Область изобретения
Данное изобретение относится к новым фармацевтически полезным соединениям, в частности к соединениям, которые полезны при лечении сердечных аритмий.
Предпосылки и уровень техники
Сердечные аритмии можно определить как отклонения от нормы в скорости, регулярности или месте возникновения сердечного импульса либо как нарушения в проводимости, которые вызывают аномальную последовательность возбуждения. С точки зрения клиники аритмии можно классифицировать по предположительному месту их возникновения (то есть как наджелудочковые, включая предсердные и предсердножелудочковые аритмии, и желудочковые аритмии) и/или по частоте сокращений (то есть брадиаритмии (замедленные) и тахиаритмии (ускоренные)).
Отрицательный результат некоторых клинических испытаний (смотри, например, результат испытания по подавлению сердечной аритмии (Cardiac Arrhythmia Suppression Trial (CAST)), описанный в New England Journal of Medicine, 321, 406 (1989)) при лечении сердечных аритмий “традиционными” антиаритмическими лекарствами, действующими главным образом путем снижения скорости проводимости (антиаритмические лекарства класса I), побудил направить разработку лекарств на соединения, которые селективно замедляют сердечную реполяризацию, тем самым удлиняя QT-интервал. Антиаритмические лекарства класса III могут быть определены как лекарства, удлиняющие продолжительность транс-мембранного потенциала действия (что может быть вызвано блокированием направленных вовне токов К+ или увеличением направленных внутрь токов ионов) и рефракторный период, не влияя на сердечную проводимость.
Известно, что одним из ключевых недостатков известных до настоящего времени лекарств, которые действуют путем замедления реполяризации (класса III или других) является то, что все они проявляют уникальную форму проаритмии, известную как torsades de pointes (полиморфная желудочковая тахиаритмия), которая иногда может заканчиваться смертельным исходом. С точки зрения безопасности сведение к минимуму этого явления (для которого также было показано, что оно проявляется как результат введения несердечных лекарств, таких как фенотиазины, трициклические антидепрессанты, антигистамины и антибиотики) представляет собой ключевую требующую решения проблему при обеспечении мер предосторожности при приеме эффективных антиаритмических лекарств.
Антиаритмические лекарства на основе биспидинов (3,7-диазабицикло[3.3.1]нонанов) известны среди прочего из международной заявки на патент WO 91/07405, европейских заявок на патент 306871, 308843 и 655228 и патентов США 3962449, 4556662, 4550112, 4459301 и 5468858, а также из журнальных статей, включая среди прочего J. Med. Chem. 39, 2559 (1996), Pharmacol. Res., 24, 149 (1991), Circulation, 90, 2032 (1994) и Anal. Sci. 9, 429 (1993). Известные антиаритмические соединения на основе биспидина включают в себя бисарамил (bisaramil) (3-метил-7-этил-9α,4'-(Сl-бензоилокси)-3,7-диазабицикло[3.3.1]нонан, тедисамил (tedisamil) (3',7'-бис(циклопропилметил)спиро(циклопентан-1,9')-3,7-диазабицикло[3.3.1]нонан), SAZ-VII-22 (3-(4-хлорбензоил)-7-изопропил-3,7-диазабицикло[3.3.1]нонан), SAZ-VII-23 (3-бензоил-7-изопропил-3,7-диазабицикло[3.3.1]нонан), GLG-V-13 (3-[4-(1Н-имидазол-1-ил)бензоил]-7-изопропил-3,7-диазабицикло[3.3.1]нонан), КМС-IV-84 (7-[4'-(1H-имидазоло-1-ил)бензолсульфонил]-3-изопропил-3,7-диазабицикло[3.3.1]нонана дигидроперхлорат) и амбасилид (3-(4-аминобензоил)-7-бензил-3,7-диазабицикло[3.3.1]нонан).
Авторы изобретения неожиданно обнаружили, что новая группа соединений на основе биспидина проявляет электрофизиологическую активность, предпочтительно электрофизиологическую активность класса III, и вследствие этого ожидается, что они могут быть полезными при лечении сердечных аритмий.
Описание изобретения
Согласно настоящему изобретению предложены соединения формулы I
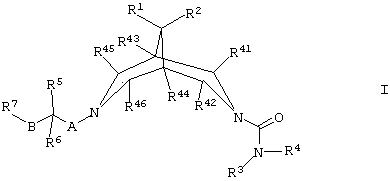
где R1 и R2 независимо представляют собой Н, С1-4алкил, OR2b или N(R2c)R2d, либо вместе образуют -O-(СН2)2-O-, -(СН2)3-, -(СН2)4- или -(СН2)5-;
R2b, R2c и R2d независимо представляют собой Н или C1-6алкил;
R3 представляет собой Н, С1-6-алкил или вместе с R4 представляет собой С3-6алкилен (причем алкиленовая группа возможно прервана атомом О и/или возможно замещена одной или более чем одной C1-3алкильной группой);
R4 представляет собой Н, С1-12алкил, C1-6алкокси (причем последние две группы обе возможно замещены и/или оканчиваются одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро, C1-4алкила и/или С1-4алкокси), -(СН2)qарил, -(CH2)q-оксиapил, -(CH2)q-Het1 (причем последние три группы возможно замещены (по-(CH2)q-части и/или арил/Неt1-части) одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро, -C(O)R10, -C(O)OR11, -N(H)S(O)2R11a, С1-6алкила и/или C1-6алкокси), -(CH2)qN(H)C(O)R8, -(CH2)qS(O)2R8, -(CH2)qC(O)R8, -(CH2)qC(O)OR8, -(CH2)qC(O)N(R9)R8 или вместе с R3 представляет собой С3-6алкилен (причем алкиленовая группа возможно прервана атомом О и/или возможно замещена одной или более чем одной C1-3алкильной группой);
q представляет собой 0, 1, 2, 3, 4, 5 или 6;
R8 представляет собой Н, С1-6алкил, арил (причем последняя группа возможно замещена и/или оканчивается одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро, -C(O)R10, -C(O)OR11, -N(H)S(O)2R11a, C1-6алкила и/или C1-6алкокси) или вместе с R9 представляет собой С3-7алкилен;
R9 представляет собой Н, С1-4алкил или вместе с R8 представляет собой С3-7алкилен;
Het1 представляет собой пяти-двенадцатичленное гетероциклическое кольцо, содержащее один или более чем один гетероатом, выбранный из кислорода, азота и/или серы, и которое также возможно включает в себя один или более чем один заместитель=O;
R41, R42, R43, R44, R45 или R46 независимо представляют собой Н или С1-3алкил;
R5 представляет собой Н, галогено, C1-3алкил, -OR12, -N(R13)R12 или вместе с R6 представляет собой=O;
R6 представляет собой Н, С1-4алкил или вместе с R5 представляет собой=O;
R12 представляет собой Н, С1-6алкил, -S(O)2-С1-4-алкил, -C(O)R14, -C(O)OR14, -C(O)N(R15)R15a или арил (причем последняя группа возможно замещена и/или оканчивается одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро, -C(O)R10, -C(O)OR11, -N(H)S(O)2R11a, C1-6алкила и/или C1-6алкокси);
R13 представляет собой Н или С1-4алкил;
R14 представляет собой Н или С1-6алкил;
R15 и R15a независимо представляют собой Н или С1-4алкил либо вместе представляют собой С3-6алкилен, возможно прерванный атомом О;
А представляет собой простую связь, С1-6алкилен, -N(R16)(CH2)r- или -O(СН2)r- (причем группа -(СН2)r- в последних двух группах присоединена к атому азота биспидина);
В представляет собой простую связь, С1-4алкилен, -(CH2)nN(R17)-, -(CH2)nS(O)p-, -(СН2)nО- (причем группа -(СН2)n- в последних трех группах присоединена к атому углерода, несущему R5 и R6), -C(O)N(R17)- (причем группа -С(О)- в последней группе присоединена к атому углерода, несущему R5 и R6), -N(R17)C(O)O(CH2)n-, -N(R17)(CH2)n- (причем группа N(R17) в последних двух группах присоединена к атому углерода, несущему R5 и R6) или -(СН2)mС(Н)(ОН)(СН2)n- (причем группа -(СH2)m- в последней группе присоединена к атому углерода, несущему R5 и R6);
m представляет собой 1, 2 или 3;
n и r независимо представляют собой 0, 1, 2, 3 или 4;
р представляет собой 0, 1 или 2;
R16 и R17 независимо представляют собой Н или С1-4алкил;
R7 представляет собой С1-6алкил, арил или Net2, каждая группа из которых возможно замещена и/или оканчивается (как подходит) одним или более чем одним заместителем, выбранным из -ОН, циано, галогено, амино, нитро, Het3, -C(O)R10, -C(O)OR11, С1-6алкила, С1-6алкокси, -N(H)S(O)2R18, -S(O)2R19, -OS(O)2R20, -N(H)C(O)N(H)R21, -C(O)N(H)R22 и/или арила (причем последняя группа возможно замещена одной или более чем одной цианогруппой);
Het2 и Het3 независимо представляют собой пяти-двенадцатичленную гетероциклическую группу, содержащую один или более чем один гетероатом, выбранный из кислорода, азота и/или серы, и которая также возможно включает в себя один или более чем один заместитель=O;
R18, R19 и R20 независимо представляют собой С1-6алкил;
R21 и R22 независимо представляют собой Н или C1-6алкил (возможно оканчивающийся циано); и
R10 и R11 независимо представляют собой, в каждом индивидуальном случае, Н или C1-6алкил;
R11a представляет собой, в каждом индивидуальном случае, С1-6алкил;
или их фармацевтически приемлемое производное;
при условии, что:
(а) когда А и В оба представляют собой простые связи, а R7 представляет собой возможно замещенный арил, тогда R5 и R6 оба не представляют собой Н;
(б) когда А представляет собой простую связь, тогда R5 и R6 вместе не представляют собой =O; и
(в) когда R5 представляет собой -OR12 или -N(R13)R12, тогда:
1) А не представляет собой -N(R16)(CH2)r- или -O(СН2)r; и/или
2) n не представляет собой 0, когда В представляет собой -(CH2)nN(R17)-, -(CH2)nS(O)p- или -(СН2)nО-, такие соединения называются здесь далее как “соединения по изобретению”.
Арильные группы, которые могут быть упомянуты, включают в себя С6-10арильные группы, такие как фенил, нафтил и им подобные. Оксиарильные группы, которые могут быть упомянуты, включают в себя С6-10оксиарильные группы, такие как оксифенил (фенокси), оксинафтил (нафтокси) и им подобные. Когда они замещены, то арильные и арилоксигруппы предпочтительно замещены заместителями в количестве от одного до трех.
Группы Het1, Het2 и Met3, которые могут быть упомянуты, включают в себя группы, содержащие от 1 до 4 гетероатомов (выбранных из группы кислорода, азота и/или серы), и в которых общее количество атомов в кольцевой системе составляет от пяти до двенадцати. Het-группы (Het1, Het2 и Het3) могут иметь полностью/частично ароматический характер и могут быть бициклическими.
Гетероциклические группы, которые могут быть упомянуты, включают в себя морфолинил, тиазолил, оксазолил, изоксазолил, циннолинил, хиназолинил, фталазинил, пуринил, бензимидазолил, пиримидинил, пиперазинил, пиразинил, пиперидинил, пиридинил, триазолил, имидазолил, хинолинил, изохинолинил, диоксанил, бенздиоксанил, бенздиоксолил, бенздиоксепанил, бензморфолинил, индолил, пиразолил, пирролил, бензтиофенил, тиофенил, хроманил, тиохроманил, бензофуранил, пиранил, тетрагидропиранил, тетрагидрофуранил, фуранил и им подобные. Значения Het1, которые могут быть упомянуты, включают в себя тетрагидропиранил, изоксазолил, бенздиоксолил, бенздиоксепанил и тиофенил. Значения Het2, которые могут быть упомянуты, включают в себя хинолинил, изохинолинил, бензморфолинил, бенздиоксанил, пиперазинил, индолил и пиразолил. Значения Het3, которые могут быть упомянуты, включают в себя имидазолил. Заместители на Het-группах (Het1, Het2 и Het3) могут, где подходит, быть локализованы на любом атоме в кольцевой системе, включая гетероатом. Точкой присоединения Het-групп (Het1, Het2 и Het3) может быть любой атом в кольцевой системе, включая (где подходит) гетероатом. Het-группы (Het1, Het2 и Het3) могут также быть в N- или S-окисленной форме.
Фармацевтически приемлемые производные включают в себя соли и сольваты. Соли, которые могут быть упомянуты, включают в себя соли присоединения кислот. Фармацевтически приемлемые производные также включают в себя, по азотам биспидина, С1-4алкильные четвертичные аммониевые соли и М-оксиды, при условии, что когда присутствует N-оксид, то:
(а) никакая Het-группа (Het1, Het2, Het3) не содержит неокисленный S-атом; и/или
(б) р не представляет собой 0, когда В представляет собой -(CH2)nS(O)p-.
Соединения по изобретению могут проявлять таутомерию. Все таутомерные формы и их смеси включены в объем данного изобретения.
Соединения по изобретению также могут содержать один или более чем один асимметричный атом углерода и поэтому могут проявлять оптическую и/или диастереоизомерию. Диастереоизомеры могут быть разделены с использованием традиционных методик, например хроматографии или фракционной кристаллизации. Различные стереоизомеры могут быть выделены путем разделения рацемической или иной смеси соединений с использованием традиционных методик, например фракционной кристаллизации или ВЭЖХ. Альтернативно, желаемые оптические изомеры могут быть получены путем взаимодействия соответствующих оптически активных исходных материалов в условиях, которые не будут вызывать рацемизацию или эпимеризацию, либо путем дериватизации, например с гомохиральной кислотой с последующим разделением диастереомерных сложных эфиров с помощью традиционных способов (например ВЭЖХ, хроматографии на диоксиде кремния). Все стереоизомеры включены в объем данного изобретения.
Алкильные группы, которыми могут быть представлены R1, R2, R2b, R2c, R2d, R3, R4, R5, R6, R7, R8, R9, R10, R11, R11a, R12, R13, R14, R15, R15a, R16, R17, R18, R19, R20, R21, R22, R41, R42, R43, R44, R45 и R46, которые может включать в себя R12 и которыми могут быть замещены R3, R4, R7, R8 и R12; и алкоксигруппы, которыми может быть представлен R4 и которыми могут быть замещены R4, R7, R8 и R12, могут быть линейными либо, когда имеется достаточное количество (то есть три) атомов углерода, могут быть разветвленными и/или циклическими. Кроме того, когда имеется достаточное количество (то есть четыре) атомов углерода, то такие алкильные и алкоксигруппы могут также быть частично циклическими/ациклическими. Кроме того, такие алкильные и алкоксигруппы могут быть насыщенными либо, когда имеется достаточное количество (то есть два) атомов углерода, могут быть ненасыщенными и/или прерванными кислородом.
Алкиленовые группы, которыми могут быть представлены R3 и R4, R8 и R9, R15 и R15a, А и В; и -(CH2)m-, -(CH2)n-, -(CH2)q- и -(СН2)r-цепи, которые могут включать в себя А, В и R4 (как подходит), могут быть линейными либо, когда имеется достаточное количество (то есть два) атомов углерода, могут быть разветвленными. Кроме того, такие алкиленовые группы и -(СН2)-содержащие цепи могут быть насыщенными либо, когда имеется достаточное количество (то есть два) атомов углерода, могут быть ненасыщенными и/или прерванными кислородом.
Галогеногруппы, которыми может быть представлен R5 и которыми могут быть замещены R4, R7, R8 и R12, включают в себя фторо, хлоро, бромо и иодо.
Во избежание сомнения каждая группа R10, R11 и R11a, идентифицированная здесь, является независимой от других групп R10, R11 и R11a, соответственно. Например, когда R4 и R7 оба представляют собой арил, замещенный -C(O)R10, данные два индивидуальных заместителя -C(O)R10 являются независимыми друг от друга и не обязательно являются идентичными (хотя такая возможность не исключена).
Сокращения приведены в виде списка в конце настоящего описания.
Согласно еще одному аспекту настоящего изобретения предложены соединения формулы I, как определено здесь ранее, но при дополнительных условиях, что:
(а) когда А представляет собой -N(R16)(CH2)r- или -O(СН2)r, тогда г не представляет собой 0 или 1; и
(б) когда R5 представляет собой -ОН или -N(R13)R12, тогда В не представляет собой -N(R17)C(O)O(CH2)n- или -N(R17)(CH2)m-.
Предпочтительные соединения по изобретению включают в себя такие соединения, где:
R1 представляет собой Н;
R2 представляет собой Н;
R3 представляет собой
- Н;
- С1-2алкил; или
- вместе с R4 представляет собой С4-5алкилен, возможно прерванный атомом О и/или возможно замещенный одной или более чем одной метильной группой;
R4 представляет собой
- Н;
- линейный либо разветвленный, и/или насыщенный либо ненасыщенный, и/или циклический, ациклический и/или частично циклический/ациклический C1-8алкил (где алкильная группа возможно замещена одной или более чем одной циано- либо галогеногруппой и/или прервана атомом О);
- C1-6алкокси;
- -(CH2)qS(O)2R8, -(CH2)qC(O)OR8, -(CH2)qN(H)C(O)R8, -(CH2)qC(O)R8 (где q в последних четырех группах представляет собой 0, 1 или 2, а R8 представляет собой линейный либо разветвленный и/илиациклический,циклическийи/иличастично циклический/ациклический С1-4алкил или фенил (причем фенильная группа возможно замещена одной или более чем одной циано- и/или C1-3алкильной группой);
- -(CH2)qC(O)N(R9)R8 (где q в последней группе представляет собой 0, 1 или 2, а R8 и R9 независимо представляют собой Н, линейный либо разветвленный и/или ациклический, циклический и/или частично циклический/ациклический С1-4лкил, или вместе представляют собой С4-6алкилен);
- -(СH2)q-фенил, -(СH2)q-оксифенил или -(CH2)q-Het1 (где q в последних трех группах представляет собой 0, 1, 2 или 3, -(CH2)q-часть возможно замещена цианогруппой, а фенильная или Net1-часть возможно замещена одним или более чем одним заместителем, выбранным из циано, нитро, линейного или разветвленного C1-4алкила, линейного или разветвленного С1-4алкокси и -N(H)S(O)2(R11a); или
- вместе с R3 представляет собой С4-5алкилен, возможно прерванный атомом О и/или возможно замещенный одной или более чем одной метильной группой;
R5 представляет собой Н;
- фторо;
- -OR12 (где R12 представляет собой Н, фенил (возможно замещенный одной или более чем одной метоксигруппой) или C(O)N(H)R15a (где R15a представляет собой линейный или разветвленный С1-4алкил));
- -N(R13)R12 (где R12 представляет собой Н, С1-2алкил, -S(O)2-C1-2алкил, -C(O)R14 (где R14 представляет собой С1-2алкил), -C(O)OR14 (где R14 представляет собой линейный или разветвленный С1-5алкил) или -С(О)N(R15)(R15a) (где R15 и R15a независимо представляют собой Н, либо линейный или разветвленный C1-3алкил, либо вместе представляют собой С4-5алкилен, причем алкиленовая группа возможно прервана атомом О), а R13 представляет собой Н или C1-2алкил); или
- вместе с R6 представляет собой=O (особенно в случае, когда R7 представляет собой алкил или Het2);
R6 представляет собой Н или С1-2алкил либо вместе с R5 представляет собой=O (особенно в случае, когда R7 представляет собой алкил или Het2);
А представляет собой простую связь, линейный или разветвленный C1-4алкилен (причем данная группа также возможно прервана атомом О), -N(H)(CH2)r- или -O(СН2)r- (где r в последних двух группах равно 1 или 2);
В представляет собой простую связь, С1-4алкилен, -(СН2)nО-, -(CH2)nS(O)2-, -(CH2)nN(H)- или -N(Н)(СН2)n- (где n в последних четырех случаях равно 0, 1, 2 или 3);
R7 представляет собой
- линейный либо разветвленный и/или ациклический, циклический и/или частично циклический/ациклический С1-6алкил (возможно замещенный и/или оканчивающийся ОН);
- Net2 (возможно замещенный одним или более чем одним заместителем, выбранным из циано, C1-3алкила, фенила (причем последняя группа возможно замещена одной или более чем одной цианогруппой),=O, -C(O)R10 (где R10 представляет собой линейный или разветвленный C1-3алкил) или -S(O)2R19 (где R19 представляет собой С1-2алкил)); или
- фенил (возможно замещенный одним или более чем одним заместителем, выбранным из циано, нитро, линейного или разветвленного C1-3алкила, линейного или разветвленного C1-3алкокси, фторо, хлоро, C(O)N(H)R22 (где R22 представляет собой линейный либо разветвленный и/или ациклический, циклический и/или частично циклический/ациклический С1-4алкил, причем данная алкильная группа возможно оканчивается циано), N(H)S(O)2R18 (где R18 представляет собой С1-2алкил) или Het3);
R41, R42, R43, R44, R45 и R46 все представляют собой Н.
Более предпочтительные соединения по изобретению включают в себя такие соединения, где:
R3 представляет собой Н;
R5 представляет собой Н, ОН или -N(H)С(О)(R15)(R15a);
R6 представляет собой Н;
А представляет собой -СН2- или -(СН2)2-;
В представляет собой простую связь, -СН2N(Н)- или -СН2О- (где, во избежание сомнения, -СН2-часть присоединена к атому углерода, несущему R5 и R6);
R7 представляет собой фенил (замещенный цианогруппой (предпочтительно по положению 4 относительно В) и одним или более чем одним возможным заместителем C(O)N(H)R22).
Предпочтительные соединения по изобретению включают в себя соединения из Примеров, описанных здесь далее.
Получение
Согласно данному изобретению также предложен способ получения соединений формулы I, при котором:
(а) для соединений формулы I, где R3 представляет собой Н, осуществляют взаимодействие соединения формулы II,
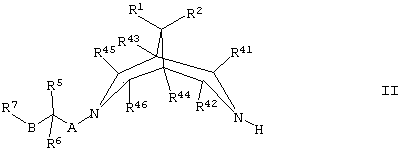
где R1, R2, R5, R6, R7, R41, R42, R43, R44, R45, R46, А и В такие, как определено здесь ранее, с соединением формулы III,
R4-N=C=O III
где R4 такой, как определено здесь ранее, например, при температуре между 0°С и температурой дефлегмации в присутствии соответствующего органического растворителя (например, дихлорметана) или посредством твердофазного синтеза в условиях, известных специалистам в данной области техники;
(б) осуществляют взаимодействие соединения формулы II, как оно определено здесь ранее, с производным карбоновой кислоты формулы IV,
(R3)(R4)NC(O)-L1 IV
где L1 представляет собой уходящую группу, такую как галогено, имидазол или R23O- (где R23 представляет собой, например, C1-10алкил, арил или C1-3алкиларил, причем данные группы возможно замещены одной или более чем одной галогено- или нитрогруппой), а R3 и R4 такие, как определено здесь ранее, например, при температуре между комнатной и температурой дефлегмации в присутствии подходящего основания (например, триэтиламина или карбоната калия) и соответствующего органического растворителя (например, дихлорметана, тетрагидрофурана (ТГФ), ацетонитрила, толуола или их смесей);
(в) осуществляют взаимодействие соединения формулы V,
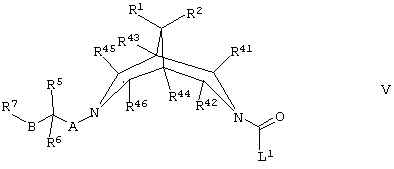
где R1, R2, R5, R6, R7, R41, R42, R43, R44, R45, R46, А, В и L1 такие, как определено здесь ранее, с соединением формулы VA,
(R3)(R4)NH VA
где R3 и R4 такие, как определено здесь ранее, например, при температуре между комнатной и температурой дефлегмации в присутствии подходящего основания (например, триэтиламина или карбоната калия) и соответствующего органического растворителя (например, дихлорметана, ТГФ, ацетонитрила, толуола или их смесей) или посредством твердофазного синтеза в условиях, известных специалистам в данной области техники;
(г) для соединений формулы I, где А представляет собой СН2, а R5 представляет собой -ОН или -N(H)R12, где R12 такой, как определено здесь ранее, осуществляют взаимодействие соединения формулы VI,
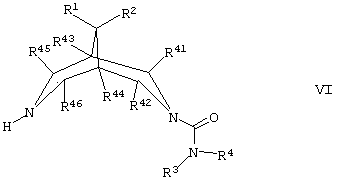
где R1, R2, R3, R4, R41, R42, R43, R44, R45 R46 такие, как определено здесь ранее, с соединением формулы VII,
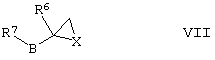
где Х представляет собой О или N(R12), a R6, R7, R12 и В такие, как определено здесь ранее, например, при повышенной температуре (например, от 60°С до температуры дефлегмации) в присутствии подходящего растворителя (например, низшего алкилового спирта (например, ИПС), ацетонитрила или смеси низшего алкилового спирта и воды);
(д) осуществляют взаимодействие соединения формулы VI, как оно определено здесь ранее, с соединением формулы VIII,
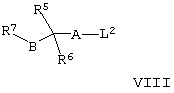
где L2 представляет собой уходящую группу (например, мезилат, тозилат или галогено), a R5, R6, R7, А и В такие, как определено здесь ранее, например, при повышенной температуре (например, между 35°С и температурой дефлегмации) в присутствии подходящего основания (например, триэтиламина или К2СО3) и соответствующего органического растворителя (например ацетонитрила или диметилсульфоксида (ДМСО));
(е) для соединений формулы I, где R5 представляет собой Н или ОН, а R6 представляет собой Н, восстанавливают соединение формулы IX,
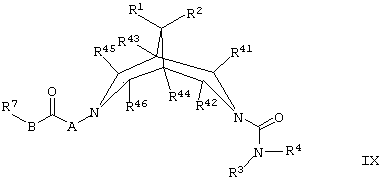
где R1, R2, R3, R4, R7, R41, R42, R43, R44, R45, R46, А и В такие, как определено здесь ранее, в присутствии подходящего восстанавливающего агента и в соответствующих реакционных условиях; например, для образования соединений формулы I, где R5 представляет собой ОН, восстановление может быть осуществлено в мягких реакционных условиях в присутствии, например, боргидрида натрия и соответствующего органического растворителя (например, ТГФ); а для образования соединений формулы I, где R5 представляет собой Н, восстановление может быть осуществлено путем активирования релевантной группы С=O с использованием соответствующего агента (такого как тозилгидразин) в присутствии подходящего восстанавливающего агента (например, боргидрида натрия или цианборгидрида натрия) и соответствующего органического растворителя (например, низшего (например, C1-6) алкилового спирта);
(ж) для соединений формулы I, где R1 и R2 оба представляют собой Н, восстанавливают соответствующее соединение формулы X,
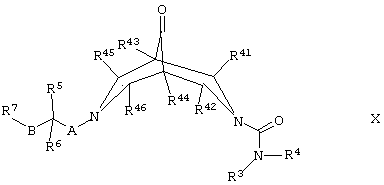
где R3, R4, R5, R6, R7, R41, R42, R43, R44, R45, R46, А и В такие, как определено здесь ранее, и где выступающая мостиковая группа С=O может быть активирована с использованием соответствующего агента, такого как тозилгидразин, в присутствии подходящего восстанавливающего агента (например, боргидрида натрия, цианборгидрида натрия) и соответствующего органического растворителя (например, низшего алкилового спирта) или в стандартных условиях Вольфа-Кишнера (Wolff-Kischner), известных специалистам в данной области техники; когда группу С=O активируют, стадия активации может быть осуществлена при температуре между комнатной и температурой дефлегмации в присутствии соответствующего органического растворителя (например, низшего алкилового спирта, такого как метанол, этанол или ИПС), после чего к реакционной смеси может быть добавлен восстанавливающий агент и восстановление осуществлено при температуре между 60°С и температурой дефлегмации, преимущественно в присутствии подходящей органической кислоты (например, уксусной кислоты);
(з) для соединений формулы I, где R1 и R2 вместе представляют собой -O(СН2)2O-, осуществляют взаимодействие соответствующего соединения формулы X, как оно определено здесь ранее, с этан-1,2-диолом в соответствующих условиях реакции, например, путем кипячения с обратным холодильником в присутствии п-TSA и соответствующего органического растворителя (например, толуола);
(и) для соединений формулы I, где В представляет собой -(CH2)nO-, осуществляют взаимодействие соединения формулы XI,
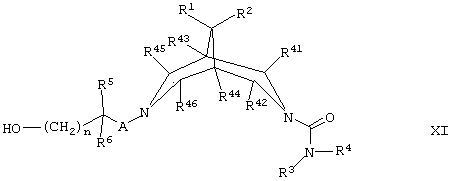
где R1, R2, R3, R4, R5, R6, R41, R42, R43, R44, R45, R46, А и n такие, как определено здесь ранее, с соединением формулы XIA,
R7OH XIA
где R7 такой, как определено здесь ранее, например, в условиях типа условий Мицунобу (Mitsunobu), например при температуре между температурой окружающей среды (например, 25°С) и температурой дефлегмации в присутствии третичного фосфина (например, трибутилфосфина или трифенилфосфина), азодикарбоксилатного производного (например, диэтилазодикарбоксилата или 1,1'-(азодикарбонил)дипиперидина) и соответствующего органического растворителя (например, дихлорметана или толуола);
(к) для соединений формулы I, которые являются N-оксидными производными по азоту биспидина, окисляют соответствующий азот биспидина соответствующего соединения формулы I в присутствии подходящего окисляющего агента (например, м-СРВА), например, при 0°С в присутствии подходящего органического растворителя (например, дихлорметана (ДХМ));
(л) для соединений формулы I, которые являются производными C1-4алкильными четвертичными аммониевыми солями, где алкильная группа присоединена к азоту биспидина, осуществляют взаимодействие по азоту биспидина соответствующего соединения формулы I с соединением формулы XII,
RbL3 XII
где Rb представляет собой С1-4алкил, a L3 является уходящей группой, такой как галогено, алкансульфонат или арилсульфонат, например, при комнатной температуре в присутствии соответствующего органического растворителя (например, ДМФ) с последующей очисткой (с использованием, например, ВЭЖХ) в присутствии подходящего донора противоиона (например, NH4OAc);
(м) для соединений формулы I, где R5 и R6 представляют собой Н, А представляет собой С1-6алкилен, а В представляет собой -N(R17)(CH2)n-, осуществляют взаимодействие соединения формулы XIII,
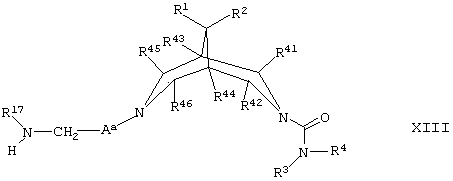
где Аa представляет собой С1-6алкилен, a R1, R2, R3, R4, R41, R42, R43, R44, R45, R46 и R17 такие, как определено здесь ранее, с соединением формулы XIV,
R7-(CH2)n-L2 XIV
где R7, n и L2 такие, как определено здесь ранее, например, при 40°С в присутствии подходящего органического растворителя (например, ацетонитрила);
(н) для соединений формулы I, где R5 представляет собой -NH2, восстанавливают соответствующее соединение формулы XV,
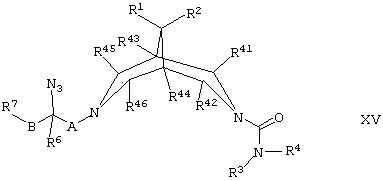
где R1, R2, R3, R4, R6, R7, R41, R42, R43, R44, R45, R46, А и В такие, как определено здесь ранее, например, путем гидрирования при соответствующем давлении в присутствии подходящего катализатора (например, палладия на угле) и соответствующего растворителя (например, водно-этанольной смеси);
(о) для соединений формулы I, где R5 представляет собой -N(R13)C(O)NH(R15), осуществляют взаимодействие соответствующего соединения формулы I, где R5 представляет собой -N(R13)H, с соединением формулы XVI,
R15N=C=O XVI
где R15 такой, как определено здесь ранее, например, при температуре окружающей среды (например, 25°С) в присутствии подходящего растворителя (например, бензола);
(п) для соединений формулы I, где R5 представляет собой -N(R13)C(O)R14, осуществляют взаимодействие соответствующего соединения формулы I, где R5 представляет собой -N(R13)H, с соединением формулы XVII,
R14C(O)RX XVII,
где RX представляет собой подходящую уходящую группу, такую как С1-4алкокси, галогено (например, Cl, Вr) или пара-нитрофенил, а R14 такой, как определено здесь ранее, например, при температуре между температурой окружающей среды и температурой дефлегмации в присутствии подходящего растворителя (например, дихлорметана или ацетонитрила) и возможно в присутствии подходящего основания (например, триэтиламина или карбоната калия);
(р) для соединений формулы I, где R5 представляет собой -N(H)R12, где R12 такой, как определено ранее, при условии, что он не представляет собой Н, осуществляют взаимодействие соответствующего соединения формулы I, где R5 представляет собой -NH2, с соединением формулы XVIII,
R12aL1 XVIII
где R12a представляет собой R12, как он определен здесь ранее, при условии, что он не представляет собой Н, a L1 такой, как определено здесь ранее, например, в условиях, известных специалистам в данной области техники;
(с) для соединений формулы I, где R5 представляет собой -OR12, где R12 представляет собой C1-6алкил или возможно замещенный арил, осуществляют взаимодействие соответствующего соединения формулы I, где R5 представляет собой -ОН, с соединением формулы XIX,
R12aOH XIX
где R12a представляет собой C1-6алкил или возможно замещенный арил, например, при температуре между температурой окружающей среды (например, 25°С) и температурой дефлегмации в условиях типа условий Мицунобу (то есть в присутствии, например, трифенилфосфина, азодикарбоксилатного производного (например, 1,1'-(азодикарбонил)дипиперидина) и подходящего органического растворителя (например, дихлорметана));
(т) для соединений формулы I, где R5 представляет собой -OR12, где R12 представляет собой C1-6алкил или возможно замещенный арил, осуществляют взаимодействие соединения формулы XX,
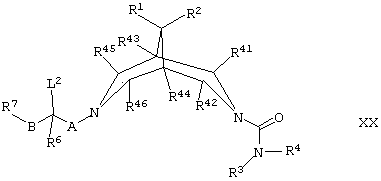
где L2, R1, R2, R3, R4, R6, R7, R41, R42, R43, R44, R45, R46, А и В такие, как определено здесь ранее, с соединением формулы XIX, как оно определено здесь ранее, например, при температуре между температурой окружающей среды (например, 25°С) и температурой дефлегмации в условиях типа условий Вильямсона (Williamson) (то есть в присутствии подходящего основания (например, КОН или NaH) и подходящего органического растворителя (например, диметилсульфоксида или ДМФ));
(у) для соединений формулы I, где R5 представляет собой OR12, и R12 представляет собой C(O)R14, a R14 такой, как он определен здесь ранее, осуществляют взаимодействие соответствующего соединения формулы I, как оно определено здесь ранее, где R5 представляет собой ОН, с соединением формулы XXI,
R14CO2H XXI
где R14 такой, как он определен здесь ранее, например, при температуре окружающей среды (например, 25°С) в присутствии подходящего агента сочетания (например, 1-(3-диметиламинопропил)-3-этилкарбодиимида), соответствующего катализатора (например, 4-диметиламинопиридина) и реакционно-инертного органического растворителя (например, ТГФ);
(ф) для соединений формулы I, где R5 представляет собой галогено, осуществляют замещение соответствующего соединения формулы I, где R5 представляет собой -ОН, с использованием подходящего галогенирующего агента (например, для соединений, где R5 представляет собой фторо, взаимодействие с диэтиламиносульфотрифторидом);
(х) для соединений формулы I, где R3 и/или R4, как подходит, представляют собой алкильные группы (например, C1-6 или С1-12алкил, как подходит), алкилируют соответствующее соединение формулы I, где R3 и/или R4 (как подходит) представляют собой Н, в условиях, хорошо известных специалистам в данной области техники;
(ц) превращают одну группу R4 в другую (например, превращение -(CH2)qC(O)OR8 в -(CH2)qC(O)N(R9)R8, где R8, R9 и q такие, как определено здесь ранее) с использованием методик, хорошо известных специалистам в данной области техники; или
(ч) для соединений формулы I, где один из R1 и R2 представляет собой Н, а другой представляет собой -ОН, восстанавливают соответствующее соединение формулы X, как оно определено здесь ранее, в присутствии мягкого восстанавливающего агента, например, боргидрида натрия, и соответствующего органического растворителя (например, низшего спирта, такого как метанол или этанол);
(ш) для соединений формулы I, где один из R2 и R3 представляет собой -NH2, а другой представляет собой Н, восстанавливают соединение формулы XXIA,
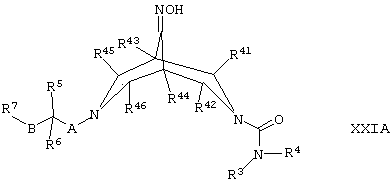
где R3, R4, R5, R6, R7, R41, R42, R43, R44, R45, R46, А и В такие, как определено здесь ранее, в присутствии подходящего восстанавливающего агента (например, LiAIH4), например, в условиях, хорошо известных специалистам в данной области техники;
(щ) для соединений формулы I, где один или оба из R1 и R2 представляют собой -N(R2c)R2d, где один или оба из R2c и R2d представляют собой C1-6алкил, алкилируют соответствующее соединение формулы I, где R1 и/или R2 представляют собой -N(R2c)R2d (как подходит), где R2c и/или R2d (как подходит) представляют собой Н, с использованием соединения формулы XXIВ.
R2eL1 XXIB
где R2e представляет собой C1-6алкил, a L1 такой, как определено здесь ранее, например, в условиях, хорошо известных специалистам в данной области техники; или
(э) превращают один заместитель на R7 в другой с использованием методик, хорошо известных специалистам в данной области техники.
Соединения формулы II могут быть получены путем осуществления взаимодействия соединения формулы XXII,
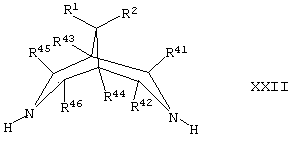
где R1, R2, R41, R42, R43, R44, R45 и R46 такие, как определено здесь ранее, с соединением формулы VIII, как определено здесь ранее, например, как описано здесь ранее для синтеза соединений формулы I (стадия (д) способа получения) или, в случае соединений формулы II, где А представляет собой CH2, и R5 представляет собой ОН или N(H)R12, с соединением формулы VII, как определено здесь ранее, например как описано здесь ранее для синтеза соединений формулы 1 (стадия (г) способа получения).
Соединения формулы II, где R1 и R2 оба представляют собой Н, могут быть получены путем восстановления соединения формулы XXIII,
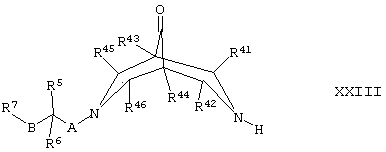
где R5, R6, R7, R41, R42, R43, R44, R45, R46, А и В такие, как определено здесь ранее, и где группа С=O может быть активирована с использованием соответствующего агента, такого как тозилгидразин, например, как описано здесь ранее для синтеза соединений формулы I (стадия (ж) способа получения).
Соединения формулы IV могут быть получены путем осуществления взаимодействия соединения формулы VA, как определено здесь ранее, с соединением формулы XXIV,
L1-C(O)-L1 XXIV
где L1 такая, как определено здесь ранее, и где две группы L1 могут быть одинаковыми или разными, например, при температуре между 0°С и температурой дефлегмации в присутствии подходящего основания (например, триэтиламина или карбоната калия) и соответствующего органического растворителя (например, толуола или дихлорметана).
Соединения формулы V могут быть получены путем осуществления взаимодействия соединения формулы II, как определено здесь ранее, с соединением формулы XXIV, как определено здесь ранее, например, как описано здесь ранее для синтеза соединений формулы IV.
Соединения формулы VI могут быть получены путем осуществления взаимодействия соединения формулы XXII, как определено здесь ранее, с соединением формулы III, как определено здесь ранее, например, как описано здесь ранее для синтеза соединений формулы I (стадия (а) способа получения), или с соединением формулы IV, как определено здесь ранее, например, как описано здесь ранее для синтеза соединений формулы I (стадия (б) способа получения).
Соединения формулы VI могут альтернативно быть получены путем осуществления взаимодействия соединения формулы XXII, как определено здесь ранее, с соединением формулы XXIV, как определено здесь ранее, например, как описано здесь ранее для синтеза соединений формулы IV, с последующим взаимодействием полученного промежуточного соединения с соединением формулы VA, как определено здесь ранее, например, как описано здесь ранее для синтеза соединений формулы I (стадия (в) способа получения).
Соединения формулы VI, где R1 и R2 представляют собой Н, могут альтернативно быть получены путем восстановления соответствующего соединения формулы XXV,
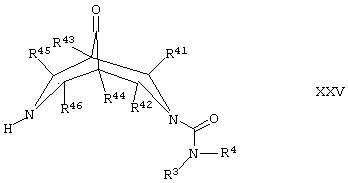
где R3, R4, R41, R42, R43, R44, R45 и R46 такие, как определено здесь ранее, и где группа С=O может быть активирована с использованием соответствующего агента, такого как тозилгидразин, например, как описано здесь ранее для соединений формулы I (стадия (ж) способа получения).
Соединения формулы VI, где один или более чем один из R41, R42, R45 и/или R46 представляет собой С1-3алкил, могут быть получены путем осуществления взаимодействия соединения формулы VI, где R41, R42, R45 и/или R46 (как подходит) представляют собой Н, с соответствующим алкилирующим агентом (например, диметилсульфатом), например, в присутствии подходящего сильного основания (например, втор-BuLi), N,N,N',N'-тетраметилэтилендиамина и реакционно-инертного растворителя (например, ТГФ).
Соединения формулы VII могут быть получены в соответствии с методиками, которые хорошо известны специалистам в данной области техники. Например, соединения формулы VII, где:
(1) В представляет собой -СН2O-, а Х представляет собой О, могут быть получены путем осуществления взаимодействия соединения формулы XIA, как определено здесь ранее, с соединением формулы XXVI,

где R6 и L2 такие, как определено здесь ранее, например, при повышенной температуре (например, между 60°С и температурой дефлегмации) в присутствии подходящего основания (например, К2СО3 или NaOH) и соответствующего органического растворителя (например, ацетонитрила или смеси толуол/вода), либо как иным образом описано в предшествующем уровне техники;
(2) R6 представляет собой Н, а Х представляет собой О, могут быть получены путем восстановления соединения формулы XXVII,
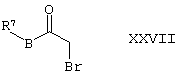
где R7 и В такие, как определено здесь ранее, например, при температуре между -15°С и комнатной температурой в присутствии подходящего восстанавливающего агента (например, NaBH4) и соответствующего органического растворителя (например, ТГФ) с осуществлением последующей реакции внутреннего замещения в полученном промежуточном соединении, например, при комнатной температуре в присутствии подходящего основания (например, К2СО3) и соответствующего органического растворителя (например, ацетонитрила);
(3) В представляет собой С1-4алкилен, -(CH2)nN(R17)-, -(CH2)nS(O)2- или -(СН2)nО- (где n в последних трех группах представляет собой 1, 2, 3 или 4) либо -(CH2)mC(H)(OH)(CH2)n-, a X представляет собой O, могут быть получены путем окисления соединения формулы XXVIII,
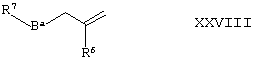
где Вa представляет собой простую связь, С1-3алкилен, -(CH2)n-1N(R17)-, -(CH2)n-1S(O)2- или -(CH2)n-1O- (где n в последних трех группах представляет собой 1, 2, 3 или 4) либо -(CH2)m-1C(H)(OH)(CH2)n- (где n в последней группе является таким, как определено здесь ранее) и во всех случаях R17 и m такие, как определено здесь ранее, в присутствии подходящего окисляющего агента (например, м-СРВА), например, путем кипячения с обратным холодильником в присутствии подходящего органического растворителя (например, ДХМ); или
(4) В представляет собой -(СН2)nО-, и Х представляет собой N(R12), a R12 представляет собой -S(O)2-С1-4алкил или -C(O)OR14, могут быть получены путем циклизации соединения формулы XXVIIIА,
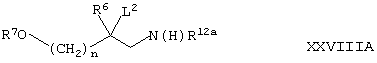
где R12a представляет собой -S(O)2-С1-4алкил или -C(O)OR14, a n, R6, R7, R14 и L2 такие, как определено здесь ранее, например, при температуре между 0°С и температурой дефлегмации в присутствии подходящего основания (например, гидроксида натрия), соответствующего растворителя (например, дихлорметана, воды или их смеси) и, при необходимости, катализатора фазового переноса (такого, как тетрабутиламмонийгидросульфат).
Соединения формулы VIII могут быть получены с помощью стандартных методик. Например, соединения формулы VIII, где:
(1) В представляет собой -(СН2)nО-, могут быть получены путем сочетания соединения формулы XIA, как определено здесь ранее, с соединением формулы XXIX,
L4-(CH2)n-C(R5)(R6)-A-L2 XXIX
где L4 представляет собой подходящую уходящую группу (например, галогено), a n, R5, R6, А и L2 такие, как определено здесь ранее; или
(2) В представляет собой -C(O)N(R17)-, могут быть получены путем сочетания соединения формулы XXX,
R7N(H)R17 XXX
где R7 и R17 такие, как определено здесь ранее, с соединением формулы XXXI,
L4-C(O)-C(R5)(R6)-A-L2 XXXI
где L4, R5, R6, А и L2 такие, как определено здесь ранее;
в обоих случаях в условиях, которые хорошо известны специалистам в данной области техники.
Соединения формулы VIII, где А представляет собой С2-алкилен, а R5 представляет собой OR12, где R12 представляет собой C1-6алкил или возможно замещенный арил, могут альтернативно быть получены путем осуществления взаимодействия соединения формулы XIX, как определено здесь ранее, с соединением формулы ХХХIА,
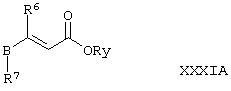
где Ry представляет собой С1-4алкил или арил (причем данные две группы возможно замещены одним или более чем одним заместителем, выбранным из С1-4алкила или галогено), a R6, R7 и В такие, как определено здесь ранее, например, между температурой окружающей среды (например, 25°С) и температурой дефлегмации в присутствии подходящего основания (например, К2СО3) и соответствующего органического растворителя (например, ацетонитрила) с последующим превращением эфирной функциональной группы в группу L2 (где L2 такая, как определено здесь ранее) в условиях, которые хорошо известны специалистам в данной области техники.
Соединения формул VII и VIII, где В представляет собой -(CH2)nS(O)- или -(CH2)nS(O)2-, могут быть получены путем окисления соответствующих соединений формул VII и VIII, где В представляет собой -(CH2)nS-, где n такой, как определено здесь ранее, в присутствии соответствующего количества подходящего окисляющего агента (например, м-СРВА) и соответствующего органического растворителя.
Соединения формул IX и XI могут быть получены способом, аналогичным для соединений формулы I (смотри, например, стадии (а), (б), (в) или (г) способа получения).
Альтернативно, соединения формулы IX, где А представляет собой C2-алкилен, могут быть получены путем осуществления взаимодействия соединения формулы VI, как определено здесь ранее, с соединением формулы XXXII,
R7-B-C(O)-CH=CH2 XXXII
где В и R7 такие, как определено здесь ранее, например, при комнатной температуре в присутствии подходящего органического растворителя (например, этанола).
Соединения формулы XIII могут быть получены путем удаления возможно замещенной бензилоксикарбонильной единицы (то есть снятия защиты) из соответствующего соединения формулы I, где R7 представляет собой возможно замещенный фенил, R5 и R6 оба представляют собой Н, В представляет собой -N(R17)С(O)O(СН2)-, А представляет собой Аa, и Аa такой, как определено здесь ранее, в условиях, которые хорошо известны специалистам в данной области техники.
Соединения формулы XV могут быть получены путем осуществления взаимодействия соответствующего соединения формулы I, как определено здесь ранее, где R5 представляет собой -ОН, с соединением формулы XXXIII,
RyS(O)2Cl XXXIII
где Ry такой, как определено здесь ранее, например, при температуре между -10°С и 25°С в присутствии подходящего растворителя (например, дихлорметана) с последующим взаимодействием с подходящим источником азид-иона (например, азидом натрия), например при температуре между температурой окружающей среды и температурой дефлегмации в присутствии соответствующего растворителя (например, ДМФ) и подходящего основания (например, NаНСО3).
Соединения формулы XV могут альтернативно быть получены путем осуществления взаимодействия соответствующего соединения формулы VI, как оно определено здесь ранее, с соединением формулы XXXIIIА,
R7-B-C(R6)(N3)-A-L2 XXXIIIA
где L2, R6, R7, А и В такие, как определено здесь ранее, например, в условиях, аналогичных тем, которые описаны здесь ранее для получения соединений формулы I (стадия (д) способа получения).
Соединения формулы XX могут быть получены путем замещения ОН-группы соединения формулы I, где R5 представляет собой ОН, L2-группой в условиях, которые хорошо известны специалистам в данной области техники.
Соединения формулы XXIA могут быть получены путем осуществления взаимодействия соответствующего соединения формулы Х с гидроксиламином, например, при повышенной температуре (например, при температуре дефлегмации) в присутствии подходящего органического растворителя (например, метанола).
Соединения формулы XXII хорошо известны по литературным данным либо легко доступны благодаря применению известных методик. Например, соединения формулы XXII, где R1 и R2 вместе представляют собой -O-(СН2)2-O-, -(СН2)3-, -(CH2)4- или -(СН2)5-, a R41, R42, R43, R44, R45 и R46 все представляют собой Н, могут быть получены путем восстановления соединения формулы XXXIV,
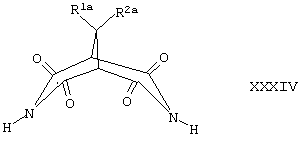
где R1a и R2a вместе представляют собой -O-(СН2)2-O-, -(СН2)3-, -(СН2)4- или -(CH2)5-, в присутствии подходящего восстанавливающего агента (например, LiAIH4) в условиях, которые хорошо известны специалистам в данной области техники.
Соединения формулы ХХХIIIА могут быть получены способом, аналогичным для соединений формулы XV (то есть из соответствующего спирта).
Соединения формул X, XXIII и XXV (где во всех случаях R45 и R46 оба представляют собой Н) могут быть получены преимущественно путем осуществления взаимодействия либо (как подходит) (1) соединения формулы XXXV,
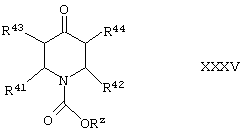
где Rz представляет собой C1-10алкил или С1-3алкиларил (например, алкилфенил, такой как бензил), a R41, R42, R43 и R44 такие, как определено здесь ранее, либо (2) 4-пиперидона (или его защищенного производного), или (как подходит) (1) с соединением формулы XXXVI,
R7-B-C(R5)(R6)-A-NH2 XXXVI
где R5, R6, R7, А и В такие, как определено здесь ранее, или (2) с NН3 (либо его защищенным (например, бензилом) производным), во всех случаях в присутствии формальдегида (то есть соответствующего источника формальдегида, такого как параформальдегид или раствор формалина), и, в случае соединений формул Х и XXV, путем превращения группы С(O)ORz в полученном промежуточном соединении в группу C(O)N(R3)(R4) с использованием методик, таких как описанные здесь методики (например, стадия (в) способа получения, указанная выше).
Образование соединений формул X, XXIII и XXV таким способом может быть осуществлено, например, при температуре между комнатной температурой и температурой дефлегмации (в зависимости от концентрации реагирующих веществ) в присутствии соответствующего растворителя (например, этанола или метанола) и, предпочтительно, в присутствии органической кислоты (например, С1-6-карбоновой кислоты, особенно уксусной кислоты).
Специалистам в данной области техники будет также ясно, что соединения формулы XXII, где R1 и R2 оба представляют собой Н, могут быть также получены посредством этого способа (то есть путем осуществления взаимодействия соединения 4-пиперидона (или его защищенного производного) с NН3 (или его защищенным производным) в присутствии формальдегида) при условии, что образованное таким образом промежуточное соединение, затем восстанавливают в соответствующих реакционных условиях.
Специалистам также будет ясно, что этот способ также может быть использован для получения соединений формулы I, где R41 и R42 представляют собой Н, а R45 и/или R46, отличные от Н, например, путем:
1) осуществления взаимодействия соединения формулы XXXV, где R41 и/или R42, отличный/отличные от Н, например, с бензиламином или его производным;
2) удаления -С(O)ОRz-единицы;
3) осуществления взаимодействия по свободному азоту биспидина полученного соединения с соединением формулы VIII, как оно определено здесь ранее;
4) удаления бензильной защитной группы; и
5) осуществления взаимодействия по свободному азоту биспидина полученного соединения, например, с соединением формулы III или IV, как оно определено здесь ранее,
в условиях, хорошо известных специалистам в данной области техники, включая условия, описанные здесь ранее. Это взаимодействие будет сопровождаться на некоторых стадиях превращением выступающей мостиковой карбонильной функциональной группы в желаемые группы R1/R2.
Соединения формулы XXXIV могут быть получены в соответствии с методиками, которые хорошо известны специалистам в данной области техники. Например, соединения формулы XXXIV, где R1a и R2a вместе представляют собой -(СН2)3-, -(СН2)4- или -(CH2)5-, могут быть получены путем осуществления взаимодействия соединения формулы XXXVII,
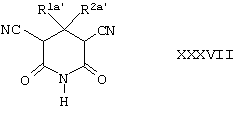
где R1а’ и R2a’ вместе представляют собой -(СН2)3-, -(СН2)4- или -(СН2)5-, со смесью фосфорной кислоты и серной кислоты, например, при 120°С.
Соединения формулы XXXVI хорошо известны по литературным данным либо легко доступны благодаря применению известных методик. Например, соединения формулы XXXVI, где R5 представляет собой ОН, R6 представляет собой Н, а А представляет собой СН2, могут быть получены путем осуществления взаимодействия соединения формулы VII, где R6 представляет собой Н, а Х представляет собой О, с гидроксидом аммония в условиях, которые хорошо известны специалистам в данной области техники.
Соединения формул III, VA, XIA, XII, XIV, XVI, XVII, XVIII, XIX, XXI, XXIB, XXIV, XXVI, XXVII, XXVIII, XXVIIIA, XXIX, XXX, XXXI, XXXIA, XXXII, XXXIII, XXXV и XXXVII и их производные либо имеются в продаже, известны по литературным данным, либо могут быть получены или аналогично описанным здесь способам, или с помощью общепринятых методов синтеза в соответствии со стандартными методиками из легко доступных исходных материалов с использованием соответствующих реагентов и реакционных условий.
Заместители на арильной (например, фенильной) и (если подходит) гетероциклической группе(ах) в соединениях, определенных здесь, могут быть превращены в другие заместители с использованием методик, хорошо известных специалистам в данной области техники. Например, нитробензол может быть восстановлен до аминобензола, гидрокси может быть превращен в алкокси, алкокси может быть гидролизован до гидрокси и так далее.
Соединения по данному изобретению могут быть выделены из их реакционных смесей с использованием общепринятых методик.
Специалистам в данной области техники будет ясно, что в способах, описанных выше, функциональные группы промежуточных соединений могут быть защищены или могут нуждаться в защите с помощью защитных групп.
Функциональные группы, которые желательно защищать, включают в себя гидрокси, амино и карбоксильную группу. Подходящие защитные группы для гидрокси включают в себя триалкилсилильные и диарилалкилсилильные группы (например, тpeт-бутилдиметилсилил, mpem-бутилдифенилсилил или триметилсилил), тетрагидропиранильные и алкилкарбонилоксигруппы (например, метил- и этилкарбонилоксигруппы). Подходящие защитные группы для амино включают в себя бензил, трет-бутилоксикарбонил, 9-флуоренилметоксикарбонил или бензилоксикарбонил. Подходящие защитные группы для карбоновой кислоты включают в себя C1-6алкиловые или бензиловые сложные эфиры.
Защита и снятие защиты с функциональных групп могут иметь место до или после любых реакционных стадий, описанных здесь ранее.
Защитные группы могут быть удалены в соответствии с методиками, которые хорошо известны специалистам в данной области техники и таким образом, как описано здесь далее.
Использование защитных групп полностью описано в “Protective Groups in Organic Chemistry”, под редакцией J.W.F.McOmie, Plenum Press (1973) и “Protective Groups in Organic Synthesis”, 2nd edition, Т W Greene & P G M Wutz, Wiley-lnterscience (1991).
Специалистам в данной области техники будет ясно, что для того, чтобы получить соединения по данному изобретению альтернативным и в некоторых случаях более удобным способом, упомянутые здесь индивидуальные стадии способа могут быть выполнены в другом порядке и/или индивидуальные реакции могут быть выполнены на другой стадии общего пути (то есть заместители могут быть добавлены к другим промежуточным соединениям и/или выполнены химические трансформации на других промежуточных соединениях по сравнению с теми, которые были здесь ранее связаны с конкретной реакцией). Это будет зависеть среди прочего от таких факторов, как природа других функциональных групп, присутствующих в конкретном субстрате, доступность ключевых промежуточных соединений и принятая стратегия (если она существует) защитных групп. Очевидно, что тип привлеченной химии будет оказывать влияние на выбор реагента, который используется в указанных стадиях синтеза, на необходимость и тип применяемых защитных групп и последовательность осуществления синтеза.
Специалистам в данной области техники также будет ясно, что несмотря на то, что определенные защищенные производные соединений формулы I, которые могут быть получены до конечной стадии снятия защиты, могут не обладать фармакологической активностью как таковой, они могут быть введены парентерально или перорально и после этого подвергаться метаболизму в организме с образованием соединений по данному изобретению, которые являются фармакологически активными. Поэтому такие производные могут быть описаны как “пролекарства”. Более того, авторами изобретения обнаружено, что определенные соединения формулы I могут действовать как пролекарства других соединений формулы I.
Все пролекарства соединений формулы I включены в объем настоящего изобретения.
Некоторые промежуточные соединения, упомянутые здесь ранее, являются новыми. Таким образом, согласно еще одному аспекту данного изобретения предложено: (а) соединение формулы II, как оно определено здесь ранее, или его защищенное производное при условии, что R7 не представляет собой возможно замещенный фенил; (б) соединение формулы V, как оно определено здесь ранее, или его защищенное производное при условии, что R7 не представляет собой возможно замещенный фенил; (в) соединение формулы X, как оно определено здесь ранее, или его защищенное производное; (г) соединение формулы XI, как оно определено здесь ранее, или его защищенное производное; (д) соединение формулы XIII, как оно определено здесь ранее, или его защищенное производное; (е) соединение формулы XV, как оно определено здесь ранее, или его защищенное производное; (ж) соединение формулы XX, как оно определено здесь ранее, или его защищенное производное; (з) соединение формулы XXIII, как оно определено здесь ранее, или его защищенное производное, при условии, что R7 не представляет собой возможно замещенный фенил; и (и) соединение формулы XXV, как оно определено здесь ранее, или его защищенное производное.
Медицинское и фармацевтическое применение
Соединения по данному изобретению являются полезными, так как они обладают фармацевтической активностью. Поэтому они показаны в качестве фармацевтических препаратов.
Таким образом, согласно еще одному аспекту данного изобретения предложены соединения по изобретению для применения в качестве фармацевтических препаратов.
В частности, соединения по изобретению проявляют миокардиальную электрофизиологическую активность, например, как продемонстрировано в тесте, описанном ниже.
Таким образом, ожидается, что соединения по изобретению будут полезны как при профилактике, так и при лечении аритмий, и в частности предсердной и желудочковой аритмий.
Таким образом, соединения по изобретению показаны при лечении или профилактике сердечных заболеваний либо при показаниях, относящихся к сердечным заболеваниям, при которых аритмии, как полагают, играют главную роль, включая ишемическую болезнь сердца, внезапный сердечный приступ, инфаркт миокарда, сердечную недостаточность, операцию на сердце и случаи тромбоэмболии.
При лечении аритмий было обнаружено, что соединения по изобретению селективно замедляют сердечную реполяризацию, таким образом увеличивая QT-интервал, и, в частности, проявляют активность класса III. Несмотря на то, что было показано, что соединения по изобретению проявляют активность класса III, в частности, при лечении аритмий, вид(ы) их активности необязательно ограничен(ы) этим классом.
Согласно еще одному аспекту данного изобретения предложен способ лечения аритмии, при котором субъекту, страдающему от такого состояния или подверженному такому состоянию, вводят терапевтически эффективное количество соединения по изобретению.
Фармацевтические препараты
Соединения по данному изобретению обычно будут вводиться перорально, подкожно, внутривенно, внутриартериально, трансдермально, интраназально, посредством ингаляции или посредством любого другого парентерального способа применения, в форме фармацевтических препаратов, содержащих активный ингредиент либо в виде свободного основания, фармацевтически приемлемого ионита, либо нетоксичной соли присоединения органической или неорганической кислоты, в фармацевтически приемлемой лекарственной форме. В зависимости от заболевания и пациента, подвергаемого лечению, а также от способа введения, композиции могут быть введены в различных дозах.
Соединения по изобретению можно также комбинировать с любыми другими лекарствами, полезными при лечении аритмий и/или других сердечно-сосудистых заболеваний.
Таким образом, согласно еще одному аспекту данного изобретения предложен фармацевтический препарат, включающий в себя соединение по изобретению в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Подходящие суточные дозы соединений по изобретению при терапевтическом лечении людей составляют приблизительно от 0,05 до 5,0 мг/кг массы тела при парентеральном введении.
Соединения по изобретению имеют преимущество в том, что они являются эффективными против сердечных аритмий.
Соединения по изобретению имеют также преимущество в том, что они могут быть более эффективными, менее токсичными, иметь более широкий интервал действия (включая проявление любой комбинации активности класса I, класса II, класса III и/или класса IV (в особенности активности класса I, класса II и/или класса IV в дополнение к активности класса III)), быть более сильнодействующими, иметь более продолжительную активность, вызывать меньше побочных эффектов (включая более низкий процент проаритмий, таких как torsades de pointes), легче всасываться, чем соединения, известные из уровня техники, или в том, что они могут обладать другими полезными фармакологическими свойствами помимо имеющихся у соединений, известных из уровня техники.
Биологические тесты
Тест А
Первичные электрофизиологические эффекты на анестезированных морских свинках
Использовали морских свинок весом между 660 и 1100 г. Животных содержали по меньшей мере в течение одной недели перед экспериментом при свободном доступе в этот период к еде и водопроводной воде.
Анестезию осуществляли путем внутрибрюшинной инъекции пентобарбитала (от 40 до 50 мг/кг), и катетеры вводили в одну сонную артерию (для регистрации кровяного давления и отбора крови) и в одну шейную вену (для инфузий лекарств). Игольчатые электроды помещали на конечности для регистрации ЭКГ (отведение II). Термистор помещали в прямую кишку, и животное помещали на электрогрелку, устанавливая ректальную температуру между 37,5 и 38,5°С.
Проводили трахеотомию, и животное подвергали искусственному вентилированию с помощью комнатного воздуха, используя небольшой вентилятор для животных, установленный так, чтобы поддерживать газы крови в нормальных пределах для данных видов. Для того чтобы снизить воздействия на вегетативную нервную систему, за 15 минут до начала эксперимента разрезали оба блуждающих нерва в области шеи и внутривенно вводили 0,5 мг/кг пропранолола.
Эпикард левого желудочка обнажили посредством левосторонней торакотомии, и к свободной стенке левого желудочка приложили изготовленный на заказ присасывающийся электрод для регистрации однофазного потенциала действия (ОПД). Электрод сохраняли в этом положении до тех пор, пока не был зарегистрирован приемлемый сигнал, в противном случае его перемещали в новое положение. Биполярный электрод для электокардиостимуляции прикрепили к левому предсердию. Электрокардиостимуляцию (длительность 2 мс, дважды диастолический порог) выполняли с помощью изготовленного на заказ стимулятора постоянного тока. Сердце стимулировали с частотой несколько выше нормального синусового ритма в течение 1 минуты каждые пять минут на протяжении всего исследования.
Кровяное давление, ОПД-сигнал и отведение II ЭКГ регистрировали на струйном самописце Mingograph (Siemens-Elema, Швеция). Все сигналы собирали (частота сбора 1000 Гц) на ПК в течение последних 10 секунд каждой последовательности стимуляции и последних 10 секунд следующей минуты синусового ритма. Сигналы обрабатывали с использованием изготовленной на заказ программы, разработанной для сбора и анализа физиологических сигналов, измеряемых на подопытных животных (смотри Axenborg and Hirsch, Comput. Methods Programs Biomed. 41, 55 (1993)).
Процедура тестирования заключалась в снятии двух базовых контрольных записей с интервалом 5 минут в продолжение как электрокардиостимуляции, так и синусового ритма. После второй контрольной записи проводили инфузию первой дозы тестируемого вещества в течение 30 секунд в объеме 0,2 мл в катетер в шейной вене. Тремя минутами позже начинали электрокардиостимуляцию и делали новую запись. Через пять минут после предыдущей дозы вводили следующую дозу тестируемого вещества. В продолжение каждого эксперимента давали от шести до десяти последовательных доз.
Анализ данных
Из многочисленных переменных, измеряемых в этом виде анализа, выбрали три как наиболее важные для сравнения и выбора активных соединений. Этими тремя выбранными переменными являлись: продолжительность ОПД при 75-ти процентной реполяризации в продолжение электрокардиостимуляции, время атриовентрикулярной (АВ) проводимости (определяемое как интервал между предсердным стимулирующим импульсом и началом желудочкового ОПД) в продолжение электрокардиостимуляции и частота сердечных сокращений (определяемая как RR-интервал в продолжение синусового ритма). Систолическое и диастолическое кровяное давление измеряли для того, чтобы оценить гемодинамический статус анестезированного животного. Далее проверяли ЭКГ на наличие аритмий и/или морфологических изменений.
Среднюю величину по двум контрольным записям принимали за нулевое значение, и эффекты, зарегистрированные после последовательных доз тестируемого вещества, выражали как отклонения от этой величины, в процентах. Построение кривых доза-ответ было осуществлено путем нанесения на график этих величин в процентах против совокупной введенной дозы перед каждой записью. Таким образом, каждый эксперимент давал три кривых доза-ответ, одну для продолжительности ОПД, одну для времени АВ-проводимости и одну для частоты синусового ритма (RR-интервал). Вычисляли среднюю кривую по всем экспериментам, проведенным с тестируемым веществом, и из средней кривой получали величины эффективности. Все кривые доза-ответ в этих экспериментах строили путем линейного соединения полученных экспериментальных точек. Совокупную дозу, увеличивающую продолжительность ОПД на 10% по сравнению с базовой линией, использовали в качестве индекса для оценки электрофизиологической эффективности класса III исследуемого агента (D10).
Тест В
Метаболическая стабильность тестируемых соединений
С целью определения метаболической стабильности соединений по изобретению проводили скрининг in vitro.
Использовали печеночную фракцию S-9 собаки, человека, кролика и крысы вместе с NADPH в качестве кофактора. Условия анализа были следующие: S-9 (3 мг/мл), NADPH (0,83 мМ), буфер трис-НСl (50 мМ) при рН 7,4 и 10 мкМ тестируемого соединения.
Реакцию начинали добавлением тестируемого соединения и заканчивали через 0, 1, 5, 15 и 30 минут путем увеличения рН в образце выше 10 (NaOH; 1 мМ). После экстракции растворителем концентрацию тестируемого соединения измеряли против внутреннего стандарта с помощью LC (детектирование флуоресценции/УФ).
Рассчитывали процент оставшегося после 30 минут тестируемого соединения (и, следовательно, t1/2) и использовали его в качестве единицы измерения метаболической стабильности.
Данное изобретение иллюстрируется следующими примерами.
Примеры
Общие экспериментальные методики
Масс-спектры регистрировали на утроенном квадрупольном масс-спектрометре Finnigan MAT TSQ 700, оборудованном электроспрей-интерфейсом (FAB-MS), и масс-спектрометре VG Platform II, оборудованном электроспрей-интерфейсом (LC-MS), газовом хроматографе Hewlett Packard модели 6890, соединенном с масс-спектрометром Hewlett-Packard модели 5973А через ГХ-колонку Hewlett Packard HP-5-MS, или ГХ/масс-спектрометре Shimadsu QP-5000 (CI, метан). 1Н ЯМР и 13С ЯМР измерения выполняли на спектрометрах BRUKER АСР 300 и Varian UNITY plus 400 и 500, работающих на 1H-частотах 300, 400 и 500 МГц, соответственно, и на 13С-частотах 75,5, 100,6 и 125,7 МГц, соответственно. Альтернативно, 13С ЯМР измерения выполняли на спектрометре BRUKER АСЕ 200 на частоте 50,3 МГц.
Ротамеры могут различаться или могут не различаться в спектре, в зависимости от легкости интерпретации спектра. Если не оговорено особо, химические сдвиги даны в миллионных долях с растворителем в качестве внутреннего стандарта.
Синтез промежуточных соединений
Пример А
4-(2-Оксиранилметокси)бензонитрил
Эпихлоргидрин (800 мл) и К2СО3 (414 г) добавляли к перемешиваемому раствору пара-цианофенола (238 г) в 2,0 л MeCN и реакционную смесь кипятили с обратным холодильником в атмосфере инертного газа в течение 2 часов. Горячий раствор фильтровали и фильтрат концентрировали, получая прозрачное масло, которое кристаллизовали из ди-изо-пропилового эфира, получая указанный в заголовке продукт с 75%-ным выходом.
13С ЯМР (CDCl3): δ 44.4, 49.7, 69.0, 104.5, 115.3, 118.9, 134.0, 161.6.
Пример Б
2(S)-Оксиранил-3-нитробензолсульфонат
мета-Нитробензолсульфонилхлорид (12,6 г; 57 ммоль) добавляли к охлажденному (-20°С) раствору (R)-(+)-глицидного спирта (5,5 г; 74 ммоль) и ТЭА (10,3 мл; 74 ммоль). Реакционную смесь перемешивали при -20°С в течение 96 ч. Раствор фильтровали и фильтрат промывали винной кислотой (10% масс/масс), соляным раствором, Н2О и концентрировали, получая указанное в заголовке соединение с 97%-ным выходом.
1H ЯМР (СDСl3): δ 2.62 (dd, 1Н), 2.84 (dd, 1H), 3.22 (m, 1Н), 4.07 (dd, 1H), 4.49 (dd, 1H), 7.80 (t, 1H), 8.25 (m, 1H), 8.52 (m, 1H), 8.78 (m, 1H).
Пример В
4-[(2S)-Оксиранилметокси]бензонитрил
Указанное в заголовке соединение было получено с 90%-ным выходом в соответствии с методикой, описанной выше в Примере А, начиная с (R)-(-)-эпихлоргидрина.
Пример Г
4-[(2R)-Оксиранилметокси]бензонитрил
Указанное в заголовке соединение было получено в соответствии с методикой, описанной выше в Примере А, начиная с (S)-(-)-эпихлоргидрина.
[α]
1H ЯМР (CDCl3): δ 2.79 (1H, m), 2.98 (1H, m), 3.39 (1H, m), 3.98 (1H, m), 4.37 (1H, m), 6.99 (2Н, d), 7.60 (2Н, d).
Пример Д
3-Бензил-3,7-диазабицикло[3.3.1]нонан
(а) 3,7-Дибензил-3,7-диазабицикло[3.3.1]нонан
Указанное в подзаголовке соединение было получено в соответствии с методикой, описанной в J.Огд. Сhem., 41, 1593, (1976), за исключением того, что вместо N-бензил-N-метилбиспидона использовали 3,7-дибензил-3,7-диазабицикло[3.3.1]нонан-9-он (также полученный в соответствии со способом, описанным в J. Org. Chem., 41, 1593, (1976)).
(б) 3-Бензил-3,7-диазабицикло[3.3.1]нонан
3,7-Дибензил-3,7-диазабицикло[3.3.1]нонан (1,97 г; 6,4 ммоль; со стадии (а), описанной выше) растворили в ЕtOН (95%) и гидрировали над 5%-ным Pd/C при давлении 98 кПа (1 атм) до тех пор, пока по данным ТСХ реакция не завершилась. Катализатор удаляли посредством фильтрования через набивку Целита® и остаток концентрировали при пониженном давлении, получая указанное в заголовке соединение с количественным выходом.
13С ЯМР (CDCl3): δ 30.1, 33.4, 36.0, 52.5, 59.6, 64.3, 126.9, 128.3, 128.7, 138.8.
Пример Е
трет-Бутил-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат
(а) трет-Бутил-7-бензил-9-окси-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат
Параформальдегид (4,00 г; 127 ммоль) добавляли к раствору бензиламина (13,7 г; 126 ммоль) в этаноле (190 мл). Раствор нагревали до 60°С и в течение 2 часов добавляли раствор уксусной кислоты (15,2 г; 252 ммоль) в этаноле (160 мл). После дополнительного перемешивания в течение 1 часа раствор охлаждали до комнатной температуры. Этот раствор добавляли (в течение 2 часов) к смеси 1-трет-бутоксикарбонил-4-пиперидона (25,5 г; 127 ммоль) и пара-формальдегида (4,80 г; 152 ммоль) в этаноле (270 мл), который был нагрет до 60°С. После кипячения с обратным холодильником в течение ночи раствор охлаждали до комнатной температуры. Этанол удаляли выпариванием. Экстрактивную обработку проводили в смеси толуол:вода и материал фильтровали через кремнезем в системе толуол:этилацетат. Выпаривание элюента давало твердый материал (37,4 г). Степень чистоты составила 90% по площади (ВЭЖХ), а выход был равен 60%. В результате проведения кристаллизации в ИПС было получено соединение со степенью чистоты 98% по площади (ВЭЖХ) и выходом 70%.
МС (EI; 70 эВ): m/z=91 (100%), m/z=57 (42%), m/z=273 (32%), m/z=330 (5%).
13С ЯМР (CDCl3): δ 28.72, 47.71, 49.91, 50.60, 58.83, 59.16. 61.96, 80.18, 127.37, 128.45, 128.89, 137.57, 154.89, 213.66 (с использованием ТМС в качестве контроля).
(б) трет-Бутил-7-бензил-9-окси-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат (альтернативный способ получения)
Бензиламин (6,51 г; 60,2 ммоль), уксусную кислоту (72,3 г; 1200 ммоль), параформальдегид (3,71 г; 120 ммоль) и 1-трет-бутоксикарбонил-4-пиперидон (12,0 г; 60,2 ммоль) добавляли к этанолу (300 мл). Раствор нагревали до 65°С и перемешивали при этой температуре в течение 2 часов. Проводили такую же процедуру обработки, как и на стадии (а), описанной выше, получая 15,78 г материала со степенью чистоты 92% по площади (ВЭЖХ) и выходом 70%. Перекристаллизация из изо-пропанола давала соединение со степенью чистоты 94% по площади (ВЭЖХ) и выходом 54%.
(в) трет-Бутил-7-бензил-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат
Смесь 4-толуолсульфонгидразида (12,4 ммоль; 2,30 г) и трет-бутил-7-бензил-9-окси-3,7-диазабицикло[3.3.1]нонан-3-карбоксилата (10,1 ммоль; 4,00 г; 83,3%; со стадии (а), описанной выше) растворяли в изо-пропаноле (30 мл) и кипятили с обратным холодильником в течение 2 часов. Добавляли уксусную кислоту (2,5 ммоль; 0,15 г) и цианборгидрид натрия (12,1 ммоль; 0,76 г) и смесь снова кипятили с обратным холодильником в течение 2 часов. Взвесь охлаждали до температуры окружающей среды и фильтровали. Фильтрат концентрировали и проводили экстрактивную обработку в смеси толуол:вода. Толуольный раствор концентрировали, получая 0,95 г указанного в подзаголовке соединения со степенью чистоты 90% по площади (ГХ) и выходом 60%.
МС (EI; 70 эВ): m/z=259 (100%), m/z=91 (95%), m/z=169 (45%), m/z=57(35%), m/z=316 (25%).
13С ЯМР (CDCl3): δ 28.67, 28.95, 31.11, 47.55, 48.38, 58.70, 58.96, 63.46, 78.71, 126.57, 128.00, 128.53, 138.94, 155.20 (с использованием ТМС в качестве контроля).
(г) трет-Бутил-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат
трет-Бутил-7-бензил-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат (со стадии (в), описанной выше) дебензилировали в соответствии со способом, описанным выше в Примере Д (б), получая указанное в заголовке соединение с количественным выходом.
13С ЯМР (CDCl3): δ 28.05, 28.29, 31.33, 48.35, 49.11, 51.53, 79.34, 155.16.
Пример Ж
4-[3-(3,7-Диазабицикло[3.3.1]нон-3-ил)-2-гидроксипропокси]бензонитрил
EtOAc, насыщенный HCI (600 мл), добавляли к раствору трет-бутил-7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксилата (62 г; смотри Пример 2 международной заявки на патент № PCT/SE98/02276) в EtOAc (600 мл) и смесь перемешивали при комнатной температуре в течение 4 ч. Растворитель удаляли при пониженном давлении, остаток растворяли в MeCN (1,3 л) и добавляли К2СО3 (100 г). Суспензию перемешивали в течение 12 ч и фильтровали. Концентрирование фильтрата давало указанное в заголовке соединение с 90%-ным выходом.
13С ЯМР (CDCl3): δ 28.9, 29.2, 32.3, 50.9, 57.7, 60.8, 62.1, 66.0, 71.2, 104.0, 115.3, 119.1, 133.9, 162.1.
(Указанное в заголовке соединение также без труда может быть превращено в соль гидрохлорид с использованием стандартных методик).
Получение соединений формулы I
Пример 1
7-[(2S)-3-(4-Цианофенокси)-2-гидроксипропил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Этилизоцианат (1,42 г; 16,6 ммоль) добавляли к раствору 4-{[(2S)-3-(3,7-диазабицикло[3.3.1]нон-3-ил)-2-гидроксипропил]окси}бензонитрила (5,0 г; 20 ммоль; смотри Пример Ж, описанный выше) в 30 мл дихлорметана. Смесь перемешивали в течение 4 часов при комнатной температуре и затем концентрировали под вакуумом и очищали колоночной хроматографией на диоксиде кремния, элюируя смесью дихлорметан:метанол (95:5), с получением 3,2 г (51%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 15.52, 29.19, 29.50, 31.89, 35.77, 48.00, 49.17, 57.21, 60.49, 61.83, 65.41, 70.71, 103.88, 115.34, 119.15, 133.78, 133.84, 158.87, 162.19.
Пример 2
7-[3-(4-Цианофенокси)-2-гидроксипропил]-N-(циклопропилметил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) Циклопропилметилизоцианат
Циклопропилметиламин (1,4 г; 19,7 ммоль) добавляли к суспензии 1,1'-карбонилдиимидазола (3,2 г; 19,7 ммоль) в ТГФ (10 мл). Полученный раствор перемешивали в течение ночи при комнатной температуре, перед тем как подвергнуть дистилляции, получая 0,4 г (21%) указанного в подзаголовке соединения.
б) 7-[3-(4-Цианофенокси)-2-гидроксипропил]-N-(циклопропилметил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Циклопропилметилизоцианат (0,4 г; 4 ммоль; со стадии (а), описанной выше) добавляли к раствору 4-[3-(3,7-диазабицикло[3.3.1]нон-3-ил)-2-гидроксипропил]бензонитрила (1,2 г; 4 ммоль; смотри пример Ж, описанный выше) в ДХМ. Раствор перемешивали в течение ночи, концентрировали под вакуумом. Полученный остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол (93:7), с получением 0,85 г (50%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 3.29, 11.21, 29.31, 29.61, 32.10, 46.11, 48.14, 49.39, 57.24, 60.58, 62.04, 65.46, 70.76, 104.03, 115.37, 119.18, 133.88, 158.97, 162.22.
Пример 3
4-({(2S)-2-Гидрокси-3-[7-(4-морфолинилкарбонил)-3,7-диазабицикло[3.3.1]нон-3-ил]пропил}окси)бензонитрил
Раствор 4-{[(2S)-3-(3,7-диазабицикло[3.3.1]нон-3-ил)-2-гидроксипропил]-окси}бензонитрила (2,0 г; 6,6 ммоль; получен аналогично способу, описанному выше в Примере Ж) в ДХМ (10 мл) обрабатывали водным NaOH (0,8 мл 10 М), затем 4-морфолинкарбонилхлоридом (1,2 г; 8 ммоль). Полученную смесь перемешивали в течение 30 мин при комнатной температуре перед тем, как добавить воду. Органический слой отделяли, промывали 2 М NaOH, затем соляным раствором перед тем, как провести разделение, высушивали (МgSO4) и концентрировали под вакуумом. Остаток дважды перекристаллизовывали, сначала из изо-пропанола, а затем из этанола, получая 0,73 г (26,5%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 23.36, 29.59, 30.05, 32.34, 47.45, 49.51, 52.18, 56.86, 60.78, 62.82, 65.35, 66.66, 70.82, 104.03, 115.33, 119.17, 133.88, 162.23, 164.99.
Пример 4
7-{3-(4-Цианофенокси)-2-[(метансульфонил)амино]пропил}-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 4-(3-Амино-2-гидроксипропокси)бензонитрил
4-(2-Оксиранилметокси)бензонитрил (100 г; 0,57 моль; смотри Пример А, описанный выше) добавляли к смеси концентрированного водного гидроксида аммония (500 мл) и изо-пропанола (300 мл). Полученную суспензию перемешивали при комнатной температуре в течение 3 дней. Реакционную смесь фильтровали для удаления нерастворимого побочного продукта и фильтрат концентрировали под вакуумом, получая неочищенный продукт, который кристаллизовали из ацетонитрила, получая 50 г (46%) указанного в подзаголовке соединения.
(б) 2-(4-Цианофенокси)-1-{[(метансульфонил)амино]метил}этилметансульфонат
Метансульфонилхлорид (17,5 г; 153 ммоль) медленно добавляли к охлажденному (-10°С) раствору 4-(3-амино-2-гидроксипропокси)бензонитрила (13,3 г; 69 ммоль; со стадии (а), описанной выше) и 4-(диметиламино)пиридина (0,2 г; 1,64 ммоль) в пиридине (100 мл). Раствор желтого цвета перемешивали при комнатной температуре в течение 1,5 часов, концентрировали под вакуумом и затем снова растворяли в ДХМ. Этот раствор дважды промывали 2М HCI и один раз раствором NaHCO3 перед тем, как отделить органическую фазу, высушивали (МgSO4) и концентрировали под вакуумом, получая 23,5 г (100%) указанного в подзаголовке соединения.
(в) 4-{[1-Метансульфонил)азиридин-2-ил]метокси}бензонитрил
Перемешиваемый раствор 2-(4-цианофенокси)-1-{[(метансульфонил)-амино]метил}этилметансульфоната (23,5 г; 67 ммоль; со стадии (б), описанной выше) в ацетонитриле (200 мл) обрабатывали карбонатом калия (30 г; 210 ммоль), с образованием плотного осадка. Через 1 час добавляли еще одну порцию К2СО3 (30 г; 210 ммоль). Перемешивание продолжали в течение 2 ч при комнатной температуре перед тем, как реакционную смесь отфильтровать, и фильтрат концентрировали под вакуумом. Полученное масло (13 г) кристаллизовали из толуола, получая 8 г (47%) указанного в подзаголовке соединения. Т.пл. 79-81°С.
(г) N-{2-(7-Бензил-3,7-диазабицикло[3.3.1]нон-3-ил)-1-[(4-цианофенокси)метил]этил}метансульфонамид
Смесь 3-бензил-3,7-диазабицикло[3.3.1]нонана (2 г; 10 ммоль; смотри Пример Д, описанный выше) и 4-{[1-(метансульфонил)азиридин-2-ил]метокси}бензонитрила (2,5 г; 10 ммоль; со стадии (в), описанной выше) в изо-пропаноле кипятили с обратным холодильником в течение ночи. Затем смесь концентрировали под вакуумом, получая остаток, который затем растворяли в воде (рН 3) и экстрагировали эфиром. Водный слой подщелачивали с помощью 2М NaOH и экстрагировали ДХМ. Дихлорметановый слой отделяли, высушивали и концентрировали под вакуумом, получая остаток, который очищали колоночной хроматографией, элюируя градиентом ДХМ:метанол:метанольный аммиак (от 98:2:0 до 97:0:3), с получением 2,5 г (53%) указанного в подзаголовке соединения.
(д) N-[2-(4-(Цианофенокси)-1-(3,7-диазабицикло[3.3.1]нон-3-илметил)-этил]метансульфонамид
Раствор N-{2-(7-бензил-3,7-диазабицикло[3.3.1]нон-3-ил)-1-[(4-цианофенокси)метил]этил}метансульфонамида (2,3 г; 4,9 ммоль; со стадии (г), описанной выше) в водном этаноле (95%; 55 мл) гидрировали над 5%-ным Pd/C при атмосферном давлении. Катализатор удаляли фильтрованием через набивку Целита® и остаток концентрировали под вакуумом, получая 1,6 г неочищенного продукта. Этот продукт перекристаллизовывали из метанола, получая 0,3 г (16%) указанного в подзаголовке соединения.
(е) 7-{3-(4-Цианофенокси)-2-[(метансульфонил)амино]пропил}-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Суспензию N-[2-(4-(цианофенокси)-1-(3,7-диазабицикло[3.3.1]нон-3-илметил)этил]метансульфонамида (0,29 г; 0,77 ммоль; со стадии (д), описанной выше) в ДХМ (10 мл) обрабатывали этилизоцианатом (66 мкл; 0,84 ммоль), получая прозрачный раствор. Смесь перемешивали в течение 1 ч при комнатной температуре, концентрировали под вакуумом и далее очищали колоночной хроматографией, элюируя 5%-ным МеОН в ДХМ, с получением указанного в заголовке соединения с 73%-ным выходом.
13С ЯМР (CDCl3): δ 15.41, 28.88. 29.18, 30.77, 35.87, 41.78, 47.93, 48.65, 49.98, 58.24, 58.51, 60.15, 68.82, 104.51, 115.28, 118.95, 134.05, 158.58, 161.55.
Пример 5
7-[(2S)-3-(4-Цианофенокси)-2-гидроксипропил]-N-изо-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 7-Бензил-N-изо-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
изо-Пропилизоцианат (1,7 г; 20 ммоль) медленно добавляли к раствору 3-бензил-3,7-диазабицикло[3.3.1]нонана (3,1 г; 14,3 ммоль; смотри Пример Д, описанный выше) в ДХМ (10 мл). Смесь перемешивали при комнатной температуре в течение ночи и затем концентрировали под вакуумом, получая 4,2 г (97%) указанного в подзаголовке соединения.
(б) N-изо-Пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Раствор
7-бензил-N-изо-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (4,2 г; 14 ммоль; со стадии (а), описанной выше) в смеси метанол/вода (17 мл смеси в соотношении 15:2) гидрировали над 5%-ным Pd/C при атмосферном давлении. Катализатор удаляли фильтрованием через набивку с Целитом® и фильтрат концентрировали под вакуумом, получая 2,6 г (87%) указанного в подзаголовке соединения.
(в) 7-[(2S)-3-(4-Цианофенокси)-2-гидроксипропил]-N-изо-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь 7-[(2S)-оксиранилметокси]бензонитрила (0,55 г; 3,14 ммоль; смотри Пример В, описанный выше) и N-изо-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (0,85 г; 4 ммоль; со стадии (б), описанной выше) в смеси изо-пропанол/вода (6,5 мл смеси в соотношении 12:1) перемешивали в течение ночи при 60°С. Смесь затем концентрировали под вакуумом и остаток снова растворяли в ДХМ. Органический раствор промывали водой, затем соляным раствором, высушивали (MgSO4) и концентрировали под вакуумом, получая указанное в заголовке соединение с 91%-ным выходом.
13С ЯМР (CDCl3): δ 23.49, 29.29, 31.78, 42.26, 47.71, 49.09, 56.92, 60.27, 61.65, 65.19, 70.61, 103.54, 115.21, 119.09, 133.65, 158.11, 162.08.
Пример 6
7-[(2R)-3-(4-Циaнo-2-{[(2-циaнoэтил)aминo]кaрбoнил}-фeнoкcи)-2-гидроксипропил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 7-Бензил-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Охлажденный (0°С) раствор 3-бензил-3,7-диазабицикло[3.3.1]нонана (32,45 г; 0,15 моль; смотри Пример Д, описанный выше) в ДХМ (300 мл) обрабатывали этилизоцианатом (11,4 г; 0,16 моль), добавляя его по каплям. Раствор перемешивали в течение 2 ч при комнатной температуре перед тем, как сконцентрировать под вакуумом. Полученный остаток очищали хроматографией на силикагеле, элюируя градиентом ДХМ:МеОН (от 100:0 до 90:10), с получением 36,4 г (84%) указанного в подзаголовке соединения.
(б) N-Этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Раствор
7-бензил-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (4,4 г; 15,3 ммоль; со стадии (а), описанной выше) в водном этаноле (25 мл 95%-ного) гидрировали над 5%-ным Pd/C при атмосферном давлении. Катализатор удаляли фильтрованием через набивку Целита® и остаток концентрировали под вакуумом, получая 2,88 г (95%) указанного в подзаголовке соединения.
(в) Метил-5-бром-2-гидроксибензоат
Вr2 (52 г) медленно добавляли к перемешиваемому раствору метилсалицилата (50 г; 330 ммоль) в 300 мл уксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 10 ч, вливали в охлажденную во льду воду и осадок перекристаллизовывали из МеОН, получая указанное в подзаголовке соединение с 83%-ным выходом.
(г) Метил-5-циано-2-гидроксибензоат
Метил-5-бром-2-гидроксибензоат (190,8 г; со стадии (в), описанной выше) и CuCN (73,9 г) кипятили с обратным холодильником в ДМФ (500 мл) в течение 7 ч. Температуре дали понизиться до 80°С и добавили HCI (500 мл) и FеСl3 (160,5 г). Реакционную смесь перемешивали в течение 30 мин, концентрировали и распределяли между H2O и ДХМ. Органический слой высушивали, концентрировали, остаток перекристаллизовывали из метилэтилкетона, получая указанное в подзаголовке соединение с 61%-ным выходом.
(д) 5-Циано-N-(2-цианоэтил)-2-гидроксибензамид
Смесь метил-5-циано-2-гидроксибензоата (20 г; 0,113 моль; со стадии (г), описанной выше), 3-аминопропаннитрила (15,4 г; 0,22 моль) и цианида натрия (1 г; 20 ммоль) в метаноле (200 мл) кипятили с обратным холодильником в течение ночи. Анализ ТСХ показал незавершенность реакции, поэтому добавили ДМСО (50 мл), и в течение следующих 5 ч продолжали кипячение с обратным холодильником. Раствор концентрировали под вакуумом, добавляли воду, затем конц. НСl до тех пор, пока не образовался осадок. Продукт отфильтровывали, промывали водой и высушивали, получая 19,4 г (80%) указанного в подзаголовке соединения.
(е) 5-Циано-N-(2-цианоэтил)-2-[(2R)-оксиранилметокси]бензамид
Смесь 5-циано-N-(2-цианоэтил)-2-гидроксибензамида (2,1 г; 9,8 ммоль; со стадии (д), описанной выше) и 10 эквивалентов (S)-эпихлоргидрина в смеси изо-пропанол:вода (55 мл смеси в соотношении 10:1) кипятили с обратным холодильником в течение ночи. Смесь концентрировали под вакуумом и остаток очищали колоночной хроматографией, элюируя этилацетатом, с получением 0,63 г (24%) указанного в подзаголовке соединения.
(ж) 7-[(2R)-3-(4-Циано-2-{[(2-цианоэтил)амино]карбонил}фенокси)-2-гидроксипропил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь 5-циано-N-(2-цианоэтил)-2-[(2R)-оксиранилметокси]бензамида (0,63 г; 2,3 ммоль; со стадии (е), описанной выше) и N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (0,59 г; 3 ммоль; со стадии (б), описанной выше) в смеси изо-пропанол:вода (33 мл смеси в соотношении 10:1) перемешивали при температуре дефлегмации в течение ночи. Реакционную смесь концентрировали под вакуумом и остаток очищали колоночной хроматографией, элюируя смесью ДХМ/МеОН (9:1), с получением 0,78 г (73%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 15.40, 15.55, 17.94, 28.04, 29.21, 29.55, 31.31, 32.03, 35.69, 35.89, 36.21, 47.93, 48.65, 49.36, 57.00, 60.47, 61.05, 65.32, 72.21, 105.39, 114.37, 118.22, 118.45, 123.28, 136.36, 136.45, 158.53, 159.20, 160.08, 163.75.
ES-MC (M+1)+ 469.0 (m/z).
Пример 7
7-((2S)-3-{4-Циано-2-[(циклопропиламино)карбонил]фенокси}-2-гидроксипропил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
а) N1-Циклопропил-5-циано-2-гидроксибензамид
Циклопропиламин (14,3 г) и Na (100 мг) добавляли к раствору метил-5-циано-2-гидроксибензоата (10,0 г; со стадии (г), описанной выше) в ДМСО (40 мл). Реакционную смесь нагревали в течение ночи при 80°С в герметизированном стальном сосуде, разбавляли Н2О, подкисляли и экстрагировали ЕtOАс, получая после концентрирования органического слоя указанное в подзаголовке соединение (11,0 г).
(б) 5-Циано-N-циклопропил-2-[(2S)-оксиранилметокси]бензамид
Смесь N1-Циклопропил-5-циано-2-гидроксибензаимида (1,56 г; 7,7 ммоль; со стадии (а), описанной выше), (2S)-оксиранилметил-3-нитробензолсульфонат (2 г; 7,7 ммоль; смотри Пример Б, описанный выше) и К2СО3 (1,16 г; 8,4 ммоль) в 2-бутаноне (15 мл) перемешивали при 60°С в течение 18 ч. Смесь концентрировали под вакуумом и остаток кристаллизовали из смеси ди-изо-пропиловый эфир:МеСN (9:1), получая 0,97 г (97%) указанного в подзаголовке соединения.
(в) 7-((2S)-3-{4-Циано-2-[(циклопропиламино)карбонил]фенокси}-2-гидроксипропил)-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь 5-циано-N-циклопропил-2-[(2S)-оксиранилметокси]бензамида (0,97 г; 3,8 ммоль; со стадии (б), описанной выше) и N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид (0,89 г; 4,5 ммоль, смотри Пример 6(б), описанный выше) в смеси изо-пропанол:вода (22 мл смеси в соотношении 10:1) кипятили с обратным холодильником в течение ночи. Растворитель удаляли под вакуумом и полученный остаток очищали колоночной хроматографией на силикагеле, элюируя смесью ДХМ:МеОН (9:1), с получением 1,37 г (79%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 6.62, 6.78, 15.81, 23.55, 29.61, 29.90, 32.48, 36.20, 48.32, 49.84, 53.68, 57.48, 60.92, 62.06, 65.61, 71.72, 105.42, 113.69, 118.64, 123.78, 136.26, 136.77, 159.70, 159.97, 164.75.
Пример 8
N-Этил-7-(4-нитрофенетил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь 1-(2-бромэтил)-4-нитробензола (1,6 г; 7,0 ммоль), N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (1,0 г; 5,1 ммоль, смотри Пример 6(б), описанный выше) и К2СО3 (1,38 г; 10 ммоль) перемешивали при комнатной температуре в течение ночи. Затем смесь фильтровали, концентрировали под вакуумом и полученный остаток очищали колоночной хроматографией, элюируя градиентом ДХМ:МеОН (от 100:0 до 90:10), с получением 1,5 г (85%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 15.71, 28.83, 30.11, 33.03, 35.67, 47.97, 59.22, 59.49, 123.34, 129.65, 146.26, 149.15, 157.95.
Пример 9
N-(Цианометил)-7-[(2(S)-3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) Цианометилизоцианат
Указанное в заголовке соединение было получено в соответствии с методикой, описанной выше в Примере 2(а), с использованием 2-аминоацетонитрила вместо циклопропилметиламина.
(б) N-(Цианометил)-7-[(2(5)-3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Указанное в заголовке соединение было получено с 26%-ным выходом (с учетом стадий (а) и (б) вместе) в соответствии с методикой, описанной выше в Примере 2(б), с использованием цианометилизоцианата (со стадии (а), описанной выше) вместо циклопропилметилизоцианата.
13С ЯМР (CDCl3): δ 28.99, 29.27, 29.47, 31.77, 48.32, 49.33, 56.88, 60.33, 61.61, 65.32, 70.63, 103.96, 115.31, 117.63, 119.21, 133.93, 157.74, 162.08.
Пример 10
N-Этил-7-{4-[(метансульфонил)амино]фенетил}-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 4-[(Метансульфонил)амино]фенетил-метансульфонат
Метансульфонилхлорид (45 г; 0,39 моль) добавляли по каплям в течение 30 минут к охлажденному (-5°С) раствору 4-аминофенетилового спирта (25,2 г; 0,18 моль) в пиридине (200 мл). Смесь перемешивали при 0°С в течение 1 ч и затем при комнатной температуре в течение ночи. Полученную суспензию красного цвета вливали в смесь льда (300 мл) и конц.НСl (60 мл). Сформировавшийся осадок розового цвета отфильтровывали, снова растворяли в ДХМ, высушивали и обрабатывали активированным углем. Полученный раствор концентрировали под вакуумом, получая остаток, который после перекристаллизации из этилацетата давал 34,5 г (64%) указанного в подзаголовке соединения. Т.пл.=133-134°С.
(б) N-Этил-7-{4-[(метансульфонил)амино]фенетил}-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид
Смесь N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (1 г; 5 ммоль; смотри Пример 6(б); описанный выше), 4-[(метансульфонил)амино]фенетил-метансульфоната (1,5 г; 5 ммоль; со стадии (а), описанной выше) и NaHCO3 (3 г; 35,7 ммоль) в MeCN (50 мл) кипятили с обратным холодильником в атмосфере азота в течение 3 ч. Реакционную смесь фильтровали и концентрировали под вакуумом, получая 2,2 г неочищенного продукта, который фильтровали через набивку диоксида кремния с МеОН/2 н. HCl. рН во фракциях увеличивали до 6,0 и экстрагировали ДХМ, получая 0,2 г указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 15.75, 28.87, 30.23, 32.58, 35.64, 35.76, 39.14, 48.18, 59.17, 60.26, 121.41, 129.85, 134.72.
Пример 11
7-[3-(4-Цианофенокси)-2-фторпропил]-N-этил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид
(а) 7-[3-(4-Цианофенокси)-2-гидроксипропил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Указанное в заголовке соединение было получено в соответствии с методикой, описанной выше в Примере 1, с использованием 4-[3-(3,7-диазабицикло[3.3.1]нон-3-ил)-2-гидроксипропокси]бензонитрила (смотри Пример Ж, описанный выше), вместо 4-{[(2S)-3-(3,7-диазабицикло[3.3.1]нон-3-ил)-2-гидроксипропил]окси}бензонитрила.
(б) 7-[3-(4-Цианофенокси)-2-фторпропил]-N-этил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид
Раствор 7-[3-(4-цианофенокси)-2-гидроксипропил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (1,0 г; 2,7 ммоль; со стадии (а), описанной выше) в ДХМ (2,5 мл) охлаждали до -78°С. Медленно, при перемешивании добавляли раствор (диэтиламино)трифторида серы в ДХМ (2,5 мл). Перемешивание продолжали в течение 35 минут, причем в течение этого времени реакционной смеси давали нагреться до комнатной температуры. Добавляли дихлорметан и затем реакционную смесь промывали NаНСО3, высушивали и концентрировали под вакуумом. Полученный остаток очищали колоночной хроматографией, элюируя смесью ДХМ:МеОН (98:2), с получением 0,68 г (67%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 15.63, 29.00, 30.33, 35.70, 47.78, 47.93, 58.36, 58.67, 59.82, 60.39, 68.60, 68.89, 89.56, 91.86, 104.15, 115.56, 119.25, 133.97, 157.61, 161.92.
Пример 12
7-[3-(4-Цианофенокси)-2-гидроксипропил]-N-[2-оксо-2-(пропиламино)этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) Этил-2-[({7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нон-3-ил}карбонил)амино]ацетат.
Охлажденный (0°С) раствор 4-[3-(3,7-диазабицикло[3.3.1]нон-3-ил)-2-гидроксипропокси]бензонитрила (23,1 г; 77 ммоль; смотри Пример Ж, описанный выше) в ДХМ (700 мл) обрабатывали этил-2-изоцианатацетатом (9,92 г; 77 ммоль) и затем перемешивали при комнатной температуре в течение 7 ч. Реакционную смесь концентрировали под вакуумом, получая 33,6 г (100%) указанного в подзаголовке соединения.
(б) 7-[3-(4-Цианофенокси)-2-гидроксипропил]-N-[2-оксо-2-(пропиламино)этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь этил-2-[({7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нон-3-ил}карбонил)амино]ацетата (0,76 г; 1,8 ммоль; со стадии (а), описанной выше), пропиламина (5 мл; 3,6 г; 69,1 ммоль) и NaCN (0,01 г; 0,2 ммоль) в метаноле (10 мл) нагревали до 75°С в герметизированной пробирке в течение ночи. Растворитель затем удаляли под вакуумом и остаток разбавляли раствором Na2CO3. Водную смесь экстрагировали ДХМ и полученный органический слой отделяли, высушивали и концентрировали под вакуумом. Полученный остаток очищали колоночной хроматографией, элюируя градиентом смеси дихлорметан:метанол (от 100:0 до 90:10), с получением указанного в заголовке соединения с 70%-ным выходом.
13С ЯМР (CDCl3): δ 11.36, 22.65, 29.12, 29.42, 31.78, 41.15, 44.75, 48.15, 49.10, 56.99, 60.40, 61.35, 65.33, 70.74, 103.99, 115.27, 119.12, 133.91, 158.71, 162.10, 170.62.
Пример 13
7-{3-(4-Цианофенокси)-2-[(4-морфолинилкарбонил)амино]пропил}-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) трет-Бутил-3-(4-цианофенокси)-2-гидроксипропилкарбамат
Охлажденный (0°С) раствор 4-(3-амино-2-гидроксипропокси)бензонитрила (44,6 г; 0,23 моль; смотри Пример 4(а), описанный выше) в смеси ТГФ:Н2O (1,5 л смеси в соотношении 1:1) обрабатывали ди-трет-бутилдикарбонатом (53 г; 0,24 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, после чего добавляли NaCl и полученный органический слой отделяли. Водный слой экстрагировали эфиром и объединенные органические экстракты высушивали и концентрировали под вакуумом. Полученное масло (70 г) фильтровали через набивку диоксида кремния и затем кристаллизовали из смеси диэтиловый эфир:ди-изо-пропиловый эфир, получая 50 г указанного в подзаголовке соединения.
(б) 2-[(трет-Бутоксикарбонил)амино]-1-[(4-цианофенокси)метил]этил-метансульфонат
Метансульфонилхлорид (22,3 г; 0,195 моль) добавляли в течение 1,5 часов к охлажденному (0°С) раствору трет-бутил-3-(4-цианофенокси)-2-гидроксипропилкарбамата (51,2 г; 0,177 моль; со стадии (а), описанной выше) и 4-(диметиламино)пиридина (1,3 г; 10,6 ммоль) в пиридине (250 мл) в атмосфере инертного газа. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре перед тем, как добавить воду и ДХМ. Органический слой отделяли, промывали водой, высушивали (МgSO4) и концентрировали под вакуумом, получая 68,1 г (100%) указанного в подзаголовке соединения.
(в) трет-Бутил-2-[(4-цианофенокси)метил]-1-азиридинкарбоксилат
Охлажденный (0°С) раствор 2-[(трет-бутоксикарбонил)амино]-1-[(4-цианофенокси)метил]этил-метансульфоната (30,6 г; 82,6 ммоль; со стадии (б), описанной выше) и тетрабутиламмония гидросульфата (3 г; 8,8 ммоль) в ДХМ (100 мл) обрабатывали 50 мас.% водного NaOH (60 мл) в атмосфере инертного газа. Полученную смесь перемешивали, и температуре дали возможность медленно подниматься до комнатной температуры в течение 4 ч, а затем экстрагировали эфиром. Органический слой промывали водой и концентрировали под вакуумом, получая остаток, который очищали колоночной хроматографией (дихлорметан в качестве элюента). Кристаллизация из смеси диэтиловый эфир:ди-изо-пропиловый эфир давала указанное в подзаголовке соединение с количественным выходом.
(г) трет-Бутил-2-(4-цианофенокси)-1-({7-[(этиламино)карбонил]-3,7-диазабицикло[3.3.1]нон-3-ил)метил)этилкарбамат
Смесь N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (2,88 г; 14,6 ммоль, смотри Пример 6(б), описанный выше) и трет-бутил-2-[(4-цианофенокси)метил]-1-азиридинкарбоксилата (4,0 г; 14,6 ммоль; со стадии (в), описанной выше) в изо-пропаноле (20 мл) кипятили с обратным холодильником в течение ночи. Реакционную смесь концентрировали под вакуумом с получением 7,4 г масла желтого цвета, которое очищали колоночной хроматографией, элюируя градиентом смеси ДХМ:МеОН (от 100:0 до 90:10), с получением 3,33 г указанного в подзаголовке соединения.
(д) 7-[2-Амино-3-(4-цианофенокси)пропил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Раствор трет-бутил-2-(4-цианофенокси)-1-({7-[(этиламино)карбонил]-3,7-диазабицикло[3.3.1]нон-3-ил}метил)этилкарбамата (2,4 г; 5,1 ммоль, со стадии (г), описанной выше) в насыщенном HCl этилацетате перемешивали в течение 1 ч при комнатной температуре. Реакционную смесь концентрировали под вакуумом и полученный остаток растворяли снова в воде. Водный раствор обрабатывали водным NаНСО3 и экстрагировали ДХМ, органический слой которого затем высушивали и концентрировали под вакуумом, получая 2 г указанного в подзаголовке соединения.
(е) 7-{3-(4-Цианофенокси)-2-[(4-морфолинилкарбонил)амино]пропил}-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Охлажденный (5°С) раствор 7-[2-амино-3-(4-цианофенокси)пропил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (0,33 г; 0,7 ммоль; со стадии (д), описанной выше) и триэтиламина (0,4 мл; 3,0 ммоль) в ДХМ (5 мл) обрабатывали 4-морфолинкарбонилхлоридом (0,11 г; 0,7 ммоль) и затем перемешивали при 5°С в течение 3 ч. После дальнейшего перемешивания при комнатной температуре в течение ночи анализ ТСХ показал, что реакция не прошла полностью, поэтому затем добавляли дополнительную порцию 4-морфолинкарбонилхлорида (40 мг; 0,27 ммоль). Перемешивание продолжали при комнатной температуре в течение ночи перед тем, как добавить раствор NаНСО3. Органический слой отделяли, высушивали и концентрировали под вакуумом, получая 400 мг неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанольный аммиак (95:5), с получением 250 мг указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 161.94, 158.26, 157.81, 133.94, 119.15, 115.37, 103.90, 67.26, 66.66, 60.66, 60.51, 57.99, 48.93, 48.37, 47.39, 44.06, 35.93, 30.71, 29.34, 29.02,15.51.
Пример 14
N-(4-Цианофенетил)-7-(4-оксогептил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 3-Бензил-7-[3-(2-пропил-1,3-диоксолан-2-ил)пропил]-3,7-диазабицикло[3.3.1]нонан
Смесь 3-бензил-3,7-диазабицикло[3.3.1]нонана (10,5 г; 48,5 ммоль; смотри Пример Д, описанный выше), 2-(3-бромпропил)-2-пропил-1,3-диоксолана (11,5 г; 48,5 ммоль; Bajrowicsz et al., Tetrahedron, 41 (1985) 1833) и К2СО3 (13,8 г; 0,1 моль) в MeCN (50 мл) кипятили с обратным холодильником в течение ночи. Реакционную смесь фильтровали и концентрировали под вакуумом, получая 18,8 г (100%) указанного в подзаголовке соединения.
(б) 3-[3-(2-Пропил-1,3-диоксолан-2-ил)пропил]-3,7-диазабицикло-[3.3.1]нонан
Раствор 3-бензил-7-[3-(2-пропил-1,3-диоксолан-2-ил)пропил]-3,7-диазабицикло[3.3.1]нонана (18,8 г; 4,85 ммоль; со стадии (а), описанной выше) в этаноле (100 мл) гидрировали над 5%-ным Pd/C при атмосферном давлении. Катализатор удаляли фильтрованием через набивку Целита® и фильтрат концентрировали под вакуумом, получая 13,7 г (100%) указанного в подзаголовке соединения.
(в) N-(4-Цианофенетил)-7-(4-оксогептил)-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид
Раствор 4-(2-аминоэтил)бензонитрила (1,0 г; 6,9 ммоль; Wiley et at., Bioorg. Med. Chem. Lett., 6 (1996) 2387) в безводном ТГФ (10 мл) обрабатывали 1,1'-карбонилдиимидазолом (1,17 г; 7,2 ммоль) и смесь перемешивали в течение 30 мин. К реакционной смеси добавляли раствор 3-[3-(2-пропил-1,3-диоксолан-2-ил)пропил]-3,7-диазабицикло[3.3.1]нонана (1,3 г; 4,6 ммоль; со стадии (б), описанной выше) в ТГФ (5 мл) и продолжали перемешивание при комнатной температуре в течение ночи. Раствор затем концентрировали под вакуумом и полученный остаток разбавляли МеОН и 2 М HCl, при этом перемешивали данный раствор в течение 2 ч при комнатной температуре. Смесь подщелачивали и экстрагировали ДХМ. Органический слой отделяли, высушивали и концентрировали под вакуумом, получая остаток, который очищали флэш-хроматографией, элюируя смесью ДХМ: МеОН (92:8), с получением 0,57 г (30%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 13.73, 17.21, 20.85, 28.79, 30.38, 36.91, 39.84, 41.83, 44.73, 47.94, 57.65, 59.05, 110.06, 118.93, 129.67, 132.20, 145.52, 157.47, 211.67.
Пример 15
N’-(4-Цианобензоил)-7-(4-оксогептил)-3,7-диазабицикло[3.3.1]нонан-3-карбогидразид
Смесь 4-цианобензогидразида (0,82 г; 5,0 ммоль) и 1,1'-карбонилдиимидазола (0,82 г; 5 ммоль) в ТГФ (15 мл) перемешивали в течение 10 мин при комнатной температуре перед тем, как добавить 3-[3-(2-пропил-1,3-диоксолан-2-ил)пропил]-3,7-диазабицикло[3.3.1]нонан (1,44 г; 5,0 ммоль; смотри Пример 14(б), описанный выше). Реакционную смесь перемешивали в течение ночи при комнатной температуре перед тем, как сконцентрировать под вакуумом. Полученный остаток растворяли в ДХМ и промывали водой. Органический слой отделяли и концентрировали под вакуумом, получая остаток, который растворяли в смеси метанол/2 М НСl. Выпаривание МеОН под вакуумом и экстракция оставшегося водного раствора дихлорметаном давала после очистки флэш-хроматографией на силикагеле (дихлорметан:метанольный аммиак в качестве элюента) 0,5 г (25%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 213.21, 164.24, 157.01, 136.31, 132.19, 128.24, 118.11, 115.11, 58.65, 57.89, 48.38, 44.31, 40.55, 31.52, 29.12, 21.60, 17.08, 13.69.
Пример 16
4-{2-Амино-3-[7-(1-пиперидинилкарбонил)-3,7-диазабицикло[3.3.1]нон-3-ил]пропокси}бензонитрил
(а) 7-Бензил-3,7-диазабицикло[3.3.1]нон-3-ил(1-пиперидинил)метанон
Указанное в подзаголовке соединение было получено путем осуществления взаимодействия между 3-бензил-3,7-диазабицикло[3.3.1]нонаном (смотри Пример Д, описанный выше) и 1-пиперидинкарбонилхлоридом (Boon, J. Chem. Soc. (1947) 307, 313).
(б) 3,7-Диазабицикло[3.3.1]нон-3-ил(1-пиперидинил)метанон Указанное в подзаголовке соединение было получено с количественным выходом в соответствии с методикой, описанной выше в Примере 14(б), с использованием 7-бензил-3,7-диазабицикло[3.3.1]нон-3-ил(1-пиперидинил)-метанона (со стадии (а), описанной выше) вместо 3-бензил-7-[3-(2-пропил-1,3-диоксолан-2-ил)пропил]-3,7-диазабицикло[3.3.1]нонана.
(в) трет-Бутил-2-(4-цианофенокси)-1-{[7-(1-пиперидинилкарбонил)-3,7-диазабицикло[3.3.1]нон-3-ил]метил}этилкарбамат
Смесь трет-бутил-2-[(4-цианофенокси)метил]-1-азиридинкарбоксилата (1,92 г; 7 ммоль; смотри Пример 13(в), описанный выше) и 3,7-диазабицикло[3.3.1]нон-3-ил(1-пиперидинил)метанона (1,85 г; 7 ммоль; со стадии (а), описанной выше) в изо-пропаноле (15 мл) кипятили с обратным холодильником в течение 30 ч. Раствор концентрировали под вакуумом, получая 3,7 г неочищенного продукта, который очищали хроматографией, с использованием 2,5%-ного МеОН в ДХМ, с получением 2,0 г (56%) указанного в подзаголовке соединения.
(г) 4-{2-Амино-3-[7-(1-пиперидинилкарбонил)-3,7-диазабицикло[3.3.1]нон-3-ил1пропокси}бензонитрил
Охлажденный (0°С) раствор трет-бутил-2-(4-цианофенокси)-1-{[7-(1-пиперидинилкарбонил)-3,7-диазабицикло[3.3.1]нон-3-ил]метил}этилкарбамата (1,9 г; 3,7 ммоль; со стадии (в), описанной выше) в этилацетате обрабатывали этилацетатом, насыщенным HCl. Смесь перемешивали в течение 4 ч перед тем, как сконцентрировать под вакуумом. Полученный остаток растворяли в воде, подщелачивали с помощью NаНСО3 и экстрагировали ДХМ. Органический слой отделяли, высушивали и концентрировали под вакуумом, получая 1,5 г (100%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 24.73, 25.72, 29.62, 29.95, 32.11, 47.44, 48.14, 49.53, 50.98, 57.87, 60.57, 62.59, 72.03, 103.90, 115.30, 119.22, 133.91, 162.23, 164.35.
Пример 17
N-Этил-7-{2-гидрокси-3-[4-(1Н-имидазол-1-ил)фенокси]пропил}-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 1-[4-(2-Оксиранилметокси)фенил]-1Н-имидазол
Смесь 4-(1-Н-имидазол-1-ил)фенола (10 г; 60 ммоль), К2СО3 (8,63 г; 60 ммоль) и 2-оксиранилметил-З-нитробензолсульфоната (15,5 г; 60 ммоль; смотри Пример Б, описанный выше) в ДМФ (140 мл) перемешивали при 40°С в течение ночи. Затем смесь концентрировали под вакуумом и полученный остаток разбавляли ДХМ, промывали водой, высушивали и затем концентрировали под вакуумом. Неочищенный остаток затем очищали флэш-хроматографией, элюируя градиентом смеси дихлорметан: метанол (от 100:0 до 70:30), получая 3,4 г (72,6%) указанного в заголовке соединения.
(б) N-Этил-7-{2-гидрокси-3-[4-(1H-имидазол-1-ил)фенокси]пропил}-3.7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь 1-[4-(2-оксиранилметокси)фенил]-1Н-имидазола (3,16 г; 14,6 ммоль; со стадии (а), описанной выше) и N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (2,88 г; 14,6 ммоль; смотри Пример 6(б), описанный выше) в смеси изо-пропанол: Н2О (18 мл смеси в соотношении 9:1) кипятили с обратным холодильником в течение 3 часов, концентрировали под вакуумом и очищали кислотно-щелочной экстракцией, получая 4,4 г (72,6%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 15.52, 29.13, 29.44, 31.84, 35.70, 47.92, 49.07, 57.21, 60.44, 61.94, 65.45, 70.76, 115.49, 118.58, 122.90, 129.86, 130.56, 135.66, 158.16, 158.78.
Пример 18
N-[3-(4-Цианофенокси)пропил]-7-(2-гидроксиэтил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а)4-(3-Бромпропокси)бензонитрил
1,3-Дибромпропан (1,02 л; 10 моль) добавляли к перемешиваемой суспензии пара-цианофенола (238 г; 2 моль), К2СО3 (276,4 г; 2 моль) в MeCN (2,7 л). Реакционную смесь кипятили с обратным холодильником в течение 4 ч, фильтровали и концентрировали. Остаток перекристаллизовывали из изо-пропилового эфира, получая указанное в подзаголовке соединение с 69%-ным выходом.
(б) 4-[3-(1,3-Диоксо-1,3-дигидро-2Н-изоиндол-2-ил)пропокси]бензонитрил
Смесь
4-(3-бромпропокси)бензонитрила (20 г; 84 ммоль; смотри стадию (а), описанную выше) и фталимида калия (15,5 г; 84 ммоль) в ДМФ (120 мл) перемешивали при 95°С в течение 4 ч. Раствор затем концентрировали под вакуумом и полученный остаток растворяли в ДХМ и промывали водой. Органический слой отделяли, высушивали (Na2SO4) и концентрировали под вакуумом, получая 25,5 г (99%) указанного в подзаголовке соединения.
(в) 4-(3-Аминопропокси)бензонитрил
Смесь 4-[3-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)пропокси]-бензонитрила (25,5 г; 83 ммоль; со стадии (б), описанной выше) и гидразина гидрата (4,15 г; 83 ммоль) в метаноле (100 мл) кипятили с обратным холодильником в течение 1 ч перед тем, как добавить воду (120 мл). Метанол выпаривали при пониженном давлении и добавляли концентрированную соляную кислоту (120 мл). Полученную смесь нагревали на паровой бане в течение 1,5 ч и затем охлаждали в холодильнике в течение ночи. Полученный осадок отфильтровывали и фильтрат концентрировали под вакуумом. К полученному остатку добавляли воду и раствор подщелачивали. Водный раствор экстрагировали ДХМ, при этом органический слой затем отделяли, высушивали и концентрировали под вакуумом, получая 6 г (41%) указанного в подзаголовке соединения.
(г) 7-Бензил-3,7-диазабицикло[3.3.1]нонан-3-этанол
Соединение было получено с 72%-ным выходом путем осуществления взаимодействия 3-бензил-3,7-диазабицикло[3.3.1]нонана (смотри Пример Д, описанный выше) с 2-бромэтанолом.
(д) 3,7-Диазабицикло[3.3.1]нонан-3-этанол
Указанное в подзаголовке соединение было получено в соответствии с методикой, описанной выше в Примере 14(б), с использованием 7-бензил-3,7-диазабицикло[3.3.1]нонан-3-этанола (со стадии (г), описанной выше) вместо 3-бензил-7-[3-(2-пропил-1,3-диоксолан-2-ил)пропил]-3,7-диазабицикло[3.3.1]-нонана.
(е) N-[3-(4-Цианофенокси)пропил]-7-(2-гидроксиэтил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Указанное в заголовке соединение было получено с 11%-ным выходом в соответствии с методикой, описанной выше в Примере 14(в), с использованием 3,7-диазабицикло[3.3.1]нонан-3-этанола (со стадии (д), описанной выше) и 4-(3-аминопропокси)бензонитрила (со стадии (в), описанной выше) вместо 3-[3-(2-пропил-1,3-диоксолан-2-ил)пропил]-3,7-диазабицикло[3.3.1]нонана и 4-(2-аминоэтил)бензонитрила, соответственно.
13С ЯМР (CDCl3): δ 162.04, 158.99, 133.66, 118.99, 115.03, 103.35, 66.55, 60.24, 57.87, 57.18, 50.02, 48.63, 37.93, 31.81, 29.26, 28.96.
Пример 19
N-{[7-[3-(4-Цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нон-3-ил]карбонил}-4-метилбензолсульфонамид
Раствор 4-[3-(3,7-диазабицикло[3.3.1]нон-3-ил)-2-гидроксипропокси]-бензонитрил (200 мг; 0,66 ммоль; смотри Пример Ж, описанный выше) в хлороформе (20 мл) обрабатывали раствором пара-толуолсульфонилизоцианата (110 мкл 96%-ной степени чистоты; 0,136 г; 0,69 ммоль) в хлороформе (4 мл), добавляя его по каплям. Немедленно образовался осадок белого цвета, и смесь затем концентрировали под вакуумом. Полученный таким образом неочищенный продукт подвергали хроматографии на силикагеле, элюируя смесью гексан:этилацетат:метанольный аммиак (75:75:50), с получением указанного в заголовке соединения с 53%-ным выходом.
13С ЯМР (CDCl3): δ 15.77, 29.18, 32.37, 36.13, 48.72, 52.27, 56.32, 109.83, 113.13, 118.27, 118.93, 120.10, 127.80, 131.39, 132.46, 132.73, 134.62, 138.75, 159.14, 167.09.
Пример 20
N-Аллил-7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь аллиламина (125 мкл; 1,66 ммоль) и 1,1'-карбонилдиимидазола (269 мг; 1,66 ммоль) в ТГФ (10 мл) перемешивали при комнатной температуре в течение 40 мин. Смесь затем обрабатывали раствором 4-[3-(3,7-диазабицикло[3.3.1]нон-3-ил)-2-гидроксипропокси]бензонитрила (смотри Пример Ж, описанный выше) в ТГФ (5 мл) и продолжали перемешивание в течение ночи. Смесь концентрировали под вакуумом и полученный остаток очищали хроматографией на силикагеле, элюируя смесью гексан:метанольный аммиак (1:1), с получением указанного в заголовке соединения с 57%-ным выходом.
13С ЯМР (MeOD): δ 29.37, 30.79, 41.95, 42.91, 58.91, 59.55, 61.12, 66.52, 70.75, 103.31, 113.81, 115.39, 118.72, 133.73, 135.57, 136.06, 158.93, 162.67.
Пример 21
7-[3-(4-Цианофенокси)-2-гидроксипропил]-N-[2-(2-тиенил)этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Указанное в заголовке соединение было получено с 83%-ным выходом в соответствии с методикой, описанной выше в Примере 19, с использованием 2-(2-изоцианатоэтил)тиофена вместо пара-толуолсульфонилизоцианата.
13С ЯМР (CDCl3): δ 29.19, 29.50, 30.59, 32.11, 42.26, 47.94, 49.37, 56.23, 60.47, 61.95, 65.32, 70.74, 103.88, 115.36, 119.52, 123.69, 125.25, 127.04, 133.90, 142.19, 158.74, 162.22.
Пример 22
7-[3-(4-Цианофенокси)-2-гидроксипропил]-N-[3-(этиламино)-3-оксопропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) Этил-3-[({7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нон-3-ил}карбонил)амино]пропаноат
Указанное в подзаголовке соединение было получено с 90%-ным выходом в соответствии с методикой, описанной выше в Примере 12(а), с использованием этил-3-изоцианатопропаноата вместо этил-2-изоцианатоацетата.
(б) 7-[3-(4-Цианофенокси)-2-гидроксипропил]-N-[3-(этиламино)-3-оксопропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Указанное в заголовке соединение было получено с 22%-ным выходом в соответствии с методикой, описанной выше в Примере 12(б), с использованием 3-[({7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нон-3-ил}карбонил)амино]пропаноата (со стадии (а), описанной выше) и этиламина вместо 2-[({7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло-[3.3.1]нон-3-ил}карбонил)амино]ацетата и пропиламина, соответственно.
13С ЯМР (CDCl3): δ 172.46, 162.17, 158.89, 133.96, 119.14, 115.37, 104,16, 65.27, 61.73, 60.58, 56.97, 49.23, 47.89, 37.51, 36.60, 34.26, 32.00, 29.54, 29.16, 14.87.
Пример 23
N-(1-Цианоэтил)-7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 2-Аминопропаннитрил
Лактонитрил (28 г; 375 ммоль) добавляли к жидкому аммиаку при -78°С в реакционной пробирке. Пробирку герметизировали и смесь перемешивали в течение ночи при комнатной температуре. Аммиак удаляли выпариванием и неочищенный материал использовали непосредственно на следующей стадии без дальнейшей очистки.
(б) N-(1-Цианоэтил)-7-[3-(4-цианофенокси)-2-гидроксипропил]-3.7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь 2-аминопропаннитрила (250 мг; 3,58 ммоль; со стадии (а), описанной выше) и N-этил-ди-изо-пропиламина (0,67 мл; 0,50 г; 3,84 ммоль) в ДХМ (9 мл) добавляли (с помощью шприцевого насоса) к раствору трифосгена (352 мг; 1,19 ммоль) в ДХМ (7 мл) в течение 1 часа. Полученную смесь перемешивали в течение 1 ч при комнатной температуре перед тем, как добавить смесь 4-[3-(3,7-диазабицикло[3.3.1]нон-3-ил)-2-гидроксипропокси]-бензонитрила (1,08 г; 3,58 ммоль; смотри Пример Ж, описанный выше) и N-этил-ди-изо-пропиламина (0,67 мл; 0,50 г; 3,84 ммоль) в ДХМ (14 мл). Перемешивание продолжали в течение следующих 20 мин перед тем, как сконцентрировать раствор под вакуумом и полученный остаток очистить флэш-хроматографией, элюируя смесью дихлорметан:метанол (95:5), с получением указанного в заголовке соединения с 65%-ным выходом.
13С ЯМР (CDCl3): δ 20.02, 20.16, 29.11, 29.32, 29.46, 31.91, 37.83, 37.89, 48.23, 48.47, 49.36, 49.61, 56.95, 60.26, 60.51, 61.58, 62.077, 65.43, 70.69, 104.06, 115.40, 119.27, 120.77, 133.96, 157.08, 162.21.
Пример 24
7-[3-(4-Цианофенокси)-2-гидроксипропил]-N-(2,2,2-трифторэтил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Указанное в заголовке соединение было получено с 46%-ным выходом в соответствии с методикой, описанной выше в Примере 23(б), с использованием 2,2,2-трифторэтиламина вместо 2-аминопропаннитрила.
13С ЯМР (CDCl3): δ 29.11, 29.42, 31.79, 42.17, 42.51, 48.36, 49.58, 57.09, 60.45, 61.77, 65.39, 70.76, 104.08, 115.39, 119.23, 123.28, 126.05, 133.93, 157.76, 162.21.
Пример 25
7-[3-(4-Цианофенокси)-2-гидроксипропил]-N-(2-оксо-2-(1-пиперидинил)этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Указанное в заголовке соединение было получено с 49%-ным выходом в соответствии с методикой, описанной выше в Примере 12(б), с использованием пиперидина вместо пропиламина.
13С ЯМР (CDCl3): δ 24.33, 25.41, 26.06, 28.74, 29.29, 29.44, 32.13, 42.67, 43.10, 45.30, 47.99, 48.09, 49.14, 49.28, 57.18, 60.42, 61.90, 65.55, 70.77, 94.22, 103.89, 115.24, 115.43, 119.24, 133.74, 134.02, 158.49, 162.20, 167.42.
Пример 26
N-(1,3-Бензодиоксол-5-ил)-7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Указанное в заголовке соединение было получено с 33%-ным выходом в соответствии с методикой, описанной выше в Примере 23(б), с использованием 1,3-бензодиоксол-5-амина вместо 2-аминопропаннитрила.
13С ЯМР (CDCl3): δ 162.22, 156.51, 147.47, 143.20, 133.98, 133.83, 119.41, 115.40, 113.68, 107.68, 103.83, 103.59, 100.96, 70.70, 65.98, 61.34, 60.34, 57.87, 49.17, 48.13, 31.52, 29.41, 29.11.
Пример 27
7-[3-(4-Цианоанилино)пропил]-N-(2-оксо-2-(пропиламино)этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 4-[(3-Гидроксипропил)амино]бензонитрил
Смесь 4-фторбензонитрила (12,0 г; 99,1 ммоль) и З-амино-1-пропанола (59,6 г; 793 ммоль) перемешивали при 80°С в атмосфере инертного газа в течение 3 часов перед тем, как добавить воду (150 мл). Смесь оставляли нагреваться до комнатной температуры и затем экстрагировали диэтиловым эфиром. Органический слой отделяли, высушивали (Na2SO4), фильтровали и концентрировали под вакуумом, получая 17 г (97%) указанного в заголовке соединения в виде масла, которое кристаллизовалось в процессе стояния.
(б) 3-(4-Цианоанилино)пропил]-4-метилбензолсульфонат
Охлажденный (0°С) раствор 4-[(3-гидроксипропил)амино]бензонитрила (17 г; 96,5 ммоль; со стадии (а), описанной выше) в безводном MeCN (195 мл) обрабатывали триэтиламином (9,8 г; 96,5 ммоль) и затем пара-толуолсульфонилхлоридом (20,2 г; 106 ммоль). Смесь перемешивали при 0°С в течение 90 минут перед тем, как сконцентрировать под вакуумом. К остатку добавляли воду (200 мл) и водный раствор экстрагировали ДХМ. Органическую фазу высушивали (Na2SO4), фильтровали и концентрировали под вакуумом. Полученный остаток очищали кристаллизацией из изо-пропанола, получая 24,6 г (77%) указанного в подзаголовке соединения.
(в) Этил-2-{[(7-бензил-3,7-диазабицикло[3.3.1]нон-3-ил)-карбонил]амино}ацетат
Указанное в подзаголовке соединение было получено с 99%-ным выходом в соответствии с методикой, описанной выше в Примере 5(а), с использованием 4-[3-(3,7-диазабицикло[3.3.1]нон-3-ил)-2-гидроксипропокси]-бензонитрила (смотри Пример Ж, описанный выше) и этил-2-изоцианатоацетата вместо 3-бензил-3,7-диазабицикло[3.3.1]нонана и изо-пропилизоцианата, соответственно.
(г) 7-Бензил-N-[2-оксо-2-(пропиламино)этил]-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид
Указанное в подзаголовке соединение было получено с 88%-ным выходом в соответствии с методикой, описанной выше в Примере 12(б), с использованием этил-2-{[(7-бензил-3,7-диазабицикло[3.3.1]нон-3-ил)-карбонил]амино}ацетата (со стадии (в), описанной выше) вместо этил-2-[({7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нон-3-ил}карбонил)амино]ацетата.
(д) N-[2-Оксо-2-(пропиламино)этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Указанное в заголовке соединение было получено в соответствии с методикой, описанной выше в Примере 5(б), с использованием 7-бензил-N-[2-оксо-2-(пропиламино)этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (со стадии (г), описанной выше) вместо 7-бензил-N-изо-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида.
(е) 7-[3-(4-Цианоанилино)пропил]-N-[2-оксо-2-(пропиламино)этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь N-[2-оксо-2-(пропиламино)этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (3,35 г; 12,5 ммоль; со стадии (д), описанной выше), К2СО3 (6,9 г; 50 ммоль) и иодида натрия (0,19 г; 1,25 ммоль) в ацетонитриле (600 мл) обрабатывали 3-(4-цианоанилино)пропил-4-метилбензолсульфонатом (4,2 г; 12,7 ммоль; со стадии (б), описанной выше) и перемешивали при температуре дефлегмации в течение 5 ч, с последующим перемешиванием в течение 21 ч при комнатной температуре. Смесь фильтровали, концентрировали под вакуумом и полученный таким образом неочищенный продукт разбавляли водой. Водный раствор экстрагировали ДХМ, при этом органический слой отделяли, высушивали и концентрировали под вакуумом. Полученный таким образом неочищенный продукт очищали хроматографией на силикагеле, элюируя смесью ДХМ:МеОН (95:5), с получением 3,08 г (58%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 11.49, 22.85, 25.11, 29.09, 31.03, 40.78, 41.40, 44.80, 48.41, 56.22, 59.32, 97.43, 111.99, 120.97, 133.74, 151.98, 157.92, 170.37.
Пример 28
7-{2-[2-(4-Цианофенокси)этокси]этил}-N-этил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид
(а) 4-[2-(2-Гидроксиэтокси)этокси]бензонитрил
Смесь пара-цианофенола (11,9 г; 100 ммоль), К2СО3 (15 г; 110 ммоль) и хлорэтоксиэтанола (12,4 г; 100 ммоль) в СН3СN кипятили с обратным холодильником в течение 24 часов, затем перемешивали при комнатной температуре в течение следующих двух суток. Реакционную смесь отфильтровывали и концентрировали под вакуумом, получая неочищенный продукт, который очищали хроматографией на силикагеле (элюент гексан:этилацетат (1:1)). Это приводило к получению 10 г (50%) указанного в подзаголовке соединения.
(б) 2-[2-(4-Цианофенокси)этокси]этил-метансульфонат
Метансульфонилхлорид (3,0 г; 26 ммоль) добавляли по каплям к охлажденной (-5°С) смеси 4-[2-(2-гидроксиэтокси)этокси]бензонитрила (5,0 г; 24 ммоль, со стадии (а), описанной выше) и триэтиламина (4 мл; 2,9 г; 29 ммоль) в ДХМ (50 мл). По окончании добавления реакционную смесь оставляли нагреваться до комнатной температуры в течение периода времени, составляющего 2 часа. Затем реакционную смесь дважды промывали водой, органический слой отделяли, высушивали (Na2CO3) и концентрировали под вакуумом, получая 7 г (100%) указанного в подзаголовке соединения.
(в) 7-{2-[2-(4-Цианофенокси)этокси]этил}-N-этил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид
Смесь 2-[2-(4-цианофенокси)этокси]этил-метансульфоната (2 г; 7,0 ммоль, со стадии (б), описанной выше), N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (1,4 г; 7,0 ммоль, смотри Пример 6(б), описанный выше) и К2СО3 (1,5 г; 10,5 ммоль) в MeCN (50 мл) перемешивали при температуре дефлегмации в течение ночи. Реакционную смесь затем концентрировали под вакуумом и полученный остаток очищали флэш-хроматографией на силикагеле, элюируя смесью дихлорметан:метанол (9:1), с получением 0,8 г (30%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 162.18, 133.92, 119.24, 115.34, 103.92, 69.07, 67.86, 59.52, 58.42, 48.18, 35.68, 30.25, 28.81, 15.69.
Пример 29
7[4-(4-Цианофенил)-4-(3,4-диметоксифенокси)бутил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 4-[1-(3,4-Диметоксифенокси)-3-бутенил]бензонитрил Охлажденную (0°С) смесь 4-(1-гидрокси-3-бутенил)бензонитрила (14,6 г; 84,3 ммоль) и 3,4-диметоксифенола (19,5 г; 125,4 ммоль) в толуоле (500 мл) обрабатывали трибутилфосфином (32,14 мл 97%-ной степени чистоты; 25,6 г; 126,4 ммоль) и затем 1,1'-(азодикарбонил)дипиперидином (31,8 г; 126,4 ммоль). По окончании добавления реакционная смесь сгущалась, и температура поднималась до 15°С. Добавляли дополнительное количество толуола (500 мл) и смесь перемешивали при комнатной температуре в течение ночи. Затем осадок оксида трибутилфосфина удаляли фильтрованием и фильтрат концентрировали под вакуумом, получая 65,8 г неочищенного продукта. Этот продукт очищали хроматографией на силикагеле, элюируя смесью толуол:метанол (98:2) с получением 17,9 г указанного в подзаголовке соединения.
(б) 4-[1-(3,4-Диметоксифенокси)-4-гидроксибутил]бензонитрил Боран-метил-сульфидный комплекс (2 М в эфире; 11 мл; 22 ммоль) добавляли по каплям в течение 15 минут к охлажденному (-5°С) раствору 4-[1-(3,4-диметоксифенокси)-3-бутенил]бензонитрила (17,6 г; 56,8 ммоль; со стадии (а), описанной выше) в безводном ТГФ (15 мл) (во время этого периода времени температура реакционной смеси повышалась до 0°С). Полученную смесь перемешивали при температуре между 0 и 10°С в течение 1,5 ч перед тем, как оставить нагреваться до комнатной температуры. Перемешивание при этой температуре продолжали в течение следующих 3,5 ч перед тем, как добавить воду (22 мл) и тетрагидрат пербората натрия (11 г; 66 ммоль). Двухфазную смесь перемешивали при комнатной температуре в течение 2 ч перед тем, как водный слой отделить и экстрагировать эфиром. Объединенные органические слои промывали соляным раствором, высушивали и концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле, элюируя смесью изо-пропанол:этилацетат:гептан (5:25:70), с получением 14,5 г (77%) указанного в подзаголовке соединения.
(в) 4-(4-Цианофенил)-4-(3,4-диметоксифенокси)бутил-метансульфонат
Раствор метансульфонилхлорида (3,4 мл; 5,0 г; 44 ммоль) в ДХМ (15 мл) медленно добавляли к охлажденной (-5°С) смеси 4-[1-(3,4-диметоксифенокси)-4-гидроксибутил]бензонитрила (11 г; 34 ммоль; со стадии (б), описанной выше) и триэтиламина (7 мл; 5,2 г; 50,6 ммоль) в ДХМ (50 мл), причем в процессе добавления температура не поднималась выше 2°С. Перемешивание продолжали при температуре между 0 и 5°С в течение следующих 2 ч перед тем, как добавить воду. Полученный органический слой отделяли и промывали водой, отделяли еще раз и затем высушивали, получая указанное в подзаголовке соединение со 100%-ным выходом.
(г) трет-Бутил-7-[4-(4-цианофенил)-4-(3,4-диметоксифенокси)бутил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат
Смесь 4-(4-цианофенил)-4-(3,4-диметоксифенокси)бутилметансульфоната (522 мг; 1,29 ммоль; со стадии (в), описанной выше), трет-бутил-3,7-диазабицикло[3.3.1]нонан-3-карбоксилата (307 мг; 1,356 ммоль; смотри Пример Е, описанный выше) и К2СО3 (216 мг; 1,56 ммоль) в смеси хлороформ:ацетонитрил (10 мл смеси в соотношении 1:1) перемешивали при 70°С в течение 23 ч. Реакционную смесь фильтровали и фильтрат концентрировали под вакуумом, получая 708 мг неочищенного продукта. Этот продукт очищали флэш-хроматографией, элюируя градиентом смеси толуол:метанол (от 97:3 до 10:1), с получением 607 мг (88%) указанного в подзаголовке соединения.
(д) 4-[4-(3,7-Диазабицикло[3.3.1]нон-3-ил)-1-(3,4-диметоксифенокси)-бутил]бензонитрил
Охлажденный (0°С) раствор трет-бутил-7-[4-(4-цианофенил)-4-(3,4-диметоксифенокси)бутил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксилата (1,92 г; 3,6 ммоль; со стадии (г), описанной выше) в этилацетате (20 мл) обрабатывали этилацетатом, насыщенным HCl (30 мл). Полученную смесь перемешивали в течение 2 ч при температуре между 0 и 5°С перед тем, как сконцентрировать под вакуумом. Полученный остаток растворяли в ацетонитриле (50 мл) и обрабатывали К2СО3 (3,5 г; 25,2 ммоль) и водой (2,25 мл). Эту смесь перемешивали в течение 3 ч при комнатной температуре и твердые частицы удаляли фильтрованием перед тем, как растворитель удалить под вакуумом (с добавлением толуола для осуществления азеотропного удаления воды), с получением 1,5 г указанного в подзаголовке соединения.
(е) 7-[4-(4-Цианофенил)-4-(3,4-диметоксифенокси)бутил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Раствор 4-[4-(3,7-диазабицикло[3.3.1]нон-3-ил)-1-(3,4-диметоксифенокси)-бутил]бензонитрила (109 мг; 0,25 ммоль; со стадии (д), описанной выше) в СНСl3 (1,43 мл) обрабатывали раствором этилизоцианата (18,6 мкл; 16,8 мг; 0,237 ммоль) в MeCN (0,5 мл). Полученную смесь перемешивали в течение 30 ч при комнатной температуре. Раствор затем наносили на ионообменную твердофазную экстракционную набивку (SiO2; 0,5 г от ISOLUTE). Набивку промывали СНСl3 (2,5 мл) и затем продукт элюировали MeCN (3×2,5 мл). В результате получили указанное в заголовке соединение (93 мг; 73%) со степенью чистоты более чем 90% (как определено по данным ВЭЖХ:УФ при 254 нм и с помощью ELS-детектирования).
МС (ES) m/z=507 (М+1)+, 505 (М-1)-.
Пример 30
7-(3-{4-Циано-2-[(циклопропиламино)карбонил]фенокси}-2-гидроксипропил)-N-фенил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 5-Циано-N-циклопропил-2-[2-оксиранилметокси]бензамид
Указанное в подзаголовке соединение было получено в соответствии со способом, описанным выше в Примере 7(б), с использованием 2-оксиранилметил-3-нитробензолсульфоната (полученный аналогично способу, описанному выше в Примере Б).
(б) 7-Бензил-N-фенил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Охлажденный (0°С) раствор 3-бензил-3,7-диазабицикло[3.3.1]нонана (10 г; 46 ммоль; смотри Пример Д, описанный выше) в ДХМ (100 мл) обрабатывали фенилизоцианатом (4,9 мл; 45 ммоль). Смесь перемешивали при комнатной температуре в течение 30 мин. Продукт образовался в виде кристаллов белого цвета, которые собирали фильтрованием, получая 10 г (66%) указанного в подзаголовке соединения.
(в) N-Фенил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Раствор 7-бензил-N-фенил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (10 г; 29,8 ммоль; со стадии (б), описанной выше) в этаноле (100 мл) подвергали гидрированию над 10%-ным Pd/C и при атмосферном давлении в течение ночи. Катализатор удаляли посредством набивки Целита® и остаток концентрировали под вакуумом, получая указанное в подзаголовке соединение с количественным выходом.
(г) 7-(3-{4-Циано-2-[(циклопропиламино)карбонил]фенокси}-2-гидроксипропил)-N-фенил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь 5-циано-N-циклопропил-2-[2-оксиранилметокси]бензамида (0,8 г; 3,1 ммоль; со стадии (а), описанной выше) и N-фенил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (0,9 г; 3,6 ммоль; со стадии (в), описанной выше) в смеси изо-пропанол:Н2O (10 мл смеси в соотношении 9:1) кипятили с обратным холодильником в течение 180 мин перед тем, как добавить дихлорметан и растворитель удалить под вакуумом. Очистка полученного остатка флэш-хроматографией с элюированием смесью ДХМ:МеОН (9:1) давала 1 г (64%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 6.33, 6.56, 23.23, 29.18, 29.51, 31.66, 48.27, 49.60, 53.44, 57.94, 60.51, 65.74, 71.28, 104.93, 113.46, 118.45, 119.54, 119.65, 122.88, 123.27, 128.84, 136.07, 156.44, 159.69, 164.53.
Пример 31
N-(4-Цианофенил)-7-[3-(этансульфонил)пропил]-3,7-диазабицикло[3.3.1]-нонан-3-карбоксамид
(а) 3-(Этансульфонил)пропил-4-метилбензолсульфонат
Триэтиламин (13,36 г; 132 ммоль) добавляли по каплям к смеси 3-(этансульфонил)-1-пропанола (13,4 г; 88 ммоль; Martin-Smith et al., J. Pharm. Pharmacol., 19 (1967) 649) и пара-толуолсульфонилхлорида (16,78 г; 88 ммоль) в ДХМ (150 мл), что приводило к умеренно-экзотермической реакции. По окончании добавления реакционную смесь дважды промывали водным раствором хлорида аммония, органический слой затем отделяли, высушивали и концентрировали под вакуумом. Полученный остаток перекристаллизовывали из смеси диэтиловый эфир:ДХМ, получая 17,9 г (65%) указанного в подзаголовке соединения.
(б) трет-Бутил-7-[(4-цианоанилино)карбонил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат
Суспензию трет-бутил-3,7-диазабицикло[3.3.1]нонан-3-карбоксилата (2,0 г; 8,8 ммоль; смотри Пример Е, описанный выше) в хлороформе (15 мл) обрабатывали 4-изоцианатбензонитрилом (1,53 г; 10,6 ммоль) Смесь перемешивали при комнатной температуре в течение 1,5 ч, причем в течение этого времени в смеси наблюдалось некоторое количество твердых частиц. С целью растворения этих частиц добавляли дополнительные 10 мл хлороформа. Масс-спектроскопический анализ смеси показал, что исходные материалы израсходованы, и ввиду этого растворитель удаляли под вакуумом. Полученный остаток очищали флэш-хроматографией, элюируя градиентом смеси ДХМ:МеСN (от 5:1 до 2:1), с получением 2,31 г (71%) указанного в подзаголовке соединения.
(в) N-(4-Цианофенил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Охлажденный (0°С) раствор трет-бутил-7-[(4-цианоанилино)карбонил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксилата (2,2 г; 5,94 ммоль; со стадии (б), описанной выше) в этилацетате (40 мл) обрабатывали в течение 30 мин этилацетатом, насыщенным HCl (65 мл). Полученную смесь перемешивали при комнатной температуре в течение следующих 4 ч перед тем, как сконцентрировать под вакуумом, получая 1,8 г (99%) соли гидрохлорида указанного в подзаголовке соединения.
(г) N-(4-Цианофенил)-7-[3-(этансульфонил)пропил]-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид
Смесь N-(4-цианофенил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (67,6 мг; 0,25 ммоль; со стадии (в), описанной выше) и К2СО3 (80 мг; 0,57 ммоль) в ДМФ (0,5 мл) обрабатывали раствором 3-(этансульфонил)пропил-4-метилбензолсульфоната (153 мг; 0,50 ммоль; со стадии (а), описанной выше) в MeCN (1,0 мл). Полученную суспензию перемешивали в течение 5 дней при 50°С перед тем, как охладить и отфильтровать. Фильтрат затем добавляли к ионнообменной твердофазной экстракционной набивке (СВА; 2 г; от ISOLUTE). Через 1 ч набивку промывали СНСl3 (3×2,5 мл) и продукт элюировали смесью СНСl3:МеОН:Еt3N (8:1:1), получая указанное в заголовке соединение (63,6 мг; 63%) со степенью чистоты более 90% (как определено по данным ВЭЖХ: УФ при 254 нм и с помощью ELS-детектирования).
МС (ES): m/z=405 (М+1)+, m/z=403 (М-1)-.
Пример 32
7-{3-[(2-Циано-1H-индол-4-ил)окси]-2-гидроксипропил}-N-фенил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь 4-(2-оксиранилметокси)-1Н-индол-2-карбонитрила (1,0 г; 4,7 ммоль; Pitha et al., J. Med. Chem., 30 (1987) 612) и N-фенил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамида (1,4 г; 5,5 ммоль; смотри Пример 30(г), описанный выше) в смеси изо-пропанол:Н2O (10 мл смеси в соотношении 9:1) перемешивали при температуре дефлегмации в течение 3 ч перед тем, как сконцентрировать под вакуумом. Полученный остаток очищали колоночной хроматографией на силикагеле, элюируя градиентом смеси ДХМ:МеОН (от 99:1 до 97:3), получая 0,8 г (37%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 29.03, 29.39, 31.27, 48.37, 49.31, 57.89, 60.42, 61.41, 66.07, 70.04, 100.72, 104.39, 105.13, 111.31, 114.95, 117.66, 120.18, 120.30, 123.00, 126.54, 128.84, 138.39, 139.16, 152.55, 156.29.
Пример 33
7-[(7-Циано-2,3-дигидро-1,4-бенздиоксин-2-ил)метил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 5-Бром-2-(3-хлор-2-гидроксипропокси)бензальдегид
Смесь 5-бром-2-гидроксибензальдегида (20,1 г; 0,1 моль), эпихлоргидрина (25 мл; 0,32 моль) и 6 капель пиперидина перемешивали при температуре дефлегмации в течение 6 ч перед тем, как сконцентрировать под вакуумом. Полученный остаток растворяли в хлороформе (25 мл) и обрабатывали концентрированной HCl (10 мл). Полученную смесь перемешивали в течение 3 ч при комнатной температуре перед тем, как органический слой промыть водой, отделить, высушить и сконцентрировать под вакуумом, получая 28,2 г (96%) указанного в подзаголовке соединения. Это соединение непосредственно использовали на следующей стадии без дальнейшей очистки.
(б) 5-Бром-2-(3-хлор-2-гидроксипропокси)фенилформиат
Раствор 5-бром-2-(3-хлор-2-гидроксипропокси)бензальдегида (28,2 г; 96 ммоль; со стадии (а), описанной выше) в ДХМ (200 мл) обрабатывали 3-хлорпероксибензойной кислотой (25 г; 70-75%-ной степени чистоты; приблизительно 100 ммоль). В результате экзотермической реакции смесь нагревалась до температуры дефлегмации в течение 20 мин. Перемешивание продолжали в течение следующих 3 дней перед тем, как смесь отфильтровать (для удаления выпавшей в осадок 3-хлорбензойной кислоты). Фильтрат промывали раствором К2СО3 и водой, высушивали и концентрировали под вакуумом, получая 26,1 г указанного в подзаголовке соединения. Это соединение непосредственно использовали на следующей стадии без какой-либо дальнейшей очистки.
(в) (7-Бром-2,3-дигидро-1,4-бенздиоксин-2-ил)метанол
Раствор 5-бром-2-(3-хлор-2-гидроксипропокси)фенилформиата (26,1 г; 84 ммоль; со стадии (б), описанной выше) в этаноле (100 мл) обрабатывали раствором гидроксида калия (6,1 г; 85%-ной степени чистоты; приблизительно 92 ммоль) в воде (10 мл). Полученную смесь кипятили с обратным холодильником в течение 1,5 ч перед тем, как профильтровать и сконцентрировать под вакуумом. Полученный остаток растворяли в этилацетате и промывали соляным раствором. Органический слой отделяли, высушивали и концентрировали под вакуумом, получая 28,8 г неочищенного продукта. Этот продукт очищали колоночной хроматографией на силикагеле, элюируя смесью диэтиловый эфир:гексан (70:30), с получением 10,0 г (49,1%) указанного в подзаголовке соединения.
(г) 3-(Гидроксиметил)-2,3-дигидро-1,4-бенздиоксин-6-карбонитрил
Смесь (7-бром-2,3-дигидро-1,4-бенздиоксин-2-ил)метанола (10,0 г; 41,2 ммоль; со стадии (в), описанной выше) и CuCN (4,0 г; 45,3 ммоль) в ДМФ (10 мл; высушен над молекулярными ситами) перемешивали при 170°С в течение 4,5 ч. Реакционную смесь выливали в теплый водный раствор цианида натрия (8,10 г; 165 ммоль NaCN в 25 мл H2O). Полученную смесь экстрагировали толуолом и ДХМ. Объединенные органические слои промывали водой и затем соляным раствором, высушивали и концентрировали под вакуумом. Полученный в результате этого остаток кристаллизовали из толуола и ДХМ, получая 2,8 г (35%) указанного в подзаголовке соединения.
(д) (7-Циано-2,3-дигидро-1,4-бенздиоксин-2-ил)метилметансульфонат
Раствор метансульфонилхлорида (1,81 г; 15,8 ммоль) в дихлорметане (5 мл) добавляли по каплям к охлажденной (0°С) смеси 3-(гидроксиметил)-2,3-дигидро-1,4-бенздиоксин-6-карбонитрила (2,75 г; 14,4 ммоль; со стадии (г), описанной выше) и пиридина (1,26 г; 16 ммоль) в ДХМ (25 мл). По окончании добавления смесь перемешивали при 0°С в течение 1 ч и затем при комнатной температуре в течение ночи. Анализ ТСХ показал на незавершенность реакции по истечении этого промежутка времени, поэтому добавляли дополнительные порции метансульфонилхлорида (0,4 г; 3,5 ммоль) и пиридина (0,5 мл; 0,49 г; 6,2 ммоль). Смесь кипятили с обратным холодильником в течение 3,5 ч, дважды промывали насыщенным раствором Na2CO3, высушивали и концентрировали под вакуумом. Полученный в результате этого неочищенный продукт (4,5 г) очищали флэш-хроматографией, элюируя ДХМ, с получением 3,5 г указанного в подзаголовке соединения, которое кристаллизовалось в процессе стояния.
(е) 7-[(7-Циано-2,3-дигидро-1,4-бенздиоксан-2-ил)метил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь (7-циано-2,3-дигидро-1,4-бенздиоксин-2-ил)метилметансульфоната (150 мг; 0,9 ммоль; со стадии (д), описанной выше), N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (186 мг; 0,94 ммоль; смотри Пример 6(б), описанный выше), К2СО3 (265 мг; 2,0 ммоль) и Nal (14 мг; 0,09 ммоль) в СН3СN кипятили с обратным холодильником в течение 20 ч. Растворитель удаляли под вакуумом и полученный остаток обрабатывали ДХМ и водой. Органический слой отделяли, высушивали (Na2SO4) и концентрировали под вакуумом. Полученный остаток очищали флэш-хроматографией, элюируя смесью ДХМ:МеОН (95:5), с получением 113,2 мг (34%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 15.61, 29.19, 30.72, 35.72, 47.78, 58.34, 59.02, 60.64, 67.01, 71.38, 71.49, 71.60, 104.10, 120.76, 120.89, 125.39, 125.79, 143.50, 147.80, 157.46.
Пример 34
7-{[(2S)-6-Циано-4-(метансульфонил)-3,4-дигидро-2Н-1,4-бензоксазин-2-ил]метил}-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) (2R)-2-(Гидроксиметил)-3,4-дигидро-2H-1,4-бензоксазин-6-карбонитрил
Смесь 3-амино-4-гидроксибензонитрила (25 г; 0,186 моль) и S-эпихлоргидрина (10,7 г; 0,22 моль) в водном этаноле (500 мл; 99%-ном) перемешивали при 60°С в течение 24 ч. Смесь концентрировали под вакуумом перед тем, как добавить этанол (500 мл) и затем К2СО3 (27 г; 0,195 моль). Полученную смесь кипятили с обратным холодильником в течение 1 ч перед тем, как отфильтровать. Фильтрат концентрировали под вакуумом, получая 61 г масла черного цвета. Это масло разбавляли водой (500 мл) и затем дважды экстрагировали ДХМ и этилацетатом. Объединенные органические экстракты высушивали и концентрировали под вакуумом, получая 20 г (57%) указанного в подзаголовке соединения в виде кристаллов желтого цвета.
(б) (2R)-6-Циано-4-(метансульфонил)-3,4-дигидро-2H-1,4-бензоксазин-2-ил]метилметансульфонат
Метансульфонилхлорид (45 г; 0,395 моль) добавляли по каплям к охлажденной (0°С) смеси (2R)-2-(гидроксиметил)-3,4-дигидро-2Н-1,4-бензоксазин-6-карбонитрила (30 г; 0,158 моль; со стадии (а), описанной выше) и пиридина (200 мл; избыток). Смесь перемешивали при комнатной температуре в течение ночи перед тем, как сконцентрировать под вакуумом. Полученный остаток обрабатывали водой и кристаллы продукта отделяли фильтрованием. Эти кристаллы подвергали перекристаллизации из МеСN, получая 29 г чистого материала. Маточную жидкость концентрировали под вакуумом, получая остаток, который кристаллизовали из хлороформа, с получением дополнительной порции (7,5 г) продукта. Общий выход указанного в подзаголовке соединения составил 36,5 г (67%).
(в) 7-{[(2S)-6-Циано-4-(метансульфонил)-3,4-дигидро-2Н-1,4-бензоксазин-2-ил]метил}-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Раствор (2R)-6-циано-4-(метансульфонил)-3,4-дигидро-2H-1,4-бензоксазин-2-ил]метилметансульфоната (1 г; 2,89 ммоль; со стадии (б), описанной выше) в MeCN (5 мл) обрабатывали триэтиламином (8 мл; 5,8 г; 57,4 ммоль), затем N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамидом (0,85 г; 4,33 ммоль; смотри Пример 6(б), описанный выше). Полученную смесь перемешивали при 70°С в течение 5 ч и затем при комнатной температуре в течение ночи. Смесь концентрировали под вакуумом и очищали кислотно/щелочной экстракцией с последующей флэш-хроматографией, элюируя смесью ДХМ:МеОН, с получением 100 мг (14%) указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 15.63, 28.87, 29.09, 30.48, 35.73, 39.50, 45.96, 47.65, 48.11, 59.03, 59.19, 60.59, 73.40, 104.15, 118.72, 119.90, 124.92, 126.51, 128.92, 150.04, 157.74.
Пример 35
7-[2-({2-[4,5-Бис(4-цианофенил)-1Н-пиразол-1-ил]ацетил}амино)этил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
(а) 4-[(Е)-1-(4-Цианобензоил)-2-(диметиламино)этенил]бензонитрил
N,N-Диметилформамид-диметилацеталь (135,2 г; 0,29 моль) добавляли по каплям в атмосфере инертного газа к нагретому (60°С) раствору 4-[2-(4-цианофенил)ацетил]бензонитрила (60,2 г; 0,24 моль, Ashley et al., J. Chem. Soc. (1942) 103, 110) в 1,2-диметоксиэтане. Полученную смесь затем фильтровали и концентрировали под вакуумом, получая остаток, который кристаллизовали из МеОН. Эта процедура давала 27,9 г (38%) указанного в подзаголовке соединения.
(б) Этил-2-[4,5-бис(4-цианофенил)-1H-пиразол-1-ил]ацетат
Раствор 4-[(Е)-1-(4-цианобензоил)-2-(диметиламино)этенил]-бензонитрила (6,2 г; 20 ммоль; со стадии (а), описанной выше) в водном этаноле (100 мл; 99%-ном) обрабатывали гидрохлоридом этил-2-гидразиноацетата (3,5 г; 22,6 ммоль). Смесь перемешивали при комнатной температуре в течение ночи перед тем, как сконцентрировать под вакуумом. Полученный остаток разбавляли водой, при этом водную смесь экстрагировали ДХМ. Органический слой затем отделяли, высушивали и концентрировали под вакуумом, получая остаток, который перекристаллизовывали из диэтилового эфира, с получением 1,7 г (23,5%) указанного в подзаголовке соединения.
(в) 2-[4,5-Бис(4-цианофенил)-1H-пиразол-1-ил]-N-(2-гидроксиэтил)-ацетамид
Смесь этил-2-[4,5-бис(4-цианофенил)-1H-пиразол-1-ил]ацетата (3,9 г; 10,9 ммоль; со стадии (б), описанной выше), 2-амино-1-этанола (1,3 г; 21,8 ммоль) и триэтиламина (0,8 г; 76 ммоль) перемешивали при 100°С в течение ночи. Добавляли воду и ДХМ, продукт кристаллизовали и выделяли фильтрованием, получая 3,53 г указанного в подзаголовке соединения.
(г) 2-[4,5-Бис(4-цианофенил)-1H-пиразол-1-ил]-N-(2-бромэтил)ацетамид
Смесь 2-[4,5-бис(4-цианофенил)-1Н-пиразол-1-ил]-N-(2-гидроксиэтил)-ацетамида (0,7 г; 1,88 ммоль; со стадии (в), описанной выше), N-бромсукцинимида (0,75 г; 5,64 ммоль) и трифенилфосфина (2,22 г; 8,4 ммоль) в ДХМ (100 мл) перемешивали при температуре дефлегмации в течение 3 ч. Реакционную смесь оставляли охлаждаться перед тем, как промыть водой. Органический слой отделяли, высушивали и концентрировали под вакуумом, получая остаток, который очищали флэш-хроматографией, элюируя смесью диэтиловый эфир:метанол (95:5), с получением 0,7 г указанного в подзаголовке соединения, загрязненного оксидом трифенилфосфина. Этот продукт непосредственно использовали на следующей стадии без какой-либо дальнейшей очистки.
(д) 7-[2-({2-[4,5-Бис(4-цианофенил)-1Н-пиразол-1-ил]ацетил}амино)-этил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид
Смесь 2-[4,5-бис(4-цианофенил)-1Н-пиразол-1-ил]-N-(2-бромэтил)-ацетамида (0,7 г; 1,6 ммоль; со стадии (г), описанной выше), N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамида (0,32 г; 1,6 ммоль; смотри Пример 6(б), описанный выше), К2СОз (0,55 г; 4 ммоль) в ацетонитриле (15 мл) перемешивали при температуре дефлегмации в течение ночи. Экстракция диэтиловым эфиром и водой давала органический слой, который отделяли, высушивали и концентрировали под вакуумом. Полученный остаток очищали хроматографией на силикагеле, элюируя смесью диэтиловый эфир:МеОН (95:5), с получением 0,27 г указанного в заголовке соединения.
13С ЯМР (CDCl3): δ 15.77, 29.18, 32.37, 36.13, 48.72, 52.27, 56.32, 109.83, 113.13, 118.27, 118.93, 120.10, 127.80, 131.39, 132.46, 132.73, 134.62, 138.75, 159.14, 167.09.
Пример 36
Используя способы, аналогичные описанным здесь, были получены следующие соединения (каждое из которых является соединением, указанным в заголовке данного Примера 36):
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-этил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид;
7-[(2R)-3-(4-цианофенокси)-2-гидроксипропил]-N-этил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-пропил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид;
7-[2-(4-цианофенокси)этил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-тетрагидро-2Н-пиран-2-ил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-(4-цианофенетил)-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-N,N-диметил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
трет-Бутил-2-(4-цианофенокси)-1-({7-[(этиламино)карбонил]-3,7-диазабицикло[3.3.1 ]нон-3-ил}метил)этилкарбамат;
7-(3-(4-цианофенокси)-2-{[(этиламино)карбонил]амино}пропил)-N-этил-3,7-диазабицикло[3.3.1 ]нонан-3-карбоксамид;
7-(3-(4-цианофенокси)-2-{[(этиламино)карбонил]амино}пропил)-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-(3-(4-цианофенокси)-2-{[(диметиламино)карбонил]амино}пропил)-N-этил-3,7-диазабицикло[3.3.1 ]нонан-3-карбоксамид;
метил-2-(4-цианофенокси)-1-({7-[(этиламино)карбонил]-3,7-диазабицикло-[3.3.1]нон-3-ил}метил)этилкарбамат;
7-[2-(ацетиламино)-3-(4-цианофенокси)пропил]-N-этил-3,7-диазабицикло-[3.3.1 ]нонан-3-карбоксамид;
7-[3-(2,4-дицианофенокси)-2-гидроксипропил]-N-этил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид;
трет-бутил-(1S)-2-(4-цианофенокси)-1-({7-[(этиламино)карбонил]-3,7-диазабицикло[3.3.1]нон-3-ил}метил)этилкарбамат;
7-[(2S)-2-[(aминoкapбoнил)aминo]-3-(4-циaнoфeнoкcи)пpoпил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
трет-бутил-(1R)-2-(4-цианофенокси)-1-({7-[(этиламино)карбонил]-3,7-диазабицикло[3.3.1]нон-3-ил}метил)этилкарбамат;
N-ацетил-7-[(2R)-3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-ацетил-7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[(2R)-3-(4-цианофенокси)-2-гидроксипропил]-N-метил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-N-метил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид;
7-[(2S)-3-(4-циано-2-{[(2-цианоэтил)амино]карбонил}фенокси)-2-гидроксипропил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
метил-(1R)-2-(4-цианофенокси)-1-({7-[(этиламино)карбонил]-3,7-диазабицикло[3.3.1]нон-3-ил}метил)этилкарбамат;
7-[(2R)-3-(4-цианофенокси)-2-гидроксипропил]-N-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-N-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-((2S)-3-{4-циано-2-[(метиламино)карбонил]фенокси}-2-гидроксипропил)-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-N-пропионил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[(2R)-3-(4-цианофенокси)-2-гидроксипропил]-N-пропионил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[2-(4-цианофенил)-2-гидроксиэтил]-N-этил-3,7-диазабицикло-[3.3.1 ]нонан-3-карбоксамид;
7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-N-(2-пропинил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-(4-цианофенетил)-N-изо-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-этил-7-[(2S)-2-гидрокси-3-(4-нитрофенокси)пропил]-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид;
мeтил-(1S)-2-(4-циaнoфeнoкcи)-1-({7-[(этилaминo)кapбoнил]-3,7-диaзaбицикло[3.3.1]нон-3-ил}метил)этилкарбамат;
7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-N-(циклопропилметил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(4-нитрофенил)-7-(4-оксогептил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[(2R)-3-(4-цианофенокси)-2-гидроксипропил]-N-(2-пропинил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-(3-{4-циано-2-[(циклопропиламино)карбонил]фенокси}-2-гидроксипропил)-N-пропионил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-N-фенил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[(2R)-3-(4-цианофенокси)-2-гидроксипропил]-N-(циклопропилметил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
трет-бутил-(1R)-2-(4-цианофенокси)-1-({7-[(пропиониламино)карбонил]-3,7-диазабицикло[3.3.1]нон-3-ил}метил)этилкарбамат;
7-(3-{4-циано-2-[(изо-пропиламино)карбонил]фенокси}-2-гидроксипропил)-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
трет-бутил-2-(4-цианофенокси)-1-({7-[(пропиониламино)карбонил]-3,7-диазабицикло[3.3.1]нон-3-ил}метил)этилкарбамат;
трет-бутил-2-(4-цианофенокси)-1-({7-[(изо-пропиламино)карбонил]-3,7-диазабицикло[3.3.1]нон-3-ил}метил)этил(метил)карбамат;
7-[3-(4-цианофенокси)-2-(метиламино)пропил]-N-изо-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-{3-(4-цианофенокси)-2-[метил(метилсульфонил)амино]пропил}-N-изо-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(трет-бутил)-7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[2-амино-3-(4-цианофенокси)пропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
трет-бутил-2-[7-(аминокарбонил)-3,7-диазабицикло[3.3.1]нон-3-ил]-1-[(4-цианофенокси)метил]этилкарбамат;
трет-бутил-2-(4-цианофенокси)-1-({7-[(тетрагидро-2Н-пиран-2-иламино)-карбонил]-3,7-диазабицикло[3.3.1]нон-3-ил}метил)этилкарбамат;
N-(4-цианофенил)-7-(4-оксогептил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(2,2-диметилпропаноил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(трет-бутокси)-7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
2-[7-(аминокарбонил)-3,7-диазабицикло[3.3.1]нон-3-ил]-1-[(4-циано-2-метилфенокси)метил]этил-трет-бутилкарбамат;
7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-N-изо-пропил-N-метил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(4-цианофенетил)-7-изо-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(трет-бутокси)-7-[3-(4-цианофенокси)-2-гидроксипропил]-N-метил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(4-цианофенетил)-7-(3,4-диметоксифенетил)-3,7-диазабицикло[3.3.1]-нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-циклопропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[2-амино-3-(4-цианофенокси)пропил]-N-(трет-бутил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-[3-(4-цианофенокси)пропил]-7-[5-(этиламино)-5-оксопентил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(3,5-диметил-4-изоксазолил)-3,7-диазабицикло[3.3.1 ]нонан-3-карбоксамид;
7-[3-(4-цианоанилино)пропил]-N-узо-пропил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид;
7-[4-(4-цианофенил)-4-гидроксибутил]-N-этил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид;
этил-{7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло-[3.3.1]нон-3-ил}карбонилкарбамат;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(2,6-диметоксифенил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(4-цианофенил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-бензил-7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-гексил-3,7-диазабицикло-[3.3.1]нонан-3-карбоксамид;
этил-3-[({7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло-[3.3.1]нон-3-ил}карбонил)амино]пропаноат;
N-(4-бутоксифенил)-7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(3-цианофенил)-3,7-диаза-бицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(3,4-диметоксифенил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
бутил-2-[({7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло-[3.3.1]нон-3-ил}карбонил)амино]ацетат;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(4-метоксифенил)-3,7-диаза-бицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(3,4-диметоксифенетил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(3,4-диметоксибензил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(3,4,5-триметоксифенил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(3,4-дигидро-2Н-1,5-бензодиоксепин-7-ил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(2,6-диметилфенил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
изо-пропил-{7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло-[3.3.1]нон-3-ил}карбонилкарбамат;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-(2-фторэтил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-{2-[(циклопропилметил)-амино]-2-оксоэтил}-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(трет-бутил)-7-{2-гидрокси-3-[(2-метил-1-оксо-1,2-дигидро-4-изохинолинил)окси]пропил}-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(1-циано-1-метилэтил)-7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[2-амино-3-(4-цианофенокси)пропил]-N-(1,3-бензодиоксол-5-илметил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-изо-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
4-(3-{7-[(2,6-диметил-4-морфолинил)карбонил]-3,7-диазабицикло-[3.3.1]нон-3-ил}-2-гидроксипропокси)бензонитрил;
N-[циано-(4-фторфенил)метил]-7-[3-(4-цианофенокси)-2-гидроксипропил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(цианометил)-7-[3-(4-цианофенокси)-2-гидроксипропил]-N-метил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[4-(4-цианофенокси)-2-гидроксибутил]-N-этил-3,7-диазабицикло[3.3.1]-нонан-3-карбоксамид;
7-[4-(4-цианофенил)бутил]-N-[2-оксо-2-(пропиламино)этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[4-(4-цианофенил)бутил]-N-пропил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[2-амино-4-(4-цианофенокси)бутил]-N-пропил-3,7-диазабицикло[3.3.1]-нонан-3-карбоксамид;
7-[4-(4-цианофенил)бутил]-N-этил-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-цианофенокси)-2-гидроксипропил]-N-[2-(2-метоксиэтокси)этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(4-цианофенил)-7-(3,3-диметил-2-оксобутил)-3,7-диазабицикло[3.3.1]-нонан-3-карбоксамид;
N-(4-цианофенил)-7-(3,4-диметоксифенил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(4-цианофенил)-7-(циклопропилметил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
N-(4-цианофенил)-7-[2-(2-метоксиэтокси)этил]-3,7-диазабицикло[3.3.1]-нонан-3-карбоксамид;
N-(4-цианофенил)-7-[2-(2,3-дигидро-1,4-бензодиоксин-6-ил)-2-оксоэтил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид;
7-[3-(4-ацетил-1-пиперазинил)пропил]-N-(4-цианофенил)-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид; и
7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-N-[2-оксо-2-(пропиламино)-этил]-3,7-диазабицикло[3.3.1]нонан-3-карбоксамид.
Пример 37
Соединения из приведенных выше Примеров протестированы в Тесте А, описанном выше, и для всех из них обнаружено, что они показывают величины D10 более чем 6,0.
Полученные в Тесте А данные приведены в следующей таблице.
Сокращения
АсОН= уксусная кислота
ADDP= 1,1'-(азодикарбонил)дипиперидин
водн.= водный
атм.= атмосфера
CBz= бензилоксикарбонил
CDI= карбонилдиимидазол
Вu= бутил
ДХМ= дихлорметан
ДМФ= диметилформамид
ДМСО= диметилсульфоксид
Et= этил
EtOAc= этилацетат
ЕtOН= этанол
ESI= электроспрей-интерфейс
экв= эквивалент
FAB= бомбардировка быстрыми атомами
ч= час
ИПС= изо-пропанол
i-РrОН= изо-пропанол
LC= жидкостная хроматография
ВЭЖХ= высокоэффективная жидкостная хроматография
м-СРВА= мета-хлорпербензойная кислота
Me= метил
MeCN= ацетонитрил
МеОН= метанол
мезил= метансульфонат
мин= минута
Ms= мезилат
МС= масс-спектроскопия
НАДФН= никотинамидадениндинуклеотидфосфат, восстановленная
форма
ЯМР= ядерный магнитный резонанс
OSu= 0-сукцинил
Pd/C= палладий на угле
п-TSA= пара-толуолсульфокислота
кт= комнатная температура
насыщ.= насыщенный
ТЭА= триэтиламин
ТГФ= тетрагидрофуран
ТСХ= тонкослойная хроматография
TMS= тетраметилсилан
Префиксы н-, втор-, и-, изо-, m- и трет- имеют обычные значения:
нормальный, изо, вторичный и третичный.
| название | год | авторы | номер документа |
|---|---|---|---|
| 3,7-ДИАЗАБИЦИКЛО[3.3.0]ОКТАНЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ СЕРДЕЧНЫХ АРИТМИЙ | 2002 |
|
RU2293085C2 |
| ПРОИЗВОДНЫЕ АЗАБИЦИКЛООКТАНА, ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ, СОЕДИНЕНИЯ | 2000 |
|
RU2262505C2 |
| 3,7-ДИАЗАБИЦИКЛО[3.3.1]-ПРЕПАРАТЫ КАК АНТИАРИТМИЧЕСКИЕ СОЕДИНЕНИЯ | 2002 |
|
RU2286993C2 |
| ПРОМЕЖУТОЧНОЕ ХИМИЧЕСКОЕ СОЕДИНЕНИЕ | 2003 |
|
RU2326122C2 |
| ЗАМЕЩЕННЫЕ 3,7-ДИАЗОБИЦИКЛО[3.3.1]НОНАНЫ, ФОКУСИРОВАННАЯ БИБЛИОТЕКА И КОМБИНАТОРНАЯ БИБЛИОТЕКА | 2003 |
|
RU2228934C1 |
| ПРОИЗВОДНЫЕ 3-ЗАМЕЩЕННОГО 1,5-ДИФЕНИЛПИРАЗОЛА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ СВ1 МОДУЛЯТОРОВ | 2005 |
|
RU2375349C2 |
| ЛИГАНД И КОМПЛЕКС ДЛЯ КАТАЛИТИЧЕСКОГО ОТБЕЛИВАНИЯ СУБСТРАТА | 2001 |
|
RU2283342C2 |
| АМИДЫ ДИАЗАБИЦИКЛОАЛКАНОВ, СЕЛЕКТИВНЫЕ В ОТНОШЕНИИ АЦЕТИЛХОЛИНОВОГО ПОДТИПА НИКОТИНОВЫХ РЕЦЕПТОРОВ | 2007 |
|
RU2517693C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНТАГОНИСТ Р2Х РЕЦЕПТОРА И ФАКТОР НЕКРОЗА ОПУХОЛИ α | 2004 |
|
RU2350354C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
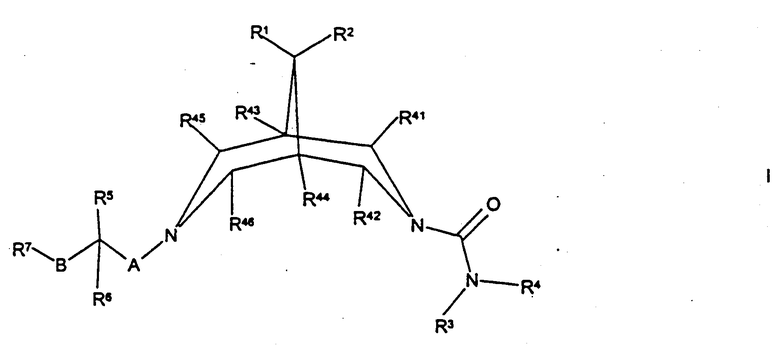
Предложены соединения формулы (I), где R1, R2, R3, R4, R5, R6, R7, R41, R42, R43, R44, R45, R46, А и В имеют значения, приведенные в описании. Технический результат: проявление электрофизиологической активности класса III в результате чего являются полезными при лечении сердечных аритмий. 13 н. и 19 з.п. ф-лы, 1 табл.
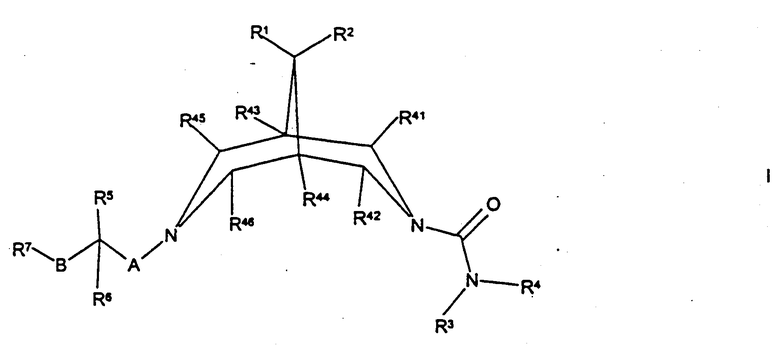
где
R1 и R2 независимо представляют собой Н, С1-4алкил, OR2b или N(R2c)R2d либо вместе образуют -O-(CH2)2-O-, -(CH2)3-, -(CH2)4- или -(CH2)5-;
R2b, R2c и R2d независимо представляют собой Н или С1-6алкил;
R3 представляет собой Н, С1-6алкил или вместе с R4 представляет собой С3-6алкилен (причем алкиленовая группа возможно прервана атомом О и/или возможно замещена одной или более чем одной С1-3алкильной группой);
R4 представляет собой Н, С1-12алкил, С1-6алкокси (причем последние две группы обе возможно замещены и/или оканчиваются одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро, С1-4алкила и/или С1-4алкокси), -(CH2)q-(С6-С10)арил, -(CH2)q-окси(С6-С10)арил, -(CH2)q-Het1 (причем последние три группы возможно замещены (по -(CH2)q-части и/или (С6-С10)арил/Het1-части) одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро, -C(O)R10, -C(O)ОR11, -N(H)S(O)2R11а, С1-6алкила и/или С1-6алкокси), -(CH2)qN(Н)С(О)R8, -(CH2)qS(O)2R8, -(CH2)qC(O)R8, -(CH2)qC(O)OR8, -(CH2)qC(O)N(R9)R8 или вместе с R3 представляет собой С3-6алкилен (причем алкиленовая группа возможно прервана атомом О и/или возможно замещена одной или более чем одной С1-3алкильной группой);
q представляет собой 0, 1, 2, 3, 4, 5 или 6;
R8 представляет собой Н, С1-6алкил, С6-С10арил (причем последняя группа возможно замещена и/или оканчивается одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро, -C(O)R10, -C(O)ОR11, -N(H)S(O)2R11а, С1-6алкила и/или С1-6алкокси) или вместе с R9 представляет собой С3-7алкилен;
R9 представляет собой Н, С1-4алкил или вместе с R8 представляет собой С3-7алкилен;
Het1 представляет собой пяти-двенадцатичленное гетероциклическое кольцо, содержащее один или более чем один гетероатом, выбранный из кислорода, азота и/или серы, и которое также возможно включает в себя один или более чем один заместитель =О;
R41, R42, R43, R44, R45 или R46 независимо представляют собой Н или С1-3алкил;
R5 представляет собой Н, галогено, С1-3алкил, -OR12,-N(R13)R12 или вместе с R6 представляет собой =О;
R6 представляет собой Н, С1-4алкил или вместе с R5 представляет собой =О;
R12 представляет собой Н, С1-6алкил, -S(O)2-С1-4алкил, -C(O)R14, -C(O)ОR14, -C(O)N(R15)R15a или С6-С10арил (причем последняя группа возможно замещена и/или оканчивается одним или более чем одним заместителем, выбранным из -ОН, галогено, циано, нитро, -C(O)R10, -C(O)ОR11, -N(H)S(O)2R11а, С1-6алкила и/или С1-6алкокси);
R13 представляет собой Н или С1-4алкил;
R14 представляет собой Н или С1-6алкил;
R15 и R15а независимо представляют собой Н или С1-4алкил либо вместе представляют собой С3-6алкилен, возможно прерванный атомом О;
А представляет собой простую связь, С1-6алкилен, -N(R16)(CH2)r- или -О(CH2)r- (причем группа -(CH2)r- в последних двух группах присоединена к атому азота биспидина);
В представляет собой простую связь, С1-4алкилен, -(CH2)nN(R17)-, -(CH2)nS(O)p-, -(CH2)nO- (причем группа -(CH2)n- в последних трех группах присоединена к атому углерода, несущему R5 и R6), -C(O)N(R17)- (причем группа -C(O)- в последней группе присоединена к атому углерода, несущему R5 и R6), -N(R17 )С(О)О(CH2)n-, -N(R17)(CH2)n- (причем группа N(R17) в последних двух группах присоединена к атому углерода, несущему R5 и R6) или -(СH2)mС(H)(OH)(СH2)n- (причем группа -(CH2)m- в последней группе присоединена к атому углерода, несущему R5 и R6);
m представляет собой 1, 2 или 3;
n и r независимо представляют собой 0, 1, 2, 3 или 4;
p представляет собой 0, 1 или 2;
R16 и R17 независимо представляют собой Н или С1-4алкил;
R7 представляет собой линейный либо разветвленный и/или ациклический, циклический и/или частично циклический/ациклический С1-6алкил (возможно замещенный и/или оканчивающийся ОН); Het2 (возможно замещенный одним или более чем одним заместителем, выбранным из циано, С1-3алкила, фенила (причем последняя группа возможно замещена одной или более чем одной цианогруппой), =О, C(O)R10 (где R10 представляет собой линейный или разветвленный С1-3алкил) или S(O)2R19 (где R19 представляет собой С1-2алкил)); или фенил (замещенный одним или более чем одним заместителем, выбранным из циано, нитро, C(O)N(H)R22 (где R22 представляет собой линейный либо разветвленный и/или ациклический, циклический и/или частично циклический/ациклический С1-4алкил, причем алкильная группа возможно оканчивается циано), N(H)S(O)2R18 (где R18 представляет собой С1-2алкил) или Het3);
Het2 и Het3 независимо представляют собой пяти-двенадцатичленную гетероциклическую группу, содержащую один или более чем один гетероатом, выбранный из кислорода, азота и/или серы, и которая также возможно включает в себя один или более чем один заместитель =О;
R10 и R11 независимо представляют собой, в каждом индивидуальном случае, Н или С1-6алкил; и
R11а представляет собой, в каждом индивидуальном случае, С1-6алкил;
или его фармацевтически приемлемое производное;
при условии, что:
(а) когда А и В оба представляют собой простые связи, а R7 представляет собой возможно замещенный (С6-С10)арил, тогда R5 и R6 оба не представляют собой Н;
(б) когда А представляет собой простую связь, тогда R5 и R6 вместе не представляют собой =О; и
(в) когда R5 представляет собой -ОR12 или -N(R13)R12, тогда:
1) А не представляет собой -N(R16)(CH2)r- или -О(CH2)r-; и/или
2) n не представляет собой 0, когда В представляет собой -(CH2)nN(R17)-, -(CH2)nS(O)p- или -(CH2)nO-.
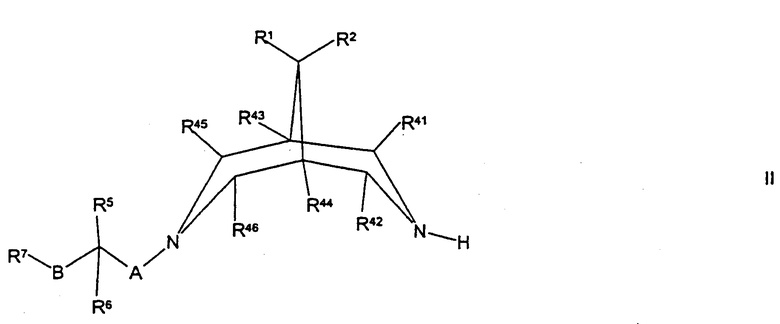
где R1, R2, R5, R6, R7, R41, R42, R43, R44, R45, R46, A и B такие, как определено в п.1, с соединением формулы III,
R4-N=C=O III
где R4 такой, как определено в п.1.
(R3)(R4)NC(O)-L1 IV
где L1 представляет собой уходящую группу, а R3 и R4 такие, как определено в п.1.
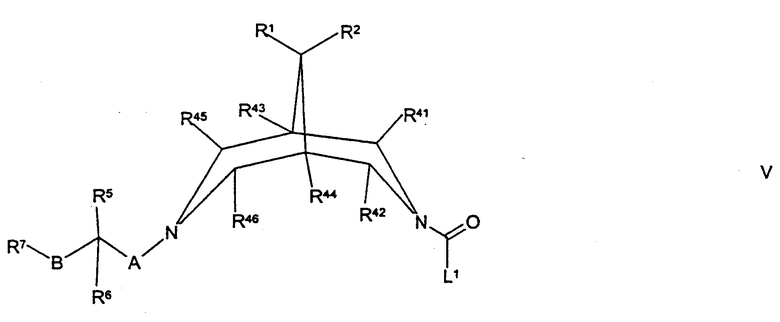
где L1 такой, как определено в п.25, и R1, R2, R5, R6, R7, R41, R42, R43, R44, R45, R46, A и B такие, как определено в п.1, с соединением формулы VA
(R3)(R4)NH VA
где R3 и R4 такие, как определено в п.1.
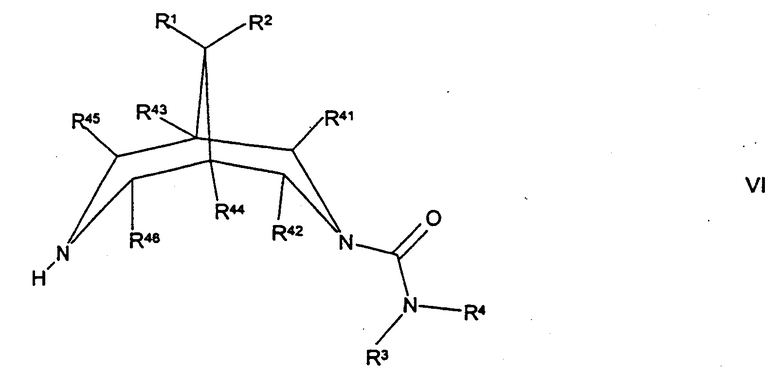
где R1, R2, R3, R4, R41, R42, R43, R44, R45 и R46 такие, как определено в п.1, с соединением формулы VII
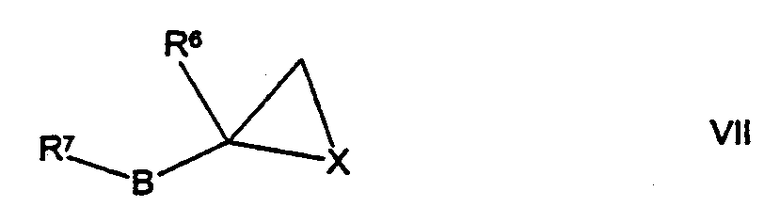
где Х представляет собой О или N(R12), а R6, R7, R12 и В такие, как определено в п.1.
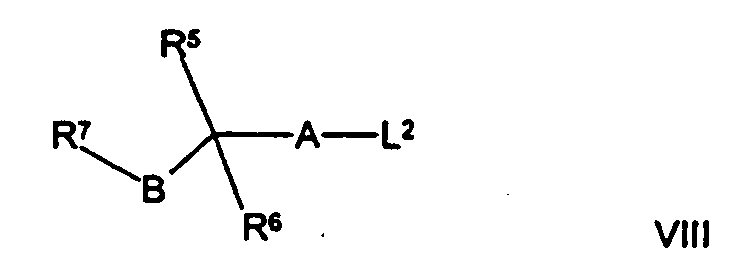
где L2 представляет собой уходящую группу, а R5, R6, R7, A и B такие, как определено в п.1.
R12аL1 XVIII
где R12a представляет собой R12, как он определен в п.1, при условии, что он не представляет собой Н, а L1 такой, как определено в п.25.
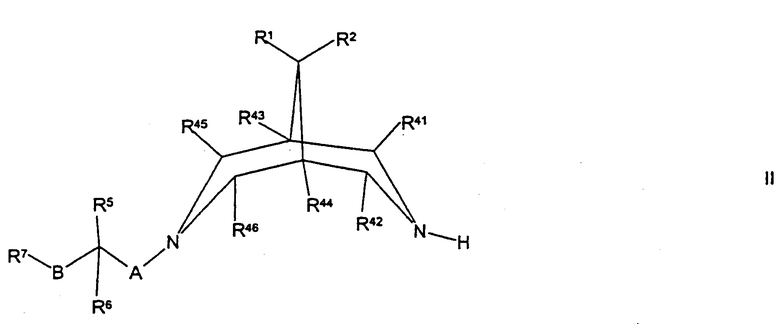
или его защищенное производное, при условии, что R7 не представляет собой возможно замещенный фенил или С1-6алкил.
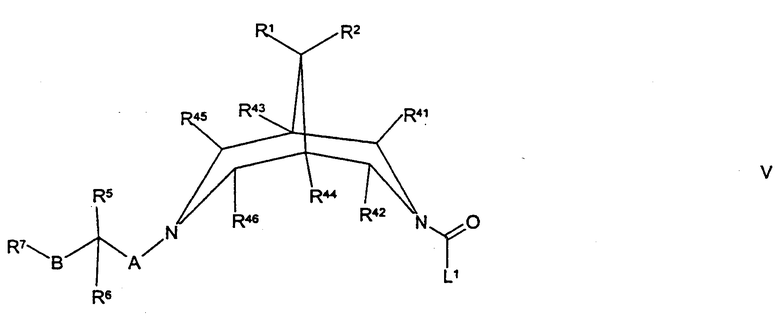
или его защищенное производное, при условии, что R7 не представляет собой возможно замещенный фенил.
| СПОСОБ АВТОМАТИЧЕСКОЙ ДУГОВОЙ СВАРКИ | 0 |
|
SU308843A1 |
| WO 9107407 A1, 30.05.1991 | |||
| ДИСКОВАЯ МЕЛЬНИЦА | 0 |
|
SU306871A1 |
| ГЕТЕРОАРИЛАМИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2100357C1 |
| 10-НИТРО-1,6,8-ТРИАЗАТРИЦИКЛО [6,3,1,1] ТРИДЕКАН-2,5-ДИОН | 1982 |
|
SU1107541A1 |

