Область техники
Изобретение относится к новому улучшенному способу получения 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамида) формулы:
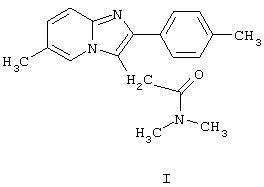
и его фармацевтически приемлемых кислотных аддитивных солей, к новым промежуточным соединениям, используемым в указанном способе, и к способу получения указанных промежуточных соединений.
Соединение Формулы I является превосходным седативным средством, продаваемым под МНН (Международным Непатентованным Названием) Золпидем.
Уровень техники
Известны два способа получения соединения Формулы I.
По способу, описанному в патенте ЕР 50563, известное соединение 6-метил-2-(4-метилфенил)имидазо[1,2-а]пиридин формулы:
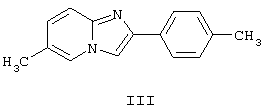
превращают по реакции Манниха в 3-(N,N-диметиламинометил)-6-метил-2-(4-метил-фенил)имидазоло[1,2-а]пиридин формулы:

Последнее соединение с помощью иодистого метила переводят в четвертичную соль, полученное таким образом триметиламмониевое производное формулы:
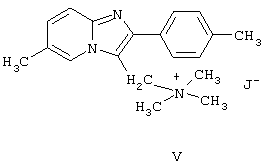
подвергают реакции с цианидом щелочного металла, образующееся при этом ацетонитрильное производное формулы:
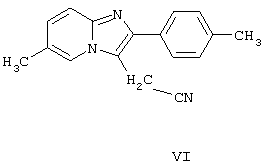
подвергают кислотному гидролизу, после чего образовавшееся ацетамидное производное формулы:

подвергают щелочному гидролизу. Полученное таким образом производное уксусной кислоты формулы:
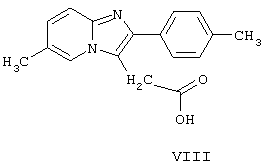
путем амидирования превращают в требуемое соединение Формулы I. Реакцию амидирования можно осуществить двумя способами. Согласно одному из способов соединение Формулы VIII подвергают реакции с хлористым тионилом и образующийся при этом хлорангидрид кислоты формулы:
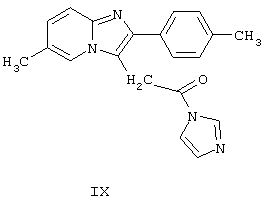
подвергают реакции с диметиламином. В патенте ЕР 50563 данный способ не поясняется примерами. При выполнении этого способа авторы настоящего изобретения обнаружили, что продукт реакции соединения Формулы VIII и хлористого тионила представляет собой смолу черного цвета, которая не может быть обработана. Таким образом, описанный выше способ не пригоден для производства в промышленном масштабе.
Согласно другому способу, производное уксусной кислоты Формулы VIII подвергают реакции с карбонилдиимидазолом в присутствии диметиламина, посредством чего требуемое соединение Формулы I получают через соединение Формулы:
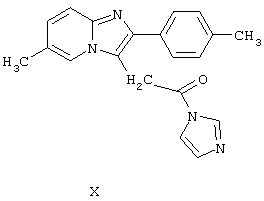
Недостаток этого способа заключается в том, что карбонилдиимидазол является дорогостоящим, токсичным, аллергенным и гигроскопичным соединением, манипуляции с которым в промышленном производстве сопряжены с большими трудностями. Еще одним недостатком является то, что требуемое соединение Формулы I загрязнено продуктами разложения карбонилдиимидазола. Таким образом, Золпидем, удовлетворяющий чрезвычайно строгим требованиям Фармакопеи, может быть получен лишь с помощью сложных методов очистки.
Целью патента ЕР 251589 является устранение перечисленных выше недостатков рассмотренного способа. В качестве исходного соединения используется 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин формулы III, который реагирует с N,N-диметил-2,2-диметоксиацетамидом формулы
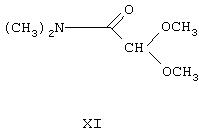
Производное α-гидрокси-N,N-диметилацетамида формулы:
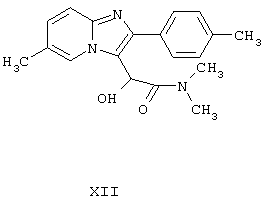
подвергают реакции с хлористым тионилом, после чего из производного α-хлор-N,N-диметилацетамида формулы:
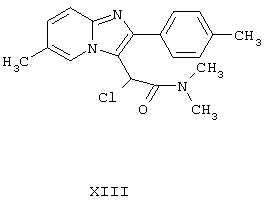
удаляют хлор путем реакции восстановления с боргидридом натрия с образованием требуемого соединения Формулы I. Недостаток данного способа заключается в том, что N,N-диметил-2,2-диметоксиацетамид Формулы XI не доступен в промышленном масштабе и может быть получен только с помощью специального оборудования. Кроме того, соединение Формулы XI чувствительно к следовым количествам влаги и кислот. Сложность получения соединения Формулы XI и чувствительность его к влаге и кислотам затрудняют использование данного способа в промышленном масштабе.
Сущность изобретения
Цель настоящего изобретения - устранить недостатки известных способов, а также создать простой способ получения высокочистого препарата Золпидем, применимый в промышленном масштабе.
Указанная выше цель достигается способом настоящего изобретения.
Согласно аспекту настоящего изобретения, представляется способ получения 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамида) Формулы I и его фармацевтически приемлемых кислотных аддитивных солей, включающий взаимодействие эфира общей формулы:
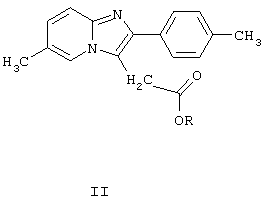
(где R - низший алкил или фенил-низший алкил) с диметиламином в полярном протонном или апротонном растворителе и, при необходимости, превращение полученного таким образом соединения Формулы I его фармацевтически приемлемую кислотную аддитивную соль.
Согласно еще одному аспекту настоящего изобретения, представляются новые эфиры общей Формулы II (где R - низший алкил, за исключением этила, или фенил-низший алкил) и их фармацевтически приемлемые кислотные аддитивные соли.
Согласно еще одному аспекту настоящего изобретения, представляются кристаллические соединения общей Формулы II и их фармацевтически приемлемые кислотные аддитивные соли (где R - низший алкил или фенил-низший алкил).
Согласно еще одному аспекту настоящего изобретения, представляется способ получения эфиров общей Формулы II (где R - низший алкил или фенил-низший алкил), включающий этерификацию производных уксусной кислоты Формулы VIII.
Подробное описание изобретения
Настоящее изобретение основывается на признании того, что соединение Формулы I может быть получено в чистом виде, без примесей, из новых соединений общей Формулы II, ранее не описанных, с помощью способа, осуществимого также в промышленном масштабе.
Термин «низший алкил» относится к прямым или разветвленным алкильным группам, содержащим 1-6 углеродных атомов (например, метил, этил,  -пропил, изопропил, н-бутил, вторичный бутил, третичный бутил, н-пентил или н-гексил). Термин «фенил-низший алкил« относится к низшим алкильным группам, определенным выше, имеющим в качестве заместителя одну или более фенильных групп (например, бензил, β-фенилэтил, β,β-дифенилэтил и т.д.).
-пропил, изопропил, н-бутил, вторичный бутил, третичный бутил, н-пентил или н-гексил). Термин «фенил-низший алкил« относится к низшим алкильным группам, определенным выше, имеющим в качестве заместителя одну или более фенильных групп (например, бензил, β-фенилэтил, β,β-дифенилэтил и т.д.).
Согласно предпочтительной реализации способа изобретения, в качестве исходного материала используются соединения общей Формулы II, в которых R представляет собой метил, этил, изопропил или бензил.
Реакция может проводиться в полярном протонном или апротонном растворителе. В качестве полярного протонного растворителя предпочтительней использовать низшие спирты (например, метанол, этанол, н-пропанол, изопропанол или н-бутанол). В качестве апротонного растворителя предпочтительней использовать ацетонитрил, диметилформамид, диметилсульфоксид или диамид гексаметилфосфорной кислоты. Предпочтительней проводить реакцию в метаноле, изопропаноле или ацетонитриле.
Реакция может осуществляться при 5-50°С, предпочтительнее при комнатной температуре.
Предпочтительнее насыщать используемый в качестве реакционной среды растворитель реагентом - диметиламином, и к полученному таким образом раствору прибавлять эфир общей Формулы II.
Реакционная смесь может быть обработана с помощью очень простых приемов. Требуемое соединение Формулы I осаждается из раствора в кристаллической форме и может быть легко отделено фильтрацией либо центрифугированием.
Согласно особенно предпочтительной реализации способа изобретения можно действовать следующим образом:
После фильтрации соединения Формулы I маточный раствор насыщают газообразным диметиламином, и таким образом непрореагировавшее соединение Формулы II, присутствующее в маточнике, превращают в требуемое соединение Формулы I. Газообразный диметиламин абсорбируют при низкой температуре, предпочтительнее при температуре от -10 до +10°С. Чистота полученной при этом второй порции соединения Формулы I идентична чистоте первой порции продукта.
Полученное таким образом соединение Формулы I может быть превращено в фармацевтически приемлемую кислотную аддитивную соль. Для образования соли могут быть использованы фармацевтически приемлемые неорганические кислоты (например, хлористый водород, бромистый водород, азотная кислота, фосфорная кислота, серная кислота и т.д.) и органические кислоты (например, винная кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, молочная кислота, n-толуолсульфокислота и т.д.). Образование соли проводят известным per se способом, например, прибавлением раствора соответствующей кислоты, образованного с органическим растворителем, к раствору соединения Формулы I в органическом растворителе. Предпочтительным является получение соли винной кислоты соединения Формулы I.
Способ настоящего изобретения имеет следующие преимущества:
- способ может быть просто и предпочтительнее выполнен;
- не требуется никакого дорогостоящего или специального оборудования;
- исключено использование исходного сырья и реагентов, трудных для обработки и/или токсичных;
- продукты реакции и маточные растворы не являются вредными для окружающей среды;
- не требуется тепловой энергии;
- требуемое соединение Формулы I легко может быть выделено в чистом виде.
Большинство исходных соединений общей Формулы II являются новыми. В журнале J. Med. Chem. 40, 3109 (1997) раскрыт общий метод получения соединения общей Формулы II, где R - этил. По этому способу 2-амино-5-метилпиридин подвергают реакции циклоконденсации с соответствующим бромкетоэфиром. Этил-[6-метил-2-(4-метил-фенил)имидазоло[1,2-а]пиридин-3-ил]ацетат получают в виде масла с выходом 10%. Физико-химические константы соединения, пригодные для его идентификации, не приводятся. С другой стороны, соединение общей Формулы II, где R - этил, полученное согласно настоящему изобретению, является кристаллическим веществом (см. Пример 4).
В способе настоящего изобретения предпочтительно использовать следующие соединения общей Формулы II:
метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат;
этил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат;
изопропил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат;
бензил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат.
Соединения общей Формулы II могут быть получены известными per se способами. Согласно способу, производное уксусной кислоты Формулы VIII этерифицируют спиртом общей Формулы:

(где R - как описано выше).
Реакция может быть предпочтительно выполнена с использованием избытка спирта общей Формулы XIV, в соответствии с чем он действует и как реагент, и как растворитель. Реакцию проводят в присутствии кислотного катализатора, предпочтительно, соляной кислоты, концентрированной серной кислоты или n-толуолсульфокислоты. Реакция может быть проведена при температуре от 20°С до точки кипения реакционной смеси; предпочтительно можно работать при температуре кипения. В качестве реакционной среды также может быть использован не смешивающийся с водой ароматический или хлорированный углеводород (например, толуол или хлороформ). В этом случае вода, образовавшаяся в ходе реакции, удаляется вместе с растворителем азеотропной отгонкой. Реакцию проводят в присутствии кислотного катализатора.
Соединение общей Формулы II выделяют из реакционной смеси известным методом. В некоторых случаях соединение общей Формулы II или его соль, образованная с катализатором, осаждается из реакционной массы. Твердое вещество отфильтровывают и в водной среде с помощью гидрокарбоната или карбоната щелочного металла переводят в основание. Основание, в большинстве случаев осаждающееся из раствора в кристаллическом виде, отфильтровывают, промывают и, при необходимости, перекристаллизовывают. Основание, которое остается в растворе, экстрагируют органическим растворителем, не смешивающимся с водой, и выделяют из экстракта выпариванием или кристаллизацией. Можно также осуществлять распределение реакционной массы между растворителем, не смешивающимся с водой, и ледяной водой для удаления катализатора, разделение слоев, сушку и выпаривание органической фазы и, наконец, очистку продукта путем перекристаллизации или хроматографии. Если образующаяся в ходе реакции вода отгоняется азеотропной перегонкой, реакционную массу разлагают ледяной водой, слои разделяют и обрабатывают, как указано выше.
Соединение общей Формулы II также может быть получено превращением производного уксусной кислоты Формулы VIII в соль щелочного металла и алкилированием полученной таким образом соли щелочного металла галогенидом общей Формулы:

(где R - как указано выше, а Х - галоген).
Солеобразование может быть выполнено с помощью гидрида щелочного металла (например, гидрида натрия или гидрида калия), амида щелочного металла (например, амида натрия или амида калия) или гидроксида щелочного металла (например, гидроксида натрия или гидроксида калия). Предпочтительней использовать гидроксид щелочного металла и работать в водной, водно-спиртовой или спиртовой среде. Особенно предпочтительно растворять производное уксусной кислоты Формулы VIII в водном, водно-спиртовом или спиртовом растворе гидроксида щелочного металла и проводить реакцию полученного таким образом раствора с галогенидом Формулы XV «in situ», т.е. без выделения соли щелочного металла соединения Формулы VIII. Реакцию проводят при температуре от -20°С до точки кипения растворителя, предпочтительнее при температуре кипения. Водно-спиртовая или спиртовая среда может быть приготовлена с использование спирта См (например, метанола, этанола, изопропанола, бутанола и т.д.).
Реакционная масса может быть обработана обычным методом. Предпочтительней экстрагировать реакционную массу органическим растворителем (целесообразно, галогенированным углеводородом, например, дихлорметаном или хлороформом) с последующей промывкой и упариванием экстракта.
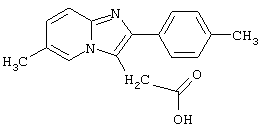
Производное уксусной кислоты Формулы VIII - известное соединение и может быть получено, как описано в ЕР 50563.
Соединения общей Формулы XIV и XV являются коммерчески доступными продуктами.
Дополнительные подробности настоящего изобретения следует искать в следующих Примерах неограничительного характера.
Пример 1
Метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат
К суспензии 28 г (0,1 моль) [6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]уксусной кислоты и 200 мл метанола по каплям прибавляют 6,9 мл (12,8 г, 0,13 моль) концентрированной серной кислоты. Реакционную массу нагревают при кипении в течение 3 часов, после чего с помощью внешнего охлаждения доводят сначала до комнатной температуры, а затем до 5-10°С и перемешивают при этой температуре в течение часа. Выпавший кристаллический продукт (сернокислую соль означенного соединения) отфильтровывают, промывают метанолом и сушат. Полученный таким образом продукт суспендируют в 500 мл воды, после чего добавлением 10%-ного раствора карбоната натрия при интенсивном перемешивании доводят рН до 8. Выпавший продукт отфильтровывают, дважды промывают водой порциями по 70 мл. Таким образом получают 27,4 г метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетата в виде кристаллического вещества белого цвета. Выход 93,3%, т. пл.: 133-136°С.
Элементный анализ: для формулы C18H18N2O2 (294,36)
вычислено: С 73,45%, Н 6,16%, N 9,52%
найдено: С 73,58%, Н 6,14%, N 9,62%
ИК(КВr) 2950, 1729, 1538, 1503, 1437, 1408, 1391, 1332, 1308, 1273, 1229, 1177, 1133, 1042, 994, 890, 823, 787, 753, 737, 706, 587, 553, 514, 417.
ПМР (CDCl3): δ, м.д. 7.84 (1Н, с, Н-5), 7.69 (2Н, д, J 8.0 Гц, Н-2',6'), 7.56 (1Н, д, J 9.2 Гц, Н-8), 7.28 (2Н, д, J 8.0 Гц, Н-3',5'), 7.06 (1Н, дд, J 1.5 и 9.2 Гц, Н-7), 4.02 (2Н, с, СН2), 3.76 (3Н, с, СН3), 2.40 (3Н, с, СН3-4'), 2.35 (3Н, с, СН3-6).
ЯМР13С (CDCl3): δ, м.д. 169.9 (С=O), 144.3 (С-8а), 143.9 (С-2), 137.5 (С-4'), 131.2 (С-1'), 129.2 (С-3',5'), 128.2 (С-2',6'), 127.5 (С-7), 122.0 (С-5), 121.1 (С-6), 116.7 (С-8), 112.1 (С-3), 52.4 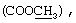 30.5 (CH2), 21.2 (СН3-4'), 18.3 (СН3-6).
30.5 (CH2), 21.2 (СН3-4'), 18.3 (СН3-6).
Пример 2
Метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат
Действуют, как описано в Примере 1, за исключением того, что серную кислоту заменяют 10 мл 20%-ного метанольного раствора хлористого водорода и реакционную массу нагревают при кипении в течение 8 часов. Метанол удаляют, к остатку прибавляют 200 мл воды, после чего рН доводят до 7 добавлением 10%-ного раствора гидрокарбоната натрия. Смесь трижды экстрагируют хлороформом порциями по 50 мл каждая.
Объединенные органические фазы промывают 50 мл воды, сушат над безводным сульфатом натрия и упаривают. Кристаллический остаток перекристаллизовывают из ацетонитрила. Таким образом получают 21,0 г метил-[6-метил-2-(4-метилфенил)-имидазоло[1,2-а]пиридин-3-ил]ацетата. Выход 71,5%. Продукт во всех отношениях идентичен соединению, полученному согласно Примеру 1.
Пример 3
Метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат
Действуют, как описано в Примере 1, за исключением того, что метанольный раствор хлористого водорода заменяют 5,0 г катализатора -  -толуолсульфокислоты - и реакционную массу нагревают при кипении в течение 6 часов. Кристаллический продукт перекристаллизовывают из ацетонитрила. Таким образом получают 22,2 г метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетата, выход 71,5%. Продукт во всех отношениях идентичен соединению, полученному согласно Примеру 1.
-толуолсульфокислоты - и реакционную массу нагревают при кипении в течение 6 часов. Кристаллический продукт перекристаллизовывают из ацетонитрила. Таким образом получают 22,2 г метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетата, выход 71,5%. Продукт во всех отношениях идентичен соединению, полученному согласно Примеру 1.
Пример 4
Этил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил)ацетат
Действуют, как описано в Примере 1, за исключением того, что метанол заменяют этанолом. Таким образом получают 24,0 г этил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетата, выход 78,3%.
Т. пл.: 101-103°С.
Элементный анализ: для формулы C19H20N2O2 (308,43)
вычислено: С 74,00%, Н 6,54%, N 9,08%
найдено: С 74,17%, Н 6,58%, N 8,97%.
ИК(КВr) 2983, 1725, 1537, 1503, 1391, 1367, 1345, 1326, 1307, 1272, 1225, 1185, 1143, 1058, 1021, 875, 824, 788, 737, 703, 584, 556, 515.
ПМР (CDCl3): δ, м.д. 7.88 (1Н, с, Н-5), 7.73 (2Н, д, J 8.0 Гц, Н-2',6'), 7.57 (1Н, д, J 9.2 Гц, Н-8), 7.28 (2Н, д, J 8.0 Гц, Н-3',5'), 7.07 (1Н, дд, J 1.5 и 9.2 Гц, Н-7), 4.00 (2Н, с, СН2), 4.22 (2Н, кв, J 7.2 Гц, 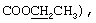 2.40 (3Н, с, СН3-4'), 2.36 (3Н, с, СН3-6), 1.28 (3Н, т, J 7.2 Гц,
2.40 (3Н, с, СН3-4'), 2.36 (3Н, с, СН3-6), 1.28 (3Н, т, J 7.2 Гц, 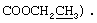
ЯМР13С (CDCl3): δ, м.д. 169.4 (С=O), 144.1 (С-8а), 143.8 (С-2), 137.5 (С-4'), 131.0 (С-1'), 129.3 (С-3',5'), 128.3 (С-2',6'), 127.6 (С-7), 122.0 (С-5), 121.3 (С-6), 116.6 (С-8), 112.3 (С-3), 61.5 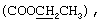 30.8 (CH2), 21.2 (СН3-4'), 18.4 (СН3-6), 14.0
30.8 (CH2), 21.2 (СН3-4'), 18.4 (СН3-6), 14.0 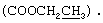
Пример 5
Изопропил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат
Действуют, как описано в Примере 1, за исключением того, что метанол заменяют изопропанолом. Таким образом получают 24,7 г изопропил-[6-метил-2-(4-метилфенил)-имидазоло[1,2-а]пиридин-3-ил]ацетата, выход 76,7%.
Т. пл.: 104-105,5°С.
Элементный анализ: для формулы C20H22N2O2 (322,41)
вычислено: С 74,51%, Н 6,88%, N 8,69%
найдено: С 74,57%, Н 6,92%, N 8,61%
ИК (КВr) 2980, 1730, 1613, 1536, 1503, 1460, 1420, 1390, 1376, 1340, 1320, 1270, 1228, 1185, 1108, 1053, 973, 945, 901, 843, 817, 796, 757, 740, 726, 704, 625, 590, 559, 514.
ПМР (CDCl3): δ, м.д. 7.91 (1Н, с, Н-5), 7.74 (2Н, д, J 8.0 Гц, Н-2',6'), 7.57 (1Н, д, J 9.0 Гц, Н-8), 7.28 (2Н, д, J 8.0 Гц, Н-3',5'), 7.07 (1Н, дд, 9.0 Гц, Н-7), 5.08 (1Н, г, J 6.2 Гц, 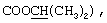 3.97 (2Н. с, CH2), 2.40 (3Н, с, СН3-4'), 2.36 (3Н, с, СН3-6), 1.26 (6Н, д, J 6.2 Гц,
3.97 (2Н. с, CH2), 2.40 (3Н, с, СН3-4'), 2.36 (3Н, с, СН3-6), 1.26 (6Н, д, J 6.2 Гц, 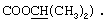
ЯМР13С (CDCl3): δ, м.д. 169.0 (С=O), 144.2 (С-8а), 143.8 (С-2), 137.5 (С-4'), 131.1 (С-1'), 129.2 (С-3',5'), 128.3 (С-2',6'), 127.5 (С-7), 121.9 (С-5), 121.3 (С-6), 116.6 (С-8), 112.5 (С-3), 69.2 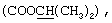 31.1 (СН2), 21.7 21.2 (СН3-4'), 18.3 (СН3-6).
31.1 (СН2), 21.7 21.2 (СН3-4'), 18.3 (СН3-6).
Пример 6
Бензил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат
К раствору 1 г (0,025 моль) гидроксида натрия в 40 мл воды добавляют 5,6 г (0,02 моль) [6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]уксусной кислоты, после чего по каплям прибавляют 2,5 мл (3,6 г, 0,021 моль) бромистого бензила. Реакционную массу нагревают при кипении в течение полутора часов, затем охлаждают до комнатной температуры и дважды экстрагируют дихлорметаном порциями по 50 мл каждая. Органическую фазу промывают 30 мл 10%-ного раствора гидроксида натрия и 50 мл воды, сушат над безводным сульфатом натрия, фильтруют и упаривают. Маслянистый остаток растирают с петролейным эфиром. Таким образом получают 5,3 г бензил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетата, выход 71,0%.
Т. пл.: 106-107°С.
Элементный анализ: для формулы C24H22N2O2 (370,45)
вычислено: С 77,81%, Н 5,99%, N 7,56%
найдено: С 77,74%, Н 6,03%, N 7,59%
ИК (КВr) 3032, 2920, 1719, 1538, 1501, 1450, 1409, 1378, 1346, 1319, 1304, 1266, 1247, 1180, 1141, 1122, 1050, 1020, 998, 970, 896, 825, 812, 740, 694, 581, 504.
ПМР (CDCl3): δ, м.д. 7.79 (1Н, с, Н-5), 7.69 (2Н, д, J 8.0 Гц, Н-2',6'), 7.55 (1Н, д, J 9.1 Гц, Н-8), ˜ 7.3 (5Н, м, АrН), 7.22 (2Н, д, J 7.8 Гц, Н-3',5'), 7.04 (1Н, дд, 9.1 Гц, Н-7), 5.18 (2Н, с, PhCH2), 4.04 (2Н, с, CH2), 2.39 (3Н, с, СН3-4'), 2.28 (3Н, с, СН3-6).
ЯМР13С (CDCl3): δ, м.д. 169.3 (С=O), 144.3 (С-8а), 143.8 (С-2), 137.5 (С-4'), 131.0 (С-1'), 129.2 (С-3',5'), 128.3 (С-2',6'), 127.6 (С-7), 122.0 (С-5), 121.3 (С-6), 116.6 (С-8), 112.1 (С-3), 67.2 (PhCH2), 30.8 (CH2), 21.2 (СН3-4'), 18.3 (СН3-6).
Пример 7
Метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат
К суспензии 28 г (0,10 моль) [6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]уксусной кислоты, 200 мл толуола и 10 мл метанола по каплям при перемешивании прибавляют 1.6 мл (2,9 г, 0,03 моль) концентрированной серной кислоты. Реакционную массу кипятят с обратным холодильником в течение 4 часов с удалением воды, охлаждают до комнатной температуры, после чего прибавляют 100 мл воды и 10%-ным раствором гидрокарбоната натрия доводят рН до 7. Слои разделяют, и водную фазу экстрагируют 50 мл толуола. Объединенные органические фракции промывают 50 мл воды, сушат над безводным сульфатом натрия, фильтруют и упаривают. Остаток перекристаллизовывают из ацетонитрила. Таким образом получают 24,1 г метил-[6-метил-2-(4-метилфенил)-имидазоло[1,2-а]пиридин-3-ил]ацетата. Продукт во всех отношениях идентичен соединению, полученному согласно Примеру 1.
Пример 8
Метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат
Действуют, как описано в Примере 7, за исключением того, что концентрированную серную кислоту заменяют 5 мл концентрированной соляной кислоты и реакционную массу кипятят с обратным холодильником в течение 5 часов. Таким образом получают 21,5 г метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетата. Продукт во всех отношениях идентичен соединению, полученному согласно Примеру 1.
Пример 9
6-Метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамид)
30,7 г (0,68 моль) газообразного диметиламина абсорбируют в 45 мл безводного метанола при температуре от -5°С до 0°С, после чего добавляют 25 г (0,085 моль) метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетата. Реакционную массу перемешивают при комнатной температуре в течение 7 дней. Выпавший кристаллический продукт отфильтровывают и перекристаллизовывают из ацетонитрила. Таким образом получают 24,1 г 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметил-ацетамида). Выход 92,2%. Т. пл.: 194-196°С; чистота, определенная с помощью ВЭЖХ: выше 99,8%.
Элементный анализ: для формулы С19Н21Н3О (307, 40)
вычислено: С 74,24%, Н 6,89%, N 13,67%
найдено: С 73,78%, Н 6,85%, N 13,73%
ИК (КВr) 2916, 1637, 1539, 1505, 1424, 1395, 1345, 1265, 1219, 1189, 1139, 1062, 824, 795, 728, 605, 518.
ПМР (CDCl3): δ, м.д. 7.96 (1Н, д, J 0.6 Гц, Н-5), 7.55 (2Н, д, J 8.0 Гц, Н-2',6'), 7.52 (1Н, д, J 9.2 Гц, Н-8), 7.25 (2Н, д, J 8.0 Гц, Н-3',5'), 7.03 (1Н, дд, J 1.5 и 9.2 Гц, Н-7), 4.05 (2Н, с, СН3), 2.93 (3Н, с, NСН3), 2.87 (3Н, с, NСН3), 2.39 (3Н, с, СН3-4'), 2.32 (3Н, с, СН3-6).
ЯМР13С (CDCl3): δ, м.д. 168.2 (С=O), 143.9 (С-8а), 143.5 (С-2), 137.4 (С-4'), 131.5 (С-1'), 129.3 (С-3',5'), 128.4 (С-2',6'), 127.6 (С-7), 122.2 (С-5), 121.8 (С-6), 116.4 (С-8), 113.6 (С-3), 37.4 (NСН3), 33.58 (NСН3), 30.2 (CH2), 21.2 (СН3-4'), 18.4 (СН3-6).
5,0 г (0,11 моль) газообразного диметиламина абсорбируют в маточнике продукта, полученного согласно Примеру 9, при температуре от -5°С до 0°С. Реакционную массу перемешивают в течение 2 дней. Таким образом получают дополнительно 1,7 г означенного соединения, выход 6,5%. Чистота этой части продукта идентична чистоте основной массы.
Пример 10
6-Метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамид)
Действуют, как описано в Примере 9, за исключением того, что реакцию проводят в закрытом аппарате при 40°С в течение 2 дней. Таким образом получают 24,5 г 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамида), выход 93,8%. Продукт во всех отношениях идентичен соединению, полученному согласно Примеру 9.
Пример 11
6-Метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамид)
Действуют, как описано в Примере 9, за исключением того, что реакционную массу перемешивают при 5°С в течение 10 дней. Полученное кристаллическое вещество перекристаллизовывают из ацетонитрила. Таким образом получают 16,1 г 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамида), выход 61,7%. Продукт во всех отношениях идентичен соединению, полученному согласно Примеру 9.
Пример 12
6-Метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамид)
Действуют, как описано в Примере 9, за исключением того, что метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат заменяют 26,2 г (0,085 моль) этил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетата, а метанол заменяют безводным этанолом. Таким образом получают 20,7 г 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамида), выход 79,3%. Продукт во всех отношениях идентичен соединению, полученному согласно Примеру 9.
Пример 13
6-Метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамид)
Действуют, как описано в Примере 9, за исключением того, что метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат заменяют 26,6 г (0,085 моль) изопропил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетата, а метанол заменяют безводным изопропанолом. Реакционную массу перемешивают в течение 48 часов при 50°С вместо комнатной температуры. Таким образом получают 13,6 г 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамида), выход 51,2%. Продукт во всех отношениях идентичен соединению, полученному согласно Примеру 9.
Пример 14
6-Метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамид)
Действуют, как описано в Примере 9, за исключением того, что метил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетат заменяют 31,5 г (0,085 моль) бензил-[6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-ил]ацетата, а метанол заменяют безводным ацетонитрилом. Таким образом получают 18,1 г 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамида), выход 69,5%. Продукт во всех отношениях идентичен соединению, полученному согласно Примеру 9.
Пример 15
6-Метил-2-(4-метилфенил)имидазоло[1.2-а]пиридин-3-(N,N-диметилацетамид)-полутартрат
10 г (0,033 моль) 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамида) растворяют в 40 мл безводного метанола, после чего при перемешивании прибавляют раствор 2,44 г L-(+)-винной кислоты в 10 мл метанола. Реакционную массу перемешивают при комнатной температуре в течение часа. Выпавшее кристаллическое вещество выдерживают в течение ночи в холодильнике, отфильтровывают и промывают небольшим количеством этанола. Таким образом получают 11,3 г 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметил-ацетамида)полутартрата в виде кристаллического вещества белого цвета. Выход 92,7%. Т. пл.: 195-197°С.
Чистота продукта по ВЭЖХ выше 99,8%; общее содержание примесей ниже 0,2%. Продукт полностью удовлетворяет требованиям Европейской Фармакопеи - Дополнение 1998, 525.
Элементный анализ: для формулы С42Н48N6О8 (764,9)
вычислено: С 65,95%, Н 6,33%, N 10,99%
найдено: С 65,71%, Н 6,23%, N 10,82%
ИК (КВr) 3542, 3456, 2921, 1638, 1513, 1405, 1264, 1199, 1124, 1072, 918, 854, 797, 683, 599, 513.
ПМР (CDCl3): δ, м.д. 8.04 (1Н, д, J 0.6 Гц, Н-5), 7.52 (2Н, д, J 8.0 Гц, Н-2',6'), 7.51 (1Н, д, J 9.2 Гц, Н-8), 7.25 (2Н, д, J 8.0 Гц, Н-3',5'), 7.12 (1Н, дд, J 1.6 и 9.2 Гц, Н-7), 4.31 (1Н, с), 4.12 (2Н, с, СН2), 3.13 (3Н, с, NСН3), 2.90 (3Н, с, NСН3), 2.34 (3Н, с, СН3-4'), 2.30 (3Н, с, СН3-6).
ЯМР13С (CDCl3): δ, м.д. 173.4 (СООН), 168.2 (С=O), 142.9 (С-8а), 142.5 (С-2), 136.7 (С-4'), 132.0 (С-1'), 129.3 (С-3',5'), 127.8 (С-2',6'), 127.3 (С-7), 122.6 (С-5), 120.9 (С-6), 115.9 (С-8), 115.4 (С-3), 72.3 (СH), 37.1 (NСН3), 35.5 (NСН3), 29.1 (CH2), 21.0 (СН3-4'), 18.0 (СН3-6).
| название | год | авторы | номер документа |
|---|---|---|---|
| N, N', N'-ТРИЗАМЕЩЕННЫЕ ИЗОСЕЛЕНОМОЧЕВИНЫ | 2010 |
|
RU2434852C1 |
| ЭНАНТИОМЕРНО ЧИСТЫЕ ОСНОВНЫЕ ЭФИРЫ АРИЛ-ЦИКЛОАЛКИЛГИДРОКСИКАРБОНОВЫХ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 1997 |
|
RU2238936C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2179554C2 |
| ЗАМЕЩЕННЫЕ МЕТИЛ-N-МЕТОКСИКАРБАМАТЫ | 2018 |
|
RU2698300C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-СУЛЬФОНИЛАМИНО-1,2,4,-ТРИАЗОЛО[1,5-a] ПИРИМИДИНОВ | 2007 |
|
RU2325390C1 |
| ЗАМЕЩЕННЫЕ ДИАЛКИЛ (ОКСИДО)-Λ-СУЛЬФАНИЛИДЕН НИКОТИНАМИД ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2014 |
|
RU2711749C2 |
| ПРОИЗВОДНЫЕ УРАЦИЛА, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ ПОДАВЛЕНИЯ РОСТА СОРНЯКОВ | 2000 |
|
RU2259359C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИН-2,4-ДИАМИНА ДЛЯ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2672916C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(АЗОЛ-1-ИЛ)ЭТАНАМИНОВ | 2006 |
|
RU2317984C2 |
| Способ получения замещенных 3-арилпирролов | 2024 |
|
RU2831117C1 |
Изобретение относится к новому улучшенному способу получения 6-метил-2-(4-метилфенил)имидазоло[1,2-а]пиридин-3-(N,N-диметилацетамида) формулы (I) или его фармацевтически приемлемых кислотных аддитивных солей, который включает взаимодействие эфира общей Формулы (II) (где R - низший алкил или фенил-низший алкил) с диметиламином в полярном протонном или апротонном растворителе и, при необходимости, превращение полученного таким образом соединения Формулы I в фармацевтически приемлемую кислотную аддитивную соль. Соединение Формулы I является известным эффективным седативным средством, применяемым в терапии. Изобретение также относится к промежуточным соединениям общей Формулы II (где R - низший алкил или фенил-низший алкил), используемым в данном способе. Способ позволяет получать в одну стадию высокочистый продукт без применения вредных и токсичных реагентов. 4 н. и 12 з.п.ф-лы.
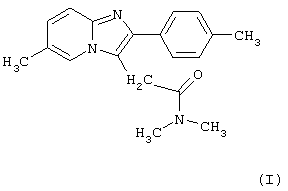
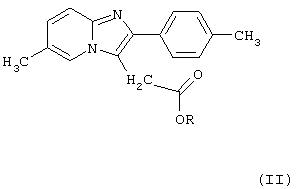
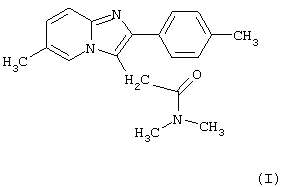
и его фармацевтически приемлемых кислотных аддитивных солей, включающий взаимодействие эфира общей формулы
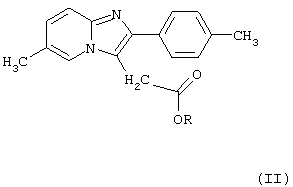
где R - низший алкил или фенил-низший алкил, с диметиламином в полярном протонном или апротонном растворителе и, при необходимости, превращение полученного таким образом соединения формулы I в его фармацевтически приемлемую кислотную аддитивную соль.
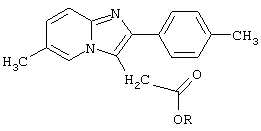
где R означает низший алкил, за исключением этила, или фенил-низший алкил.
где R означает низший алкил или фенил - низший алкил.
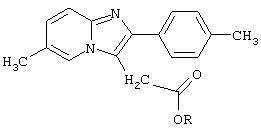
где R означает низший алкил или фенил-низший алкил, характеризующийся тем, что этерифицируют производное уксусной кислоты формулы VIII

где R имеет то же значение, что указано в п.10.

где R - как указано в п.10, а Х означает галоген.
| TRAPANT G | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ofmed | |||
| Chem | |||
| Приспособление с иглой для прочистки кухонь типа "Примус" | 1923 |
|
SU40A1 |
| КОМНАТНАЯ ПЕЧЬ С ПЛИТОЙ | 1926 |
|
SU8021A1 |
| ЕР 1104765, А 06.06.2001. | |||

