Техническая область
Данное изобретение касается новых кристаллов производных таксана, обладающих противоопухолевой активностью, и способа их получения.
Обоснование созданияизобретения
Предполагается, что таксол, соединение, проявляющее противоопухолевую активность, в основе которой лежит подавление деполимеризации микротрубочек во время клеточного деления, эффективно в клинической практике в качестве противоопухолевого агента, имеющего механизм действия, отличный от механизма действия обычных противоопухолевых агентов. Различные типы производных таксола соответственно были описаны в публикациях. Например, описаны соединения, в которых заместитель введен в пиридиновое кольцо боковой цепи, находящееся в 13 положении, для подавления модификации (преобразования) данных соединений в ходе метаболизма (Международная публикация WO 01/27115). В частности, соединение, описанное в примере 7 упомянутой выше международной публикации, (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат с трудом метаболизируется микросомами печени человека и, как предполагается, является противоопухолевым агентом, который может быть введен перорально.
Что касается соединения, описанного в примере 7 упомянутой выше международной публикации, способ получения данного соединения описан также и в примере 9 публикации, как и в примере 7. Пример 9 описывает целевое соединение, полученное с помощью «проведения операций, сходных с теми, что описаны на стадии 4 в примере 8». Что касается объяснения, рассматривается тот факт, что растворитель в экстракте реакционной смеси, содержащей целевое соединение, был упарен, остаток очищен с использованием хроматографии на силикагеле и перекристаллизован из смеси воды и этанола с получением твердого вещества, представляющего собой, как определено, кристаллы целевого соединения. Однако никакой физико-химической оценки не приведено в примерах 7 и 9 упомянутой выше международной публикации, что подтверждает тот факт, что упомянутое выше соединение было выделено в форме кристаллов.
Описание изобретения
Авторы данного изобретения повторили большинство экспериментов, относящихся к способу примера 9 упомянутой выше международной публикации. В результате авторы в конечном итоге пришли к выводу, что получить кристаллическую субстанцию посредством осаждения, даже с использованием смеси воды с этанолом в качестве растворителя, абсолютно невозможно, а целевая субстанция была неожиданно получена в виде масла или аморфной субстанции. Ввиду данных обстоятельств авторы данного изобретения провели различные исследования с целью получения упомянутого выше соединения в виде кристаллической субстанции, и авторами впервые были успешно получены кристаллы упомянутого выше соединения с использованием растворителя, отличного от смеси воды и этанола. Авторы также обнаружили, что существует по крайней мере два типа полиморфизма кристаллов упомянутого выше соединения, и получили смеси двух типов кристаллов в зависимости от условий, таких как тип растворителя для кристаллизации кристаллов.
С точки зрения постоянного применения продукта, стабильности качества и т.д. в качестве активного ингредиента лекарственного препарата желательно использовать кристаллическую, а не аморфную субстанцию, и желательно, чтобы кристаллическая субстанция представляла собой один тип кристаллов. Ввиду указанных причин авторы данного изобретения провели различные исследования для получения одного типа кристаллов упомянутого выше соединения и в результате обнаружили, что кристаллическое вещество (субстанцию) упомянутого выше соединения, состоящее из одного типа кристаллов, можно получить при использовании ацетона, смеси ацетона и воды или смеси ацетонитрила и воды в качестве органического растворителя, используемого для кристаллизации упомянутого выше соединения. Данное изобретение было создано на основе этих открытий.
Данное изобретение, таким образом, обеспечивает получение кристаллов (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионата, которые демонстрируют характеристические пики при углах дифракции (2θ), равных 6,2°, 10,3°, 10,7°, 11,4° и 12,0°в картине дифракции рентгеновских лучей в порошке.
Данное изобретение также обеспечивает способ получения упомянутых выше кристаллов, который включает этап проведения кристаллизации с использованием органического растворителя, выбранного из группы, состоящей из растворителя типа кетона, растворителя типа нитрила и их смеси или смеси указанных органических растворителей и воды. Предпочтительно в качестве органических растворителей, выбранных из группы, состоящей из растворителя кетонного типа, растворителя типа нитрила, и их смеси, могут быть использованы ацетон или ацетонитрил и смесь ацетона и воды или смесь ацетонитрила и воды в качестве предпочтительного растворителя для кристаллизации. При использовании упомянутой выше смеси органического растворителя и воды содержание воды в упомянутой выше смеси составляет предпочтительно 60% по массе или меньше, более предпочтительно в интервале значений от 40 до 50% по массе.
В соответствии с особенно предпочтительным воплощением упомянутого выше способа выполнение упомянутого выше способа обеспечивается, когда смесь ацетона и воды, содержащая от 40 до 50% воды по массе, или смесь ацетонитрила и воды, содержащая от 40 до 50% воды по массе, используются в качестве кристаллизационных растворителей в количестве от 20 до 25 мас. частей от массы соединения, соединение кристаллизуется при температуре в интервале значений от 0 до 45°С, и получающиеся кристаллы высушивают при пониженном давлении с перемешиванием при температуре в интервале значений от 30 до 60°С.
Данное изобретение, кроме того, представляет противоопухолевый агент, содержащий в качестве активного ингредиента упомянутый выше кристалл; противоопухолевый агент в форме фармацевтической композиции, которая содержит в качестве активного ингредиента упомянутый выше кристалл и один или более типов фармацевтических добавок; использование упомянутого выше кристалла для производства упомянутого выше противоопухолевого агента; и способ терапевтического воздействия на опухоль (лечения), который включает этап введения пациенту терапевтически эффективного количества упомянутого выше кристалла. Данное изобретение также обеспечивает использование упомянутого выше кристалла для производства лекарственного препарата, содержащего в качестве активного ингредиента (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат.
Краткое пояснение к фигурам
Фиг.1 показывает картину дифракции рентгеновских лучей в порошке кристаллов данного изобретения (β кристаллы).
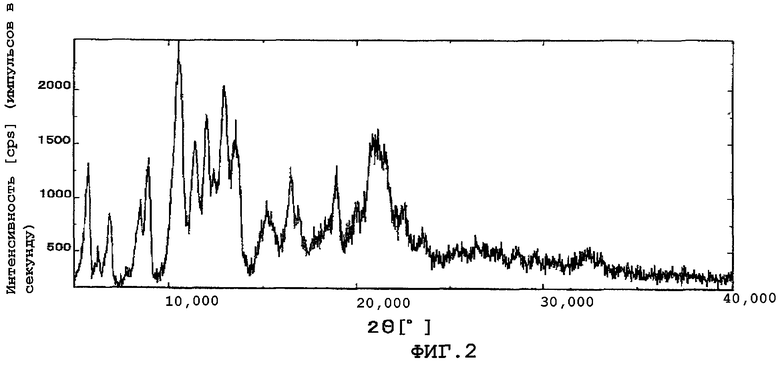
Фиг.2 показывает картину дифракции рентгеновских лучей в порошке смеси полиморфных кристаллов (α кристаллы), которые отличаются от кристаллов данного изобретения, и кристаллов по данному изобретению (β кристаллы).
Фиг.3 показывает кривую термогравиметрического/дифференциального температурного анализа (TG/DTA) кристаллов данного изобретения (β кристаллы).
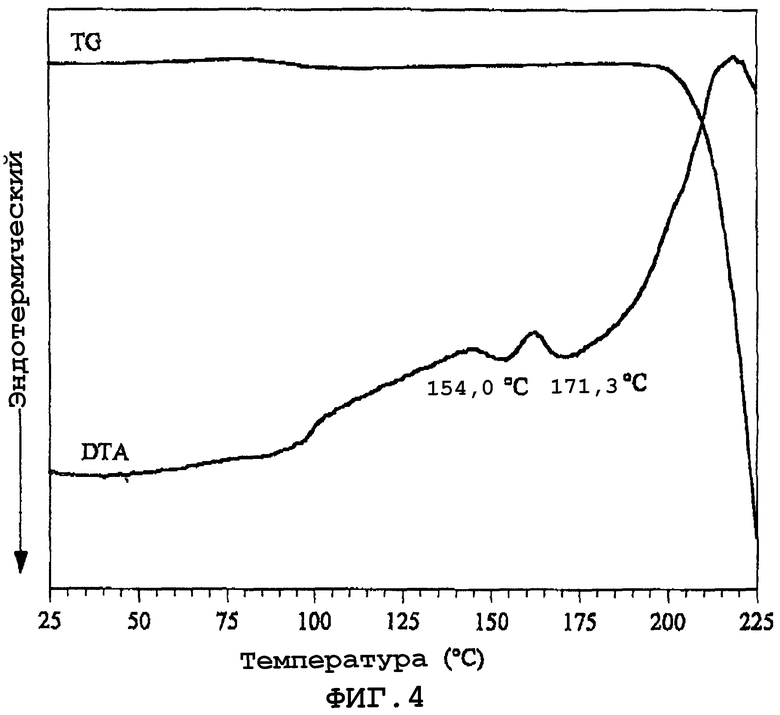
Фиг.4 показывает кривую термогравиметрического/дифференциального температурного анализа (TG/DTA) смеси полиморфных кристаллов (α кристаллы), которые отличаются от кристаллов данного изобретения, и кристаллов данного изобретения (β кристаллы).
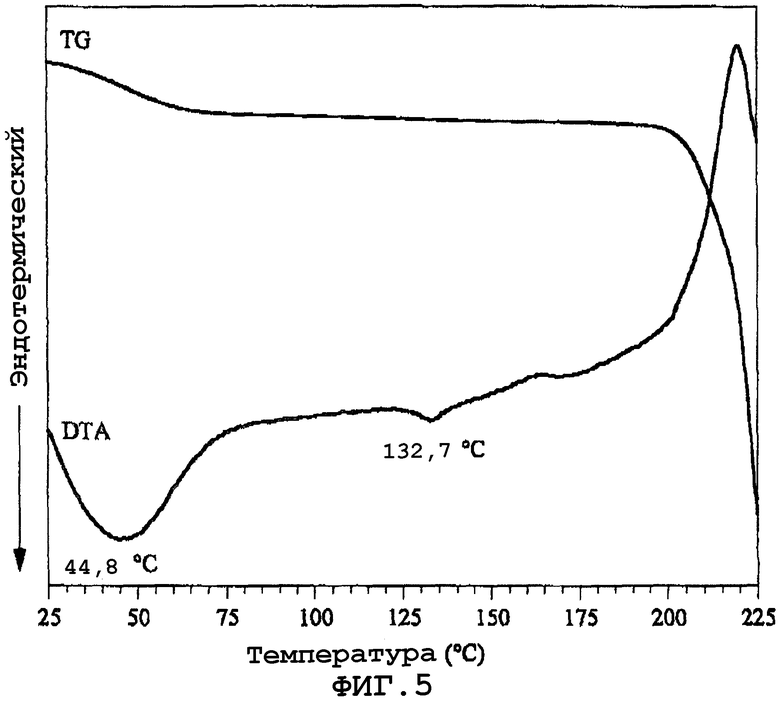
Фиг.5 показывает кривую термогравиметрического/дифференциального температурного анализа (TG/DTA) аморфного твердого вещества.
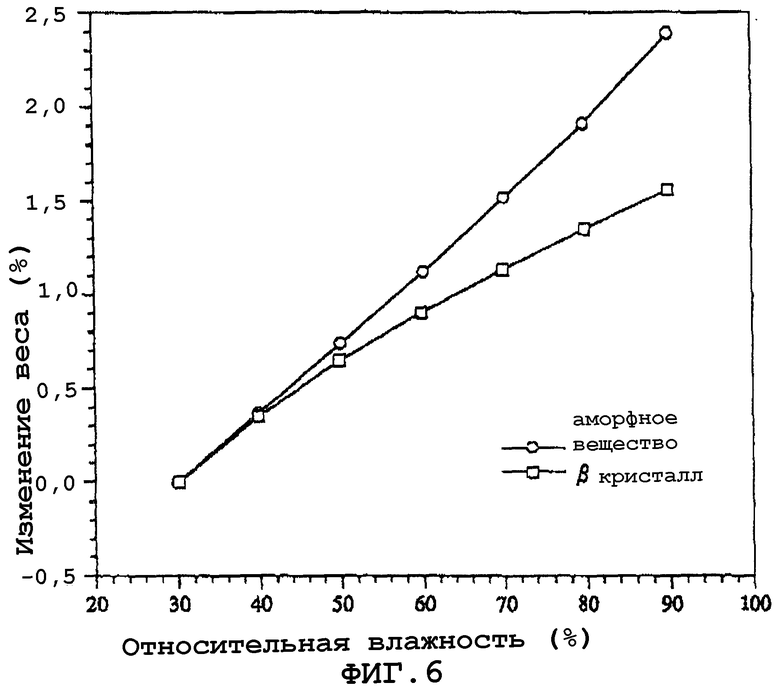
Фиг.6 показывает результаты измерения характера поглощения влаги кристаллами данного изобретения (β кристаллы) и аморфной субстанцией, измеренной при относительной влажности от 30 до 90%.
Наилучший способ воплощения изобретения
(1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат является соединением, представленным следующей формулой, и может быть легко получен квалифицированными специалистами в данной области в соответствии со способом, описанным в примере 7 международной публикации WO 01/27115.
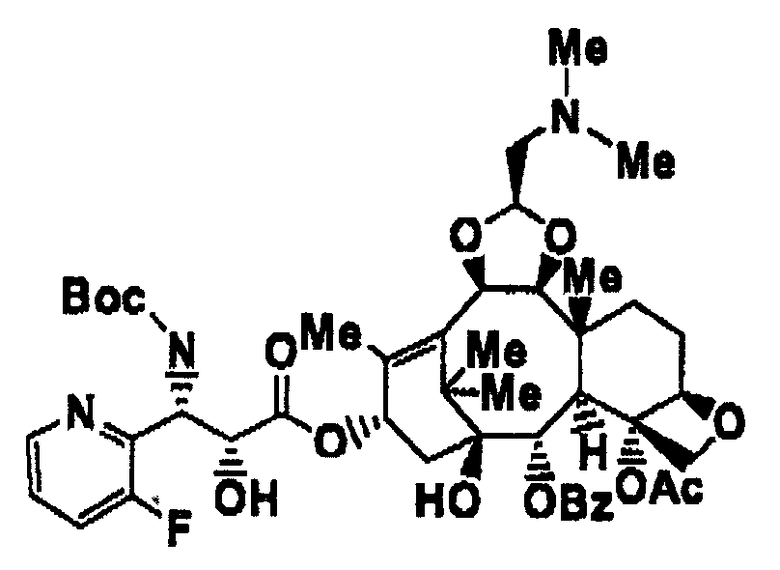
(В формуле Ме представляет собой метильную группу, Ас представляет собой ацетильную группу, Bz представляет собой бензоильную группу, и Вос представляет собой трет-бутоксикарбонильную группу.)
Кристаллы данного изобретения представляют собой кристаллы (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионата, который имеет характеристические пики при углах дифракции (2θ), равных 6,2°, 10,3°, 10,7°, 11,4° и 12,0°в картине дифракции рентгеновских лучей в порошке (эти кристаллы также названы «β кристаллами» в описании). Ввиду того что дифракционные углы (2θ) в картине дифракции рентгеновских лучей в порошке в основном имеют экспериментальную ошибку в ряду значений менее чем 5%, каждый из упомянутых выше дифракционных углов должен идентифицироваться как множественный ряд значений, включающий погрешность менее чем 5%. Следовательно, кристаллы, имеющие углы дифракционных пиков дифракции рентгеновских лучей в порошке, идентичные выше упомянутым углам с экспериментальной ошибкой менее чем 5% кристаллов, так же как кристаллы, имеющие полностью идентичные указанным углы дифракционных пиков, попадают в объем данного изобретения.
Кристаллы данного изобретения могут быть получены с помощью исследуемого соединения, руководствуясь способом, описанным в примере 7 международной публикации WO 01/27115, кристаллизацией из растворителя, в котором органический растворитель выбран из группы, состоящей из растворителя типа кетона, растворителя типа нитрила и их смеси или смеси упомянутого выше органического растворителя и воды. Что касается растворителя типа кетона, ацетона, метилэтилкетона и подобных, они могут быть использованы, и ацетонитрил и подобные могут быть использованы в качестве растворителя типа нитрила. Среди данных растворителей предпочтительными являются ацетон и ацетонитрил.
При использовании для кристаллизации в качестве растворителя смеси органического растворителя и воды содержание воды в упомянутой выше смеси предпочтительно составляет 60% по массе или меньше, более предпочтительно от 40 до 50% по массе. Что касается смеси органического растворителя и воды, смесь ацетона и воды и смесь ацетонитрила и воды предпочтительны. Особенно предпочтительным растворителем для кристаллизации является смесь ацетона и воды, содержащая от 40 до 50% воды по массе, или смесь ацетонитрила и воды, содержащая от 40 до 50% воды по массе. Для кристаллического осаждения упомянутое выше соединение может быть непосредственно растворено в смеси ацетона и воды, смеси ацетонитрила и воды и подобных. Альтернативно соединение может быть растворено в растворителе, таком как ацетон или ацетонитрил, и далее раствор с соответствующим содержанием воды может быть добавлен для проведения кристаллизации.
Количество растворителя, используемого в кристаллизации, специально не ограничено. Количество в основном составляет примерно от 5 до 50 частей по массе, предпочтительно от примерно 20 до 25 частей по массе, от веса упомянутого выше соединения. Температура кристаллизации особенно не ограничена. Температура находится предпочтительно, например, в интервале значений от 0 до 45°С. Время кристаллизации в основном от приблизительно 8 часов до 1 дня. Кристаллы после кристаллизации могут быть собраны фильтрованием и затем высушены обычным способом для получения кристаллов данного изобретения. Когда кристаллы высушены, желательно не понижать резко температуру кристаллов. Высушивание предпочтительно проводится на воздухе или при пониженном давлении с перемешиванием, при температуре в интервале значений от комнатной до 60°С. Высушивание предпочтительно проводится при температуре в интервале значений от 30 до 60°С. Если температура высушивания низка, кристаллы могут быть загрязнены кристаллами других кристаллических форм (α кристаллы). Получают ли кристаллы данного изобретения кристаллизацией и высушиванием при несложном контроле на основе информации или нет, каждый из дифракционных углов полученного кристалла в дифракции рентгеновских лучей в порошке является идентичным любому из упомянутых выше дифракционных углов. Фиг.1 показывает картину дифракции рентгеновских лучей в порошке кристаллов данного изобретения (β кристаллы), и фиг.2 показывает картину дифракции для смеси α и β кристаллов. Кристаллическая субстанция данного изобретения обладает более высокой стабильностью, чем аморфная субстанция. Например, как показано в примерах, гигроскопичность кристаллического вещества (субстанции) данного изобретения ниже, чем у аморфного вещества, что обнаруживает высокую стабильность кристаллической субстанции. Кроме того, повышенная стабильность кристаллической субстанции данного изобретения может быть также подтверждена с помощью других методик, таких как эксперимент по облучению светом.
Использование (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионата в качестве противоопухолевого агента в деталях описано в международной публикации WO 01/27115, и кристалл данного изобретения может быть использован в качестве активного ингредиента упомянутого выше противоопухолевого агента. Полное описание международной публикации WO 01/27115 включено в данное изобретение в виде ссылки. Ссылаясь на описание упомянутой выше международной публикации, квалифицированные специалисты могут использовать лекарственный препарат, содержащий кристалл данного изобретения в качестве активного ингредиента противоопухолевого агента. Кристаллы данного изобретения могут быть также использованы для производства лекарственного препарата, содержащего упомянутое выше соединение в качестве активного ингредиента. Например, кристалл данного изобретения может быть использован для производства инъекций или растворов, лекарственных форм в виде растворов.
Лекарственный препарат, содержащий кристалл данного изобретения в качестве активного ингредиента, предпочтительно предлагается в форме фармацевтической композиции, содержащей кристаллы данного изобретения в качестве активного ингредиента и один или более типов фармацевтических добавок. Путь введения лекарственного препарата данного изобретения жестко не регламентирован, и лекарственный препарат может быть введен перорально или парентерально. (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат, при применении его в качестве активного ингредиента лекарственного препарата данного изобретения, характеризуется тем, что проявляет противоопухолевый эффект даже при пероральном применении. Таким образом, пероральный путь введения является предпочтительным.
Примеры фармакологически и фармацевтически приемлемых добавок, используемых для производства упомянутой выше фармацевтической композиции, включают, но не ограничиваются эксципиентами, разрыхляющими агентами или разрыхляющими добавками, связывающими веществами, смазывающими веществами, пленочными покрытиями, красителями, разбавителями, основными материалами, растворяющими агентами или растворяющими добавками, изотоническими агентами, регуляторами pH, стабилизаторами, диспергаторами, веществами, повышающими клейкость, и тому подобными.
Примеры получения подходящих лекарственных форм для перорального введения включают, например, таблетки, порошки, гранулы, капсулы и тому подобные. Примеры получения подходящих лекарственных форм для парентерального введения включают, например, инъекции, fusion drips, суппозитории, ингаляционные формы, пластыри и тому подобные. Среди них капсулы и таблетки являются предпочтительными. Для приготовления подходящих для перорального применения форм могут использоваться, например, эксципиенты, такие как глюкоза, лактоза, Д-маннит, крахмал и кристаллическая целлюлоза; разрыхляющие агенты или разрыхляющие добавки, такие как карбоксиметилцелюлоза, крахмал и кальций карбоксиметилцеллюлоза; связующие вещества, такие как гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, поливинилпирролидон и желатин; смазывающие вещества, такие как стеарат магния и тальк; пленочные агенты, такие как гидроксипропилметилцеллюлоза, сахароза, полиэтиленгликоль и оксид титана; основы, такие как вазелин, жидкий парафин, полиэтиленгликоль, желатин, каолин, глицерин, очищенная вода и твердый жир, в качестве фармакологически и фармацевтически приемлемых добавок.
Доза лекарственного препарата данного изобретения строго не регламентируется и может быть подходящим образом подобрана в зависимости от различных условий, таких как тип опухоли, возраст, вес, симптомы и особенности пациента и тому подобные. Лекарственный препарат обычно вводят в количестве, варьирующем от 0,5 мг до 50 мг, предпочтительно от примерно 1 мг до 20 мг на 1 м2 площади поверхности тела.
Примеры
Далее изобретение конкретно поясняется и иллюстрируется с помощью примеров. Однако объем изобретения не ограничивается данными примерами.
Пример 1
(1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат (30,0 г, 34 моль) добавляют к ацетону (45 мл) и растворяют с перемешиванием при нагревании на водяной бане примерно при 45°С. К раствору по каплям добавляют воду (30 мл) примерно при 45°С при перемешивании и по завершении добавления перемешивают в течение 2 часов. Затем водяную баню охлаждают до примерно 23°С и раствор перемешивают в течение ночи. Выпавшие кристаллы собирают фильтрацией и промывают смесью ацетона и воды (30 мл), содержащей 60% воды.
Кристаллы сушат при пониженном давлении, равном примерно 600 мм рт. ст., при примерно 60°С при перемешивании в течение 3 часов. Затем кристаллы 1,5 часа сушат при пониженном давлении, равном примерно 150 мм рт. ст., и 1 час при давлении, равном примерно 30 мм рт. ст., получая 27 г (90%) белых кристаллов.
Полученные кристаллы подвергают анализу дифракции рентгеновских лучей в порошке. В результате наблюдали характеристические пики при углах дифракции (2θ), равных 6,18°, 10,30°, 10,68°, 11,38° и 11,96°.
Результаты анализа дифракции рентгеновских лучей в порошке показаны на фиг.1. Кроме того, результаты термогравиметрического анализа и дифференциального температурного анализа (TG/DTA) кристаллов приведены на фиг. 3.
Пример 2
(1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат (30,0 г, 34 моль) добавляют к ацетону (45 мл) и растворяют с перемешиванием при нагревании на водяной бане примерно при 45°С. К раствору по каплям добавляют воду (80 мл) примерно при 45°С при перемешивании и по завершении добавления перемешивают в течение 2 часов. Затем водяную баню охлаждают до примерно 23°С и раствор перемешивают в течение ночи. Выпавшие кристаллы собирают фильтрацией и промывают смесью ацетона и воды (80 мл), содержащей 60% воды.
Кристаллы сушат при пониженном давлении, равном примерно 60 мм рт. ст., при примерно 60°С, получая 27 г (90%) белых кристаллов.
Полученные кристаллы подвергают анализу дифракции рентгеновских лучей в порошке. В результате наблюдали характеристические пики, схожие с теми, что были получены в примере 1.
Пример 3
(1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат (2,5 г, 2,8 моль) добавляют к ацетону (38 мл) и перемешивают при нагревании на водяной бане примерно при 45°С. К раствору по каплям добавляют воду (25 мл) примерно при 45°С при перемешивании и по завершении добавления продолжают перемешивание в течение 2 часов. Затем водяную баню охлаждают до примерно 23°С и раствор перемешивают в течение ночи. Выпавшие кристаллы собирают фильтрацией и промывают смесью ацетона и воды (25 мл), содержащей 60% воды.
Кристаллы сушат при пониженном давлении, равном примерно 600 мм рт. ст., при 60°С при перемешивании в течение 3 часов. Затем кристаллы сушат на воздухе при комнатной температуре (примерно 19°С), получая 2,3 г (92%) белых кристаллов.
Полученные кристаллы подвергают анализу дифракции рентгеновских лучей на образце порошка. В результате наблюдали характеристические пики, близкие к тем, что получены в примере 1.
Пример 4
(1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1-гидрокситакс-11-ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионат (2,5 г, 2,8 моль) добавляют к ацетонитрилу (5 мл) и перемешивают при нагревании на водяной бане примерно при 45°С. К раствору затем добавляют воду (3 мл) примерно при 45°С и оставляют стоять при комнатной температуре в течение 1 часа. Затем раствор оставляют стоять примерно при 5°С в течение 22 часов. Выпавшие кристаллы собирают фильтрацией и промывают холодной смесью ацетонитрил:вода (1:2,5 мл).
Кристаллы сушат при пониженном давлении при 40°С в течение 22 часов, получая 0,37 г (74%) белых игольчатых кристаллов.
Полученные кристаллы подвергают анализу дифракции рентгеновских лучей в порошке. В результате наблюдали характеристические пики, близкие к тем, что получены в примере 1.
Пример 5
Было измерено влагопоглощение кристаллов данного изобретения (β кристаллы) и аморфного вещества при относительной влажности от 30 до 90%. Были использованы образцы с весом каждого примерно 20 мг, и относительную влажность изменяли начиная с 30%, шагом в 10% с использованием микровесов (автоматический прибор для измерения поглощения влаги). Температура измерения была равна 25°С. Количественно изменение было определено, когда образец давал изменение, равное 0,03% или менее, в течение 30 мин (максимальное время выдерживания 180 минут). Результаты показаны на фиг.6. Аморфное вещество при высокой влажности поглощает влаги больше на 1% в сравнении с кристаллами данного изобретения.
Промышленная применимость
Данное изобретение обеспечивает кристаллическое вещество, состоящее из одного вида кристаллов соединения, полезного в качестве противоопухолевого агента. Применение кристаллов данного изобретения позволит обеспечить стабильное качество лекарственного препарата.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕНТАЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ ТАКСАНА И ПРОТИВООПУХОЛЕВЫЕ АГЕНТЫ НА ЕГО ОСНОВЕ | 2000 |
|
RU2257387C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЭНДОТОКСИНА | 2006 |
|
RU2408023C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТАКСАНА И ПРОИЗВОДНЫЕ ТАКСАНА | 1991 |
|
RU2095356C1 |
| АНГИДРИДЫ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ ТАКСОЛА ИЛИ ТАКСОТЕРА | 1993 |
|
RU2104274C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСАЗОЛИДИНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ ПРОИЗВОДНЫХ, ОКСАЗОЛИДИНКАРБОНОВАЯ КИСЛОТА В КАЧЕСТВЕ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ТАКСОИДОВ | 1994 |
|
RU2128652C1 |
| Иммуномодулирующие азалиды на основе мочевины | 2021 |
|
RU2811591C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ ТАКСОИДОВ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2157200C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА | 1993 |
|
RU2188198C2 |
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА СОЕДИНЕНИЯ-БЛОКАТОРА ПРОНИЦАЕМОСТИ СОСУДОВ | 2020 |
|
RU2809162C2 |

Изобретение относится к новой кристаллической форме (1S,2S,3R,4S,5R,8R,9S,10R,13S)-4-ацетокси-2-бензоилокси-9,10-[(1S)-2-(диметиламино)этилидендиокси]-5,20-эпокси-1 -гидрокситакс-11 -ен-13-ил(2R,3S)-3-(трет-бутоксикарбониламино)-3-(3-фтор-2-пиридил)-2-гидроксипропионата, которая демонстрирует картину дифракции рентгеновских лучей в порошке с характеристическими пиками при углах дифракции (2θ), равных 6,2°, 10,3°, 10,7°, 11,4° и 12,0°, и способу ее получения, который включает этап проведения кристаллизации с использованием органического растворителя, выбранного из группы, состоящей из растворителя типа кетона, растворителя типа нитрила и их смеси или смеси указанного органического растворителя и воды. Изобретение также относится к противоопухолевому агенту на основе полученной кристаллической формы. Технический результат - обеспечение стабильного качества лекарственного препарата за счет более низкой гигроскопичности. 3 н. и 5 з.п. ф-лы, 6 ил.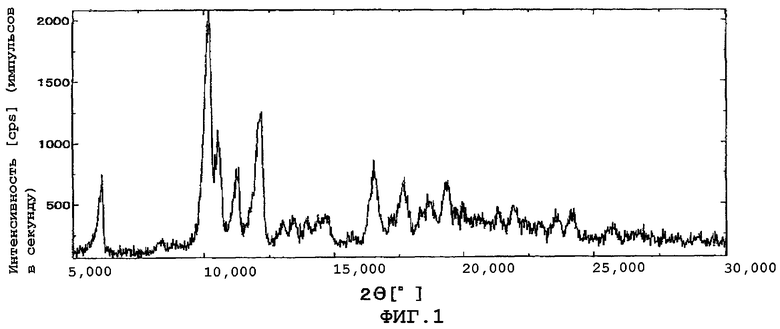
| Электромагнитный двухступенчатый регулятор скорости к прядильной машине | 1959 |
|
SU127115A1 |
| ЕР 0826688 А1, 04.03.1998 | |||
| US 6075140 А, 13.06.2000 | |||
| ПРОИЗВОДНЫЕ ТАКСАНА Ш, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2119485C1 |
| ПРОИЗВОДНЫЕ ТАКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2137764C1 |

