ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к производному таксола, которое может быть введено перорально и имеет противоопухолевую активность.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Таксол является природным веществом, представленным следующей химической структурной формулой, которое может быть получено в небольших количествах из коры или других частей Taxus brevifolia.
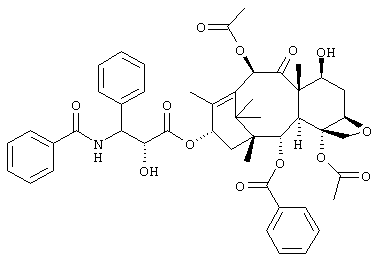
Известно, что таксол обладает противоопухолевой активностью, и считается, что механизм его действия состоит в ингибировании деполимеризации межклеточных микротрубок; таким образом, ожидают, что в клинике его можно применять в качестве противоопухолевого агента, имеющего механизм действия, отличный от обычных противоопухолевых агентов.
Таксол до сих пор получают из природных источников и в весьма незначительных количествах. Однако сообщается о производных таксола, синтезированных с использованием предшественника таксола 10-O-деацетилбаккатина III, представленного следующей формулой, который может быть получен из листьев и других частей растений семейства Taxus в относительно больших количествах.
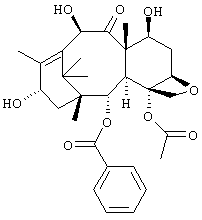
Среди указанных соединений внимание привлекает соединение (таксотер, далее обозначаемое как "соединение А"), имеющее структуру, представленную формулой ниже, и имеющее противоопухолевую активность, равную или выше активности таксола, и его исследование в качестве противоопухолевого агента в настоящее время широко развивается.
Авторы настоящего изобретения сообщают, что соединение, полученное превращением гидроксильной группы, образованной восстановлением 9 положения кетона, и гидроксильной группы в положении 10, в циклическую ацетальную форму имеет высокую противоопухолевую активность (JP-A-9-12578, приведенное здесь обозначение "JP-A" означает и опубликованную, не прошедшую экспертизу заявку на патент Японии).
Таксол, таксотер и соединение, раскрытое в JP-A-9-12578, являются перспективными в качестве противоопухолевых агентов. Однако соединения, раскрытые в Примерах заявки JP-A-9-12578, имеют недостаток с точки зрения их токсичности, а их эффективность при пероральном введении неизвестна. Например, с точки зрения удобства для пациента в процессе приема препарата, а также с точки зрения медицинской экономики существует потребность в производном таксола, которое можно вводить перорально.
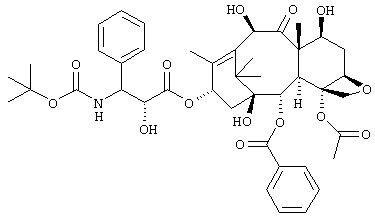
В результате интенсивных исследований с целью получения производного таксола, которое может обеспечить высокую безопасность при пероральном введении, одновременно сохраняя высокую противоопухолевую активность и уменьшенное с точки зрения проблем, связанных с токсичностью, авторы настоящего изобретения провели интенсивные исследования и получили соединение (далее обозначенное как "соединение В"), имеющее следующую формулу и демонстрирующее высокую противоопухолевую активность даже при пероральном введении, например в тесте на противоопухолевую активность на мышах.
Проблема токсичности для этого соединения уменьшена по сравнению с соединением, раскрытым в Примерах заявки JP-A-9-12578. Однако не удалось достичь гарантии в его применимости при пероральном введении человеку, поскольку было обнаружено с применением метаболического теста in vitro с использованием микросом печени человека, что метаболизм соединения в микросоме печени человека протекает очень быстро.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
С целью ингибирования модификации соединения из-за его метаболизма авторы провели исследование новой модификации лекарственного средства и обнаружили, что соединение, в котором вводится заместитель в пиридиновое кольцо в положении 13 боковой цепи, плохо подвергается процессу метаболизма в микросоме печени человека и способно обеспечить безопасность, необходимую при пероральном введении, и при этом сохранить противоопухолевую активность, а также снизить проблемы с токсичностью, что приводит к достижению целей изобретения.
Соответственно, изобретение относится к соединению, представленному приведенной ниже формулой, или его соли, медикаменту, включающему соединение с приведенной ниже формулой или его соль, а также противоопухолевому препарату, который включает соединение следующей формулы или его соль.
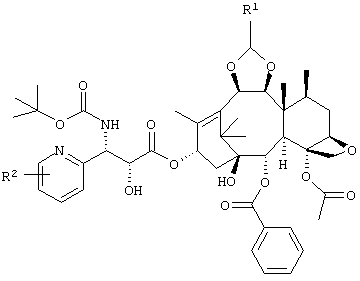
Изобретение также включает промежуточное соединение (далее обозначенное как "промежуточное соединение по изобретению"), представленное приведенной ниже формулой, применяемое для получения производного таксола, а также его применение.
В приведенной ниже формуле R1 представляет собой диметиламинометил или морфолинометил, и R2 представляет собой атом галогена или алкокси группу, содержащую от 1 до 6 атомов углерода. Предпочтительные примеры R2 включают метокси группу, атом фтора или атом хлора, более предпочтительно атом фтора и метокси группу.
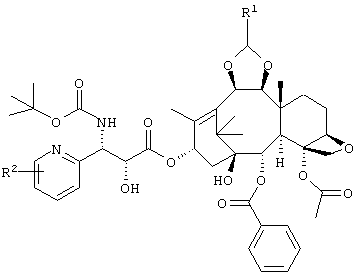
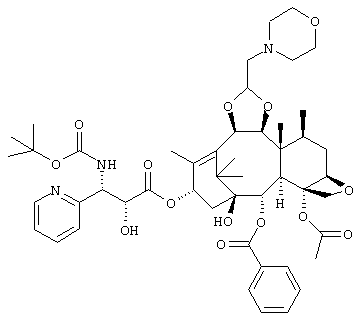
Особенно предпочтительным является соединение, представленное следующей формулой, а именно:
(1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокси-такс-11-ен-13-ил(2R, 3S)-3-(трет.-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат,
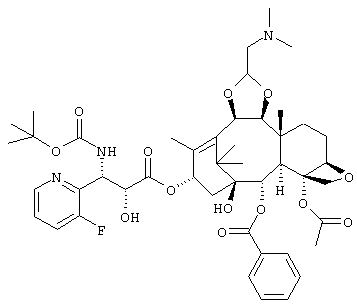
или его соль.
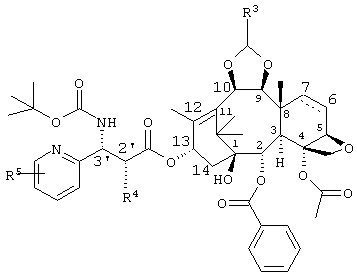
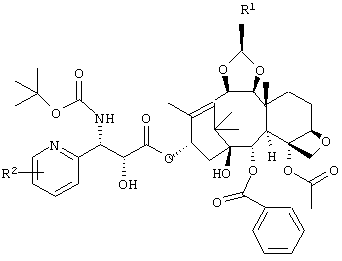
Также, в вышеупомянутом промежуточном соединении по изобретению R3 представляет собой диметиламинометил, морфолинометил или винил, R4 представляет собой гидроксильную группу, которая может иметь защитную группу, и R5 представляет собой алкокси группу, содержащую от 1 до 6 атомов углерода, или атом галогена. Кроме того, часть пунктирной линии между позициями 6 и 7 частичной структуры в промежуточном соединении по изобретению, представленной следующей формулой
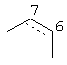
означает, что связь в этой части может быть двойной связью.
В промежуточном соединении по настоящему изобретению примеры защитных групп для R4 включают триалкилсилильную группу, бензильную группу, замещенную бензильную группу, 1-этоксиэтил, бензилоксикарбонил и 2,2,2-трихлорэтоксикарбонильную группу. Предпочтительными являются триалкилсилильная группа, такая как триизопропилсилильная группа, трет. бутилдиметилсилильная группа или триэтилсилильная группа и бензильная группа, наиболее предпочтительными являются триизопропилсилильная группа и бензильная группа.
Промежуточный продукт получения производного таксола по настоящему изобретению может быть использован путем необязательного выбора в соответствии с конечным интересующим продуктом. Например, для получения соединения следующей формулы
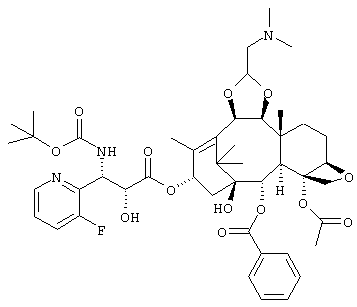
или его соли желательно использовать соединение следующей формулы
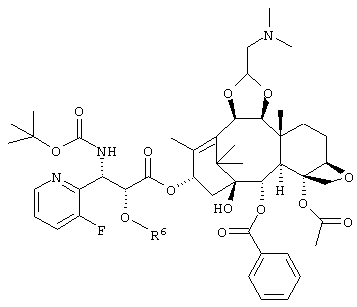
(где R6 является триизопропилсилильной группой, трет. бутилдиметилсилильной группой, триэтилсилильной группой, или бензильной группой) или его соль, соединение формулы
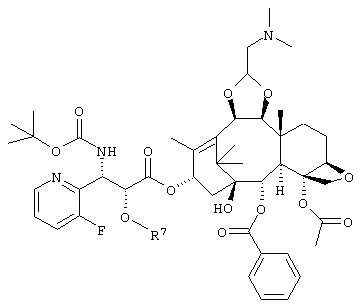
(где R7 является триизопропилсилильной группой, трет. бутилдиметилсилильной группой, триэтилсилильной группой или бензильной группой) или его соль или соединение следующей формулы:
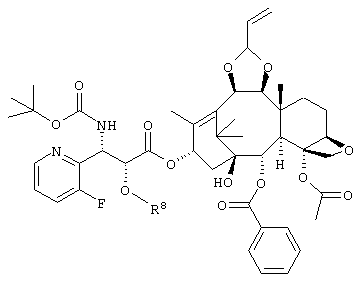
(где R8 представляет собой триизопропилсилил, трет. бутилдиметилсилил, триэтилсилил или бензил) или его соль.
Соединение по настоящему изобретению может находиться в свободной форме или в форме кислотно-аддитивной соли. Примеры кислотно-аддитивных солей включают соли неорганических кислот, таких как гидрохлорид, сульфат, нитрат, гидробромид, гидройодид и фосфат, или соли органических кислот, таких как ацетат, метансульфонат, бензолсульфонат, толуолсульфонат, цитрат, малеат, фумарат и лактат. Оно также может находиться в форме сольвата, и примеры растворителя включают воду, метанол, этанол, пропанол, бутанол, ацетон, ацетонитрил, бензол, толуол, тетрагидрофуран и N,N-диметилформамид.
Соединение по настоящему изобретению может быть синтезировано в соответствии со способом, описанным в JP-A-9-12578, например, по последующим методам синтеза. В этой связи реакция может быть проведена посредством защиты замещающих групп с помощью необходимых в каждом случае защитных групп, однако последовательность снятия защиты не столь существенна.
Способ Синтеза 1:
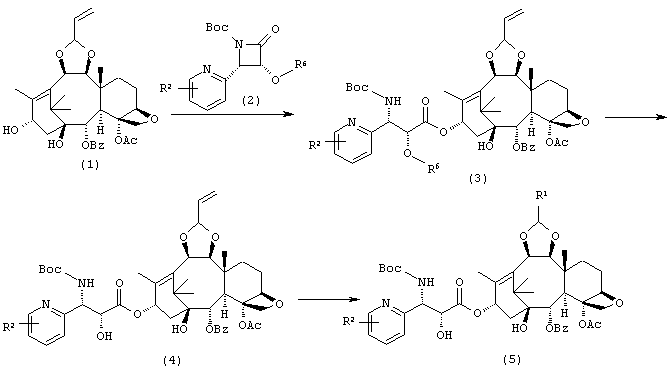
Соединение (3) получают конденсацией соединения (1) с соединением (2) в присутствии основания. Далее защитную группу на гидроксильной группе полученного таким образом соединения (3) удаляют с образованием соединения (4). Его терминальную олефиновую группу превращают в диол действием окисляющего агента, такого как N-метилморфолин-N-оксид, в присутствии катализатора тетраоксида осмия, и затем подвергают окислительному расщеплению действием перйодата натрия или подобного реагента с образованием альдегида. Затем проводят восстановительную реакцию с соответствующим амином с получением соединения (5).
Способ Синтеза 2:
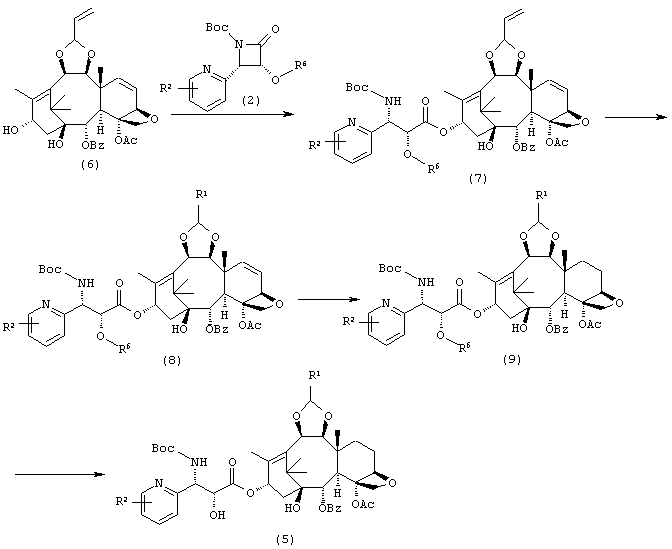
Соединение (7) получено конденсацией соединения (6) с соединением (2) аналогично Способу Синтеза 1. Далее соединение (8) может быть получено превращением его терминальной олефиновой группы аналогично Способу Синтеза 1. Затем соединение (9) получают восстановлением олефина при 6- и 7-положениях гидрированием, и затем наконец удаляют защитные группы с гидроксильной группы, получая таким образом соединение (5).
Исходные вещества (1) и (6) в описанном выше Способе Синтеза 1 могут быть синтезированы в соответствии с описанием, представленным в JP-A-9-12578. Соединение (2) также может быть синтезировано в соответствии с известным способом синтеза для β -лактамов, описанным в литературе (см., например, J. Org. Сhem., 61, 2664-2676 (1996)).
В указанных выше способах синтеза R1, R2 и R6 определены, как описано ниже. Что касается сокращений, Воc обозначает третичную бутоксикарбонильную группу, Ас обозначает ацетильную группу, и Bz обозначает бензоильную группу.
Кроме того, медикамент по изобретению может использоваться при лечении раковых заболеваний на основе его противоопухолевого действия, и примеры объектов для лечения включают различные виды рака, такие как рак легких, рак желудочно-кишечного тракта, рак яичников, рак матки, рак груди, рак печени, рак головы и шеи, рак крови, рак почек и опухоли яичек.
Соединение по настоящему изобретению может вводиться посредством различных инъекций, таких как внутривенная инъекция, внутримышечная инъекция и подкожная инъекция, либо с помощью различных методов, таких как пероральное или подкожное введение. Пероральное введение среди указанных способов является желательным с точки зрения достижения эффекта, который будет описан ниже. В случае перорального введения может применяться любое из свободных соединений или солей.
При проведении теста с использованием нераковых мышей соединение по данному изобретению не показало почечной токсичности.
Применимость соединения по изобретению в виде пероральных препаратов может быть предсказана на основе тестов in vitro с использованием микросомы печени человека. В случае перорального введения лекарство растворяется в желудочно-кишечном тракте, претерпевает метаболизм в пищеварительном тракте и печени и затем попадает в систему кровообращения. Соответственно, считается, что метаболизм лекарства в печени показывает влияние на выражение эффективности лекарства. В особенности предсказывается, что соединение по изобретению и аналогичные ему соединения претерпевают метаболизм под действием CYP3A, являющегося ферментом, распределенным в микросоме печени. Таким образом, предсказание метаболизма в тестах in vitro с использованием микросомы печени является важным при рассмотрении возможного клинического применения на практике. Сообщалось, например, в Pharm. Tech. Japan, 13, 17-39, 1997 и J. Pharmacol. Exp. Ther., 283, 46-58, 1997, что предсказанные величины метаболизма при in vitro тестировании с использованием микросомы печени практически совпадают с величинами, измеренными в клинических тестах на человеке. Микросома печени человека поставляется, например, компанией Xenotech LLC, и измерение скорости метаболизма может проводиться с учетом вышеуказанных журналов.
После измерения скорости метаболизма в микросоме печени можно рассчитать биодоступность лекарства как теоретическую величину (J. Pharmacol. Exp. Ther., 283, 46-58, 1997). Биодоступность определяется как количество и уровень лекарства, которое достигает системы кровообращения по отношению к количеству введенного лекарства (Pharmacokinetic Studies on Drug Development, edited by Yuichi Sugiyama, стр.15, издано Yakuji Jino). В случае перорального введения существует много препятствий до попадания лекарства в систему кровообращения, такие как растворение в желудочно-кишечном тракте, прохождение через слизистую мембрану кишечного тракта, а также метаболизм в кишечном тракте и печени. Таким образом, считается, что диапазон изменения его конечной концентрации в крови, а именно биодоступность, у индивидуума становится выше по сравнению со случаем прямого введения в систему кровообращения. Hellriegel и др. изучили величину биодоступности и ее индивидуальные изменения (величина CV) для 149 наименований различных лекарств, имеющихся на рынке, и сообщили, что между ними существует отрицательная корреляция (Clin. Pharmacol. Ther., 60, 601-607, 1996). Таким образом, известно, что диапазон изменчивости биодоступности среди индивидуумов становится больше, в то время как величина биодоступности становится меньше.
Что касается противоопухолевых агентов, их вводят в количествах, практически близких к максимально переносимой дозе, с целью усиления ответной реакции, таким образом, что терапевтический диапазон и диапазон токсичности становятся очень близкими, это в результате приводит к сужению безопасного диапазона. Таким образом, становится затруднительным применять в качестве противоопухолевого агента лекарство с широким диапазоном изменчивости индивидуальной биодоступности.
Что касается соединения по настоящему изобретению, скорость его метаболизма в микросоме печени человека была снижена, и теоретическая величина биодоступности его неизмененной формы также повысилась. Таким образом, было предсказано, что диапазон изменчивости величин биодоступности неизмененного соединения для индивидуумов будет небольшим. Вследствие указанного эффекта вполне возможно провести пероральное введение соединения по изобретению с точки зрения безопасности в увеличенном диапазоне безопасности, а также с точки зрения увеличения эффективности лекарства. В этой связи теоретическая величина биодоступности неизмененного соединения предпочтительно равна 0,4 или выше, более предпочтительно 0,7 или выше.
Кроме того, применимость соединения по изобретению в качестве препаратов для перорального введения может быть предсказана на основе теста биодоступности (БП) на обезьянах. Метаболизм соединения (В) микросомами печени мыши и собаки является медленным, и свойство его пероральной абсорбции является действительно отличным. С другой стороны, его метаболизм под действием микросомы печени обезьяны является быстрым, аналогично случаю с микросомой печени человека. В этом случае свойство пероральной абсорбции соединения (В) для обезьян является низким. С другой стороны, метаболизм соединения по изобретению под действием микросомы печени обезьяны является медленным аналогично случаю с микросомами печени мыши и собаки. Таким образом, когда биодоступность (БП) измеряли с использованием обезьян с целью получить подтверждение улучшения абсорбции при пероральном введении посредством ингибирования метаболизма, было подтверждено, что пероральная абсорбция у обезьян была значительно улучшена для соединения по изобретению по сравнению с данными для соединения (В).
Что касается способа получения фармацевтического состава медикаментов и противоопухолевых препаратов, они могут быть получены выбором приемлемого фармацевтического препарата на основе требуемого способа ведения и с использованием обычно применяемых способов получения. Примеры дозированных форм противоопухолевого агента по данному изобретению для перорального введения могут включать таблетки, порошки, гранулы и капсулы. Примеры других дозированных форм включают растворы, сиропы, эликсиры и водные или масляные суспензии. Желательными из перечисленных выше являются капсулы, таблетки и растворы. В случае инъекций в процессе получения могут быть использованы добавки, такие как стабилизаторы, антисептические агенты и агенты, способствующие солюбилизации. Если раствор, содержащий указанные вспомогательные вещества, превращают в твердую препаративную форму сушкой вымораживанием или другим подобным способом, его можно использовать как фармацевтический препарат, который растворяют перед употреблением.
Растворы, суспензии и эмульсии могут быть представлены как жидкие препараты, и в процессе их получения могут использоваться дополнительные агенты, такие как суспендирующие агенты и эмульгаторы.
Соединение по данному изобретению может применяться для лечения рака у млекопитающих, особенно человека, и в случае введения человеку желательно вводить его один раз в день и повторять через соответствующие интервалы.
Что касается дозы, желательно вводить препарат в диапазоне от примерно 0,5 мг до 50 мг, предпочтительно от примерно 1 мг до 20 мг, но 1 м2 площади поверхности тела.
Изобретение подробно описано в следующих примерах. При описании примеров будут использованы следующие сокращения. Вое обозначает трет.-бутоксикарбонильную группу. Ас обозначает ацетильную группу, Bz обозначает бензоильную группу, и TIPS означает триизопропилсилильную группу.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На чертеже представлен график, показывающий изменения во времени количества метаболита, образовавшегося из соответствующих соединений.
ПРЕДПОЧТИТЕЛЬНЫЙ СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
ПРИМЕР 1
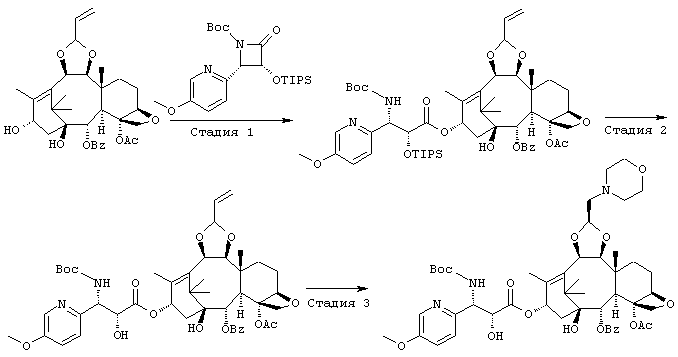
СТАДИЯ 1: (1S, 2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси)такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(5-метокси-2-пиридил)-2-триизопропилсилилоксипропионат
300 мг образца (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-5,20-эпокси-1,13-дигидрокси-9,10-(2-пропенил-идендиокси)такс-11-ена растворили в 10 мл сухого тетрагидрофурана, и раствор смешивали с 0,63 мл гексаметилдисилазида лития (1М раствор в тетрагидрофуране) при -60° и перемешивали в течение 25 минут. К реакционной смеси при той же температуре добавили 5 мл раствора тетрагидрофурана, содержащего 280 мг (3R,4S)-1-(трет.-бутоксикарбонил)-4-(5-метокси-2-пиридил)-3-триизопропилсилилокси-2-азетидинона, и смесь перемешивали при охлаждении льдом в течение 40 минут. К реакционной смеси добавили насыщенный водный раствор хлорида аммония и этилацетат для разделения слоев, водный слой экстрагировали этилацетатом. Органические слои объединяли, промывали насыщенным солевым раствором и затем сушили безводным сульфатом натрия. Растворитель упаривали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле (элюент гексан:этилацетат = 1:1 (об/об)), получили 540 мг требуемого соединения.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
0,89-0,95 (21 Н, м), 1,32 (3Н, с), 1,33-1,62 (3 Н, м), 1,41 (9 Н, с), 1,52 (3 Н, с), 1,65 (3 Н, с), 1,82 (3 Н, с), 1,92-2,32 (3 Н, м), 2,49 (3 Н, с), 2,98 (1 Н, д J=4,9 Гц), 3,85 (3 Н, с), 4,20 (1 Н, д, J=7,4 Гц), 4,22 (1 Н, д, J=6,8 Гц), 4,32 (1 Н, д, J=8,3 Гц), 4,95 (1 Н, с), 5,21 (1 Н, д, J=5,8 Гц), 5,26-5,29 (2 Н, м), 5,39-5,47 (3 Н, м), 5,57 (1 Н, д, J=17,6 Гц), 5,96-6,02 (2 Н, м), 6,11 (1 Н, тр.- образный, J=8,3 Гц), 7,15 (1 Н, дд, J=2,4, 8,8 Гц),7,31 (1 Н, д, J=8,8 Гц), 7,44 (2 Н, т, J=7,8 Гц), 7,56 (1 Н, т, J=7,8 Гц), 8,13 (2 Н, д J=7,8 Гц), 8,26 (1 Н, д J=3,0 Гц).
СТАДИЯ 2: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Aцетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси)такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-2-гидрокси-3-(5-метокси-2-пиридил)пропионат
530 мг соединения, полученного на стадии 1, растворили в 10 мл сухого тетрагидрофурана, при охлаждении льдом добавили 1,0 мл тетрабутиламмоний фторида (1М раствор в тетрагидрофуране), и смесь перемешивали при этой температуре в течение 30 минут. К реакционной смеси добавили воду и этилацетат для разделения слоев, и водный слой экстрагировали этилацетатом. Органические слои объединили, промыли насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором в указанном порядке, затем сушили безводным сульфатом натрия. Растворитель упарили при пониженном давлении, полученный остаток очищали колоночной хроматографией на силикагеле (элюент гексан:этилацетат = 1:1 (по объему)), получили 410 мг требуемого соединения.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,26 (3 Н, с), 1,43 (9 Н, с), 1,50 (3 Н, с), 1,60-1,91 (3 Н, м), 1,64 (3 Н, с), 1,74 (3 Н, С), 1,91 (1 Н, с), 2,04-2,16 (2 Н, м), 2,32-2,37 (1 Н, М), 2,34 (3 Н, с), 2,93 (1 Н, д, J=5,3 Гц), 3,85 (3 Н, с), 4,18 (1 Н, д, J=7,3 Гц), 4,22 (1 Н, д, J=8,3 Гц), 4,33 (1 Н, д, J=8,3 Гц), 4,79 (1 Н, шир. с.), 4,85 (1 Н, шир. с.), 4,92 (1 Н, шир. с.), 5,23 (1 Н, д, J=5,8 Гц), 5,29-5,30 (2 Н, м), 5,46 (1 Н, д, J=10,3 Гц), 5,58 (1 Н, д, J=17,1 Гц), 5,90 (1 Н, д, J=9.7 Гц), 5,96-6.03 (2 Н, м), 6.09 (1 Н, тр.-образный, J=8,4 Гц), 7,22 (1 Н, дд, J=2,4, 8,8 Гц), 7,34 (1 Н, д, J=8,8 Гц), 7,47 (2 Н, т, J=7,8 Гц), 7,60 (1 Н, т, J=7,8 Гц), 8,13 (2 Н, д, J=7,8 Гц), 8,22 (1 Н, д, J=2,4 Гц).
СТАДИЯ 3: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Aцетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-[(1S)-2-(морфолино)этилидендиокси]такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-2-гидрокси-3-(5-метокси-2-пиридил)пропионат
400 мг соединения, полученного на предыдущей стадии 2, растворили в 5 мл тетрагидрофурана, раствор смешали с 5 мл ацетона, 5 мл воды, 5,9 мг тетраоксида осмия и 270 мг N-метилморфолин-N-оксида, и перемешивали при комнатной температуре в течение 4,5 часов. К реакционной смеси добавили этил-ацетат и 10% водный раствор тиосульфата натрия для разделения слоев, и водный слой экстрагировали этилацетатом. Органические слои объединили, промыли насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором в указанном порядке, затем сушили безводным сульфатом натрия. Растворитель упарили при пониженном давлении, полученный остаток растворили в 5 мл тетрагидрофурана, затем раствор смешали с 5 мл метанола, 5 мл воды и 990 мг метапериодата натрия и перемешивали при комнатной температуре в течение 1,5 часов. К реакционной смеси добавили этилацетат и воду для разделения слоев, и водный слой экстрагировали этилацетатом. Органические слои объединили, промыли насыщенным водным солевым раствором, затем сушили безводным сульфатом натрия. Растворитель упарили при пониженном давлении, полученный остаток растворили в 30 мл этанола, и затем раствор смешали с 0,2 мл морфолина, 0,13 мл уксусной кислоты и 140 мг цианоборгидрида натрия и перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавили насыщенный водный раствор бикарбоната натрия, этилацетат и воду для разделения слоев, и водный слой экстрагировали этилацетатом. Органические слои объединили, промыли насыщенным солевым раствором и затем сушили безводным сульфатом натрия. Растворитель упарили при пониженном давлении, полученный остаток очищали колоночной хроматографией на силикагеле (элюент хлороформ:метанол 50:1 (об./об.), получили 220 мг требуемого соединения.
Температура плавления: 160-161° С
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,27 (3 Н, с), 1,43 (9 Н, с), 1,48 (3 Н, с), 1,60 (3 Н, с),1,72 (3 Н, с), 1,78-2,12 (6 Н, м), 2,31-2,38 (1 Н, м), 2,34 (3 Н, с), 2,58-2,68 (4 Н, м), 2,71 (1 Н, дд, J=5,4, 13,2 Гц), 2,79 (1 Н, дд, J=3,9, 13,2 Гц), 2,93 (1 Н, д, J=5,3 Гц), 3,75 (4 Н, т, J=4,9 Гц), 3,86 (3 Н, с), 4,12 (1 Н, д, J=7,3 Гц), 4,21 (1 Н, д, J=8,3 Гц), 4,33 (1 Н, д, J=8,3 Гц), 4,76 (1 Н, шир. с.), 4,85 (1 Н, шир. с), 4,92 (1 Н, с), 5,04 (1 Н, т, J=4,6 Гц), 5,23 (1 Н, д, J=6,9 Гц), 5,29 (1 Н, д, J=8,8 Гц), 5,90 (1 Н, д, J=9,3 Гц), 5,98 (1 Н, д, J=4,9 Гц), 6,08 (1 Н, тр.-образный, J=8,3 Гц), 7,22 (1 Н, дд, J=2,9, 8,8 Гц), 7,34 (1 Н, д, J=8,8 Гц), 7,47 (2 Н, т, J=7,8 Гц), 7,60 (1 Н, т, J=7,8 Гц), 8,13 (2 Н, д, J=7,8 Гц), 8,22 (1 Н, д, J=2,9 Гц).
Элементный анализ (для C49H65N3O15)
Вычислено: С 62,87; Н 7,00; N 4,49
Получено: С 62,66; Н 7,08; N 4,28
ПРИМЕР 2
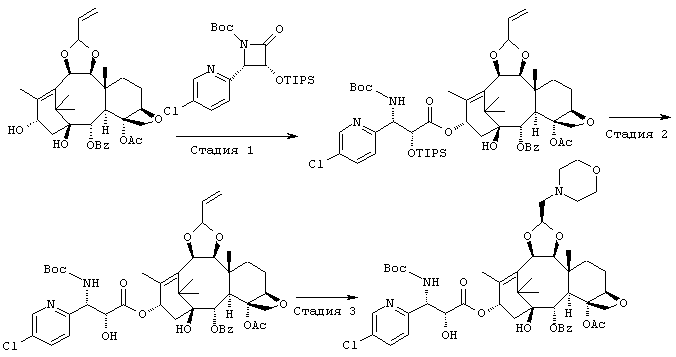
СТАДИЯ 1: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Aцетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси)такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(5-хлор-2-пиридил)- 2 -триизопропилсилилоксипропионат
Указанное соединение было получено по той же методике, что и на стадии 1 Примера 1, за исключением того, что использовали (3R,4S)-1-(трет.-бутоксикарбонил)-4-(5-хлор-2-пиридил)-3-триизопропилсилилокси-2-азетидинон вместо (3R,4S)-1-(трет.-бутоксикарбонил)-4-(5-метокси-2-пиридил)-3-триизопропилсилилокси-2-азетидинона.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
0,87-0,94 (21 Н, м), 1,18-1,69 (2 Н, м), 1,31 (3 Н, с), 1,41 (9 Н, с), 1,52 (3 Н, с), 1,65 (3 Н, с), 1,82 (3 Н, с), 1,72-2,05 (2 Н, м), 2,24-2,34 (2 Н, м), 2,48 (3 Н, с), 2,97 (1 Н, д, J=5,4 Гц), 4,19-4,23 (2 Н, м), 4,33 (1 Н, д, J=7,8 Гц), 4,95 (1 Н, с), 5,21 (1 Н, д, J=5,8 Гц), 5,27-5,31 (2 Н, м), 5,42-5,47 (3 Н, м), 5,58 (1 Н, д, J=17,5 Гц), 5,96-6,04 (2 Н, м), 6,11 (1 Н, т, J=8,8 Гц), 7,38 (1 Н, д, J=8,3 Гц), 7,44 (2 Н, т, J=7,3 Гц), 7,57 (1 Н, т, J=7,3 Гц), 7,65 (1 Н, дд, J=8,3 Гц, 2,5 Гц), 8,13 (2 Н, д, J=7,3 Гц), 8,53 (1 Н, д, J=2,5 Гц).
СТАДИЯ 2: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси)такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(5-хлор-2-пиридил)-2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 2 Примера 1, за исключением того, что в качестве исходного было использовано соединение, полученное на предыдущей стадии 1.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,26 (3 Н, с), 1,22-1,65 (2 Н, м), 1,43 (9 Н, с), 1,49 (3 Н, с), 1,64 (3 Н, с), 1,74 (3 Н, с), 1,75-2,09 (2 Н, м), 2,30-2,39 (2 Н, м), 2,33 (3 Н, с), 2,94 (1 Н, д, J=4,9 Гц), 4,18 (1 Н, д, J=5,3 Гц), 4,22 (1 Н, д, J=8,3 Гц), 4,32 (1 Н, д, J=8,3 Гц), 4,61 (1 Н, шир. с.), 4,92 (2 Н, м), 5,24 (1 Н, д, J=6,3 Гц), 5,30 (1 Н, д, J=6,8 Гц), 5,36 (1 Н, д, J=9,3 Гц), 5,46 (1 Н, д, J=10,5 Гц), 5,58 (1 Н, д, J=17,5 Гц), 5,87 (1 Н, д, J=9,3 Гц), 5,96-6,05 (2 Н, м), 6,11 (1 Н, т, J=7,8 Гц), 7,39 (1 Н, д, J=8,3 Гц), 7,47 (2 Н, т, J=7,3 Гц), 7,60 (1 Н, т, J=7,3 Гц), 7,69 (1 Н, дд, J=8,3 Гц, 2,4 Гц), 8,12 (2 Н, д, J=7,3 Гц), 8,51 (1 Н, д, J=2,4 Гц).
СТАДИЯ 3: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-[(1S)-2-(морфолино)этилиден-диокси]такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(5-хлор-2-пиридил)-2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 3 Примера 1, за исключением того, что в качестве исходного было использовано соединение, полученное на предыдущей стадии 2.
Температура плавления: 146-150° С
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,26 (3 Н, с), 1,20-1,72 (2 Н, м), 1,43 (9 Н, с), 1,48 (3 Н, с), 1,63 (3 Н, с), 1,73 (3 Н, с), 1,75-2,03 (2 Н, м), 2,33 (3 Н, с), 2,30-2,38 (2 Н, м), 2,59-2,69 (4 Н, м), 2,72 (1 Н, дд, J=5,4, 13,2 Гц), 2,79 (1 Н, дд, J=3,9, 13,2 Гц), 2,92 (1 Н, д, J=4,9 Гц), 3,74 (4 Н, т, J=4,9 Гц), 4,12 (1 Н, д, J=7,9 Гц), 4,22 (1 Н, д, J=8,8 Гц), 4,32 (1 Н, д, J=8,8 Гц), 4,59 (1 Н, шир. с.), 4,91 (2 Н, м), 5,05 (1 Н, т, J=4,4 Гц), 5,24 (1 Н, д, J=6,8 Гц), 5,35 (1 Н, д, J=9,3 Гц), 5,87 (1 Н, д, J = 9,8 Гц), 5,99 (1 Н, д, J = 4,9 Гц), 6,10 (1 Н, т, J = 8,0 Гц), 7,39 (1 Н, д, J = 8,3 Гц), 7,47 (2 Н, т, J = 7,3 Гц), 7,60 (1 Н, т, J = 7,3 Гц), 7,69 (1 Н, дд, J = 8,3 Гц, 2,4 Гц), 8,12 (2 Н, д, J = 7,3 Гц), 8,50 (1 Н, д, J = 2,5 Гц).
Элементный анализ (для С48H62СlN3О14·Н2О)
Вычислено: С 60,15; Н 6,73; N 4,38; Cl 3,70
Получено: С 60,15; Н 6,74; N 4,20; Cl 3,63
ПРИМЕР 3
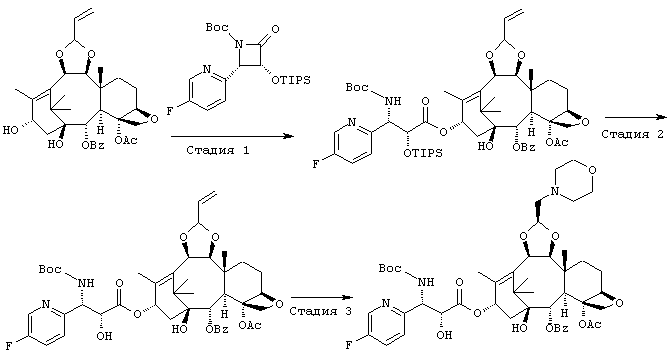
СТАДИЯ 1: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси)такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(5-фтор-2-пиридил)-2-триизопропилсилилоксипропионат
Указанное соединение было получено по той же методике, что и на стадии 1 Примера 1, за исключением того, что использовали (3R,4S)-1-(трет.-бутоксикарбонил)-4-(5-фтор-2-пиридил)-3-триизопропилсилилокси-2-азетидинон вместо (3R,4S)-1-(трет.-бутоксикарбонил)-4-(5-метокси-2-пиридил)-3-триизопропилсилилокси-2-азетидинона.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
0,87 - 0,94 (21 Н, м), 1,20 - 1,70 (2 Н, м), 1,31 (3 Н, с), 1,41 (9 Н, с), 1,52 (3 Н, с), 1,65 (3 Н, с), 1,82 (ЗН, с), 1,75 - 2,07 (2 Н, м), 2,26 - 2,32 (2 Н, м), 2,49 (3 Н, с), 2,97 (1 Н, д, J = 5,4 Гц), 4,19 - 4,23 (2 Н, м), 4,33 (1 Н, д, J = 8 Гц), 4,96 (1 Н, с), 5,21 (1 Н, д, J = 5,9 Гц), 5,27 -5,32 (2 Н, м), 5,43 - 5,49 (3 Н, м), 5,58 (1 Н, д, J = 17,5 Гц), 5,96 - 6,04 (2 Н, м), 6,12 (1 Н, т, J = 8 Гц), 7,36 -7,47 (4 Н, м), 7,57 (1 Н, т, J = 7,3 Гц), 8,13 (2 Н, д, J =7,3 Гц), 8,43 (1 Н, д, J = 2,4 Гц).
СТАДИЯ 2: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Aцетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси)такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(5-фтор-2-пиридил)-2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 2 Примера 1, за исключением того, что в качестве исходного было использовано соединение, полученное на предыдущей стадии 1.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,27 (3 Н, с), 1,20 - 1,68 (2 Н, м), 1,44 (9 Н, с), 1,49 (3 Н, с), 1,64 (3 Н, с), 1,74 (3 Н, с), 1,75 - 2,05 (2 Н, м), 2,30 - 2,39 (2 Н, м), 2,34 (3 Н, с), 2,93 (1 Н, д, J = 4,9 Гц), 4,18 (1 Н, д, J = 6,8 Гц), 4,23 (1 Н, д, J = 8,3 Гц), 4,33 (1 Н, д, J = 8,3 Гц), 4,62 (1 Н, д, J = 2,5 Гц), 4,90 - 4,92 (2 Н, м), 5,24 (1 Н, д, J = 5,8 Гц), 5,30 (1 Н, д, J = 6,8 Гц), 5,37 (1 Н, д, J = 9,3 Гц), 5,46 (1 Н, д, J = 10,2 Гц), 5,58 (1 Н, д, J= 17 Гц), 5,90 (1 Н, д, J = 10,2 Гц), 5,96 - 6,05 (2 Н, м), 6,10 (1 Н, т, J = 7,8 Гц), 7,40 - 7,49 (4 Н, м), 7,60 (1 Н, т, J = 7,3 Гц), 8,12 (2 Н, д, J = 7,3 Гц), 8,41 (1 Н, с).
СТАДИЯ 3: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-[(1S)-2-(морфолино)этилиден-диокси]такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(5-фтор-2-пиридил)-2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 3 Примера 1, за исключением того, что в качестве исходного было использовано соединение, полученное на предыдущей стадии 2.
Температура плавления: 148-152° С
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,26 (3 Н, с), 1,20 - 1,69 (2 Н, м), 1,43 (9 Н, с), 1,48 (3 Н, с), 1,62 (3 Н, с), 1,72 (3 Н, с), 1,75 - 2,02 (2 Н, м), 2,33 (3 Н, с), 2,30 - 2,39 (2 Н, м), 2,59 - 2,69 (4 Н, м), 2,71 (1 Н, дд, J = 5,4, 13,2 Гц), 2,79 (1 Н, дд, J = 3,9, 13,2 Гц), 2,92 (1 Н, д, J = 4,9 Гц), 3,74 (4 Н, т, J = 4,9 Гц), 4,12 (1 Н, д, J = 7,3 Гц), 4,22 (1 Н, д, J = 8,3 Гц), 4,32 (1 Н, д, J = 8,3 Гц), 4,60 (1 Н, шир. с.), 4,90 - 4,92 (2 Н, м), 5,04 (1 Н, т, J = 4,9 Гц), 5,24 (1 Н, д, J = 6,8 Гц), 5,36 (1 Н, д, J=9,3 Гц), 5,89 (1 Н, д, J = 9,8 Гц), 5,99 (1 Н, д, J = 4,9 Гц), 6,09 (1 Н, т, J = 8,0 Гц), 7,42 - 7,49 (3 Н, м), 7,60 (1 Н, т, J = 7,3 Гц), 7,60 (1 Н, т, J = 7,3 Гц), 8,12 (2 Н, д, J = 7,3 Гц), 8,40 (1 Н, с).
Элементный анализ (для C48H62FN3O14·H2O)
Вычислено: С 61,19; Н 6,85; N 4,46; F 2,02
Получено: С 61,16; Н 6,85; N 4,36; F 2,05
ПРИМЕР 4
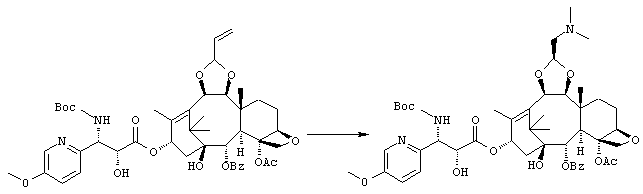
(1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокси-такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-2-гидрокси-3-(5-метокси-2-пиридил)пропионат
Указанное соединение было получено по той же методике, что и на стадии 3 Примера 1, за исключением того, что в качестве исходного было использовано соединение, полученное на стадии 2 Примера 1, и вместо морфолина использовали диметиламин (2М раствор в метаноле).
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,26 (3 Н, с), 1,43 (9 Н, с), 1,48 (3 Н, с), 1,61 (3 Н, с), 1,73 (3 Н, с), 1,83 - 1,97 (3 Н, м), 2,04 - 2,12 (2 Н, м), 2,31 - 2,38 (2 Н, м), 2,34 (3 Н, с), 2,38 (6 Н, с), 2,64 -2,76 (2 Н, м), 2,93 (1 Н, д, J = 4,9 Гц), 3,85 (3 Н, с), 4,13 (1 Н, д, J = 7,4 Гц), 4,21 (1 Н, д, J = 8,3 Гц), 4,33 (1 Н, д, J = 8,3 Гц), 4,84 (1 Н, д, J = 2,4 Гц), 4,92 (1 Н, с), 5,01 (1 Н, т, J = 4,9 Гц), 5,24 (1 Н, д, J = 6,8 Гц), 5,29 (1 Н, д, J=8,8 Гц), 5,91 (1 Н, д, J = 9,3 Гц), 5,99 (1 Н, д, J = 5,4 Гц), 6,08 (1 Н, т, J = 7,8 Гц), 7,23 (1 Н, дд, J = 3,0, 8,3 Гц), 7,34 (1 Н, д, J = 8,8 Гц), 7,47 (2 Н, т, J = 7,8 Гц), 7,60 (1 Н, т, J = 7,8 Гц), 8,12 (2 Н, д, J = 7,8 Гц), 8,22 (1 Н, д, J=3,0 Гц).
ПРИМЕР 5
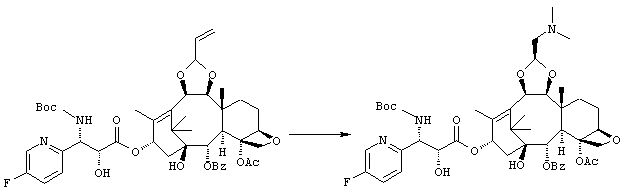
(1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокси-такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(5-фтор-2-пиридил)-2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 3 Примера 1, за исключением того, что в качестве исходного было использовано соединение, полученное на стадии 2 Примера 3, и вместо морфолина использовали диметиламин (2М раствор в метаноле).
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,26 (3 Н, с), 1,20 - 1,70 (2 Н, м), 1,43 (9 Н, с), 1,48 (3 Н, с), 1,62 (3 Н, с), 1,73 (3 Н, с), 1,75 - 2,01 (3 Н, м), 2,33 (3 Н, с), 2,38 (6 Н, с), 2,32 - 2,39 (2 Н, м), 2,66 (1 Н, дд, J = 5,4, 13,2 Гц), 2,74 (1 Н, дд, J = 4,0, 13,2 Гц), 2,93 (1 Н, д, J = 4,9 Гц), 4,12 (1 Н, д, J = 7,3 Гц), 4,22 (1 Н, д, J=8,3 Гц), 4,32 (1 Н, д, J = 8,3 Гц), 4,90 - 4,92 (2 Н, м), 5,02 (1 Н, т, J = 5,4 Гц), 5,25 (1 Н, д, J = 6,8 Гц), 5,36 (1 Н, д, J = 6,8 Гц), 5,90 (1 Н, д, J = 8,8 Гц), 5,99 (1 Н, д, J = 4,9 Гц), 6,09 (1 Н, т, J = 8,1 Гц), 7,42 - 7,49 (4 Н, м), 7,60 (1 Н, т, J = 7,3 Гц), 8,12 (2 Н, д, J = 7,3 Гц), 8,41 (1 Н, с).
ПРИМЕР 6
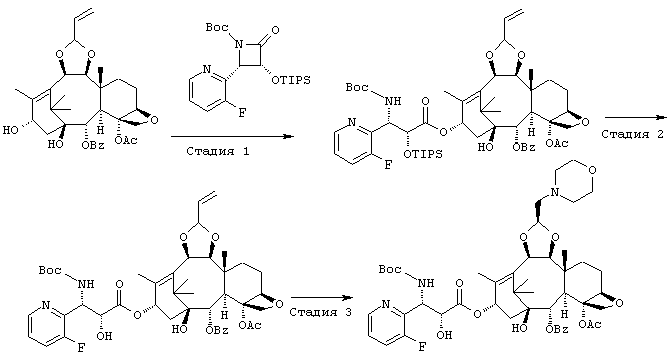
СТАДИЯ 1: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Aцетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси]такс-11-ен-13-ил(2R,3S)-3-(трет.-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-триизопропилсилилоксипропионат
Указанное соединение было получено по той же методике, что и на стадии 1 Примера 1, за исключением того, что в качестве исходного был использован (3R,4S)-1-(трет.-бутокси-карбонил)-4-(3-фтор-2-пиридил)-3-триизопропилсилилокси-2-азетидинон вместо (3R,43)-1-(трет.-бутоксикарбонил)-4-(5-метокси-2-пиридил)-3-триизопропилсилилокси-2-азетидинона.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
0,89 - 0,93 (21 Н, м), 1,28 (3 Н, с), 1,39 (9 Н, с), 1,54 (3 Н, с), 1,66 (3 Н, с), 1,82 (3 Н, с), 1,61 - 1,64 (3 Н, м),1,89 - 1,96 (2 Н, м), 2,33 - 2,39 (2 Н, м), 2,49 (3 Н, с), 2,98 (1 Н, д, J = 4,8 Гц), 4,21 - 4,23 (2 Н, м), 4,36 (1 Н, д, J = 7,8 Гц), 4,96 (2 Н, шир. с.), 5,20 (1 Н, д, J = 5,9 Гц), 5,27 (1 Н, д, J = 6,8 Гц), 5,46 (1 Н, д, J = 9,8 Гц), 5,58 (1 Н, д, J = 17,1 Гц), 5,61 (1 Н, д, J = 6,8 Гц), 5,96 - 6,03 (2 Н, м), 6,08 - 6,12 (2 Н, м), 7,25 - 7,29 (1 Н, м), 7,40 (1 Н, т, J = 8,3 Гц), 7,47 (1 Н, т, J = 7,8 Гц), 7,59 (1 Н, т, J=7,8 Гц), 8,16 (2 Н, д, J = 7,8 Гц), 8,39 (1 Н, д, J = 3,4 Гц).
СТАДИЯ 2: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Aцетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси)такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(3-фтор-2-пиридил) -2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 2 Примера 1, за исключением того, что в качестве исходного было использовано соединение, полученное на предыдущей стадии 1.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,30 (3 Н, с), 1,41 (9 Н, с), 1,51 (3 Н, с), 1,65 (3 Н, с), 1,81 (3 Н, с), 1,57 - 1,63 (3 Н, м), 1,89 - 1,95 (2 Н, м), 2,03 - 2,10 (1 Н, м), 2,35 (3 Н, с), 2,43 - 2,49 (1 Н, м), 2,95 (1 Н, д, J = 4,9 Гц), 4,20 (1 Н, д, J = 7,4 Гц), 4,23 (1 Н, д, J = 8,8 Гц), 4,33 (1 Н, д, J = 8,3 Гц), 4,68 (1 Н, д, J=2,5 Гц), 4,92 (1 Н, с), 5,24 (1 Н, д, J = 6,4 Гц), 5,31 (1 Н, д, J = 6,8 Гц), 5,46 (1 Н, д, J = 9,8 Гц), 5,58 (1 Н, д, J = 17,1 Гц), 5,65 (1 Н, д, J = 18,3 Гц), 5,97 - 6,05 (2 Н, м), 6,10 (1 Н, т, J = 8,8 Гц), 6,21 (1 Н, д, J = 8,3 Гц), 7,29 -7,32 (1 Н, м), 7,43 - 7,49 (3 Н, м), 7,60 (1 Н, т, J = 7,3 Гц), 8,14 (2 Н, д, J = 7,3 Гц), 8,41 (1 Н, д, J = 4,9 Гц).
СТАДИЯ 3: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Aцетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-[(1S)-2-(морфолино)этилиден-диокси]такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 3 Примера 1, за исключением того, что в качестве исходного было использовано соединение, полученное на предыдущей стадии 2.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,29 (3 Н, с), 1,40 (9 Н, с), 1,49 (3 Н, с), 1,61 (3 Н, с), 1,79 (3 Н, с), 1,70 - 2,03 (5 Н, м), 2,30 - 2,44 (2 Н, м), 2,35 (3 Н, с), 2,61 - 2,65 (4 Н, м), 2,70 - 2,82 (2 Н, м), 2,94 (1 Н, д, J = 4,8 Гц), 3,75 (4 Н, т, J = 4,9 Гц), 4,14 (1 Н, д, J = 7,3 Гц), 4,23 (1 Н, д, J = 8,3 Гц), 4,33 (1 Н, д, J= 7,8 Гц), 4,67 (1 Н, с), 4,92 (1 Н, с), 5,05 (1 Н, т, J =4,9 Гц), 5,25 (1 Н, д, J = 7,3 Гц), 5,65 (1 Н, д, J = 7,8 Гц), 5,99 (1 Н, д, J = 5,4 Гц), 6,09 (1 Н, т, J = 7,8 Гц), 6,20 (1 Н, д, J = 8,3 Гц), 7,29 - 7,33 (1 Н, м), 7,43 - 7,49 (3 Н, м), 7,60 (1 Н, т, J = 7,3 Гц), 8,13 (2 Н, д, J = 7,3 Гц), 8,40 (1 Н, д, J = 4,9 Гц).
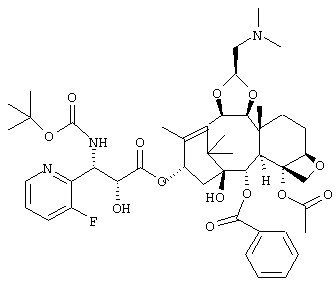
ПРИМЕР 7
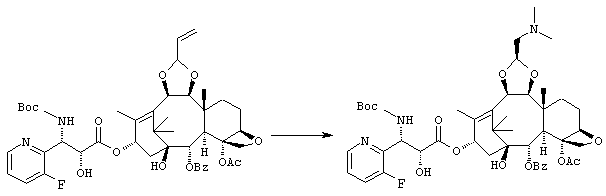
(1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокси-такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 3 Примера 1, за исключением того, что в качестве исходного было использовано соединение, полученное на стадии 2 Примера 6, и вместо морфолина использовали диметиламин (2М раствор в метаноле).
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,29 (3 Н, с), 1,41 (9 Н, с), 1,49 (3 Н, с), 1,63 (3 Н, с), 1,79 (3 Н, с), 1,86 - 2,08 (5 Н, м), 2,32 - 2,38 (2 Н, м), 2,34 (3 Н, с), 2,38 (6 Н, с), 2,66 (1 Н, дд, J = 5,4, 13,6 Гц), 2,75 (1 Н, дд, J = 3,9, 13,6 Гц), 2,94 (1 Н, д, J = 4,9 Гц), 4,14 (1 Н, д, J = 6,9 Гц), 4,23 (1 Н, д, J = 8,3 Гц), 4,33 (1 Н, д, J = 8,3 Гц), 4,68 (1 Н, д, J = 2,9 Гц), 4,92 (1 Н, с), 5,02 (1 Н, т, J = 4,9 Гц), 5,25 (1 Н, д, J = 6,8 Гц), 5,65 (1 Н, д, J = 8,3 Гц), 6,00 (1 Н, д, J = 4,9 Гц), 6,09 (1 Н, т, J = 7,8 Гц), 6,21 (1 Н, д, J = 8,3 Гц), 7,28 - 7,33 (1 Н, м), 7,43 -7,49 (3 Н, м), 7,60 (1 Н, т, J = 7,3 Гц), 8,14 (2 Н, д, J = 7,3 Гц), 8,40 (1 Н, д, J = 4,4 Гц).
ПРИМЕР 8
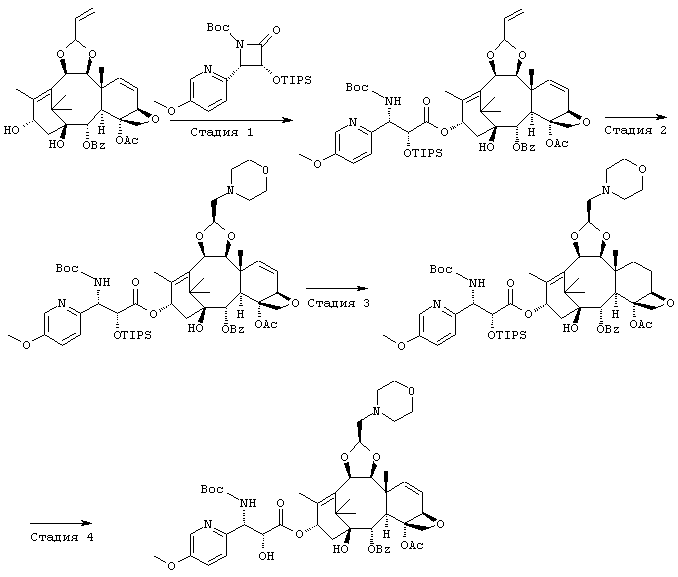
СТАДИЯ 1: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Aцетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси]такс-6,11-диен-13-ил(2R, 3S)-3-(трет.-бутоксикарбониламино) -3- (5-метокси-2-пиридил)-2-триизопропилсилилоксипропионат
300 мг (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-5,20-эпокси-1,13-дигидрокси-9,10-(2-пропенилидендиокси)такс-6,11-диена растворили в 10 мл сухого тетрагидрофурана, и раствор смешали с 0,63 мл гексаметилдисилазида лития (1М раствор в тетрагидрофуране) при -60° С и перемешивали в течение 20 минут. К реакционной смеси при той же температуре добавили 5 мл раствора тетрагидрофурана, содержащего 280 мг (3R,4S)-1-(трет.-бутоксикарбонил)-4-(5-метокси-2-пиридил)-3-триизопропилсилилокси-2-азетидинона, и смесь перемешивали при охлаждении льдом в течение 30 минут. К реакционной смеси добавили насыщенный водный раствор хлорида аммония и этилацетат для разделения слоев, и водный слой экстрагировали этилацетатом. Органические слои объединяли, промывали насыщенным солевым раствором и затем сушили безводным сульфатом натрия. Растворитель упаривали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле (элюент гексан:этил-ацетат = 5:1 (об./об.)), получили 530 мг требуемого соединения.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
0,87 - 0,93 (21 Н, м), 1,29 (3 Н, с), 1,41 (9 Н, с), 1,54 (3 Н, с), 1,69 (3 Н, с), 1,75 (3 Н, с), 1,82 (1 Н, с), 2,29 (1 Н, дд, J = 9,8, 15,1 Гц), 2,40 (1 Н, дд, J = 8,8, 15,1 Гц), 2,53 (3 Н, с), 3,13 (1 Н, д, J = 5,8 Гц), 3,85 (3 Н, с), 4,04 (1 Н, д, J = 7,3 Гц), 4,30 (2 Н, шир. с.), 4,90 (1 Н, д, J = 3,9 Гц), 5,20 - 5,23 (2 Н, м), 5,28 (1Н, д, J = 9,8 Гц), 5,38 (1 Н, с), 5,47 - 5,49 (2 Н, м), 5,60 (1 Н, д, J = 17,0 Гц), 5,71 (1 Н, дд, J = 4,4, 10,2 ГЦ), 5,96 - 6,06 (2 Н, м), 6,09 - 6,14 (2 Н, м), 7,16 (1 Н, дд, J = 2,9, 8,3 Гц), 7,31 (1 Н, д, J =8,3 Гц), 7,47 (2 Н, т, J = 7,8 Гц), 7,58 (1 Н, т, J = 7,8 Гц), 8,3 Гц), 7,47 (2 Н, т, J = 7,8 Гц), 7,58 (1 Н, т, J = 7,8 Гц), 8,14 (2 Н, д, J = 7,8 Гц), 8,26 (1 Н, д, J = 2,9 Гц).
СТАДИЯ 2: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-[(1S)-2-(морфолино)этилиден-диокси]такс-6,11-диен-13-ил- (2R,3S)-3-(трет.-бутоксикарбонил-амино)-3-(5-метокси-2-пиридил)-2-триизопропилсилилокси-пропионат
520 мг соединения, полученного выше на стадии 1, растворили в 5 мл тетрагидрофурана, раствор смешали с 5 мл ацетона, 5 мл воды, 13 мг тетраоксида осмия и 300 мг N-метилморфолин-N-оксида и перемешивали при комнатной температуре в течение 7,5 часов. К реакционной смеси добавили этилацетат и 10% водный раствор тиосульфата натрия для разделения слоев, и водный слой экстрагировали этилацетатом. Органические слои объединили, промыли насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором в указанном порядке, затем сушили безводным сульфатом натрия. Растворитель упарили при пониженном давлении, полученный остаток растворили в 5 мл тетрагидрофурана, затем раствор смешали с 5 мл метанола, 5 мл воды и 1,1 г метапериодата натрия, и перемешивали при комнатной температуре в течение 1,5 часов. К реакционной смеси добавили этил-ацетат и воду для разделения слоев, и водный слой экстрагировали этилацетатом. Органические слои объединили, промыли насыщенным водным солевым раствором, затем сушили безводным сульфатом натрия. Растворитель упарили при пониженном давлении, полученный остаток растворили в 30 мл этанола, и затем раствор при охлаждении льдом смешали с 0,22 мл морфолина, 0,15 мл уксусной кислоты и 160 мг цианоборгидрида натрия, и перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавили насыщенный водный раствор бикарбоната натрия, этилацетат и воду для разделения слоев, и водный слой экстрагировали этилацетатом. Органические слои объединили, промыли насыщенным солевым раствором и затем сушили безводным сульфатом натрия. Растворитель упарили при пониженном давлении, полученный остаток очищали колоночной хроматографией на силикагеле (элюент гексан:этилацетат 3:2 (об./об.)), получили 290 мг требуемого соединения
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
0,87 - 0,93 (21 Н, м), 1,28 (3 Н, с), 1,41 (9 Н, с), 1,53 (3 Н, с), 1,55 (3 Н, с), 1,73 (3 Н, с), 1,80 (1 Н, с), 2,26 (1 Н, дд, J = 8,8, 15,1 Гц), 2,39 (1 Н, дд, J = 9,8, 15,1 Гц), 2,53 (3 Н, с), 2,60 - 2,68 (4 Н, м), 2,74 (1 Н, дд, J = 4,9, 13,7 Гц), 2,81 (1 Н, дд, J = 4,9, 13,7 Гц), 3,12 (1 Н, д, J = 5,4 Гц), 3,76 (1 Н, т, J = 4,8 Гц), 3,85 (3 Н, с), 3,99 (1 Н, д, J = 7,9 Гц), 4,30 (2 Н, с), 4,89 (1 Н, д, J = 3,9 Гц), 5,02 (1 Н, т, J = 3,9 Гц), 5,14 (1 Н, д, J = 7,3 Гц), 5,27 (1 Н, д, J=9,8 Гц), 5,37 (1 Н, д, J = 1,5 Гц), 5,47 (1 Н, д, J = 9,8 Гц), 5,69 (1 Н, дд, J = 3,9, 10,5 Гц), 5,94 (1 Н, д, J = 5,3 Гц), 6,07 - 6,13 (2 Н, м), 7,16 (1 Н, дд, J = 2,9, 6,3 Гц), 7,30 (1 Н, д, J = 6,3 Гц), 7,47 (2 Н, т, J = 7,8 Гц), 7,58 (1 Н, т, J=7,8 Гц), 8,15 (2 Н, д, J = 7,8 Гц), 8,26 (1 Н, д, J = 2,9 Гц).
СТАДИЯ 3: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Aцетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-[(1S)-2-(морфолино)этилиден-диокси]такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(5-метокси-2-пиридил)-2-триизопропилсилилоксипропионат
235 мг соединения, полученного выше на стадии 2, растворили в 10 мл этанола, и раствор смешали с 235 мг катализатора 5% палладия на угле (влажный) и встряхивали 10 часов под давлением водорода (392 кПа). После удаления катализатора фильтрованием фильтрат концентрировали с получением 230 мг требуемого соединения.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
0,88 - 0,94 (21 Н, м), 1,30 (3 Н, с), 1,42 (9 Н, с), 1,50 (3 Н, с), 1,60 (3 Н, с), 1,79 (3 Н, с), 1,84 - 2,30 (7 Н, м), 2,50 (3 Н, с), 2,60 - 2,84 (4 Н, м), 2,85 - 2,92 (2 Н, м), 2,95 (1 Н, д, J = 4,4 Гц), 3,80 (4 Н, т, J = 4,4 Гц), 3,85 (3 Н, с), 4,17 (1 Н, д, J = 7,3 Гц), 4,19 (1 Н, д, J = 8,7 Гц), 4,33 (1 Н, д, J = 8,3 Гц), 4,96 (1 Н, с), 5,10 (1 Н, шир. с.), 5,22 - 5,28 (2 Н, м), 5,40 (1 Н, с), 5,48 (1 Н, д, J = 10,3 Гц), 5,96 (1 Н, д, J = 4,9 Гц), 6,10 (1 Н, т, J = 8,3 Гц), 7,12 - 7,17 (1 Н, м), 7,31 (1 Н, д, J = 8,3 Гц), 7,45 (2 Н, т, J = 7,8 Гц), 7,57 (1 Н, т, J = 7,8 Гц), 8,13 (2 Н, д, J = 7,8 Гц), 8,26 (1 Н, д, J = 2,9 Гц).
СТАДИЯ 4: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-[(1S)-2-(морфолино)этилиден-диокси]такс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-2-гидрокси-3-(5-метокси-2-пиридил)пропионат
230 мг соединения, полученного выше на стадии 3, растворили в 5 мл сухого тетрагидрофурана, при охлаждении льдом добавили 0,43 мл тетрабутиламмоний фторида (1М раствор в тетрагидрофуране), и смесь перемешивали при этой температуре в течение 30 минут. К реакционной смеси добавили насыщенный солевой раствор и этилацетат для разделения слоев, и водный слой экстрагировали этилацетатом. Органические слои объединили, промыли насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором в указанном порядке, затем сушили безводным сульфатом натрия. Растворитель упарили при пониженном давлении, полученный остаток очищали колоночной хроматографией на силикагеле (элюент хлороформ:метанол = 50:1 (об./об.)), затем перекристаллизовали из водного этанола, получили 110 мг требуемого соединения. Его аналитические данные совпали с таковыми для соединения, полученного на стадии 3 Примера 1.
ПРИМЕР 9
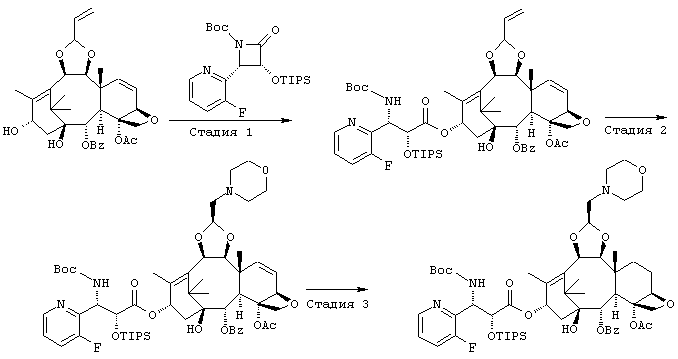
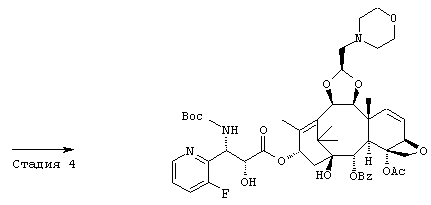
СТАДИЯ 1: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси)такс-6,11-диен-13-ил (2R, 3S) -3- (трет.-бутоксикарбониламино) -3- (3-фтор-2-пиридил)-2-триизопропилсилилоксипропионат
Указанное соединение было получено по той же методике, что и на стадии 1 Примера 8, за исключением того, что в качестве исходного был использован (3R,4S)-1-(трет.-бутоксикарбонил)-4-(5-фтор-2-пиридил)-3-триизопропилсилилокси-2-азетидинон вместо (3R, 4S)-1-(трет.-бутоксикарбонил)-4-(5-метокси-2-пиридил)-3-триизопропилсилилокси-2-азетидинона.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
0,88 - 0,92 (21 Н, м), 1,33 (3 Н, с), 1,38 (9 Н, с), 1,56 (3 Н, с), 1,76 (3 Н, с), 2,41 - 2,45 (2 Н, м), 2,51 (3 Н, с), 3,14 (1 Н, д, J = 5,8 Гц), 4,06 (1 Н, д, J = 7,8 Гц), 4,33 (2 Н, с), 4,90 (1 Н, д, J = 4,4 Гц), 4,94 (1 Н, д, J = 2,4 Гц), 5,19 - 5,22 (2 Н, м), 5,48 (1 Н, д, J = 10,3 Гц), 5,58 - 5,64 (2 Н, м), 5,70 (1 Н, дд, J = 10,3, 4,4 Гц), 5,96 - 6,14 (5 Н, м), 7,26 - 7,30 (1 Н, м), 7,41 (1 Н, т, J = 8,5 Гц), 7,49 (2 Н, т, J = 7,5 Гц), 7,59 (1 Н, т, J = 7,5 Гц), 8,17 (2 Н, д, J=7,5 Гц), 8,40 (1 Н, д, J = 4,4 Гц).
СТАДИЯ 2: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Aцетокси-2-бензоил-окси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-6,11-диен-13-ил (2R,3S)-3-(трет.-бутокси-карбониламино)-3-(3-фтор-2-пиридил)-2-триизопропилсилилокси-пропионат
Указанное соединение было получено по той же методике, что и на стадии 2 Примера 8, за исключением того, что в качестве исходного было использовано соединение, полученное на предыдущей стадии, и вместо морфолина использовали диметиламин (2М раствор в метаноле).
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
0,87 - 0,92 (21 Н, м), 1,32 (3 Н, с), 1,38 (9 Н, с), 1,55 (3 Н, с), 1,57 (3 Н, с), 1,75 (3 Н, с), 2,39 (6 Н, с), 2,42 -2,45 (2 Н, м), 2,51 (3 Н, с), 2,66 (1 Н, дд, J = 5,1, 13,2 Гц), 2,74 (1 Н, дд, J = 4,2, 13,2 Гц), 3,14 (1 Н, д, J = 5,8 Гц), 4,01 (1 Н, д, J = 7,9 Гц), 4,32 (2 Н, с), 4,90 - 4,94 (2 Н, м), 5,00 (1 Н, т, J = 4,9 Гц), 5,15 (1 Н, д, J = 7,9 Гц), 5,63 (1 Н, д, J = 9,8 Гц), 5,69 (1 Н, дд, J = 9,8, 4,4 Гц), 5,95 (1 Н, д, J = 5,8 Гц), 6,07 - 6,13 (3 Н, м), 7,26 - 7,28 (1 Н, м), 7,41 (1 Н, т, J = 9,2 Гц), 7,49 (2 Н, т, J = 7,5 Гц), 7,59 (1 Н, т, J = 7,5 Гц), 8,17 (2 Н, д, J = 7,5 Гц), 8,40 (1 Н, д, J = 4,4 Гц).
СТАДИЯ 3: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбонил-амино)-3-(3-фтор-2-пиридил)-2-триизопропилсилилоксипропионат
Указанное соединение было получено по той же методике, что и на стадии 3 Примера 8, за исключением того, что в качестве исходного было использовано соединение, полученное на предыдущей стадии 2.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
0,83 - 0,93 (21 Н, м), 1,35 (3 Н, с), 1,38 (9 Н, с), 1,52 (3 Н, с), 1,56 - 2,07 (5 Н, м), 1,62 (3 Н, с), 1,81 (3 Н, с), 2,34 - 2,43 (2 Н, м), 2,38 (6 Н, с), 2,49 (3 Н, с), 2,66 (1 Н, дд, J = 5,4, 13,2 Гц), 2,74 (1 Н, дд, J = 3,4, 13,2 Гц), 2,98 (1 Н, д, J = 5,4 Гц), 4,17 (1 Н, д, J = 7,3 Гц), 4,22 (1 Н, д, J = 7,8 Гц), 4,36 (1 Н, д, J = 8,3 Гц), 4,96 (2 Н, с), 5,00 (1 Н, т, J = 4,8 Гц), 5,22 (1 Н, д, J = 7,3 Гц), 5,60 (1 Н, д, J=8,8 Гц), 5,98 (1 Н, д, J = 4,9 Гц), 6,08 - 6,10 (2 Н, м), 7,26-7,28 (1 Н, м), 7,40 (1 Н, т, J = 9,2 Гц), 7,48 (2 Н, т, J= 7,5 Гц), 7,59 (1 Н, т, J = 7,5 Гц), 8,16 (2 Н, д, J = 7,5 Гц), 8,40 (1 Н, д, J = 3,9 Гц).
СТАДИЯ 4: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбонил-амино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 4 Примера 8, за исключением того, что в качестве исходного было использовано соединение, полученное на предыдущей стадии 3. Его аналитические данные совпали с таковыми для соединения, полученного на стадии 3 Примера 7.
ПРИМЕР 10
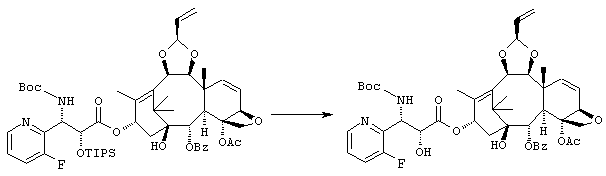
(1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоилокси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси]такс-6,11-диен-13-ил (2R,3S)-3-(трет.-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 2 Примера 1, за исключением того, что в качестве исходного было использовано соединение, полученное на стадии 1 Примера 9.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,29 (3 Н, с), 1,39 (9 Н, с), 1,54 (3 Н, с), 1,60 (3 Н, с), 1,74 (3 Н, с), 1,91 (1 Н, с), 2,35 - 2,48 (2 Н, м), 2,41 (3 Н, с), 3,11 (1 Н, д, J = 5,4 Гц), 3,92 (1 Н, шир. с.), 4,03 (1 Н, д, J = 7,6 Гц), 4,27 (1 Н, д, J = 8,1 Гц), 4,33 (1 Н, д, J=8,2 Гц), 4,67 (1 Н, шир. с.), 4,87 (1 Н, д, J = 4,1 Гц), 5,22 -5,25 (2 Н, м), 5,48 (1 Н, д, J = 10,8 Гц), 5,60 (1 Н, д, J = 17,3 Гц), 5,62 - 5,64 (1 Н, м), 5,69 (1 Н, дд, J = 4,1, 10,3 Гц), 5,98 - 6,13 (4 Н, м), 6,21 (1 Н, д, J = 8,3 Гц), 7,29 -7,33 (1 Н, м), 7,43 - 7,50 (3 Н, м), 7,60 (1 Н, т, J = 7,3 Гц), 8,15 (2 Н, д, J=7,6 ГЦ), 8,39 (1Н, д, J=4,6 Гц).
ПРИМЕР 11
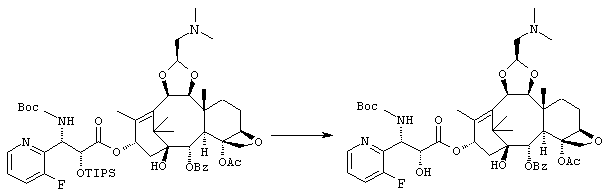
(1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокси-такс-6,11-диен-13-ил (2R, 3S)-3- (трет.-бутоксикарбониламино) -3-(3-фтор-2-пиридил)-2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 2 Примера 1, за исключением того, что в качестве исходного было использовано соединение, полученное на стадии 2 Примера 9.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,28 (3 Н, с), 1,39 (9 Н, с), 1,52 (3 Н, с), 1,57 (3 Н, с), 1,72 (3 Н, с), 1,86 (1 Н, с), 2,27 - 2,46 (2 Н, м), 2,39 (6 Н, с), 2,41 (3 Н, с), 2,69 (1 Н, дд, J = 5,2, 13,2 Гц), 2,79 (1 Н, дд, J = 4,2, 13,2 Гц), 3,11 (1 Н, д, J = 5,9 Гц), 3,98 (1 Н, д, J = 7,6 Гц), 4,28 (1 Н, д, J = 8,1 Гц), 4,33 (1 Н, д, J = 8,3 Гц), 4,66 (1 Н, д, J = 2,5 Гц), 4,87 (1 Н, д, J = 4,1 Гц), 5,02 (1 Н, дд, J = 4,2, 4,8 Гц), 5,17 (1 Н, д, J = 7,8 Гц), 5,62 (1 Н, д, J = 8,5 Гц), 5,68 (1 Н, дд, J = 4,1, 10,3 Гц), 5,96 (1 Н, м), 6,10 (2 Н, м), 6,20 (1 Н, д, J = 6,9 Гц), 7,27 - 7,60 (6 Н, м), 8,15 (2 Н, д, J = 7,3 Гц), 8,40 (1 Н, д, J = 4,6 Гц).
ПРИМЕР 12
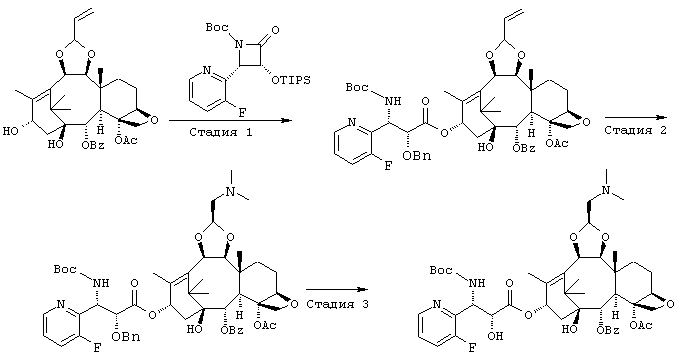
СТАДИЯ 1: (1S, 2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-5,20-эпокси-1-гидрокси-9,10-(2-пропенилидендиокси)такс-6,11-диен-13-ил (2R,3S)-2-бензилокси-3-(трет.-бутоксикарбонил-амино)-3-(3-фтор-2-пиридил)пропионат
Указанное соединение было получено по той же методике, что и на стадии 1 Примера 8, за исключением того, что в качестве исходного был использован (3R,4S)-3-бензилокси-1-(трет.-бутоксикарбонил)-4-(3-фтор-2-пиридил)-2-азетидинон вместо (3R,4S)-1-(трет.-бутоксикарбонил)-4-(5-метокси-2-пиридил)-3-триизопропилсилилокси-2-азетидинона.
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,32 (3 Н, с), 1,39 (9 Н, с), 1,56 (3 Н, с), 1,59 (3 Н, с), 1,77 (3 Н, с), 1,85 (1 Н, с), 2,31 (3 Н, с), 2,39 (2 Н, м), 3,13 (1 Н, д, J = 6,1 Гц), 4,07 (1 Н, д, J = 7,6 Гц), 4,18 (1 Н, д, J = 12,0 Гц), 4,31 (3 Н, м), 4,68 (1 Н, д, J = 12,2 Гц), 4,90 (1 Н, д, J = 4,2 Гц), 5,23 (2 Н, т, J = 7,1 Гц), 5,48 (1 Н, д, J = 11,0 Гц), 5,59 (2 Н, м), 5,70 (1 Н, дд, J = 4,4, 10,5 Гц), 6,02 (1 Н, м), 6,13 (2 Н, д, J = 10,2 Гц), 6,26 (1 Н, д, J = 9,0 Гц), 6,88 (2 Н, д, J = 7,1 Гц), 7,19 (3 Н, м), 7,29 (2 Н, т, J = 6,8 Гц), 7,49 (2 Н, т, J = 7,8 Гц), 7,60 (1 Н, т, J = 7,3 Гц), 8,16 (2 Н, д, J = 7,3 Гц), 8,42 (1 Н, м).
СТАДИЯ 2: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Aцетокси-2-бензоил-окси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-6,11-диен-13-ил (2R,3S)-2-бензилокси-3-(трет.-бутоксикарбониламино)-3-(3-фтор-2-пиридил)пропионат
Указанное соединение было получено по той же методике, что и на стадии 2 Примера 8, за исключением того, что в качестве исходного было использовано соединение, полученное на предыдущей стадии 1, и вместо морфолина использовали диметиламин (2М раствор в метаноле).
1H-ЯМР (400 МГц, CDCl3, TMC) δ :
1,26 (3 Н, с), 1,39 (9 Н, с), 1,54 (3 Н, с), 1,57 (3 Н, с), 1,75 (3 Н, с), 1,82 (1 Н, с), 2,31 (3 Н, с), 2,36 -2,39 (2 Н, м), 2,38 (6 Н, с), 2,71 (1 Н, дд, J = 5,2, 13,2 Гц), 2,77 (1 Н, дд, J = 4,1, 13,2 Гц), 3,12 (1 Н, д, J = 5,6 Гц), 4,02 (1 Н, д, J = 7,8 Гц), 4,19 (1 Н, д, J = 12,2 Гц), 4,31 (2 Н, м), 4,36 (1 Н, д, J = 2,9 Гц), 4,68 (1 Н, д, J = 12,7 Гц), 4,88 (1 Н, д, J = 4,1 Гц), 5,01 (1 Н, т, J = 4,7 Гц), 5,16 (1 Н, д, J = 7,8 Гц), 5,60 (1 Н, д, J = 8,8 Гц), 5,69 (1 Н, дд, J = 4,2, 10,3 Гц), 5,93 (1 Н, д, J = 5,6 Гц), 6,11 (2 Н, м), 6,23 (1 Н, д, J = 9,3 Гц), 6,88 (2 Н, д, J = 6,6 Гц), 7,16 - 7,31 (5 Н, м), 7,48 (1 Н, т, J = 7,8 Гц), 7,59 (1 Н, т, J = 7,3 Гц), 8,15 (2 Н, дд, J = 1,5, 7,1 Гц), 8,41 (1 Н, д, J = 2,9 Гц).
СТАДИЯ 3: (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-Ацетокси-2-бензоил-окси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил (2R,3S)-3-(трет.-бутоксикарбонил-амино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат
Указанное соединение было получено по той же методике, что и на стадии 3 Примера 8, за исключением того, что в качестве исходного было использовано соединение, полученное на предыдущей стадии 2. Его аналитические данные совпали с таковыми для соединения, полученного на стадии 3 Примера 7,
ПРИМЕР ТЕСТИРОВАНИЯ 1
Фибросаркома мыши Meth А была подкожно трансплантирована мышам (название линии: Balb/c), и каждое из соединений, растворенных в смеси растворителей - этанол, Твин 80 и 5% глюкоза (5:5:90 (об./об.)) вводили внутривенной инъекцией через 6, 8 или 10 дней (либо только через 6 дней) после трансплантации. Каждое животное анатомировали на 17-й день для определения веса опухоли, количества тромбоцитов и почечной токсичности. В каждой группе использовали шесть мышей.
Противоопухолевый эффект рассчитывали по следующей формуле: {1-(вес опухоли в группе с введенным соединением/вес опухоли в группе с введенным растворителем)} × 100
Количество тромбоцитов выражали как (тромбоциты в группе с введенным соединением/тромбоциты в группе с введенным растворителем) } × 100
Почечную токсичность обозначали как "измененная" в том случае, когда во время анатомирования наблюдали такое макроскопическое изменение, как увядание, либо такое изменение, как осаждение прозрачных капелек вещества в цитоплазме почечных цилиндрических клеток при гистологическом исследовании.
Соединение (Б)
22,5× 1
69
124
-
ПРИМЕР ТЕСТИРОВАНИЯ 2
В16 Меланому BL6 подкожно трансплантировали мышам (C57BL/6), и через 4 дня вводили каждое из соединений. В случае внутривенного введения соединение формулы А вводили после его растворения в смеси растворителей этанол, Твин 80 и 5% глюкоза (5:15:80 (об./об.)), соединение Примера 7 после растворения в смеси тех же растворителей при соотношении 5:5:90 (об./об.). В случае перорального введения каждое из соединений суспендировали в 0,5% водном растворе натриевой соли карбоксиметилцеллюлозы. Каждые 2 или 3 дня после введения измеряли вес тела, каждое животное анатомировали через 15 дней после трансплантации для измерения веса опухоли. Противоопухолевый эффект рассчитывали по следующей формуле: {1-(вес опухоли в группе с введенным соединением/вес опухоли в группе с введенным растворителем)} × 100
В каждой группе содержалось шесть мышей.
600,0
Пероральный
6,2
13,3
Пероральный
90,5
7,9
Внутривенный
91,5
7,9
Пероральный
91,5
ПРИМЕР ТЕСТИРОВАНИЯ 3
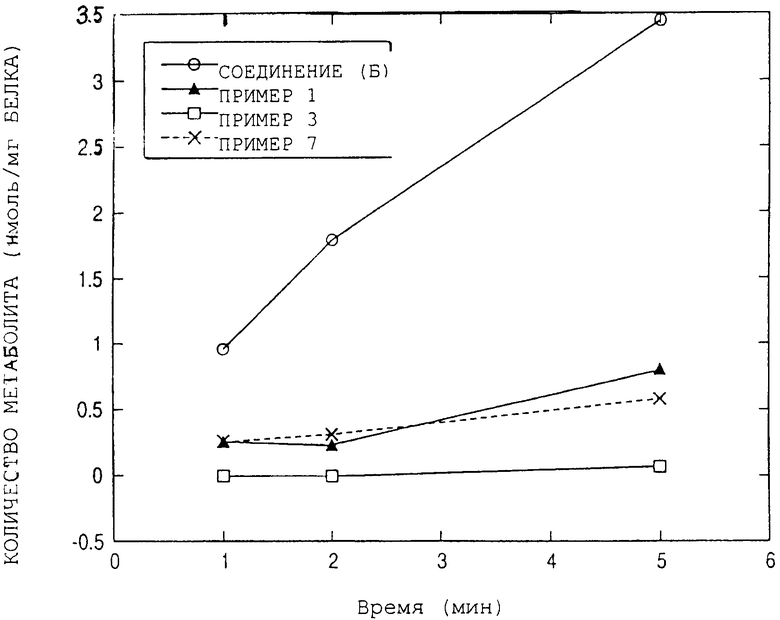
МЕТАБОЛИЗМ В МИКРОСОМЕ Р450 ЧЕЛОВЕКА
Каждый из испытуемых образцов растворяли в смеси ацето-нитрил/вода (1:1 об./об.) до концентрации 500 мкМ, и раствор смешивали с микросомой печени человека (Xenotech LLC) и другими компонентами, такими как различные коферменты и буферные растворы, и позволяли пройти реакции метаболизма при 37° С.
Реакционный раствор состоял из фосфатного буфера (0,076 М; конечная концентрация, то же самое будет применяться далее), испытуемый образец (10 мкМ), микросома печени человека (1 мг/мл), глюкоза-6-фосфат (10 мМ), глюкоза-6-фосфат дегидрогеназа (1 единица/мл), хлорид магния (4 мМ) и восстановленный фосфат никотинамид аденин динуклеотида (β -НАДФ, 1 мМ), и 500 мкл раствора использовались в одной реакции. В этом случае реакционный раствор, исключая β -НАДФ, предварительно инкубировали при 37° С 2 минуты, затем реакцию инициировали добавлением водного раствора β -НАДФ (50 мМ, 10 мкл).
Реакцию останавливали добавлением 1 мл ледяного ацетонитрила через 1, 2 или 5 минут после начала реакции.
В этой связи образец 0 минут после начала реакции был получен добавлением воды вместо водного раствора β -НАДФ и немедленным добавлением 1 мл ацетонитрила. К каждому из этих образцов добавляли 100 мкл вещества - внутреннего стандарта, и реакционный раствор центрифугировали 15 минут. Полученную надосадочную жидкость вводили в колонку для высокоэффективной жидкостной хроматографии (ВЭЖХ) для измерения искомой концентрации в образце. Количество, уменьшившееся по сравнению с концентрацией при 0 минутах от начала реакции, принимали как образовавшееся количество метаболита (нмоль/мг белка). Количество метаболита, образовавшегося за 1 минуту (константа скорости метаболизма: k (нмоль/мин/мг белка)) рассчитывали по наклону графика линейной зависимости количества образовавшегося метаболита от времени реакции, обсчитанной по методу наименьших квадратов.
Из полученной таким способом константы скорости метаболизма k (нмоль/мин/мг белка) вычисляли специфический для печени клиренс (CLint) по следующей формуле. CLint (мл/мин/кг веса тела) = k × (г веса печени)/(кг веса тела) × (45 мг белка микросомы)/(г веса печени), где вес печени на 1 кг веса тела составляет 20 г.
Кроме того, вычисляли клиренс печени (CLh) из величины Clint в соответствии с моделью идеального смешения (J. Pharmacol. Exp. Ther., 283, 46-58, 1997).
CLh (мл/мин/кг веса тела) = Q × Clint/(Q + Clint), где Q представляет собой поток крови через печень человека, определенный как 20 мл/мин/кг.
Теоретическая величина биодоступность (F) была рассчитана из CLh по следующей формуле: F = (1-CLh/Q).
Кроме того, теоретическую величину биодоступности неизмененного соединения рассчитывали по формуле 1 - F.
Результаты представлены на чертеже 1 и в Таблице 1. Теоретические величины F неизменной формы соединений по изобретению превышали теоретическую величину F 0,27 неизмененной формы контрольного соединения (соединение формулы В), что означает, что диапазон изменения биодоступности уменьшается, разделение терапевтического диапазона и диапазона токсичности может быть достигнуто более точно, и, следовательно, возможно использование перорального введения.
ПРИМЕР ТЕСТИРОВАНИЯ 4
Соединение формулы (В) или Примера 1 вводили обезьяне внутривенно или перорально в виде одной дозы, и измеряли изменения его концентрации в крови для вычисления величины ППКо-∞ . ППКо-∞ означает площадь под кривой концентрации лекарства в крови во времени за период от времени введения (0 ч) до бесконечности, и она может быть вычислена в соответствии с методом (правило трапеции), описанным в Kiyoshi Yamaoka и Yusuke Tanigawara, Yakubutsu Sokudoron Nyumon (Руководство по Фармакокинетике), стр. 116-117, Кроме того, отношение ППК при пероральном введении к ППК при внутривенном введении рассчитывали как пероральное ВА. Тест для соединения формулы (В) проводили на разных особях одинаковых обезьян при внутривенном или пероральном введении, а также тест для соединения Примера 7 проводили на 4 обезьянах тех же особей при внутривенном и пероральном введении для расчета средней величины ППК.
Животное: женская особь Масаса irus, способ введения (соединение формулы (В)) [внутривенно] этанол: Твин 80: 5% глюкоза = 5:5:90, [перорально] 0,1 Н раствор НСl, (соединение Примера 7) [внутривенно] 10% β -CyD-SBE7 (pH 3,5 в физиологическом растворе), [перорально] 40 мМ ацетатного буфера (pH 4,0).
Примера 7
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Соединение по изобретению было улучшено в смысле его токсичности и не проявило почечной токсичности. Соединение по изобретению показало высокую противоопухолевую эффективность на мышах при пероральном введении. Поскольку соединение по изобретению имеет высокую теоретическую величину F для его неизмененной формы, диапазон изменения биодоступности уменьшается, и может быть достигнуто разделение терапевтического диапазона и диапазона токсичности. Соединение по изобретению показало отличные свойства абсорбции при пероральном введении на обезьянах. Соответственно, соединение по изобретению может быть использовано в качестве противоопухолевого агента, который может вводиться перорально.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛЫ ПРОИЗВОДНЫХ ТАКСАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2284328C2 |
| СПОСОБ ИДЕНТИФИКАЦИИ ПРОТИВООПУХОЛЕВЫХ ЦЕЛЕВЫХ ФЕРМЕНТОВ | 2002 |
|
RU2319482C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЭНДОТОКСИНА | 2006 |
|
RU2408023C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| РЕГУЛЯТОРЫ-ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2797408C2 |
| РЕГУЛЯТОРЫ ПРОИЗВОДНЫХ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2803499C1 |
| Иммуномодулирующие азалиды на основе мочевины | 2021 |
|
RU2811591C1 |
| ПРОИЗВОДНЫЕ КАРБАПЕНЕМА, ПРИМЕНЕНИЕ ИХ ДЛЯ ИЗГОТОВЛЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ ИНФЕКЦИОННЫХ ЗАБОЛЕВАНИЙ | 2001 |
|
RU2276154C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОПИРАНА В КАЧЕСТВЕ ПРОТИВОАРИТМИЧЕСКИХ АГЕНТОВ | 2005 |
|
RU2380370C2 |
| ПРОЛЕКАРСТВО ПРОИЗВОДНОГО АМИНОКИСЛОТЫ | 2017 |
|
RU2739318C2 |
Настоящее изобретение относится к новому пентациклическому соединению таксана, представленному следующей формулой:
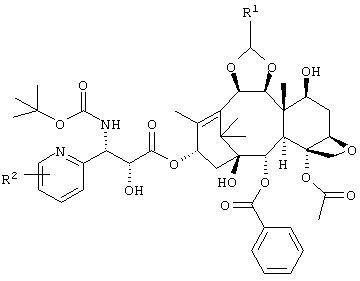
где R1 представляет собой диметиламинометил или морфолинометил, и R2 представляет собой атом галогена или алкокси группу, имеющую от 1 до 6 атомов углерода, или его соли, обладающему противоопухолевым действием, а также к медикаменту на его основе. Технический результат - получение новых производных таксана, обладающих ценным биологическим действием. 6 н. и 7 з.п. ф-лы., 1 ил., 4 табл.
где R1 представляет собой диметиламинометил или морфолинометил, и R2 представляет собой атом галогена или алкоксигруппу, имеющую от 1 до 6 атомов углерода,
или его соль.
или его соль.
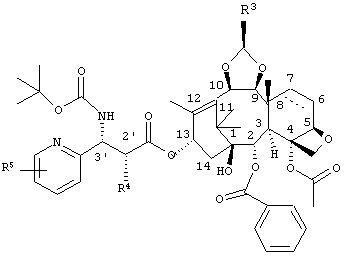
где R3 представляет собой диметиламинометил, морфолинометил или винил, R4 представляет собой гидроксильную группу, которая может иметь защитную группу, и R5 представляет собой атом галогена или алкоксигруппу, содержащую от 1 до 6 атомов углерода, и часть пунктирной линии между позициями 6 и 7 частичной структуры, представленной следующей формулой
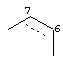
означает, что связь в этой части может быть двойной связью,
или его соль.
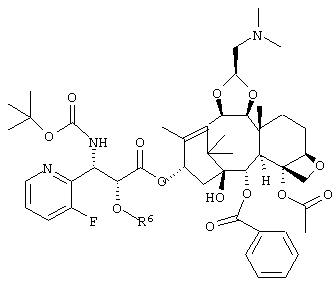
где R6 представляет собой триизопропилсилильную группу, трет-бутилдиметилсилильную группу, триэтилсилильную группу или бензильную группу,
или его соль.
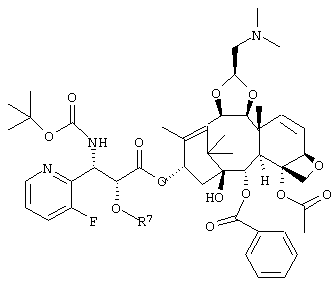
где R7 представляет собой триизопропилсилильную группу, трет-бутилдиметилсилильную группу, триэтилсилильную группу или бензильную группу,
или его соль.
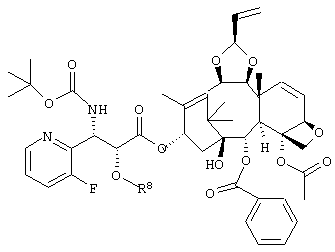
где R8 представляет собой триизопропилсилильную группу, третичную бутилдиметилсилильную группу, триэтилсилильную группу или бензильную группу,
или его соль.
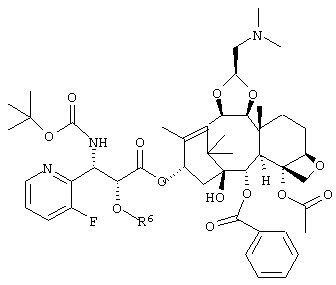
где R6 представляет собой триизопропилсилильную группу, трет-бутилдиметилсилильную группу, триэтилсилильную группу или бензильную группу,
или его соль для получения соединения, представленного следующей формулой:
или его соли.
где R7 представляет собой триизопропилсилильную группу, трет-бутилдиметилсилильную группу, триэтилсилильную группу или бензильную группу,
или его соль для получения соединения, представленного следующей формулой:
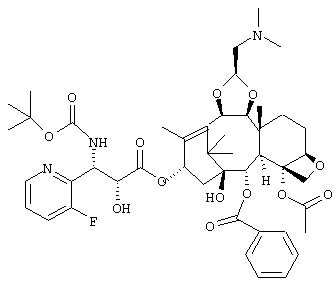
или его соли.
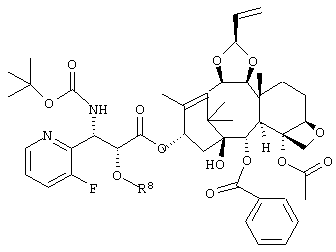
где R8 представляет собой триизопропилсилильную группу, третичную бутилдиметилсилильную группу, триэтилсилильную группу или бензильную группу,
или его соль для получения соединения, представленного следующей формулой:
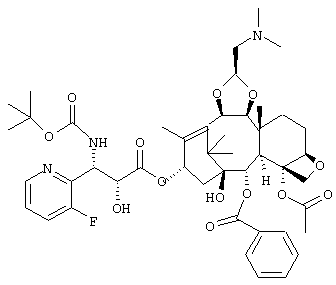
или его соли.
| EP 0826688 A1, 31.10.1996 | |||
| WO 9321173 A, 28.10.1995 | |||
| ПРОИЗВОДНЫЕ ТАКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2137764C1 |
| RU 2059631 C1, 10.05.1996 | |||
| ТАКСОИДЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2139864C1 |

