Область изобретения
Настоящее изобретение относится к классу аналогов пиридино[1,2-а]пиримидинона, используемых в качестве ингибиторов mTOR/PI3K, и, в частности, настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли.
Уровень техники
Путь PI3K представляет собой участок в раковых клетках человека, где мутации происходят наиболее часто и могут приводить к пролиферации клеток, активации и усилению сигнала. PI3K и mTOR представляют собой две наиболее важные киназы в сигнальном пути PI3K.
Киназа PI3 (фосфатидилинозитол-3-киназа, PI3K) принадлежит к семейству липидных киназ и может фосфорилировать 3'-ОН-конец инозитольного кольца фосфатидилинозитола. Фосфатидилинозитол-3-киназа (PI3K) представляет собой липидную киназу, состоящую из регуляторной субъединицы р85 или р101, и каталитической субъединицы p110, и играет ключевую роль в пролиферации клеток, выживании и метаболизме, и т.д., катализируя фосфорилирование фосфатидилинозитол-4,5-дифосфата (PIP2) с образованием фосфатидилинозитол-3,4,5-трифосфата (PIP3), активируя тем самым нижерасположенную АКТ (протеинкиназу В) и тому подобные. Следовательно, ингибирование фосфатидилинозитол-3-киназы может влиять на путь PI3K и ингибировать таким образом пролиферацию и активацию раковых клеток.
Ген-супрессор опухолевого роста PTEN (фосфотаза с гомологом тензина, отсутствующая на 10-ой хромосоме) дефосфорилирует PIP3 с образованием PIP2, приводя таким образом к отрицательной регуляции Р13K/АKТ сигнального пути, ингибируя пролиферацию клеток и способствуя апоптозу. Частые случаи мутации и амплификации гена PI3K, а также потеря PTEN в раковых клетках и тому подобное указывает на то, что PI3K тесно связана с онкогенезом.
Белок-мишень рапамицина у млекопитающего mTOR представляет собой серин-треониновую протеинкиназу, находящуюся в цитоплазме, которая принадлежит к семейству киназ, родственному семейству фосфатидилинозитол-3-киназ, и играет важную роль в регуляции сигнальной трансдукции многих путей. Было установлено, что mTOR представляет собой нижерасположенную мишень PI3K/AKT. В настоящее время установлено, что в клетках присутствуют два различных комплекса mTOR, т.е. mTORC1 и mTORC2. Они отдельно друг от друга выполняют различные функции, при этом основной функцией mTORC1 является стимулирование клеточного роста и пролиферации, тогда как mTORC2 регулирует клеточное выживание и цитоскелет посредством активации АКТ, РКС (протеинкиназы С) и других киназ. Исследования показали, что сигнальный путь mTOR связан с возникновением рака, и одновременное ингибирование активностей двух комплексов mTOR в раковых клетках обладает более продолжительным и эффективным противораковым действием.
Двойной ингибитор PI3K-mTOR может одновременно блокировать множество участков сигнальной трансдукции и будет более эффективно предотвращать киназную сигнальную трансдукцию, тем самым преодолевая или задерживая возникновение устойчивости к лекарственным средствам.
В патентных заявках W02008163636 (Novartis) и W02008144463 (GSK) раскрыта серия соединений, обладающих ингибирующим эффектом как в отношении PI3K, так и в отношении mTOR, которые обладают хорошей терапевтической активностью в отношении опухолей. Тем не менее, в настоящий момент на рынке не существует лекарственного средства, обладающего ингибирующим эффектом как в отношении PI3K, так и в отношении mTOR. Следовательно, существует необходимость в разработке лекарственных средств мультинаправленного действия, обладающих ингибирующим эффектом как в отношении PI3K, так и в отношении mTOR, способствующих лечению рака.
Краткое описание изобретения
Целью настоящего изобретения является получение соединения формулы (I) или его фармацевтически приемлемой соли,
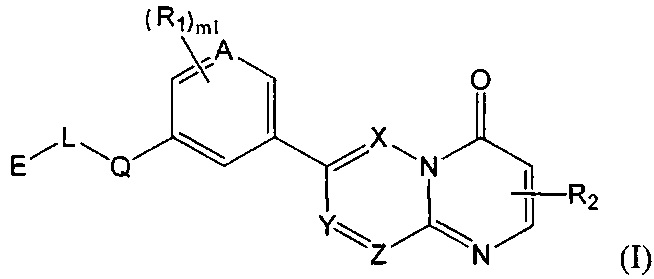
где
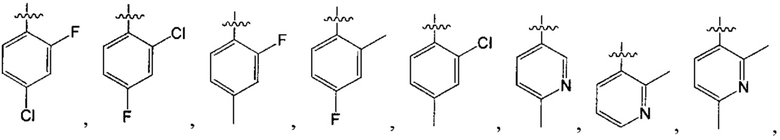
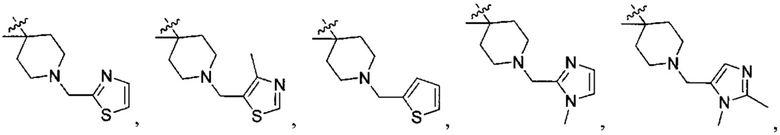
структурная единица 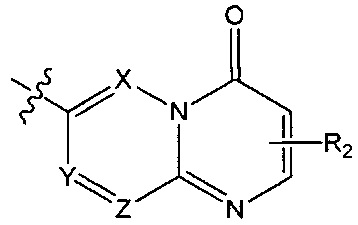 может быть заменена на
может быть заменена на 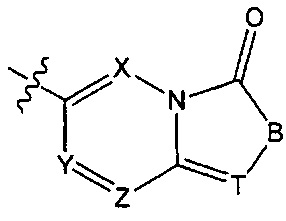 ,
, 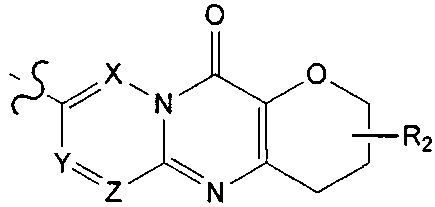 ;
;
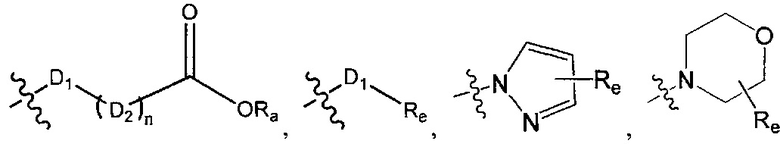
Е выбран из группы, состоящей из С1-6 алкила, 3-10-членного циклогидрокарбила и гетероциклогидрокарбила, необязательно замещенного 1, 2 или 3 R3;
один из L и Q выбран из группы, состоящей из -С(R3)(R3)-, -C(=O)N(Ra)-, -N(Ra)-, -C(=NRa)-, -S(=O)2N(Ra)-, -S(=O)N(Ra)-, -O-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(Ra)C(=O)N(Ra)-, а другой выбран из группы, состоящей из одинарной связи и -C(R3)(R3)-;
А и Т независимо выбраны из группы, состоящей из N и C(R3);
ноль или один из X, Y, и Z выбран из группы, состоящей из N, а другие представляют собой C(R3);
В выбран из группы, состоящей из -C(R3)(R3)-, -C(=O)N(Ra)-, -N(Ra)-, -C(=NRa)-, -S(=O)2N(Ra)-, -S(=O)N(Ra)-, -O-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(Ra)C(=O)N(Ra)-;
Гетероатом или гетероатомная группа независимо выбрана из группы, состоящей из -C(O)N(Ra)-, -N(Ra)-, -C(=NRa)-, -S(=O)2N(Ra)-, -S(=O)N(Ra)-, -О-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(Ra)C(=O)N(Ra)-;
каждый m1 независимо выбран из группы, состоящей из 0, 1, 2 или 3;
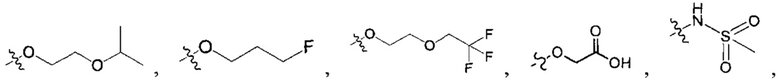
R1-3 независимо выбраны из группы, состоящей из Н, F, Cl, Br, I, CN, ORa, N(Rb)(Rc), С1-3 алкила, необязательно замещенного Rd, 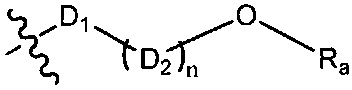 ,
,  ;
;
D1 выбран из группы, состоящей из одинарной связи, -C(Re)(Re)-, -C(=O)N(Ra)-, -N(Ra)-, -C(=NRa)-, -S(=O)2N(Ra)-, -S(=O)N(Ra)-, -O-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(-O)2- и -N(Ra)C(=O)N(Ra)-;
D2 выбран из -C(Ra)(Ra)-;
n выбран из группы, состоящей из 1, 2, 3, 4, 5 или 6;
Ra, Rb и Rc независимо выбраны из группы, состоящей из Н и С3-6 циклоалкила или С1-6 алкила, необязательно замещенного Rd;
Re выбран из группы, состоящей из Н, C1-6 алкила или алкокси, необязательно замещенного Rd, С3-6 циклоалкила или циклоалкокси, необязательно замещенного Rd;
Rd выбран из группы, состоящей из F, Cl, Br, I, CN, ОН, СНО, СООН, СН3, CF3, СН3O и СН3СН2О, и число Rd выбрано из группы, состоящей из 0, 1, 2 или 3;
необязательно, любые два из R1, Ra и Ra в одном и том же D2, два D2, или Ra и один D2, вместе с тем же атомом углерода или атомом кислорода, к которому они оба присоединены, образуют одно или два 3-, 4-, 5- или 6-членных карбоциклических колец или кислородсодержащих гетероциклических колец, где число атомов кислорода составляет 1 или 2.
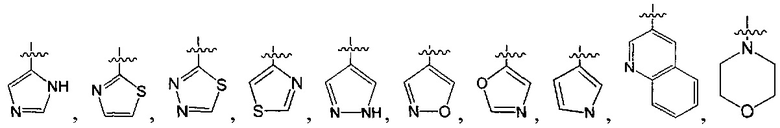
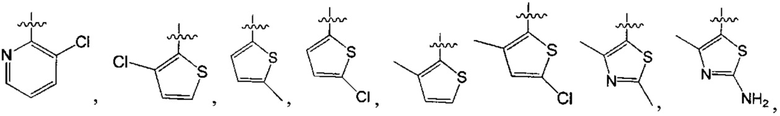
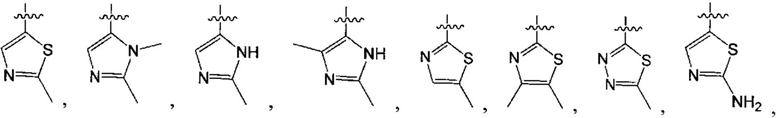
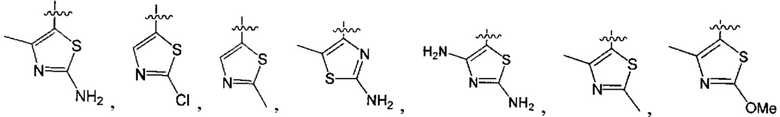
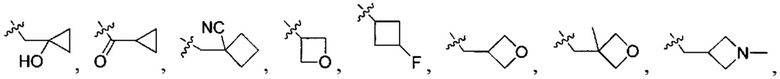
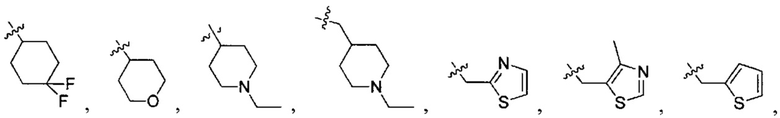
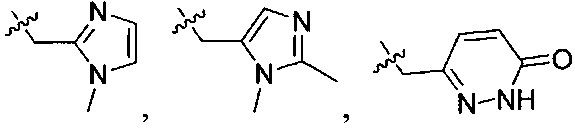
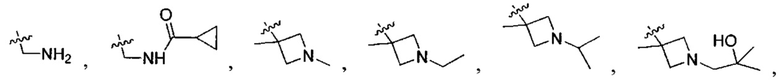
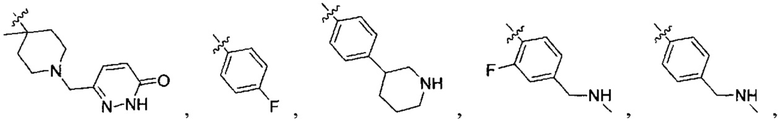
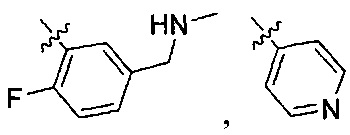
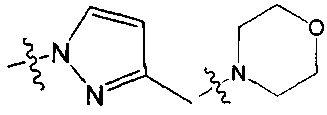
В одном из вариантов осуществления настоящего изобретения указанный Е выбран из С3-6 циклоалкила или C1-6 алкила, замещенного R3, число R3 составляет 0, 1, 2 или 3, или Е выбран из группы, состоящей из 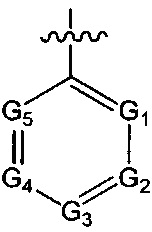 ,
,  ,
,  ,
,  или
или  ,
,
где ноль, один, два или три из G1~5 выбраны из N, а остальные выбраны из C(R3);
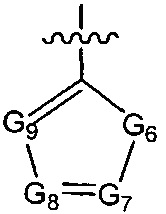
G6 выбран из группы, состоящей из -C(R3)(R3)-, -C(=O)N(R3)-, -N(R3)-, -C(=NR3)-, -S(=O)2N(R3)-, -S(=O)N(R3)-, -O-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(R3)C(=O)N(R3)-;
ноль, один или два из G7~9 выбраны из N, а остальные выбраны из C(R3);
ноль, один, два, три или четыре из G10~16 выбраны из N, а остальные выбраны из C(R3);
G17 выбран из N или C(R3);
ноль, один, два или три из G18~22 выбраны из -C(=O)N(R3)-, -N(R3)-, -C(=NR3)-, -S(=O)2N(R3)-, -S(=O)N(R3)-, -O-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(R3)C(=O)N(R3)-, а остальные выбраны из -C(R3)(R3)-; и
остальные переменные являются такими, как определено выше.
В одном из вариантов осуществления настоящего изобретения указанный Е выбран из группы, состоящей из метила, этила, пропила,  ,
, 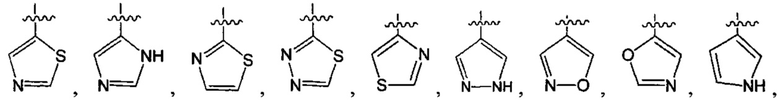
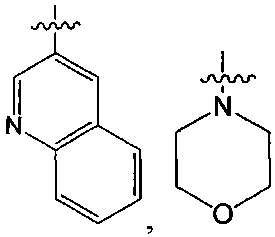 и
и 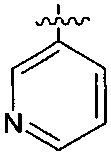 , который необязательно замещен 1, 2 или 3 R3.
, который необязательно замещен 1, 2 или 3 R3.
В одном из вариантов осуществления настоящего изобретения указанный Е выбран из группы, состоящей из 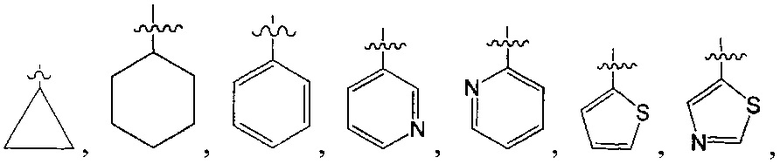
 и С1-3 алкила, который необязательно замещен 1, 2, или 3 атомами галогена, ОН, OC1-3 алкилом, CN, NH2, NH(C1-3 алкилом), N(C1-3 алкилом)2, С1-3 алкилом, трифторметилом, трифторэтилом, C(=O)NH2, С1-3 алкилС(=O), C1-3 алкилС(=O)NH, С1-3 алкилS(=O), C1-3 алкилS(=O)NH, С1-3 алкилS(=O)2 или С1-3 из алкилS(=O)2NH.
и С1-3 алкила, который необязательно замещен 1, 2, или 3 атомами галогена, ОН, OC1-3 алкилом, CN, NH2, NH(C1-3 алкилом), N(C1-3 алкилом)2, С1-3 алкилом, трифторметилом, трифторэтилом, C(=O)NH2, С1-3 алкилС(=O), C1-3 алкилС(=O)NH, С1-3 алкилS(=O), C1-3 алкилS(=O)NH, С1-3 алкилS(=O)2 или С1-3 из алкилS(=O)2NH.
В одном из вариантов осуществления настоящего изобретения указанный Е выбран из группы, состоящей из 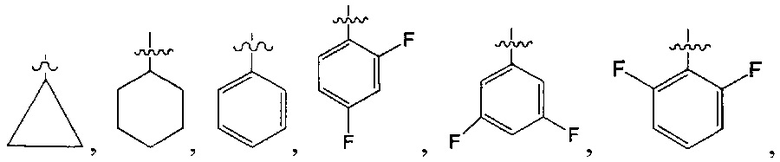
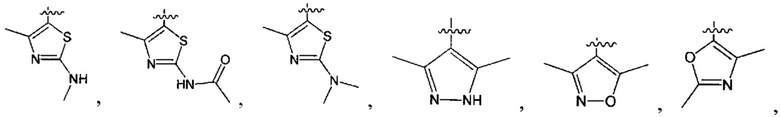
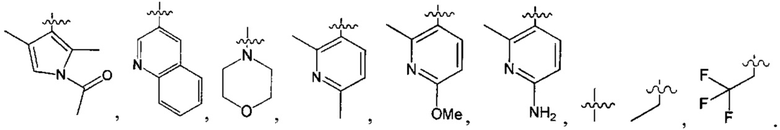
В одном из вариантов осуществления настоящего изобретения один из L и Q выбран из группы, состоящей из -S(=O)2NH-, -S(=O)2-, -NH- и -NHC(=O)NH-, а другой выбран из одинарной связи или -СН2-.
В одном из вариантов осуществления настоящего изобретения ноль или один из X, Y и Z выбран из N, и остальные выбраны из группы, состоящей из СН, С(СН3), С(СF3), ССl и CF.
В одном из вариантов осуществления настоящего изобретения А и Т независимо выбраны из группы, состоящей из N, СН, С(СН3), С(CF3), ССl и CF; или В выбран из группы, состоящей из NH, N(СН3) и N(CF3).
В одном из вариантов осуществления настоящего изобретения кольцо, образованное любыми двумя R1, Ra и Ra в одном и том же D2, двумя D2, или Ra и одним D2, выбрано из группы, состоящей из циклопропила, циклобутила, циклопентила, циклогексила, оксетанила, 1,3-диоксоланила.
В одном из вариантов осуществления настоящего изобретения указанный R1-3 выбран из группы, состоящей из Н, F, Cl, Br, I, CN, ОН, NH2, метила, этила, пропила, метокси, этокси, метиламино, диметиламино, галогенметила, галогенэтила, галогенпропила, аминометила, аминоэтила, аминопропила, циклопропила, 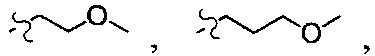

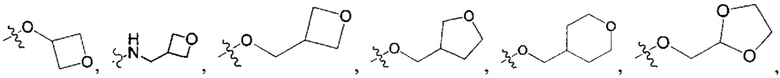
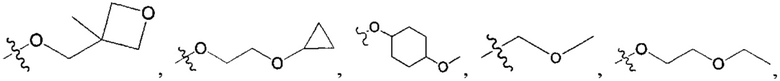

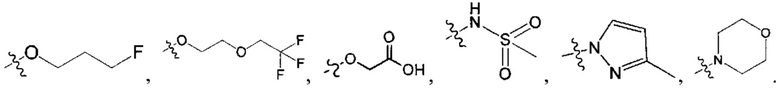
В одном из вариантов осуществления настоящего изобретения вышеуказанные соединения или их фармацевтически приемлемые соли выбраны из группы, состоящей из Соединений 1-284.
Связанные определения:
Если не указано иное, термины и фразы, используемые в тексте настоящего описания, подразумевают следующие значения. Конкретный термин или фразу не следует понимать как неясную или неточную в отсутствие конкретного определения, а следует понимать в соответствии с общепринятым значением. В случае, когда в тексте указан торговое наименование, подразумевается, что оно относится к соответствующему продукту или его активному ингредиенту.
C1-10 выбран из группы, состоящей из С1, С2, С3, С4, С5, С6, С7, C8, С9 и С10; С3-10 выбран из группы, состоящей из С3, С4, С5, С6, С7, C8, С9 и С10.
C1-10 алкил или гетероалкил, С3-10 циклогидрокарбил или гетероциклогидрокарбил, С1-10 алкил или гетероалкил, замещенный С3-10 циклогидрокарбилом или гетероциклогидрокарбилом включают, но не ограничиваясь указанными:
C1-10 алкил, C1-10 алкиламино, N,N-ди(C1-10 алкил)амино, C1-10 алкокси, C1-10 алканоил, C1-10 алкоксикарбонил, C1-10 алкилсульфонил, C1-10 алкилсульфинил, С3-10 циклоалкил, С3-10 циклоалкиламино, С3-10 гетероциклоалкиламино, С3-10 циклоалкилокси, С3-10 циклоалкилацил, С3-10 циклоалкоксикарбонил, С3-10 циклоалкилсульфонил, С3-10 циклоалкилсульфинил;
метил, этил, н-пропил, изопропил, -СН2С(СН3)(СН3)(ОН), циклопропил, циклобутил, пропилметилен, циклопропионил, бензилокси, трифторметил, аминометил, гидроксиметил, метокси, формил, метоксикарбонил, метилсульфонил, метилсульфинил, этокси, ацетил, этансульфонил, этоксикарбонил, диметиламино, диэтиламино, диметиламинокарбонил, диэтиламинокарбонил;
N(CH3)2, NH(CH3), -CH2CF3, -CH2CH2CF3, -CH2CH2F, -CH2CH2S(=O)2CH3, -CH2CH2CN, 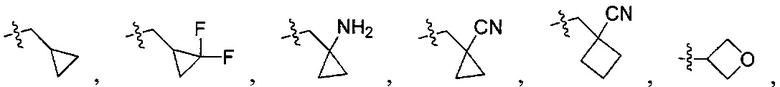 -CH2CH(OH)(CH3)2, -CH2CH(F)(CH3)2, -CH2CH2F, -CH2CF3, -CH2CH2CF3, -CH2CH2NH2, -CH2CH2OH, -CH2CH2OCH3, -CH2CH2CH2OCH3, -CH2CH2N(CH3)2, -S(=O)2CH3, -CH2CH2S(=O)2CH3,
-CH2CH(OH)(CH3)2, -CH2CH(F)(CH3)2, -CH2CH2F, -CH2CF3, -CH2CH2CF3, -CH2CH2NH2, -CH2CH2OH, -CH2CH2OCH3, -CH2CH2CH2OCH3, -CH2CH2N(CH3)2, -S(=O)2CH3, -CH2CH2S(=O)2CH3, 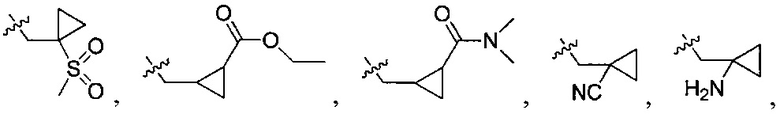
 ;
;
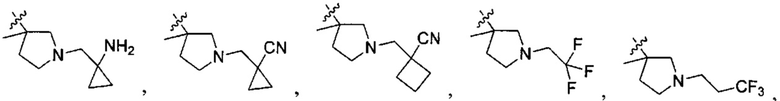
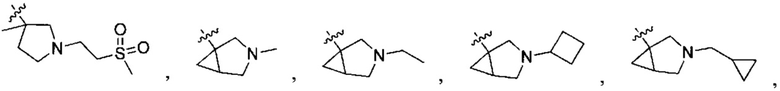
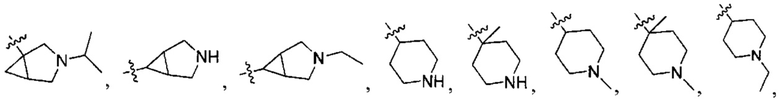
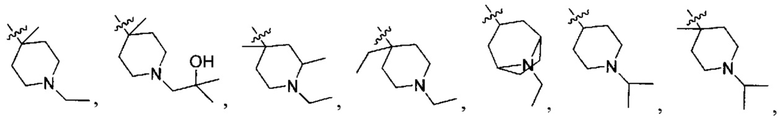
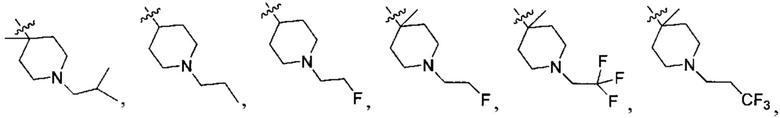
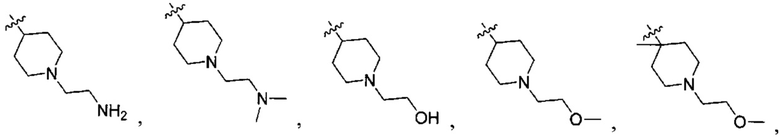
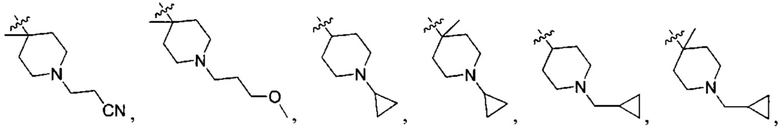
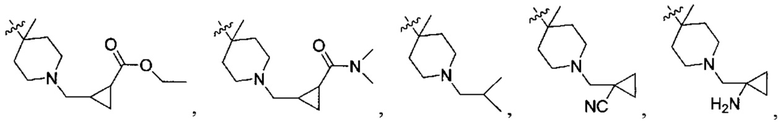
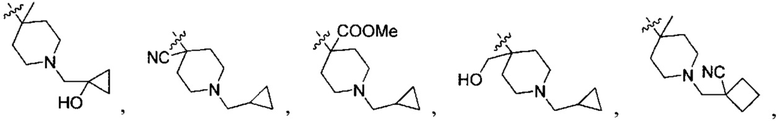
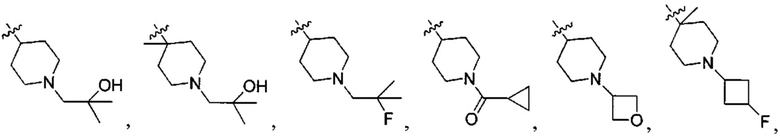
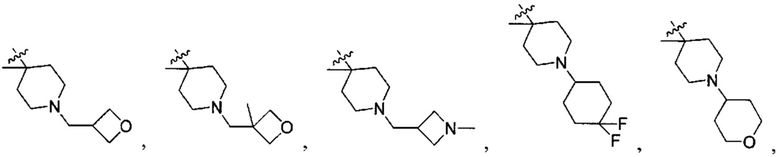
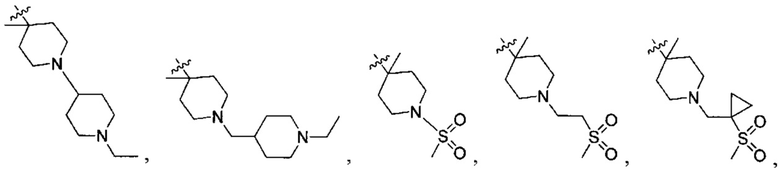
 ;
;
фенил, тиазолил, бифенил, нафтил, циклопентил, фурил, 3-пирролинил, пирролидинил, 1,3-диоксоланил, пиразолил, 2-пиразолинил, пиразолидинил, имидазолил, оксазолил, тиазолил, 1,2,3-азолил, 1,2,3-триазолил, 1,2,4-триазолил, 1,3,4-тиадиазолил, 4Н-пиранил, пиридинил, пиперидинил, 1,4-диоксанил, морфолинил, пирадазинил, пиримидинил, пиразинил, пиперазинил, 1,3,5-тритианил, 1,3,5-триазинил, бензофуранил, бензотиенил, индолил, бензимидазолил, бензотиазолил, пиринил, хинолил, изохинолил, циннолинил или хиноксалинил; и
метил, этил, пропил, метокси, этокси, метиламино, диметиламино, галогенметил, галогенэтил, галогенпропил, аминометил, аминоэтил, аминопропил, циклопропил, 


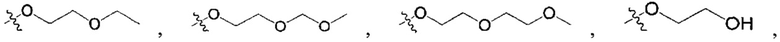
 .
.
Термин "фармацевтически приемлемый" при использовании в настоящем описании относится к таким соединениям, материалам, композициям и/или лекарственным формам, которые, в объеме достоверного медицинского суждения, подходят для применения к тканям человека и животного без избыточной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, и могут соответствовать разумному соотношению польза/риск.
Термин "фармацевтически приемлемая соль" относится к солям соединений по настоящему изобретению, получаемым из соединений, имеющих определенные замещающие группировки, обнаруженных в настоящем изобретении, и относительно нетоксичных кислот или оснований. Когда соединения по настоящему изобретению содержат относительно кислотные функциональные группы, соли присоединения основания могут быть получены путем взаимодействия достаточного количества основания с нейтральной формой таких соединений в беспримесном растворе или в подходящем инертном растворителе. Фармацевтически приемлемые соли присоединения основания включают натриевую, калиевую, кальциевую, аммонийную, органическую аминную или магниевую соль, или схожую соль. Когда соединения по настоящему изобретению содержат относительно основные функциональные группы, соли присоединения кислоты могут быть получены путем взаимодействия достаточного количества кислоты с нейтральной формой таких соединений в беспримесном растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемых солей присоединения кислоты включают соли, полученные из таких неорганических кислот, как соляная, бромоводородная, азотная, угольная, одноосновная угольная, фосфорная, одноосновная фосфорная, двухосновная фосфорная, серная, одноосновная серная, йодистоводородная, фосфористая кислота и тому подобные; а также соли, полученные из органических кислот, таких как уксусная, пропионовая, изомасляная, малеиновая, малоновая, бензойная, янтарная, субериновая, фумаровая, молочная, миндальная, фталевая, бензолсульфоновая, пара-толуолсульфоновая, лимонная, винная, метансульфоновая, и тому подобные. Также включены соли аминокислот, такие как аргинат и тому подобные, и соли органических кислот, таких как глюкуроновая кислота и тому подобные (см. Berge et al, "Pharmaceutical salts", Journal of Pharmaceutical Science 66: 1-19 (1977)). Некоторые конкретные соединения по настоящему изобретению содержат как основные, так и кислотные функциональные группы, что позволяет таким соединениям быть преобразованными в соли присоединения основания или кислоты.
Нейтральные формы соединений предпочтительно регенерируют путем взаимодействия соли с основанием или кислотой и выделения исходного соединения общепринятым способом. Исходная форма соединений отличается от различных форм солей этих соединений определенными физическими свойствами, такими как разная растворимость в полярных растворителях.
Термин "фармацевтически приемлемые соли", используемый в настоящем описании, относится к производным раскрытых соединений, в которых исходное соединение модифицировано путем образования солей с кислотой или основанием. Примеры фармацевтически приемлемых солей включают, но не ограничиваясь указанными, соли неорганических или органических кислот с основными остатками, такими как амины; щелочные или органические соли с кислотными остатками, такими как карбоновые кислоты; и тому подобные. Фармацевтически приемлемые соли включают общепринятые нетоксичные соли или четвертичные аммониевые соли исходных соединений, образованные, например, из нетоксичных неорганических или органических кислот. Общепринятые нетоксичные соли включают, но не ограничиваются указанными, соли, полученные из неорганических кислот и органических кислот, где неорганические кислоты или органические кислоты выбраны из группы, состоящей из 2-ацетоксибензойной, 2-гидроэтансульфоновой, уксусной, аскорбиновой, бензолсульфоновой, бензойной, одноосновной угольной, угольной, лимонной, этилендиаминтетрауксусной, этандисульфоновой, этансульфоновой, фумаровой, глюкогептоновой, глюконовой, глютаминовой, гликолевой, бромоводородной, соляной, йодистоводородной, гидроксильной, гидроксинафтойной, изэтионовой, молочной, лактозной, додецилсульфоновой, малеиновой, яблочной, миндальной, метансульфоновой, азотной, щавелевой, памоевой, пантотеновой, фенилуксусной, фосфорной, полигалактуроновой, пропионовой, салициловой, стеариновой, этиленуксусной, янтарной, аминосерной, сульфаниловой, серной, таниновой, винной, и пара-толуолсульфоновой кислот.
Фармацевтически приемлемые соли по настоящему изобретению могут быть получены из исходных соединений, содержащих кислотный остаток или основание, путем общепринятых химических способов. В общем, такие соли получают путем взаимодействия свободных кислотных или основных форм этих соединений со стехиометрическим количеством подходящей кислоты или основания в воде или в органическом растворителе, или в смеси двух растворителей. В общем, предпочтительной является неводная среда, такая как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил и тому подобные.
Помимо форм солей, соединения, предлагаемые в настоящем изобретении, имеют формы пролекарств. Пролекарства описанных здесь соединений в физиологических условиях легко претерпевают химические изменения с получением соединений по настоящему изобретению. Кроме того, пролекарства могут быть превращены в соединения по настоящему изобретению с помощью химических или биохимических методов в среде in vivo.
Конкретные соединения по настоящему изобретению могут быть представлены в несольватированных или сольватированных формах, в том числе в гидратированной форме. В общем, несольватированная форма эквивалентна сольватированной форме, и обе формы входят в объем настоящего изобретения. Конкретные соединения по настоящему изобретению могут существовать в разнообразных кристаллических или аморфных формах.
Конкретные соединения по настоящему изобретению могут также иметь асимметрический атом углерода (оптический центр) или двойную связь. Рацематы, диастереомеры, геометрические изомеры и индивидуальные изомеры входят в объем настоящего изобретения.
Используемые в настоящем описании графические изображения рацемических, амбискалемических и скалемических или энантиометрно чистых соединений взяты из Maehr J. Chem. Ed. 62, 114-120 (1985). Если не указано иное, клиновидная связь и пунктирная связь используются для обозначения абсолютной конфигурации хирального центра. Если не указано иное, когда соединения по настоящему изобретению содержат олефиновую двойную связь или любой другой центр геометрической асимметрии, они включают Е- и Z-геометрические изомеры. Аналогично, все таутомерные формы входят в объем настоящего изобретения.
Соединения по настоящему изобретению могут иметь определенные геометрические или стереоизомерные формы. Настоящее изобретение рассматривает все такие соединения, включая цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереоизомеры, (О)-изомеры, (b)-изомеры и рацемические смеси, и другие смеси, например смеси, обогащенные энантиомерами или диастереоизомерами, все из которых входят в объем настоящего изобретения. Заместители, такие как алкил и т.д., могут иметь дополнительные асимметрические атомы углерода. Все эти изомеры и их смеси входят в объем настоящего изобретения.
Оптически активные (R)- и (S)-изомеры, или (D)- и (L)-изомеры могут быть получены с использованием хирального синтеза, хиральных реагентов или других общепринятых способов. Если необходимо получить один вид энантиомера конкретного соединения по настоящему изобретению, чистый целевой энантиомер может быть получен путем ассиметрического синтеза или дериватизации со вспомогательным хиральным агентом с последующим разделением полученной смеси диастереомеров и отщеплением вспомогательной группы. Альтернативно, когда молекула содержит основную функциональную группу (такую как аминогруппа) или кислотную функциональную группу (такую как карбоксильная), соединение взаимодействует с подходящей оптически активной кислотой или основанием с образованием соли диастереоизомера, после чего диастереоизомер подвергают разделению путем фракционной кристаллизации или хроматографии, хорошо известных в уровне техники, и выделяют с получением чистого энантиомера. Кроме того, энантиомеры и диастереомеры обычно разделяют с использованием хроматографии, в которой используется хиральная неподвижная фаза, и необязательно при помощи способа химических производных (например, карбамат, получаемый из амина).
Соединения по настоящему изобретению также могут иметь неестественные соотношения атомных изотопов для одного или нескольких атомов, которые входят в состав таких соединений. Например, эти соединения могут быть помечены радиоактивными изотопами, например такими как тритий (3Н), йод-125 (125I) или углерод-14 (14С). Все изотопные разновидности соединений по настоящему изобретению, радиоактивные или нет, входят в объем настоящего изобретения.
Термин "фармацевтически приемлемый носитель" относится к любому веществу или несущей среде, которые могут доставить эффективное количество активных веществ по настоящему изобретению, не препятствует биологической активности активных веществ и не оказывает токсических побочных эффектов на хозяина или пациента. Примеры носителей включают воду, растительное и минеральное масло, основу в виде крема, основу в виде лосьона, основу в виде мази и тому подобные. Эти основы включают суспензии, загустители, усилители проникновения и тому подобные. Их составы хорошо известны специалистам в косметической области или области фармацевтики местного применения. За дополнительной информацией о носителях можно обратиться к Remington: The Science and Practice of Pharmacy, 21st Ed, Lippincott, Williams & Wilkins (2005), включенному в настоящее описание посредством ссылки.
Термин "эксципиент" обычно относится к носителю, разбавителю и/или наполнителю, необходимому для составления эффективных фармацевтических композиций.
Для лекарственного средства или фармацевтически активного агента термин "эффективное количество" или "терапевтически эффективное количество" относится к нетоксичному, но достаточному количеству, необходимому для достижения желаемого эффекта лекарственного средства или агента. Для лекарственных форм для перорального применения по настоящему изобретению "эффективное количество" активного вещества в композиции относится к количеству, необходимому для достижения желаемого эффекта при объединении с другим активным веществом в композиции. Эффективное количество различается для разных людей и определяется в зависимости от возраста и общего состояния пациентов, а также от конкретного активного вещества. Подходящее активное количество в индивидуальных случаях может быть определено специалистом на основании стандартного эксперимента.
Термин "активный ингредиент", "терапевтический агент", "активное вещество" или "активный агент" относится к химическому соединению, которое может эффективно лечить целевое расстройство, заболевание или состояние.
Термин "замещенный" означает, что один или более атомов водорода на определенном атоме замещены заместителем(ями), включая дейтерий и варианты водорода, при условии что валентное состояние определенного атома является нормальным и замещенное соединение стабильно. Когда заместитель представляет собой кето- (т.е. =O), это означает, что замещены два атома водорода. Кето-замещение не происходит на ароматической группе. Термин "необязательно замещенный" означает, что замещение может происходить или может не происходить, и если не указано иное, вид и количество заместителей может быть произвольным, при условии что это может быть достигнуто химически.
Когда любая переменная (например, R) встречается в композиции или структуре соединения более одного раза, ее значение в каждом случае является независимым. Так, например, если группа замещена 0-2 R, это означает, что группа может необязательно быть замещена до двух R, и в каждом случае R выбирается независимо. Более того, комбинация заместителей и/или их вариантов возможна только в том случае, если такая комбинация обусловит стабильное соединение.
Когда одна из переменных выбрана из одинарной связи, она представляет собой две группы, связанные напрямую. Например, когда L в A-L-Z представляет собой одинарную связь, структура A-L-Z на самом деле представляет собой A-Z.
Когда связь заместителя может перекрестно связываться с двумя атомами кольца, такой заместитель может быть связан с любым атомом кольца. Когда не указано, через какой атом указанный заместитель связан с соединением, представленным общей химической структурной формулой, но не определенным конкретно, такой заместитель может быть связан через любой из его атомов. Комбинация заместителей и/или их вариантов возможна только в том случае, если такая комбинация обусловит стабильное соединение. Например, структурная единица  или
или  обозначает, что любая позиция в циклогексиле или циклогексадиене может быть замещена.
обозначает, что любая позиция в циклогексиле или циклогексадиене может быть замещена.
Заместители алкильных и гетероалкильных радикалов (включая группы, обычно называемые алкиленовой, алкенильной, гетероалкиленовой, гетероалкенильной, алкинильной, циклоалкильной, гетероциклоалкильной, циклоалкенильной и гетероциклоалкенильной) известны под названием "алкильные заместители", которые могут быть выбраны, но не ограничиваясь указанными, из одной или более из следующих групп: -R', -OR', =O, =NR', =N-OR', -NR'R'', -SR', галогена, -SiR'R''R''', OC(O)R', -C(O)R', -CO2R', -CONR'R'', -OC(O)NR'R'', -NR''C(O)R', NR'C(O)NR''R''', -NR''C(O)2R', -NR'''''-C(NR'R''R''')=NR'''', NR''''C(NR'R'')=NR''', -S(O)R', -S(O)2R', -S(O)2NR'R'', NR''SO2R', -CN, -NO2, -N3, -CH(Ph)2 и фтор(С1-С4) алкила; число заместителей составляет от 0 до 2m' плюс 1, где m' представляет собой общее число атомов углерода в таком радикале. Каждый из R', R'', R''', R'''' и R''''' каждый независимо и предпочтительно представляет собой водород, замещенный или незамещенный гетероалкил, замещенный или незамещенный арил (например, арил, замещенный 1-3 галогенами), замещенный или незамещенный алкил, алкокси, тиоалкокси или аралкил. Когда соединение по настоящему изобретению включает более чем один R, например, каждый R выбирается независимо, так же как каждый из R', R'', R''', R'''' и R''''', когда присутствует более чем один R', R'', R''', R'''' и R'''''. Когда R' и R'' присоединены к одному и тому же атому азота, они могут образовывать 5-, 6- или 7-членное кольцо вместе с атомом азота. Например, подразумевается, что -NR'R'' включает, но не ограничиваясь указанными, 1-пирролидинил и 4-морфолинил. В соответствии с вышеприведенным обсуждением заместителей специалисту ясно, что подразумевается, что термин "алкил" включает группу, образованную связью атома углерода с неводородной группой, такой как галогеналкильная (например, -CF3, -CH2CF3) и ацильная (например, -С(O)СН3, -C(O)CF3, -С(O)СН2ОСН3 и т.д.).
Аналогично алкильному заместителю, арильный и гетероарильный заместители обычно оба называют "арильными заместителями", и они выбраны из группы, состоящей из, например, -R', -OR', -NR'R'', -SR', -галогена, -SiR'R''R''', OC(O)R', -C(O)R, -CO2R', -CONR'R'', -OC(O)NR'R'', -NR''C(O)R', NR'C(O)NR''R''', -NR''C(O)2R', -NR'''''-C(NR'R''R''')=NR'''', NR''''C(NR'R'')=NR''', -S(O)R', -S(O)2R', -S(O)2NR'R'', NR''SO2R', -CN, -NO2, -N3, -CH(Ph)2, фтор(С1-С4)алкокси и фтор(С1-С4)алкила и тому подобных, и число заместителей составляет от 0 до общего числа свободных валентностей ароматического кольца; где каждый из R', R'', R''', R'''' и R''''' независимо и предпочтительно выбран из группы, состоящей из водорода, замещенного или незамещенного алкила, замещенного или незамещенного гетероалкила, замещенного или незамещенного арила и замещенного или незамещенного гетероарила. Когда соединение по настоящему изобретению включает более чем один R, например, каждый R выбирается независимо, так же как каждый из R', R'', R''', R'''' и R''''', когда присутствует более чем одна из этих групп.
Два заместителя соседних атомов арильного или гетероарильного кольца могут необязательно быть замещены заместителем формулы -T-C(O)-(CRR')q-U-, где Т и U независимо выбраны из группы, состоящей из -NR-, -О-, CRR'- и одинарной связи, и q представляет собой целое число от 0 до 3. Альтернативно, два заместителя соседних атомов арильного или гетероарильного кольца могут необязательно быть замещены заместителем формулы -А(СН2)rВ-, где А и В независимо выбраны из группы, состоящей из -CRR'-, -О-, -NR-, -S-, -S(O)-, S(O)2-, -S(O)2NR'- и одинарной связи, и г представляет собой целое число от 1 до 4. Необязательно, одна одинарная связь образованного таким образом нового кольца может быть заменена двойной связью. Альтернативно, два заместителя соседних атомов арильного или гетероарильного кольца могут необязательно быть замещены заместителем формулы -А(СН2)rВ-, где s и d независимо выбраны из целого числа от 0 до 3, X выбран из группы, состоящей из -О-, -NR', -S-, -S(O)-, -S(O)2- и -S(O)2NR'-. Каждый из заместителей R, R', R'' и R''' независимо и предпочтительно выбран из группы, состоящей из водорода и замещенного или незамещенного (C1-С6) алкила.
Если не указано иное, термин "галоген" или "гало", сам по себе или как часть другого заместителя, относится к атому фтора, хлора, брома или йода. Кроме того, термин "галогеналкил" включает моногалогеналкил и полигалогеналкил. Например, подразумевается, что термин "галоген(С1-С4)алкил" включает, но не ограничиваясь указанными, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и тому подобные.
Примеры галогенкалкила включают, но не ограничиваясь указанными: трифторметил, трихлорметил, пентафторэтил и пентахлорэтил. "Алкокси" означает вышеуказанный алкил, имеющий определенное количество атомов углерода, присоединенных к кислороду. С1-6 алкокси включает С1, С2, С3, С4, С5 и С6 алкокси. Примеры алкокси включают, но не ограничиваясь указанными: метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентокси. "Циклоалкил" включает насыщенные кольцевые группы, такие как циклопропил, циклобутил или циклопентил. 3-7 циклоалкил включает С3, С4, С5, С6 и С7 циклоалкил. "Алкенил" относится к прямому или разветвленному углеводороду, где в любом стабильном месте цепи присутствует одна или более двойных связей углерод-углерод, такому как винил и пропенил.
Термин "гало" или "галоген" относится к фтор-, хлор-, бром- и йод-.
Если не указано иное, термин "гетеро" означает гетероатом или гетероатомную группу (т.е. группу атомов, содержащую гетероатом), включающую атомы, отличные от углерода (С) и водорода (Н), и группы атомов, содержащие эти гетероатомы, включая, например, кислород (О), азот (N), серу (S), кремний (Si), германий (Ge), алюминий (Аl), бор (В), -О-, -S-, =O, =S, -С(=O)O-, -С(=O)-, -C(S)-, -S(=O), -S(=O)2- и необязательно замещенные -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- или -S(=O)N(H)-.
Если не указано иное, "кольцо" означает замещенный или незамещенный циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил, арил или гетероарил. Так называемое кольцо включает монокольцо, связанное кольцо, спиро-кольцо, конденсированное кольцо или мостиковое кольцо. Число атомов в кольце обычно определяется как число членов кольца, например, "5-7-членное кольцо" означает, что от 5 до 7 атомов расположены в кольце. Если не указано иное, кольцо необязательно содержит от 1 до 3 гетероатомов. Следовательно, "5-7-членное кольцо" включает, например, фенилпиридин и пиперидинил; с другой стороны, термин "5-7-членное гетероциклоалкильное кольцо" включает пиридил и пиперидил, но не включает фенил. Термин "кольцо" также включает систему колец, содержащую по меньшей мере одно кольцо, где каждое кольцо независимо соответствует вышеуказанному определению.
Если не указано иное, термин "гетероцикл", "гетероциклическое кольцо" или "гетероцикло" означает стабильный моноцикл, бицикл или трицикл, содержащий гетероатом или гетероатомную группу, который может быть насыщенным, частично ненасыщенным или ненасыщенным (ароматическим) и содержит атомы углерода и 1, 2, 3 или 4 гетероатома кольца, независимо выбранных из N, О и S, где указанный гетероцикл может быть необязательно конденсирован с бензольным кольцом с образованием бицикла. Гетероатомы азота и серы могут необязательно быть окисленными (т.е. NO и S(O)p). Атом азота может быть замещенным или незамещенным (т.е. N или NR, где R представляет собой Н или другие заместители, уже определенные выше). Гетероциклическое кольцо может быть присоединено к боковой группе любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное таким образом соединение стабильно, описанный здесь гетероцикл может быть замещен в положении углерода или азота. Атом азота в гетероцикле необязательно является кватернизованным. В предпочтительном варианте осуществления, когда общее число атомов S и О в гетероцикле составляет более 1, эти гетероатомы не являются соседними. В другом предпочтительном варианте осуществления общее число атомов S и О в гетероцикле составляет не более 1. При использовании в настоящем описании, термин "ароматический гетероцикл" или "гетероарил" относится к стабильному арильному кольцу, представляющему собой 5-, 6-, 7- членный моноцикл или бицикл, или 7-, 8-, 9- или 10-членный бициклический гетероцикл, содержащий атомы углерода и 1, 2, 3 или 4 гетероатомов кольца, независимо выбранных из N, О и S. Атом азота может быть замещенным или незамещенным (т.е. N или NR, где R представляет собой Н или другие заместители, уже определенные выше). Гетероатомы азота и серы могут необязательно быть окисленными (т.е. NO и S(O)p). Следует отметить, что общее число атомов S и О в ароматическом гетероцикле составляет не более одного. Мостиковое кольцо также входит в определение гетероцикла. Мостиковое кольцо образуется, когда один или более атомов (т.е. С, О, N или S) связаны с двумя несмежными атомами углерода или азота. Предпочтительное мостиковое кольцо включает, но не ограничиваясь указанными: один атом углерода, два атома углерода, один атом азота, два атома азота и одну углерод-азотную группу. Следует отметить, что мостик всегда преобразует моноцикл в трицикл. Заместитель также может присутствовать на мостике мостикового кольца.
Примеры гетероциклических соединений включают, но не ограничиваясь указанными: акридинил, азоцинил, бензимидазолил, бензофуранил, бензомеркаптофуранил, бензомеркаптофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензоизоксазолил, бензоизотиазолил, бензоимидазолинил, карбазолил, 4аН-карбазолил, карболинил, хроманил, хромен, циннолинил, декагидрохинолинил, 2Н,6Н-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1Н-индазолил, индоленил, индолинил, индолизинил, индолил, 3Н-индолил, изатиногруппу, изобензофуранил, изоиндолил, изоиндолинил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, оксиндолил, пиримидинил, фенантридинил, фенантролинил, феназин, фенотиазин, бензоксантинил, фенолоксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пирролидинил, пирролинил, 2Н-пирролил, пирролил, хиназолинил, хинолинил, 4Н-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6Н-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, изотиазолилтиенил, тиенооксазолил, тиенотиазолил, тиеноимидазолил, тиенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил. Также включены соединения с конденсированными кольцами и спиро-кольцами.
Если не указано иное, термин "гидрокарбил" или его любые конкретные воплощения (такие как алкил, алкенил, алкинил, фенил и т.д.), сам по себе или как часть другого заместителя, означает углеводородные радикалы с прямой или разветвленной цепью или циклические углеводородные радикалы, или их комбинации, которые могут быть полностью насыщенными, моно- или полиненасыщенными, могут быть моно-, ди- или полизамещенными, могут включать бивалентный или мультивалентный радикал и обладают определенным числом атомов углерода (например, C1-С10 означает от 1 до 10 атомов углерода). "Гидрокарбил" включает, но не ограничиваясь указанными, алифатический гидрокарбил и ароматический гидрокарбил, где алифатический гидрокарбил включает линейный и циклический гидрокарбил, в частности включая, но не ограничиваясь указанными, алкил, алкенил и алкинил, и ароматический гидрокарбил включает, но не ограничиваясь указанными, 6-12-членный ароматический гидрокарбил, такой как фенил, нафтил и тому подобные. В некоторых вариантах осуществления термин "алкил" означает прямой или разветвленный радикал или комбинацию радикалов, который может быть полностью насыщенным, моно- или полиненасыщенным, и может включать бивалентный или мультивалентный радикал. Примеры насыщенного углеводородного радикала включают, но не ограничиваясь указанными, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, изобутил, циклогексил, (циклогексил)метил, циклопропилметил и гомологи или изомеры радикалов, такие как н-пентил, н-гексил, н-гептил, н-октил и тому подобные. Ненасыщенный алкил имеет одну или более двойных или тройных связей, и их примеры включают, но не ограничиваясь указанными, этенил, 2-пропенил, бутенил, кротил, 2-изопентил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1-й 3-пропинил, 3-бутинил и высшие гомологи и изомеры.
Если не указано иное, термин "гетерогидрокарбил" или любые его конкретные воплощения (такие как гетероалкил, гетероалкенил, гетероалкинил, гетероарил и т.д.), сам по себе или как часть другого заместителя, представляет собой углеводородные радикалы с прямой или разветвленной цепью или циклические углеводородные радикалы, или их комбинации, имеющие определенное число атомов углерода и по меньшей мере один гетероатом. В некоторых вариантах осуществления термин "гетероалкил" сам по себе или в сочетании с другим термином означает стабильный углеводородный радикал с прямой или разветвленной цепью, или их комбинации, имеющие определенное число атомов углерода и по меньшей мере один гетероатом. В одном иллюстративном варианте осуществления гетероатом выбран из В, О, N и S, где атомы азота и серы необязательно окислены, и атом азота необязательно кватернизован. Гетероатом В, О, N и S может быть расположен в любом внутреннем положении гетерогидрокарбила (включая положение, в котором гидрокарбил присоединяется к остальной части молекулы). Примеры включают, но не ограничиваясь указанными, -СН2-СН2-О-СН3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -СН2-СН2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3, и -CH=CH-N(CH3)-CH3. Подряд может быть расположено до двух гетероатомов, как например в -CH2-NH-OCH3.
Термины "алкокси", "алкиламино" и "алкилтио" (или тиоалкокси) принадлежат к идиоматическим выражениям и относятся к алкильным группам, присоединенным к оставшейся молекуле через атом кислорода, аминогруппу или атом серы, соответственно.
Если не указано иное, термин "циклогидрокарбил", "гетероциклогидрокарбил" или любые его конкретные воплощения (такие как арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил и т.д.), сам по себе или в сочетании с другим термином, означает циклизованный "гидрокарбил" и "гетерогидрокарбил". Кроме того, для гетерогидрокарбила или гетероциклогидрокарбила (например, гетероалкила, гетероциклоалкила), гетероатом может занимать положение, в котором гетероцикл присоединяется к остальной части молекулы. Примеры циклоалкила включают, но не ограничиваясь указанными, циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и тому подобные. Неограничивающие примеры гетероциклических групп включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофураниндол-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-шшеразинил.
Если не указано иное, термин "арил" означает полиненасыщенные ароматические углеводородные заместители, которые могут быть моно-, ди- или полизамещенными, и могут представлять собой одно кольцо или несколько колец (предпочтительно 1-3 кольца), где кольца могут быть конденсированными или ковалентно связанными. Термин "гетероарил" относится к арилу (или кольцу), содержащему от одного до четырех гетероатомов. В одном иллюстративном варианте осуществления гетероатом выбран из В, О, N и S, где атомы азота и серы необязательно окислены, и атом азота необязательно кватернизован. Гетероарил может быть присоединен к остальной части молекулы через гетероатом. Неограничивающие примеры арила или гетероарила включают фенил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместитель любой из вышеуказанных арильных и гетероарильных кольцевых систем выбран из приемлемых заместителей, описанных ниже.
Для удобства при использовании в сочетании с другим термином (например, арилокси, арилтио, арилалкил), термин "арил" включает арильное и гетероарильное кольцо, определенные выше. Так, подразумевается, что термин "аралкил" включает радикалы, образованные из арила, присоединенного к алкилу (например, бензил, фенэтил, пиридилметил и т.д.), включая такие алкилы, в которых атом углерода (например, метилен) заменен, например, атомом кислорода, например феноксиметил, 2-пиридилоксиметил, 3-(1-нафтилокси)пропил и тому подобные.
Термин "уходящая группа" относится к функциональной группе или атому, который может быть замещен другой функциональной группой или атомом через реакцию замещения (такую как реакция нуклеофильного замещения). Например, типичные уходящие группы включают трифталат; хлор, бром и йод; сульфонатные группы, такие как мезилатная, тозилатная, брозилатная, тозилатная и тому подобные; ацилокси, такая как ацетокси, трифторацетокси и тому подобные.
Термин "защитная группа" включает, но не ограничиваясь указанными, "аминозащитную группу", "гидроксизащитную группу" или "меркаптозащитную группу." Термин "аминозащитная группа" означает защитную группу, подходящую для блокировки побочных реакций по азоту аминогруппы. Типичные аминозащитные группы включают, но не ограничиваясь указанными: формил; ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (Вое); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-фторфенил метоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), тритил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) и тому подобные. Термин "гидроксизащтная группа" относится к защитной группе, подходящей для блокировки побочных реакций по гидроксилу. Типичные гидроксизащитные группы включают, но не ограничиваясь указанными: алкил, такой как метил, этил и трет-бутил; ацил, например алканоил (например, ацетил); арилметил, такой как бензил (Bn), пара-метоксибензил (РМВ), 9-фторенилметил (Fm) и дифенилметил (бензогидрил, DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS), и тому подобные.
Соединения по настоящему изобретению могут быть получены путем различных способов синтеза, хорошо известных специалистам в данной области техники, включая конкретные варианты осуществления, проиллюстрированные ниже, варианты осуществления, полученные в сочетании с другими способами синтеза, и эквиваленты, хорошо известные специалистам в данной области техники. Предпочтительные варианты осуществления включают, но не ограничиваясь указанными, примеры по настоящему изобретению.
Все растворители, использованные в настоящем изобретении, коммерчески доступны и были использованы без дополнительной очистки. Реакцию обычно проводили в атмосфере инертного азота в безводном растворителе. Данные протонного ядерного магнитного резонанса получали на спектрометре Bruker Avance III 400 (400 МГц), где химические сдвиги указаны как млн-1 (миллионные доли) тетраметилсилана в слабом поле. Масс-спектры измеряли на Agilent 1200 Series plus 6110 (& 1956А). Жидкостная хроматомасс-спектрометр (ЖХ/МС) или масс-спектрометр (МС) Shimadzu содержал DAD (диодноматричный детектор): SPD-M20A (ЖХ) и детектор Shimadzu Micromass 2020. Масс-спектрометр был оборудован источником электрораспылительной ионизации (ЭРИ), работающим в положительном или отрицательном режиме.
В настоящем изобретении использованы следующие аббревиатуры: aq обозначает воду; HATU обозначает O-(7-аза-бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат; EDC означает N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорид; m-СРВА обозначает 3-хлорпероксибензойную кислоту; экв. обозначает эквивалент, эквивалентное количество; CDI обозначает карбонилдиимидазол; DCM обозначает дихлорметан; РЕ обозначает петролейный эфир; DIAD обозначает диизопропилазодикарбоксилат; DMF обозначает N,N-диметилформамид; DMSO обозначает диметилсульфоксид; EtOAc обозначает этилацетат; ЕtOН обозначает этанол; МеОН обозначает метанол; CBz обозначает бензилоксикарбонил, который представляет собой аминозащитную группу; ВОС обозначает трет-бутилкарбонил, который представляет собой аминозащитную группу; НОАс обозначает уксусную кислоту; NaCNBFD обозначает цианоборгидрид натрия; r.t. обозначает комнатную температуру; O/N означает в течение ночи; THF обозначает тетрагидрофуран; Вос2О обозначает ди-трет-бутилдикарбонат; TFA обозначает трифторуксусную кислоту; DIPEA обозначает диизопропилэтиламин; SOCl2 обозначает тионилхлорид; CS2 обозначает дисульфид углерода; TsOH обозначает пара-толу олсульфоновую кислоту; NFSI обозначает N-фтор-N-(фенилфульфонил)бензолсульфонамид; NCS обозначает 1-хлорпирролидин-2,5-дион; n-Bu4NF обозначает фторид тетрабутиламмония; iPrOH обозначает 2-пропанол; и т. плав., обозначает точку плавления.
Соединения были названы вручную или с помощью программного обеспечения ChemDraw®. Для коммерчески доступных соединений использованы названия из каталога производителя.
Краткое описание графических материалов
Фиг. 1 демонстрирует результаты фармакодинамического исследования in vivo тестируемых лекарственных средств на подкожных ксенотрансплантатных опухолевых моделях клеток рака яичника человека SK-OV-3, где
1) число мышей на группу составило 6;
2) объем введения: 10 мкл/г массы тела мыши. Если потеря массы превышает 15%, режим введения следует скорректировать соответственно;
3) наполнитель, используемый для тестируемых соединений, и Группа, получавшая наполнитель, представляли собой: 1% МС (метилцеллюлозы), перорально (п/о), ежедневно в течение 19 дней.
Фиг. 2-1 демонстрирует результаты фармакодинамического исследования in vivo (I) тестируемых лекарственных средств на подкожных ксенотрансплантатных опухолевых моделях клеток рака предстательной железы человека РС-3М, где
1) число мышей на группу составило 7;
2) объем введения: 10 мкл/г массы тела мыши. Если потеря массы превышает 15%, режим введения следует скорректировать соответственно.
Фиг. 2-2 демонстрирует результаты фармакодинамического исследования in vivo (II) тестируемых лекарственных средств на подкожных ксенотрансплантатных опухолевых моделях клеток рака предстательной железы человека РС-3М, где
1) число мышей на группу составило 6;
2) введение: 10 мкл/г массы тела мыши. Если потеря массы превышает 15%, режим введения следует скорректировать соответственно;
3) наполнитель, используемый для тестируемых соединений, и Группа, получавшая наполнитель, представляли собой: 1% DMSO плюс 99% (1% МС), п/о, ежедневно в течение 2 недель.
Фиг. 2-3а демонстрирует результаты фармакодинамического исследования in vivo (III) тестируемых лекарственных средств на подкожных ксенотрансплантатных опухолевых моделях клеток рака предстательной железы человека РС-3М, где
1) число мышей на группу составило 6;
2) объем введения: 10 мкл/г массы тела мыши. Если потеря массы превышает 15%, режим введения следует скорректировать соответственно;
3) наполнитель, используемый для PF0512384: 30% пропиленгликоль плюс 5%Ttween 80 плюс 65% D5W (5% водный раствор декстрозы), в/в, раз в неделю в течение 2 недель;
4) наполнитель, используемый для тестируемых соединений, и Группа, получавшая наполнитель, представляли собой: 5% DMSO плюс 60% PEG400 плюс 35% воды, п/о, ежедневно в течение 2 недель.
Фиг. 2-3b демонстрирует результаты фармакодинамического исследования in vivo (III) тестируемых лекарственных средств на подкожных ксенотрансплантатных опухолевых моделях клеток рака предстательной железы человека РС-3М, где,
1) число мышей на группу составило 6;
2) объем введения: 10 мкл/г массы тела мыши. Если потеря массы превышает 15%, режим введения следует скорректировать соответственно;
3) наполнитель, используемый для PF0512384: 30% пропиленгликоль плюс 5% Tween 80 плюс 65% D5W, в/в, раз в неделю в течение 2 недель; 4) наполнитель, используемый для тестируемых соединений, и Группа, получавшая наполнитель, представляли собой: 5% DMSO плюс 60% PEG400 плюс 35% воды, п/о, ежедневно в течение 2 недель.
Подробное описание изобретения
Для более подробного иллюстрирования настоящего изобретения приведены следующие примеры, не ограничивающие объем настоящего изобретения.
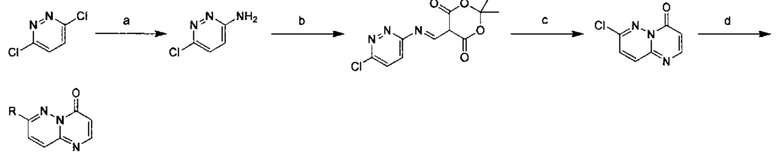
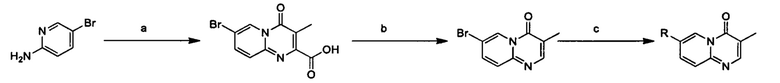
Схема 1:
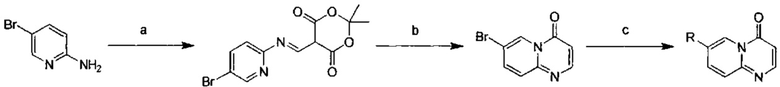
Условия реакции: а) триэтилортоформиат, 2,2-диметил-1,3-диоксан-4,6-дион, при нагревании; EtOH, при нагревании; b) дифениловый эфир, при нагревании с обратным холодильником; с) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 1
2,4-дифтор-N-(2-метокси-5-(4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
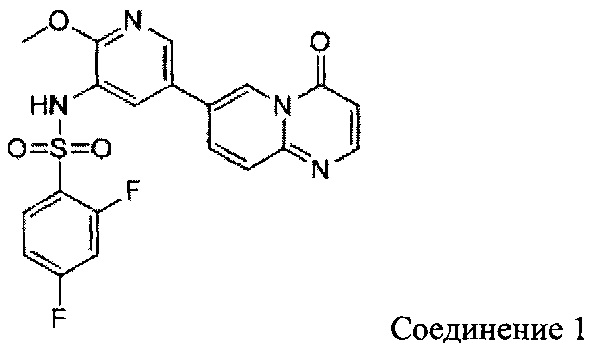
a) (Е)-5-(((5-бромпиридин-2-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион
Триэтилортоформиат (25,8 г, 0,174 моль) и 2,2-диметил-1,3-диоксан-4,6-дион (25,1 г, 0,174 моль) помещали в трехгорлую кругло донную колбу и проводили реакцию при перемешивании в течение 2 часов при 60°С. К этой смеси по каплям добавляли 2-амино-5-бромпиридин (30 г, 0,174 моль) в этаноле (150 мл). Реакционный раствор перемешивали при 60°С в течение 2 часов, и затем охлаждали до 25°С и фильтровали. Фильтрационный осадок промывали этанолом (200 мл × 3) с получением целевого соединения в виде белого твердого вещества (40 г, 70%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 1,77 (s, 6Н), 6,93-7,04 (m, 1Н), 8,44-8,53 (m, 1Н), 7,85-7,91 (m, 1Н), 9,31-9,42 (m, 1Н), 11,28-11,40 (m, 1Н).
b) 7-бром-4Н-пиридо[1,2-а]пиримидин-4-он
(Е)-5-(((5-бромпиридин-2-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион (18 г, 0,056 ммоль) и дифениловый эфир (180 мл) помещали в кругло донную колбу объемом 250 мл и перемешивали при 220°С в течение 1 часа. Окончание реакции определяли с помощью ТСХ (тонкослойной хроматографии). Реакционный раствор охлаждали до комнатной температуры и очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения (10 г, 80%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 6,46 (d, 1Н), 7,53 (d, 1Н), 7,75 (dd, 1Н), 8,27 (d, 1Н), 9,19 (d, 1H).
c) 2,4-дифтор-N-(2-метокси-5-(4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
7-бром-4Н-пиридо[1,2-а]пиримидин-4-он (0,28 ммоль) растворяли в диоксане (2 мл) и воде (0,4 мл) и добавляли 2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)бензсульфамид (0,28 ммоль), карбонат калия (0,56 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (20 мг) в атмосфере азота. Реакционную смесь подвергали воздействию микроволнового излучения при 100°С в течение 2 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и концентрировали с получением неочищенного продукта, который затем очищали с помощью препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) с получением целевого продукта.
1H ЯМР (400 МГц, CDCl3) млн-1 δ 3,87 (s, 3Н), 6,53 (d, 1Н), 7,12 (t, 1Н), 7,24 (t, 1Н), 7,83 (d, 1Н), 7,87-7,97 (m, 1Н), 8,10 (s, 1Н), 8,26 (d, 1Н), 8,31-8,40 (m, 2Н), 9,21 (s, 1Н).
Следующие 37 соединений были также синтезированы согласно способу получения Соединения 1.
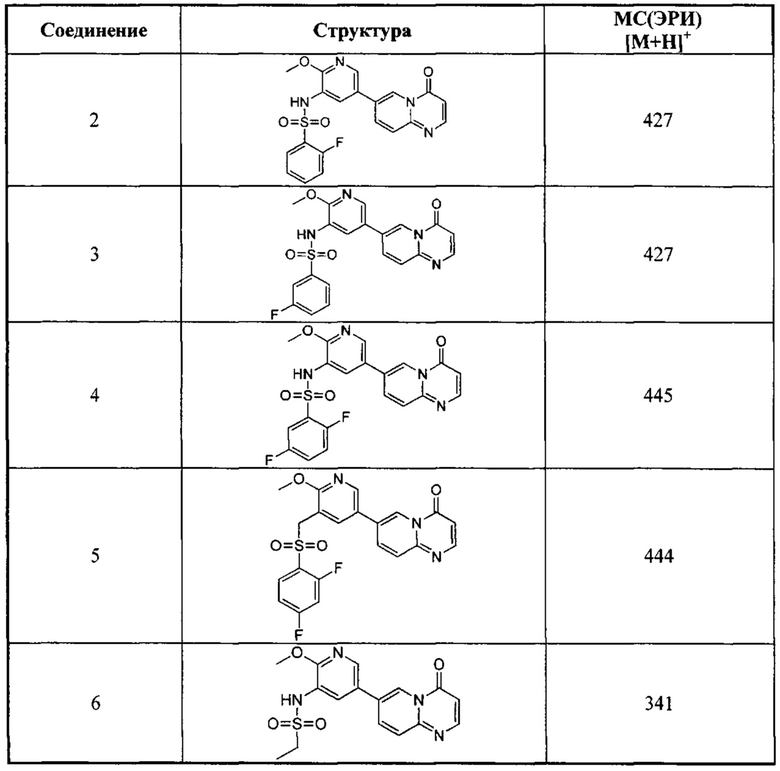
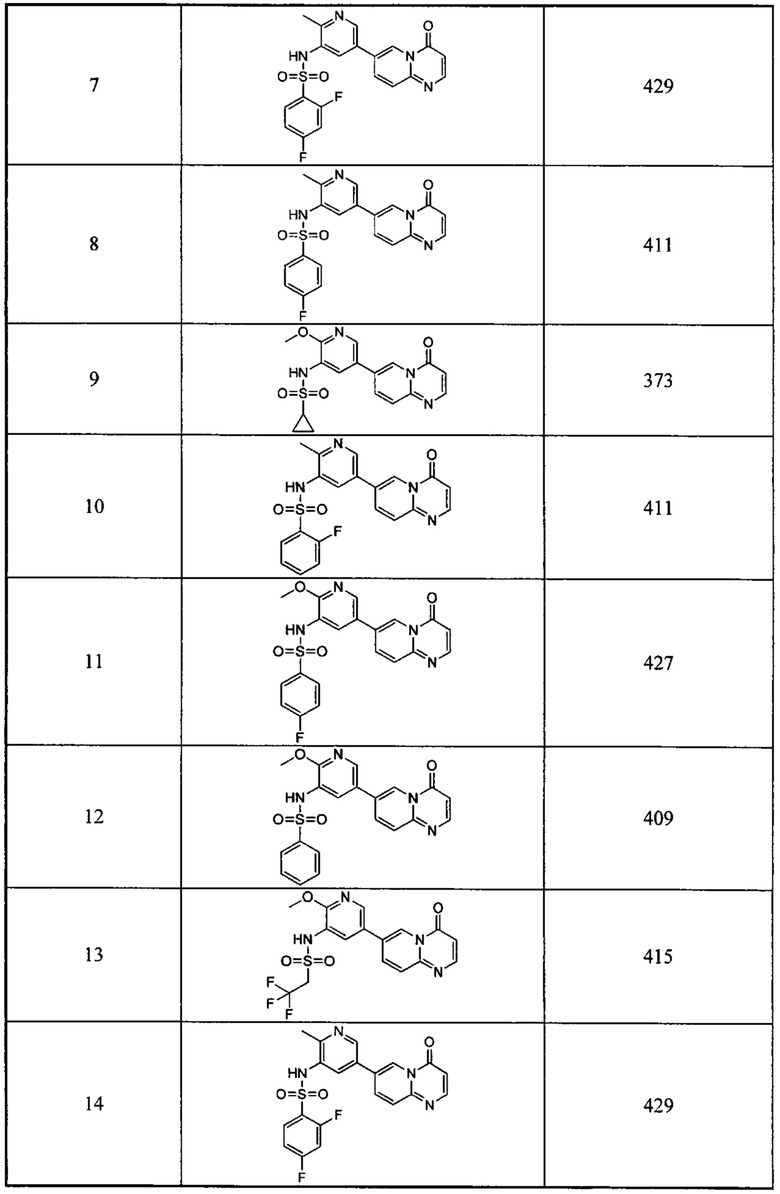
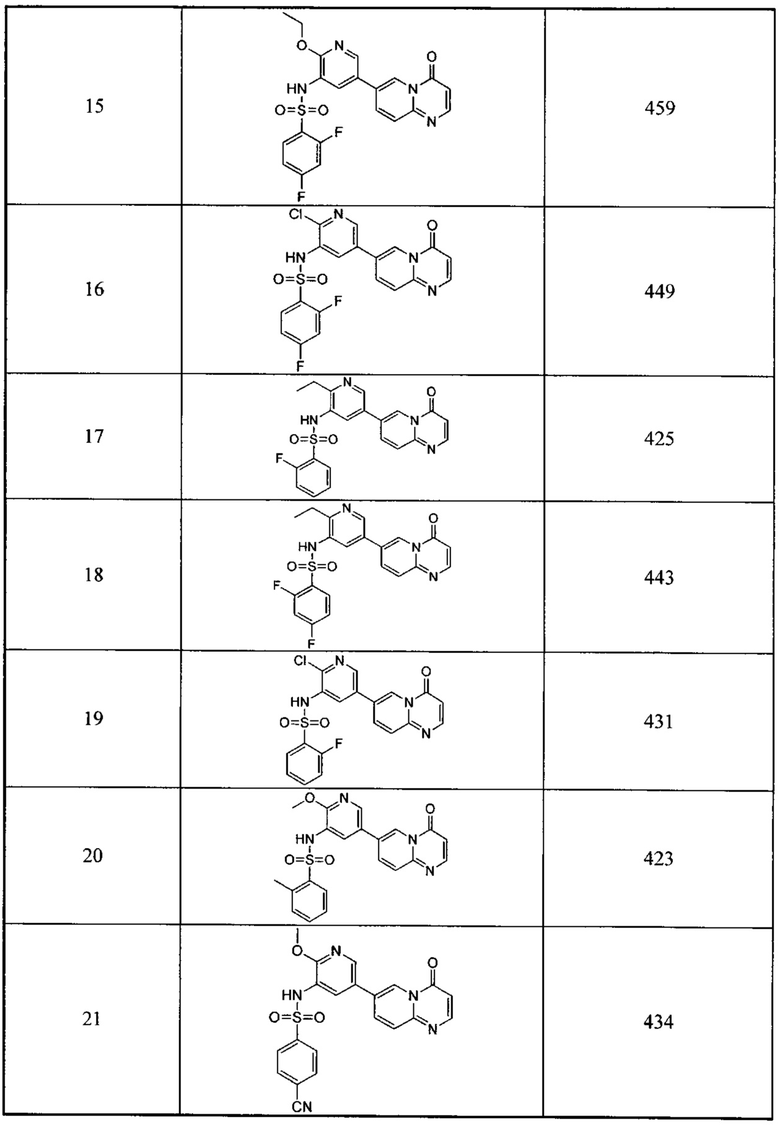
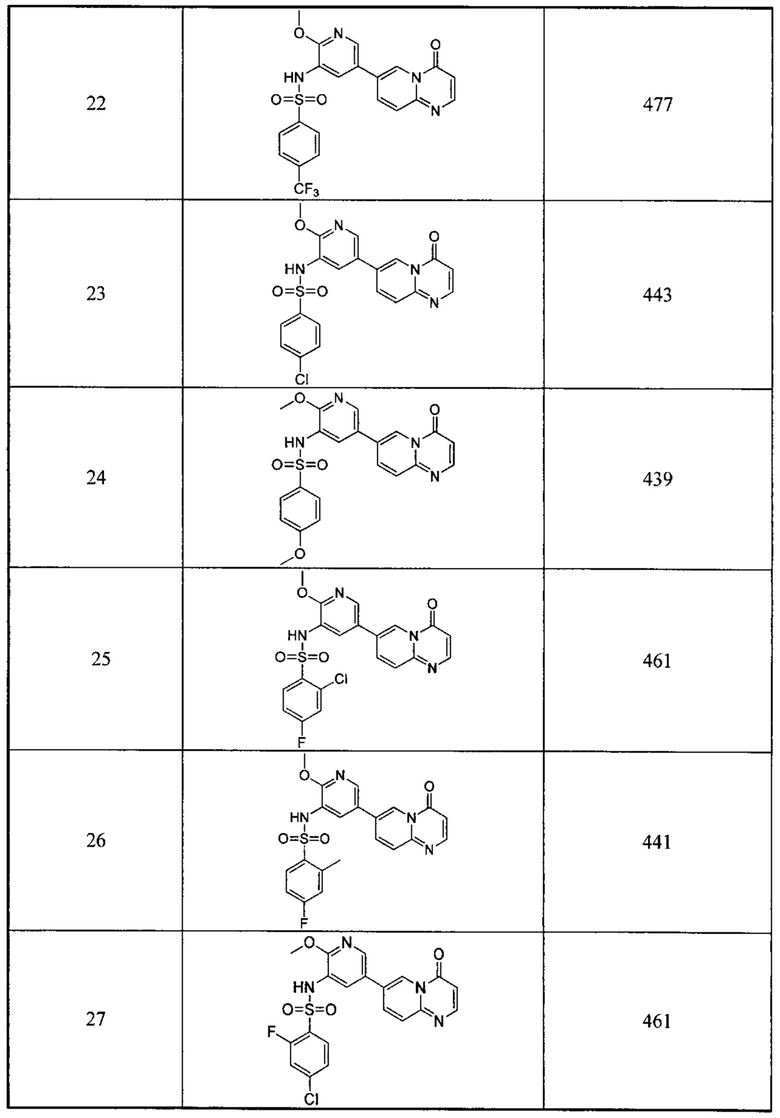
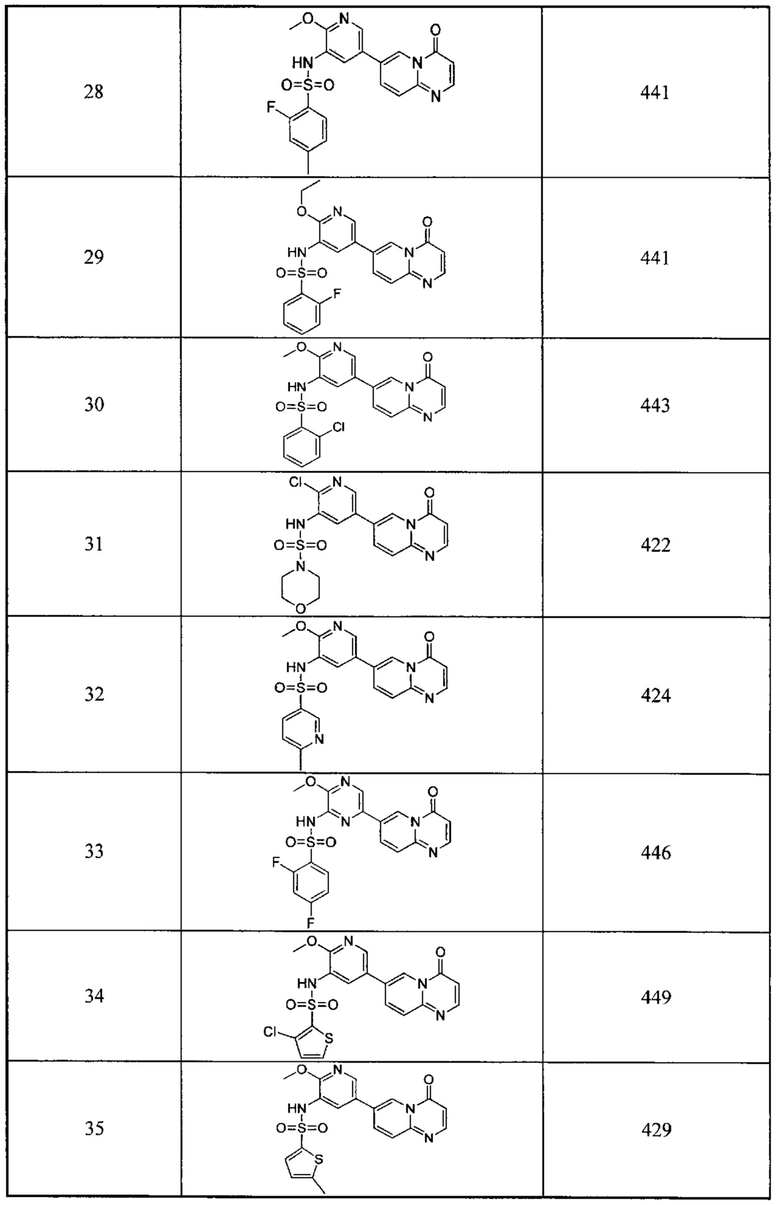
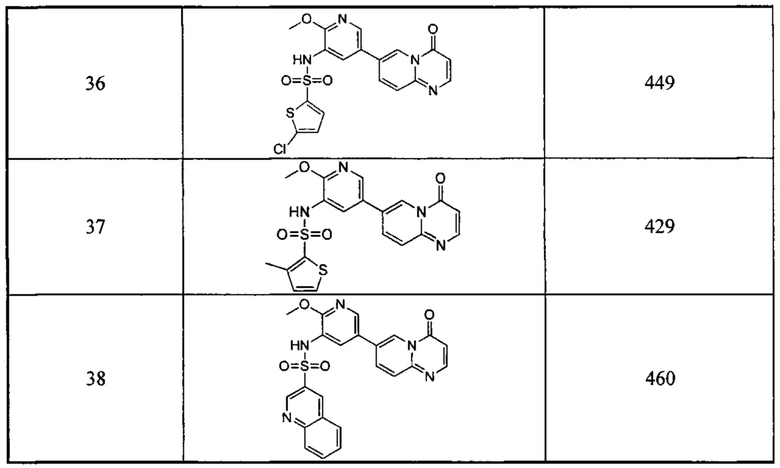
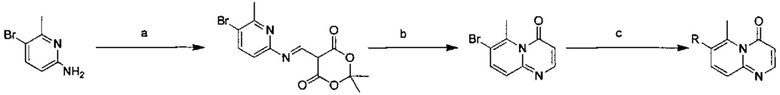
Схема 2:
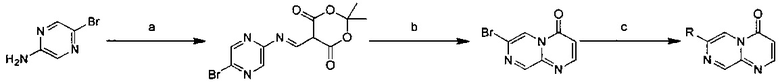
Условия реакции: а) триэтилортоформиат, 2,2-диметил-1,3-диоксан-4,6-дион, при нагревании; EtOH, при нагревании; b) дифениловый эфир, при нагревании с обратным холодильником; с) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид, и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 39
2,4-дифтор-N-(2-метокси-5-(4-оксо-4Н-пиразино[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
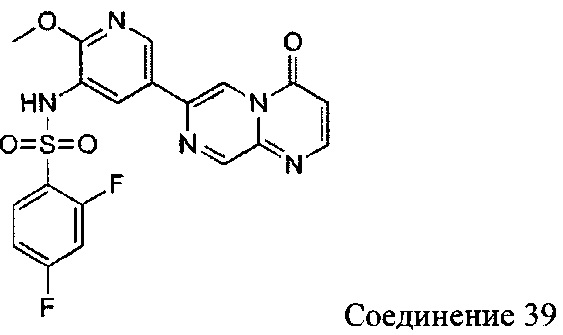
а) (Е)-5-(((5-бромпиримидин-2-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион
Триэтилортоформиат (9,9 г, 0,0689 моль) и 2,2-диметил-1,3-диоксан-4,6-дион (10,8 г, 0,073 моль) помещали в трехгорлую круглодонную колбу и проводили реакцию при перемешивании в течение 2 часов при 60°С. К этой смеси по каплям добавляли 5-бром-2-аминопиразин (12 г, 0,0689 моль) в этаноле (50 мл). Реакционный раствор перемешивали при 60°С в течение 2 часов. Смесь охлаждали до 25°С и фильтровали, и затем фильтрационный осадок промывали этанолом (200 мл × 3) с получением целевого соединения в виде белого твердого вещества (12,5 г, 55,3%).
1Н ЯМР (400 МГц, DMSO-D6) млн-1 δ 11,601 (s, 1Н), 9,039 (s, 1Н), 8,825 (s, 1Н), 8,712 (s, 1Н), 1,690 (s, 6Н).
b) 7-бром-4Н-пиразино[1,2-а]пиримидин-4-он
(Е)-5-(((5-бромпиримидин-2-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион (12 г, 0,0368 моль) и дифениловый эфир (50 мл) помещали в круглодонную колбу объемом 500 мл и проводили реакцию при перемешивании при 220°С в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде оранжевого твердого вещества (2 г, 24,4%).
1H ЯМР(400 МГц, DMSO-D6) млн-1 δ 8,944-8,919 (d, 2Н), 8,485-8,399 (s, 1Н), 6,687-6,672 (d, 1Н).
c) 2,4-дифтор-N-(2-метокси-5-(4-оксо-4Н-пиразино[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
7-бром-4Н-пиразино[1,2-а]пиримидин-4-он (0,22 ммоль) растворяли в диоксане (0,22 мл) и воде (0,44 мл) и добавляли 2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)бензсульфамид (0,22 ммоль), карбонат калия (0,56 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (20 мг) в атмосфере азота. Реакционную смесь подвергали воздействию микроволнового излучения при 100°С в течение 2 часов. Завершение реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и концентрировали оранжевую органическую фазу с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ с получением целевого соединения.
1Н ЯМР (400 МГц, CD3OD) млн-1 δ 9,169 (s, 1Н), 8,999 (s, 1Н), 8,473 (s, 1Н), 8,439-8,423 (d, 1Н), 8,197 (s, 1Н), 7,941-7,922 (d, 1Н), 7,145-7,098 (m, 1Н), 6,684-6,669 (d, 1Н), 3,884 (s, 3Н).
Следующие 12 соединений были также синтезированы согласно способу получения Соединения 39.

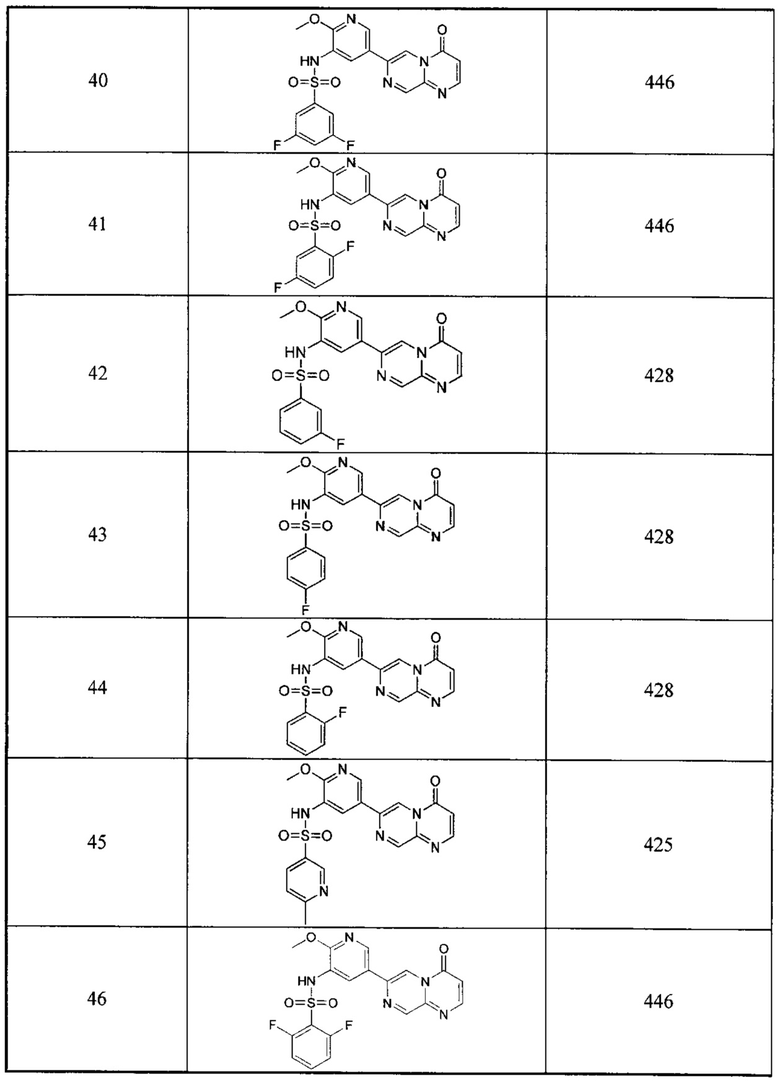
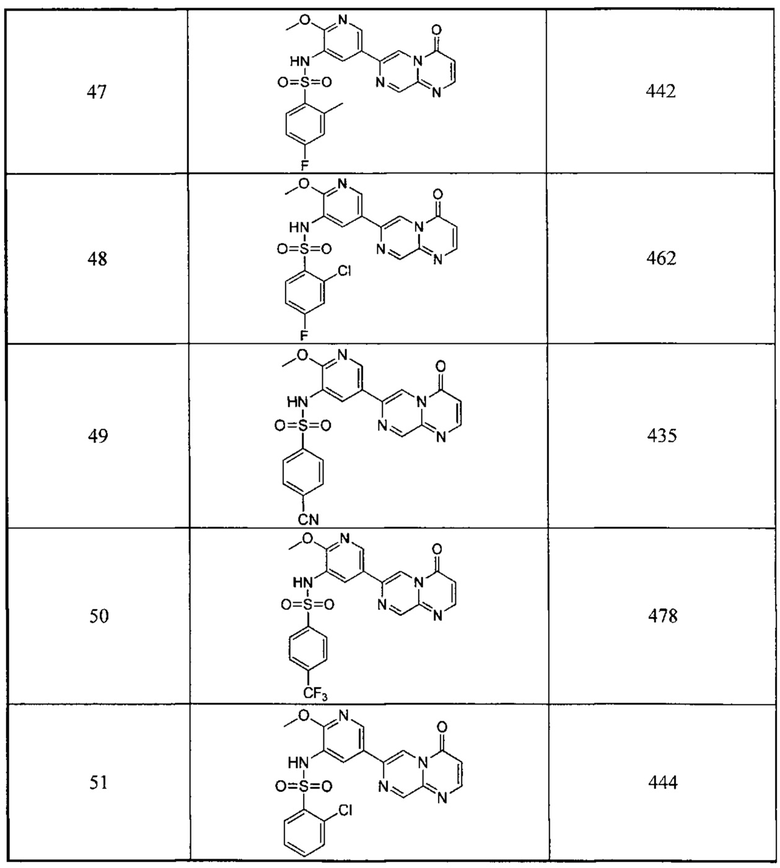
Схема 3:
Условия реакции: а) триэтилортоформиат, 2,2-диметил-1,3-диоксан-4,6-дион, при нагревании; этанол, при нагревании; b) дифениловый эфир, при нагревании с обратным холодильником; с) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 52
2,4-дифтор-N-(2-метокси-5-(6-метил-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
a) (Е)-5-(((5-бром-6-метилпиридин-2-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион
Трметилортоформиат (4,39 г, 0,03 ммоль) и 2,2-диметил-1,3-диоксан-4,6-дион (4,03 г, 0,028 ммоль) помещали в трехгорлую круглодонную колбу, оборудованную механической мешалкой. Полученную суспензию перемешивали при 60°С в течение 2 часов. К этой смеси по каплям добавляли 2-амино-5-бромпиразин (5 г, 0,027 ммоль) в этаноле (50 мл). Реакционный раствор перемешивали при 60°С в течение 2 часов и затем охлаждали до 25°С и фильтровали. Фильтрационный осадок промывали этанолом (200 мл × 3) с получением целевого соединения в виде белого твердого вещества (6 г, 65,6%).
1Н ЯМР (400 МГц, DMSO-D6) млн-1 δ 11,344-11,378 (d, 1Н), 9,143-9,177 (d, 1Н), 8,066-8,087 (d, 1Н), 7,457-7,479 (d, 1Н), 2,578 (s, 3Н), 1,678 (s, 6Н).
b) 7-бром-6-метил-4Н-пиридо[1,2-а]пиримидин-4-он
((Е)-5-(((5-бром-6-метилпиридин-2-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион (200 мг, 0,59 ммоль) и дифениловый эфир (4 мл) в круглодонной колбе объемом 50 мл перемешивали при 220°С под воздействием микроволнового излучения в течение 0,5 часа. Реакционный раствор охлаждали до комнатной температуры. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде оранжевого твердого вещества (60,7 мг, 43,2%).
1H ЯМР (400 МГц, CDCl3) млн-1 δ 8,075-8,090 (d, 1Н), 7,625-7,649 (d, 1Н), 7,246 (d, 1Н), 6,337-6,352 (d, 1Н), 3,026 (s, 3Н).
с) 2,4-дифтор-N-(2-метокси-5-(6-метил-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)пиридин-3-ил)бензсульфамид (0,28 ммоль), карбонат калия (0,5 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (20 мг) добавляли к раствору 7-бром-6-метил-4Н-пиридо[1,2-а]пиримидин-4-она (0,25 ммоль) в диоксане (0,2 мл) и воде (0,4 мл) в атмосфере азота. Реакционную смесь нагревали до 100°С под воздействием микроволнового излучения и перемешивали в течение 2 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ с получением целевого соединения.
1H ЯМР (400 МГц, CDCl3) млн-1 δ 8,120-8,135 (d, 1Н), 7,878-7,914 (m, 1Н), 7,865-7,870 (d, 1Н), 7,742-7,748 (d, 1Н), 7,441-7,464 (d, 1Н), 7,368-7,391 (d, 1Н), 6,947-6,986 (m, 2Н), 6,362-6,377 (d, 1Н), 3,995 (s, 3Н), 2,701 (s, 3Н).
Следующие 8 соединений были также синтезированы согласно способу получения Соединения 52.
Схема 4:
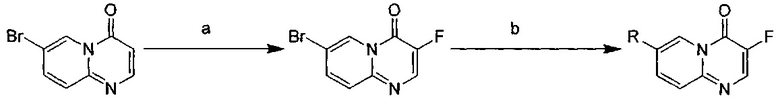
Условия реакции: а) реагент Selectflour, ацетонитрил, при нагревании; b) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин) палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 61
2,4-дифтор-N-(5-(3-фтор-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)бензсульфамид
а) 7-бром-3-фтор-4Н-пиридо[1,2-а]пиримидин-4-он
7-бром-4Н-пиридо[1,2-а]пиримидин-4-он (1 г, 4,6 ммоль), Selectflour (1,6 г, 4,46 ммоль) и ацетонитрил (15 мл) помещали в кругло донную колбу объемом 100 мл и перемешивали при 80°С в течение 2 дней. Реакционный раствор концентрировали и добавляли воду (15 мл). Смесь трижды экстрагировали дихлорметаном (20 мл). Органическую фазу концентрировали с получением неочищенного продукта, который очищали с помощью колоночной хроматографии с получением целевого соединения в виде желтого твердого вещества (200 мг, 18,5%).
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 9,195 (s, 1Н), 8,404 (s, 1Н), 7,763-7,739 (d, 1Н), 7,606-7,582 (d, 1Н).
b) 2,4-дифтор-N-(5-(3-фтор-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)бензсульфамид
2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)пиридин-3-ил)бензсульфамид (0,28 ммоль), карбонат калия (0,6 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (20 мг) добавляли к раствору 7-бром-3-фтор-4Н-пиридо[1,2-а]пиримидин-4-она (0,28 ммоль) в диоксане (0,2 мл) и воде (0,4 мл) в атмосфере азота. Реакционную смесь нагревали до 100°С под воздействием микроволнового излучения и перемешивали в течение 2 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и концентрировали органическую фазу с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ с получением целевого соединения.
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 10,438 (s, 1Н), 9,049 (s, 1Н), 8,636-8,628 (d, 1Н), 8,489 (s, 1Н), 8,282-8,259 (d, 1Н), 8,030 (s, 1Н), 7,872-7,848 (d, 1Н), 7,796-7,780 (d, 1Н), 7,611-7,562 (m, 1Н), 7,250-7,231 (m, 1Н), 3,691 (s, 3Н).
Следующие 9 соединений были также синтезированы согласно способу получения Соединения 61.
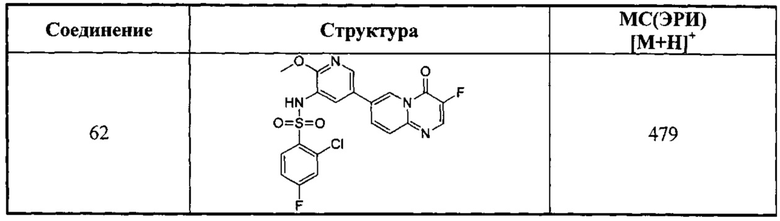
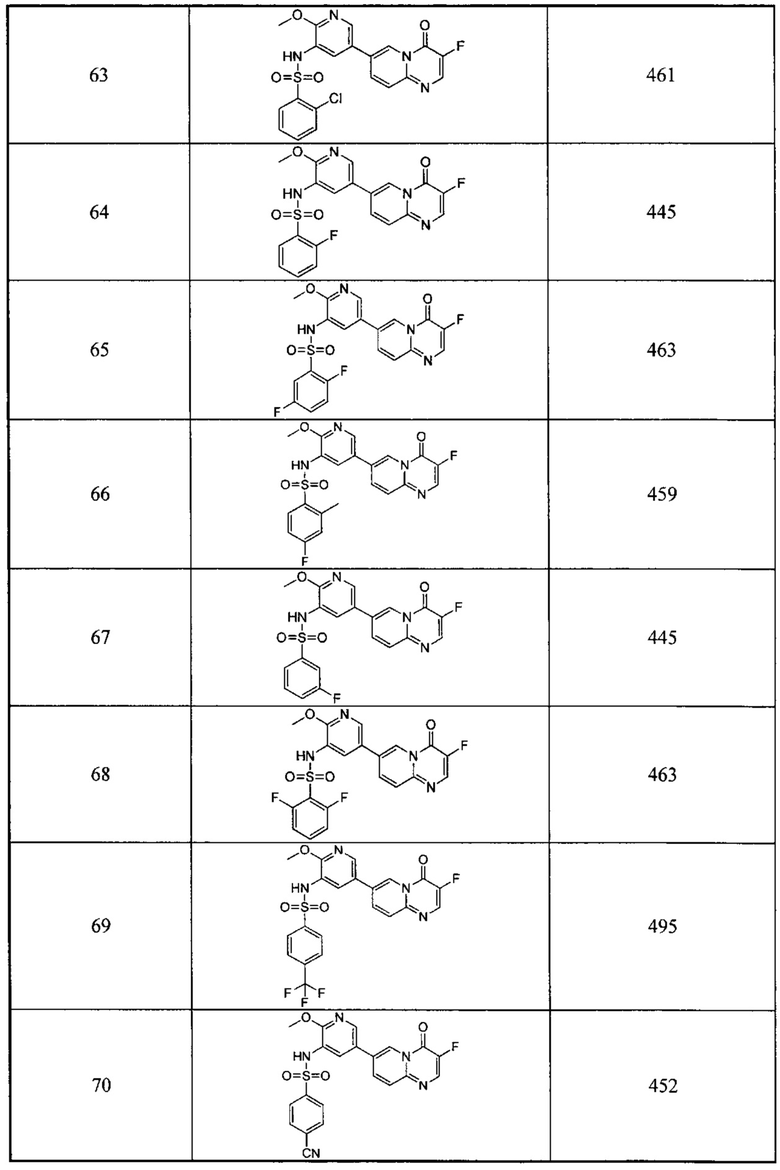
Схема 5:
Условия реакции: а) гидроксид аммония, хлорид аммония, при нагревании; b) триэтоксиметан, 2,2-диметил-1,3-диоксан-4,6-дион, при нагревании; этанол, при нагревании; с) дифениловый эфир, при нагревании с обратным холодильником; d) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 71
2,4-дифтор-N-(2-метокси-5-(4-оксо-4Н-пиримидо[1,2-b]пиридазин-7-ил)пиридин-3-ил)бензсульфамид
a) 6-хлор-пиридазин-3-амин
3,6-дихлорпиридазин (20 г, 0,134 моль) и раствор гидроксида аммония (140 мл), хлорид аммония (11,47 г, 0,214 моль) и воду (80 мл) добавляли в круглодонную колбу объемом 100 мл и затем перемешивали при 90°С в течение 20 часов. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Затем фильтрационный осадок промывали водой (100 мл) с получением продукта в виде белого твердого вещества (14,3 г, 82,7%).
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 7,365-7,361 (d, 1Н), 6,853-6,830 (d, 1Н), 6,614 (s, 1Н).
b) (Е)-5-(((6-хлорпиридазин-3-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион
Триэтоксиметан (16,3 г, 0,110 моль) и 2,2-диметил-1,3-диоксан-4,6-дион (14,5 г, 0,1 ммоль) помещали в круглодонную колбу объемом 3 л и перемешивали при 60°С в течение 2 часов. К реакционному раствору по каплям добавляли раствор 3-амино-6-хлорпиридазина (13 г, 100,3 ммоль) в этаноле (100 мл). Затем реакционный раствор перемешивали при 60°С в течение еще 2 часов. Реакционную смесь охлаждали до 25°С и фильтровали. Фильтрационный осадок промывали этанолом (50 мл × 3) с получением продукта в виде белого твердого вещества (16 г, 56%).
1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 11,521-11,484 (d, 1Н), 9,219-9,185 (d, 1Н), 8,100-7,984 (m, 2Н).
c) 7-хлор-4Н-пиримидо[1,2-b]пиридазин-4-он
(Е)-5-(((6-хлорпиридазин-3-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион (15 г, 52,9 ммоль) и дифениловый эфир (70 мл) добавляли в круглодонную колбу объемом 250 мл и перемешивали при 220°С в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением продукта в виде оранжевого твердого вещества (2,4 г, 25,3%).
1Н ЯМР (400 МГц, CD3OD) млн-1 δ 8,329-8,313 (d, 1Н), 8,003-8,979 (d, 1Н), 7,788-7,764 (d, 1Н), 6,713-6,696 (d, 1Н).
d) 2,4-дифтор-N-(2-метокси-5-(4-оксо-4Н-пиримидо[1,2-b]пиридазин-7-ил)пиридин-3-ил)бензсульфамид
2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)пиридин-3-ил)бензсульфамид (0,22 ммоль), карбонат калия (0,44 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (22 мг) последовательно добавляли к смешанному раствору 7-хлор-4Н-пиримидо[1,2-b]пиридазин-4-она (0,22 ммоль) в 1,4-диоксане (0,2 мл) и воде (0,4 мл) в атмосфере азота. Реакционный раствор нагревали до 100°С под воздействием микроволнового излучения и перемешивали в течение 2 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и концентрировали фильтрат с получением неочищенного продукта, который разделяли с помощью препаративной ВЭЖХ с получением продукта.
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 8,37-7,89 (m, 6Н), 7,30-7,02 (m, 2Н), 6,67-6,54 (m, 1Н), 3,85 (m, 3Н).
Следующие 9 соединений были также синтезированы согласно способу получения Соединения 71.

Схема 6:
Условия реакции: а) этил-2-метил-3-оксосукцинат, этанол, при нагревании; b) дифениловый эфир, при нагревании; с) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, 1,4-дикосан, вода, при нагревании.
Пример 81
2,4-дифтор-N-(2-метокси-5-(3-метил-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
а) 7-бром-3-метил-4-оксо-4Н-пиридо[1,2-а]пиримидин-2-карбоновая кислота
2-амино-5-бромпиридин (6 г, 0,035 моль), диэтил-2-метил-3-диоксоборан (7 г, 0,035 моль) и этанол (165 мл) добавляли в круглодонную колбу объемом 250 мл. Реакционный раствор перемешивали при 100°С в течение 30 часов. Реакционный раствор охлаждали до комнатной температуры и промывали твердое вещество холодным этанолом с получением продукта в виде белого твердого вещества (3 г, 30,6%).
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 8,936 (s, 1Н), 7,963-7,940 (d, 1Н), 7,577-7,553 (d, 1Н), 2,111 (s, 3H).
b) 7-бром-3-метил-4Н-пиридо[1,2-а]пиримидин-4-он
Смешанный раствор 7-бром-3-метил-4-он-4Н-пиридо[1,2-а]пиримидин-2-карбоновой кислоты (1,5 г, 5,2 ммоль) и дифенилового эфира (20 мл) перемешивали при 220°С в течение 1,5 часов. Реакционный раствор охлаждали до комнатной температуры. Неочищенный продукт разделяли с помощью колоночной хроматографии с получением продукта в виде оранжевого твердого вещества (550 мг, 44%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 9,174 (s, 1Н), 8,233 (s, 1Н), 7,688-7,665 (d, 1Н), 7,505-7,482 2,279 (s, 1Н).
с) 2,4-дифтор-N-(2-метокси-5-(3-метил-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)пиридин-3-ил)бензсульфамид (0,22 ммоль), карбонат калия (0,44 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (22 мг) последовательно добавляли к смешанному раствору 7-бром-3-метил-4Н-пиридо[1,2-а]пиримидин-4-она (0,22 ммоль) в 1,4-диоксане (2 мл) и воде (0,4 мл) в атмосфере азота. Реакционный раствор перемешивали при 90°С в течение 1 часа под воздействием микроволнового излучения. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и сушили фильтрат посредством вращательного испарения с получением неочищенного продукта, который разделяли с помощью препаративной высокоэффективной жидкостной хроматографии с получением продукта в виде белого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 8,935 (s, 1Н), 8,274 (s, 1Н), 8,150 (s, 1Н) 8,096-8,074 (d, 1Н), 7,797-7,761 (d, 2Н), 7,683-7,660 (d, 1Н), 7,392 (s, 1Н), 7,157-7,115 (d, 2Н), 3,676, 2,127 (s, 3H).
Следующие 5 соединений были также синтезированы согласно способу получения Соединения 81.

Схема 7:

Условия реакции: а) концентрированная серная кислота, азотная кислота; b) железные опилки, хлорид аммония, при нагревании; с) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен] палладий хлорид и т.д.), карбонат калия, 1,4-диоксан, вода, при нагревании.
Пример 87
N-(5-(3-амино-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)-2,4-дифторбензсульфамид
a) 7-бром-3-нитро-4Н-пиридо[1,2-а]пиримидин-4-он
7-бром-4Н-пиридо[1,2-а]пиримидин-4-он (10 г, 0,045 моль) и концентрированную серную кислоту (50 мл) добавляли в трехгорлую колбу и медленно по каплям добавляли азотную кислоту (8,65 г, 98%) при 0°С. Смесь перемешивали при 0°С в течение одного часа. Затем реакционный раствор выливали в воду (200 мл) и добавляли гидроксид натрия для доведения рН до 9. Водную фазу экстрагировали этилацетатом (200 мл × 3) и органические фазы объединяли, сушили и концентрировали с получением неочищенного продукта. Неочищенный продукт разделяли с помощью колоночной хроматографии на силикагеле с получением продукта в виде белого твердого вещества (1,3 г, 10,8%).
1H ЯМР (400 МГц, CDCl3) млн-1 δ 9,489-9,485 (d, 1Н), 9,368 (s, 1Н), 8,178-8,150 (m, 1Н), 7,843-7,820 (d, 1Н).
b) 3-амино-7-бром-4Н-пиридо[1,2-а]пиримидин-4-он
Хлорид аммония (1,2 г, 0,019 моль) и железные опилки (1,0 г) добавляли к смешанному раствору 7-бром-3-нитро-4Н-пиридо[1,2-а]пиримидин-4-она (1 г, 0,0037 моль) в этаноле (10 мл) и воде (2 мл) и перемешивали реакционную смесь при 70°С в течение 4 часов. Реакционную смесь фильтровали, фильтрационный осадок промывали этилацетатом (30 мл × 3) и концентрировали фильтрат с получением неочищенного продукта. Неочищенный продукт растворяли в этилацетате (50 мл) и промывали водой (20 мл) и затем органическую фазу концентрировали с получением продукта в виде коричневого твердого вещества (0,8 г, 89,9%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 9,013 (s, 1Н), 7,974 (s, 1Н), 7,395 (s, 2Н), 4,235 (s, 2Н).
c) N-(5-(3-амино-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)-2,4-дифторбензсульфамид
2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)пиридин-3-ил)бензсульфамид (0,22 ммоль), карбонат калия (0,44 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (22 мг) добавляли к смешанному раствору 3-амино-7-бром-4Н-пиридо[1,2-а]пиримидин-4-она (0.22 ммоль) в 1,4-диоксане (2 мл) и воде (0,4 мл) в атмосфере азота. Реакционный раствор перемешивали при 90°С в течение 1 часа под воздействием микроволнового излучения. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и фильтрат концентрировали с получением неочищенного продукта, который разделяли с помощью препаративной ВЭЖХ с получением продукта в виде белого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 8,763 (s, 1Н), 8,013-7,687 (m, 6Н), 7,549-7,526 (d, 1Н), 7,372-7,175 (m, 1Н), 5,284 (s, 2Н), 3,758 (s, 3Н).
Следующие 7 соединений были также синтезированы согласно способу получения Соединения 87.
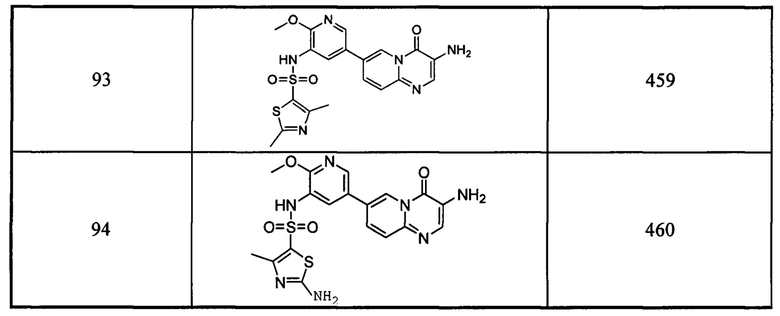
Схема 8:
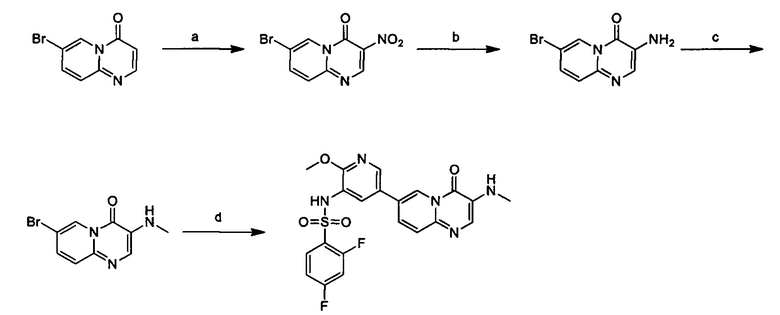
Условия: а) азотная кислота, концентрированная серная кислота; b) хлорид аммония, железные опилки, при нагревании; с) карбонат калия, йодистый метил, при нагревании; d) под воздействием микроволнового излучения, палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 95
2,4-дифтор-N-(2-метокси-5-(3-(метиламино)-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
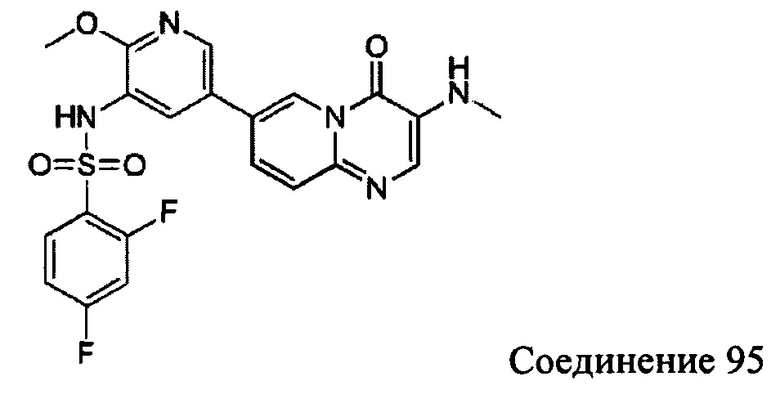
а) 7-бром-3-нитро-4Н-пиридо[1,2-а]пиримидин-4-он
7-бром-4Н-пиридо[1,2-а]пиримидин-4-он (5 г, 22,2 ммоль) растворяли в концентрированной серной кислоте (11,2 мл) и помещали в трехгорлую кругло донную колбу. По каплям добавляли азотную кислоту (5,2 мл) при температуре от 5 до 10°С. Реакционную смесь перемешивали при 20°С в течение 3 часов и затем медленно выливали в ледяную воду. Добавляли 1 экв. водного раствора гидроксида натрия для доведения рН до 8. Реакционную смесь фильтровали и фильтрационный осадок промывали водой и сушили с получением целевого соединения в виде желтого твердого вещества (4,0 г, 66,7%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 9,47 (d, 1Н), 9,35 (s, 1H), 8,14 (dd, 1H), 7,81 (d, 1H).
b) 3-амино-7-бром-4Н-пиридо[1,2-а]пиримидин-4-он
7-бром-3-нитро-4Н-пиридо[1,2-а]пиримидин-4-он (1,6 г, 5,93 ммоль) растворяли в этаноле (20 мл) и воде (4 мл) и добавляли хлорид аммония (3,17 г, 59,25 ммоль) и железные опилки (3,17 г, 59,25 ммоль). Смесь перемешивали при 70°С в течение 16 часов. Реакционную смесь фильтровали, фильтрационный осадок промывали дихлорметаном, органическую фазу полученного фильтрата промывали насыщенным солевым раствором (50 мл), сушили над сульфатом натрия и концентрировали с получением неочищенного целевого соединения (3,56 г).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 8,99 (s, 1H), 7,96 (s, 1H), 7,38 (s, 2Н), 4,13 (br. s., 2Н).
c) 7-бром-3-(метиламино)-4Н-пиридо[1,2-а]пиримидин-4-он
3-амино-7-бром-4Н-пиридо[1,2-а]пиримидин-4-он (0,8 г, 3,33 ммоль) растворяли в ацетоне (30 мл) и добавляли карбонат калия (1,38 г, 10,0 ммоль) и йодистый метил (7,1 г, 49,99 ммоль). Смесь перемешивали при 80°С в течение 3 часов в атмосфере азота. Реакционный раствор фильтровали и промывали фильтрационный осадок дихлорметаном. Фильтрат концентрировали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения (250 мг, 29,5%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 8,91 (d, 1Н), 7,66 (s, 1Н), 7,36-7,32 (m, 1H), 7,28 (d, 1Н), 4,72 (br. s., 1H), 2,97 (d, 3Н).
d) 2,4-дифтор-N-(2-метокси-5-(3-(метиламино)-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
7-бром-3-(метиламино)-4Н-пиридо[1,2-а]пиримидин-4-он (100 мг, 0,39 ммоль) растворяли в диоксане (2 мл) и воде (0,4 мл) и добавляли 2,4-дихлор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил) бензсульфамид (168 мг, 0,39 ммоль), карбонат калия (109 мг, 0,78 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (16 мг, 0,02 ммоль) в атмосфере азота. Смесь реагировала при 100°С под воздействием микроволнового излучения в течение 1 часа. Окончание реакции определяли с помощью ЖХ/МС. Реакционный раствор фильтровали и концентрировали органическую фазу с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением желтого целевого продукта.
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 8,84 (br. s., 1Н), 8,12 (br. s., 1Н), 7,97-7,87 (m, 2Н), 7,70 (s, 1H), 7,55 (d, 1H), 7,39 (d, 1H), 7,04-6,89 (m, 2Н), 4,70 (br. s., 1Н), 3,97 (s, 3Н), 2,99 (d, 3Н).
Следующие 5 соединений были также синтезированы согласно способу получения Соединения 95.
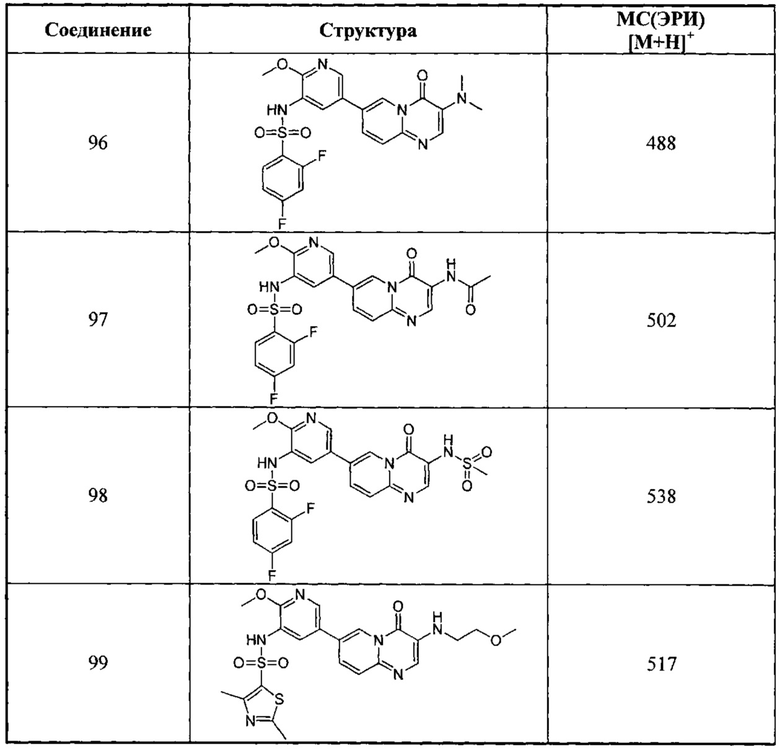
Схема 9:
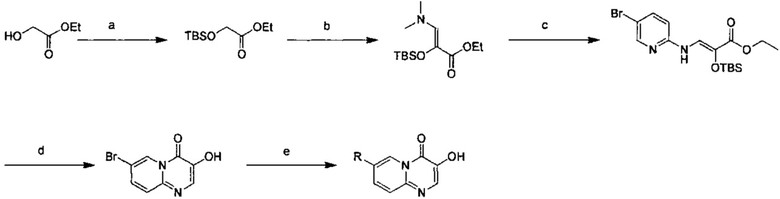
Условия реакции: а) трет-бутилдиметилсилил хлорид, 1Н-имидазол; b) 1-t-бутокси-N,N,N',N'-тетраметилдиаминометан, при нагревании; с) 2-амино-5-бромпиридин, уксусная кислота, при нагревании; d) уксусная кислота, под воздействием микроволнового излучения; е) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 101
2,4-дифтор-N-(5-(3-гидрокси-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)бензсульфамид
а) этил-2-((трет-бутилдиметилсилил)окси)ацетат
Этилгликолят (10 г, 96,1 ммоль) и 1Н-имидазол (13 г, 0,19 моль) растворяли в дихлорметане (100 мл) и помещали в трехгорлую круглодонную колбу. При 0°С добавляли трет-бутилдиметилсилил хлорид (15,8 г, 0,1 моль) и перемешивали смесь при комнатной температуре в течение 8 часов, затем промывали водой (100 мл × 3), сушили над сульфатом натрия и концентрировали с получением целевого соединения в виде желтого масла (18 г, 85,8%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 4,14-4,09 (m, 4Н), 1,20-1,16 (t, 3Н), 0,83 (s, 9Н), 0,01 (s, 6Н).
b) (Z)-этил-2-((трет-бутилдиметилсилил)окси)-3-(диметиламино)акрилат
Этил-2-((трет-бутилдиметилсилил)окси)ацетат (52 г, 0,24 моль) и 1-трет-бутокси-N,N,N',N'-тетраметилдиаминометан (50 г, 0,58 моль) перемешивали при нагревании с обратным холодильником в течение 24 часов. Смесь концентрировали и очищали остаток с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде желтого масла (45 г, 47,1%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 6,68 (s, 1Н), 4,13-4,11 (q, 2Н), 2,96 (s, 6Н), 1,28-1,24 (t, 3Н), 0,95 (s, 9H), 0,14 (s, 6Н).
c) (Z)-этил-3-((5бpoмпиpидин-2-ил)aминo)-2-((тpeт-бyтилдимeтилcилил)oкcи)aкpилaт
(Z)-этил-3-((5-бромпиридин-2-ил)амино)-2-((трет-бутилдиметилсилил)окси)акрилат (15 г, 54,9 ммоль) и 2-амино-5-бромпиридин (9,4 г, 54,9 ммоль) растворяли в уксусной кислоте (150 мл) и перемешивали при 80°С в течение 2 часов. Смесь концентрировали. Затем остаток растворяли в этилацетате (100 мл), промывали раствором карбоната натрия (100 мл) и насыщенным солевым раствором (100 мл), сушили над сульфатом натрия и концентрировали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде желтого масла (14 г, 63,7%). 1Н ЯМР (400 МГц, CDCl3) млн-1 δ 8,24 (s, 1Н), 7,75-7,72 (d, 1Н), 7,63-7,60 (d, 1Н), 6,75-6,72 (d, 1Н), 6,57-6,54 (d, 1Н), 4,25-4,20 (q, 2Н), 1,34-1,30 (t, 3Н), 1,02 (s, 9Н), 0,22 (s, 6Н).
d) 7-бром-3-гидрокси-4Н-пиридо[1,2-а]пиримидин-4-он
(Z)-этил-3-((5-бромпиридин-2-ил)амино)-2-((трет-бутидиметилсилил)окси)акрилат (200 мг × 50, 29 ммоль) растворяли в уксусной кислоте (5 мл × 50) и перемешивали при 140°С под воздействием микроволнового излучения в течение 3 часов. Смесь концентрировали. Остаток растворяли в этилацетате (100 мл), промывали раствором карбоната натрия (100 мл) и насыщенным солевым раствором (100 мл), сушили над сульфатом натрия и концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле с получением целевого соединения (3,2 г, 46,4%). 1Н ЯМР (400 МГц, CDCl3) млн-1 δ 8,98 (s, 1Н), 8,14 (s, 1Н), 8,00-7,98 (d, 1Н), 7,79-7,77 (d, 1Н).
е) 2,4-дифтор-N-(5-(3-гидрокси-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)бензсульфамид
7-бром-3-гидрокси-4Н-пиридо[1,2-а]пиримидин-4-он (0,22 ммоль) растворяли в диоксане (2 мл) и воде (0,4 мл), добавляли 2,4-дихлор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)бензсульфамид (0,22 ммоль), карбонат калия (0,44 ммоль) и 1,1'-бис(дифенилфосфино)ферроценпалладий хлорид (22 мг) в атмосфере азота. Смесь реагировала при 90°С под воздействием микроволнового излучения в течение 1 часа. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и концентрировали органическую фазу с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением белого целевого продукта.
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 8,93 (s, 1Н), 8,41 (s, 1Н), 8,08 (s, 1Н), 7,96 (s, 2Н), 7,79-7,78 (m, 1Н), 7,68-7,66 (m, 1Н), 7,56 (m, 1Н), 7,24-7,20 (m, 1Н), 3,68 (s, 3Н).
Следующее соединение было также синтезировано согласно способу получения Соединения 101.
Схема 10:
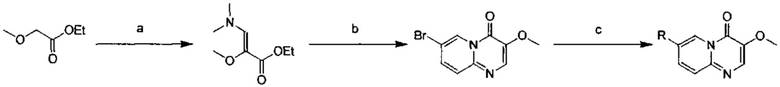
Условия реакции: а) 1-трет-бутокси-N,N,N',N'-тетраметилдиаминометан, при нагревании; b) 2-амино-5-бромпиридин, уксусная кислота, при нагревании; с) R борная кислота (борат), палладиевый реагент (тетракистрифенилфосфин палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид, и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 103
2,4-дифтор-N-(2-метокси-5-(3-метокси-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
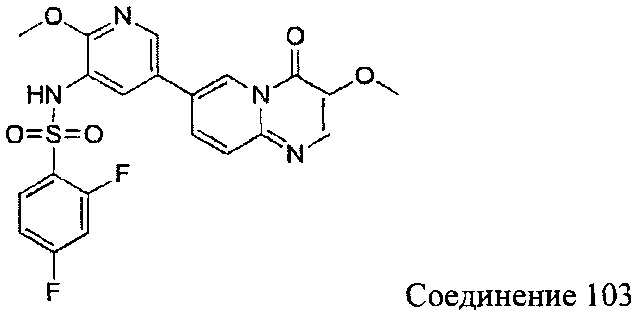
a) (Z)-этил-3-(диметиламино)-2-метоксиакрилат
Этил-2-метоксиацетат (2 г, 16,9 ммоль) и 1-трет-бутокси-N,N,N',N'-тетраметилдиаминометан (3,5 г, 20,1 ммоль) помещали в круглодонную колбу и перемешивали при нагревании с обратным холодильником в течение ночи. Смесь концентрировали и очищали остаток с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде желтого масла (2 г, 67,8%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 6,78 (s, 1Н), 4,18-4,16 (t, 2Н), 3,55 (s, 3Н), 3,02 (s, 6Н), 1,29-1,26 (q, 3H).
b) 7-бром-3-метокси-4Н-пиридо[1,2-а]пиримидин-4-он
(Z)-этил-3-(диметиламино)-2-метоксиаркилат (2,5 г, 14,4 ммоль) и 2-амино-5-бромпиридин (2,5 г, 14,4 ммоль) растворяли в уксусной кислоте (25 мл) и перемешивали при 80°С в течение 2 часов. Смесь концентрировали. Остаток растворяли в этил ацетате (30 мл), промывали раствором карбоната натрия (50 мл) и насыщенным солевым раствором (30 мл), сушили над сульфатом натрия, концентрировали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения (1,3 г, 35,1%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 9,10 (s, 1Н), 8,05 (s, 1Н), 7,52 (d, 1Н), 7,46 (d, 1Н), 4,00 (s, 3 Н).
с) 2,4-дифтор-N-(2-метокси-5-(3-метокси-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
7-бром-3-метокси-4Н-пиридо[1,2-а]пиримидин-4-он (0,27 ммоль) растворяли в диоксане (3,5 мл) и воде (0,7 мл) и добавляли 2,4-дихлор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)бензсульфамид (0,33 ммоль), карбонат калия (0,41 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (20 мг) в атмосфере азота. Смесь реагировала при 100°С под воздействием микроволнового излучения в течение 2 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и концентрировали органическую фазу с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого продукта.
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 8,99 (s, 1Н), 8,12 (s, 1Н), 8,09 (s, 1Н), 7,97-7,93 (m, 2Н), 7,66 (s, 2Н), 7,08-7,04 (q, 1Н), 6,97-6,93 (q, 1Н), 4,02-3,98 (d, 6Н).
Следующие 8 соединений были также синтезированы согласно способу получения Соединения 103.
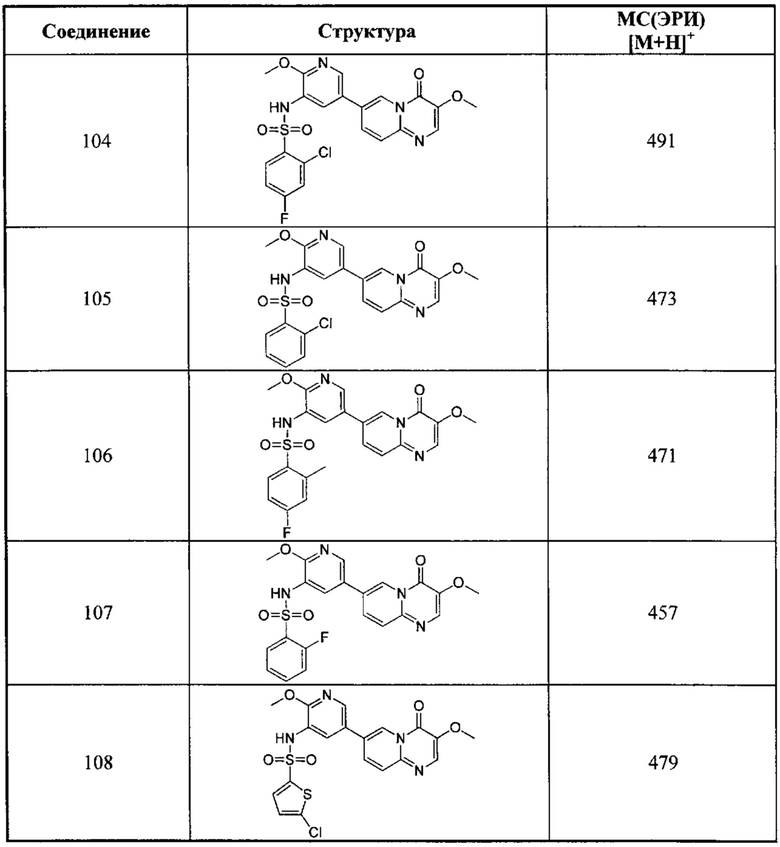
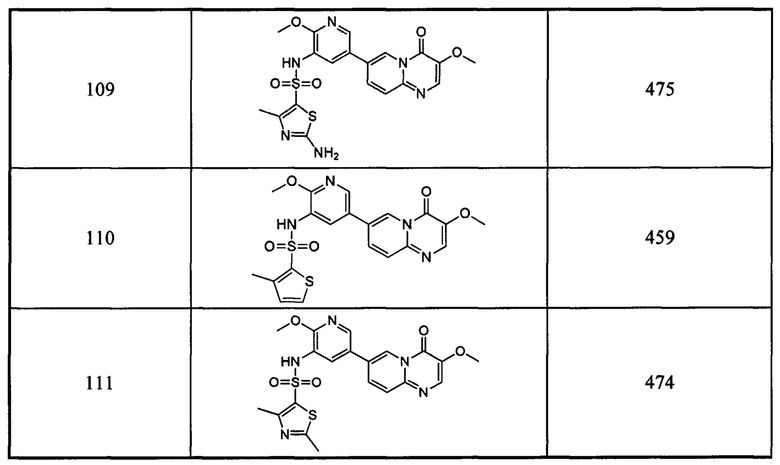
Схема 11:
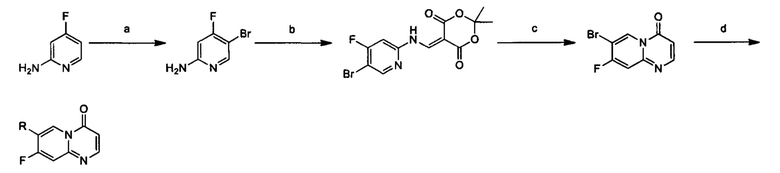
Условия реакции: a) NBS, MeCN; 2) триэтилортоформиат, 2,2-диметил-1,3-диоксан-4,6-дион, при нагревании; ETOН, при нагревании; с) дифениловый эфир, при нагревании с обратным холодильником; d) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 112
2,4-дифтор-N-(5-(8-фтор-4-оксо-4Н-пиридо[1,2-а])пиримидин-7-ил)-2-метоксипиридин-3-ил)бензсульфамид
a) 5-бром-4-фторпиридин-2-амин
NBS (28,6 г, 0,16 моль) порциями добавляли к раствору 4-фторпиридин-2-амино-2,2,2-трифторацетата (18 г, 0,16 моль) в ацетонитриле (200 мл). Реакционный раствор перемешивали при 25°С в темноте в течение 4 часов. Растворитель удаляли при пониженном давлении и неочищенный продукт очищали с помощью колоночной флэш-хроматографии на силикагеле с получением целевого соединения в виде белого твердого вещества (15 г, 49%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 8,155-8,131 (d, 1Н), 6,301-6,276 (d, 1Н), 4,638 (s, 2Н)
b) (Е)-5-(((5-бром-4-фторпиридин-2-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион
Триэтилортоформиат (7,3 г, 0,05 моль) и 2,2-диметил-1,3-диоксан-4,6-дион (7,5 г, 0,05 моль) добавляли в трехгорлую круглодонную колбу, оборудованную мешалкой. Суспензию перемешивали при 70°С в течение 1 часа. К смеси по каплям добавляли раствор 5-бромпиридин-2-амина (8 г, 0,042 моль) в этаноле (100 мл). Реакционный раствор перемешивали при 70°С в течение 0,5 часа и охлаждали до 25°С и фильтровали. Фильтрационный осадок промывали этанолом (100 мл × 3) с получением целевого соединения в виде белого твердого вещества (11,6 г, 80%).
1H ЯМР (400 МГц, DMSO-d6) млн-1 δ 11,477-11,442 (d, 1Н), 9,190-9,156 (d, 1Н), 8,728-8,705 (d, 1Н), 7,854-7,830 (d, 1Н), 1,694 (s, 6Н).
c) 7-бром-8-фтор-4Н-пиридо[1,2-а]пиримидин-4-он
(Е)-5-(((5-бром-4-фторпиридин-2-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион (11,6 г, 0,034 моль) и дифениловый эфир (50 мл) помещали в круглодонную колбу объемом 100 мл, оборудованную мешалкой, и оставляли при 220°С на 1 час. Окончание реакции определяли с помощью ТСХ. Реакционный раствор охлаждали до 100°С и затем выливали в петролейный эфир (100 мл). Добавляли смесь соляной кислоты и этилацетата (50 мл) и фильтровали смесь с получением твердого вещества. Твердое вещество растворяли в метаноле (50 мл) и добавляли насыщенный раствор NaНСО3 для доведения рН до 7. Смесь концентрировали при пониженном давлении, затем добавляли воду (50 мл). Смесь экстрагировали дихлорметаном (100 мл × 2). Органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения (4 г, 50%).
1H ЯМР (400 МГц, CDCl3) млн-1 δ 9,335-9,317 (d, 1Н), 8,272-8,256 (d, 1Н), 7,371-7,350 (d, 1Н), 6,461-6,445 (d, 1Н).
d) 2,4-дифтор-N-(5-(8-фтор-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-7-ил)-2-метоксипиридин-3-ил)бензсульфамид
7-бром-8-фтор-4Н-пиридо[1,2-а]пиримидин-4-он (0.28 ммоль) растворяли в диоксане (2 мл) и воде (0,4 мл) и добавляли 2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)бензсульфамид (0.28 м моль), карбонат калия (0,56 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (20 мг) в атмосфере азота. Реакционную смесь подвергали воздействию микроволнового излучения при 100°С в течение 2 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и концентрировали с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ с получением целевого соединения.
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 10,44 (s, 1Н), 9,03-8,96 (m, 1Н), 8,30 (br. s., 2Н), 7,93-7,85 (m, 1Н), 7,83-7,71 (m, 2Н), 7,65-7,55 (m, 1Н), 7,30-7,21 (m, 1Н), 6,46-6,40 (m, 1Н), 3,71 (s, 3Н).
Следующие 7 соединений были также синтезированы согласно способу получения Соединения 112.
Схема 12:
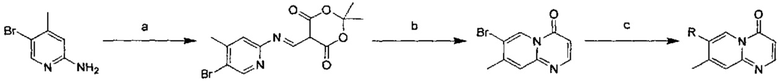
Условия реакции: а) триэтилортоформиат, 2,2-диметил-1,3-диоксан-4,6-дион, при нагревании; EtOH, при нагревании; b) дифениловый эфир, при нагревании с обратным холодильником; с) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 121
2,4-дифтор-N-(2-метокси-5-(8-метил-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
a) (Е)-5-(((5-бром-4-метилпиридин-2-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион
Триэтилортоформиат (1,75 г, 0,01 моль) и 2,2-диметил-1,3-диоксан-4,6-дион (1,61 г, 0,014 моль) помещали в трехгорлую круглодонную колбу, оборудованную мешалкой, и при перемешивании проводили реакцию в течение 2 часов при 60°С. К вышеуказанной смеси по каплям добавляли раствор 5-бромпиридин-2-амина (2 г, 0,017 моль) в этаноле (20 мл). Реакционный раствор перемешивали при 60°С в течение 2 часов. Реакционный раствор охлаждали до 25°С и фильтровали, и фильтрационный осадок промывали этанолом (20 мл × 3) с получением целевого соединения в виде белого твердого вещества (2,1 г, 61,76%).
1H ЯМР (400 МГц, CDCl3) млн-1 δ 9,342-9,308 (d, 1Н), 8,420 (s, 1Н), 6,946 (s, 1Н).
b) 7-бром-8-метил-4Н-пиридо[1,2-а]пиримидин-4-он
(Е)-5-(((5-бром-4-фторпиридин-2-ил)имино)метил)-2,2-диметил-1,3-диоксан-4,6-дион (1,2 г, 0,0035 моль) и дифениловый эфир (18 мл) помещали в круглодонную колбу объемом 100 мл и перемешивали при 220°С в течение 1 часа. Окончание реакции определяли с помощью ТСХ. Реакционный раствор охлаждали до 100°С и выливали в петролейный эфир (20 мл). Добавляли смешанный раствор соляной кислоты и этилацетата (20 мл) и смесь фильтровали с получением твердого вещества. Твердое вещество растворяли в метаноле (20 мл) и добавляли насыщенный раствор NaHСO3 для доведения рН до 7. Смесь концентрировали и добавляли воду (20 мл). Смесь экстрагировали дихлорметаном (20 мл × 2). Органическую фазу сушили над Na2SO4, концентрировали при пониженном давлении и очищали неочищенный продукт с помощью колоночной хроматографии на силикагеле с получением целевого соединения (700 мг, 83,3%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 9,236 (s, 1Н), 8,277-8,262 (d, 1Н), 7,527 (s, 1Н), 6,421-6,406 (s, 1Н), 2,550 (s, 3Н).
с) 2,4-дифтор-N-(2-метокси-5-(8-метил-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
7-бром-8-метил-4Н-пиридо[1,2-а]пиримидин-4-он (0,28 ммоль) растворяли в диоксане (2 мл) и воде (0,44 мл) и добавляли 2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил) бензсульфамид (0,28 ммоль), карбонат калия (0,56 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (20 мг) в атмосфере азота. Реакционную смесь подвергали воздействию микроволнового излучения при 100°С в течение 2 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали, фильтрат концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения.
Следующие 10 соединений были также синтезированы согласно способу получения Соединения 121.
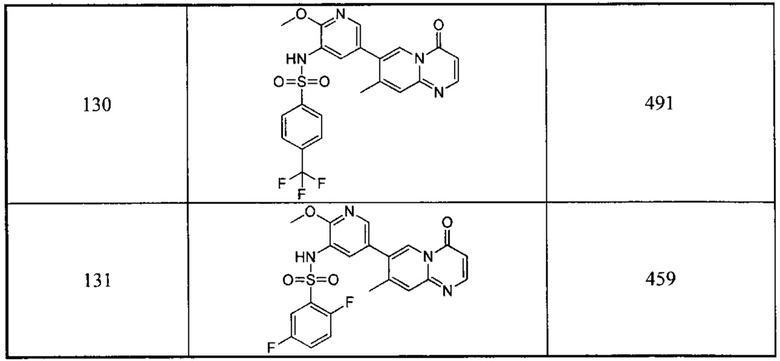
Схема 13:
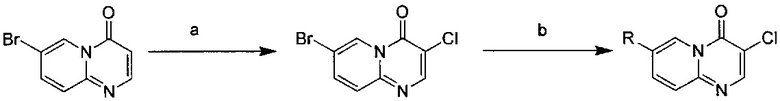
Условия реакции: а) 1-хлорпирролидин-2,5-дион, N,N-диметилформамид; b) R борная кислота (борат), карбонат калия, палладиевый катализатор (тетракис(трифенилфосфин)палладий и т.д.), диоксан, вода, при нагревании.
Пример 132
N-(5-(3-хлор-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)-2,4-дифторбензсульфамид
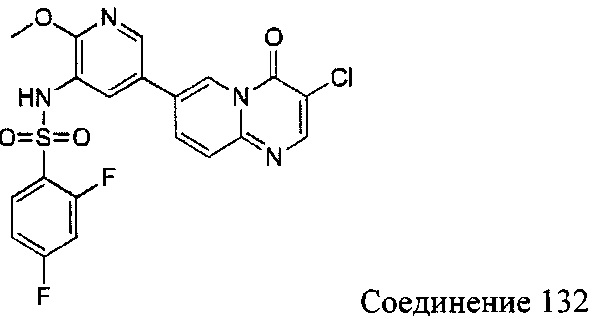
а) 7-бром-3-хлор-4Н-пиридо [1,2-а]пиримидин-4-он
1-хлор-пирролидин-2,5-дион (500 мг, 3,75 ммоль) добавляли к раствору 7-бром-4Н-пиридо[1,2-а]пиримидин-4-она (800 мг, 3,57 ммоль) в N,N-диметилформамиде (10 мл). Реакционную смесь перемешивали при 25°С в течение 14 часов. Реакционный раствор выливали в воду (10 мл) и трижды экстрагировали дихлорметаном (15 мл). Полученную органическую фазу дихлорметана концентрировали с получением неочищенного продукта, который разделяли на колонке с силикагелем с получением беловатого твердого вещества (600 мг, 65%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 9,228-9,224 (d, 1Н), 8,496 (s, 1Н), 7,831-7,801 (m, 1Н), 7,616-7,581 (m, 1Н).
b) N-(5-(3-хлор-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)-2,4-дифторбензсульфамид
2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)бензсульфамид (0,28 ммоль), карбонат калия (0,56 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (20 мг) добавляли к раствору 7-бром-8-метил-4Н-пиридо[1,2-а]пиримидин-4-она (0,28 ммоль) в диоксане (2 мл) и воде (0,4 мл) в атмосфере азота. Реакционный раствор подвергали воздействию микроволнового излучения при 100°С в течение двух часов, за ходом реакции следили при помощи жидкостной МС. После завершения реакции реакционный раствор фильтровали и концентрировали фильтрат с получением неочищенного продукта, который выделяли с помощью препаративной колоночной ЖХ с получением целевого соединения.
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 10,415 (s, 1Н), 9,087-9,084 (d, 2Н), 8,618 (s, 1Н), 8,487-8,481 (d, 1Н), 8,361-8,357 (d, 1Н), 8,338 (s, 1Н), 8,028-8,022 (s, 1Н), 7,883-7,860 (d, 1Н), 7,792-7,775 (d, 1Н), 7,597-7,575 (d, 1Н), 7,242-7,226 (d, 1Н), 3,687 (s, 3Н).
Следующие 37 соединений были также синтезированы согласно способу получения Соединения 132.
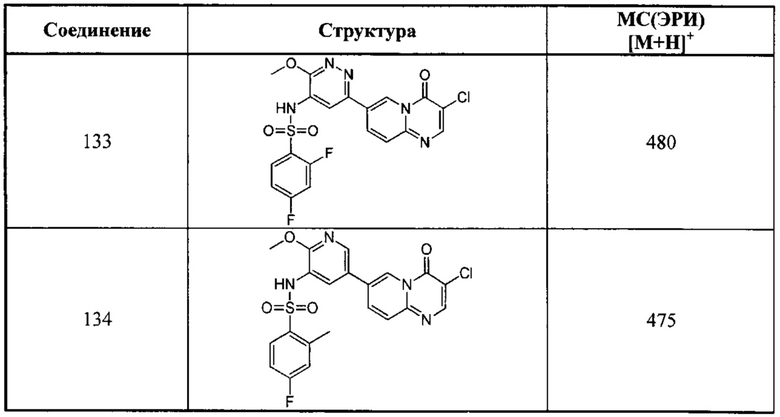
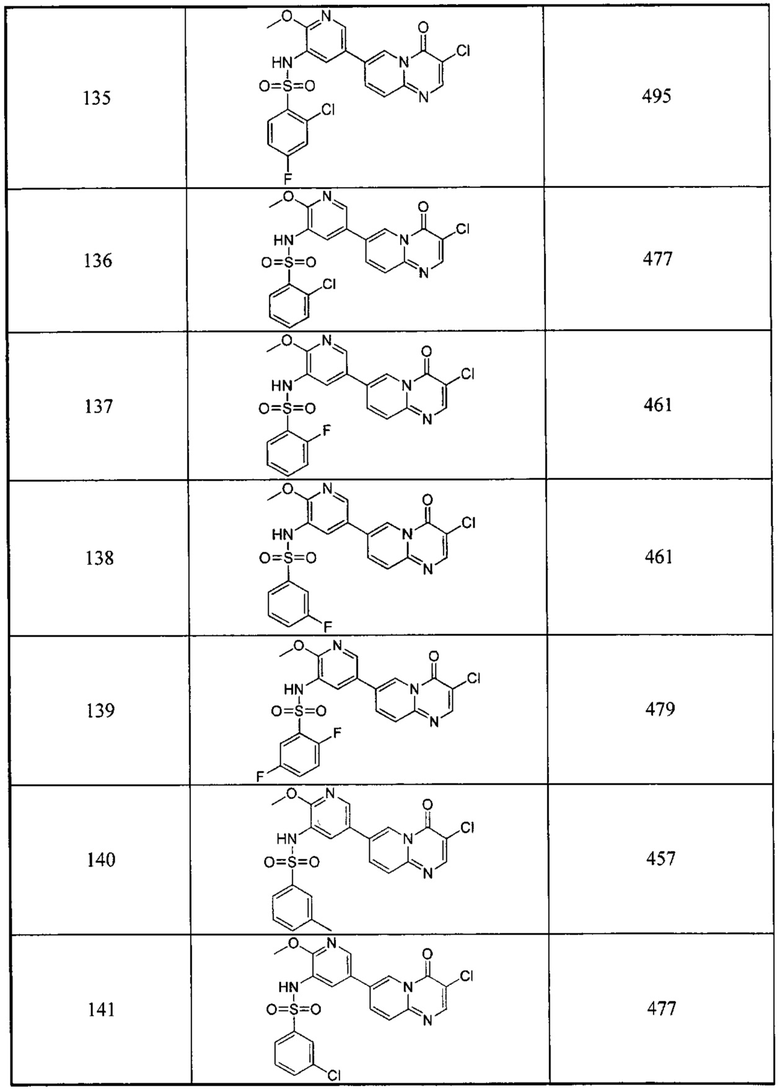
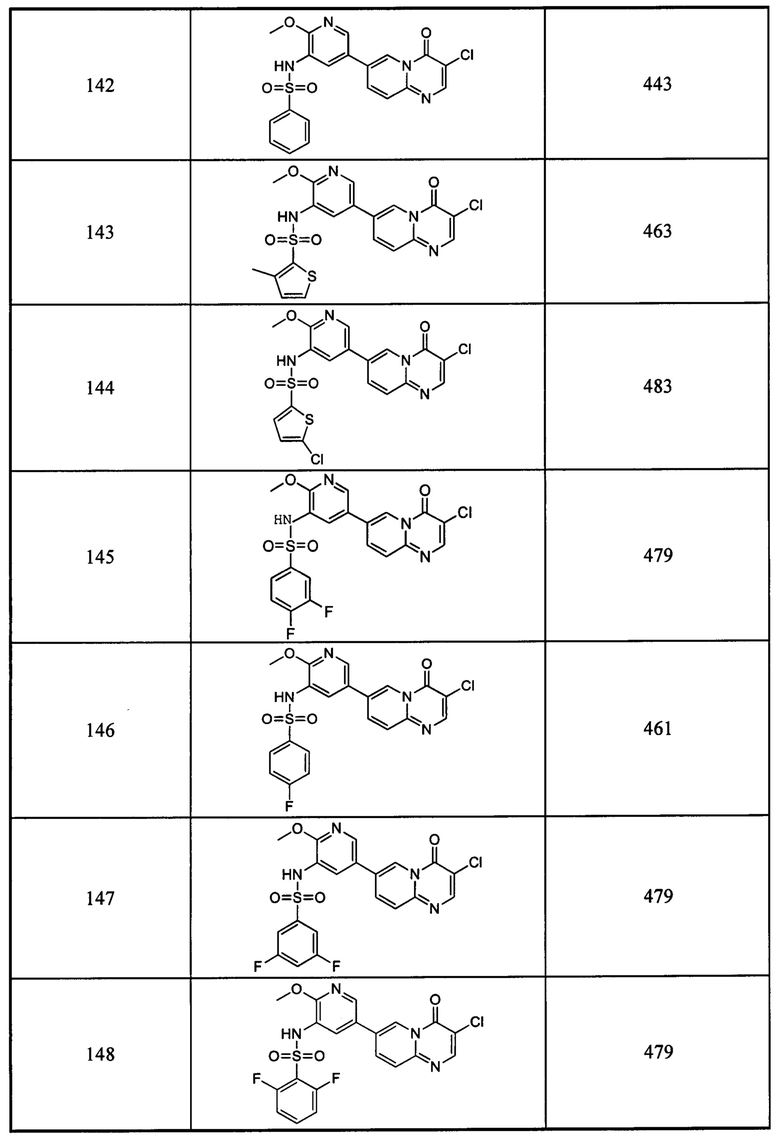
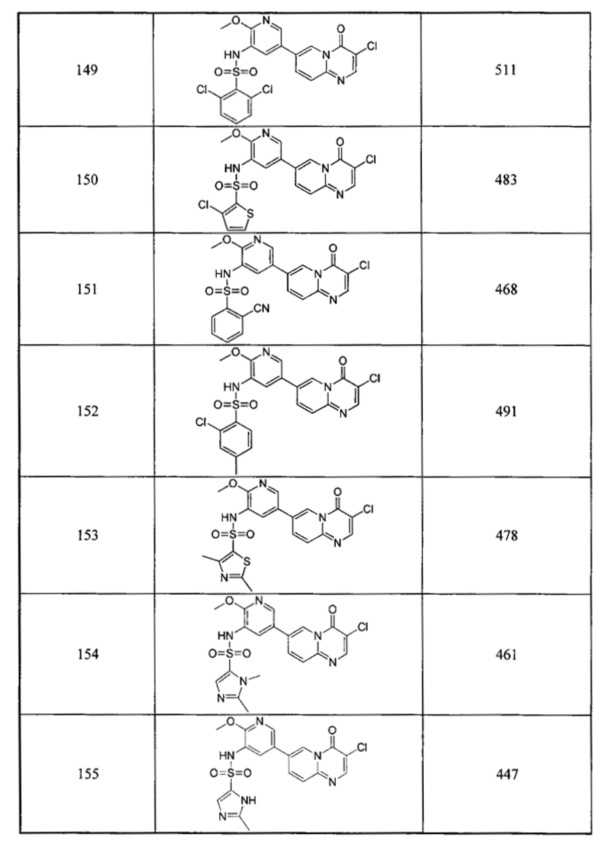
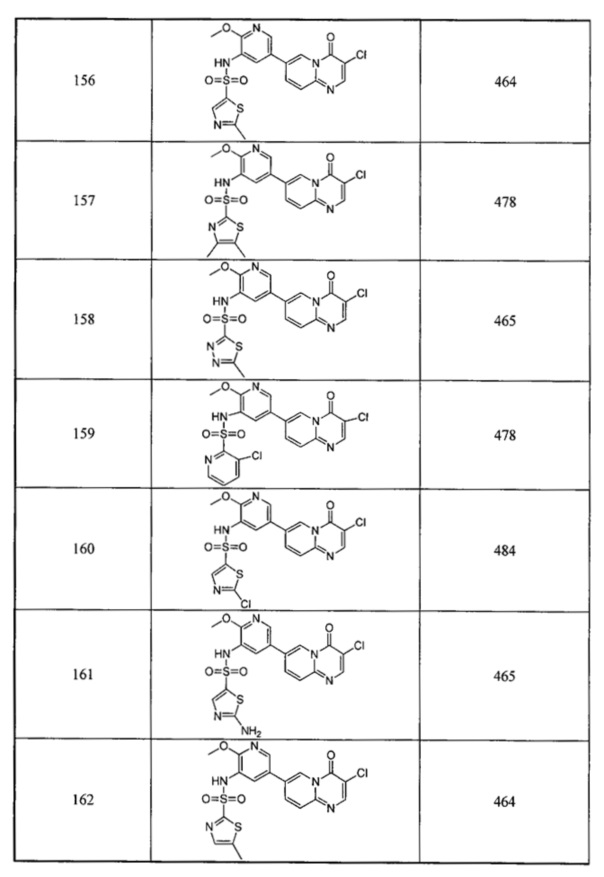
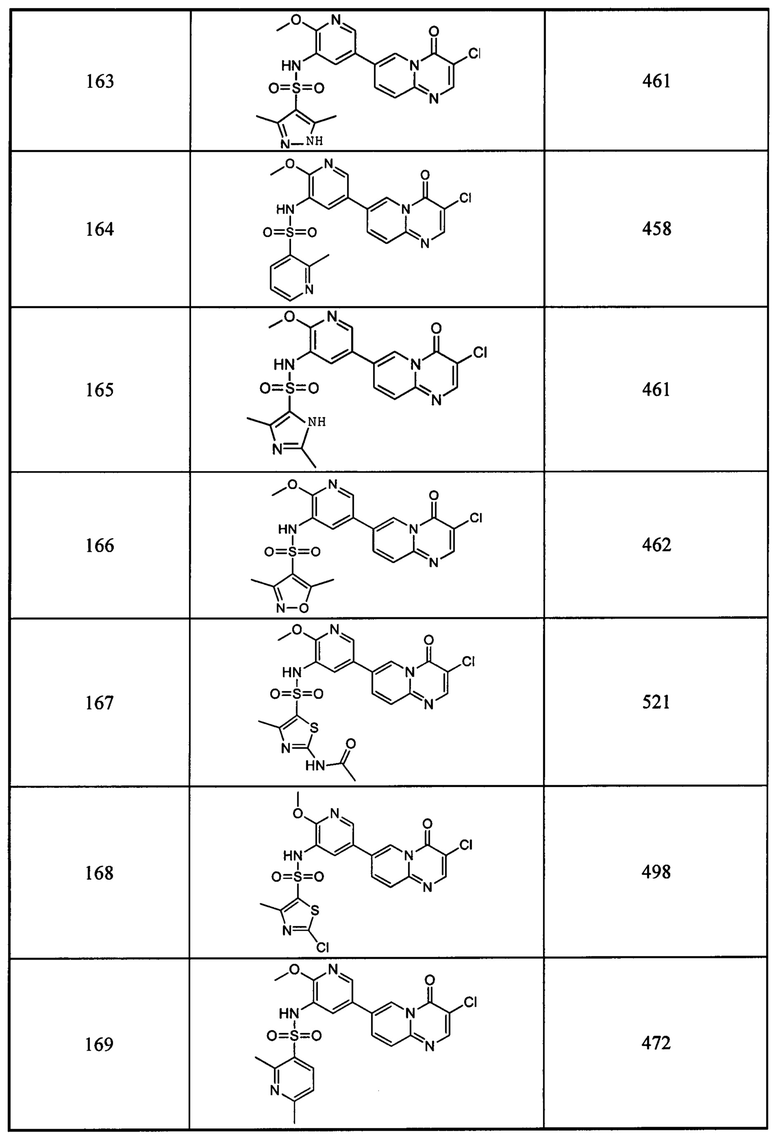
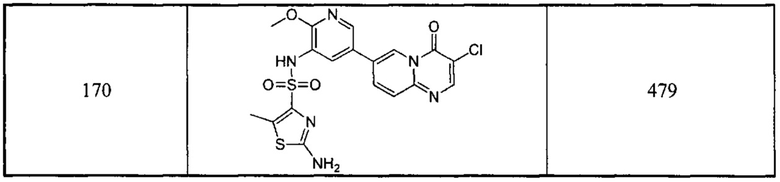
Схема 14:
Условия реакции: a) серная кислота, этанол, при нагревании; b) 1-трет-бутокси-N,N,N',N'-тетраэтилдиамин, при нагревании; с) 5-бромпиридин-2-амин, уксусная кислота, при нагревании; d) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 171
N-(5-(3-этокси-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)-2,4-дифторбензсульфамид
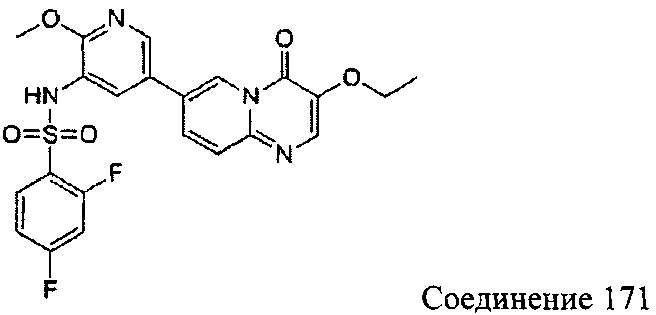
a) этил-2-этоксиацетат
Серную кислоту (10 мл) добавляли к раствору 2-этоксиуксусной кислоты (20 г, 0,19 моль) в этаноле (200 мл). Полученный реакционный раствор реагировал при 100°С в течение двух часов. После завершения реакции реакционный раствор концентрировали и разбавляли этилацетатом. Полученную органическую фазу дважды промывали водой, сушили над безводным сульфатом натрия и концентрировали с получением желтой маслянистой жидкости (19,5 г, 78%).
b) (Z)-этил-3-(диметиламино)-2-метоксиакрилат
Смешивали 1-трет-бутокси-N,N,N',N'-тетраэтилдиамин (2,0 г, 0,011 моль) и этил-2-этоксиацетат (1,5 г, 0,011 моль), нагревали до 80°С, перемешивали в течение 12 часов и концентрировали с получением желтого твердого вещества (420 мг, 20,4%).
1Н ЯМР (400 МГц, CDCl3) δ 6,80 (s, 1Н), 4,19-4,13 (q, 2Н), 4,05 (s, 1Н), 3,71-3,76 (s, 1Н), 3,03 (s, 6H), 1,26-1,29 (t, 6Н).
c) 7-бром-3-этокси-4Н-пиридо[1,2-а]пиримидин-4-он
Раствор (Z)-этил-3-(диметиламино)-2-метоксиакрилата (50 мг, 0,267 ммоль) и 5-бромпиридин-2-амина (46 мг, 0,267 ммоль) в уксусной кислоте нагревали до 90°С и перемешивали в течение ночи. После завершения реакции реакционный раствор концентрировали и добавляли воду (2 мл) для разбавления концентрированного раствора. рН доводили до 7 насыщенным раствором карбоната натрия и экстрагировали смесь дихлорметаном. Органическую фазу концентрировали с получением неочищенного продукта. Неочищенный продукт выделяли с помощью колоночной флэш-хроматографии с получением целевого соединения в виде желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,11-9,12 (d, 1Н), 8,08 (s, 1Н), 7,54-7,56 (d, 1Н), 7,46-7,48 (d, 1Н), 4,2-4,25 (q, 2Н), 2,11 (s, 3Н).
d) N-(5-(3-этокси-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)-2,4-дифторбензсульфамид
2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)бензсульфамид (0,28 ммоль), карбонат калия (0,56 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (20 мг) добавляли к смешанному раствору 7-бром-8-метил-4Н-пиридо[1,2-а]пиримидин-4-она (0,28 ммоль) в диоксане (2 мл) и воде (0,4 мл) в атмосфере азота. Реакционный раствор подвергали воздействию микроволнового излучения при 100°С в течение двух часов. За ходом реакции следили посредством ЖХ/МС. После завершения реакции реакционную смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта, который разделяли с помощью препаративной жидкостной хроматографией с получением целевого соединения.
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 10,4 (s, 1Н), 8,95 (s, 1Н), 8,44 (s, 1Н), 8,22 (s, 1Н), 8,04-8,07 (d, 1Н), 7,98 (s, 1Н), 7,77-7,8 (t, 1Н), 7,98 (s, 1Н), 7,55-7,60 (t, 1Н), 7,21-7,25 (t, 1Н), 4,16-4,21 (q, 2Н), 3,69 (q, 3Н), 1,35-1,39 (t, 3Н).
Следующие 3 соединения были также синтезированы согласно способу получения Соединения 171.

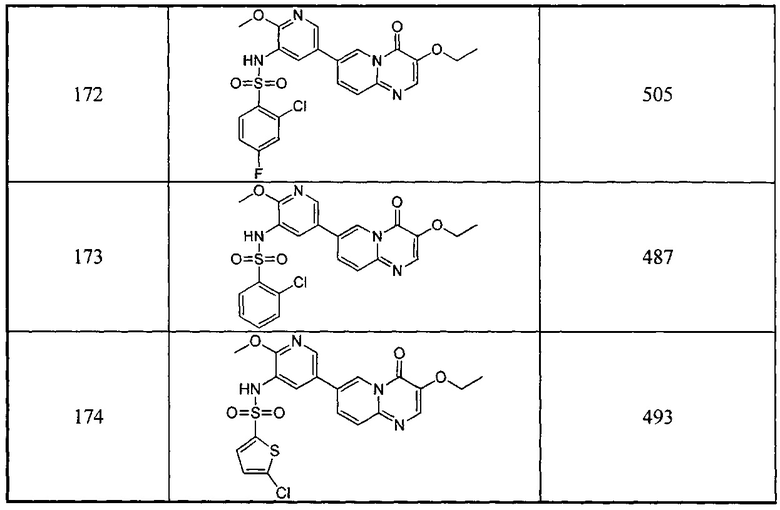
Пример 175
Схема 15:
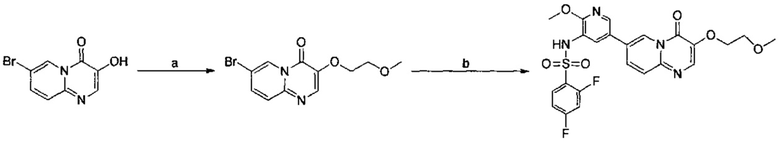
Условия: а) 2-бромэтилметиловый эфир, карбонат калия, N,N-диметилформамид, при нагревании; b) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
2,4-дифтор-N-(2-метокси-5-(3-(2-метоксиэтокси)-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
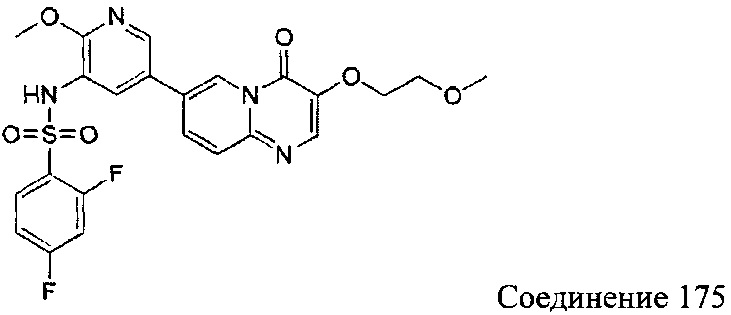
а) 7-бром-3-(2-метоксиэтокси)-4Н-пиридо[1,2-а]пиримидин-4-он
7-бром-3-гидрокси-4Н-пиридо[1,2-а]пиримидин-4-он (500 мг, 2,08 ммоль), 2-бромэтилметиловый эфир (350 мг, 2,45 ммоль) и карбонат калия (830 мг, 6,24 ммоль) растворяли в N,N-диметилформамиде (10 мл), помещали в трехгорлую круглодонную колбу и перемешивали при 110°С в течение 3 часов. Смесь промывали водой (10 мл), экстрагировали дихлорметаном (20 мл × 6), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением целевого соединения в виде желтого твердого вещества (250 мг, 40,4%).
b) 2,4-дифтор-N-(2-метокси-5-(3-(2-метоксиэтокси)-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
7-бром-3-(2-метоксиэтокси)-4Н-пиридо[1,2-а]пиримидин-4-он (200 мг, 0,67 ммоль), 2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)бензсульфамид (257 мг, 0,60 ммоль) и карбонат калия (185 мг, 1,34 ммоль) растворяли в диоксане (2,5 мл) и воде (0,5 мл), помещали в трехгорлую круглодонную колбу и добавляли [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (49 мг, 0,067 ммоль) в атмосфере азота при комнатной температуре. Смесь помещали в условия микроволнового излучения и перемешивали при 100°С в течение 2 часов. К смеси добавляли воду (5 мл), экстрагировали ее дихлорметаном (5 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения в виде зеленого твердого вещества (25 мг, 24,8%).
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 8,944 (s, 1Н), 8,413 (s, 1H), 8,231 (s, 1Н), 8,069-8,046 (d, 1H), 7,962 (s, 1H), 7,791-7,755 (t, 1H), 7,723-7,700 (d, 2Н), 7,562-7,539 (d, 1H), 7,235-7,197 (t, 1H), 4,248 (s, 2Н), 3,680 (s, 5Н).
Следующее 41 соединение было также синтезировано согласно способу получения Соединения 175.
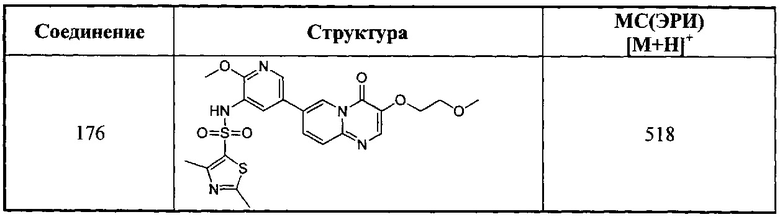
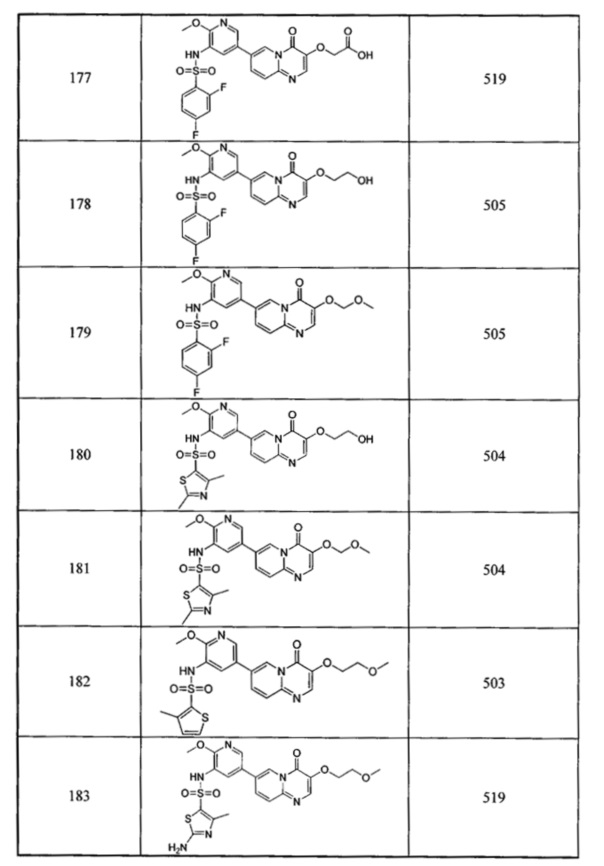
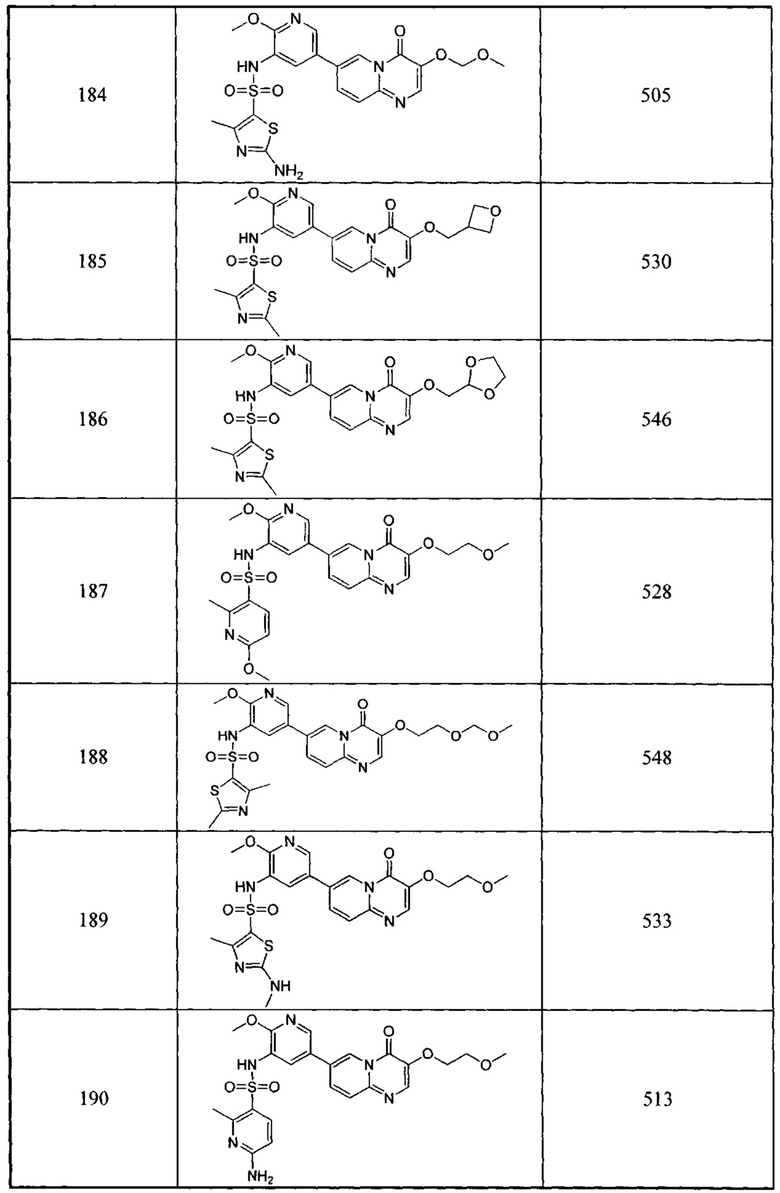
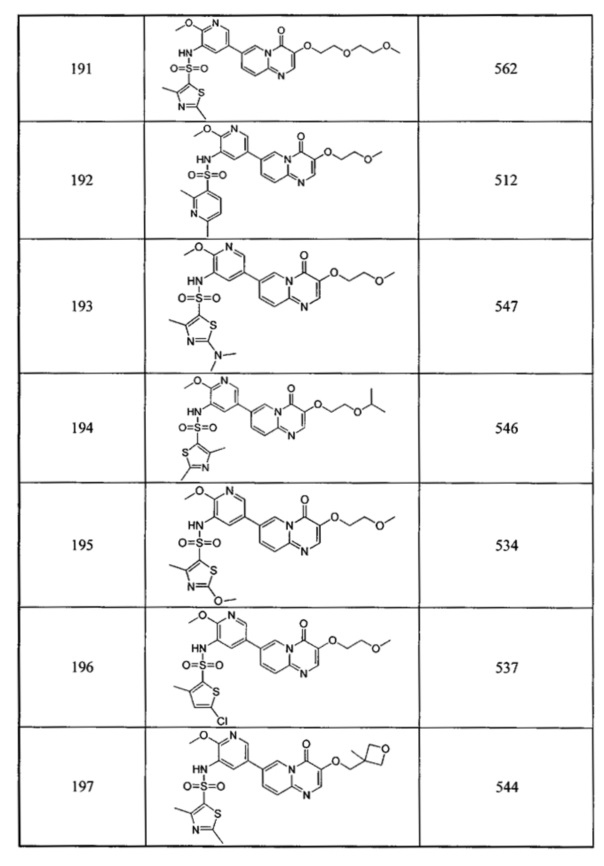
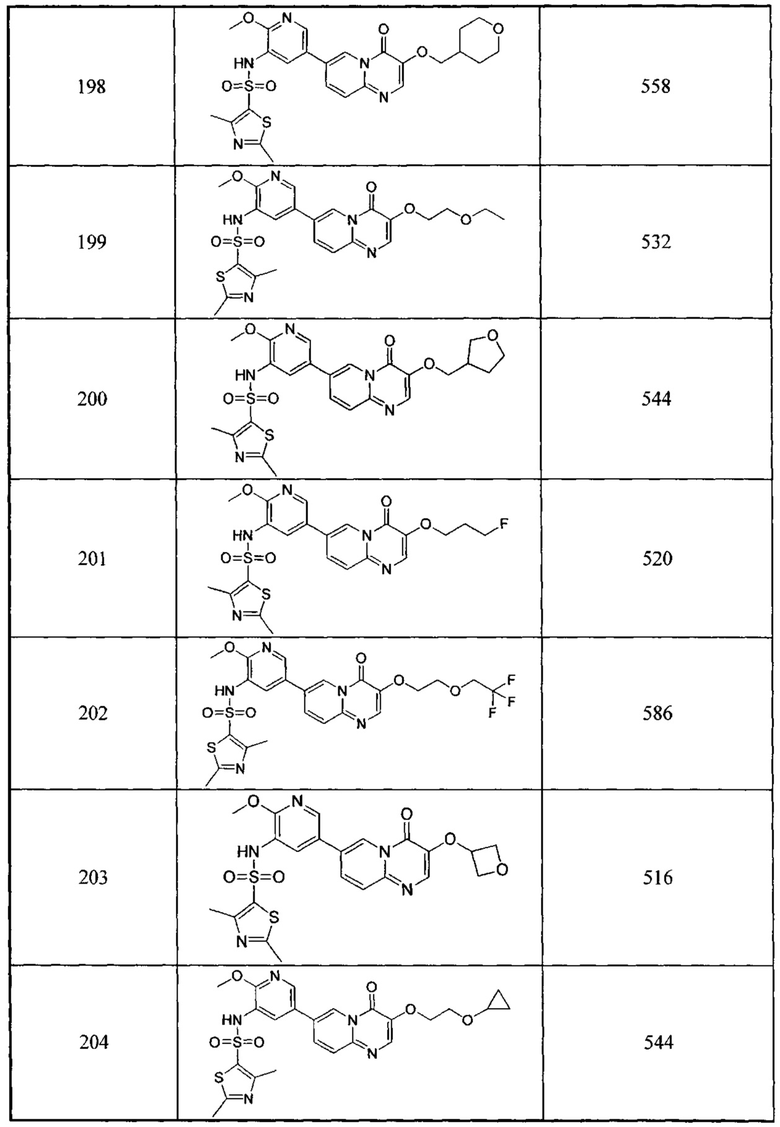
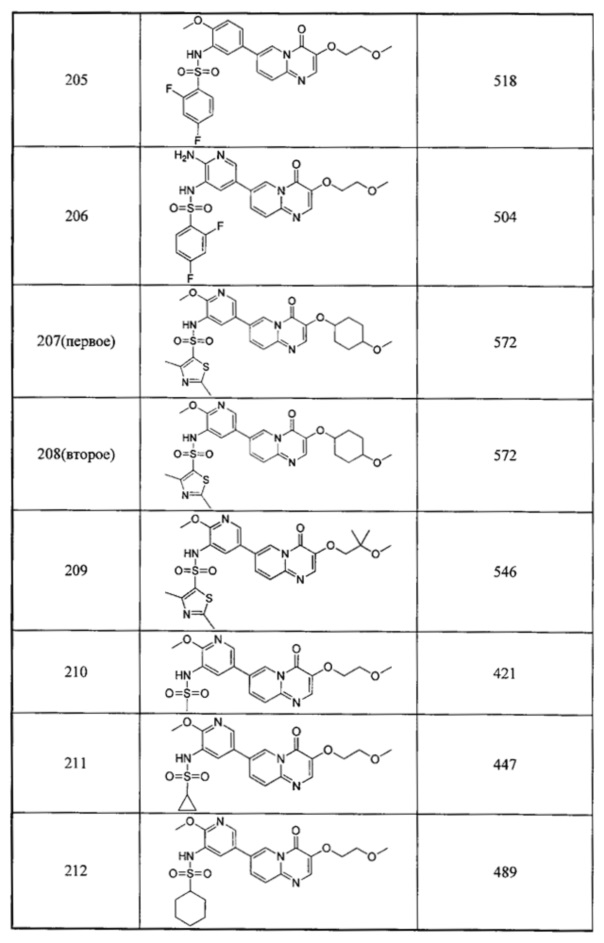
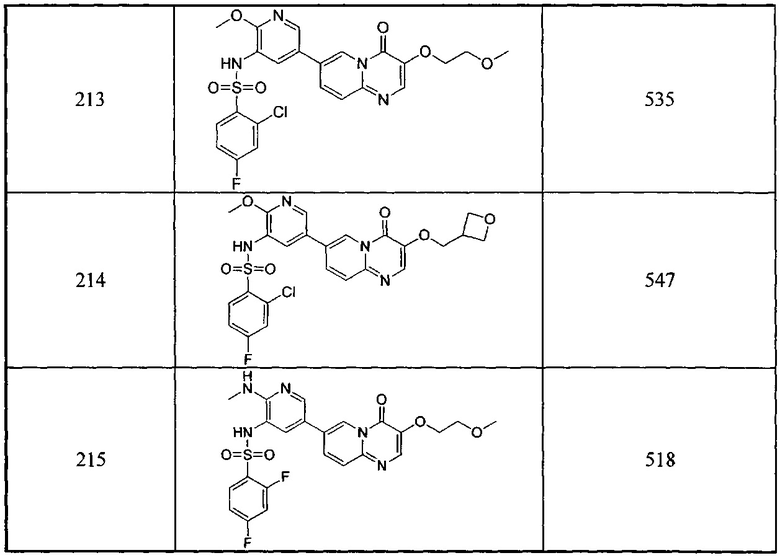
Схема 16:
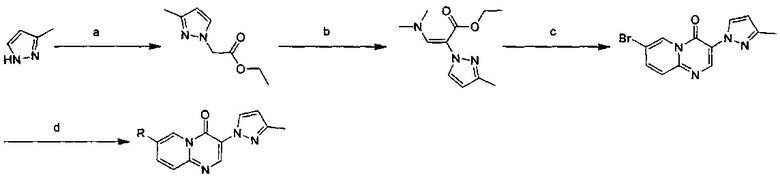
Условия реакции: а) карбонат калия, N,N-диметилформамид; b) 1-трет-бутокси-N,N,N',N'-тетраэтилдиамин; с) 5-бромпиридин-2-амин, уксусная кислота, при нагревании; 4) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид, и т.д.), диоксан, вода, при нагревании.
Пример 216
2,4-дифтор-N-(2-метокси-5-(3-(3-метил-1Н-пиразол-1-ил)-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
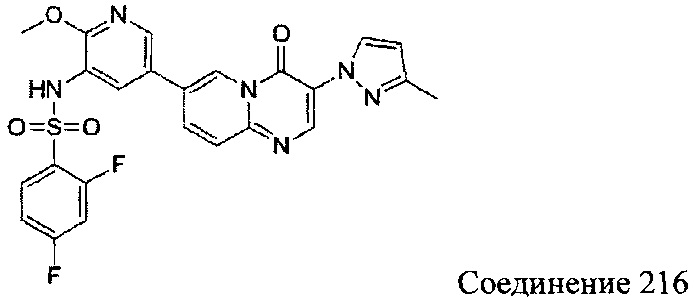
a) этил-2-(3-метил-1Н-пиразол-1-ил)ацетат
Раствор 3-метил-1-Н-пиразола (30 г, 365,9 ммоль), этилбромацетата (66,8 г, 402,4 ммоль) и карбоната калия (101 г, 731,8 ммоль) в N,N-диметилформамиде (300 мл) нагревали в круглодонной колбе с обратным холодильником в течение ночи. Реакционный раствор охлаждали и затем разбавляли 100 мл воды и экстрагировали дихлорметаном (100 мл × 3). Полученную дихлорметановую органическую фазу сушили над сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт выделяли с помощью колоночной хроматографии на силикагеле с получением желтой маслянистой жидкости (11 г, 18,03%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 7.43-7,35 (q, 1Н), 6,10-6,07 (q, 1Н), 4,84-4,82 (d, 2Н), 4,25-4,20 (s, 2Н), 2,36-2,26 (q, 3Н), 1,29-1,26 (q, 3Н).
b) (Е)-этил-3-(диметиламино)-2-(3-метил-1Н-пиразол-1-ил)акрилат
Этил-2-(3-метил-1Н-пиразол-1-ил)ацетат (4 г, 23,8 ммоль) и 1-трет-бутокси-N,N,N',N'-тетраэтилдиамин (4,1 г, 23,8 ммоль) смешивали и перемешивали при 100°С в течение ночи. Реакционную смесь концентрировали с получением коричневого маслянистого неочищенного продукта (5 г, 94,34%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 7,442 (s, 1Н), 6,080 (s, 2Н), 4,872-4,853 (d, 4Н), 3,778-3,729 (t, 6Н), 2,294-2,269 (d, 2Н).
c) 7-бром-3-(3-метил-1Н-пиразол-1-ил)-4Н-пиридо[1,2-а]пиримидин-4-он
Раствор (Е)-этил-3-(диметиламино)-2-(3-метил-1Н-пиразол-1-ил)акрилата (3,5 г, 150,2 ммоль) и 5-бромпиридин-2-амина (2,6 г, 150,2 ммоль) в уксусной кислоте (30 мл) в круглодонной колбе подвергали воздействию микроволнового излучения при 100°С в течение двух часов. Реакционную смесь концентрировали, разбавляли 50 мл воды и экстрагировали дихлорметаном (50 мл). Полученную органическую фазу дихлорметана сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт выделяли с помощью колоночной хроматографии на силикагеле с получением желтого твердого вещества (1,3 г, 28,4%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 9,160 (s, 1Н), 8,622-8,616 (d, 1Н), 8,327-8,322 (d, 1Н), 7,772-7,743 (m, 1Н), 7,640-7,628 (m, 1Н), 6,297-6,291 (s, 1Н), 2,405 (s, 3Н).
d) 2,4-дифтор-N-(2-метокси-5-(3-(3-метил-1Н-пиразол-1-ил)-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
2,4-дифтор-N-(2-метокси-5(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)бензсульфамид (0,28 ммоль), карбонат калия (0,56 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (20 мг) добавляли к смешанному раствору 7-бром-3-(3-метил-1Н-пиразол-1-ил)-4Н-пиридо[1,2-а]пиримидин-4-она (0,28 ммоль) в воде (0,4 мл) и диоксана (2 мл) в атмосфере азота. Реакционный раствор подвергали воздействию микроволнового излучения при 100°С в течение двух часов. За ходом реакции следили посредством ЖХ/МС. После завершения реакции реакционный раствор фильтровали и концентрировали с получением неочищенного продукта, который затем разделяли с помощью препаративной жидкостной хроматографии с получением целевого соединения.
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 9,159 (s, 1H), 8,945 (s, 1Н), 8,582-8,577 (d, 1H), 8,334-8,277 (t, 2Н), 7,939-7,889 (t, 2Н), 7,837-7,820 (d, 1Н), 7,519-7,473 (t, 1H), 7,236-7,195 (t, 1Н), 6,376-6,371 (d, 1H), 3,736 (s, 3Н), 2,314 (s, 1H).
Следующие 4 соединения были также синтезированы согласно способу получения Соединения 216.
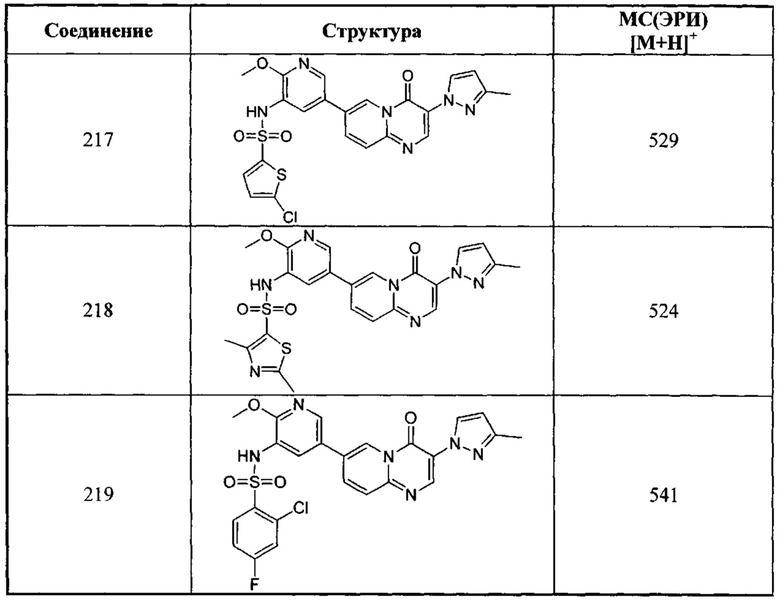
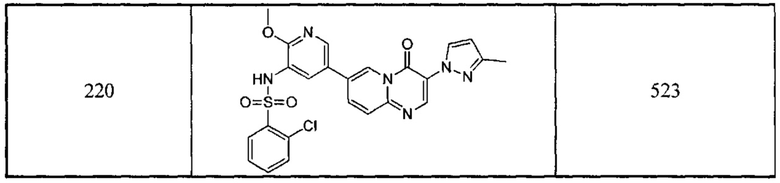
Схема 17:
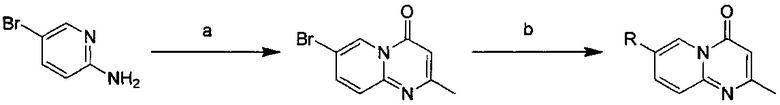
Условия реакции: а) этилацетоацетат, полифосфорная кислота; b) R борная кислота (борат), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 221
2,4-дифтор-N-(2-метокси-5-(2-метил-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
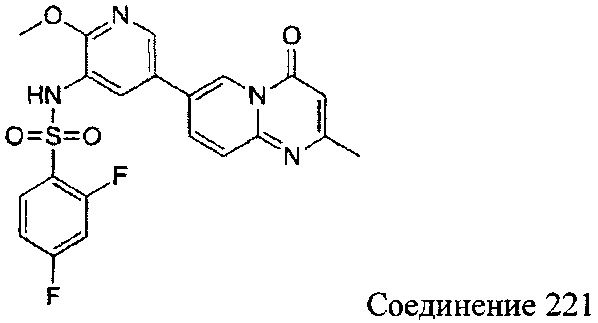
а) 7-бром-2-метил-4Н-пиридо[1,2-а]пиримидин-4-он
Смесь, содержащую 5-бромпиридин-2-амин гидрохлорид (2 г, 11,63 ммоль) и этилацетоацетат (2,3 г, 17,44 ммоль), растворяли в полифосфорной кислоте (10 мл) и перемешивали при 150°С в течение 30 минут. Смесь промывали этилацетатом и затем добавляли раствор гидроксида натрия для доведения значения рН смешанной системы до более чем 9. Раствор этилацетата разделяли и водную фазу экстрагировали этилацетатом (20 мл × 3). Объединенные органические фазы сушили над безводным сульфатом натрия, сушили вращательным испарением и выделяли на колонке с получением целевого соединения в виде желтого твердого вещества (3,2 г, 70%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 9,15 (s, 1Н), 7,76-7,78 (d, 1Н), 7,50-7,52 (d, 2Н), 6,36 (s, 1Н), 2,47 (s, 3Н).
b) 2,4-дифтор-N-(2-метокси-5-(2-метил-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
7-бром-2-метил-4Н-пиридо[1,2-а]пиримидин-4-он (0,28 ммоль) растворяли в диоксане (2 мл) и воде (0,4 мл) и затем к смешанному раствору добавляли 2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил) бензсульфамид (0,28 ммоль), карбонат калия (0,56 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (20 мг) в атмосфере азота. Реакционную смесь подвергали воздействию микроволнового излучения и перемешивали при 100°С в течение двух часов. Окончание реакции определяли с помощью ЖХ/МС. Смесь фильтровали и затем сушили вращательным испарением с получением неочищенного продукта, который разделяли с помощью препаративной ВЭЖХ с получением целевого соединения.
1H ЯМР (400 МГц, CD3OD) млн-1 δ 9,13 (s, 1Н), 8,31 (s, 1Н), 8,21-8,23 (d, 1Н), 8,01 (s, 1Н), 7,91-7,93 (m, 1Н), 7,73-7,75 (d, 1Н), 7,22-7,27 (m, 1Н), 7,10-7,14 (m, 1Н), 6,43 (s, 1Н), 3,87 (s, 3Н), 2,50 (s, 3Н).
Следующие 9 соединений были также синтезированы согласно способу получения Соединения 221.
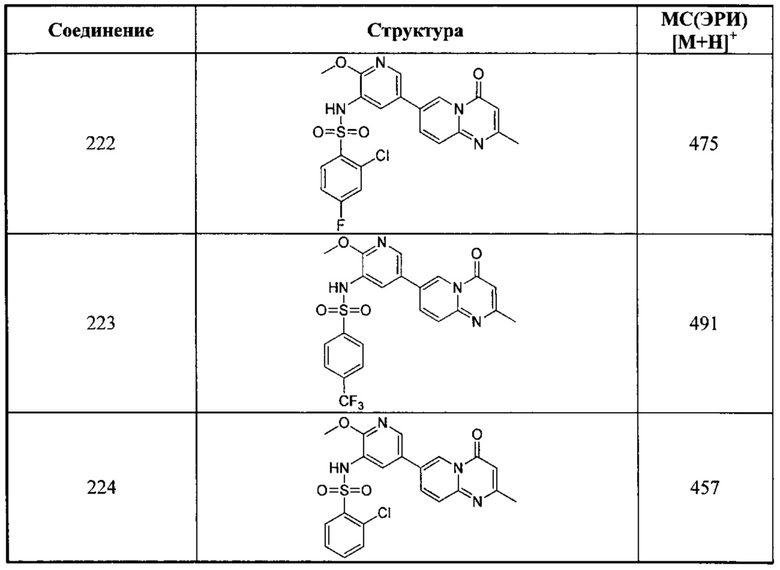
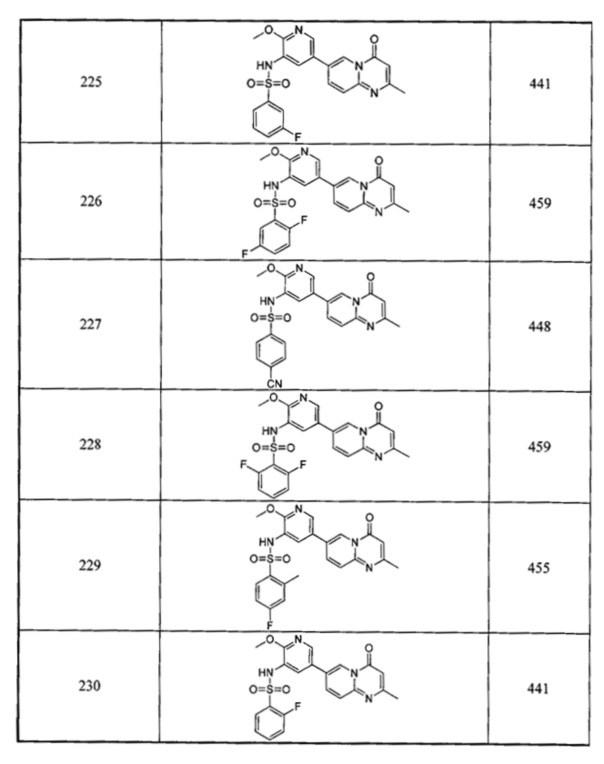
Схема 18:
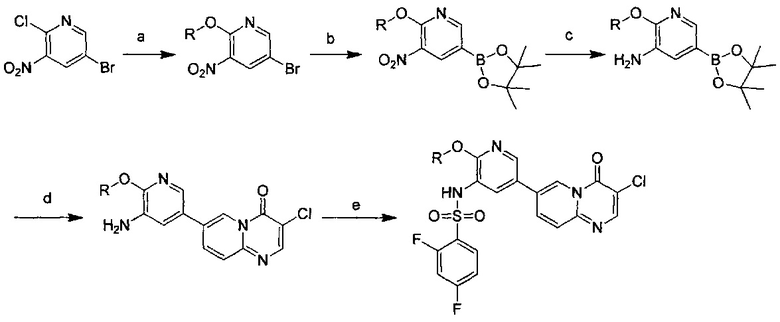
Условия: а) 5-бром-2-хлор-3-нитропиридин, R спирт, гидроксид калия, карбонат калия, 2-(2-метоксиэтокси)-N,N-бис[2-(2-метоксиэтокси)этил]этанамин, толуол; b) 4,4,5,5-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)-1,3,2-диоксоборан, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид, ацетат калия, диоксан, при нагревании; с) Pd/C (палладиевая чернь), метанол; d) 7-бром-3-хлор-пиридо[1,2-а]пиримидин-4-он, 1,1'-бис(дифенилфосфино)ферроценпалладий хлорид, карбонат калия, диоксан, вода, при нагревании; е) 2,4-дифторбензолсульфохлорид, пиридин.
Пример 231
N-[5-(3-хлор-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-(2-метокси-этокси)пиридин-3-ил]-2,4-дифтор-бензсульфамид
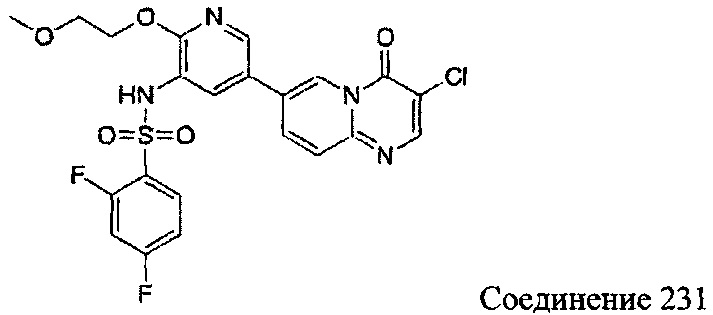
а) 5-бром-2-(2-метоксиэтокси)-3-нитропиридин
5-бром-2-хлор-3-нитропиридин (3,00 г, 12,63 ммоль, 1,00 экв.), 2-метоксиэтанол (1,15 г, 15,16 ммоль, 1,20 экв.) и 2-(2-метоксиэтокси)-N,N-ди[2-(2-метоксиэтокси)этил]этанамин (816,96 мг, 2,53 ммоль, 0,20 экв.) добавляли к смешанному раствору гидроксида калия (1,20 г, 21,47 ммоль, 1,70 экв.) и карбоната калия (2,97 г, 21,47 ммоль, 1,70 экв.) в толуоле (30 мл). Смесь перемешивали при 15°С в атмосфере азота в течение 18 часов. После завершения реакции реакционный раствор фильтровали и фильтрат концентрировали и очищали с помощью колоночной хроматографии на силикагеле (РЕ:ЕА составляет от 20:1 до 4:1) с получением целевого соединения в виде желтого твердого вещества (1,60 г, 5,77 ммоль, 45,72%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 8,41 (d, J=2,4 Гц, 1Н), 8,38 (d, J=2,2 Гц, 1H), 4,70-4,53 (m, 2Н), 3,85-3,72 (m, 2Н), 3,43 (s, 3Н).
b) 2-(2-метоксиэтокси)-3-нитро-5-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)пиридин
[1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (42,22 мг, 57,70 мкмоль, 0,01 экв.) добавляли к смешанному раствору 5-бром-2-(2-метоксиэтокси)-3-нитропиридина (1,60 г, 5,77 ммоль, 1,00 экв.), 4,4,5,5-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)-1,3,2-диоксоборана (1,76 г, 6,92 ммоль, 1,20 экв.) и ацетата калия (1,70 г, 17,31 ммоль, 3,00 экв.) в диоксане (20 мл). Смесь перемешивали при 90°С в атмосфере азота в течение 18 часов. После завершения реакции реакционный раствор фильтровали и концентрировали фильтрат с получением коричневого маслянистого неочищенного продукта (2,50 г, 5,55 ммоль, выход: 96,24%, чистота: 72%).
1H ЯМР (400 МГц, CDCl3) млн-1 δ 8,65 (d, J=1,5 Гц, 1H), 8,57 (d, J=1,5 Гц, 1H), 4,71-4,63 (m, 2Н), 3,86-3,75 (m, 2Н), 3,44 (s, 3Н), 1,34 (s, 12Н).
c) 2-(2-метоксиэтокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)пиридин-3-амин
Pd/C (150,00 мг) добавляли к раствору 2-(2-метоксиэтокси)-3-нитро-5-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)пиридина (1,50 г, 3,33 ммоль, 1,00 экв.) в метаноле (30 мл). Эту смесь перемешивали при 18°С в атмосфере водорода в течение 2 часов. После завершения реакции реакционный раствор фильтровали и концентрировали фильтрат с получением желтого маслянистого неочищенного продукта (1,40 г, 2,38 ммоль, выход: 71,46%, чистота: 50%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 7,94 (d, J=1,5 Гц, 1Н), 7,23 (d, J=1,5 Гц, 1H), 4,57-4,53 (m, 2Н), 3,78-3,75 (m, 2Н), 3,42 (s, 3Н), 1,32 (s, 12Н).
d) 7-(5-амино-6-(2-метоксиэтокси)пиридин-3-ил)-3-хлор-4Н-пиридо[1,2-а]пиримидин-4-он
[1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (5,64 мг, 7,71 мкмоль, 0,01 экв.) и воду (1 мл) добавляли к смешанному раствору 7-бром-3-хлор-пиридо[1,2-а]пиримидин-4-она (200,00 мг, 770,74 мкмоль, 1,00 экв.), 2-(2-метоксиэтокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксоборан-2-ил)пиридин-3-амина (453,43 мг, 770,74 мкмоль, 1,00 экв.) и карбоната калия (319,57 мг, 2,31 ммоль, 3,00 экв.) в диоксане (5 мл). Эту смесь перемешивали в атмосфере азота при 90°С в течение 18 часов. После завершения реакции реакционный раствор сушили над безводным сульфатом натрия и фильтровали и концентрировали фильтрат при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (DCM:МеОН составляет от 1% до 5%) с получением целевого соединения в виде желтого твердого вещества (270,00 мг, 622,89 мкмоль, выход: 80,82%, чистота: 80%).
1H ЯМР (400 МГц, CDCl3) δ 9,17 (d, J=1,7 Гц, 1Н), 8,47 (s, 1H), 7,97 (dd, J=2,1, 9,2 Гц, 1H), 7,79 (d, J=2,0 Гц, 1H), 7,75 (d, J=9,3 Гц, 1H), 7,13 (d, J=2,2 Гц, 1H), 4,62-4,53 (m, 2H), 4,06 (br. s., 2H), 3,82-3,76 (m, 2H), 3,44 (s, 3H).
e) N-[5-(3-хлор-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-(2-метокси-этокси)-пиридин-3-ил]-2,4-дифторбензсульфамид
2,4-дифторбензолсульфохлорид (91,96 мг, 432,57 мкмоль, 1,50 экв.) добавляли к смешанному раствору 7-[5-амино-6-(2-метоксиэтокси)пиридин-3-ил]-3-хлор-пиридо[1,2-а]пиримидин-4-она (125,00 мг, 288,38 мкмоль, 1,00 экв.) в пиридине (3 мл). Смесь реагировала при 15°С в течение 4 часов. После завершения реакции реакционный раствор концентрировали. Остаток растворяли в дихлорметане и промывали водой и солевым раствором. Органическую фазу сушили над безводным сульфатом натрия и концентрировали. Полученный остаток очищали с помощью препаративной тонкослойной хроматографии с получением целевого соединения в виде желтого твердого вещества (51,32 мг, 98,14 мкмоль, 34,03%).
1Н ЯМР (400 МГц, CDCl3) δ 9,12 (d, J=1,7 Гц, 1H), 8,49 (s, 1H), 8,11 (d, J=2,2 Гц, 1H), 7,98 (d, J=2,2 Гц, 1H), 7,96-7,85 (m, 2H), 7,78 (d, J=9,3 Гц, 1H), 7,47 (s, 1H), 7,01 (t, J=8,2 Гц, 1H), 6,97-6,89 (m, 1H), 4,56-4,47 (m, 2H), 3,76-3,67 (m, 2H), 3,42 (s, 3H). Следующие 2 соединения были также синтезированы согласно способу получения Соединения 232.
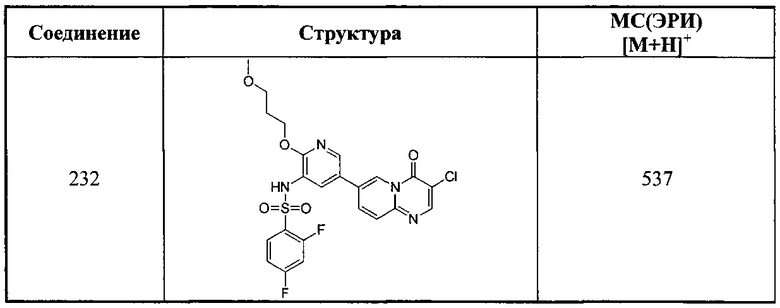
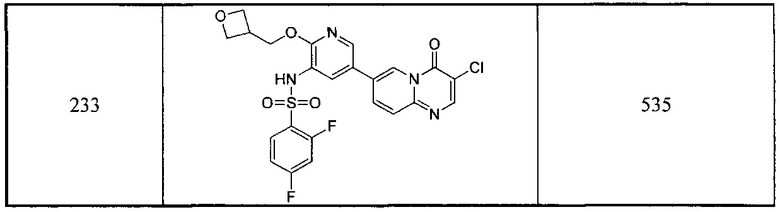
Схема 19:
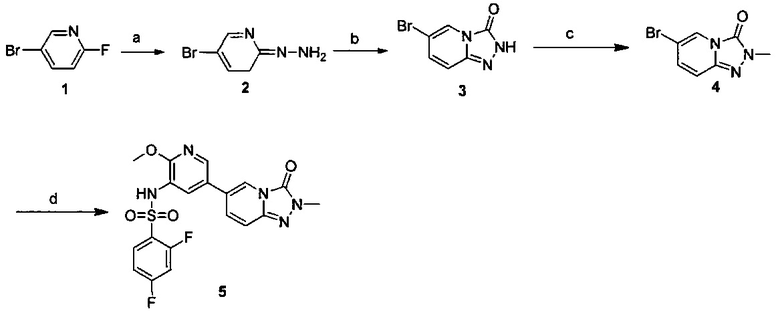
Условия: а) гидразингидрат, этанол, при нагревании; b) 1,1-карбонилдиимидазол, ацетонитрил, при нагревании; с) карбонат цезия, йодистый метил, N,N-диметилформамид; d) R борат (борная кислота), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), карбонат калия, диоксан, вода, при нагревании.
Пример 234
2,4-дифтор-N-(2-метокси-5-(2-метил-3-оксо-2,3-2Н-[1,2,4-]триазоло[4,3-а]пиридин-6-ил)пиридин)-3-ил)бензсульфамид
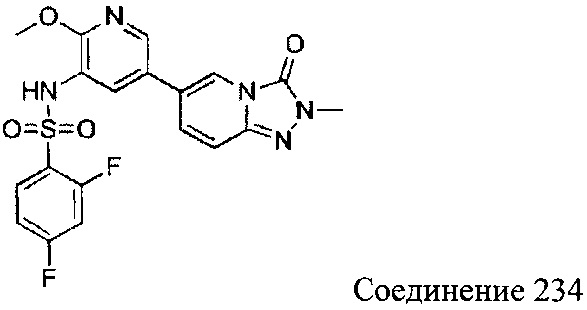
а) 5-бром-2-гидразо-2,3-дигидропиридин
Гидразингидрат (8 г) добавляли к раствору 5-бром-2-фторпиридина (2 г, 11,36 ммоль) в этаноле (25 мл). Реакционный раствор нагревали до 80°С и перемешивали в течение 16 часов. Реакционный раствор охлаждали до комнатной температуры, концентрировали при пониженном давлении для удаления половины растворителя, фильтровали и собирали фильтрационный осадок. Неочищенный целевой продукт получали вакуумной сушкой.
b) 6-бром-[1,2,4]триазоло[4,3-а]пиридин-3(2Н)-он
Раствор 6-бром-[1,2,4]триазоло[4,3-а]пиридин-3(2Н)-она (1 г, 5,32 ммоль) и 1,1-карбонилдиимидазола (948 мг, 8,85 ммоль) в ацетонитриле (10 мл) нагревали до 85°С и перемешивали смесь с обратным холодильником в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры и перемешивали в течение еще 16 часов. Реакционной смеси давали отстояться и фильтровали, собирали фильтрационный осадок и сушили с получением неочищенного целевого продукта.
c) 6-бром-2-метил-[1,2,4]триазоло[4,3-а]пиридин-3(2Н)-он
Карбонат цезия (685 мг, 2,1 ммоль) и йодистый метил (0,26 мл, 4,2 ммоль) добавляли к раствору 6-бром-[1,2,4]триазоло[4,3-а]пиридин-3(2Н)-она (150 мг, 0,7 ммоль) в безводном DMF (3 мл). Реакционный раствор перемешивали при 20°С в течение 16 часов. Реакционный раствор разбавляли этилацетатом, фильтровали для удаления твердого вещества. Фильтрат промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением целевого продукта в виде желтого твердого вещества (140 мг, 87,5%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 3,67 (s, 3Н) 6,97-7,03 (m, 1Н) 7,07-7,13 (m, 1Н) 7,92 (s, 1Н).
d) 2,4-дифтор-N-(2-метокси-5-(2-метил-3-оксо-2,3-2Н-[1,2,4-]триазоло[4,3-а]пиридин-6-ил)пиридин)-3-ил)бензсульфамид
В атмосфере азота [1,1-бис(дифенилфосфино)ферроцен]палладий хлорид (10 мг) добавляли к смешанному раствору 6-бром-2-метил-[1,2,4]триазоло[4,3-а]пиридин-3(2Н)-она (112 мг, 0,49 ммоль), 2,4-дифтор-N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил) бензсульфамида (200 мг, 0,47 ммоль) и карбоната натрия (124 мг, 1,17 ммоль) в 1,4-диоксане (3 мл) и воде (1,2 мл). Реакционный раствор подогревали до 80°С и перемешивали в течение 16 часов. Реакционную смесь фильтровали. Фильтрат разбавляли водой и экстрагировали водную фазу этилацетатом (15 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали и очищали остаток на пластинке для препаративной хроматографии (DCM:МеОН составляет 15:1) с получением целевого соединения в виде белого порошка (50 мг, 23,81%).
1Н ЯМР (400 МГц, CDCl3) млн-1 3,71 (s, 3Н) 3,96 (s, 3Н) 6,91-7,01 (m, 2Н) 7,17-7,23 (m, 2Н) 7,79-7,87 (m, 2Н) 7,88-7,93 (m, 1Н) 8,01 (d, J=2,20 Гц, 1Н).
Следующее соединение было также синтезировано согласно способу получения Соединения 234.
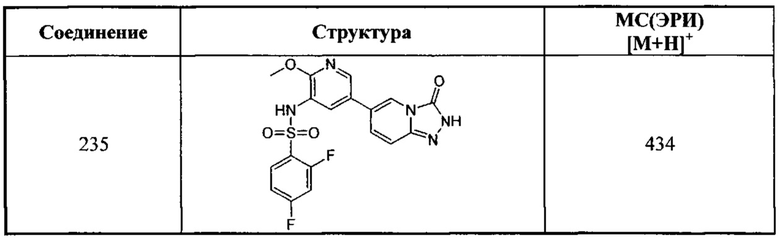
Схема 20:
Условия: a) малонилдихлорид, дихлорметан, при комнатной температуре; b) оксихлорид фосфора, при нагревании с обратным холодильником; c) R1 амин, при нагревании; d) R2 бор, палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), K2СО3, диоксан, вода, при нагревании.
Пример 236
N-(5-(2-амино-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)-2,4-диметилтиазолил-5-сульфамид
а) 7-бром-2-гидрокси-4Н-пиридо[1,2-а]пиримидин-4-он
2-амино-5-бромпиридин (10,0 г, 57,8 ммоль) растворяли в дихлорметане (100 мл) и помещали в круглодонную колбу объемом 250 мл. Малонилхлорид (9,78 г, 69,36 ммоль) добавляли по каплям при 0°С, после чего реакционный раствор нагревали до 15°С и перемешивали в течение 48 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционную смесь фильтровали и фильтрационный осадок промывали дихлорметаном (200 мл) с получением целевого соединения в виде желтого твердого вещества (13 г, 84%).
b) 7-бром-2-хлор-4Н-пиридо[1,2-а]пиримидин-4-он
7-бром-2-гидрокси-4Н-пиридо[1,2-а]пиримидиин-4-он (7 г, 29 ммоль) растворяли в оксихлориде фосфора (50 мл) и помещали в круглодонную колбу объемом 100 мл. Смесь перемешивали при 120°С в течение 18 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор охлаждали до комнатной температуры и медленно выливали в воду (1 л) комнатной температуры для остановки реакции. Смесь экстрагировали этилацетатом (300 мл × 6). Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде желтого твердого вещества (2,9 г, 37%).
1H ЯМР (400 МГц, DMSO-d6) млн-1 δ 9,01 (d, 1Н), 8,23 (dd, 1Н), 7,67 (d, 1Н), 6,58 (s, 1Н).
c) 2-амино-7-бром-4Н-пиридо[1,2-а]пиримидин-4-он
7-бром-2-хлор-4Н-пиридо[1,2-а]пиримидин-4-он (1 г, 3,85 ммоль), растворенный в смеси жидкого аммиака и этанола (30 мл и 15 мл), помещали в герметичную емкость объемом 100 мл и оставляли реагировать при 80°С в течение 48 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде фиолетового твердого вещества (90 мг, 9,7%).
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 8,78 (d, 1Н), 7,85 (dd, 1Н), 7,17 (d, 1Н), 6,86 (br. s., 2Н), 5,26 (s, 1Н).
d) N-(5-(2-амино-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)-2-метоксипиридин-3-ил)-2,4-диметилтиазолил-5-сульфамид
2-амино-7-бром-4Н-пиридо[1,2-а]пиримидин-4-он (72 мг, 210 мкмоль) растворяли в диоксане (5 мл) и воде (1 мл) и добавляли N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)-2,4-диметилтиазолил-5-сульфамид (89 мг, 210 мкмоль), карбонат калия (87 мг, 630 мкмоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (10 мг). Реакционный раствор перемешивали при 100°С в течение 18 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого продукта в виде белого твердого вещества (30 мг, 30%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 9,11 (d, 1Н), 8,16 (d, 1Н), 7,99 (d, 1Н), 7,81 (dd, 1Н), 7,42 (d, 1Н), 7,17 (br. s., 1Н), 5,59 (s, 1Н), 4,87 (br. s., 2Н), 3,97 (s, 3Н), 2,65 (s, 3Н), 2,57 (s, 3Н).
Следующие 2 соединения были также синтезированы согласно способу получения Соединения 236.
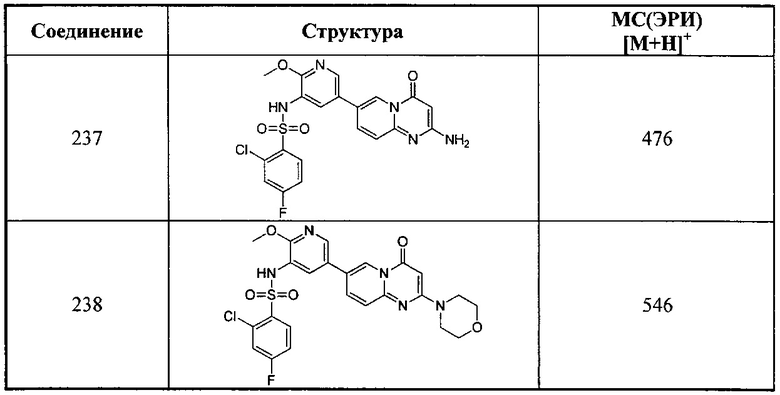
Схема 21:
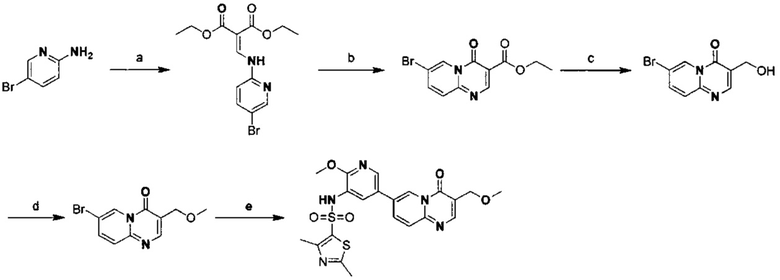
Условия: а) диметил-2-(метоксиметилен)малонат, этанол, при нагревании; b) оксибромид фосфора, при нагревании; с) DIBAL-H (гидрид диизобутилалюминия), тетрагидрофуран, 0°С; d) йодистый метил, гидрид натрия, тетрагидрофуран, 0-15°С; е) R борат (борная кислота), палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), K2СО3, диоксан, вода, при нагревании.
Пример 239
N-(2-метокси-5-(3-(метоксиметил)-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)-2,4-диметилтиазолил-5-сульфамид
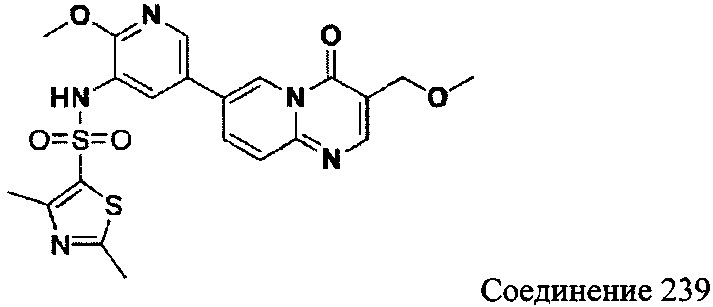
a) диэтил-2-(((5-бромпиридин-2-ил)амино)метенил)малонат
5-бромпиридин-2-амин (5 г, 28,9 ммоль) и диметил-2-(метоксиметенил)малонат (5,84 г, 28,9 ммоль) растворяли в этаноле (50 мл) и перемешивали при 80°С в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрационный осадок промывали петролейным эфиром и сушили с получением целевого соединения в виде белого твердого вещества (8,0 г, 81%).
1H ЯМР (400 МГц, CDCl3) млн-1 δ 11,12 (d, 1Н), 9,08 (d, 1Н), 8,40 (d, 1Н), 7,76 (dd, 1Н), 6,78 (d, 1Н), 4,41-4,19 (m, 4Н), 1,37 (td, 6Н).
b) метил-7-бром-4-оксо-4Н-пиридо[1,2-а]пиримидин-3-карбоксилат
Диэтил-2-(((5-бромпиридин-2-ил)амино)метенил)малонат (3,0 г, 8,74 ммоль) и оксибромид фосфора (9,27 г, 32,35 ммоль) помещали в круглодонную колбу и перемешивали смесь при 80°С в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, медленно выливали в ледяную воду, доводили рН до 8 насыщенным раствором карбоната натрия и экстрагировали дихлорметаном. Полученную органическую фазу промывали насыщенным солевым раствором (100 мл), сушили над сульфатом натрия и концентрировали с получением целевого соединения в виде желтого твердого вещества (2,0 г, 76,9%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 9,36 (d, 1Н), 9,05-8,98 (m, 1Н), 7,96 (dd, 1Н), 7,65 (d, 1Н), 4,42 (q, 2Н), 1,47-1,37 (m, 3H).
c) 7-бром-3-(гидроксиметил)-4Н-пиридо[1,2-а]пиримидн-4-он
Метил-7-бром-4-оксо-4Н-пиридо[1,2-а]пиримидин-3-карбоксилат (800 мг, 2,69 ммоль) растворяли в тетрагидрофуране (20 мл) и добавляли DIBAL-H (4 мл) при 0°С. Смесь оставляли при 0°С в течение 3 часов. К реакционному раствору добавляли насыщенный раствор хлорида аммония (20 мл) для остановки реакции. Полученную смесь экстрагировали этилацетатом (20 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения (110 мг, 16%).
d) 7-бром-3-(метоксиметил)-4Н-пиридо[1,2-а]пиримидин-4-он
7-бром-3-(гидроксиметил)-4Н-пиридо[1,2-а]пиримидин-4-он (110 мг, 431 мкмоль) растворяли в тетрагидрофуране (3 мл) и добавляли гидрид натрия (26 мг, 647 мкмоль, чистота 60%) при 0°С. Реакционный раствор перемешивали при 20°С в течение одного часа и затем добавляли йодистый метил (183 мг, 1,29 ммоль). Реакционный раствор перемешивали при 20°С в течение 6 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционную смесь выливали в ледяную воду (30 мл) для остановки реакции и затем экстрагировали этилацетатом (20 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали и фильтрат концентрировали с получением неочищенного продукта, который очищали с помощью препаративной тонкослойной хроматографии с получением целевого соединения (23 мг, 19,8%).
е) N-(2-метокси-5-(3-(метоксиметил)-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)-2,4-диметилтиазолил-5-сульфамид
7-бром-3-(метоксиметил)-4Н-пиридо[1,2-а]пиримидин-4-он (23 мг, 85 мкмоль) растворяли в диоксане (2,5 мл) и воде (0,5 мл) и добавляли N-(2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)-2,4-диметилтиазолил-5-сульфамид (36 мг, 85 мкмоль), карбонат калия (24 мг, 170 мкмоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (10 мг). Смесь реагировала при 100°С под воздействием микроволнового излучения в течение 1 часа. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого продукта в виде бледно-желтого твердого вещества (18 мг, 43,2%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 9,20 (s, 1Н), 8,42 (s, 1Н), 8,18 (d, 1Н), 8,03 (s, 1Н), 7,98-7,85 (m, 2Н), 7,20 (s, 1Н), 4,57 (s, 2Н), 3,99 (s, 3Н), 3,50 (s, 3Н), 2,64 (s, 3Н), 2,57 (s, 3Н), 1,23 (s, 2H).
Схема 22:
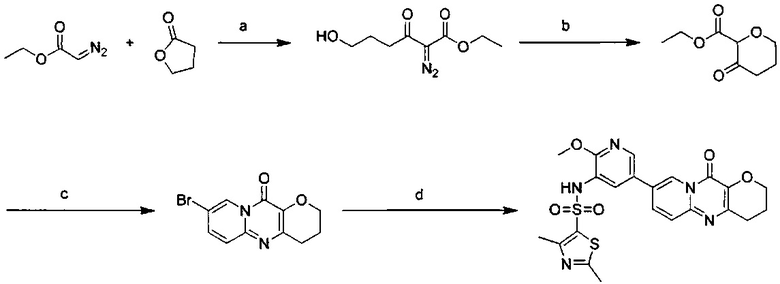
Условия: a) LDA (диизопропиламид лития), тетрагидрофуран, -78°С; b) родиевый реагент, толуол, при нагревании; с) 5-бромпиридин-2-амин, уксусная кислота, 110°С; d) R борат, палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), K3РО4, тетрагидрофуран, вода, при нагревании.
Пример 240
2-метокси-5-(11-оксо-2,3,4,11-тетрагидропирано[3,2-d]пиридо[1,2-а]пиримидин-8-ил)пиридин-3-ил)-2,4-диметилтиазолил-5-сульфамид
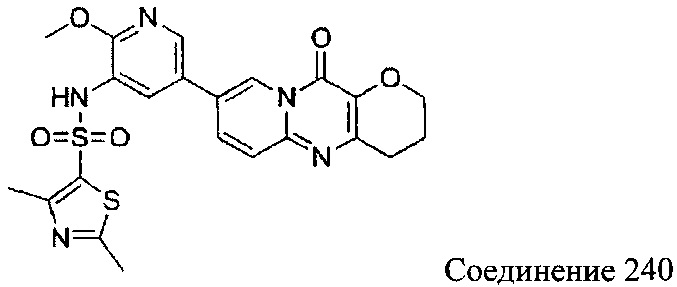
а) этил-2-диазо-6-гидрокси-3-оксогексаноат
Этилдиазоацетат (6,00 г, 52,59 ммоль) и тетрагидрофуран (60 мл) помещали в трехгорлую круглодонную колбу и медленно по каплям добавляли диизопропиламид лития (5,63 г, 52,59 ммоль) при -78°С в атмосфере азота. Смесь перемешивали при -78°С в течение 0,5 часов. Медленно по каплям добавляли тетрагидрофуран-2-он (4,07 г, 47,33 ммоль) при -78°С в атмосфере азота и перемешивали при -78°С в течение 2 часов. Окончание реакции определяли с помощью ТСХ. К смеси добавляли насыщенный раствор хлорида аммония (300 мл) и экстрагировали этилацетатом (200 мл × 3). Объединенные органические фазы промывали насыщенным солевым раствором (200 мл × 2), сушили над безводным сульфатом натрия, фильтровали, концентрировали и выделяли при помощи хроматографии на силикагеле с получением целевого соединения (4,00 г, 38%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 1,34 (t, 3Н), 1,90-1,97 (m, 2Н), 3,00 (t, 2Н), 3,70 (t, 2Н), 4.32 (q, 2Н).
b) этил-3-оксотетрагидро-2Н-пиран-2-карбоксилат
Этил-2-диазо-6-гидрокси-3-оксогексаноат (316,00 мг, 1,58 ммоль) и толуол (40 мл) помещали в круглодонную колбу объемом 250 мл и медленно по каплям добавляли раствор димера ацетата родия (6,29 мг, 14,22 мкмоль) в толуоле (40 мл) при 80°С в атмосфере азота и перемешивали при 80°С в течение 1 часа. Окончание реакции определяли с помощью ТСХ. Реакционный раствор охлаждали до комнатной температуры и очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения (180 мг, выход: 67%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 1,34 (t, 3Н), 1,94-1,98 (m, 2Н), 2,38 (t, 2H), 3,95 (t, 2Н), 4.33 (q, 2H), 10,36 (s, 1Н).
c) 8-бром-3,4-дигидро-пирано[3,2-D]пиридо[1,2-а]пиримидин11(2Н)-он
Этил-3-оксотетрагидро-2Н-пиран-2-карбоксилат (90 мг, 0,53 ммоль) и 2-амино-5-бромпиридин (90,44 мг, 0,53 ммоль) растворяли в уксусной кислоте (2 мл). Смесь реагировала при 110°С в течение 5 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор концентрировали с получением неочищенного продукта, который очищали с помощью препаративной тонкослойной хроматографией с получением целевого продукта (25 мг, выход: 17%).
d) 2-метокси-5-(11-оксо-2,3,4,11-тетрагидро-пирано[3,2-d]пиридо [1,2-а]пиримидин-8-ил)пиридин-3-ил)-2,4-диметилтиазолил-5-сульфамид
[5-[(2,4-диметилтиазол-5-ил)сульфонил]-6-метокси-3-пиридил]борную кислоту (18,31 мг, 0,054 ммоль), К3РО4 (33,98 мг, 0,16 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (3,48 мг, 0,0054 ммоль) добавляли к раствору 8-бром-3,4-дигидро-пирано-[3,2-D]пиридо[1,2-а]пиримидин11(2Н)-она (15,00 мг, 0,054 ммоль) в тетрагидрофуране (4 мл) и воде (1 мл), смесь реагировала при 80°С в течение 5 часов. Окончание реакции определяли с помощью жидкостной масс-спектрометрии. Реакционный раствор фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого продукта (12,00 мг, выход: 27%).
1Н ЯМР (400 МГц, CDCl3) млн-1 δ 2,14-2,20 (m, 2Н), 2,45 (s, 3Н), 2,61 (s, 3Н), 2,91 (t, 2Н), 3,85 (s, 3Н), 4,30 (t, 2Н), 7,58 (dd, 1Н), 7,91 (dd, 1Н), 8,06 (d, 1Н), 8,30 (d, 1Н), 8,96 (d, 1Н).
Схема 23:
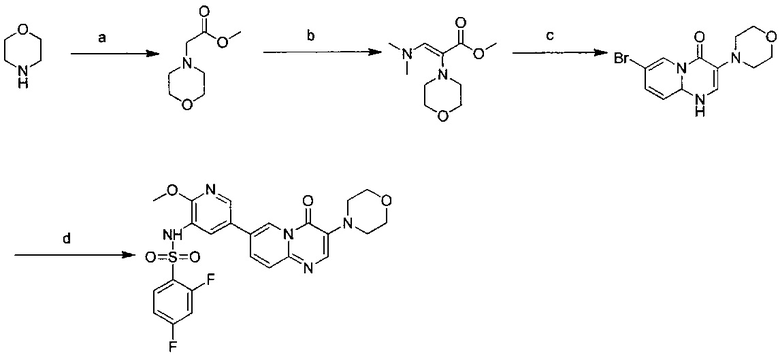
Условия: а) метилбромацетат, гидроксид калия, карбонат калия, дихлорметан, 40°С; b) 1-трет-бутокси-N,N,N',N'-тетраметилметандиамин, толуол, при нагревании; с) 5-бромпиридин-2-амин, уксусная кислота, 110°С; d) R борат, палладиевый реагент (тетракис(трифенилфосфин)палладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид и т.д.), K3РО4, тетрагидрофуран, вода, при нагревании.
Пример 241
2,4-дифтор-N-(2-метокси-5-(3-морфолинил-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
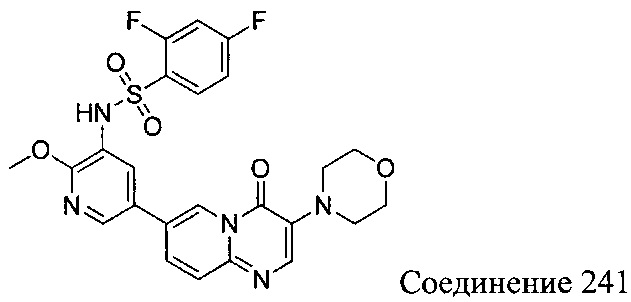
а) метил-2-морфолиноацетат
Морфолин (2,00 г, 22,9 ммоль), метилбромацетат (4,80 г, 31,4 ммоль), гидроксид калия (1,33 г, 23,6 ммоль), карбонат калия (3,30 г, 23,9 ммоль) и дихлорметан (50 мл) помещали в круглодонную колбу объемом 100 мл и перемешивали при комнатной температуре в течение 12 часов и затем перемешивали при 40°С в течение 6 часов. Окончание реакции определяли с помощью ТСХ. Реакционный раствор охлаждали до комнатной температуры и промывали насыщенным солевым раствором (10 мл × 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат, 1/3) с получением целевого соединения (2,80 г, 77%).
1H ЯМР (400 МГц, CDCl3) млн-1 δ 2,56-2,59 (m, 4Н), 3,22 (s, 2Н), 3,73 (s, 3Н), 3,74-3,76 (m, 4Н).
b) метил-7-метил-(Е)-3-(диметиламино)-2-морфолино-проп-2-еноат
Метил-2-морфолиноацетат (1,80 г, 11,3 ммоль), 1-трет-бутокси-N,N,N',N'-тетраметилметандиамин (2,36 г, 13,6 ммоль) и толуол (50 мл) помещали в круглодонную колбу объемом 100 мл и перемешивали при 120°С в течение 10 часов. Окончание реакции определяли с помощью ТСХ. Реакционный раствор концентрировали с получением целевого соединения (2,08 г, 86%), которое использовали на следующей стадии без дополнительной очистки.
c) 7-бром-3-морфолино-2,3-дигидропиридо[1,2-а]пиримидин-4-он
Метил-7-метил-(Е)-3-(диметиламино)-2-морфолино-проп-2-еноат (300 мг, 1,40 ммоль), 5-бром-2-аминопиридин (484 мг, 2,80 ммоль) и уксусную кислоту (5 мл) помещали в круглодонную колбу объемом 10 мл и нагревали с обратным холодильником в течение 4 часов. Окончание реакции определяли с помощью ЖХ/МС. Реакционный раствор концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир/этилацетат, от 5/1 до 1/1) с получением целевого соединения (80 мг, 18%).
1Н ЯМР (400 МГц, CD3OD) млн-1 δ 3,23-3,25 (m, 4Н), 3,88-3,90 (m, 4Н), 7,52 (d, 1Н), 7,77-7,80 (m, 1Н), 8,02 (s, 1Н), 9,09 (s, 1Н).
d) 2,4-фтор-N-(2-метокси-5-(3-морфолинил-4-оксо-4Н-пиридо[1,2-а]пиримидин-7-ил)пиридин-3-ил)бензсульфамид
7-бром-3-морфолино-2,3-дигидропиридо[1,2-а]пиримидин-4-он (50 мг, 0,160 ммоль), 2,4-дифтор-N-[2-метокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3-пиридинил]бензсульфамид (68 мг, 0,160 ммоль), фосфат калия (68 мг, 0,320 ммоль), тетрагидрофуран (1 мл) и воду (0,1 мл) помещали в круглодонную колбу объемом 10 мл и добавляли [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий хлорид (10 мг) в атмосфере азота. Смесь перемешивали при 70°С в течение 2 часов. Окончание реакции определяли с помощью ЖХ/МС. Реакционный раствор концентрировали и очищали остаток с помощью препаративной ВЭЖХ с получением целевого продукта (20 мг, 24%).
1Н ЯМР (400 МГц, DMSO-d6) млн-1 δ 3,16-3,25 (m, 4Н), 3,69 (s, 3Н), 3,75-3,83 (m, 4Н), 7,17-7,27 (m, 1Н), 7,55-7,13 (m, 1Н), 7,70 (d, 1Н), 7,78-7,80 (m, 1Н), 7,98 (s, 1Н), 8,03 (s, 1Н), 8,44 (s, 1Н), 8,98 (s, 1Н).
Следующие 43 соединения были также синтезированы согласно способу получения Соединения 175.
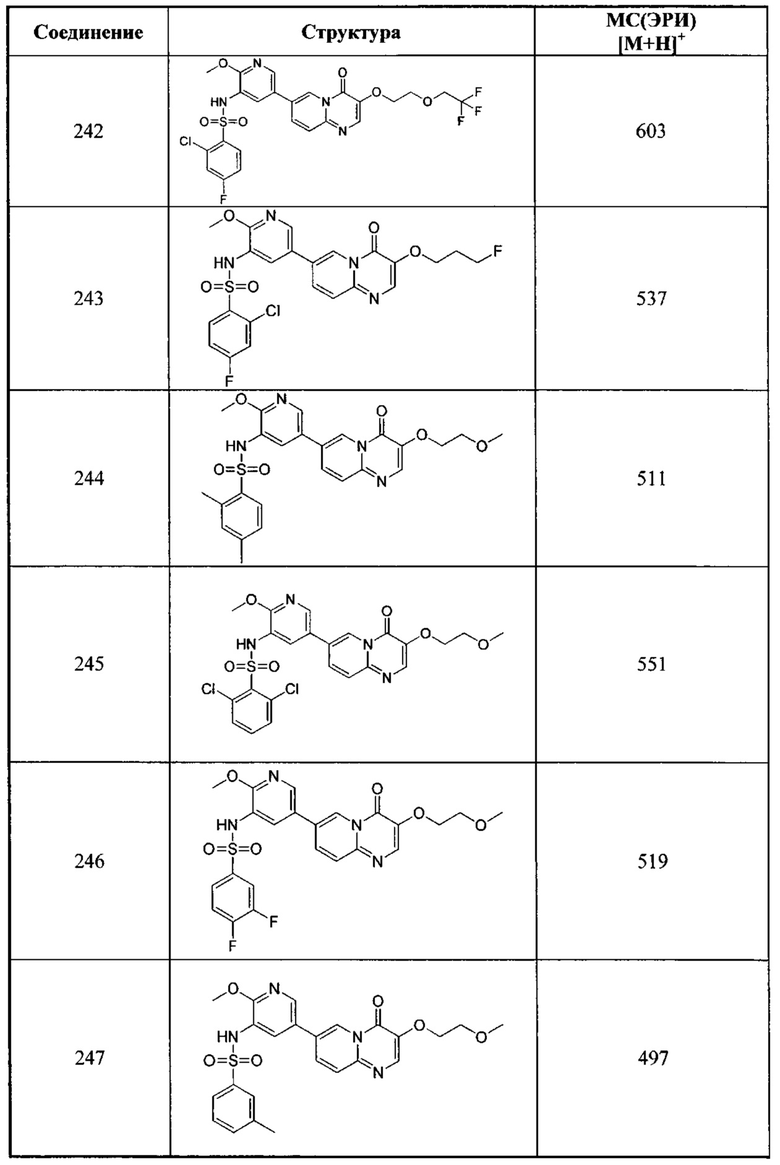
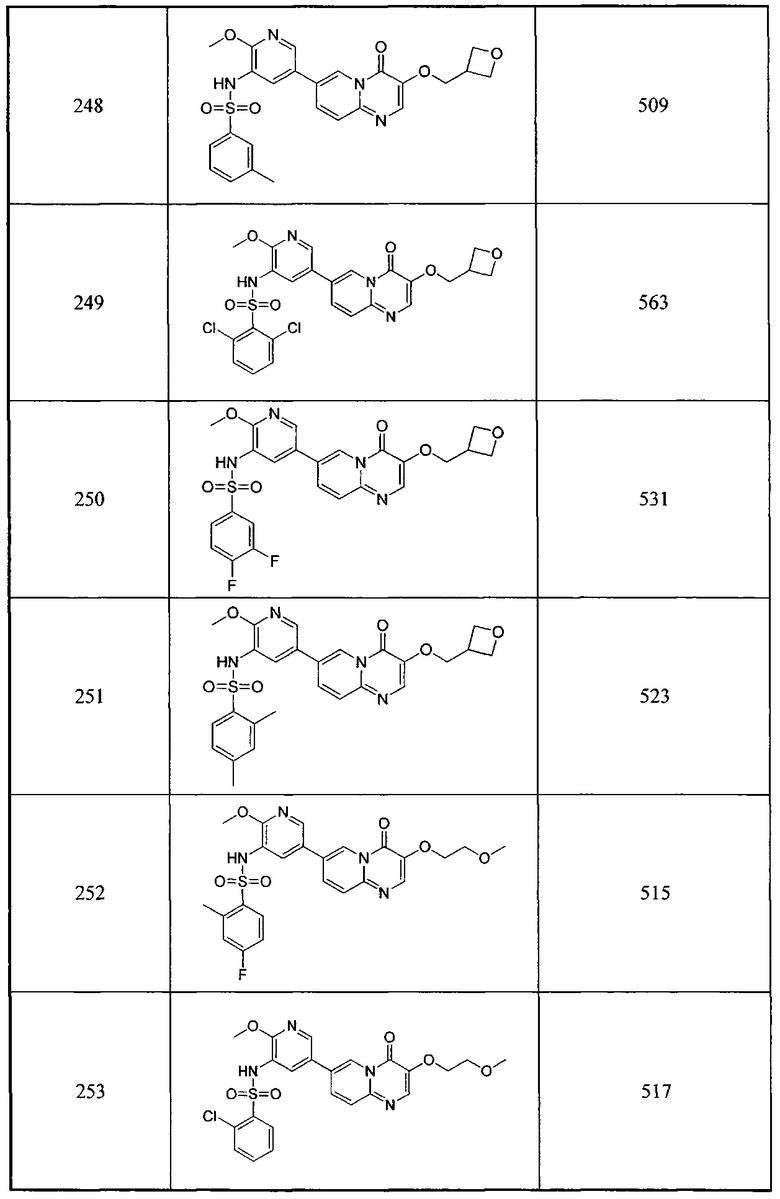
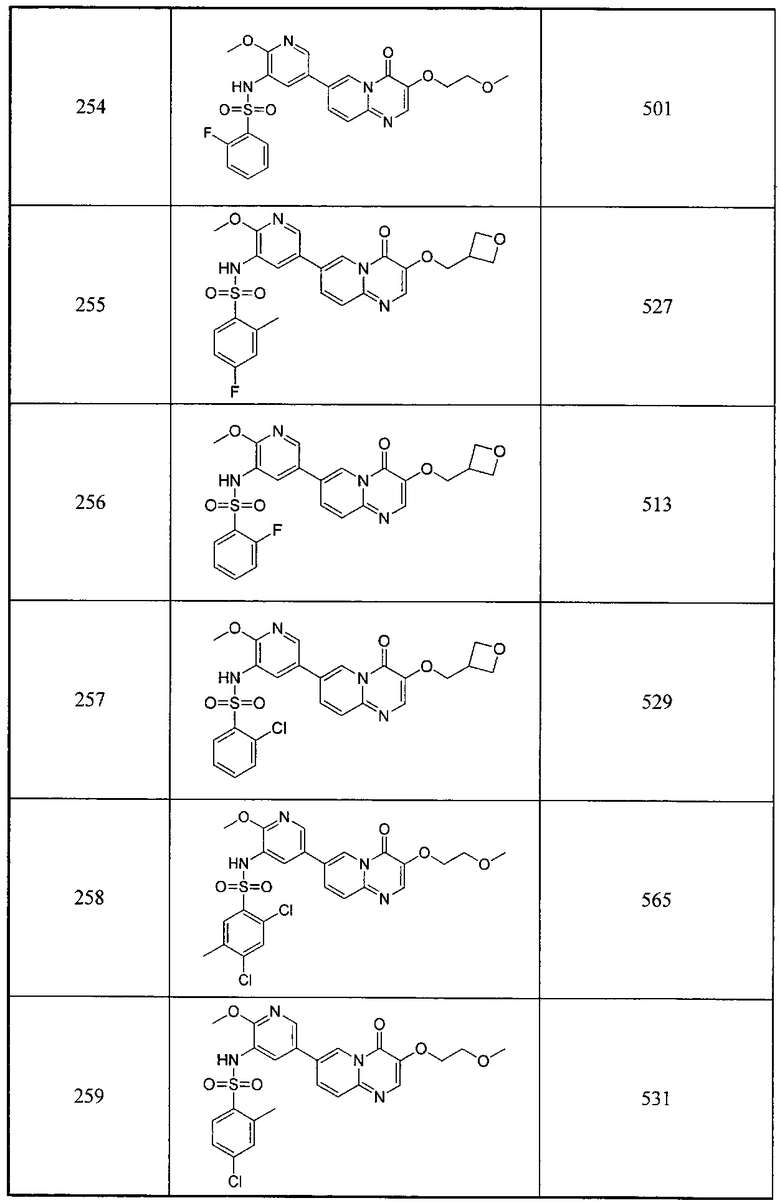
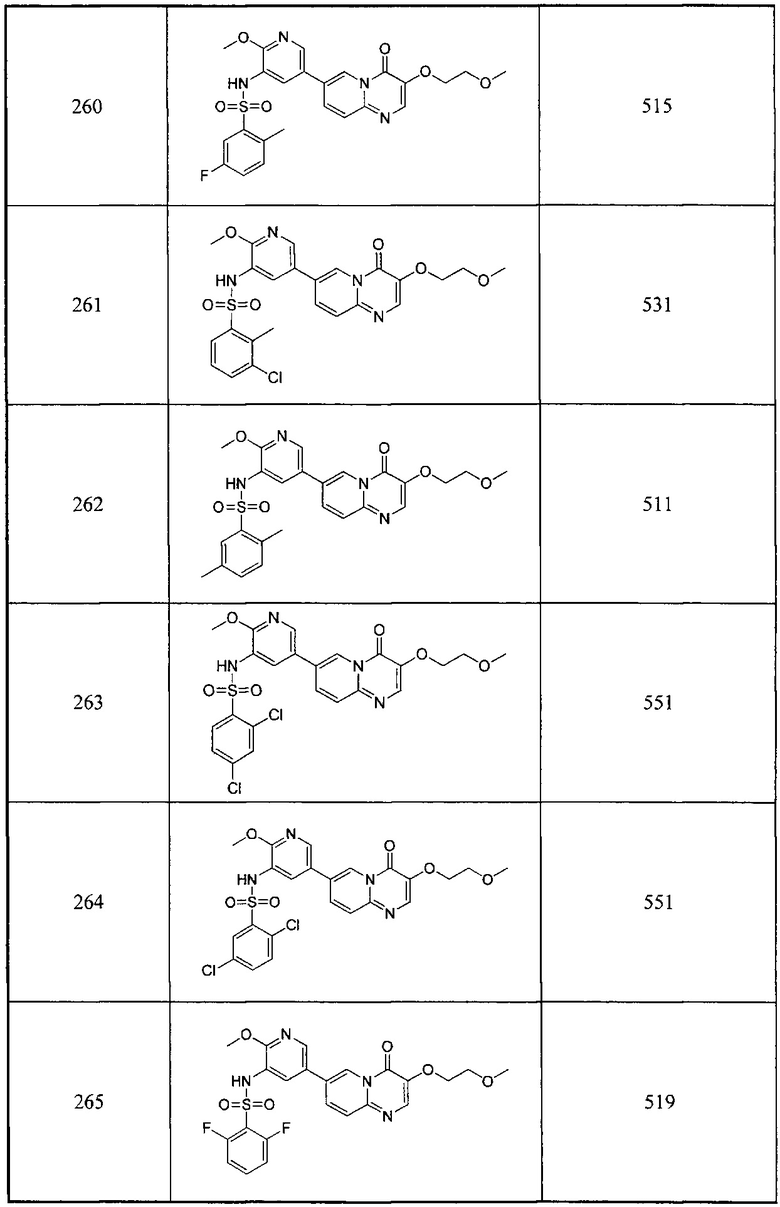
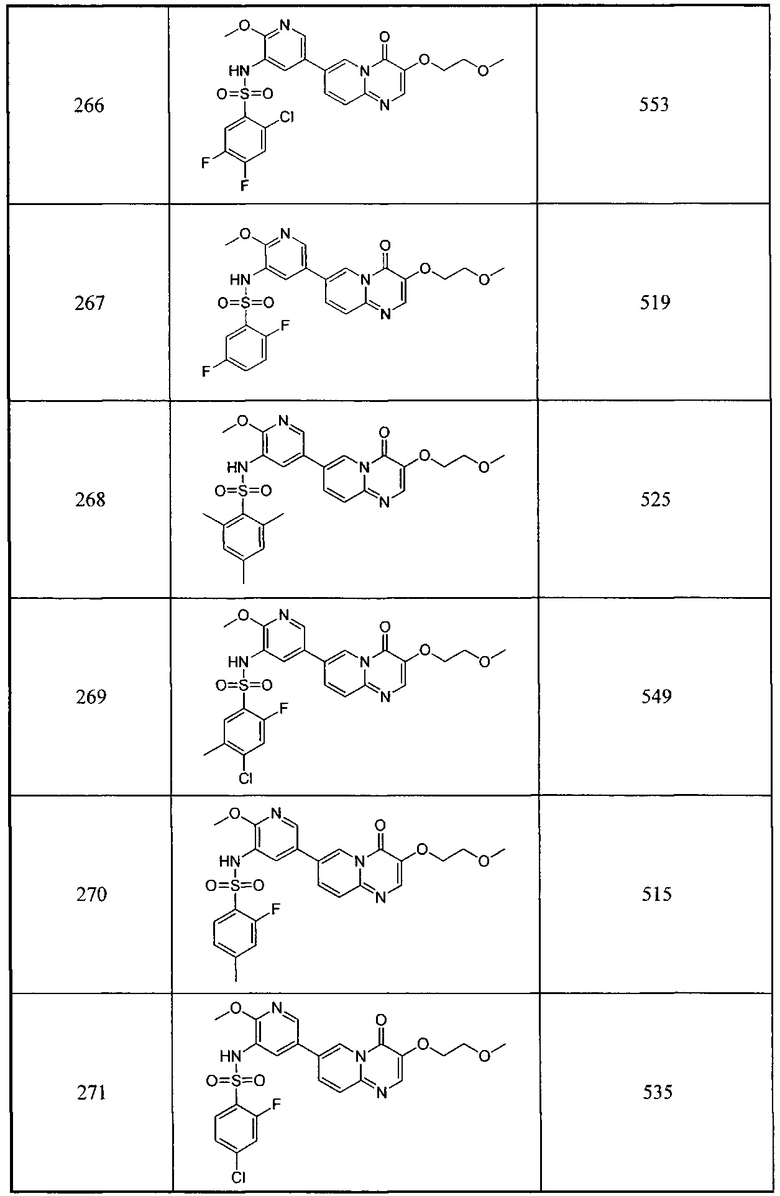
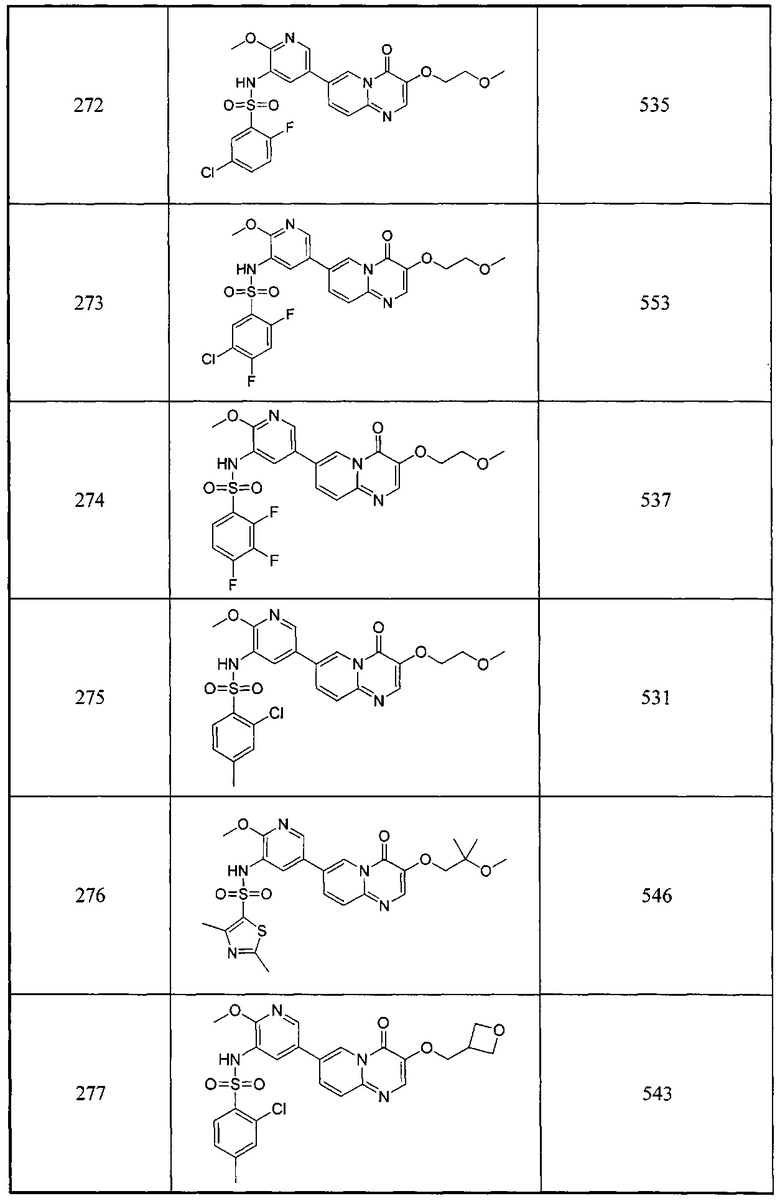
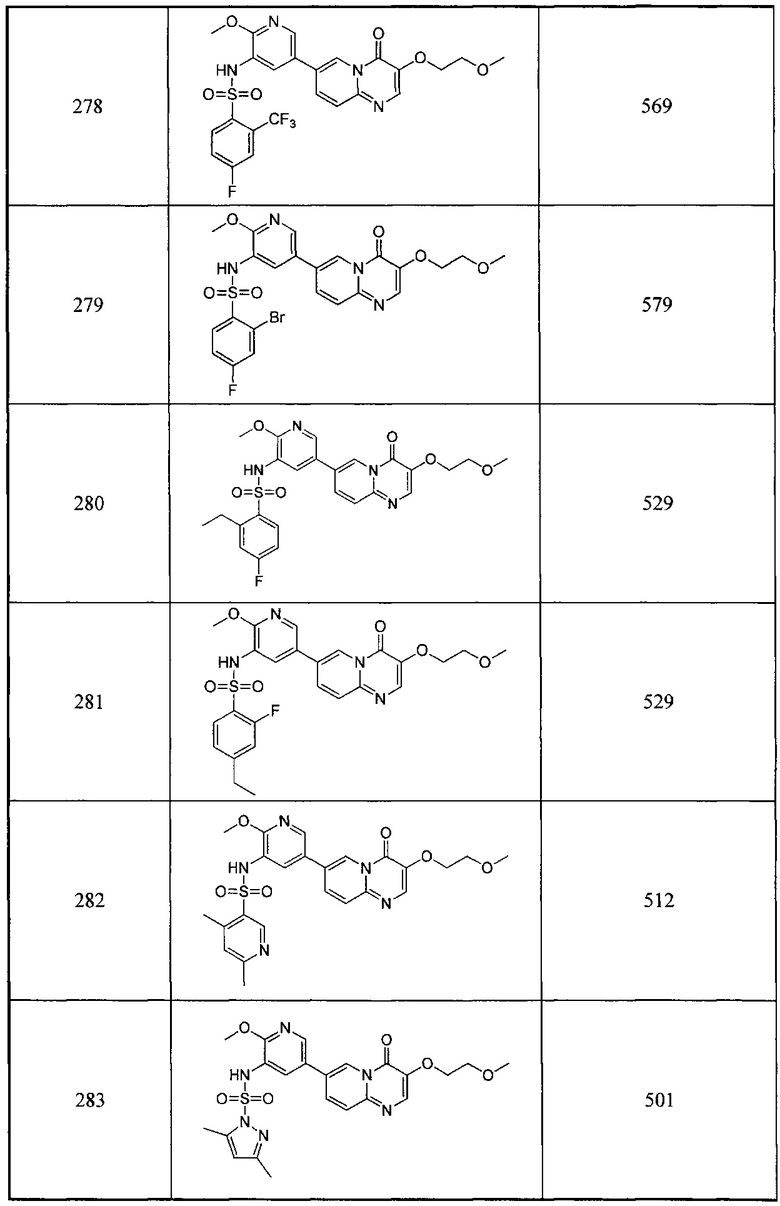
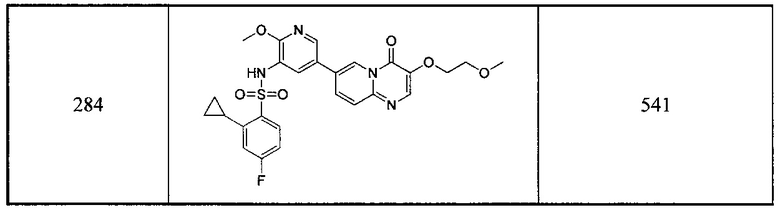
Пример исследований клеточных активностей in vitro
Стадии и методика проведения эксперимента:
1) клетки MCF-7 были посеяны в количестве 2,5×104 клеток/лунка в 96-луночные планшеты (используемая культуральная среда должна быть полной культуральной средой, содержащей 10% FBS (фетальной бычьей сыворотки)).
2) На следующий день среду удаляли из каждой лунки. Определенную концентрацию (предварительный отбор) или серию концентраций (исследование IC50) соединений растворяли в культуральной среде без сыворотки и добавляли в 96-луночные планшеты для культивирования клеток в течение 2 часов.
3) В культуральной среде без сыворотки растворяли инсулин, добавляли к культивируемым клеткам и инкубировали в течение 30 минут, при этом конечная концентрация инсулина составила 10 мг/мл.
4) В период ожидания реакции был приготовлен лизирующий раствор в соответствии со следующим способом:
a) Усиливающий раствор заранее доставали из холодильника, чтобы он растаял.
b) Усиливающий раствор десятикратно разбавляли 5Х лизирующим буфером с получением концентрированного лизирующего раствора.
c) Концентрированный лизирующий раствор пятикратно разбавляли дважды дистиллированной водой с получением лизирующего раствора.
5) Из каждой лунки полностью удаляли среду и быстро однократно промывали каждую лунку PBS (натрий-фосфатным буфером).
6) 150 мкл свежеприготовленного лизирующего раствора добавляли в каждую лунку и встряхивали при комнатной температуре в течение 10 минут.
7) Убедившись в том, что все клетки были отделены, лизирующий раствор вместе с фрагментами клеток переносили в пробирку объемом 1,5 мл.
8) Пробирку встряхивали на вихревом миксере, чтобы полностью смешать клетки и лизирующий раствор, и затем центрифугировали смесь при 4°С при 12000 g в течение 10 минут.
9) Рассчитывали необходимое число стрипов микролуночного планшета ELISA-one. Ненужные стрипы были удалены из рамки и упакованы обратно в пакет. Перед использованием стрипов микролуночного планшета использовали 200 мкл дважды дистиллированной воды для промывки каждой лунки, чтобы удалить консервант.
10) В каждую лунку добавляли 50 мкл смеси антитела. (Раствор смеси антитела получали смешиванием питательной среды антитела и реагента-антитела, меченого ферментом, в равных долях. Для получения смеси антитела не требовалось перемешивание вихревым способом.)
11) В каждую лунку микролуночного планшета ELISA-One добавляли 25 мкл клеточных лизатов. Микролуночный планшет герметично закрывали клейкой пленкой и инкубировали на микропланшетном шейкере при комнатной температуре в течение 1 часа.
12) Каждую лунку трижды промывали 150 мкл 1X промывающего буфера. После последнего промывания промывающий буфер полностью удаляли из лунки. При необходимости 1X промывающий буфер можно оставить в микролуночном планшете до 30 минут, чтобы в течение этого периода времени можно было приготовить смешанный раствор субстрата.
13) Смешанный раствор субстрата должен быть приготовлен непосредственно перед каждым применением. 10 мкл смешанного раствора субстрата добавляли в каждую лунку, герметично закрывали оловянной фольгой и инкубировали на микропланшетном шейкере при комнатной температуре в течение 10 минут.
14) В каждую лунку добавляли 10 мкл стоп-раствора и слегка перемешивали (5-10 секунд) на микропланшетном шейкере.
15) Устанавливали соответствующую группу фильтров ELISA-One и использовали ее для определения интенсивности сигнала флуоресценции.
Результаты исследования приведены в таблице 1.
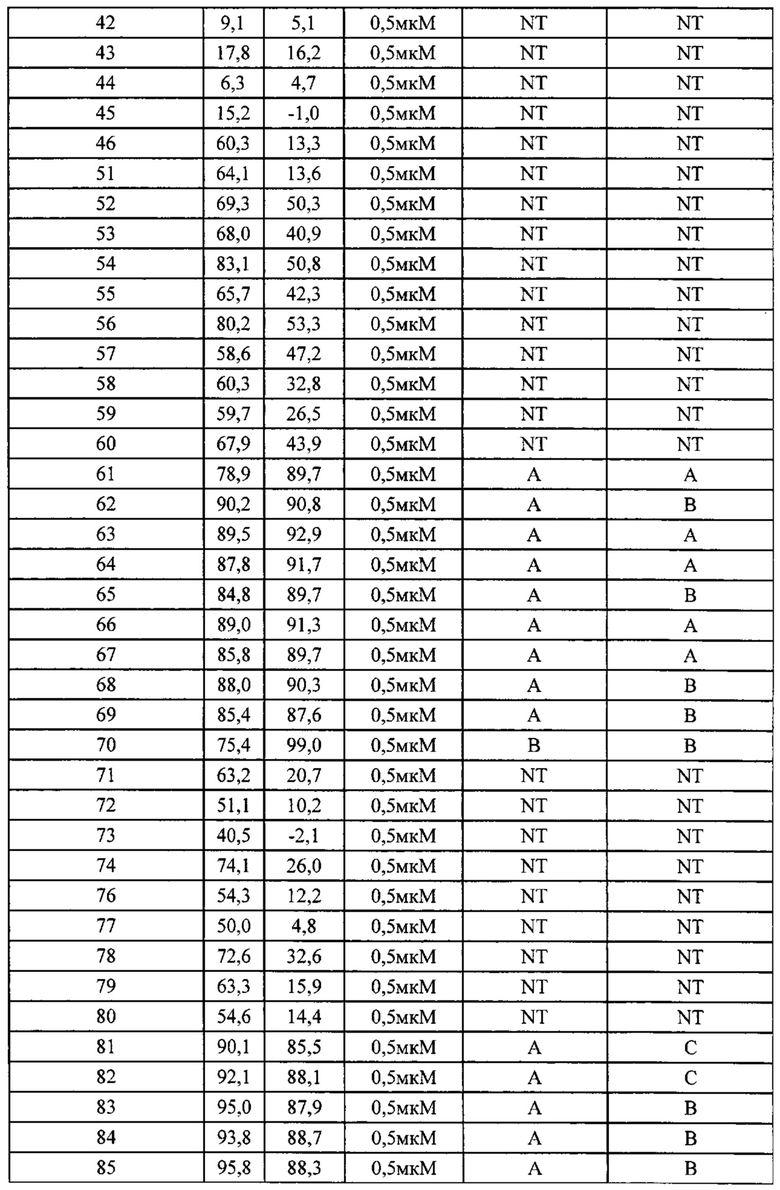
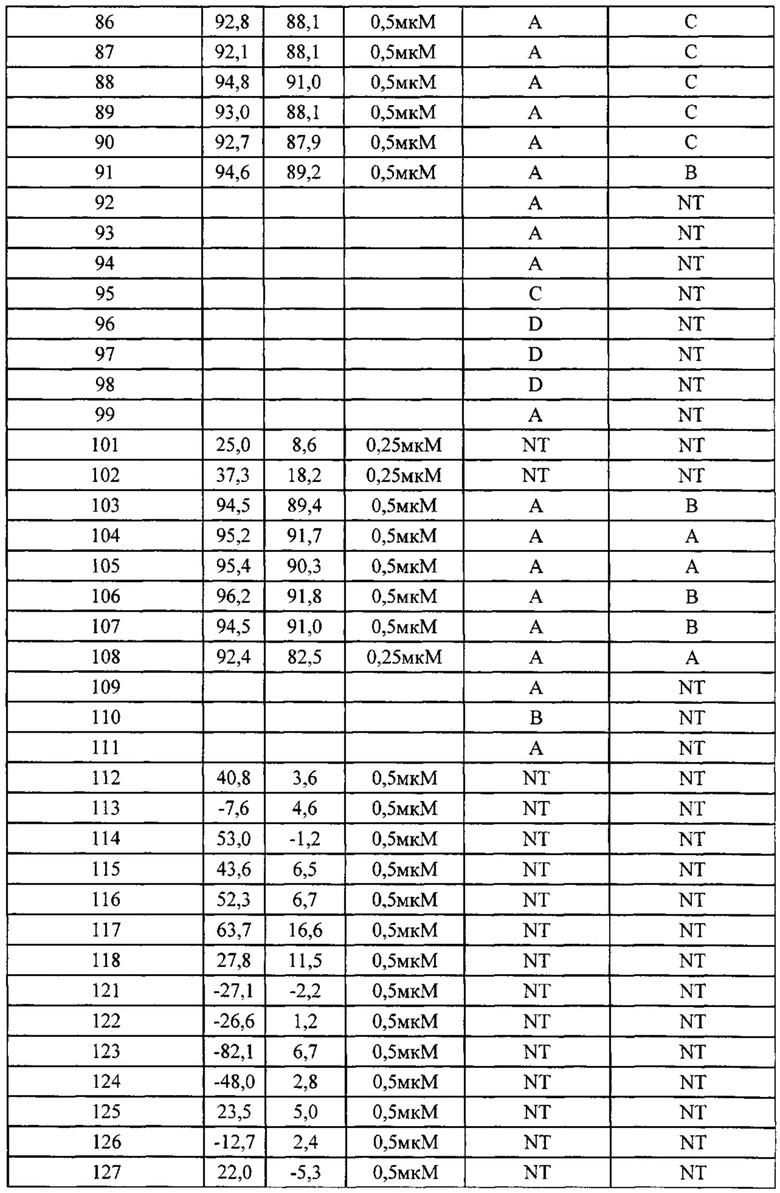
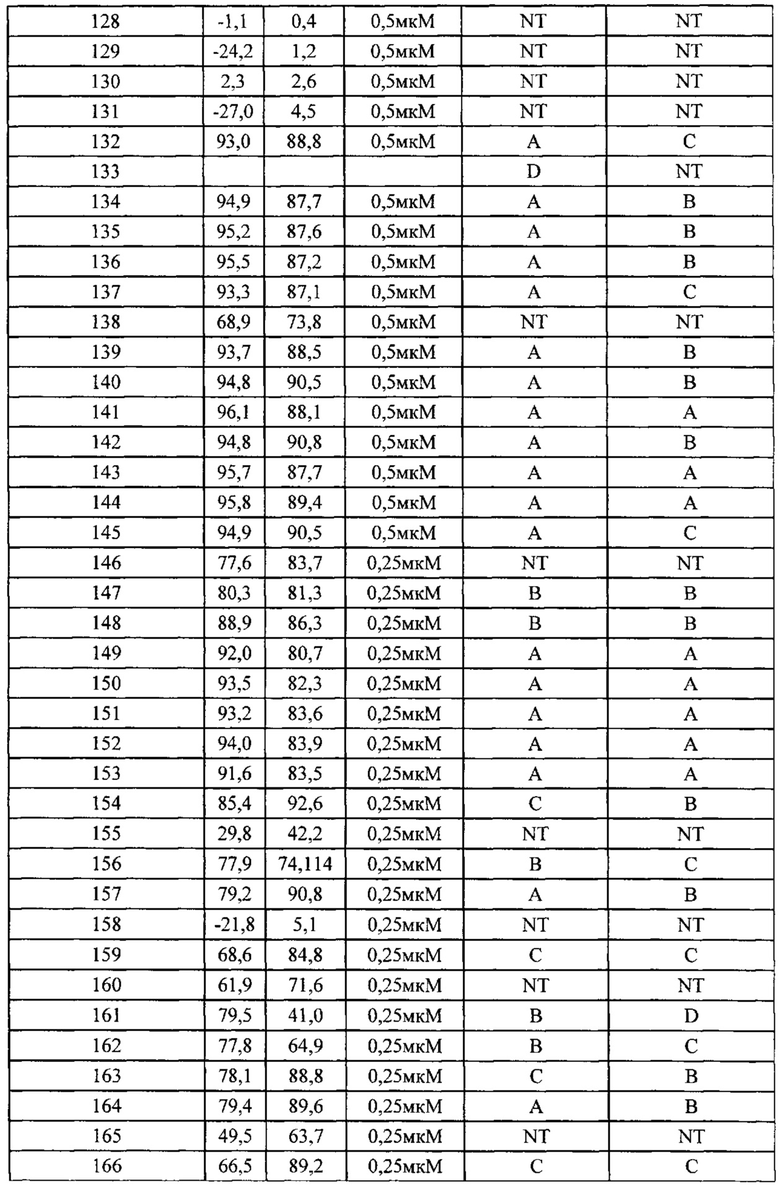
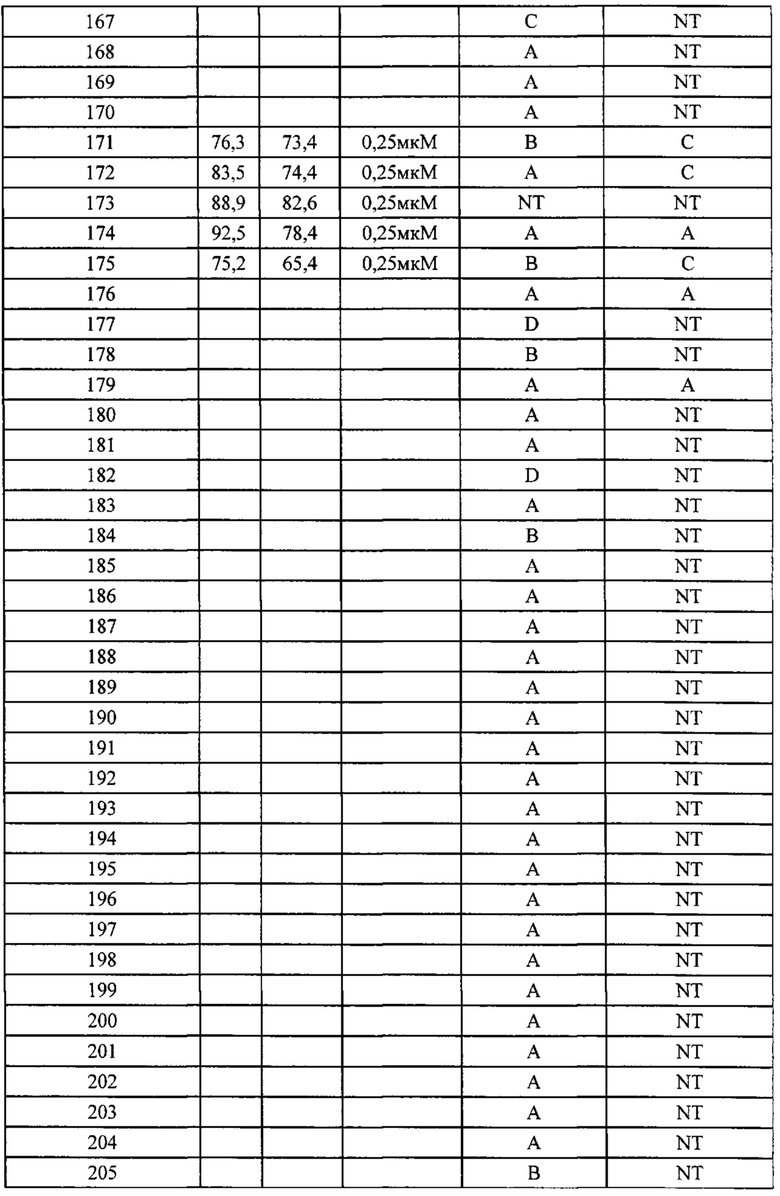
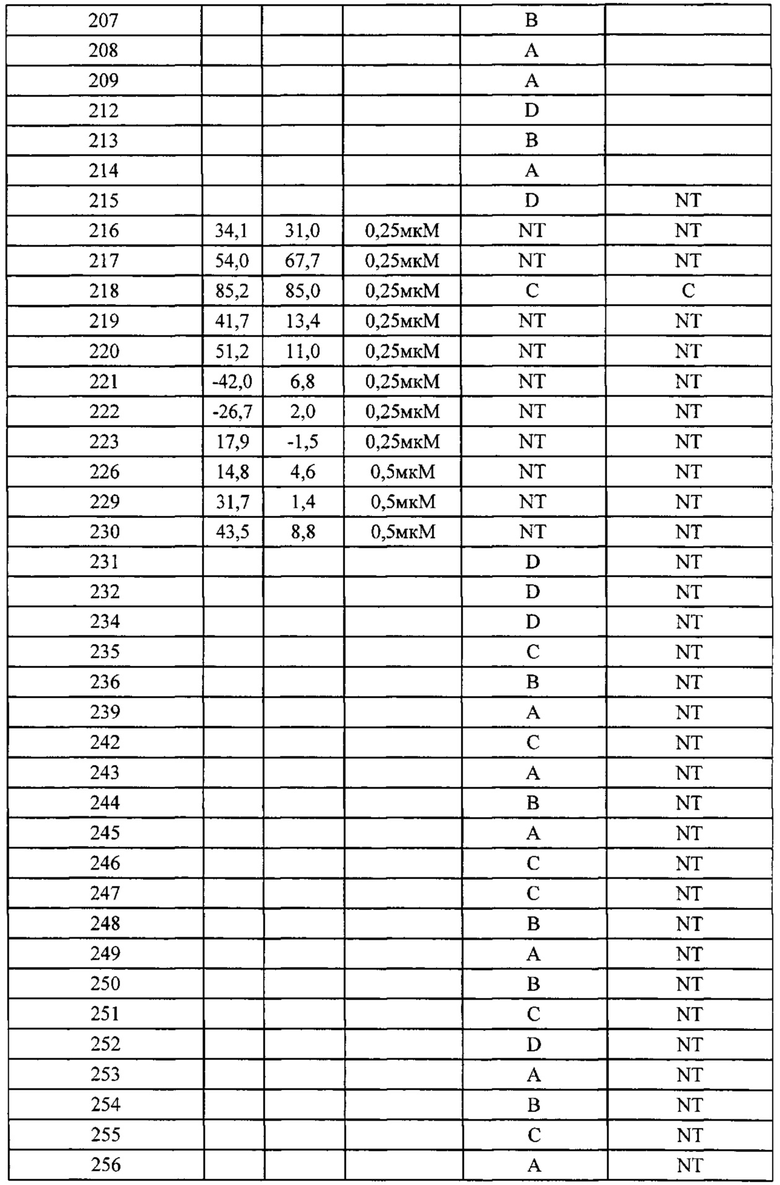
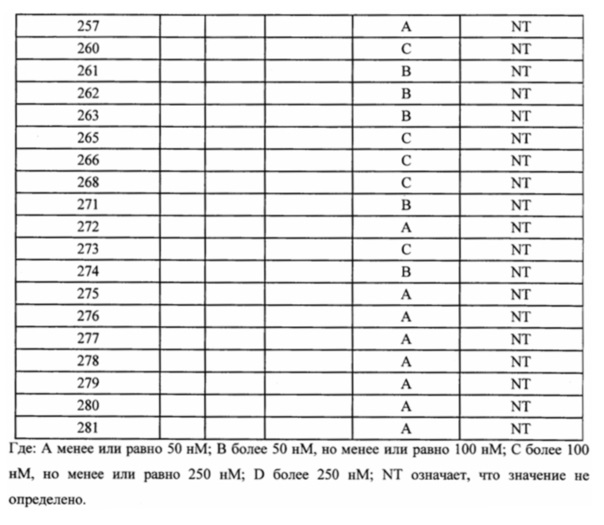
Вывод: соединения по настоящему изобретению обладают существенным ингибирующим воздействием на mTOR/PBK.
Пример исследования ферментативной активности in vitro
1. Порядок проведения эксперимента и способы исследования киназы PI3K (p110α):
1) Цель эксперимента
Цель эксперимента состояла в оценке ингибирующего воздействия исследуемых образцов на активность киназы PI3K (p110α) на молекулярном уровне.
2) Методика проведения эксперимента
исследование PI3K методом гомогенной флюоресценции с временным разрешением (HTRF)
a) Основной инструмент
Ридер PerkinElmer Envision 2104 Multilabel Reader.
b) Основные реагенты
Набор для исследования PI 3 киназы методом HTRF (384 лунки) был приобретен у компании Upstate (Millipore); киназа PI3K (p110α) была изготовлена в лаборатории заявителя.
с) Порядок проведения эксперимента
Каждый раствор готовили в соответствии с инструкциями в наборе для исследования PI 3 киназы методом HTRF. Киназную реакцию проводили в белых 384-луночных планшетах (Proxiplate-384 plus). 0,5 мкл DMSO (концентрация соответствовала самой высокой концентрации DMSO для исследуемых соединений) добавляли в каждую из двух контрольных лунок с ферментом и без него, затем в каждую лунку добавляли 0,5 мкл серии различных концентраций исследуемых соединений. Киназную реакционную жидкость (10 мкМ субстрата PIP2, 0,5 нг PI3K (p110α)) добавляли в контрольную лунку, содержащую фермент, и исследуемые лунки, в то время как в контрольную лунку, не содержащую фермента, добавляли только рабочую реакционную жидкость (10 мкМ субстрата PIP2), и, наконец, добавляли 5 мкМ АТФ рабочей реакционной жидкости для активации реакции. После проведения реакции при комнатной температуре в течение 30 минут в каждую лунку добавляли стоп-раствор для остановки киназной реакции. После полного смешивания смеси в каждую лунку добавляли детектирующую жидкость и полностью смешивали. Планшет герметично закрывали, используя Parafilm, и помещали в темноту. После инкубирования в течение ночи проводили измерение смеси. Заданные условия детектора приведены в таблице 2.

Данные HTRF (гомогенной флуоресценции с временным разрешением) были рассчитаны в соответствии со следующей формулой:
Отношение HTRF = Эмиссия при 665 нм / Эмиссия при 620 нм × 10000
Относительная степень ингибирования (%) = (значение HTRF исследуемой лунки - значение HTRF контрольной лунки, содержащей фермент) / (значение HTRF контрольной лунки, не содержащей фермент - значение HTRF контрольной лунки, содержащей фермент) × 100
Значение IC50 было рассчитано с использованием программного обеспечения GraphPad после нанесения на график зависимости относительной степени ингибирования от концентрации.
2. Порядок проведения эксперимента и способы исследования киназы mTOR
1) Цель эксперимента
Цель эксперимента состояла в оценке ингибирующего воздействия исследуемых образцов в отношении активности киназы mTOR на молекулярном уровне.
2) Методика эксперимента
Исследование киназы mTOR
a) Основной инструмент
Ридер PerkinElmer Envision 2104 Multilabel Reader.
b) Основные реагенты
Набор для исследования mTOR Kinase Assay (384 лунок) был приобретен у компании PerkinElmer; киназа mTOR была изготовлена в лаборатории заявителя.
c) Порядок проведения эксперимента
Каждый раствор был приготовлен в соответствии с инструкциями в наборе mTOR Kinase Assay. Киназную реакцию проводили в белых 384-луночных планшетах (Proxiplate-384 plus). 2,5 мкл DMSO (концентрация соответствовала самой высокой концентрации DMSO для исследуемых соединений) добавляли в каждую из двух контрольных лунок с ферментом и без него, затем в каждую исследуемую лунку добавляли 2,5 мкл серии различных концентраций исследуемых соединений. Смесь пептида ULight-4E-BP1 (Thr37/46)/АТФ (конечная концентрация АТФ составила 100 мкМ) и 5 мкл киназы mTOR добавляли в контрольную лунку, содержащую фермент, а также в лунки с исследуемыми соединениями, и полностью смешивали. Планшет герметично закрывали Parafilm и инкубировали в течение 2 часов. Затем добавляли 5 мкл стоп-раствора и инкубировали в течение 5 минут. После этого добавляли 5 мкл смеси для детектирования (антитело Eu-anti-phospho-4E-BP1 (Thr37/46), конечная концентрация составила 2 нМ) и инкубировали в течение 1 часа, и затем проводили детектирование смеси. Заданные условия детектора приведены в таблице 3.

Данные HTRF (гомогенной флуоресценции с временным разрешением) были рассчитаны в соответствии со следующей формулой:
Отношение HTRF = Эмиссия при 665 нм / Эмиссия при 620 нм × 10000
Относительная степень ингибирования (%) = {1 - (значение HTRF исследуемой лунки - значение HTRF контрольной лунки, содержащей фермент) / (значение HTRF контрольной лунки, содержащей фермент - значение HTRF контрольной лунки, не содержащей фермент)} × 100%
Значение IC50 было рассчитано с использованием программного обеспечения GraphPad после нанесения на график зависимости относительной степени ингибирования от концентрации.
Результаты исследования приведены в таблице 4.
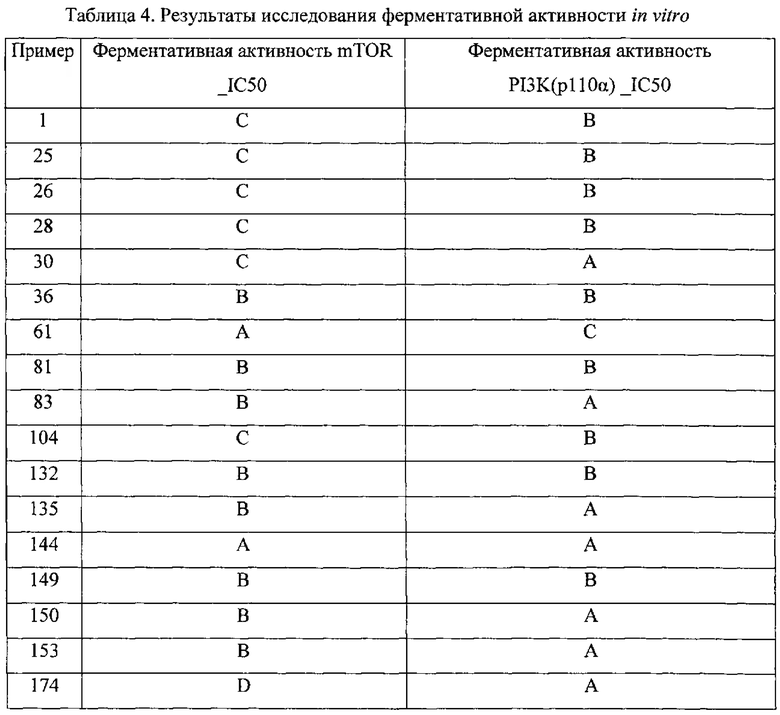
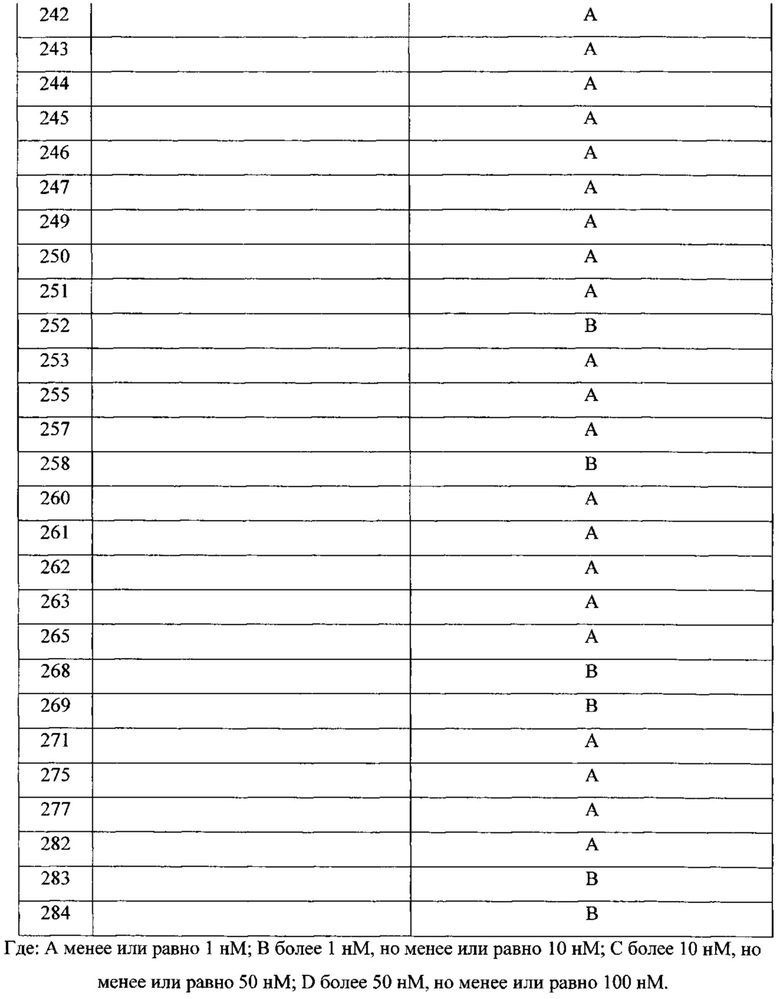
Фармакодинамическое исследование in vivo:
Исследования проводились, чтобы определить, обладают ли исследуемые лекарственные средства эффективностью in vivo на животной модели рака яичника SK-OV-3 и животной модели рака предстательной железы РС-3М. Описание кормления животных, компонентов корма, наблюдений в ходе эксперимента, экспериментальных критериев, прекращения эксперимента, а также анализа данных приведены ниже.
Кормление животных: Животных доставляли к месту эксперимента и кормили в течение 3-7 дней до начала эксперимента. Животных держали в клетках, снабженных IVF (системой независимой подачи воздуха), 5 животных на клетку, в комнатах для животных, свободных от патогенных микроорганизмов. Все клетки, подстилки и питьевая вода обязательно стерилизовались перед использованием, учетные данные по стерилизации приведены в приложении. При работе весь персонал лаборатории в комнате для животных находился в защитной одежде и латексных перчатках. На каждой клетке была информация, указывающая на число животных в клетке, пол, линию, дату получения, режим введения, экспериментальный номер, группу и дату вступления животного в эксперимент. Клетки, корм и воду заменяли два раза в неделю. Условия кормления и освещения были следующими:
температура: 20~26°С;
влажность: 40~70%;
длина светового дня: 12 ч со светом, 12 ч без света.
Компоненты корма: Корм соответствовал опознавательным стандартам корма для экспериментальных животных. Максимальное содержание загрязняющих веществ находилось в контролируемом диапазоне и контролировалось изготовителем. Для питья использовали питьевую воду, стерилизованную высоким давлением.
Группы животных: перед введением доз животных взвешивали и измеряли объем опухоли. Животных случайно группировали в соответствии с объемом опухоли (схема рандомизированных блоков).
Наблюдения: Осуществление разработки протокола и любые модификации были оценены и одобрены комитетом по этике по отношению к животным (IACUC) WuXi АРР Тес (Шанхай). Использование животных и качество их жизни регулировались правилами Международной ассоциации по оценке и аккредитации условий содержания лабораторных животных (AAALAC). За состоянием здоровья и статусом смертности животных следили ежедневно. Плановые осмотры включали наблюдения за ростом опухолей и влиянием лечения лекарственными средствами на повседневное поведение животных, такое как активность, прием пищи и воды, изменения в весе (измерялся дважды в неделю), внешний вид или другие отклонения от нормы. Число смертей животных и побочные эффекты в каждой группе регистрировались на основании числа животных в каждой группе. Соответствующие учетные данные прилагаются.
Экспериментальные критерии: Определение ингибирования, задержки роста опухоли или излечения проводилось с помощью экспериментальных критериев. Диаметр опухоли измеряли дважды в неделю с использованием штангенциркуля. Объем опухоли измеряли в соответствии со следующей формулой: V=0,5а×b2, где а и b обозначают больший диаметр и меньший диаметр опухоли, соответственно. Ингибирование роста опухоли (TGI) соединением оценивали в Т-С (сутки) и Т/С (%). Т-С (сутки) обозначает задержку роста опухоли. Т представляет собой среднее количество суток, в течение которых опухоль в группе введения достигала заранее определенного объема (например, 1,000 мм3), С представляет собой среднее число суток, в течение которых опухоль в контрольной группе достигала того же объема. Процент Т/С (%) обозначает отношение ингибирования роста опухоли, и Т и С представляют собой массу опухоли (объем опухоли) в группе введения и в контрольной группе в заданный день, соответственно.
Отношение ингибирования роста опухоли рассчитывали по следующей формуле:
TGI (%)=[1-(Ti-T0)/(Vi-V0)]×100,
где Ti представляет собой средний объем опухолей в определнной группе введения в заданный день, ТО представляет собой средний объем опухолей в этой группе введения в начале введения. Vi представляет собой средний объем опухолей в контрольной группе, получавшей наполнитель, в заданный день (тот же день, что и для Ti), V0 представляет собой средний объем опухолей в контрольной группе, получавшей носитель, в начале введения. В конце эксперимента измеряли массу опухоли и рассчитывали процент Т/С. Т и С представляют собой массу опухоли в группе введения и в контрольной группе, получавшей наполнитель, соответственно.
Прекращение эксперимента: Если состояние здоровья животного продолжало ухудшаться, или объем опухоли превышал 2,000 мм3, или животное имело серьезное заболевание или боль, животное усыпляли. Ветеринаров извещали и усыпляли животное в следующих случаях:
Животные становились очень худыми, и потеря массы составляла более 20%.
Животные не могли есть и пить без затруднений.
Средний объем опухолей в контрольной группе достигал 2,000 мм3, и эксперимент прекращали.
Животные демонстрировали следующие клинические проявления, и их состояние продолжало ухудшаться:
Пилоэрекция
Выгнутая спина
Бледное ухо, нос, глаз или ступня
Затрудненное дыхание
Судорожные припадки
Продолжительная диарея
Обезвоживание
Медленные движения
Звук
Анализ данных: Три или более групп сравнивали посредством однофакторного дисперсионного анализа. Если значение F существенно различалось, после однофакторного дисперсионного анализа проводили несколько сравнений. Весь анализ данных был проведен с использованием статистического программного обеспечения SPSS 17.0. p менее 0,05 считали существенным различием.
Фармакодинамическое исследование in vivo тестируемых лекарственных средств на моделях подкожной ксенотрансплантатной модели опухоли с клетками рака яичника человека SK-OV-3:
Планирование эксперимента
Клеточная культура: Клетки рака яичника человека SK-OV-3 (АТСС, Manassas, VA, номер партии: НТВ-77) культивировали в монослое in vitro в среде McCoy 5А, обогащенной 10% FBS, 100 ЕД/мл пенициллина и 100 мкг/мл стрептомицина при 37°С в инкубаторе с 5% СО2. Для периодического отделения и пассирования использовали трипсин-ЭДТА дважды в неделю. Когда клетки достигали 80%-90% конфлюэнтности, и их число достигало требуемого значения, клетки собирали, считали и высевали.
Животные: бестимусные мыши BALB/c, самки, 4-х недельного возраста, массой тела 12-14 г, были предоставлены Shanghai Sippr - ВК Laboratory Animal Co., Ltd.
Посев опухоли: 0,2 мл (1×107) клеток SK-OV-3 (с матриксным гелем, объемное отношение 1:1) сеяли подкожно в правую часть спины каждой мыши. Когда средний объем опухолей достигал приблизительно 100-200 мм3, мышей разделяли на группы и вводили каждой группе лекарственные средства.
Результаты фармакодинамики in vivo: см. Фиг. 1.
Экспериментальное фармакодинамическое исследование in vivo тестируемых лекарственных средств на моделях подкожной ксенотрансплантатной модели опухоли с клетками рака предстательной железы человека РС-3М:
Цель эксперимента: фармакодинамическое исследование in vivo тестируемых соединений в подкожных ксенотрансплантатных опухолевых моделях клеток рака предстательной железы человека РС-3М.
Планирование эксперимента
Клеточная культура: Клетки рака предстательной железы человека РС-3М культивировали в среде RPMI-1640, обогащенной 10% FBS, 100 ЕД/мл пенициллина и 100 мкг/мл стрептомицина при 37°С в инкубаторе с 5% СО2. Для периодического отделения и пассирования использовали трипсин-ЭДТА дважды в неделю. Когда клетки достигали 80%-90% конфлюэнтности, и их число достигало требуемого значения, клетки собирали, считали и высевали.
Животные: бестимусные мыши BALB/c, самцы, 4-х недельного возраста, массой тела 12-14 г, были предоставлены Shanghai Sippr - ВК Laboratory Animal Co., Ltd.
Посев опухоли: 0,2 мл (1×107) клеток РС-3М сеяли подкожно в правую часть спины каждой мыши. Когда средний объем опухолей достигал приблизительно 150-200 мм3, мышей разделяли на группы и вводили каждой группе лекарственные средства.
Результаты фармакодинамики in vivo: см. Фиг. 2-1, Фиг. 2-2, Фиг. 2-3а и Фиг. 2-3b.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ГЕТЕРОАРИЛ[4,3-с]ПИРИМИДИН-5-АМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2018 |
|
RU2764655C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2667498C2 |
| ИМИДАЗО[1,2-А]ПИРИДИНОВЫЕ И ПИРАЗОЛ[2,3-А]ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2000 |
|
RU2248976C2 |
| ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ 2Н-ПИРАЗОЛА | 2016 |
|
RU2722363C2 |
| ИНГИБИТОР ГЕКСОН-ГЛЮКОКИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2797185C2 |
| Новые соединения пиридопиримидинона для модулирования каталитической активности гистонлизиндеметилаз (KDMS) | 2015 |
|
RU2684396C2 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2015 |
|
RU2681537C2 |
| ПРОИЗВОДНЫЕ ПИРИДИЛКЕТОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2014 |
|
RU2667892C2 |
| ИНГИБИТОР EGFR И ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2702631C2 |
| ПРОИЗВОДНОЕ ИНДАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2817737C2 |
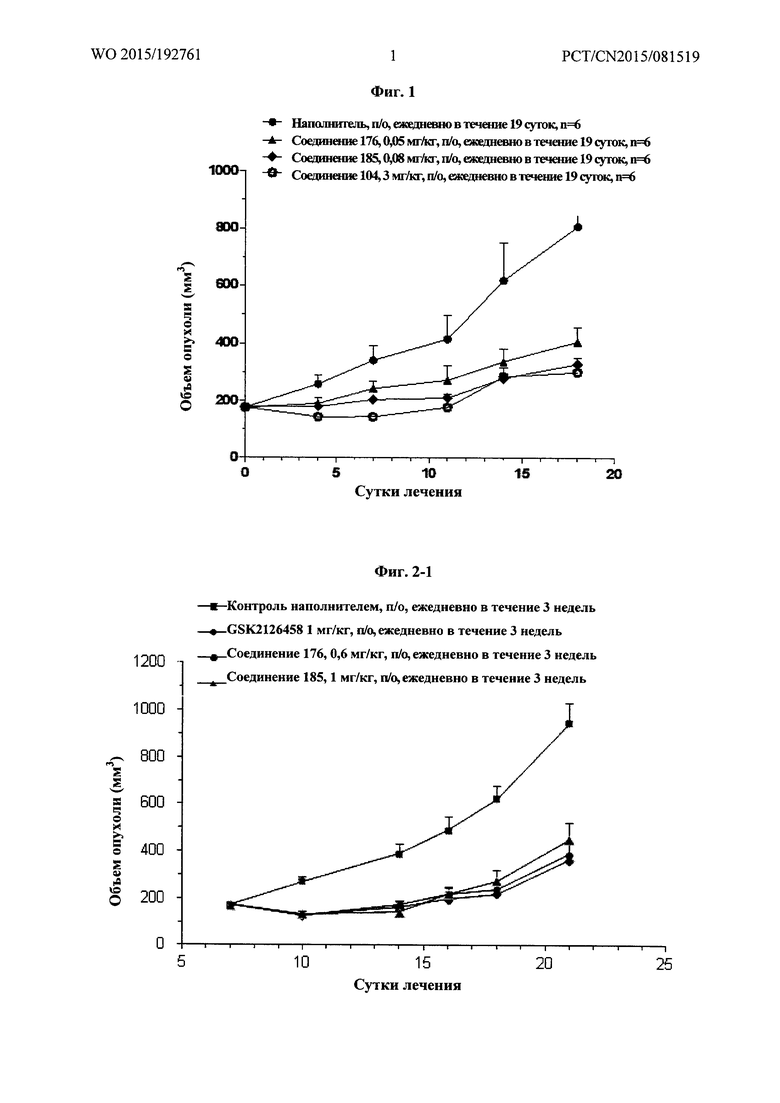
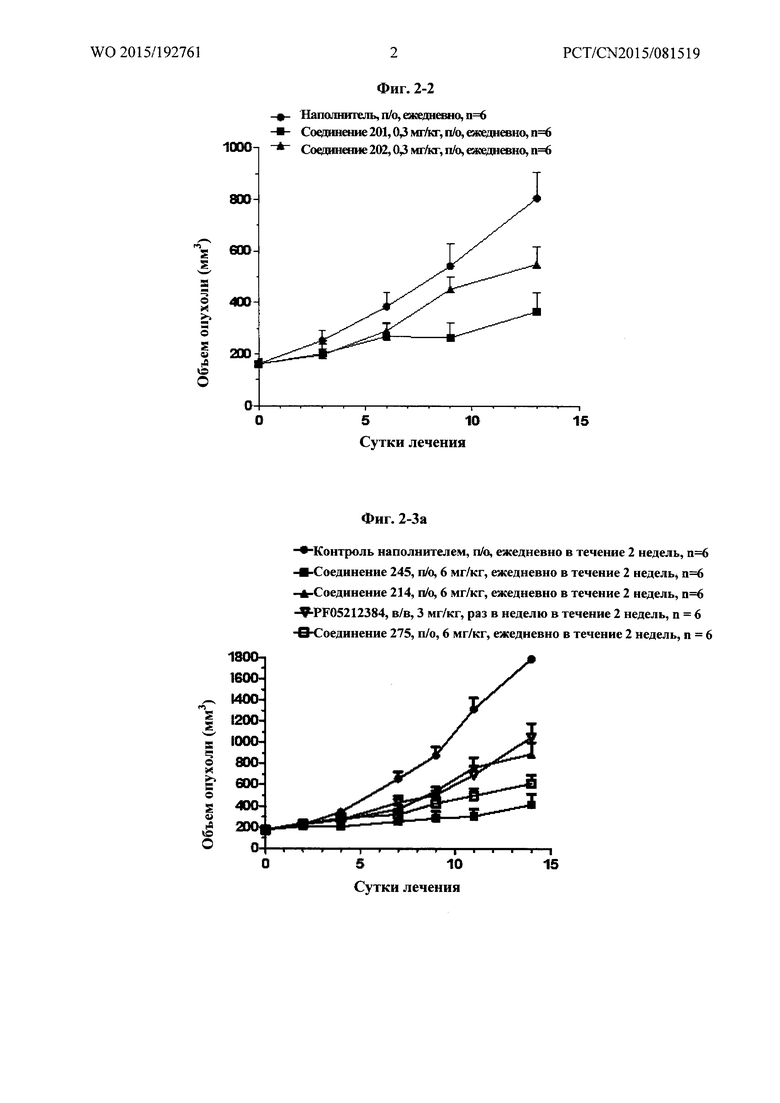
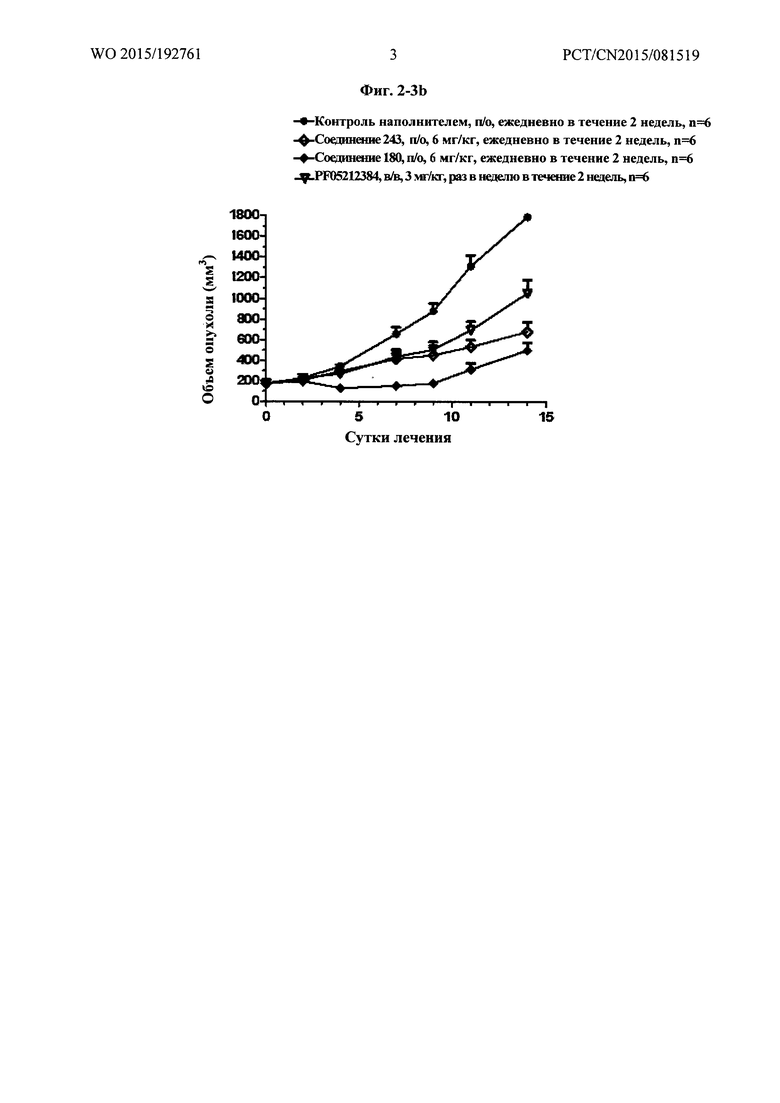
Изобретение относится к новому соединению формулы (I) или его фармацевтически приемлемой соли, обладающим свойствами ингибитора mTOR/РI3K. Соединения могут найти применение для лечения рака. Действие двойного ингибитора позволяет одновременно блокировать множество участков сигнальной трансиндукции и позволяет более эффективно предотвращать сигнальную трансиндукцию, тем самым преодолевая или задерживая возникновение устойчивости к лекарственным средствам, и обладает более продолжительным и эффективным действием при лечении рака. В соединении формулы (I)
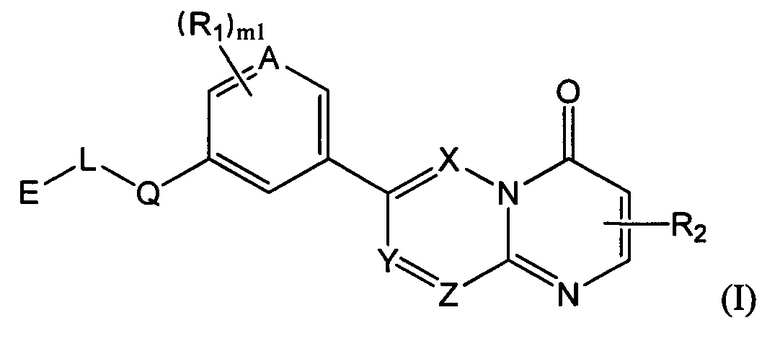
структурная единица  может быть заменена на
может быть заменена на 
или  ;
;
Е выбран из группы, состоящей из С3-6 циклоалкила или C1-6 алкила, замещенного R3, и число R3 составляет 0, 1, 2 или 3, или Е выбран из группы, состоящей из 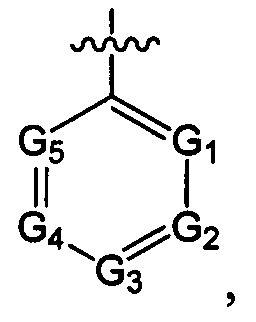
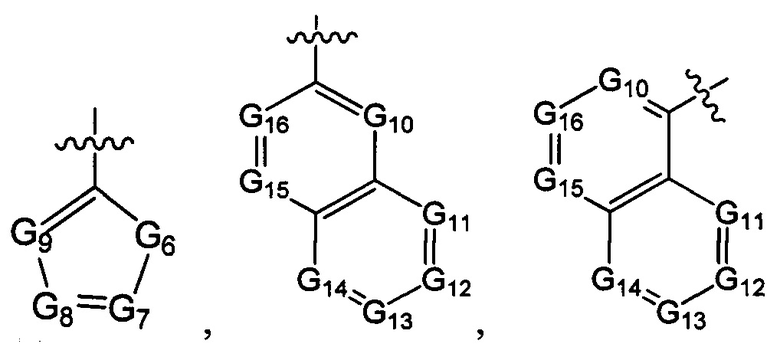 и
и 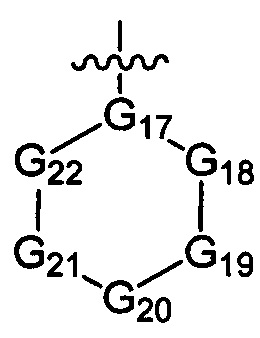 , или Е выбран из группы, состоящей из
, или Е выбран из группы, состоящей из  и
и 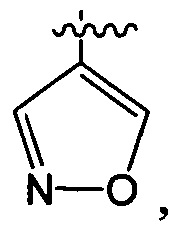 необязательно замещенного 1, 2 или 3
необязательно замещенного 1, 2 или 3
R3; где ноль, один, два или три из G1~5 выбраны из N, а остальные выбраны из C(R3); G6 выбран из группы, состоящей из -C(R3)(R3)-, -C(=O)N(R3)-, -N(R3)-, -C(=NR3)-, -S(=O)2N(R3)-, -S(=O)N(R3)-, -O-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(R3)C(=O)N(R3)-; ноль, один или два из G7~9 выбраны из N, а остальные выбраны из C(R3); ноль, один, два, три или четыре из G10~16 выбраны из N, и остальные выбраны из C(R3); G17 выбран из N или C(R3); ноль, один, два или три из G18~22 выбраны из группы, состоящей из -C(=O)N(R3)-, -N(R3)-, -C(=NR3)-, -S(=O)2N(R3)-, -S(=O)N(R3)-, -O-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(R3)C(=O)N(R3)-, и остальные выбраны из -C(R3)(R3)-; один из L и Q представляет собой -S(=O)2NH- и другой представляет собой одинарную связь; или один из L и Q представляет собой -S(=O)2- и другой представляет собой СН2; А и Т независимо выбраны из N или СН; один из X и Y выбран из N и другой выбран из C(R3) и Z выбран из СН; или X и Y выбран из C(R3)
и Z выбран из СН; В выбран из группы, состоящей из -N(Ra)-; гетероатом или гетероатомная группа независимо выбраны из группы, состоящей из -C(=O)N(Ra)-, -N(Ra)-, -C(=NRa)-, -S(=O)2N(Ra)-, -S(=O)N(Ra)-, -О-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(Ra)C(=O)N(Ra)-; каждый m1 независимо выбран из 0, 1, 2 или 3; R1 независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ORa, N(Rb)(Rc), С1-3 алкила, 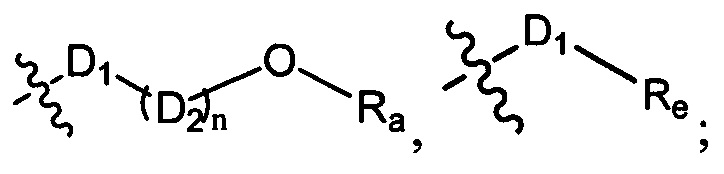
R2 и R3 независимо выбраны из группы, состоящей из Н, F, Cl, Br, I, CN, ORa, N(Rb)(Rc), С1-3 алкила, необязательно замещенного Rd, 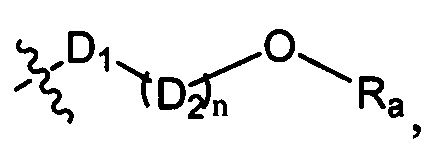
D1 выбран из группы, состоящей из одинарной связи, -C(Re)(Re)-, -C(=O)N(Ra)-, -N(Ra)-, -C(=NRa)-, -S(=O)2N(Ra)-, -S(=O)N(Ra)-, -О- и -S-; D2 выбран из -C(Ra)(Ra)-; n выбран из 1, 2, 3, 4, 5 или 6; Ra, Rb и Rc независимо выбраны из группы, состоящей из Н, С3-6 циклоалкила или С1-6 алкила, необязательно замещенного Rd; Re выбран из группы, состоящей из Н, C1-6 алкила или алкокси, необязательно замещенного Rd, С3-6 циклоалкила, необязательно замещенного Rd; Rd выбран из группы, состоящей из F, Cl, Br, I, CN, ОН, СНО, СООН, СН3, CF3, СН3O и СН3СН2О, и число Rd выбрано из 0, 1, 2 или 3; необязательно, Ra и Ra в одном и том же D2, два D2, или Ra и один D2 вместе с одним и тем же атомом углерода или атомом кислорода, к которому они оба присоединены, образуют одно 3-, 4-, 5- или 6-членное карбоциклическое кольцо или кислородсодержащее гетероциклическое кольцо, где число атомов кислорода составляет 1 или 2. 2 н. и 6 з.п. ф-лы, 3 ил., 4 табл., 241 пр.
.
1. Соединение формулы (I) или его фармацевтически приемлемая соль,
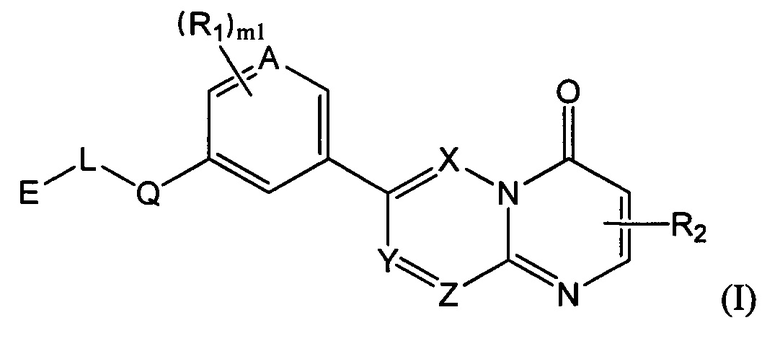
где
структурная единица 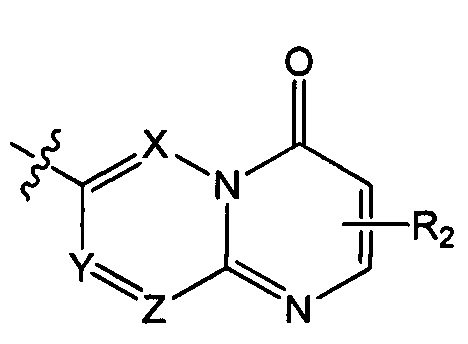 может быть заменена на
может быть заменена на 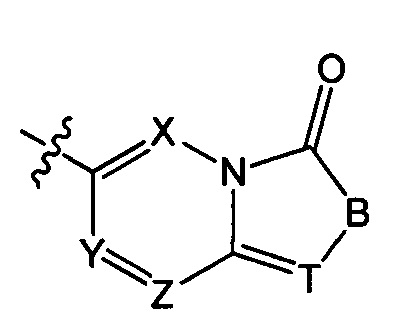
или 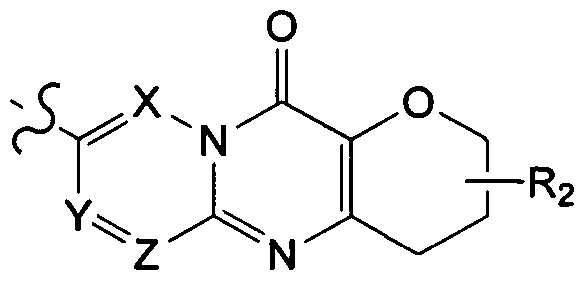 ;
;
Е выбран из группы, состоящей из С3-6 циклоалкила или C1-6 алкила, замещенного R3, и число R3 составляет 0, 1, 2 или 3, или Е выбран из группы, состоящей из 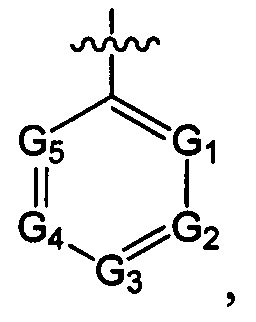
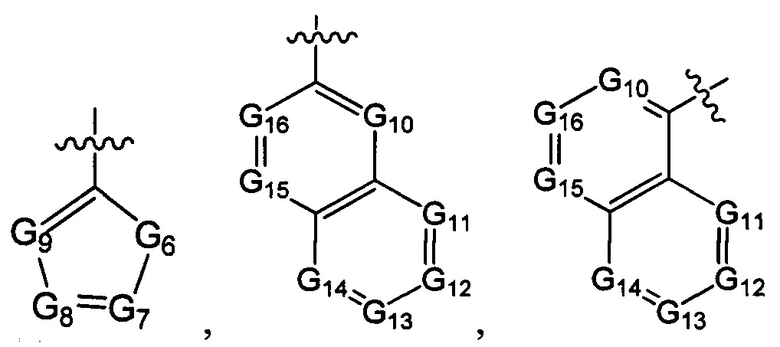 и
и 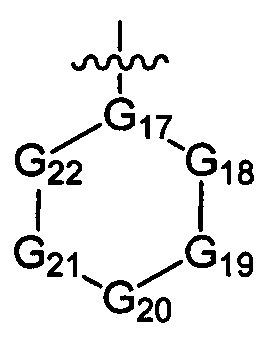 , или Е выбран из группы, состоящей из
, или Е выбран из группы, состоящей из  и
и 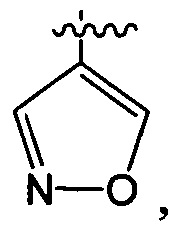 необязательно замещенного 1, 2 или 3
необязательно замещенного 1, 2 или 3
R3; где
ноль, один, два или три из G1~5 выбраны из N, а остальные выбраны из C(R3);
G6 выбран из группы, состоящей из -C(R3)(R3)-, -C(=O)N(R3)-, -N(R3)-, -C(=NR3)-, -S(=O)2N(R3)-, -S(=O)N(R3)-, -O-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(R3)C(=O)N(R3)-;
ноль, один или два из G7~9 выбраны из N, а остальные выбраны из C(R3);
ноль, один, два, три или четыре из G10~16 выбраны из N и остальные выбраны из C(R3);
G17 выбран из N или C(R3);
ноль, один, два или три из G18~22 выбраны из группы, состоящей из -C(=O)N(R3)-, -N(R3)-, -C(=NR3)-, -S(=O)2N(R3)-, -S(=O)N(R3)-, -O-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(R3)C(=O)N(R3)-, и остальные выбраны из -C(R3)(R3)-;
один из L и Q представляет собой -S(=O)2NH- и другой представляет собой одинарную связь; или один из L и Q представляет собой -S(=O)2- и другой представляет собой СН2;
А и Т независимо выбраны из N или СН;
один из X и Y выбран из N и другой выбран из C(R3)
и Z выбран из СН; или X и Y выбран из C(R3)
и Z выбран из СН;
В выбран из группы, состоящей из -N(Ra)-;
гетероатом или гетероатомная группа независимо выбраны из группы, состоящей из -C(=O)N(Ra)-, -N(Ra)-, -C(=NRa)-, -S(=O)2N(Ra)-, -S(=O)N(Ra)-, -О-, -S-, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- и -N(Ra)C(=O)N(Ra)-;
каждый m1 независимо выбран из 0, 1, 2 или 3;
R1 независимо выбран из группы, состоящей из Н, F, Cl, Br, I, ORa, N(Rb)(Rc), С1-3 алкила, 
R2 и R3 независимо выбраны из группы, состоящей из Н, F, Cl, Br, I, CN, ORa, N(Rb)(Rc), С1-3 алкила, необязательно замещенного Rd, 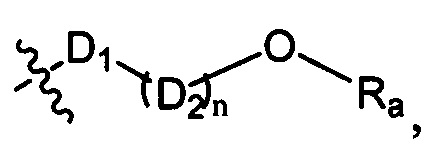
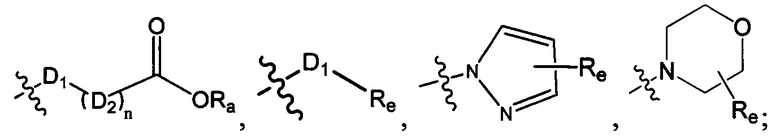
D1 выбран из группы, состоящей из одинарной связи, -C(Re)(Re)-, -C(=O)N(Ra)-, -N(Ra)-, -C(=NRa)-, -S(=O)2N(Ra)-, -S(=O)N(Ra)-, -О- и -S-; D2 выбран из -C(Ra)(Ra)-; n выбран из 1, 2, 3, 4, 5 или 6;
Ra, Rb и Rc независимо выбраны из группы, состоящей из Н, С3-6 циклоалкила или С1-6 алкила, необязательно замещенного Rd;
Re выбран из группы, состоящей из Н, C1-6 алкила или алкокси, необязательно замещенного Rd, С3-6 циклоалкила, необязательно замещенного Rd;
Rd выбран из группы, состоящей из F, Cl, Br, I, CN, ОН, СНО, СООН, СН3, CF3, СН3O и СН3СН2О, и число Rd выбрано из 0, 1, 2 или 3;
необязательно, Ra и Ra в одном и том же D2, два D2 или Ra и один D2 вместе с одним и тем же атомом углерода или атомом кислорода, к которому они оба присоединены, образуют одно 3-, 4-, 5- или 6-членное карбоциклическое кольцо или кислородсодержащее гетероциклическое кольцо, где число атомов кислорода составляет 1 или 2.
2. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где G6 выбран из группы, состоящей из -C(R3)(R3)-, -N(R3)-, -О- и -S-;
ноль или один из G10~16 выбраны из N, а остальные выбраны из C(R3);
ноль или один из G18~22 выбраны из -О- и остальные выбраны из -C(R3)(R3)-; и
остальные переменные являются такими, как определено в п.1.
3. Соединение формулы (I) или его фармацевтически приемлемая соль по п.1, где Е выбран из группы, состоящей из метила, этила, пропила, 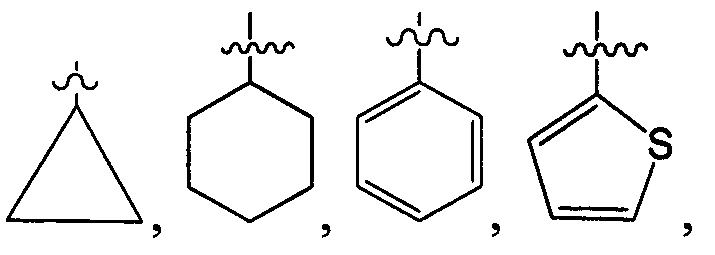
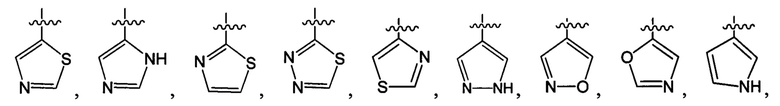
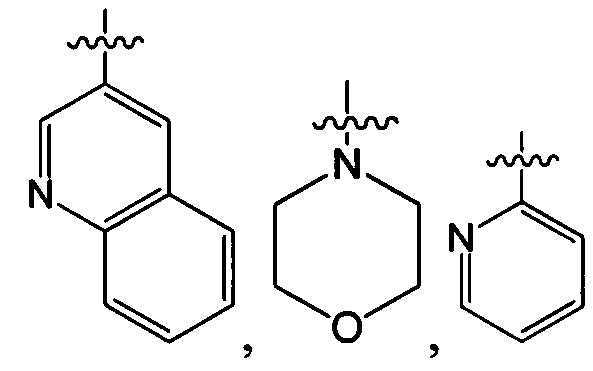 и
и  , необязательно замещенного 1, 2 или 3 R3.
, необязательно замещенного 1, 2 или 3 R3.
4. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 3, где Е выбран из группы, состоящей из 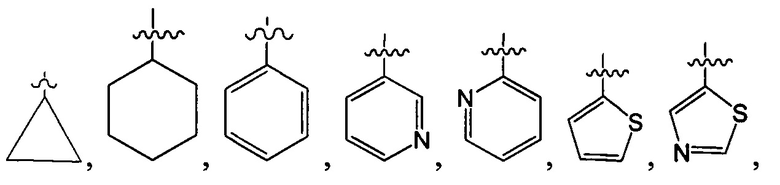
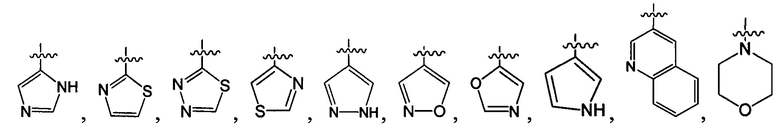 и С1-3 алкила, причем вышеуказанная группа необязательно замещена 1, 2 или 3 галогенами, ОН, ОС1-3 алкилом, CN, NH2, NH(C1-3 алкилом), N(C1-3 алкилом)2, С1-3 алкилом, трифторметилом, трифторэтилом, С1-3 алкилС(=O)NН, C1-3 алкилS(=O)NH.
и С1-3 алкила, причем вышеуказанная группа необязательно замещена 1, 2 или 3 галогенами, ОН, ОС1-3 алкилом, CN, NH2, NH(C1-3 алкилом), N(C1-3 алкилом)2, С1-3 алкилом, трифторметилом, трифторэтилом, С1-3 алкилС(=O)NН, C1-3 алкилS(=O)NH.
5. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где Е выбран из группы, состоящей из 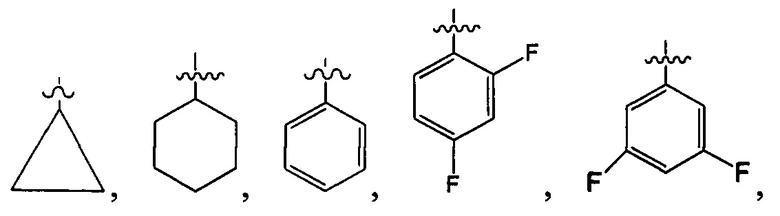
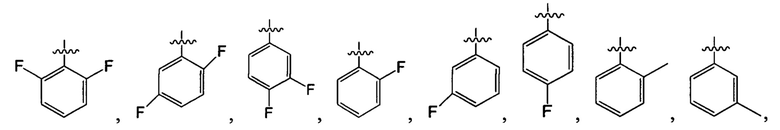
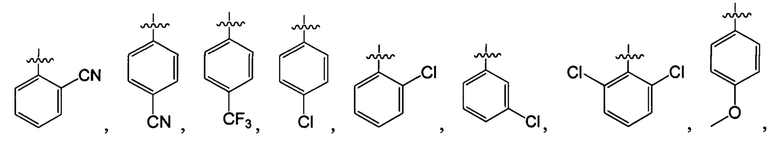
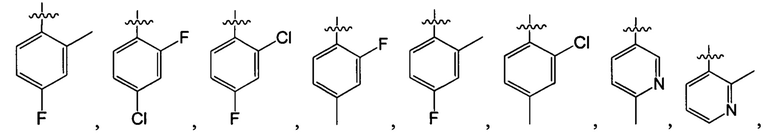
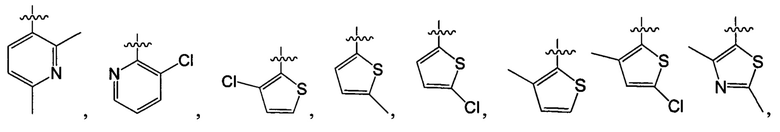
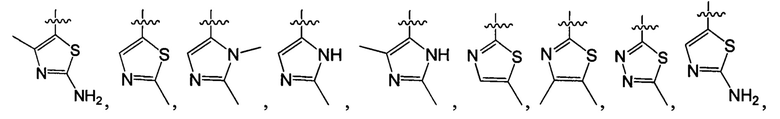
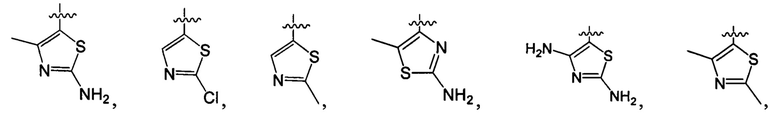
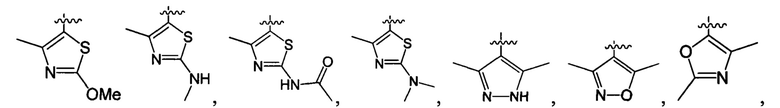
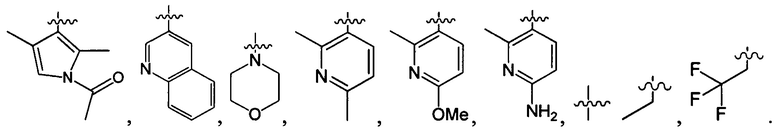
6. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где один из X и Y выбран из N и другой выбран из группы, состоящей из СН, С(СН3), С(СF3), ССl и CF, или X и Y выбран из группы, состоящей из СН, С(СН3), С(СF3), ССl и CF.
7. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где В выбран из группы, состоящей из NH, N(СН3) и N(CF3).
8. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где кольцо, образованное между Ra и Ra в одном и том же D2, двумя D2 или Ra и одним D2, выбрано из группы, состоящей из циклопропила, циклобутила, циклопентила, циклогексила, оксетанила, 1,3-диоксоланила.
9. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где R1, R2 и R3 независимо выбраны из группы, состоящей из Н, F, Cl, Br, I, CN, ОН, NH2, метила, этила, пропила, метокси, этокси, метиламино, диметиламино, галогенметила, галогенэтила, галогенпропила, аминометила, аминоэтила, аминопропила, циклопропила, 
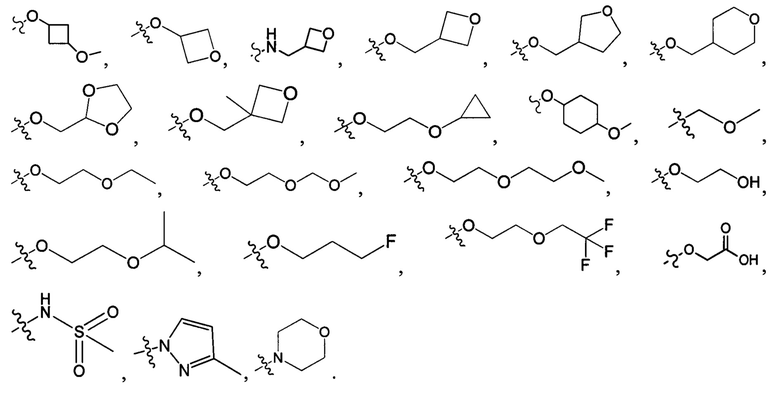
10. Соединение, выбранное из группы, состоящей из:
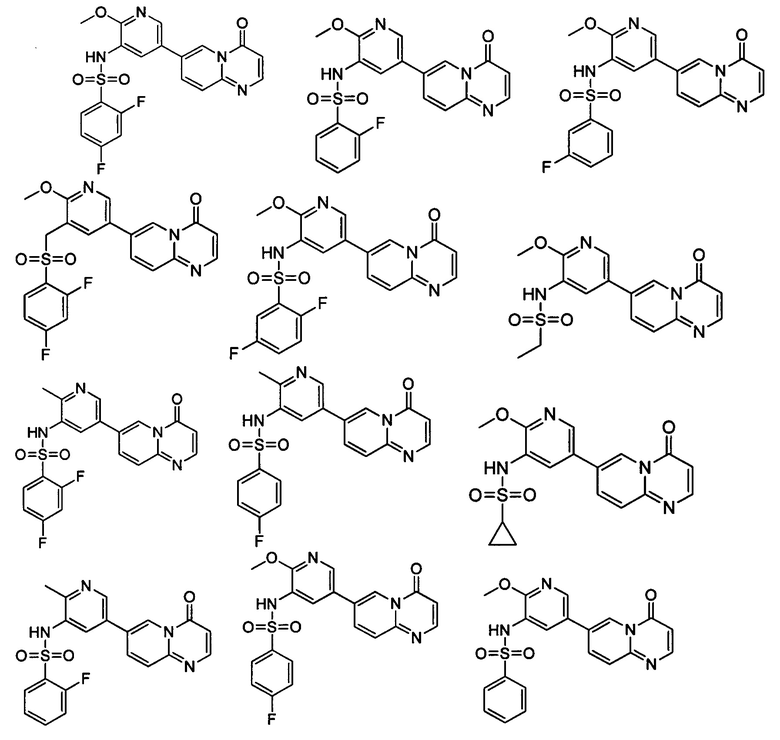
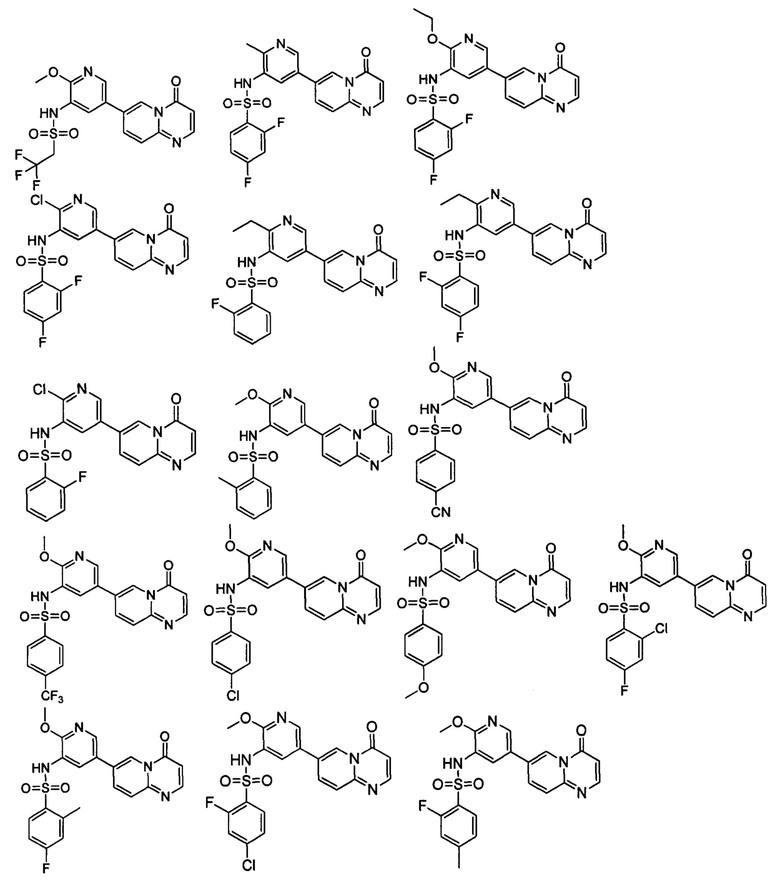
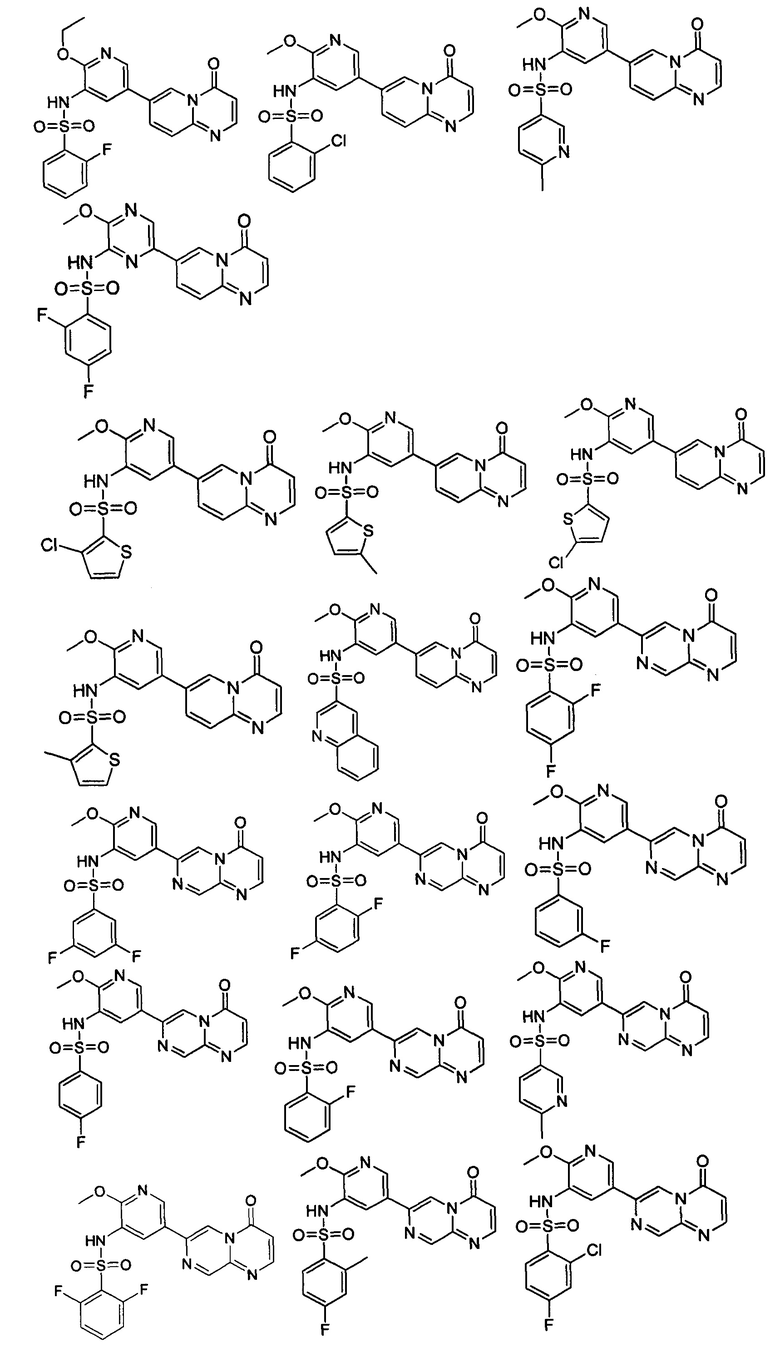
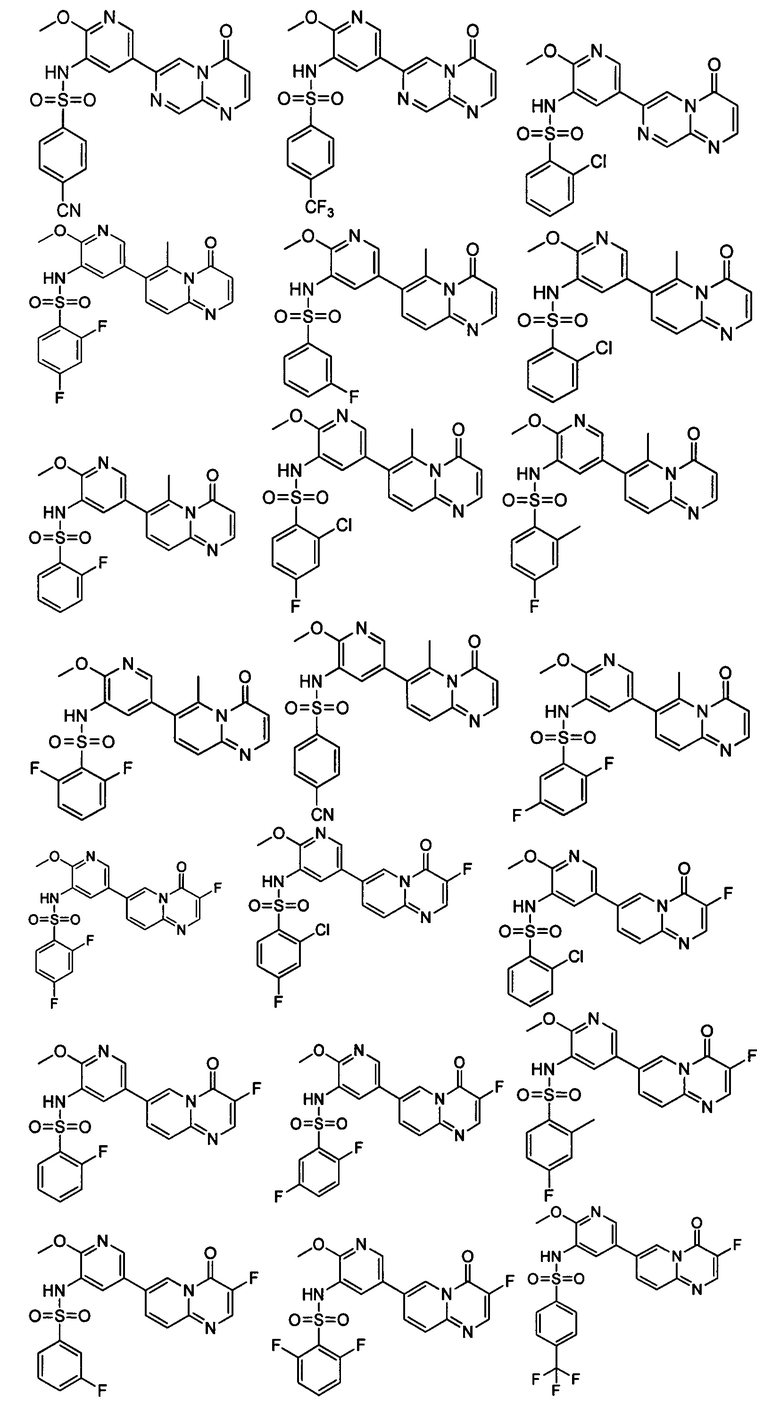
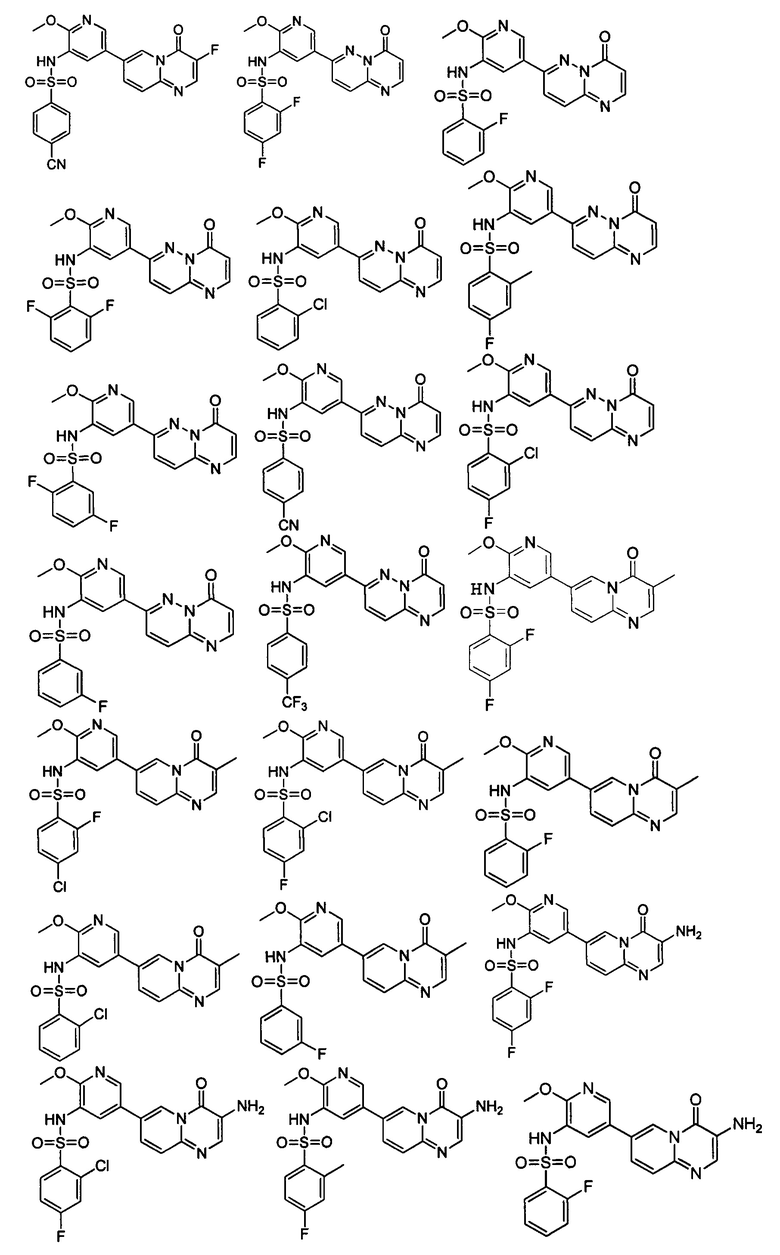
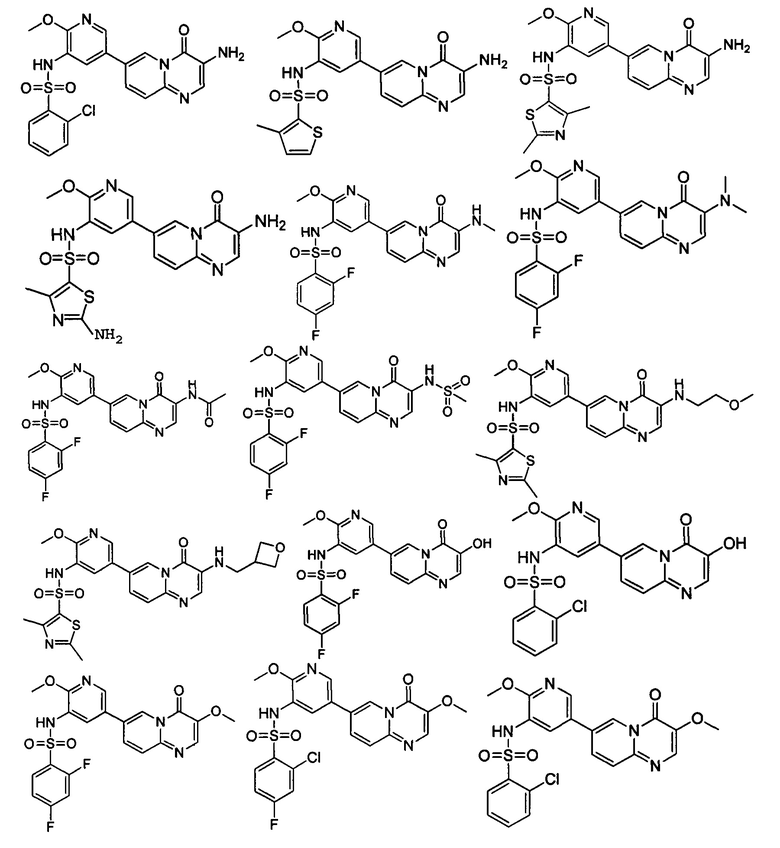
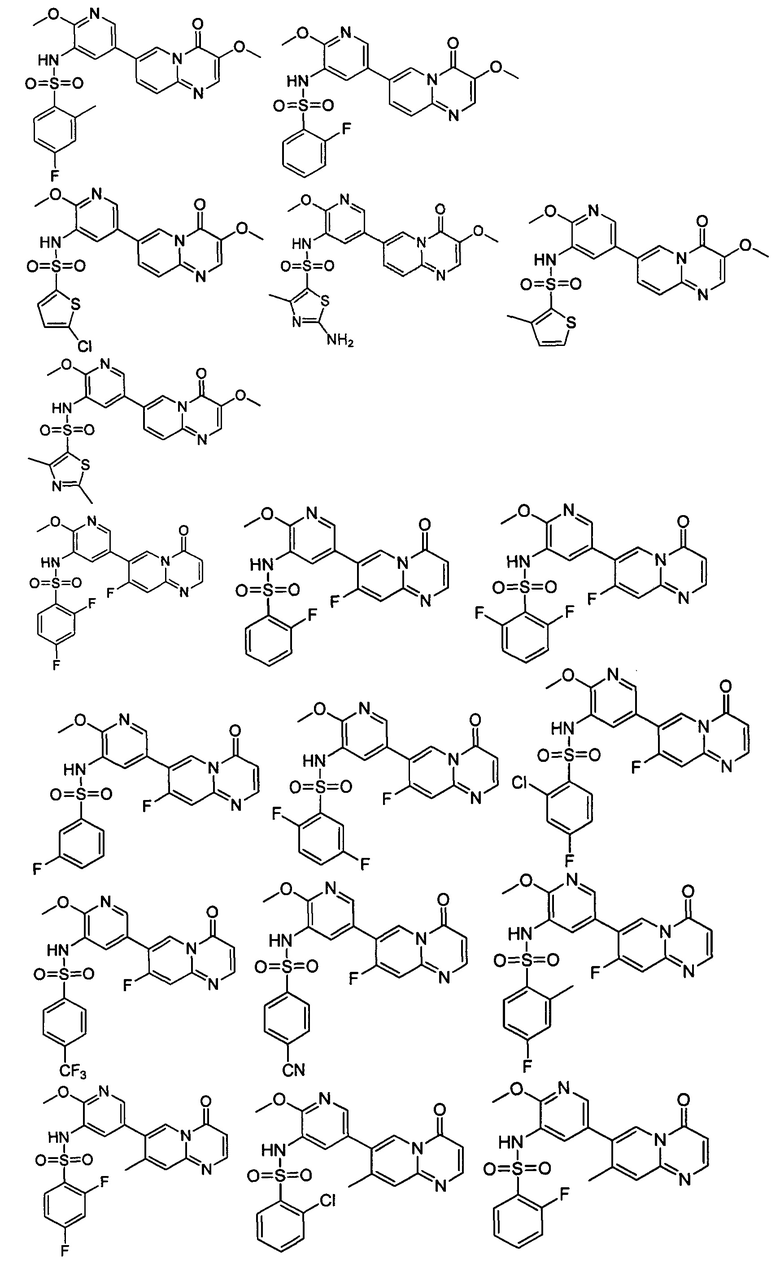
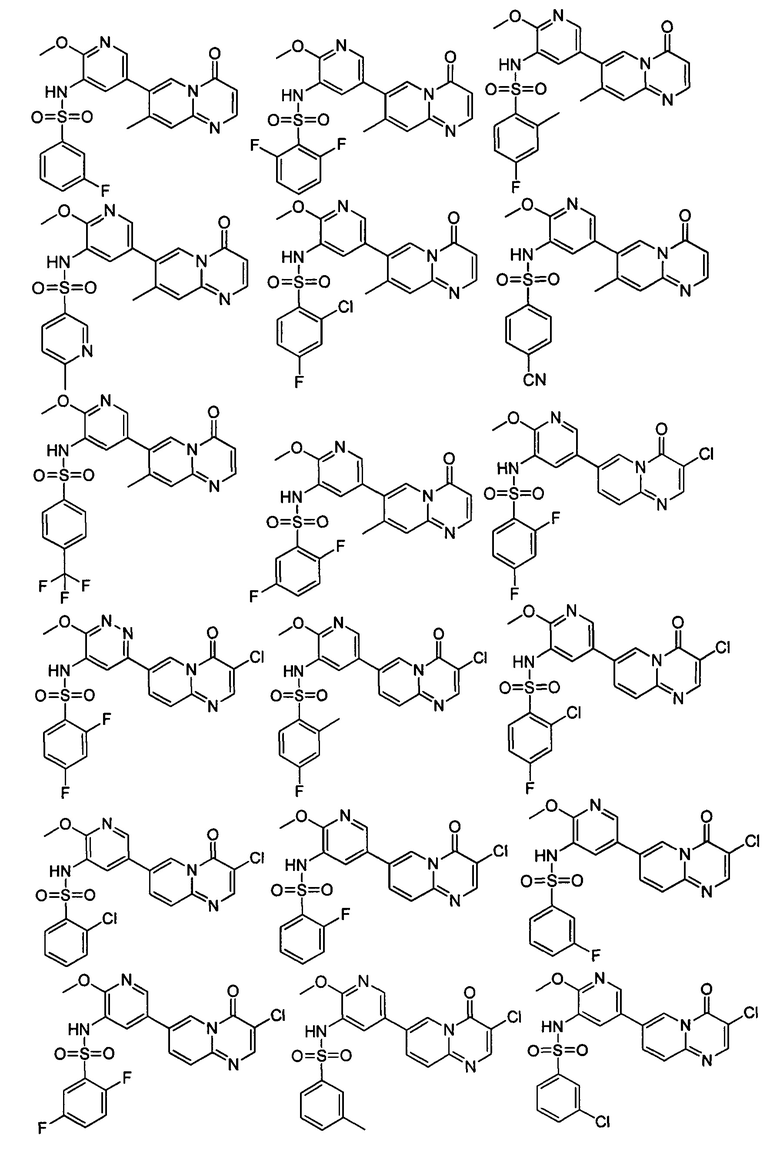
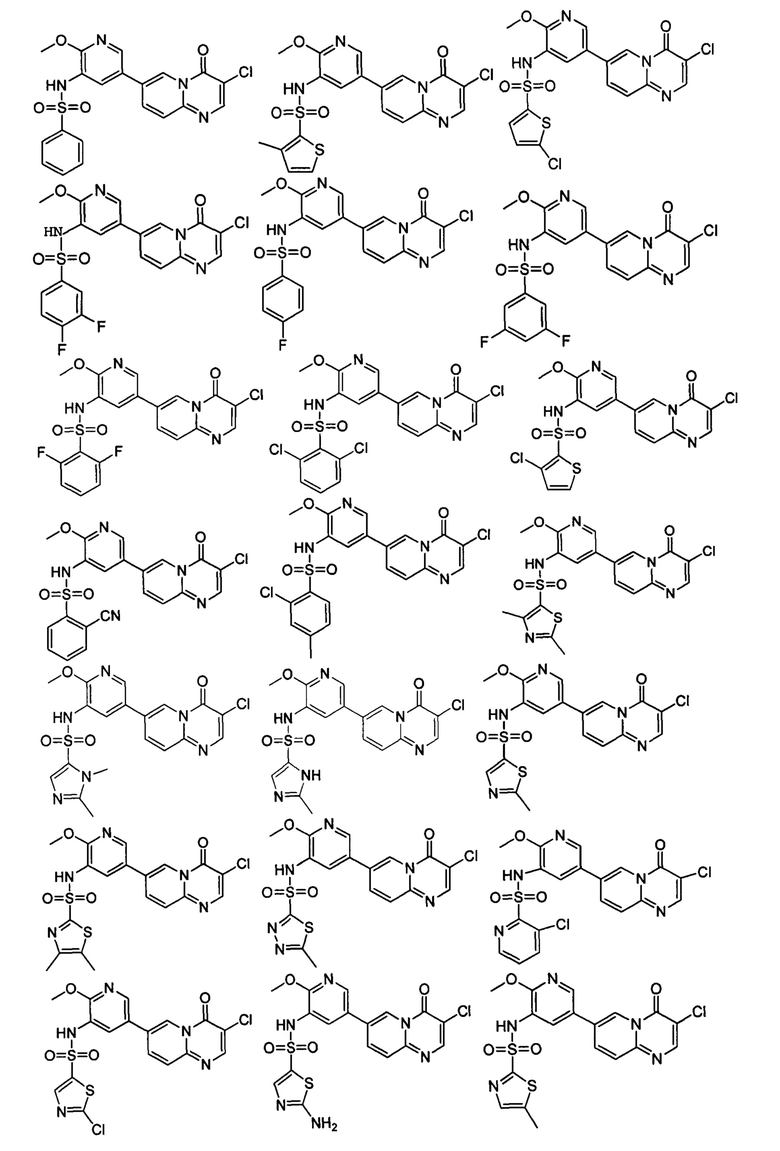
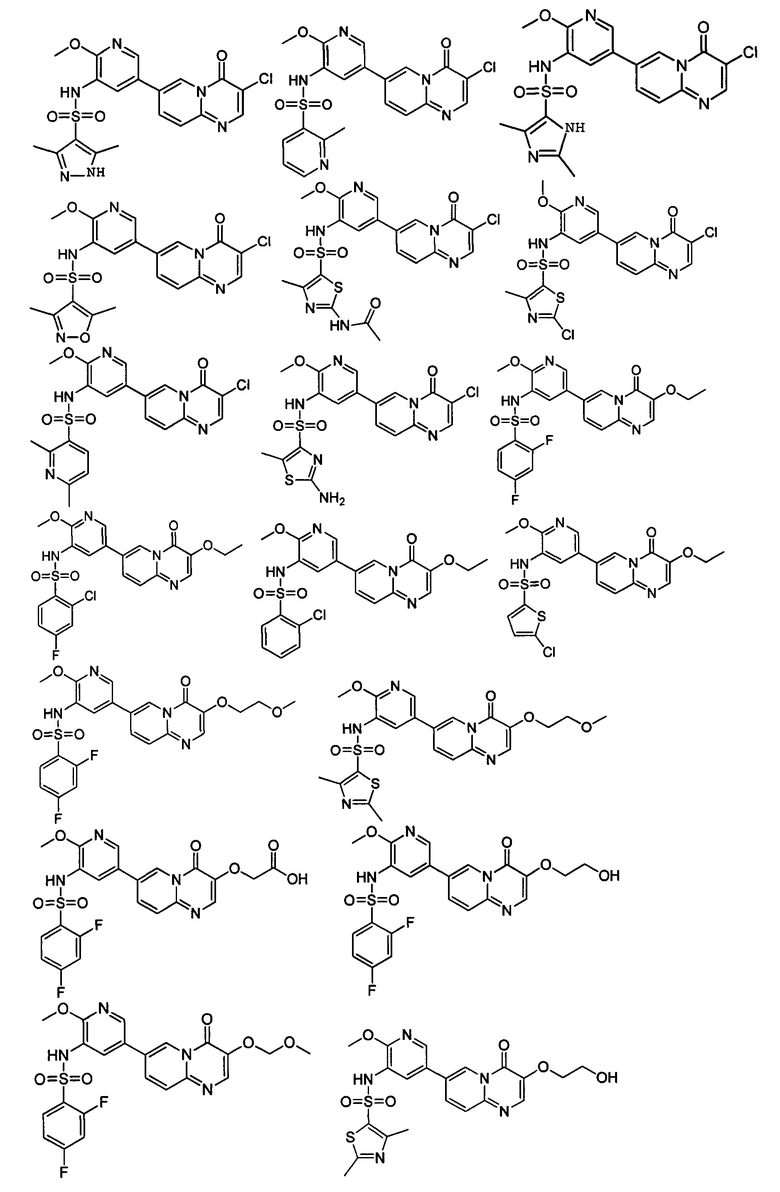
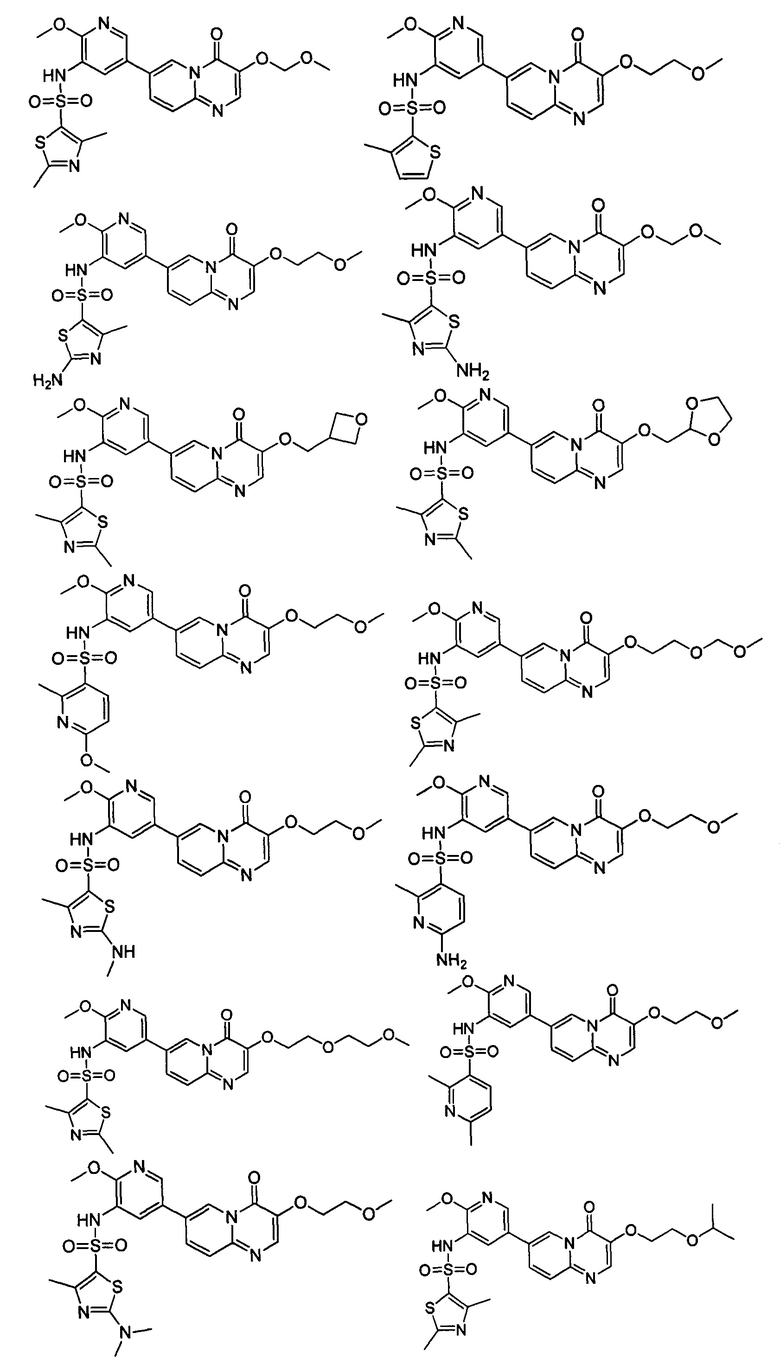
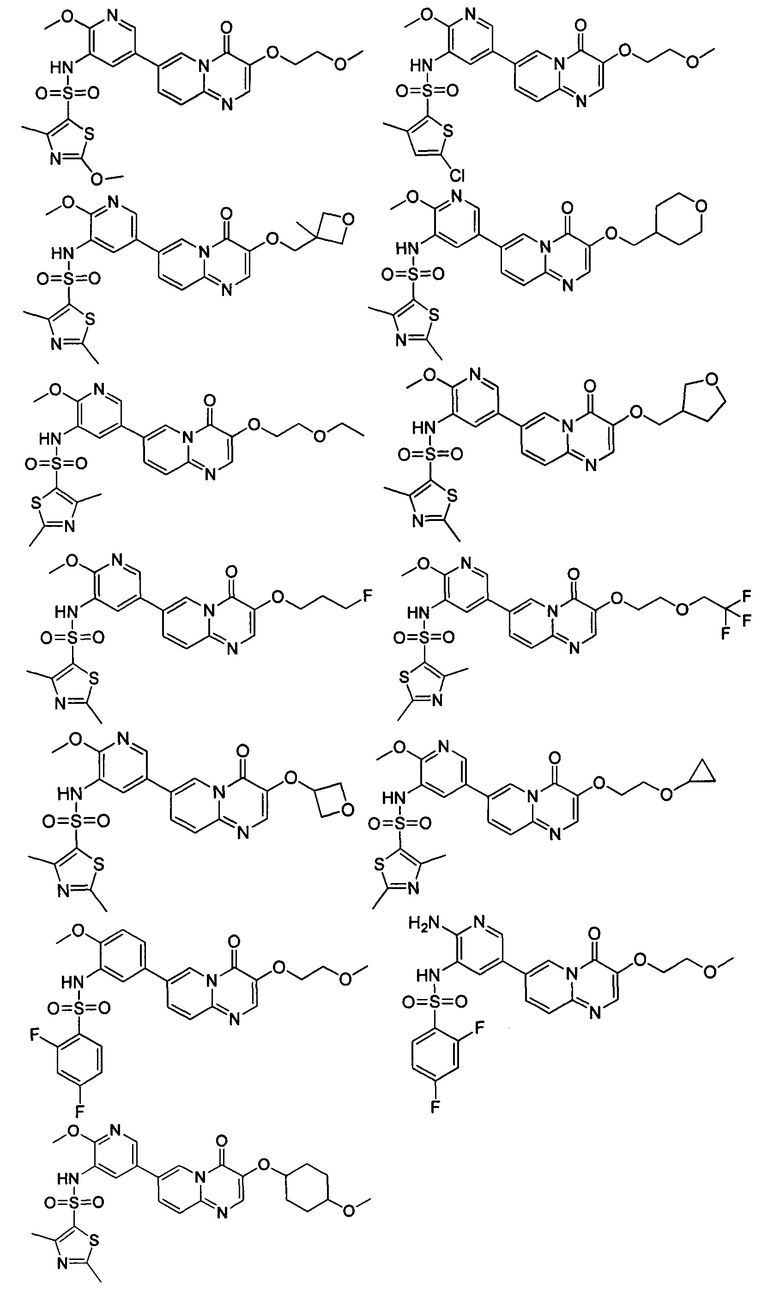
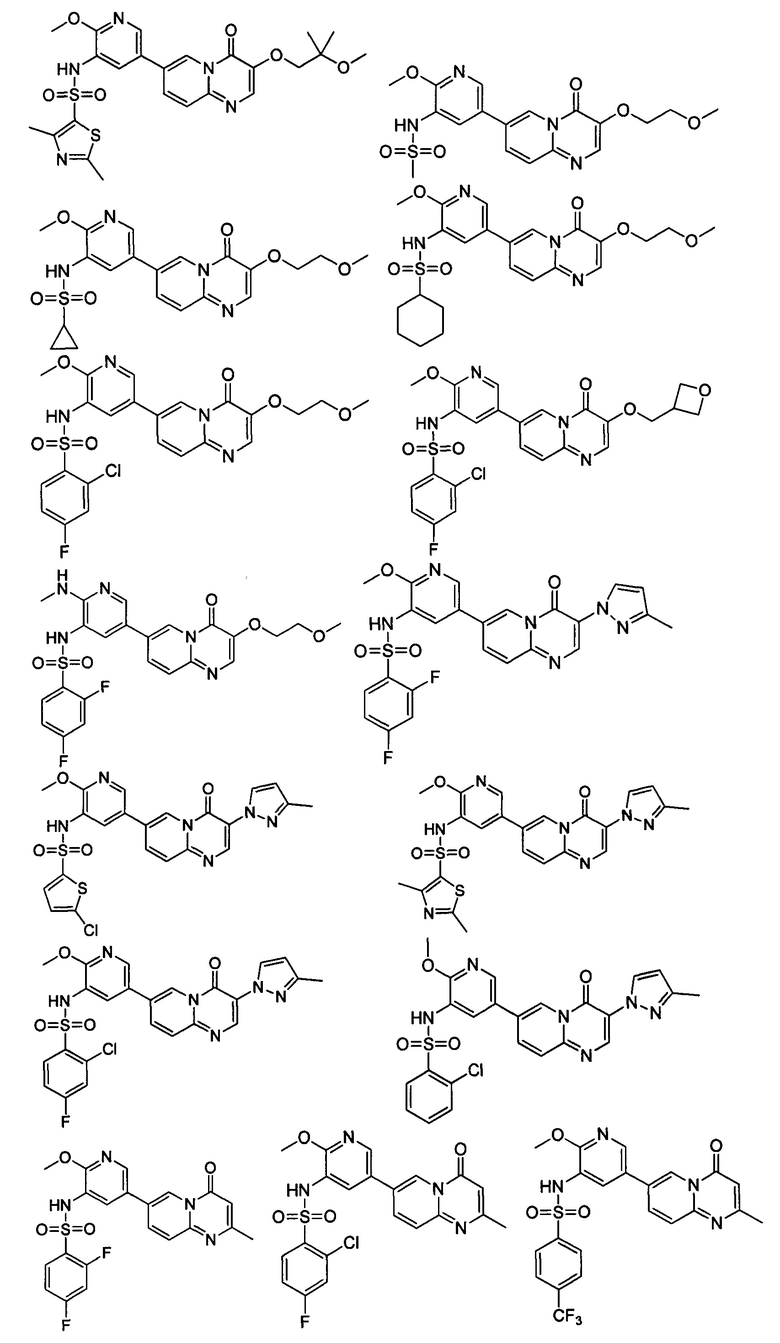
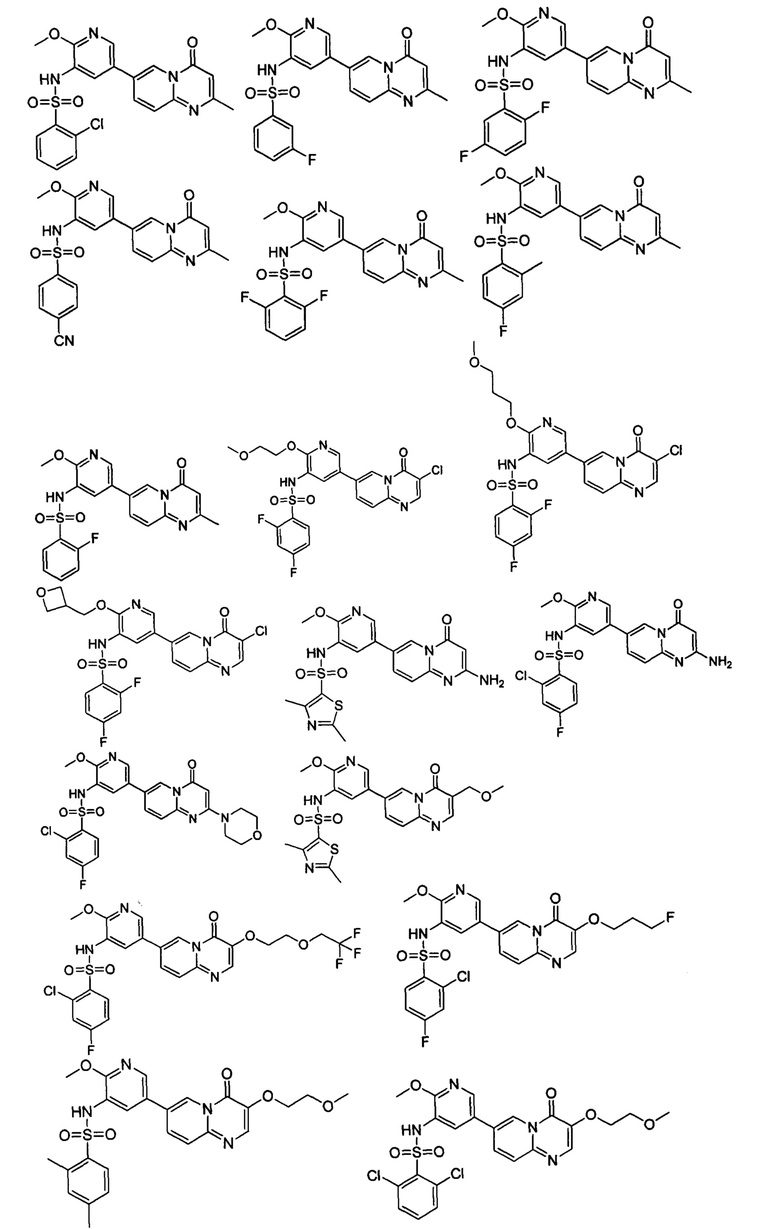
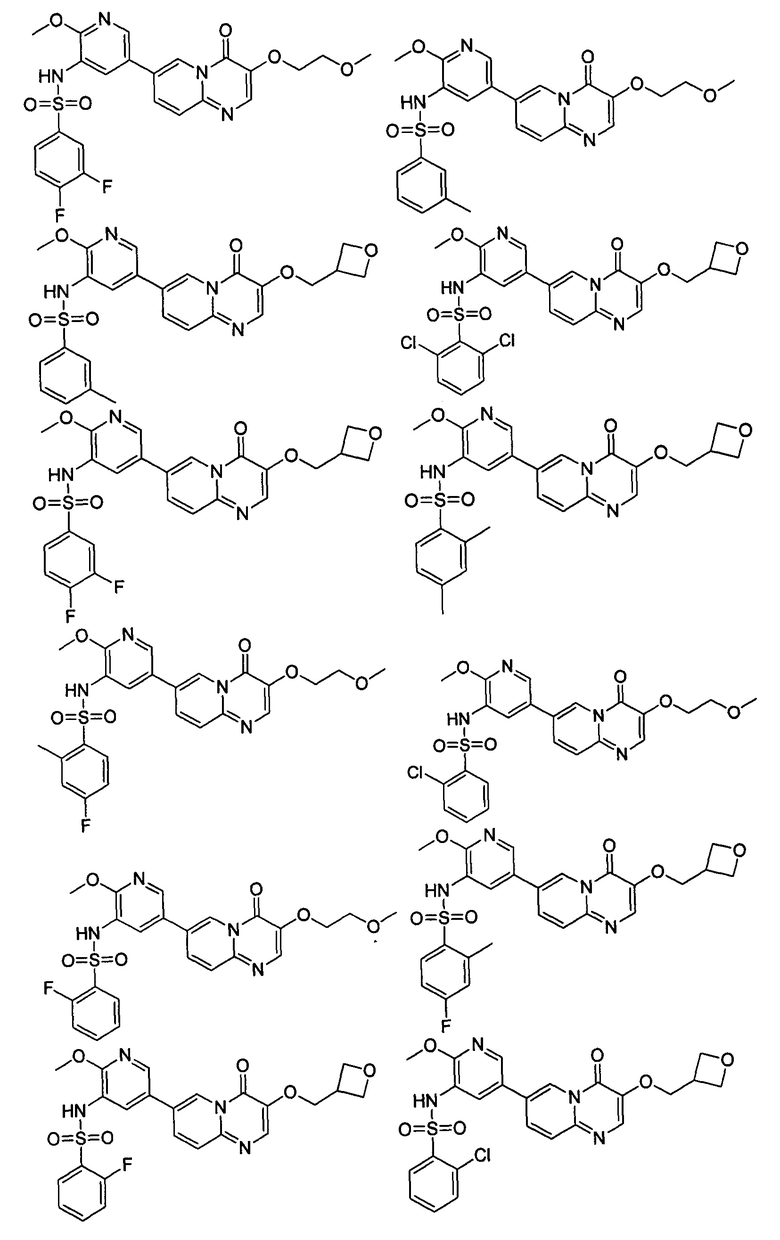
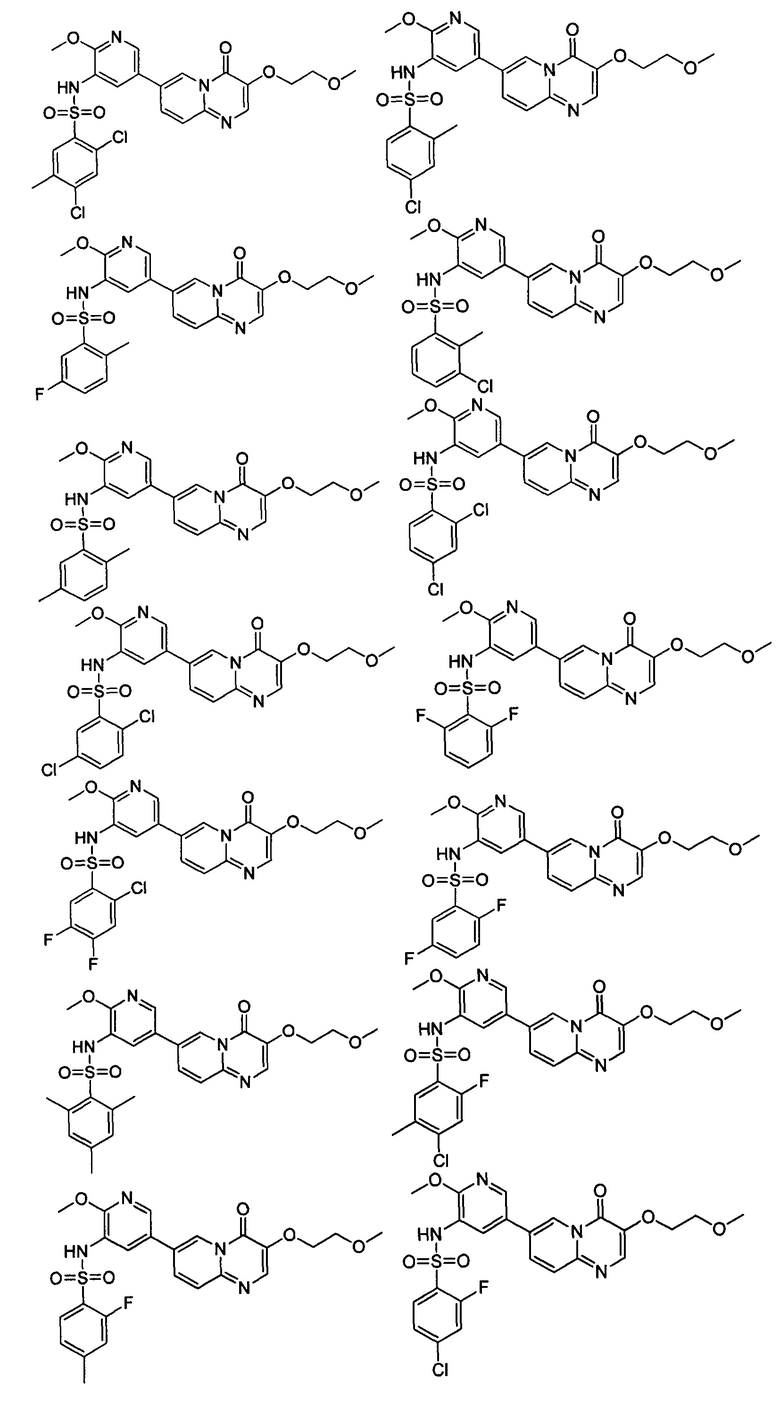
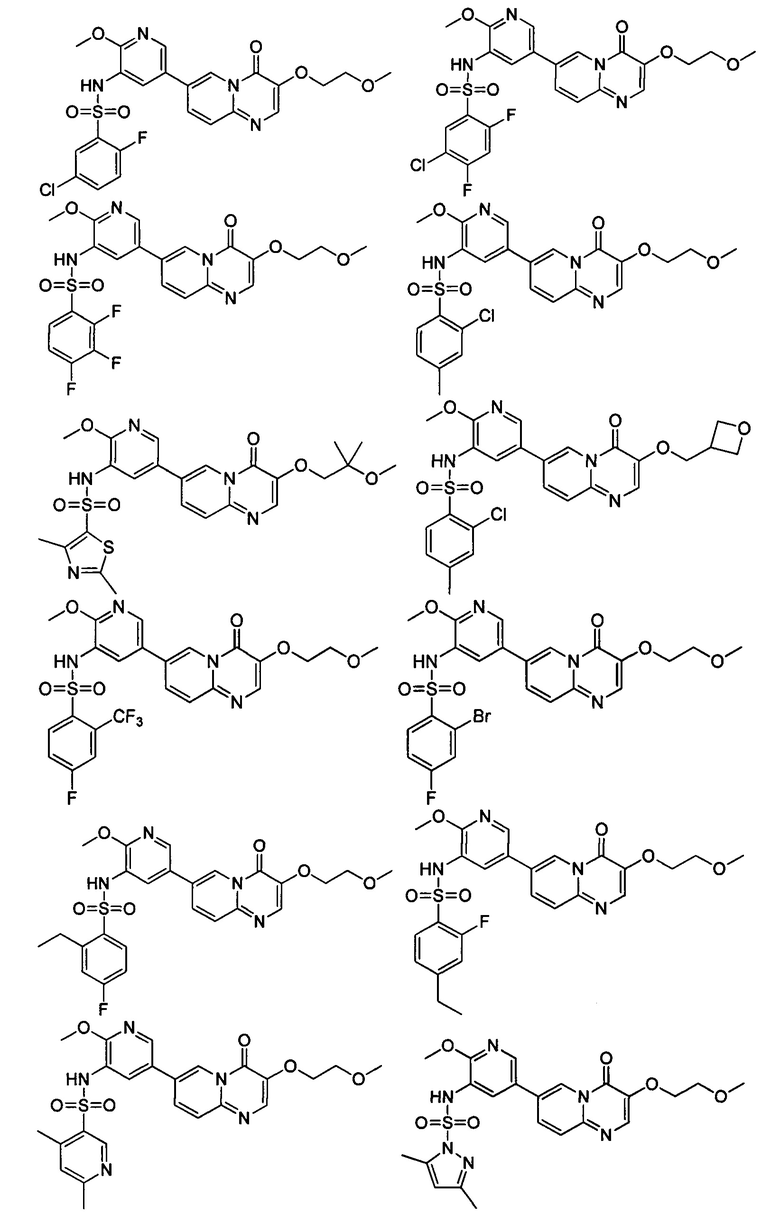
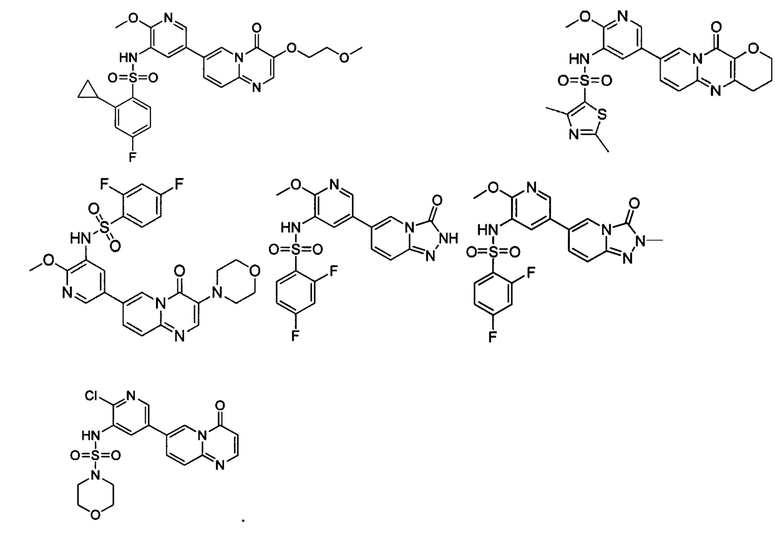
| CN 103539777 A, 29.01.2014 | |||
| WO 2013071698 A1, 23.05.2013 & EP 2781520 B1, 04.05.2016 | |||
| WO 2014022128 A1, 06.02.2014 | |||
| Способ приготовления черной типографской краски | 1935 |
|
SU48943A1 |
| Цоколь к электрической лампе накаливания | 1928 |
|
SU16312A1 |
| WO 2008005457 A2, 10.01.2008 | |||
| CN 103539777 A (Guangdong HEC Pharmaceutical Co., Ltd.) 29.01.2014 (реферат, абзацы [0092], 0106 - 0108, примеры, пункты 1, 8 формулы изобретения) | |||
| WO 2013071698 A1 (XUANZHU PHARMA CO., LTD), 23.05.2013 & EP 2781520 B1, 04.05.2016 (реферат, пункт 1 формулы изобретения, таблица 1, соединения 1-262 и др., примеры, соединения BGT226 b BEZ-235) | |||
| WO 2014022128 A1 (CALITOR SCIENCES LLC), 05.02.2014 (абзац [019], [020], [073]-[082], [0145], примеры, формула изобретения) | |||
| Способ приготовления черной типографской краски | 1935 |
|
SU48943A1 |
| Цоколь к электрической лампе накаливания | 1928 |
|
SU16312A1 |
| WO 2008005457 A2 (Sunesis Pharmaceuticals), 10.01.2008 (реферат, формула изобретения, абзац [0109], [0485], примеры 105, 127). | |||

