Область изобретения
Данное изобретение относится к новым, твердым по состоянию формам определенных антиаритмических лекарственных средств, к содержащим их фармацевтическим композициям и к способам их получения.
Предпосылки изобретения
При изготовлении лекарственных композиций важно, чтобы лекарственное вещество находилось в форме, в которой ее можно удобно подвергать обработке. Это важно не только с точки зрения достижения промышленно эффективного способа производства, но также и с точки зрения последующего производства фармацевтических препаратов, содержащих активное соединение.
Кроме того, при производстве лекарственных композиций важно, чтобы после введения пациенту был обеспечен достоверный, воспроизводимый и постоянный профиль концентрации в плазме лекарственного средства.
Химическая стабильность, стабильность в твердом состоянии и "срок годности" активных ингредиентов также являются очень важными факторами. Лекарственное вещество и содержащие его композиции предпочтительно должны быть способны к эффективному хранению в продолжение значительных периодов времени без существенного изменения в физико-химических характеристиках активного компонента (например, в его химическом составе, плотности, гигроскопичности и растворимости).
Более того, также важно уметь изготовить лекарственное средство в такой химически чистой форме, насколько возможно.
В этом отношении значительные проблемы могут представлять аморфные материалы. Например, такие материалы обычно трудно поддаются обработке и изготовлению из них препаратов, обуславливают недостоверную растворимость и зачастую проявляют нестабильность и химическую неоднородность.
Специалисту очевидно, что если лекарственное средство можно без труда получить в стабильной кристаллической форме, то вышеупомянутые проблемы могут быть разрешены.
Таким образом, при производстве коммерчески жизнеспособных и фармацевтически приемлемых лекарственных композиций, таких как композиции с модифицированным высвобождением, важно, где это возможно, получить лекарственное средство по существу в кристаллической и стабильной форме.
Следует отметить, однако, что эта цель не всегда достижима. Действительно, обычно невозможно, исходя только из молекулярной структуры, предсказать, как будет вести себя соединение при кристаллизации. В большинстве случаев это может быть определено только эмпирически.
Предшествующий уровень техники
В публикации международной заявки WO 01/28992 раскрыт ряд оксабиспидиновых соединений, включающих в себя:
(а) 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил]пропил}амино)бензнитрил:
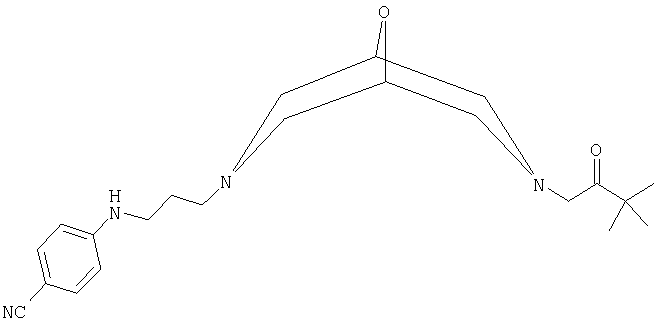
где данное соединение обозначено ниже как Соединение А. Соединение А раскрыто, в частности, в WO 01/28992 как в форме свободного основания, так и в форме бензолсульфонатной соли;
(б) трет-бутил-2-{7-[3-(4-цианоанилино)пропил]-9-окса-3,7-диаза-бицикло[3.3.1]нон-3-ил}этилкарбамат:
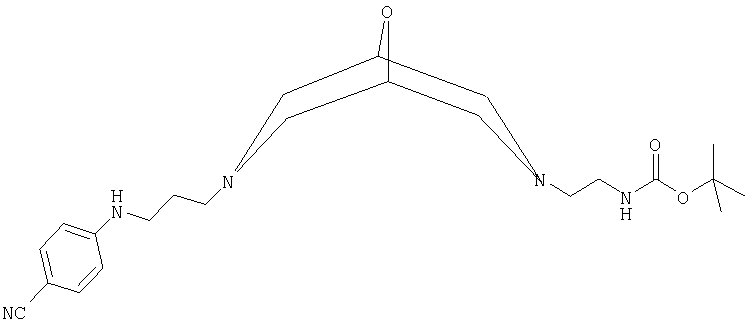
в форме свободного основания, где данное соединение обозначено ниже как Соединение В;
в) трет-бутил-2-{7-[4-(4-цианофенил)бутил]-9-окса-3,7-диазабицикло-[3.3.1]нон-3-ил}этилкарбамат:
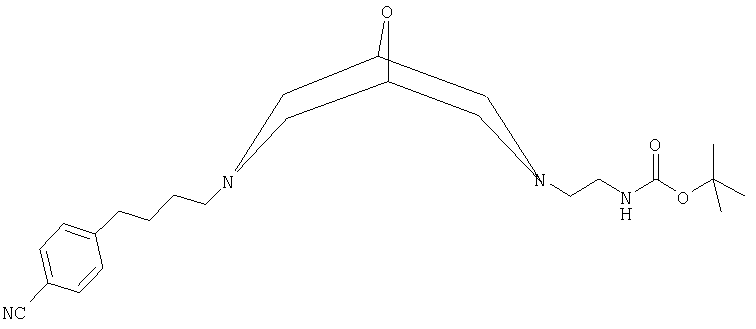
в форме свободного основания, где данное соединение обозначено ниже как Соединение С; и
г) трет-бутил-2-{7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил}этилкарбамат:

в форме свободного основания, где данное соединение обозначено ниже как Соединение D.
Показано, что соединения из публикации международной заявки WO 01/28992 являются полезными при лечении сердечных аритмий.
Способы синтеза Соединений А, В, С и D описаны в Примерах 3, 7, 8 и 9 (соответственно) WO 01/28992.
Конкретные фармацевтически приемлемые соли Соединений В, С и D не раскрыты в WO 01/28992. Кроме того, не представлено никакой информации относительно различных кристаллических форм любых Соединений А, В, С или D либо их солей, которые могут быть получены.
Описание изобретения
Согласно первому аспекту данного изобретения предложена фармацевтически приемлемая соль Соединения А, Соединения В, Соединения С или Соединения D, при условии, что данная соль не представляет собой соль Соединения А с бензолсульфоновой кислотой.
Согласно второму аспекту изобретения предложено Соединение А, Соединение В, Соединение С или Соединение D либо фармацевтически приемлемая соль любого из этих соединений в по существу кристаллической форме.
Соединения А, В, С и D и их соли согласно первому и второму аспектам изобретения обозначены в данном описании как "соединения по изобретению".
Несмотря на то, что авторами изобретения обнаружено, что существует возможность получения соединений по изобретению в формах, которые являются кристаллическими более чем на 80%, в термин "по существу кристаллические" авторы включают более чем на 20%, предпочтительно более чем на 30% и, более предпочтительно, более чем на 40% кристаллические. Степень (%) кристалличности может быть определена специалистом путем использования дифракции рентгеновских лучей на порошке (XRPD). Кроме того, могут быть использованы и другие техники, такие как ЯМР для веществ в твердом состоянии, FT-IR (инфракрасная спектроскопия с фурье-преобразованием), спектроскопия комбинационного рассеяния, дифференциальная сканирующая калориметрия (DSC) и микрокалориметрия.
Предпочтительно, что когда соединение по изобретению представляет собой соль Соединения А с пара-толуолсульфоновой кислотой, оно предложено в по существу кристаллической форме.
Когда соединение по изобретению предложено в виде кристаллической формы соли Соединения А с бензолсульфоновой кислотой, предпочтительно, чтобы кристаллическая форма не являлась формой, конкретно описанной ниже в Примере 4 (и/или в п.8 формулы изобретения).
Когда соединение по изобретению предложено в виде кристаллической формы Соединения А (свободного основания), предпочтительно, чтобы кристаллическая форма не являлась формой, конкретно описанной ниже в любом из Примеров: Примере 1 (и/или в п.4 формулы изобретения), Примере 2 (и/или в п.5 формулы изобретения) или Примере 3 (и/или в п.6 формулы изобретения).
Когда соединение по изобретению предложено в виде кристаллической формы Соединения А, предпочтительно, чтобы данное соединение не было предложено в форме свободного основания или в форме бензолсульфонатной соли, когда оно предложено в форме соли.
Когда соединение по изобретению предложено в виде кристаллической формы Соединения С (свободного основания), предпочтительно, чтобы кристаллическая форма не являлась формой, конкретно описанной ниже в Примере 12 (и/или в п.25 формулы изобретения).
Когда соединение по изобретению предложено в виде кристаллической формы Соединения С, предпочтительно, чтобы данное соединение не было предложено в форме свободного основания.
Когда соединение по изобретению предложено в виде кристаллической формы Соединения D (свободного основания), предпочтительно, чтобы кристаллическая форма не являлась формой, конкретно описанной ниже в Примере 9 (и/или в п.19 формулы изобретения).
Когда соединение по изобретению предложено в виде кристаллической формы Соединения D, предпочтительно, чтобы данное соединение не было предложено в форме свободного основания.
Когда соединение по изобретению предложено в виде кристаллической формы Соединения В, предпочтительно, чтобы данное соединение не было предложено в форме свободного основания.
Соединения по изобретению могут находиться в форме сольвата, гидрата или смеси сольват/гидрат. Сольваты могут быть образованы с одним или более чем одним органическим растворителем, таким как низшие алкиловые (например, С1-4алкил) спирты (например, метанол, этанол или изопропанол), кетоны (такие как ацетон), сложные эфиры (такие как этилацетат) или их смеси.
Соединения по изобретению могут обладать улучшенной стабильностью по сравнению с соединениями/солями, раскрытыми в WO 01/28992.
Термин "стабильность", как он определен здесь, включает в себя химическую стабильность и стабильность твердого состояния.
В термин "химическая стабильность" авторы включают возможность хранения соединений по изобретению в изолированной форме или в форме препарата, в котором они предложены в смеси с фармацевтически приемлемыми носителями, разбавителями или адъювантами (например, в пероральной лекарственной форме, такой как таблетка, капсула и так далее), при нормальных условиях хранения, с незначительной степенью химической деградации или деструкции.
В термин "стабильность твердого состояния" авторы включают возможность хранения соединений по изобретению в изолированной твердой форме или в форме твердого препарата, в которой они предложены в смеси с фармацевтически приемлемыми носителями, разбавителями или адъювантами (например, в пероральной лекарственной форме, такой как таблетка, капсула и так далее), при нормальных условиях хранения, с незначительной степенью трансформации твердого состояния (например, кристаллизации, перекристаллизации, фазового перехода твердого состояния, гидратации, дегидратации, сольватации или десольватации).
Примерами "нормальных условий хранения" являются температура от минус 80 до плюс 50°С (предпочтительно от 0 до 40°С, более предпочтительна комнатная температура, такая как от 15 до 30°С), давление от 10 до 200 кПа (от 0,1 до 2 бар) (предпочтительно атмосферное давление), относительная влажность от 5 до 95% (предпочтительно от 10 до 75%) и/или воздействие Уф/видимым светом до 460 люкс, для длительных периодов времени (то есть более чем шесть месяцев, или периодов, равных шести месяцам). Может быть обнаружено, что при таких условиях соединения по изобретению менее чем на 15%, более предпочтительно менее чем на 10% и в особенности менее чем на 5% химически деградированы/деструктурированы или имеют трансформированное твердое состояние, как подходит. Специалисту очевидно, что вышеупомянутые верхние и нижние пределы для температуры, давления и относительной влажности представляют собой крайние значения нормальных условий хранения, и что некоторые сочетания этих крайних значений не будут испытываться в продолжение нормального хранения (например, температура 50°С и давление 10 кПа (0,1 бар)).
Предпочтительные соли Соединений А, В, С и D включают в себя соли присоединения основания или, предпочтительно, кислоты, причем эти соли могут быть образованы путем добавления соответствующего количества соответствующих кислоты или основания перед выделением (которое может включать в себя кристаллизацию). Например, в случае соединений по изобретению в по существу кристаллической форме кислота или основание могут быть добавлены к кристаллизационной смеси перед началом кристаллизации.
Предпочтительные соли присоединения включают в себя соли присоединения неорганических и, в особенности, органических кислот, предпочтительно соли карбоновых кислот, таких как гиппуровая кислота, нафтойная кислота и гидроксизамещенная нафтойная кислота (например, 1-гидрокси-2-нафтойная кислота), аспарагиновая кислота, малеиновая кислота, янтарная кислота, малоновая кислота, уксусная кислота, фумаровая кислота, бензойная кислота, терефталевая кислота, памовая кислота и гидроксибензойная кислота; соли оксикислот, таких как салициловая кислота, гликолевая кислота, яблочная кислота, аскорбиновая кислота, лимонная кислота, глюконовая кислота, молочная кислота, а также винная кислота и ее производные, такие как O,O'-дибензоилвинная кислота (например, O,O'-дибензоил-D-винная кислота или O,O'-дибензоил-L-винная кислота) и O,O'-ди-пара-толуоилвинная кислота (например, O,O'-ди-пара-толуоил-D-винная кислота или O,O'-ди-паратолуоил-L-винная кислота); соли других двухосновных кислот, таких как 2,2,3,3-тетраметил-1,4-дибутановая кислота и 1,2-циклопентандикарбоновая кислота; и соли алкил-, арил- и алкиларилсульфокислот, например С1-8алкил-, С6-10арил-, и С1-4алкил-С6-10арил-сульфоновых кислот (причем арил- и алкиларилсульфоновые кислоты могут быть замещены по арильной части, например, одной или более чем одной метильной, метокси, гидрокси или галогеногруппой), включая соли бензолсульфоновой кислоты, толуолсульфоновой кислоты, гидроксизамещенной бензолсульфоновой кислоты, нафталинсульфоновой кислоты, нафталиндисульфоновой кислоты, мезитиленсульфоновой кислоты, метансульфоновой кислоты, этансульфоновой кислоты и 2-гидроксиэтансульфоновой кислоты.
Соли присоединения кислоты, в частности, Соединения D, которые также могут быть упомянуты, включают в себя соли, где кислота представляет собой производное гиппуровой кислоты, например кислоту формулы I,

где
Ar1 представляет собой фенил или нафтил, оба из которых возможно замещены одним или более чем одним заместителем, выбранным из галогено (например, хлоро), нитро, С1-6алкила, С1-6алкокси, гидрокси и фенила; и
R1, R2 и R3 независимо представляют собой Н или C1-3алкил.
Специалисту очевидно, что когда Ar1 представляет собой фенил, и R1, R2 и R3 все представляют собой H, тогда кислота формулы I является гиппуровой кислотой.
Предпочтительные группы Ar1 включают в себя фенил, причем эта фенильная группа возможно замещена фенилом (например, по положению 4 относительно точки присоединения группы С(O)), хлоро (например, по положениям 3 и/или 4 относительно группы С(O)), нитро (например, по положению 4 относительно группы С(O)) и/или С1-4алкилом, таким как метил (например, по положениям 2 и/или 4 относительно группы С(O)); и нафтил. Более предпочтительные значения Ar1 включают в себя фенил, 4-фенилфенил (дифенил), 3,4-дихлорфенил, 2-нафтил, 4-нитрофенил и 2,4,6-триметилфенил.
Предпочтительные группы R1 и R2 включают в себя Н и метил. Предпочтительно, когда R1 и R2 либо оба представляют собой Н, либо оба представляют собой метил.
Предпочтительные группы R3 включают в себя Н.
Когда R1 и R2 оба представляют собой метил, предпочтительно, чтобы Ar1 представлял собой фенил. Когда R1 и R2 оба представляют собой Н, предпочтительно, чтобы Ar1 представлял собой 4-нитрофенил, 2,4,6-триметилфенил или, в особенности, 3,4-дихлорфенил, 2-нафтил или 4-фенилфенил (дифенил).
Кислоты формулы I имеются в продаже (например, гиппуровая кислота, 4-нитрогиппуровая кислота и 2-, 3- или 4-метилгиппуровая кислота) или могут быть получены в соответствии со стандартными методиками.
Например, кислоты формулы I могут быть получены путем осуществления взаимодействия соединения формулы II,
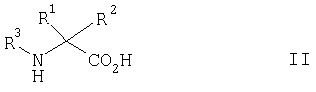
где R1, R2 и R3 такие, как они определены выше, с хлорангидридом формулы III,

где Ar1 такой, как он определен выше, например, в присутствии основания, например водного NaOH, в соответствии с классическими методиками Шоттена-Баумана (смотри, например, J. Med. Chem., 1989, 32, 1033). Нейтрализация кислотой, например концентрированной соляной кислотой, может осадить кислоту формулы I, которая может быть перекристаллизована при необходимости из различных растворителей, например изопропилового спирта, метанола, этанола, ацетона и воды или смеси этих растворителей.
Альтернативно, эфирные производные (например, низших алкиловых эфиров) соединений формулы II, возможно в форме соли, например соли гидрохлорида, могут быть приведены во взаимодействие с хлорангидридом формулы III в присутствии основания, например триэтиламина, в подходящем растворителе, например дихлорметане, с получением эфир-амида формулы IV,
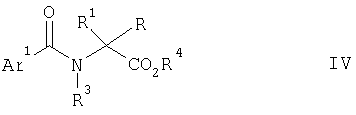
где R4 представляет собой низший алкил (такой как С1-6алкил) или низший алкилфенил (например, С1-3лкилфенил), и Ar1, R1, R2 и R3 такие, как они определены выше (смотри, например, J. Heterocyclic Chem., 1973, 10, 935; Tetrahedron, 1989, 45, 1691 и J. Org. Chem., 1999, 64, 8929). Эфир-амиды формулы IV могут быть твердыми веществами при комнатной температуре и поэтому могут быть очищены кристаллизацией после их образования, если подходит. Соединения формулы IV затем могут быть превращены в соединения формулы I путем обычного гидролиза, например водным раствором гидроксида натрия, с последующим добавлением кислоты, например соляной кислоты, для осаждения продукта. При необходимости затем может быть проведена перекристаллизация.
Соединения формул I, II и IV, где R3 представляет собой C1-3алкил, могут быть получены путем обычного алкилирования соответствующего соединения формулы I, II или IV, где R3 представляет собой Н.
Соединения формул II (и эфирные производные) и III имеются в продаже или могут быть без труда получены с помощью стандартных методик.
Согласно еще одному аспекту данного изобретения предложен способ получения соединения по изобретению, при котором:
(а) к Соединению А, В, С или D добавляют соответствующее количество кислоты или основания с образованием соли присоединения кислоты или основания; и/или
(б) осуществляют кристаллизацию Соединений А, В, С или D либо соли Соединения А, В, С или D.
Возможно осуществить кристаллизацию Соединений А, В, С и D и их фармацевтически приемлемых солей в присутствии или в отсутствие системы растворителей (например, кристаллизация может быть осуществлена из расплава в сверхкритических условиях или достигнута посредством сублимации). Однако авторы предпочитают кристаллизацию из соответствующей системы растворителей.
Кристаллические соли присоединения кислоты или основания Соединений А, В, С или D могут быть получены путем добавления соответствующего количества подходящей кислоты или основания к кристаллизационной смеси (например, системе растворителей, включающей в себя Соединение А, В, С или D в виде свободного основания) перед осуществлением кристаллизации. Например, могут быть добавлены органические кислоты (возможно в форме раствора, содержащего подходящий полярный растворитель (например, низший алкиловый спирт, такой как метанол или этанол) или ацетат, такой как этилацетат) к Соединению А, В, С или D (возможно в форме раствора, где данное свободное основание находится в соответствующем растворителе для кристаллизации). (Специалисту очевидно, что в данном контексте термин "свободное основание" означает "свободную форму" Соединений А, В, С или D (то есть формы, которые не являются формой солей присоединения кислоты или основания)).
Специалисту очевидно, что кислота или основание могут быть объединены в таком способе с Соединением А, В, С или D посредством растворения соответствующих материалов в соответствующих растворителях, как описано выше, по меньшей мере частичного удаления этих растворителей и затем повторного растворения полученной смеси перед осуществлением кристаллизации, как описано выше.
Если соединения по изобретению находятся в форме солей присоединения кислоты, подходящие стехиометрические отношения кислоты к свободному основанию находятся в интервале от 0,25:1,5 до 1,5:1, таких как от 0,45:1,25 до 1,25:1, включая от 0,50:1 до 1:1.
Система растворителей может быть гетерогенной или гомогенной и может таким образом содержать один или более чем один органический растворитель, такой как алкилацетат (например, линейный или разветвленный C1-6алкилацетат, такой как этилацетат, изопропилацетат и бутилацетат); низший (например, линейный или разветвленный C1-6) алкиловый спирт, такой как гексан-1-ол, 3-метилбутан-1-ол, пентан-1-ол, пентан-2-ол, 4-метил-2-пентанол и 2-метил-1-пропанол, или С1-4алкиловый спирт, такой как метанол, этанол, н-пропанол, изопропанол и бутанол (например, н-бутанол); алифатический (например, С6-12, такой как С7-12алифатический) углеводород (например, изогексан, изооктан и н-гептан) и ароматический углеводород (например, толуол); хлорированный алкан (например, хлороформ и дихлорметан); диалкилкетон (например, ацетон, метил-изобутилкетон), ацетонитрил, диалкиловый эфир (например, диэтиловый эфир, ди-изопропиловый эфир, ди-н-пропиловый эфир, ди-н-бутиловый эфир и трет-бутилметиловый эфир); и/или водный растворитель, такой как вода. Могут быть использованы смеси любых из упомянутых выше растворителей.
Различные кристаллические формы могут обладать различной растворимостью в каждом данном растворителе при каждой данной температуре. В связи с этим вышеупомянутые растворители могут быть использованы как "антирастворители" (то есть растворители, в которых соединения по изобретению слабо растворимы) и поэтому могут способствовать процессу кристаллизации. Так, антирастворители включают в себя углеводороды и диалкиловые эфиры, перечисленные выше.
Таким образом, подходящие растворители включают в себя алкилацетаты (такие как этилацетат или изопропилацетат), низшие алкиловые спирты (такие как метанол, этанол и изопропанол), хлорированные метаны (такие как дихлорметан), алканы (такие как н-гептан), простые эфиры (такие как диэтиловый эфир), кетоны (такие как ацетон), воду и так далее.
Кристаллизация соединений по изобретению из соответствующей системы растворителей может быть выполнена путем достижения сверхнасыщения в системе растворителей, которая содержит Соединение А, В, С или D либо его соль (например, путем охлаждения, путем выпаривания растворителей и/или посредством добавления антирастворителя (то есть растворителя, в котором соединения по изобретению слабо растворимы (например, углеводорода, такого как изооктан, н-гептан или изогексан, либо диалкилового эфира, такого как диизопропиловый эфир, ди-н-бутиловый эфир и так далее))), или путем уменьшения растворимости вещества посредством добавления соли (такой как NaCl), или, в случае соединений по изобретению, которые представляют собой соли присоединения кислот, путем добавления избытка соответствующей кислоты.
Температура кристаллизации и время кристаллизации зависят от соединения или соли, которые нужно кристаллизовать, концентрации соединения/соли в растворе и используемой системы растворителей.
Кроме того, кристаллизация может быть инициирована и/или выполнена согласно стандартным методикам, например с использованием или без использования затравки кристаллов соответствующего кристаллического соединения по изобретению и/или доведением рН.
Конкретный процесс кристаллизации, который может быть использован для получения Соединений А, В, С и, в особенности D, и их солей, включает в себя растворение соединения по изобретению в системе растворителей, содержащей С3-7алкиловый спирт и соответствующий ди-С3-5алкиловый эфир в качестве антирастворителя.
Таким образом, согласно еще одному аспекту данного изобретения предложен способ получения по существу кристаллической формы Соединения А, Соединения В, Соединения С или, в особенности, Соединения D либо фармацевтически приемлемой соли любого из этих соединений, при котором осуществляют кристаллизацию релевантного соединения из системы растворителей, содержащей комбинацию С3-7алкилового спирта и ди-С3-5алкилового эфира.
Предпочтительные эфиры включают в себя ди-С3-5алкиловые эфиры, такие как ди-н-пропиловый эфир, диизопропиловый эфир и ди-н-бутиловый эфир. Предпочтительные спирты включают в себя н-пропанол, изопропанол, н-бутанол, 4-метил-2-пентанол, 3-метил-1-бутанол, 2-метил-1-пропанол и пентан-1-ол.
Предпочтительные комбинации растворителей включают в себя:
н-пропанол и ди-н-пропиловый эфир;
изопропанол и диизопропиловый эфир;
н-бутанол и ди-н-бутиловый эфир;
4-метил-2-пентанол и ди-н-бутиловый эфир;
изопропанол и ди-н-бутиловый эфир;
4-метил-2-пентанол и диизопропиловый эфир; и
пентан-1-ол и диизопропиловый эфир.
Соединение/соль могут быть добавлены к системе растворителей спирт/эфир, причем последняя находится в предварительно смешанной форме. Альтернативно, соединение/соль могут быть растворены в соответствующем спирте и затем к полученному раствору может быть добавлен эфир. Соли также могут быть получены in situ, как описано выше.
Предпочтительно соединение/соль растворяют в комбинации растворителей путем нагревания при повышенной температуре (например, от 50 до 100°С, в частности от 65 до 90°С, например от 75 до 85°С) для обеспечения полного растворения. Затем полученный раствор оставляют охлаждаться для осуществления кристаллизации.
Такой процесс кристаллизации из спирта/эфира предпочтительно применяют для получения кристаллических форм Соединения D и его солей. Предпочтительно он может быть использован для получения кристаллического Соединения D в форме свободного основания.
Авторами данного изобретения обнаружено, что когда соединения по изобретению (и, в частности, Соединение D) получают посредством кристаллизации из этой конкретной системы растворителей, то эффективным образом получают высококристаллический материал, с высокой степенью извлечения кристаллического материала (с хорошим выходом) и за предсказуемый промежуток времени.
Кроме того, соединения по изобретению могут быть получены в форме сольвата (в понятие которого авторы изобретения включают форму гидрат, такую как моногидрат) или в другой форме (например, в форме ангидрата).
Чтобы обеспечить образование ангидрата, растворитель, из которого осуществляется кристаллизация, следует предпочтительно обезвоживать, либо перед процессом кристаллизации, либо во время процесса кристаллизации для того, чтобы уменьшить содержание воды ниже критического уровня, который предпочтительно не следует превышать в продолжение кристаллизации. Растворитель может быть обезвожен в продолжение процесса кристаллизации, например, путем уменьшения содержания воды в смеси соединение/соль, которую нужно кристаллизовать, и в соответствующей системе органический растворитель/водный растворитель (например, путем увеличения количества присутствующего органического растворителя и/или удаления воды в результате образования азеотропной смеси с последующими перегонками).
Чтобы обеспечить образование гидратов (например, моногидратов), в растворителе, из которого осуществляется кристаллизация, должна присутствовать вода. В продолжение кристаллизации содержание воды предпочтительно следует поддерживать выше критического уровня, упомянутого выше.
"Критический уровень" воды зависит от таких факторов, как температура, концентрация в растворе соединения, которое нужно кристаллизовать, примесный профиль и используемая система растворителей, но может быть определен неизобретательно.
Так, кристаллические гидраты могут быть получены путем кристаллизации соединений по изобретению из системы растворителей, содержащей воду или комбинацию воды и одного либо более чем одного органического растворителя, включая органические растворители, которые способны к растворению воды (например, метанол, этанол, изопропанол и так далее).
Кристаллические ангидраты, наоборот, могут быть получены путем кристаллизации соединений по изобретению из подходящей системы органических растворителей, которые могут уже быть обезвоженными и/или могут быть обезвожены в продолжение процесса кристаллизации, так чтобы содержание воды было ниже вышеупомянутого критического уровня. Таким образом, ангидрат может быть образован путем кристаллизации из системы растворителей, которая по существу свободна от воды.
В понятие "по существу свободный от воды" авторы включают содержание воды в системе растворителей ниже такого содержания воды, которое будет приводить к образованию максимум 10% гидрата (например, моногидрата) для любой конкретной системы растворителей и совокупности условий кристаллизации.
Соединения по изобретению, представляющие собой ангидраты, содержат не более чем 3%, предпочтительно 2%, более предпочтительно 1% и более предпочтительно 0,5% (по массе) воды, находится ли такая вода в связанном состоянии (кристаллизационная вода или иная) или нет. Гидраты содержат не менее чем 0,5 моль воды на моль соединения по изобретению.
Будут ли ангидраты или гидраты кристаллизоваться, связано с кинетикой и с условиями равновесия соответствующих форм в конкретных условиях.
Если соединение по изобретению представлено в кристаллической форме соли Соединения А с бензолсульфоновой кислотой, предпочтительно, чтобы эта соль была представлена в форме моногидрата.
Кроме того, как очевидно специалисту, соединения по изобретению, находящиеся в одной и той же химической форме (свободное основание/соль), также могут быть получены в различных физических формах (например, различных кристаллических формах) при различных условиях кристаллизации. Получаемая кристаллическая форма соединения по изобретению зависит как от кинетики, так и от термодинамики процесса кристаллизации. При определенных термодинамических условиях (система растворителей, температура, давление и концентрация соединения по изобретению) одна кристаллическая форма может быть более стабильна, чем другая (или, несомненно, любая иная). Однако кристаллические формы, имеющие относительно низкую термодинамическую стабильность, могут иметь кинетические преимущества. Таким образом, в дополнение к этому, кинетические факторы, такие как время, примесный профиль, перемешивание, присутствие или отсутствие затравки и так далее, также могут оказывать влияние на то, какая форма образуется. Таким образом, раскрытые в данном описании методики могут быть адаптированы специалистом, как подходит, для получения различных кристаллических форм.
Различные кристаллические формы соединений по изобретению без труда могут быть охарактеризованы с использованием методов дифракции рентгеновских лучей на порошке (XRPD), например, как описано здесь далее.
Чтобы обеспечить получение конкретной кристаллической формы при отсутствии других кристаллических форм, кристаллизацию предпочтительно осуществляют путем введения затравки из зародышевых и/или затравочных кристаллов желаемой кристаллической формы по существу при полном отсутствии зародышевых и/или затравочных кристаллов других кристаллических форм. Затравочные кристаллы соответствующих соединения/соли могут быть получены, например, посредством медленного выпаривания растворителя из части раствора соответствующих соединения/соли.
Соединения по изобретению могут быть выделены с использованием методик, которые хорошо известны специалистам в данной области техники, например, декантацией, фильтрованием или центрифугированием.
Соединения могут быть высушены с использованием стандартных методик. Специалисту очевидно, что температура сушки и продолжительность сушки могут оказывать влияние на свойства твердого состояния соединений (или солей), находящихся в форме сольватов, таких как гидраты (например, при повышенных температурах и/или пониженном давлении может иметь место дегидратация). Например, после образования кристаллического гидрата критическая влажность может иметь значение, ниже которого высушивание не должно проводиться, поскольку кристаллизационная вода может быть потеряна, и может иметь место трансформация твердого состояния, то есть кристаллизационная вода может быть потеряна, если кристаллы высушены при высоких температурах или при очень низких давлениях в течение более продолжительного периода.
Авторами изобретения обнаружено, что с помощью используемых процессов кристаллизации, как они раскрыты в данном описании, возможно получить соединения по изобретению с более высокой химической чистотой, чем у соединения по изобретению, которое должно быть выделено вначале.
Дальнейшая очистка соединений по изобретению может быть выполнена с использованием методик, которые хорошо известны специалистам в данной области техники. Например, примеси могут быть удалены посредством перекристаллизации из системы соответствующих растворителей (например, этилацетата, изопропилацетата, дихлорметана, изопропанола, н-гептана, метанола, этанола, метилэтилкетона, ацетонитрила, пентан-2-ола, 3-метилбутан-1-ола, гексан-1-ола, воды или комбинации этих растворителей). Подходящие температуры и время перекристаллизации зависят от концентрации соединения или соли в растворе и от используемой системы растворителей.
Когда соединения по изобретению кристаллизованы или перекристаллизованы, как здесь описано, полученное соединение или соль может находиться в форме, имеющей улучшенную химическую стабильность и/или стабильность твердого состояния, как они упомянуты здесь выше.
Фармацевтические препараты и терапевтическое применение.
Соединения по изобретению полезны, так как они обладают фармакологической активностью. Поэтому они показаны в качестве фармацевтических средств.
Таким образом, согласно еще одному аспекту изобретения предложены соединения по изобретению для применения в качестве фармацевтических средств.
В частности, соединения по изобретению проявляют миокардиальную электрофизиологическую активность, например, как продемонстрировано в тестах, таких как тесты, описанные среди прочего в публикациях международных заявок WO 99/31100, WO 00/77000 и WO 01/28992, релевантные описания которых включены посредством ссылки.
Таким образом, ожидается, что соединения по изобретению будут полезны как при профилактике, так и при лечении аритмий, и в частности, предсердной и желудочковой аритмий. Конкретные болезненные состояния, которые могут быть упомянуты, включают в себя мерцательную аритмию (например, трепетание предсердий).
Таким образом, соединения по изобретению показаны при лечении или профилактике сердечных заболеваний либо при показаниях, относящихся к сердечным заболеваниям, при которых, как полагают, аритмии играют главную роль, включая ишемическую болезнь сердца, внезапный сердечный приступ, инфаркт миокарда, сердечную недостаточность, операцию на сердце и случаи тромбоэмболии.
При лечении аритмий было обнаружено, что соединения по изобретению селективно замедляют сердечную реполяризацию. Несмотря на то, что было обнаружено, что соединения по изобретению замедляют сердечную реполяризацию, в частности, при лечении аритмий, способ(ы) их действия необязательно ограничен(ы) этим способом действия.
Согласно еще одному аспекту данного изобретения предложен способ лечения аритмии, при котором субъекту, страдающему от такого состояния или подверженному такому состоянию, вводят терапевтически эффективное количество соединения по изобретению.
Во избежание неопределенности, в термин "лечение" авторы включают терапевтическое лечение состояния, а также профилактику состояния.
Соединения по изобретению обычно будут вводить перорально, подкожно, внутривенно, внутриартериально, трансдермально, интраназально, посредством ингаляции или посредством любого другого парентерального пути введения, в форме фармацевтических препаратов, содержащих активный ингредиент (либо в виде свободного основания, либо в виде соли), в фармацевтически приемлемой лекарственной форме. В зависимости от заболевания и пациента, которого лечат, а также от способа введения, композиции могут быть введены в различных дозах.
Кроме того, соединения по изобретению можно объединять с любыми другими лекарствами, полезными при лечении аритмий и/или других сердечно-сосудистых заболеваний.
Таким образом, согласно еще одному аспекту изобретения предложен фармацевтический препарат, включающий в себя соединение по изобретению в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Соединения по изобретению могут быть подвергнуты дальнейшей обработке перед изготовлением препарата в форме подходящего фармацевтического препарата. Например, кристаллическую форму можно измельчить или размолоть до частиц меньшего размера.
Предпочтительные фармацевтические препараты включают в себя препараты в форме гелеобразующей матричной системы, в которой соединения по изобретению представлены в ассоциации с полимером, который набухает в водной среде, такой как обнаруживаемая в желудочно-кишечном тракте. Такие системы хорошо известны специалистам в данной области техники (смотри, например, Advanced Drug Delivery Reviews, 11, 37 (1993)).
Количество соединения по изобретению, которое используют в таком препарате, будет зависеть от состояния и пациента, которого лечат, а также от используемого(ых) соединения(ий), но может быть определено неизобретательно.
Подходящие суточные дозы соединений по изобретению при (например, терапевтическом) лечении людей составляют приблизительно от 0,005 до 25,0 мг/кг массы тела при пероральном введении и приблизительно от 0,005 до 10,0 мг/кг массы тела при парентеральном введении для свободного основания (то есть, в случае соли, за вычетом любой массы, получающейся в результате присутствия противоиона). Предпочтительные интервалы суточных доз соединений по изобретению (в форме свободного основания, как показано выше) при (например, терапевтическом) лечении людей составляют приблизительно от 0,005 до 10,0 мг/кг массы тела при пероральном введении и приблизительно от 0,005 до 5,0 мг/кг массы тела при парентеральном введении.
Соединения по изобретению имеют преимущество в том, что они эффективны против сердечных аритмий.
Кроме того, соединения по изобретению имеют преимущество в том, что они могут быть более эффективны, менее токсичны, иметь более широкий интервал действия (включая проявление любой комбинации активности класса I, класса II, класса III и/или класса IV (в особенности активности класса I и/или класса IV в дополнение к активности класса III)), оказывать более сильное действие, иметь более продолжительное действие, вызывать меньше побочных эффектов (включая более низкий процент проаритмий, таких как torsades de pointes), легче всасываться, чем соединения, известные из уровня техники, или в том, что они могут обладать другими полезными фармакологическими свойствами сверх имеющихся у соединений, известных из уровня техники.
Соединения по изобретению имеют преимущество в том, что они находятся в форме, которая обеспечивает повышенное удобство в обращении. Кроме того, соединения по изобретению имеют преимущество в том, что они могут быть изготовлены в форме, которая обладает улучшенной химической стабильностью и стабильностью твердого состояния (включая, например, пониженную гигроскопичность). Таким образом, данные соединения могут быть стабильны при хранении в продолжение длительных периодов времени.
Соединения по изобретению могут также иметь преимущество в том, что они могут кристаллизоваться с хорошими выходами, с более высокой степенью чистоты, за меньший период времени, более удобным образом и при более низкой стоимости, чем формы соединений А, В, С и D и их солей, полученные ранее.
Изобретение проиллюстрировано, но никоим образом этим не ограничено, следующими далее примерами со ссылкой на прилагаемые фигуры, где:
на Фиг.1 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы Соединения А (формы В), полученной способом из Примера 1;
на Фиг.2 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы Соединения А (формы А), полученной способом из Примера 2;
на Фиг.3 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы Соединения А (формы С), полученной способом из Примера 3;
на Фиг.4 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения А с бензолсульфоновой кислотой, полученной способом из Примера 4;
на Фиг.5(а) показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения А с паратолуолсульфоновой кислотой (формы А), полученной способом из Примера 5;
на Фиг.5(б) показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения А с паратолуолсульфоновой кислотой (формы В), полученной способом из Примера 5;
на Фиг.6 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения А с 1-гидрокси-2-нафтойной кислотой, полученной способом из Примера 6;
на Фиг.7 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения А с 1,5-нафталинсульфоновой кислотой, полученной способом из Примера 7;
на Фиг.8 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения А с 2-мезитиленсульфоновой кислотой, полученной способом из Примера 8;
на Фиг.9 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы Соединения D, полученной способом из Примера 9;
на Фиг.10 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения D с метансульфоновой кислотой, полученной способом из Примера 10;
на Фиг.11 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения D с гиппуровой кислотой, полученной способом из Примера 11;
на Фиг.12 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы Соединения С, полученной способом из Примера 12;
на Фиг.13 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения С с метансульфоновой кислотой, полученной способом из Примера 13;
на Фиг.14 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения С с паратолуолсульфоновой кислотой, полученной способом из Примера 14;
на Фиг.15 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения D с [(дифенил-4-карбонил)-амино]уксусной кислотой, полученной способом из Примера 15;
на Фиг.16 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения D с гемиянтарной кислотой, полученной способом из Примера 16;
на Фиг.17 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения D с (3,4-дихлорбензоиламино)-уксусной кислотой, полученной способом из Примера 17;
на Фиг.18 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения D с [(нафталин-2-карбонил)-амино]уксусной кислотой, полученной способом из Примера 18;
на Фиг.19 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения D с 2,2,3,3-тетраметил-1,4-дибутановой кислотой, полученной способом из Примера 19;
на Фиг.20 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения D с транс-D,L-1,2-циклопентан-дикарбоновой кислотой, полученной способом из Примера 20;
на Фиг.21 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения D с (+)-O,O'-дибензоил-D-винной кислотой, полученной способом из Примера 21;
на Фиг.22 показана дифрактограмма рентгеновских лучей на порошке для кристаллической формы соли Соединения D с (+)-O,O'-ди-паратолуоил-D-винной кислотой, полученной способом из Примера 22.
Общие методики
Анализ дифракции рентгеновских лучей на порошке (XRPD) выполняли с использованием вариабельных щелей на образцах, приготовленных согласно стандартным способам, например, как описано в Giacovazzo, С. et al (1995), Fundamentals of Crystallography, Oxford University Press; Jenkins, R. and Snyder, R.L. (1996), Introduction to X-Ray Powder Diffractometry, John Wiley & Sons, New York; Bunn, C.W. (1948), Chemical Crystallography, Clarendon Press, London; или Klug, H.P. & Alexander, L.E. (1974), X-ray Diffraction Procedures, John Wiley & Sons, New York. Рентгеновский анализ выполняли с использованием дифрактометра Siemens D5000.
Дифференциальную сканирующую калориметрию (DSC) выполняли с использованием прибора Mettler DSC820 согласно стандартным способам, например, как описано в Höhne, G. W. H. et al (1996), Differential Scanning Calorimetry, Springer, Berlin.
Термогравиметрический анализ (TGA) выполняли с использованием прибора Mettler Toledo TGA850.
Специалисту очевидно, что кристаллические формы соединений по изобретению могут быть получены аналогично способам, описанным здесь, и/или в соответствии с приведенными ниже Примерами, и могут показывать по существу одни и те же картины дифракции XRPD и/или DSC и/или термограммы TGA, как те, что представлены здесь. В понятие "по существу одни и те же" картины дифракции XRPD и/или DSC и/или термограммы TGA авторы включают такие случаи, когда из релевантных картин и/или термограмм (допуская ошибку эксперимента) ясно, что образована по существу одна и та же кристаллическая форма. Если предусмотрено, то точки начала в DSC могут изменяться в интервале ±5°С (например, ±2°C), и значения XRPD-расстояний (d) могут изменяться в интервале ±2 последнего десятичного разряда. Специалисту очевидно, что интенсивности XRPD могут изменяться при измерении по существу одной и той же кристаллической формы по ряду причин, включая, например, предпочтительную ориентацию.
Подготовительный пример А
Получение соединения А и его бензолсульфонатной соли
(1) 4-[(3-Гидроксипропил)амино]бензонитрил
Вариант 1. Смесь 4-фторбензонитрила (12,0 г; 99,1 ммоль) и 3-амино-1-пропанола (59,6 г; 793 ммоль) перемешивали при 80°С в атмосфере инертного газа в течение 3 часов перед добавлением воды (150 мл). Смесь оставляли охлаждаться до комнатной температуры и затем экстрагировали диэтиловым эфиром. Органический слой отделяли, сушили (Na2SO4), отфильтровывали и концентрировали под вакуумом, получая 17 г (97%) соединения, указанного в подзаголовке, в виде масла, которое кристаллизовалось при стоянии.
Вариант 2. 4-Фторбензонитрил (24,6 г; 0,203 моль, Aldrich 99%) добавляли к 3-амино-1-пропанолу (122,0 г; 1,625 моль; 8 экв.; Aldrich 99%) и смесь нагревали до 80°С в течение 5 часов в атмосфере азота. Раствор оставляли охлаждаться до 22°С и добавляли воду (300 мл). Мутный раствор дважды экстрагировали метиленхлоридом (300 мл и 200 мл) и объединенные метиленхлоридные экстракты промывали водой (300 мл; анализ ГХ (газовая хроматография) органического слоя показал ˜1% площади остаточного аминопропанола).
Вариант 3. К 4-фторбензонитрилу (30,29 г; 247,7 ммоль; 1,0 экв.) добавляли 3-амино-1-пропанол (150 мл; 148,8 г; 1981,5 ммоль; 8,0 экв.). Смесь перемешивали в атмосфере азота при комнатной температуре (27°С) до полного растворения твердой фазы. Раствор нагревали (масляная баня) до 77°С и выдерживали при этой температуре в течение 7 часов, перед тем как перемешивать при температуре окружающей среды в течение ночи (14 часов). Добавляли воду (365 мл) и полученный в результате мутный раствор экстрагировали дихлорметаном (365 мл, затем 245 мл). Объединенные органические слои промывали водой (365 мл). Раствор продукта в дихлорметане (ДХМ) сушили путем перегонки: растворитель удаляли (200 мл) и замещали свежим ДХМ (200 мл). Избыток растворителя (250 мл) удаляли, доводя общий объем растворителя до 365 мл.
(2) 3-(4-Цианоанилино)пропил-4-метилбензолсульфонат
Вариант 1. Охлажденный раствор (0°С) 4-[(3-гидроксипропил)-амино]бензонитрила (с вышеуказанной стадии (1) (Вариант 1); 17 г; 96,5 ммоль) в безводном MeCN (195 мл) обрабатывали триэтиламином (9,8 г; 96,5 ммоль) и затем паратолуолсульфонилхлоридом (20,2 г; 106 ммоль). Смесь перемешивали при 0°С в течение 90 минут перед концентрированием под вакуумом. К остатку добавляли воду (200 мл) и водный раствор экстрагировали ДХМ. Органическую фазу сушили (Na2SO4), фильтровали и концентрировали под вакуумом. Получающийся остаток очищали посредством кристаллизации из изопропанола с получением 24,6 г (77%) соединения, указанного в заголовке.
Вариант 2. Раствор неочищенного 4-[(3-гидроксипропил)амино]-бензонитрила (с вышеуказанной стадии (1) (Вариант 2)) концентрировали до объема 300 мл посредством перегонки, добавляли дополнительные 200 мл метиленхлорида и повторно перегоняли до 300 мл (водный раствор по Карлу-Фишеру. 0,07%). Добавляли триэтиламин (20,55 г; 0,203 моль) с последующим добавлением 4-(N,N-диметиламино)пиридина (248 мг; 2,0 ммоль) и охлаждали раствор до 0°С. Добавляли раствор тозилхлорида (38,70 г; 0,203 моль) в метиленхлориде (150 мл) в течение примерно 30 минут при охлаждении и достаточном перемешивании, давая температуре подняться до 5°С. Реакционную смесь перемешивали в течение 23 часов в интервале температур от 3 до 5°С в атмосфере азота. (После 5 часов происходило осаждение гидрохлорида триэтиламина. ТСХ (тонкослойная хроматография) показала очень незначительное, если таковое вообще имелось, дальнейшее превращение остаточного цианоспирта на 20-23 часа.) Добавляли воду (300 мл) и энергично перемешивали слои в течение 15 минут. Органический раствор концентрировали путем перегонки при температуре от 35 до 40°С до объема примерно от 60 до 70 мл. Добавляли изопропанол (100 мл) в течение 5 минут. (На этой стадии перед добавлением изопропанола происходило незначительное осаждение продукта в виде зернистого осадка. При добавлении изопропанола кристаллизация происходила быстро). Перегонку продолжали, используя вакуум, для того чтобы удалить остатки метиленхлорида. (Удалили еще ˜30 мл и дистиллят проверяли посредством ГХ на отсутствие метиленхлорида). Суспензию кристаллов охлаждали до температуры в пределах от 0 до 5°С в течение примерно 1 часа при медленном перемешивании и выдерживали в течение одного часа при 0-5°С. Кристаллы отфильтровывали через фильтрующий материал и плотный влажный осадок на фильтре осторожно промывали холодным (0°С) изопропанолом (80 мл). Осадок на фильтре сушили под вакуумом и в потоке азота в течение ночи. Выход: 52,6 г; 78,4 моль%; ВЭЖХ (высокоэффективная жидкостная хроматография): 99,64% площади.
Микроанализ: обнаружено (теоретически): % С: 61,60 (61,67); % Н: 5,41 (5,49); % N: 8,44 (8,47); % S: 9,71 (9,70).
(3) N,N-бис(2-Оксиранилметил)бензолсульфонамид
К бензолсульфонамиду (250 г; 1 экв.) добавляли воду (2,5 л; 10 об.) с последующим добавлением эпихлоргидрина (500 мл; 4 экв.). Реагенты нагревали до 40°С. Водный гидроксид натрия (130 г в 275 мл воды) добавляли так, чтобы температура реакционной смеси оставалась между 40 и 43°С. Это занимало приблизительно 2 часа. (Необходимо, чтобы скорость добавления гидроксида натрия была медленнее в начале добавления, чем в конце, для того чтобы поддерживать установленные пределы температур). После того как добавление гидроксида натрия было закончено, реакционную смесь перемешивали при 40°С в течение 2 часов, а затем при температуре окружающей среды в течение ночи. Избыток эпихлоргидрина удаляли в виде водной азеотропной смеси путем вакуумной перегонки (примерно 4000 Па (40 мбар); внутренняя температура 30°С) до полной отгонки эпихлоргидрина. Добавляли дихлорметан (1 л) и эту смесь быстро перемешивали в течение 15 минут. Фазы оставляли расслаиваться (это занимало 10 минут, хотя абсолютно чистые фазы получали после отстаивания в течение ночи). Фазы разделяли и раствор дихлорметана использовали на следующей стадии, приведенной ниже.
1H ЯМР (400 МГц, CDCl3): δ 2.55-2.65 (2Н, m), 2.79 (2Н, t, J4.4), 3.10-3.22 (4H, m), 3.58-3.73 (2Н, m), 7.50-7.56 (2Н, m), 7.58-7.63 (1H, m), 7.83-7.87 (2Н, m).
(4) 5-Бензил-3,7-дигидрокси-1-фенилсульфонил-1,5-диазациклооктан
IMS (промышленный метилированный спирт) (2,5 л; 10 об.) добавляли к раствору дихлорметана, полученному на приведенной выше стадии (3). Раствор перегоняли до тех пор, пока внутренняя температура не достигла 70°С. Собирали примерно 1250 мл растворителя. Добавляли избыток IMS (2,5 л; 10 об.) с последующим добавлением бензиламина (120 мл; 0,7 экв.) в виде одной порции (выделения тепла не наблюдалось) и нагревали реакционную смесь с обратным холодильником в течение 6 часов (никаких изменений, начиная с пробной точки через 2 часа). Добавляли избыток бензиламина (15 мл) и нагревали раствор в течение еще 2 часов. IMS отгоняли (примерно 3,25 л) и добавляли толуол (2,5 л). Избыток растворителя перегоняли (примерно 2,4 л) и затем добавляли дополнительное количество толуола (1 л). Температура в верхней части дистиллятора теперь составляла 110°С. Собирали еще 250 мл растворителя при 110°С. Теоретически, после этого оставался продукт в примерно 2,4 л толуола при 110°. Этот раствор использовали на следующей стадии.
1H ЯМР (400 МГц, CDCl3): δ 7.83-7.80 (4H, m, ArH), 7.63-7.51 (6Н, m, ArH), 7.30-7.21 (10Н, ArH), 3.89-3.80 (4H, m, CH(a)+CH(b)), 3.73 (2Н, s, CH2Ph(a)), 3.70 (2Н, s, CH2Ph(b)), 3.59 (2Н, dd, CHHNSO2Ar(a)), 3.54 (2Н, dd, CHHNSO2Ar(b)), 3.40 (2Н, dd, CHHNSO2Ar(b)), 3.23 (2Н, dd, CHHNSCO2Ar(a)), 3.09-2.97 (4H, m, CHHNBn(a) + CHHNBn(b)), 2.83 (2Н, dd, CHHNBn(b)), 2.71 (2Н, dd, CHHNBn(a)).
(Данные получены на очищенном материале, содержащем смесь 1:1 транс- (а) и цис-диола (b)).
(5) 3-Бензил-7-(фенилсульфонил)-9-окса-3,7-диазабицикло[3.3.1]нонан
Раствор толуола с предыдущей стадии (4), приведенной выше, охлаждали до 50°С. Добавляли безводную метансульфоновую кислоту (0,2 л). Это вызывало повышение температуры от 50 до 64°С. Через 10 минут добавляли метансульфоновую кислоту (1 л) и реакционную смесь нагревали до 110°С в течение 5 часов. Затем отгоняли толуол из реакционной смеси; собрали 1,23 л. (Обратите внимание, что внутренняя температура не должна превышать 110°С на любой стадии, в противном случае выход будет снижаться). Реакционную смесь затем охлаждали до 50°С и использовали вакуум для удаления остатков толуола. Нагревание до 110°С под давлением 65000 Па (650 мбар) позволило удалить еще 0,53 л. (Если толуол может быть удален при более низкой температуре и давлении, это выгодно). Реакционную смесь затем оставляли для охлаждения до 30°С и добавляли деионизированную воду (250 мл). Это вызывало повышение температуры от 30 до 45°С. Добавляли избыток воды (2,15 л) в течение общего периода времени 30 минут, так чтобы температура была меньше чем 54°С. Раствор охлаждали до 30°С и затем добавляли дихлорметан (2 л). При внешнем охлаждении и быстром перемешивании реакционную смесь подщелачивали посредством добавления водного гидроксида натрия (10 М; 2 л) с такой скоростью, чтобы поддерживать внутреннюю температуру ниже 38°С. Это занимало 80 минут. Перемешивание прекращали и разделяли фазы в течение 3 минут. Слои отделяли друг от друга. К раствору дихлорметана добавляли IMS (2 л) и начинали перегонку. Растворитель (2,44 л) собирали до тех пор, пока температура в верхней части дистиллятора не достигла 70°С. Теоретически, после этого оставался продукт в 1,56 л IMS. Этот раствор затем оставляли охлаждаться до температуры окружающей среды в течение ночи при медленном перемешивании. Осажденный твердый продукт отфильтровывали и промывали IMS (0,5 л) с получением продукта желтовато-коричневого цвета, который после сушки при 50°С под вакуумом давал 50,8 г (8,9% по 3 стадиям). 20,0 г этого продукта растворяли в ацетонитриле (100 мл) при температуре дефлегмации, получая бледно-желтый раствор. После охлаждения до температуры окружающей среды образовавшиеся кристаллы собирали путем фильтрации и промывали ацетонитрилом (100 мл). Продукт сушили под вакуумом при 40°С в течение 1 часа с получением 17,5 г (87%) соединения, указанного в подзаголовке.
1H ЯМР (400 МГц, CDCl3): δ 7.18-7.23 (10Н, m), 3.86-3,84 (2Н, m), 3.67 (2H, d), 3.46 (2H, s), 2.91 (2H, d), 2.85 (2H, dd), 2.56 (2H, dd).
(6) 3-Бензил-9-окса-3,7-диазабицикло[3.3.1]нонан × 2HCl
Концентрированную бромистоводородную кислоту (1,2 л; 3 отн. (rel) об.) добавляли к твердому 3-бензил-7-(фенилсульфонил)-9-окса-3,7-диазабицикло[3.3.1]нонану (400 г; смотри выше стадию (5)) и эту смесь нагревали до температуры дефлегмации в атмосфере азота. Твердое вещество растворяли в кислоте при 95°С. После нагревания реакционной смеси в течение 8 часов анализ ВЭЖХ показал, что реакция завершена. Содержимое охлаждали до комнатной температуры. Добавляли толуол (1,2 л; 3 отн. об.) и эту смесь энергично перемешивали в течение 15 минут. Перемешивание прекращали и фазы отделяли друг от друга. Фазу толуола вместе с небольшим количеством материала с межфазной поверхности раздела отбрасывали. Кислотную фазу возвращали в исходный реакционный сосуд и добавляли гидроксид натрия (10 M; 1,4 л; 3,5 отн. об.) в виде одной порции. Внутреннюю температуру поднимали от 30 до 80°С. Контролировали рН, чтобы убедиться, что оно составляет >14. Добавляли толуол (1,6 л; 4 отн. об.) и понижали температуру от 80 до 60°С. После энергичного перемешивания в течение 30 минут фазы отделяли друг от друга. Водный слой вместе с небольшим количеством материала с межфазной поверхности раздела отбрасывали. Фазу толуола возвращали в исходный реакционный сосуд и добавляли 2-пропанол (4 л; 10 отн. об.). Температуру устанавливали между 40 и 45°С. Добавляли концентрированную соляную кислоту (200 мл) в течение 45 минут, так чтобы температура оставалась между 40 и 45°С. Образовывался белый осадок. Смесь перемешивали в течение 30 минут и затем охлаждали до 7°С. Продукт собирали путем фильтрации, промывали 2-пропанолом (0,8 л; 2 отн. об.), сушили посредством отсасывания жидкости и затем дополнительно сушили в вакуум-сушильном шкафу при 40°С. Выход = 297 г (91%).
1H ЯМР (CD3OD+4 капли D2O): δ 2.70 (br d, 2H), 3.09 (d, 2H), 3.47 (br s, 4H), 3.60 (s, 2H), 4.12 (br s, 2H), 7.30-7.45 (m, 5H).
API MS: m/z=219 [C13H18+N2O+H]+.
(7) 3,3-Диметил-1-[9-окса-7-(фенилметил)-3,7-диазабицикло[3.3.1]нон-3-ил]-2-бутанон
К бикарбонату натрия (114,2 г; 4 экв.) добавляли воду (500 мл; 5 об.) с последующим добавлением 1-хлорпинаколина (45,8 мл; 1 экв.). Медленно добавляли раствор 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонана × 2HCl (100,0 г; смотри выше стадию (6)) в воде (300 мл; 3 об.), так чтобы контролировать выделение диоксида углерода (20 минут). Реакционную смесь нагревали при температуре от 65 до 70°С в течение 4 часов. После охлаждения до температуры окружающей среды добавляли дихлорметан (400 мл; 4 об.) и после перемешивания в течение 15 минут фазы разделяли. Водную фазу промывали дихлорметаном (400 мл; 4 об.), а органические экстракты объединяли. Раствор перегоняли и собирали растворитель (550 мл). Добавляли этанол (1 л) и продолжали перегонку. Продолжали собирать растворитель (600 мл). Добавляли этанол (1 л) и продолжали перегонку. Продолжали собирать растворитель (500 мл) (температура в верхней части дистиллятора теперь составляла 77°С). Этот раствор (теоретически содержащий 1150 мл этанола) непосредственно использовали на следующей стадии.
1H ЯМР (400 МГц, CDCl3): δ 1.21 (9Н, s), 2.01-2.59 (2Н, m), 2.61-2.65 (2H, m), 2.87-2.98 (4H, m), 3.30 (2H, s), 3.52 (2H, s), 3.87 (2H, br s), 7.26 (2H, d, J 7.6), 7.33 (1H, dd, J 7.6, 7.6), 7.47 (2H, d, J 7.6).
(8) 3,3-Диметил-1-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)-2-бутанон
К этанольному раствору с предыдущей стадии (7), приведенной выше, добавляли палладий на угле (44 г; 0,4 вес. экв. 61%-ного влажного катализатора; Johnson Matthey Type 440L). Смесь гидрировали под давлением 400000 Па (4 бар). Реакцию считали завершенной через 5 часов. Катализатор удаляли путем фильтрования и промывали этанолом (200 мл). Объединенные этанольные фильтраты использовали на стадии (9), приведенной ниже. Анализ раствора показал наличие 61,8 г указанного в заголовке продукта в этаноле (теоретически 1,35 л; при измерении 1,65 л). Часть продукта выделяли и очищали. Анализ проводили на очищенном продукте.
1H ЯМР (300 МГц, CDCl3): δ 1.17 (9Н, s), 2.69 (2H, dt, J 11.4, 2.4), 2.93 (2H, d, J 10.8), 3.02 (2H, d, J 13.8), 3.26 (2H, s), 3.32 (2H, dt, J 14.1), 3.61 (2H, br s).
Эту реакцию можно также проводить, используя более низкое массовое отношение катализатора к бензилированному исходному материалу. Это может быть достигнуто несколькими различными способами, например с использованием различных катализаторов (таких как Pd/C с металлическим наполнением, отличающимся от того, которое присутствует в катализаторе Type 440 L, используемом выше, или Rh/C) и/или путем улучшения характеристик массообмена реакционной смеси (специалисту в данной области техники будет понятно, что улучшения массообмена можно добиться, например, посредством проведения гидрирования в более крупном масштабе чем тот, который описан в вышеприведенной реакции). При использовании таких методов массовое отношение катализатора к исходному материалу можно снизить до отношения ниже 4:10 (например, между 4:10 и 1:20).
(9) Моногидрат соли Соединения А с бензолсульфоновой кислотой
Способ 1
Карбонат калия (56,6 г; 1,5 экв.) и 3-(4-цианоанилино)пропил-4-метилбензолсульфонат (смотри выше стадию (2); 90,3 г; 1 экв.) добавляли к этанольному раствору 3,3-диметил-1-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)-2-бутанона (смотри выше стадию (8); 61,8 г из анализа в 1,65 л). Реакционную смесь нагревали при 80°С в течение 4 часов. Анализ показал, что остается некоторое количество реагента (8,3 г), поэтому добавляли избыток 3-(4-цианоанилино)пропил-4-метилбензолсульфоната (12,2 г) и нагревали полученную в результате смесь при 80°С в течение 4 часов. Растворитель (1,35 л) перегоняли, затем добавляли изопропилацетат (2,5 л). Растворитель (2,51 л) удаляли. Добавляли изопропилацетат (2,5 л). Растворитель (0,725 л) удаляли. Внутренняя температура теперь составляла 88°С. Растворитель (0,825 л) удаляли, оставляя продукт в виде раствора в изопропилацетате (теоретически в 2,04 л). После охлаждения до 34°С добавляли воду (0,5 л). Смесь содержала черную суспензию, возможно палладия. рН водной фазы составлял 11. Гидроксид натрия (1 М; 0,31 л) добавляли так, чтобы температура была меньше чем 25°С, и энергично перемешивали эту смесь в течение 5 минут. рН водной фазы составлял 12. Фазы разделяли и водную фазу отбрасывали. Добавляли избыток воды (0,5 л) и разделяли фазы. Водную фазу отбрасывали. Оставшийся эфирный раствор фильтровали для удаления взвешенных частиц и затем объем фильтрата доводили в точности до 2 л. Затем этот раствор разделяли на порции 2×1 л.
(Для того чтобы избежать получения указанного в подзаголовке продукта с высоким содержанием палладия, можно провести следующую обработку: смолу Deloxan® (12,5 г; 25 мас.%) добавляли к раствору свободного основания (1 л) и эту смесь нагревали с обратным холодильником при энергичном перемешивании в течение 5 часов. Затем раствор охлаждали до комнатной температуры и перемешивали в течение 2 дней. Смолу удаляли путем фильтрации).
Чтобы рассчитать требуемое количество бензолсульфоновой кислоты для получения соли бензолсульфоната, проводили анализ.
Раствор бензолсульфоновой кислоты (20,04 г; 1 экв.; принимая, что кислота представляла собой чистый моногидрат) в изопропилацетате (200 мл) добавляли в течение 5 минут (лучше добавлять медленнее, если возможно) при энергичном перемешивании к раствору свободного основания (1 л) и образовывался бледно-желтый осадок. Температура повышалась от 18 до 22°С. Через 10 минут смесь охлаждали до 10°С и продукт собирали путем фильтрации. Продукт промывали изопропилацетатом (250 мл), затем сушили на фильтре посредством отсасывания жидкости под вакуумом при 40°С в течение 2 дней, получая 59,0 г (61% от 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонана × 2HCl).
(Альтернативно, неочищенную соль бензолсульфонат получали путем добавления 70%-ного (мас./мас.) водного раствора бензолсульфоновой кислоты к этанольному раствору свободного основания).
Неочищенный продукт, указанный в подзаголовке, выделяют в виде моногидрата.
К неочищенному соединению (50,0 г), указанному в подзаголовке, добавляли этанол (500 мл) и воду (250 мл). Раствор нагревали до 75°С. Материал полностью растворялся при 55°С. Раствор выдерживали при 75°С в течение 5 минут, затем охлаждали до 5°С в течение 1 часа. Осаждение начиналось при 18°С. Холодный раствор фильтровали и фильтрат промывали смесью этанол:вода (2:1; 150 мл), сушили на фильтре посредством отсасывания жидкости и затем сушили под вакуумом при 40°С, получая чистый продукт, указанный в подзаголовке (41,2 г; 82%).
(Эту перекристаллизацию можно проводить с использованием больших объемов растворителя, если необходимо, подбирая реакционные сосуды, например:
EtOH:вода, 2:1, 45 об. (давал выход 62%),
EtOH:вода, 6:1, 35 об. (давал выход 70%)).
Продукт, указанный в подзаголовке, выделяли в виде моногидрата после перекристаллизации (как определено путем рентгеновской дифрактометрии на монокристалле).
Способ 2
(а) 3-(4-Цианоанилино)пропилбензолсульфонат
К раствору 4-[(3-гидроксипропил)амино]бензонитрила (со стадии (1) вышеуказанного Варианта 3; исходные 43,65 г; 247,7 ммоль; 1,0 экв.) в дихлорметане (360 мл общего объема раствора) последовательно добавляли триэтиламин (52 мл; 37,60 г; 371,55 ммоль; 1,5 экв.) и гидрохлорид триметиламина (11,89 г; 123,85 ммоль; 0,5 экв.) в виде одной порции. Желтый раствор охлаждали до -20°С (используя баню ацетон/сухой лед или охлаждающую пластину) и обрабатывали раствором бензолсульфонилхлорида (32 мл; 43,74 г; 247,7 ммоль; 1,0 экв.) в дихлорметане (220 мл; 5 об. относительно цианоспирта) с помощью капельной воронки для выравнивания давления. Раствор добавляли порциями, так чтобы внутренняя температура не превышала -14°С. Чтобы закончить добавление, потребовалось 25 минут. Затем смесь перемешивали в течение 35 минут при температуре между -15 и -10°С. Добавляли воду (365 мл), и температура поднималась до 10°С. Смесь снова охлаждали до 0°С и энергично перемешивали в течение 15 минут. Органический слой (объем 570 мл) собирали и перегоняли при атмосферном давлении для удаления ДХМ (450 мл; температура куба 40-42°С; температура в верхней части дистиллятора 38-39°С). Добавляли этанол (250 мл) и раствор оставляли охлаждаться до температуры ниже 30°С, перед тем как создать вакуум. Избыток растворителя удаляли (40 мл собирали; давление 5,2 кПа (52 мбар); температура куба и в верхней части дистиллятора составляла 21-23°С) и продукт постепенно выпадал из раствора. В этот момент перегонку прекращали и добавляли избыток этанола (50 мл). Эту смесь нагревали (горячая водяная баня при 50°С) до 40°С для растворения всего твердого вещества и медленно добавляли воду (90 мл) с помощью капельной воронки. Раствор медленно перемешивали при комнатной температуре (20°С) в течение ночи (15 часов), к этому времени некоторая часть продукта выкристаллизовывалась из раствора. Смесь охлаждали до -5°С (баня лед/метанол) и перемешивали при этой температуре в течение 20 минут, перед тем как путем фильтрации собрать бледно-желтое твердое вещество. Это твердое вещество промывали смесью этанол/вода (42 мл EtOH; 8 мл Н2О) и сушили посредством отсасывания жидкости в течение 30 минут перед сушкой до постоянного веса в вакуум-сушильном шкафу (40°С; 72 часа). Масса полученного неочищенного продукта составляла 47,42 г (149,9 ммоль; 60%). К неочищенному продукту (20,00 г; 63,22 ммоль; 1,0 экв.) добавляли этанол (160 мл; 8 об.). Эту смесь перемешивали в атмосфере азота и нагревали до 40°С, используя горячую водяную баню. При достижении этой температуры все твердое вещество растворялось, давая прозрачный желтый раствор. Воду (60 мл; 3 об.) добавляли по каплям в течение периода 10 минут и в то же время поддерживали внутреннюю температуру в пределах 38-41°С. Убирали водяную баню и раствор оставляли охлаждаться до 25°С в течение 40 минут, к этому времени начиналась кристаллизация. Смесь охлаждали до -5°С в течение 10 минут, затем выдерживали при этой температуре в течение еще 10 минут. Бледно-желтое твердое вещество собирали путем фильтрации, сушили посредством отсасывания жидкости в течение 10 минут, затем сушили до постоянного веса в вакуум-сушильном шкафу (40°С; 15 часов). Масса полученного соединения, указанного в заголовке, составляла 18,51 г (58,51 ммоль; 93% (от неочищенного продукта)).
(б) Моногидрат соли Соединения А с бензолсульфоновой кислотой
К этанольному раствору (общий объем 770 мл; приблизительно 20 об. относительно амина) 3,3-диметил-1-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)-2-бутанона (исходные 34,97 г (подтверждено анализом); 154,5 ммоль; 1,0 экв.; смотри выше стадию (8)) добавляли 3-(4-цианоанилино)пропил-бензолсульфонат (49,05 г; 154,52 ммоль; 1,0 экв.; смотри выше стадию (а)) в виде одной порции. Полученную в результате смесь нагревали при 74°С в течение 6 часов, затем перемешивали при комнатной температуре (20°) в течение 65 часов (в течение выходных дней; специалисту в данной области техники будет понятно, что реакция будет также успешно протекать и без этого длительного перемешивания при комнатной температуре). Этанол (370 мл) удаляли и добавляли воду (200 мл) (получилась смесь EtOH:Н2О, 2:1, общий объем 600 мл). При добавлении воды температура куба снижалась с 80 до 61°С. Раствор вновь нагревали до 70°С, затем оставляли охлаждаться естественным путем до температуры окружающей среды в течение ночи (19 часов), медленно перемешивая при этом. На этой стадии наблюдали образование твердого вещества. Смесь охлаждали до 0°С и затем перемешивали при этой температуре в течение 15 минут, перед тем как собрать путем фильтрации желтоватое твердое вещество. Это твердое вещество промывали холодной смесью этанол:вода, 2:1 (150 мл), сушили посредством отсасывания жидкости в течение 1,25 часа, затем сушили в вакуум-сушильном шкафу (40°С; 20 часов). Масса полученного неочищенного продукта составляла 57,91 г (103,3 ммоль; 60%).
Установили, что неочищенный продукт имеет степень чистоты 98,47% (как определено путем ВЭЖХ-анализа), и перекристаллизовывали его (используя методику, подробно описанную ниже) с получением соединения, указанного в заголовке, с чистотой 99,75% (выход 84%).
Методика перекристаллизации:
К неочищенному продукту, полученному выше (56,2 г), добавляли этанол (562 мл) и воду (281 мл). Раствор нагревали до 75°С. Весь материал растворялся при 55°С. Раствор выдерживали при 75°С в течение 5 минут, перед тем как охлаждать до 5°С в течение 1,5 часов. Осаждение начиналось при 35°С. Холодный раствор отфильтровывали и собранный осадок промывали смесью этанол:вода (2:1; 168 мл). Твердый материал высушивали на фильтре посредством отсасывания жидкости, перед тем как сушить под вакуумом при 40°С, с получением продукта (47,1 г; 84%).
(10) Соединение А (свободное основание)
Способ I
Неочищенную бензолсульфонатную соль (50,0 г; 1,0 экв., со стадии (9), описанной выше; Способ 1) добавляли к водному гидроксиду натрия (1М; 500 мл), промывая дихлорметаном (1,0 л; 20 объемов). Объединенную смесь перемешивали в течение 15 минут. Затем слои разделяли и небольшое количество межфазного материала отбирали вместе с верхним водным слоем. К дихлорметановому раствору добавляли этанол (500 мл; 10 объемов) и затем растворитель удаляли перегонкой (1,25 л). Температура головного погона составляла на тот момент 78°С. Раствор оставляли охлаждаться до температуры ниже температуры дефлегмации и добавляли этанол (250 мл; 5 объемов). Растворитель удаляли (250 мл). Этот теплый раствор разбавляли этанолом до 890 мл, 17,8 объемов (25 объемов в расчете на 100%-ную конверсию в свободное основание). После нагревания до температуры дефлегмации раствор медленно охлаждали. При 5°С добавляли затравку указанного в заголовке соединения. Начиналась кристаллизация, и смесь перемешивали при 5°С в течение 30 минут. Продукт собирали фильтрованием и промывали этанолом (2×50 мл; 2×1 объем). Далее продукт высушивали в вакуумной печи при 40°С в течение 60 часов с получением порошка беловатой окраски (26,3 г; 74%).
1H ЯМР (400 МГц; CDCl3): δ 7.86-7.82 (2Н, m), 7.39-7.32 (3H, m), 7.30-7.26 (2H, m), 6.47 (2Н, m), 4.11-4.07 (4H, m), 3.70 (2Н, s), 3.36-3.33 (4H, m), 3.26 (2Н, t), 3.12 (2Н, d), 2.90 (2Н, d), 2.28-2.21 (2Н, m), 1.06 (9H, s).
13C ЯМР (CDCl3): δ 24.07, 26.38, 41.52, 43.52, 56.17, 56.47, 63.17, 68.46, 96.61, 111.64, 121.03,133.43.
MS (ES): m/z=385.1 (M+H)+.
Способ II
Смесь 4-{[3-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)пропил]амино}-бензнитрила (смотри Подготовительный пример B(l)(6) ниже; 5,73 г; 0,02 моль), К2СО3 (11,05 г; 0,08 моль) в MeCN (300 мл) обрабатывали 1-хлорпинаколином (4,44 г; 0,032 моль). Смесь перемешивали при 50°С в течение ночи перед тем, как добавить DCM и профильтровать смесь. Осадок на фильтре далее промывали смесью DCM и MeCN перед тем, как из фильтрата выпарить растворитель. Полученный остаток очищали хроматографией на диоксиде кремния, элюируя градиентом этилацетат/метанол/аммиачный метанол (от 95:5:0 до 95:0:5) с получением указанного в заголовке соединения (5,8 г; 73,9%).
Подготовительный пример В(I)
Получение Соединения В (Способ I)
(1) трет-Бутил-2-бромэтилкарбамат
Бикарбонат натрия (6,15 г; 0,073 моль) и ди-трет-бутил-дикарбонат (11,18 г; 0,051 моль) растворяли в смеси Н2О (50 мл) и дихлорметана (150 мл), затем охлаждали до 0°С. Медленно добавляли твердый 2-бромэтиламина гидробромид (10,0 г; 0,049 моль) и реакционную смесь перемешивали в течение ночи при 25°С. Дихлорметановый слой отделяли, промывали Н2O (200 мл) и промывали раствором гидросульфата натрия (150 мл; рН 3,5). Органический слой высушивали (Na2SO4) и концентрировали под вакуумом. Неочищенное масло хроматографировали на силикагеле, элюируя дихлорметаном, с получением 7,87 г (72%) указанного в подзаголовке соединения в виде прозрачного бесцветного масла.
1H ЯМР (300 МГц; CDCl3): δ 4.98 (bs, 1Н), 3.45-3.57 (m, 4H), 1.47 (s, 9H).
API-MS: (М+1-С5Н8CO2) 126 m/z.
(2) 3-Бензил-9-окса-3,7-диазабицикло[3.3.1]нонан × HCl
Этот способ получения является альтернативным описанному выше в Подготовительном примере А(6). Трехгорлую колбу вместимостью 3 л оборудовали магнитной мешалкой, термометром и парциальным конденсатором. Водную бромистоводородную кислоту (48%-ную; 0,76 л; 4,51 моль) добавляли к твердому 3-бензил-7-(фенилсульфонил)-9-окса-3,7-диазабицикло[3.3.1]нонану (190 г; 0,53 моль; смотри Подготовительный пример А(5) выше) и смесь нагревали до температуры дефлегмации в атмосфере азота. Твердое вещество растворялось при 90°С. После нагревания смеси в течение 12 часов ГХ-анализ показывал, что реакция полностью завершена. Содержимое реакционной смеси охлаждали до комнатной температуры. Добавляли толуол (0,6 л) и смесь перемешивали в течение нескольких минут. Фазы разделяли. Водную фазу возвращали в исходный реакционный сосуд и добавляли водный гидроксид натрия (10 М; 0,85 л; 8,5 моль) в виде одной порции. Внутренняя температура повышалась до 80°С и смесь становилась сильноосновной. Когда внутренняя температура опускалась до 55°С, добавляли толуол (0,8 л). После энергичного перемешивания в течение 30 минут толуольную фазу отделяли и возвращали в исходный реакционный сосуд. Добавляли 2-пропанол (1,9 л) и внутреннюю температуру поддерживали между 40°С и 50°С. Добавляли концентрированную соляную кислоту (до тех пор, пока смесь не становилась кислой) с такой скоростью, чтобы поддерживать температуру между 40 и 50°С. Образовался осадок белого цвета. Смесь перемешивали в течение 30 минут и затем охлаждали до 7°С. Порошок белого цвета собирали фильтрованием, промывали 2-пропанолом (0,4 л), высушивали, пропуская воздух через образец в течение десяти минут, и затем дополнительно высушивали в вакуумном шкафу при 40°С. Выход: 130 г (84%).
(3) трет-Бутил-7-бензил-9-окса-3,7-диазабицикло[3.3.1]нонан-3-карбоксилата гидрохлорид
Трехгорлую колбу вместимостью 5 л оборудовали подвесной мешалкой, термометром и азотным барботером. В колбу поочередно загружали воду (1,4 л), дихлорметан (1,4 л), бикарбонат натрия (150 г; 1,79 моль) и 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонан × 2 HCl (130 г; 0,447 моль; со стадии (2), выше). Смесь быстро перемешивали в течение десяти минут и затем медленно добавляли ди-трет-бутил-дикарбонат (0,113 л; 0,491 моль). Смесь быстро перемешивали в течение трех часов при комнатной температуре. Органический слой отделяли, высушивали с сульфатом магния, фильтровали и концентрировали с получением 160 г твердого вещества беловатой окраски. Твердое вещество беловатой окраски загружали в трехгорлую колбу вместимостью 3 л, оборудованную подвесной мешалкой, термометром и капельной воронкой. Загружали этилацетат (0,6 л) и прозрачный раствор охлаждали до -10°С. По каплям добавляли раствор HCl в диоксане (4М) до тех пор, пока рН не станет меньше 4. Соль гидрохлорид выпадала в осадок и смесь перемешивали в течение дополнительного часа. Продукт собирали фильтрованием, промывали этилацетатом (0,1 л) и высушивали в течение ночи в вакуумном шкафу. Масса белого кристаллического продукта составляла 146 г (92%-ный выход).
(4) трет-Бутил-9-окса-3,7-диазабицикло[3.3.1]нонан-3-карбоксилата гидрохлорид
Соль гидрохлорид со стадии (3), описанной выше, (146 г, 0,411 моль) и 20%-ный Pd(OH)2-C (7,5 г) загружали в сосуд Парра для гидрирования. Добавляли метанол (0,5 л) и сосуд энергично встряхивали в атмосфере водорода при 350 кПа (3,5 бар). За ходом реакции следили с помощью ГХ-анализа и установили, что реакция полностью завершилась через один час. Катализатор отфильтровывали и фильтрат концентрировали с получением кристаллического продукта беловатой окраски. Неочищенный продукт растворяли в горячем ацетонитриле (1,2 л) и затем фильтровали в горячем состоянии. Фильтрат разбавляли этилацетатом (1,2 л). Прозрачный раствор оставляли на ночь при комнатной температуре. Собирали первую партию кристаллов и сушили ее под вакуумом с получением 52 г указанного в подзаголовке соединения в виде твердого вещества белого цвета. Фильтрат концентрировали почти до сухого состояния, затем растворяли в горячем ацетонитриле (0,4 л) и разбавляли этилацетатом (0,4 л). Вторую партию кристаллов (38 г) получили после охлаждения раствора до 10°С. Обе партии по данным ГХ-анализа и 1H ЯМР-анализа были сопоставимы. Суммарный выход: 90 г (83%).
(5) трет-Бутил-7-[3-(4-цианоанилино)пропил]-9-окса-3,7-диазабицикло-[3.3.1]нонан-3-карбоксилат
Соль гидрохлорид трет-бутил-9-окса-3,7-диазабицикло[3.3.1]нонан-3-карбоксилата (смотри стадию (4), описанную выше; 1,1 г; 4,15 ммоль) смешивали с MeCN (46 мл), водой (2,5 мл) и К2СО3 (3,5 г; 25 ммоль). Смесь перемешивали в течение 4 ч перед тем, как добавить CHCl3, и смесь фильтровали через Целит®. Фильтрат концентрировали под вакуумом с получением 0,933 г свободного основания. Затем его смешивали с 3-(4-цианоанилино)пропил-4-метилбензолсульфонатом (смотри Подготовительный пример А(2), описанный выше; 2,1 г; 6,2 ммоль) и К2СО3 (0,86 г; 6,2 ммоль) в MeCN (18 мл). Полученную смесь перемешивали в течение ночи при 60°С перед тем, как сконцентрировать под вакуумом. Остаток обрабатывали DCM (250 мл) и 1М NaOH (50 мл). Слои разделяли и DCM-слой дважды промывали водным NaHCO3 перед тем, как высушить (Na2SO4) и сконцентрировать под вакуумом. Продукт очищали флэш-хроматографией, элюируя градиентом смеси толуол: этилацетат; триэтиламин (от 2:1:0 до 1000:1000:1), с получением 1,47 г (91%) указанного в подзаголовке соединения.
(6) 4-{[3-(9-Окса-3,7-диазабицикло[3.3.1]нон-3-ил)пропил]амино}-бензнитрил
Указанное в подзаголовке соединение получили с 96%-ным выходом, применяя методику, аналогичную описанной ниже в Подготовительных примерах С(5) и D(3), и используя трет-бутил-7-[3-(4-цианоанилино)пропил]-9-окса-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат (со стадии (5), описанной выше).
(7) Соединение В
К раствору трет-бутил-2-бромэтилкарбамата (4,21 г; 0,019 моль; смотри стадию (1), описанную выше) в DMF (65 мл) добавляли 4-{[3-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)пропил]амино}бензнитрил (смотри стадию (6), описанную выше; 4,48 г; 0,016 моль) и триэтиламин (3,27 мл; 0,024 моль). Смесь перемешивали в течение ночи при 35°С и затем концентрировали под вакуумом. Остаток растворяли в дихлорметане (80 мл) и промывали насыщенным хлоридом натрия. Водный слой экстрагировали дихлорметаном (1×150 мл). Объединенные органические экстракты высушивали (Na2SO4) и концентрировали под вакуумом. Неочищенное масло красно-коричневой окраски хроматографировали (×2) на силикагеле, элюируя смесью хлороформ: метанол: концентрированный NH4OH (9:1:0,02), с получением 3,75 г (56%) указанного в заголовке соединения.
1H ЯМР (300 МГц; CDI3OD): δ 7.37-7.40 (d, J=8.8 Гц, 2Н), 6.64-6.67 (d, J=8.8 Гц, 2Н). 3.94 (bs, 2Н), 3.21-3.31 (m, 4H), 3.01 (bs, 4H), 2.47-2.59 (m, 8H), 1.90 (bs, 2H), 1.39(s, 9H).
13C ЯМР (75 МГц, CD3OD): δ 158.5, 134.7, 121.9, 113.2, 97.7, 80.3, 69.2, 58.8, 58.1, 57.5, 57.3, 41.9, 38.3, 28.9, 26.2.
API-MS:(M+1)=430 m/z.
Подготовительный пример B (II)
Получение соединения В (Способ II)
(1) трет-Бутиловый эфир [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]-нон-3-ил)этил]карбаминовой кислоты
Вариант 1
(а) 2-трет-Бутилоксикарбониламино)этилтозилат
Раствор паратолуолсульфонилхлорида (28,40 г; 148 ммоль) в дихлорметане (100 мл) добавляли по каплям в течение 30 минут при 0°С к смеси трет-бутил-N-(2-гидроксиэтил)карбамата (20 г; 120 ммоль), триэтиламина (18,80 г; 186 ммоль) и хлорида триметиламмония (1,18 г; 12,4 ммоль) в дихлорметане (120 мл). Смесь перемешивали при 0°С в течение 1 часа, затем фильтровали, промывали дихлорметаном (100 мл). Фильтрат промывали 10%-ной лимонной кислотой (3×100 мл) и соляным раствором (100 мл). Органический слой высушивали с сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении с получением масла. Масло растворяли в этилацетате (40 мл) и затем медленно добавляли изогексан (160 мл). Полученную суспензию перемешивали при комнатной температуре в течение 17 часов и затем фильтровали. Собранный твердый материал промывали изогексаном (240 мл) с получением указанного в подзаголовке соединения в виде бесцветного порошка (25 г; 64%).
Т.пл. 64-66°С.
1H ЯМР (300 МГц; CDCl3): δ 1.40 (9Н, s), 2.45 (3H, s), 3.38 (2H, q), 4.07 (2H, t), 4.83 (1H, bs), 7.34 (2H, d), 7.87 (2H, d).
MS: m/z=216 (МН+(316)-Вос).
(б) трет-Бутиловый эфир [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]-нон-3-ил)этил]карбаминовой кислоты
Раствор 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонана дигидрохлорида (смотри Подготовительный пример А(6), описанный выше; 10 г; 34 ммоль) в воде (25 мл) медленно добавляли к раствору бикарбоната натрия (10 г; 119 ммоль) в воде (10 мл). Добавляли дополнительное количество воды (5 мл) и смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли раствор 2-(трет-бутилоксикарбониламино)этилтозилата (смотри стадию (а), описанную выше; 11,92 г; 37 ммоль) в толуоле (40 мл). Эту смесь затем нагревали при 65-70°С в течение 7 часов перед тем, как оставить при перемешивании на ночь при комнатной температуре. Реакционную смесь повторно нагревали до 50°С и производили разделение фаз. Водный слой экстрагировали толуолом (40 мл) при 50°С. Объединенные органические слои промывали насыщенным бикарбонатом натрия (25 мл). Растворители выпаривали при пониженном давлении, получая смесь масла и твердого вещества (13 г; >100%). К части маслянистого твердого вещества (5 г; 138 ммоль) добавляли этилацетат (50 мл) и лимонную кислоту (10%-ную; 25 мл). Водный слой отделяли и органический слой еще раз промывали лимонной кислотой (10%-ной; 20 мл). Водные слои объединяли и обрабатывали твердым бикарбонатом натрия до нейтральной реакции. Водную фазу экстрагировали этилацетатом (2×50 мл), высушивали над сульфатом магния и фильтровали. Фильтрат упаривали до сухого состояния при пониженном давлении с получением указанного в подзаголовке соединения в виде бесцветного полутвердого вещества, которое полностью отвердевало при хранении в холодильнике (4,86 г; 93%).
Т.пл. 58-60°С.
1H ЯМР (300 МГц; CDCl3): δ 1.46 (9Н, s), 2.38-2.57 (4H, m), 2.6-2.68 (2H, m), 2.75-2.85 (4H, m), 3.22 (2H, q), 3.26 (2H, s), 3.83 (2H, bs), 6.17 (1H, bs), 7.2-7.4 (5H, m).
MS: m/z=362 (MH+).
Вариант 2
(а) 3-(7-Бензил-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]пропионамид
Триэтиламин (3,60 г; 35,7 ммоль) медленно добавляли к раствору 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонана дигидрохлорида (смотри Подготовительный пример А(6), описанный выше; 5 г; 17 ммоль) в этаноле (50 мл). К этой смеси добавляли акриламид (1,34 г; 18 ммоль) и эту смесь затем нагревали при температуре дефлегмации в течение 7 часов. Реакционную смесь затем концентрировали при пониженном давлении. К остатку добавляли воду (50 мл) и гидроксид натрия (1М; 150 мл) и смесь экстрагировали этилацетатом (2×200 мл). Объединенные органические экстракты высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая бесцветное твердое вещество. Его перекристаллизовывали из этилацетата (50 мл) с получением указанного в подзаголовке соединения (3,80 г; 76%).
Т.пл. 157-159°С.
1H ЯМР (300 МГц; CDCl3): δ 2.39 (2H, t), 2.42-2.61 (6Н, m), 2.82-2.95 (4H, m), 3.39 (2H, s), 3.91 (2H, bs), 5.07 (1H, bs), 7.18-7.21 (2H, m), 7.25-7.39 (3H, m), 9.5(1H, bs).
MS: m/z=290 (MH+).
(6) трет-Бутиловый эфир [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]-нон-3-ил)этил]карбаминовой кислоты
N-Бромсукцинимид (6,0 г; 33 ммоль) порциями в течение 1 минуты добавляли к раствору 3-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]-пропионамида (смотри стадию (а), описанную выше; 5 г; 12 ммоль) в трет-бутилате калия в трет-бутаноле (1М; 81 мл) и трет-бутанола (20 мл). Смесь далее нагревали при 60-65°С в течение 30 минут. Реакционную смесь оставляли охлаждаться до комнатной температуры и затем добавляли воду (100 мл). Смесь экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты промывали соляным раствором (50 мл), высушивали над сульфатом магния, фильтровали (промывая осадок на фильтре этилацетатом (50 мл)) и затем фильтрат концентрировали при пониженном давлении с получением указанного в подзаголовке соединения в виде коричневого масла (6,5 г; >100%).
1H ЯМР (300 МГц; CDCl3): δ 1.46 (9Н, s), 2.4-2.58 (4Н, m), 2.58-2.7 (2H, m), 2.75-2.91 (4Н, m), 3.22 (2H, q), 3.28 (2H, s), 3.83 (2H, bs), 6.19 (1H, bs), 7.2-7.42 (5H, m).
MS: m/z=316 (MH+).
Вариант 3
(а) 3-Бензил-9-окса-3.7-диазабицикло[3.3.1]нонан
Величины всех объемов и эквивалентов измеряют относительно используемого количества 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонана дигидрохлорида (смотри Подготовительный пример А(6), описанный выше). К дигидрохлориду 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонана (60,07 г; 206,03 ммоль; 1 экв.; смотри Подготовительный пример А(6), описанный выше) добавляли толуол (420 мл; 7 объемов) и водный раствор гидроксида натрия (2М; 420 мл; 7 объемов; 4 экв.). Смесь перемешивали в атмосфере азота, нагревали до 60°С и выдерживали при этой температуре в течение 30 минут, в течение которых формировались два прозрачных слоя. Нижний водный слой удаляли и толуоловый раствор указанного в подзаголовке соединения (свободного основания) азеотропно высушивали при атмосферном давлении (суммарный объем удаленного растворителя = 430 мл; суммарный объем добавленного толуола = 430 мл), затем концентрировали до объема 240 мл (4 объема). Анализ по Карлу Фишеру (Karl Fischer) на этой стадии показал наличие 0,06% воды в растворе. Высушенный раствор указанного в подзаголовке соединения (теоретически 44,98 г; 206,03 ммоль; 1,0 экв.) непосредственно использовали на следующей стадии.
(б) 2-(трет-Бутилоксикарбониламино)этил-2,4.6-триметилбензол-сульфонат
Триэтиламин (65 мл; 465,3 ммоль; 1,5 экв.) добавляли в виде одной порции к раствору трет-бутил-N-(2-гидроксиэтил)карбамата (50,11 г; 310,2 ммоль; 1,0 экв.) в дихлорметане (250 мл; 5 объемов). Раствор охлаждали до -10°С и добавляли в виде одной порции триметиламина гидрохлорид (14,84 г; 155,1 ммоль; 0,5 экв.). Полученную смесь охлаждали далее до -15°С, перемешивали в течение 5 минут, затем обрабатывали раствором мезитиленсульфонилхлорида (74,74 г; 341,2 ммоль; 1,1 экв.) в дихлорметане (250 мл; 5 объемов) в продолжение 28 минут, так что внутренняя температура оставалась ниже -10°С. По завершении добавления образовывался осадок, и смесь перемешивали при -10°С в течение следующих 30 минут. Добавляли воду (400 мл; 8 объемов) и весь осадок растворяли. Смесь быстро перемешивали в течение 5 минут, и затем смесь разделялась на два слоя. Осуществляли замену растворителя дихлорметана на изопропанол посредством перегонки при пониженном давлении. Растворитель удаляли (450 мл) и заменяли изопропанолом (450 мл) (начальное давление 45 кПа (450 мбар), т.кип. 24°С; конечное давление 11 кПа (110 мбар), т.кип. 36°С). В конце перегонки растворитель (150 мл) удаляли, что приводило к понижению объема до 350 мл (7 объемов по отношению к количеству используемого трет-бутил-N-(2-гидроксиэтил)карбамата). Раствор охлаждали до 25°С, затем медленно при перемешивании добавляли воду (175 мл), в результате чего раствор постепенно мутнел. На этой стадии не образовалось никакого твердого осадка. Добавляли дополнительное количество воды (125 мл), и после добавления приблизительно 75 мл начиналось образование твердого осадка. Внутренняя температура поднималась от 25 до 31°С. Смесь медленно перемешивали и охлаждали до 7°С. Твердое вещество собирали фильтрованием, промывали смесью изопропанол: вода (1:1, 150 мл) и высушивали под вакуумом при 40°С в течение 21 часа с получением указанного в подзаголовке соединения в виде кристаллического вещества белого цвета (92,54 г; 87%).
Т.пл. 73,5°С.
1H ЯМР (300 МГц; CDCl3): δ 1.42 (9H, s), 2.31 (3H, s), 2.62 (6H, s), 3.40 (2H, q), 4.01 (2H, t), 4.83 (1H, bs), 6.98 (2H, s).
(в) Соль трет-бутилового эфира [2-(7-бензил-9-окса-3,7-диазабицикло-[3.3.1]нон-3-ил)этил]карбаминовой кислоты с 2,4,6-триметил-бензолсульфоновой кислотой
Теплый (28°С) раствор 2-трет-бутилоксикарбониламино)этил-2,4,6-триметилбензолсульфоната (70,93 г; 206,03 ммоль; 1,0 экв.; смотри стадию (б), описанную выше) в толуоле (240 мл, 4 объема) добавляли к раствору 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонана (44,98 г; 206,03 ммоль; 1,0 экв.) в толуоле (240 мл, 4 объема) (смотри стадию (а), описанную выше). Полученный раствор медленно перемешивали в атмосфере азота с нагреванием при 68°С в течение 8 часов. Реакционную смесь оставляли перемешиваться при температуре окружающей среды в течение 84 часов. В бледно-желтом растворе образовался плотный твердый осадок белого цвета. Смесь охлаждали до +9°С и указанное в подзаголовке соединение собирали фильтрованием. Реакционный сосуд промывали толуолом (100 мл) и добавляли на фильтр. Осадок на фильтре промывали толуолом (150 мл). Твердый продукт белого цвета высушивали отсасыванием в течение 15 минут, затем высушивали до постоянной массы под вакуумом при 40°С в течение 23 часов. Выход полученного указанного в подзаголовке соединения составил 79,61 г; 141,7 ммоль; 69%. Объединенные фильтрат и промывки (670 мл) промывали водным раствором гидроксида натрия (2М; 200 мл; 3,3 объема). Смесь нагревали до 60°С и выдерживали при этой температуре в течение 20 минут при быстром перемешивании. Затем два слоя разделяли. Толуоловый раствор концентрировали до 200 мл путем вакуумной перегонки (т.кип. 50-54°С при 65-70 кПа (650-700 мбар); т.кип. 46°С при 12 кПа (120 мбар) в конце). По мере осуществления перегонки раствор становился мутным вследствие образования указанного в подзаголовке соединения. Предполагали, что 20% первоначального количества 3-бензил-9-окса-3,7-диазабицикло[3.3.1]нонана остается в фильтрате, и поэтому дополнительно в виде одной порции добавляли 2-(трет-бутилоксикарбониламино)этил-2,4,6-триметилбензол-сульфонат (14,20 г; 41,21 ммоль; 0,2 экв.) (загрузку лучше осуществлять в виде твердого вещества, чем в виде раствора в толуоле). Мутный раствор нагревали при 67°С в течение 8 часов при быстром перемешивании и затем оставляли перемешиваться при температуре окружающей среды в течение 11 часов. Смесь охлаждали до +8°С и указанное в подзаголовке соединение собирали фильтрованием. Реакционный сосуд еще раз промывали толуолом (2×30 мл) и добавляли на фильтр. Твердый продукт белого цвета высушивали отсасыванием в течение 15 минут, затем высушивали до постоянной массы под вакуумом при 40°С в течение 7 часов. Выход полученного указанного в подзаголовке соединения составил 23,25 г; 41,39 ммоль; 20%. Суммарный выход указанного в подзаголовке соединения (в виде твердого вещества белого цвета) составил 102,86 г; 183,11 ммоль; 89%.
Т.пл. 190-190,5°С.
1H ЯМР (300 МГц; CDCl3): δ 1.43 (9Н, s), 2.17 (3H, s), 2.51 (6H, s), 2.73-2.80 (2H, m), 2.90-2.94 (4H, m), 3.14-3.22 (4H, m), 3.37 (2H, bm), 3.89 (2H, bs), 4.13 (2H, bs), 6.74 (2H, s), 7.12 (1H, bt), 7.42-7.46 (5H, m).
(2) трет-Бутиловый эфир [2-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)-этил]карбаминовой кислоты
Способ 1. Бикарбонат натрия (0,058 г; 0,069 ммоль) и 5%-ный Pd/C (0,250 г, паста Johnson Mattey Type 440) добавляли к раствору трет-бутилового эфира [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]карбаминовой кислоты (смотри стадию (1), Вариант 1, описанный выше; 1 г; 2,77 ммоль) в этаноле (10 мл). Затем смесь гидрировали при 500 кПа (5 бар) в течение 18 часов. Реакционную смесь фильтровали через Целит® и затем промывали этанолом (20 мл). Раствор концентрировали при пониженном давлении с получением масла. Его растворяли в дихлорметане (20 мл) и промывали гидроксидом натрия (1М; 10 мл). Органическую фазу отделяли, высушивали над сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в подзаголовке соединения в виде твердого вещества желтого цвета (0,67 г; 87%).
Т.пл.91-93°С.
1H ЯМР (300 МГц; CDCl3): δ 1.46 (9Н, s), 2.25 (2H, t), 2.58-2.65 (2H, m), 2.95-3.06 (4H, m), 3.2-3.38 (4H, m), 3.64 (2H, bs), 4.65 (1H, bs).
MS: m/z=272 (MH+).
Способ 2. Соль трет-бутилового эфира [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]карбаминовой кислоты с 2,4,6-триметилбензолсульфоновой кислотой (320 г; 1,0 моль-экв.; 1,0 отн. об./масса; смотри стадию (1), Вариант 3, описанный выше), толуол (640 мл; 2,0 об.) и водный гидроксид натрия (1М, 1,6 л; 5,0 объемов) перемешивали вместе в течение 15 минут и затем слои разделяли. Органический слой, содержащий трет-бутиловый эфир [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]карбаминовой кислоты, разбавляли этанолом (690 мл; 2,16 объема) и водой (130 мл; 0,4 объема). Добавляли лимонную кислоту (32,83 г; 0,3 моль-экв.) и 5%-ный Pd/C (20,8 г; 0,065 мас.-экв. катализатора Johnson Mattey Type 440L с 61%-ным содержанием влаги). Объединенную смесь далее гидрировали в течение 24 часов при давлении водорода 400 кПа (4 бар). Ход реакции контролировали с помощью ТСХ, используя пластинку диоксида кремния с подвижной фазой Х:DCM (1:1 (об./об.); Х представляет собой смесь хлороформ: метанол: концентрированный аммиак 80:18:2 (об./об.)). Визуализацию осуществляли с помощью УФ- света (254 нм) и путем окрашивания водным перманганатом калия. Продемонстрировано полное исчезновение исходного материала и присутствие указанного в подзаголовке соединения. Реакционную смесь фильтровали через кизельгур и промывали этанолом (590 мл; 1,84 объема). Полученный раствор указанного в подзаголовке соединения (предположительно 154,85 г; 100%) использовали непосредственно в последующей реакции.
Способ 3. Соль mpem-бутилового эфира [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]карбаминовой кислоты с 2,4,6-триметилбензолсульфоновой кислотой (50 г; 1,0 моль-экв.; 1,0 отн. об./мас.; смотри стадию (1), Вариант 3, описанный выше), толуол (100 мл; 2,0 объема) и водный гидроксид натрия (1М, 100 мл; 2,0 объема) перемешивали вместе в течение 20 минут, затем при 30°С в течение 10 минут, после чего слои разделяли. Органический слой, содержащий трет-бутиловый эфир [2-(7-бензил-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]карбаминовой кислоты, разбавляли этанолом (100 мл; 2,0 объема). К нему добавляли раствор лимонной кислоты (5,14 г; 0,3 моль-экв.) в воде (5 мл; 0,1 объема) с последующим добавлением 5%-ного Pd/C (1,50 г; 0,03 мас.-экв. катализатора Johnson Mattey Type 440L с 61%-ным содержанием влаги). Объединенную смесь далее гидрировали в течение 24 часов при давлении водорода 400 кПа (4 бар). Ход реакции контролировали с помощью ТСХ, используя пластинку диоксида кремния с подвижной фазой Х:DCM (1:1 (об./об.); Х представлял собой смесь хлороформ: метанол: концентрированный аммиак 80:18:2 (об./об.)). Визуализацию осуществляли с помощью УФ-света (254 нм) и путем окрашивания водным перманганатом калия. Продемонстрировано полное исчезновение исходного материала и присутствие указанного в подзаголовке соединения. Реакционную смесь подщелачивали водным гидроксидом натрия (10М; 8 мл; 0,9 моль-экв.), затем фильтровали через кизельгур. Осадок на фильтре промывали этанолом (100 мл; 2,0 объема). Получившийся раствор указанного в подзаголовке соединения (предположительно 24,15 г; 100%) использовали непосредственно в последующей реакции.
(3) Соединение В
Способ I
3-(4-Цианоанилино)пропил-4-метилбензолсульфонат (смотри Подготовительный пример А(2), описанный выше; 0,30 г; 0,92 ммоль) и карбонат калия (0,2 г; 1,38 ммоль) добавляли к раствору трет-бутилового эфира [2-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]карбаминовой кислоты (смотри стадию (2), Способ 1, описанный выше; 0,250 г; 0,92 ммоль) в этаноле (5 мл). Реакционную смесь нагревали до 70°С в течение 10 часов перед концентрированием смеси при пониженном давлении. Остаток распределяли между этилацетатом (20 мл) и гидроксидом натрия (1М, 10 мл). Водную фазу повторно экстрагировали этилацетатом (20 мл). Объединенные органические фазы концентрировали при пониженном давлении с получением твердого вещества желтого цвета (0,290 г). Твердое вещество растворяли в этилацетате (10 мл) и этот раствор промывали раствором лимонной кислоты (0,250 г) в воде (10 мл). Водную фазу отделяли, подщелачивали гидроксидом натрия (1М; 10 мл) и экстрагировали этилацетатом (2×10 мл). Объединяли все органические фазы, высушивали над сульфатом магния и затем фильтровали (промывая отфильтрованные твердые вещества этилацетатом (10 мл)). Фильтрат концентрировали при пониженном давлении с получением твердого вещества желтого цвета (0,160 г). Его суспендировали в этилацетате (0,2 мл) и затем фильтровали с получением указанного в заголовке соединения (0,050 г; 12%).
Т.пл. 113-115°С.
1H ЯМР (400 МГц; DMSO-D6): δ 1.32 (9Н, s), 1.7 (2Н, qt), 2.20 (2Н, t), 2.22-2.3 (4H, m), 2.38-3.1 (2Н, m), 2.8-2.85 (4H, m), 3.05 (2Н, q), 3.19 (2Н, q), 3.79 (2Н, bs), 6.47 (1H, t), 6.66 (2Н, d), 6.69 (1H, t), 7.41 (2Н, d).
MS: m/z=430 (MH+).
Способ II
К раствору трет-бутилового эфира [2-(9-окса-3,7-диазабицикло-[3.3.1]нон-3-ил)этил]карбаминовой кислоты, полученного на стадии (2) (Способ 3), описанной выше (предположительно 24,15 г; 1,0 моль-экв.; 1,0 мас./об.) в смеси толуола (приблизительно 100 мл), этанола (приблизительно 200 мл) и воды (приблизительно 14 мл), добавляли безводный карбонат калия (18,58 г; 1,5 моль-экв.). Добавляли твердый 3-(4-цианоанилино)пропилбензолсульфонат (28,17 г; 1,0 моль-экв.; смотри Подготовительный пример А(9), Способ 2, стадию (а), описанную выше) и объединенную смесь нагревали до 70°С в течение шести часов. За ходом реакции следили с помощью ТСХ, используя пластинку с диоксидом кремния с подвижной фазой Х:DCM (1:1 (об./об.); где Х представлял собой смесь хлороформ: метанол: концентрированный аммиак в отношении 80:18:2 (об./об.)). Визуализацию осуществляли в УФ-свете (254 нм) и с помощью окрашивания водным перманганатом калия. Этот анализ показал полное исчезновение исходного материала и присутствие указанного в заголовке соединения. Реакционную смесь охлаждали и растворитель концентрировали под вакуумом. Остаток распределяли между толуолом (200 мл) и водой (200 мл). Слои разделяли и органическую фазу концентрировали под вакуумом с получением твердого вещества желтого цвета (38,6 г).
Подготовительный пример С
Получение Соединения С
(1) 4-(4-Цианофенил)бут-3-ин-1-ол
Карбонат калия (376,7 г; 2,5 моль-экв.) растворяли в смеси 1,2-диметоксиэтана (DME; 1,2 л; 6 объемов) и воды (1,2 л; 6 объемов). Добавляли палладий на угле (20 г; 0,01 моль-экв.; 10%-ный Johnson Matthey Type 87L; 60% воды), трифенилфосфин (11,5 г; 0,04 моль-экв.) и иодид меди(1) (4,2 г; 0,02 моль-экв.). Затем добавляли 4-бромбензнитрил (200 г; 1 моль-экв.), промывали смесью DME (200 мл, 1 объем) и воды (200 мл; 1 объем). Эту смесь быстро перемешивали в атмосфере азота в течение как минимум тридцати минут. По каплям в течение пяти минут добавляли раствор бут-3-ин-1-ола (92,1 мл; 1,1 моль-экв.) в DME (200 мл; 1 объем) и воде (200 мл; 1 объем). Объединенную смесь затем нагревали до 80°С в течение трех часов. За ходом реакции следили с помощью ВЭЖХ по исчезновению арилбромида и образованию указанного в подзаголовке соединения. Когда весь исходный материал израсходовался, реакционную смесь охлаждали до 25°С и фильтровали через кизельгур. Осадок на фильтре отдельно промывали толуолом (1,6 л; 8 объемов). Смесь DME: вода частично концентрировали под вакуумом для удаления основной части DME. Далее ее распределяли с помощью толуольной промывки. Толуольный слой концентрировали под вакуумом, получая указанный в подзаголовке алкин в виде твердого вещества желтого цвета, которое высушивали в вакуумном шкафу в течение ночи при 40°С. Выход 182,88 г; 97%.
1H ЯМР (300 МГц, CDCl3): δ 7.599-7.575 (d, J=7.2 Гц, 2Н, СН), 7.501-7.476 (d, J=7.5 Гц, 2Н, СН), 3.880-3.813 (q, 2Н, CH2), 2.751-2.705 (t, 2Н, CH2), 1.791-1.746(t, 1H, OH).
Т.пл. 79,6-80,5°C.
(2) 4-(4-Гидроксибутил)бензнитрил
4-(4-Цианофенил)бут-3-ин-1-ол (40 г; 1 мас.-экв.; смотри стадию (1), описанную выше) в этаноле (200 мл; 5 объемов) и палладий на угле (20 г; 0,5 мас.-экв.; 10%-ный Johnson Matthey Type 487; 60% воды) быстро перемешивали под давлением водорода 500 кПа (пять бар) в течение пяти часов. За ходом реакции следили с помощью ВЭЖХ по исчезновению исходного материала и образованию указанного в подзаголовке соединения. Реакционную смесь фильтровали через кизельгур и промывали этанолом (80 мл; 2 объема). Этанольный раствор концентрировали под вакуумом с получением указанного в подзаголовке спирта в виде масла желто-коричневой окраски. Выход 36,2 г; 88,5%.
1H ЯМР (300 МГц, CDCl3): δ 7.550-7.578 (d, J=8.4 Гц, 2Н), 7.271-7.298 (d, J=8.1 Гц, 2Н), 3.646-3.688 (t, 2Н), 2.683-2.733 (t, 2Н), 1.553-1.752 (m, 4H).
13С ЯМР (300 МГц, CDCl3): δ 148.04 (С), 132.16 (С), 119.1 (С), 109.64 (С), 62.46 (С), 35.77 (С), 32.08 (С), 27.12 (С).
(3) 4-(4-Цианофенил)бутилтолуолсульфонат
Указанное в подзаголовке соединение получали путем добавления толуолсульфонилхлорида к 4-(4-гидроксибутил)бензнитрилу (смотри стадию (2), описанную выше).
(4) трет-Бутил-7-[4-(4-цианофенил)бутил]-9-окса-3,7-диазабицикло-[3.3.1]нонан-3-карбоксилат
Трехгорлую колбу вместимостью 2 л оснащали магнитной мешалкой, термометром и парциальным конденсатором. В колбу загружали раствор 4-(4-цианофенил)бутилтолуолсульфоната (72 г; 0,218 моль; смотри стадию (3), описанную выше) в диметилформамиде (0,55 л). Добавляли трет-бутил-9-окса-3,7-диазабицикло[3.3.1]нонан-3-карбоксилата гидрохлорид (48,2 г; 0,182 моль; смотри Подготовительный пример B(I)(4), описанный выше) с последующим добавлением карбоната калия (62,9 г; 0,455 моль). Гетерогенную смесь перемешивали в течение 22 часов при 85°С. ТСХ-анализ показал полное расходование исходного материала. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой (0,5 л). Смесь экстрагировали этилацетатом (3×0,4 л) и органические фракции объединяли. После промывания водой (2×200 мл) и соляным раствором (200 мл) органический слой высушивали с сульфатом магния, фильтровали и концентрировали под вакуумом. Неочищенное коричневое масло очищали с помощью хроматографии на силикагеле, элюируя смесью гексаны/этилацетат (3:2), с получением 34 г (48%-ный выход) указанного в подзаголовке соединения в виде твердого вещества беловатой окраски.
(5) 4-[4-(9-Окса-3,7-диазабицикло[3.3.1]нон-3-ил)бутил]бензнитрил
Трехгорлую колбу вместимостью 2 л оснащали магнитной мешалкой, термометром и капельной воронкой. В колбу загружали трет-бутил-7-[4-(4-цианофенил)бутил]-9-окса-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат (34 г; 88 ммоль; со стадии (4), описанной выше) и дихлорметан (440 мл). Медленно при комнатной температуре добавляли трифторуксусную кислоту (132 мл). Раствор перемешивали в течение трех часов, по прошествии которых ТСХ-анализ показал полное расходование исходного материала. Содержимое переносили в одногорлую колбу и концентрировали под вакуумом. Остаток растворяли в дихлорметане (500 мл) и промывали насыщенным раствором бикарбоната натрия. Водный слой отделяли и экстрагировали дихлорметаном (2×200 мл). Объединенные органические слои промывали соляным раствором (200 мл), высушивали над сульфатом магния и концентрировали под вакуумом с получением 25,8 г (100%-ный выход) указанного в подзаголовке соединения в виде твердого вещества беловатой окраски. Неочищенный материал использовали на следующей стадии без дальнейшей очистки.
(6) Соединение С
Трехгорлую колбу вместимостью 3 л оснащали магнитной мешалкой, термометром и парциальным конденсатором. В колбу загружали неочищенный 4-[4-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)бутил]бензнитрил (25,8 г; 88 ммоль; со стадии (5), описанной выше), дихлорметан (0,88 л) и трет-бутил-2-бромэтилкарбамат (смотри Подготовительный пример В(I)(1), описанный выше; 27,7 г; 123 ммоль). Затем добавляли триэтиламин (0,0197 л; 0,141 моль). Прозрачный раствор кипятили с обратным холодильником в течение 12 часов в атмосфере азота и затем охлаждали до комнатной температуры. За развитием реакции следили с помощью ТСХ-анализа и обнаружили, что к тому моменту времени реакция полностью завершена. Реакционную смесь переносили в делительную воронку и последовательно промывали водой (200 мл), 15%-ным водным гидроксидом натрия (200 мл), водой (200 мл) и соляным раствором (200 мл). Органический слой высушивали над сульфатом магния и концентрировали под вакуумом. Получающееся вязкое желтое масло хроматографировали на силикагеле, элюируя сначала смесью дихлорметан/метанол (9:1), затем смесью дихлорметан/метанол/28%-ный водный гидроксид аммония (9:1:0,02), с получением указанного в заголовке соединения (25,1 г; 66%-ный выход) в виде твердого вещества беловатой окраски. Было обнаружено, что более ранние фракции (5,1 г) с хроматографии содержали небольшое количество менее полярной примеси (по данным ТСХ-анализа), которая элюировалась смесью дихлорметан/метанол/28%-ный водный гидроксид аммония (9:1:0,05), в то время как более поздние фракции (20 г) представляли собой одно пятно по данным ТСХ-анализа. Более ранние фракции (5,1 г) объединяли с другой партией указанного в заголовке соединения (7,1 г, содержащих небольшое количество примеси) и хроматографировали на силикагеле, элюируя сначала смесью дихлорметан/метанол (19:1), затем смесью дихлорметан/метанол (9:1), с получением порошка бледно-желтой окраски (5,5 г). Порошок растворяли в дихлорметане (200 мл). Полученный раствор последовательно промывали 25%-ным водным гидроксидом натрия (50 мл), водой (50 мл) и соляным раствором (40 мл). Затем материал высушивали над сульфатом магния и концентрировали под вакуумом с получением указанного в заголовке соединения в виде порошка беловатой окраски (5 г). Фракцию массой 20 г растворяли в дихлорметане (500 мл). Органический слой последовательно промывали 25%-ным водным гидроксидом натрия (100 мл), водой (100 мл) и соляным раствором (100 мл). Затем материал высушивали над сульфатом магния и концентрировали под вакуумом с получением указанного в заголовке соединения в виде порошка беловатой окраски (19 г). Партии порошка смешивали вместе.
Подготовительный пример D
Получение соединения D
(1) 4-[(2S)-Оксиранилметокси]бензнитрил
Карбонат калия (414 г) и (R)-(-)-эпихлоргидрин (800 мл) добавляли к перемешиваемому раствору парацианофенола (238 г) в 2,0 л MeCN и реакционную смесь кипятили с обратным холодильником в атмосфере инертного газа в течение 2 ч. Раствор фильтровали в горячем состоянии и фильтрат концентрировали, получая прозрачное масло, которое кристаллизовалось из диизопропилового эфира с получением продукта с 90%-ным выходом.
(2) трет-Бутил-7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-9-окса-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат
В трехгорлую колбу вместимостью 3 л, оборудованную магнитной мешалкой и термометром, загружали трет-бутил-9-окса-3,7-диазабицикло[3.3.1]нонан-3-карбоксилат в виде свободного основания (53,7 г; 0,235 моль, полученный из соли гидрохлорида в Подготовительном примере B(I)(4), описанном выше), 4-[(2S)-оксиранилметокси]бензнитрил (41,2 г; 0,235 моль; смотри стадию (1), описанную выше) и раствор 2-пропанол/вода (0,94 л; 10:1 (об./об.)). Смесь перемешивали при 60°С в течение 20 часов, в продолжение которых исходные материалы постепенно расходовались (по данным ТСХ-анализа). Смесь охлаждали и концентрировали под вакуумом с получением 100 г (выход >100%) указанного в подзаголовке соединения в виде твердого вещества белого цвета. Неочищенный материал использовали на следующей стадии.
(3) 4-{[(2S)-2-Гидрокси-3-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)пропил]-окси}бензнитрил
В трехгорлую колбу вместимостью 3 л, оборудованную магнитной мешалкой, термометром и капельной воронкой, загружали неочищенный трет-бутил-7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-9-окса-3,7-диазабицикло-[3.3.1]нонан-3-карбоксилат (100 г; со стадии (2), описанной выше) и дихлорметан (1,15 л). Медленно при комнатной температуре добавляли трифторуксусную кислоту (0,352 л) и полученный раствор перемешивали в течение трех часов, по истечении которых ТСХ-анализ показал полное завершение реакции. Содержимое переносили в одногорлую колбу и концентрировали под вакуумом. Остаток растворяли в дихлорметане (1,2 л) и промывали насыщенным раствором бикарбоната натрия. Водный слой отделяли и экстрагировали дихлорметаном (2×0,2 л). Объединенные органические слои промывали соляным раствором (0,25 л), высушивали над сульфатом магния и концентрировали под вакуумом с получением 73 г (выход >100%) указанного в подзаголовке соединения в виде твердого вещества беловатой окраски. Неочищенный материал использовали на следующей стадии.
(4) Соединение D
Способ I. Трехгорлую колбу вместимостью 2 л оборудовали магнитной мешалкой, термометром и парциальным конденсатором. В колбу загружали неочищенный 4-{[(2S)-2-гидрокси-3-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)-пропил]окси}бензнитрил (73 г; со стадии (3), описанной выше), дихлорметан (0,7 л) и трет-бутил-2-бромэтилкарбамат (смотри Подготовительный пример В(I)(1), описанный выше); 74 г; 0,330 моль). Затем добавляли триэтиламин (52 мл; 0,359 моль). Прозрачный раствор кипятили с обратным холодильником в течение 16 часов, затем охлаждали до комнатной температуры. Реакционную смесь переносили в делительную воронку и последовательно промывали водой (100 мл) и соляным раствором (100 мл). Органический слой высушивали над сульфатом магния, фильтровали и концентрировали под вакуумом. Получающееся вязкое масло желтого цвета очищали хроматографией на силикагеле, элюируя сначала смесью дихлорметан/метанол (9:1), затем смесью дихлорметан/метанол/28%-ный водный гидроксид аммония (9:1:0,02) с получением пенящегося твердого вещества беловатой окраски (40 г). Твердое вещество растворяли в дихлорметане (200 мл) и последовательно промывали 20%-ным водным гидроксидом натрия (100 мл) и водой (100 мл). Органический слой высушивали над сульфатом магния и концентрировали под вакуумом с получением указанного в заголовке соединения в виде твердого вещества беловатой окраски (35,4 г; 67%-ный выход на трех стадиях).
Способ II. Изопропанол (5 мл) и воду (0,5 мл) добавляли к трет-бутиловому эфиру [2-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]-карбаминовой кислоты (смотри Подготовительный пример В(II)(2), Способ I, описанный выше; 0,43 г; 1,6 ммоль) и добавляли 4-[(2S)-оксиранилметокси]бензнитрил (0,280 г; 1,6 ммоль; смотри стадию (1), описанную выше). Смесь нагревали при 66°С в течение 19 часов (реакция завершалась за 2 часа). Растворитель выпаривали до сухого состояния при пониженном давлении, получая указанное в заголовке соединение в виде твердого вещества беловатой окраски (0,71 г; 100%).
1H ЯМР (300 МГц; CDCl3): δ 1.41 (9Н, s), 2.3-2.75 (6H, m), 2.75-3.0 (5H, m), 3.1-3.38 (3H, m), 3.88 (2H, s), 3.95-4.19 (3H, m), 5.85 (1H, bs), 6.99 (2H, d), 7.6 (2H, d).
1H ЯМР (300 МГц; DMSO-D6): δ 1.35 (9Н, s), 2.12-2.59 (7H, m), 2.63-2.78 (1H, m), 2.78-2.9 (4H. m), 3.2 (2H, q), 3.78 (2H, m), 4-4.1 (2H, m), 4.12-4.19 (1H, m), 5.3 (1H, bs), 6.61 (1H, t), 7.15 (2H, d), 7.76 (2H, d).
MS: m/z=447 (MH+).
Способ III. Раствор трет-бутилового эфира [2-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)этил]карбаминовой кислоты, полученного выше в Подготовительном примере В(II)(2), Способ 2 (предположительно 154,85 г; 1,0 моль-экв.; 1,0 мас./об.), в смеси толуола (приблизительно 640 мл), этанола (приблизительно 1280 мл) и воды (приблизительно 130 мл) подщелачивали водным гидроксидом натрия (10 M; 51 мл; 0,9 моль-экв.). Добавляли твердый 4-[(2S)-оксиранилметокси]бензнитрил (99,80 г; 1,0 моль-экв.; смотри стадию (1), описанную выше) и объединенную смесь нагревали до 70°С в течение четырех часов. Ход реакции контролировали с помощью ТСХ, используя пластинку с диоксидом кремния с подвижной фазой Х: DCM (1:1 (об./об.), где Х представлял собой смесь хлороформ: метанол: концентрированный аммиак 80:18:2 (об./об.)). Визуализацию осуществляли с помощью УФ-света (254 нм) и путем окрашивания водным перманганатом калия. Продемонстрировано полное исчезновение исходного материала и присутствие указанного в заголовке соединения. Реакционную смесь охлаждали, фильтровали через кизельгур и промывали этанолом (620 мл; 4,0 объема). Это давало раствор указанного в заголовке соединения (предположительно 254,38 г; 100% (теоретически); 2,4 л; 1,0 мас./об. для завершенной реакции). Этот раствор загружали в колбу, подготовленную для проведения перегонки при пониженном давлении. На боковой поверхности этой колбы делали градуировочную отметку. Растворитель (1250 мл) удаляли при температуре от 50 до 35°С, давлении 32 кПа (320 мбар) и 10 кПа (100 мбар). Затем добавляли 4-метилпентан-2-ол (1500 мл) до градуировочной отметки. Растворитель (1250 мл) удаляли при температуре от 35 до 80°С, давлении 22 кПа (220 мбар) и 40 кПа (400 мбар). Добавляли дополнительное количество 4-метилпентан-2-ола (1500 мл) до градуировочной отметки. Растворитель (1250 мл) удаляли при температуре от 62 до 76°С, давлении 10 кПа (100 мбар) и 9 кПа (90 мбар). Объединенную смесь охлаждали до температуры ниже 25°С и добавляли водный гидроксид натрия (2М; 1,27 л; 5,0 объемов). Разделяли слои и органический слой фильтровали через кизельгур, получая прозрачный раствор (1,2 л). Этот раствор загружали в чистую колбу, подготовленную для проведения перегонки при пониженном давлении. Растворитель (450 мл) удаляли при температуре от 52 до 55°С, давлении 9 кПа (90 мбар) и 3,5 кПа (35 мбар). Теоретически на данный момент продукт оставался в 2 объемах 4-метилпентан-2-ола. Добавляли ди-н-бутиловый эфир (1,27 л; 5 объемов) и раствор оставляли медленно охлаждаться до комнатной температуры, что вызвало образование осадка. Смесь охлаждали от комнатной температуры до приблизительно 10°С. Продукт собирали фильтрованием и промывали предварительно смешанным раствором ди-н-бутилового эфира (320 мл; 1,25 объемов) и 4-метилпентан-2-ола (130 мл; 0,50 объема). Влажный продукт высушивали под вакуумом при 55°С до постоянной массы, получая указанное в заголовке соединение в виде твердого вещества белого цвета (193,6 г; 76%).
Т.пл.99-101°С.
1H ЯМР (300 МГц; CDCl3): δ 1.41 (9Н, s), 2.3-2.75 (6Н, m), 2.75-3.0 (5H, m), 3.1-3.38 (3H, m), 3.88 (2H, s), 3.95-4.19 (3H, m), 5.85 (1H, bs), 6.99 (2H, d), 7.6 (2H, d).
Пример 1
Кристаллизация Соединения А (Способ (I): форма В)
Соединение А (246,58 г; полученное аналогично методикам, описанным выше в Подготовительном примере А, хотя в этом случае материал повторно объединяли с маточными растворами, промывали щелочью NaOH и затем упаривали, получая твердое вещество) суспендировали в этилацетате (500 мл) и затем нагревали для растворения твердого вещества. Получили прозрачный раствор. Этилацетат (80 мл) удаляли перегонкой для того, чтобы азеотропно высушить соединение (оставляя 1,7 объема этилацетата). Данный раствор затем переносили горячим в предварительно подогретую колбу вместимостью 1 л с фланцем (температура при переносе составляла 68°С). Затем раствор оставляли охлаждаться естественным образом (полностью удаляли источник тепла) в атмосфере азота с перемешиванием воздухом. Через 40 минут охлаждения начиналась кристаллизация (внутренняя температура составляла 38°С). Температура раствора в течение следующих 1,5 часа составляла 38-40°С. Затем раствор охлаждали до 20°С с помощью бани вода/лед и оставляли на 1 час. Продукт отфильтровывали, промывали холодным (0°С) этилацетатом (330 мл) и затем высушивали под вакуумом в течение ночи при 40°С с получением 126,83 г (выход 51%, предполагая, что исходный материал сухой).
Кристаллы анализировали с помощью XRPD, и результаты представлены ниже в таблице (Таблица 1) и показаны на Фиг.1.
Элементарную ячейку определили по данным дифракции рентгеновских лучей на монокристалле. Она является орторомбической, с пространственной группой P212121 и следующими размерами: а=9,096 Å, b=11,077 Å, с=22,136 Å, α=β=γ=90°, и V=2230,3 Å3.
DSC-анализ показал две эндотермы с экстраполированными точками начала приблизительно 121 и 126°С. TGA-анализ показал уменьшение в массе приблизительно 0,1% (по массе) между 110 и 130°С.
Пример 2
Кристаллизация Соединения А (Способ (II); форма А)
Этилацетат (250 мл) добавляли к Соединению А (188,10 г; полученному аналогично способам, описанным выше в Подготовительном примере А) и смесь нагревали. Полученный горячий раствор (нет никакого осадка) фильтровали. В продолжение фильтрования из фильтруемого раствора выпадало некоторое количество твердого осадка. Его повторно растворяли в этилацетате (50 мл) и фильтровали. Объединенные фильтраты концентрировали путем удаления этилацетата (120 мл). Оставшийся раствор оставляли охлаждаться до комнатной температуры (кристаллизация начиналась при 40°С). Затем суспензию охлаждали до 10°С в течение 1 часа. Твердый продукт отфильтровывали, промывали холодным этилацетатом (100 мл) и затем высушивали под вакуумом при 40°С с получением 126,7 г (68%).
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 2) и показаны на Фиг.2.
DSC-анализ показал эндотерму с экстраполированной точкой начала приблизительно 125°С. TGA-анализ показал уменьшение в массе приблизительно 0,1% (по массе) между 120 и 130°С.
Пример 3
Кристаллизация Соединения А (Способ (III): форма С)
Моногидрат соли Соединения А с бензолсульфоновой кислотой (4,5 кг; полученный аналогично способам, описанным выше в Подготовительном примере А) добавляли к дихлорметану (89,7 кг). Добавляли водный гидроксид натрия (1М; 46,9 кг) и реакционную смесь перемешивали в течение 45 минут. Конечная температура содержимого составляла 16,9°С. Две фазы оставляли разделяться в течение 68 минут и водный слой (48,8 кг) отбрасывали. Органический слой переносили в чистый сосуд и фильтровали. Добавляли этанол (72 кг). Растворитель (101,1 кг) отгоняли (3 часа 20 мин). Конечная температура содержимого составляла 76,0°С. Добавляли этанол (25,4 кг) и отгоняли дополнительные 11,6 кг растворителя (1 час 30 мин), конечная температура содержимого составляла 79,3°С. Раствор охлаждали до -17°С в течение ночи, но никакой кристаллизации не наблюдалось. Отгоняли еще 32,7 кг растворителя и реакционную смесь охлаждали до 15°С в течение ночи, в продолжение которой продукт кристаллизовался. Реакционную смесь охлаждали до 0°С и перемешивали в течение 30 минут. Продукт отфильтровывали и промывали этанолом (7,3 кг) с получением влажного твердого вещества (2,7 кг). Продукт высушивали при комнатной температуре в течение 2 часов 16 мин (4,4 кПа (33 мм Hg)), затем при 40-45°С в течение 15 часов 24 минут (3,87 кПа (29 мм Hg)) с получением 2,18 кг продукта 99,76%-ной чистоты (по площади), содержащего 0,3% мас./мас. этанола.
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 3) и показаны на Фиг.3.
Элементарную ячейку определили по данным дифракции рентгеновских лучей на монокристалле. Она является орторомбической, с пространственной группой C2cb и следующими размерами: а=10,685 Å, b=19,391 Å, с=21,071 Å, α=β=γ=90°, и V=4365,8 Å3.
DSC-анализ показал эндотерму с экстраполированной точкой начала приблизительно 122°С. TGA-анализ показал уменьшение в массе приблизительно 0,3% (по массе) между 25 и 140°С.
Пример 4
Кристаллизация моногидрата соли Соединения А с бензолсульфоновой кислотой
1,88 г (4,9 ммоль) Соединения А полученного аналогично методикам, описанным выше в Подготовительном примере А) растворяли в 23 мл этилацетата. 0,807 г (5,1 ммоль) бензолсульфоновой кислоты растворяли в 4,5 мл метанола. Раствор бензолсульфоновой кислоты добавляли к перемешиваемому раствору Соединения А. Бензолсульфонатная соль выпадала в осадок приблизительно через 10 минут. Осадок выдерживали при 4°С в течение ночи. Кристаллы отфильтровывали, промывали этилацетатом и высушивали под вакуумом в течение ночи. Выход 1,6 г (60%). Продукт перекристаллизовывали из смеси EtOH:Н2O (1:1).
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 4) и показаны на Фиг.4.
Элементарную ячейку определили по данным дифракции рентгеновских лучей на монокристалле. Она является моноклинной, с пространственной группой P21/c и следующими размерами: а=18,833 Å, b=9,293 Å, с=16,271 Å, α=90°, β =94,94°, γ=90°, и V=2837,1 Å3.
Продукт представляет собой моногидрат без четко определенной точки плавления. DSC-анализ показал эндотерму с экстраполированной точкой начала в интервале приблизительно 85-110°С. TGA-анализ показал уменьшение в массе приблизительно 3% (по массе) между 60 и 160°С.
Пример 5
Кристаллизация соли Соединения А с паратолуолсульфоновой кислотой (Формы А и В)
Соединение А (1,14 г; 2,96 ммоль; полученное аналогично методикам, описанным выше в Подготовительном примере А (например, перед добавлением бензолсульфоновой кислоты в методике, описанной здесь для образования соли Соединения А с бензолсульфоновой кислотой)) растворяли в этилацетате (20 мл). По каплям добавляли раствор паратолуолсульфоновой кислоты (PTSA) (0,57 г; 2,99 ммоль) в метаноле (0,3 мл). Проводили промывку PTSA в 0,1 мл метанола и добавляли дополнительные 5 мл этилацетата. Через несколько минут образовалась суспензия белого цвета. Ее перемешивали в течение 1 ч. Твердое вещество отфильтровывали и промывали 5 мл этилацетата. Выход 1,4 г (84,8%). Твердую соль толуолсульфоновой кислоты (1,40 г) смешивали с 15 мл этилацетата и нагревали до температуры дефлегмации. К суспензии добавляли 0,85 мл метанола. В пределах нескольких секунд все твердое вещество перешло в раствор. Его оставляли охлаждаться, и через несколько минут наблюдалась кристаллизация. Вскоре он полностью затвердевал. К нему еще добавляли этилацетат (10 мл) и метанол (0,15 мл). Густую суспензию затем перемешивали при 0°С в течение 30 минут. Твердые вещества отфильтровывали и промывали этилацетатом (5 мл). Их оставляли высушиваться на вакуум-отсосе. Выход 1,10 г (66,6%). Часть (500 мг) высушивали при 50°С при пониженном давлении, получая 0,440 г.
Часть соли толуолсульфоновой кислоты перекристаллизовывали из ацетона. Кристаллы (форма А) анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 5(а)) и показаны на Фиг.5(а). Часть соли толуолсульфоновой кислоты суспендировали в фосфатном буфере (ионная сила 0,1М при рН 3) с последующей декантацией. Кристаллы (форма В) анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 5(б)) и показаны на Фиг.5(б).
Для формы А DSC-анализ показал эндотерму с экстраполированной точкой начала приблизительно 145°С. TGA-анализ показал уменьшение в массе приблизительно 3,3% (по массе) между 20 и 120°С.
Для формы В DSC-анализ показал эндотерму с экстраполированной точкой начала приблизительно 153°С. TGA-анализ показал уменьшение в массе приблизительно 4,4% (по массе) между 25 и 120°С.
Пример 6
Кристаллизация соли Соединения А с 1-гидрокси-2-нафтойной кислотой
К Соединению А (0,60 г; 1,56 ммоль; полученному аналогично методикам, описанным выше в Подготовительном примере А (например, перед добавлением бензолсульфоновой кислоты в методике, описанной здесь для образования соли Соединения А с бензолсульфоновой кислотой)) в этилацетате (10 мл) добавляли 1-гидрокси-2-нафтойную кислоту (0,323 г; 1,7 ммоль) в метаноле (1 мл). Раствор не образовался. Смесь перемешивали при комнатной температуре в течение 30 минут. Не отмечалось никакой кристаллизации. Затем раствор охлаждали в течение 30 минут. Наблюдали образование небольших количеств кристаллов. Растворители выпаривали до сухого состояния, твердые вещества переводили в этилацетат (5 мл) и нагревали до температуры дефлегмации. К части раствора добавляли метанол (0,2 мл). Полученную суспензию охлаждали смесью лед/вода, фильтровали и промывали этилацетатом (2 мл). Твердое вещество затем высушивали в сушильном шкафу при 50°С при пониженном давлении в течение 24 ч. Выход 0,510 г (57%).
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 6) и показаны на Фиг.6.
Пример 7
Кристаллизация соли Соединения А с 1,5-нафталинсульфоновой кислотой
Соединение А (0,490 г; 1,27 ммоль; полученное аналогично методикам, описанным выше в Подготовительном примере А (например, перед добавлением бензолсульфоновой кислоты в методике, описанной здесь для образования соли Соединения А с бензолсульфоновой кислотой)) растворяли в этилацетате (10 мл). Добавляли раствор 1,5-нафталинсульфоновой кислоты в метаноле (0,5 мл). Через несколько минут появилось твердое белое вещество. Все перемешивали при комнатной температуре в течение 30 минут и затем охлаждали смесью лед/вода. В суспензии отмечалось некоторое количество больших комков твердого белого вещества. Твердые вещества отфильтровывали с получением 0,34 г твердого вещества белого цвета. Его переводили в метанол (50 мл) и воду (100 мл) и нагревали до температуры дефлегмации до тех пор, пока не получился полностью прозрачный раствор. Раствор оставляли до достижения комнатной температуры и затем охлаждали смесью лед/вода в течение 30 минут. Твердые вещества отфильтровывали с получением порошка белого цвета (0,150 г). Твердое вещество высушивали в сушильном шкафу при 50°С и пониженном давлении в течение 5 ч.
1H ЯМР показал отношение кислоты к основанию, равное 1:2. Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 7) и показаны на Фиг.7.
Пример 8
Кристаллизация соли Соединения А с 2-мезитиленсульфоновой кислотой
Раствор 2-мезитиленсульфоновой кислоты (0,276 г) в метаноле (0,3 мл) добавляли к перемешиваемому раствору Соединения А (0,45 г; полученного аналогично методикам, описанным выше в Подготовительном примере А (например, перед добавлением бензолсульфоновой кислоты в методике, описанной здесь для образования соли Соединения А с бензолсульфоновой кислотой)) в этилацетате (10 мл). Образование осадка происходило немедленно. Суспензию перемешивали при комнатной температуре в течение 30 минут и затем охлаждали смесью лед/вода. Продукт отфильтровывали и промывали этилацетатом (3 мл) с получением указанного в заголовке соединения (0,60 г; 88%). Полученную соль растворяли в этилацетате (10 мл) и нагревали до температуры дефлегмации. Добавляли метанол (3 мл) с образованием прозрачного раствора. Раствор оставляли охлаждаться до комнатной температуры и затем перемешивали при 0-5°С в течение 1 ч. Суспензию фильтровали с получением бесцветного твердого вещества (0,370 г; 54% на двух стадиях).
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 8) и показаны на Фиг.8.
Пример 9
Кристаллизация Соединения D
Способ I. Соединение D (полученное аналогично методикам, описанным здесь ранее) сначала очищали колоночной хроматографией на силикагеле, используя смесь дихлорметан/метанол/28%-ный водный аммиак (9:1:0,5) в качестве элюента. Полученный порошок светло-желтого цвета (203 г) растворяли в дихлорметане (400 мл) и затем желтый раствор разбавляли н-гептаном (2 л) до тех пор, пока смесь не стала мутной. Смесь энергично перемешивали при комнатной температуре и вносили затравку кристаллов Соединения D (полученных медленным выпариванием растворителя из небольшой порции раствора указанного в заголовке соединения, полученного аналогичным этой методике способом). Большая часть продукта выпадала в осадок после перемешивания в течение двух часов. Получающуюся суспензию фильтровали и затем высушивали под вакуумом при 40°С с получением 179 г указанного в заголовке соединения в виде порошка беловатой окраски. Вторая часть (44 г) очищенного на колонке материала давала дополнительные 25 г указанного в заголовке соединения после подобной перекристаллизации.
Способ II. Смесь Соединения D (полученного аналогично методикам, описанным здесь ранее (главным образом смотри Подготовительный пример D(4), Способ III, описанный выше); 14,29 г), изопропанола (28 мл) и диизопропилового эфира (140 мл) нагревали до 80°С. Раствор фильтровали горячим до прозрачного состояния и затем повторно нагревали до 80°С. Затем раствор оставляли охлаждаться до комнатной температуры, после чего начиналось образование осадка. После перемешивания в течение двух часов осадок собирали фильтрованием, промывали смесью изопропанол: диизопропиловый эфир (1:6, 70 мл) и затем высушивали отсасыванием на фильтре. Влажный продукт высушивали под вакуумом при 70°С в течение ночи с получением указанного в заголовке соединения в виде твердого вещества белого цвета (10,1 г; 70%).
1H ЯМР (300 МГц, CDCl3) δ 1.41 (9Н, s), 2.3-2.75 (6Н, m), 2.75-3.0 (5Н, m), 3.1-3.38 (3H, m), 3.88 (2H, s), 3.95-4.19 (3H, m), 5.85 (1H, bs), 6.99 (2H, d), 7.6 (2H, d).
Способ III. Соединение D (полученное аналогично методикам, описанным здесь ранее; 1,0 г) растворяли в горячем метилизобутилкетоне (10 мл). Добавляли изогексан (30 мл) и раствор отделяли от небольшого количества нерастворенного материала декантацией в чистую колбу. Раствор охлаждали в течение ночи в холодильнике, что приводило к образованию гранулированного осадка. Раствор оставляли нагреваться до комнатной температуры и затем твердое вещество собирали фильтрованием. Твердое вещество высушивали отсасыванием на фильтре, затем высушивали под вакуумом при 40°С с получением твердого вещества беловатой окраски (0,73 г).
Способ IV. Соединение D (полученное аналогично методикам, описанным здесь ранее; 1,0 г) растворяли в горячем изопропилацетате (10 мл). Добавляли изооктан (20 мл) и раствор отделяли от небольшого количества нерастворенного материала декантацией в чистую колбу. Раствор охлаждали в течение ночи в холодильнике, что приводило к образованию гранулированного осадка. Раствор оставляли нагреваться до комнатной температуры и затем твердое вещество собирали фильтрованием. Твердое вещество высушивали отсасыванием на фильтре, затем высушивали под вакуумом при 40°С с получением твердого вещества беловатой окраски (0,82 г).
Способ V. Соединение D (полученное аналогично методикам, описанным здесь ранее; 1,0 г) растворяли в горячем водном этаноле (50%-ном, 20 мл). Раствор охлаждали в течение выходных дней в холодильнике, что приводило к образованию кристаллов игольчатой формы. Кристаллы собирали фильтрованием и высушивали отсасыванием на фильтре. Высушивание под вакуумом при 40°С давало бесцветное твердое вещество (0,48 г).
Способ VI. Соединение D (полученное аналогично методикам, описанным здесь ранее; 100 мг) растворяли в толуоле (1 мл). Добавляли изооктан (2 мл) и мутный раствор охлаждали в течение выходных дней в холодильнике, что приводило к образованию осадка в виде мелких гранул. Раствор оставляли нагреваться до комнатной температуры и затем твердое вещество собирали фильтрованием. Твердое вещество высушивали отсасыванием на фильтре, затем высушивали под вакуумом при 40°С с получением твердого вещества беловатой окраски (62 мг).
Кристаллы (из Способов I-VI) анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 9) и показаны на Фиг.9.
Элементарную ячейку определили по данным дифракции рентгеновских лучей на монокристалле. Она является орторомбической, с пространственной группой P212121 и следующими размерами: а=5,870 Å, b=9,098 Å, с=45,101 Å, α=β=γ=90°, и V=2408,6 Å3.
DSC-анализ показал эндотерму с экстраполированной точкой начала приблизительно 100-102°С. TGA-анализ показал уменьшение в массе приблизительно 0,4% (по массе) между 90 и 110°С.
Пример 10
Кристаллизация соли Соединения D с метансульфоновой кислотой
Способ I. Соединение D (250 мг; полученное аналогично способам, описанным выше в Подготовительном примере D) растворяли в метаноле (10 мл). К раствору добавляли метансульфоновую кислоту (56 мг; 38 мкл). Метанол удаляли при пониженном давлении и остающуюся смолу снова растворяли в минимальном, насколько возможно, количестве горячего этилацетата. В раствор вносили затравку предварительно полученных кристаллов соли (полученных медленным выпариванием растворителя из небольшой порции раствора указанного соединения, полученного аналогичным этой методике способом) и оставляли в холодильнике на несколько суток для кристаллизации. Получающиеся кристаллы собирали фильтрованием и промывали небольшим количеством холодного этилацетата (с выходом 280 мг, 98%).
Способ II. Соединение D (4,00 г, полученное аналогично способам, описанным выше в Подготовительном примере D) растворяли в этилацетате (40 мл). Добавляли раствор метансульфоновой кислоты (0,87 г) в этилацетате (40 мл). Через 3 минуты в раствор вносили затравку кристаллов предварительно полученной соли (14 мг, из Способа I, описанного выше), что вызывало немедленное образование осадка. Через 5 минут твердое вещество собирали фильтрованием и промывали этилацетатом перед тем, как высушить отсасыванием на фильтре. Высушивание под вакуумом при 40°С в течение 3,5 часа давало твердое кристаллическое вещество белого цвета (4,71 г; 97%).
Т.пл. 170-2°С (разл.).
Способ III. Соль Соединения D с метансульфоновой кислотой (0,8 г; полученную аналогично способу, описанному выше в Способе II) нагревали в ацетонитриле (8 мл, 10 объемов). Раствор образовался при 70°С. Его оставляли охлаждаться до температуры окружающей среды. Продукт собирали фильтрованием и промывали небольшим количеством холодного ацетонитрила. Сначала его высушивали под вакуумом. при 40°С (точка плавления (разложения) от 169,4 до 169,9°С; содержание остаточного растворителя ацетонитрила 0,08 мас.% (ГХ)). Затем его высушивали под вакуумом при 80°С; содержание остаточного растворителя ацетонитрила менее чем 0,02 мас.% (ГХ). Чистота 99,92% по ВЭЖХ-площади (пика).
Способ IV. Смесь соли Соединения D с метансульфоновой кислотой (2,0 г; 1,0 моль-экв., полученной аналогично способу, описанному выше в Способе II) и ацетонитрила (20 мл, 10 отн. об.) нагревали до 80°С. При этой температуре образовался раствор. Раствор оставляли охлаждаться до 25°С±5°С и затем охлаждали еще до 5°С±5°С. Продукт собирали фильтрованием и промывали холодным ацетонитрилом (4 мл, 2 отн. об.). Влажное твердое вещество высушивали под вакуумом при 80°С. Это давало указанное в заголовке соединение (0,15 г; 7,5%) в виде твердого вещества беловатой окраски. 99,97% ВЭЖХ-площадь. Содержание остаточного растворителя ацетонитрила менее чем 0,02 мас.% (ГХ).
Способ V. Смесь соли Соединения D с метансульфоновой кислотой (10,0 г; 1,0 моль-экв., полученной аналогично способу, описанному выше в Способе II за исключением того, что в этом случае вместо этилацетата в качестве растворителя использовали бутилацетат)) и пентан-2-ола (100 мл, 10 отн. об.) нагревали до 85°С, получая раствор. Раствор оставляли охлаждаться до 25°С±5°С и затем охлаждали еще до 5°С±5°С. Продукт собирали фильтрованием и промывали холодным пентан-2-олом (20 мл, 2 отн. об.). Влажное твердое вещество высушивали под вакуумом при 40°С в течение двадцати часов. Это давало указанное в заголовке соединение (6,72 г; 67,2%) в виде твердого вещества беловатой окраски. 99,47% ВЭЖХ-площадь.
Способ VI. Смесь соли Соединения D с метансульфоновой кислотой (3,0 г; 1,0 моль-экв., полученной аналогично способу, описанному выше в Способе V) и 3-метилбутан-1-ола (15 мл, 5 отн. об.) нагревали до 90°С, получая раствор. Раствор оставляли охлаждаться до 25°С±5°С и затем охлаждали еще до 5°С±5°С. Продукт собирали фильтрованием и промывали холодным 3-метилбутан-1-олом (6 мл, 2 отн. об.). Влажное твердое вещество высушивали под вакуумом при 40°С. Это давало указанное в заголовке соединение в виде твердого вещества беловатой окраски (2,43 г; 81%).
Способ VII. Смесь соли Соединения D с метансульфоновой кислотой (2,0 г; 1,0 моль-экв., полученной аналогично способу, описанному выше в Способе V) и гексан-1-ола (15 мл, 5 отн. об.) нагревали до 80°С, получая раствор. Раствор оставляли охлаждаться до 25°С±5°С и затем охлаждали еще до 5°С±5°С. Продукт собирали фильтрованием и промывали холодным гексан-1-олом (6 мл, 2 отн. об.). Влажное твердое вещество высушивали под вакуумом при 40°С. Это давало указанное в заголовке соединение в виде твердого вещества беловатой окраски (2,43 г; 81%).
Кристаллы (Способы I-VII) анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 10) и показаны на Фиг.10.
Элементарную ячейку определили по данным дифракции рентгеновских лучей на монокристалле. Она является орторомбической, с пространственной группой P212121 и следующими размерами: а=7,772 Å, b=14,250 Å, с=25,750 Å, α=β=γ=90°, и V=1717,2 Å3.
DSC-анализ показал эндотерму с экстраполированной точкой начала приблизительно 167°С. TGA-анализ показал уменьшение в массе приблизительно 1,2% (по массе) между 25 и 100°С.
Пример 11
Кристаллизация соли Соединения D с гиппуровой кислотой
Раствор гиппуровой кислоты (0,8 г) в метаноле (20 мл) добавляли к раствору Соединения D (2,00 г, полученного аналогично способам, описанным выше в Примере 9) в метаноле (5 мл). Затем раствор концентрировали под вакуумом с получением масла. Добавляли диэтиловый эфир (20 мл) и смесь повторно концентрировали с получением пены. Перемешивание в эфире (100 мл) в течение ночи и фильтрование давали 2,22 г указанного в заголовке соединения (79%). Соль высушивали под вакуумом при 34°С, получая 2,19 г (78%).
Наличие соли гиппуровой кислоты подтверждено ЯМР.
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 11) и показаны на Фиг.11.
Пример 12
Кристаллизация Соединения С
(1) Соединение С получили аналогично методике, описанной выше в Подготовительном примере С, за исключением того, что выполняли следующую конечную стадию способа: трехгорлую колбу вместимостью 3 л оборудовали магнитной мешалкой, термометром и парциальным конденсатором. В колбу загружали неочищенный 4-[4-(9-окса-3,7-диазабицикло[3.3.1]нон-3-ил)бутил]-бензнитрил (смотри Подготовительный пример С(5), описанный выше; 25,8 г; 88 ммоль), дихлорметан (0,88 л) и трет-бутил-2-бромэтилкарбамат (смотри Подготовительный пример В(I)(1), описанный выше; 27,7 г; 123 ммоль). Затем добавляли триэтиламин (0,0197 л; 0,141 моль). Прозрачный раствор кипятили с обратным холодильником в течение 12 часов в атмосфере азота и затем охлаждали до комнатной температуры. Ход реакции контролировали с помощью ТСХ-анализа и обнаружили, что она полностью завершилась к этому моменту времени.
(2) Реакционную смесь переносили в делительную воронку и промывали последовательно водой (200 мл), 15%-ным водным гидроксидом натрия (200 мл), водой (200 мл) и соляным раствором (200 мл). Органический слой высушивали над сульфатом магния и концентрировали под вакуумом. Получающееся желтое вязкое масло хроматографировали на силикагеле, элюируя сначала смесью дихлорметан/метанол (9:1), затем смесью дихлорметан/метанол/28%-ный водный гидроксид аммония (9:1:0,02), с получением неочищенного Соединения С (25,1 г; 66%-ный выход) в виде твердого вещества беловатой окраски. Обнаружено, что более ранние хроматографические фракции (5,1 г) содержали небольшое количество менее полярных примесей (по ТСХ-анализу с элюированием смесью дихлорметан/метанол/28%-ный водный гидроксид аммония (9:1:0,05)), в то время как более поздние фракции (20 г) давали одно пятно по данным ТСХ-анализа. Более ранние фракции (5,1 г) объединяли с полученной ранее партией Соединения С (7,1 г, содержащей незначительные примеси) и хроматографировали на силикагеле, элюируя сначала смесью дихлорметан/метанол (19:1), затем смесью дихлорметан/метанол (9:1), с получением порошка бледно-желтой окраски (5,5 г). Порошок растворяли в дихлорметане (200 мл). Получающийся раствор промывали последовательно 25%-ным водным гидроксидом натрия (50 мл), водой (50 мл) и соляным раствором (40 мл). Затем материал высушивали над сульфатом магния и концентрировали под вакуумом с получением неочищенного указанного в заголовке соединения в виде порошка беловатой окраски (5 г). Фракцию в 20 г растворяли в дихлорметане (500 мл). Органический слой промывали последовательно 25%-ным водным гидроксидом натрия (100 мл), водой (100 мл) и соляным раствором (100 мл). Затем материал высушивали над сульфатом магния и концентрировали под вакуумом с получением порошка беловатой окраски (19 г). Партии смешивали вместе.
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 12) и показаны на Фиг.12.
DSC-анализ показал эндотермический пик с экстраполированной точкой начала приблизительно 97°С. TGA-анализ показал уменьшение в массе приблизительно 0,6% (по массе) между 25 и 120°С.
Пример 13
Кристаллизация соли Соединения С с метансульфоновой кислотой
Соединение С (250 мг; полученное аналогично способу, описанному выше в Примере 12) растворяли в метаноле (10 мл). К раствору добавляли метансульфоновую кислоту (56 мг; 38 мкл). Метанол удаляли при пониженном давлении и оставшуюся смолу снова растворяли в минимальном, насколько возможно, количестве горячего изопропанола. В раствор вносили затравку предварительно полученной соли (полученной медленным выпариванием растворителя из небольшой порции раствора указанного в заголовке соединения, полученного аналогичным этой методике способом) и оставляли в холодильнике на несколько суток для кристаллизации. Получающиеся кристаллы собирали фильтрованием и промывали небольшим количеством холодного изопропанола (с выходом 220 мг, 72%).
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 13) и показаны на Фиг.13.
DSC-анализ показал эндотермические пики с экстраполированными точками начала приблизительно 145°С (минорный) и 170°С (основной). TGA-анализ показал уменьшение в массе приблизительно 0,9% (по массе) между 25 и 105°С.
Пример 14 Кристаллизация соли Соединения С с паратолуолсульфоновой кислотой
Соединение С (250 мг; полученное аналогично способу, описанному выше в Примере 12) растворяли в метаноле (10 мл). К раствору добавляли толуолсульфоновую кислоту (110 мг). Метанол удаляли при пониженном давлении и оставшуюся смолу снова растворяли в минимальном, насколько возможно, количестве горячего этилацетата. В раствор вносили затравку предварительно полученной соли (полученной медленным выпариванием растворителя из небольшой порции раствора указанного в заголовке соединения, полученного аналогичным этой методике способом) и оставляли в холодильнике на несколько суток для кристаллизации. Получающиеся кристаллы собирали фильтрованием и промывали небольшим количеством холодного этилацетата (с выходом 280 мг, 78%).
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 14) и показаны на Фиг.14.
DSC-анализ показал эндотермический пик с экстраполированной точкой начала приблизительно 138°С. TGA-анализ показал уменьшение в массе приблизительно 0,2% (по массе) между 25 и 150°С.
Пример 15
Соль Соединения D с [(дифенил-4-карбонил)амино]уксусной кислотой
(а) Метиловый эфир [(дифенил-4-карбонил)амино]уксусной кислоты
Дихлорметан (50 мл) и затем триэтиламин (11,2 мл; 79,6 ммоль, 2,0 экв.) добавляли к гидрохлориду метилового эфира глицина (5,0 г; 39,8 ммоль; 1,0 экв.) Смесь перемешивали и охлаждали до -5°С, используя баню лед/метанол. В продолжение 22 минут добавляли суспензию дифенил-4-карбонилхлорида (8,26 г; 39,8 ммоль; 1,0 экв.) в дихлорметане (25 мл). Смесь перемешивали в течение 3 ч при -5°С и затем оставляли перемешиваться при комнатной температуре в течение ночи (16 часов). Добавляли воду (75 мл) и смесь быстро перемешивали в течение 30 минут при комнатной температуре. Слои разделяли. Органический слой промывали водой (75 мл) и затем упаривали до сухого состояния с использованием роторного испарителя с получением твердого вещества беловатой окраски (6,58 г; 62%).
1H ЯМР (300 МГц, CDCl3) δ 3.82 (s, 3Н), 4.29 (d, J=5.1 Гц, 2Н), 6.68 (s, 1H), 7.3-7.5 (m, 3Н), 7.62 (d, J=4.8 Гц, 2Н), 7.68 (d, J=8.1 Гц, 2Н), 7.90 (d, J=8.4 Гц, 2Н).
Т.пл. 127-128°С.
(б) [(Дифенил-4-карбонил)амино]уксусная кислота
Метиловый эфир [(дифенил-4-карбонил)амино]уксусной кислоты (6,58 г; 25 ммоль; 1,0 экв.; со стадии (а), описанной выше) добавляли в колбу с последующим добавлением водного гидроксида натрия (1М, 84 мл, 50 ммоль; 2,0 экв.). Смесь нагревали до 50°С в течение 5 часов, используя масляную баню. Затем раствор перемешивали в течение ночи (16 часов) при комнатной температуре. По мере охлаждения образовывался осадок белого цвета. Смесь охлаждали еще до 5°С, используя баню лед/вода. К охлажденному раствору очень медленно добавляли концентрированную соляную кислоту (8 мл), стараясь, чтобы температура не поднималась выше 10°С. Смесь перемешивали в течение 15 минут и затем фильтровали. Твердое вещество белого цвета высушивали на воздухе в течение 30 минут и затем высушивали под вакуумом при 40°С в течение 16 часов с получением твердого вещества беловатой окраски (5,75 г; 93%).
1H ЯМР (300 МГц, DMSO-d6) δ 3.95 (d, J=5.7 Гц, 2Н), 7.35-7.5 (m, 3H), 7.7-7.8 (m, 4H), 7.97 (d, J=6.9 Гц, 2Н), 8.89 (t, J=6.0 Гц, 1Н), 12.58 (s, 1H).
Т.пл. 217-217,5°С.
(в) Перекристаллизация [(дифенил-4-карбонил)амино]уксусной кислоты
К [(дифенил-4-карбонил)амино]уксусной кислоте (5,0 г; со стадии (б), описанной выше) добавляли метанол (100 мл, 20 объемов). Смесь нагревали до 62°С, используя масляную баню, при постоянном перемешивании. Получающийся бледно-оранжевый раствор выдерживали при этой температуре в течение 10 минут. Раствор оставляли охлаждаться до комнатной температуры и затем охлаждали еще до 5°С, используя баню лед/вода. Кристаллизация начиналась приблизительно при 30°С. Осадок собирали фильтрованием, высушивали на воздухе в течение 15 минут, затем высушивали под вакуумом при 40°С в течение 26 часов с получением бесцветных кристаллов (2,9 г; 58%).
1H ЯМР (300 МГц, DMSO-d6) δ 3.95 (d, J=5.7 Гц, 2Н), 7.35-7.5 (m, 3H), 7.7-7.8 (m, 4H), 7.97 (d, J=6.9 Гц, 2Н), 8.89 (t, J=6.0 Гц, 2Н), 12.58 (s, 1H).
(г) Соль Соединения D с [(дифенил-4-карбонил)амино]уксусной кислотой
[(Дифенил-4-карбонил)амино]уксусную кислоту (1,14 г; смотри стадии (б) или (в), описанные выше) и Соединение D (2 г; полученное аналогично способам, описанным здесь ранее) растворяли в горячем изопропаноле (40 мл). По мере охлаждения до комнатной температуры образовывался кристаллический осадок, который отфильтровывали, промывали изопропанолом (2×20 мл) и высушивали отсасыванием на фильтре. Высушивание в течение 6 часов под вакуумом при 40°С давало соль в виде бесцветного кристаллического твердого вещества (2,50 г; 80%).
1H ЯМР (300 МГц, DMSO-d6+) δ 1.34 (9Н, s), 2.25 (2H, t), 2.3-2.5 (4H, m), 2.6-2.7 (1H, m), 2.7-2.8 (1H, m), 2.85-3.0 (4H, m), 3.0-3.1 (2H, m), 3.82 (2H, s), 3.88 (2H, d), 3.95-4.05 (2H, m), 4.1-4.2 (1H, m), 6.65 (1H, t), 7.14 (2H, d), 7.35-7.55 (3Н, m), 7.7-7.85 (6H, m), 7.96 (2H, d), 8.75 (1H, t).
Т.пл. 143-143,5°С.
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 15) и показаны на Фиг.15.
Пример 16
Соль Соединения D с гемиянтарной кислотой
В горячий раствор Соединения D (5,35 г; полученного аналогично методикам, описанным здесь ранее) и янтарной кислоты (0,71 г) в изопропаноле (20 мл) вносили затравку соли Соединения D с гемиянтарной кислотой (полученной медленным выпариванием растворителя из небольшой порции раствора указанного в заголовке соединения, полученного аналогичным этой методике способом) и затем оставляли стоять в холодильнике. На следующий день твердый осадок собирали фильтрованием и промывали изопропанолом (20 мл). Твердое вещество высушивали отсасыванием на фильтре и затем высушивали под вакуумом при 40°С в течение 2 часов с получением указанного в заголовке соединения в виде твердого вещества белого цвета. Фильтрат оставляли для полного выпаривания посредством выдерживания открытым на воздухе. Это давало твердое вещество, которое суспендировали в изопропаноле (20 мл) и затем фильтровали, промывая на фильтре изопропанолом (70 мл). Твердое вещество высушивали отсасыванием на фильтре и затем высушивали под вакуумом при 40°С в течение 90 минут с получением указанного в заголовке соединения в виде твердого вещества белого цвета (2,8 г).
Т.пл. 112-114°С.
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 16) и показаны на Фиг.16.
Пример 17
Соль Соединения D с (3,4-дихлорбензоиламино)уксусной кислотой
(а) Метиловый эфир (3.4-дихлорбензоиламино)уксусной кислоты
Дихлорметан (150 мл) и затем триэтиламин (33,0 мл; 234 ммоль, 2,0 экв.) добавляли к гидрохлориду метилового эфира глицина (14,7 г; 117 ммоль; 1,0 экв.) Смесь перемешивали и охлаждали до 2°С, используя баню лед/вода. В продолжение 7 минут добавляли раствор 3,4-дихлорбензоилхлорида (24,55 г; 117 ммоль; 1,0 экв.) в дихлорметане (75 мл). Смесь перемешивали в течение 1 ч при 2°С и затем оставляли перемешиваться при комнатной температуре в течение ночи (16 часов). Добавляли воду (225 мл) и смесь быстро перемешивали в течение 30 минут при комнатной температуре. Слои разделяли. Органические слои промывали водой (225 мл) и затем упаривали до сухого состояния с использованием роторного испарителя с получением твердого вещества беловатой окраски. Выделенное твердое вещество (26,18 г; 85%) добавляли к дихлорметану (300 мл, 10 объемов) с 1М раствором гидроксида натрия (300 мл, 10 объемов). Нижний органический слой концентрировали до сухого состояния под вакуумом (25,91 г; 84%).
Т.пл. 133,2-134,3°С.
δН (300 МГц, CDCl3) 3.66 (1Н, s, СН3), 4.03 (2Н, d, J=6 Гц, CH2), 7.78-7.87 (2H, m, CH), 8.100 (1Н, s, CH), 9.18 (1Н, t, J=5.7 Гц, NH).
(б) (3,4-Дихлорбензоиламино)уксусная кислота
Метиловый эфир (3,3-дихлорбензоиламино)уксусной кислоты (25,91 г; 100 ммоль; 1,0 экв.; смотри стадию (а), описанную выше) добавляли в колбу с последующим добавлением водного гидроксида натрия (1М, 198 мл, 200 ммоль; 2,0 экв.). Смесь нагревали до 50°С в течение 2 часов, используя масляную баню. По мере охлаждения образовывался осадок белого цвета. Смесь дополнительно охлаждали до 5°С, используя баню лед/вода. К охлажденному раствору очень медленно добавляли концентрированную соляную кислоту (60 мл), следя за тем, чтобы температура не превышала 10°С. Смесь перемешивали в течение 10 минут и затем фильтровали. Твердое вещество белого цвета высушивали на воздухе в течение 15 минут и затем высушивали под вакуумом при 40°С в течение 16 часов, получая твердое вещество беловатой окраски (19,15 г; 78%).
Т.пл. 140,0-140,3°С.
δН (300 МГц, DMSO-D6) 3.94 (2Н, d, J=6 Гц, CH2), 7.77-7.87 (2Н, m, CH), 8.10 (1Н, s, CH), 9.06 (1H, t, J=6 Гц), 12.66 (1H, bs, ОН).
(в) Соль Соединения D с (3,4-дихлорбензоиламино)уксусной кислотой
(3,4-Дихлорбензоиламино)уксусную кислоту (0,56 г; смотри стадию (б), описанную выше) и Соединение D (1,02 г; полученное согласно описанным здесь ранее методикам) растворяли в горячем этилацетате (4 мл). По мере охлаждения до комнатной температуры образовывался кристаллический осадок, который отфильтровывали, промывали этилацетатом (15 мл) и высушивали отсасыванием на фильтре. Высушивание в течение ночи под вакуумом при 40°С давало указанную в заголовке соль в виде бесцветного кристаллического твердого вещества (0,92 г; 58%).
Т.пл. 128,5-130,5°С.
1H ЯМР (400 МГц, DMSO-D6) δ 1.34 (9Н, s), 2.26 (2Н, t), 2.3-2.5 (3Н, m), 2.5-2.6 (1H, m), 2.6-2.7 (1H, m), 2.7-2.8 (1H, m), 2.85-3.0 (4H, m), 3.0-3.1 (2Н, m), 3.8-3.9 (4H, m), 4.01 (2Н, d), 4.1-4.2 (1H, m), 6.69 (1H, t), 7.12 (2Н, d), 7.7-7.8 (3Н, m), 7.84 (1H, dd), 8.09 (1H, dd), 8.92 (1H, t).
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 17) и показаны на Фиг.17.
Пример 18
Соль Соединения D с [(нафталин-2-карбонил)амино]уксусной кислотой
(а) Метиловый эфир [нафталин-2-карбонил)амино]уксусной кислоты
Дихлорметан (66 мл) и затем триэтиламин (14,6 мл; 105 ммоль, 2,0 экв.) добавляли к гидрохлориду метилового эфира глицина (6,61 г; 52,5 ммоль; 1,0 экв.) По мере добавления триэтиламина появлялся осадок белого цвета, и раствор становился более густым. Смесь перемешивали и охлаждали до 2°С, используя баню лед/вода. В продолжение 15 минут добавляли раствор 2-нафтоилхлорида (10,07 г; 52,5 ммоль; 1,0 экв.) в дихлорметане (33 мл). Смесь бледно-коричневой окраски перемешивали в течение 25 часов при 5°С. Добавляли воду (100 мл) и смесь быстро перемешивали в течение 30 минут при комнатной температуре. Слои разделяли. Органический слой промывали гидроксидом натрия (1М; 100 мл) и затем упаривали до сухого состояния с использованием роторного испарителя с получением твердого вещества беловатой окраски (12,21 г; 96%).
Т.пл. 117,7-118,1°С.
δН (400 МГц, DMSO-D6) 3.68 (3Н, s, СН3), 4.08 (2Н, d, J=4,5 Гц, CH2), 7.59-7.66 (2Н, m, CH), 7.935-8.015 (4Н, m, СН), 8.491 (1Н, s, CH), 9.124 (1Н, t, J=45,6 Гц, NH).
(б) [(Нафталин-2-карбонил)амино]уксусная кислота
Метиловый эфир [(нафталин-2-карбонил)амино]уксусной кислоты (10,03 г; 41 ммоль; 1,0 экв.; смотри стадию (а), описанную выше) добавляли в колбу с последующим добавлением водного гидроксида натрия (1М, 120 мл, 123 ммоль; 3,0 экв.). Смесь нагревали до 55°С в течение 2 часов, используя масляную баню. Смесь охлаждали до 5°С, используя баню лед/вода. К охлажденному раствору очень медленно добавляли концентрированную соляную кислоту (50 мл), следя за тем, чтобы температура не превышала 10°С. Образовался плотный осадок желтого цвета. Смесь перемешивали в течение 10 минут и затем фильтровали. Твердое вещество желтого цвета высушивали на воздухе в течение 15 минут и затем высушивали под вакуумом при 40°С в течение 16 часов (8,73 г; 93%). К части указанного в подзаголовке соединения (5,0 г; 22 ммоль) добавляли метанол (50 мл; 10 объемов) и воду (100 мл; 20 объемов). Смесь нагревали до 70°С, используя масляную баню, при перемешивании. Раствор выдерживали при этой температуре в течение 10 минут и затем оставляли охлаждаться еще до 5°С, используя баню лед/вода. Кристаллизация начиналась приблизительно при 30°С. Осадок собирали фильтрованием, высушивали на воздухе в течение 15 минут, затем высушивали под вакуумом при 40°С в течение 2 часов (3,2 г; 64%). Выделенное указанное в подзаголовке соединение (3,2 г; 0,014 моль; 64%) добавляли к воде (100 мл; 20 объемов) и метанолу (50 мл; 10 объемов). Смесь нагревали до 70°С для растворения твердого вещества. Раствор оставляли охлаждаться до комнатной температуры, по мере охлаждения происходила кристаллизация. Смесь охлаждали еще до 2°С и затем фильтровали, используя керамическую воронку. Твердое вещество высушивали на воздухе в течение 10 минут, затем высушивали под вакуумом при 40°С в течение 16 часов (2,21 г; 44%).
Т.пл. 167,1-167,4°С.
δН (400 МГц, DMSO-D6) 3.98 (2Н, d, J=5.6 Гц, СН2), 7.58-7.65 (2Н, m, CH), 7.95-8.05 (4Н, m, CH), 8.49 (1H, s, CH), 8.99 (1H, t, J=5.6 Гц, NH), 12.63 (1H, bs, ОН).
(в) Соль Соединения D с [(нафталин-2-карбонил)амино]уксусной кислотой
[(Нафталин-2-карбонил)амино]уксусную кислоту (0,51 г; смотри стадию (б), описанную выше) и Соединение D (1,01 г; полученное согласно описанным здесь ранее методикам) растворяли в метилизобутилкетоне (30 мл) при 100°С. По мере охлаждения до комнатной температуры образовывался кристаллический осадок, который отфильтровывали, промывали ацетоном (25 мл) и высушивали отсасыванием на фильтре. Высушивание в течение выходных дней под вакуумом при 40°С давало указанную в заголовке соль в виде бесцветного кристаллического твердого вещества (1,17 г; 77%).
Т.пл. 138,5-140°С.
1H ЯМР (300 МГц, DMSO-D6) δ 1.34 (9Н, s), 2.25 (2Н, t), 2.3-2.5 (4Н, m), 2.6-2.7 (1H, m), 2.7-2.8 (1H, m), 2.85-3.0 (4Н, m), 3.0-3.1 (2Н, m), 3.81 (2Н, s), 3.92 (2Н, d), 3.95-4.05 (2Н, m), 4.1-4.2 (1H, m), 6.68 (1H, t), 7.11 (2Н, d), 7.5-7.7 (2Н, m), 7.7-7.8 (2Н, m), 7.9-8.1 (4Н, m), 8.47 (1H, d), 8.85 (1H, t).
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 18) и показаны на Фиг.18.
Пример 19
Соль Соединения D с2,2,3,3-тетраметил-1.4-дибутановой кислотой
Соединение D (200 мг; полученное аналогично описанным здесь ранее методикам) растворяли в этилацетате (10 мл). 2,2,3,3-Тетраметил-1,4-дибутановую кислоту (38,3 мг; 0,5 экв.) растворяли в 1 мл метанола и добавляли к раствору Соединения D. Растворители выпаривали, добавляли этилацетат и затем этот растворитель медленно выпаривали путем выдерживания полученной смеси в открытой колбе при комнатной температуре в течение нескольких суток. Образующиеся кристаллы отфильтровывали. ЯМР-анализ показал образование соли в отношении 1:1 (˜100 мг, 42%).
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 19) и показаны на Фиг.19.
Пример 20
Соль Соединения D транс-D,L-1,2-циклопентандикарбоновой кислотой
Соединение D (100 мг; полученное аналогично описанным здесь ранее методикам) растворяли в этилацетате (5 мл). транс-D,L-1,2-Циклопентан-дикарбоновую кислоту (17,4 мг; 0,5 экв.) растворяли в метаноле и затем добавляли к раствору Соединения D. Растворители выпаривали и затем растворяли в этилацетате. Через некоторый промежуток времени образовался осадок белого цвета. Образованные кристаллы отфильтровывали. ЯМР-анализ показал образование соли в отношении 1:1 (˜60 мг).
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 20) и показаны на Фиг.20.
Пример 21
Соль Соединения D с (+)-O,O'-дибензоил-D-винной кислотой
Соединение D (100 мг; полученное аналогично описанным здесь ранее методикам) и 0,5 эквивалента (+)-O,O'-дибензоил-D-винной кислоты смешивали согласно методике, описанной выше в Примере 20. Соль кристаллизовалась из этилацетата. ЯМР показал, что образовалась соль с соотношением лекарственное средство: кислота, равным 2:1, и с ˜70%-ным выходом.
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 21) и показаны на Фиг.21.
Пример 22
Соль Соединения D с (+)-O,O'-ди-паратолуоил-D-винной кислотой
Соединение D (1,0 г; полученное аналогично описанным здесь ранее методикам) растворяли в этилацетате (15 мл). (+)-O,O'-Ди-паратолуоил-D-винную кислоту (0,43 г) растворяли в этилацетате (20 мл). Растворы смешивали. Через несколько минут образовался осадок белого цвета. Смесь выдерживали при 4°С в течение ночи. Кристаллы отфильтровывали и высушивали под вакуумом, получая 1,1 г (76%) соли с соотношением 2:1 (подтверждено ЯМР).
Кристаллы анализировали с помощью XRPD. Результаты представлены ниже в таблице (Таблица 22) и показаны на Фиг.22.
Пример 23
Кристаллизация Соединения В
Неочищенный материал из Подготовительного примера В, Варианта II, стадии (3), Способа II, растворяли при 60°С в изопропаноле (190 мл, 5,0 отн. об.) и горячий раствор фильтровали. Фильтрат перемешивали и оставляли охлаждаться до комнатной температуры. Кристаллизовалось твердое вещество белого цвета. Смесь охлаждали от комнатной температуры до приблизительно 8°С. Продукт собирали фильтрованием и промывали изопропанолом (50 мл, 2,0 об.). Влажный продукт высушивали под вакуумом при 40°С до постоянной массы с получением указанного в заголовке соединения в виде твердого вещества белого цвета (30,96 г; 81%).
Т.пл. 113,5°С.
1H ЯМР (400 МГц, CD3OD) δ 1.40 (9Н, s), 1.81-1.90 (2Н, m), 2.35-2.54 (8H, m), 2.93 (4H, t), 3.18-3.27 (4H, m), 3.87 (2Н, bs), 6.66 (2Н, d), 7.39 (2Н, d).
MS: m/z=(MH+, 430).
Сокращения
API - ионизация при атмосферном давлении (относительно масс-спектроскопии (MS))
br - уширенный (относительно ЯМР)
d - дублет (относительно ЯМР)
DCM - дихлорметан
DMF - N,N-диметилформамид
DMSO - диметилсульфоксид
dd - дублет дублетов (относительно ЯМР)
Et - этил
экв. - эквивалент(ы)
ГХ - газовая хроматография
ч - час(ы)
HCl - соляная кислота
ВЭЖХ - высокоэффективная жидкостная хроматография
IMS - промышленный метилированный спирт
IPA - изопропиловый спирт
KF - Карл-Фишер
m - мультиплет (относительно ЯМР)
Me - метил
MeCN - ацетонитрил
мин - минута(ы)
т.пл. - точка плавления
MS - масс-спектроскопия
Pd/C - палладий на угле
q - квартет (относительно ЯМР)
кг - комнатная температура
s - синглет (относительно ЯМР)
t - триплет (относительно ЯМР)
ТСХ - тонкослойная хроматография
УФ - ультрафиолет
Префиксы н-, втор-, изо- и трет- имеют свои обычные значения:
нормальный, вторичный, изо и третичный.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОМЕЖУТОЧНОЕ ХИМИЧЕСКОЕ СОЕДИНЕНИЕ | 2003 |
|
RU2326122C2 |
| (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид | 2012 |
|
RU2610091C2 |
| НОВЫЕ БИСПИДИНОВЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ СЕРДЕЧНЫХ АРИТМИЙ | 2000 |
|
RU2250903C2 |
| 2-АМИНОБЕНЗОКСАЗОЛКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-НТ3 | 2007 |
|
RU2448105C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ВКЛЮЧАЯ ТРАНС-7-ОКСО-6-(СУЛЬФОКСИ)-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИД И ЕГО СОЛИ | 2012 |
|
RU2769076C2 |
| ЗАМЕЩЕННЫЕ 3,7-ДИАЗОБИЦИКЛО[3.3.1]НОНАНЫ, ФОКУСИРОВАННАЯ БИБЛИОТЕКА И КОМБИНАТОРНАЯ БИБЛИОТЕКА | 2003 |
|
RU2228934C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| ПРОИЗВОДНЫЕ 2-АМИНО-7-(CHRR)-3Н,5Н-ПИРРОЛО[3,2-D]-ПИРИМИДИН-4-ОНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ ПРОЛИФЕРАЦИИ Т-ЛИМФОЦИТОВ МЛЕКОПИТАЮЩЕГО И НЕ ОКАЗЫВАЮЩИЙ ВОЗДЕЙСТВИЯ НА B-ЛИМФОЦИТЫ | 1992 |
|
RU2097384C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2323006C2 |
| ЛЕЧЕНИЕ НЕВРОЛОГИЧЕСКИХ РАССТРОЙСТВ | 2017 |
|
RU2765868C2 |

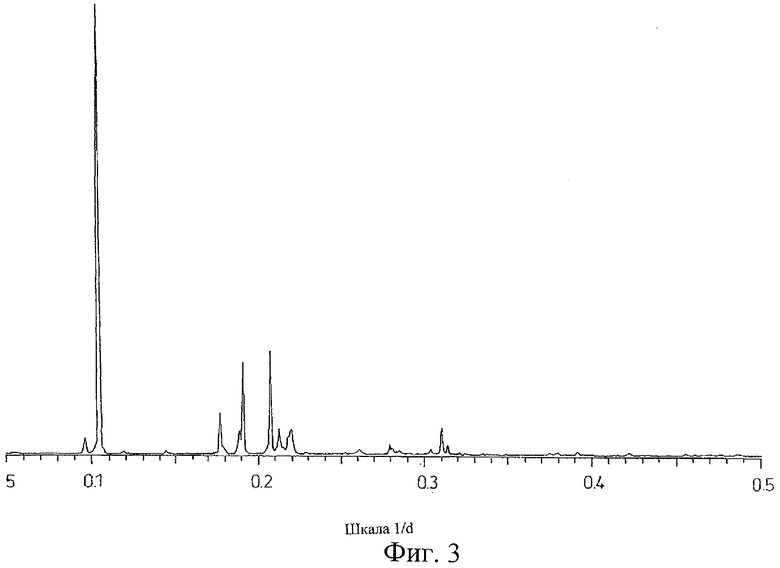
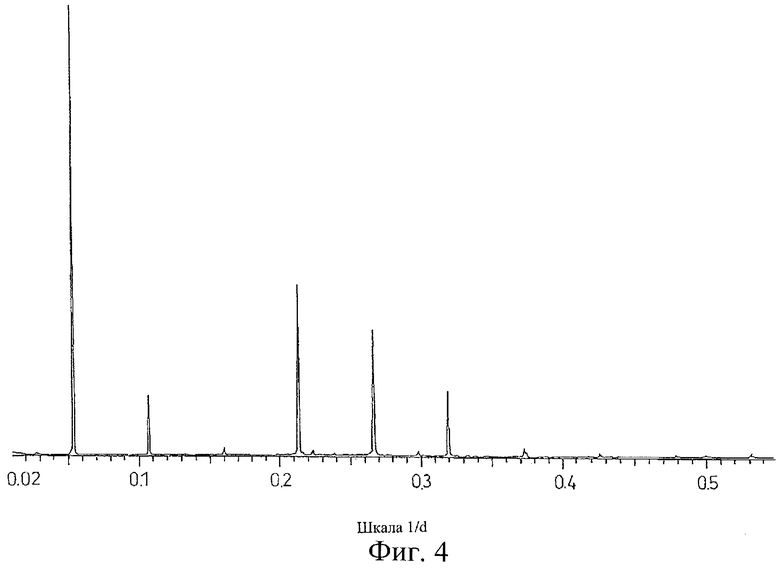


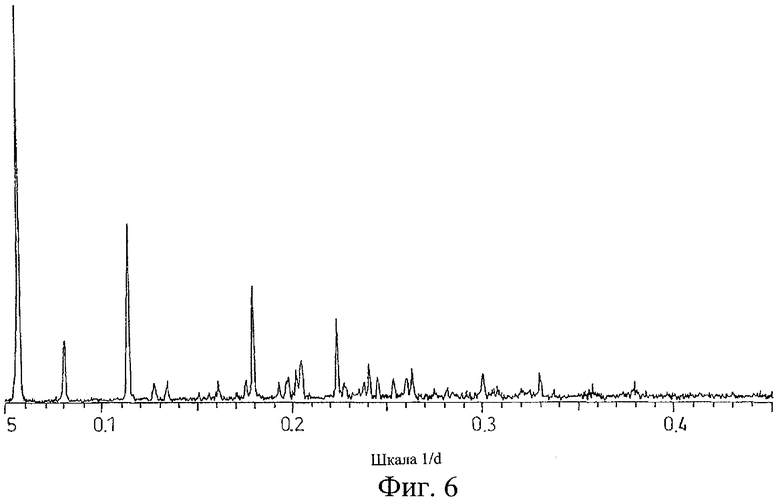
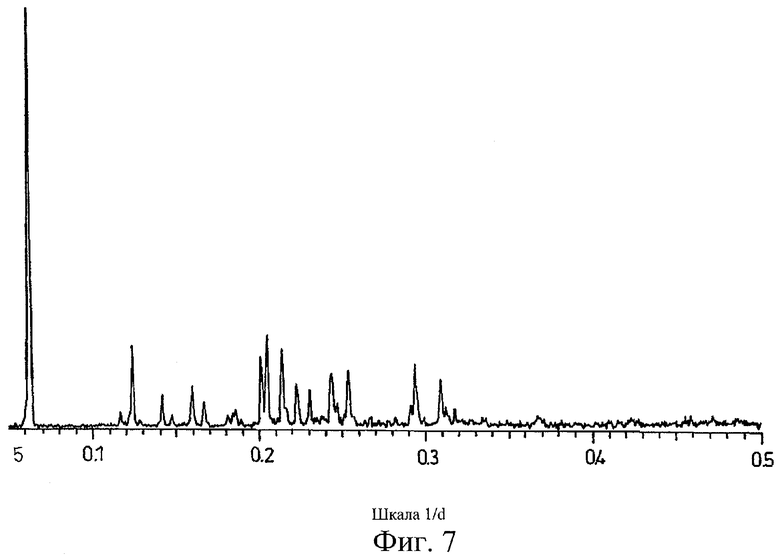

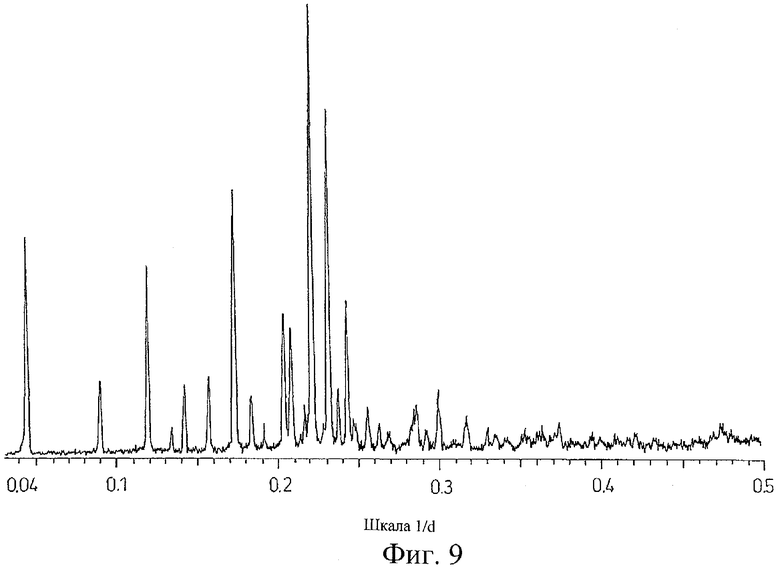
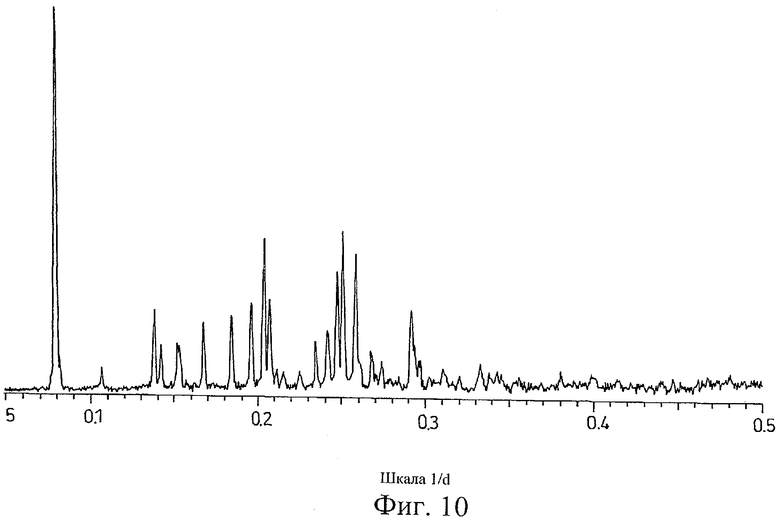
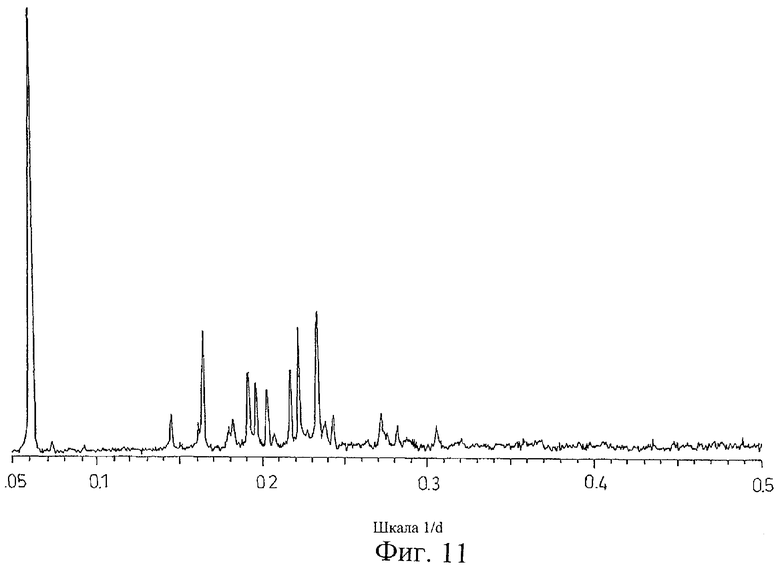

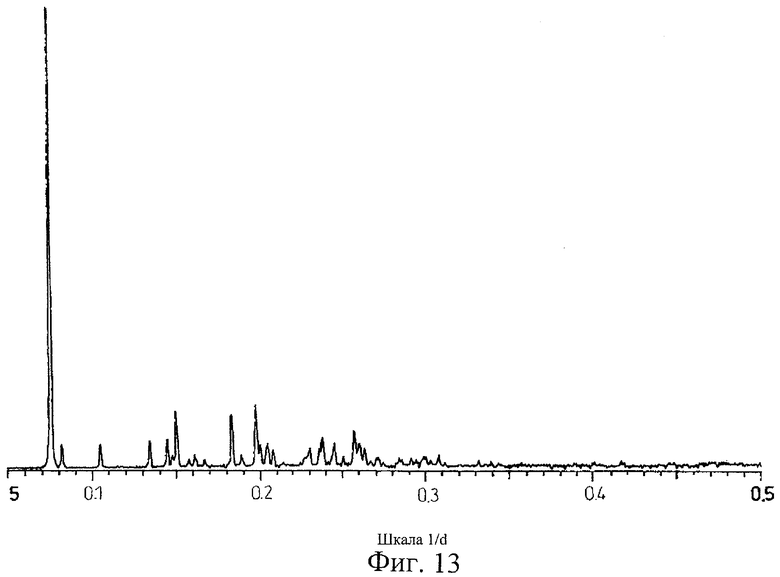

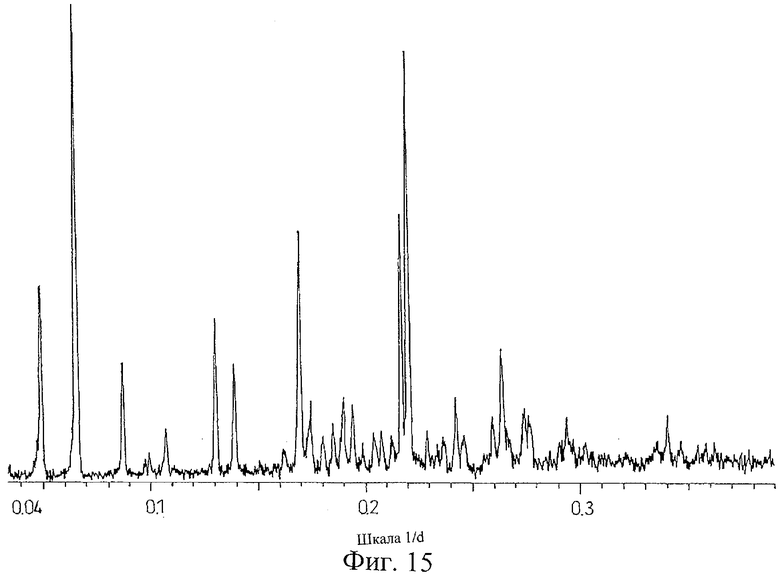




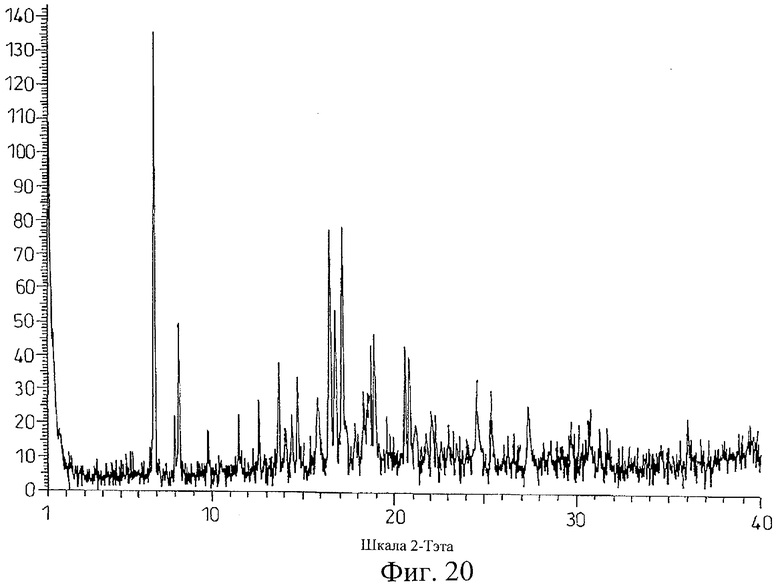
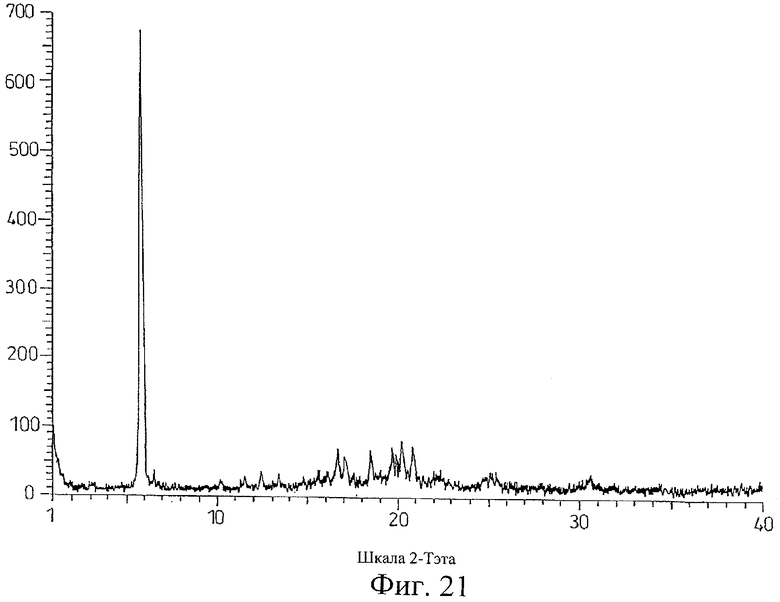
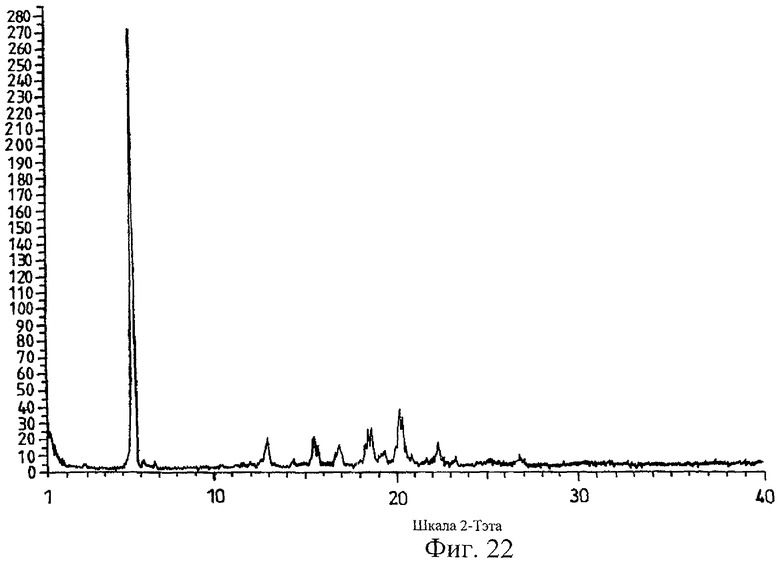
Изобретение относится к новым, твердым по состоянию формам антиаритмических лекарственных средств. Описываются по существу кристаллические формы 4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил]пропил}амино)-бензнитрила; трет-бутил-2-{7-[3-(4-цианоанилино)пропил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил}этилкарбамата; трет-бутил-2-{7-[4-(4-цианофенил)бутил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил}этилкарбамата; или трет-бутил-2-{7-[(25)-3-(4-цианофенокси)-2-гидроксипропил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил}этилкарбамата и их фармацевтически приемлемых солей. Также описываются способы их получения, фармацевтический препарат на их основе, способ профилактики или лечения аритмии и их применение. 10 н. и 63 з.п. ф-лы, 22 ил., 22 табл.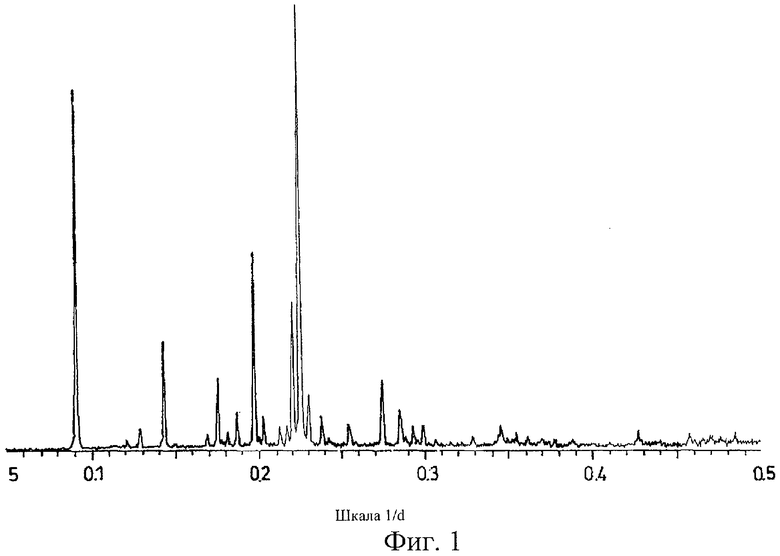
4-({3-[7-(3,3-диметил-2-оксобутил)-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил]пропил} амино)бензнитрила;
трет-бутил-2-{7-[3-(4-цианоанилино)пропил]-9-окса-3,7-диазабицикло-[3.3.1]нон-3-ил}этилкарбамата;
трет-бутил-2-{7-[4-(4-цианофенил)бутил]-9-окса-3,7-диазабицикло-[3.3.1]нон-3-ил}этилкарбамата; или
трет-бутил-2-{7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил}этилкарбамата,
при условии, что данная соль не представляет собой соль 4-({3-(7-(3,3-диметил-2-оксобутил)-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил]пропил}амино)-бензнитрила с бензолсульфоновой кислотой.
трет-бутил-2-{7-[3-(4-цианоанилино)пропил]-9-окса-3,7-диазабицикло-[3.3.1]нон-3-ил}этилкарбамат;
трет-бутил-2-{7-[4-(4-цианофенил)бутил]-9-окса-3,7-диазабицикло-[3.3.1]нон-3-ил}этилкарбамат; или
трет-бутил-2-{7-[(2S)-3-(4-цианофенокси)-2-гидроксипропил]-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил}этилкарбамат
либо фармацевтически приемлемая соль любого из этих соединений в по существу кристаллической форме.
| Огнетушитель | 0 |
|
SU91A1 |
| СПОСОБ АВТОМАТИЧЕСКОЙ ДУГОВОЙ СВАРКИ | 0 |
|
SU308843A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

