Предлагаемое изобретение относится к области антивирусной терапии, в частности, предлагает ненуклеозидные ингибиторы ревертазы (обратной транскриптазы) для лечения заболеваний, вызванных вирусом иммунодефицита человека (ВИЧ). Изобретение предлагает новые соединения пиразола, фармацевтические композиции, включающие эти соединения, методы лечения или профилактики заболеваний, вызванных ВИЧ, при использовании указанных соединений в монотерапии или в комплексной терапии.
Вирус иммунодефицита человека (ВИЧ) является возбудителем синдрома приобретенного иммунодефицита (СПИД), заболевания, характеризующегося нарушением иммунной системы, в частности, клеток CD4+Т с сопровождающейся подверженностью инфекциям. ВИЧ инфекция является также предшественницей комплекса заболеваний, связанных со СПИД (КЗСС), синдромом, характеризующимся такими проявлениями, как устойчивая генерализованная лимфоаденопатия, лихорадка и потеря веса. Вместе с другими ретровирусами геном ВИЧ кодирует протеиновые предшественники, известные как ген вируса гэг (gag) и комбинация генов гэг-пол, которые производятся вирусной протеазой для создания протеазы, ревертазы (Р), эндонуклеазы/интегразы и сформировавшихся структурных белков ядра вируса. Прерывание этого процесса предотвращает производство нормальных вирусов инфекции. Предпринимаются значительные усилия для ограничения ВИЧ путем ингибирования ферментов, кодируемых вирусом.
Современная химиотерапия «бьет» по следующим ключевым вирусным ферментам: ВИЧ протеазе и ВИЧ ревертазе (J.S.G. Montaner и др., Antiretroviral therapy: "The State of the Art", Biomed. и Pharmacother, 53, 1999, сс. 63-72; R.W.Shafer и D.A.Vuitton, Highly active antiretroviral therapy (HAART) for the treatment of infection with human immunodeficiency virus type 1, Biomed. & Pharmacother, 53, 1999, cc.73-86; E. De Clercq, New Developments in Anti-HIV Chemotherap., Curr. Med. Chem., 8, 2001, cc.1543-1572). Идентифицированы два основных класса ингибиторов ревертазы (ИР): нуклеозидные ингибиторы ревертазы (НИР) и ненуклеозидные ингибиторы ревертазы (ННИР).
Типичными представителями НИР являются аналоги 2′,3′-дидеоксинуклеозида (ддН), которые должны быть фосфорилированы до взаимодействия с вирусной Р. Соответствующие трифосфаты действуют как конкурентные ингибиторы или альтернативные субстраты для вирусной Р. После внедрения в нуклеиновые кислоты нуклеозидные аналоги встраиваются в конец цепи процесса удлинения. ВИЧ ревертаза имеет ДНК, имеющую такие характеристики, которые вызывают устойчивость к подавлению блокады, создаваемой присоединяющимся нуклеозидным аналогом, и способность к продолжению цепи. Современные используемые НИР включают зидовудин (AZT), диданозин (ddI), зальцитабин (ddC), ставудин (d4T), ламивудин (3ТС) и тенофовир (РМРА).
ННИР впервые были открыты в 1989 г. ННИР являются аллостерическими ингибиторами, которые обратимо привязываются к несубстратному связывающему месту в ВИЧ ревертазе, изменяя таким образом форму активного места связывания или блокируя полимеразную активность (R.W.Buckheit, мл., Non-nucleoside reverse transcriptase inhibitors: perspectives for novel therapeutic compounds and strategies for treatment of HIV infection, Expert Opin. Investig. Drugs, 10, №8, 2001, cc.1423-1442; E. De Clercq, The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antiviral Res., 38, 1998, cc.153-179; G. Moyle, The Emerging Roles of Non-Nucleoside Reverse Transcriptase Inhibitors in Antiviral Therapy, Drugs, 61, №1, 2001, cc.19-26). В лабораториях были идентифицированы более тридцати структурных классов ННИР, но только три соединения опробованы в лечении ВИЧ: эфавиренз, невирапин и делавирдин. Заявленные вначале как обещающие, многие классы соединений ННИР при изучениях in vitro и in vivo проявляли низкий барьер к возникновению устойчивых к лекарствам штаммов ВИЧ при использовании в монотерапии и к тому же имели высокую класс-специфическую токсичность. Устойчивость к действию лекарства часто проявляется уже при единичной мутации в Р.
При комбинированной терапии с применением НИР и ННИР вирусная нагрузка существенно снижается, и развитие заболевания замедляется, однако, остаются значительные терапевтические проблемы. Смеси лекарств не являются эффективными для всех пациентов, часто возникают острые негативные реакции, доказана также быстрая репродукция ВИЧ вирусов, умело создающая мутантные, устойчивые к лекарствам варианты дикого типа протеазы и ревертазы.
Остается необходимость создания безопасных лекарств против дикого типа и часто возникающих устойчивых штаммов ВИЧ.
WO 02/100852 (В.W.Dymock и др.) описывает новые производные пиразола, процессы получения новых пиразолов, фармацевтические композиции, содержащие пиразолы, и применение пиразолов в качестве ингибиторов ревертазного фермента вируса ВИЧ, который включен в репликацию вируса. WO 02/30907 (В.W.Dymock и др.) также описывает новые пиразолы, пригодные для ингибирования ревертазы ВИЧ. Эти патенты полностью включены в предлагаемое изобретение в виде ссылок.
US 6,005,109 (W.S.Faraci) EP 0691128 (G.M.Bright и др.) и ЕР 0959074 (G.M.Bright и др.) описывают производные пиразола, которые обладают активностью антагонистов высвобождения кортикотропина.
ЕР 1072597 (Banks, B.J. и др.) описывает производные пиразола с активностью антагонистов эндотелина. WO 97/04773 (J.I.Luengo и др.) описывает фенилпиразолы в качестве антагонистов рецепторов эндотелина для лечения сердечно-сосудистых или почечных нарушений.
WO 02/04424 (R.G.Corbau и др.) описывает использование производных пиразола для получения ингибитора или модулятора ревертазы, новые производные пиразола и процессы получения производных пиразола и композиций, включающих новые производные пиразола. WO 02/085860 (L.Н.Jones и др.) описывает пиразольные соединения, процессы получения пиразольных соединений и применение этих соединений для ингибирования или модулирования вирусного фермента ревертазы. Описано также применение пиразолов для лечения заболеваний, вызванных вирусом иммунодефицита человека (ВИЧ).
WO 00/66562 (V.В.Lohray и др.) описывает фенилсульфенил-, фенилсульфонил- и фенилтиозамещенные соединения пиразола, которые ингибируют r-hu COX-2, что является полезным для ингибирования биосинтеза простагландинов и лечения боли, вызванной лихорадкой или воспалением. WO 01/16138 (Т.Kolasta и М.V.Patel) и WO 01/64669 (Н. Cheng и др.) также описывает сульфонилфенилзамещенные пиразолы, ингибирующие СОХ-2.
Описаны гидроксипиразолы, имеющие пестицидную активность. WO 99/33813 (Р.Desbordes и др.) описывает арилоксипиразолы с фунгицидной активностью.
Предлагаемое изобретение включает соединения формулы I, методы лечения заболеваний, вызванных вирусом иммунодефицита человека, путем введения соединений формулы I и фармацевтических композиций, предназначенных для лечения заболеваний, вызванных вирусом иммунодефицита человека, и содержащих соединение формулы I,
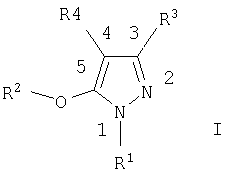
в котором
R1 выбирают из ряда, содержащего C1-С6алкил, C1-С6галоалкил, С3-С6алкенил, С3-С6алкинил, С3-С7циклоалкил, С1-С3алкокси-С1-С3алкил, фенил и бензил, в которых указанные фенил и бензил необязательно замещены одним-тремя заместителями, независимо выбранными из ряда, содержащего группы C1-С6алкил, C1-С6галоидалкил, C1-С6алкокси-, C1-С6галоидалкокси-, C1-С6алкилтио-, нитро-, циано-группу или галоид;
R2 означает фенил или пиридил, необязательно замещенные одним или тремя заместителями, независимо выбранными из ряда, содержащего группы: галоид, циано, C1-С6алкил, C1-С6алкокси, C1-С6алкоксикарбонил и CONR6R7;
R3 означает замещенный С1-С6алкил, замещенный C1-С3алкокси-С1-С3алкил, замещенный С3-С6алкенил, С3-С7циклоалкил, необязательно замещенную C1-С6алкокси-группу, -(CH2)nR5, -CH(OH)R5, -(CH2)o-O-(CH2)pR5, NR6R7, C(=Y)Z, -X(C=Y)Z или группы IIa-c,
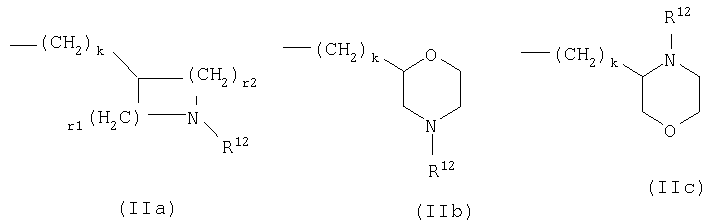
в которых
указанный алкил, С1-С3алкокси-С1-С3алкил и алкенил замещены группами -ОН,
-NR6R7, -C(=Y)Z, -X(C=Y)Z, CN, -S(O)q-С1-С6алкил; -SO2NR6R7, -SO2NHNH2 или NR6SO2-C1-C6алкил;
указанная алкокси-группа необязательно замещена группами -ОН, -NR6R7,
-C(=Y)Z, -X(C=Y)Z, CN, -S(O)q-C1-C6 алкил, -SO2NR6R7 или -SO2NHNH2;
R12 означает водород, C1-С6алкил или -C(=Y)Z;
R5 означает фенил или гетероарильное кольцо в соответствии с формулами IIIa-IIIh,
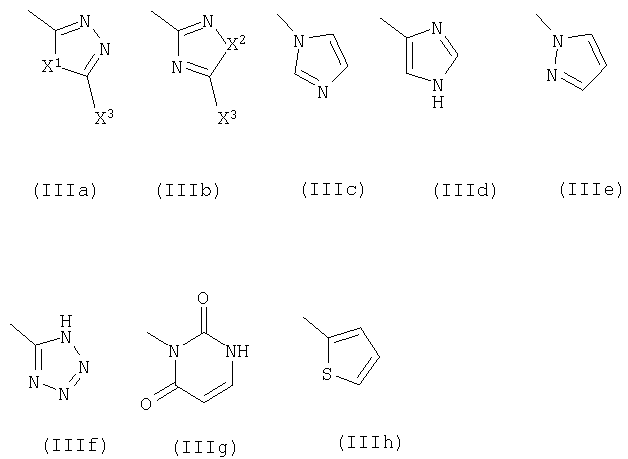
в которых
Х1 выборочно означает группы, состоящие из -R10C=CR10а-, -О-, -S-, -NR6- и -CHR6;
Х2 выборочно означает группы, состоящие из -R10C=CR10а-, -О-, -S- и -CHR6;
Х3 выборочно означает группы, состоящие из водорода, гидроксила и тиола; указанные фенил и гетероарильное кольцо необязательно замещены галоидом, группами OR6, NR6R7, C(=O)Z, -X(C=O)Z;
R10 и R10a независимо выбираются из ряда, состоящего из водорода или C1-С6алкила, необязательно замещенного одним или двумя заместителями, независимо выбранными из ряда, содержащего группы: гидро-, C1-С6алкокси-, тиол, C1-С6алкилтио-, C1-С6алкилсульфонил, галоид, амино-, C1-С6алкиламино-, C1-С3диалкиламино-, амино-С1-С3алкил, С1-С3алкиламино-C1-С3алкил и C1-С3диалкиламино-С1-С3алкил;
R4 означает C1-С6алкил, С2-С6алкенил, С2-С6алкинил, С3-С7циклоалкил, C1-С3 алкокси-C1-С3алкил, (CH2)nR11 или -(CH2)o-O-(CH2)pR11, где
указанные алкил, алкенил, алкинил и циклоалкил могут быть выборочно замещены группами -ОН, -OR6, -NR8R9, -C(-Y)Z, -X(C=Y)Z, -S(O)q-C1-С6алкил, -SO2NR6R7 или -SO2NHNH2;
R11 означает фенил или гетероарильное кольцо, выбранное из ряда, содержащего: пиридинил, пиримидинил, пиразинил, пиррол, имидазол, пиразол и тиофен; указанные гетероарильное кольцо и фенил могут быть выборочно замещены одной -тремя независимо выбранными группами, представляющими собой галоид, циано-, C1-С3алкил, C1-С3галоидалкил и C1-С3алкокси; или R11 означает N[(CH2)2]2W, где W выбирают из ряда, содержащего группы: NR6, (CH2)s, -N(C=O)Z, CHOR6, CHR6 CHNHC(=O)Z и CHNR6R7;
n, о и р определены ниже, a s означает 0 или 1;
R6, R7 и R9 (а) независимо выбраны из ряда, состоящего из групп: водород, C1-С6 алкил, C1-С6гидроксиалкил, С1-С3алкокси-С1-С3алкил, C1-С3алкиламино-C1-С3алкил и С1-С3диалкиламино-С1-С3алкил или (б) R6 и R7 вместе с атомом азота, к которому они присоединены, образуют пирролидин, пиперидин, пиперазин или морфолин;
Х и Y независимо друг от друга означают О или NR6;
Z означает водород, гидроксил, C1-С6алкоксигруппу, NR6R13, C1-С6алкил, C1-С3алкокси-С1-С3алкил, где R13 означает R7 или фенил, необязательно замещенный одной -тремя группами, независимо выбранными из ряда: галоид, циано-, C1-С3 алкил, C1-С3галоидалкил и C1-С3алкоксигруппа;
n означает от 0 до 3;
о и р независимо означают от 0 до 4 и о+р≤5;
q означает от 0 до 2;
k, r1 и r2 независимо означают от 0 до 4, и 5≥(r1+r2)≥2;
а также их кислотно-аддитивные соли, гидраты и сольваты;
с условием, что в тех случаях, когда R4 означает (CH2)nR11, n равно 1, а R11 означает замещенный фенил, R2 не означает незамещенный фенил.
Одна часть предлагаемого изобретения относится к соединению, соответствующему формуле I,
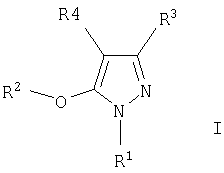
в которой R1, R2, R3 и R4 определены выше, а также его гидратам, сольватам и кислотно-аддитивным солям.
Другая часть предлагаемого изобретения относится к соединению, соответствующему формуле I, в которой R1 выбрано из ряда, состоящего из групп: C1-С6алкил, C1-С6галоидалкил, С3-С7циклоалкил, С1-С3алкокси-С1-С3алкил и необязательно замещенный фенил; R2 означает необязательно замещенный фенил; R4 означает C1-С6алкил, Сз-С7циклоалкил, -(CH2)nR11 или -(CH2)о-О-(CH2)pR11, где
указанные алкил и циклоалкил выборочно замещены группами -ОН, -OR6, -NR8R9, -C(=Y)Z, -X(C=Y)Z; R11 означает необязательно замещенный фенил; а R2 и другие группы определены выше.
Другая часть предлагаемого изобретения относится к соединению, соответствующему формуле I, в которой R1 выбрано из ряда, состоящего из групп: C1-С6алкил, C1-С6галоидалкил, С3-С7циклоалкил, C1-С3алкокси-С1-С3алкил и необязательно замещенный фенил; R2 означает необязательно замещенный фенил; R3 означает C1-С6алкил, -(CH2)nR5, где R5 соответствует формулам IIIa-IIIh или IIa-с, R4 означает C1-С6алкил, С3-С7циклоалкил, -(CH2)nR11 или -(CH2)o-O-(CH2)pR11, где указанные алкил и циклоалкил выборочно замещены группами -ОН, -OR6, -NR8R9, -C(=Y)Z, -X(C=Y)Z; R11 означает необязательно замещенный фенил; а другие группы определены выше.
Другая часть предлагаемого изобретения относится к соединению, соответствующему формуле I, в которой R1 выбрано из ряда, состоящего из C1-С6алкила, C1-С6галоидалкила, С3-С7циклоалкила, С1-С3алкокси-С1-С3алкила и фенила; R2 означает необязательно замещенный фенил; R3 означает -(CH2)nNR6R7, -(CH2)nC(=O)Z или -(CH2)nXC(=O)Z; R4 означает C1-С6алкил, С3-С7циклоалкил, -(CH2)nR1 или -(CH2)o-O-(CH2)pR11, где указанные алкил и циклоалкил выборочно замещены группами -ОН, -OR6, -NR8R9, -C(=Y)Z, -X(C=Y)Z; R11 означает необязательно замещенный фенил; а другие группы определены выше.
Другая часть предлагаемого изобретения относится к соединению, соответствующему формуле I, в которой R1 выбрано из ряда, состоящего из C1-С6алкила, C1-С6галоидалкила, С3-С7циклоалкила, С1-С3алкокси-С1-С3алкила и необязательно замещенного фенила; R2 означает необязательно замещенный фенил; R4 означает C1-С6алкил, С3-С7циклоалкил, -(CH2)nR11 или -(СН2)о-O-(CH2)pR11, где указанные алкил и циклоалкил выборочно замещены группами -ОН, -OR6, -NR8R9, -C(=Y)Z, -X(C=Y)Z; R11 означает необязательно замещенное гетероарильное кольцо, выбранное из ряда, состоящего из групп: пиридинил, пиримидинил, пиразинил, пиррол, имидазол, пиразол и тиофен; а R3 и другие группы определены выше.
Другая часть предлагаемого изобретения относится к соединению, соответствующему формуле I, в которой R1 выбрано из ряда, состоящего из C1-С6алкила, C1-С6галоидалкила, С3-С7циклоалкила, C1-С3алкокси-С1-С3алкила и необязательно замещенного фенила; R2 означает необязательно замещенный фенил; R3 означает C1-С6алкил, -(CH2)nR5, где R5 соответствует формулам IIIa-IIIh или IIa-с, R4 означает C1-С6алкил, С3-С7циклоалкил, -(CH2)nR11 или -(СН2)о-O-(CH2)pR11, где указанные алкил и циклоалкил выборочно замещены группами -ОН, -OR6, -NR8R9, -C(=Y)Z, -X(C=Y)Z; R11 означает необязательно замещенное гетероарильное кольцо, а другие группы определены выше.
Другая часть предлагаемого изобретения относится к соединению, соответствующему формуле I, в которой R1 выбрано из ряда, состоящего из C1-С6 алкила, C1-С6галоидалкила, С3-С7циклоалкила, С1-С3алкокси-С1-С3алкила и необязательно замещенного фенила; R2 означает необязательно замещенный фенил; R3 означает -(CH2)nNR6R7, -(CH2)nC(=O)Z или -(СН2)nХС(=O)Z; R4 означает C1-С6алкил, С3-С7циклоалкил, -(CH2)nR11 или -(CH2)o-O-(CH2)pR11, где указанные алкил и циклоалкил необязятельно замещены группами -ОН, -OR6, -NR8R9, -C(=Y)Z, -X(C=Y)Z; R11 означает необязательно замещенное гетероарильное кольцо, выбранное из ряда, состоящего из пиридинила, пиримидинила, пиразинила, пиррола, имидазола, пиразола и тиофена, а другие группы определены выше.
Другая часть предлагаемого изобретения относится к соединению, соответствующему формуле I, в которой R1 выбрано из ряда, состоящего из C1-С6алкила, C1-С6галоидалкила, С3-С7циклоалкила, С1-С3алкокси-С1-С3алкила и необязательно замещенного фенила; R2 означает необязательно замещенный фенил; R4 означает C1-С6алкил, С3-С7циклоалкил, -(CH2)nR11, или -(СН2)о-O-(CH2)pR11, где указанные алкил и циклоалкил выборочно замещены группами -ОН, -OR6, -NR8R9, -C(=Y)Z, -X(C=Y)Z; R11 означает N[(CH2)2]2W, где W выбирают из ряда, состоящего из групп: NR6, (CH2)s, NC(=O)Z, CHOR6, CHR6, CHNHC(=O)Z и CHNR6R7; a R3 и другие группы определены выше.
Другая часть предлагаемого изобретения относится к соединению, соответствующему формуле I, в которой R1 выбрано из ряда, состоящего из С1-С6алкила, C1-С6галоидалкила, С3-С7циклоалкила, С1-С3алкокси-С1-С3алкила и необязательно замещенного фенила; R2 означает необязательно замещенный фенил; R3 означает C1-С6алкил, -(CH2)nR5, где R5 соответствует формулам IIIa-IIIh или IIa-c; R4 означает C1-С6алкил, С3-С7циклоалкил, -(CH2)nR11 или -(СН2)о-O-(CH2)pR11, где указанные алкил и циклоалкил выборочно замещены группами -ОН, -OR6, -NR8R9, -C(=Y)Z, -X(C=Y)Z; R11 означает N[(CH2)2]2W, где W выбирают из ряда, состоящего из групп: -NR6, (CH2)s, NC(=O)Z, CHOR6, CHR6, CHNHC(=O)Z и CHNR6R7; а другие группы определены выше.
Другая часть предлагаемого изобретения относится к соединению, соответствующему формуле I, в которой R1 выбрано из ряда, состоящего из C1-С6алкила, C1-С6галоидалкила, С3-С7циклоалкила, С1-С3алкокси-С1-С3алкила и необязательно замещенного фенила; R2 означает необязательно замещенный фенил; R3 означает -(CH2)nNR6R7, -(CH2)nC(=O)Z или -(CH2)nXC(=O)Z; R4 означает C1-С6алкил, С3-С7циклоалкил, -(CH2)nR11, или -(CH2)o-O-(CH2)pR11, где указанные алкил и циклоалкил необязательно замещены группами -ОН, -OR6, -NR8R9, -C(=Y)Z, -X(C=Y)Z; R11 означает N[(CH2)2]2W, где W выбирают из ряда, состоящего из групп: -NR6, (CH2)s, NC(=O)Z, CHOR6, CHR6, CHNHC(=O)Z и CHNR6R7; а другие группы определены выше.
В другой части изобретения предлагается метод лечения ВИЧ инфекции или профилактики ВИЧ инфекции или лечения СПИД или КЗСС, включающий введение пациенту, нуждающемуся в лечении, терапевтически активного количества соединения формулы I, в которой R1, R2, R3 и R4 определены выше, и его гидратов, сольватов или кислотно-аддитивных солей.
В другой части изобретения предлагается метод лечения ВИЧ инфекции или профилактики ВИЧ инфекции или лечения СПИД или КЗСС, включающий введение пациенту, нуждающемуся в лечении, терапевтически активного количества соединения формулы I, в которой R1, R2, R3 и R4 определены выше, и его гидратов, сольватов или кислотно-аддитивных солей; при этом по крайней мере одно соединение выбрано из ряда, состоящего из ингибиторов ВИЧ протеазы, нуклеозидных ингибиторов ревертазы, ненуклеозидных ингибиторов ревертазы, CCR5 ингибиторов и ингибиторов слияния вирусов.
В другой части изобретения предлагается метод лечения ВИЧ инфекции или профилактики ВИЧ инфекции или лечения СПИД или КЗСС, включающий введение пациенту, нуждающемуся в лечении, терапевтически активного количества соединения формулы I, в которой R1, R2, R3 и R4 определены выше, и его гидратов, сольватов или кислотно-аддитивных солей, а также ингибиторов ревертазы, выбранных из группы, состоящей из зидовудина, ламивудина, диданозина, зальцитабина и ставудина, рескриптора, сустива и вирамуна и/или ингибитора протеазы, выбранного из группы, состоящей из сакъюнавира, ритонавира, нельфинавира, индинавира, ампренавира, лопинавирата и атаназавира.
В другой части изобретения предлагается метод ингибирования ретровируса ревертазы, включающий введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества соединения формулы I, в которой R1, R2, R3 и R4 определены выше, а также его гидратов, сольватов или кислотно-аддитивных солей.
В другой части изобретения предлагается метод лечения ВИЧ инфекции или профилактики ВИЧ инфекции или лечения СПИД или КЗСС, при которых больной инфицирован штаммом ВИЧ, выделяющим ревертазу с, по крайней мере, одной мутацией, включающий введение больному, нуждающемуся в лечении, терапевтически эффективного количества соединения формулы I, в которой R1, R2, R3 и R4 определены выше, а также его гидратов, сольватов или кислотно-аддитивных солей.
В другой части изобретения предлагается метод лечения ВИЧ инфекции или профилактики ВИЧ инфекции или лечения СПИД или КЗСС, при которых указанный штамм ВИЧ проявляет пониженную восприимчивость к эфаверенцу, делавирдину или невирапину, включающий введение больному, нуждающемуся в лечении, терапевтически эффективного количества соединения формулы I, в которой R1, R2, R3 и R4 определены выше, а также его гидратов, сольватов или кислотно-аддитивных солей.
В другой части изобретения предлагается терапевтическая композиция, включающая терапевтически эффективное количество соединения формулы I, в которой R1, R2, R3 и R4 определены выше, а также его гидратов, сольватов или кислотно-аддитивных солей в виде смеси с, по крайней мере, одним фармацевтически приемлемым носителем или разбавителем, достаточным для введения больному разовой или многократной дозы, для лечения заболеваний, вызванных вирусом иммунодефицита, или для ингибирования ВИЧ.
Артикль единственного числа, используемый в изобретении, относится к одному или более объектам, например, если указано «соединение», то это может означать одно или более соединений, по крайней мере, одно. Выражения «одно», «одно или более» и «по крайней мере, одно» могут быть в тексте изобретения взаимозаменяемы.
Выражение «как указано (определено) выше» относится к первому упоминанию в кратком описании сущности изобретения.
Термин «алкил» означает неразветвленную или разветвленную цепь насыщенного моновалентного углеводородного остатка, содержащего 1-6 атомов углерода. Примеры алкильных групп включают, но не ограничиваются ими, низшие алкильные группы, включающие метил, этил, пропил, изо-пропил, н-бутил, изо-бутил, трет-бутил или пентил, изо-пентил, нео-пентил и гексил.
Термин «алкилен» означает бивалентный неразветвленный или разветвленный насыщенный углеводородный радикал, состоящий только из атомов углерода и водорода и имеющий 1-6 атомов углерода, если не указано другое. Примеры алкиленовых радикалов включают, но не исчерпываются ими, метилен, этилен, пропилен, 2-метилэтилен, 3-метилпропилен, 2-этилэтилен, пентилен, гексилен и тому подобное.
Термин «галоидалкил» используется для определения алкильной группы с неразветвленной или разветвленной цепью, как указано выше, в которой 1, 2, 3 или более атомов водорода замещены галогеном. Примерами являются 1-фторметил, 1-хлорметил, 1-бромметил, 1-йодметил, трифторметил, трихлорметил, трибромметил, трийодметил, 1-фторэтил, 1-хлорэтил, 1-бромэтил, 1-йодэтил, 2-фторэтил, 2-хлорэтил, 2-бромэтил, 2-йодэтил, 2,2-дихлорэтил, 3-бромпропил или 2,2,2-трифторэтил. Термин «фторалкил» означает галоидалкил, в котором галоидом является фтор.
Термин «циклоалкил» используется для обозначения насыщенного карбоциклического кольца, включающего 3-7 атомов углерода, т.е. циклопропил, циклобутил, циклопентил, циклогексил или циклогептил.
Термин «алкенил» означает радикал с ненасыщенной углеводородной цепью, имеющей 2-6 атомов углерода и одну или две олефиновые двойные связи. Примерами являются винил, 1-пропенил, 2-пропенил (аллил) или 2-бутенил (кротил).
Термин «алкинил» означает радикал с ненасыщенной углеводородной цепью, имеющей 2-6 атомов углерода и одну, а там где возможно, две тройные связи. Примерами являются этинил, 1-пропинил, 2-пропинил, 1-бутинил, 2-бутинил или 3-бутинил.
Термин «алкокси» используется для обозначения ненасыщенной или насыщенной цепи алкоксигруппы, где термин «алкил» определен выше, такой как группы: метокси, этокси, н-пропокси, изо-пропокси, н-бутокси, изо-бутокси, трет-бутокси, пентилокси и гексилокси, включая их изомеры.
Термин «галоидалкокси» означает O-галоидалкильную группу, где галоидалкил определен выше. Примеры галоидалкильных групп включают, но не ограничиваются ими, 2,2,2-трифторэтокси, дифторметокси и 1,1,1,3,3,3-гексафтор-изо-пропокси-группы.
Термин «тиоалкил» или «алкилтио» означает группу -SR, в которой R означает алкильную группу, как это определено выше, такую как метилтио, этилтио, н-пропилтио, изо-пропилтио и н-бутилтио-группу, включая их изомеры.
Термин «алкоксиалкил» используется для обозначения радикала R′R′′- где R′ радикал означает алкокси-группу, как указано выше, а R′′ означает алкеновый радикал, как определено выше, имея в виду, что прикрепление алкоксиалкеновой группы происходит по алкеновому радикалу. Примерами являются группы метоксиметильная, метоксиэтильная, метоксипропильная, этоксиметильная, этоксиэтильная, этоксипропильная, пропоксипропильная, метоксибутильная, этоксибутильная, пропоксибутильная, бутоксибутильная, трет-бутоксибутильная, метоксипентильная, этоксипентильная, пропоксипентильная, включая их изомеры.
Термин «гидроксиалкил» означает радикал R′R′′, в котором R′ означает гидроксильный радикал, а R′′ означает алкеновый, как определено выше; при этом местом прикрепления гидроксиалкильного радикала является алкеновый радикал.
Термин «ацил» используется для определения группы с формулой C(=O)R («алкилкарбонил»), в которой R означает водород, разветвленный или неразветвленный алкил с числом атомов углерода 1-6, циклоалкил с числом атомов углерода 3-7, арил, алкокси-группу или группу NR′R′′. Термин ацил включает также группу формулы C(=O)OR6 («алкоксикарбонил») или C(=O)NR6R7(«карбамоил»), где R означает алкильную группу, а R6 и R7 определены выше.
Термин «ацилирующий агент» используется для определения реагента, который способен переносить ацильную группу, определенную ранее, к другой функциональной группе, способной реагировать с ацилирующим агентом. Как правило, алкилкарбонил вводится при взаимодействии с ангидридом или ацилгалогенидом. Термин «ангидрид» означает соединения общей формулы RC(O)-O-C(O)R, что определено в предыдущем параграфе. Термин «ацилгалогенид» используется для обозначения группы RC(O)X, где Х означает бром или хлор. Как правило, алкоксикарбонильная группа вводится при взаимодействии с алкоксикарбонилхлоридом. Термин «алкоксикарбонилхлорид» означает соединения с общей формулой RC(O)Cl. Как правило, карбамоильная группа вводится взаимодействием с изоцианатом. Термин «изоцианат» означает соединения с общей формулой RN=C=O.
Функциональная группа, описываемая как «-XC(=Y)Z», в которой Х и Y независимо друг от друга означают О или NR6, a Z означает C1-С6алкокси, NR6R7, алкил или алкоксиалкил, преимущественно относится к «гуанидинам» (-NR6(=NR6) NR6R7), «имидатам» (-OC(=NR6)алкил), «амидинам» (-NR6C(=NR6)алкил), «карбонатам» (-OC(=O)OR), «карбаматам» (-ОС(=O)NR6R7 или -NR6C(=O)OR), «мочевинам» (-NR6C(=O)NR6R7), «амидам» (-NR6С(=O)алкил) или «эфирам» (-ОС(=O)алкил), где R6 и R7 определены выше, а R означает алкильную группу.
Функциональная группа «C(=Y)Z» относится к эфирам, амидам, имидатам и амидинам.
Термин «гетероциклоалкил» означает радикал -R′R′′, в котором R′ означает алкиленовый радикал, а R′′ означает гетероциклический радикал. Примерами гетероциклоалкильных радикалов являются, но не исчерпываются ими, тетрагидропиран-2-илметил, 2-пиперидинилметил, 3-пиперидинилметил, морфолин-1-илпропил и тому подобные.
Термин «алкиламино» означает радикал NR′R′′, в котором R′ означает водород, а R′′ означает алкильный радикал. Термин «диалкиламино» означает радикал NR′R′′, в котором R′ и R′′ означают алкильные радикалы. Примеры алкиламино радикалов включают, но не исчерпываются ими, радикалы: метиламино, этиламино, циклопропилметиламино, дициклопропилметиламино, диметиламино, метилэтиламино, диэтиламино, ди-(1-метилэтил)амино и тому подобные.
Термин «арил» определяет необязательно замещенную моно- или полициклическую ароматическую группу, содержащую атомы углерода и водорода. Примеры подходящих арильных групп включают, но не ограничиваются ими, фенил и нафтил (например, 1-нафтил или 2-нафтил). Подходящие заместители для арильного радикала могут быть выбраны из ряда, содержащего С1-С6алкил, C1-С6галоидалкил, C1-С6алкокси, С1-С6галоидалкокси, C1-С6алкилтио, алкоксикарбонил, CONR6R7, нитро-, галоид- или циано-группу. Необязательно замещенный фенил в R2 может быть, например, 2-хлорфенилом, 3-хлорфенилом, 4-хлорфенилом, 2,3-дихлорфенилом, 2,4-дихлорфенилом, 2,5-дихлорфенилом, 2,6-дихлорфенилом, 3,4-дихлорфенилом, 3,5-дихлорфенилом, 2,3,4-трихлорфенилом, 3,4,5-трихлорфенилом, 2,3,4,5,6-пентахлорфенилом, 2-цианофенилом, 3-цианофенилом, 4-цианофенилом, 2,3-дицианофенилом, 2,4-дицианофенилом, 2,5-дицианофенилом, 2,6-дицианофенилом, 3,4-дицианофенилом, 3,5-дицианофенилом, 3,6-дицианофенилом, 2-бромфенилом, 3-бромфенилом, 4-бромфенилом, 2,3-дибромфенилом, 2,4-дибромфенилом, 2,5-дибромфенилом, 2,6-дибромфенилом, 3,4-дибромфенилом, 3,5-дибромфенилом, 3,6-дибромфенилом или 3-хлор-5-цианофенилом.
Термин «гетероарил» или «гетероароматический» означает моно- или бициклический радикал с числом атомов в кольце 5-12, имеющий, по крайней мере, одно ароматическое кольцо, содержащее 4-8 атомов на одно кольцо, включающий один или более гетероатомов N, О или S с остальными углеродными атомами в кольце; при этом подразумевается, что присоединение указанного гетероарильного радикала происходит по указанному ароматическому кольцу. Как известно специалистам, гетероарильные кольца обладают меньшей ароматичностью, чем их полностью углеродные противоположные части. Для целей изобретения необходимо, чтобы гетероарильная группа имела только хоть какую-нибудь степень ароматичности. Примеры гетероарильных единиц включают моноциклические ароматические гетероциклы с числом атомов кольца 5 или 6 и с числом гетероатомов 1-3, включая, но не ограничиваясь ими, пиридинил, пиримидинил, пиразинил, пиридазинон, пирролил, пиразолил, имидазолил, триазолин и оксадиаксолин, которые могут быть необязательно замещены одним или более, предпочтительно одним или двумя, заместителями, выбранными из ряда, содержащего группы: гидрокси, циано, алкил, алкокси, тио, низший галоидалкокси, алкилтио, галоидалкил, алкилсульфинил, алкилсульфонил, галоид, амино, алкиламино, диалкиламино, аминоалкил, алкиламиноалкил и диалкиламиноалкил, нитро, алкоксикарбонил и карбамоил, алкилкарбамоил и диалкилкарбамоил.
Термин «гетероциклоалкил» означает радикал R′R′′, в котором R′ означает алкиленовый радикал, а R′′ означает гетероциклический радикал, как это определено выше. Примеры гетероциклалкильных радикалов включают, но не ограничиваются ими, 2-пиперидинилметил, 3-пиперидинилметил, морфолин-1-илпропил и тому подобное.
Термин «гетероцикл» или «гетероциклический» означает здесь неароматическое моно- или полициклическое кольцо, включающее атомы углерода и водорода и один или более гетероатомов N, S или О. Гетероциклическая группа может иметь в кольце одну или более двойных связей углерод-углерод или углерод-гетероатом, не вызывающих своим присутствием ароматического характера. Примерами таких групп являются пирролидинил, пирролидино, пиперидинил, пиперидино, пиперазинил, пиперазино, морфолинил, морфолино, тиоморфолинил, тиоморфолино. Гетероциклическая группа может быть незамещенной или замещенной подходящими одним-тремя заместителями, выбранными из ряда групп: гидрокси, циано, алкил, алкокси, тио, низший галоидалкокси, алкилтио, галоидалкил, алкилсульфинил, алкилсульфонил, галоид, амино, алкиламино, диалкиламино, аминоалкил, алкиламиноалкил и диалкиламиноалкил, нитро, алкоксикарбонил и карбамоил, алкилкарбамоил и диалкилкарбамоил.
Термины «амино», «алкиламино» и «диалкиламино» относятся к группам -NH2, -NHR и NR2, соответственно, где R означает алкил, как определено выше. Две алкильные группы, соединенные с атомом азота в диалкиламино-группе, могут быть одинаковыми или различными. Термины «аминоалкил», «алкиламиноалкил» и «диалкиламиноалкил» относятся здесь к группам NH2(CH2)n-, RHN(CH2)n- и R2N(СН2)n- соответственно, где n равно 1-6, a R означает алкил, как определено выше.
Термин «ациламино» означает радикал формулы -NH-C(=O)R, где R означает водород, неразветвленный или разветвленный алкил, содержащий 1-6 атомов углерода, циклоалкил, содержащий 3-7 атомов углерода или арил.
Термин «галоген» означает фтор, хлор, бром или йод.
Термин «алкилтио» или «тиоалкил» означает группу -S-алкил, в которой алкил определен выше, такую как метилтио, этилтио, н-пропилтио, изо-пропилтио, н-бутилтио, гексилтио и их изомеры.
Термин «алкилсульфинил» означает радикал -S(O)R′, в котором R′ означает алкил, как определено выше. Примеры алкилсульфинильной группы включают, но не ограничиваются ими, группы метилсульфинил и изо-пропилсульфинил.
Термин «алкилсульфонил» означает радикал -S(O)2R′, в котором R′ означает алкил, как определено выше. Примеры алкилсульфонилов включают, но не ограничиваются, группы метилсульфонил и изо-пропилсульфонил.
Термин «сульфонилирующий агент» здесь означает реагент, способный трансформировать алкилсульфонильную группу, как она определена выше, в другую функциональную группу, способную взаимодействовать с сульфонилирующим агентом, таким как Cl-SO2-R.
Приставка «карбамоил» означает здесь радикал -CONH2. Приставка «N-алкилкарбамоил» и «N,N-диалкилкарбамоил» здесь означает радикал CONHR′ или CONR′R′′ соответственно, где R′ и R′′ группы независимо друг от друга означают алкил, как определено выше.
Термин «гомологичный» относится здесь к серии родственных соединений, структура которых в какой-то части молекулы отличается от других членов серии только группами -(СН2)- или -(СН2)n.
Соединения формулы I проявляют таутомерию. Таутомерные соединения могут существовать в двух или более взаимопревращающихся формах. Прототропные таутомеры являются следствием миграции ковалентно связанных атомов водорода между двумя атомами. В общем случае существует равновесие между таутомерами, и попытки выделить индивидуальные таутомеры обычно приводят к получению смеси, химические и физические свойства которой соответствуют смеси соединений. Положение равновесия зависит от химического строения молекулы. Например, для многих алифатических альдегидов и кетонов, таких как ацетальдегид, преобладает кето-форма, в то время как для фенолов преобладает енольная форма. Типичными таутомерами являются такие таутомеры, как кето/енолы (-С(=O)-СН- -C(-OH)=CH-), амиды/имидокислоты (-C(=O)-NH--C(-OH)=N-) и амидины (-C(=NR)-NH--C(-NHR)=N-). Два последних, в частности, характерны для гетероарильных и гетероциклических колец, и настоящее изобретение охватывает все таутомерные формы соединений.
-C(-OH)=CH-), амиды/имидокислоты (-C(=O)-NH--C(-OH)=N-) и амидины (-C(=NR)-NH--C(-NHR)=N-). Два последних, в частности, характерны для гетероарильных и гетероциклических колец, и настоящее изобретение охватывает все таутомерные формы соединений.
Соединения формулы I, которые являются основными, могут образовывать фармацевтически приемлемые кислотно-аддитивные соли с неорганическими кислотами, такими как галоидводородные кислоты (например, хлористоводородная и бромистоводородная кислоты), серная кислота, азотная кислота, фосфорная кислота и подобные кислоты, а также с органическими кислотами (например, с уксусной кислотой, винной кислотой, янтарной кислотой, фумаровой кислотой, малеиновой кислотой, яблочной кислотой, салициловой кислотой, лимонной кислотой, метансульфоновой кислотой, п-толуолсульфоновой кислотой и подобными кислотами).
Термин «сольват» означает здесь соединение предлагаемого изобретения или его соль, которое включает стехиометрическое или нестехиометрическое количество растворителя, связанного нековалентными внутримолекулярными связями. Предпочтительными растворителями являются летучие, нетоксичные растворители и/или приемлемые для введения людям в следовых количествах.
Термин «гидрат» означает соединение предлагаемого изобретения или его соль, которое включает стехиометрическое или нестехиометрическое количество воды, связанной нековалентными внутримолекулярными связями.
Термин «дикий тип» здесь относится к ВИЧ вирусу, который обладает доминантным генотипом, обычно встречающимся в нормальной популяции, и который не был подвержен действию ингибиторов ревертазы. Термин «ревертаза дикого типа» здесь относится к ревертазе с номером Р 03366 в базе данных SwissProt.
Термин «подверженность» означает здесь изменение в 10 и более раз в чувствительности отдельного вирусного изолята по сравнению с чувствительностью, проявляемой вирусом дикого типа в той же экспериментальной системе.
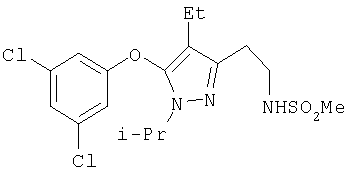
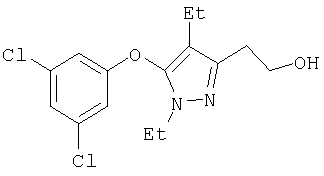
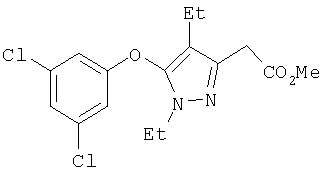
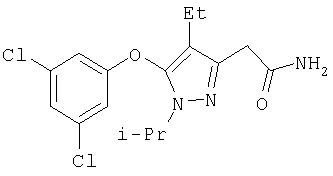
Соединения
Примеры репрезентативных соединений предлагаемого изобретения представлены в следующей таблице 1. Эти примеры и препараты дают возможность специалистам более ясно понять и применить предлагаемое изобретение. Их не следует рассматривать как ограничивающие весь объем изобретения, но скорее как иллюстративный и репрезентативный материал.
В общем случае, номенклатура, используемая в этой таблице, основывается на компьютеризированной системе Института Бельштейна AUTONOM™ т.4.0 для создания систематической номенклатуры IUPAC.
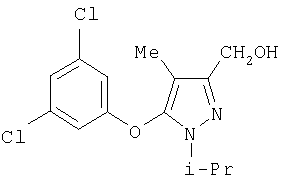
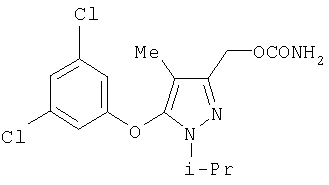
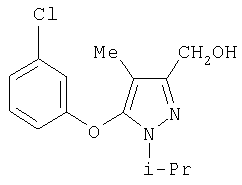
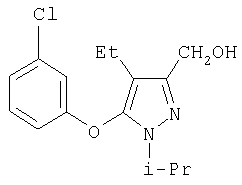
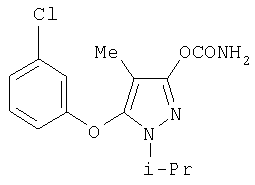
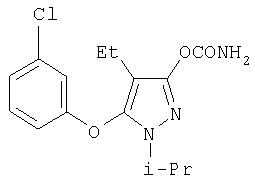
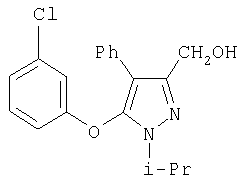
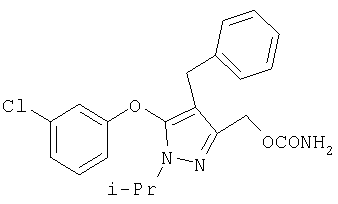
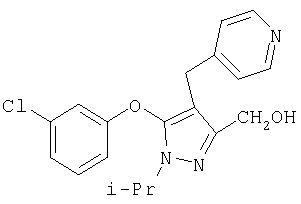
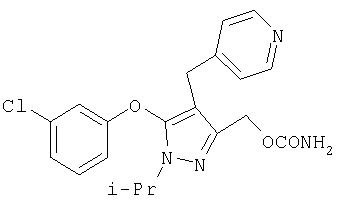
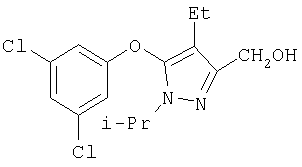
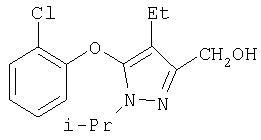
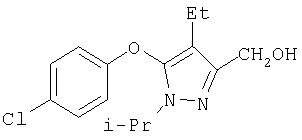
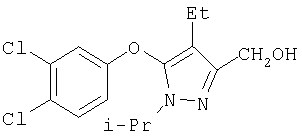
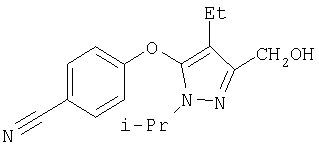
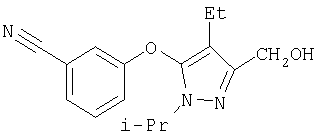
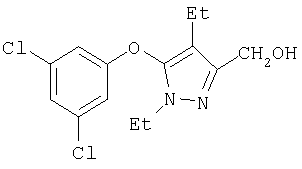
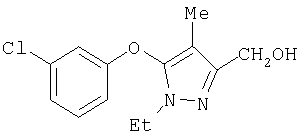
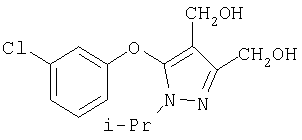
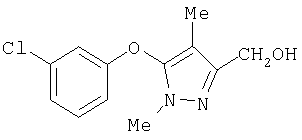
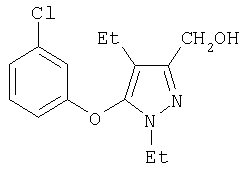
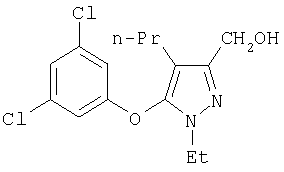
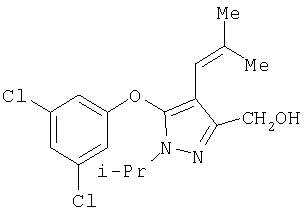
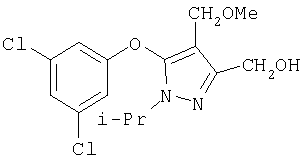
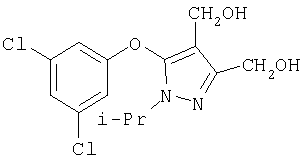
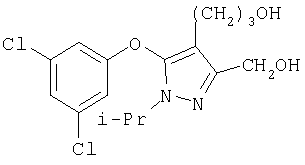
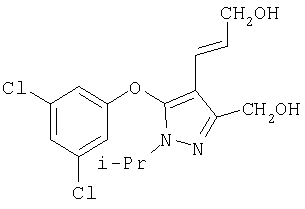
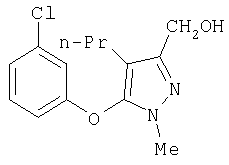
281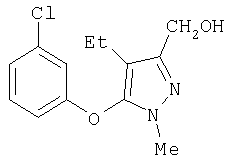
267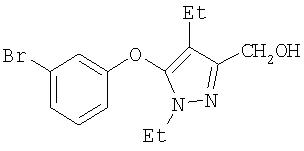
325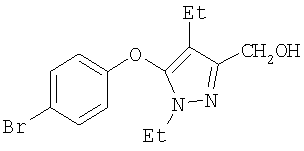
325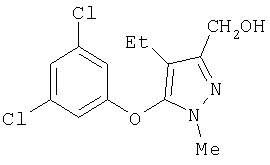
М+=300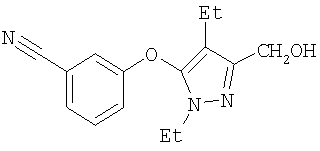
272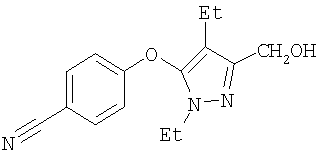
272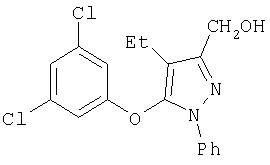
363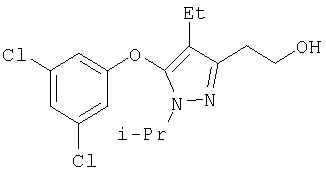
343
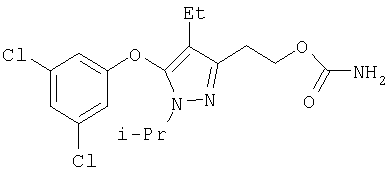
386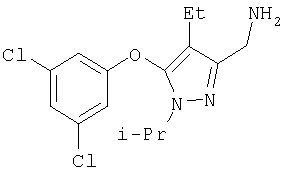
328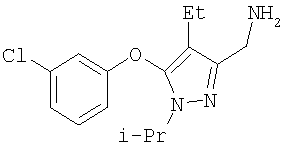
294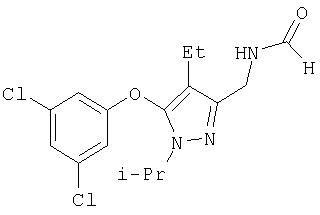
356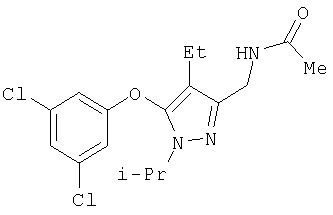
370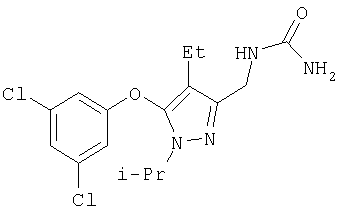
371
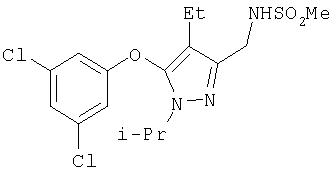
406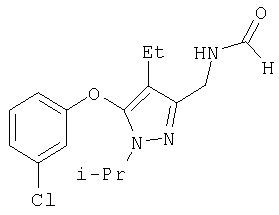
322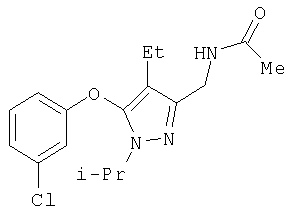
336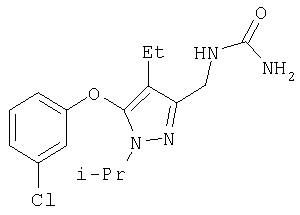
337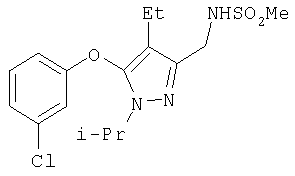
372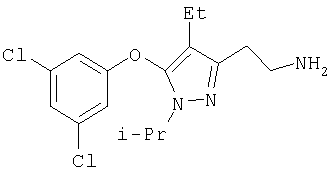
342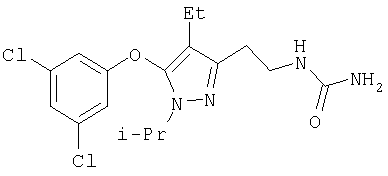
385
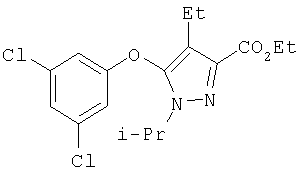
371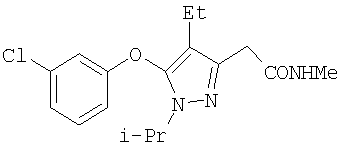
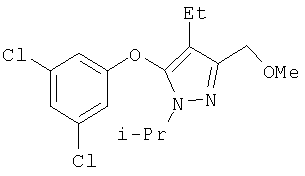
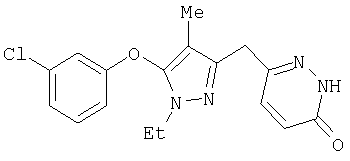
345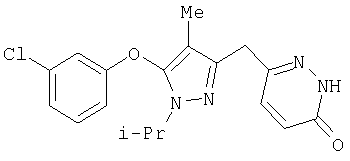
359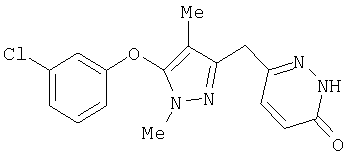
331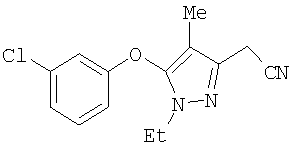
276
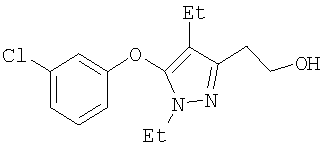
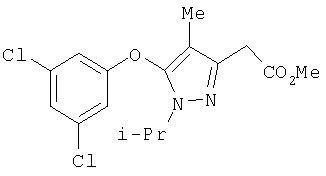
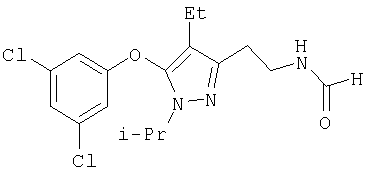
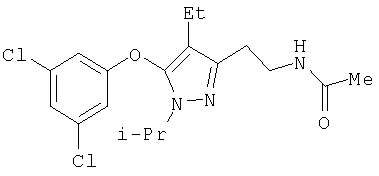
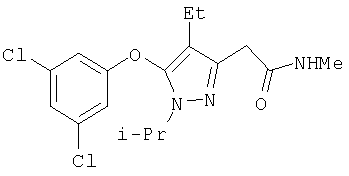
385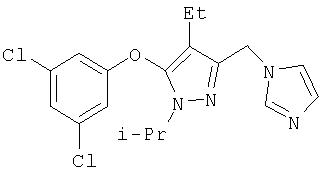
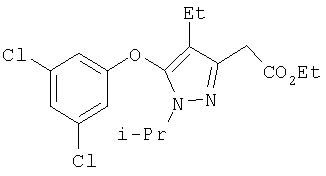
379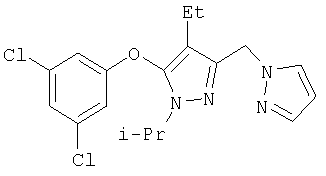
379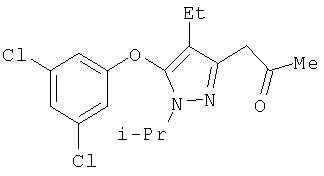
355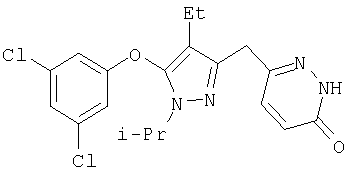
407
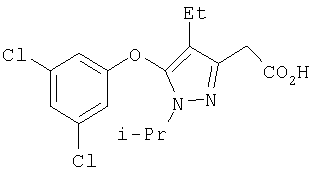
357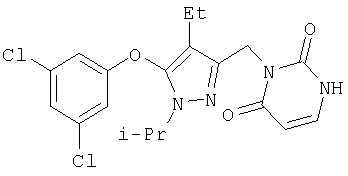
М+=423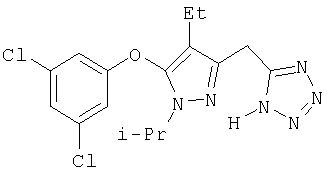
381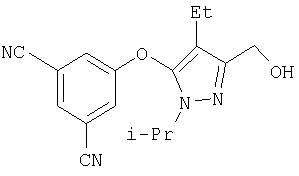
311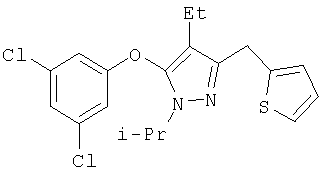
М+=395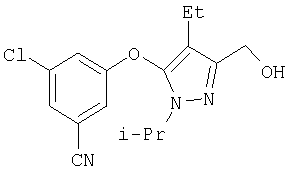
320
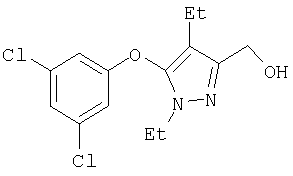
306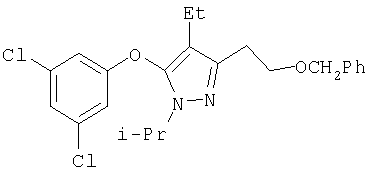
433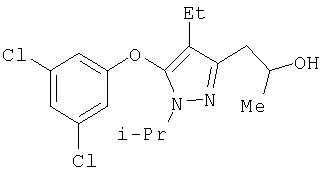
357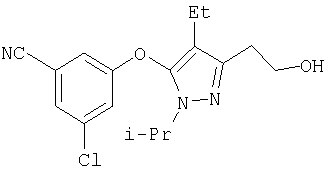
334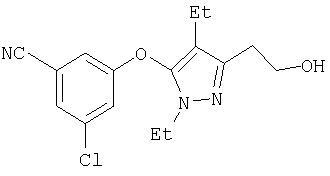
320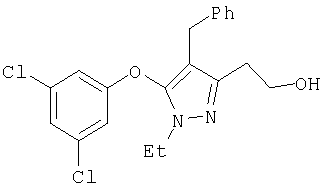
391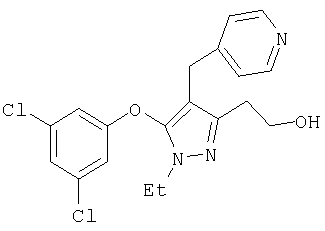
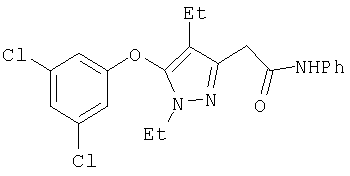
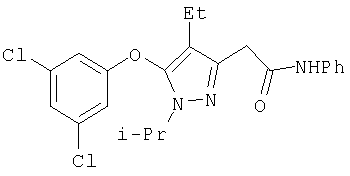
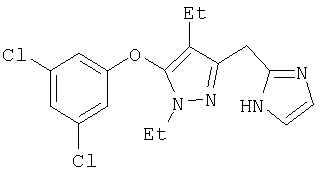
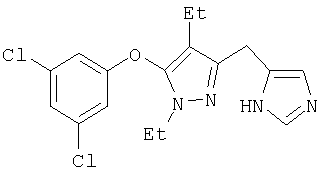
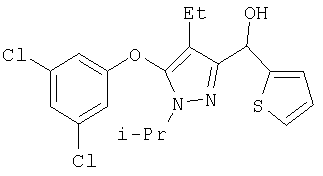
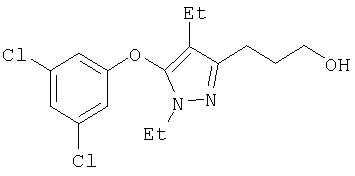
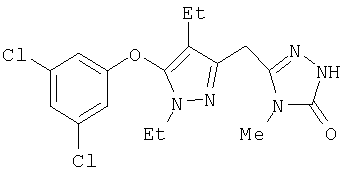
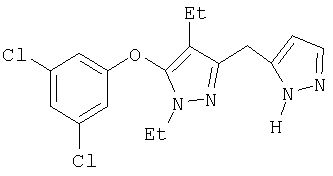
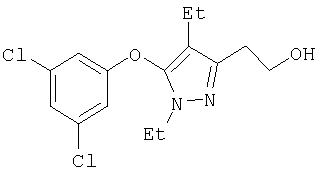
Получение соединений
2Н-пиразол-3-олы, используемые в качестве синтетических предшественников соединений предлагаемого изобретения, получают циклизацией N-замещенных гидразинов и необязательно замещенного β-кетоэфира (схема 1) (R.H.Wiley и Р.Wiley, Pyrazolines, Pyrazolidines and Derivatives в The Chemistry of Heterocyclic Compounds, т.20, A. Weissberger (ред.), изд-во J. Wiley and Sons, New-York, 1964, cc.18-31 и cc.95-97; К.Kirschke, 1H-Pyrazoles, в Houben-Weyl Methoden der Organischen Chemie E8B Hetarene III, Ч.2, изд-во George Thieme Verlag, Stuttgart, 1994, cc.433-448).
Схема 1
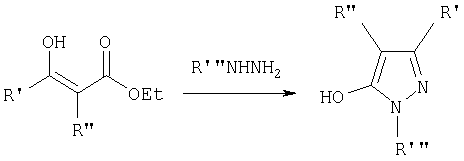
С-3 замещенные карбоэтоксипиразолы получали реакцией 1,2-бис-этоксикарбонилэтеноксида натрия и замещенного гидразина или гидразингидрата в кипящем бензоле, получая соединения 2а и 3, соответственно (схема 2). Алкилирование пиразола 3 в положение N-1 проводили путем защиты гидроксильного заместителя, что может быть легко выполнено в виде силильного эфира, например 5 (другие защитные группы описаны в Т.W.Greene и Р.G.M. Wuts, Protective Groups in Organic Synthesis, изд-во Wiley Interscience, New-York, 3 изд., 1999), с последующим алкилированием и снятием защиты; в результате получали соединение 6. Алкилирование по атому азота проводят обычно последовательной обработкой 5 основанием и алкилирующим агентом. Типичными основаниями для подобного превращения являются карбонат натрия, карбонат калия, гидрид натрия, гидрид калия, трет-бутилат калия в растворителе, подобном диметилформамиду (ДМФ), диметилсульфоксиду (ДМСО), N-метилпирролидону (N-МП), ацетонитрилу или тетрагидрофурану (ТГФ). Альтернативно циклизация может быть проведена гидразином, замещенным лабильной защитной группой (например, п-СН2С6Н4ОМе), которая затем удаляется с получением соединения 3.
Схема 2
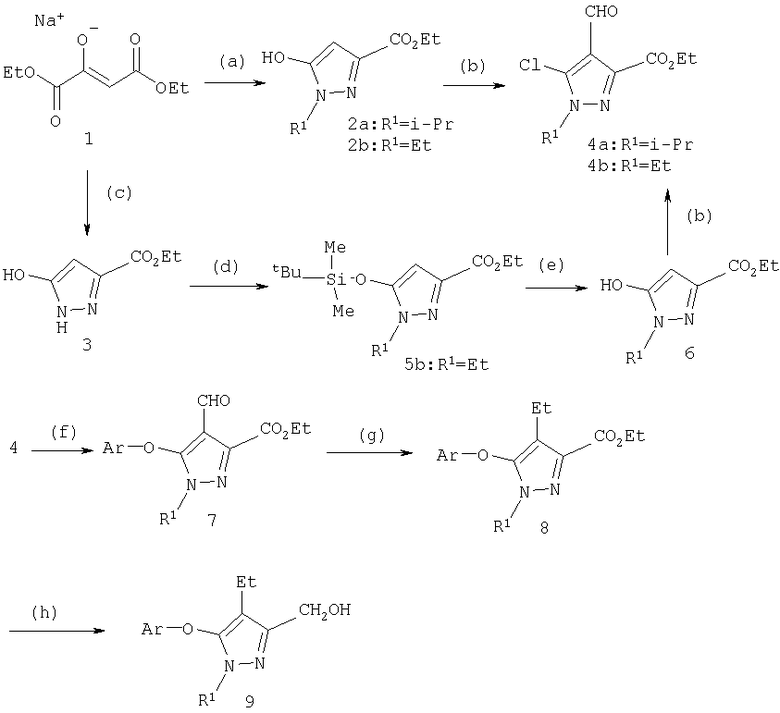
"а" серии: R1=изо-Pr, Ar=3,5-ди-Cl-С6Н3-
"b" серии: R1=Et, Ar=3,5-ди-Cl-С6Н3-
"c" серии: R1=изо-Pr, Ar=3-Cl-C6H4-
(a) R1-NHNH2, PhH, кипячение; (b) POCl3, ДМФ; (с) NH2NH2, PhH, кипячение; (d) Me2(трет-Bu)SiCl, имидазол, ДМФ; (е) (i) R1Br, Na2CO3 (ii) Bu4N+F-, СН2Cl2; (f) Ar-OH, NaH, ДМФ; (g) (i) MeMgBr, ТГФ-Et2О, (ii) Et3SiH, ТФУ, CH2Cl2; (h) LiEt3BH, ТГФ
Введение формильной группы в положение 4 в условиях реакции Вильсмайера сопровождается заменой гидроксильной группы на хлорид с получением функционализированного пиразольного интермедиата 4 (G.Jones и S.Р.Stanforth, Organic Reactions, изд-во Wiley & Sons, New-York, т.49, 4.1, 1997). С-5 гидроксильный заместитель легко замещается на хлор даже в отсутствие С-4 формильного заместителя путем взаимодействия с POCl3 (К.Kirschke, 1H-Pyrazoles в Houben-Weyl Methoden der Organischen Chemie E8B Hetarene III Ч.2, изд-во George Thieme Verlag, Stuttgart, 1994, cc.638-641).
Замена хлора необязательно замещенным фенолятом натрия или пиридиноксидом натрия в ДМФ приводит к получению 5-арилоксипиразола 7. Реакцию проводят в тетрагидрофуране (ТГФ) или других диполярных апротонных растворителях, таких как диметилсульфоксид (ДМСО), диметилацетамид (ДМА) или N,N-диметилформамид (ДМФ) в присутствии основания, такого как н-бутиллитий, гидрид натрия или трет-бутилат натрия. Обычно реакцию проводят в инертной среде, например, в атмосфере азота или аргона при температуре реакции от 0°С до температуры кипения реакционной смеси, преимущественно при температуре между примерно 10°С и примерно 180°С.
4-Алкилпиразолы получали реакцией альдегида с алкильным реагентом Гриньяра с образованием вторичного карбинола 8 и последующим восстановлением этого вторичного карбинола триэтилсиланом с образованием соединения 9 (схема 3). Специалистам должно быть понятно, что хотя на схеме указан метильный реагент Гриньяра, другие алкильные и алкенильные реагенты Гриньяра, также как другие металлоорганические производные, обычно используемые в органическом синтезе, включая, но не ограничиваясь ими, литий, цинк, кадмий, цирконий, натрий, калий, тоже могут быть применены. Реакцию проводят при температуре от -78°С до 0°С в инертных растворителях, таких как диэтиловый эфир, тетрагидрофуран, 1,2-диметоксиэтан, гексан.
Восстановление альдегида 7 до карбинола 12 осуществляют с помощью гидридного восстанавливающего агента. Типичные восстанавливающие агенты включают боргидрид натрия, боргидрид лития и триацетоксиборгидрид натрия. Альтернативно может быть использовано каталитическое гидрирование или другие восстанавливающие агенты, применяемые в синтезе. Восстановление с помощью NaBH4 легко осуществляется в органическом растворителе, например, спиртовых растворителях, таких как метанол, этанол, пропанол или эфирах, таких как тетрагидрофуран, диэтиловый эфир или диметоксиэтан, или в смесях упомянутых растворителей. Для более высокой реакционной способности реагентов гидридного переноса требуются апротонные растворители. Реакцию проводят при температуре между примерно -10°С и примерно 60°С, преимущественно при комнатной температуре. Восстановление может быть также проведено в соответствии с примерами, описанными в учебниках по органической химии, например в книге J. March "Advanced Organic Chemistry: Reactions, Mechanisms, and Structure", 4 изд., изд-во John Wiley & Sons, 1992.
Затем получают производное карбинола 13 (R13=ацил, алкил, аралкил, арил, карбамоил).
Схема 3
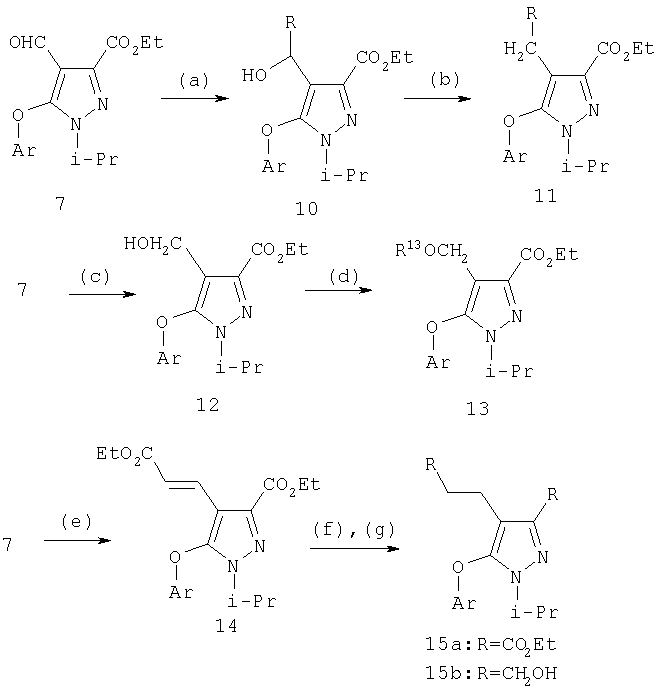
(a) RMgBr, ТГФ; (b) Et3SiH, ТФУ, CH2Cl2; (c) NaBH4, MeOH; (d) алкилирующий или ацилирующий агент; (е) Ph3P+CH2CO2Me, NaH, ТГФ, 0°С; (g) LiEt3BH, ТГФ, -40°С до комн. темп.
Альтернативно С-4 альдегид может быть превращен в алкен 14 (схема 3) или в замещенный алкен с помощью реагента Виттига или реагента Эммонса-Вадсворта (J.W.Schulenberger и S.Archer, Organic Reactions, изд-во Wiley & Sons, New-York, т.14, Ч.1, 1965, cc.1-51; J. March, Advanced Organic Chemistry, 4 изд., изд-во John Wiley & Sons, New-York, 1992, cc.956-963). Введение двойной связи проводят в соответствии с методиками, подобными описанным в литературе, например, в присутствии сильного основания, такого как н-бутиллитий, или предпочтительно действием гидрида натрия в органическом растворителе, таком как безводный эфир, например, диэтиловый эфир, дибутиловый эфир, диоксан, предпочтительно безводный тетрагидрофуран, в инертной атмосфере, такой как атмосфера азота или аргона, при температуре от 0°С до 80°С, преимущественно при температуре реакции между примерно 5°С и примерно 50°С. Введение двойной связи является эффективным методом получения гомологов с С-4 заместителем.
Полученный алкен может быть восстановлен в соединение 15а путем каталитического гидрирования со стандартным платиновым, палладиевым или рутениевым катализатором на носителях, таких как активированный уголь или глинозем, или по методам, описанным в учебниках по органической химии (J.March, Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 4 изд., изд-во John Wiley & Sons, New-York, 1992, cc.771-780), при давлении 1-40 атм; или восстановлением с растворенным металлом (Yuon и др., Tetrahedron Lett., 27, 1986, с.2409; Hudlicky и др., Tetrahedron Lett, 28, 1987, с.5287), если это необходимо. Подходящими растворителями для гидрирования являются такие органические растворители, как спирты (например, метанол, этанол), простые эфиры (например, тетрагидрофуран, 1,2-диметоксиэтан), сложные эфиры (например, этилацетат), галогенированные углеводороды (например, дихлорметан) или углеводороды (например, гексан, циклогексан или толуол). Восстановление с растворенным металлом проводят магнием в метаноле. Восстановление 15а гидридом диизобутилалюминия (ДИБАЛ-Н), алюмогидридом лития или триэтилборгидридом лития приводит к диолу 15б.
Введение заместителей к С-4 может также сопровождаться ацилированием гидроксипиразола (схема 4). Ацильное производное 16 (путь а), где R15 представляет собой алкил, арил или аралкил, образуется реакцией соответствующего хлорангидрида кислоты с 5-гидроксипиразолом-2. Реакцию удобно проводить в условиях, известных для реакций ацилирования, например, в инертном растворителе, таком как эфиры, например, безводный тетрагидрофуран, диэтиловый эфир, дибутиловый эфир, диоксан или смесь упомянутых растворителей, при температуре реакции от комнатной до температуры кипения реакционной смеси, в присутствии катализатора, такого как Са(ОН)2, К2СО3, AlCl3, BF3, FeCl3, SnCl4 или ZnCl2.
5-Гидроксипиразол 16 легко превращается в производное 5-хлорпиразола 17 действием хлорирующего агента, такого как (COCl)2, HCl, PCl5, PCl3, SOCl2 или POCl3. Реакцию удобно проводить в инертной атмосфере, например, в атмосфере азота или аргона при температуре от комнатной до температуры кипения реакционной смеси. Предпочтительно проводить реакцию в присутствии хлорокиси фосфора (POCl3) при температуре реакции между примерно 50°С и примерно 180°С. Реакцию можно проводить в таких органических растворителях, как галогенированные углеводороды (например, дихлорметан или хлороформ), углеводороды (например, циклогексан, метилциклогексан, декалин, бензол, толуол, о-ксилол, м-ксилол или п-ксилол) или смесь упомянутых растворителей.
Схема 4
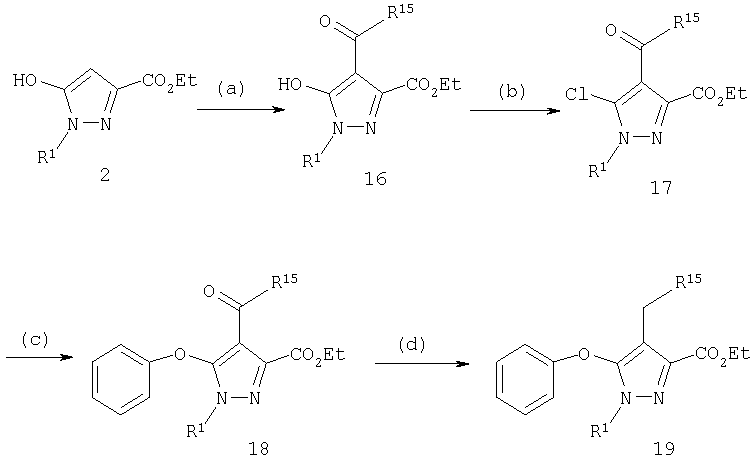
(a) R15COCl, Ca(OH)2, Et2O; (b) POCl3, СН2Cl2; (с) Ar-OH, NaH,
ДМФ; (d) Et3SiH, ТФУ, CH2Cl2
Восстановление карбонила 18 в алкан 19 (схема 4, стадия d) проводят алкилсиланами в присутствии протонной кислоты или кислоты Льюиса. Реакцию проводят с триметилсиланом, триэтилсиланом или трипропилсиланом. Предпочтительной протонной кислотой является трифторуксусная кислота (ТФУ), а предпочтительной кислотой Льюиса - SnCl4 (D.L.Comins и др., Tetrahedron. Lett., 27, 1986, с.1869) при температуре реакции от 0°С до 80°С, предпочтительно между примерно 5°С и 50°С. Необязательно, но выборочно оксопроизводное 18 прямо восстанавливают в соответствующий метилен 19 с использованием других методик, известных специалистам, например, восстановлением по Клемменсену, восстановлением по Вольфу-Кижнеру и гидрогенолизом тиоацетатов или их восстановлением.
Схема 5
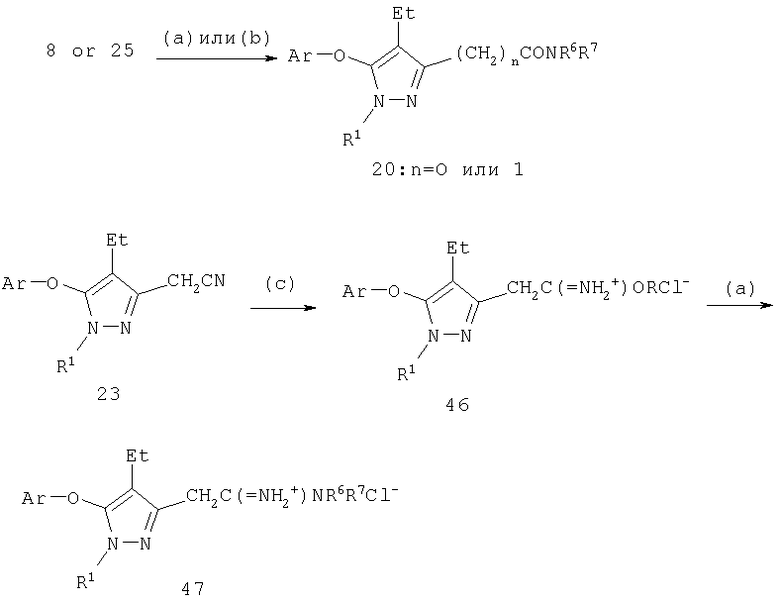
(a) R15COCl, Ca(OH)2, Et2O; (b) POCl3, СН2Cl2; (с) Ar-ОН, NaH,
ДМФ; (d) Et3SiH, ТФУ, CH2Cl2
С-3 эфир или пиразолы 8 и 25 (схема 5) превращают в соответствующие амиды 45 переамидированием или омылением эфира, который затем может быть превращен в амид по стандарной методике (J.March, Advanced Organic Chemistry, 4 изд., изд-во J.Wiley & Sons, New-York, 1991, cc.419-424). Пиразол с нитрильной группой 23 переводят в соответствующий имидат 46 обработкой нитрила в спирте в присутствии хлористоводородной кислоты (R.Sandier и W.Karo, Organic Functional Group Preparations, 2 изд., изд-во Academic Press, New-York, т.III, 1986, cc.314-330). Амидины 47 получают обработкой имидатов аммиаком или замещенными аминами или альтернативно последовательной обработкой амида 45 хлорокисью фосфора и аммиаком или замещенным амином.
С-3 карбинол в соединении 9 (схема 6) может быть превращен в эфиры (20; R13=C(=O)R6), карбонаты (20; R13=C(=O)OR6) и карбаматы (20; C(=O)NHR6) конденсацией 9 с хлорангидридами или ангидридами кислот, алкилхлорформиатами и изоцианатами, соответственно (J.March, Advanced Organic Chemistry, 4 изд., изд-во J. Wiley & Sons, New-York, 1991, сс.392-396 и cc.891-892; S.R.Sandier и W.Karo, Organic Functional Group Preparations, 2 изд., изд-во Academic Press, New-York, t.I, 1983, cc.299-304; т.II, 1986, cc.260-271). Эфиры (20, R13=алкил или аралкил) могут быть получены по методу синтеза эфиров Вильямсона или по реакции Мицуноби (J.March, Advanced Organic Chemistry, 4 изд., изд-во J.Wiley & Sons, New-York, 1991, cc.386-387; S.R.Sandler и W.Karo, Organic Functional Group Preparations, 2 изд., изд-во Academic Press, New-York, т.I, 1983, cc.129-133). Синтез эфира по Вильямсону может быть предпочтительно проведен в органическом растворителе, таком как диполярные апротонные растворители, подобные N,N-диметилацетамиду или N,N-диметилфорамиду (ДМФ), ацетонитрилу или тетрагидрофурану, с использованием основания, такого как гидрид натрия, гидрид лития, гидрид калия, трет-бутилат калия, карбонат лития, карбонат натрия, карбонат калия, или органических аминов, таких как триэтиламин, или N-алкилморфолинов, таких как N-метилморфолин, при температуре реакции между примерно -10°С и примерно 60°С, предпочтительно при комнатной температуре. Альтернативно карбинол может быть превращен в галоидный алкил с последующим взаимодействием с фенолятом щелочного металла.
Амины 21 были получены из спирта 9 конденсацией по Мицуноби (J.March, Advanced Organic Chemistry, 4 изд., изд-во J.Wiley & Sons, New-York, 1991, cc.414-415). Обработка 21 ацилирующими агентами приводит к амидам (22, R13=COR6), к карбаматам (22, R13=CO2R6) и мочевинам (22, R13=C(=O)NHR6). Гуанидины (22, R13=C(=NH)NR6R7) получают из тиомочевины (22, R13=C(=S)NHR6) последовательной обработкой диметилсульфатом и амином (Y.Yamamoto и др., Synthesis and Chemistry of Guanidines в The Chemistry of Amidines and Imidates, S.Patai и Z.Rappoport (ред.), изд-во Wiley & Sons, Chichester, 1991, Ч.10, cc.489-492). Конденсация амина с сульфонилирующим агентом приводит к соответствующему сульфонамиду (22, R13=SO2R6).
Схема 6
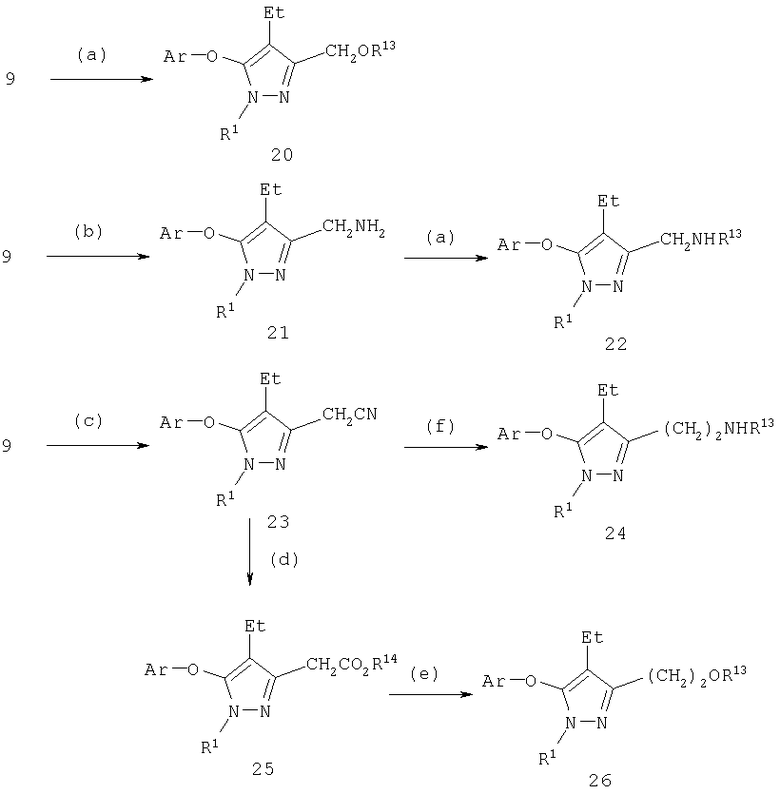
(а) ацилирующий или алкилирующий агент; (b) (i) ДЭАД, Ph3Р, фталимид, (ii) NH2NH2; (с) (i) SOCl2, (ii) NaCN, ДМФ; (d) (i) HCl, HOAc, Н2О, (ii) MeOH, HCl; (e) (i) LiEt3BH, ТГФ, (ii) NaBH4, МеОН, СН2Cl2
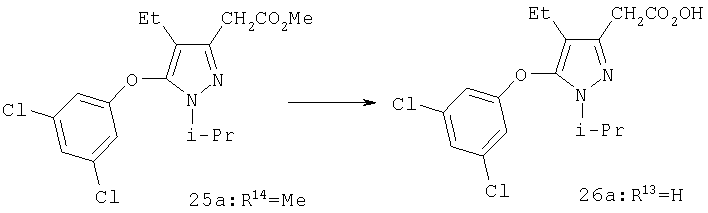
Производные гомологичных аминов и карбинолов получают двухступенчатым синтезом, включающим превращение первичного спирта в алкилгалогенид и замещение галоида цианидом натрия. Полученный нитрил 23 может быть восстановлен в амин 24 (R13=Н) путем последовательной обработки гидридом диизобутилалюминия и боргидридом натрия. Полученный амин 24 (R13=Н) может быть обработан ацилирующим, алкилирующим и сульфонилирующим агентами. Гидролиз и этерификация 23 приводили к соответствующему эфиру 25 (R14=Me), который был восстановлен в спирт 26 (R13=Н), из которого в дальнейшем были получены производные действием алкилирующих и ацилирующих агентов, как описано выше.
Схема 7
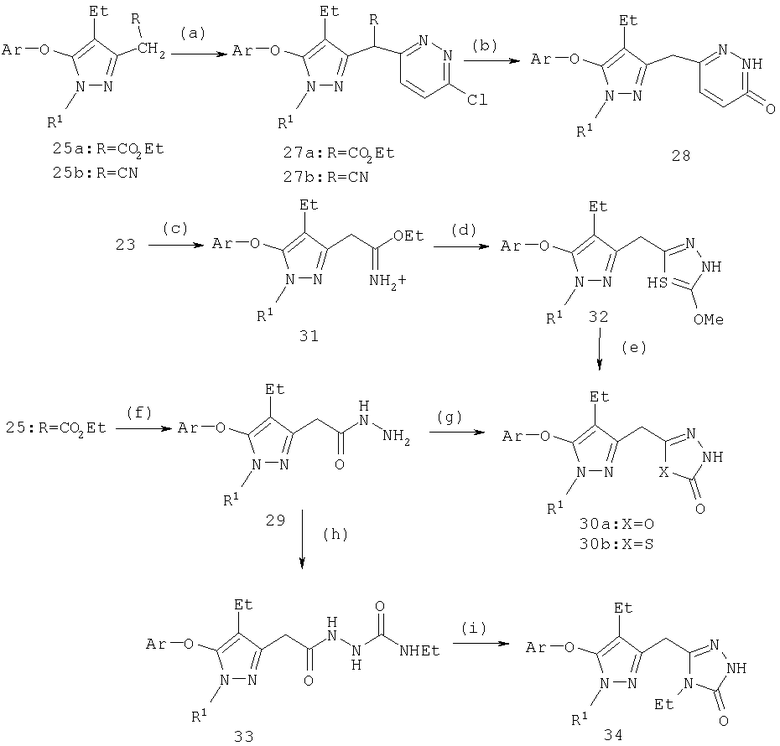
(а) 3,6-дихлорпиразин, NaH, ДМФ; (b) HOAc, HCl, H2O; (с) HCl, EtOH; (d) H2NNHC(=S)OMe, диоксан; (e) HOAc, (f) H2NNH2, EtOH; (g) ClC(=O)Cl, пиридин, СН2Cl2; (h) EtNCO, ТГФ; (i) KOH, MeOH
Введение гетероциклилалкильных заместителей в положение С-3 пиррола осуществляли модификацией нитрила 23 или эфира 25. Пиридазиноны 28 получали катализируемой основанием конденсацией эфира или нитрила с подходящим заместителем и 3,6-дихлорпиридазина (схема 7). Конденсация протекает успешно с гидридом натрия и ДМФ. Обработка 27a или 27б водной хлористоводородной и уксусной кислотами вызывает гидролиз, декарбоксилирование и сопутствующий гидролиз хлорпиридазина с получением пиридазинона 28.
2-Оксо-2,3-дигидро-1,3,4-оксадиазол 30а получали циклизацией ацилгидразида 29 фосгеном (или эквивалентными реагентами, такими как карбонилдиимидазол, алкилхлорформиаты и подобные), прямо получая требуемый оксадиазол (A.Hetzheim, 1,3,4 Oxadiazoles в Houben-Weyl Methoden der Organischen Chemie, Hetarene III/Часть 3, Том Е8с; изд-во Verlag, Stuttgart, 1994, cc.531-536) (схема 7). 2-Оксо-2,3-дигидро-1,3,4-тиадиазолы получают конденсацией O-алкилимидата 31 и метокситиокарбонилгидразида, приводящей к производному 2-метокси-3,4-тиадиазола 32, который был гидролизован в кислотных условиях до соответствующего 2-оксо-2,3-дигидро-1,3,4-тиадиазола 30б (Н.Kristinsson и др., Helv. Chim. Acta, 65, 1982, с.2606). Альтернативно циклизация N-ацил-N-алкоксикарбонилгидразидов с реагентом Лоуссона может прямо приводить к тиадиазолу (В. Р. Rasmussen и др., Bull. Soc. Chim. Fr., 1985, с.62). Триазолоны 34 могут быть получены карбамоилированием ацилгидразида 29 этилизоцианатом, приводящим к получению N-ацил-N′-карбамоилгидразида 33, циклизующегося в триазолон 34 путем обработки метанольным раствором гидроксида калия.
Другие содержащие гетероарил боковые цепи могут быть получены при использовании вариантов синтеза, при которых легко доступно положение 3. Галоидметильные соединения (например, 37) подвержены нуклеофильной замене гетероатомами, что приводит к имидазол-1-илметилу (67), пиразол-1-илметилу (68) и N-замещенным урацилам (72) (примеры 41 и 42). Присоединение к атому углерода гетероарильных заместителей может быть проведено добавлением металлоорганического соединения с подходящей защитной группой к пиразолу, содержащему альдегидную группу в боковой цепи (например, 105), с последующим восстановительным удалением карбинольной группы и, если нужно, снятием защитной группы. Гетероарилы и гетероциклы могут быть также введены путем [1,3]диполярного циклоприсоединения 1,3-диполярных соединений к кратным связям (например, J.March, Advanced Organic Chemistry, 4 изд., изд-во J.Wiley & Sons, New-York, 1991, cc.836-839). Такое циклоприсоединение азидов к нитрилам приводит к тетразолам 73 (пример 36).
Дозы и введение
Соединения предлагаемого изобретения эффективны при использовании различных методов введения, включая длительное (внутривенное капельное) местное парентеральное, внутримышечное, внутривенное, подкожное, чрезкожное (которое может включать использование агента, повышающего проницаемость), буккальное, насальное введение и введение в виде суппозиториев, а также другие методы введения. Оральное применение может быть в виде таблеток, таблеток с покрытием, драже, твердых и мягких капсул, растворов, эмульсий, сиропов и суспензий.
При получении фармацевтических форм соединения, так же как их фармацевтически используемые соли, могут быть скомпонованы с терапевтически инертными неорганическими или органическими наполнителями для получения таблеток, таблеток с покрытием, драже, твердых и мягких капсул, растворов, эмульсий, или суспензий. Соединения формулы I могут быть соединены с фармацевтически приемлемыми носителями. Соединения предлагаемого изобретении могут быть, например, введены орально в виде фармакологически приемлемых солей. Поскольку соединения предлагаемого изобретения в основном водорастворимы, они могут быть введены внутривенно в физиологическом солевом растворе (то-есть в буфере с рН примерно 7,2-7,5). Удобные буферы, такие как фосфаты, бикарбонаты или цитраты, могут быть применены в предлагаемых композициях. Подходящими наполнителями для таблеток, таблеток с покрытием, драже и твердых желатиновых капсул являются, например, лактоза, кукурузный крахмал и его производные, тальк и стеариновая кислота или ее соли. Если нужно, таблетки или капсулы могут быть изготовлены по стандартным технологиям таким образом, что являются или кишечно-обволакивающими, или постепенно освобождающими активное соединение. Подходящими наполнителями для мягких желатиновых капсул являются, например, растительные масла, парафины, жиры, полутвердые и жидкие полиолы. Подходящими наполнителями растворов для инъекций являются, например, вода, солевой раствор, спирты, полиолы, глицерин или растительные масла. Подходящими наполнителями для суппозиториев являются, например, натуральные или отвержденные масла, парафины, жиры, полужидкие или жидкие полиолы. Подходящими наполнителями для растворов и сиропов для энтерального применения являются, например, вода, полиолы, сахароза, инвертный сахар и глюкоза. Фармацевтические композиции могут также содержать консерванты, солюбилизаторы, стабилизаторы, увлажняющие агенты, эмульгаторы, смягчители, красители, отдушки, соли для создания осмотического давления, буферы, маскирующие агенты или антиоксиданты. Фармацевтические формы могут также содержать другие известные терапевтически активные агенты.
Другие подходящие фармацевтические носители и их формы описаны в Remington: The Science and Practice of Pharmacy, ред. E.W.Martin, изд-во Mack Publishing Company, 19 изд., Easton, Pensylvania, 1995. Репрезентативные фармацевтические формы, содержащие соединение предлагаемого изобретения, описаны в примерах 6-8. Квалифицированный ученый в этой области может модифицировать формы внутри спецификации для того, чтобы получить многочисленные формы для особого способа введения без придания композициям предлагаемого изобретения неустойчивости или уничтожения их терапевтической активности.
В частности, модификация предлагаемых соединений с целью повышения их растворимости в воде или другой среде, например, может быть легко достигнута небольшими изменениями (образованием соли, этерификацией и т.д.), которые являются обычными для специалистов. Хорошо также с помощью обычных приемов модифицировать способ введения или режим приема определенного соединения с тем, чтобы отрегулировать фармакокинетику предлагаемых соединений с целью достижения максимального успеха лечения пациентов.
Термин «терапевтически эффективное количество», используемый здесь, означает такое количество, которое требуется для уменьшения симптомов заболевания у пациента. Доза может варьироваться в широких пределах и удовлетворять индивидуальным требованиям в каждом конкретном случае. Для орального применения дневную дозу между примерно 0,01 и примерно 100 мг/кг веса тела в день следует применять в монотерапии и/или комбинированной терапии. Предпочтительная дневная доза составляет от примерно 0,1 и до примерно 500 мг/кг веса тела, более часто от 0,1 до примерно 100 мг/кг веса тела, наиболее предпочтительно от 1,0 до примерно 100 мг/кг веса/тела в день. Типичная форма будет содержать от примерно 5% до примерно 95% активного соединения (по весу). Дневная доза может быть введена в виде единичной дозы или в виде дробных доз, обычно 1-5 доз в день.
В предлагаемом изобретении активное соединение или его соль может быть введено в комбинации с другим антивирусным агентом, таким как нуклеозидный ингибитор ревертазы, другие ненуклеозидные ингибиторы ревертазы или ингибитор ВИЧ протеазы. При введении активного соединения или его производного или соли в комбинации с другим антивирусным агентом его активность может повышаться по сравнению с самим соединением. При комбинированной терапии такое введение может быть одновременным или последовательным по отношению к нуклеозидным производным. Используемое здесь выражение «одновременное введение» включает прием агентов в то же самое или в разное время.
Упоминаемое здесь лечение включает профилактику заболеваний, также как лечение существующих состояний, а лечение животных включает лечение людей, так же как других живых существ. Кроме того, лечение ВИЧ инфекции, предлагаемое здесь, включает лечение или профилактику заболевания или состояния, сопутствующего или связанного с ВИЧ инфекцией, или клинических симптомов.
Фармацевтические формы готовят преимущественно в виде единичных форм. В такой форме препарат разделен на дозы, содержащие подходящее количество активного соединения. Формой единичной дозы может быть распакованный препарат, упаковка, содержащая дискретные количества препарата, такая как упаковка таблеток, капсул и порошков в пузырьках или ампулах. Также единичная доза может представлять собой капсулу, таблетку, облатку или лепешку, или таких форм может быть несколько в упаковке.
Соединения формулы I могут быть получены различными методами, известными специалистам в области органической химии. Исходные вещества для синтеза или легко доступны из коммерческих источников, или сами могут быть получены по известным методикам. Следующие примеры дают возможность специалистам более легко понять и применить предлагаемое изобретение. Их не следует рассматривать как исчерпывающие весь объем изобретения, а только как иллюстративный и репрезентативный материал.
Пример 1
Этиловый эфир 5-гидрокси-1Н-пиразол-3-карбоновой кислоты
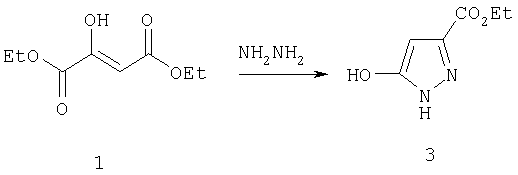
Натриевую соль диэтилоксалацетата (14,53 г, 69,15 ммол) растворяли в 100 мл бензола и перемешивали в течение 20 мин. К раствору добавляли 100 мл уксусной кислоты, и реакционную смесь перемешивали в течение еще 30 мин, после чего добавляли моногидрохлорид гидразина (9,47 г, 138 ммол) и перемешивали еще 30 мин. Затем реакционную смесь нагревали при кипении при 100°С в течение 24 час, после чего охлаждали до комнатной температуры, экстрагировали этилацетатом, промывали 10%-ной хлористоводородной кислотой, насыщенным раствором бикарбоната натрия, водой и солевым раствором. Растворитель удаляли в вакууме, получая маслянистое твердое вещество, которое растирали в порошок со смесью (2:1) диэтилового эфира и гексана, выделяя 3 (10,00 г, 92%) в виде не совсем белого твердого вещества. МСНР (электрораспыление): m/z [M+H]+=157
Пример 2
Этиловый эфир 5-(трет-бутилдиметилсиланилокси)-1Н-пиразол-3-карбоновой кислоты
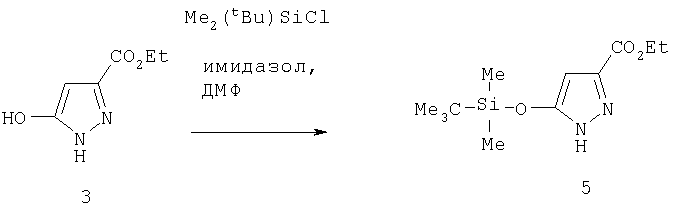
Раствор гидроксипиразола 3 (1,00 г, 6,40 ммол) в 10 мл диметилформамида охлаждали до 0°С и продували азотом, добавляли 12,8 мл (12,8 ммол) силилирующего реагента BDCS (Aldrich), и реакционную смесь перемешивали 24 час при комнатной температуре, после чего добавляли воду и экстрагировали этилацетатом. Объединенные органические фазы затем промывали водой и солевым раствором, сушили MgSO4 и фильтровали. Растворитель удаляли в вакууме, получая темное масло. Сырой продукт очищали хроматографией на силикагеле смесью гексан:этилацетат (9:1), получая желаемый силиловый эфир 5 (1,64 г, 94%). МСНР (электрораспыление): m/z [М+Н]+=271
Пример 3
Этиловый эфир 5-(трет-бутилдиметилсиланилокси)-1-(2,2,2-трифторэтил)-1H-пиразол-3-карбоновой кислоты
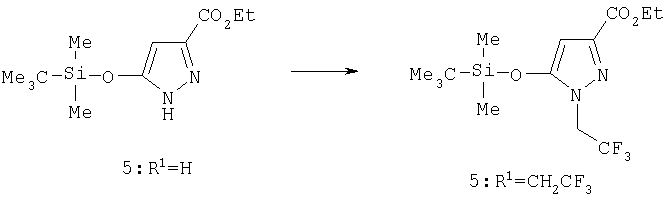
Эфир силиленола 5 (R1=Н) (1,64 г, 6,06 ммол) растворяли в 15 мл диметилформамида в атмосфере азота и охлаждали до 0°С, добавляли карбонат натрия и перемешивали в течение 15 мин, продувая смесь азотом. Затем к смеси добавляли 2-бром-1,1,1-трифторэтан (1,00 г, 6,06 ммол) и перемешивали в течение 24 час, после чего нагревали смесь при кипении еще 24 час, разбавляли водой, экстрагировали этилацетатом и промывали насыщенным раствором бикарбоната натрия, водой и солевым раствором. Смесь сушили MgSO4 и фильтровали, растворитель удаляли в вакууме, получая масло. Сырой продукт очищали колоночной хроматографией на силикагеле элюированием смесью гексан: этилацетат (85:15), выделяя 5 (R1=СН3; 1,84 г, 85%).
Пример 4
Этиловый эфир 5-гидрокси-1-(2,2,2-трифторэтил)-1Н-пиразол-3-карбоновой кислоты
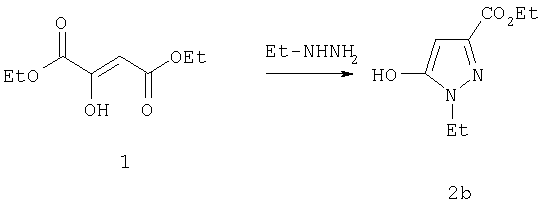
Эфир силиленола 5 (1,84 г, 5,22 ммол) растворяли в 10 мл дихлорметана и перемешивали в атмосфере азота. Смесь охлаждали до 0°С и перемешивали в течение 15 мин, после чего добавляли гидрат тетрабутиламмоний фторида (1,36 г, 5,22 ммол) и перемешивали в течение 24 час. В реакционную смесь добавляли насыщенный раствор бикарбоната натрия и экстрагировали дихлорметаном. Объединенные органические фазы промывали водой, солевым раствором, сушили MgSO4 и фильтровали. Растворитель удаляли в вакууме, получая бледно-желтое масло. Сырой продукт очищали хроматографией на силикагеле смесью гексан: этилацетат (3:1), получая желаемый продукт 6 (R1=СН2CF3; 1,14 г, 91%).
Пример 5
Этиловый эфир 1-этил-5-гидрокси-1Н-пиразол-3-карбоновой кислоты
Уксусную кислоту (100 мл) добавляли через капельную воронку к раствору натриевой соли диэтилоксалацетата (12,8 г, 60,9 ммол) в 175 мл бензола при комнатной температуре. После окончания добавления по каплям при перемешивании добавляли раствор оксалатной соли этилгидразина (9,1 г, 60,9 ммоль) в 40 мл теплой воды. После нагревания при кипении в течение 36 час реакционную смесь охлаждали до комнатной температуры, выливали в воду и экстрагировали этилацетатом. Комбинированные органические экстракты промывали солевым раствором, и растворитель отгоняли в вакууме, получая сырой продукт в виде твердого коричневого маслянистого вещества. Этот остаток растирали со смесью 2:1 этиловый эфир:гексан, получая 2б (7,7 г) в виде твердого белого вещества. МСНР (электрораспыление): m/z [M+H]+=185
Пример 6
Этиловый эфир 5-хлор-4-формил-1-изопропил-1Н-пиразол-3-карбоновой кислоты
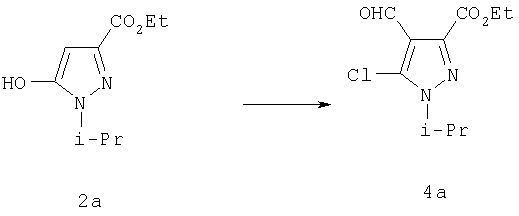
Круглодонную колбу, содержащую 100 мл 1,2-дихлорэтана, охлаждали до 0°С и заполняли азотом. Добавляли диметилформамид (14,75 г, 201 ммол) и оставляли перемешиваться 5 мин при 0°С. Медленно добавляли хлорокись фосфора (155 г, 1,0 ммоль), поддерживая температуру реакции 0°С. Раствор гидроксипиразола (20,0 г, 100 ммол) в 100 мл 1,2-дихлорэтана медленно добавляли к смеси диметилформамида и хлорокиси фосфора при 0°С. После полного добавления гидроксипиразола баню со льдом удаляли, и смесь перемешивали при комнатной температуре в течение 30 мин. В заключение реакционную смесь нагревали при 110°С в течение 24 час. Затем нагревание прекращали, и смесь доводили до комнатной температуры. Удаляли в вакууме избыток 1,2-дихлорэтана и хлорокиси фосфора, получая черное масло. Масло медленно растворяли в избытке насыщенного раствора бикарбоната натрия и перемешивали еще 6 час. Раствор экстрагировали смесью 1:1 тетрагидрофурана и этилацетата и промывали водой и солевым раствором. Органические вытяжки сушили MgSO4 и упаривали, получая темное масло. Продукт очищали хроматографией на силикагеле смесью гексан:этилацетат (9:1), получая продукт (20,34 г, 80%).
Пример 7
Этиловый эфир (3-хлорфенокси)-4-формил-1-изопропил-1Н-пиразол-3-карбоновой кислоты
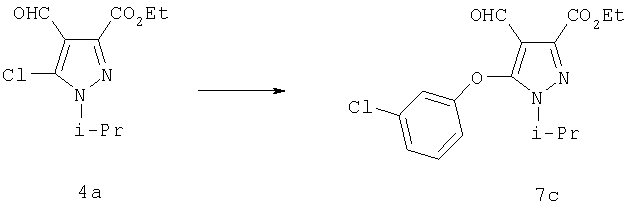
Гидрид натрия (60% в минеральном масле, 0,48 г, 12 ммоль) добавляли небольшими порциями к 3-хлорфенолу (1,54 г, 12 ммоль) в 40 мл безводного диметилформамида при комнатной температуре. Раствор перемешивали 15 мин, добавляли 4a (2,0 г, 8,2 ммоль) в одну порцию, и реакцию нагревали при 110°С в атмосфере азота в течение 1 час. Затем смесь охлаждали до комнатной температуры и выливали в 0,5 н. раствор бисульфата натрия. Экстрагировали сырой продукт смесью 1:1 гексан:этилацетат, комбинированные вытяжки промывали 0,1 н. NaOH и солевым раствором, и затем растворитель удаляли в вакууме. Сырой продукт очищали методом гель-хроматографии на силикагеле (10:1, затем 5:1 гексан:этилацетат), получая 7 с (2,3 г) в виде белого твердого вещества. МСНР (электрораспыление): m/z [M+H]+=337.
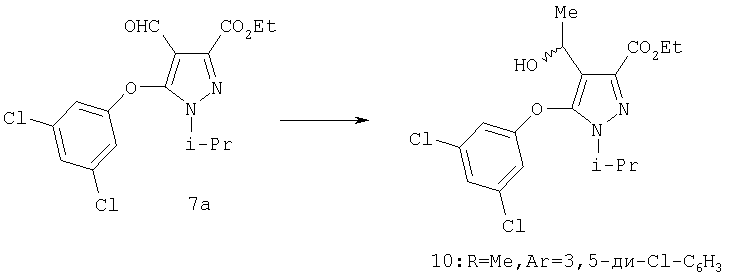
Пример 8
Этиловый эфир 5-(3,5-дихлорфенокси)-4-(1-гидроксиэтил)-1-изопропил-1Н-пиразол-3-карбоновой кислоты
Метилмагнийбромид (3,0 М в тетрагидрофуране, 0,9 мл, 2,7 ммоль) медленно добавляли к раствору 7a в смеси ТТФ:диэтиловый эфир (1:6, 30 мл) при -30°С. После окончания добавления реакционную смесь перемешивали при 0°С в течение 2 час, затем добавляли еще 0,3 мл раствора реактива Гриньяра и дополнительно перемешивали 1 час. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония. Продукт экстрагировали этилацетатом, и комбинированные органические вытяжки промывали солевым раствором. Сырой продукт очищали хроматографией на силикагеле (10:1 гексан:этилацетат), получая целевой продукт (0,93 г) в виде бесцветного масла. МСНР (электрораспыление): m/z [M+Na]+=409.
Пример 9
Этиловый эфир 5-(3,5-дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-карбоновой кислоты
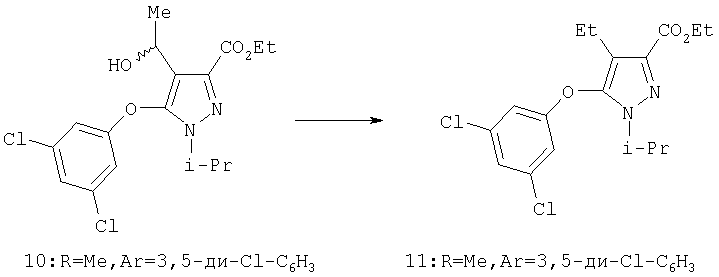
К раствору спирта (0,61 г, 1,6 ммоль) и трифторуксусной кислоты (1,3 мл, 17 ммол) в 20 мл дихлорметана добавляли при комнатной температуре триэтилсилан (0,28 мл, 1,7 ммоль). Через 2 час добавляли дополнительно 0,28 мл триэтилсилана, и реакцию перемешивали в течение ночи. После этого добавляли еще 0,3 мл триэтилсилана, и реакционную смесь оставляли на 5 час для завершения реакции. Растворитель удаляли в вакууме, остаток смешивали с этилацетатом и промывали насыщенным раствором бикарбоната натрия и солевым раствором. Сырой продукт очищали хроматографией на силикагеле (20:1 гексан:этилацетат), получая целевой продукт (0.55 г). МСНР (электрораспыление): m/z [M+Na]+=371.
Пример 10
[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-ил]метанол
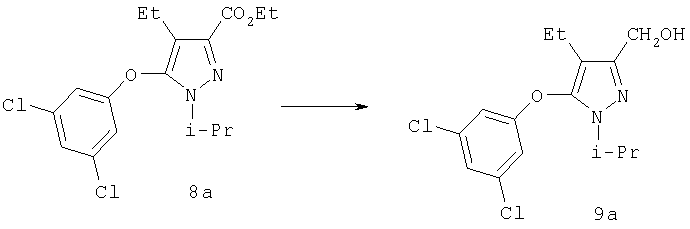
Триэтилборгидрид лития (1,0 М в ТГФ, 3,0 мл, 3,0 ммоль) медленно добавляли к эфиру 8а (0,54 г, 1,5 ммоль) в 10 мл тетрагидрофурана при -20°С. Реакцию перемешивали при -20°С в течение 30 мин, затем дополнительно 1 час при 0°С, затем к реакционной смеси добавляли 4 мл 10% раствора уксусной кислоты в этаноле. Через 10 мин растворители отгоняли в вакууме, к остатку добавляли 1 М HCl, и экстрагировали продукт этилацетатом. Комбинированные органические вытяжки промывали насыщенным водным раствором бикарбоната натрия и солевым раствором, и растворитель удаляли в вакууме. Очистка методом хроматографии на силикагеле (2:1 гексан:этилацетат) приводила к получению 9а (0,42 г) в виде белого твердого вещества. МСНР (электрораспыление): m/z [M+Na]+=329.
Пример 11
3-Хлорметил-5-(3,5-дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол
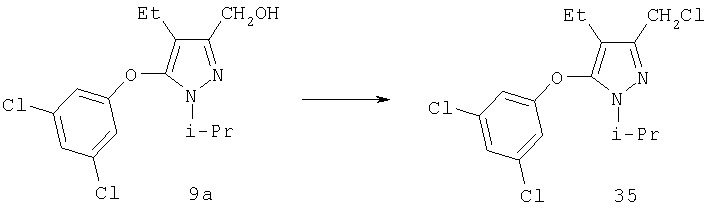
Тионилхлорид (0,13 мл, 1,8 ммоль) добавляли по каплям в охлаждаемый льдом раствор 9a (0,35 г, 1,1 ммоль) в 10 мл хлористого метилена. Через 1 час растворитель удаляли в вакууме, остаток обрабатывали насыщенным водным раствором бикарбоната натрия, и продукт экстрагировали этилацетатом. Комбинированные органические фазы промывали солевым раствором, и растворитель удаляли в вакууме, получая 35 (0,37 г) достаточно чистым настолько, что он не требовал дальнейшей очистки. МСНР (электрораспыление);
m/z [M+H]+=347.
Пример 12
[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-ил]ацетонитрил
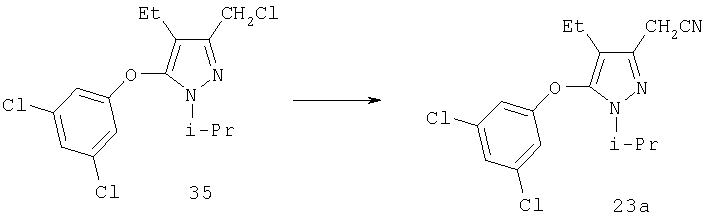
Раствор 35 (0,37 г, 1,1 ммоль) в 2 мл диметилсульфоксида добавляли к перемешиваемой смеси цианида натрия (0,11 г, 2,2 ммоль) в 10 мл диметилсульфоксида при комнатной температуре. Через 4 час реакционную смесь выливали в водный раствор 0,1 н. гидроксида натрия, и продукт экстрагировали этилацетатом. Комбинированные органические фазы разбавляли равным объемом гексана, три раза промывали водой, а затем солевым раствором. Растворитель удаляли в вакууме, и после очистки хроматографией на силикагеле (8:1, затем 5:1 гексан:этилацетат) получали 23а (0,345 г). МСНР (электрораспыление); m/z [M+H]+=338.
Пример 13
2-[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1H-пиразол-3-ил]этиламин
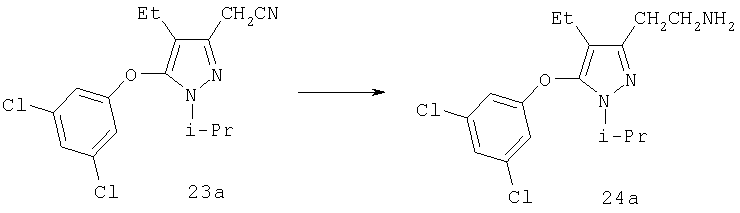
Гидрид диизобутилалюминия (1,5 М в толуоле, 0,88 мл, 1,3 ммоль) медленно добавляли к раствору 23а (0,15 г, 0,44 ммоль) в 5 мл толуола при -10°С, перемешивали при этой температуре в течение 30 мин, затем добавляли в один прием весь боргидрид натрия (0,10 г, 2,7 ммоль) с последующим добавлением по каплям 10 мл метанола. После окончания добавления охлаждение прекращали, и реакционную смесь перемешивали при комнатной температуре в течение 30 мин, после чего выливали в водный раствор натрий, калий тартрата и экстрагировали эфиром. Комбинированные эфирные вытяжки промывали солевым раствором и сушили над карбонатом калия. После очистки хроматографией на силикагеле (95:5:0,5 хлористый метилен:метанол:насыщенный водный раствор гидроксида аммония) получали 24а (0,11 г). МСНР (электрораспыление); m/z [М+Н]+=342
Пример 14
[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-ил]уксусная кислота
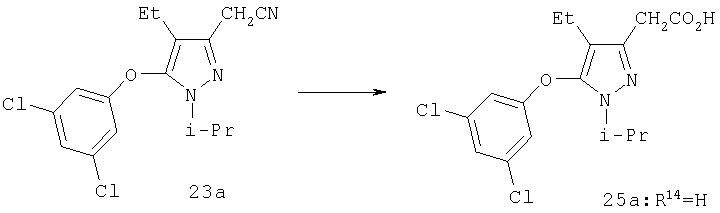
Нитрил 23а (0,19 г, 0,56 ммоль) нагревали при 100°С в течение 1,5 час в смеси 3 мл ледяной уксусной кислоты, 3 мл воды и 6 мл концентрированной хлористоводородной кислоты. Реакционную смесь выливали в 50 мл воды, и продукт экстрагировали этилацетатом. Комбинированные органические фазы промывали солевым раствором, и растворитель удаляли в вакууме, получая 25а (0,19 г). МСНР (электрораспыление); m/z [M+H]+=357.
Пример 15
Метиловый эфир [5-(3,5-дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-ил] уксусной кислоты
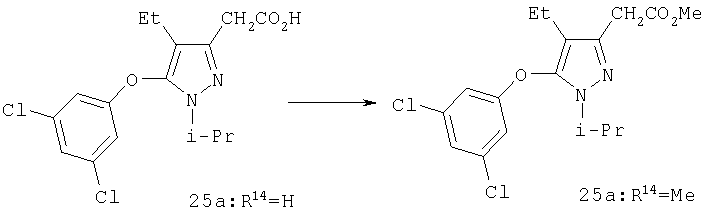
Раствор 25а (R14=Н; 0,19 г, 0,53 ммоль) в 10 мл 3М метанольного раствора хлористого водорода перемешивали в течение ночи при комнатной температуре. Реакционную смесь затем концентрировали в вакууме, остаток выливали в этилацетат и промывали насыщенным раствором бикарбоната натрия и солевым раствором. После удаления растворителя в вакууме выделяли 25 a (R14=СН3; 0,19 г), который не нуждался в дальнейшей очистке. МСНР (электрораспыление); m/z [M+H]+=371.
Пример 16
2-[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-ил] этанол
Раствор триэтилборгидрида лития (1,0 М в ТГФ, 1,5 мл, 1,5 ммоль) добавляли медленно к раствору 25а (0,19 г, 0,51 ммоль) в 5 мл ТГФ при -20°С, после чего продолжали перемешивание при -20°С в течение 30 мин и затем при 0°С в течение 1 час. Затем в реакционную смесь добавляли 5 мл 10% раствора уксусной кислоты в этаноле. После перемешивания в течение 30 мин растворитель удаляли в вакууме, остаток выливали в 1 н. хлористоводородную кислоту, и продукты (смесь альдегида и спирта) экстрагировали этилацетатом. Комбинированные органические фазы промывали солевым раствором, и растворитель удаляли в вакууме. Смесь сырых продуктов затем растворяли в 10 мл метанола, и в один прием добавляли при 0°С весь боргидрид натрия (0,10 г, 2,6 ммоль). Смесь перемешивали в течение 30 мин и добавляли 10 мл насыщенного водного раствора хлорида аммония. Смесь разбавляли 50 мл воды, и продукт экстрагировали этилацетатом. После очистки хроматографией на силикагеле (2:1 гексан:этилацетат) выделяли 26а (0,14 г) в виде бесцветного масла. МСНР (электрораспыление); m/z [M+H]+=343.
Пример 17
5-(3,5-Дихлорфенокси)-1-изопропил-4-метил-1Н-пиразол-3-илметиловый эфир карбаминовой кислоты
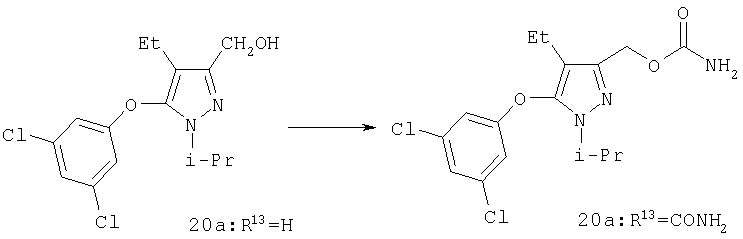
К раствору 20a (R13=H; 0,20 г, 0,64 ммоль) в 5 мл хлористого метилена при 0°С по каплям добавляли трихлорацетилизоцианат (91 мкл, 0,77 ммоль). Через 30 мин удаляли растворитель в вакууме, к остатку добавляли 4 мл метанола и обрабатывали 2 мл воды и 200 мг карбоната калия. Реакцию перемешивали при комнатной температуре в течение 2 час, затем реакционную смесь выливали в 50 мл воды, и продукт экстрагировали этилацетатом. Комбинированные органические фазы промывали солевым раствором, и удаляли в вакууме растворитель. После очистки хроматографией на силикагеле (2:1 гексан:этилацетат) с последующей перекристаллизацией из смеси хлористый метилен/гексан получали 20a (R13=CONH2; 0,21 г) в виде твердого белого вещества. МСНР (электрораспыление); m/z [M+H]+=358
Пример 18
Этиловый эфир 5-(3-хлорфенокси-4-гидроксиметил-1-изопропил-1Н-пиразол-3-карбоновой кислоты
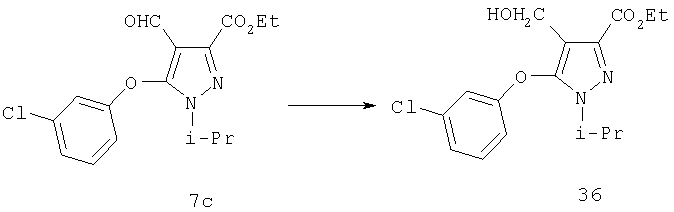
Боргидрид натрия (80 мг, 2,1 ммоль) добавляли в одну порцию к раствору 7с (0,72 г, 2,1 ммоль) в 20 мл метанола при 0°С. После перемешивания в течение 30 мин к смеси добавляли 4 мл насыщенного водного раствора хлорида аммония и отгоняли в вакууме все растворители. К остатку добавляли воду и экстрагировали продукт этилацетатом. Комбинированные органические фазы промывали солевым раствором и удаляли в вакууме растворители. После очистки хроматографией на силикагеле (6:1, затем 4:1 гексан:этил ацетат) получали соединение 36 (0,64 г) в виде бесцветного масла. МСНР (электрораспыление); m/z [M+Na]+=361.
Пример 19
Этиловый эфир 5-(3-хлорфенокси-4-иодметил-1-изопропил-1Н-пиразол-3-карбоновой кислоты
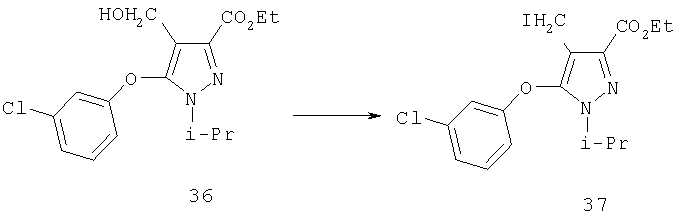
Раствор тетрайодида дифосфина (0,62 г, 1,1 ммоль) в 40 мл толуола нагревали в темноте при 85°С в течение 10 мин, добавляли сразу в одну порцию раствор 36 (0,62 г, 1,8 ммоль) в 4 мл толуола и перемешивали в течение 10 мин, затем в реакцию добавляли 40 мл 10% водного раствора бисульфита натрия, и перемешивали реакционную смесь до обесцвечивания, затем разделяли слои. Органический слой промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Сырой продукт 37 использовали непосредственно на следующей стадии синтеза.
Пример 20
[5-(3-Хлорфенокси)-1-изопропил-4-метил-1Н-пиразол-3-ил]метанол
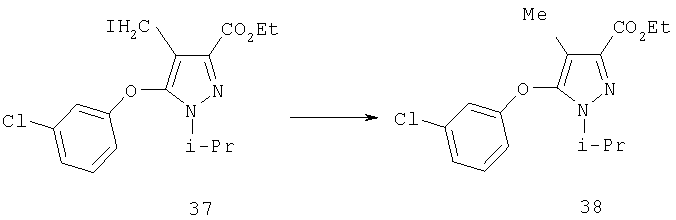
Раствор триэтилборгидрида лития (1,0 М в ТГФ, 5,4 мл, 5,4 ммоль) медленно добавляли к сырому йодиду 37 (1,8 ммоль) в 10 мл тетрагидрофурана при -20°С. Через 30 мин температуру реакции доводили до 0°С и перемешивали 1 час, после чего добавляли дополнительно 2,7 мл раствора триэтилборгидрида лития и перемешивали еще 30 мин. Затем в реакционную смесь добавляли 5 мл 10% раствора уксусной кислоты в этаноле, и смесь концентрировали в вакууме. К полученному остатку добавляли 1 н. HCl и экстрагировали продукт этилацетатом. Комбинированные органические фазы промывали насыщенным водным раствором бикарбоната натрия и солевым раствором, растворитель удаляли в вакууме. После очистки хроматографией на силикагеле (2:1 гексан:этилацетат) получали 38 (0,46 г) в виде бесцветного масла. МСНР (электрораспыление); m/z [M+H]+=281.
Пример 21
6-[5-(3-Хлорфенокси)-1-изопропил-4-метил-1Н-пиразол-3-илметил]-2Н-пиридазин-3-он
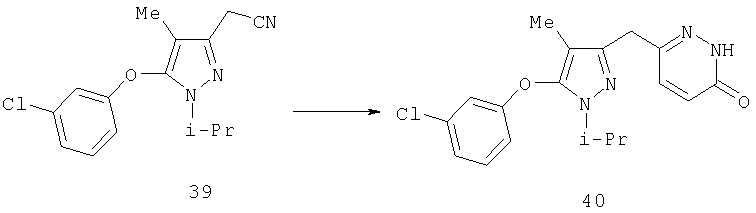
Гидрид натрия (60% дисперсия в минеральном масле, 0,14 г, 3,5 ммоль) добавляли одной порцией к раствору 39 (0,40 г, 1,4 ммоль) и 3,6-дихлорпиридазина (0,42 г, 2,8 ммоль) в 10 мл ДМФ при комнатной температуре. Реакционную смесь перемешивали 1 час и выливали при интенсивном перемешивании в 100 мл 0,5 н. водного раствора бисульфата натрия. Полученное красное маслянистое вещество отделяли фильтрованием и промывали водой. Его затем растворяли в этилацетате, промывали солевым раствором, и растворитель удаляли в вакууме. К остатку добавляли смесь 4 мл уксусной кислоты, 8 мл 12 н. HCl и 4 мл воды и нагревали в атмосфере аргона при 100°С в течение 1 час. Реакционную смесь охлаждали и осторожно добавляли к водному карбонату калия. Продукт экстрагировали этилацетатом, и после очистки препаративной тонкослойной хроматографией (95:5 хлористый метилен: метанол) получали 40 (0,35 г) в виде твердого белого вещества. МСНР (электрораспыление); m/z [M+H]+=358.
Пример 22
2-[5-(3-Хлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-илметил]изоиндол-1,3-дион
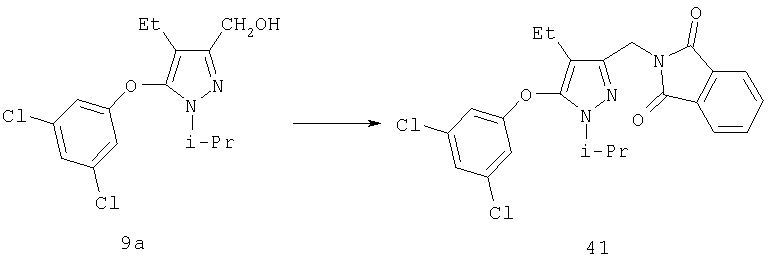
К смеси 9а (220 мг, 0,746 ммоль), трифенилфосфина (391 мг, 1,49 ммоль) и фталимида (220 мг, 1,49 ммоль) в тетрагидрофуране (20 мл) добавляли по каплям в атмосфере аргона при комнатной температуре диэтилазодикарбоксилат (260 мг, 1,492 ммоль). Полученный желтый раствор перемешивали в атмосфере аргона при комнатной температуре в течение 24 час и добавляли метанол (3 мл). Все растворители удаляли в вакууме, остаток очищали хроматографией на силикагеле в смеси гексан:этилацетат (4:1), получая твердое белое вещество 41 (310 мг, 98%). МСНР (электрораспыление); m/z [M+Н]+=424.
Пример 23
2-[5-(3-Хлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-ил]метиламин
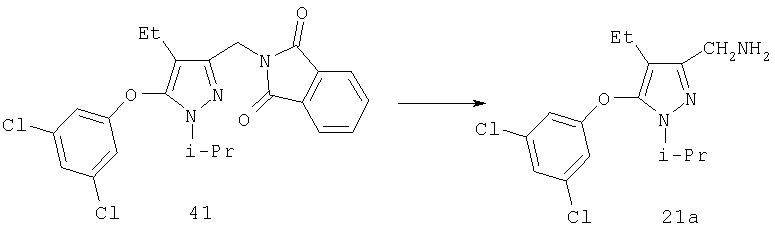
К раствору 32 (310 мг, 0,731 ммоль) в метаноле (10 мл) и тетрагидрофуране (10 мл) добавляли безводный гидразин (243 мг, 0,24 мл, 7,31 ммоль) при комнатной температуре. Реакционную смесь нагревали при кипении в атмосфере азота в течение 2 час, затем охлаждали до комнатной температуры и добавляли 10% раствор NaOH (30 мл). Сырой продукт экстрагировали хлористым метиленом (4×25 мл), растворители удаляли в вакууме. Остаток очищали хроматографией на силикагеле со смесью этилацетат: метанол (4:1), получая бледно-желтое масло 21 а (182 мг, 85%). МСНР (электрораспыление); m/z [М+Н]+=294.
Пример 24
N-[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-илметил]формамид
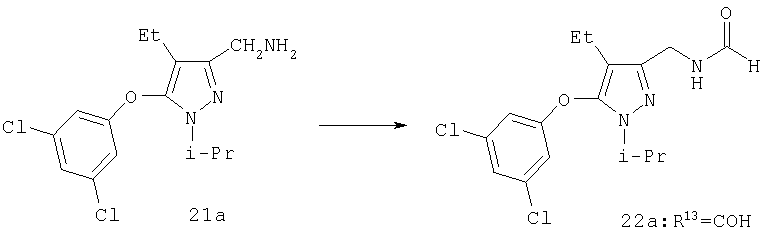
Раствор амина 21a (71 мг, 0,21 ммоль) в этилформиате (6 мл) нагревали при кипении в течение 5 час. Растворитель удаляли в вакууме, остаток очищали хроматографией на силикагеле со смесью гексан:этилацетат (2:1), получая твердое белое вещество 22а (R13=COH; 73 мг, выход 95%). МСНР (электрораспыление); m/z [M+H]+=356.
Пример 25
N-[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-илметил]ацетамид
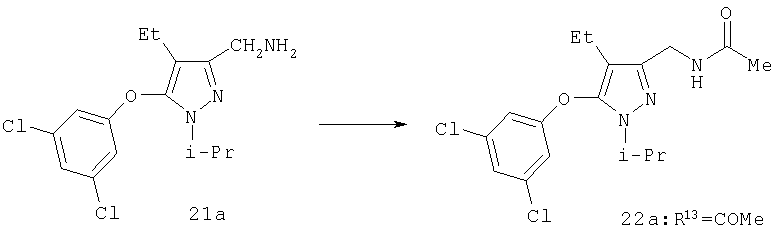
Раствор амина 21a (71 мг, 0,22 ммоль) в уксусном ангидриде (5 мл) перемешивали при комнатной температуре 2,5 час. Добавляли к реакционной смеси метанол (10 мл) и удаляли растворители в вакууме. Остаток обрабатывали 10% NaHCO3 (20 мл) и перемешивали в течение 20 мин. Сырой продукт экстрагировали хлористым метиленом (3×20 мл). Органическую фазу собирали и промывали солевым раствором, после чего растворитель удаляли в вакууме. Остаток очищали хроматографией на силикагеле со смесью гексан:этилацетат (2:1), получая твердое белое вещество 22a (R13=СОМе; 70 мг, выход 87,5%). МСНР (электрораспыление); m/z [M+H]+=370.
Пример 26
N-[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-илметил]метансульфонамид
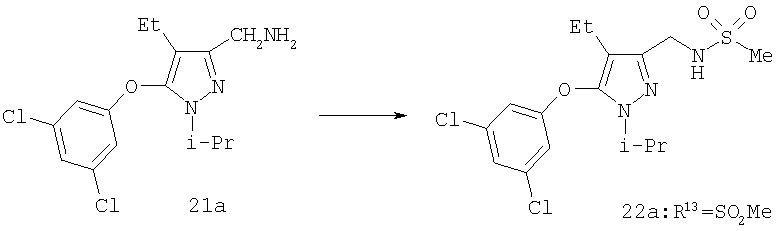
К раствору 21a (83 мг, 0,25 ммоль) и триэтиламина (76 мг, 0,75 ммоль) в безводном хлористом метилене (5 мл) добавляли метансульфонилхлорид (41 мг, 0,35 ммоль). Полученную желтую смесь перемешивали в атмосфере азота при комнатной температуре в течение 20 час, после чего добавляли воду (10 мл). Сырой продукт экстрагировали хлористым метиленом (3×10 мл). Органические фазы объединяли и промывали солевым раствором, удаляли растворитель в вакууме. Остаток очищали хроматографией на силикагеле со смесью гексан:этилацетат (3:1), получая твердое белое вещество 22а (R13=SO2Ме; 98 мг, выход 96,5%). МСНР (электрораспыление); m/z [M+H]+=406.
Пример 27
[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-илметил]мочевина
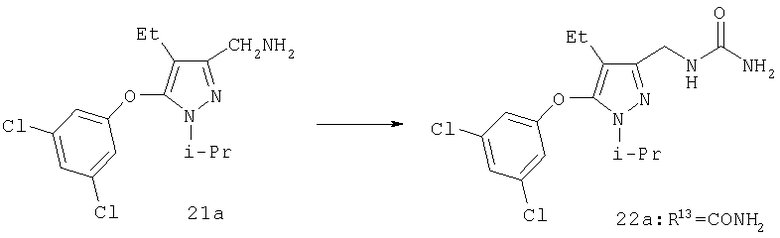
К раствору 21 а (85 мг, 0,26 ммоль) в тетрагидрофуране (5 мл) добавляли триметилсилилизоцианат (53 мг, 0,39 ммоль) в одну порцию. Реакционную смесь перемешивали при комнатной температуре в атмосфере азота 10 час, затем разбавляли метанолом (10 мл), и все растворители удаляли в вакууме. Остаток очищали хроматографией на силикагеле со смесью гексан:этилацетат (2:1), получая твердое белое вещество 22а (R13=CONH2; 80 мг, 83%). МСНР (электрораспыление); m/z [M+H]+=371.
Пример 28
[5-(3-Хлорфенокси)-3-гидроксиметил-1-изопропил-1Н-пиразол-4-ил]метанол
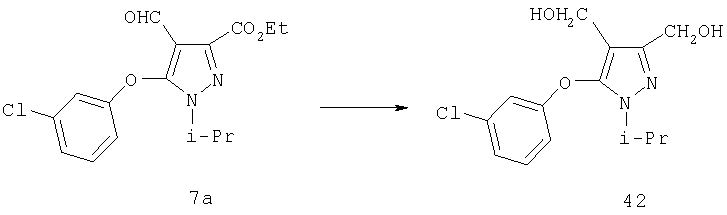
К раствору 7а (110 мг, 0,327 ммоль) в тетрагидрофуране (10 мл), охлажденному до -78°С, добавляли раствор алюмогидрида лития (1,0 М в тетрагидрофуране, 0,72 мл, 0,72 ммоль). Реакционную смесь перемешивали в атмосфере аргона при -78°С в течение 30 мин и затем при 0°С еще 45 мин. К реакционной смеси добавляли метанол (0,5 мл) и перемешивали с насыщенным раствором натрий, калий тартрата (15 мл) в течение 2 час. Сырой продукт экстрагировали диэтиловым эфиром (4×25 мл) и объединенные органические фазы промывали солевым раствором. Удаляли растворители в вакууме, остаток очищали хроматографией на силикагеле со смесью гексан:этилацетат (1:2), получая продукт 42 (95 мг, выход 97,8%). МСНР (электрораспыление); m/z [М+Н]+=297.
Пример 29
Этиловый эфир 5-(3,5-дихлорфенокси)-1-изопропил-4-(2-метоксикарбонилвинил)-1Н-пиразол-3-карбоновой кислоты
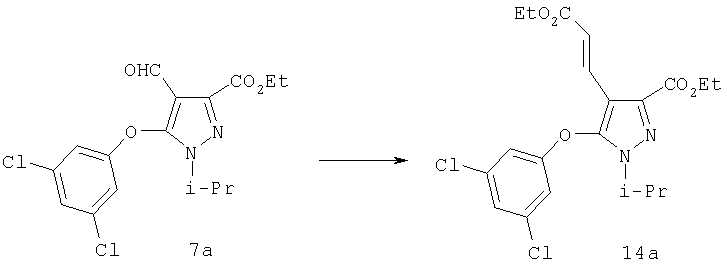
К раствору 7a (200 мг, 0,54 ммоль) в тетрагидрофуране (10 мл) при 0°С добавляли метил(трифенилфосфоранилиден)ацетат (1,30 г, 3,89 ммоль). Реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 7 час, после чего добавляли воду (40 мл). Сырой продукт экстрагировали этилацетатом (3×35 мл). Объединенные органические фазы промывали солевым раствором. Растворители удаляли в вакууме. Остаток очищали хроматографированием на силикагеле смесью гексан: этилацетат (4:1), получая 14а (220 мг, выход 95%). МСНР (электрораспыление); m/z [M+H]+=427.
Пример 30
Этиловый эфир 5-(3,5-дихлорфенокси)-1-изопропил-4-(2-метоксикарбонилэтил)-1Н-пиразол-3-карбоновой кислоты
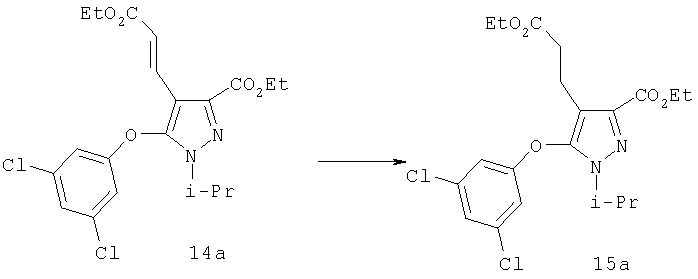
К смеси предварительно высушенной стружки магния (50 мг, 2,10 ммоль) и безводного метанола (30 мл) при 0°С добавляли раствор 14 (180 мг, 0,42 ммоль) в метаноле (2 мл) и наблюдали выделение газа. Реакционную смесь перемешивали 5 час при 0°С и затем 10 час при комнатной температуре. Затем реакционную смесь фильтровали через целлиты CELITE®. Объединенный фильтрат обрабатывали 10% раствором бисульфата натрия. Сырой продукт экстрагировали этилацетатом (3×25 мл). Объединенные органические фазы промывали солевым раствором. Растворитель удаляли в вакууме. Остаток очищали хроматографированием на силикагеле смесью гексан: этилацетат (4:1), получая продукт 15а в виде бесцветного масла (157 мг, выход 90%). МСНР (электрораспыление); m/z [M+H]+=429.
Пример 31
3-[5-(3,5-Дихлорфенокси)-3-гидроксиметил-1-изопропил-1Н-пиразол-4-ил]пропан-1-ол
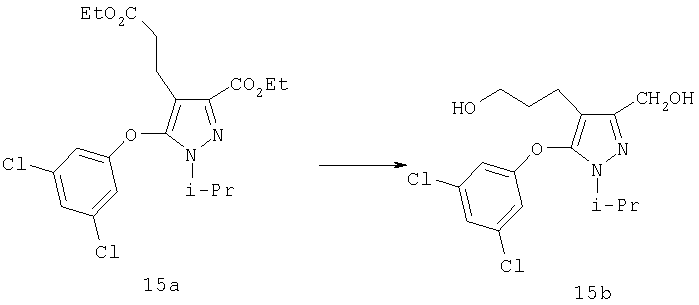
К раствору 15а (100 мг, 0,24 ммоль) в тетрагидрофуране (15 мл) при -40°С медленно добавляли раствор триэтилборгидрида лития (1,0 М в тетрагидрофуране, 1,25 мл, 1,25 ммоль), затем перемешивали в атмосфере азота при -40°С в течение 10 мин и при 0°С еще 45 мин. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 30 мин с 1 н. HCl (20 мл). Сырой продукт экстрагировали диэтиловым эфиром (3×25 мл). Объединенные органические фазы промывали солевым раствором. Растворители удаляли в вакууме. Остаток очищали хроматографированием на силикагеле смесью гексан:этилацетат (1:1), получая продукт 156 (52 мг, выход 60%). МСНР (электрораспыление); m/z [M+H]+=359
Пример 32
3-(2,4-Диэтил-5-гидроксиметил-2Н-пиразол-3-илокси)бензонитрил
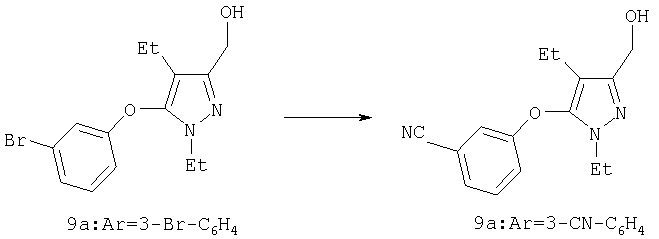
К раствору арилбромида (96 мг, 0,30 ммоль) в диметилформамиде (8 мл) добавляли тетракис(трифенилфосфин)палладий (0) (0,173 мг, 0,15 ммоль) и цианид цинка (32 мг, 0,27 ммоль) при комнатной температуре. Смесь нагревали в атмосфере аргона при 90°С в течение 6 час и выливали в насыщенный раствор бикарбоната натрия (50 мл). Сырой продукт экстрагировали этилацетатом (3×30 мл). Комбинированные органические фазы промывали солевым раствором, и растворители удаляли в вакууме. Остаток очищали хроматографированием на силикагеле смесью гексан: этилацетат (1:1), получая целевой продукт (50 мг, 61,5%). МСНР (электрораспыление); m/z [M+H]+=272.
Пример 33
Этиловый эфир 5-(3,5-дихлорфенокси)-4-гидроксиметил-1-изопропил-1Н-пиразол-3-карбоновой кислоты
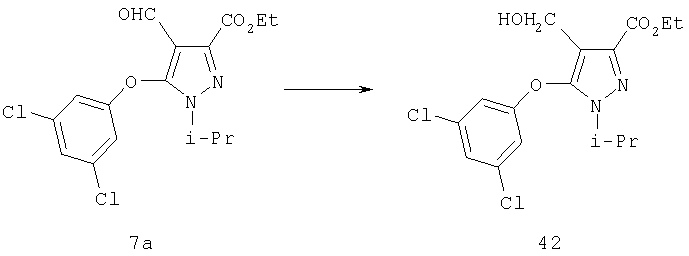
К раствору 7а (559 мг, 1.51 ммоль) в тетрагидрофуране (5 мл) и метаноле (15 мл) при 0°С в одну порцию добавляли боргидрид натрия (58 мг, 1,52 ммоль). Реакционную смесь перемешивали в атмосфере азота при 0°С в течение 30 мин, после чего добавляли насыщенный раствор хлористого аммония (25 мл). Отделяли органическую фазу, водную фазу экстрагировали этилацетатом (3×20 мл). Объединенные органические фазы промывали солевым раствором и концентрировали в вакууме, остаток очищали хроматографией на силикагеле смесью гексан:этилацетат (4:1), получая спирт 42 (494 мг, выход 87%). МСНР (электрораспыление); m/z [M+H]+=373.
Пример 34
Этиловый эфир 5-(3,5-дихлорфенокси)-1-изопропил-4-метоксиметил-1Н-пиразол-3-карбоновой кислоты
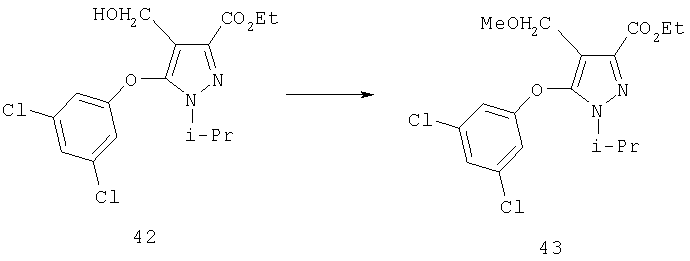
К раствору 42 (87 мг, 0,233 ммоль) в безводном диметилформамиде (5 мл) при 0°С добавляли гидрид натрия (60% дисперсия в минеральном масле, 12 мг, 0,280 ммоль). Реакционную смесь перемешивали в атмосфере аргона при 0°С в течение 30 мин, после чего добавляли йодистый метил (50 мг, 0,35 ммоль). Полученную реакционную смесь нагревали до комнатной температуры и перемешивали 2 час, затем добавляли 10% раствор бисульфата натрия (10 мл). Сырой продукт экстрагировали этилацетатом (3×10 мл), объединенные органические фазы промывали солевым раствором, и растворитель удаляли в вакууме. Остаток очищали хроматографией на силикагеле в системе гексан:этилацетат (6:1), получая 43 (50 мг, выход 56%). МСНР (электрораспыление); m/z [M+H]+=387.
Пример 35
[5-(3,5-Дихлорфенокси)-1-изопропил-4-метоксиметил-1Н-пиразол-3-ил]метанол
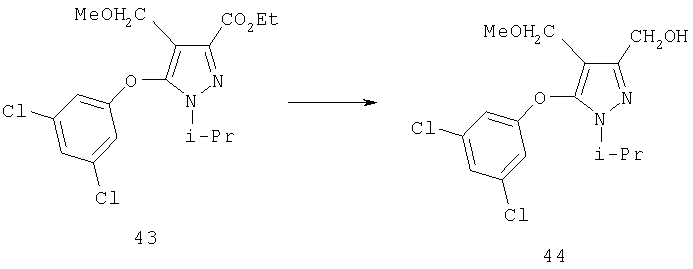
К раствору 43 (47 мг, 0,12 ммоль) в тетрагидрофуране (15 мл) при -40°С медленно добавляли триэтилборгидрид лития (1,0 М в ТГФ, 0,25 мл, 0,25 ммоль). Смесь перемешивали в атмосфере аргона при -40°С в течение 10 мин и затем при 0°С в течение 45 мин, после чего нагревали до комнатной температуры и обрабатывали 1 н. хлористоводородной кислотой (20 мл) в течение 30 мин. Сырой продукт экстрагировали диэтиловым эфиром (3×25 мл). Объединенные органические слои промывали солевым раствором, и растворители удаляли в вакууме. Остаток очищали хроматографированием на силикагеле смесью гексан: этилацетат (1:1), получая 44 (26 мг, выход 63%). МСНР (электрораспыление); m/z [M+H]+=345
Пример 36
5-[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-илметил]-2Н-тетразол
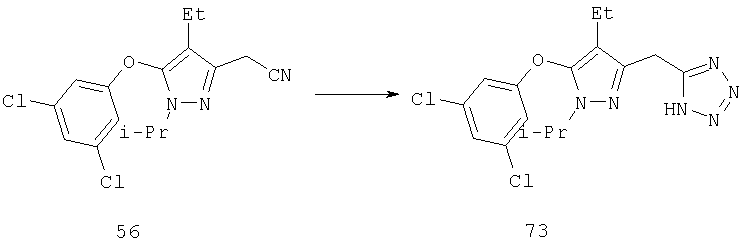
К раствору нитрила (56; 0,065 г, 0,192 ммоль) в 3 мл ксилола добавляли азидотрибутилолово (0,058 мл, 0,221 ммоль), и реакционную смесь нагревали при 130°С в течение 12 час, затем концентрировали в вакууме. Остаток экстрагировали этилацетатом и промывали водным раствором хлористого аммония. Органический слой сушили над сульфатом магния, фильтровали и затем концентрировали в вакууме. Сырой продукт очищали флэш-хроматографией на силикагеле (1:1 гексан:этилацетат, затем 9:1 этилацетат:метанол), получая целевой продукт (73; 3,4 мг, 5%). МСНР (электрораспыление); m/z [MH]=381.
Пример 37
1-[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-ил]пропан-2-он
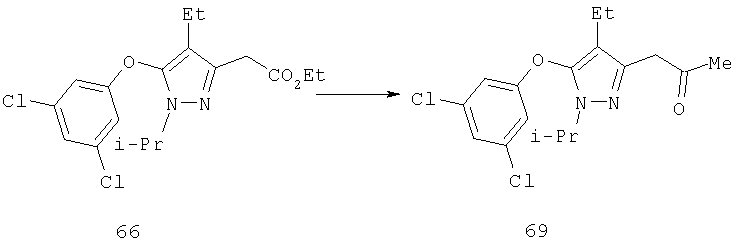
К раствору эфира 66 (0,054 г, 0,140 ммоль) в 5 мл тетрагидрофурана при 0°С в атмосфере аргона добавляли раствор метилмагнийбромида (1 М в диэтиловом эфире, 1,26 мл, 1,26 ммоль). Реакционную смесь доводили до комнатной температуры и перемешивали в течение ночи. Затем к реакционной смеси добавляли по каплям воду с последующим подкислением 1 н. водной хлористоводородной кислотой. Продукт экстрагировали этилацетатом, высушивали над сульфатом магния и удаляли растворители в вакууме. После очистки флеш-хроматографией на силикагеле (3:1 гексан:этилацетат) получали продукт в виде масла (8 мг, 16%). МСНР (электрораспыление); m/z [MH]=355.
Пример 38
1-[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-ил]пропан-2-ол
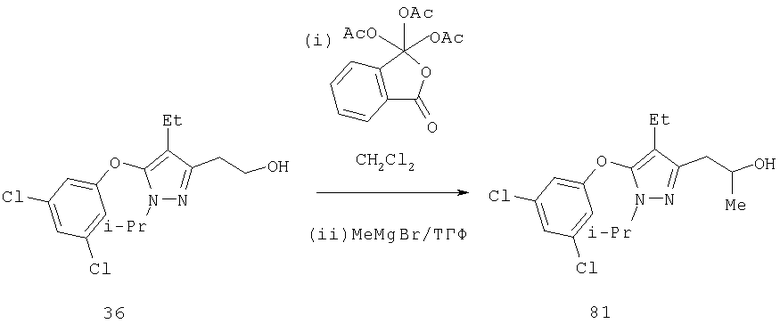
К раствору спирта 36 (0,080 г, 0,233 ммоль) в 7 мл дихлорметана по каплям добавляли раствор перйодинана (реактив Десс-Мартина, 1,1,1-триацетокси-1,1-дигидро-1,2-бензйодоксол-3(1Н)-он; 0,09 г, 0,233 ммоль) в 0,7 мл дихлорметана. Через 30 мин добавляли смесь воды (0,005 мл, 0,256 ммоль) и 5 мл хлористого метилена и перемешивали в течение ночи при комнатной температуре. Реакционную смесь экстрагировали хлористым метиленом и 10% водным раствором бисульфита/карбоната натрия. Органический слой высушивали над сульфатом магния и концентрировали в вакууме. Сырой продукт (альдегид) растворяли в тетрагидрофуране, охлаждали до -24°С и затем добавляли по каплям метилмагнийбромид (1 М в тетрагидрофуране, 0,26 мл, 0,26 ммоль). После перемешивания в течение 72 час к реакционной смеси добавляли по каплям воду, и полученную смесь концентрировали в вакууме. Остаток экстрагировали этилацетатом и водой, органический слой сушили над сульфатом магния. После очистки флэш-хроматографией на силикагеле (7:3 гексан: этилацетат) получали вторичный спирт 81 в виде масла (11,4 мг, 14%).
Пример 39
2-[5-(3,5-Дихлорфенокси)-1,4-диэтил-1Н-пиразол-3-ил]-N-фенилацетамид
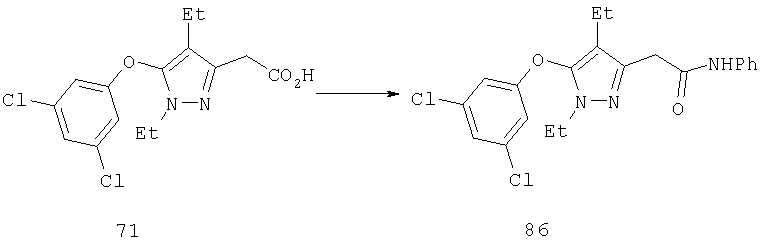
К раствору карбоновой кислоты 71 (0,15 г, 0,44 ммоль) в 5 мл тетрагидрофурана добавляли 1,1'-карбонилдиимидазол (0,70 г, 0,44 ммоль) и нагревали смесь при 50°С в течение 30 мин. К смеси добавляли анилин (0,040 мл, 0,44 ммоль), и реакционную смесь выдерживали при 50°С еще 3 час, а затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в 30 мл этилацетата, и промывали этот раствор 1 н. хлористоводородной кислотой, насыщенным раствором бикарбоната натрия и солевым раствором, удаляли в вакууме растворитель, и сырой продукт очищали препаративной тонкослойной хроматографией на силикагеле (4:1 гексан:этилацетат), получая амид 86 в виде белого твердого вещества (0,174 г, 95%); t.пл. 112,2-115,9°С. МСНР (электрораспыление); m/z [MH]=418.
Пример 40
5-(3,5-Дихлорфенокси)-1,3,4-триэтил-1Н-пиразол
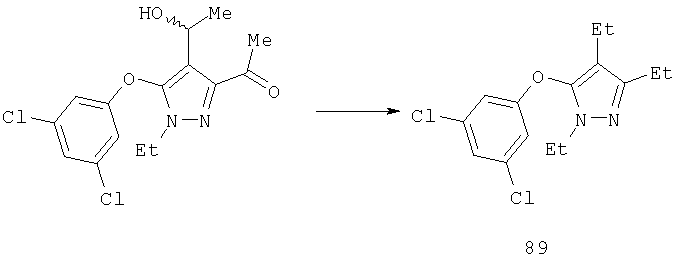
Раствор кетоспирта (0,16 г, 0,52 ммоль) в 0,5 мл триэтилсилана и 0,5 мл трифторуксусной кислоты перемешивали при 35°С в течение ночи. Реакционную смесь концентрировали в вакууме, и полученный сырой продукт очищали препаративной тонкослойной хроматографией на силикагеле (10:1 гексан:этилацетат), получая 89 в виде масла (94 мг, 64%). МСНР (электрораспыление); m/z [MH]=313.
Пример 41
5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-3-пиразол-1-илметил-1Н-пиразол
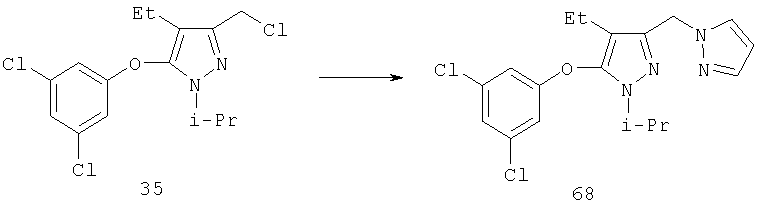
Одногорлую круглодонную колбу на 10 мл продували азотом, помещали в нее хлорметилпиразол (0,100 г, 0,288 ммоль) и растворяли его в 1 мл диметилформамида. Затем в колбу последовательно добавляли карбонат калия и пиразол (0,029 г, 0,431 ммоль). Реакционную смесь перемешивали в течение 24 час и экстрагировали водой и этилацетатом. Объединенные органические экстракты промывали водой и солевым раствором, высушивали над сульфатом натрия и фильтровали. Раствор концентрировали в вакууме, получая сырой продукт, который очищали флэш-хроматографией на силикагеле (85:15 гексан:этилацетат), выделяя целевой продукт (68; 90%). МСНР (электрораспыление); m/z [MH]=379.
Соответствующее производное имидазола 67 получали аналогичным способом, заменяя пиразол имидазолом в примере 41. Целевой продукт выделяли с выходом 83%. МСНР (электрораспыление); m/z [MH]=379.
Пример 42
3-[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-илметил]-1Н-пиримидин-2,4-дион
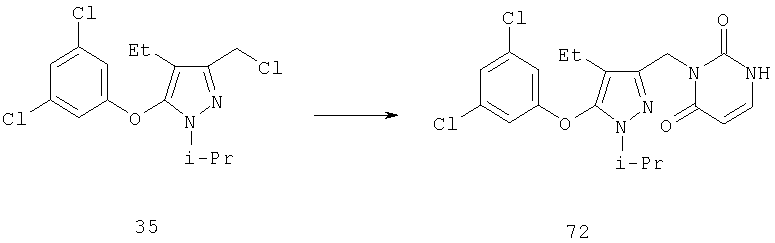
Одногорлую круглодонную колбу на 10 мл продували азотом, помещали в нее хлорметилпиразол (0,100 г, 0,288 ммоль) и растворяли его в 1 мл диметилформамида. Затем в колбу последовательно добавляли карбонат калия и урацил (0,050 г, 0,43 ммоль). Реакционную смесь перемешивали в течение 24 час и экстрагировали водой и этилацетатом. Объединенные органические экстракты промывали водой и солевым раствором, высушивали над сульфатом натрия и фильтровали. Раствор концентрировали в вакууме, получая сырой продукт, который очищали флэш-хроматографией на силикагеле (9:1 гексан:этилацетат), выделяя 72 с выходом 85%. МСНР (электрораспыление); m/z [MH]=423.
Пример 43
[5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-1Н-пиразол-3-ил]тиофен-2-ил-метанол
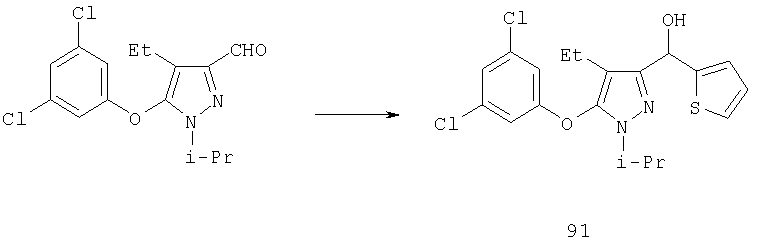
Трехгорлую круглодонную колбу на 100 мл продували азотом, заполняли хлопьями магния (0,074 г, 3,067 ммоль), нагревали, продувая азотом. Затем добавляли в колбу тетрагидрофуран (5 мл) и 2-йодтиофен (0,500 г, 2,384 ммоль) и нагревали. Когда магний весь растворился, аликвоту (0,61 мл, 0,611 ммоль) добавляли к раствору альдегида (0,200 г, 0,611 ммоль) в тетрагидрофуране при 0°С. Реакцию нагревали до комнатной температуры, затем охлаждали до 0°С и добавляли насыщенный раствор хлористого аммония, после чего экстрагировали водой и этилацетатом. Объединенные экстракты в этилацетате промывали хлористым аммонием и насыщенным солевым раствором, органическую фазу (в этилацетате) сушили над сульфатом натрия и фильтровали, раствор концентрировали в вакууме. Сырой продукт очищали флэш-хроматографией на силикагеле (80:20 гексан: этилацетат), получая 91 с выходом 75%. МСНР М+=411.
Пример 44
5-(3.5-Дихлорфенокси)-4-этил-1-изопропил-3-тиофен-2-илметил-1Н-пиразол
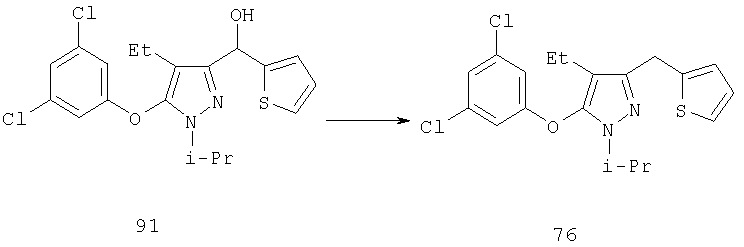
Раствор гидроксиметилтиофена 91 (0,100 г, 2,431 ммоль) и 3 мл трифторуксусной кислоты охлаждали до 0°С, добавляли триэтилсилан (0,58 мл, 3,65 ммоль), и реакционную смесь перемешивали при 0°С в атмосфере азота, доводили смесь до комнатной температуры и перемешивали еще 24 час, после чего смесь охлаждали до 0°С и медленно добавляли насыщенный раствор бикарбоната натрия. Реакционную смесь экстрагировали этилацетатом, и комбинированные органические фазы промывали насыщенным раствором бикарбоната натрия, водой и солевым раствором. Экстракт в этилацетате сушили над сульфатом натрия, фильтровали, концентрировали в вакууме, и сырой продукт очищали флэш-хроматографией на силикагеле (90:10 гексан:этилацетат), получая 76 с выходом 90%. МСНР М+=395
Пример 45
5-(3,5-Дихлорфенокси)-1,4-диэтил-1Н-пиразол-3-карбальдегид
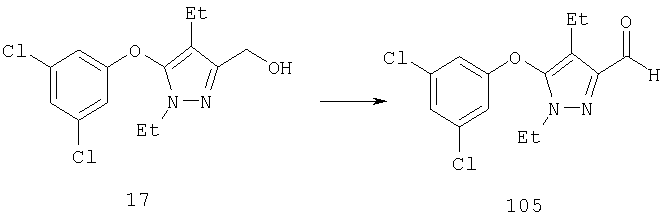
Твердый перрутенат тетрапропиламмония (118 мг, 0,33 ммоль) добавляли в одну порцию к перемешиваемой смеси спирта 17 (2,12 г, 6,72 ммоль), N-оксида N-метилморфолина (1,18 г, 10,1 ммоль) и молекулярных сит 4Å (3,36 г) в хлористом метилене (66 мл) и ацетонитриле (8 мл) при комнатной температуре в атмосфере аргона. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 час, фильтровали через целит, и фильтрат концентрировали в вакууме. Сырой продукт очищали флэш-хроматографией на силикагеле (4:1 гексан:этилацетат), получая 1,79 г (85%) 105 в виде бледно-желтого масла. МСНР (электрораспыление); m/z [MH]=313.
Пример 46
5-(3,5-Дихлорфенокси)-1,4-диэтил-3-(1Н-имидазол-2-илметил)-1Н-пиразол
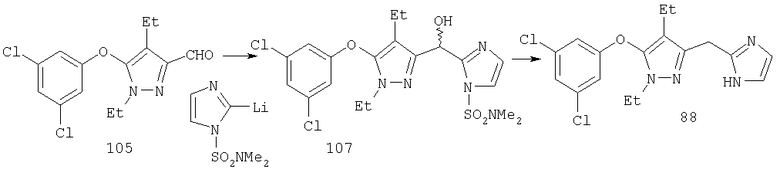
Стадия 1.
Диметилхлорсульфонамид (3,8 г, 26,5 ммоль) перемешивали с имидазолом (2,0 г, 29,4 ммоль) и триэтиламином (2,97 г, 29,4 ммоль) в бензоле (35 мл) при комнатной температуре в течение 16 час, затем смесь фильтровали, и твердое вещество промывали бензолом (50 мл). Комбинированные фильтраты концентрировали в вакууме. Сырой продукт очищали флэш-хроматографией на силикагеле (4:1 гексан:этилацетат), получая сульфонамид 106 в виде бесцветного масла (3,6 г, 69%).
Стадия 2.
К раствору имидазолилсульфонамида 106 (146 мг, 0,834 ммоль) в тетрагидрофуране (8 мл) при -78°С по каплям добавляли н-бутиллитий (1,6 М в гексане, 0,521 мл, 0,834 ммоль) и перемешивали реакционную смесь при этой температуре в атмосфере аргона в течение 45 мин, затем медленно добавляли раствор альдегида 105 (201 мг, 0,642 ммоль) в тетрагидрофуране (1 мл). Полученную реакционную смесь доводили до комнатной температуры и перемешивали 19 час, добавляли насыщенный раствор хлорида аммония (10 мл), и сырой карбинол 107 экстрагировали этилацетатом (3×10 мл). Объединенные фильтраты сушили над сульфатом магния, фильтровали и выпаривали. Сырой продукт очищали флэш-хроматографией на силикагеле (4:1 гексан:этилацетат), получая 107 в виде бледно-желтого масла (156 мг, 50%).
Стадия 3.
Карбинол 107 смешивали при комнатной температуре с трифторуксусной кислотой (1,0 мл) и триэтилсиланом (0,6 мл), реакционную смесь нагревали при кипении 3 час, затем охлаждали до комнатной температуры, и удаляли в вакууме трифторуксусную кислоту и триэтилсилан. Остаток очищали флэш-хроматографией на силикагеле (5% метанола в хлористом метилене), получая 88 в виде твердого белого вещества (90 мг, 80%). МСНР (электрораспыление); m/z (МН)=365, t пл. 145-148°С.
Пример 47
5-(3,5-Дихлорфенокси)-1,4-диэтил-3-(3Н-имидазол-4-илметил)-1Н-пиразол
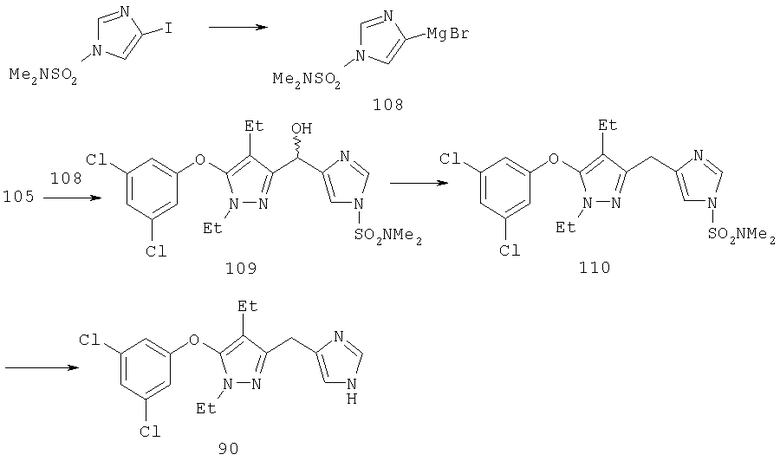
Стадия 1
К раствору N,N-диметил-4-йод-1Н-имидазол-1-сульфонамида (193 мг, 0,64 ммоль) в хлористом метилене (3 мл) добавляли этилмагнийбромид (3 М в диэтиловом эфире, 0,18 мл, 0,60 ммоль) при комнатной температуре в атмосфере аргона, после чего перемешивали реакционную смесь при комнатной температуре в течение 30 мин. Затем к ранее образовавшемуся реактиву Гриньяра по каплям добавляли раствор альдегида (100 мг, 0,32 ммоль) в хлористом метилене (0,7 мл) при комнатной температуре и перемешивали в течение 16 час. В реакцию добавляли насыщенный водный раствор хлористого аммония (10 мл). Сырой карбинол экстрагировали этилацетатом (3х10 мл), сушили комбинированные экстракты этилацетата над сульфатом магния, фильтровали и упаривали. Сырой продукт очищали флеш-хроматографией на силикагеле (5% метанола в хлористом метилене), получая карбинол 109 в виде бесцветного масла (120 мг, 76,8%).
Стадия 2.
Карбинол 109 растворяли в трифторуксусной кислоте (1,0 мл) и триэтилсилане (0,4 мл) при комнатной температуре, и затем смесь нагревали 3 час при 80°С. Сырое дезоксипроизводное 110 выделяли выпариванием летучих реагентов в вакууме.
Стадия 3.
Сырое N-замещенное дезоксипроизводное 110 обрабатывали хлористоводородной кислотой (1 М). Реакционную смесь нагревали при кипении в течение 3 час, а затем 48 час перемешивали при комнатной температуре. К реакционной смеси добавляли насыщенный раствор бикарбоната натрия до рН 8, и сырой продукт экстрагировали этилацетатом (3×10 мл). Объединенные органические экстракты промывали водой (1×10 мл) и солевым раствором (1×10 мл), и затем растворитель удаляли в вакууме. Сырой продукт очищали флеш-хроматографией на силикагеле (5% метанола в хлористом метилене), получая 90 в виде твердого белого вещества (60 мг, 67% после двух стадий). МСНР (электрораспыление); m/z (МН)=365, t пл. 142-145,2°С.
Пример 48
3-[5-(3,5-Дихлорфенокси)-1,4-диэтил-1Н-пиразол-3-ил]пропан-1-ол
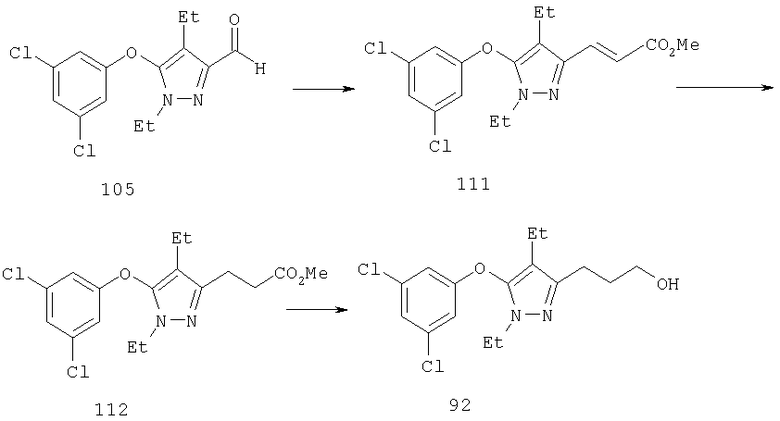
К раствору 105 (102 мг, 0,326 ммоль) в тетрагидрофуране (10 мл) добавляли метил(трифенилфосфоранилиден)ацетат (1,09 г, 3,26 ммоль) при комнатной температуре в атмосфере аргона, перемешивали реакционную смесь при этой температуре 24 час и затем концентрировали в вакууме. α,β-Ненасыщенный эфир 111 очищали флеш-хроматографией на силикагеле (5:1 гексан:этилацетат), получая 111 в виде белого твердого вещества (107 мг, 88,9%).
Раствор 111 в метаноле (1,0 мл) добавляли к перемешиваемой смеси порошка магния (42 мг, 1,74 ммоль) и метанола (15 мл) при 0°С. Реакцию выдерживали при 0°С 3 час, затем доводили до комнатной температуры в течение 16 час и выливали в 1 М водный раствор бисульфата натрия (20 мл). Сырой продукт экстрагировали этилацетатом (3×10 мл), отгоняли этилацетат в вакууме и очищали сырой продукт флеш-хроматографией на силикагеле (5:1 гексан:этилацетат), получая 112 в виде бесцветного масла (75 мг, 70%).
К раствору 112 (75 мг, 0,202 ммоль) в тетрагидрофуране (10 мл) при -30°С добавляли триэтилборгидрид лития (1М в тетрагидрофуране, 0,606 мл, 0,606 ммоль) в течение 5 мин. Реакционную смесь доводили до 0°С и перемешивали 3 час, затем выливали в 2 н. хлористоводородную кислоту (50 мл), и отгоняли в вакууме тетрагидрофуран. Оставшийся раствор перемешивали 6 час при комнатной температуре. Экстрагировали сырой продукт хлористым метиленом (4×10 мл), сушили объединенные органические вытяжки над сульфатом магния, фильтровали и упаривали. Сырой продукт очищали флеш-хроматографией на силикагеле (2:1 гексан:этилацетат), получая карбинол 92 в виде бесцветного масла (58 мг, 85%). МСНР (электрораспыление); m/z (MH)=342.
Пример 49
5-[5-(3,5-Дихлорфенокси)-1,4-диэтил-1Н-пиразол-3-илметил]-4-метил-2,4-дигидро[1,2,4]триазол-3-он
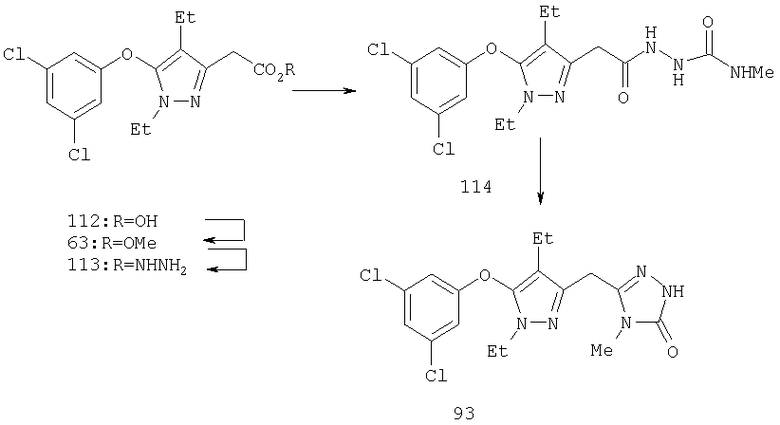
Стадия 1.
К раствору 112 (100 мг, 0,292 ммоль) в метаноле (25 мл) добавляли три капли концентрированной серной кислоты. Реакционную смесь нагревали при кипении в течение 3 час, затем метанол удаляли в вакууме. К остатку добавляли насыщенный водный раствор бикарбоната натрия до рН 8, и сырой продукт экстрагировали этилацетатом (3×10 мл). После удаления растворителя в вакууме получали сырой метиловый эфир 63 в виде бесцветного масла (102 мг, 97,8%).
Стадия 2
К раствору 63 в абсолютном этаноле (20 мл) добавляли гидразин моногидрат (2 мл). Реакционную смесь нагревали при кипении в течение 4 час, и удаляли этанол в вакууме. Полученный остаток растворяли в этилацетате (20 мл), и смесь промывали водой (3×10 мл) и солевым раствором (1×10 мл) и сушили над сульфатом магния. Растворитель удаляли в вакууме, и сырой гидразид (113, 95 мг, 93,1%) использовали без дальнейшей очистки.
Стадия 3
К раствору 113 (95 мг, 0,27 ммоль) в тетрагидрофуране (8 мл) добавляли метилизоцианат (25 мг, 0,40 ммоль) при комнатной температуре, и перемешивали смесь в атмосфере аргона в течение 16 час. Затем к реакционной смеси добавляли метанол (10 мл), и отгоняли в вакууме летучие реагенты. Сырой продукт 114 использовали далее без очистки.
Стадия 4.
Из раствора 114 в метаноле (25 мл) вытесняли кислород пробулькиванием через раствор аргона в течение 20 мин. К раствору добавляли гидроксид калия (149 мг, 2,66 ммоль), и полученную реакционную смесь нагревали при кипении в течение 19 час. Реакционную смесь выливали в 20 мл 10% водного раствора бисульфата натрия, и затем сырой продукт экстрагировали этилацетатом (3×10 мл). Объединенные экстракты затем упаривали и очищали флэш-хроматографией на силикагеле (5% метанол в хлористом метилене), выделяя 93 в виде белого твердого вещества (89 мг, 77% на 4 стадии). МСНР (электрораспыление); m/z (МН)=396.
Пример 50
5-(3,5-Дихлорфенокси)-1,4-диэтил-3-(2Н-пиразол-3-илметил)-1Н-пиразол
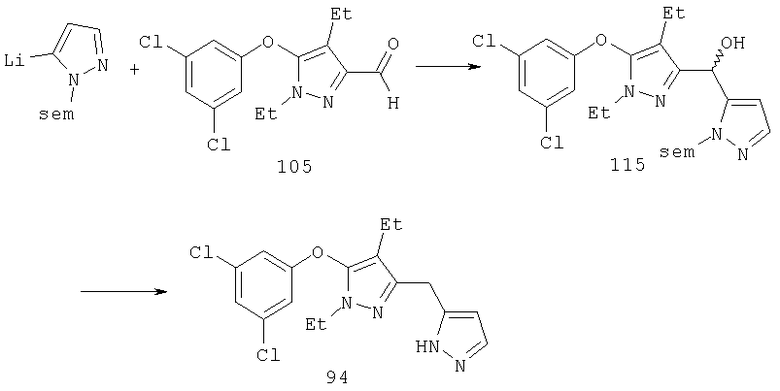
К раствору вышеуказанного sem-защищенного (sem=2-(триметилсилил)этоксиметил) пиразола (190 мг, 0,958 ммоль) в тетрагидрофуране (5 мл) при -78°С добавляли по каплям раствор н-бутиллития (1,6 М в гексане, 0,56 мл, 0,896 ммоль). Реакционную смесь перемешивали при -78°С в атмосфере аргона в течение 45 мин, после чего медленно добавляли раствор 105 в тетрагидрофуране (1 мл), и смесь перемешивали при -78°С в течение 2 час. Затем в реакционную смесь добавляли насыщенный раствор хлористого аммония (10 мл), и сырой карбинол 115 экстрагировали этилацетатом (3×10 мл). Объединенные экстракты упаривали и очищали флэш-хроматографией на силикагеле (4:1 гексан:этилацетат), выделяя 115 в виде бледно-желтого масла (112 мг, 68%).
Карбинол (115, 112 мг, 0,219 ммоль) смешивали с тетрайоддифосфином (124 мг, 0,219 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 30 мин, затем охлаждали до комнатной температуры и интенсивно перемешивали с 10% водным раствором бисульфита натрия (20 мл) до обесцвечивания органического слоя. Сырой продукт экстрагировали этилацетатом (3×10 мл), и растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией на силикагеле (5% метанол в хлористом метилене), выделяя 96 в виде бледно-желтого масла (68 мг, 85%).
Пример 51
3-Хлор-5-[2,4-диэтил-5-(2-гидроксиэтил)-2Н-пиразол-3-илокси]бензонитрил
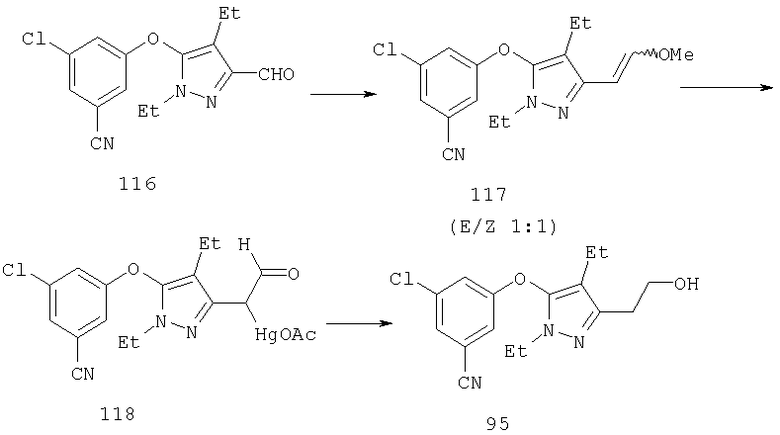
Стадия 1
К раствору хлористого (метоксиметил)трифенилфосфония (928 мг, 2,7 ммоль) в тетрагидрофуране (15 мл) добавляли бис-(триметилсилил)амид калия (0,5 М в толуоле, 5,4 мл, 2,7 ммоль) в течение 10 мин при -78°С. Полученную красную смесь перемешивали при -78°С в течение 20 мин, после чего медленно в течение 10 мин добавляли раствор альдегида (116: 82 мг, 0,27 ммоль) в тетрагидрофуране (1,5 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали 16 час, после чего добавляли уксусную кислоту (5 мл), и доводили смесь до рН 7 10% водным раствором бикарбоната натрия. Сырой продукт экстрагировали этилацетатом (3×20 мл), и растворитель удаляли в вакууме. После очистки сырого продукта флэш-хроматографией на силикагеле (4:1 гексан: этилацетат) получали смесь (1:1) эфиров енола 117 (74 мг, 83%).
Стадия 2
К раствору 117 (74 мг, 0,223 ммоль) в ацетонитриле (5 мл) и воде (5 мл) добавляли в один прием порошок ацетата ртути (II) (92 мг, 0,29 ммоль) при комнатной температуре. Реакция заканчивалась через 1,5 час. Ацетонитрил удаляли из реакционной среды в вакууме, получая водный раствор ртутного аддукта 118.
Стадия 3
Этанол (5 мл) добавляли к вышеуказанному водному раствору 118 с последующим добавлением при 0°С боргидрида натрия (34 мг, 0,90 ммоль). Мутную реакционную смесь перемешивали при 0°С в течение 1,5 час и затем выливали в 20 мл 10% водного раствора бисульфата натрия. Полученную смесь затем нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия. Сырой продукт экстрагировали этилацетатом (3×10 мл) и затем очищали флэш-хроматографией на силикагеле (4:1 гексан:этилацетат), выделяя 97 в виде бесцветного масла (60 мг, 84,3% на две стадии). МСНР (электрораспыление); m/z (МН)=319.
Пример 52
5-(3,5-Дихлорфенокси)-4-этил-1-изопропил-3-метоксиметил-1Н-пиразол
К раствору 11 (150 мг, 0,456 ммоль) в N,N-диметилформамиде (5 мл) при комнатной температуре добавляли гидрид натрия (60% дисперсия в минеральном масле, 22 мг, 0,547 ммоль). Реакционную смесь перемешивали до прекращения образования пузырьков. Затем добавляли метилйодид (97 мг, 0.684 ммоль), перемешивали реакционную смесь при комнатной температуре 20 мин и выливали в 20 мл 10% водного бисульфата натрия. Сырой продукт экстрагировали этилацетатом (3×10 мл), комбинированные органические вытяжки промывали водой (2×10 мл) и солевым раствором (1×10 мл), и удаляли растворитель в вакууме. Сырой продукт очищали флеш-хроматографией на силикагеле (5:1 гексан:этилацетат), получая чистый продукт 52 в виде бесцветного масла (110 мг, 70%). МСНР (электрораспыление); m/z (MH)=343.
Пример 53
Опыты с ВИЧ-ревертазой: определение величины IC50 ингибитора
Опыты с ВИЧ-1 ревертазой проводили на пластинах MultiScreen MADVNOB50 с 96 ячейками, используя очищенный рекомбинантный фермент и поли(гА)/олиго(dT)16 матричный праймер в общем объеме 50 мкл. Состав образца включал 50 мМ трис/HCl, 50 мМ NaCl, 1 мМ ЭДТА, 6 мМ MgCl2, 5 мкМ ди-ТТФ, 0,15 мкCi(дюйм3 ?) [3Н] d-ТТФ, предварительно полученный гибрид, состоящий из 5 мкг/мл поли(гА) и 2,5 мкг/мл олиго(dT)16, в интервале концентраций ингибитора в конечной концентрации ДМСО 10%. Реакции инициировали добавлением 4 нМ ВИЧ-1 ревертазы, выдерживали при 37°С в течение 30 мин, останавливали добавлением 50 мкл охлажденной во льду 20% трихлоруксусной кислоты (ТХУ) и оставляли выпадать осадок при 4°С в течение 30 мин. Осадки собирали, помещая пластины в вакуум и последовательно промывая 3×200 мкл 10% ТХУ и 2×200 мкл 70% этанолом. Затем пластины высушивали и измеряли радиоактивность на счетчике Packard TopCounter после добавления 25 мкл сцинциляционной жидкости в каждую ячейку. Значения IC50 рассчитывали, строя зависимость % ингибирования от log10 концентрации ингибитора. Отдельные значения IC50 включены в таблицу 2.
Метод проведения измерений по антивирусной активности
Антивирусную ВИЧ активность измеряли, применяя измененный метод Pauwels и др. (Pauwels и др., J.Virol. Methods, 20, 1988, cc.309-321). Этот метод основан на способности соединений защищать ВИЧ-инфицированные лимфобластоидные клетки (МТ4 клетки) от гибели, вызванной ВИЧ инфекцией. Результат опыта определяли по концентрации соединения, при которой наблюдалась 50% выживаемость культур («50% ингибиторная концентрация», IC50). Выживаемость клеток культуры определяли по расходу растворимой желтой соли - 3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолийбромиду (МТТ) - и ее восстановлению до красной нерастворимой соли формазана. После растворения последнего соединения использовали спектрофотометрический метод определения концентрации производного формазана.
МТ4 клетки готовили так, чтобы они возрастали в логарифмической зависимости до общего количества 2×106 клеток, инфицированных штаммом НХВ2 ВИЧ, при единичной дозе 0,0001 инфицированной единицы вируса на клетку и общем объеме между 200-500 микролитров. Клетки инкубировали с вирусом в течение одного часа при 37°С до удаления вируса. Затем клетки промывали 0,01 М фосфатным буфером с рН 7,2, ресуспензировали в среде культуры и инкубировали в культуре с набором разбавлений тестируемого соединения. В качестве культуральной среды использовали RPMI 1640 без фенола красного, с добавками пенициллина, стрептомицина, L-глутамина и 10% эмбриональной сыворотки крови теленка (GM10).
Тестируемые соединения готовили в виде 2 мМ растворов в диметилсульфоксиде (ДМСО). Затем готовили в четырех повторах серию двукратных разведений в GM10 и 50 мкл помещали в пластины с 96 ячейками до конечного интервала наномолярной концентрации в интервале 625-1,22. Затем в каждую ячейку добавляли пятьдесят микролитров GM10 и 3,5×104 инфицированных клеток. Также готовили контрольные культуры, не содержащие клеток («пустые»), содержащие неинфицированные клетки (100% выживаемость, 4 повтора), и инфицированные клетки без соединения (тотальная гибель клеток, вызванная вирусом, 4 повтора). Культуры затем инкубировали при 37°С во влажной атмосфере с 5% СО2 в воздухе в течение 5 дней.
Готовили свежий раствор 5 мг/мл МТТ в 0,01 М фосфатном буфере с рН 7,2 и добавляли по 20 микролитров к каждой культуре. Культуры затем инкубировали как и раньше в течение 2 час. Растворы затем перемешивали, наполняя и спуская пипетку, и добавляли 170 микролитров Тритона Х-100 в подкисленном изопропаноле (10% по объему Тритона Х-100 в смеси 1:250 концентрированной HCl в изопропаноле). После перемешивания и полного растворения образующегося формазана измеряли поглощение (OD) образцов культур при длинах волн 540 нм и 690 нм (показания при 690 нм использовали как контроль за артефактами между ячейками). Процент протекции для каждой обработанной культуры рассчитывали затем по следующему уравнению:

Значения IC50 получали из графика зависимости процента протекции от log10 концентрации лекарства.
В обоих случаях соединения формулы I имеют интервал активности IC50 от примерно 0,5 до примерно 10000 нМ или от 0,5 до примерно 5000 нМ с преобладанием соединений, имеющих интервал активности от примерно 0,5 до примерно 750 нМ, с большим преобладанием соединений с интервалом активности от 0,5 до 300 нМ и наибольшим преобладанием соединений с интервалом активности от 0,5 до 50 нМ.
Пример 54
Фармацевтические композиции
Ингредиенты смешивают и вводят в капсулы, содержащие каждая примерно 100 мг; одна капсула приблизительно соответствует общей дневной дозе.
Ингредиенты смешивают и гранулируют, используя растворители, подобные метанолу. Смесь затем высушивают и формируют в таблетки (содержащие примерно 20 мг активного соединения), используя подходящее таблетирующее оборудование.
Ингредиенты смешивают для получения суспензии для орального введения.
Активный ингредиент растворяют в порции воды для инъекции. Затем при перемешивании добавляют хлористый натрий для получения изотонического раствора. Раствор доводят до веса остатком воды, фильтруют через 0,2-микронную фильтрующую мембрану и расфасовывают в стерильных условиях.
Ингредиенты сплавляют вместе, перемешивают на паровой бане и выливают в формочки, содержащие 2,5 г общего веса смеси.
Все ингредиенты, за исключением воды, объединяют и нагревают при 60°С при перемешивании. Затем при интенсивном перемешивании и температуре 60°С добавляют соответствующее количество воды для получения эмульсии, а затем добавляют воду до получения 100 г.
Формула для назального спрея.
Несколько водных суспензий, содержащих примерно 0,025-0,5 процента активного соединения, готовят в виде спрея для назального применения. Формулы необязательно содержат неактивные компоненты, такие как, например, микрокристаллическая целлюлоза, натрий карбоксиметилцеллюлоза, декстроза и тому подобное. Хлористоводородная кислота может быть добавлена для создания подходящего рН. Формулы для назального спрея могут быть снабжены мерными пульверизаторами, обычно впрыскивающими примерно 50-100 микролитров смеси на прием. Типичное дозовое назначение составляет 2-4 впрыскивания каждые 4-12 час.
Характерные черты, выраженные выше в описании или в дальнейшей формуле изобретения или сопровождающие рисунки, представленные в специфических формах или терминах для представления изображаемой функции или метода или процесса для достижения желаемого результата, могут, отдельно или в комбинации с другими такими чертами, быть использованы для реализации предлагаемого изобретения в разнообразных формах.
Предлагаемое изобретение описано в деталях путем иллюстрации или примера для того, чтобы достигнуть полной ясности и понимания. Специалистам понятно, что могут быть внесены изменения и модификации внутри общей структуры изобретения. Понятно также, что приведенное выше описание предназначено быть иллюстративным, а не исчерпывающим. Общий состав изобретения следует определять не по ссылкам на приведенное выше описание, но по ссылкам на последующую прилагаемую формулу изобретения вместе с полным объемом изобретения, к которому формула изобретения относится.
Все патенты, патентные заявки и публикации, цитируемые в предложенном изобретении, таким образом, включены ссылками во всей их полноте для всех целей в такой степени, как если бы каждый индивидуальный патент, патентная заявка или публикация были индивидуально указаны.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЦИКЛИЧЕСКИЕ ДИОНЫ В КАЧЕСТВЕ ГЕРБИЦИДНЫХ СОЕДИНЕНИЙ | 2020 |
|
RU2822391C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ АГЕНТОВ | 2000 |
|
RU2242474C2 |
| СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ, ОПОСРЕДУЕМОГО ЦИТОКИНАМИ | 2000 |
|
RU2298008C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА | 2020 |
|
RU2836442C2 |
| НОВЫЕ ТРИАРИЛИМИДАЗОЛЫ | 2004 |
|
RU2372346C2 |
| ГЕРБИЦИДНЫЕ СОЕДИНЕНИЯ | 2020 |
|
RU2839723C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИКЛИЧЕСКИХ ДИКЕТОНОВ | 2003 |
|
RU2316544C2 |
| АЗАИНДОЛИЛПИРИДОНЫ И ДИАЗАИНДОЛИЛПИРИДОНЫ | 2018 |
|
RU2788659C2 |
| ГЕРБИЦИДНЫЕ ЦИКЛОГЕКСАНДИОНОВЫЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2830286C2 |
| ПИРИДИЛПИРИДОНЫ | 2018 |
|
RU2805334C2 |
Изобретение относится к новым производным пиразола формулы (I),
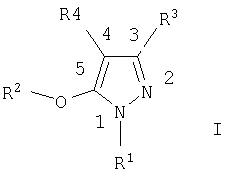
в котором R1 выбирают из ряда, содержащего C1-С6алкил, фенил и бензил; R2 означает фенил, необязательно замещенный одним - тремя заместителями, независимо выбранными из группы, содержащей галоид, циано; R3 означает замещенный C1-C6алкил, замещенный С1-С3алкокси-С1-С3алкил, необязательно замещенную С1-С6алкоксигруппу, группы -(CH2)nR5, -(СН2)О-О-(CH2)pR5, где алкил, C1-C3алкокси-C1-C3алкил замещены группами -ОН, -NR6R7, -C(=Y)Z, -X(C=Y)Z, CN или NR6SO2-C1-C6алкил; алкоксигруппа необязательно замещена группами -C(=Y)Z, -X(C=Y)Z; R5 означает фенил или гетероарильное кольцо в соответствии с формулами IIIa-IIIh,
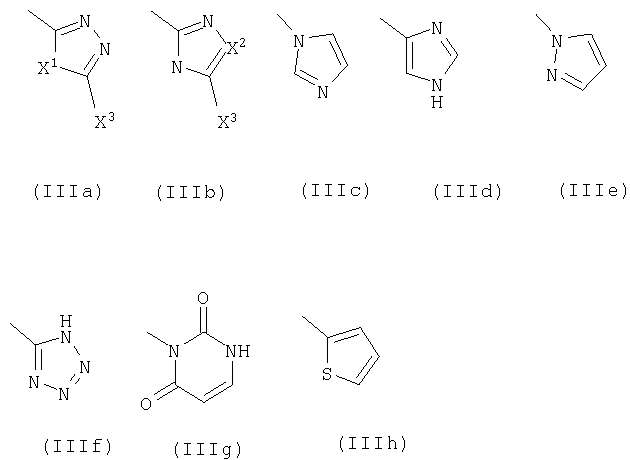
в которых X1, X2 означают -CHR6; X3 выбирают из ряда, содержащего водород, гидроксил; R4 означает C1-С6алкил, С2-С6алкенил, С1-С3алкокси-С1-С3алкил, -(CH2)nR11; где алкил, алкенил необязательно замещены группами: -ОН, -OR6, С1-С6алкил; R11 означает фенил или пиридинил; R6, R7 и R9 независимо выбраны из ряда, содержащего водород, C1-C6алкил, С1-С6гидроксиалкил, С1-С3алкокси-С1-С3алкил; X и Y независимо друг от друга означают О или NR6; Z означает водород, гидроксил, С1-С6алкокси, NR6R13, C1-С6алкил, С1-С3алкокси-С1-С3алкил, где R13 означает R7 или фенил; n означает от 0 до 3; о и р независимо означают от 0 до 4 и о+p≤5; q означает от 0 до 2; а также их кислотно-аддитивные соли, гидраты и сольваты; при условии, что в тех случаях, когда R4 означает (CH2)nR11, n равно 1, a R11 означает замещенный фенил, R2 не означает незамещенный фенил. Изобретение также относится к фармацевтической композиции и к применению соединений на основе формулы I. Технический результат - получение новых биологически активных соединений и фармацевтических композиций на их основе, предназначенных для ингибирования ВИЧ. 3 н. и 10 з.п. ф-лы, 2 табл.
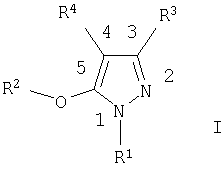
в котором R1 выбирают из ряда, содержащего С1-С6алкил, фенил и бензил;
R2 означает фенил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, содержащей галоид, циано;
R3 означает замещенный С1-С6алкил, замещенный С1-С3алкокси-С1-С3алкил, необязательно замещенную C1-С6алкоксигруппу, группы -(CH2)nR5, -(СН2)o-О-(CH2)pR5,
где алкил, С1-С3алкокси-С1-С3алкил замещены группами -ОН, -NR6R7, -C(=Y)Z, -X(C=Y)Z, CN или NR6SO2-C1-C6алкил; алкоксигруппа необязательно замещена группами -C(=Y)Z, -X(C=Y)Z;
R5 означает фенил или гетероарильное кольцо в соответствии с формулами IIIa-IIIh
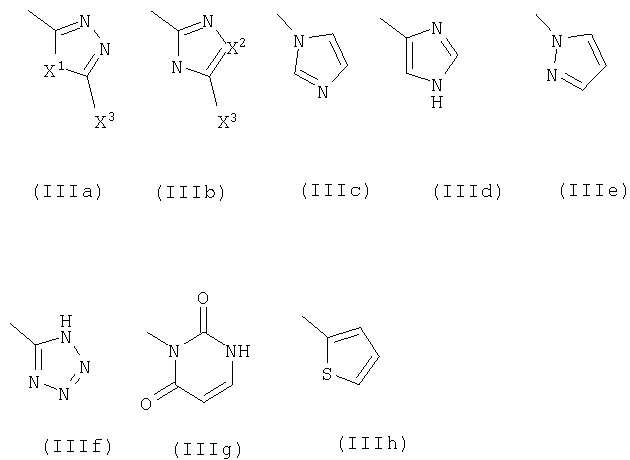
в которых Х1, Х2 означают -CHR6;
X3 выбирают из ряда, содержащего водород, гидроксил;
R4 означает С1-С6алкил, С2-С6алкенил, С1-С3алкокси-С1-С3алкил, -(CH2)nR11,
где алкил, алкенил, необязательно замещены группами: -ОН, -OR6, C1-С6алкил;
R11 означает фенил или пиридинил;
R6 и R7 независимо выбраны из ряда, содержащего водород, С1-С6алкил, С1-С6гидроксиалкил, С1-С3алкокси-С1-С3алкил;
X и Y независимо друг от друга означают О или NR6;
Z означает водород, гидроксил, С1-С6алкокси, NR6R13, С1-С6алкил, С1-С3алкокси-С1-С3алкил,
где R13 означает R7 или фенил;
n означает от 0 до 3;
о и р независимо означают от 0 до 4 и о+р≤5;
q означает от 0 до 2;
а также их кислотно-аддитивные соли, гидраты и сольваты;
при условии, что в тех случаях, когда R4 означает (CH2)nR11, n равно 1, а R11 означает замещенный фенил, R2 не означает незамещенный фенил.
R1 выбрано из ряда, содержащего С1-С6алкил и фенил;
R2 означает необязательно замещенный фенил;
R4 означает C1-С6алкил, -(CH2)nR11, где указанный алкил необязательно замещен группами -ОН, -OR6;
R11 означает фенил.
R1 выбрано из ряда, содержащего С1-С6алкил и фенил;
R2 означает необязательно замещенный фенил;
R4 означает С1-С6алкил, -(CH2)nR11, где алкил необязательно замещен группами -ОН, -OR6;
R11 означает пиридинил.
R1 выбирают из ряда, содержащего С1-С6алкил, фенил и бензил;
R2 означает фенил, необязательно замещенный одним-тремя заместителями, независимо выбранными из ряда, содержащего галоид и циано;
R3 означает замещенный С1-С6алкил, замещенный С1-С3алкокси-С1-С3алкил, необязательно замещенную C1-С6алкоксигруппу, группы -(CH2)nR5, -(CH2)o-O-(CH2)pR5,
в которых алкил, С1-С3алкокси-С1-С3алкил замещены группами -ОН, -NR6R7, -C(=Y)Z, -X(C=Y)Z, CN или NR6SO2-С1-С6алкил;
алкоксигруппа необязательно замещена группами -C(=Y)Z, -X(C=Y)Z;
R5 означает фенил или гетероарильное кольцо формул IIIa-IIIh
в которых Х1, Х2 означает -CHR6;
X3 выбирают из ряда, содержащего водород, гидроксил;
R4 означает С1-С6алкил, С2-С6алкенил, С1-С3алкокси-С1-С3алкил, -(CH2)nR11,
где алкил, алкенил необязательно замещены группами: -ОН, -OR6;
R11 означает фенил или пиридинил;
R6 и R7 независимо выбраны из ряда, содержащего водород, С1-С6алкил, C1-С6гидроксиалкил, С1-С3алкокси-С1-С3алкил;
X и Y независимо друг от друга означают О или NR6;
Z означает С1-С6алкокси, NR6R13, С1-С6алкил, С1-С3алкокси-С1-С3алкил,
где R13 означает R7 или фенил;
n означает от 0 до 3;
о и р независимо означают от 0 до 4 и о+р≤5;
q означает от 0 до 2,
а также их кислотно-аддитивные соли, гидраты и сольваты, при условии, что в тех случаях, когда R4 означает (CH2)nR11, n = 1, а R11 означает замещенный фенил, R2 не означает незамещенный фенил.
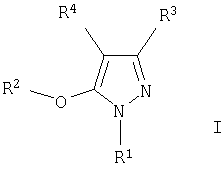
в которой R1 выбирают из ряда, содержащего C1-С6алкил, фенил и бензил;
R2 означает фенил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, содержащей галоид, циано;
R3 означает замещенный С1-С6алкил, замещенный С1-С3алкокси-С1-С3алкил, необязательно замещенную С1-С6алкоксигруппу, группы -(CH2)nR5, -(CH2)o-O-(CH2)pR5,
где алкил, С1-С3алкокси-С1-С3алкил замещены группами -ОН, -NR6R7, -C(=Y)Z, -X(C=Y)Z, CN или NR6SO2-C1-C6алкил; алкоксигруппа необязательно замещена группами -C(=Y)Z, -X(C=Y)Z;
R5 означает фенил или гетероарильное кольцо в соответствии с формулами IIIa-IIIh
в которых X1, X2 означают -CHR6;
X3 выбирают из ряда, содержащего водород, гидроксил;
R4 означает С1-С6алкил, С2-С6алкенил, С1-С3алкокси-С1-С3алкил, -(СН2)nR11, где алкил, алкенил необязательно замещены группами -ОН, -OR6, С1-С6алкил;
R11 означает фенил или пиридинил;
R6 и R7 независимо выбраны из ряда, содержащего водород, С1-С6алкил, С1-С6гидроксиалкил, С1-С3алкокси-С1-С3алкил;
X и Y независимо друг от друга означают О или NR6;
Z означает водород, гидроксил, С1-С6алкокси, NR6R13, С1-С6алкил, С1-С3алкокси-С1-С3алкил,
где R13 означает R7 или фенил;
n означает от 0 до 3;
о и р независимо означают от 0 до 4 и о+р≤5;
q означает от 0 до 2;
а также их кислотно-аддитивные соли, гидраты и сольваты;
при условии, что в тех случаях, когда R4 означает (CH2)nR11, n = 1, a R11 означает замещенный фенил, R2 не означает незамещенный фенил,
при приготовлении лекарственного средства для лечения или профилактики ВИЧ-инфекции или для лечения СПИДа или КЗСС.
в котором R1 выбирают из ряда, содержащего С1-С6алкил, фенил и бензил;
R2 означает фенил, необязательно замещенный одним-тремя заместителями, независимо выбранными из группы, содержащей галоид, циано;
R3 означает замещенный С1-С6алкил, замещенный С1-С3алкокси-С1-С3алкил, необязательно замещенную С1-С6алкоксигруппу, группы -(CH2)2R5, -(CH2)o-O-(CH2)pR5,
где алкил, С1-С3алкокси-С1-С3алкил замещены группами -ОН, -NR6R7, -C(=Y)Z, -X(C=Y)Z, CN или -NR5SO2-С1-С6алкил; алкоксигруппа необязательно замещена группами -C(=Y)Z, -X(C=Y)Z;
R5 означает фенил или гетероарильное кольцо в соответствии с формулами IIIa-IIIh
в которых X1, X2 означают -CHR6;
X3 выбирают из ряда, содержащего водород, гидроксил;
R4 означает С1-С6алкил, С2-С6алкенил, С1-С3алкокси-С1-С3алкил, -(CH2)nR11,
где алкил, алкенил необязательно замещены следующими группами: -ОН, -OR6, С1-С6алкил;
R11 означает фенил или пиридинил;
R6 и R7 независимо выбраны из ряда, содержащего водород, С1-С6алкил, С1-С6гидроксиалкил, С1-С3алкокси-С1-С3алкил;
X и Y независимо друг от друга означают О или NR6;
Z означает водород, гидроксил, С1-С6алкокси, NR6R13, С1-С6алкил, С1-С3алкокси-С1-С3алкил,
где R13 означает R7 или фенил;
n означает от 0 до 3;
о и р независимо означают от 0 до 4 и о+р≤5;
q означает от 0 до 2,
а также их кислотно-аддитивные соли, гидраты и сольваты,
при условии, что в тех случаях, когда R4 означает (CH2)nR11, n равно 1, a R11 означает замещенный фенил, R не означает незамещенный фенил, в смеси с по крайней мере одним фармацевтически приемлемым носителем или разбавителем, в приемлемой для введения единичной или дробной дозах.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| RU 99109971 А, 20.03.2001. | |||

