Область техники, к которой относится изобретение
Настоящее изобретение относится к новому соединению, обладающему ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), его стереоизомерам, его фармацевтически приемлемым солям, его применению при приготовлении терапевтического лекарственного средства, содержащей его фармацевтической композиции, терапевтическому способу с использованием этой композиции и способу ее приготовления.
Уровень техники
В клетках посттрансляционная модификация, такая как ацетилирование, служит очень важным регуляторным модулем в центре биологических процессов, а также строго контролируется рядом ферментов. В качестве корового белка, составляющего хроматин, гистон выполняет функцию оси, вокруг которой закручивается ДНК, и, таким образом, способствует конденсации ДНК. Кроме того, баланс между ацетилированием и деацетилированием гистона играет очень важную роль в экспрессии генов.
Как фермент для удаления ацетильной группы из лизинового остатка гистонового белка, который составляет хроматин, гистондеацетилаза (HDAC), как известно, связана с сайленсингом генов и индуцирует остановку клеточного цикла, ингибирование ангиогенеза, иммунорегуляцию, апоптоз и т.д. (Hassig et al., Curr. Opin. Chem. Biol. 1997, 1, 300-308). Также сообщается, что ингибирование функций фермента HDAC индуцирует апоптоз раковых клеток за счет снижения активности факторов, связанных с выживанием раковых клеток, и активации факторов, связанных с гибелью раковых клеток в организме (Warrell et al., J. Natl. Cancer Inst. 1998, 90, 1621-1625).
Для человека известно 18 HDAC, которые разделяются на четыре класса в соответствии с гомологией с дрожжевой HDAC. При этом одиннадцать HDAC, использующих цинк в качестве кофактора, можно разделить на три группы: класс I (HDAC1, 2, 3, 8), класс II (IIa: HDAC4, 5, 7, 9; IIb: HDAC6, 10) и класс IV (HDAC11). Кроме того, семь HDAC класса III (SIRT 1-7) используют NAD+ в качестве кофактора вместо цинка (Bolden et al., Nat. Rev. Drug Discov. 2006, 5(9), 769-784).
Различные ингибиторы HDAC в настоящее время находятся на доклинической или клинической стадиях разработки, однако до настоящего времени только неселективные ингибиторы HDAC известны в качестве противоракового средства. Вориностат (SAHA) и ромидепсин (FK228) получили одобрение в качестве
терапевтического средства при кожной Т-клеточной лимфоме, в то время как панобиностат (LBH-589) получил одобрение в качестве терапевтического средства при множественной миеломе. Однако известно, что неселективные ингибиторы HDAC обычно вызывают побочные эффекты, такие как усталость, тошнота и тому подобное, при применении в высоких дозах (Piekarz et al., Pharmaceuticals 2010, 3, 2751-2767). Сообщается, что побочные эффекты вызваны ингибированием HDAC класса I. Из-за побочных эффектов и т.п.неселективные ингибиторы HDAC подвергаются ограничениям при разработке лекарственных препаратов в других областях, отличных от противораковых агентов (Witt et al., Cancer Letters 277 (2009) 8.21).
В то же время, сообщается, что селективное ингибирование HDAC класса II не приведет к токсичности, которая имела бы место при ингибировании HDAC класса I. В случае разработки селективных ингибиторов HDAC это, вероятно, позволило бы устранить побочные эффекты, такие как токсичность и т.д., вызванные неселективным ингибированием HDAC. Соответственно, существует вероятность того, что селективные ингибиторы HDAC могут разрабатываться в качестве эффективного терапевтического средства для лечения различных заболеваний (Matthias et al., Mol. Cell. Biol. 2008, 28, 1688-1701).
Известно, что HDAC6, одна из HDAC класса lib, в основном присутствует в цитоплазме и содержит белок тубулин, соответственно, участвуя в деацетилировании ряда негистоновых субстратов (HSP90, кортактин и т.д.) (Yao et al., Mol. Cell 2005, 18, 601-607). HDAC6 имеет два каталитических домена, где домен цинкового пальца С-конца может связываться с убиквитинированным белком. Известно, что HDAC6 имеет ряд негистоновых белков в качестве субстрата, и таким образом, играет важную роль в различных заболеваниях, таких как рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания, нейродегенеративные нарушения и тому подобное (Santo et al., Blood 2012 119: 2579-258; Vishwakarma et al., International Immunopharmacology 2013, 16, 72-78; Hu et al., J. Neurol. Sci. 2011, 304, 1-8).
Структурная особенность, общая для различных ингибиторов HDAC, включает в себя кэп-группу, линкерную группу и цинксвязывающую группу (ZBG), как показано в следующей структуре вориностата. Многие исследователи провели изучение ингибирующей активности и селективности в отношении ферментов посредством структурной модификации кэп-группы и линкерной группы. Кроме этих групп известно, что цинксвязывающая группа играет более важную роль в ингибирующей активности и селективности фермента (Wiest et al., J. Org. Chem. 2013 78: 5051-5065; Methot et al., Bioorg. Med. Chem. Lett. 2008, 18, 973-978).
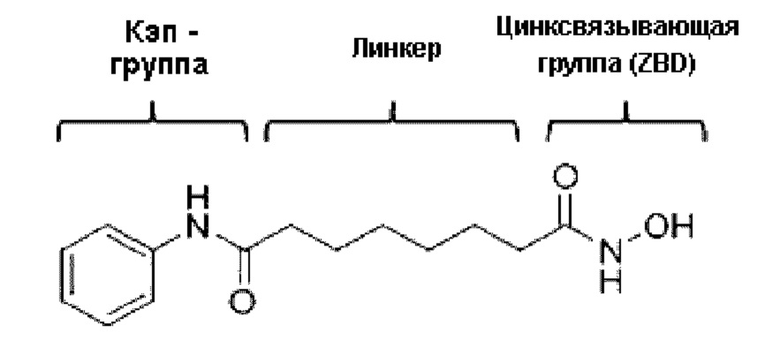
Большая часть указанной цинксвязывающей группы состоит из гидроксамовой кислоты или бензамида, из который производные гидроксамовой кислоты проявляют сильное ингибирующее HDAC действие, но имеют проблемы с низкой биодоступностью и серьезной побочной активностью. Бензамид содержит анилин и, таким образом, имеет проблему, заключающуюся в том, что он может продуцировать токсичные метаболиты in vivo (Woster et al., Med. Chem. Commun. 2015, публикация в интернете).
Соответственно, в отличие от неселективных ингибиторов, обладающих побочными эффектами, существует необходимость в разработке селективного ингибитора HDAC6, который имеет цинксвязывающую группу, с улучшенной биодоступностью, не вызывающего при этом побочных эффектов, для лечения рака, воспалительных заболеваний, аутоиммунных заболеваний, неврологических заболеваний, нейродегенеративных нарушений и тому подобного.
[Ссылка на известный уровень техники]
[Патентный документ]
Международная патентная публикация WO 2011/091213 (опубликована 28 июля 2011): ACY-1215
Международная патентная публикация WO 2011/011186 (опубликована 27 января 2011): Tubastatin
Международная патентная публикация WO 2013/052110 (опубликована 11 апреля 2013): Sloan-K
Международная патентная публикация WO 2013/0414 07 (опубликована 28 марта 2013): Cellzome
Международная патентная публикация WO 2013/134 4 67 (опубликована 12 сентября 2013): Kozi
Международная патентная публикация WO 2013/008162 (опубликована 17 января 2013): Novartis
Международная патентная публикация WO 2013/080120 (опубликована 06 июня 2013): Novartis
Международная патентная публикация WO 2013/066835 (опубликована 10 мая 2013): Tempero
Международная патентная публикация WO 2013/066838 (опубликована 10 мая 2013): Tempero
Международная патентная публикация WO 2013/066833 (опубликована 10 мая 2013): Tempero
Международная патентная публикация WO 2013/066839 (опубликована 10 мая 2013): Tempero
Подробное описание изобретения
Техническая задача
Одной из целей настоящего изобретения является предложить новое соединение, обладающее селективной ингибирующей
активностью в отношении HDAC6, его стереоизомеры или его фармацевтически приемлемые соли.
Другой целью настоящего изобретения является предложить фармацевтическую композицию, содержащую новое соединение, обладающее селективной ингибирующей активностью в отношении HDAC6, его стереоизомеры или его фармацевтически приемлемые соли.
Еще одной целью настоящего изобретения является предложить способ их получения.
Еще одной целью настоящего изобретения является предложить фармацевтическую композицию для предупреждения или лечения заболеваний, связанных с активностью HDAC6, содержащую соединение, его стереоизомеры или его фармацевтически приемлемые соли в качестве эффективного ингредиента.
Еще одной целью настоящего изобретения является предложить применение соединения, его стереоизомеров или его фармацевтически приемлемых солей; или содержащей их фармацевтической композиции в качестве эффективного ингредиента для предупреждения или лечения заболеваний, связанный с активностью HDAC6.
Еще одной целью настоящего изобретения является предложить применение соединения, его стереоизомеров или его фармацевтически приемлемый солей; или содержащей их фармацевтической композиции в качестве эффективного ингредиента для приготовления лекарственного средства для предупреждения или лечения заболеваний, связанных с активностью HDAC6.
Еще одной целью настоящего изобретения является предложить способ для предупреждения или лечения заболеваний, связанных с активностью HDAC6, включающий введение терапевтически эффективного количества соединения, его стереоизомеров или его фармацевтически приемлемых солей; или содержащей их фармацевтической композиции в качестве эффективного ингредиента нуждающемуся в этом субъекту.
Техническое решение
Авторы настоящего изобретения обнаружили соединение производное оксадиазола, обладающее ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), и использовали его для ингибирования или лечения заболеваний, связанных с активностью HDAC6, что позволило осуществить настоящее изобретение.
Соединение, представленное формулой I
Настоящее изобретение предлагает новое соединение, обладающее селективной ингибирующей активностью в отношении HDAC6, представленное следующей химической формулой I, его стереоизомеры или его фармацевтически приемлемые соли:
[Химическая формула I]

где
каждый из Z1 - Z4 независимо представляет собой N или CR0 (здесь R0 является Н или галогеном);
R1 представляет собой СХ3 или СХ2Н (здесь X является галогеном);
 является
является 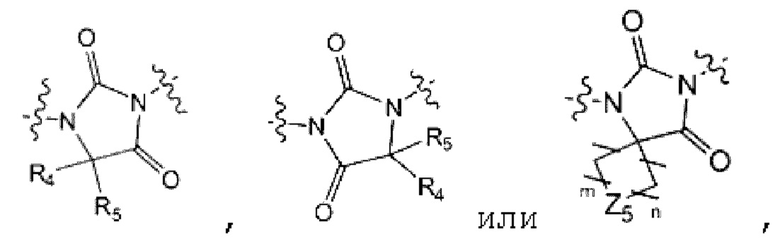
каждый из R4 и R5 независимо представляет собой Н или С1-С4 алкил,
Z5 представляет собой N-R6 или СН2,
R6 представляет собой Н, С1-С4 алкил, -С(=О)-(C1-C4 алкил), -С(=О)-О-(С1-С4 алкил) или 4-6-членный петероциклоалкил, имеющий один О;
L1 представляет собой -(С1-С2 алкилен)-;
 представляет собой С6-С12 арил, 5-9-членный петероарил, имеющий по меньшей мере один N, или
представляет собой С6-С12 арил, 5-9-членный петероарил, имеющий по меньшей мере один N, или 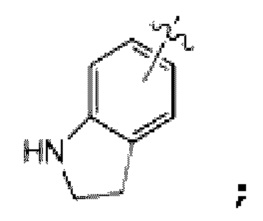
каждый из R2 и R3 независимо представляет собой Н, галоген, С1-С4 алкил, С6-С12 арил, 5- или 6-членный гетероарил, имеющий N или 0, 5- или 6-членный гетероциклоалкил, имеющий N, 5- или 6-членный гетероциклоалкенил, имеющий N, -С(=О)-О-(С1-С4 алкил), -С(=О)-(С1-С4 алкил), -NH-C(=О)- (С1-С4 алкил), -NO2 или -NH2,
по меньшей мере один Н из вышеуказанных R2 и R3 может быть в каждом случае независимо замещен галогеном или С1-С4 алкилом; и
n и m в каждом случае независимо равно 1 или 2.
В одном варианте осуществления, в приведенной выше химической формуле I,
каждый из R2 и R3 независимо представляет собой Н, галоген, С1-С4 алкил, фенил, фуранил, пиридинил, С1-С4 алкилзамещенный или незамещенный пиперидинил, С1-С4 алкилзамещенный или незамещенный тетрагидропиридинил, -С(=О)-О-(С1-С4 алкил), С(=О)-(С1-С4 алкил), -NH-C(=О)-(С1-С4 алкил), -NO2 или -NH2,
 представляет собой фенил, индол или
представляет собой фенил, индол или 
Если  представляет собой индол или
представляет собой индол или  Н из его NH может быть замещен -С(=О)-О-(С1-С4 алкилом) или -С(=О)-(С1-С4 алкилом), и если
Н из его NH может быть замещен -С(=О)-О-(С1-С4 алкилом) или -С(=О)-(С1-С4 алкилом), и если  представляет собой фенил, по меньшей мере один Н фенила может быть в каждом случае независимо замещен галогеном, и
представляет собой фенил, по меньшей мере один Н фенила может быть в каждом случае независимо замещен галогеном, и
n и m могут быть в каждом случае независимо равны 1 или 2.
В настоящем изобретении термин «замещенный» может обозначать фрагмент, имеющий заместитель, который замещает по меньшей мере один водород на углероде основной цепи. Выражение «замещение», «может быть замещен ~» или «замещен ~» может быть определено как включающее неявные условия, в которых замещение соответствует допустимой валентности замещенного атома и заместителя и образует соединение, стабилизированное замещением, например, соединение, которое не модифицируется естественным образом перегруппировкой, циклизацией, удалением и т.д.
В настоящем изобретении «Сх-у» может относиться к наличию атомов углерода в диапазоне от х до у.
В настоящем изобретении «алкил» может относиться к линейной (или прямоцепочечной) насыщенной углеводородной группе или разветвленной (или боковой) насыщенной углеводородной группе, и включает метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил и т.д.
В настоящем изобретении «алкилен» может относиться к двухвалентной функциональной группе, которая образуется из алкильной группы, как определено выше.
В настоящем изобретении «арил» может включать моноциклическую ароматическую структуру или полициклическую ароматическую структуру, а также структуру, в которой насыщенное углеводородное кольцо конденсировано в моноциклическую или полициклическую ароматическую группу. Арил может включать фенильную группу, нафталенил, тетрагидронафталенил, антраценил, фенантренил, пиренил, и т.д.
В настоящем изобретении «гетероарил» может означать моноциклическое или полициклическое гетерокольцо, в котором по меньшей мере один атом углерода замещен по меньшей мере одним гетероатомом, который является по меньшей мере одним из азота (N) и кислорода (О) в ариле, как определено выше. Гетероарил может включать пиридинил, триазолил, тетразолил, индолил, изоиндолил, фуранил, пирролил, имидазолил, оксазолил, изоксазолил, пиразинил, пиридазинил, пиримидинил, и т.д., но этим не ограничивается.
В настоящем изобретении «циклоалкил» может относиться к насыщенному углеводородному кольцу, обычно имеющему указанное число атомов углерода в составе кольца, и может включать циклогексил, циклогептанил, циклооктанил и т.д. В настоящем изобретении «гетероциклоалкил» может относиться к насыщенной кольцевой структуре, содержащей от одного до четырех гетероатомов, представляющих собой по меньшей мере одно из азота (N) и кислорода (О).
В настоящем изобретении «гетероциклоалкенил» может относиться к структуре, имеющей по меньшей мере одну двойную углерод-углеродную связь, содержащей от одного до четырех гетероатомов, представляющих собой по меньшей мере одно из азота (N) и кислорода (О).
В настоящем изобретении «галоген» может относится к F, Cl или Br.
В настоящем изобретении «стереоизомер» может включать диастереомер и оптический изомер (энантиомер), при этом оптический изомер может включать не только энантиомер, но также смесь энантиомера и даже рацемата.
В настоящем изобретении фармацевтически приемлемые соли могут относиться к солям, традиционно используемым в фармацевтической промышленности, например, к неорганическим ионным солям, полученным из кальция, калия, натрия, магния и тому подобного; солям неорганических кислот, полученным из соляной кислоты, азотной кислоты, фосфорной кислоты, бромноватой кислоты, йодноватой кислоты, перхлорной кислоты, серной кислоты и тому подобного; солям органических кислот, полученным из уксусной кислоты, трифторуксусной кислоты, лимонной кислоты, малеиновой кислоты, янтарной кислоты, щавелевой кислоты, бензойной кислоты, винной кислоты, фумаровой кислоты, миндальной кислоты, пропионовой кислоты, молочной кислоты, гликолевой кислоты, глюконовой кислоты, галактуроновой кислоты,
глутаминовой кислоты, глутаровой кислоты, глюкуроновой кислоты, аспарагиновой кислоты, аскорбиновой кислоты, угольной кислоты, ванилиновой кислоты, иодистоводородной кислоты и т.д.; солям сульфоновой кислоты, полученным из метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, п-толуолсульфоновой кислоты, нафталинсульфоновой кислоты и тому подобного; солям аминокислот, полученным из глицина, аргинина, лизина и т.д.; аминным солям, полученным из триметиламина, триэтиламина, аммиака, пиридина, пиколина и т.д.; и тому подобному, однако типы солей, подразумеваемые в настоящем изобретении, не ограничиваются перечисленными солями.
В настоящем изобретении предпочтительные соли могут включать гидрохлорид, фосфат, сульфат, трифторацетат, цитрат, бромат, малеат или тартрат.
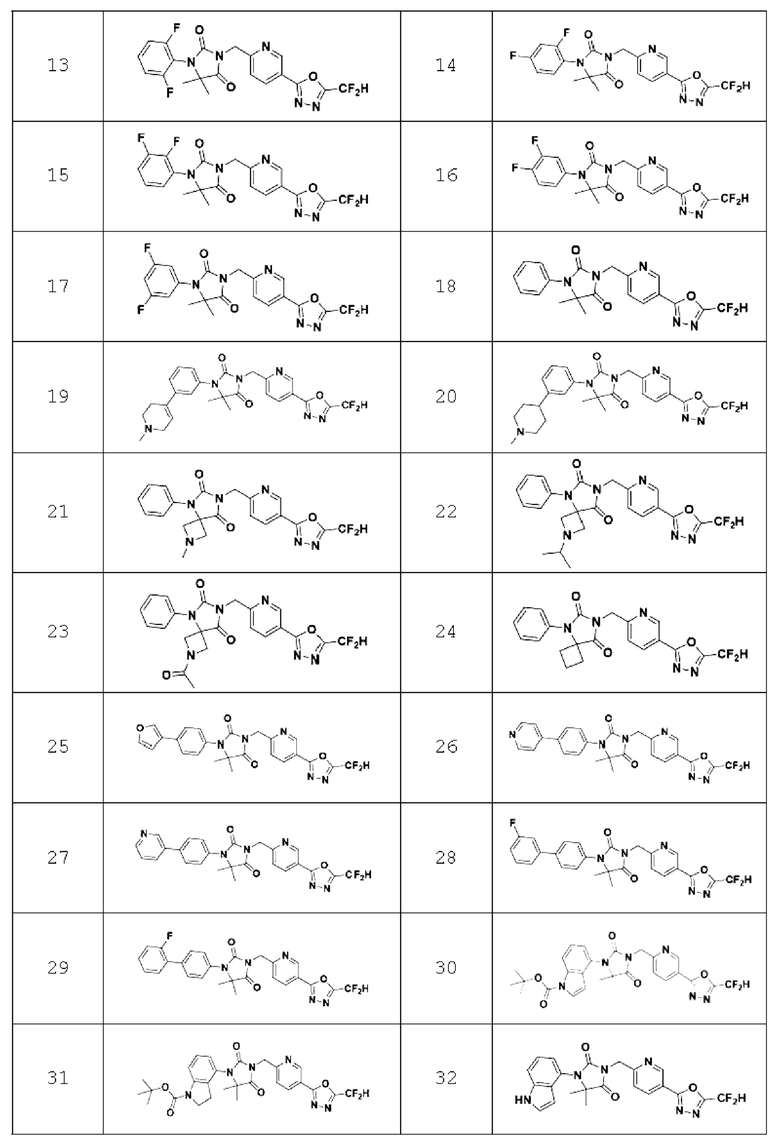
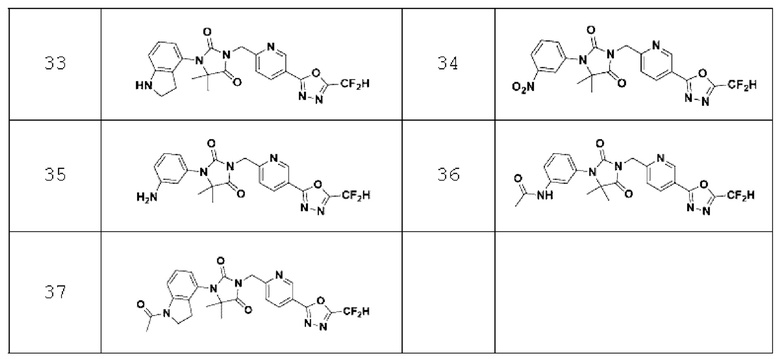
В одном варианте осуществления соединение, представленное химической формулой I настоящего изобретения, его стереоизомеры или его фармацевтически приемлемые соли могут включать соединения, показанные в таблице 1 ниже.

Способ получения соединения, представленного химической формулой I
Настоящее изобретение может предложить способ получения соединения, представленного химической формулой I, его стереоизомеров или его фармацевтически приемлемых солей.
Предпочтительный способ получения соединения,
представленного химической формулой I, его стереоизомеров или его фармацевтически приемлемых солей является таким, как показано в формулах реакций 1, 1-1 и с 2 по 7, и даже способ получения, модифицированный на уровне, очевидном специалистам в данной области, также в него включен.
Б каждой из формул реакции 1, 1-1 и с 2 по 7, R1 - R5, Z1 - Z4, L1, m, n и X являются, по существу, такими же, как определено в химической формуле I. Б формулах реакции 1, 1-1 и с 2 по 7 «гало» может относиться к галогену F, Cl или Br. В дополнение к этому, в формулах реакции 1, 1-1 и с 2 по 7, «PG» может относиться к защитной группе атома азота и может включать трет-бутилоксикарбонил (Вос), бензилоксикарбонил (Cbz) или тому подобное.
[Формула реакции 1]
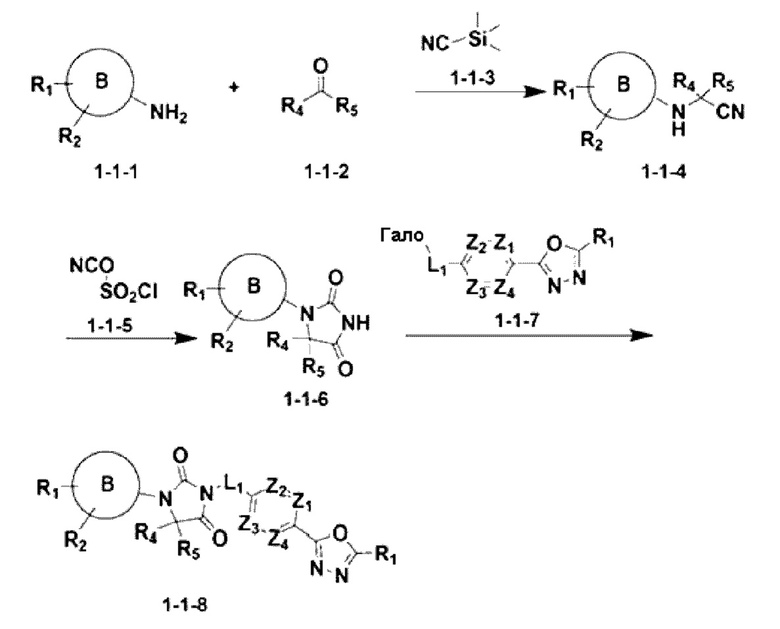
Приведенная выше формула реакции 1 показывает способ синтеза соединения, имеющего структуру имидазолидин-2,4-диона, где соединение химической формулы 1-1-1 может реагировать с соединением химической формулы 1-1-2 и соединением химической формулы 1-1-3 для получения соединения химической формулы 1-1-4, имеющего структуру аминонитрила. После этого полученное соединение может реагировать с соединением химической формулы 1-1-5 с получением соединения химической формулы 1-1-6, имеющего структуру имидазолидин-2,4-диона, и затем реагировать с соединением химической формулы 1-1-7 для получения соединения химической формулы 1-1-8.
В настоящем изобретении соединение, полученное в соответствии с приведенной выше формулой реакции 1, может включать 3, 4, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 и тому подобное.
[Формула реакции 1-1]

Приведенная выше формула реакции 1-1 относится, по существу, к такой же химической реакции, что и приведенная выше формула реакции 1, в которой соединение химической формулы 1-1-1 может реагировать с соединением химической формулы l-2-la и соединением химической формулы 1-1-3 для получения соединения химической формулы 1-2-2а. После этого полученное соединение может реагировать с соединением химической формулы 1-1-5 с получением соединения химической формулы 1-2-За, имеющего структуру имидазолидин-2,4-диона, и затем реагировать с соединением химической формулы 1-1-7 для получения соединения химической формулы 1-2-4а. В настоящем изобретении соединение, полученное в соответствии с приведенной выше формулой реакции 1-1, может включать 24 и тому подобное.
[Формула реакции 2]
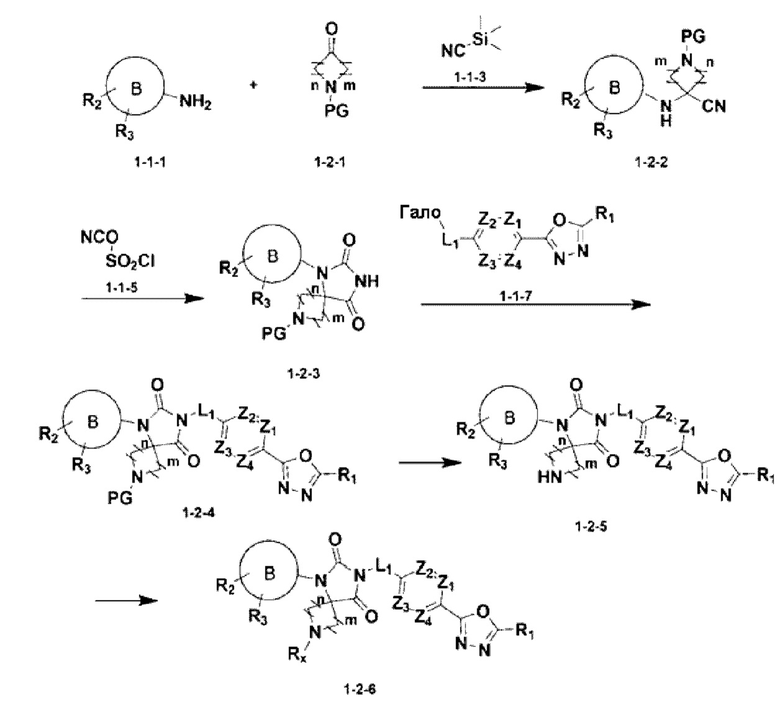
В приведенной выше формуле реакции 2 «Rx» может представлять собой С1-С4 алкил, 4-6-членный гетероциклоалкил, имеющий один О, или С(=О)-(С1-С4 алкил). Приведенная выше формула реакции 2 показывает способ синтеза соединения, имеющего структуру имидазолидин-2,4-диона, где соединение химической формулы 1-1-1 может реагировать с соединением химической формулы 1-2-1, к которому добавлена защитная группа, и с соединением химической формулы 1-1-3 для получения соединения химической формулы 1-2-2, имеющего структуру аминонитрила. После этого полученное соединение может реагировать с соединением химической формулы 1-1-5 с получением соединения химической формулы 1-2-3, имеющего структуру имидазолидин-2,4-диона, и затем реагировать с соединением химической формулы 1-1-7 для получения соединения химической формулы 1-2-4. Защитная группа может быть удалена из соединения химической формулы 1-2-4 для получения соединения химической формулы 1-2-5, и затем может быть проведена реакция восстановительного аминирования или реакция замещения для получения соединения химической формулы 1-2-6.
В настоящем изобретении соединение, полученное в соответствии с приведенной выше формулой реакции 2, может включать 5, 6, 7, 8, 21, 22, 23 и тому подобное.
[Формула реакции 3]

Приведенная выше формула реакции 3 показывает способ синтеза соединения, имеющего структуру имидазолидин-2,4-диона, где соединение химической формулы 1-3-1, полученное в формуле реакции 1, может быть подвергнуто С-С сочетанию (реакция Сузуки) с соединением химической формулы 1-3-2 для получения соединения химической формулы 1-3-3.
В настоящем изобретении соединение, полученное в соответствии с приведенной выше формулой реакции 3, может включать 25, 26, 27, 28, 29 и тому подобное.
[Формула реакции 4]
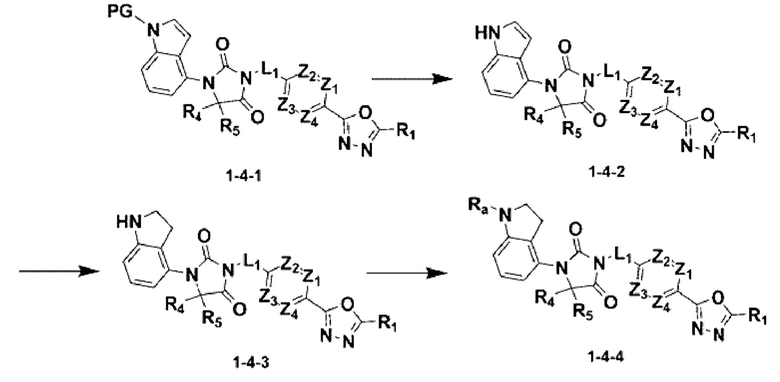
В приведенной выше формуле реакции 4 «Ra» может быть С(=О)-(С1-С4 алкилом).
Приведенная выше формула реакции 4 показывает способ синтеза соединения, имеющего структуру имидазолидин-2,4-диона, где защитная группа может быть удалена из соединения химической формулы 1-4-1, имеющего защитную группу, полученного в формуле реакции 1, для получения соединения химической формулы 1-4-2, после чего может быть проведена реакция восстановления для получения соединения химической формулы 1-4-3. После этого может быть проведена реакция восстановительного аминирования или реакция замещения для получения соединения химической формулы 1-4-4.
В настоящем изобретении соединение, полученное в соответствии с приведенной выше формулой реакции 4, может включать 30, 31, 32, 33, 37 и тому подобное.
[Формула реакции 5]
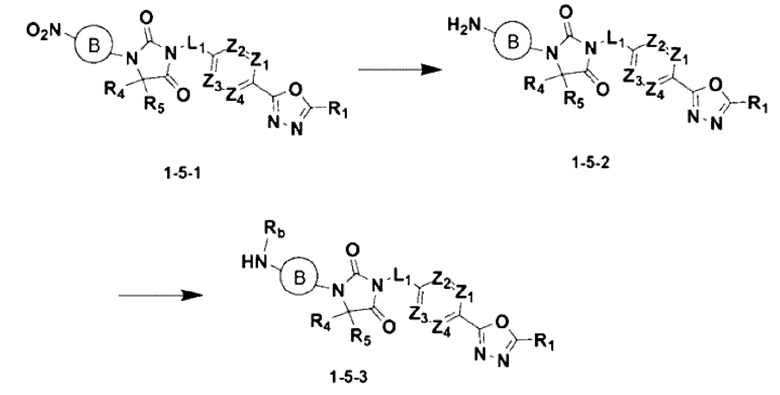
В приведенной выше формуле реакции 5 «Rb» может быть -С(=О)-(С1-С4 алкилом).
Приведенная выше формула реакции 5 показывает способ синтеза соединения, имеющего структуру имидазолидин-2,4-диона, где реакция восстановления может осуществляться с соединением химической формулы 1-5-1, полученным в формуле реакции 1, с добавлением нитро, для получения соединения химической формулы 1-5-2, после чего может быть проведена реакция
восстановительного аминирования или реакция замещения для получения соединения химической формулы 1-5-3.
В настоящем изобретении соединение, полученное в соответствии с приведенной выше формулой реакции 5, может включать 34, 35, 36 и тому подобное.
[Формула реакции 6]
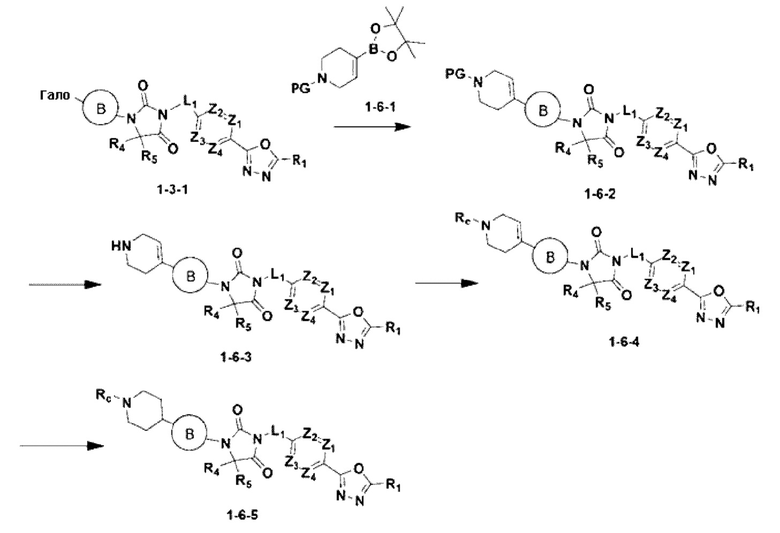
В приведенной выше формуле реакции 6 Rc может быть С1-С4 алкилом.
Приведенная выше формула реакции 6 показывает способ синтеза соединения, имеющего структуру имидазолидин-2,4-диона, где соединение химической формулы 1-3-1, полученное в формуле реакции 1, может быть подвергнуто С-С сочетанию (реакция Сузуки) с соединением химической формулы 1-6-1, имеющим защитную группу, для получения соединения химической формулы 1-6-2. Защитная группа может быть удалена из соединения химической формулы 1-6-2 для получения соединения химической формулы 1-6-3, и затем может быть проведена реакция восстановительного аминирования или реакция замещения для получения соединения химической формулы 1-6-4. После этого может быть проведена реакция восстановления для получения соединения химической формулы 1-6-5.
В настоящем изобретении соединение, полученное в соответствии с приведенной выше формулой реакции 6, может включать 19, 20 и тому подобное.
[Формула реакции 7]
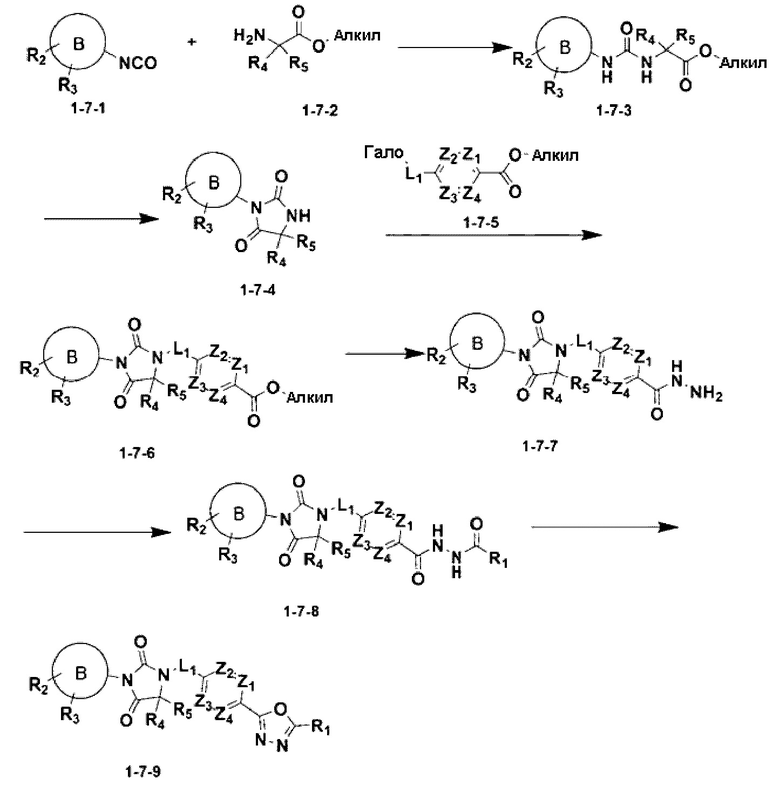
В приведенной выше формуле реакции 7 алкил может быть С1-С4 алкилом.
Приведенная выше формула реакции 7 иллюстрирует способ синтеза соединения, имеющего структуру имидазолидин-2,4-диона, где соединение химической формулы 1-7-1 может реагировать с соединением химической формулы 1-7-2 с получением соединения химической формулы 1-7-3, и реакция циклизации может быть проведена для получения соединения химической формулы 1-7-4. Соединение химической формулы 1-7-6 может быть получено посредством реакции замещения между соединением химической формулы 1-7-4 и соединением химической формулы 1-7-5, после чего соединение химической формулы 1-7-6 может вступать в реакцию с гидразином с получением соединения химической формулы 1-7-7, и затем реагировать с дифторуксусным ангидридом и трифторуксусным ангидридом с получением соединения химической формулы 1-7-8. После этого соединение химической формулы 1-7-8 может быть подвергнуто реакции циклизации с реагентом Бургесса для получения соединения химической формулы 1-7-9.
В настоящем изобретении соединение, полученное в соответствии с приведенной выше формулой реакции 7, может включать 1, 2 и тому подобное.
Композиция, содержащая соединение, представленное химической формулой I, ее применение и терапевтический способ с ее использованием
Настоящее изобретение может предложить фармацевтическую композицию для предупреждения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC), содержащую соединение, представленное химической формулой I, его стереоизомеры или его фармацевтически приемлемые соли в качестве эффективного ингредиента. Предпочтительно настоящее изобретение может предложить фармацевтическую композицию для предупреждения или лечения заболеваний, связанных с активностью HDAC6. Указанная выше химическая формула I такая же, как определено выше.
Фармацевтическая композиция настоящего изобретения может селективно ингибировать HDAC6, тем самым проявляя замечательный эффект в предупреждении или лечении заболеваний, связанных с активностью гистондеацетилазы 6.
Заболевания, опосредованные гистондеацетилазой (HDAC), в частности заболевания, связанные с активностью HDAC6, могут включать инфекционные заболевания, такие как прионная болезнь; новообразование, такое как доброкачественная опухоль (например, миелодиспластический синдром) или злокачественная опухоль (например, множественная миелома, лимфома, лейкемия, рак легких, колоректальный рак, рак толстой кишки, рак предстательной железы, уротелиальная карцинома, рак молочной железы, меланома, рак кожи, рак печени, рак мозга, рак желудка, рак яичников, рак поджелудочной железы, рак головы и шеи, рак ротовой полости или глиома); эндокринопатию, алиментарные и метаболические заболевания, такие как болезнь Вильсона, амилоидоз или диабет; психические и поведенческие нарушения, такие как депрессия или синдром Ретта; неврологические заболевания, такие как атрофия центральной нервной системы (например, болезнь Гентингтона, спинальная мышечная атрофия (SMA), спиноцеребеллярная атаксия (SCA)), нейродегенеративное заболевание (например, болезнь Альцгеймера), двигательное нарушение (например, болезнь Паркинсона), нейропатию (например, наследственная нейропатия (болезнь Шарко-Мари-Тута), спорадическая нейропатия, воспалительная нейропатия, лекарственно-индуцированная нейропатия), моторная нейропатия (например, амиотрофический боковой склероз (ALS)), демиелинизирующее заболевание центральной нервной системы (например, рассеянный склероз (MS)) или тому подобное; глазные и глазные аднексальные заболевания, такие как увеит; заболевания системы кровообращения, такие как мерцание предсердий, инсульт или тому подобное; респираторные заболевания, такие как астма; заболевания органов пищеварения, такие как алкогольная болезнь печени, воспалительная болезнь кишечника, болезнь Крона, язвенная болезнь кишечника или тому подобное; заболевания кожи и подкожной клетчатки, такие как псориаз; заболевания опорно-двигательного аппарата и соединительной ткани, такие как ревматоидный артрит, остеоартрит, системная красная волчанка (SLE) или тому подобное; или тератоз, деформации и хромосомная аберрация, такие как аутосомно-доминантный поликистоз почек, а также могут включать и другие симптомы или заболевания, связанные с нарушением функций гистондеацетилазы.
Фармацевтически приемлемые соли такие же, как описано в фармацевтически приемлемых солях соединения настоящего изобретения.
Для введения фармацевтическая композиция настоящего изобретения может также содержать по меньшей мере один тип фармацевтически приемлемого носителя в дополнение к соединению, его стереоизомерам или его фармацевтически приемлемым солям. Используемый фармацевтически приемлемый носитель может включать физиологический раствор, стерилизованную воду, раствор Рингера, забуференный физиологический раствор, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол и смесь из по меньшей мере одного из их ингредиентов, а также другие обычные добавки, такие как антиоксиданты, буферные растворы, бактериостатические средства и т.д, могут быть добавлены к ним при необходимости. Кроме того, могут быть добавлены разбавители, диспергирующие вещества, поверхностно-активные вещества, связующие и лубриканты для приготовления лекарственных форм для инъекций, таких как водные растворы, суспензии, эмульсии и т.д., драже, капсулы, гранулы или таблетки. Таким образом, композицией по настоящему изобретению могут быть пластыри, жидкие лекарственные средства, драже, капсулы, гранулы, таблетки, суппозитории и т.д. Такие препараты могут быть получены традиционным способом, используемым для приготовления в данной области техники, или способом, описанным в Remington's Pharmaceutical Science (последнее издание), Mack Publishing Company, Easton PA, и такая композиция может быть приготовлена в виде различных препаратов в зависимости от каждого заболевания или компонента.
Композицию по настоящему изобретению можно вводить перорально или парентерально (например, применять внутривенно, подкожно, интраперитонеально или местно) в соответствии с целевым способом, при этом ее доза варьирует в определенном диапазоне в зависимости от веса пациента, возраста, пола, состояния здоровья и диеты, времени введения, способа введения, скорости выведения, тяжести заболевания и тому подобного. Суточная доза соединения настоящего изобретения, его стереоизомеров или его фармацевтически приемлемых солей может составлять примерно 1-1000 мг/кг, предпочтительно 5-100 мг/кг, и может вводиться один раз в день или несколько раз в день путем деления суточной дозы соединения.
Фармацевтическая композиция настоящего изобретения может дополнительно содержать по меньшей мере один эффективный ингредиент, который проявляет такой же или аналогичный лечебный эффект, в дополнение к соединению, его стереоизомерам или его фармацевтически приемлемым солям.
Настоящее изобретение может предложить способ
предупреждения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC), включающий введение терапевтически эффективного количества соединения, представленного приведенной выше химической формулой I, его стереоизомеров или его фармацевтически приемлемых солей; или содержащей их фармацевтической композиции в качестве эффективного ингредиента нуждающемуся в этом субъекту. Заболевания, опосредованные гистондеацетилазой (HDAC), могут быть заболеваниями, связанными с активностью HDAC6.
Используемый в настоящем описании термин «терапевтически эффективное количество» может относиться к количеству соединения, его стереоизомеров или его фармацевтически приемлемых солей, которое эффективно в профилактике или лечении заболеваний, опосредованных гистондеацетилазой (HDAC), в частности заболеваний, связанных с активностью HDAC6.
В настоящем изобретении термин «субъект» может относиться к млекопитающим, включая человека, и термин «введение» может относиться к предоставлению субъекту заданного вещества любым подходящим способом. Специалистам в данной области очевидно, что терапевтически эффективная доза и число введений для эффективного ингредиента настоящего изобретения могут варьировать в зависимости от желаемого эффекта.
В настоящем изобретении термин «предупреждение» может относиться к задержке возникновения заболевания, нарушения или состояния. Если появление заболевания, нарушения или состояния откладывается на ожидаемый период времени, предупреждение можно считать состоявшимся.
В настоящем изобретении термин «лечение» может относиться к лечению, которое частично или полностью уменьшает, улучшает, смягчает, ингибирует или задерживает появление определенного заболевания, нарушения и/или состояния, уменьшает его тяжесть или снижает появление по меньшей мере одного его симптома или свойства.
В дополнение к этому, настоящее изобретение может предложить способ селективного ингибирования HDAC6 с помощью введения терапевтически эффективного количества соединения, представленного приведенной выше химической формулой I, его стереоизомеров или его фармацевтически приемлемых солей; или содержащей их фармацевтической композиции в качестве эффективного ингредиента млекопитающим, включая человека.
Способ предупредения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC), в частности, заболеваний, связанных с активностью HDAC6, согласно настоящему изобретению может включать не только борьбу с самими заболеваниями до проявления их симптомов, но также ингибирование или устранение таких симптомов путем введения соединения, его стереоизомеров или его фармацевтически приемлемых солей. При лечении заболевания профилактическая или терапевтическая доза определенного активного ингредиента может варьировать в зависимости от природы и тяжести заболевания или состояния и способа введения активного компонента. Доза и частота ее применения могут варьировать в зависимости от возраста, веса и реакций конкретного пациента. Подходящая доза и способ применения могут быть легко выбраны специалистами, естественно учитывающими такие факторы. В дополнение к этому, способ предупреждения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC), в частности, заболеваний, связанных с активностью HDAC6, согласно настоящему изобретению может также включать введение терапевтически эффективного количества дополнительного активного агента, который полезен при лечении заболеваний, наряду с соединением, представленным указанной выше химической формулой I, и дополнительный активный агент может проявлять синергетический эффект или аддитивный эффект вместе с данным соединением, его стереоизомерами или его фармацевтически приемлемыми солями.
Настоящее изобретение может также предложить применение соединения, представленного химической формулой I, его стереоизомеров или его фармацевтически приемлемых солей; или содержащей их фармацевтической композиции в качестве эффективного ингредиента для предупреждения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC).
Заболевания, опосредованные гистондеацетилазой (HDAC), могут быть заболеваниями, связанными с активностью HDAC6.
Настоящее изобретение может также предложить применение соединения, представленного указанной выше химической формулой I, его стереоизомеров или его фармацевтически приемлемых солей; или содержащей их фармацевтической композиции в качестве эффективного ингредиента для приготовления лекарственного средства для предупреждения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC). Заболевания, опосредованные гистондеацетилазой (HDAC), могут быть заболеваниями, связанными с активностью HDAC6.
Для приготовления лекарственного средства соединение, его стереоизомеры или его фармацевтически приемлемые соли могут быть объединены с приемлемыми адъювантами, разбавителями, носителями и т.д., и могут быть приготовлены в виде комплексного препарата вместе с другими активными агентами и, таким образом, обладать синергическим действием активных компонентов.
Вещества, упомянутые в применении, композиции и терапевтическом способе настоящего изобретения, применяются в равной степени, если не являются взаимоисключающими.
Полезные эффекты
В соответствии с настоящим изобретением соединение, представленное указанной выше химической формулой I, его стереоизомеры или его фармацевтически приемлемые соли обладают не только ингибирующей HDAC6 активностью, но также на удивление превосходным эффектом предупреждения или лечения заболеваний, связанных с активностью HDAC6, путем селективного ингибирования HDAC6.
К тому же, соединение по изобретению, обладающее селективной ингибирующей активностью в отношении HDAC6, его стереоизомеры или его фармацевтически приемлемые соли может быть успешно использовано для предупреждения или лечения заболеваний, связанных с активностью HDAC6, таких как рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания или нейродегенеративные нарушения и т.д.
Наилучший вариант осуществления изобретения
Далее настоящее изобретение будет описано более подробно с помощью следующих примеров и экспериментальных примеров. Однако эти примеры приведены только с целью иллюстрации настоящего изобретения, и, соответственно, объем настоящего изобретения ими не ограничивается.
Получение соединения
Конкретный способ получения соединения, представленного химической формулой I, является таким, как показано ниже.
Пример 1: Синтез соединения 1, 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-5,5-диметил-3-фенилимидазолидин-2,4-диона
[Стадия 1] Синтез N'-(2,2-дифторацетил)-4-((5,5-диметил-2,4-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фторбензогидразида

4-((5,5-Диметил-2,4-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фторбензогидразид (0,119 г, 0,321 ммоль) и триэтиламин (0,067 мл, 0,482 ммоль) растворяли в дихлорметане (4 мл) при комнатной температуре, после чего 2,2-дифторуксусный ангидрид (0,036 мл, 0,289 ммоль) добавляли в полученный раствор и перемешивали при той же температуре. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 4 г; этилацетат/гексан=20-70%) и концентрировали с получением указанного в заголовке соединения (0,053 г, 36,8%) в форме бесцветного масла.
[Стадия 2] Синтез соединения 1
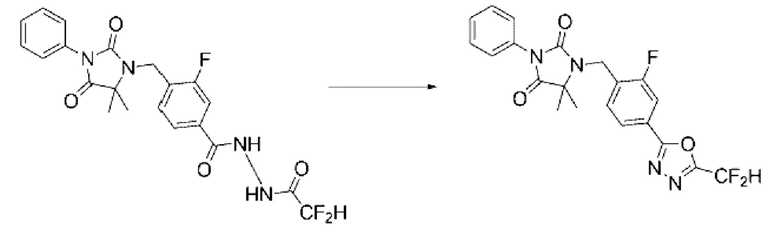
N'-(2,2-дифторацетил)-4-((5,5-диметил-2,4-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фторбензогидразид (0,053 г, 0,118 ммоль), полученный на стадии 1, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,042 г, 0,177 ммоль) смешивали в тетрагидрофуране (4 мл) при комнатной температуре, после чего полученную смесь облучали микроволнами, затем нагревали при 150°С в течение 30 мин, и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором гидрокарбоната, обезвоживали с использованием безводного сульфата магния, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 4 г; этилацетат/гексан=0-40%) и концентрировали с получением указанного в заголовке соединения (0,009 г, 17,7%) в форме бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 7, 94 (д, J=8,2 Гц, 1Н), 7,88 (д, J=10,0 Гц, 1Н), 7,53 ~ 7,47 (м, 4Н), 7,43 ~ 7,39 (м, 1Н), 6,95 (т, J=51,6 Гц, 1Н), 4,77 (с, 2Н); МСНР (ES) m/z 431,0 (М++1).
Синтез соединения 2, 1-(2-фтор-4-(5-(трифторметил)-1,3,4-оксадиазол-2-ил)бензил)-5,5-диметил-3-фенилимидазолидин-2,4-диона
[Стадия 1] Синтез метил-2-метил-2-(3-фенилуреидо)пропаноата

Изоцианатобензол (1,000 г, 8,395 ммоль), метил-2-амино-2-метилпропаноат (1,418 г, 9,234 ммоль) и триэтиламин (1,280 мл, 9,234 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 8 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего полученный продукт использовали без дополнительного процесса очистки (1,900 г, 95,8%, белое твердое вещество).
[Стадия 2] Синтез 5,5-диметил-3-фенилимидазолидин-2, 4-диона

Метил-2-метил-2-(3-фенилуреидо)пропаноат (1,900 г, 8,028 ммоль), полученный на стадии 1, и 4М водный раствор соляной кислоты (4,00 М раствор в диоксане, 8,028 мл, 32,112 ммоль) растворяли в метаноле (20 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 12 ч. Б реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата магния, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-30%) и концентрировали с получением указанного в заголовке целевого соединения (1,300 г, 79,3%) в виде белого твердого вещества.
[Стадия 3] Синтез метил-4-((5,5-диметил-2,4-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фторбензоата

5,5-Диметил-3-фенилимидазолидин-2,4-дион (0,413 г, 2,022 ммоль), полученный на стадии 2, растворяли в N, N-диметилформамиде (15 мл) при 0°С, после чего гидрид натрия (0,073 г, 3,033 ммоль) добавляли в полученный раствор и перемешивали при той же температуре в течение 30 мин. Метил-4-(бромметил)-3-фторбензоат (0,500 г, 2,022 ммоль) добавляли в реакционную смесь и далее перемешивали при комнатной температуре в течение 12 ч. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата магния, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-30%) и концентрировали с получением указанного в заголовке целевого соединения (0,265 г, 35,4%) в виде бесцветного масла.
[Стадия 4] Синтез 4-((5,5-диметил-2,4-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фторбензогидразида
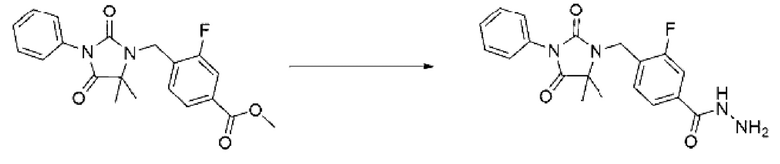
Метил-4-((5,5-диметил-2,4-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фторбензоат (0,265 г, 0,715 ммоль), полученный на стадии 3, и гидразинмоногидрат (0,676 мл, 14,310 ммоль) смешивали в этаноле (10 мл), затем нагревали при 120°С в течение 1 ч путем облучения микроволнами, и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата магния, фильтровали и концентрировали при пониженном давлении. Полученный продукт использовали без дополнительного процесса очистки (0,220 г, 83,0%, белое пенообразное твердое вещество).
[Стадия 5] Синтез 4 -((5,5-диметил-2,4-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фтор-N'-(2,2,2-трифторацетил)бензогидразида
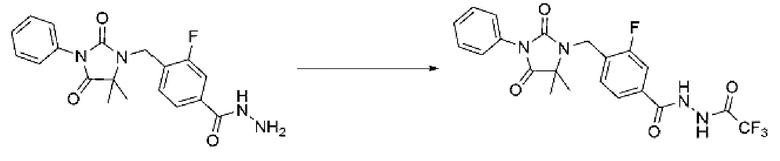
4-((5,5-Диметил-2,4-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фторбензогидразид (0,108 г, 0,292 ммоль), полученный на стадии 4, трифторуксусный ангидрид (0,037 мл, 0,262 ммоль) и триэтиламин (0,061 мл, 0,437 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 1 ч. В реакционную смесь вливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата магния, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; дихлорметан/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке целевого соединения (0,084 г, 61,8%) в виде бесцветного масла.
[Стадия 6] Синтез соединения 2

4-((5,5-Диметил-2,4-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фтор-N'-(2,2,2-трифторацетил)бензогидразид (0, 084 г, 0,180 ммоль), полученный на стадии 5, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,064 г, 0,270 ммоль) смешивали в тетрагидрофуране (10 мл), после чего полученную смесь облучали микроволнами, затем нагревали при 150°С в течение 30 мин, и далее реакцию завершали путем понижения температуры до комнатной температуры. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата магния, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением целевого соединения (0,040 г, 49,5%) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 7,95 ~ 7,92 (м, 1Н), 7,88 (дд, J=9,9, 1,5 Гц, 1Н), 7,78 (т, J=7,7 Гц, 1Н), 7,54 ~ 7,38 (м, 5Н), 4,77 (с, 2Н), 1,48 (с, 9Н).
Синтез соединения 3, 3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1-фенилимидазолидин-2,4-диона
[Стадия 1] Синтез 4-((2,5-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фторбензогидразида
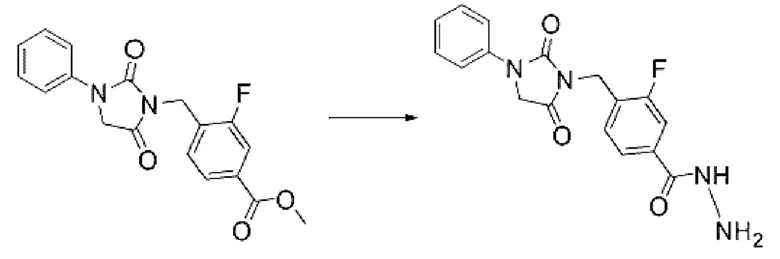
Метил-4-((4,4-диметил-2,5-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фторбензоат (0,175 г, 0,511 ммоль) и гидразинмоногидрат (0,497 мл, 10,224 ммоль) растворяли в этаноле (3 мл) при комнатной температуре, после чего полученный раствор перемешивали при 120°С в течение 1 ч, и далее реакцию завершали путем понижения температуры до комнатной температуры. Осажденное твердое вещество фильтровали, промывали этанолом и сушили с получением указанного в заголовке соединения (0,100 г, 57,1%) в виде белого твердого вещества.
[Стадия 2] Синтез N'-(2,2-дифторацетил)-4 -((2,5-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фторбензогидразида

4-((2,5-Диоксо-3-фенилимидазолидин-1-ил)метил) -3-фторбензогидразид (0,100 г, 0,292 ммоль), полученный на стадии 1, и триэтиламин (0,061 мл, 0,438 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего 2,2-дифторуксусный ангидрид (0,029 мл, 0,263 ммоль) добавляли в полученный раствор и перемешивали при той же температуре в течение 17 ч. Осажденное твердое вещество фильтровали, промывали дихлорметаном и сушили с получением указанного в заголовке соединения (0,100 г, 81,4%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 3
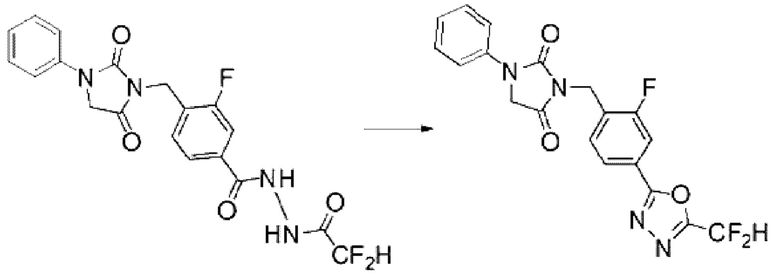
N'-(2,2-дифторацетил)-4-((2,5-диоксо-3-фенилимидазолидин-1-ил)метил)-3-фторбензогидразид (0,100 г, 0,238 ммоль), полученный на стадии 2, и 1-метокси-П-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,085 г, 0,357 ммоль) смешивали в тетрагидрофуране (3 мл) при комнатной температуре, после чего полученную смесь облучали микроволнами, затем нагревали при 150°С в течение 30 мин, и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и экстракцию проводили дихлорметаном, фильтровали через пластиковый фильтр для удаления оттуда твердого остатка и слоя водного раствора, и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 4 г; этилацетат/гексан=5-70%) и концентрировали с получением указанного в заголовке соединения (0,019 г, 19,9%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 7,89 (дд, J=4,8, 1,3 Гц, 1Н), 7,87 (т, J=2,2 Гц, 1Н), 7,69 ~ 7,38 (м, 6Н), 7,14 (т, J=7,4 Гц, 1Н), 4,79 (с, 2Н), 4,61 (с, 2Н).
Синтез соединения 4, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-фенилимидазолидин-2,4-диона
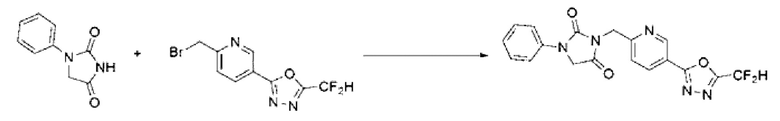
1-Фенилимидазолидин-2,4-дион (0,200 г, 1,135 ммоль) растворяли в N, N-диметилформамиде (10 мл) при 0°С, после чего гидрид натрия (60,00%, 0,068 г, 1,703 ммоль) добавляли в полученный раствор и перемешивали при той же температуре в течение 30 мин. 2-(6-(Бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,329 г, 1,135 ммоль) добавляли в реакционную смесь и далее перемешивали при комнатной температуре в течение 3 ч. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-30%) и концентрировали с получением указанного в заголовке соединения (0,100 г, 22,9%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,27 ~ 9,26 (м, 1Н), 8,39 (дд, J=8,2, 2,2 Гц, 1Н), 7,61 (дд, J=7,8, 1,1 Гц, 2Н), 7,54 (дд, J=8,2, 0,7 Гц, 1Н), 7,44 ~ 7,40 (м, 2Н), 7,20 ~ 7,18 (м, 1Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,03 (с, 2Н), 4,47 (с, 2Н); МСНР (ES) m/z 386, 4 (М++1).
Синтез соединения 5, трет-бутил-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3-фторфенил)-2,4-диоксо-1, 3, 8-триазаспиро[4.5]декан-8-карбоксилата
[Стадия 1] Синтез трет-бутил-4-((3-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-3-фенилуреидо)метил)пиперидин-1-карбоксилата
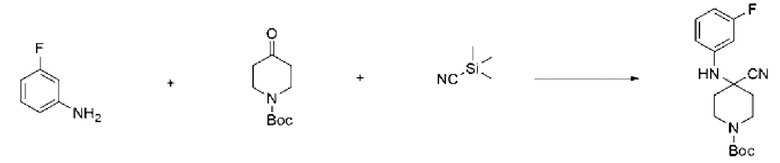
3-Фторанилин (1,000 г, 8,999 ммоль), трет-бутил-4-оксопиперидин-1-карбоксилат (1,793 г, 8,999 ммоль) и триметилсилакарбонитрил (0,893 г, 8,999 ммоль) растворяли в уксусной кислоте (30 мл), после чего полученный раствор перемешивали при 0°С в течение 30 мин и далее перемешивали при комнатной температуре в течение 18 ч. Б реакционную смесь вливали насыщенный водный раствор хлорида аммония и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-30%) и концентрировали с получением указанного в заголовке соединения (1,850 г, 64,4%) в виде белого твердого вещества.
[Стадия 2] Синтез трет-бутил-1-(3-фторфенил)-2,4-диоксо-1,3,8-триазаспиро[4.5]декан-8-карбоксилата
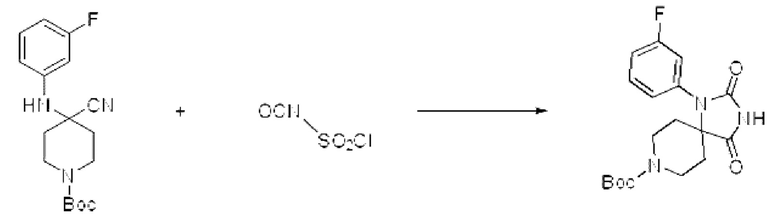
Трет-бутил-4-циано-4-((3-фторфенил)амино)пиперидин-1-карбоксилат (1,850 г, 5,792 ммоль), полученный на стадии 1, растворяли в дихлорметане (5 мл), после чего в полученный раствор добавляли хлорсульфонилизоцианат (1,230 г, 8,689 ммоль) при 0°С и перемешивали в течение 30 мин. Б реакционную смесь вливали 1 н. водный раствор соляной кислоты (10 мл), после чего растворитель концентрировали при пониженном давлении и затем добавляли этанол (15 мл). Полученную смесь снова перемешивали при температуре 80°С в течение 30 мин, после чего растворитель концентрировали при пониженном давлении. После этого полученную смесь растворяли в THF (20 мл) и рН доводили до 8 10% раствором карбоната калия, после чего добавляли ди-трет-бутилдикарбонат (1,896 г, 8,689 ммоль), растворенный в THF (20 мл), и перемешивали в течение 18 ч. После этого твердый осадок отфильтровывали с получением указанного в заголовке целевого соединения (1,23 г, 59,9%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 5
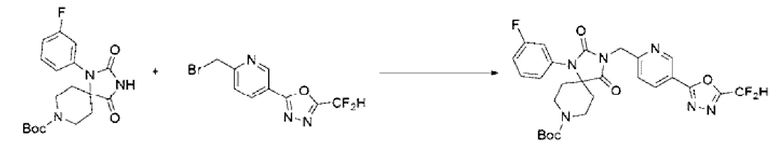
Трет-бутил-1-(3-фторфенил)-2,4-диоксо-1,3,8-триазаспиро[4.5]декан-8-карбоксилат (0,600 г, 1,651 ммоль), полученный на стадии 2, растворяли в N, N-диметилформамиде (10 мл) при 0°С, после чего гидрид натрия (60,00%, 0,099 г, 2,477 ммоль) добавляли в полученный раствор и перемешивали при той же температуре в течение 30 мин. 2-(6-(Бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,718 г, 2,477 ммоль) добавляли в реакционную смесь и далее перемешивали при комнатной температуре в течение 3 ч. Б реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,257 г, 27,2%) в форме бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 9,22 (т, J=1,1 Гц, 1Н), 8,34 (дд, J=8,2, 2,2 Гц, 1Н), 7,48 ~ 7,39 (м, 23.), 7,20 ~ 7,15 (м, 1Н), 7,06 (с, 0,25Н), 7,01 ~ 7,00 (м, 1Н), 6,98 (с, 0,5Н), 6,99 ~ 6,94 (м, 1Н), 6,92 (с, 0,25Н), 4,95 (с, 2Н), 4,10 - 3,95 (м, 2Н), 3,50 ~ 3,40 (м, 2Н), 1,99 ~ 1,95 (м, 2Н), 1,80 ~ 1,75 (м, 2Н), 1,36 (с, 9Н); МСНР (ES) m/z 573,4 (М++1).
Синтез соединения 6, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3-фторфенил)-8-метил-1,3,8-триазаспиро[4.5]декан-2,4-диона
[Стадия 1] Синтез 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3-фторфенил)-1,3,8-триазаспиро[4.5]декан-2,4-дион-2,2,2-трифторацетата
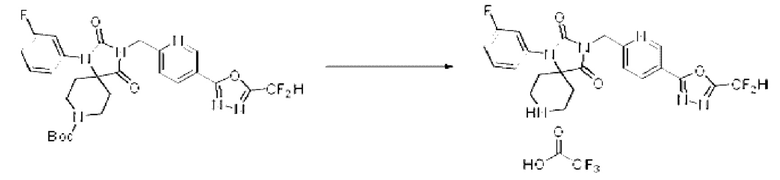
Трет-бутил-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3-фторфенил)-2,4-диоксо-1,3,8-триазаспиро[4.5]декан-8-карбоксилат (0,257 г, 0,449 ммоль) и трифторуксусную кислоту (0,344 мл, 4,489 ммоль) растворяли в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 12 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего полученный продукт использовали без дополнительного процесса очистки (0,250 г, 95,0%, желтое масло).
[Стадия 2] Синтез соединения 6

3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3-фторфенил)-1,3,8-триазаспиро[4.5]декан-2,4-дион-2,2,2-трифторацетат (0,200 г, 0,341 ммоль), полученный на стадии 1, формальдегид (0,020 г, 0,682 ммоль), N, N-диизопропилэтиламин (0,059 мл, 0,341 ммоль) и триацетоксиборгидрид натрия (0,145 г, 0,682 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 2 ч. В реакционную смесь вливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; метанол/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке соединения (0,100 г, 60,3%) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 9,28 (д, J=1,4 Гц, 1Н), 8,41 (дд, J=8,2, 2,2 Гц, 1Н), 7,51 (д, J=8,2 Гц, 1Н), 7,47 ~ 7,41 (м, 1Н), 7,18 ~ 7,14 (м, 1Н), 7,08 (с, 0,25Н), 7,05 ~ 7,03 (м, 1Н), 7,00 ~ 6,97 (м, 1Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,00 (с, 2Н), 3,20 ~ 2,90 (м, 4Н), 2,48 (с, 3Н), 2,23 ~ 2,20 (м, 2Н), 2,08 ~ 2,05 (м, 2Н); МСНР (ES) m/z 487, 5 (М++1).
Синтез соединения 7, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3-фторфенил)-8-(оксетан-3-ил)-1,3,8-триазаспиро[4.5]декан-2,4-диона
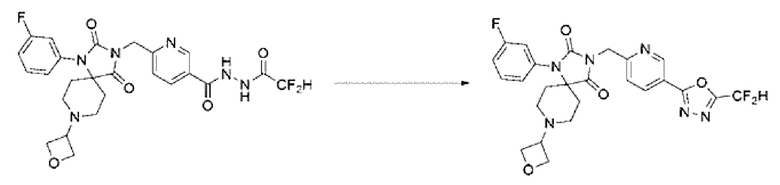
N'-(2, 2-дифторацетил)-6-((1-(3-фторфенил)-8-(оксетан-3-ил)-2,4-диоксо-1,3,8-триазаспиро[4,5]декан-3-ил)метил)никотиногидразид (0,060 г, 0,110 ммоль) и 1-метокси-1\[-триэтиламмониосульфонил-метанимидат (реагент Бургесса, 0,052 г, 0,220 ммоль) растворяли в тетрагидрофуране (5 мл) при 80°С, после чего полученный раствор перемешивали при той же температуре в течение 13 ч, и далее реакцию завершали путем понижения температуры до комнатной температуры. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-100%) и концентрировали с получением указанного в заголовке соединения (0,020 г, 34,5%) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 9,28 (дд, J=2,2, 0,8 Гц, 1Н), 8,39 (дд, J=8,2, 2,2 Гц, 1Н), 7,51 ~ 7,49 (м, 1Н), 7,47 ~ 7,43 (м, 1Н), 7,21 ~ 7,16 (м, 1Н), 7,08 ~ 7,05 (м, 1Н), 7,08 (с, 0,25Н), 7,01 ~ 6,98 (м, 1Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 4,99 (с, 2Н), 4,66 (т, J=6,6 Гц, 2Н), 4,55 (т, J=5,0 Гц, 2Н), 3,61 ~ 3,58 (м, 1Н), 2,74 ~ 2,66 (м, 4Н), 2,11 ~ 2,04 (м, 4Н).
Синтез соединения 8, 3-((5-(5-(дифторметил)-1, 3, 4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3-фторфенил)-8-изопропил-1,3,8-триазаспиро[4.5]декан-2,4-диона
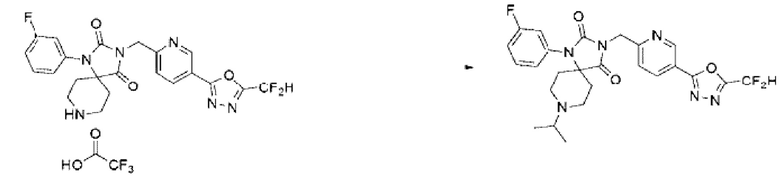
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3-фторфенил)-1,3,8-триазаспиро[4.5]декан-2,4-дион-2,2,2-трифторацетат (0,200 г, 0,341 ммоль), ацетон (0,051 мл, 0,682 ммоль), N, N-диизопропилэтиламин (0,059 мл, 0,341 ммоль) и триацетоксиборгидрид натрия (0,145 г, 0,682 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; метанол/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке соединения (0,110 г, 62,8%) в виде белого пенообразного твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,29 (дд, J=2,l, 0,7 Гц, 1Н), 8,41 (дд, J=8,2, 2,2 Гц, 1Н), 7,53 ~ 7,50 (м, 1Н), 7,47 ~ 7,43 (м, 1Н), 7,19 ~ 7,16 (м, 1Н), 7,08 (с, 0,25Н), 7,01 ~ 7,00 (м, 1Н), 6,99 ~ 6,98 (м, 1Н), 6,96 (с, 0,5Н), 6,83 (с, 0,25Н), 5,00 (с, 2Н), 3,28 ~ 3,25 (м, 1Н), 3,10 ~ 3,08 (м, 4Н), 2,33 ~ 2,30 (м, 2Н), 2,09 ~ 2,06 (м, 2Н), 1,18 ~ 1,13 (м, 6Н); МСНР (ES) m/z 515, 5 (М++1).
Синтез соединения 9, 3-((5-(5-(дифторметил)-1, 3, 4-оксадиазол-2-ил)пиримидин-2-ил)метил)-1-фенилимидазолидин-2,4-диона
1-Фенилимидазолидин-2,4-дион (0,300 г, 1,703 ммоль), 2-(2-(бромметил)пиримидин-5-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,496 г, 1,703 ммоль) и карбонат калия (0,353 г, 2,554 ммоль) растворяли в N, N-диметилформамиде (5 мл) при 80°С, после чего полученный раствор перемешивали при той же температуре в течение 12 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,110 г, 16,7%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,7 (с, 2Н), 7,65 ~ 7,62 (м, 2Н), 7,46 ~ 7,42 (м, 2Н), 7,23 ~ 7,19 (м, 1Н), 7,10 (с, 0,25Н), 6,97 (с, 0,5Н), 6,84 (с, 0,25Н), 5,18 (с, 2Н), 4,52 (с, 2Н); МСНР (ES) m/z 387,3 (М++1).
Синтез соединения 10, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3-фторфенил)-5,5-диметилимидазолидин-2,4-диона
[Стадия 1] Синтез 2-((3-фторфенил)амино)-2-метилпропаннитрила

3-Фторанилин (1,000 г, 8,999 ммоль), триметилсилакарбонитрил (0,893 г, 8,999 ммоль) и пропан-2-он (0,523 г, 8,999 ммоль) растворяли в ацетоне (20 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 12 ч. В реакционную смесь вливали насыщенный водный раствор хлорида аммония и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 40 г; этилацетат/гексан=0-20%) и концентрировали с получением указанного в заголовке соединения (1,240 г, 77,3%) в виде белого твердого вещества.
[Стадия 2] Синтез 1-(3-фторфенил)-5,5-диметилимидазолидин-2,4-диона

2-(3-Фторфенил)-2-метилпропаннитрил (1,240 г, 7, 598 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (1,613 г, 11,397 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 1 ч. В реакционную смесь вливали 1 н. водный раствор соляной кислоты (10 мл), после чего растворитель концентрировали при пониженном давлении и затем добавляли этанол (15 мл). Полученную смесь снова перемешивали при температуре 80°С в течение 30 мин, после чего растворитель концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-30%) и концентрировали с получением указанного в заголовке соединения (0,880 г, 52,1%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 10
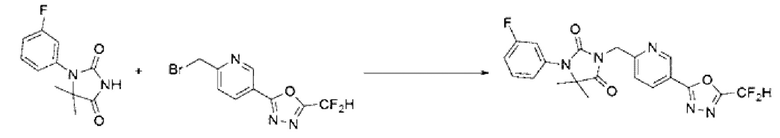
1-(3-Фторфенил)-5,5-диметилимидазолидин-2,4-дион (0,100 г, 0,450 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,144 г, 0,495 ммоль) и карбонат калия (0,124 г, 0,900 ммоль) растворяли в N, N-диметилформамиде (10 мл), после чего полученный раствор перемешивали при 50°С в течение 18 ч и затем дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-80%) и концентрировали с получением указанного в заголовке соединения (0,130 г, 67,0%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,26 (т, J=1,1 Гц, 1Н), 8,37 (дд, J=8,2, 2,2 Гц, 1Н), 7,49 (д, J=8,2 Гц, 1Н), 7,43 ~ 7,40 (м, 1Н), 7,14 ~ 7,06 (м, 3Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,00 (с, 2Н), 1,55 (с, 6Н); МСНР (ES) m/z 432,3 (М++1).
Синтез соединения 11, 1-(3-бромфенил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметилимидазолидин-2,4-диона
[Стадия 1] Синтез 2-((3-бромфенил)амино)-2-метилпропаннитрила

3-Броманилин (2,000 г, 11,626 ммоль), триметилсилакарбонитрил (1,153 г, 11,626 ммоль) и пропан-2-он (0,675 г, 11,626 ммоль) растворяли в ацетоне (20 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 12 ч. В реакционную смесь вливали насыщенный водный раствор хлорида аммония и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 40 г; этилацетат/гексан=0-20%) и концентрировали с получением указанного в заголовке соединения (2,200 г, 79,1%) в виде коричневого масла.
[Стадия 2] Синтез 1-(3-бромфенил)-5,5-диметилимидазолидин-2,4-диона
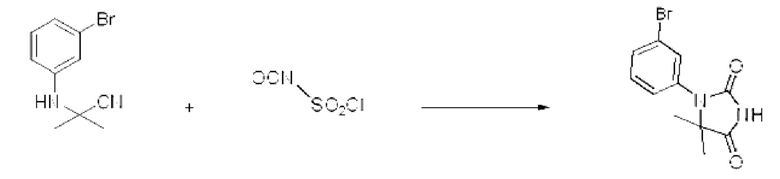
2-(3-Бромфенил)-2-метилпропаннитрил (2,200 г, 9,817 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (2,084 г, 14,726 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 1 ч. В реакционную смесь вливали 1 н. водный раствор соляной кислоты (10 мл), после чего растворитель концентрировали при пониженном давлении и затем добавляли этанол (15 мл). Полученную смесь снова перемешивали при температуре 80°С в течение 30 мин, после чего растворитель концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-30%) и концентрировали с получением указанного в заголовке соединения (1,500 г, 54,0%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 11
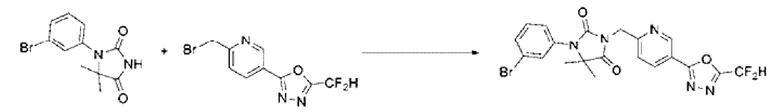
1-(3-Бромфенил)-5,5-диметилимидазолидин-2,4-дион (0,892 г, 3,150 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,005 г, 3,466 ммоль) и карбонат калия (0,871 г, 6,301 ммоль) растворяли в N, N-диметилформамиде (10 мл), после чего полученный раствор перемешивали при 50°С в течение 18 ч и затем дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-80%) и концентрировали с получением указанного в заголовке соединения (1,100 г, 70,9%) в виде желтого масла.
1Н ЯМР (400 МГц, CDCl3) δ 9,30 (т, J=1,1 Гц, 1Н), 8,41 (дд, J=8,2, 2,2 Гц, 1Н), 7,57 ~ 7,55 (м, 1Н), 7,51 ~ 7,49 (м, 1Н), 7,36 (т, J=8,0 Гц, 1Н), 7,30 ~ 7,27 (м, 2Н), 7,09 (с, 0,25Н), 6,95 (с, 0,5Н), 6,83 (с, 0,25Н), 5,03 (с, 2Н), 1,59 (с, 6Н); МСНР (ES) m/z 494, 2 (М++1).
Синтез соединения 12, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(4-фторфенил)-5,5-диметилимидазолидин-2,4-диона
[Стадия 1] Синтез 2-((4-фторфенил)амино)-2-метилпропаннитрила
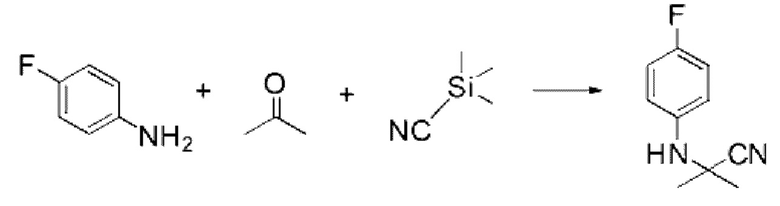
4-Фторанилин (1,000 г, 8,999 ммоль), триметилсилакарбонитрил (0,893 г, 8,999 ммоль) и пропан-2-он (0,523 г, 8,999 ммоль) растворяли в ацетоне (20 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 12 ч. В реакционную смесь вливали насыщенный водный раствор хлорида аммония и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 40 г; этилацетат/гексан=0-20%) и концентрировали с получением указанного в заголовке соединения (0,535 г, 33,4%) в виде белого твердого вещества.
[Стадия 2] Синтез 1-(4-фторфенил)-5,5-диметилимидазолидин-2,4-диона

2-(4-Фторфенил)-2-метилпропаннитрил (0,530 г, 3,248 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (0,689 г, 4,871 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 1 ч. В реакционную смесь вливали 1 н. водный раствор соляной кислоты (10 мл), после чего растворитель концентрировали при пониженном давлении и затем добавляли этанол (15 мл). Полученную смесь снова перемешивали при температуре 80°С в течение 30 мин, после чего растворитель концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-30%) и концентрировали с получением указанного в заголовке соединения (0,330 г, 45,7%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 12

1-(4-Фторфенил)-5,5-диметилимидазолидин-2,4-дион (0,100 г, 0,450 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,144 г, 0,495 ммоль) и карбонат калия (0,124 г, 0,900 ммоль) растворяли в N, N-диметилформамиде (10 мл), после чего полученный раствор перемешивали при 50°С в течение 18 ч и затем дополнительно перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-80%) и концентрировали с получением указанного в заголовке соединения (0,110 г, 56,7%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,27 (т, J=1,1 Гц, 1Н), 8,38 (дд, J=8,2, 2,2 Гц, 1Н), 7,49 (д, J=8,2 Гц, 1Н), 7,30 ~ 7,26 (м, 2Н), 7,17 ~ 7,13 (м, 2Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,01 (с, 2Н), 1,52 (с, 6Н); МСНР (ES) m/z 432, 3 (М++1).
Синтез соединения 13, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(2,б-дифторфенил)-5,5-диметилимидазолидин-2, 4-диона
[Стадия 1] Синтез 2-((2,6-дифторфенил)амино)-2-метилпропаннитрила
2,6-Дифторанилин (0,781 мл, 7,745 ммоль), триметилсилакарбонитрил (0,973 мл, 7,745 ммоль) и пропан-2-он (0,569 мл, 7,745 ммоль) растворяли в уксусной кислоте (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли гексан (20 мл) и этилацетат (10 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (0,330 г, 21,7%) в виде белого твердого вещества.
[Стадия 2] Синтез 1-(2,6-дифторфенил)-5,5-диметилимидазолидин-2,4-диона

2-((2,6-Дифторфенил)амино)-2-метилпропаннитрил (0,330 г, 1,682 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (0,357 г, 2,523 ммоль) растворяли в дихлорметане (50 мл), после чего полученный раствор перемешивали при 0°С в течение 30 мин и далее перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь вливали 1 н. водный раствор соляной кислоты (10 мл), после чего растворитель концентрировали при пониженном давлении и затем добавляли этанол (30 мл). Полученную смесь снова перемешивали при температуре 80°С в течение 30 мин, после чего растворитель концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,100 г, 24,8%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 13

1-(2,6-Дифторфенил)-5,5-диметилимидазолидин-2,4-дион (0,100 г, 0,416 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,121 г, 0,416 ммоль) и карбонат калия (0,115 г, 0,833 ммоль) растворяли в N, N-диметилформамиде (5 мл) при 80°С, после чего полученный раствор перемешивали при той же температуре в течение 18 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,150 г, 80,2%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,30 (дд, J=2,2, 0,8 Гц, 1Н), 8,40 (дд, J=8,2, 2,2 Гц, 1Н), 7,48 ~ 7,39 (м, 2Н), 7,09 ~ 7,05 (м, 2Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,04 (с, 2Н), 1,54 (с, 6Н); МСНР (ES) m/z 450, 2 (М++1).
Синтез соединения 14, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(2,4-дифторфенил)-5,5-диметилимидазолидин-2, 4-диона
[Стадия 1] Синтез 2-((2,4-дифторфенил)амино)-2-метилпропаннитрила
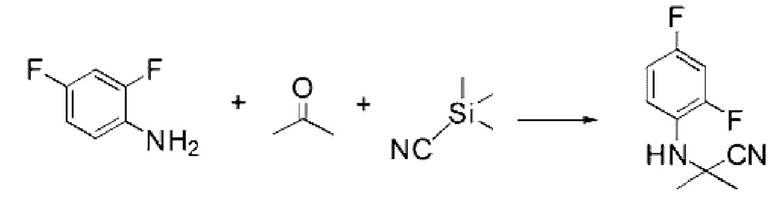
2,4-Дифторанилин (1,000 г, 7,745 ммоль), триметилсилакарбонитрил (0,768 г, 7,745 ммоль) и пропан-2-он (0,450 г, 7,745 ммоль) растворяли в уксусной кислоте (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли гексан (20 мл) и этилацетат (10 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (1,000 г, 65,8%) в виде белого твердого вещества.
[Стадия 2] Синтез 1-(2,4-дифторфенил)-5,5-диметилимидазолидин-2,4-диона
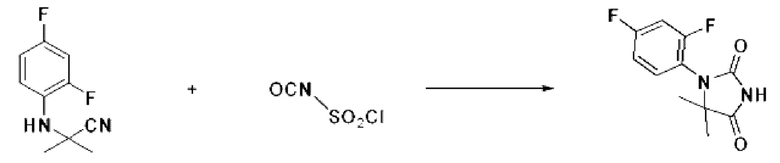
2-((2,4-Дифторфенил)амино)-2-метилпропаннитрил (1,000 г, 5,097 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (1,082 г, 7,645 ммоль) растворяли в дихлорметане (50 мл), после чего полученный раствор перемешивали при 0°С в течение 30 мин и далее перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь вливали 1 н. водный раствор соляной кислоты (10 мл), после чего растворитель концентрировали при пониженном давлении и затем добавляли этанол (30 мл). Полученную смесь снова перемешивали при температуре 80°С в течение 30 мин, после чего растворитель концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/пексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,700 г, 57,2%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 14

1-(2,4-Дифторфенил)-5,5-диметилимидазолидин-2,4-дион (0,100 г, 0,416 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,121 г, 0,416 ммоль) и карбонат калия (0,115 г, 0,833 ммоль) растворяли в N, N-диметилформамиде (5 мл) при 80°С, после чего полученный раствор перемешивали при той же температуре в течение 18 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,130 г, 69,5%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,30 (д, J=2,2 Гц, 1Н), 8,40 (дд, J=8,2, 2,2 Гц, 1Н), 7,49 (д, J=8,2 Гц, 1Н), 7,31 ~ 7,28 (м, 1Н), 7,08 (с, 0,25Н), 7,04 ~ 6,99 (м, 2Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,03 (с, 2Н), 1,52 (с, 6Н); МСНР (ES) m/z 450, 2 (М++1).
Синтез соединения 15, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(2,3-дифторфенил)-5,5-диметилимидазолидин-2,4-диона
[Стадия 1] Синтез 2-((2,3-дифторфенил)амино)-2-метилпропаннитрила

2,3-Дифторанилин (1,000 г, 7,745 ммоль), триметилсилакарбонитрил (0,768 г, 7,745 ммоль) и пропан-2-он (0,450 г, 7,745 ммоль) растворяли в уксусной кислоте (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли пексан (20 мл) и этилацетат (10 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (1,100 г, 72,4%) в виде белого твердого вещества.
[Стадия 2] Синтез 1-(2,3-дифторфенил)-5,5-диметилимидазолидин-2,4-диона

2-((2,3-Дифторфенил)амино)-2-метилпропаннитрил (1,100 г, 5,607 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (1,190 г, 8,410 ммоль) растворяли в дихлорметане (50 мл), после чего полученный раствор перемешивали при 0°С в течение 30 мин и далее перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь вливали 1 н. водный раствор соляной кислоты (10 мл), после чего растворитель концентрировали при пониженном давлении и затем добавляли этанол (30 мл). Полученную смесь снова перемешивали при температуре 80°С в течение 30 мин, после чего растворитель концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,800 г, 59,4%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 15

1-(2,3-Дифторфенил)-5,5-диметилимидазолидин-2,4-дион (0,100 г, 0,416 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,121 г, 0,416 ммоль) и карбонат калия (0,115 г, 0,833 ммоль) растворяли в N, N-диметилформамиде (5 мл) при 80°С, после чего полученный раствор перемешивали при той же температуре в течение 18 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/пексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,150 г, 80,2%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,30 (дд, J=3,0, 1,7 Гц, 1Н), 8,41 (дд, J=8,2, 2,2 Гц, 1Н), 7,50 (дд, J=8,2, 0,7 Гц, 1Н), 7,31 ~ 7,28 (м, 1Н), 7,21 ~ 7,19 (м, 1Н), 7,12 ~ 7,08 (м, 1Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,04 (с, 2Н), 1,54 (с, 6Н); МСНР (ES) m/z 450, 2 (М++1).
Синтез соединения 16, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3,4-дифторфенил)-5,5-диметилимидазолидин-2,4-диона
[Стадия 1] Синтез 2-((3,4-дифторфенил)амино)-2-метилпропаннитрила
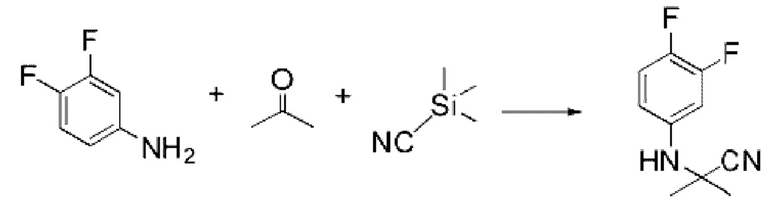
3,4-Дифторанилин (1,000 г, 7,745 ммоль), триметилсилакарбонитрил (0,768 г, 7,745 ммоль) и пропан-2-он (0,450 г, 7,745 ммоль) растворяли в уксусной кислоте (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли гексан (20 мл) и этилацетат (10 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (0,700 г, 46,1%) в виде белого твердого вещества.
[Стадия 2] Синтез 1-(3,4-дифторфенил)-5,5-диметилимидазолидин-2,4-диона
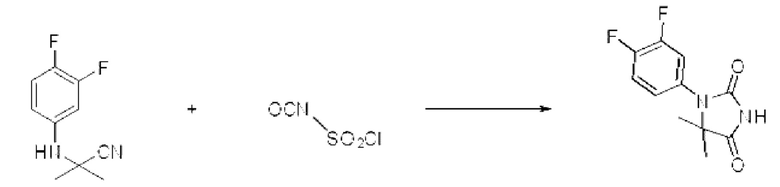
2-((3,4-Дифторфенил)амино)-2-метилпропаннитрил (0,700 г, 3,568 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (0,757 г, 5,352 ммоль) растворяли в дихлорметане (50 мл), после чего полученный раствор перемешивали при 0°С в течение 30 мин и далее перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь вливали 1 н. водный раствор соляной кислоты (10 мл), после чего растворитель концентрировали при пониженном давлении и затем добавляли этанол (30 мл). Полученную смесь снова перемешивали при температуре 80°С в течение 30 мин, после чего растворитель концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,450 г, 52,5%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 16
1-(3,4-Дифторфенил)-5,5-диметилимидазолидин-2,4-дион (0,100 г, 0,416 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,121 г, 0,416 ммоль) и карбонат калия (0,115 г, 0,833 ммоль) растворяли в N, N-диметилформамиде (5 мл) при 80°С, после чего полученный раствор перемешивали при той же температуре в течение 18 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,130 г, 69,5%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,28 (т, J=1,1 Гц, 1Н), 8,40 (дд, J=8,2, 2,2 Гц, 1Н), 7,51 ~ 7,49 (м, 1Н), 7,28 ~ 7,19 (м, 2Н), 7,09 ~ 7,08 (м, 1Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,01 (с, 2Н), 1,55 (с, 6Н); МСНР (ES) m/z 450, 4 (М++1).
Синтез соединения 17, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3,5-дифторфенил)-5,5-диметилимидазолидин-2,4-диона
[Стадия 1] Синтез 2-((3,5-дифторфенил)амино)-2-метилпропаннитрила

3,5-Дифторанилин (1,000 г, 7,745 ммоль), триметилсилакарбонитрил (0,768 г, 7,745 ммоль) и пропан-2-он (0,450 г, 7,745 ммоль) растворяли в уксусной кислоте (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли гексан (20 мл) и этилацетат (10 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (1,100 г, 72,4%) в виде белого твердого вещества.
[Стадия 2] Синтез 1-(3,5-дифторфенил)-5,5-диметилимидазолидин-2, 4-диона
2-((3,5-Дифторфенил)амино)-2-метилпропаннитрил (1,100 г, 5,607 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (1,190 г, 8,410 ммоль) растворяли в дихлорметане (50 мл), после чего полученный раствор перемешивали при 0°С в течение 30 мин и далее перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь вливали 1 н. водный раствор соляной кислоты (10 мл), после чего растворитель концентрировали при пониженном давлении и затем добавляли этанол (30 мл). Полученную смесь снова перемешивали при температуре 80°С в течение 30 мин, после чего растворитель концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/пексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,800 г, 59,4%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 17
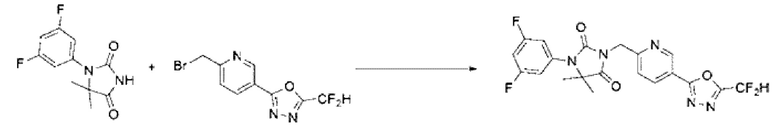
1-(3,5-Дифторфенил)-5,5-диметилимидазолидин-2,4-дион (0,100 г, 0,416 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,121 г, 0,416 ммоль) и карбонат калия (0,115 г, 0,833 ммоль) растворяли в N, N-диметилформамиде (5 мл) при 80°С, после чего полученный раствор перемешивали при той же температуре в течение 18 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,110 г, 58,8%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,28 (т, J=1,1 Гц, 1Н), 8,41 (дд, J=8,2, 2,2 Гц, 1Н), 7,51 (д, J=8,2 Гц, 1Н), 7,09 ~ 7,08 (м, 1Н), 7,08 (с, 0,25Н), 6,97 ~ 6,94 (м, 2Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,02 (с, 2Н), 1,61 (с, 6Н); МСНР (ES) m/z 450, 2 (М++1).
Синтез соединения 18, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-1-фенилимидазолидин-2, 4-диона
[Стадия 1] Синтез 2-метил-2-(фениламино)пропаннитрила

Анилин (0,980 мл, 10,738 ммоль), триметилсилакарбонитрил (1,065 г, 10,738 ммоль) и пропан-2-он (0,624 г, 10,738 ммоль) растворяли в уксусной кислоте (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли гексан (20 мл) и этилацетат (10 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (1,100 г, 63,9%) в виде белого твердого вещества.
[Стадия 2] Синтез 5,5-диметил-1-фенилимидазолидин-2,4-диона
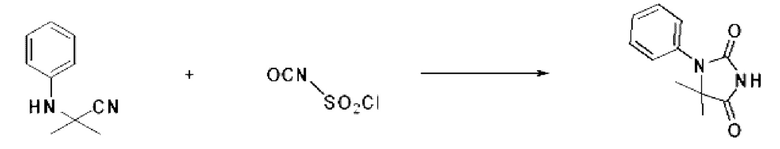
2-Метил-2-(фениламино)пропаннитрил (1,100 г, 6,866 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (1,458 г, 10,298 ммоль) растворяли в дихлорметане (50 мл), после чего полученный раствор перемешивали при 0°С в течение 30 мин и далее перемешивали при комнатной температуре в течение 18 ч. Б реакционную смесь вливали 1 н. водный раствор соляной кислоты (10 мл), после чего растворитель концентрировали при пониженном давлении и затем добавляли этанол (30 мл). Полученную смесь снова перемешивали при температуре 80°С в течение 30 мин, после чего растворитель концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,600 г, 42,8%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 18
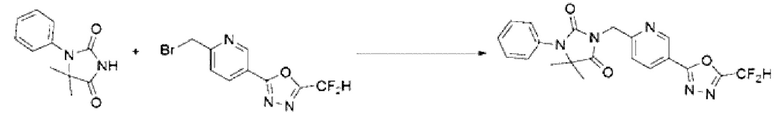
5,5-Диметил-1-фенилимидазолидин-2,4-дион (0,100 г, 0,490 ммоль), полученный на стадии 2,2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,142 г, 0,490 ммоль) и карбонат калия (0,135 г, 0,979 ммоль) растворяли в N, N-диметилформамиде (5 мл) при 80°С, после чего полученный раствор перемешивали при той же температуре в течение 18 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/пексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,080 г, 39,5%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,26 ~ 9,25 (м, 1Н), 8,37 ~ 8,34 (м, 1Н), 7,49 ~ 7,38 (м, 4Н), 7,31 ~ 7,23 (м, 2Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,02 (с, 2Н), 1,52 (с, 6Н); МСНР (ES) m/z 414,2 (М++1).
Синтез соединения 19, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-1-(3-(1-метил-1,2,3,б-тетрагидропиридин-4-ил)фенил)имидазолидин-2,4-диона
[Стадия 1] Синтез трет-бутил-4-(3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-2,4-диоксоимидазолидин-1-ил)фенил)-3,6-дигидропиридин-1(2Н)-карбоксилата
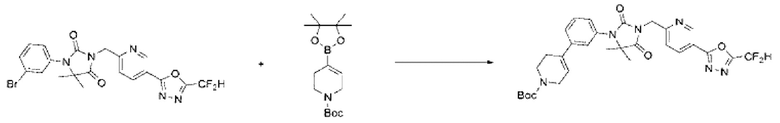
1-(3-Бромфенил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметилимидазолидин-2,4-дион (0,200 г, 0,406 ммоль), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (0,251 г, 0,813 ммоль), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II) дихлорид (0,026 г, 0,041 ммоль) и карбонат цезия (0,199 г, 0,609 ммоль) смешивали в смеси 1,2-дихлорэтан (6 мл)/вода(2 мл), после чего полученную смесь облучали микроволнами, нагревали при 100°С в течение 20 мин и реакцию завершали путем понижения температуры до комнатной температуры. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-70%) и концентрировали с получением указанного в заголовке соединения (0,140 г, 58,0%) в виде бесцветного масла.
[Стадия 2] Синтез 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-1-(3-(1,2,3,6-тетрагидропиридин-4-ил)фенил)имидазолидин-2, 4-дион-2, 2, 2-трифторацетата
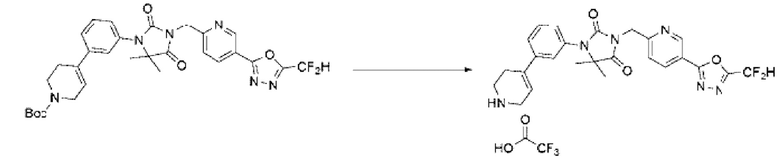
Трет-бутил-4-(3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-2,4-диоксоимидазолидин-1-ил)фенил)-3,6-дигидропиридин-1(2Н)-карбоксилат (0,140 г, 0,235 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,180 мл, 2,354 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего полученный продукт использовали без дополнительного процесса очистки (0,140 г, 97,7%, коричневое масло).
[Стадия 3] Синтез соединения 19

3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-1-(3-(1,2,3,6-тетрагидропиридин-4-ил)фенил)имидазолидин-2,4-дион-2,2,2-трифторацетат (0,080 г, 0,131 ммоль), полученный на стадии 2, формальдегид (0,008 г, 0,263 ммоль), N, N-диизопропилэтиламин (0,023 мл, 0,131 ммоль) и триацетоксиборгидрид натрия (0,056 г, 0,263 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; метанол/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке соединения (0,040 г, 59,8%) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 9,29 (т, J=1,1 Гц, 1Н), 8,39 (дд, J=8,2, 2,2 Гц, 1Н), 7,50 ~ 7,48 (м, 1Н), 7,41 ~ 7,39 (м, 2Н), 7,29 ~ 7,28 (м, 1Н), 7,20 ~ 7,18 (м, 1Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 6,10 ~ 6,08 (м, 1Н), 5,02 (с, 2Н), 3,16 - 3,15 (м, 2Н), 2,70 ~ 2,69 (м, 2Н), 2,61 ~ 2,60 (м, 2Н), 2,43 (с, 3Н), 1,53 (с, 6Н).
Синтез соединения 20, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-1-(3-(1-метилпиперидин-4-ил)фенил)имидазолидин-2,4-диона
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-1-(3-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)фенил)имидазолидин-2,4-дион (0,034 г, 0,067 ммоль) растворяли в метаноле (10 мл) при комнатной температуре, после чего медленно добавляли 10% Pd/C (60 мг) и перемешивали в течение 18 ч в присутствии присоединенного баллона с водородом при той же температуре. Реакционную смесь фильтровали через слой целита для удаления из нее твердого вещества, после чего растворитель удаляли из полученного фильтрата при пониженном давлении, и далее полученный продукт использовали без дополнительного процесса очистки (0,028 г, 82,0%, бесцветное масло).
1Н ЯМР (400 МГц, CDCl3) δ д 9,30 ~ 9,29 (м, 1Н), 8,40 (дд, J=8,2, 2,2 Гц, 1Н), 7,50 (д, J=8,2 Гц, 1Н), 7,41 ~ 7,37 (м, 1Н), 7,29 ~ 7,27 (м, 1Н), 7,16 ~ 7,14 (м, 2Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,02 (с, 2Н), 3,20 ~ 3,18 (м, 2Н), 2,62 ~ 2,55 (м, 1Н), 2,45 (с, 3Н), 2,29 ~ 2,25 (м, 2Н), 2,02 ~ 1,90 (м, 4Н), 1,52 (с, 6Н); МСНР (ES) m/z 511,4 (М++1).
Синтез соединения 21, 7-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-метил-5-фенил-2,5,7-триазаспиро[3.4]октан-б,8-диона
[Стадия 1] Синтез бензил-3-циано-3-(фениламино)азетидин-1-карбоксилата
Анилин (1,961 мл, 21,475 ммоль), триметилсилакарбонитрил (2,131 г, 21,475 ммоль) и бензил-3-оксоазетидин-1-карбоксилат (4,407 г, 21,475 ммоль) растворяли в уксусной кислоте (20 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли этилацетат (10 мл) и гексан (20 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (4,700 г, 71,2%) в виде белого твердого вещества.
[Стадия 2] Синтез бензил-6,8-диоксо-5-фенил-2,5,7-триазаспиро[3.4]октан-2-карбоксилата

Бензил-3-циано-3-(фениламино)азетидин-1-карбоксилат (4,700 г, 15,292 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (3,246 г, 22,938 ммоль) растворяли в дихлорметане (50 мл), после чего полученный раствор перемешивали при 0°С в течение 30 мин и далее перемешивали при комнатной температуре в течение 18 ч. В реакционную смесь вливали 1 н. водный раствор соляной кислоты (10 мл), после чего растворитель концентрировали при пониженном давлении и затем добавляли этанол (30 мл). Полученную смесь снова перемешивали при температуре 80°С в течение 30 мин, после чего растворитель концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (2,200 г, 40,9%) в виде белого твердого вещества.
[Стадия 3], Синтез бензил-7-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6,8-диоксо-5-фенил-2,5,7-триазаспиро[3.4]октан-2-карбоксилата

Бензил-6,8-диоксо-5-фенил-2,5,7-триазаспиро[3,4] октан-2-карбоксилат (0,500 г, 1,423 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,413 г, 1,423 ммоль) и карбонат калия (0,393 г, 2,846 ммоль) растворяли в N, N-диметилформамиде (5 мл) при 80°С, после чего полученный раствор перемешивали при той же температуре в течение 18 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,600 г, 75,2%) в виде белого твердого вещества.
[Стадия 4] Синтез 7-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фенил-2,5,7-триазаспиро[3.4]октан-6,8-диона

Бензил-7-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-6,8-диоксо-5-фенил-2,5,7-триазаспиро[3.4]октан-2-карбоксилат (0,600 г, 1,070 ммоль), полученный на стадии 3, растворяли в метаноле (10 мл) при комнатной температуре, после чего 10% Pd/C (60 мг) медленно добавляли и перемешивали при той же температуре в течение 18 ч в присутствии присоединенного баллона с водородом. Реакционную смесь фильтровали через слой целита для удаления из нее твердого вещества, после чего растворитель удаляли из полученного фильтрата при пониженном давлении, и далее полученный продукт использовали без дополнительного процесса очистки (0,450 г, 98,6%, бесцветное масло).
[Стадия 5] Синтез соединения 21
7-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фенил-2,5,7-триазаспиро[3.4]октан-6,8-дион (0,150 г, 0,352 ммоль), полученный на стадии 4, формальдегид (37,00% раствор, 0,053 мл, 0,704 ммоль) и триацетоксиборгидрид натрия (0,149 г, 0,704 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; метанол/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке соединения (0,100 г, 64,5%) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 9,24 (д, J=2,2 Гц, 1Н), 8,36 (дд, J=8,2, 2,2 Гц, 1Н), 7,53 ~ 7,42 (м, 6Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 4,99 (с, 2Н), 3,80 ~ 3,73 (м, 4Н), 2,31 (с, 3Н).
Синтез соединения 22, 7-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-2-изопропил-5-фенил-2,5, 7-триазаспиро[3.4]октан-6,8-диона

7-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фенил-2,5,7-триазаспиро[3,4]октан-б,8-дион (0,100 г, 0,235 ммоль), ацетон (0,035 мл, 0,469 ммоль) и триацетоксиборгидрид натрия (0,099 г, 0,469 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; метанол/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке соединения (0,070 г, 63,7%) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 9, 24 (т, J=1,1 Гц, 1Н), 8,36 (дд, J=8,2, 2,2 Гц, 4Н), 7,51 ~ 7,39 (м, 6Н), 7,08 (с, 1Н), 6,95 (с, 1Н), 6,82 (с, 1Н), 4,98 (с, 2Н), 3,84 (с, 4Н), 2,58 ~ 2,55 (м, 1Н), 0,91 ~ 0,87 (м, 6Н).
Синтез соединения 23, 2-ацетил-7-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фенил-2,5,7-триазаспиро[3.4]октан-б,8-диона

7-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фенил-2,5,7-триазаспиро[3,4]октан-6,8-дион (0,124 г, 0,291 ммоль), ацетилхлорид (0,041 мл, 0,582 ммоль) и N, N-диизопропилэтиламин (0,101 мл, 0,582 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; метанол/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке соединения (0,100 г, 73,4%) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3) δ 9, 26 (т, J=1,1 Гц, 1Н), 8,40 (дд, J=8,2, 2,2 Гц, 1Н), 7,55 ~ 7,39 (м, 6Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,03 (с, 2Н), 4,62 ~ 4,48 (м, 2Н), 4,42 ~ 4,33 (м, 2Н), 1,85 (с, 3Н).
Синтез соединения 24, 7-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5-фенил-5,7-диазаспиро[3.4]октан-6,8-диона
[Стадия 1] Синтез 1-(фениламино)циклобутан-1-карбонитрила
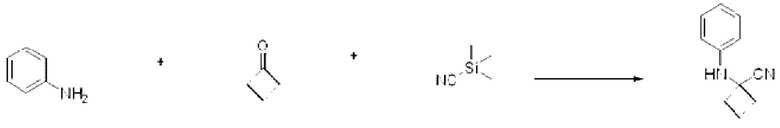
Анилин (0,980 мл, 10,738 ммоль), циклобутанон (0,753 г, 10,738 ммоль) и триметилсилакарбонитрил (1,065 г, 10,738 ммоль) растворяли в уксусной кислоте (20 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-30%) и концентрировали с получением указанного в заголовке соединения (1,200 г, 64,9%) в виде белого твердого вещества.
[Стадия 2] Синтез 5-фенил-5,7-диазаспиро[3.4] октан-6,8-диона

1-(Фениламино)циклобутан-1-карбонитрил (1,000 г, 5,806 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (0,761 мл, 8,709 ммоль) растворяли в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 12 ч. После этого к реакционной смеси добавляли 1М HCl (10 мл), после чего завершали реакцию с удалением растворителя. К реакционной смеси добавляли этанол (10 мл) и перемешивали при 80°С в течение 1 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли этилацетат (20 мл) и пексан (10 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (0,950 г, 75,7%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 24

5-Фенил-5,7-диазаспиро[3,4]октан-6,8-дион (0,100 г, 0,462 ммоль), полученный на стадии 2, растворяли в N, N-диметилформамиде (20 мл) при 0°С, после чего гидрид натрия (60,00%, 0,022 г, 0,555 ммоль) добавляли в полученный раствор и перемешивали при той же температуре в течение 30 мин. 2-(6-(Бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,134 г, 0,462 ммоль) добавляли в реакционную смесь и далее перемешивали при комнатной температуре в течение 2 ч. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 40 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,130 г, 66,1%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3) δ 9,30 ~ 9,26 (м, 1Н), 8,37 (дд, J=8,2, 2,2 Гц, 1Н), 7,53 ~ 7,43 (м, 4Н), 7,33 ~ 7,31 (м, 2Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,00 (с, 2Н), 2,59 ~ 2,42 (м, 4Н), 2,23 ~ 2,04 (м, 2Н), 1,69 ~ 1,63 (м, 2Н).
Синтез соединения 25, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(4-(фуран-3-ил)фенил)-5,5-диметилимидазолидин-2,4-диона
[Стадия 1] Синтез 2-((4-бромфенил)амино)-2-метилпропаннитрила
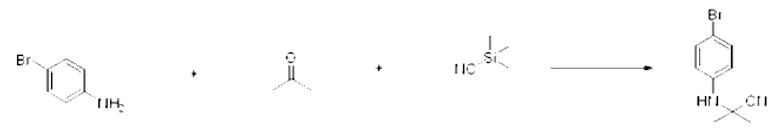
4-Броманилин (3,000 г, 17,439 ммоль), пропан-2-он (1,013 г, 17,439 ммоль) и триметилсилакарбонитрил (1,730 г, 17,439 ммоль) растворяли в уксусной кислоте (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 12 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли этилацетат (10 мл) и гексан (20 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (2,700 г, 64,7%) в виде белого твердого вещества.
[Стадия 2] Синтез 1-(4-бромфенил)-5,5-диметилимидазолидин-2,4-диона

2-((4-Бромфенил)амино)-2-метилпропаннитрил (2,740 г, 11,459 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (1,502 мл, 17,188 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. К реакционной смеси добавляли 1М HCl (10 мл) для концентрирования органического слоя, после чего добавляли этанол (20 мл) и перемешивали при 80°С в течение 1 ч. После этого, растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли этилацетат (20 мл) и гексан (30 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением целевого соединения (2,5 г, 77,1%) в виде белого твердого вещества.
[Стадия 3] Синтез 1-(4-бромфенил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметилимидазолидин-2,4-диона 
1-(4-Бромфенил)-5,5-диметилимидазолидин-2,4-дион (1,000 г, 3,532 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,025 г, 3,532 ммоль) и карбонат калия (0,976 г, 7,064 ммоль) растворяли в N, N-диметилформамиде (30 мл) при 50°С, после чего полученный раствор перемешивали при той же температуре в течение 12 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. Б реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 40 г; этилацетат/пексан=0-50%) и концентрировали с получением указанного в заголовке соединения (1,300 г, 74,8%) в виде желтого твердого вещества.
[Стадия 4] Синтез соединения 25

1-(4-Бромфенил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметилимидазолидин-2,4-дион (0,150 г, 0,305 ммоль), полученный на стадии 3, фуран-3-илбороновую кислоту (0,051 г, 0,457 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II, 0,020 г, 0,030 ммоль) и карбонат цезия (0,149 г, 0,457 ммоль) смешивали в смеси 1,4-диоксан (6 мл)/вода (2 мл), после чего полученную смесь облучали микроволнами, затем нагревали при 100°С в течение 20 мин и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; метанол/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке соединения (0,022 г, 15,1%) в виде коричневого масла.
1Н ЯМР (400 МГц, CDCl3) δ 9,30 (дд, J=2,2, 0,8 Гц, 1Н), 8,40 (дд, J=8,2, 2,2 Гц, 1Н), 7,77 ~ 7,74 (м, 2Н), 7,52 ~ 7,49 (м, 2Н), 7,35 ~ 7,32 (м, 2Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 6,72 (дд, J=3,4, 0,7 Гц, 1Н), 6,51 (дд, J=3,4, 1,8 Гц, 1Н), 5,04 (с, 2Н), 1,56 (с, 6Н); МСНР (ES) m/z 480, 3 (М++1).
Синтез соединения 26, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-1-(4-(пиридин-4-ил)фенил)имидазолидин-2,4-диона

1-(4-Бромфенил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметилимидазолидин-2,4-дион (0,150 г, 0,305 ммоль), пиридин-4-илбороновую кислоту (0,056 г, 0,457 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II, 0,020 г, 0,030 ммоль) и карбонат цезия (0,149 г, 0,457 ммоль) смешивали в смеси 1,4-диоксан (6 мл)/вода(2 мл), после чего полученную смесь облучали микроволнами, затем нагревали при 100°С в течение 20 мин и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; метанол/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке соединения (0,015 г, 10,0%) в виде коричневого масла.
1Н ЯМР (400 МГц, CDCl3) δ 9,29 (дд, J=2,2, 0,8 Гц, 1Н), 8,70 (дд, J=4,5, 1,6 Гц, 2Н), 8,39 (дд, J=8,2, 2,2 Гц, 1Н), 7,73 ~ 7,71 (м, 2Н), 7,52 ~ 7,50 (м, 3Н), 7,47 ~ 7,45 (м, 2Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,04 (с, 2Н), 1,60 (с, 6Н); МСНР (ES) m/z 491,2 (М++1).
Синтез соединения 27, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-1-(4-(пиридин-3-ил) фенил)имидазолидин-2,4-диона
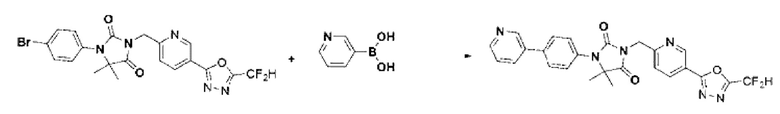
1-(4-Бромфенил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметилимидазолидин-2, 4-дион (0,150 г, 0,305 ммоль), пиридин-3-илбороновую кислоту (0,056 г, 0,457 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II, 0,020 г, 0,030 ммоль) и карбонат цезия (0,149 г, 0,457 ммоль) смешивали в смеси 1,4-диоксан (6 мл)/вода(2 мл), после чего полученную смесь облучали микроволнами, затем нагревали при 100°С в течение 20 мин и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г;
метанол/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке соединения (0,030 г, 20,1%) в виде коричневого масла.
1Н ЯМР (400 МГц, CDCl3) δ 9,30 ~ 9,29 (м, 1Н), 8,87 ~ 8,86 (м, 1Н), 8,64 (дд, J=4,8, 1,6 Гц, 1Н), 8,40 (дд, J=8,2, 2,2 Гц, 1Н), 7,91 ~ 7,88 (м, 1Н), 7, 68 ~ 7, 65 (м, 2Н), 7,52 (д, J=8,2 Гц, 1Н), 7,46 ~ 7,38 (м, 3Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,04 (с, 2Н), 1,60 (с, 6Н); МСНР (ES) m/z 491,3 (М++1).
Синтез соединения 28, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(3'-фтор-[1,1'-бифенил]-4-ил)-5,5-диметилимидазолидин-2,4-диона
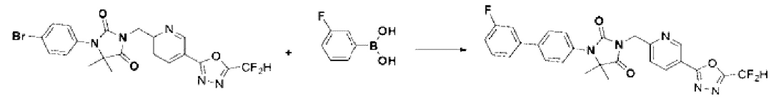
1-(4-Бромфенил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметилимидазолидин-2,4-дион (0,150 г, 0,305 ммоль), (3-фторфенил) бороновую кислоту (0, 064 г, 0,457 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II, 0,020 г, 0,030 ммоль) и карбонат цезия (0,149 г, 0,457 ммоль) смешивали в смеси 1,4-диоксан (6 мл)/вода(2 мл), после чего полученную смесь облучали микроволнами, затем нагревали при 100°С в течение 20 мин и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; метанол/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке соединения (0,060 г, 38,8%) в виде коричневого масла.
1Н ЯМР (400 МГц, CDCl3) δ 9,31 ~ 9,30 (м, 1Н), 8,40 (дд, J=8,2, 2,2 Гц, 1Н), 7,67 ~ 7,64 (м, 2Н), 7,52 (д, J=8,2 Гц, 1Н), 7,45 ~ 7,28 (м, 5Н), 7,11 ~ 7,07 (м, 1Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,05 (с, 2Н), 1,60 (с, 6Н); МСНР (ES) m/z 508,2 (М++1).
Синтез соединения 29, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(2'-фтор-[1,1'-бифенил]-4-ил)-5,5-диметилимидазолидин-2,4-диона

1-(4-Бромфенил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметилимидазолидин-2,4-дион (0,150 г, 0,305 ммоль), (2-фторфенил)бороновую кислоту (0,064 г, 0,457 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II, 0,020 г, 0,030 ммоль) и карбонат цезия (0,149 г, 0,457 ммоль) смешивали в смеси 1,4-диоксан (6 мл)/вода (2 мл), после чего полученную смесь облучали микроволнами, затем нагревали при 100°С в течение 20 мин и далее реакцию завершали путем понижения температуры до комнатной температуры. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего воду вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; метанол/дихлорметан=0-10%) и концентрировали с получением указанного в заголовке соединения (0,060 г, 38,8%) в виде коричневого масла.
1Н ЯМР (400 МГц, CDCl3) δ 9,30 ~ 9,29 (м, 1Н), 8,40 (дд, J=8,2, 2,2 Гц, 1Н), 7,66 ~ 7,63 (м, 2Н), 7,52 ~ 7,33 (м, 5Н), 7,26 ~ 7,17 (м, 2Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,04 (с, 2Н), 1,60 (с, 6Н); МСНР (ES) m/z 508, 2 (М++1).
Синтез соединения 30, трет-бутил-4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-2,4-диоксоимидазолидин-1-ил)-1Н-индол-1-карбоксилата
[Стадия 1] Синтез трет-бутил-4-((2-цианопропан-2-ил)амино)-1Н-индол-1-карбоксилата

Трет-бутил-4-амино-1Н-индол-1-карбоксилат (1,680 г, 7,233 ммоль), пропан-2-он (0,420 г, 7,233 ммоль) и триметилсилакарбонитрил (0,718 г, 7,233 ммоль) растворяли в уксусной кислоте (30 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли этилацетат (10 мл) и гексан (20 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (1,930 г, 89,1%) в виде белого твердого вещества.
[Стадия 2] Синтез трет-бутил-4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-1Н-индол-1-карбоксилата

Трет-бутил-4-((2-цианопропан-2-ил)амино)-1Н-индол-1-карбоксилат (1,930 г, 6,447 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (1,369 г, 9,670 ммоль) растворяли в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. В реакционную смесь вливали 1М HCl (10 мл) и растворитель концентрировали при пониженном давлении. После этого полученный концентрат растворяли в этаноле (20 мл) и затем перемешивали при 80°С в течение 1 ч. Температуру реакции понижали до комнатной температуры, после чего растворитель удаляли при пониженном давлении. Затем реакционную смесь растворяли в THF (20 мл), после чего добавляли 10% раствор K2CO3 (10 мл) для доведения рН до 8, и затем добавляли ди-трет-бутилдикарбонат (2,111 г, 9,670 ммоль) и перемешивали в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-30%) и концентрировали с получением указанного в заголовке соединения (0,250 г, 11,3%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 30

Трет-бутил-4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)-1Н-индол-1-карбоксилат (0,200 г, 0,582 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,169 г, 0,582 ммоль) и карбонат калия (0,161 г, 1,165 ммоль) растворяли в N, N-диметилформамиде (10 мл) при 45°С, после чего полученный раствор перемешивали при той же температуре в течение 18 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,110 г, 34,2%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) 9,33 (д, J=1,6 Гц, 1Н), 8,38 (дд, J=8,2, 2,2 Гц, 1Н), 8,25 ~ 8,23 (м, 1Н), 7,66 (д, J=3,8 Гц, 1Н), 7,52 ~ 7,50 (м, 1Н), 7,38 (т, J=8,0 Гц, 1Н), 7,16 (дд, J=7,6, 0,6 Гц, 1Н), 7,09 (с, 0,25Н), 6,96 (с, 0,5Н), 6,83 (с, 0,25Н), 6,56 (дд, J=3,8, 0,5 Гц, 1Н), 5,06 (с, 2Н), 1,67 (с, 9Н), 1,53 (с, 6Н).
Синтез соединения 31, трет-бутил-4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-2,4-диоксоимидазолидин-1-ил)индолин-1-карбоксилата
[Стадия 1] Синтез трет-бутил-4-((2-цианопропан-2-ил)амино)индолин-1-карбоксилата

Трет-бутил-4-аминоиндолин-1-карбоксилат (2,300 г, 9,816 ммоль), пропан-2-он (0,570 г, 9,816 ммоль) и триметилсилакарбонитрил (0,974 г, 9,816 ммоль) растворяли в уксусной кислоте (30 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли этилацетат (10 мл) и гексан (20 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (2,800 г, 94,6%) в виде белого твердого вещества.
[Стадия 2] Синтез трет-бутил-4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)индолин-1-карбоксилата
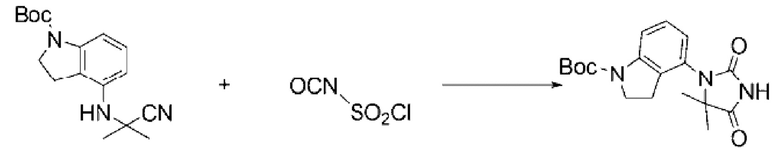
Трет-бутил-4-((2-цианопропан-2-ил)амино)индолин-1-карбоксилат (2,800 г, 9,290 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (1,972 г, 13,935 ммоль), и ди-трет-бутилдикарбонат (3,041 г, 13,935 ммоль) растворяли в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-30%) и концентрировали с получением указанного в заголовке соединения (0,490 г, 15,3%) в виде белого твердого вещества.
[Стадия 3] Синтез соединения 31

Трет-бутил-4-(5,5-диметил-2,4-диоксоимидазолидин-1-ил)индолин-1-карбоксилат (0,485 г, 1,404 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,407 г, 1,404 ммоль) и карбонат калия (0,388 г, 2,808 ммоль) растворяли в N, N-диметилформамиде (10 мл) при 45°С, после чего полученный раствор перемешивали при той же температуре в течение 18 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. В реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/пексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,660 г, 84,8%) в виде желтого пенообразного твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,28 (дд, J=2,2, 0,7 Гц, 1Н), 8,40 (дд, J=8,2, 2,2 Гц, 1Н), 7,95 (ушир. с, 1Н), 7,50 (дд, J=8,2, 0,7 Гц, 1Н), 7,28 ~ 7,26 (м, 1Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 6,85 ~ 6,83 (м, 1Н), 5,02 (с, 2Н), 4,04 ~ 4,00 (м, 2Н), 3,10 ~ 3,00 (м, 2Н), 1,58 ~ 1,54 (м, 15Н).
Синтез соединения 32, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(1Н-индол-4-ил) -5, 5-диметилимидазолидин-2,4-диона 
Трет-бутил-4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-2,4-диоксоимидазолидин-1-ил)-1Н-индол-1-карбоксилат (0,258 г, 0,467 ммоль) и трифторуксусную кислоту (0,358 мл, 4,669 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор пидрокарбоната натрия вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный продукт использовали без дополнительного процесса очистки (0,200 г, 94,7%, серое твердое вещество).
1Н ЯМР (400 МГц, ДМСО-d6) 9,21 (д, J=1,9 Гц, 1Н), 8,48 (дд, J=8,2, 2,2 Гц, 1Н), 7,72 (с, 0,25Н), 7,69 (д, J=8,2 Гц, 1Н), 7,59 (с, 0,5Н), 7,50 (д, J=8,1 Гц, 1Н), 7,46 (с, 0,25Н), 7,40 (т, J=2,8 Гц, 1Н), 6,99 (д, J=7,0 Гц, 1Н), 6,42 (т, J=2,2 Гц, 1Н), 5,75 (с, 1Н), 4,96 (с, 2Н), 1,42 (с, 6Н).
Синтез соединения 33, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(индолин-4-ил)-5,5-диметилимидазолидин-2,4-диона

Трет-бутил-4-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-2,4-диоксоимидазолидин-1-ил)индолин-1-карбоксилат (0,670 г, 1,208 ммоль) и трифторуксусную кислоту (0,925 мл, 12,082 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия вливали в полученный концентрат и затем проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный продукт использовали без дополнительного процесса очистки (указанное в заголовке соединение, 0,500 г, 91,1%, желтое пенообразное твердое вещество).
1Н ЯМР (400 МГц, CDCl3) δ 9,28 (т, J=1,1 Гц, 1Н), 8,39 (дд, J=8,2, 2,2 Гц, 1Н), 7,50 ~ 7,47 (м, 1Н), 7,08 (дд, J=8,1, 7,6 Гц, 1Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 6,67 (д, J=7,8 Гц, 1Н), 6,58 ~ 6,55 (м, 1Н), 5,04 (с, 2Н), 3,62 ~ 3,56 (м, 2Н), 3,01 ~ 2,97 (м, 2Н), 1,58 (с, 6Н).
Синтез соединения 34, 3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-1-(3-нитрофенил)имидазолидин-2,4-диона
[Стадия 1] Синтез 2-метил-2-((3-нитрофенил)амино)пропаннитрила

3-Нитроанилин (3,000 п, 21,719 ммоль), пропан-2-он (1,261 г, 21,719 ммоль) и триметилсилакарбонитрил (2,155 г, 21,719 ммоль) растворяли в уксусной кислоте (30 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. Растворитель удаляли из реакционной смеси при пониженном давлении, после чего в полученный концентрат добавляли этилацетат (10 мл) и гексан (20 мл) и перемешивали для отфильтровывания осажденного твердого вещества, промывали гексаном и сушили с получением указанного в заголовке соединения (4,000 г, 89,7%) в виде желтого твердого вещества.
[Стадия 2] Синтез 5,5-диметил-1-(3-нитрофенил)имидазолидин-2,4-диона

2-Метил-2-((3-нитрофенил)амино)пропаннитрил (4,000 г, 19,491 ммоль), полученный на стадии 1, и хлорсульфонилизоцианат (4,138 г, 29,237 ммоль) растворяли в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. В реакционную смесь вливали 1М HCl (10 мл) и растворитель концентрировали при пониженном давлении. После этого полученный концентрат растворяли в этаноле (20 мл) и затем перемешивали при 80°С в течение 1 ч. Осажденное твердое вещество фильтровали, промывали гексаном и сушили с получением указанного в заголовке соединения (4,300 г, 88,5%) в виде желтого твердого вещества.
[Стадия 3] Синтез соединения 34

5,5-Диметил-1-(3-нитрофенил)имидазолидин-2, 4-дион (1,000 г, 4,012 ммоль), полученный на стадии 2, 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,164 г, 4,012 ммоль) и карбонат калия (1,109 г, 8,025 ммоль) растворяли в N, N-диметилформамиде (20 мл) при 45°С, после чего полученный раствор перемешивали при той же температуре в течение 18 ч и далее реакцию завершали путем понижения температуры до комнатной температуры. Б реакционную смесь вливали воду и проводили экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 40 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (1,400 г, 76,1%) в виде пенообразного белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,30 (д, J=1,6 Гц, 1Н), 8,42 (дд, J=8,2, 2,2 Гц, 1Н), 8,28 ~ 8,25 (м, 2Н), 7,76 ~ 7,73 (м, 1Н), 7,69 ~ 7,65 (м, 1Н), 7,53 ~ 7,51 (м, 1Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 5,05 (с, 2Н), 1,63 (с, 6Н).
Синтез соединения 35, 1-(3-аминофенил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметилимидазолидин-2,4-диона

3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-1-(3-нитрофенил)имидазолидин-2,4-дион (1,400 г, 3,054 ммоль) растворяли в метаноле (30 мл) при комнатной температуре, после чего медленно добавляли никель Ренея и перемешивали в течение 12 ч в присутствии присоединенного баллона с водородом при той же температуре. Реакционную смесь фильтровали через слой целита для удаления из нее твердого вещества, после чего растворитель удаляли из полученного фильтрата при пониженном давлении и далее полученный продукт использовали без дополнительного процесса очистки (1,200 г, 91,7%, белое твердое вещество).
1Н ЯМР (400 МГц, CDCl3) δ 9, 30 (д, J=1,7 Гц, 1Н), 8,40 (дд, J=8,2, 2,2 Гц, 1Н), 7,50 (д, J=8,2 Гц, 1Н), 7,23 (т, J=8,0 Гц, 1Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,82 (с, 0,25Н), 6,73 ~ 6,68 (м, 2Н), 6,62 ~ 6,61 (м, 1Н), 5,02 (с, 2Н), 3,78 (ушир. с, 2Н), 1,55 (с, 6Н); МСНР (ES) m/z 429,3 (М++1).
Синтез соединения 36, N-(3-(3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметил-2,4-диоксоимидазолидин-1-ил)фенил)ацетамида

1-(3-Аминофенил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметилимидазолидин-2,4-дион (0,140 г, 0,327 ммоль), уксусный ангидрид (0,031 мл, 0,327 ммоль) и N, N-диизопропилэтиламин (0,114 мл, 0,654 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,110 г, 71,6%) в виде пенообразного белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,26 (дд, J=2,l, 0,7 Гц, 1Н), 8,38 (дд, J=8,2, 2,2 Гц, 1Н), 8,22 (с, 1Н), 7,68 (т, J=1,9 Гц, 1Н), 7,50 (дд, J=8,2, 0,5 Гц, 1Н), 7,25 (т, J=8,0 Гц, 1Н), 7,17 ~ 7,14 (м, 1Н), 7,08 (с, 0,25Н), 6,95 (с, 0,5Н), 6,97 ~ 6,95 (м, 1Н), 6,82 (с, 0,25Н), 5,02 (с, 2Н), 1,99 (с, 3Н), 1,52 (с, 6Н).
Синтез соединения 37, 1-(1-ацетилиндолин-4-ил)-3-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-5,5-диметилимидазолидин-2,4-диона
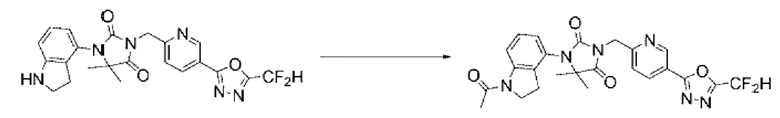
3-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1-(индолин-4-ил)-5,5-диметилимидазолидин-2,4-дион (0,100 г, 0,220 ммоль), уксусный ангидрид (0,021 мл, 0,220 ммоль) и N, N-диизопропилэтиламин (0,077 мл, 0,440 ммоль) растворяли в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивали при той же температуре в течение 18 ч. В реакционную смесь вливали воду и проводили экстракцию дихлорметаном. Органический слой промывали насыщенным водным раствором хлорида натрия, обезвоживали с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный концентрат очищали с помощью колоночной хроматографии (SiO2, картридж 12 г; этилацетат/гексан=0-50%) и концентрировали с получением указанного в заголовке соединения (0,080 г, 73,2%) в виде пенообразного белого твердого вещества.
1Н ЯМР (400 МГц, CDCl3) δ 9,24 ~ 9,23 (м, 1Н), 8,37 (дд, J=8,2, 2,2 Гц, 1Н), 8,27 (д, J=8,1 Гц, 1Н), 7,49 (дд, J=8,2, 0,6 Гц, 1Н), 7,29 ~ 7,25 (м, 1Н), 7,08 (с, 1Н), 6,95 (с, 1Н), 6,92 (д, J=7,6 Гц, 1Н), 6,82 (с, 1Н), 5,00 (с, 2Н), 4,14 ~ 4,07 (м, 2Н), 3,16 ~ 3,14 (м, 2Н), 2,22 (с, 3Н), 1,52 (с, 6Н).
Протокол измерения и анализа активности соединения по изобретению
<Экспериментальный пример 1> Идентификация ингибирования активности фермента HDAC (in vitro)
Селективный ингибитор HDAC6 важен для селективности ингибирования HDAC1, что является причиной побочных эффектов, и соответственно, была идентифицирована селективность фермента HDAC1/6 и клеточная селективность (HDAC1: ацетилирование гистонов/НСАС6: ацетилирование тубулина).
1. Экспериментальный метод
Ингибирующую способность исследуемого материала фермента HDAC измеряли с использованием набора для анализа лекарственных средств на основе флуоресценции (HDAC1 Fluorimetrie Drug Discovery Assay Kit (Enzolifesciences: BML-AK511) и рекомбинантной HDAC6 человека (Calbiochem: 382180). Для анализа HDAC1 образцы обрабатывали при концентрации 100, 1000 и 10000 нМ. Для анализа HDAC6 образцы обрабатывали при концентрации 0,1, 1, 10, 100 и 1000 нМ. После вышеуказанной обработки образца реакцию продолжали при 37°С в течение 60 мин, обрабатывали проявителем и подвергали реакции при 37°С в течение 30 мин, после чего измеряли интенсивность флуоресценции (Ех 390, Em 460) с использованием FlexStatin3 (Molecular Device).
2. Результаты эксперимента
Результаты поиска ингибирования активности фермента HDAC, полученные в соответствии с указанным выше экспериментальным методом, приведены в таблице 2.

Ссылаясь на таблицу 2, можно подтвердить, что соединения -производные 1,3,4-оксадиазола по настоящему изобретению проявляют превосходную селективность в отношении фермента HDAC1/6.
Настоящее изобретение относится к новому соединению, представленному химической формулой I, или его фармацевтически приемлемой соли, где 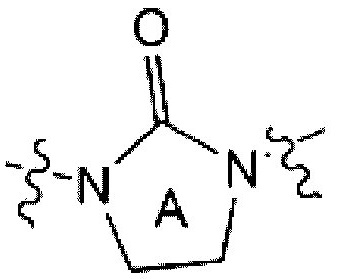 представляет собой
представляет собой 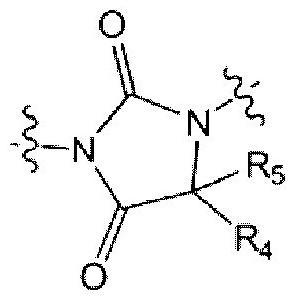 ; каждый из R4 и R5 независимо представляет собой С1-С4 алкил; Z5 представляет собой N-R6 или СН2; R6 представляет собой С1-С4 алкил, -С(=O)-(С1-С4 алкил), -С (=O)-О-(С1-С4 алкил) или 4-6-членный гетероциклоалкил, имеющий один О; L1 представляет собой -(С1-С2 алкилен)-;
; каждый из R4 и R5 независимо представляет собой С1-С4 алкил; Z5 представляет собой N-R6 или СН2; R6 представляет собой С1-С4 алкил, -С(=O)-(С1-С4 алкил), -С (=O)-О-(С1-С4 алкил) или 4-6-членный гетероциклоалкил, имеющий один О; L1 представляет собой -(С1-С2 алкилен)-;  представляет собой С6-С12 арил, 5-9-членный гетероарил, имеющий по меньшей мере один N, или
представляет собой С6-С12 арил, 5-9-членный гетероарил, имеющий по меньшей мере один N, или  ; каждый из R2 и R3 независимо представляет собой Н, галоген, С6-С12 арил, 5- или 6-членный гетероарил, имеющий N или О, 5- или 6-членный гетероциклоалкил, имеющий N, 5- или 6-членный гетероциклоалкенил, имеющий N, -С(=O)-О-(С1-С4 алкил), -С(=O)-(С1-С4 алкил), -NH-C(=O)-(С1-С4 алкил), -NO2 или -NH2, по меньшей мере один Н из вышеуказанных R2 и R3 может быть в каждом случае независимо замещен галогеном или С1-С4 алкилом; и n, и m в каждом случае независимо равно 1 или 2. Также изобретение относится к фармацевтической композиции, содержащей соединение, представленное химической формулой I, к способу предупреждения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC), и к применению соединения, представленного химической формулой I, для предупреждения или лечения заболеваний, опосредованных HDAC. Технический результат изобретения заключается в разработке селективного ингибитора HDAC6, который имеет цинксвязывающую группу, с улучшенной биодоступностью, не вызывающего при этом побочных эффектов, для лечения рака, воспалительных, аутоиммунных и неврологических заболеваний, нейродегенеративных нарушений. 5 н. и 5 з.п. ф-лы, 2 табл., 38 пр.
; каждый из R2 и R3 независимо представляет собой Н, галоген, С6-С12 арил, 5- или 6-членный гетероарил, имеющий N или О, 5- или 6-членный гетероциклоалкил, имеющий N, 5- или 6-членный гетероциклоалкенил, имеющий N, -С(=O)-О-(С1-С4 алкил), -С(=O)-(С1-С4 алкил), -NH-C(=O)-(С1-С4 алкил), -NO2 или -NH2, по меньшей мере один Н из вышеуказанных R2 и R3 может быть в каждом случае независимо замещен галогеном или С1-С4 алкилом; и n, и m в каждом случае независимо равно 1 или 2. Также изобретение относится к фармацевтической композиции, содержащей соединение, представленное химической формулой I, к способу предупреждения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC), и к применению соединения, представленного химической формулой I, для предупреждения или лечения заболеваний, опосредованных HDAC. Технический результат изобретения заключается в разработке селективного ингибитора HDAC6, который имеет цинксвязывающую группу, с улучшенной биодоступностью, не вызывающего при этом побочных эффектов, для лечения рака, воспалительных, аутоиммунных и неврологических заболеваний, нейродегенеративных нарушений. 5 н. и 5 з.п. ф-лы, 2 табл., 38 пр.
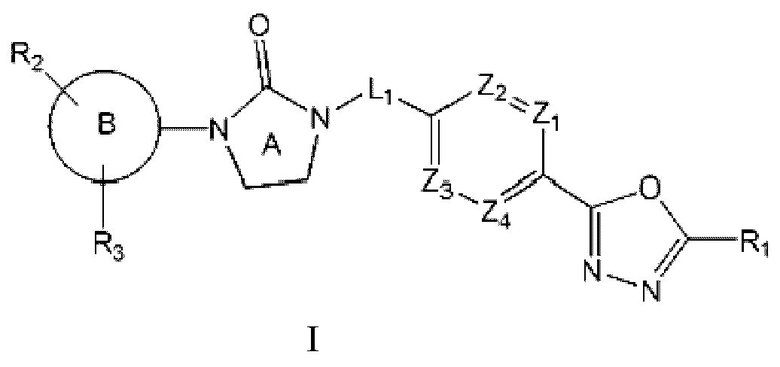
1. Соединение, представленное следующей химической формулой I, или его фармацевтически приемлемые соли:
[Химическая формула I]
где
каждый из Z1-Z4 независимо представляет собой N или CR0, где R0 является Н или галогеном;
R1 представляет собой СХ3 или СХ2Н, где X является галогеном;
 является
является 
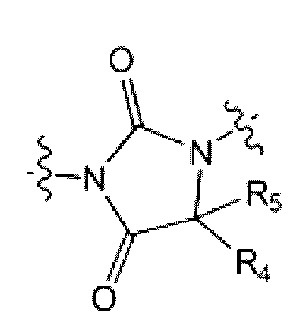 или
или 
где представляет собой 
каждый из R4 и R5 независимо представляет собой Н или С1-С4 алкил,
где
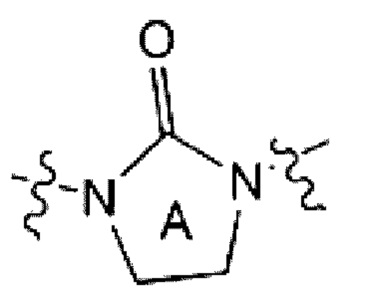 представляет
представляет 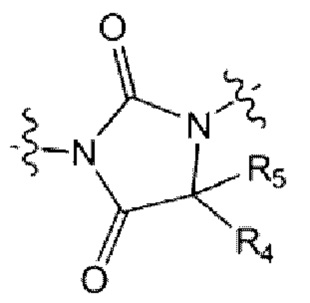 собой
собой
каждый из R4 и R5 независимо представляет собой С1-С4 алкил,
Z5 представляет собой N-R6 или СН2,
R6 представляет собой С1-С4 алкил, -С(=O)-(С1-С4 алкил), -С (=O)-О-(С1-С4 алкил) или 4-6-членный гетероциклоалкил, имеющий один О;
L1 представляет собой -(С1-С2 алкилен)-;
 представляет собой С6-С12 арил, 5-9-членный гетероарил, имеющий по меньшей мере один N, или
представляет собой С6-С12 арил, 5-9-членный гетероарил, имеющий по меньшей мере один N, или  ;
;
каждый из R2 и R3 независимо представляет собой Н, галоген, С6-С12 арил, 5- или 6-членный гетероарил, имеющий N или О, 5- или 6-членный гетероциклоалкил, имеющий N, 5- или 6-членный гетероциклоалкенил, имеющий N, -С(=O)-О-(С1-С4 алкил), -С(=O)-(С1-С4 алкил), -NH-C(=O)-(С1-С4 алкил), -NO2 или -NH2,
по меньшей мере один Н из вышеуказанных R2 и R3 может быть в каждом случае независимо замещен галогеном или С1-С4 алкилом; и
n и m в каждом случае независимо равно 1 или 2.
2. Соединение, представленное химической формулой I, или его фармацевтически приемлемые соли по п. 1, где
каждый из Z1 и Z2 независимо представляет собой N, СН или CF, и каждый из Z3 и Z4 представляет собой СН;
R1,  , R4-R6, Z5, L1,
, R4-R6, Z5, L1,  n и m являются такими же как в химической формуле I п. 1; и
n и m являются такими же как в химической формуле I п. 1; и
каждый из R2 и R3 независимо представляет собой Н, галоген, галогензамещенный или незамещенный фенил, фуранил, пиридинил, С1-С4 алкилзамещенный или незамещенный пиперидинил, С1-С4 алкилзамещенный или незамещенный тетрагидропиридинил, -С(=O)-O-(С1-С4 алкил), -С(=O)-(С1-С4 алкил), -NH-C(=O)-(С1-С4 алкил), -NO2 или -NH2.
3. Соединение, представленное химической формулой I, или его фармацевтически приемлемые соли по п. 1, где
каждый из Z1 и Z2 независимо представляет собой N, СН или CF, и каждый из Z3 и Z4 представляет собой СН; 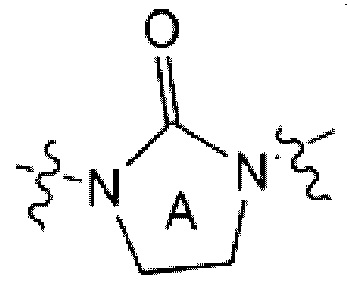
Z5, R1, R4-R6, L1, n и m такие же, как в химической формуле I по п. 1;
представляет собой фенил, индол или 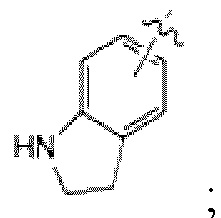 и
и
R2 и R3 каждый независимо представляет собой Н, галоген, галогензамещенный или незамещенный фенил, фуранил, пиридинил, С1-С4 алкил замещенный или незамещенный пиперидинил, С1-С4 алкил замещенный или незамещенный тетрагидропиридинил, -С(=O)-O-(С1-С4 алкил), -С(=O)-(С1-С4 алкил), -NH-C(=O)-(С1-С4 алкил), -NO2 или -NH2.
4. Соединение, представленное химической формулой I, или его фармацевтически приемлемые соли по п. 1, где
соединение, представленное химической формулой 1, представляет собой соединение, выбранное из группы, состоящей из следующих соединений 1-37: 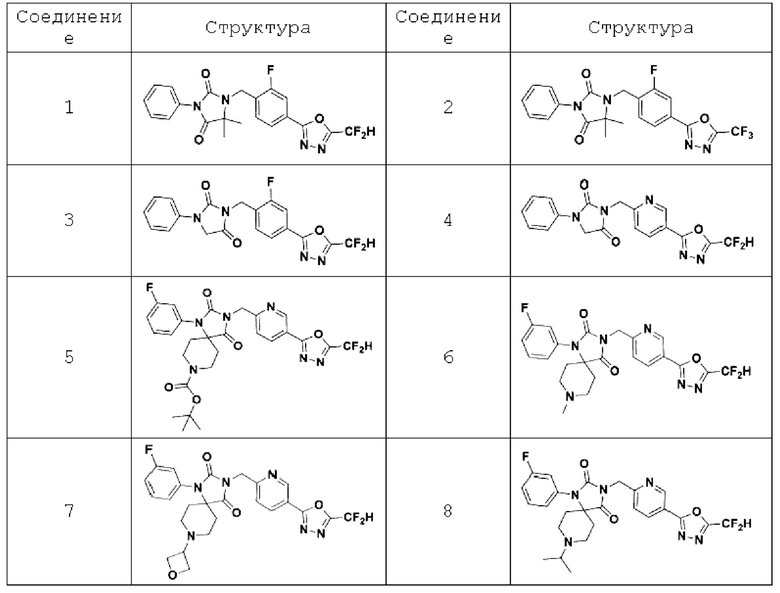
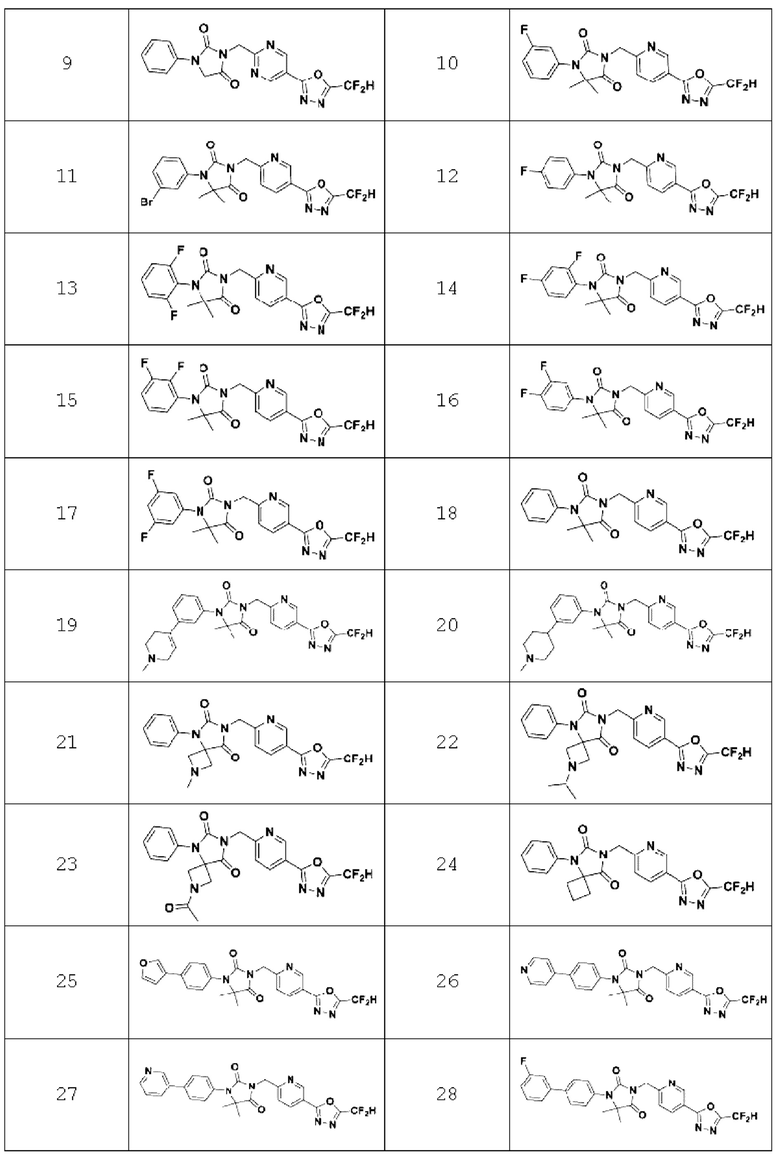
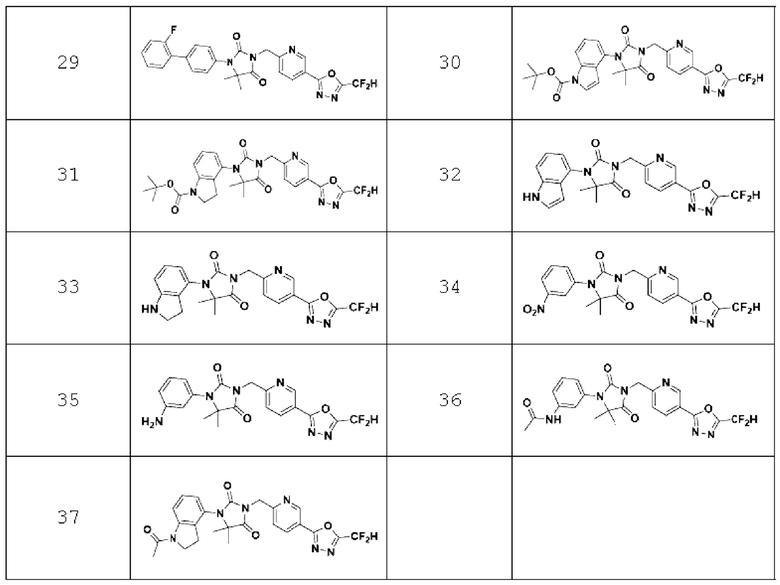
5. Фармацевтическая композиция, содержащая соединение, представленное химической формулой I по любому из пп. 1-4, или его фармацевтически приемлемые соли в качестве эффективного ингредиента для предупреждения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC).
6. Фармацевтическая композиция по п. 5, где заболевания, опосредованные гистонодеацетилазой, включают инфекционные заболевания, новообразование, эндокринопатию, алиментарные и метаболические заболевания, психические и поведенческие нарушения, неврологические заболевания, глазные и глазные аднексальные заболевания, заболевания системы кровообращения, респираторные заболевания, заболевания органов пищеварения, заболевания кожи и подкожной клетчатки, заболевания опорно-двигательного аппарата и соединительной ткани или тератоз, деформации и хромосомную аберрацию.
7. Фармацевтическая композиция по п. 6, где
эндокринопатия, алиментарные и метаболические заболевания представляют болезнь Вильсона, амилоидоз или диабет;
психические и поведенческие нарушения представляют депрессию или синдром Ретта;
неврологические заболевания представляют атрофию центральной нервной системы, нейродегенеративное заболевание, двигательное нарушение, нейропатию, заболевание двигательных нейронов или демиелинизирующее заболевание центральной нервной системы;
глазные и глазные аднексальные заболевания представляют увеит;
заболевания кожи и подкожной клетчатки представляют псориаз;
заболевания опорно-двигательного аппарата и соединительной ткани представляют ревматоидный артрит, остеоартрит или системную красную волчанку;
тератоз, деформации и хромосомная аберрация представляют аутосомно-доминантный поликистоз почек;
инфекционные заболевания представляют прионную болезнь;
новообразование представляет доброкачественную опухоль или злокачественную опухоль;
заболевания системы кровообращения представляют мерцание предсердий или инсульт;
респираторные заболевания представляют астму; и
заболевания органов пищеварения представляют алкогольную болезнь печени, воспалительную болезнь кишечника, болезнь Крона или язвенную болезнь кишечника.
8. Способ предупреждения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC), предусматривающий введение терапевтически эффективного количества соединения, представленного химической формулой I по любому из пп. 1-4, или его фармацевтически приемлемых солей; или содержащей их фармацевтической композиции в качестве эффективного ингредиента нуждающемуся в этом субъекту.
9. Применение соединения, представленного химической формулой I по любому из пп. 1-4, или его фармацевтически приемлемых солей; или содержащей их фармацевтической композиции в качестве эффективного ингредиента для предупреждения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC).
10. Применение соединения, представленного химической формулой I по любому из пп. 1-4, или его фармацевтически приемлемых солей; или содержащей их фармацевтической композиции в качестве эффективного ингредиента для приготовления лекарственного средства для предупреждения или лечения заболеваний, опосредованных гистондеацетилазой (HDAC).
| WO 2017222951 A1, 28.12.2017 | |||
| WO 2017023133 A2, 09.02.2017 | |||
| RU 2018121499 A, 20.12.2019 | |||
| WO 2019110663 A1, 13.06.2019 | |||
| СПОСОБ ВОССТАНОВЛЕНИЯ НИЖНЕЙ ЧАСТИ ЛИЦА | 2009 |
|
RU2411009C1 |
