Область изобретения
Данное изобретение относится к новым оксатиепино[6,5-b]дигидропиридинам, полезным в качестве блокаторов кальциевых каналов. Данные соединения и связанные с ними фармацевтические композиции полезны для лечения и предотвращения или профилактики ряда расстройств, таких как повышенная или гиперчувствительность, аллергия, астма, бронхоспазм, дисменорея, эзофагеальный спазм, глаукома, преждевременные роды, расстройства мочевых путей, двигательной функции желудочно-кишечного тракта и сердечно-сосудистые расстройства.
Предпосылки создания изобретения
Тиациклоалкено[3,2-b]пиридины являются ингибиторами включения ионов кальция в ткань гладких мышц. Они действуют, расслабляя или предотвращая сокращение ткани, опосредуемое кальциевыми механизмами (Dodd et al., Drug Des. Discov. 1997, 15:135-48). Данные соединения являются активными гипотензивными и бронхорасширяющими средствами.
Тиациклоалкено[3,2-b]пиридины также полезны для лечения сердечно-сосудистых расстройств, включая гипертензию, ишемию, стенокардию, застойную сердечную недостаточность, мигрень, инфаркт миокарда и удар (инсульт). Такие соединения полезны также для лечения других расстройств, таких как повышенная чувствительность, аллергия, астма, дисменорея, эзофагеальный спазм, нарушения двигательной функции желудочно-кишечного тракта, глаукома, преждевременные роды и расстройства мочевого тракта.
Dodd et al. оценили ряд тиациклоалкено[3,2-b]пиридинов, имеющих размер сульфонового кольца от пяти- до девятичленного на их антагонистическую активность в отношении кальция. Было обнаружено, что увеличение размера сульфонового кольца от 5 до 8-членного ведет in vitro к увеличению активности на два порядка. Было обнаружено, что характером ароматического замещения, который способствует эффекту, в отношении трахей, по сравнению с действием на аорту, является 2-NO2 и 2-Сl, 6-F. Найдено, что сложноэфириой боковой цепью, которая максимально повышает in vivo активность, является N-бензил-N-метиламиноэтиловая группа (Dodd. et al., Drug Des. Discov. 1997, 15:135-48, and Drug. Des. Discov. 1993, 10:65-75).
Известны многочисленные соединения, относящиеся к тиациклоалкенопиридинам, примеры которых даются в следующих публикациях. В патенте США №5708177 на имя Straub описан способ получения оптически активных ортозамещенных 4-арил- или -гетероарил-1,4-дигидропиридикоз окислением и последующим восстановлением из их противоположных энантиомеров. В патенте №5075440 Wustrow et al. описаны пиридо[2,3-f] [1,4]тиазепины и пиридо[3,2-b][1,5]бензотиазепины, которые полезны в качестве антагонистов кальциевых каналов и обладают сердечно-сосудистой, антиастматической и антибронхосужающей активностью. В патентах США №4879384 и 4845225, оба на имя Schwender и Dodd, описаны замещенные тиациклоалкено[3,2-b]-пиридины, которые полезны также в качестве антагонистов кальциевых каналов и обладают сердечно-сосудистой, антиастматической и антибронхосужающей активностью. В патентах США №№4285955 и 4483985 описаны замещенные ациклическим сульфоном дигидропиридины, которые обладают антагонистической активностью в отношении кальциевых каналов. В патенте США №4532248 описывается обширный род дигидропиридинов, включающих циклические сульфоны, сконденсированные с дигидропиридиновым ядром. Кардиотоническая активность описана для всего рода замещенных таким образом дигидропиридинов. Наконец, в публикации Pagani, G.P.A., J. Chem. Soc. Perkin Trans. 2, 1392 (1974), описываются 10-фенил-2Н-тиопирано[3,2-b]хинолины. Однако ни одно из этих соединений не является антагонистом кальциевых каналов.
"Мягкие лекарственные средства" (известные также в качестве предлекарственных средств) являются биологически активными лекарствами, которые метаболически инактивируются после достижениями ими своей терапевтической роли в предназначенном для них месте действия. Использование мягких лекарственных средств вместо их неинактивируемых аналогов позволяет избежать нежелательных побочных действий. Мягкие лекарственные средства в общем известны (см., например, Biggadike et al., 2000, J. Med. Chem. 43:19-21; Lee et al., 1998, Curr. Opin. Drug Disc. Dev. 1:235-44). Однако не известны дигидропиридиновые мягкие лекарственные средства.
Краткое изложение сущности изобретения
Данное изобретение предоставляет новые соединения, классифицированные формулой I, определенной в данном описании ниже, а также способы их получения. Данное изобретение предоставляет также фармацевтическую композицию, включающую настоящее соединение и фармацевтически приемлемый носитель.
Данное изобретение предоставляет далее способ лечения субъекта, страдающего от расстройств, облегчение которых опосредуется снижением притока ионов кальция в клетки, действие которых усугубляет расстройства, который включает введение субъекту терапевтически эффективной дозы данной фармацевтической композиции.
Данное изобретение далее предоставляет способ ингибирования у субъекта начала расстройства или нарушения, облегчение которого опосредуется уменьшением притока ионов кальция в клетки, действие которых способствует нарушению, который включает введение субъекту профилактически эффективной дозы данной фармацевтической композиции.
Наконец, данное изобретение предоставляет устройство для введения субъекту данной фармацевтической композиции, включающее контейнер и фармацевтическую композицию в нем, при этом контейнер имеет средство для доставки субъекту терапевтической и/или профилактической дозы фармацевтической композиции.
Подробное описание изобретения
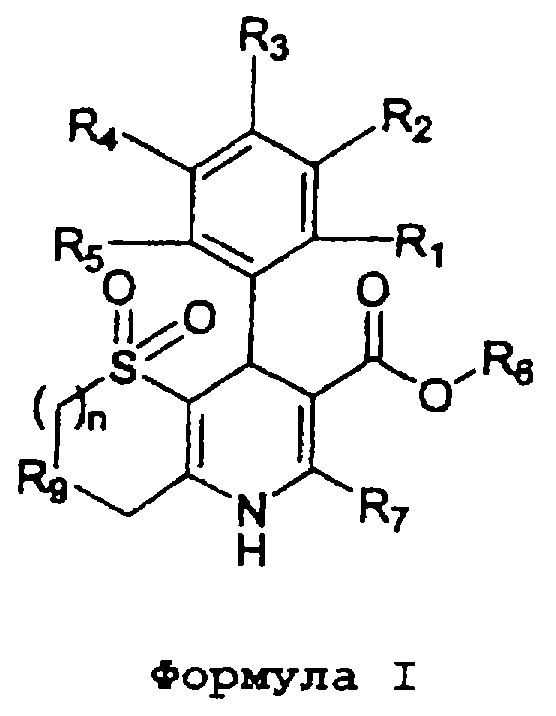
Данное изобретение предоставляет соединения формулы I,
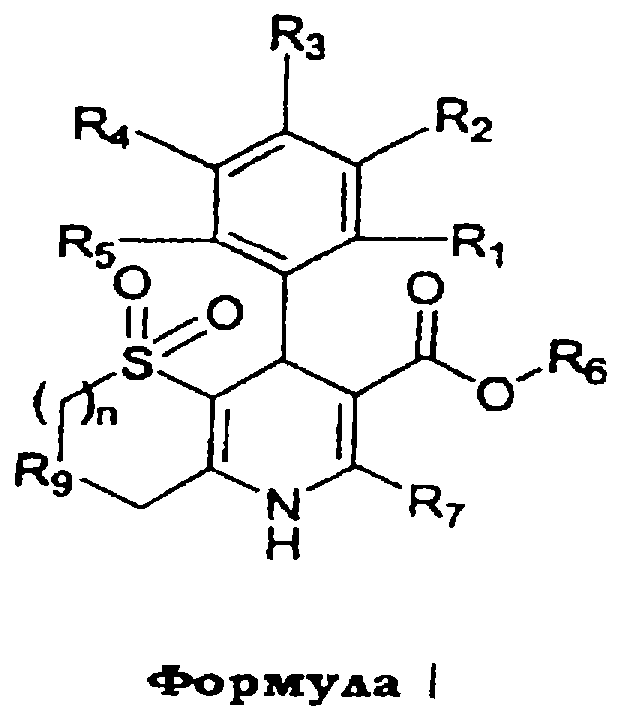
в которой
(a) R1, R2, R3, R4 и R5, независимо, выбраны из группы, состоящей из Н, ОН, галогена, циано, NO2, алкила, C1-8-алкокси, C1-8-алкилсульфонила, C1-4-карбоалкокси, C1-8-алкилтио, дифторметокси, дифторметилтио, трифторметила и оксадиазола (образованного R1 и R2);
(b) R6 выбран из группы, состоящей из Н, неразветвленного или разветвленного C1-5-алкила, арила, 3-пиперидила, N-замещенного 3-пиперидила, N-замещенного 2-пирролидинилметилена и замещенного алкила, из которых
указанный N-замещенный 3-пиперидил и N-замещенный 2-пирролидинилметилен могут быть замещены C1-8-алкилом с неразветвленной или разветвленной цепью или бензилом, а указанный замещенный алкил может быть замещен C1-8-алкокси, С1-8-алканоилокси, фенилацетилокси, бензоилокси, гидрокси, галогеном, п-тозилокси, мезилокси, амино, карбоалкокси или группой NR’R’’,
в которой (i) R’ и R’’, независимо, выбраны из группы, состоящей из Н, неразветвленного или разветвленного C1-8-алкила, С3-7-циклоалкила, фенила, бензила и фенетила или (ii) R’ и R’’ вместе образуют гетероциклическое кольцо, выбранное из группы, состоящей из пиперидино, пирролидино, морфолино, тиоморфолино, пиперазино, 2-тиено, 3-тиено и N-замещенных производных указанных гетероциклических колец, причем указанные N-замещенные производные замещены Н, неразветвленным или разветвленным C1-8-алкилом, бензилом, бензгидрилом, фенилом и/или замещенным фенилом (замещенным группой NО2, галогеном, C1-8-алкилом с неразветвленной или разветвленной цепью, C1-8-алкокси и/или трифторметилом);
(с) R7 выбран из группы, состоящей из Н, амино, алкила, арила, трифторметила, алкоксиметила, 2-тиено и 3-тиено;
(d) R9 представляет кислород или серу; и
(e) n равно целому числу от 1 до 4;
или их фармацевтически приемлемые соли.
В одном воплощении настоящего соединения R6 представляет -(CH2)2N(СН3)CH2Ph или метил. В другом воплощении R7 представляет метил. В следующем воплощении R1, R2, R3, R4 и R5, независимо, выбраны из группы, состоящей из Н, галогена и NO2. В предпочтительном воплощении R9 представляет кислород.
Следующие соединения являются примерами соединений настоящего изобретения.
Соединение 1: 1,1-диоксид 2-[метил(фенилметил)амино]этилового эфира 2,3,6,9-тетрагидро-7-метил-9-(3-нитрофенил) - 5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты.
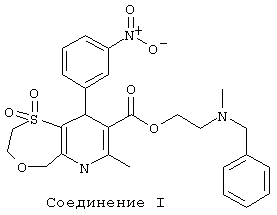
Соединение 2: 1,1-диоксид метилового эфира 9-(2-хлорфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты.
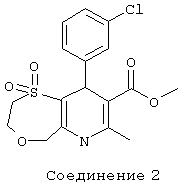
Соединение 3: 1,1-диоксид 2-[метил(фенилметил)амино]-этилового эфира 9-(2-хлорфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты.
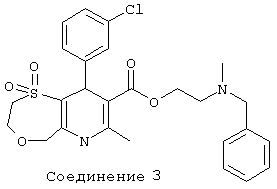
Соединение 4: 1,1-диоксид метилового эфира 2,3,6,9-тетрагидро-7-метил-9-(3-нитрофенил)-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты.
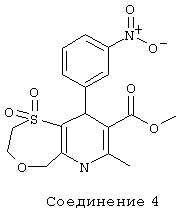
Соединение 5: 1,1-диоксид метилового эфира 2,3,6,9-тетрагидро-7-метил-9-(2-нитрофенил)-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты.
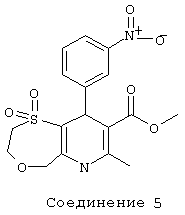
Соединение 6: 1,1-диоксид метилового эфира 9-(3-хлорфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты.
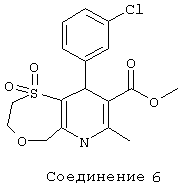
Соединение 7: 1,1-диоксид 2[метил(фенилметил)-амино]этилового эфира 9-(3-хлорфенил)-2,3,6,9-тетрагидро-7-метил-5H-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты.
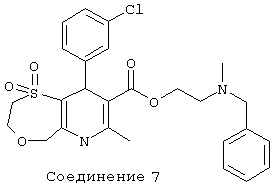
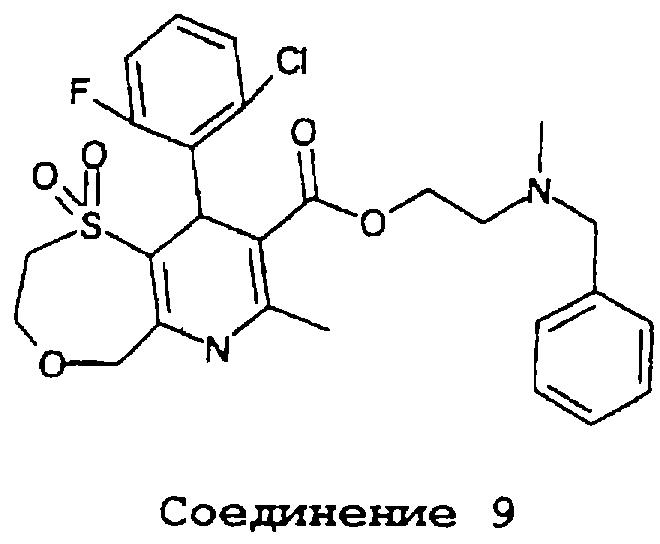
Соединение 8: 1,1-диоксид 2-[метил(фенилметил)-амино]этилового эфира 2,3,6,9-тетрагидро-7-метил-9-(2-нитрофенил)-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты.
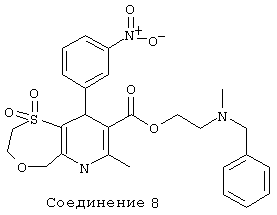
Соединение 9: 1,1-диоксид 2-[метил(фенилметил)-амино]этилового эфира 9-(2-хлор-6-фторфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты.
Данное изобретение предоставляет также мягкие лекарственные аналоги соединений формулы I. Данные мягкие лекарственные средства характеризуются химически лабильной частью, связанной со сложноэфирной группой, в свою очередь, связанной со структурой кольца дигидропиридина. Мягкие лекарственные средства позволяют данным лекарственным средствам проявлять их действие локально и впоследствии метаболизироваться в кровотоке, тем самым снижая нежелательные системные действия (например, низкое кровяное давление). Использование таких мягких лекарственных аналогов позволяет вводить более высокие дозы заявленных соединений дигидропиридина, не подвергая при этом субъекта непереносимым уровням нежелательных системных действий.
В частности, данное изобретение предоставляет соединения формулы II,
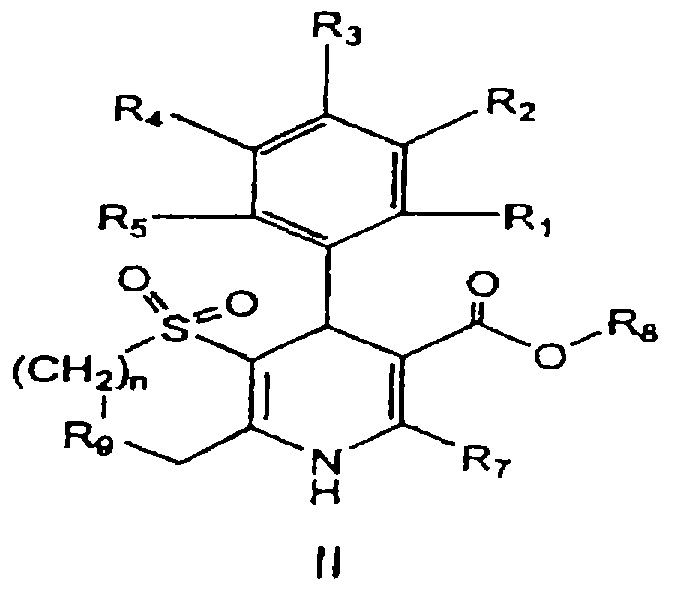
в которой
(a) R1, R2, R3, R4 и R5, независимо, выбраны из группы, состоящей из водорода, ОН, галогена, циано, NO2, алкила, C1-8-алкокси, C1-8-алкилсульфонила, C1-4-карбоалкокси, C1-8-алкилтио, дифторметокси, дифторметилтио, трифторметила и оксадиазола (образованного R1 и R2);
(b) R7 выбран из группы, состоящей из водорода, амино, алкила, арила, трифторметила, алкоксиметила, 2-тиено и 3-тиено;
(c) R8 выбран из группы, состоящей из -алкил-ОН, алкиламина, лактона, циклического карбоната, алкилзамещенного циклического карбоната, арилзамешенного циклического карбоната, -арил-С(O)ОR’, -алкиларил-С(O)ОR’, -алкил-ОС(О)R’, -алкил-С(O)R’, -алкил-С(O)OR’, -алкил-N(R’’)С(О)R’ и -алкил-N(R’’)C(O)OR’, в которых
R’ и R’’, независимо, выбраны из группы, состоящей из водорода, амино, алкила, арила, арилсконденсированного циклоалкила и гетероциклила, причем амино, алкил, арил, арилсконденсированный циклоалкил и гетероциклил, необязательно, замещены галогеном, циано, NO2, лактоном, амино, алкиламино, арилзамещенным алкиламино, aмидoм, карбаматом, карбамоилом, циклическим карбонатом, алкилом, галогензамещенным алкилом, арилалкилом, алкокси, гетероциклилом и/или арилом (причем арил, необязательно, замещен ОН, галогеном, циано, NO2, алкилом, амино, диметиламино, алкокси, алкилсульфонилом, C1-4-карбоалкокси, алкилтио и/или трифторметилом); и
(d) R9 представляет кислород или серу;
или их фармацевтически приемлемые соли.
Каждое из предпочтительных воплощений соединений формулы I, указанных выше, рассматриваются также в качестве воплощения соединений формулы II. Кроме того, в предпочтительном воплощении соединения формулы II R8 выбран из -алкил-ОН, лактона, циклического карбоната, алкилзамещенного циклического карбоната, арилзамещенного циклического карбоната и -алкил-ОС(О)R’, в котором R’ имеет указанные выше значения.
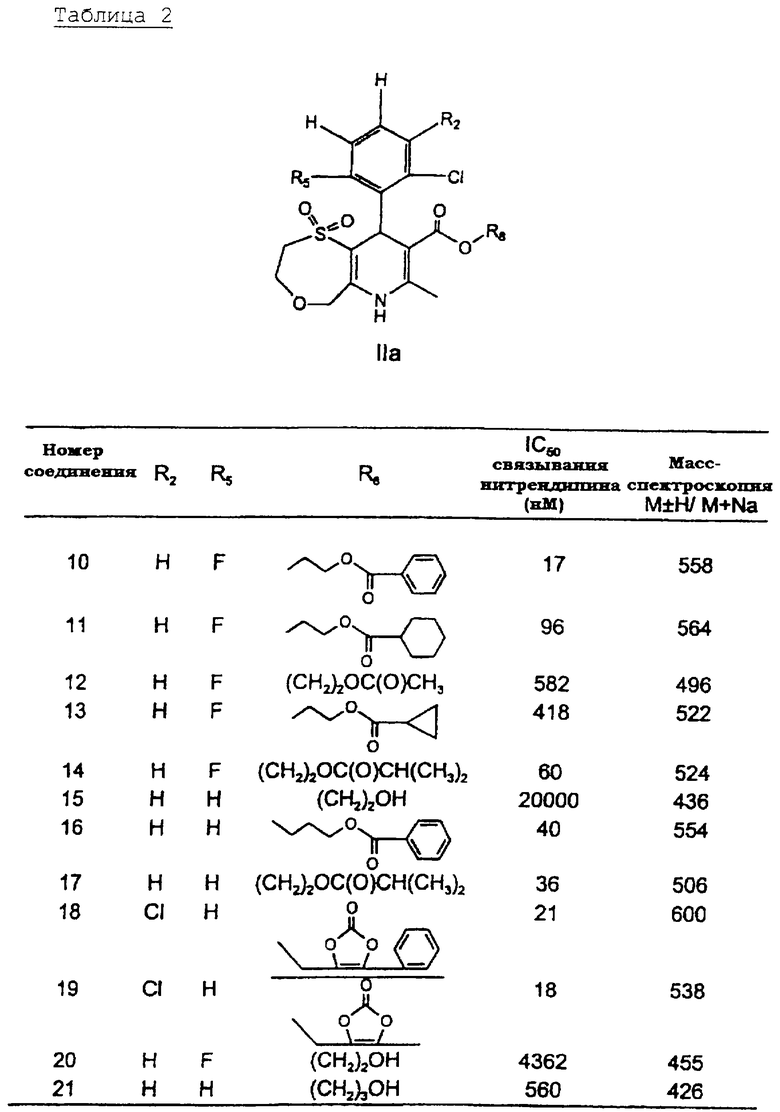
Следующие соединения (указываются здесь как соединения №№10-19) также являются предпочтительными воплощениями соединений настоящего изобретения:
1,1-диоксид(5-метил-2-оксо-1,3-диоксол-4-ил)метилового эфира 9-(2,3-дихлорфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты;
1,1-диоксид(2-оксо-5-фенил-1,3-диоксол-4-ил)метилового эфира 9-(2,3-дихлорфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты;
1,1-диоксид 2-гидроксиэтилового эфира 9-(2-хлорфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино-[6,5-b]пиридин-8-карбоновой кислоты;
1,1-диоксид 2-(2-метил-1-оксопропокси)этилового эфира 9-(2-хлорфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты;
1,1-диоксид 2-(2-метил-1-оксопропокси)этилового эфира 9-(2-хлор-6-фторфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]-оксатиепино[6,5-b]пиридин-8-карбоновой кислоты;
1,1-диоксид 2-[(циклопропилкарбонил)окси]этилового эфира 9-(2-хлор-6-фторфенил)-1,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты;
1,1-диоксид 2-(ацетилокси)этилового эфира 9-(2-хлор-6-фторфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино-[6,5-b]пиридин-8-карбоновой кислоты;
1,1-диоксид 2-[(циклогексилкарбонил)окси]этилового эфира 9-(2-хлор-6-фторфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты;
1,1-диоксид 2-(бензоилокси)этилового эфира 9-(2-хлор-6-фторфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты и
1,1-диоксид 3-(бензоилокси)пропилового эфира 9-(2-хлорфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино-[6,5-b]пиридин-8-карбоновой кислоты.
Если не оговорено особо, термин "алкил" относится к неразветвленному, разветвленному или циклическому заместителю, состоящему только из углерода и Н без ненасыщенностей. Термин "алкокси" относится к О-алкилу, в котором алкил имеет указанные выше значения. Иллюстративные арильные заместители включают, например, фенил, нафтил, дифенил, фторфенил, дифторфенил, бензил, бензоилоксифенил, карбоэтоксифенил, ацетилфенил, этоксифенил, феноксифенил, гидроксифенил, карбоксифенил, трифторметилфенил, метоксиэтилфенил, ацетамидофенил, толил, ксилил, диметилкарбамилфенил, -(СН2)2N(СН3)СН2Рh, -CH2CH2-N(Me)-СН2-гетероарил и аналогичные. Термин "галоген" означает фтор, хлор, бром и иод. Символ "Ph" относится к фенилу. "Независимо" означает, что, когда имеется более чем один заместитель, заместители могут быть различными.
Соединения настоящего изобретения являются асимметрическими в 4-положении кольца дигидропиридина и, таким образом, существуют в виде оптических антиподов. Как таковые, все возможные оптические изомеры, антиподы, энантиомеры или диастереомеры, являющиеся результатом дополнительных асимметрических центров, которые могут существовать в оптических антиподах, рацематах и их рацемических смесях, также являются частью данного изобретения. Антиподы можно разделять способами, известными специалистам в данной области, такими как, например, фракционная перекристаллизация диастереомерных солей энантиомерно чистых кислот. Альтернативно антиподы могут быть разделены с помощью хроматографии на колонке типа Pirkle.
Используемая в данном описании фраза "фармацевтически приемлемая соль" означает соль свободного основания, которая обладает требуемой фармакологической активностью свободного основания и которая ни в биологическом, ни в каком-либо другом отношении не является нежелательной. Данные соли могут происходить из неорганических или органических кислот. Примерами неорганических кислот являются хлористовородная, азотная, бромистоводородная, серная и фосфорная кислоты. Примерами органических кислот являются уксусная, пропионовая, гликолевая, молочная, пировиноградная, малоновая, янтарная, яблочная, малеиновая, фумаровая, винная, лимонная, бензойкал, коричная, миндальная, метансульфоновая, этансульфоновая, п-толуолсульфоновая, метилсульфоновая, салициловая кислоты и аналогичные.
Данные соединения могут быть получены с использованием свободно доступных исходных материалов. Первая стадия синтеза, как показано ниже на схеме 1, хорошо известна в данной области (Shibata et al., Fuji Photo Film Co., Ltd, Jpn. Kokai Tokkyo Koho, p. 47; патент Японии 62253161, 1987; заявка на патент Японии 86-39760 (860224); заявка на патент Канады №429975, 1988).
Данное изобретение предоставляет также фармацевтическую композицию, включающую настоящее соединение и фармацевтически приемлемый носитель.
Фармацевтические композиции, содержащие соединение настоящего изобретения в качестве активного ингредиента в тесной смеси с фармацевтическим носителем, можно получать по общепринятым фармацевтическим методикам. Носитель может иметь большое число форм в зависимости от формы препарата, требуемого для введения, такого как системное введение, включая, но не ограничиваясь ими, внутривенное, пероральное, назальное или парентеральное. При получении композиций в пероральной лекарственной форме можно использовать любой из обычных фармацевтических носителей, таких как вода, гликоли, масла, спирты, корригенты, консерванты, красящие агенты, сироп и аналогичные в случае пероральных жидких препаратов (например, суспензий, эликсиров и растворов), или такие носители, как крахмалы, сахара, разбавители, гранулирующие агенты, смазывающие вещества, связующие, дезинтегрирующие агенты к аналогичные в случае пероральных твердых препаратов (например, порошков, капсул и таблеток).
В особом воплощении соединения настоящего изобретения вводят ингаляцией. Для ингаляционной терапии соединение может быть в растворе, полезном для введения с помощью ингаляторов с подачей измеренной дозы, или в форме, подходящей для ингалятора или инсуффлятора с сухим порошком. Более конкретно, соединения для использования в соответствии с настоящим изобретением удобно доставляют в форме аэрозольного спрея из контейнера под давлением с помощью тампона или распылителя, например, с использованием подходящего пропеллента, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа, внутри такого контейнера. Единичную дозу можно определить или ограничить снабжением контейнера клапаном для доставки отмеренного количества. Можно сформулировать капсулы и картриджи, изготовленные из фармацевтически приемлемого материала, такого как желатин, для использования в ингаляторе или инсуффляторе, содержащие порошкообразную смесь соединения и подходящей порошкообразной основы, такой как лактоза или крахмал.
Вследствие их легкости введения, таблетки и капсулы представляют собой преимущественную пероральную форму единичкой дозы, в которой используют твердые фармацевтические носители. При желании таблетки могут быть покрыты сахаром или энтерическим покрытием с помощью стандартных приемов. Для парентеральных введений носитель обычно включает стерильную воду, хотя могут быть включены также другие ингредиенты для целей растворимости или консервации. Можно также приготовить инъецируемые суспензии, в которых можно использовать подходящие жидкие носители, суспендирующие агенты и аналогичные. Соединения можно также вводить в форме аэрозоля.
Данные фармацевтические композиции обычно содержат на единичную дозу (например, таблетку, капсулу, порошок, инъекцию, полную чайную ложку и т.д.) примерно от 0,001 до 100 мг/кг и, предпочтительно, примерно от 0,01 до 20 мг/кг настоящего соединения. В данной области известны методы определения терапевтически и профилактически эффективных доз для настоящей фармацевтической композиции. Эффективную дозу для введения фармацевтической композиции человеку, например, можно определить математически по результатам исследований на животных.
Соединения настоящего изобретения ингибируют захват ионов кальция гладкими мышцами и, следовательно, действуют, расслабляя или предотвращая опосредованное ионами кальция сокращение ткани гладких мышц.
Taким образом, данное изобретение далее предоставляет способ лечения субъекта, страдающего от расстройств, облегчение которых опосредуется снижением притока в клетки ионов кальция, действие которых способствует нарушению, который включает введение субъекту терапевтически эффективной дозы данной фармацевтической композиции. В качестве примера у субъекта, страдающего от астмы, дыхательные пути сжимаются вследствие сокращения клеток гладких мышц дыхательных путей ("SMC"). Можно ожидать, что снижение притока в SMC кальция, действие которого способствует нарушению, облегчит симптомы расстройства.
Данное изобретение далее предоставляет способ ингибирования у субъекта начала расстройства, облегчение которого опосредуется снижением притока в клетки ионов кальция, действие которых способствует нарушению, который включает введение субъекту профилактически эффективной дозы данной фармацевтической композиции.
В одном воплощении нарушение относится к группе, состоящей из повышенной чувствительности, аллергии, астмы, бронхоспазма, дисменореи, эзофагеального спазма, глаукомы, преждевременных родов, нарушений мочевых путей, нарушений двигательной функции желудочно-кишечного тракта и сердечно-сосудистых нарушений. В предпочтительном воплощении нарушением является астма. Сердечно-сосудистым нарушением может быть, например, гипертензия, ишемия, стенокардия, застойная сердечная недостаточность, инфаркт миокарда или удар.
Используемый в данном описании термин "лечение" нарушения означает устранение или иным образом ослабление причины его и/или его действия. "Ингибирование" начала нарушения означает предотвращение, замедление или снижение вероятности такого начала.
Термин "субъект" включает, без ограничения, любое животное или искусственно модифицированное животное. В предпочтительном воплощении субъектом является человек.
Данное изобретение далее предоставляет устройство для введения субъекту настоящей фармацевтической композиции, включающей контейнер и фармацевтическую композицию в нем, при этом контейнер имеет средство для доставки субъекту терапевтической и/или профилактической дозы фармацевтической композиции. В предпочтительном воплощении устройством является устройство с аэрозольным спреем для лечения и/или профилактики астмы с помощью местного респираторного введения.
Наконец, как изложено более подробно ниже, данное изобретение предоставляет способ получения соединения формулы I:
Данное изобретение можно лучше понять со ссылкой на экспериментальные подробности, которые приводятся ниже, но специалист в данной области очевидно свободно понимает, что они являются лишь иллюстративными для изобретения, описываемого более полно в формуле изобретения, которая приводится ниже. Кроме того, на протяжении данной заявки цитируются различные публикации. Содержание данных публикаций включено для сведения в данную заявку для более полного описания состояния области техники, к которой относится данное изобретение.
Экспериментальные подробности
А. Схемы и синтезы
Процедуры получения дигидропиридинов широко описаны в данной области, как показано в публикациях Eistert et al. (Chem. Bеr. 110, 1069-1085, 1977), G.A.Pagani (J.Chem. Soc., Perkin Trans, 2, 1392-7, 1974), Mason et al. (J.Chem. Soc. (C) 2171-76, 1967), E.A.Fehnel (J.Amer. Chem. Soc. 74, 1569-74, 1952) and M.Seiyaku (заявка на патент Японии №58201764, 1984).
Схема I показывает получение соединений формулы I:
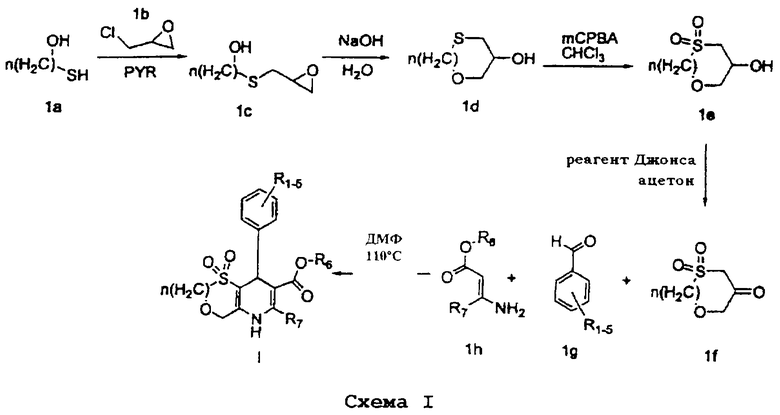
Соединения формулы II могут быть получены в соответствии со схемой II (на которой соединение 2а можно получить с помощью стадий, аналогичных стадиям на схеме I, и R1-9 имеют указанные выше значения), предпочтительно, в присутствии К2СО3 или СsСО3, в органическом растворителе, таком как диметилформамид (ДМФ).
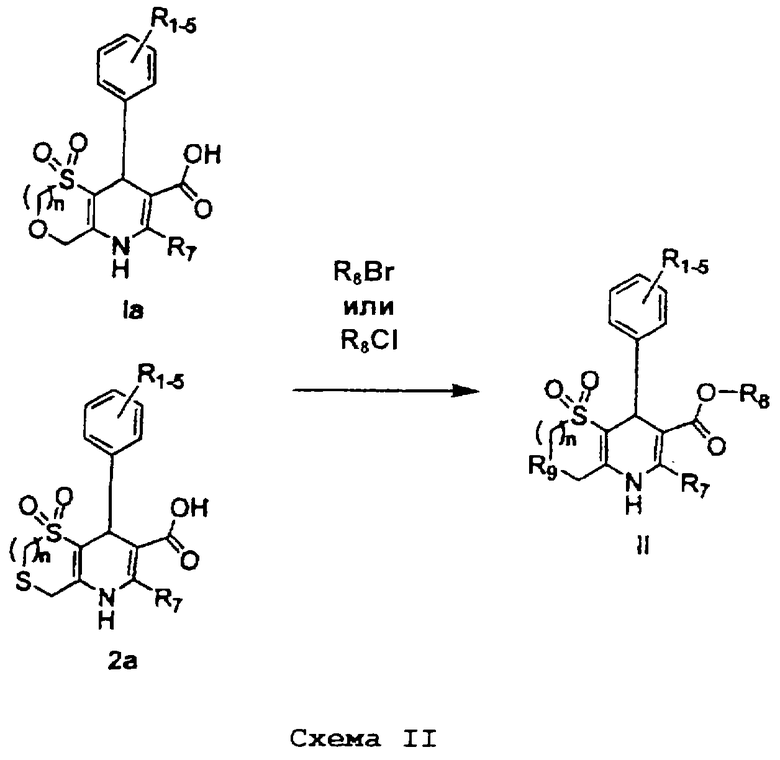
Соединения формулы II можно также получить в соответствии со схемой III (на которой соединение 3а можно получить с помощью стадий, аналогичных стадиям на схеме I, и R1-9 имеют указанные выше значения), предпочтительно, в присутствии муравьиной кислоты или NaOH (водный), соответственно.
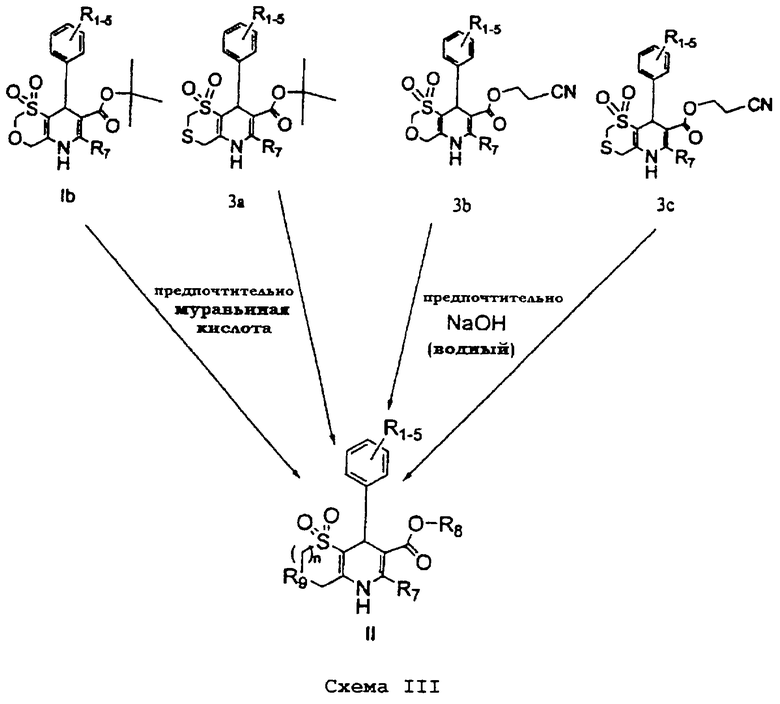
Следующие примеры описывают более подробно химический синтез характерных представителей соединений настоящего изобретения. Остальные описываемые соединения можно получить аналогично в соответствии с одним или более из данных способов. Не предпринималось никаких попыток для оптимизации выходов, полученных в данных реакциях, но специалисту в данной области, конечно, ясно, что варьирование времени, температур, растворителей и/или реагентов реакций может увеличить выходы.
Пример 1
1,1-Диоксид 2-[метил(фенилметил)амино]этилового эфира 9-(2-хлорфенил)-2,3,6,9-тетрагидро-7-метил-5Н-[1,4]оксатиепино[6,5-b]пиридин-8-карбоновой кислоты
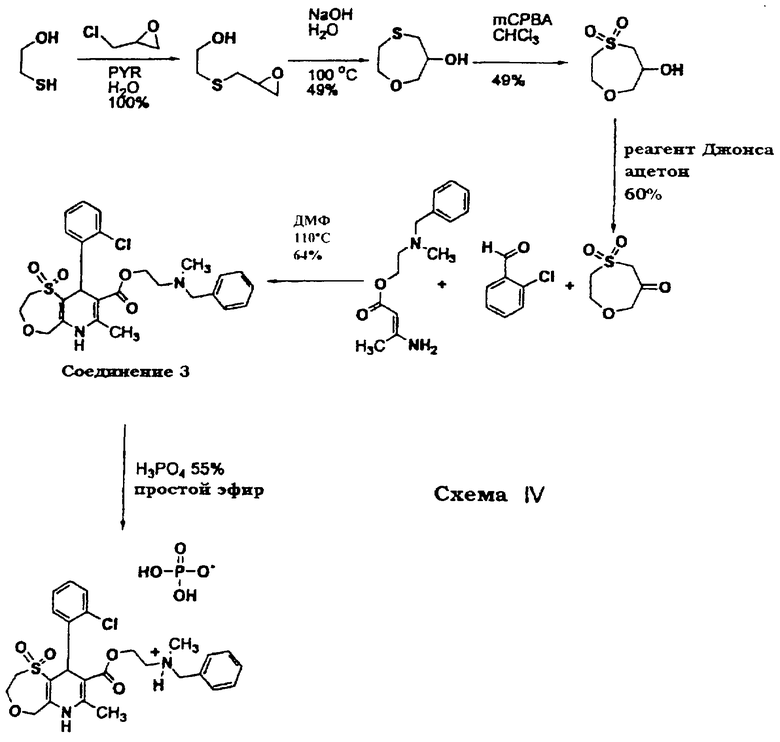
Синтез соединения 3, который показан на приведенной выше схеме IV, проводят следующим образом:
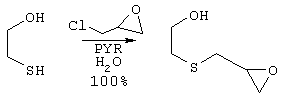
24,95 г (269,7 ммоль) эпихлоргидрина добавляют по каплям через капельную воронку к раствору 21,07 г (269,7 ммоль) 2-меркаптоэтанола в 100 мл воды и 21,33 г (269,7 ммоль) пиридина при 0°С. После завершения добавления охлаждающую баню удаляют и раствор перемешивают при комнатной температуре в течение 6 часов. Реакционную смесь затем подкисляют 1 н. раствором НСl и экстрагируют 4×200 мл EtOAc. Органические слои отделяют, объединяют, сушат над MgSO4, фильтруют и концентрируют в вакууме с получением 40,8 г продукта в виде бесцветного масла (выход >100%). Синтез данного продукта, (2-[(оксиранилметил)тио]этанола), описан подробно в литературе (Benzyl alcohol-free rapid processing of silver halide color photographic print paper, Shibata et al. (Fuji Photo Film Co., Ltd., Japan); Jpn. Kokai Tokkyo Koho, pp.47; патент Японии 62253161, 1987; заявка на патент Японии 96-39760, 860224; заявка на патент Канады №429975, 1988).
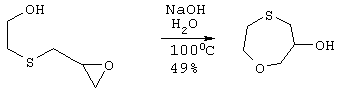
12,16 г (304 ммоль) гидроксида натрия растворяют в 120 мл воды. По каплям через капельную воронку добавляют 40,8 г (304 ммоль) сырого эпоксида. Реакционную смесь нагревают до кипения с обратным холодильником в течение 5 часов (в течение данного времени реакционная смесь становится очень темной), охлаждают до комнатной температуры, подкисляют 6 н. раствором НСl и экстрагируют 4×400 мл EtOAc. Органические слои объединяют, сушат над MgSO4, фильтруют и концентрируют в вакууме с получением 20,08 г (150 ммоль) коричневого масла, которое передвигается слегка быстрее, чем исходный материал, при ТСХ с использованием смеси 1:1 гексан/этилацетат для элюирования.
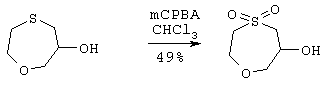
В 3-горлую 1 л колбу, снабженную термометром, капельной воронкой и приводимой в движение воздухом мешалкой, загружают 43,5 г (214,1 ммоль) 85% 3-хлорпероксибензойной кислоты и 260 мл СНСl3 и затем колбу охлаждают в ледяной ванне. По каплям через капельную воронку на протяжении 1 часа добавляют 13,06 г (97,32 ммоль) сырого сульфида в 200 мл СНСl3. Охлаждающую баню затем удаляют и реакционную суспензию перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь затем фильтруют и фильтрат концентрируют в вакууме. Остаток обрабатывают эфиром и декантируют. Получающееся в результате масло затем обрабатывают теплым толуолом и декантируют с получением 9,07 г светло-коричневого масла. Колоночная хроматография с использованием 1% МеОH в EtOAc дает 7,9 г (47,53 ммоль) сульфона в виде светло-желтого масла.

К 7,9 г (47,53 ммоль) спирта в 125 мл ацетона при 0°C добавляют по каплям через капельную воронку 20 мл (54 ммоль, 1,1 эквивалент) свежеприготовленного 2,7 М реагента Джонса. Реагент Джонса получают осторожным растворением 5,34 г триоксида хрома в 4,6 мл концентрированной серной кислоты и затем осторожным разбавлением водой до 20 мл общего объема. Охлаждающую баню удаляют и получающуюся в результате суспензию перемешивают при комнатной температуре в течение ночи. Реакционную суспензию затем разбавляют 200 мл золы и экстрагируют 4×200 мл EtOAc. Органические слои отделяют, объединяют, промывают 2×200 мл воды, сушат над MgSO4, фильтруют и концентрируют в вакууме с получением белого остатка. Остаток растирают со смесью эфир/этилацетат и фильтруют с получением 4,67 г (28,44 ммоль) требуемого продукта в виде белого твердого вещества.
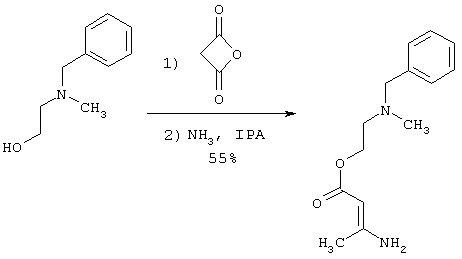
Раствор 25,43 г (153,9 ммоль) N-бензил-N-метилэтаноламина и 0,2 мл триэтиламина нагревают на масляной бане до 60°С. По каплям через капельную воронку добавляют 13,6 г (161,79 ммоль) дикетена при поддержании температуры взаимодействия между 60-85°С. После завершения добавления дикетена реакционную смесь перемешивают еще 30 минут, охлаждают до комнатной температуры и затем охлаждают на ледяной бане. Добавляют 20 мл 2-пропанола. В течение 2 ч через реакционную смесь барботируют газообразный аммиак. Оранжевую реакционную смесь закрывают пробкой и оставляют ее на ночь при 5°С. Реакционную смесь затем перемешивают на ледяной бане и добавляют 10 мл гептана. Начинает образовываться осадок. Спустя один час реакционную суспензию фильтруют и осадок промывают 3×40 мл 10% объем/объем смеси 2-пропанол/гептан с получением 21,15 г (85,17 ммоль) белого твердого вещества.
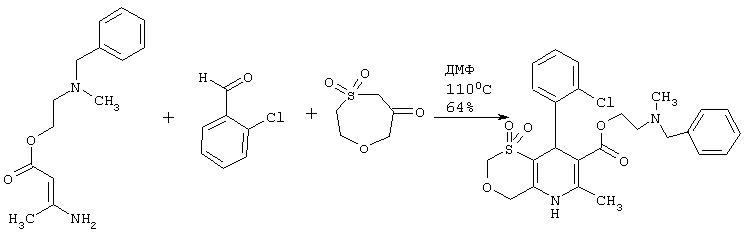
2,4 г (14,62 ммоль) циклического простого β-кетосульфонэфира, 2,06 г (14,62 ммоль) 2-хлорбензальдегида и 3,63 г (14,62 ммоль) 2-(N-бензил-N-метиламино)этил-3-амино-кротоната нагревают до 110°С в 50 мл ДМФ в течение 3,5 час. Реакционную смесь затем охлаждают, разбавляют 500 мл ЕtОАс, промывают 4×200 мл воды, 1×100 мл насыщенного раствора соли, сушат над Na2SO4, фильтруют и концентрируют в вакууме с получением коричневого масла. Колоночная хроматография с использованием смеси 3:2 EtOAc/гексан дает 4,97 г (9,42 ммоль) требуемого продукта (соединение 3) в виде желтой пены.
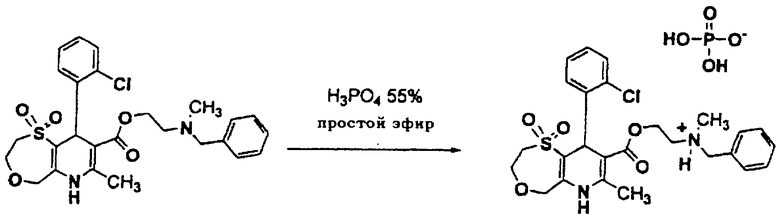
4,87 г (9,42 ммоль) дигидропиридина растворяют в 150 мл эфира, содержащего небольшое количество этилацетата. Через капельную воронку в течение 90 минут добавляют 1,08 г (9,42 ммоль) 85% ортофосфорной кислоты в 75 мл эфира. Получающуюся в результате белую суспензию перемешивают в течение 4 ч и затем фильтруют. Получающееся в результате белое твердое вещество промывают избытком эфира и сушат с получением 2,68 г (5,18 ммоль) фосфатной соли.
В. Анализы
Пример 2
Анализ на ингибирование связывания нитрендипина
Самок Новозеландских белых кроликов (1-2 кг) умерщвляют цервикальным смещением и сразу же извлекают сердце, очищают и нарезают на мелкие кусочки. Ткань гомогенизируют в 5х- объеме 0,05 буфера Hереs, рН 7,4. Гомогенат центрифугируют при 4000 г в течение 10 минут и супернатант повторно центрифугируют при 42000 g в течение 90 минут. Получающийся мембранный осадок снова суспендируют (0,7 мл/г массы) в 0,05 М Hереs, рН 7,4 и хранят при 70°С до использования. Каждая пробирка для анализа связывания содержит 3H-нитрендипин (0,05-0,50 нМ), буфер, мембраны (0,10 мл) и испытуемое соединение в общем объеме 1,0 мл. После выдерживания 90 минут при 4°С связанный нитрендипин отделяют от несвязанного фильтрованием на фильтрах Whatman GF/C. После прополаскивания фильтры сушат и осуществляют подсчет с использованием жидкостного сцинтилляционного счетчика.
Неспецифически связанный 3H-нитрендипин (т.е. количество, связанное в присутствии избыточного немеченого нитрендипина) вычитают из общего связанного количества для получения специфически связанного меченого радиоактивным изотопом нитрендипина. Количество специфически связанного нитрендипина в присутствии испытуемого соединения сравнивают с количеством связанного нитрендипина в отсутствии соединения. Затем можно получить процент вытеснения (или ингибирования).
Пример 3
Испытание на ингибирование кальцийзависимого сокращения гладкой мышцы
Трахею и аорту собак, умерщвленных инъекцией избытка КСl, хранят в течение ночи при 4°С в оксигенированном буфере Кребса-Хенселейта. Трахеальные кольца, ширина одного сегмента хряща (5-10 мм), разрезают, начиная с бронхиального конца. Приготавливают также кольца ткани аорты такой же ширины. После нарезки хряща ткань мышцы трахеи и ткань аорты суспендируют в оксигенированном буфере Кребса-Хенселейта при 37°С в 25 мл ванне для ткани. После 60-минутного периода установления равновесия в ткани вводят 10 мкМ карбахол. Спустя 5 минут ткани споласкивают и оставляют в покое на 50 минут. Ткани затем обрабатывают 50 мМ КСl и спустя 30 минут количественно оценивают сокращения. Ткани затем споласкивают и снова приводят в равновесное состояние в течение 50 минут. Затем в течение 10 минут добавляют испытуемые соединения и ткань снова обрабатывают 50 мМ КСl. Через 30 минут регистрируют сокращение и используют для определения % ингибирования контроля. Процентное ингибирование сокращения гладкой мышцы вычисляют следующим образок по данным ответной реакции до и после обработки лекарственным средством:

В приведенной ниже таблице 1 представлены данные масс-спектров, ингибирование связывания нитрендипина и ингибирование кальцийзависимого сокращения гладкой мышцы в виде процента ингибирования для выбранных соединений формулы I.
В таблице 2, представленной ниже, приводятся данные масс-спектров и ингибирования связывания нитрендипина для выбранных соединений формулы IIа.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ДИТИЕПИНО[6,5-B]ПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ИНГИБИРОВАНИЯ И ЛЕЧЕНИЯ РАССТРОЙСТВ | 2000 |
|
RU2230746C2 |
| ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА У МЛЕКОПИТАЮЩЕГО | 2000 |
|
RU2263109C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2277909C2 |
| Бициклические производные пиридина, полезные в качестве ингибитора белков, связывающих жирные кислоты (FABP) 4 и/или 5 | 2013 |
|
RU2648247C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИЙ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2290179C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ | 2001 |
|
RU2286343C2 |
| НИТРАТНЫЕ СОЛИ ГИПОТЕНЗИВНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 1999 |
|
RU2235097C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ПРИМЕНЕНИЕ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ КАТЕПСИНА S | 2001 |
|
RU2317988C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ КАТЕПСИНА S | 2001 |
|
RU2278863C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
Настоящее изобретение относится к новым оксатиепино[6,5-b]дигидропиридинам формулы I
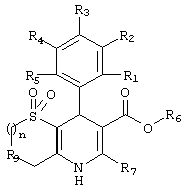
в которой
(a) R1, R2, R3, R4 и R5, независимо, выбраны из группы, состоящей из Н, галогена, NO2;
(b) R6 выбран из группы, состоящей из неразветвленного или разветвленного С1-5-алкила, причем указанный алкил может быть замещен фенилацетилокси, гидрокси, карбоалкокси или группой NR'R", в которой
R' и R", независимо, выбраны из группы, состоящей из Н, неразветвленного или разветвленного C1-8-алкила, бензила;
(c) R7 выбран из группы, состоящей из Н, алкила;
(d) R9 представляет кислород; и
(e) n равно целому числу от 1 до 2;
или его фармацевтически приемлемая соль.
Соединения полезны в качестве антагонистов кальциевых каналов и обладают сердечно-сосудистой, антиастматической и антибронхосужающей активностью. Описана также фармацевтическая композиция. 3 н. и 25 з.п. ф-лы, 2 табл.
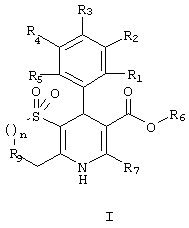
в которой
(a) R1, R2, R3, R4 и R5 независимо выбраны из группы, состоящей из Н, галогена, NО2;
(b) R6 выбран из группы, состоящей из неразветвленного или разветвленного C1-5-алкила, причем указанный алкил может быть замещен фенилацетилокси, гидрокси, карбоалкокси или группой NR'R", в которой R' и R" независимо выбраны из группы, состоящей из Н, неразветвленного или разветвленного C1-8-алкила, бензила;
(c) R7 выбран из группы, состоящей из Н, алкила;
(d) R9 представляет кислород и
(e) n равно целому числу от 1 до 2;
или его фармацевтически приемлемая соль.
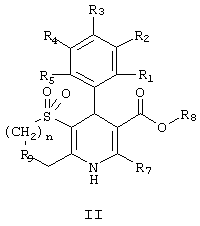
в которой
(a) R1, R2, R3, R4 и R5 независимо выбраны из группы, состоящей из водорода, галогена, NО2;
(b) R7 выбран из группы, состоящей из Н, алкила;
(c) R8 выбран из группы, состоящей из -алкил- ОН, -алкил-ОС(O)R’, в котором R' выбран из группы, состоящей из алкила, фенила;
(d) R9 представляет кислород.
| ПРОИЗВОДНЫЕ АНЕЛЛИРОВАННОГО ДИГИДРОПИРИДИНА ИЛИ ИХ СОЛИ С ФИЗИОЛОГИЧЕСКИ ПЕРЕНОСИМЫМИ КИСЛОТАМИ И СРЕДСТВО, БЛОКИРУЮЩЕЕ НЕСЕЛЕКТИВНЫЕ КАТИОННЫЕ КАНАЛЫ | 1993 |
|
RU2127736C1 |
| Станок для разрезания штучного проката | 1973 |
|
SU462696A1 |
| ВИБРОБУНКЕР | 0 |
|
SU241281A1 |

