Предлагаемое изобретение относится к области синтеза 1,3-дикарбонильных соединений, конкретно к способу получения 4-замещенных алкил 3-оксобутаноатов формулы:
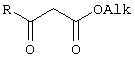
где
R=С6Н5СН2, 2-F-6-СlС6Н3СН2, 2,6-Сl2С6Н3СН2, 1-С10Н7СН2, Ph2CH; Alk=Me.
R=1-AdCH2, Alk=i-Pr,
которые находят применение как предшественники противовирусных средств пиримидинового ряда.
Известны способы получения 4-замещенных алкил 3-оксобутаноатов путем ацилирования 2,2-диметил-1,3-диоксан-4,6-диона в виде натриевой соли, в абсолютном ДМФА, ангидридами карбоновых кислот с последующим алкоголизом продукта ацилирования [Houghton R.P., Lapham D.J. /A modified preparation of β-ketoesters //Synthesis -1982. -P. 451-452].
Недостатком этого метода является необходимость предварительного получения натриевой соли 2,2-диметил-1,3-диоксан-4,6-диона, использование дорогостоящего растворителя (без регенерации) и дорогостоящих ацилирующих агентов. Кроме этого, выход целевых продуктов в этом случае составляет 25-83%.
Известны способы получения 4-замещенных алкил 3-оксобутаноатов путем ацилирования 2,2-диметил-1,3-диоксан-4,6-диона ацилимидазолами в присутствии безводного пиридина [Long-acting dihydropyridine calcium antagonists. 3. Synthesis and structure-activity relationships for a series of 2-[(heterocyclylmethoxy)methyl]derivatives /Alker D., Campbell S.F., Cros P.E., et al. //J. Med. Chem. -1989. -Vol. 32, №10 -P. 2381-2388] или 4-(диметиламино)пиридина в абсолютном дихлорметане с последующим алкоголизом продукта ацилирования [A practical synthesis of thienamycin /Melillo D.G., Shinkai I., Liu Т., et al. //Tetrahedron Lett. -1980. -Vol. 21 -P. 2783-2786].
Недостатком этих способов является использование дорогостоящих ацилирующих агентов, наряду с дорогостоящим основанием (4-(диметиламино)пиридином). Выход целевых продуктов не превышает 12% (при использовании пиридина) и 60-72% (при использовании 4-(диметиламино)пиридина).
Известен способ получения 4-замещенных алкил 3-оксобутаноатов, заключающийся в ацилировании 2,2-диметил-1,3-диоксан-4,6-диона ацилхлоридами в присутствии безводного пиридина в абсолютном дихлорметане с последующим расщеплением продуктов ацилирования алкоголизом [Oikawa Y., Sugano К., Yonemitsu О. /Meldrum's acid in organic synthesis. 2. A general and versatile synthesis of β-ketoesters //J. Org. Chem. -1978. -Vol. 43, №10 -P. 2087-2088].
Несмотря на то, что в случае простых алифатических алкил 3-оксобутаноатов этот метод позволяет получить целевые продукты с выходом до 92% [Oikawa Y., Sugano К., Yonemitsu О. /Meldrum's acid in organic synthesis. 2. A general and versatile synthesis of β-ketoesters //J. Org. Chem. -1978. -Vol. 43, №10 - P. 2087-2088], в случае 4-арилзамещенных алкил 3-оксобутаноатов он не дает стабильного выхода целевых продуктов и, в ряде случаев, позволяет получить целевые продукты с выходом лишь 27-48% [Studies on cerebral protective agents. VI. Synthesis of novel 4-(4-nitrobenzoyl)pyrimidine and related compounds with anti-anoxic activity //Chem. Pharm. Bull. -1994. - Vol. 42, №6 - P. 1279-1285; Synthesis and antiviral activity of new 3,4-dihydro-2-alkoxy-6-benzyl-4-oxopyrimidines (DABOs), specific inhibitors of human immunodeficiency virus type 1 / Massa S., Mat A., Artico M., et al. // Antivir. Chem. Chemother. -1995. -Vol. 6, N 1 -P. 1-8.].
Недостатком данного метода является также относительно низкая степень чистоты целевых продуктов.
Известен способ получения 4-замещенных алкил 3-оксобутаноатов путем ацилирования 2,2-диметил-1,3-диоксан-4,6-диона ацилхлоридами в безводном дихлорметане в присутствии основания Хюнига (N-этил-N,N-диизопропиламина) с последующим алкоголизом продукта ацилирования [Moody C.J., Rahimtoola K.F. / Diels-Alder reactivity of pyrano[4,3-b]indol-3-ones, indole 2,3-quinodimethane analogues // J. Chem. Soc., Perkin. Trans. I -1990.-P. 673-679.].
Недостатком данного метода является использование дорогостоящего основания, в то время как выход целевого продукта не превышает 85%.
Наиболее близким является способ получения 4-замещенных алкил 3-оксобутаноатов путем ацилирования 2,2-диметил-1,3-диоксан-4,6-диона ацилхлоридами в присутствии безводного триэтиламина в безводном дихлорметане с последующим акоголизом продуктов ацилирования [Навроцкий М.Б. Синтез, противовирусная и цитотоксическая активность 6-бензгидрил-2-(алкилтио)-4(3H)-пиримидинонов // Автореферат Канд. Дисс.-Пятигорск, стр.8-10, 2002].
Недостатком этого способа является то, что выход 4-арилзамещенных алкил 3-оксобутаноатов не превышает 78-87%, а выход 4-(1-адамантил)-3-оксобутаноатов - 62%.
Задачей предлагаемого технического решения является разработка нового технологичного способа получения 4-замещенных алкил 3-оксобутаноатов, позволяющего проводить синтез в мягких условиях, с использованием дешевых основания и растворителя и получением целевых продуктов с высокими выходом и степенью чистоты.
Техническим результатом является повышение выхода и чистоты заявляемых соединений.
Предлагаемый технический результат достигается в способе получения 4-замещенных алкил 3-оксобутаноатов общей формулы:
где
R=С6Н5СН2, 2-F-6-СlС6Н3СН2, 2,6-Сl2С6Н3СН2, 1-C10H7CH2, Рh2СН; Аlk=Me;
R=1-AdCH2, Alk=i-Pr,
заключающемся в ацилировании в дихлорметане 2,2-диметил-1,3-диоксан-4,6-диона ацилхлоридами в присутствии триэтиламина с последующим алкоголизом 5-(1-гидроксиэтилиден)-2,2-диметил-1,3-диоксан-4,6-диона, причем ацилирование осуществляется ацилхлоридами в присутствии триметилсилилхлорида, при мольном соотношении 2,2-диметил-1,3-диоксан-4,6-дион: ацилхлорид: триметилсилилхлорид: триэтиламин, равном 1-2:1:1,1:3,5, с образованием промежуточного продукта 5-[1-(триметилсилилокси)этилиден]-2,2-диметил-1,3-диоксан-4,6-диона, который подвергается гидролизу с образованием 5-(1-гидроксиэтилиден)-2,2-диметил-1,3-диоксан-4,6-диона, с его последующим алкоголизом и образованием целевого продукта.
В качестве ацилхлоридов используются 2-фенилацетилхлорид, 2-(2-фтор-6-хлорфенил)ацетилхлорид, 2-(2,6-дихлорфенил)ацетилхлорид, 2-(1-нафтил)ацетилхлорид, 2,2-дифенилацетилхлорид или 2-(1-адамантил)ацетилхлорид.
Сущностью предлагаемого способа является ацилирование 2,2-диметил-1,3-диоксан-4,6-диона с сопутствующим силилированием образующегося 5-(1-гидроксиэтилиден)-2,2-диметил-1,3-диоксан-4,6-диона, гидролизом продукта силилирования и алкоголизом гидролизата:
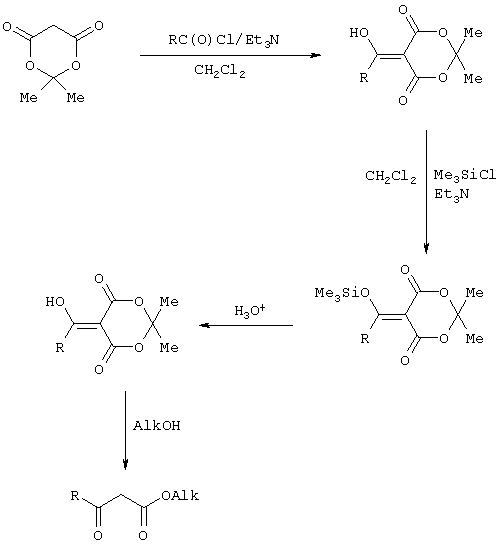
где
R=С6Н5СН2, 2-F-6-СlС6Н3СН2, 2,6-Сl2С6Н3СН2, 1-С10Н7СН2, Рh2СН; Alk=Me.
R=1-AdCH2, Alk=i-Pr
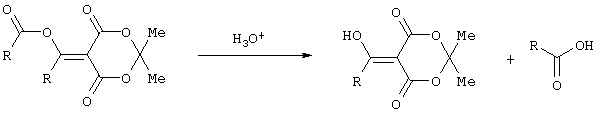
Применение триметилсилилхлорида при проведении ацилирования 2,2-диметил-1,3-диоксан-4,6-диона не является традиционным. Эта модификация процесса ацилирования направлена на превращение образующегося 2,2-диметил-5-(2-гидроксиэтилиден)-2,2-диметил-1,3-диоксан-4,6-диона в его триметилсилиловый эфир. Это позволяет полностью предотвратить протекание процесса O-ацилирования 2,2-диметил-5-(2-гидроксиэтилиден)-2,2-диметил-1,3-диоксан-4,6-диона, что существенно повышает выход целевого 4-замещенного 3-оксобутаноата и практически исключает образование побочных продуктов (соответствующей исходному ацилхлориду кислоты и ее алкилового эфира), образующихся по следующим схемам.
Схема образования примеси алкилового эфира кислоты, соответствующей исходному ацилхлориду:
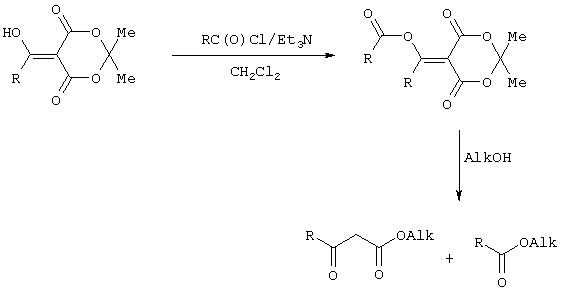
Схема образования кислоты, соответствующей исходному ацилхлориду:
Необходимость применения избытка триметилсилилхлорида обусловлена гидролитической нестабильностью реагента и его частичным гидролизом при проведении манипуляций. В связи с этим применение стехиометрического количества триметилсилилхлорида приводит к снижению выхода целевого 4-замещенного алкил 3-оксобутаноата. Применение избытка триметилсилилхлорида свыше 1,1 не приводит к повышению выхода целевого продукта.
Триэтиламин берется в избытке в связи с тем, что 2 эквивалента основания расходуются для связывания двух эквивалентов хлористого водорода, отщепляющегося при ацилировании и силилировании, а еще 1,5 эквивалента служат для эффективного депротонирования 2,2-диметил-1,3-диоксан-4,6-диона.
2,2-Диметил-1,3-диоксан-4,6-дион также берется в избытке по отношению к ацилхлориду для обеспечения полной конверсии последнего в целевой продукт. При применении стехиометрического количества 2,2-диметил-1,3-диоксан-4,6-диона приводит к снижению выхода 4-замещенных алкил-3-оксобутаноатов. Применение избытка 2,2-диметил-1,3-диоксан-4,6-диона свыше 1,03 также приводит к снижению выхода целевых продуктов за счет осложнения процедуры его очистки.
Преимуществом данного способа является возможность получения практически любых 4-замещенных алкил 3-оксобутаноатов с выходом, близким количественному, и высокой степенью чистоты.
Предлагаемый способ осуществляется следующим образом.
Получение 4-замещенных 3-оксобутаноатов.
В трехгорлый реактор с магнитной мешалкой, снабженный внутренним термометром, капельной воронкой с компенсатором давления и влагозащитной трубкой, помещается безводный дихлорметан и переосажденный 2,2-диметил-1,3-диоксан-4,6-дион. Полученная смесь перемешивается при охлаждении льдом. Когда температура смеси достигает 0°С, к реакционной массе по каплям, в течение 30 минут прибавляется безводный триэтиламин. После прибавления примерно половины триэтиламина температура реакционной массы достигает 10-15°С, а к концу прибавления триэтиламина вновь опускается до 0°С. Затем к реакционной массе при перемешивании по каплям прибавляется триметилсилилхлорид. При этом также происходит незначительное повышение температуры реакционной массы и последующее ее понижение до 0°С. При этой температуре к реакционной смеси по каплям при интенсивном перемешивании прибавляется раствор ацилхлорида в безводном дихлорметане. Скорость прибавления раствора ацилхлорида регулируется таким образом, чтобы внутренняя температура не превышала 2°С.
После прибавления всего раствора ацилхлорида реакционная масса перемешивается еще 1 час при 0°С. Затем ледяная баня убирается, и реакционная масса перемешивается до тех пор, пока ее температура не достигнет 22°С. При этой температуре реакционная масса перемешивается еще 1-24 часа. Затем реакционная смесь выливается в смесь 2N серной кислоты и льда. Полученная двухфазная система интенсивно встряхивается в делительной воронке. Органический слой отделяется, а водный извлекается дихлорметаном. Объединенные органические вытяжки промываются 2N водным раствором холодной серной кислоты, водой и сушатся безводным сульфатом натрия. После фильтрования от осушителя растворитель удаляется в вакууме водоструйного насоса при температуре бани не выше 40°С. К остатку прибавляется абсолютный алканол, и реакционная смесь кипятится с обратным холодильником и защитой от влаги в течение 3-х часов. После удаления растворителя в вакууме водоструйного насоса остаток очищается вакуумной перегонкой.
Изобретение иллюстрируется следующими примерами:
Пример 1. Метил 3-оксо-4-фенилбутаноат.
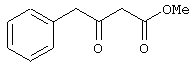
В трехгорлый реактор на 100 мл с магнитной мешалкой, снабженный внутренним термометром, капельной воронкой с компенсатором давления и влагозащитной трубкой, помещается безводный дихлорметан (20 мл) и переосажденный 2,2-диметил-1,3-диоксан-4,6-дион (7,06 грамма, 49,0 ммоль). Полученная смесь перемешивается при охлаждении льдом. Когда температура смеси достигает 0°С, к реакционной массе по каплям, в течение 30 минут прибавляется безводный триэтиламин (16,82 г, 166,3 ммоль). После прибавления примерно половины триэтиламина температура реакционной массы достигает 10-15°С, а к концу прибавления триэтиламина вновь опускается до 0°С. Затем к реакционной массе при перемешивании по каплям прибавляется триметилсилилхлорид (5,68 г, 52,3 ммоль). При этом также происходит незначительное повышение температуры реакционной массы и последующее ее понижение до 0°С. При этой температуре к реакционной массе по каплям при интенсивном перемешивании прибавляется раствор свежеперегнанного 2-фенилацетилхлорида (7,35 г, 47,5 ммоль) в безводном дихлорметане (15 мл). Скорость прибавления раствора хлорангидрида регулируется таким образом, чтобы внутренняя температура не превышала 2°С (на это требуется около 2,5 часов). После прибавления всего раствора хлорангидрида реакционная масса перемешивается еще 1 час при 0°С. Затем ледяная баня убирается, и реакционная масса перемешивается до тех пор, пока ее температура не достигнет 22°С. При этой температуре реакционная масса перемешивается еще 1 час. Затем реакционная смесь выливается в смесь 2N серной кислоты (65 мл) и 100 г льда. Полученная двухфазная система интенсивно встряхивается в делительной воронке. Органический слой отделяется, а водный -извлекается дихлорметаном (3·15 мл). Объединенные органические вытяжки промываются 2N водным раствором холодной серной кислоты (50 мл), водой (50 мл) и сушатся безводным сульфатом натрия. После фильтрования от осушителя растворитель удаляется в вакууме водоструйного насоса при температуре бани не выше 40°С. К остатку прибавляется абсолютный метанол (75 мл), и реакционная смесь кипятится с обратным холодильником и защитой от влаги в течение 3-х часов. После удаления растворителя в вакууме водоструйного насоса остаток очищается вакуумной перегонкой. Выход метил 3-оксо-4-фенилбутаноата - 9,04 г (99%). Т. кип.111-112°С/3 мм рт. ст.
Соотношения исходных реагентов: 2,2-диметил-1,3-диоксан-4,6-дион:ацилхлорид:триметилсилилхлорид:триэтиламин составляют 1,03:1:1,1:3,5.
Пример 2. Метил 3-оксо-4-фенилбутаноат.
Выполняется аналогично примеру 1, за исключением соотношения исходных реагентов: 2,2-диметил-1,3-диоксан-4,6-дион:ацилхлорид:триметилсилилхлорид:триэтиламин составляют 1:1:1,1:3,5.
Выход метил 3-оксо-4-фенилбутаноата - 93%.
Пример 3. Метил 3-оксо-4-фенилбутаноат.
Выполняется аналогично примеру 1, за исключением соотношения исходных реагентов: 2,2-диметил-1,3-диоксан-4,6-дион:ацилхлорид:триметилсилилхлорид:триэтиламин составляют 2:1:1,1:3,5.
Выход метил 3-оксо-4-фенилбутаноата - 96%.
Пример 4. Метил 4-(2-фтор-6-хлорфенил)-3-оксобутаноат.
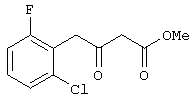
Выполняется аналогично примеру 1, за исключением использования 2-(2-фтор-6-хлорфенил)ацетилхлорида в качестве ацилхлорида. Соотношения исходных реагентов: 2,2-диметил-1,3-диоксан-4,6-дион:ацилхлорид:триметилсилилхлорид:триэтиламин составляют 1,03:1:1,1:3,5.
Выход метил 3-оксо-4-(2-фтор-6-хлорфенил)бутаноата -94%.
Т. кип.131-132°С/3 мм рт. ст. Т. пл. 51.5-53°С (из петр. эфира 40-70).
Масс-спектр, m/z (Iотн, %): 244 (11.1%) [M+], 213 (1.6%), 185 (2.4%), 171 (9.5%), 143 (91.3%), 115 (19.8%), 101 (81.7%), 59 (100%)
Пример 5. Метил 4-(2,6-дихлорфенил)-3-оксобутаноат.
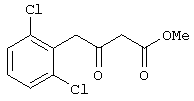
Выполняется аналогично примеру 1, за исключением использования 2-(2,6-дихлорфенил)ацетилхлорида в качестве ацилхлорида. Соотношения исходных реагентов: 2,2-диметил-1,3-диоксан-4,6-дион:ацилхлорид:триметилсилилхлорид:триэтиламин составляют 1,03:1:1,1:3,5.
Выход метил 3-оксо-4-(2,6-дихлорфенил)бутаноата - 94%.
Т. кип.145-146°С/1-2 мм рт. ст. Т. пл. 82,5-84°С (из петр. эфира 40-70).
Масс-спектр, m/z (Iотн, %): 260 (8.9%) [М+], 229 (1.4%), 201 (5.5%), 159 (40.4%), 123 (21.2%), 101 (100%), 73 (13.0%), 59 (76.7%)
Пример 6. Метил 4-(1-нафтил)-3-оксобутаноат.
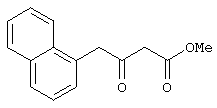
Выполняется аналогично примеру 1, за исключением использования 2-(1-нафтил)ацетилхлорида в качестве ацилхлорида. Соотношения исходных реагентов: 2,2-диметил-1,3-диоксан-4,6-дион:ацилхлорид:триметилсилилхлорид:триэтиламин составляют 1,03:1:1,1:3,5.
Выход метил 3-оксо-4-(1-нафтил)бутаноата - 97%.
Т. кип.183-184°С/2 мм рт. ст. n24 D=1,588
Масс-спектр, m/z (Iотн, %): 242 (11.5%) [M+], 211 (2.7%), 183 (1.8%), 169 (22.1%), 141 (100%), 115 (33.6%), 101 (22.1%), 59 (23.9%)
Пример 7. Метил 4,4-дифенил-3-оксобутаноат.
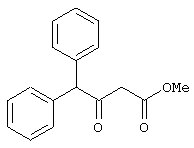
Выполняется аналогично примеру 1, за исключением использования 2,2-дифенилацетилхлорида в качестве ацилхлорида. Соотношения исходных реагентов: 2,2-диметил-1,3-диоксан-4,6-дион:ацилхлорид:триметилсилилхлорид:триэтиламин составляют 1,03:1:1,1:3,5.
Выход метил 3-оксо-4,4-дифенилбутаноата - 98%.
Т. кип.172-174°С/1-2 мм рт. ст. n28.5 D=1,562
Масс-спектр, m/z (Iотн, %): 268 (5.4%) [M+], 195 (2.3%), 167 (100%), 101 (31.5%), 91 (4.6%), 77 (10%), 73 (17.7%), 59 (15.4%)
Пример 8. Изопропил 4-(1-адамантил)-3-оксобутаноат.
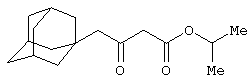
Выполняется аналогично примеру 1, за исключением использования 2-(1-адамантил)ацетилхлорида в качестве ацилхлорида, времени перемешивания реакционной массы - 24 часа и применения изопропанола, вместо метанола при алкоголизе. Соотношения исходных реагентов: 2,2-диметил-1,3-диоксан-4,6-дион:ацилхлорид:триметилсилилхлорид:триэтиламин составляют 1,03:1:1,1:3,5.
Выход изопропил 4-(1-адамантил)-3-оксобутаноата -90%.
Т. кип.163-165°С/1-2 мм рт. ст.
Масс-спектр, m/z (Iотн, %): 278 (1.4%) [M+], 235 (1.5%), 219 (1.5%), 191 (1.3%), 177 (5.7%), 149 (3.6%), 135 (100%), 107 (7.1%)
Как следует из представленных примеров, предложенный нами способ получения 4-замещенных алкил 3-оксобутаноатов является технологичным, позволяет получать широкий спектр указанных соединений с высоким выходом и высокой чистотой.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОМОЧЕВИНЫ, ИХ ПОЛУЧЕНИЕ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2004 |
|
RU2341523C2 |
| ОКСИМНОЕ ПРОИЗВОДНОЕ ЦИКЛОГЕКС-2-ЕН-1-ОНА И СПОСОБ ПОДАВЛЕНИЯ РОСТА НЕЖЕЛАТЕЛЬНЫХ РАСТЕНИЙ | 1988 |
|
RU2009138C1 |
| АНАЛОГ ПИРИДИНО[1,2-А]ПИРИМИДОНА, ИСПОЛЬЗУЕМЫЙ В КАЧЕСТВЕ ИНГИБИТОРА mTOR/PI3K | 2015 |
|
RU2658912C1 |
| ПРИМЕНЕНИЕ ХИРАЛЬНЫХ АРИЛКЕТОНОВ В ЛЕЧЕНИИ НЕЙТРОФИЛ-ЗАВИСИМЫХ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2003 |
|
RU2345759C2 |
| ГОМОФТАЛИМИДНЫЕ ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА КАК ИНГИБИТОР ГИСТОНДЕЗАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2020 |
|
RU2793331C1 |
| ПРОИЗВОДНЫЕ 2-АЦИЛАМИНО-4-ФЕНИЛТИАЗОЛА, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2348630C2 |
| Способ получения кислородсодержащих гетероциклических соединений или их металлических солей | 1975 |
|
SU577999A3 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2814288C1 |
| НОВЫЕ ИМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2016 |
|
RU2772429C2 |
| БИФЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЯ, КОСМЕТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ПРИМЕНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2280033C2 |
Изобретение относится к области синтеза 1,3-дикарбонильных соединений, к новому способу получения 4-замещенных алкил 3-оксобутаноатов:
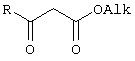
где R=С6H5СН2, 2-F-6-ClC6Н3СН2, 2,6-Cl2С6Н3СН2, 1-C10H7CH2, Ph2СН; Alk=Me;R=1-AdCH2, Alk=i-Pr, которые находят применение в качестве предшественников противовирусных средств пиримидинового ряда. Способ заключается в ацилировании в дихлорметане 2,2-диметил-1,3-диоксан-4,6-диона ацилхлоридами в присутствии триэтиламина с последующим алкоголизом 5-(1-гидроксиэтилиден)-2,2-диметил-1,3-диоксан-4,6-диона, причем ацилирование осуществляется ацилхлоридами в присутствии триметилсилилхлорида, при мольном соотношении 2,2-диметил-1,3-диоксан-4,6-дион:ацилхлорид:триметилсилилхлорид:триэтиламин, равном 1-2:1:1,1:3,5, с образованием промежуточного продукта 5-[1-(триметилсилилокси)этилиден]-2,2-диметил-1,3-диоксан-4,6-диона, который подвергается гидролизу с образованием 5-(1-гидроксиэтилиден)-2,2-диметил-1,3-диоксан-4,6-диона, с его последующим алкоголизом и образованием целевого продукта. Техническим результатом является повышение выхода и чистоты заявляемых соединений. 1 з.п. ф-лы.
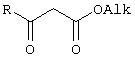
где R=С6H5СН2, 2-F-6-ClC6Н3СН2, 2,6-Cl2С6Н3СН2, 1-C10H7CH2, Ph2СН; Alk=Me;
R=1-AdCH2, Alk=i-Pr
заключающийся в ацилировании в дихлорметане 2,2-диметил-1,3-диоксан-4,6-диона ацилхлоридами в присутствии триэтиламина с последующим алкоголизом 5-(1-гидроксиэтилиден)-2,2-диметил-1,3-диоксан-4,6-диона, отличающийся тем, что ацилирование осуществляется ацилхлоридами в присутствии триметилсилилхлорида, при мольном соотношении 2,2-диметил-1,3-диоксан-4,6-дион:ацилхлорид:триметилсилилхлорид:триэтиламин, равном 1-2:1:1,1:3,5, с образованием промежуточного продукта 5-[1-(триметилсилилокси)этилиден]-2,2-диметил-1,3-диоксан-4,6-диона, который подвергается гидролизу с образованием 5-(1-гидроксиэтилиден)-2,2-диметил-1,3-диоксан-4,6-диона, с его последующим алкоголизом и образованием целевого продукта.
| Навроцкий М.Б | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| дисс | |||
| Пятигорск, 2002 с | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| DE 19730940 C1 (The Associated Octel Co., Ltd.), 24.09.1998 | |||
| DE 4116906 A1 (Wacker-Chemie GmbH), 26.11.1992 | |||
| Навроцкий М.Б. | |||

