Область техники, к которой относится изобретение
Настоящее изобретение относится к гомофталимидным производным 1,3,4-оксадиазола, обладающим ингибирующей активностью по отношению к гистондезацетилазе 6 (HDAC6), к их стереоизомерам, их фармацевтически приемлемым солям, к их применению для приготовления лекарственного средства, содержащей их фармацевтической композиции, к терапевтическому способу с использованием композиции, и способу их получения.
Уровень техники
В клетках посттрансляционная модификация, такая как ацетилирование, служит в качестве очень важного регулирующего модуля для многих биологических процессов и также строго регулируется целым рядом ферментов. В качестве основного белка, образующего хроматин, гистон действует, как ось, вокруг которой закручивается DNA, и это способствует конденсации DNA. Кроме того, баланс между ацетилированием и дезацетилированием гистона играет очень важную роль в экспрессии генов.
Известно, что в качестве фермента для удаления ацетильной группы их лизинового остатка белка гистона, который образует хроматин, гистондезацетилаза (HDAC) связана с молчанием генов и индуцирует остановку клеточного цикла, ангиогенное ингибирование, иммунорегуляцию, апоптоз и т. п. (Hassig et al., Curr. Opin. Chem. Biol. 1997, 1, 300-308). Кроме того, сообщали, что ингибирование функций фермента HDAC индуцирует проявление апоптоза самих раковых клеток путем снижения активности связанных с выживанием раковых клеток факторов и активации связанных с гибелью раковых клеток факторов в организме (Warrell et al., J. Natl. Cancer Inst. 1998, 90, 1621-1625).
Для людей известны 18 HDAC и они разделены на четыре класса в соответствии с гомологией с HDAC дрожжей. В этом случае одиннадцать HDAC, использующих цинк в качестве кофактора, можно разделить на три группы: класс I (HDAC1, 2, 3, 8), класс II (IIa: HDAC4, 5, 7, 9; IIb: HDAC6, 10) и класс IV (HDAC11). Кроме того, семь HDAC класса III (SIRT 1-7) используют NAD+ в качестве кофактора вместо цинка (Bolden et al., Nat. Rev. Drug Discov. 2006, 5(9), 769-784).
Разные ингибиторы HDAC в настоящее время находятся на стадии доклинической или клинической разработки, но только неселективные ингибиторы HDAC до настоящего времени были известны, как противораковое средство. Вориностат (SAHA) и ромидепсин (FK228) утверждены в качестве терапевтического средства для лечения кожной T-клеточной лимфомы, а панобиностат (LBH-589) утвержден в качестве терапевтического средства для лечения множественной миеломы. Однако известно, что неселективные ингибиторы HDAC в высоких дозах обычно приводят к побочным эффектам, таким как усталость, тошнота и т. п. (Piekarz et al., Pharmaceuticals 2010, 3, 2751-2767). Сообщали, что побочные эффекты вызваны ингибированием HDAC класса I. Вследствие побочных эффектов и т. п., на неселективные ингибиторы HDAC наложены ограничения при разработке лекарственных средств в других областях, кроме противораковых средств. (Witt et al., Cancer Letters 277 (2009) 8,21).
Кроме того, сообщали, что селективное ингибирование HDACs класса II не приводит к токсичности, которая проявлялась при ингибировании HDACs класса I. При разработке селективных ингибиторов HDAC было бы желательно устранить побочные эффекты, такие как токсичность и т. п., вызванные неселективным ингибированием HDACs. Соответственно, есть вероятность того, что селективные ингибиторы HDAC можно разработать в качестве эффективного терапевтического средства для разных заболеваний (Matthias et al., Mol. Cell. Biol. 2008, 28, 1688-1701).
Известно, что HDAC6 одного из классов IIb HDACs в основном содержится в цитоплазме и содержит белок тубулин и таким образом участвует с дезацетилировании целого ряда негистонных субстратов (HSP90, кортактин и т. п.) (Yao et al., Mol. Cell 2005, 18, 601-607). HDAC6 содержит два каталитических домена, в которых домен цинковый палец C-конца может связываться с убиквинированным белком. Известно, что HDAC6 содержит целый ряд негистонных белков в качестве субстрата и поэтому играет важную роль в разных заболеваниях, таких как рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания, нейродегенеративные нарушения и т. п. (Santo et al., Blood 2012 119: 2579-2589; Vishwakarma et al., International Immunopharmacology 2013, 16, 72-78; Hu et al., J. Neurol. Sci. 2011, 304, 1-8).
Структурная особенность, которой обладают разные ингибиторы HDAC, включает кэп-группы, линкер и связывающие цинк группы (ZBG), как показано в следующей структуре вориностата. Многие исследователи изучали ингибирующую активность по отношению к ферментам и селективность по отношению к структурной модификации кэп-групп и линкера. Известно, что кроме этих групп связывающая цинк группа играет более важную роль в ингибирующей активности и селективности по отношению к ферменту (Wiest et al., J. Org. Chem. 2013 78: 5051-5065; Methot et al., Bioorg. Med. Chem. Lett. 2008, 18, 973-978).
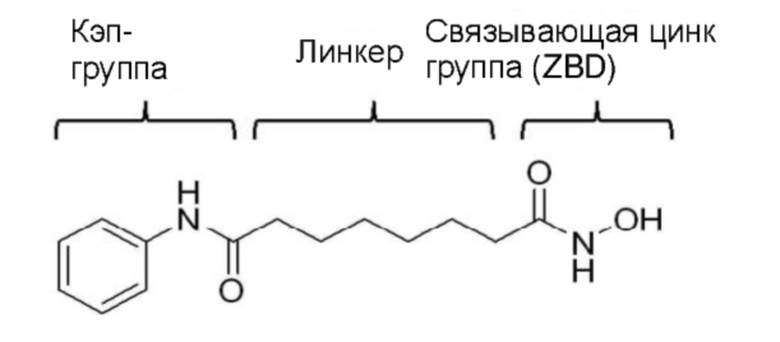
Большинство из указанных связывающих цинк групп состоит из гидроксамовой кислоты или бензамида, из-за которые производные гидроксамовой кислоты оказывают сильное ингибирующее воздействие на HDAC, но характеризуются низкой биодоступностью и сильной нецелевой активностью. Бензамид содержит анилин и поэтому приводит к затруднениям, поскольку бензамид может образовывать токсичные метаболиты in vivo (Woster et al., Med. Chem. Commun. 2015, online publication).
Соответственно, в отличие от неселективных ингибиторов, характеризующихся побочными эффектами, необходимо разработать селективный ингибитор HDAC, который содержит связывающую цинк группу, с улучшенной биодоступностью, но без побочных эффектов, для лечения рака, воспалительных заболеваний, аутоиммунных заболеваний, неврологических заболеваний, нейродегенеративных нарушений и т. п.
[Литература предшествующего уровня техники]
(Патентный документ 1) International Patent Publication No. WO 2011/091213 (publicized on Jul. 28, 2011): ACY-1215
(Патентный документ 2) International Patent Publication No. WO 2011/011186 (publicized on Jan. 27, 2011): Tubastatin
(Патентный документ 3) International Patent Publication No. WO 2013/052110 (publicized on Apr. 11, 2013): Sloan-K
(Патентный документ 4) International Patent Publication No. WO 2013/041407 (publicized on Mar. 28, 2013): Cellzome
(Патентный документ 5) International Patent Publication No. WO 2013/134467 (publicized on Sep. 12, 2013): Kozi
(Патентный документ 6) International Patent Publication No. WO 2013/008162 (publicized on Jan. 17, 2013): Novartis
(Патентный документ 7) International Patent Publication No. WO 2013/080120 (publicized on Jun. 06, 2013): Novartis
(Патентный документ 8) International Patent Publication No. WO 2013/066835 (publicized on May 10, 2013): Tempero
(Патентный документ 9) International Patent Publication No. WO 2013/066838 (publicized on May 10, 2013): Tempero
(Патентный документ 10) International Patent Publication No. WO 2013/066833 (publicized on May 10, 2013): Tempero
(Патентный документ 11) International Patent Publication No. WO 2013/066839 (publicized on May 10, 2013): Tempero
Подробное описание изобретения
Техническая задача
Задачей настоящего изобретения является получение гомофталимидных производных 1,3,4-оксадиазола, обладающих селективной ингибирующей активностью по отношению к HDAC6, их стереоизомеров или их фармацевтически приемлемых солей.
Другой задачей настоящего изобретения является разработка способа получения гомофталимидных производных 1,3,4-оксадиазола, их стереоизомеров или их фармацевтически приемлемых солей.
Еще одной задачей настоящего изобретения является получение фармацевтической композиции, содержащей гомофталимидные производные 1,3,4-оксадиазола, обладающие селективной ингибирующей активностью по отношению к HDAC6, их стереоизомеры или их фармацевтически приемлемые соли.
Еще одной задачей настоящего изобретения является получение фармацевтической композиции для предупреждения или лечения связанных с активностью HDAC6 заболеваний, включая рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания или нейродегенеративные нарушения, содержащей гомофталимидные производные 1,3,4-оксадиазола, их стереоизомеры или их фармацевтически приемлемые соли.
Еще одной задачей настоящего изобретения является разработка способа предупреждения или лечения связанных с активностью HDAC6 заболеваний, включающего введение терапевтически эффективного количества фармацевтической композиции, содержащей гомофталимидные производные 1,3,4-оксадиазола, их стереоизомеры или их фармацевтически приемлемые соли.
Еще одной задачей настоящего изобретения является разработка способа селективного ингибирования HDAC6 путем введения гомофталимидных производных 1,3,4-оксадиазола, их стереоизомеров или их фармацевтически приемлемых солей млекопитающим, включая людей.
Еще одной задачей настоящего изобретения является применение гомофталимидных производных 1,3,4-оксадиазола, их стереоизомеров или их фармацевтически приемлемых солей для предупреждения или лечения связанных с активностью HDAC6 заболеваний.
Еще одной задачей настоящего изобретения является применение гомофталимидных производных 1,3,4-оксадиазола, их стереоизомеров или их фармацевтически приемлемых солей для приготовления лекарственного средства для предупреждения или лечения связанных с активностью HDAC6 заболеваний.
Техническое решение
Авторы настоящего изобретения обнаружили гомофталимидные производные 1,3,4-оксадиазола, обладающие ингибирующей активностью по отношению к гистондезацетилазе 6 (HDAC6), и использовали их для предупреждения или лечения связанных с активностью HDAC6 заболеваний, и таким образом завершили настоящее изобретение.
Гомофталимидные производные 1,3,4-оксадиазола
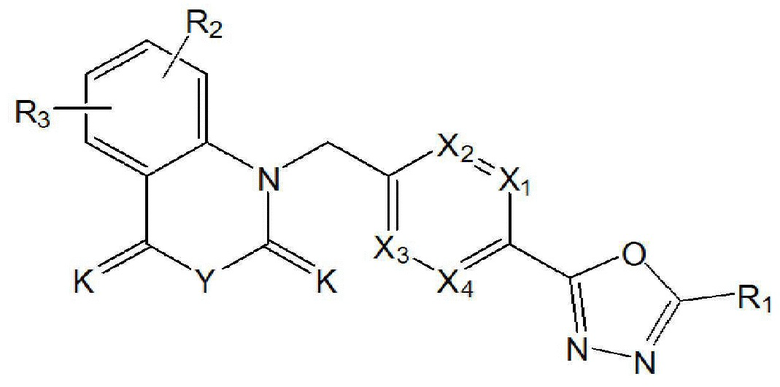
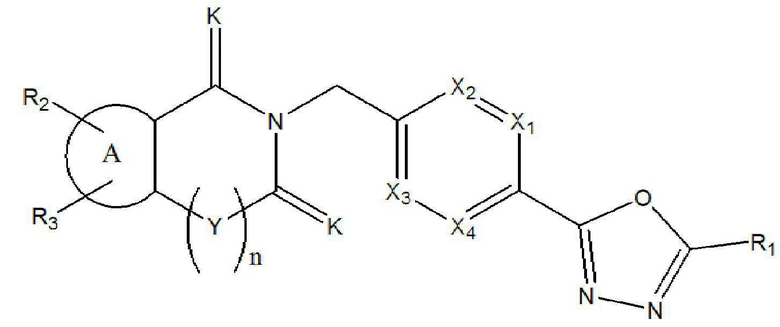
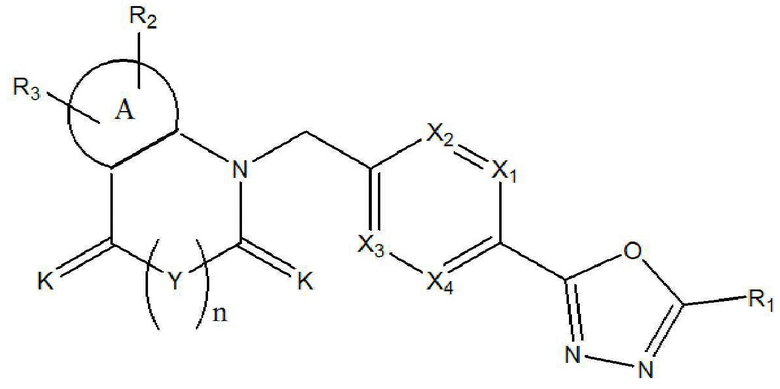
Настоящее изобретение относится к гомофталимидным производным 1,3,4-оксадиазола, описывающимся следующей химической формулой I, их стереоизомерам или их фармацевтически приемлемым солям:
Химическая формула I]
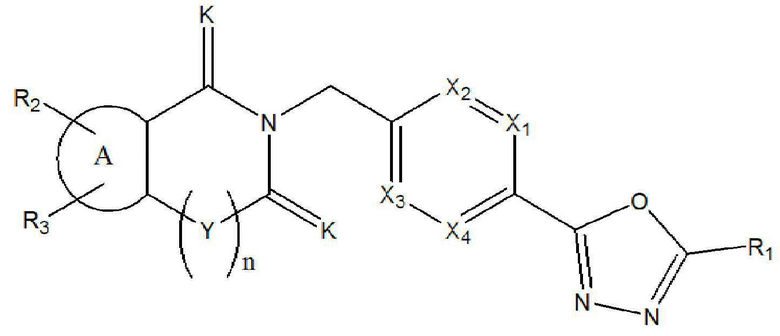
где
X1 - X4 все независимо означают CR0 или N,
где каждый R0 независимо означает водород, галоген, линейный или разветвленный -C1-7 алкил или линейный или разветвленный -O-C1-7 алкил, где по меньшей мере два из X1 - X4 означают CR0,
R1 означает линейный или разветвленный -C1-5 галогеналкил,
R2 и R3 все независимо означают H, галоген, 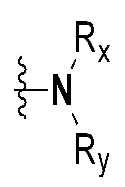 , 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 
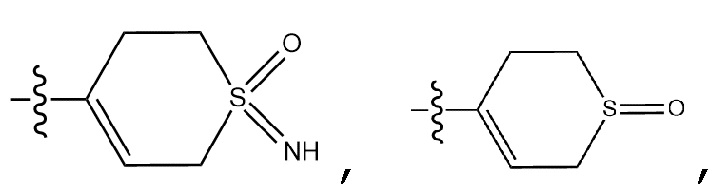 -C1-7 алкил, 3-7-членный циклоалкил, 3- - 7-членный циклоалкенил, циклопента-1,3-диен, фенил, индолил,
-C1-7 алкил, 3-7-членный циклоалкил, 3- - 7-членный циклоалкенил, циклопента-1,3-диен, фенил, индолил, 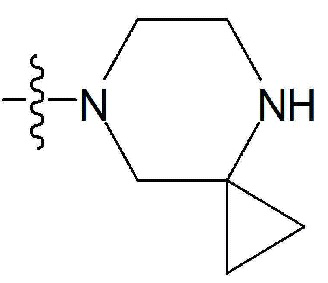 или
или 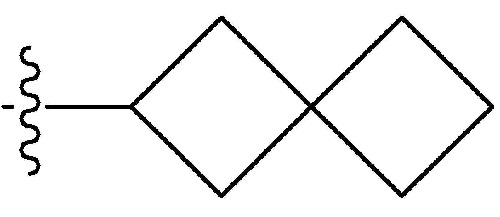 ,
,
{где по меньшей мере один водород указанного 3-7-членного гетероциклоалкила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членного гетероциклоалкенила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкил, 3-7-членный циклоалкил, 3-7-членный циклоалкенил, циклопента-1,3-диен, фенила, индолила, или может быть замещен с помощью R4,
R4 означает галоген, -C1-7 алкил, -C1-7 галогеналкил, -O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-OH, -C(=O)-O-C1-7 алкил, -S(=O)2-C1-7 алкил, 3- - 7-членный циклоалкил, 3- - 7-членный галогенциклоалкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 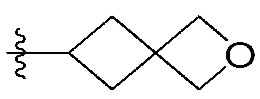 , -C1-7 алкил-C(=O)-R5, -C1-7 алкил-C(=O)-O-R6, -C1-7 алкил-R7, -C1-7 алкил-O-R8, -NR9R10, -C(=O)-NR11R12 или -C1-7 алкил-NR13R14,
, -C1-7 алкил-C(=O)-R5, -C1-7 алкил-C(=O)-O-R6, -C1-7 алкил-R7, -C1-7 алкил-O-R8, -NR9R10, -C(=O)-NR11R12 или -C1-7 алкил-NR13R14,
где R5 означает -C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, циклопента-1,3-диен или фенил,
R6 означает -C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, циклопента-1,3-диен или фенил,
R7 представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, циклопента-1,3-диен или фенил,
R8 означает -C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, циклопента-1,3-диен или фенил,
R9 и R10 все независимо означают H или -C1-7 алкил,
R11 и R12 все независимо означают H или -C1-7 алкил, и
R13 и R14 все независимо означают H или -C1-7 алкил},
Rx и Ry все независимо означают -C1-7 алкил, -C1-7 алкил-NR15R16, H, -C1-7 алкил-O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-гетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-гетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-циклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил], -C1-7 алкил-O-гетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S] или -C1-7 алкилциклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил],
{где по меньшей мере один водород группы -C1-7 алкил, -C1-7 алкил-O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-гетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-гетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-циклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил], -C1-7 алкил-O-гетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S] или -C1-7 алкилциклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил] может быть замещен следующим заместителем: -C1-7 алкил, галоген, -O-C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, -S(=O)2-C1-7 алкил, -CF3, 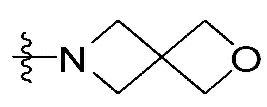 или , и
или , и
R15 и R16 все независимо означают H или -C1-7 алкил},
K означает O или S,
Y означает CRaRb, NRc или ординарную связь,
Ra и Rb все независимо означают водород, -C1-7 алкил, 3- - 7-членный циклоалкил, -C1-7 алкил-O-C1-7 алкил, -C1-7 алкил-NR17R18, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкил-C(=O)-C1-7 алкил или -C1-7 алкил-C(=O)-O-C1-7 алкил, или Ra и Rb связаны друг с другом и образуют 3- - 7-членный циклоалкил, {где по меньшей мере один водород группы C1-7 алкил, 3- - 7-членного циклоалкил, -C1-7 алкил-O-C1-7 алкил, -C1-7 алкил-NR17R18, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкил-C(=O)-C1-7 алкил или -C1-7 алкил-C(=O)-O-C1-7 алкил может быть замещен следующим заместителем: -C1-7 алкил, галоген, -O-C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, -S(=O)2-C1-7 алкил, -CF3, или , и
R17 и R18 все независимо означают H или -C1-7 алкил},
Rc означает водород, -C1-7 алкил, -C1-7 алкилгетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкилфенил, -C1-7 алкилгетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкил-O-C1-7 алкил, -C1-7 алкил-NR19R20, -C1-7 алкилциклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил], 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, циклопента-1,3-диен, фенил, -C(=O)-гетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-циклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил], -C(=O)-гетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-фенил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-O-C1-7 алкил или -C(=O)-C1-7 алкил-NR21R22,
{где по меньшей мере один водород группы -C1-7 алкил, -C1-7 алкилгетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкилфенил, -C1-7 алкилгетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкил-O-C1-7 алкил, -C1-7 алкил-NR19R20, -C1-7 алкилциклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил], 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, циклопента-1,3-диен, фенил, -C(=O)-гетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-циклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил], -C(=O)-гетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-фенил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-O-C1-7 алкил или -C(=O)-C1-7 алкил-NR19R20 может быть замещен следующим заместителем: -C1-7 алкил, галоген, -O-C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C(=O)-O-C1-7 алкил, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, гетероарил-C1-5 галогеналкил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], 3- - 7-членный циклоалкил, -S(=O)2-C1-7 алкил, -CF3, или , и
R19 и R20 все независимо означают H или -C1-7 алкил},
 означает фенилен или 5- или 6-членный гетероарилен, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
означает фенилен или 5- или 6-членный гетероарилен, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
галоген представляет собой F, Cl, Br или I, и
n равно 0 или 1.
В настоящем описании термины, использующиеся в определении заместителей гомофталимидных производных 1,3,4-оксадиазола, предлагаемых в настоящем изобретении, их стереоизомеров или их фармацевтически приемлемых солей, являются следующими.
В настоящем изобретении термин "замещение" означает, что атом водорода, связанный с атомом углерода соединения, заменен другим заместителем, и положение замещения не ограничивается определенным положением, если атом водорода является замещенным, т. е. является положением, в котором заместитель может быть замещенным. Если имеются два или большее количество заместителей, эти два или большее количество заместителей могут быть одинаковыми или разными.
В настоящем изобретении термин "галоген" означает элемент группы галогенов и включает, например, фтор (F), хлор (Cl), бром (Br) или йод (I).
В настоящем изобретении термин "алкил" означает линейный или разветвленный насыщенный углеводород, содержащий указанное количество атомов углерода если не указано иное.
В настоящем изобретении термин "галогеналкил" означает, что по меньшей мере один атом водорода, связанный с линейным или разветвленным насыщенным углеводородом, обладающим заданным количеством атомов углерода, замещен галогеном, если не указано иное.
В настоящем изобретении термин "гетероциклоалкил" означает циклический насыщенный углеводород, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S. Примеры гетероциклоалкила включают, без наложения ограничений, азетидинил, пирролидинил, пиперидинил, пиперазинил, пирролидонил, пиперидонил, морфолидинил, имидазолидинил, пиразолидинил, оксетанил, тетрагидро-2H-пиранил, морфолинил, тиоморфолинил, оксазолидинонил и тиазолидинонил.
В настоящем изобретении термин "гетероциклоалкенил" включает по меньшей мере одну двойную связь и означает циклический ненасыщенный углеводород, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S. Примеры гетероциклоалкенила включают, без наложения ограничений, тетрагидропиридинил, дигидрофуранил, и 2,5-дигидро-1H-пирролил.
В настоящем изобретении термин "гетероарил" означает гетероциклическую ароматическую группу, содержащую от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S. Примеры гетероарила включают, без наложения ограничений, фуранил, пирролил, тиофенил, тиазолил, изотиазолил, имидазолил, триазолил, тетразолил, пиразолил, оксазолил, изоксазолил, пиридинил, пиразинил, пиридазинил, пиримидинил и триазинил.
В настоящем изобретении термин "циклоалкил" означает циклический насыщенный углеводород, содержащий заданное количество атомов углерода. Примеры циклоалкила включают, без наложения ограничений, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
В настоящем изобретении термин "галогенциклоалкил" означает, что по меньшей мере один атом водорода, связанный с циклическим насыщенным углеводородом, содержащим заданное количество атомов углерода замещен галогеном, если не указано иное.
В настоящем изобретении термин "циклоалкенил" означает циклический ненасыщенный углеводород, который содержит заданное количество атомов углерода и включает по меньшей мере одну двойную связь. Примеры циклоалкенила включают, без наложения ограничений, циклопропенил, циклобутенил, циклопентенил, циклогексенил и циклогептенил.
В настоящем изобретении термин "ординарная связь" означает, что атом не содержится в соответствующем положении. Например, если Y означает ординарную связь в структуре X-Y-Z, X и Z непосредственно связаны друг с другом и образуют структуру X-Z.
В настоящем изобретении кроме указанных заместителей "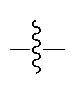 " означает положение присоединения атома, который связан с остальной частью молекулы или с остальной частью фрагмента молекулы в химической структуре.
" означает положение присоединения атома, который связан с остальной частью молекулы или с остальной частью фрагмента молекулы в химической структуре.
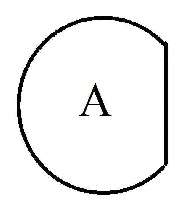
В настоящем изобретении означает структуру , сконденсированную путем использования двух общих с другим кольцом атомов, и два общих/сконденсированных атома углерода являются двумя последовательными атомами. Например, означает фенилен или 5- или 6-членный гетероарилен, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S. "5- или 6-членный гетероарилен" указанного означает фуранилен, пирролилен, тиофенилен, тиазолилен, изотиазолилен, имидазолилен, триазолилен, тетразолилен, пиразолилен, оксазолилен, изооксазолилен, пиридинилен, пиразинилен, пиридазинилен, пиримидинилен, триазинилен и т. п., который содержит от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S. В этом случае указанный фенилен и указанный гетероарилен сконденсированы путем обобщения двух атомов углерода с другим кольцом (кольцом, содержащим Y химической формулы I, обладающим структурой 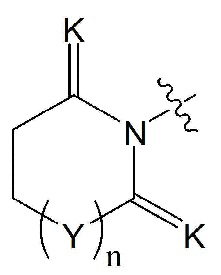 ). В этом случае два атома углерода, сконденсрованные путем обобщения в фенилен или 5- или 6-членный гетероарилен, являются двумя последовательными атомами углерода, образующих другое кольцо (кольцо, содержащее Y химической формулы I). В качестве примера, если означает фенилен, химическая формула I может содержать структуру
). В этом случае два атома углерода, сконденсрованные путем обобщения в фенилен или 5- или 6-членный гетероарилен, являются двумя последовательными атомами углерода, образующих другое кольцо (кольцо, содержащее Y химической формулы I). В качестве примера, если означает фенилен, химическая формула I может содержать структуру 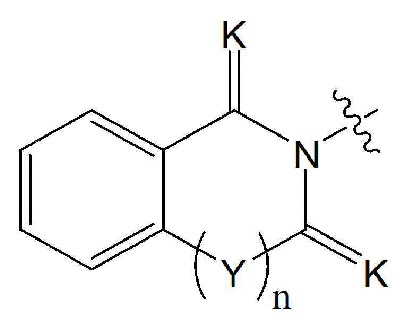 .
.
Одним вариантом осуществления настоящего изобретения является соединение, описывающееся приведенной выше химической формулой I, где:
X1 - X4 все независимо означают CR0 или N,
где R0 означает водород, галоген или -O-C1-7 алкил,
R1 означает -C1-5 галогеналкил,
R2 и R3 все независимо означают H, галоген, 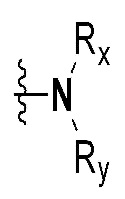 , 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, фенил, индолил,
, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, фенил, индолил,  или -C1-7 алкил,
или -C1-7 алкил,
{где по меньшей мере один водород указанного 3- - 7-членного гетероциклоалкила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членного гетероциклоалкенила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, фенила, индолила, или -C1-7 алкила может быть замещен с помощью R4,
R4 означает галоген, -C1-7 алкил, -C1-7 галогеналкил, -O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-OH, -C(=O)-O-C1-7 алкил, -S(=O)2-C1-7 алкил, 3- - 7-членный циклоалкил, 3- - 7-членный галогенциклоалкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,  , -C1-7 алкил-C(=O)-R5, -C1-7 алкил-C(=O)-O-R6, -C1-7 алкил-R7, -C1-7 алкил-O-R8, -NR9R10, -C(=O)-NR11R12 или -C1-7 алкил-NR13R14,
, -C1-7 алкил-C(=O)-R5, -C1-7 алкил-C(=O)-O-R6, -C1-7 алкил-R7, -C1-7 алкил-O-R8, -NR9R10, -C(=O)-NR11R12 или -C1-7 алкил-NR13R14,
где R5 означает -C1-7 алкил или 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
R6 означает -C1-7 алкил,
R7 представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S или 3- - 7-членный циклоалкил,
R8 означает -C1-7 алкил,
R9 и R10 все независимо означают H или -C1-7 алкил,
R11 и R12 все независимо означают H или -C1-7 алкил, и
R13 и R14 все независимо означают H или -C1-7 алкил},
Rx и Ry все независимо означают -C1-7 алкил, -C1-7 алкил-NR15R16, H, -C1-7 алкил-O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-гетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-гетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S] или -C(=O)-циклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил],
{где по меньшей мере один водород группы -C1-7 алкил, -C1-7 алкил-O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-гетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-гетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S] или -C(=O)-циклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил] может быть замещен следующим заместителем: -C1-7 алкил, галоген, -O-C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, -S(=O)2-C1-7 алкил, -CF3,  или , и
или , и
R15 и R16 все независимо означают H или -C1-7 алкил},
K означает O или S,
Y означает CRaRb, NRc или ординарную связь,
Ra и Rb все независимо означают водород, -C1-7 алкил, 3- - 7-членный циклоалкил, -C1-7 алкил-O-C1-7 алкил, -C1-7 алкил-NR17R18, или Ra и Rb связаны друг с другом и образуют 3- - 7-членный циклоалкил,
{где по меньшей мере один водород группы -C1-7 алкил, 3- - 7-членного циклоалкил, -C1-7 алкил-O-C1-7 алкил или -C1-7 алкил-NR17R18 может быть замещен следующим заместителем: -C1-7 алкил, галоген, -O-C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, -S(=O)2-C1-7 алкил, -CF3, или , и
R17 и R18 все независимо означают H или -C1-7 алкил},
Rc означает водород, -C1-7 алкил, -C1-7 алкилгетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкилфенил, -C1-7 алкилгетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкил-O-C1-7 алкил, -C1-7 алкил-NR19R20, -C1-7 алкилциклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил], 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, циклопента-1,3-диен, фенил, -C(=O)-гетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-циклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил], -C(=O)-гетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-фенил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-O-C1-7 алкил или -C(=O)-C1-7 алкил-NR21R22,
{где по меньшей мере один водород группы -C1-7 алкил, -C1-7 алкилгетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкилфенил, -C1-7 алкилгетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкил-O-C1-7 алкил, -C1-7 алкил-NR19R20, -C1-7 алкилциклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил], 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный циклоалкил, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, циклопента-1,3-диен, фенил, -C(=O)-гетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-циклоалкил [в этом случае циклоалкил представляет собой 3- - 7-членный циклоалкил], -C(=O)-гетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C(=O)-фенил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-O-C1-7 алкил или -C(=O)-C1-7 алкил-NR19R20 может быть замещен следующим заместителем: -C1-7 алкил, галоген, -O-C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C(=O)-O-C1-7 алкил, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, гетероарил-C1-5 галогеналкил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], 3- - 7-членный циклоалкил, -S(=O)2-C1-7 алкил, -CF3, или  , и
, и
R19 и R20 все независимо означают H или -C1-7 алкил},
означает фенилен или 5- или 6-членный гетероарилен, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
галоген представляет собой F, Cl, Br или I, и
n равно 0 или 1.
Также конкретным вариантом осуществления настоящего изобретения является соединение, описывающееся приведенной выше химической формулой I, где:
X1 - X4 все независимо означают CR0 или N,
R0 означает водород или галоген,
R1 означает -C1-5 галогеналкил,
R2 и R3 все независимо означают H, галоген, 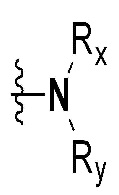 , 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 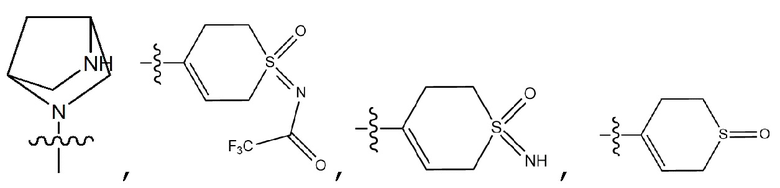 , фенил, индолил,
, фенил, индолил, 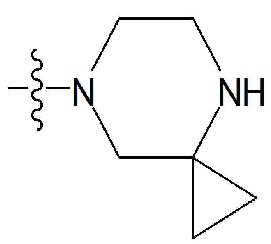 или
или 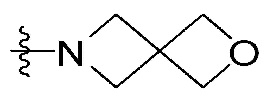 ,
,
{где по меньшей мере один водород указанного 3- - 7-членного гетероциклоалкила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членного гетероциклоалкенила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , фенила, индолила, или может быть замещен с помощью R4,
R4 означает галоген, -C1-7 алкил, -C1-7 галогеналкил, -O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-OH, -C(=O)-O-C1-7 алкил, -S(=O)2-C1-7 алкил, 3- - 7-членный циклоалкил, 3- - 7-членный галогенциклоалкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , -C1-7 алкил-C(=O)-R5, -C1-7 алкил-R7, -C1-7 алкил-O-R8, -NR9R10 или -C(=O)-NR11R12,
где R5 представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
R7 представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S или 3- - 7-членный циклоалкил,
R8 означает -C1-7 алкил,
R9 и R10 все независимо означают -C1-7 алкил, и
R11 и R12 все независимо означают H или -C1-7 алкил},
Rx и Ry все независимо означают -C1-7 алкил или -C1-7 алкил-NR15R16,
{где R15 и R16 все независимо означают -C1-7 алкил},
K означает O,
Y означает CRaRb, NRc или ординарную связь,
Ra и Rb все независимо означают водород или -C1-7 алкил, или Ra и Rb связаны друг с другом и образуют 3- - 7-членный циклоалкил,
Rc означает водород, -C1-7 алкил, -C1-7 алкилгетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкилфенил, -C1-7 алкилгетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкил-O-C1-7 алкил или -C1-7 алкил-NR19R20,
{где по меньшей мере один водород группы -C1-7 алкил, -C1-7 алкилгетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкилфенил, -C1-7 алкилгетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкил-O-C1-7 алкил или -C1-7 алкил-NR19R20 может быть замещен следующим заместителем: -C1-7 алкил, -O-C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, гетероарил-C1-5 галогеналкил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S] или -C(=O)-O-C1-7 алкил, и
R19 и R20 все независимо означают -C1-7 алкил},
означает фенилен,
галоген представляет собой F или Br, и
n равно 0 или 1.
Более предпочтительным вариантом осуществления настоящего изобретения является соединение, описывающееся приведенной выше химической формулой I, где:
X1 - X4 все независимо означают CR0 или N,
R0 означает водород или F,
R1 означает CF2H,
R2 и R3 все независимо означают H, F, Br, 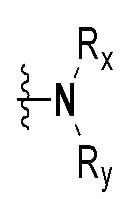 , 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 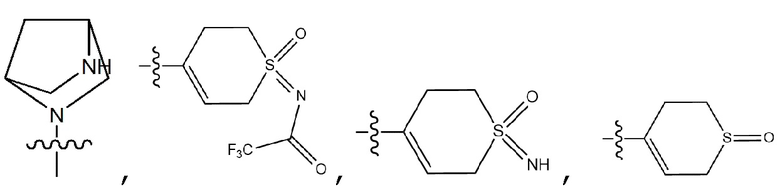 , фенил, индолил,
, фенил, индолил, 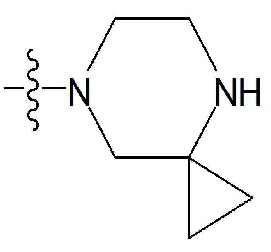 или
или 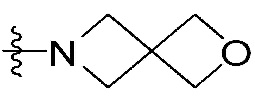 ,
,
{где по меньшей мере один водород указанного 3- - 7-членного гетероциклоалкила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3- - 7-членного гетероциклоалкенила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , фенила, индолила, или может быть замещен с помощью R4,
R4 означает F, -C1-7 алкил, -C1-7 галогеналкил, -O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-OH, -C(=O)-O-C1-7 алкил, -S(=O)2-C1-7 алкил, 3- - 7-членный циклоалкил, 3- - 7-членный галогенциклоалкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , -C1-7 алкил-C(=O)-R5, -C1-7 алкил-R7, -C1-7 алкил-O-R8, -NR9R10 или -C(=O)-NR11R12,
где R5 представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
R7 представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S или 3- - 7-членный циклоалкил,
R8 означает -C1-7 алкил,
R9 и R10 все независимо означают -C1-7 алкил, и
R11 и R12 все независимо означают H или -C1-7 алкил},
Rx и Ry все независимо означают -C1-7 алкил или -C1-7 алкил-NR15R16,
{где R15 и R16 все независимо означают -C1-7 алкил},
K означает O,
Y означает CRaRb, NRc или ординарную связь,
Ra и Rb все независимо означают водород или -C1-7 алкил, или Ra и Rb связаны друг с другом и образуют 3- - 7-членный циклоалкил,
Rc означает водород, -C1-7 алкил, -C1-7 алкилгетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкилфенил, -C1-7 алкилгетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкил-O-C1-7 алкил или -C1-7 алкил-NR19R20,
{где по меньшей мере один водород группы -C1-7 алкил, -C1-7 алкилгетероциклоалкил [в этом случае гетероциклоалкил представляет собой 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкилфенил, -C1-7 алкилгетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], -C1-7 алкил-O-C1-7 алкил или -C1-7 алкил-NR19R20 может быть замещен следующим заместителем: -C1-7 алкил, -O-C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, гетероарил-C1-5 галогеналкил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S] или -C(=O)-O-C1-7 алкил, и
R19 и R20 все независимо означают -C1-7 алкил},
означает фенилен,
галоген представляет собой F или Br, и
n равно 0 или 1.
В конкретном варианте осуществления настоящего изобретения соединение, описывающееся приведенной выше химической формулой I, может представлять собой соединение, описывающееся следующей химической формулой I-1:
Химическая формула I-1]
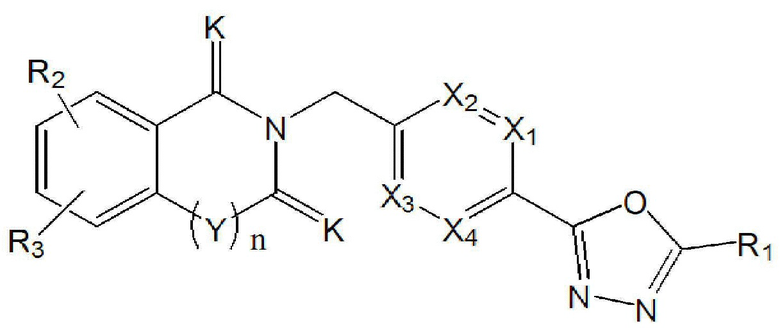
где
X1 - X4, R1 - R3, Y, K и n являются такими же, как определено для химической формулы I.
Настоящее изобретение относится к гомофталимидным производным 1,3,4-оксадиазола, описывающимся следующей химической формулой II, их стереоизомерам или их фармацевтически приемлемым солям:
Химическая формула II]
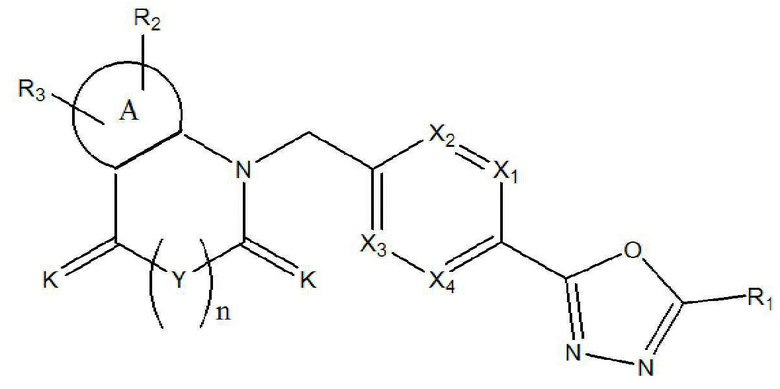
где
A, X1 - X4, R1 - R3, Y, K и n являются такими же, как определено для химической формулы I.
Конкретным вариантом осуществления настоящего изобретения является соединение, описывающееся приведенной выше химической формулой II, где:
X1 - X4 все независимо означают CR0 или N,
R0 означает водород,
R1 означает CF2H,
R2 и R3 означают H,
K означает O,
Y означает NRc,
Rc означает -C1-7 алкилфенил, -C1-7 алкилгетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S] или -C1-7 алкил-O-C1-7 алкил,
{где по меньшей мере один водород группы -C1-7 алкилфенил, -C1-7 алкилгетероарил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S] или -C1-7 алкил-O-C1-7 алкил может быть замещен следующим заместителем: гетероарил-C1-5 галогеналкил [в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S]},
означает фенилен,
галоген представляет собой F, и
n равно 1.
В конкретном варианте осуществления настоящего изобретения соединение, описывающееся приведенной выше химической формулой II, может представлять собой соединение, описывающееся следующей химической формулой II-1:
Химическая формула II-1]
где
X1 - X4, R1 - R3, Y, K и n являются такими же, как определено для химической формулы I.
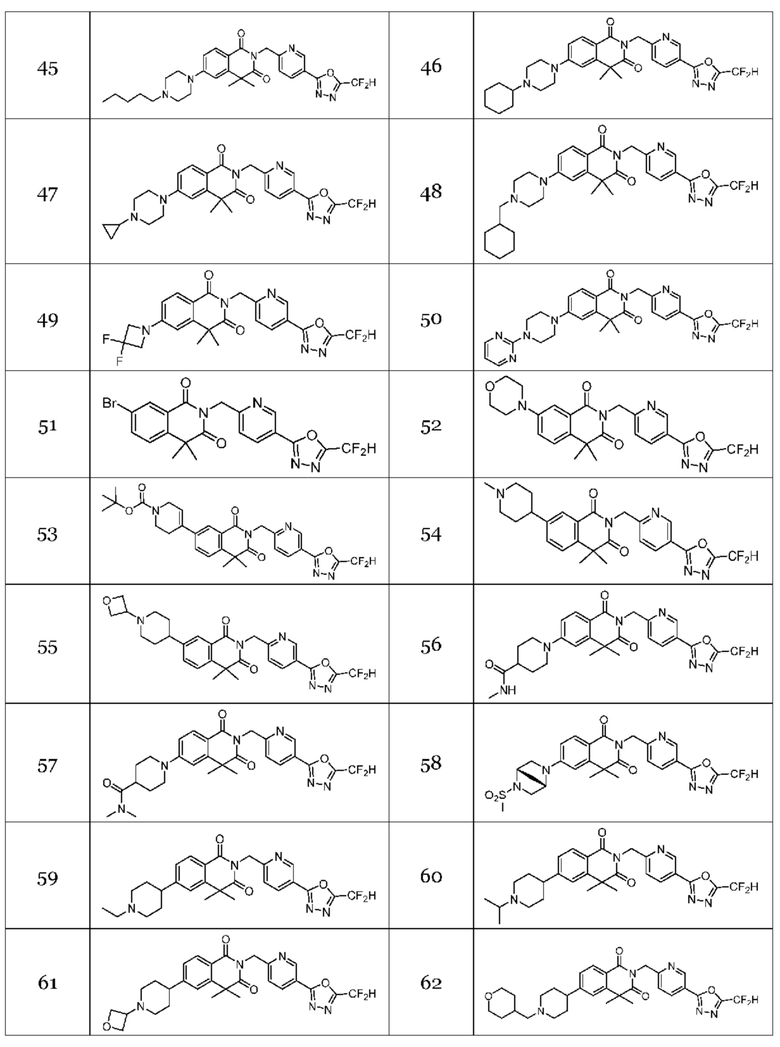
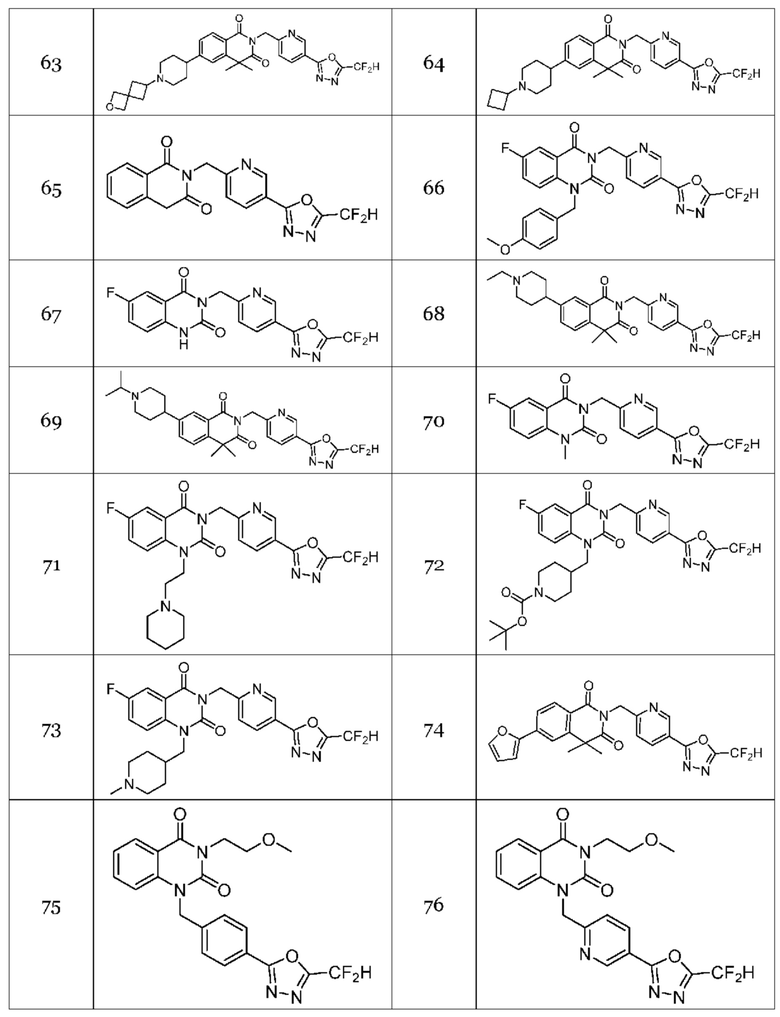
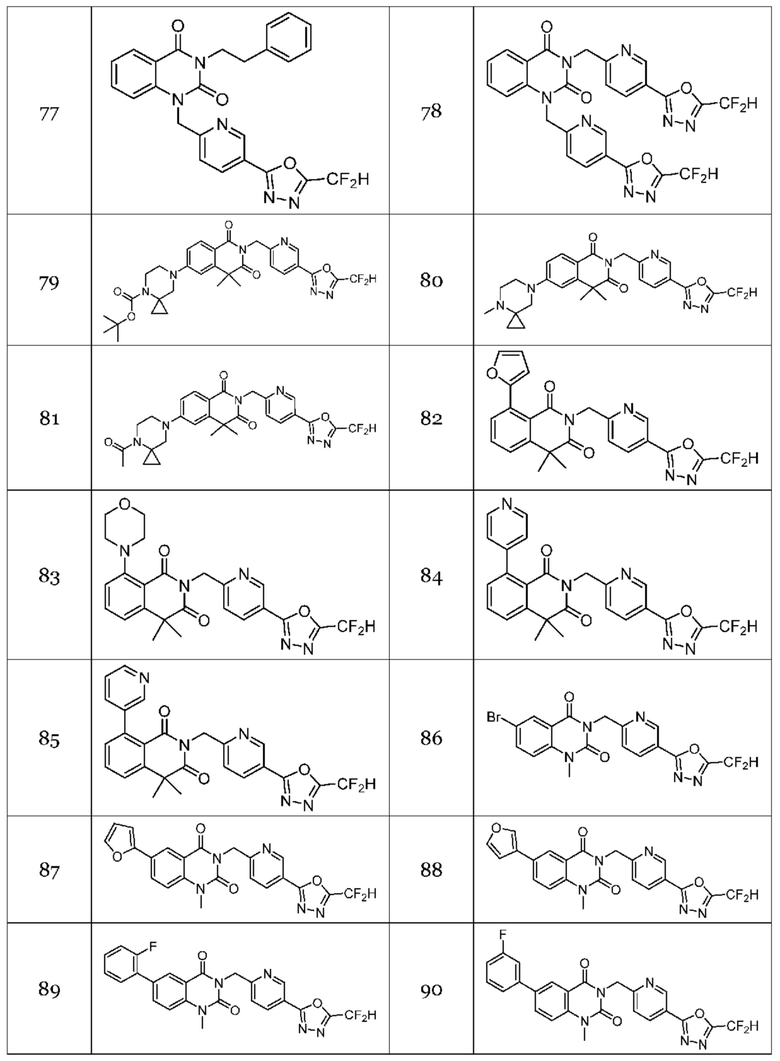
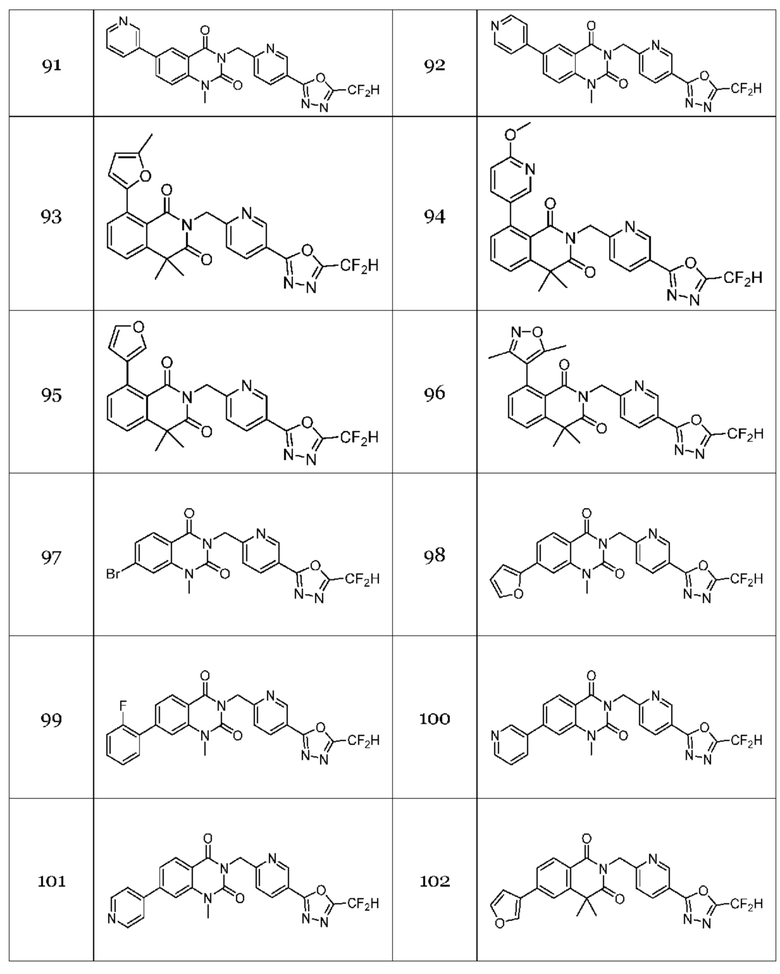
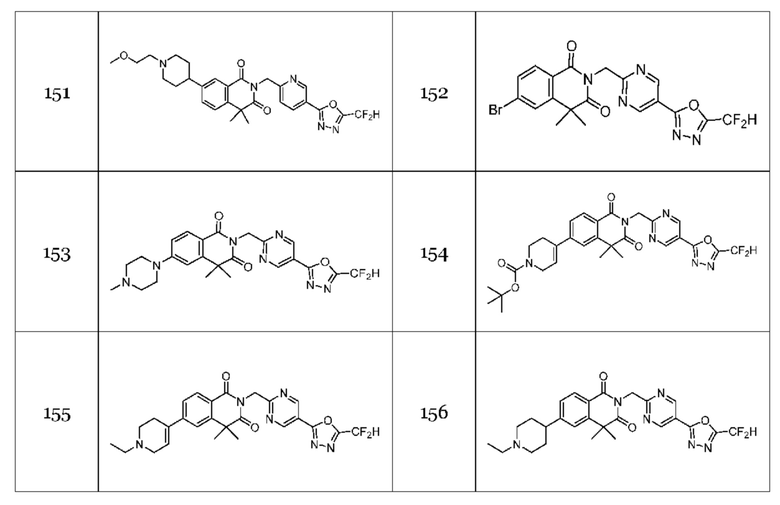
Настоящее изобретение относится к гомофталимидным производным 1,3,4-оксадиазола, описанным в следующей таблице 1, их стереоизомерам или их фармацевтически приемлемым солям.
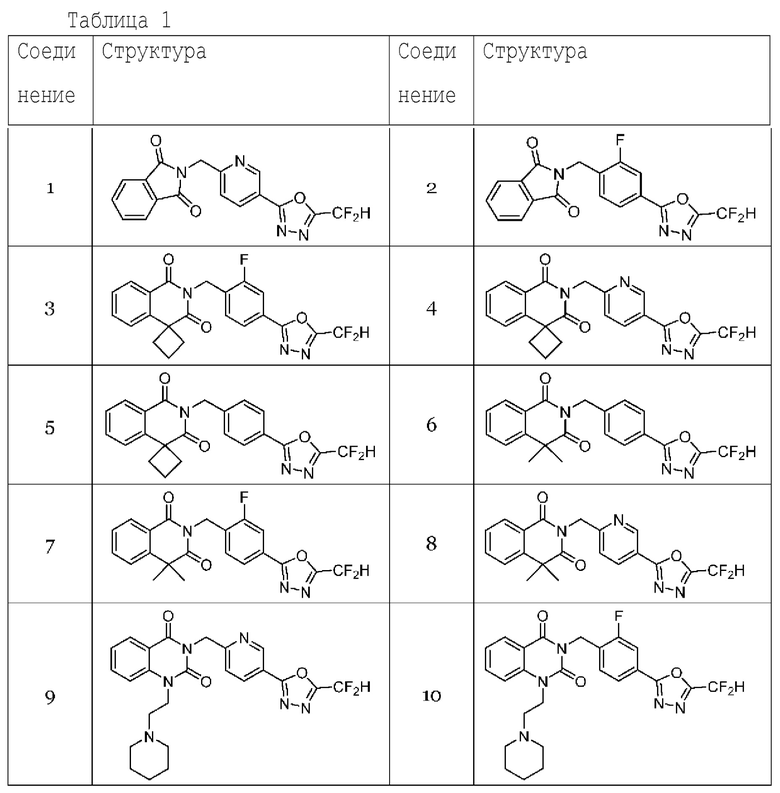
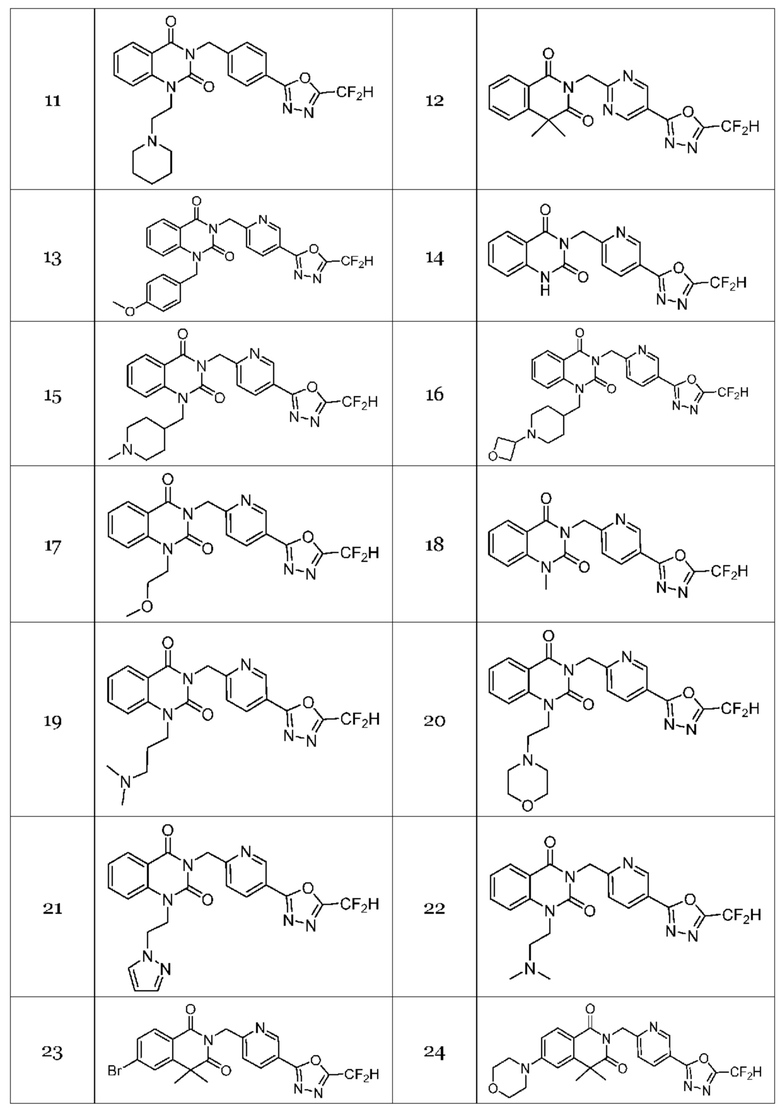
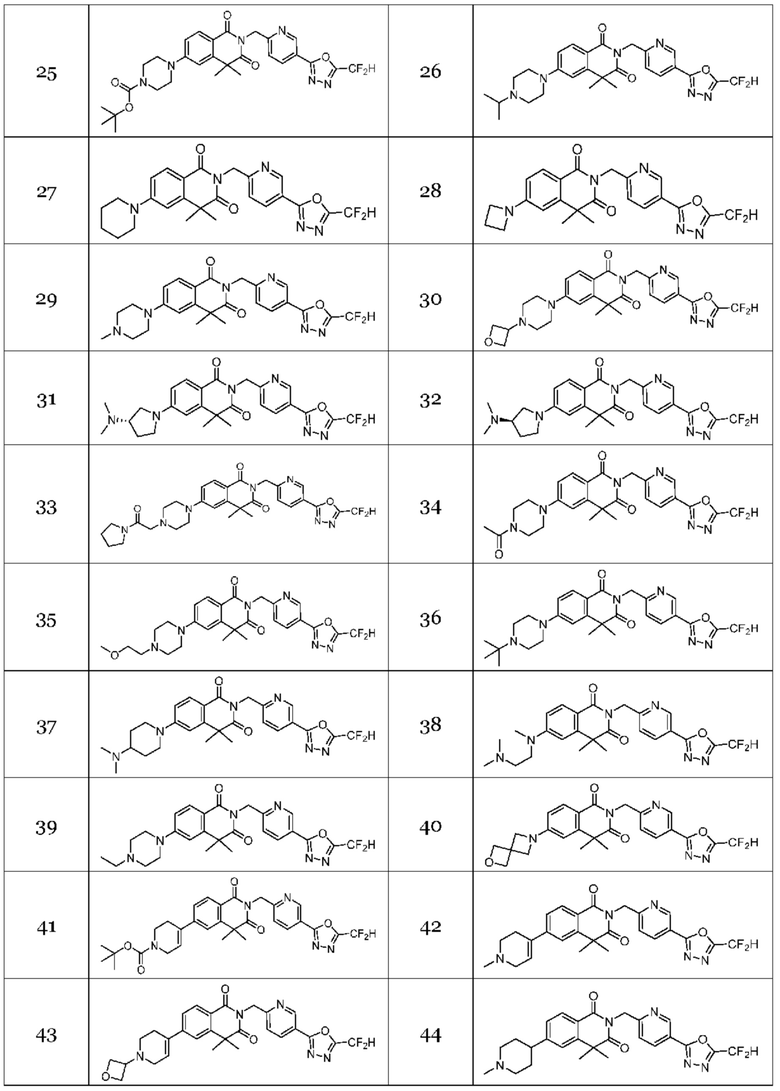
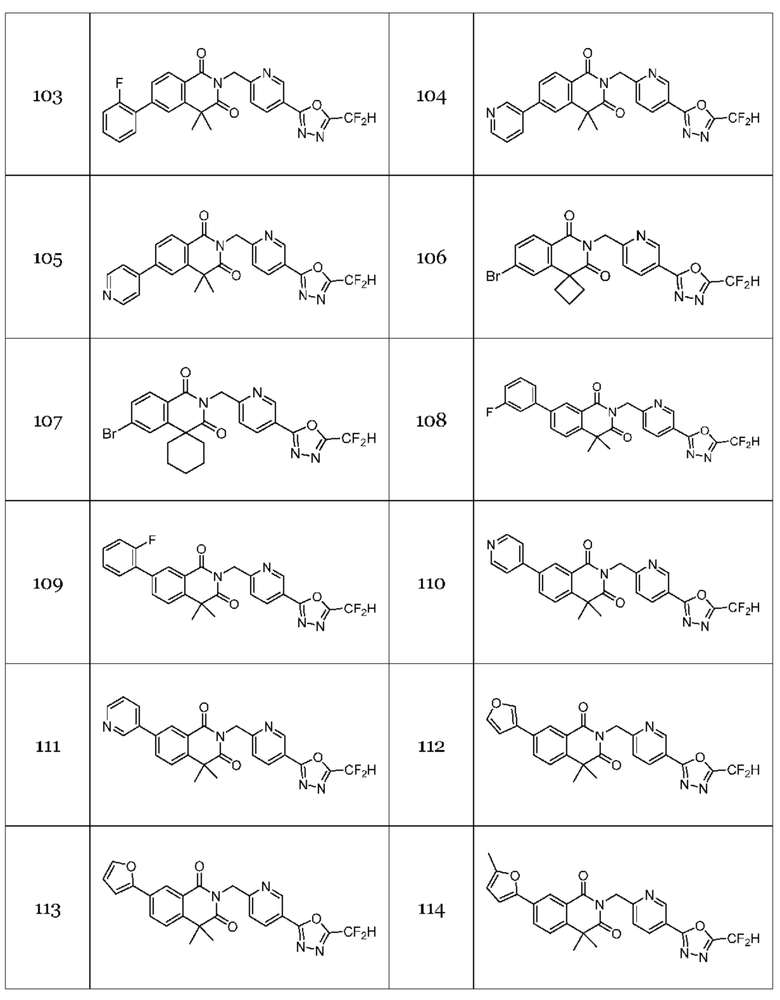
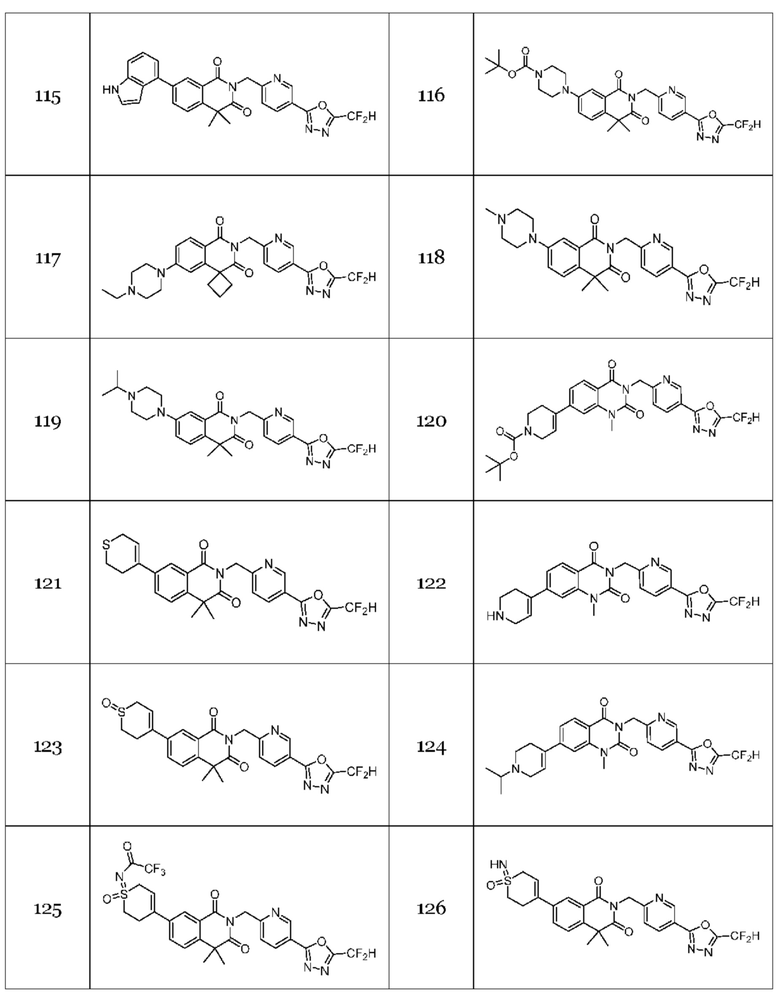
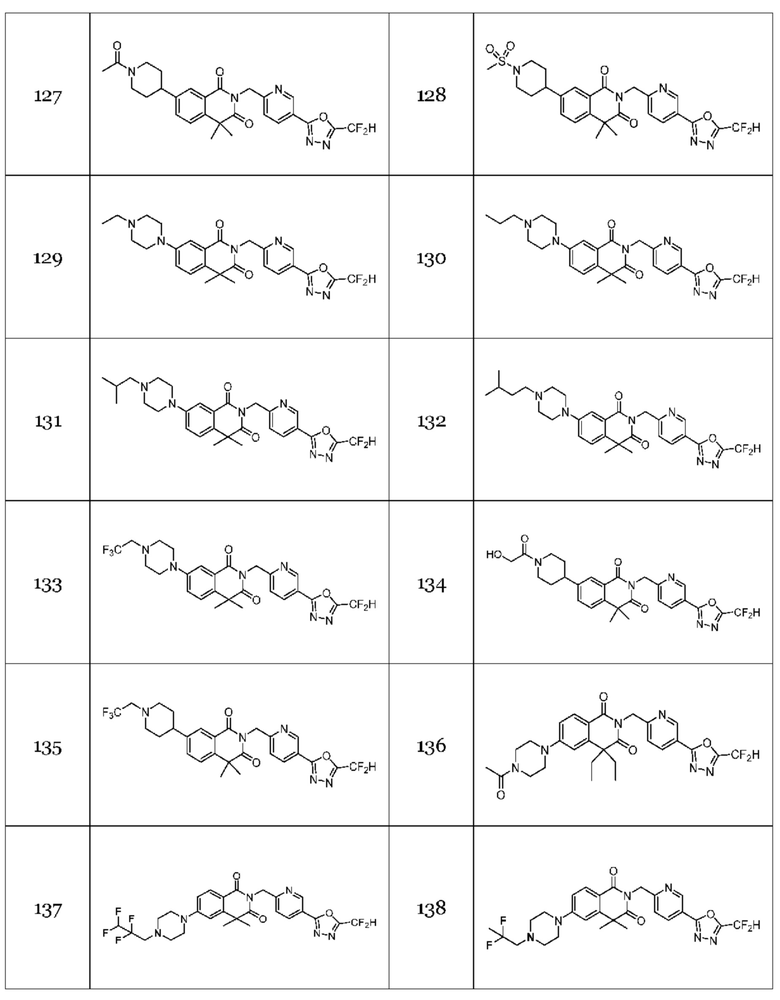
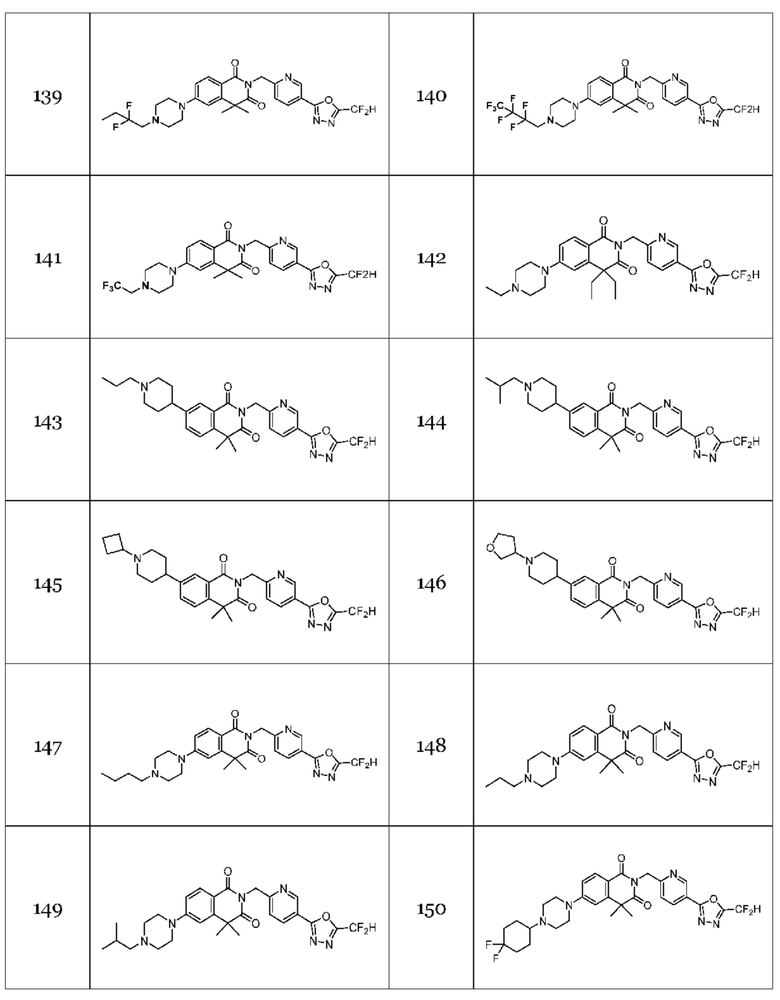
Гомофталимидные производные 1,3,4-оксадиазола, предлагаемые в настоящем изобретении, могут содержать по меньшей мере один асимметрический углерод и, таким образом, могут содержаться в виде рацемата, рацемической смеси, одного энантиомера (оптического изомера), смеси диастереоизомеров и соответствующих его диастереоизомеров. Стереоизомеры можно разделить по методикам родственной области техники, например, колоночной хроматографии, HPLC и т. п. Альтернативно, соответствующие стереоизомеры гомофталимидных производных 1,3,4-оксадиазола, предлагаемые в настоящем изобретении, можно стереоспецифически синтезировать с помощью общеизвестной группы оптически чистых исходных веществ и/или реагентов.
В настоящем изобретении термин "фармацевтически приемлемый" означает являющийся физиологически приемлемым и обычно не приводящий к аллергической реакции, такой как желудочно-кишечное заболевание и головокружение или другим аналогичным реакциям при введении человеку, и термин "соль" означает соль, полученную по обычной методике в виде соли присоединения с кислотой, образованную фармацевтически приемлемой свободной кислотой, и способ получения фармацевтически приемлемой соли обычно известен специалистам в данной области техники. Фармацевтически приемлемые соли включают, например, соли неорганического иона, полученные из кальция, калия, натрия, магния и т. п.; соли неорганической кислоты, полученные из хлористоводородной кислоты, азотной кислоты, фосфорной кислоты, бромноватой кислоты, йодноватой кислоты, иодистоводоролной кислоты, хлорной кислоты, серной кислоты и т. п.; соли органической кислоты, полученные из уксусной кислоты, трифторуксусной кислоты, лимонной кислоты, малеиновой кислоты, янтарной кислоты, щавелевой кислоты, бензойной кислоты, винной кислоты, фумаровой кислоты, миндальной кислоты, пропионовой кислоты, лимонной кислоты, молочной кислоты, гликолевой кислоты, глюконовой кислоты, галактуроновой кислоты, глутаминовой кислоты, глутаровой кислоты, глюкуроновой кислоты, аспарагиновой кислоты, аскорбиновой кислоты, угольной кислоты, ванилиновой кислоты и т. п.; соли сульфоновой кислоты, полученные из метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, п-толуолсульфоновой кислоты, нафталинсульфоновой кислоты и т. п.; соли аминокислот, полученные из глицина, аргинина, лизина и т. п.; соли аминов, полученные получали из триметиламина, триэтиламина, аммиака, пиридина, пиколина и т. п.; и т. п., но типы солей, использующихся в настоящем изобретении, не ограничиваются перечисленными солями. В настоящем изобретении предпочтительные соли включают соли хлористоводородной кислоты, трифторуксусной кислоты, лимонной кислоты, бромноватой кислоты, малеиновой кислоты, фосфорной кислоты, серной кислоты и винной кислоты.
Способ получения гомофталимидных производных 1,3,4-оксадиазола
Настоящее изобретение относится к способу получения гомофталимидных производных 1,3,4-оксадиазола, их стереоизомеров или их фармацевтически приемлемых солей.
В настоящем изобретении предпочтительный способ получения гомофталимидных производных 1,3,4-оксадиазола, их стереоизомеров или их фармацевтически приемлемых солей является таким, как показано на схемах реакций 1-14, и в настоящее изобретение даже также включен способ получения, модифицированный в степени, очевидной для специалистов в данной области техники.
Схема реакции 1
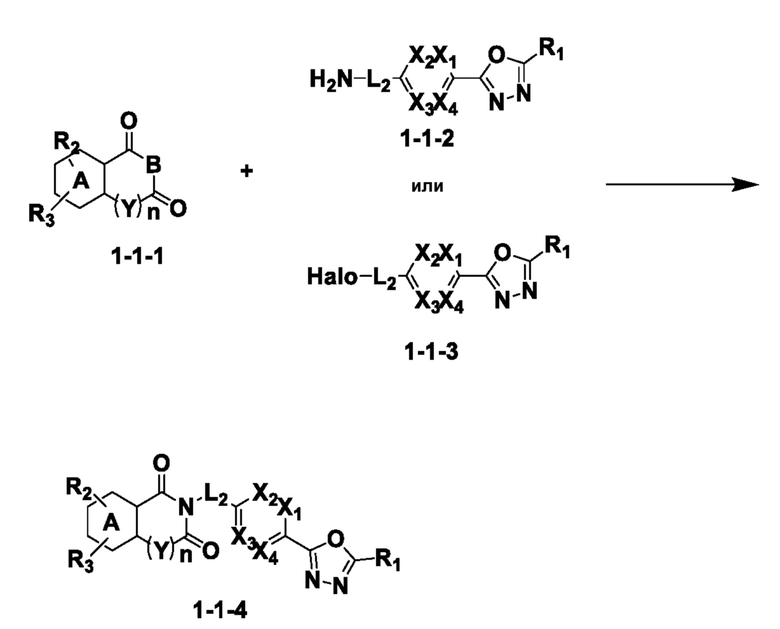
На приведенной выше схеме реакции 1, A, X1 - X4, R1 - R3, Y и n являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 1, A означает фенил, X1 - X4 все независимо означают CH, CF или N, L2 означает метилен (CH2), B означает N, R1 означает CF2H, R2 и R3 означают H, Y означает метилен (CH2) или C (C1-7 алкил)2, Halo означает галоген и n равно 0 или 1.
На приведенной выше схеме реакции 1 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-1-1 взаимодействует с соединением химической формулы 1-1-2 или химической формулы 1-1-3 и получают соединения химической формулы 1-1-4, обладающее структурой 1,3,4-оксадиазола.
В настоящем изобретении соединения, полученные по приведенным выше схемам реакций, включают 1, 2, 12, 65 и т. п.
Схема реакции 2
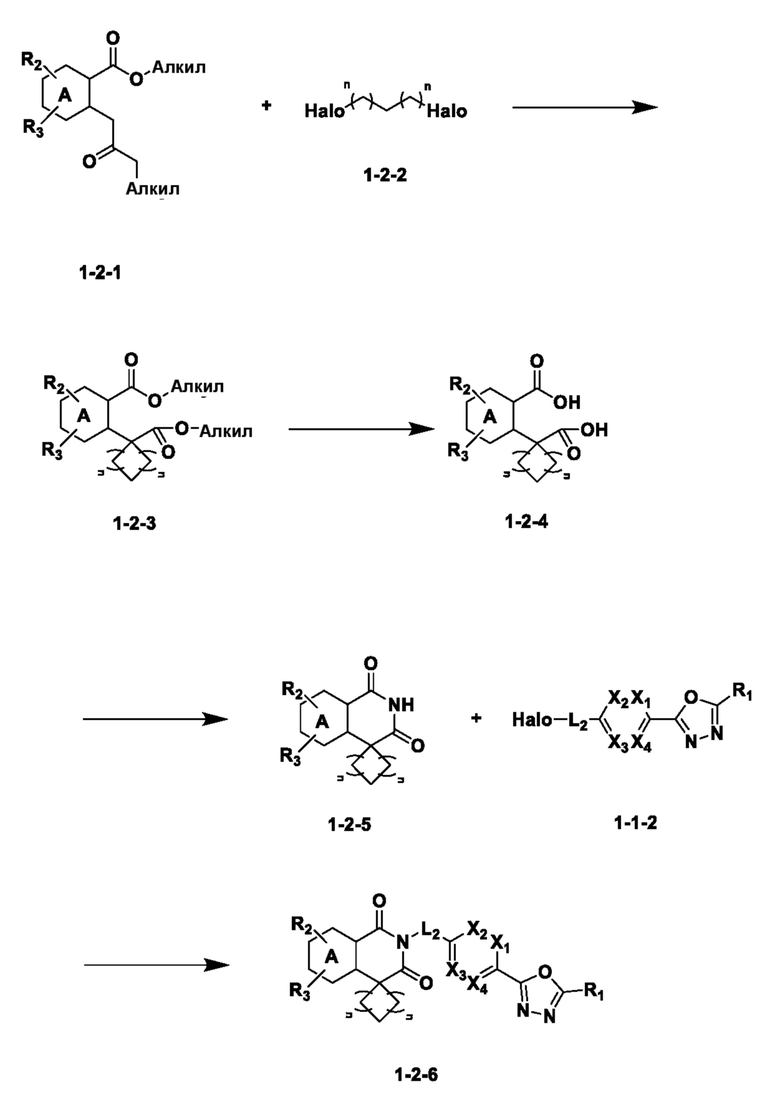
На приведенной выше схеме реакции 2, A, X1 - X4 и R1 - R3 являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 2, A означает фенил, X1 - X4 все независимо означают CH, CF или N, L2 означает метилен (CH2), R1 означает CF2H, R2 и R3 означают H, Y означает CRaRb (Ra и Rb образуют циклобутан), Halo означает галоген и алкил представляет собой C1-7 алкил.
На приведенной выше схеме реакции 2 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-2-1 вводят в реакцию замещения с соединением химической формулы 1-2-2 и получают соединения химической формулы 1-2-3 и затем вводят в реакцию гидролиза и получают соединения химической формулы 1-2-4. Затем соединение химической формулы 1-2-4 взаимодействует с мочевиной и получают соединения химической формулы 1-2-5 и затем вводят в реакцию замещения с соединением химической формулы 1-1-2 и получают соединения химической формулы 1-2-6.
В настоящем изобретении соединения, полученные по приведенным выше схемам реакций, включают 3, 4, 5, 106, 107 и т. п.
Схема реакции 3
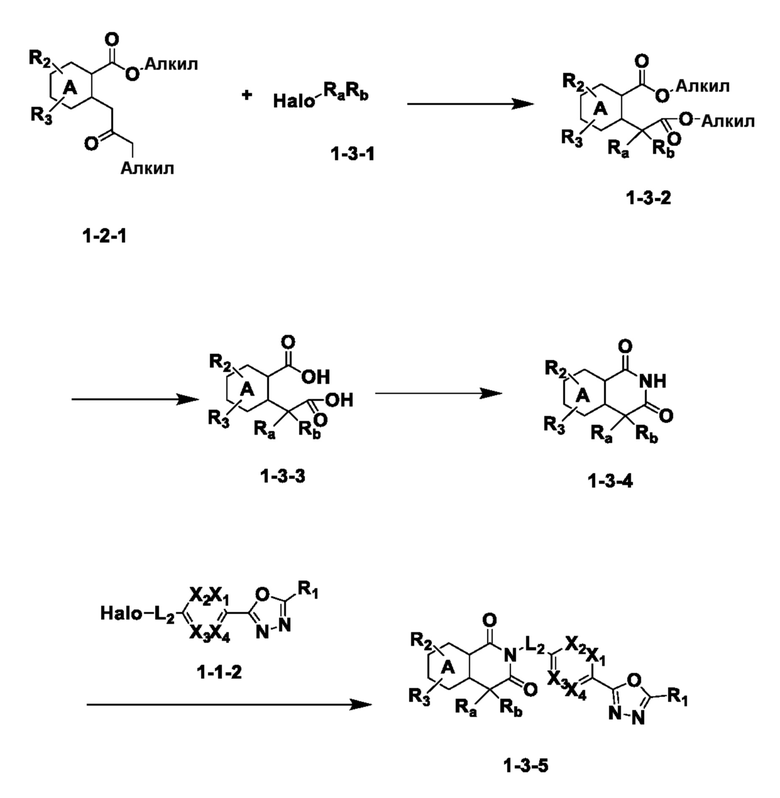
На приведенной выше схеме реакции 3, A, X1 - X4, R1 - R3 и Ra - Rb являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 3, A означает фенил, X1 - X4 все независимо означают CH, CF или N, L2 означает метилен (CH2), R1 означает CF2H, R2 и R3 все независимо означают H или галоген, Ra и Rb означают C1-7 алкил, Halo означает галоген и алкил представляет собой C1-7 алкил.
На приведенной выше схеме реакции 3 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-2-1 вводят в реакцию замещения с соединением химической формулы 1-3-1 и получают соединения химической формулы 1-3-2 и затем вводят в реакцию гидролиза и получают соединения химической формулы 1-3-3. Затем соединение химической формулы 1-3-3 взаимодействует с мочевиной и получают соединения химической формулы 1-3-4 и затем вводят в реакцию замещения с соединением химической формулы 1-1-2 и получают соединения химической формулы 1-3-5.
В настоящем изобретении соединения, полученные по приведенным выше схемам реакций, включают 6, 7, 8, 23, 51, 152 и т. п.
Схема реакции 4
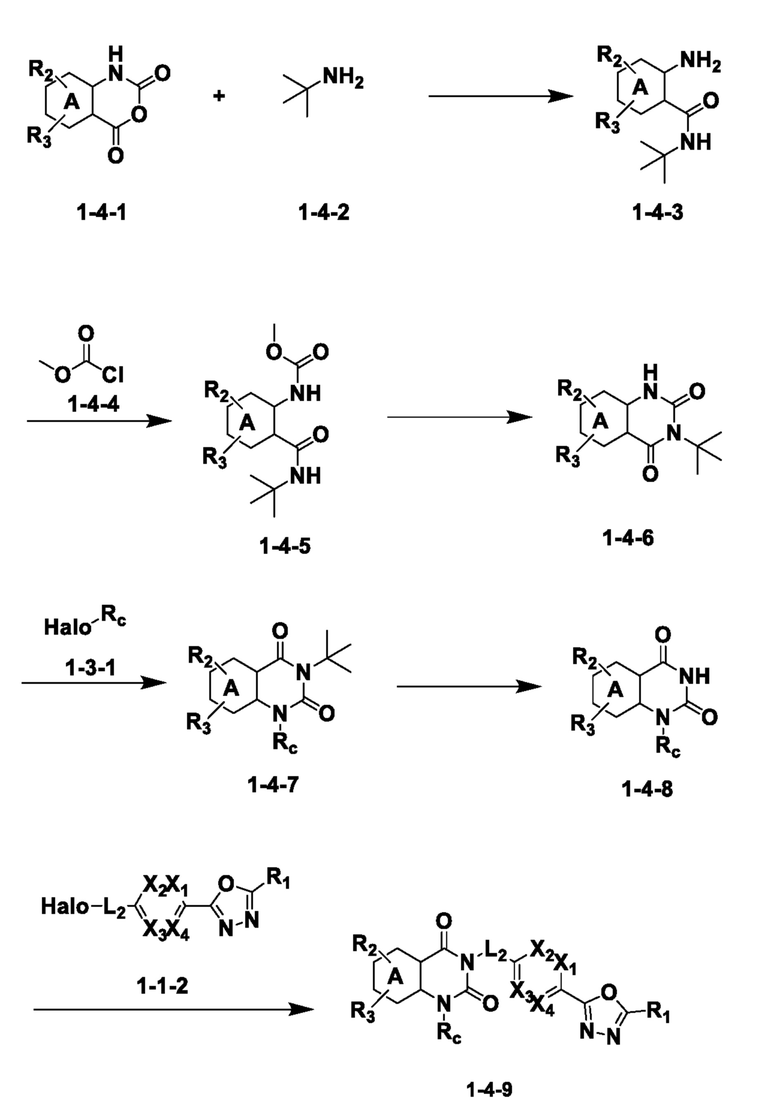
На приведенной выше схеме реакции 4, A, X1 - X4, R1 - R3 и Rc являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 4, A означает фенил, X1 - X4 все независимо означают CH, CF или N, L2 означает метилен (CH2), R1 означает CF2H, R2 и R3 все независимо означают H или галоген, Rc означает C1-7 алкилгетероциклоалкил, C1-7 алкилфенил или C1-7 алкил, Halo означает галоген и алкил представляет собой C1-7 алкил.
На приведенной выше схеме реакции 4 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-4-1 взаимодействует с соединением химической формулы 1-4-2 и получают соединения химической формулы 1-4-3 и затем вводят в реакцию замещения с соединением химической формулы 1-4-4 и получают соединения химической формулы 1-4-5. Затем соединение химической формулы 1-4-5 взаимодействует с гидроксидом калия и получают соединения химической формулы 1-4-6 и затем вводят в реакцию замещения с соединением химической формулы 1-3-1 и получают соединения химической формулы 1-4-7. Соединение химической формулы 1-4-7 взаимодействует с водным раствором хлористоводородной кислоты и получают соединения химической формулы 1-4-8 и затем вводят в реакцию замещения с соединением химической формулы 1-1-2 и получают соединения химической формулы 1-4-9.
В настоящем изобретении соединения, полученные по приведенным выше схемам реакций, включают 9, 10, 11, 13, 66, 86, 97 и т. п.
Схема реакции 5
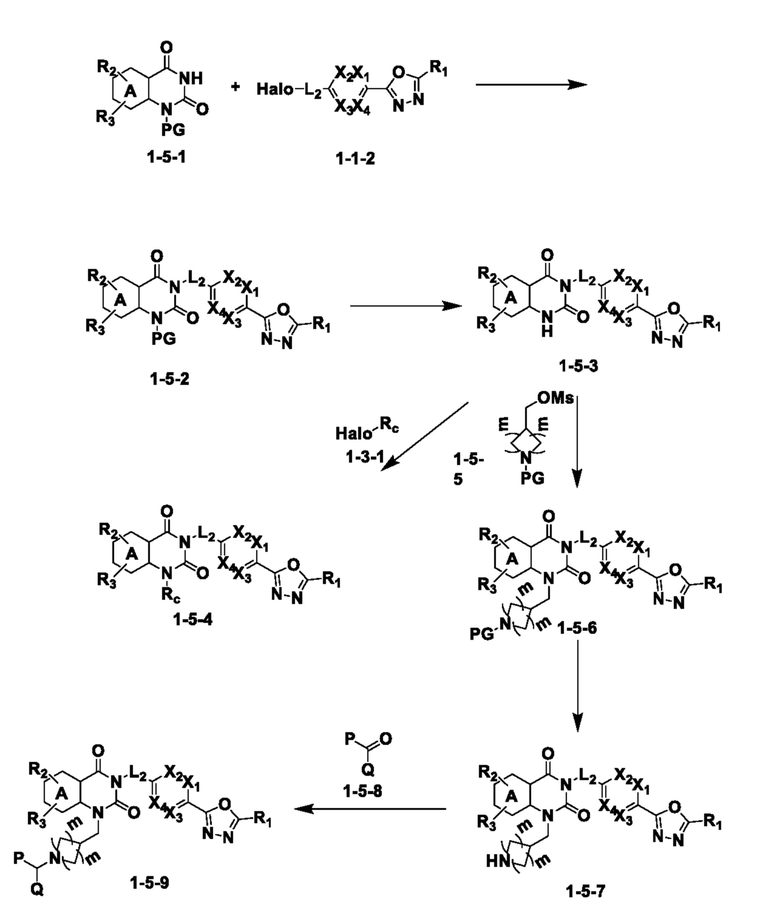
На приведенной выше схеме реакции 5, A, X1 - X4, R1 - R3 и Rc являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 5, A означает фенил, X1 - X4 все независимо означают CH или N, L2 означает метилен (CH2), R1 означает CF2H, R2 и R3 все независимо означают H или галоген, Rc означает C1-7 алкилгетероциклоалкил, C1-7 алкил-O-C1-7 алкил, C1-7 алкил, C1-7 алкил-N(C1-7 алкил)2 или C1-7 алкилгетероарил, Halo означает галоген, алкил представляет собой C1-7 алкил, OMs означает мезилат, PG означает защитную группу, m равно 2 и P и Q означают водород.
На приведенной выше схеме реакции 5 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-5-1, которое получают по схеме реакций 4 и в которое введена защитная группа, вводят в реакцию замещения с соединением химической формулы 1-1-2 и получают соединения химической формулы 1-5-2 и затем защитную группу удаляют и получают соединения 14, 67 и т. п. химической формулы 1-5-3. Затем соединение химической формулы 1-5-3 вводят в реакцию замещения с соединением химической формулы 1-3-1 и получают соединения химической формулы 1-5-4.
Также соединение химической формулы 1-5-3 вводят в реакцию замещения с соединением химической формулы 1-5-5, в которое введена защитная группа, и получают соединения химической формулы 1-5-6 и затем защитную группу удаляют и получают соединения химической формулы 1-5-7. Затем проводят реакцию восстановительного аминирования с соединением химической формулы 1-5-8 и получают соединения химической формулы 1-5-9.
В настоящем изобретении соединения, полученные по приведенным выше схемам реакций, включают 15, 16, 17, 18, 19, 20, 21, 22, 70, 71, 72, 73 и т. п.
Схема реакции 6
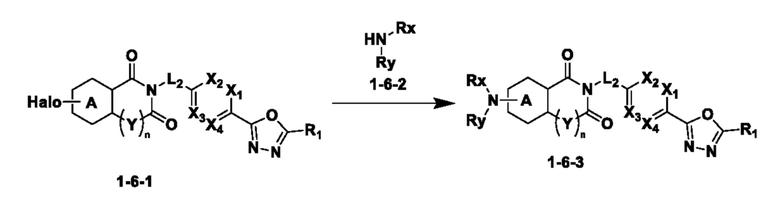
На приведенной выше схеме реакции 6, A, X1 - X4, R1 - R3 и Rx - Ry являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 6, A означает фенил, X1 - X4 все независимо означают CH или N, L2 означает метилен (CH2), R1 означает CF2H, R2 и R3 все независимо означают H или -NRxRy, Rx и Ry связаны друг с другом с образованием кольца вместе со связанным с ними атомом азота {в этом случае образованное кольцо может дополнительно содержать один гетероатом N или O, и по меньшей мере один водород образованного кольца, с которым связаны Rx и Ry и связаны с атомом азота, может содержать в качестве заместителя C1-7 алкил, C(=O)-C1-7 алкил, 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, N(C1-7 алкил)2, C1-7 алкил-C(=O)-3- - 7-членный гетероциклоалкил [в этом случае гетероциклоалкил содержит от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], C(=O)-C1-7 алкил, C1-7 алкил-O-C1-7 алкил, C(=O)-O-C1-7 алкил, 3- - 7-членный циклоалкил, C1-7 алкил-3- - 7-членный циклоалкил, галоген, 5- или 6-членный гетероарил [в этом случае гетероарил содержит от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], C(=O)-NH-C1-7 алкил, C(=O)-N(C1-7 алкил)2 или S(=O)2-C1-7 алкил}, Y означает C(C1-7 алкил)2, n равно 1 и Halo означает галоген.
На приведенной выше схеме реакции 6 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-6-1 вводят в C-N сочетание (реакция Бухвальда) с соединением химической формулы 1-6-2 и получают соединения химической формулы 1-6-3.
В настоящем изобретении соединения, полученные по приведенным выше схемам реакций, включают 24, 27, 28, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 45, 46, 47, 48, 49, 50, 52, 56, 57, 58, 117, 153 и т. п.
Схема реакции 7
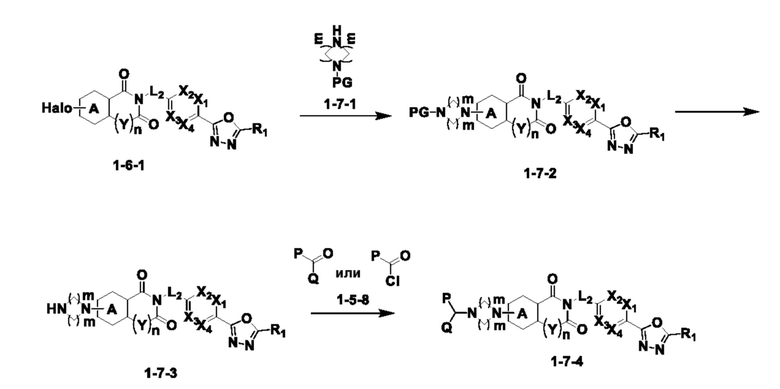
На приведенной выше схеме реакции 7, A, X1 - X4, R1 - R3, Y и n являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 7, A означает фенил, X1 - X4 все независимо означают CH или N, L2 означает метилен (CH2), R1 означает CF2H, R2 и R3 все независимо означают H или 3- - 7-членный гетероциклоалкил [в этом случае гетероциклоалкил содержит от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S], Y означает C(C1-7 алкил)2, n равно 1, Halo означает галоген, алкил представляет собой C1-7 алкил, PG означает защитную группу, m равно 2 P и Q означают C1-7 алкил, или P и Q связаны друг с другом с образованием кольца вместе со связанным с ними атомом углерода, где образованное кольцо может дополнительно содержать один гетероатом N или O.
На приведенной выше схеме реакции 7 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-6-1 вводят в C-N сочетание (реакция Бухвальда) с соединением химической формулы 1-7-1, содержащим защитную группу, и получают соединения 25, 79 и т. п. химической формулы 1-7-2. Затем защитную группу удаляют и получают соединения химической формулы 1-7-3, и проводят реакцию восстановительного аминирования и реакцию ацилирования с соединением химической формулы 1-5-8 и получают соединения 26, 30, 80, 81, 136, 141, 142, 147, 148, 149, 150 и т. п. химической формулы 1-7-4.
Схема реакции 8
На приведенной выше схеме реакции 8, A, X1 - X4, R1 - R3, Y и n являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 8, A означает фенил, X1 - X4 все независимо означают CH или N, L2 означает метилен (CH2), R1 означает CF2H, R2 и R3 все независимо означают H или 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, Y означает C(C1-7 алкил)2, n равно 1, Halo означает галоген, PG означает защитную группу, P и Q все независимо означают H, C1-7 алкил или 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, или P и Q связаны друг с другом с образованием кольца вместе со связанным с ними атомом углерода, где образованное кольцо может дополнительно содержать один гетероатом N или O.
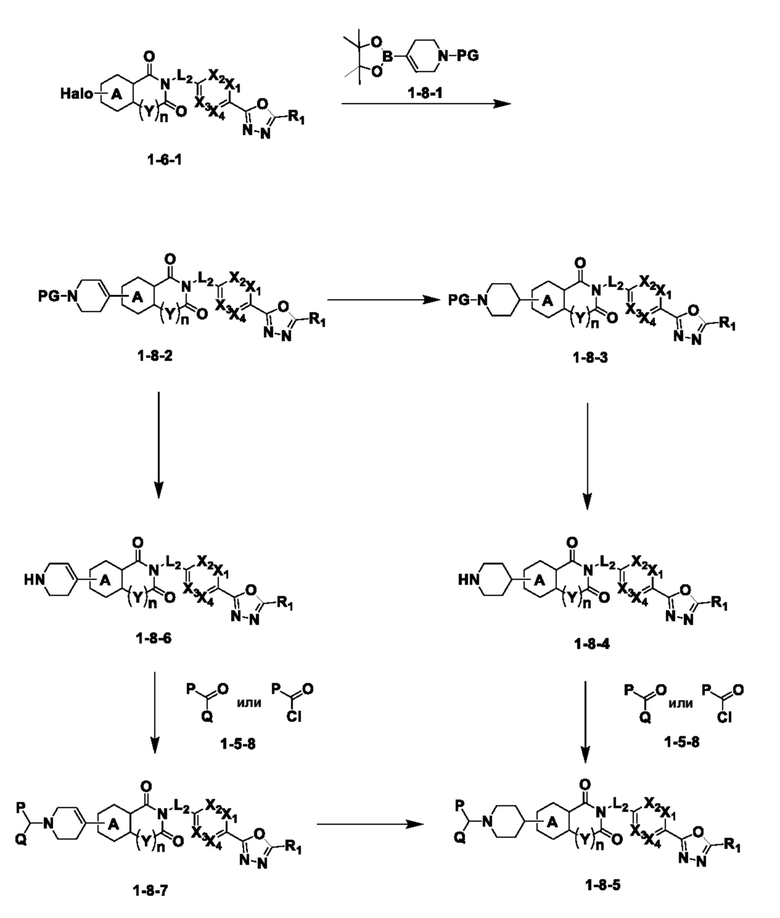
На приведенной выше схеме реакции 8 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-6-1 вводят в C-C сочетание (реакция Судзуки) с соединением химической формулы 1-8-1, содержащим защитную группу, и получают соединения 41, 53, 120, 154 и т. п. химической формулы 1-8-2. Проводят реакцию восстановления и получают соединения химической формулы 1-8-3 и затем защитную группу удаляют и получают соединение 122 и т. п. химической формулы 1-8-4. Затем соединения химической формулы 1-5-8 добавляют к соединению химической формулы 1-8-4 и вводят в реакцию восстановительного аминирования и получают соединения химической формулы 1-8-5.
Кроме того защитную группу удаляют из соединения химической формулы 1-8-2 и получают соединения химической формулы 1-8-6 и затем вводят в реакцию восстановительного аминирования и реакцию ацилирования и получают соединения 42, 43, 124, 155 и т. п. химической формулы 1-8-7. Затем проводят реакцию восстановления с соединением химической формулы 1-8-7 и получают соединения химической формулы 1-8-5.
В настоящем изобретении соединения, полученные по приведенным выше схемам реакций, включают 44, 54, 55, 59, 60, 61, 62, 63, 64, 68, 69, 127, 128, 134, 135, 143, 144, 145, 146, 151, 156 и т. п.
Схема реакции 9
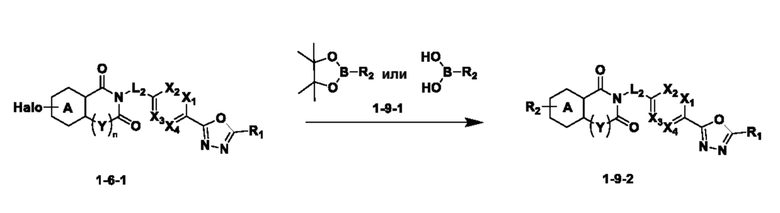
На приведенной выше схеме реакции 9, A, X1 - X4, R1, R2, Y и n являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 9, A означает фенил, X1 - X4 все независимо означают CH или N, L2 означает метилен (CH2), R1 означает CF2H, R2 означает 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, или 3- - 7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, Y означает C(C1-7 алкил)2, Halo означает галоген, и n равно 1.
На приведенной выше схеме реакции 9 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-6-1 вводят в C-C сочетание (реакция Судзуки) с соединением химической формулы 1-9-1 и получают соединения химической формулы 1-9-2.
В настоящем изобретении соединения, полученные по приведенным выше схемам реакций, включают 74, 82, 83, 84, 85, 93, 94, 95, 96, 98, 99, 100, 101, 102, 103, 104, 105, 108, 109, 110, 111, 112, 113, 114, 115 и т. п.
Схема реакции 10
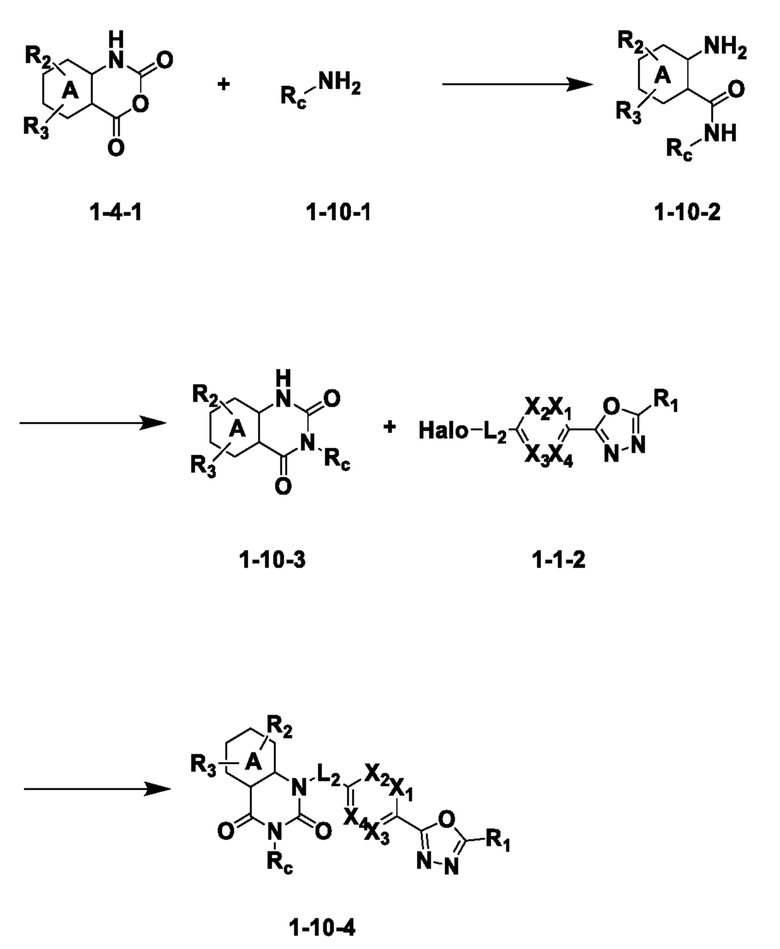
На приведенной выше схеме реакции 10, A, X1 - X4, R1 - R3 и Rc являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 10, A означает фенил, X1 - X4 все независимо означают CH или N, L2 означает метилен (CH2), R1 означает CF2H, R2 и R3 означают H, Rc означает -C1-7 алкил-O-C1-7 алкил, -C1-7 алкилфенил или -C1-7 алкил-5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, и Halo означает галоген.
На приведенной выше схеме реакции 10 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-4-1 вводят в реакцию с соединением химической формулы 1-10-1 и получают соединения химической формулы 1-10-2 и затем вводят в реакцию циклизации и получают соединения химической формулы 1-10-3. Затем проводят реакцию замещения с соединением химической формулы 1-1-2 и получают соединения 75, 77, 78 и т. п. химической формулы 1-10-4.
Схема реакции 11
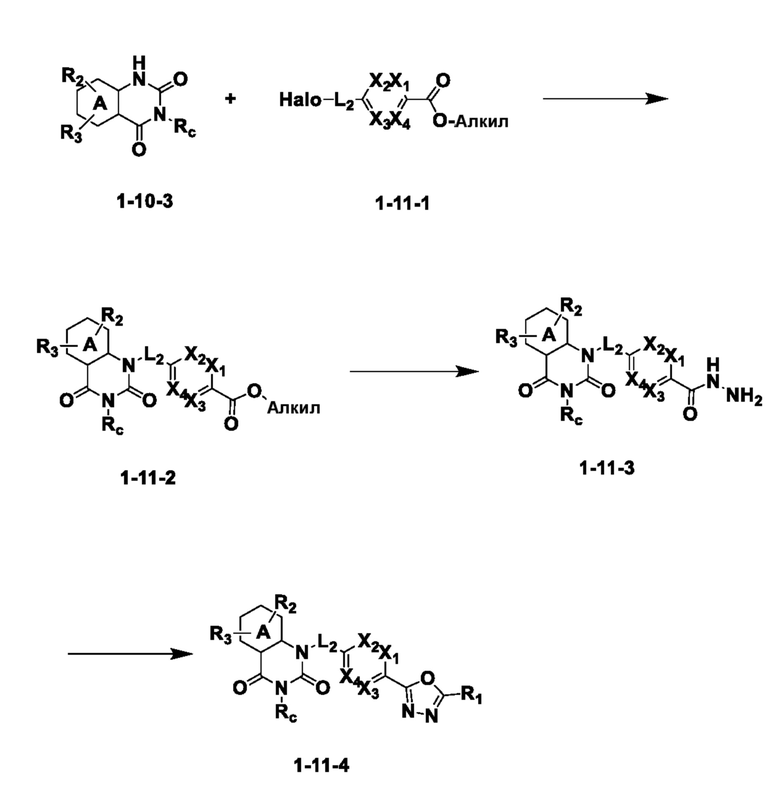
На приведенной выше схеме реакции 11, A, X1 - X4, R1 - R3 и Rc являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 11, A означает фенил, X1 - X4 все независимо означают CH или N, L2 означает метилен (CH2), R1 означает CF2H, R2 и R3 означают H, Rc означает -C1-7 алкил-O-C1-7 алкил и Halo означает галоген.
На приведенной выше схеме реакции 11 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-10-3 вводят в реакцию замещения с соединением химической формулы 1-11-1 и получают соединения химической формулы 1-11-2, затем вводят в реакцию с гидразином и получают соединения химической формулы 1-11-3 и затем вводят в реакцию с дифторуксусным ангидридом и получают соединение 76 и т. п. химической формулы 1-11-4.
Схема реакции 12
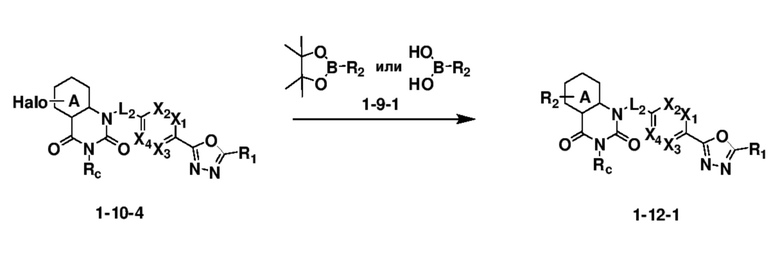
На приведенной выше схеме реакции 12, A, X1 - X4, R1, R2 и Rc являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 12, A означает фенил, X1 - X4 все независимо означают CH или N, L2 означает метилен (CH2), R1 означает CF2H, R2 означает H, фенил или 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, Rc означает -C1-7 алкил и Halo означает галоген.
На приведенной выше схеме реакции 12 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-10-4 вводят в C-C сочетание (реакция Судзуки) с соединением химической формулы 1-9-1 и получают соединения химической формулы 1-12-1.
В настоящем изобретении соединения, полученные по приведенным выше схемам реакций, включают 87, 88, 89, 90, 91, 92 и т. п.
Схема реакции 13
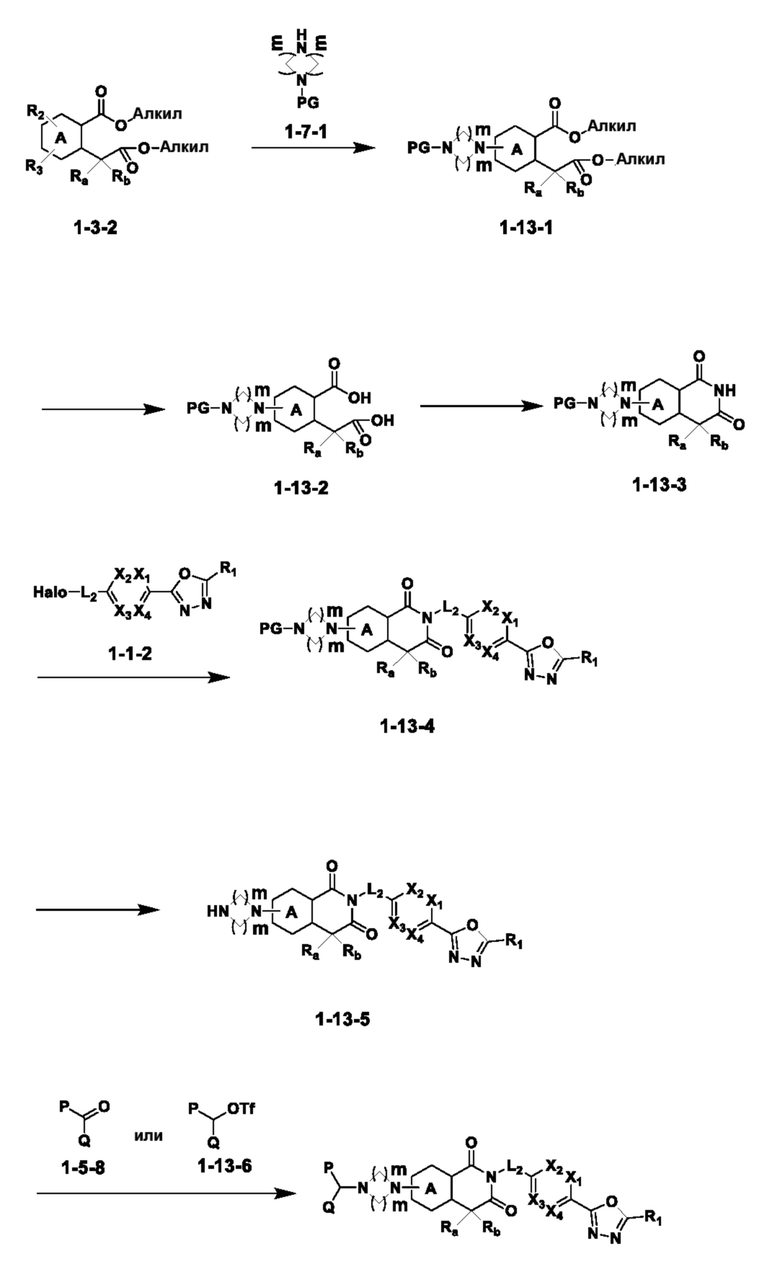
На приведенной выше схеме реакции 13, A, X1 - X4, R1, Ra и Rb являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 13, A означает фенил, X1 - X4 все независимо означают CH или N, L2 означает метилен (CH2), R1 означает CF2H, Ra и Rb означают -C1-7 алкил, Halo означает галоген, алкил представляет собой C1-7 алкил, PG означает защитную группу, m равно 2 и P и Q все независимо означают водород, C1-7 алкил или C1-7 галогеналкил.
На приведенной выше схеме реакции 13 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-3-2 вводят в C-N сочетание (реакция Бухвальда) с соединением химической формулы 1-7-1, содержащим защитную группу, и получают соединения химической формулы 1-13-1 и затем вводят в реакцию гидролиза и получают соединения химической формулы 1-13-2. Затем соединение химической формулы 1-13-2 взаимодействует с мочевиной и получают соединения химической формулы 1-13-3 и затем вводят в реакцию замещения с соединением химической формулы 1-1-2 и получают соединение 116 и т. п. химической формулы 1-13-4. Кроме того защитную группу удаляют из соединения химической формулы 1-13-4 и получают соединения химической формулы 1-13-5 и затем проводят реакцию восстановительного аминирования и реакцию замещения и получают соединения химической формулы 1-13-7.
В настоящем изобретении соединения, полученные по приведенным выше схемам реакций, включают 118, 119, 129, 130, 131, 132, 133, 137, 138, 139, 140 и т. п.
Схема реакции 14
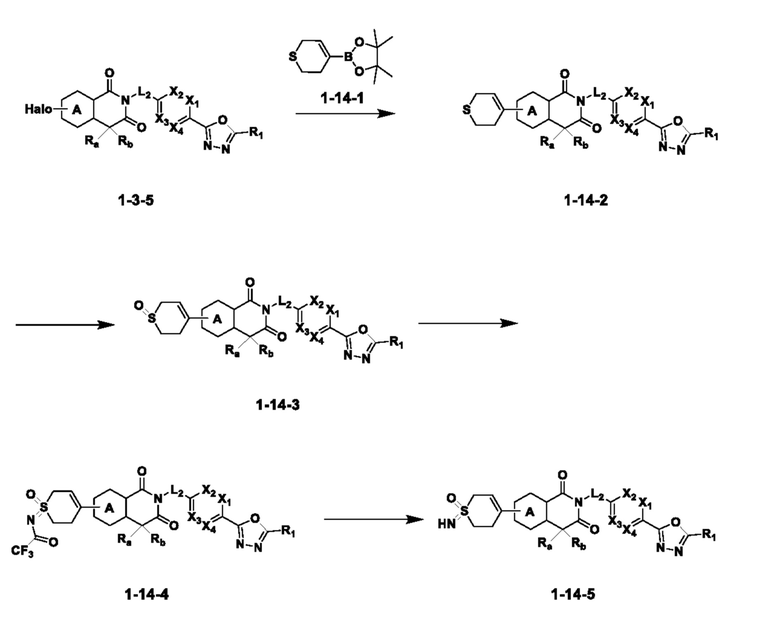
На приведенной выше схеме реакции 14, A, X1 - X4, R1, Ra и Rb являются такими же, как описано для химической формулы I. В частности, на приведенной выше схеме реакции 14, A означает фенил, X1 - X4 все независимо означают CH или N, L2 означает метилен (CH2), R1 означает CF2H, Ra и Rb означают -C1-7 алкил и Halo означает галоген.
На приведенной выше схеме реакции 14 представлена методика синтеза 1,3,4-оксадиазола, обладающего гетероциклической кольцевой структурой, и соединение химической формулы 1-3-5 вводят в C-C сочетание (реакция Судзуки) с соединением химической формулы 1-14-1 и получают соединение 121 и т. п. химической формулы 1-14-2. Затем проводят реакцию окисления с соединением химической формулы 1-14-2 и получают соединение 123 и т. п. химической формулы 1-14-3 и затем 2,2,2-трифторацетамид используют для получения соединения 125 и т. п. химической формулы 1-14-3. Затем удаляют трифторацетильный заместитель и получают соединение 126 и т. п. химической формулы 1-14-5.
Применение в медицине гомофталимидных производных 1,3,4-оксадиазола
Настоящее изобретение относится к применению в медицине гомофталимидных производных 1,3,4-оксадиазола, их стереоизомеров или их фармацевтически приемлемых солей.
Одним вариантом осуществления настоящего изобретения является фармацевтическая композиция для предупреждения или лечения связанных с активностью гистондезацетилазы 6 заболеваний, содержащая соединение, описывающееся следующей химической формулой I, его стереоизомеры или его фармацевтически приемлемые соли в качестве эффективного компонента.
Химическая формула I
Приведенная выше химическая формула I является такой же, как определено выше.
Одним вариантом осуществления настоящего изобретения является фармацевтическая композиция для предупреждения или лечения связанных с активностью гистондезацетилазы 6 заболеваний, содержащая соединение, описывающееся следующей химической формулой II, его стереоизомеры или его фармацевтически приемлемые соли в качестве эффективного компонента.
Химическая формула II
Приведенная выше химическая формула II является такой же, как определено выше.
Фармацевтическая композиция, предлагаемая в настоящем изобретении, селективно ингибирует гистондезацетилазу 6 и тем самым проявляет заметное влияние при предупреждении или лечении связанных с активностью гистондезацетилазы 6 заболеваний.
В настоящем изобретении связанные с активностью гистондезацетилазы 6 заболевания включают по меньшей мере одно, выбранное из группы, включающей инфекционные заболевания; неоплазму; эндокринопатии; связанные с питанием и метаболические заболевания; психические и поведенческие расстройства; неврологические заболевания; аднексальные заболевания глаз; заболевания кровообращения; респираторные заболевания; пищеварительные заболевания; заболевания кожи и подкожных тканей; заболевания скелетно-мышечной системы и соединительной ткани; и порок развития или уродства, и хромосомную аберрацию.
Указанные фармацевтически приемлемые соли являются такими же, как описанные для фармацевтически приемлемых солей гомофталимидных производных 1,3,4-оксадиазола, предлагаемые в настоящем изобретении.
Для введения фармацевтическая композиция, предлагаемая в настоящем изобретении, может дополнительно содержать по меньшей мере один тип фармацевтически приемлемого носителя в дополнение к гомофталимидным производным 1,3,4-оксадиазола, предлагаемым в настоящем изобретении, их стереоизомерам или их фармацевтически приемлемым солям. В качестве фармацевтически приемлемого носителя можно использовать следующие: физиологический раствор, стерилизованную воду, раствор Рингера, забуференный физиологический раствор, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол и смесь по меньшей мере одного такого компонента и можно при необходимости использовать добавление других обычных добавок, таких как антиоксиданты, буферные растворы, бактериостатические средства и т. п. Кроме того, такую фармацевтическую композицию можно приготовить в виде дозированных форм для инъекции, таких как водные растворы, суспензии, эмульсии и т. п., пилюли, капсулы, гранулы или таблетки таким образом, что к ним дополнительно добавляют разбавители, диспергирующие агенты, поверхностно-активные вещества, связующие и смазывающие вещества. Таким образом, композиция, предлагаемая в настоящем изобретении, может представлять собой пластыри, жидкости и растворы, пилюли, капсулы, гранулы, таблетки, суппозитории и т. п. Эти препараты можно приготовить по обычной методике, использующейся для приготовления препаратов в данной области техники, или по методике раскрытой в Remington's Pharmaceutical Science (latest edition), Mack Publishing Company, Easton PA, и композицию можно приготовить в виде разных препаратов в соответствии с каждым заболеванием или компонентом.
Композицию, предлагаемую в настоящем изобретении, можно вводить перорально или парентерально (например, вводить внутривенно, подкожно, внутрибрюшинно или местно) в соответствии с назначенной методикой, в которой ее доза меняется в некотором диапазоне в зависимости от массы тела, возраста, пола, состояния здоровья и диеты пациента, времени введения, методики введения, скорости выведения, тяжести заболевания и т. п. Суточная доза гомофталимидных производных 1,3,4-оксадиазола, предлагаемых в настоящем изобретении, их стереоизомеров или их фармацевтически приемлемых солей может составлять примерно от 1 до 1000 мг/кг, предпочтительно от 5 до 100 мг/кг, и ее можно вводить один раз в сутки или несколько раз в сутки путем разделения суточной дозы соединения.
В дополнение к гомофталимидным производным 1,3,4-оксадиазола, предлагаемым в настоящем изобретении, их стереоизомерам или их фармацевтически приемлемым солям указанная фармацевтическая композиция, предлагаемая в настоящем изобретении, может дополнительно содержать по меньшей мере один эффективный компонент, который оказывает такое же или аналогичное медицинское воздействие.
Настоящее изобретение относится к способу предупреждения или лечения связанных с активностью гистондезацетилазы 6 заболеваний, включающему введение терапевтически эффективного количества гомофталимидных производных 1,3,4-оксадиазола, предлагаемых в настоящем изобретении, их стереоизомеров или их фармацевтически приемлемых солей.
В настоящем изобретении термин "терапевтически эффективное количество" означает количество гомофталимидных производных 1,3,4-оксадиазола, предлагаемых в настоящем изобретении, их стереоизомеров или их фармацевтически приемлемых солей, которое эффективно для предупреждения или лечения связанных с активностью гистондезацетилазы 6 заболеваний.
В настоящем изобретении термин "предупреждение" означает задержку возникновения заболевания, нарушения или патологического состояния. Если возникновение заболевания, нарушения или патологического состояния задержано на ожидаемый период времени, предупреждение можно считать полным.
В настоящем изобретении термин "лечение" означает частичное или полное уменьшение, улучшение протекания, облегчение, подавление или задержку возникновения некоторого заболевания, нарушения и/или патологического состояния, уменьшение его тяжести или ослабление по меньшей мере его одного симптома или признака.
Способ предупреждения или лечения связанных с активностью гистондезацетилазы 6 заболеваний, предлагаемый в настоящем изобретении, включает не только оказание помощи в связи с самими заболеваниями до проявления симптомов, но также подавление или устранение симптомов путем введения гомофталимидных производных 1,3,4-оксадиазола, предлагаемых в настоящем изобретении. При оказание помощи при заболевании предупредительная или терапевтическая доза некоторого активного компонента может меняться в зависимости от характера и тяжести заболевания или патологического состояния и пути введения активного компонента. Доза и ее частота могут меняться в зависимости от возраста, массы тела и реакций отдельного пациента. Подходящую дозу и применение могут легко выбрать специалисты в данной области техники, разумеется, с учетом таких факторов. Кроме того, способ предупреждения или лечения связанных с активностью гистондезацетилазы 6 заболеваний, предлагаемый в настоящем изобретении, может дополнительно включать введение терапевтически эффективного количества дополнительного активного средства, которое полезно для лечения заболеваний, вместе с гомофталимидными производными 1,3,4-оксадиазола, предлагаемыми в настоящем изобретении, причем дополнительное активное средство может проявлять синергетический эффект или дополнительный эффект вместе с гомофталимидными производными 1,3,4-оксадиазола, предлагаемыми в настоящем изобретении.
Настоящее изобретение также относится к применению гомофталимидных производных 1,3,4-оксадиазола, предлагаемых в настоящем изобретении, их стереоизомеров или их фармацевтически приемлемых солей для предупреждения или лечения связанных с активностью гистондезацетилазы 6 заболеваний.
Настоящее изобретение также относится к применению гомофталимидных производных 1,3,4-оксадиазола, предлагаемых в настоящем изобретении, их стереоизомеров или их фармацевтически приемлемых солей для приготовления лекарственного средства для лечения связанных с активностью гистондезацетилазы 6 заболеваний. Для приготовления лекарственного средства гомофталимидные производные 1,3,4-оксадиазола, предлагаемые в настоящем изобретении, можно смешать с приемлемым вспомогательным веществом, разбавителем, носителем и т. п., и можно ввести ив комплексный препарат вместе с другими активными средствами и таким образом оказать синергетические воздействие.
Кроме того, настоящее изобретение относится к способу селективного ингибирования HDAC6 путем введения гомофталимидных производных 1,3,4-оксадиазола, предлагаемых в настоящем изобретении, их стереоизомеров или их фармацевтически приемлемых солей млекопитающим, включая людей.
В настоящем изобретении термин "млекопитающее, включая людей" означает млекопитающих, таких как обезьяна, корова, лошадь, собака, кошка, кролик, крыса, мышь и т. п., и предпочтительно включает людей.
В настоящем изобретении термин "подавление" означает ослабление или задержку данного патологического состояния, симптома, нарушения или заболевания, или значительное уменьшение биологической активности или базовой активности биологического процесса.
Положения, отмеченные для применения, композиции и терапевтического способа, предлагаемых в настоящем изобретении, применимы в равной степени, если они не противоречат друг другу.
Полезные эффекты
В контексте настоящего изобретения гомофталимидные производные 1,3,4-оксадиазола, их стереоизомеры или их фармацевтически приемлемые соли могут селективно ингибировать HDAC6 и, таким образом, проявляют удивительно превосходное воздействие при предупреждении или лечении связанных с активностью гистондезацетилазы 6 заболеваний.
Наилучший вариант осуществления настоящего изобретения
Ниже настоящее изобретение описано подробнее с помощью последующих примеров и экспериментальных примеров. Однако последующие примеры и т. п. приведены только для иллюстрации настоящего изобретения и поэтому объем настоящего изобретения не ограничивается только ими.
Синтез соединения 1, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)изоиндолин-1,3-дион
[Стадия 1] Синтез соединения 1
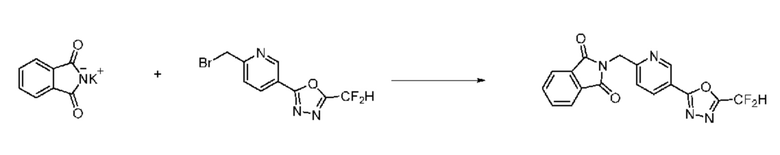
1,3-Диоксоизоиндолин-2-ид калия (0,100 г, 0,540 ммоля) и 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,157 г, 0,540 ммоля) растворяли в N, N-диметилформамиде (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 2 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем этилацетат (20 мл) и гексан (10 мл) вводили в полученный концентрат и перемешивали и отфильтровывали осадившееся твердое вещество, затем промывали гексаном и затем сушили и получали искомое соединение (0,160 г, 83,2%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,23 (d, J=2,2 Hz, 1H), 8,30 (dd, J=44,4, 12,6 Hz, 1H), 7,94 ~ 7,90 (m, 2H), 7,81 ~ 7,77 (m, 2H), 7,52 ~ 7,49 (m, 1H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 5,12 (s, 2H).; LRMS (ES) m/z 357,2 (M+ + 1).
Синтез соединения 2, 2-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)изоиндолин-1,3-дион
[Стадия 1] Синтез соединения 2
1,3-Диоксоизоиндолин-2-ид калия (0,100 г, 0,540 ммоля), 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,166 г, 0,540 ммоля) и карбонат калия (0,112 г, 0,810 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 2 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,100 г, 49,6%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 7,91 ~ 7,75 (m, 6H), 5,12 (s, 2H), 7,53 (t, J=7,7 Hz, 1H), 7,05 (s, 0,25H), 6,92 (s, 0,5H), 6,79 (s, 0,25H), 5,01 (s, 2H).
Синтез соединения 3, 2'-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1'H-спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-дион
[Стадия 1] Синтез метил-2-(1-(метоксикарбонил)циклобутил)бензоата
Метил-2-(2-метокси-2-оксоэтил)бензоат (3,000 г, 14,409 ммоля) растворяли в N, N-диметилформамиде (30 мл) при 0°C, затем гидрид натрия (60,00%, 1,441 г, 36,021 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. К реакционной смеси добавляли 1,3-дибромпропан (2,909 г, 14,409 ммоля) и дополнительно перемешивали при комнатной температуре в течение 8 ч. В полученную реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (2,220 г, 62,1%) в виде бесцветного масла.
[Стадия 2] Синтез 2-(1-карбоксициклобутил)бензойной кислоты
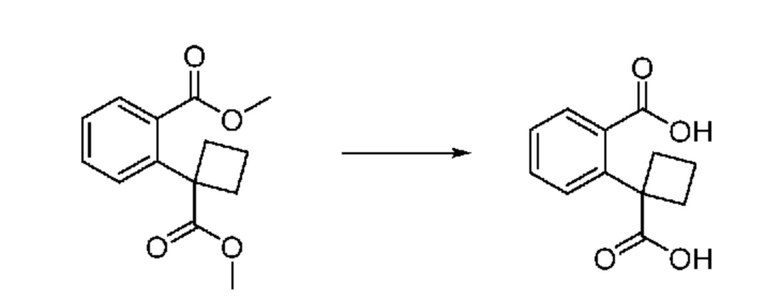
Метил-2-(1-(метоксикарбонил)циклобутил)бензоат (2,220 г, 8,942 ммоля), полученный на стадии 1, и гидроксид натрия (3,576 г, 89,415 ммоля) растворяли в смеси метанол (25 мл)/вода (25 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали 1 н. водный раствор хлористоводородной кислоты и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (1,900 г, 96,5%, белое твердое вещество).
[Стадия 3] Синтез 1'H-спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-диона
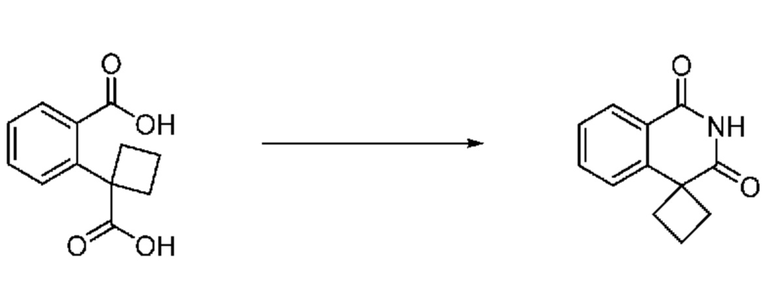
2-(1-Карбоксициклобутил)бензойную кислоту (0,820 г, 3,724 ммоля), полученную на стадии 2, перемешивали в дихлорбензоле (10 мл), затем облучали микроволновым излучением, затем нагревали при 175°C в течение 1 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (0,660 г, 88,1%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 3
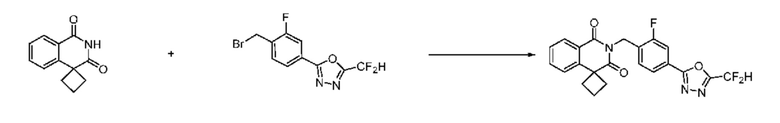
1'H-Спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-дион (0,150 г, 0,745 ммоля), полученный на стадии 3, 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,229 г, 0,745 ммоля) и карбонат калия (0,206 г, 1,491 ммоля) растворяли в N, N-диметилформамиде (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 2 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,100 г, 31,4%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 8,21 ~ 8,18 (m, 1H), 7,85 ~ 7,72 (m, 4H), 7,47 ~ 7,43 (m, 1H), 7,38 (t, J=7,9 Hz, 1H), 7,04 (s, 0,25H), 6,92 (s, 0,5H), 6,79 (s, 0,25H), 5,33 (s, 2H), 2,99 ~ 2,91 (m, 2H), 2,50 ~ 2,30 (m, 4H).; LRMS (ES) m/z 428,4 (M+ + 1).
Синтез соединения 4, 2'-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1'H-спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-дион
[Стадия 1] Синтез соединения 4
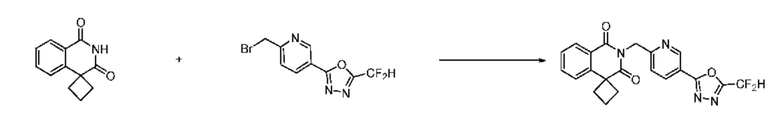
1'H-Спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-дион (0,150 г, 0,745 ммоля), 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,216 г, 0,745 ммоля) и карбонат калия (0,206 г, 1,491 ммоля) растворяли в N, N-диметилформамиде (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 2 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,070 г, 22,9%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,18 (dd, J=2,2, 0,9 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,22 ~ 8,20 (m, 1H), 7,87 ~ 7,84 (m, 1H), 7,77 ~ 7,73 (m, 1H), 7,48 ~ 7,44 (m, 2H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,44 (s, 2H), 3,04 ~ 2,97 (m, 2H), 2,55 ~ 2,27 (m, 4H).; LRMS (ES) m/z 411,3 (M+ + 1).
Синтез соединения 5, 2'-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1'H-спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-дион
[Стадия 1] Синтез соединения 5
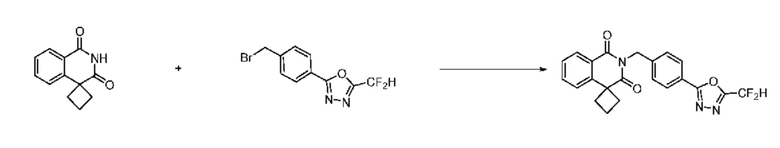
1'H-Спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-дион (0,150 г, 0,745 ммоля), 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,215 г, 0,745 ммоля) и карбонат калия (0,206 г, 1,491 ммоля) растворяли в N, N-диметилформамиде (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 2 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,100 г, 32,8%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 8,19 ~ 8,17 (m, 1H), 8,02 ~ 8,00 (m, 2H), 7,81 ~ 7,79 (m, 1H), 7,73 ~ 7,68 (m, 1H), 7,60 ~ 7,57 (m, 2H), 7,45 ~ 7,41 (m, 1H), 7,03 (s, 0,25H), 6,90 (s, 0,5H), 6,77 (s, 0,25H), 5,24 (s, 2H), 2,94 ~ 2,87 (m, 2H), 2,47 ~ 2,25 (m, 4H).; LRMS (ES) m/z 410,3 (M+ + 1).
Синтез соединения 6, 2-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез метил-2-(1-метокси-2-метил-1-оксопропан-2-ил)бензоата
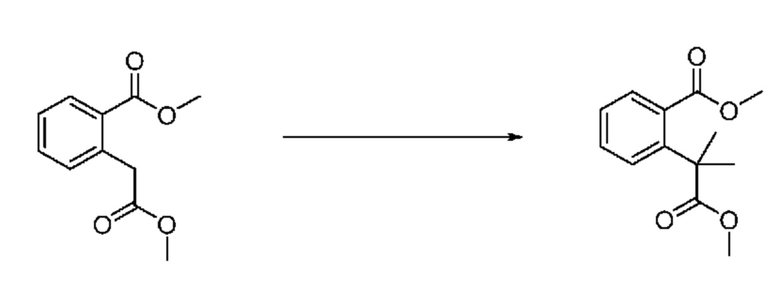
Метил-2-(2-метокси-2-оксоэтил)бензоат (3,270 г, 15,705 ммоля) растворяли в N, N-диметилформамиде (30 мл) при 0°C, затем гидрид натрия (60,00%, 1,884 г, 47,116 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. К реакционной смеси добавляли йодметан (2,933 мл, 47,116 ммоля) и дополнительно перемешивали при комнатной температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 15%) и концентрировали и получали искомое соединение (3,000 г, 80,8%) в виде бесцветного масла.
[Стадия 2] Синтез 2-(2-карбоксипропан-2-ил)бензойной кислоты
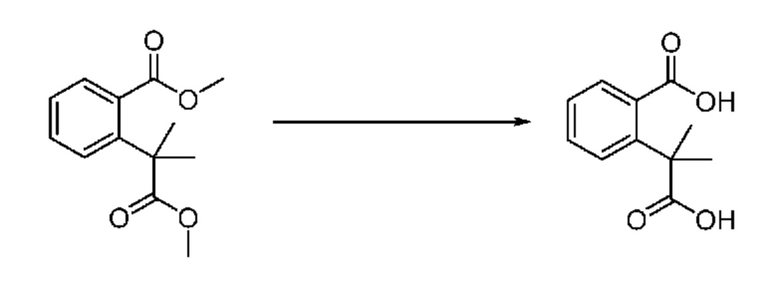
Метил-2-(1-метокси-2-метил-1-оксопропан-2-ил)бензоат (3,000 г, 12,697 ммоля), полученный на стадии 1, и гидроксид лития (3,041 г, 126,973 ммоля) растворяли в смеси метанол (15 мл)/вода (15 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В полученную реакционную смесь выливали 1 н. водный раствор хлористоводородной кислоты и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (2,500 г, 94,6%) в виде белого твердого вещества.
[Стадия 3] Синтез 4,4-диметилизохинолин-1,3(2H,4H)-диона
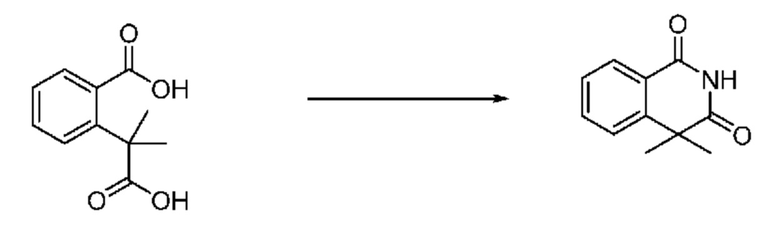
2-(2-Карбоксипропан-2-ил)бензойную кислоту (2,500 г, 12,007 ммоля), полученную на стадии 2, перемешивали в 1,2-дихлорбензоле (10 мл), затем облучали микроволновым излучением, затем нагревали при 175°C в течение 1 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (1,700 г, 74,8%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 6
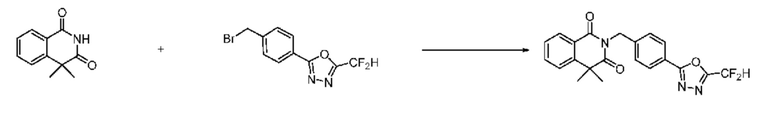
4,4-Диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,529 ммоля), полученный на стадии 3, 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,153 г, 0,529 ммоля) и карбонат калия (0,146 г, 1,057 ммоля) растворяли в N, N-диметилформамиде (10 мл), затем полученный раствор перемешивали при 80°C в течение 2 ч и затем дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,120 г, 57,1%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 8,25 ~ 8,22 (m, 1H), 8,04 ~ 8,02 (m, 2H), 7,65 ~ 7,63 (m, 1H), 7,59 ~ 7,57 (m, 2H), 7,50 ~ 7,42 (m, 2H), 7,04 (s, 0,25H), 6,91 (s, 0,5H), 6,78 (s, 0,25H), 5,24 (s, 2H), 1,63 (s, 6H).; LRMS (ES) m/z 398,3 (M+ + 1).
Синтез соединения 7, 2-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 7
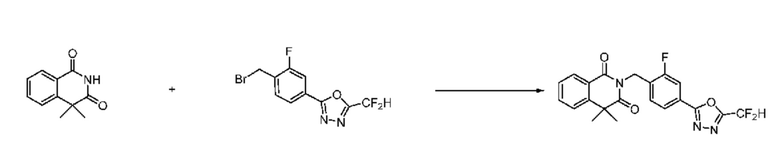
4,4-Диметилизохинолин-1,3(2H,4H)-дион (0,200 г, 1,057 ммоля), 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,325 г, 1,057 ммоля) и карбонат калия (0,292 г, 2,114 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 3 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; / = от 0 до 30%) и концентрировали и получали искомое соединение (0,100 г, 22,8%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 8,24 (dd, J=7,9, 1,4 Hz, 1H), 7,83 ~ 7,78 (m, 2H), 7,68 ~ 7,65 (m, 1H), 7,52 ~ 7,50 (m, 1H), 7,47 ~ 7,45 (m, 1H), 7,40 ~ 7,38 (m, 1H), 7,04 (s, 0,25H), 6,91 (s, 0,5H), 6,78 (s, 0,25H), 5,33 (s, 2H), 1,66 (s, 6H); LRMS (ES) m/z 416,4 (M+ + 1).
Синтез соединения 8, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 8
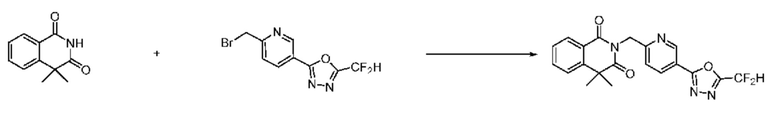
4,4-Диметилизохинолин-1,3(2H,4H)-дион (0,200 г, 1,057 ммоля), 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,307 г, 1,057 ммоля) и карбонат калия (0,292 г, 2,114 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 3 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; / = от 0 до 30%) и концентрировали и получали искомое соединение (0,180 г, 42,7%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,17 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,24 (dd, J=7,9, 1,3 Hz, 1H), 7,68 ~ 7,65 (m, 1H), 7,53 ~ 7,51 (m, 1H), 7,47 ~ 7,43 (m, 2H), 7,05 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 1,69 (s, 6H).; LRMS (ES) m/z 399,4 (M+ + 1).
Синтез соединения 9, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(2-(пиперидин-1-ил)этил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез 2-амино-N-(трет-бутил)бензамида
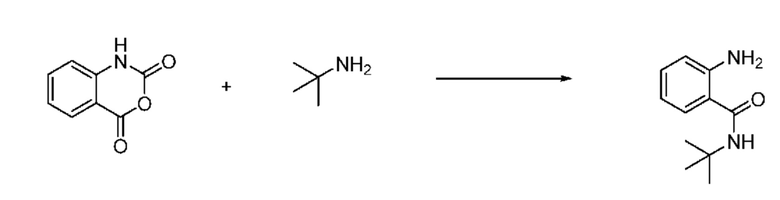
2H-Бензо[d][1,3]оксазин-2,4(1H)-дион (15,300 г, 93,790 ммоля), 2-метилпропан-2-амин (8,232 г, 112,548 ммоля) и N, N-диметилпиридин-4-амин (DMAP, 1,146 г, 9,379 ммоля) растворяли в N, N-диметилформамиде (100 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре. К реакционной смеси добавляли воду (20 мл) и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали водой и затем сушили и получали искомое соединение (9,500 г, 52,7%) в виде светло-коричневого твердого вещества.
[Стадия 2] Синтез метил-(2-(трет-бутилкарбамоил)фенил)карбамата
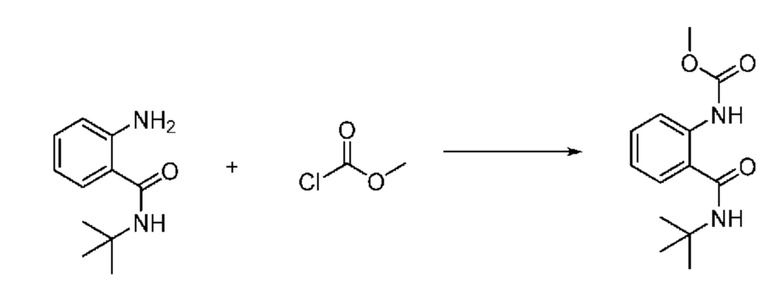
2-Амино-N-(трет-бутил)бензамид (9,500 г, 49,412 ммоля), полученный на стадии 1, метилхлорформиат (7,003 г, 74,118 ммоля) и гидроксид натрия (1,00 M раствор, 98,825 мл, 98,825 ммоля) растворяли в 1,4-диоксане (50 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 3 ч. 1M Водный раствор хлористоводородной кислоты (100 мл) добавляли к реакционной смеси и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали водой и затем сушили и получали искомое соединение (8,700 г, 70,3%) в виде белого твердого вещества.
[Стадия 3] Синтез 3-(трет-бутил)хиназолин-2,4(1H,3H)-диона
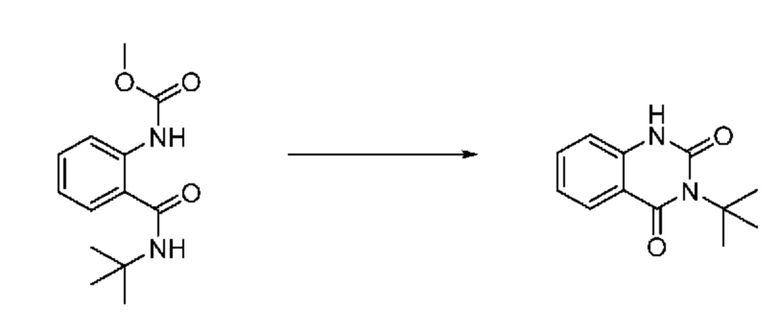
Метил-(2-(трет-бутилкарбамоил)фенил)карбамат (8,400 г, 33,560 ммоля), полученный на стадии 2, и гидроксид калия (18,829 г, 335,597 ммоля) растворяли в этаноле (100 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. 2M Водный раствор хлористоводородной кислоты (20 мл) добавляли к реакционной смеси и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали водой и затем сушили и получали искомое соединение (6,000 г, 81,9%) в виде бежевого твердого вещества.
[Стадия 4] Синтез 3-(трет-бутил)-1-(2-(пиперидин-1-ил)этил)хиназолин-2,4(1H,3H)-диона
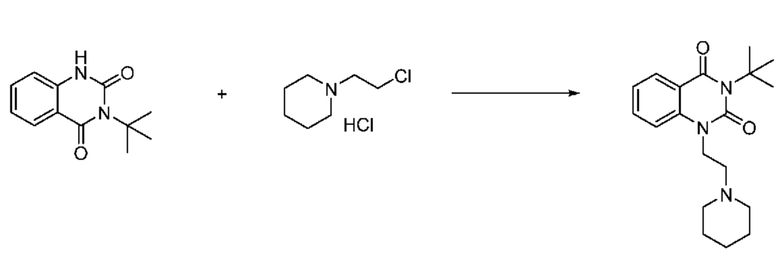
3-(Трет-бутил)хиназолин-2,4(1H,3H)-дион (3,000 г, 13,745 ммоля), полученный на стадии 3, растворяли в N, N-диметилформамиде (30 мл) при 0°C, затем гидрид натрия (60,00%, 1,374 г, 34,363 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. К реакционной смеси добавляли 1-(2-хлорэтил)пиперидингидрохлорид (3,037 г, 16,494 ммоля) и дополнительно перемешивали при комнатной температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (1,700 г, 37,5%) в виде желтого твердого вещества.
[Стадия 5] Синтез 1-(2-(пиперидин-1-ил)этил)хиназолин-2,4(1H,3H)-дионгидрохлорида
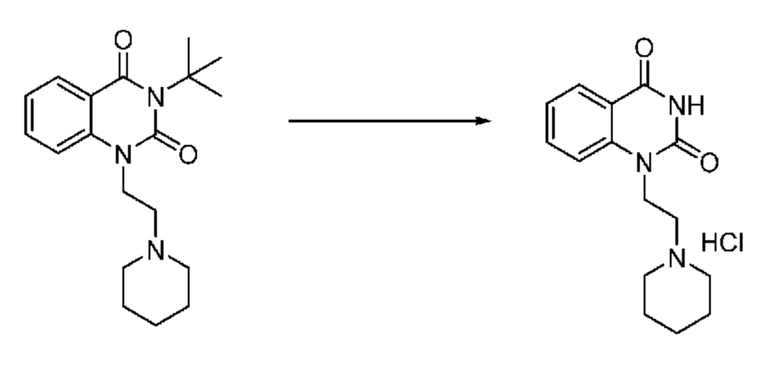
3-(Трет-бутил)-1-(2-(пиперидин-1-ил)этил)хиназолин-2,4(1H,3H)-дион (1,700 г, 5,160 ммоля), полученный на стадии 4, и хлористоводородную кислоту (4,00 M раствор в диоксане, 12,901 мл, 51,603 ммоля) смешивали при комнатной температуре, затем полученную смесь кипятили с обратным холодильником в течение 12 ч и охлаждали до комнатной температуры. Затем, растворитель удаляли из реакционной смеси при пониженном давлении, затем полученный продукт использовали без проведения дополнительной очистки (1,500 г, 93,8%, белое твердое вещество).
[Стадия 6] Синтез соединения 9
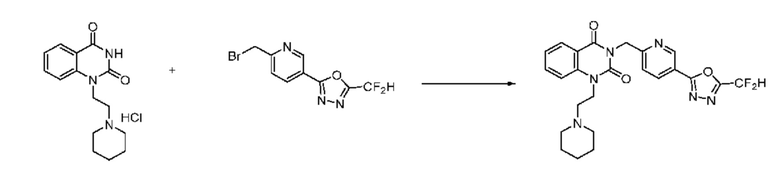
1-(2-(Пиперидин-1-ил)этил)хиназолин-2,4(1H,3H)-дионгидрохлорид (0,180 г, 0,581 ммоля), полученный на стадии 5, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,219 г, 0,755 ммоля) и карбонат калия (0,161 г, 1,162 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 30 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 80%) и концентрировали и получали искомое соединение (0,200 г, 71,3%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 7,45 ~ 7,43 (m, 1H), 8,29 (dd, J=8,2, 2,2 Hz, 1H), 8,20 (dd, J=7,9, 1,6 Hz, 1H), 7,68 ~ 7,65 (m, 1H), 7,45 ~ 7,43 (m, 1H), 7,32 ~ 7,28 (m, 1H), 7,25 ~ 7,21 (m, 1H), 7,04 (s, 0,25H), 6,91 (s, 0,5H), 6,79 (s, 0,25H), 5,47 (s, 2H), 4,28 ~ 4,24 (m, 2H), 2,62 ~ 2,58 (m, 2H), 2,50 ~ 2,45 (m, 4H), 1,53 ~ 1,49 (m, 4H), 1,39 ~ 1,38 (m, 2H).; LRMS (ES) m/z 483,6 (M+ + 1).
Синтез соединения 10, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-(2-(пиперидин-1-ил)этил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 10
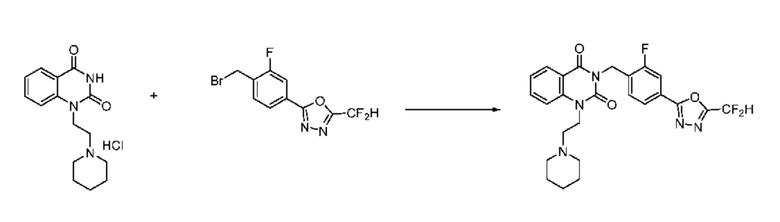
1-(2-(пиперидин-1-ил)этил)хиназолин-2,4(1H,3H)-дионгидрохлорид (0,200 г, 0,646 ммоля), 2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,258 г, 0,839 ммоля) и карбонат калия (0,178 г, 1,291 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 30 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 80%) и концентрировали и получали искомое соединение (0,200 г, 62,0%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 8,25 (dd, J=7,9, 1,5 Hz, 1H), 7,81 ~ 7,78 (m, 2H), 7,73 ~ 7,68 (m, 1H), 7,43 ~ 7,40 (m, 1H), 7,34 ~ 7,25 (m, 2H), 7,04 (s, 0,25H), 6,91 (s, 0,5H), 6,78 (s, 0,25H), 5,41 (s, 2H), 4,31 ~ 4,27 (m, 2H), 2,64 ~ 2,61 (m, 2H), 2,60 ~ 2,45 (m, 4H), 1,57 ~ 1,52 (m, 4H), 1,44 ~ 1,41 (m, 2H).; LRMS (ES) m/z 458,0 (M+ + 1).
Синтез соединения 11, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1-(2-(пиперидин-1-ил)этил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 11
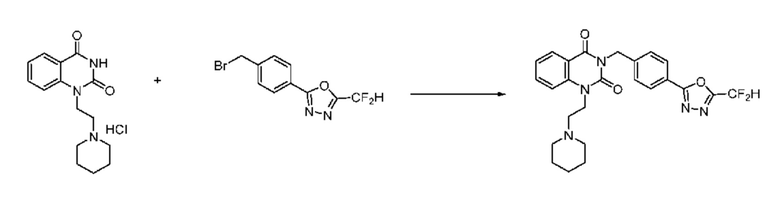
1-(2-(Пиперидин-1-ил)этил)хиназолин-2,4(1H,3H)-дионгидрохлорид (0,190 г, 0,613 ммоля), 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,230 г, 0,797 ммоля) и карбонат калия (0,170 г, 1,227 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 30 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 80%) и концентрировали и получали искомое соединение (0,150 г, 50,8%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 8,23 (dd, J=7,9, 1,5 Hz, 1H), 8,04 ~ 7,99 (m, 2H), 7,69 ~ 7,63 (m, 3H), 7,30 ~ 7,22 (m, 2H), 7,03 (s, 0,25H), 6,90 (s, 0,5H), 6,78 (s, 0,25H), 5,32 (s, 2H), 4,29 ~ 4,25 (m, 2H), 2,62 ~ 2,58 (m, 2H), 2,55 ~ 2,48 (m, 4H), 1,57 ~ 1,52 (m, 4H), 1,44 ~ 1,40 (m, 2H).
Синтез соединения 12, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 12
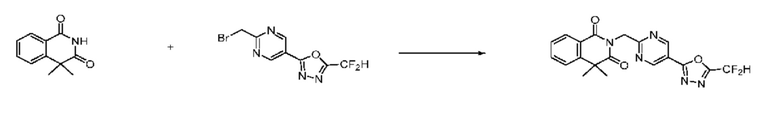
4,4-Диметилизохинолин-1,3(2H,4H)-дион (0,200 г, 1,057 ммоля), 2-(2-(бромметил)пиримидин-5-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,308 г, 1,057 ммоля) и карбонат калия (0,219 г, 1,586 ммоля) растворяли в N, N-диметилформамиде (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,150 г, 35,5%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,30 (s, 2H), 8,24 (dd, J=7,9, 1,5 Hz, 1H), 7,71 ~ 7,67 (m, 1H), 7,55 ~ 7,53 (m, 1H), 7,48 ~ 7,44 (m, 1H), 7,08 (s, 0,25H), 6,95 (s, 0,5H), 6,82 (s, 0,25H), 5,55 (s, 2H), 1,72 (s, 6H).; LRMS (ES) m/z 400,3 (M+ + 1).
Синтез соединения 13, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(4-метоксибензил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез 3-(трет-бутил)-1-(4-метоксибензил)хиназолин-2,4(1H,3H)-диона
3-(Трет-бутил)хиназолин-2,4(1H,3H)-дион (2,800 г, 12,829 ммоля) растворяли в N, N-диметилформамиде (30 мл) при 0°C, затем гидрид натрия (60,00%, 1,026 г, 25,657 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. К реакционной смеси добавляли 1-(хлорметил)-4-метоксибензол (2,210 г, 14,112 ммоля) и дополнительно перемешивали при комнатной температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 15%) и концентрировали и получали искомое соединение (3,400 г, 78,3%) в виде желтого твердого вещества.
[Стадия 2] Синтез 1-(4-метоксибензил)хиназолин-2,4(1H,3H)-диона
3-(Трет-бутил)-1-(4-метоксибензил)хиназолин-2,4(1H,3H)-дион (3,400 г, 10,047 ммоля), полученный на стадии 1, и хлористоводородную кислоту (6,00 M раствор в H2O, 10,047 мл, 60,282 ммоля) смешивали в 1,4-диоксане (15 мл) при комнатной температуре, затем полученную смесь кипятили с обратным холодильником в течение 12 ч и охлаждали до комнатной температуры. Затем, осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (2,250 г, 79,3%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 13
1-(4-Метоксибензил)хиназолин-2,4(1H,3H)-дион (2,250 г, 7,970 ммоля), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (3,006 г, 10,361 ммоля) и карбонат калия (2,203 г, 15,940 ммоля) растворяли в N, N-диметилформамиде (30 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 3 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (3,200 г, 81,7%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,24 (d, J=1,6 Hz, 1H), 8,37 (dd, J=8,2, 2,2 Hz, 1H), 8,27 (dd, J=7,9, 1,5 Hz, 1H), 7,63 ~ 7,59 (m, 1H), 7,53 (d, J=8,2 Hz, 1H), 7,26 ~ 7,22 (m, 4H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,89 ~ 6,87 (m, 2H), 6,81 (s, 0,25H), 5,60 (s, 2H), 5,36 (s, 2H), 3,79 (s, 3H).
Синтез соединения 14, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 14
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(4-метоксибензил)хиназолин-2,4(1H,3H)-дион (1,000 г, 2,035 ммоля) и нитрат церия-аммония (3,347 г, 6,104 ммоля) растворяли в смеси ацетонитрил (10 мл)/вода (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 3 ч. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (0,680 г, 90,0%) в виде желтого твердого вещества.
1H NMR (400 MHz, CDCl3) δ T11,59 (s, 1H), 9,09 (dd, J=2,2, 0,8 Hz, 1H), 8,37 (dd, J=8,3, 2,3 Hz, 1H), 7,95 (dd, J=8,2, 1,3 Hz, 1H), 7,73 ~ 7,69 (m, 1H), 7,67 (s, 0,25H), 7,61 (dd, J=8,3, 0,8 Hz, 1H), 7,54 (s, 0,5H), 7,41 (s, 0,25H), 7,26 ~ 7,22 (m, 2H), 5,32 (s, 2H).
Синтез соединения 15, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-((1-метилпиперидин-4-ил)метил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез трет-бутил-4-((3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2,4-диоксо-3,4-дигидрохиназолин-1(2H)-ил)метил)пиперидин-1-карбоксилата
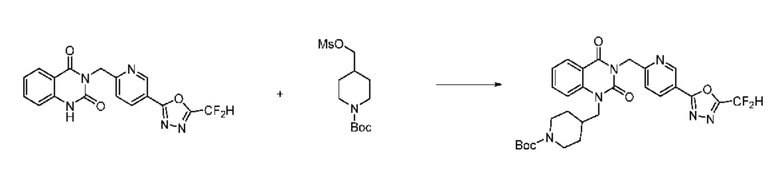
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)хиназолин-2,4(1H,3H)-дион (0,680 г, 1,831 ммоля), трет-бутил-4-(((метилсульфонил)окси)метил)пиперидин-1-карбоксилат (0,645 г, 2,198 ммоля) и карбонат калия (0,506 г, 3,663 ммоля) растворяли в N, N-диметилформамиде (15 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,500 г, 48,0%) в виде белого вспененного твердого вещества.
[Стадия 2] Синтез 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-илметил)хиназолин-2,4(1H,3H)-диона
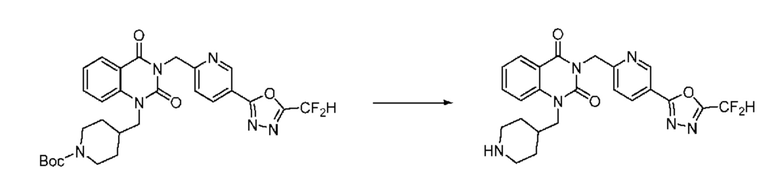
Трет-бутил-4-((3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2,4-диоксо-3,4-дигидрохиназолин-1(2H)-ил)метил)пиперидин-1-карбоксилат (0,500 г, 0,879 ммоля), полученный на стадии 1, и трифторуксусную кислоту (0,337 мл, 4,397 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 3 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем насыщенный водный раствор гидрокарбоната натрия выливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (0,200 г, 48,5%, желтое масло).
[Стадия 3] Синтез соединения 15
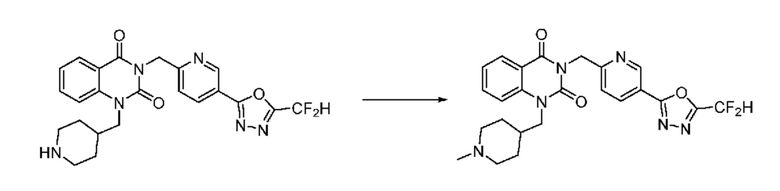
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-илметил)хиназолин-2,4(1H,3H)-дион (0,100 г, 0,213 ммоля), полученный на стадии 2, формальдегид (0,013 г, 0,427 ммоля) и триацетоксиборогидрид натрия (0,090 г, 0,427 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,037 г, 35,9%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,17 ~ 9,16 (m, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,27 (dd, J=7,9, 1,5 Hz, 1H), 7,74 ~ 7,72 (m, 1H), 7,51 ~ 7,48 (m, 1H), 7,32 ~ 7,28 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,51 (s, 2H), 4,13 ~ 4,11 (m, 2H), 3,29 ~ 3,15 (m, 3H), 2,48 (s, 3H), 2,29 ~ 2,26 (m, 2H), 1,81 ~ 1,70 (m, 4H).; LRMS (ES) m/z 483,6 (M+ + 1).
Синтез соединения 16, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-((1-(оксетан-3-ил)пиперидин-4-ил)метил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 16
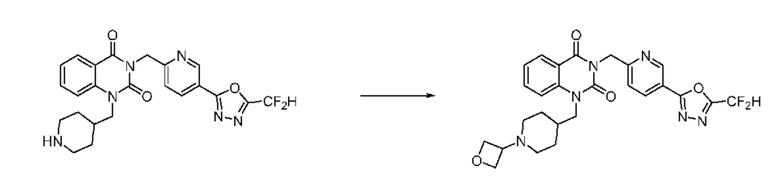
3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(пиперидин-4-илметил)хиназолин-2,4(1H,3H)-дион (0,100 г, 0,213 ммоля), оксетан-3-он (0,025 мл, 0,427 ммоля) и триацетоксиборогидрид натрия (0,090 г, 0,427 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,040 г, 35,7%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,19 (dd, J=2,2, 0,8 Hz, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 8,29 (dd, J=7,9, 1,5 Hz, 1H), 7,73 ~ 7,70 (m, 1H), 7,51 ~ 7,48 (m, 1H), 7,33 ~ 7,26 (m, 2H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,53 (s, 2H), 4,67 ~ 4,60 (m, 4H), 4,16 ~ 4,11 (m, 1H), 3,45 ~ 3,40 (m, 1H), 2,85 ~ 2,75 (m, 2H), 2,02 ~ 1,74 (m, 4H), 1,60 ~ 1,50 (m, 2H).; LRMS (ES) m/z 525,6 (M+ + 1).
Синтез соединения 17, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(2-метоксиэтил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 17
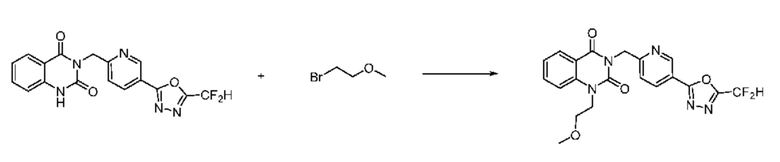
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)хиназолин-2,4(1H,3H)-дион (0,150 г, 0,404 ммоля), 1-бром-2-метоксиэтан (0,112 г, 0,808 ммоля) и карбонат калия (0,112 г, 0,808 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 3 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,080 г, 46,1%) в виде коричневого масла.
1H NMR (400 MHz, CDCl3) δ 9,21 (dd, J=2,2, 0,7 Hz, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,25 (dd, J=7,9, 1,5 Hz, 1H), 7,72 ~ 7,68 (m, 1H), 7,49 ~ 7,43 (m, 2H), 7,30 ~ 7,26 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,52 (s, 2H), 4,37 (t, J=5,8 Hz, 2H), 3,74 (t, J=5,8 Hz, 2H), 3,36 (s, 2H).; LRMS (ES) m/z 430,5(M+ + 1).
Синтез соединения 18, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 18
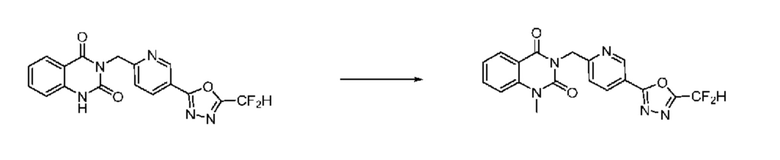
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)хиназолин-2,4(1H,3H)-дион (0,150 г, 0,404 ммоля), йодметан (0,050 мл, 0,808 ммоля) и карбонат калия (0,112 г, 0,808 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,080 г, 51,4%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,21 ~ 9,20 (m, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,26 ~ 8,24 (m, 1H), 7,76 ~ 7,71 (m, 1H), 7,50 (d, J=8,2 Hz, 1H), 7,32 ~ 7,26 (m, 2H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,28 (s, 2H), 3,64 (s, 3H).; LRMS (ES) m/z 386,5 (M+ + 1).
Синтез соединения 19, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3-(диметиламино)пропил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 19
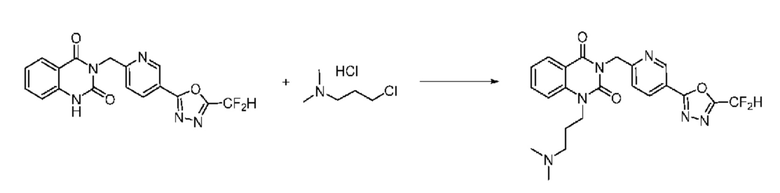
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)хиназолин-2,4(1H,3H)-дион (0,150 г, 0,404 ммоля), 3-хлор-N, N-диметилпропан-1-амингидрохлорид (0,096 г, 0,606 ммоля) и карбонат калия (0,195 г, 1,414 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,060 г, 32,5%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,22 ~ 9,21 (m, 1H), 8,35 (dd, J=8,2, 2,3 Hz, 1H), 8,28 (dd, J=7,9, 1,6 Hz, 1H), 7,75 ~ 7,71 (m, 1H), 7,50 (d, J=8,2 Hz, 1H), 7,41 ~ 7,36 (m, 2H), 7,32 ~ 7,30 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,53 (s, 2H), 4,24 (t, J=7,5 Hz, 2H), 2,48 (t, J=7,0 Hz, 2H), 2,31 (s, 6H), 2,01 ~ 1,93 (m, 2H).; LRMS (ES) m/z 457,6 (M+ + 1).
Синтез соединения 20, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(2-морфолиноэтил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 20
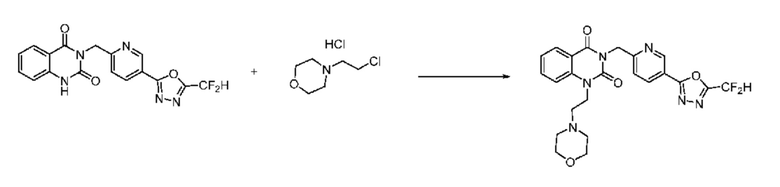
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)хиназолин-2,4(1H,3H)-дион (0,150 г, 0,404 ммоля), 4-(2-хлорэтил)морфолингидрохлорид (0,113 г, 0,606 ммоля) и карбонат калия (0,195 г, 1,414 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,070 г, 35,8%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,22 (d, J=1,8 Hz, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 8,28 (dd, J=7,8, 1,6 Hz, 1H), 7,75 ~ 7,71 (m, 1H), 7,51 ~ 7,48 (m, 1H), 7,33 ~ 7,28 (m, 2H), 7,06 (s, 1H), 6,93 (s, 1H), 6,80 (s, 1H), 5,52 (s, 2H), 4,33 (t, J=7,2 Hz, 2H), 4,33 (t, J=7,2 Hz, 2H), 3,68 (t, J=4,6 Hz, 4H), 2,71 (t, J=7,2 Hz, 2H), 2,58 (t, J=4,5 Hz, 4H).; LRMS (ES) m/z 485,5 (M+ + 1).
Синтез соединения 21, 1-(2-(1H-пиразол-1-ил)этил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 21
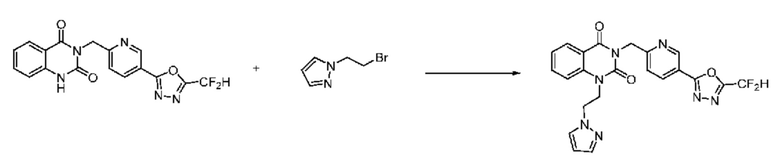
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)хиназолин-2,4(1H,3H)-дион (0,150 г, 0,404 ммоля), 1-(2-бромэтил)-1H-пиразол (0,106 г, 0,606 ммоля) и карбонат калия (0,112 г, 0,808 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,030 г, 16,0%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,23 ~ 9,22 (m, 1H), 8,38 (dd, J=8,2, 2,3 Hz, 1H), 8,23 (dd, J=7,9, 1,4 Hz, 1H), 7,61 ~ 7,52 (m, 3H), 7,33 (dd, J=2,2, 0,6 Hz, 1H), 7,26 ~ 7,22 (m, 1H), 7,07 (s, 0,25H), 7,03 (d, J=8,5 Hz, 1H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 6,14 ~ 6,12 (m, 1H), 5,49 (s, 2H), 4,59 ~ 4,52 (m, 4H).; LRMS (ES) m/z 466,5 (M+ + 1).
Синтез соединения 22, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(2-(диметиламино)этил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 22
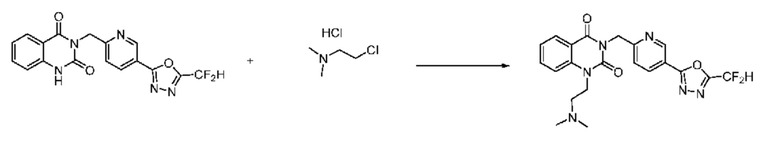
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)хиназолин-2,4(1H,3H)-дион (0,150 г, 0,404 ммоля), 2-хлор-N, N-диметилэтан-1-амингидрохлорид (0,087 г, 0,606 ммоля) и карбонат калия (0,195 г, 1,414 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,040 г, 22,4%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,21 (dd, J=2,2, 0,8 Hz, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 8,28 ~ 8,25 (m, 1H), 7,80 ~ 7,73 (m, 1H), 7,50 ~ 7,48 (m, 1H), 7,33 ~ 7,29 (m, 2H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,5H), 5,52 (s, 2H), 4,31 (t, J=7,5 Hz, 2H), 2,66 (t, J=7,5 Hz, 2H), 2,36 (s, 6H).; LRMS (ES) m/z 443,5 (M+ + 1).
Синтез соединения 23, 6-бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез метил-4-бром-2-(1-метокси-2-метил-1-оксопропан-2-ил)бензоата

Метил-4-бром-2-(2-метокси-2-оксоэтил)бензоат (9,500 г, 33,088 ммоля) растворяли в N, N-диметилформамиде (50 мл) при 0°C, затем гидрид натрия (60,00%, 3,970 г, 99,265 ммоля) добавляли к полученному раствору и перемешивали в течение 30 мин. К реакционной смеси медленно добавляли йодметан (6,180 мл, 99,265 ммоля) и дополнительно перемешивали при комнатной температуре в течение 12 ч. В реакционную смесь выливали 1 н. водный раствор хлористоводородной кислоты (20 мл) и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (7,290 г, 69,9%) в виде белого твердого вещества.
[Стадия 2] Синтез 4-бром-2-(2-карбоксипропан-2-ил)бензойной кислоты
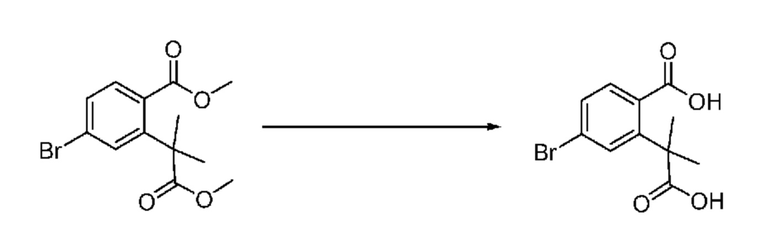
Метил-4-бром-2-(1-метокси-2-метил-1-оксопропан-2-ил)бензоат (7,290 г, 23,131 ммоля), полученный на стадии 1, и гидроксид калия (12,978 г, 231,311 ммоля) растворяли в смеси метанол (30 мл)/вода (60 мл) при 100°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали 1 н. водный раствор хлористоводородной кислоты и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (6,000 г, 90,3%, белое твердое вещество).
[Стадия 3] Синтез 6-бром-4,4-диметилизохинолин-1,3(2H,4H)-диона
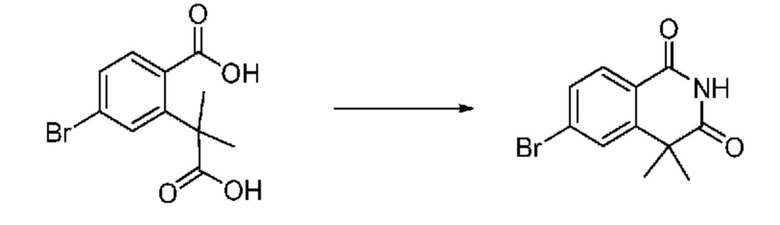
4-Бром-2-(2-карбоксипропан-2-ил)бензойную кислоту (7,460 г, 25,983 ммоля), полученную на стадии 2, и мочевину (1,717 г, 28,581 ммоля) смешивали в хлорбензоле (30 мл), затем облучали микроволновым излучением, затем нагревали при 150°C в течение 45 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (5,500 г, 79,0%) в виде желтого твердого вещества.
[Стадия 4] Синтез соединения 23
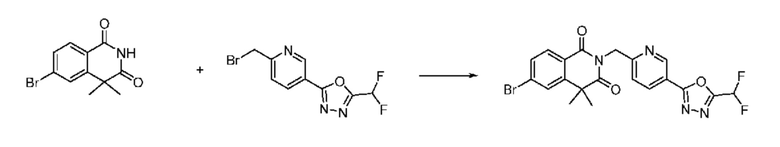
6-Бром-4,4-диметилизохинолин-1,3(2H,4H)-дион (1,400 г, 5,222 ммоля), полученный на стадии 3, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (2,272 г, 7,833 ммоля) и карбонат калия (1,443 г, 10,443 ммоля) растворяли в N, N-диметилформамиде (30 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (2,200 г, 88,3%) в виде желтого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,18 (dd, J=2,2, 0,8 Hz, 1H), 8,36 (dd, J=8,2, 2,2 Hz, 1H), 8,13 (d, J=8,4 Hz, 1H), 7,68 (d, J=1,8 Hz, 1H), 7,62 (dd, J=8,4, 1,9 Hz, 1H), 7,47 (dd, J=8,2, 0,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 1,70 (s, 6H).
Синтез соединения 24, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-морфолиноизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 24
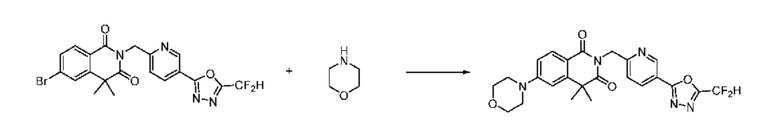
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,470 г, 0,985 ммоля), морфолин (0,170 мл, 1,970 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,057 г, 0,098 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,090 г, 0,098 ммоля) и карбонат цезия (0,963 г, 2,954 ммоля) растворяли в толуоле (5 мл) при 65°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 70%) и концентрировали и получали искомое соединение (0,220 г, 46,2%) в виде желтого твердого вещества.
1H NMR (400 MHz, DMSO-d6) δ 9,07 (dd, J=2,2, 0,8 Hz, 1H), 8,37 (dd, J=8,3, 2,3 Hz, 1H), 7,92 (d, J=8,9 Hz, 1H), 7,68 (s, 1H), 7,56 (s, 1H), 7,53 (d, J=8,3 Hz, 1H), 7,43 (s, 1H), 7,10 (d, J=2,3 Hz, 1H), 7,06 (dd, J=8,9, 2,5 Hz, 1H), 5,26 (s, 2H), 3,76 (t, J=4,8 Hz, 4H), 3,38 (t, J=4,8 Hz, 4H), 1,61 (s, 6H).
Синтез соединения 25, трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-6-ил)пиперазин-1-карбоксилат
[Стадия 1] Синтез соединения 25
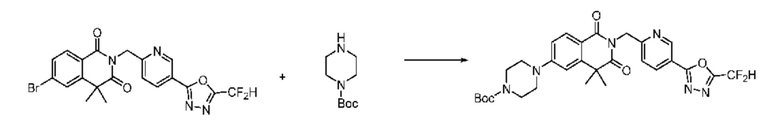
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,893 г, 1,871 ммоля), трет-бутилпиперазин-1-карбоксилат (1,046 г, 5,613 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,108 г, 0,187 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,171 г, 0,187 ммоля) и карбонат цезия (1,829 г, 5,613 ммоля) растворяли в толуоле (5 мл) при 65°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 70%) и концентрировали и получали искомое соединение (0,300 г, 27,5%) в виде желтого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,18 (dd, J=2,2, 0,8 Hz, 1H), 8,31 (dd, J=8,2, 2,2 Hz, 1H), 8,11 (d, J=8,9 Hz, 1H), 7,41 (dd, J=8,2, 0,8 Hz, 1H), 7,05 (s, 0,25H), 6,92 (s, 0,5H), 6,92 ~ 6,90 (m, 1H), 6,83 (d, J=2,4 Hz, 1H), 6,79 (s, 0,25H), 5,40 (s, 2H), 3,63 (t, J=5,2 Hz, 4H), 3,39 (t, J=5,2 Hz, 4H), 1,67 (s, 6H), 1,49 (s, 9H).; LRMS (ES) m/z 583,6 (M+ + 1).
Синтез соединения 26, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(4-изопропилпиперазин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 26
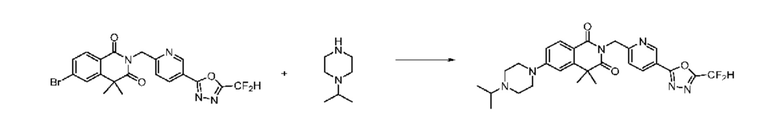
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), 1-изопропилпиперазин (0,060 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,087 г, 52,8%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,7 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,10 (d, J=8,9 Hz, 1H), 7,42 ~ 7,39 (m, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,91 (m, 1H), 6,92 (s, 0,5H), 6,84 (d, J=2,4 Hz, 0,25H), 6,80 (s, 1H), 5,40 (s, 2H), 3,43 (t, J=5,1 Hz, 4H), 2,78 ~ 2,69 (m, 5H), 1,68 (s, 6H), 1,12 ~ 1,10 (m, 6H).
Синтез соединения 27, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперидин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 27
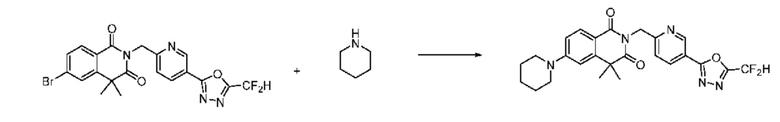
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), пиперидин (0,040 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,080 г, 52,9%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,21 (d, J=1,5 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,08 (d, J=8,9 Hz, 1H), 7,41 (dd, J=8,2, 0,7 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,93 ~ 6,90 (m, 1H), 6,83 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,41 (s, 2H), 3,42 ~ 3,40 (m, 4H), 1,71 ~ 1,68 (m, 12H).; LRMS (ES) m/z 458,0 (M+ + 1).
Синтез соединения 28, 6-(азетидин-1-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 28
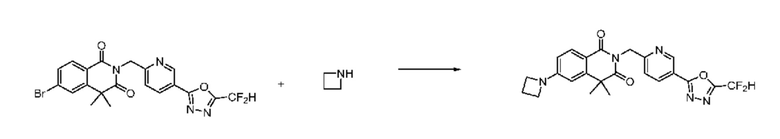
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), азетидин (0,027 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,050 г, 35,1%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,21 (dd, J=2,2, 0,8 Hz, 1H), 8,32 (dd, J=9,2, 1,3 Hz, 1H), 8,07 (d, J=8,6 Hz, 1H), 7,41 (dd, J=8,2, 0,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,42 (dd, J=8,7, 2,2 Hz, 1H), 6,29 (d, J=2,2 Hz, 1H), 5,41 (s, 2H), 4,07 (t, J=7,4 Hz, 4H), 2,50 ~ 2,46 (m, 2H), 1,70 (s, 6H).
Синтез соединения 29, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4-метилпиперазин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетата
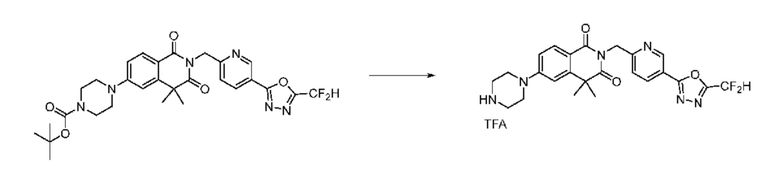
Трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-6-ил)пиперазин-1-карбоксилат (0,300 г, 0,515 ммоля) и трифторуксусную кислоту (0,394 мл, 5,149 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 5 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем полученный продукт использовали без проведения дополнительной очистки (0,300 г, 97,7%, желтое масло).
[Стадия 2] Синтез соединения 29
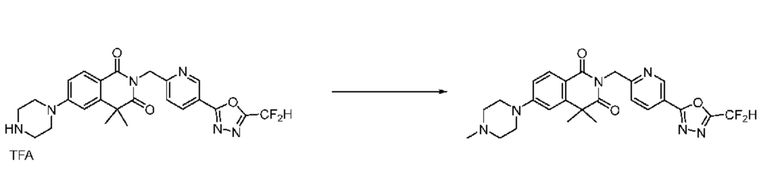
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,178 г, 0,298 ммоля), полученный на стадии 1, и N, N-диизопропилэтиламин (0,052 мл, 0,298 ммоля) растворяли в дихлорметане (10 мл), затем полученный раствор перемешивали при комнатной температуре в течение 30 мин и затем добавляли формальдегид (0,018 г, 0,597 ммоля) и триацетоксиборогидрид натрия (0,126 г, 0,597 ммоля) и дополнительно перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/гексан=от 0 до 10%) и концентрировали и получали искомое соединение (0,090 г, 60,7%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,18 (d, J=1,6 Hz, 1H), 8,31 (dd, J=8,2, 2,2 Hz, 1H), 8,09 (d, J=9,0 Hz, 1H), 7,40 (d, J=8,2 Hz, 1H), 7,05 (s, 0,25H), 6,93 ~ 6,90 (m, 1H), 6,92 (s, 0,5H), 6,83 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,39 (s, 2H), 3,43 (t, J=5,1 Hz, 4H), 2,63 (t, J=5,1 Hz, 4H), 2,38 (s, 3H), 1,66 (s, 6H).; LRMS (ES) m/z 497,5 (M+ + 1).
Синтез соединения 30, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4-(оксетан-3-ил)пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 30
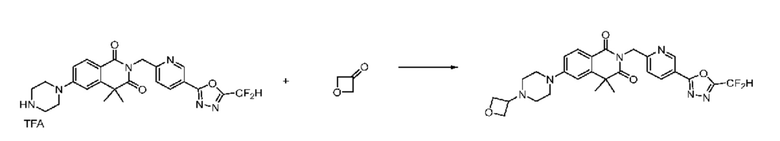
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,182 г, 0,305 ммоля) и N, N-диизопропилэтиламин (0,053 мл, 0,305 ммоля) растворяли в дихлорметане (10 мл), затем полученный раствор перемешивали при комнатной температуре в течение 30 мин и затем добавляли оксетан-3-он (0,044 г, 0,610 ммоля) и триацетоксиборогидрид натрия (0,129 г, 0,610 ммоля) и дополнительно перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/гексан=от 0 до 10%) и концентрировали и получали искомое соединение (0,100 г, 60,9%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=2,0 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,10 (d, J=8,9 Hz, 1H), 7,41 (d, J=8,2 Hz, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,91 (m, 1H), 6,92 (s, 0,5H), 6,84 (d, J=2,2 Hz, 1H), 6,80 (s, 0,25H), 5,40 (s, 2H), 4,74 ~ 4,65 (m, 4H), 3,59 ~ 3,56 (m, 1H), 3,45 (t, J=4,9 Hz, 4H), 2,53 (t, J=4,9 Hz, 4H), 1,67 (s, 6H).; LRMS (ES) m/z 539,7 (M+ + 1).
Синтез соединения 31, (S)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(3-(диметиламино)пирролидин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 31
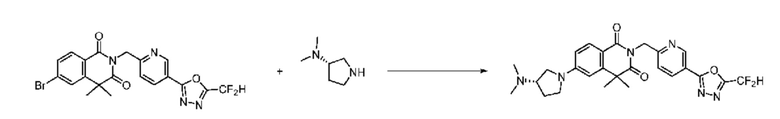
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), (S)-N, N-диметилпирролидин-3-амин (0,054 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,079 г, 49,2%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,7 Hz, 1H), 8,31 (dd, J=8,2, 2,2 Hz, 1H), 8,08 (d, J=8,7 Hz, 1H), 7,41 ~ 7,38 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,59 (dd, J=8,9, 2,3 Hz, 1H), 5,40 (s, 2H), 3,63 ~ 3,57 (m, 2H), 3,45 ~ 3,43 (m, 1H), 3,27 (t, J=8,8 Hz, 2H), 2,95 ~ 2,85 (m, 1H), 2,35 (s, 6H), 2,31 ~ 2,27 (m, 1H), 2,05 ~ 1,99 (m, 1H), 1,68 (s, 6H).; LRMS (ES) m/z 511,6 (M+ + 1).
Синтез соединения 32, (R)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(3-(диметиламино)пирролидин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 32
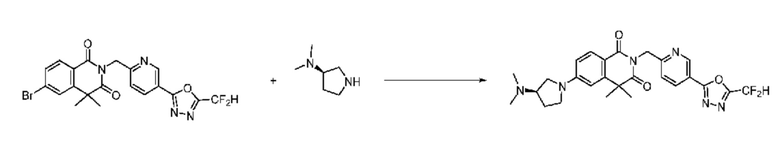
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), (R)-N, N-диметилпирролидин-3-амин (0,054 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,050 г, 31,2%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,7 Hz, 1H), 8,31 (dd, J=8,2, 2,2 Hz, 1H), 8,08 (d, J=8,7 Hz, 1H), 7,41 ~ 7,38 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,59 (dd, J=8,9, 2,3 Hz, 1H), 5,40 (s, 2H), 3,63 ~ 3,57 (m, 2H), 3,45 ~ 3,43 (m, 1H), 3,27 (t, J=8,8 Hz, 2H), 2,95 ~ 2,85 (m, 1H), 2,35 (s, 6H), 2,31 ~ 2,27 (m, 1H), 2,05 ~ 1,99 (m, 1H), 1,68 (s, 6H).; LRMS (ES) m/z 511,6 (M+ + 1).
Синтез соединения 33, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4-(2-оксо-2-(пирролидин-1-ил)этил)пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 33
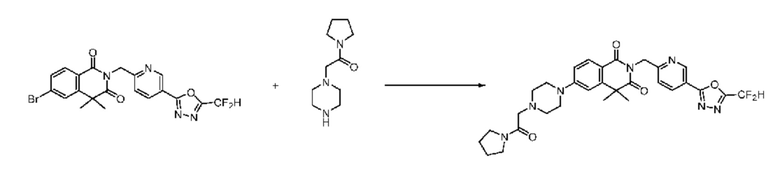
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), 2-(пиперазин-1-ил)-1-(пирролидин-1-ил)этан-1-он (0,093 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,100 г, 53,6%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,18 (dd, J=2,2, 0,8 Hz, 1H), 8,30 (dd, J=8,2, 2,3 Hz, 1H), 8,08 (d, J=8,9 Hz, 1H), 7,40 (dd, J=8,3, 0,8 Hz, 1H), 7,06 (s, 0,25H), 6,92 ~ 6,90 (m, 1H), 6,92 (s, 0,5H), 6,82 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,40 (s, 1H), 3,51 ~ 3,42 (m, 8H), 3,20 (s, 2H), 2,75 (t, J=4,4 Hz, 4H), 1,98 ~ 1,85 (m, 4H), 1,67 (s, 6H).; LRMS (ES) m/z 594,7 (M+ + 1).
Синтез соединения 34, 6-(4-ацетилпиперазин-1-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 34
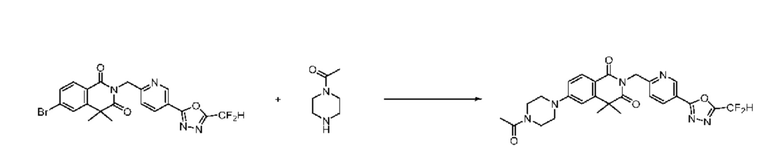
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), 1-(пиперазин-1-ил)этан-1-он (0,060 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,090 г, 54,6%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,18 (dd, J=2,2, 0,7 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,13 (d, J=8,8 Hz, 1H), 7,40 ~ 7,38 (m, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,91 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,40 (s, 2H), 3,83 ~ 3,81 (m, 2H), 3,70 ~ 3,67 (m, 2H), 3,46 ~ 3,39 (m, 4H), 2,17 (s, 3H), 1,68 (s, 6H).; LRMS (ES) m/z 525,6 (M+ + 1).
Синтез соединения 35, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(4-(2-метоксиэтил)пиперазин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 35
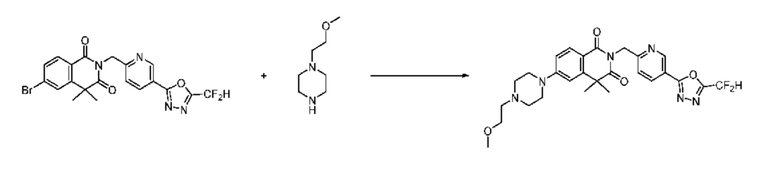
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), 1-(2-метоксиэтил)пиперазин (0,068 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,100 г, 58,9%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,19 ~ 9,19 (m, 1H), 8,31 (dd, J=8,3, 2,2 Hz, 1H), 8,09 (d, J=8,9 Hz, 1H), 7,40 (d, J=8,3 Hz, 1H), 7,06 (s, 1H), 6,93 ~ 6,90 (m, 1H), 6,93 (s, 1H), 6,80 (s, 1H), 5,40 (s, 2H), 3,58 ~ 3,56 (m, 2H), 3,47 ~ 3,42 (m, 3H), 3,39 (s, 3H), 2,70 ~ 2,65 (m, 6H), 1,67 (s, 6H).; LRMS (ES) m/z 541,7 (M+ + 1).
Синтез соединения 36, 6-(4-(трет-бутил)пиперазин-1-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 36
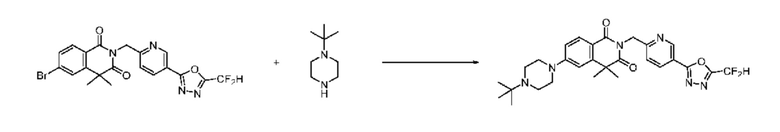
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), 1-(трет-бутил)пиперазин (0,067 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,088 г, 52,0%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,21 ~ 9,19 (m, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,10 (d, J=8,9 Hz, 1H), 7,41 (d, J=8,2 Hz, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,91 (m, 1H), 6,92 (s, 0,5H), 6,84 ~ 6,83 (m, 1H), 6,80 (s, 0,25H), 5,41 (s, 2H), 3,42 (t, J=5,0 Hz, 4H), 2,77 (t, J=5,0 Hz, 4H), 1,68 (s, 6H), 1,14 (s, 9H).; LRMS (ES) m/z 539,7 (M+ + 1).
Синтез соединения 37, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(4-(диметиламино)пиперидин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 37
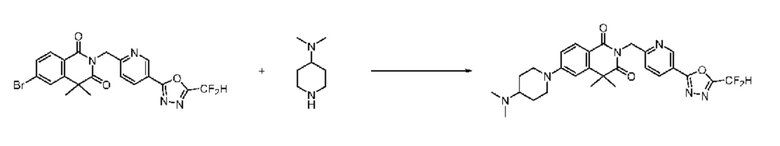
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), N, N-диметилпиперидин-4-амин (0,060 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,080 г, 48,5%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,20 ~ 9,19 (m, 1H), 8,31 (dd, J=8,2, 2,2 Hz, 1H), 8,08 (d, J=8,9 Hz, 1H), 7,41 (d, J=8,3 Hz, 1H), 7,06 (s, 0,25H), 6,93 ~ 6,91 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,40 (s, 2H), 3,99 ~ 3,95 (m, 2H), 2,98 ~ 2,92 (m, 2H), 2,45 ~ 2,36 (m, 9H), 2,02 ~ 1,99 (m, 2H), 1,67 (s, 6H).; LRMS (ES) m/z 525,6 (M+ + 1).
Синтез соединения 38, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-((2-(диметиламино)этил)(метил)амино)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 38
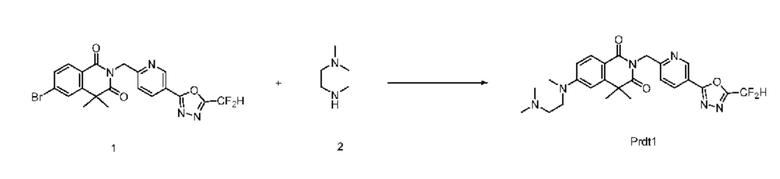
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), N1,N1,N2-триметилэтан-1,2-диамин (0,048 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,110 г, 70,2%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,19 (dd, J=2,2, 0,7 Hz, 1H), 8,31 (dd, J=8,3, 2,3 Hz, 1H), 8,07 (d, J=9,0 Hz, 1H), 7,40 ~ 7,38 (m, 1H), 7,06 (s, 1H), 6,93 (s, 1H), 6,80 (s, 1H), 6,72 (dd, J=9,0, 2,5 Hz, 1H), 6,63 (d, J=2,5 Hz, 1H), 5,40 (s, 2H), 3,58 (t, J=7,5 Hz, 2H), 3,10 (s, 3H), 2,53 (t, J=7,5 Hz, 2H), 2,33 (s, 6H), 1,67 (s, 6H).; LRMS (ES) m/z 499,6 (M+ + 1).
Синтез соединения 39, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(4-этилпиперазин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 39
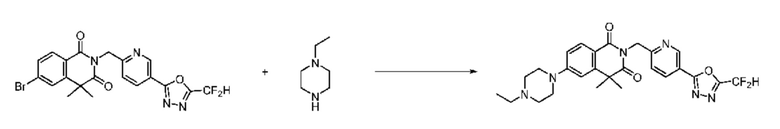
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), 1-этилпиперазин (0,054 г, 0,471 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,080 г, 49,9%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,19 (dd, J=2,2, 0,8 Hz, 1H), 8,31 (dd, J=8,2, 2,2 Hz, 1H), 8,09 (d, J=8,9 Hz, 1H), 7,41 (dd, J=8,7, 1,2 Hz, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,91 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,40 (s, 2H), 3,43 (t, J=5,1 Hz, 4H), 2,63 (t, J=5,1 Hz, 4H), 2,51 (q, J=7,2 Hz, 2H), 1,67 (s, 6H), 1,15 (t, J=7,2 Hz, 3H).; LRMS (ES) m/z 511,6 (M+ + 1).
Синтез соединения 40, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(2-окса-6-азаспиро[3.3]гептан-6-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 40
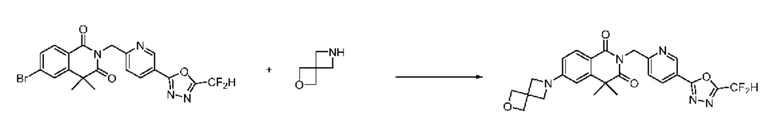
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), 2-окса-6-азаспиро[3.3]гептан (0,031 г, 0,314 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,018 г, 0,031 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,029 г, 0,031 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,049 г, 31,5%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,09 (d, J=8,6 Hz, 1H), 7,42 (dd, J=8,3, 0,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,44 (dd, J=8,7, 2,3 Hz, 1H), 6,33 (d, J=2,2 Hz, 1H), 5,40 (s, 2H), 4,90 (s, 4H), 4,21 (s, 4H), 1,67 (s, 6H).; LRMS (ES) m/z 496,6 (M+ + 1).
Синтез соединения 41, трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-6-ил)-3,6-дигидропиридин-1(2H)-карбоксилат
[Стадия 1] Синтез соединения 41
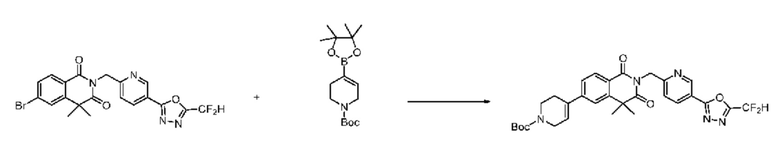
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,700 г, 1,467 ммоля), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,544 г, 1,760 ммоля), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (Pd(dppf)Cl2, 0,107 г, 0,147 ммоля) и карбонат натрия (0,311 г, 2,933 ммоля) растворяли в 1,2-диметоксиэтан (8 мл)/вода (4 мл) при 90°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 70%) и концентрировали и получали искомое соединение (0,450 г, 52,9%) в виде коричневого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 ~ 9,19 (m, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 8,22 (d, J=8,0 Hz, 1H), 7,48 ~ 7,45 (m, 3H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,23 (br s, 1H), 5,44 (s, 2H), 4,16 ~ 4,13 (m, 2H), 3,70 (t, J=5,6 Hz, 2H), 2,59 (s, 2H), 1,71 (s, 6H), 1,52 (s, 9H).; LRMS (ES) m/z 580,5 (M+ + 1).
Синтез соединения 42, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(1,2,3,6-тетрагидропиридин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетата
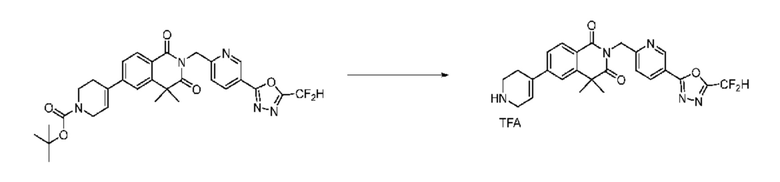
Трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-6-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,450 г, 0,776 ммоля) и трифторуксусную кислоту (0,595 мл, 7,764 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 2 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем полученный продукт использовали без проведения дополнительной очистки (0,460 г, 99,8%, коричневое масло).
[Стадия 2] Синтез соединения 42
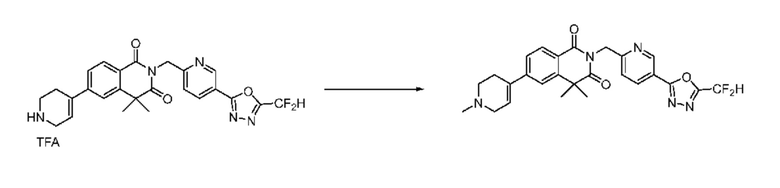
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(1,2,3,6-тетрагидропиридин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,200 г, 0,337 ммоля), полученный на стадии 1, формальдегид (0,020 г, 0,674 ммоля), N, N-диизопропилэтиламин (0,059 мл, 0,337 ммоля) и триацетоксиборогидрид натрия (0,143 г, 0,674 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,080 г, 48,1%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,17 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,19 ~ 8,17 (m, 1H), 7,48 ~ 7,42 (m, 3H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,24 ~ 6,22 (m, 1H), 5,41 (s, 2H), 3,27 ~ 3,25 (m, 2H), 2,81 ~ 2,79 (m, 2H), 2,69 ~ 2,67 (m, 2H), 2,48 (s, 3H), 1,70 (s, 6H).; LRMS (ES) m/z 494,6 (M+ + 1).
Синтез соединения 43, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(1-(оксетан-3-ил)-1,2,3,6-тетрагидропиридин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 43
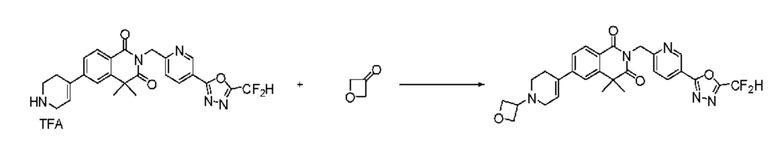
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(1,2,3,6-тетрагидропиридин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,200 г, 0,337 ммоля), оксетан-3-он (0,049 г, 0,674 ммоля), N, N-диизопропилэтиламин (0,059 мл, 0,337 ммоля) и триацетоксиборогидрид натрия (0,143 г, 0,674 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,030 г, 16,6%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,17 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,19 ~ 8,17 (m, 1H), 7,48 ~ 7,42 (m, 3H), 7,06 (s, 0,25H), 6,92 (s, 0,5H), 6,80 (s, 0,25H), 6,23 (t, J=3,5 Hz, 1H), 5,42 (s, 2H), 4,76 ~ 4,70 (m, 4H), 3,74 ~ 3,67 (m, 1H), 3,13 ~ 3,12 (m, 2H), 2,65 (s, 4H), 1,69 (s, 6H).; LRMS (ES) m/z 536,6 (M+ + 1).
Синтез соединения 44, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(1-метилпиперидин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 44
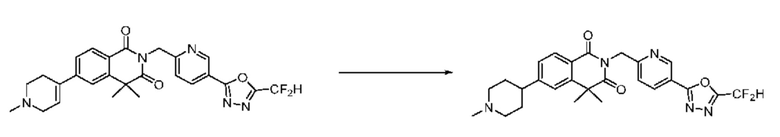
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)изохинолин-1,3(2H,4H)-дион (0,050 г, 0,101 ммоля) растворяли в метаноле (5 мл) при комнатной температуре, затем медленно добавляли 10%-Pd/C (5 мг) и перемешивали в течение 12 ч с подачей водорода из присоединенного баллона при такой же температуре. Реакционную смесь фильтровали через слой целита для удаления твердых веществ, затем растворитель удаляли из полученного фильтрата без твердых веществ при пониженном давлении. Затем полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,018 г, 35,9%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,18 (dd, J=1,8, 1,3 Hz, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,19 (d, J=8,5 Hz, 1H), 7,44 (dd, J=8,2, 0,8 Hz, 1H), 7,38 ~ 7,33 (m, 2H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,43 (s, 2H), 3,18 ~ 3,15 (m, 2H), 2,70 ~ 2,65 (m, 1H), 2,28 ~ 2,22 (m, 2H), 2,05 ~ 1,90 (m, 4H), 1,69 (s, 6H).; LRMS (ES) m/z 496,8 (M+ + 1).
Синтез соединения 45, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4-пентилпиперазин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 45
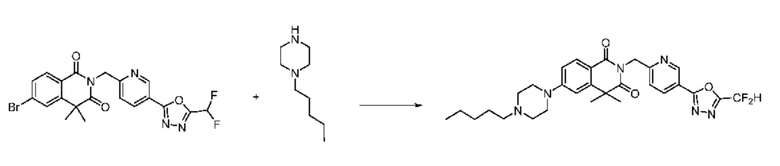
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), 1-пентилпиперазин (0,049 г, 0,314 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,012 г, 0,021 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,019 г, 0,021 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) растворяли в толуоле (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,010 г, 8,6%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,21 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,3, 2,3 Hz, 1H), 8,11 (d, J=8,9 Hz, 1H), 7,42 (dd, J=8,3, 0,8 Hz, 1H), 7,06 (s, 0,25H), 6,95 ~ 6,92 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,41 (s, 2H), 3,46 (t, J=4,8 Hz, 4H), 2,68 ~ 2,67 (m, 4H), 2,45 ~ 2,43 (m, 2H), 1,69 (s, 6H), 1,39 ~ 1,32 (m, 6H), 1,00 ~ 0,95 (m, 3H).; LRMS (ES) m/z 553,6 (M+ + 1).
Синтез соединения 46, 6-(4-циклогексилпиперазин-1-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 46
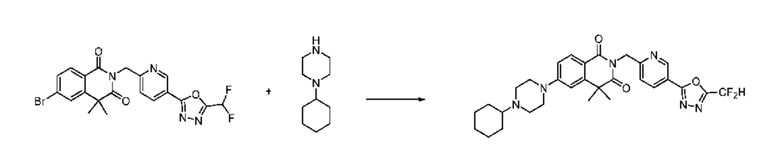
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), 1-циклогексилпиперазин (0,053 г, 0,314 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,012 г, 0,021 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,019 г, 0,021 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) растворяли в толуоле (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,020 г, 16,9%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,8 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,10 (d, J=8,9 Hz, 1H), 7,41 ~ 7,38 (m, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,91 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,41 (s, 2H), 3,41 (t, J=5,1 Hz, 4H), 2,75 (t, J=5,1 Hz, 4H), 2,60 ~ 2,55 (m, 2H), 2,45 ~ 2,35 (m, 1H), 2,05 ~ 1,82 (m, 2H), 1,68 (s, 6H), 1,29 ~ 1,24 (m, 2H).; LRMS (ES) m/z 565,7 (M+ + 1).
Синтез соединения 47, 6-(4-циклопропилпиперазин-1-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 47
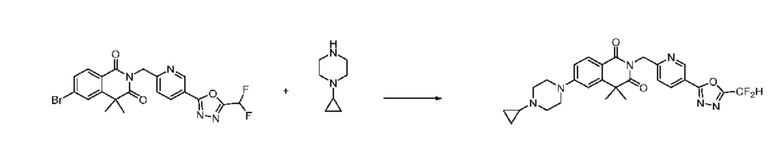
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), 1-циклопропилпиперазин (0,040 г, 0,314 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,012 г, 0,021 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,019 г, 0,021 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) растворяли в толуоле (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,020 г, 18,3%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 ~ 9,19 (m, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,10 (d, J=8,9 Hz, 1H), 7,41 (d, J=8,2 Hz, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,91 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,41 (s, 2H), 3,38 (t, J=5,1 Hz, 4H), 2,80 (t, J=5,1 Hz, 4H), 1,71 ~ 1,67 (m, 7H), 0,53 ~ 0,49 (m, 4H).; LRMS (ES) m/z 523,6 (M+ + 1).
Синтез соединения 48, 6-(4-(циклогексилметил)пиперазин-1-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 48
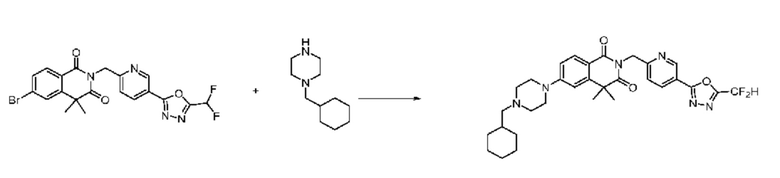
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), 1-(циклогексилметил)пиперазин (0,057 г, 0,314 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,012 г, 0,021 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,019 г, 0,021 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) растворяли в толуоле (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,030 г, 24,7%) в виде желтого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,7 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,10 (d, J=8,9 Hz, 1H), 7,42 ~ 7,40 (m, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,91 (m, 1H), 6,93 (s, 0,5H), 6,83 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,41 (s, 2H), 3,41 (t, J=5,1 Hz, 4H), 2,57 (t, J=5,1 Hz, 4H), 2,21 (d, J=7,2 Hz, 2H), 1,83 ~ 1,71 (m, 4H), 1,67 ~ 1,71 (m, 6H), 1,60 ~ 1,55 (m, 1H), 1,32 ~ 1,27 (m, 4H), 1,00 ~ 0,80 (m, 2H). ; LRMS (ES) m/z 579,6 (M+ + 1).
Синтез соединения 49, 6-(3,3-дифторазетидин-1-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 49
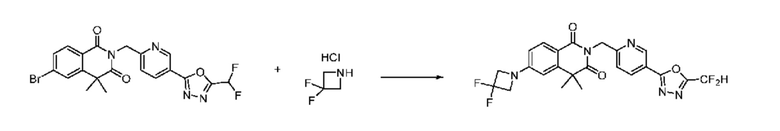
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), 3,3-дифторазетидингидрохлорид (0,041 г, 0,314 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,012 г, 0,021 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,019 г, 0,021 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) растворяли в толуоле (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,020 г, 19,5%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=5,5, 4,1 Hz, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,15 (d, J=8,6 Hz, 1H), 7,43 (dd, J=8,2, 0,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,52 (dd, J=8,6, 2,3 Hz, 1H), 6,41 (d, J=2,2 Hz, 1H), 5,41 (s, 2H), 4,39 (t, J=11,6 Hz, 4H), 1,68 (s, 6H).; LRMS (ES) m/z 490,3 (M+ + 1).
Синтез соединения 50, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4-(пиримидин-2-ил)пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 50
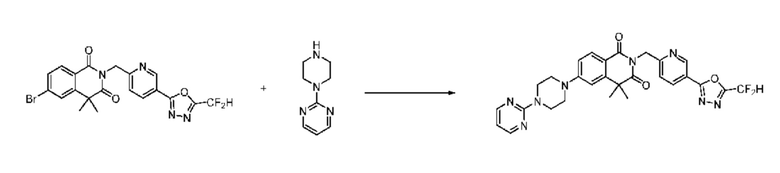
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), 2-(пиперазин-1-ил)пиримидин (0,052 г, 0,314 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,012 г, 0,021 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,019 г, 0,021 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) растворяли в толуоле (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,020 г, 17,0%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,8 Hz, 1H), 8,37 (d, J=4,8 Hz, 2H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,13 (d, J=8,9 Hz, 1H), 7,43 ~ 7,40 (m, 1H), 7,06 (s, 0,25H), 6,97 (dd, J=8,9, 2,5 Hz, 1H), 6,93 (s, 0,5H), 6,87 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 6,58 (t, J=4,8 Hz, 1H), 5,41 (s, 2H), 4,04 (t, J=5,3 Hz, 4H), 3,52 (t, J=5,3 Hz, 4H), 1,68 (s, 6H) .; LRMS (ES) m/z 561,5 (M+ + 1).
Синтез соединения 51, 7-бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез метил-5-бром-2-(1-метокси-2-метил-1-оксопропан-2-ил)бензоата
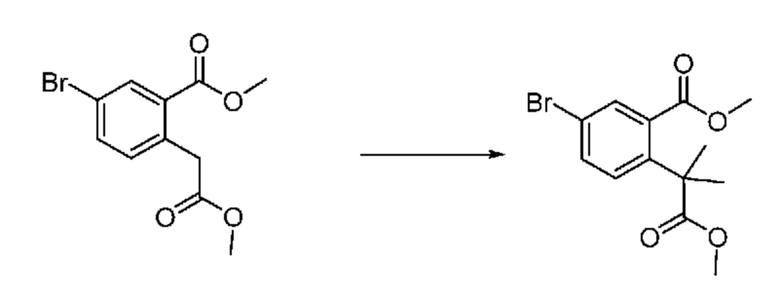
Метил-5-бром-2-(2-метокси-2-оксоэтил)бензоат (6,260 г, 21,803 ммоля) растворяли в N, N-диметилформамиде (50 мл) при 0°C, затем гидрид натрия (60,00%, 2,616 г, 65,410 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. К реакционной смеси добавляли йодметан (4,072 мл, 65,410 ммоля) и дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 10%) и концентрировали и получали искомое соединение (5,300 г, 77,1%) в виде бесцветного масла.
[Стадия 2] Синтез 5-бром-2-(2-карбоксипропан-2-ил)бензойной кислоты
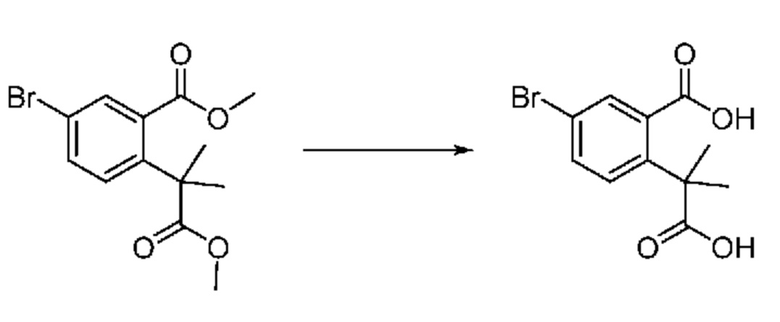
Метил-5-бром-2-(1-метокси-2-метил-1-оксопропан-2-ил)бензоат (5,300 г, 16,817 ммоля), полученный на стадии 1, и гидроксид калия (9,435 г, 168,169 ммоля) растворяли в смеси метанол (30 мл)/вода (60 мл) при 100°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали 1 н. водный раствор хлористоводородной кислоты и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (4,800 г, 99,4%, белое твердое вещество).
[Стадия 3] Синтез 7-бром-4,4-диметилизохинолин-1,3(2H,4H)-диона
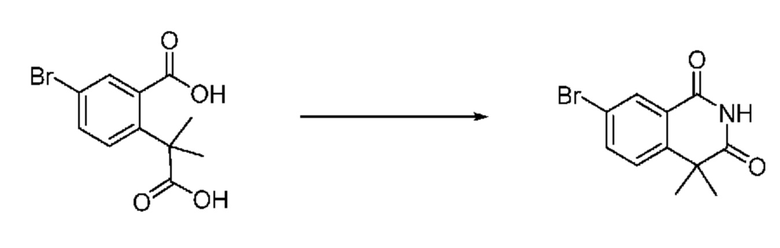
5-Бром-2-(2-карбоксипропан-2-ил)бензойную кислоту (4,800 г, 16,718 ммоля), полученную на стадии 2, и мочевину (1,105 г, 18,390 ммоля) смешивали в хлорбензоле (30 мл), затем облучали микроволновым излучением, затем нагревали при 150°C в течение 1 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (4,480 г, 99,9%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 51
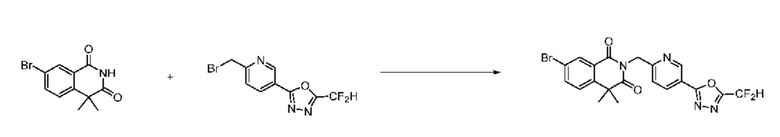
7-Бром-4,4-диметилизохинолин-1,3(2H,4H)-дион (4,480 г, 16,710 ммоля), полученный на стадии 3, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (7,270 г, 25,064 ммоля) и карбонат калия (4,619 г, 33,419 ммоля) растворяли в N, N-диметилформамиде (50 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (4,500 г, 56,4%) в виде желтого твердого вещества.
1H NMR (400 MHz, DMSO-d6) δ 9,06 ~ 9,05 (m, 1H), 8,37 (dd, J=8,3, 2,3 Hz, 1H), 8,16 (d, J=2,2 Hz, 1H), 7,95 ~ 7,93 (m, 1H), 7,75 (d, J=8,5 Hz, 1H), 7,68 (s, 0,25H), 7,63 (d, J=8,3 Hz, 1H), 7,55 (s, 0,5H), 7,42 (s, 0,25H), 5,30 (s, 2H), 1,61 (s, 6H).
Синтез соединения 52, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-морфолиноизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 52
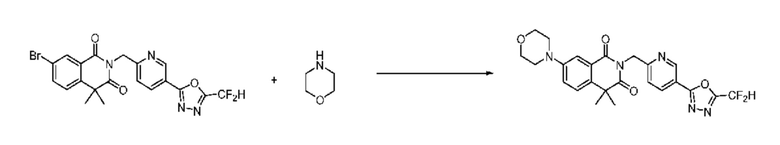
7-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), морфолин (0,027 мл, 0,314 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,012 г, 0,021 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,019 г, 0,021 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) растворяли в толуоле (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,015 г, 14,8%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,21 ~ 9,20 (m, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 7,72 (d, J=2,8 Hz, 1H), 7,43 ~ 7,40 (m, 2H), 7,26 ~ 7,25 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,41 (s, 2H), 3,89 (t, J=4,9 Hz, 4H), 3,26 ~ 3,23 (m, 4H), 1,67 (s, 6H).; LRMS (ES) m/z 484,6 (M+ + 1).
Синтез соединения 53, трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-7-ил)-3,6-дигидропиридин-1(2H)-карбоксилат
[Стадия 1] Синтез соединения 53
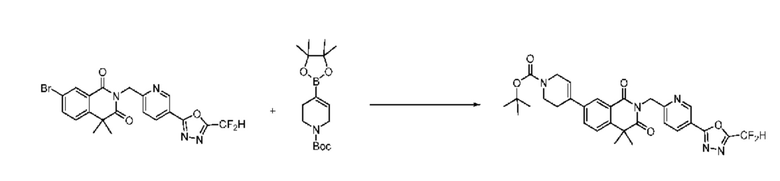
7-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (1,000 г, 2,095 ммоля), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,777 г, 2,514 ммоля), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (Pd(dppf)Cl2, 0,153 г, 0,210 ммоля) и карбонат натрия (0,444 г, 4,191 ммоля) растворяли в 1,2-диметоксиэтан (10 мл)/вода (5 мл)при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 80%) и концентрировали и получали искомое соединение (0,490 г, 40,3%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=2,0 Hz, 1H), 8,36 (dd, J=8,2, 2,2 Hz, 1H), 8,24 (s, 1H), 7,71 (dd, J=8,2, 2,0 Hz, 1H), 7,51 ~ 7,45 (m, 2H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,17 (s, 1H), 5,45 (s, 2H), 4,16 ~ 4,11 (m, 2H), 3,67 (t, J=5,6 Hz, 2H), 2,60 ~ 2,56 (m, 2H), 1,67 (s, 6H), 1,51 (s, 9H).
Синтез соединения 54, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(1-метилпиперидин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-7-ил)пиперидин-1-карбоксилата
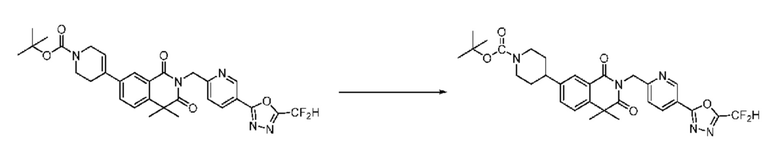
Трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-7-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,490 г, 0,845 ммоля) растворяли в метаноле (10 мл) при комнатной температуре, затем медленно добавляли 10%-Pd/C (50 мг) и перемешивали в течение 12 ч с подачей водорода из присоединенного баллона при такой же температуре. Полученный продукт использовали без проведения дополнительной очистки (0,480 г, 97,6%, бесцветное масло).
[Стадия 2] Синтез 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетата
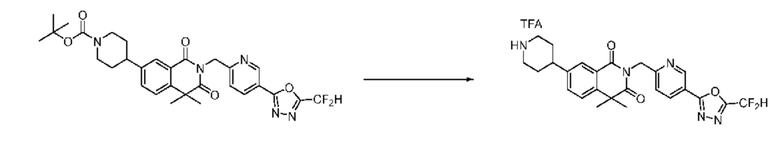
Трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-7-ил)пиперидин-1-карбоксилат (0,488 г, 0,839 ммоля), полученный на стадии 1, и трифторуксусную кислоту (0,642 мл, 8,390 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 2 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем полученный продукт использовали без проведения дополнительной очистки (0,490 г, 98,1%, желтое масло).
[Стадия 3] Синтез соединения 54
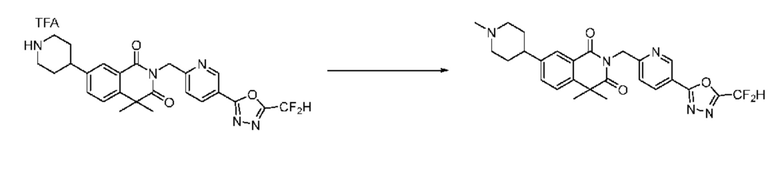
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,150 г, 0,252 ммоля), полученный на стадии 2, формальдегид (0,015 г, 0,504 ммоля), N, N-диизопропилэтиламин (0,044 мл, 0,252 ммоля) и триацетоксиборогидрид натрия (0,107 г, 0,504 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,050 г, 40,1%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,14 (dd, J=2,2, 0,8 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,06 (d, J=2,1 Hz, 1H), 7,58 (dd, J=8,2, 2,1 Hz, 1H), 7,45 (d, J=31,8 Hz, 1H), 7,44 (dd, J=8,0, 1,0 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,40 (s, 2H), 3,62 (d, J=12,0 Hz, 2H), 2,88 ~ 2,81 (m, 6H), 2,27 ~ 2,25 (m, 2H), 2,06 ~ 2,03 (m, 2H), 1,67 (s, 6H).; LRMS (ES) m/z 496,6 (M+ + 1).
Синтез соединения 55, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(1-(оксетан-3-ил)пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 55
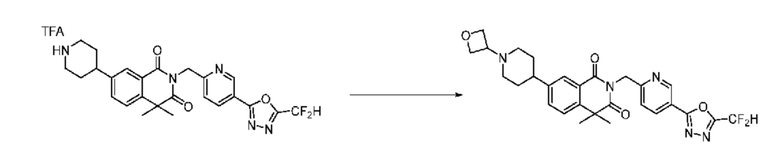
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,150 г, 0,252 ммоля), оксетан-3-он (0,036 г, 0,504 ммоля), N, N-диизопропилэтиламин (0,044 мл, 0,252 ммоля) и триацетоксиборогидрид натрия (0,107 г, 0,504 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,030 г, 22,2%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,18 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,2, 2,3 Hz, 1H), 8,11 (d, J=2,0 Hz, 1H), 7,56 (dd, J=8,3, 2,0 Hz, 1H), 7,47 ~ 7,43 (m, 2H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 4,69 ~ 4,67 (m, 4H), 3,55 ~ 3,52 (m, 1H), 2,93 ~ 2,90 (m, 2H), 2,70 ~ 2,60 (m, 1H), 1,99 ~ 1,98 (m, 2H), 1,90 ~ 1,87 (m, 4H), 1,68 (s, 6H).; LRMS (ES) m/z 538,6 (M+ + 1).
Синтез соединения 56, 1-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-6-ил)-N-метилпиперидин-4-карбоксамид
[Стадия 1] Синтез соединения 56
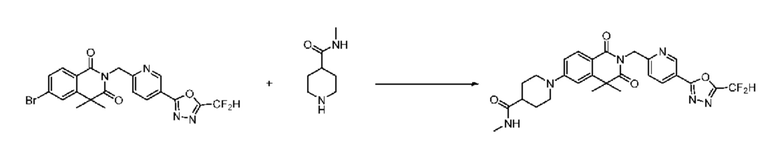
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), N-метилпиперидин-4-карбоксамид (0,030 г, 0,210 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,019 г, 0,021 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,012 г, 0,021 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,030 г, 26,6%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,21 ~ 9,20 (m, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,11 ~ 8,10 (m, 1H), 7,42 ~ 7,40 (m, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,92 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,60 ~ 5,55 (m, 1H), 5,41 (s, 2H), 4,00 ~ 3,97 (m, 2H), 3,02 ~ 2,96 (m, 2H), 2,87 ~ 2,85 (m, 3H), 2,42 ~ 2,38 (m, 1H), 2,19 ~ 1,88 (m, 4H), 1,68 (s, 6H).
Синтез соединения 57, 1-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-6-ил)-N, N-диметилпиперидин-4-карбоксамид
[Стадия 1] Синтез соединения 57
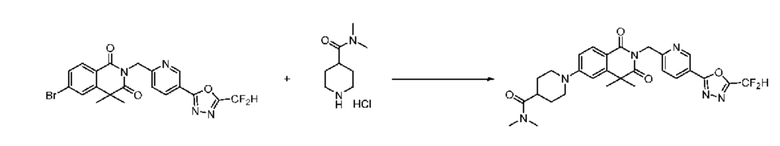
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), N, N-диметилпиперидин-4-карбоксамидгидрохлорид (0,040 г, 0,210 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,019 г, 0,021 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,012 г, 0,021 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,020 г, 17,3%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,6 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,08 (d, J=8,9 Hz, 1H), 7,40 (dd, J=8,3, 0,5 Hz, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,91 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,41 (s, 2H), 4,00 ~ 3,96 (m, 2H), 3,12 (s, 3H), 3,05 ~ 2,98 (m, 5H), 2,80 ~ 2,75 (m, 1H), 1,97 ~ 1,83 (m, 4H), 1,67 (s, 6H).; LRMS (ES) m/z 553,6 (M+ + 1).
Синтез соединения 58, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-((1S,4S)-5-(метилсульфонил)-2,5-диазабицикло[2.2.1]гептан-2-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 58
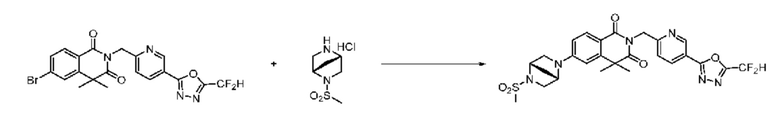
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), (1S,4S)-2-(метилсульфонил)-2,5-диазабицикло[2.2.1]гептангидрохлорид (0,045 г, 0,210 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,019 г, 0,021 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,012 г, 0,021 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) растворяли в толуоле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,050 г, 41,7%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,21 ~ 9,20 (m, 1H), 8,33 (dd, J=8,2, 2,3 Hz, 1H), 8,11 (d, J=8,8 Hz, 1H), 7,42 (d, J=8,3 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,63 (dd, J=8,8, 2,4 Hz, 1H), 6,49 (d, J=2,3 Hz, 1H), 5,41 (s, 2H), 4,67 (s, 2H), 3,69 ~ 3,66 (m, 1H), 3,58 ~ 3,50 (m, 3H), 2,92 (s, 3H), 2,50 ~ 2,04 (m, 2H), 1,67 (s, 6H).; LRMS (ES) m/z 573,6 (M+ + 1).
Синтез соединения 59, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-этилпиперидин-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетата
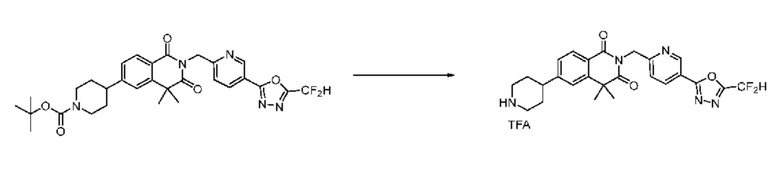
Трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-6-ил)пиперидин-1-карбоксилат (1,340 г, 2,304 ммоля) и трифторуксусную кислоту (1,764 мл, 23,039 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 3 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем полученный продукт использовали без проведения дополнительной очистки (1,300 г, 94,7%, коричневое масло).
[Стадия 2] Синтез соединения 59
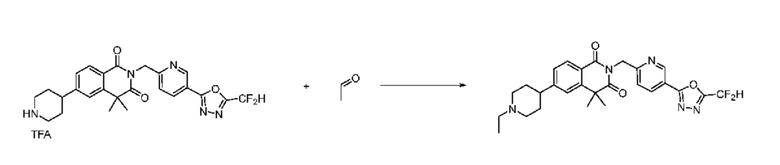
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,200 г, 0,336 ммоля), полученный на стадии 1, и N, N-диизопропилэтиламин (0,058 мл, 0,336 ммоля) растворяли в дихлорметане (10 мл), затем полученный раствор перемешивали при комнатной температуре в течение 30 ч и затем добавляли ацетальдегид (0,030 г, 0,672 ммоля) и триацетоксиборогидрид натрия (0,142 г, 0,672 ммоля) и дополнительно перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,080 г, 46,7%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,18 ~ 9,17 (m, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,19 (d, J=8,1 Hz, 1H), 7,45 ~ 7,41 (m, 2H), 7,36 ~ 7,33 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 3,53 ~ 3,49 (m, 2H), 2,92 ~ 2,86 (m, 2H), 2,77 ~ 2,76 (m, 1H), 2,53 ~ 2,47 (m, 2H), 2,24 ~ 2,20 (m, 2H), 2,02 ~ 1,98 (m, 2H), 1,67 (s, 6H), 1,33 ~ 1,30 (m, 3H).; LRMS (ES) m/z 510,6 (M+ + 1).
Синтез соединения 60, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(1-изопропилпиперидин-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 60
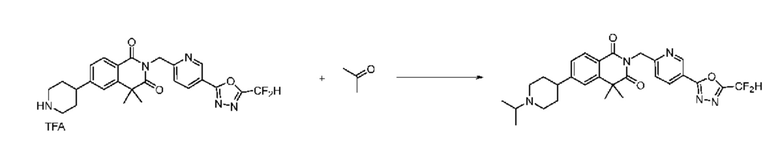
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,200 г, 0,336 ммоля) и N, N-диизопропилэтиламин (0,058 мл, 0,336 ммоля) растворяли в дихлорметане (10 мл), затем полученный раствор перемешивали при комнатной температуре в течение 30 ч и затем добавляли ацетон (0,039 г, 0,672 ммоля) и триацетоксиборогидрид натрия (0,142 г, 0,672 ммоля) и дополнительно перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,050 г, 28,4%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,19 ~ 9,18 (m, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,21 (d, J=8,1 Hz, 1H), 7,45 ~ 7,43 (m, 2H), 7,35 (dd, J=8,1, 1,5 Hz, 1H), 7,06 (s, 1H), 6,93 (s, 1H), 6,80 (s, 1H), 5,42 (s, 2H), 3,69 ~ 3,50 (m, 3H), 2,87 ~ 2,82 (m, 3H), 2,53 ~ 2,49 (m, 2H), 2,09 ~ 2,06 (m, 2H), 1,67 (s, 6H), 1,42 ~ 1,38 (m, 6H).; LRMS (ES) m/z 524,6 (M+ + 1).
Синтез соединения 61, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(1-(оксетан-3-ил)пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 61
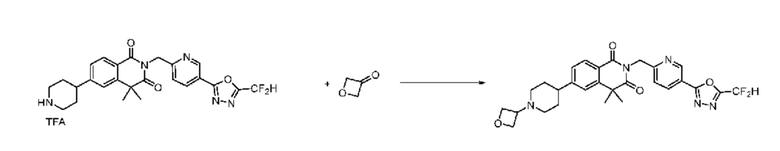
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,200 г, 0,336 ммоля) и N, N-диизопропилэтиламин (0,058 мл, 0,336 ммоля) растворяли в дихлорметане (10 мл), затем полученный раствор перемешивали при комнатной температуре в течение 30 ч и затем добавляли оксетан-3-он (0,048 г, 0,672 ммоля) и триацетоксиборогидрид натрия (0,142 г, 0,672 ммоля) и дополнительно перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,100 г, 55,4%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,19 ~ 9,18 (m, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,20 (d, J=8,1 Hz, 1H), 7,45 (d, J=8,2 Hz, 1H), 7,38 ~ 7,33 (m, 2H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,43 (s, 2H), 4,73 ~ 4,67 (m, 4H), 3,58 ~ 3,54 (m, 1H), 2,96 ~ 2,93 (m, 2H), 1,70 ~ 1,60 (m, 1H), 2,09 ~ 2,00 (m, 2H), 1,93 ~ 1,88 (m, 4H), 1,67 (s, 6H).; LRMS (ES) m/z 538,6 (M+ + 1).
Синтез соединения 62, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(1-((тетрагидро-2H-пиран-4-ил)метил)пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 62
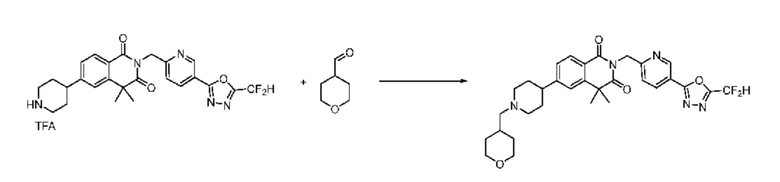
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,200 г, 0,336 ммоля) и N, N-диизопропилэтиламин (0,058 мл, 0,336 ммоля) растворяли в дихлорметане (10 мл), затем полученный раствор перемешивали при комнатной температуре в течение 30 ч и затем добавляли тетрагидро-2H-пиран-4-карбальдегид (0,077 г, 0,672 ммоля) и триацетоксиборогидрид натрия (0,142 г, 0,672 ммоля) и дополнительно перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,060 г, 30,8%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,19 ~ 9,18 (m, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,19 (d, J=8,1 Hz, 1H), 7,45 (d, J=8,2 Hz, 1H), 7,40 (d, J=1,3 Hz, 1H), 7,36 (dd, J=8,2, 1,6 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,43 (s, 2H), 4,16 ~ 4,11 (m, 2H), 3,46 ~ 4,41 (m, 2H), 3,05 ~ 2,85 (m, 1H), 2,69 ~ 2,68 (m, 1H), 2,48 ~ 2,47 (m, 2H), 2,34 ~ 2,28 (m, 2H), 2,08 ~ 2,05 (m, 2H), 1,93 ~ 1,90 (m, 2H), 1,73 ~ 1,70 (m, 2H), 1,67 (s, 6H), 1,42 ~ 1,39 (m, 2H).; LRMS (ES) m/z 580,6 (M+ + 1).
Синтез соединения 63, 6-(1-(2-оксаспиро[3.3]гептан-6-ил)пиперидин-4-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 63
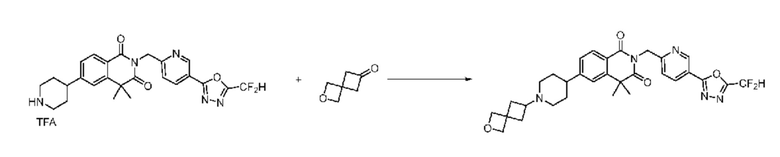
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,200 г, 0,336 ммоля) и N, N-диизопропилэтиламин (0,058 мл, 0,336 ммоля) растворяли в дихлорметане (10 мл), затем полученный раствор перемешивали при комнатной температуре в течение 30 ч и затем добавляли 2-оксаспиро[3.3]гептан-6-он (0,075 г, 0,672 ммоля) и триацетоксиборогидрид натрия (0,142 г, 0,672 ммоля) и дополнительно перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,020 г, 10,3%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,18 (dd, J=2,2, 0,8 Hz, 1H), 8,34 (dd, J=8,2, 2,3 Hz, 1H), 8,18 (d, J=8,1 Hz, 1H), 7,44 (dd, J=8,2, 0,8 Hz, 1H), 7,37 (d, J=1,4 Hz, 1H), 7,32 (dd, J=8,2, 1,4 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 4,74 ~ 4,63 (m, 8H), 4,16 ~ 4,12 (m, 1H), 3,15 ~ 3,13 (m, 2H), 2,68 ~ 2,61 (m, 3H), 2,47 ~ 2,45 (m, 2H), 2,30 ~ 2,28 (m, 2H), 1,68 (s, 6H) .; LRMS (ES) m/z 578,6 (M+ + 1).
Синтез соединения 64, 6-(1-циклобутилпиперидин-4-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 64
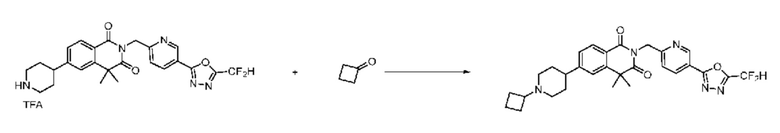
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,200 г, 0,336 ммоля) и N, N-диизопропилэтиламин (0,058 мл, 0,336 ммоля) растворяли в дихлорметане (10 мл), затем полученный раствор перемешивали при комнатной температуре в течение 30 ч и затем добавляли циклобутанон (0,047 г, 0,672 ммоля) и триацетоксиборогидрид натрия (0,142 г, 0,672 ммоля) и дополнительно перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,100 г, 55,6%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,19 ~ 9,18 (m, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,19 (d, J=8,1 Hz, 1H), 7,44 (d, J=8,3 Hz, 1H), 7,40 (d, J=1,4 Hz, 1H), 7,34 (dd, J=8,2, 1,6 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,43 (s, 2H), 3,43 ~ 3,38 (m, 2H), 2,99 ~ 2,93 (m, 1H), 2,72 ~ 2,68 (m, 1H), 2,24 ~ 2,01 (m, 8H), 1,98 ~ 1,71 (m, 4H), 1,68 (s, 6H).; LRMS (ES) m/z 536,6 (M+ + 1).
Синтез соединения 65, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола
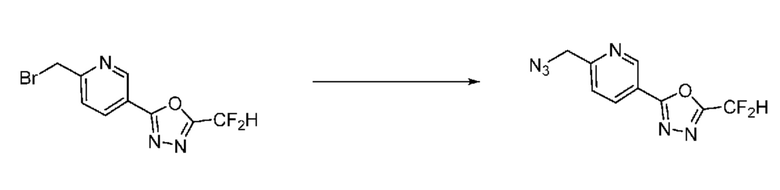
2-(6-(Бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (3,000 г, 10,342 ммоля) и азид натрия (1,009 г, 15,513 ммоля) растворяли в N, N-диметилформамиде (5 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (2,310 г, 88,6%, белое твердое вещество).
[Стадия 2] Синтез (5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метанамина

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,500 г, 5,948 ммоля), полученный на стадии 1, растворяли в метаноле (20 мл) при комнатной температуре, затем медленно добавляли 10%-Pd/C (100 мг) и перемешивали в течение 12 ч с подачей водорода из присоединенного баллона при такой же температуре. Реакционную смесь фильтровали через слой целита для удаления твердых веществ, затем растворитель удаляли из полученного фильтрата при пониженном давлении и затем полученный продукт использовали без проведения дополнительной очистки (1,300 г, 96,6%, коричневое твердое вещество).
[Стадия 3] Синтез соединения 65
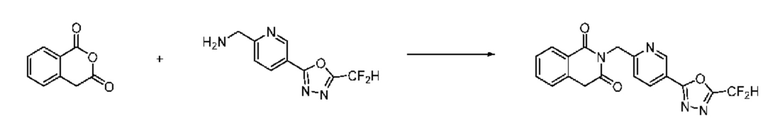
(5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метанамин (1,235 г, 5,458 ммоля), полученный на стадии 2, и изохромен-1,3-дион (0,590 г, 3,639 ммоля) растворяли в толуоле (10 мл) при 100°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,150 г, 11,1%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,22 ~ 9,21 (m, 1H), 8,36 (dd, J=8,2, 2,2 Hz, 1H), 8,25 (d, J=7,3 Hz, 1H), 7,67 ~ 7,63 (m, 1H), 7,52 ~ 7,50 (m, 1H), 7,38 ~ 7,36 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,45 (s, 2H), 4,20 (s, 2H).; LRMS (ES) m/z 371,4 (M+ + 1).
Синтез соединения 66, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-1-(4-метоксибензил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез 2-амино-N-(трет-бутил)-5-фторбензамида
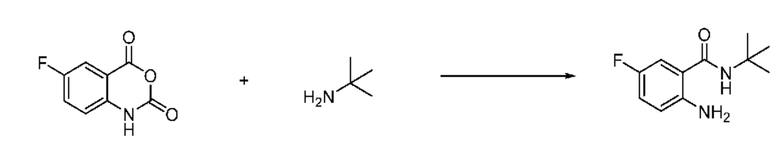
6-Фтор-2H-бензо[d][1,3]оксазин-2,4(1H)-дион (5,000 г, 27,606 ммоля), 2-метилпропан-2-амин (2,423 г, 33,127 ммоля) и N, N-диметилпиридин-4-амин (DMAP, 0,337 г, 2,761 ммоля) растворяли в N, N-диметилформамиде (30 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (2,700 г, 46,5%) в виде желтого твердого вещества.
[Стадия 2] Синтез метил-(2-(трет-бутилкарбамоил)-4-фторфенил)карбамата
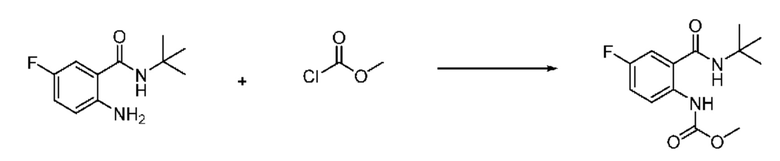
2-Амино-N-(трет-бутил)-5-фторбензамид (2,700 г, 12,842 ммоля), полученный на стадии 1, метилхлорформиат (1,456 г, 15,410 ммоля) и гидроксид натрия (1,00 M раствор в H2O, 25,684 мл, 25,684 ммоля) растворяли в 1,4-диоксане (20 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали 1 н. водный раствор хлористоводородной кислоты (10 мл) и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (2,570 г, 74,6%) в виде белого твердого вещества.
[Стадия 3] Синтез 3-(трет-бутил)-6-фторхиназолин-2,4(1H,3H)-диона
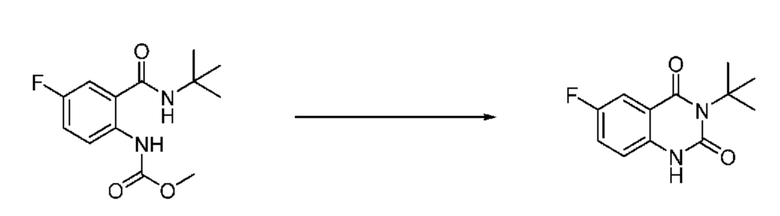
Метил-(2-(трет-бутилкарбамоил)-4-фторфенил)карбамат (2,570 г, 9,579 ммоля), полученный на стадии 2, и гидроксид калия (5,374 г, 95,792 ммоля) растворяли в этаноле (50 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. К реакционной смеси добавляли воду (10 мл) и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (1,520 г, 67,2%) в виде белого твердого вещества.
[Стадия 4] Синтез 3-(трет-бутил)-6-фтор-1-(4-метоксибензил)хиназолин-2,4(1H,3H)-диона
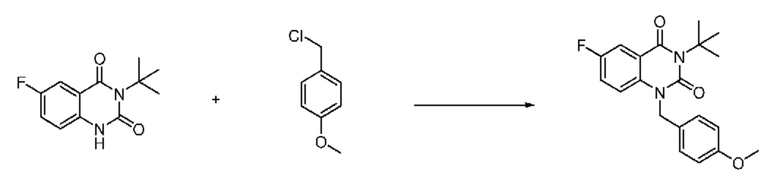
3-(Трет-бутил)-6-фторхиназолин-2,4(1H,3H)-дион (1,520 г, 6,434 ммоля), полученный на стадии 3, растворяли в N, N-диметилформамиде (20 мл) при 0°C, затем гидрид натрия (60,00%, 0,386 г, 9,651 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. К реакционной смеси добавляли 1-(хлорметил)-4-метоксибензол (1,310 г, 8,364 ммоля) и дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (1,660 г, 72,4%) в виде белого твердого вещества.
[Стадия 5] Синтез 6-фтор-1-(4-метоксибензил)хиназолин-2,4(1H,3H)-диона
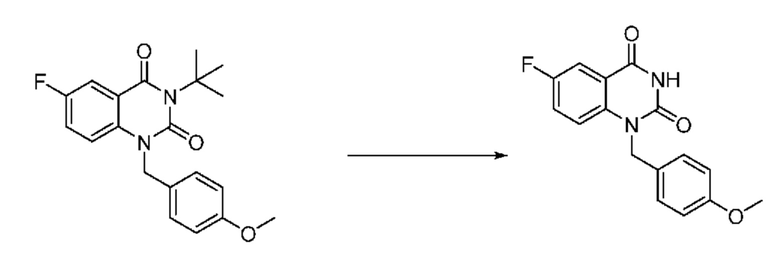
3-(Трет-бутил)-6-фтор-1-(4-метоксибензил)хиназолин-2,4(1H,3H)-дион (1,660 г, 4,658 ммоля), полученный на стадии 4, и хлористоводородную кислоту (4,00 M раствор в диоксане, 23,288 мл, 93,154 ммоля) смешивали при 100°C, затем полученную реакционную смесь перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. К реакционной смеси добавляли воду (10 мл) и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (1,250 г, 89,4%) в виде белого твердого вещества.
[Стадия 6] Синтез соединения 66
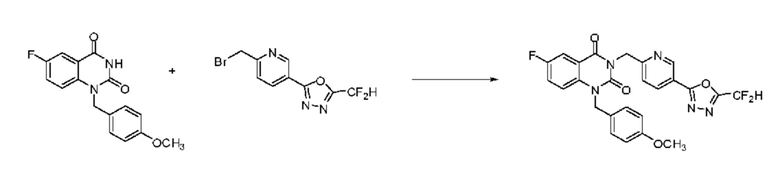
6-Фтор-1-(4-метоксибензил)хиназолин-2,4(1H,3H)-дион (1,250 г, 4,163 ммоля), полученный на стадии 5, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,570 г, 5,411 ммоля) и карбонат калия (1,151 г, 8,325 ммоля) растворяли в N, N-диметилформамиде (20 мл) при 90°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (1,600 г, 75,4%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,25 ~ 9,24 (m, 1H), 8,39 (dd, J=8,2, 2,2 Hz, 1H), 7,94 (dd, J=8,1, 3,1 Hz, 1H), 7,54 (d, J=8,2 Hz, 1H), 7,34 ~ 7,30 (m, 1H), 7,23 ~ 7,19 (m, 3H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,90 ~ 6,88 (m, 2H), 6,81 (s, 0,25H), 5,60 (s, 2H), 5,35 (s, 2H), 3,80 (s, 3H).; LRMS (ES) m/z 510,6 (M+ + 1).
Синтез соединения 67, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фторхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 67
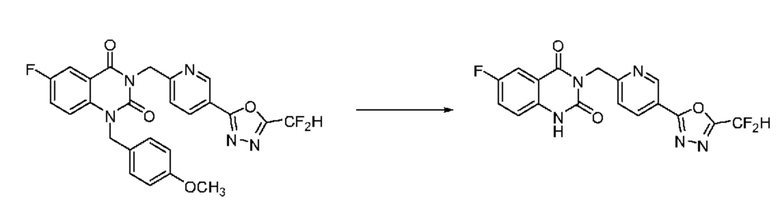
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-1-(4-метоксибензил)хиназолин-2,4(1H,3H)-дион (1,600 г, 3,141 ммоля) и нитрат церия-аммония (5,165 г, 9,422 ммоля) растворяли в смеси ацетонитрил (20 мл)/вода (20 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (0,900 г, 73,6%) в виде желтого твердого вещества.
1H NMR (400 MHz, DMSO-d6) δ 9,09 ~ 9,08 (m, 1H), 8,38 (dd, J=8,3, 2,3 Hz, 1H), 7,69 (s, 0,25H), 7,67 ~ 7,61 (m, 3H), 7,56 (s, 0,5H), 7,43 (s, 0,25H), 7,31 ~ 7,28 (m, 1H), 7,12 ~ 6,99 (m, 1H), 5,31 (s, 2H).; LRMS (ES) m/z 390,5 (M+ + 1).
Синтез соединения 68, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(1-этилпиперидин-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 68
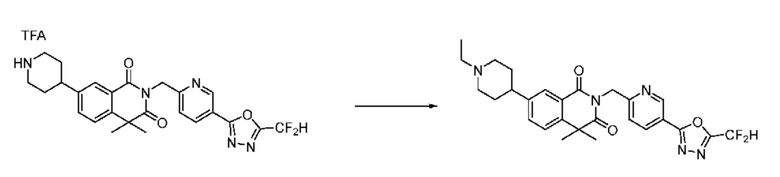
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,120 г, 0,202 ммоля) и N, N-диизопропилэтиламин (0,035 мл, 0,202 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем ацетальдегид (0,018 г, 0,403 ммоля) и триацетоксиборогидрид натрия (0,085 г, 0,403 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; дихлорметан/метанол=от 0 до 10%) и концентрировали и получали искомое соединение (0,023 г, 22,4%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,17 (dd, J=2,2, 0,7 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,09 (d, J=2,0 Hz, 1H), 7,60 (dd, J=8,2, 2,0 Hz, 1H), 7,50 (d, J=8,2 Hz, 1H), 7,45 (dd, J=8,2, 0,6 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 3,52 (d, J=11,7 Hz, 1H), 2,94 ~ 2,88 (m, 2H), 2,82 ~ 2,75 (m, 1H), 2,59 ~ 2,53 (m, 2H), 2,21 ~ 2,18 (m, 2H), 2,02 ~ 2,00 (m, 2H), 1,68 (s, 6H), 1,34 ~ 1,30 (m, 3H).; LRMS (ES) m/z 510,6 (M+ + 1).
Синтез соединения 69, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(1-изопропилпиперидин-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 69
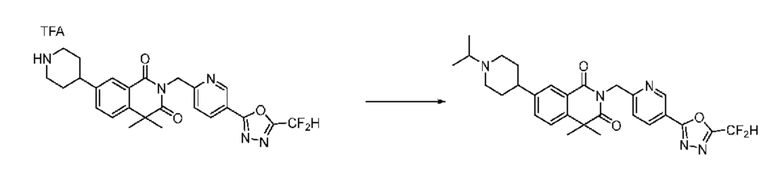
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,120 г, 0,202 ммоля) и N, N-диизопропилэтиламин (0,035 мл, 0,202 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем ацетон (0,030 мл, 0,403 ммоля) и триацетоксиборогидрид натрия (0,085 г, 0,403 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; дихлорметан/метанол=от 0 до 10%) и концентрировали и получали искомое соединение (0,040 г, 37,9%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,17 ~ 9,16 (m, 1H), 8,34 ~ 8,31 (m, 1H), 8,06 (s, 1H), 7,63 ~ 7,62 (m, 1H), 7,52 ~ 7,50 (m, 1H), 7,45 ~ 7,43 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 3,54 ~ 3,51 (m, 3H), 2,83 ~ 2,80 (m, 3H), 2,45 ~ 2,35 (m, 2H), 2,08 ~ 2,02 (m, 2H), 1,67 (s, 6H), 1,38 ~ 1,36 (m, 6H).; LRMS (ES) m/z 524,6 (M+ + 1).
Синтез соединения 70, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-1-метилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 70
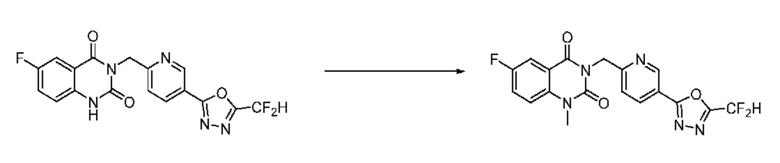
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фторхиназолин-2,4(1H,3H)-дион (0,100 г, 0,257 ммоля), йодметан (0,032 мл, 0,514 ммоля) и карбонат калия (0,071 г, 0,514 ммоля) растворяли в N, N-диметилформамиде (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,030 г, 29,0%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,22 ~ 9,21 (m, 1H), 8,36 (dd, J=8,2, 2,2 Hz, 1H), 7,94 (dd, J=8,0, 3,0 Hz, 1H), 7,53 ~ 7,43 (m, 2H), 7,28 ~ 7,25 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,52 (s, 2H), 3,65 (s, 3H).; LRMS (ES) m/z 404,4 (M+ + 1).
Синтез соединения 71, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-1-(2-(пиперидин-1-ил)этил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 71
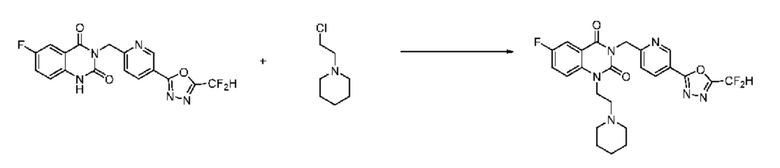
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фторхиназолин-2,4(1H,3H)-дион (0,100 г, 0,257 ммоля), 1-(2-хлорэтил)пиперидин (0,076 г, 0,514 ммоля) и карбонат калия (0,124 г, 0,899 ммоля) растворяли в N, N-диметилформамиде (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 50%) и концентрировали и получали искомое соединение (0,020 г, 15,6%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 7,91 (dd, J=8,1, 3,0 Hz, 1H), 7,49 ~ 7,33 (m, 3H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,50 (s, 2H), 4,28 (t, J=7,4 Hz, 2H), 2,64 (t, J=6,3 Hz, 2H), 2,55 ~ 2,45 (m, 4H), 1,58 ~ 1,53 (m, 4H), 1,47 ~ 1,40 (m, 2H).; LRMS (ES) m/z 501,5 (M+ + 1).
Синтез соединения 72, Трет-бутил-4-((3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-2,4-диоксо-3,4-дигидрохиназолин-1(2H)-ил)метил)пиперидин-1-карбоксилат
[Стадия 1] Синтез соединения 72
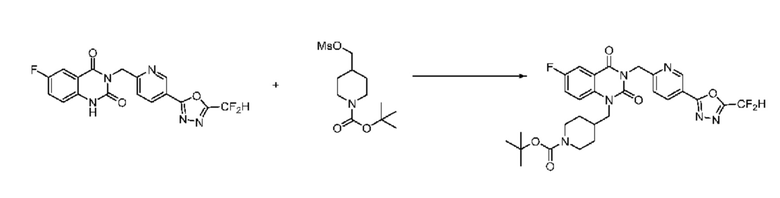
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фторхиназолин-2,4(1H,3H)-дион (0,283 г, 0,727 ммоля), трет-бутил-4-(((метилсульфонил)окси)метил)пиперидин-1-карбоксилат (0,427 г, 1,454 ммоля) и карбонат калия (0,201 г, 1,454 ммоля) растворяли в N, N-диметилформамиде (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,166 г, 38,9%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,16 ~ 9,15 (m, 1H), 8,35 (dd, J=8,1, 2,1 Hz, 1H), 7,93 (dd, J=8,0, 3,1 Hz, 1H), 7,50 ~ 7,44 (m, 2H), 7,23 ~ 7,20 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,50 (s, 2H), 4,14 ~ 4,08 (m, 4H), 2,65 ~ 2,60 (m, 2H), 2,05 ~ 2,03 (m, 1H), 1,68 ~ 1,65 (m, 2H), 1,45 (s, 9H), 1,27 ~ 1,25 (m, 2H).; LRMS (ES) m/z 587,5 (M+ + 1).
Синтез соединения 73, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-1-((1-метилпиперидин-4-ил)метил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-1-(пиперидин-4-илметил)хиназолин-2,4(1H,3H)-дион-2,2,2-трифторацетата
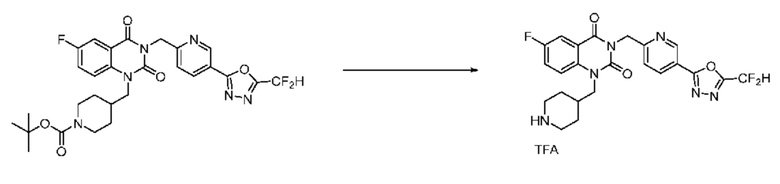
Трет-бутил-4-((3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-2,4-диоксо-3,4-дигидрохиназолин-1(2H)-ил)метил)пиперидин-1-карбоксилат (0,166 г, 0,283 ммоля) и трифторуксусную кислоту (0,217 мл, 2,830 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 3 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем полученный продукт использовали без проведения дополнительной очистки (0,160 г, 94,2%, коричневое масло).
[Стадия 2] Синтез соединения 73
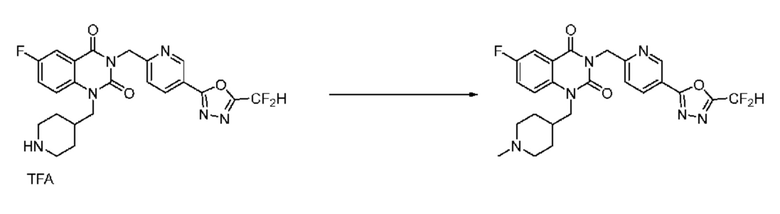
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-фтор-1-(пиперидин-4-илметил)хиназолин-2,4(1H,3H)-дион-2,2,2-трифторацетат (0,160 г, 0,266 ммоля), полученный на стадии 1, формальдегид (0,016 г, 0,533 ммоля), триацетоксиборогидрид натрия (0,113 г, 0,533 ммоля) и N, N-диизопропилэтиламин (0,046 мл, 0,266 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,080 г, 60,0%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,17 ~ 9,16 (m, 1H), 8,38 (dd, J=8,2, 2,1 Hz, 1H), 7,96 (dd, J=7,9, 3,0 Hz, 1H), 7,54 (d, J=8,2 Hz, 1H), 7,48 ~ 7,45 (m, 1H), 7,38 ~ 7,37 (m, 1H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 5,51 (s, 2H), 3,78 ~ 3,77 (m, 2H), 3,77 ~ 3,76 (m, 1H), 3,60 ~ 3,50 (m, 2H), 2,76 (s, 3H), 2,65 ~ 2,55 (m, 2H), 2,13 ~ 2,06 (m, 2H), 1,90 ~ 1,85 (m, 2H).; LRMS (ES) m/z 501,5 (M+ + 1).
Синтез соединения 74, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(фуран-2-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 74
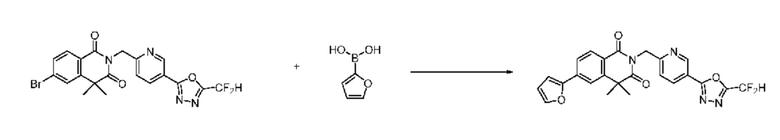
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), фуран-2-илбороновую кислоту (0,053 г, 0,471 ммоля), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (Pd(dppf)Cl2, 0,023 г, 0,031 ммоля) и карбонат натрия (0,067 г, 0,629 ммоля) растворяли в 1,2-диметоксиэтан (6 мл)/вода (3 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,003 г, 20,6%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,21 (dd, J=2,2, 0,8 Hz, 1H), 8,36 (dd, J=8,2, 2,2 Hz, 1H), 8,27 (dd, J=8,3, 0,3 Hz, 1H), 7,82 (d, J=1,5 Hz, 1H), 7,74 (dd, J=8,3, 1,6 Hz, 1H), 7,59 (dd, J=1,8, 0,7 Hz, 1H), 7,47 (dd, J=8,2, 0,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,89 (dd, J=3,4, 0,7 Hz, 1H), 6,80 (s, 0,25H), 6,58 ~ 6,57 (m, 1H), 5,45 (s, 2H), 1,76 (s, 2H).
Синтез соединения 75, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-3-(2-метоксиэтил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез 2-амино-N-(2-метоксиэтил)бензамида
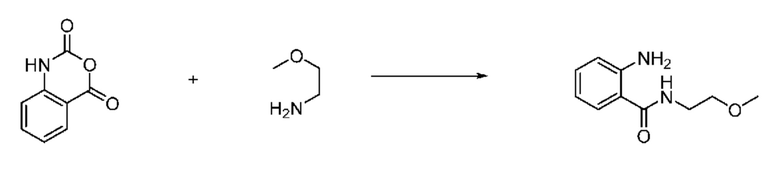
2H-Бензо[d][1,3]оксазин-2,4(1H)-дион (10,000 г, 61,301 ммоля), 2-метоксиэтан-1-амин (4,604 г, 61,301 ммоля) и триэтиламин (8,544 мл, 61,301 ммоля) растворяли в этаноле (50 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (9,800 г, 82,3%) в виде бесцветного масла.
[Стадия 2] Синтез 3-(2-метоксиэтил)хиназолин-2,4(1H,3H)-диона
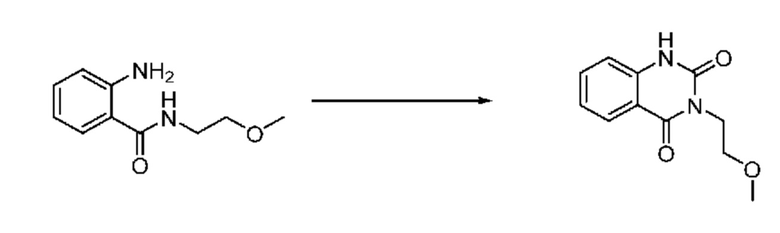
2-Амино-N-(2-метоксиэтил)бензамид (1,500 г, 7,723 ммоля), полученный на стадии 1, и 1,1'-карбонилдиимидазол (CDI, 1,252 г, 7,723 ммоля) растворяли в тетрагидрофуране (20 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (1,300 г, 76,4%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 75
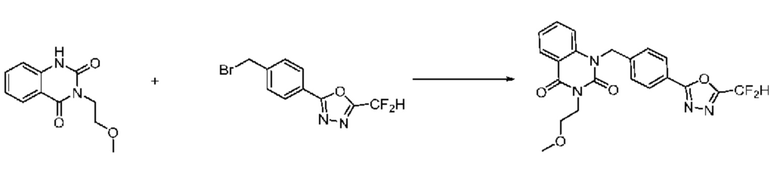
3-(2-Метоксиэтил)хиназолин-2,4(1H,3H)-дион (0,100 г, 0,454 ммоля), полученный на стадии 2, растворяли в N, N-диметилформамиде (10 мл) при 0°C, затем гидрид натрия (60,00%, 0,027 г, 0,681 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. К реакционной смеси добавляли 2-(4-(бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,131 г, 0,454 ммоля) и дополнительно перемешивали при комнатной температуре в течение 2 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,050 г, 25,7%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 8,29 (dd, J=7,9, 1,4 Hz, 1H), 8,11 (dd, J=6,7, 1,8 Hz, 2H), 7,59 ~ 7,55 (m, 1H), 7,47 (d, J=8,6 Hz, 2H), 7,29 ~ 7,25 (m, 1H), 7,06 ~ 7,04 (m, 1H), 7,06 (s, 0,25H), 6,92 (s, 0,5H), 6,79 (s, 0,25H), 5,48 (s, 2H), 4,45 (t, J=5,7 Hz, 2H), 3,77 (t, J=5,7 Hz, 2H), 3,42 (s, 3H).; LRMS (ES) m/z 429,3 (M+ + 1).
Синтез соединения 76, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-(2-метоксиэтил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез метил-6-((3-(2-метоксиэтил)-2,4-диоксо-3,4-дигидрохиназолин-1(2H)-ил)метил)никотината
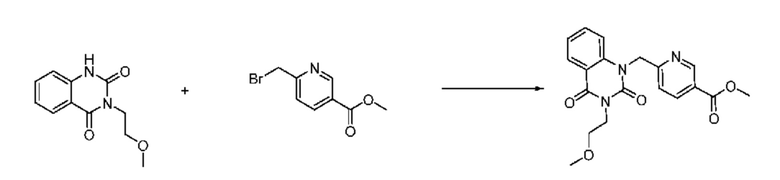
3-(2-Метоксиэтил)хиназолин-2,4(1H,3H)-дион (0,300 г, 1,362 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 0°C, затем гидрид натрия (60,00%, 0,109 г, 2,724 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. К реакционной смеси добавляли метил-6-(бромметил)никотинат (0,313 г, 1,362 ммоля) и дополнительно перемешивали при комнатной температуре в течение 2 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,300 г, 59,6%) в виде бесцветного масла.
[Стадия 2] Синтез 6-((3-(2-метоксиэтил)-2,4-диоксо-3,4-дигидрохиназолин-1(2H)-ил)метил)никотиногидразида
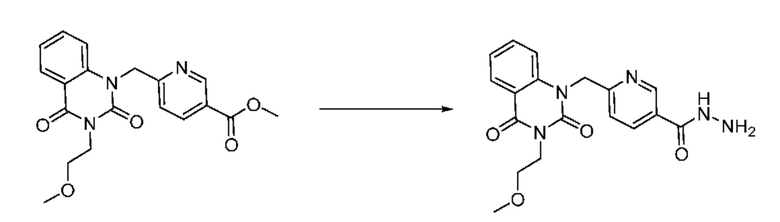
Метил-6-((3-(2-метоксиэтил)-2,4-диоксо-3,4-дигидрохиназолин-1(2H)-ил)метил)никотинат (0,090 г, 0,244 ммоля), полученный на стадии 1, и гидразинмоногидрат (0,237 мл, 4,873 ммоля) растворяли в этаноле (20 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем полученный продукт использовали без проведения дополнительной очистки (0,090 г, 100,0%, белое твердое вещество).
[Стадия 3] Синтез соединения 76
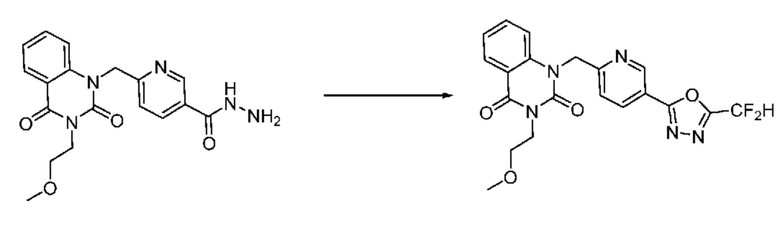
6-((3-(2-Метоксиэтил)-2,4-диоксо-3,4-дигидрохиназолин-1(2H)-ил)метил)никотиногидразид (0,090 г, 0,244 ммоля), полученный на стадии 2, 2,2-дифторуксусный ангидрид (0,091 мл, 0,731 ммоля) и имидазол (0,050 г, 0,731 ммоля) растворяли в дихлорметане (10 мл) при 45°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,030 г, 28,7%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,32 ~ 9,30 (m, 1H), 8,36 (dd, J=8,2, 2,2 Hz, 1H), 8,26 (dd, J=7,9, 1,6 Hz, 1H), 7,62 ~ 7,58 (m, 1H), 7,49 (d, J=8,2 Hz, 1H), 7,28 ~ 7,20 (m, 2H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 5,58 (s, 2H), 4,43 (t, J=5,7 Hz, 2H), 3,76 (t, J=5,7 Hz, 2H), 3,40 (s, 3H).; LRMS (ES) m/z 430,4 (M+ + 1).
Синтез соединения 77, 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-3-фенетилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез 2-амино-N-фенетилбензамида
2H-Бензо[d][1,3]оксазин-2,4(1H)-дион (3,000 г, 18,390 ммоля), 2-фенилэтан-1-амин (2,674 г, 22,068 ммоля) и N, N-диметилпиридин-4-амин (DMAP, 0,225 г, 1,839 ммоля) растворяли в N, N-диметилформамиде (30 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (4,000 г, 90,5%) в виде коричневого масла.
[Стадия 2] Синтез метил-(2-(фенетилкарбамоил)фенил)карбамата
2-Амино-N-фенетилбензамид (4,000 г, 16,645 ммоля), полученный на стадии 1, метилхлорформиат (1,887 г, 19,974 ммоля) и гидроксид натрия (1,00 M раствор в H2O, 33,290 мл, 33,290 ммоля) растворяли в 1,4-диоксане (30 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали 1 н. водный раствор хлористоводородной кислоты и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,790 г, 15,9%) в виде бесцветного масла.
[Стадия 3] Синтез 3-фенетилхиназолин-2,4(1H,3H)-диона
Метил-(2-(фенетилкарбамоил)фенил)карбамат (0,790 г, 2,648 ммоля), полученный на стадии 2, и гидроксид калия (1,486 г, 26,480 ммоля) растворяли в этаноле (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,500 г, 70,9%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 77
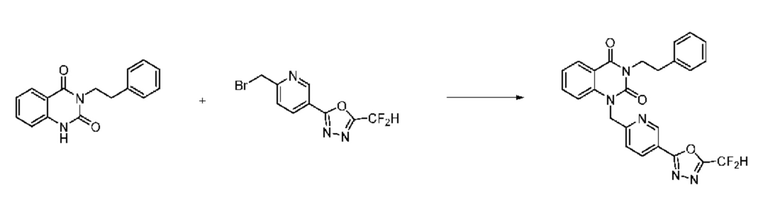
3-Фенетилхиназолин-2,4(1H,3H)-дион (0,150 г, 0,563 ммоля), полученный на стадии 3, растворяли в N, N-диметилформамиде (10 мл) при 0°C, затем гидрид натрия (60,00%, 0,034 г, 0,845 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. К реакционной смеси добавляли 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,196 г, 0,676 ммоля) и дополнительно перемешивали при комнатной температуре в течение 2 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,130 г, 48,5%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,32 (dd, J=2,2, 0,8 Hz, 1H), 8,35 (dd, J=5,9, 2,4 Hz, 1H), 8,29 (dd, J=7,9, 1,3 Hz, 1H), 7,62 ~ 7,58 (m, 1H), 7,37 ~ 7,26 (m, 8H), 7,08 (s, 0,25H), 6,95 (s, 0,5H), 6,82 (s, 0,25H), 5,56 (s, 2H), 4,44 ~ 4,40 (m, 2H), 3,10 ~ 3,06 (m, 2H).; LRMS (ES) m/z 475,9 (M+ + 1).
Синтез соединения 78, 1,3-бис((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 78
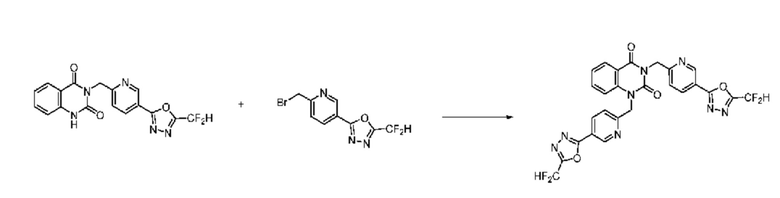
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)хиназолин-2,4(1H,3H)-дион (0,060 г, 0,162 ммоля), 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,052 г, 0,178 ммоля) и карбонат калия (0,045 г, 0,323 ммоля) растворяли в N, N-диметилформамиде (10 мл), затем полученный раствор перемешивали при 50°C в течение 18 ч и затем дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 80%) и концентрировали и получали искомое соединение (0,050 г, 53,3%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,31 (d, J=2,2 Hz, 1H), 9,23 (d, J=2,1 Hz, 1H), 8,39 ~ 8,36 (m, 2H), 8,28 (dd, J=8,0, 1,2 Hz, 1H), 7,63 ~ 7,61 (m, 1H), 7,56 ~ 7,51 (m, 2H), 7,31 ~ 7,28 (m, 2H), 7,07 (s, 0,5H), 6,95 ~ 6,94 (m, 1H), 6,82 ~ 6,81 (m, 0,5H), 5,61 ~ 5,60 (m, 4H), 2,18 (s, 6H).
Синтез соединения 79, трет-бутил 7-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-6-ил)-4,7-диазаспиро[2.5]октан-4-карбоксилат
[Стадия 1] Синтез соединения 79
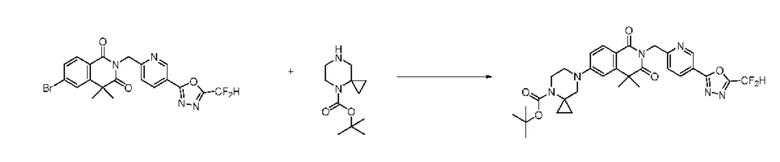
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,500 г, 1,048 ммоля), трет-бутил-4,7-диазаспиро[2.5]октан-4-карбоксилат (0,334 г, 1,571 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,096 г, 0,105 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,061 г, 0,105 ммоля) и карбонат цезия (1,024 г, 3,143 ммоля) растворяли в толуоле (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,230 г, 36,1%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,16 (d, J=1,8 Hz, 1H), 8,29 (dd, J=8,2, 2,2 Hz, 1H), 8,07 (d, J=8,9 Hz, 1H), 7,39 (d, J=8,3 Hz, 1H), 7,05 (s, 0,25H), 6,92 (s, 0,5H), 6,86 (dd, J=9,0, 2,3 Hz, 1H), 6,79 (s, 0,25H), 6,76 (d, J=2,3 Hz, 1H), 5,37 (s, 2H), 3,73 (t, J=5,2 Hz, 2H), 3,38 (t, J=5,2 Hz, 2H), 3,15 (s, 2H), 1,65 (s, 6H), 1,47 (s, 9H), 1,08 ~ 1,07 (m, 2H), 0,87 ~ 0,86 (m, 2H).
Синтез соединения 80, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4-метил-4,7-диазаспиро[2.5]октан-7-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4,7-диазаспиро[2.5]октан-7-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетата
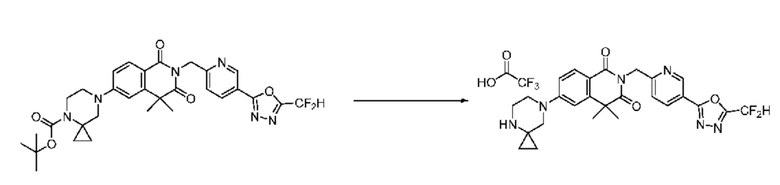
Трет-бутил 7-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-6-ил)-4,7-диазаспиро[2.5]октан-4-карбоксилат (0,230 г, 0,378 ммоля) и трифторуксусную кислоту (0,289 мл, 3,779 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем полученный продукт использовали без проведения дополнительной очистки (0,220 г, 93,5%, коричневое масло).
[Стадия 2] Синтез соединения 80
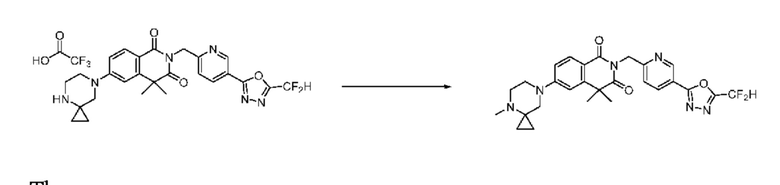
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4,7-диазаспиро[2.5]октан-7-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,100 г, 0,161 ммоля), полученный на стадии 1, N, N-диизопропилэтиламин (0,028 мл, 0,161 ммоля), формальдегид (0,010 г, 0,321 ммоля) и триацетоксиборогидрид натрия (0,068 г, 0,321 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,050 г, 59,6%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=2,2 Hz, 1H), 8,31 (dd, J=8,2, 2,2 Hz, 1H), 8,09 (d, J=8,9 Hz, 1H), 7,41 (d, J=8,2 Hz, 1H), 7,05 (s, 0,25H), 6,96 (s, 0,5H), 6,88 (dd, J=9,2, 2,2 Hz, 1H), 6,80 ~ 6,78 (m, 1,25H), 5,41 (s, 2H), 3,47 ~ 3,39 (m, 2H), 3,17 (s, 2H), 3,15 ~ 3,12 (m, 2H), 2,45 (s, 3H), 1,69 (s, 6H), 0,87 (t, J=5,7 Hz, 2H), 0,61 (t, J=5,8 Hz, 2H).
Синтез соединения 81, 6-(4-ацетил-4,7-диазаспиро[2.5]октан-7-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 81
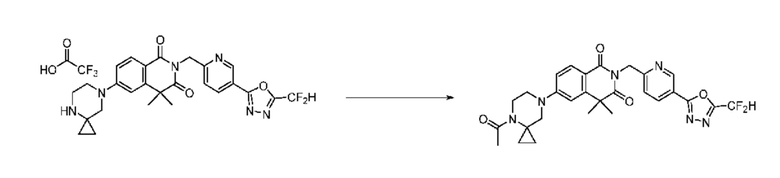
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4,7-диазаспиро[2.5]октан-7-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,100 г, 0,161 ммоля), ацетилхлорид (0,023 мл, 0,321 ммоля), и N, N-диизопропилэтиламин (0,084 мл, 0,482 ммоля) растворяли в дихлорметане (5 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 100%) и концентрировали и получали искомое соединение (0,060 г, 67,8%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,18 ~ 9,17 (m, 1H), 8,31 (dd, J=8,2, 2,2 Hz, 1H), 7,41 (d, J=8,1 Hz, 1H), 7,05 (s, 0,25H), 6,92 (s, 0,5H), 6,86 (dd, J=9,0, 2,4 Hz, 1H), 6,79 (s, 0,25H), 6,76 (d, J=2,4 Hz, 1H), 5,39 (s, 2H), 4,00 ~ 3,80 (m, 2H), 3,47 ~ 3,43 (m, 2H), 3,20 (s, 2H), 2,23 (s, 3H), 1,66 (s, 6H), 1,14 ~ 1,08 (m, 4H).
Синтез соединения 82, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-8-(фуран-2-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез 2-бром-6-(карбоксиметил)бензойной кислоты
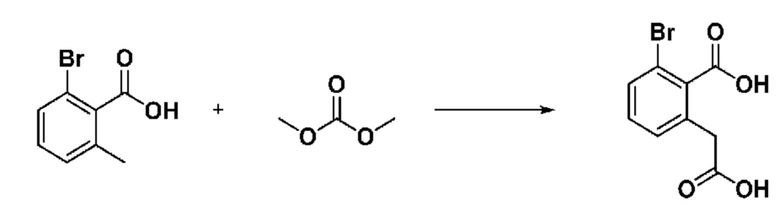
Диизопропиламин (27,691 мл, 186,003 ммоля) растворяли в тетрагидрофуране (300 мл) при -78°C, затем н-бутиллитий (2,50 M раствор, 74,401 мл, 186,003 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 1 ч и затем перемешивали при комнатной температуре в течение 10 мин. К реакционной смеси добавляли 2-бром-6-метилбензойную кислоту (10,000 г, 46,501 ммоля) и диметилкарбонат (7,830 мл, 93,002 ммоля) при -78°C и дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. В слой водного раствора добавляли 1 н. водный раствор хлористоводородной кислоты (10 мл) и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом магния, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (7,700 г, 63,9%, желтое масло).
[Стадия 2] Синтез метил-2-бром-6-(2-метокси-2-оксоэтил)бензоата
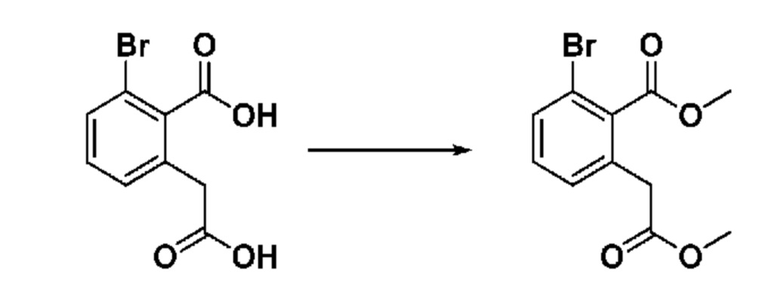
2-Бром-6-(карбоксиметил)бензойную кислоту (7,700 г, 29,723 ммоля), полученную на стадии 1, диметилсульфат (11,247 г, 89,169 ммоля) и карбонат калия (12,324 г, 89,169 ммоля) растворяли в 1,4-диоксане (150 мл) при комнатной температуре, затем полученный раствор перемешивали при 80°C в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали 1 н. водный раствор хлористоводородной кислоты и затем проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом магния, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (8,500 г, 99,6%, желтое масло).
[Стадия 3] Синтез метил-2-бром-6-(1-метокси-2-метил-1-оксопропан-2-ил)бензоата
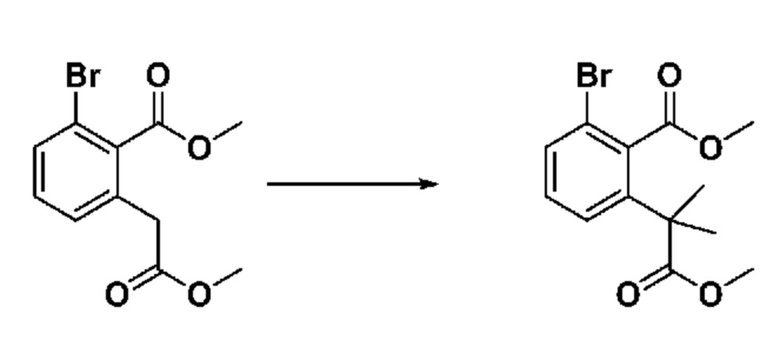
Метил-2-бром-6-(2-метокси-2-оксоэтил)бензоат (8,500 г, 29,605 ммоля), полученный на стадии 2, и гидрид натрия (60,00%, 0,059 г, 1,480 ммоля) растворяли в N, N-диметилформамиде (200 мл) при 0°C, затем йодметан (2,212 мл, 35,526 ммоля) добавляли к полученному раствору и перемешивали при комнатной температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом магния, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 80 г картридж; этилацетат/гексан=от 0 до 10%) и концентрировали и получали искомое соединение (3,600 г, 38,6%) в виде белого твердого вещества.
[Стадия 4] Синтез 2-бром-6-(2-карбоксипропан-2-ил)бензойной кислоты
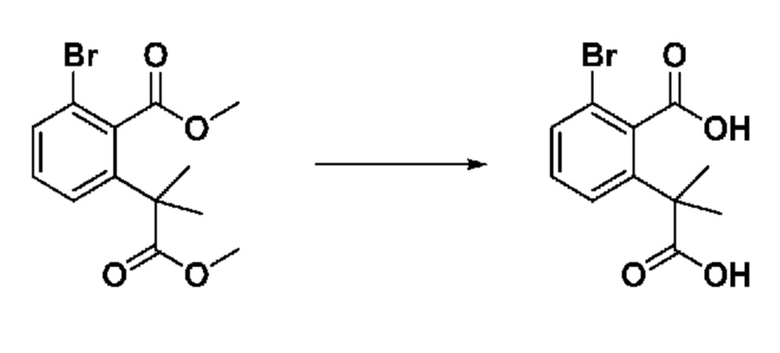
Метил-2-бром-6-(1-метокси-2-метил-1-оксопропан-2-ил)бензоат (3,600 г, 11,423 ммоля), полученный на стадии 3, и гидроксид калия (6,409 г, 114,228 ммоля) растворяли в смеси метанол (15 мл)/вода (30 мл) при комнатной температуре, затем полученный раствор кипятили с обратным холодильником в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, в полученный концентрат добавляли 1 н. водный раствор хлористоводородной кислоты и перемешивали и отфильтровывали осадившееся твердое вещество, затем промывали водой и затем сушили и получали искомое соединение (3,250 г, 90,3%) в виде светло-желтого твердого вещества.
[Стадия 5] Синтез 8-бром-4,4-диметилизохинолин-1,3(2H,4H)-диона
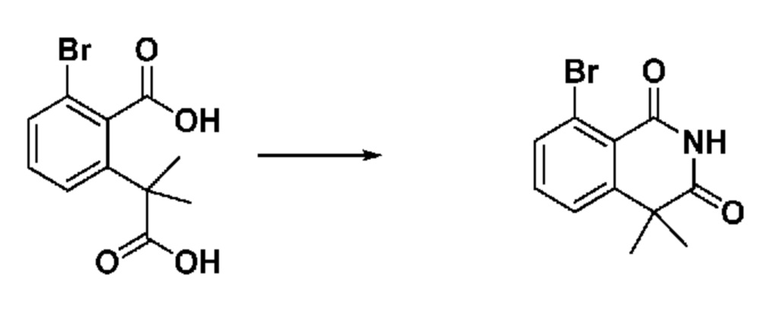
2-Бром-6-(2-карбоксипропан-2-ил)бензойную кислоту (3,250 г, 11,320 ммоля), полученную на стадии 4, и мочевину (0,680 г, 11,320 ммоля) смешивали в 1,2-дихлорбензоле (20 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 150°C в течение 45 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили, затем полученный фильтрат перекристаллизовывали из гексана при -10°C и фильтровали и получали твердое вещество. Затем твердое вещество промывали гексаном и сушили и получали искомое соединение (2,670 г, 88,0%) в виде светло-желтого твердого вещества.
[Стадия 6] Синтез 8-бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-диона
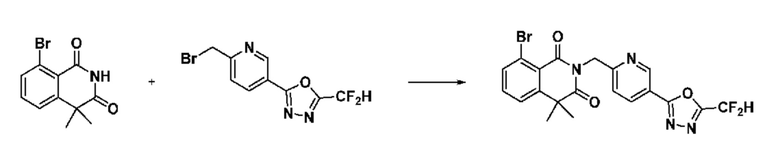
8-Бром-4,4-диметилизохинолин-1,3(2H,4H)-дион (2,000 г, 7,460 ммоля), полученный на стадии 5, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (2,380 г, 8,206 ммоля), карбонат калия (3,093 г, 22,379 ммоля) и йодид калия (0,124 г, 0,746 ммоля) растворяли в N, N-диметилформамиде (30 мл) при комнатной температуре, затем полученный раствор перемешивали при 80°C в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем насыщенный водный раствор гидрокарбоната натрия выливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом магния, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 10 до 40%) и концентрировали и получали искомое соединение (1,640 г, 46,1%) в виде желтого твердого вещества.
[Стадия 7] Синтез соединения 82
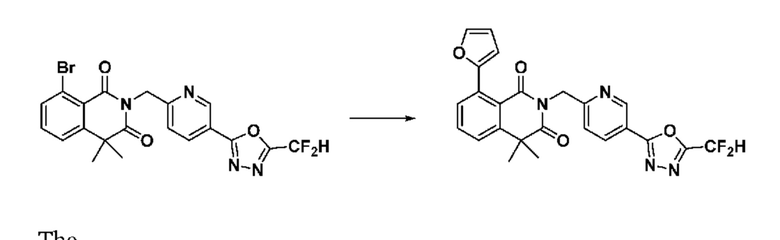
8-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,200 г, 0,419 ммоля), полученный на стадии 6, фуран-2-илбороновую кислоту (0,056 г, 0,503 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,014 г, 0,021 ммоля) и карбонат цезия (0,410 г, 1,257 ммоля) смешивали в смеси 1,4-диоксан (3 мл)/вода (1 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 10 до 30%) и концентрировали и получали искомое соединение (0,046 г, 23,6%) в виде светло-желтого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,18 (d, J=1,6 Hz, 1H), 8,32 (dd, J=8,2, 2,0 Hz, 1H), 7,70-7,66 (m, 1H), 7,60 (dd, J=8,0, 1,2 Hz, 1H), 7,53-7,50 (m, 2H), 7,42 (d, J=8,2 Hz, 1H), 7,06-6,80 (m, 1H), 6,52 (d, J=1,2 Hz, 2H), 5,40 (s, 2H), 1,76 (s, 6H).; LRMS (ES) m/z 465,2 (M+ + 1).
Синтез соединения 83, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-8-морфолиноизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 83
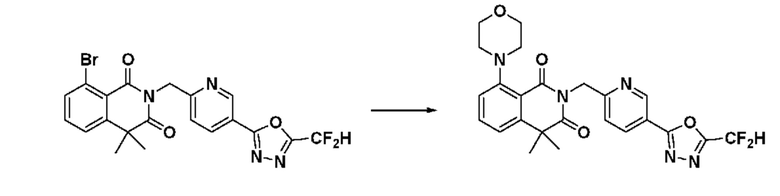
8-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,068 г, 0,142 ммоля), полученный на стадии 6, соединение 82, трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,013 г, 0,014 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,008 г, 0,014 ммоля) и карбонат цезия (0,139 г, 0,427 ммоля) растворяли в 1,4-диоксане (2 мл) при комнатной температуре, затем полученный раствор перемешивали при 80°C в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 0 до 40%) и концентрировали и получали искомое соединение (0,005 г, 7,3%) в виде желтого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,17 (d, J=1,5 Hz, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 7,60 (t, J=8,0 Hz, 1H), 7,50 (d, J=8,5 Hz, 1H), 7,23 (d, J=7,8 Hz, 1H), 7,13 (d, J=8,0 Hz, 1H), 7,07-6,81 (m, 1H), 5,44 (s, 2H), 3,97-3,95 (m, 4H), 3,24-3,23 (m, 4H), 1,71 (s, 6H).; LRMS (ES) m/z 484,3 (M+ + 1).
Синтез соединения 84, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-8-(пиридин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 84
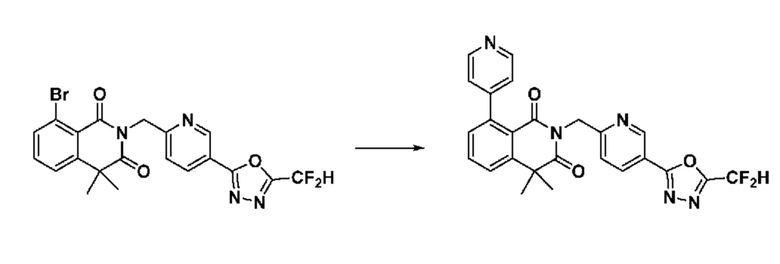
8-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), полученный на стадии 6, соединение 82, пиридин-4-илбороновую кислоту (0,046 г, 0,377 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,010 г, 0,016 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) смешивали в смеси 1,4-диоксан (3 мл)/вода (1 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 10 до 60%) и концентрировали и получали искомое соединение (0,042 г, 28,1%) в виде серого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,14 (d, J=1,5 Hz, 1H), 8,60-8,59 (m, 2H), 8,29 (dd, J=8,2, 2,2 Hz, 1H), 7,72-7,64 (m, 2H), 7,39 (d, J=8,7 Hz, 1H), 7,23-7,21 (m, 3H), 7,05-6,80 (m, 1H), 5,30 (s, 2H), 1,76 (s, 6H).; LRMS (ES) m/z 476,3 (M+ + 1).
Синтез соединения 85, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-8-(пиридин-3-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 85
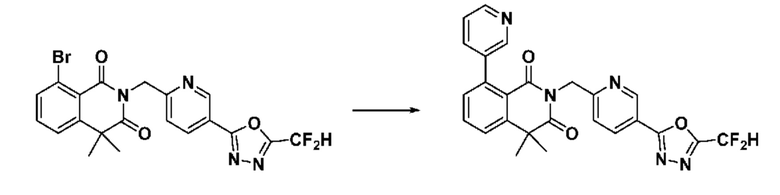
8-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), полученный на стадии 6, соединение 82, пиридин-3-илбороновую кислоту (0,046 г, 0,377 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,010 г, 0,016 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) смешивали в смеси 1,4-диоксан (3 мл)/вода (1 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 10 до 60%) и концентрировали и получали искомое соединение (0,047 г, 31,5%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,16 (dd, J=2,1, 0,6 Hz, 1H), 8,59 (dd, J=4,9, 1,4 Hz, 1H), 8,53 (d, J=1,7 Hz, 1H), 8,31 (dd, J=8,2, 2,2 Hz, 1H), 7,74-7,65 (m, 3H), 7,40-7,33 (m, 3H), 7,30-7,27 (m, 1H), 7,06-6,80 (m, 1H), 5,31 (s, 2H), 1,78 (s, 6H).; LRMS (ES) m/z 476,2 (M+ + 1).
Синтез соединения 86, 6-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез 2-амино-5-бром-N-(трет-бутил)бензамида
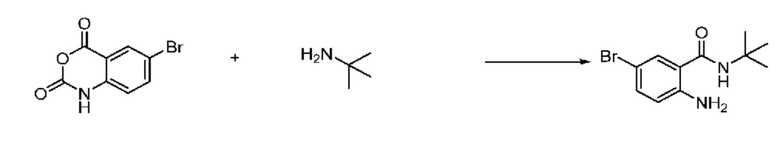
6-Бром-2H-бензо[d][1,3]оксазин-2,4(1H)-дион (8,000 г, 33,054 ммоля), 2-метилпропан-2-амин (2,901 г, 39,665 ммоля) и N, N-диметилпиридин-4-амин (DMAP, 0,404 г, 3,305 ммоля) растворяли в N, N-диметилформамиде (30 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. К реакционной смеси добавляли воду (20 мл) и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (5,500 г, 61,4%) в виде белого твердого вещества.
[Стадия 2] Синтез метил-(4-бром-2-(трет-бутилкарбамоил)фенил)карбамата
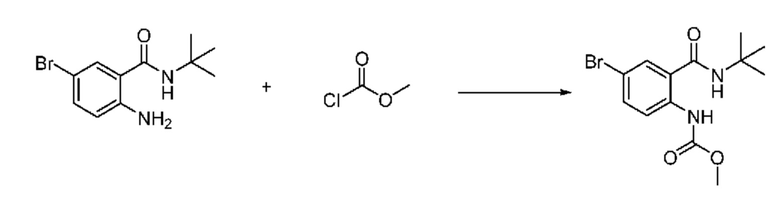
2-Амино-5-бром-N-(трет-бутил)бензамид (4,300 г, 15,858 ммоля), полученный на стадии 1, метилхлорформиат (1,498 г, 15,858 ммоля) и N, N-диизопропилэтиламин (4,143 мл, 23,787 ммоля) растворяли в дихлорметане (50 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 12 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (2,280 г, 43,7%) в виде желтого твердого вещества.
[Стадия 3] Синтез 6-бром-3-(трет-бутил)хиназолин-2,4(1H,3H)-диона
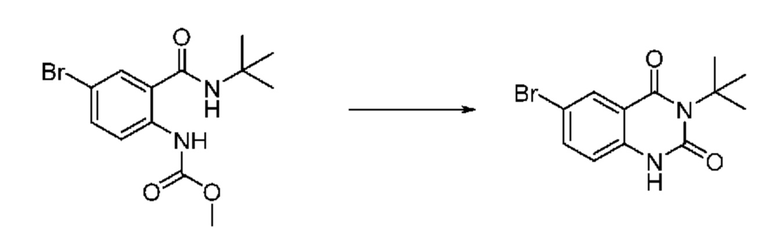
Метил-(4-бром-2-(трет-бутилкарбамоил)фенил)карбамат (2,280 г, 6,926 ммоля), полученный на стадии 2, и гидроксид калия (3,886 г, 69,261 ммоля) растворяли в этаноле (20 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 12 ч и затем реакцию завершали путем снижения температуры до комнатной температуры К реакционной смеси добавляли хлористоводородную кислоту (20 мл) и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (1,830 г, 88,9%) в виде белого твердого вещества.
[Стадия 4] Синтез 6-бром-3-(трет-бутил)-1-метилхиназолин-2,4(1H,3H)-диона
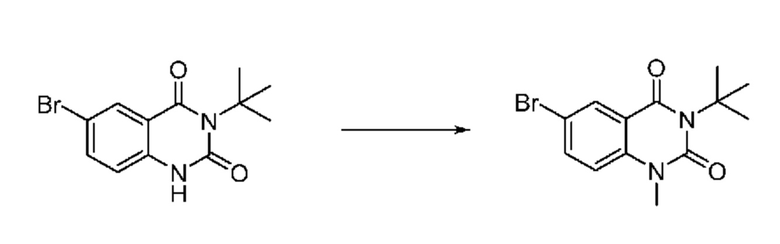
6-Бром-3-(трет-бутил)хиназолин-2,4(1H,3H)-дион (1,830 г, 6,159 ммоля), полученный на стадии 3, растворяли в N, N-диметилформамиде (20 мл) при 0°C, затем гидрид натрия (60,00%, 0,369 г, 9,238 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. К реакционной смеси добавляли йодметан (0,575 мл, 9,238 ммоля) и дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (1,440 г, 75,1%) в виде бесцветного масла.
[Стадия 5] Синтез 6-бром-1-метилхиназолин-2,4(1H,3H)-диона
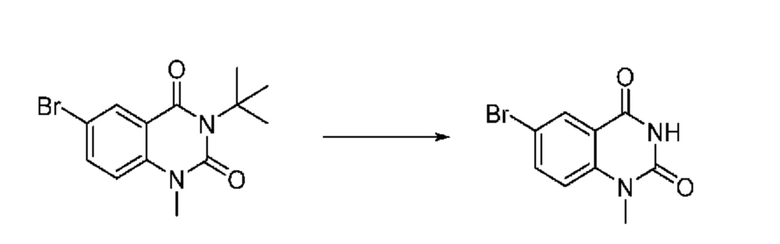
6-Бром-3-(трет-бутил)-1-метилхиназолин-2,4(1H,3H)-дион (1,300 г, 4,178 ммоля), полученный на стадии 4, и хлористоводородную кислоту (6,00 M раствор в H2O, 17,407 мл, 104,441 ммоля) растворяли в 1,4-диоксане (25 мл) при 100°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (0,980 г, 92,0%) в виде белого твердого вещества.
[Стадия 6] Синтез соединения 86
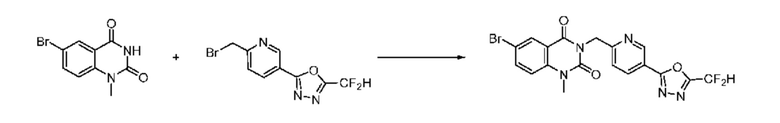
6-Бром-1-метилхиназолин-2,4(1H,3H)-дион (0,980 г, 3,842 ммоля), полученный на стадии 5, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,226 г, 4,226 ммоля) и карбонат калия (1,062 г, 7,684 ммоля) растворяли в N, N-диметилформамиде (20 мл) при 45°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (1,600 г, 89,7%) в виде белого твердого вещества.
LRMS (ES) m/z 465,4 (M+ + 1).
Синтез соединения 87, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(фуран-2-ил)-1-метилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 87
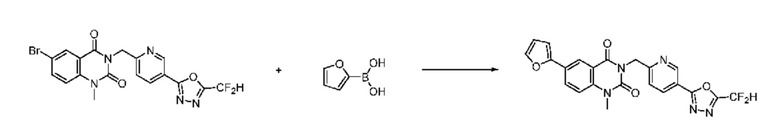
6-Бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион (0,100 г, 0,215 ммоля), фуран-2-илбороновую кислоту (0,036 г, 0,323 ммоля), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий (II, 0,014 г, 0,022 ммоля) и карбонат цезия (0,105 г, 0,323 ммоля) смешивали в смеси 1,4-диоксан (9 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,020 г, 20,6%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,24 (d, J=1,6 Hz, 1H), 8,52 (d, J=2,1 Hz, 1H), 8,37 (dd, J=8,2, 2,2 Hz, 1H), 8,05 (dd, J=8,7, 2,2 Hz, 1H), 7,53 ~ 7,51 (m, 2H), 7,31 (d, J=8,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,75 (dd, J=3,4, 0,7 Hz, 1H), 6,53 (dd, J=3,4, 1,8 Hz, 1H), 5,55 (s, 2H), 3,68 (s, 3H).; LRMS (ES) m/z 452,2 (M+ + 1).
Синтез соединения 88, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(фуран-3-ил)-1-метилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 88
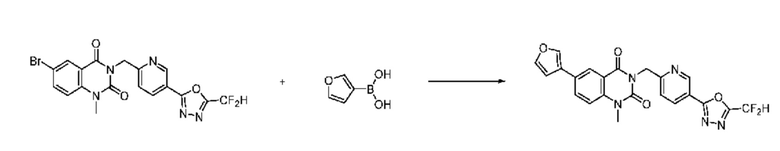
6-Бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион (0,100 г, 0,215 ммоля), фуран-3-илбороновую кислоту (0,036 г, 0,323 ммоля), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий (II, 0,014 г, 0,022 ммоля) и карбонат цезия (0,105 г, 0,323 ммоля) смешивали в смеси 1,4-диоксан (9 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,030 г, 30,9%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,23 ~ 9,22 (m, 1H), 8,37 ~ 8,34 (m, 2H), 7,86 ~ 7,81 (m, 2H), 7,30 ~ 7,28 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,77 (dd, J=1,9, 0,9 Hz, 1H), 5,55 (s, 2H), 3,67 (s, 3H).
Синтез соединения 89, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(2-фторфенил)-1-метилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 89
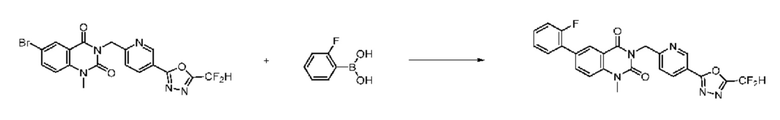
6-Бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион (0,100 г, 0,215 ммоля), (2-фторфенил)бороновую кислоту (0,045 г, 0,323 ммоля), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий (II, 0,014 г, 0,022 ммоля) и карбонат цезия (0,105 г, 0,323 ммоля) смешивали в смеси 1,4-диоксан (9 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,020 г, 19,4%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,24 (d, J=1,8 Hz, 1H), 8,45 (d, J=1,8 Hz, 1H), 8,37 (dd, J=8,3, 2,2 Hz, 1H), 7,98 (dt, J=8,6, 2,0 Hz, 1H), 7,54 ~ 7,49 (m, 2H), 7,41 ~ 7,35 (m, 2H), 7,28 ~ 7,26 (m, 1H), 7,24 ~ 7,17 (m, 1H), 7,06 (s, 1H), 6,93 (s, 1H), 6,80 (s, 1H), 5,56 (s, 2H), 3,70 (s, 3H).; LRMS (ES) m/z 480,2 (M+ + 1).
Синтез соединения 90, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(3-фторфенил)-1-метилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 90
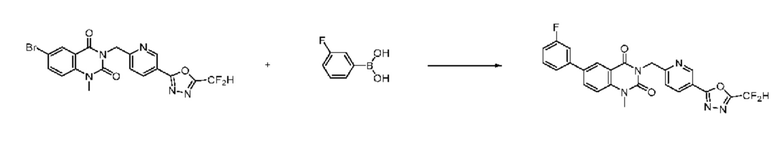
6-Бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион (0,100 г, 0,215 ммоля), (3-фторфенил)бороновую кислоту (0,045 г, 0,323 ммоля), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий (II, 0,014 г, 0,022 ммоля) и карбонат цезия (0,105 г, 0,323 ммоля) смешивали в смеси 1,4-диоксан (9 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,030 г, 29,0%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,24 (dd, J=2,2, 0,8 Hz, 1H), 8,50 (d, J=2,2 Hz, 1H), 8,37 (dd, J=8,2, 2,2 Hz, 1H), 7,97 (dd, J=8,7, 2,3 Hz, 1H), 7,54 (dd, J=8,2, 0,7 Hz, 1H), 7,46 ~ 7,44 (m, 1H), 7,38 ~ 7,33 (m, 1H), 7,12 ~ 7,07 (m, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,57 (s, 2H), 3,70 (s, 3H).
Синтез соединения 91, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метил-6-(пиридин-3-ил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 91
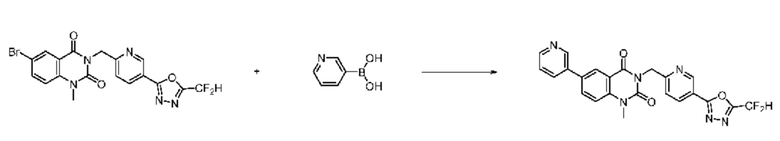
6-Бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион (0,100 г, 0,215 ммоля), пиридин-3-илбороновую кислоту (0,040 г, 0,323 ммоля), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий (II, 0,014 г, 0,022 ммоля) и карбонат цезия (0,105 г, 0,323 ммоля) смешивали в смеси 1,4-диоксан (9 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,025 г, 25,1%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,24 (d, J=2,2 Hz, 1H), 8,93 (d, J=2,4 Hz, 1H), 8,66 (dd, J=4,6, 1,3 Hz, 1H), 8,51 (d, J=2,2 Hz, 1H), 8,38 (dd, J=8,4, 2,4 Hz, 1H), 7,55 (d, J=8,2 Hz, 1H), 7,46 ~ 7,40 (m, 2H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,57 (s, 2H), 3,71 (s, 3H).; LRMS (ES) m/z 463,2 (M+ + 1).
Синтез соединения 92, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метил-6-(пиридин-4-ил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 92
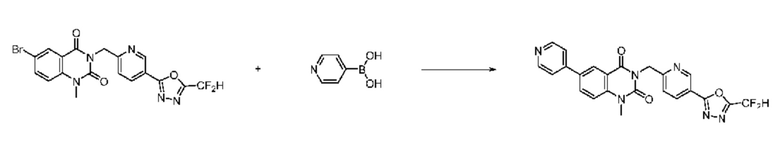
6-Бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион (0,100 г, 0,215 ммоля), пиридин-4-илбороновую кислоту (0,040 г, 0,323 ммоля), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий (II, 0,014 г, 0,022 ммоля) и карбонат цезия (0,105 г, 0,323 ммоля) смешивали в смеси 1,4-диоксан (9 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,030 г, 30,1%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,23 (d, J=1,6 Hz, 1H), 8,72 ~ 8,71 (m, 2H), 8,58 (d, J=2,2 Hz, 1H), 8,37 (dd, J=8,2, 2,2 Hz, 1H), 8,04 (dd, J=8,7, 2,3 Hz, 1H), 7,59 ~ 7,53 (m, 3H), 7,42 (d, J=8,7 Hz, 1H), 7,06 (s, 1H), 6,93 (s, 1H), 6,80 (s, 1H), 5,56 (s, 2H), 3,71 (s, 2H).
Синтез соединения 93, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-8-(5-метилфуран-2-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 93
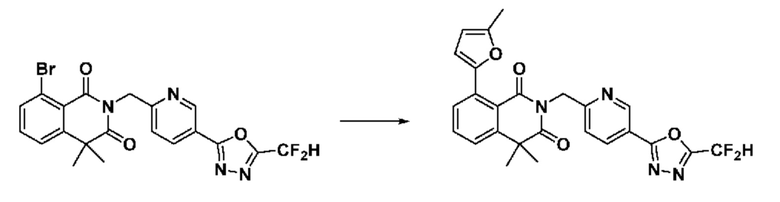
8-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), полученный на стадии 6, соединение 82, 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан (0,078 г, 0,377 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,010 г, 0,016 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) смешивали в смеси 1,4-диоксан (3 мл)/вода (1 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,020 г, 13,3%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,19 (dd, J=2,1, 0,7 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 7,67-7,63 (m, 1H), 7,56-7,51 (m, 2H), 7,43 (d, J=8,2 Hz, 1H), 7,06-6,80 (m, 1H), 6,44 (d, J=3,1 Hz, 1H), 6,09 (dd, J=2,1, 1,0 Hz, 1H), 5,40 (s, 2H), 2,31 (s, 3H), 1,74 (s, 6H).; LRMS (ES) m/z 479,2 (M+ + 1).
Синтез соединения 94, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-8-(6-метоксипиридин-3-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 94
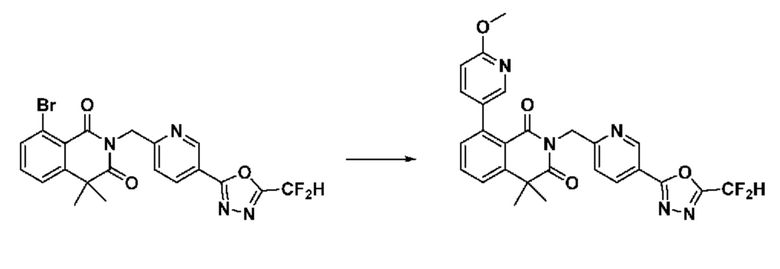
8-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), полученный на стадии 6, соединение 82, (6-метоксипиридин-3-ил)бороновую кислоту (0,058 г, 0,377 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,010 г, 0,016 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) смешивали в смеси 1,4-диоксан (3 мл)/вода (1 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,016 г, 10,1%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,17 (d, J=1,6 Hz, 1H), 8,31 (dd, J=8,2, 2,2 Hz, 1H), 8,08 (d, J=2,4 Hz, 1H), 7,71-7,67 (m, 1H), 7,61 (dd, J=8,0, 1,3 Hz, 1H), 7,55-7,52 (m, 1H), 7,40 (d, J=8,2 Hz, 1H), 7,29-7,27 (m, 1H), 7,06-6,80 (m, 1H), 6,75 (d, J=8,5 Hz, 1H), 5,33 (s, 2H), 3,98 (s, 3H), 1,78 (s, 6H).; LRMS (ES) m/z 506,2 (M+ + 1).
Синтез соединения 95, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-8-(фуран-3-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 95
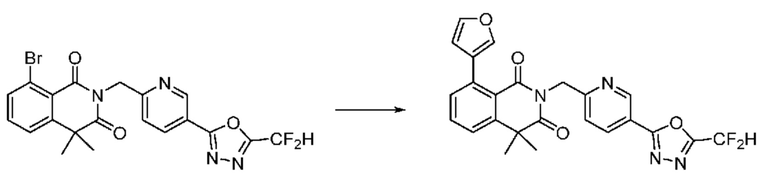
8-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), фуран-3-илбороновую кислоту (0,042 г, 0,377 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,010 г, 0,016 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) смешивали в смеси 1,4-диоксан (3 мл)/вода (1 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали продукт, затем полученный продукт повторно очищали с помощью хроматографии (пластина с SiO2, 20×20×1 мм; этилацетат/гексан водный раствор=25%) и концентрировали и получали искомое соединение (0,046 г, 31,5%) в виде светло-коричневого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,18 (d, J=1,5 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 7,64 (t, J=7,7 Hz, 1H), 7,56 (dd, J=8,0, 1,3 Hz, 1H), 7,52 ~ 7,52 (m, 1H), 7,45 ~ 7,42 (m, 2H), 7,36 (dd, J=7,5, 1,3 Hz, 1H), 7,06 ~ 6,80 (m, 1H), 6,48 ~ 6,47 (m, 1H), 5,32 (s, 2H), 1,76 (s, 6H).; LRMS (ES) m/z 465,0 (M+ + 1).
Синтез соединения 96, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-8-(3,5-диметилизооксазол-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 96
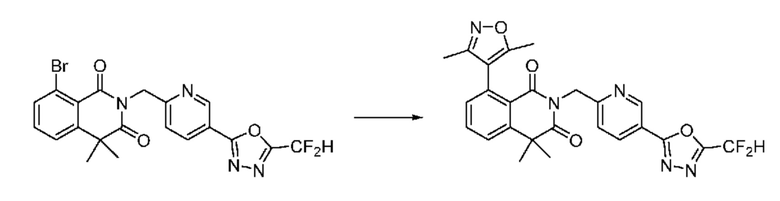
8-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,150 г, 0,314 ммоля), (3,5-диметилизооксазол-4-ил)бороновую кислоту (0,053 г, 0,377 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,010 г, 0,016 ммоля) и карбонат цезия (0,307 г, 0,943 ммоля) смешивали в смеси 1,4-диоксан (3 мл)/вода (1 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,092 г, 59,3%) в виде коричневого масла.
1H NMR (400 MHz, CDCl3) δ 9,13 (d, J=1,6 Hz, 1H), 8,32 (dd, J=8,2, 2,0 Hz, 1H), 7,71 (t, J=7,7 Hz, 1H), 7,64 (d, J=7,5 Hz, 1H), 7,43 (d, J=8,2 Hz, 1H), 7,20 (d, J=7,0 Hz, 1H), 7,06 ~ 6,80 (m, 1H), 5,34 (s, 2H), 2,24 (s, 3H), 1,99 (s, 3H), 1,77 (d, J=5,4 Hz, 6H).; LRMS (ES) m/z 494,2 (M+ + 1).
Синтез соединения 97, 7-бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез 2-амино-4-бром-N-(трет-бутил)бензамида
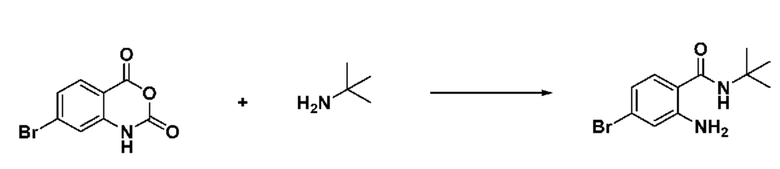
7-Бром-2H-бензо[d][1,3]оксазин-2,4(1H)-дион (10,000 г, 41,317 ммоля), 2-метилпропан-2-амин (3,626 г, 49,581 ммоля) и N, N-диметилпиридин-4-амин (DMAP, 0,505 г, 4,132 ммоля) растворяли в N, N-диметилформамиде (30 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. К реакционной смеси добавляли воду (20 мл) и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (7,700 г, 68,7%) в виде белого твердого вещества.
[Стадия 2] Синтез метил-(5-бром-2-(трет-бутилкарбамоил)фенил)карбамата
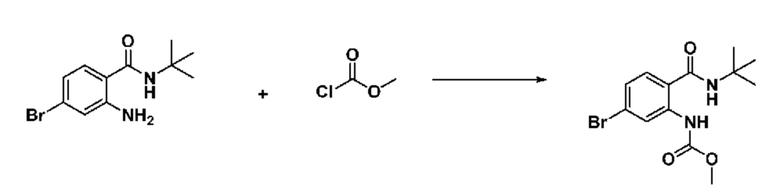
2-Амино-4-бром-N-(трет-бутил)бензамид (7,700 г, 28,397 ммоля), полученный на стадии 1, метилхлорформиат (2,683 г, 28,397 ммоля) и N, N-диизопропилэтиламин (7,419 мл, 42,595 ммоля) растворяли в дихлорметане (30 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (3,720 г, 39,8%) в виде коричневого твердого вещества.
[Стадия 3] Синтез 7-бром-3-(трет-бутил)хиназолин-2,4(1H,3H)-диона
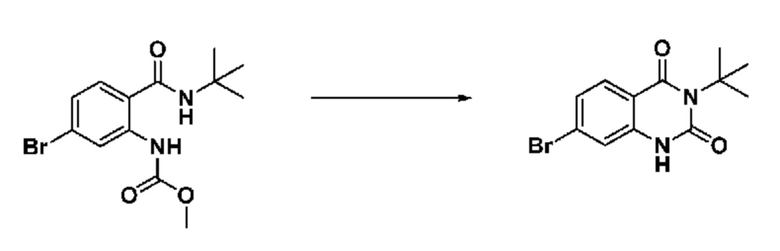
Метил-(5-бром-2-(трет-бутилкарбамоил)фенил)карбамат (3,720 г, 11,300 ммоля), полученный на стадии 2, и гидроксид калия (6,340 г, 113,005 ммоля) растворяли в этаноле (30 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (2,000 г, 59,6%, коричневое масло).
[Стадия 4] Синтез 7-бром-3-(трет-бутил)-1-метилхиназолин-2,4(1H,3H)-диона
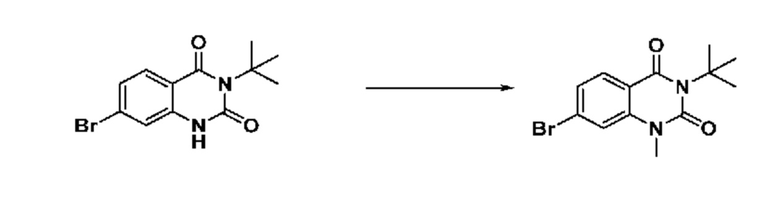
7-Бром-3-(трет-бутил)хиназолин-2,4(1H,3H)-дион (2,000 г, 6,731 ммоля), полученный на стадии 3, растворяли в N, N-диметилформамиде (30 мл) при 0°C, затем йодметан (0,629 мл, 10,096 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. Гидрид натрия (60,00%, 0,404 г, 10,096 ммоля) к реакционной смеси добавляли, и дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (2,000 г, 95,5%) в виде белого твердого вещества.
[Стадия 5] Синтез 7-бром-1-метилхиназолин-2,4(1H,3H)-диона
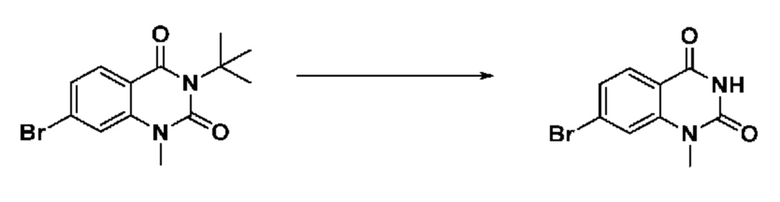
7-Бром-3-(трет-бутил)-1-метилхиназолин-2,4(1H,3H)-дион (2,000 г, 6,427 ммоля), полученный на стадии 4, и хлористоводородную кислоту (6,00 M раствор в H2O, 16,068 мл, 96,407 ммоля) растворяли в 1,4-диоксане (20 мл) при 100°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (1,500 г, 91,5%) в виде коричневого твердого вещества.
[Стадия 6] Синтез соединения 97
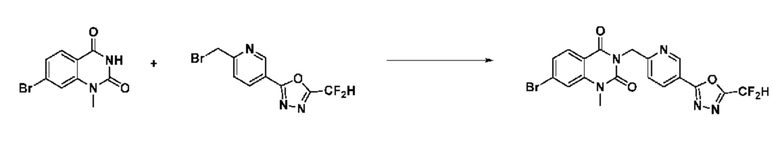
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), полученный на стадии 5, пиридин-4-илбороновую кислоту (0,039 г, 0,314 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,014 г, 0,021 ммоля) и карбонат цезия (0,102 г, 0,314 ммоля) смешивали в смеси 1,4-диоксан (6 мл)/вода (2 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,040 г, 40,2%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,22 (d, J=1,5 Hz, 1H), 8,36 (dd, J=8,2, 2,2 Hz, 1H), 8,12 (d, J=8,6 Hz, 1H), 7,51 (d, J=8,2 Hz, 1H), 7,45 ~ 7,42 (m, 2H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,51 (s, 2H), 3,63 (s, 3H).
Синтез соединения 98, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(фуран-2-ил)-1-метилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 98
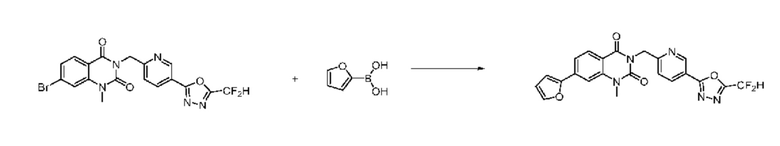
7-Бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион (0,100 г, 0,215 ммоля), полученный на стадии 6, соединение 97, фуран-2-илбороновую кислоту (0,036 г, 0,323 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,014 г, 0,022 ммоля) и карбонат цезия (0,105 г, 0,323 ммоля) смешивали в смеси 1,4-диоксан (10 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,020 г, 20,6%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,24 (d, J=1,7 Hz, 1H), 8,36 (dd, J=8,2, 2,2 Hz, 1H), 8,25 (d, J=8,6 Hz, 1H), 7,60 ~ 7,50 (m, 4H), 7,06 (s, 0,25H), 6,94 ~ 6,92 (m, 1H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,59 (dd, J=3,3, 1,7 Hz, 1H), 5,54 (s, 2H), 3,72 (s, 3H).; LRMS (ES) m/z 452,4 (M+ + 1).
Синтез соединения 99, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(2-фторфенил)-1-метилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 99
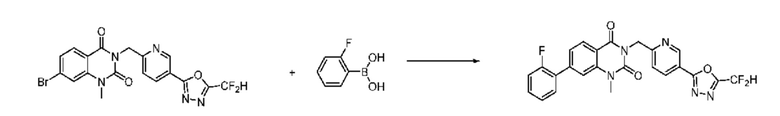
7-Бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион (0,100 г, 0,215 ммоля), полученный на стадии 6, соединение 97, (2-фторфенил)бороновую кислоту (0,045 г, 0,323 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,014 г, 0,022 ммоля) и карбонат цезия (0,105 г, 0,323 ммоля) смешивали в смеси 1,4-диоксан (10 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,023 г, 22,3%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,24 ~ 9,23 (m, 1H), 8,37 ~ 8,32 (m, 2H), 7,54 ~ 7,42 (m, 5H), 7,32 ~ 7,21 (m, 2H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 5,56 (s, 2H), 3,69 (s, 3H).; LRMS (ES) m/z 480,4 (M+ + 1).
Синтез соединения 100, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метил-7-(пиридин-3-ил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 100
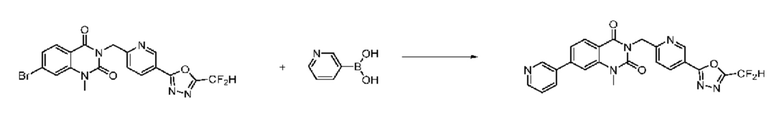
7-Бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион (0,100 г, 0,215 ммоля), полученный на стадии 6, соединение 97, пиридин-3-илбороновую кислоту (0,040 г, 0,323 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,014 г, 0,022 ммоля) и карбонат цезия (0,105 г, 0,323 ммоля) смешивали в смеси 1,4-диоксан (10 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,026 г, 26,1%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) 9,22 (s, 1H), 8,93 (s, 1H), 8,73 (d, J=4,3 Hz, 1H), 8,38 ~ 8,35 (m, 2H), 7,98 ~ 7,96 (m, 1H), 7,62 ~ 7,42 (m, 4H), 7,07 (s, 1H), 6,94 (s, 1H), 6,81 (s, 1H), 5,56 (s, 2H), 3,73 (s, 3H).; LRMS (ES) m/z 463,4 (M+ + 1).
Синтез соединения 101, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метил-7-(пиридин-4-ил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 101
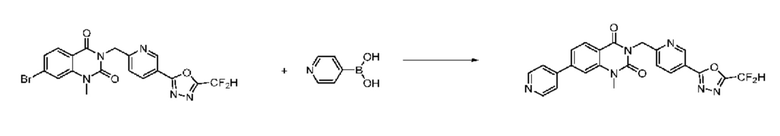
7-Бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион (0,100 г, 0,215 ммоля), полученный на стадии 6, соединение 97, пиридин-4-илбороновую кислоту (0,040 г, 0,323 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,014 г, 0,022 ммоля) и карбонат цезия (0,105 г, 0,323 ммоля) смешивали в смеси 1,4-диоксан (10 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,030 г, 30,1%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,24 ~ 9,20 (m, 1H), 8,77 (dd, J=4,4, 1,6 Hz, 1H), 8,38 ~ 8,35 (m, 1H), 7,58 ~ 7,52 (m, 4H), 7,46 (d, J=1,4 Hz, 1H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 5,55 (s, 2H), 3,73 (s, 3H).; LRMS (ES) m/z 463,4 (M+ + 1).
Синтез соединения 102, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(фуран-3-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 102
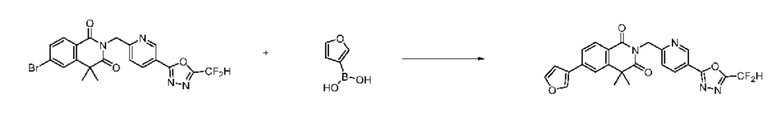
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), полученный на стадии 6, соединение 97, фуран-3-илбороновую кислоту (0,035 г, 0,314 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,014 г, 0,021 ммоля) и карбонат цезия (0,102 г, 0,314 ммоля) смешивали в смеси 1,4-диоксан (30 мл)/вода (10 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,034 г, 34,9%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,8 Hz, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 8,26 (dd, J=7,6, 1,2 Hz, 1H), 7,89 (dd, J=1,5, 0,9 Hz, 1H), 7,59 ~ 7,56 (m, 3H), 7,47 (dd, J=8,2, 0,8 Hz, 1H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 6,79 (dd, J=1,9, 0,9 Hz, 1H), 5,45 (s, 2H), 1,15 (s, 6H).; LRMS (ES) m/z 465,4 (M+ + 1).
Синтез соединения 103, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(2-фторфенил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 103
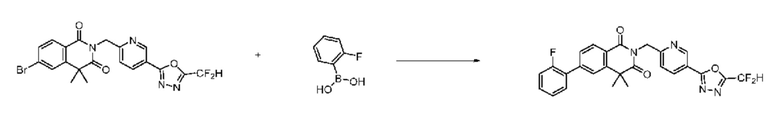
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), полученный на стадии 6, соединение 97, (2-фторфенил)бороновую кислоту (0,044 г, 0,314 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,014 г, 0,021 ммоля) и карбонат цезия (0,102 г, 0,314 ммоля) смешивали в смеси 1,4-диоксан (30 мл)/вода (10 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,035 г, 33,9%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,21 (dd, J=2,2, 0,6 Hz, 1H), 8,37 ~ 8,32 (m, 2H), 7,72 ~ 7,71 (m, 1H), 7,66 ~ 7,63 (m, 1H), 7,53 ~ 7,42 (m, 3H), 7,32 ~ 7,29 (m, 1H), 7,25 ~ 7,20 (m, 1H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 5,42 (s, 2H), 1,76 (s, 6H).; LRMS (ES) m/z 493,4 (M+ + 1).
Синтез соединения 104, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиридин-3-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 104
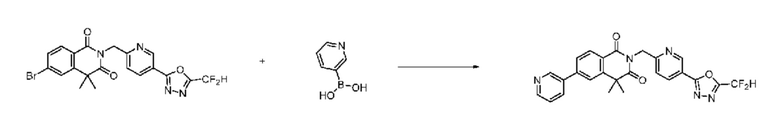
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), полученный на стадии 6, соединение 97, пиридин-3-илбороновую кислоту (0,039 г, 0,314 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,014 г, 0,021 ммоля) и карбонат цезия (0,102 г, 0,314 ммоля) смешивали в смеси 1,4-диоксан (30 мл)/вода (10 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,010 г, 10,0%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,21 (d, J=1,6 Hz, 1H), 8,93 (dd, J=2,3, 0,7 Hz, 1H), 8,72 (dd, J=4,8, 1,6 Hz, 1H), 8,39 ~ 8,35 (m, 2H), 7,97 ~ 7,94 (m, 1H), 7,71 ~ 7,69 (m, 2H), 7,50 ~ 7,45 (m, 2H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 5,47 (s, 2H), 1,78 (s, 6H).; LRMS (ES) m/z 476,3 (M+ + 1).
Синтез соединения 105, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиридин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 105
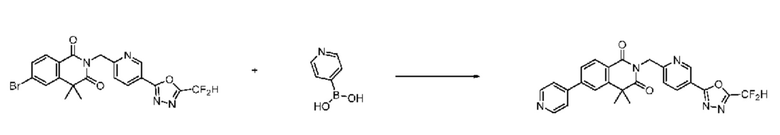
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), полученный на стадии 6, соединение 97, пиридин-4-илбороновую кислоту (0,039 г, 0,314 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,014 г, 0,021 ммоля) и карбонат цезия (0,102 г, 0,314 ммоля) смешивали в смеси 1,4-диоксан (6 мл)/вода (2 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,040 г, 40,2%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,18 (dd, J=2,2, 0,7 Hz, 1H), 8,75 (d, J=6,0 Hz, 1H), 8,38 ~ 8,33 (m, 2H), 7,74 ~ 7,70 (m, 2H), 7,56 ~ 7,55 (m, 2H), 7,48 (dd, J=8,2, 0,6 Hz, 1H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 5,45 (s, 2H), 1,78 (s, 6H).; LRMS (ES) m/z 476,4 (M+ + 1).
Синтез соединения 106, 6'-бром-2'-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1'H-спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-дион
[Стадия 1] Синтез метил-4-бром-2-(1-(метоксикарбонил)циклобутил)бензоата
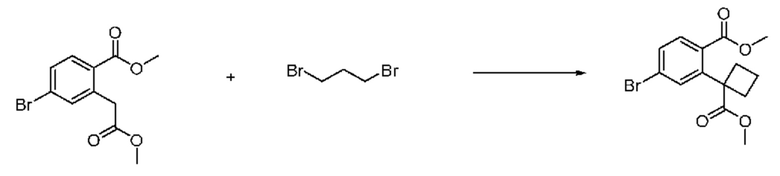
Метил-4-бром-2-(2-метокси-2-оксоэтил)бензоат (2,500 г, 8,707 ммоля) растворяли в N, N-диметилформамиде (30 мл) при 0°C, затем гидрид натрия (60,00%, 1,045 г, 26,122 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. К реакционной смеси добавляли 1,3-дибромпропан (1,758 г, 8,707 ммоля) и дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (1,100 г, 38,6%) в виде белого твердого вещества.
[Стадия 2] Синтез 4-бром-2-(1-карбоксициклобутил)бензойной кислоты

Метил-4-бром-2-(1-(метоксикарбонил)циклобутил)бензоат (1,100 г, 3,362 ммоля), полученный на стадии 1, и гидроксид калия (1,886 г, 33,622 ммоля) растворяли в смеси метанол (10 мл)/вода (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали 1 н. водный раствор хлористоводородной кислоты (20 мл) и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (0,840 г, 83,5%) в виде белого твердого вещества.
[Стадия 3] Синтез 6'-бром-1'H-спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-диона
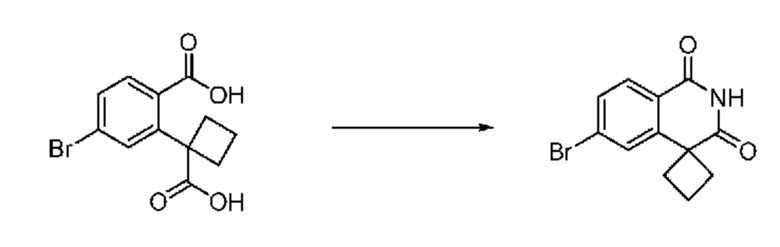
4-Бром-2-(1-карбоксициклобутил)бензойную кислоту (0,840 г, 2,808 ммоля), полученную на стадии 2, и мочевину (0,186 г, 3,089 ммоля) смешивали в N, N-диметилформамиде (10 мл), затем облучали микроволновым излучением, затем нагревали при 150°C в течение 45 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (0,700 г, 89,0%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 106
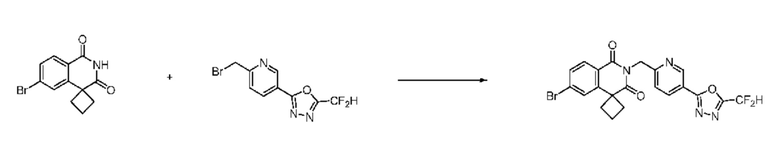
6'-Бром-1'H-спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-дион (0,500 г, 1,785 ммоля), полученный на стадии 3, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,518 г, 1,785 ммоля) и карбонат калия (0,370 г, 2,677 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 90°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,440 г, 50,4%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=2,1 Hz, 1H), 8,36 (dd, J=8,2, 2,2 Hz, 1H), 8,09 (d, J=8,4 Hz, 1H), 8,00 ~ 7,98 (m, 1H), 7,62 (dd, J=8,4, 1,8 Hz, 1H), 7,48 (d, J=8,2 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,45 (s, 2H), 3,06 ~ 2,99 (m, 2H), 2,55 ~ 2,45 (m, 2H), 2,44 ~ 2,29 (m, 2H).
Синтез соединения 107, 6'-бром-2'-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1'H-спиро[циклогексан-1,4'-изохинолин]-1',3'(2'H)-дион
[Стадия 1] Синтез метил-4-бром-2-(1-(метоксикарбонил)циклогексил)бензоата
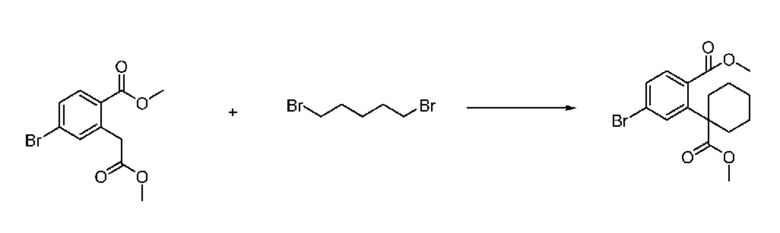
Метил-4-бром-2-(2-метокси-2-оксоэтил)бензоат (2,500 г, 8,707 ммоля) растворяли в N, N-диметилформамиде (30 мл) при 0°C, затем гидрид натрия (60,00%, 1,045 г, 26,122 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 30 мин. 1,5-дибромпентан (2,002 г, 8,707 ммоля) к реакционной смеси добавляли, и дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (1,000 г, 32,3%) в виде бесцветного масла.
[Стадия 2] Синтез 4-бром-2-(1-карбоксициклогексил)бензойной кислоты
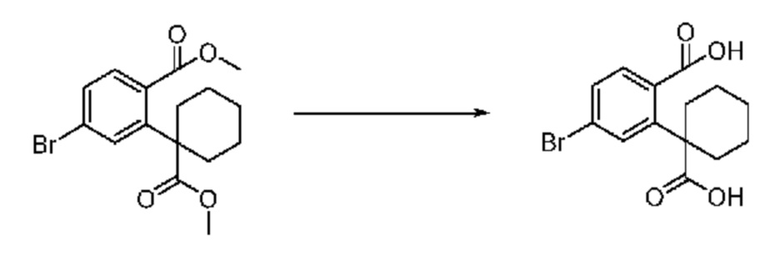
Метил-4-бром-2-(1-(метоксикарбонил)циклогексил)бензоат (1,000 г, 2,815 ммоля), полученный на стадии 1, и гидроксид калия (1,579 г, 28,151 ммоля) растворяли в смеси метанол (10 мл)/вода (10 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали 1 н. водный раствор хлористоводородной кислоты (20 мл) и перемешивали, затем осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (0,894 г, 97,1%) в виде белого твердого вещества.
[Стадия 3] Синтез 6'-бром-1'H-спиро[циклогексан-1,4'-изохинолин]-1',3'(2'H)-диона
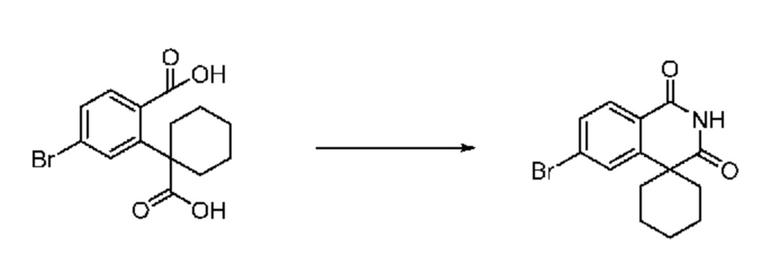
4-Бром-2-(1-карбоксициклогексил)бензойную кислоту (0,890 г, 2,720 ммоля), полученную на стадии 2, и мочевину (0,180 г, 2,992 ммоля) смешивали в N, N-диметилформамиде (10 мл), затем облучали микроволновым излучением, затем нагревали при 150°C в течение 45 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Осадившееся твердое вещество отфильтровывали, затем промывали гексаном и затем сушили и получали искомое соединение (0,347 г, 41,4%) в виде белого твердого вещества.
[Стадия 4] Синтез соединения 107
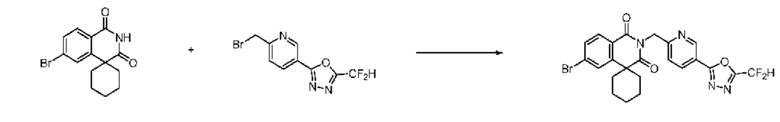
6'-Бром-1'H-спиро[циклогексан-1,4'-изохинолин]-1',3'(2'H)-дион (0,370 г, 1,201 ммоля), полученный на стадии 3, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,348 г, 1,201 ммоля) и карбонат калия (0,249 г, 1,801 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 90°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,200 г, 32,2%) в виде желтого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,18 ~ 9,17 (m, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 8,09 (d, J=8,4 Hz, 1H), 7,77 (d, J=1,8 Hz, 1H), 7,59 (dd, J=8,4, 1,8 Hz, 1H), 7,47 (dd, J=8,2, 0,5 Hz, 1H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 5,37 (s, 2H), 2,17 ~ 2,14 (m, 2H), 2,07 ~ 1,80 (m, 6H), 1,79 ~ 1,66 (m, 2H).
Синтез соединения 108, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(3-фторфенил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 108
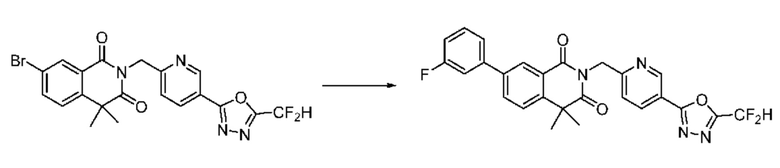
7-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), (3-фторфенил)бороновую кислоту (0,035 г, 0,251 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,007 г, 0,010 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) смешивали в смеси 1,4-диоксан (1,5 мл)/вода (0,5 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 0 до 40%) и концентрировали и получали искомое соединение (0,066 г, 64,0%) в виде светло-коричневого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=1,3 Hz, 1H), 8,47 (d, J=1,8 Hz, 1H), 8,35 (dd, J=8,2, 2,1 Hz, 1H), 7,89 (dd, J=8,2, 2,0 Hz, 1H), 7,62 (d, J=8,2 Hz, 1H), 7,50 ~ 7,43 (m, 3H), 7,34 (d, J=10,1 Hz, 1H), 7,11 ~ 6,81 (m, 2H), 5,47 (s, 2H), 1,75 (s, 6H).; LRMS (ES) m/z 493,3 (M+ + 1).
Синтез соединения 109, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(2-фторфенил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 109
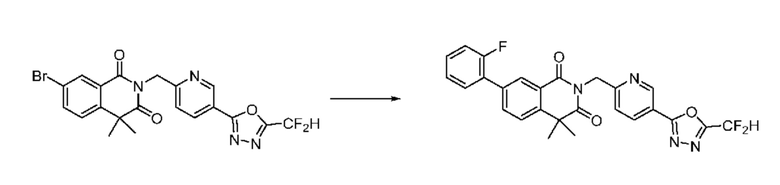
7-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), (2-фторфенил)бороновую кислоту (0,035 г, 0,251 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,007 г, 0,010 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) смешивали в смеси 1,4-диоксан (1,5 мл)/вода (0,5 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 0 до 40%) и концентрировали и получали искомое соединение (0,057 г, 55,2%) в виде светло-коричневого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=1,8 Hz, 1H), 8,43 (s, 1H), 8,35 ~ 8,33 (m, 1H), 7,91 ~ 7,88 (m, 1H), 7,61 (d, J=8,2 Hz, 1H), 7,52 ~ 7,46 (m, 2H), 7,38 ~ 7,35 (m, 1H), 7,28 ~ 7,16 (m, 2H), 7,07 ~ 6,81 (m, 1H), 5,46 (s, 2H), 1,75 (s, 6H).; LRMS (ES) m/z 493,3 (M+ + 1).
Синтез соединения 110, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиридин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 110
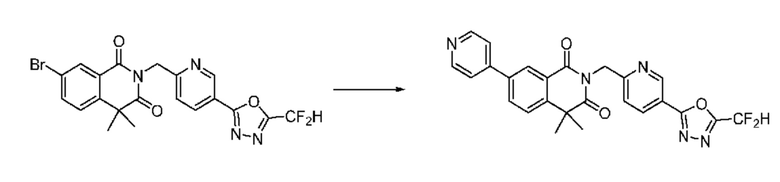
7-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), пиридин-4-илбороновую кислоту (0,031 г, 0,251 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,007 г, 0,010 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) смешивали в смеси 1,4-диоксан (1,5 мл)/вода (0,5 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 10 до 60%) и концентрировали и получали искомое соединение (0,047 г, 47,2%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,18 (d, J=2,0 Hz, 1H), 8,72 (d, J=4,6 Hz, 2H), 8,55 (d, J=2,0 Hz, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 7,96 (dd, J=8,2, 2,1 Hz, 1H), 7,67 (d, J=8,2 Hz, 1H), 7,59 (d, J=4,9 Hz, 2H), 7,49 (d, J=8,2 Hz, 1H), 7,06 ~ 6,80 (m, 1H), 5,47 (s, 2H), 1,75 (s, 6H).; LRMS (ES) m/z 476,2 (M+ + 1).
Синтез соединения 111, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиридин-3-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 111
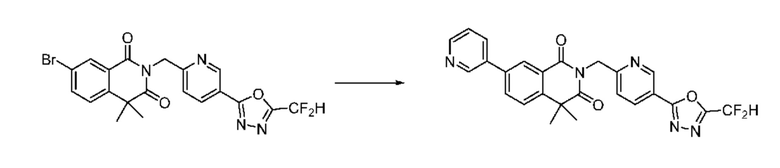
7-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), пиридин-3-илбороновую кислоту (0,031 г, 0,251 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,007 г, 0,010 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) смешивали в смеси 1,4-диоксан (1,5 мл)/вода (0,5 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 10 до 60%) и концентрировали и получали искомое соединение (0,042 г, 42,2%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,17 (d, J=2,0 Hz, 1H), 8,90 (d, J=1,3 Hz, 1H), 8,64 (d, J=4,1 Hz, 1H), 8,47 (d, J=2,0 Hz, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 7,96 ~ 7,89 (m, 2H), 7,65 (d, J=8,2 Hz, 1H), 7,48 (d, J=8,2 Hz, 1H), 7,43 ~ 7,39 (m, 1H), 7,06 ~ 6,80 (m, 1H), 5,45 (s, 2H), 1,74 (s, 6H).; LRMS (ES) m/z 476,4 (M+ + 1).
Синтез соединения 112, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(фуран-3-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 112
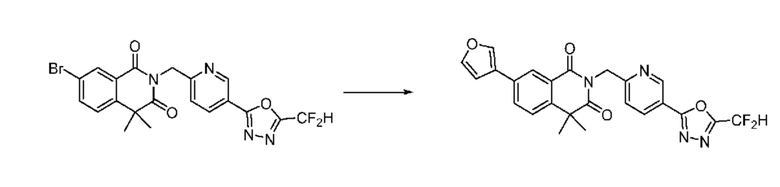
7-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), фуран-2-илбороновую кислоту (0,028 г, 0,251 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,007 г, 0,010 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) смешивали в смеси 1,4-диоксан (1,5 мл)/вода (0,5 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 0 до 40%) и концентрировали и получали искомое соединение (0,050 г, 51,4%) в виде коричневого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=1,8 Hz, 1H), 8,37 ~ 8,34 (m, 2H), 7,84 (s, 1H), 7,80 (dd, J=8,2, 2,0 Hz, 1H), 7,55 ~ 7,52 (m, 2H), 7,47 (d, J=8,2 Hz, 1H), 7,06 ~ 6,78 (m, 2H), 5,46 (s, 2H), 1,72 (s, 6H).; LRMS (ES) m/z 465,2 (M+ + 1).
Синтез соединения 113, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(фуран-2-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 113
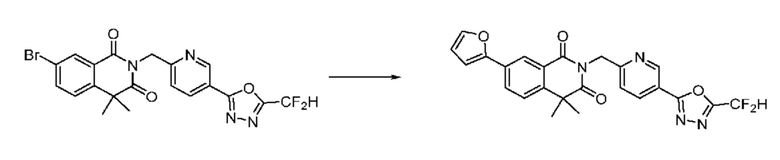
7-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), фуран-3-илбороновую кислоту (0,028 г, 0,251 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,007 г, 0,010 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) смешивали в смеси 1,4-диоксан (1,5 мл)/вода (0,5 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 0 до 40%) и концентрировали и получали искомое соединение (0,050 г, 51,4%) в виде светло-коричневого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (d, J=1,7 Hz, 1H), 8,52 (d, J=1,9 Hz, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 7,98 (dd, J=8,3, 2,0 Hz, 1H), 7,56 ~ 7,46 (m, 2H), 7,47 (d, J=8,2 Hz, 1H), 7,06 ~ 6,78 (m, 2H), 6,53 ~ 6,52 (m, 1H), 5,46 (s, 2H), 1,72 (s, 6H).; LRMS (ES) m/z 465,3 (M+ + 1).
Синтез соединения 114, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(5-метилфуран-2-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 114
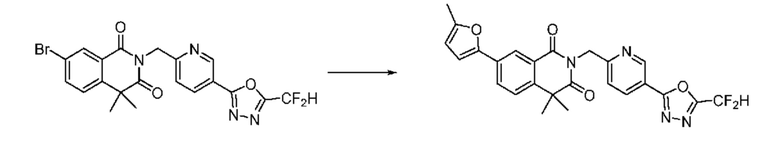
7-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан (0,052 г, 0,251 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,007 г, 0,010 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) смешивали в смеси 1,4-диоксан (1,5 мл)/вода (0,5 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 0 до 40%) и концентрировали и получали искомое соединение (0,053 г, 52,9%) в виде светло-коричневого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=1,7 Hz, 1H), 8,46 (d, J=1,9 Hz, 1H), 8,35 (dd, J=8,2, 2,1 Hz, 1H), 7,92 (dd, J=8,3, 2,0 Hz, 1H), 7,51 (d, J=8,3 Hz, 1H), 7,47 (d, J=8,2 Hz, 1H), 7,06 ~ 6,80 (m, 1H), 6,67 (d, J=3,2 Hz, 1H), 6,10 ~ 6,09 (m, 1H), 5,46 (s, 2H), 2,39 (s, 3H), 1,71 (s, 6H).; LRMS (ES) m/z 479,2 (M+ + 1).
Синтез соединения 115, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(1H-индол-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 115
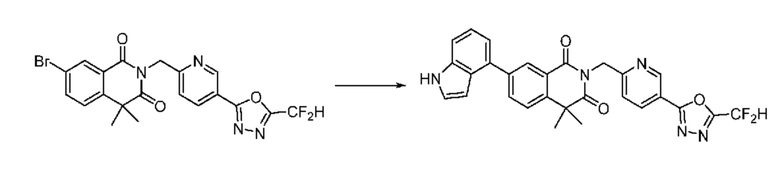
7-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,210 ммоля), (1H-индол-4-ил)бороновую кислоту (0,040 г, 0,251 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,007 г, 0,010 ммоля) и карбонат цезия (0,205 г, 0,629 ммоля) смешивали в смеси 1,4-диоксан (1,5 мл)/вода (0,5 мл) при комнатной температуре, затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,045 г, 41,8%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,22 (d, J=1,7 Hz, 1H), 8,62 (d, J=1,9 Hz, 1H), 8,49 (brs, 1H), 8,34 (dd, J=8,2, 2,1 Hz, 1H), 8,05 (dd, J=8,2, 2,0 Hz, 1H), 7,65 (d, J=8,2 Hz, 1H), 7,48 ~ 7,44 (m, 2H), 7,32 ~ 7,24 (m, 3H), 7,06 ~ 6,80 (m, 1H), 6,75 ~ 6,74 (m, 1H), 5,49 (s, 2H), 1,79 (s, 6H).; LRMS (ES) m/z 514,3 (M+ + 1).
Синтез соединения 116, трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-7-ил)пиперазин-1-карбоксилат
[Стадия 1] Синтез трет-бутил-4-(4-(1-метокси-2-метил-1-оксопропан-2-ил)-3-(метоксикарбонил)фенил)пиперазин-1-карбоксилата
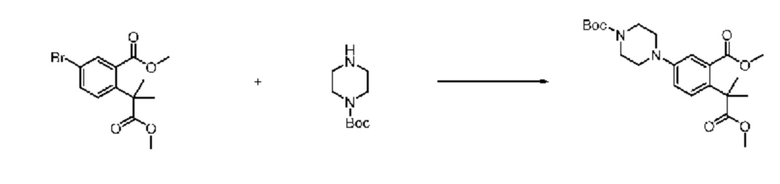
Метил-5-бром-2-(1-метокси-2-метил-1-оксопропан-2-ил)бензоат (4,990 г, 15,833 ммоля), трет-бутилпиперазин-1-карбоксилат (3,834 г, 20,583 ммоля), бис(три-трет-бутилфосфин)палладий (0, 0,809 г, 1,583 ммоля) и карбонат цезия (12,897 г, 39,583 ммоля) растворяли в толуоле (20 мл) при 100°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 80 г картридж; этилацетат/дихлорметан=от 0 до 30%) и концентрировали и получали искомое соединение (2,020 г, 30,3%) в виде желтого твердого вещества.
[Стадия 2] Синтез 5-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-2-(2-карбоксипропан-2-ил)бензойной кислоты
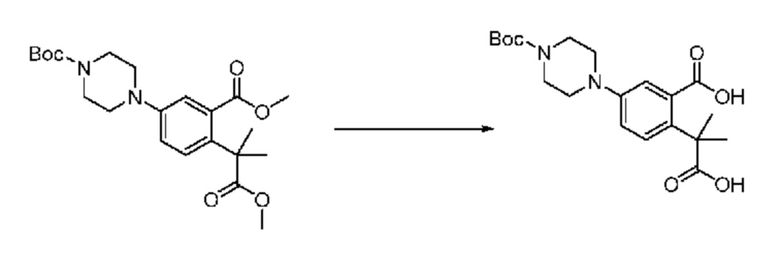
Трет-бутил-4-(4-(1-метокси-2-метил-1-оксопропан-2-ил)-3-(метоксикарбонил)фенил)пиперазин-1-карбоксилат (2,000 г, 4,756 ммоля), полученный на стадии 1, и гидроксид калия (2,668 г, 47,561 ммоля) растворяли в смеси метанол (30 мл)/вода (30 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали 1 н. водный раствор хлористоводородной кислоты и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (1,500 г, 80,4%, белое твердое вещество).
[Стадия 3] Синтез трет-бутил-4-(4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-7-ил)пиперазин-1-карбоксилата
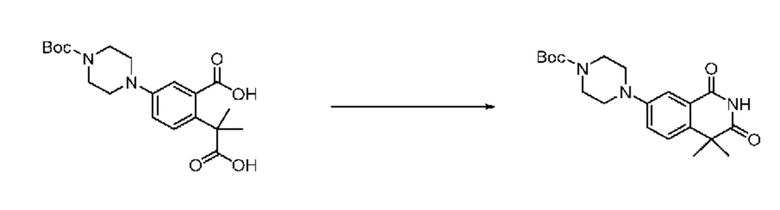
5-(4-(Трет-бутоксикарбонил)пиперазин-1-ил)-2-(2-карбоксипропан-2-ил)бензойную кислоту (1,500 г, 3,822 ммоля), полученную на стадии 2, и мочевину (0,253 г, 4,204 ммоля) растворяли в N, N-диметилформамиде (20 мл), затем полученный раствор перемешивали при 150°C в течение 18 ч, затем дополнительно перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 30%) и концентрировали и получали искомое соединение (0,530 г, 37,1%) в виде желтого твердого вещества.
[Стадия 4] Синтез соединения 116
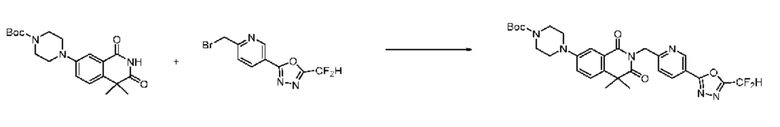
Трет-бутил-4-(4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-7-ил)пиперазин-1-карбоксилат (0,420 г, 1,125 ммоля), полученный на стадии 3, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,359 г, 1,237 ммоля) и карбонат калия (0,311 г, 2,249 ммоля) растворяли в N, N-диметилформамиде (10 мл) при 90°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,400 г, 61,0%) в виде желтого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,8 Hz, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 7,73 (d, J=2,8 Hz, 1H), 7,45 ~ 7,40 (m, 2H), 7,28 ~ 7,27 (m, 1H), 7,07 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,45 (s, 2H), 3,62 ~ 3,59 (m, 4H), 3,24 ~ 3,22 (m, 4H), 1,66 (s, 6H), 1,50 (s, 9H).
Синтез соединения 117, 2'-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6'-(4-этилпиперазин-1-ил)-1'H-спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-дион
[Стадия 1] Синтез соединения 117
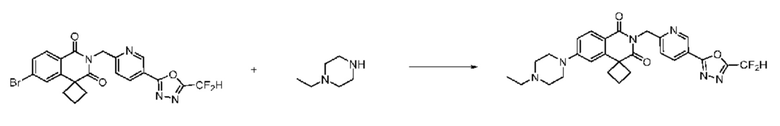
6'-Бром-2'-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1'H-спиро[циклобутан-1,4'-изохинолин]-1',3'(2'H)-дион (0,138 г, 0,282 ммоля), полученный на стадии 4, соединение 106, 1-этилпиперазин (0,064 г, 0,564 ммоля), ацетат палладия (II, 0,006 г, 0,028 ммоля), ruphos (0,013 г, 0,028 ммоля) и карбонат калия (0,230 г, 0,705 ммоля) растворяли в толуоле (10 мл) при 100°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,020 г, 13,6%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,21 (d, J=1,8 Hz, 1H), 8,32 (dd, J=8,2, 2,3 Hz, 1H), 8,08 (d, J=8,9 Hz, 1H), 7,42 (d, J=8,2 Hz, 1H), 7,19 (d, J=2,3 Hz, 1H), 7,06 (s, 0,25H), 6,95 (dd, J=9,7, 3,0 Hz, 1H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 3,51 ~ 3,48 (m, 4H), 3,03 ~ 2,96 (m, 2H), 2,68 ~ 2,63 (m, 4H), 2,55 ~ 2,21 (m, 6H), 1,17 ~ 1,13 (m, 3H).; LRMS (ES) m/z 523,3 (M+ + 1).
Синтез соединения 118, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(4-метилпиперазин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетата
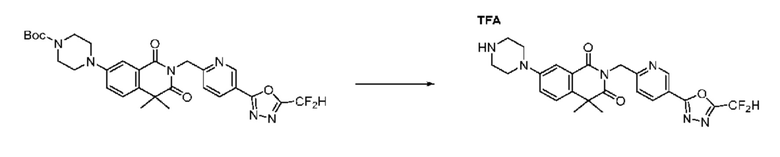
Трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-7-ил)пиперазин-1-карбоксилат (0,400 г, 0,687 ммоля) и трифторуксусную кислоту (0,526 мл, 6,866 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем полученный продукт использовали без проведения дополнительной очистки (0,400 г, 97,7%, желтое масло).
[Стадия 2] Синтез соединения 118
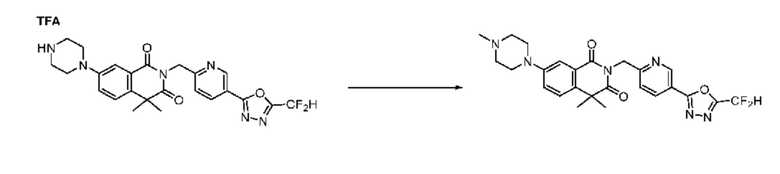
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,200 г, 0,335 ммоля), полученный на стадии 1, формальдегид (0,020 г, 0,671 ммоля), триацетоксиборогидрид натрия (0,142 г, 0,671 ммоля) и N, N-диизопропилэтиламин (0,058 мл, 0,335 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,110 г, 66,1%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,18 (d, J=2,1 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 7,71 (d, J=2,8 Hz, 1H), 7,43 ~ 7,37 (m, 2H), 7,25 (dd, J=8,7, 2,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 3,30 (t, J=5,0 Hz, 4H), 2,61 (t, J=5,0 Hz, 4H), 2,36 (s, 3H), 1,64 (s, 6H).; LRMS (ES) m/z 497,4 (M+ + 1).
Синтез соединения 119, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(4-изопропилпиперазин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 119
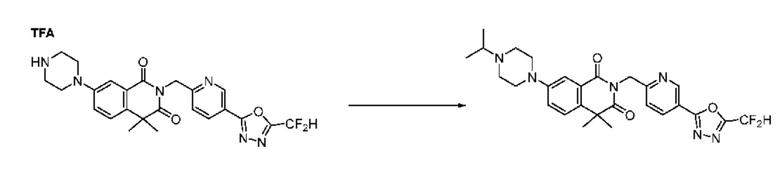
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион-2,2,2-трифторацетат (0,200 г, 0,335 ммоля), ацетон (0,039 г, 0,671 ммоля), триацетоксиборогидрид натрия (0,142 г, 0,671 ммоля) и N, N-диизопропилэтиламин (0,058 мл, 0,335 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,130 г, 73,9%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 ~ 9,19 (m, 1H), 8,33 (dd, J=8,2, 2,3 Hz, 1H), 7,72 (d, J=2,8 Hz, 1H), 7,44 ~ 7,38 (m, 2H), 7,26 (dd, J=8,7, 2,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 3,32 (t, J=5,0 Hz, 4H), 2,81 ~ 2,78 (m, 1H), 2,75 (t, J=5,0 Hz, 4H), 1,65 (s, 6H), 1,13 (d, J=6,5 Hz, 6H).; LRMS (ES) m/z 525,4 (M+ + 1).
Синтез соединения 120, трет-бутил-4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метил-2,4-диоксо-1,2,3,4-тетрагидрохиназолин-7-ил)-3,6-дигидропиридин-1(2H)-карбоксилат
[Стадия 1] Синтез соединения 120
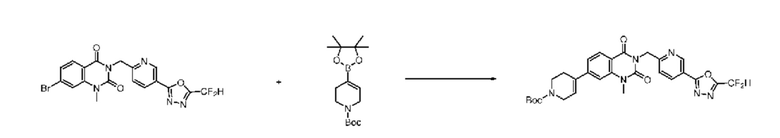
7-Бром-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метилхиназолин-2,4(1H,3H)-дион (0,729 г, 1,570 ммоля), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,728 г, 2,356 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,102 г, 0,157 ммоля) и карбонат цезия (0,767 г, 2,356 ммоля) смешивали в смеси 1,4-диоксан (9 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 80%) и концентрировали и получали искомое соединение (0,700 г, 78,7%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,24 ~ 9,20 (m, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 8,21 (d, J=8,3 Hz, 1H), 7,50 (dd, J=8,2, 0,8 Hz, 1H), 7,35 ~ 7,31 (m, 1H), 7,24 (d, J=2,2 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,25 ~ 6,20 (m, 1H), 5,53 (s, 2H), 4,16 ~ 4,11 (m, 2H), 3,70 ~ 3,65 (m, 2H), 2,62 ~ 2,58 (m, 2H), 1,63 (s, 3H), 1,52 (s, 9H).
Синтез соединения 121, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(3,6-дигидро-2H-тиопиран-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 121
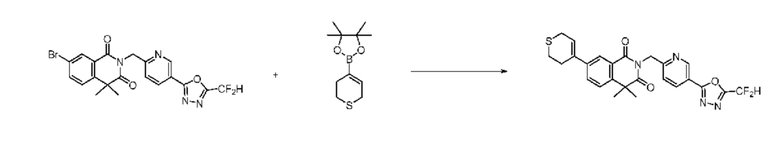
7-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (1,000 г, 2,095 ммоля), 2-(3,6-дигидро-2H-тиопиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,711 г, 3,143 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,137 г, 0,210 ммоля) и карбонат цезия (1,024 г, 3,143 ммоля) смешивали в смеси 1,4-диоксан (9 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 20 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 70%) и концентрировали и получали искомое соединение (0,840 г, 80,7%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,20 (d, J=1,4 Hz, 1H), 8,34 ~ 8,33 (m, 1H), 8,22 (d, J=2,1 Hz, 1H), 7,70 ~ 7,63 (m, 1H), 7,50 ~ 7,47 (m, 2H), 7,03 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,40 ~ 6,35 (m, 1H), 5,44 (s, 2H), 3,38 ~ 3,37 (m, 2H), 2,92 ~ 2,90 (m, 2H), 2,80 ~ 2,75 (m, 2H), 1,70 (s, 6H).; LRMS (ES) m/z 497,0 (M+ + 1).
Синтез соединения 122, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метил-7-(1,2,3,6-тетрагидропиридин-4-ил)хиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 122
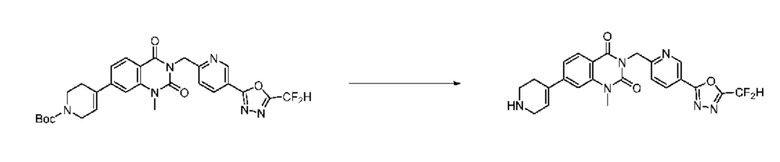
Трет-бутил-4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метил-2,4-диоксо-1,2,3,4-тетрагидрохиназолин-7-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,720 г, 1,271 ммоля) и трифторуксусную кислоту (0,973 мл, 12,708 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (0,700 г, 94,9%, белое твердое вещество).
LRMS (ES) m/z 467,3 (M+ + 1).
Синтез соединения 123, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(1-оксидо-3,6-дигидро-2H-тиопиран-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 123

2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(3,6-дигидро-2H-тиопиран-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,730 г, 1,470 ммоля) и 3-хлорпербензойную кислоту (77,00%, 0,329 г, 1,470 ммоля) растворяли в дихлорметане (10 мл) при 0°C, затем полученный раствор перемешивали при такой же температуре в течение 1 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 70%) и концентрировали и получали искомое соединение (0,300 г, 39,8%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,1, 0,7 Hz, 1H), 8,36 (dd, J=8,2, 2,2 Hz, 1H), 8,27 (d, J=2,0 Hz, 1H), 7,71 (dd, J=8,3, 2,2 Hz, 1H), 7,53 (d, J=8,3 Hz, 1H), 7,48 (dd, J=8,2, 0,7 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,07 ~ 6,05 (m, 1H), 5,45 (s, 2H), 3,63 ~ 3,54 (m, 2H), 3,30 ~ 3,20 (m, 2H), 3,00 ~ 2,97 (m, 1H), 2,85 ~ 2,80 (m, 1H), 1,71 (s, 6H).; LRMS (ES) m/z 513,3 (M+ + 1).
Синтез соединения 124, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(1-изопропил-1,2,3,6-тетрагидропиридин-4-ил)-1-метилхиназолин-2,4(1H,3H)-дион
[Стадия 1] Синтез соединения 124
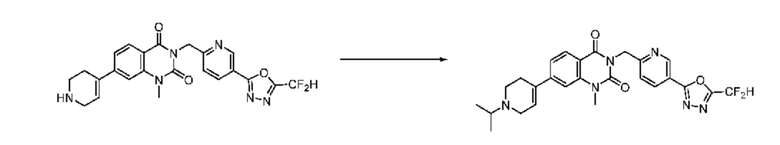
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-метил-7-(1,2,3,6-тетрагидропиридин-4-ил)хиназолин-2,4(1H,3H)-дион-2,2,2-трифторацетат (0,450 г, 0,775 ммоля), ацетон (0,090 г, 1,550 ммоля), триацетоксиборогидрид натрия (0,329 г, 1,550 ммоля) и N, N-диизопропилэтиламин (0,135 мл, 0,775 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,200 г, 50,7%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,21 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,18 (d, J=8,3 Hz, 1H), 7,49 (dd, J=8,3, 0,8 Hz, 1H), 7,32 (dd, J=8,3, 1,5 Hz, 1H), 7,25 (d, J=38,7 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 6,28 ~ 6,27 (m, 1H), 5,51 (s, 2H), 3,65 (s, 3H), 3,48 ~ 3,46 (m, 2H), 3,12 ~ 3,09 (m, 1H), 2,98 ~ 2,95 (m, 2H), 2,74 ~ 2,72 (m, 2H), 1,22 (d, J=6,6 Hz, 6H).; LRMS (ES) m/z 509,4 (M+ + 1).
Синтез соединения 125, N-(4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-7-ил)-1-оксидо-3,6-дигидро-2H-1λ6-тиопиран-1-илиден)-2,2,2-трифторацетамид
[Стадия 1] Синтез соединения 125
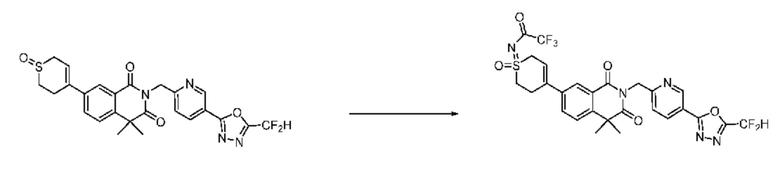
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(1-оксидо-3,6-дигидро-2H-тиопиран-4-ил)изохинолин-1,3(2H,4H)-дион (0,157 г, 0,306 ммоля), 2,2,2-трифторацетамид (0,069 г, 0,613 ммоля), йодбензолдиацетат (0,148 г, 0,459 ммоля), оксид магния (0,049 г, 1,225 ммоля) и димер ацетата родия(II) (0,014 г, 0,031 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,100 г, 52,4%) в виде фиолетового масла.
1H NMR (400 MHz, CDCl3) δ 9,20 (s, 1H), 8,38 (dd, J=8,2, 2,2 Hz, 1H), 8,26 (d, J=2,1 Hz, 1H), 7,68 (dd, J=8,3, 2,2 Hz, 1H), 7,57 (d, J=8,2 Hz, 1H), 7,49 (dd, J=8,3, 0,7 Hz, 1H), 7,07 (s, 0,25H), 6,94 (s, 0,5H), 6,81 (s, 0,25H), 6,05 ~ 6,03 (m, 2H), 5,46 (s, 2H), 4,58 ~ 4,56 (m, 1H), 4,22 ~ 4,19 (m, 1H), 3,84 ~ 3,82 (m, 1H), 3,68 ~ 3,64 (m, 1H), 3,28 ~ 3,26 (m, 2H), 1,76 (s, 6H).; LRMS (ES) m/z 624,3 (M+ + 1).
Синтез соединения 126, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(1-имино-1-оксидо-1,2,3,6-тетрагидро-1λ6-тиопиран-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 126
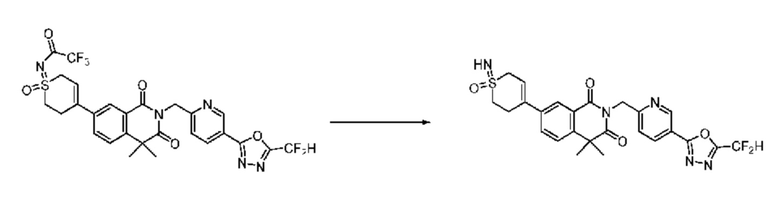
N-(4-(2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-7-ил)-1-оксидо-3,6-дигидро-2H-1λ6-тиопиран-1-илиден)-2,2,2-трифторацетамид (0,100 г, 0,160 ммоля) и карбонат калия (0,066 г, 0,481 ммоля) растворяли в метаноле (5 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 3 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,010 г, 11,8%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,33 ~ 9,31 (m, 1H), 8,49 ~ 8,45 (m, 1H), 8,31 ~ 8,22 (m, 1H), 7,74 ~ 7,69 (m, 1H), 7,56 ~ 7,42 (m, 2H), 7,17 (s, 1H), 7,07 (s, 1H), 6,92 (s, 1H), 6,08 ~ 6,07 (m, 1H), 5,56 (s, 2H), 4,30 ~ 4,25 (m, 1H), 4,05 ~ 4,01 (m, 1H), 3,94 (s, 1H), 3,71 ~ 3,67 (m, 1H), 3,50 ~ 3,47 (m, 1H), 3,26 ~ 3,22 (m, 2H), 1,68 (s, 6H).; LRMS (ES) m/z 528,22 (M+ + 1).
Синтез соединения 127, 7-(1-ацетилпиперидин-4-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 127
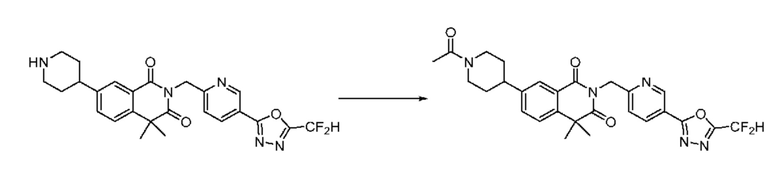
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,208 ммоля) и триэтиламин (0,058 мл, 0,415 ммоля) растворяли в дихлорметане (4 мл) при 0°C, затем уксусный ангидрид (0,029 мл, 0,312 ммоля) добавляли к полученному раствору и перемешивали при комнатной температуре в течение 18 ч. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 40 до 90%) и концентрировали и получали искомое соединение (0,042 г, 38,6%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=1,6 Hz, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 8,10 (d, J=1,8 Hz, 1H), 7,54 ~ 7,45 (m, 3H), 7,06 ~ 6,81 (m, 1H), 5,44 (s, 2H), 4,83 (d, J=11,4 Hz, 1H), 3,98 (d, J=11,7 Hz, 1H), 3,21 (td, J=13,0, 2,2 Hz, 1H), 2,90 ~ 2,84 (m, 1H), 2,70 ~ 2,63 (m, 1H), 2,16 (s, 3H), 1,95 (t, J=14,7 Hz, 2H), 1,73 ~ 1,66 (m, 8H).; LRMS (ES) m/z 524,4 (M+ + 1).
Синтез соединения 128, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(1-(метилсульфонил)пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 128
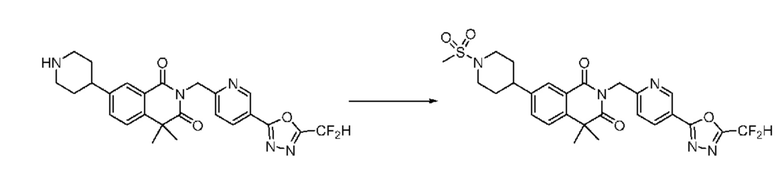
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,208 ммоля) и триэтиламин (0,058 мл, 0,415 ммоля) растворяли в дихлорметане (4 мл) при 0°C, затем метансульфонилхлорид (0,024 мл, 0,312 ммоля) добавляли к полученному раствору и перемешивали при комнатной температуре в течение 18 ч. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 30 до 70%) и концентрировали и получали искомое соединение (0,036 г, 31,0%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=1,6 Hz, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 8,11 (d, J=1,9 Hz, 1H), 7,56 ~ 7,46 (m, 3H), 7,06 ~ 6,81 (m, 1H), 5,45 (s, 2H), 3,99 (d, J=11,9 Hz, 2H), 2,85 ~ 2,72 (m, 6H), 2,03 ~ 2,00 (m, 2H), 1,95 ~ 1,88 (m, 2H), 1,70 (s, 6H).; LRMS (ES) m/z 560,4 (M+ + 1).
Синтез соединения 129, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(4-этилпиперазин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 129
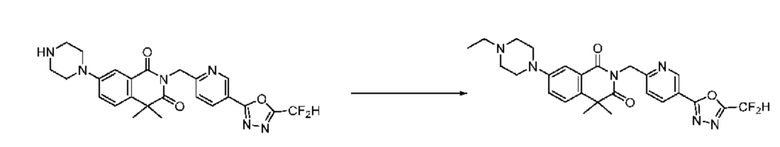
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,116 г, 0,240 ммоля), ацетальдегид (0,021 г, 0,481 ммоля) и триацетоксиборогидрид натрия (0,102 г, 0,481 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,060 г, 48,9%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,18 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 7,71 (d, J=2,8 Hz, 1H), 7,43 ~ 7,37 (m, 2H), 7,25 (dd, J=8,7, 2,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 3,33 (t, J=5,1 Hz, 4H), 2,70 (t, J=5,1 Hz, 4H), 2,56 ~ 2,54 (m, 2H), 1,16 (t, J=7,2 Hz, 3H).;LRMS (ES) m/z 511,3 (M+ + 1).
Синтез соединения 130, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(4-propylpiperazine-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 130
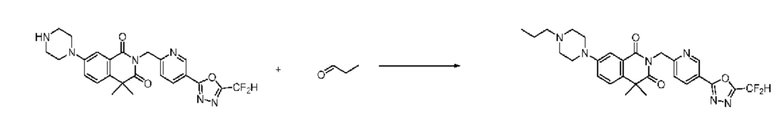
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), пропионовый альдегид (0,024 г, 0,415 ммоля) и триацетоксиборогидрид натрия (0,088 г, 0,415 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,050 г, 46,0%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (dd, J=2,2, 0,6 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 7,71 (d, J=2,8 Hz, 1H), 7,44 ~ 7,38 (m, 2H), 7,25 (dd, J=8,7, 2,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 3,32 (t, J=5,1 Hz, 4H), 2,68 (t, J=5,0 Hz, 4H), 2,40 ~ 2,40 (m, 2H), 1,66 (s, 6H), 1,65 ~ 1,57 (m, 2H), 0,94 (t, J=7,4 Hz, 3H).; LRMS (ES) m/z 525,5 (M+ + 1).
Синтез соединения 131, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(4-изобутилпиперазин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 131
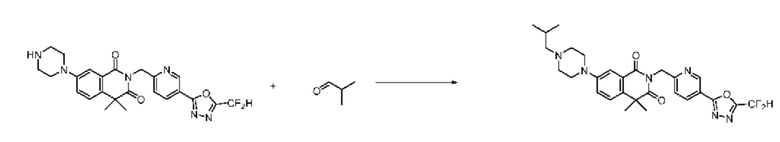
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), изомасляный альдегид (0,030 г, 0,415 ммоля) и триацетоксиборогидрид натрия (0,088 г, 0,415 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,060 г, 53,7%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 7,71 (d, J=2,8 Hz, 1H), 7,43 ~ 7,37 (m, 2H), 7,25 (dd, J=8,8, 2,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,42 (s, 2H), 3,28 (t, J=5,0 Hz, 4H), 2,58 (t, J=5,0 Hz, 4H), 2,17 ~ 2,15 (m, 2H), 1,90 ~ 1,85 (m, 1H), 1,66 (s, 6H), 0,94 ~ 0,91 (m, 6H).; LRMS (ES) m/z 539,5 (M+ + 1).
Синтез соединения 132, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(4-изопентилпиперазин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 132
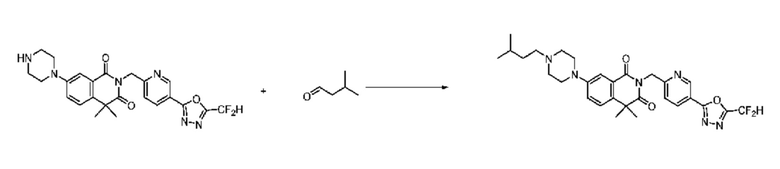
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), 3-метилбутаналь (0,036 г, 0,415 ммоля) и триацетоксиборогидрид натрия (0,088 г, 0,415 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,060 г, 52,4%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,18 (d, J=2,2 Hz, 1H), 8,32 (dd, J=8,2, 2,3 Hz, 1H), 7,70 (d, J=2,8 Hz, 1H), 7,43 ~ 7,37 (m, 2H), 7,24 (dd, J=8,7, 2,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (s, 0,5H), 6,80 (s, 0,25H), 5,41 (s, 2H), 3,33 (t, J=5,0 Hz, 4H), 2,73 (t, J=5,0 Hz, 4H), 2,51 ~ 2,47 (m, 2H), 1,66 (s, 6H), 1,48 ~ 1,46 (m, 2H), 0,94 ~ 0,91 (m, 6H).; LRMS (ES) m/z 553,4(M+ + 1).
Синтез соединения 133, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(4-(2,2,2-трифторэтил)пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 133
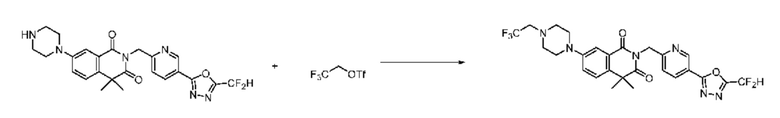
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,130 г, 0,269 ммоля), 2,2,2-трифторэтилтрифторметансульфонат (0,081 г, 0,350 ммоля) и карбонат калия (0,074 г, 0,539 ммоля) растворяли в ацетонитриле (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,100 г, 65,7%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,8 Hz, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 7,73 (d, J=2,8 Hz, 1H), 7,45 ~ 7,40 (m, 2H), 7,27 ~ 7,25 (m, 1H), 7,06 (s, 1H), 6,93 (s, 1H), 6,80 (s, 1H), 5,43 (s, 2H), 3,32 (t, J=5,0 Hz, 4H), 3,07 (dd, J=19,1, 9,5 Hz, 2H), 2,88 (t, J=5,0 Hz, 4H), 1,67 (s, 6H).; LRMS (ES) m/z 565,5 (M+ + 1).
Синтез соединения 134, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(1-(2-гидроксиацетил)пиперидин-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 134
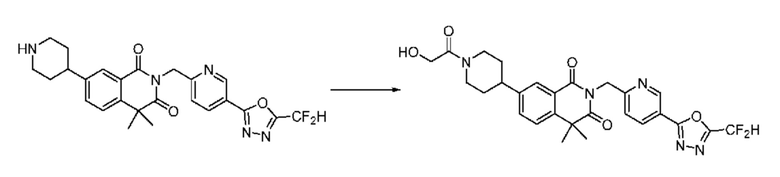
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,208 ммоля), 2-гидроксиуксусную кислоту (0,032 г, 0,415 ммоля), 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний 3-оксид гексафторфосфат (HATU, 0,158 г, 0,415 ммоля) и N, N-диизопропилэтиламин (0,181 мл, 1,038 ммоля) растворяли в N, N-диметилформамиде (4 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 30 до 80%) и концентрировали и получали продукт, затем полученный продукт повторно очищали с помощью хроматографии (пластина с SiO2, 20×20×1 мм; этилацетат=100%) и концентрировали и получали искомое соединение (0,036 г, 32,1%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=1,6 Hz, 1H), 8,35 (dd, J=8,2, 2,2 Hz, 1H), 8,10 (d, J=1,8 Hz, 1H), 7,54 ~ 7,46 (m, 3H), 7,06 ~ 6,81 (m, 1H), 5,44 (s, 2H), 4,80 (d, J=11,4 Hz, 1H), 4,24 ~ 4,15 (m, 2H), 3,76 ~ 3,64 (m, 2H), 3,16 (td, J=13,1, 2,3 Hz, 1H), 2,94 ~ 2,80 (m, 2H), 1,99 (d, J=12,8 Hz, 2H), 1,77 ~ 1,66 (m, 8H).; LRMS (ES) m/z 540,5 (M+ + 1).
Синтез соединения 135, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(1-(2,2,2-трифторэтил)пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 135
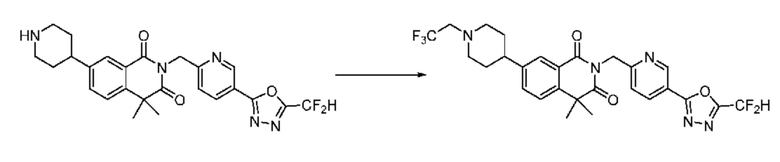
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,208 ммоля), 2,2,2-трифторэтилтрифторметансульфонат (0,072 г, 0,312 ммоля) и N, N-диизопропилэтиламин (0,109 мл, 0,623 ммоля) растворяли в дихлорметане (4 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. Насыщенный водный раствор хлорида натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 10 до 50%) и концентрировали и получали искомое соединение (0,032 г, 27,3%) в виде бесцветного масла.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=1,8 Hz, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,12 (d, J=1,9 Hz, 1H), 7,56 (dd, J=8,2, 2,0 Hz, 1H), 7,48 ~ 7,44 (m, 2H), 7,06 ~ 6,80 (m, 1H), 5,44 (s, 2H), 3,12 (d, J=11,6 Hz, 2H), 3,05 (q, J=9,7 Hz, 2H), 2,62 ~ 2,61 (m, 1H), 2,56 ~ 2,49 (m, 2H), 1,90 ~ 1,85 (m, 4H), 1,69 (s, 6H).; LRMS (ES) m/z 564,5 (M+ + 1).
Синтез соединения 136, 6-(4-ацетилпиперазин-1-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диэтилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез метил-4-бром-2-(3-(метоксикарбонил)пентан-3-ил)бензоата
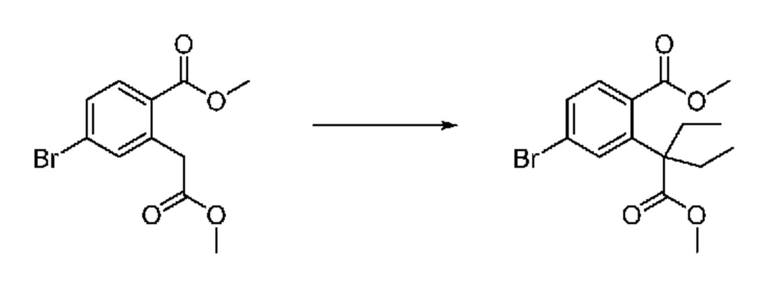
Метил-4-бром-2-(2-метокси-2-оксоэтил)бензоат (3,000 г, 10,449 ммоля) и гидрид натрия (60,00%, 1,672 г, 41,796 ммоля) растворяли в N, N-диметилформамиде (150 мл) при 0°C, затем йодэтан (3,360 мл, 41,796 ммоля) добавляли к полученному раствору и перемешивали при комнатной температуре в течение 18 ч. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом магния, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 10%) и концентрировали и получали искомое соединение (2,800 г, 78,1%) в виде белого твердого вещества.
[Стадия 2] Синтез 4-бром-2-(3-карбоксипентан-3-ил)бензойной кислоты
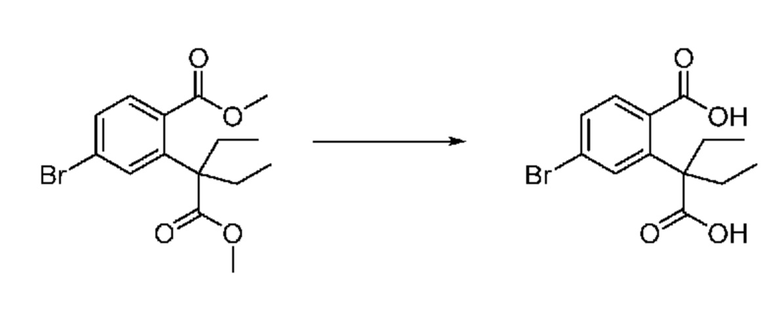
Метил-4-бром-2-(3-(метоксикарбонил)пентан-3-ил)бензоат (2,800 г, 8,158 ммоля), полученный на стадии 1, и гидроксид калия (4,577 г, 81,580 ммоля) растворяли в смеси метанол (25 мл)/вода (50 мл) при комнатной температуре, затем полученный раствор перемешивали при 100°C в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В полученную реакционную смесь выливали 1 н. водный раствор хлористоводородной кислоты и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом магния, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (2,550 г, 99,2%, белое твердое вещество).
[Стадия 3] Синтез 6-бром-4,4-диэтилизохинолин-1,3(2H,4H)-диона
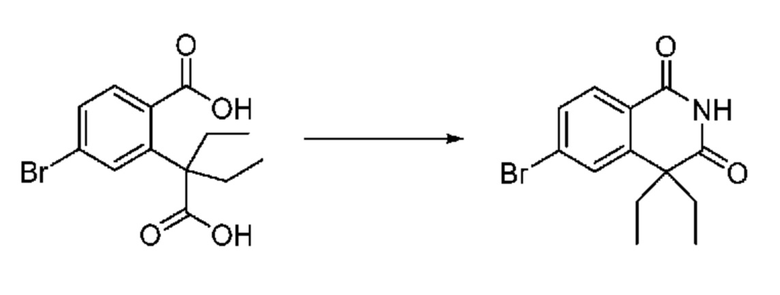
4-Бром-2-(3-карбоксипентан-3-ил)бензойную кислоту (2,550 г, 8,091 ммоля), полученную на стадии 2, и мочевину (0,486 г, 8,091 ммоля) растворяли в N, N-диметилформамиде (150 мл) при комнатной температуре, затем полученный раствор перемешивали при 150°C в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 10%) и концентрировали и получали искомое соединение (0,301 г, 12,6%) в виде белого твердого вещества.
[Стадия 4] Синтез N-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-N-(3,4-дифторфенил)-4-метилпиперазин-1-карбоксамида
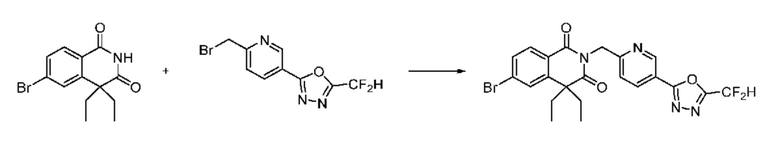
6-Бром-4,4-диэтилизохинолин-1,3(2H,4H)-дион (0,300 г, 1,013 ммоля), полученный на стадии 3, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,353 г, 1,216 ммоля), карбонат калия (0,420 г, 3,039 ммоля) и йодид калия (0,017 г, 0,101 ммоля) растворяли в N, N-диметилформамиде (5 мл) при комнатной температуре, затем полученный раствор перемешивали при 100°C в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 20%) и концентрировали и получали искомое соединение (0,419 г, 81,9%) в виде светло-желтого твердого вещества.
[Стадия 5] Синтез соединения 136
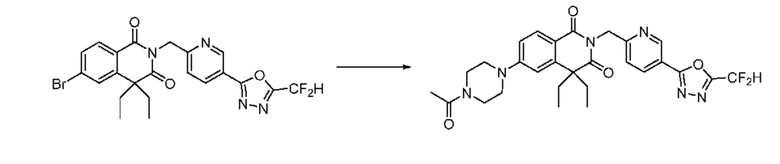
N-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-N-(3,4-дифторфенил)-4-метилпиперазин-1-карбоксамид (0,100 г, 0,198 ммоля), полученный на стадии 4, 1-ацетилпиперазин (0,028 мл, 0,237 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,018 г, 0,020 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,011 г, 0,020 ммоля) и карбонат цезия (0,129 г, 0,396 ммоля) растворяли в 1,4-диоксане (4 мл) при комнатной температуре, затем полученный раствор перемешивали при 100°C в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; этилацетат/гексан=от 60 до 100%) и концентрировали и получали искомое соединение (0,034 г, 31,1%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,19 (d, J=1,6 Hz, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,17 (d, J=8,9 Hz, 1H), 7,48 (d, J=8,2 Hz, 1H), 7,06 ~ 6,80 (m, 2H), 6,73 (d, J=2,4 Hz, 1H), 5,44 (s, 2H), 3,84 (t, J=5,3 Hz, 2H), 3,71 (t, J=5,2 Hz, 2H), 3,46 (t, J=5,2 Hz, 2H), 3,41 (t, J=5,3 Hz, 2H), 2,38 ~ 2,32 (m, 2H), 2,18 (s, 3H), 1,92 ~ 1,87 (m, 2H), 0,64 (t, J=7,4 Hz, 6H).; LRMS (ES) m/z 553,5 (M+ + 1).
Синтез соединения 137, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4-(2,2,3,3-тетрафторпропил)пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 137
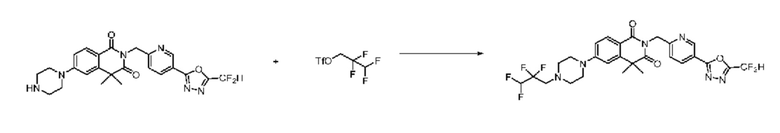
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), 2,2,3,3-тетрафторпропилтрифторметансульфонат (0,071 г, 0,269 ммоля) и карбонат калия (0,057 г, 0,415 ммоля) растворяли в ацетонитриле (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,060 г, 48,5%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,21 (dd, J=2,2, 0,7 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,13 (d, J=8,9 Hz, 1H), 7,44 ~ 7,41 (m, 1H), 7,06 (s, 0,25H), 6,95 ~ 6,92 (m, 1H), 6,93 (s, 0,5H), 6,85 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 6,18 (t, J=4,7 Hz, 0,25H), 6,04 (t, J=4,9 Hz, 0,5H), 5,91 (t, J=4,9 Hz, 0,25H), 5,42 (s, 2H), 3,42 (t, J=5,1 Hz, 4H), 3,03 (t, J=14,1 Hz, 2H), 2,86 (t, J=5,0 Hz, 4H), 1,69 (s, 6H).; LRMS (ES) m/z 597,5 (M+ + 1).
Синтез соединения 138, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(4-(2,2-дифторпропил)пиперазин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 138
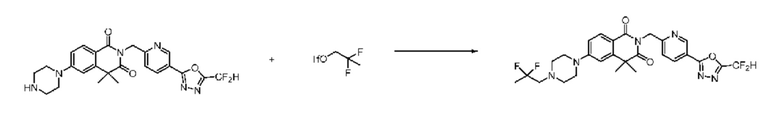
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), 2,2-дифторпропилтрифторметансульфонат (0,057 г, 0,249 ммоля) и карбонат калия (0,057 г, 0,415 ммоля) растворяли в ацетонитриле (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,050 г, 43,0%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,7 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,11 (d, J=8,9 Hz, 1H), 7,42 (dd, J=8,2, 0,6 Hz, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,91 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,41 (s, 2H), 3,42 (t, J=5,1 Hz, 4H), 2,81 ~ 2,74 (m, 6H), 1,75 ~ 1,65 (m, 9H).
Синтез соединения 139, 6-(4-(2,2-дифторбутил)пиперазин-1-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 139
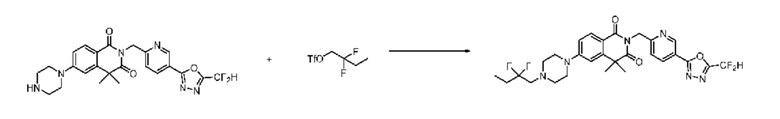
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), 2,2-дифторбутилтрифторметансульфонат (0,065 г, 0,269 ммоля) и карбонат калия (0,057 г, 0,415 ммоля) растворяли в ацетонитриле (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,050 г, 42,0%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,2, 2,3 Hz, 1H), 8,11 (d, J=8,9 Hz, 1H), 7,42 (dd, J=8,2, 0,8 Hz, 1H), 7,06 (s, 0,25H), 6,93 (dd, J=8,9, 2,5 Hz, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,41 (s, 2H), 3,42 (t, J=5,1 Hz, 4H), 2,81 ~ 2,74 (m, 6H), 2,05 ~ 1,99 (m, 2H), 1,68 (s, 6H), 1,06 (t, J=7,5 Hz, 3H).
Синтез соединения 140, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(4-(2,2,3,3,4,4,4-гептафторбутил)пиперазин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 140
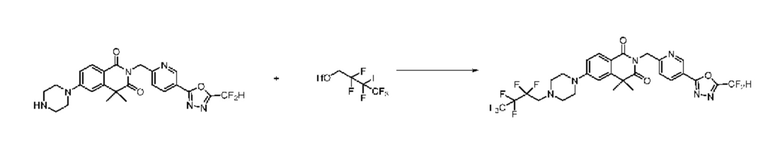
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), 2,2,3,3,4,4,4-гептафторбутилтрифторметансульфонат (0,089 г, 0,269 ммоля) и карбонат калия (0,057 г, 0,415 ммоля) растворяли в ацетонитриле (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,040 г, 29,0%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,21 (dd, J=2,2, 0,8 Hz, 1H), 8,33 (dd, J=8,2, 2,3 Hz, 1H), 8,12 (d, J=8,9 Hz, 1H), 7,42 (dd, J=8,3, 0,8 Hz, 1H), 7,06 (s, 0,25H), 6,94 (dd, J=8,5, 2,9 Hz, 1H), 6,93 (s, 0,5H), 6,85 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,42 (s, 2H), 3,43 (t, J=5,0 Hz, 4H), 3,14 (t, J=15,6 Hz, 2H), 2,88 (t, J=5,0 Hz, 4H), 1,68 (s, 6H).; LRMS (ES) m/z 665,4 (M+ + 1).
Синтез соединения 141, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4-(2,2,2-трифторэтил)пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 141
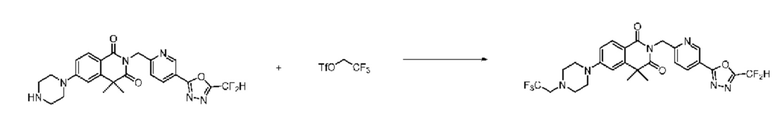
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), 2,2,2-трифторэтилтрифторметансульфонат (0,063 г, 0,269 ммоля) и карбонат калия (0,057 г, 0,415 ммоля) растворяли в ацетонитриле (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,070 г, 59,8%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (dd, J=2,2, 0,8 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,10 (d, J=8,9 Hz, 1H), 7,41 (dd, J=8,2, 0,7 Hz, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,91 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,40 (s, 2H), 3,43 (t, J=5,0 Hz, 4H), 3,11 ~ 3,03 (m, 1H), 2,87 (t, J=5,0 Hz, 4H), 1,67 (s, 6H).; LRMS (ES) m/z 564,52 (M+ + 1).
Синтез соединения 142, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диэтил-6-(4-этилпиперазин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 142
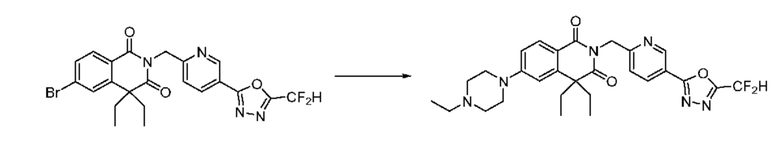
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диэтилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,198 ммоля), 1-этилпиперазин (0,027 г, 0,237 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,018 г, 0,020 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,011 г, 0,020 ммоля) и карбонат цезия (0,129 г, 0,396 ммоля) растворяли в 1,4-диоксане (3 мл) при комнатной температуре, затем полученный раствор перемешивали при 100°C в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем насыщенный водный раствор гидрокарбоната натрия выливали в полученный концентрат и проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; метанол/дихлорметан=от 0 до 5%) и концентрировали и получали продукт, затем полученный продукт повторно очищали с помощью хроматографии (пластина с SiO2, 20×20×1 мм; метанол/дихлорметан=5%) и концентрировали и получали искомое соединение (0,019 г, 17,8%) в виде розового твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,10 (d, J=1,6 Hz, 1H), 8,41 (dd, J=8,3, 2,1 Hz, 1H), 8,06 (d, J=9,0 Hz, 1H), 7,57 (d, J=8,3 Hz, 1H), 7,37 ~ 7,07 (m, 2H), 6,95 (d, J=2,0 Hz, 1H), 5,39 (s, 2H), 3,48 (t, J=4,9 Hz, 4H), 2,66 (t, J=4,8 Hz, 4H), 2,53 (q, J=7,2 Hz, 2H), 2,27 ~ 2,22 (m, 2H), 2,06 ~ 2,01 (m, 2H), 1,18 (t, J=7,2 Hz, 3H), 0,62 (t, J=7,3 Hz, 6H).; LRMS (ES) m/z 539,5 (M+ + 1).
Синтез соединения 143, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(1-propylpiperidine-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 143
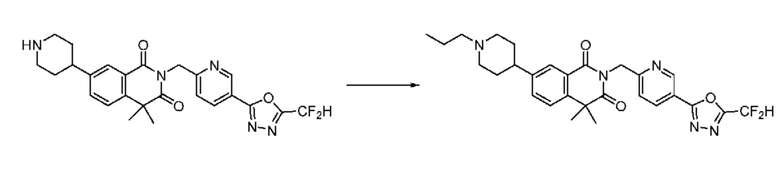
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,208 ммоля) и пропионовый альдегид (0,018 г, 0,312 ммоля) растворяли в дихлорметане (4 мл) при комнатной температуре, затем триацетоксиборогидрид натрия (0,088 г, 0,415 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 18 ч. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; метанол/дихлорметан=от 0 до 5%) и концентрировали и получали искомое соединение (0,042 г, 38,6%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,18 (s, 1H), 8,33 (d, J=8,2 Hz, 1H), 8,12 (s, 1H), 7,57 (d, J=8,1 Hz, 1H), 7,45 (t, J=7,7 Hz, 2H), 7,06 ~ 6,80 (m, 1H), 5,43 (s, 2H), 3,12 (d, J=11,0 Hz, 2H), 2,65 ~ 2,61 (m, 1H), 2,38 (t, J=7,7 Hz, 2H), 2,14 ~ 2,05 (m, 2H), 1,88 ~ 1,87 (m, 4H), 1,68 (s, 6H), 1,63 ~ 1,55 (m, 2H), 0,93 (t, J=7,3 Hz, 3H).; LRMS (ES) m/z 524,5 (M+ + 1).
Синтез соединения 144, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(1-isobutylpiperidine-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 144
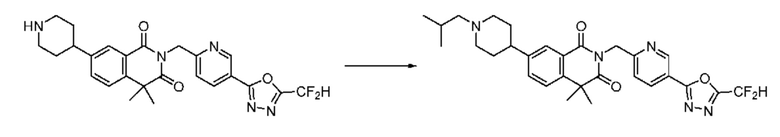
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,208 ммоля) и изомасляный альдегид (0,022 г, 0,312 ммоля) растворяли в дихлорметане (4 мл) при комнатной температуре, затем триацетоксиборогидрид натрия (0,088 г, 0,415 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 18 ч. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; метанол/дихлорметан=от 0 до 5%) и концентрировали и получали искомое соединение (0,055 г, 49,3%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,19 (s, 1H), 8,34 (d, J=8,2 Hz, 1H), 8,13 (s, 1H), 7,56 (d, J=8,0 Hz, 1H), 7,46 ~ 7,44 (m, 2H), 7,06 ~ 6,80 (m, 1H), 5,43 (s, 2H), 3,01 (d, J=10,6 Hz, 2H), 2,61 ~ 2,57 (m, 1H), 2,13 (d, J=7,0 Hz, 2H), 2,05 ~ 1,99 (m, 2H), 1,83 ~ 1,79 (m, 5H), 1,69 (s, 6H), 0,93 (d, J=6,1 Hz, 6H).; LRMS (ES) m/z 538,3 (M+ + 1).
Синтез соединения 145, 7-(1-циклобутилпиперидин-4-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 145
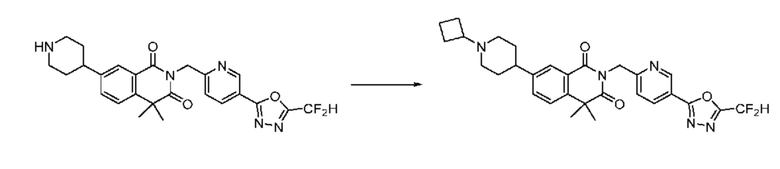
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,208 ммоля) и циклобутанон (0,016 г, 0,228 ммоля) растворяли в дихлорметане (4 мл) при комнатной температуре, затем триацетоксиборогидрид натрия (0,066 г, 0,312 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 18 ч. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; метанол/дихлорметан=от 0 до 5%) и концентрировали и получали искомое соединение (0,053 г, 47,6%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,17 (s, 1H), 8,32 (d, J=8,2 Hz, 1H), 8,12 (s, 1H), 7,56 (d, J=8,1 Hz, 1H), 7,44 (t, J=7,5 Hz, 2H), 7,05 ~ 6,79 (m, 1H), 5,42 (s, 2H), 3,04 ~ 3,03 (m, 2H), 2,77 ~ 2,73 (m, 1H), 2,60 ~ 2,59 (m, 1H), 2,07 ~ 2,05 (m, 2H), 1,95 ~ 1,69 (m, 10H), 1,67 (s, 6H).; LRMS (ES) m/z 536,3 (M+ + 1).
Синтез соединения 146, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(1-(тетрагидрофуран-3-ил)пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 146
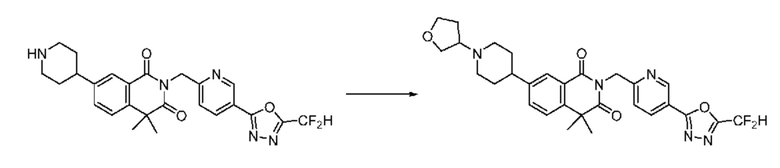
N-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-фторанилин (0,500 г, 1,482 ммоля) и дигидрофуран-3(2H)-он (0,191 г, 2,224 ммоля) растворяли в дихлорметане (4 мл) при комнатной температуре, затем триацетоксиборогидрид натрия (0,628 г, 2,965 ммоля) добавляли к полученному раствору и перемешивали при такой же температуре в течение 18 ч. Насыщенный водный раствор гидрокарбоната натрия выливали в реакционную смесь, затем проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; метанол/дихлорметан=от 0 до 5%) и концентрировали и получали искомое соединение (0,062 г, 7,6%) в виде белого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,16 (d, J=2,0 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,10 (d, J=1,8 Hz, 1H), 7,55 (dd, J=8,2, 1,9 Hz, 1H), 7,44 (t, J=8,7 Hz, 2H), 7,05 ~ 6,79 (m, 1H), 5,41 (s, 2H), 3,96 ~ 3,88 (m, 2H), 3,82 ~ 3,71 (m, 2H), 3,17 (d, J=11,3 Hz, 1H), 3,11 ~ 3,08 (m, 1H), 2,97 (d, J=12,2 Hz, 1H), 2,65 ~ 2,64 (m, 1H), 2,26 ~ 2,22 (m, 2H), 2,10 ~ 2,07 (m, 1H), 1,97 ~ 1,86 (m, 5H), 1,66 (s, 6H).; LRMS (ES) m/z 552,5 (M+ + 1).
Синтез соединения 147, 6-(4-бутилпиперазин-1-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 147
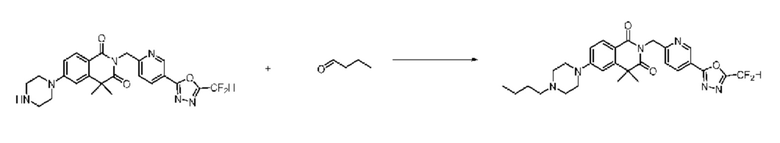
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), масляный альдегид (0,030 г, 0,415 ммоля) и триацетоксиборогидрид натрия (0,088 г, 0,415 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,060 г, 53,7%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,21 (dd, J=2,1, 0,6 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,11 (d, J=8,9 Hz, 1H), 7,41 (dd, J=8,3, 0,5 Hz, 1H), 7,06 (s, 0,25H), 6,95 ~ 6,92 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,42 (s, 2H), 3,45 ~ 3,43 (m, 4H), 2,65 ~ 2,63 (m, 4H), 2,44 (t, J=7,5 Hz, 2H), 1,68 (s, 6H), 1,57 ~ 1,54 (m, 2H), 1,41 ~ 1,36 (m, 2H), 0,98 ~ 0,95 (m, 3H).; LRMS (ES) m/z 539,5 (M+ + 1).
Синтез соединения 148, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(4-propylpiperazine-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 148
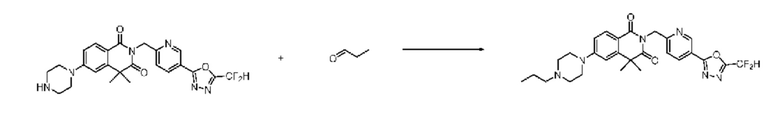
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), пропионовый альдегид (0,016 г, 0,269 ммоля) и триацетоксиборогидрид натрия (0,088 г, 0,415 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,050 г, 46,0%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (dd, J=2,2, 0,6 Hz, 1H), 8,32 (dd, J=8,2, 2,2 Hz, 1H), 8,11 (d, J=9,1 Hz, 1H), 7,41 (dd, J=8,2, 0,6 Hz, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,92 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,41 (s, 2H), 3,44 (t, J=5,0 Hz, 4H), 2,64 (t, J=4,8 Hz, 4H), 2,42 ~ 2,38 (m, 2H), 1,68 (s, 6H), 1,61 ~ 1,55 (m, 2H), 0,96 (t, J=7,4 Hz, 3H).; LRMS (ES) m/z 525,5 (M+ + 1).
Синтез соединения 149, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6-(4-изобутилпиперазин-1-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 149
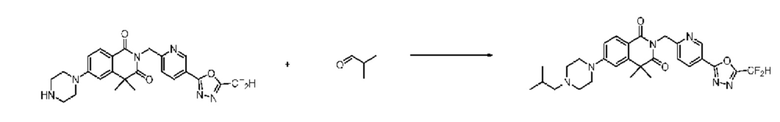
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), изомасляный альдегид (0,019 г, 0,269 ммоля) и триацетоксиборогидрид натрия (0,088 г, 0,415 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,050 г, 44,8%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,21 ~ 9,20 (m, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,11 ~ 8,09 (m, 1H), 7,41 (d, J=8,2 Hz, 1H), 7,06 (s, 0,25H), 6,94 ~ 6,92 (m, 1H), 6,93 (s, 0,5H), 6,84 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,43 (s, 2H), 3,43 ~ 3,40 (m, 4H), 2,60 ~ 2,55 (m, 4H), 2,18 ~ 2,16 (m, 2H), 1,86 ~ 1,81 (m, 1H), 1,68 (s, 6H), 0,98 ~ 0,96 (m, 6H).; LRMS (ES) m/z 539,5 (M+ + 1).
Синтез соединения 150, 6-(4-(4,4-дифторциклогексил)пиперазин-1-ил)-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 150
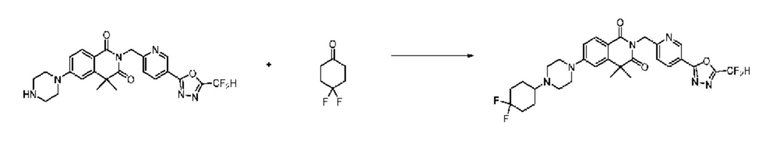
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-6-(пиперазин-1-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,207 ммоля), 4,4-дифторциклогексан-1-он (0,036 г, 0,269 ммоля) и триацетоксиборогидрид натрия (0,088 г, 0,415 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,090 г, 72,3%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,21 (d, J=1,5 Hz, 1H), 8,33 (dd, J=8,2, 2,2 Hz, 1H), 8,12 (d, J=8,8 Hz, 1H), 7,42 (d, J=8,3 Hz, 1H), 7,06 (s, 0,25H), 6,94 (dd, J=8,8, 2,5 Hz, 1H), 6,93 (s, 0,5H), 6,85 (d, J=2,4 Hz, 1H), 6,80 (s, 0,25H), 5,42 (s, 2H), 3,44 ~ 3,40 (m, 4H), 2,77 ~ 2,73 (m, 4H), 2,55 ~ 2,45 (m, 1H), 2,00 ~ 1,40 (m, 8H), 1,69 (s, 6H).; LRMS (ES) m/z 601,5 (M+ + 1).
Синтез соединения 151, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-7-(1-(2-метоксиэтил)пиперидин-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 151
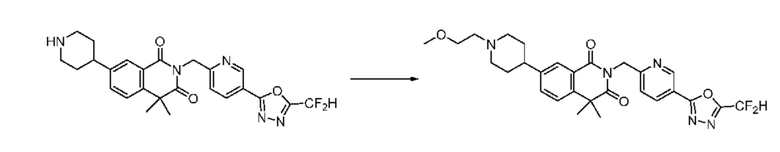
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4,4-диметил-7-(пиперидин-4-ил)изохинолин-1,3(2H,4H)-дион (0,100 г, 0,208 ммоля), 1-хлор-2-метоксиэтан (0,028 мл, 0,312 ммоля) и карбонат калия (0,057 г, 0,415 ммоля) растворяли в ацетонитриле (4 мл) при комнатной температуре, затем полученный раствор перемешивали при 80°C в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, затем в полученный концентрат выливали воду и проводили экстракцию дихлорметаном, затем фильтровали через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 4 г картридж; метанол/дихлорметан=от 0 до 5%) и концентрировали и получали продукт, затем полученный продукт повторно очищали с помощью хроматографии (пластина с SiO2, 20×20×1 мм; метанол/дихлорметан водный раствор=3%) и концентрировали и получали искомое соединение (0,010 г, 8,9%) в виде оранжевого твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,20 (d, J=2,1 Hz, 1H), 8,34 (dd, J=8,2, 2,2 Hz, 1H), 8,13 (d, J=2,0 Hz, 1H), 7,57 (dd, J=8,2, 1,9 Hz, 1H), 7,46 (t, J=8,0 Hz, 2H), 7,06 ~ 6,80 (m, 1H), 5,44 (s, 2H), 3,60 (t, J=5,6 Hz, 2H), 3,39 (s, 3H), 3,18 (d, J=11,4 Hz, 2H), 3,20 ~ 2,64 (m, 3H), 2,21 (t, J=10,6 Hz, 2H), 1,96 ~ 1,87 (m, 4H), 1,69 (s, 6H).; LRMS (ES) m/z 506,2 (M+ + 1).
Синтез соединения 152, 6-бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 152
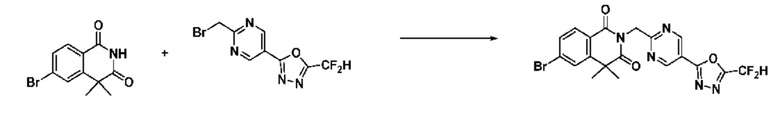
6-Бром-4,4-диметилизохинолин-1,3(2H,4H)-дион (1,700 г, 6,341 ммоля), 2-(2-(бромметил)пиримидин-5-ил)-5-(дифторметил)-1,3,4-оксадиазол (2,399 г, 8,243 ммоля) и карбонат калия (1,753 г, 12,681 ммоля) растворяли в N, N-диметилформамиде (20 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 40 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (1,900 г, 62,7%) в виде желтого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,31 (s, 2H), 8,13 (d, J=8,4 Hz, 1H), 7,70 (d, J=1,6 Hz, 1H), 7,63 (dd, J=8,4, 1,7 Hz, 1H), 7,08 (s, 0,25H), 6,95 (s, 0,5H), 6,82 (s, 0,25H), 5,55 (s, 2H), 1,73 (s, 6H).
Синтез соединения 153, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-4,4-диметил-6-(4-метилпиперазин-1-ил)изохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 153
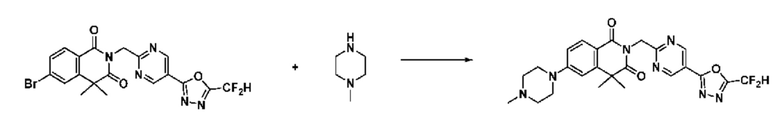
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,100 г, 0,209 ммоля), 1-метилпиперазин (0,047 мл, 0,418 ммоля), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,019 г, 0,021 ммоля), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (Xantphos, 0,012 г, 0,021 ммоля) и карбонат цезия (0,204 г, 0,627 ммоля) растворяли в толуоле (5 мл) при 80°C, затем полученный раствор перемешивали при такой же температуре в течение 18 ч и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,010 г, 9,4%) в виде коричневого масла.
1H NMR (400 MHz, CDCl3) δ 9,30 (s, 2H), 8,13 ~ 8,10 (m, 1H), 7,08 (s, 0,25H), 6,96 ~ 6,93 (m, 1H), 6,94 (s, 0,5H), 6,87 (d, J=2,4 Hz, 1H), 6,82 (s, 0,25H), 5,55 (s, 2H), 3,48 ~ 3,45 (m, 4H), 2,68 ~ 2,64 (m, 4H), 2,43 (s, 3H), 1,71 (s, 6H).; LRMS (ES) m/z 498,5 (M+ + 1).
Синтез соединения 154, трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-6-ил)-3,6-дигидропиридин-1(2H)-карбоксилат
[Стадия 1] Синтез соединения 154
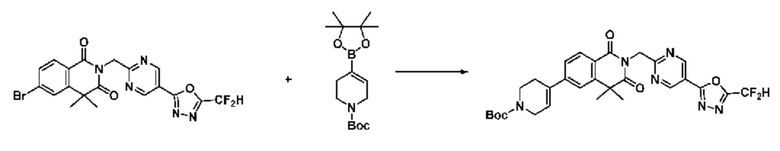
6-Бром-2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,800 г, 1,673 ммоля), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,672 г, 2,175 ммоля), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II)дихлорид (Pd(dtbpf)Cl2, 0,109 г, 0,167 ммоля) и карбонат цезия (0,818 г, 2,509 ммоля) смешивали в смеси 1,4-диоксан (9 мл)/вода (3 мл), затем полученную смесь облучали микроволновым излучением, затем нагревали при 100°C в течение 25 мин и затем реакцию завершали путем снижения температуры до комнатной температуры. В реакционную смесь выливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; этилацетат/гексан=от 0 до 50%) и концентрировали и получали искомое соединение (0,381 г, 39,2%) в виде желтого масла.
1H NMR (400 MHz, CDCl3) δ 9,30 (s, 2H), 8,22 (d, J=2,5 Hz, 1H), 7,49 ~ 7,43 (m, 2H), 7,08 (s, 0,25H), 6,95 (s, 0,5H), 6,82 (s, 0,25H), 6,22 (s, 1H), 5,55 (s, 2H), 4,15 ~ 4,09 (m, 2H), 3,70 ~ 3,66 (m, 2H), 2,59 (s, 2H), 1,72 (s, 6H), 1,50 (s, 9H).
Синтез соединения 155, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-6-(1-этил-1,2,3,6-тетрагидропиридин-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-4,4-диметил-6-(1,2,3,6-тетрагидропиридин-4-ил)изохинолин-1,3(2H,4H)-диона
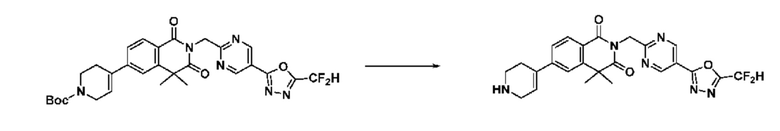
Трет-бутил-4-(2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-4,4-диметил-1,3-диоксо-1,2,3,4-тетрагидроизохинолин-6-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,381 г, 0,656 ммоля) и трифторуксусную кислоту (0,503 мл, 6,562 ммоля) растворяли в дихлорметане (10 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 5 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, затем насыщенный водный раствор гидрокарбоната натрия выливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный продукт использовали без проведения дополнительной очистки (0,241 г, 76,4%, желтое масло).
[Стадия 2] Синтез соединения 155
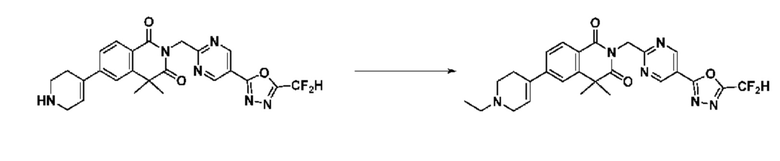
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-4,4-диметил-6-(1,2,3,6-тетрагидропиридин-4-ил)изохинолин-1,3(2H,4H)-дион (0,241 г, 0,502 ммоля), полученный на стадии 1, ацетальдегид (0,056 мл, 1,003 ммоля) и триацетоксиборогидрид натрия (0,213 г, 1,003 ммоля) растворяли в дихлорметане (20 мл) при комнатной температуре, затем полученный раствор перемешивали при такой же температуре в течение 18 ч. В реакционную смесь выливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, затем дегидратировали безводным сульфатом натрия, затем фильтровали и затем концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,150 г, 58,8%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,30 (s, 2H), 8,20 (d, J=8,2 Hz, 1H), 7,50 ~ 7,47 (m, 2H), 7,08 (s, 0,25H), 6,95 (s, 0,5H), 6,82 (s, 0,25H), 6,25 (s, 1H), 5,56 (s, 2H), 3,40 ~ 3,39 (m, 2H), 2,95 ~ 2,92 (m, 2H), 2,77 ~ 2,72 (m, 4H), 1,72 (s, 6H), 1,25 (t, J=7,2 Hz, 3H).
Синтез соединения 156, 2-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-6-(1-этилпиперидин-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион
[Стадия 1] Синтез соединения 156
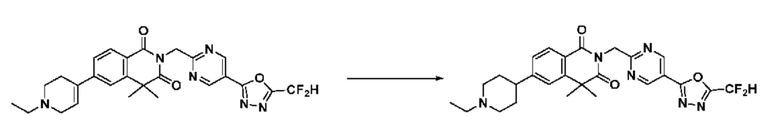
2-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиримидин-2-ил)метил)-6-(1-этил-1,2,3,6-тетрагидропиридин-4-ил)-4,4-диметилизохинолин-1,3(2H,4H)-дион (0,125 г, 0,246 ммоля) растворяли в метаноле (10 мл) при комнатной температуре, затем медленно добавляли 10%-Pd/C (10 мг) и перемешивали в течение 18 ч с подачей водорода из присоединенного баллона при такой же температуре. Реакционную смесь фильтровали через слой целита для удаления твердых веществ, затем растворитель удаляли из полученного фильтрата без твердых веществ при пониженном давлении. Затем полученный концентрат очищали с помощью колоночной хроматографии (SiO2, 12 г картридж; метанол/дихлорметан=от 0 до 10%) и концентрировали и получали искомое соединение (0,100 г, 79,7%) в виде белого вспененного твердого вещества.
1H NMR (400 MHz, CDCl3) δ 9,30 (s, 2H), 8,19 (d, J=8,1 Hz, 1H), 7,42 (d, J=1,4 Hz, 1H), 7,36 (dd, J=8,2, 1,5 Hz, 1H), 7,08 (s, 0,25H), 6,95 (s, 0,5H), 6,82 (s, 0,25H), 5,55 (s, 2H), 3,40 ~ 3,37 (m, 2H), 2,78 ~ 2,72 (m, 3H), 2,39 ~ 2,33 (m, 2H), 2,18 ~ 2,15 (m, 2H), 1,99 ~ 1,95 (m, 2H), 1,71 (s, 6H), 1,30 ~ 1,26 (m, 3H).; LRMS (ES) m/z 511,4 (M+ + 1).
Протокол для исследования и анализа активности соединения, предлагаемого в настоящем изобретении
<Пример 1> Выявление активности ингибирования фермента HDAC (in vitro)
Селективный ингибитор HDAC важен для селективности ингибирования HDAC1, который является причиной побочных эффектов, и поэтому выявлены селективность фермента HDAC1/6 и клеточная селективность (HDAC1: ацетилирование гистона/HDAC6: ацетилирование тубулина).
1. Экспериментальная методика
Способность исследуемого вещества ингибировать фермент HDAC определяли с помощью набора для исследования HDAC1 Fluorimetric Drug Discovery Assay Kit (Enzolifesciences: BML-AK511) и рекомбинантного HDAC6 человека (Calbiochem: 382180). Для исследования HDAC1 образцы обрабатывали при концентрациях 100, 1000 и 10000 нМ. Для исследования HDAC6 образцы обрабатывали при концентрациях 0,1, 1, 10, 100 и 1000 нМ. После такой обработки образцов реакцию проводили при 37°C в течение 60 мин, затем обрабатывали проявителем и затем вводили в реакцию при 37°C в течение 30 мин, затем интенсивность флуоресценции определяли (возбуждение при 390 нм, испускание при 460 нм) с помощью FlexStatin3 (Molecular device).
2. Результаты эксперимента
Его результаты приведены в следующей таблице 2.
Таблица 2. Результаты исследования активности ингибирования фермента HDAC
Как показано в приведенной выше таблице 2, из результатов исследования активности ингибирования HDAC1 и HDAC6, можно заключить, что гомофталимидные производные 1,3,4-оксадиазола, предлагаемые в настоящем изобретении, их стереоизомеры или их фармацевтически приемлемые соли обладают не только превосходной ингибирующей активностью по отношению к HDAC6, но и также превосходной селективной ингибирующей активностью по отношению к HDAC6 - HDAC1.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА, КАК ИНГИБИТОР ГИСТОНДЕЗАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2763936C1 |
| 1,3,4-ОКСАДИАЗОЛТИОКАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2022 |
|
RU2831346C2 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2822180C1 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛСУЛЬФАМИДА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2695227C1 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2810081C1 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛСУЛЬФОНАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ ДЕАЦЕТИЛАЗЫ ГИСТОНОВ 6 И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ | 2016 |
|
RU2697665C1 |
| 1,3,4,-ОКСАДИЗОЛАМИДНОЕ ПРОИЗВОДНОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2700696C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ | 2021 |
|
RU2817736C1 |
| ИНГИБИТОРЫ GCN2 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2811408C2 |
| ПРОИЗВОДНЫЕ ОКСАДИАЗОЛАМИНА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2695133C1 |
Группа изобретений относится к фармацевтической химии и включает соединение формулы (I), (II), конкретные соединения, указанные в формуле изобретения, их применение, а также фармацевтическую композицию и способ ингибирования на их основе. В формуле (I) X1 - X4 независимо означают CR0 или N, где по меньшей мере два из X1 - X4 означают CR0, R0 независимо означает H или галоген, R1 означает линейный или разветвленный -C1-5 галогеналкил, R2 и R3 все независимо означают H, галоген, 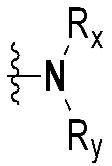 , 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3-7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
, 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3-7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,  ,
, 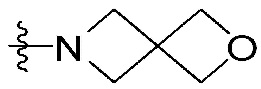 ,
, 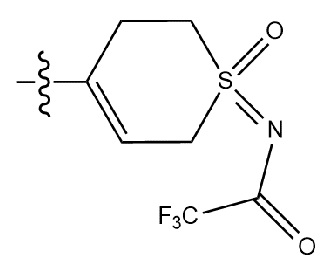 ,
, 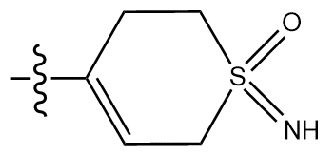 ,
, 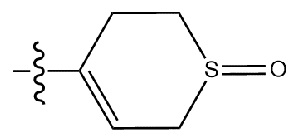 , фенил, индолил или
, фенил, индолил или  , K означает O или S, Y означает CRaRb, NRc или ординарную связь, Ra и Rb все независимо означают водород или -C1-7 алкил или Ra и Rb связаны друг с другом и образуют 3-7-членный циклоалкил, кольцо А означает фенилен, n равно 0 или 1, остальные заместители являются такими, как определено в формуле изобретения. В формуле (II) X1 - X4 независимо означают CR0 или N, где по меньшей мере два из X1 - X4 означают CR0, где R0 означает H, R1 означает линейный или разветвленный -C1-5 галогеналкил, R2 и R3 все независимо означают H, K означает O, Y означает NRc, Rc означает -C1-7 алкилфенил, -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, или -C1-7 алкил-O-C1-7 алкил, где один атом H группы -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, может быть замещен следующим заместителем: гетероарил-C1-5 галогеналкил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, кольцо А означает фенилен, n равно 0 или 1. Технический результат: производные 1,3,4-оксадиазола, обладающие свойствами высокоселективного ингибитора HDAC6. 7 н. и 5 з.п. ф-лы, 2 табл., 1 пр.
, K означает O или S, Y означает CRaRb, NRc или ординарную связь, Ra и Rb все независимо означают водород или -C1-7 алкил или Ra и Rb связаны друг с другом и образуют 3-7-членный циклоалкил, кольцо А означает фенилен, n равно 0 или 1, остальные заместители являются такими, как определено в формуле изобретения. В формуле (II) X1 - X4 независимо означают CR0 или N, где по меньшей мере два из X1 - X4 означают CR0, где R0 означает H, R1 означает линейный или разветвленный -C1-5 галогеналкил, R2 и R3 все независимо означают H, K означает O, Y означает NRc, Rc означает -C1-7 алкилфенил, -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, или -C1-7 алкил-O-C1-7 алкил, где один атом H группы -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, может быть замещен следующим заместителем: гетероарил-C1-5 галогеналкил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, кольцо А означает фенилен, n равно 0 или 1. Технический результат: производные 1,3,4-оксадиазола, обладающие свойствами высокоселективного ингибитора HDAC6. 7 н. и 5 з.п. ф-лы, 2 табл., 1 пр.
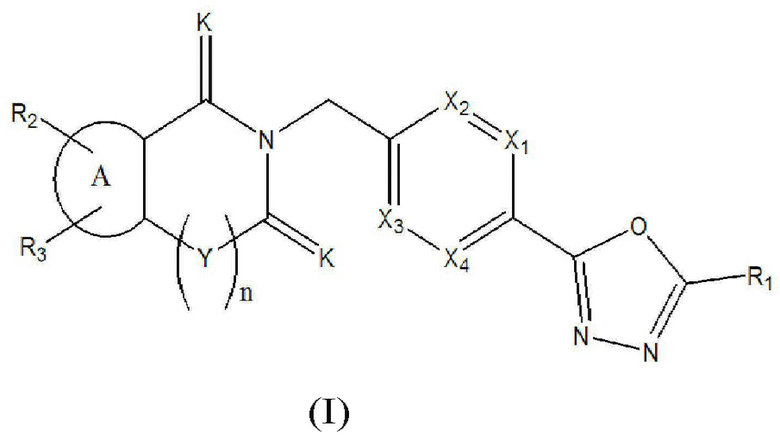
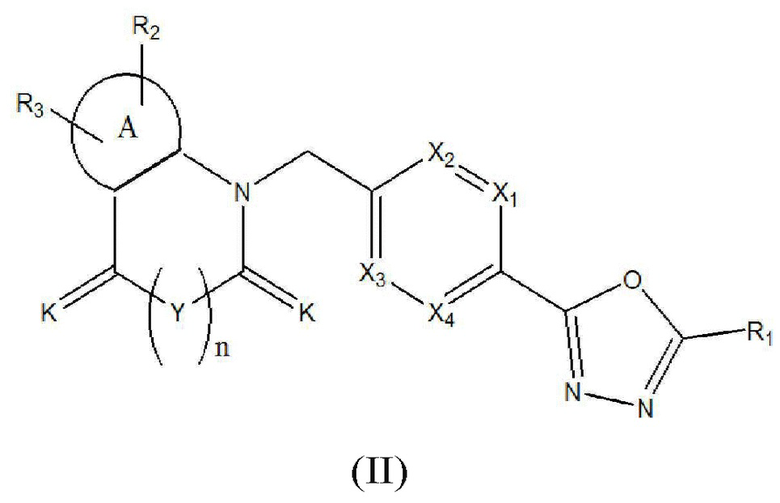
1. Соединение, описывающееся следующей химической формулой I, его стереоизомеры или его фармацевтически приемлемые соли:
[Химическая формула I]
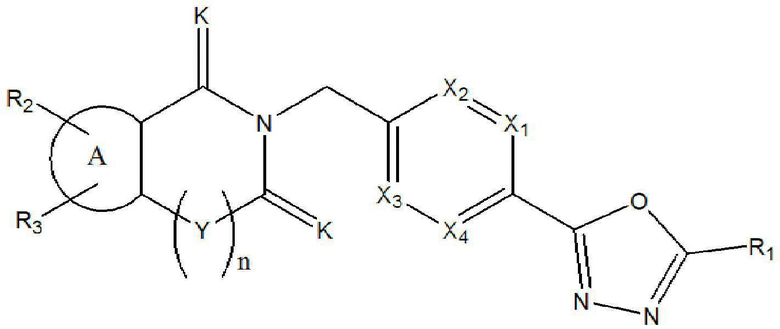 ,
,
где X1-X4 все независимо означают CR0 или N, где по меньшей мере два из X1-X4 означают CR0,
где каждый R0 независимо означает водород или галоген,
R1 означает линейный или разветвленный -C1-5 галогеналкил,
R2 и R3 все независимо означают H, галоген, 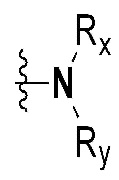 , 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3-7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
, 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3-7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 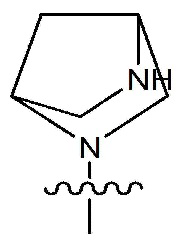 ,
, 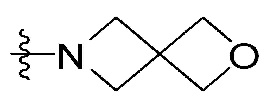 ,
, 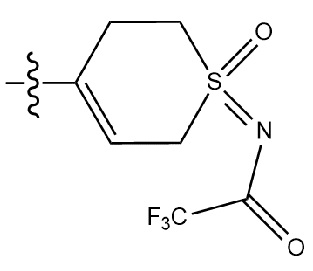 ,
, 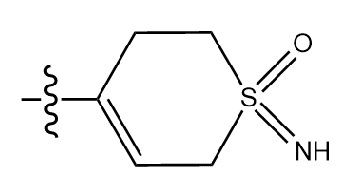 ,
, 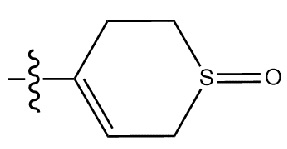 , фенил, индолил или
, фенил, индолил или 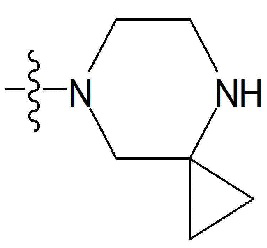 ,
,
где один и два атома водорода указанного 3-7-членного гетероциклоалкила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3-7-членного гетероциклоалкенила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , , , , , фенил, индолил или , может быть замещен с помощью R4,
R4 означает галоген, -C1-7 алкил, -C1-7 галогеналкил, -O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-OH, -C(=O)-O-C1-7 алкил, -S(=O)2-C1-7 алкил, 3-7-членный циклоалкил, 3-7-членный галогенциклоалкил, 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 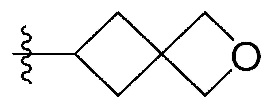 , -C1-7 алкил-C(=O)-R5, -C1-7 алкил-R7, -C1-7 алкил-O-R8, -NR9R10 или -C(=O)-NR11R12,
, -C1-7 алкил-C(=O)-R5, -C1-7 алкил-R7, -C1-7 алкил-O-R8, -NR9R10 или -C(=O)-NR11R12,
где R5 означает 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
R7 представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, или 3-7-членный циклоалкил,
R8 означает -C1-7 алкил,
R9 и R10 все независимо означают H или -C1-7 алкил и
R11 и R12 все независимо означают H или -C1-7 алкил,
Rx и Ry все независимо означают -C1-7 алкил или -C1-7 алкил-NR15R16,
где R15 и R16 все независимо означают -C1-7 алкил,
K означает O или S,
Y означает CRaRb, NRc или ординарную связь,
Ra и Rb все независимо означают водород или -C1-7 алкил или Ra и Rb связаны друг с другом и образуют 3-7-членный циклоалкил,
Rc означает водород, -C1-7 алкил, -C1-7 алкилгетероциклоалкил, в этом случае гетероциклоалкил представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкилфенил, -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкил-O-C1-7 алкил или -C1-7 алкил-NR19R20,
где один атом водорода группы -C1-7 алкил, -C1-7 алкилгетероциклоалкил, в этом случае гетероциклоалкил представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкилфенил или -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, может быть замещен следующим заместителем: -C1-7 алкил, -O-C1-7 алкил, 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C(=O)-O-C1-7 алкил или гетероарил-C1-5 галогеналкил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, и
R19 и R20 все независимо означают H или -C1-7 алкил,
 означает фенилен,
означает фенилен,
галоген представляет собой F, Cl, Br или I и
n равно 0 или 1.
2. Соединение, описывающееся химической формулой I, его стереоизомеры или его фармацевтически приемлемые соли по п. 1, где
X1-X4 все независимо означают CR0 или N, где по меньшей мере два из X1-X4 означают CR0,
где R0 означает водород или галоген,
R1 означает -C1-5 галогеналкил,
R2 и R3 все независимо означают H, галоген, , 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3-7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , , , , , фенил, индолил или ,
где один и два атома водорода указанного 3-7-членного гетероциклоалкила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3-7-членного гетероциклоалкенила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , , , , , фенила, индолила или , может быть замещен с помощью R4,
R4 означает галоген, -C1-7 алкил, -C1-7 галогеналкил, -O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-OH, -C(=O)-O-C1-7 алкил, -S(=O)2-C1-7 алкил, 3-7-членный циклоалкил, 3-7-членный галогенциклоалкил, 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , -C1-7 алкил-C(=O)-R5, -C1-7 алкил-R7, -C1-7 алкил-O-R8, -NR9R10 или -C(=O)-NR11R12,
где R5 означает 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
R7 представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, или 3-7-членный циклоалкил,
R8 означает -C1-7 алкил,
R9 и R10 все независимо означают -C1-7 алкил и
R11 и R12 все независимо означают H или -C1-7 алкил,
Rx и Ry все независимо означают -C1-7 алкил или -C1-7 алкил-NR15R16,
где R15 и R16 все независимо означают -C1-7 алкил,
K означает O или S,
Y означает CRaRb, NRc или ординарную связь,
Ra и Rb все независимо означают водород или -C1-7 алкил или Ra и Rb связаны друг с другом и образуют 3-7-членный циклоалкил,
Rc означает водород, -C1-7 алкил, -C1-7 алкилгетероциклоалкил, в этом случае гетероциклоалкил представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкилфенил, -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкил-O-C1-7 алкил или -C1-7 алкил-NR19R20,
где один атом водорода группы -C1-7 алкил, -C1-7 алкилгетероциклоалкил, в этом случае гетероциклоалкил представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкилфенил или -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, может быть замещен следующим заместителем: -C1-7 алкил, -O-C1-7 алкил, 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C(=O)-O-C1-7 алкил или гетероарил-C1-5 галогеналкил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, и
R19 и R20 все независимо означают H или -C1-7 алкил,
означает фенилен,
галоген представляет собой F, Cl, Br или I и
n равно 0 или 1.
3. Соединение, описывающееся химической формулой I, его стереоизомеры или его фармацевтически приемлемые соли по п. 1, где
X1-X4 все независимо означают CR0 или N, где по меньшей мере два из X1-X4 означают CR0,
R0 означает водород или галоген,
R1 означает -C1-5 галогеналкил,
R2 и R3 все независимо означают H, галоген, , 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3-7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , , , , фенил, индолил, или ,
где один и два атома водорода указанного 3-7-членного гетероциклоалкила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3-7-членного гетероциклоалкенила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , , , , фенила, индолила, или , может быть замещен с помощью R4,
R4 означает галоген, -C1-7 алкил, -C1-7 галогеналкил, -O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-OH, -C(=O)-O-C1-7 алкил, -S(=O)2-C1-7 алкил, 3-7-членный циклоалкил, 3-7-членный галогенциклоалкил, 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , -C1-7 алкил-C(=O)-R5, -C1-7 алкил-R7, -C1-7 алкил-O-R8, -NR9R10 или -C(=O)-NR11R12,
где R5 представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
R7 представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S или 3-7-членный циклоалкил,
R8 означает -C1-7 алкил,
R9 и R10 все независимо означают -C1-7 алкил и
R11 и R12 все независимо означают H или -C1-7 алкил,
Rx и Ry все независимо означают -C1-7 алкил или -C1-7 алкил-NR15R16,
где R15 и R16 все независимо означают -C1-7 алкил,
K означает O,
Y означает CRaRb, NRc или ординарную связь,
Ra и Rb все независимо означают водород или -C1-7 алкил или Ra и Rb связаны друг с другом и образуют 3-7-членный циклоалкил,
Rc означает водород, -C1-7 алкил, -C1-7 алкилгетероциклоалкил, в этом случае гетероциклоалкил представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкилфенил, -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкил-O-C1-7 алкил или -C1-7 алкил-NR19R20,
где один атом водорода группы -C1-7 алкил, -C1-7 алкилгетероциклоалкил, в этом случае гетероциклоалкил представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкилфенил или -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, может быть замещен следующим заместителем: -C1-7 алкил, -O-C1-7 алкил, 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, гетероарил-C1-5 галогеналкил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, или -C(=O)-O-C1-7 алкил, и
R19 и R20 все независимо означают -C1-7 алкил,
означает фенилен,
галоген представляет собой F или Br и
n равно 0 или 1.
4. Соединение, описывающееся химической формулой I, его стереоизомеры или его фармацевтически приемлемые соли по п. 1, где
X1-X4 все независимо означают CR0 или N, где по меньшей мере два из X1-X4 означают CR0,
R0 означает водород или F,
R1 означает CF2H,
R2 и R3 все независимо означают H, F, Br, , 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3-7-членный гетероциклоалкенил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , , , , фенил, индолил, или ,
где один и два атома водорода указанного 3-7-членного гетероциклоалкила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 3-7-членного гетероциклоалкенила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членного гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , , , , фенила, индолила, или , может быть замещен с помощью R4,
R4 означает F, -C1-7 алкил, -C1-7 галогеналкил, -O-C1-7 алкил, -C(=O)-C1-7 алкил, -C(=O)-C1-7 алкил-OH, -C(=O)-O-C1-7 алкил, -S(=O)2-C1-7 алкил, 3-7-членный циклоалкил, 3-7-членный галогенциклоалкил, 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, , -C1-7 алкил-C(=O)-R5, -C1-7 алкил-R7, -C1-7 алкил-O-R8, -NR9R10 или -C(=O)-NR11R12,
где R5 представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
R7 представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, или 3-7-членный циклоалкил,
R8 означает -C1-7 алкил,
R9 и R10 все независимо означают -C1-7 алкил и
R11 и R12 все независимо означают H или -C1-7 алкил,
Rx и Ry все независимо означают -C1-7 алкил или -C1-7 алкил-NR15R16,
где R15 и R16 все независимо означают -C1-7 алкил,
K означает O,
Y означает CRaRb, NRc или ординарную связь,
Ra и Rb все независимо означают водород или -C1-7 алкил или Ra и Rb связаны друг с другом и образуют 3-7-членный циклоалкил,
Rc означает водород, -C1-7 алкил, -C1-7 алкилгетероциклоалкил, в этом случае гетероциклоалкил представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкилфенил, -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкил-O-C1-7 алкил или -C1-7 алкил-NR19R20,
где один атом водорода группы -C1-7 алкил, -C1-7 алкилгетероциклоалкил, в этом случае гетероциклоалкил представляет собой 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, -C1-7 алкилфенил, -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, может быть замещен следующим заместителем: -C1-7 алкил, -O-C1-7 алкил, 3-7-членный гетероциклоалкил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, гетероарил-C1-5 галогеналкил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, или -C(=O)-O-C1-7 алкил, и
R19 и R20 все независимо означают -C1-7 алкил,
означает фенилен,
галоген представляет собой F или Br и
n равно 0 или 1.
5. Соединение, описывающееся следующей химической формулой II, его стереоизомеры или его фармацевтически приемлемые соли:
[Химическая формула II]
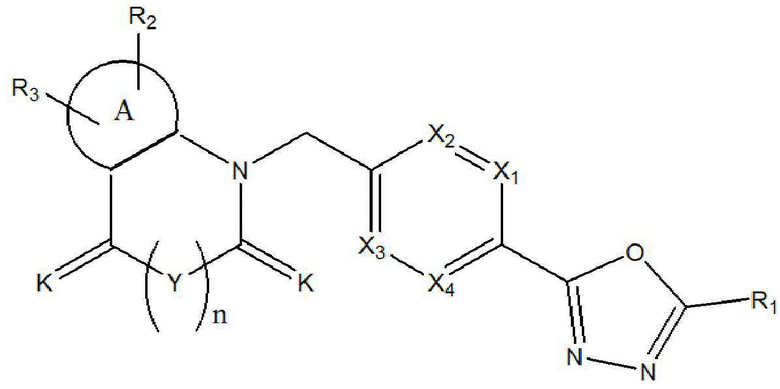 ,
,
где X1-X4 все независимо означают CR0 или N, где по меньшей мере два из X1-X4 означают CR0,
где R0 означает водород,
R1 означает линейный или разветвленный -C1-5 галогеналкил,
R2 и R3 все независимо означают H,
K означает O,
Y означает NRc,
Rc означает -C1-7 алкилфенил, -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, или -C1-7 алкил-O-C1-7 алкил,
где один атом водорода группы -C1-7 алкилгетероарил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S, может быть замещен следующим заместителем: гетероарил-C1-5 галогеналкил, в этом случае гетероарил представляет собой 5- или 6-членный гетероарил, содержащий от 1 до 3 гетероатомов, выбранных из группы, включающей N, O или S,
означает фенилен,
галоген представляет собой F, Cl, Br или I и
n равно 0 или 1.
6. Соединение, описанное в следующей таблице, его стереоизомеры или его фармацевтически приемлемые соли:
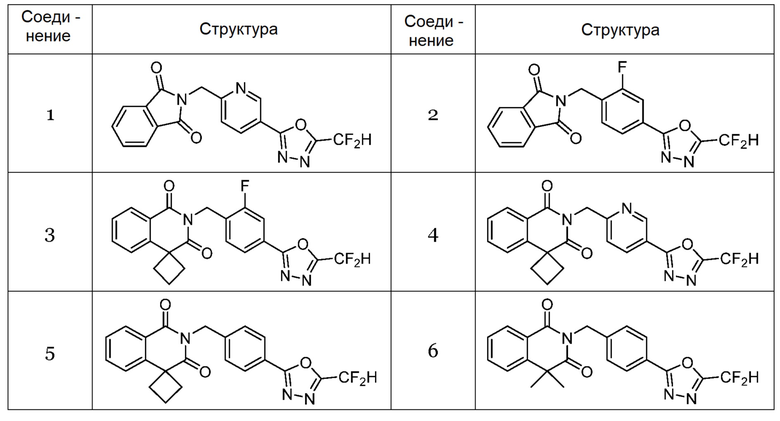
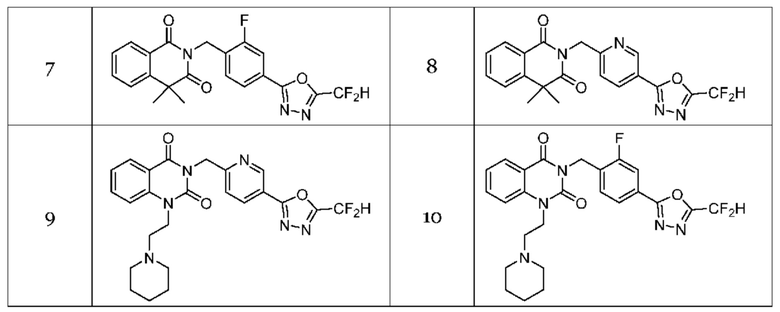
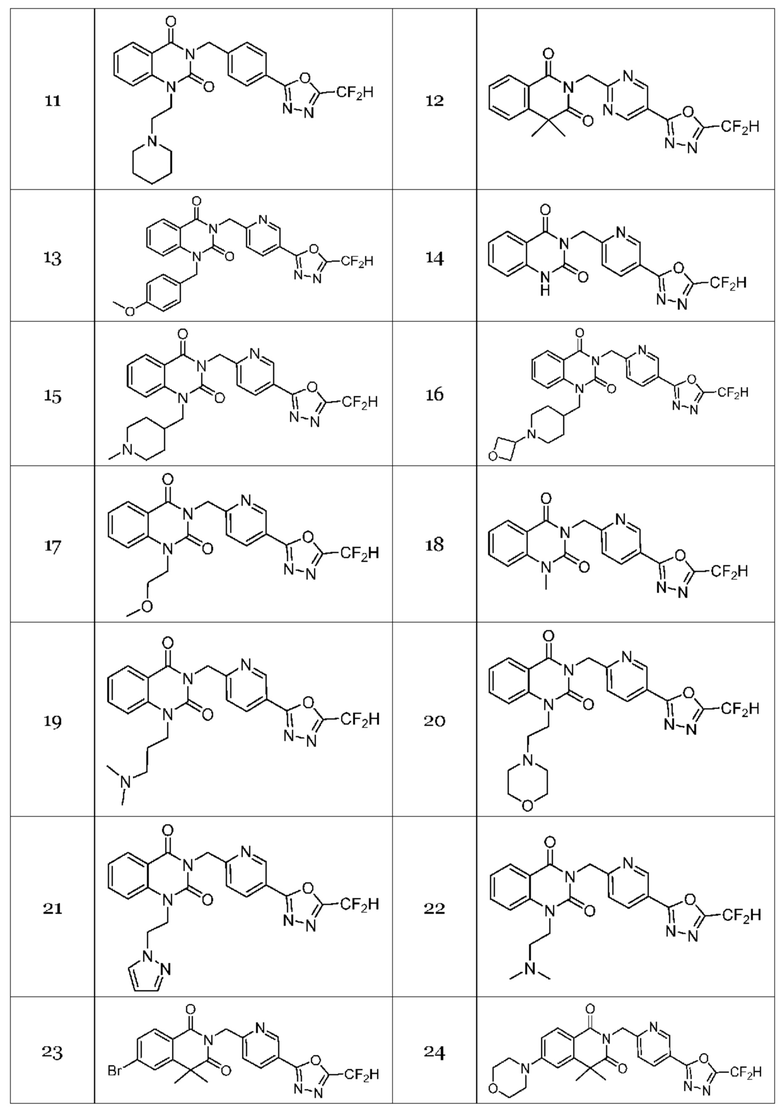
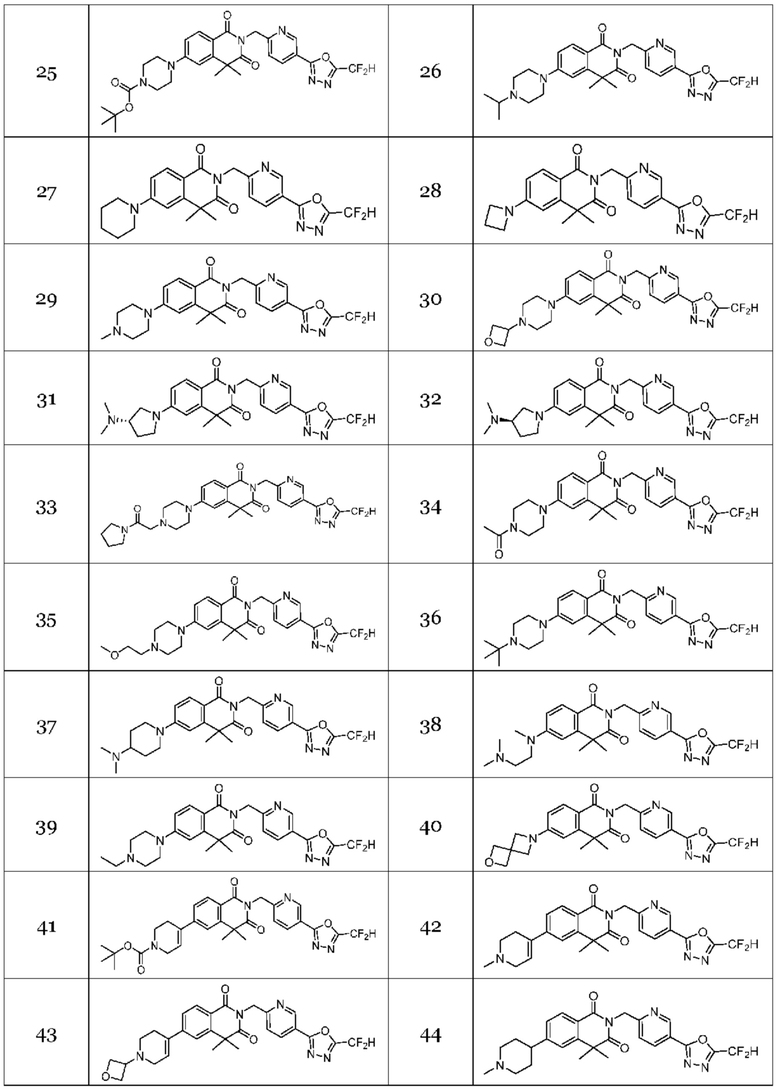
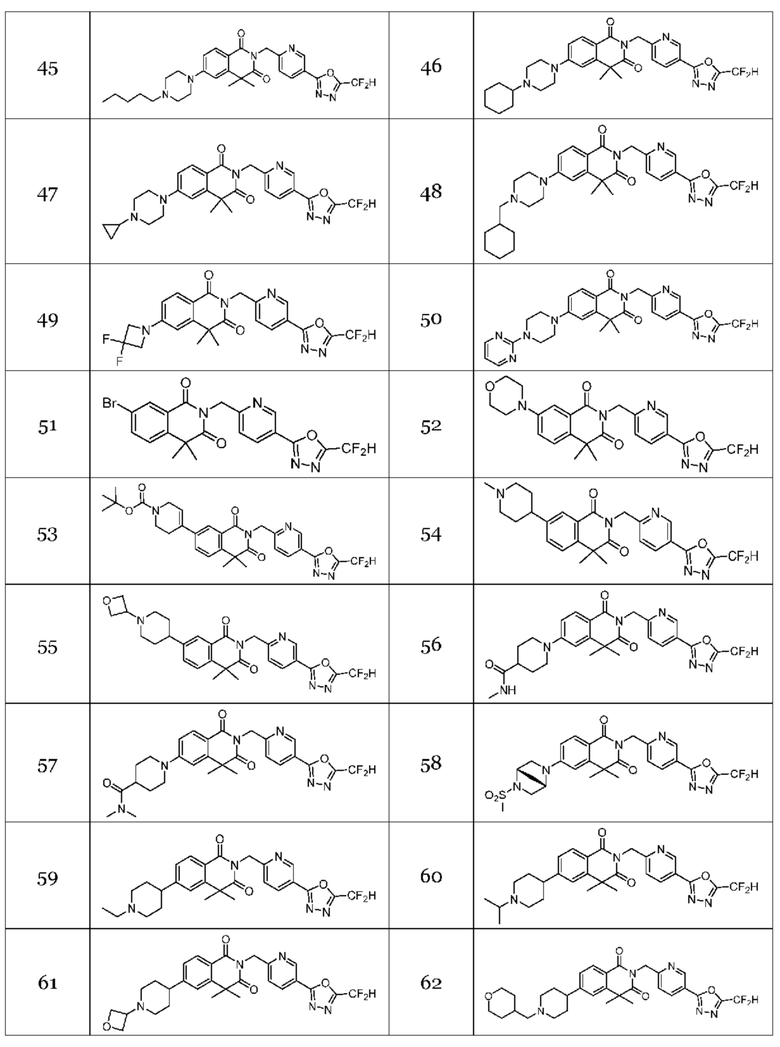
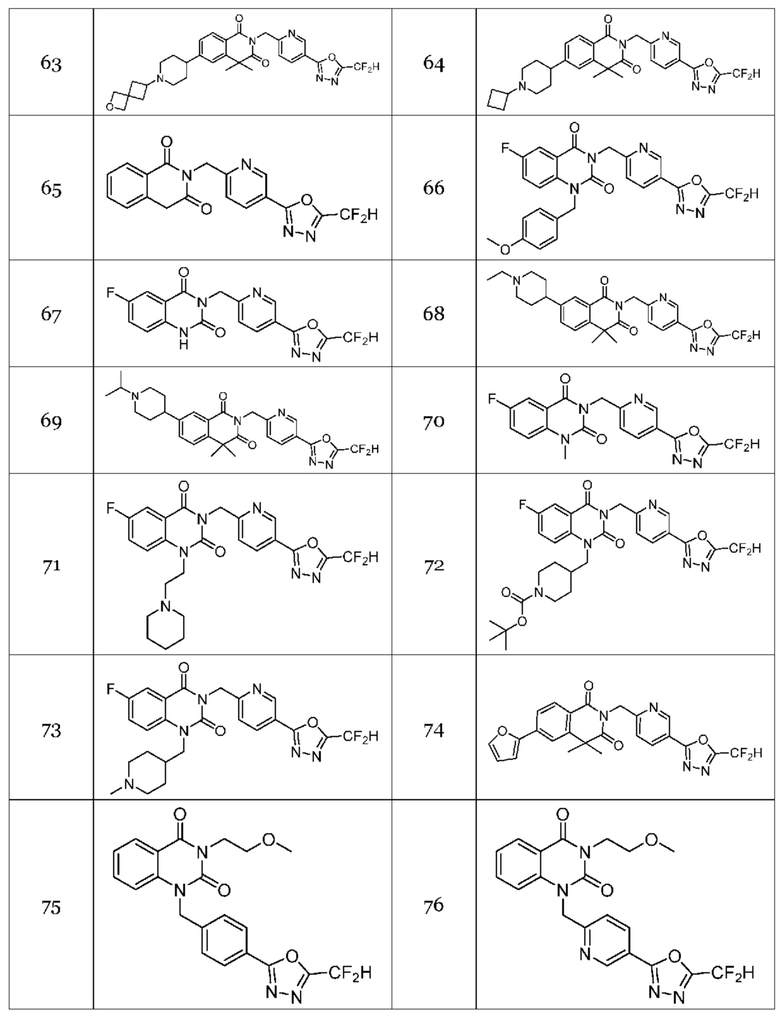
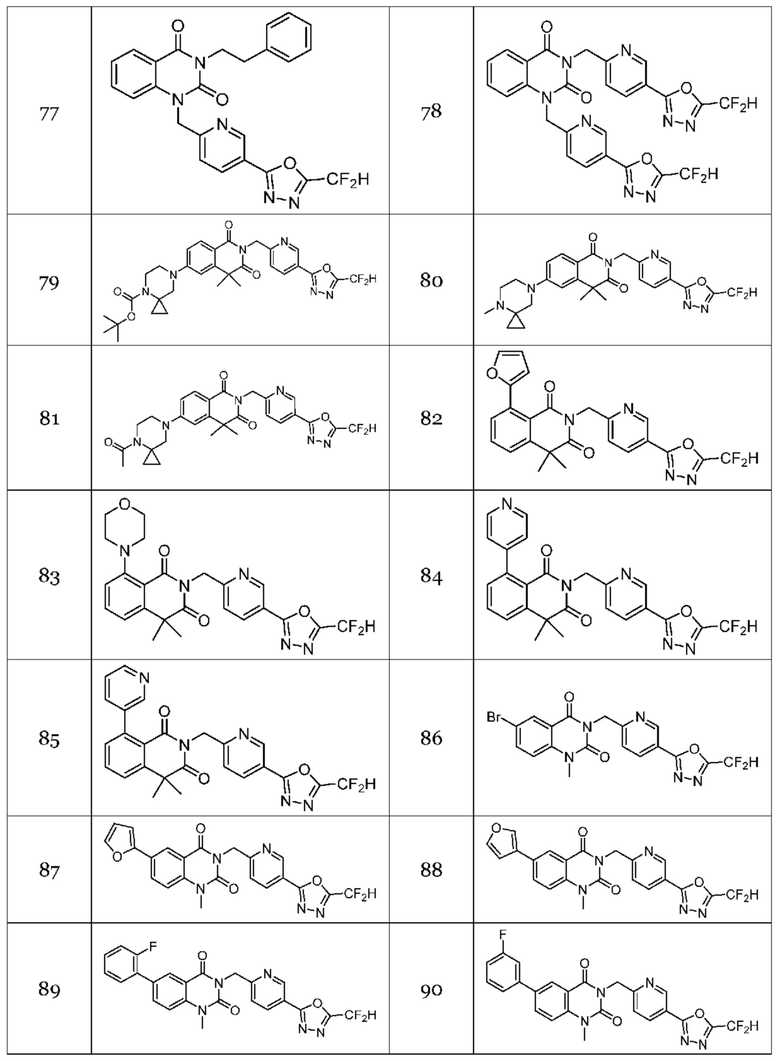
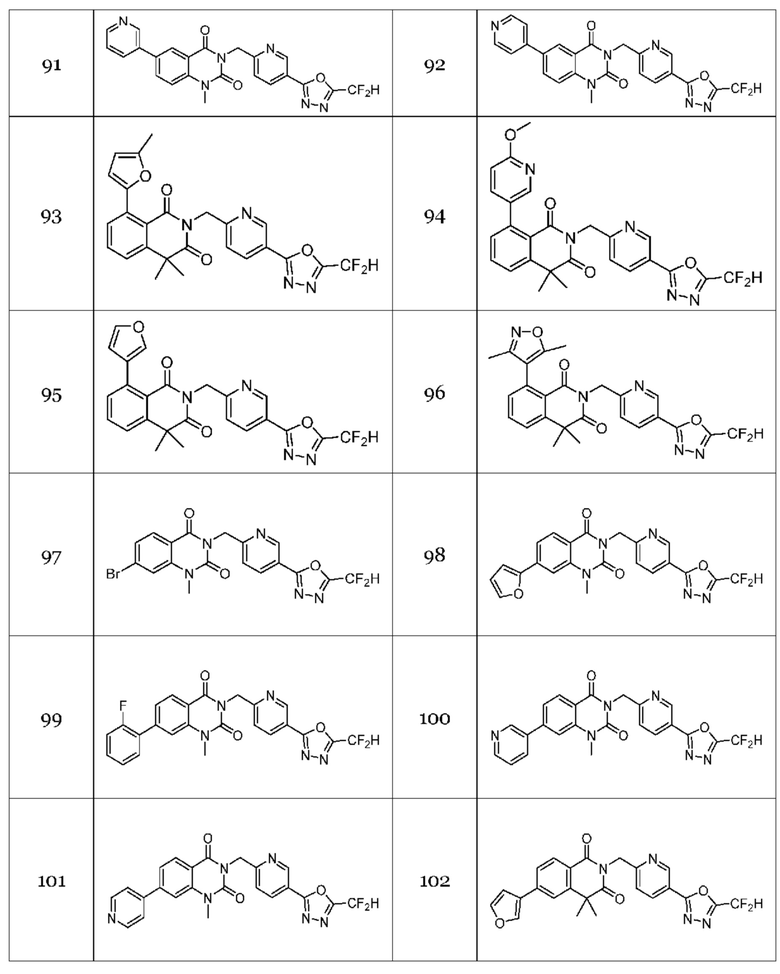
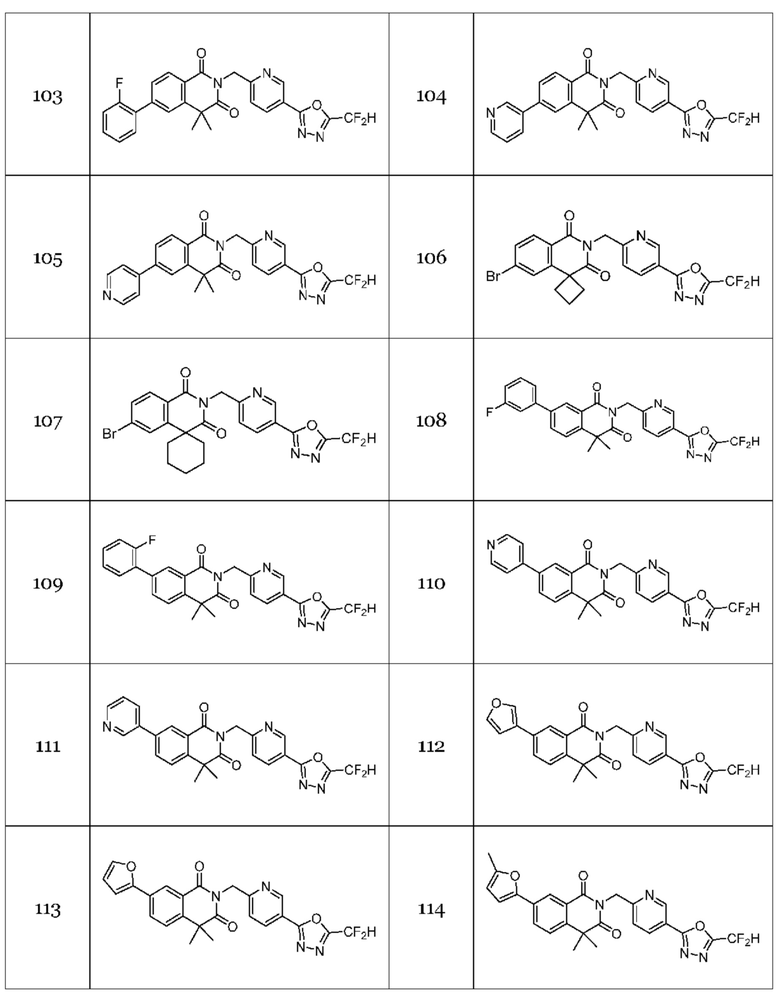
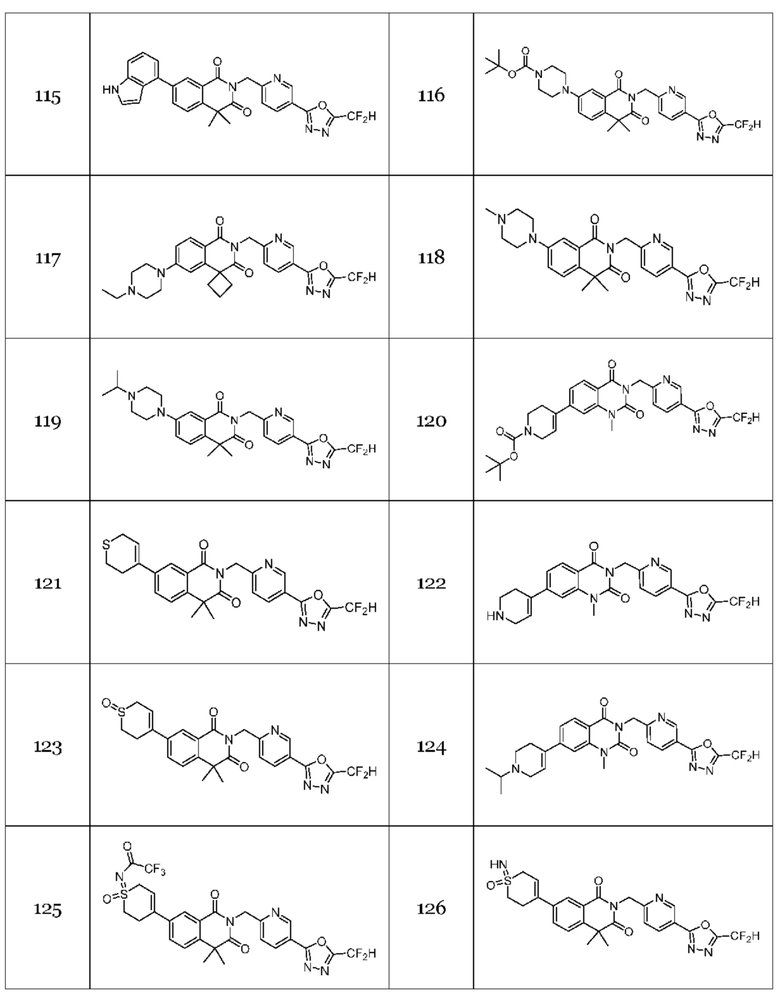
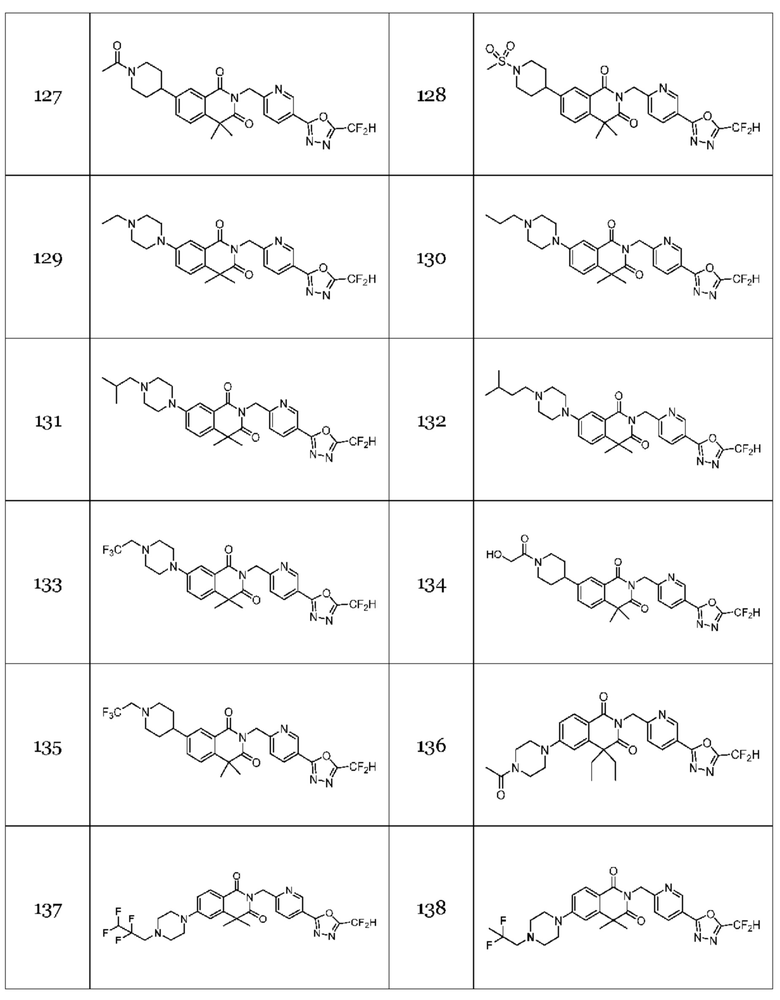
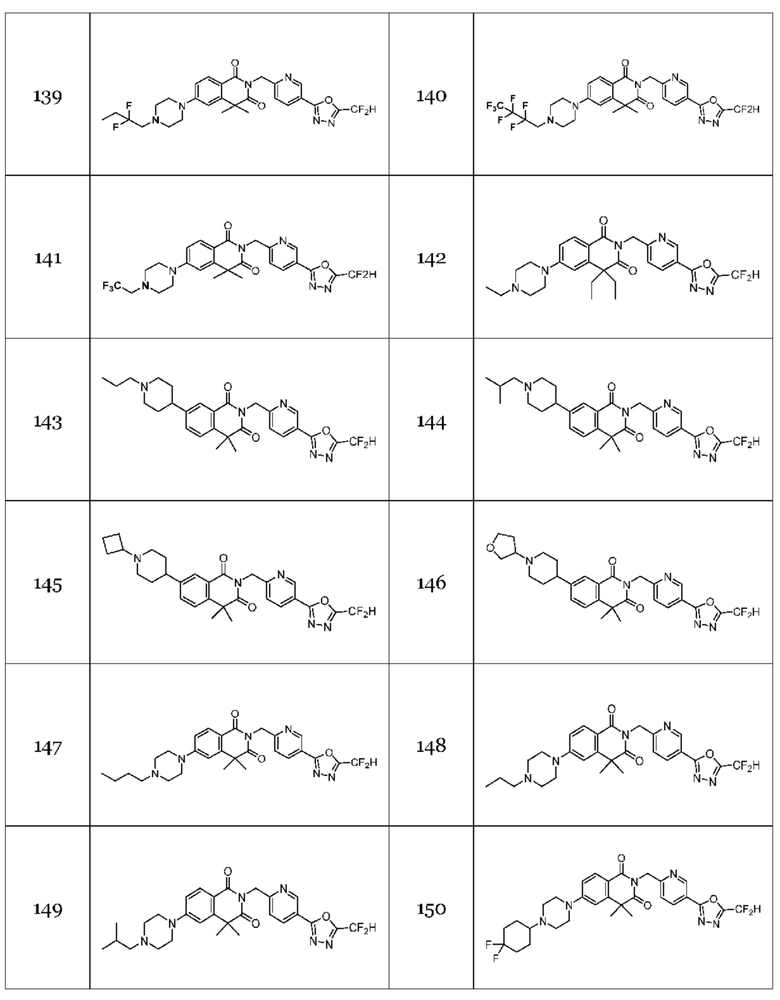
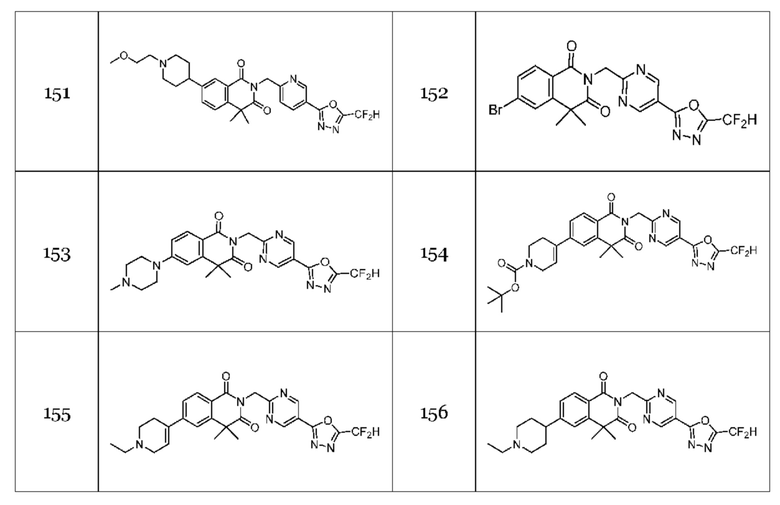
7. Фармацевтическая композиция, обладающая ингибирующей активностью по отношению к HDAC6, где композиция содержит терапевтически эффективное количество соединения по любому из пп. 1-6, его стереоизомеры или его фармацевтически приемлемые соли в качестве эффективного компонента и фармацевтически приемлемый носитель.
8. Фармацевтическая композиция по п. 7, где указанная фармацевтическая композиция предназначена для предупреждения или лечения связанных с активностью гистондезацетилазы 6 заболеваний.
9. Фармацевтическая композиция по п. 8, где связанные с активностью гистондезацетилазы 6 заболевания представляют собой по меньшей мере одно, выбранное из группы, включающей инфекционные заболевания; неоплазму; эндокринопатии; связанные с питанием и метаболические заболевания; психические и поведенческие расстройства; неврологические заболевания; аднексальные заболевания глаз; заболевания кровообращения; респираторные заболевания; пищеварительные заболевания; заболевания кожи и подкожных тканей; заболевания скелетно-мышечной системы и соединительной ткани; и порок развития или уродства, и хромосомную аберрацию.
10. Способ ингибирования гистондезацетилазы 6, включающий введение терапевтически эффективного количества соединения по любому из пп. 1-6, его стереоизомеров или его фармацевтически приемлемых солей.
11. Применение соединения по любому из пп. 1-6, его стереоизомеров или его фармацевтически приемлемых солей для ингибирования гистондезацетилазы 6.
12. Применение соединения по любому из пп. 1-6, его стереоизомеров или его фармацевтически приемлемых солей для приготовления лекарственного средства для ингибирования гистондезацетилазы 6.
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Токарный резец | 1924 |
|
SU2016A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| EA 201491060 A1, 30.09.2014. | |||

