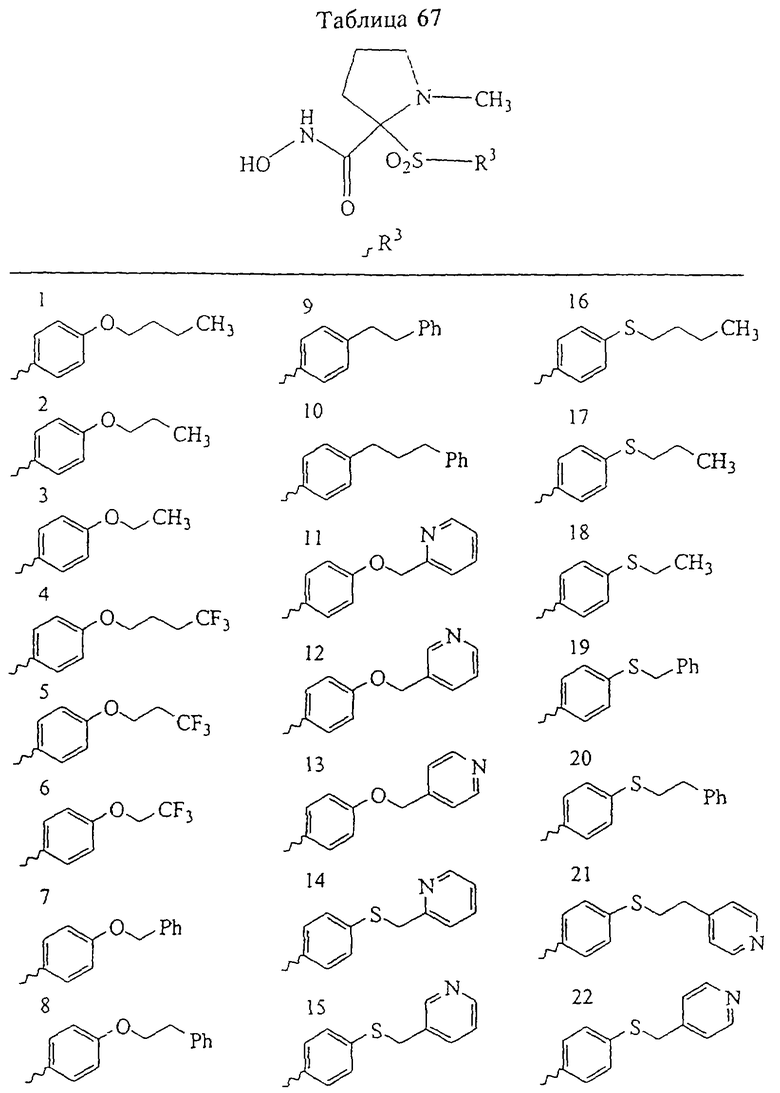
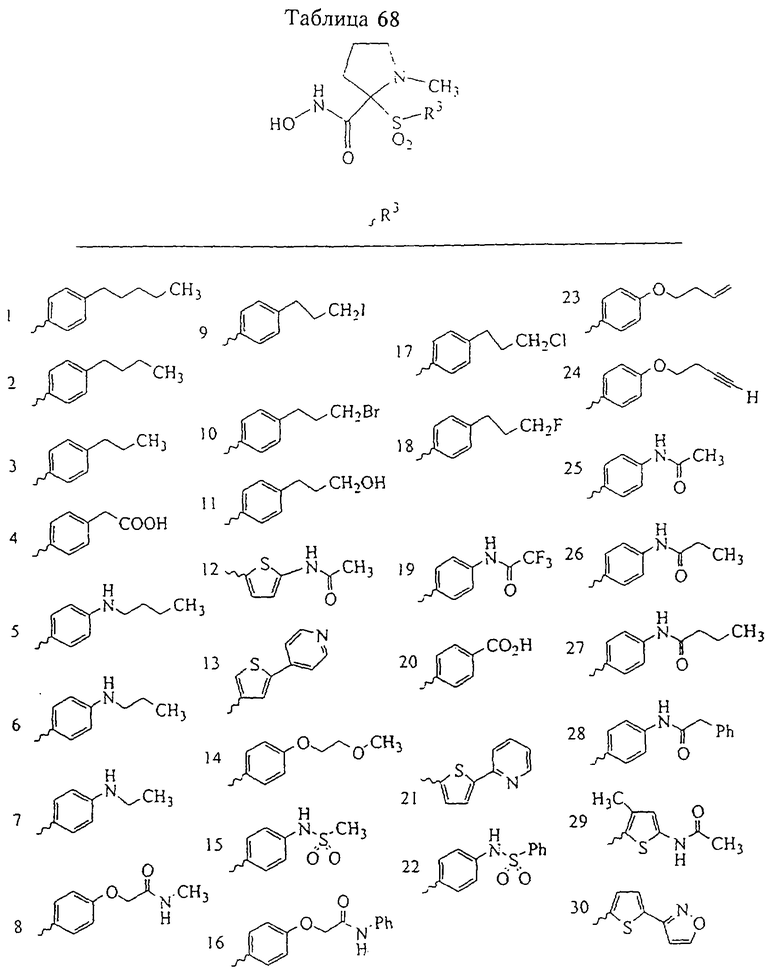
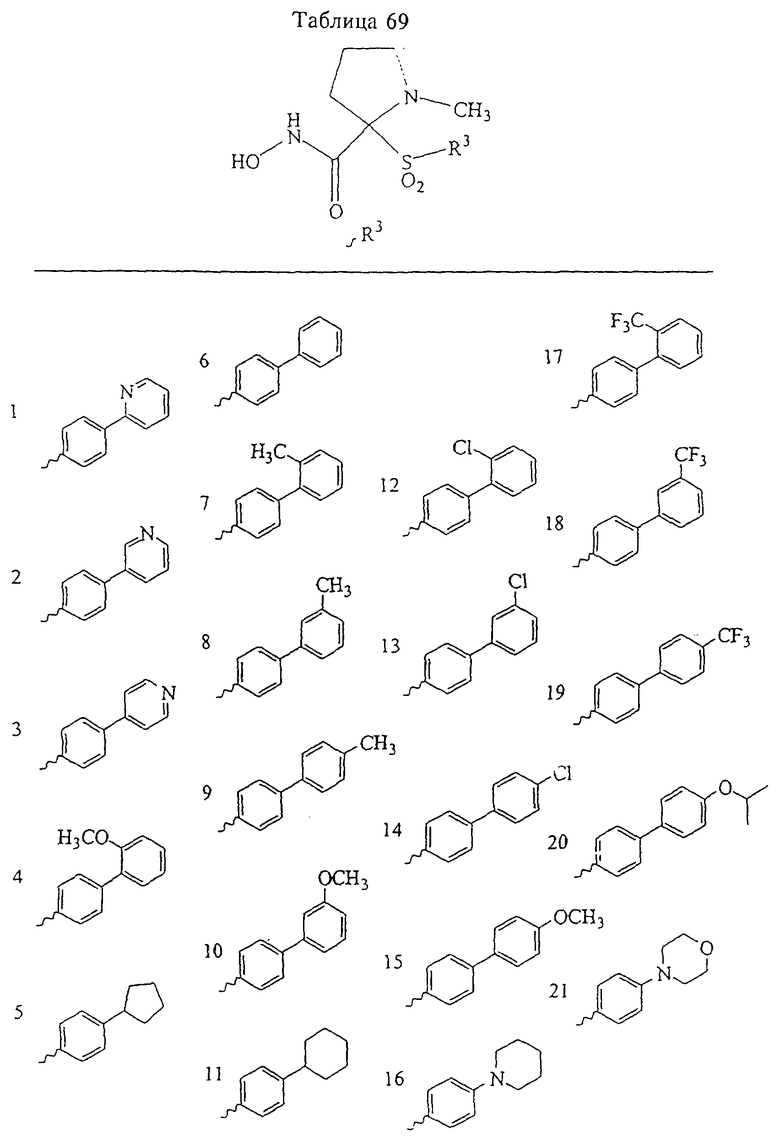
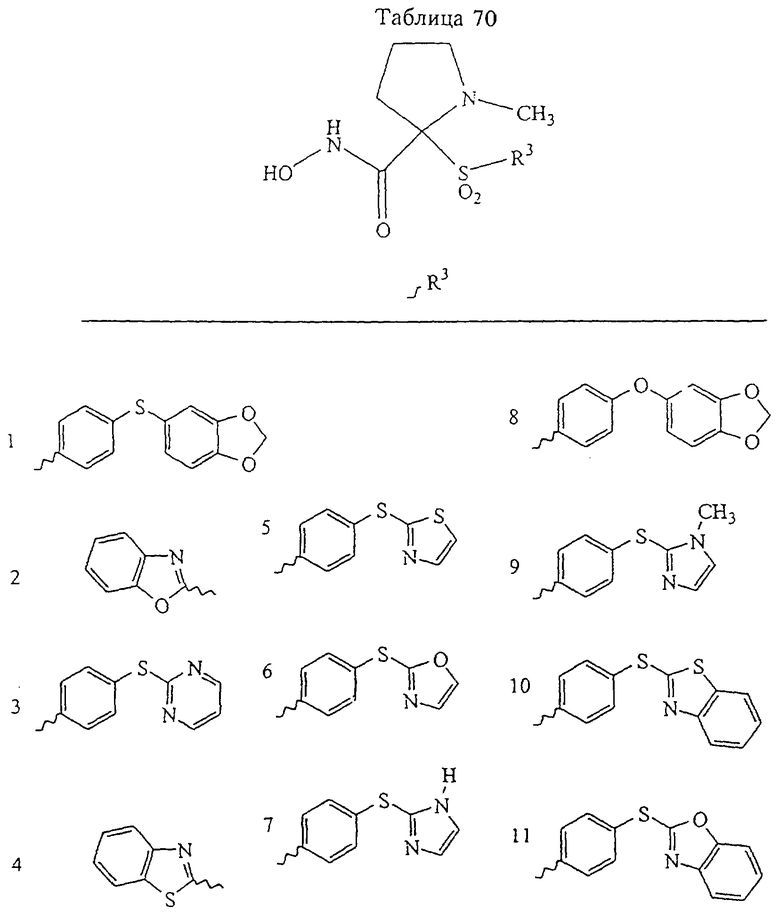
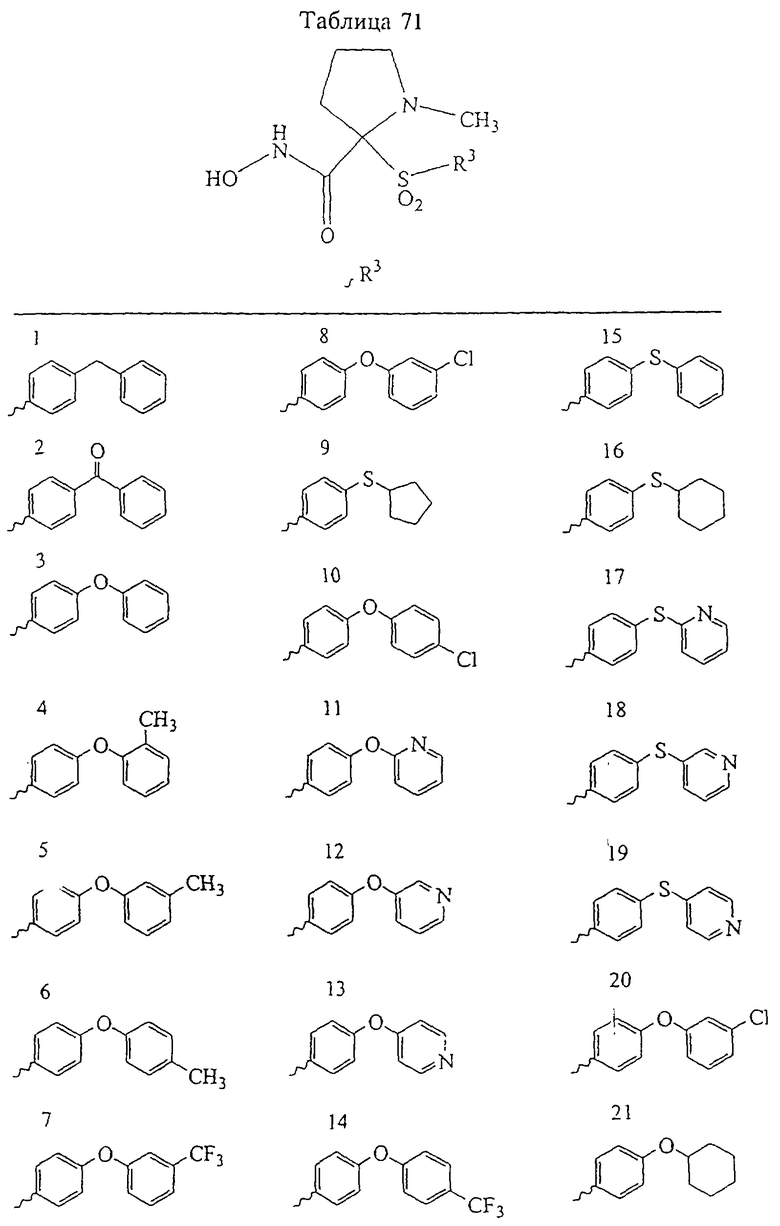
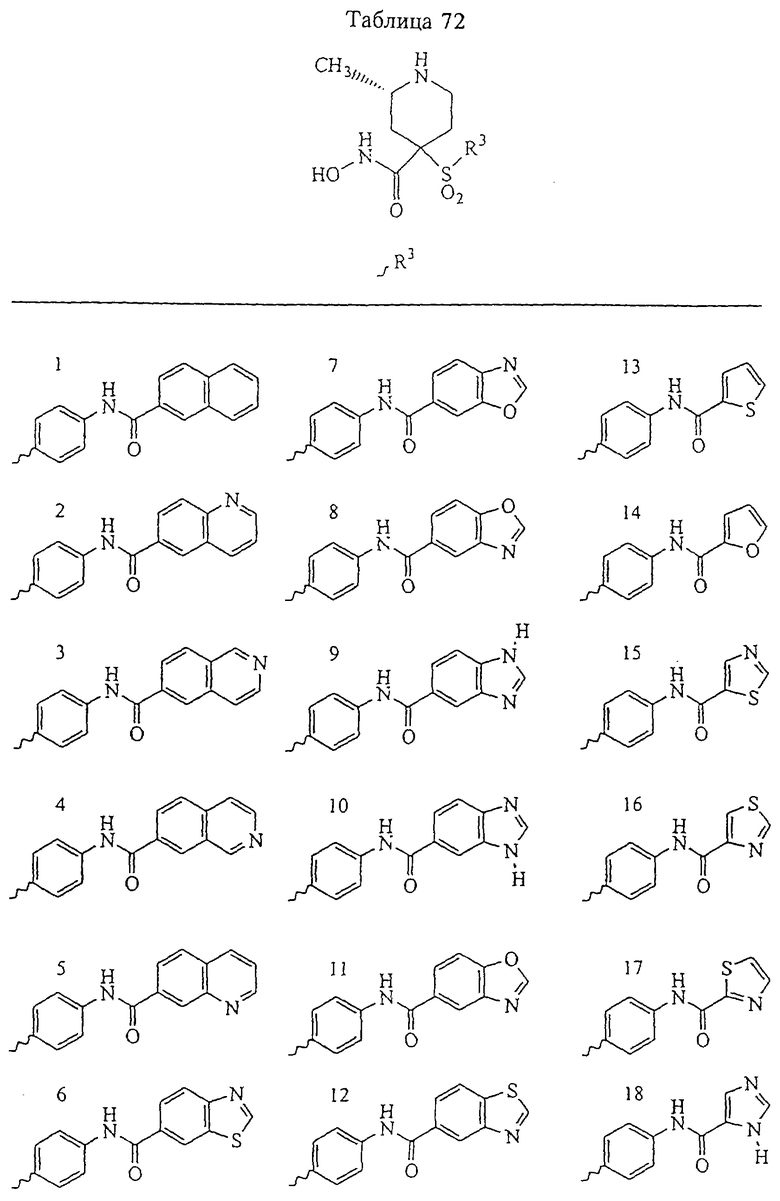
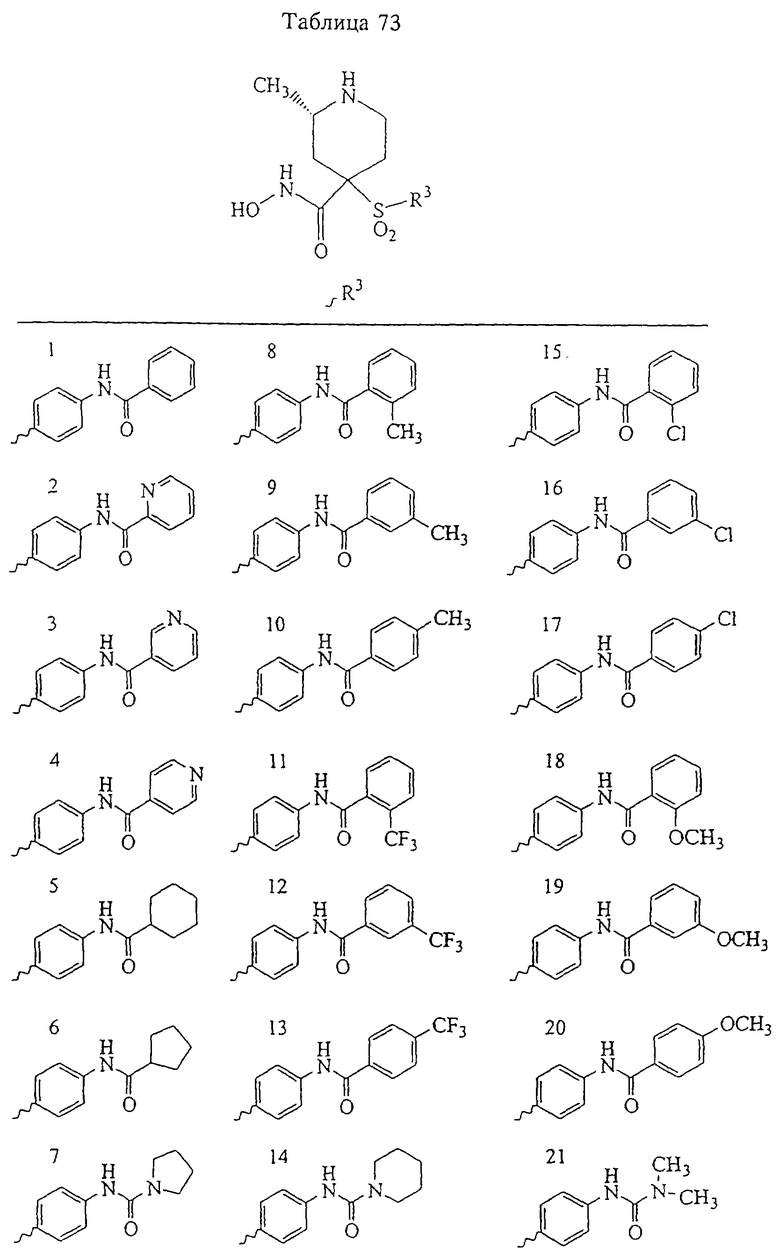
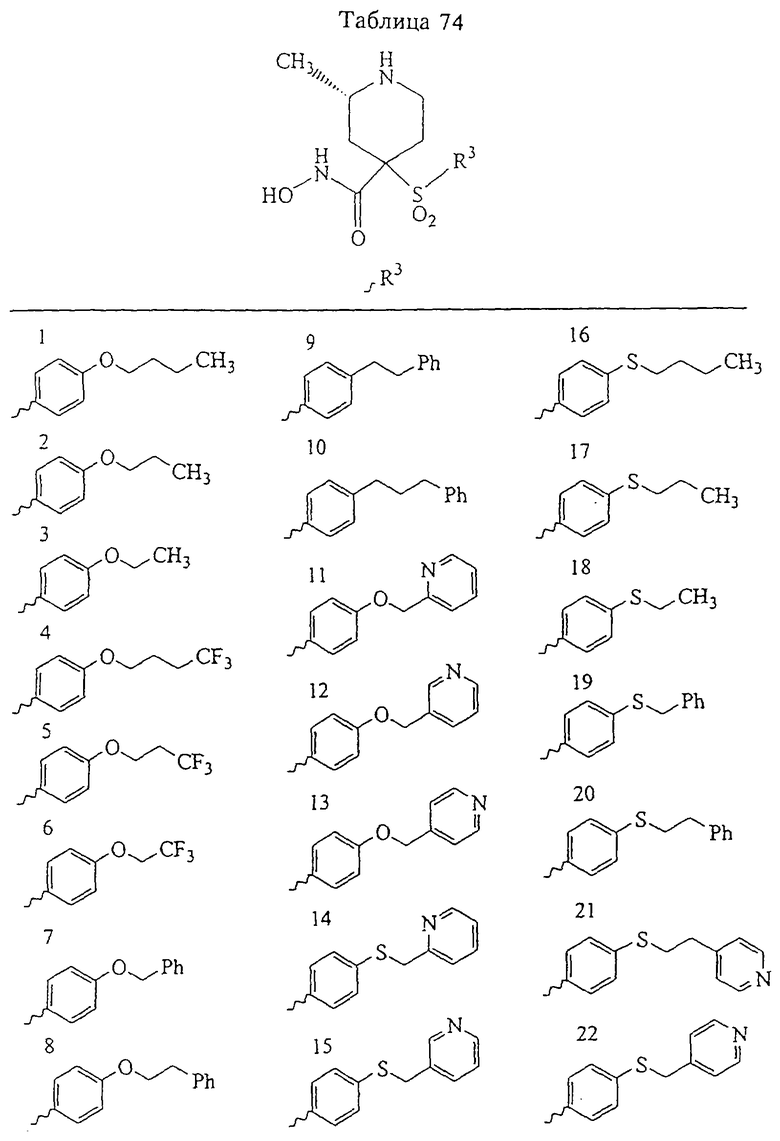

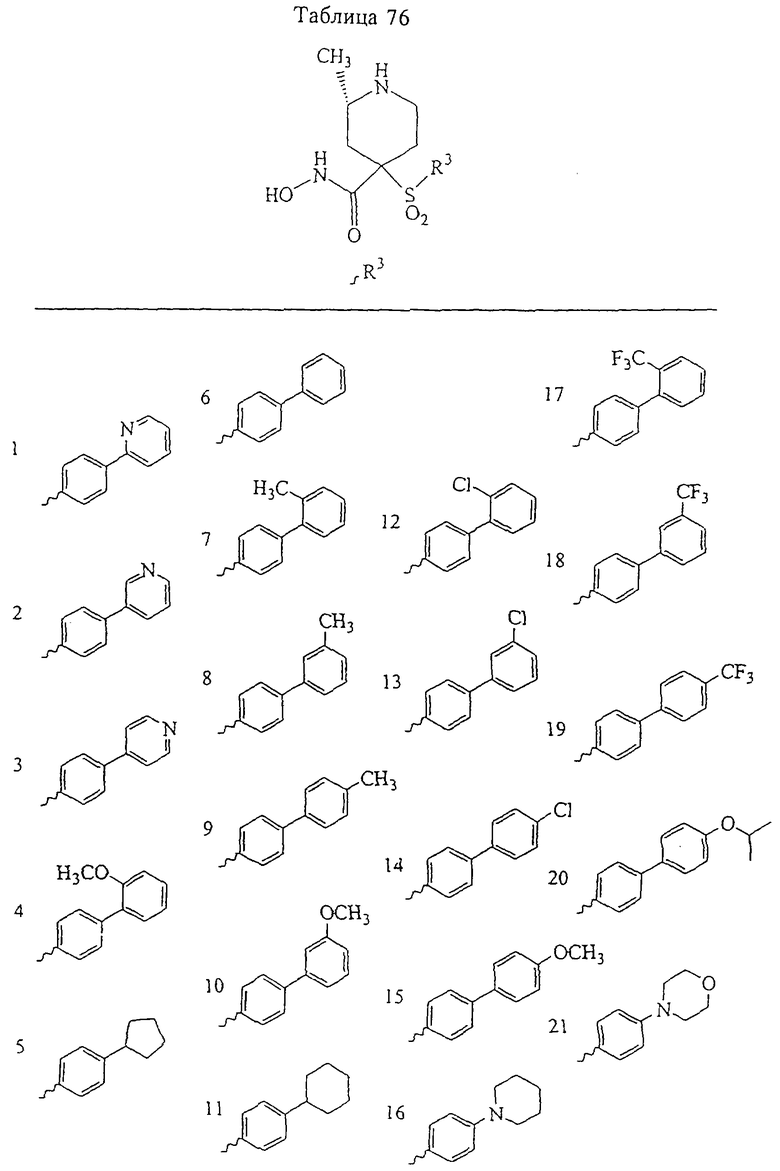
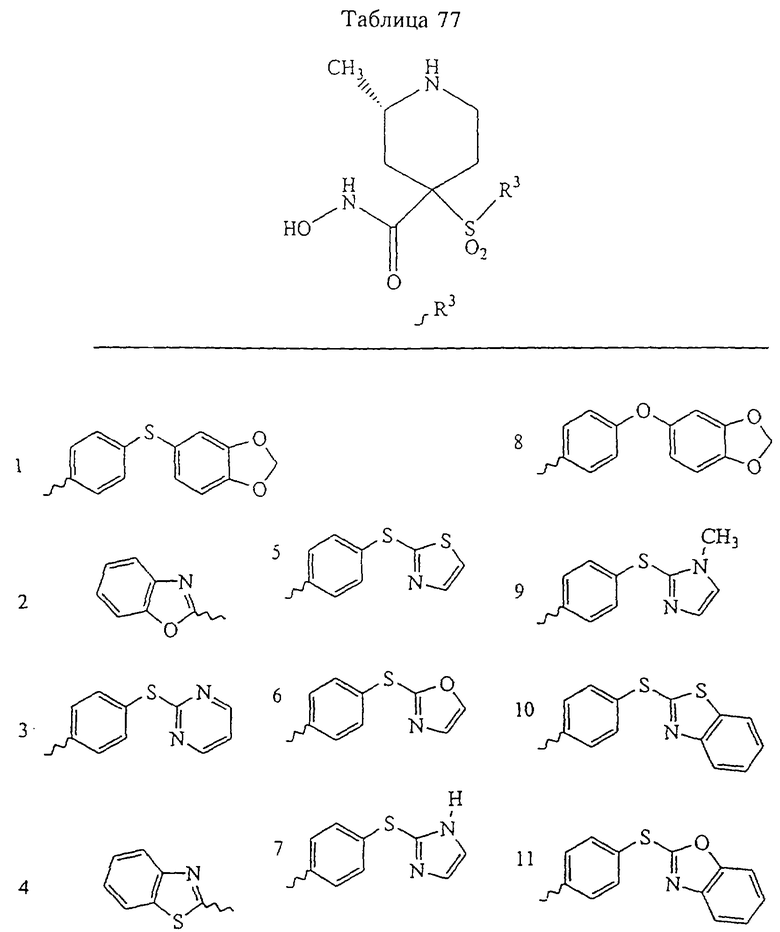
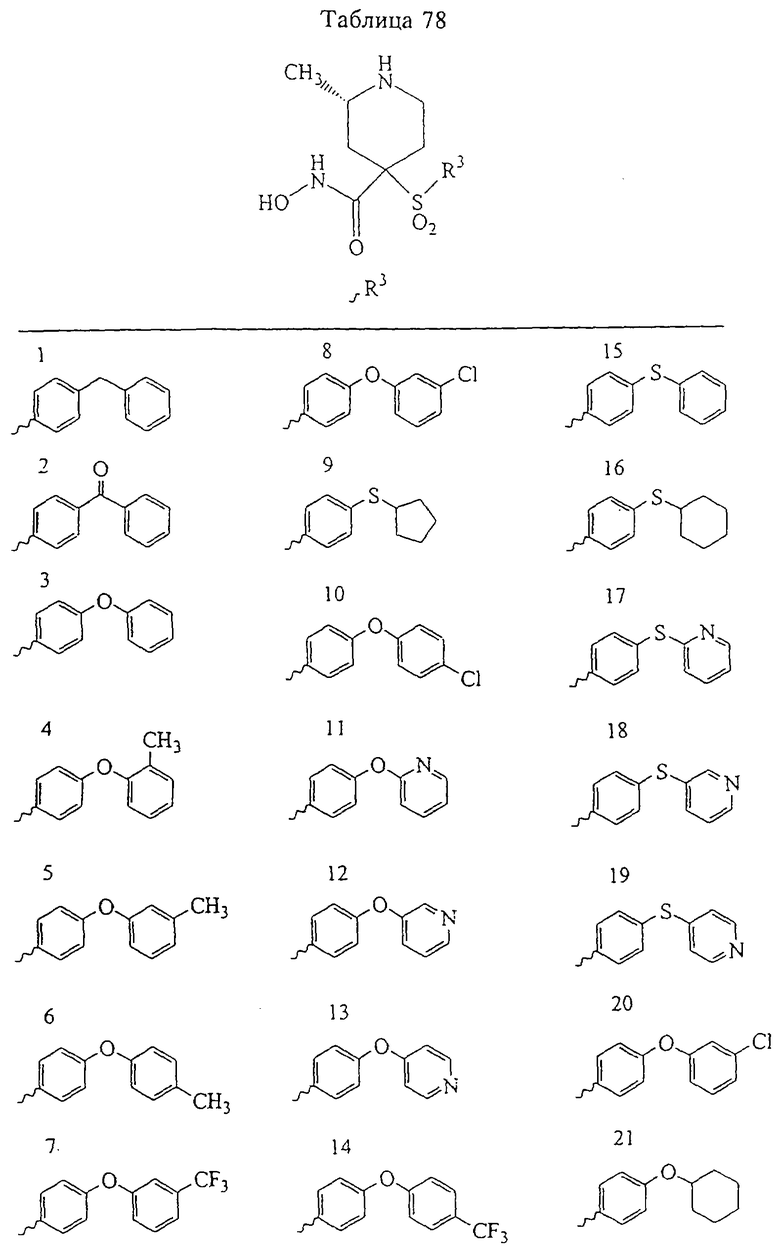
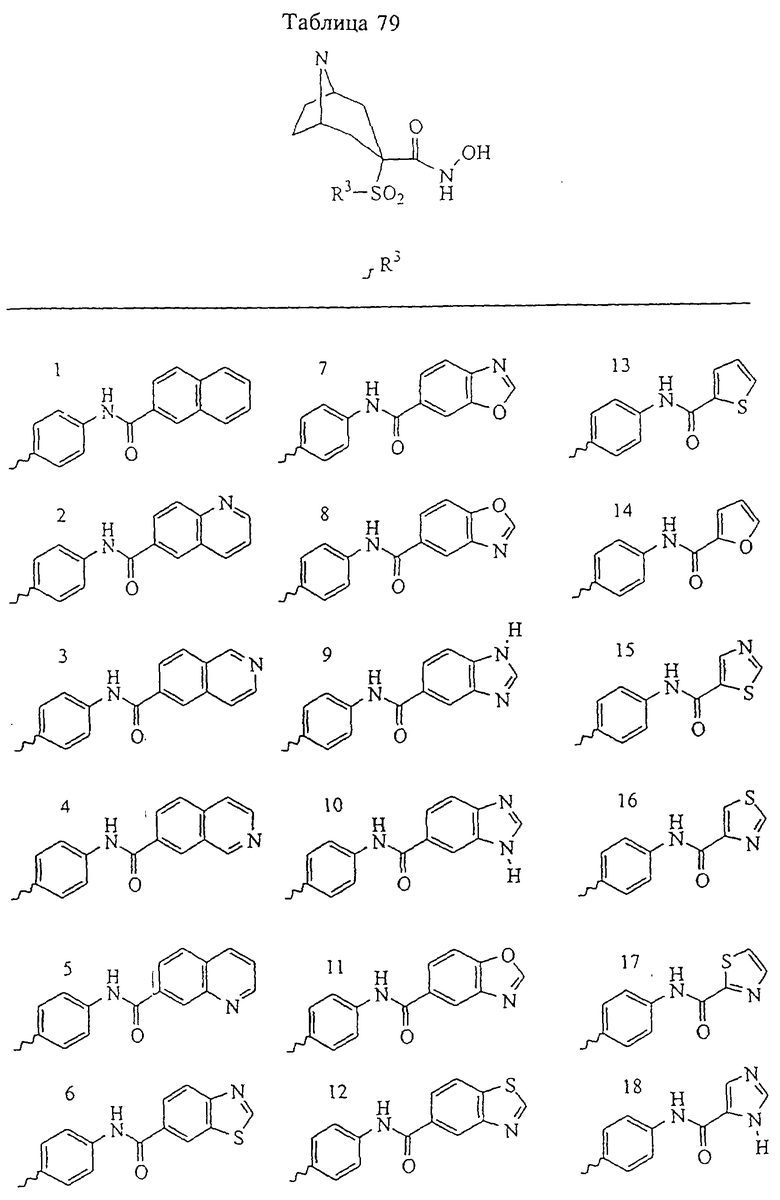
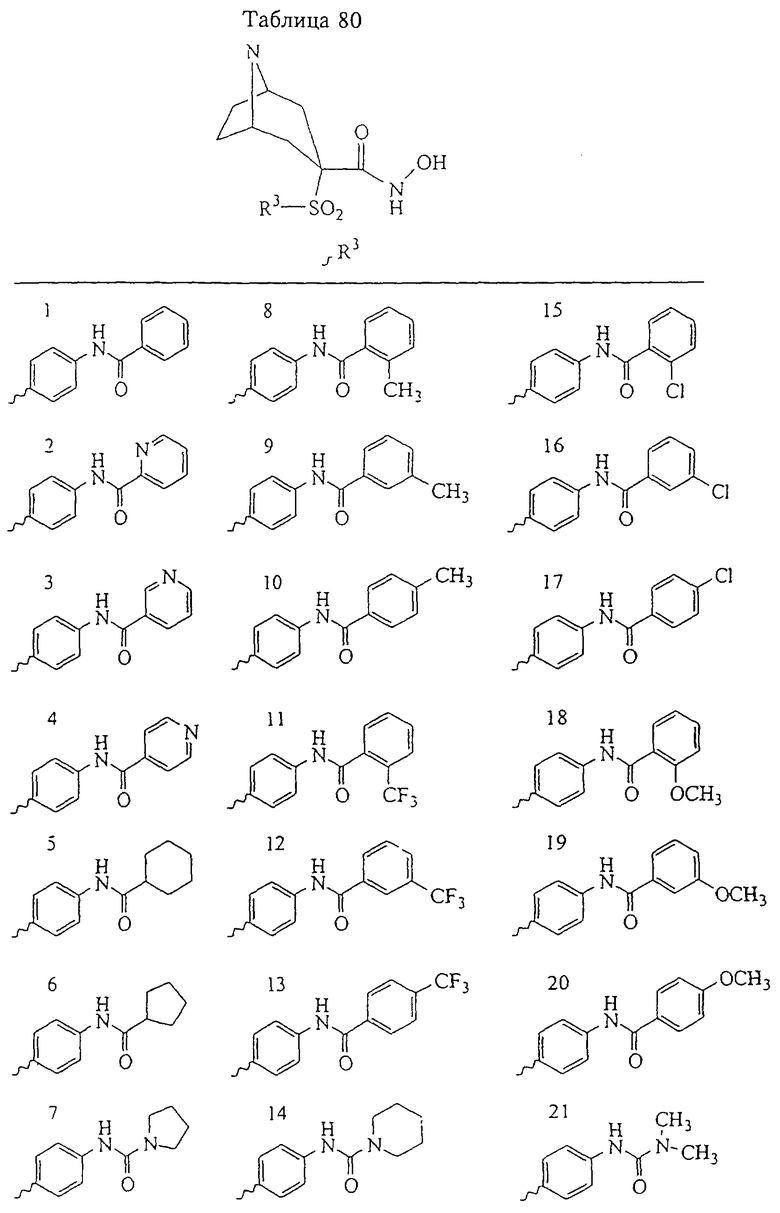
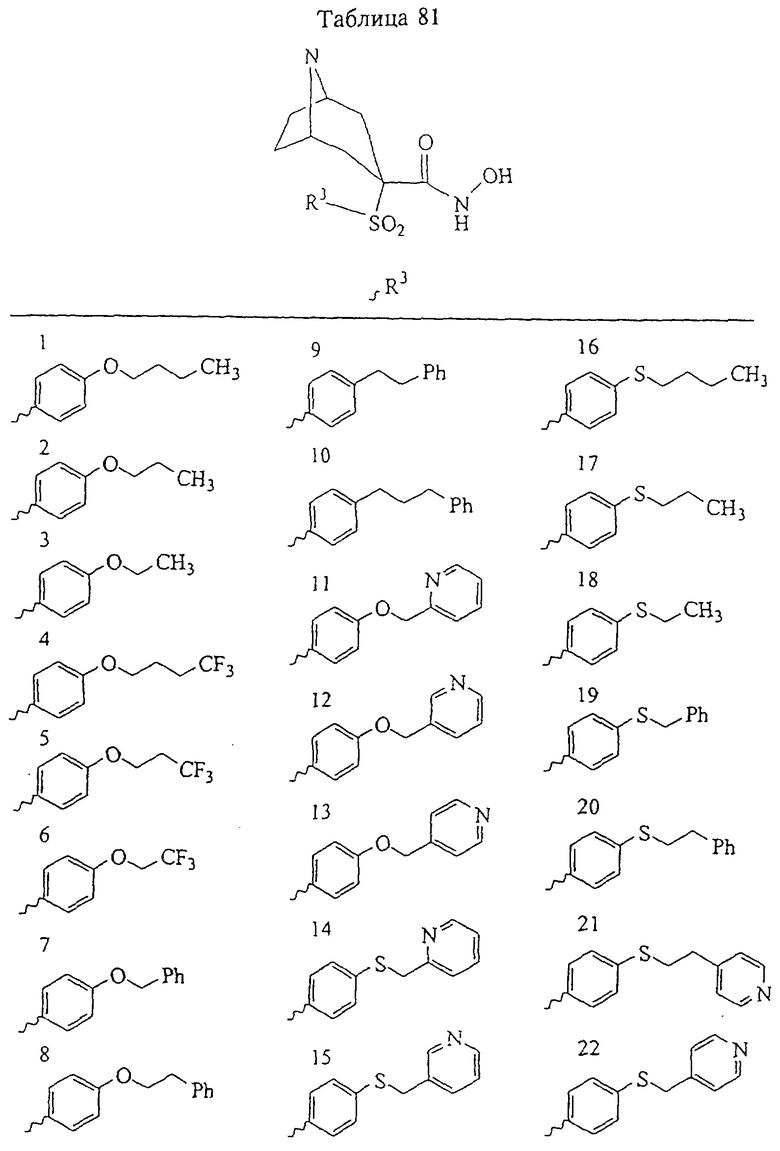
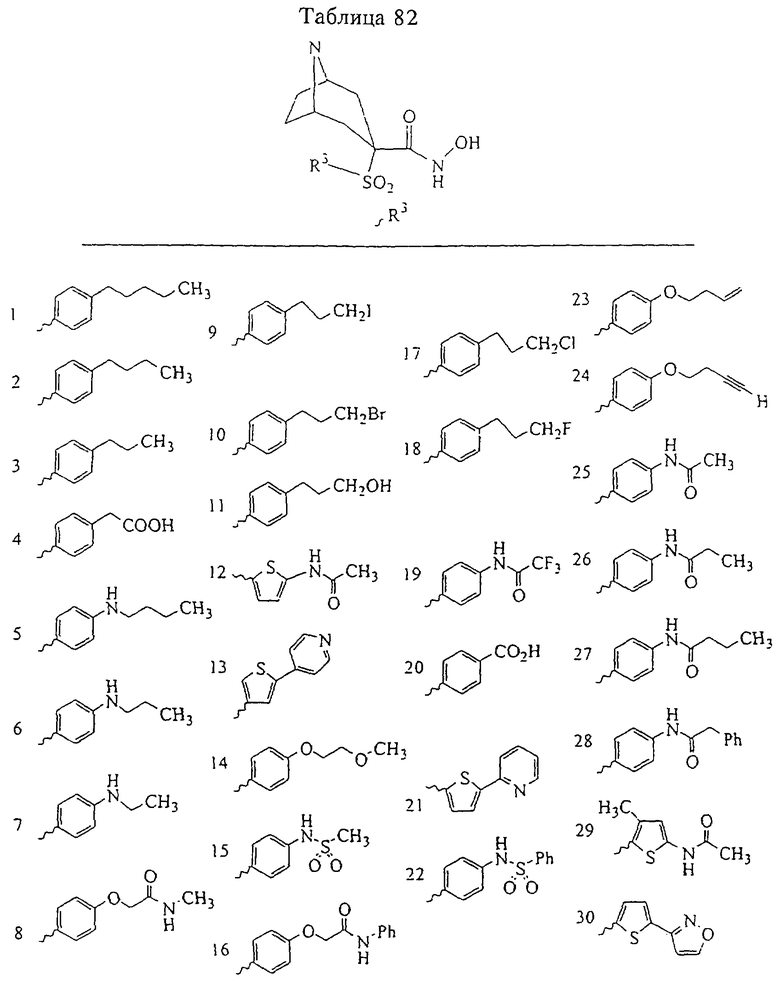
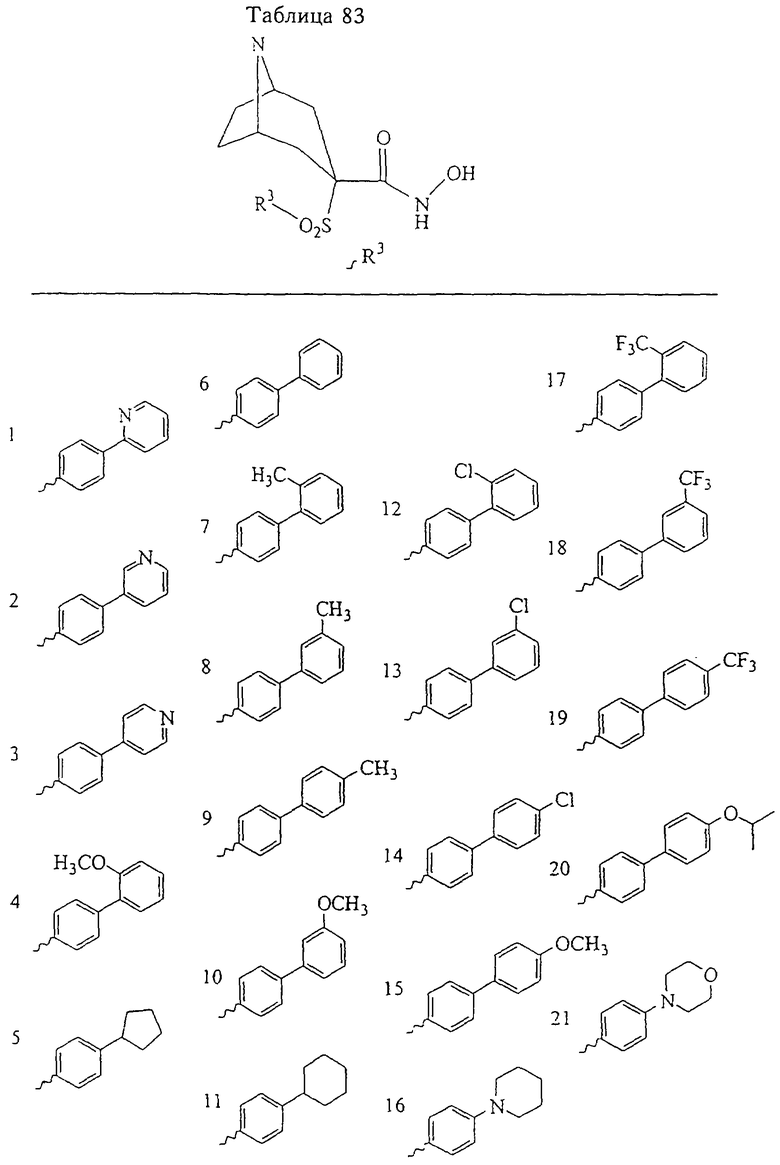
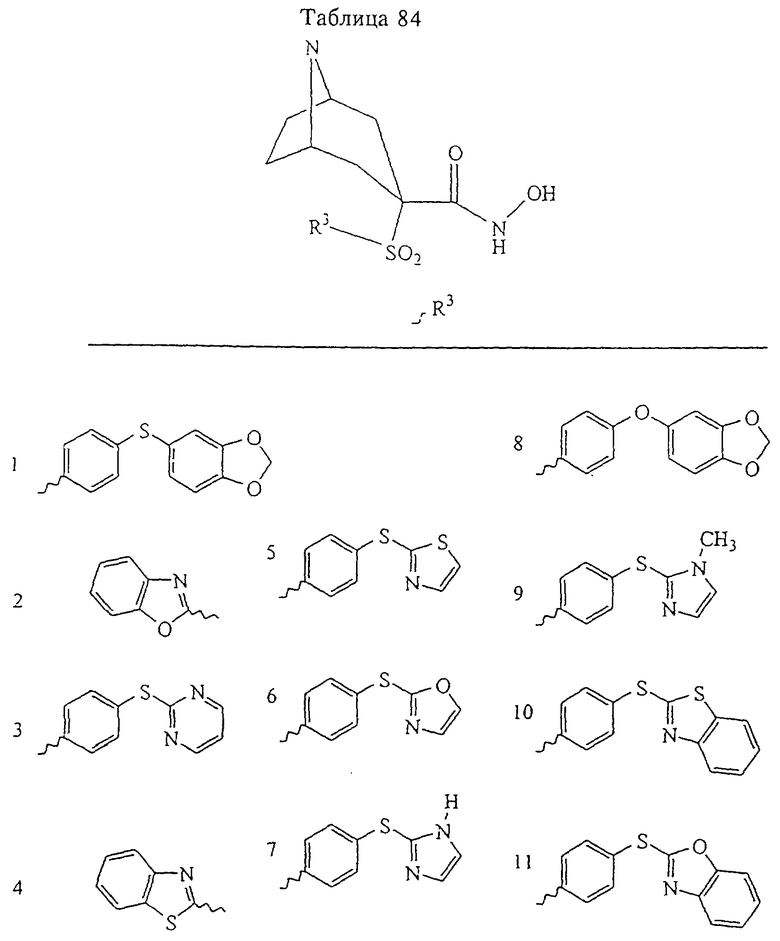
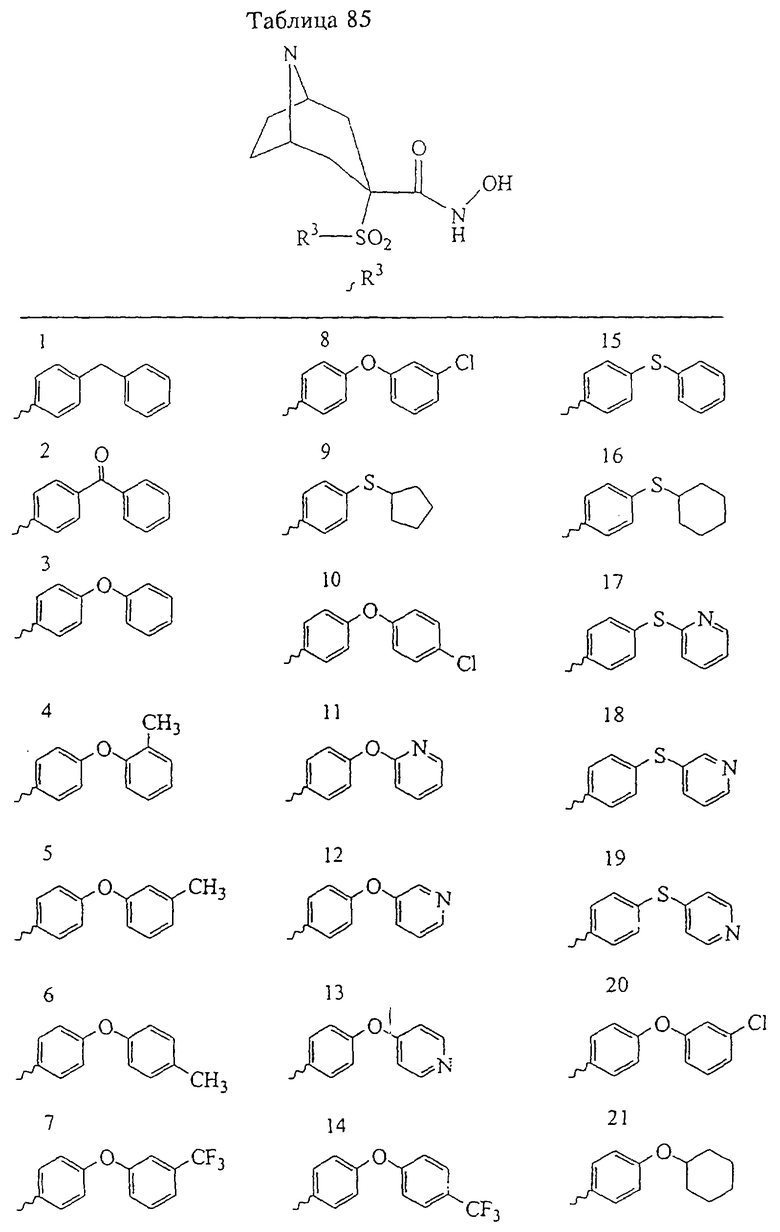
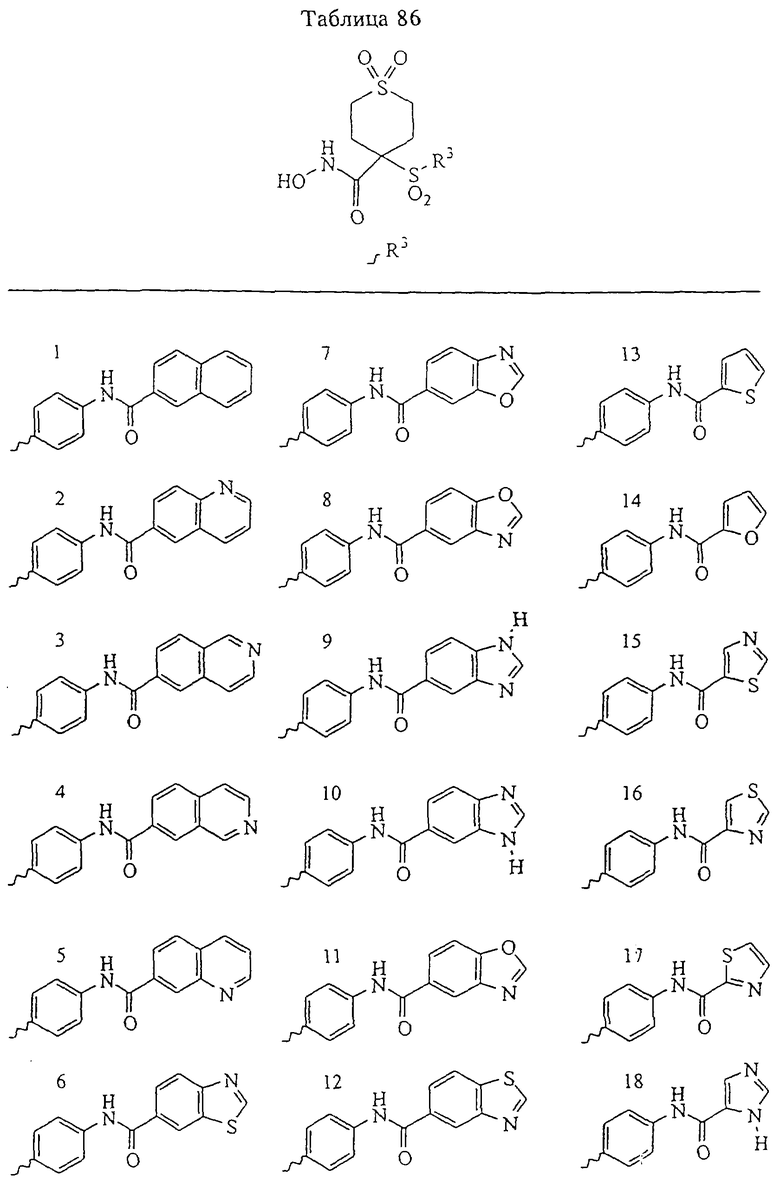
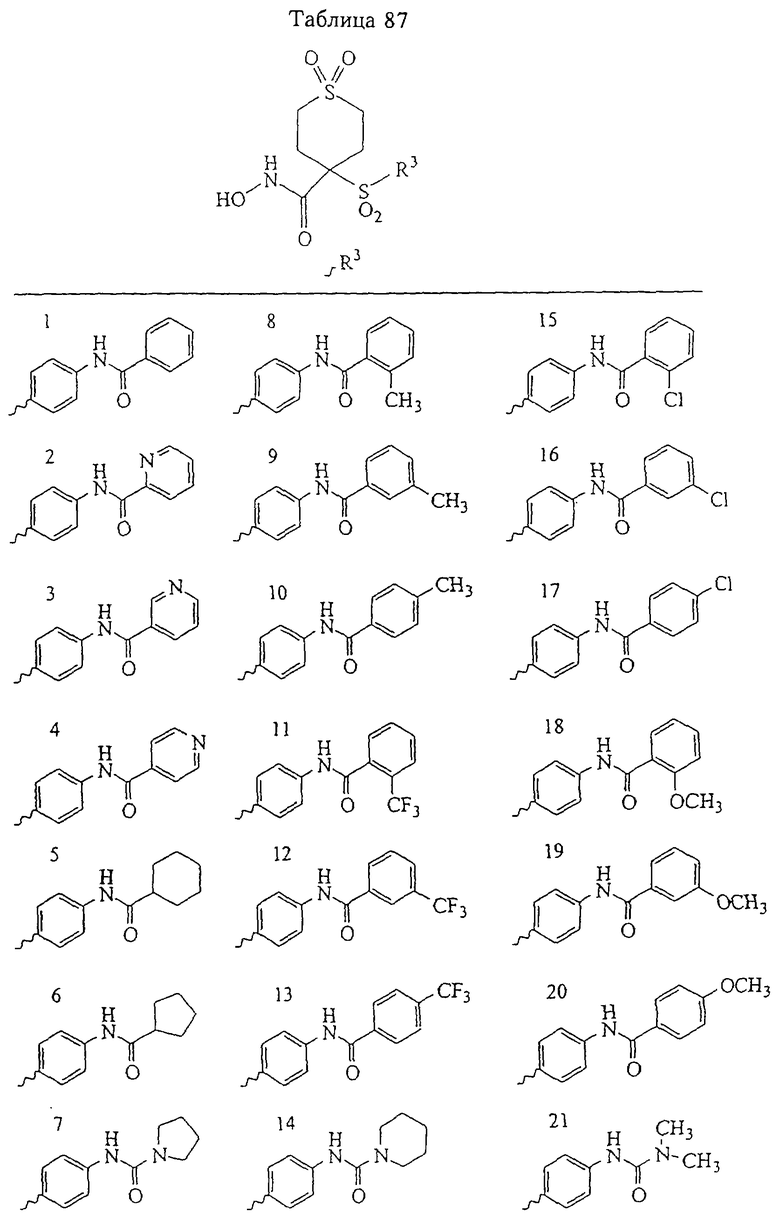
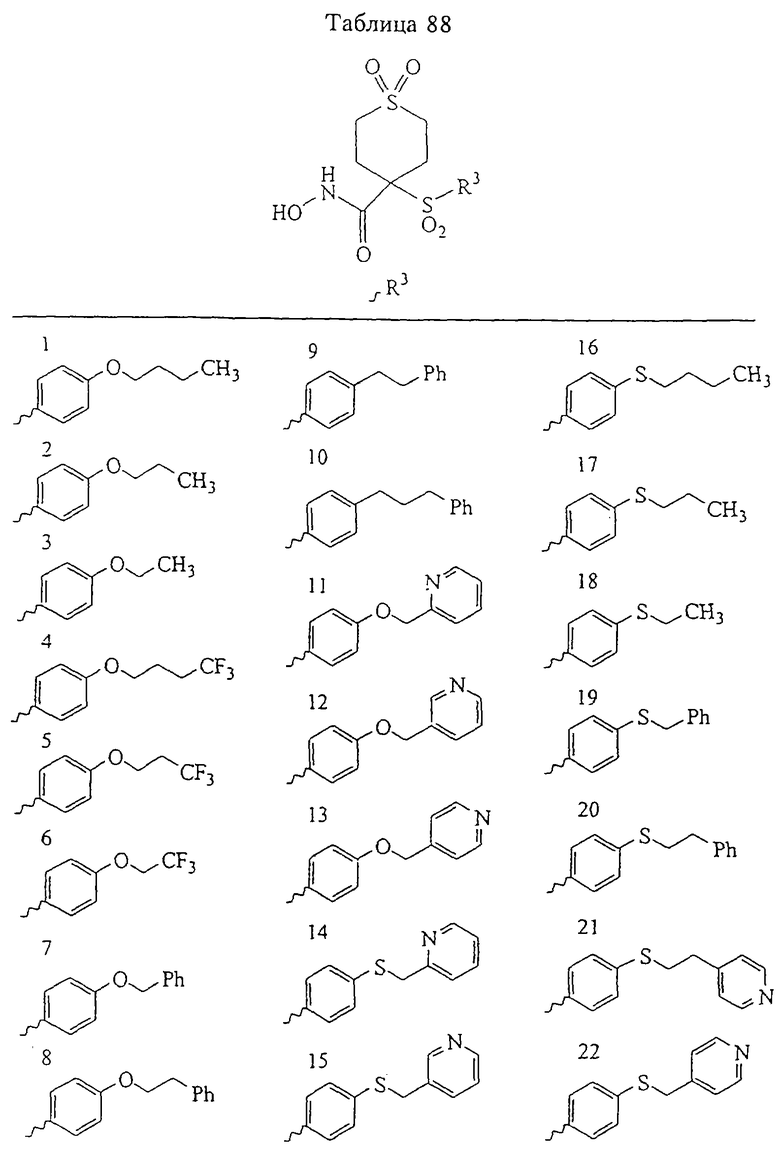
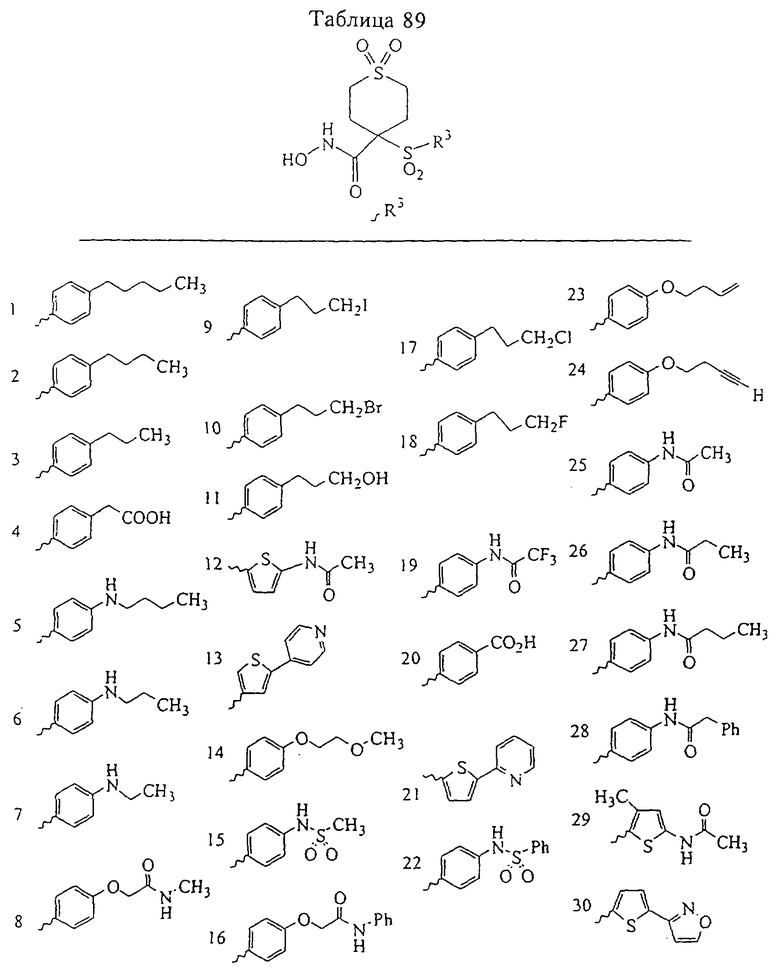
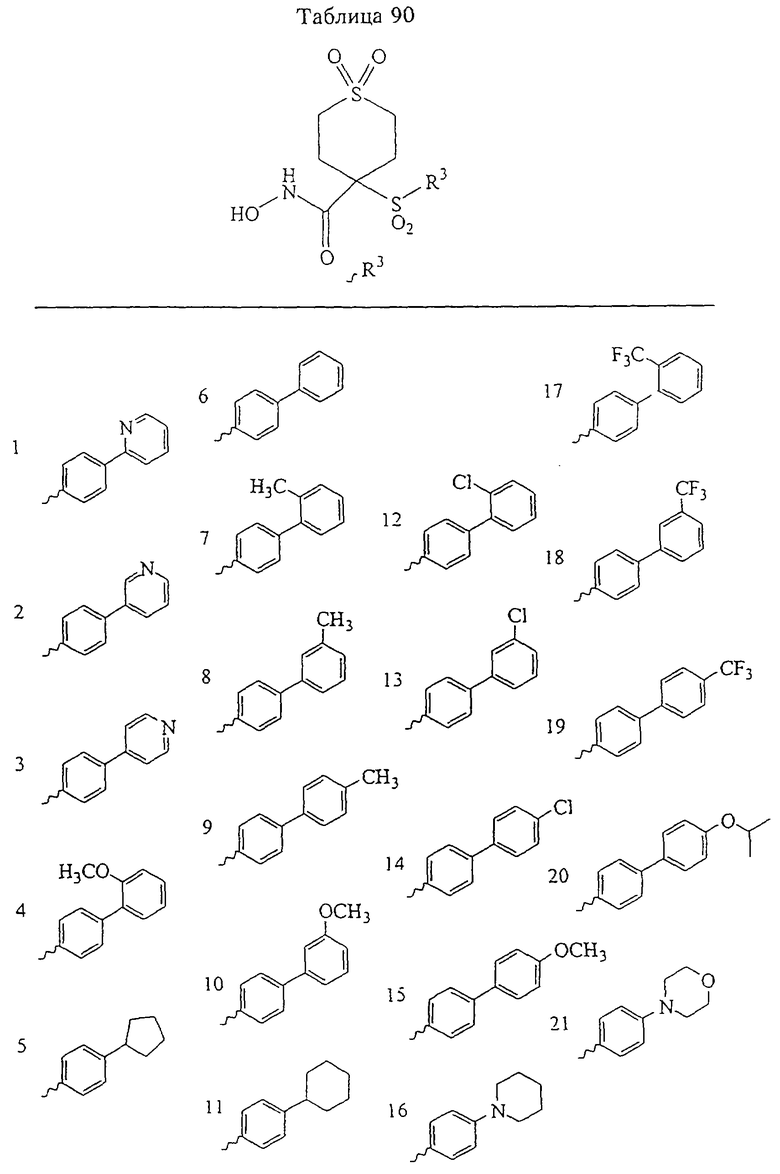

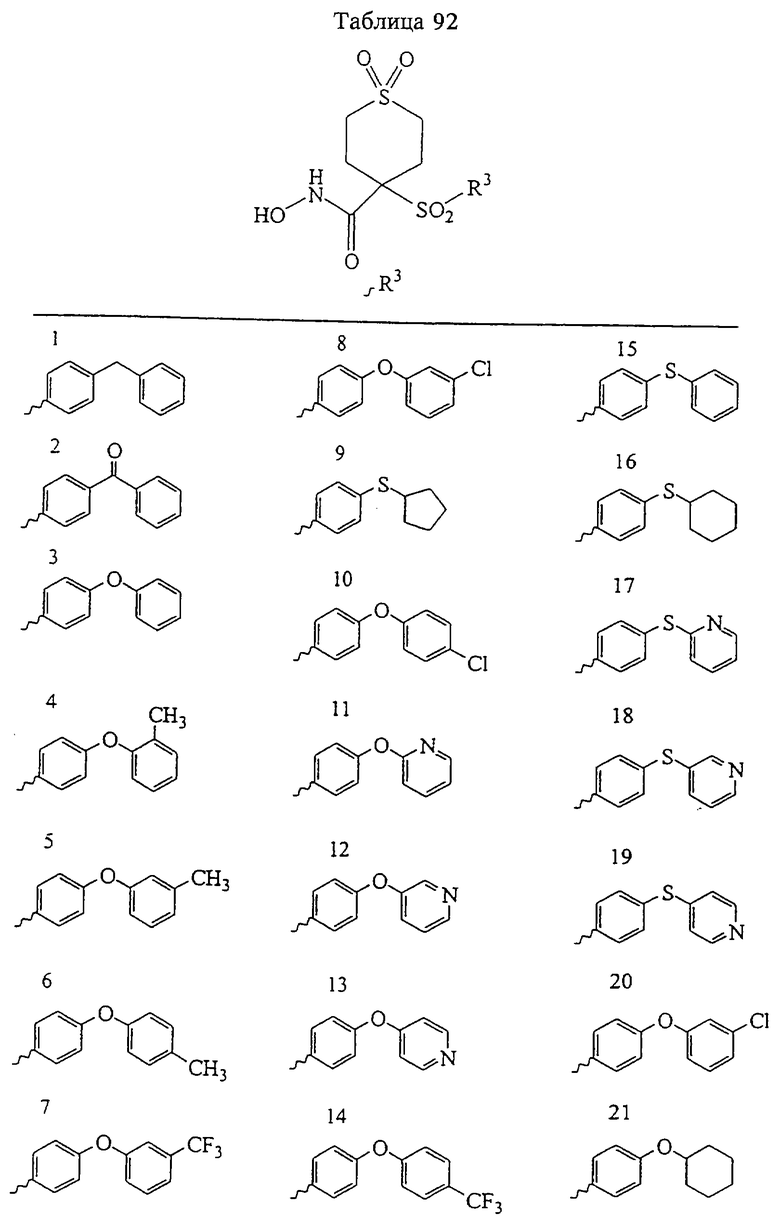
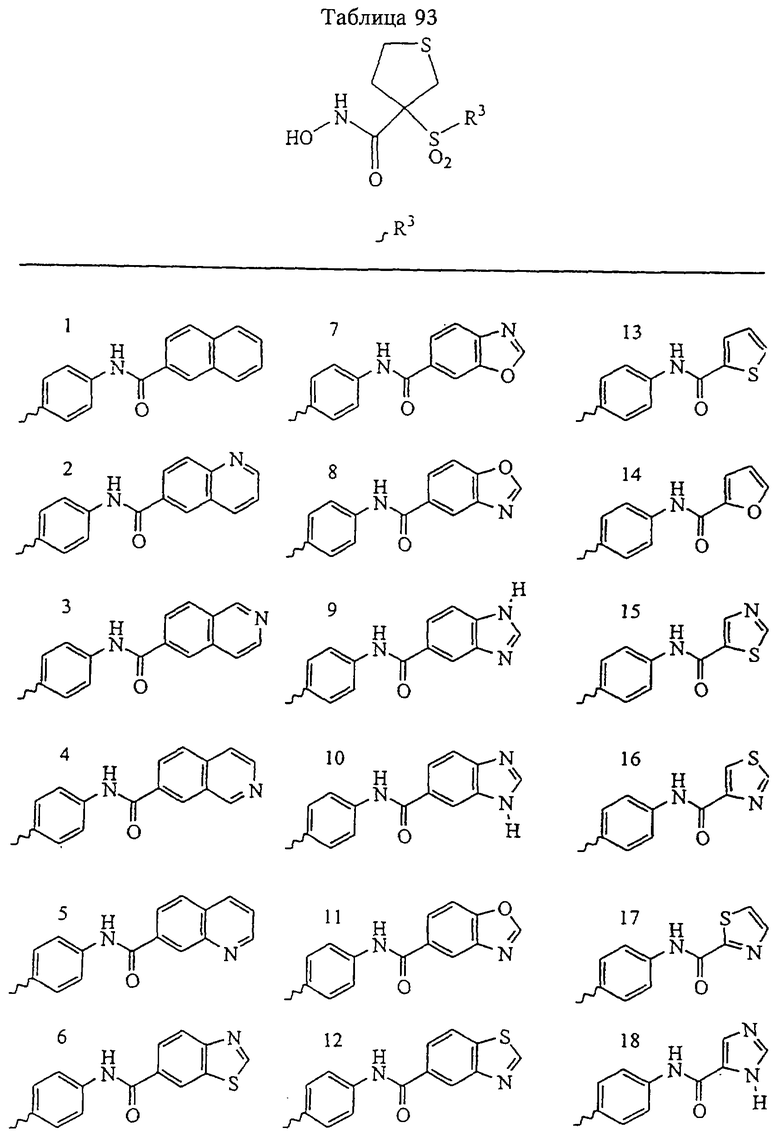

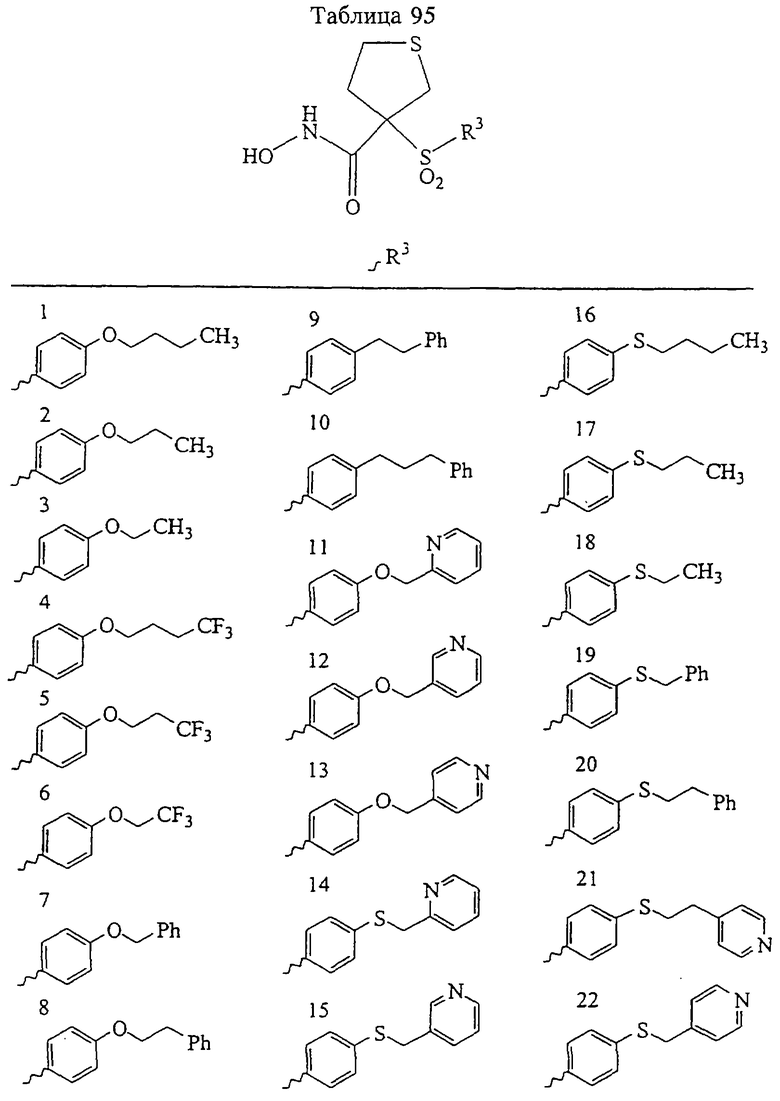
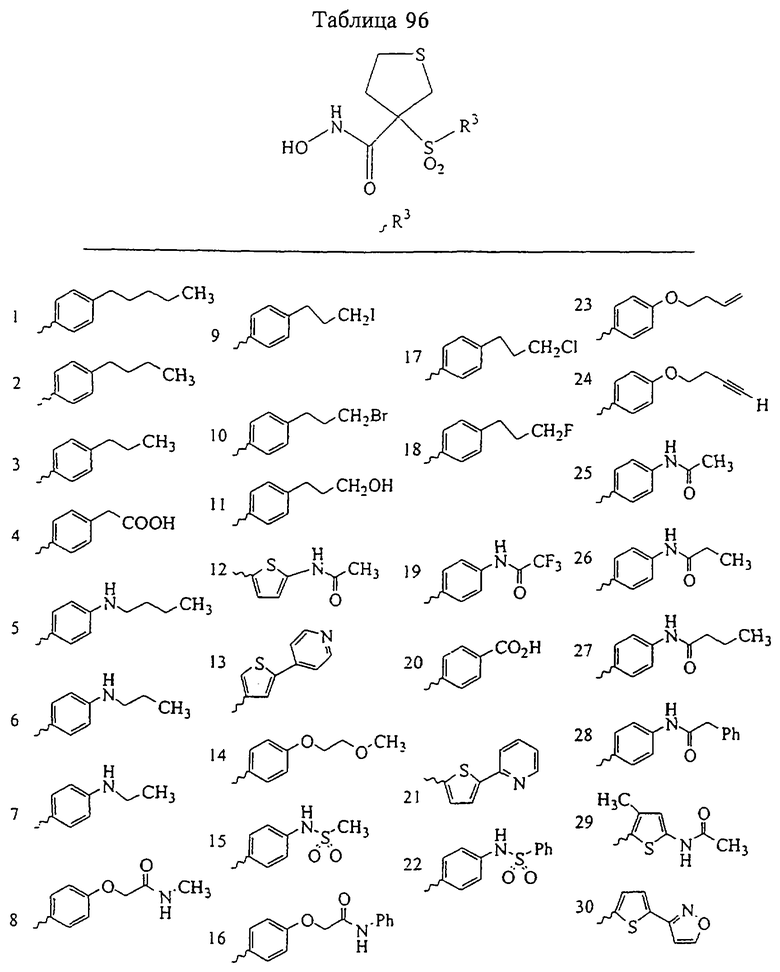
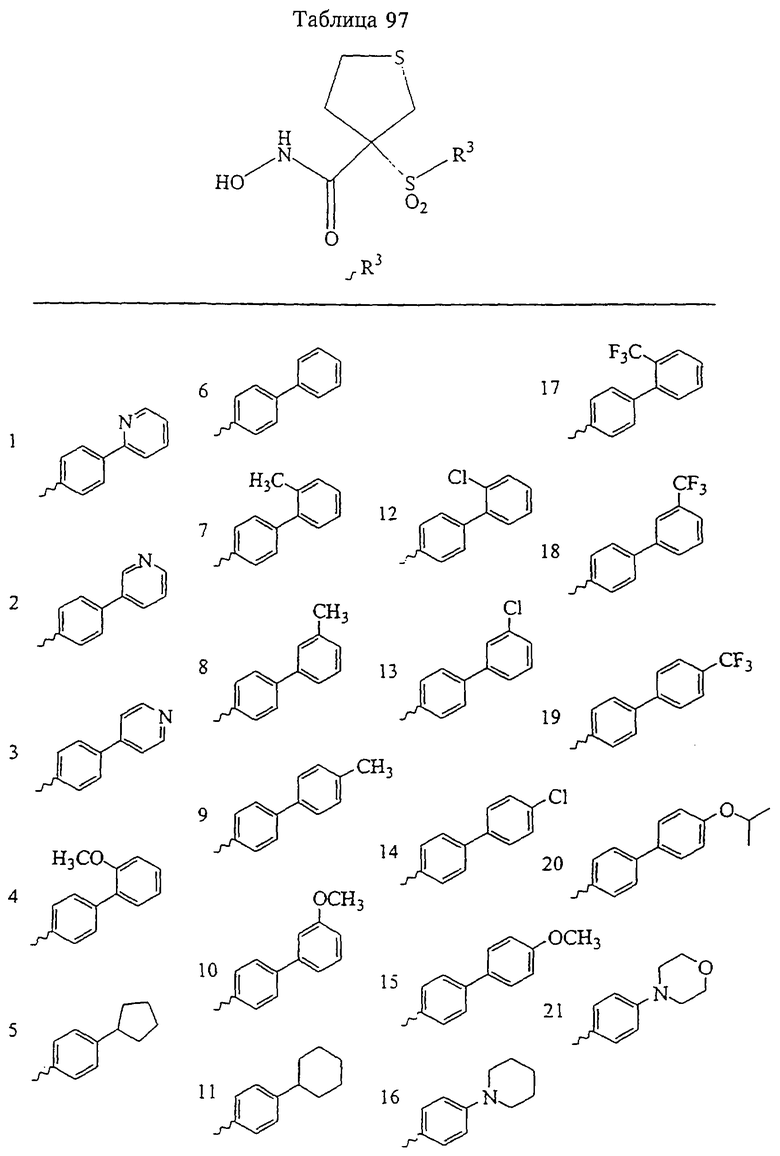
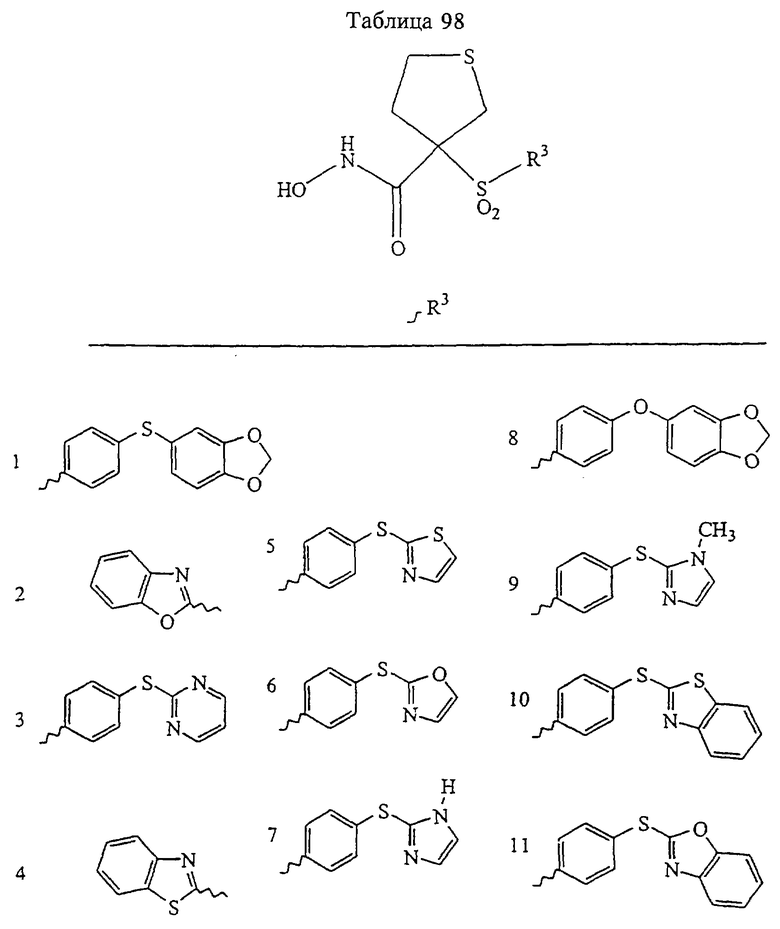
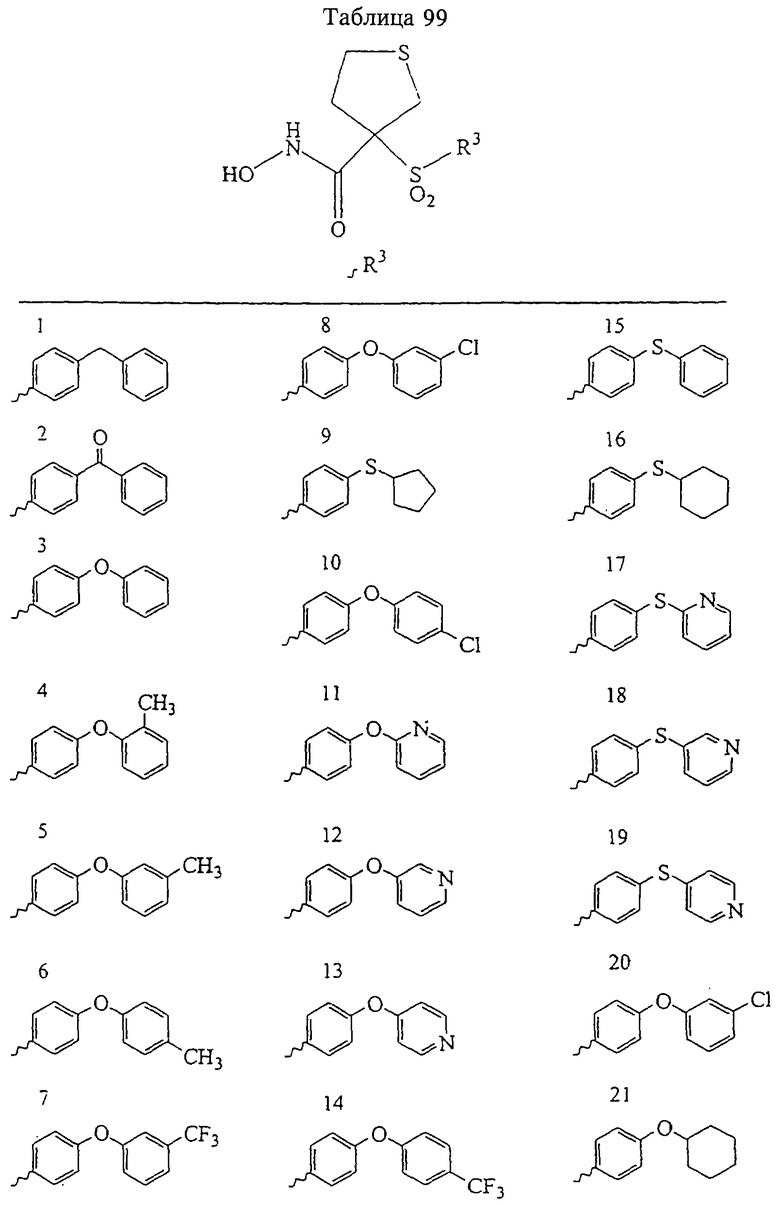
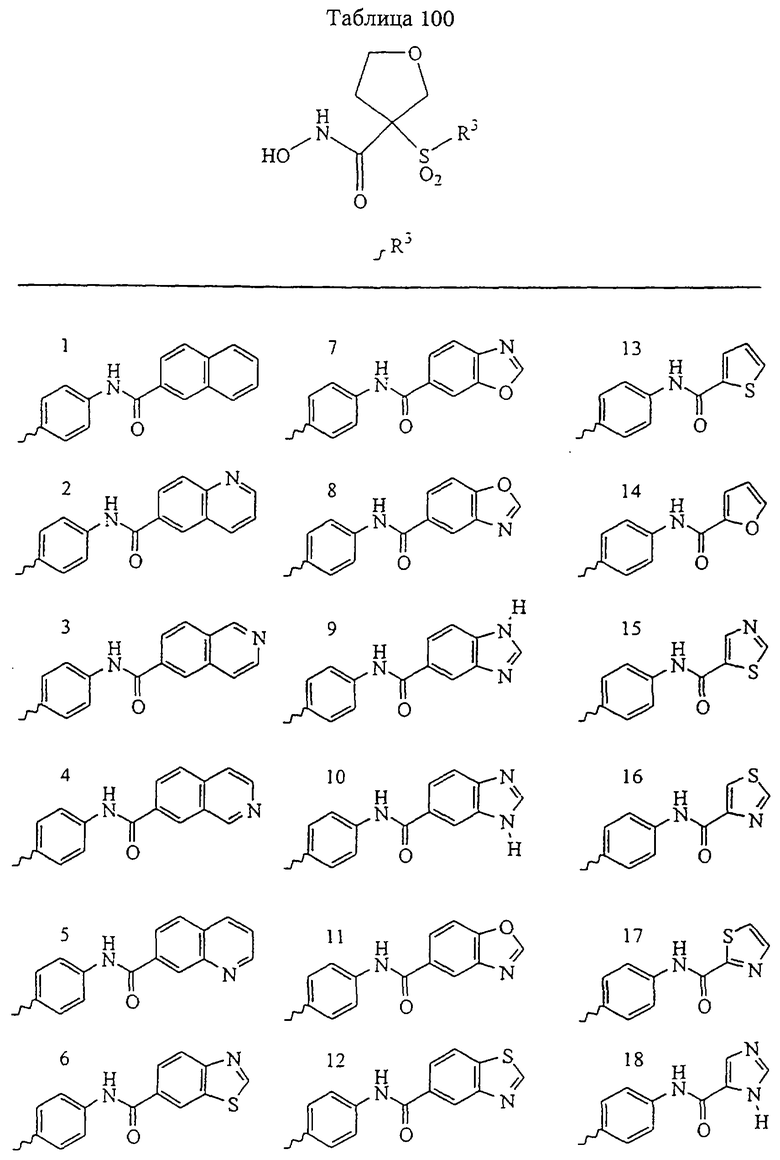
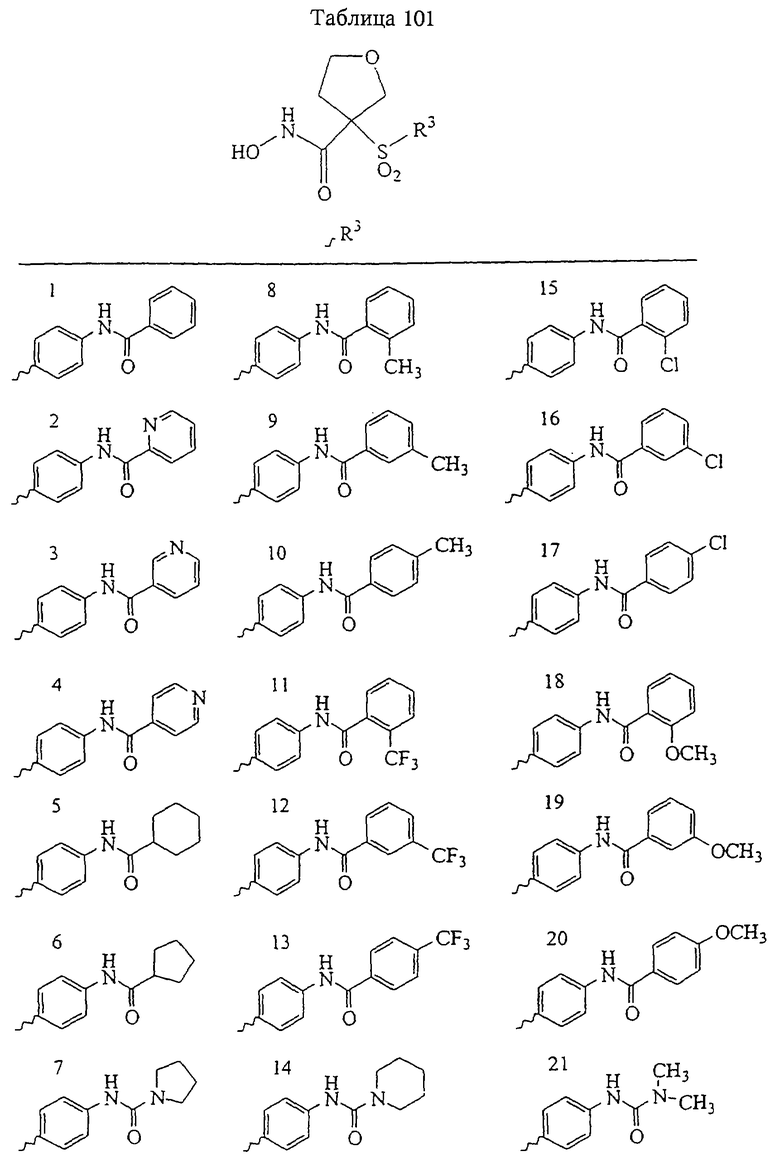
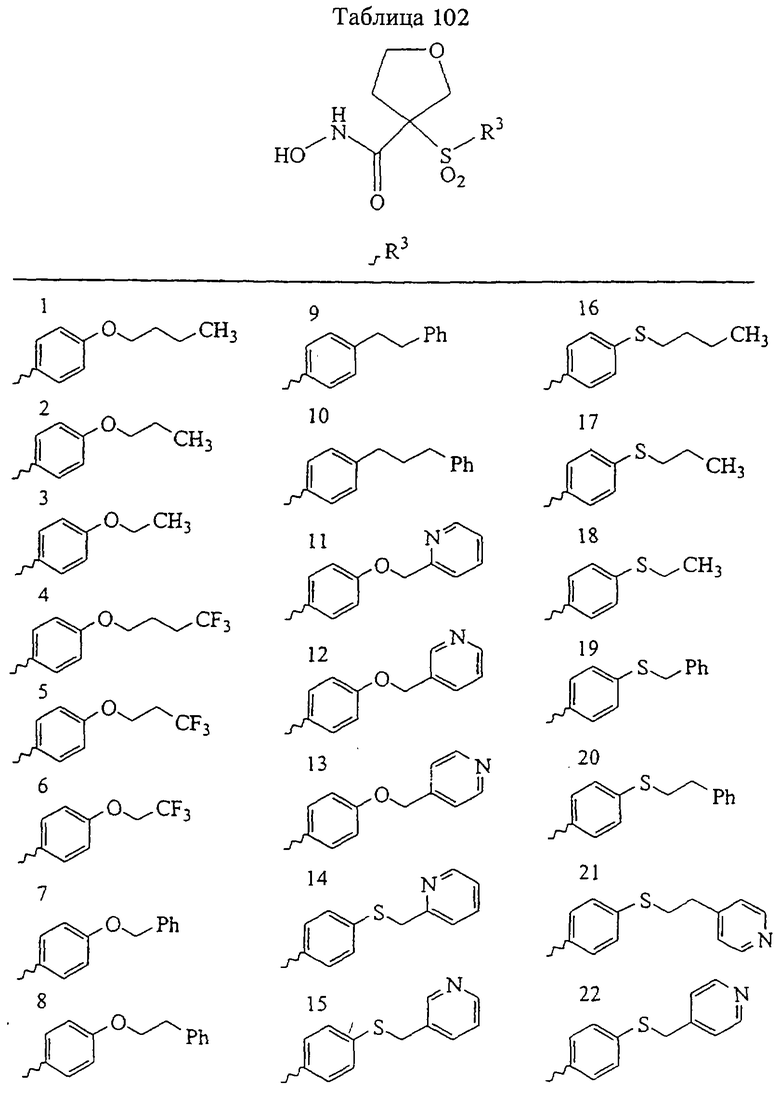
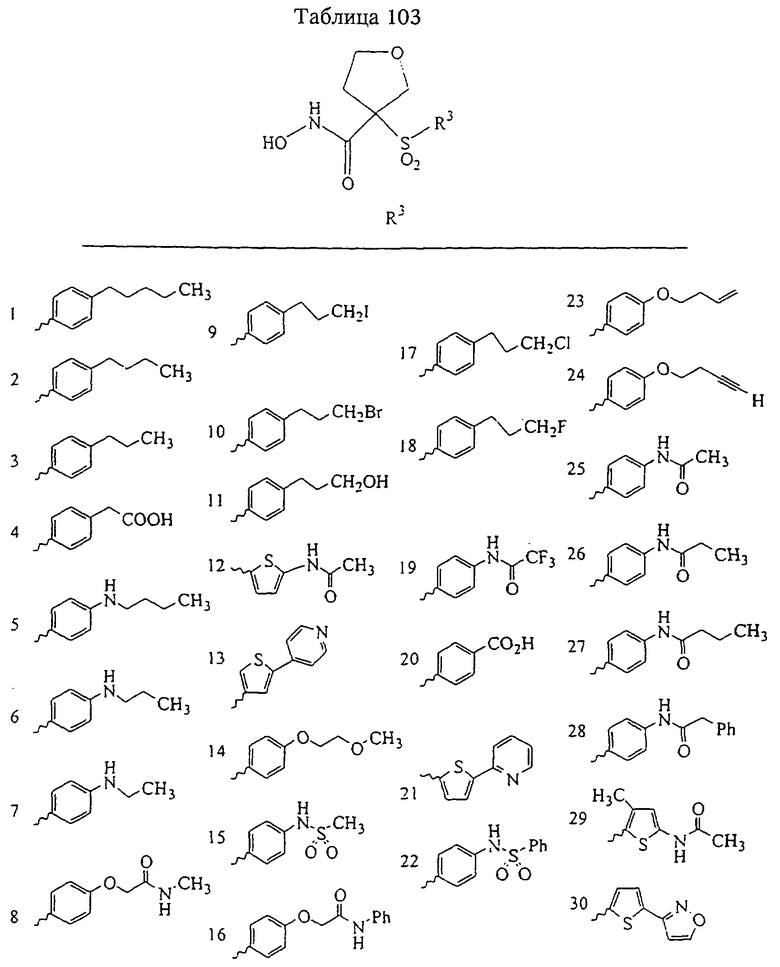
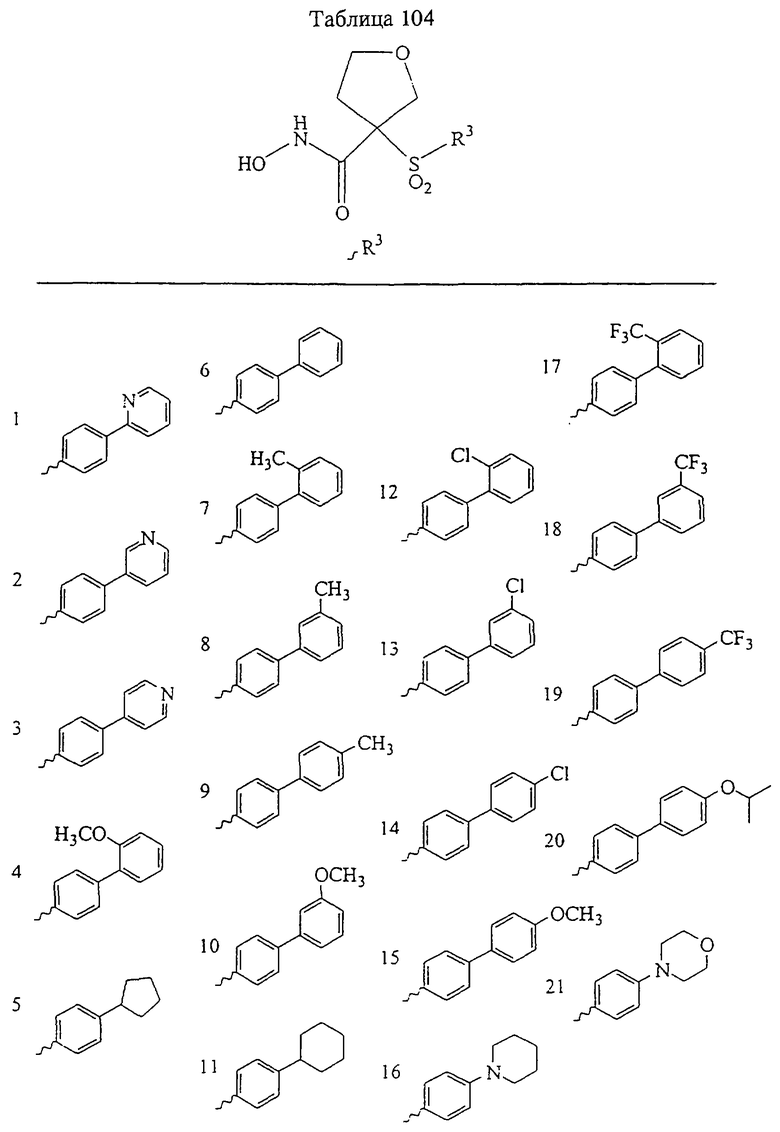
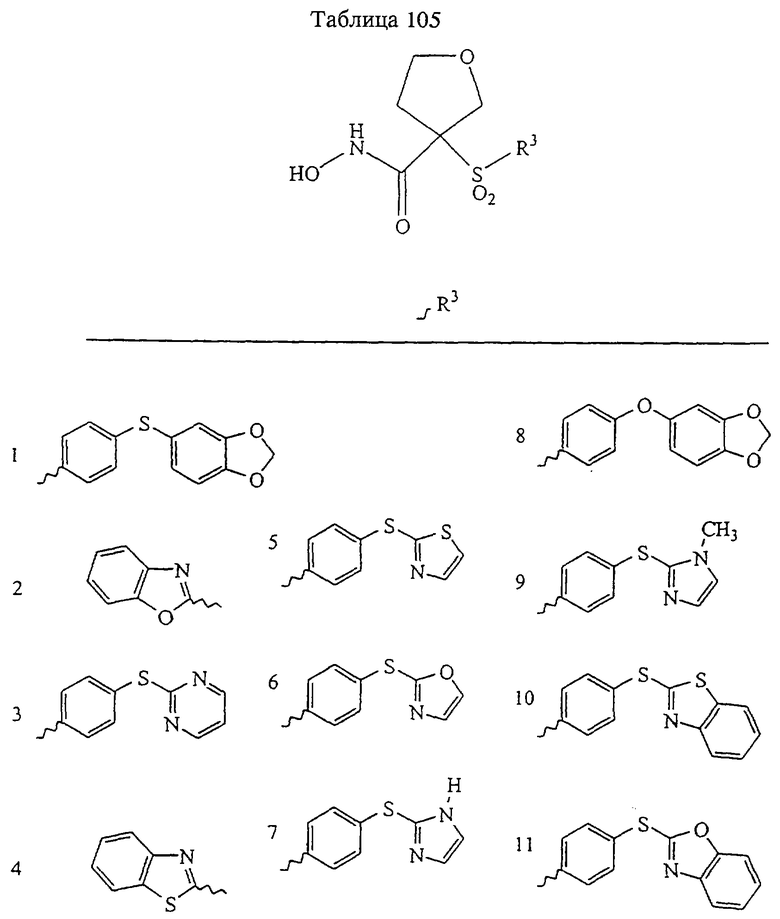
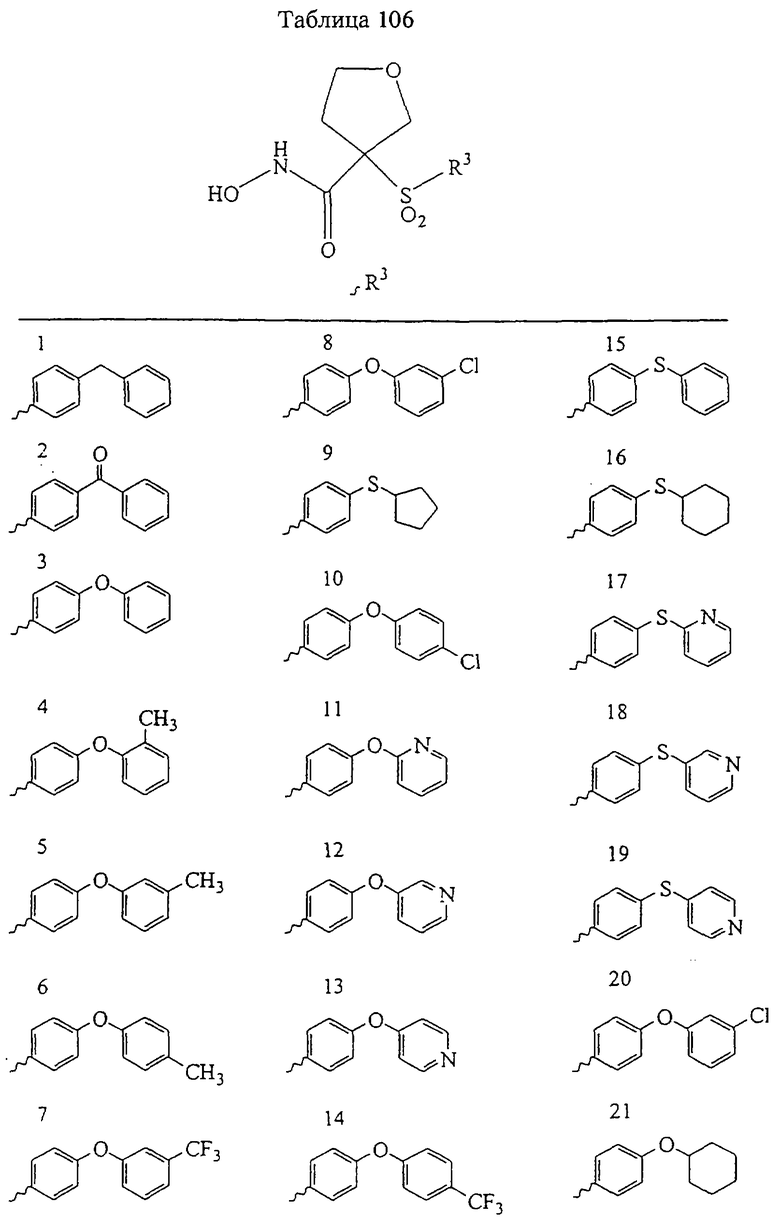
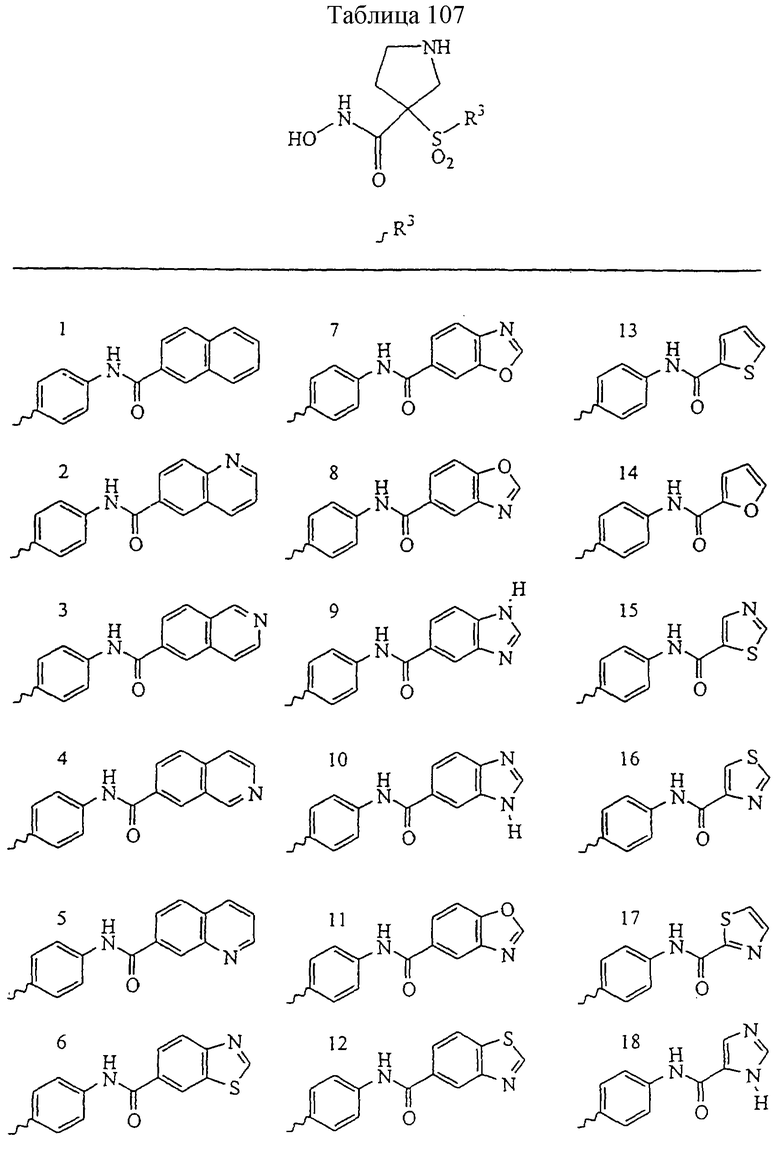
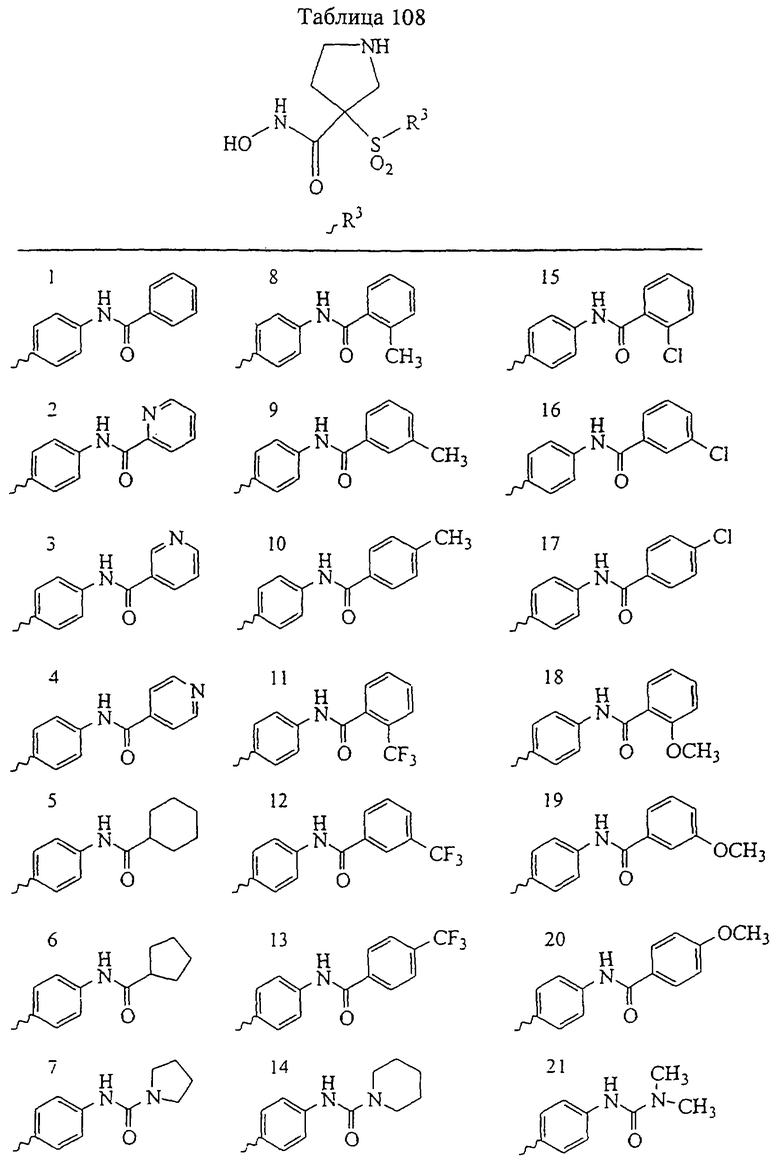
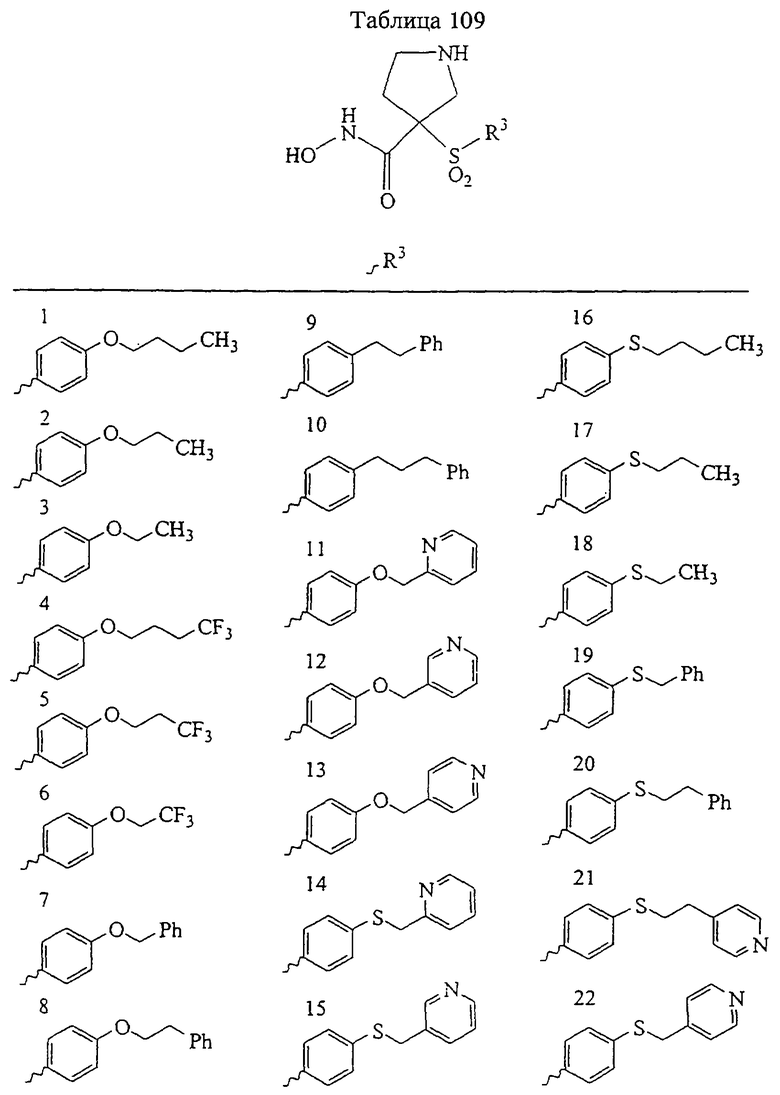
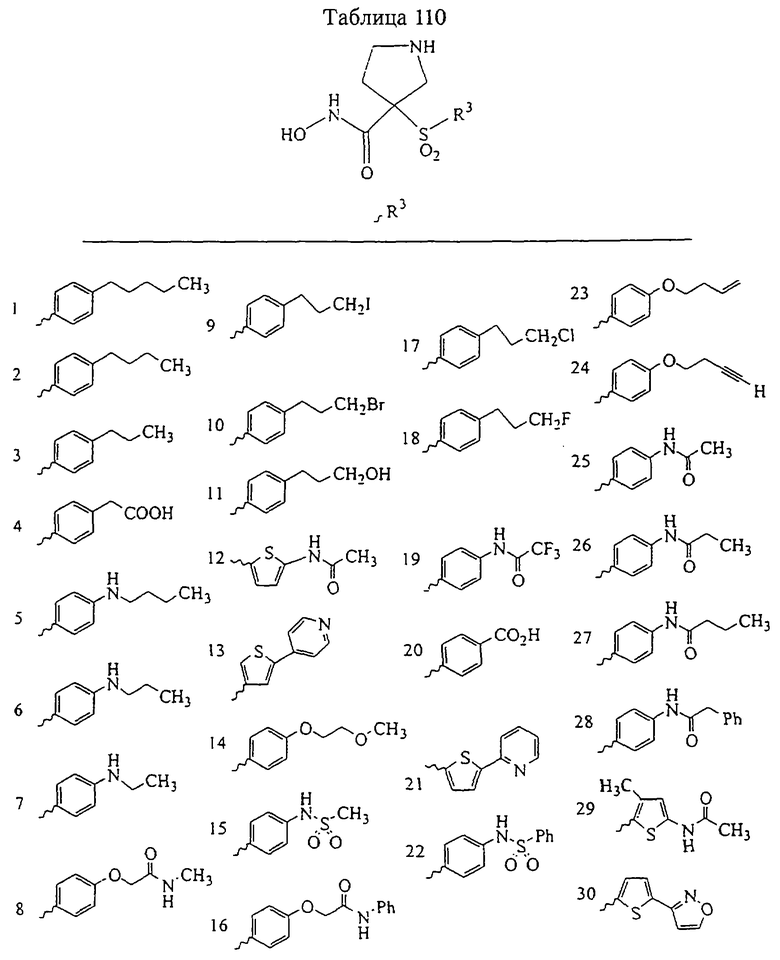
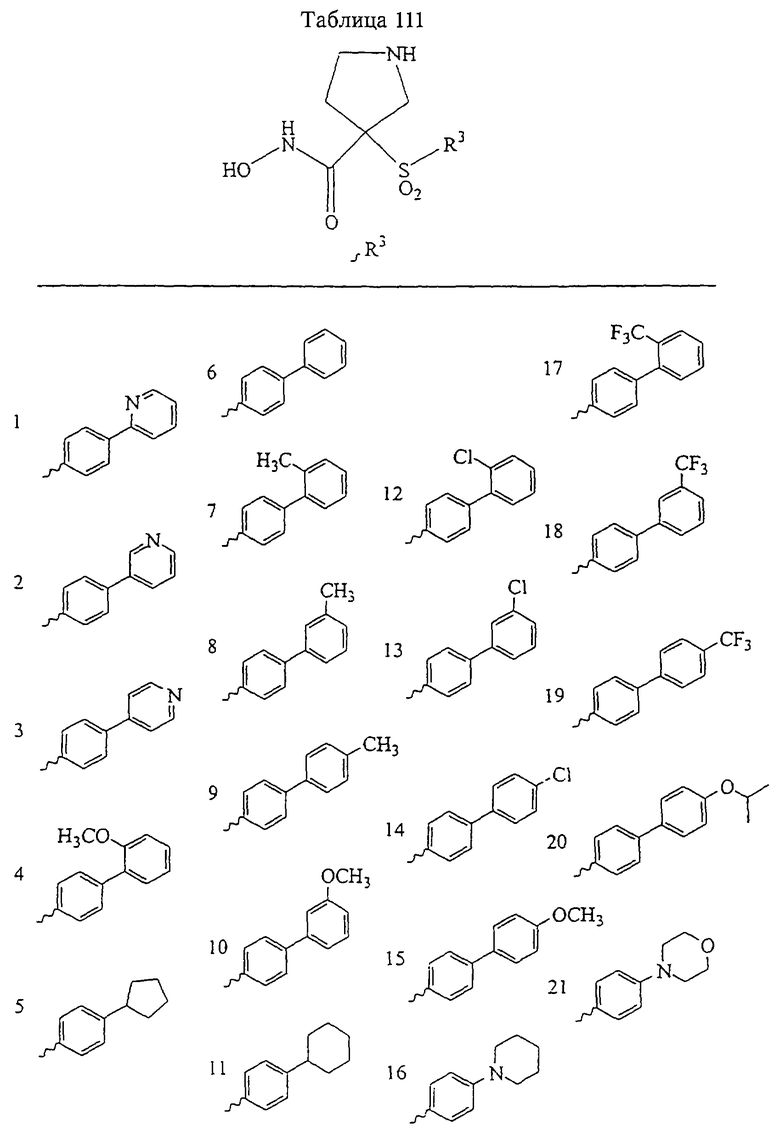
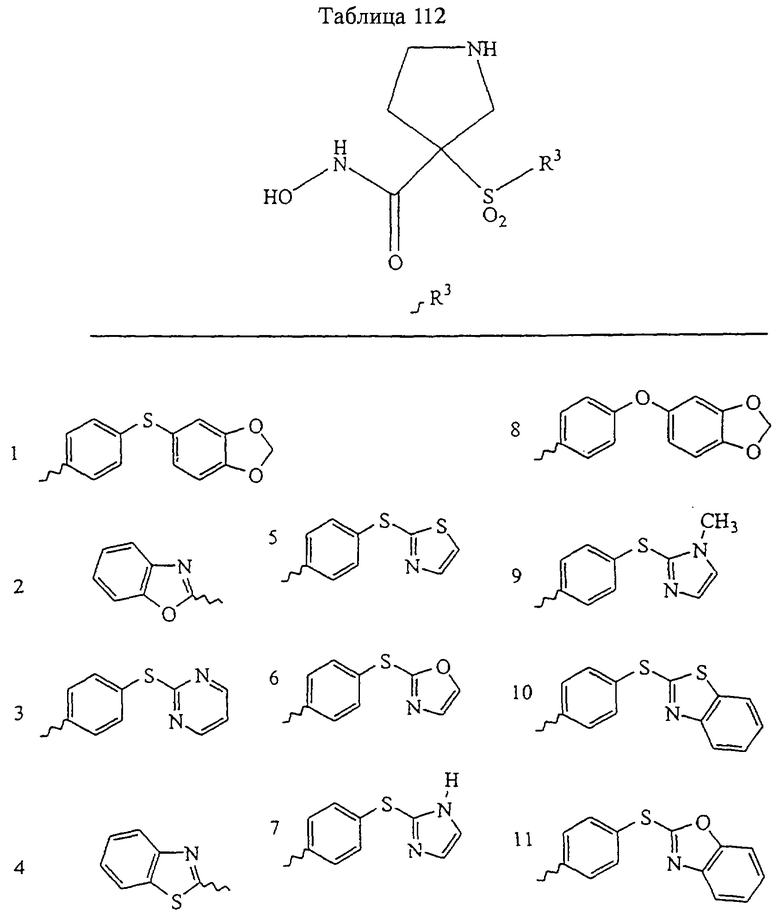
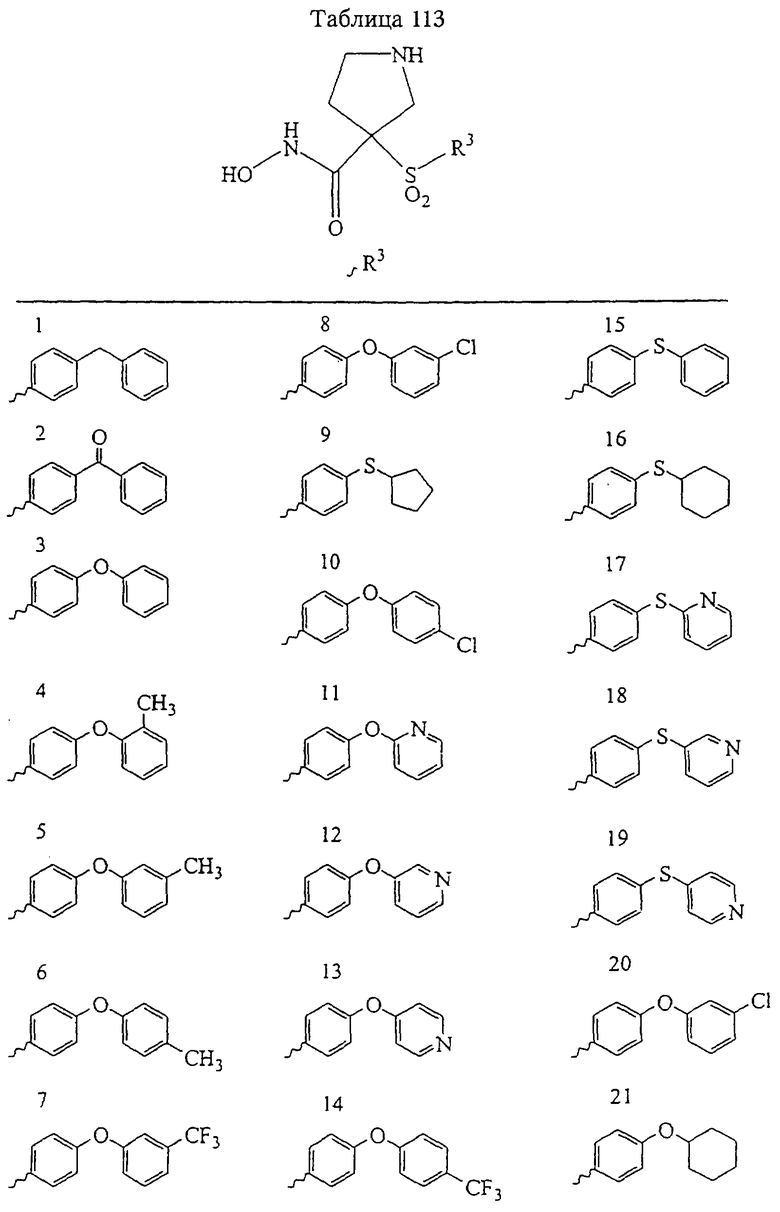
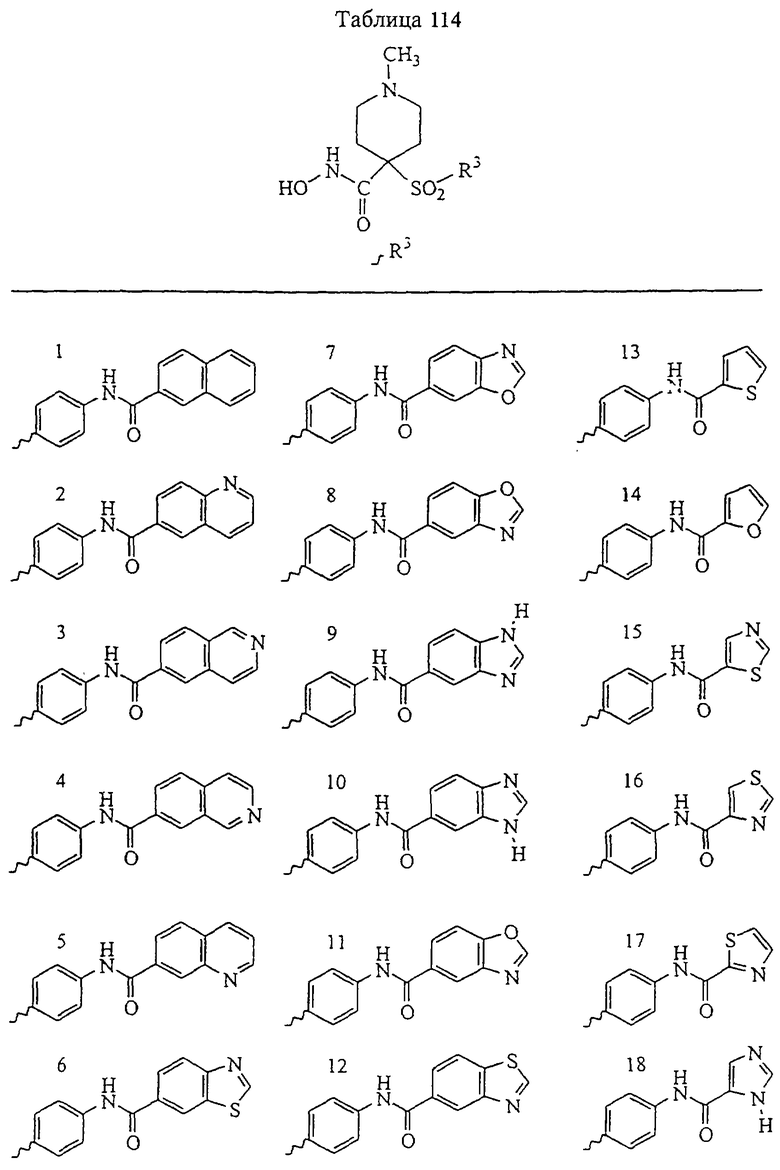
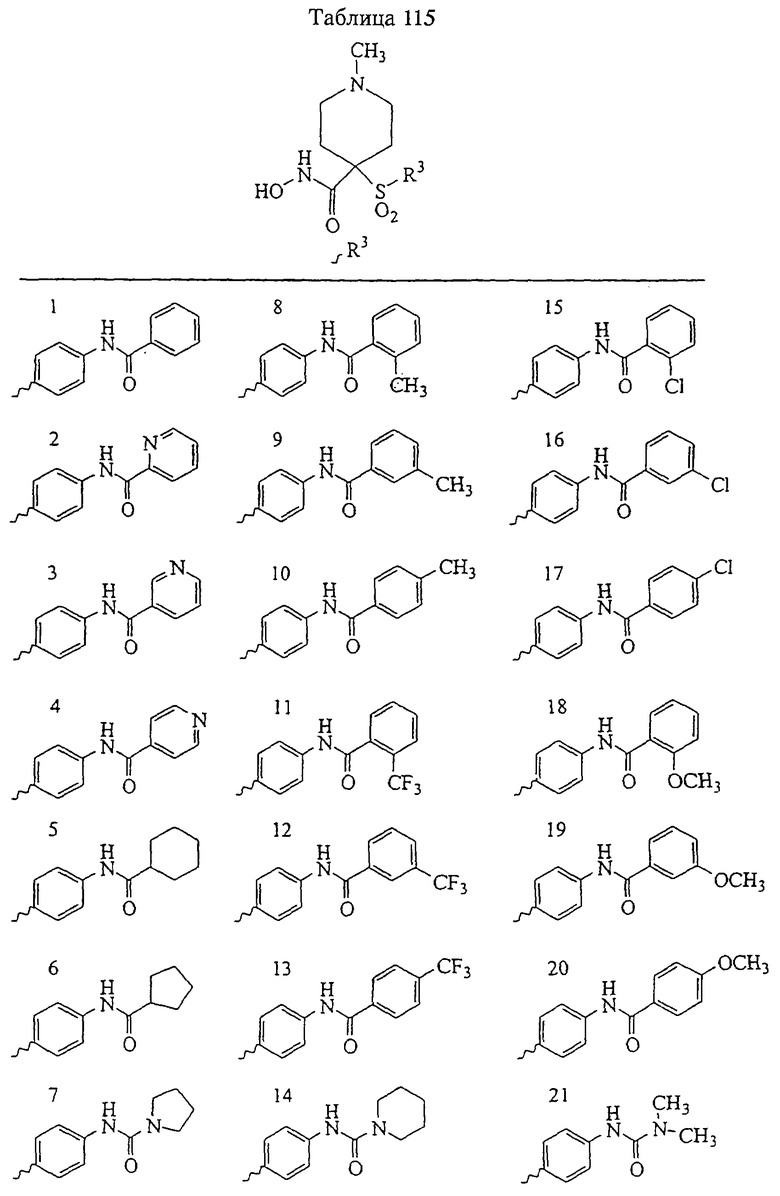
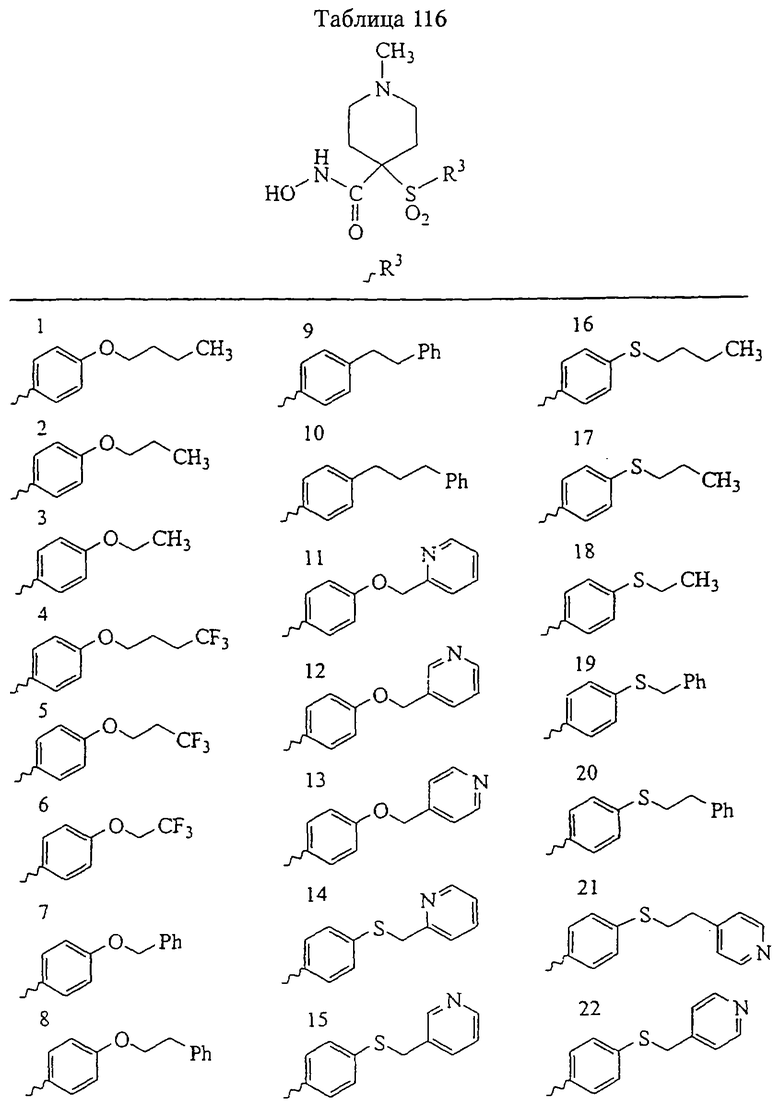
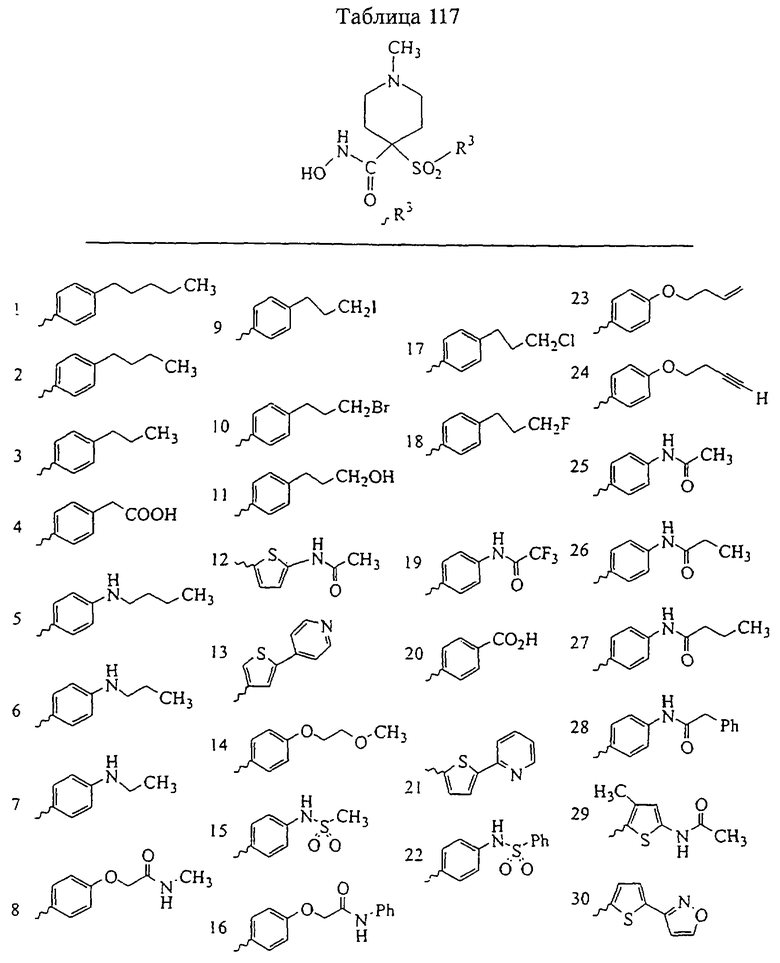
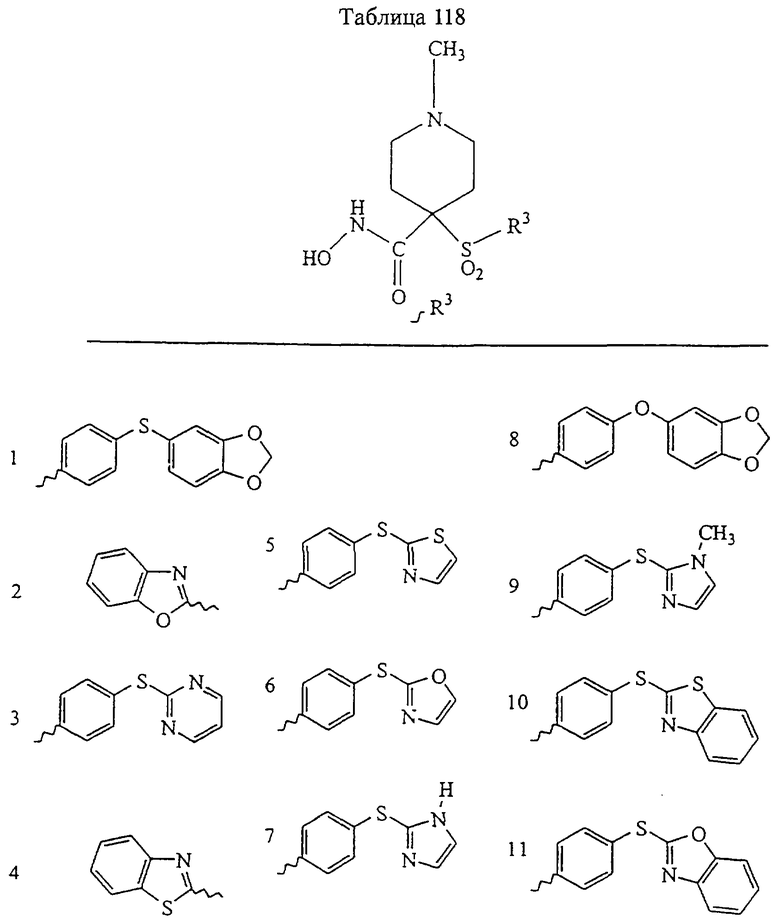
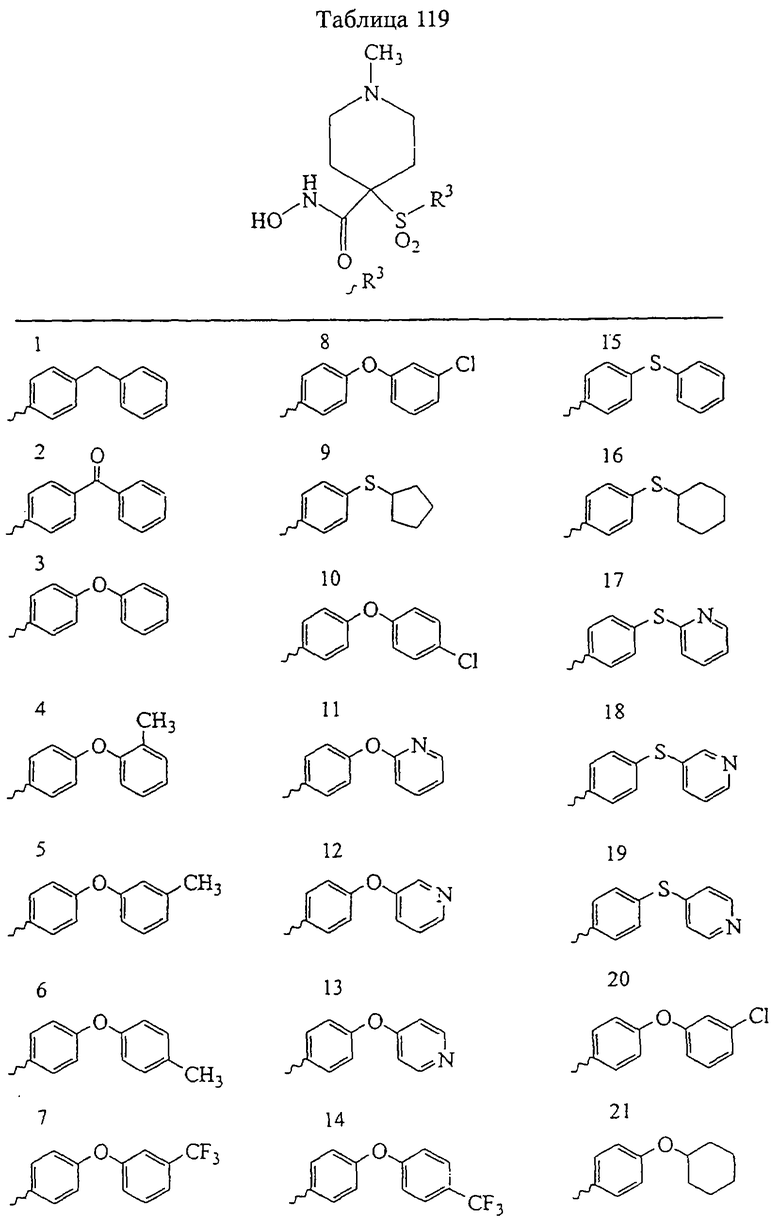
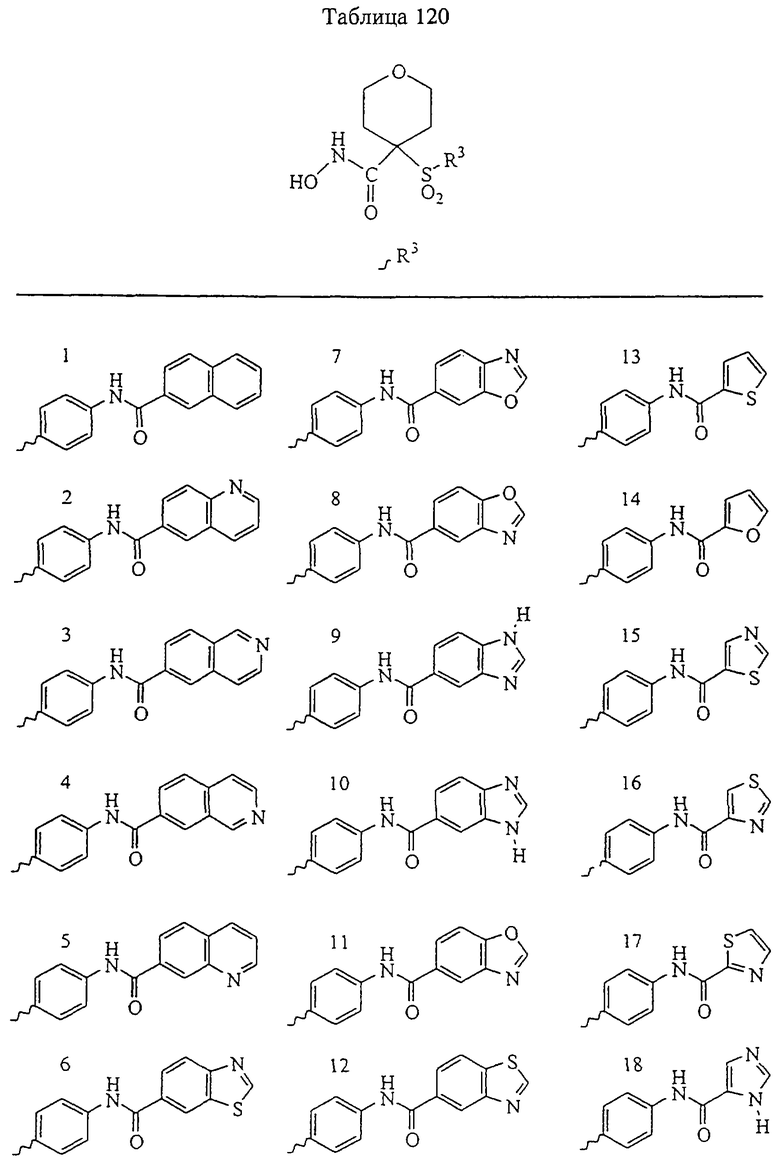
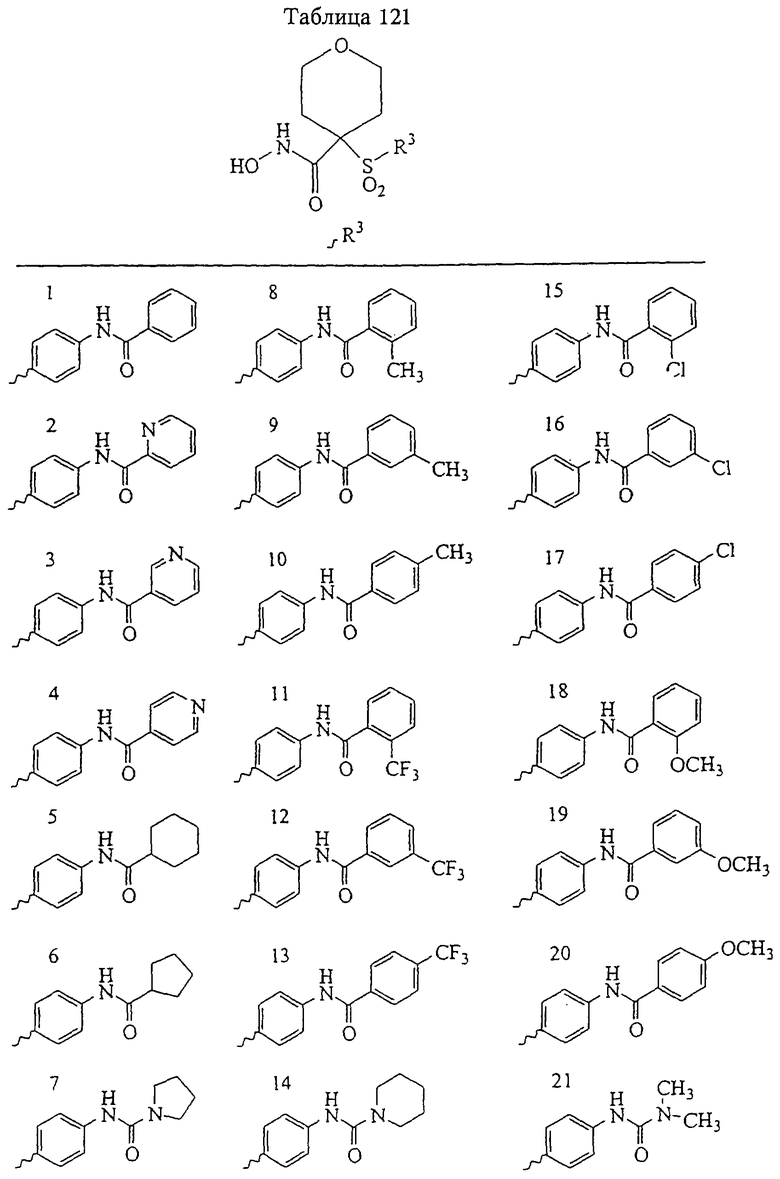
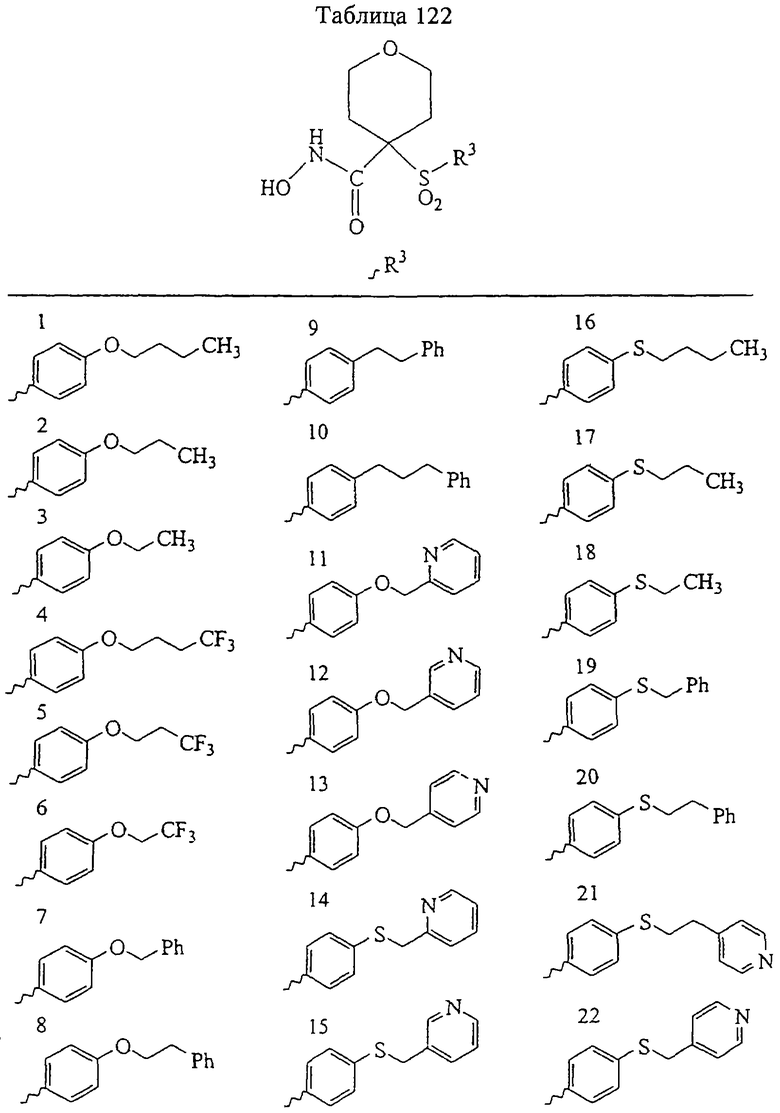
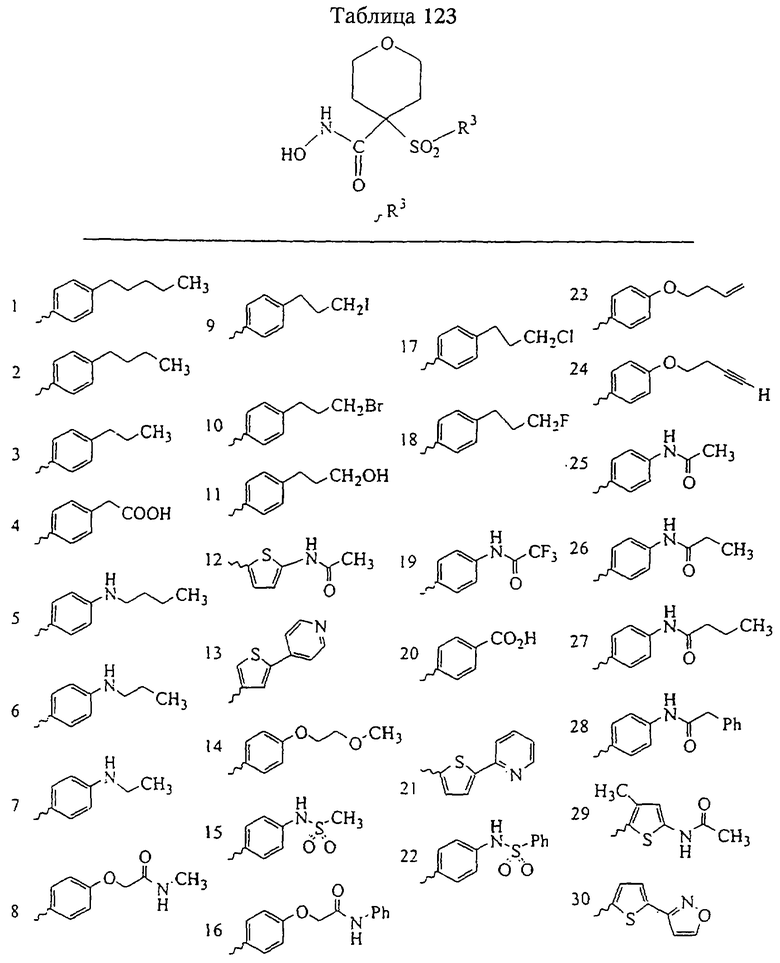
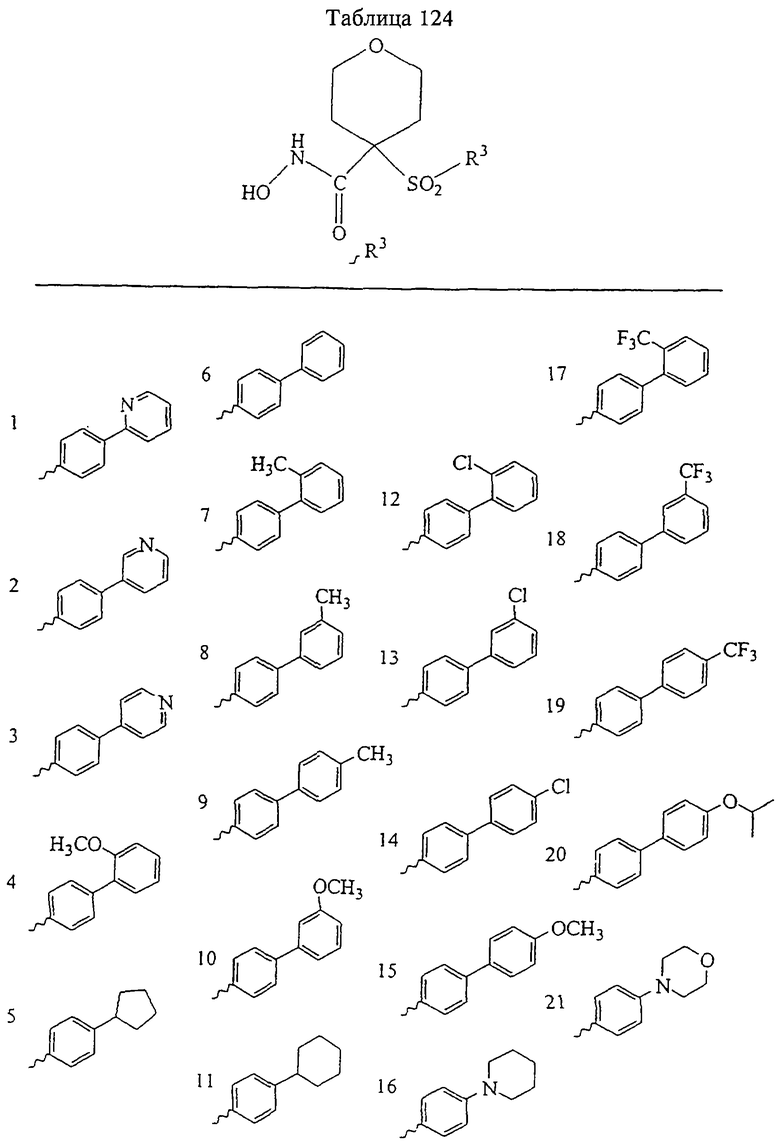
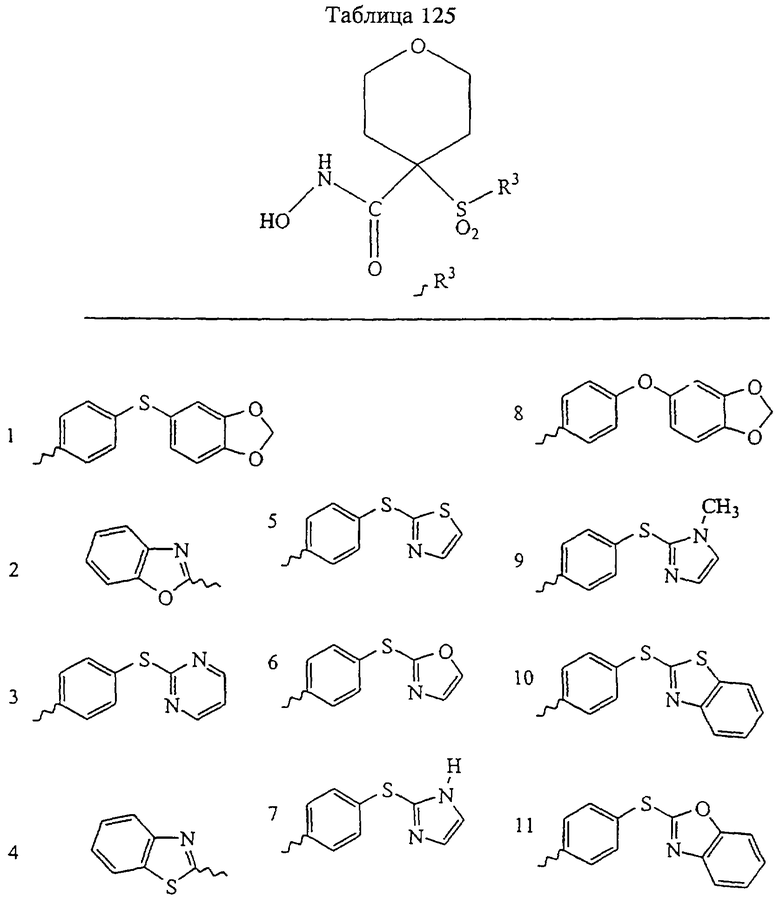
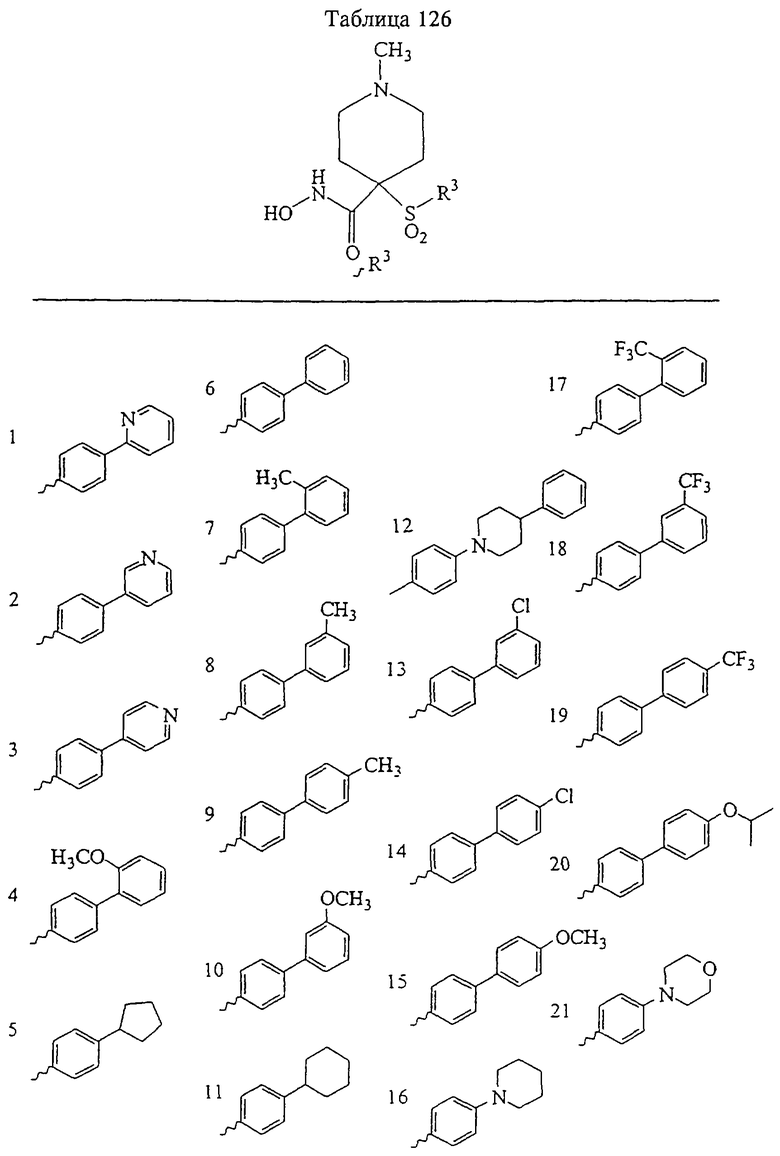

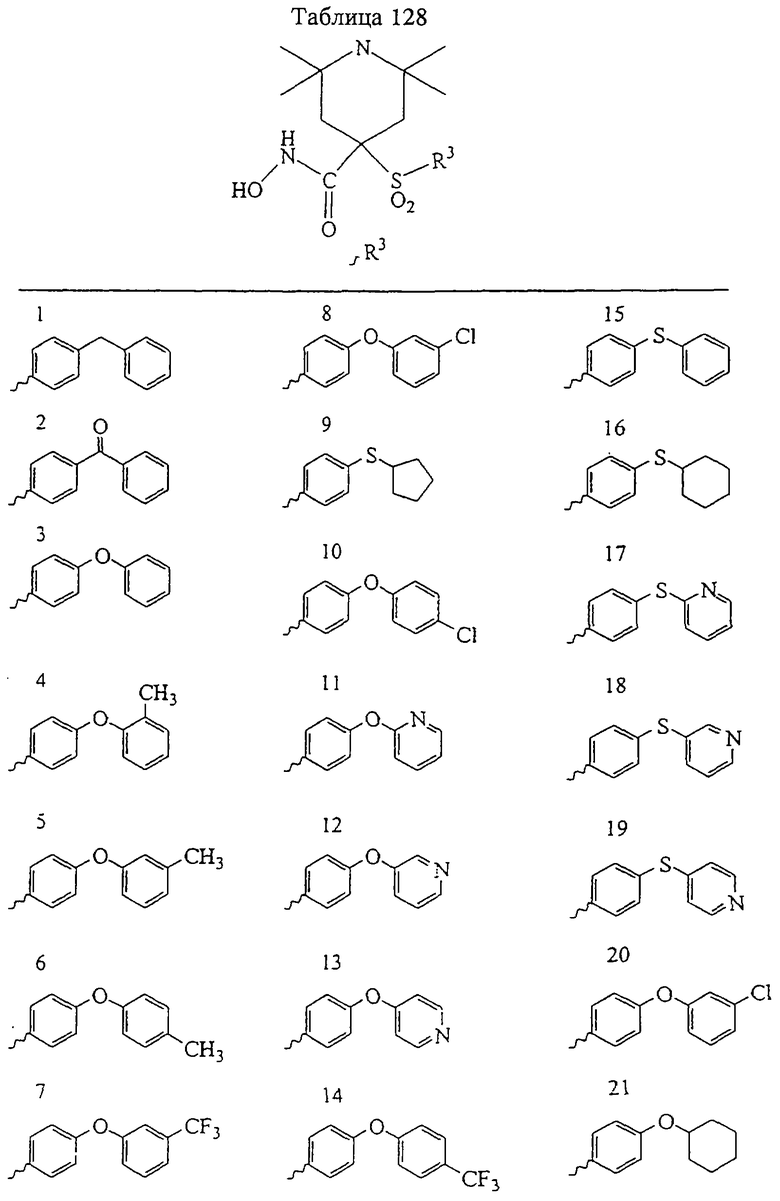
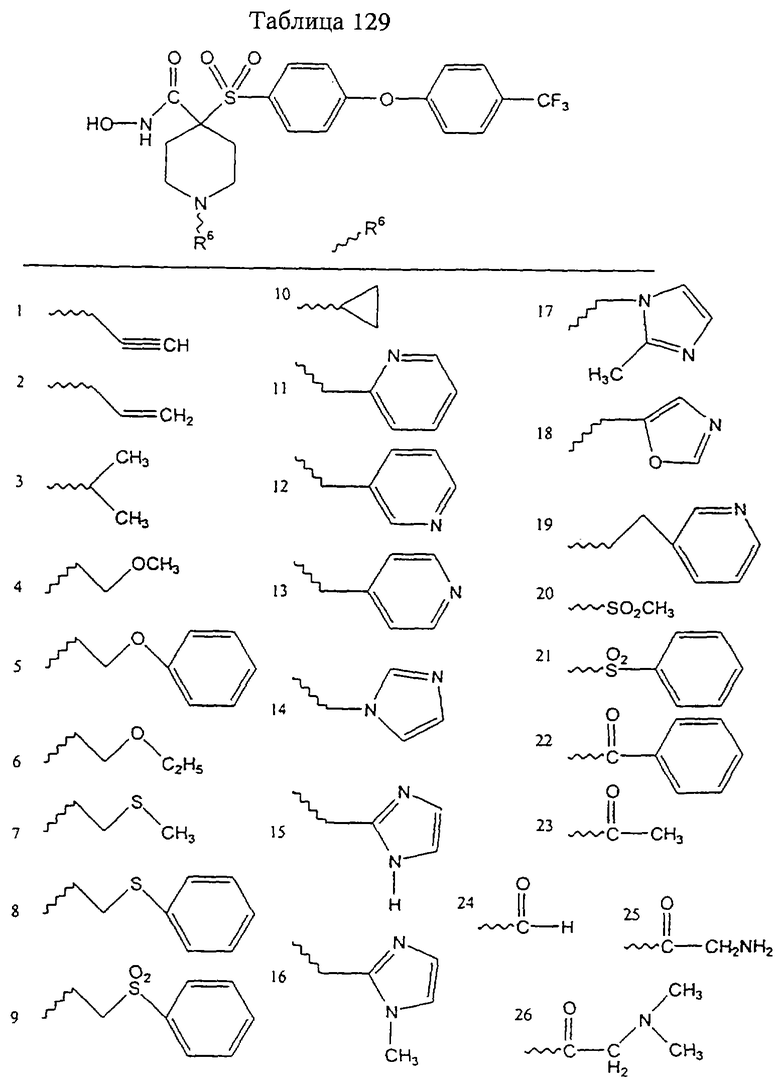
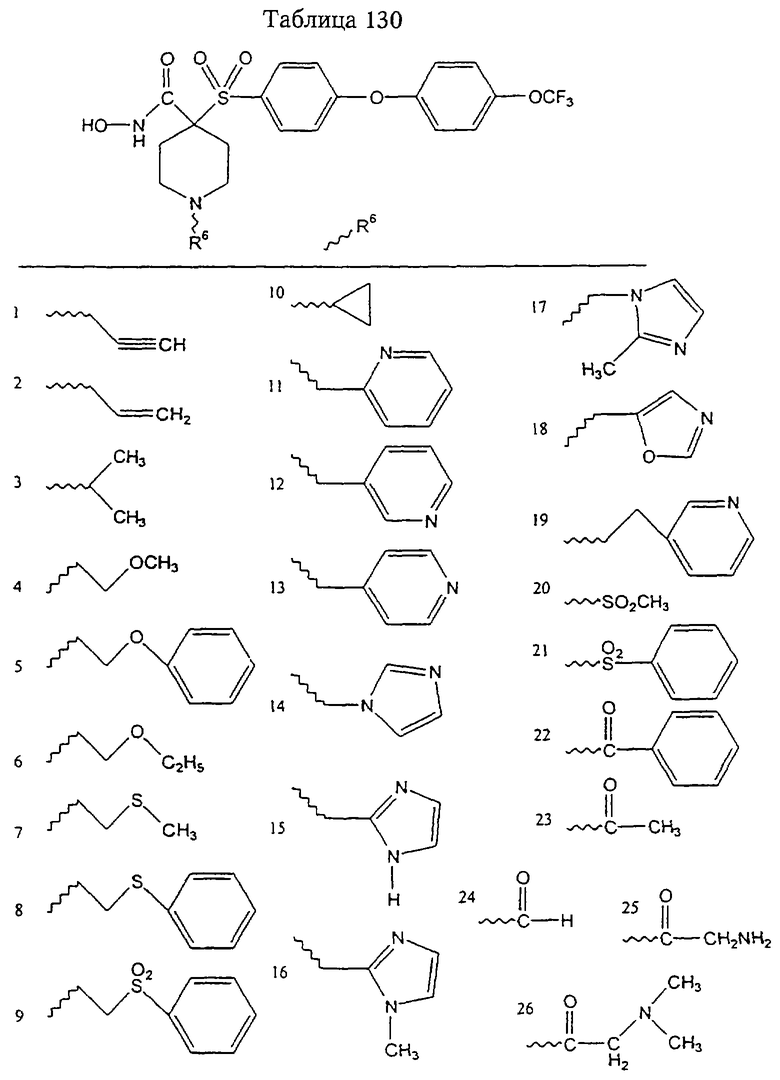
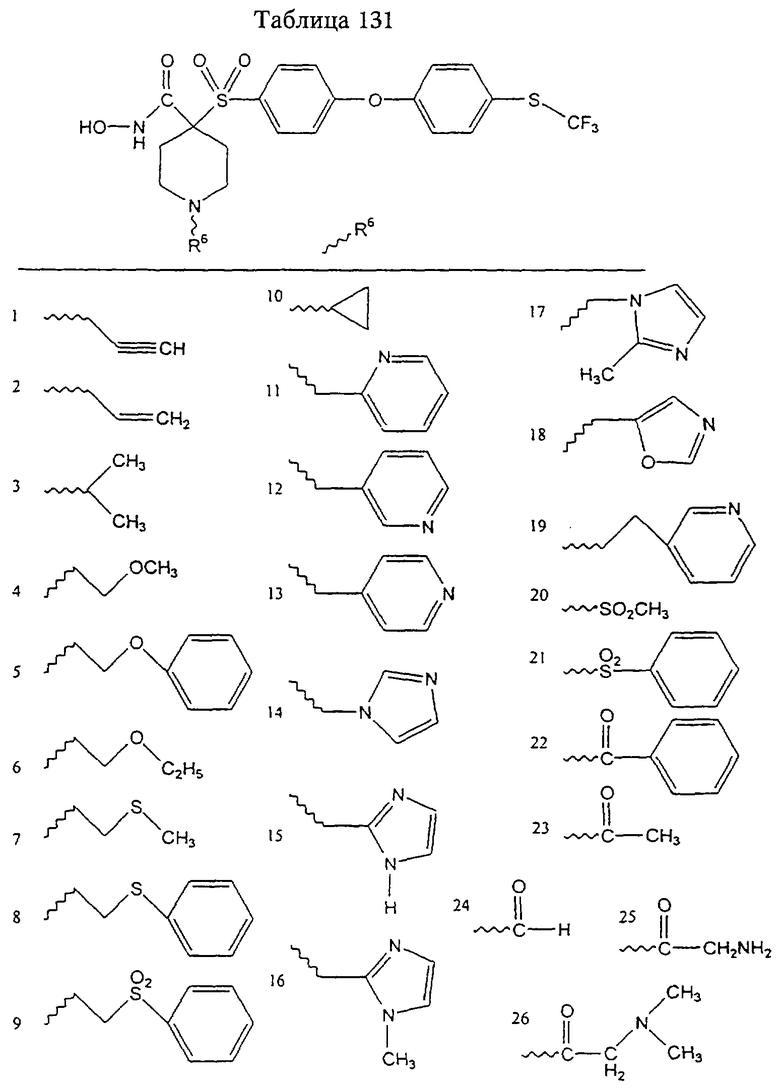
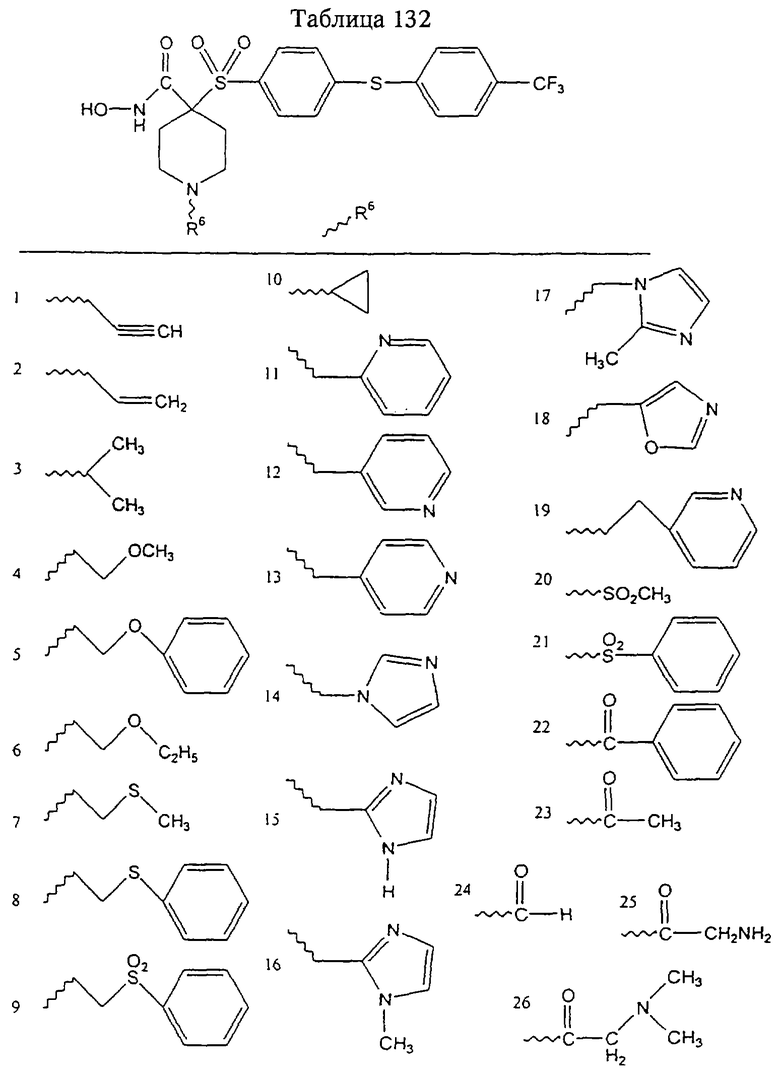
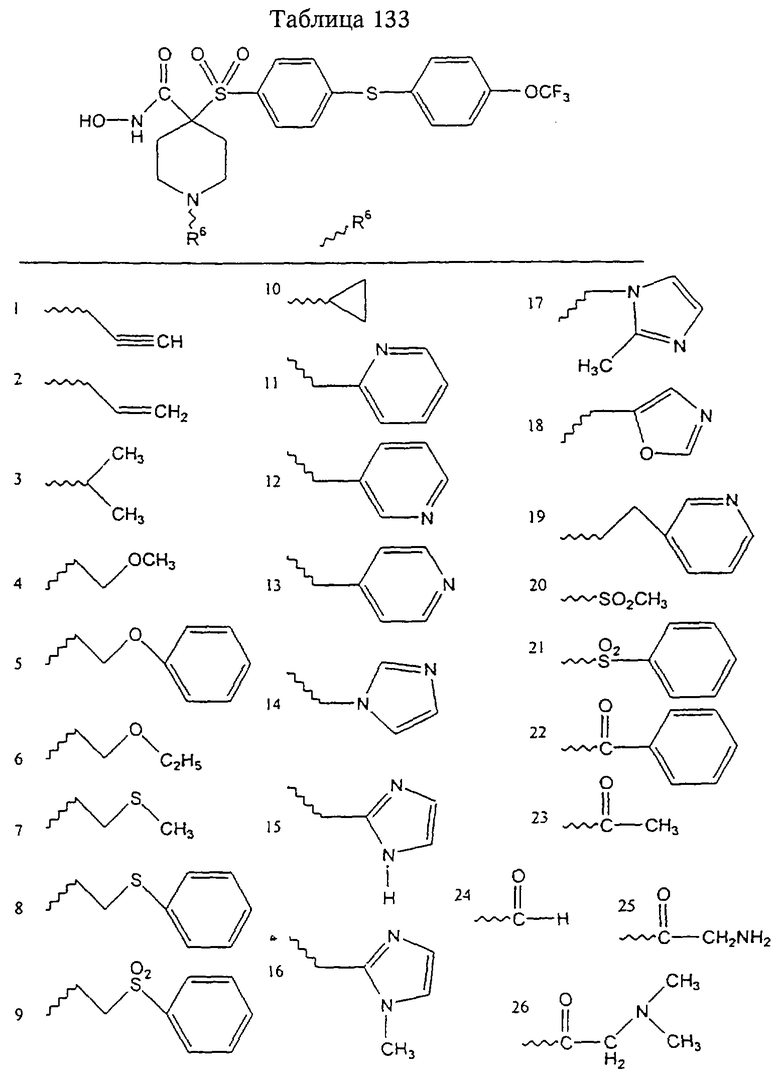
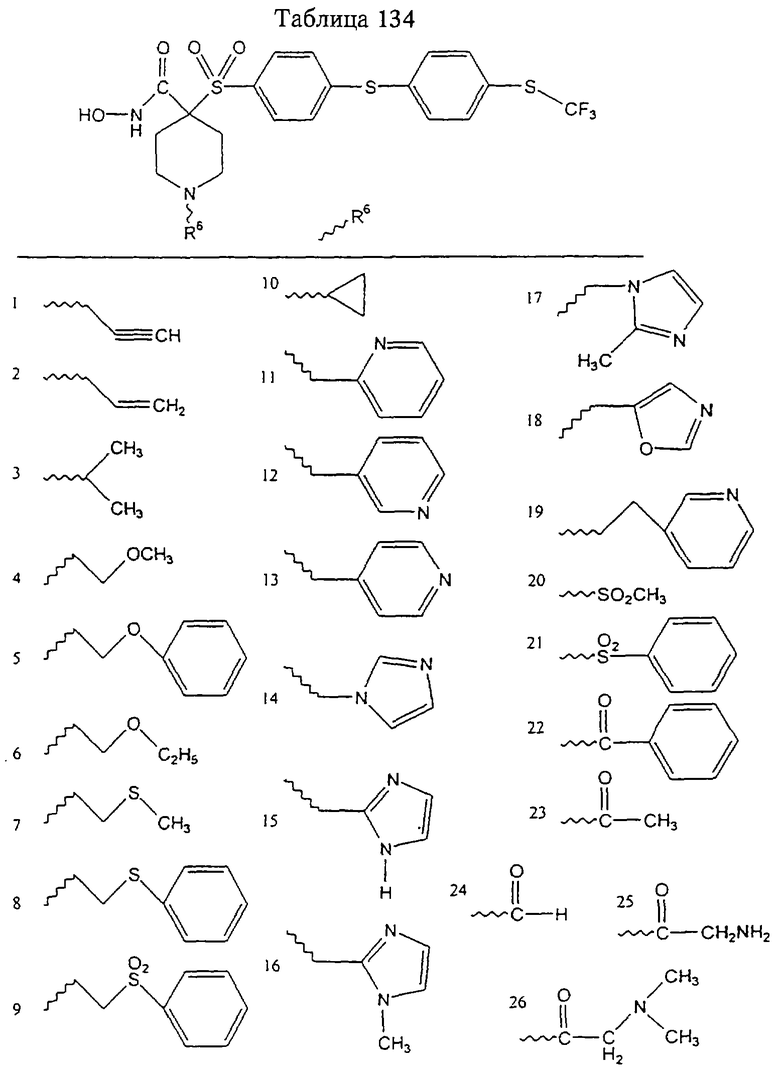
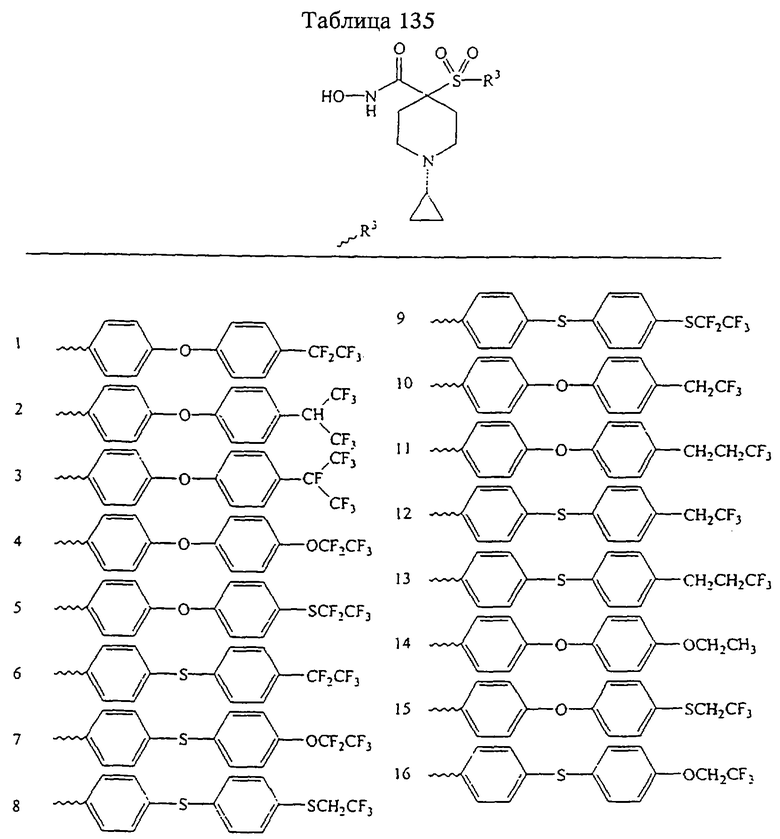
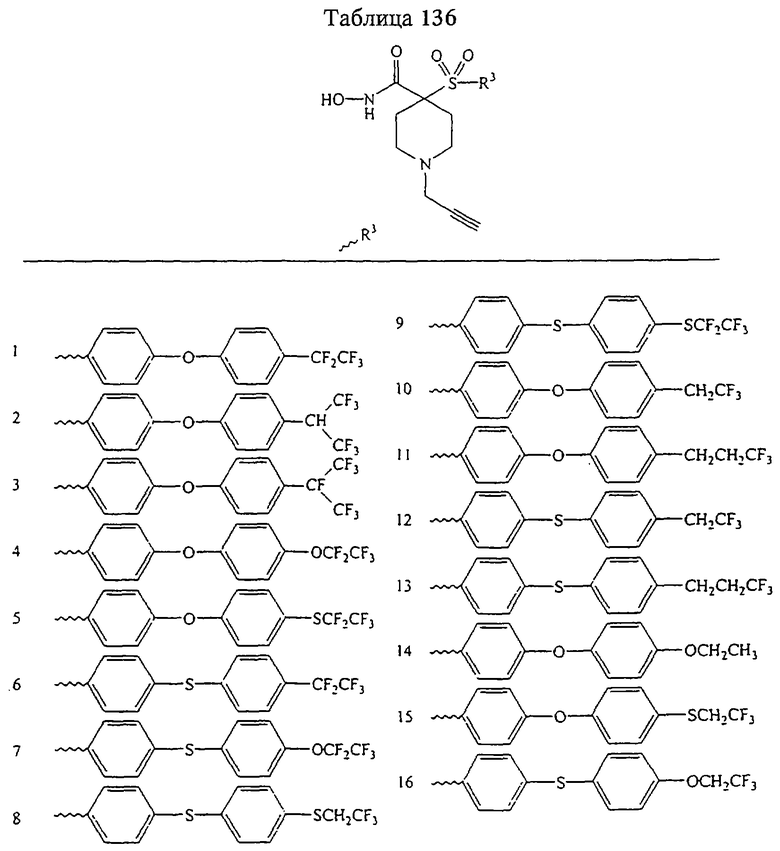

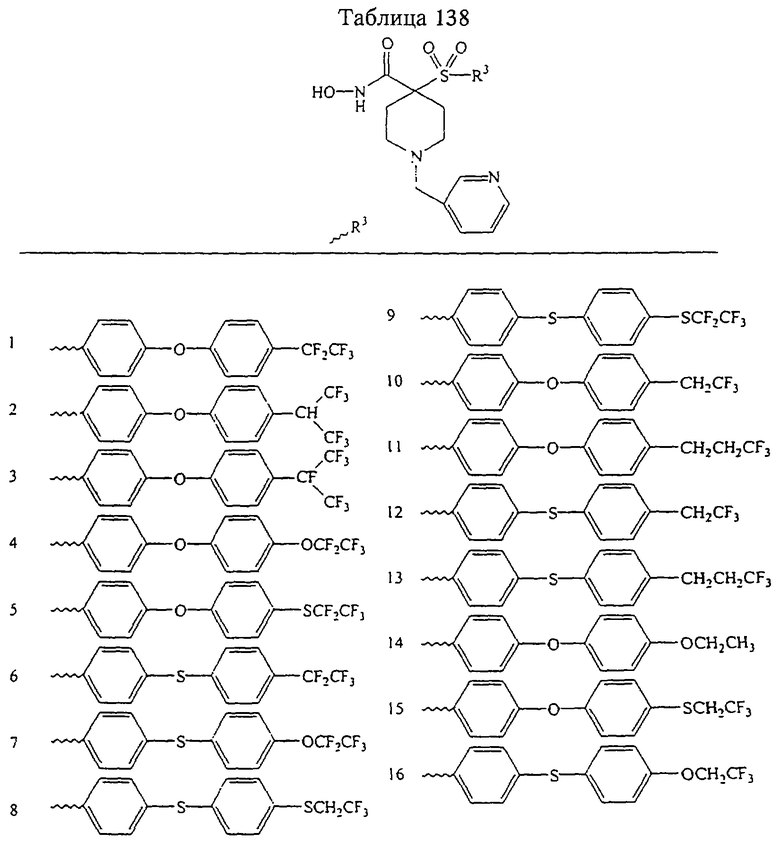
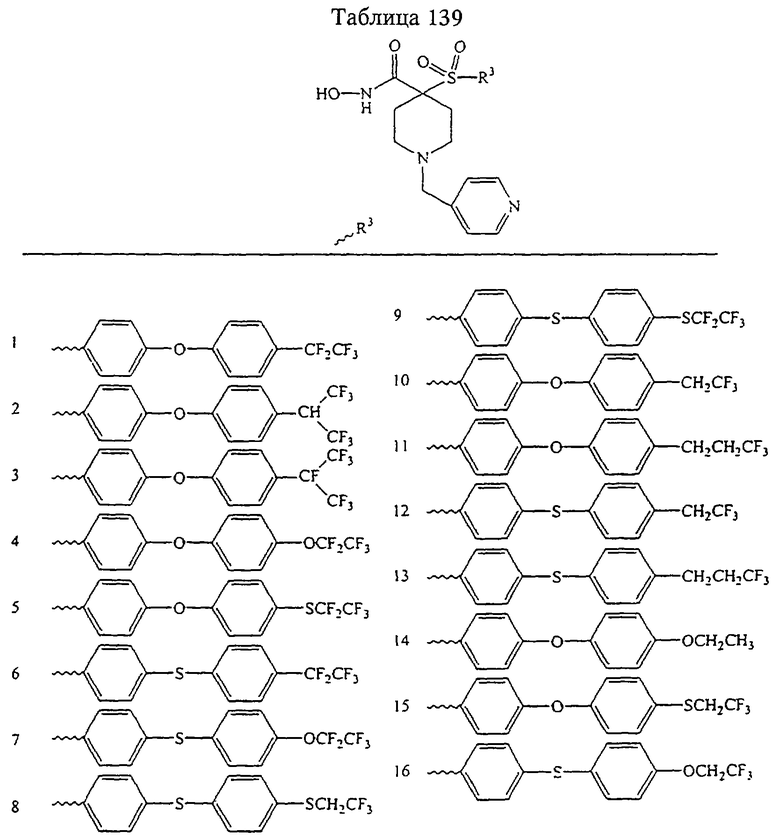
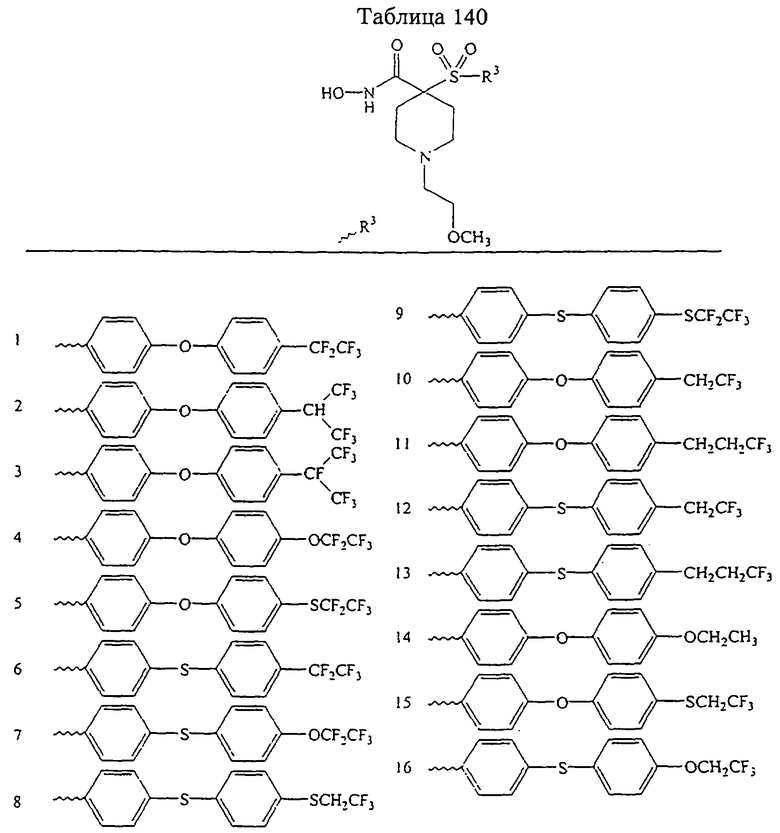
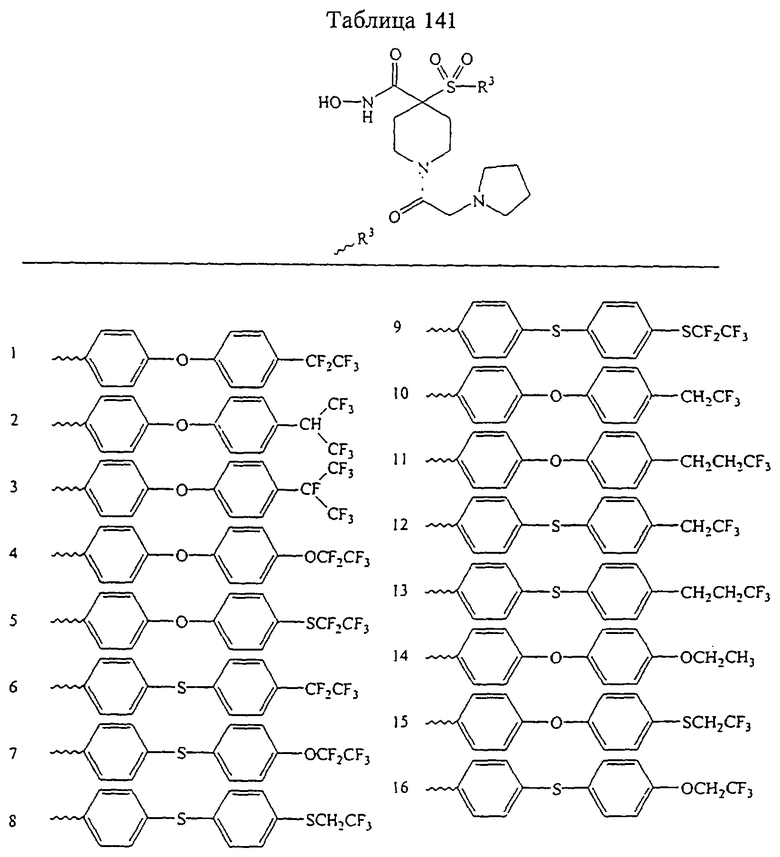
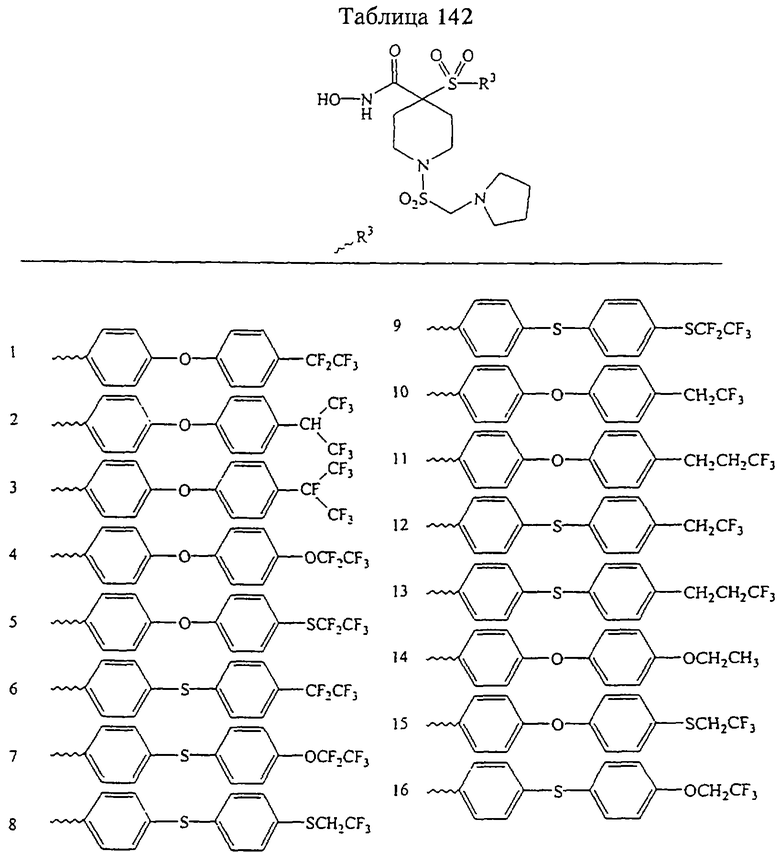
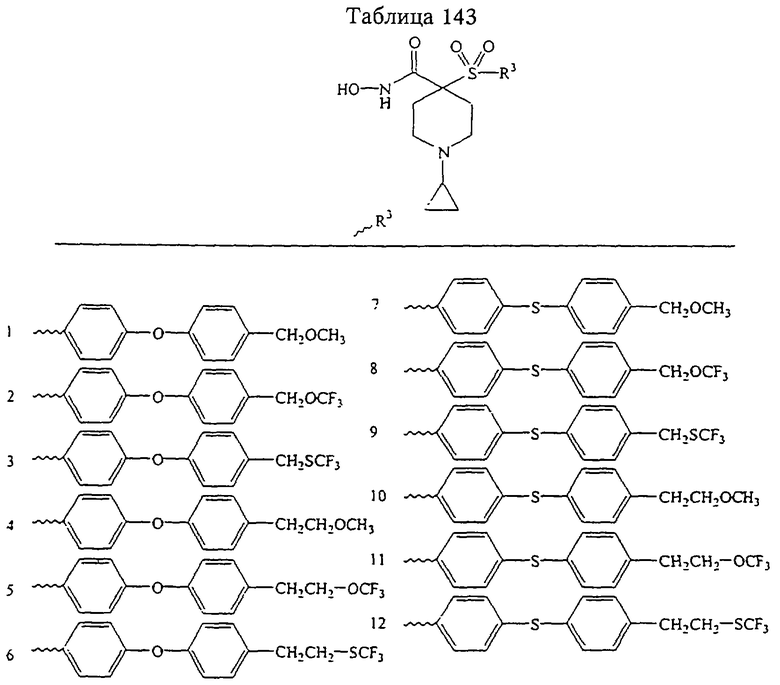
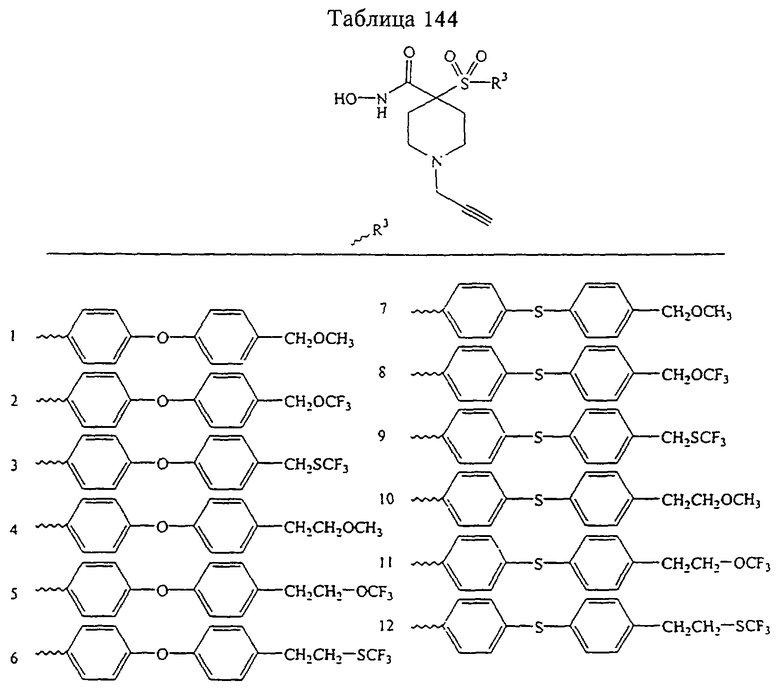
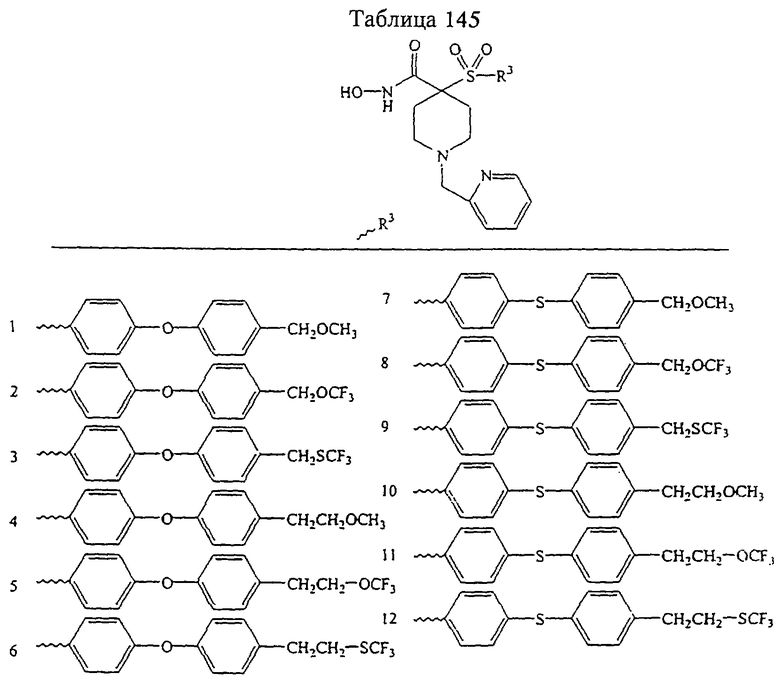
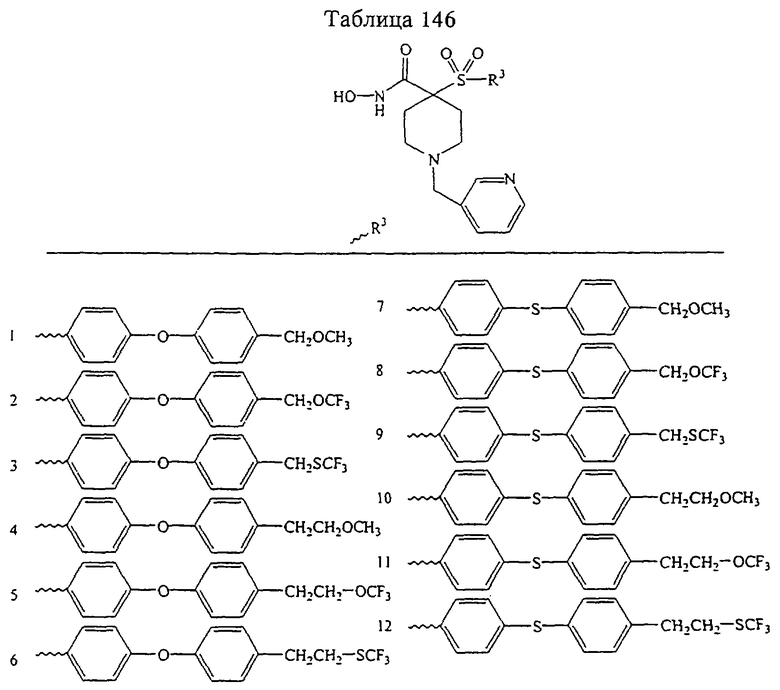
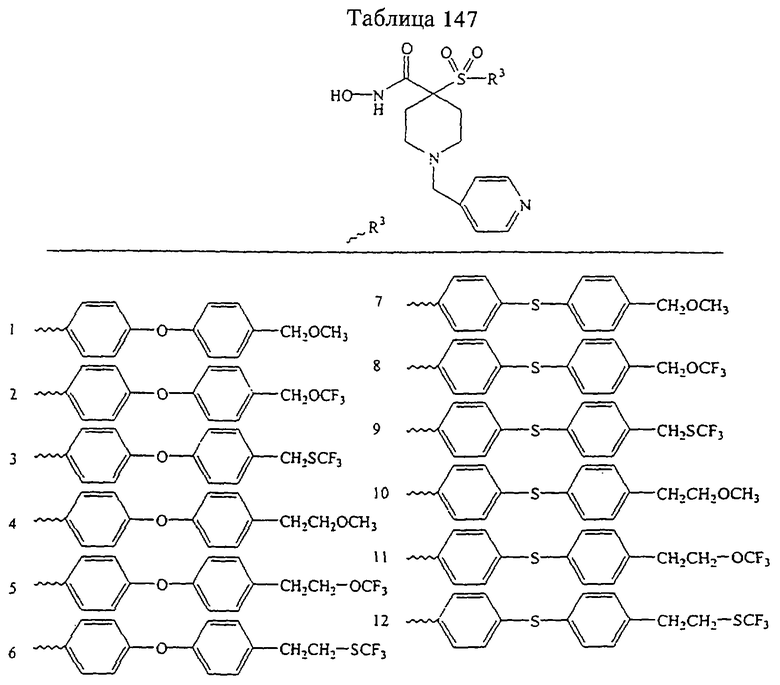
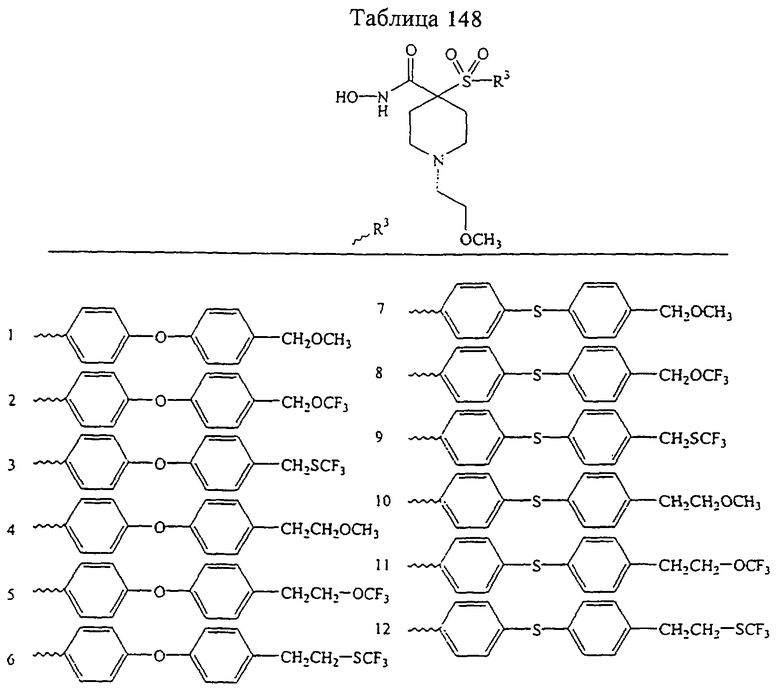
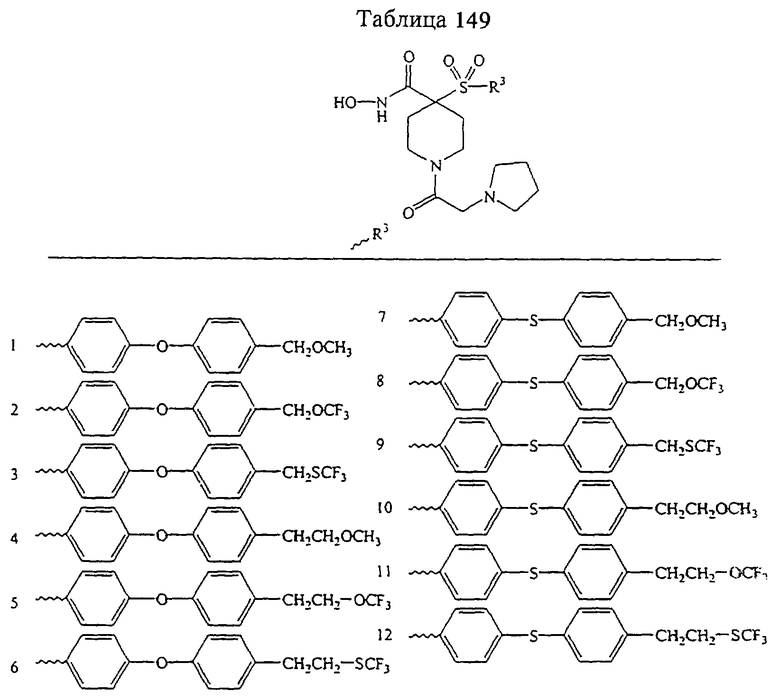
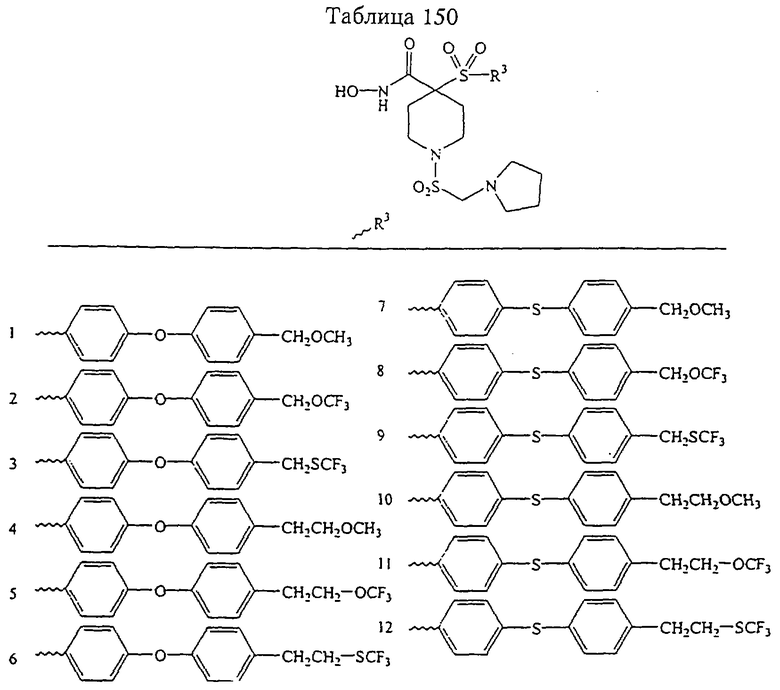
Рассматриваемое соединение с ингибирующей активностью применяют для лечения млекопитающего, такого как мышь, крыса, кролик, собака, лошадь, примат, такой как обезьяна, шимпанзе или человек, который имеет состояние, связанное с патологической активностью матриксных металлопротеаз.
Также рассматривается применение указанного соединения, являющегося ингибитором металлопротеаз, для лечения болезненного состояния, на которое может оказывать влияние активность такой металлопротеазы, как TNF-α -конвертаза.
Примерами таких болезненных состояний являются острая фаза реакций на шок и сепсис, реакции на коагуляцию, кровотечение и сердечно-сосудистые нарушения, лихорадка и воспаление, анорексия и кахексия.
Для лечения болезненного состояния, связанного с патологической активностью матриксных металлопротеиназ, рассматриваемое соединение, являющееся ингибитором ММП, может применяться в форме соли амина, полученной из неорганической или органической кислоты. Примеры таких солей включают, но не ограничены только ими, ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, циклопентанпропионат, додецилсульфат, этансульфонат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гиройодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат, мезилат и ундеканоат.
Кроме того, основная азотсодержащая группа с целью повышения растворимости в воде может быть кватернизована с помощью таких агентов, как (низш.)алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлориды, бромиды и йодиды, диалкилсульфаты, типа диметил-, диэтил-, дибутил- и диамилсульфатов, галогениды с длинной цепью, такие как децил-, лаурил-, миристил- и стеарилхлориды, бромиды и йодиды, аралкилгалогениды, типа бензил- и фенилэтилбромидов, и другие. Таким образом, при необходимости могут быть получены водо- или маслораствормые или диспергируемые продукты. Соли получают взаимодействием основных соединений с требуемой кислотой.
Другие применяемые в настоящем изобретении соединения представляют собой кислоты, которые также могут образовывать соли. Примеры солей включают соли щелочных металлов или щелочноземельных металлов, таких как натрий, калий, кальций или магний, или соли органических оснований или основного четвертичного аммония.
В некоторых случаях соли также могут применяться для выделения, очистки или разделения соединений по изобретению.
Общая суточная доза, вводимая млекопитающему в виде однократной или разделенных доз, может составлять, например, от 0,001 до 30 мг/кг веса тела в день, более предпочтительно от 0,01 до 10 мг. Композиции стандартных доз могут содержать такие количества или меньшие кратные количества, в сумме дающие необходимую суточную дозу. Приемлемая суточная доза может быть введена в виде нескольких более мелких доз. С помощью нескольких мелких доз также можно увеличить общую суточную дозу, если этого требует специалист, назначивший лекарство.
Схему приема лекарственного средства для лечения болезненного состояния с помощью соединения и/или композиции по настоящему изобретению выбирают в зависимости от многочисленных факторов, включая тип, возраст, вес, пол, диету и медицинское состояние пациента, серьезность болезни, путь введения, фармакологические особенности, такие как активность, эффективность, фармакокинетический профиль и профиль токсичности конкретного применяемого соединения, в зависимости от того, используется ли система введения лекарства и вводят ли соединение в виде одной из частей комбинации лекарственных средств. Таким образом, конкретно используемая схема приема лекарственного средства может варьироваться в значительной степени и в результате этого может отклоняться от предпочтительной изложенной выше схемы.
Соединения по изобретению могут использоваться для изготовления фармацевтической композиции. Такая композиция затем может быть введена орально, парентерально, с помощью аэрозоля для ингаляции, ректально или местно в виде композиций, содержащих суточную дозу, в состав которых при необходимости входят обычные нетоксичные фармацевтически приемлемые наполнители, адъюванты и носители. Местное нанесение также может включать использование таких методов трансдермального применения, как трансдермальные бляшки или устройства для ионофореза. Понятие "парентеральный" в контексте настоящего описания включает подкожные инъекции, внутривенную, внутримышечную, внутрибрюшинную, интраназальную инъекцию или инфузию. Композиции лекарственных средств описаны, например, у Hoover John E., в Remington’s Pharmaceutical Science, Mack Publishing Co. Easton, Pennsylvania, 1975, и у Liberman H.A. и Lachman L. (ред.), Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980.
Инъецируемые препараты, например стерильные инъецируемые водные или маслянистые суспензии, могут быть изготовлены по известной в данной области технологии с использованием приемлемых диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный инъецируемый препарат также может представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном приемлемом для парентерального применения разбавителе или растворителе, например раствор в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые можно использовать, следует назвать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют нелетучие масла. Для этой цели может применяться любое чистое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для приготовления инъецируемых форм находят применение жирные кислоты, такие как олеиновая кислота. Могут применяться диметилацетамид, поверхностно-активные вещества, включая ионогенные и неионогенные детергенты, полиэтиленгликоли. Также могут использоваться смеси растворителей и смачивающих агентов, таких как указанные выше.
Суппозитории для ректального введения лекарственного средства могут быть приготовлены смешением лекарственного средства с приемлемым не вызывающим раздражение эксципиентом, таким как масло какао, синтетические моно-, ди- или триглицериды, жирные кислоты и полиэтиленгликоли, которые являются твердыми при обычных температурах, но могут расплавляться в прямой кишке и высвобождать лекарство.
Твердые дозируемые формы для орального введения могут включать капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых дозируемых формах соединения по настоящему изобретению, как правило, объединяют с одним или с несколькими адъювантами, приемлемыми для соответствующего пути введения. При пероральном введении рассматриваемое производное ароматического сульфонгидроксамата, являющееся ингибитором, может быть смешено с лактозой, сахарозой, порошкообразным крахмалом, эфирами целлюлозы и алкановых кислот, алкиловыми эфирами целлюлозы, тальком, стеариновой кислотой, стеаратом магния, оксидом магния, натриевыми и кальциевыми солями фосфорной и серной кислот, желатином, аравийской камедью, альгинатом натрия, поливинилпирролидоном и/или поливиниловым спиртом, а затем таблетировано или капсулировано для обычного применения. Такие капсулы или таблетки могут содержать композицию с контролируемым высвобождением, которая может быть получена в виде дисперсии действующего вещества в гидроксипропилметилцеллюлозе. В случае капсул, таблеток и пилюль дозируемые формы могут также включать забуферивающие агенты, такие как цитрат натрия, карбонат или бикарбонат магния или кальция. Кроме того, таблетки и пилюли могут быть изготовлены с энтеросолюбильными покрытиями.
Для терапевтических целей композиции для парентерального введения могут находиться в форме водных или неводных изотоничных стерильных инъецируемых растворов или суспензий. Эти растворы и суспензии могут быть получены из растворимых порошков или гранул, имеющих один или несколько носителей или разбавителей, которые, как указано выше, могут применяться в композициях для орального введения. Рассматриваемое производное ароматического сульфонгидроксамата, являющегося ингибитором, может быть растворено в воде, полиэтиленгликоле, пропиленгликоле, этаноле, кукурузном масле, хлопковом масле, арахисовом масле, кунжутном масле, бензиловом спирте, хлориде натрия и/или в различных буферах. Другие адъюванты и методы введения являются хорошо известными и широко применяются в области фармацевтики.
Жидкие дозируемые формы для орального введения могут включать фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно применяемые в данной области, такие как вода. Такие композиции также могут включать адъюванты, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, а также подслащивающие вещества, корригенты и отдушки.
Количество действующего вещества, которое может быть объединено с носителями для получения однократной дозируемой формы, варьируется в зависимости от подлежащего терапии млекопитающего и конкретного пути введения.
Предпочтительные варианты осуществления изобретения
Приведенное выше описание позволяет реализовать настоящее изобретние без дополнительных исследований в полном объеме. Таким образом, приведенные ниже конкретные предпочтительные варианты осуществления служат только с целью иллюстрации изобретения и, следовательно, не ограничивают объем изобретения.
Пример 1: Получение N-гидрокси-2-[(4-феноксифенил)сульфонил]ацетамида
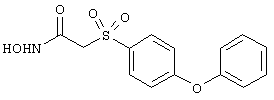
Стадия А: К раствору гидрата 3-бромпировиноградной кислоты (1,95 г, 11,7 ммоль), охлажденному до 0° С, в метаноле (50 мл) добавляли 4-(фенокси)бензолтиол (2,35 г, 11,7 ммоль). Раствор перемешивали в течение 15 мин, после чего концентрировали в вакууме. Остаток распределяли между этилацетатом и Н2О и органический слой сушили над сульфатом магния. После концентрирования в вакууме получали неочищенный сульфид в виде твердого вещества желтого цвета, который использовали без дополнительной очистки.
Стадия Б: К раствору полученного на стадии А неочищенного сульфида (1,2 г, <2,6 ммоль) в смеси метанол/Н2О, охлажденному до 0° С, добавляли Oxone® (3,5 г, 5,72 ммоль). Раствор перемешивали в течение 1 ч, после чего избыток Oxone® удаляли фильтрацией. Фильтрат концентрировали и остаток растворяли в этилацетате, промывали насыщенным раствором NаНСО3 и насыщенным раствором NaCl и сушили над сульфатом магния. После концентрирования в вакууме образовавшийся остаток растворяли в метаноле и добавляли тионилхлорид (1,9 мл, 26 ммоль). После хроматографии (на силикагеле, этилацетат/гексан) получали сульфон в виде твердого вещества (350 мг, 44%). МС (CI) MH+: рассчитано для С15Н14О5S: 307, обнаружено 307.
Стадия В: К раствору сульфона (350 мг, 1,1 ммоль) в метаноле (2 мл) и ТГФ (ТГФ, 2 мл) добавляли 50%-ный водный раствор гидроксиламина (1 мл). Раствор перемешивали в течение ночи. После растирания с этилацетатом получали указанное в заголовке соединение в виде твердого вещества белого цвета (270 мг, 77%). Чистота по данным ЖХВР: >97%. МС (CI) MH+: рассчитано для C14H13NO5S: 308, обнаружено 308.
Пример 2: Получение N-гидрокси-2-метил-2-[(4-феноксифенил)сульфонил]пропанамида
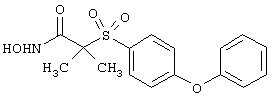
Стадия А: К раствору 4-(фенокси)бензолтиола (3,8 г, 18,8 ммоль) в метаноле (60 мл), охлажденному до 0° С, добавляли трет-бутилбромацетат (2,8 мл, 18,8 ммоль) и триэтиламин (2,6 мл, 19,0 ммоль). Раствор перемешивали в течение 30 мин и затем концентрировали в вакууме. Остаток распределяли между этилацетатом и Н2О, органический слой промывали насыщенным раствором NaCl и сушили над сульфатом магния. После концентрирования в вакууме получали сульфид в виде масла. К раствору сульфида в дихлорметане (85 мл) в течение 15 мин добавляли мета-хлорпербензойную кислоту (13,8 г, 43,2 ммоль). Раствор перемешивали при температуре окружающей среды в течение 2 ч. Реакцию прекращали добавлением водного раствора Na2SO3. Через 30 мин раствор фильтровали через Celite®. Фильтрат промывали 25%-ным водным раствором гидроксиламина, 1 н. НСl и насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфон в виде твердого вещества белого цвета (4,0 г, 68%).
Стадия Б: К раствору полученного на стадии А сульфона (3,2 г, 9,2 ммоль) в ТГФ (65 мл), охлажденному до 0° С, добавляли гидрид натрия (730 мг в виде 60%-ной дисперсии в минеральном масле, 18,4 ммоль). Через 10 мин по каплям добавляли метилйодид (2,28 мл, 36,8 ммоль) и смесь перемешивали в течение 18 ч при температуре окружающей среды. Реакцию прекращали, добавляя Н2О и реакционную смесь концентрировали в вакууме. Водный остаток разбавляли этилацетатом и органическую фазу промывали Н2О и сушили над Na2SO4. После концентрирования в вакууме получали диметильное производное в виде твердого вещества беловатого цвета (3,2 г, 92%). Чистота по данным ЖХВР: 95%.
Стадия В: К раствору полученного на стадии Б диметильного производного (3,2 г, 8,5 ммоль) в анизоле (10 мл) добавляли трифторуксусную кислоту (30 мл) и раствор перемешивали в течение 30 мин. После концентрирования в вакууме и последующего растирания (диэтиловый эфир) получали кислоту в виде твердого вещества белого цвета (750 мг, 28%). Чистота по данным ЖХВР: 99%. МС (CI) MH+: рассчитано для C16H16O5S: 321, обнаружено 321.
Стадия Г: К раствору полученной на стадии В кислоты (723 мг, 2,26 ммоль) в ДМФ (ДМФ, 4,5 мл) добавляли N-гидроксибензотриазол· Н2О (ГОБТ, 366 мг, 2,71 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (ЭДК, 476 мг, 2,49 ммоль). После перемешивания раствора в течение 1 ч при температуре окружающей среды добавляли 50%-ный водный гидроксиламин (0,40 мл, 6,8 ммоль). Через 15 мин раствор распределяли между этилацетатом и Н2О. Органический слой промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали указанное в заголовке соединение в виде пены белого цвета (434 мг, 57%). Чистота по данным ЖХВР: 99%. МС (CI) M+Li-: рассчитано для C16H17NO5O: 342, обнаружено 342.
Пример 3: Получение 1,1-диметилового эфира 4-[(гидроксиамино)карбонил]-4-[(феноксифенил)сульфонил]-1-пиперидинкарбоновой кислоты
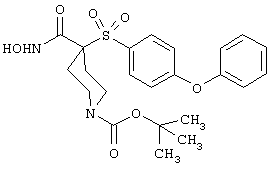
Стадия А: Раствор 4-(фенокси)бензолтиола (2,03 г, 10,0 ммоль) в диметилсульфоксиде (ДМСО, 20 мл) нагревали до 65° С в течение 5 ч. Раствору давали выстояться в течение 18 ч при температуре окружающей среды. Раствор экстрагировали этилацетатом и объединенные органические слои промывали Н2О и насыщенным NaCl и сушили над сульфатом магния. После концентрирования в вакууме получали дисульфид в виде масла желтого цвета (2,3 г, количественный выход).
Стадия Б: К раствору этилизонипекотата (15,7 г, 0,1 моль) в ТГФ (100 мл) по каплям в течение 20 мин добавляли раствор ди-трет-бутилдикарбоната (21,8 г, 0,1 моль) в ТГФ (5 мл). Раствор перемешивали в течение ночи при температуре окружающей среды и концентрировали в вакууме, получая светлое масло. Масло фильтровали через силикагель (этилацетат/гексан, 7:3) и концентрировали в вакууме, получая ВОС-защищенное пиперидиновое производное (26,2 г, количественный выход) в виде прозрачного бесцветного масла.
Стадия В: К раствору диизопропиламина (2,8 мл, 20 ммоль) в ТГФ (30 мл), охлажденному до -78° С, по каплям добавляли н-бутиллитий (12,5 мл, 20 ммоль). Через 15 мин по каплям добавляли полученное на стадии Б ВОС-защищенное пиперидиновое производное (2,6 г, 10 ммоль) в ТГФ (10 мл). Через 1,5 ч раствор охлаждали до -60° С и добавляли полученный на стадии А дисульфид (2,0 г, 10 ммоль) в ТГФ (7 мл). Раствор перемешивали в течение 2 ч при температуре окружающей среды. Раствор разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали Н2О и насыщенным NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфид в виде масла (1,8 г, 40%).
Стадия Г: К раствору полученного на стадии В сульфида (1,8 г, 3,95 ммоль) в дихлорметане (75 мл), охлажденному до 0° С, добавляли мета-хлорпербензойную кислоту (1,7 г, 7,9 ммоль). Раствор перемешивали в течение 1,5 ч, после чего разбавляли водой и экстрагировали дихлорметаном. Органический слой промывали 10%-ным раствором Na2SO4, Н2О и насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфон в виде твердого вещества (1,15 г, 59%).
Стадия Д: К раствору полученного на стадии Г сульфона (800 мг, 1,63 ммоль) в ТГФ (9 мл) и этаноле (9 мл) добавляли NaOH (654 мг, 16,3 ммоль) в Н2О (3 мл). Раствор выдерживали в течение 18 ч при 65° С. Раствор концентрировали в вакууме и остаток растворяли в Н2О. После подкисления с помощью 2 н. НСl до рН 4 раствор экстрагировали этилацетатом и органический слой промывали насыщенным раствором NaCl и сушили над сульфатом магния. После концентрирования в вакууме получали кислоту в виде пены белого цвета (790 мг, количественный выход). Элементный анализ: рассчитано для C23H27NO7S: С 59,86; Н 5,90; N 3,04; S 6,95. Обнаружено: С 59,49; Н 6,37; N 2,81; S 6,59.
Стадия Е: К раствору полученной на стадии Д кислоты (730 мг, 1,58 ммоль) в ДМФ (9 мл) добавляли ГОБТ (256 мг, 1,90 ммоль), а затем ЭДК (424 мг, 2,21 ммоль), 4-метилморфолин (0,521 мл, 4,7 ммоль) и 50%-ный водный раствор гидроксиламина (1,04 мл, 15,8 ммоль). Раствор перемешивали в течение 20 ч, после чего добавляли в виде дополнительной порции N-гидроксибензотриазол· Н2О (256 мг), ЭДК (424 мг) и 50%-ный водный раствор гидроксиламина (1,04 мл). После перемешивания еще в течение 24 ч раствор разбавляли Н2О и экстрагировали этилацетатом, органический слой промывали насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали указанное в заголовке соединение в виде твердого вещества белого цвета (460 мг, 61%).
Чистота по данным ЖХВР: >99%. Элементный анализ: рассчитано для C23H28N2O7S: C 57,97; H 5,92; N 5,88; S 6,73. Обнаружено: С 57,95; Н 6,02; N 5,81; S 6,85.
Пример 4: Получение моногидрохлорида N-гидрокси-4-[(4-феноксифенил)сульфонил]-4-пиперидинкарбоксамида
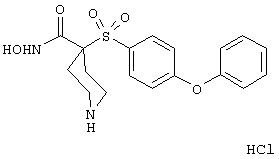
Стадия А: Раствор 4-(фенокси)бензолтиола (2,03 г, 10,0 ммоль) в ДМСО (20 мл) нагревали до 65° С в течение 5 ч. Раствору давали выстояться в течение 18 ч при температуре окружающей среды. Раствор экстрагировали этилацетатом и объединенные органические слои промывали Н2О и насыщенным NaCl и сушили над сульфатом магния. После концентрирования в вакууме получали дисульфид в виде масла желтого цвета (2,3 г, количественный выход).
Стадия Б: К раствору этилизонипекотата (15,7 г, 0,1 моль) в ТГФ (100 мл) по каплям в течение 20 мин добавляли раствор ди-трет-бутилдикарбоната (21,8 г, 0,1 моль) в ТГФ (5 мл). Раствор перемешивали в течение ночи при температуре окружающей среды и концентрировали в вакууме, получая светлое масло. Масло фильтровали через силикагель (на силикагеле, этилацетат/гексан) и концентрировали в вакууме, получая ВОС-защищенное пиперидиновое производное (26,2 г, количественный выход) в виде прозрачного бесцветного масла.
Стадия В: К раствору диизопропиламина (2,8 мл, 20 ммоль) в ТГФ (30 мл), охлажденному до -78° С, по каплям добавляли н-бутиллитий (12,5 мл, 20 ммоль). Через 15 мин по каплям добавляли полученное на стадии Б ВОС-защищенное пиперидиновое производное (2,6 г, 10 ммоль) в ТГФ (10 мл). Через 1,5 ч раствор охлаждали до -60° С и добавляли полученный на стадии А дисульфид (2,0 г, 10 ммоль) в ТГФ (7 мл). Раствор перемешивали в течение 2 ч при температуре окружающей среды. Раствор разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали Н2О и насыщенным NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфид в виде масла (1,8 г, 40%).
Стадия Г: К раствору полученного на стадии В сульфида (1,8 г, 3,95 ммоль) в дихлорметане (75 мл), охлажденному до 0° С, добавляли мета-хлорпербензойную кислоту (1,7 г, 7,9 ммоль). Раствор перемешивали в течение 1,5 ч, после чего разбавляли Н2О и экстрагировали дихлорметаном. Органический слой промывали 10%-ным раствором Na2SO3, Н2О и насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфон в виде твердого вещества (1,15 г, 59%).
Стадия Д: К раствору полученного на стадии Г сульфона (800 мг, 1,63 ммоль) в ТГФ (9 мл) и этаноле (9 мл) добавляли NaOH (654 мг, 16,3 ммоль) в Н2О (3 мл). Раствор выдерживали в течение 18 ч при 65° С. Раствор концентрировали в вакууме и остаток растворяли в Н2О. После подкисления с помощью 2 н. НСl до рН 4 раствор экстрагировали этилацетатом и органический слой промывали насыщенным раствором NaCl и сушили над сульфатом магния. После концентрирования в вакууме получали кислоту в виде пены белого цвета (790 мг, количественный выход). Элементный анализ: рассчитано для C23H27NO7S: С 59,86; Н 5,90; N 3,04; S 6,95; обнаружено: С 59,49; Н 6,37; N 2,81; S 6,59.
Стадия Е: К раствору полученной на стадии Д кислоты (730 мг, 1,58 ммоль) в ДМФ (9 мл) добавляли ГОБТ (256 мг, 1,90 ммоль), а затем ЭДК (424 мг, 2,21 ммоль), 4-метилморфолин (0,521 мл, 4,7 ммоль) и 50%-ный водный раствор гидроксиламина (1,04 мл, 15,8 ммоль). Раствор перемешивали в течение 20 ч, после чего добавляли в виде дополнительной порции N-гидроксибензотриазол· Н2О (256 мг), ЭДК (424 мг) и 50%-ный водный раствор гидроксиламина (1,04 мл). После перемешивания еще в течение 24 ч раствор разбавляли Н2О и экстрагировали этилацетатом, органический слой промывали насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали гидроксамат в виде твердого вещества белого цвета (460 мг, 61%). Чистота по данным ЖХВР: >99%. Элементный анализ: рассчитано для C23H28N2O7S: С 57,97; Н 5,92; N 5,88; S 6,73; обнаружено: С 57,95; Н 6,02; N 5,81; S 6,85.
Стадия Ж: Раствор полученного на стадии Е гидроксамата (385 мг, 0,808 ммоль) в этилацетате (25 мл), охлажденный до 0° С, барботировали в течение 5 мин газообразным НСl. После выстаивания в течение 30 мин раствор концентрировали в вакууме. После растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (330 мг, количественный выход). МС (CI) MH+: рассчитано для С18Н20O5S: 377, обнаружено 377. МСВР (масс-спектроскопия высокого разрешения): рассчитано для C18H20N2O5S: 377,1171 обнаружено 377,1170. Элементный анализ: рассчитано для C18H20N2O5S· 1HCl· 0,25Н2О: С 51,35; Н 5,17; N 6,65; S 7,62; Cl 9,26; обнаружено: С 51,58; Н 5,09; N 6,55; S 8,02; Cl 9,09.
Пример 5: Получение (Е)-N-гидрокси-2-[(4-феноксифенил)сульфонил]-3-фенил-2-пропенамида
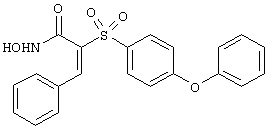
Стадия А: К раствору 4-(фенокси)бензолтиола (5,00 г, 24,7 ммоль) в метаноле (100 мл), охлажденному до 0° С, добавляли трет-бутилбромацетат (3,99 мл, 24,7 ммоль). После добавления триэтиламина (3,60 мл, 25,8 ммоль) раствор перемешивали в течение 40 мин. Раствор концентрировали в вакууме и образовавшийся остаток растворяли в этилацетате и промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали сульфид в виде масла (7,9 г, количественный выход).
Стадия Б: К раствору полученного на стадии А сульфида (7,9 г, 24,7 ммоль) в метаноле (180 мл) и Н2О (20 мл) добавляли Охоnе® (38,4 г, 62,5 ммоль) и смесь перемешивали в течение 22 ч. Смесь подкисляли с помощью 2,5 н. NaOH до рН 4 и сливали, удаляя нерастворимые соли. Полученный сливанием продукт концентрировали до половинного объема и распределяли между этилацетатом и Н2О. Органический слой промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфон в виде твердого вещества желтого цвета (5,79 г, 67%).
Стадия В: К раствору, содержащему полученный на стадии Б сульфон (2,5064 г, 7,20 ммоль) и бензальдегид (0,748 мл, 7,36 ммоль) в бензоле (20 мл), добавляли уксусную кислоту (0,15 мл) и пиперидин (0,05 мл). Раствор нагревали в течение 2 ч до температуры дефлегмации и конденсат собирали с помощью ловушки Дина-Старка. После выдерживания еще в течение 1,5 ч при температуре дефлегмации раствор охлаждали до температуры окружающей среды и перемешивали в течение 18 ч. Раствор разбавляли этилацетатом, промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) и последующего растирания (диэтиловый эфир/гексан) получали ненасыщенный сульфон в виде твердого вещества белого цвета (1,97 г, 73%). Чистота по данным ЖХВР: >98%.
Стадия Г: Раствор полученного на стадии В ненасыщенного сульфона (0,5053 г, 1,16 ммоль) барботировали газообразным НСl в течение 1 ч. Раствор концентрировали в вакууме и остаток растворяли в этилацетате, промывали Н2О и сушили над Na2SO4. После концентрирования в вакууме получали кислоту в виде масла (0,41 г, 93%).
Стадия Д: К раствору полученной на стадии Г кислоты (461 мг, 1,21 ммоль) добавляли тионилхлорид (3,0 мл) и раствор нагревали в течение 1 ч до 100° С. После концентрирования в вакууме получали хлорангидрид в виде масла янтарного цвета (380 мг, 79%).
Стадия Е: К раствору полученного на стадии Д хлорангидрида (380 мг, 0,95 ммоль) в ТГФ (20 мл) добавляли 50%-ный водный раствор гидроксиламина (1,7 мл, 9,5 ммоль). Раствор перемешивали в течение 1 ч при 0° С. Раствор разбавляли этилацетатом, промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) и последующего растирания (диэтиловый эфир/гексан) получали указанное в заголовке соединение в виде твердого вещества белого цвета (131 мг, 35%). Чистота по данным ЖХВР: >97%.
Пример 6: Получение моногидрохлорида N-гидрокси-4-[(4-феноксифенил)сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида

Стадия А: Раствор 4-(фенокси)бензолтиола (2,03 г, 10,0 ммоль) в ДМСО (20 мл) нагревали в течение 5 ч до 65° С. Раствор выдерживали в течение 18 ч при температуре окружающей среды. Раствор экстрагировали этилацетатом, объединенные органические слои промывали Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После концентрирования в вакууме получали дисульфид в виде масла желтого цвета (2,3 г, количественный выход).
Стадия Б: К раствору этилизонипекотата (15,7 г, 0,1 моль) в ТГФ (100 мл) по каплям в течение 20 мин добавляли раствор ди-трет-бутилдикарбоната (21,8 г, 0,1 моль) в ТГФ (5 мл). Раствор перемешивали в течение ночи при температуре окружающей среды и концентрировали в вакууме, получая светлое масло. Масло фильтровали через силикагель (на силикагеле, этилацетат/гексан) и концентрировали в вакууме, получая ВОС-защищенное пиперидиновое производное (26,2 г, количественный выход) в виде прозрачного бесцветного масла.
Стадия В: К раствору диизопропиламина (2,8 мл, 20 ммоль) в ТГФ (30 мл), охлажденному до -78° С, по каплям добавляли н-бутиллитий (12,5 мл, 20 ммоль). Через 15 мин по каплям добавляли полученное на стадии Б ВОС-защищенное пиперидиновое производное (2,6 г, 10 ммоль) в ТГФ (10 мл). Через 1,5 ч раствор охлаждали до -60° С и добавляли полученный на стадии А дисульфид (2,0 г, 10 ммоль) в ТГФ (7 мл). Раствор перемешивали в течение 2 ч при температуре окружающей среды. Раствор разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали Н2О и насыщенным NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфид в виде масла (1,8 г, 40%).
Стадия Г: К раствору полученного на стадии В сульфида (1,8 г, 3,95 ммоль) в дихлорметане (75 мл), охлажденному до 0° С, добавляли мета-хлорпербензойную кислоту (1,7 г, 7,9 ммоль). Раствор перемешивали в течение 1,5 ч, после чего разбавляли водой и экстрагировали дихлорметаном. Органический слой промывали 10%-ным раствором Na2SO4, Н2О и насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфон в виде твердого вещества (1,15 г, 59%).
Стадия Д: Раствор полученного на стадии Г сульфона (3,56 г, 7,0 ммоль) в этилацетате (100 мл), охлажденный до 0° С, барботировали в течение 5 мин газообразным НСl. После концентрирования в вакууме и последующего растирания с диэтиловым эфиром получали гидрохлорид амина в виде твердого вещества белого цвета (3,5 г, количественный выход). МС (CI) MH+: рассчитано для С30Н23NO5S: 390, обнаружено 390.
Стадия Е: К раствору, содержащему полученный на стадии Д гидрохлорид амина (2,6 г, 6 ммоль) и K2CO3 (1,66 г, 12 ммоль) в ДМФ (50 мл), добавляли пропаргилбромид (892 мг, 6 ммоль) и раствор перемешивали в течение 4 ч при температуре окружающей среды. Раствор разбавляли Н2О и экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали пропаргиламин в виде твердого вещества белого цвета (2,15 г, 82%).
Стадия Ж: К раствору полученного на стадии Е пропаргиламина (2,15 г, 5 ммоль) в ТГФ (30 мл) и этаноле (30 мл) добавляли NaOH (2,0 г, 50 ммоль) и раствор в течение 48 ч выдерживали при 65° С. Раствор концентрировали в вакууме и водный остаток подкисляли до значения рН 5. После вакуумной фильтрации образовавшегося осадка получали кислоту в виде твердого вещества белого цвета (2,04 г, количественный выход).
Стадия 3: К раствору полученной на стадии Ж кислоты (559 мг, 1,4 ммоль) в дихлорметане (5 мл) добавляли триэтиламин (0,585 мл, 4,2 ммоль) и 50%-ный водный раствор гидроксиламина (0,925 мл, 14,0 ммоль), а затем гексафторфосфат бромтрис(пирролидино)фосфония (РуВrоР®, 718 мг, 1,54 ммоль). Раствор перемешивали в течение 4 ч при температуре окружающей среды. Раствор разбавляли Н2О и экстрагировали дихлорметаном. Органический слой промывали насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали гидроксамат в виде твердого вещества белого цвета (140 мг, 25%). Элементный анализ: рассчитано для C21H22N2O5S: С 60,85; Н 5,37; N 6,76; S 7,74; обнаружено: С 60,47; Н 5,35; N 6,61; S 7,46.
Стадия И: К раствору полученного на стадии 3 гидроксамата (121 мг, 0,292 ммоль) в метаноле (2 мл), охлажденному до 0° С, добавляли ацетилхлорид (0,228 мл, 0,321 ммоль) в метаноле (1 мл). После перемешивания в течение 30 мин при температуре окружающей среды раствор концентрировали в потоке N2 После растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (107 мг, 81%). Элементный анализ: рассчитано для С21Н22N2О5S· НСl· 0,3Н2О: С 55,27; Н 5,21; N 6,14; обнаружено: С 54,90; Н 5,37; N 6,07.
Пример 7: Получение N-[4-[[2-(гидроксиамино)-2-оксоэтил]сульфонил]фенил]бензамида
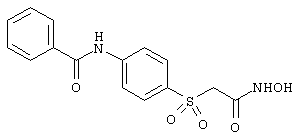
Стадия А: К суспензии 2-(4-аминофенилтио) уксусной кислоты (20,00 г, 0,109 ммоль) в метаноле (100 мл), охлажденной до 0° С, по каплям добавляли тионилхлорид (24,0 мл, 0,327 ммоль). Добавляли еще одну порцию метанола (100 мл) и суспензию нагревали в течение 2 ч до температуры дефлегмации. Раствор концентрировали в вакууме и остаток растворяли в Н2О и нейтрализовали насыщенным раствором NаНСО3. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали метиловый эфир в виде масла темно-пурпурного цвета (22,75 г, количественный выход). Чистота по данным ЖХВР: 99%.
Стадия Б: К раствору, содержащему полученный на стадии А метиловый эфир (5,00 г, 25,35 ммоль) и триэтиламин (7,07 мл, 50,70 ммоль) в дихлорметане (50 мл), добавляли бензоилхлорид (3,24 мл, 27,89 ммоль) и раствор перемешивали в течение 2 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток распределяли между этилацетатом, ТГФ и Н2О. Органический слой промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали бензамид в виде твердого вещества пурпурного цвета (7,06 г, 92%). Чистота по данным ЖХВР: 98%. МС (CI) M+Li+: рассчитано для C16H15NO3S: 308, обнаружено 308.
Стадия В: К раствору полученного на стадии Б бензамида (4,00 г, 13,27 ммоль) в ТГФ (100 мл) и Н2О (10 мл), охлажденному до 0° С, добавляли Охоnе® (моноперсульфат калия, 24,47 г, 39,81 ммоль). Суспензию перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды. Смесь фильтровали для удаления избытка Охоnе® и фильтрат концентрировали в вакууме. Остаток растворяли в этилацетате и промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали сульфон в виде твердого вещества розового цвета (4,11 г, 93%). Чистота по данным ЖХВР: 98%. МС (CI) М+Li+: рассчитано для C16H15NO5S: 340, обнаружено 340.
Стадия Г: К раствору полученного на стадии В сульфона (400 мг, 1,2 ммоль) в ТГФ (9 мл) добавляли 50%-ный водный раствор гидроксиламина (5,0 мл). Раствор перемешивали в течение 8 ч и концентрировали в вакууме. После растирания с горячим диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества беловатого цвета (348 мг, 78%). Чистота по данным ЖХВР: 97%. МС (CI) MH+: рассчитано для С17Н14N2O5S: 335, обнаружено 335.
Пример 8: Получение моногидрохлорида N-[4-[[2-(гидроксиамино)-2-оксо-1-(пиперидин-4-ил)этил]сульфонил]фенил]бензамида
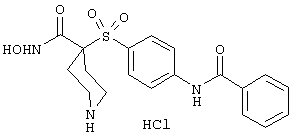
Стадия А: К раствору диэтаноламина (22,16 г, 0,211 моль) в ТГФ (100 мл), охлажденному до 0° С, добавляли ди-трет-бутилдикарбонат (46,0 г, 0,211 моль) и раствор перемешивали в течение 20 ч при температуре окружающей среды. Раствор концентрировали в вакууме и образовавшийся остаток фильтровали через подушку из силикагеля (5% метанол/95% дихлорметан), получая диол в виде прозрачного масла (45,06 г, количественный выход). МС (CI) MH+: рассчитано для C9H19O4S: 206, обнаружено 206.
Стадия Б: К суспензии 2-(4-аминофенилтио)уксусной кислоты (20,00 г, 0,109 ммоль) в метаноле (100 мл), охлажденной до 0° С, добавляли по каплям тионилхлорид (24,0 мл, 0,327 ммоль). После добавления еще одной порции метанола (100 мл) суспензию в течение 2 ч нагревали до температуры дефлегмации. Смесь концентрировали в вакууме, остаток растворяли в Н2О и нейтрализовали насыщенным раствором NaHCO3. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали метиловый эфир в виде масла темно-пурпурного цвета (22,75 г, количественный выход). Чистота по данным ЖХВР: 99%.
Стадия В: К раствору, содержащему полученный на стадии Б метиловый эфир (5,00 г, 25,35 ммоль) и триэтиламин (7,07 мл, 50,70 ммоль) в дихлорметане (50 мл), добавляли бензоилхлорид (3,24 мл, 27,89 ммоль) и раствор перемешивали в течение 2 ч при температуре окружающей среды.
Раствор концентрировали в вакууме и остаток распределяли между этилацетатом, ТГФ и Н2О. Органический слой промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали бензамид в виде твердого вещества пурпурного цвета (7,06 г, 92%). Чистота по данным ЖХВР: 98%.
Стадия Г: К раствору полученного на стадии В бензамида (4,00 г, 13,27 ммоль) в ТГФ (100 мл) и Н2О (10 мл), охлажденному до 0° С, добавляли Охоnе® (24,47 г, 39,81 ммоль). Суспензию перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды. Смесь фильтровали для удаления избытка Охоnе® и фильтрат концентрировали в вакууме. Остаток растворяли в этилацетате и промывали Н2О, насыщенным раствором NaCl и сушили над Nа2SO4. После концентрирования в вакууме получали сульфон в виде твердого вещества розового цвета (4,11 г, 93%). Чистота по данным ЖХВР: 98%.
Стадия Д: К раствору, содержащему полученный на стадии А диол (1,03 г, 5,00 ммоль) и полученный на стадии Г метиловый эфир (2,00 г, 6,00 ммоль) в ТГФ (100 мл), добавляли 1,1’-(азодикарбонил)дипиперидин (5,05 г, 20,00 ммоль). К этой суспензии добавляли триметилфосфин (20,00 мл, 1,0 М раствор в ТГФ, 20,00 ммоль). Смесь перемешивали в течение 1 ч при температуре окружающей среды, а затем выдерживали при 40° С в течение 18 ч. После охлаждения суспензии до температуры окружающей среды добавляли диэтиловый эфир и удаляли фильтрацией нерастворимые твердые частицы. Фильтрат концентрировали в вакууме и образовавшийся остаток растворяли в этилацетате, промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали пиперидин в виде твердого вещества желтого цвета (600 мг, 24%). МС (CI) МН+: рассчитано для C25H30N2O7S: 503, обнаружено 503.
Стадия Е: К раствору полученного на стадии Д пиперидина (950 мг, 1,89 ммоль) в ТГФ (10 мл) добавляли силанолат калия (970 мг, 7,56 ммоль) и раствор перемешивали в течение 72 ч при температуре окружающей среды. Раствор промывали Н2О, подкисляли 1 М НСl до рН 2 и экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали кислоту в виде твердого вещества желтого цвета (772 мг, 84%).
Стадия Ж: К раствору полученной на стадии Е кислоты (772 мг, 1,48 ммоль) в ДМФ (9 мл) добавляли ГОБТ (240 мг, 1,77 ммоль), 4-метилморфолин (0,488 мл, 4,44 ммоль), O-тетрагидропиранилгидроксиамин (538 мг, 4,54 ммоль) и ЭДК (397 мг, 2,07 ммоль). Раствор перемешивали в течение 2 ч при температуре окружающей среды. После концентрирования в вакууме остаток распределяли между этилацетатом и Н2О. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксиламин в виде твердого вещества белого цвета (608 мг, 70%). Чистота по данным ЖХВР: >99%.
Стадия 3: К раствору полученного на стадии Ж защищенного гидроксиламина (596 мг, 1,01 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли 4 М HCl в диоксане (2,50 мл, 10,14 ммоль) и раствор перемешивали в течение 50 мин при температуре окружающей среды. После растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (433 мг, 98%). Чистота по данным ЖХВР: 98%. МС (CI) МН+: рассчитано для С19Н21N3О5S: 404, обнаружено 404.
Пример 9: Получение моногидрохлорида N-гидрокси-4-[[4-(фенилтио)фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида
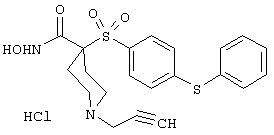
Стадия А: К раствору этилизонипекотата (15,7 г, 0,1 моль) в ТГФ (100 мл) по каплям в течение 20 мин добавляли раствор ди-трет-бутилдикарбоната (21,8 г, 0,1 моль) в ТГФ (5 мл). Раствор перемешивали в течение ночи 15 (приблизительно в течение 18 ч) при температуре окружающей среды и концентрировали в вакууме, получая светлое масло. Масло фильтровали через силикагель (этилацетат/гексан) и концентрировали в вакууме, получая ВОС-защищенное пиперидиновое производное (26,2 г, количественный выход) в виде прозрачного бесцветного масла.
Стадия Б: Раствор 4-фтортиофенола (50,29 г, 390 ммоль) в ДМСО (500 мл) нагревали в течение 6 ч до 65° С. Реакцию прекращали с помощью тающего льда и образовавшееся твердое вещество собирали вакуумной фильтрацией, получая дисульфид в виде твердого вещества белого цвета (34,4 г, 68,9%).
Стадия В: К раствору полученного на стадии А ВОС-защищенного пиперидинового производного (16 г, 62 ммоль) в ТГФ (300 мл), охлажденному до -50° С, добавляли диизопропиламид лития (ДАЛ) (41,33 мл, 74 ммоль) и раствор перемешивали в течение 1,5 ч при 0° С. К этому раствору добавляли дисульфид, полученный на стадии Б (15,77 г, 62 ммоль) и образовавшийся раствор перемешивали в течение 20 ч при температуре окружающей среды. Реакцию прекращали добавлением Н2О и раствор концентрировали в вакууме. Водный остаток экстрагировали этилацетатом и органический слой промывали 0,5 н. раствором КОН, Н2О и насыщенным раствором NaCl. После хроматографии (на силикагеле, гексан/этилацетат) получали сульфид в виде масла (18,0 г, 75%).
Стадия Г: К раствору полученного на стадии В сульфида (16,5 г, 43 ммоль) в дихлорметане (500 мл), охлажденному до 0° С, добавляли 3-хлорпербен-зойную кислоту (18,0 г, 86 ммоль) и раствор перемешивали в течение 20 ч. Раствор разбавляли Н2О и экстрагировали дихлорметаном. Органический слой промывали 10%-ным раствором Nа2SО3, Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфон в виде твердого вещества (10,7 г, 60%).
Стадия Д: Раствор полученного на стадии Г сульфона (10 г, 24,0 ммоль) в этилацетате (250 мл) барботировали в течение 10 мин газообразным HCl, после чего перемешивали в течение 4 ч при температуре окружающей среды. После концентрирования в вакууме получали гидрохлорид амина в виде твердого вещества белого цвета (7,27 г, 86%).
Стадия Е: К раствору полученого на стадии Д гидрохлорида амина (5,98 г, 17,0 ммоль) в ДМФ (120 мл) добавляли карбонат калия (4,7 г, 34,0 ммоль), а затем пропаргилбромид (2,02 г, 17,0 ммоль) и раствор перемешивали в течение 4 ч при температуре окружающей среды. Раствор распределяли между этилацетатом и Н2О, органический слой промывали Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали пропаргиламин в виде масла желтого цвета (5,2 г, 86%).
Стадия Ж: К раствору полученого на стадии Е пропаргиламина в ДМФ (15 мл) добавляли тиофенол (0,80 мл, 7,78 ммоль) и СsСО3 (2,79 г, 8,56 ммоль) и раствор нагревали в течение 6 ч до 70° С. Раствор распределяли между диэтиловым эфиром и Н2О. Органический слой промывали Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали S-феноксифенильное производное в виде масла (1,95 г, 56%).
Стадия З: К раствору полученого на стадии Ж S-феноксифенильного производного (1,81 г, 4,06 ммоль) в этаноле (21 мл) и Н2О (3,5 мл) добавляли КОН (1,37 г, 24,5 ммоль) и раствор нагревали в течение 4,5 ч до 105° С. Раствор подкисляли с помощью концентрированного раствора HCl до значения рН 1 и затем концентрировали, получая кислоту в виде остатка желтого цвета, которую использовали без дополнительной очистки (1,82 г).
Стадия И: К раствору полученой на стадии 3 кислоты (1,82 г, 4,06 ммоль) в ацетонитриле (20 мл) добавляли O-тетрагидро-2Н-пиран-2-илгидроксиламин (723 мг, 6,17 ммоль) и триэтиламин (0,67 мл, 4,86 ммоль). К этому раствору при перемешивании добавляли ЭДК (1,18 г, 6,17 ммоль) и раствор перемешивали в течение 18 ч. Раствор распределяли между Н2О и этилацетатом. Органический слой промывали Н2О, насыщенным раствором NaHCO3, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества белого цвета (1,32 г, 63%).
Стадия К: К раствору полученого на стадии И защищенного гидроксамата (9,65 г, 18,7 ммоль) в метаноле (148 мл), охлажденному до 0° С, добавляли ацетилхлорид (4,0 мл, 56,2 ммоль) и раствор перемешивали в течение 45 мин при температуре окружающей среды. После концентрирования в вакууме и последующего растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (8,10 г, 94%). МС (CI) MH+: рассчитано для C21H22N2O4S2: 431, обнаружено 431.
Пример 10: Получение моногидрохлорида 4-[[4-(1,3-бензодиоксол-5-илокси)фенил]сульфонил]-N-гидрокси-1-(2-пропинил)-4-пиперидинкарбоксамида

Стадия А: К раствору полученного на стадии Е примера 9 пропаргиламина 10 (7,0 г, 19,8 ммоль) в ДМФ (30 мл) добавляли сезамол (5,52 г, 40 ммоль) и карбонат калия (5,52 г, 40 ммоль) и раствор нагревали в течение 48 ч до 85° С. Раствор распределяли между этилацетатом и Н2О. Органический слой сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфид в виде масла (9,38 г, количественный выход).
Стадия Б: К раствору полученного на стадии А сульфида (2,72 г, 5,92 ммоль) в этаноле (30 мл) и Н2О (5 мл) добавляли гидроксид калия (2,0 г, 36 ммоль) и раствор нагревали в течение 4 ч до температуры дефлегмации. Раствор подкисляли до рН 3 с помощью концентрированной HCl. Раствор концентрировали в вакууме и остаток растворяли в ацетонитриле (30 мл). К раствору добавляли O-тетрагидро-2Н-пиран-2-илгидроксиламин (1,05 г, 9,0 ммоль), триэтиламин (1 мл) и ЭДК (1,72 г, 9,0 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме, разбавляли насыщенным раствором NaHCO3 и экстрагировали этилацетатом. Органический слой сушили над сульфатом магния.
После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде масла (2,86 г, 93%).
Стадия В: К раствору полученого на стадии Б защищенного гидроксамата (2,86 г, 5,27 ммоль) в метаноле (40 мл) добавляли ацетилхлорид (1,13 г, 15,8 ммоль) и раствор перемешивали в течение 3 ч. Раствор концентрировали в вакууме. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О (HCl)) получали указанное в заголовке соединение в виде твердого вещества белого цвета (2,2 г, 84%). МС (CI) MH+: рассчитано для C22H22N2O7S: 459, обнаружено 459.
Пример 11: Получение моногидрохлорида тетрагидро-N-гидрокси-4-[[4-(4-фенил-1-пиперидинил)фенил]сульфонил]-2Н-пиран-4-карбоксамида
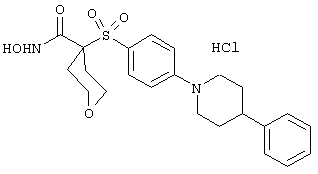
Стадия А: К раствору Na (8,97 г, 390 ммоль) в метаноле (1 л) при 0° С добавляли 4-фтортиофенол (50 г, 390 ммоль) и метилхлорацетат (34,2 мл, 390 ммоль) и раствор перемешивали в течение 4 ч при температуре окружающей среды. Раствор фильтровали для удаления солей и фильтрат концентрировали в вакууме, получая сульфид в виде бесцветного масла (75,85 г, 97%).
Стадия Б: К раствору полученного на стадии А сульфида (75,85 г, 380 ммоль) в метаноле (1 л) и Н2О (100 мл) добавляли Охоnе® (720 г, 1,17 моль) и раствор перемешивали в течение 2 ч. Реакционную смесь фильтровали для удаления избытка солей и фильтрат концентрировали в вакууме. Остаток растворяли в этилацетате, промывали Н2О, насыщенным раствором NаНСО3, насыщенным раствором NaCl и затем сушили над сульфатом магния. После концентрирования в вакууме, получали сульфон в виде твердого вещества белого цвета (82,74 г, 94%).
Стадия В: К раствору полученного на стадии Б сульфона (28,5 г, 123 ммоль) в N,N-диметилацетамиде (200 мл) добавляли карбонат калия (37,3 г, 270 ммоль), простой бис(2-бромэтиловый) эфир (19,3 мл, 147 ммоль), 4-диметиламинопиридин (750 мг, 6 ммоль) и бромид тетрабутиламмония (1,98 г, 6 ммоль) и раствор перемешивали в течение 72 ч при температуре окружающей среды. Раствор сливали на 1 н. HCl (300 мл) и образовавшийся осадок собирали вакуумной фильтрацией. После перекристаллизации (этилацетат/гексан) получали тетрагидропирановое производное в виде твердого вещества бежевого цвета (28,74 г, 77%).
Стадия Г: К раствору полученного на стадии В тетрагидропиранового производного (1,21 г, 4,0 ммоль) в ДМСО (10 мл) добавляли Сs2СО3 (3,26 г, 10,0 ммоль) и 4-фенилпиперидин (640 мг, 4,0 ммоль) и раствор нагревали в течение 2 ч до 90° С. Раствор разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали 5%-ным водным раствором KHSO4, насыщенным раствором NaHCO3, насыщенным раствором NaCl и затем сушили над сульфатом магния. После концентрирования в вакууме получали амин в виде твердого вещества белого цвета (1,2 г, 67%).
Стадия Д: К раствору полученного на стадии Г амина (815 мг, 1,84 ммоль) в метаноле (5 мл) и ТГФ (5 мл) добавляли 50%-ный водный раствор NaOH (2 мл) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток разбавляли Н2О и подкисляли до рН 7. Образовавшийся осадок собирали вакуумной фильтрацией, получая кислоту в виде твердого вещества белого цвета (680 мг, 86%).
Стадия Е: К раствору полученной на стадии Д кислоты (620 мг, 1,44 ммоль) в дихлорметане (10 мл) и ДМФ (3 мл) добавляли РуВrоР (810 мг, 1,73 ммоль), N-метилморфолин (0,5 мл, 4,3 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (190 мг, 1,59 ммоль) и раствор перемешивали в течение 4 ч при температуре окружающей среды. Раствор концентрировали в вакууме, остаток растворяли в этилацетате и промывали Н2О и насыщенным раствором NaCl и сушили над Na2SО4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества белого цвета (630 мг, 83%). МС (CI) MH+: рассчитано для С28Н36N2О6S: 529, обнаружено 529.
Стадия Ж: К раствору полученного на стадии Е защищенного гидроксамата (600 мг, 1,14 ммоль) в диоксане (1,5 мл) и метаноле (1,5 мл) добавляли 4 н. HCl в диоксане (1,5 мл) и раствор перемешивали в течение 2 ч. Раствор сливали на диэтиловый эфир и образовавшийся осадок собирали вакуумной фильтрацией, получая указанное в заголовке соединение в виде твердого вещества бежевого цвета (500 мг, 91%). МС (CI) М+Li+: рассчитано для C23H28N2O5S: 445, обнаружено 445.
Пример 12: Получение 1-ацетил-N-гидрокси-4-[(4-феноксифенил)сульфонил]-4-пиперидинкарбоксамида
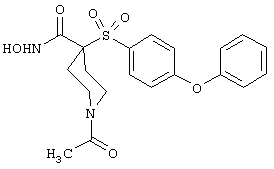
Стадия А: К раствору полученного на стадии Г примера 6 сульфона (2,75 г, 5,6 ммоль) в ТГФ (10 мл) и этаноле (10 мл) добавляли NaOH (2,25 г, 56 ммоль) и раствор нагревали до 70° С в течение 18 ч. Раствор концентрировали в вакууме, остаток растворяли в Н2О и экстрагировали диэтиловым эфиром. Водный раствор подкисляли до значения рН 2 и экстрагировали этилацетатом. Органический слой сушили над сульфатом магния. После концентрирования в вакууме получали неочищенную кислоту в виде твердого вещества. Раствор кислоты в дихлрметане (6 мл) и трифторуксусной кислоте (6 мл) перемешивали в течение 1 ч при температуре окружающей среды. После концентрирования в вакууме получали гидрохлорид амина в виде твердого вещества (2,3 г, количественный выход).
Стадия Б: К раствору полученного на стадии А гидрохлорида амина (2,3 г, <5,6 ммоль) в ацетоне (10 мл) и Н2О (10 мл), охлажденному до 0° С, добавляли триэтиламин (1,17 мл, 8,4 ммоль) и ацетилхлорид (0,60 мл, 8,4 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме для удаления ацетона и водный раствор экстрагировали диэтиловым эфиром. Водный раствор подкисляли до значения рН 2 и экстрагировали этилацетатом. Органический слой сушили над сульфатом магния и после концентрирования в вакууме получали N-ацетильное производное в виде твердого вещества белого цвета (1,5 г, 65,2%).
Стадия В: К раствору полученного на стадии Б N-ацетильного производного (0,6 г, 1,49 ммоль) в ДМФ (10 мл) добавляли ЭДК (401 мг, 2,1 ммоль), а затем 50%-ный водный раствор гидроксиламина (0,9 мл) и 4-метилморфолин (0,7 мл, 6,4 ммоль), и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток растворяли в этилацетате. Органический слой промывали Н2О и сушили над сульфатом магния. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали указанное в заголовке соединение в виде твердого вещества белого цвета (101 мг, 16%). МС (CI) MH+: рассчитано для C20H22N2O6S: 419, обнаружено 419.
Пример 13: Получение моногидрохлорида 4-[[4-(циклогексилтио)фенил]сульфонил]-N-гидрокси-1-(2-пропинил)-4-пиперидинкарбоксамида
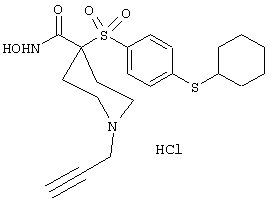
Стадия А: К раствору полученного на стадии Е примера 9 пропаргиламина (6,5 г, 18,4 ммоль) в ДМФ (10 мл) добавляли карбонат калия (3,81 г, 27,6 ммоль) и циклогексилмеркаптан (3,37 мл, 27,6 ммоль). Раствор нагревали в течение 6,5 ч до 100° С. Раствор разбавляли Н2О и экстрагировали этилацетатом. Органические слои сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфид в виде масла желтого цвета (6,05 г, 73%).
Стадия Б: К раствору полученного на стадии А сульфида (612 мг, 1,4 ммоль) в этаноле (8,4 мл) и Н2О (1,4 мл) добавляли гидроксид калия (470 мг, 8,4 ммоль) и раствор кипятили с обратным холодильником в течение 3 ч. Раствор подкисляли до значения рН 3 и концентрировали в вакууме. Остаток растворяли в ацетонитриле (10 мл) и к этому раствору добавляли O-тетрагидро-2Н-пиран-2-илгидроксиламин (230 мг, 2,0 ммоль) и триэтиламин (0,5 мл), а затем ЭДК (380 мг, 2,0 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток разбавляли насыщенным раствором NaHCO3 и экстрагировали этилацетатом. Органический слой сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде масла (246 мг, 34%).
Стадия В: К раствору полученного на стадии Б защищенного гидроксамата (246 мг, 0,47 ммоль) в метаноле (4 мл) добавляли ацетилхлорид (0,11 мл, 1,5 ммоль) и раствор перемешивали в течение 3 ч при температуре окружающей среды. После концентрирования в вакууме и хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О(HCl)) получали указанное в заголовке соединение в виде твердого вещества белого цвета (223 мг, количественный выход).
Пример 14: Получение моногидрохлорида N-гидрокси-1-метил-4-[(феноксифенил)сульфонил]-4-пиперидинкарбоксамида
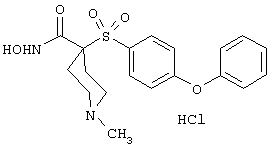
Стадия А: К раствору полученного на стадии Г примера 6 сульфона (2,67 г, 5,5 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (5 мл) и раствор перемешивали в течение 2 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток растирали с диэтиловым эфиром, получая неочищенный трифторацетат амина. К раствору неочищенного трифторацетата амина в метаноле (10 мл) добавляли формальдегид (37%-ный водный раствор, 2,0 мл, 27,5 ммоль) и боранпиридин (2,2 мл, 22 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме. Остаток растворяли в этилацетате, промывали Н2О и сушили над сульфатом магния. После концентрирования в вакууме получали N-метильное производное в виде масла желтого цвета (2,17 г, 98%).
Стадия Б: К раствору полученного на стадии А N-метильного производного (2,17 г, 5,4 ммоль) в этаноле (10 мл) и ТГФ (10 мл) добавляли NaOH (2,0 г, 50 ммоль) и реакционную смесь перемешивали в течение 18 ч при -65° С. Раствор концентрировали в вакууме. Остаток растворяли в Н2О и экстрагировали диэтиловым эфиром. Водный раствор подкисляли до значения рН 2 и образовавшееся твердое вещество собирали вакуумной фильтрацией, получая кислоту в виде твердого вещества белого цвета (1,8 г, 90%).
Стадия В: К раствору полученной на стадии Б кислоты (0,5 г, 1,3 ммоль) в ДМФ (10 мл) добавляли ЭДК (1,06 г, 5,5 ммоль), а затем O-тетрагидро-2Н-пиран-2-илгидроксиламин (490 мг, 4,2 ммоль) и 4-метилморфолин (0,76 мл) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток растворяли в этилацетате, промывали Н2О и сушили над сульфатом магния. После концентрирования в вакууме получали неочищенный защищенный гидроксамат. К раствору неочищенного гидроксамата в метаноле (10 мл) добавляли ацетилхлорид (0,28 мл, 3,9 ммоль) и раствор перемешивали в течение 3 ч при температуре окружающей среды. Раствор концентрировали в вакууме. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О (0,0125% HCl)) получали указанное в заголовке соединение в виде твердого вещества белого цвета (261 мг, 46%). МС (CI) MH+: рассчитано для C19H22N2S: 391, обнаружено 391.
Пример 15: Получение моногидрохлорида N-гидрокси-4-[[4-(4-метоксифенокси)фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида
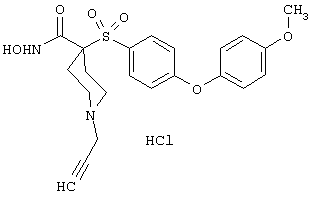
Стадия А: К раствору полученного на стадии Е примера 9 пропаргиламина (2,00 г, 5,66 ммоль) в ДМФ (10 мл) добавляли карбонат цезия (4,7 г, 14,5 ммоль) и 4-метокситиофенол (1,80 г, 14,5 ммоль) и раствор нагревали в течение 24 ч до 95° С. Раствор разбавляли этилацетатом и промывали 1 н. NaOH и насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали феноксипроизводное в виде твердого вещества белого цвета (2,67 г, количественный выход).
Стадия Б: К раствору полученного на стадии А феноксипроизводного (2,40 г, 5,25 ммоль), в этаноле (30 мл) и Н2О (6 мл) добавляли гидроксид калия (2,0 г, 31,37 ммоль) и раствор нагревали до температуры дефлегмации в течение 4 ч. Раствор подкисляли концентрированной HCl до значения рН 3 и остаток собирали вакуумной фильтрацией, получая неочищенную кислоту, которую использовали без дополнительной очистки.
Стадия В: К раствору полученной на стадии Б кислоты (2,25 г, 5,25 ммоль) в ацетонитриле (30 мл) добавляли триэтиламин (1 мл) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (1,34 г, 9,0 ммоль). После перемешивания раствора в течение 15 мин добавляли ЭДК (1,72 г, 9,0 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток растворяли в этилацетате. Этилацетатный раствор промывали насыщенным NaHCO3, Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества белого цвета (0,93 г, 33%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (0,93 г, 1,7 ммоль) в метаноле (15 мл) добавляли ацетилхлорид (0,36 мл, 5,1 ммоль) и раствор перемешивали в течение 3 ч. Раствор концентрировали в вакууме, получая указанное в заголовке соединение в виде твердого вещества белого цвета (650 мг, 82%). Элементный анализ: рассчитано для С22Н24N2О6·НСl: С 54,84; Н 5,24; N 5,82; S 6,67; Cl 6,67; обнаружено: С 53,10; Н 5,07; N 5,59; S 7,04; Cl 6,32.
Пример 16: Получение моногидрохлорида 4-[[4-(4-бутокси-1-пиперидинил)фенил]сульфонил]-тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
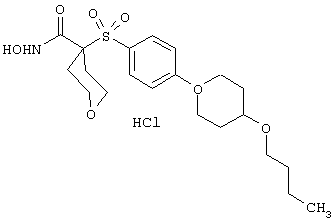
Стадия А: К раствору полученного на стадии В примера 11 тетрагидропиранового производного (1,95 г, 6,46 ммоль) в диметилсульфоксиде (ДМСО, 25 мл) добавляли Сs2СО3 (7,4 г, 22,6 ммоль) и 4-бутоксипиперидин (1,25 г, 6,46 ммоль) и раствор нагревали в течение 1 ч до 90° С. Реакцию в растворе прекращали добавлением Н2О и экстрагировали этилацетатом. Органический слой промывали 5%-ным водным раствором KHSO4, насыщенным раствором NаНСО3, насыщенным раствором NaCl и затем сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/дихлорметан) получали амин в виде масла желтого цвета (1,85 г, 65%).
Стадия Б: К раствору полученного на стадии А амина (1,65 г, 3,76 ммоль) в ТГФ (10 мл) добавляли триметилсиланолат калия (530 мг, 4,13 ммоль) и раствор перемешивали в течение 22 ч при температуре окружающей среды. Раствор концентрировали в вакууме и неочищенный остаток использовали в последующей реакции без дополнительной очистки.
Стадия В: К раствору полученной на стадии Б неочищенной кислоты (1,74 г, 3,76 ммоль) в дихлорметане (10 мл) добавляли РуВrоР (2,10 г, 4,51 ммоль), N-метилморфолин (1,24 мл, 11,3 ммоль) и О-тетрагидро-2Н-пиран-2-илгидроксиламин (484 мг, 4,14 ммоль), и раствор перемешивали в течение 30 мин при температуре окружающей среды. Раствор концентрировали в вакууме. Остаток растворяли в этилацетате, промывали Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан/метанол) получали защищенный гидроксамат в виде бесцветного масла (1,5 г, общий выход по двум стадиям 76%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (1,25 г, 2,4 ммоль) в диоксане (3 мл) добавляли 4 н. раствор HCl в диоксане (3 мл) и раствор перемешивали в течение 15 мин. После добавления метанола (3 мл) раствор перемешивали в течение 5 ч при температуре окружающей среды. Раствор сливали на диэтиловый эфир и образовавшийся осадок собирали вакуумной фильтрацией, получая указанное в заголовке соединение в виде твердого вещества белого цвета (1,0 г, 88%). МС (CI) MH+: рассчитано для С21Н32N2О6S: 441, обнаружено 441.
Пример 17: Получение моногидрохлорида 1-циклопропил-N-гидрокси-4-[(4-феноксифенил)сульфонил]-4-пиперидинкарбоксамида
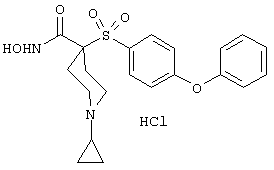
Стадия А: К раствору полученного на стадии Д примера 6 гидрохлорида амина (2,13 г, 5,0 ммоль) в метаноле (25 мл) добавляли молекулярные сита размером 3Е, уксусную кислоту (2,86 мл, 50 ммоль) и раствор перемешивали в течение 5 мин. К этому раствору добавляли ((1-этиоксициклопропил)окси)триметилсилан (6,08 мл, 30 ммоль), а затем цианборогидрид натрия (1,41 г, 22,0 ммоль) и раствор нагревали до температуры дефлегмации в течение 18 ч. Избыток солей и молекулярные сита собирали фильтрацией и фильтрат концентрировали в вакууме. Остаток разбавляли этилацетатом и промывали 1 н. раствором NaOH, Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали циклопропиламин в виде твердого вещества белого цвета (1,90 г, 86%).
Стадия Б: К раствору полученного на стадии А циклопропиламина (1,9 г, 4,2 ммоль) в ТГФ (12 мл) и этаноле (12 мл) добавляли NaOH (1,71 г, 4,3 ммоль) в Н2О (10 мл) и раствор в течение 20 ч нагревали до 62° С. Раствор концентрировали в вакууме, остаток разбавляли Н2О и подкисляли 1 н. раствором HCl до значения рН 5. Образовавшееся твердое вещество собирали вакуумной фильтрацией, получая кислоту в виде твердого вещества белого цвета (1,49 г, 82%). МС (CI) MH+: рассчитано для C21H23NO5S: 402, обнаружено 402. МСВР: рассчитано для С21Н23NО5S: 402,1375, обнаружено 402,1350.
Стадия В: К раствору полученной на стадии Б кислоты (1,49 г, 3,4 ммоль) в дихлорметане (50 мл) добавляли триэтиламин (1,42 мл, 10,21 ммоль), а затем 50%-ный водный раствор гидроксиламина (2,25 мл, 34,0 ммоль) и РуВrоР (3,17 г, 6,8 ммоль), и раствор перемешивали в течение 72 ч. Смесь разбавляли Н2О, органический слой отделяли, промывали насыщенным раствором NaCl и сушили над сульфатом магния. После концентрирования в вакууме и последующей хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали гидроксамат.
Гидрохлорид получали путем растворения свободного основания (830 мг, 2,0 ммоль) в метаноле (20 мл) с последующим добавлением ацетилхлорида (0,17 мл, 2,0 ммоль). Раствор перемешивали в течение 10 мин при 0° С. Образовавшееся твердое вещество собирали вакуумной фильтрацией и промывали холодным диэтиловым эфиром, получая указанное в заголовке соединение (595 мг, 66%). МСВР: рассчитано для С21Н24N2О5S: 416,1407, обнаружено 416,1398. Элементный анализ: рассчитано для C21H24N2O5S: С 55,68; Н 5,56; N 6,18; S 7,08; Сl 7,83; обнаружено: С 55,39; Н 5,72; N 6,15; S 7,29; Сl 8,17.
Пример 18: Получение N-гидрокси-1-(метилсульфонил)-4-(феноксифенил)сульфонил]-4-пиперидинкарбоксамида
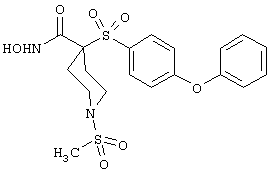
Стадия А: К раствору полученного на стадии Д примера 6 гидрохлорида амина (1,06 г, 2,5 ммоль) в дихлорметане (10 мл) добавляли триэтиламин (0,76 мл, 5,5 ммоль) и метансульфонилхлорид (0,23 мл, 3,0 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток распределяли между этилацетатом и Н2О. Органический слой промывали Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали метансульфонамид в виде твердого вещества (2,1 г, 58%).
Стадия Б: К раствору полученного на стадии А метансульфонамида (2,0 г, 4,15 ммоль) в этаноле (12 мл) и Н2О (12 мл) добавляли NaOH (1,66 г, 41,5 ммоль) и раствор нагревали в течение 18 ч до 65° С. Раствор концентрировали в вакууме и оставшийся водный раствор подкисляли до рН 4. Раствор экстрагировали этилацетатом и органический слой промывали насыщенным раствором NaCl и сушили над сульфатом магния. После концентрирования в вакууме получали кислоту в виде пены желтого цвета (1,46 г, 80%).
Стадия В: К раствору полученной на стадии Б кислоты (1,46 г, 3,38 ммоль) в дихлорметане (50 мл) добавляли триэтиламин (1,41 мл, 10,1 ммоль), 50%-ный водный раствор гидроксиламина (2,2 мл, 33,8 ммоль) и РуВrоР (3,16 г, 6,76 ммоль), и раствор перемешивали в течение 72 ч при температуре окружающей среды. Раствор разбавляли Н2О, органический слой отделяли, промывали насыщенным раствором NaCl и затем сушили над сульфатом магния. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) и последующего растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (160 мг, 11%). Элементный анализ: рассчитано для C19H22N2O7S2: С 50,21; Н 4,88; N 6,16; S 14,11; обнаружено: С 48,72; Н 5,36; N 5,61; S 12,81.
Пример 19: Получение моногидрохлорида 4-[[4-(циклогексилтио)фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
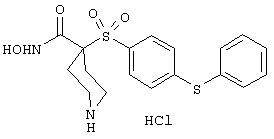
Стадия А: К раствору полученного на стадии Г примера 9 сульфона (10,1 г, 24,0 ммоль) в ДМФ (20 мл) добавляли К2СО3 (5,0 г, 36,0 моль) и циклогексилмеркаптан (4,4 мл, 36,0 ммоль) и раствор нагревали в течение 6,5 ч до 85° С. Раствор распределяли между этилацетатом и Н2О. Органический слой промывали насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфид в виде масла (8,2 г, 67%).
Стадия Б: К раствору сульфида (2,32 г, 4,5 ммоль) в этаноле (10 мл) и ТГФ (10 мл) добавляли NaOH (1,81 г, 45 ммоль) в Н2О (10 мл) и раствор нагревали в течение 18 ч до 65° С. Раствор концентрировали в вакууме и водный остаток подкисляли до значения рН 2. Раствор экстрагировали дихлорметаном и сушили над сульфатом магния. После концентрирования в вакууме получали кислоту в виде твердого вещества белого цвета (830 мг, 38%).
Стадия В: К раствору полученной на стадии Б кислоты (2,0 г, 4,0 ммоль) в дихлорметане (25 мл) добавляли N-метилморфолин (1,32 мл, 12,0 ммоль), РуВrоР (2,12 г, 2,12 ммоль) и 50%-ный водный раствор гидроксиламина (2,6 мл, 40 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли Н2О и слои разделяли. Органический слой промывали насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/метанол) получали гидроксамат в виде твердого вещества белого цвета (1,4 г, 70%).
Стадия Г: Раствор полученного на стадии В гидроксамата (1,31 г, 2,63 ммоль) в этилацетате (70 мл), охлажденный до 0° С, барботировали газообразным HCl в течение 30 мин. Раствор концентрировали в вакууме. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О(НСl)) получали указанное в заголовке соединение в виде твердого вещества белого цвета (378 мг, 33%). Элементный анализ: рассчитано для C18H26N2O4S2: С 49,70; Н 6,26; N 6,44; S 14,74; Cl 8,15; обнаружено: С 48,99; Н 6,34; N 6,24; S 14,66; Cl 8,56.
Пример 20: Получение дигидрохлорида тетрагидро-N-гидрокси-4-[[4-(4-фенил-1-пиперазинил)фенил]сульфонил]-2Н-пиран-4-карбоксамида
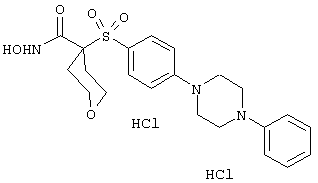
Стадия А: К раствору полученного на стадии В примера 11 тетрагидропиранового производного (1,96 г, 6,5 ммоль) в ДМСО (20 мл) добавляли Cs2CO3 (4,9 г, 15 ммоль) и 4-фенилпиперазин (1,1 г, 7,15 ммоль) и раствор нагревали в течение 45 мин до 90° С. Реакцию прекращали добавлением Н2О и смесь экстрагировали этилацетатом. Органический слой промывали 5%-ным водным раствором KHSO4, насыщенным раствором NаНСО3, насыщенным раствором NaCl и затем сушили над сульфатом магния. После концентрирования в вакууме, получали амин в виде твердого вещества бежевого цвета (1,7 г, 59%).
Стадия Б: К раствору полученного на стадии А амина (1,5 г, 3,38 ммоль) в ТГФ (20 мл) добавляли триметилсиланолат калия (480 мг, 3,72 ммоль) и раствор перемешивали в течение 22 ч при температуре окружающей среды. После концентрирования в вакууме получали неочищенную кислотно-аддитивную соль, которую можно использовать на следующей стадии без очистки.
Стадия В: К раствору полученной на стадии Б кислотно-аддитивной соли (1,58 г, 3,38 ммоль) в дихлорметане (10 мл) и ДМФ (3 мл) добавляли РуВrоР (1,89 г, 4,06 ммоль), N-метилморфолин (1,1 мл, 10,1 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (435 мг, 3,72 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток распределяли между этилацетатом и Н2О, органический слой промывали Н2О и насыщенным раствором NaCl и затем сушили над Na2SO4. После хроматографии (на силикагеле, дихлорметан/метанол) получали защищенный гидроксамат в виде пены белого цвета (1,7 г, общий выход по двум стадиям 95%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (1,28 г, 2,4 ммоль) в диоксане (5 мл) и метаноле (5 мл) добавляли 4 н. HCl в диоксане (5 мл) и раствор перемешивали в течение 2 ч при температуре окружающей среды. Раствор сливали на диэтиловый эфир и образовавшийся осадок собирали вакуумной фильтрацией, получая указанное в заголовке соединение в виде твердого вещества белого цвета (900 мг, 73%). МС (CI) МН+: рассчитано для C22H27N3O5S: 446, обнаружено 446.
Пример 21: Получение моногидрохлорида 4-[[4-(циклогексилтио)фенил]сульфонил]-1-циклопропил-N-гидрокси-4-пиперидинкарбоксамида

Стадия А: К раствору полученного на стадии Г примера 9 сульфона (10,1 г, 24,0 ммоль) в ДМФ (20 мл) добавляли К2СО3 (5,0 г, 36,0 моль) и циклогексилмеркаптан (4,4 мл, 36,0 ммоль) и раствор нагревали в течение 6,5 ч до 85° С. Раствор распределяли между этилацетатом и Н2О. Органический слой промывали насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфид в виде масла (8,2 г, 67%).
Стадия Б: Раствор полученного на стадии А сульфида (8,2 г, 17,0 ммоль) в этилацетате (100 мл), охлажденном до 0° С, барботировали в течение 30 мин газообразным HCl. Раствор концентрировали в вакууме, получая амин в виде твердого вещества белого цвета (5,99 г, 79%). МС (CI) MH+: рассчитано для C20H29NO4S: 412, обнаружено 412.
Стадия В: К раствору полученного на стадии Б амина (2,24 г, 5,0 ммоль) в метаноле (20 мл) добавляли уксусную кислоту (2,86 мл, 50 ммоль), а затем (1-этоксициклопропил)окситриметилсилан (6,03 мл, 30 ммоль) и борогидрид натрия (1,41 г, 22,5 ммоль) и раствор кипятили с обратным холодильником в течение 18 ч. Раствор концентрировали в вакууме, остаток растворяли в этилацетате и промывали 1н. раствором NaOH, Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали циклопропиламин в виде твердого вещества белого цвета (1,97 г, 87%).
Стадия Г: К раствору полученного на стадии В циклопропиламина (1,9 г, 4,2 ммоль) в этаноле (10 мл) и ТГФ (10 мл) добавляли NaOH (1,68 г, 42,0 ммоль) в Н2О (10 мл) и раствор нагревали в течение 18 ч до 68° С. Раствор концентрировали в вакууме и водный остаток подкисляли до значения рН 2. Собирали образовавшееся твердое вещество и промывали диэтиловым эфиром, получая кислоту в виде твердого вещества белого цвета (1,61 г, 81%). МСВР: рассчитано для C21H29NO4S2: 424,1616, обнаружено 424,1615.
Стадия Д: К раствору полученной на стадии Г кислоты (1,61 г, 3,0 ммоль) в дихлорметане (30 мл) добавляли N-метилморфолин (1,0 г, 9,0 ммоль), РуВrоР (1,54 г, 3,3 ммоль) и 50%-ный водный раствор гидроксиламина (2,0 мл, 30 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме. Остаток распределяли между этилацетатом и Н2О, органический слой промывали Н2О и насыщенным раствором NaCl и сушили над сульфатом магния. После фильтрации через подушку из двуокиси кремния (этилацетат/метанол) получали гидроксамат в виде твердого вещества белого цвета (1,07 г, 80%).
Стадия Е: К раствору полученного на стадии Д гидроксамата (1,07 г, 2,4 ммоль) в холодном метаноле (2 мл) добавляли ацетилхлорид (0,27 мл, 3,6 ммоль) и раствор перемешивали в течение 30 мин. Раствор концентрировали в вакууме. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О(HCl)) получали указанное в заголовке соединение в виде твердого вещества белого цвета (245 мг, 21%).
Пример 22: Получение моногидрохлорида 4-[[4-[(4-фторфенил)тио]фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
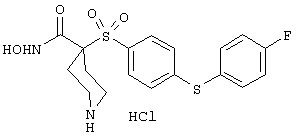
Стадия А: К раствору полученного на стадии Г примера 9 сульфона (6,0 г, 14,4 ммоль) в ДМФ (30 мл) добавляли карбонат калия (2,39 мг, 17,3 ммоль) и 4-фтортиофенол (3,0 мл, 28,1 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли этилацетатом и промывали 1 н. раствором NaOH и насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфид в виде твердого вещества (6,6 г, 87%).
Стадия Б: К раствору полученного на стадии А сульфида (6,6 г, 12,6 ммоль) в этаноле (90 мл) и Н2О (20 мл) добавляли гидроксид натрия (5,04 г, 126 ммоль) и раствор выдерживали в течение 18 ч при 70° С. Смесь подкисляли до значения рН 4 и раствор экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/этанол) получали кислоту в виде твердого вещества (4,8 г, 79%).
Стадия В: К раствору полученной на стадии Б кислоты (4,8 г, 10,0 ммоль) в ДМФ (30 мл) добавляли 4-метилморфолин (3,03 г, 30,0 ммоль), а затем O-тетрагидро-2Н-пиран-2-илгидроксиламин (7,45 г, 50,0 ммоль) и РуВrоР (5,59 г, 12,0 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме. Остаток растворяли в этилацетате и промывали Н2О и насыщенным раствором NaCl и затем сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества белого цвета (4,0 г, 67%).
Стадия Г: Раствор полученного на стадии Г защищенного гидроксамата (4,0 г, 6,7 ммоль) в этилацетате (120 мл) барботировали в течение 5 мин газообразным HCl, а затем перемешивали в течение 1,5 ч при температуре окружающей среды. Образовавшееся твердое вещество собирали вакуумной фильтрацией, получая указанное в заголовке соединение в виде твердого вещества белого цвета (1,90 г, 64%). МС (CI) MH+: рассчитано для C18H19N2O4S2F: 411, обнаружено 411.
Пример 23: Получение дигидрохлорида N-гидрокси-4-[[4-[4-(1Н-имидазол-1-ил)фенокси]фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида
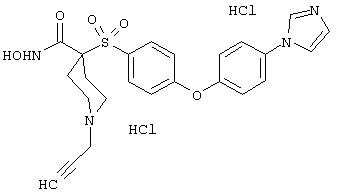
Стадия А: К раствору полученного на стадии Е примера 9 гидрохлорида амина (3,00 г, 8,49 ммоль) в ДМФ (13 мл) добавляли К2СО3 (2,35 г, 17,0 ммоль) и 4-(имидазол-1-ил)фенол (2,72 г, 17,0 ммоль) и раствор нагревали в течение 64 ч до 85° С. Раствор концентрировали в вакууме и остаток распределяли между этилацетатом и Н2О. Органический слой промывали Н2О и насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, хлороформ/метанол) получали сложный этиловый эфир в виде пены белого цвета (2,36 г, 56%).
Стадия Б: К раствору полученного на стадии А сложного этилового эфира (2,36 г, 5,33 ммоль) в этаноле (2,8 мл) и Н2О (4,6 мл) добавляли КОН (1,80 г, 32,1 ммоль) и раствор нагревали в течение 4,5 ч до 100° С. Раствор подкисляли концентрированным раствором HCl до значения рН 1 и затем концентрировали, получая кислоту в виде твердого вещества рыжевато-коричневого цвета, которую использовали без дополнительной очистки (2,87 г).
Стадия В: К раствору полученной на стадии Б кислоты (2,87 г, 5,33 ммоль) в ацетонитриле (24 мл) добавляли O-тетрагидро-2Н-пиран-2-илгидроксиламин (870 мг, 7,45 ммоль), ЭДК (1,43 г, 7,45 ммоль) и N-метилморфолин (1,21 мл, 11,0 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали и остаток разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (хлороформ/метанол) получали защищенный гидроксиламин в виде твердого вещества белого цвета (1,62 г, 53%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксиламина (1,60 г, 2,83 ммоль) в метаноле (23 мл) добавляли ацетилхлорид (0,61 мл, 8,52 ммоль) и раствор перемешивали в течение 1 ч. Раствор концентрировали в вакууме. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О (HCl)) получали указанное в заголовке соединение в виде твердого вещества белого цвета (975 мг, 62%). МС (Cl) МН+: рассчитано для C24H25N4O5S: 481, обнаружено 481. Элементный анализ: рассчитано для C24H25N4O5S· HCl: С 52,08; Н 4,73; N 10,12; S 5,79; Cl 12,81; обнаружено: С 51,59; Н 4,84; N 10,93; S 5,51; Cl 11,98.
Пример 24: Получение миногидрохлорида 4-[[4-(4-фторфенил)тиофенил]сульфонил]-N-гидрокси-1-(2-пропинил)-4-пиперидинкарбоксамида
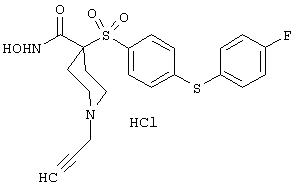
Стадия А: К раствору полученного на стадии Е примера 9 пропаргиламина (4,06 г, 11,49 ммоль) в ДМФ (20 мл) добавляли карбонат калия (3,18 г, 22,98 ммоль) и 4-фтортиофенол (2,95 г, 22,98 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли этилацетатом, промывали 1н. NaOH и насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфид в виде твердого вещества (4,46 г, 84%).
Стадия Б: К раствору полученного на стадии А сульфида (4,46 г, 9,7 ммоль) в тетрагидропиране (90 мл), Н2О (30 мл) и этаноле (30 мл) добавляли NaOH (3,86 г, 97,0 ммоль) и раствор нагревали в течение 2 ч до 65° С. Раствор концентрировали в вакууме и остаток растворяли в Н2О и подкисляли 2 н. HCl до значения рН 4. Образовавшийся остаток собирали вакуумной фильтрацией, получая кислоту в виде твердого вещества белого цвета (4,0 г, 95%).
Стадия В: К раствору полученной на стадии Б кислоты (4,0 г, 9,2 ммоль) в ДМФ (50 мл) и 4-метилморфолине (2,8 г, 27,7 ммоль) добавляли O-тетрагидро-2Н-пиран-2-илгидроксиламин (6,88 г, 46,1 ммоль) и РуВrоР (5,16 г, 11,1 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток растворяли в этилацетате. Раствор промывали Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества белого цвета (2,8 г, 56%).
Стадия Г: Раствор полученного на стадии В защищенного амина (2,8 г, 5,1 ммоль) в этилацетате (100 мл) барботировали в течение 10 мин газообразным HCl и раствор перемешивали в течение 1 ч. Раствор концентрировали в вакууме и твердое вещество перекристаллизовывали (этанол), получая указанное в заголовке соединение в виде твердого вещества белого цвета (1,12 г, 45%). МС (CI) MH+: рассчитано для C21H21N2O4S2F: 449, обнаружено 449.
Пример 25: Получение 4-[[4-[(4-хлорфенил)тио]фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
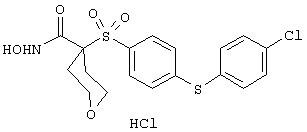
Стадия А: К раствору полученного на стадии В примера 11 тетрагидропиранового производного (8,0 г, 26,5 ммоль) в ТГФ (250 мл) добавляли триметилсилонат калия (10,2 мг, 79,5 ммоль) и раствор перемешивали в течение 1,5 ч. Реакцию прекращали добавлением Н2О, раствор подкисляли до значения рН 2,5 и раствор экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали кислотно-аддитивную соль в виде твердого вещества белого цвета (5,78 г, 76%).
Стадия Б: К раствору полученной на стадии А кислотно-аддитивной соли (5,4 г, 18,7 ммоль) в ДМФ (35 мл) добавляли ГОБТ (3,04 г, 22,5 ммоль), N-метилморфолин (6,2 мл, 56,2 ммоль), O-тетрагидро-2Н-пиран-2-илгидроксиламин (6,8 г, 58,1 ммоль) и ЭДК (5,0 г, 26,2 ммоль) и раствор перемешивали в течение 3 ч при температуре окружающей среды. Раствор концентрировали в вакууме, остаток распределяли между этилацетатом и Н2О и органический слой промывали 5%-ным водным раствором KHSO4, Н2О, насыщенным раствором NаНСО3, насыщенным раствором NaCl и затем сушили над Na2SO4. После концентрированна в вакууме получали защищенный гидроксамат в виде твердого вещества белого цвета (6,34 г, 87%).
Стадия В: К раствору пара-хлортиофенола (2,71 г, 18,7 ммоль) в ДМФ (10 мл) добавляли К2СО3 (2,6 г, 18,7 ммоль), а затем полученный на стадии Б защищенный гидроксамат (2,9 г, 7,5 ммоль) и раствор выдерживали в течение 5 ч при 75° С. Раствор концентрировали в вакууме, остаток распределяли между этилацетатом и Н2О, органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан/метанол) получали сульфид в виде пены белого цвета (3,56 г, 93%). МС (CI) MH+: рассчитано для C23H26ClNO6S2: 512, обнаружено 512.
Стадия Г: К раствору полученного на стадии В сульфида (3,5 г, 6,8 ммоль) в диоксане (10 мл) добавляли 4н. раствор HCl в диоксане (10 мл). После перемешивания в течение 10 мин добавляли в течение 1 ч при непрерывном перемешивании метанол (10 мл). Раствор концентрировали в вакууме. После перекристаллизации (ацетон/гексан) получали указанное в заголовке соединение в виде твердого вещества белого цвета (2,4 г, 83%). МС (CI) MH+: рассчитано для С18Н18СlNО5S: 428, обнаружено 428.
Пример 26: Получение моногидрохлорида тетрагидро-N-гидрокси-4-[[4-[4-(1Н-1,2,4-триазол-1-ил)фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида

Стадия А: К раствору полученного на стадии Б примера 25 защищенного карбоксамата (2,9 г, 7,5 ммоль) в ДМФ (10 мл) добавляли 4-(1,2,4-триазол-1-ил)фенол (2,47 г, 15 ммоль) в ДМФ (5 мл), а затем Сs2СО3 (7,33 г, 22,5 ммоль), и раствор выдерживали при 95° С в течение 5 ч. Раствор концентрировали в вакууме и остаток распределяли между этилацетатом и Н2О. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан/метанол) получали фенол в виде твердого вещества белого цвета (3,16 г, 80%).
Стадия Б: К раствору полученного на стадии А фенола (2,8 г, 5,3 ммоль) в диоксане (10 мл) добавляли 4н. раствор HCl в диоксане (10 мл). После перемешивания в течение 5 мин добавляли метанол (10 мл) и перемешивание продолжали в течение 1 ч. Затем раствор сливали на диэтиловый эфир и образовавшийся осадок собирали вакуумной фильтрацией, получая твердое вещество белого цвета (2,44 г, 96%). МС (CI) МН+: рассчитано для C20H20N4О6S:445, обнаружено 445.
Пример 27: Получение моногидрохлорида 1-циклопропил-4-[[4-[(4-фторфенил)тио]фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
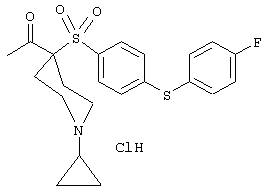
Стадия А: Раствор полученного на стадии Г примера 9 сульфида (7,06 г, 13,5 ммоль) в этилацетате (150 мл) барботировали в течение 7 мин газообразным HCl и раствор перемешивали в течение 15 мин при 0° С. Раствор концентрировали в вакууме, получая амин в виде твердого вещества белого цвета (6,43 г, количественный выход).
Стадия Б: К раствору полученного на стадии А амина (6,4 г, 13,9 ммоль) в метаноле (65 мл) добавляли уксусную кислоту (7,96 мл, 139 ммоль) и порцию молекулярных сит размером 3ЕС, входящую в лодочку для взвешивания. К этой смеси добавляли (1-этоксициклопропил)окситриметилсилан (16,8 мл, 84 ммоль), а затем цианборогидрид натрия (3,9 г, 62 ммоль). Раствор нагревали до температуры дефлегмации в течение 6 ч. Раствор фильтровали и фильтрат концентрировали в вакууме. Остаток растворяли в этилацетате, промывали Н2О, 2н. NaOH, насыщенным раствором NaCl и сушили над сульфатом магния. После фильтрации через подушку из двуокиси кремния (гексан/этилацетат) получали циклопропиламин в виде твердого вещества белого цвета (6,49 г, количественный выход).
Стадия В: К раствору полученного на стадии Б циклопропиламина (6,4 г, 13,8 ммоль) в этаноле (30 мл) и ТГФ (30 мл) добавляли NaOH (5,5 г, 138 ммоль) в Н2О (23 мл) и раствор нагревали в течение 12 ч до 65° С. Раствор концентрировали в вакууме и водный слой подкисляли 2н. HCl до значения рН 2. Образовавшийся осадок белого цвета собирали фильтрацией, получая кислоту в виде твердого вещества белого цвета (5,2 г, 97%). МС (CI) MH+: рассчитано для C21H22NO4S2F: 436, обнаружено 436.
Стадия Г: К раствору полученной на стадии В кислоты (2,27 г, 5,2 ммоль) в ДМФ (60 мл) добавляли ГОБТ (845 мг, 6,2 ммоль), а затем N-метилморфолин (1,71 мл, 15,6 ммоль), ЭДК (1,40 г, 7,28 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (913 мг, 7,8 ммоль) и раствор перемешивали в течение 72 ч при температуре окружающей среды. Раствор концентрировали в вакууме, остаток растворяли в дихлорметане и промывали Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества белого цвета (1,95 г, 70%).
Стадия Д: К раствору полученного на стадии Г защищенного гидроксамата (3,2 г, 6,0 ммоль) в холодном метаноле (100 мл) добавляли ацетилхлорид (1,3 мл, 18,0 ммоль) в метаноле (30 мл) и раствор перемешивали в течение 4 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток растирали с диэтиловым эфиром, получая указанное в заголовке соединение в виде твердого вещества белого цвета (2,86 г, 98%). МС (CI) MH+: рассчитано для C21H23N2O4S2F: 451, обнаружено 451. Элементный анализ: рассчитано для C21H23N2O4S2F· 0,25Н2О· HCl: С 51,32; Н 5,02; N 5,70; S 13,05; Cl 7,21; обнаружено: С 50,99; Н 4,91; N 5,65; S 13,16; Cl 7,83.
Пример 28: Получение моногидрохлорида N-гидрокси-4-[[4-(фенилтио)фенил]сульфонил]-1-(2-пропенил)-4-пиперидинкарбоксамида
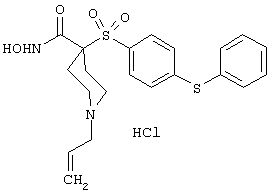
Стадия А: К раствору полученного на стадии Д примера 9 гидрохлорида амина (4,78 г, 10,8 ммоль) в ДМФ (25 мл) добавляли К2СО3 (2,98 г, 21,6 ммоль) и аллилбромид (0,935 мл, 10,8 ммоль) и раствор перемешивали в течение 5 ч при температуре окружающей среды. Остаток распределяли между этилацетатом и Н2О, органический слой промывали Н2О и насыщенным раствором NaCl и сушили над сульфатом магния. После фильтрации через подушку из двуокиси кремния (гексан/этилацетат) получали аллиламин в виде масла (4,80 г, количественный выход).
Стадия Б: К раствору полученного на стадии А аллиламина (4,8 г, 10,8 ммоль) в этаноле (25 мл) и ТГФ (25 мл) добавляли NaOH (4,3 г, 108 ммоль) в Н2О (20 мл) и раствор нагревали в течение 18 ч до 65° С. Раствор концентрировали в вакууме и разбавляли Н2О. Водный раствор подкисляли до значения рН 3. Образовавшийся осадок собирали вакуумной фильтрацией, получая кислоту в виде твердого вещества бежевого цвета (4,1 г, 84%). МС (Cl) МН+: рассчитано для C21H23NO4S2: 418, обнаружено 418.
Стадия В: К раствору полученной на стадии Б кислоты (4,1 г, 9,0 ммоль) в ДМФ (90 мл) добавляли ГОБТ (1,46 г, 11,0 ммоль), а затем N-метилморфолин (2,97 мл, 2,7 ммоль), O-тетрагидро-2Н-пиран-2-илгидроксиламин (1,58 г, 13,5 ммоль) и ЭДК (2,42 г, 13,0 ммоль) и раствор перемешивали в течение 72 ч. Раствор концентрировали в вакууме. Остаток растворяли в дихлорметане и промывали Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/метанол) получали защищенный гидроксиламин в виде твердого вещества белого цвета (4,11 г, 88%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксиламина (4,11 г, 8,0 ммоль) в этилацетате (100 мл), охлажденному до 0° С, добавляли ацетилхлорид (1,71 мл, 24,0 ммоль) и раствор перемешивали в течение 4 ч при температуре окружающей среды. Раствор концентрировали в вакууме и после растирания с диэтиловым эфиром, получая указанное в заголовке соединение в виде твердого вещества белого цвета (3,53 г, 95%). Элементный анализ: рассчитано для C21H24N4O4S2·HCl· 0,5Н2О: С 52,76; Н 5,48; N 5,86; S 13,42; Cl 7,42; обнаружено: С 52,57; Н 5,69; N 6,29; S 12,59; Cl 7,80.
Пример 29: Получение моногидрохлорида 1-циклопропил-N-гидрокси-4-[(4-феноксифенил)сульфонил]-4-пиперидинкарбоксамида
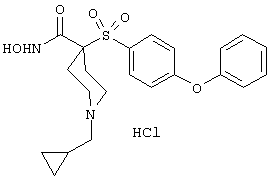
Стадия А: Раствор полученного на стадии Д примера 6 гидрохлорида амина (2,13 г, 5,0 ммоль) в ДМФ (10 мл) добавляли К2СО3 (1,4 г, 10,0 ммоль) и бромметилциклопропан (0,48 мл, 5,0 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор распределяли между этилацетатом и Н2О, органический слой промывали Н2О и насыщенным раствором NaCl и затем сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали циклопропилметиламин в виде твердого вещества (2,09 г, 91%).
Стадия Б: К раствору полученного на стадии А циклопропилметиламина (2,0 г, 4,4 ммоль) в этаноле (12 мл) и ТГФ (12 мл) добавляли NaOH (1,75 г, 44 ммоль) в Н2О (10 мл) и раствор нагревали в течение 18 ч до 65° С. Раствор концентрировали в вакууме и водный остаток подкисляли до значения рН 5. Образовавшийся осадок собирали вакуумной фильтрацией, получая кислоту в виде твердого вещества белого цвета (1,58 г, 79%). МСВР: рассчитано для C22H25NO5S: 414,1375, обнаружено 414,1334.
Стадия В: К раствору полученной на стадии Б кислоты (1,58 г, 3,5 ммоль) в дихлорметане (50 мл) добавляли триэтиламин (1,46 мл, 10,5 ммоль), а затем 50%-ный водный раствор гидроксиламина (2,3 мл, 35 ммоль) и РуВrоР (3,26 г, 6,99 ммоль) и раствор перемешивали в течение 72 ч при температуре окружающей среды. Раствор промывали Н2О, насыщенным раствором NaCl и сушили над сульфатом магния. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали гидроксамат в виде твердого вещества белого цвета (3,2 г, количественный выход).
Стадия Г: К раствору полученного на стадии В гидроксамата (1,5 г, 3,5 ммоль) в холодном метаноле (20 мл) добавляли ацетилхлорид (0,25 мл, 3,5 ммоль) в метаноле (5 мл) и раствор перемешивали в течение 15 мин при 0° С. После перемешивания раствора еще в течение 30 мин при температуре окружающей среды его концентрировали в вакууме. После растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (229 мг, 7%).
Пример 30: Получение моногидрохлорида N-гидрокси-1-(2-метоксиэтил)-4-[(4-феноксифенил)сульфонил]-4-пиперидинкарбоксамида

Стадия А: Раствор полученного на стадии Д примера 6 гидрохлорида амина (2,5 г, 5,87 ммоль) и К2СО3 (1,6 г, 11,57 ммоль) в N,N-диметилформамиде (25 мл) добавляли метил-2-бромэтиловый эфир (0,66 мл, 7,0 ммоль) и затем перемешивали в течение 18 ч при температуре окружающей среды. После этого N,N-диметилформамид выпаривали в глубоком вакууме и остаток разбавляли этилацетатом. Органический слой промывали водой и сушили над MgSO4. После концентрирования в вакууме получали метоксилэтиламин в виде геля светло-желтого цвета (2,63 г, количественный выход).
Стадия Б: Раствор полученного на стадии А метоксилэтиламина (2,63 г, 5,87 ммоль) в тетрагидрофуране (18 мл) и этаноле (18 мл) добавляли NaOH (2,1 г, 5,25 ммоль) в воде (6 мл). Раствор нагревали до температуры дефлегмации в течение 12 ч. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали простым эфиром (2× 100 мл) и подкисляли до рН 2. После вакуумной фильтрации образовавшегося осадка получали кислоту в виде твердого вещества белого цвета (2,4 г, количественный выход).
Стадия В: К раствору полученной на стадии Б кислоты (2,0 г, 4,33 ммоль), также содержащему N-метилморфолин (1,8 мл, 16,4 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,767 г, 6,44 ммоль), в N,N-диметилформамиде (20 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (3,1 г, 16,2 ммоль) и раствор перемешивали в течение 20 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали Н2О и сушили над MgSO4. После концентрирования в вакууме получали амид в виде пены беловатого цвета (1,60 г, 71,1%).
Стадия Г: К раствору полученного на стадии В амида (1,58 г, 3,05 ммоль) в метаноле (20 мл), охлажденном до 0° С, добавляли ацетилхлорид (0,65 мл, 9,15 ммоль) и образовавшийся раствор перемешивали при этой же температуре в течение 3 ч. Раствор концентрировали и после хроматографии с обращенной фазой (на колонках С-18 с двуокисью кремния, ацетонитрил/Н2О с добавлением 0,01% HCl) получали гидрохлорид гидроксамата в виде твердого вещества белого цвета (0,65 г, 45,5%). Элементный анализ: рассчитано для С21Н26N2О6S· HCl· 0,75Н2О: С 52,06; Н 5,93; N 5,78; S 6,62; обнаружено: С 51,94; Н 5,67; N 5,91; S 6,66. МСВР: рассчитано для С21H26N2О6S: 435,1590, обнаружено 435,1571.
Пример 31: Получение моногидрохлорида N-гидрокси-4-[(4-феноксифенил)сульфонил]-1-(1-пирролидинилацетил)-4-пиперидинкарбоксамида
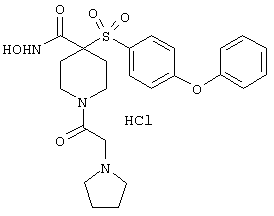
Стадия А: К раствору полученного на стадии Г примера 6 сульфона (2,75 г, 5,6 ммоль) в тетрагидрофуране (10 мл) и этаноле (10 мл) добавляли NaOH (2,25 г, 56 ммоль) в Н2О (20 мл) и раствор нагревали в течение 20 ч до 70° С. Раствор концентрировали в вакууме и сухой остаток растворяли в Н2О. Водный слой экстрагировали простым эфиром и подкисляли до рН 2, после чего экстрагировали этилацетатом. Объединенные органические слои еще раз промывали Н2О и сушили над MgSO4. После концентрирования в вакууме получали ВОС-защищенную кислоту в виде пены белого цвета (2,3 г, 88,8%).
Стадия Б: К раствору полученой на стадии А ВОС-защищенной кислоты (2,3 г, 4,98 ммоль) в дихлорметане (6 мл) добавляли трифторуксусную кислоту (6 мл, 77,8 ммоль) и образовавшийся раствор перемешивали в течение 1 ч при температуре окружающей среды. После концентрирования в вакууме получали амин в виде пены белого цвета (2,44 г, количественный выход).
Стадия В: К раствору, содержащему полученный на стадии Б амин (2,4 г, 4,9 ммоль) и триэтиламин (3,5 мл, 24,4 ммоль) в ацетоне (15 мл) и Н2О (15 мл), добавляли хлорацетилхлорид (1,2 мл, 14,7 ммоль) и раствор перемешивали в течение 20 ч при температуре окружающей среды. Ацетон выпаривали и водный слой подкисляли до рН 2. Водный слой экстрагировали этилацетатом и органический слой промывали водой и сушили над MgSO4. После концентрирования в вакууме получали хлорацетиламид в виде геля светло-желтого цвета (2,78 г, количественный выход).
Стадия Г: К раствору, содержащему получении на стадии В хлорацетиламид (2,78 г, 4,93 ммоль) и К2СО3 (5 г, 36 ммоль) в N,N-диметилформамиде (20 мл) добавляли пирролидин (3 мл, 36 ммоль). Затем раствор перемешивали в течение 18 ч при температуре окружающей среды. После этого N,N-диметилфомамид выпаривали в глубоком вакууме и после хроматографии с обращенной фазой (на колонках С-18 с двуокисью кремния, ацетонитрил/Н2О с добавлением 0,01% HCl) получали пирролидинацетиламид (0,25 г, 10,7%).
Стадия Д: К раствору полученного на стадии Г амида пирролидинацетила (0,25 г, 0,53 ммоль), также содержащему N-метилморфолин (0,14 мл, 1,27 ммоль), 1-гидроксибензотриазол (0,17 г, 1,2 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,15 г, 1,26 ммоль) в N,N-диметилформамиде (4 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,23 г, 1,2 ммоль). Затем раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным раствором NaHCO3, Н2О и сушили над MgSO4. После концентрирования в вакууме получали ТГП-защищенный амид в виде пены белого цвета (0,25 г, 83,3%).
Стадия Е: К раствору полученного на стадии Д амида (0,25 г, 0,437 ммоль) в метаноле (4 мл), охлажденному до 0° С, добавляли ацетилхлорид (0,075 мл, 1,05 ммоль) и образовавшийся раствор перемешивали в течение 2,5 ч при температуре окружающей среды. Раствор концентрировали в вакууме и после хроматографии с обращенной фазой (на колонках С-18 с двуокисью кремния, ацетонитрил/Н2О с добавлением 0,01% HCl) получали гидрохлорид гидроксамата в виде твердого вещества белого цвета (80 мг, 29%). Элементный анализ: рассчитано для C24H29N3O6S· HCl· 0,9Н2О: С 53,36; Н 5,98; N 7,78; обнаружено: С 53,61; Н 5,71; N 7,94. МСВР: рассчитано для С24Н29N3О6S: 488,1855, обнаружено: 488,1835.
Пример 32: Получение моногидрохлорида 1-циклопропил-N-гидрокси-4-[[4-(фенилтио)фенил]сульфонил]-4-пиперидинкарбоксамида
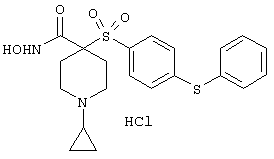
Стадия А: Раствор 4-фтортиофенола (50,29 г, 0,39 ммоль) в диметилсульфоксиде (500 мл) нагревали в течение 5 ч до 65° С. Раствор охлаждали до температуры окружающей среды и сливали на ледяную воду при интенсивном перемешивании. Осадок фильтровали и дважды промывали водой. После сушки в глубоком вакууме при температуре окружающей среды получали дисульфид в виде масла желтого цвета (34,39 г, 68,9%).
Стадия Б: Раствор ди-трет-бутилдикарбоната (21,8 г, 0,1 моль) в тетрагидрофуране (5 мл) добавляли по каплям в течение 20 мин к раствору этилизонипекотата (15,7 г, 0,1 моль) в тетрагидрофуране (100 мл). Образовавшийся раствор перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды и концентрировали в вакууме, получая прозрачое масло. Масло фильтровали через силикагель (этилацетат/гексан) и концентрировали в вакууме, получая ВОС-защищенное пиперидиновое производное в виде прозрачного бесцветного масла (26,2 г, количественный выход).
Стадия В: К раствору полученного на стадии Б ВОС-защищенного пиперидинового производного (15,96 г, 62 ммоль) в тетрагидрофуране (300 мл), охлажденному до -40° С, добавляли диизопропиламид лития (41,33 мл, 74 ммоль). Затем раствор перемешивали в течение 1 ч при -40° с и в течение 0,5 ч при 0° С. Затем раствор снова охлаждали до -40° С и добавляли полученный на стадии А дисульфид (15,77 г, 62 ммоль) в тетрагидрофуране (20 мл). Образовавшийся раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали Н2О, насыщенным раствором NaCl и сушили над MgSO4. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфид в виде масла (18,0 г, 75%).
Стадия Г: К раствору полученного на стадии В сульфида (16,5 г, 43 ммоль) в дихлорметане (500 мл), охлажденному до 0° С, добавляли мета-хлорпербензойную кислоту (18,5 г, 107 ммоль). Через 2 ч раствор разбавляли дихлорметаном и промывали 1 н. КОН, Н2О и сушили над MgSО4. После концентрирования в вакууме получали сульфон в виде твердого вещества (21 г, количественный выход)
Стадия Д: К раствору, содержащему полученный на стадии Г сульфон (40 г, 96 ммоль) и порошкообразный К2СО3 (26 г, 188 ммоль) в N,N-диметилформамиде (200 мл), охлажденному до 0° С, добавляли тиолфенол (19,8 мл, 192 ммоль) и затем образовавшуюся смесь перемешивали в течение 36 ч при температуре окружающей среды. Этот раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали Н2О и сушили над сульфатом магния. После хроматографии (на силикагеле, этилацетат/гексан) получали ВОС-защищенный сульфон фенилтиофенила в виде твердого вещества белого цвета (44,34 г, 91%).
Стадия Е: К раствору полученного на стадии Д ВОС-защищенного сульфона фенилтиофенила (8,6 г, 17 ммоль) в дихлорметане (30 мл), охлажденному до 0° С, добавляли трифторуксусную кислоту (ТФК, 30 мл) и образовавшийся раствор перемешивали в течение 2 ч при температуре окружающей среды. После концентрирования в вакууме получали трифторацетат амина в виде геля светло-желтого цвета (8,7 г, количественный выход).
Стадия Ж: К раствору полученного на стадии Е трифторацетата амина (6 г, 11,9 ммоль) добавляли уксусную кислоту (6,8 мл, 119 ммоль). После перемешивания в течение 5 мин при температуре окружающей среды добавляли (1-этоксилциклопропил)окситриометилсилан (14,3 мл, 71,4 ммоль), а затем спустя 5 мин добавляли гидрат цианборана натрия (3,35 г, 53,55 ммоль). После этого раствор нагревали до температуры дефлегмации в течение 18 ч. Метанол выпаривали и остаток растворяли в этилацетате. Органический слой промывали 1 н. раствором NaOH, Н2О и сушили над MgSO4. После концентрирования в вакууме получали циклопропиламин в виде порошка беловатого цвета (4,9 г, 92,6%).
Стадия 3: К раствору полученного на стадии Ж циклопропиламина (4,88 г, 10,95 ммоль) в тетрагидрофуране (12,5 мл) и этаноле (12,5 мл) добавляли NaOH (4,3 г, 100 ммоль) в воде (25 мл). Затем раствор нагревали в течение 12 ч до 50-55° С и перемешивали в течение 18 ч при температуре окружающей среды. Раствор подкисляли до рН 2 и после концентрирования в вакууме получали кислоту в виде твердого вещества белого цвета в смеси с NaCl. К раствору этой смеси в ацетонитриле (50 мл) последовательно добавляли O-тетрагидропирони-ламин (1,95 г, 16,3 ммоль), N-метилморфолин (2,4 мл, 21,9 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (3,14 г, 16,3 ммоль). Затем раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме и остаток растворяли в этилацетате. Органический слой промывали Н2О и сушили над MgSO4. После концентрирования в вакууме получали тетрагидропиранил (ТГП)-защищенный амид в виде твердого вещества белого цвета (3,0 г, 53,1%).
Стадия И: К раствору полученного на стадии 3 ТГП-защищенного амида (3 г, 5,8 ммоль) в метаноле (45 мл), охлажденному до 0° С, добавляли ацетилхлорид (1,5 мл, 21,1 ммоль) и раствор перемешивали в течение 2,5 ч при температуре окружающей среды. После вакуумной фильтрации осадка получали гидрохлорид гидроксамата в виде твердого вещества белого цвета (1,844 г, 68,3%). Элементный анализ: рассчитано для C21H24N2O4S2·HCl: С 53,78; Н 5,37; N 5,97; S 13,67; обнаружено: С 53,40; Н 5,26; N 5,95; S 13,68.
Пример 33: Получение моногидрохлорида N-гидрокси-1-метил-4-[[4-(фенилтио)фенил]сульфонил]-4-пиперидинкарбоксамида
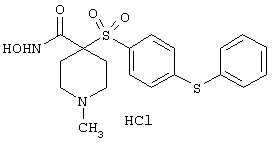
Стадия А: К раствору, содержащему полученный на стадии Е примера 32 трифторацетат амина (2,67 г, 5,14 ммоль) и 37%-ный водный раствор формальдегида (2,0 мл, 25,7 ммоль) в метаноле (20 мл), добавляли при температуре окружающей среды боранпиридин (2,6 мл, 25,7 ммоль). Затем раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор подкисляли для разложения избытка реактива. Метанол выпаривали и остаток распределяли между водным раствором NaHCO3 и этилацетатом. Слой водного раствора NaHCO3 экстрагировали этилацетатом. Объединенные органические слои промывали Н2О и сушили над MgSO4. После концентрирования в вакууме получали метиламин в виде пены беловатого цвета (1,6 г, 76%).
Стадия Б: К раствору полученного на стадии А метиламина (1,63 г, 3,88 ммоль) в этаноле (20 мл) добавляли раствор КОН (1,31 г, 23,2 ммоль) в воде (4 мл) и образовавшийся раствор нагревали в течение 8 ч до 50° С, в течение 4 ч до 70° С и перемешивали в течение 18 ч при температуре окружающей среды. Раствор подкисляли и концентрировали в вакууме, получая кислоту в виде твердого вещества белого цвета в смеси с NaCl. К раствору этой смеси в N,N-диметилформамиде (50 мл) последовательно добавляли O-тетрагидропирониламин (0,92 г, 7,76 ммоль), N-метилморфолин (1,05 мл, 7,76 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (1,5 г, 7,76 ммоль). Раствор перемешивали в течение 72 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным раствором NaHCO3, Н2О и сушили над MgSO4. После концентрирования в вакууме и хроматографии
(на силикагеле, дихлорметан/метанол) получали ТГП-защищенный амид в виде твердого вещества белого цвета (0,46 г, 24,2%).
Стадия В: К раствору полученного на стадии Б ТГП-защищенного амида (0,22 г, 0,45 ммоль) в метаноле (5 мл), охлажденному до 0° С, добавляли ацетилхлорид (0,096 мл, 13,5 ммоль) и образовавшийся раствор перемешивали в течение 3 ч при температуре окружающей среды. Раствор концентрировали в вакууме и после хроматографии с обращенной фазой (на колонках С-18 с двуокисью кремния, ацетонитрил/Н2О с добавлением 0,01% HCl) получали гидрохлорид гидроксамата в виде твердого вещества белого цвета (0,12 г, 60,6%). МСВР: рассчитано для C19H22N2O4S2: 407,1099, обнаружено 407,1105.
Пример 34: Получение моногидрохлорида N-гидрокси-1-(1-метилэтил)-4-[[4-(фенилтио)фенил]сульфонил]-4-пиперидинкарбоксамида
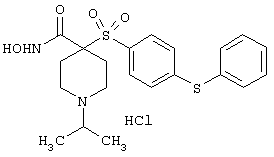
Стадия А: Раствор полученного на стадии Г примера 32 ВОС-защищенного сульфона (11,19 г, 22,12 ммоль) в этилацетате (150 мл), охлажденный до 0° С, барботировали в течение 20 мин газообразным HCl. Раствор перемешивали при этой же температуре еще в течение 40 мин. После концентрирования в вакууме и растирания с простым эфиром получали гидрохлорид амина (9,88 г, количественный выход).
Стадия Б: К раствору, содержащему полученный на стадии А гидрохлорид амина (4,7 г, 10,6 ммоль), триэтиламин (2,0 мл, 14,4 ммоль) и ацетон (2,0 мл, 27,2 ммоль) в дихлорметане (100 мл), добавляли при температуре окружающей среды триацетоксилборогидрид натрия (5,7 г, 26,9 ммоль), а затем уксусную кислоту (1,5 мл, 26,9 ммоль). Раствор перемешивали в течение 18 ч, а затем распределяли между 1 н. раствором NaOH и простым эфиром. Водный слой экстрагировали простым эфиром и объединенные органические слои промывали 1 н. раствором NaOH, Н2О и сушили над MgSO4. После концентрирования в вакууме получали изопропиламин в виде пены белого цвета (4,58 г, 96%).
Стадия В: К раствору полученного на стадии Б изопропиламина (4,58 г, 10,2 ммоль) в тетрагидрофуране (10 мл) и этаноле (10 мл) добавляли раствор NaOH (2,1 г, 5,25 ммоль) в воде (20 мл). Раствор нагревали в течение 13,5 ч до 60° С, затем перемешивали в течение 18 ч при температуре окружающей среды. Раствор подкисляли и концентрировали в вакууме, получая кислоту в виде твердого вещества белого цвета в смеси с NaCl. К раствору этой смеси в N,N-диметилформамиде (75 мл) последовательно добавляли 1-гидроксибензотриазол (1,94 г, 14,4 ммоль), O-тетрагидропирониламин (1,8 г, 15,1 ммоль), N-метилморфолин (3,37 мл, 30,7 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (2,74 г, 14,3 ммоль). Затем раствор перемешивали в течение 48 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным NaHCO3, Н2О и сушили над MgSO4. После концентрирования в вакууме хроматографии (на силикагеле, дихлорметан/метанол) получали ТГП-защищенный амид в виде твердого вещества белого цвета (3,78 г, 71,3%).
Стадия Г: К раствору полученного на стадии В ТГП-защищенного амида (1,15 г, 2,2 ммоль) в метаноле (20 мл) добавляли ацетилхлорид (0,096 мл, 13,5 ммоль) и образовавшийся раствор перемешивали в течение 2,5 ч при температуре окружающей среды. Раствор концентрировали в вакууме и после хроматографии с обращенной фазой (на колонках С-18 с двуокисью кремния, ацетонитрил/Н2О с добавлением 0,01% HCl) получали гидрохлорид гидроксамата в виде твердого вещества белого цвета (0,69 г, 66,3%). Элементный анализ: рассчитано для С21Н26N2O4S2·НСl· Н2О: С 51,58; Н 5,98; N 5,73; S 13,11; обнаружено: С 51,76; Н 5,47; N 5,72; S 12,68.
Пример 35: Получение моногидрохлорида N-гидрокси-1-(2-метоксиэтил)-4-[[4-(фенилтио)фенил]сульфонил]-4-пиперидинкарбоксамида
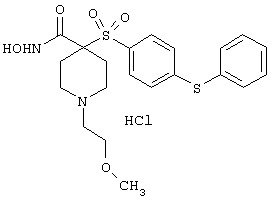
Стадия А: К раствору, содержащему полученный на стадии А примера 34 гидрохлорид амина (463 г, 9,43 ммоль) и K2CO3 (2,62 г, 19,0 ммоль) в N,N-диметилформамиде (40 мл), добавляли метил-2-бромэтиловый эфир (1,9 мл, 20,2 ммоль). Раствор перемешивали в течение 48 ч при температуре окружающей среды. Затем N,N-диметилформамид выпаривали в глубоком вакууме и остаток разбавляли этилацетатом. Органический слой промывали водой и сушили над MgSO4. После концентрирования в вакууме получали метоксилэтиламин в виде пены белого цвета (4,26 г, 95,3%).
Стадия Б: К раствору полученного на стадии А метоксилэтиламина (4,26 г, 9,2 ммоль) в тетрагидрофуране (5 мл) и этаноле (5 мл) добавляли раствор NaOH (3,7 г, 92,5 ммоль) в воде (9 мл). Образовавшийся раствор нагревали в течение 12 ч до 60° С и перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали простым эфиром (2× 100 мл) и подкисляли до рН 2. После вакуумной фильтрации образовавшегося осадка получали кислоту в виде твердого вещества белого цвета (3,5 г, 87,5%).
Стадия В: К раствору полученной на стадии Б кислоты (3,4 г, 7,8 ммоль) в N,N-диметилформамиде (20 мл), также содержащему N-метилморфолин (2,6 мл, 23,4 ммоль), 1-гидроксибензотриазол (3,16 г, 23,4 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (1,85 г, 15,5 ммоль), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (4,47 г, 23,4 ммоль). Раствор перемешивали в течение 36 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным раствором NaHCO3, Н2О и сушили над MgSO4. После концентрирования в вакууме получали амид в виде твердого вещества беловатого цвета (2,98 г, 71,5%).
Стадия Г: К раствору полученного на стадии В амида (2,98 г, 5,6 ммоль) в метаноле (40 мл), охлажденному до 0° С, добавляли ацетилхлорид (1,19 мл, 16,8 ммоль) и образовавшийся раствор перемешивали в течение 3 ч при температуре окружающей среды. Раствор концентрировали в вакууме и после хроматографии с обращенной фазой (на колонках С-18 с двуокисью кремния, ацетонитрил/Н2О с добавлением 0,01% HCl) получали гидрохлорид гидроксамата в виде твердого вещества белого цвета (2,29 г, 84,6%). Элементный анализ: рассчитано для C21H26N2O6S· HCl· 0,9Н2О: С 50,12; Н 5,77; N 5,57; S 12,74; обнаружено: С 50,41; Н 5,85; N 5,73; S 12,83.
Пример 36: Получение моногидрохлорида 1-ацетил-N-гидрокси-4-[[4-(фенилтио)фенил]сульфонил]-4-пиперидинкарбоксамида
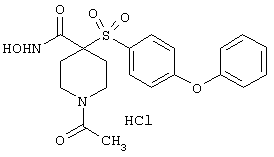
Стадия А: К раствору полученного на стадии Д примера 32 ВОС-защищенного сульфона фенилтиофенила (7 г, 1,29 ммоль) в тетрагидрофуране (25 мл) и этаноле (25 мл) добавляли NaOH (5,1 г, 12,9 ммоль) в Н2О (50 мл). Раствор нагревали в течение 20 ч до температуры дефлегмации. После охлаждения раствор концентрировали в вакууме и сухой остаток растворяли в Н2О. Водный слой экстрагировали простым эфиром и подкисляли до рН 2, а затем экстрагировали этилацетатом. Объединенные органические слои снова промывали Н2О и сушили над MgSO4. После концентрирования в вакууме получали ВОС-защищенную кислоту в виде пены белого цвета (3,9 г, 60%).
Стадия Б: К раствору полученной на стадии А ВОС-защищенной кислоты (2,3 г, 4,98 ммоль) в дихлорметане (6 мл) добавляли трифторуксусную кислоту (6 мл, 77,8 ммоль) и раствор перемешивали в течение 1 ч при температуре окружающей среды. После концентрирования в вакууме получали амин в виде пены белого цвета (2,44 г, количественный выход).
Стадия В: К раствору, содержащему полученный на стадии Б амин (5,0 г, 12,08 ммоль) и триэтиламин (8,7 мл, 60,4 ммоль) в ацетоне (20 мл) и Н2О (20 мл), охлажденному до 0° С, добавляли ацетилхлорид (4,6 мл, 36 ммоль) и раствор перемешивали в течение 40 ч при температуре окружающей среды. Ацетон выпаривали и водный слой подкисляли до рН 2. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали водой и сушили над Mg2SO4. После концентрирования в вакууме получали ацетиламид в виде пены светло-желтого цвета (5 г, количественный выход).
Стадия Г: К раствору полученного на стадии В ацетиламида (5 г, 11,9 ммоль) в N,N-диметилформамиде (50 мл), также содержащему N-метилморфолин (5,3 мл, 11,9 ммоль), 1-гидроксибензотриазол (4,8 г, 35,7 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (2,8 г, 23,5 ммоль), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (6,8 г, 35,7 ммоль) и раствор перемешивали в течение 20 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным раствором NaHCO3, KHSO4, Н2О и сушили над MgSO4. После концентрирования в вакууме получали ТГП-амид в виде пены белого цвета (6,07 г, 98,2%).
Стадия Д: К раствору полученного на стадии Г ТГП-амида (6,07 г, 11,7 ммоль) в метаноле (100 мл), охлажденному до 0° С, добавляли ацетилхлорид (2,5 мл, 35,1 ммоль) и образовавшийся раствор перемешивали в течение 3 ч при температуре окружающей среды. Раствор концентрировали и после хроматографии (на силикагеле, метанол/дихлорметан) получали гидрохлорид гидроксамата в виде твердого вещества белого цвета (3,3 г, 65%). Элементный анализ: рассчитано для C24H29N3O6S· HCl· 0,9Н2О: С 53,36; Н 5,98; N 7,78; обнаружено: С 53,61; Н 5,71; N 7,94. МСВР: рассчитано для С24Н29N3О6S: 488,1855, обнаружено 488,1835.
Пример 37: Получение моногидрохлорида 1-ацетил-4-[[4-(1,3-бензодиоксол-5-илокси)фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
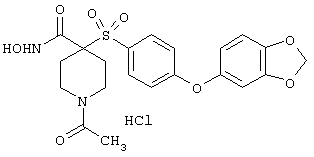
Стадия А: К раствору, содержащему полученный на стадии Г примера 32 сульфона (25 г, 67,3 ммоль) и порошкообразный K2CO3 (23,3 г, 16,9 ммоль) в N,N-диметилформамиде, добавляли при температуре окружающей среды сезамол (23,24 г, 16,8 ммоль) и раствор нагревали в течение 24 ч до 90° С. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали 1 н. NaOH, Н2О и сушили над MgSO4. После хроматографии (на силикагеле, этилацетат/гексан) получали ВОС-защищенный сульфон сезамола в виде пены белого цвета (33,6 г, 93,6%).
Стадия Б: К раствору полученного на стадии А ВОС-защищенного сульфона сезамола (29,31 г, 54,93 ммоль) в этаноле (60 мл) и тетрагидрофуране (60 мл) добавляли через дополнительную воронку в течение 20 мин при температуре окружающей среды NaOH (21,97 г, 544 ммоль). Раствор нагревали в течение 9 ч до 60° С, а затем выдерживали в течение 12 ч при температуре окружающей среды. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали простым эфиром и подкисляли до рН 2. Затем его экстрагировали этилацетатом и объединенные органические слои промывали Н2О и сушили над MgSO4. После концентрирования в вакууме получали кислоту в виде твердого вещества белого цвета (25,3 г, 91%).
Стадия В: Раствор полученной на стадии Б кислоты (20,3 г, 40,15 ммоль) в этилацетате, охлажденный до 0° С, барботировали газообразным HCl. Через 1,5 ч путем вакуумной фильтрации осадка белого цвета получали гидрохлорид амина в виде твердого вещества белого цвета (16 г, 93,6%).
Стадия Г: К раствору, содержащему полученный на стадии В гидрохлорид амина (8,1 г, 19,01 ммоль) и триэтиламин (13,2 мл, 95,05 ммоль) в ацетоне (150 мл) и Н2О (150 мл), охлажденному до 0° С, добавляли ацетилхлорид (5,4 мл, 76 ммоль). Раствор перемешивали в течение 18 ч при температуре окружающей среды. Ацетон выпаривали и водный слой подкисляли до рН 2. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали Н2О и сушили над MgSO4. После концентрирования в вакууме получали ацетиламид в виде пены светло-желтого цвета (9,24 г, количественный выход).
Стадия Д: К раствору, содержащему полученный на стадии Г ацетиламид (9,1 г, 20,33 ммоль), N-метилморфолин (6,7 мл, 61 ммоль), 1-гидроксибензотриазол (8,2 г, 60 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (4,85 г, 40 ммоль) в N,N-диметилформамиде (40 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (11,65 г, 60 ммоль). Образовавшийся раствор перемешивали в течение 20 ч при температуре окружающей среды. Затем раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным раствором NaHCO3, KHSO4, Н2О и сушили над MgSO4. После концентрирования в вакууме и хроматографии (на силикагеле, этилацетат/гексан) получали ТГП-амид в виде пены белого цвета (10 г, 89,7%).
Стадия Е: К раствору 4н. HCl в диоксане (20 мл) добавляли раствор полученного на стадии Д амида (5,0 г, 9,1 ммоль) в метаноле (5 мл) и диоксане (15 мл). Этот раствор перемешивали в течение 30 мин при температуре окружающей среды. После вакуумной фильтрации осадка белого цвета получали гидрохлорид гидроксамата в виде твердого вещества белого цвета (3,3 г, 65%). Элементный анализ: рассчитано для C21H22N2O8S· HCl: С 54,34; Н 5,15; N 5,49; S 6,43; обнаружено: С 54,54; Н 4,79; N 6,06; S 6,93. МСВР: рассчитано для C21H22N2O8S: 463,1175, обнаружено 463,118.
Пример 38: Получение моногидрохлорида 4-[[4-(3,4-диметоксифенокси)фенил]сульфонил]-N-гидрокси-1-(2-пропинил)-4-пиперидинкарбоксамида
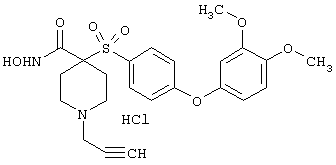
Стадия А: Раствор полученного на стадии Г примера 32 сульфона (10 г, 24 ммоль) в этилацетате, охлажденный до 0° С, барботировали газообразным HCl. Через 4 ч путем вакуумной фильтрации осадка белого цвета получали гидрохлорид амина в виде твердого вещества белого цвета (7,27 г, 86%).
Стадия Б: К раствору, содержащему полученный на стадии А гидрохлорид амина (5,98 г, 17 ммоль) и порошкообразный К2СО3 (4,7 г, 34 ммоль) в N,N-диметилформамиде (120 мл), добавляли при температуре окружающей среды пропаргилбромид (2,02 г, 17 ммоль), после чего перемешивали в течение 4 ч. Раствор разбавляли этилацетатом и промывали Н2О, насыщенным раствором NaCl и сушили над MgSO4. После концентрирования в вакууме и хроматографии (на силикагеле, этилацетат/гексан) получали пропаргиламин в виде твердого вещества белого цвета (5,2 г, 86%).
Стадия В: К раствору, содержащему полученный на стадии Б пропаргиламин (8 г, 22,63 ммоль) и порошкообразный К2СО3 (8,8 г, 56,6 ммоль) в N,N-диметилформамиде (150 мл), добавляли при температуре окружающей среды 3,4-диметоксифенол (6,98 г, 45 ммоль). Смесь нагревали в течение 36 ч до 90° С. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали 1 н. раствором NаНСО3, Н2О и сушили над MgSO4. После хроматографии (на силикагеле, этилацетат/гексан) получали феноксипропаргиламин в виде геля светло-желтого цвета (10 г, 90,9%).
Стадия Г: Раствор NaOH (8,2 г, 200 ммоль) в Н2О (30 мл) добавляли через капельную воронку при температуре окружающей среды к раствору полученного на стадии В феноксипропаргиламина (10 г, 20,5 ммоль) в этаноле (15 мл) и тетрагидрофуране (15 мл). Образовавшийся раствор нагревали в течение 48 ч до 60° С и выдерживали в течение 48 ч при температуре окружающей среды. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали простым эфиром и подкисляли до рН 2. После вакуумной фильтрации осадка белого цвета получали кислоту в виде твердого вещества белого цвета (9,4 г, количественный выход).
Стадия Д: К раствору, содержащему полученную на стадии Г кислоту (9,4 г, 20,5 ммоль), N-метилморфолин (6,8 мл, 62 ммоль), 1-гидроксибензотриазол (8,3 г, 60 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (4,8 г, 40 ммоль) в N,N-диметилформамиде (50 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (11,7 г, 60 ммоль). Затем образовавшийся раствор перемешивали в течение 20 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным раствором NaHCO3, Н2О и сушили над MgSO4. После концентрирования в вакууме и хроматографии (на силикагеле, этилацетат/гексан) получали ТГП-защищенный амид в виде пены белого цвета (10 г, 89,7%).
Стадия Е: К раствору 4 н. HCl в диоксане (38 мл, 152 ммоль) добавляли раствор полученного на стадии Д амида (8,5 г, 15,2 ммоль) в метаноле (8 мл) и диоксане (24 мл). Образовавшуюся смесь перемешивали в течение 80 мин при температуре окружающей среды. После концентрирования в вакууме и растирания с простым эфиром получали гидрохлорид гидроксамата в виде твердого вещества белого цвета (7,7 г, количественный выход). МСВР: рассчитано для C23H26N2O7S: 475,1461, обнаружено 475,1539.
Пример 39: Получение моногидрохлорида 4-[[4-(3,5-диметоксифенокси)фенил]сульфонил]-N-гидрокси-1-(2-пропинил)-4-пиперидинкарбоксамида
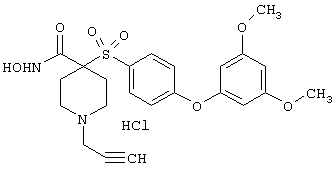
Стадия А: К раствору, содержащему полученный на стадии Б примера 38 пропаргиламин (2 г, 5,6 ммоль) и порошкообразный К2СО3 (1,9 г, 13,7 ммоль) в N,N-диметилформамиде (20 мл), добавляли при температуре окружающей среды 3,5-диметоксифенол (2,18 г, 13,7 ммоль). Смесь нагревали в течение 36 ч до 90° С. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали 1н. раствором NаНСО3, Н2О и сушили над MgSO4. После хроматографии (на силикагеле, этилацетат/гексан) получали феноксипропаргиламин в виде геля светло-желтого цвета (2,76 г, количественный выход).
Стадия Б: К раствору полученного на стадии А феноксипропаргиламина (2,75 г, 5,6 ммоль) в этаноле (5 мл) и тетрагидрофуране (5 мл) добавляли при температуре окружающей среды NaOH (2,3 г, 56 ммоль) в Н2О (10 мл). Затем раствор нагревали в течение 18 ч до 60° С. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали простым эфиром и подкисляли до рН 2. После вакуумной фильтрации осадка белого цвета получали кислоту в виде твердого вещества белого цвета (2 г, 77,2%).
Стадия В: К раствору, содержащему полученную на стадии Б кислоту (2 г, 4,3 ммоль), N-метилморфолин (1,9 мл, 17,2 ммоль), 1-гидроксибензотриазол (1,74 г, 13,2 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (1,02 г, 8,6 ммоль) в N,N-диметилформамиде (20 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (2,47 г, 12,9 ммоль). Образовавшуюся смесь перемешивали в течение 20 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным раствором NaHCO3, Н2О и сушили над MgSO4. После концентрирования в вакууме и хроматографии (на силикагеле, этилацетат/гексан) получали ТГП-амид в виде пены белого цвета (2,4 г, количественный выход).
Стадия Г: К раствору 4 н. HCl в диоксане (13 мл, 52 ммоль) добавляли раствор полученного на стадии В ТГП-защищенного амида (2,43 г, 4,35 ммоль) в метаноле (2 мл) и диоксане (6 мл) и смесь перемешивали в течение 80 мин при температуре окружающей среды. После вакуумной фильтрации осадка и промывки простым эфиром получали гидрохлорид гидроксамата в виде твердого вещества белого цвета (1,25 г, 56,3%). Элементный анализ: рассчитано для C23H26N2O7S· 1,5HCl: С 52,20; Н 5,24; N 5,29; обнаружено: С 52,00; Н 5,05; N5,17.
Пример 40: Получение моногидрохлорида 4-[[4-(1,3-бензодиоксол-5-илокси)фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
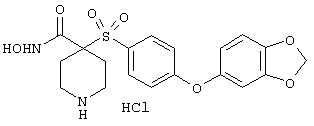
Стадия А: К раствору, содержащему полученное на стадии Б примера 37 N-ВОС-защищенное производное карбоновой кислоты (1,25 г, 2,47 ммоль), N- метилморфолин (1,00 г, 9,89 ммоль) и гидрат 1-гидроксибензотриазола (0,40 г, 2,96 ммоль) в N,N-диметилформамиде (8 мл), добавляли при температуре окружающей среды гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,616 г, 3,21 ммоль). Через 5 мин добавляли раствор O-тетрагидро-2Н-пиран-2-илпидроксиламина (0,39 г, 3,33 ммоль) в N,N-диметилформамиде (2 мл). Через 2 дня раствор светло-желтого цвета концентрировали в вакууме, получая остаток, который растворяли в этилацетате и последовательно промывали водой (3х) и соляным раствором и сушили над сульфатом магния. После концентрирования получали остаток, который хроматографировали на силикагеле, элюируя этилацетатом/гексаном (20/80), с получением ТГП-защищенного гидроксамата в виде масла (1,54 г, 100%).
Стадия Б: К раствору полученного на стадии А ТГП-защищенного гидроксамата (1,49 г, 2,46 ммоль) в диоксане (9 мл) и метаноле (3 мл) добавляли 4н. HCl в диоксане (10 мл, 40 ммоль). После выдерживания в течение 1,5 ч при температуре окружающей среды суспензию обрабатывали диэтиловым эфиром (15 мл) и фильтровали, получая указанный в заголовке гидроксамат (1,00 г, 89%) в виде бесцветного порошка. МС (CI) MH+: рассчитано для C19H20N2SO7: 421, обнаружено 421. Элементный анализ: рассчитано для C19H20N2SO7·HCl: С 49,95; Н 4,63; N 6,13; Cl 7,76; S 7,02; обнаружено: С 49,82; Н 4,60; N 5,98; Cl 17,38; S 7,10.
Пример 41: Получение моногидрохлорида N-гидрокси-4-[[4-(3-метилфенокси)фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида
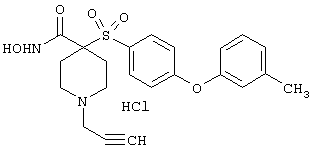
Стадия А: К раствору, содержащему полученный на стадии Е примера 9 пропаргиламин (8,0 г, 22,6 ммоль) и K2CO3 в N,N-диметилформамиде (30 мл), добавляли мета-крезол (3,5 г, 33,9 ммоль) и раствор перемешивали в течение 18 ч при 90° С. Раствор разбавляли Н2О и экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором NaCl и сушили над MgSO4. После хроматографии (на силикагеле, элюируя смесью 10%-ный этилацетат/гексан) получали 3-метилфеноксифенильное производное в виде твердого вещества (10,3 г, 98%). МС: рассчитано для C24H28NSO5: 441,1688, обнаружено 442,1697.
Стадия Б: К раствору полученного на стадии А 3-метилфеноксифенильного производного (10,3 г, 22,0 ммоль) в тетрагидрофуране (50 мл) и этаноле (50 мл) добавляли NaOH (8,9 г, 22,3 моль) и раствор выдерживали при температуре 65° С в течение 24 ч. Раствор концентрировали в вакууме и водный остаток подкисляли до рН 3. После вакуумной фильтрации образовавшегося осадка получали кислоту в виде твердого вещества белого цвета (9,0 г, 91%). МС: рассчитано для C22H24NSO5: 414,1375, обнаружено 414,1389.
Стадия В: К раствору полученной на стадии Б кислоты (9,0 г, 19,5 ммоль) добавляли 1-гидроксибензотриазол (3,24 г, 23,9 ммоль), N-метилморфолин (6,58 мл, 59,9 ммоль) и O-тетрагидро-2Н-пиран-2-илпидроксиламин (3,5 г, 29,9 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (5,35 г, 27,9 ммоль). Затем раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4. После хроматографии (на силикагеле, элюируя смесью 40%-ный этилацетат/гексан) получали требуемый ТГП-защищенный гидроксамат в виде твердого вещества (6,9 г, 67%). Элементный анализ: рассчитано для C27H33N2SO6·0,1Н2О: С 62,92; Н 6,49; N 5,43; S 6,23; обнаружено: С 62,69; Н 6,47; N 5,57; S 6,33. МС: рассчитано для С27H33N2SО6: 513,2059, обнаружено 513,2071.
Стадия Г: К раствору полученного на стадии В ТГП-защищенного гидроксамата (6,4 г, 12,5 ммоль) в диоксане (56 мл) и метаноле (19 мл) добавляли смесь 4 н. HCl/диоксан (40 мл). После перемешивания в течение 1 ч при температуре окружающей среды раствор концентрировали в вакууме. После растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (5,66 г, 97,4%). МС: рассчитано для С22Н24N2SO5: 429,1484, обнаружено М+1: 429,1493.
Пример 42: Получение 4-[[4-(1,3-бензодиоксол-5-илокси)фенил]сульфонил]-N-гидрокси-1-(метилсульфонил)-4-пиперидинкарбоксамида
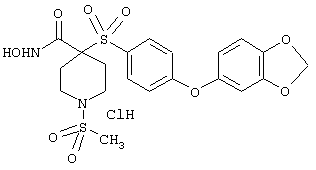
Стадия А: К раствору полученного на стадии Г примера 32 сульфона (25, г, 67,3 ммоль) в N,N-диметилформамиде добавляли карбонат калия (23,3 г, 0,169 моль) и сезамол (23,2 г, 0,164 моль). Раствор помещали в масляную баню при 90° С и перемешивали в течение 25 ч. К раствору добавляли этилацетат и органическую фазу промывали водой, 1н. раствором NaOH и водой, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. После хроматографии на двуокиси кремния с элюированием смесью этилацетат/гексан (15/85) получали соединение, представляющее собой сложный этиловый эфир, в виде масла (29,3 г, 82%).
Стадия Б: К раствору полученного на стадии А сложного этилового эфира (29,3 г, 54,93 ммоль) в этаноле (60 мл) и тетрагидрофуране (60 мл) добавляли раствор NaOH (21,9 г, 0,549 моль) в воде (120 мл) и раствор выдерживали в течение 10 ч при 65° С. Раствор концентрировали в вакууме и водный остаток подкисляли до рН 3. Раствор экстрагировали этилацетатом. Раствор сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получая кислоту в виде пены желтого цвета (25,6 г, 92,1%).
Стадия В: Раствор полученной на стадии Б кислоты (20,3 г, 40,15 ммоль) в этилацетате барботировали в течение 20 мин при 0° С газообразным HCl. Раствор перемешивали в течение 1,5 ч при 0° С. Образовавшийся осадок фильтровали и промывали простым эфиром, получая гидрохлорид амина в виде твердого вещества белого цвета (16,0 г, 93,5%).
Стадия Г: К раствору полученного на стадии В гидрохлорида амина (7,5 г, 17,0 ммоль) в метиленхлориде (200 мл) добавляли метансульфонилхлорид (2,0 г, 25,0 моль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор промывали Н2О и насыщенным раствором NaCl, сушили над сульфатом магния и концентрировали в вакууме, получая кислоту в виде твердого вещества белого цвета (6,97 г, 85%).
Стадия Д: К раствору полученной на стадии Г кислоты (7,37 г, 15,0 ммоль) добавляли 1-гидроксибензотриазол (2,43 г, 18,0 ммоль), N-метилморфолин (4,94 мл, 45,0 ммоль), O-тетрагидро-2Н-пиранилгидроксиламин (2,65 г, 22,5 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (4,02 г, 21,0 ммоль). Раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4. После хроматографии (на силикагеле, элюируя смесью 50%-ный этилацетат/гексан) получали требуемый ТГП-защищенный гидроксамат в виде твердого вещества белого цвета (7,54 г, 85%).
Стадия Е: К раствору полученного на стадии Д ТГП-защищенного гидроксамата (6,32 г, 10,8 ммоль) в диоксане (75 мл) и метаноле (25 мл) добавляли смесь 4н. HCl/диоксан (30 мл). После перемешивания в течение 1 ч при температуре окружающей среды раствор концентрировали в вакууме. После растирания с диэтиловым эфиром получали указанное в заголовке соединение. После хроматографии (на силикагеле, элюируя смесью 5%-ный метанол/этилацетат) получали гидроксамат в виде твердого вещества белого цвета (4,32 г, 80%). МС: рассчитано для C22H22N2S2O9+1: 499,0845, обнаружено: 499,0848.
Пример 43: Получение моногидрохлорида 4-[[4-(3,4-диметилфенокси)фенил]сульфонил]-N-гидрокси-1-(2-пропинил)-4-пиперидинкарбоксамида
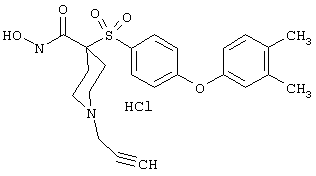
Стадия А: Смесь, включающую полученное на стадии Е примера 9 фторсодержащее соединение (2,0 г, 5,66 ммоль), 3,4-диметилфенол (2,0 г, 16,5 ммоль) и карбонат калия (2,3 г, 16,5 ммоль) в N,N-диметилформамиде (15 мл), выдерживали в течение ночи (приблизительно 18 ч) при 90° С в атмосфере азота. Смесь коричневого цвета концентрировали в вакууме и очищали с помощью хроматографии (на силикагеле, этилацетат/гексан), получая 3,4-диметилфеноксифенильное производное в виде прозрачного масла желтого цвета (2,0 г, выход 79%). Элементный анализ: рассчитано для C25H29NO5S: С 65,9; Н 6,42; N 3,04; S 7,04; обнаружено: С 65,76; Н 6,37; N 3,03; S 7,00.
Стадия Б: Раствор, содержащий полученное на стадии А 3,4-диметилфеноксифенильное производное (2,0 г, 4,93 ммоль) и гидроксид калия (1,7 г, 29,7 ммоль) в смеси этанола (25 мл) и воды (4 мл), перемешивали в течение 4 ч при температуре дефлегмации в атмосфере азота. Раствор охлаждали с помощью ледяной бани, затем подкисляли концентрированной соляной кислотой и концентрировали, получая неочищенный остаток. Смесь, содержащую неочищенный остаток, O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,88 г, 7,50 ммоль), триэтиламин (0,81 мл, 5,81 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида в ацетонитриле (24 мл), перемешивали в течение ночи при температуре окружающей среды. Смесь разбавляли водой и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором бикарбоната натрия, водой и насыщенным соляным раствором. После сушки над сульфатом магния фильтрат, представляющий ТГП-защищенный гидроксамат, концентрировали, получая пену желтого цвета.
Стадия В: Полученный на стадии Б ТГП-защищенный гидроксамат (920 мг, 1,75 ммоль) растворяли в метаноле (16 мл). Добавляли ацетилхлорид (0,37 мл, 5,3 ммоль). Через 3 ч путем концентрирования и последующей ЖХВР с обращенной фазой получали указанное в заголовке соединение в виде твердого вещества белого цвета (611 мг, 79%). МС (ЭУ) МН+: рассчитано для С23Н26N2О5S: 443, обнаружено 443.
Пример 44: Получение моногидрохлорида 4-[[4-(4-хлорфенил)тиолфенил]сульфонил]-1-(пропинил)-4-пиперидинкарбоновой кислоты и моногидрохлорида-4-[[4-(4-хлорфенил)тиолфенил]сульфонил]-N-гидрокси-1-(пропинил)-4-пиперидинкарбоксамида
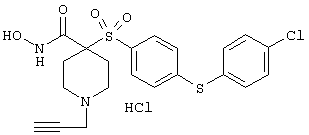
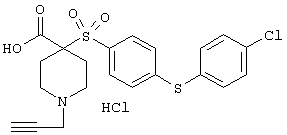
Стадия А:. Смесь, включающую полученное на стадии Е примера 9 фторсодержащее соединение (2,0 г, 5,66 ммоль), 4-хлортиофенол (1,0 г, 6,94 ммоль) и карбонат калия (1,1 г, 8,0 ммоль) в N,N-диметилформамиде (12 мл), перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды в атмосфере азота. Смесь концентрировали в вакууме. Остаток разбавляли водой и экстрагировали этилацетатом. Органический слой промывали водой и насыщенным соляным растовором, сушили над сульфатом магния и концентрировали в вакууме, получая масло желтого цвета. Масло очищали с помощью хроматографии (на силикагеле, этилацетат/гексан), получая 4-хлорфенилтиолфенильное производное в виде твердого вещества белого цвета (2,0 г, выход 75%). Элементный анализ: рассчитано для C23H24NO4S2Cl: С 57,791; Н 5,06; N 2,93; S 13,42; Cl 7,42; обнаружено: С 57,57; Н 5,11; N 2,94; S 13,19; Cl 7,73.
Стадия Б: Полученное на стадии А хлорфенилтиофенильное производное (2,04 г, 4,27 ммоль) разбавляли этанолом (30 мл) и водой (5 мл). Добавляли гидроксид калия (1,55 г, 27,7 ммоль) и смесь выдерживали в течение 3 ч при температуре дефлегмации. После завершения реакции раствор охлаждали и подкисляли концентрированной HCl до рН 1-3. Растворитель удаляли на роторном испарителе и остаток высушивали досуха в азеотропных условиях при повторном добавлении ацетонитрила. Затем хлорангидрид подвергали дальнейшей сушке с помощью вакуумной установки, после чего использовали в реакции сочетания без дополнительной очистки. Предполагалось, что омыление является количественным.
Стадия В: Полученный на предыдущей стадии хлорангидрид карбоновой кислоты (4,27 ммоль) суспендировали в ацетонитриле (20 мл). Добавляли N-метилморфолин (приблизительно 1,0 мл), а затем O-тетрагидро-2Н-пиран-2-илгидроксиламин (585 мг, 5 ммоль). Через 5 мин добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (ЭДК, 955 мг, 5 ммоль). Смесь перемешивали в течение ночи (приблизительно 18 ч), затем растворитель удаляли на роторном испарителе, остаток разбавляли полу насыщенным раствором NaHCO3 (50 мл) и продукт экстрагировали этилацетатом (2× 100 мл). В данном примере плохо вступающая в реакцию эмульсия осложняла выделение соединения. Объединенные органические слои сушили над MgSO4, фильтровали через силикагель, концентрировали и подвергали хроматографии (экспресс-хроматография, силикагель, этилацетат/гексан), получая после концентрирования указанный в заголовке О-ТГП-защищенный гидроксамат (162 мг, 7%, из сложного эфира) в виде пены. МС (ЭУ) МН+: рассчитано для C21H22N2O4S2Cl: 450 обнаружено 450. Поскольку массовый выход был малым, то осадок на фильтре из двуокиси кремния экстрагировали смесью метанол : этилацетат (1:1), получая моногидрохлорид 4-[[4-(4-хлорфенил)тиолфенил]сульфонил]-1-(пропинил)-4-пиперидинкарбоновой кислоты (540 мг, 26%).
Стадия Г: Полученный на стадии В О-ТГП-защищенный гидроксамат (441 мг, 0,80 ммоль) растворяли в метаноле (2 мл). Добавляли ацетилхлорид (0,2 мл, 3 ммоль). Через 3 ч путем концентрирования и последующей ЖХВР с обращенной фазой получали указанное в заголовке соединение, представляющее собой гидроксамат, в виде твердого вещества розового цвета (162 мг, 44%). МС (ЭУ) МН+: рассчитано для C21H22N2O4S2: 465, обнаружено 465.
Пример 45: Получение моногидрохлорида 4-[[4-(циклопентилтио)фенил]сульфонил]-N-гидрокси-1-(2-пропинил)-4-пиперидинкарбоксамида
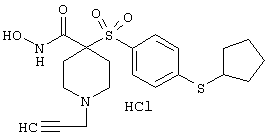
Стадия А: Полученный на стадии Е примера 9 пропаргиламин (3,09 г, 8,5 ммоль) смешивали с К2СО3 (1,38 г, 10 ммоль), N,N-диметилформамидом (6 мл) и циклопентилмеркаптаном (1,02 мл, 10 ммоль). Смесь нагревали в течение 4 ч до 80° С и в течение 2,5 ч до 95° С, контролируя реакцию с помощью ТСХ. Водную обработку осуществляли с использованием воды (10 мл) и этилацетата (2× 100 мл). Объединенные органические слои сушили над сульфатом магния, концентрировали и хроматографировали (экспресс-хроматография, элюент - этилацетат/гексан), получая циклопентилмеркаптильное производное в виде масла (3,2 г, 86%).
Стадия Б: Полученное на стадии А циклопентилмеркаптильное производное (3,12 г, 7,13 ммоль) разбавляли этанолом (50 мл) и водой (8 мл). Добавляли гидроксид калия (2,59 г, 46,3 ммоль) и смесь выдерживали в течение 3,5 ч при температуре дефлегмации. После завершения реакции раствор охлаждали и подкисляли концентрированной HCl до рН 1-3. Растворитель удаляли на роторном испарителе и остаток высушивали досуха в азеотропных условиях при повторном добавлении ацетонитрила. Хлорангидрид карбоновой кислоты подвергали дальнейшей сушке с помощью вакуумной установки, после чего использовали в реакции сочетания без дополнительной очистки. Предполагалось, что омыление является количественным.
Стадия В: Полученный на стадии Б хлорангидрид карбоновой кислоты (7,13 ммоль) суспендировали в ацетонитриле (50 мл). Добавляли N-метилморфолин (приблизительно 2,0 мл), а затем O-тетрагидро-2Н-пиран-2-илгидроксиламин (1,05 г, 9 ммоль). Через 5 мин добавляли ЭДК (1,72 г, 9 ммоль). Смесь перемешивали в течение ночи (приблизительно 18 ч), затем растворитель удаляли на роторном испарителе, остаток разбавляли полунасыщенным раствором NaHCO3 (50 мл) и продукт экстрагировали этилацетатом (2× 100 мл). Объединенные органические слои сушили над MgSO4, фильтровали через силикагель, концентрировали и подвергали хроматографии (экспресс-хроматография, на силикагеле, этилацетат/гексан), получая после концентрирования О-ТГП-защищенный гидроксамат (2,0 г, 51%, из сложного эфира) в виде пены.
Стадия Г: Полученный на стадии В О-ТГП-защищенный гидроксамат (2,0 г, 3,95 ммоль) растворяли в метаноле (16 мл). Через 2 мин добавляли ацетилхлорид (0,86 мл, 12 ммоль). Реакционную смесь перемешивали в течение 4 ч при температуре окружающей среды, затем концентрировали, повторно добавляя хлороформ и ацетонитрил для осуществления сушки. Указанное в заголовке соединение осаждалось в виде твердого вещества белого цвета (1,77 г, 98%). МС (ЭУ) МН+: рассчитано для C20H26N2O4S2: 422, обнаружено 422.
Пример 47: Получение 1-оксида N-гидрокси-4-[[4-(фенилтио)фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида и N-гидрокси-4-[[4-(фенилсульфинил)фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида

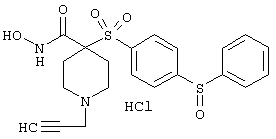
К раствору N-гидрокси-4-[[4-(фенилтио)фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида (указанное в заголовке соединение, пример 9) (215 мг, 0,5 ммоль) в метаноле (5 мл) добавляли при 0° С мета-хлорпербензойную кислоту (120 мг, 57-86%). Реакционной смеси давали медленно нагреться до температуры окружающей среды и через 16 ч смесь пропускали через фильтр с размерами пор порядка микрометра и концентрировали. С помощью ЖХВР с обращенной фазой (колонка типа Delta Pak 50× 300 мм; 15 мк C18 100 Е; метод с использованием 30-минутного градиента, начиная с соотношения в смеси: разбавленная HCl (0,5 мл/4 л) : ацетонитрил, равного 80:20, и заканчивая соотношением 50:50) разделяли 5 основных компонентов. После концентрирования первого и второго пика, элюированных из колонки, получали 14 мг (6%) и 16 мг (7%) двух соединений, которые были идентифицированы на основе их ЯМР-спектров как диастереоизомеры N-гидрокси-4-[[4-(фенилсульфинил)фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида. Третий пик не был идентифицирован. Четвертый пик на основе ЯМР-спектра был идентифицирован как 1-оксид N-гидрокси-4-[[4-(фенилтио)фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида (147 мг, 66%). МС (ЭУ) МН+: рассчитано для C21H22N2O5S2: 447, обнаружено 447. Последний пик содержал 73 мг восстановленной 3-хлорбензойной кислоты.
Пример 48: Получение N-гидрокси-2,2-диметил-5-[(4-феноксифенил)сульфонил]–1,3-диоксан-4-карбоксамида
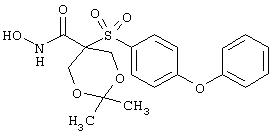
Стадия А: Приготавливали свежий раствор метоксида натрия путем медленного добавления при 0° С промытых гексаном гранул натрия (9,4 г, 410 ммоль) к метанолу (1,0 л). К этому охлажденному раствору добавляли 4-фтортиофенол (50,0 г, 390 ммоль), а затем метил-2-хлорацетат (42,3 г, 390 ммоль). После нагрева до температуры окружающей среды реакционную смесь перемешивали в течение ночи (приблизительно 18 ч). Метанол удаляли в вакууме и остаток растворяли в этилацетате (300 мл). Органический слой промывали водой (2× 200 мл) и сушили над MgSO4. После концентрирования получали продукт, представляющий собой сульфид метилового эфира, в виде прозрачного масла (71,8 г, 92%).
Стадия Б: К раствору полученного на стадии А сульфида метилового эфира (71,8 г, 358 ммоль) в смеси 70%-ный метанол/Н2О (1,0 л) медленно добавляли Охоnе™ (660 г, 1,08 моль). Смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды. Избыток Охоnе™ отфильтровывали и метанол удаляли из фильтрата в вакууме. Оставшийся водный раствор экстрагировали этилацетатом (3× 300 мл). Органические слои промывали водой (2× 300 мл) и сушили над MgSO4. После концентрирования получали сульфон в виде масла рыжевато-коричневого цвета (82 г, 98%).
Стадия В: К приготовленной суспензии бикарбоната калия (1,0 г, 9,8 ммоль) в 37%-ном растворе формальдегида добавляли полученный на стадии Б сульфон (28,6 г, 123 ммоль). Реакционную смесь перемешивали в течение 1 ч и затем добавляли насыщенный раствор сульфата натрия (20 мл). После перемешивания в течение 30 мин смесь экстрагировали диэтиловым эфиром (4× 100 мл). Органические слои сушили над MgSO4. После хроматографии (на силикагеле, этилацетат/гексан) получали продукт, представляющий собой сульфониол, в виде прозрачного масла (15,3 г, 42%).
Стадия Г: Полученный на стадии В сульфондиол (1,3 г, 4,5 ммоль) растворяли вместе с 2,2-диметоксипропаном (1,1 мл, 9,0 ммоль) и моногидратом пара-толуолсульфоновой кислоты (0,03 мг, 0,14 ммоль) в ацетоне (40 мл) и образовавшуюся смесь кипятили с обратным холодильником в течение 6 ч. После охлаждения смесь нейтрализовали с помощью твердого Na2CO3 (рН~7), фильтровали и концентрировали. Остаток растворяли в хлороформе (50 мл) и промывали водой (2× 30 мл). После сушки над MgSO4 и концентрирования получали продукт, представляющий собой диметилкеталь, в виде темного масла (1,4 г, 94%).
Стадия Д: К раствору полученного на стадии Г диметилкеталя (1,4 г, 4,2 ммоль) в N,N-диметилформамиде (20 мл) добавляли фенол (0,6 г, 6,3 ммоль) и карбонат цезия (2,0 г, 6,3 ммоль). Смесь выдерживали в течение 5 ч при 90° С, разбавляли водой (20 мл) и экстрагировали этилацетатом (4× 100 мл). Органические слои промывали соляным раствором (1х100 мл) и водой (1× 100 мл). После концентрирования получали фенол-O-фенолдиметилкеталь в виде темно-коричневого масла (1,51 г, 88%).
Стадия Е: К раствору полученного на стадии Д фенол-O-фенолдиметилкеталя (1,5 г, 3,4 ммоль) в тетрагидрофуране (10 мл) добавляли водный раствор гидроксида лития (0,34 г, 14,8 ммоль, в 5 мл Н2О). Реакционную смесь перемешивали в течение 2 ч и затем разбавляли водой (15 мл) и подкисляли с помощью 30%-ного водного раствора HCl до рН 3. Кислый раствор экстрагировали диэтиловым эфиром (3× 100 мл). После сушки над MgSO4 и концентрирования получали продукт, представляющий собой карбоновую кислоту, в виде масла коричневого цвета (1,5 г, количественный выход).
Стадия Ж: К раствору, содержащему полученную на стадии Е карбоновую кислоту (1,3 г, 3,3 ммоль) и гидрат N-гидроксибензотриазола (0,54 г, 4,0 ммоль) в ДМФ (15 мл), последовательно добавляли 4-метилморфолин (1,67 г, 16,5 ммоль), O-тетрагидро-2Н-пиран-2-илгидроксиламин (1,2 г, 10,2 ммоль) и ЭДК (0,88 г, 4,6 ммоль). После перемешивания в течение ночи ДМФ удаляли в вакууме и остаток растворяли в смеси этилацетат/вода (1:1, 50 мл). Органический слой промывали соляным раствором (1× 20 мл) и водой (1× 20 мл) и сушили над MgSO4. После хроматографии (на силикагеле, этилацетат/гексан) получали ТГП-защищенный гидроксиламин в виде твердого вещества белого цвета (0,36 г, 22%), а также декарбоксилированный побочный продукт (0,27 г, 24%).
Стадия З: К раствору полученного на стадии Ж ТГП-защищенного гидроксиламина (0,36 г, 0,73 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли 4 н. раствор HCl в диоксане (2 мл). Реакционную смесь перемешивали в течение 5 мин и затем растворители удаляли в вакууме. После хроматографии (обращенная фаза, колонка С-18, ацетонитрил/вода) получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,13 г, 44%). МС (FAB) MH+: рассчитано для C19H21NO7S: 408, обнаружено 408.
Пример 49: Получение тетрагидро-N-гидрокси-4-[[4-(фенилтио)фенил]сульфонил]-2Н-тиопиран-4-карбоксамида
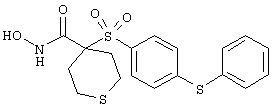
Стадия А: К раствору метил-2-хлорацетата (322 г, 2,96 моль) в N,N-диметилацетамиде (1,0 л) добавляли тиофенол (400 г, 3,12 моль) и карбонат калия (408 мг, 2,96 моль). Реакционную смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды. После разбавления минимальным количеством воды (800 мл) смесь экстрагировали этилацетатом (4× 1 л). Органические слои промывали водой (1× 800 мл), сушили над MgSO4 и концентрировали, получая продукт, представляющий собой сульфид, в виде прозрачного масла (614 г, количественный выход).
Стадия Б: К раствору полученного на стадии А сульфида (75,85 г, 0,38 моль) в метаноле (1000 мл) добавляли при 20° С воду (100 мл) и Охоnе® (720 г, 1,17 моль). Происходила экзотермическая реакция, в результате которой температура повышалась до 67° С. Через 2 ч реакционную смесь фильтровали и остаток на фильтре тщательно промывали метанолом. Фильтрат концентрировали в вакууме. Остаток растворяли в этилацетате и промывали соляным раствором, сушили над MgSO4 и концентрировали в вакууме, получая сульфон в виде кристаллического твердого вещества (82,74 г, 94%).
Стадия В: К раствору полученного на стадии Б сульфона (60 г, 258 ммоль) в ДМА (350 мл) добавляли дибромэтилтиоэфир (76,9 г, 310 ммоль), а затем карбонат калия (78,3 г, 568 моль). Смесь перемешивали в течение 5 мин, после чего добавляли каталитические количества 4-диметиламинопиридина и бромида тетрабутиламмония. Реакционную смесь перемешивали в течение ночи (приблизительно 18 ч), после чего сливали при перемешивании на 10%-ный водный раствор HCl (2,5 л). Образовавшийся осадок фильтровали и промывали гексаном для удаления избытка тиоэфира. После сушки в вакууме в течение ночи (приблизительно 18 ч) получали метиловый эфир тиопирана -Ph-p-F в виде порошка желтого цвета (76,1 г, 93%).
Стадия Г: К раствору полученного на стадии В метилового эфира тиопирана -Ph-p-F (4,0 г, 12,6 моль) в N,N-диметилацетамиде (25 мл) добавляли карбонат цезия (6,1 г, 18,9 ммоль) и тиофенол (2,1 г, 18,9 ммоль). Смесь перемешивали в течение 2 ч при 90° С. Смесь разбавляли водой (30 мл) и экстрагировали этилацетатом (3× 100 мл). Органические слои промывали соляным раствором (1× 75 мл) и водой (1× 75 мл), после чего сушили над MgSO4. После хроматографии (на силикагеле, этилацетат/гексан) получали метилфенил-S-фениловый эфир в виде твердого вещества желтоватого цвета (3,6 г, 71%).
Стадия Д: К раствору полученного на стадии Г метилфенил-З-фенилового эфира (3,6 г, 8,8 ммоль) в тетрагидрофуране (15 мл) добавляли триметилсилонат калия (1,24 г, 9,7 ммоль). Смесь перемешивали в течение 2-3 ч при температуре окружающей среды или до тех пор, пока не образовывался твердый осадок. После завершения гидролиза добавляли N-метилморфолин (2,9 мл, 26,4 ммоль), а затем РуВrоР (4,9 г, 10,6 ммоль). Раствор перемешивали в течение 10 мин. Добавляли водный раствор гидроксиламина (0,32 г, 9,7 ммоль) и смесь перемешивали еще в течение 2 ч. После завершения реакции растворитель удаляли в вакууме. После хроматографии (обращенная фаза, колонка С-18, ацетонитрил/вода) остатка получали указанное в заголовке соединение в виде твердого вещества беловатого цвета (0,82 г, 23%). МС (FAB) MH+: рассчитано для С18Н19NO4S3: 410, обнаружено 410.
Пример 50: Получение 4-[4-фторфенил)сульфонил]тетрагидро-N-[(тетрагидро-2Н-пиран-2-ил)окси]-2Н-тиопиран-4-карбоксамида
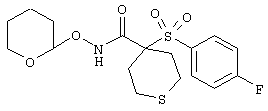
Стадия А: К раствору метил-2-хлорацетата (322 г, 2,96 моль) в N,N-диметилацетамиде (1,0 л) добавляли тиофенол (400 г, 3,12 моль) и карбонат калия (408 г, 2,96 моль). Реакционную смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды. После разбавления минимальным количеством воды (800 мл) смесь экстрагировали этилацетатом (4× 1 л). Органические слои промывали водой (1× 800 мл), сушили над MgSO4 и концентрировали, получая продукт, представляющий собой сульфид, в виде прозрачного масла (614 г, количественный выход).
Стадия Б: К раствору полученного на стадии А сульфида (75,85 г, 0,38 моль) в метаноле (1000 мл) добавляли при 20° С воду (100 мл) и Охоnе® (720 г, 1,17 моль). Происходила экзотермическая реакция, в результате которой температура повышалась до 67° С. Через 2 ч реакционную смесь фильтровали и осадок на фильтре тщательно промывали метанолом. Фильтрат концентрировали в вакууме. Остаток растворяли в этилацетате и промывали соляным раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме, получая сульфон сложного метилового эфира в виде кристаллического твердого вещества (82,74 г, 94%).
Стадия В: К раствору полученного на стадии Б сульфона сложного метилового эфира (60,0 г, 258 ммоль) в N,N-диметилацетамиде (350 мл) добавляли 2,2-дибромэтилтиоэфир (76,9 г, 310 ммоль), а затем карбонат калия (78,3 г, 568 моль). Смесь перемешивали в течение 5 мин, после чего добавляли каталитические количества 4-диметиламинопиридина и бромида тетрабутиламмония. Реакционную смесь перемешивали в течение ночи (приблизительно 18 ч), после чего сливали при перемешивании на 10%-ный водный раствор HCl (2,5 л). Образовавшийся осадок фильтровали и промывали гексаном для удаления избытка тиоэфира. После сушки в вакууме в течение ночи (приблизительно 18 ч) получали метиловый эфир тиопирана в виде порошка желтого цвета (76,1 г, 93%).
Стадия Г: К раствору полученного на стадии В метилового эфира тиопирана (30,0 г, 94 ммоль) в тетрагидрофуране (250 мл) добавляли триметилсилонат калия (28,9 г, 226 ммоль). Смесь перемешивали в течение 2-3 ч при температуре окружающей среды или до тех пор, пока не образовывался твердый осадок. После завершения гидролиза растворитель удаляли в вакууме. Добавляли воду (200 мл) и смесь промывали диэтиловым эфиром (1× 200 мл). Водный слой охлаждали до 0° С и медленно добавляли 10%-ный водный раствор HCl до тех пор, пока не образовывался осадок. Твердое вещество собирали и сушили в вакууме в присутствии пентоксида фосфора, получая тиопиранкарбоновую кислоту в виде твердого вещества желтого цвета (17,8 г, 62%).
Стадия Д: К раствору полученной на стадии Г тиопиранкарбоновой кислоты (17,8 г, 58,5 ммоль) в N,N-диметилформамиде (100 мл) добавляли N-метилморфолин (19,3 мл, 176 ммоль), а затем гидрат N-гидроксибензотриазола (9,5 г, 70,2 ммоль), O-тетрагидро-2Н-пиран-2-илгидроксиламин (10,3 г, 87,8 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (16,8 г, 87,8 моль). Смесь перемешивали в течение 3 ч и затем разбавляли водой (100 мл). Смесь экстрагировали этилацетатом (4× 200 мл). Органические слои промывали насыщенным водным раствором карбоната калия (1× 200 мл), 1%-ным водным раствором HCl и соляным раствором (1× 200 мл). После сушки над MgSO4 и концентрирования в вакууме получали указанное в заголовке соединение в виде твердого вещества беловатого цвета (30,8 г, количественный выход). МС (FAB) MH+: рассчитано для C17H22FNO5S2: 404, обнаружено 404.
Пример 51: Получение тетрагидро-N-гидрокси-4-[[4-[4-метоксифенил)тио]фенил]сульфонил]-2Н-тиопиран-4-карбоксамида
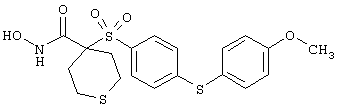
Стадия А: К раствору указанного в заголовке примера 50 соединения (6,0 г, 14,9 ммоль) в N,N-диметилацетамиде (25 мл) добавляли 4-метокситиофенол (2,5 г, 17,8 мл), а затем карбонат калия (6,2 г, 44,7 ммоль). Реакционную смесь выдерживали в течение 3 ч при 60° С. Реакционную смесь разбавляли водой (25 мл) и экстрагировали этилацетатом (4× 100 мл). Органические слои промывали водой (2× 50 мл) и сушили над MgSO4. После концентрирования в вакууме получали ТГП-защищенное фенил-S-пара-фенил-ОМе-производное в виде твердого вещества желтоватого цвета (9,2 г, количественный выход).
Стадия Б: К раствору полученного на стадии А ТГП-защищенного фенил -S-пара-фенил-ОМе-производного (9,2 г, 14,9 ммоль) в диоксане медленно добавляли 4н. раствор HCl в диоксане (10 мл). После перемешивания в течение ночи (приблизительно 18 ч) растворитель удаляли. После хроматографии образовавшегося остатка (обращенная фаза, колонка С-18, ацетонитрил/вода) получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,84 г, 28,3%). МС (FAB) MH+: рассчитано для С19Н21О5S3: 440, обнаружено 440.
Пример 52: Получение 1,1-диоксида тетрагидро-N-гидрокси-4-[[(4-(фенилтио)фенил]сульфонил]-2Н-тиопиран-4-карбоксамида
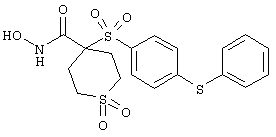
Стадия А: К раствору указанного в заголовке примера 50 соединения (13,0 г, 24,5 ммоль) в метиленхлориде (100 мл), охлажденному до 0° С, медленно добавляли 50-60%-ную мета-хлорпербензойную кислоту (17,1 г, 49,5 ммоль). Смесь перемешивали в течение 1 ч при 0° С, а затем еще в течение 3 ч, при этом температура повышалась до температуры окружающей среды. Добавляли воду (200 мл) и смесь нейтрализовали 10%-ным раствором гидроксида аммония (100 мл). Органический слой промывали водой (1× 200 мл) и сушили над MgSO4. После концентрирования в вакууме получали масло оранжеватого цвета (3,5 г, 33%). Раствор продукта в смеси вода/10%-ный гидроксид аммония насыщали хлоридом натрия и экстрагировали этилацетатом (2× 400 мл). Органический слой сушили над MgSO4 и концентрировали, получая ТГП-защищенное сульфонтиопиран-пара-F-производное в виде пены оранжевого цвета (6,1 г, 57%).
Стадия Б: К раствору полученного на стадии А ТГП-защищенного сульфонтиопиран-пара-F-производного (9,6 г, 22 ммоль) в N,N-диметилацета-миде (120 мл) добавляли тиофенол (2,9 г, 26,4 мл), а затем карбонат калия (9,1 г, 66 ммоль). Реакционную смесь выдерживали в течение 4 ч при 60° С. Реакционную смесь разбавляли водой (25 мл) и экстрагировали этилацетатом (4× 100 мл). Органические слои промывали водой (2× 50 мл) и сушили над MgSO4. После хроматографии (на силикагеле, этилацетат/гексан) получали ТГП-защищенное фенил-S-фенильное производное в виде масла оранжевого цвета (5,1 г, 43%).
Стадия В: К раствору полученного на стадии Б ТГП-защищенного фенил-S-фенильного производного (5,1 г, 9,4 ммоль) в диоксане медленно добавляли 4н. раствор HCl в диоксане (10 мл). После перемешивания в течение ночи (приблизительно 18 ч) растворитель удаляли. После хроматографии образовавшегося остатка (обращенная фаза, колонка С-18, ацетонитрил/вода) получали указанное в заголовке соединение в виде твердого вещества розового цвета (1,2 г, 29%). МС (FAB) MH+: рассчитано для C18H19NO6S3: 442, обнаружено 442.
Пример 53: Получение моногидрохлорида тетрагидро-N-гидрокси-4-[[4-[4-(1Н-1,2,4-триазол-1-ил)фенокси]фенил]сульфонил]-2Н-тиопиран-4-карбоксамид-1,1’-диоксида
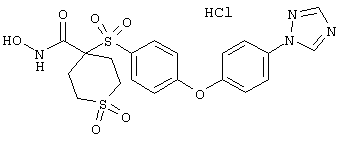
Стадия А: К раствору указанного в заголовке примера 50 соединения (13,0 г, 24,5 ммоль) в метиленхлориде (100 мл), охлажденному до 0° С, медленно добавляли 50-б0%-ную мета-хлорпербензойную кислоту (17,1 г, 49,5 ммоль). Смесь перемешивали в течение 1 ч при 0° С, а затем еще в течение 3 ч, при этом температура повышалась до температуры окружающей среды. Добавляли воду (200 мл) и смесь нейтрализовали 10%-ным раствором гидроксида аммония (100 мл). Органический слой промывали водой (1× 200 мл) и сушили над MgSO4. После концентрирования в вакууме получали масло оранжеватого цвета (3,5 г, 33%). Раствор продукта в смеси вода/10%-ный гидроксид аммония насыщали хлоридом натрия и экстрагировали этилацетатом (2× 400 мл). Органический слой сушили над MgSO4 и концентрировали, получая ТГП-защищенное сульфонтиопиран-пара-F-производное в виде пены оранжевого цвета (6,1 г, 57%).
Стадия Б: К раствору полученого на стадии А ТГП-защищенного сульфонтиопиран-пара-F-производного (6,0 г, 13,8 ммоль) в N,N-диметилформамиде (25 мл) добавляли 4-(1Н-1,2,4-триазол-1-ил)фенол (4,4 г, 27,5 ммоль), а затем карбонат цезия (13,4 г, 41,4 ммоль). Реакционную смесь выдерживали в течение 5 ч при 95° С. Реакционную смесь разбавляли водой (25 мл) и экстрагировали этилацетатом (4× 100 мл). Органические слои промывали водой (2× 50 мл) и сушили над MgSO4. После концентрирования в вакууме получали ТГП-защищенное фенил-O-фенилтриазольное производное в виде твердого вещества рыжевато-коричневого цвета (9,7 г, количественный выход).
Стадия В: К раствору полученного на стадии Б неочищенного ТГП-защищенного фенил-O-фенилтриазольного производного (8,0 г, 13,8 ммоль) в ацетонитриле медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил удаляли. Образовавшийся осадок собирали, получая указанное в заголовке соединение в виде твердого вещества рыжевато-коричневого цвета (1,3 г, 18%). МС (FAB) MH+: рассчитано для C20H21ClN4O7S2: 493, обнаружено 493.
Пример 54: Получение моногидрохлорида 4-[[4-[4-(2-амино-этил)фенокси]фенил]сульфонил]тетрагидро-N-гидрокси-2Н-тиопиран-4-карбоксамид-1,1’-диоксида
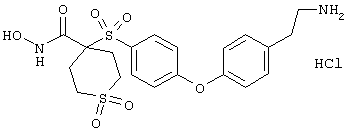
Стадия А: К раствору указанного в заголовке примера 50 соединения (13,0 г, 24,5 ммоль) в метиленхлориде (100 мл), охлажденному до 0° С, медленно добавляли 50-60%-ную мета-хлорпербензойную кислоту (17,1 г, 49,5 ммоль). Смесь перемешивали в течение 1 ч при 0° С, а затем еще в течение 3 ч, при этом температура повышалась до температуры окружающей среды. Добавляли воду (200 мл) и смесь нейтрализовали 10%-ным раствором гидроксида аммония (100 мл). Органический слой промывали водой (1× 200 мл) и сушили над MgSO4. После концентрирования в вакууме получали масло оранжеватого цвета (3,5 г, 33%). Раствор продукта в смеси вода/10%-ный гидроксид аммония насыщали хлоридом натрия и экстрагировали этилацетатом (2× 400 мл). Органический слой сушили над MgSO4 и концентрировали, получая ТГП-защищенное сульфонтиопиран-пара-Р-производное в виде пены оранжевого цвета (6,1 г, 57%).
Стадия Б: К раствору полученого на стадии А ТГП-защищенного сульфонтиопиран-пара-F-производного (6,0 г, 13,8 ммоль) в N,N-диметилацетамиде (25 мл) добавляли тирамин (3,8 г, 28 ммоль), а затем карбонат цезия (13,6 г, 42 ммоль). Реакционную смесь выдерживали в течение 5 ч при 95° С. После удаления N,N-диметилацетамида в вакууме получали твердое вещество коричневого цвета (20 г). После хроматографии (обращенная фаза, колонка С-18, ацетонитрил/вода) получали ТГП-защищенный тирамин в виде масла рыжевато-коричневого цвета (1,0 г, 13%).
Стадия В: К раствору полученного на стадии Б неочищенного ТГП-защищенного тирамина (1,0 г, 1,8 ммоль) в ацетонитриле медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил удаляли. Образовавшийся осадок собирали, получая указанное в заголовке соединение в виде твердого вещества рыжевато-коричневого цвета (0,9 г, 99%). МС (FAB) MH+: рассчитано для C20H25CIN2O7S2: 469 обнаружено 469.
Пример 55: Получение 4-[(4-фторфенил)сульфонил]тетрагидро-N-[(тетрагидро-2Н-пиран-2-ил)окси]-2Н-пиран-4-карбоксамида
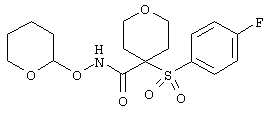
К метанолу (1000 мл), находящемуся в сухом сосуде добавляли в атмосфере азота при 2° С металлический натрий (8,97 г, 0,39 моль). Реакционную смесь перемешивали в течение 45 мин при температуре окружающей среды, в течение этого времени натрий растворялся. Раствор охлаждали до 5° С и добавляли пара-фтортиофенол (41,55 мл, 0,39 ммоль), а затем метил-2-хлорацетат (34,2 мл, 0,39 моль). Реакционную смесь перемешивали в течение 4 ч при температуре окружающей среды, фильтровали и концентрировали в вакууме, получая сульфид в виде прозрачного бесцветного масла (75,85 г, 97%).
Стадия Б: К раствору полученного на стадии А сульфида (75,85 г, 0,38 моль) в метаноле (1000 мл) добавляли при 20° С воду (100 мл) и Охоnе® (720 г, 1,17 моль). При этом происходила экзотермическая реакция, в результате которой температура повышалась до 67° С. Через 2 ч реакционную смесь фильтровали и осадок на фильтре тщательно промывали метанолом. Фильтрат концентрировали в вакууме. Остаток растворяли в этилацетате и промывали соляным раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме, получая сульфон в виде кристаллического твердого вещества (82,74 г, 94%).
Стадия В: К раствору полученного на стадии Б сульфона (28,5 г, 0,123 моль) в N,N-диметилацетамиде (200 мл) добавляли карбонат калия (37,3 г, 0,27 моль), простой бис(2-бромэтиловый) эфир (19,3 мл, 0,147 моль), 4-диметиламинопиридин (0,75 г, 6 ммоль) и бромид тетрабутиламмония (1,98 г, 6 ммоль). Реакционную смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды. Реакционную смесь медленно сливали на 1 н. раствор HCl (300 мл), отфильтровывали образовавшийся твердый продукт и осадок на фильтре тщательно промывали гексаном. Твердое вещество перекристаллизовывали из этилацетата/гексана, получая пиран в виде твердого вещества бежевого цвета (28,74 г, 77%). МС (ES+) МН+: рассчитано для C13H15O5S1F1: 303, обнаружено 303.
Стадия Г: В сухом сосуде в атмосфере азота полученный на стадии В пиран (8,0 г, 26,5 ммоль) растворяли в безводном тетрагидрофуране (250 мл) и при температуре окружающей среды добавляли раствор триметилсилоната калия (10,2 г, 79,5 ммоль) в безводном тетрагидрофуране (15 мл). Через 90 мин добавляли воду (100 мл) и раствор концентрировали в вакууме. Остаток растворяли в воде и экстрагировали этилацетатом для удаления непрореагировавшего исходного продукта. Водный раствор обрабатывали 6н. раствором HCl до достижения рН 1. Суспензию экстрагировали этилацетатом и объединенные экстракты промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток нагревали в диэтиловом эфире, твердый продукт фильтровали и сушили, получая карбоновую кислоту в виде кристаллического твердого вещества (5,78 г, 76%). МСВР (ES-) М-Н: рассчитано для C12H13O5S1F1: 287,04, обнаружено 287,04.
Стадия Д: Полученную на стадии Г карбоновую кислоту (9,1 г, 31,6 ммоль) растворяли в сухом сосуде в атмосфере азота в безводном N,N-диметилформамиде (70 мл) и затем к раствору добавляли остальные реактивы в следующем порядке: гидрат N-гидроксибензотриазола (5,1 г, 37,9 ммоль), N-метилморфолин (10,4 мл, 94,8 ммоль), O-тетрагидро-2Н-пиран-2-илгидроксиламин (11,5 г, 98 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (8,48 г, 44,2 ммоль). После выдерживания в течение 3 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали водой, 5%-ным KHSO4, насыщенным раствором NаНСО3, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали указанное в заголовке соединение в виде кристаллического твердого вещества (9,7 г, 80%). МСВР (ES+) МН+: рассчитано для C17H22NO6S1F1: 388,12, обнаружено 388,12.
Пример 56: Получение 4-[[4-(3,4-дифторфенокси)фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
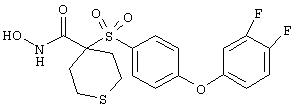
Стадия А: К раствору указанного в заголовке примера 55 соединения (2,0 г, 5,2 ммоль) в N,N-диметилацетамиде (6 мл) добавляли 3,4-дифторфенол (1,0 г, 7,7 ммоль), а затем карбонат цезия (6,6 г, 20,2 ммоль). Реакционную смесь выдерживали в течение 5 ч при 95° С. После удаления N,N-диметилацетамида в вакууме получали твердое вещество коричневого цвета (8,3 г, количественный выход). После хроматографии (обращенная фаза, колонка С-18, ацетонитрил/вода) получали ТГП-защищенный дифторсодержащий продукт в растворе.
Стадия Б: К собранному полученному на стадии А ТГП-защищенному дифторсодержащему продукту в смеси ацетонитрил/вода (50 мл) медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил удаляли. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества белого цвета (1,02 г, 48,6%). МС (FAB) M+H: рассчитано для C18H17FNO6S: 414, обнаружено 414.
Пример 57: Получение тетрагидро-N-гидрокси-4-[[4-(4-йодфенокси)фенил]сульфонил]-2Н-пиран-4-карбоксамида
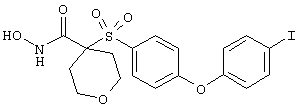
Стадия А: К раствору указанного в заголовке примера 55 соединения (2,0 г, 5,2 ммоль) в N,N-диметилацетамиде (6 мл) добавляли 4-йодфенол (1,7 г, 7,8 ммоль), а затем карбонат цезия (6,6 г, 20,2 ммоль). Реакционную смесь выдерживали в течение 5 ч при 95° С. После удаления N,N-диметилацетамида в вакууме получали твердое вещество коричневого цвета (5,7 г, количественный выход). После хроматографии (обращенная фаза, колонка С-18, ацетонитрил/вода) получали ТГП-защищенный йодсодержащий продукт в растворе.
Стадия Б: К раствору полученного на стадии А ТГП-защищенного йодсодержащего продукта в смеси ацетонитрил/вода (40 мл) медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил удаляли. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества белого цвета (2,6 г, 99%). МС (FAB) M+H: рассчитано для C18H18INO6S: 504, обнаружено 504.
Пример 58: Получение тетрагидро-N-гидрокси-4-[[4-(2,4,5-трифторфенокси)фенил]сульфонил]-2Н-пиран-4-карбоксамида
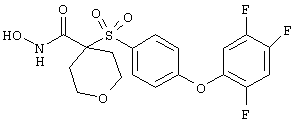
Стадия А: К раствору указанного в заголовке примера 55 соединения (2,0 г, 5,2 ммоль) в N,N-диметилацетамиде (6 мл) добавляли 2,4,5-трифторфенол (1,2 г, 7,8 ммоль), а затем карбонат цезия (10,1 г, 31,0 ммоль). Реакционную смесь выдерживали в течение 32 ч при 95° С. После удаления N,N-диметилацетамида в вакууме получали твердое вещество коричневого цвета (5,7 г, количественный выход). После хроматографии (обращенная фаза, колонка С-18, ацетонитрил/вода) получали ТГП-защищенный фенол (1,2 г, 44%).
Стадия Б: К раствору полученного на стадии А неочищенного ТГП-защищенного фенола (1,2 г, 2,3 ммоль) в смеси ацетонитрил/вода (40 мл) медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил удаляли. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,79 г, 79%). МС (FAB) M-H: рассчитано для С18Н16F3NO6S: 430 обнаружено 430.
Пример 59: Получение 4-[[4-(3,5-дихлорфенокси)фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
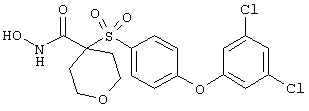
Стадия А: К раствору указанного в заголовке примера 55 соединения (2,0 г, 5,2 ммоль) в N,N-диметилацетамиде (6 мл) добавляли 3,5-дихлорфенол (1,3 г, 7,8 ммоль), а затем карбонат цезия (6,6 г, 20,2 ммоль). Реакционную смесь выдерживали в течение 12 ч при 95° С. После удаления N,N-диметилацетамида в вакууме получали твердое вещество коричневого цвета (5,7 г, количественный выход). Остаток растворяли в смеси ацетонитрил/вода (20 мл) и подкисляли до рН 6. Образовывался осадок белого цвета, который собирали, получая ТГП-защищенное вещество в виде осадка на фильтре белого цвета (1,8 г, 64%).
Стадия Б: К раствору полученого на стадии А ТГП-защищенного вещества (1,8 г, 3,4 ммоль) в смеси ацетонитрил/вода (20 мл) медленно добавляли 10%-ный водный раствор HCl (40 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил удаляли. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,71 г, 47%). МС (FAB) M+H: рассчитано для C18H17Cl2NO6S: 447, обнаружено 447.
Пример 59а: Получение моногидрохлорида тетрагидро-N-гидрокси-4-[[4-[[5-(трифторметил)-2-пиридинил]тио]фенил]сульфонил]-2Н-пиран-4-карбоксамида
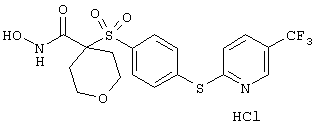
Стадия А: К раствору указанного в заголовке примера 55 соединения (2,0 г, 5,2 ммоль) в N,N-диметилацетамиде (6 мл) добавляли 5-(трифторметил)-2-пиридинилтиофенол (1,4 г, 7,8 ммоль), а затем карбонат калия (2,2 г, 15,6 ммоль). Реакционную смесь выдерживали в течение 12 ч при 65° С. После удаления N,N-диметилацетамида в вакууме получали твердое вещество коричневого цвета (5,4 г, количественный выход). После хроматографии (обращенная фаза, колонка С-18, ацетонитрил/вода) получали ТГП-защищенное вещество в растворе.
Стадия Б: К раствору полученного на стадии А неочищенного ТГП-защищенного вещества в смеси ацетонитрил/вода (40 мл) медленно добавляли 10%-ный водный раствор HCl (40 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил удаляли. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,20 г, 8%). МС (FAB) M+H: рассчитано для C18H17F3N2O5S2: 463, обнаружено 463.
Пример 60: Получение 4-[[4-(3,4-дихлорфенил)тио]фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
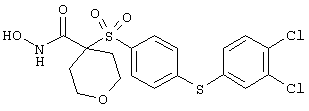
Стадия А: К раствору указанного в заголовке примера 55 соединения (2,0 г, 5,2 ммоль) в N,N-диметилацетамиде (6 мл) добавляли 3,4-дихлортиофенол (1,4 г, 7,8 ммоль), а затем карбонат калия (2,2 г, 15,6 ммоль). Реакционную смесь выдерживали в течение 6 ч при 70° С. После удаления N,N-диметилацетамида в вакууме получали твердое вещество коричневого цвета (5,6 г, количественный выход). После хроматографии (обращенная фаза, колонка С-18, ацетонитрил/вода) получали ТГП-защищенное вещество в растворе.
Стадия Б: К раствору полученного на стадии А неочищенного ТГП-защищенного вещества в смеси ацетонитрил/вода (40 мл) медленно добавляли 10%-ный водный раствор HCl (40 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил удаляли. Собирали образовавшийся осадок, получая указанное в заголовке соединение 6 виде твердого вещества белого цвета (1,50 г, 62%). МС (FAB) M+H: рассчитано для C18H17Cl2NO5S: 463, обнаружено 463.
Пример 61: Получение моногидрохлорида 4-[[4-[[2-амино-4-(трифторметил)фенил]тио]фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
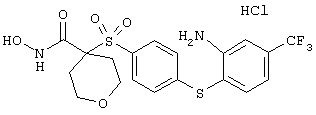
Стадия А: К раствору указанного в заголовке примера 55 соединения (2,0 г, 5,2 ммоль) в N,N-диметилацетамиде (6 мл) добавляли гидрохлорид 2-амино-4-(трифторметил)тиофенола (1,8 г, 7,8 ммоль), а затем карбонат калия (3,6 г, 26 ммоль). Реакционную смесь выдерживали в течение 8 ч при 70° С. После удаления N,N-диметилацетамида в вакууме получали твердое вещество коричневого цвета (14 г, количественный выход). После хроматографии (обращенная фаза, колонка С-18, ацетонитрил/вода) получали ТГП-защищенное вещество в растворе.
Стадия Б: К раствору полученного на стадии А неочищенного ТГП-защищенного вещества в смеси ацетонитрил/вода (40 мл) медленно добавляли 10%-ный водный раствор HCl (40 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил удаляли. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества белого цвета (1,3 г, 52%). МС (FAB) M+H: рассчитано для C18H17Cl2NO6S: 477, обнаружено 477.
Пример 62: Получение моногидрохлорида тетрагидро-4-[[4-(4-фенил-1-пиперидинил)фенил]сульфонил]-2Н-пиран-4-карбоксамида
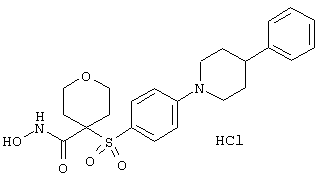
Стадия А: К метанолу (1000 мл), находящемуся в сухом сосуде добавляли в атмосфере азота при 2° С металлический натрий (8,97 г, 0,39 моль). Реакционную смесь перемешивали в течение 45 мин при температуре окружающей среды, в течение этого времени натрий растворялся. Раствор охлаждали до 5° С и добавляли пара-фтортиофенол (41,55 мл, 0,39 ммоль), а затем метил-2-хлорацетат (34,2 мл, 0,39 моль). Реакционную смесь перемешивали в течение 4 ч при температуре окружающей среды, фильтровали и концентрировали в вакууме, получая сульфид в виде прозрачного бесцветного масла (75,85 г, 97%).
Стадия Б: К раствору полученного на стадии А сульфида (75,85 г, 0,38 моль) в метаноле (1000 мл) добавляли при 20° С воду (100 мл) и Охоnе® (720 г, 1,17 моль). При этом происходила экзотермическая реакция, в результате которой температура повышалась до 67° С. Через 2 ч реакционную смесь фильтровали и осадок на фильтре тщательно промывали метанолом. Фильтрат концентрировали в вакууме. Остаток растворяли в этилацетате и промывали соляным раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме, получая сульфон в виде кристаллического твердого вещества (82,74 г, 94%).
Стадия В: К раствору полученного на стадии Б сульфона (28,5 г, 0,123 моль) в N,N-диметилацетамиде (200 мл) добавляли карбонат калия (37,3 г, 0,27 моль), простой бис(2-бромэтиловый) эфир (19,3 мл, 0,147 моль), 4-диметиламинопиридин (0,75 г, 6 ммоль) и бромид тетрабутиламмония (1,98 г, 6 ммоль). Реакционную смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды. Реакционную смесь медленно сливали на 1 н. раствор HCl (300 мл), фильтровали образовавшийся твердый продукт и осадок на фильтре тщательно промывали гексаном. Твердое вещество перекристаллизовывали из этилацетата/гексана, получая пиран в виде твердого вещества бежевого цвета (28,74 г, 77%). МС (ES+) МН+: рассчитано для С13Н15О5S1F1: 303, обнаружено 303.
Стадия Г: К раствору полученного на стадии В пирана (1,21 г, 4,0 ммоль) в диметилсульфоксиде (10 мл) добавляли карбонат цезия (3,26 г, 10 ммоль) и 4-фенилпиперидин (0,64 г, 4,0 ммоль) в метилсульфоксиде (10 мл). Суспензию перемешивали в течение 2 ч при 90° С. Реакционную смесь охлаждали, разбавляли водой и экстрагировали этилацетатом. Объединенные органические слои промывали 5%-ным раствором KHSO4, насыщенным раствором NaHCO3, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Образовавшееся твердое вещество суспендировали в диэтиловом эфире, фильтровали и сушили, получая N-замещенный пиперидин в виде твердого вещества белого цвета (1,2 г, 67%). МС (FAB+) МН+: рассчитано для C24H29N1O5S1: 444, обнаружено 444.
Стадия Д: К суспензии, содержащей полученное на стадии Г N-замещенный пиперидин (815 г, 1,84 ммоль) в метаноле (5 мл) и тетрагидрофуране (5 мл) добавляли 50%-ный раствор гидроксида натрия (3 мл). После выдерживания в течение 24 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме. Суспензию разбавляли водой (10 мл) и добавляли 6 н. раствор HCl до получения значения рН 7. После вакуумной фильтрации образовавшегося осадка получали кислоту в виде твердого вещества белого цвета (705 мг, 89%). МС (FAB+) МН+: рассчитано для C23H27N1O5S1: 430, обнаружено 430.
Стадия Е: Полученную на стадии Д карбоновую кислоту (620 мг, 1,44 ммоль) суспендировали в сухом сосуде в атмосфере азота в метиленхлориде (10 мл) и N,N-диметилформамиде (3 мл) и затем к суспензии добавляли остальные реактивы в следующем порядке: гексафторфосфат бром-трис-пирролидинофосфония (810 мг, 1,73 ммоль), N-метилморфолин (0,5 мл, 4,34 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (190 мг, 1,59 ммоль) После выдерживания в течение 4 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали водой, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали ТГП-защищенный гидроксамат в виде твердого вещества белого цвета (630 мг, 83%). МС (FAB+) МН+: рассчитано для С28Н22N2O6S1: 529, обнаружено 529.
Стадия Ж: К суспензии полученного на стадии Е ТГП-защищенного гидроксамата (600 мг, 1,14 ммоль) в диоксане (1,5 мл) добавляли 4н. раствор HCl в диоксане (1,5 мл) и метанол (1,5 мл). После выдерживания в течение 2 ч при температуре окружающей среды реакционную смесь сливали на диэтиловый эфир (100 мл). После вакуумной фильтрации образовавшегося осадка получали указанное в заголовке соединение в виде твердого вещества светло-бежевого цвета (500 мг, 91%). МС (FAB+) M+Li: рассчитано для C23H28N2O5S1: 445, обнаружено 445.
Пример 63: Получение моногидрохлорида 4-[[4-[4-(1,3-бензодиоксол-5-илокси)-1-пиперидинил)фенил]сульфонил]тетрагидро-N-тидрокси-2Н-пиран-4-карбоксамида
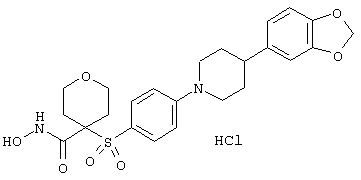
Стадия А: В сухом сосуде в атмосфере азота растворяли 4-гидроксипиперидин (20,2 г, 0,2 моль) в тетрагидрофуране (200 мл) и триэтиламине (29 мл, 0,21 моль). Добавляли раствор ди-трет-бутилдикарбоната (43,65 г, 0,2 моль) с такой скоростью, чтобы температура не превышали 30° С. После перемешивания в течение 4 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали водой, 5%-ным раствором KHSO4, насыщенным раствором NaHCO3, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая ВОС-защищенный пиперидин в виде твердого вещества белого цвета (37,7 г, 94%).
Стадия Б: В сухом сосуде в атмосфере азота полученный на стадии А ВОС-защищенный пиперидин (5,0 г, 24,8 ммоль), растворенный в безводном тетрагидрофуране (100 мл), охлаждали до 0° С и добавляли трифенилфосфин (9,77 г, 37,3 ммоль). После перемешивания в течение 15 мин при 0° С к реакционной смеси добавляли сезамол (5,15 г, 37,3 ммоль), а затем по каплям диэтилазодикарбоксилат (5,87 мл, 37,7 ммоль). Реакционную смесь перемешивали в течение 30 мин при 0° С, а затем в течение 12 ч при температуре окружающей среды. Реакционную смесь концентрировали в вакууме. Остаток суспендировали в диэтиловом эфире, отфильтровывали оксид трифенилфосфина и фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ВОС-защищенный пиперидин в виде твердого вещества белого цвета (3,14 г, 39%).
Стадия В: К суспензии полученного на стадии Б замещенного ВОС-защищенного пиперидина (3,14 г, 9,8 ммоль) в диоксане (15 мл) добавляли 4 н. раствор HCl в диоксане (15 мл). После выдерживания в течение 3 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме. Остаток суспендировали в диэтиловом эфире и после вакуумной фильтрации образовавшегося осадка получали гидрохлорид в виде твердого вещества белого цвета (2,3 г, 100%).
Стадия Г: К суспензии полученного на стадии В гидрохлорида (0,93 г, 3,6 ммоль) в N,N-диметилформамиде (10 мл) добавляли карбонат цезия (2,93 г, 9 ммоль) и указанное в заголовке примера 55 соединение (1,16 г, 3,0 ммоль). Суспензию перемешивали в течение 24 ч при 90° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали водой, 5%-ным раствором KHSO4, насыщенным раствором NаНСО3, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде твердого вещества белого цвета (640 мг, 36%). МС (FAB+) МН+: рассчитано для C29H36N2O9S1: 589, обнаружено 589.
Стадия Д: К суспензии полученного на стадии Г ТГП-защищенного гидроксамата (600 мг, 1,02 ммоль) в диоксане (3 мл) добавляли 4н. раствор HCl в диоксане (3 мл) и метанол (3 мл). После выдерживания в течение 1 ч при температуре окружающей среды реакционную смесь сливали на диэтиловый эфир (100 мл). После вакуумной фильтрации образовавшегося осадка получали указанное в заголовке соединение в виде твердого вещества светло-бежевого цвета (440 мг, 80%). МСВР (ES+) МН+: рассчитано для C24H28N2O8S1: 505,16, обнаружено 505,16.
Пример 64: Получение тетрагидро-N-гидрокси-4-[[4-(4-метоксифенокси)фенил]сульфонил]-2Н-пиран-4-карбоксамида
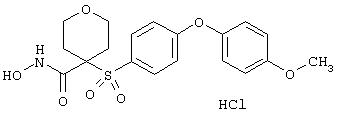
Стадия А: К раствору указанного в заголовке примера 55 соединения (3,48 г, 9 ммоль) в N,N-диметилформамиде (20 мл) добавляли карбонат цезия (8,8 г, 27 ммоль) и пара-метоксифенол (2,23 г, 18 ммоль). Суспензию перемешивали в течение 24 ч при 95° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены бежевого цвета (3,82 г, 86%). МС (FAB+) МН+: рассчитано для C24H29N1O8S1: 492, обнаружено 492.
Стадия Б: К суспензии полученного на стадии А ТГП-защищенного гидроксамата (3,6 г, 7,33 ммоль) в диоксане (18 мл) добавляли 4н. раствор HCl в диоксане (18 мл) и метанол (18 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом, промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Продукт перекристаллизовывали (ацетон/гексан), получая указанное в заголовке соединение в виде твердого вещества светло-бежевого цвета (2,1 г, 70%). МСВР (ES+) МН+: рассчитано для C19H21N1O7S1: 408,11, обнаружено 408,11.
Пример 65: Получение тетрагидро-N-гидрокси-4-[[4-(4-метоксифенилтио)фенил]сульфонил]-2Н-пиран-4-карбоксамида
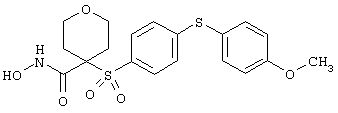
Стадия А: К раствору указанного в заголовке примера 55 соединения (3,1 г, 8 ммоль) в N,N-диметилформамиде (20 мл) добавляли карбонат калия (1,33 г, 9,6 ммоль) и пара-метоксибензолтиол (1,48 г, 12 ммоль). Суспензию перемешивали в течение 24 ч при 65° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены белого цвета (4,1 г, 100%). МСВР (ES+) M+NH
Стадия Б: К суспензии полученного на стадии А ТГП-защищенного гидроксамата (4,0 г, 7,9 ммоль) в диоксане (20 мл) добавляли 4н. раствор HCl в диоксане (20 мл) и метанол (20 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом, промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Продукт перекристаллизовывали (ацетон/гексан), получая указанное в заголовке соединение в виде твердого вещества белого цвета (2,21 г, 67%). МСВР (ES+) МН+: рассчитано для С19Н21N1О6S2: 424,09, обнаружено: 424,09.
Пример 66: Получение 4-[(4-фторфенил)сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
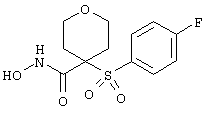
Стадия А: К суспензии указанного в заголовке примера 55 соединения (530 мг, 1,38 ммоль) в диоксане (5 мл) добавляли 4 н. раствор HCl в диоксане (5 мл) и метаноле (5 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь концентрировали в вакууме. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/вода) получали указанное в заголовке соединение в виде твердого вещества бежевого цвета (140 мг, 34%). МСВР (ES+) M+NH4+: рассчитано для C12H14N1O5S1F1: 321,09, обнаружено: 321,09.
Пример 67: Получение моногидрохлорида тетрагидро-N-гидрокси-4-[[4-(4-пиперидинилокси)фенил]сульфонил]-2Н-пиран-4-карбоксамида
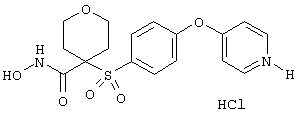
Стадия А: В сухом сосуде в атмосфере азота при 0° С к 60%-ному раствору гидрида натрия (210 мг, 5,25 ммоль) в безводном N,N-диметилформамиде (10 мл) добавляли 4-гидрокси-N-трет-(бутоксикарбонил)пиперидин (844 мг, 4,2 ммоль). Суспензию перемешивали в течение 2 ч при температуре окружающей среды. При 5° С добавляли указанное в заголовке примера 55 соединение (1,35 г, 3,5 ммоль) и реакционную смесь нагревали в течение 3 ч до 50° С. Реакционную смесь охлаждали, прекращали реакцию путем добавления воды и концентрировали в вакууме. Остаток разбавляли этилацетатом, промывали соляным раствором, сушили над Nа2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены белого цвета (283 мг, 14%). МС (FAB+) МН+: рассчитано для C27H40N2O9S1: 569, обнаружено: 569.
Стадия Б: К суспензии полученного на стадии А ТГП-защищенного гидроксамата (530 мг, 0,93 ммоль) в диоксане (5 мл) добавляли 4 н. раствор HCl в диоксане (5 мл) и метаноле (5 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь концентрировали в вакууме. После хроматографии с обращенной фазой (на силикагеле, смесь ацетонитрил/вода, забуференная 0,01%-ным раствором HCl) получали указанное в заголовке соединение в виде твердого вещества бежевого цвета (240 мг, 62%). МСВР (ES+) МН+: рассчитано для С17H24N2О6S1: 385,14, обнаружено: 385,14.
Пример 68: Получение моногидрохлорида тетрагидро-N-гидрокси-4-[[4-[(4-фенилметил)амино]фенил]сульфонил]-2Н-пиран-4-карбоксамида
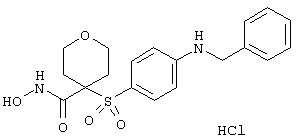
Стадия А: В сосуде для реакции в твердой фазе к смоле II (полученной способом, описанным ниже, 5,0 г, 4,55 ммоль), расплавленной в безводном 1-метил-2-пирролидиноне (40 мл), добавляли бензиламин (11,0 мл, 100 ммоль). Реакционную смесь нагревали в течение 48 ч до 100° С при интенсивном встряхивании. Смолу переносили на фритту и промывали 4 раза N,N-диметилформамидом (30 мл), 4 раза метанолом (30 мл), 4 раза метиленхлоридом (30 мл) и сушили. Высушенную смолу переносили в колбу и добавляли смесь 95% уксусной кислоты/5% воды (50 мл). Суспензию перемешивали в течение 1 ч, фильтровали и осадок на фильтре промывали метиленхлоридом. Объединенные фильтраты концентрировали в вакууме. Остаток растворяли в этилацетате и добавляли насыщенный раствор бикарбоната натрия до получения значения рН 7. Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии с обращенной фазой (на силикагеле, смесь ацетонитрил/вода, забуференная 0,01%-ным раствором HCl) получали указанное в заголовке соединение в виде твердого вещества красноватого цвета (1,01 г, 52%). МСВР (ES+) M+NH
Пример 69: Получение тетрагидро-N-гидрокси-4-[[4-(4-трифторметокси)фенокси)фенил]сульфонил]-2Н-пиран-4-карбоксамида
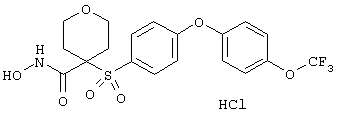
Стадия А: К раствору указанного в заголовке примера 55 соединения (3,1 г, 8 ммоль) в N,N-диметилацетамиде (20 мл) добавляли карбонат цезия (8,8 г, 27 ммоль) и пара - (трифторметокси) фенол (2,1 мл, 16 ммоль). Суспензию перемешивали в течение 19 ч при 95° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены белого цвета (4,2 г, 96%). МСВР (ES+) МН+: рассчитано для C24H26N1O8S1F3: 546,14, обнаружено: 546,14.
Стадия Б: К суспензии полученного на стадии А ТГП-защищенного гидроксамата (4,0 г, 7,3 ммоль) в диоксане (20 мл) добавляли 4 н. раствор HCl в диоксане (20 мл) и метанол (20 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом, промывали водой, сушили над Nа2SO4, фильтровали и концентрировали в вакууме. Продукт перекристаллизовывали (ацетон/гексан), получая указанное в заголовке соединение в виде твердого вещества белого цвета (2,2 г, 65%). МСВР (ES+) M+NH
Пример 70: Получение 4-[[4-(3,5-дифторфенокси)фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
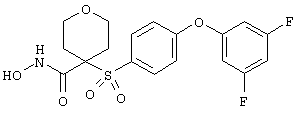
Стадия А: К раствору указанного в заголовке примера 55 соединения (3,1 г, 8 ммоль) в N,N-диметилацетамиде (20 мл) добавляли карбонат цезия (8,8 г, 27 ммоль) и 3,5-дифторфенол (2,1 г, 16 ммоль). Суспензию перемешивали в течение 48 ч при 95° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены бежевого цвета (3,23 г, 81%). МСВР (ES+) МН+: рассчитано для C23H25N1O7S1F2: 498,14, обнаружено: 498,14.
Стадия Б: К суспензии полученного на стадии А ТГП-защищенного гидроксамата (3,2 г, 6,3 ммоль) в диоксане (20 мл) добавляли 4н. раствор HCl в диоксане (20 мл) и метанол (20 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом, промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток суспендировали в диэтиловом эфире и после вакуумной фильтрации образовавшегося осадка получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,5 г, 57%). МСВР (ES+) M+NH
Пример 71: Получение 4-[[4-(3,4-дихлорфенокси)фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
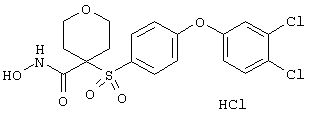
Стадия А: К раствору указанного в заголовке примера 55 соединения (3,1 г, 8 ммоль) в N,N-диметилацетамиде (20 мл) добавляли карбонат цезия (8,8 г, 27 ммоль) и 3,4-дихлорфенол (2,61 г, 16 ммоль). Суспензию перемешивали в течение 41 ч при 95° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены белого цвета (4,17 г, 98%). МСВР (ES+) M+NH
Стадия Б: К суспензии полученного на стадии А ТГП-защищенного гидроксамата (3,5 г, 6,6 ммоль) в диоксане (20 мл) добавляли 4 н. раствор HCl в диоксане (20 мл) и метанол (20 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом, промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток суспендировали в диэтиловом эфире и после вакуумной фильтрации образовавшегося осадка получали указанное в заголовке соединение в виде твердого вещества белого цвета (2,98 г, 100%). МСВР (ES+) M+NH
Пример 72: Получение тетрагидро-N-гидрокси-4-[[4-[4-[(фенилметил)окси]фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
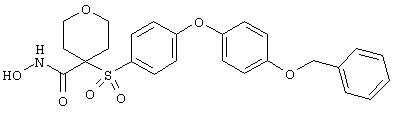
Стадия А: К раствору указанного в заголовке примера 55 соединения (2,7 г, 7 ммоль) в N,N-диметилацетамиде (20 мл) добавляли карбонат цезия (6,84 г, 21 ммоль) и 4-(бензилокси)фенол (2,8 г, 14 ммоль). Суспензию перемешивали в течение 6 ч при 95° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены белого цвета (3,94 г, 99%). МСВР (ES+) M+NH
Стадия Б: К суспензии полученного на стадии А ТГП- защищенного гидроксамата (1,42 г, 2,5 ммоль) в диоксане (6,3 мл) добавляли 4 н. раствор HCl в диоксане (6,3 мл) и метанол (6,3 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом, промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Продукт перекристаллизовывали (ацетон/гексан), получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,56 г, 46%). МСВР (ES+) МН+: рассчитано для C25H25N1O7S1: 484,14, обнаружено: 484,14.
Пример 73: Получение тетрагидро-N-гидрокси-4-[[4-[4-(трифторметокси)фенилтио]фенил]сульфонил]-2Н-пиран-4-карбоксамида

Стадия А: К раствору указанного в заголовке примера 55 соединения (3,1 г, 8 ммоль) в N,N-диметилформамиде (20 мл) добавляли карбонат калия (2,21 г, 16 ммоль) и пара-(трифторметокси)бензолтиол (2,33 г, 12 ммоль). Суспензию перемешивали в течение 2 ч при 70° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде твердого вещества белого цвета (4,4 г, 98%). МСВР (ES+) M+NH
Стадия Б: К суспензии полученного на стадии А ТГП-защищенного гидроксамата (4,15 г, 7,4 ммоль) в диоксане (20 мл) добавляли 4н. раствор HCl в диоксане (20 мл) и метанол (20 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом, промывали водой, сушили над Nа2SO4, фильтровали и концентрировали в вакууме. Продукт перекристаллизовывали (ацетон/гексан), получая указанное в заголовке соединение в виде твердого вещества белого цвета (3,0 г, 85%). МСВР (ES+) M+NH
Пример 74: Получение фенилметил-[4-[[2-(гидроксиамино)-2-оксоэтил]сульфонил]фенил]карбамата
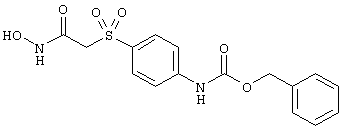
Стадия А: К суспензии 2-(4-аминофенилтио) уксусной кислоты (20,0 г, 0,11 моль) в метаноле (100 мл), охлажденной до 0° С, медленно добавляли тионилхлорид (24,0 мл, 0,33 моль). Добавляли дополнительную порцию метанола (100 мл) и удаляли охлаждающую баню. Затем образовавшуюся смесь выдерживали в течение 2 ч при температуре дефлегмации. Затем реакционную смесь охлаждали до температуры окружающей среды и концентрировали в вакууме. Остаток растворяли в Н2О и нейтрализовали насыщенным раствором NaHCO3. Водную реакционную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали сульфид метилового эфира в виде масла темно-пурпурного цвета (22,75 г, количественный выход).
Стадия Б: К раствору полученного на стадии А сульфида метилового эфира (10,0 г, 50,7 ммоль) в дихлорметане (100 мл) добавляли N-метилморфолин (11,2 мл, 101,4 ммоль), а затем N-(бензилоксикарбонилокси)сукцинимид (12,6 г, 50,7 ммоль). Образовавшуюся смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды и затем концентрировали в вакууме. Остаток растворяли в этилацетате и затем промывали Н2О, 5%-ным раствором KHSO4, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали сульфид бензилоксикарбамата в виде темного масла (16,2 г, 96%).
Стадия В: К раствору, содержащему полученный на стадии Б сульфид бензилоксикарбамата (16,2 г, 48,7 ммоль) в тетрагидрофуране (100 мл) и Н2О (10 мл), добавляли Охоnе® (90,0 г, 146,4 ммоль) и образовавшуюся смесь перемешивали в течение 16 ч при температуре окружающей среды. Затем реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Остаток растворяли в этилацетате, промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали сульфон бензилоксикарбамата в виде твердого вещества рыжевато-коричневого цвета (15,6 г, 88%).
Стадия Г: К раствору полученного на стадии В сульфона бензилоксикарбамата (0,25 г, 0,69 ммоль) в тетрагидрофуране (3 мл) добавляли 50%-ный водный раствор гидроксиламина (1,5 мл). Образовавшуюся смесь перемешивали в течение 24 ч при температуре окружающей среды. Затем смесь разбавляли этилацетатом (30 мл), промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме и последующей промывки горячим диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества светло-розового цвета (0,20 г, 80%). МС МН+: рассчитано для C16H17O6N2S: 365, обнаружено: 365.
Пример 75: Получение N-гидрокси-2-[[4-[[(фениламино)карбонил]амино]фенил]сульфонил]ацетамида
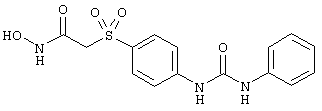
Стадия А: Газообразным водородом барботировали суспензию, содержащую полученный на стадии В примера 74 сульфон бензилоксикарбамата (13,4 г, 36,8 ммоль) и 4%-ный Pd/C в тетрагидрофуране (100 мл). После прекращения поглощения Н2 смесь продували N2 и затем фильтровали через подушку из Celite®, промывая тетрагидрофураном. Фильтрат концентрировали в вакууме, получая анилин в виде твердого вещества коричневого цвета (8,1 г, 96%).
Стадия Б: К суспензии полученного на стадии А анилина (0,50 г, 2,2 ммоль) в дихлорметане (4 мл) добавляли фенилизоцианат (0,36 мл, 3,3 ммоль). Смесь перемешивали при температуре окружающей среды в течение ночи (приблизительно 18 ч) и затем разбавляли дихлорметаном (50 мл). Затем смесь промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали мочевину в виде твердого вещества белого цвета (0,59 г, 78%).
Стадия В: К раствору полученной на стадии Б мочевины (0,32 г, 0,92 ммоль) в тетрагидрофуране (3 мл) добавляли 50%-ный водный раствор гидроксиламина (1,5 мл). Образовавшуюся смесь перемешивали в течение 24 ч при температуре окружающей среды. Затем смесь разбавляли этилацетатом (30 мл), промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме и последующей промывки горячим диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества светло-розового цвета (0,24 г, 76%). МС МН+: рассчитано для C15H16O5N3S: 350, обнаружено: 350.
Пример 78: Получение дигидрохлорида 5-[4-(3,4-диметилфенокси)фенил]сульфонил-N5-гидрокси-1,3-диметилгексагидро-5-пиримидинкарбоксамида
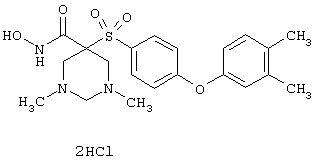
Стадия А: К раствору, содержащему полученное на стадии Б примера 55 соединение (2,00 г, 8,61 ммоль) и 1,3,5-триметилгексагидро-1,3,5-триазин (1,21 мл, 8,61 ммоль) в бензоле (20 мл), медленно добавляли трифторуксусную кислоту (0,66 мл, 8,61 ммоль). Образовавшуюся смесь выдерживали при температуре дефлегмации в течение 1 ч и затем охлаждали до температуры окружающей среды. После этого смесь экстрагировали 2н. HCl. Водный слой нейтрализовали насыщенным раствором NaHCO3 и затем экстрагировали диэтиловым эфиром. Органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали тетрагидропиримидин в виде прозрачного масла (2,31 г, 81%).
Стадия Б: К раствору полученного на стадии А тетрагидропиримидина (1,26 г, 3,81 ммоль) в N,N-диметилформамиде (5,0 мл) добавляли 3,4-диметилфенол (0,559 г, 4,58 ммоль) и Cs2CO3 (3,72 г, 11,43 ммоль). Образовавшуюся смесь выдерживали при 90° С в течение 16 ч. После охлаждения до температуры окружающей среды реакционную смесь разбавляли Н2О и экстрагировали этилацетатом. Органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат) получали простой биариловый эфир в виде масла светло-янтарного цвета (1,40 г, 85%).
Стадия В: К раствору полученного на стадии Б простого биарилового эфира (0,936 г, 2,16 ммоль) в тетрагидрофуране (5,0 мл) добавляли триметилсиланолат калия (0,360 г, 2,81 ммоль). Образовавшуюся смесь перемешивали в течение 48 ч при температуре окружающей среды и затем удаляли растворитель. Образовавшийся остаток растворяли в дихлорметане (5,0 мл), после чего добавляли N-метилморфолин (0,712 мл, 6,48 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,278 г, 2,38 ммоль). После перемешивания в течение 10 мин при температуре окружающей среды добавляли РуВrоР® (1,21 г, 2,59 ммоль). Образовавшуюся смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды, затем разбавляли дихлорметаном (50 мл) и промывали Н2О. Отделяли органический слой и промывали насыщенным раствором NaCl и сушили над Nа2SO4. После хроматографии (на силикагеле, этилацетат) получали гидроксамат в виде твердого вещества белого цвета (0,970 г, 87%).
Стадия Г: К раствору полученного на стадии В гидроксамата (0,667 г, 1,29 ммоль) в диоксане (3,0 мл) и метаноле (1,0 мл) добавляли раствор 4 н. HCl в диоксане (3,22 мл, 12,9 ммоль). После перемешивания в течение 30 мин при температуре окружающей среды реакционную смесь концентрировали в вакууме. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О/трифторуксусная кислота) получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,379 г, 58%). МС МН +: рассчитано для С21Н28О5N3S: 434, обнаружено: 434.
Пример 79: Получение моногидрохлорида 4-[[4-(4-хлор-3-метилфенокси)фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
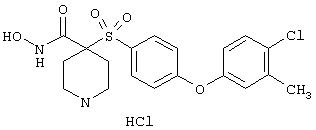
Стадия А: К суспензии изонипекотиновой кислоты (50,0 г, 0,39 моль) в метаноле (300 мл), охлажденной до 0° С, медленно по каплям добавляли тионилхлорид (85 мл, 1,16 моль). После завершения добавления удаляли охлаждающую баню и смесь выдерживали при температуре дефлегмации в течение 2 ч. После охлаждения до температуры окружающей среды реакционную смесь концентрировали в вакууме. Образовавшиеся твердые частицы суспендировали в этилацетате и затем промывали насыщенным раствором NaHCO3. Водный слой концентрировали в вакууме и образовавшиеся твердые частицы растворяли в горячем этилацетате и сливали из раствора солей. После этого органические слои концентрировали в вакууме, получая метиловый эфир в виде твердого вещества белого цвета (55,4 г, количественный выход).
Стадия Б: К раствору ди-трет-бутилдикарбоната (15,3 г, 70,0 ммоль) в тетрагидрофуране (100 мл) добавляли полученный на стадии А метиловый эфир (10,0 г, 70,0 ммоль). Образовавшуюся смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды и затем концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали метиловый эфир Воc-защищенного пиперидина в виде масла светло-желтого цвета (10,1 г, 59%).
Стадия В: К раствору полученного на стадии Б метилового эфира Вос-защищенного пиперидина (23,31 г, 0,096 моль) в тетрагидрофуране (500 мл), охлажденному до -40° С, медленно добавляли диизопропиламид лития (57,5 мл, 2,0 М в ТГФ, 0,115 моль). Образовавшуюся смесь перемешивали в течение 1 ч при -40° С и затем в течение 30 мин при 0° °С. Затем смесь повторно охлаждали до -40° С и медленно добавляли раствор полученного на стадии А примера 6 дисульфида (24,37 г, 0,096 моль) в тетрагидрофуране (60 мл). Образовавшуюся смесь медленно нагревали в течение ночи (приблизительно 18 ч) до температуры окружающей среды, после чего добавляли Н2О (200 мл). Затем смесь концентрировали в вакууме и водный слой экстрагировали этилацетатом. Органические слои промывали 0,5 М раствором NaOH, Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфид в виде масла янтарного цвета (28,1 г, 79%).
Стадия Г: К раствору полученного на стадии В сульфида (28,2 г, 0,076 моль) в дихлорметане (250 мл), охлажденному до 0° С, добавляли мета-хлорпероксибензойную кислоту (48 г, 0,152 моль). Образовавшуюся смесь перемешивали в течение 1 ч при 0° С, а затем в течение 2,5 ч при температуре окружающей среды. После этого смесь разбавляли Н2О и 10%-ным раствором NH4OH. Органический слой промывали 10%-ным раствором NH4OH, Н2О и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали сульфон в виде твердого вещества белого цвета (24,7 г, 81%).
Стадия Д: К раствору полученного на стадии Г сульфона (3,00 г, 7,47 ммоль) в N,N-диметилформамиде (15 мл) добавляли 4-хлор-3-метилфенол (1,28 г, 8,96 ммоль) и Сs2СО3 (7,30 г, 22,42 ммоль). Образовавшуюся смесь выдерживали в. течение 8 ч при 80° С. Затем смесь концентрировали в вакууме и остаток распределяли между Н2О и этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали простой биариловый эфир в виде прозрачного масла (3,26 г, 83%).
Стадия Е: К раствору полученного на стадии Д простого биарилового эфира (3,17 г, 6,05 ммоль) в тетрагидрофуране (30 мл) добавляли триметилсиланолат калия (1,01 г, 7,87 ммоль). Образовавшуюся смесь перемешивали в течение 20 ч при температуре окружающей среды. Добавляли дополнительную порцию тетрагидрофурана (40 мл) и смесь перемешивали в течение 36 ч при температуре окружающей среды. Добавляли дополнительную порцию триметилсиланолата калия (0,233 г, 1,82 ммоль) и смесь перемешивали в течение 23 ч при температуре окружающей среды. Удаляли тетрагидрофуран и образовавшийся остаток суспендировали в дихлорметане (30 мл). К суспензии добавляли N-метилморфолин (2,00 мл, 18,15 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,780 г, 6,66 ммоль), а затем РуВrоР® (3,38 г, 7,26 ммоль). Смесь перемешивали в течение 24 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органический слой промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали гидроксамат в виде пены беловатого цвета (2,98 г, 81%).
Стадия Ж: К раствору полученного на стадии Е гидроксамата (2,98 г, 4,89 ммоль) в диоксане (14 мл) и метаноле (6 мл) добавляли раствор 4 н. HCl в диоксане (10 мл). Образовавшуюся смесь перемешивали в течение 3,5 ч при температуре окружающей среды, затем добавляли диэтиловый эфир (40 мл) и осадок собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества светло-розового цвета (2,00 г, 88%). МС МН+: рассчитано для С19Н22O5N2ClS: 425, обнаружено: 425.
Пример 80: Получение моногидрохлорида 4-[[4-(4-хлор-3-метилфенокси)фенил]сульфонил]-4-(гидроксиамино)карбонил]-1-пиперидинуксусной кислоты

Стадия А: К суспензии указанного в заголовке примера 79 соединения (0,250 г, 0,542 ммоль) в ацетонитриле (4,0 мл) добавляли трет-бутилбромацетат (0,088 мл, 0,542 ммоль) и К2СО3 (0,150 г, 1,08 ммоль). Образовавшуюся смесь перемешивали в течение 18 ч при температуре окружающей среды, после чего фильтровали через подушку из Celite®, промывая этилацетатом. Затем фильтрат концентрировали в вакууме. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О/трифторуксусная кислота) получали сложный трет-бутиловый эфир в виде твердого вещества белого цвета (0,156 г, 53%).
Стадия Б: Полученный на стадии А сложный трет-бутиловый эфир (0,156 г, 0,289 ммоль) обрабатывали раствором 4 н. HCl в диоксане (1,5 мл) и образовавшуюся смесь перемешивали в течение 3,5 ч при температуре окружающей среды, после чего добавляли дополнительную порцию диоксана (2 мл). После перемешивания в течение 8 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме. Остаток вновь обрабатывали в течение 4 ч при температуре окружающей среды раствором 4 н. HCl в диоксане (1,5 мл). К реакционной смеси добавляли диэтиловый эфир и осадок собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества беловатого цвета (0,111 г, 74%). МС МН+: рассчитано для С21Н24O7N2SCl: 483, обнаружено: 483.
Пример 81: Получение моногидрохлорида 4-[[4-(4-хлор-3-метилфенокси)фенил]сульфонил]-N-гидрокси-1-(2-пропинил)-4-пиперидинкарбоксамида

Стадия А: К суспензии указанного в заголовке примера 79 соединения (0,500 г, 1,08 ммоль) в ацетонитриле (8,0 мл) добавляли пропаргилбромид (0,126 мл, 80%-ный раствор в толуоле, 1,13 ммоль) и К2СО3 (0,300 г, 2,17 ммоль). Образовавшуюся смесь перемешивали в течение 24 ч при температуре окружающей среды, после чего фильтровали через подушку из Celite®, промывая метанолом, и затем фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат) получали N-пропаргилгидроксамат в виде твердого вещества рыжевато-коричневого цвета (0,200 г, 40%).
Стадия Б: К раствору полученного на стадии А N-пропаргилгидроксамата (0,200 г, 0,432 ммоль) в ацетонитриле (3,0 мл) и Н2О (0,5 мл) добавляли концентрированную HCl (0,05 мл). Образовавшуюся смесь перемешивали в течение 5 мин при температуре окружающей среды и концентрировали в вакууме, получая указанное в заголовке соединение в виде твердого вещества розового цвета (0,200 г, 93%). МС МН+: рассчитано для C22H24O5N2SCl: 463, обнаружено: 463.
Пример 82: Получение моногидрохлорида 4-[[4-(4-хлор-3-метилфенокси)фенил]сульфонил]-N-гидрокси-1-(2-пропенил)-4-пиперидинкарбоксамида
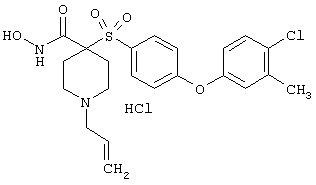
Стадия А: К суспензии указанного в заголовке примера 79 соединения (0,500 г, 1,08 ммоль) в ацетонитриле (8,0 мл) добавляли аллилбромид (0,093 мл, 1,08 ммоль) и К2СО3 (0,300 г, 2,17 ммоль). Образовавшуюся смесь перемешивали в течение 22 ч при температуре окружающей среды. Добавляли дополнительную порцию аллилбромида (0,054 мл, 1 М раствор в ацетонитриле, 0,054 ммоль) и продолжали перемешивание в течение 6 ч при температуре окружающей среды. Образовавшуюся смесь фильтровали через подушку из Celite®, промывая этилацетатом, и затем фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, метанол/этилацетат) получали N-аллилгидроксамат в виде твердого вещества беловатого цвета (0,080 г, 15%).
Стадия Б: К раствору полученного на стадии А N-аллилгидроксамата (0,080 г, 0,172 ммоль) в ацетонитриле (3,0 мл) и Н2О (1,0 мл) добавляли концентрированную HCl (0,05 мл). Образовавшуюся смесь перемешивали в течение 10 мин при температуре окружающей среды и затем концентрировали в вакууме, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,100 г, количественный выход). МС МН+: рассчитано для С22Н26О5N2SCl: 465, обнаружено: 465.
Пример 83: Получение моногидрохлорида 4-[[4-(4-фтор-3-метилфенокси)фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
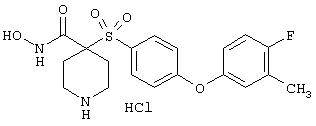
Стадия А: К раствору полученного на стадии Г примера 79 сульфона (5,00 г, 12,45 ммоль) в тетрагидрофуране (100 мл) добавляли триметилсиланолат калия (4,79 г, 37,36 ммоль). Образовавшуюся смесь перемешивали в течение 1,5 ч при температуре окружающей среды, разбавляли Н2О и диэтиловым эфиром (100 мл). Водный слой экстрагировали диэтиловым эфиром и объединенные органические слои промывали Н2О. Водные слои объединяли, подкисляли 2 н. HCl (до рН 2) и затем экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4, получая кислоту в виде твердого вещества беловатого цвета (4,61 г, 96%).
Стадия Б: К суспензии полученной на стадии А кислоты (0,830 г, 2,14 ммоль) в дихлорметане (10 мл) добавляли N-метилморфолин (0,706 мл, 6,42 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,276 г, 2,35 ммоль). После перемешивания в течение 5 мин при температуре окружающей среды добавляли РуВrоР® (1,20 г, 2,57 ммоль) и образовавшуюся смесь перемешивали в течение 19 ч при температуре окружающей среды. Смесь концентрировали в вакууме и остаток распределяли между Н2О и этилацетатом. Затем водный слой экстрагировали этилацетатом и объединенные органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали пара-фторсульфон в виде кристаллического твердого вещества белого цвета (0,993 г, 95%).
Стадия В: К раствору полученного на стадии Б пара-фторсульфона (0,485 г, 0,996 ммоль) в N,N-диметилформамиде (5 мл) добавляли 4-фтор-3-метилфенол (0,133 мл, 1,20 ммоль) и Cs2CO3 (0,973 г, 2,99 ммоль). Образовавшуюся смесь выдерживали при 60° С в течение 17 ч. Добавляли дополнительную порцию 4-фтор-3-метилфенола (0,055 мл, 0,498 ммоль) и температуру реакционной смеси повышали до 80° С в течение 4 ч и до 100° С в течение 3 ч. Добавляли дополнительную порцию 4-фтор-3-метилфенола (0,133 мл, 1,20 ммоль) и реакционную смесь выдерживали в течение 7,5 ч при 100° С. Добавляли дополнительную порцию Cs2CO3 и смесь продолжали выдерживать при 100° С в течение 17 ч. Реакционную смесь охлаждали до температуры окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества беловатого цвета (0,490 г, 83%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (0,479 г, 0,808 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли раствор 4 н. HCl в диоксане (2,02 мл, 8,08 ммоль). Образовавшуюся смесь перемешивали в течение 1,5 ч при температуре окружающей среды. Добавляли диэтиловый эфир (5 мл) и осадок собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества беловатого цвета (0,323 г, 90%). МС МН+: рассчитано для С19Н22О5N2SF: 409, обнаружено: 409.
Пример 84: Получение моногидрохлорида 4-[[4-(3-хлор-4-фторфенокси)фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
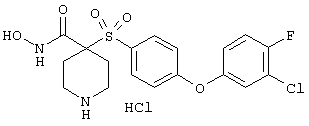
Стадия А: К раствору полученного на стадии Б примера 83 пара-фторсульфона (0,485 г, 0,996 ммоль) в N,N-диметилформамиде (5,0 мл) добавляли 4-фтор-3-хлорфенол (0,176 г, 1,20 ммоль) и Cs2CO3 (0,973 г, 2,99 ммоль). Образовавшуюся смесь выдерживали в течение 17 ч при 60° С, затем добавляли дополнительную порцию 4-фтор-3-хлорфенола (0,073 г, 0,498 ммоль) и реакционную смесь выдерживали в течение 24 ч при 80° С, а затем температуру повышали до 90° С. После выдерживания в течение 7 ч при 90° С добавляли дополнительную порцию 4-фтор-3-хлорфенола (0,176 г, 1,20 ммоль) и продолжали выдерживать при 90° С в течение 7,5 ч. Добавляли дополнительную порцию Cs2CO3 (0,973 г, 2,99 ммоль) и смесь продолжали выдерживать при 90° С в течение 24 ч. После охлаждения до температуры окружающей среды реакционную смесь концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества беловатого цвета (0,550 г, 90%).
Стадия Б: К раствору полученного на стадии А защищенного гидроксамата (0,530 г, 0,864 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли раствор 4 н. HCl в диоксане (2,00 мл, 8,00 ммоль). Образовавшуюся смесь перемешивали в течение 1,5 ч при температуре окружающей среды. Добавляли диэтиловый эфир (5 мл) и осадок собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества беловатого цвета (0,377 г, 94%). МС МН+: рассчитано для С19Н19О5N2SFСl: 429, обнаружено: 429.
Пример 85: Получение моногидрохлорида 4-[[4-(4-хлорфенокси)фенил]сульфонил]-N-гидрокси-1-(2-пропинил)-4-пиперидинкарбоксамида
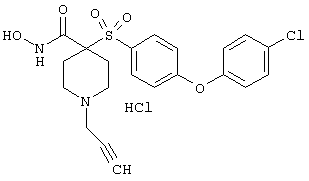
Стадия А: К раствору полученного на стадии Г примера 79 сульфона (4,53 г, 11,28 ммоль) в N,N-диметилформамиде (20 мл) добавляли 4-хлорфенол (4,41 г, 13,54 ммоль) и Cs2CO3 (11,03 г, 33,85 ммоль). Образовавшуюся смесь выдерживали в течение 5 ч при 90° С. После охлаждения до температуры окружающей среды реакционную смесь концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали простой биариловый эфир в виде твердого вещества белого цвета (4,60 г, 78%).
Стадия Б: К раствору полученного на стадии А простого биарилового эфира (4,57 г, 8,96 ммоль) в диоксане (10 мл) добавляли раствор 4 н. HCl в диоксане (10 мл). Образовавшуюся смесь перемешивали в течение 2,5 ч при температуре окружающей среды, после чего добавляли дополнительную порцию диоксана (10 мл). После перемешивания в течение 1,5 ч при температуре окружающей среды смесь концентрировали в вакууме. Образовавшееся твердое вещество суспендировали в диоксане (20 мл) и повторно обрабатывали раствором 4 н. HCl в диоксане (10 мл). Смесь перемешивали в течение 1 ч при температуре окружающей среды, добавляли метанол (1 мл) и продолжали перемешивание при температуре окружающей среды. Через 1 ч смесь концентрировали в вакууме, получая амин в виде твердого вещества белого цвета (4,09 г, количественный выход).
Стадия В: К суспензии полученного на стадии Б амина (4,00 г, 8,96 ммоль) в ацетонитриле (20 мл) добавляли пропаргилбромид (1,05 мл, 80%-ный раствор в толуоле, 9,41 ммоль) и К2СО3 (2,60 г, 18,82 ммоль). Образовавшуюся смесь перемешивали в течение 18 ч при температуре окружающей среды, фильтровали через подушку из Celite®, промывая этилацетатом, и затем фильтрат концентрировали в вакууме, получая N-пропаргиламин в виде клейкой пены (4,14 г, количественный выход).
Стадия Г: К суспензии полученного на стадии В N-пропаргиламина (4,14 г, 8,96 ммоль) в тетрагидрофуране (20 мл) добавляли триметилсиланолат калия (1,26 г, 9,86 ммоль). Образовавшуюся смесь перемешивали в течение 17 ч при температуре окружающей среды, после чего добавляли дополнительные порции тетрагидрофурана (5 мл) и триметилсиланолата калия (0,350 г, 2,73 ммоль). После перемешивания в течение 4 ч при температуре окружающей среды добавляли еще одну порцию тетрагидрофурана (5 мл) и продолжали перемешивание при температуре окружающей среды в течение 24 ч. Затем добавляли еще одну порцию триметилсиланолата калия (0,115 г, 0,896 ммоль) и смесь перемешивали в течение 24 ч при температуре окружающей среды, после чего добавляли дополнительную порцию триметилсиланолата калия и образовавшуюся смесь перемешивали при температуре окружающей среды еще в течение 24 ч. Удаляли тетрагидрофуран и остаток суспендировали в дихлорметане (20 мл).
Стадия Д: К дихлорметановой суспензии добавляли N-метилморфолин (2,96 мл, 26,9 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (1,15 г, 9,86 ммоль), а затем РуВrоР® (5,01 г, 10,75 ммоль). Образовавшуюся смесь перемешивали при температуре окружающей среды в течение ночи и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде пены беловатого цвета (3,29 г, 69%).
Стадия Е: К раствору полученного на стадии Д защищенного гидроксамата (3,27 г, 6,13 ммоль) в диоксане (21 мл) и метаноле (7 мл) добавляли раствор 4 н. HCl в диоксане (10 мл). Образовавшуюся смесь перемешивали в течение 4 ч при температуре окружающей среды и затем добавляли диэтиловый эфир (75 мл). Твердые частицы собирали фильтрацией, промывая диэтиловым эфиром, в результате чего получали указанное в заголовке соединение в виде твердого вещества беловатого цвета (2,95 г, 99%). МС МН+: рассчитано для C21H22O5N2SCl: 449, обнаружено: 449.
Пример 86: Получение моногидрохлорида 4-[[4-(фенилтио)фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида

Стадия А: К раствору полученного на стадии Г примера 79 сульфона (0,500 г, 1,25 ммоль) в N,N-диметилформамиде (3,0 мл) добавляли тиофенол (0,154 мл, 1,50 ммоль) и К2СО3 (0,518 г, 3,75 ммоль). Образовавшуюся смесь перемешивали в течение 24 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали биариловый тиоэфир в виде прозрачного клейкого масла (0,480 г, 78%).
Стадия Б: К раствору полученного на стадии А биарилового тиоэфира (2,01 г, 4,09 ммоль) в тетрагидрофуране (40 мл) добавляли триметилсиланолат калия (0,682 г, 5,31 ммоль). Образовавшуюся смесь перемешивали в течение 23 ч при температуре окружающей среды и затем концентрировали в вакууме. Затем остаток суспендировали в дихлорметане (20 мл), после чего добавляли N-метилморфолин (1,35 мл, 12,27 ммоль) и 50%-ный водный раствор гидроксиламина (0,265 мл, 4,50 ммоль), а затем РуВrоР® (2,29 г, 4,91 ммоль). Образовавшуюся смесь перемешивали в течение 16 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. Часть образца подвергали хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О/трифторуксусная кислота) получая гидроксамат в виде твердого вещества беловатого цвета (0,190 г).
Стадия В: К раствору полученного на стадии А гидроксамата (0,181 г, 0,367 ммоль) в диоксане (5 мл) и метаноле (1 мл) добавляли раствор 4 н. HCl в диоксане (3 мл). Образовавшуюся смесь перемешивали в течение 3 ч при температуре окружающей среды и затем концентрировали в вакууме, получая указанное в заголовке соединение в виде твердого вещества беловатого цвета (0,170 г, количественный выход). МС МН+: рассчитано для С18Н21O4N2S2: 393, обнаружено:393.
Пример 87: Получение моногидрохлорида 4-[(гидроксиамино)карбонил]4-[[4-(фенилтио)фенил]сульфонил]-1-пиперидинуксусной кислоты
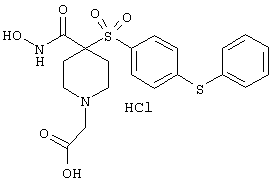
Стадия А: К раствору полученного в примере 86 соединения (0,322 г, 0,751 ммоль) в ацетонитриле (4,0 мл) добавляли трет-бутилбромацетат (0,121 мл, 0,751 ммоль) и K2CO3 (0,207 г, 1,50 ммоль). Образовавшуюся смесь перемешивали в течение 18 ч при температуре окружающей среды, фильтровали через подушку из Celite®, промывали этилацетатом и фильтрат концентрировали в вакууме. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О/трифторуксусная кислота) получали сложный трет-бутиловый эфир в виде твердого вещества беловатого цвета (0,150 г, 40%).
Стадия Б: Полученный на стадии А сложный трет-бутиловый эфир (0,145 г, 0,286 ммоль) обрабатывали раствором 4 н. HCl в диоксане (3,0 мл). Образовавшуюся смесь перемешивали в течение 7 ч при температуре окружающей среды, добавляли диэтиловый эфир и осадок собирали фильтрацией. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О/НСl) получали указанное в заголовке соединение в виде твердого вещества беловатого цвета (0,060 г, 43%). МС МН+: рассчитано для C20H23O6N2S2:451, обнаружено: 451.
Пример 88: Получение моногидрохлорида 4-[[4-(4-хлорфенокси)фенил]сульфонил]-4-[(гидроксиамино)карбонил]-1-пиперидинуксусной кислоты
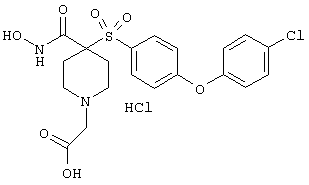
Стадия А: К суспензии гидробромида 4-бромпиперидина (40,0 г, 0,16 моль) в тетрагидрофуране (200 мл) медленно добавляли триэтиламин (45,4 мл, 0,33 моль), а затем ди-трет-бутилдикарбонат (37,4 г, 0,17 моль), который добавляли в в виде нескольких порций. Образовавшуюся смесь перемешивали в течение 17 ч при температуре окружающей среды, затем фильтровали и концентрировали в вакууме. Твердые частицы промывали гексаном и затем собирали фильтрацией, получая Воc-защищенное пиперидиновое производное в виде масла янтарного цвета (45,8 г, >100%).
Стадия Б: К раствору 4-фторфенола (25,0 г, 0,20 моль) в ацетоне (150 мл), дегазированному с помощью N2, добавляли Cs2CO3 (79,7 г, 0,25 моль). После дегазации в течение 5 мин образовавшейся смеси с помощью N2 добавляли полученное на стадии А Воc-защищенное пиперидиновое производное (43,1 г, 0,16 моль). Образовавшуюся смесь перемешивали в течение 22 ч при температуре окружающей среды и затем фильтровали через подушку из Celite®, промывая ацетоном. Остаток промывали диэтиловым эфиром и твердые частицы собирали фильтрацией, получая сульфид в виде масла желтого цвета (47,6 г, 93%).
Стадия В: К раствору полученного на стадии Б сульфида (47,3 г, 0,15 моль) в дихлорметане (350 мл), охлажденному до 0° С, добавляли мета-хлорпероксибензойную кислоту (80 г, 57-86%). Добавляли дополнительную порцию дихлорметана (50 мл) и смесь перемешивали в течение 1 ч при 0° С и затем в течение 1,5 ч при температуре окружающей среды. Реакционную смесь разбавляли Н2О и добавляли водный раствор мета-бисульфита натрия (4,0 г в 50 мл). Смесь концентрировали в вакууме и затем экстрагировали диэтиловым эфиром и этилацетатом. Объединенные органические слои промывали 10%-ным раствором NH4OH, насыщенным раствором NaCl и сушили над Na2SO4. После перекристаллизации из этилацетата получали сульфон в виде твердого вещества белого цвета (18,9 г, 36%).
Стадия Г: К раствору полученного на стадии В сульфона (8,00 г, 23,3 моль) в N,N-диметилформамиде (40 мл) добавляли 4-хлорфенол (3,59 г, 27,96 ммоль) и К2СО3 (22,77 г, 69,90 ммоль). Образовавшуюся смесь выдерживали в течение 4 ч при 60° С, а затем в течение 7 ч повышали температуру до 80° С. Реакционную смесь охлаждали до температуры окружающей среды и затем концентрировали в вакууме. К остатку добавляли Н2О (100 мл) и твердые частицы собирали фильтрацией, получая простой биариловый эфир в виде твердого вещества беловатого цвета (10,5 г, 99%).
Стадия Д: К раствору полученного на стадии Г простого биарилового эфира (5,00 г, 11,1 ммоль) в тетрагидрофуране (50 мл), охлажденному до 0° С, добавляли бис (триметилсилил)амид лития (13,3 мл, 1 М раствор в тетрагидрофуране, 13,3 ммоль) с такой скоростью, чтобы температура реакционной смеси ни разу не поднималась выше 2° С. Образовавшуюся смесь перемешивали в течение 30 мин при 0° С, затем медленно добавляли диметилкарбонат (1,40 мл, 16,6 ммоль) с такой скоростью, чтобы температура реакционной смеси ни разу не поднималась выше 2° С. После этого реакционной смеси давали меделенно нагреться до температуры окружающей среды.
Через 17 ч реакционную смесь повторно охлаждали до 0° С и добавляли дополнительную порцию бис(триметилсилил)амида лития (5,50 мл, 1 М раствор в тетрагидрофуране, 5,50 ммоль) с такой скоростью, чтобы температура реакционной смеси ни разу не поднималась выше 2° С. После перемешивания в течение 30 мин добавляли диметилкарбонат (0,048 мл, 0,570 ммоль) и продолжали перемешивание в течение 45 мин при 0° С. Медленно добавляли дополнительную порцию бис(триметилсилил)амида лития (0,500 мл, 1 М раствор в тетрагидрофуране, 0,500 ммоль), а через 1 ч добавляли дополнительную порцию диметилкарбоната (0,010 мл, 0,119 ммоль). После перемешивания в течение 20 мин при 0° С добавляли насыщенный раствор NH4Cl и затем реакционную смесь концентрировали в вакууме. Остаток разбавляли Н2О и экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным NaCl и сушили над Na2SO4. После перекристаллизации из метанола получали сложный метиловый эфир в виде кристаллического твердого вещества белого цвета (3,56 г, 63%).
Стадия Е: К полученному на стадии Д сложному метиловому эфиру (3,54 г, 6,94 ммоль) в диоксане (18 мл) и метаноле (6 мл) добавляли раствор 4 н. HCl в диоксане (10,0 мл). Образовавшуюся смесь перемешивали в течение 5 ч при температуре окружающей среды и затем концентрировали в вакууме, получая амин в виде твердого вещества беловатого цвета (3,10 г, количественный выход).
Стадия Ж: К раствору полученного на стадии Е амина (1,50 г, 3,36 ммоль) в ацетонитриле (15 мл) добавляли трет-бутилбромацетат (0,570 мл, 3,53 ммоль) и К2СО3 (1,16 г, 8,40 ммоль). Образовавшуюся смесь перемешивали в течение 3 ч при температуре окружающей среды, затем фильтровали через подушку из Celite®, промывая этилацетатом. Фильтрат концентрировали в вакууме, получая сложный трет-бутиловый эфир в виде масла светло-желтого цвета (1,83 г, >100%).
Стадия З: К раствору полученного на стадии Ж сложного трет-бутилового эфира (1,76 г, 3,36 ммоль) в тетрагидрофуране (15 мл) добавляли триметилсиланолат калия (0,475 г, 3,70 ммоль). Образовавшуюся смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды, затем добавляли дополнительную порцию тетрагидрофурана (10 мл). После перемешиваний в течение ночи (приблизительно 18 ч) при температуре окружающей среды добавляли дополнительную порцию триметилсиланолата калия (0,475 г, 3,70 ммоль). Образовавшуюся смесь перемешивали в течение 4 ч при температуре окружающей среды, после чего разбавляли Н2О. Реакционную смесь подкисляли (рН 7) с помощью 1н. раствора HCl и затем концентрировали в вакууме. Твердые частицы промывали диэтиловым эфиром и затем Н2О, получая кислоту в виде твердого вещества беловатого цвета (0,597 г, 32%).
Стадия И: К суспензии полученной на стадии З кислоты (0,597 г, 1,17 ммоль) в дихлорметане (5 мл) добавляли N-метилморфолин (0,386 мл, 3,51 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,151 г, 1,29 ммоль), а затем РуВrоР® (0,655 г, 1,40 ммоль). Образовавшуюся смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде пены белого цвета (0,510 г, 72%).
Стадия К: Полученный на стадии И защищенный гидроксамат (0,510 г, 0,837 ммоль) обрабатывали 4 н. раствором HCl в диоксане (10 мл). Образовавшуюся смесь перемешивали в течение 24 ч при температуре окружающей среды, затем добавляли диэтиловый эфир (20 мл) и твердые частицы собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,370 г, 87%). МС МН+: рассчитано для C20H22O7N2SCl: 469, обнаружено: 469.
Пример 89: Получение дигидрохлорида 4-[[4-(4-хлорфенокси)фенил]сульфонил]-N-гидрокси-1-[2-(4-морфолинил)этил]-4-пиперидинкарбоксамида
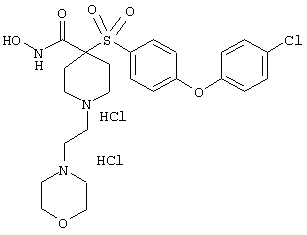
Стадия А: К раствору полученного на стадии Е примера 88 амина (1,00 г, 2,24 ммоль) в ацетонитриле (10 мл) добавляли 4-(2-хлорэтил)морфолин (0,438 г, 2,35 моль) и К2СО3 (1,24 г, 8,96 ммоль). Образовавшуюся смесь перемешивали в течение 1,5 ч при температуре окружающей среды, после чего добавляли каталитическое количество NaI и продолжали перемешивание в течение 21 ч при температуре окружающей среды. Затем температуру реакционной смеси повышали в течение 29 ч до 60° С. После охлаждения до температуры окружающей среды реакционную смесь фильтровали через подушку из Celite®, промывая этилацетатом. Фильтрат концентрировали в вакууме, получая сложный эфир в виде маслянистого твердого вещества (1,15 г, 98%).
Стадия Б: К раствору полученного на стадии А сложного эфира (1,15 г, 2,20 ммоль) в тетрагидрофуране (10 мл) добавляли триметилсиланолат калия (0,579 г, 4,51 ммоль). Смесь перемешивали в течение 4 ч при температуре окружающей среды, затем добавляли дополнительную порцию тетрагидрофурана (10 мл) и продолжали перемешивание в течение ночи (приблизительно 18 ч) при температуре окружающей среды. Реакционную смесь разбавляли Н2О (10 мл) и подкисляли (до рН 7) с помощью 1н. раствора HCl. Образовавшийся осадок собирали фильтрацией, получая кислоту в виде твердого вещества серого цвета (0,753 г, 72%).
Стадия В: К суспензии полученной на стадии Б кислоты (0,750 г, 1,47 ммоль) в дихлорметане (7 мл) добавляли N-метилморфолин (0,500 мл, 4,55 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,198 г, 1,62 ммоль), а затем РуВrоР® (0,822 г, 1,76 ммоль). Образовавшуюся смесь перемешивали в течение 24 ч при температуре окружающей среды и затем добавляли дополнительные порции N-метилморфолина (0,242 мл, 2,21 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламина (0,052 г, 0,441 ммоль), а затем РуВгоР® (0,343 г, 0,735 ммоль). Образовавшуюся смесь перемешивали в течение 23 ч при температуре окружающей среды и затем добавляли дополнительные порции O-тетрагидро-2Н-пиран-2-илгидроксиламина (0,017 г, 0,145 ммоль) и РуВrоР® (0,073 г, 0,157 ммоль). Образовавшуюся смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Nа2SO4. После хроматографии (на силикагеле, метанол/хлороформ) получали защищенный гидроксамат в виде твердого вещества беловатого цвета (0,750 г, 84%).
Стадия Г: Полученный на стадии В защищенный гидроксамат (0,730 г, 1,20 ммоль) обрабатывали 4 н. раствором HCl в диоксане (10 мл) и метаноле (10 мл). Образовавшуюся смесь перемешивали в течение 1 ч при температуре окружающей среды, затем добавляли диэтиловый эфир (20 мл) и твердые частицы собирали фильтрацией, получая указанное в заголовке соедиение в виде твердого вещества светло-желтого цвета (0,625 г, 87%). МС МН+: рассчитано для C24H31O6N3SCl: 525, обнаружено 525.
Пример 90: Получение 4-[[4-(4-хлорфенокси)фенил]сульфонил]-N-гидрокси-N-1-(метилэтил)-1,4-пиперидиндикарбоксамида
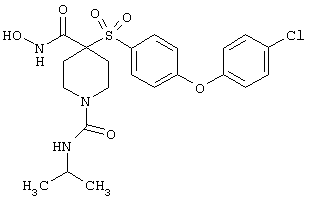
Стадия А: К суспензии полученного на стадии Е примера 88 амина (0,600 г, 1,34 ммоль) в дихлорметане (5 мл) добавляли триэтиламин (0,411 мл, 2,95 ммоль) и изопропилизоцианат (0,198 мл, 2,01 ммоль). Образовавшуюся смесь перемешивали в течение 2 ч при температуре окружающей среды и затем разбавляли дихлорметаном (50 мл). Смесь промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4, получая мочевину в виде твердого вещества беловатого цвета (0,670 г, >100%).
Стадия Б: К раствору полученной на стадии А мочевины (0,640 г, 1,29 ммоль) в тетрагидрофуране (10 мл) добавляли триметилсиланолат калия (0,199 г, 1,55 ммоль). Образовавшуюся смесь перемешивали в течение 17 ч при температуре окружающей среды, после чего добавляли дополнительную порцию триметилсиланолата калия (0,015 г, 0,117 ммоль). Образовавшуюся смесь перемешивали еще в течение 24 ч, затем удаляли тетрагидрофуран, продувая смесь N2. К суспензии остатка в дихлорметане (5 мл) добавляли N-метилморфолин (0,426 мл, 3,87 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,181 г, 1,55 ммоль), а затем РуВrоР® (0,902 г, 1,94 ммоль).
Образовавшуюся смесь перемешивали в течение 7 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества беловатого цвета (0,330 г, 44%).
Стадия В: К раствору полученного на стадии Б защищенного гидроксамата (0,330 г, 0,569 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли 4 н. раствор HCl в диоксане (10 мл). Образовавшуюся смесь перемешивали в течение 3,5 ч при температуре окружающей среды, затем добавляли диэтиловый эфир. Твердые частицы собирали фильтрацией, получая указанное в заголовке соедиенние в виде твердого вещества белого цвета (0,259 г, 92%). МС МН+: рассчитано для C22H27O6N3SCl: 496, обнаружено: 496.
Пример 91: Получение моногидрохлорида 4-[(4’-хлор[1,1’-бифенил]-4-ил)сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
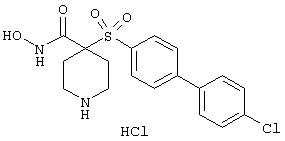
Стадия А: К раствору 4-бромтиофторфенола (16,98 г, 89,80 моль) в ацетоне (200 мл), дегазированному с помощью N2, добавляли К2СО3 (12,41 г, 89,80 моль). После дегазации образовавшейся смеси в течение 5 мин с помощью N2 добавляли полученное на стадии А примера 88 Воc-защищенное пиперидиновое производное (21,57 г, 81,64 моль). Образовавшуюся смесь перемешивали в течение 19 ч при температуре окружающей среды и затем фильтровали через подушку из Celite®, промывая ацетоном. Остаток промывали диэтиловым эфиром и твердые частицы собирали фильтрацией, получая сульфид в виде масла зеленого цвета (31,7 г, >100%).
Стадия Б: К раствору полученного на стадии А сульфида (31,68 г, 81,64 моль) в дихлорметане (200 мл), охлажденному до 0° С, добавляли мета-хлорпероксибензойную кислоту (56,35 г, 50-60%, 163,28 ммоль). Образовавшаяся смесь становилась очень густой, и добавляли дополнительную порцию дихлорметана (100 мл). Смесь перемешивали в течение 1,5 ч при 0° С и затем в течение 1,5 ч при температуре окружающей среды. Реакционную смесь разбавляли Н2О (300 мл) и добавляли водный раствор мета-бисульфита натрия (8,0 г, 42,08 ммоль в 50 мл Н2О). Дихлорметан удаляли в вакууме и водную реакционную смесь экстрагировали этилацетатом. Объединенные органические слои промывали 10%-ным раствором NH4OH, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали сульфон в виде масла желтого цвета (33,42 г, >100%).
Стадия В: К раствору полученного на стадии Б сульфона (7,80 г, 19,34 моль) в тетрагидрофуране (100 мл), охлажденному до 0° С, добавляли бис(триметилсилил)амид лития (23,8 мл, 1 М раствор в тетрагидрофуране, 23,8 ммоль) с такой скоростью, чтобы температура реакционной смеси ни разу не поднималась выше 2° С. После перемешивания в течение 30 мин при 0° С добавляли раствор метилхлорформиата (2,30 мл, 29,8 ммоль) в тетрагидрофуране (5 мл) с такой скоростью, чтобы температура реакционной смеси ни разу не поднималась выше 2° С. После этого реакционной смеси давали медленно нагреться до температуры окружающей среды. Затем смесь разбавляли насыщенным раствором NH4Cl и тетрагидрофуран удаляли в вакууме. Водный слой экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали сложный эфир в виде твердого вещества желтого цвета (6,33 г, 69%).
Стадия Г: К раствору полученного на стадии В сложного эфира (4,74 г, 10,28 ммоль) в диметоксиэтане (50 мл) добавляли 4-хлорфенилбороновую кислоту (1,77 г, 11,30 ммоль), водный раствор Cs2CO3 (25 мл, 2,0 М, 50,0 ммоль) и тетракис(трифенилфосфин) палладий (0) (1 г). Образовавшуюся смесь перемешивали в течение 3 дней при температуре окружающей среды. Реакционную смесь фильтровали через подушку из Celite®, промывая этилацетатом, и фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали бифенильное производное в виде твердого вещества беловатого цвета (4,16 г, 82%).
Стадия Д: К раствору полученного на стадии Г бифенильного производного (1,50 г, 3,04 ммоль) в тетрагидрофуране (10 мл) добавляли триметилсиланолат калия (0,468 г, 3,65 ммоль). Образовавшуюся смесь перемешивали в течение 1 ч при температуре окружающей среды, после чего добавляли дополнительную порцию тетрагидрофурана (5 мл) и реакционную смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды. Добавляли дополнительную порцию тетрагидрофурана (15 мл) и смесь перемешивали еще в течение 26 ч при температуре окружающей среды. Добавляли дополнительную порцию триметилсиланолата калия (0,040 г, 0,304 ммоль), смесь перемешивали в течение ночи (приблизительно 18 ч) при температуре окружающей среды и затем растворитель удаляли, продувая реакционную смесь N2.
К суспензии остатка в дихлорметане (20 мл) добавляли N-метилморфолин (1,00 мл, 9,12 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламина (0,427 г, 3,65 ммоль), а затем РуВrоР® (2,13 г, 4,56 ммоль). Образовавшуюся смесь перемешивали в течение 24 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества белого цвета (1,25 г, 71%).
Стадия Е: К раствору полученного на стадии Д защищенного гидроксамата (1,25 г, 2,16 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли 4н. раствор HCl в диоксане (10 мл). Образовавшуюся смесь перемешивали в течение 3,5 ч при температуре окружающей среды, затем добавляли диэтиловый эфир (20 мл). Твердые частицы собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,900 г, 97%). МС МН+: рассчитано для C18H20O4N2SCl: 395, обнаружено: 395.
Пример 92: Получение моногидрохлорида N-гидрокси-4-[[4-(метилфениламино)фенил]сульфонил]-4-пиперидинкарбоксамида
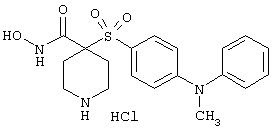
Стадия А: К раствору полученного на стадии В примера 91 сложного эфира (1,00 г, 2,17 ммоль) в толуоле (4 мл) добавляли N-метиланилин (0,282 мл, 2,60 ммоль), Cs2CO3 (0,990 г, 3,04 ммоль), трис(дибензилиденацетон)дипалладий(0) (0,018 г, 0,02 ммоль) и (R)-(+)-2,2’-бис(дифенилфосфино)-1,1’-бинафтил (БИНАФ, 0,021 г, 0,033 ммоль). Образовавшуюся смесь выдерживали в течение 20 ч при 100° С. После охлаждения до температуры окружающей среды добавляли диэтиловый эфир, смесь фильтровали через подушку из Celite®, промывая диэтиловым эфиром, и фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали анилин в виде клейкой смолы желтого цвета (0,930 г, 88%).
Стадия Б: К раствору полученного на стадии А анилина (0,930 г, 1,90 ммоль) в тетрагидрофуране (10 мл) добавляли триметилсиланолат калия (0,293 г, 2,28 ммоль). Образовавшуюся смесь перемешивали в течение 19 ч при температуре окружающей среды, после чего добавляли дополнительную порцию триметилсиланолата калия (0,024 г, 0,190 ммоль). После перемешивания при температуре окружающей среды в течение ночи (приблизительно 18 ч) растворитель удаляли, продувая смесь N2.
К суспензии остатка в дихлорметане (10 мл) добавляли N-метилморфолин (0,627 мл, 5,70 ммоль) и O-тетрагидро-2Н-пиран-2-илгидроксиламина (0,267 г, 2,85 ммоль), а затем РуВrоР® (1,33 г, 2,28 ммоль). Образовавшуюся смесь перемешивали в течение 2 дней при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества белого цвета (0,860 г, 79%).
Стадия В: К раствору полученного на стадии Б защищенного гидроксамата (0,890 г, 1,55 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли 4 н. раствор HCl в диоксане (5 мл). Образовавшуюся смесь перемешивали в течение 1 ч при температуре окружающей среды, затем добавляли диэтиловый эфир (15 мл). Твердые частицы собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,529 г, 80%). МС МН+: рассчитано для C19H24O4N3S: 390, обнаружено: 390.
Пример 93: Получение моногидрохлорида 4-[[4-(4-хлорфенокси)фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
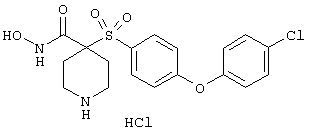
Стадия А: К суспензии смолы I (4,98 г, 5,87 ммоль) в 1-метил-2-пирролидиноне (45 мл) в пептидной колбе добавляли полученную на стадии А примера 83 кислоту (4,55 г, 11,74 ммоль), гексафторфосфат бензотриазол-1-илокситриспирролидинофосфония (6,11 г, 11,74 ммоль), N-метилморфолин (2,58 мл, 23,48 ммоль). Образовавшуюся смесь перемешивали в течение 14 ч при температуре окружающей среды. Затем смолу собирали фильтрацией, фильтрат удаляли и откладывали, смолу промывали N,N-диметилформамидом, Н2О, N,N-диметилформамидом, метанолом, дихлорметаном и в завершение диэтиловым эфиром. Смолу сушили в вакууме при температуре окружающей среды, получая связанный со смолой пара-фторсульфон в виде твердого вещества желтого цвета (6,72 г, 95%).
Фильтрат разбавляли Н2О и экстрагировали этилацетатом. Водный слой подкисляли (до рН 2,0) с помощью 2н. HCl и затем экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. Образовавшийся остаток растворяли в 1-метил-2-пирролидиноне (40 мл), добавляли полученную выше смолу, а затем N-метилморфолин (1,50 мл, 13,64 ммоль) и гексафторфосфат бензотриазол-1-илокситриспирролидинофосфония (3,05 г, 5,86 ммоль). Образовавшуюся смесь перемешивали в течение 3,5 ч при температуре окружающей среды. Затем смолу собирали фильтрацией и смолу промывали N,N-диметилформамидом, Н2О, N,N-диметилформамидом, метанолом, дихлорметаном и в завершение диэтиловым эфиром. Смолу сушили в вакууме при температуре окружающей среды, получая связанный со смолой пара-фторсульфон в виде твердого вещества светло-оранжевого цвета (6,34 г, 89%). Загрузку (составляющую 0,78 ммоль/г) определяли путем расщепления небольшой порции связанного со смолой пара-фторсульфона с помощью смеси 95%-ная трифторуксусная кислота/Н2О.
Стадия Б: К суспензии связанного со смолой пара-фторсульфона (0,700 г, 0,546 ммоль) в 1-метил-2-пирролидиноне (3 мл) добавляли пара-хлорфенол (0,702 г, 5,46 ммоль) и Cs2CO3 (1,78 г, 5,46 ммоль). Образовавшуюся смесь нагревали в течение 13 ч до 110° С. Затем смолу собирали фильтрацией и последовательно промывали N,N-диметилформамидом, Н2О, N,N-диметилформамидом, 2 н. HCl, N,N-диметилформамидом, метанолом и дихлорметаном. Образовавшуюся смолу повторно подвергали реакции в течение 3 ч в указанных выше условиях. После этого смолу собирали фильтрацией и последовательно промывали N,N-диметилформамидом, Н2О, N,N-диметилформамидом, 2 н. HCl, N,N-диметилформамидом, метанолом и дихлорметаном. Твердое вещество сушили в вакууме при температуре окружающей среды, получая связанный со смолой гидроксамат в виде твердого вещества оранжевого цвета (0,692 г, 91%).
Стадия В: Полученный на стадии Б связанный со смолой гидроксамат (0,692 г, 0,540 ммоль) обрабатывали в течение 1 ч при температуре окружающей среды смесью 95%-ная трифторуксусная кислота/Н2О (3 мл). Смолу фильтровали и промывали смесью 95%-ная трифторуксусная кислота/Н2О (3 мл), а затем дихлорметаном (2х3 мл). После этого фильтрат выпаривали. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О/трифторуксусная кислота) получали гидроксамат. Образовавшееся твердое вещество растворяли в ацетонитрила (5 мл) и Н2О (0,5 мл) и обрабатывали концентрированной HCl. Образовавшуюся смесь перемешивали в течение 5 мин при температуре окружающей среды и концентрировали в вакууме, получая указанное в заголовке соединение в виде твердого вещества беловатого цвета (0,220 г, 91%). МС МН+: рассчитано для С18Н20О5N2SCl: 411, обнаружено: 411.
Пример 94: Получение тетрагидро-N-гидрокси-4-[(4-феноксифенил)сульфонил]-2Н-пиран-4-карбоксамида
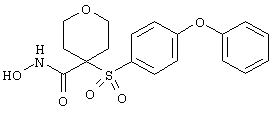
Стадия А: К раствору полученного на стадии В примера 55 сложного метилового эфира (0,96 г, 3,2 ммоль) в N,N-диметилформамиде (30 мл) при перемешивании добавляли фенол (0,3 г, 3,2 ммоль), а затем карбонат цезия (3,2 г, 10 ммоль). Образовавшуюся композицию нагревали в течение 5 ч до 70° С. Раствору давали выстояться в течение 18 ч при температуре окружающей среды, после чего разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали полунасыщенным раствором NaCl и сушили над сульфатом натрия. Растворитель удаляли на роторном испарителе, получая требуемое феноксипроизводное (1,1 г, 92%).
Стадия Б: К раствору полученного на стадии А феноксипроизводного (1,1 г, 2,9 ммоль) в ТГФ (10 мл) и этаноле (10 мл) добавляли гидроксид натрия (1 г, 25 ммоль). Образовавшийся раствор перемешивали в течение 1 ч при температуре окружающей среды. Затем раствор нагревали в течение 1 ч до 80° С. Растворитель удаляли на роторном испарителе, образовавшуюся натриевую соль подкисляли 1 н. HCl (50 мл) и экстрагировали этилацетатом. Органический слой сушили над Na2SO4. Растворитель удаляли на роторном испарителе, получая требуемую феноксикарбоновую кислоту (1,1 г, 99%).
Стадия В: К раствору полученной на стадии Б феноксикарбоновой кислоты (1,1 г, 3 ммоль) в ДМФ (7 мл) добавляли при перемешивании N-гидроксибензотриазол· Н2О (0,623 г, 4,6 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,634 г, 3,3 ммоль). Через 10 мин добавляли 50%-ный водный раствор гидроксиламина (2 мл, 30 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли насыщенным бикарбонатом натрия и экстрагировали этилацетатом. Органический слой промывали Н2О, затем полунасыщенным раствором NaCl, после чего сушили над Na2SO4. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,37 г, 33%). МСВР (ES+) МН+: рассчитано для C18H19NO6S: 378,1011 обнаружено: 378,0994.
Пример 95: Получение тетрагидро-N-гидрокси-4-[[4-(фенилтио)фенил]сульфонил]-2Н-пиран-4-карбоксамида

Стадия А: К раствору полученного на стадии В примера 55 сложного метилового эфира (1,02 г, 3,4 ммоль) в N,N-диметилформамиде (20 мл) при перемешивании в атмосфере азота добавляли тиофенол (0,37 г, 3,4 ммоль), а затем карбонат цезия (3,3 г, 10,1 ммоль) и раствор нагревали в течение 17 ч до 70° С. Раствору давали выстояться в течение 1 ч при температуре окружающей среды, после чего разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали полунасыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали S-фенильное производное (0,6 г, 41%).
Стадия Б: К раствору полученного на стадии А S-фенильного производного (0,6 г, 1,4 ммоль) в ТГФ (10 мл) и этаноле (10 мл) добавляли при перемешивании NaOH (0,8 г, 20 ммоль). Раствор нагревали в течение 1 ч до 80° С. Раствору давали выстояться в течение 18 ч при температуре окружающей среды. Растворитель удаляли на роторном испарителе, образовавшуюся натриевую соль подкисляли 1 н. HCl (25 мл), экстрагировали этилацетатом и органический слой сушили над сульфатом натрия. Растворитель удаляли на роторном испарителе, получая требуемую S-фенилкарбоновую кислоту (0,6 г, количественный выход).
Стадия В: К раствору полученной на стадии Б S-фенилкарбоновой кислоты (0,6 г, 1,5 ммоль) в ДМФ (6 мл) добавляли при перемешивании N-гидроксибензотриазол· Н2О (0,30 г, 2,2 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,32 г, 1,6 ммоль). Через 10 мин добавляли 50%-ный водный раствор гидроксиламина (1,5 мл, 22 ммоль) и раствор перемешивали в течение 42 ч при температуре окружающей среды. Раствор разбавляли насыщенным бикарбонатом натрия и экстрагировали этилацетатом. Органический слой промывали Н2О, затем полунасыщенным раствором NaCl, после чего сушили над сульфатом натрия. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,15 г, 26%). МСВР (ES+) МН+: рассчитано для C18H19NO5S2 392,0783 обнаружено: 394,0753.
Пример 96: Получение 4-[[4-(3,4-диметилфенокси)фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
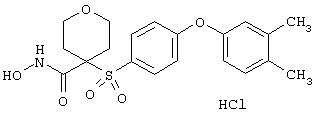
Стадия А: К раствору полученного на стадии В примера 55 сложного метилового эфира (1,04 г, 3,3 ммоль) в N,N-диметилформамиде (30 мл) при перемешивании добавляли 3,4-диметилфенол (0,4 г, 3,3 ммоль), а затем карбонат цезия (3,2 г, 10 ммоль). Образовавшийся раствор нагревали в течение 5 ч до 88° С. Раствор концентрировали на роторном испарителе, разбавляли Н2О и экстрагировали этилацетатом. Органический слой сушили над MgSO4. Растворитель удаляли на роторном испарителе, получая требуемое диметилфеноксипроизводное (1,2 г, 91%).
Стадия Б: К раствору полученного на стадии А диметилфеноксипроизводного (1,2 г, 3 ммоль) в ТГФ (10 мл) и этаноле (10 мл) добавляли NaOH (1 г, 25 ммоль). Образовавшийся раствор нагревали в течение 1 ч до 80° С. Растворитель удаляли на роторном испарителе, образовавшуюся натриевую соль подкисляли 1 н. HCl (50 мл) и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия. Растворитель удаляли на роторном испарителе, получая требуемую диметилфеноксикарбоновую кислоту (1,2 г, количественный выход).
Стадия В: К раствору полученной на стадии Б диметилфеноксикарбоновой кислоты (1,2 г, 3 ммоль) в ДМФ (7 мл) добавляли при перемешивании N-гидроксибензотриазол-Н2О (0,623 г, 4,6 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,634 г, 3,3 ммоль). Через 10 мин добавляли 50%-ный водный раствор гидроксиламина (2 мл, 30 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли насыщенным бикарбонатом натрия и экстрагировали этилацетатом. Органический слой промывали Н2О, затем полунасыщенным раствором NaCl, после чего сушили над Na2SO4. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,52 г, 43%). МСВР (ES+) МН+: рассчитано для С20Н23NO6S: 406,1324 обнаружено: 406,1302.
Пример 97: Получение моногидрохлорида тетрагидро-N-гидрокси-4-[[4-[(6-метил-3-пиридинил)окси]фенил]сульфонил]-2Н-пиран-4-карбоксамида

Стадия А: К раствору полученного на стадии В примера 55 сложного метилового эфира (1,02 г, 3,4 ммоль) в N,N-диметилформамиде (20 мл) при перемешивании добавляли 5-гидрокси-2-метилпиридин (0,54 г, 5 ммоль), а затем карбонат цезия (3,2 г, 10 ммоль). Образовавшийся раствор нагревали в течение 5 ч до 70° С. Раствору давали выстояться в течение 4 дней при температуре окружающей среды, после чего разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали полунасыщенным раствором NaCl и сушили над Na2SO4. Растворитель удаляли на роторном испарителе, получая густое масло, из которого в вакууме при температуре окружающей среды кристаллизовали требуемое метилпиридиновое производное белого цвета (1,2 г, 94%).
Стадия Б: К раствору полученного на стадии А метилпиридинового производного (1,2 г, 3,2 ммоль) в ТГФ (13 мл) добавляли триметилсиланолат калия (0,5 г, 3,5 ммоль). Образовавшийся раствор перемешивали в течение 18 ч при температуре окружающей среды, в результате чего образовывался гель. Растворитель удаляли на роторном испарителе, получая требуемую метилпиридинкарбоновую кислоту (1,4 г, количественный выход).
Стадия В: К раствору полученной на стадии Б метилпиридинкарбоновой кислоты (1,4 г, 3,2 ммоль) в метиленхлориде (10 мл) добавляли при перемешивании гексафторфосфат бромтриспирролидинофосфония (1,79 г, 3,8 ммоль), затем 4-метилморфолин (0,79 г, 9,6 ммоль), а потом O-тетрагидро-2Н-пиран-2-идгидроксиламин (0,41 г, 3,5 ммоль) и раствор перемешивали в течение 1,5 ч при температуре окружающей среды. Раствор фильтровали для удаления осадка и растворитель удаляли на роторном испарителе. После хроматографии (на силикагеле, этилацетат/гексан) получали O-тетрагидропиранметилпиридин в виде твердого вещества белого цвета (1,48 г, 97%).
Стадия Г: К раствору полученного на стадии В O-тетрагидропиранметилпиридина (1,48 г, 3,1 ммоль) в 4 н. растворе HCl в диоксане (5 мл) добавляли при перемешивании метанол (3 мл). Раствор перемешивали в течение 3 ч при температуре окружающей среды и сливали на диэтиловый эфир. Образовавшийся осадок собирали вакуумной фильтрацией. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О/НСl) получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,64 г, 53%). МСВР (ES+) МН+: рассчитано для C18H20N2O6S: 393,1120 обнаружено: 393,1110.
Пример 98: Получение тетрагидро-N-гидрокси-4-[[4-[(6-метил-2-пиридинил)окси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
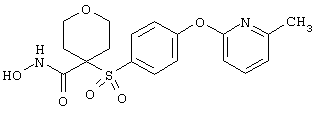
Стадия А: К раствору полученного на стадии В примера 55 сложного метилового эфира (1,0 г, 3,3 ммоль) в N,N-диметилформамиде (20 мл) при перемешивании добавляли 2-гидрокси-6-метилпиридин (0,54 г, 5 ммоль), а затем карбонат цезия (3,2 г, 10 ммоль). Образовавшийся раствор нагревали в течение 5 ч до 70° С. Раствору давали выстояться в течение 11 ч при температуре окружающей среды, после чего к раствору при перемешивании добавляли дополнительную порцию 2-гидрокси-6-метилпиридин (0,3 г, 2,7 ммоль) и образовавшийся раствор нагревали в течение 3 ч до 70° С. Раствор концентрировали на роторном испарителе, разбавляли насыщенным раствором NaCl в Н2О и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия. Растворитель удаляли на роторном испарителе и после хроматографий (на силикагеле, этилацетат/метанол) получали требуемый метилпиридин в виде твердого вещества белого цвета (0,63 г, 49%).
Стадия Б: К раствору полученного на стадии А метилпиридина (0,63 г, 1,6 ммоль) в ТГФ (13 мл) добавляли триметилсиланолат калия (0,5 г, 3,5 ммоль). Образовавшийся раствор перемешивали в течение 18 ч при температуре окружающей среды. Образовавшийся осадок удаляли фильтрацией, промывали метиленхлоридом и сушили в вакууме, получая калиевую соль метилпиридин-карбоновой кислоты (0,4 г, 55%).
Стадия В: К раствору полученной на стадии Б калиевой соли метилпиридинкарбоновой кислоты (0,4 г, 0,9 ммоль) в N,N-диметилформамиде (5 мл) добавляли при перемешивании гексафторфосфат бромтриспирролидинофосфония (0,5 г, 1 ммоль), затем 4-метилморфолин (0,27 г, 2,6 ммоль), а потом 50%-ный водный раствор гидроксиламина (0,6 г, 9 ммоль). Раствор перемешивали в течение 32 ч при температуре окружающей среды. Раствор концентрировали на роторном испарителе и после хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,162 г, 47%). МСВР (ES+) МН+: рассчитано для C18H20N2O6S: 393,1120 обнаружено: 393,1119.
Пример 99: Получение моногидрохлорида тетрагидро-N-гидрокси-4-[[4-[4-(1Н-имидазол-1-ил)фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
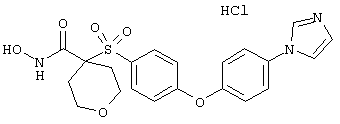
Стадия А: К раствору полученного на стадии В примера 55 ТГП-защищенного пиранфторпроизводного (2,0 г, 5,2 ммоль) в N,N-диметилацетамиде (6 мл) добавляли 4-(1,3-имидазол) фенол (12,9 г, 33,3 ммоль), а затем карбонат цезия (32,5 г, 99,9 ммоль). Реакционную смесь выдерживали в течение 12 ч при 65° С. После удаления диметилацетамида в вакууме получали твердое вещество коричневого цвета. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/вода) получали ТГП-защищенное соединение в растворе.
Стадия Б: К раствору полученного на стадии А неочищенного ТГП-защищенного соединения в смеси ацетонитрил/вода (100 мл) добавляли водный 10%-ный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) удаляли ацетонитрил. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества коричневого цвета (6,0 г, 41%). МС (FAB) М+Н: рассчитано для C21H21N3O6S1: 444, обнаружено: 444.
Пример 100: Получение 4-[[4-(4-хлорфенокси)фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
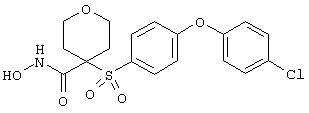
Стадия А: К раствору полученного на стадии В примера 55 ТГП-защищенного пиранфторпроизводного (2,9 г, 7,5 ммоль) в N,N-диметилформамиде (15 мл) добавляли при перемешивании пара-хлорфенол (1,93 г, 15 ммоль), а затем карбонат цезия (7,3 г, 22,5 ммоль). Образовавшуюся смесь нагревали в течение 1,5 ч до 90° С. Раствор выдерживали при перемешивании в течение 18 ч при температуре окружающей среды, после чего к раствору добавляли при перемешивании диметилформамид (20 мл), а затем карбонат цезия (2 г, 6,2 ммоль). Образовавшуюся смесь нагревали в течение 3 ч до 95° С. Затем раствор выдерживали в течение 20 ч при температуре окружающей среды, после чего его разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали полунасыщенным раствором NaCl и сушили над сульфатом натрия. Растворитель удаляли на роторном испарителе. После хроматографии (на силикагеле, этилацетат/гексан) получали пара-хлорфеноксифенил-ТГП-защищенный гидроксамат (2,9 г, 78%).
Стадия Б: К раствору полученного на стадии А пара-хлорфеноксифенил-ТГП-защищенного гидроксамата (2,9 г, 5,7 ммоль) в диоксане (5 мл) добавляли 4н. раствор HCl в диоксане (5 мл, 20 ммоль), а затем метанол (7,5 мл). Образовавшийся раствор перемешивали в течение 1 ч при температуре окружающей среды. Растворитель удаляли на роторном испарителе. После хроматографии с обращенной фазой (на силикагеле, этилацетат/гексан) получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,35 г, 58%). МС (FAB) М+H: рассчитано для C18H18NO6SCl: 412, обнаружено: 412.
Пример 101: Получение 4-[[4-(3-хлорфенокси)фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
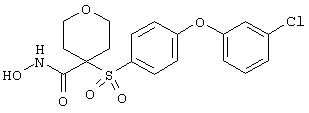
Стадия А: К раствору полученного на стадии В примера 55 ТГП-защищенного пиранфторпроизводного (5,0 г, 13 ммоль) в N,N-диметилформамиде (20 мл) добавляли при перемешивании пара-хлорфенол (5 г, 39 ммоль), а затем карбонат цезия (17 г, 52 ммоль). Образовавшийся раствор нагревали в течение 7 ч до 95° С. Раствор выдерживали при перемешивании в течение 7 ч при температуре окружающей среды, разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали полунасыщенным раствором NaCl и сушили над сульфатом натрия. Раствор концентрировали на роторном испарителе. После хроматографии (на силикагеле, этилацетат/гексан) получали мета-хлорфеноксифенил-ТГП-защищенный гидроксамат (5,2 г, 82%).
Стадия Б: К раствору полученного на стадии А пара-хлорфеноксифенил-ТГП-защищенного гидроксамата (5,2 г, 10 ммоль) в диоксане (5 мл) добавляли 4 н. раствор HCl в диоксане (5 мл, 20 ммоль), а затем метанол (10 мл). Образовавшийся раствор перемешивали в течение 1 ч при температуре окружающей среды. Растворитель удаляли на роторном испарителе, получая указанное в заголовке соединение в виде твердого вещества белого цвета (3,4 г, 79%). МСВР (ES+) М+Н+: рассчитано для C18H18NO6SCl: 429,0887, обнаружено: 429,0880.
Пример 102: Получение метил-4-[4-[(тетрагидро-4-[(гидроксиамино)карбонил]-2Н-пиран-4-ил]сульфонил]фенокси]бензолпропаноата
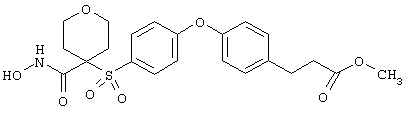
Стадия А: К раствору полученного на стадии В примера 55 ТГП-защищенного пиранфторсодержащего соединения (5,0 г, 13 ммоль) в N,N-диметилформамиде (45 мл) добавляли при перемешивании метил-3-(4-гидроксифенил)пропаноат (7 г, 39 ммоль), а затем карбонат цезия (17 г, 52 ммоль). Образовавшуюся смесь нагревали в течение 7 ч до 95° С. Затем раствор выдерживали в течение 7 ч при температуре окружающей среды. После этого раствор разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали полунасыщенным раствором NaCl и сушили над сульфатом натрия. Раствор концентрировали на роторном испарителе. После хроматографии (на силикагеле, этилацетат/гексан) получали метилпропаноатфеноксифенил-ТГП-защищенный гидроксамат (5,6 г, 79%).
Стадия Б: К раствору полученного на стадии А метилпропаноатфеноксифенил-ТГП-защищенного гидроксамата (5,6 г, 10 ммоль) в метаноле (5 мл) добавляли 4 н. раствор HCl в диоксане (5 мл, 20 ммоль). Образовавшийся раствор перемешивали в течение 0,5 ч при температуре окружающей среды. Растворитель удаляли на роторном испарителе. Остаток растворяли в смеси метиленхлорид/этилацетат и соединение осаждали гексаном. Осадок промывали гексаном и сушили в вакууме, получая указанное в заголовке соединение в виде твердого вещества белого цвета (3,8 г, 80%). МСВР (ES+) М+: рассчитано для C22H25NO8S: 464,138, обнаружено: 464,135.
Пример 103: Получение 4-[[4-[(4-фторфенил)тио]фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
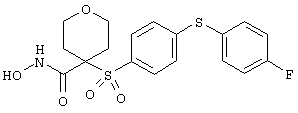
Стадия А: К раствору полученного на стадии В примера 55 ТГП-защищенного пиранфторпроизводного (2,9 г, 7,5 ммоль) в N,N-диметилформамиде (25 мл) добавляли при перемешивании в атмосфере азота карбонат цезия (4,9 г, 15 ммоль), а затем 4-фтортиофенол (1,9 г, 15 ммоль). Образовавшуюся смесь нагревали в течение 7 ч до 95° С. После нагревания в течение 1 ч добавляли карбонат цезия (1,2 г, 3,8 ммоль), а затем через 2 ч еще одну порцию этого соединения. Раствор после выдерживания в течение 9 ч при температуре окружающей среды концентрировали на роторном испарителе, разбавляли 30%-ным водным соляным раствором и экстрагировали этилацетатом. Органический слой промывали полунасыщенным раствором NaCl и сушили над сульфатом натрия. Раствор концентрировали на роторном испарителе. После хроматографии (на силикагеле, этилацетат/гексан) и последующей хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали пара-фторфенфенил-S-фенил-ТГП-защищенный гидроксамат (1,9 г, 55%).
Стадия Б: К раствору полученного на стадии А пара-фторфенил-S-фенил-ТГП-защищенного гидроксамата (1,9 г, 4 ммоль) в метаноле (5 мл) добавляли 4н. раствор HCl в диоксане (5 мл, 20 ммоль), а затем метанол (10 мл). Образовавшийся раствор перемешивали в течение 0,5 ч при температуре окружающей среды. Растворитель удаляли на роторном испарителе, остаток растворяли в метиленхлориде и осаждали гексаном. Образовывался осадок, который сушили в вакууме, получая указанное в заголовке соединение в виде твердого вещества белого цвета (1,5 г, 89%). МСВР (ES+) M+NH
Пример 104: Получение моногидрохлорида тетрагидро-N-гидрокси-4-[[4-(4-пиридинилтио]фенил]сульфонил]-2Н-пиран-4-карбоксамида
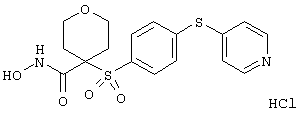
Стадия А: К раствору полученного на стадии В примера 55 ТГП-защищенного пиранфторпроизводного (2,9 г, 7,5 ммоль) в N,N-диметилформамиде (20 мл) добавляли при перемешивании карбонат калия (2,6 г, 19 ммоль), а затем 4-меркаптопиридин (1,7 г, 15 ммоль). Образовавшуюся смесь нагревали в течение 5 ч до 75° С. После нагревания в течение 1 ч добавляли карбонат калия (0,26 г, 1,9 ммоль), а затем через 2 ч еще одну порцию этого соединения. Раствору давали выстояться в течение 14 ч при температуре окружающей среды. Раствор концентрировали на роторном испарителе, разбавляли 30%-ным водным соляным раствором и экстрагировали этилацетатом. Органический слой промывали полунасыщенным раствором NaCl и сушили над Na2SO4. Раствор концентрировали на роторном испарителе. После хроматографии (на силикагеле, этилацетат/гексан) получали меркаптопиридин-ТГП-защищенный гидроксамат (1,2 г, 33%).
Стадия Б: К раствору полученного на стадии А меркаптопиридин-ТГП-защищенного гидроксамата (1,2 г, 2,5 ммоль) в ацетонитриле (20 мл) добавляли 12,5н. раствор HCl в диоксане (0,4 мл, 5 ммоль), а затем метанол (3 мл). Образовавшийся раствор перемешивали в течение 1 ч при температуре окружающей среды. Осадок фильтровали, промывали метанолом, а затем диэтиловым эфиром и сушили в вакууме, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,92 г, 86%). МСВР (ES+) M+NH
Пример 105: Получение 4-[4-[[тетрагидро-4-[(гидроксиамино)карбонил]2Н-пиран-4-ил]сульфонил]фенокси]бензолпропановой кислоты
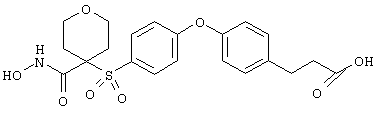
Стадия А: К раствору указанного в заголовке примера 102 соединения (0,1 г, 0,2 ммоль) в метаноле (0,5 мл) добавляли при перемешивании 1 М водный раствор Li(OH)2 (0,43 мл, 0,43 ммоль). После выстаивания в течение 24 ч при температуре окружающей среды раствор кипятили с обратным холодильником в течение 20 ч. Раствор лиофилизировали досуха и после хроматографии с обращенной фазой получали указанное в заголовке соединение в виде твердого вещества белого цвета (9 мг, 9%). МС (FAB) М+Li+: рассчитано для C21H23NO8S: 456, обнаружено: 456.
Пример 106: Получение моногидрохлорида тетрагидро-N-гидрокси-4-[[4-[[1-(2-пропинил)-4-пиперидинил]окси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
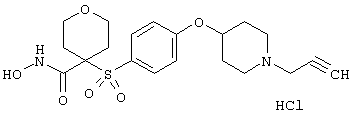
Стадия А: В высушенную нагреванием трехгорлую колбу в атмосфере азота добавляли NaH (1,59 г, 60%-ный, 40 ммоль), суспендированный в N,N-диметилформамиде (50 мл). Суспензию охлаждали до 0° С с помощью ледяной бани и добавляли N-Воc-защищенный гидроксипиперидин (8 г, 40 ммоль), а затем промывали N,N-диметилформамидом (10 мл). Ледяную баню удаляли и раствору давали нагреться в течение 2 ч при премешивании до температуры окружающей среды. Раствор снова охлаждали при перемешивании до 0° С и добавляли полученный на стадии В примера 55 сложный метиловый эфир (10 г, 33 ммоль), растворенный в N,N-диметилформамиде (40 мл). Ледяную баню удаляли и раствор перемешивали в течение 48 ч при температуре окружающей среды. Раствор концентрировали на роторном испарителе. Раствор разбавляли Н2О и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия. После хроматографии (на силикагеле, этилацетат/гексан/метанол) неочищенный N-Boc-защищенный сложный метиловый эфир обрабатывали 1н. раствором HCl в метаноле. Растворитель удаляли на роторном испарителе. Затем остаток растворяли в ацетонитриле (21 мл), к которому добавляли Н2О (21 мл). После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали очищенный гидрохлорид сложного пиперидинметилового эфира (4,9 г, 35%).
Стадия Б: К суспензии полученного на стадии А гидрохлорида сложного пиперидинметилового эфира (1,8 г, 4 ммоль) в ацетонитриле (24 мл) добавляли карбонат калия (1,8 г, 13 ммоль), а затем пропаргилбромид (0,58 мл 80%-ный раствор, 5,2 ммоль). Смесь перемешивали в течение 18 ч при температуре окружающей среды. Раствор концентрировали на роторном испарителе, разбавляли Н2О и экстрагировали этилацетатом. Органический слой сушили над Na2SO4 и концентрировали на роторном испарителе. После хроматографии (на силикагеле, метиленхлорид/метанол) получали сложный пропаргилпиперидин-метиловый эфир (1,1 г, 63%).
Стадия В: К раствору полученного на стадии Б сложного пропаргилпипери-динметилового эфира (1,1 г, 2,7 ммоль) в ТГФ (3 мл) добавляли триметилсиланолат калия (0,57 г, 4 ммоль). Через 5 мин добавляли ТГФ (12 мл), а еще через 10 мин добавляли вторую порцию ТГФ (15 мл). Образовавшийся раствор перемешивали в течение 18 ч при температуре окружающей среды, в результате чего образовывался гель. Растворитель удаляли на роторном испарителе и остаток разбавляли Н2О и промывали этилацетатом. Водный слой подкисляли и хроматографировали (на силикагеле, ацетонитрил/Н2О), получая после лиофилизации требуемую пропаргилпиперидинкарбоновую кислоту (0,64 г, 59%).
Стадия Г: К раствору полученной на стадии В пропаргилпиперидинкарбоновой кислоты (0,64 г, 1,64 ммоль) в N,N-диметилформамиде (5 мл) добавляли при перемешивании 1-гидроксибензотриазол (0,3 г, 2,3 ммоль), затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,33 г, 1,7 ммоль), а после этого O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,57 г, 4,8 ммоль). Раствор перемешивали в течение 42 ч при температуре окружающей среды, концентрировали на роторном испарителе, разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором бикарбоната натрия, затем соляным раствором и сушили над Na2SO4. Раствор концентрировали на роторном испарителе и после хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) и последующей лиофилизации получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,2 г, 30%). МСВР (ES+) MH+: рассчитано для C20H26N2O6S: 423,159, обнаружено: 423,159.
Пример 107: Получение 4-[[4-[1-ацетил-4-пиперидинил)окси]фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
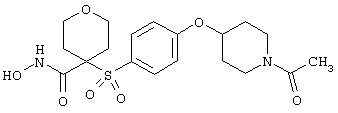
Стадия А: К суспензии полученного на стадии А примера 106 гидрохлорида сложного пиперидинметилового эфира (1,8 г, 4 ммоль) в пиридине (2 мл) добавляли при перемешивании уксусный ангидрид (1,7 г, 16 ммоль). Смесь перемешивали в течение 20 мин при температуре окружающей среды. Раствор концентрировали на роторном испарителе и после хроматографии (на силикагеле, этилацетат/метанол) получали соединение, представляющее собой сложный ацетилпиперидинметиловый эфир (1,5 г, 83%).
Стадия Б: К раствору полученного на стадии Б сложного ацетилпиперидин-метилового эфира (1,5 г, 3,3 ммоль) в ТГФ (5 мл) добавляли триметилсиланолат калия (0,86 г, 6 ммоль). Через 5 мин добавляли ТГФ (15 мл), а еще через 10 мин добавляли вторую порцию ТГФ (15 мл). Образовавшийся раствор перемешивали в течение 18 ч при температуре окружающей среды. Осадок выделяли фильтрацией, получая требуемую ацетилпиперидинкарбоновую кислоту (1,5 г, 98%).
Стадия В: К раствору полученной на стадии Б ацетилпиперидинкарбоновой кислоты (0,9 г, 2 ммоль) в диметилацетамиде (5 мл) добавляли гексафторфосфат бромтриспирролидинофосфония (1 г, 2,3 ммоль), затем 4-метилморфолин (0,6 г, 6 ммоль), а после этого водный раствор O-тетрагидро-2Н-пиран-2-илгидроксиламина (1,5 г, 23 ммоль) и раствор перемешивали в течение 48 ч при температуре окружающей среды. После хроматографии с обращенной фазой (на силикагеле, ацетонитрил/Н2О) получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,1 г, 12%). МС (FAB) MH+: рассчитано для C19H26N2O7S: 427, обнаружено: 427.
Пример 108: Получение 4-[[4-(3-хлор-4-фторфенокси)фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
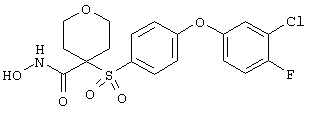
Стадия А: К раствору полученного на стадии В примера 55 пиранфторпроизводного (3,2 г, 7,7 ммоль) в N,N-диметилацетамиде (15 мл) добавляли при перемешивании 3-хлор-4-фторфенол (1,7 мл, 12 ммоль), а затем карбонат цезия (5 г, 15,5 ммоль). Реакционную смесь выдерживали в течение 2 ч при 95° С. Добавляли карбонат цезия (2,5 г, 8 ммоль) и реакционную смесь выдерживали в течение 6 ч при 95° С. Раствору давали выстояться в течение 8 ч при температуре окружающей среды. Затем неочищенную реакционную смесь фильтровали для удаления хлорида цезия и осаждали продукт. Остаток на фильтре суспендировали в Н2О и подкисляли с помощью HCl до рН 6. После прекращения пенообразования осадок удаляли фильтрацией, промывали Н2О, растворяли в смеси Н2О/ацетонитрил и хроматографировали на колонке для ЖХВР с обращенной фазой (Н2О/ацетонитрил), получая 3-хлор-4-фторфенокси-ТГП-защищенный гидроксамат (1,4 г, 35%).
Стадия Б: К раствору полученного на стадии А 3-хлор-4-фторфенокси-ТГП-защищенного гидроксамата (1,4 г, 2,7 ммоль) в ацетонитриле (10 мл) добавляли 1н. водный раствор HCl (10 мл). Раствор перемешивали в течение 1 ч при температуре окружающей среды. Ацетонитрил выпаривали при температуре окружающей среды в постоянном потоке азота до тех пор, пока не образовывался густой осадок. Осадок фильтровали и осадок на фильтре промывали Н2О, а затем диэтиловым эфиром и сушили в вакууме, получая указанное в заголовке соединение в виде твердого вещества белого цвета (12,5 г, 96%). Соединение перекристаллизовывали из смеси ацетон/гексан, получая кристаллы белого цвета (10,9 г, 86%). МСВР (ES) M+NH
Пример 109: Получение тетрагидро-N-гидрокси-4-[[4-(4-фенокси)фенил]сульфонил]-2Н-тиопиран-4-карбоксамида
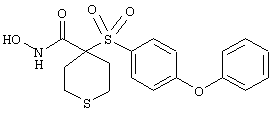
Стадия А: К раствору полученного на стадии В примера 50 сложного метилового эфира тиопирана (ММ (молекулярная масса) 318, 3 г, 1,0 экв.) в N,N-диметилацетамиде (ДМА, 40 мл) добавляли карбонат цезия (12 г, 1,5 экв.) и фенол (1,5 г). Смесь нагревали до 95° С в течение б ч. После охлаждения реакционной смеси до температуры окружающей среды реакционную смесь фильтровали и затем удаляли N,N-диметилацетамид на роторном испарителе. Остаток растворяли в 10%-ном водном растворе HCl (100 мл) и дважды экстрагировали этилацетатом. Этилацетатный экстракт сушили над сульфатом натрия и упаривали при пониженном давлении, получая масло. Масло очищали на силикагеле, получая 2 г сложного метилового эфира. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: К раствору полученного на стадии А сложного метилового эфира (ММ 392, 2 г) в ТГФ (20 мл) добавляли триметилсиланолат калия (ММ 128, 1,6 г, 1,2 экв.). Смесь перемешивали в течение 2-3 ч при температуре окружающей среды до тех пор, пока не образовывался твердый осадок. После завершения гидролиза добавляли N-метилморфолин (2 мл), а затем РуВrоР (2,3 г, 1,2 экв.). Раствор перемешивали в течение 10 мин, затем добавляли водный раствор гидроксиламина и продолжали перемешивание еще в течение 2 ч. После завершения реакции (2 ч) растворитель удаляли на роторном испарителе. Остаток растворяли в смеси вода/ацетонитрил, подкисляли с помощью ТФК (до рН 2), затем очищали с помощью препаративной ЖХВР с обращенной фазой (ЖХВРОФ), получая 1 г указанного в заголовке соединения в виде твердого вещества белого цвета. Результаты анализа с помощью IH-ЯMP, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) М+Н: рассчитано для C18H19NO5S2: 393, обнаружено: 393.
Пример 110: Получение тетрагидро-N-гидрокси-4-[[4-(4-фенок-си)фенил]сульфонил]-2Н-пиран-4-карбоксамида
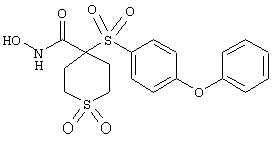
Стадия А: К раствору полученного на стадии А примера 109 соединения (2 г) в тетрагидрофуране (50 мл) добавляли воду (50 мл). К этой смеси при интенсивном перемешивании добавляли Охоnе® 8 г, 3 экв.). Процесс реакции контролировали с помощью ЖХВРОФ. Через 3 ч добавляли воду и продукт дважды экстрагировали этилацетатом (100 мл). Этилацетатный слой сушили над сульфатом натрия. После удаления растворителя при пониженном давлении получали 1,8 г сложного феноксиметилового эфира в виде твердого вещества белого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: К раствору полученного на стадии А сложного феноксиметилового эфира (ММ 590, 2 г) в тетрагидрофуране (20 мл) добавляли триметилсиланолат калия (ММ 128, 1,2 г, 1,2 экв.). Смесь перемешивали в течение 2-3 ч при температуре окружающей среды до тех пор, пока не образовывался твердый осадок. После завершения гидролиза добавляли N-метилморфолин (2 мл), а затем РуВrоР (2,3 г, 1,2 экв.). Раствор перемешивали в течение 10 мин, затем добавляли водный раствор гидроксиламина и продолжали перемешивание еще в течение 2 ч. После завершения реакции (2 ч) растворитель удаляли на роторном испарителе. Остаток растворяли в смеси вода/ацетонитрил, подкисляли с помощью ТФК (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 500 мг указанного в заголовке соединения в виде твердого вещества белого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) M+H: рассчитано для C18H19NO7S2:425, обнаружено: 425.
Пример 111: Получение тетрагидро-N-гидрокси-4-[[4-(4-фенок-си)фенил]сульфонил]-2Н-сульфоксилпиран-4-карбоксамида

Стадия А: К раствору полученного на стадии А примера 109 метилового эфира (2 г) в смеси уксусная кислота/вода (25/5 мл) добавляли перикись водорода (2 мл, 30%-ный раствор). Процесс реакции в этом интенсивно перемешиваемом растворе контролировали с помощью ЖХВРОФ. Через 3 ч добавляли воду и продукт дважды экстрагировали этилацетатом (100 мл). Этилацетатный слой сушили над сульфатом натрия. После удаления растворителя при пониженном давлении получали 2,1 г соединения, представляющего собой сложный метиловый эфир сульфоксидпиранфенил-O-фенила в виде твердого вещества белого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: К раствору полученного на стадии А сложного метилового эфира сульфоксидпиранфенил-О-фенила (ММ 578, 1,8 г) в тетрагидрофуране (25 мл) добавляли триметилсиланолат калия (ММ 128, 1,2 г, 1,2 экв.). Смесь перемешивали в течение 2-3 ч до тех пор, пока не образовывался твердый осадок. После завершения гидролиза добавляли N-метилморфолин (2 мл), а затем РуВrоР (2,3 г, 1,2 экв.). Раствор перемешивали в течение 10 мин, затем добавляли водный раствор гидроксиламина и продолжали перемешивание еще в течение 2 ч. После завершения реакции (12 ч) растворитель удаляли на роторном испарителе. Остаток растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 500 мг указанного в заголовке соединения в виде твердого вещества белого цвета. Результаты анализа с помощью Н-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) М+Н: рассчитано для C18H19NO6S2: 409, обнаружено: 409.
Пример 112: Получение тетрагидро-N-гидрокси-4-[[4-(1-ацетил-4-(4-пиперазинфенокси)фенил]сульфонил-2Н-тиопиран-4-карбоксамида
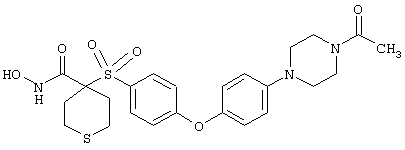
Стадия А: К раствору полученного на стадии В примера 50 сложного метилового эфира тиопирана (ММ 318, 5 г, 1,0 экв.) в N,N-диметилацетамиде (70 мл) добавляли карбонат цезия (ММ 5,5 г, 1,5 экв.), тетрагидробутиламмонийфторид (2 мл, 2 М раствор в ТГФ) и 1-ацетил-4-(4-гидроксифенил)пиперазин (4,9 г). Смесь перемешивали и выдерживали при 90° С в течение 6 ч. Реакционную смесь фильтровали и затем удаляли N,N-диметилацетамид с помощью роторного испарителя. Остаток растворяли в воде (100 мл) и экстрагировали дважды этилацетатом. Этилацетатный слой сушили над сульфатом натрия и этилацетат удаляли при пониженом давлении, получая масло. Масло очищали на силикагеле, получая 3 г сложного метилового эфира. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: К раствору полученного на стадии А сложного метилового эфира (ММ 433, 3 г) в тетрагидрофуране (50 мл) добавляли триметилсиланолат калия (ММ 128, 0,9 г, 1,2 экв.). Смесь перемешивали в течение 2-3 ч до тех пор, пока не образовывался твердый осадок. После завершения гидролиза добавляли N-метилморфолин (2 мл), а затем РуВrоР (3,5 г, 1,2 экв.). Раствор перемешивали в течение 10 мин, затем еще в течение 2 ч добавляли при перемешивании водный раствор гидроксиламина. После завершения реакции (2 ч) растворитель удаляли на роторном испарителе. Остаток растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 1,2 г указанного в заголовке соединения в виде твердого вещества белого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) M+H: рассчитано для C24H29N3O6S2: 519, обнаружено: 519.
Пример 113: Получение тетрагидро-N-гидрокси-4-[[4-(4-тиофенокси)фенил]сульфонил-2Н-тиопиран-4-карбоксамида
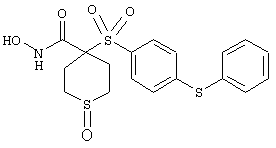
Стадия А: К раствору полученного на стадии В примера 50 сложного метилового эфира тиопирана (5 г) в уксусной кислоте (40 мл) добавляли смесь вода/перекись водорода (8 мл, 4 мл/4 мл, 30%-ный раствор). Процесс реакции в этом интенсивно перемешиваемом растворе контролировали с помощью ЖХВРОФ. После выдерживания при температуре окружающей среды в течение 3 ч добавляли воду и продукт дважды экстрагировали этилацетатом (100 мл). Этилацетатный слой сушили над сульфатом натрия. После удаления растворителя при пониженном давлении получали 4,5 г сложного метилового эфира сульфоксидпиран-Ph-пapa-F в виде твердого вещества белого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: К раствору полученного на стадии А сложного метилового эфира сульфоксидпиран-Ph-пapa-F (ММ 318, 5 г, 1,0 экв.) в ДМА (70 мл) добавляли карбонат цезия (ММ 4,5 г, 1,1 экв.) и тиофенол (1,5 г, 1,05 экв.). Смесь перемешивали в течение 2 ч при комнатной температуре. Реакционную смесь фильтровали и затем удаляли N,N-диметилацетамид с помощью роторного испарителя. Остаток растворяли в воде (100 мл) и экстрагировали дважды этилацетатом. Этилацетатный слой сушили над сульфатом натрия и этилацетат удаляли при пониженном давлении, получая масло. Масло очищали с помощью ЖХВРОФ на силикагеле, получая 2 г сложного метилового эфира сульфоксидпиранфенил-S-Ph. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия В: К раствору полученного на стадии Б сложного метилового эфира сульфоксидпиранфенил-S-Ph (ММ 590, 5 г) в тетрагидрофуране (100 мл) добавляли триметилсиланолат калия (ММ 128, 1,5 г, 2 экв.). Смесь перемешивали в течение 2-3 ч при температуре окружающей среды до тех пор, пока не образовывался твердый осадок. После завершения гидролиза добавляли N-метилморфолин (6 мл), а затем РуВrоР (4 г, 1,1 экв.). Раствор перемешивали в течение 10 мин, затем еще в течение 2 ч добавляли при перемешивании водный раствор гидроксиламина. После завершения реакции (12 ч) растворитель удаляли на роторном испарителе. Остаток растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 1,9 г указанного в заголовке соединения в виде твердого вещества белого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) M+H: рассчитано для С18Н19NО5S3: 425, обнаружено: 425.
Пример 114: Получение тетрагидро-N-гидрокси-4-[[4-[4-(4-гидроксифенил)тиофенокси]фенил]сульфонил-2Н-тиопиран-4-карбоксамида
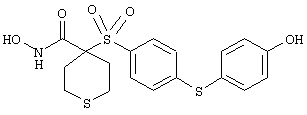
Стадия А: К раствору указанного в заголовке примера 50 соединения (ММ 402, 5 г, 1,0 экв.) в N,N-диметилацетамиде (70 мл) добавляли 4-гидрокситио-фенол (ММ 126, 1,6 г, 1,3 экв.), а затем карбонат калия (ММ 138, 5 г, 2,0 экв.). Реакционную смесь выдерживали при 65° С в течение 3 ч, пока результаты анализа с помощью ЖХВР не показали, что реакция завершена. Реакционную смесь фильтровали в вакууме, N,N-диметилацетамид удаляли в вакууме. Остаток растворяли в воде (100 мл) и экстрагировали дважды этилацетатом. Этилацетатный слой сушили над сульфатом натрия и этилацетат удаляли при пониженном давлении, получая пара-ОН-тиофеноксипроизводное в виде неочищенного масла. Результаты анализа с помощью lH-ЯMP, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: Полученное на стадии А пара-ОН-тиофеноксипроизводное, перемешивали в течение 2 ч в смеси HCl/диоксан (50 мл). Растворитель удаляли, остаток сушили и растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 2,1 г указанного в заголовке соединения в виде твердого вещества желтого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) М+Н: рассчитано для C18H19NO5S3: 425, обнаружено: 425.
Пример 115: Получение тетрагидро-N-гидрокси-4-[[4-(4-аминофенил)тиофенокси]фенил]сульaонил-2Н-тиопиран-4-карбоксамида
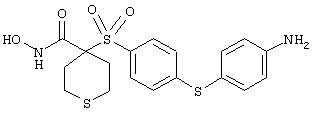
Стадия А: К раствору указанного в заголовке примера 50 соединения (ММ 402, 5 г, 1,0 экв.) в N,N-диметилацетамиде (70 мл) добавляли 4-аминотиофенол (ММ 126, 1,6 г, 1,3 экв.), а затем карбонат калия (ММ 138, 5 г, 2,0 экв.). Реакционную смесь выдерживали при 65° С в течение 3 ч, пока результаы анализа с помощью ЖХВР не показали, что реакция завершена. Реакционную смесь фильтровали и N,N-диметилацетамид удаляли в вакууме. Остаток растворяли в воде (100 мл) и экстрагировали дважды этилацетатом. Этилацетатный слой сушили над сульфатом натрия и этилацетат удаляли при пониженом давлении, получая пара-NН2-тиофеноксипроизводное в виде неочищенного масла. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: Полученное на стадии А пара-NН2-тиофеноксипроизводное перемешивали в течение 2 ч в смеси HCl/диоксан (50 мл). Растворитель удаляли, остаток сушили и растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 2,1 г указанного в заголовке соединения в виде твердого вещества желтого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) М+Н: рассчитано для С18Н20N2O4S3·С2НF3О2: 538, обнаружено: 538.
Пример 116: Получение тетрагидро-N-гидрокси-4-[[4-(4-тирамин)фенокси]фенил]сульфонил-2Н-тиопиран-4-карбоксамида
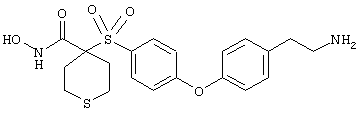
Стадия А: К раствору указанного в заголовке примера 50 соединения (ММ 402, 5 г, 1,0 экв.) в N,N-диметилацетамиде (50 мл) добавляли трипамин (3 г, 2 экв.), а затем карбонат цезия (10 г, 2,0 экв.). Реакционную смесь выдерживали при 95° С в течение 5 ч, пока результаты анализа с помощью ЖХВР не показали, что реакция завершена: Реакционную смесь фильтровали и N,N-диметилацетамид удаляли в вакууме. Растворитель удаляли, остаток сушили и растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (ТФК, рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 2,5 г неочищенного сложного метилового эфира в виде твердого вещества желтого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: Полученный на стадии А неочищенный сложный метиловый эфир перемешивали в течение 1 ч в водном растворе HCl (50 мл). Растворитель удаляли, остаток сушили и растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 2,2 г трифторацетата указанного в заголовке соединения в виде пены желтого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) M+H: рассчитано для C20H24N2O5S2·C2HF3O2: 550, обнаружено: 550.
Пример 117: Получение тетрагидро-N-гидрокси-4-[[4-(4-гидроксифенил-глицин)]фенил]сульфонил-2Н-тиопиран-4-карбоксамида

Стадия А: К раствору указанного в заголовке примера 50 соединения (ММ 402, 5 г, 1,0 экв.) в N,N-диметилацетамиде (50 мл) добавляли гидроксифенил-глицин (3 г, 2 экв.), а затем карбонат цезия (10 г, 2,0 экв.). Реакционную смесь выдерживали при 95° С в течение 5 ч, пока результаты анализа с помощью ЖХВР не показали, что реакция завершена. Реакционную смесь фильтровали и N,N-диметилацетамид удаляли в вакууме. Растворитель удаляли, остаток сушили и растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 2,0 г неочищенного сложного метилового эфира в виде твердого вещества рыжевато-корчневого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: Полученный на стадии А неочищенный сложный метиловый эфир перемешивали в течение 1 ч в водном растворе HCl (50 мл). Растворитель удаляли, остаток сушили и растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 2,2 г трифторацетата указанного в заголовке соединения в виде пены/твердого вещества рыжевато-коричневого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) M+H: рассчитано для С20Н22N2O7S2·С2НF3О2: 580, обнаружено: 580.
Пример 118: Получение тетрагидро-N-гидрокси-4-[[4-(4-гидроксифенил-глицин)]фенил]сульфонил-2Н-тиопиран-4-карбоксамида
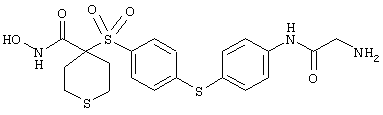
Стадия А: Раствор указанного в заголовке примера 115 соединения (ММ 518, 2,5 г, 1,0 экв.) в ТГФ (25 мл) и N-Вос-защищенном N-гидроксисукцинилглицине (2,1 г, 2 экв.), содержащий N-метилморфолин (2 мл) и 4-диметиламинопиридин (250 мг), перемешивали в течение 12 ч. После того, как по данным анализа с помощью ЖХВРОФ реакция завершилась, растворитель удаляли при пониженном давлении, получая масло. Добавляли при перемешивании в течение еще 1-2 ч 10%-ный водный раствор соляной кислоты. Затем раствор очищали с помощью препаративной ЖХВРОФ, получая 1,2 г пены/твердого вещества белого цвета в виде трифторацетата. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. Твердое вещество сушили при пониженном давлении, затем суспендировали в диэтиловом эфире, после чего добавляли смесь 4 н. HCl/диоксан (20 мл). Фильтровали гидрохлорид и промывали диэтиловым эфиром, получая указанное в заголовке соединение в виде твердого вещества рыжевато-коричневого цвета (1,1 г). Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) М+Н: рассчитано для C20H23N3O5s2·C2HF3o2: 595, обнаружено: 595.
Пример 119: Получение моногидрохлорида тетрагидро-N-гидрокси-4-[[4-(4-пиридинилтио)]фенил]сульфонил-2Н-тиопиран-4-карбоксамида
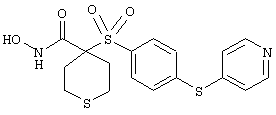
Стадия А: К раствору указанного в заголовке примера 50 соединения (ММ 402, 5 г, 1,0 экв.) в N,N-диметилацетамиде (50 мл) добавляли 4-тиопиридин (3 г, 2 экв.), а затем карбонат цезия (10 г, 2,0 экв.). Реакционную смесь выдерживали при 75° С в течение 5 ч, пока результаты анализа с помощью ЖХВР не показывали, что реакция завершена. Реакционную смесь фильтровали и N,N-диметилацетамид удаляли в вакууме. Растворитель удаляли, остаток сушили и растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 2,0 г неочищенного S-пиридил-ТГП-защищенного тиопирана в виде твердого вещества коричневого цвета. Результаты анализа с помощью 1H-ЯMP, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: Полученный на стадии А неочищенный S-пиридил-ТГП-защищенный тиопиран перемешивали в течение 1 ч в водном растворе HCl (50 мл). Растворитель удаляли, остаток сушили и растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 1,8 г трифторацетата указанного в заголовке соединения в виде пены/стекла рыжевато-коричневого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) M+H: рассчитано для C17H18N2O4S3·HCl: 447, обнаружено: 447.
Пример 120: Получение 4-[[4-[(4-аминофенил)тио)]фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
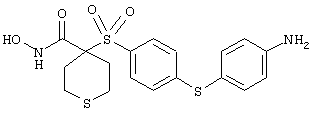
Стадия А: К раствору указанного в заголовке примера 55 соединения (ММ 387, 5 г, 1,0 экв.) в N,N-диметилацетамиде (50 мл) добавляли 4-аминотиофенол (3 г, 2 экв.), а затем карбонат калия (10 г, 2,0 экв.). Реакционную смесь выдерживали при 60° С в течение 5 ч, пока результаты анализа с помощью ЖХВР не показывали, что реакция завершена. Реакционную смесь фильтровали и ДМА удаляли в вакууме. Растворитель удаляли, остаток сушили и растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 2,0 г неочищенного соединения, представляющего собой 4-амино-S-Рh-ТГП-защищенный тиопиран в виде твердого вещества коричневого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: Полученный на стадии А неочищенный 4-амино-S-Рh-ТГП-защищенный тиопиран перемешивали в течение 1 ч в водном растворе HCl (50 мл). Растворитель удаляли, остаток сушили и растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 1,4 г трифторацетата указанного в заголовке соединения в виде пены/стекла рыжевато-коричневого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) M+H: рассчитано для C18H20N2O5S2: 408, обнаружено: 408.
Пример 121: Получение тетрагидро-N-гидрокси-4-[[4-[(2-метил-5-бензотиазолил)окси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
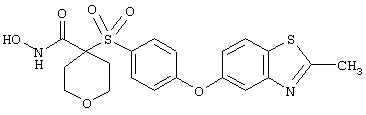
Стадия А: К раствору указанного в заголовке примера 55 соединения (ММ 387, 10 г, 1,0 экв.) в ДМА (50 мл) добавляли гидроксиметилбензотиазол (8 г, 1,5 экв.), а затем карбонат цезия (20 г, 2,0 экв.). Реакционную смесь выдерживали при 90° С в течение 5 ч, пока результаты анализа с помощью ЖХВР не показывали, что реакция завершена. Реакционную смесь охлаждали, затем фильтровали и отбрасывали N,N-диметилацетамид. Осадок на фильтре растворяли в 10%-ном водном растворе HCl и перемешивали в течение 30 мин для удаления солей цезия. Требуемое твердое вещество выделяли из раствора в виде смолы. Эту смолу растворяли в этилацетате (100 мл), промывали водой и сушили над сульфатом натрия. Растворитель удаляли в вакууме, получая масло, которое растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 2-метил-5-бензотиазолилоксипроизводное. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: Полученное на стадии А 2-метил-5-бензотиазолилоксипроизводное перемешивали в течение 1 ч в смеси водный раствор HCl (20 мл)/ацетонитрил (20 мл). Растворитель концентрировали и выделившееся твердое вещество фильтровали, получая 6,5 г указанного в заголовке соединения. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) M+H: рассчитано для C20H20N2O6S2:448, обнаружено: 448.
Пример 122: Получение 4-[[4-(4-хлор-3-фторфенокси)фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
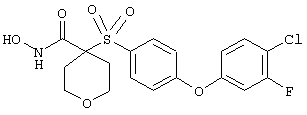
Стадия А: К раствору указанного в заголовке примера 55 соединения (ММ 387, 10 г, 1,0 экв.) в N,N-диметилацетамиде (50 мл) добавляли 4-хлор-3-фторфенол (7 г, 1,4 экв.), а затем карбонат цезия (20 г, 2,0 экв.). Реакционную смесь выдерживали при 90° С в течение 5 ч, пока результаты анализа с помощью ЖХВР не показывали, что реакция завершена. Реакционную смесь охлаждали, затем фильтровали и отбрасывали ДМА. Осадок на фильтре растворяли в 10%-ном водном растворе HCl и перемешивали в течение 30 мин для удаления солей цезия. Требуемое 4-хлор-3-фторфеноксипроизводное выделяли из раствора и фильтровали. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: Полученное на стадии А 4-хлор-3-фторфеноксипроизводное (3,4 г), перемешивали в течение 1 ч в смеси водный раствор HCl (20 мл) /ацетонитрил (20 мл). Растворитель концентрировали и выделившееся
твердое вещество фильтровали, получая 2,0 г указанного в заголовке соединения. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) M+H: рассчитано для C18H17ClFNO6S: 429, обнаружено: 429.
Пример 123: Получение трифторацетата 4-[[4-[4-(4-ацетил-1-пиперазинил)фенокси]фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
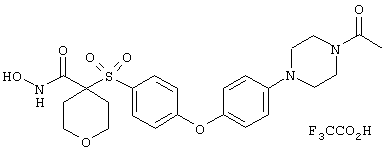
Стадия А: К раствору указанного в заголовке примера 55 соединения (ММ 387, 10 г, 1,0 экв.) в ДМА (50 мл) добавляли 1-ацетил-4-(4-гидроксифенил)пиперазин (3 г, 2 экв.), а затем карбонат цезия (10 г, 2,0 экв.). Реакционную смесь выдерживали при 90° С в течение 5 ч, пока результаты анализа с помощью ЖХВР не показывали, что реакция завершена. Реакционную смесь фильтровали и удаляли ДМА в вакууме. Остаток растворяли в смеси вода/ацетонитрил, подкисляли с помощью трифторуксусной кислоты (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 3,1 г неочищенного 4-ацетил-1-пиперазинилфеноксипроизводного. Результаты анализа с помощью 1Н-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: Полученное на стадии А 4-ацетил-1-пиперазинилфеноксипроиз-водное перемешивали в течение 1 ч в водном растворе HCl (50 мл). Растворитель удаляли и остаток растворяли в смеси вода/ацетонитрил, подкисляли с помощью ТФК (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 2,0 г трифторацетата указанного в заголовке соединения в виде пены рыжевато-коричневого цвета. Результаты анализа с помощью 1Н-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) M+H: рассчитано для C24H29N3O7S· C2HF3O2: 617, обнаружено: 617.
Пример 124: Получение трифторацетата N,N-диметил-5-[4-[[тетрагидро-4-[(гидроксиамино)карбонил]-2Н-пиран-4-ил]сульфонил]фенокси]-1Н-индол-2-карбоксамида
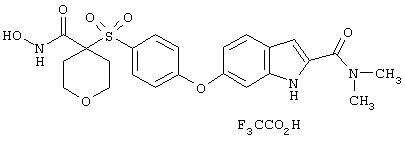
Стадия А: К раствору указанного в заголовке примера 55 соединения (ММ 387, 5 г, 1,0 экв.) в ДМА (50 мл) добавляли 5-гидрокси-2-индолдиметилкарбоксилат (3 г, 2 экв.), а затем карбонат цезия (10 г, 2,0 экв.). Реакционную смесь выдерживали при 90° С в течение 5 ч, пока результаты анализа с помощью ЖХВР не показывали, что реакция завершена. Реакционную смесь фильтровали и отделяли ДМА в вакууме. Остаток растворяли в смеси вода/ацетонитрил, подкисляли с помощью ТФК (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 2,1 г неочищенного ТГП-защищенного пирангидроксамата в виде твердого вещества коричневого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: Полученный на стадии А ТГП-защищенный пирангидроксамат перемешивали в течение 1 ч в водном растворе HCl (50 мл). Растворитель удаляли, остаток сушили и растворяли в смеси вода/ацетонитрил, подкисляли с помощью ТФК (до рН 2), затем очищали с помощью препаративной ЖХВРОФ, получая 1,5 г трифторацетата указанного в заголовке соединения в виде твердого вещества рыжевато-коричневого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) М+Н: рассчитано для C23H25N3O7S: 487, обнаружено: 487.
Пример 125: Получение тетрагидро-N-гидрокси-4-[[4-[4-(1-метилэтил)фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
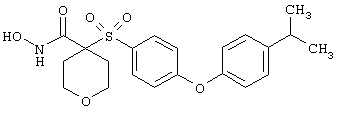
Стадия А: К раствору указанного в заголовке примера 55 соединения (ММ 387, 5 г, 1,0 экв.) в ДМА (50 мл) добавляли 4-изопропилфенол (3 г, 2 экв.), а затем карбонат цезия (10 г, 2,0 экв.). Реакционную смесь выдерживали при 90° С в течение 8 ч, пока результаты анализа с помощью ЖХВР не показывали, что реакция завершена. Реакционную смесь фильтровали и отбрасывали часть, содержащую ДМА. Остаток на фильтре растворяли в 10%-ном водном растворе HCl и перемешивали в течение 30 мин для удаления солей цезия. Отделяли и фильтровали твердое вещество (3,5 г), представляющее собой изопропилфеноксифенил-ТГП-защищенный гидроксамат. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения.
Стадия Б: К раствору, содержащему водный раствор HCl (20 мл) и ацетонитрил (20 мл), добавляли при перемешивании полученный на стадии А неочищенный изопропилфеноксифенил-ТГП-защищенный гидроксамат и образовавшуюся смесь перемешивали в течение 1-2 ч. Растворитель концентрировали, пропуская поток азота над поверхностью раствора. Твердое вещество фильтровали и сушили, получая 2,2 г указанного в заголовке соединения в виде твердого вещества рыжевато-коричневого цвета. Результаты анализа с помощью 1H-ЯМР, МС и ЖХВР подтверждали получение требуемого соединения. МС (CI) M+H: рассчитано для C21H25NO6S: 419, обнаружено: 419.
Пример 126: Получение смолы II
Стадия 1: Связывание полученного на стадии Г примера 55 соединения со смолой I
В круглодонную колбу объемом 500 мл загружали смолу I [Ployd и др., Tetrahedron Lett. 1996, 37, 8045-8048] (8,08 г, 9,7 ммоль) и 1-метил-2-пирролидинон (50 мл). Присоединяли магнитную мешалку и медленно перемешивали суспензию смолы. К суспензии добавляли отдельно приготовленный раствор полученного на стадии Г примера 55 соединения (5,58 г, 19,4 ммоль) в 1-метил-2-пирролидиноне (35 мл), а затем в виде одной порции гексафторфосфат бензотриазол-1-илокситриспирролидинофосфония (10,1 г, 19,4 ммоль). После растворения гексафторфосфата по каплям добавляли 4-метилморфолин (4,26 мл, 39 ммоль). Реакционную суспензию перемешивали в течение 24 ч при комнатной температуре, затем смолу собирали в дисковую воронку из оплавленного стекла и промывали N,N-диметилформамидом, метанолом, метиленхлоридом и диэтиловым эфиром (3× 30 мл каждого растворителя). Смолу сушили в вакууме, получая 10,99 г связанного с полимером гидроксамата в виде полимерного твердого вещества рыжевато-коричневого цвета. Теоретическая загрузка полимера составляла 0,91 ммоль/г. Анализ с помощью инфракрасной спектроскопии с фурье-преобразованием (ИКФП) показал наличие полос при 1693 и 3326 см-1, являющихся характерными признаками присутствия соответственно гидроксаматкарбонильных и азот-водородных связей.
Стадия 2: Получение смолы III
Взаимодействие смолы II с нуклеофилами
Смолу II (50 мг, 0,046 ммоль) загружали в стеклянный флакон и в сосуд добавляли 0,5М раствор нуклеофила в 1-метил-2-пирродиноне (1 мл). В случае фенольных и тиофенольных нуклеофилов добавляли карбонат цезия (148 мг, 0,46 ммоль), а в случае замещенных пиперазиновых нуклеофилов добавляли карбонат калия (64 мг, 0,46 ммоль). Флакон закрывали крышкой и нагревали в течение 24-48 ч до 70-155° С, после чего охлаждали до комнатной температуры. Смолу сливали и промывали 1-метил-2-пирродиноном, смесью 1-метил-2-пирродинон/вода, водой, смесью 10%-ная уксусная кислота/вода, метанолом и метиленхлоридом (3× 3 мл каждого растворителя).
Стадия 3: Отщепление гидроксамовых кислот от полимерной подложки
Смолу III обрабатывали в течение 1 ч при комнатной температуре смесью трифторуксусная кислота/вода (19:1, 1 мл). В течение этого времени смола приобретала темно-красный цвет. После этого смолу сливали и промывали смесью трифторуксусная кислота/вода (19:1) и метиленхлоридом (2× 1 мл каждого растворителя), собирая объединенные фильтраты в мерный флакон. Летучие компоненты удаляли в вакууме, после чего к остатку добавляли смесь толуол/метиленхлорид (по 2 мл каждого). Смесь снова концентрировали в вакууме. Характеристики продукта определяли с помощью масс-спектроскопии с ионизацией электронным ударом МС (ES).
Перечисленные ниже гидроксамовые кислоты синтезировали из смолы II, используя описанные для стадии 2 условия с применением указанных нуклеофилов, после чего отделяли от полимера с использованием реакционных условий, указанных для стадии 3.
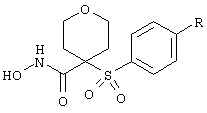
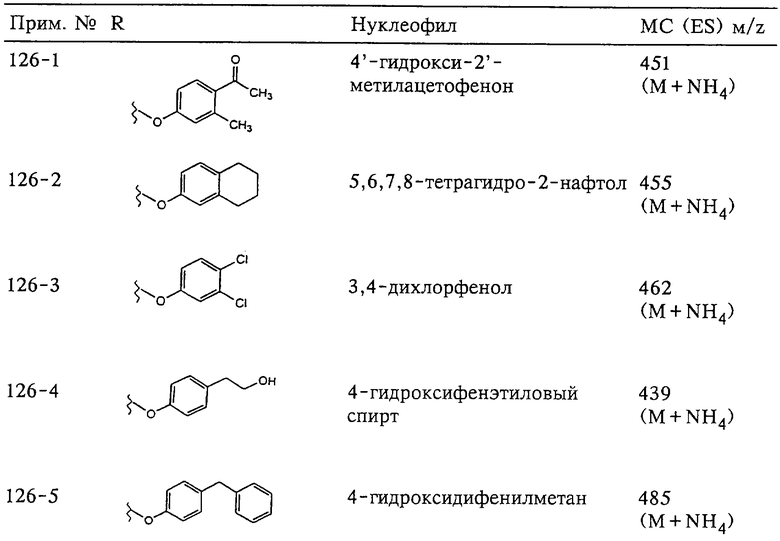
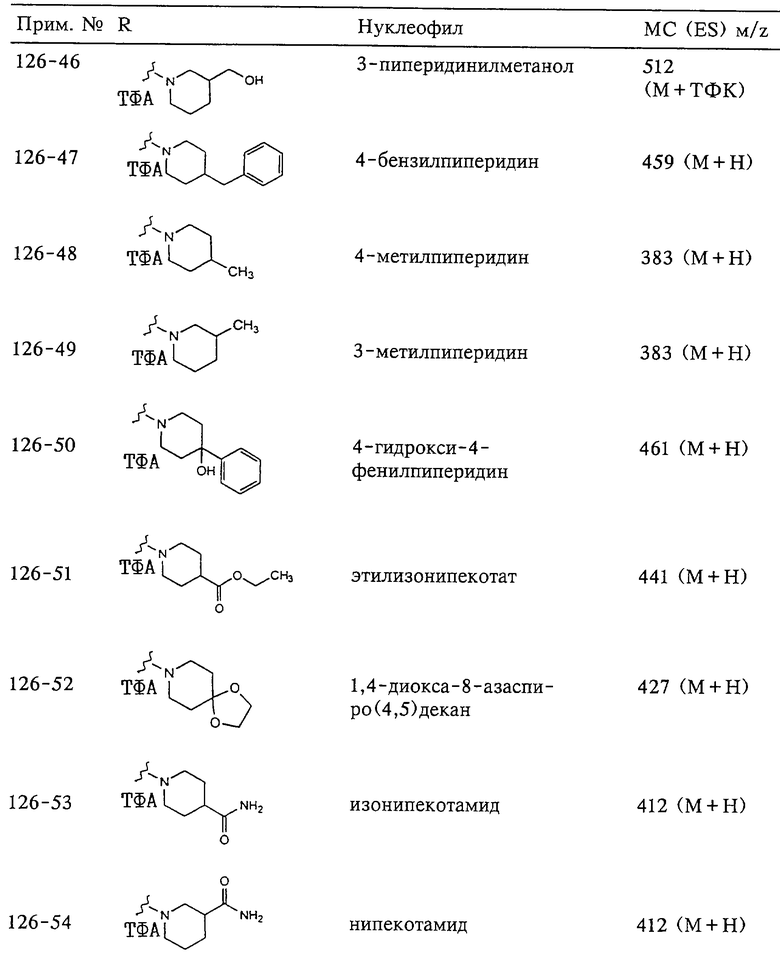
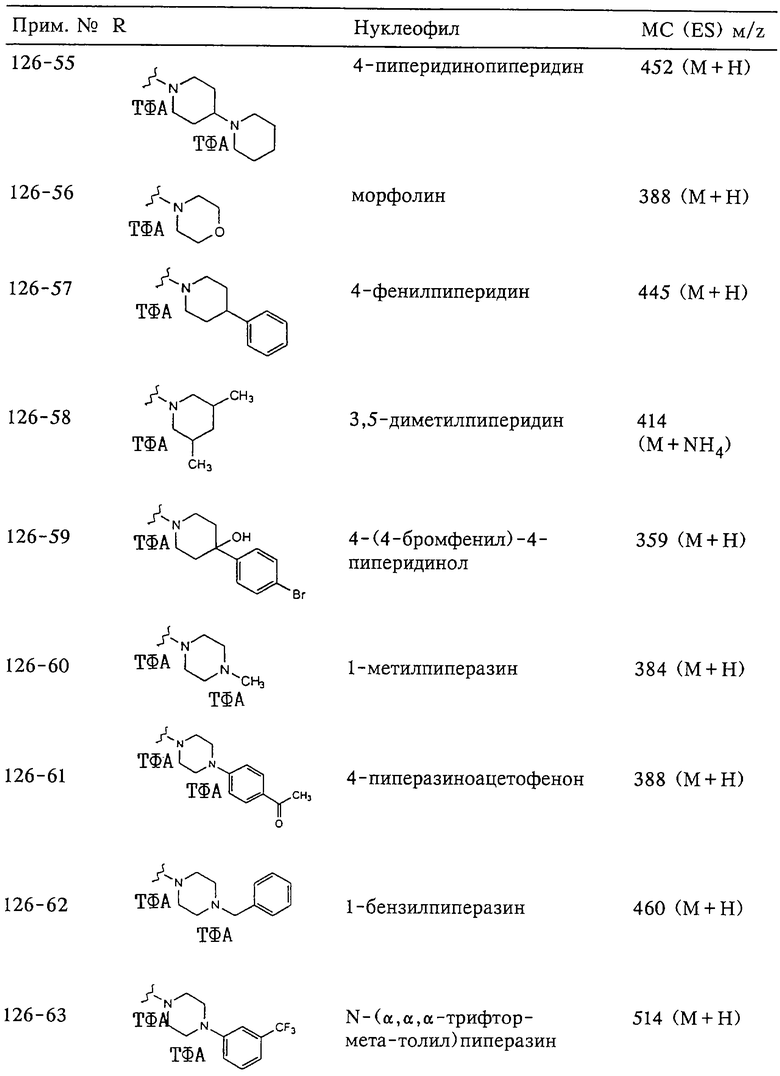
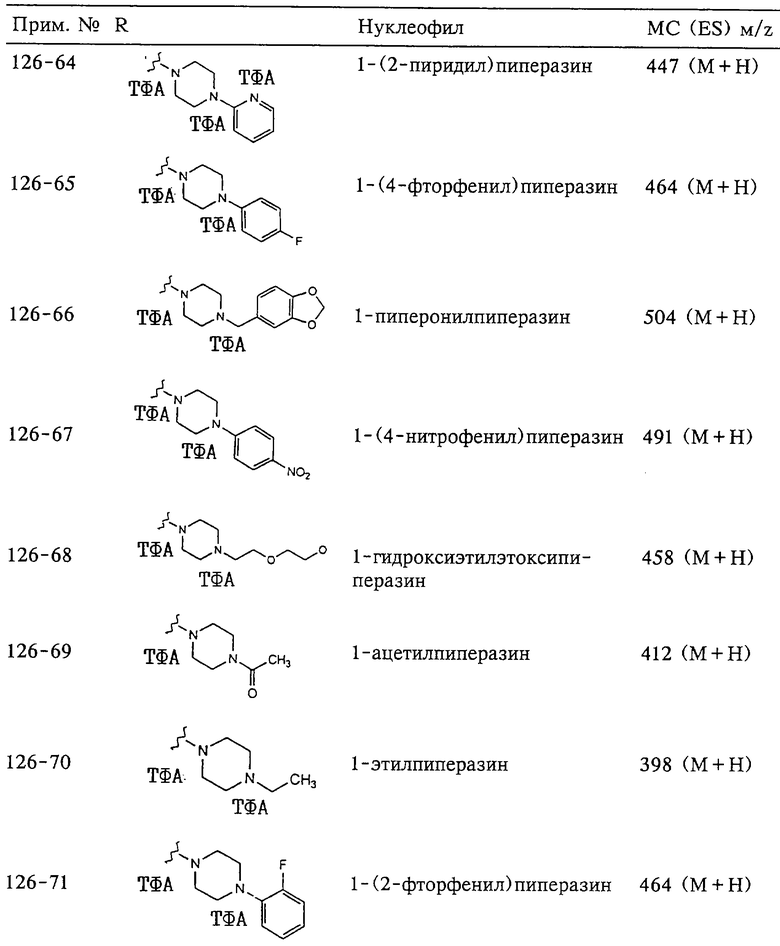
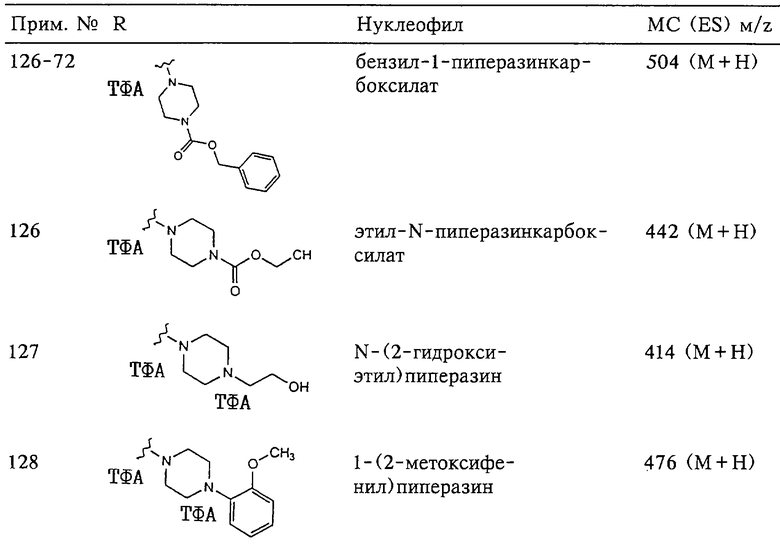
Пример XX: Крупномасштабный способ получения смолы IIIa
В высушенную в печи трехгорлую круглодонную колбу, снабженную датчиком температуры, воздушной лопастной мешалкой и впускным клапаном для азота, загружали смолу II (5 г, 0,91 ммоль). В колбу добавляли безводный 1-метил-2-пирролидинон (35 мл), а затем этилизонипекотат (7,0 мл, 45,5 ммоль). Суспензию смолы медленно перемешивали воздушной мешалкой и смесь нагревали в течение 65 ч до 80° С с помощью нагревательного кожуха. После этого колбу охлаждали до комнатной температуры.
Смолу собирали в дисковую воронку из оплавленного стекла и промывали N,N-диметилформамидом, метанолом и метиленхлоридом (3× 30 мл каждого растворителя). Смолу сушили в вакууме, получая 5,86 г смолы IIIa в виде смоляных шариков беловатого цвета. Теоретическая загрузка полимера составляла 0,81 ммоль/г. Путем расщепления с помощью ТФК 50 мг смолы IIIa согласно методу, описанному для стадии 3, получали 10,4 мг твердого вещества беловатого цвета, неотличимого спектроскопическим методом от продукта реакции, полученного в примере 211 с использованием этилизонипекотата.
Пример YY: Крупномасштабный способ получения смолы IIIb
Получение смолы IIIb осуществляли по методу, описанному для получения смолы Ilia, за исключением того, что вместо этилизонипекотата использовали этилнипекотат. После сушки в вакууме получали смолу IIIb в виде смоляных шариков светло-желтого цвета с выходом 5,77 г. Теоретическая загрузка полимера составляла 0,81 ммоль/г. Путем расщепления с помощью ТФК 50 мг смолы IIIb по методу, описанному для стадии 3, получали 14,7 мг твердого вещества беловатого цвета, неотличимого спектроскопическим методом от продукта реакции, полученного в примере 212 с использованием этилнипекотата.
Стадия 4: Гидролиз связанного с полимером сложного эфира
Получение смолы IVa
В трехгорлую круглодонную колбу, снабженную воздушной лопастной мешалкой, загружали смолу IIIa (5,8 г, 4,5 ммоль). В колбу добавляли 1,4-диоксан и суспензию смолы перемешивали в течение 15 мин. Затем добавляли 4 М раствор КОН (5 мл, 20 ммоль) и смесь перемешивали в течение 44 ч. После этого смолу собирали в дисковую воронку из оплавленого стекла и промывали смесью диоксан/вода (9:1), водой, смесью 10%-ная уксусная кислота/вода, метанолом и метиленхлоридом (3× 30 мл каждого растворителя). Смолу сушили в вакууме, получая 5,64 г смолы IVa в виде полимерных шариков беловатого цвета. Анализ с помощью ИКФП показал наличие полос при 1732 и 1704 см-1 и широкой полосы при 2500-3500 см-1. Теоретическая загрузка связанной с полимером кислотой составляла 0,84 ммоль/г.
Получение смолы IVb
Работая по методу, описанному для стадии 4, смолу IIIb (5,71 г, 4,5 ммоль) превращали в смолу IVb (5,61 г). Анализ с помощью ИКФП показал наличие полос при 1731 и 1705 см-1 и широкой полосы при 2500-3500 см. Теоретическая загрузка связанной с полимером кислотой составляла 0,84 ммоль/г.
Стадия 5: Образование амидной связи: получение смолы V В снабженный фриттой реакционый сосуд загружали либо смолу IVa, либо смолу IVb (50 мг, 0,042 ммоль) и сосуд закрывали крышкой в атмосфере азота. Добавляли 0,5 М раствор гидроксибензотриазола в 1-метил-2-пирролидиноне (0,3 мл, 0,15 ммоль), а затем 0,5 М раствор диизопропилкарбодиимида в 1-метил-2-пирролидиноне (0,3 мл, 0,15 ммоль). Смолу перемешивали в течение 15 мин с помощью мешалки верхнего крепления, затем добавляли 0,7 М раствор амина в 1-метил-2-пирролидиноне (0,3 мл, 0,21 ммоль). Реакционную смесь перемешивали в течение 6 ч, затем смолу сливали и промывали 1-метил-2-пирролидиноном (3× 1 мл). Реакцию повторяли с использованием указанных выше количеств реагентов. Реакционную смесь перемешивали в течение 16 ч, затем смолу сливали и промывали 1-метил-2-пирролидиноном, метанолом и метиленхлоридом (3× 1 мл каждого растворителя).
Перечисленные ниже гидроксамовые кислоты синтезировали с использованием указанной связанной с полимером кислоты и указанного амина в реакционных условиях, описанных для стадии 5, с последующим отделением от полимера в реакционных условиях, описанных для стадии 3.
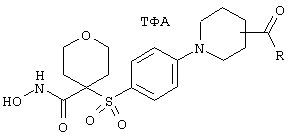
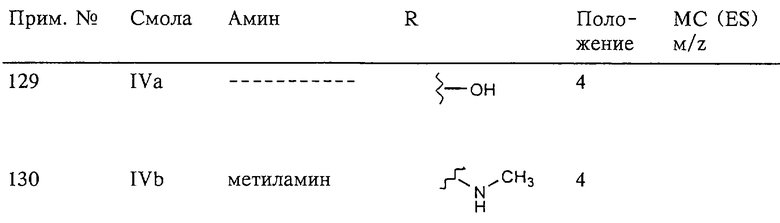

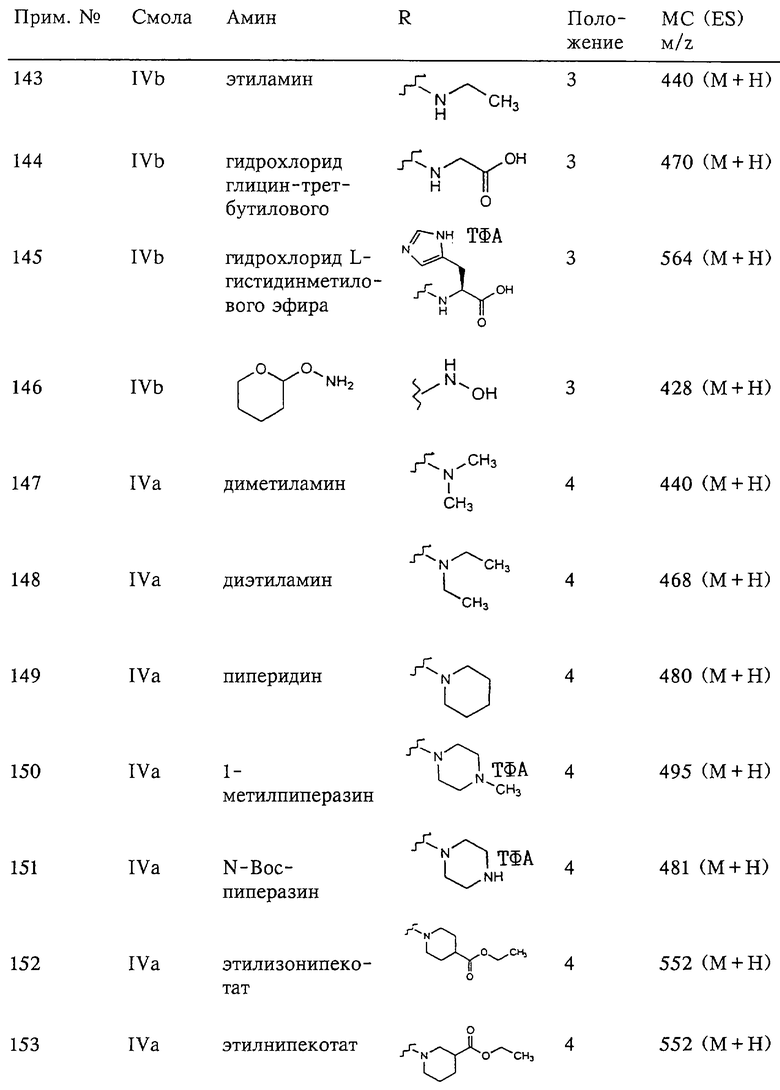
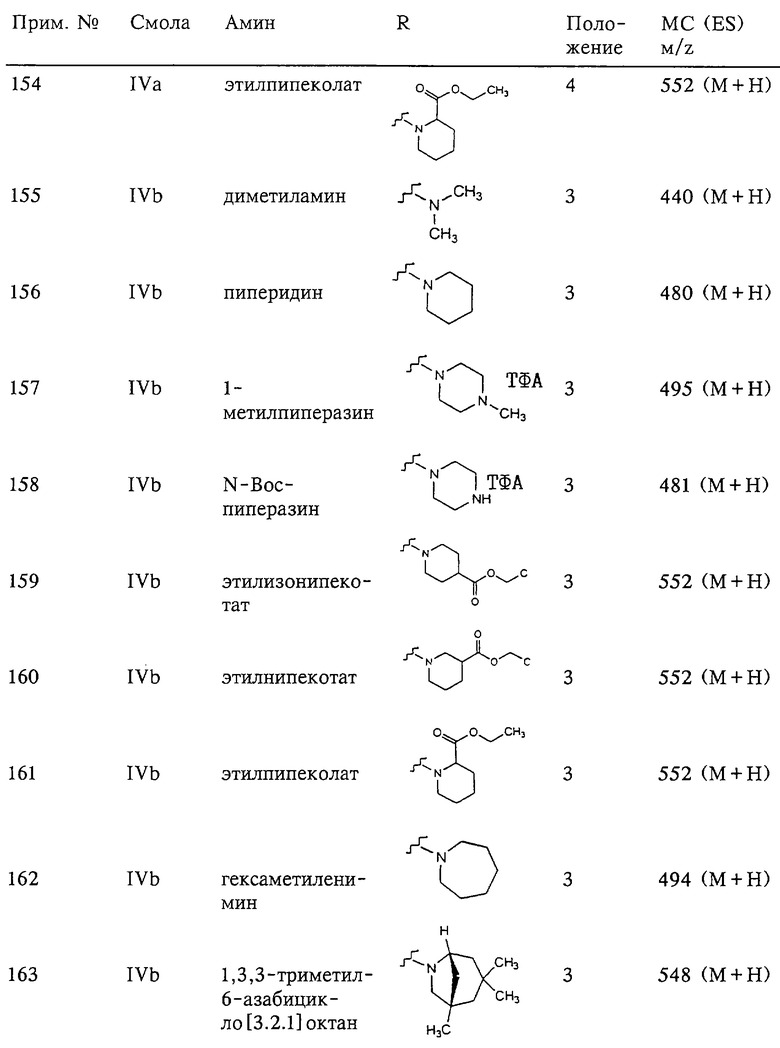
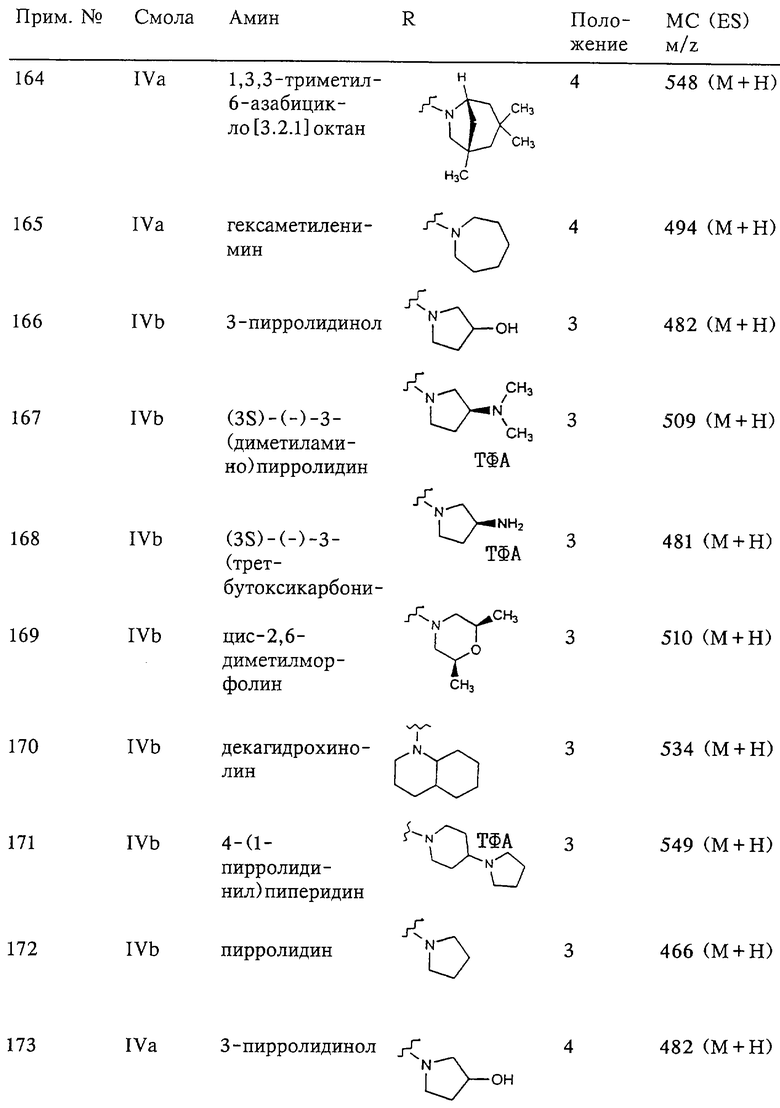
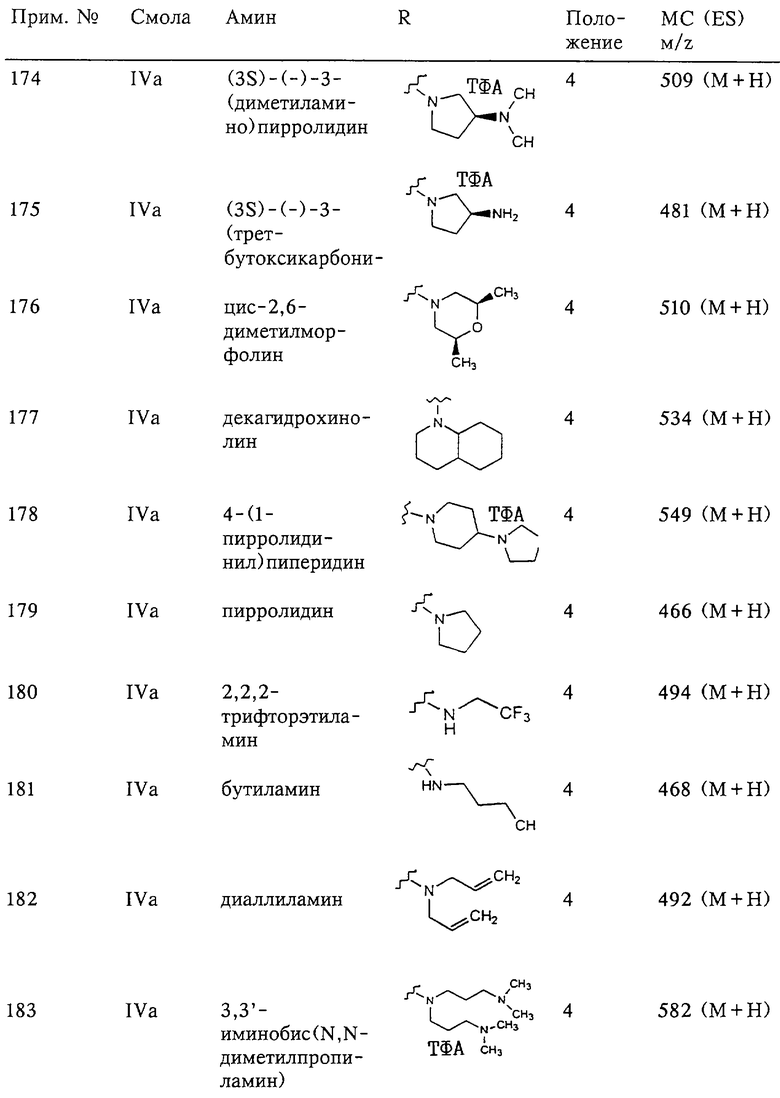
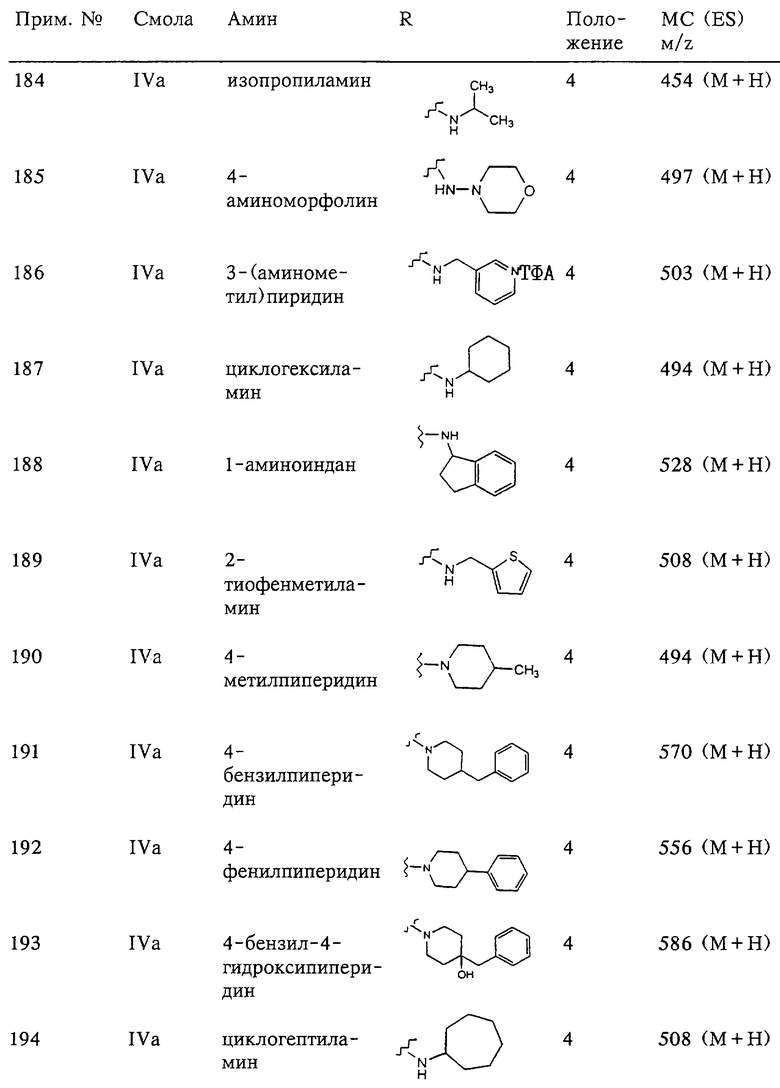
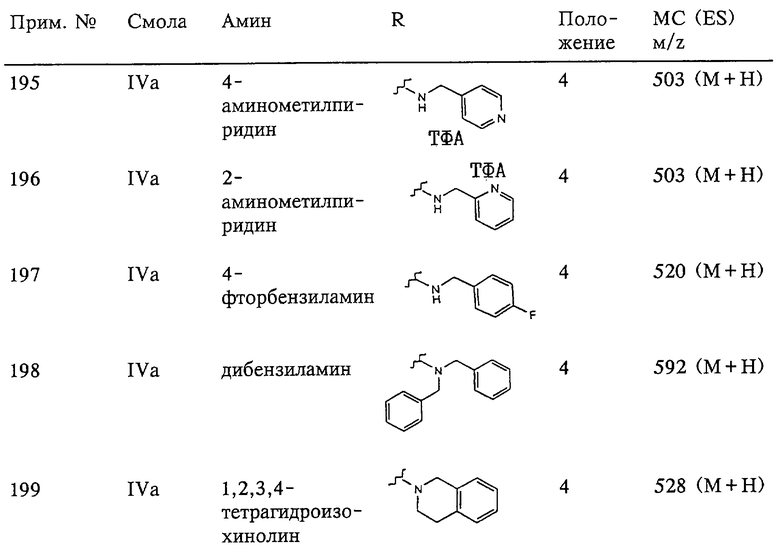
Крупномасштабный способ получения смолы IIIc
В высушенную в печи трехгорлую круглодонную колбу, снабженную воздушной лопастной мешалкой, датчиком температуры и впускным клапаном для азота, загружали смолу II (3,01 г, 2,74 ммоль). Добавляли 1-метил-2-пирролидинон (25 мл), а затем пиперазин (2,36 г, 27,4 ммоль) и карбонат цезия (8,93 г, 27,4 ммоль). Добавляли дополнительную порцию 1-метил-2-пирролидинона (10 мл) и реакционную смесь нагревали до 100° С и перемешивали в течение 18 ч. Колбу охлаждали до комнатной температуры и собирали смолу в дисковую воронку из оплавленного стекла и промывали смесью N,N-диэтилформамид/вода (1:1), водой, смесью 10%-ная уксусная кислота/вода, метанолом и метиленхлоридом (3× 30 мл каждого растворителя). После сушки в вакууме получали 3,14 г смолы IIb в виде смоляных шариков светло-желтого цвета. Теоретическая загрузка полимера составляла 0,86 ммоль/г. Путем расщепления с помощью ТФК 50 мг смолы IIIb по методу, описанному для стадии 3, получали 21 мг твердого вещества беловатого цвета, не отличимого спектроскопическим методом от продукта, приведенного в примере 209.
Стадия 6: Образование амидной связи со смолой IIIc: получение смолы VI
В снабженный фриттой реакционый сосуд загружали карбоновую кислоту (0,215 ммоль) и 1-гидроксибензотриазол (44 мг, 0,326 ммоль). Сосуд закрывали крышкой в атмосфере азота и добавляли 1-метил-2-пирролидинон, а затем диизопропилкарбодиимид (0,034 мл, 0,215 ммоль). Раствор перемешивали в течение 15 мин с помощью мешалки верхнего крепления, затем добавляли в виде одной порции смолу IIIc (50 мг, 0,043 ммоль). Реакционную смесь перемешивали в течение 16 ч, затем смолу сливали и промывали 1-метил-2-пирролидиноном, метанолом и метиленхлоридом (3× 1 мл каждого растворителя). В случае N-9-флуоренилметоксикарбонил-защищенных аминокислот смолу дополнительно обрабатывали в течение 30 мин раствором пиперидин/N,N-диметилформамид (1:4, 1 мл). Смолу сливали и промывали N,N-диметилформамидом, метанолом и метиленхлоридом (3× 1 мл каждого растворителя).
Перечисленные ниже гидроксамовые кислоты синтезировали из смолы IIIc по методу, описанному для стадии 6, с использованием указанной карбоновой кислоты с последующим отделением от полимера в реакционных условиях, описанных для стадии 3.
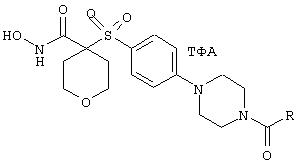
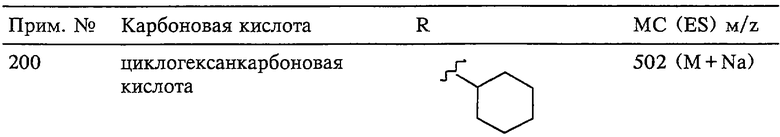
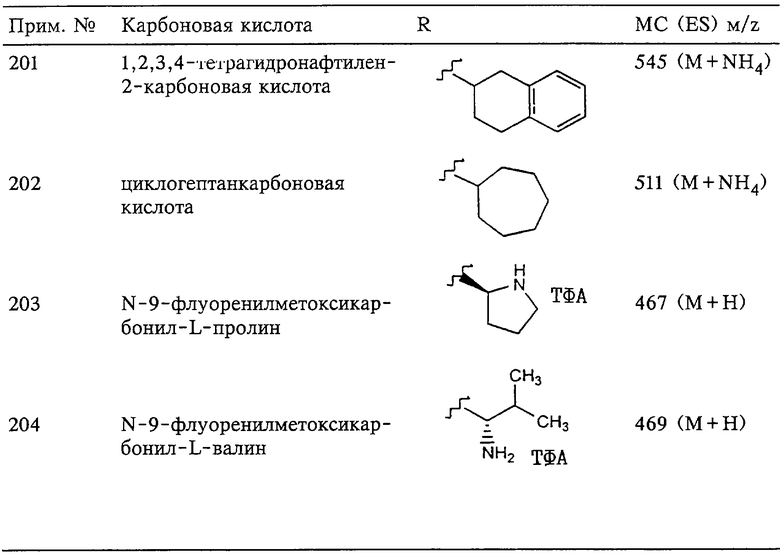
Стадия 7: Получение смолы VII
Смолу IIIс (1,0 г, 0,86 ммоль) загружали в высушенную в печи круглодонную колбу объемом 100 мл и присоединяли магнитную мешалку и мембрану с иглой для впуска азота. Добавляли метиленхлорид (10 мл) и суспензию смолы медленно перемешивали. Добавляли в виде одной порции пара-нитрофенилхлорформиат (0,867 г, 4,3 ммоль), а затем по каплям добавляли диизопропилэтиламин (0,75 мл, 4,3 ммоль). При добавлении наблюдалось небольшое повышение температуры. Реакционную смесь перемешивали в течение 18 ч при комнатной температуре, после чего смолу собирали в дисковую воронку из оплавленного стекла и промывали метиленхлоридом, метанолом и метиленхлоридом (3× 10 мл каждого растворителя).
Связанный с полимером продукт сушили в вакууме, получая 1,25 г смолы VII в виде смоляных шариков коричневого цвета. Анализ с помощью ИКФП-микроскопии показал наличие полос при 1798, 1733, 1696 и 1210 см-1. Теоретическая загрузка полимера составляла 0,75 ммоль/г.
Стадия 8: Взаимодействие смолы VII со смолой VIII. обработанной аминами
Во флакон объемом 8 мл загружали смолу VII (50 мг, 0,038 ммоль), к нему присоединяли небольшую магнитную мешалку и добавляли 0,5 М раствор амина в 1-метил-2-пирролидиноне (1 мл). Флакон закрывали крышкой и нагревали до 50° С. Суспензию смолы осторожно перемешивали в течение 15 ч, затем флакон охлаждали до комнатной температуры. Смолу собирали в реакционный сосуд с фриттой и промывали 1-метил-2-пирролидиноном, метанолом и метиленхлоридом (3× 10 мл каждого растворителя).
Перечисленные ниже гидроксамовые кислоты синтезировали из смолы VII в реакционных условиях, описанных для стадии 8, с использованием указанного амина с последующим отделением от полимера в реакционных условиях, описанных для стадии 3.

Пример XXX: Получение 4-[(4-бромфенил)сульфонил]тетрагидро-2Н-пиран-4-карбоновой кислоты
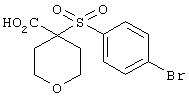
Стадия А: Получение
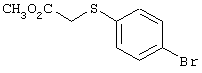
В высушенную в печи трехгорлую круглодонную колбу объемом 500 мл загружали в камере, заполненной азотом, 60%-ную масляную дисперсию гидрида натрия (4,0 г, 0,1 моль) и к колбе присоединяли впускной клапан для азота, датчик температуры, воздушную лопастную мешалку и резиновую мембрану. В колбу добавляли безводный тетрагидрофуран (200 мл), затем ее охлаждали в ледяной бане. По каплям добавляли 4-бромтиофенол (18,91 г, 0,1 моль), поддерживая температуру ниже 7° С. Во время добавления наблюдали интенсивное выделение газа. После завершения добавления смесь перемешивали при охлаждении в течение 10 мин. Затем по каплям добавляли метилбромацетат (9,5 мл, 0,1 моль), поддерживая температуру ниже 7° С. Реакционную смесь перемешивали при охлаждении в течение 10 мин, после чего ледяную баню удаляли и смесь перемешивали еще в течение 30 мин. Реакцию прекращали добавлением 5 мл воды, после чего растворитель удаляли на роторном испарителе. Остаток в виде масла распределяли между этилацетатом (200 мл) и водой (200 мл). Органический слой промывали смесью 5%-ная соляная кислота/вода (1× 200 мл), насыщенным раствором бикарбоната натрия (1× 200 мл) и соляным раствором (1× 200 мл). Органическую фазу сушили над сульфатом магния и концентрировали, получая 24,53 г продукта в виде масла желтого цвета (94%). Результаты анализа с помощью lH-ЯMP подтверждали, что вещество имело требуемое строение. Анализ с помощью масс-спектроскопии показал, что величина м/z равна 260 (М+Н).
Стадия Б: Получение
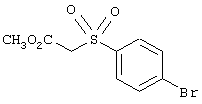
Полученное выше на стадии А соединение (24,5 г, 0,094 ммоль) загружали в круглодонную колбу объемом 1,0 л, снабженную воздушной лопастной мешалкой и датчиком температуры, после этого добавляли 550 мл метанола, а затем 55 мл воды, в результате чего раствор становился слегка мутным. Колбу погружали в ледяную баню и когда температура опускалась ниже 5° С, добавляли порциями в течение 5 мин Охоnе® (144,5 г, 0,235 моль). Наблюдали небольшое повышение температуры до 8° С. Реакционную смесь перемешивали при охлаждении в течение 10 мин, после чего ледяную баню удаляли. Через 4 ч анализ с помощью жидкостной хроматографии высокого давления с обращенной фазой показал наличие одного компонента с временем удерживания 13,6 мин. Реакционную смесь фильтровали и твердое вещество тщательно промывали метанолом. Объединенные фильтраты концентрировали на роторном испарителе и остаток распределяли между этилацетатом (300 мл) и водой (200 мл). Органический слой промывали водой (3× 200 мл), насыщенным раствором бикарбоната натрия (1× 200 мл) и соляным раствором (1× 200 мл), после этого органическую фазу сушили над сульфатом магния и концентрировали, получая 25 г продукта в виде твердого вещества рыжевато-коричневого цвета. После растирания с гексаном получали 24,3 г чистого сульфона в виде твердого вещества беловатого цвета (88%). Результаты анализа с помощью 1H-ЯМР подтверждали, что вещество имело требуемое строение.
Анализ с помощью масс-спектроскопии показал, что величина м/z равна 293 (М+Н).
Стадия В: Получение
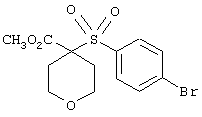
В высушенную в печи трехгорлую круглодонную колбу объемом 1,0 л загружали в камере, заполненной азотом, 60%-ную масляную дисперсию гидрида натрия (5,76 г, 0,144 моль) и к колбе присоединяли впускной клапан для азота, датчик температуры, воздушную лопастную мешалку и резиновую мембрану. В колбу добавляли безводный N,N-диметилформамид (250 мл), начинали механическое перемешивание и смесь нагревали до 50° С. К суспензии гидрида натрия добавляли по каплям раствор, содержащий полученное выше на стадии Б соединение (17,59 г, 0,06 моль) и простой дибромдиэтиловый эфир (14,5 г, 0,06 моль) в 40 мл N,N-диметилформамида, поддерживая температуру в диапазоне 50-55° С, при этом происходило постоянное выделение водорода. После завершения добавления температуру реакционной смеси повышали до 65° С и смесь перемешивали в течение 2 ч. Затем колбу охлаждали до комнатной температуры и колбу погружали в ледяную баню. Когда температура опускалась ниже 20° С, добавляли 0,5 л ледяной воды.
Смесь переносили в делительную воронку объемом 4,0 л, добавляли еще 1,0 л воды и смесь экстрагировали этилацетатом (3× 200 мл). Объединенные органические слои промывали смесью 5%-ная соляная кислота/вода (1× 200 мл), насыщенным раствором карбоната натрия (1× 200 мл) и соляным раствором (1× 200 мл), сушили над сульфатом магния и концентрировали в вакууме, получая 18,2 г неочищенного продукта в виде полутвердого вещества желтого цвета. После перекристаллизации из смеси этилацетат/гексан получали 6,53 г чистого продукта в виде кристаллов рыжевато-коричневого цвета (30%). Результаты анализа с помощью 1H-ЯМР подтверждали, что вещество имело требуемое строение. Анализ с помощью масс-спектроскопии показал, что величина м/z равна 363 (М+Н).
Стадия Г: Получение указанного в заголовке соединения
Раствор полученного выше на стадии В соединения (4,57 г, 12,6 ммоль) в 50 мл безводного тетрагидрофурана, помещенный в высушенную в печи круглодонную колбу объемом 100 мл, перемешивали в атмосфере азота при комнатной температуре и добавляли в виде одной порции 4,84 г триметилсиланолата калия (37,7 ммоль). Смесь перемешивали в течение 2 ч, затем по каплям добавляли 10 мл воды. Летучие компоненты удаляли в вакууме и остаток распределяли между 100 мл диэтилового эфира и 100 мл воды. Водный слой подкисляли до значения рН меньшего 2 с использованием концентрированной соляной кислоты, в результате чего выпадал осадок. Эту смесь экстрагировали этилацетатом (3× 75 мл), объединенные этилацетатные слои сушили над сульфатом магния и концентрировали в вакууме, получая 4,15 г чистого продукта в виде твердого вещества белого цвета (94%). 1H-ЯМР (CDCl3/CD3OD) 2,10 (m, 4Н), 3,28 (m, 2Н), 3,90 (m, 2H), 7,60 (m, 4H). Анализ с помощью масс-спектроскопии показал, что величина м/z равна 349 (М+Н).
Стадия 9: Связывание со смолой I
Получение смолы IX
Работая по методу, описанному выше для стадии 1, подвергали взаимодействию 3,13 г указанного выше в заголовке соединения с 3,73 г смолы I, получая 5,19 г связанного с полимером гидроксимата в виде полимерного твердого вещества рыжевато-коричневого цвета. Теоретическая загрузка полимера составляла 0,86 ммоль/г. Анализ с помощью ИКФП-микроскопии показал наличие полос при 1693 и 3332 см-1, являющихся характерными признаками присутствия соответственно гидроксаматкарбонильных и азот-водородных связей.
Стадия 10: Катализируемое палладием взаимодействие смолы IX с бороновыми кислотами
Получение смолы VII
В стеклянный сосуд для твердофазной реакции объемом 8 мл загружали смолу IX (50 мг, 0,043 ммоль). Смолу промывали безводным диметоксиэтаном (2× 3 мл). В сосуд добавляли 0,017 М раствор тетракистрифенилфосфинпалладия (0,6 мл, 0,01 ммоль), а затем 0,6 М раствор бороновой кислоты в смеси (1:1) диметоксиэтан/этанол (0,6 мл, 0,36 ммоль) и 2 М раствор гидроксида калия в воде (0,4 мл, 0,8 ммоль). В сосуде поддерживали избыточное давление аргона и его выдерживали в течение 16 ч при 90° С. Сосуд охлаждали до комнатной температуры, затем смолу сливали и промывали 1-метил-2-пирролидиноном, смесью 1-метил-2-пирролидинон/вода (1:1), водой, смесью уксусная кислота/вода (1:9), метанолом и метиленхлоридом (3× 3 мл каждого растворителя).
Перечисленные ниже гидроксамовые кислоты синтезировали из смолы IX в реакционных условиях, описанных для стадии 10, с использованием указанной бороновой кислоты с последующим отделением от полимера в реакционных условиях, описанных для стадии 3.
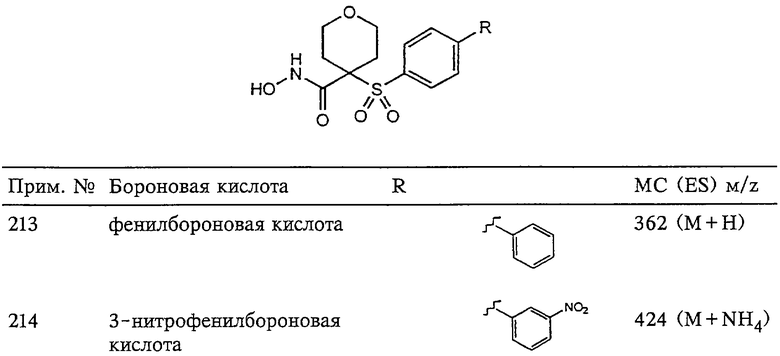
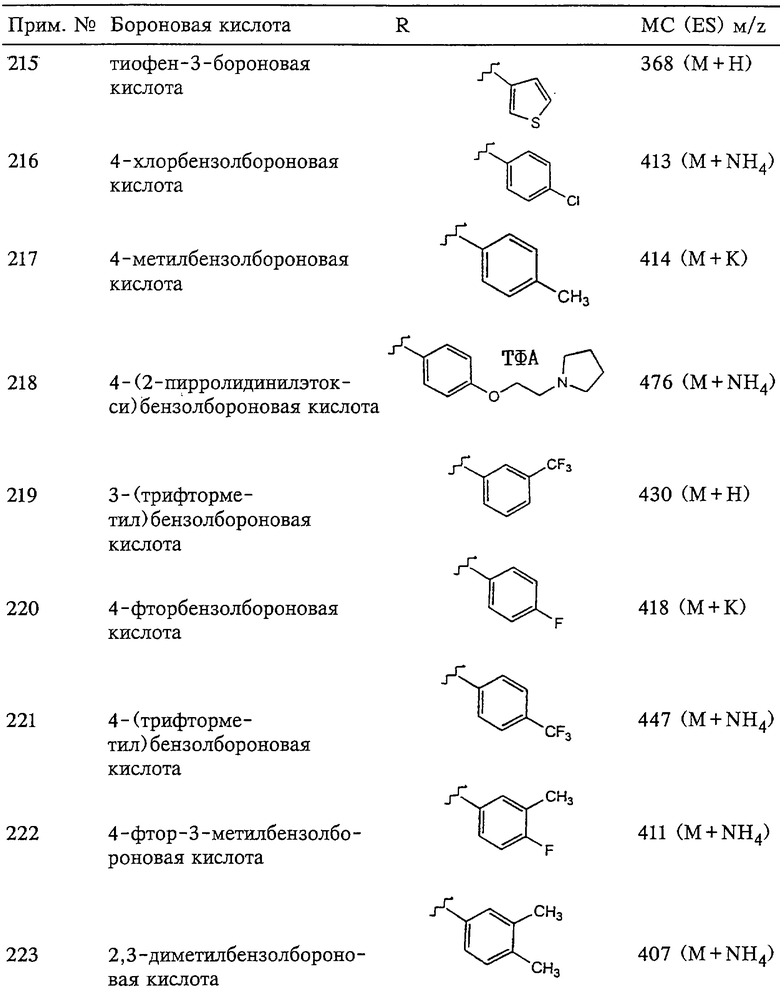
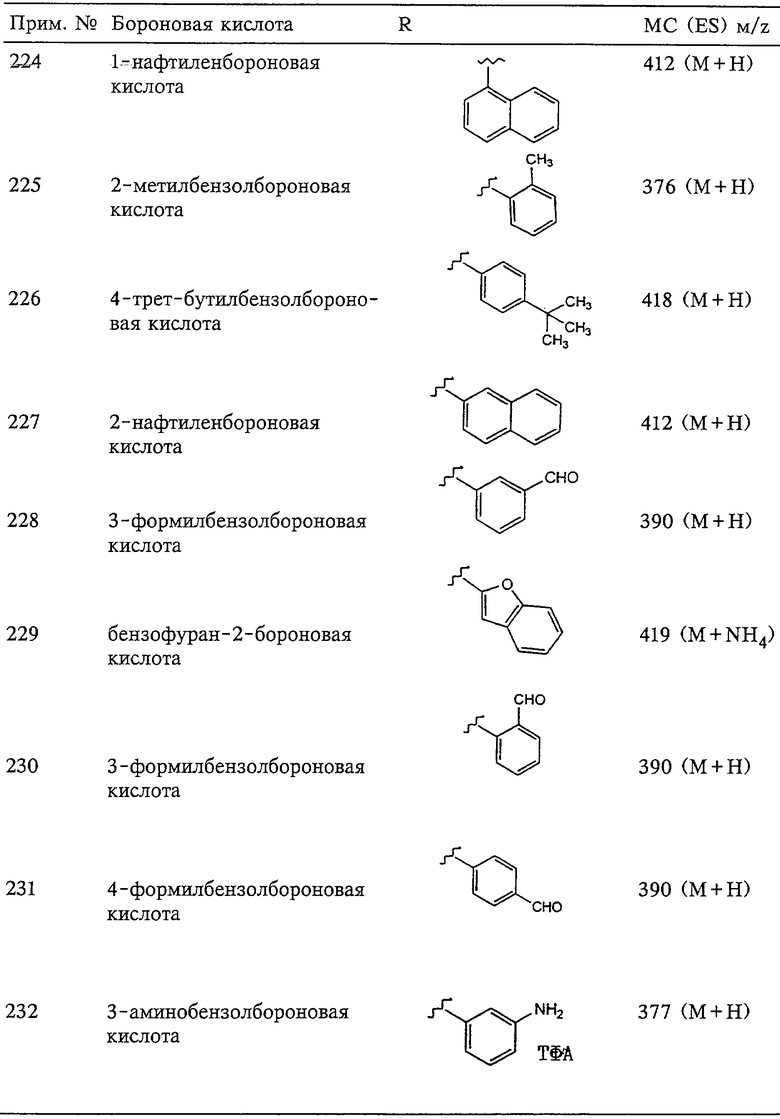
Пример 233: Получение монометансульфонатных солей: монометансульфонат N-гидрокси-4-[[4-(фенилтио)фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида
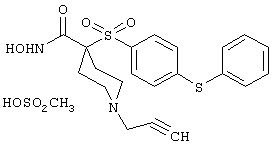
Первый способ получения
Стадия А: Раствор полученного на стадии К примера 9 соединения (2,1 г, 4,5 ммоль) в теплой Н2О (200 мл) смешивали с NаНСО3 при температуре окружающей среды. После перемешивания в течение 20 мин образовавшееся твердое вещество белого цвета выделяли фильтрацией, промывали водой и сушили в вакуумной печи при 37° С, получая указанное в заголовке соединение в форме свободного основания в виде твердого вещества белого цвета (1,7 г, 86%). Элементный анализ: рассчитано для С21Н22N2О4S2·3%Н2О: С 57,86; Н 5,23; N 6,43; S 14,71; обнаружено: С 57,84; Н 4,96; N 6,39; S 14,89.
Стадия Б: К раствору полученного на стадии А свободного основания (1,6 г, 3,7 ммоль) в метаноле (10 мл) добавляли при температуре окружающей среды метансульфоновую кислоту (0,28 мл, 4,1 ммоль). Через 3 ч образовавшееся твердое вещество выделяли фильтрацией, промывали метанолом и сушили в вакуумной печи при температуре окружающей среды, получая указанное в заголовке соединение, представляющее собой монометансульфонат, в виде твердого вещества белого цвета (1,6 г, 81%). Элементный анализ: рассчитано для C2lH22N2O4S2·CH4O3: C 48,51; H 5,18; N 5,14; S 17,66; обнаружено: С 48,88; Н 5,15; N 5,23; S 17,81.
Второй способ получения
Стадия А: Раствор полученного на стадии И примера 9 защищенного гидроксамата (6,0 г, 12 ммоль) в метаноле (37 мл) добавляли в атмосфере азота метансульфоновую кислоту (0,91 мл, 14 ммоль). Через 1 ч осадок выделяли фильтрацией, промывали метанолом и сушили в вакуумной печи в течение 1 дня при 40° С, получая указанное в заголовке соединение, представляющее собой метансульфонат, в виде твердого вещества белого цвета (5,5 г, 89%), идентичное продукту, полученному согласно описанному в примере 233 первому способу получения.
Приведенные в настоящем описании метансульфонаты других соединений, представляющих собой циклические амины, могут быть получены аналогичным путем с использованием указанных выше двух способов получения.
Примеры 234-280
Соединения из примеров 234-280 получали по методу, аналогичному методу получения соединений из примеров 129-199.
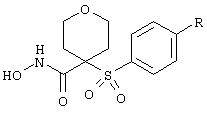
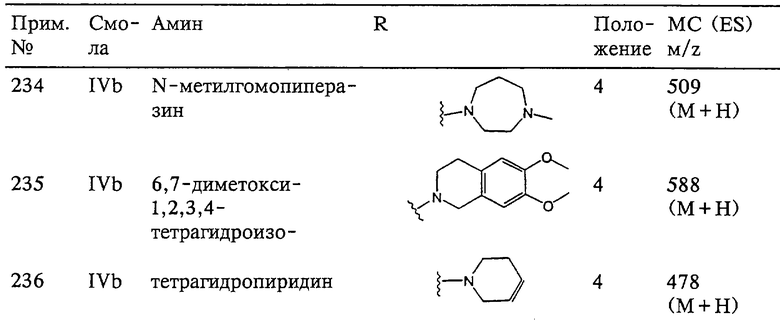
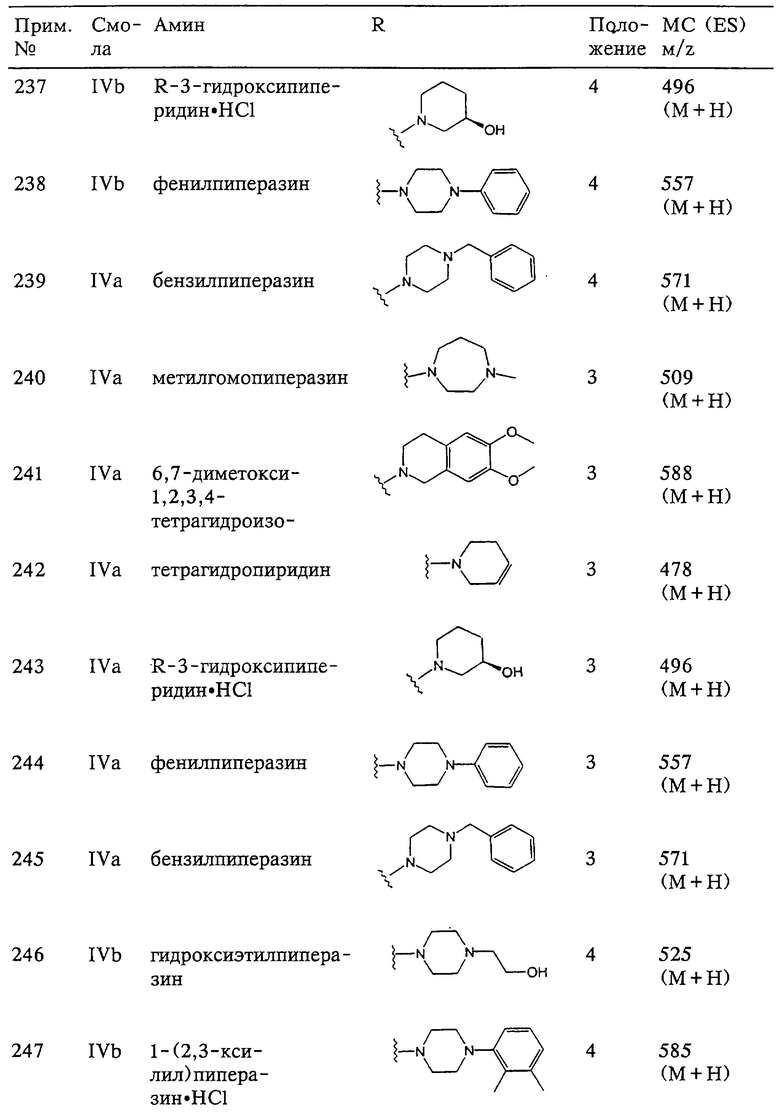
Пример 281-288
Перечисленные ниже гидроксамовые кислоты синтезировали аналогично примерам 213-232 из смолы IX по методу, изложенному в описании стадии 10, с использованием указанной бороновой кислоты с последующим отделением от полимера методом, изложенным в описании стадии 3.
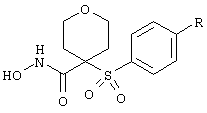
Примеры 289-294
Стадия 11: Получение смолы XI
В реакционный сосуд, снабженный фриттой, загружали смолу IIIc (50 мг, 0,043 ммоль). Добавляли 0,43 М раствор изоцианата в 1-метил-2-пирролидиноне (1 мл, 0,43 ммоль), а затем диизопропилэтиламин (75 мкл, 0,43 ммоль). Сосуд закрывали крышкой в атмосфере азота, перемешивали на настольном шейкере и нагревали в течение 48 ч до 50° С. После этого сосуд охлаждали до комнатной температуры, смолу сливали и промывали 1-метил-2-пирролидиноном, смесью 1-метил-2-пирролидинон/вода (1:1), водой, смесью уксусная кислота/вода (1:9), метанолом и метиленхлоридом (31 мл каждого растворителя).
Перечисленные ниже гидроксамовые кислоты синтезировали из смолы IIIc по методу, описанному для стадии 11, с использованием указанного изоцианата с последующим отделением от полимера в реакционных условиях, описанных для стадии 3.
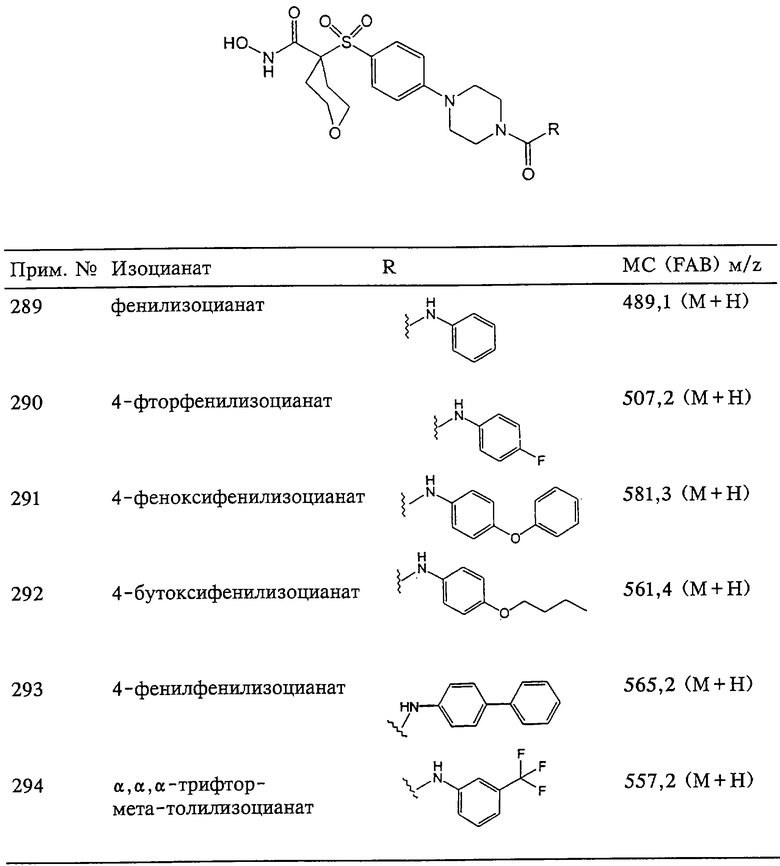
Примеры 295-300
Стадия 12: Синтез смолы XII
В реакционный сосуд, снабженный фриттой, загружали смолу VII (50 мг, 0,038 ммоль) и карбонат цезия (122 мг, 0,38 ммоль). Добавляли 0,43 М раствор фенола в 1-метил-2-пирролидиноне (1 мл, 0,43 ммоль), затем сосуд закрывали крышкой в атмосфере азота. Реакционную смесь перемешивали на настольном шейкере и нагревали в течение 48 ч до 50° С. После этого сосуд охлаждали до комнатной температуры, смолу сливали и промывали 1-метил-2-пирролидиноном, смесью 1-метил-2-пирролидинон/вода (1:1), водой, смесью уксусная кислота/вода (1:9), метанолом и метиленхлоридом (3× 1 мл каждого растворителя).
Перечисленные ниже гидроксамовые кислоты синтезировали из смолы IIIс по методу, описанному для стадии 11, с использованием указанного фенола с последующим отделением от полимера в реакционных условиях, описанных для стадии 3.
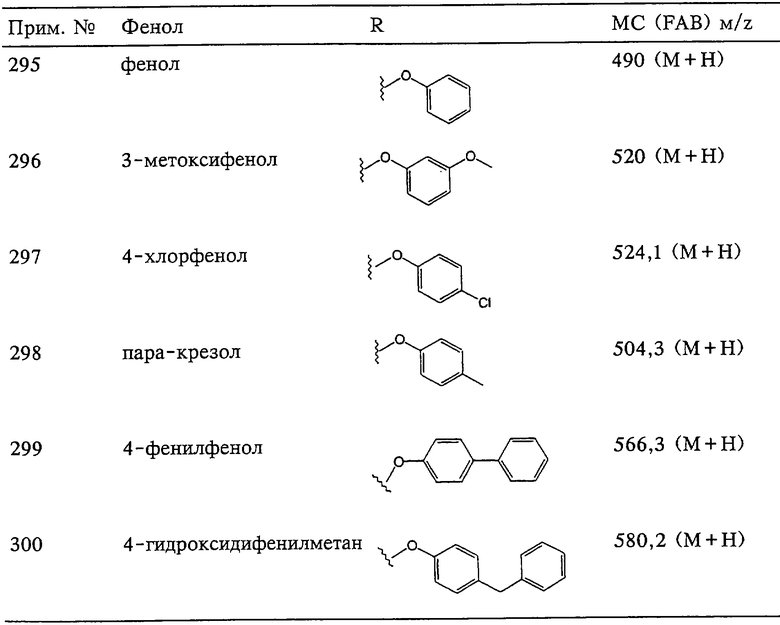
Примеры 301-323
Крупномасшабное получение смолы Ха
В реакционный сосуд, снабженный фриттой, загружали смолу IX (1 г, 0,86 ммоль) и 0,008 М раствор тетракис(трифенилфосфин) палладия (0) в диметиловом эфире этиленгликоля (5 мл, 0,04 ммоль). Добавляли 1М раствор 2-формилбензолбороновой кислоты в смеси (1:1) этанола и диметилового эфира этиленгликоля (6 мл, 6 ммоль), а затем 1 М раствор карбоната цезия в воде (2 мл, 2 ммоль). Сосуд закрывали крышкой в атмосфере аргона и нагревали в течение 16 ч до 90° С. После этого сосуд охлаждали до комнатной температуры, смолу сливали и промывали следующими растворителями: диметилформамидом, смесью диметилформамид/вода (1:1), диметилформамидом, водой, метанолом и метиленхлоридом (3× 5 мл каждого растворителя). Смолу сушили в вакууме, получая 1,025 г продукта в виде полимерного твердого вещества рыжевато-коричневого цвета. Теоретическая загрузка полимера составляла 0,84 ммоль/г. Путем расщепления с помощью ТФК 35 мг смолы Ха по методу, описанному для стадии 3, получали 11,2 мг твердого вещества рыжевато-коричневого цвета.
Крупномасшабное получение смолы Хb
Смолу Хb получали аналогично методу получения смолы Ха за исключением того, что вместо 2-формилбензолбороновой кислоты использовали 3-формилбензолбороновую кислоту. После сушки в вакууме выход смолы Хb в виде смоляных шариков рыжевато-коричневого цвета составлял 1,052 г. Теоретическая загрузка полимера составляла 0,84 ммоль/г. Путем расщепления с помощью ТФК 20 мг смолы Хb по методу, описанному для стадии 3, получали 6,5 мг твердого вещества рыжевато-коричневого цвета.
Крупномасшабное получение смолы Хс
Смолу Хс получали аналогично методу получения смолы Ха за исключением того, что вместо 2-формилбензолбороновой кислоты использовали 4-формилбензолбороновую кислоту. После сушки в вакууме выход смолы Хс в виде смоляных шариков рыжевато-коричневого цвета составлял 1,03 г. Теоретическая загрузка полимера составляла 0,84 ммоль/г. Путем расщепления с помощью ТФК 28 мг смолы Хс по методу, описанному для стадии 3, получали 9,4 мг твердого вещества рыжевато-коричневого цвета.
Стадия 13: Синтез смолы XIII:
В реакционный сосуд, снабженный фриттой, загружали смолу Ха, Хb или Хс (50 мг, 0,042 ммоль). Добавляли 0,2М раствор амина в триметилортоформиате (1 мл, 0,2 ммоль) и затем сосуд закрывали крышкой в атмосфере азота. Реакционную смесь перемешивали на настольном шейкере в течение 3 ч. Затем в сосуд добавляли 0,5 М раствор триацетоксиборогидрида натрия в 1-метил-2-пирролидиноне (0,8 мл, 0,4 ммоль) и смесь перемешивали еще в течение 40 ч. После этого смолу сливали и промывали растворителями в следующей последовательности: 1-метил-2-пирролидиноном, метанолом, водой, метанолом и метиленхлоридом (3× 1 мл каждого растворителя).
Перечисленные ниже гидроксамовые кислоты синтезировали с использованием указанного связанного со смолой альдегида и амина по методу, описанному для стадии 13, с последующим отделением от полимера в реакционных условиях, описанных для стадии 3.
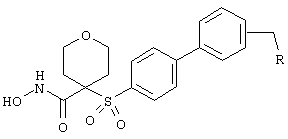

Крупномасшабное получение смолы Xd
Смолу Xd получали аналогично методу получения смолы Ха, за исключением того, что вместо 2-формилбензолбороновой кислоты использовали 4-карбоксибензолбороновую кислоту. После сушки в вакууме выход смолы Xd в виде полимерного твердого вещества рыжевато-коричневого цвета составлял 1,07 г. Теоретическая загрузка полимера составляла 0,83 ммоль/г. Путем расщепления с помощью ТФК 23,5 мг смолы Xd по методу, описанному для стадии 3, получали 4,9 мг твердого вещества рыжевато-коричневого цвета.
Стадия 14: Синтез смолы XIV:
В реакционный сосуд, снабженный фриттой, загружали смолу Xd (50 мг, 0,042 ммоль). Смолу промывали 1-метил-2-пирролидиноном (2× 3 мл), после этого добавляли 1,0 М раствор гексафторфосфата бензотриазол-1-илокситрис-пирролидинофосфония в 1-метил-2-пирролидиноне (0,2 мл, 0,2 ммоль), а затем 0,7 М раствор амина в 1-метил-2-пирролидиноне (0,3 мл, 0,21 ммоль) и 1,0 М раствор диизопропилэтиламина в 1-метил-2-пирролидиноне (0,4 мл, 0,4 ммоль). Сосуд закрывали крышкой в атмосфере азота и реакционную смесь перемешивали на настольном шейкере в течение 24 ч. Затем смолу сливали и промывали 1-метил-2-пирролидиноном (3× 1 мл). Повторно осуществляли взаимодействие с амином путем добавления 1,0 М раствора гексафторфосфата бензотриазол-1-илокситриспирролидинофосфония в 1-метил-2-пирролидиноне (0,2 мл, 0,2 ммоль), 0,7 М раствор амина в 1-метил-2-пирролидиноне (0,3 мл, 0,21 ммоль) и 1,0 М раствор диизопропилэтиламина в 1-метил-2-пирролидиноне (0,4 мл, 0,4 ммоль). Сосуд закрывали крышкой в атмосфере азота и реакционную смесь перемешивали еще в течение 8 ч. После этого смолу сливали и промывали растворителями в следующей последовательности: 1-метил-2-пирролидиноном, смесью 1-метил-2-пирролидинон/вода (1:1), водой, смесью уксусная кислота/вода (1:9), метанолом, метиленхлоридом (3х1 мл каждого растворителя).
Перечисленные ниже гидроксамовые кислоты синтезировали с использованием смолы Xd и указанного амина по методу, описанному для стадии 14, с последующим отделением от полимера в реакционных условиях, описанных для стадии 3.
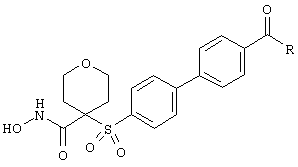
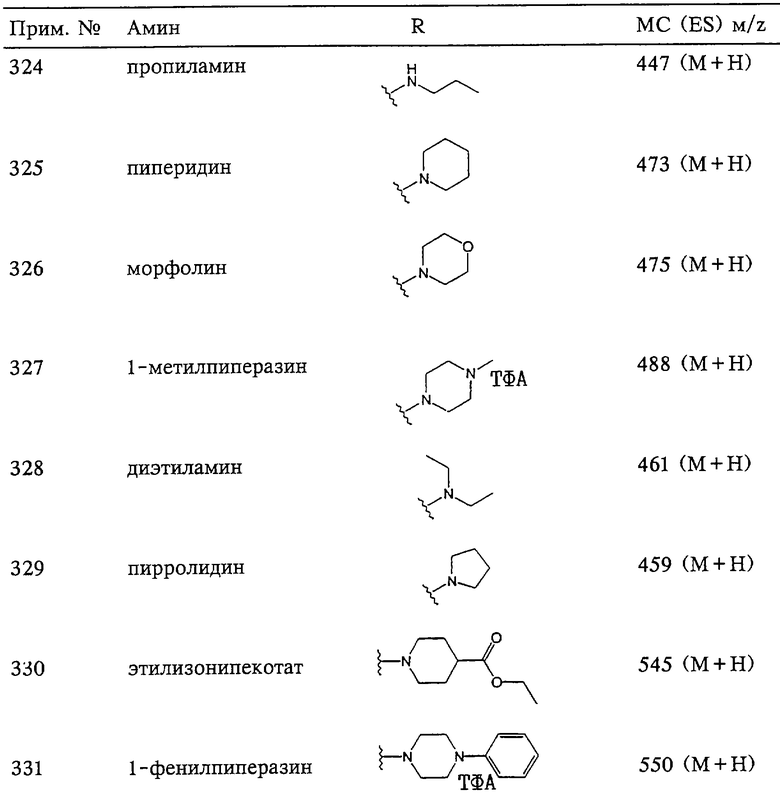
Пример 336: Получение 4-[[4-[4-[[(9Н-флуорен-9-илметокси)карбонил]амино]-1-пиперидинил]фенил]сульфонил]тетрагидро-2Н-пиран-4-карбоновой кислоты
Стадия А: К раствору полученного на стадии Б примера 11 продукта (10,0 г, 34,7 ммоль) в 1-метил-2-пирролидиноне (70 мл) добавляли 4-(N-трет-бутоксикарбониламино) пиперидин (10,43 г, 52,1 ммоль), а затем диизопропилэтиламин (6,0 мл, 34,7 ммоль). Образовавшуюся смесь нагревали в течение 24 ч до 80° С и затем охлаждали до комнатной температуры. Неочищенную смесь сливали на 700 мл воды и мутный водный слой экстрагировали этилацетатом (3× 150 мл). Объединенные органические слои промывали 5%-ным раствором бисульфата калия (2× 150 мл) и соляным раствором (2× 150 мл), сушили над сульфатом магния и концентрировали в вакууме, получая неочищенный сложный эфир в виде пенообразного твердого вещества белого цвета (13,04 г, 78%).
Стадия Б: К раствору полученного на стадии А сложного эфира (5,74 г, 11,9 ммоль) в смеси этанола (80 мл) и тетрагидрофурана (40 мл) добавляли 2н. гидроксид натрия (60 мл, 120 ммоль). Образовавшийся раствор нагревали в течение 1 ч до 60° С и затем охлаждали до комнатной температуры. Раствор концентрировали в вакууме и остаток распределяли между водой (300 мл) и этилацетатом (200 мл). Водный слой отделяли и подкисляли концентрированной соляной кислотой до рН 2. Образовывался осадок белого цвета, который собирали вакуумной фильтрацией и сушили в вакууме, получая карбоновую кислоту в виде твердого вещества белого цвета (4,88 г, 88%).
Стадия В: К суспензии полученной на стадии Б карбоновой кислоты (4,88 г, 10,4 ммоль) в метиленхлориде (35 мл) добавляли трифторуксусную кислоту (35 мл), в результате чего происходило растворение твердого вещества. После выдерживания в течение 15 мин при температуре окружающей среды раствор концентрировали в вакууме. Продукт растирали с диэтиловым эфиром, получая аминокислоту в виде твердого вещества белого цвета (4,92 г, 98%).
Стадия Г: Суспензию полученной на стадии В аминокислоты (4,92 г, 10,21 ммоль) в смеси, содержащей 10%-ный раствор карбоната калия/воду (35 мл), воду (100 мл) и диоксан (100 мл), охлаждали в ледяной бане. К охлажденной суспензии добавляли по каплям раствор 9-флуоренилметилсукцинимидилкарбонат (3,79 г, 11,23 ммоль) в диоксане (50 мл). После завершения добавления ледяную баню удаляли и смесь нагревали до комнатной температуры. Через 1 ч раствор концентрировали в вакууме и остаток распределяли между водой (300 мл) и этилацетатом (200 мл). Водный слой отделяли и подкисляли концентрированной соляной кислотой до рН 2. Образовывался осадок белого цвета, который собирали вакуумной фильтрацией, промывали гексаном и сушили в вакууме, получая указанное в заголовке соединение в виде твердого вещества белого цвета (5,46 г, 91%).
Стадия 15: Получение смолы XVI
Стадия А: Работая по методу, описанному выше для стадии 1, полученный в примере 336 продукт (2,4 г, 4,06 ммоль) подвергали взаимодействию со смолой I (1,7 г, 2,03 ммоль), получая смолу XV в виде полимерного твердого вещества рыжевато-коричневого цвета (2,82 г). Теоретическая загрузка полимера составляла 0,71 ммоль/г.
Стадия Б: Полученную выше на стадии А смолу XV (2,76 г, 1,96 ммоль) суспендировали в снабженном фриттой реакционном сосуде в растворе (1:4) пиперидин/диметилформамид (20 мл) и перемешивали на настольном шейкере в течение 5 мин. Смолу сливали и в сосуд добавляли дополнительную порцию смеси (1:4) пиперидин/диметилформамид (20 мл). Суспензию перемешивали в течение 30 мин при комнатной температуре. После этого смолу сливали и промывали диметилформамидом, метанолом и метиленхлоридом (3× 20 мл каждого растворителя). После сушки в вакууме получали указанную смолу в виде полимерного твердого вещества рыжевато-коричневого цвета (2,30 г).
Стадия 16: Адилирование/сульфонилирование смолы XVI В снабженном фриттой реакционном сосуде смолу XVI (50 мг, 0,043 ммоль) промывали 1-метил-2-пирродидиноном (2× 1 мл). После этого к смоле добавляли 0,22 М раствор ацилирующего или сульфонилирующего реагента в 1-метил-2-пирродидиноне (1 мл, 0,22 ммоль), а затем диизопропилэтиламин (40 мкл, 0,22 ммоль). Сосуд закрывали крышкой в атмосфере азота и перемешивали на настольном шейкере в течение 16 ч при комнатной температуре. Затем смолу сливали и промывали 1-метил-2-пирродидиноном, водой, смесью уксусная кислота/вода (1:9), метанолом и метиленхлоридом (3× 1 мл каждого растворителя).
Перечисленные ниже гидроксамовые кислоты синтезировали из смолы XVI по методу, описанному для стадии 16, с использованием указанного ацилирующего или сульфонилирующего реагента с последующим отделением от полимера в реакционных условиях, описанных для стадии 3.
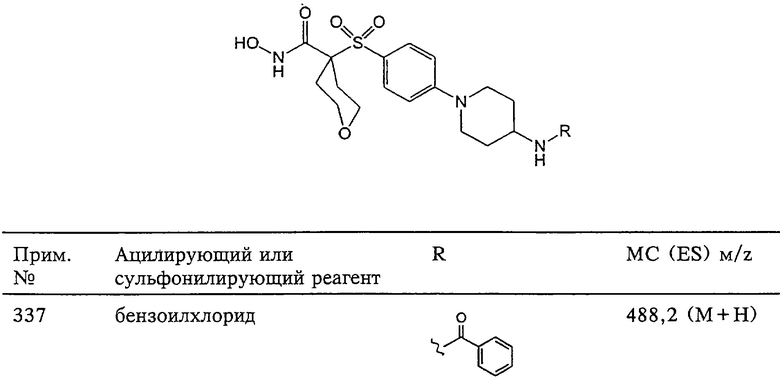
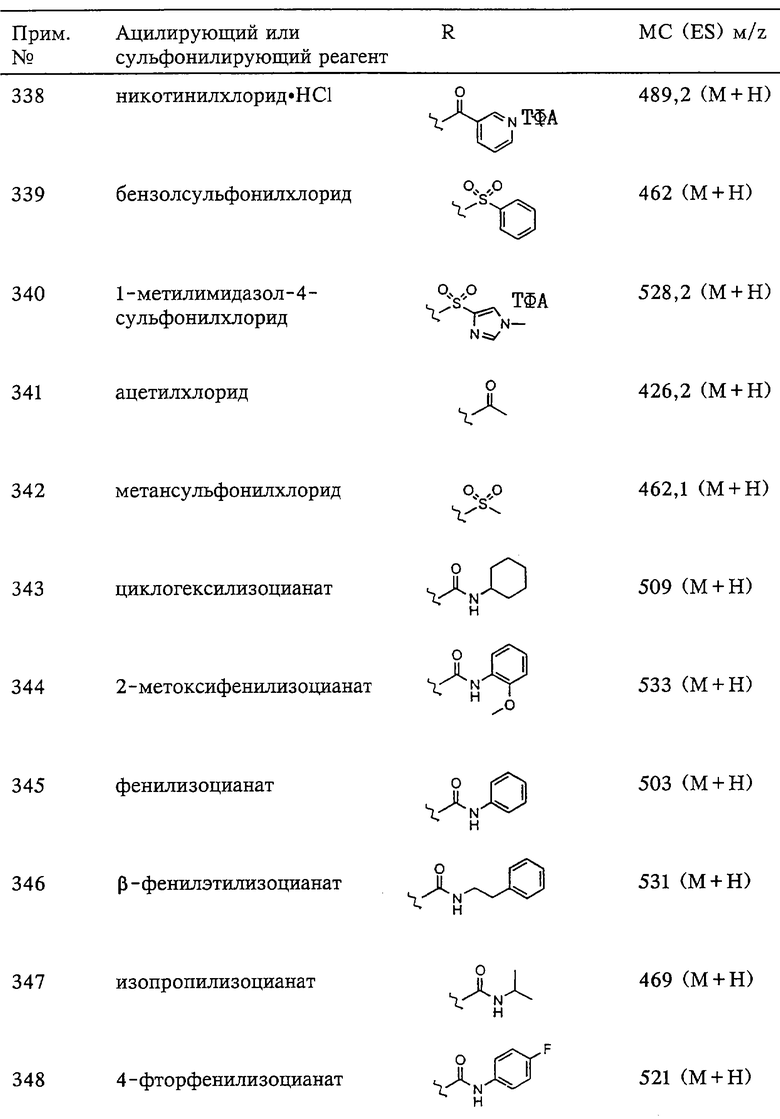


Примеры 365-371
Стадия 17: Восстановительное алкилирование смолы XVI В снабженном фриттой реакционном сосуде смолу XVI (50 мг, 0,043 ммоль) промывали метиленхлоридом (2× 1 мл). После этого к смоле добавляли 1 М раствор альдегида или кетона в метиленхлориде (1 мл, 1 ммоль). Сосуд закрывали крышкой в атмосфере азота и перемешивали на настольном шейкере в течение 3 ч при комнатной температуре. Смолу сливали и промывали метиленхлоридом (3× 1 мл). После этого смолу повторно обрабатывали 1 М раствором альдегида или кетона в метиленхлориде (1 мл, 1 ммоль). Смолу сливали и промывали метиленхлоридом (3× 1 мл). После этого к смоле добавляли 1М раствор триацетоксиборогидрида натрия в 1-метил-2-пирролидиноне (1 мл, 1 ммоль) и реакционную смесь перемешивали в течение ночи. После этого смолу сливали и промывали 1-метил-2-пирролидиноном, метанолом, водой, смесью уксусная кислота/вода (1:9), метанолом и метиленхлоридом (3× 1 мл каждого растворителя).
Перечисленные ниже гидроксамовые кислоты синтезировали из смолы XVI по методу, описанному для стадии 17, с использованием указанного альдегида или кетона с последующим отделением от полимера в реакционных условиях, описанных для стадии 3.
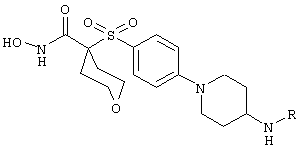
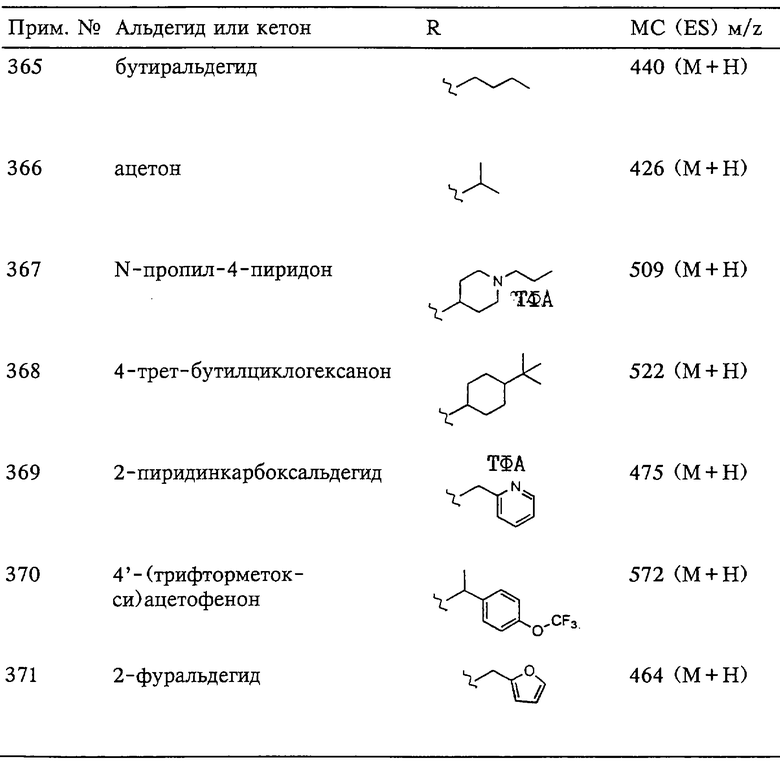
Пример 372: Получение 4-[[4-(4-бутоксифенокси)фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
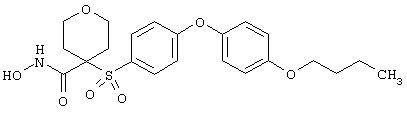
Стадия А: К раствору полученного в примере 55 продукта (3,1 г, 8 ммоль) в диметилацетамиде (20 мл) добавляли карбонат цезия (7,28 г, 24 ммоль) и 4-бутоксифенол (2,66 г, 16 ммоль). Суспензию перемешивали в течение 19 ч при 95° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Nа2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены беловатого цвета (3,96 г, 93%). МСВР (ES+) M+NH
Стадия Б: К раствору полученного на стадии А ТГП-защищенного гидроксамата (3,9 г, 7,3 ммоль) в 1,4-диоксане (20 мл) добавляли 4н. раствор HCl в диоксане (20 мл) и метанол (20 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом и промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После перекристаллизации продукта (ацетон/гексан) получали указанное в заголовке соединение в виде твердого вещества белого цвета (2,75 г, 84%). МСВР (ES+) М+Н+: рассчитано для C22H27N1O7S1:450,16, обнаружено: 450,16.
Пример 373: Получение тетрагидро-N-гидрокси-4-[[4-[3-(трифторметил)фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
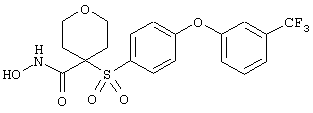
Стадия А: К раствору полученного в примере 55 продукта (3,1 г, 8 ммоль) в диметилацетамиде (20 мл) добавляли карбонат цезия (7,28 г, 24 ммоль) и мета - (трифторметил) фенол (1,95 мл, 16 ммоль). Суспензию перемешивали в течение 19 ч при 95° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены белого цвета (4,1 г, 97%). МСВР (ES+) М+Н+: рассчитано для C24H26N1O7S1F3: 530,15, обнаружено: 530,14.
Стадия Б: К раствору полученного на стадии А ТГП-защищенного гидроксамата (3,9 г, 7,4 ммоль) в 1,4-диоксане (20 мл) добавляли 4 н. раствор HCl в диоксане (20 мл) и метанол (20 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом и промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После перекристаллизации продукта (ацетон/гексан) получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,9 г, 58%). МСВР (ES+) М+Н+: рассчитано для C19H18N1O6S1F3: 446,09, обнаружено: 446,09.
Пример 374: Получение тетрагидро-N-гидрокси-4-[[4-[4-(метилтио)фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
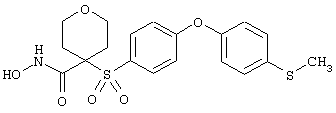
Стадия А: К раствору полученного в примере 55 продукта (3,1 г, 8 ммоль) в диметилацетамиде (20 мл) добавляли карбонат цезия (7,28 г, 24 ммоль) и 4-(метилтио) фенол (2,24 г, 16 ммоль). Суспензию перемешивали в течение 24 ч при 95° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены белого цвета (4,1 г, 100%). МСВР (ES+) М+Н+: рассчитано для C24H29N1O7S2: 508,15, обнаружено: 508,14.
Стадия Б: К раствору полученного на стадии А ТГП-защищенного гидроксамата (4,0 г, 7,9 ммоль) в 1,4-диоксане (20 мл) добавляли 4 н. раствор HCl в диоксане (20 мл) и метанол (20 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом и промывали водой, сушили над Nа2SO4, фильтровали и концентрировали в вакууме. После перекристаллизации продукта (ацетон/гексан) получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,9 г, 57%). МСВР (ES+) М+Н+: рассчитано для C19H21N1O6S2: 424,09, обнаружено: 424,09.
Пример 375: Получение тетрагидро-N-гидрокси-4-[[4-[4-(фенилметил)фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
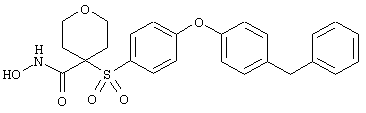
Стадия А: К раствору полученного в примере 55 продукта (2,7 г, 7 ммоль) в диметилацетамиде (15 мл) добавляли карбонат цезия (6,84 г, 21 ммоль) и 4-гидроксидифенилметан (2,8 г, 14 ммоль). Суспензию перемешивали в течение 19 ч при 90° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены светло-желтого цвета (3,7 г, 96%). МСВР (ES+) М+Н+: рассчитано для С30Н33N1O7S1: 552,21, обнаружено: 552,21.
Стадия Б: К раствору полученного на стадии А ТГП-защищенного гидроксамата (3,5 г, 6,4 ммоль) в 1,4-диоксане (16 мл) добавляли 4н. раствор HCl в диоксане (16 мл) и метанол (16 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом и промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После перекристаллизации продукта (ацетон/гексан) получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,95 г, 67%). МСВР (ES+) М+Н+: рассчитано для С25Н25N1О6S1: 468,15, обнаружено: 468,15.
Пример 376: Получение тетрагидро-N-гидрокси-4-[[4-(4-гидроксифенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
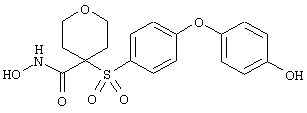
Стадия А: К раствору полученного в примере 55 продукта (2,7 г, 7 ммоль) в диметилацетамиде (20 мл) добавляли карбонат цезия (6,84 г, 21 ммоль) и 4-(бензилокси) фенол (2,8 г, 14 ммоль). Суспензию перемешивали в течение 6 ч при 95° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены белого цвета (3,94 г, 99%). МСВР (ES+) M+H+: рассчитано для С30Н33N1О8S1: 585,23, обнаружено: 585,23.
Стадия Б: К раствору полученного на стадии А ТГП-защищенного гидроксамата (1,5 г, 2,64 ммоль) в ледяной уксусной кислоте (5 мл) добавляли концентрированную HCl (5 мл) и реакционную смесь нагревали в течение 20 мин до 60° С. Реакционную смесь охлаждали, разбавляли водой (100 мл) и экстрагировали этилацетатом. Этилацетатный экстракт трижды промывали водой, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После перекристаллизации продукта (ацетон/гексан) получали указанное в заголовке соединение в виде твердого вещества белого цвета (810 мг, 78%). МСВР (ES+) M+NH4+: рассчитано для C18H19N1O7S1: 468,15, обнаружено: 468,15.
Пример 377: Получение тетрагидро-N-гидрокси-4-[[4-[4-[(1-метилэтил)тио]фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
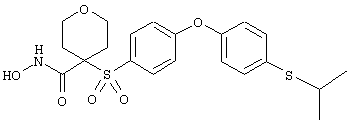
Стадия А: К суспензии, содержащей 4-гидрокситиофенол (5,0 г, 40 ммоль) и карбонат калия (8,0 г, 58 ммоль) в диметилформамиде (70 мл), добавляли 2-йодпропан (7,0 г, 41 ммоль). Суспензию перемешивали в течение 1 ч при температуре окружающей среды. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали дважды водой, 10%-ным раствором HCl, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный фенол в виде прозрачного бесцветного масла (5,1 г, 76%).
Стадия Б: К раствору полученного в примере 55 продукта (3,1 г, 8 ммоль) в диметилацетамиде (20 мл) добавляли карбонат цезия (7,28 г, 24 ммоль) и полученный на стадии А фенол (2,7 г, 16 ммоль). Суспензию перемешивали в течение 15 ч при 95° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены белого цвета (4,15 г, 97%). МСВР (ES+) M+H+: рассчитано для C26H33N1O7S2: 536,18, обнаружено: 536,17.
Стадия В: К раствору полученного на стадии Б ТГП-защищенного гидроксамата (3,9 г, 7,3 ммоль) в 1,4-диоксане (18 мл) добавляли 4н. раствор HCl в диоксане (18 мл) и метанол (18 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом и промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После перекристаллизации продукта (ацетон/гексан) получали указанное в заголовке соединение в виде твердого вещества беловатого цвета (2,32 г, 71%). МСВР (ES+) М+Н+: рассчитано для C21H25N1O6S2: 452,12, обнаружено: 452,12.
Пример 378: Получение тетрагидро-N-гидрокси-4-[[4-[4-(1-метилэтокси)фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
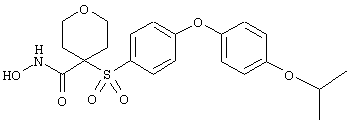
Стадия А: К раствору 4-гидроксифенилового эфира бензойной кислоты (8,57 г, 40 ммоль) в диметилацетамиде (65 мл) добавляли карбонат калия (8,3 г, 60 ммоль) и 2-йодпропан (5 мл, 50 ммоль). Суспензию перемешивали в течение 1 ч при 65° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали трижды водой, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая изопропоксипроизводное в виде твердого вещества светло-серого цвета (9,7 г, 95%).
Стадия Б: К суспензии полученного на стадии А изопропоксипроизводного (9,7 г, 38 ммоль) в 1,4-диоксане (20 мл) и воде (20 мл) добавляли 2,5 н. раствор гидроксида натрия (26 мл, 65 ммоль). Суспензию перемешивали в течение 4 ч при 60° С. Реакционную смесь охлаждали и добавляли 6н. раствор соляной кислоты до достижения значения рН 5. Реакционную смесь экстрагировали метиленхлоридом. Органический слой промывали 4 раза 5%-ным раствором гидроксида аммония, водой, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая фенол в виде масла янтарного цвета (5,4 г, 94%).
Стадия В: К суспензии полученного в примере 55 продукта (3,1 г, 8 ммоль) в диметилацетамиде (20 мл) добавляли карбонат цезия (7,28 г, 24 ммоль) и полученный на стадии Б фенол (2,4 г, 16 ммоль). Суспензию перемешивали в течение 21 ч при 95° С. Реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали трижды водой, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали замещенный ТГП-защищенный гидроксамат в виде пены беловатого цвета (3,65 г, 88%). МСВР (ES+) М+Н+: рассчитано для С26Н33N1О8S1: 520,20, обнаружено: 520,20.
Стадия Г: К раствору полученного на стадии В ТГП-защищенного гидроксамата (3,5 г, 6,7 ммоль) в 1,4-диоксане (17 мл) добавляли 4 н. раствор HCl в диоксане (17 мл) и метанол (17 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом и промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После перекристаллизации продукта (ацетон/гексан) получали указанное в заголовке соединение в виде твердого вещества беловатого цвета (2,2 г, 80%). МСВР (ES+) М+Н+: рассчитано для C21H25N1O7S1: 436,14, обнаружено: 436,14.
Пример 379: Получение тетрагидро-N-гидрокси-4-[[4-[4-[(трифторметил]фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
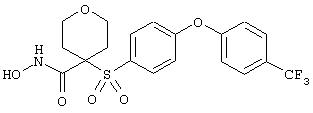
Стадия А: В сухом сосуде в атмосфере азота при 0° С к раствору [4-(трифторметил) фенокси] фенола (50,0 г, 0,197 моль) в безводном диметилформамиде (150 мл) добавляли гидрид натрия (60%-ная дисперсия в масле) (11,0 г, 0,275 моль). Через 15 мин добавляли раствор диметилтиокарбамоилхлорида (32 г, 0,259 моль) в безводном диметилформамиде (100 мл). Реакционную смесь перемешивали в течение 16 ч при температуре окружающей среды. Реакционную смесь сливали на 10%-ный раствор HCl (1 л). После вакуумной фильтрации образовавшегося осадка получали тионопроизводное в виде твердого вещества белого цвета (67,0 г, 100%).
Стадия Б: Полученное на стадии А тионопроизводное (70 г, 0,2 моль) нагревали с использованием защитного экрана в течение 30 мин до 317° С. В результате экзотермической реакции температура повышалась до 330° С. Нагрев прекращали и реакционная смесь охлаждалась до температуры окружающей среды, в результате чего получали тиокарбамат в виде твердого вещества коричневого цвета (70 г, 100%).
Стадия В: К раствору полученного на стадии Б тиокарбамата (65,0 г, 0,19 моль) в метаноле (510 мл) добавляли 2,5 н. раствор гидроксида натрия (160 мл, 0,4 моль), пропуская через раствор струю азота. Суспензию перемешивали в течение 2 ч при 74° С. Реакционную смесь охлаждали и метанол удаляли в вакууме. Остаток разбавляли водой (100 мл) и 4 раза экстрагировали диэтиловым эфиром. Через водный раствор пропускали струю азота и добавляли хлорацетат натрия (22,2 г, 0,19 моль). Реакционную смесь перемешивали при температуре окружающей среды и через 30 мин прекращали подачу азота. Через 12 ч раствор охлаждали и добавляли 6 н. раствор соляной кислоты до достижения значения рН 1. Суспензию 4 раза экстрагировали этилацетатом. Объединенные этилацетатные экстракты промывали 0,1н. соляной кислотой, водой, соляным раствором, сушили над Na2SO4, фильтровали и сушили в вакууме, получая тиоуксуснуто кислоту в виде твердого вещества рыжевато-коричневого цвета (61,0 г, 98%).
Стадия Г: К раствору полученной на стадии В тиоуксусной кислоты (54,45 г, 0,166 моль) в тетрагидрофуране (370 мл) добавляли при 20° С воду (45 мл) и Охоnе® (306 г, 0,498 моль). Наблюдали повышение температуры вследствие экзотермической реакции до 42° С. Через 2 ч реакционную смесь фильтровали и осадок на фильтре тщательно промывали тетрагидрофураном, после чего к фильтрату добавляли воду (250 мл). Фильтрат концентрировали в вакууме. Суспензию 4 раза экстрагировали этилацетатом. Объединенные экстракты промывали трижды водой, соляным раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме, получая сульфон в виде твердого вещества бежевого цвета (60,0 г, 100%).
Стадия Д: Раствор, содержащий полученный на стадии Г сульфон (119,52 г, 0,332 моль) в метаноле (660 мл) и 4 н. раствор соляной кислоты в диоксане (20 мл), перемешивали в течение 12 ч при температуре окружающей среды. Реакционную смесь нагревали до кипения и медленно охлаждали до температуры окружающей среды. Образовавшиеся кристаллы отфильтровывали, тщательно промывали холодным метанолом и сушили, получая сложный метиловый эфир в виде твердого вещества белого цвета (89,4 г, 72%).
Стадия Е: К раствору полученного на стадии Д сложного метилового эфира (64,5 г, 0,180 моль) в диметилацетамиде (360 мл) добавляли карбонат калия (66,8 г, 0,48 моль), простой бис(2-бромэтиловый)эфир (40 мл, 0,305 моль), 4-диметиламинопиридин (1,1 г, 9 ммоль) и бромид тетрабутиламмония (2,9 г, 9 ммоль). Реакционную смесь перемешивали в течение ночи при температуре окружающей среды. Реакционную смесь медленно сливали на 1 н. HCl (500 мл). Образовавшийся осадок фильтровали, промывали водой, затем гексаном. Твердое вещество перекристаллизовывали из метанола, получая пиран в виде твердого вещества белого цвета (62,8 г, 79%). МСВР (ES+) M+NH
Стадия Ж: Полученный на стадии Е пиран (64,0 г, 0,144 моль) растворяли в сухом сосуде в атмосфере азота в безводном тетрагидрофуране (250 мл) и добавляли при температуре окружающей среды раствор триметилсиланолата калия (55,9 г, 0,432 моль) в безводном тетрагидрофуране (40 мл). Через 2 ч добавляли воду (200 мл) и раствор концентрировали в вакууме. Суспензию экстрагировали этилацетатом для удаления непрореагировавшего исходного продукта. Водный раствор обрабатывали 6 н. HCl до достижения значения рН 1. Суспензию экстрагировали этилацетатом и объединенные экстракты промывали водой, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток нагревали в диэтиловом эфире, образовавшееся твердое вещество фильтровали и сушили, получая карбоновую кислоту в виде твердого вещества белого цвета (56,3 г, 91%). МСВР (ES+) M+NH
Стадия З: Полученную на стадии Ж карбоновую кислоту (49,0 г, 0,114 моль) растворяли в сухом сосуде в атмосфере азота в безводном диметилформамиде (280 мл) и затем к раствору добавляли остальные реактивы в следующем порядке: гидрат N-гидроксибензотриазола (18,5 г, 0,137 моль), N-метилморфолин (37,5 мл, 0,342 моль), O-тетрагидро-2Н-пиран-2-илгидроксиламин (41,3 г, 0,353 моль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (30,6 г, 0,160 моль). После выдерживания в течение 4 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате, промывали водой, 5%-ным KHSO4, насыщенным раствором NаНСО3, соляным раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая ТГП-защищенный гидроксамат в виде пены белого цвета (62,6 г, 100%). МСВР (ES+) M+NH
Стадия И: К раствору полученного на стадии 3 ТГП-защищенного гидроксамата (58,5 г, 0,11 моль) в 1,4-диоксане (280 мл) добавляли 4 н. раствор HCl в диоксане (280 мл) и метанол (280 мл). После выдерживания в течение 15 мин при температуре окружающей среды реакционную смесь разбавляли этилацетатом и промывали водой, сушили над Na2SO4, фильтровали и концентрировали в вакууме. После перекристаллизации продукта (ацетон/гексан) получали указанное в заголовке соединение в виде твердого вещества белого цвета (42,79 г, 87%). МСВР (ES+) М+NH
Пример 380: Получение 4-[[4-([1,1’-бифенил]-4-илокси]фенил)сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
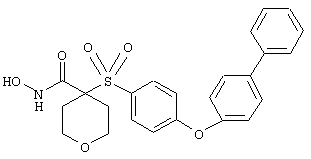
Стадия А: К раствору полученного в примере 55 продукта (2,0 г, 5,2 ммоль) в диметилацетамиде (8 мл) добавляли 4-фенилфенол (фирма Aldrich, 1,3 г, 7,8 ммоль), а затем карбонат цезия (6,8 г, 20,8 ммоль). Реакционную смесь выдерживали в течение 5 ч при 95° С. После выпаривания диметилацетамида в вакууме получали твердое вещество коричневого цвета (5,3 г, количественный выход). После хроматографии (обращенная фаза, колонка типа С-18, ацетонитрил/вода) получали ТГП-защищенный бифенил в растворе.
Стадия Б: К собранному полученному на стадии А ТГП-защищенному бифенилу в смеси ацетонитрил/вода (50 мл) медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил выпаривали. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества белого цвета (2,0 г, 83%). МС (FAB) M+H: рассчитано для C24H23NO6S: 454, обнаружено:454.
Пример 381: Получение тетрагидро-N-гидрокси-4-[[4-[[4-(трифторметил)фенил]тио]фенил]сульфонил]-2Н-пиран-4-карбоксамида
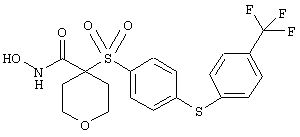
Стадия А: К раствору полученного в примере 55 продукта (2,0 г, 5,2 ммоль) в диметилацетамиде (6 мл) добавляли 4-трифторметилтиофенол (фирма Maybridge, 2,0 г, 11,2 ммоль), а затем карбонат калия (2,9 г, 20,8 ммоль). Реакционную смесь выдерживали в течение 12 ч при 65° С. После выпаривания диметилацетамида в вакууме получали твердое вещество коричневого цвета (6,5 г, количественный выход). После хроматографии (обращенная фаза, колонка типа С-18, ацетонитрил/вода) получали ТГП-защищенный трифторметил в растворе.
Стадия Б: К раствору неочищенного ТГП-защищенного трифторметила в смеси ацетонитрил/вода (40 мл) медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил выпаривали. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества рыжевато-коричневого цвета (0,75 г, 31%). МС (FAB) М+H: рассчитано для С19Н18F3NО5S2: 462, обнаружено: 462.
Пример 382: Получение тетрагидро-N-гидрокси-4-[[4-[4-[(трифторметил)тио]фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида

Стадия А: К раствору полученного в примере 55 продукта (2,0 г, 5,2 ммоль) в диметилацетамиде (6 мл) добавляли 4-(трифторметилтио)тиофенол (фирма Aldrich, 1,5 г, 7,8 ммоль), а затем карбонат цезия (6,8 г, 20,8 ммоль). После добавления каталитического количества фторида калия реакционную смесь выдерживали в течение 12 ч при 95° С. После выпаривания диметилацетамида в вакууме получали твердое вещество коричневого цвета (7,2 г, количественный выход). После хроматографии (обращенная фаза, колонка типа С-18, ацетонитрил/вода) получали ТГП-защищенное трифторметилтиопроизводное в растворе.
Стадия Б: К раствору полученного на стадии А неочищенного ТГП-защищенного трифторметилтиопроизводного в смеси ацетонитрил/вода (40 мл) медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил выпаривали. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества рыжевато-коричневого цвета (0,60 г, 24%). МС (FAB) М-Н: рассчитано для С19Н18F3NО6S2: 476, обнаружено: 476.
Пример 383: Получение 4-[[4-[4-хлор-3-(трифторметил)фенокси]фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
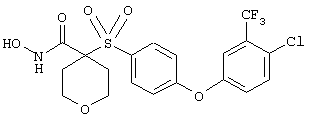
Стадия А: К раствору полученного в примере 55 продукта (2,0 г, 5,2 ммоль) в диметилацетамиде (6 мл) добавляли 4-хлор-3-трифторметилфенол (фирма Avocado, 1,5 г, 7,8 ммоль), а затем карбонат цезия (6,8 г, 20,8 ммоль). Реакционную смесь выдерживали в течение 12 ч при 95° С. После выпаривания диметилацетамида в вакууме получали твердое вещество коричневого цвета (7,6 г, количественный выход). После хроматографии (обращенная фаза, колонка типа С-18, ацетонитрил/вода) получали ТГП-защищенное соединение в растворе.
Стадия Б: К раствору полученного на стадии А неочищенного ТГП-защищенного соединения в смеси ацетонитрил/вода (40 мл) медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил выпаривали. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества рыжевато-коричневого цвета (0,92 г, 37%). МС (FAB) М+Н: рассчитано для C19H17CIF3NO6S: 480, обнаружено: 480.
Пример 384: Получение 4-[[4-[4-(1,1-диметилэтил)фенокси]фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
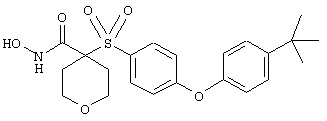
Стадия А: К раствору полученного в примере 55 продукта (5,0 г, 12,9 ммоль) в диметилацетамиде (25 мл) добавляли 4-трет-бутилфенол (фирма Avocado, 2,9 г, 19,4 ммоль), а затем карбонат цезия (20,4 г, 62,5 ммоль). Реакционную смесь выдерживали в течение 12 ч при 95° С. После выпаривания диметилацетамида в вакууме получали твердое вещество коричневого цвета (9,4 г, количественный выход). После хроматографии (обращенная фаза, колонка типа С-18, ацетонитрил/вода) получали ТГП-защищенное соединение в растворе.
Стадия Б: К раствору полученного на стадии А неочищенного ТГП-защищенного соединения в смеси ацетонитрил/вода (60 мл) медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил выпаривали. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,28 г, 5%). МС (FAB) М+H: рассчитано для C22H27NO6S: 434, обнаружено: 434.
Пример 385: Получение 4-[[4-[3,5-бис(трифторметил)фенокси]фенил]сульфонил]тетрагидро-N-гидрокси-2Н-пиран-4-карбоксамида
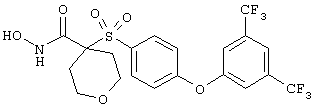
Стадия А: К раствору полученного в примере 55 продукта (3,0 г, 7,7 ммоль) в диметилацетамиде (15 мл) добавляли 3,5-дитрифторметилфенол (2,9 г, 19,4 ммоль), а затем карбонат цезия (20,4 г, 62,5 ммоль). Реакционную смесь выдерживали в течение 12 ч при 95° С. После выпаривания диметилацетамида в вакууме получали твердое вещество коричневого цвета (14,7 г, количественный выход). После хроматографии (обращенная фаза, колонка типа С-18, ацетонитрил/вода) получали ТГП-защищенное соединение в растворе.
Стадия Б: К раствору полученного на стадии А неочищенного ТГП-защищенного соединения в смеси ацетонитрил/вода (60 мл) медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил выпаривали. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества белого цвета (1,2 г, 31%). МС (FAB) M+H: рассчитано для С20Н17F6NO6S:514, обнаружено: 514.
Пример 386: Получение тетрагидро-N-гидрокси-4-[[4-[3-метил-4-(1-метилэтил)фенокси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
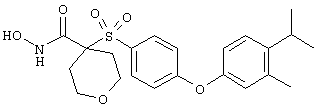
Стадия А: К раствору полученного в примере 55 продукта (4,0 г, 10,3 ммоль) в диметилацетамиде (20 мл) добавляли 4-изопропил-3-метилфенол (фирма Aldrich, 2,3 г, 15,5 ммоль), а затем карбонат цезия (16,8 г, 51,5 ммоль). Реакционную смесь выдерживали в течение 12 ч при 95° С. После выпаривания диметилацетамида в вакууме получали твердое вещество коричневого цвета (18,3 г, количественный выход). После хроматографии (обращенная фаза, колонка типа С-18, ацетонитрил/вода) получали ТГП-защищенное соединение в растворе.
Стадия Б: К раствору полученного на стадии А неочищенного ТГП-защищенного соединения в смеси ацетонитрил/вода (40 мл) медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил выпаривали. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества рыжевато-коричневого цвета (1,8 г, 40%). МС (FAB) М-Н: рассчитано для C22H27F3NO6S: 432, обнаружено: 432.
Пример 387: Получение тетрагидро-N-гидрокси-4-[[4-[(2,2,3,3-тетрафтор-2,3-дигидро-1,4-бензодиоксин-6-ил)окси]фенил]сульфонил]-2Н-пиран-4-карбоксамида
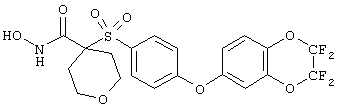
Стадия А: К раствору полученного в примере 55 продукта (5,0 г, 12,9 ммоль) в диметилацетамиде (25 мл) добавляли 2,2,3,3-тетрафтор-6-гидроксибензодиоксен (фирма Oakwood, 4,3 г, 19,4 ммоль), а затем карбонат цезия (21,0 г, 64,5 ммоль). Реакционную смесь выдерживали в течение 5 ч при 95° С. После выпаривания диметилацетамида в вакууме получали твердое вещество коричневого цвета (11,3 г, количественный выход). После хроматографии (обращенная фаза, колонка типа С-18, ацетонитрил/вода) получали ТГП-защищенное соединение в растворе.
Стадия Б: К собранному полученному на стадии А неочищенному ТГП-защищенного соединения в смеси ацетонитрил/вода (50 мл) медленно добавляли 10%-ный водный раствор HCl (100 мл). После перемешивания в течение ночи (приблизительно 18 ч) ацетонитрил выпаривали. Собирали образовавшийся осадок, получая указанное в заголовке соединение в виде твердого вещества белого цвета (3,5 г, 54%). МС (FAB) М-Н: рассчитано для C20H17F4NO8S: 506, обнаружено: 506.
Пример 388: Получение дигидрохлорида N-гидрокси-1-[2-(4-морфолинил)этил]-4-[[4-[4-(трифторметил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
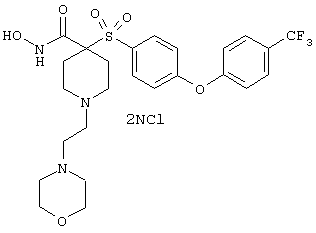
Стадия А: К суспензии гидробромида 4-бромпиперидина (107,0 г, 0,436 моль) в тетрагидрофуране (1 л) медленно добавляли триэтиламин (122 мл, 0,872 моль), а затем ди-трет-бутилдикарбонат (100 г, 0,458 моль), который добавляли в виде нескольких порций. Образовавшуюся смесь перемешивали в течение 22 ч при температуре окружающей среды, после чего фильтровали и концентрировали в вакууме. Твердые частицы промывали гексаном и затем собирали фильтрацией, получая Воc-защищенный пиперидин в виде масла янтарного цвета (124 г, >100%).
Стадия Б: К раствору 4-фторфенола (50,0 г, 0,390 моль) в ацетоне (400 мл), дегазированному с помощью N2, добавляли Cs2CO3 (159 г, 0,488 моль). После дегазации образовавшейся смеси в течение 5 мин с помощью N2 добавляли полученный на стадии А Воc-защищенный пиперидин (85,9 г, 0,325 моль). Образовавшуюся смесь перемешивали в течение 18 ч при температуре окружающей среды, после чего фильтровали через подушку из Celite®, промывая ацетоном. Фильтрат концентрировали в вакууме, получая сульфид в виде остатка рыжевато-коричневого цвета (98,5 г, 97%).
Стадия В: К раствору полученного на стадии Б сульфида (8,00 г, 25,7 ммоль) в дихлорметане (90 мл) и метаноле (15 мл) добавляли в виде двух порций гексагидрат магниевой соли монопероксифталевой кислоты (19,1 г, 38,6 ммоль). Образовавшуюся смесь перемешивали в течение 1,5 ч при температуре окружающей среды и затем фильтровали. Фильтрат промывали насыщенным раствором NaHCO3 и затем насыщенным раствором NaCl. Объединенные водные слои экстрагировали дихлорметаном (100 мл). Объединенные органические слои сушили над Na2SO4 и затем концентрировали в вакууме. Образовавшиеся твердые частицы промывали гексаном, затем их растворяли в дихлорметане и фильтровали через подушку из Celite®, промывая дихлорметаном. Фильтрат концентрировали в вакууме перекристаллизовывали из этилацетата, получая сульфон в виде кристаллического твердого вещества белого цвета (4,45 г, 50%).
Стадия Г: К раствору полученного на стадии В сульфона (7,00 г, 20,4 ммоль) в N,N-диметилформамиде (40 мл) добавляли Cs2CO3 (19,9 г, 61,2 ммоль) и α ,α ,α -трифтор-пара-крезол (3,97 г, 24,5 ммоль). Образовавшуюся смесь выдерживали в течение 16 ч при 80° С. После охлаждения до температуры окружающей среды реакционную смесь концентрировали в вакууме. Образовавшийся остаток обрабатывали Н2О и твердые частицы собирали фильтрацией. После этого твердые частицы промывали гексаном, затем метанолом, получая простой биариловый эфир в виде твердого вещества рыжевато-коричневого цвета (8,60 г, 87%).
Стадия Д: К раствору полученного на стадии Г простого биарилового эфира (8,59 г, 17,7 ммоль) в тетрагидрофуране (100 мл), охлажденному до 0° С, медленно добавляли бис(триметилсилил)амид лития (22,0 мл, 1,0 М раствор в тетрагидрофуране, 22,0 ммоль) с такой скоростью, чтобы температура реакционной смеси ни разу не превысила 1° С. Образовавшуюся смесь перемешивали в течение 1 ч при 0° С, после чего медленно добавляли раствор метилхлорформиата (2,05 мл, 26,6 ммоль) в тетрагидрофуране (5,0 мл) с такой скоростью, чтобы температура реакционной смеси ни разу не превысила 4° С. После завершения добавления смеси давали медленно нагреться до температуры окружающей среды. Добавляли насыщенный раствор NH4Cl (50 мл), после чего тетрагидрофуран удаляли в вакууме. К остатку добавляли воду (50 мл) и затем экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4. После перекристаллизации из метанола получали сложный метиловый эфир в виде кристаллического твердого вещества светло-желтого цвета (7,66 г, 80%).
Стадия Е: К раствору полученного на стадии Д сложного метилового эфира (7,66 г, 14,1 ммоль) в диоксане (30 мл) и метаноле (10 мл) добавляли 4 н. раствор HCl в диоксане (10 мл, 40 ммоль). После перемешивания в течение 2 ч при температуре окружающей среды добавляли дополнительную порцию 4 н. раствора HCl в диоксане (10 мл, 40 ммоль). После перемешивания в течение 2,5 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме, получая амин в виде твердого вещества беловатого цвета (6,80 г, >100%).
Стадия Ж: К суспензии полученного на стадии Е амина (3,00 г, 6,25 ммоль) в ацетонитриле (20 мл) добавляли K2CO3 (3,46 г, 25,0 ммоль), гидрохлорид 4-(2-хлорэтил)морфолина (1,22 г, 6,56 моль) и каталитическое количество NaI. Образовавшуюся смесь выдерживали в течение 22 ч при температуре дефлегмации. После охлаждения до температуры окружающей среды реакционную смесь фильтровали через подушку из Celite®, промывая этилацетатом. Фильтрат концентрировали в вакууме, получая морфолинилэтиламин в виде твердого вещества рыжевато-коричневого цвета (3,45 г, >100%).
Стадия З: К раствору полученного на стадии Ж морфолинилэтиламина (3,45 г, 6,25 ммоль) в тетрагидрофуране (60 мл) добавляли триметилсиланолат калия (1,60 г, 12,50 ммоль). После перемешивания в течение 25 ч при температуре окружающей среды добавляли Н2О. Затем реакционную смесь нейтрализовали (до рН 7) с помощью 1 н. раствора HCl. Тетрагидрофуран удаляли в вакууме и образовавшийся осадок собирали фильтрацией и промывали диэтиловым эфиром, получая аминокислоту в виде твердого вещества беловатого цвета (2,87 г, 85%).
Стадия И: К суспензии полученной на стадии 3 аминокислоты (2,87 г, 5,29 ммоль) в дихлорметане (25 мл) добавляли N-метилморфолин (1,74 мл, 15,9 ммоль), O-(тетрагидропиранил)гидроксиламин (0,682 г, 5,82 ммоль) и РуВrоР® (2,96 г, 6,35 ммоль). После перемешивания в течение 19 ч при температуре окружающей среды добавляли дополнительные порции N-метилморфолина (0,872 мл, 7,94 ммоль), O-(тетрагидропиранил)гидроксиламина (0,310 г, 2,65 ммоль) и РуВrоР® (1,48 г, 3,17 ммоль). Образовавшуюся смесь перемешивали в течение 3 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между этилацетатом и Н2О. Органический слой промывали насыщенным раствором NaCl и сушили над Nа2SO4. После хроматографии (на силикагеле, метанол/хлороформ) получали защищенный гидроксамат в виде твердого вещества беловатого цвета (2,62 г, 77%).
Стадия К: К раствору полученного на стадии И защищенного гидроксамата (2,62 г, 4,08 ммоль) в диоксане (9 мл) и метаноле (3 мл) добавляли 4 н. раствор HCl в диоксане (10 мл, 40,0 ммоль). Образовавшуюся смесь перемешивали в течение 2 ч при температуре окружающей среды, затем добавляли диэтиловый эфир (20 мл). Образовавшиеся твердые частицы собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества беловатого цвета (2,31 г, 90%). МС МН+: рассчитано для С25Н31O6N3SF3: 558, обнаружено: 558.
Пример 389: Получение дигидрохлорида N-гидрокси-1-(4-пиридинилметил)-4-[[4-[4-(трифторметил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
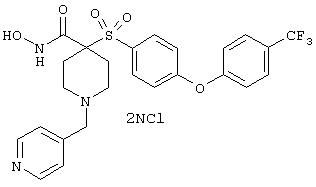
Стадия А: К суспензии полученного на стадии Е примера 388 амина (1,50 г, 3,13 ммоль) в ацетонитриле (10 мл) добавляли К2СО3 (1,73 г, 12,5 ммоль) и гидрохлорид 4-пиколилхлорида (0,565 г, 3,44 ммоль). После перемешивания в течение 21,5 ч при температуре дефлегмации реакционную смесь фильтровали через подушку из Celite®, промывая этилацетатом. Фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали пиколиламин в виде прозрачной смолы (1,44 г, 86%).
Стадия Б: К раствору полученного на стадии А пиколиламина (1,44 г, 2,69 ммоль) в тетрагидрофуране (20 мл) добавляли триметилсиланолат калия (0,690 г, 5,38 ммоль). Образовавшуюся смесь перемешивали в течение 20 ч при температуре окружающей среды и затем тетрагидрофуран удаляли, продувая N2 через реакционную смесь. Добавляли воду и реакционную смесь нейтрализовали (до рН 7) с помощью 2 н. раствора HCl. Образовавшийся осадок собирали фильтрацией, получая аминокислоту в виде твердого вещества белого цвета (1,31 г, 94%).
Стадия В: К суспензии полученной на стадии Б аминокислоты (1,31 г, 2,52 ммоль) в N,N-диметилформамиде (10 мл) добавляли 1-гидроксибензотриазол (0,408 г, 3,02 ммоль), N-метилморфолин (0,832 мл, 7,56 ммоль), O-(тетрагидропиранил)гидроксиламин (0,443 г, 3,78 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,676 г, 3,53 ммоль). Образовавшуюся смесь перемешивали в течение 3 дней при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Объединенные органические слои промывали насыщенным раствором NaHCO3, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде пены белого цвета (1,24 г, 79%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (1,24 г, 2,00 ммоль) в диоксане (6 мл) и метаноле (2 мл) добавляли 4 н. раствор HCl в диоксане (5,00 мл, 20,0 ммоль). После перемешивания в течение 2,5 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме. Образовавшуюся пену вновь обрабатывали в течение 15 мин 4 н. раствором HCl в диоксане (3,00 мл), затем добавляли диэтиловый эфир и образовавшийся осадок собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества беловатого цвета (1,04 г, 85%). МС МН+: рассчитано для С25Н25O5N3SF3: 536, обнаружено: 536.
Пример 390: Получение дигидрохлорида N-гидрокси-1-(3-пиридинилметил)-4-[[4-[4-(трифторметил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
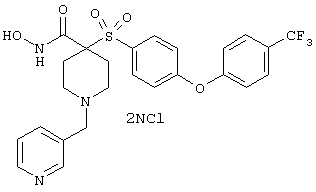
Стадия А: К суспензии полученного на стадии Е примера 388 амина (1,00 г, 2,08 ммоль) в ацетонитриле (10 мл) добавляли К2СО3 (1,15 г, 8,33 ммоль) и гидрохлорид 3-пиколилхлорида (0,375 г, 2,29 ммоль). После перемешивания в течение 12 ч при температуре дефлегмации реакционную смесь фильтровали через подушку из Celite®, промывая этилацетатом. Фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали пиколиламин в виде пены светло-желтого цвета (0,740 г, 67%).
Стадия Б: К раствору полученного на стадии А пиколиламина (0,740 г, 1,38 ммоль) в тетрагидрофуране (10 мл) добавляли триметилсиланолат калия (0,355 г, 2,77 ммоль). Образовавшуюся смесь перемешивали в течение 17 ч при температуре окружающей среды, затем добавляли дополнительную порцию триметилсиланолата калия (0,044 г, 0,343 ммоль) и образовавшуюся смесь перемешивали в течение 2 ч при температуре окружающей среды. Тетрагидрофуран удаляли, продувая N2 через реакционную смесь. Добавляли воду (5 мл) и реакционную смесь нейтрализовали (до рН 7) с помощью 2н. раствора HCl. Образовавшийся осадок собирали фильтрацией и сушили путем концентрирования в вакууме с ацетоном, получая аминокислоту в виде твердого вещества беловатого цвета (0,700 г, 97%).
Стадия В: К суспензии полученной на стадии Б аминокислоты (0,700 г, 1,34 ммоль) в N,N-диметилформамиде (10 мл) добавляли 1-гидроксибензотриазол (0,218 г, 1,61 моль), N-метилморфолин (0,442 мл, 4,02 ммоль), O-(тетрагидропиранил)гидроксиламин (0,235 г, 2,01 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,360 г, 1,88 ммоль). Образовавшуюся смесь перемешивали в течение 23 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Объединенные органические слои промывали Н2О, насыщенным раствором NaHCO3, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде пены беловатого цвета (0,500 г, 60%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (0,500 г, 0,807 ммоль) в диоксане (1,5 мл) и метаноле (0,5 мл) добавляли 4 н. раствор HCl в диоксане (3,00 мл, 12,0 ммоль). После перемешивания в течение 2 ч при температуре окружающей среды добавляли диэтиловый эфир и образовавшийся осадок собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества желтого цвета (0,363 г, 74%). МС МН+: рассчитано для C25H25O5N3SF3: 536, обнаружено: 536.
Пример 391: Получение дигидрохлорида N-гидрокси-1-(2-пиридинилметил)-4-[[4-[4-(трифторметил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
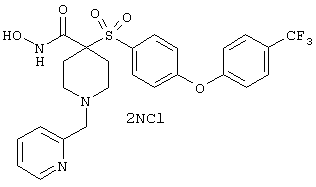
Стадия А: К суспензии полученного на стадии Е примера 388 амина (1,26 г, 2,63 ммоль) в ацетонитриле (10 мл) добавляли К2СО3 (1,45 г, 10,5 ммоль) и гидрохлорид 2-пиколилхлорида (0,475 г, 2,89 моль). После перемешивания в течение 12 ч при температуре дефлегмации реакционную смесь фильтровали через подушку из Celite®, промывая этилацетатом. Фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали пиколиламин в виде смолы янтарного цвета (1,40 г, 99%).
Стадия Б: К раствору полученного на стадии А пиколиламина (1,40 г, 2,62 ммоль) в тетрагидрофуране (20 мл) добавляли триметилсиланолат калия (0,672 г, 5,24 ммоль). Образовавшуюся смесь перемешивали в течение 15 ч при температуре окружающей среды. Тетрагидрофуран удаляли, продувая N2 через реакционную смесь. Добавляли Н2О (5 мл) и реакционную смесь нейтрализовали (до рН 7) с помощью 2 н. раствора HCl. Образовавшийся осадок собирали фильтрацией и сушили путем концентрирования в вакууме с ацетонитрилом, получая аминокислоту в виде твердого вещества беловатого цвета (1,07 г, 79%).
Стадия В: К суспензии полученной на стадии Б аминокислоты (1,07 г, 2,06 ммоль) в N,N-диметилформамиде (10 мл) добавляли 1-гидроксибензотриазол (0,333 г, 2,47 моль), N-метилморфолин (0,679 мл, 6,18 ммоль), O-(тетрагидропиранил)гидроксиламин (0,362 г, 3,09 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимид а (0,553 г, 2,88 ммоль). Образовавшуюся смесь перемешивали в течение 19 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Объединенные органические слои промывали Н2О, насыщенным раствором NаНСО3, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, метанол/дихлорметан) получали защищенный гидроксамат в виде твердого вещества белого цвета (1,03 г, 81%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (1,03 г, 1,66 ммоль) в диоксане (3,0 мл) и метаноле (1,0 мл) добавляли 4 н. раствор HCl в диоксане (3,00 мл, 12,0 ммоль). После перемешивания в течение 1,5 ч при температуре окружающей среды добавляли диэтиловый эфир и образовавшийся осадок собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества светло-розового цвета (0,970 г, 96%). МС МН+: рассчитано для С25Н25O5N3SF3:536, обнаружено: 536.
Пример 392: Получение моногидрохлорида N-гидрокси-4-[[4-[(4-метоксифенил)амино]фенил]сульфонил]-4-пиперидинкарбоксамида
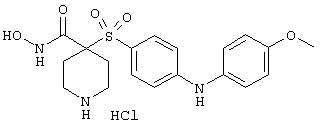
Стадия А: К полученному на стадии В примера 91 сложного эфира (1,00 г, 2,17 ммоль) добавляли Сs2СО3 (0,990 г, 3,04 ммоль), BINAP (0,061 г, 0,098 ммоль), трис(дибензилиденацетон)дипалладий(0) (0,060 г, 0,07 моль), пара-анизидин (0,320 г, 2,60 ммоль) и толуол (4 мл). Образовавшуюся смесь нагревали в течение 22 ч до 100° С. После охлаждения до температуры окружающей среды добавляли диэтиловый эфир и смесь фильтровали через подушку из Celite®, промывая этилацетатом. Фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали анилин в виде пены оранжевого цвета (0,810 г, 74%).
Стадия Б: К раствору полученного на стадии А анилина (0,780 г, 1,55 ммоль) в тетратидрофуране (4,0 мл) добавляли триметилсиланолат калия (0.238 г, 1,86 ммоль). Образовавшуюся смесь перемешивали в течение 17 ч при температуре окружающей среды и затем добавляли дополнительную порцию триметилсиланолата калия (0,020 г, 0,1955 ммоль). После перемешивания в течение 24 ч при температуре окружающей среды добавляли дополнительную порцию триметилсиланолата калия (0,040 г, 0,310 ммоль). После перемешивания в течение 26 ч при температуре окружающей среды растворитель удаляли, продувая N2 через смесь. К суспензии остатка в дихлорметане (10 мл) добавляли N-метилморфолин (0,511 мл, 4,65 ммоль), O-(тетрагидропиранил)гидроксиламин (0,218 г, 1,86 ммоль), а затем РуВrоР® (1,08 г, 2,33 ммоль). Образовавшуюся смесь перемешивали в течение 2 дней при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Объединенные органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества беловатого цвета (0,600 г, 66%).
Стадия В: К раствору полученного на стадии Б защищенного гидроксамата (0,580 г, 0,984 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли 4 н. раствор HCl в диоксане (2,5 мл, 10,0 ммоль). Образовавшуюся смесь перемешивали в течение 1 ч при температуре окружающей среды, затем добавляли диэтиловый эфир (10 мл). Твердые частицы собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,437 г, 100%). МС МН+: рассчитано для С19H24O5N3S: 406, обнаружено: 406.
Пример 393: Получение моногидрохлорида N-гидрокси-4-[[4-[[4-(трифторметокси)фенил]амино]фенил]сульфонил]-4-пиперидинкарбоксамида
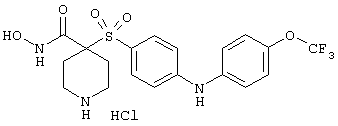
Стадия А: К раствору полученного на стадии В примера 91 сложного эфира (3,27 г, 7,09 ммоль) добавляли Сs2СО3 (3,23 г, 9,92 ммоль), BINAP (0,066 г, 0,107 ммоль), трис(дибензилиденацетон)дипалладий(0) (0,065 г, 0,071 моль), 4-трифторметоксианилин (1,15 мл, 8,51 ммоль) и толуол (14 мл). Образовавшуюся смесь нагревали в течение 22 ч до 100° С. После охлаждения до температуры окружающей средь" смесь фильтровали через подушку из Celite®, промывая этилацетатом, и фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали анилин в виде твердого вещества рыжевато-коричневого цвета (3,59 г, 91%).
Стадия Б: К раствору полученного на стадии А анилина (1,03 г, 1,84 ммоль) в тетрагидрофуране (10 мл) добавляли триметилсиланолат калия (0,331 г, 2,58 ммоль). Образовавшуюся смесь перемешивали в течение 24 ч при температуре окружающей среды и затем добавляли дополнительную порцию триметилсиланолата калия (0,118 г, 0,092 ммоль). После перемешивания в течение 26 ч при температуре окружающей среды растворитель удаляли, продувая N2 через смесь. Добавляли Н2О и реакционную смесь подкисляли (до рН 3) с помощью 1 н. раствора HCl. Водную реакционную смесь экстрагировали этилацетатом и объединенные органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали кислоту в виде твердого вещества рыжевато-коричневого цвета (1,01 г, 100%).
Стадия В: К суспензии полученной на стадии Б кислоты (1,00 г, 1,84 ммоль) в N,N-диметилформамиде (10 мл) добавляли 1-гидроксибензотриазол (0,298 г, 2,21 моль), N-метилморфолин (0,607 мл, 5,52 ммоль), O-(тетрагидропиранил)гидроксиламин (0,323 г, 2,76 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,494 г, 2,58 ммоль). Образовавшуюся смесь перемешивали в течение 17 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Объединенные органические слои промывали Н2О, насыщенным раствором NaHCO3, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества белого цвета (0,960 г, 81%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (0,960 г, 1,49 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли 4н. растворHCl в диоксане (4,0 мл, 16,0 ммоль). Образовавшуюся смесь перемешивали в течение 2,5 ч при температуре окружающей среды. Затем растворитель удаляли, продувая N2 через реакционную смесь. Добавляли диэтиловый эфир (20 мл) и осадок собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества светло-розового цвета (0,716 г, 100%). МС МН+: рассчитано для C19H21O5N3SF3: 460, обнаружено: 460.
Пример 394: Получение моногидрохлорида N-гидрокси-1-(2-метоксиэтил)-4-[[4-[[4-(трифторметокси)фенил]амино]фенил]сульфонил]-4-пиперидинкарбоксамида
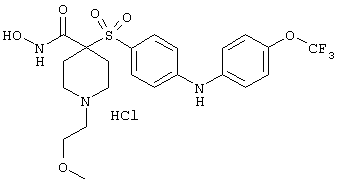
Стадия А: К раствору полученного на стадии А примера 392 анилина (2,55 г, 4,57 ммоль) в диоксане (9,0 мл) и метаноле (3,0 мл) добавляли 4 н. раствор HCl в диоксане (10 мл, 40 ммоль). После перемешивания в течение 2 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме, получая амин в виде твердого вещества рыжевато-коричневого цвета (2,36 г, >100%).
Стадия Б: К суспензии полученного на стадии А амина (1,50 г, 3,03 ммоль) в ацетонитриле (12 мл) добавляли К2СО3 (1,26 г, 9,09 ммоль) и простой метил-2-бромэтиловый эфир (0,313 мл, 3,33 ммоль). После перемешивания в течение 23 ч при температуре дефлегмации добавляли Cs2CO3 (2,96 г, 9,09 ммоль). После выдерживания в течение 6 ч при температуре дефлегмации реакционную смесь фильтровали через подушку из Celite®, промывая дихлорметаном. Фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, метанол/дихлорметан) получали метоксиэтиламин в виде твердого вещества рыжевато-коричневого цвета (1,13 г, 72%).
Стадия В: К раствору полученного на стадии Б метоксиэтиламина (1,13 г, 2,19 ммоль) в тетрагидрофуране (20 мл) добавляли триметилсиланолат калия (0,561 г, 4,38 ммоль). Образовавшуюся смесь перемешивали в течение 18 ч при температуре окружающей среды и затем добавляли дополнительную порцию триметилсиланолата калия (0,140 г, 1,09 ммоль). После перемешивания в течение 5 ч при температуре окружающей среды растворитель удаляли, продувая N2 через смесь. Добавляли воду (8 мл) и реакционную смесь нейтрализовали (до рН 7) с помощью 1 н. раствора HCl. Твердые частицы собирали фильтрацией и сушили путем концентрирования в вакууме с ацетонитрилом, получая аминокислоту в виде твердого вещества беловатого цвета (0,900 г, 82%).
Стадия Г: К суспензии полученной на стадии В аминокислоты (0,900 г, 1,79 ммоль) в N,N-диметилформамиде (8,0 мл) добавляли 1-гидроксибензотриазол (0,290 г, 2,15 моль), N-метилморфолин (0,590 мл, 5,37 ммоль), O-(тетрагидропиранил)гидроксиламин (0,315 г, 2,69 ммоль) и гидрохлорид 1-[3-(диметиламино) пропил] -3-этилкарбодиимида (0,480 г, 2,51 ммоль). Образовавшуюся смесь перемешивали в течение 16 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Объединенные органические слои промывали Н2О, насыщенным раствором NаНСО3, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, метанол/дихлорметан) получали защищенный гидроксамат в виде твердого вещества беловатого цвета (0,870 г, 81%).
Стадия Д: К раствору полученного на стадии Г защищенного гидроксамата (0,870 г, 1,45 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли 4 н. раствор HCl в диоксане (10,0 мл, 40,0 ммоль). Образовавшуюся смесь перемешивали в течение 2,0 ч при температуре окружающей среды. Реакционную смесь концентрировали в вакууме и затем еще раз обрабатывали в течение 30 мин 4 н. раствором HCl (3 мл). Затем растворитель удаляли, продувая N2 через реакционную смесь. Добавляли диэтиловый эфир (30 мл) и осадок собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества светло-розового цвета (0,771 г, 96%). МС МН+: рассчитано для С22Н27О6N3SF3: 518, обнаружено: 518.
Пример 395: Получение моногидрохлорида N-гидрокси-4-[[4-[[4-(трифтоометил)фенил]амино]фенил]сульфонил]-4-пиперидинкарбоксамида
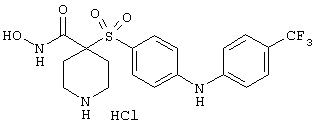
Стадия А: К раствору полученного на стадии В примера 91 сложного эфира (3,16 г, 6,85 ммоль) добавляли Cs2CO3 (3,13 г, 9,59 ммоль), BINAP (0,064 г, 0,103 ммоль), трис(дибензилиденацетон)дипалладий(0) (0,063 г, 0,069 моль), α ,α ,α -трифторметиланилин (1,03 мл, 8,22 ммоль) и толуол (14 мл). Образовавшуюся смесь нагревали в течение 17 ч до 100° С. После охлаждения до температуры окружающей среды смесь фильтровали через подушку из Celite®, промывая дихлорметаном, и фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, этилацетат/гексан) получали анилин в виде пены светло-оранжевого цвета (3,08 г, 83%).
Стадия Б: К раствору полученного на стадии А анилина (1,00 г, 1,84 ммоль) в тетрагидрофуране (10 мл) добавляли триметилсиланолат калия (0,473 г, 3,69 ммоль). Образовавшуюся смесь перемешивали в течение 25 ч при температуре окружающей среды, затем растворитель удаляли, продувая N2 через смесь. Добавляли воду и реакционную смесь подкисляли (до рН 3) с помощью 1 н. раствора HCl. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали кислоту в виде пены оранжевого цвета (1,00 г, >100%).
Стадия В: К суспензии полученной на стадии Б кислоты (0,972 г, 1,84 ммоль) в N,N-диметилформамиде (10 мл) добавляли 1-гидроксибензотриазол (0,298 г, 2,21 моль), N-метилморфолин (0,607 мл, 5,52 ммоль), O-(тетрагидропиранил)гидроксиламин (0,323 г, 2,76 ммоль) и гидрохлорид 1-13-(диметиламино)пропил]-3-этилкарбодиимида (0,494 г, 2,58 ммоль). Образовавшуюся смесь перемешивали в течение 18 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Объединенные органические слои промывали Н2О, насыщенным раствором NaHCO3, насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества белого цвета (0,970 г, 84%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (0,950 г, 1,51 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли 4н. растворHCl в диоксане (4,0 мл, 16,0 ммоль). Образовавшуюся смесь перемешивали в течение 1,5 ч при температуре окружающей среды. Добавляли диэтиловый эфир (20 мл) и осадок собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,630 г, 87%). МС МН+: рассчитано для C19H21O4N3SF3: 444, обнаружено: 444.
Пример 396: Получение моногидрохлорида N-гидрокси-1-(2-метоксиэтил)-4-[[4-[[4-(трифторметил)фенил]амино]фенил]сульфонил]-4-пиперидинкарбоксамида

Стадия А: К раствору полученного на стадии А примера 395 анилина (2,07 г, 3,82 ммоль) в диоксане (9,0 мл) и метаноле (3,0 мл) добавляли 4 н. раствор HCl в диоксане (10 мл, 40 ммоль). После перемешивания в течение 2 ч при температуре окружающей среды реакционную смесь концентрировали в вакууме, получая амин в виде твердого вещества желтого цвета (1,89 г, >100%).
Стадия Б: К суспензии полученного на стадии А амина (1,83 г, 3,82 ммоль) в ацетонитриле (20 мл) добавляли К2СО3 (1,58 г, 11,46 ммоль) и метил-2-бромэтиловый эфир (0,395 мл, 4,20 ммоль). После перемешивания в течение 18 ч при температуре дефлегмации реакционную смесь фильтровали через подушку из Celite®, промывая дихлорметаном и фильтрат концентрировали в вакууме. После хроматографии (на силикагеле, метанол/дихлорметан) получали метоксиэтиламин в виде твердого вещества беловатого цвета (1,58 г, 83%).
Стадия В: К раствору полученного на стадии Б метоксиэтиламина (1,58 г, 3,15 ммоль) в тетрагидрофуране (30 мл) добавляли триметилсиланолат калия (0,810 г, 6,31 ммоль). Образовавшуюся смесь перемешивали в течение 3 дней при температуре окружающей среды и затем растворитель удаляли, продувая N2 через смесь. Добавляли воду (10 мл) и реакционную смесь нейтрализовали (до рН 7) с помощью 1 н. раствора HCl. Твердые частицы собирали фильтрацией и сушили путем концентрирования в вакууме с ацетонитрилом, получая аминокислоту в виде твердого вещества розового цвета (1,32 г, 86%).
Стадия Г: К суспензии полученной на стадии В аминокислоты (1,32 г, 2,71 ммоль) в N,N-диметилформамиде (12 мл) добавляли 1-гидроксибензотриазол (0,439 г, 3,25 моль), N-метилморфолин (0,894 мл, 8,13 ммоль), O-(тетрагидропиранил)гидроксиламин (0,476 г, 4,07 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,727 г, 3,79 ммоль). Образовавшуюся смесь перемешивали в течение 20 ч при температуре окружающей среды и затем концентрировали в вакууме. Остаток распределяли между Н2О и этилацетатом. Объединенные органические слои промывали Н2О, насыщенным раствором NaHCO3, насыщенным раствором NaCl и сушили над Nа2SO4. После хроматографии (на силикагеле, метанол/этилацетат) получали защищенный гидроксамат в виде твердого вещества беловатого цвета (1,39 г, 88%).
Стадия Д: К раствору полученного на стадии Г защищенного гидроксамата (1,40 г, 2,39 ммоль) в диоксане (3 мл) и метаноле (1 мл) добавляли 4 н. раствор HCl в диоксане (5,98 мл, 23,9 ммоль). Образовавшуюся смесь перемешивали в течение 2,5 ч при температуре окружающей среды. Реакционную смесь концентрировали почти досуха, продувая N2 через реакционную смесь. Добавляли диэтиловый эфир (25 мл) и осадок собирали фильтрацией. Образовавшееся твердое вещество растворяли в метаноле (1 мл) и обрабатывали 4 н. раствором HCl в диоксане (1,5 мл). После перемешивания в течение 1,5 ч при температуре окружающей среды реакционную смесь медленно добавляли к диэтиловому эфиру (50 мл). Образовавшийся осадок собирали фильтрацией, получая указанное в заголовке соединение в виде твердого вещества беловатого цвета (1,08 г, 84%). МС МН+: рассчитано для С22Н27O5N3SF3: 502, обнаружено: 502.
Пример 397: Получение этил-1-(2-метоксиэтил)-3-фенилпропокси)фенил]сульфонил]-4-пиперидинкарбоксилата
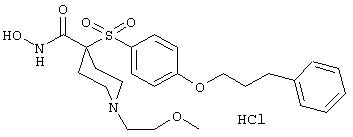
Стадия А: Смесь, содержащую метоксиэтиламин этил-4-[(4-фторфенилсульфонил)]-1-(2-метоксиэтил)-4-пиперидинкарбоксилата (1,5 г, 4,0 ммоль), 3-фенил-1-пропанол (2,2 мл, 16 ммоль) и К2СО3 (2,2 г, 16 ммоль) в ДМАц (6 мл), выдерживали в течение 1 дня при 125° С и в течение 3 дней при 135° С. После этого смесь концентрировали в вакууме, разбавляли водой и экстрагировали этилацетатом. Органический слой промывали водой и соляным раствором, сушили над сульфатом магния и концентрировали в вакууме, получая неочишенное масло. Масло очишали экспресс-хроматографией (смесь гексан/этилацетат, 20:80), получая простой эфир в виде масла коричневого цвета (1,35 г, 67%).
Стадия Б: Смесь, содержащую полученный на стадии А простой эфир (1,3 г, 2,7 ммоль) и 50%-ный водный раствор NaOH (2,1 г, 27 ммоль) в ТГФ (23 мл), ЕtOН (23 мл) и Н2О (12 мл), выдерживали в течение 24 ч при 60° С в атмосфере азота. Продукт концентрировали в вакууме и растирали с диэтиловым эфиром, получая твердое вещество. Твердое вещество растворяли в воде, охлаждали в ледяной бане и подкисляли концентрированной соляной кислотой. Осадок выделяли фильтрацией, промывали холодной водой и сушили в вакуумной печи в течение 3 дней при температуре окружающей среды, получая неочищенную кислоту.
Смесь, содержащую полученную выше неочищенную кислоту (1,1 г), N-гидроксибензотриазол (0,36 г, 2,7 ммоль), 4-метилморфолин (0,74 мл, 6,7 ммоль), O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,39 г, 3,3 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,60 г, 3,1 ммоль) в ДМФ (11 мл), перемешивали в течение 18 ч при температуре окружающей среды в атмосфере азота. Смесь концентрировали в вакууме и растворяли в растворе, содержащем насыщенный раствор NaHCO3 (90 мл), этилацетат (25 мл) и несколько капель 2н. раствора NaOH. Водный слой экстрагировали дополнительной порцией этилацетата. Объединенные этилацетатные слои промывали насыщенным раствором NaHCO3, водой и соляным раствором. После сушки над сульфатом магния фильтрат концентрировали в вакууме, получая масло темно-желтого цвета. Масло очищали экспресс-хроматографией (смесь ацетонитрил/толуол, 40:60), получая защищенный гидроксамат в виде масла желтого цвета (0,32 г, 25%). МС МН+: рассчитано для С29Н40N2O7S: 561, обнаружено: 561.
Стадия В: К раствору полученного на стадии Б защищенного гидроксамата (0,28 г, 0,50 ммоль) в метаноле (4,0 мл) добавляли ацетилхлорид (0,11 мл, 1,5 ммоль) и раствор перемешивали в течение 2,5 ч при температуре окружающей среды в атмосфере азота. Раствор разбавляли диэтиловым эфиром и концентрировали. Твердое вещество растирали с диэтиловым эфиром и сушили в вакуумной печи при 40° С, получая указанное в заголовке соединение в виде твердого вещества беловатого цвета (0,15 г, 20%). МС МН+: рассчитано для C24H32N2O6S: 477, обнаружено: 477.
Пример 398: Получение моногидрохлорида 1-циклопропил-N-гидрокси-4-[[4-(2-феноксиэтокси)фенил]сульфонил]-4-пиперидинкарбоксамида
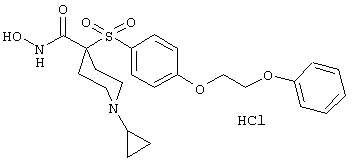
Стадия А: К раствору полученного на стадии Д примера 9 соединения (14,36 г, 40 ммоль) в метаноле (50 мл) добавляли уксусную кислоту (24,5 г, 400 ммоль), порцию (приблизительно 2 г) молекулярных сит размером 4 Е, (1-этоксициклопропил)окситриметилсилан (25,8 мл, 148 ммоль) и цианборогидрид натрия (7,05 г, 112 ммоль). Раствор выдерживали в течение 8 ч при температуре дефлегмации. Осадившиеся твердые частицы удаляли фильтрацией и фильтрат концентрировали в вакууме. Остаток разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. Твердое вещество фильтровали, промывали смесью Н2О/диэтиловый эфир, получая требуемый циклопропиламин этил-4-[(4-фторфенилсульфонил)]-1-циклопропил-4-пиперидинкарбоксилата в виде твердого вещества белого цвета (11,83 г, 81,5%). МС МН+: рассчитано для C17H22NO4SF: 356, обнаружено: 356.
Стадия Б: Раствор, содержащий полученный на стадии А циклопропиламин (2,0 г, 5,6 ммоль), фениловый эфир этиленгликоля (2,8 мл, 23 ммоль) и карбонат цезия (7,3 г, 23 ммоль) в ДМАц (10 мл), выдерживали в течение 18 ч при 125-135° С в атмосфере азота. Смесь концентрировали в вакууме, разбавляли водой и экстрагировали этилацетатом. Объединенные этилацетатные слои промывали водой и соляным раствором, сушили над сульфатом магния, концентрировали в вакууме, растворяли в диэтиловом эфире, осаждали в виде гидрохлорида и сушили в вакуумной печи при 40° С. Твердое вещество растворяли в смеси воды, ацетонитрила и этанола и затем значение рН доводили до 12 с помощью 1 н. раствора NaOH. Смесь концентрировали в вакууме для удаления этанола и ацетонитрила. Твердое вещество выделяли фильтрацией, промывали водой и сушили в вакуумной печи при 50° С, получая простой эфир в виде твердого вещества белого цвета (1,8 г, 68%). МС+: рассчитано для C25H31NO6S: 474, обнаружено: 474. Элементный анализ: рассчитано для C25H31NO6S: С 63,40; Н 6,60; N 2,96; S 6,77; обнаружено: С 63,35; Н 6,59; N 2,99; S 6,61.
Стадия В: Смесь, содержащую полученный на стадии Б простой эфир (1,8 г, 3,7 ммоль) и 50%-ный водный раствор NaOH (3,0 г, 37 ммоль) в ТГФ (32 мл), EtOH (32 мл) и Н2О (16 мл), выдерживали в течение 24 при 60° С в атмосфере азота. Продукт концентрировали в вакууме и растирали с диэтиловым эфиром, получая твердое вещество. Твердое вещество рыжевато-коричневого цвета растворяли в смеси воды, этанола и ТГФ, осаждали путем доведения значения рН до 3 с помощью концентрированной соляной кислоты, концентрировали в вакууме, растирали с водой и сушили в вакуумной печи при 50° С, получая неочищенную кислоту в виде твердого вещества белого цвета (2,3 г).
Смесь, содержащую полученную выше неочищенную кислоту в виде твердого вещества белого цвета (2,3 г), N-гидроксибензотриазол (1,9 г, 14 ммоль), 4-метилморфолин (1,6 мл, 14 ммоль), O-тетрагидро-2Н-пиран-2-илгидроксиламин (1,1 г, 9,4 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (2,7 г, 14 ммоль) в ДМФ (90 мл), перемешивали в течение 2 дней при температуре окружающей среды в атмосфере азота. Смесь концентрировали в вакууме, разбавляли водой и экстрагировали этилацетатом. Органический слой промывали 1 н. раствором NaOH, водой и соляным раствором, сушили над сульфатом магния, концентрировали в вакууме и очищали экспресс-хроматографией (смесь этилацетат/толуол, от 20:80 до 40:60), получая защищенный гидроксамат в виде твердого вещества белого цвета (0,43 г, 21%). МС МН+: рассчитано для C28H36N2O7S: 545, обнаружено: 545. Элементный анализ: рассчитано для С28Н36N2О7S: С 61,74; Н 6,66; N 5,14; S 5,89; обнаружено: С 61,72; Н 6,75; N 5,06; S 5,91.
Дополнительную порцию соединения выделяли путем подкисления водного слоя до рН 3, сбора твердого вещества фильтрацией и сушки с получением твердого вещества белого цвета (0,80 г).
Стадия Г: К расгвору ацетилхлорида (0,31 мл, 4,4 ммоль) в метаноле (11 мл), находящемуся при температуре окружающей среды, добавляли в атмосфере азота полученный на стадии В защищенный гидроксамат (0,80 г, 1,5 ммоль). После перемешивания в течение 2,5 ч осадок собирали фильтрацией, промывали диэтиловым эфиром и сушили в вакуумной печи при 45° С, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,58 г, 79%). МС МН+: рассчитано для C23H28N2O6S: 461, обнаружено: 461. Элементный анализ: рассчитано для C23H28N2O6S· 1,5HCl: С 53,62; Н 5,77; N 5,44; S 6,22; обнаружено: С 53,47; Н 5,79; N 5,41; S 6,16.
Пример 399: Получение дигидрохлорида гидрокси-1-(3-пиридинилметил)-4-[[4-[4-трифторметокси)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
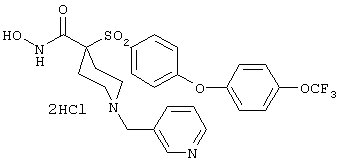
Стадия А: Раствор, содержащий полученный в примере 410 гидрохлорид амина (2,4 г, 4,6 ммоль), 3-пиколилхлорид (1,5 г, 8,8 ммоль) и карбонат калия (4,3 г, 31 ммоль) в ДМФ (12 мл), выдерживали в течение 1 дня при 50° С в атмосфере азота. Смесь концентрировали в вакууме, растворяли в воде и экстрагировали этилацетатом. Органические слои промывали водой и соляным раствором, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали экспресс-хроматографией (смесь гексан/этилацетат, 50:50), получая 3-пиколиламин в виде масла янтарного цвета (1,6 г, 60%). МС МН+: рассчитано для С27Н27N2О6SF3: 565, обнаружено: 565. Элементный анализ: рассчитано для C27H27N2O6SF3: С 57,44; Н 4,82; N 4,96; S 5,68; обнаружено: С 57,49; Н 5,10; N 4,69; S 5,67.
Стадия Б: Смесь, содержащую полученный на стадии А пиколиламин (1,5 г, 2,6 ммоль) и 50%-ный водный раствор NaOH (2,1 г, 26 ммоль) в ТГФ (22 мл), EtOH (22 мл) и Н2О (11 мл), выдерживали в течение 24 ч при 65° С в атмосфере азота. Продукт концентрировали в вакууме и растирали с диэтиловым эфиром, получая твердое вещество. Твердое вещество рыжевато-коричневого цвета растворяли в воде растворяли в воде и значение рН доводили до 1 с помощью концентрированной соляной кислоты. Смесь концентрировали в вакууме и сушили в вакуумной печи при 45° С, получая неочищенную кислоту в виде твердого вещества белого цвета (2,5 г). МС МН: рассчитано для С25Н23N2О6SF3: 537, обнаружено: 537.
Стадия В: Смесь, содержащую полученную на стадии Б неочищенную кислоту белого цвета (2,5 г), N-гидроксибензотриазол (1,0 г, 7,7 ммоль), 4-метилморфолин (0,64 мл, 7,7 ммоль), O-тетрагидро-2Н-пиран-2-илгидроксиламин (0,60 г, 5,1 ммоль) и гидрохлорид 1-[3-(диметиламино) пропил]-3-этилкарбодиимида (1,5 г, 7,7 ммоль) в ДМФ (40 мл), перемешивали в течение 5 дней при температуре окружающей среды в атмосфере азота. Смесь концентрировали в вакууме, разбавляли этилацетатом и промывали водой и соляным раствором. Органический слой сушили над сульфатом магния, концентрировали в вакууме и очищали экспресс-хроматографией (смесь метанол/хлороформ, 5:95), получая защищенный гидроксамат в виде пены белого цвета (1,1 г, 66%). МС МН+: рассчитано для C30H32N3O7SF3: 636, обнаружено: 636.
Стадия Г: Раствор, содержащий полученный на стадии В защищенный гидроксамат (1,0 г, 1,6 ммоль) и ацетилхлорид (0,34 мл, 4,7 ммоль) в метаноле (11 мл), находящийся при температуре окружающей среды, перемешивали в течение 2,5 ч в атмосфере азота и затем сливали на диэтиловый эфир. Твердое вещество выделяли фильтрацией и сушили в вакуумной печи при 46° С, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,85 г, 87%). Элементный анализ: рассчитано для С25Н24N3О6SF3·2,2HCl: С 47,53; Н 4,18; N 6,65; S 5,08; обнаружено: С 47,27; Н 4,34; N 6,60; S 5,29. МС МН+: рассчитано для С25Н24N3О6SF3: 552, обнаружено: 552.
Пример 400: Получение дигидрохлорида N-гидрокси-4-[4-(4-метоксифенокси)фенил]сульфонил]-1-(2-пиридинилметил)-4-пиперидинкарбоксамида

Стадия А: Гидрохлорид этил-4-[(4-фторфенилсульфонил)]-4-пиперидинкарбоксилата (2,02 г, 5,76 ммоль) объединяли с порошкообразным карбонатом калия (2,48 г, 18 ммоль) и N,N-диметилформамидом (12 мл).
Добавляли гидрохлорид 2-пиколила (1,0 г, 6,1 ммоль) и смесь перемешивали в течение 24 ч при 40° С. Реакционную смесь разбавляли водой (80 мл) и экстрагировали этилацетатом (3× 50 мл). Объединенные органические слои сушили над сульфатом магния, концентрировали и подвергали хроматографии (этилацетат), получая требуемый сложный эфир пиридина в виде масла (2,30 г, количественный выход).
Стадия Б: Полученный на стадии А сложный этиловый эфир пиридина (2,30 г, 5,76 ммоль) объединяли с порошкообразным карбонатом калия (1,29 г, 9 ммоль), 4-метоксифенолом (1,12 г, 9,0 ммоль) и N,N-диметилформамидом (3 мл), и смесь выдерживали в течение 24 ч при 75-80° С. Добавляли дополнительную порцию 4-метоксифенола (300 мг) и карбоната калия (350 мг) и смесь перемешивали еще в течение 3 ч при 90° С. Смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (3× 50 мл). Объединенные органические слои сушили над сульфатом магния, концентрировали и хроматографировали, получая требуемый сложный эфир в виде масла (2,85 г, количественный выход).
Стадия В: Полученный на стадии Б сложный эфир (2,85 г) объединяли с этанолом (18 мл), водой (6 мл) и гидроксидом калия (2,24 г, 40 ммоль). Смесь нагревали до температуры дефлегмации и выдерживали в течение 4,5 ч. Смесь охлаждали до 0° С и подкисляли с помощью концентрированного водного раствора соляной кислоты. Растворитель удаляли и образовавшиеся твердые частицы сушили путем азеотропии с ацетонитрилом. Продукт выдерживали в вакууме до достижения постоянной массы.
Неочищенный хлорангидрид перемешивали с N-метилморфолином (1 мл), 1-гидроксибензотриазолом (0,945 г, 7 ммоль), O-тетрагидропиранилгидроксиламином (0,82 г, 7 ммоль) и N,N-диметилформамидом (21 мл). Через 10 мин добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (1,34 г, 7 ммоль) и смесь перемешивали в течение ночи. Затем реакционную смесь разбавляли полунасыщенным водным раствором бикарбоната натрия 100 мл) и экстрагировали этилацетатом (200 мл, а затем 50 мл). Объединенные органические слои сушили над сульфатом магния, концентрировали и хроматографировали (этилацетат/гексан, 9:1), получая требуемый O-тетрагидропиранил-защищенный гидроксамат в виде масла желтого цвета (2,82 г, 88%).
Стадия Г: Полученный на стадии Б О-тетрагидропиранил-защищенный гидроксамат (2,82 г, 5 ммоль) разбавляли метанолом (20 мл). В течение 2 мин добавляли ацетилхлорид (2,1 мл, 30 ммоль). Реакционную смесь перемешивали в течение 4 ч при температуре окружающей среды, после чего концентрировали, получая 2,59 г неочищенного дигидрохлорида, который перекристаллизовывали из смеси этанол/вода, получая в первом выходе 525 мг (18%) указанного в заголовке гидроксамата. МС (ES) МН+: рассчитано для C25H27N3O6S: 498, обнаружено: 498.
Пример 401: Получение гидрохлорида N-гидрокси-4-[4-(4-циклогексилтио)фенил]сульфонил]-1-(2-метоксиэтил)-4-пиперидинкарбоксамида

Стадия А: Этил-4-[(4-фторфенилсульфонил)]-1-(2-метоксиэтил)-4-пиперидинкарбоксилат (5,5 г, 14 ммоль) объединяли с порошкообразным карбонатом калия (2,76 г, 20 ммоль), N,N-диметилформамидом (7 мл) и циклогексилмеркаптаном (2,4 мл, 20 ммоль) и перемешивали в течение 2 дней при температуре окружающей среды. Температуру повышали до 45-50° С и перемешивание продолжали еще в течение 24 ч. Добавляли дополнительные порции карбоната калия (1,0 г) и циклогексилмеркаптана (1,0 мл) и реакционную смесь нагревали еще в течение 16 ч. Смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (100 мл, затем 25 мл). Объединенные органические слои сушили, концентрировали и хроматографировали (этилацетат), получая требуемый сульфид в виде масла желтого цвета (3,59 г, 53%).
Стадия Б: Полученный на стадии А сульфид (3,59 г, 7,4 ммоль) превращали в тетрагидропиранил-защищенный гидроксамат путем омыления и последующего сочетания с гидрохлоридом 1-[3-(диметиламино)пропил]-3-этилкарбодиимида согласно способу, описанному на стадии В примера 401, получая 2,16 г (54%) требуемого тетрагидропиранил-защищенного гидроксамата в виде масла.
Стадия В: Полученный на стадии Б тетрагидропиранил-защищенный гидроксамат (2,16 г, 4 ммоль) разбавляли метанолом (16 мл). В течение 1 мин добавляли ацетилхлорид (1,1 мл, 16 ммоль). Реакционную смесь перемешивали в течение 4 ч, затем концентрировали и подвергали азеотропии с ацетонитрилом, получая 1,11 г неочищенного продукта, который перекристаллизовывали из абсолютного этанола, получая в первом выходе 804 мг указанного в заголовке соединения (41%). МС (ES) MH+: рассчитано для С21H32N2О5S2: 457, обнаружено: 457.
Пример 402: Получение N-гидроксил-1-(2-метоксиэтил)-4-[[(фенилметокси)фенил]сульфонил]-4-пиперидинкарбоксамида
Стадия А: Этил-4-[(4-фторфенилсульфонил)]-1-(2-метоксиэтил)-4-пиперидинкарбоксилат (1,58 г, 4,5 ммоль) объединяли с порошкообразным карбонатом калия (2,42 г, 18 ммоль), N,N-диметилацетамидом (5 мл) и бензиловым спиртом (1,94 мл, 18 ммоль) и перемешивали в течение 16 ч при 140° С. Смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (125 мл, затем 25 мл). Объединенные органические слои сушили, концентрировали и хроматографировали (этилацетат), получая требуемый сложный этиловый эфир в виде масла (1,16 г, 56%).
Стадия Б: Полученный на стадии А сложный этиловый эфир (1,16 г, 2,5 ммоль) превращали в тетрагидропиранил-защищенный гидроксамат путем омыления и последующего сочетания с гидрохлоридом 1-[3-(диметиламино) пропил] -3-этилкарбодиимида согласно способу, описанному на стадии В примера 401, получая 880 мг (80%) требуемого тетрагидропиранил-защищенного гидроксамата в виде масла.
Стадия В: Полученный на стадии Б тетрагидропиранил-защищенный гидроксамат (880 мг, 2,0 ммоль) разбавляли метанолом (8 мл). В течение 1 мин добавляли ацетилхлорид (0,68 мл, 10 ммоль). Реакционную смесь перемешивали в течение 3 ч, затем концентрировали и подвергали азеотропии с ацетонитрилом, получая неочищенный продукт, который превращали в свободное основание путем добавления насыщенного водного раствора бикарбоната натрия (25 мл), достаточного для нейтрализации соляной кислоты, после чего экстрагировали этилацетатом (100 мл, затем 50 мл). Органическую фазу сушили над сульфатом магния, концентрировали и хроматографировали (дихлорметан/метанол, 9:1, 1%-ный гидроксид аммония), получая указанный в заголовке гидроксамат в виде стекла (327 мг, 36%). МС (ES) МН+: рассчитано для C22H28N2O6S: 447, обнаружено: 447.
Пример 403: Получение N-гидроксил-1-(1-метилэтил)-4-[[4-(2-фенилэтокси)фенил]сульфонил]-4-пиперидинкарбоксамида

Стадия А: Этил-4-[(4-фторфенилсульфонил)]-1-(1-метилэтил)-4-пиперидинкарбоксилат (2,75 г, 7,7 ммоль) объединяли с порошкообразным карбонатом калия (2,62 г, 19 ммоль), N,N-диметилформамидом (10 мл) и 2-фенилэтанолом (2 мл, 19 ммоль) и перемешивали в течение 24 ч при 85° С. Добавляли дополнительную порцию карбоната калия (1,3 г) и 2-фенилэтанола и температуру повышали в течение 48 ч до 110° С, а затем в течение 4 ч до 135° С. Смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (200 мл, затем 25 мл). Объединенные органические слои сушили, концентрировали и хроматографировали (этилацетат), получая требуемый сложный этиловый эфир в виде масла (3,19 г, 90%).
Стадия Б: Полученный на стадии А сложный этиловый эфир (3,19 г, 6,9 ммоль) превращали в тетрагидропиранил-защищенный гидроксамат путем омыления и последующего сочетания с гидрохлоридом 1-[3-(диметиламино)пропил]-3-этилкарбодиимида согласно способу, описанному на стадии В примера 401, получая 2,27 г (64%) указанного в заголовке соединения в виде масла.
Стадия В: Полученный на стадии Б тетрагидропиранил-защищенный гидроксамат (2,27 г, 4,4 ммоль) разбавляли метанолом (16 мл). В течение 1 мин добавляли ацетилхлорид (0,68 мл, 10 ммоль). Реакционную смесь перемешивали в течение 3 ч, затем концентрировали и подвергали азеотропии с ацетонитрилом, получая неочищенный продукт, который превращали в свободное основание путем добавления насыщенного водного раствора бикарбоната натрия (25 мл), достаточного для нейтрализации соляной кислоты, после чего экстрагировали этилацетатом (100 мл, затем 50 мл). Органическую фазу сушили над сульфатом магния, концентрировали и хроматографировали (дихлорметан/метанол, 9:1, 1%-ный гидроксид аммония), получая требуемый гидроксамат в виде стекла (812 мг, 42%). МС (ES) MH+: рассчитано для C23H30N2O5S: 449, обнаружено: 449.
Пример 404: Получение фосфата N-гидрокси-4-[(4-фенилтиофенил)сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида
Стадия А: N-Гидрокси-4-[(4-фенилтиофенил)сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамид (430 мг, 1,0 ммоль) растворяли в метаноле (15 мл). Добавляли концентрированную фосфорную кислоту (67 мкл) и затем раствор концентрировали в вакууме. Остаток перекристаллизовывали из метанола, выделяли фильтрацией и затем вторично перекристаллизовывали из смеси метанол/метил-трет-бутиловый эфир, получая указанный в заголовке фосфат в виде твердого вещества (215 мг, 41%). Элементный анализ: рассчитано для C21H22N2O4·3H3PO4: С 47,72; Н 4,77; N 5,30; обнаружено: С 47,63; Н 5,04; N 4,82.
Пример 405: Получение пара-толуолсульфоната N-гидрокси-4-[(4-фенилтиофенил)сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида
Стадия А: N-Гидрокси-4-[(4-фенилтиофенил)сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамид (516 мг, 1,0 ммоль) объединяли с моногидратом пара-толуолсульфоновой кислоты (200 мг, 1,05 ммоль) и смесь растворяли в метаноле (3 мл). Через 4 ч образовавшийся осадок белого цвета собирали фильтрацией, получая 488 мг (81%) указанного в заголовке тозилата, характеристики которого определяли спектроскопическим путем.
Пример 406: Получение моногидрохлорида 4-[[4-[(2,3-дигидро-1Н-инден-2-ил)амино]Фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
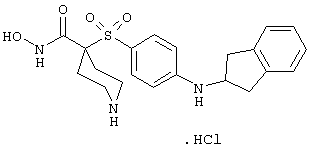
Стадия А: Раствор, содержащий полученный на стадии Г примера 9 продукт (0,979 г, 2,36 ммоль), гидрохлорид 2-аминоиндана (1,00 г, 5,89 ммоль) и карбонат цезия (1,92 г, 5,89 ммоль) в N,N-диметилформамиде (8 мл), нагревали в течение 22 ч до 95° С. Затем реакционную смесь охлаждали, разбавляли этилацетатом (50 мл) и промывали трижды водой и однократно соляным раствором, после чего сушили над сульфатом натрия. После концентрирования получали остаток, который хроматографировали на силикагеле. После элюирования смесью этилацетат/гексан (30/70) получали требуемое 4-аминосульфоновое производное (450 мг, 36%). МС (ES) МН+:рассчитано для С28Н36N2О6S: 529, обнаружено: 529. МСВР М+: рассчитано для C28H36N2O6S: 528,2294, обнаружено: 528,2306.
Стадия Б: К раствору полученного на стадии А сложного этилового эфира (450 мг, 0,85 ммоль) в этаноле (3 мл), воде (2 мл) и тетрагидрофуране (3 мл) добавляли гидроксид натрия (340 мг, 8,5 ммоль) и раствор нагревали в течение 26 ч до 60° С. Раствор охлаждали, после чего разбавляли водой (10 мл) и затем 10%-ным водным раствором соляной кислоты (3 мл), доводя значение рН до 2. Образовавшийся раствор экстрагировали этилацетатом. Органические экстракты объединяли и промывали водой и соляным раствором и сушили над сульфатом натрия, получая требуемую карбоновую кислоту в виде пены светло-коричневого цвета (376 мг, 88%). Элементный анализ: рассчитано для С26Н32N2О6S: С 62,38; Н 6,44; N 5,60; S 6,40; обнаружено: С 62,48; Н 6,69; N 5,42; S 6,27.
Стадия В: К суспензии полученной на стадии Б карбоновой кислоты (305 мг, 0,609 ммоль) в N,N-диметилформамиде (2 мл) добавляли 4-метилморфолин (247 мг, 2,44 ммоль), N-гидроксибензотриазол (99 мг, 0,73 моль), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (152 г, 0,79 ммоль), а затем O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (97 мг, 0,82 ммоль). После перемешивания в течение 2 дней при температуре окружающей среды раствор концентрировали, получая масло. Добавляли воду и смесь экстрагировали этилацетатом. Органические экстракты промывали водой и соляным раствором и сушили над сульфатом натрия. После концентрирования получали пену коричневого цвета, которую хроматографировали на силикагеле. После элюирования смесью этилацетат/гексан (40/60) получали защищенный гидроксамат в виде бесцветного стекла (0,38 г, 100%). МС (ES) MH+:рассчитано для С31Н41Н3O7S: 600, обнаружено: 600.
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (350 мг, 0,584 ммоль) в метаноле (3 мл) и 1,4-диоксане (1,5 мл) добавляли смесь 4 н. раствор HCl/1,4-диоксан (1,5 мл, 6 ммоль) и раствор перемешивали в течение 3 ч при температуре окружающей среды. После концентрирования получали остаток, который растирали с диэтиловым эфиром, получая указанное в заголовке соединение в виде твердого вещества, которое фильтровали и сушили в течение 40 ч при 51° С (249 мг, 94%). МСВР (ESI) МН+: рассчитано для C21H25N3O4S: 416,1644, обнаружено: 416,1647.
Пример 407: Получение моногидрохлорида 4-[[4-(диметиламино)фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
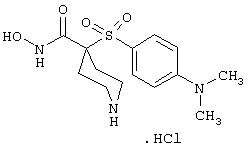
Стадия А: Раствор, содержащий полученный на стадии Г примера 9 продукт (0,979 г, 2,36 ммоль), гидрохлорид 2-аминоиндана (1,00 г, 5,89 ммоль) и карбонат цезия (1,92 г, 5,89 ммоль) в N,N-диметилформамиде (8 мл), нагревали в течение 22 ч до 95° С. Затем реакционную смесь охлаждали, разбавляли этилацетатом (50 мл) и промывали трижды водой и однократно соляным раствором, после чего сушили над сульфатом натрия. После концентрирования получали остаток, который хроматографировали на силикагеле. После элюирования смесью этилацетат/гексан (30/70) получали требуемый 4-N,N-диметиламиносульфон (590 мг, 57%) наряду с продуктом из примера 406. МС (ES) MH+: рассчитано для C21H32N2O6S: 441, обнаружено: 441. МСВР: рассчитано для С21Н32N2О6S: 440,1981, обнаружено: 440,1978.
Стадия Б: К раствору полученного на стадии А сложного этилового эфира (580 мг, 1,3 ммоль) в этаноле (4 мл), воде (3 мл) и тетрагидрофуране (4 мл) добавляли гидроксид натрия (520 мг, 13 ммоль) и раствор нагревали в течение 5 ч до 62° С. Раствор охлаждали, после чего разбавляли водой (5 мл) и затем 10%-ным водным раствором соляной кислоты (5 мл), подкисляя до значения рН 2. Образовавшийся раствор экстрагировали этилацетатом. Органические экстракты объединяли и промывали водой и соляным раствором и сушили над сульфатом натрия, получая требуемую карбоновую кислоту в виде пены светло-коричневого цвета (520 мг, 97%). МС МН+: рассчитано для C19H28N2O6S: 413, обнаружено:413.
Стадия В: К раствору полученной на стадии Б карбоновой кислоты (500 мг, 1,21 ммоль) в N,N-диметилформамиде (4 мл) добавляли 4-метилморфолин (490 мг, 4,8 ммоль), N-гидроксибензотриазол (197 мг, 1,45 моль) и гидрохлорид 1-[3-(диметиламино) пропил]-3-этилкарбодиимида (302 г, 1,57 ммоль), а затем O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (192 мг, 1,63 ммоль). После перемешивания в течение 2 дней при температуре окружающей среды раствор концентрировали, получая масло. Добавляли воду (25 мл) и смесь экстрагировали этилацетатом. Органические экстракты промывали водой и соляным раствором и сушили над сульфатом натрия. После концентрирования получали масло коричневого цвета, которое кристаллизовали из смеси этилацетата, гексана и метиленхлорида (1:1:2), получая защищенный гидроксамат в виде бесцветного твердого вещества (506 мг, 82%). МС МН+: рассчитано для C24H37N3O7S: 512, обнаружено: 512.
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (477 мг, 0,932 ммоль) в метаноле (3 мл) и 1,4-диоксане (3 мл) добавляли смесь 4 н. раствор HCl/1,4-диоксан (2,3 мл, 9,3 ммоль) и раствор перемешивали в течение 3 ч при температуре окружающей среды. После концентрирования получали остаток, который растирали с диэтиловым эфиром, получая указанное в заголовке соединение в виде твердого вещества, которое фильтровали и сушили в течение 40 ч при 51° С (372 мг, 100%). МСВР (ESI) МН+: рассчитано для C14H21N3O4S: 328,1331, обнаружено: 328,1343.
Пример 408: Получение моногидрохлорида 1-циклопропил-4-[[4-[(2,3-дигидро-1,4-бензодиоксин-6-ил)окси]фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
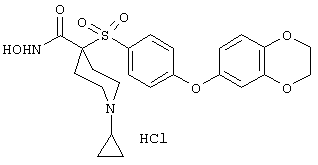
Стадия А: К раствору полученного на стадии А примера 398 продукта (1,36 г, 3,47 моль) в N,N-диметилформамиде (8 мл) добавляли 6-гидроксибензо-1,4-диоксан (792 мг, 5,21 ммоль), а затем карбонат цезия (2,83 г, 8,69 ммоль) и раствор выдерживали в течение 20 ч при 100° С. Раствор распределяли между этилацетатом и Н2О. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали Н2О и насыщенным раствором NaCl и сушили над Na2SO4. После фильтрации через подушку из силикагеля (этилацетат/гексан) получали феноксифенил, в виде масла оранжевого цвета (1,81 г, количественный выход). МС (CI) MH+: рассчитано для C25H29NO7S: 488, обнаружено: 488.
Стадия Б: К раствору полученного на стадии А феноксифенола (1,81 г, <3,47 ммоль), в тетрагидрофуране (10 мл) и этаноле (10 мл) добавляли гидроксид натрия (1,39 г, 34,7 ммоль) в Н2О (5 мл). Раствор нагревали в течение 20 ч до 60° С. Раствор концентрировали в вакууме и водный остаток подкисляли до рН 2 с помощью 10%-ной HCl. Образовавшийся твердый продукт собирали вакуумной фильтрацией, получая кислоту в виде твердого вещества желтого цвета (1,23 г, 72%). МС (CI) MH+: рассчитано для С23Н25NO7S: 460, обнаружено: 460. МСВР: рассчитано для C23H25NO7S: 460,1430, обнаружено: 460,1445.
Стадия В: К суспензии полученной на стадии Б кислоты (1,21 г, 2,46 ммоль) в N,N-диметилформамиде (20 мл) добавляли N-гидроксибензотриазол (399 мг, 2,95 моль), 4-метилморфолин (0,81 мл, 7,38 ммоль) и O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (432 мг, 3,69 ммоль). После перемешивания в течение 1 ч добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (660 мг, 3,44 ммоль) и раствор перемешивали в течение 20 ч при температуре окружающей среды. Раствор распределяли между этилацетатом и Н2О и водный слой экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде масла желтого цвета (940 мг, 70%). МС (CI) MH+:рассчитано для C28H34N2O2S: 559, обнаружено: 559.
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (920 мг, 1,68 ммоль) в 1,4-диоксане (15 мл) добавляли 4 н. раствор HCl в 1,4-диоксане (10 мл). После перемешивания в течение 2 ч при температуре окружающей среды образовавшийся осадок собирали вакуумной фильтрацией и промывали диэтиловым эфиром, получая указанное в заголовке соединение в виде твердого вещества белого цвета (510 мг, 60%). МС (CI) MH+: рассчитано для C23H26N2O7S: 475, обнаружено: 475. МСВР: рассчитано для C23H26NO7S: 475,1539, обнаружено: 475,1553. Элементный анализ: рассчитано для С23Н26N2O7S· 1,15HCl· 0,5Н2О: С 52,57; Н 5,40; N 5,33; Cl 7,76; обнаружено: С 52,62; Н 5,42; N 5,79; Cl 7,71.
Пример 409: Получение моногидрохлорида N-гидрокси-4-[[4-[4-(трифторметил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
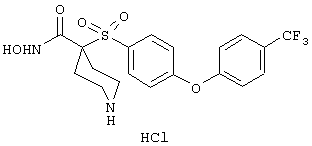
Стадия А: К раствору полученного на стадии Г примера 9 продукта (1,5 г, 3,61 ммоль) в N,N-диметилформамиде (10 мл) добавляли карбонат цезия (2,94 г, 9,03 ммоль) и α ,α ,α -трифтор-пара-крезол (877 мг, 5,41 ммоль). Раствор нагревали в течение 20 ч до 90° С. Раствор распределяли между этилацетатом и Н2О и органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После фильтрации через подушку из силикагеля (этилацетат) получали простой диариловый эфир в виде масла желтого цвета (2,30 г, количественный выход). МС (CI) MH+: рассчитано для С26Н30NO7SF3: 558, обнаружено: 558.
Стадия Б: К раствору полученного на стадии А простого диарилового эфира (2,30 г, <3,61 ммоль) в тетрагидрофуране (10 мл) и этаноле (10 мл) добавляли гидроксид натрия (1,44 г, 36,1 ммоль) в Н2О (5 мл) и раствор нагревали в течение 18 ч до 60° С. Раствор концентрировали и водный остаток подкисляли до рН 2 с помощью 10%-ной HCl и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали кислоту в виде твердого вещества (2,11 г, количественный выход). МС (CI) MH+: рассчитано для C24H25NO7SF3: 530, обнаружено: 530.
Стадия В: К раствору полученной на стадии Б кислоты (2,11 г, <3,61 ммоль) в N,N-диметилформамиде (10 мл) добавляли N-гидроксибензотриазол (586 мг, 4,33 моль), 4-метилморфолин (1,19 мл, 10,83 ммоль) и O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (634 мг, 5,41 ммоль). После перемешивания в течение 1 ч добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (969 мг, 5,05 ммоль) и раствор перемешивали в течение 18 ч. Раствор распределяли между этилацетатом и Н2О. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали Н2О, насыщенным раствором NaCl и сушили над MgSO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде прозрачного бесцветного масла (1,40 г, 62%). МС (CI) MH+: рассчитано для C29H35N2O8SF3: 629, обнаружено: 629.
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (1,40 г, 2,23 ммоль) в 1,4-диоксане (10 мл) добавляли 4 н. раствор HCl в 1,4-диоксане (15 мл) и раствор перемешивали в течение 2 ч. Раствор разбавляли диэтиловым эфиром и образовавшийся осадок собирали вакуумной фильтрацией, получая указанное в заголовке соединение в виде твердого вещества белого цвета (747 мг, 70%). Чистота по данным ЖХВР: 97,5%. МС (CI) MH+: рассчитано для C19H19N2O5SF3: 445, обнаружено: 445. МСВР: рассчитано для C19H19N2O5SF3: 445,1045, обнаружено: 445,1052. Элементный анализ: рассчитано для С19Н19N2О5SF3·0,5Н2О· 0,1НСl: С 46,58; Н 4,32; N 5,72; S 6,55; Cl 7,24; обнаружено: С 46,58; Н 3,82; N 5,61; S 6,96; С1 7,37.
Пример 410: Получение моногидрохлорида N-гидрокси-4-[[4-[(трифторметокси)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
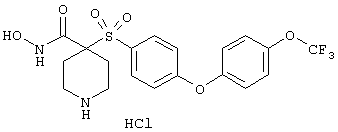
Стадия А: К раствору полученного на стадии Г примера 9 продукта (1,5 г, 3,61 ммоль) в N,N-диметилформамиде (10 мл) добавляли карбонат цезия (2,94 г, 9,03 ммоль) и 4-(трифторметокси)фенол (0,70 г, 5,41 ммоль). Раствор нагревали в течение 20 ч до 90° С. Раствор распределяли между этилацетатом и Н2О и органический слой промывали насыщенным раствором NaCl и сушили над Na2SO4. После фильтрации через подушку из силикагеля (этилацетат) получали феноксифенол в виде масла желтого цвета (2,11 г, количественный выход). МС (CI) MNa+: рассчитано для C26H30NO8SF3: 596, обнаружено: 596.
Стадия Б: К раствору полученного на стадии А феноксифенола (2,11 г, <3,61 ммоль) в тетрагидрофуране (10 мл) и этаноле (10 мл) добавляли гидроксид натрия (1,44 г, 36,1 ммоль) в Н2О (5 мл) и раствор нагревали в течение 18 ч до 60° С. Раствор концентрировали и водный остаток подкисляли до рН 2 с помощью 10%-ной HCl и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над Nа2SO4. После концентрирования в вакууме получали кислоту в виде твердого вещества (2,2 г, количественный выход). МС (CI) MH+: рассчитано для C24H26NO8SF3: 546, обнаружено: 546.
Стадия В: К раствору полученной на стадии Б кислоты (2,2 г) в N,N-диметилформамиде (10 мл) добавляли N-гидроксибензотриазол (586 мг, 4,33 моль), 4-метилморфолин (1,19 мл, 10,83 ммоль) и O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (634 мг, 5,41 ммоль). После перемешивания в течение 30 мин добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (969 мг, 5,05 ммоль) и раствор перемешивали в течение 96 ч. Раствор распределяли между этилацетатом и Н2О. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали Н2О, насыщенным раствором NaCl и сушили над MgSO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде прозрачного бесцветного масла (1,26 г, 53%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (1,26 г, 1,96 ммоль) в 1,4-диоксане (10 мл) добавляли 4н. раствор HCl в 1,4-диоксане (10 мл) и раствор перемешивали в течение 2 ч. Раствор разбавляли диэтиловым эфиром и образовавшийся осадок собирали вакуумной фильтрацией, получая указанное в заголовке соединение в виде твердого вещества белого цвета (455 мг, 47%). Чистота по данным ЖХВР: 98%. МС (CI) МН+:рассчитано для C19H19N2O6SF3: 461, обнаружено: 461. МСВР: рассчитано для C19H19N2O6SF3: 461,0994, обнаружено: 461,0997. Элементный анализ:рассчитано для C19H19N2O6SF3·1,0НСl: С 45,93; Н 4,06; N 5,64; S 6,45; Cl 6,45; обнаружено: С 46,23; Н 4,07; N 5,66; S 6,59; Cl 7,03.
Пример 411: Получение моногидрохлорида 1-циклопропил-4-[[4-[(2,3-дигидро-1,4-бензодиоксин-6-ил)амино]фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
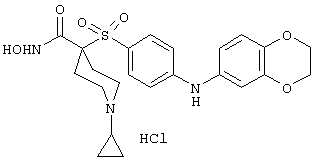
Стадия А: К раствору полученного на стадии В примера 91 сложного эфира (1,57 г, 3,40 ммоль) в 1,4-диоксане (5 мл) добавляли 4М раствор HCl в диоксане (10 мл). После перемешивания в течение 1 ч образовавшийся осадок собирали вакуумной фильтрацией, получая гидрохлорид амина в виде твердого вещества белого цвета (1,16 г, 86%).
Стадия Б: К суспензии полученного на стадии А гидрохлорида амина (1,16 г, 2,91 ммоль) в метаноле (10 мл) добавляли уксусную кислоту (1,68 мл, 29,1 ммоль), а затем (1-этилоксициклопропил)окситриметилсилан (3,51 мл, 17,5 ммоль) и цианборогидрид натрия (823 мг, 13,1 ммоль). Раствор нагревали до температуры дефлегмации в течение 6 ч. Раствор фильтровали и фильтрат концентрировали в вакууме. Остаток растворяли в этилацетате и промывали Н2О, водным раствором гидроксида натрия и насыщенным раствором NaCl и сушили над MgSO4. После концентрирования в вакууме получали N-циклопропильное производное в виде твердого вещества белого цвета (1,03 г, 88%).
Стадия В: К раствору полученного на стадии Б N-циклопропильного производного (1,0 г, 2,49 ммоль) в толуоле (6 мл) добавляли карбонат цезия (1,14 г, 3,49 ммоль), трис(дибензилиденацетон)дипалладий(0) (69 мг, 0,075 ммоль), К-(+)-2,2’-бис(дифенилфосфино)-1,1’-бинафтил (69 мг, 0,112 ммоль) и 1,4-бензодиоксан-6-амин (451 мг, 2,99 ммоль) и раствор нагревали в течение 19 ч до 100° С. Раствор разбавляли диэтиловым эфиром и фильтровали через Super Cel®. Фильтрат концентрировали и после хроматографии (на силикагеле, этилацетат/гексан) получали анилин в виде масла оранжевого цвета (561 мг, 48%). МС (CI) MH+: рассчитано для C24H28N2O6S: 473, обнаружено: 473.
Стадия Г: К раствору полученного на стадии В анилина (550 мг, 1,16 ммоль) в тетрагидрофуране (10 мл) добавляли триметилсиланолат калия (297 мг, 3,48 ммоль) и раствор перемешивали течение 18 ч при температуре окружающей среды. Раствор концентрировали и образовавшийся остаток суспендировали в Н2О. Твердое вещество собирали вакуумной фильтрацией, получая неочищенную кислоту (282 мг).
Стадия Д: К раствору полученной на стадии Г неочищенной кислоты (282 мг, 0,62 ммоль) в N,N-диметилформамиде (10 мл) добавляли N-гидроксибензотриазол (100 мг, 0,74 моль), 4-метилморфолин (0,20 мл, 1,86 ммоль) и O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (108 мг, 0,93 ммоль). После перемешивания в течение 30 мин добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (166 г, 0,87 ммоль) и раствор перемешивали в течение 72 ч. Раствор распределяли между этилацетатом и Н2О и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали Н2О и насыщенным раствором NaCl и сушили над Na2SO4. После хроматографии (на силикагеле, этилацетат/гексан) получали защищенный гидроксамат в виде твердого вещества белого цвета (150 мг, 43%). МС (CI) MH+: рассчитано для С28H35N3O7S: 558, обнаружено: 558.
Стадия Е: К раствору полученного на стадии Д защищенного гидроксамата (133 мг, 0,24 ммоль) в 1,4-диоксане (5 мл) добавляли 4н. раствор HCl в 1,4-диоксане (10 мл) и раствор перемешивали в течение 1,5 ч. Раствор разбавляли диэтиловым эфиром и образовавшийся осадок собирали вакуумной фильтрацией, получая указанный в заголовке гидроксамат в виде твердого вещества белого цвета (80 мг, 66%). МС (CI) MH+: рассчитано для С23Н27N3О6S: 474, обнаружено: 474. МСВР: рассчитано для C23H27N3O6S: 474,1699, обнаружено: 474,1715. Элементный анализ: рассчитано для C23H27N3O6S· 1,5HCl· 1,5H2O: С 49,75; Н 5,72; N 7,57; S 5,77; Cl 9,58; обнаружено: С 49,78; Н 5,52; N 8,05; S 9,16; Cl 5,76.
Пример 412: Получение тригидрохлорида 1-циклопропил-4-[[4-[4-[[4-(2,3-диметилфенил)-1-пиперазинил]карбонил]-1-пиперидинил]фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
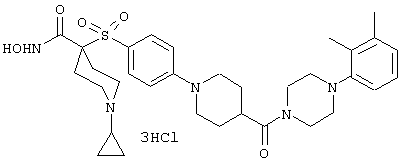
Стадия А: К раствору изонипекотиновой кислоты (10,5 г, 81,3 ммоль) в Н2О (325 мл) добавляли карбонат натрия (8,37 г, 81,3 ммоль) и раствор перемешивали до тех пор, пока он не становился гомогенным. К этому раствору по каплям добавляли ди-трет-бутилдикарбонат (18,22 г, 83,5 ммоль) в 1,4-диоксане (77 мл) и образовавшийся раствор перемешивали в течение 72 ч при температуре окружающей среды. Раствор концентрировали в вакууме и образовавшийся водный раствор промывали диэтиловым эфиром. Водный раствор подкисляли до рН 2 с помощью концентрированной HCl. Раствор экстрагировали диэтиловым эфиром и концентрировали в вакууме, получая твердое вещество белого цвета. После перекристаллизации (этилацетат) получали N-Boc-изонипекотиновую кислоту в виде твердого вещества белого цвета (10 г, 54%).
Стадия Б: К раствору полученной на стадии А N-Boc-изонипекотиновой кислоты (2,14 г, 9,33 ммоль) в дихлорметане (19 мл) добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (1,82 г, 9,49 ммоль), N-гидроксибензотриазол (1,32 г, 9,77 ммоль) и моногидрохлорид 1-(2,3-ксилил)пиперазина (2,47 г, 10,89 ммоль). Через 30 мин добавляли диизопропилэтиламин (0,74 мл, 20,7 ммоль) и раствор перемешивали в течение 18 ч. Раствор концентрировали в вакууме и остаток растворяли в этилацетате и промывали 1 М HCl, насыщенным раствором NаНСО3 и насыщенным раствором NaCl. Раствор сушили над MgSO4. После перекристаллизации (этилацетат/гексан) получали амид в виде твердого вещества беловатого цвета (2,65 г, 71%).
Стадия В: К раствору полученного на стадии Б амида (1,0 г, 3,75 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (5 мл) и раствор перемешивали в течение 15 мин. Раствор концентрировали в вакууме и образовавшееся масло растворяли в N,N-диметилацетамиде (10 мл). К этому раствору добавляли полученный на стадии А примера 398 продукт (979 мг, 2,50 ммоль) и карбонат цезия (3,67 г, 11,25 ммоль) и раствор выдерживали в течение 17 ч до 110° С. Раствор распределяли между этилацетатом и Н2О. Органический слой промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали пиперидин в виде твердого вещества белого цвета (1,89 г, количественный выход). МС (CI) MH+:рассчитано для C35H48N4O5S: 637, обнаружено: 637.
Стадия Г: К раствору полученного на стадии В пиперидина (1,89 г), в этаноле (8 мл) и тетрагидрофуране (8 мл) добавляли гидроксид натрия (1,0 г, 25 ммоль) в Н2О (5 мл). Раствор нагревали в течение 8 ч до 50° С и выдерживали в течение 8 ч при 62° С. Раствор концентрировали в вакууме и остаток разбавляли Н2О и подкисляли до рН 3 с помощью 3 М HCl.
Образовавшийся осадок собирали вакуумной фильтрацией, получая кислоту в виде твердого вещества белого цвета (1,16 г, 65%). МС (CI) MH+: рассчитано для C33H44N4O5S: 609, обнаружено: 609.
Стадия Д: К суспензии полученной на стадии Г кислоты (1,16 г, 1,62 ммоль) в N,N-диметилформамиде (10 мл) добавляли N-гидроксибензотриазол (262 мг, 1,94 моль), 4-метилморфолин (0,90 мл, 8,2 ммоль) и O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (282 мг, 2,4 ммоль). После перемешивания в течение 45 мин добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (334 мг, 2,2 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор распределяли между этилацетатом и Н2О и органический слой промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После растирания (дихлорметан) получали защищенный гидроксамат в виде твердого вещества белого цвета (850 мг, 75%). МС (CI) MH+: рассчитано для С38Н53N5О6S: 708, обнаружено: 708. Элементный анализ: рассчитано для C38H53N5O6S· 0,5Н2О: С 63,66; Н 7,59; N 9,77; S 4,47; обнаружено: С 63,68; Н 7,54; N 9,66; S 4,67.
Стадия Е: К раствору полученного на стадии Д защищенного гидроксамата (746 мг, 1,07 ммоль) в метаноле (10 мл) добавляли 4М раствор HCl в 1,4-диоксане (10 мл) и раствор перемешивали в течение 1 ч. Образовавшееся твердое вещество собирали вакуумной фильтрацией и промывали диэтиловым эфиром, получая указанное в заголовке соединение в виде твердого вещества белого цвета (650 мг, 83%). МС (CI) MH+: рассчитано для C33H45N5O5S: 624, обнаружено: 624. МСВР: рассчитано для C33H49N5O5S: 624,3220, обнаружено: 624,3253. Элементный анализ: рассчитано для С33H45N5O5S· 3,5HCl· Н2О: С 51,82; Н 6,59; N 9,16; обнаружено: С 52,04; Н 6,30; N 8,96.
Пример 413: Получение тригидрохлорида 4-[[4-[4-[[4-(2,3-диметилфенил)-1-пиперазинил]карбонил]-1-пиперидинил]фенил]сульфонил]-N-гидрокси-1-(2-метоксиэтил)-4-пиперидинкарбоксамида
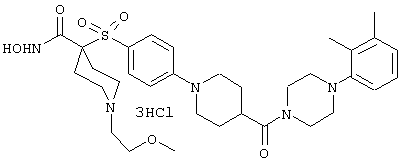
Стадия А: К раствору изонипекотиновой кислоты (10,5 г, 81,3 ммоль) в Н2О (325 мл) добавляли карбонат натрия (8,37 г, 81,3 ммоль) и раствор перемешивали до тех пор, пока он не становился гомогенным. К этому раствору по каплям добавляли ди-трет-бутилдикарбонат (18,22 г, 83,5 ммоль) в 1,4-диоксане (77 мл) и образовавшийся раствор перемешивали в течение 72 ч при температуре окружающей среды. Раствор концентрировали в вакууме и образовавшийся водный раствор промывали диэтиловым эфиром. Водный раствор подкисляли до рН 2 с помощью концентрированной HCl. Раствор экстрагировали диэтиловым эфиром и концентрировали в вакууме, получая твердое вещество белого цвета. После перекристаллизации (этилацетат) получали N-Boc-изонипекотиновую кислоту в виде твердого вещества белого цвета (10 г, 54%).
Стадия Б: К раствору полученной на стадии А N-Boc-изонипекотиновой кислоты (2,14 г, 9,33 ммоль) в дихлорметане (19 мл) добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (1,82 г, 9,49 ммоль), N-гидроксибензотриазол (1,32 г, 9,77 ммоль) и моногидрохлорид 1-(2,3-ксилил)пиперазина (2,47 г, 10,89 ммоль). Через 30 мин добавляли диизопропилэтиламин (0,74 мл, 20,7 ммоль) и раствор перемешивали в течение 18 ч. Раствор концентрировали в вакууме и остаток растворяли в этилацетате и промывали 1 М HCl, насыщенным раствором NаНСО3 и насыщенным раствором NaCl. Раствор сушили над MgSO4. После перекристаллизации (этилацетат/гексан) получали амид в виде твердого вещества беловатого цвета (2,65 г, 71%).
Стадия В: К раствору полученного на стадии Б амида (0,965 мг, 2,41 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (5 мл) и раствор перемешивали в течение 15 мин. Раствор концентрировали в вакууме и образовавшееся масло растворяли в N,N-диметилацетамиде (10 мл). К этому раствору добавляли этил-4-[(4-фторфенилсульфонил)]-1-(2-метоксиэтил)-4-пиперидинкарбоксилат (600 мг, 1,61 ммоль) и карбонат цезия (2,75 г, 8,43 ммоль) и раствор выдерживали в течение 20 ч до 110° С. Раствор распределяли между этилацетатом и Н2О. Органический слой промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали пиперидин в виде твердого вещества белого цвета (1,26 г, количественный выход). МС (CI) MH+: рассчитано для C35H50N4O6S: 655, обнаружено: 655.
Стадия Г: К раствору полученного на стадии В пиперидина (1,26 г), в этаноле (5 мл) и тетрагидрофуране (5 мл) добавляли гидроксид натрия (644 мг, 16 ммоль) в Н2О (5 мл). Раствор нагревали в течение 8 ч до 60° С и выдерживали в течение 8 ч при 62° С. Раствор концентрировали в вакууме и остаток разбавляли Н2О и подкисляли до рН 3 с помощью 3 М HCl. Образовавшийся осадок собирали вакуумной фильтрацией, получая кислоту в виде твердого вещества белого цвета (650 мг, 65%). МС (CI) MH+: рассчитано для C33H46N4O6S: 627, обнаружено: 627.
Стадия Д: К суспензии полученной на стадии Г кислоты (620 мг, 0,94 ммоль) в N,N-диметилформамиде (10 мл) добавляли N-гидроксибензотриазол (152 мг, 1,13 моль), 4-метилморфолин (0,52 мл, 4,7 ммоль) и O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (165 мг, 1,4 ммоль). После перемешивания в течение 45 мин добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (252 мг, 1,32 ммоль) и раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор распределяли между этилацетатом и Н2О и органический слой промывали Н2О, насыщенным раствором NaCl и сушили над Na2SO4. После концентрирования в вакууме получали защищенный гидроксамат в виде твердого вещества белого цвета (641 мг, 94%,). МС (CI) МН +: рассчитано для C38H55N5O7S: 726, обнаружено: 726.
Стадия Е: К раствору полученного на стадии Д защищенного гидроксамата (630 мг, 0,87 ммоль) в метаноле (8 мл) добавляли 4М раствор HCl в 1,4-диоксане (10 мл) и раствор перемешивали в течение 1 ч. Образовавшееся твердое вещество собирали вакуумной фильтрацией и промывали диэтиловым эфиром, получая указанное в заголовке соединение в виде твердого вещества белого цвета (624 мг, 83%). МС (CI) MH+: рассчитано для C33H47N5O6S: 642, обнаружено: 642.
Пример 414: Получение моногидрохлорида N-гидрокси-4-[[4-[4-(1-метилэтил)фенокси]фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида
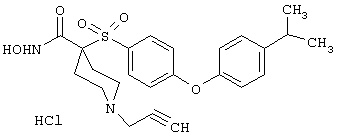
Стадия А: К раствору, содержащему полученный на стадии Г примера 9 продукт (6,0 г, 15,4 ммоль) и порошкообразный К2СО3 (8,0 г, 38,5 ммоль) в N,N-диметилформамиде (70 мл), добавляли при температуре окружающей среды 4-изопропилфенол (5,24 г, 38,5 ммоль) и раствор нагревали в течение 32 ч до 90° С.
Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали 1 н. NaOH, Н2О и сушили над MgSO4. После хроматографии на силикагеле с элюированием этилацетатом/гексаном получали простой диариловый эфир в виде геля светло-желтого цвета (6,89 г, 87%).
Стадия Б: К раствору полученного на стадии А простого диарилового эфира (6,89 г, 14,7 ммоль) в этаноле (14 мл) и тетрагидрофуране (14 мл) добавляли при температуре окружающей среды с помощью капельной воронки NaOH (5,88 г, 147 ммоль) в Н2О (28 мл). Затем раствор нагревали в течение 17 ч до 60° С и в течение 24 ч при температуре окружающей среды. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали простым эфиром и подкисляли до рН 2. После вакуумной фильтрации осадка белого цвета получали кислоту в виде твердого вещества белого цвета (6,56 г, количественный выход).
Стадия В: К раствору, содержащему полученную на стадии Б кислоту (6,56 г, 14,86 ммоль), N-метилморфолин (6,5 мл, 59,4 ммоль), 1-гидроксибензотриазол (6,0 г, 44,6 ммоль) и O-(тетрагидропиранил)гидроксиламин (3,5 г, 29,7 ммоль) в N,N-диметилформамиде (50 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (8,5 г, 44,6 ммоль) и раствор перемешивали в течение 20 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным водным раствором NаНСО3, Н2О и сушили над MgSO4. После концентрирования в вакууме и хроматографии на силикагеле с элюированием этилацетатом/гексаном получали тетрагидропиранил-защищенный гидроксамат в виде пены белого цвета (8,03 г, количественный выход).
Стадия Г: К раствору 4н. HCl в диоксане (37 мл, 149 ммоль) добавляли раствор полученного на стадии В тетрагидропиранил-защищенного гидроксамата (8,03 г, 14,9 ммоль) в метаноле (5 мл) и диоксане (15 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. После концентрирования в вакууме и растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (5,0 г, 71,1%). Элементный анализ: рассчитано для C24H28N2O5S· HCl· 0,9Н2О: С 56,61; Н 6,10; N 5,50; S 6,30; обнаружено: С 56,97;Н 6,05; N 5,41; S 5,98. MCBP МН+: рассчитано для C24H28N2O5S: 457,1797, обнаружено: 457,1816.
Пример 415: Получение моногидрохлорида 4-[[4-(1,3-бензодиоксол-5-илокси)фенил]сульфонил]-N-гидрокси-1-(2-метоксиэтил)-4-пиперидинкарбоксамида
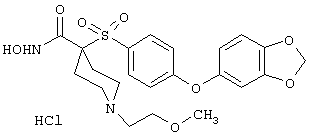
Стадия А: К раствору, содержащему полученный на стадии Г примера 9 продукт (25 г, 67,3 ммоль) и порошкообразный K2CO3 (23,3 г, 169 ммоль) в N,N-диметилформамиде (150 мл), добавляли при температуре окружающей среды сезамол (23,2 г, 168 ммоль) и раствор нагревали в течение 25 ч до 90° С.
Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали 1 н. NaOH, Н2О и сушили над MgSO4. После хроматографии на силикагеле с элюированием этилацетатом/гексаном получали требуемый простой диариловый эфир в виде геля светло-желтого цвета (33,6 г, 93,6%).
Стадия Б: К раствору полученного на стадии А простого диарилового эфира (4,0 г, 7,4 ммоль) в дихлорметане (7 мл), охлажденному до 0° С, добавляли трифторуксусную кислоту (7 мл) и раствор перемешивали при температуре окружающей среды в течение 2 ч. После концентрирования в вакууме получали трифторацетат амина в виде геля светло-желтого цвета. К раствору, содержащему трифторацетат и К2СО3 (3,6 г, 26 ммоль) в N,N-диметилформамиде (50 мл), добавляли простой метил-2-бромэтиловый эфир (1,8 мл, 18,7 ммоль) и раствор перемешивали в течение 36 ч при температуре окружающей среды. N,N-диметилформамид выпаривали в глубоком вакууме и остаток разбавляли этилацетатом. Органический слой промывали водой и сушили над MgSO4. После концентрирования в вакууме получали метоксиэтиламин в виде геля светло-желтого цвета (3,7 г, количественный выход).
Стадия В: К раствору полученного на стадии Б метоксиэтиламина (3,7 г, 7,5 ммоль) в этаноле (7 мл) и тетрагидрофуране (7 мл) добавляли через капельную воронку при температуре окружающей среды NaOH (3,0 г, 75 ммоль) в Н2О (15 мл). Затем раствор нагревали в течение 19 ч до 60° С и выдерживали в течение 12 ч при температуре окружающей среды. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали простым эфиром и подкисляли до рН 2. После вакуумной фильтрации белого осадка получали кислоту в виде твердого вещества белого цвета (4,0 г, количественный выход).
Стадия Г: К раствору, содержащему полученную на стадии В кислоту (4,0 г, 7,5 ммоль), N-метилморфолин (3,3 мл, 30 ммоль), 1-гидроксибензотриазол (3,0 г, 22,5 ммоль) и O-(тетрагидропиранил)гидроксиламин (1,8 г, 15 ммоль) в N,N-диметилформамиде (100 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (4,3 г, 22,5 ммоль) и раствор перемешивали в течение 4 дней при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным водным раствором NaHCO3, Н2О и сушили над MgSO4. После концентрирования в вакууме и хроматографии на силикагеле с элюированием этилацетатом/гексаном получали тетрагидропиранил-защищенный гидроксамат в виде пены белого цвета (2,40 г, 57,1%).
Стадия Д: К раствору 4н. HCl в диоксане (11 мл, 43 ммоль) добавляли раствор полученного на стадии Г тетрагидропиранил-защищенного гидроксамата (2,4 г, 4,3 ммоль) в метаноле (2 мл) и диоксане (6 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. После концентрирования в вакууме и растирания с простым эфиром получали гидрохлорид гидроксамата в виде твердого вещества белого цвета (1,88 г, 85,8%). Элементный анализ:рассчитано для C22H26N2O8S· HCl· Н2О: С 49,58; Н 5,48; N 5,26; S 6,02; обнаружено: С 49,59; Н 5,53; N 5,06; S 5,71. МСВР МН+: рассчитано для C22H26N2O8S: 479,1488, обнаружено: 479,1497.
Пример 416: Получение моногидрохлорида N-гидрокси-1-(2-метоксиэтил)-4-[[4-[4-(трифторметоксиэтил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
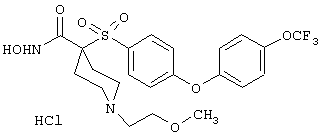
Стадия А: К раствору полученного на стадии Г примера 9 продукта (30 г, 161 ммоль) в дихлорметане (50 мл), охлажденному до 0° С, добавляли трифторуксусную кислоту (25 мл) и раствор перемешивали при температуре окружающей среды в течение 1 ч. После концентрирования в вакууме получали трифторацетат амина в виде геля светло-желтого цвета. К раствору, содержащему трифторацетат и K2CO3 (3,6 г, 26 ммоль) в N,N-диметилформамиде (50 мл), охлажденному до 0° С, добавляли простой метил-2-бромэтиловый эфир (19 мл, 201 ммоль) и раствор перемешивали в течение 36 ч при температуре окружающей среды. N,N-диметилформамид выпаривали в глубоком вакууме и остаток рабавляли этилацетатом. Органический слой промывали водой и сушили над MgSO4. После концентрирования в вакууме получали метоксиэтиламин в виде геля светло-желтого цвета (26,03 г, 86,8%).
Стадия Б: К раствору, содержащему полученный на стадии А метоксиэтиламин (6,0 г, 16,0 ммоль) и порошкообразный К2СО3 (4,44 г, 32 ммоль) в N,N-диметилформамиде (30 мл), добавляли при температуре окружающей среды 4-(трифторметокси)фенол (5,72 г, 32 ммоль) и раствор нагревали в течение 25 ч до 95° С. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали 1 н. раствором NaOH, Н2О и сушили над MgSO4. После хроматографии на силикагеле с элюированием этилацетатом/гексаном получали трифторметоксифеноксифенилсульфон в виде геля светло-желтого цвета (7,81 г, 91,5%).
Стадия В: К раствору полученного на стадии Б трифторметоксифеноксифенилсульфона (7,81 г, 14,7 ммоль) в этаноле (14 мл) и тетрагидрофуране (14 мл) добавляли при температуре окружающей среды с помощью капельной воронки NaOH (5,88 г, 147 ммоль) в Н2О (28 мл). Затем раствор нагревали в течение 18 ч до 60° С. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали простым эфиром и подкисляли до рН 2. После вакуумной фильтрации осадка белого цвета получали кислоту в виде твердого вещества белого цвета (5,64 г, 73,3%).
Стадия Г: К раствору, содержащему полученную на стадии В кислоту (5,64 г, 10,8 ммоль), N-метилморфолин (4,8 мл, 43,1 ммоль), 1-гидроксибензотриазол (4,38 г, 32,4 ммоль) и O-(тетрагидропиранил)гидроксиламин (2,5 г, 21,6 ммоль) в N,N-диметилформамиде (50 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (6,2 г, 32,4 ммоль) и раствор перемешивали в течение 24 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным водным раствором NaHCO3, Н2О и сушили над MgSO4. После концентрирования в вакууме и хроматографии на силикагеле с элюированием этилацетатом/гексаном получали тетрагидропиранил-защищенный гидроксамат в виде пены белого цвета (6,65 г, количественный выход).
Стадия Д: К раствору 4 н. HCl в диоксане (28 мл, 110 ммоль) добавляли раствор полученного на стадии Г тетрагидропиранил-защищенного гидроксамата (6,65 г, 11,03 ммоль) в метаноле (3 мл) и диоксане (9 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. После концентрирования в вакууме и растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (4,79 г, 78,2%). Элементный анализ: рассчитано для С22Н25N2О7SF3·НСl· 0,5Н2О: С 46,85; Н 4,83; N 4,97; S 5,69; обнаружено: С 46,73; Н 4,57; N 4,82; S 5,77.
Пример 417: Получение моногидрохлорида N-гидрокси-1-(2-метоксиэтил)-4-[[4-[4-(1-метилэтил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида

Стадия А: К раствору, содержащему этил-4-[(4-фторфенилсульфонил)]-1-(2-метоксиэтил)-4-пиперидинкарбоксилат (1,47 г, 3,9 ммоль) и порошкообразный К2СО3 (1,6 г, 11,7 ммоль) в N,N-диметилформамиде (15 мл), добавляли при температуре окружающей среды 4-изопропилфенол (1,07 г, 7,8 ммоль) и раствор нагревали в течение 24 ч до 90° С. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали 1 н. NaOH, Н2О и сушили над MgSO4. После хроматографии на силикагеле с элюированием этилацетатом/гексаном получали простой диариловый эфир в виде геля светло-желтого цвета (1,77 г, 92,2%).
Стадия Б: К раствору полученного на стадии А прсотого диарилового эфира (1,77 г, 3,6 ммоль) в этаноле (3,5 мл) и тетрагидрофуране (3,5 мл) добавляли при температуре окружающей среды NaOH (1,46 г, 36 ммоль) в Н2О (7 мл). Затем раствор нагревали в течение 18 ч до 60° С. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали диэтиловым эфиром и подкисляли до рН 2. После вакуумной фильтрации осадка белого цвета получали кислоту в виде твердого вещества белого цвета (1,39 г, 83,7%).
Стадия В: К раствору, содержащему полученную на стадии Б кислоту (1,39 г, 3,0 ммоль), N-метилморфолин (1 мл, 9 ммоль), 1-гидроксибензотриазол (1,22 г, 9 ммоль) и O-(тетрагидропиранил)гидроксиламин (0,72 г, 6,0 ммоль) в N,N-диметилформамиде (90 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (1,72 г, 9,0 ммоль) и раствор перемешивали в течение 48 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным водным раствором NaHCO3, Н2О и сушили над MgSO4. После концентрирования в вакууме и хроматографии на силикагеле с элюированием этилацетатом/гексаном получали тетрагидропиранил-защищенный гидроксамат в виде пены белого цвета (1,65 г, 98,2%).
Стадия Г: К раствору 4 н. HCl в диоксане (7,35 мл, 29,4 ммоль) добавляли раствор полученного на стадии В тетрагидропиранил-защищенного гидроксамата (1,65 г, 2,94 ммоль) в метаноле (1 мл) и диоксане (3 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. После концентрирования в вакууме и растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,2 г, 79,5%). Элементный анализ: рассчитано для C24H32N2O6S· HCl· 0,5Н2О: С 55,22; Н 6,56; N 5,37; S 6,14; обнаружено: С 55,21; Н 6,41; N 5,32; S 6,18.
Пример 418: Получение моногидрохлорида N-гидрокси-1-(2-метоксиэтил)-4-[[4-[4-(трифторметил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
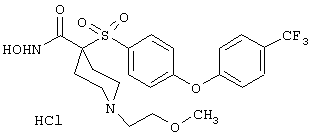
Стадия А: К раствору, содержащему этил-4-[(4-фторфенилсульфонил)]-1-(2-метоксиэтил)-4-пиперидинкарбоксилат (6 г, 16,0 ммоль) и порошкообразный К2СО3 (4,44 г, 32 ммоль) в N,N-диметилформамиде (50 мл), добавляли при температуре окружающей среды 4-трифторметилфенол (5,72 г, 32 ммоль) и раствор нагревали в течение 48 ч до 90° С. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали 1 н. NaOH, Н2О и сушили над MgSO4. После хроматографии на силикагеле с элюированием этилацетатом/гексаном получали простой диариловый эфир в виде геля светло-желтого цвета (2,66 г, 32,1%).
Стадия Б: К раствору полученного на стадии А простого диарилового эфира (1,5 г, 2,9 ммоль) в этаноле (3 мл) и тетрагидрофуране (3 мл) добавляли при температуре окружающей среды NaOH (1,22 г, 29 ммоль) в Н2О (6 мл). Затем раствор нагревали в течение 18 ч до 60° С. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали диэтиловым эфиром и подкисляли до рН 2. После вакуумной фильтрации осадка белого цвета получали требуемую кислоту в виде твердого вещества белого цвета (1,0 г, 70,9%).
Стадия В: К раствору, содержащему полученную на стадии Б кислоту (1,0 г, 2,05 ммоль), N-метилморфолин (0,68 мл, 6,1 ммоль), 1-гидроксибензотриазол (0,84 г, 6,15 ммоль) и O-(тетрагидропиранил)гидроксиламин (0,5 г, 4,1 ммоль) в N,N-диметилформамиде (20 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (1,18 г, 6,0 ммоль) и раствор перемешивали в течение 24 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным водным раствором NaHCO3, Н2О и сушили над MgSO4. После концентрирования в вакууме и хроматографии на силикагеле с элюированием этилацетатом/гексаном получали тетрагидропиранил-защищенный гидроксамат в виде пены белого цвета (1,16 г, 96,7%).
Стадия Г: К раствору 4 н. HCl в диоксане (5 мл, 20 ммоль) добавляли раствор полученного на стадии В тетрагидропиранил-защищенного гидроксамата (1,16 г, 2 ммоль) в метаноле (1 мл) и диоксане (3 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. После концентрирования в вакууме и растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,79 г, 74,5%). Элементный анализ: рассчитано для С22Н25О6SF3·НСl: С 49,03; Н 4,86; N 5,20; S 5,95; обнаружено: С 48,85; Н 4,60; N 5,22; S 6,13.
Пример 419: Получение моногидрохлорида N-гидрокси-1-(2-метоксиэтил)-4-[[4-[4-[(трифторметил)тио]фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
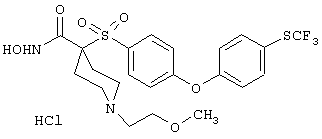
Стадия А: К раствору, содержащему этил-4-[(4-фторфенилсульфонил)]-1-(2-метоксиэтил)-4-пиперидинкарбоксилат (5 г, 13,4 ммоль) и порошкообразный К2СО3 (3,7 г, 27 ммоль) в N,N-диметилформамиде (20 мл), добавляли при температуре окружающей среды 4-(трифторметилтио)фенол (3,9 г, 20 ммоль) и раствор нагревали в течение 24 ч до 90° С. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали 1 н. NaOH, Н2О и сушили над MgSO4. После хроматографии на силикагеле с элюированием этилацетатом/гексаном получали требуемый простой диариловый эфир в виде геля светло-желтого цвета (5,94 г, 81,04%).
Стадия Б: К раствору полученного на стадии А простого диарилового эфира (5,94 г, 210 ммоль) в этаноле (10 мл) и тетрагидрофуране (10 мл) добавляли по каплям при температуре окружающей среды NaOH (4,34 г, 108 ммоль) в Н2О (20 мл). Затем раствор нагревали в течение 24 ч до 60° С и выдерживали в течение еще 24 ч при температуре окружающей среды. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали диэтиловым эфиром и подкисляли до рН 2. После вакуумной фильтрации осадка белого цвета получали кислоту в виде твердого вещества белого цвета (5,5 г, количественный выход).
Стадия В: К раствору, содержащему полученную на стадии Б кислоту (5,5 г, 10,8 ммоль), N-метилморфолин (3,6 мл, 32,4 ммоль), 1-гидроксибензотриазол (4,4 г, 32,4 ммоль) и O-(тетрагидропиранил)гидроксиламин (2,6 г, 21,8 ммоль) в N,N-диметилформамиде (200 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (6,2 г, 32,4 ммоль) и раствор перемешивали в течение 24 ч при температуре окружающей среды. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали насыщенным водным раствором NаНСО3, Н2О и сушили над MgSO4. После концентрирования в вакууме и хроматографии на силикагеле с элюированием этилацетатом/гексаном получали тетрагидропиранил-защищенный гидроксамат в виде пены белого цвета (4,66 г, 69,8%).
Стадия Г: К раствору 4 н. HCl в диоксане (20 мл, 79 ммоль) добавляли раствор полученного на стадии В тетрагидропиранил-защищенного гидроксамата (4,65 г, 7,9 ммоль) в метаноле (2,5 мл) и диоксане (8 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. После концентрирования в вакууме и растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (3,95 г, 92,1%). Элементный анализ: рассчитано для C22H25N2O6S2F3·HCl: С 46,27; Н 4,59; N 4,91; S 11,23; обнаружено: С 46,02; Н 4,68; N 4,57; S 11,11.
Пример 420: Получение моногидрохлорида N-гидрокси-1-(1-метилэтил)-4-[[4-[4-(1-метилэтил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
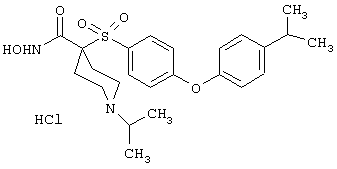
Стадия А: К раствору полученного на стадии Г примера 9 продукта (30 г, 161 ммоль) в дихлорметане (40 мл), охлажденному до 0° С, добавляли трифторуксусную кислоту (30 мл) и раствор перемешивали при температуре окружающей среды в течение 1 ч. После концентрирования в вакууме получали трифторацетат в виде геля светло-желтого цвета. К раствору, содержащему трифторацетат и триэтиламин (28 мл, 201 ммоль) в дихлорметане (250 мл), охлажденному до 0° С, добавляли в виде небольших порций ацетон (24 мл, 320 ммоль) и триацетоксиборогидрид натрия (68 г, 201 ммоль), а затем уксусную кислоту (18,5 мл, 320 ммоль) и раствор перемешивали в течение 48 ч при температуре окружающей среды. Затем дихлорметан выпаривали в глубоком вакууме и остаток разбавляли диэтиловым эфиром. Органический слой промывали 1 н. раствором NaOH, водой и сушили над MgSO4. После концентрирования в вакууме получали изопропиламин в виде геля светло-желтого цвета (21,03 г, 72,8%).
Стадия Б: К раствору, содержащему полученный на стадии А изопропиламин (4 г, 11,2 ммоль) и порошкообразный К2СО3 (3,09 г, 22,4 ммоль) в N,N-диметилформамиде (30 мл), добавляли при температуре окружающей среды 4-изопропилфенол (3,05 г, 22 ммоль) и раствор нагревали в течение 25 ч до 90° С. Раствор концентрировали в глубоком вакууме и остаток растворяли в этилацетате. Органический слой промывали 1 н. раствором NaOH, Н2О и сушили над MgSO4. После хроматографии на силикагеле с элюированием этилацетатом/гексаном получали требуемый простой диариловый эфир в виде геля светло-желтого цвета (5,10 г, 96,2%).
Стадия В: К раствору полученного на стадии Б простого диарилового эфира (5,10 г, 10,77 ммоль) в этаноле (10 мл) и тетрагидрофуране (10 мл) добавляли при температуре окружающей среды с помощью капельной воронки NaOH (4,3 г, 108 ммоль) в Н2О (20 мл). Затем раствор нагревали в течение 24 ч до 60° С и выдерживали еще в течение 24 ч при температуре окружающей среды. Раствор концентрировали в вакууме и разбавляли водой. Водный слой экстрагировали диэтиловым эфиром и подкисляли до рН 2. После вакуумной фильтрации осадка белого цвета получали требуемую кислоту в виде твердого вещества белого цвета (4,80 г, количественный выход).
Стадия Г: К раствору, содержащему полученную на стадии В кислоту (4,80 г, 10,8 ммоль), N-метилморфолин (3,6 мл, 32,4 ммоль), 1-гидроксибензотриазол (4,4 г, 32,4 ммоль) и O-(тетрагидропиранил)гидроксиламин (2,6 г, 21,6 ммоль) в N,N-диметилформамиде (100 мл), добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (6,17 г, 32,4 ммоль) и раствор перемешивали в течение 7 дней при температуре окружающей среды. Раствор фильтровали для удаления непрореагировавшего исходного продукта и фильтрат концентрировали в глубоком вакууме. Остаток растворяли в этилацетате и органический слой промывали насыщенным водным раствором NaHCO3, Н2О и сушили над MgSO4. После концентрирования в вакууме и хроматографии на силикагеле с элюированием этилацетатом/гексаном получали тетрагидропиранил-защищенный гидроксамат в виде пены белого цвета (2,45 г, 41,7%).
Стадия Д: К раствору 4н. HCl в диоксане (11,2 мл, 45 ммоль) добавляли раствор полученного на стадии Г тетрагидропиранил-защищенного гидроксамата (2,45 г, 11,03 ммоль) в метаноле (4 мл) и диоксане (8 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. После концентрирования в вакууме и растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (2,01 г, 89,7%). Элементный анализ: рассчитано для C24H32N2O5S· HCl· 0,5Н2О: С 56,96; Н 6,77; N 5,54; S 6,34; обнаружено: С 56,58; Н 6,71; N 5,44; S 6,25.
Пример 421: Получение моногидрохлорида 4-[[4-(1,3-бензодиоксол-5-илокси)фенил]сульфонил]-1-циклопропил-N-гидрокси-4-пиперидинкарбоксамида
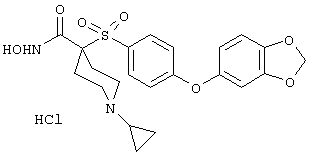
Стадия А: К раствору полученного на стадии Г примера 9 продукта (9,0 г, 22,0 ммоль) в ДМФ (30 мл) добавляли К2СО3 (4,55 г, 33 ммоль) и сезамол (4,55 г, 33 ммоль). Раствор перемешивали в течение 24 ч при 90° С. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 10%-ный этилацетат/гексан получали требуемый сложный эфир в виде масла (9,3 г, 79%). МСВР МН+: рассчитано для С26Н31NSO9: 534,1798, обнаружено: 534,1796.
Стадия Б: Раствор полученного на стадии А сложного эфира (9,3 г, 17 ммоль) в этилацетате (100 мл), охлажденный до 0° С, барботировали в течение 10 мин газообразным HCl. Реакционную смесь перемешивали при этой температуре в течение 0,5 ч. Раствор концентрировали в вакууме, получая гидрохлорид (7,34 г, 92%). МСВР МН+: рассчитано для C21H23NSO7: 434,1273, обнаружено: 434,1285.
Стадия В: К раствору полученного на стадии Б гидрохлорида (7,34 г, 15,6 ммоль) в метаноле (60 мл) добавляли уксусную кислоту (8,94 мл, 156 ммоль), порцию (приблизительно 2 г) молекулярных сит размером 4 Е, (1-этоксициклопропил)окситриметилсилан (18,82 мл, 93,6 ммоль) и цианборогидрид натрия (4,41 г, 70,2 ммоль). Раствор кипятили с обратным холодильником в течение 8 ч. Осадок удаляли фильтрацией и фильтрат концентрировали в вакууме. Остаток разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием 100%-ным этилацетатом получали требуемый циклопропиламин в виде твердого вещества (7,9 г, 100%). МСВР МН+: рассчитано для C24H27NSO7: 474,1586, обнаружено: 474,1599.
Стадия Г: К раствору полученного на стадии В циклопропиламина (7,9 г, 16,7 ммоль) в этаноле (50 мл) и тетрагидрофуране (50 мл) добавляли раствор NaOH (6,68 г, 166,8 ммоль) в воде (30 мл) и раствор выдерживали в течение 18 ч при 60° С. Раствор концентрировали в вакууме и водный остаток подкисляли до рН 3. Образовавшийся осадок фильтровали, получая требуемую карбоновую кислоту (6,14 г, 76%). МСВР МН+: рассчитано для C22H25NSO7: 446,1273, обнаружено: 446,1331.
Стадия Д: К раствору полученной на стадии Г карбоновой кислоты (6,14 г, 12,7 ммоль) в ДМФ (60 мл) добавляли 1-гидроксибензотриазол (2,06 г, 15,2 ммоль), N-метилморфолин (4,2 мл, 38,0 ммоль) и O-(тетрагидропиранил)гидроксиламин (2,23 г, 19,0 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (3,41 г, 17,8 ммоль). Раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 40%-ный этилацетат/гексан получали требуемый тетрагидропиранил-защищенный гидроксамат в виде твердого вещества (6,67 г, 96%).
Стадия Е: К раствору полученного на стадии Д тетрагидропиранил-защищенного гидроксамата (6,67 г, 12,0 ммоль) в диоксане (70 мл) добавляли смесь 4н. HCl/диоксан (6,6 мл). После перемешивания в течение 3 ч при температуре окружающей среды раствор концентрировали в вакууме. После хроматографии с обращенной фазой на колонке С18 с элюированием смесью ацетонитрил/(HCl) вода получали твердое вещество белого цвета (4,21 г, 69%). МСВР МН+: рассчитано для C22H24N2SO7: 461,1382, обнаружено: 461,1386.
Пример 422: Получение моногидрохлорида 1-циклопропил-4-[[4-(4-этоксифенокси) фенил] сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
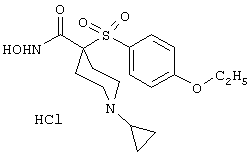
Стадия А: К раствору полученного на стадии Г примера 9 продукта (8,0 г, 19,2 ммоль) в ДМФ (30 мл) добавляли К2СО3 (4,00 г, 28,8 ммоль) и 4-этоксифенол (3,99 г, 28,8 ммоль). Раствор перемешивали в течение 24 ч при 90° С. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 10%-ный этилацетат/гексан получали требуемый сложный эфир в виде масла (9,62 г, 94%). МСВР МН +: рассчитано для C27H35NSO8: 534,2162, обнаружено: 534,2175.
Стадия Б: Раствор полученного на стадии А сложного эфира (9,62 г, 18 ммоль) в этилацетате (100 мл), охлажденный до 0° С, барботировали в течение 5 мин газообразным HCl. Реакционную смесь перемешивали при этой температуре в течение 0,5 ч. Затем раствор концентрировали в вакууме, получая гидрохлорид (8,1 г, 96%). МСВР MH+: рассчитано для С22Н27NSО6: 434,1637, обнаружено: 434,1637.
Стадия В: К раствору полученного на стадии Б гидрохлорида (8,1 г, 17,2 ммоль) в метаноле (70 мл) добавляли уксусную кислоту (9,86 мл, 172 ммоль), порцию (приблизительно 2 г) молекулярных сит размером 4 Е, (1-этоксициклопропил)окситриметилсилан (20,7 мл, 103 ммоль) и цианборогидрид натрия (4,86 г, 77,4 ммоль). Раствор кипятили с обратным холодильником в течение 8 ч. Осадок удаляли фильтрацией и фильтрат концентрировали в вакууме. Остаток разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали 1 н. раствором NaOH, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После растирания с диэтиловым эфиром получали требуемый циклопропиламин в виде твердого вещества белого цвета (6,84 г, 84%).
Стадия Г: К раствору полученного на стадии В циклопропиламина (6,84 г, 14,0 ммоль) в этаноле (50 мл) и тетрагидрофуране (50 мл) добавляли раствор NaOH (5,60 г, 140 ммоль) в воде (30 мл) и раствор выдерживали в течение 18 ч при 60° С. Раствор концентрировали в вакууме и подкисляли до рН 3. После фильтрации получали требуемую кислоту (6,07 г, 88%). МС МН+: рассчитано для C22H27NSO6: 446, обнаружено: 446.
Стадия Д: К раствору полученной на стадии Г кислоты (6,07 г, 12,6 ммоль) в ДМФ (60 мл) добавляли 1-гидроксибензотриазол (2,04 г, 15,1 ммоль), N-метилморфолин (4,15 мл, 37,8 ммоль) и O-(тетрагидропиранил)гидроксиламин (2,21 г, 18,9 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (3,38 г, 17,6 ммоль). Раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 60%-ный этилацетат/гексан получали требуемый тетрагидропиранил-защищенный гидроксамат в виде пены белого цвета (6,29 г, 92%). МСВР МН+: рассчитано для С28Н36N2SO7: 545,2321, обнаружено: 545,2316.
Стадия Е: К раствору полученного на стадии Д тетрагидропиранил-защищенного гидроксамата (2,84 г, 5,0 ммоль) в диоксане (40 мл) добавляли смесь 4 н. HCl/диоксан (30 мл). После перемешивания в течение 2,5 ч при температуре окружающей среды раствор концентрировали в вакууме. После растирания образовавшегося твердого вещества с диэтиловым эфиром и фильтрации получали требуемый гидроксамат в виде твердого вещества белого цвета (2,33 г, 90%). МСВР МН+: рассчитано для С23Н28N2SO6: 460,1677, обнаружено: 460,1678.
Пример 423: Получение 4-[[4-(циклогексилтио)фенил]сульфонил]-N-гидрокси-1-(метилсульфонил)-4-пиперидинкарбоксамида
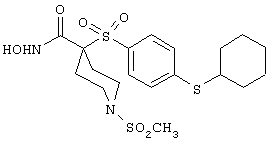
Стадия А: К раствору полученного на стадии Г примера 9 продукта (10,0 г, 24,0 ммоль) в ДМФ (20 мл) добавляли К2СО3 (4,99 г, 36,0 ммоль) и циклогексилмеркаптан (4,40 г, 36,0 ммоль). Раствор перемешивали в течение 48 ч при 90° С. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После растиранием с этанолом получали требуемый сульфид в виде твердого вещества белого цвета (7,16 г, 58%).
Стадия Б: К раствору полученного на стадии А сульфида (9,46 г, 18,5 ммоль) в этаноле (30 мл) и тетрагидрофуране (30 мл) добавляли раствор NaOH (7,39 г, 185 ммоль) в воде (15 мл) и раствор выдерживали в течение 18 ч при 65° С. Раствор концентрировали в вакууме и водный остаток подкисляли до рН 3,5. Образовавшееся твердое вещество белого цвета собирали фильтрацией, промывали Н2О и диэтиловым эфиром, получая требуемую карбоновую кислоту (8,57 г, 95%).
Стадия В: Раствор полученной на стадии Б карбоновой кислоты (8,3 г, 17,0 ммоль) в этилацетате (200 мл), охлажденный до 0° С, барботировали в течение 15 мин газообразным HCl. Затем реакционную смесь перемешивали при этой температуре в течение 0,5 ч. Раствор концентрировали в вакууме, получая остаток, который растирали с диэтиловым эфиром, получая требуемый гидрохлорид в виде твердого вещества белого цвета (7,03 г, 98%). МСВР МН+: рассчитано для С18Н25NS2O4: 384,1303, обнаружено: 384,1318.
Стадия Г: К раствору полученного на стадии В гидрохлорида (1,0 г, 2,4 ммоль) добавляли N-метилморфолин (654 мл, 5,9 ммоль), а затем мезилхлорид (280 мл, 3,6 ммоль) в метиленхлориде (20 мл). Раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали метиленхлоридом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме, получая требуемый метансульфонамид в виде пены (1,0 г, количественный выход).
Стадия Д: К раствору полученного на стадии Г метансульфонамида (1,3 г, 2,9 ммоль) в ДМФ (30 мл) добавляли 1-гидроксибензотриазол (474 мг, 3,5 ммоль), N-метилморфолин (956 мл, 8,7 ммоль), O-(тетрагидропиранил)гидроксиламин (509 мг, 4,3 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (778 мг, 4,06 ммоль). Раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли Н2О(400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 30%-ный этилацетат/гексан получали требуемый тетрагидропиранил-защищенный гидроксамат в виде пены белого цвета (1,05 г, 82%).
Стадия Е: К раствору полученного на стадии Д тетрагидропиранил-защищенного гидроксамата (1,05 г, 1,97 ммоль) в диоксане (30 мл) добавляли смесь 4 н. HCl/диоксан (10 мл). После перемешивания в течение 2,5 ч при температуре окружающей среды раствор концентрировали в вакууме. После хроматографии с обращенной фазой на колонке С18 с элюированием смесью ацетонитрил / HCl / вода получали твердое вещество белого цвета (602 мг, 64%). МС М+: рассчитано для C19H28N2S3O6: 477, обнаружено: 477.
Пример 424: Получение N-гидрокси-1-(метилсульфонил)-4-[[4-(фенилтио)фенил]сульфонил]-4-пиперидинкарбоксамида
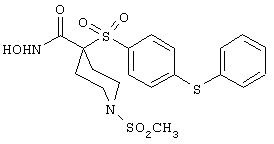
Стадия А: К раствору полученного на стадии Г примера 9 продукта (40,0 г, 96,0 ммоль) в ДМФ (200 мл) добавляли К2СО3 (20 г, 144 ммоль) и тиофенол (22,2 г, 144 ммоль). Раствор перемешивали в течение 24 ч при температуре окружающей среды. Затем раствор разбавляли Н2О (1 л) и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии (на силикагеле, элюирование смесью 15%-ный этилацетат/гексан) получали требуемый сульфид в виде твердого вещества белого цвета (44,4 г, 91%).
Стадия Б: Раствор полученного на стадии А сульфида (31,2 г, 6,6 ммоль) в этилацетате (500 мл), охлажденный до 0° С, барботировали в течение 30 мин газообразным HCl. Реакционную смесь перемешивали при этой температуре в течение 1,5 ч. Раствор концентрировали в вакууме и образовавшееся твердое вещество растирали с диэтиловым эфиром, получая гидрохлорид в виде твердого вещества белого цвета (26,95 г, 96%).
Стадия В: К раствору полученного на стадии Б гидрохлорида (2,0 г, 4,7 ммоль) добавляли N-метилморфолин (1,29 мл, 11,7 ммоль), а затем мезилхлорид (550 мл, 7,05 ммоль) в метиленхлориде (35 мл). Раствор перемешивали в течение 48 ч при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали метиленхлоридом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме, получая требуемый метансульфонамид в виде твердого вещества белого цвета (2,17 г, 96%).
Стадия Г: К раствору полученного на стадии В метансульфонамида (2,1 г, 4,3 ммоль) в этаноле (25 мл) и тетрагидрофуране (25 мл) добавляли раствор NaOH (1,72 г, 43 ммоль) в воде (10 мл) и раствор выдерживали в течение 18 ч при 60° С. Раствор концентрировали в вакууме и водный остаток подкисляли до рН 3,5. Образовавшийся осадок фильтровали, получая требуемую карбоновую кислоту в виде твердого вещества белого цвета (2,1 г, количественный выход).
Стадия Д: К раствору полученной на стадии Г карбоновой кислоты (1,98 г, 4,3 ммоль) в ДМФ (30 мл) добавляли 1-гидроксибензотриазол (705 мг, 5,2 ммоль), N-метилморфолин (1,54 мл, 12,9 ммоль) и гидрохлорид O-(тетрагидропиранил)гидроксиламина (755 мг, 6,5 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (1,17 г, 6,1 ммоль). Раствор перемешивали в течение 5 дней при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии с обращенной фазой на колонке С18 с элюированием смесью ацетонитрил/(HCl) вода получали требуемый тетрагидропиранил-защищенный гидроксамат в виде твердого вещества белого цвета (1,86 г, 80%). МСВР МН+: рассчитано для С24H30N2S3О7: 555,1293, обнаружено: 555,1276.
Стадия Е: К раствору полученного на стадии Д тетрагидропиранил-защищенного гидроксамата (1,86 г, 3,5 ммоль) в диоксане (30 мл) и метаноле (10 мл) добавляли смесь 4 н. HCl/диоксан (20 мл). После перемешивания в течение 2,5 ч при температуре окружающей среды раствор концентрировали в вакууме. После хроматографии с обращенной фазой на колонке С18 с элюированием смесью ацетонитрил/(НСl) вода получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,48 г, 91%). МСВР МН+: рассчитано для C19H22N2S3O6: 471,0718, обнаружено: 471,0728.
Пример 425: Получение моногидрохлорида 1-циклопропил-N-гидрокси-4-[[4-[4-[(трифторметокси)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
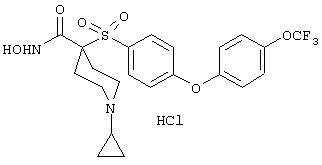
Стадия А: К раствору полученного на стадии А примера 398 продукта (6,97 г, 19,6 ммоль) в ДМФ (500 мл) добавляли К2СО3 (3,42 г, 18,0 ммоль) и 4-(трифторметокси) фенол (3,7 г, 24,8 ммоль). Раствор перемешивали в течение 40 ч при 90° С. Раствор разбавляли Н2О и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме, получая требуемый простой диариловый эфир в виде масла (8,5 г, количественный выход). МСВР МН+: рассчитано для C24H26NSO6F3: 514,1511, обнаружено: 514,1524.
Стадия Б: К раствору полученного на стадии А простого диарилового эфира (8,4 г, 16,4 ммоль) в этаноле (50 мл) и тетрагидрофуране (50 мл) добавляли раствор NaOH (6,54 г, 164 ммоль) в Н2О (20 мл) и раствор выдерживали в течение 18 ч при 60° С. Раствор концентрировали в вакууме для удаления большей части органических растворителей и водный остаток подкисляли до рН 4,0. Образовавшийся осадок фильтровали, получая требуемый гидрохлорид в виде твердого вещества белого цвета (5,01 г, 63%). МСВР МН+: рассчитано для C22H22NSO6F3: 486,1198, обнаружено: 486,1200.
Стадия В: К раствору полученного на стадии Б гидрохлорида (5,0 г, 10,3 ммоль) в ДМФ (80 мл) добавляли 1-гидроксибензотриазол (1,65 г, 12,3 ммоль), N-метилморфолин (3,4 мл, 30,9 ммоль) и гидрохлорид O-(тетрагидропиранил)гидроксиламина (1,8 г, 15,4 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (1,60 г, 12,3 ммоль). Раствор перемешивали в течение 42 ч при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 30%-ный этилацетат/гексан получали требуемый тетрагидропиранил-защищенный гидроксамат в виде твердого вещества белого цвета (5,41 г, 89%).
Стадия Г: К раствору полученного на стадии В тетрагидропиранил-защищенного гидроксамата (5,4 г, 9,2 ммоль) в диоксане (80 мл) и метаноле (20 мл) добавляли раствор 4н. HCl/диоксан (50 мл). Реакционную смесь перемешивали в течение 2,5 ч при температуре окружающей среды и раствор концентрировали в вакууме. После растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (4,02 г, 81%). МСВР МН+: рассчитано для C22H23N2SO6F3: 501,1307, обнаружено:501,1324.
Пример 426: Получение моногидрохлорида 1-циклопропил-4-[(4-этоксифенил)сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
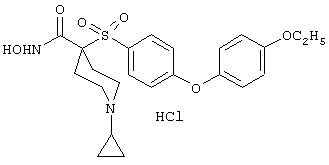
Стадия А: К раствору полученного на стадии А примера 398 продукта (5,87 г, 16,5 ммоль) в ДМФ (50 мл) добавляли К2СО3 (3,42 г, 24,7 ммоль) и α ,α ,α -(трифторметил)-пара-крезол (4,01 г, 24,7 ммоль). Раствор перемешивали в течение 48 ч при 90° ° С. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме, получая неочищенный продукт, содержащий большой процент исходного продукта (8,39 г). К раствору этого продукта (8,39 г) в этаноле (50 мл) и тетрагидрофуране (50 мл) добавляли раствор NaOH (6,75 г, 169 ммоль) в воде (20 мл) и раствор выдерживали в течение 18 ч при 60° С. Раствор концентрировали в вакууме и водный остаток подкисляли до рН 3,5. Образовавшийся осадок фильтровали, получая требуемый гидрохлорид в виде воскообразного твердого вещества (5,04 г, 64%).
Стадия Б: К раствору полученного на стадии А гидрохлорида (5,0 г, 10,3 ммоль) в ДМФ (80 мл) добавляли 1-гидроксибензотриазол (1,73 г, 12,8 ммоль), N-метилморфолин (3,5 мл, 31,8 ммоль) и гидрохлорид O-(тетрагидропиранил)гидроксиламина (1,86 г, 15,9 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (2,84 г, 14,8 ммоль). Раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 30%-ный этилацетат/гексан получали требуемый тетрагидропиранил-защищенный гидроксамат в виде твердого вещества белого цвета (1,5 г, 32%).
Стадия В: К раствору полученного на стадии Б тетрагидропиранил-защищенного гидроксамата (1,5 г, 3,3 ммоль) в диоксане (30 мл) и метаноле (15 мл) добавляли раствор 4 н. HCl/диоксан (50 мл). Реакционную смесь перемешивали в течение 2 ч при температуре окружающей среды и раствор концентрировали в вакууме. После растирания остатка с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,09 г, 81%). МС МН+: рассчитано для C17H24N2SO5: 369, обнаружено:369.
Пример 427: Получение моногидрохлорида 1-циклопропил-N-гидрокси-4-[[4-[4-(трифторметил)фенокси]фенил)сульфонил]-4-пиперидинкарбоксамида
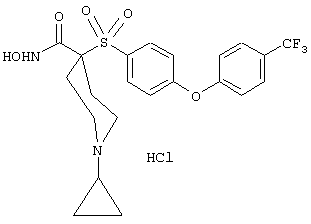
Стадия А: К раствору полученного на стадии А примера 398 продукта (5,96 г, 15,0 ммоль) в ДМФ (100 мл) добавляли К2СО3 (12,34 г, 38,0 ммоль) и α ,α ,α -трифторметилфенол (3,65 г, 22,5 ммоль). Раствор перемешивали в течение 28 ч до 90° С. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме, получая требуемый простой ариловый эфир в виде масла (7,54 г, количественный выход).
Стадия Б: К раствору полученного на стадии А простого арилового эфира (7,54 г, 15,0 ммоль) в этаноле (40 мл) и тетрагидрофуране (40 мл) добавляли раствор NaOH (6,06 г, 151,0 ммоль) в воде (20 мл) и раствор выдерживали в течение 18 ч при 60° С. Раствор концентрировали в вакууме и водный остаток подкисляли до рН 2. Образовавшийся осадок фильтровали, получая требуемый гидрохлорид в виде твердого вещества белого цвета (7,98 г, количественный выход). МС МН+: рассчитано для С22Н22NSO5F3: 470, обнаружено: 470.
Стадия В: К раствору полученного на стадии Б гидрохлорида (7,60 г, 15,0 ммоль) в ДМФ (100 мл) добавляли 1-гидроксибензотриазол (2,44 г, 18,0 ммоль), N-метилморфолин (3,4 мл, 30,9 ммоль) и гидрохлорид O-(тетрагидропиранил)гидроксиламина (2,63 г, 22,5 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (4,02 г, 21,0 ммоль). Раствор перемешивали в течение 96 ч при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 30%-ный этилацетат/гексан получали требуемый тетрагидропиранил-защищенный гидроксамат в виде твердого вещества белого цвета (5,93 г, 69%).
Стадия Г: К раствору полученного на стадии В тетрагидропиранил-защищенного гидроксамата (3,8 г, 6,7 ммоль) в диоксане (100 мл) добавляли смесь 4 н. HCl/диоксан (30 мл). Реакционную смесь перемешивали в течение 2 ч при температуре окружающей среды, после чего раствор концентрировали в вакууме. После растирания остатка с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (3,33 г, 96%). МС МН+: рассчитано для (С22Н23N2SO5F3: 485, обнаружено: 485.
Пример 428: Получение моногидрохлорида N-гидрокси-1-(1-метилэтил)-4-[[4-[4-(трифторметил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
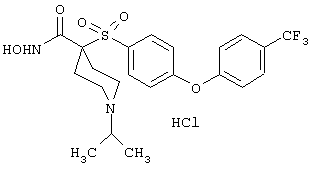
Стадия А: К раствору полученного на стадии Г примера 9 продукта (30,0 г, 80,8 ммоль) в метиленхлориде (100 мл) добавляли трифторуксусную кислоту (30 мл) в метиленхлориде (40 мл). Раствор перемешивали в течение 2 ч при температуре окружающей среды. Раствор концентрировали в вакууме. К остатку, растворенному в метиленхлориде (150 мл), при 0° С добавляли триэтиламин (28,0 мл, 277 ммоль), ацетон (24,0 мл, 413 ммоль), цианборогидрид натрия (68 г, 323,1 ммоль) и уксусную кислоту (18,5 мл, 308 ммоль). Реакционную смесь перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли 1 н. раствором NaOH и экстрагировали диэтиловым эфиром. Органический слой промывали 1н. раствором NaOH, водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме, получая требуемый изопропиламин (21,03 г, 72%).
Стадия Б: К раствору полученного на стадии А изопропиламина (4,04 г, 11,0 ммоль) в ДМФ (50 мл) добавляли Cs2CO3 (10,75 г, 33,3 ммоль) и α ,α ,α -трифтор-пара-крезол (2,67 г, 16,5 ммоль). Раствор перемешивали в течение 40 ч при 90° С. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 30%-ный этилацетат/гексан получали требуемый простой диариловый эфир в виде масла (5,35 г, 97%). МСВР: рассчитано для C24H28NO5SF3: 500,1640, обнаружено: 500,1678.
Стадия В: К раствору полученного на стадии Б простого диарилового эфира (5,3 г, 10,6 ммоль) в этаноле (50 мл) и тетрагидрофуране (50 мл) добавляли раствор NaOH (4,2 г, 106,0 ммоль) в воде (25 мл) и раствор выдерживали в течение 18 ч при 60° С. Раствор концентрировали в вакууме и водный остаток подкисляли до рН 3,0. Образовавшийся осадок фильтровали, получая требуемый гидрохлорид в виде твердого вещества белого цвета (5,38 г, количественный выход). МСВР МН+: рассчитано для C22H24NSO5F3: 472,1406, обнаружено: 472,1407.
Стадия Г: К раствору полученного на стадии В гидрохлорида (5,4 г, 10,6 ммоль) в ДМФ (90 мл) добавляли 1-гидроксибензотриазол (1,72 г, 12,3 ммоль), N-метилморфолин (3,5 мл, 32,0 ммоль) и гидрохлорид O-(тетрагидропиранил)гидроксиламина (1,87 г, 15,9 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (2,8 г, 15,0 ммоль). Раствор перемешивали в течение 144 ч при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 2%-ный метанол/этилацетат получали требуемый тетрагидропиранил-защищенный гидроксамат в виде твердого вещества белого цвета (2,74 г, 45%). МСВР МН+: рассчитано для С27Н33N2SO5F3: 571,2090, обнаружено: 571,2103.
Стадия Д: К раствору полученного на стадии Г тетрагидропиранил-защищенного гидроксамата (2,7 г, 4,7 ммоль) в диоксане (50 мл) добавляли смесь 4 н. HCl/диоксан (20 мл). Реакционную смесь перемешивали в течение 2 ч при температуре окружающей среды. После фильтрации получали указанное в заголовке соединение в виде твердого вещества белого цвета (2,08 г, 84%). МС МН+: рассчитано для С22Н25N2SO5F3: 487, обнаружено: 487.
Пример 429: Получение моногидрохлорида 1-этил-N-гидрокси-4-[[4-[4-(трифторметил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
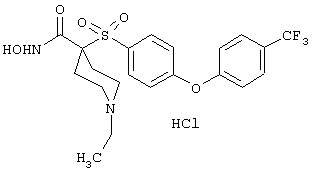
Стадия А: Раствор полученного на стадии Г примера 9 продукта (48 г, 115,0 ммоль) в этилацетате (750 мл), охлажденный до 0° С, барботировали в течение 45 мин газообразным HCl и перемешивали при этой температуре в течение 7 ч. Раствор концентрировали в вакууме, получая остаток, который растирали с диэтиловым эфиром, получая требуемый гидрохлорид в виде твердого вещества белого цвета (32,76 г, 81%).
Стадия Б: К раствору полученного на стадии А гидрохлорида (15,8 г, 45,0 ммоль) в ДМФ (75 мл) добавляли К2СО3 (12,4 г, 90,0 ммоль) и бромэтан (3,4 мл, 45,0 ммоль). Раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли Н2О (200 мл) и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме, получая требуемый этиламин в виде масла (15,4 г, количественный выход).
Стадия В: К раствору полученного на стадии Б этиламина (5,2 г, 15,0 ммоль) в ДМФ (50 мл) добавляли СsСО3 (12,21 г, 37,5 ммоль) и α ,α ,α -трифтор-пара-крезол (3,65 г, 23,0 ммоль). Раствор перемешивали в течение 25 ч при 90° С. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 20%-ный этилацетат/гексан получали требуемый простой диариловый эфир в виде масла (7,3 г, количественный выход).
Стадия Г: К раствору полученного на стадии В простого диарилового эфира (7,3 г, 15,0 ммоль) в этаноле (40 мл) и тетрагидрофуране (40 мл) добавляли раствор NaOH (6,0 г, 150 ммоль) в воде (30 мл) и раствор выдерживали в течение 16 ч при 60° С. Раствор концентрировали в вакууме и водный остаток подкисляли до рН 4,0. Образовавшийся осадок фильтровали, получая требуемый гидрохлорид в виде твердого вещества белого цвета (5,96 г, 80%). МСВР МН+: рассчитано для C21H22NSO5F3: 458,1249, обнаружено: 458,1260.
Стадия Д: К раствору полученного на стадии Г гидрохлорида (5,96 г, 12,0 ммоль) в ДМФ (80 мл) добавляли 1-гидроксибензотриазол (1,96 г, 14,0 ммоль), N-метилморфолин (3,9 мл, 36,0 ммоль) и гидрохлорид O-(тетрагидропиранил)гидроксиламина (2,11 г, 18,0 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (3,24 г, 17,0 ммоль). Раствор перемешивали в течение 168 ч при температуре окружающей среды. Нерастворившийся продукт удаляли фильтрацией и фильтрат разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 70%-ный этилацетат/гексан получали требуемый тетрагидропиранил-защищенный гидроксамат в виде твердого вещества белого цвета (2,80 г, 41%).
Стадия Е: К раствору полученного на стадии Д тетрагидропиранил-защищенного гидроксамата (2,8 г, 5,0 ммоль) в диоксане (80 мл) добавляли смесь 4 н. HCl/диоксан (20 мл). Реакционную смесь перемешивали в течение 5 ч при температуре окружающей среды и раствор концентрировали в вакууме. После растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (2,08 г, 84%). МС МН+: рассчитано для C21H23N2SO5F3: 473, обнаружено: 473.
Пример 430: Получение моногидоохлорида 1-этил-N-гидрокси-4-[[4-[4-(1-метилэтил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида

Стадия А: Раствор полученного на стадии Г примера 9 продукта (48 г, 115,0 ммоль) в этилацетате (750 мл), охлажденный до 0° С, барботировали в течение 45 мин газообразным HCl. Реакционную смесь перемешивали при этой температуре в течение 7 ч. Раствор концентрировали в вакууме, получая остаток, который растирали с диэтиловым эфиром, получая требуемый гидрохлорид в виде твердого вещества белого цвета (32,8 г, 81%).
Стадия Б: К раствору полученного на стадии А гидрохлорида (15,8 г, 45,0 ммоль) в ДМФ (75 мл) добавляли K2CO3 (12,4 г, 90,0 ммоль) и бромэтан (3,4 мл, 45,0 ммоль). Раствор перемешивали в течение 18 ч при температуре окружающей среды. Раствор разбавляли Н2О (200 мл) и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме, получая требуемый этиламин в виде масла (15,4 г, количественный выход).
Стадия В: К раствору полученного на стадии Б этиламина (5,2 г, 15,0 ммоль) в ДМФ (50 мл) добавляли СsСО3 (12,21 г, 37,5 ммоль) и 4-изопропилфенол (3,15 г, 23,0 ммоль). Раствор перемешивали в течение 5 ч при 90° С. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 20%-ный этилацетат/гексан получали требуемый простой диариловый эфир в виде масла (6,2 г, 95%). МСВР МН+: рассчитано для C25H33N3SO5: 460,2158, обнаружено: 460,2160.
Стадия Г: К раствору полученного на стадии В простого диарилового эфира (6,2 г, 13,0 ммоль) в этаноле (40 мл) и тетрагидрофуране (40 мл) добавляли раствор NaOH (5,2 г, 130 ммоль) в воде (30 мл) и раствор выдерживали в течение 16 ч при 60° С. Раствор концентрировали в вакууме и водный остаток подкисляли до рН 4,0. Образовавшийся осадок фильтровали и промывали Н2О и диэтиловым эфиром, получая требуемый гидрохлорид (6,0 г, количественный выход). МСВР МН+: рассчитано для C23H29NSO5: 432,1845, обнаружено: 432,1859.
Стадия Д: К раствору полученного на стадии Г гидрохлорида (6,08 г, 13,0 ммоль) в ДМФ (80 мл) добавляли 1-гидроксибензотриазол (2,11 г, 15,6 ммоль), N-метилморфолин (4,3 мл, 39,0 ммоль) и гидрохлорид O-(тетрагидропиранил)гидроксиламина (2,28 г, 19,5 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (3,49 г, 18,2 ммоль). Раствор перемешивали в течение 168 ч при температуре окружающей среды. Нерастворившийся продукт удаляли фильтрацией и фильтрат разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 50%-ный этилацетат/гексан получали требуемый тетрагидропиранил-защищенный гидроксамат в виде твердого вещества белого цвета (1,7 г, 25%). МСВР МН+: рассчитано для С28H38N2SО6: 531,2529, обнаружено: 531,2537.
Стадия Е: К раствору полученного на стадии Д тетрагидропиранил-защищенного гидроксамата (1,7 г, 3,0 ммоль) в диоксане (60 мл) добавляли смесь 4 н. HCl/диоксан (10 мл). Реакционную смесь перемешивали в течение 4 ч при температуре окружающей среды и раствор концентрировали в вакууме. После хроматографии с обращенной фазой на колонке С18 с элюированием смесью ацетонитрил/(HCl) вода получали указанное в заголовке соединение в виде твердого вещества белого цвета (860 мг, 59%). МСВР МН+: рассчитано для С23Н30N2SO5: 447,1954, обнаружено: 447,1972.
Пример 431: Получение моногидрохлорида 1-циклопропил-N-гидрокси-4-[[4-[4-(1-метилэтил)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
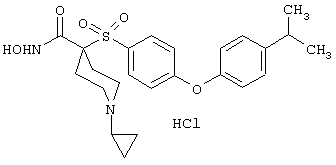
Стадия А: К раствору полученного на стадии А примера 398 продукта (4,0 г, 10,2 ммоль) в ДМФ (40 мл) добавляли К2СО3 (12,46 г, 38,0 ммоль) и 4-изопропилфенол (4,99 г, 15,3 ммоль). Раствор перемешивали в течение 24 ч при 90° С. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали водой, насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 30%-ный этилацетат/гексан получали требуемый простой диариловый эфир в виде твердого вещества белого цвета (3,89 г, 76%). МСВР МН+: рассчитано для C26H33NSO5: 472,2158, обнаружено: 472,2171.
Стадия Б: К раствору полученного на стадии А простого диарилового эфира (3,89 г, 8,20 ммоль) в этаноле (40 мл) и тетрагидрофуране (40 мл) добавляли раствор NaOH (3,30 г, 82,5 ммоль) в воде (25 мл) и раствор выдерживали в течение 18 ч при 60° С. Раствор концентрировали в вакууме для удаления большей части органических растворителей и водный остаток подкисляли до рН 3,0. Образовавшийся осадок фильтровали и промывали Н2О и диэтиловым эфиром, получая требуемый гидрохлорид в виде твердого вещества белого цвета (7,98 г, количественный выход). МС МН+: рассчитано для C24H29NSO5: 444, обнаружено: 444.
Стадия В: К раствору полученного на стадии Б гидрохлорида (3,6 г, 7,0 ммоль) в ДМФ (70 мл) добавляли 1-гидроксибензотриазол (1,22 г, 9,0 ммоль), N-метилморфолин (2,3 мл, 21,0 ммоль) и гидрохлорид O-(тетрагидропиранил)гидроксиламина (1,23 г, 10,5 ммоль), а затем гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (2,01 г, 10,4 ммоль). Раствор перемешивали в течение 15 дней при температуре окружающей среды. Раствор разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 15%-ный этилацетат/гексан получали требуемый тетрагидропиранил-защищенный гидроксамат в виде твердого вещества белого цвета (3,51 г, 92%). МС МН+: рассчитано для C29H38N2SO6: 543,2529, обнаружено: 543,2539.
Стадия Г: К раствору полученного на стадии В тетрагидропиранил-защищенного гидроксамата (3,51 г, 6,0 ммоль) в метаноле (10 мл) и диоксане (200 мл) добавляли смесь 4н. HCl/диоксан (30 мл). После перемешивания в течение 2,5 ч при температуре окружающей среды раствор концентрировали в вакууме. После растирания с диэтиловым эфиром получали указанное в заголовке соединение в виде твердого вещества белого цвета (2,56 г, 86%). МС МН+: рассчитано для С24Н30N2SО5: 459,1875, обнаружено: 459,1978.
Пример 432: Получение моногидрохлорида N-гидрокси-4-[[4-[4-(1-метилэтокси)фенокси]фенил]сульфонил]-1-(1-метилэтил)-4-пиперидинкарбоксамида
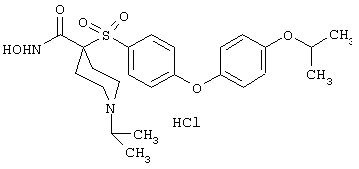
Стадия А: К раствору этил-4-[(4-фторфенилсульфонил)]-1-(1-метилэтил)-4-пиперидинкарбоксилата (2,0 г, 5,4 ммоль) в N,N-диметилформамиде (10 мл) добавляли 4-изопропилоксифенол, который может быть получен согласно способу, описанному в J. Indian Chem. Soc., 73. 1996, 507-511, (1,63 г, 10,7 ммоль) и карбонат цезия (7 г, 21,5 ммоль) и образовавшуюся суспензию выдерживали в течение 16 ч при 60° С. Затем реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате и промывали 1 н. раствором гидроксида натрия, водой и соляным раствором и сушили над сульфатом магния. После концентрирования органической фазы получали остаток, который очищали хроматографией на силикагеле с элюированием этилацетатом/гексаном, получая требуемый простой ариловый эфир (1,06 г, 39%).
Стадия Б: К раствору полученного на стадии Б простого арилового эфира (1,06 г, 2,1 ммоль) в этаноле (20 мл) и воде (20 мл) добавляли гидроксид натрия (0,84 г, 21 ммоль) и смесь нагревали в течение 16 ч до 65° С. Затем растворители удаляли в вакууме. Добавляли воду (50 мл) и смесь снова концентрировали в вакууме и образовавшуюся смесь подкисляли с помощью 2 н. HCl до рН 4-5. Твердый осадок собирали фильтрацией и промывали диэтиловым эфиром, получая требуемую карбоновую кислоту (3,13 г, 100%).
Стадия В: Раствор полученной на стадии Б карбоновой кислоты (1,0 г, 2,0 ммоль) в тионилхлориде (5 мл) кипятили с обратным холодильником в течение 2 ч. Растворитель удаляли в вакууме. К образовавшемуся остатку в ДМФ (10 мл) добавляли N-метилморфолин (0,66 мл, 6,0 ммоль) и гидрохлорид O-тетрагидропиранилгидроксиламина (351 мг, 3,0 ммоль). Раствор перемешивали в течение 18 ч при температуре окружающей среды. Суспензию фильтровали и фильтрат разбавляли Н2О (400 мл) и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaCl и сушили над MgSO4, фильтровали и концентрировали в вакууме. После хроматографии на силикагеле с элюированием смесью 90%-ный этилацетат/гексан получали требуемый тетрагидропиранил-защищенный гидроксамат в виде твердого вещества белого цвета (280 мг, 23%). МСВР МН+: рассчитано для C29H40N2SO7: 561,2634, обнаружено: 561,2653.
Стадия Г: К раствору полученного на стадии В тетрагидропиранил-защищенного гидроксамата (275 мг, 0,48 ммоль) в диоксане (15 мл) добавляли смесь 4 н. HCl/диоксан (5 мл). После перемешивания в течение 2 ч при температуре окружающей среды раствор концентрировали в вакууме. После растирания с диэтиловым эфиром и фильтрации образовавшегося твердого вещества получали указанное в заголовке соединение в виде твердого вещества белого цвета (193 мг, 76%). МС МН+: рассчитано для C24H32N2SO6: 477, обнаружено: 477.
Пример 433: Получение моногидрохлорида 4-[[4-[(2-фторфенил)тио]фенил]сульфонил]-N-гидрокси-4-пиперидинкарбоксамида
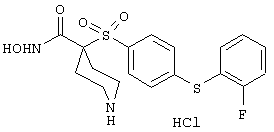
Стадия А: К раствору полученного на стадии Г примера 9 продукта (6,0 г, 14,4 ммоль) в N,N-диметилформамиде (30 мл) добавляли 2-фтортиофенол (2,22 г, 17,3 ммоль) и карбонат калия (2,40 г, 17,3 ммоль) и образовавшуюся суспензию перемешивали в течение 48 ч при температуре окружающей среды. Затем реакционную смесь разбавляли этилацетатом (200 мл) и промывали 1 н. раствором гидроксида натрия (200 мл) и соляным раствором (3х). После концентрирования органической фазы получали остаток, который очищали с помощью хроматографии на силикагеле с элюированием смесью этилацетат/гексан (1:4), получая требуемый арилсульфид в виде твердого вещества белого цвета (8,0 г, 100%).
Стадия Б: К раствору полученного на стадии А сложного этилового эфира (8,00 г, 15 ммоль) в этаноле (90 мл) и воде (20 мл) добавляли гидроксид натрия (6,1 г, 152 ммоль) и смесь нагревали в течение 16 ч до 65° С. Летучие органические компоненты удаляли в вакууме и образовавшуюся водную смесь подкисляли с помощью 2 н. HCl до рН 3-4. Добавляли твердый хлорид натрия и смесь экстрагировали этилацетатом. Объединенные органические экстракты промывали соляным раствором и сушили над сульфатом магния. После удаления растворителя получали требуемую карбоновую кислоту (4,92 г, 68%).
Стадия В: К раствору полученного на стадии Б карбоновой кислоты (4,92 г, 9,93 ммоль) в N,N-диметилформамиде (100 мл) добавляли 4-метилморфолин (1,52 г, 15,0 ммоль), N-гидроксибензотриазол (1,62 г, 12,0 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (2,70 г, 14,1 ммоль), а затем O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (2,24 г, 15,0 ммоль). После перемешивания в течение 16 ч при температуре окружающей среды реакционную смесь концентрировали, получая остаток, который растворяли в этилацетате (200 мл) и промывали водой и соляным раствором. После концентрирования и очистки с помощью хроматографии на силикагеле получали защищенный гидроксамат (4,9 мг, 83%).
Стадия Г: Охлажденный в ледяной бане раствор полученного на стадии В защищенного гидроксамата (4,9 г, 8,24 ммоль) в этилацетате (30 мл) барботировали в течение 10 мин газообразным хлористым водородом. Затем смеси давали выстояться в течение 2 ч при температуре окружающей среды, после чего растворитель удаляли в вакууме. Добавляли порцию свежего этилацетата (30 мл) и затем его удаляли в вакууме, после чего эту процедуру повторяли. Затем добавляли этилацетат (50 мл) и твердое вещество собирали фильтрацией, получая твердое вещество, которое очищали с помощью хроматографии с обращенной фазой, элюируя смесью ацетонитрил/вода (градиент от 20/80 до 100%-ного ацетонитрила), в результате чего получали указанное в заголовке соединение (1,9 г, 43%). Элементный анализ: рассчитано для C18H19FN2O4S2·HCl: С 48,37; Н 4,51; N 6,27; Cl 7,93; обнаружено: С 48,14; Н 4,33; N 6,21; Cl 8,64. MCBP (ESI) MH+: рассчитано для C18H19FN2S2: 411,0849, обнаружено: 411,0844.
Пример 434: Получение моногидрохлорида 4-[[4-[(2-фторфенил)тио]фенил]сульфонил]-N-гидрокси-1-(2-пропинил)-4-пиперидинкарбоксамида
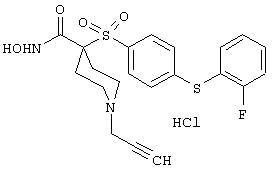
Стадия А: К раствору полученного на стадии Е примера 9 продукта (4,46 г, 12,6 ммоль) в N,N-диметилформамиде (30 мл) добавляли 2-фтортиофенол (1,94 г, 15,1 ммоль) и карбонат калия (2,09 г, 15,1 ммоль) и образовавшуюся суспензию перемешивали в течение 48 ч при температуре окружающей среды. Затем реакционную смесь разбавляли этилацетатом (200 мл) и промывали 1 н. раствором гидроксида натрия (200 мл) и соляным раствором (3х). После концентрирования органической фазы получали требуемый арилсульфид (5,2 г, 90%).
Стадия Б: К раствору полученного на стадии А сложного этилового эфира (5,1 г, 11,4 ммоль) в этаноле (90 мл) и воде (30 мл) добавляли гидроксид натрия (5,0 г, 125 ммоль) и смесь нагревали в течение 16 ч до 65° С. Органические компоненты удаляли в вакууме и образовавшуюся водную смесь подкисляли с помощью 2 н. HCl до рН 3-4. Добавляли твердый хлорид натрия и смесь экстрагировали этилацетатом. Объединенные органические экстракты промывали соляным раствором и сушили над сульфатом магния. После удаления растворителя получали требуемую карбоновую кислоту (4,5 г, 94%).
Стадия В: К раствору полученной на стадии Б карбоновой кислоты (4,5 г, 11,0 ммоль) в N,N-диметилформамиде (50 мл) добавляли 4-метилморфолин (1,62 мл, 16,0 ммоль), N-гидроксибензотриазол (1,73 г, 12,8 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (2,87 г, 14,9 ммоль), а затем O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (2,39 г, 16,0 ммоль). После перемешивания в течение 16 ч при температуре окружающей среды реакционную смесь концентрировали, получая остаток, который растворяли в этилацетате (200 мл) и промывали водой и соляным раствором. После концентрирования и очистки с помощью хроматографии на силикагеле получали защищенный гидроксамат, который непосредственно использовали на следующей стадии.
Стадия Г: Охлажденный в ледяной бане раствор полученного на стадии В защищенного гидроксамата в этилацетате (30 мл) барботировали в течение 10 мин газообразным хлористым водородом. Затем смеси давали выстояться в течение 2 ч при температуре окружающей среды, после чего растворитель удаляли в вакууме. Добавляли порцию свежего этилацетата (30 мл) и затем его удаляли в вакууме, после чего эту процедуру повторяли. Затем добавляли этилацетат (50 мл) и твердое вещество собирали фильтрацией, получая твердое вещество, которое очищали с помощью хроматографии с обращенной фазой, элюируя смесью ацетонитрил/вода (градиент от 20/80 до 100%-ного ацетонитрила), в результате чего получали указанное в заголовке соединение (1,85 г, 35% для стадий В и Г). МСВР (ESI) MH+: рассчитано для C21H21FN2O4S2: 449,1005, обнаружено: 449,1023.
Пример 435: Получение моногидрохлорида 4-[[4-(4-этоксифенокси)фенил]сульфонил]-N-гидрокси-1-(2-пропинил)-4-пиперидинкарбоксамида
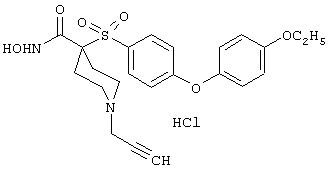
Стадия А: К раствору полученного на стадии Е примера 9 продукта (8,00 г, 22,6 ммоль) в N,N-диметилформамиде (50 мл) добавляли 4-этоксифенол (9,38 г, 70 ммоль) и карбонат цезия (22,8 г, 70 ммоль) и образовавшуюся суспензию выдерживали в течение 20 ч при 75° С. Затем реакционную смесь разбавляли этилацетатом (1000 мл) и промывали 1н. раствором гидроксида натрия, водой и соляным раствором. После концентрирования органической фазы получали остаток, который очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетат/гексан (1:2), в результате чего получали требуемый простой диариловый эфир (10,5 г, 99%).
Стадия Б: К раствору полученного на стадии А простого диарилового эфира (10,5 г, 22,3 ммоль) в этаноле (70 мл) и воде (60 мл) добавляли гидроксид натрия (8,9 г, 222 ммоль) и смесь нагревали в течение 16 ч до 65° С. Летучие органические компоненты удаляли в вакууме и образовавшуюся водную смесь подкисляли с помощью 2 н. HCl до рН 3-4. Добавляли твердый хлорид натрия и смесь экстрагировали этилацетатом. Объединенные органические экстракты промывали соляным раствором и сушили над сульфатом магния. После удаления растворителя получали требуемую карбоновую кислоту (10 г, 100%).
Стадия В: К раствору полученной на стадии Б карбоновой кислоты (10 г, 22,5 ммоль) в N,N-диметилформамиде (50 мл) добавляли 4-метилморфолин (3,42 мл, 33,8 ммоль), N-гидроксибензотриазол (3,66 г, 27,1 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (6,05 г, 31,6 ммоль), а затем O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (5,05 г, 33,8 ммоль). После перемешивания в течение 16 ч при температуре окружающей среды реакционную смесь концентрировали, получая остаток, который растворяли в этилацетате (200 мл) и промывали водой и соляным раствором. После концентрирования и очистки с помощью хроматографии на силикагеле с элюированием смесью этилацетат/гексан (1:1) получали защищенный гидроксамат (6,5 г, 53%), который непосредственно использовали на следующей стадии.
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата в смеси метанол/1,4-диоксан (1:3, 70 мл) добавляли смесь 4 н. HCl/1,4-диоксан (30 мл) и раствор перемешивали в течение 4 ч при температуре окружающей среды. Затем растворитель удаляли в вакууме. Добавляли метанол (40 мл) и затем его удаляли в вакууме. Добавляли диэтиловый эфир (100 мл) и образовавшееся твердое вещество собирали фильтрацией, получая указанное в заголовке соединение (4,3 г, 72%). Элементный анализ: рассчитано для C23H26N2O6S· HCl· Н2О: С 53,85; Н 5,70; N 5,46; Cl 6,91; S 6,25; обнаружено: С 53,65; Н 5,62; N 5,41; Cl 6,86; S 6,48. MC (ESI) MH+: рассчитано для C23H26N2O6S: 459, обнаружено: 459.
Пример 436: Получение моногидрохлорида N-гидрокси-4-[[4-[4-(метилсульфонил)фенокси]фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида
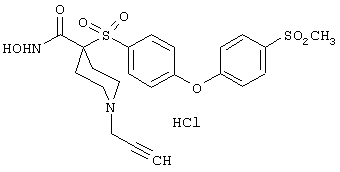
Стадия А: К раствору полученного на стадии Е примера 9 продукта (2,5 г, 6,4 ммоль) в N,N-диметилформамиде (15 мл) добавляли 4-метилсульфонилфенол (3,5 г, 20,3 ммоль) и карбонат цезия (8,7 г, 27 ммоль) и образовавшуюся суспензию выдерживали в течение 16 ч при 90° С. Затем реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате (500 мл) и промывали 1 н. раствором гидроксида натрия, водой и соляным раствором. После концентрирования органической фазы получали остаток, который очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетат/гексан (1:1), в результате чего получали требуемый простой диариловый эфир (2,5 г, 77%).
Стадия Б: К раствору полученного на стадии А простого диарилового эфира (2,5 г, 4,9 ммоль) в этаноле (50 мл) и воде (30 мл) добавляли гидроксид натрия (2,0 г, 49 ммоль) и смесь нагревали в течение 8 ч до 65° С. Растворители удаляли в вакууме. Добавляли воду (50 мл), смесь снова концентрировали в вакууме и образовавшуюся смесь подкисляли с помощью 2 н. HCl до рН 4-5. Добавляли твердый хлорид натрия и смесь экстрагировали этилацетатом. Твердый осадок собирали фильтрацией, получая требуемую карбоновую кислоту (1,57 г, 67%).
Стадия В: К раствору полученной на стадии Б карбоновой кислоты (1,57 г, 3,3 ммоль) в N,N-диметилформамиде (15 мл) добавляли 4-метилморфолин (0,5 г, 4,9 ммоль), N-гидроксибензотриазол (0,53 г, 3,9 ммоль) и гидрохлорид 1-[3-(диметиламино) пропил] -3-этилкарбодиимида (0,88 г, 4,6 ммоль), а затем O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (0,74 г, 4,9 ммоль). После перемешивания в течение 16 ч при температуре окружающей среды реакционную смесь концентрировали, получая остаток, который растворяли в этилацетате (200 мл) и промывали водой и соляным раствором. После концентрирования и очистки с помощью хроматографии на силикагеле с элюированием смесью этилацетат/гексан (1:1) получали защищенный гидроксамат (1,5 г, 79%), который непосредственно использовали на следующей стадии.
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (1,5 г, 2,60 ммоль) в смеси метанол/1,4-диоксан (1:3, 40 мл) добавляли смесь 4 н. HCl/1,4-диоксан (10 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. Затем растворитель удаляли в вакууме. Добавляли метанол (30 мл) и затем его удаляли в вакууме. Добавляли диэтиловый эфир (100 мл) и образовавшееся твердое вещество собирали фильтрацией, получая указанное в заголовке соединение (1,35 г, 98%). Элементный анализ: рассчитано для C22H24N2O7S2·HCl: С 49,95; Н 4,76; N 5,30; Cl 6,70; S 12,12; обнаружено: С 49,78; Н 4,56; N 5,25; Cl 6,98; S 11,98. MCBP (ESI) MH+: рассчитано для С22Н24N2O7S2: 493,1103, обнаружено: 493,1116.
Пример 437: Получение моногидрохлорида N-гидрокси-4-[[4-[(фенилметил)амино]фенил]сульфонил]-1-(2-пропинил)-4-пиперидинкарбоксамида
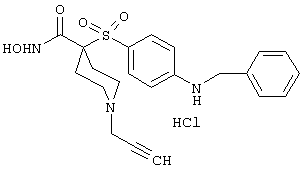
Стадия А: К раствору полученного на стадии Е примера 9 продукта (2,5 г, 6,4 ммоль) в N,N-диметилформамиде (30 мл) добавляли бензиламин (3,44 г, 32,1 ммоль) и карбонат цезия (10,5 г, 32,3 ммоль) и образовавшуюся суспензию выдерживали в течение 16 ч при 100° С. Затем реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате (500 мл) и промывали водой и соляным раствором и сушили над сульфатом магния. После концентрирования органической фазы получали остаток, который очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетат/гексан (1:1), в результате чего получали требуемый бензиланилин (2,5 г, 88%).
Стадия Б: К раствору полученного на стадии А бензиланилина (2,5 г, 5,67 ммоль) в этаноле (50 мл) и воде (30 мл) добавляли гидроксид натрия (2,27 г, 56,7 ммоль) и смесь нагревали в течение 8 ч до 65° С. Растворители удаляли в вакууме. Добавляли воду (50 мл), смесь снова концентрировали в вакууме и образовавшуюся смесь подкисляли с помощью 2 н. HCl до рН 4-5. Твердый осадок собирали фильтрацией и промывали диэтиловым эфиром, получая требуемую карболовую кислоту (2,3 г, 98%).
Стадия В: К раствору полученной на стадии Б карбоновой кислоты (2,3 г, 5,57 ммоль) в N,N-диметилформамиде (15 мл) добавляли 4-метилморфолин (0,85 г, 8,36 ммоль), N-гидроксибензотриазол (0,9 г, 6,69 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (1,5 г, 7,8 ммоль), а затем O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (1,25 г, 8,36 ммоль). После перемешивания в течение 16 ч при температуре окружающей среды реакционную смесь концентрировали, получая остаток, который растворяли в этилацетате и промывали водой и соляным раствором. После концентрирования и очистки с помощью хроматографии на силикагеле с элюированием смесью этилацетат/гексан получали защищенный гидроксамат, который непосредственно использовали на следующей стадии.
Стадия Г: Раствор полученного на стадии В защищенного гидроксамата в этилацетате (50 мл), охлажденный в ледяной бане, барботировали в течение 10 мин газообразным хлористым водородом. Затем растворитель удаляли в вакууме. Добавляли этилацетат (100 мл) и затем его удаляли в вакууме. После этого добавляли этилацетат (100 мл) и образовавшееся твердое вещество собирали фильтрацией, получая указанное в заголовке соединение (1,6 г, 62% для стадий В и Г). МСВР (ESI) МН+: рассчитано для С22Н25N3O4S: 428,1644, обнаружено: 428,1652.
Пример 438: Получение моногидрохлорида 1-этил-N-гидрокси-4-[[4-[[4-(трифторметил)фенил]метокси]фенил]сульфонил]-4-пиперидинкарбоксамида
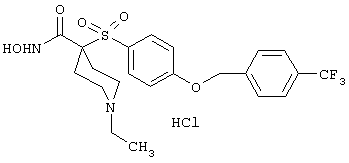
Стадия А: К раствору полученного на стадии Б примера 429 продукта (1,0 г, 2,9 ммоль) в N,N-диметилацетамиде (30 мл) добавляли 4-(трифторметил) бензиловый спирт (1,53 г, 8,74 ммоль) и карбонат цезия (2,85 г, 8,74 ммоль) и образовавшуюся суспензию выдерживали в течение 8 ч при 95-100° С. Затем реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате и промывали 1н. раствором гидроксида натрия, водой и соляным раствором. После концентрирования органической фазы получали остаток, который очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетат/гексан, в результате чего получали требуемый простой ариловый эфир (0,8 г, 54%).
Стадия Б: К раствору полученного на стадии А простого арилового эфира (0,8 г, 1,5 ммоль) в этаноле (50 мл) и воде (50 мл) добавляли гидроксид натрия (1,0 г, 25 ммоль) и смесь нагревали в течение 16 ч до 60° С. Растворители удаляли в вакууме. Добавляли воду (50 мл) и смесь подкисляли с помощью 2 н. HCl до рН 4. Твердый осадок собирали фильтрацией, получая требуемую карбоновую кислоту (0,75 г, 99%).
Стадия В: К раствору полученной на стадии Б карбоновой кислоты (0,75 г, 1,54 ммоль) в N,N-диметилформамиде (10 мл) добавляли 4-метилморфолин (0,47 г, 4,6 ммоль), N-гидроксибензотриазол (0,25 г, 1,85 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,41 г, 2,16 ммоль), а затем O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (0,35 г, 2,3 ммоль). После перемешивания в течение 16 ч при температуре окружающей среды реакционную смесь концентрировали, получая остаток, который растворяли в этилацетате (200 мл) и промывали водой и соляным раствором. После концентрирования и очистки с помощью хроматографии на силикагеле с элюированием смесью этилацетат/гексан получали защищенный гидроксамат (250 мг, 57%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (250 мг, 0,43 ммоль) в смеси метанол/1,4-диоксан (1:3, 20 мл) добавляли смесь 4 н. HCl/1,4-диоксан (5 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. Затем растворитель удаляли в вакууме. Добавляли дополнительную порцию этилацетата и затем его удаляли в вакууме. Добавляли диэтиловый эфир (100 мл) и образовавшееся твердое вещество собирали фильтрацией, получая указанное в заголовке соединение (190 мг, 82%). МС (CI) MH+: рассчитано для С22Н25F3N2O5S: 487, обнаружено: 487.
Пример 439: Получение моногидрохлорида 1-циклопропил-N-гидрокси-4-[[4-[4-(1-метилэтокси)фенокси]фенил]сульфонил]-4-пиперидинкарбоксамида
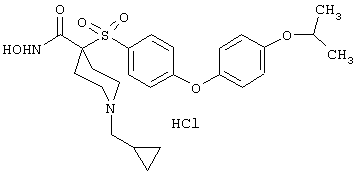
Стадия А: К раствору полученного на стадии А примера 398 продукта (2,49 г, 7,0 ммоль) в N,N-диметилацетамиде (30 мл) добавляли 4-изопропоксифенол (1,28 г, 8,4 ммоль), который может быть получен согласно способу, описанному в J. Indian Chem. Soc. 73, 1996, 507-511, и карбонат цезия (5,48 г, 16,8 ммоль) и образовавшуюся суспензию выдерживали в течение 16 ч при 60° С. Затем реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате и промывали 1 н. раствором гидроксида натрия, водой и соляным раствором. После концентрирования органической фазы получали остаток, который очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетат/гексан, в результате чего получали требуемый простой ариловый эфир (2,8 г, 82%).
Стадия Б: К раствору полученного на стадии А простого арилового эфира (2,8 г, 5,7 ммоль) в этаноле (50 мл) и воде (50 мл) добавляли гидроксид натрия (2,3 г, 57 ммоль) и смесь нагревали в течение 16 ч до 60° С. Растворители удаляли в вакууме. Добавляли воду (50 мл) и смесь подкисляли с помощью 2 н. HCl до рН 4. Твердый осадок собирали фильтрацией, получая требуемую карбоновую кислоту (1,4 г, 53%).
Стадия В: К раствору полученной на стадии Б карбоновой кислоты (1,4 г, 3,1 ммоль) в N,N-диметилформамиде (15 мл) добавляли 4-метилморфолин (0,92 г, 9,1 ммоль), N-гидроксибензотриазол (0,49 г, 3,66 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,82 г, 4,26 ммоль), а затем O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (0,68 г, 4,5 ммоль). После перемешивания в течение 16 ч при температуре окружающей среды реакционную смесь концентрировали, получая остаток, который растворяли в этилацетате и промывали водой и соляным раствором. После концентрирования и очистки с помощью хроматографии на силикагеле с элюированием смесью этилацетат/гексан получали защищенный гидроксамат, который непосредственно использовали на следующей стадии.
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата в смеси метанол/1,4-диоксан (1:3, 20 мл) добавляли смесь 4 н. HCl/1,4-диоксан (10 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. Затем растворитель удаляли в вакууме. Добавляли дополнительную порцию этилацетата и затем его удаляли в вакууме. Добавляли диэтиловый эфир и образовавшееся твердое вещество собирали фильтрацией, получая указанное в заголовке соединение (0,3 г, 19% для стадий В и Г в сумме). Элементный анализ: рассчитано для C24H30N2O6S· HCl: С 56,41; Н 6,11; N 5,48; обнаружено: С 56,04; Н 5,82; N 5,44. МС (CI) MH+: рассчитано для С24Н30N2О6S: 475, обнаружено: 475.
Пример 440: Получение моногидрохлорида 4-[[4-[[2-(4-хлорфенил)этил]амино]фенил]сульфонил]-1-этил-N-гидрокси-4-пиперидинкарбоксамида
Стадия А: К раствору полученного на стадии Б примера 429 продукта (1,0 г, 2,91 ммоль) в N,N-диметилацетамиде (20 мл) добавляли 4-хлорфенэтиламин (0,91 г, 5,8 ммоль) и карбонат цезия (3,80 г, 11,6 ммоль) и образовавшуюся суспензию выдерживали в течение 24 ч при 90° С. Затем реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате и промывали 1 н. раствором гидроксида натрия, водой и соляным раствором. После концентрирования органической фазы получали остаток, который очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетат/гексан, в результате чего получали требуемый простой ариловый эфир (0,8 г, 58%).
Стадия Б: К раствору полученного на стадии А простого арилового эфира (0,8 г, 1,7 ммоль) в этаноле (50 мл) и воде (50 мл) добавляли гидроксид натрия (1,0 г, 25 ммоль) и смесь нагревали в течение 16 ч до 60° С. Растворители удаляли в вакууме. Добавляли воду (50 мл) и смесь подкисляли с помощью 2 н. HCl до рН 4. Твердый осадок собирали фильтрацией, получая требуемую карбоновую кислоту (0,75 г, 92%).
Стадия В: К раствору полученной на стадии Б карбоновой кислоты (0,75 г, 1,7 ммоль) в N,N-диметилформамиде (20 мл) добавляли 4-метилморфолин (0,51 г, 5,1 ммоль), N-гидроксибензотриазол (0,27 г, 2,0 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,45 г, 2,3 ммоль), а затем O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (0,37 г, 2,5 ммоль). После перемешивания в течение 16 ч при температуре окружающей среды реакционную смесь концентрировали, получая остаток, который растворяли в этилацетате и промывали водой и соляным раствором. После концентрирования и очистки с помощью хроматографии на силикагеле с элюированием смесью этилацетат/гексан получали защищенный гидроксамат, который непосредственно использовали на следующей стадии.
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата в смеси метанол/1,4-диоксан (1:3, 20 мл) добавляли смесь 4 н. HCl/1,4-диоксан (10 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. Затем растворитель удаляли в вакууме. Добавляли дополнительную порцию этилацетата и затем его удаляли в вакууме. Добавляли диэтиловый эфир и образовавшееся твердое вещество собирали фильтрацией, получая указанное в заголовке соединение (30 мг, 4% в сумме для стадий В и Г).
Пример 441: Получение моногидрохлорида N-гидрокси-1-(2-метоксиэтил)-4-[[4-[[[4-(трифторметокси)фенил]метил]амино]фенил]сульфонил]-4-пиперидинкарбоксамида
Стадия А: К раствору этил-4-[(4-фторфенилсульфонил)]-1-(2-метоксиэтил)-4-пиперидинкарбоксилата (1,38 г, 3,7 ммоль) в N,N-диметилформамиде (20 мл) добавляли 4-(трифторметилокси)бензиламин (1,0 г, 5,2 ммоль) и карбонат цезия (1,7 г, 5,2 ммоль) и образовавшуюся суспензию выдерживали в течение 24 ч при 90° С. Затем реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате и промывали 1н. раствором гидроксида натрия, водой и соляным раствором. После концентрирования органической фазы получали остаток, который очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетат/гексан, в результате чего получали требуемое трифторметоксипроизводное (0,6 г, 30%).
Стадия Б: К раствору полученного на стадии А трифторметоксипроизводного (0,6 г, 1,1 ммоль) в этаноле (30 мл), воде (30 мл) и тетрагидрофуране (15 мл) добавляли гидроксид натрия (0,44 г, 11 ммоль) и смесь нагревали в течение 16 ч до 60° С. Растворители удаляли в вакууме. Добавляли воду (50 мл) и смесь подкисляли с помощью 2 н. HCl до рН 4. Твердый осадок собирали фильтрацией, получая требуемую карбоновую кислоту (0,5 г, 88%).
Стадия В: К раствору полученной на стадии Б карбоновой кислоты (0,50 г, 0,98 ммоль) в N,N-диметилформамиде (10 мл) добавляли 4-метилморфолин (0,15 г, 1,5 ммоль), N-гидроксибензотриазол (0,16 г, 1,2 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,27 г, 1,4 ммоль), а затем O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (0,22 г, 1,5 ммоль). После перемешивания в течение 16 ч при температуре окружающей среды реакционную смесь концентрировали, получая остаток, который растворяли в этилацетате и промывали водой и соляным раствором. После концентрирования и очистки с помощью хроматографии на силикагеле с элюированием смесью этилацетат/гексан получали защищенный гидроксамат (110 мг, 18%).
Стадия Г: К раствору полученного на стадии В защищенного гидроксамата (110 мг, 0,18 ммоль) в смеси метанол/1,4-диоксан (1:4, 20 мл) добавляли смесь 4 н. HCl/1,4-диоксан (7 мл) и раствор перемешивали в течение 3 ч при температуре окружающей среды. Затем растворитель удаляли в вакууме. Добавляли дополнительную порцию метанола (20 мл) и затем его удаляли в вакууме. Добавляли диэтиловый эфир и образовавшееся твердое вещество собирали фильтрацией, получая указанное в заголовке соединение (30 мг, 31%). МСВР (ESI) МН+: рассчитано для C23H28F3N3O6S: 532, обнаружено: 532.
Пример 442: Получение моногидрохлорида N-гидрокси-4-[[4-[4-(1-метилэтокси)фенокси]фенил]сульфонил]-1-(2-метоксиэтил)-4-пиперидинкарбоксамида
Стадия А: К раствору этил-4-[(4-фторфенилсульфонил)]-1-(2-метоксиэтил)-4-пиперидинкарбоксилата (2,0 г, 5,4 ммоль) в N,N-диметилформамиде (20 мл) добавляли 4-изопропоксифенол (1,63 г, 10,7 ммоль), который может быть получен по методу, описанному в J. Indian Chem. Soc. 73, 1996, 507-511, и карбонат цезия (7 г, 21,5 ммоль) и образовавшуюся суспензию выдерживали в течение 16 ч при 60° С. Затем реакционную смесь концентрировали в вакууме. Остаток растворяли в этилацетате и промывали 1н. раствором гидроксида натрия, водой и соляным раствором и сушили над сульфатом магния. После концентрирования органической фазы получали остаток, который очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетат/гексан, в результате чего получали требуемый простой ариловый эфир (1,37 г, 50%).
Стадия Б: К раствору полученного на стадии А простого арилового эфира (1,37 г, 2,7 ммоль) в этаноле (30 мл) и воде (30 мл) добавляли гидроксид натрия (1,08 г, 27 ммоль) и смесь нагревали в течение 16 ч до 65° С. Растворители удаляли в вакууме. Добавляли воду (50 мл) и смесь снова концентрировали в вакууме, затем образовавшуюся смесь подкисляли с помощью 2 н. HCl до рН 4-5. Твердый осадок собирали фильтрацией и промывали диэтиловым эфиром, получая требуемую карбоновую кислоту (1,25 г, 100%).
Стадия В: К суспензии полученной на стадии Б карбоновой кислоты (1,25 г, 2,7 ммоль) в N,N-диметилформамиде (15 мл) добавляли 4-метилморфолин (0,82 г, 8,1 ммоль), O-(тетрагидро-2Н-пиран-2-ил)гидроксиламин (0,61 г, 4,1 ммоль), а затем гексафторфосфат бромтриспирролидинофосфония (РуВrоР, 1,51 г, 3,3 ммоль). После перемешивания в течение 16 ч при температуре окружающей среды реакционную смесь концентрировали, получая остаток, который растворяли в этилацетате и промывали водой и соляным раствором. После концентрирования и очистки с помощью хроматографии на силикагеле с элюированием смесью этилацетат/гексан получали защищенный гидроксамат (1,0 г, 63%).
Стадия Г: Охлажденный в ледяной бане раствор полученного на стадии В защищенного гидроксамата (1,0 г, 1,7 ммоль) в этилацетате (20 мл) барботировали в течение 5 мин газообразным хлористым водородом. После перемешивания в течение 5 ч при температуре окружающей среды растворитель удаляли в вакууме. Добавляли этилацетат (30 мл) и затем его удаляли в вакууме. Еще раз добавляли этилацетат (30 мл) и образовавшееся твердое вещество собирали фильтрацией, получая указанное в заголовке соединение (0,5 г, 56%). Элементный анализ: рассчитано для C24H32N2O7S· HCl· 1,5Н2О: С 51,84; Н 6,53; N 5,04; Cl 6,38; S 5,77; обнаружено: С 51,87; Н 6,12; N 4,92; Cl 6,38; S 5,84. МС МН+: рассчитано для C24H32N2O7S: 493, обнаружено: 493.
Пример 443: Получение дигидрохлорида N-гидрокси-1-(2-пиридинилметил)-4-[4-(4-трифторметоксифенокси)фенил]сульфонил]-4-пиперидинкарбоксамида

Стадия А: Полученный на стадии Г примера 9 арилфторид (6,22 г, 15 ммоль) смешивали с карбонатом калия (3,04 г, 22 ммоль), 4-(трифторметокси)фенолом (3,92 г, 322 ммоль) и N,N-диметилфомамидом (7 мл) и смесь перемешивали в течение 16 ч при 90° С. Добавляли дополнительные порции 4-(трифторметокси) фенола (1 г) и карбоната калия (800 мг) и реакцию проводили еще в течение 20 ч при 115° С. Смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (100 мл, затем 2× 25 мл). Объединенные органические слои сушили над сульфатом магния, концентрировали и хроматографировали, получая требуемый простой ариловый эфир в виде масла (6,9 г, почти количественный выход).
Стадия Б: Полученный на стадии А простой ариловый эфир (9,6 г, приблизительно 15 ммоль) растворяли в этилацетате (45 мл). Добавляли раствор HCl в диоксане (4 н., 12 мл) и смесь перемешивали в течение 3 ч при температуре окружающей среды. После этого анализ с помощью тонкослойной хроматографии показал, что удаление защитных групп было проведено не полностью. Добавляли концентрированный водный раствор HCl (4 мл) и реакционную смесь несколько раз нагревали до температуры дефлегмации с помощью фена. Раствор концентрировали и затем подвергали азеотропии с ацетонитрилом, получая требуемый гидрохлорид пиперидина в виде пены (9,6 г). Спектроскопический анализ методом ядерного магнитного резонанса показал наличие небольшого количества примеси в виде 4-(трифторметокси) фенола, который добавляли на стадии А.
Гидрохлорид пиперидина (6,0 г) растворяли в этилацетате (125 мл) и промывали водным раствором гидроксида натрия (2 г NaOH в 50 мл воды). Органический слой сушили над сульфатом магния и фильтровали через подушку из силикагеля. Таким образом элюировали фенольные примеси. После этого требуемый пиперидин выделяли из осадка на фильтре путем элюирования метанолом, содержащим 1%-ный водный раствор гидроксида аммония (приблизительно 100 мл). Фильтрат концентрировали и подвергали азеотропии с ацетонитрилом, получая 3,3 г продукта (7,3 ммоль).
Стадия В: Полученный на стадии Б пиперидин (1,24 г, 2,7 ммоль) объединяли с порошкообразным карбонатом калия (828 мг, 6,0 ммоль), гидрохлоридом 2-пиколила (492 мг, 3,0 ммоль) и N,N-диметилфомамидом (3 мл), после чего смесь перемешивали в течение 2 ч при температуре окружающей среды, затем выдерживали еще в течение 2 ч при 50° С. Смесь разбавляли водой (40 мл) и экстрагировали этилацетатом (150 мл, затем 50 мл). Объединенные органические слои сушили над сульфатом магния, концентрировали и хроматографировали, получая требуемый сложный эфир в виде масла (1,13 г, 74%).
Стадия Г: Полученный на стадии В сложный эфир (1,1 г, 2,0 ммоль) смешивали с этанолом (6 мл), водой (2 мл) и гидроксидом калия (0,90 г, 16 ммоль). Смесь нагревали до температуры дефлегмации и выдерживали в течение 4,5 ч. Затем раствор охлаждали до 0° С и подкисляли с помощью концентрированного водного раствора соляной кислоты. Растворитель удаляли и образовавшиеся твердые частицы сушили путем азеотропии с ацетонитрилом. Продукт выдерживали в вакууме до достижения постоянной массы.
После этого неочищенный гидрохлорид перемешивали с 4-метилморфолином (приблизительно 0,5 мл), 1-гидроксибензотриазолом (0,405 г, 3 ммоль), O-(тетрагидропиранил)гидроксиламином (0,35 г, 3,0 ммоль) и N,N-диметилформамидом (9 мл). Через 10 мин добавляли гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (0,57 г, 3,0 ммоль) и смесь перемешивали в течение ночи. Затем реакционную смесь разбавляли полунасыщенным водным раствором бикарбоната натрия (50 мл) и экстрагировали этилацетатом (100 мл, затем 25 мл). Объединенные органические слои сушили над сульфатом магния, концентрировали и хроматографировали (смесь этилацетат: метанол, 9:1), получая требуемый тетрагидропиранил-защищенный гидроксамат в виде масла желтого цвета (1,20 г, 95%).
Стадия Д: Полученный на стадии В тетрагидропиранил-защищенный гидроксамат (1,20 г, 1,90 ммоль) разбавляли метанолом (9 мл). В течение 2 мин добавляли ацетилхлорид (0,78 мл, 11 ммоль). Реакционную смесь перемешивали в течение 2 ч при температуре окружающей среды, затем концентрировали, получая требуемый дигидрохлорид (1,20 г, количественный выход) в виде кристаллического вещества белого цвета. Элементный анализ: рассчитано для С25H24F3N3O6S· 0,2НСl· 1/3Н2О:C 47,58; H 4,07; N 6,66; обнаружено: С 47,31; Н 4,14; N 6,80.
Пример 444: Ингибирование металлопротеаз in vitro
Активность соединений, полученных описанными выше в примерах способами, оценивали с помощью опыта in vitro по методам, описанным Knight и др., FEBS Lett. 296(3): 263 (1992). В целом этот опыт состоял в том, что 4-аминофенилртутьацетат (АФРА) или активируемые трипсином ММП инкубировали с различными концентрациями соединения-ингибитора при комнатной температуре в течение 5 мин.
В частности, рекомбинантные человеческие ферменты ММП-13, ММП-1, ММП-2 и ММП-9 получали в лабораторных условиях по обычным лабораторным методам. ММП-13 из полноразмерного клона кДНК экспрессировали в виде профермента с использованием бакуловирусов, как это описано у V.A. Luckow, Insect Cell Expression Technology, стр. 183-218 в Protein Engineering: Principles and Practice, под ред. J.L. Cleland и др., Wiley-Liss Inc. (1996). Более подробно применение систем экспрессии бакуловирусов описано также у Luckow и др., J. Virol. 67, 4566-4579 (1993); у O’Reilly и др., Baculovirus Expression Vectors: A Laboratory Manual, W.H. Freeman и Companym New York, (1992); у King и др. в The Baculovirus Expression Vectors: A Laboratory Guide, Chapman и Hall, London (1992). Экспрессированный фермент очищали сначала с помощью гепариновой агарозной колонки и затем с помощью хелатирующей колонки с хлоридом цинка. Для использования в опыте профермент активировали АФРА.
МММ-1, экспрессированная в трансфектированных клетках линии НТ-1080, была предоставлена доктором Harold Welgus из Вашингтонского университета, С.-Луис, шт. Миссури. Фермент также активировали с помощью АФРА и затем очищали с помощью колонки с гидроксамовой кислотой. Доктором Welgus также были предоставлены трансфектированные клетки линии НТ-1080, которые экспрессировали ММП-9. Трансфектированные клетки, экспрессирующие ММП-2, были получены от доктора Gregory Golgberg также из Вашингтонского университета. Данные исследований, проведенных с использованием ММП в присутствии 0,02%-ного меркаптоэтанола, приведены ниже в таблице и помечены звездочкой. Более подробно получение и применение этих ферментов описано в научной литературе, касающейся этих ферментов (см., например, Enzyme Nomenclature, Academic Press, San Diego, CA (1992) и в указанных в этой публикации ссылках, а также у Frije и др., J. Biol. Chem., 26(24): 16766-16773 (1994). Субстратом для ферментов является полипептид, содержащий метоксикумарин, который имеет следующую последовательность:
MCA-ProLeuGlyLeuDpaAlaArgNH2, где МСА обозначает метоксикумарин, а Dpa обозначает 3-(2,4-динитрофенил)-L-2,3-диаминопропионилаланин. Этот субстрат поставляется на рынок фирмой Baychem как продукт М-1895.
Используемый для опытов буфер содержал 100 мМ Трис-HCl, 100 мМ NaCl, 10 мМ СаСl2 и 0,5% лаурилового эфира полиэтиленгликоля(23), рН 7,5. Опыты проводили при комнатной температуре и для растворения соединения-ингибитора использовали диметилсульфоксид (ДМСО) в конечной концентрации 1%.
Активность соединения-ингибитора в растворе ДМСО/буфер оценивали с помощью планшетов типа Microfluor™ White Plates (фирма Dynatech) в сравнении с не обладающим ингибирующим действием контролем, в качестве которого использовали такое же количество раствора ДМСО/буфер. Раствор с ингибитором или контрольный раствор выдерживали в планшете в течение 10 мин и субстрат добавляли в таком количестве, чтобы получить конечную концентрацию 4 мкМ.
В отсутствие ингибирующей активности флуорогенный пептид расщеплялся по пептидной связи Gly-Leu, а отделение пептида с выраженной флуорогенной активностью от соединения-гасителя 2,4-динитрофенола приводило к увеличению интенсивности флуоресценции (возбуждение при 328 нм/испускание при 415 нм). Ингибирование оценивали по уменьшению интенсивности флуоресценции в виде функции от концентрации ингибитора, используя планшет-ридер типа L550 фирмы Perkin Elmer. Из этих данных рассчитывали значения IC50. Результаты в виде значений IC50, полученные соответственно по трем достоверным повторностям, приведены ниже в таблицах А и Б, в которых обобщены данные об ингибировании.
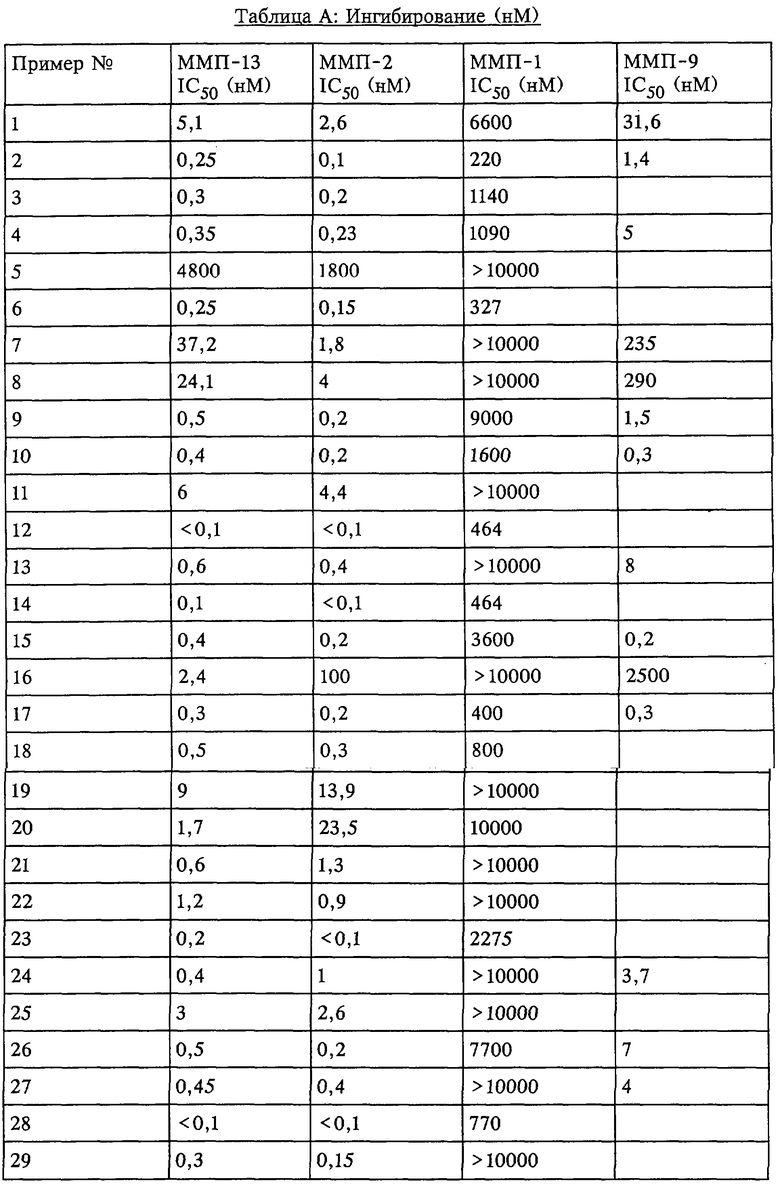
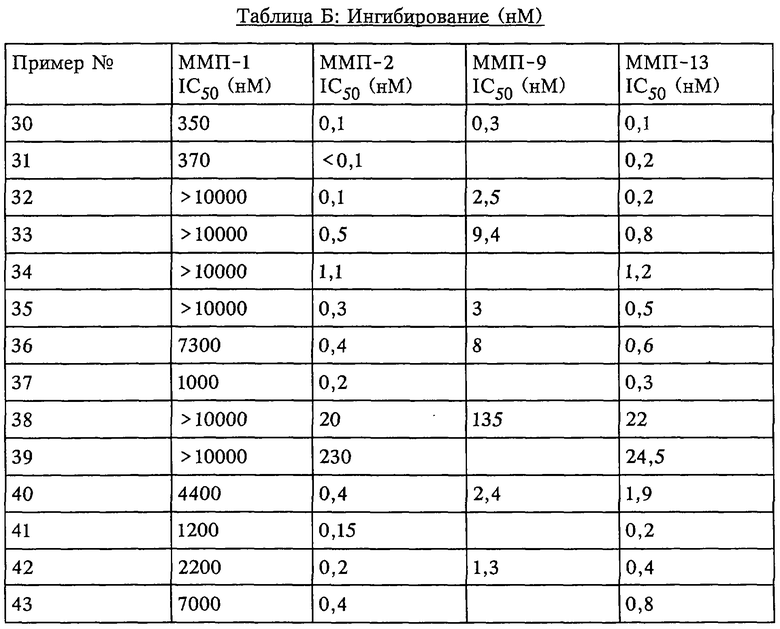
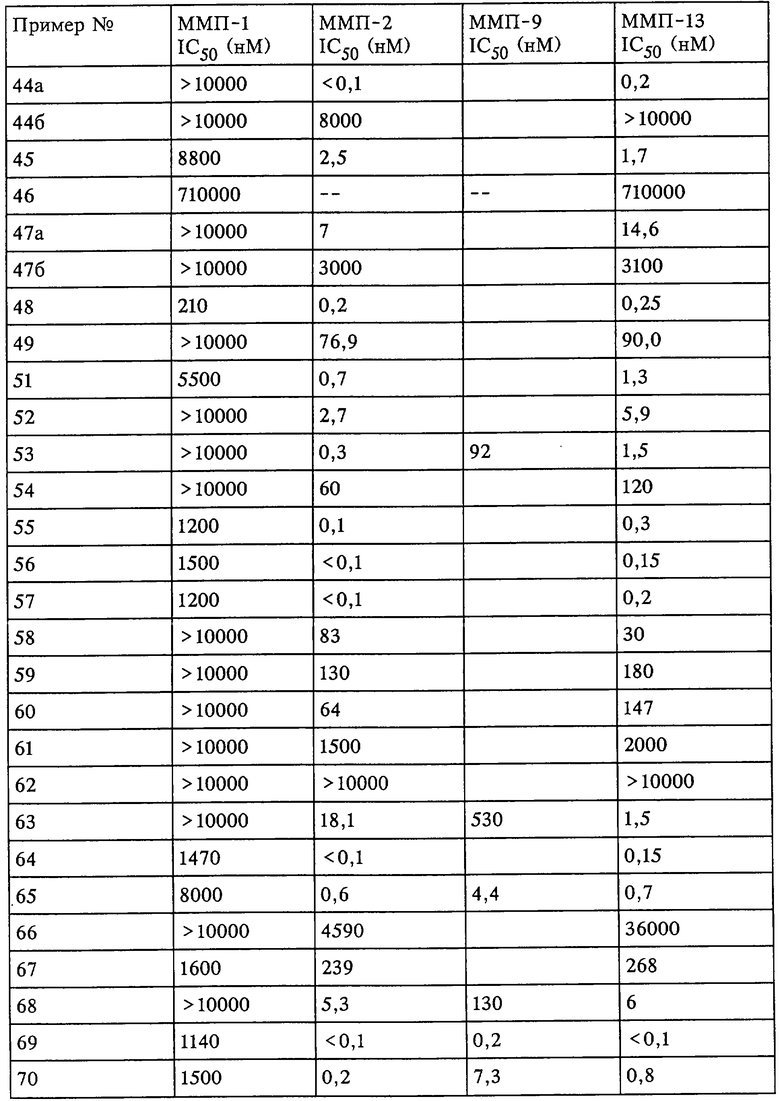
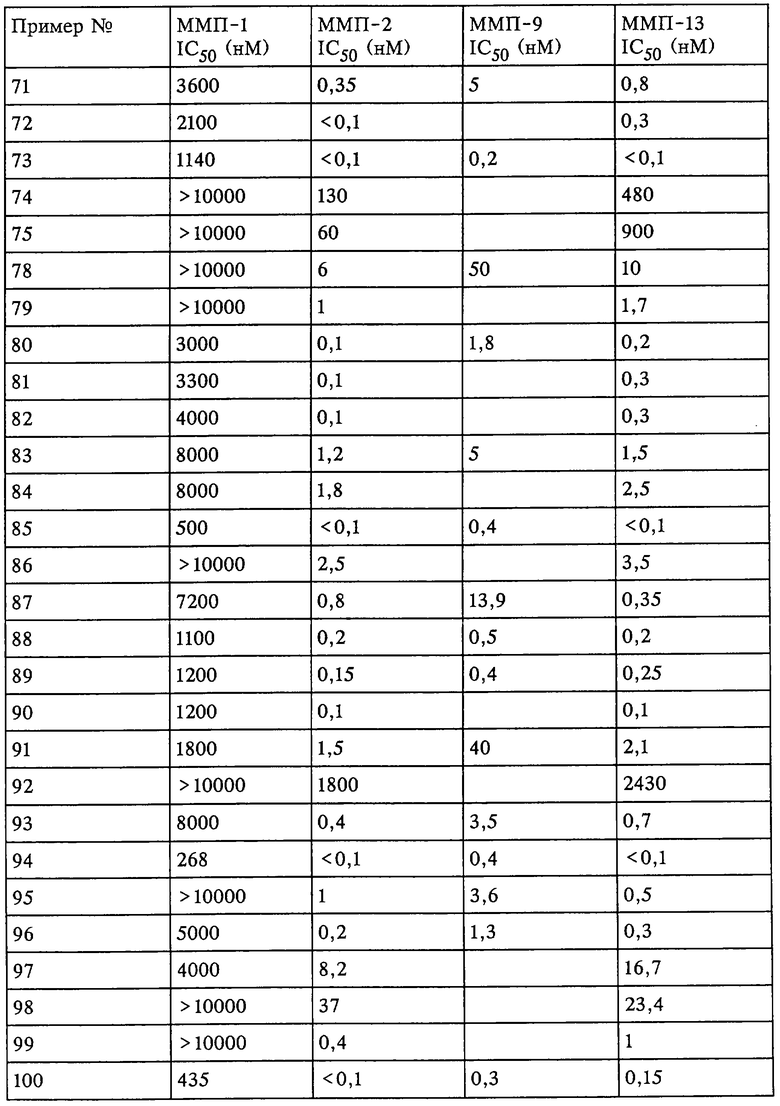
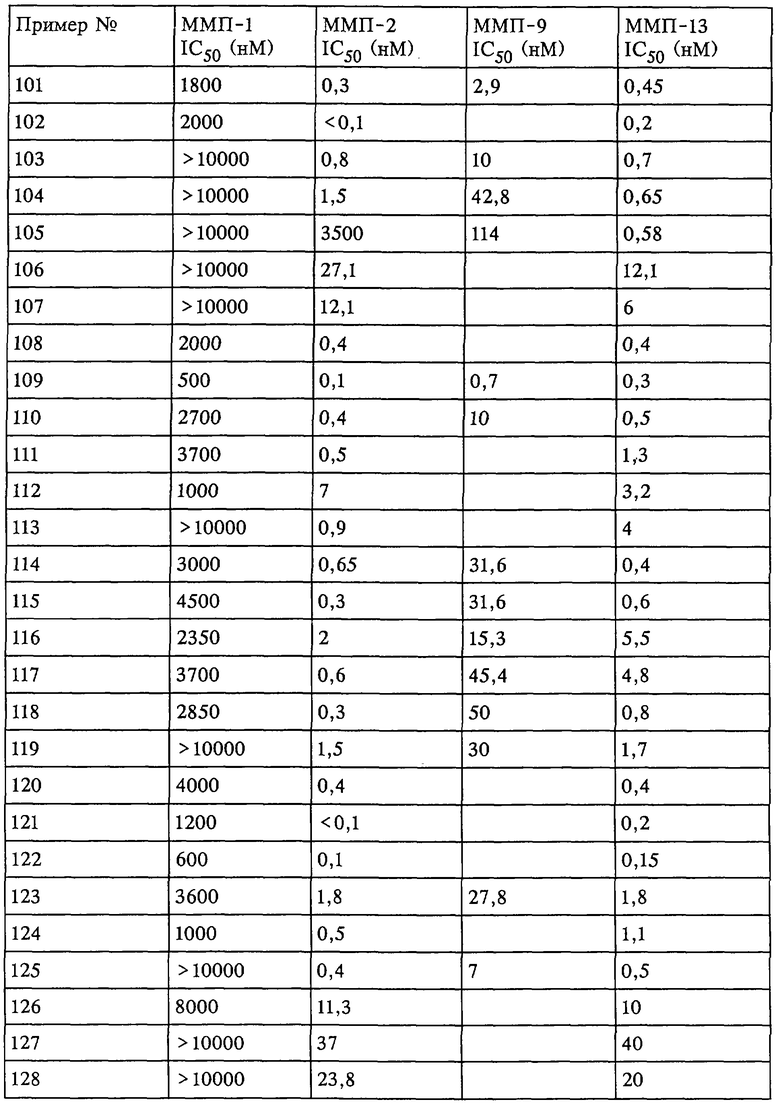
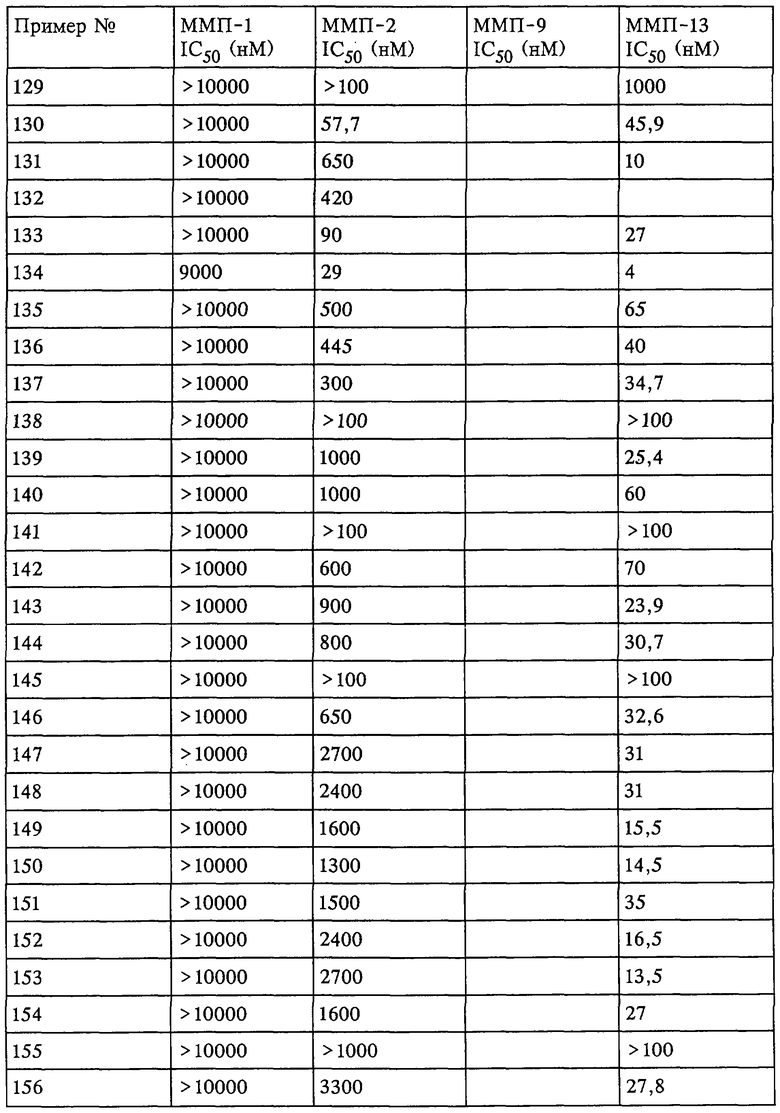
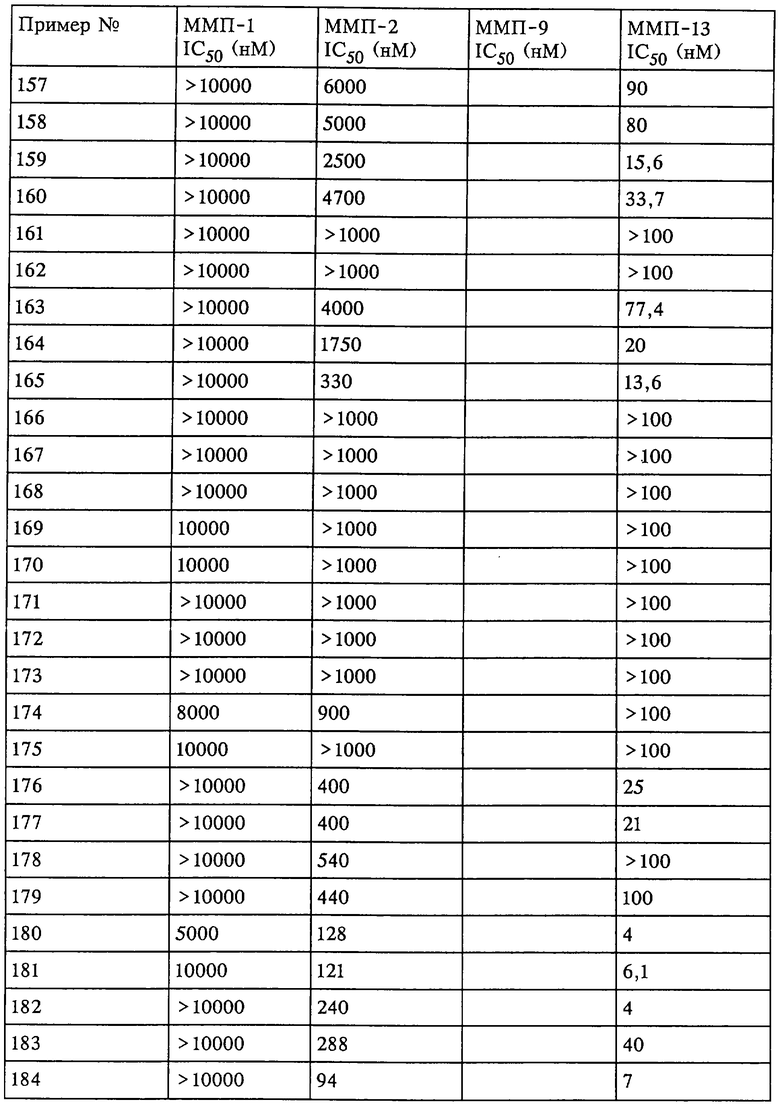
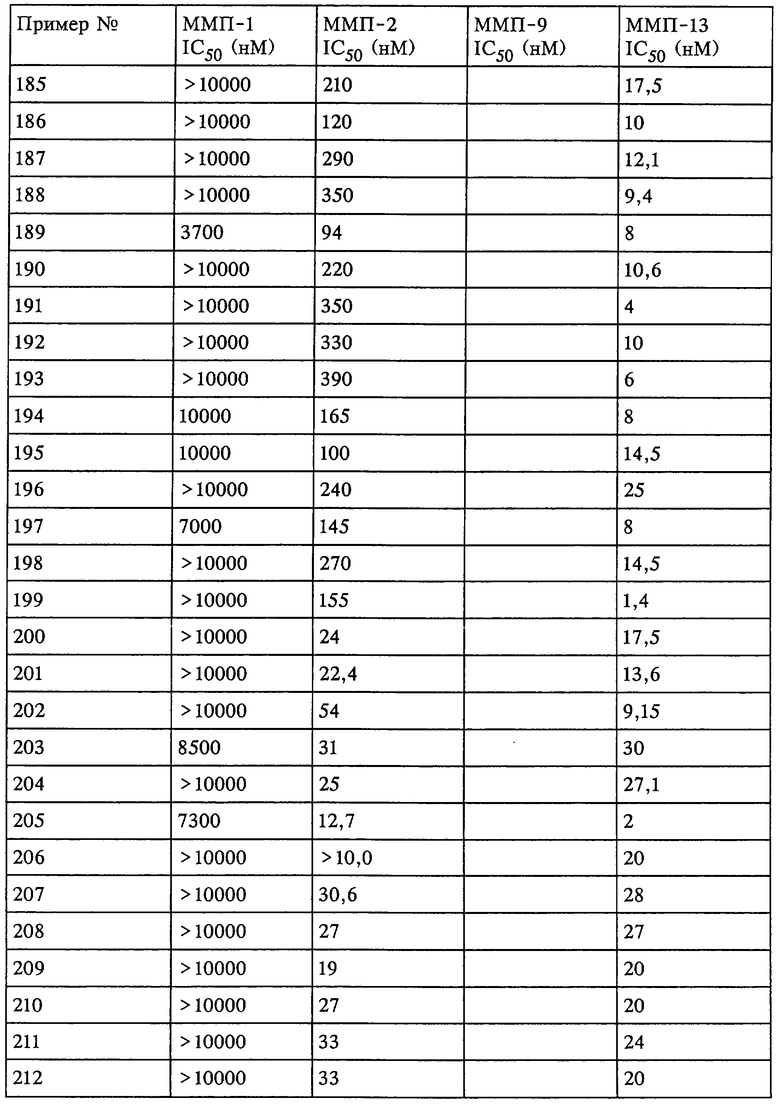
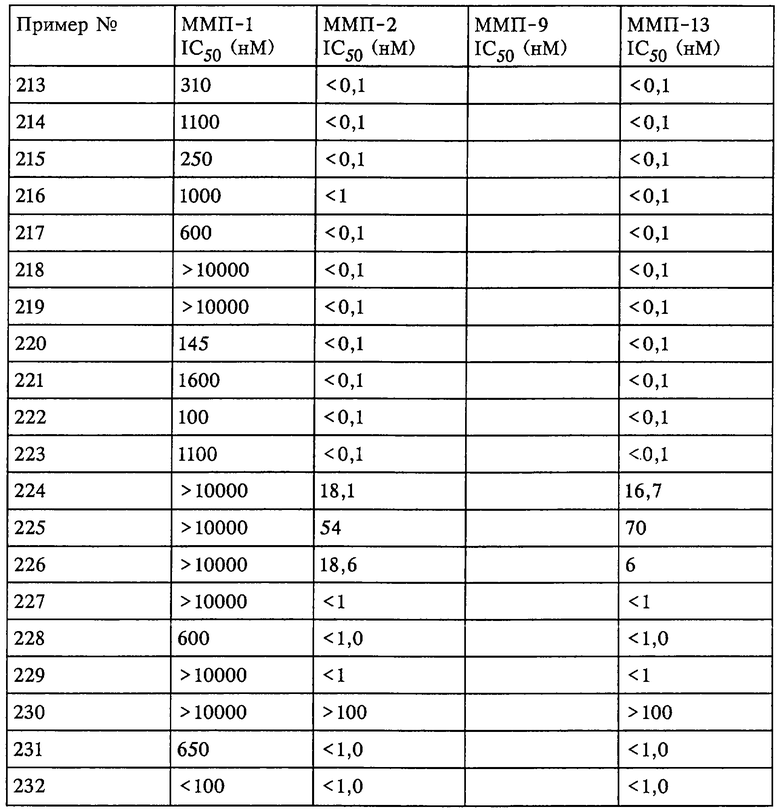
Пример 445: Оценка ангиогенеза in vivo
Исследование ангиогенеза зависит от надежности и воспроизводимости модели стимуляции и ингибирования неоваскулярной реакции. Такой моделью, позволяющей изучать ангиогенез, является микрокарманчик в роговице мыши (см. Kenyon B.M. и др., A Model of Angiogenesis in the Mouse Cornea, Investigative Ophthalmology & Visual Science, июль 1996, том 37, №8).
В этом опыте получали гранулы одинакового размера на основе Hydron™ , содержащие bFGF и сукралфат (комплекс гидросульфата октакиссахарозы с алюминием), и хирургическим путем имплантировали в строму роговицы мыши по соседству с височным лимбом. Для получения гранул готовили суспензию, содержащую в 20 мкл стерильного физиологического раствора 10 мкг рекомбинантного bFGF, 10 мг сукралфата и 10 мкл 12%-ного Hydron™ в этаноле. Затем суспензию наносили на кусок стерильной найлоновой сетки размером 10× 10 мм. После сушки найлоновые волокна сетки разделяли, высвобождая гранулы.
Кармашек роговицы получали, выдвигая вперед с помощью ювелирных пинцетов глазное яблоко у предварительного подвергнутых анестезии 7-недельных самок мышей линии С57В1/6. Используя препаровальную лупу с помощью хирургического лезвия №15 делали центральную внутристромальную линейную кератотомию роговицы длиной примерно 0,6 мм параллельно постановки латеральной прямой мышцы головы. С помощью модифицированного скальпеля для удаления катаракты ламеллярный микрокарманчик рассекали по направлению к височному лимбу. Карманчик на 1,0 мм входил в область височного лимба. С помощью ювелирных пинцетов одну гранулу помещали на поверхность роговицы в основание карманчика. Затем гранулу перемещали в прилегающий к височной части конец карманчика. После этого на глаз наносили антибактериальную мазь.
Мышам вводили лекарство каждый день на протяжении всего эксперимента. Вводимые животным дозы зависели от биологической доступности и общей эффективности соединения, и приведенные в качестве примера дозы составляли 10 или 50 мг/кг, дважды в день, перорально. Неоваскуляризация стромы роговицы начиналась примерно через 3 дня и ей давали возможность продолжаться до пятого дня, при этом животных подвергали обработке исследуемым соединением. На пятый день определяли уровень ингибирования ангиогенеза, оценивая образование новых сосудов с помощью щелевого лампового микроскопа.
Мышей подвергали анестезии и обследовали глаз, снова выдвигая вперед глазное яблоко. Измеряли максимальную длину вновь образовавшегося сосуда, расположенного от сосудистого сплетения лимба до гранулы. Кроме того, измеряли соседнюю периферическую зону неоваскуляризации, представляя роговицу в виде циферблата часов, где дугу, равную 30 градусам, принимали за один час. Зону ангиогенеза рассчитывали по из следующей формуле
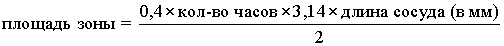
Для оценки каждого соединения в каждом опыте использовали по 5-6 мышей. Затем опытных мышей сравнивали с контрольными мышами и различие в площади неоваскуляризации вычисляли в виде усредненного значения. Каждая исследованная группа мышей была обозначена как "n=1", поэтому значения "n" больше единицы означают, что было проведено большее количество опытов, усредненный результат которых приведен в таблице. Как правило, рассматриваемое соединение вызывало ингибирование примерно на 25-75%, в то время как в обработанном носителем контроле процент ингибирвания был равен 0.
Ниже представлены данные для четырех соединений из приведенных выше примеров, которые применяли в дозах 10 и 50 мг/кг.
Ингибирование ангиогенеза
Пример 446: Уменьшение опухоли, вызванной клетками линии PC-3, in vivo
Линию клеток рака поджелудочной железы человеа PC-3 (ATCC CRL 1435) выращивали до 90%-ной конфлуэнции в среде F12/MEM (фирмы Gibco), содержащий 7% фетальной телячьей сыворотки (ФТС) (фирмы Gibco). Клетки механически собирали с помощью резинового скребка и затем дважды промывали холодной средой. Полученные в результате клетки повторно суспендировали в холодной среде с 30% матригеля (фирма Collaborative Research) и содержащую клетки среду сохраняли на льду до использования.
Мышей линии Balb/c nu/nu возрастом 7-9 недель подвергали анестезии с помощью авертина [смесь 2,2,2-трибромметанол/трет-амиловый спирт (1 г/1 мл), разбавленная из расчета 1:60 забуференным фосфатом физиологическим раствором (ЗФР)] и в левый бок каждой мыши инъецировали 3-5× 106 вышеуказанных клеток в 0,2 мл среды. Клетки инъецировали утром, а введение ингибиторов начинали в 6 ч вечера. Введение осуществляли через желудочный зонд дважды в день, начиная со дня 0 (день инъекции клеток) до дня 25-30, когда животных подвергали автаназии и взвешивали опухоли.
Соединения вводили в дозе 10 мг/мл в смеси 0,5% метилцеллюлоза/0,1% полисорбат 80, получая при этом дозу, составляющую 50 мг/кг дважды в день, или разбавляли, получая дозу в 10 мг/кг дважды в день. Измерение опухолей начинали на 7-ой день и продолжали через каждые 3-4 дня до завершения эксперимента. В каждом опыте использовали группы из 10 мышей, 9-10 из которых выживали. Каждая исследованная группа мышей была обозначена как n=1, поэтому значения n больше единицы означают, что было проведено большее количество опытов, усредненный результат которых приведен в таблице. Результаты, полученные в этом опыте с использованием некоторых из ранее описанных соединений, приведены ниже в виде средних значений уменьшения веса опухолей.
Средний процент уменьшения веса опухолей
Пример 447: Опыты по оценке влияния на фактор некроза опухоли
Культура клеток
Клетки, которые использовали в опыте, представляли собой линию клеток моноцитов человека U-937 (ATCC CRL-1593). Клетки выращивали в среде RPMI, содержащей 10 мас.% ФТС и добавку PSG (R-10), не давая им перерастать.
Опыт проводили следующим образом.
1. Подсчитывали количество клеток, затем собирали их центрифугированием. Повторно суспендировали дебрис в добавке R-10 до концентрации 1,540× 10 клеток/мл.
2. В соответствующие лунки 96-луночного плоскодонного планшета для культуры тканей добавляли тестируемое соединение в 65 мкл R-10. Из исходного маточного раствора в ДМСО (концентрация соединения 100 мМ) брали 400 мкл раствора, из которого получали 5 дополнительных трехкратных серийных разведении. В каждом разведении объемом 65 мкл (взятом в трех повторностях) концентрации тестируемых соединений составляли 100 мкМ, 33,3 мкМ, 11,1 мкМ, 3,7 мкМ, 1,2 мкМ и 0,4 мкМ.
3. В каждую лунку добавляли подсчитанные, промытые и повторно суспендированые клетки (200000 клеток/лунку) в 130 мкл.
4. Инкубировали в течение промежутка времени, составлявшего от 45 мин до 1 ч при 37° С в 5% СО2 в насыщенном водой контейнере.
5. В каждую лунку добавляли среду R-10 (65 мкл), содержащую 160 нг/мл ФМА (форболмиристатацетат) (фирма Sigma).
6. Тестируемую систему инкубировали при 37° С в 5% СО2 в течение ночи (18-20 ч) при 100%-ной влажности.
7. Из каждой лунки осторожно удаляли надосадочную жидкость (150 мкл) для оценки с помощью твердофазного иммуноферментного анализа (ELISA-анализа).
8. Для оценки токсичности аликвоты объемом по 50 мкл рабочего раствора, содержащие 5 мл R-10, 5 мл раствора MTS [CellTiter 96 AQueous One Solution Cell Proliferation Assay, каталожный №G358/0,1 (фирма Promega Biotech)] и 250 мкл раствора СЖК (сыворотка жеребой кобылы), добавляли в каждую лунку, содержащую оставшуюся надосадочную жидкость и клетки, далее клетки инкубировали при 37° С и 5% СО2 до появления окраски. Анализ системы проводили при длине волны возбуждения 570 нм, а регистрацию проводили при длине волны 630 нм.
Анализ TNF-редептора II (TNFrII) методом ELISA
1. Вносили 100 мкл/лунку 2 мкг/мл мышиного антитела к человеческому TNFrII (фирма R&D System №МАВ22б) в однократном (1× ) ЗФР (рН 7,1, фирма Gibco) на планшет типа NUNC-Immuno Maxisorb. Инкубировали планшет при 4° С в течение ночи (примерно 18-20 ч).
2. Промывали планшет с помощью смеси ЗФР-Твин (1× ЗФР/0,05 мас.% Твина).
3. Добавляли 200 мкл 5%-ного БСА в ЗФР и блокировали реакцию, выдерживая при 37° С в насыщенной водой атмосфере в течение 2 ч.
4. Промывали планшет с помощью смеси ЗФР-Твин.
5. В каждую лунку добавляли образец и контроль (по 100 мкл каждого). В качестве стандартов использовали 0, 50, 100, 200, 300 и 500 пг рекомбинантного человеческого TNFrII (фирма R&D System №226-В2) в 100 мкл 0,5%-ного БСА в ЗФР. Анализируемая зависимость была линейной до концентрации стандарта в 400-500 пг.
6. Инкубировали при 37° С в насыщенной атмосфере в течение 1,5 ч.
7. Промывали планшет с помощью смеси ЗФР-Твин.
8. Добавляли 100 мкл козьего поликлонального антитела к человеческому TNFrII (1,5 мкг/мл фирма R&D System №АВ22б)-РВ в 0,5%-ном БСА в ЗФР).
9. Инкубировали при 37° С в насыщенной атмосфере в течение 1 ч.
10. Промывали планшет с помощью смеси ЗФР-Твин.
11. Добавляли 100 мкл антикозьего IgG, конъюгированного с пероксидазой (1:50000 в 0,5%-ном БСА в ЗФР, фирма Sigma, №А5420).
12. Инкубировали при 37° С в насыщенной атмосфере в течение 1 ч.
13. Промывали планшет с помощью смеси ЗФР-Твин.
14. Добавляли 10 мкл реактива KPL ТМВ для осуществления реакции, проводили реакцию при комнатной температуре (обычно в течение 10 мин), затем прекращали ее с помощью фосфорной кислоты и анализировали при длине волны возбуждения 450 нм, а регистрацию проводили при длине волны 570 нм.
Анализ TNFα методом ELISA
Плашпеты типа Immulon® 2 сенсибилизировали с помощью 0,1 мл/лунку МАт фирмы Genzyme (1 мкг/мл) в 0,1М буфере NaHCO3, pH 8,0, в течение ночи (примерно 18-20 ч) при 4° С, плотно завернув их в обертку типа Saran®.
Стряхивали покрывающий раствор и блокировали планшеты, используя по 0,3 мл/лунку блокирующего буфера в течение ночи при 4° С, завернув их в обертку типа Saran®.
Тщательно четырежды промывали лунки с помощью буфера для промывки и полностью удаляли весь буфер для промывки. Добавляли по 0,1 мл/лунку либо образцов, либо стандартов rhTNFα . При необходимости разводили образцы приемлемым разбавителем (например, средой для культуры ткани). Разводили стандарт этим же разбавителем. Стандарты и образцы были взяты в трех повторностях.
Инкубировали при 37° С в течение 1 ч в увлажняющем контейнере.
Промывали планшеты по описанному выше методу. Добавляли по 0,1 мл/лунку разбавления 1:200 кроличьего антитела к hTNF фирмы Genzyme.
Повторяли инкубацию.
Повторяли промывку. Добавляли по 0,1 мл/лунку козьего антитела к кроличьему IgG (тяжелая цепь + легкая цепь), конъюгированного с пероксидазой (0,1 мкг/мл; фирма Jackson).
Инкубировали при 37° С в течение 30 мин.
Повторяли промывку. Добавляли по 0,1 мл/лунку раствора пероксида 2,2’-азинобис(3-этил-2,3-дигидробензотиазолсульфоната) (АБТС).
Инкубировали при комнатной температуре в течение 5-20 мин.
Определяли ОП при длине волны 405 нм.
Двенадцать указанных реагентов представляли собой следующие:
- мышиное моноклональное антитело к человеческому TNFα фирмы Genzyme (каталожный №80-3399-01);
- кроличье поликлональное антитело к человеческому TNFα фирмы Genzyme (каталожный №IP-300);
- рекомбинантный человеческий TNFα фирмы Genzyme (каталожный №TNF-H);
- козий антикроличий IgG (тяжелая цепь + легкая цепь), конъюгированный с пероксидазой, фирмы Jackson Immunoresearch (каталожный № 111-035-144);
- раствор пероксида АБТС Киркергаарда/Перри (каталожный №50-66-01);
- титрационные 96-луночные микропланшеты типа Immulon 2;
- блокирующий раствор представляет собой раствор, содержащий 1 мг/мл желатина в ЗФР с 1-кратным тимеразолом;
- буфер для промывки представляет собой 0,5 мл Tween® 20 в 1 л ЗФР.
Результаты
Пример 448: Фармакокинетическая (ФК) оценка ингибиторов MMIL проведенная на крысах
После анестезии метафаном выделяли бедренную артерию (у всех 8 подопытных крыс) и бедренную вену (только у 4 из 8 подопытных крыс) и осуществляли катетеризацию зондом типа РЕ50, после чего рану закрывали с помощью шелкового шовного материала номер 3.0. Для следующего исследования требовалась 2 катетера, причем венозную линию использовали для инфузии соединения (в группе крыс, которая получала соединение внутривенным (в.в.) путем), а артериальную линию использовали для сбора образцов крови. Затем крыс помещали в изолированные клетки, которые позволяли осуществлять минимальное движение, и давали животным в течение примерно 30 мин восстановиться после анестезии. Через артериальный катетер в момент, обозначенный как "время 0" (до обработки соединениями), брали образцы крови (400 мкл).
Одна группа крыс (4 крысы в группе) получала соединение оральным путем в виде объемной дозы 2 мл/кг (10 мг/мл, растворенное в 0,5%-ной метилцеллюлозе, 0,1% Tween® 20), а вторая группа крыс получала через внутривенный катетер в виде объемной дозы 2 мл/кг (10 мг/мл, растворенное в смеси, содержащей 10% EtOH, 50% ПЭГ 400, 40% физиологического раствора). Образцы крови собирали через артериальный катетер через 15, 30, 60, 120, 240 и 360 мин у животных из группы, которую подвергали оральной обработке, а у животных из группы IV, которую подвергали в.в. обработке, брали образцы через эти же промежутки времени и, кроме того, дополнительно брали образец крови через 3 мин после обработки. После взятия каждого образца катетеры промывали ЗФР, содержащем 10 ЕД/мл гепарина. Через 6 ч, когда эксперимент завершался, животных подвергали автаназии с помощью избыточной анестезии или путем асфиксии монооксидом углерода. В образцах крови, полученных в каждый момент времени, анализировали ингибирующую активность в отношении фермента ММП-13 и на основе стандартной кривой определяли концентрацию в кровотоке соединения плюс активных метаболитов.
Фармакокинетические (ФК) параметры рассчитывали с помощью программы CSTRIP на компьютере типа Vax. Определяли те же параметры, которые описаны в таких руководствах, как руководство Goodman и Gilman: The Pharmacological Basis of Therapeutics, 8-е изд., McGraw-Hill, Inc. New York (1993) и приведенные в этой публикации ссылки.
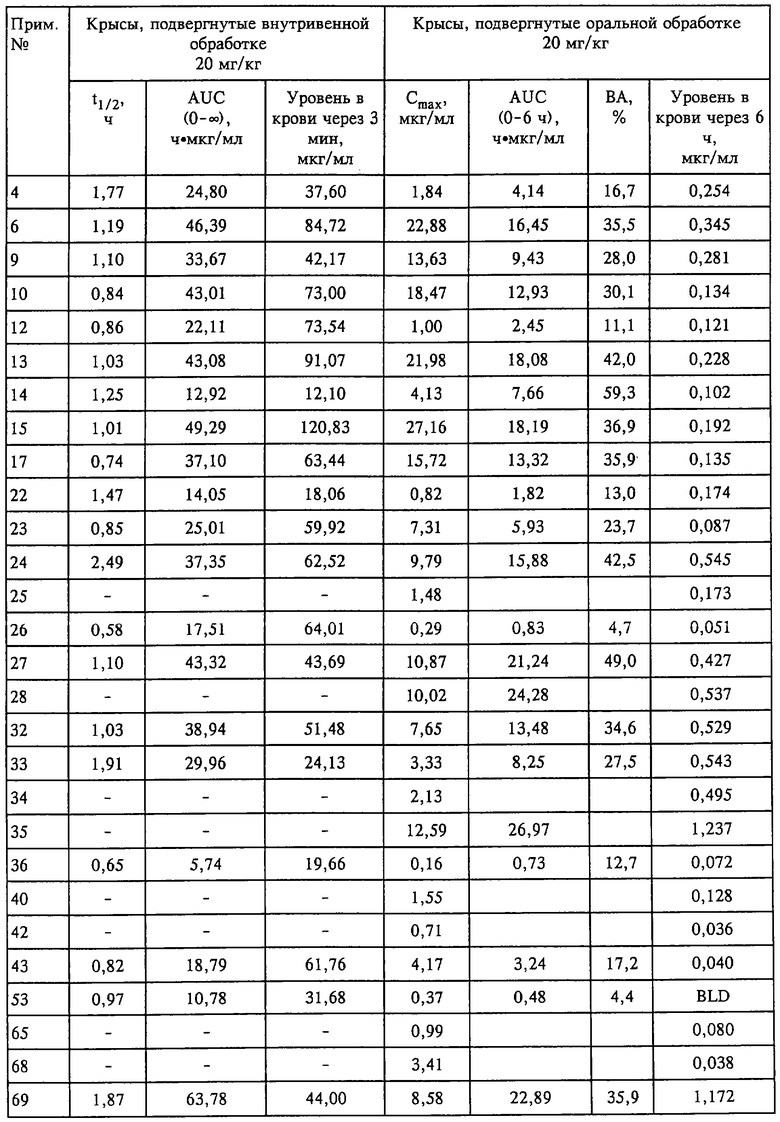
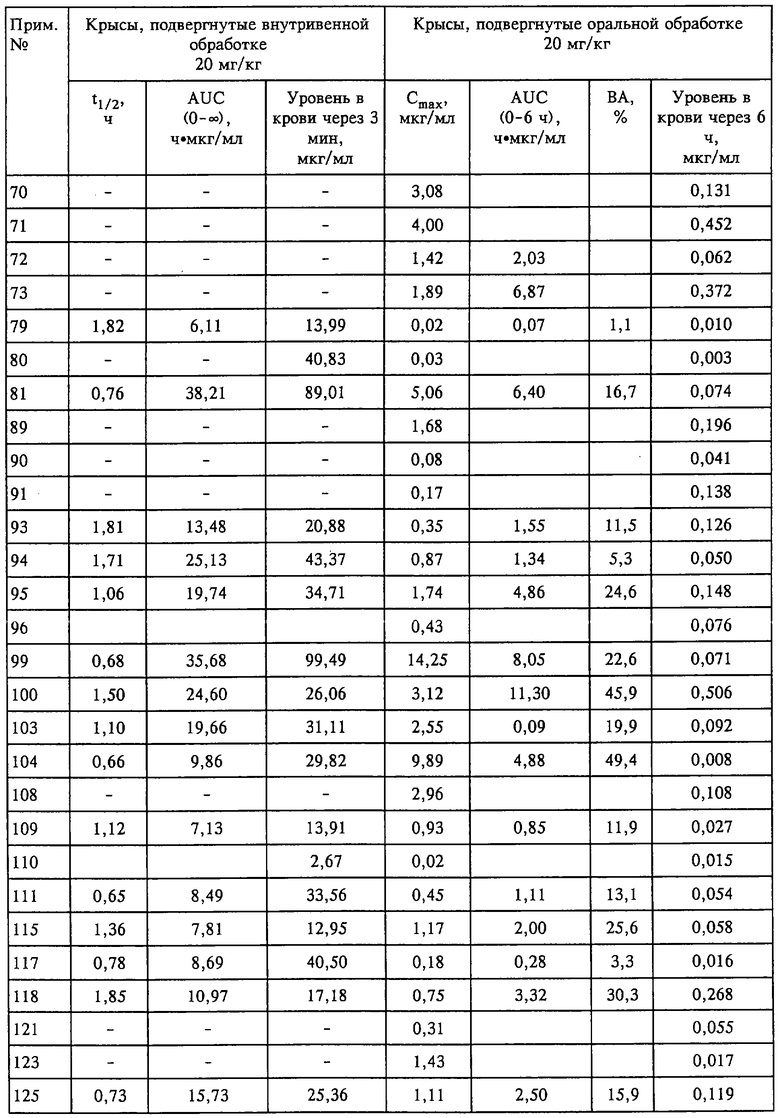

Из всего вышесказанного очевидно, что в изобретение могут быть внесены различные модификации и изменения, не влияющие на сущность и объем новых концепций, предложенных в изобретении. Очевидно также, что объем изобретения не ограничен конкретными вариантами его осуществления, представленными в описании. Подразумевается, что приведенная ниже формула изобретения включает все такие модификации и изменения, которые подпадают под объем изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЛЕЧЕНИЯ ОСТРОГО, ХРОНИЧЕСКОГО И ПОДОСТРОГО КАШЛЯ И НЕПРЕОДОЛИМОГО ЖЕЛАНИЯ ОТКАШЛЯТЬСЯ | 2014 |
|
RU2650118C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ МИМЕТИКОВ ТРО | 2006 |
|
RU2385865C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАК ИНГИБИТОРЫ КАТЕПСИНА S | 2005 |
|
RU2424239C2 |
| МАКРОЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ ВИРУСА ГЕПАТИТА C | 2000 |
|
RU2247126C2 |
| АЛЬТЕРНАТИВНЫЕ СПОСОБЫ СИНТЕЗА ИНГИБИТОРОВ РЕНИНА И ИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2005 |
|
RU2411230C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2006 |
|
RU2411242C2 |
| ИНГИБИТОРЫ СЕРИНПРОТЕАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2252935C2 |
| АРИЛСУЛЬФОНИЛЬНЫЕ ПРОИЗВОДНЫЕ, ОБЛАДАЮЩИЕ СРОДСТВОМ К РЕЦЕПТОРУ 5-HT, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2268884C2 |
| 6-АМИНО-2-ЗАМЕЩЕННЫЕ-5- ВИНИЛСИЛИЛПИРИМИДИН-4-КАРБОНОВЫЕ КИСЛОТЫ И СЛОЖНЫЕ ЭФИРЫ И 4-АМИНО-6-ЗАМЕЩЕННЫЕ-3-ВИНИЛСИЛИЛПИРИДИН-ПИКОЛИНОВЫЕ КИСЛОТЫ И СЛОЖНЫЕ ЭФИРЫ КАК ГЕРБИЦИДЫ | 2012 |
|
RU2556000C2 |
| НЕНУКЛЕОЗИДНЫЕ ИНГИБИТОРЫ ОБРАТНОЙ ТРАНСКРИПТАЗЫ, ИХ ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ ВИЧ | 2004 |
|
RU2305680C2 |
Изобретение относится к области медицины и касается способа лечения заболеваний, связанных с патологической активностью матриксных металлопротеаз, с помощью соединений формулы I, соединений формулы I, а также способа получения указанных соединений. Изобретение повышает эффективность лечения. 25 с. и 111 з.п. ф-лы, 152 табл.
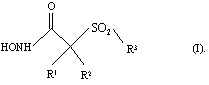
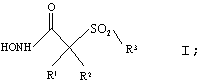
R1 и R2 оба обозначают гидрид или R1 и R2 вместе с атомами, к которым они присоединены, образуют 5-8-членное кольцо, содержащее в кольце один, два или три гетероатома, независимо выбранных из группы, содержащей кислород, серу и азот;
R3 выбирают из группы, содержащей арил и гетероарил, где этот арил или гетероарил является замещенным заместителем, выбранным из группы, включающей циклоалкил, гетероциклоалкил, арил, гетероарил, аралкил, гетероаралкил, аралокси-, гетероаралкоксигруппу, аралкоксиалкил, арилоксиалкил, аралканоилалкил, арилкарбонилалкил, аралкиларил, арилоксиалкиларил, аралкоксиарил, арилазоарил, арилгидразиноарил, алкилтиоарил, арилтиоалкил, алкилтиоаралкил, аралкилтиоалкил, аралкилтиоарил, сульфоксид любого из тиозаместителей, сульфон любого из тиозаместителей, и сконденсированную кольцевую структуру, включающую два или большее количество 5-6-членных колец, выбранных из группы, включающей арил, гетероарил, циклоалкил и гетероциклоалкил, где любой заместитель сам необязательно замещен одним или двумя заместителями, независимо друг от друга выбранными из группы, включающей цианогруппу, перфторалкил, трифторметокси-, трифторметилтиогруппу, галоалкил, трифторметилалкил, аралкоксикарбонил, арилоксикарбонил, гидроксигруппу, галоген, алкил, алкокси-, нитрогруппу, тиол, гидроксикарбонил, арилокси-, арилтиогруппу, аралкил, арил, арилкарбониламино-, гетероарилокси-, гетероарилтиогруппу, гетероаралкил, циклоалкил, гетероциклоокси-, гетероциклотио-, гетероциклоамино-, циклоалкилокси-, циклоалкилтио-, гетероаралкокси-, гетероаралкилтио-, аралкокси-, аралкилтио-, аралкиламино-, гетероциклогруппу, гетероарил, арилазо-, гидроксикарбонилалкокси-, алкоксикарбонилалкоксигруппу, алканоил, арилкарбонил, аралканоил, алканоилокси-, аралканоилоксигруппу, гидроксиалкил, гидроксиалкокси-, алкилтио-, алкоксиалкилтиогруппу, алкоксикарбонил, арилоксиалкоксиарил, арилтиоалкилтиоарил, арилоксиалкилтиоарил, арилтиоалкилтиоарил, арилтиоалкоксиарил, гидроксикарбонилалкокси-, гидроксикарбонилалкилтио-, алкоксикарбонилалкокси-, алкоксикарбонилалкилтио-, амино-, карбоксамидогруппу и аминоалкил, причем что касается аминогруппы, то азот аминогруппы является необязательно замещенным не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, гетероарил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил, арилкарбонил, аралканоил, гетероарилкарбонил, гетероаралканоил и алканоил, или
где азот аминогруппы необязательно замещен двумя заместителями, которые вместе с атомом азота аминогруппы образуют 5-8-членное гетероцикло- или гетероарильное кольцо,
необязательно содержащее до двух гетероатомов в дополнение к уже имеющемуся атому азота, представляющих собой азот, кислород и серу, и
необязательно замещенное не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей арил, алкил, гетероарил, аралкил, гетероаралкил, гидрокси-, алкоксигруппу, алканоил,
циклоалкил, гетероциклоалкил, алкоксикарбонил, гидроксиалкил, трифторметил, сконденсированный с бензольным кольцом гетероциклоалкил, гидроксиалкоксиалкил, аралкоксикарбонил, гидроксикарбонил, арилоксикарбонил, сконденсированную с бензольным кольцом гетероциклоалкоксигруппу, сконденсированный с бензольным кольцом циклоалкилкарбонил, гетероциклоалкилкарбонил и циклоалкилкарбонил, что касается карбоксамидогруппы, то
азот карбоксамидогруппы представляет собой реакционноспособный амин аминокислоты, или
является необязательно замещенным не более чем двумя заместителями, независимо выбранными из группы, включающей алкил, гидроксиалкил, гидроксигетероаралкил, циклоалкил, аралкил, трифторметилалкил, гетероциклоалкил, сконденсированный с бензольным кольцом гетероциклоалкил, сконденсированный с бензольным кольцом циклоалкил и N,N-диалкилзамещенную алкиламиноалкильную группу, или
азот карбоксамидогруппы необязательно замещен двумя заместителями, которые вместе с азотом карбоксамидогруппы образуют 5-8-членное гетероцикло-, гетероарильное или сконденсированное с бензольным кольцом гетероциклоалкильное кольцо, необязательно замещенное не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, алкоксикарбонил, нитрогруппу, гетероциклоалкил, гидроксигруппу, гидроксикарбонил, арил, аралкил, гетероаралкил и аминогруппу,
где азот аминогруппы необязательно
замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил и гетероарил, или
азот аминогруппы необязательно замещен двумя заместителями, которые вместе в азотом аминогруппы образуют 5-8-членное гетероцикло- или гетероарильное кольцо, и что касается аминоалкила, то
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил и алканоил, или
азот аминоалкила необязательно замещен двумя заместителями, которые вместе с этим азотом аминоалкила образуют 5-8-членное гетероцикло- или гетероарильное кольцо, причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
введение такому млекопитающему эффективного количества соединения или его фармацевтически приемлемой соли, проявляющего избирательную ингибирующую активность в отношении металлопротеаз ММП-2, ММП-9 и/или ММП-13 по сравнению с металлопротеазой ММП-1, охарактеризованного формулой (I)
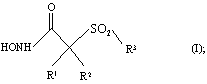
где
R1 и R2 вместе с атомами, к которым они присоединены, образуют 5-8-членное кольцо, содержащее в кольце один, два или три гетероатома, независимо выбранных из группы, содержащей кислород, серу или азот;
R3 выбирают из группы, содержащей фенил и 5- или 6-членный моноциклический гетероарил, где фенил или гетероарил замещены в положении 4 в случае 6-членного кольца или в положении 3 или 4 в случае 5-членного кольца заместителем, выбранным из группы, включающей фенил, моноциклический гетероарил, N-пиперидил, N-пиперазин, феноксигруппу, тиофеноксигруппу, 4-тиопиридил, фенилазогруппу и бензамидогруппу.
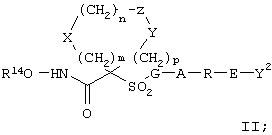
где
R14 выбран из группы, включающей гидрид, фармацевтически приемлемый катион и C(W)R15, где W выбран из группы, включающей О и S, и
R15 выбирают из группы, включающей С1-С6алкил, арил, С1-С6алкоксигруппу, гетероарилС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, арилокси-, арС1-С6алкоксигруппу, арС1-С6алкил, гетероарил и аминоС1-С6алкил, где азот аминоалкила может быть замещен
не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арил, арС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, apС1-С6алкоксикарбонил, С1-С6алкоксикарбонил и С1-С6алканоил, или
двумя заместителями, образующими с азотом аминоалкила 5-8-членное гетероцикло- или гетероарильное кольцо,
m выбирают из группы, включающей 0, 1 и 2,
n выбирают из группы, включающей 0, 1 и 2,
р выбирают из группы, включающей 0, 1 и 2,
сумму m+n+p выбирают из группы, включающей 1, 2, 3 и 4,
X, Y и Z имеют следующие значения:
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя из группы, включающей X, Y и Z, обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей Х и Y, обозначает CR8R9, или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей
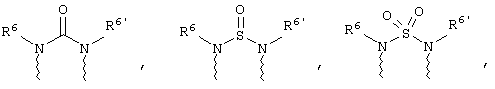
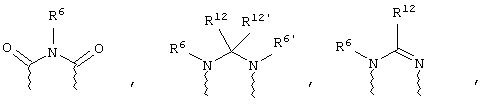
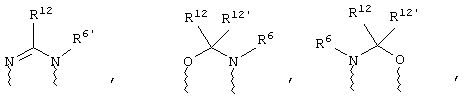
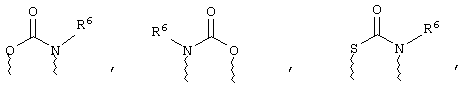
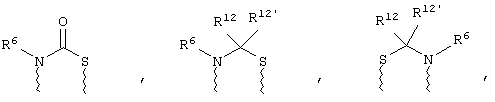

где волнистые линии обозначают связи с атомами изображенного кольца, R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкилкарбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкокси-С1-С6алкил, гетероарилтиоС1-С6алкил, С6арилсульфонил, С1-С6алкилсульфонил, С5-С6 гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4алкоксикарбонил-С1-С6 алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С5-С6гетероциклоиминокарбонил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5алкилкарбонил, гидроксиС1-С5алкил, аминокарбонил, гидроксиаминокарбонил, аминосульфонил, аминоС1-С6алкилсульфонил и аминоС1-С6алкил, где
азот аминокарбонила, аминосульфонила, аминоалкилсульфонила и аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы,
включающей С1-С6алкил, apС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил, R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
R8 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С1-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R8 и R10 вместе с атомом(ами) углерода, к которым они присоединены,
образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R9 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6 алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6 алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R10 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламинoС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R10 и R11 вместе с атомом(ами) углерода, к которым они присоединены,
образуют 5-8-членное карбоциклическое кольцо или
5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R11 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R10 и R11 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу,
при условии, что только один из R8 и R9 или R10 и R11 может обозначать гидроксигруппу,
R12 и R12' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероаралкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, C1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6 алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С2-С6алкинил, С2-С6алкенил и С1-С6гидроксиалкил, и
G-A-R-E-Y2 представляет собой заместитель, который предпочтительно имеет длину, большую, чем длина пентильной группы, и меньшую, чем длина икозильной группы, где
G обозначает арильную или гетероарильную группу,
А выбирают из группы, включающей
(1) -О-,
(2) -S-,
(3) -NR17-,
(4) -CO-NR17-,
(5) -NR17-CO-,
(6) -СО-O-,
(7) -O-СО-,
(8) -O-СО-О-,
(9) -НС=СН-,
(10) -NH-CO-NH-,
(11) -С=С-,
(12) -NH-CO-O-,
(13) -O-CO-NH-,
(14) -N=N-,
(15) -NH-NH-,
(16) -CS-N(R18)-,
(17) -N(R18)-СS- и
(18) связь;
R17 выбирают из группы, включающей гидрид, С1-С4алкил или фенил, R18 выбирают из группы, включающей гидрид, С1-С4алкил или фенил, R обозначает фрагмент, выбранный из группы, включающей алкил, алкоксиалкил, арил, гетероарил, циклоалкил, гетероциклоалкил, аралкил, гетероаралкил, гетероциклоалкилалкил, циклоалкилалкил, циклоалкоксиалкил, гетероциклоалкоксиалкил, арилоксиалкил, гетероарилоксиалкил, арилтиоалкил, гетероарилтиоалкил, циклоалкилтиоалкил и гетероциклоалкилтиоалкил, где арил, гетероарил, циклоалкил или гетероциклоалкил необязательно замещены не более чем двумя заместителями, независимо выбранными из группы, включающей галоген, алкил, перфторалкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, аминогруппу, алкоксикарбонилалкил, алкокси-, С1-С2алкилендиоксигруппу, гидроксикарбонилалкил, гидроксикарбонилалкиламино-, нитро-, гидроксигруппу, гидроксиалкил, алканоиламиногруппу и алкоксикарбонил, и
R не обозначает алкил или алкоксиалкил, когда А обозначает -О- или -S-, Е выбирают из группы, включающей
(1) -CO(R19)-,
(2) -(R19)CO-,
(3) -CONH-,
(4) -HNCO-,
(5) -СО-,
(6) -SO2-R19-,
(7) -R19-SO2-,
(8) -SO2-,
(9) -NH-SO2-,
(10) -SO2-NH и
(11) связь;
R19 выбирают из группы, включающей гетероциклоалкил или циклоалкил,
Y2 выбирают из группы, включающей гидрид, алкил, алкоксигруппу, галоалкил, арил, аралкил, циклоалкил, гетероарил, гидрокси-, арилокси-, аралкокси-, гетероарилоксигруппу, гетероаралкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, алкенил, гетероциклоалкил, циклоалкил, трифторметил, алкоксикарбонил и аминоалкил, где
арил, гетероарил или гетероциклоалкил необязательно замещены не более чем двумя радикалами, независимо друг от друга выбранными из группы, включающей алканоил, галоген, нитрогруппу, аралкил, арил, алкокси- и аминогруппу, где
азот в аминогруппе необязательно замещен не более чем двумя группами, независимо друг от друга выбранными из группы, включающей алкил и аралкил; причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
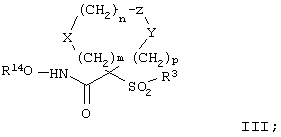
где
R3 выбирают из группы, включающей арил и гетероарил, где арил или гетероарил обозначает 5- или 6-членную арильную или гетероарильную группу, замещенную в положении 4, когда она представляет собой 6-членное кольцо, и в положении 3 или 4, когда она представляет собой 5-членное кольцо, заместителем, выбранным из группы, включающей тиофенокси-, 4-хлорфенокси-, 3-хлорфенокси-, 4-метоксифенокси-, 3-бензодиоксол-5-илокси-, 3,4-диметилфенокси-, 4-фторфенокси-, 4-фтортиофенокси-, фенокси-, 4-трифторметоксифенокси-, 4-трифторметилфенокси-, 4-(трифторметилтио)фенокси-, 4-(трифторметилтио)тиофенокси-, 4-хлор-3-фторфенокси-, 4-изопропоксифенокси-, 4-изопропилфенокси-, (2-метил-1,3-бензотиазол-5-ил)окси-, 4-(1Н-имидазол-1-ил)фенокси-, 4-хлор-3-метилфенокси-, 3-метилфенокси-, 4-этоксифенокси-, 3,4-дифторфенокси-, 4-хлор-3-метилфенокси-, 4-фтор-3-хлорфенокси-, 4-(1Н-1,2,4-триазол-1-ил)фенокси-, 3,5-дифторфенокси-, 3,4-дихлорфенокси-, 4-циклопентилфенокси-, 4-бром-3-метилфенокси-, 4-бромфенокси-, 4-метилтиофенокси-, 4-фенилфенокси-, 4-бензилфенокси-, 6-хинолинилокси-, 4-амино-3-метилфенокси-, 3-метоксифенокси-, 5,6,7,8-тетрагидро-2-нафталинилокси-, 3-гидроксиметилфенокси- и 4-бензилоксифеноксигруппу,
R14 выбирают из группы, включающей гидрид, фармацевтически приемлемый катион и C(W)R15, где W обозначает О или S, и
R15 выбирают из группы, включающей С1-С6алкил, арил, С1-С6алкокси, гетероарилС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, арилокси, арС1-С6 алкокси, арС1-С6алкил, гетероарил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арил, арС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, арС1-С6алкоксикарбонил, С1-С6алкоксикарбонил и С1-С6алканоил, или
двумя заместителями, образующими с азотом аминоалкила 5-8-членное гетероцикло- или гетероарильное кольцо,
m выбирают из группы, включающей 0, 1 и 2,
n выбирают из группы, включающей 0, 1 и 2,
р выбирают из группы, включающей 0, 1 и 2,
сумму m+n+p выбирают из группы, включающей 1, 2, 3 и 4,
X, Y и Z имеют следующие значения:
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя из группы, включающей X, Y и Z, обозначают CR8R9 и CR10R11, или
(б) X и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей Х и Y, обозначает CR8R9, или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей

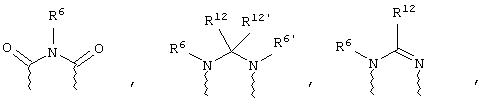
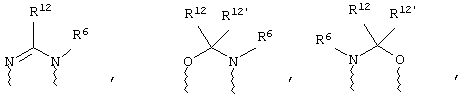
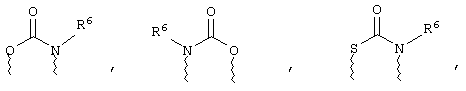
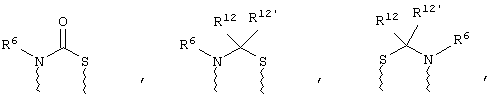
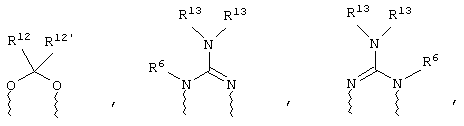
где волнистые линии обозначают связи с атомами изображенного кольца,
R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкилкарбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкокси-С1-С6алкил, гетероарилтиоС1-С6алкил, С6арилсульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4алкоксикарбонил-С1-С6алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С5-С6гетероциклоиминокарбонил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С8алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5алкилкарбонил, гидроксиС1-С5алкил, аминокарбонил, гидроксиаминокарбонил, аминосульфонил, аминоС1-С6алкилсульфонил и аминоС1-С6алкил, где
азот аминокарбонила, аминосулъфонила, аминоалкилсульфонила и аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
R8 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из
этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или,
R8 и R9 или R8 и R10 вместе с атомом(ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R9 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R10 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R10 и R11 вместе с атомом(ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R11 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R10 и R11 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу,
при условии, что только один из R8 и R9 или R10 и R11 может обозначать гидроксигруппу,
R12 и R12′ независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероаралкил, С2-С6алкинил, С2-С6 алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С2-С6алкинил, С2-С6алкенил и С1-С6гидроксиалкил,
причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
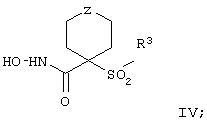
где
R3 выбирают из группы, включающей арил и гетероарил, где арил или гетероарил замещены заместителем, выбирают из группы, включающей циклоалкил, гетероциклоалкил, арил, гетероарил, аралкил, гетероаралкил, аралокси-, гетероаралкоксигруппу, аралкоксиалкил, арилоксиалкил, аралканоилалкил, арилкарбонилалкил, аралкиларил, арилоксиалкиларил, аралкоксиарил, арилазоарил, арилгидразиноарил, алкилтиоарил, арилтиоалкил, алкилтиоаралкил, аралкилтиоалкил, аралкилтиоарил, сульфоксид любого из тиозаместителей, сульфон любого из тиозаместителей, и сконденсированную кольцевую структуру, включающую два или большее количество 5-6-членных колец, выбранных из группы, включающей арил, гетероарил, циклоалкил и гетероциклоалкил, и где
любой из заместителей необязательно может быть замещен одним или более заместителями, независимо друг от друга выбранными из группы, включающей цианогруппу, перфторалкил, трифторметокси-, трифторметилтиогруппу, галоалкил, трифторметилалкил, аралкоксикарбонил, арилоксикарбонил, гидроксигруппу, галоген, алкил, алкокси-, нитрогруппу, тиол, гидроксикарбонил, арилокси-, арилтиогруппу, аралкил, арил, арилкарбониламино-, гетероарилокси-, гетероарилтиогруппу, гетероаралкил, циклоалкил, гетероциклоокси-, гетероциклотио-, гетероциклоамино-, циклоалкилокси-, циклоалкилтио-, гетероаралкокси-, гетероаралкилтио-, аралкокси-, аралкилтио-, аралкиламино-, гетероциклогруппу, гетероарил, арилазо-, гидроксикарбонилалкокси-, алкоксикарбонилалкоксигруппу, алканоил, арилкарбонил, аралканоил, алканоилокси-,
аралканоилоксигруппу, гидроксиалкил, гидроксиалкокси-, алкилтио-, алкоксиалкилтиогруппу, алкоксикарбонил, арилоксиалкоксиарил, арилтиоалкилтиоарил, арилоксиалкилтиоарил, арилтиоалкилтиоарил, арилтиоалкоксиарил, гидроксикарбонилалкокси-, гидроксикарбонилалкилтио-, алкоксикарбонилалкокси-, алкоксикарбонилалкилтио-, аминогруппу, карбоксамидогруппу и аминоалкил, где
азот аминогруппы необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, гетероарил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил, арилкарбонил, аралканоил, гетероарилкарбонил, гетероаралканоил и алканоил, или где
азот аминогруппы необязательно замещен двумя заместителями, которые вместе с этим азотом аминогруппы образуют 5-8-членное гетероцикло- или гетероарильное кольцо,
необязательно содержащее не более двух гетероатомов в дополнение к атому азота аминогруппы, которые выбирают из группы, включающей азот, кислород и серу, и
необязательно замещенное не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей арил, алкил, гетероарил, аралкил, гетероаралкил, гидрокси-, алкоксигруппу, алканоил, циклоалкил, гетероциклоалкил, алкоксикарбонил, гидроксиалкил, трифторметил, сконденсированный с бензольным кольцом гетероциклоалкил, гидроксиалкоксиалкил, аралкоксикарбонил, гидроксикарбонил, арилоксикарбонил,
сконденсированную с бензольным кольцом гетероциклоалкоксигруппу, сконденсированный с бензольным кольцом циклоалкилкарбонил, гетероциклоалкилкарбонил и циклоалкилкарбонил, где
азот карбоксамидогруппы представляет собой реакционноспособный амин аминокислоты, или
азот карбоксамидогруппы необязательно замещен не более чем двумя заместителями, выбранными из группы, включающей алкил, гидроксиалкил, гидроксигетероаралкил, циклоалкил, аралкил, трифторметилалкил, гетероциклоалкил, сконденсированный с бензольным кольцом гетероциклоалкил, сконденсированный с бензольным кольцом циклоалкил и N,N-диалкилзамещенную алкиламиноалкильную группу, или
азот карбоксамидогруппы необязательно замещен двумя заместителями, которые вместе с этим азотом карбоксамидогруппы образуют 5-8-членное гетероцикло-, гетероарильное или сконденсированное с бензольным кольцом гетероциклоалкильное кольцо, которое необязательно замещено не более чем двумя радикалами, независимо друг от друга выбранными из группы, включающей алкил, алкоксикарбонил, нитрогруппу, гетероциклоалкил, гидроксигруппу, гидроксикарбонил, арил, аралкил, гетероаралкил и аминогруппу, где азот аминогруппы необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил и гетероарил, или
где азот аминогруппы необязательно замещен двумя заместителями, которые вместе с этим азотом аминогруппы образуют 5-8-членное гетероцикло- или гетероарильное кольцо,
где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил и алканоил, или где азот аминоалкила необязательно замещен двумя заместителями, которые вместе с этим азотом аминоалкила образуют 5-8-членное гетероцикло- или гетероарильное кольцо, и
Z выбирают из группы, включающей О, S, NR6, SO, SO2 и NSO2R7, где R6 выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алканоил, бензил, бензоил, С3-С5алкинил, С3-С5алкенил, С1-С3алкокси-С1-С4алкил, С3-С6циклоалкил, гетероарилС1-С6алкил, С1-С5гидроксиалкил, С1-С5карбоксиалкил, С1-С5алкокси-С1-С5алкилкарбонил, NR8R9-С1-С6алкилкарбонил и NR8R9-С1-С5алкил, где
R8 и R9 независимо друг от друга выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алкоксикарбонил или арилС1-С5алкокси-карбонил, или
NR8R9 вместе образуют гетероциклическое кольцо, содержащее 5-8 атомов в кольце,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
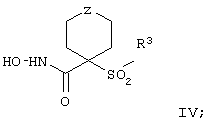
где R3 выбирают из группы, включающей арил и гетероарил, где арил или гетероарил представляет собой 5- или 6-членную группу, замещенную в положении 4, когда она представляет собой 6-членное кольцо, или в положении 3 или 4, когда она представляет собой 5-членное кольцо, заместителем, выбранным из группы, включающей моноциклический арил или моноциклический гетероарил, N-пиперидил, N-пиперазинил, феноксигруппу, тиофеноксигруппу, 4-тиопиридильную группу, фенилазогруппу и бензамидогруппу;
Z выбирают из группы, включающей О, S, NR6, SO, SO2 и NSO2R7, где R6 выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алканоил, бензил, бензоил, С3-С5алкинил, С3-С5алкенил, С1-С3алкокси-С1-С4алкил, С3-С6циклоалкил, гетероарил С1-С6алкил, С1-С5гидроксиалкил, С1-С5карбоксиалкил, С1-С5алкокси-С1-С5алкилкарбонил, NR8R9-С1-С6алкилкарбонил и NR8R9-С1-С5алкил, где
R8 и R9 независимо друг от друга выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алкоксикарбонил или арилС1-С5алкоксикарбонил, или
NR8R9 вместе образуют гетероциклическое кольцо, содержащее 5-8 атомов в кольце,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
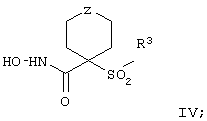
R3 обозначает заместитель G-A-R-E-Y2, где
G выбирают из группы, включающей арил или гетероарил,
А выбирают из группы, включающей
(1) -О-,
(2) -S-,
(3) -NR17-,
(4) -CO-NR17-,
(5) -NR17-CO-,
(6) -СО-O-,
(7) -O-СО-,
(8) -O-СО-О-,
(9) -НС=СН-,
(10) -NH-CO-NH-,
(11) -С=С-,
(12) -NH-CO-O-,
(13) -O-CO-NH-,
(14) -N=N-,
(15) -NH-NH-,
(16) -CS-N(R18)-,
(17) -N(R18)-CS- и
(18) связь;
R17 выбирают из группы, включающей гидрид, С1-С4алкил или фенил, R18 выбирают из группы, включающей гидрид, С1-С4алкил или фенил, или
R выбирают из группы, включающей алкил, алкоксиалкил, арил, гетероарил, циклоалкил, гетероциклоалкил, аралкил, гетероаралкил, гетероциклоалкилалкил, циклоалкилалкил, циклоалкоксиалкил, гетероциклоалкоксиалкил, арилоксиалкил, гетероарилоксиалкил, арилтиоарил, гетероарилтиоалкил, циклоалкилтиоалкил и гетероциклоалкилтиоалкил, где
арил, гетероарил, циклоалкил или гетероциклоалкил необязательно замещены не более чем двумя радикалами, выбранными из группы, включающей галоген, алкил, перфторалкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, аминогруппу, алкоксикарбонилалкил, алкокси-, С1-С2алкилендиоксигруппу, гидроксикарбонилалкил, гидроксикарбонилалкиламино-, нитро-, гидроксигруппу, гидроксиалкил, алканоиламиногруппу и алкоксикарбонил, и
R не обозначает алкил или алкоксиалкил, когда А обозначает -О- или -S-, Е выбирают из группы, включающей
(1) -CO(R19)-,
(2) -(R19)CO-,
(3) -CONH-,
(4) -HNCO-,
(5) -СО-,
(6) -SO2-R19-,
(7) -R19-SO2-,
(8) -SO2-,
(9) -NH-SO2-,
(10) -SO2-NH и
(11) связь;
R19 выбирают из группы, включающей гетероциклоалкил или циклоалкил, Y2 выбирают из группы, включающей гидрид, алкил, алкоксигруппу, галоалкил, арил, аралкил, циклоалкил, гетероарил, гидрокси-, арилокси-, аралкокси-, гетероарилоксигруппу, гетероаралкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, алкенил, гетероциклоалкил, циклоалкил, трифторметил, алкоксикарбонил и аминоалкил, где
арил, гетероарил или гетероциклоалкил необязательно замещены не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алканоил, галоген, нитрогруппу, аралкил, арил, алкокси- и аминогруппу, где
азот в аминогруппе необязательно замещен не более чем двумя группами, независимо друг от друга выбранными из группы, включающей алкил и аралкил;
Z выбирают из группы, включающей О, S, NR6, SO, SO2 и NSO2R7, где R6 выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алканоил, бензил, бензоил, С3-С5алкинил, С3-С5алкенил, С1-С3алкокси-С1-С4алкил, С3-С6циклоалкил, гетероарилС1-С6алкил, С1-С5гидроксиалкил, С1-С6карбоксиалкил, С1-С5алкокси-С1-С5алкилкарбонил, NR8R9-С1-С5алкилкарбонил и NR8R9-С1-С5алкил, где
R8 и R9 независимо друг от друга выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алкоксикарбонил или арилС1-С5алкоксикарбонил, или
NR8R9 вместе образуют гетероциклическое кольцо, содержащее 5-8 атомов в кольце,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С1-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил, причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
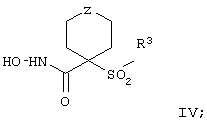
R3 выбирают из группы, включающей арил и гетероарил, где арил или гетероарил представляет собой 5- или 6-членную группу, замещенную в положении 4, когда она представляет собой 6-членное кольцо, или в положении 3 или 4, когда она представляет собой 5-членное кольцо, заместителем, выбранным из группы, включающей тиофенокси-, 4-хлорфенокси-, 3-хлорфенокси-, 4-метоксифенокси-, 3-бензодиоксол-5-илокси-, 3,4-диметилфенокси-, 4-фторфенокси-, 4-фтортиофенокси-, фенокси-, 4-трифторметоксифенокси-, 4-трифторметилфенокси-, 4-(трифторметилтио)фенокси-, 4-(трифторметилтио)тиофенокси-, 4-хлор-3-фторфенокси-, 4-изопропоксифенокси-, 4-изопропилфенокси-, (2-метил-1,3-бензотиазол-5-ил)окси-, 4-(1Н-имидазол-1-ил)фенокси-, 4-хлор-3-метилфенокси-, 3-метилфенокси-, 4-этоксифенокси-, 3,4-дифторфенокси-, 4-хлор-3-метилфенокси-, 4-фтор-3-хлорфенокси-, 4-(1Н-1,2,4-триазол-1-ил)фенокси-, 3,5-дифторфенокси-, 3,4-дихлорфенокси-, 4-циклопентилфенокси-, 4-бром-3-метилфенокси-, 4-бромфенокси-, 4-метилтиофенокси-, 4-фенилфенокси-, 4-бензилфенокси-, 6-хинолинилокси-, 4-амино-3-метилфенокси-, 3-метоксифенокси-, 5,6,7,8-тетрагидро-2-нафталинилокси-, 3-гидроксиметилфенокси- и 4-бензилоксифеноксигруппу;
Z выбирают из группы, включающей О, S, NR6, SO, SO2 и NSO2R7, где R6 выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алканоил, бензил, бензоил, С3-С5алкинил, С3-С5алкенил, С1-С3алкокси-С1-С4алкил, С3-С6циклоалкил, гетероарилС1-С6алкил, С1-С5гидроксиалкил, С1-С5карбоксиалкил, С1-С5алкокси-С1-С5алкилкарбонил, NR8R9-С1-С5алкилкарбонил и NR8R9-С1-С5алкил, где
R8 и R9 независимо друг от друга выбирают из группы, включающей гидрид, С1-С5-алкил, С1-С5алкоксикарбонил или арилС1-С5алкоксикарбонил, или
NR8R9 вместе образуют гетероциклическое кольцо, содержащее 5-8 атомов в кольце,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
фармацевтически приемлемой соли, проявляющего избирательную ингибирующую активность в отношении металлопротеаз ММП-2, ММП-9 и/или ММП-13 по сравнению с металлопротеазой ММП-1, охарактеризованного формулой V

где
Z-О, S и NR6,
W и Q независимо друг от друга обозначают кислород (О), NR6 или серу (S),
R6 выбирают из группы, включающей С3-С6циклоалкил, С1-С6алкил, С3-С6алкенил, С3-С6алкинил, С1-С6алкокси-С1-С6алкил, аминоС1-С6алкил, аминосульфонил, гетероарилС1-С6алкил, арилоксикарбонил и С1-С6алкоксикарбонил, и
q равно 0 или 1,
причем когда q равно 0, Q отсутствует;
причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
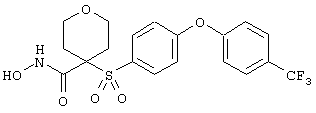
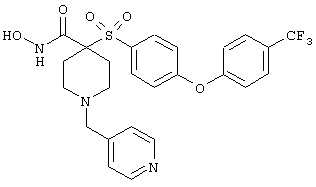
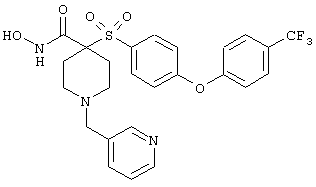
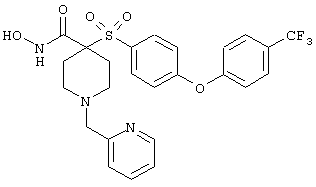
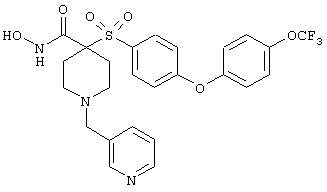
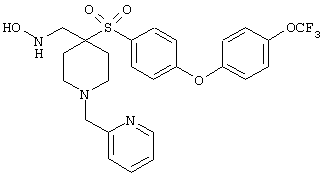

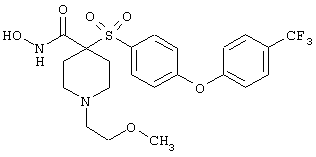
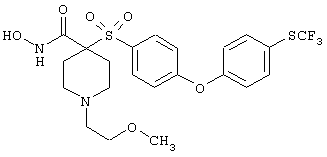
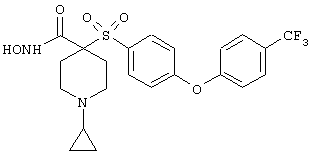
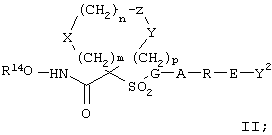
где
R14 выбирают из группы, включающей гидрид, фармацевтически приемлемый катион и C(W)R15,
W выбирают из группы, включающей О и S, и
R15 выбирают из группы, включающей С1-С6алкил, арил, С1-С6алкоксигруппу, гетероарилС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, арилокси-, арС1-С6алкоксигруппу, арС1-С6алкил, гетероарил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арил, арС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, арС1-С6алкоксикарбонил, С1-С6алкоксикарбонил и С1-С6алканоил, или
двумя заместителями, которые вместе с этим азотом аминоалкила образуют 5-8-членное гетероцикло- или гетероарильное кольцо,
m выбирают из группы, включающей 0, 1 и 2,
n выбирают из группы, включающей 0, 1 и 2,
р выбирают из группы, включающей 0, 1 и 2,
сумму m+n+p выбирают из группы, включающей 1, 2, 3 и 4,
X, Y и Z имеют следующие значения:
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя из группы, включающей X, Y и Z, обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей Х и Y, обозначает CR8R9, или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей
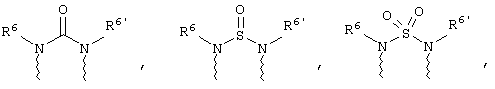
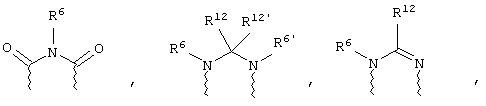
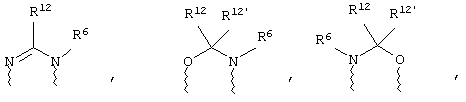
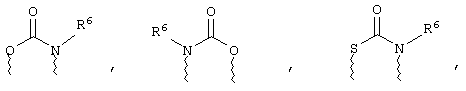
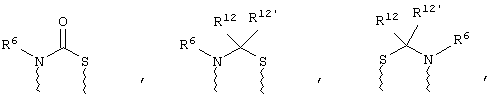
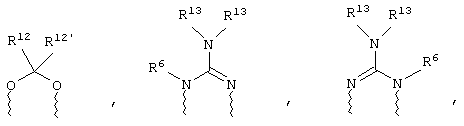
где волнистые линии обозначают связи с атомами изображенного кольца,
R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6 перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкилкарбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкокси-С1-С6алкил, гетероарилтиоС1-С6алкил, С6арилсульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4алкоксикарбонил-С1-С6алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С5-С6гетероциклоиминокарбонил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5алкилкарбонил, гидроксиС1-С5алкил, аминокарбонил, гидроксиаминокарбонил, аминосульфонил, аминоС1-С6 алкилсульфонил и аминоС1-С6алкил, где
азот аминокарбонила, аминосульфонила, аминоалкилсульфонила и аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
R8 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R8 и R10 вместе с атомом(ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R9 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетеpoapС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламинoС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R10 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6 алкенил тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R10 и R11 вместе с атомом (ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R11 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидpoкcиС1-С6aлкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R10 и R11 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу,
при условия, что только один из R8 и R9 или R10 и R11 может обозначать гидроксигруппу,
R12 и R12' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероаралкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6 алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С2-С6алкинил, С2-С6алкенил и С1-С6гидроксиалкил, и G-A-R-E-Y2 представляет собой заместитель, который предпочтительно имеет длину, большую, чем длина пентильной группы, и меньшую, чем длина икозильной группы, где
G обозначает арильную или гетероарильную группу, А выбирают из группы, включающей
(1) -О-,
(2) -S-,
(3) -NR17-,
(4) -CO-NR17-,
(5) -NR17-CO-,
(6) -СО-O-,
(7) -O-СО-,
(8) -O-СО-О-,
(9) -НС=СН-,
(10) -NH-CO-NH-,
(11) -С=С-,
(12) -NH-CO-O-,
(13) -O-CO-NH-,
(14) -N=N-,
(15) -NH-NH-,
(16) -CS-N(R18)-,
(17) -N(R18)-CS- и
(18) связь;
R17 выбирают из группы, включающей гидрид, С1-С4алкил или фенил,
R18 выбирают из группы, включающей гидрид, С1-С4алкил или фенил,
R обозначает фрагмент, выбранный из группы, включающей алкил, алкоксиалкил, арил, гетероарил, циклоалкил, гетероциклоалкил, аралкил, гетероаралкил, гетероциклоалкилалкил, циклоалкилалкил, циклоалкоксиалкил, гетероциклоалкоксиалкил, арилоксиалкил, гетероарилоксиалкил, арилтиоалкил, гетероарилтиоалкил, циклоалкилтиоалкил и гетероциклоалкилтиоалкил, где арил, гетероарил, циклоалкил или гетероциклоалкил необязательно замещены не более чем двумя заместителями, независимо выбранными из группы, включающей галоген, алкил, перфторалкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, аминогруппу, алкоксикарбонилалкил, алкокси-, С1-С2алкилендиоксигруппу, гидроксикарбонилалкил, гидроксикарбонилалкиламино-, нитро-, гидроксигруппу, гидроксиалкил, алканоиламиногруппу и алкоксикарбонил, и
R не обозначает алкил или алкоксиалкил, когда А обозначает -О- или -S-,
Е выбирают из группы, включающей
(1) -CO(R19)-,
(2) -(R19)CO-,
(3) -CONH-,
(4) -HNCO-,
(5) -СО-,
(6) -SO2-R19-,
(7) -R19-SO2-,
(8) -SO2-,
(9) -NH-SO2-,
(10) -SO2-NH и
(11) связь;
R19 выбирают из группы, включающей гетероциклоалкил или циклоалкил,
Y2 выбирают из группы, включающей гидрид, алкил, алкоксигруппу, галоалкил, арил, аралкил, циклоалкил, гетероарил, гидрокси-, арилокси-, аралкокси-, гетероарилоксигруппу, гетероаралкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, алкенил, гетероциклоалкил, циклоалкил, трифторметил, алкоксикарбонил и аминоалкил, где
арил, гетероарил или гетероциклоалкил необязательно замещены не более чем двумя радикалами, независимо друг от друга выбранными из группы, включающей алканоил, галоген, нитрогруппу, аралкил, арил, алкокси- и аминогруппу, где
азот в аминогруппе необязательно замещен не более чем двумя группами, независимо друг от друга выбранными из группы, включающей алкил и аралкил;
причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
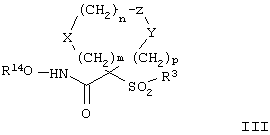
где
R3 выбирают из группы, включающей арил и гетероарил, где арил или гетероарил представляет собой 5- или 6-членную группу, замещенную в положении 4, когда она представляет собой 6-членное кольцо, или в положении 3 или 4, когда она представляет собой 5-членное кольцо, заместителем, выбранным из группы, включающей тиофенокси-, 4-хлорфенокси-, 3-хлорфенокси-, 4-метоксифенокси-, 3-бензодиоксол-5-илокси-, 3,4-диметилфенокси-, 4-фторфенокси-, 4-фтортиофенокси-, фенокси-, 4-трифторметоксифенокси-, 4-трифторметилфенокси-, 4-(трифторметилтио)фенокси-, 4-(трифторметилтио) тиофенокси-, 4-хлор-3-фторфенокси-, 4-изопропоксифенокси-, 4-изопропилфенокси-, (2-метил-1,3-бензотиазол-5-ил)окси-, 4-(1Н-имидазол-1-ил)фенокси-, 4-хлор-3-метилфенокси-, 3-метилфенокси-, 4-этоксифенокси-, 3,4-дифторфенокси-, 4-хлор-3-метилфенокси-, 4-фтор-3-хлорфенокси-, 4-(1Н-1,2,4-триазол-1-ил)фенокси-, 3,5-дифторфенокси-, 3,4-дихлорфенокси-, 4-циклопентилфенокси-, 4-бром-3-метилфенокси-, 4-бромфенокси-, 4-метилтиофенокси-, 4-фенилфенокси-, 4-бензилфенокси-, 6-хинолинилокси-, 4-амино-3-метилфенокси-, 3-метоксифенокси-, 5,6,7,8-тетрагидро-2-нафталинилокси-, 3-гидроксиметилфенокси- и 4-бензилоксифеноксигруппу,
R14 выбирают из группы, включающей гидрид, фармацевтически приемлемый катион и C(W)R15,
W выбирают из группы, включающей О и S, и
R15 выбирают из группы, включающей С1-С6алкил, арил, С1-С6алкоксигруппу, гетероарилС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, арилокси-, арС1-С6алкоксигруппу, арС1-С6алкил, гетероарил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арил, арС1-С6алкил, С3-С8циклоалкил-С1-С6алкил, арС1-С6алкоксикарбонил, С1-С6алкоксикарбонил и С1-С6алканоил, или
двумя заместителями, которые вместе с этим азотом аминоалкила образуют 5-8-членное гетероцикло- или гетероарильное кольцо, m выбирают из группы, включающей 0, 1 и 2, n выбирают из группы, включающей 0, 1 и 2, р выбирают из группы, включающей 0, 1 и 2, сумму m+n+p выбирают из группы, включающей 1, 2, 3 и 4, X, Y и Z имеют следующие значения:
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя из группы, включающей X, Y и Z, обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, которой выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей Х и Y, обозначает CR8R9 или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей
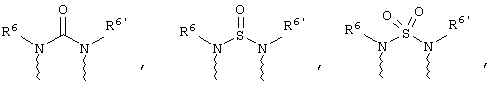
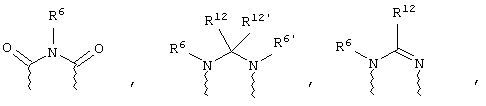
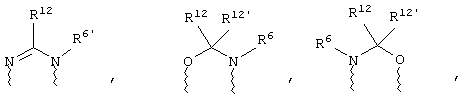

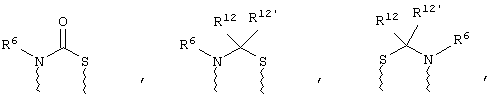

где волнистые линии обозначают связи с атомами изображенного кольца, R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкилкарбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарил С1-С6алкокси-С1-С6алкил, гетероарилтиоС1-С6алкил, С6арилсульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4алкоксикарбонил-С1-С6-алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С5-С6гетероциклоиминокарбонил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С6алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5aлкилкapбoнил, гидроксиС1-С5алкил, аминокарбонил, гидроксиаминокарбонил, аминосульфонил, аминоС1-С6алкилсульфонил и аминоС1-С6алкил, где
азот аминокарбонила, аминосульфонила, аминоалкилсульфонила и аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
R8 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламинoС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы,
включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R8и R10 вместе с атомом(ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R9 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R10 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6 алканоил, или R8 и R9 или R10 и R11 вместе с атомом (ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R11 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R10 и R11 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или
5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу,
при условии, что только один из R8 и R9 или R10 и R11 может обозначать гидроксигруппу,
R12 и R12' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероаралкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С2-С6алкинил, С2-С6алкенил и С1-С6гидроксиалкил, причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и
во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
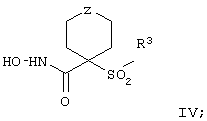
где
R3 выбирают из группы, включающей арил и гетероарил, где арил или гетероарил представляет собой 5- или 6-членную группу, замещенную в положении 4, когда она представляет собой 6-членное кольцо, или в положении 3 или 4, когда она представляет собой 5-членное кольцо, заместителем, выбранным из группы, включающей фенил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидинил, фурил, тиенил, триазолил, оксазолил, оксадиазолил, тиазолил, тиадиазолил, N-пиперидил, N-пиперазинил, феноксигруппу, тиофеноксигруппу, 4-тиопиридил, фенилазогруппу и бензамидогруппу, и
Z выбирают из группы, включающей О, S, NR6, SO, SO2 и NSO2R7, R6 выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алканоил, бензил, бензоил, С3-С6алкинил, С3-С5алкенил, С1-С3алкокси-С1-С4алкил, С3-С6циклоалкил, гетероарилС1-С6алкил, С1-С5гидроксиалкил, С1-С5 карбоксиалкил, С1-С5алкокси-С1-С5алкилкарбонил и NR8R9-С1-С5алкилкарбонил и NR8R9-С1-С5алкил, где
R8 и R9 независимо друг от друга выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алкоксикарбонил и арилС1-С5алкоксикарбонил, или
NR8R9 вместе образуют гетероциклическое кольцо, содержащее 5-8 атомов в кольце, а
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6 карбоксиалкил и С1-С6гидроксиалкил, причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
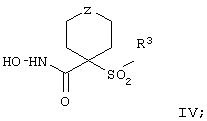
где
R3 обозначает заместитель G-A-R-E-Y2, G выбирают из группы, включающей арил и гетероарил,
А выбирают из группы, включающей
(1) -О-,
(2) -S-,
(3) -NR17-,
(4) -CO-NR17-,
(5) -NR17-CO-,
(6) -СО-O-,
(7) -O-СО-,
(8) -O-СО-О-,
(9) -НС=СН-,
(10) -NH-CO-NH-,
(11) -С=С-,
(12) -NH-CO-O-,
(13) -O-CO-NH-,
(14) -N=N-,
(15) -NH-NH-,
(16) -CS-N(R18)-,
(17) -N(R18)-CS- и
(18) связь;
R17 выбирают из группы, включающей гидрид, С1-С4алкил или фенил,
R18 выбирают из группы, включающей гидрид, С1-С4алкил или фенил,
R обозначает фрагмент, выбранный из группы, включающей алкил, алкоксиалкил, арил, гетероарил, циклоалкил, гетероциклоалкил, аралкил, гетероаралкил, гетероциклоалкилалкил, циклоалкилалкил, циклоалкоксиалкил, гетероциклоалкоксиалкил, арилоксиалкил, гетероарилоксиалкил, арилтиоалкил, гетероарилтиоалкил, циклоалкилтиоалкил и гетероциклоалкилтиоалкил, где арил, гетероарил, циклоалкил или гетероциклоалкил необязательно замещены не более чем двумя заместителями, независимо выбранными из группы, включающей галоген, алкил, перфторалкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, аминогруппу, алкоксикарбонилалкил, алкокси-, С1-С2алкилендиоксигруппу, гидроксикарбонилалкил, гидроксикарбонилалкиламино-, нитро-, гидроксигруппу, гидроксиалкил, алканоиламиногруппу и алкоксикарбонил, и
R не обозначает алкил или алкоксиалкил, когда А обозначает -О- или -S-,
Е выбирают из группы, включающей
(1) -CO(R19)-,
(2) -(R19)CO-,
(3) -CONH-,
(4) -HNCO-,
(5) -СО-,
(6) -SO2-R19-,
(7) -R19-SO2-,
(8) -SO2-,
(9) -NH-SO2-,
(10) -SO2-NH и
(11) связь;
R19 выбирают из группы, включающей гетероциклоалкил или циклоалкил,
Y2 выбирают из группы, включающей гидрид, алкил, алкоксигруппу, галоалкил, арил, аралкил, циклоалкил, гетероарил, гидрокси-, арилокси-, аралкокси-, гетероарилоксигруппу, гетероаралкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, алкенил, гетероциклоалкил, циклоалкил, трифторметил, алкоксикарбонил и аминоалкил, где
арил, гетероарил или гетероциклоалкил необязательно замещены не более чем двумя радикалами, независимо друг от друга выбранными из группы, включающей алканоил, галоген, нитрогруппу, аралкил, арил, алкокси- и аминогруппу, где
азот в аминогруппе необязательно замещен не более чем двумя группами, независимо друг от друга выбранными из группы, включающей алкил и аралкил;
Z выбирают из группы, включающей О, S, NR6, SO, SO2 и NSO2R7,
где R6 выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алканоил, бензил, бензоил, С3-С5алкинил, С3-С5алкенил, С1-С3алкокси-С1-С4алкил, С3-С6циклоалкил, гетероарилС1-С6алкил, С1-С5гидроксиалкил, С1-С5 карбоксиалкил, С1-С5алкокси-С1-С5алкилкарбонил, NR8R9-С1-С5алкилкарбонил и NR8R9-С1-С5алкил, где
R8 и R9 независимо друг от друга выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алкоксикарбонил или арилС1-С5алкоксикарбонил, или
NR8R9 вместе образуют гетероциклическое кольцо, содержащее 5-8 атомов в кольце,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил, причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
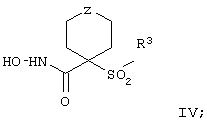
где
R3 выбирают из группы, включающей арил и гетероарил, где арил или гетероарил представляет собой 5- или 6-членную группу, замещенную в положении 4, когда она представляет собой 6-членное кольцо, или в положении 3 или 4, когда она представляет собой 5-членное кольцо, заместителем, выбранным из группы, включающей тиофенокси-, 4-хлорфенокси-, 3-хлорфенокси-, 4-метоксифенокси-, 3-бензодиоксол-5-илокси-, 3,4-диметилфенокси-, 4-фторфенокси-, 4-фтортиофенокси-, фенокси-, 4-трифторметоксифенокси-, 4-трифторметилфенокси-, 4-(трифторметилтио)фенокси-, 4-(трифторметилтио)тиофенокси-, 4-хлор-3-фторфенокси-, 4-изопропоксифенокси-, 4-изопропилфенокси-, (2-метил-1,3-бензотиазол-5-ил)окси-, 4-(1Н-имидазол-1-ил)фенокси-, 4-хлор-3-метилфенокси-, 3-метилфенокси-, 4-этоксифенокси-, 3,4-дифторфенокси-, 4-хлор-3-метилфенокси-, 4-фтор-3-хлорфенокси-, 4-(1Н-1,2,4-триазол-1-ил)фенокси-, 3,5-дифторфенокси-, 3,4-дихлорфенокси-, 4-циклопентилфенокси-, 4-бром-3-метилфенокси-, 4-бромфенокси-, 4-метилтиофенокси-, 4-фенилфенокси-, 4-бензилфенокси-, 6-хинолинилокси-, 4-амино-3-метилфенокси-, 3-метоксифенокси-, 5,6,7,8-тетрагидро-2-нафталинилокси-, 3-гидроксиметилфеноксигруппу, N-пиперидил, N-пиперазинил и 4-бензилоксифеноксигруппу, Z выбирают из группы, включающей О, S, NR6, SO, SO2 и NSO2R7, где R6 выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алканоил, бензил, бензоил, С3-С5алкинил, С3-С5алкенил, С1-С3алкокси-С1-С4алкил, С3-С6циклоалкил, гетероарилС1-С6алкил, С1-С5гидроксиалкил, С1-С5карбоксиалкил, С1-С5алкокси-С1-С5алкилкарбонил, NR8R9-С1-С5алкилкарбонил и NR8R9-С1-С5алкил, где
R8 и R9 независимо друг от друга выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алкоксикарбонил или арилС1-С5алкоксикарбонил, или
NR8R9 вместе образуют гетероциклическое кольцо, содержащее 5-8 атомов в кольце,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6 карбоксиалкил и С1-С6гидроксиалкил, причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или
сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
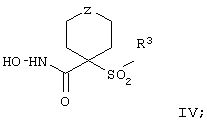
где R3 обозначает фенил, замещенный в положении 4 группой R23, и
R23 выбирают из группы, включающей фенил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидинил, фурил, тиенил, триазолил, оксазолил, оксадиазолил, тиадиазолил, пиперидил, пиперазинил, феноксигруппу, тиофеноксигруппу, фенилазогруппу и бензамидогруппу, где
фенил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидицил, фурил, тиенил, триазолил, оксазолил, оксадиазолил, тиадиазолил, пиперидил, пиперазинил, феноксигруппа, тиофеноксигруппа, фенилазогруппа и бензамидогруппа либо
замещены заместителем, выбранным из группы, включающей галоген, С1-С4алкоксигруппу, С1-С6алкильную группу, диметиламиногруппу, карбоксилС1-С3алкиленовую группу, С1-С4алкоксикарбонил-С1-С3алкиленовую группу, трифторметилтиогруппу, трифторметоксигруппу, трифторметильную группу и карбоксамидоС1-С3алкиленовую группу, либо
замещены в мета- и в пара-положениях метилендиоксигруппой, и
Z выбирают из группы, включающей О, S, NR6, SO, SO2 и NSO2R7, где R6 выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алканоил, бензил, бензоил, С3-С5алкинил, С3-С5алкенил, С1-С3алкокси-С1-С4алкил, С3-С6циклоалкил, гетероарилС1-С6алкил, С1-С5гидроксиалкил, С1-С5карбоксиалкил, С1-С5алкокси-С1-С5алкилкарбонил, NR8R9-С1-С5алкилкарбонил и NR8R9-С1-С5алкил, где
R8 и R9 независимо друг от друга выбирают из группы, включающей гидрид, С1-С5алкил, С1-С5алкоксикарбонил или арилС1-С5алкоксикарбонил, или
NR8R9 вместе образуют гетероциклическое кольцо, содержащее 5-8 атомов в кольце,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
R23 выбирают из группы, включающей фенил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидинил, фурил, тиенил, триазолил, оксазолил, оксадиазолил, тиазолил, тиадиазолил, N-пиперидил, N-пиперазинил, феноксигруппу, тиофеноксигруппу, фенилазогруппу и бензамидогруппу.
замещены в пара-положении заместителем, выбранным из группы, включающей галоген, С1-С4 алкоксигруппу, С1-С4алкильную группу, диметиламиногруппу, карбоксилС1-С3алкиленовую группу, С1-С4алкоксикарбонил-С1-С3алкиленовую группу, трифторметилтиогруппу, трифторметоксигруппу, трифторметильную группу и карбоксамидоС1-С3алкиленовую группу, либо замещены в мета- и в пара-положениях метилендиоксигруппой.
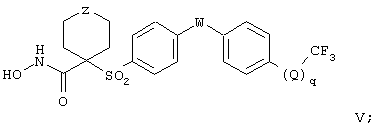
где
Z выбирают из группы, включающей О, S и NR6,
W и Q независимо друг от друга выбирают из группы, включающей О, NR6 и S,
R6 выбирают из группы, включающей С3-С6циклоалкил, С1-С6алкил, С3-С6алкенил, С3-С6алкинил, С1-С6алкокси-С1-С6алкил, аминоС1-С6алкил, аминосульфонил, гетероарилС1-С6алкил, арилоксикарбонил и С1-С6алкоксикарбонил, и
q равно 0 или 1,
причем когда q равно 0, Q отсутствует, и
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до
примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.


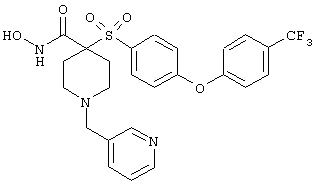
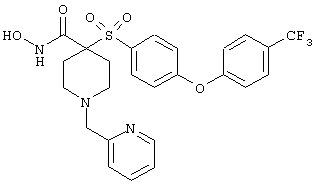
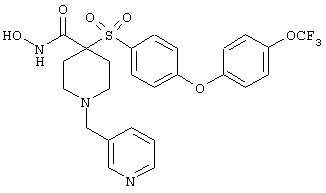
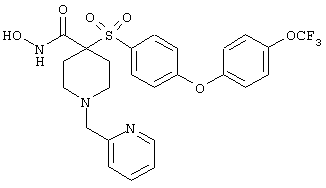
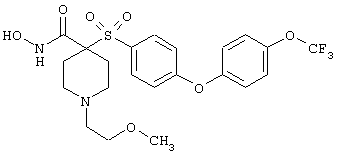
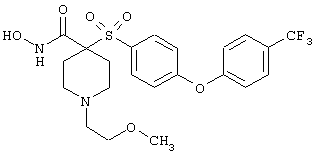
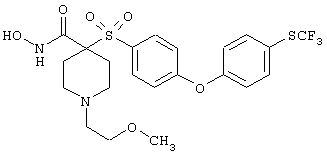
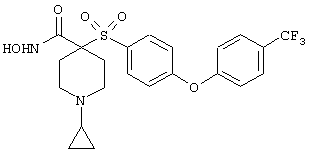
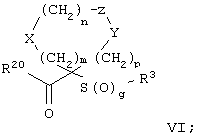
где
g выбирают из группы, включающей 0, 1 и 2,
R3 выбирают из группы, включающей арил и гетероарил, где
арил или гетероарил замещены способной к нуклеофильному замещению уходящей группой, или
арил и гетероарил необязательно замещены заместителем, который выбирают из группы, включающей циклоалкил, гетероциклоалкил, арил, гетероарил, аралкил, гетероаралкил, аралокси-, гетероаралкоксигруппу, аралкоксиалкил, арилоксиалкил, аралканоилалкил, арилкарбонилалкил, аралкиларил, арилоксиалкиларил, аралкоксиарил, арилазоарил, арилгидразиноарил, алкилтиоарил, арилтиоалкил, алкилтиоаралкил, аралкилтиоалкил, аралкилтиоарил, сульфоксид любого из тиозаместителей, сульфон любого из тиозаместителей, и сконденсированную кольцевую структуру, включающую два или большее количество 5-6-членных колец, выбранных из группы, включающей арил, гетероарил, циклоалкил и гетероциклоалкил, где любой из этих заместителей необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей цианогруппу, перфторалкил, трифторметокси-, трифторметилтиогруппу, галоалкил, трифторметилалкил, аралкоксикарбонил, арилоксикарбонил, гидроксигруппу, галоген, алкил, алкокси-, нитрогруппу, тиол, гидроксикарбонил, арилокси-, арилтиогруппу, аралкил, арил, арилкарбониламино-, гетероарилокси-, гетероарилтиогруппу, гетероаралкил, циклоалкил, гетероциклоокси-, гетероциклотио-, гетероциклоамино-, циклоалкилокси-, циклоалкилтио-, гетероаралкокси-, гетероаралкилтио-, аралкокси-, аралкилтио-, аралкиламино-, гетероциклогруппу, гетероарил, арилазо-, гидроксикарбонилалкокси-, алкоксикарбонилалкоксигруппу, алканоил, арилкарбонил, аралканоил, алканоилокси-, аралканоилоксигруппу, гидроксиалкил, гидроксиалкокси-, алкилтио-, алкоксиалкилтиогруппу, алкоксикарбонил, арилоксиалкоксиарил, арилтиоалкилтиоарил, арилоксиалкилтиоарил, арилтиоалкилтиоарил, арилтиоалкоксиарил, гидроксикарбонилалкокси-, гидроксикарбонилалкилтио-, алкоксикарбонилалкокси-, алкоксикарбонилалкилтио-, аминогруппу, карбоксамидогруппу и аминоалкил,
где азот аминогруппы необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, гетероарил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил, арилкарбонил, аралканоил, гетероарилкарбонил, гетероаралканоил и алканоил, или
азот аминогруппы необязательно замещен двумя заместителями, которые с этим азотом аминогруппы образуют 5-8-членное гетероцикло- или гетероарильное кольцо, необязательно содержащее не более двух гетероатомов в дополнение к этому атому азота аминогруппы, независимо выбранных из группы, включающей азот, кислород и серу, и необязательно замещенное не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей арил, алкил, гетероарил, аралкил, гетероаралкил, гидрокси-, алкоксигруппу, алканоил, циклоалкил, гетероциклоалкил, алкоксикарбонил, гидроксиалкил, трифторметил, сконденсированный с бензольным кольцом гетероциклоалкил, гидроксиалкоксиалкил, аралкоксикарбонил, гидроксикарбонил, арилоксикарбонил, сконденсированную с бензольным кольцом гетероциклоалкоксигруппу, сконденсированный с бензольным кольцом циклоалкилкарбонил, гетероциклоалкилкарбонил и циклоалкилкарбонил,
где азот карбоксамидогруппы представляет собой реакционноспособный амин аминокислоты, или
азот карбоксамидогруппы необязательно замещен не более чем двумя заместителями, выбранными из группы, включающей алкил, гидроксиалкил, гидроксигетероаралкил, циклоалкил, аралкил, трифторметилалкил, гетероциклоалкил, сконденсированный с бензольным кольцом гетероциклоалкил, сконденсированный с бензольным кольцом циклоалкил и N,N-диалкилзамещенную алкиламиноалкильную группу, или
азот карбоксамидогруппы необязательно замещен двумя заместителями, образующими с этим азотом карбоксамидогруппы 5-8-членное гетероцикло-, гетероарильное или сконденсированное с бензольным кольцом гетероциклоалкильное кольцо, необязательно замещенное не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, алкоксикарбонил, нитрогруппу, гетероциклоалкил, гидроксигруппу, гидроксикарбонил, арил, аралкил, гетероаралкил и аминогруппу,
где азот аминогруппы необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил и гетероарил, или азот аминогруппы необязательно замещен двумя заместителями, которые вместе с этим азотом аминогруппы образуют 5-8-членное гетероцикло- или гетероарильное кольцо, и
где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил и алканоил, или где азот аминоалкила необязательно замещен двумя заместителями, которые вместе с этим азотом аминоалкила образуют 5-8-членное гетероцикло- или гетероарильное кольцо,
m выбирают из группы, включающей 0, 1 и 2,
n выбирают из группы, включающей 0, 1 и 2,
р выбирают из группы, включающей 0, 1 и 2,
сумму m+n+p выбирают из группы, включающей 1, 2, 3 и 4,
X, Y и Z имеют следующие значения:
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя из группы, включающей X, Y и Z, обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей Х и Y, обозначает CR8R9, или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей
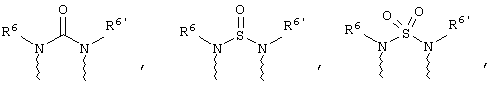
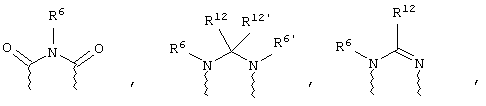
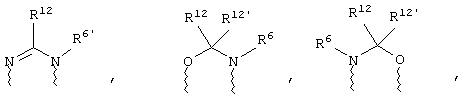
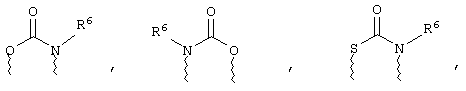
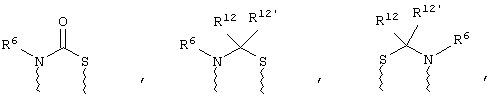
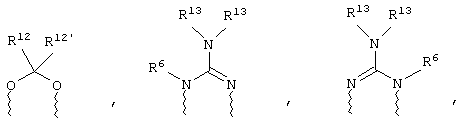
где волнистые линии обозначают связи с атомами изображенного кольца, R6 и R6′ независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкилкарбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкокси-С1-С6алкил, гетероарилтиоС1-С6алкил, С6арилсульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4алкоксикарбонил-С1-С6алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С5-С6гетероциклоиминокарбонил, С6apилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5алкилкарбонил, гидроксиС1-С5алкил, аминокарбонил, гидроксиаминокарбонил, аминосульфонил, аминоС1-С6 алкилсульфонил и аминоС1-С6алкил, где
азот аминокарбонила, аминосульфонила, аминоалкилсульфонила и аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6 карбоксиалкил и С1-С6гидроксиалкил,
R8 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R8 и R10 вместе с атомом (ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R9 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламинoС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R10 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R10 и R11 вместе с атомом(ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R11 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R10 и R11 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу,
при условии, что только один из R8 и R9 или R10 и R11 может обозначать гидроксигруппу,
R12 и R12′ независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероаралкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С2-С6алкинил, С2-С6алкенил и С1-С6гидроксиалкил, и R20 выбирают из группы, включающей -O-R и -NH-O-R22;
R21 выбирают из группы, включающей гидрид, С1-С6алкил, арил, apС1-С6алкил и фармацевтически приемлемый катион, и
R22 обозначает избирательно удаляемую защитную группу, причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
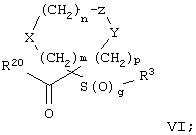
где
R3 обозначает заместитель G-A-R-E-Y2, где
G выбирают из группы, включающей арил и гетероарил,
А выбирают из группы, включающей
(1) -О-,
(2) -S-,
(3) -NR17-,
(4) -CO-NR17-,
(5) -NR17-CO-,
(6) -СО-O-,
(7) -O-СО-,
(8) -O-СО-О-,
(9) -НС=СН-,
(10) -NH-CO-NH-,
(11) -С=С-,
(12) -NH-CO-O-,
(13) -O-CO-NH-,
(14) -N=N-,
(15) -NH-NH-,
(16) -CS-N(R18)-,
(17) -N(R18)-CS - и
(18) связь;
R17 выбирают из группы, включающей гидрид, С1-С4алкил или фенил,
R18 выбирают из группы, включающей гидрид, С1-С4алкил или фенил, R обозначает фрагмент, выбранный из группы, включающей алкил, алкоксиалкил, арил, гетероарил, циклоалкил, гетероциклоалкил, аралкил, гетероаралкил, гетероциклоалкилалкил, циклоалкилалкил, циклоалкоксиалкил, гетероциклоалкоксиалкил, арилоксиалкил, гетероарилоксиалкил, арилтиоалкил, гетероарилтиоалкил, циклоалкилтиоалкил и гетероциклоалкилтиоалкил, где арил, гетероарил, циклоалкил или гетероциклоалкил необязательно замещены не более чем двумя заместителями, независимо выбранными из группы, включающей галоген, алкил, перфторалкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, аминогруппу, алкоксикарбонилалкил, алкокси-, С1-С2алкилендиоксигруппу, гидроксикарбонилалкил, гидроксикарбонилалкиламино-, нитро-, гидроксигруппу, гидроксиалкил, алканоиламиногруппу и алкоксикарбонил, и
R не обозначает алкил или алкоксиалкил, когда А обозначает -О- или -S-, Е выбирают из группы, включающей
(1) -CO(R19)-,
(2) -(R19)CO-,
(3) -CONH-,
(4) -HNCO-,
(5) -СО-,
(6) -SO2-R19-,
(7) -R19-SO2-,
(8) -SO2-,
(9) -NH-SO2-,
(10) -SO2-NH и
(11) связь;
R19 выбирают из группы, включающей гетероциклоалкил или циклоалкил,
Y2 выбирают из группы, включающей гидрид, алкил, алкоксигруппу, галоалкил, арил, аралкил, циклоалкил, гетероарил, гидрокси-, арилокси-, аралкокси-, гетероарилоксигруппу, гетероаралкил, перфторалкокси-, перфторалкилтиогруппу, трифторметилалкил, алкенил, гетероциклоалкил, циклоалкил, трифторметил, алкоксикарбонил и аминоалкил, где
арил, гетероарил или гетероциклоалкил необязательно замещены не более чем двумя радикалами, независимо друг от друга выбранными из группы, включающей алканоил, галоген, нитрогруппу, аралкил, арил, алкокси- и аминогруппу, где
азот в аминогруппе необязательно замещен не более чем двумя группами, независимо друг от друга выбранными из группы, включающей алкил и аралкил;
m выбирают из группы, включающей 0, 1 и 2,
n выбирают из группы, включающей 0, 1 и 2,
р выбирают из группы, включающей 0, 1 и 2,
сумму m+n+p выбирают из группы, включающей 1, 2, 3 и 4,
X, Y и Z имеют следующие значения:
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя из группы, включающей X, Y и Z, обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей Х и Y, обозначает CR8R9, или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей
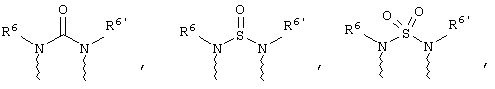
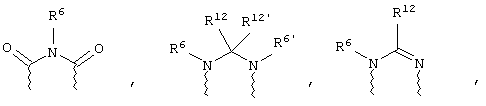
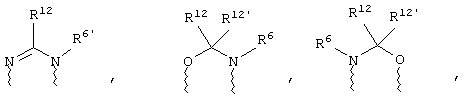

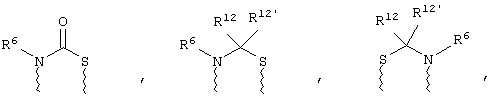
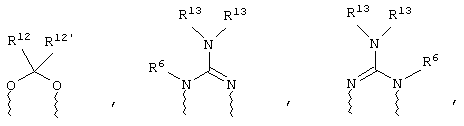
где волнистые линии обозначают связи с атомами изображенного кольца,
R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкилкарбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкокси-С1-С6алкил, гетероарилтиоС1-С6алкил, С6арилсульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4алкоксикарбонил-С1-С6алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С5-С6гетероциклоиминокарбонил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5алкилкарбонил, гидроксиС1-С5алкил, аминокарбонил, гидроксиаминокарбонил, аминосульфонил, аминоС1-С6 алкилсульфонил и аминоС1-С6алкил, где
азот аминокарбонила, аминосульфонила, аминоалкилсульфонила и аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
R8 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R8 и R10 вместе с атомом (ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R9 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R10 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R10 и R11 вместе с атомом(ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R11 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил, или
R10 и R11 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу,
при условии, что только один из R8 и R9 или R10 и R11 может обозначать гидроксигруппу,
R12 и R12' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероаралкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С2-С6алкинил, С2-С6алкенил и С1-С6гидроксиалкил, и R20 выбирают из группы, включающей -O-R21 и -NH-O-R22;
R21 выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил и фармацевтически приемлемый катион, и
R22 обозначает избирательно удаляемую защитную группу,
причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
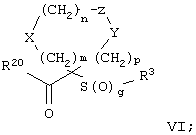
R3 выбирают из группы, включающей арил и гетероарил, где арил или гетероарил представляют собой 5- или 6-членную группу, замещенную в положении 4, когда она представляет собой 6-членное кольцо, или в положении 3 или 4, когда она представляет собой 5-членное кольцо, заместителем, выбранным из группы, включающей тиофенокси-, 4-хлорфенокси-, 3-хлорфенокси-, 4-метоксифенокси-, 3-бензодиоксол-5-илокси-, 3,4-диметилфенокси-, 4-фторфенокси-, 4-фтортиофенокси-, фенокси-, 4-трифторметоксифенокси-, 4-трифторметилфенокси-, 4-(трифторметилтио) фенокси-, 4 - (трифторметилтио) тиофенокси-, 4-хлор-3-фторфенокси-, 4-изопропоксифенокси-, 4-изопропилфенокси-, (2-метил-1,3-бензотиазол-5-ил)окси-, 4-(1Н-имидазол-1-ил)фенокси-, 4-хлор-3-метилфенокси-, 3-метилфенокси-, 4-этоксифенокси-, 3,4-дифторфенокси-, 4-хлор-3-метилфенокси-, 4-фтор-3-хлорфенокси-, 4-(1Н-1,2,4-триазол-1-ил)фенокси-, 3,5-дифторфенокси-, 3,4-дихлорфенокси-, 4-циклопентилфенокси-, 4-бром-3-метилфенокси-, 4-бромфенокси-, 4-метилтиофенокси-, 4-фенилфенокси-, 4-бензилфенокси-, 6-хинолинилокси-, 4-амино-3-метилфенокси-, 3-метоксифенокси-, 5,6,7,8-тетрагидро-2-нафталинилокси-, 3-гидроксиметилфенокси- и 4-бензилоксифеноксигруппу,
m выбирают из группы, включающей 0, 1 и 2,
n выбирают из группы, включающей 0, 1 и 2,
р выбирают из группы, включающей 0, 1 и 2,
сумму m+n+p выбирают из группы, включающей 1, 2, 3 и 4,
X, Y и Z имеют следующие значения:
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя из группы, включающей X, Y и Z, обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей Х и Y, обозначает CR8R9 или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей
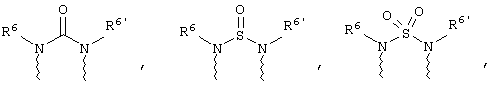
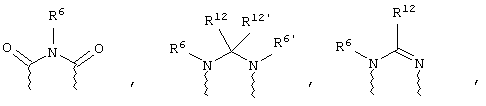
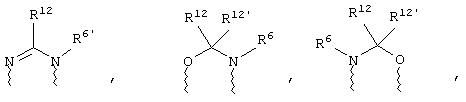

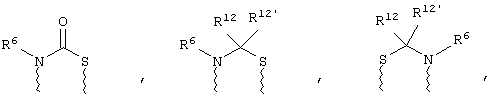

где волнистые линии обозначают связи с атомами изображенного кольца, R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С8циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкилкарбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкокси-С1-С6алкил, гетероарилтиоС1-С6алкил, С6арилсульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4алкоксикарбонил-С1-С6алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С5-С6гетероциклоиминокарбонил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5aлкилкapбoнил, гидроксиС1-С5алкил, аминокарбонил, гидроксиаминокарбонил, аминосульфонил, аминоС1-С6алкилсульфонил и аминоС1-С6алкил, где
азот аминокарбонила, аминосульфонила, аминоалкилсульфонила и аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
R8 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилокси С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R8 и R10 вместе с атомом(ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R9 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R10 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6 алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6 алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R10 и R11 вместе с атомом(ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R11 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6 алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R10 и R11 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу,
при условии, что только один из R8 и R9 или R10 и R11 может обозначать гидроксигруппу,
R12 и R12' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероаралкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С2-С6алкинил, С2-С6алкенил и
С1-С6гидроксиалкил, и
R20 выбирают из группы, включающей -O-R21 и -NH-O-R22;
R21 выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил и фармацевтически приемлемый катион, и
R22 обозначает избирательно удаляемую защитную группу,
причем
во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
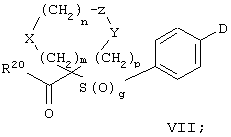
где
g выбирают из группы, включающей 0, 1 и 2,
D обозначает способную к нуклеофильному замещению уходящую группу,
m выбирают из группы, включающей 0, 1 и 2,
n выбирают из группы, включающей 0, 1 и 2,
р выбирают из группы, включающей 0, 1 и 2,
сумму m+n+p выбирают из группы, включающей 1, 2, 3 и 4,
X, Y и Z имеют следующие значения:
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя из группы, включающей X, Y и Z, обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС (О), при этом один оставшийся заместитель из группы, включающей Х и Y, обозначает CR8R9, или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей

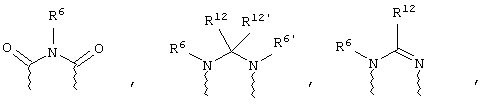
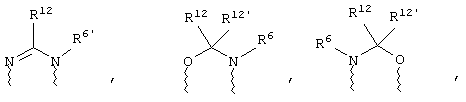

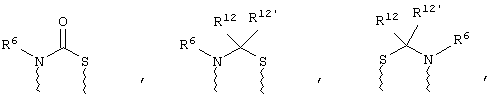
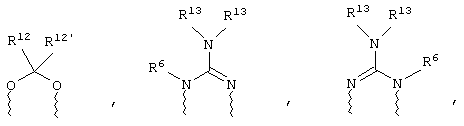
где волнистые линии обозначают связи с атомами изображенного кольца, R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкилкарбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкокси-С1-С6алкил, гетероарилтиоС1-С6алкил, С6арилсульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4алкоксикарбонил-С1-С6алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С5-С6гетероциклоиминокарбонил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5алкилкарбонил, гидроксиС1-С5алкил, аминокарбонил, гидроксиаминокарбонил, аминосульфонил, аминоС1-С6алкилсульфонил и аминоС1-С6алкил, где
азот аминокарбонила, аминосульфонила, аминоалкилсульфонила и аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С3-С8алкил, apС1-С6алкил, С1-С6циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
R8 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R8и R10 вместе с атомом(ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R9 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R10 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6 алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R10 и R11 вместе с атомом (ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R11 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R10 и R11 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу,
при условии, что только один из R8 и R9 или R10 и R11 может обозначать гидроксигруппу,
R12 и R12' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероаралкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфтор С1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С2-С6алкинил, С2-С6алкенил и С1-С6гидроксиалкил, и
R20 выбирают из группы, включающей -O-R21 и -NH-O-R22;
R21 выбирают из группы, включающей гидрид, С1-С6алкил, арил, арС1-С6алкил и фармацевтически приемлемый катион, и
R22 обозначает избирательно удаляемую защитную группу,
причем во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и
во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
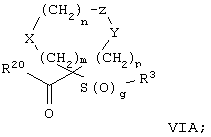
включающий стадию сочетания промежуточного соединения, представленного формулой VIB
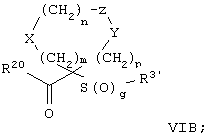
с подходящим для получения целевого продукта соединением, где в приведенных выше формулах VIA и VIB
g выбирают из группы, включающей 0, 1 и 2,
R3' выбирают из группы, включающей арил и гетероарил, где арил или гетероарил замещены способной к нуклеофильному замещению уходящей группой, выбранной из ряда, включающего галоген, нитро-, азидо-, фенилсульфоксидо-, арилокси-, С1-С6алкоксигруппу, С1-С6алкилсульфонат или арилсульфонат и тризамещенную аммонийную группу, где заместители тризамещенной аммонийной группы независимо друг от друга выбирают из группы, включающей арил, apС1-С6алкил и С1-С6алкил;
R3 выбирают из группы, включающей арил и гетероарил, где арил или гетероарил замещены заместителем, выбранным из группы, включающей циклоалкил, гетероциклоалкил, арил, гетероарил, аралкил, гетероаралкил, аралокси-, гетероаралкоксигруппу, аралкоксиалкил, арилоксиалкил, аралканоилалкил, арилкарбонилалкил, аралкиларил, арилоксиалкиларил, аралкоксиарил, арилазоарил, арилгидразиноарил, алкилтиоарил, арилтиоалкил, алкилтиоаралкил, аралкилтиоалкил, аралкилтиоарил, сульфоксид любого из тиозаместителей, сульфон любого из тиозаместителей, и сконденсированную кольцевую структуру, включающую по крайней мере два 5-6-членных кольца, выбранных из группы, включающей арил, гетероарил, циклоалкил и гетероциклоалкил, где
каждый такой заместитель необязательно замещен одним или более заместителями, независимо друг от друга выбранными из группы, включающей цианогруппу, перфторалкил, трифторметокси-, трифторметилтиогруппу, галоалкил, трифторметилалкил, аралкоксикарбонил, арилоксикарбонил, гидроксигруппу, галоген, алкил, алкокси-, нитрогруппу, тиол, гидроксикарбонил, арилокси-, арилтиогруппу, аралкил, арил, арилкарбониламино-, гетероарилокси-, гетероарилтиогруппу, гетероаралкил, циклоалкил, гетероциклоокси-, гетероциклотио-, гетероциклоамино-, циклоалкилокси-, циклоалкилтио-, гетероаралкокси-, гетероаралкилтио-, аралкокси-, аралкилтио-, аралкиламино-, гетероциклогруппу, гетероарил, арилазо-, гидроксикарбонилалкокси-, алкоксикарбонилалкоксигруппу, алканоил, арилкарбонил, аралканоил, алканоилокси-, аралканоилоксигруппу, гидроксиалкил, гидроксиалкокси-, алкилтио-, алкоксиалкилтиогруппу, алкоксикарбонил, арилоксиалкоксиарил, арилтиоалкилтиоарил, арилоксиалкилтиоарил, арилтиоалкилтиоарил, арилтиоалкоксиарил, гидроксикарбонилалкокси-, гидроксикарбонилалкилтио-, алкоксикарбонилалкокси-, алкоксикарбонилалкилтио-, аминогруппу, карбоксамидогруппу и аминоалкил,
где азот аминогруппы необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, гетероарил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил, арилкарбонил, аралканоил, гетероарилкарбонил, гетероаралканоил и алканоил, или
азот аминогруппы необязательно замещен двумя заместителями, которые с этим азотом аминогруппы образуют 5-8-членное гетероцикло- или гетероарильное кольцо, необязательно содержащее не более двух гетероатомов в дополнение к этому атому азота аминогруппы, независимо выбранных из группы, включающей азот, кислород и серу, и необязательно замещенное не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей арил, алкил, гетероарил, аралкил, гетероаралкил, гидрокси-, алкоксигруппу, алканоил, циклоалкил, гетероциклоалкил, алкоксикарбонил, гидроксиалкил, трифторметил, сконденсированный с бензольным кольцом гетероциклоалкил, гидроксиалкоксиалкил, аралкоксикарбонил, гидроксикарбонил, арилоксикарбонил, сконденсированную с бензольным кольцом гетероциклоалкоксигруппу, сконденсированный с бензольным кольцом циклоалкилкарбонил, гетероциклоалкилкарбонил и циклоалкилкарбонил,
где азот карбоксамидогруппы представляет собой реакционноспособный амин аминокислоты, или
азот карбоксамидогруппы необязательно замещен не более чем двумя заместителями, выбранными из группы, включающей алкил, гидроксиалкил, гидроксигетероаралкил, циклоалкил, аралкил, трифторметилалкил, гетероциклоалкил, сконденсированный с бензольным кольцом гетероциклоалкил, сконденсированный с бензольным кольцом циклоалкил и N,N-диалкилзамещенную алкиламиноалкильную группу, или
азот карбоксамидогруппы необязательно замещен двумя заместителями, образующими с этим азотом карбоксамидогруппы 5-8-членное гетероцикло-, гетероарильное или сконденсированное с бензольным кольцом гетероциклоалкильное кольцо, необязательно замещенное не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, алкоксикарбонил, нитрогруппу, гетероциклоалкил, гидроксигруппу, гидроксикарбонил, арил, аралкил, гетероаралкил и аминогруппу,
где азот аминогруппы необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил и гетероарил, или азот аминогруппы необязательно замещен двумя заместителями, которые вместе с этим азотом аминогруппы образуют 5-8-членное гетероцикло- или гетероарильное кольцо, и
где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей алкил, арил, аралкил, циклоалкил, аралкоксикарбонил, алкоксикарбонил и алканоил, или где азот аминоалкила необязательно замещен двумя заместителями, которые вместе с этим азотом аминоалкила образуют 5-8-членное гетероцикло- или гетероарильное кольцо,
m выбирают из группы, включающей 0, 1 и 2,
n выбирают из группы, включающей 0, 1 и 2,
р выбирают из группы, включающей 0, 1 и 2,
сумму m+n+p выбирают из группы, включающей 1, 2, 3 и 4,
X, Y и Z имеют следующие значения:
(а) один из заместителей X, Y и Z выбирают из группы, включающей С(O), NR6, О, S, S(O), S(O)2 и NS(O)2R7, а два остальных заместителя из группы, включающей X, Y и Z, обозначают CR8R9 и CR10R11, или
(б) Х и Z или Z и Y вместе образуют фрагмент, который выбирают из группы, включающей NR6C(O), NR6S(O), NR6S(O)2, NR6S, NR6O, SS, NR6NR6 и ОС(O), при этом один оставшийся заместитель из группы, включающей Х и Y, обозначает CR8R9, или
(в) n равно 0 и X, Y и Z вместе образуют фрагмент, выбранный из группы, включающей
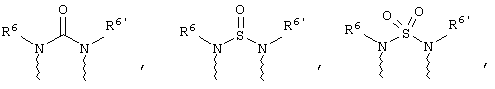
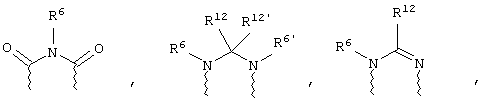
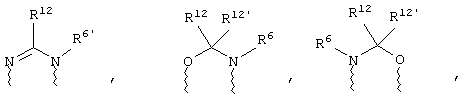
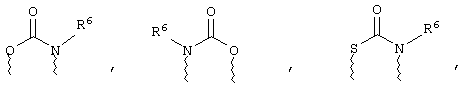
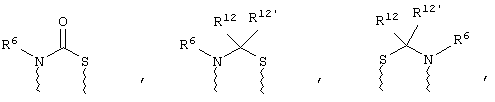
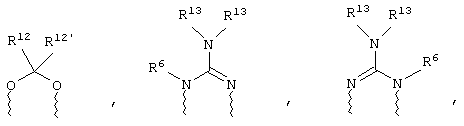
где волнистые линии обозначают связи с атомами изображенного кольца, R6 и R6' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алканоил, С6арил-С1-С6алкил, ароил, бис(С1-С6алкокси-С1-С6алкил)-С1-С6алкил, С1-С6алкил, С1-С6галоалкил, С1-С6перфторалкокси-С1-С6алкил, С1-С6алкокси-С1-С6алкил, С3-С6циклоалкил, С3-С8гетероциклоалкил, С3-С8гетероциклоалкилкарбонил, С6арил, С5-С6гетероциклогруппу, С5-С6гетероарил, С3-С8циклоалкил-С1-С6алкил, С6арилокси-С1-С6алкил, гетероарилоксиС1-С6алкил, гетероарилС1-С6алкокси-С1-С6алкил, гетероарилтиоС1-С6алкил, С6арилсульфонил, С1-С6алкилсульфонил, С5-С6гетероарилсульфонил, карбоксиС1-С6алкил, С1-С4алкоксикарбонил-С1-С6алкил, аминокарбонил, С1-С6алкилиминокарбонил, С6арилиминокарбонил, С5-С6гетероциклоиминокарбонил, С6арилтио-С1-С6алкил, С1-С6алкилтио-С1-С6алкил, С6арилтио-С3-С6алкенил, С1-С4алкилтио-С3-С6алкенил, С5-С6гетероарил-С1-С6алкил, галоС1-С6алканоил, гидроксиС1-С6алканоил, тиолС1-С6алканоил, С3-С6алкенил, С3-С6алкинил, С1-С5алкоксикарбонил, арилоксикарбонил, NR8R9-С1-С5алкилкарбонил, гидроксиС1-С5алкил, аминокарбонил, гидроксиаминокарбонил, аминосульфонил, аминоС1-С6алкилсульфонил и аминоС1-С6алкил, где
азот аминокарбонила, аминосульфонила, аминоалкилсульфонила и аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, С3-С8циклоалкил и С1-С6алканоил,
R7 выбирают из группы, включающей арилалкил, арил, гетероарил, гетероциклогруппу, С1-С6алкил, С3-С6алкинил, С3-С6алкенил, С1-С6карбоксиалкил и С1-С6гидроксиалкил,
R8 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламинoС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R8 и R 10 вместе с атомом (ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R9 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламинoС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R10 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил, или
R8 и R9 или R10 и R11 вместе с атомом(ами) углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу, и
R11 выбирают из группы, включающей гидрид, гидроксигруппу, С1-С6алкил, арил, арС1-С6алкил, гетероарил, гетероарС1-С6алкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, С1-С6алкилтио-С1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, гало, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где
азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, apС1-С6алкил, циклоалкил и С1-С6алканоил, или
R10 и R11 вместе с атомом углерода, с которым они связаны, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один или два гетероатома, независимо выбранные из группы, включающей азот, кислород и серу,
при условии, что только один из R8 и R9 или R10 и R11 может обозначать гидроксигруппу,
R12 и R12' независимо друг от друга выбирают из группы, включающей гидрид, С1-С6алкил, арил, apС1-С6алкил, гетероарил, гетероаралкил, С2-С6алкинил, С2-С6алкенил, тиолС1-С6алкил, циклоалкил, циклоалкилС1-С6алкил, гетероциклоалкилС1-С6алкил, С1-С6алкокси-С1-С6алкил, аралкоксиС1-С6алкил, аминоС1-С6алкил, С1-С6алкокси-С1-С6алкокси-С1-С6алкил, гидроксиС1-С6алкил, гидроксикарбонилС1-С6алкил, гидроксикарбониларС1-С6алкил, аминокарбонилС1-С6алкил, арилоксиС1-С6алкил, гетероарилоксиС1-С6алкил, С1-С6алкилтио-С1-С6алкил, арилтиоС1-С6алкил, гетероарилтиоС1-С6алкил, сульфоксид любого из этих тиозаместителей, сульфон любого из этих тиозаместителей, перфторС1-С6алкил, трифторметилС1-С6алкил, галоС1-С6алкил, алкоксикарбониламиноС1-С6алкил и аминоС1-С6алкил, где азот аминоалкила необязательно замещен не более чем двумя заместителями, независимо друг от друга выбранными из группы, включающей С1-С6алкил, арС1-С6алкил, циклоалкил и С1-С6алканоил,
R13 выбирают из группы, включающей гидрид, бензил, фенил, С1-С6алкил, С2-С6алкинил, С2-С6алкенил и С1-С6гидроксиалкил, и
R20 выбирают из группы, включающей -O-R21 и -NH-O-R22;
R21 выбирают из группы, включающей гидрид, С1-С6алкил, арил, apС1-С6алкил и фармацевтически приемлемый катион, и
R22 обозначает избирательно удаляемую защитную группу,
причем во всех случаях за исключением специально оговоренных циклоалкил представляет собой циклический алкильный радикал, содержащий от трех до примерно восьми атомов углерода;
во всех случаях за исключением специально оговоренных арил выбирают из группы, содержащей фенил, инденил и нафтил; и во всех случаях за исключением специально оговоренных гетероарил обозначает 5- или 6-членный ароматический содержащий кольцо фрагмент или сконденсированную кольцевую систему, содержащую два или три кольца, которые содержат в цикле атомы углерода, а также один или несколько гетероатомов, таких как сера, кислород и азот.
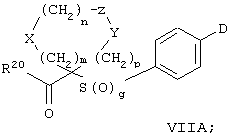
где D обозначает замещаемую нуклеофилом уходящую группу, которую выбирают из ряда, включающего галоген, нитро-, азидо-, фенилсульфоксидо-, арилокси-, С1-С6алкоксигруппу, С1-С6алкилсульфонат или арилсульфонат и тризамещенную аммонийную группу, где заместители тризамещенной аммонийной группы независимо друг от друга выбирают из группы, включающей арил, apС1-С6алкил и С1-С6алкил.
| RU 95109920, А1, 27.10.1996 | |||
| WO 9727117, A1, 10.07.1997 | |||
| ЕР 780386, А1, 25.06.1997. |

