Область техники
Настоящее изобретение относится к промежуточным соединениям, которые полезны при получении антибактериальных соединений, и способам их получения. Предшествующий уровень техники
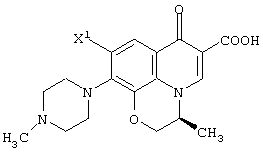
(3S)-(-)-9-Фтор-3-метил-10-(4-метил-1-пиперазинил)-7-оксо-2,3-дигидро-7Н-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота (левофлаксацин, LVFX: JP-A-62-252790, термин "JP-A", используемый в данном описании, означает не прошедшую экспертизу опубликованную заявку на патент Японии)
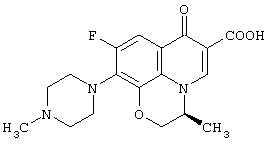
известна в качестве превосходного синтетического антибактериального агента.
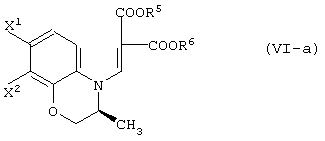
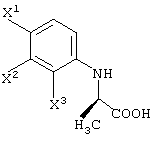
В качестве промежуточных соединений при получении данного левофлоксацина также полезны соединения, представленные формулой (VI-a) (далее называемые здесь соединениями (VI-a);
то же самое применимо к соединениям, представленным другими формулами):
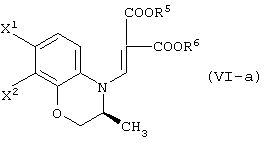
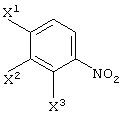
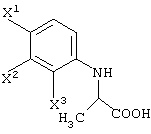
(в которой Х1 и X2, каждый независимо, представляет атом галогена).
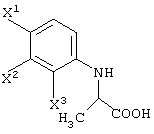
В качестве промежуточных соединений для рацемической 9-фтор-3-метил-10-(4-метил-1-пиперазинил)-7-оксо-2,3-дигидро-7Н-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновой кислоты (офлаксацин, OFLX):
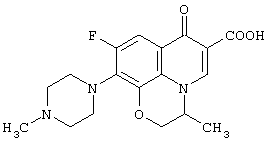
полезны соединения, представленные формулой (VI):
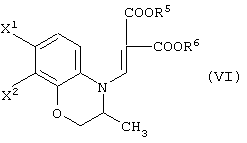
(в которой X1 и X2, каждый независимо, представляет атом галогена, и R5 и R6, каждый независимо, представляет алкильную группу). Обычными способами получения соединения (VI-а) являются следующие.

Способ получения, о котором сообщается в патенте Японии №2612327, показанный выше на схеме, страдает в результате проблемы возникновения эпимеризации в основных или кислых условиях и таким образом выход оптически активного (R)-NPNB снижается.
В способе, о котором сообщается в патенте Японии №2771871, который является способом микробного восстановления, трудности заключаются в очистке продукта, поскольку физические свойства продукта не сильно отличаются от свойств исходного вещества.
Далее, способ, о котором сообщается в патенте Японии №2573269, требует значительного усовершенствования для использования в качестве промышленного процесса, поскольку в качестве восстанавливающего агента здесь используют дорогой асимметрический ацилоксиборгидрид щелочного металла.
В способе оптического разделения, о котором сообщается в JP-B-7-20946 (термин "JP-B", используемый в данном описании, означает «публикацию прошедшей экспертизу заявки на патент Японии), кроме того, возникает потребность в рассмотрении вопроса повторного использования ненужного изомера, который образуется теоретически в 50% соотношении.
Способ получения, о котором сообщается в патенте США 5644056, относится к реакции рацемата. Следовательно, для получения левофлоксацина данным способом требуется оптически разделить полученный продукт, а ненужный изомер следует рацемизировать или инвертировать. Кроме того, в описании данного патента нет никаких экспериментальных примеров получения оптически активного соединения.
Способ, о котором сообщается в китайском документе (Chinese Chemical Letters, Vol.6, No.10, 57-860 (1995)), страдает в результате той проблемы, что необходима дополнительная стадия снятия используемой в качестве защитной группы п-толуолсульфонилоксигруппы.
Описание изобретения
Настоящее изобретение относится к способам, с помощью которых соединение (VI-a), важное в качестве промежуточного соединения при получении левофлоксацина, может экономически синтезироваться в течение короткого времени, и которые, таким образом, представляют собой промышленно благоприятные способы получения. В результате интенсивных исследований авторы настоящего изобретения установили, что цель может быть достигнута с помощью получения промежуточного соединения левофлоксацина в соответствии со следующими путями синтеза, завершив, таким образом, настоящее изобретение. На следующей схеме показаны способы получения соединения (VI) из соединения (I) согласно настоящему изобретению.
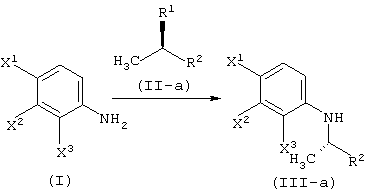
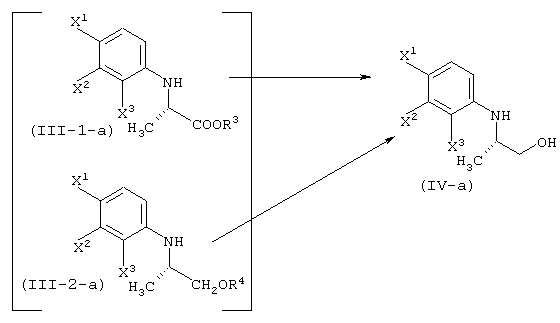
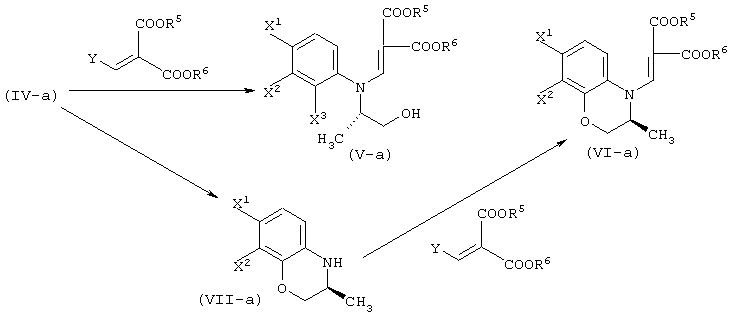 Соответственно, настоящее изобретение предоставляет процессы промышленно выгодного получения соединения, представленного формулой (VI-a), которое является промежуточным соединением для промышленно выгодного получения левофлоксацина:
Соответственно, настоящее изобретение предоставляет процессы промышленно выгодного получения соединения, представленного формулой (VI-a), которое является промежуточным соединением для промышленно выгодного получения левофлоксацина:
А именно, настоящее изобретение относится к следующим способам.
Способ А:
Способ, включающий реакцию соединения, представленного формулой (I):
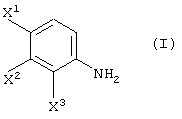
с соединением, представленным формулой (II-1-a), в присутствии основания:
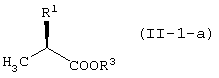
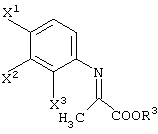
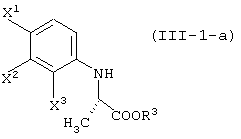
давая соединение, представленное формулой (III-1-а):
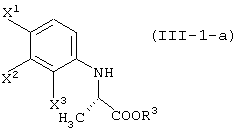
восстановление данного соединения в соединение, представленное формулой (IV-a):
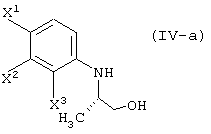
реакцию данного соединения с соединением, представленным следующей формулой:

давая соединение, представленное формулой (V-a):
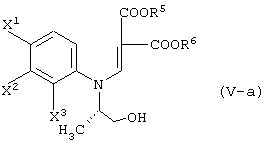
а затем обработку данного соединения в присутствии основания.
Способ В
Способ, включающий реакцию соединения, представленного формулой (I):
с соединением, представленным формулой (II-2-a), в присутствии основания:
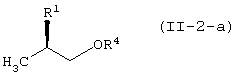
давая соединение, представленное формулой (III-2-a):
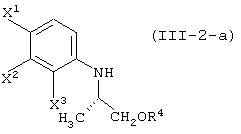
элиминирование защитной группы гидроксила (заместитель R4) данного соединения, с получением соединения, представленного формулой (IV-a):
взаимодействие данного соединения с соединением, представленным следующей формулой:
давая соединение, представленное формулой (V-a):
а затем обработку данного соединения в присутствии основания.
Способ С:
Способ, включающий реакцию соединения, представленного формулой (I):
с соединением, представленным формулой (II-1-a), в присутствии основания
давая соединение, представленное формулой (III-1-a):
восстановление данного соединения в соединение, представленное формулой (IV-a):
обработку данного соединения в присутствии основания с получением соединения, представленного формулой (VII-a):
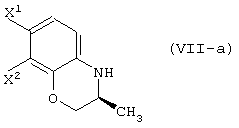
и реакцию данного соединения с соединением, представленным следующей формулой:
Способ D:
Способ, включающий реакцию соединения, представленного формулой (I):
с соединением, представленным формулой (II-2-a), в присутствии основания:
давая соединение, представленное формулой (III-2-a):
элиминирование защитной группы гидроксила (заместитель R4) данного соединения, с получением соединения, представленного формулой (IV-a):
обработку данного соединения в присутствии основания с получением соединения, представленного формулой (VII-a):
а затем взаимодействие данного соединения с соединением, представленным следующей формулой:
Способ Е:
Способ, включающий реакцию соединения, представленного формулой (I):
с соединением, представленным формулой (II-1), в присутствии основания:
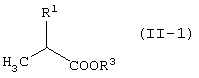
давая соединение, представленное формулой (III-1):
а затем подвергают данное соединение следующим методам 1 или 2;
Метод 1:
в случае соединения, представленного формулой (III-1), в которой R3 не является атомом водорода, метод включает обработку данного соединения ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, и после завершения данной обработки выделение продукта из обработанной жидкой смеси;
Метод 2:
в случае соединения, представленного формулой (III-1), в которой R3 представляет атом водорода, метод, который включает оптическое разделение данного соединения по реакции с оптически активным органическим основанием;
с получением соединения карбоновой кислоты, представленного следующей формулой:
этерификацию данного соединения в присутствии спирта, представленного следующей формулой:
R7-OH
с получением сложноэфирного соединения, представленного следующей формулой:
восстановление данного соединения в соединение, представленное формулой (IV-a):
реакцию данного соединения с соединением, представленным следующей формулой:
давая соединение, представленное формулой (V-a):
а затем обработку данного соединения в присутствии основания.
Способ F:
Способ, включающий реакцию соединения, представленного формулой (I):
с соединением, представленным формулой (II-1), в присутствии основания:
давая соединение, представленное формулой (III-1):
а затем подвергают данное соединение следующим методам 1 или 2;
Метод 1:
в случае соединения, представленного формулой (III-1), в которой R3 не является атомом водорода, метод включает обработку данного соединения ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, и после завершения данной обработки выделение продукта из обработанной жидкой смеси;
Метод 2:
в случае соединения, представленного формулой (III-1), в которой R3 представляет атом водорода, метод включает оптическое разделение данного соединения по реакции с оптически активным органическим основанием;
с получением соединения карбоновой кислоты, представленного следующей формулой:
этерификацию данного соединения в присутствии спирта, представленного следующей формулой:
R7-OH
с получением сложноэфирного соединения, представленного следующей формулой:
восстановление данного соединения в соединение, представленное формулой (IV-a):
обработку данного соединения в присутствии основания, давая соединение, представленное формулой (VII-a):
а затем реакцию данного соединения с соединением, представленным следующей формулой:
Способ G:
Способ, включающий реакцию соединения, представленного следующей формулой:
или следующей формулой:
с соединением, представленным следующей формулой, в присутствии металлического катализатора в атмосфере газообразного водорода, необязательно в присутствии дегидратирующего агента или кислоты:
СН3COCOOR3,
давая соединение, представленное формулой (III-1):
а затем подвергают данное соединение следующим методам 1 или 2;
Метод 1:
в случае соединения, представленного формулой (III-1), в которой R3 не является атомом водорода, метод включает обработку данного соединения ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, и после завершения данной обработки выделение продукта из обработанной жидкой смеси.
Метод 2:
в случае соединения, представленного формулой (III-1), в которой R3 представляет атом водорода, метод включает оптическое разделение данного соединения по реакции с оптически активным органическим основанием;
с получением соединения карбоновой кислоты, представленного следующей формулой:
этерификацию данного соединения в присутствии спирта, представленного следующей формулой:
R7-OH
с получением сложноэфирного соединения, представленного следующей формулой:
восстановление данного соединения в соединение, представленное формулой (IV-a):
взаимодействие данного соединения с соединением, представленным следующей формулой:
давая соединение, представленное формулой (V-a):
а затем обработку данного соединения в присутствии основания.
Способ Н:
Способ, включающий реакцию соединения, представленного следующей формулой:
или следующей формулой:
с соединением, представленным следующей формулой, в присутствии металлического катализатора в атмосфере газообразного водорода, необязательно в присутствии дегидратирующего агента или кислоты:
СН3COCOOR3,
давая соединение, представленное формулой (III-1):
а затем подвергают данное соединение следующим методам 1 или 2.
Метод 1:
в случае соединения, представленного формулой (III-1), в которой R3 не является атомом водорода, метод включает обработку данного соединения ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, и после завершения данной обработки выделение продукта из обработанной жидкой смеси.
Метод 2:
в случае соединения, представленного формулой (III-1), в которой R3 представляет атом водорода, метод включает оптическое разделение данного соединения по реакции с оптически активным органическим основанием;
с получением соединения карбоновой кислоты, представленного следующей формулой:
этерификацию данного соединения в присутствии спирта, представленного следующей формулой:
R7-OH
с получением сложноэфирного соединения, представленного следующей формулой:
восстановление данного соединения в соединение, представленное формулой (IV-a):
обработку данного соединения в присутствии основания, давая соединение, представленное формулой (VII-a):
а затем реакцию данного соединения с соединением, представленным следующей формулой:
Способ I
Способ, включающий реакцию соединения, представленного следующей формулой:
с соединением, представленным следующей формулой:
СН3COCOOR3,
давая соединение, представленное следующей формулой:
асимметрическое восстановление данного соединения в соединение, представленное формулой (III-1-a):
восстановление данного соединения в соединение, представленное формулой (IV-a):
реакцию данного соединения с соединением, представленным следующей формулой:
давая соединение, представленное формулой (V-a):
а затем обработку данного соединения в присутствии основания.
Способ J:
Способ, включающий реакцию соединения, представленного следующей формулой:
с соединением, представленным следующей формулой:
СН3COCOOR3,
давая соединение, представленное следующей формулой:
асимметрическое восстановление данного соединения в соединение, представленное формулой (III-1-a):
восстановление данного соединения в соединение, представленное формулой (IV-a):
обработку данного соединения в присутствии основания, давая соединение, представленное формулой (VII-а):
а затем реакцию данного соединения с соединением, представленным следующей формулой:
[в каждой из вышеуказанных формул, X1, X2 и X3, каждый независимо, представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила;
R5 и R6, каждый независимо, представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила; и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными, и каждая представляет собой алкильную группу, содержащую 1-6 атомов углерода); и заместители, которые будут использованы далее, имеют, соответственно, такие же значения, как определено выше].
Настоящее изобретение дополнительно относится к следующим способам, составляющим каждый из способов, как описано выше.
Относительно способов получения соединения, представленного формулой (III-a) в способах G и Н;
способ получения соединения, представленного формулой (III-1)
отличающийся реакцией соединения, представленного формулой (I-0):
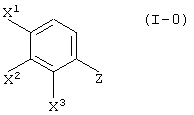
(где Z представляет нитрогруппу или аминогруппу; а другие группы являются такими, как определено выше)
с соединением, представленным следующей формулой:
СН3COCOOR3,
необязательно в присутствии акцептора кислоты или кислоты, в присутствии металлического катализатора в атмосфере газообразного водорода;
вышеуказанный способ получения соединения, представленного формулой (III-1), где R3 представляет атом водорода;
вышеуказанный способ получения соединения, представленного формулой (III-1), где R3 представляет метильную группу;
вышеуказанный способ получения соединения, представленного формулой (III-1), где R3 представляет этильную группу;
вышеуказанный способ получения соединения, представленного формулой (III-1), где Z представляет аминогруппу;
вышеуказанный способ получения соединения, представленного формулой (III-1), где Z представляет нитрогруппу;
вышеуказанный способ получения соединения, представленного формулой (III-1), где Z представляет аминогруппу и R3 представляет атом водорода;
вышеуказанный способ получения соединения, представленного формулой (III-1), где Z представляет аминогруппу и R3 представляет метильную группу;
вышеуказанный способ получения соединения, представленного формулой (III-1), где Z представляет аминогруппу и R3 представляет этильную группу;
вышеуказанный способ получения соединения, представленного формулой (III-1), где Z представляет нитрогруппу и R3 представляет атом водорода;
вышеуказанный способ получения соединения, представленного формулой (III-1), где Z представляет нитрогруппу и R3 представляет метильную группу;
вышеуказанный способ получения соединения, представленного формулой (III-1), где Z представляет нитрогруппу и R представляет этильную группу.
Относительно способов, включающих выделение единственного оптического изомера в способах Е, F, G и Н;
способ получения соединения карбоновой кислоты, представленной следующей формулой:
отличающийся обработкой сложноэфирного соединения в ряду соединений, представленных формулой (III-1):
ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, а затем выделение продукта из обработанной жидкой среды;
способ получения соединения карбоновой кислоты, представленной следующей формулой:
отличающийся обработкой сложноэфирного соединения в ряду соединений, представленных формулой (III-1):
ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, а затем удаление соединения, представленного формулой (III-1-b) из обработанной жидкой среды;
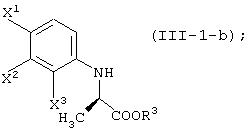
способ получения сложноэфирного соединения среди соединений, представленных формулой (III-1-a):
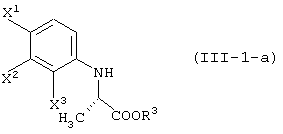
отличающийся обработкой сложноэфирного соединения в ряду соединений, представленных формулой (III-1):
ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, а затем выделение соединения из обработанной жидкой среды;
способ получения сложноэфирного соединения среди соединений, представленных формулой (III-1-a):
отличающийся обработкой сложноэфирного соединения в ряду соединений, представленных формулой (III-1):
ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, а затем удаление соединения карбоновой кислоты, представленного следующей формулой из обработанной жидкой среды;
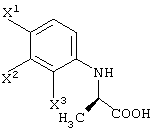
каждый из вышеописанных способов получения, где R3 представляет метильную группу;
каждый из вышеописанных способов получения, где R3 представляет этильную группу;
каждый из вышеописанных способов получения, где используемый при обработке фермент представляет собой эстеразу, протеазу или химотрипсин;
каждый из описанных выше способов получения, где микроорганизм представляет собой микроорганизм, выбранный из бактерий, принадлежащих к роду Bacillus, Micrococcus и Actinomyces:
каждый из описанных выше способов получения, где микроорганизм представляет собой микроорганизм, выбранный из грибков, принадлежащих к роду Aspergillus, Rhizopus, Nannizia и Penicillium; и
каждый из описанных выше способов получения, где микроорганизм представляет собой микроорганизм, выбранный из дрожжей, принадлежащих к роду Candida, Saccharomyces и Zygoascus.
Относительно способов, включающих выделение единственного оптического изомера в способах Е, F, G и Н;
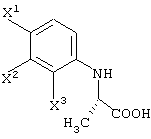
способ получения 2-(2,3,4-тригалогенанилино)пропионовой кислоты, состоящей из единственного оптического изомера, отличающийся оптическим разделением соединения, представленного следующей формулой:
с использованием оптически активного органического основания;
способ получения 2-(2,3,4-тригалогенанилино)пропионовой кислоты, состоящей из единственного оптического изомера, отличающийся обработкой соединения, представленного следующей формулой:
оптически активным органическим основанием, давая диастереомерную соль одного из оптических изомеров 2-(2,3,4-тригалогенанилино)пропионовой кислоты и оптически активного органического основания, а затем обработкой данной диастереомерной соли кислотой;
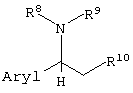
вышеописанные способы получения единственного оптического изомера, где оптически активное органическое основание представляет собой соединение, представленное следующей формулой:
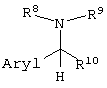
(где Aryl представляет арильную группу, необязательно содержащую атом галогена, нитрогруппу, цианогруппу, карбамоильную группу, алкильную группу, содержащую 1-6 атомов углерода, или алкоксигруппу, содержащую 1-6 атомов углерода; и
R8, R9 и R10, каждый независимо, представляют собой:
(1) фенильную группу, необязательно содержащую атом галогена, алкильную группу, содержащую 1-6 атомов углерода, галогеналкильную группу, имеющую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, нитрогруппу, карбамоильную группу или цианогруппу;
(2) бензильную группу, необязательно содержащую атом галогена, алкильную группу, содержащую 1-6 атомов углерода, галогеналкильную группу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, нитрогруппу, карбамоильную группу или цианогруппу;
(3) алкильную группу, содержащую 1-6 атомов углерода;
или
(4) атом водорода);
вышеописанные способы получения единственного оптического изомера, где оптически активное органическое основание представляет собой 1-фенилэтиламин;
вышеописанные способы получения единственного оптического изомера, где оптически активное органическое основание представляет собой 1-(п-толил)этиламин; и
вышеописанные способы получения единственного оптического изомера, где оптически активное органическое основание представляет собой 1-фенил-2-(толил)этиламин.
Относительно способов получения, включающих выделение единственного оптического изомера в способах Е, F, G и Н;
способ получения сложноэфирного соединения, представленного следующей формулой:
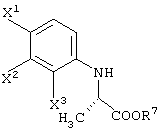
отличающийся обработкой соединения карбоновой кислоты, представленного следующей формулой:

в присутствии соединения, представленного следующей формулой:
R7-OH
и кислотного катализатора; и
способ получения сложноэфирного соединения, представленного следующей формулой:
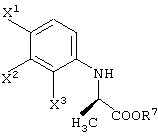
отличающийся обработкой соединения карбоновой кислоты, представленного следующей формулой:
в присутствии соединения, представленного следующей формулой:
R7-OH
и кислотного катализатора.
Относительно способов получения, включающих выделение единственного оптического изомера в способах Е, F, G и Н;
способ получения сложноэфирного соединения в виде рацемата, представленного формулой (III-1):
отличающийся обработкой сложноэфирного соединения в ряду соединений, представленных формулой (III-1-b):
в присутствии основания;
способ получения сложноэфирного соединения, как описано выше, где основание представляет собой азотсодержащее гетероциклическое соединение;
способ получения сложноэфирного соединения, как описано
выше, где основание представляет собой 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ) или 1,8-диазабицикло[4.3.0]ундец-5-ен (ДБН);
способ получения сложноэфирного соединения, как описано выше, где основание представляет собой карбонат щелочного металла или щелочноземельного металла; и
способ получения сложноэфирного соединения, как описано выше, где основание представляет собой карбонат калия.
Относительно способов получения, включающих выделение единственного оптического изомера в способах Е, F, G и Н;
способ получения рацемического соединения карбоновой кислоты, представленного следующей формулой:
отличающийся рацемизацией сложноэфирного соединения в ряду соединений, представленных следующей формулой (III-1-b), обработкой в присутствии основания:
а затем гидролизом;
способ получения рацемического соединения карбоновой кислоты, как описано выше, где основание представляет собой алкоксид металла;
способ получения рацемического соединения карбоновой кислоты, как описано выше, где основание представляет собой третичный-бутоксид калия;
способ получения рацемического соединения карбоновой кислоты, как описано выше, где основание представляет собой карбонат щелочного металла или щелочноземельного металла;
способ получения рацемического соединения карбоновой кислоты, как описано выше, где основание представляет собой карбонат калия.
Относительно способов получения соединения (V-a) в способах А, В, Е, G и I;
способ получения соединения, представленного формулой (V-a):
отличающийся реакцией соединения, представленного формулой (IV-a):
с соединением, представленным следующей формулой, в основных условиях:
Относительно способов получения соединения (VI-a) в способах А, В, Е, G и I;
способ получения соединения, представленного формулой (VI-a):
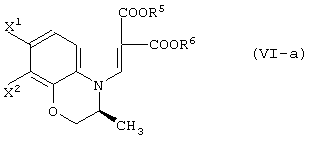
отличающийся реакцией соединения, представленного формулой (V-a):
в основных условиях;
способ получения соединения, представленного формулой (VI-a), как описано выше, где основные условия представляют собой основные условия при совместном существовании основания и катализатора межфазного переноса;
способ получения соединения, представленного формулой (VI-a), как описано выше, где основание представляет собой гидроксид щелочного металла или гидроксид щелочноземельного металла;
способ получения соединения, представленного формулой (VI-a), как описано выше, где основание представляет собой гидроксид калия;
способ получения соединения, представленного формулой (VI-a), как описано выше, где катализатор межфазного переноса представляет собой четвертичную соль аммония или краун-эфир;
способ получения соединения, представленного формулой (VI-a), как описано выше, где катализатор межфазного переноса представляет собой четвертичную соль аммония;
способ получения соединения, представленного формулой (VI-a), как описано выше, где четвертичная соль аммония представляет собой хлорид тетра(нормальный-гексил)аммония, хлорид триметилбензиламмония, хлорид триэтилбензиламмония, хлорид триметилфениламмония или гидросульфат тетрабутиламмония.
Относительно стадий восстановления сложноэфирного соединения в способах А, С, Е, F, G, Н, I и J;
способ получения соединения, представленного формулой (IV-a):
отличающийся обработкой соединения, представленного формулой (III-1-a):
или соединения, представленного следующей формулой:
в апротонном растворителе соединением боргидрида металла и спиртом;
способ получения соединения, представленного формулой (IV-a), как описано выше, где соединение, представленное формулой (III-1-a), представляет собой сложноэфирное соединение;
способ получения соединения, представленного формулой (IV-a), как описано выше, где R3 и R7, каждый, представляет алкильную группу, содержащую от 1 до 6 атомов углерода;
способ получения соединения, представленного формулой (IV-a), как описано выше, где R3 и R7, каждый, представляет метильную группу;
способ получения соединения, представленного формулой (IV-a), как описано выше, где R3 и R7, каждый, представляет этильную группу;
каждый из способов получения соединения, представленного формулой (IV-a), как описано выше, где апротонный растворитель представляет собой растворитель, выбранный из соединений группы, состоящей из ароматических углеводородов, алканов, циклоалканов, простых эфиров, галогенированных углеводородов и эфиров уксусной кислоты;
каждый из способов получения соединения, представленного формулой (IV-a), как описано выше, где апротонный растворитель представляет собой ароматический углеводород;
каждый из способов получения соединения, представленного формулой (IV-a), как описано выше, где апротонный растворитель представляет собой алкан;
каждый из способов получения соединения, представленного формулой (IV-a), как описано выше, где апротонный растворитель представляет собой циклоалкан;
каждый из способов получения соединения, представленного формулой (IV-a), как описано выше, где апротонный растворитель представляет собой простой эфир;
каждый из способов получения соединения, представленного формулой (IV-a), как описано выше, где апротонный растворитель представляет собой галогенированный углеводород;
каждый из способов получения соединения, представленного формулой (IV-a), как описано выше, где апротонный растворитель представляет собой эфир уксусной кислоты;
каждый из способов получения соединения, представленного формулой (IV-a), как описано выше, где спирт представляет собой первичный спирт;
каждый из способов получения соединения, представленного формулой (IV-a), как описано выше, где первичный спирт представляет собой метанол;
каждый из способов получения соединения, представленного формулой (IV-a), как описано выше, где соединение боргидрида металла представляет собой боргидрид натрия; и
каждый из способов получения соединения, представленного формулой (IV-a), как описано выше, где X1, X2 и X3, каждый, представляет собой атом фтора.
Относительно стадий восстановления сложноэфирного соединения в способах А, В, Е, G и I;
способ получения соединения, представленного формулой
отличающийся реакцией соединения, представленного формулой (III-1-a):
или соединения, представленного следующей формулой:
с соединением боргидрида металла в апротонном растворителе в присутствии спирта, давая соединение, представленное формулой (IV-a):
затем реакцией данного соединения с соединением, представленным следующей формулой, в основных условиях:
давая соединение, представленное формулой (V-a)
и обработкой данного соединения в основных условиях. Кроме того, настоящее изобретение относится к следующим соединениям, касающимся указанных выше способов и стадий.
Соединения, представленные формулой (III-1):
(где X1, X2 и X3, каждый, независимо, представляет атом галогена; и R3 представляет атом водорода или защитную группу карбоксила) ;
соединения, представленные формулой (III-1-a):
(где X1, X2 и X3, каждый, независимо, представляет атом галогена; и R3 представляет атом водорода или защитную группу карбоксила);
соединения, представленные формулой (III-1-b):
(где X1, X2 и X3, каждый, независимо, представляет собой атом галогена; и R3 представляет атом водорода или алкильную группу) ;
каждое из соединений формулы (III-1), (III-1-a) или (III-1-b), где R3 представляет атом водорода;
каждое из соединений формулы (III-1), (III-1-a) или (III-1-b), где R3 представляет метильную группу;
каждое из соединений формулы (III-1), (III-1-a) или (III-1-b), где R3 представляет этильную группу;
соединения, представленные формулой (V):
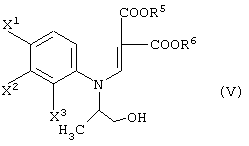
соединения, представленные формулой (V-a):
каждое из соединений формулы (III-1), (III-1-a), (III-1-b), (V) или (V-a), где X1, X2 и X3, каждый, представляет собой атом фтора;
соли соединений карбоновых кислот, представленных следующей формулой:
с оптически активным органическим основанием;
соли соединений, представленных следующей формулой:
с оптически активным органическим основанием;
вышеописанные соли, в которых оптически активное органическое основание представляет собой соединение, представленное следующей формулой:
(где Aryl представляет арильную группу, необязательно содержащую атом галогена, нитрогруппу, цианогруппу, карбамоильную группу, алкильную группу, содержащую 1-6 атомов углерода, или алкоксигруппу, содержащую 1-6 атомов углерода; и
R8, R9 и R10, каждый независимо, представляет:
(1) фенильную группу, необязательно содержащую атом галогена, алкильную группу, содержащую 1-6 атомов углерода, галогеналкильную группу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, нитрогруппу, карбамоильную группу или цианогруппу;
(2) бензильную группу, необязательно содержащую атом галогена, алкильную группу, содержащую 1-6 атомов углерода, галогеналкильную группу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, нитрогруппу, карбамоильную группу или цианогруппу;
(3) алкильную группу, содержащую 1-6 атомов углерода;
или
(4) атом водорода);
вышеописанные соли, где оптически активное основание представляет собой 1-фенилэтиламин;
вышеописанные соли, где 1-фенилэтиламин представляет собой (R)-(+)-1-фенилэтиламин;
вышеописанные соли, где оптически активное органическое основание представляет собой 1-(п-толил)этиламин;
вышеописанные соли, где 1-(п-толил)этиламин представляет собой (R)-(+)-(п-толил)этиламин;
вышеописанные соли, где оптически активное органическое основание представляет собой 1-фенил-2-(п-толил)этиламин;
вышеописанные соли, где 1-фенил-2-(п-толил)этиламин представляет собой (S)-(+)-1-фенил-2-(п-толил)этиламин;
каждая из вышеописанных солей, где X1, X2 и X3, каждый, представляет атом фтора.
Настоящее изобретение, кроме того, относится к способу получения соединения (левофлоксацина), представленного следующей формулой:
с использованием соединения, представленного формулой (IV-a), которое было получено с помощью любого из способов и любого из соединений, как описано выше, отличающемуся тем, что включает следующие стадии получения соединения, представленного формулой (VI-a), по любому из способов A-J:
обработку данного соединения соединением трехфтористого бора с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
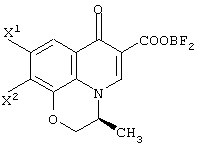
реакцию данного соединения с 4-метилпиперазином, дающую соединение, представленное следующей формулой:
а затем отщепление хелата бора в данном соединении.
Настоящее изобретение, кроме того, относится к следующим способам получения:
способу получения левофлоксацина, как описано выше, где способ А используют в качестве способа получения соединения, представленного формулой (VI-a);
способу получения левофлоксацина, как описано выше, где способ В используют в качестве способа получения соединения, представленного формулой (VI-a);
способу получения левофлоксацина, как описано выше, где способ С используют в качестве способа получения соединения, представленного формулой (VI-a);
способу получения левофлоксацина, как описано выше, где способ D используют в качестве способа получения соединения, представленного формулой (VI-a);
способу получения левофлоксацина, как описано выше, где способ Е используют в качестве способа получения соединения, представленного формулой (VI-a);
способу получения левофлоксацина, как описано выше, где способ F используют в качестве способа получения соединения, представленного формулой (VI-a);
способу получения левофлоксацина, как описано выше, где способ G используют в качестве способа получения соединения, представленного формулой (VI-a);
способу получения левофлоксацина, как описано выше, где способ Н используют в качестве способа получения соединения, представленного формулой (VI-a);
способу получения левофлоксацина, как описано выше, где способ I используют в качестве способа получения соединения, представленного формулой (VI-a);
способу получения левофлоксацина, как описано выше, где способ J используют в качестве способа получения соединения, представленного формулой (VI-a);
способу получения левофлоксацина, как описано выше, где каждый X1 и X2 представляет атом фтора;
способу получения левофлоксацина, как описано выше, где соединение трехфтористого бора представляет собой соединение трехфтористого бора, состоящее из трехфтористого бора и простого эфирного соединения;
способу получения левофлоксацина, как описано выше, где соединение трехфтористого бора представляет собой комплекс трехфтористого бора и диэтилового эфира или тетрагидрофурановый комплекс трехфтористого бора;
способу получения левофлоксацина, как описано выше, где реакция с 4-метилпиперазином представляет собой реакцию, проводимую в присутствии триалкиламина;
способу получения левофлоксацина, как описано выше, где триалкиламин представляет собой триэтиламин или трибутиламин и т.д.
Теперь изобретение будет проиллюстрировано более подробно. Во-первых, будут описаны заместители, использованные в настоящем описании.
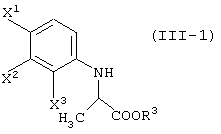
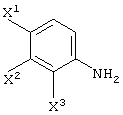
X1, X2 и X3, каждый независимо представляет атом галогена, предпочтительно атом фтора.
R1 представляет уходящую группу. В качестве уходящей группы можно назвать атомы галогена, необязательно замещенные алкилсульфонилоксигруппы, необязательно замещенные арилсульфонилоксигруппы и т.д.
В качестве примеров необязательно замещенной алкилсульфонилоксигруппы можно отметить метансульфонилоксигруппу, этансульфонилоксигруппу, пропансульфонилоксигруппу, бутансульфонилоксигруппу, изобутансульфонилоксигруппу, трет-бутансульфонилоксигруппу и трифторметансульфонилоксигруппу.
В качестве примеров необязательно замещенных арилсульфонилоксигрупп можно отметить бензолсульфонилоксигруппу, п-толуолсульфонилоксигруппу, м-толуолсульфонилоксигруппу, п-нитробензолсульфонилоксигруппу, м-нитробензолсульфонилоксигруппу, п-метоксибензолсульфонилоксигруппу, п-хлорбензолсульфонилоксигруппу, м-хлорбензолсульфонилоксигруппу, 2, 4-диметилбензолсульфонилоксигруппу и 3,5-динитробензолсульфонилоксигруппу.
В качестве уходящей группы замещенные сульфонилоксигруппы и атомы галогена являются предпочтительными и еще более предпочтительными являются трифторметансульфонилоксигруппа, метансульфонилоксигруппа, п-толуолсульфонилоксигруппа, атом хлора и т.д.
R1 представляет -COOR3 или -CH2OR4.
R3 представляет атом водорода или защитную группу карбоксила.
Карбоксильная группа может представлять собой такие группы, обычно используемые в данной области. Конкретные примеры включают аралкильные группы, алкильные группы и т.д.
Термин аралкильные группы означает группы, состоящие из алкильной группы, имеющей 1-6 атомов углерода, и арильной группы. Конкретные примеры включают бензильную группу, нафтилметильную группу и т.д. Алкильная группа может быть линейной, разветвленной или циклической алкильной группой, содержащей 1-6 атомов углерода. Конкретные примеры включают метильную группу, этильную группу, пропильную группу, циклобутильную группу, циклопентильную группу, циклогексильную группу и т.д.
В качестве R3 предпочтительны алкильные группы, содержащие 1-6 атомов углерода, и особенно предпочтительными являются метильная группа, этильная группа и изопропильная группа.
R4 представляет защитную группу гидроксила. В качестве защитной группы гидроксила можно отметить необязательно замещенные алкильные группы, необязательно замещенные арильные группы, необязательно замещенные аралкильные группы, необязательно замещенные ацильные группы и т.д.
В качестве необязательно замещенных алкильных групп можно отметить метоксиметильную группу, метоксиэтильную группу и т.д.
В качестве необязательно замещенных арильных групп можно отметить фенильную группу, диметоксифенильную группу, п-метоксифенильную группу и т.д.
В качестве необязательно замещенных аралкильных групп можно отметить α-фенилэтильную группу, бензильную группу, тритильную группу, толильную группу и т.д.
В качестве необязательно замещенных ацильных групп можно отметить ацетильную группу, метоксиацетильную группу, трифторацетильную группу, хлорацетильную группу, пивалоильную группу, формильную группу, бензоильную группу, п-метоксибензильную группу, п-нитробензоильную группу и т.д.
В качестве R4 предпочтительны необязательно замещенные ацильные группы, и особенно предпочтительной является п-нитробензоильная группа.
R5 и R6 независимо представляют алкильную группу, и предпочтительными в этом случае являются метильная группа и этильная группа.
R7 представляет защитную группу карбоксила, которая может быть аналогична группам, указанным выше в случае R3.
Y представляет алкоксигруппу, атом галогена или диалкиламиногруппу (где алкильные группы могут быть либо одинаковыми, либо различными (предпочтительно одинаковые), и каждая содержит 1-6 атомов углерода). Среди указанного, алкоксигруппы являются предпочтительными. Алкоксигруппы могут представлять собой алкильные группы, содержащие 1-6 атомов углерода, и предпочтительными среди них являются метоксигруппа и этоксигруппа.
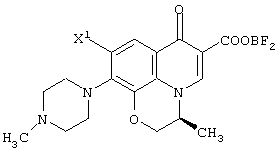
На вышеуказанной реакционной схеме представлен исключительно способ получения одного из изомеров. Однако, другой изомер может быть синтезирован аналогично с использованием соединения, имеющего противоположную конфигурацию относительно соединения (II-а). При использовании рацемического соединения (II) также можно получить соединение (VI) в форме рацемата.
Теперь каждая стадия настоящего изобретения будет проиллюстрирована подробно.
Стадия из соединения (I) в соединение (III)

Соединение (III) может быть получено реакцией соединения (I) с соединением (II) в присутствии основания. Данную реакцию обычно осуществляют в растворителе.
Соединение (II) находится либо в виде соединения (II-1), либо соединения (II-2) в зависимости от определения заместителя R2. В соединении (II) оптически активное соединение используется при получении LVFX. Говоря более конкретно, один из изомеров, то есть соединение (II-а), необходим для получения LVFX. То же самое относится к соединению (II-1) и соединению (II-2). То есть, соединение (II-1-a) и соединение (II-2-a) необходимы для получения LVFX. Данные соединения представлены следующими формулами:
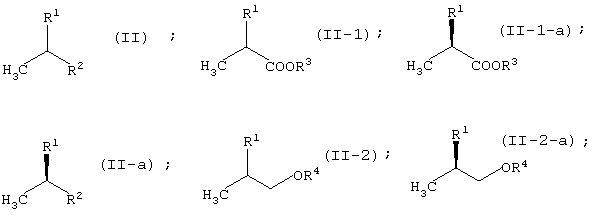
Соединение (II) может быть получено различными способами. Его можно получить превращением сложноэфирного соединения молочной кислоты.
Например, соединение (II-1-a) может быть получено превращением гидроксильной группы сложноэфирного соединения D-молочной кислоты в уходящую группу. То есть, гидроксильная группа может быть превращена в ацетоксигруппу или трифторацетоксигруппу обработкой соединения уксусным ангидридом или трифторуксусным ангидридом, соответственно; или в замещенную сульфонилоксигруппу, такую как трифторметансульфонилоксигруппа, метансульфонилоксигруппа или п-толуолсульфонилоксигруппа, обработкой соединения соответственно трифторметансульфонилхлоридом, метансульфонилхлоридом, п-толуолсульфонилхлоридом или трифторметансульфоновым ангдиридом в присутствии основания.
Соединение (II-2-a) получают с использованием защиты гидроксильной группы сложноэфирного соединения D-молочной кислоты, затем восстановления сложноэфирного фрагмента в гидроксиметильную группу, защиты полученной таким образом гидроксильной группы, элиминирования защитной группы из первоначально защищенной гидроксильной группы для восстановления таким образом гидроксильной группы и затем превращения ее в уходящую группу таким же способом, как описано выше.
Альтернативно, соединение (II-2) может быть получено из 1,2-пропандиола. А именно, терминальную гидроксильную группу первоначально защищают с использованием различия в реакционной способности между первичной и вторичной гидроксильной группами. Затем, оставшуюся гидроксильную группу преобразовывают в уходящую группу. При использовании оптически активного пропандиола может быть получено соединение (II-2-a).
Соединение (III) может быть получено из соединения (I) и соединения (II). Соединение (III-а) получают реакцией с соединением (II-а); соединение (III-1) получают реакцией с соединением (II-1); соединение (III-2) получают реакцией с соединением (II-2); соединение (III-1-a) получают реакцией с соединением (II-1-а); и соединение (III-2-a) получают реакцией с соединением (II-2-a).
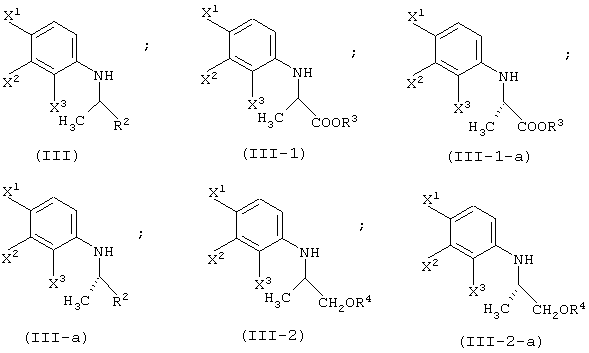
Реакцию соединения (I) с соединением (II-1) или соединением (II-2) можно осуществлять при почти одинаковых условиях. Указанные реакции будут описаны далее.
Соединение (II) можно использовать в количестве от 1 до 2-кратного (по молям), предпочтительно от 1,0 до 1,1-кратного, от числа молей соединения (I).
В качестве основания можно использовать либо неорганическое, либо органическое основание. Примеры неорганических оснований включают карбонаты и гидрокарбонаты щелочных и щелочноземельных металлов, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия и гидрокарбонат калия; и галогениды щелочных и щелочноземельных металлов, такие как фторид калия, фторид цезия и иодид калия.
Примеры органического основания включают триалкиламины, такие как триэтиламин и этилдиизопропиламин; производные N,N-диалкиланилина, содержащие 1-4 атомов углерода, такие как N,N-диметиланилин и N,N-диэтиланилин, и производные пиридина, необязательно замещенные алкильной группой, содержащей 1-4 атомов углерода, такие как пиридин и 2,6-лутидин.
В том случае, когда R1 представляет трифторметансульфонилоксигруппу, предпочтительно проводить реакцию в присутствии органического основания, еще предпочтительнее, 2,6-лутидина. В том случае, когда R1 представляет атом галогена, метансульфонилоксигруппу или п-толуолсульфонилоксигруппу, предпочтительно проводить реакцию в присутствии карбоната или гидрокарбоната щелочного или щелочноземельного металла, еще более предпочтительно, карбоната калия.
Основание можно использовать в количестве от 1 до 3-кратного (по молям), предпочтительно от 1,1 до 2-кратного, из расчета от числа молей соединения (I).
В качестве растворителя можно использовать любой растворитель, который не оказывает влияния на реакцию. Примеры включают ароматические углеводородные растворители, такие как толуол и ксилол; простые эфирные растворители, такие как диэтиловый эфир, тетрагидрофуран (ТГФ) и 1,4-диоксан; кетонные растворители, такие как ацетон и метилэтилкетон; амидные растворители, такие как N,N-диметилформамид (ДМФ) и N,N-диметилацетамид (ДМА.); галогенированные углеводородные растворители, такие как дихлорметан и хлороформ; сложноэфирные растворители, такие как метилацетат и этилацетат; и спиртовые растворители, такие как метанол, этанол и изопропанол (IPA).
В том случае, когда R1 представляет трифторметансульфонилоксигруппу, предпочтительно использовать дихлорметан, хлороформ и т.д. В том случае, когда R1 представляет атом галогена, метансульфонилоксигруппу или п-толуолсульфонил-оксигруппу, предпочтительно использовать N,N-диметилформамид, N,N-диметилацетамид, толуол, ацетон, дихлорметан и т.д.
Растворитель можно использовать в количестве от 5-кратного или более, предпочтительно от 10 до 15-кратного из расчета на соединение (I) (использование 1 мл растворителя на грамм соединения (I) обозначено как 1-кратное количество).
В том случае, когда R1 представляет атом галогена, метансульфонилоксигруппу или п-толуолсульфонилоксигруппу, выход можно повысить при использовании добавки. Примеры добавки включают катализаторы межфазного переноса, молекулярные сита и т.д.
Примеры межфазных катализаторов включают четвертичные соли аммония, такие как хлорид тетра(нормальный гексил)аммония и иодид тетра(нормальный гексил)аммония; и краун эфиры, такие как 18-краун-6, 15-краун-5.
В качестве добавки предпочтительным является катализатор межфазного переноса. Среди всех еще более предпочтительной является липофильная четвертичная соль аммония.
Добавку можно использовать в количестве от 1 до 100%, предпочтительно от 5 до 30% от числа молей соединения (I).
В случае реакции соединения (II-1) температура реакции не ограничена конкретно при условии, что она не превышает температуру кипения используемого растворителя. Обычно она колеблется от -5°С до 50°С, предпочтительно от -5°С до комнатной температуры. В случае реакции соединения (II-2) температура реакции обычно колеблется от -78°С до 50°С, предпочтительно от -50°С до 0°С и еще более предпочтительно от -50°С до -30°С.
Хотя время реакции зависит от температуры реакции, взаимодействие обычно завершается в течение от 30 минут до 5 дней.
В том случае, когда продукт представляет собой соединение (III-1), продукт может использоваться как таковой на последующей стадии без выделения. То есть, стадии от соединения (I) до соединения (IV) могут быть выполнены непрерывно.
На стадии получения соединения (IV) из соединения (III) необходимо выбирать различные способы в зависимости от соединения (III), то есть либо соединения (III-1), либо соединения (III-2).
Соединение (III) также может быть получено следующим способом.
Соединение (III-1) может быть получено реакцией соединения (I-0):
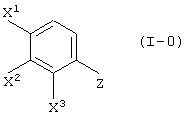
(где X1, X2 и X3, каждый, независимо представляет атом галогена; и Z представляет аминогруппу или нитрогруппу); с пировиноградной кислотой (кислотой или сложным эфиром):
CH3COCOOR3
(где R3 представляет атом водорода или алкильную группу);
в растворителе в присутствии металлического катализатора в атмосфере газообразного водорода.
Металлический катализатор для использования в данном способе получения не является конкретно ограниченным при условии, что он применяется в реакции каталитического гидрирования. Среди таких катализаторов предпочтительными являются палладий на угле, никель Ренея и кобальт Ренея.
В данной реакции для промотирования взаимодействия может быть добавлен дегидратирующий агент. Дегидратирующий агент не является конкретно ограниченным при условии, что он не оказывает влияния на реакцию. Например, можно использовать безводный сульфат магния, безводный сульфат натрия, молекулярные сита и т.д. Среди данных дегидратирующих агентов безводный сульфат магния и безводный сульфат натрия являются предпочтительными.
Реакцию между соединением (I-0) и пировиноградной кислотой можно осуществить более легко при добавлении каталитического количества кислоты и проведении реакции гидрирования при повышенной температуре. Добавляемая кислота может представлять собой или органическую кислоту, или неорганическую кислоту. Примеры кислот включают неорганические кислоты, такие как хлористоводородная кислота, азотная кислота, серная кислота и фосфорная кислота; и органические кислоты, такие как соединения замещенных карбоновых кислот и соединения замещенных сульфоновых кислот. Примеры замещенных карбоновых кислот включают уксусную кислоту, трифторуксусную кислоту и фумаровую кислоту. Примеры замещенных сульфоновых кислот включают метансульфоновую кислоту, трифторметансульфоновую кислоту, бензолсульфоновую кислоту и толуолсульфоновую кислоту. В качестве добавляемой неорганической кислоты предпочтительными являются хлористоводородная кислота и серная кислота.
В качестве добавляемой кислоты может быть добавлена такая кислота, как описано выше. Альтернативно, также возможно выбрать саму пировиноградную кислоту (СН3СОСООН) в качестве производного пировиноградной кислоты, используемого в качестве реагента, что приводит к тому, что пировиноградная кислота служит как в качестве реагента, так и кислоты для промотирования реакции.
Кислота может быть добавлена в каталитическом количестве. В случае использования кислоты, отличной от пировиноградной кислоты, она может быть добавлена в количестве от 1 до 30% (мол.) от числа молей соединения (I-0). При использовании самой пировиноградной кислоты в качестве промотора реакции, ее можно добавлять в эквимолярном количестве в соответствии с количеством молей соединения (I-0). Однако эффект промотирования реакции может достигаться путем дополнительного добавления кислоты в небольшом количестве. Для достижения каталитического действия пировиноградная кислота может быть добавлена в количестве от примерно 1 до примерно 5 мол.%.
В качестве растворителя без ограничения можно использовать любой растворитель, не оказывающий влияния на реакцию. Примеры включают спиртовые растворители, такие как метанол, этанол, пропанол и изопропанол, простые эфирные растворители, такие как диэтиловый эфир, тетрагидрофуран и 1,4-диоксан; галогенированные углеводородные растворители, такие как дихлорметан и хлороформ; амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид; диметилсульфоксид, ацетонитрил, ацетон, сложные эфиры уксусной кислоты, вода и т.д. Также можно использовать смеси данных растворителей.
Среди данных растворителей предпочтительными являются спиртовые растворители, и еще более предпочтительны метанол, этанол и изопропанол.
Хотя температура реакции изменяется в зависимости от используемого растворителя, она обычно колеблется от -78°С до температуры кипения растворителя, предпочтительно от комнатной температуры до температуры кипения растворителя.
Время реакции колеблется от 1 до 24 часов. Обычно реакция завершается в течение 1-16 часов.
Данный способ осуществляют в атмосфере газообразного водорода. Давление газообразного водорода может колебаться от 0,1 до 10 МПа, предпочтительно от 0,1 до 5 МПа.
В том случае, когда данную реакцию осуществляют с использованием производного нитробензола (Z=NO2), нитрогруппа первоначально восстанавливается в аминогруппу (производное анилина). Затем данная аминногруппа взаимодействует с карбонильной группой в пировиноградной кислоте, давая иминосоединение:
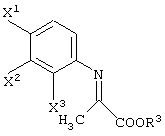
Затем иминогруппа данного иминосоединения гидрируется в аминогруппу. (При этом присутствует исключительно один из геометрических изомеров иминосоединения). Следовательно, не приходится и говорить, что анилиновое соединение, имеющее восстановленную нитрогруппу, используется в качестве исходного вещества в данной реакции. Данное иминосоединение получают либо в виде одного геометрического изомера, либо в виде смеси изомеров. Каждый случай пригоден для асимметрического восстановления.
При использовании способа получения, основанного на реакции соединения (I-0) с соединением пировиноградной кислоты в восстановительных условиях, соединение (III-1) обычно получают в виде рацемата. Для получения оптически активного соединения (III-1-a) иминосоединение, образованное при взаимодействии соединения (I-0) с соединением пировиноградной кислоты, восстанавливают в условиях асимметрического восстановления.
Реакция асимметрического восстановления имина может быть осуществлена при использовании следующих условий реакции:
(1) реакций восстановления с использованием соединений бора и алюминия, о которых сообщалось в K.Yamada, J.Chem.Soc., Perkin Trans.1, 265 (1983); S.Ituno, Bull. Chem.Soc. Jpn., 60, 395(1987); S. Ituno, J.Chem.Soc., Perkin Trans.l, 1859 (1990); B.T.Cho, Tetrahedron Asymmetry, 3, 1583 (1992); T.Sakai, Synlett., 753 (1995); M.Shimizu, Tetrahedron Lett., 36, 8607 (1995); C.Bolm., Synlett., 655 (1994); J.M.Brunel, Synlett., 177 (1996); R.O.Hutchins, J.Org.Chem., 52, 704 (1987), и т.д.
(2) реакций гидросилилирования, о которых сообщалось в N.Langlois, Tetrahedron Lett., 4865 (1973); H.B.Kagan, J.Organomet.Chem., 90, 353 (1975); X.Verdaguer, J.Am.Chem. Soc., 118, 6784 (1996), и т.д.
(3) реакций каталитического гидрирования, о которых сообщалось в следующих документах и им подобным (Rh катализатор: A.Levi, J.Chem.Soc., Chem.Commun., 6 (1975); S.Vastag, J.Mol.Catal., 22, 283 (1984); G.-K. Kang, J.Chem.Soc., Chem.Commun., 1466 (1988); W.R.Cullen, J.Mol.Catal., 62, 243 (1990); A.G. Becalski, Inorg.Chem., 30, 5002 (1991); J.Bakos, J.Organomet. Chem., 279, 23 (1985); J.Bakos, J.Organomet. Chem., 370, 263 (1989); J.Bakos, J.Chem.Soc., Chem.Commun., 1684 (1991); C.Lensink, Tetrahedron Asymmetry, 3, 235 (1992); C.Lensink, Tetrahedron Asymmetry, 4, 215 (1993); J.M.Buriak, Organometallics, 15, 3161 (1996); M.J.Murk, J.Am.Chem.Soc., 114, 6266 (1992); M.J.Burk, Tetrahedron, 50, 4399 (1994); Ir катализатор: F.Spindler, Angew.Chem.Int.Ed.Engl., 29, 558 (1990); A.Togni., Angew. Chem.Int.Ed.Engl., 35, 1475 (1996); T.Morimoto, Chem.Pharm.Bull., 42, 1951 (1994); T.Morimoto, Tetrahedron Asymmetry, 6, 2661 (1995); T.Morimoro, Synlett., 748 (1995); K.Tani, Chem.Lett., 955 (1995); K.Satoh. Tetrahedron Asymmetry, 9, 2657 (1998); Y.Ng C.Chan, J.Chem.Soc.Chem.Commun., 869 (1990); Y.Ng C.Chan, J.Am.Chem.Soc., 112, 9400 (1990); R.Sablong, Tetrahedron Lett., 37, 4937 (1996); Ti катализатор: C.A.Willoughby, J.Am.Chem.Soc., 114, 7562 (1992); C.A. Willoughby, J.Org.Chem.,. 58, 7627 (1993); C.A.Willoughby, J.Am.Chem.Soc., 116, 8952 (1994); C.A.Willoughby, J.Am.Chem.Soc., 116, 11703 (1994); Ru катализатор: C.Botteghi, Chimia, 29, 256 (1975); W.Oppolzer, Tetrahedron Lett., 31, 4117 (1990); D.E.Fogg, Inorg.Chim.Acta., 222, 85 (1984); и
(4) реакций восстановления водородного переноса, о которых сообщалось в S.Hashiguchi, J.Am.Chem.Soc., 117, 7562 (1995); A.Fujii, J.Am.Chem.Soc., 118, 2521 (1996); N.Uematsu, J.Am.Chem.Soc., 118, 4916 (1996) и т.д.
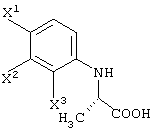
Среди оптически активных соединений (III-1-a) соединение карбоновой кислоты (смотри следующую структурную формулу):
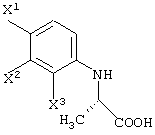
может быть получено обработкой сложноэфирного соединения соответствующего соединения ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма.
Для осуществления асимметрического гидролиза сложного эфира сложноэфирное соединение (рацемат) соединения (III-1) суспендируют в подходящем буфере и затем добавляют фермент, способный асимметрически гидролизовать сложный эфир, или жидкую культуральную среду микроорганизма, клетки данного микроорганизма или переработанные клетки данного микроорганизма с последующим перемешиванием. Фермент и т.д., которые можно использовать в данной реакции, не являются конкретно ограниченными при условии, что они споосбны асимметрически гидролизовать сложный эфир. Примеры фермента включают продаваемые препараты ферментов, получаемые из микроорганизмов, животных и растений. Например, можно использовать различные эстеразы, протеазы или химотрипсины. В качестве микроорганизмов можно использовать бактерии, принадлежащие к родам Bacillus, Micrococcus и Actinomyces; грибки, принадлежащие к родам Aspergillus, Rhizopus, Nannizia и Penicillium; и дрожжи, принадлежащие к родам Candida, Saccharomyces и Zygoascus.
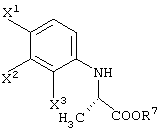
При обработке описанным выше ферментом, микробными клетками и т.д. сложноэфирный фрагмент одного из изомеров (энантиомеров) соединения (III-1) гидролизуется, давая карбоновую кислоту. Далее данный продукт преобразовывают в соль карбоновой кислоты и таким образом растворяют в обрабатываемом маточном растворе. В этот момент, обрабатываемый маточный раствор экстрагируют органическим растворителем, таким как этилацетат, хлороформ, диизопропиловый эфир (IPE) или метил-трет-бутиловым эфиром. Таким образом, сложноэфирное соединение (см. следующую структурную формулу), которое представляет собой ненужный изомер (этантиомер) соединения (III-1-b):
может быть выделено и собрано.
Перед экстракцией соединения (III-1-b) выигрышным является удаление фермента, микробных клеток и т.д., например, фильтрованием. После экстракции соединения (III-1-b) обработанный маточный раствор подкисляют и затем экстрагируют органическим растворителем, таким как диизопропиловый эфир, метил-трет-бутиловый эфир или этилацетат. Таким образом, можно получить карбоксильное соединение соединения (III-1-a), которое представляет собой свободное соединение.
Обработка ферментом, микробными клетками и т.д. может быть выполнена обычно при температуре от 5°С до 60°С, предпочтительно от 20°С до 40°С.
Значение рН данного обрабатываемого маточного раствора может колебаться от 4 до 9, предпочтительно от 6 до 8.
Обработку ферментом, микробными клетками и т.д. можно проводить в течение от 4 часов до 7 дней, предпочтительно от 8 часов до 50 часов.
Концентрация соединения (III-1) в обрабатываемом маточном растворе обычно колеблется от 0,1% до 20% по массе, предпочтительно от 0,5% до 5%.
Количество фермента или жидкой культуральной среды микроорганизма, клеток микроорганизма или переработанных клеток микроорганизма не являются ограниченными конкретно. Обычно, их предпочтительно используют в количестве от 0,05 до 0,5-кратного по массе от массы сухого соединения (III-1).
Также возможно получать соединение карбоновой кислоты соединения (III-1-b) с использованием фермента или жидкой культуральной среды микроорганизма, имеющего способность обратного асимметрического распознавания для расщепления сложного эфира в соединении (III-1-a) и превращения в карбоксильную группу, клеток данного микроорганизма или переработанных клеток данного микроорганизма.
Соединение карбоновой кислоты типа рацемического соединения (III-1) может быть получено путем разделения изомеров (энантиомеров) с помощью образования диастереомерных солей с оптически активным органическим основанием и их кристаллизации. При дополнительной перекристаллизации полученной таким образом соли с использованием подходящего растворителя может быть получена соль, имеющая более высокую стереоизомерную чистоту.
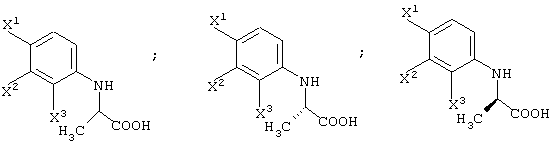
При обработке образованных таким образом диастереомерных солей кислотой могут быть выделены карбоксильные соединения типа соединения (III-1-a) и соединения (III-1-b).
Термин «включает единственный (оптический) изомер», используемый в данном описании, означает не только случай, при котором изомер совсем не содержит другого (оптического) изомера, но также и тот случай, при котором другой изомер может присутствовать в такой степени, что он не оказывает влияния на физические константы.
Термин «стереоизомерно чистая соль», используемый в данном описании, имеет следующее значение. В том случае, когда кислота и основание, составляющие соль, имеют стереоизомеры, то именно, соль, образованная кислотой, включающей единственный стереоизомер, и основанием, аналогично включающим единственный стереоизомер, приводится как стереоизомерно чистая соль. То есть, это означает соль, в которой каждый из ее составляющих кислота и основание - включает единственный стереоизомер. Термин «включает единственный стереоизомер», используемый в данном описании, может рассматриваться в качестве состояния, по существу, не содержащего другого изомера.
Примеры оптически активных органических оснований, которые можно использовать при образовании таких солей, включают оптически активные производные этиламина, замещенные арилом в 1-положении (производные 1-арилэтиламина), представленные следующей формулой:
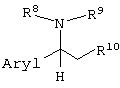
(где Aryl представляет арильную группу, необязательно содержащую атом галогена, нитрогруппу, цианогруппу, карбамоильную группу, алкильную группу, содержащую 1-6 атомов углерода, или алкоксигруппу, содержащую 1-6 атомов углерода; и
R8, R9 и R10, каждый независимо, представляют:
(1) фенильную группу, необязательно содержащую атом галогена, алкильную группу, содержащую 1-6 атомов углерода, галогеналкильную группу, имеющую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, нитрогруппу, карбамоильную группу или цианогруппу;
(2) бензильную группу, необязательно содержащую атом галогена, алкильную группу, содержащую 1-6 атомов углерода, галогеналкильную группу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, нитрогруппу, карбамоильную группу или цианогруппу;
(3) алкильную группу, содержащую 1-6 атомов углерода;
или
(4) атом водорода);
Примеры арильной группы включают фенильную группу и нафтильную группу. Ароматические кольца данных арильных групп могут иметь один или несколько заместителей, таких как атомы галогена, нитрогруппа, цианогруппа, карбамоильная группа, алкильные группы, содержащие 1-6 атомов углерода, и алкоксигруппы, содержащие 1-6 атомов углерода, или один или несколько типов данных заместителей.
В качестве примеров таких оптически активных оснований можно отметить 1-фенилэтиламин, 1-(п-толил)этиламин и 1-фенил-2-(п-толил)этиламин.
Среди таких оснований примеры оптически активных оснований, способных к преимущественному образованию соли в сочетании с соединением карбоновой кислоты типа соединений (III-1-a), включают (R)-(+)-1-фенилэтиламин, (R)-(+)-(п-толил)этиламин и (S)-(+)-1-фенил-2-(п-толил)этиламин.
Примеры оптически активных оснований, способных к преимущественному образованию соли в сочетании с соединением карбоновой кислоты типа соединений (III-1-b), включают (S)-(+)-1-фенилэтиламин, (S)-(+)-1-(п-толил)этиламин и (R)-(+)-1-фенил-2-(п-толил)этиламин.
С другой стороны, ароматические кольца производных 1-арилэтиламина не ограничены углеводородными ароматическими кольцами, но также включают гетероциклы, содержащие атом серы, атом азота, атом кислорода и тому подобное. Примеры включают тиофен, бензотиофен, пиридин, хинолин, изохинолин, фуран, бензофуран и тому подобное.
Оптически активное основание обычно можно использовать в эквимолярном количестве или менее относительно числа молей соединения карбоновой кислоты.
В качестве растворителя для кристаллизации или перекристаллизации целевой соли можно использовать различные растворители. Примеры растворителей, которые пригодны в данном случае, включают алифатические или ароматические углеводородные растворители, такие как н-гексан, н-пентан, бензол, толуол и ксилол; спиртовые растворители, такие как метанол, этанол, пропанол, изопропанол, н-бутанол и трет-бутанол; простые эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран, 1,2-диметоксиэтан и 1,4-диоксан; амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид; и галогенированные углеводородные растворители, такие как хлороформ, хлористый метилен и 1,2-дихлорэтан (ДХЭ). Кроме того, также можно использовать воду, ацетонитрил, сложные эфиры уксусной кислоты, ацетон и т.д. Можно использовать либо один из данных растворителей, либо смесь их нескольких типов.
Растворитель обычно можно использовать в количестве от 1 до 100 кратного по массе, предпочтительно от примерно 2 до 50-кратного по массе.
Хотя температура кристаллизации или перекристаллизации целевой соли не является определяющей, обычно можно выбрать температурные условия. Говоря более конкретно, ее можно проводить в температурном диапазоне от охлаждения льдом до температуры кипения используемого растворителя.
Время реакции обычно колеблется от 1 до 24 часов.
Соль карбоновой кислоты может быть превращена в свободную карбоновую кислоту при обработке кислотой. А именно, соль карбоновой кислоты обрабатывают неорганической кислотой, такой как хлористоводородная кислота или серная кислота с последующим выделением, например, с помощью экстракции органическим растворителем.
Поскольку изомер (энантиомер) для применения при получении левофлоксацина представляет собой соединение (III-1-a), другое соединение (III-1-b) само по себе не находит применения. Сложноэфирное соединение данного соединения (III-1-b) может быть рацемизовано обработкой в присутствии основания. Таким образом, данным способом ненужный изомер может быть превращен в необходимый изомер.
В качестве растворителя, применяемого в данной реакции изомеризации, можно привести различные растворители. Их примеры включают алифатические или ароматические углеводородные растворители, такие как н-гексан, н-пентан, бензол, толуол и ксилол; спиртовые растворители, такие как метанол, этанол, пропанол, изопропанол, н-бутанол и трет-бутанол; простые эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран, 1,2-диметоксиэтан и 1,4-диоксан; амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид; и галогенированные углеводородные растворители, такие как хлороформ, хлористый метилен и 1,2-дихлорэтан. Кроме того, также можно использовать воду, ацетонитрил, эфиры уксусной кислоты, ацетон и т.д. Можно использовать либо один из данных растворителей, либо смесь их нескольких типов.
Среди данных растворителей ароматические углеводороды, такие как толуол, и амиды, такие как N,N-диметилформамид и N,N-диметилацетамид, являются предпочтительными.
Хотя температура реакции изменяется в зависимости от используемого растворителя, она обычно колеблется от -78°С до температуры кипения растворителя, предпочтительно от комнатной температуры до температуры кипения растворителя.
Время реакции колеблется от 1 до 24 часов, предпочтительно от 1 до 16 часов.
Основание может представлять собой или органическое основание, или неорганическое основание. Например, можно использовать гидроксиды, карбонаты, гидрокарбонаты и алкоксиды щелочных металлов и щелочноземельных металлов, таких как натрий, калий, литий, магний и кальций; гидриды металлов, такие как гидрид натрия, гидрид калия и гидрид лития; алкиллитиевые реагенты, такие как н-бутиллитий, метиллитий и диизопропиламид лития; третичные амины, такие как триэтиламин и N,N-диизопропилэтиламин; азотсодержащие гетероциклические соединения, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ), 1,8-диазабицикло[4.3.0]нон-5-ен (ДБН) и N-метилморфолин; N,N-диалкиланилины, такие как диметиланилин и диэтиланилин; и т.д.
Среди данных оснований предпочтительно использовать азотсодержащие гетероциклические соединения, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ); карбонаты щелочных металлов или щелочноземельных металлов, такие как карбонат калия; или алкоксиды щелочных металлов или щелочноземельных металлов, такие как третичный-бутоксид калия (t-BuOK).
Основание можно использовать в количестве от 0,1 до 15-кратного (по молям), предпочтительно от 1 до 5-кратного, от числа молей сложноэфирного соединения (III-1-b).
Для промотирования реакции взаимодействие можно осуществлять в присутствии четвертичной соли аммония, такой как бромид тетрабутиламмония или хлорид бензилтриэтиламмония; иодида щелочного металла или щелочноземельного металла, такого как иодид калия или иодид натрия, краун-эфира и т.д.
Соединение (III-1-b) может быть превращено в соединение карбоновой кислоты (II-1) рацемизацией при обработке основанием, а затем гидролизом.
В качестве растворителя можно использовать различные растворители, например, алифатические или ароматические углеводородные растворители, такие как н-гексан, н-пентан, бензол, толуол и ксилол; спиртовые растворители, такие как метанол, этанол, пропанол, изопропанол, н-бутанол и трет-бутанол; простые эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран, 1,2-диметоксиэтан и 1,4-диоксан; амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид; и галогенированные углеводородные растворители, такие как хлороформ, хлористый метилен и 1,2-дихлорэтан. Кроме того, также можно использовать воду, ацетонитрил, эфиры уксусной кислоты, ацетон и т.д. Можно использовать либо один из данных растворителей, либо смесь их нескольких типов.
Среди данных растворителей ароматические углеводородные растворители, такие как толуол, и N,N-диметилформамид и N,N-диметилацетамид являются предпочтительными.
Хотя температура реакции изменяется в зависимости от используемого растворителя, она обычно колеблется от -78°С до температуры кипения растворителя, предпочтительно от комнатной температуры до температуры кипения растворителя.
Время реакции колеблется от 1 до 24 часов. Обычно реакция завершается в течение 1-16 часов.
Основание может представлять собой или органическое основание, или неорганическое основание. Например, можно использовать гидроксиды, карбонаты, гидрокарбонаты и алкоксиды щелочных металлов и щелочноземельных металлов, таких как натрий, калий, литий, магний и кальций; гидриды металлов, такие как гидрид натрия, гидрид калия и гидрид лития; алкиллитиевые реагенты, такие как н-бутиллитий, метиллитий и диизопропиламид лития; третичные амины, такие как триэтиламин и N,N-диизопропилэтиламин; азотсодержащие гетероциклические соединения, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ), 1,8-диазабицикло[4.3.0]нон-5-ен (ДБН) и N-метилморфолин; N,N-диалкиланилины, такие как диметиланилин и диэтиланилин; и т.д.
Среди данных оснований предпочтительным является использование алкоксидов щелочных металлов, таких как третичный бутоксид калия, или карбонатов щелочных или щелочноземельных металлов, таких как карбонат калия.
Основание можно использовать в количестве от 0,1 до 15-кратного (по молям), предпочтительно от 1 до 5-кратного, от числа молей сложноэфирного соединения (III-1-b).
Для промотирования реакции взаимодействие можно осуществлять в присутствии четвертичной соли аммония, такой как бромид тетрабутиламмония или хлорид бензилтриэтиламмония; иодида щелочного металла или щелочноземельного металла, такого как иодид калия или иодид натрия, краун-эфира и т.д.
Сложный эфир гидролизуют с использованием кислоты или основания. При кислотном гидролизе используют кислоту, такую как хлористоводородная кислота или серная кислота. При основном гидролизе используют основание, например, гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия; карбонат щелочного металла, такой как карбонат натрия или карбонат калия; или гидрокарбонат щелочного металла, такой как гидрокарбонат натрия или гидрокарбонат калия. Основание обычно используют в виде водного раствора.
Соединение карбоновой кислоты в соединении (III-a), которое получают гидролизом с использованием фермента, жидкой культуральной среды микроорганизма, микробных клеток или переработанных микробных клеток или гидролизом в кислотных или основных условиях, может быть превращено в сложноэфирное соединение обычным образом. А именно, оно может быть подвергнуто взаимодействию со следующим спиртом в присутствии кислотного катализатора:
R7-OH
Примеры подходящего спирта включают метанол, этанол, пропанол, изопропанол и н-бутанол. При использовании такого спирта протекает этерификация в сложный эфир, соответствующий спирту. Хотя температура реакции зависит от используемого спирта, обычно она колеблется от -78°С до температуры кипения спирта, предпочтительно от комнатной температуры до температуры кипения спирта. Примеры используемой в данном случае кислоты включают хлористоводородную кислоту, серную кислоту и фосфорную кислоту. В качестве другого способа этерификации также можно использовать этерификацию с помощью получения хлорангидрида кислоты с последующей обработкой спиртом.
Соединение карбоновой кислоты среди соединений (III-a), полученное асимметрическим гидролизом сложного эфира или гидролизом сложного эфира в присутствии кислоты или основания, может быть очищено образованием солей с различными аминами. В качестве амина, подходящего для такой очистки, предпочтительным является высоко липофильный амин и его примеры включают циклические алкиламины, такие как циклогексиламин; и аралкиламины, такие как бензиламин и фенэтиламин. Среди данных аминов предпочтительными являются циклогексиламин и бензиламин, и еще более предпочтительным является циклогексиламин. Соль такого амина может быть очищена перекристаллизацией обычным способом. В качестве условий для очистки подходящим образом можно использовать описанные выше условия оптического разделения. Соль амина соединения карбоновой кислоты в ряду полученных таким образом соединений (III-1) может быть превращена в свободное соединение при обработке кислотой. Впоследствии оно может быть этерифицировано вышеуказанным методом. Также возможно проводить этерификацию, пропуская процедуру получения чистого соединения с использованием кислоты, взятой в избытке от числа молей соли карбоновой кислоты.
Стадия из соединения (III-1) в соединение (IV)
Соединение (IV) может быть получено восстановлением соединения (III-1). Данная реакция может быть осуществлена обработкой соединения (III-1) в растворителе в присутствии восстанавливающего агента. В качестве соединения (III-1), используемого в данном восстановлении, соединение, в котором фрагмент COOR3 представляет собой сложный эфир, является особенно предпочтительным.
Примеры восстанавливающего агента включают боргидридные восстанавливающие агенты, такие как боргидрид натрия, боргидрид лития, боргидрид кальция, боргидрид цинка, боргидрид магния и цианид цианоборгидрида натрия; и алюмогидридные восстанавливающие агенты, такие как алюмогидрид лития. В качестве восстанавливающего агента предпочтительными являются боргидридные восстанавливающие агенты, и особенно предпочтительным является боргидрид натрия.
Восстанавливающий агент можно использовать в количестве от 1,1 до 2,5-кратного по молям, предпочтительно от 1,1 до 1,5-кратного, от числа молей соединения (III-1).
Растворитель, используемый здесь, не является конкретно ограниченным при условии, что он не оказывает влияния на реакцию. Примеры включают спиртовые растворители, такие как метанол, этанол, изопропанол и трет-бутанол; простые эфирные растворители, такие как диэтиловый эфир, тетрагидрофуран и т.д. В качестве растворителя предпочтительными являются спиртовые растворители, и еще более предпочтительным является изопропанол. При использовании изопропанола реакция может быть промотирована добавлением метанола в количестве от 0,5 до 5-кратного по молям, предпочтительно от 0,5 до 2-кратного, от числа молей соединения (III-1).
Температурой реакции может являться температура, не оказывающая нежелательного влияния на реакцию. Предпочтительно она находится в пределах от 0 до 60°С, еще предпочтительнее от комнатной температуры до 50°С. Время реакции может находиться в пределах от 1 часа до 20 часов.
В результате исследования данной реакции восстановления, проведенного авторами данного изобретения, было установлено, что в том случае, когда реакции восстановления подвергают оптически активное соединение в ряду соединений формулы (III-1), предпочтительным в качестве растворителя является выбор неспиртового растворителя (апротонного растворителя) и использование соединения гидрида металла в качестве восстанавливающего агента для данной реакции. А именно, выяснено, что в данном способе при осуществлении реакции восстановления оптически активного соединения в протонном растворителе стереохимическая структура частично претерпевает инверсию, и вследствие этого снижается оптическая чистота.
В качестве соединения гидрида металла можно использовать соединение боргидрида металла или соединение алюмогидрида металла. Конкретные примеры включают соединения боргидрида металла, такие как боргидрид натрия, боргидрид лития, боргидрид кальция, боргидрид калия, боргидрид цинка, боргидрид магния и цианид цианоборгидрида натрия и соединения алюмиогидрида металла, такие как алюмогидрид лития. Среди данных соединений предпочтительными являются соединения боргидридов металлов, и особенно предпочтительным является боргидрид натрия.
Восстанавливающий агент можно использовать в количестве от 1 до 5-кратного по молям, предпочтительно от примерно 1,1 до 2-кратного из расчета от числа молей соединения (III-1-a) или (III-1-b).
На данной стадии особенно предпочтительным является использование апротонного растворителя. Примеры апротонного растворителя, который можно использовать в данном случае, включают линейные и разветвленные алифатические углеводородные растворители, такие как н-гексан, н-пентан, циклогексан и циклопентан; ароматические углеводородные растворители, такие как бензол, толуол и ксилол; простые эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран, 1,2-диметоксиэтан и 1,4-диоксан; и галогенированные углеводородные растворители, такие как хлороформ, хлористый метилен и 1,2-дихлорэтан. Кроме того, также можно использовать эфиры уксусной кислоты и т.д. Можно использовать либо один из данных растворителей, либо смесь нескольких их типов.
Среди данных растворителей алифатические углеводородные растворители, такие как н-гексан и циклогексан, простые эфирные растворители, такие как диизопропиловый эфир и метил-трет-бутиловый эфир и ароматические углеводородные растворители, такие как толуол, являются предпочтительными.
В качестве добавляемого в данном случае спирта предпочтительными являются первичные спирты, и особенно предпочтительным является метанол. Спирт можно использовать в количестве от 3 до 20-кратного, предпочтительно от 4 до 15-кратного из расчета на соединение (III-1-a) или (III-1-b).
Хотя температура реакции изменяется в зависимости от используемого растворителя, обычно она находится в пределах от -78°С до температуры кипения растворителя, предпочтительно от 10°С до температуры кипения растворителя.
Время реакции находится в пределах от 1 до 24 часов. Обычно реакция завершается в течение примерно 2-16 часов.
Для осуществления реакции восстановления на данной стадии без изомеризации оптически активного соединения предпочтительным является добавление соединения (III-1-a) или (III-1-b) и восстанавливающего агента к апротонному растворителю, а затем добавление туда спирта (при перемешивании).
Стадия из соединения (III-2) в соединение (IV)
Соединение (IV) может быть получено снятием защитной группы в соединении (III-2).
Хотя способ снятия защитной группы изменяется в зависимости от типа R4, используемого в качестве защитной группы гидроксила, он может быть осуществлен способом, подходящим для этого типа R4, обычно применяемым в данной области. В том случае, когда R4 представляет аралкильную группу (арилметил) или аралкилоксикарбонильную группу, можно использовать реакцию каталитического гидрирования. В том случае, когда R4 представляет ацильную группу, можно применять реакцию гидролиза кислотой или щелочью. В том случае, когда R4 представляет алкоксикарбонильную группу или простой эфир, можно использовать разложение кислотой или обработку цинком в уксусной кислоте.
Стадия из соединения (IV) в соединение (V)
На данной стадии соединение (V) может быть получено добавлением к соединению (IV) производного диалкилового эфира метиленмалоновой кислоты, представленного следующей формулой:
(где R5 и R6, каждый, независимо представляет алкильную группу; и Y представляет алкоксигруппу, атом галогена или диалкиламиногруппу); с последующим нагреванием или обработкой соединения (IV) и производного диалкилового эфира метиленмалоновой кислоты в растворителе в присутствии основания и катализатора межфазного переноса.
(1) Способ добавления производного диалкилового эфира метиленмалоновой кислоты к соединению (IV)
Производное диалкилового эфира метиленмалоновой кислоты можно использовать в количестве от 1 до 3-кратного по молям, предпочтительно от 1,05 до 1,2-кратного, от числа молей соединения (IV).
Реакцию можно осуществлять или без использования растворителя, или в растворителе. В качестве растворителя можно использовать любой растворитель при условии, что он не оказывает влияния на реакцию. Примеры включают ароматические углеводородные растворители, такие как толуол и ксилол.
Предпочтительным является проведение реакции без использования растворителя или с использованием ароматического углеводородного растворителя, такого как толуол или ксилол.
Температура реакции не ограничена конкретно при условии, что она не превышает температуру кипения растворителя. Предпочтительно она колеблется от 100°С до температуры кипения растворителя. Хотя время реакции изменяется в зависимости от температуры реакции, обычно она завершается в течение от 1 часа до 1 дня.
(2) Способ обработки соединения (IV) и производного диалкилового эфира метиленмалоновой кислоты в растворителе в присутствии основания и катализатора межфазного переноса.
Производное диалкилового эфира метиленмалоновой кислоты можно использовать в количестве от 1 до 3-кратного по молям, предпочтительно от 1,05 до 1,2-кратного, от числа молей соединения (IV).
Растворитель не ограничен конкретно при условии, что он не оказывает нежелательного действия на реакцию. Примеры включают алифатические углеводородные растворители, такие как н-гексан и н-пентан; ароматические углеводородные растворители, такие как бензол, толуол и ксилол; простые эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран и 1,4-диоксан; кетонные растворители, такие как ацетон и метилэтилкетон, амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид; галогенированные углеводородные растворители, такие как дихлорметан и хлороформ; сложноэфирные растворители, такие как метилацетат и этилацетат; спиртовые растворители, такие как метанол, этанол, изопропанол, н-бутанол и трет-бутанол; и галогенированные углеводороды, такие как хлороформ, хлористый метилен и 1,2-дихлорэтан. Среди данных растворителей предпочтительными являются ароматические углеводородные растворители, амидные растворители, кетонные растворители и галогенированные растворители, и еще более предпочтительными являются толуол, N,N-диметилформамид, N,N-диметилацетамид, ацетон и дихлорметан. Среди данных растворителей дополнительно предпочтительными являются амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид.
Основание может представлять собой или неорганическое основание, или органическое основание. Примеры неорганических оснвоаний включают гидриды щелочных металлов, такие как гидрид натрия и гидрид лития; гидриды щелочноземельных металлов, такие как гидрид кальция; гидроксиды щелочных металлов, такие как гидроксид натрия и гидроксид калия; карбонаты или гидрокарбонаты щелочных металлов или щелочноземельных металлов, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия и гидрокарбонат калия; галогениды щелочных металлов или щелочноземельных металлов, такие как фторид калия, фторид цезия и иодид калия.
Примеры органических оснований включают алкоксиды щелочных металлов, такие как метоксид натрия, метоксид лития, этоксид натрия, этоксид лития, третичный-бутоксид натрия и третичный -бутоксид калия; триалкиламины, такие как триэтиламин и этилдиизопропиламин; производные анилина, содержащие алкильные группы, имеющие 1-4 атомов углерода, такие как N,N-диметиланилин и N,N-диэтиланилин; производные пиридина, необязательно замещенные алкильными группами, имеющими 1-4 атомов углерода, такие как пиридин и 2,6-лутидин; и азотсодержащие гетероциклические соединения, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен.
Среди данных оснований предпочтительным является использование алкоксидов щелочных металлов, азотсодержащих гетероциклических соединений и гидроксидов щелочных металлов или щелочноземельных металлов. Третичный-бутоксид калия, 1,8-диазабицикло[5.4.0]ундец-7-ен и гидроксиды щелочных металлов являются предпочтительными, и еще более предпочтительным является гидроксид калия. Гидроксиды щелочных металлов, в частности, гидроксид калия, можно использовать подходящим образом, поскольку в таком случае во время реакции изомеризация не протекает.
Основание можно использовать в количестве от 1 до 15-кратного по молям, предпочтительно от 1 до 3-кратного, от числа молей сложноэфирного соединения соединения (IV).
В данной реакции выход может быть повышен при добавлении добавки. Примеры добавок включают катализаторы межфазного переноса и молекулярные сита.
Примеры катализаторов межфазного переноса включают четвертичные соли аммония, такие как хлорид тетра(нормальный-гексил)аммония, хлорид триметилбензиламмония, хлорид триэтилбензиламмония, хлорид триметилфениламмония и гидросульфат тетрабутиламмония; и краун-эфиры, такие как 18-краун-6, 15-краун-5.
В качестве добавки предпочтительным является катализатор межфазного переноса. Среди всех еще более предпочтительными являются липофильные четвертичные соли аммония.
Среди данных катализаторов межфазного переноса четвертичные соли аммония, такие как хлорид тетра(нормальный-гексил)аммония, хлорид триметилбензиламмония, хлорид триэтилбензиламмония, хлорид триметилфениламмония и гидросульфат тетрабутиламмония, являются предпочтительными.
Катализатор межфазного переноса можно использовать в количестве от 1% до 100%, еще предпочтительнее от примерно 3% до 30%, от числа молей соединения (II).
Хотя температура реакции изменяется в зависимости от используемого растворителя, обычно она находится в пределах от -78°С до температуры кипения растворителя, предпочтительно от комнатной температуры до 60°С и еще предпочтительнее в пределах комнатной температуры.
Время реакции находится в пределах от 1 до 24 часов. Обычно реакция завершается в течение от 1 до 12 часов.
Соединение (V), которое представляет собой получаемый продут, можно использовать как таковое без выделения на последующей стадии. А именно, стадии от соединения (IV) до соединения (VI) можно проводить непрерывно.
Стадия из соединения (V) в соединение (VI)
Соединение, представленное формулой (VI), может быть получено внутримолекулярной циклизацией соединения, представленного формулой (V). Данную стадию можно проводить обработкой в растворителе в присутствии основания и катализатора межфазного переноса.
Основание может представлять собой или органическое основание, или неорганическое основание. Примеры неорганических оснований включают гидриды щелочных металлов, такие как гидрид натрия и гидрид лития; гидриды щелочноземельных металлов, такие как гидрид кальция; гидроксиды щелочных металлов, такие как гидроксид натрия и гидроксид калия; карбонаты или гидрокарбонаты щелочных металлов или щелочноземельных металлов, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия и гидрокарбонат калия; галогениды щелочных металлов или щелочноземельных металлов, такие как фторид калия, фторид цезия и иодид калия.
Примеры органических оснований включают алкоксиды щелочных металлов, такие как метоксид натрия, метоксид лития, этоксид натрия, этоксид лития, третичный-бутоксид натрия и третичный -бутоксид калия; триалкиламины, такие как триэтиламин и этилдиизопропиламин; производные N,N-диалкиланилина, содержащие алкильные группы, имеющие 1-4 атомов углерода, такие как N,N-диметиланилин и N,N-диэтиланилин; производные пиридина, необязательно замещенные алкильными группами, имеющими 1-4 атомов углерода, такие как пиридин и 2,6-лутидин; и азотсодержащие гетероциклические соединения, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен.
Предпочтительным является использование в качестве основания гидроксидов щелочных металлов или щелочноземельных металлов или алкоксидов щелочных металлов. Еще предпочтительнее гидроксид калия или третичный-бутоксид калия, и еще более предпочтителен гидроксид калия.
Основание можно использовать в количестве от 0,1 до 15-кратного по молям, предпочтительно от 1 до 3-кратного, от числа молей сложноэфирного соединения (V).
На данной стадии реакция может быть промотирована при ее проведении в присутствии катализатора межфазного переноса.
Примеры катализаторов межфазного переноса включают четвертичные соли аммония, такие как хлорид тетра(нормальный-гексил)аммония, хлорид триметилбензиламмония, хлорид триэтилбензиламмония, хлорид триметилфениламмония и гидросульфат тетрабутиламмония; и краун-эфиры, такие как 18-краун-6, 15-краун-5.
Среди данных катализаторов межфазного переноса четвертичные соли аммония, такие как хлорид тетра(нормальный-гексил)аммония, хлорид триметилбензиламмония, хлорид триэтилбензиламмония, хлорид триметилфениламмония и гидросульфат тетрабутиламмония, являются предпочтительными.
Катализатор межфазного переноса можно использовать в количестве от 1% до 100%, еще предпочтительнее от примерно 3% до 30%, от числа молей соединения (IV).
Растворитель не является ограниченным конкретно при условии, что он не оказывает нежелательного влияния на реакцию. Примеры включают алифатические углеводородные растворители, такие как н-гексан и н-пентан; ароматические углеводородные растворители, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как хлороформ, хлористый метилен и 1,2-дихлорэтан; простые эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран и 1,4-диоксан; кетонные растворители, такие как ацетон и метилэтилкетон; амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид; галогенированные углеводородные растворители, такие как дихлорметан и хлороформ; сложноэфирные растворители, такие как метилацетат и этилацетат; и спиртовые растворители, такие как метанол, этанол, пропанол, изопропанол, н-бутанол и трет-бутанол. Кроме того, также можно использовать воду, ацетонитрил, эфиры уксусной кислоты, ацетон и т.д. Можно использовать либо один из данных растворителей, либо смесь нескольких их типов.
Предпочтительными в качестве растворителя являются ароматические углеводородные растворители, амидные растворители, кетонные растворители и галогенированные углеводородные растворители, и еще более предпочтительными являются толуол, N,N-диметилформамид, N,N-диметилацетамид, ацетон и дихлорметан. Среди данных растворителей еще более предпочтительными являются амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид.
Хотя температура реакции изменяется в зависимости от используемого растворителя, обычно она находится в пределах от -78°С до температуры кипения растворителя, предпочтительно от 40°С до 80°С и еще предпочтительнее составляет примерно 60°С.
Время реакции находится в пределах от 1 до 24 часов. Обычно реакция завершается в течение от 1 до 16 часов.
Непрерывная стадия из соединения (IV) в соединение (VI)
Соединение (VI) может быть сразу же получено смешиванием соединения (IV) с производным диалкилового эфира метиленмалоновой кислоты и обработкой в присутствии основания. Именно таким способом соединение (VI) синтезируют из соединения (IV) сразу же без выделения соединения (V). В обеих стадиях взаимодействие может быть осуществлено с использованием катализатора межфазного переноса. Каждый из продуктов соответстующих стадий может быть получен с высоким выходом и высокой чистотой при проведении стадии получения соединения (V) при комнатной температуре и проведении стадии циклизации соединения (V) при нагревании до примерно 60°С.
Производное диалкилового эфира метиленмалоновой кислоты можно использовать в количестве 1-4-кратного (по молям), предпочтительно от 1,5 до 3-кратного, от числа молей соединения (IV).
Растворитель не является ограниченным конкретно при условии, что он не оказывает нежелательного влияния на реакцию. Примеры включают ароматические углеводородные растворители, такие как бензол, толуол и ксилол; простые эфирные растворители, такие как диэтиловый эфир, тетрагидрофуран и 1,4-диоксан; кетонные растворители, такие как ацетон и метилэтилкетон; амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид; галогенированные углеводородные растворители, такие как дихлорметан и хлороформ; сложноэфирные растворители, такие как метилацетат и этилацетат; и спиртовые растворители, такие как метанол, этанол и изопропанол.
Предпочтительными в качестве растворителя являются ароматические углеводородные растворители, амидные растворители, кетонные растворители и галогенированные растворители, и еще более предпочтительными являются толуол, N,N-диметилформамид, N,N-диметилацетамид, ацетон и дихлорметан.
Основание может представлять собой или органическое основание, или неорганическое основание. Примеры неорганических оснований включают гидриды щелочных металлов, такие как гидрид натрия и гидрид лития; гидриды щелочноземельных металлов, такие как гидрид кальция; гидроксиды щелочных металлов, такие как гидроксид натрия и гидроксид калия; карбонаты или гидрокарбонаты щелочных металлов или щелочноземельных металлов, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия и гидрокарбонат калия; и галогениды щелочных металлов или щелочноземельных металлов, такие как фторид калия, фторид цезия и иодид калия.
Примеры органических оснований включают алкоксиды щелочных металлов, такие как метоксид натрия, метоксид лития, этоксид натрия, этоксид лития, третичный-бутоксид натрия и третичный -бутоксид калия; триалкиламины, такие как триэтиламин и этилдиизопропиламин; производные N,N-диалкиланилина, содержащие алкильные группы, имеющие 1-4 атомов углерода, такие как N,N-диметиланилин и N,N-диэтиланилин; производные пиридина, необязательно замещенные алкильными группами, имеющими 1-4 атомов углерода, такие как пиридин и 2,6-лутидин; и азотсодержащие гетероциклические соединения, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен.
Среди данных оснований предпочтительно использование алкоксидов щелочных металлов, азотсодержащих гетероциклических соединений и гидроксидов щелочных металлов или щелочноземельных металлов. Еще предпочтительнее третичный-бутоксид калия, 1,8-диазабицикло[5.4,0]ундец-7-ен и щелочные гидроксиды, и еще более предпочтителен гидроксид калия.
Основание можно использовать в количестве от 2 до 6-кратного по молям, предпочтительно от 2 до 4-кратного, от числа молей сложноэфирного соединения (IV).
В данной реакции выход может быть повышен при добавлении добавки. Примеры добавок включают катализаторы межфазного переноса и молекулярные сита.
Примеры катализаторов межфазного переноса включают четвертичные соли аммония, такие как хлорид тетра(нормальный-гексил)аммония и иодид тетра(нормальный-гексил)аммония, и краун-эфиры, такие как 18-краун-6, 15-краун-5.
В качестве добавки предпочтительным является катализатор межфазного переноса. Среди всех еще более предпочтительными являются липофильные четвертичные соли аммония.
Катализатор межфазного переноса можно использовать в количестве от 1% до 100%, еще предпочтительнее от примерно 3% до 30%, от числа молей соединения (II).
Хотя температура реакции не является конкретно ограниченной при условии, что она не превышает температуру кипения растворителя, предпочтительно она находится в пределах от комнатной температуры до 60°С.
Хотя время реакции изменяется в зависимости от температуры реакции, оно находится в пределах от 1 часа до 3 дней.
В том случае, когда две стадии осуществляют непрерывно, например, катализатор межфазного переноса добавляют в присутствии основания (гидроксида калия и т.д., 1,5-кратное количество по молям от числа молей соединения (IV)) и смесь перемешивают при комнатной температуре в течение примерно 1 часа. Затем жидкую реакционную смесь нагревают до 60°С и добавляют основание в том же количестве, как описано выше. После перемешивания в течение примерно 5 часов можно получить целевое соединение. То есть, первоначально при перемешивании при комнатной температуре образуется соединение (V), а затем добавляют основание и повышают температуру. Таким образом превращение вплоть до реакции циклизации может быть выполнено в одну стадию.
Стадия из соединения (IV) в соединение (VII)
Соединение (VII) может быть получено обработкой соединения (IV) в присутствии основания для осуществления внутримолекулярной циклизации.
Основание для использования в данном случае может представлять собой либо неорганическое основание, либо органическое основание. Примеры неорганических оснований включают гидриды щелочных металлов, такие как гидрид натрия и гидрид лития; гидриды щелочноземельных металлов, такие как гидрид кальция; гидроксиды щелочных металлов, такие как гидроксид натрия и гидроксид калия; карбонаты или гидрокарбонаты щелочных металлов или щелочноземельных металлов, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия и гидрокарбонат калия; и галогениды щелочных металлов или щелочноземельных металлов, такие как фторид калия, фторид цезия и иодид калия.
Примеры органических оснований включают алкоксиды щелочных или щелочноземельных металлов, такие как метоксид натрия, метоксид лития, метоксид магния, этоксид натрия, этоксид лития, третичный-бутоксид натрия и третичный-бутоксид калия; алкиллитиевые реагенты, такие как н-бутиллитий, метиллитий и диизопропиламид лития; триалкиламины, такие как триэтиламин и этилдиизопропиламин; производные анилина, содержащие алкильные группы, имеющие 1-4 атомов углерода, такие как N,N-диметиланилин и N,N-диэтиланилин; производные пиридина, необязательно замещенные алкильными группами, имеющими 1-4 атомов углерода, такие как пиридин и 2,6-лутидин; и азотсодержащие гетероциклические соединения, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ) и 1,8-диазабицикло[4.3.0]нона-5-ен (ДБН).
Среди данных оснований предпочтительным является использование карбонатов щелочных металлов или щелочноземельных металлов, гидроксидов щелочных металлов, алкоксидов щелочных металлов и гидридов металлов. Более конкретно, еще более предпочтительными являются карбонат калия, гидроксид натрия, третичный-бутоксид калия, третичный -бутоксид натрия (t-BuONa) и гидрид натрия.
Основание можно использовать в количестве от 1 до 15-кратного по молям, предпочтительно от 1 до 3-кратного, от числа молей сложноэфирного соединения соединения (IV).
При использовании карбоната щелочного металла или карбоната щелочного металла, или гидроксида щелочного металла, предпочтительно применять добавку. Примеры добавки включают катализаторы межфазного переноса и молекулярные сита. Примеры катализаторов межфазного переноса включают четвертичные соли аммония, такие как хлорид тетра(нормальный-гексил)аммония, иодид тетра(нормальный-гексил)аммония, бромид тетрабутиламмония и хлорид бензилтриэтиламмония. Также возможно осуществлять изобретение в присутствии иодида щелочного металла или щелочноземельного металла, такого как иодид калия или иодид натрия, и краун-эфира, такого как 18-краун-6, 15-краун-5.
В качестве добавки предпочтительным является катализатор межфазного переноса. Среди всех еще более предпочтительной является липофильная четвертичная соль аммония.
Добавку можно использовать в количестве от 1% до 100%, предпочтительно, от 5% до 30%, от числа молей соединения (IV).
Растворитель не является ограниченным конкретно при условии, что он не оказывает нежелательного влияния на реакцию. Примеры включают ароматические углеводородные растворители, такие как бензол, толуол и ксилол; ароматические углеводородные растворители, такие как н-гексан, н-пентан и циклогексан; простые эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, 1,2-диметоксиэтан, метил-трет-бутиловый эфир (МТВЕ), тетрагидрофуран и 1,4-диоксан; кетонные растворители, такие как ацетон и метилэтилкетон; амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид; галогенированные углеводородные растворители, такие как дихлорметан и хлороформ; сложноэфирные растворители, такие как метилацетат и этилацетат; и спиртовые растворители, такие как метанол, этанол, изопропанол, н-бутанол и трет-бутанол.
В качестве растворителя предпочтительными являются амидные растворители и еще более предпочтительными являются N,N-диметилформамид и N,N-диметилацетамид.
Температура реакции не ограничена конкретно, но обычно она находится в пределах от -78°С до температуры кипения растворителя. Предпочтительно она находится в пределах от комнатной температуры до температуры кипения растворителя.
Хотя время реакции изменяется в зависимости от температуры реакции, оно находится в пределах от 15 минут до 12 часов.
Полученное таким образом соединение (VII) может быть очищено с помощью образования соли с соединением, представленным следующей формулой:
R11-SO3Н
[где R11 представляет фенильную группу (которая может содержать одну или более групп одного или нескольких типов, выбранную из группы, состоящей из атомов галогена, алкильных групп, содержащих 1-6 атомов углерода, галогеналкильных групп, содержащих 1-6 атомов углерода, алкоксигрупп, содержащих 1-6 атомов углерода, нитрогруппы, карбамоильной группы и цианогруппы), группу камфоры (которая может содержать одну или более групп одного или нескольких типов, выбранную из группы, состоящей из атомов галогена, нитрогруппы, карбамоильной группы и цианогруппы, алкильных групп, содержащих 1-6 атомов углерода, галогеналкильных групп, содержащих 1-6 атомов углерода, и алкоксигрупп, содержащих 1-6 атомов углерода), алкильную группу, содержащую 1-6 атомов углерода или галогеналкильную группу, содержащую 1-6 атомов углерода].
Поскольку оптически активный изомер соединения (VII) представляет собой маслянистое вещество, чистота конечного продукта левофлоксацина может быть повышена очисткой с помощью образования такой соли, как описано выше.
Среди данных сульфоновых кислот предпочтительными являются метансульфоновая кислота, пара-толуолсульфоновая кислота и камфорсульфоновая кислота.
Примеры растворителя, используемого при образовании соли, включают углеводородные растворители, такие как н-гексан и н-пентан; ароматические углеводородные растворители, такие как бензол, толуол и ксилол; спиртовые растворители, такие как метанол, этанол, пропанол. изопропанол, н-бутанол и трет-бутанол; простые эфирные растворители, такие как диэтиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран, 1,2-диметоксиэтан и 1,4-диоксан; амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид; галогенированные углеводородные растворители, такие как хлороформ, хлористый метилен и 1,2-дихлорэтан. Кроме того, также можно использовать воду, ацетонитрил, эфиры уксусной кислоты, ацетон и т.д. Можно использовать либо один из данных растворителей, либо смесь их нескольких типов.
Среди данных растворителей предпочтительными являются ароматические углеводородные растворители, такие как толуол, эфиры уксусной кислоты и ацетон.
Обычно растворитель можно использовать в количестве от 1 до 100-кратного по массе, предпочтительно от 2 до 50-кратного по массе.
Хотя температура для кристаллизации целевой соли не является постоянной, здесь можно использовать температурные условия, обычно применяемые в данной области. Говоря более конкретно, кристаллизацию можно проводить в диапазоне от температуры охлаждения льдом до температуры кипения используемого растворителя. Соль может быть образована следующим образом. После завершения реакции циклизации, дающей соединение (VII), растворитель заменяют на другой растворитель, используемый при образовании соли, и затем добавляют сульфоновую кислоту. Не приходится и говорить о том, что жидкая реакционная смесь после кристаллизации может быть обработана и выделена обычным образом для образования, таким образом, соли.
Образованная таким образом соль может быть преобразована в свободное соединение обработкой щелочью. Например, можно использовать основания, включая гидроксиды щелочных металлов, такие как гидроксид натрия и гидроксид калия, карбонат щелочных металлов, такие как карбонат натрия и карбонат калия, и гидрокарбонат щелочного металла, такой как гидрокарбонат натрия и гидрокарбонат калия. Такое основание обычно используют в виде водного раствора и свободное соединение можно выделять экстракцией и т.д.
Стадия из соединения (VII) в соединение (VI)
Соединение (VI) может быть получено взаимодействием соединения (VII) с производным диалкилового эфира метиленмалоновой кислоты.
На данной стадии соединение (VI) может быть получено добавлением производного диалкилового эфира метиленмалоновой кислоты к соединению (VII) и нагреванием, или обработкой соединения (VII) и производного диалкилового эфира метиленмалоновой кислоты в растворителе в присутствии основания.
(1) Способ добавления производного диалкилового эфира метиленмалоновой кислоты к соединению (VII) с последующим нагреванием.
Производное диалкилового эфира метиленмалоновой кислоты можно использовать в количестве от 1 до 3-кратного по молям, предпочтительно от 1,1 до 1,6-кратного, от числа молей соединения (VII).
Реакцию можно осуществлять или без использования растворителя, или в растворителе. В качестве растворителя можно использовать любой растворитель при условии, что он не оказывает влияния на реакцию. Примеры включают ароматические углеводородные растворители, такие как толуол и ксилол.
Предпочтительно проводить реакцию без использования растворителя или с использованием ароматического углеводородного растворителя, такого как толуол или ксилол.
Температура реакции не ограничена конкретно при условии, что она не превышает температуру кипения растворителя. Предпочтительно она находится в пределах от 100°С до 160°С. Хотя время реакции изменяется в зависимости от температуры реакции, обычно она завершается в течение от 1 часа до 1 дня.
(2) Способ обработки соединения (VII) и производного диалкилового эфира метиленмалоновой кислоты в растворителе в присутствии основания и катализатора межфазного переноса.
Производное диалкилового эфира метиленмалоновой кислоты можно использовать в количестве от 1 до 3-кратного по молям, предпочтительно от 1,05 до 2-кратного, от числа молей соединения (VII).
Растворитель не ограничен конкретно при условии, что он не оказывает нежелательного действия на реакцию. Примеры включают ароматические углеводородные растворители, такие как бензол, толуол и ксилол; простые эфирные растворители, такие как диэтиловый эфир, тетрагидрофуран и 1,4-диоксан; кетонные растворители, такие как ацетон и метилэтилкетон, амидные растворители, такие как N,N-диметилформамид и N,N-диметилацетамид; галогенированные углеводородные растворители, такие как дихлорметан и хлороформ; сложноэфирные растворители, такие как метилацетат и этилацетат; и спиртовые растворители, такие как метанол, этанол и изопропанол.
Среди данных растворителей предпочтительными являются ароматические углеводородные растворители, амидные растворители, кетонные растворители и галогенированные растворители, и еще более предпочтительными являются толуол, N,N-диметилформамид, ацетон и дихлорметан.
Основание может представлять собой или органическое основание, или неорганическое основание. Примеры неорганических оснований включают гидриды щелочных металлов, такие как гидрид натрия и гидрид лития; гидриды щелочноземельных металлов, такие как гидрид кальция; алкоксиды щелочных металлов, такие как метоксид натрия, метоксид лития, этоксид натрия, этоксид лития, третичный-бутоксид натрия и третичный-бутоксид калия; гидроксиды щелочных металлов, такие как гидроксид натрия и гидроксид калия; карбонаты или гидрокарбонаты щелочных металлов или щелочноземельных металлов, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия и гидрокарбонат калия; и галогениды щелочных металлов или щелочноземельных металлов, такие как фторид калия, фторид цезия и иодид калия.
Примеры органических оснований включают триалкиламины, такие как триэтиламин и этилдиизопропиламин; производные анилина, содержащие алкильные группы, имеющие 1-4 атомов углерода, такие как N,N-диметиланилин и N,N-диэтиланилин; производные пиридина, необязательно замещенные алкильными группами, имеющими 1-4 атомов углерода, такие как пиридин и 2,6-лутидин; и азотсодержащие гетероциклические соединения, такие как 1,8-диазабицикло[5.4.0]ундец-7-ен.
В качестве основания алкоксиды щелочных металлов являются предпочтительными, и еще более предпочтительным является третичный-бутоксид калия.
Основание можно использовать в количестве от 1 до 3-кратного по молям, предпочтительно от 1 до 2-кратного, от числа молей сложноэфирного соединения (VII).
Хотя время реакции изменяется в зависимости от температуры реакции, она обычно завершается в течение от 1 часа до 1 дня.
При осуществлении способов, как описано выше, соединение (VI) может быть получено из соединения (I). Предполагается, что вследствие этого также можно использовать следующую стадию в дополнение к данным способам.

Полученное таким образом соединение (VI-a) можно превратить в левофлаксацин известным способом. Теперь способ будет кратко описан. А именно, соединение (VI-a) подвергают циклизации нагреванием вместе с полифосфорной кислотой или ее сложным эфиром с получением сложноэфирного соединения трициклической карбоновой кислоты. Затем данный сложный эфир карбоновой кислоты гидролизуют в основных или кислотных условиях, получая соединение трициклической карбоновой кислоты. Данное соединение трициклической карбоновой кислоты затем подвергают взаимодействию с 4-метилпиперазином в присутствии основания и таким образом может быть получен левофлоксацин. Основание может представлять собой или неорганическое основание, или органическое основание. Примеры неорганического основания включают карбонаты и гидрокарбонаты щелочных металлов или щелочноземельных металлов. Примеры органических оснований включают триалкиламины и азотсодержащие гетероциклические соединения. Говоря более конкретно, можно использовать в избытке триэтиламин, трибутиламин, этилдиизопропиламин и т.д. или 4-метилморфолин, диметиламинопиридин и т.д., или 4-метилпиперазин, которые, таким образом, служат также в качестве основания. Предпочтительным является использование растворителя в данной реакции и в качестве растворителя можно использовать диметилсульфоксид. При реакции 4-метилпиперазина более эффективным является использование не соединения трициклической карбоновой кислоты, а хелатного соединения дигалогенбора данной карбоновой кислоты. Такое хелатное соединение дигалогенбора может быть получено взаимодействием соединения трициклической карбоновой кислоты с соединением тригалогенбора. При этом удобно использование комплекса соединения тригалогенбора с соединением простого эфира, например, комплекс диэтилового эфира или комплекс тетрагидрофурана. В качестве атома галогена предпочтительным является атом фтора. Перемешиванием данного эфирного комплекса с карбоновой кислотой в различных простых эфирных растворителях может быть получено хелатное соединение дигалогенбора карбоновой кислоты. Взаимодействие с 4-метилпиперазином может быть осуществлено в растворителе в присутствии основания, аналогично описанному выше случаю. Хелатное соединение дигалогенбора карбоновой кислоты может быть получено в одну стадию нагреванием соединения (VI-a), соединения дигалогенбора (предпочтительно комплекса с соединением простого эфира) в растворителе (например, уксусном ангидриде). По завершении реакции с 4-метилпиперазином необходимо удалить (гидролизовать) хелат. Это можно осуществить при нагревании в апротонном растворителе в присутствии основания, что приводит к расщеплению и элиминированию. Например, можно привести нагревание в спиртовом растворителе в присутствии триалкиламина. Говоря более конкретно, соединение можно нагревать при перемешивании в этаноле в присутствии триэтиламина.
ЛУЧШИЙ СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Теперь настоящее изобретение будет проиллюстрировано более подробно со ссылкой на следующие примеры. Однако, не следует понимать, что настоящее изобретение ограничено данными примерами.
Пример 1: Метил (2S)-2-(2,3,4-трифторанилино)пропионат
При охлаждении льдом метил D-лактат (8,5 г) и 2,6-лутидин (11,4 г) растворяли в дихлорметане (100 мл). После добавления по каплям безводной трифторметансульфоновой кислоты (25,4 г), смесь нагревали до комнатной температуры и перемешивали в течение 30 минут. Затем опять охлаждали до 0°С и добавляли к смеси по каплям раствор (30 мл) 2,3,4-трифторанилина (12,0 г) в дихлорметане. Смесь перемешивали при той же температуре в течение 17 часов. К полученному раствору добавляли хлористоводородную кислоту (0,5 моль/л) и смесь экстрагировали дихлорметаном. Экстракт промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния и фильтровали. После упаривания растворителя полученный таким образом остаток подвергали колончоной хроматографии на силикагеле. Таким образом, получали 17,1 г (90%) указанного в заголовке соединения в виде маслянистого вещества. Оптическая чистота, определенная по ВЭЖХ, составляла 97% эи.
1H-ЯМР (CDCl3, 270 МГц) δ: 1,51 (д, J=6,9 Гц, 3Н), 3,73 (с, 3Н), 4,07-4,13 (м, 1Н), 4,22 (ушир.с, 1Н), 6,22-6,31 (м, 1Н), 6,73-6,85 (м, 1Н)
ИК спектр (светлое нефтяное масло глубокой очистки): 3407, 2994, 2956, 1739 см-1
Масс-спектр; m/z: 233 (М+)
Пример 2: Метил (2S)-2-(2,3,4-трифторанилино)пропионат
2,3,4-Трифторанилин (100 мг) растворяли в толуоле (1 мл). После добавления карбоната калия (188 мг), метил (2R)-2-[[(4-метилфенил)сульфонил]окси]пропионата (193 мг) и хлорида тетрагексиламмония (40 мг) смесь перемешивали при нагревании и кипении с обратным холодильником в течение 15,5 часов. После обработки как в примере 1 полученный продукт анализировали ВЭЖХ с обращенной фазой с использованием соединения примера 1 в качестве образца. В результате продукт соответствовал 41 мг (26%) указанного в заголовке соединения.
Пример 3: Метил (2S)-2-(2,3,4-трифторанилино)пропионат
В соответствии со способом примера 2 реакцию конденсации проводили с использованием 2,3,4-трифторанилина (100 мг), карбоната калия (188 мг), метил (2R)-2-[(метансульфонил)-окси)]пропионата (78 мг) и хлорида тетрагексиламмония (40 мг), получая указанное в заголовке соединение в виде маслянистого вещества. По данным анализа ВЭЖХ с обращенной фазой с использованием соединения примера 1 в качестве образца продукт соответствовал 38 мг (24%) указанного в заголовке соединения.
Пример 4: Метил (2S)-2-(2,3,4-трифторанилино)пропионат
В соответствии со способом примера 2 реакцию конденсации проводили с использованием 2,3,4-трифторанилина (100 мг), карбоната калия (188 мг), метил (2R)-хлорпропионата (92 мг) и хлорида тетрагексиламмония (40 мг). По данным анализа ВЭЖХ с обращенной фазой с использованием соединения примера 1 в качестве образца продукт соответствовал 56 мг (36%) указанного в заголовке соединения.
Пример 5: Метил 2-(2,3,4-трифторанилино)пропионат
2,3,4-Трифторнитробензол (100 г) и метилпируват (57,6 г) растворяли в метаноле (1000 мл). После добавления 5% Pd-C (20,0 г) и безводного сульфата магния (90 г), смесь перемешивали при комнатной температуре в атмосфере водорода в течение 16 часов. Затем жидкую реакционную смесь фильтровали через целит для удаления, таким образом, Pd-C и сульфата магния.
Полученный фильтрат концентрировали при пониженном давлении и Florisil (100 г) и диэтиловый эфир (700 мл) добавляли к остатку. После перемешивания в течение 2 часов жидкую реакционную смесь фильтровали. Полученный органический слой упаривали и выпавшие при этом в осадок кристаллы отфильтровывали, промывая гексаном. Таким образом, указанное в заголовке соединение (128,2 г) получали в виде слегка желтоватых кристаллов. Температура плавления: от 41 до 43°С.
1H-ЯМР (CDCl3) δ: 1,51 (д, J=6,9 Гц, 3Н), 3,74 (с, 3Н), 4,0-4,3 (м, 2Н), 6,2-6,4 (м, 1Н), 6,7-6,9 (м, 1Н).
ИК спектр (KBr): 3357, 1719, 1510 см-1
Элементный анализ для С10Н10NO2F3
Вычислено (%): С, 51,51; Н, 4,32; N, 6,01
Найдено (%): С, 51,65; Н, 4,31; N, 5,99
Пример 6: Метил 2-(2,3,4-трифторанилино)пропионат
2,3,4-Трифторанилин (2,94 г) и метилпируват (2,04 г) растворяли в метаноле (30 мл). После добавления 5% Pd-C (2,0 г) и безводного сульфата магния (2,65 г), смесь перемешивали при 50°С в атмосфере водорода в течение 16 часов. После отфильтровывания Pd-C и сульфата магния, полученный фильтрат концентрировали при пониженном давлении. Выпавшие при этом в осадок кристаллы отфильтровывали, промывая гексаном. Таким образом, указанное в заголовке соединение (4,44 г) получали в виде слегка желтоватых кристаллов. Различные спектральные данные продукта были идентичны данным, полученным в примере 5.
Пример 7: Этил 2-(2,3,4-трифторанилино)пропионат
2,3,4-Трифторнитробензол (3,54 г) и метилпируват (2,32 г) растворяли в этаноле (30 мл). После добавления 5% Pd-C (2,0 г) и безводного сульфата магния (2,65 г), смесь перемешивали при 50°С в атмосфере водорода в течение 16 часов. После отфильтровывания Pd-C и сульфата магния полученный фильтрат концентрировали при пониженном давлении. Полученный при этом остаток подвергали колоночной хроматографии на силикагеле (этилацетат-нормальный гексан=1:4), получая указанное в заголовке соединение (4,84 г) в виде бледно-желтого маслянистого вещества. 1Н-ЯМР (CDCl3) δ: 1,25 (т, J=7,1 Гц, 3Н), 1,50 (д, J=7,1 Гц, 3Н), 4,0-4,3 (м, 2Н), 4,19 (дд, J=7,3, 10,9 Гц, 3Н), 6,2-6,4 (м, 1Н), 6,7-6,9 (м, 1Н).
ИК спектр (см-1): 1737, 1524, 909
Пример 8: Этил 2-(2,3,4-трифторанилино)пропионат
2,3,4-Трифторанилин (2,94 г) и этилпируват (2,32 г) растворяли в метаноле (30 мл). После добавления 5% Pd-C (2,0 г) и безводного сульфата магния (2,65 г) смесь перемешивали при 50°С в атмосфере водорода в течение 16 часов. После отфильтровывания Pd-C и сульфата магния, полученный фильтрат концентрировали при пониженном давлении. Полученный остаток подвергали колоночной хроматографии на силикагеле (этилацетат-нормальный гексан=1:4), получая таким образом указанное в заголовке соединение (4,69 г) в виде бледно-желтого маслянистого вещества. Различные спектральные данные продукта были идентичны данным, полученным в примере 7.
Пример 9: Метил 2-(2,3,4-трифторанилино)пропионат
2,3,4-Трифторанилин (1,01 г) и метилпируват (0,87 г) растворяли в метаноле (8 мл). После добавления 5% Pd-C (0,11 г) и конц. хлористоводородной кислоты (0,03 г) смесь перемешивали при 40°С под давлением газообразного водорода 2,94 МПа (преобразовано из 30 кгс/см2) в течение 2 часов. После отфильтровывания Pd-C полученный фильтрат концентрировали при пониженном давлении. Таким образом, указанное в заголовке соединение (1,31 г) получали в виде слегка желтоватых кристаллов. Различные спектральные данные продукта были идентичны данным, полученным в примере 5.
Пример 10: Этил 2-(2,3,4-трифторанилино)пропионат
2,3,4-Трифторнитробензол (1,01 г) и этилпируват (1,15 г) растворяли в этаноле (8 мл). После добавления 5% Pd-C (0,11 г) и конц. хлористоводородной кислоты (0,03 г) смесь перемешивали при 40°С под давлением газообразного водорода 2,94 МПа в течение 3 часов. После отфильтровывания Pd-C полученный фильтрат концентрировали при пониженном давлении. Таким образом, указанное в заголовке соединение (1,38 г) получали в виде бледно-желтого маслянистого вещества. Различные спектральные данные продукта были идентичны данным, полученным в примере 7.
Пример 11: Метил 2-(2,3,4-трифторанилино)пропионат
2,3,4-Трифторанилин (0,83 г) и метилпируват (0,87 г) растворяли в метаноле (8 мл). После добавления 5% Pd-C (0,11 г) и конц. хлористоводородной кислоты (0,03 г) смесь перемешивали при 40°С под давлением газообразного водорода 2,94 МПа в течение 2 часов. После отфильтровывания Pd-C полученный фильтрат концентрировали при пониженном давлении. Таким образом, указанное в заголовке соединение (1,26 г) получали в виде слегка желтоватых кристаллов. Различные спектральные данные продукта были идентичны данным, полученным в примере 5.
Пример 12: Этил 2-(2,3,4-трифторанилино)пропионат
2,3,4-Трифторанилин (0,83 г) и этилпируват (1,15 г) растворяли в этаноле (8 мл). После добавления 5% Pd-C (0,11 г) и конц. хлористоводородной кислоты (0,03 г) смесь перемешивали при 40°С под давлением газообразного водорода 2,94 МПа в течение 3 часов. После отфильтровывания Pd-C полученный фильтрат концентрировали при пониженном давлении. Таким образом, указанное в заголовке соединение (1,32 г) получали в виде бледно-желтого маслянистого вещества. Различные спектральные данные продукта были идентичны данным, полученным в примере 7.
Пример 13: 2-(2,3,4-Трифторанилино)пропионовая кислота
2,3,4-Трифторнитробензол (5,03 г) и пировиноградную кислоту (2,75 г) растворяли в изопропаноле (IPA; 40 мл). После добавления 10% Pd-C (0,21 г) смесь перемешивали при 40°С при атмосферном давлении в атмосфере водорода в течение 3 часов. После отфильтровывания Pd-C полученный фильтрат концентрировали при пониженном давлении. Таким образом, указанное в заголовке соединение (6,11 г) получали в виде бесцветных кристаллов. Различные спектральные данные продукта были идентичны данным для образца, синтезированного отдельно.
Пример 14: 2-(2,3,4-Трифторанилино)пропионовая кислота
2,3,4-Трифторнитробензол (5,03 г) и пировиноградную кислоту (2,75 г) растворяли в IPA (40 мл). После добавления 10% Pd-C (0,21 г) смесь перемешивали при 40°С под давлением газообразного водорода 2,94 МПа в течение 3 часов. После отфильтровывания Pd-C полученный фильтрат концентрировали при пониженном давлении. Таким образом, указанное в заголовке соединение (6,09 г) получали в виде бесцветных кристаллов. Различные спектральные данные продукта были идентичны данным для образца, синтезированного отдельно.
Пример 15: 2-(2,3,4-Трифторанилино)пропионовая кислота
2,3,4-Трифторнитробензол (1,01 г) и пировиноградную кислоту (0,75 г) растворяли в метаноле (8 мл). После добавления 5% Pd-C (0,11 г) смесь перемешивали при 40°С под давлением газообразного водорода 4,9 МПа (преобразовано из 50 кгс/см2) в течение 5 часов. После отфильтровывания Pd-C полученный фильтрат концентрировали при пониженном давлении. Таким образом, указанное в заголовке соединение (1,20 г) получали в виде бесцветных кристаллов. Различные спектральные данные продукта были идентичны данным для образца, синтезированного отдельно.
Пример 16: 2-(2,3,4-Трифторанилино)пропионовая кислота
2,3,4-Трифторанилин (4,18 г) и пировиноградную кислоту (2,75 г) растворяли в IPA (40 мл). После добавления 10% Pd-C (0,21 г) смесь перемешивали при 40°С при атмосферном давлении в атмосфере водорода в течение 3 часов. После отфильтровывания Pd-C, полученный фильтрат концентрировали при пониженном давлении. Таким образом, указанное в заголовке соединение (5,69 г) получали в виде бесцветных кристаллов. Различные спектральные данные продукта были идентичны данным для образца, синтезированного отдельно.
Пример 17: N-(1-Метоксикарбонилэтилиден)-2,3,4-трифторанилин
Трифторанилин (1 г) и сульфат магния (1,36 г) перемешивали в метаноле (5 мл) при комнатной температуре. После добавления метилпирувата (1,27 г) смесь нагревали до 40°С и перемешивали в течение 20 часов. По завершении реакции, сульфат магния отфильтровывали. Полученный таким образом фильтрат концентрировали при пониженном давлении и остаток подвергали колоночной хроматографии на силикагеле (гексан-диэтиловый эфир=1:3), получая, таким образом, указанное в заголовке соединение (552 мг) в виде кристаллов с метанолом.
1Н-ЯМР (CDCl3) δ: 6,92-6,74 (м, 2Н), 5,09 (ушир.с, 1Н), 3,84 (с, 3Н), 3,24 (с, 3Н), 1,65 (с, 3Н).
Пример 18: Метил (2S)-2-(2,3,4-трифторанилино)пропионат
Димер хлор-1,5-циклооктадиениридия (12,8 мг) и (2S,4S)-BCPM (23,6 мг) растворяли в IPA (2 мл) в токе газообразного аргона и перемешивали при комнатной температуре в течение 1 часа. К жидкой реакционной смеси добавляли раствор монометанольного кристаллического N-(1-метоксикарбонил-этилиден)-2,3,4-трифторанилина (50 мг) в IPA (2 мл). Жидкую реакционную смесь переносили в автоклав и создавали давление водорода 50 кг/см2. Затем жидкую реакционную смесь перемешивали при 10°С в течение 15 часов. Химический выход и оптическая чистота указанного в заголовке соединения, содержащегося в жидкой реакционной смеси, измеренные с использованием высокоэффективной жидкостной хроматографии, составляли 70% и 50% эи (S-соединение), соответственно.
Примеры 19-22:
Изменяя оптическиактивный лиганд, иминосоединения асимметрически восстанавливали таким же способом, как в описанной выше реакции. Результаты данных примеров приведены в таблице 1.
(2S,4S)-BCPM:
(2S,4S)-N-(трет-бутоксикарбонил)-4-(дициклогексилфосфино)-2-[(дифенилфосфино)метил]пирролидин
(S)-(R)-JOSIPHOS:
(S)-1-[(R)-2-(дифенилфосфино)ферроценил]этилдициклогексилфосфин (4R,5R)-MOD-DIOP:
(4R,5R)-4,5-бис[[бис(4'-метокси-3',5'-диметилфенил)-фосфино]метил]-2,2-диметил-1,3-диоксолан
Пример 23: 2-(2,3,4-Трифторанилино)пропионовая кислота
Метил 2-(2,3,4-трифторанилино)пропионат (46,64 г) растворяли в метаноле (130 мл) и медленно добавляли к нему при 0°С водный раствор (3 моль/л; 100 мл) гидроксида лития. После перемешивания при комнатной температуре в течение 3 часов растворитель выпаривали. После добавления воды остаток промывали хлороформом. Затем хлористоводородную кислоту (6 моль/л) медленно добавляли к водному слою до достижения значения рН 1. Затем водный слой экстрагировали диизопропиловым эфиром (IPE). Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали, получая указанное в заголовке соединение (43,7 г) в виде бесцветных кристаллов.
Температура плавления: от 114 до 119°С
1H-ЯМР (CDCl3) δ: 1,57 (д, J=6,9 Гц, 3Н), 4,11 (дд, J=6,9, 10,3 Гц, 1Н), 6,2-6,4 (м, 1H), 6,7-6,9 (м, 1Н)
ИК спектр (см-1): 3357, 1725, 1524, 1195
Элементный анализ для С9Н8NO2F3
Вычислено (%): С, 49,32; Н, 3,68; N, 6,39
Найдено (%): С, 49,33; Н, 3,65; N, 6,34
Пример 24: 2-(2,3,4-Трифторанилино)пропионовая кислота
Этил 2-(2,3,4-Трифторанилино)пропионат (2,47 г) растворяли в этаноле (40 мл) и при 0°С медленно добавляли к нему водный раствор (3 моль/л; 10 мл) гидроксида натрия. После перемешивания при комнатной температуре в течение 3 часов растворитель выпаривали. После добавления воды остаток промывали хлороформом. Затем к водному слою медленно добавляли хлористоводородную кислоту (6 моль/л) до достижения значения рН 1. Затем водный слой экстрагировали IPE. Органический слой сушили над безводным сульфатом магния и затем растворитель выпаривали, давая указанное в заголовке соединение (2,19 г) в виде бесцветных кристаллов. Различные спектральные данные продукта были идентичны данным, полученным в примере 23.
Пример 25: Соль (2S)-2-(2,3,4-трифторанилино)пропионовая кислота·(R)-1-фенилэтиламин
2-(2,3,4-Трифторанилино)пропионовую кислоту (1,1 г) растворяли в смеси растворителей (15 мл; метанол-IPE=1:20). При комнатной температуре медленно добавляли раствор (15 мл) (R)-1-фенилэтиламина (333,2 мг) в смеси растворителей (метанол-IPE=1:20). Полученную суспензию перемешивали при комнатной температуре дополнительно в течение 2 часов и затем фильтровали, промывая IPE. Таким образом, указанное в заголовке соединение получали в виде бесцветных кристаллов (802 мг). Оптическая чистота данного продукта составляла 80% эи. Впоследствии к полученной соли добавляли хлороформ и смесь перемешивали при 50°С в течение 18 часов. Затем суспензию фильтровали, промывая IPE, получая 703 мг указанного в заголовке соединения в виде бесцветных кристаллов.
Оптическая чистота данного продукта составляла 99% эи.
[α]D=5,7°(с=0,386, метанол)
Температура плавления (разложение): от 189 до 197°С
1H-ЯМР (CD3OD) δ: 1,41 (д, J=6,9 Гц, 3Н), 1,61 (д, J=6,9 Гц, 3Н), 3,80 (дд, J=6,9, 15,4Гц, 1Н), 4,42 (дд, J=6,9, 10,0 Гц, 1Н), 6,3-6,5 (м, 1Н), 6,7-6,9(м, 1Н), 7,3-7,5 (м, 5Н)
Элементный анализ для C17H19NO2F3
Вычислено (%): С, 60,86; Н, 5,96; N, 7,91
Найдено (%): С, 61,01; Н, 5,97; N, 7,85
Пример 26: Соль (2S)-2-(2,3,4-трифторанилино)пропионовая кислота·(R)-1-триэтиламин
2-(2,3,4-Трифторанилино)пропионовую кислоту (1,1 г) растворяли в смеси растворителей (15 мл; метанол-IPE=1:20). При комнатной температуре медленно добавляли раствор (15 мл) (R)-1-толилэтиламина (371,8 мг) в смеси растворителей (метанол-IPE=1:20). Полученную суспензию перемешивали при комнатной температуре дополнительно в течение 2 часов и затем фильтровали, промывая IPE. Таким образом, указанное в заголовке соединение получали в виде бесцветных кристаллов (860 мг). Оптическая чистота данного продукта составляла 52% эи. Впоследствии к полученной соли добавляли хлороформ и смесь перемешивали при 50°С в течение 18 часов. Затем суспензию фильтровали, промывая IPE, получая 591 мг указанного в заголовке соединения в виде бесцветных кристаллов. Оптическая чистота данного продукта составляла 99% эи.
[α]D=-2,0° (с=0,197, метанол)
Температура плавления (разложение): от 190 до 197°С
1H-ЯМР (CD3OD) δ: 1,41 (д, J=6,9 Гц, 3Н), 1,59 (д, J=6,9 Гц, 3Н), 2,35 (с, 3Н), 3,80 (дд, J=6,9, 12,0 Гц, 1Н), 4,38 (дд, J=6,9, 12,0 Гц, 1Н), 6,3-6,5 (м, 1Н), 6,7-6,9 (м, 1Н), 7,2-7,3 (м, 4Н)
Элементный анализ для C18H21NO2F3
Вычислено (%): С, 59,99; Н, 5,63; N, 8,23
Найдено (%): С, 59,96; Н, 5,67; N, 8,16
Пример 27: Соль (2S)-2-(2,3,4-трифторанилино)пропионовая кислота·(S)-1-фенил-2-п-триэтиламин
2-(2,3,4-Трифторанилино)пропионовую кислоту (1,1 г) растворяли в смеси растворителей (15 мл; метанол-IPE=1:20). При комнатной температуре медленно добавляли раствор (15 мл) (R)-1-п-толилэтиламина (581,8 мг) в смеси растворителей (метанол-IPE=1:20). Полученную суспензию перемешивали при комнатной температуре дополнительно в течение 2 часов и затем фильтровали, промывая IPE. Таким образом, указанное в заголовке соединение получали в виде 1,1 г бесцветных кристаллов. Оптическая чистота данного продукта составляла 79% эи. Впоследствии к полученной соли добавляли хлороформ и смесь перемешивали при 50°С в течение 18 часов. Затем суспензию фильтровали, промывая IPE, получая 923 мг указанного в заголовке соединения в виде бесцветных кристаллов. Оптическая чистота данного продукта составляла 99% эи.
[α]D=-5,6° (с=0,386, метанол)
Температура плавления (разложение): от 187 до 193°С
1Н-ЯМР (CD3OD) δ: 1,41 (д, J=6,9 Гц, 3Н), 2,26 (с 3Н), 3,0-3,3 (м, 2Н), 3,81 (дд, J=6,9, 11,7 Гц, 1Н),4,43 (дд, J=6,6, 8,3 Гц), 6,3-6,5 (м, 1Н), 6,7-6,9 (м, 1Н), 7,00 (дд, J=7,9, 21,0 Гц), 7,2-7,3 (м, 5Н)
Элементный анализ для С23Н23O2F3
Вычислено (%): С, 66,96; Н, 5,85; N, 6,51
Найдено (%): С, 56,85; Н, 5,89; N, 6,44
Пример 28: (2S)-2-(2,3,4-Трифторанилино)пропионовая кислота
К соли (2S)-2-(2,3,4-трифторанилино)пропионовая кислота·(S)-1-фенилэтиламин (1,0 г; 99% эи) добавляли IPE (20 мл) и хлористоводородную кислоту (1 моль/л) до достижения значения рН 1 и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Органический слой сушили над безводным сульфатом магния. После упаривания растворителя получали 618 мг указанного в заголовке соединения в виде бесцветных кристаллов. Оптическая чистота данного продукта составляла 99% эи. 1H-ЯМР и ИК спектральные данные для продукта были идентичны данным соединения, полученного в примере 23.
Пример 29: (2S)-2-(2,3,4-Трифторанилино)пропионовая кислота
К соли (2S)-2-(2,3,4-трифторанилино)пропионовая кислота·(S)-1-триэтиламин (1,0 г; 99% эи) добавляли IPE (22 мл) и хлористоводородную кислоту (1 моль/л) до достижения значения рН 1 и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Органический слой сушили над безводным сульфатом магния. После упаривания растворителя получали 645 мг указанного в заголовке соединения в виде бесцветных кристаллов. Оптическая чистота данного продукта составляла 99% эи. 1H-ЯМР и ИК спектральные данные для продукта были идентичны данным соединения, полученного в примере 23.
Пример 30: (2S)-2-(2,3,4-Трифторанилино)пропионовая кислота
К соли (2S)-2-(2,3,4-трифторанилино)пропионовая кислота·(R)-1-фенил-2-п-триэтиламин (1,0 г; 99% эи) добавляли IPE (25 мл) и хлористоводородную кислоту до достижения значения рН 1 и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Органический слой сушили над безводным сульфатом магния. После упаривания растворителя получали 510 мг указанного в заголовке соединения в виде бесцветных кристаллов. Оптическая чистота данного продукта составляла 99% эи. 1H-ЯМР и ИК спектральные данные для продукта были идентичны данным соединения, полученного в примере 23.
Пример 31: Метил (2S)-2-(2,3,4-трифторанилино)пропионат
(2S)-2-(2,3,4-трифторанилино)пропионовую кислоту (1,1 г; 99% эи) растворяли в метаноле (10 мл) и добавляли хлористоводородную кислоту (5 моль/л; 1 мл) при комнатной температуре. Жидкую реакционную смесь нагревали при кипении с обратным холодильником в течение 6 часов и затем растворитель выпаривали. К полученному остатку добавляли хлороформ (10 мл). Затем органический слой промывали насыщенным водным раствором хлорида натрия и водой и сушили над безводным сульфатом магния. После упаривания растворителя полученный остаток подвергали колоночной хроматографии на силикагеле (этилацетат-нормальный гексан=1:4), давая указанное в заголовке соединение (1,17 г) в виде маслянистого вещества. Оптическая чистота продукта составляла 99% эи. 1H-ЯМР и ИК спектральные данные для продукта были идентичны данным соединения, полученного в примере 5.
[α]D=-49,4° (с=0,119, метанол)
Пример 32: Метил (2R)-2-(2,3,4-трифторанилино)пропионат
(2R)-2-(2,3,4-трифторанилино)пропионовую кислоту (1,1 г; 98% эи) растворяли в метаноле (10 мл) и добавляли хлористоводородную кислоту (5 моль/л; 1 мл) при комнатной температуре. Жидкую реакционную смесь нагревали при кипении с обратным холодильником в течение 6 часов и затем растворитель выпаривали. К полученному остатку добавляли хлороформ (10 мл). Затем органический слой промывали насыщенным водным раствором хлорида натрия и водой и сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле (этилацетат-нормальный гексан=1:4), давая указанное в заголовке соединение (1,17 г) в виде маслянистого вещества. Оптическая чистота продукта составляла 99% эи. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 5.
Пример 33: Этил (2S)-2-(2,3,4-трифторанилино)пропионат
(2S)-2-(2,3,4-Трифторанилино)пропионовую кислоту (219 мг; 99% эи) растворяли в этаноле (2 мл) и добавляли хлористоводородную кислоту (5 моль/л; 0,2 мл) при комнатной температуре. Жидкую реакционную смесь нагревали при кипении с обратным холодильником в течение 6 часов и затем растворитель выпаривали. К полученному остатку добавляли хлороформ. Затем, органический слой промывали насыщенным водным раствором хлорида натрия и водой и сушили над безводным сульфатом магния. После упаривания растворителя полученный остаток подвергали колоночной хроматографии на силикагеле (этилацетат-нормальный гексан=1:4), получая указанное в заголовке соединение (246 мг) в виде бледно-желтого маслянистого вещества. Оптическая чистота продукта составляла 99% эи. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 7.
[α]D=-57,2° (с=0,352, метанол)
Пример 34: Этил (2R)-2-(2,3,4-трифторанилино)пропионат
(2R)-2-(2,3,4-трифторанилино)пропионовую кислоту (219 мг; 99% эи) растворяли в этаноле (2 мл) и добавляли к раствору хлористоводородную кислоту (5 моль/л; 0,2 мл) при комнатной температуре. Жидкую реакционную смесь нагревали при кипении с обратным холодильником в течение 6 часов и затем растворитель выпаривали. К полученному остатку добавляли хлороформ (10 мл). Затем органический слой промывали насыщенным водным раствором хлорида натрия и водой и сушили над безводным сульфатом магния. После упаривания растворителя полученный остаток подвергали колоночной хроматографии на силикагеле (этилацетат-нормальный гексан=1:4), получая указанное в заголовке соединение (245 мг) в виде бледно-желтого маслянистого вещества. Оптическая чистота продукта составляла 98% эи. 1Н-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 7.
Пример 35: (2S)-2-(2,3,4-трифторанилино)пропионовая кислота
Метил 2-(2,3,4-трифторанилино)пропионат (2,0 г) суспендировали в 0,1 М фосфатном буферном растворе (рН 6,5; 400 мл). После добавления Протеазы N (полученной Amano Seiyaku, из бактерий, принадлежащих к Bacillus; 0,4 г), смесь осторожно перемешивали. Смесь дополнительно перемешивали в течение 14 часов, поддерживая температуру 30°С. После добавления хлористого метилена жидкую реакционную смесь фильтровали через целит для удаления денатурированного белка и затем разделяли. Органический слой промывали 5%-ным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Затем растворитель выпаривали при пониженном давлении, получая таким образом метил (2R)-2-(2,3,4-трифторанилино)пропионат (0,94 г). Оптическая чистота данного продукта составляла 98% эи. С другой стороны, все водные слои, полученные при разделении, объединяли и доводили до рН 2 10%-ной хлористоводородной кислотой с последующей экстракцией IPE. Органический слой сушили над безводным сульфатом магния и упаривали. Таким образом, указанное в заголовке соединение получали в виде неочищенного продукта (0,96 г). Оптическая чистота данного продукта составляла 96% эи. Далее неочищенный продукт перекристаллизовывали из смеси растворителей, состоящей из изопропилового эфира и гексана. Таким образом, получали указанное в заголовке соединение со 100% эи. 1Н-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 28.
Пример 36: (2R)-2-(2,3,4-трифторанилино)пропионовая кислота
Метил 2-(2,3,4-трифторанилино)пропионат (1,0 г) суспендировали в 0,1 М фосфатном буферном растворе (рН 6,5; 200 мл). После добавления α-химотрипсина (полученного от Sigma; 0,2 г), смесь осторожно перемешивали. Смесь дополнительно перемешивали в течение 16 часов, поддерживая температуру 30°С. После добавления хлористого метилена жидкую реакционную смесь фильтровали через целит для удаления денатурированного белка и затем разделяли. Органический слой промывали 5%-ным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом магния. Затем растворитель выпаривали при пониженном давлении, получая таким образом метил (2S)-2-(2,3,4-трифторанилино)пропионат (0,43 г). Оптическая чистота данного продукта составляла 98% эи. С другой стороны все водные слои, полученные при разделении, объединяли и доводили до рН 2 10%-ной хлористоводородной кислотой с последующей экстракцией IPE. Органический слой сушили над безводным сульфатом магния и упаривали. Таким образом, указанное в заголовке соединение получали в виде неочищенного продукта (0,47 г). Оптическая чистота данного продукта составляла 92% эи. Далее неочищенный продукт перекристаллизовывали из смеси растворителей, состоящей из изопропилового эфира и гексана. Таким образом, получали указанное в заголовке соединение со 100% эи. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 29.
Примеры 37-42:
Реакции осуществляли, как в примере 36, но используя различные субстраты и катализаторы (ферменты и микроорганизмы), подвергнутые реакции асимметрического гидролиза (см. табл.2).
Пример 43: (2S)-2-(2,3,4-Трифторанилино)пропионовая кислота
Микробные клетки (JAM-1623; Bacillus subtilis) культивировали в среде бульона (рН 7,0; 50 мл) при 30°С в течение 14 часов. После удаления среды из полученной таким образом культуры центрифугированием клетки подвергали сублимационной сушке, получая высушенные сублимированные клетки. Метил 2-(2,3,4-трифторанилино)пропионат (2,0 г) суспендировали в фосфатном буферном растворе (рН 6,5; 100 мл). Затем добавляли вышеописанные сублимированные микробные клетки (0,2 г) и осторожно перемешивали. Смесь перемешивали дополнительно в течение 6 часов, поддерживая температуру 30°С. После добавления хлористого метилена жидкую реакционную смесь фильтровали через целит для удаления таким образом денатурированного белка и затем отделяли. Органический слой промывали 5% водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Затем растворитель выпаривали при пониженном давлении, получая таким образом метил (2R)-2-(2,3,4-трифторанилино)пропионовую кислоту (0,92 г). Оптическая чистота данного продукта составляла 97% эи. С другой стороны, все водные слои, полученные при разделении, объединяли и доводили до рН 2 10%-ной хлористоводородной кислотой с последующей экстракцией IPE. Органический слой сушили над безводным сульфатом магния и упаривали, получая таким образом указанное в заголовке соединение в виде 0,97 г бесцветных кристаллов неочищенного продукта. Оптическая чистота данного продукта составляла 96% эи. Далее неочищенный продукт перекристаллизовывали из смеси растворителей, состоящей из изопропилового эфира и гексана. Таким образом, получали указанное в заголовке соединение со 100% эи. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 28.
Пример 44: (2S)-2-(2,3,4-Трифторанилино)пропионовая кислота
Микробные клетки (IFO-1575; Zygoascus hellenicus) культивировали в среде MY (pH 6,0; 50 мл) при 30°С в течение 48 часов. Метил 2-(2,3,4-трифторанилино)пропионат (1,0 г) суспендировали в 0,1 М фосфатном буферном растворе (pH 6,5; 90 мл). Затем добавляли к нему описанную выше жидкую культуру (10 мл) и осторожно перемешивали. Смесь перемешивали дополнительно в течение 16 часов, поддерживая температуру при 30°С. Затем ее обрабатывали, как в примере 43, получая таким образом метил (2R)-2-(2,3,4-трифторанилино)пропионат (0,39 г, оптическая чистота 91% эи) и указанное в заголовке соединение (0,45 г, оптическая чистота 84% эи).
Когда такую же, как описано выше, реакцию асимметрического гидролиза проводили с использованием в качестве микробных клеток IF08306:Nannizia gypsea, то указанное в заголовке соединение получали при реакционном соотношении 55% (карбоновая кислота 80% эи (S), сложный эфир 80% эи (R)).
Аналогично, указанное в заголовке соединение получали при реакционном соотношении 42% (карбоновая кислота 92% эи (S), сложный эфир 60% эи (R)) с использованием IFO-12883:Actinomyces leporis. Также указанное в заголовке соединение получали при реакционном соотношении 37% (карбоновая кислота 91% эи (S), сложный эфир 50% эи (R)) с использованием NRIC1271:Penicillium chrysogenum. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 28.
Пример 45: Метил 2-(2,3,4-трифторанилино)пропионат
Метил (2R)-2-(2,3,4-трифторанилино)пропионат (100 мг, 38% эи) растворяли в толуоле (2 мл) и добавляли к раствору 1,8-диазабицикло[5,4,0]ундец-7-ен (ДБУ; 71,8 мг) при комнатной температуре. Затем жидкую реакционную смесь перемешивали при 110°С в течение 16 часов. После добавления хлористоводородной кислоты (1 моль/л; 1 мл) к жидкой реакционной смеси водный слой экстрагировали толуолом. Органический слой промывали водой и насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. После упаривания растворителя полученный остаток подвергали колоночной хроматографии на силикагеле (этилацетат-нормальный гексан=1:4), получая, таким образом, указанное в заголовке соединение (86,8 мг) в виде бесцветных кристаллов. Оптическая чистота данного продукта составляла 0% эи. 1Н-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 5.
Пример 46: Метил 2-(2,3,4-трифторанилино)пропионат
Метил (2R)-2-(2,3,4-трифторанилино)пропионат (50 мг, 57% эи) растворяли в N,N-диметилформамиде (ДМФ; 1 мл) и добавляли при комнатной температуре карбонат калия (63,2 мг). Затем жидкую реакционную смесь перемешивали при 110°С в течение 19 часов. После добавления воды к жидкой реакционной смеси водный слой экстрагировали этилацетатом. Органический слой промывали водой и сушили над безводным сульфатом магния. После упаривания растворителя полученный остаток подвергали колоночной хроматографии на силикагеле (этилацетат-нормальный гексан =1:4), получая, таким образом, указанное в заголовке соединение (42,5 мг) в виде бесцветных кристаллов. Оптическая чистота данного продукта составляла 0% эи. 1Н-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 5.
Пример 47: Метил 2-(2,3,4-трифторанилино)пропионат
Метил (2R)-2-(2,3,4-трифторанилино)пропионат (200 мг, 57% эи) растворяли в диметилацетамиде (ДМА; 3 мл) и добавляли карбонат калия (474,1 мг) при комнатной температуре. Затем жидкую реакционную смесь перемешивали при 95°С в течение 19 часов. После добавления воды к жидкой реакционной смеси водный слой экстрагировали этилацетатом. Органический слой промывали водой и сушили над безводным сульфатом магния. После упаривания растворителя полученный остаток подвергали колоночной хроматографии на силикагеле (этилацетат-нормальный гексан =1:4), получая, таким образом, указанное в заголовке соединение (179 мг) в виде бесцветных кристаллов. Оптическая чистота данного продукта составляла 0% эи. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 5.
Пример 48: 2-(2,3,4-Трифторанилино)пропионовая кислота
трет-Бутоксид калия (123,4 мг) суспендировали в ДМА (2 мл). При охлаждении льдом добавляли к суспензии раствор метил (2R)-2-(2,3,4-трифторанилино)пропионата (223 мг, 91% эи) в ДМА (2 мл). Жидкую реакционную смесь перемешивали при той же температуре в течение 1 часа. Затем добавляли водный раствор гидроксида натрия (3 моль/л; 2 мл) и смесь перемешивали в течение 1 часа. Жидкую реакционную смесь доводили до рН 2 водным раствором хлористоводородной кислоты (3 моль/л) и затем экстрагировали IPE. Органический слой сушили над безводным сульфатом магния и упаривали. Полученный таким образом неочищенный продукт перекристаллизовывали из смеси растворителей, состоящей из хлористого метилена и нормального гексана, получая, таким образом, указанное в заголовке соединение (206 мг) в виде бесцветных кристаллов. Оптическая чистота данного продукта составляла 0% эи. Различные спектральные данные продукта были идентичны данным, полученным в примере 23.
Пример 49: 2-(2,3,4-Трифторанилино)пропионовая кислота
Метил (2R)-2-(2,3,4-трифторанилино)пропионат (223 мг, 91% эи) растворяли в ДМА (3 мл) и добавляли карбонат калия (474,1 мг) при комнатной температуре. Жидкую реакционную смесь перемешивали при 95°С в течение 19 часов. После добавления водного раствора гидроксида натрия (3 моль/л) жидкую реакционную смесь перемешивали в течение 1 часа и затем доводили до рН 2 хлористоводородной кислотой (3 моль/л) с последующей экстракцией IPE. Затем смесь сушили над безводным сульфатом магния. После упаривания растворителя, полученный таким образом неочищенный продукт перекристаллизовывали из смеси растворителей, состоящей из хлористого метилена и нормального гексана, получая, таким образом, указанное в заголовке соединение (198 мг) в виде бесцветных кристаллов. Оптическая чистота данного продукта составляла 0% эи. Различные спектральные данные продукта были идентичны данным, полученным в примере 23.
Пример 50: 2-(2,3,4-Трифторанилино)пропионовая кислота
Карбонат калия (1,66 г) суспендировали в ДМА (18 мл). Затем добавляли к суспензии по каплям раствор (5 мл) метил (2R)-2-(2,3,4-трифторанилино)пропионата (2,33 г, 54% эи) в ДМА. Жидкую реакционную смесь перемешивали при той же температуре в течение 2 часов. Затем добавляли водный раствор гидроксида калия (3 моль/л; 2 мл) и смесь перемешивали в течение 15 минут. Жидкую реакционную смесь доводили до рН 2 хлористоводородной кислотой (6 моль/л), затем экстрагировали метил-трет-бутиловым эфиром и сушили над безводным сульфатом магния. После упаривания растворителя полученный таким образом неочищенный продукт растворяли в этилацетате (12 мл) и добавляли по каплям к раствору (10 мл) циклогексиламина (991,8 мг) в этилацетате при 60°С в течение 30 минут. Затем жидкую реакционную смесь перемешивали при той же температуре в течение 2 часов и выпавшую при этом в осадок циклогексиламиновую соль 2-(2,3,4-трифторанилино)пропионовой кислоты (2,74 г) отфильтровывали.
Данные циклогексиламиновой соли 2-(2,3,4-трифторанилино)-пропионовой кислоты следующие:
Элементный анализ C15H21F3N2O2
Вычислено (%): С, 56,59; Н, 6,65; N, 8,80
Найдено (%): С, 56,52; Н, 6,67; N, 8,77
1H-ЯМР (270 МГц, CDCl3) δ (м.д.): 1,11-2,05 (м, 16Н), 2,90-3,13 (м, 1Н), 3,73-3,86 (м, 1H), 6,30-6,47 (м, 1Н), 6,75-6,89 (м, 1H).
Впоследствии к данной циклоаминовой соли добавляли хлористоводородную кислоту (6 моль/л) и смесь экстрагировали метил-трет-бутиловым эфиром (МТВЕ) и сушили над безводным сульфатом магния. После упаривания растворителя, указанное в заголовке соединение (1,92 г) получали в виде бесцветных кристаллов. Его оптическая чистота составляла 0% эи.
Пример 51: (2S)-2-(2,3,4-Трифторанилино)-1-пропанол
При охлаждении льдом боргидрид натрия (1,2 г) растворяли в IPA (50 мл). После добавления метанола (5 мл) добавляли по каплям раствор соединения (5,0 г), полученного в примере 1, в IPA. Затем жидкую реакционную смесь нагревали до 50°С и перемешивали в течение 1 часа. Затем добавляли хлористоводородную кислоту (1 моль/л) и смесь перемешивали некоторое время. Затем добавляли насыщенный водный раствор гидрокарбоната натрия и смесь экстрагировали этилацетатом. Экстракт промывали водой, сушили над безводным сульфатом магния и фильтровали. После упаривания растворителя полученный остаток подвергали колоночной хроматографии на силикагеле, получая 3,7 г (84%) указанного в заголовке соединения в виде маслянистого вещества.
1H-ЯМР (CDCl3, 270 МГц,) δ: 1,21 (д, J=6,3 Гц, 3Н), 1,77 (ушир.с, 1Н), 3,55-3,71 (м, 4Н), 6,39-6,48 (м, 1Н), 6,75-6,87 (м, 1Н).
ИК спектр: 3394, 2967, 2933 см-1. Масс-спектр; m/z: 205 (М+)
Пример 52: (2S)-2-(2,3,4-Трифторанилино)пропанол
При комнатной температуре боргидрид натрия (35,7 мг) суспендировали в толуоле (0,2 мл). Затем к раствору добавляли раствор (0,8 мл) метил (2S)-2-(2,3,4-трифторанилино)-пропионата (200 мг, 99,8% эи) в толуоле. После добавления метанола (137,4 мг) жидкую реакционную смесь перемешивали в течение 6 часов. Затем к жидкой реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. После упаривания растворителя полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 162,9 мг (99,8% эи) указанного в заголовке соединения в виде маслянистого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 51.
Пример 53: (2S)-2-(2,3,4-Трифторанилино)пропанол
При комнатной температуре боргидрид натрия (35,7 мг) суспендировали в хлорбензоле (0,2 мл). Затем к раствору добавляли раствор (0,8 мл) метил (2S)-2-(2,3,4-трифтор-анилино)пропионата (200 мг, 99,8% эи) в хлорбензоле. После добавления метанола (137,4 мг) жидкую реакционную смесь перемешивали в течение 6 часов. Затем к жидкой реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. После упаривания растворителя полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 162,9 мг (99,8% эи) указанного в заголовке соединения в виде маслянистого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 51.
Пример 54: (2S)-2-(2,3,4-Трифторанилино)пропанол
При комнатной температуре боргидрид натрия (35,7 мг) суспендировали в гексане (0,2 мл). Затем к раствору добавляли раствор (0,8 мл) метил (2S)-2-(2,3,4-трифторанилино)пропионата (200 мг, 99,8% эи) в гексане. После добавления метанола (137,4 мг), жидкую реакционную смесь перемешивали в течение 1 часа. Затем к жидкой реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. После упаривания растворителя полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 176 мг (99,8% эи) указанного в заголовке соединения в виде маслянистого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 51.
Пример 55: (2S)-2-(2,3,4-Трифторанилино)пропанол
При комнатной температуре боргидрид натрия (35,7 мг) суспендировали в циклогексане (0,2 мл). Затем к раствору добавляли раствор (0,8 мл) метил (2S)-2-(2,3,4-трифтор-анилино)пропионата (200 мг, 99,8% эи) в циклогексане. После добавления метанола (137,4 мг) жидкую реакционную смесь перемешивали в течение 6 часов. Затем к жидкой реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 176 мг (99,8% эи) указанного в заголовке соединения в виде маслянистого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 51.
Пример 56: (2S)-2-(2,3,4-Трифторанилино)пропанол
При комнатной температуре боргидрид натрия (35,7 мг) суспендировали в IPE (0,2 мл). Затем к раствору добавляли раствор (0,8 мл) метил (2S)-2-(2,3,4-трифторанилино)пропионата (200 мг, 99,8% эи) в IPE. После добавления метанола (137,4 мг) жидкую реакционную смесь перемешивали в течение 2 часов. Затем к жидкой реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 176 мг (99,8% эи) указанного в заголовке соединения в виде маслянистого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 51.
Пример 57: (2S)-2-(2,3,4-Трифторанилино)пропанол
При комнатной температуре боргидрид натрия (35,7 мг) суспендировали в метил-трет-бутиловом эфире (0,2 мл). Затем к раствору добавляли раствор (0,8 мл) метил (2S)-2-(2,3,4-трифторанилино)пропионата (200 мг, 99,8% эи) в метил-трет-бутиловом эфире. После добавления метанола (137,4 мг) жидкую реакционную смесь перемешивали в течение 1 часа. Затем к жидкой реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 176 мг (99,8% эи) указанного в заголовке соединения в виде маслянистого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 51.
Пример 58: (2S)-2-(2,3,4-Трифторанилино)пропанол
При комнатной температуре боргидрид натрия (35,7 мг) суспендировали в ТГФ (0,2 мл). Затем к раствору добавляли раствор (0,8 мл) метил (2S)-2-(2,3,4-трифторанилино)пропионата (200 мг, 99,8% эи) в ТГФ. После добавления метанола (137,4 мг) жидкую реакционную смесь перемешивали в течение 1 часа. Затем к жидкой реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 176 мг (99,8% эи) указанного в заголовке соединения в виде маслянистого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 51.
Пример 59: (2S)-2-(2,3,4-Трифторанилино)пропанол
При комнатной температуре боргидрид натрия (35,7 мг) суспендировали в 1,2-диметоксиэтане (0,2 мл). Затем к раствору добавляли раствор (0,8 мл) метил (2S)-2-(2,3,4-трифторанилино)пропионата (200 мг, 99,8% эи) в DME. После добавления метанола (137,4 мг) жидкую реакционную смесь перемешивали в течение 1 часа. Затем к жидкой реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 176 мг (99,8% эи) указанного в заголовке соединения в виде маслянистого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 51.
Пример 60: (2S)-2-(2,3,4-Трифторанилино)пропанол
При комнатной температуре боргидрид натрия (35,7 мг) суспендировали в хлороформе (0,2 мл). Затем к раствору добавляли раствор (0,8 мл) метил (2S)-2-(2,3,4-трифтор-анилино)пропионата (200 мг, 99,8% эи) в хлороформе. После добавления метанола (137,4 мг) жидкую реакционную смесь перемешивали в течение 6 часов. Затем к жидкой реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 176 мг (99,8% эи) указанного в заголовке соединения в виде маслянистого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 51.
Пример 61: (2S)-2-(2,3,4-Трифторанилино)пропанол
При комнатной температуре боргидрид натрия (35,7 мг) суспендировали в хлористом метилене (0,2 мл). Затем к раствору добавляли раствор (0,8 мл) метил (2S)-2-(2,3,4-трифторанилино)пропионата (200 мг, 99,8% эи) в хлористом метилене. После добавления метанола (137,4 мг) жидкую реакционную смесь перемешивали в течение 1 часа. Затем к жидкой реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 159,8 мг (99,8% эи) указанного в заголовке соединения в виде маслянистого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 51.
Пример 62: (2S)-2-(2,3,4-Трифторанилино)пропанол
При комнатной температуре боргидрид натрия (35,7 мг) суспендировали в 1,2-дихлорэтане (EDC, 0,2 мл). Затем к раствору добавляли раствор (0,8 мл) метил (2S)-2-(2,3,4-трифторанилино)пропионата (200 мг, 99,8% эи) в EDC. После добавления метанола (137,4 мг) жидкую реакционную смесь перемешивали в течение 1 часа. Затем к жидкой реакционной смеси добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и насыщенным водным раствором хлорида аммония и сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 159,8 мг (99,8% эи) указанного в заголовке соединения в виде маслянистого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 51.
Пример 63: Диэтил [2,3,4-трифтор[(1S)-2-гидрокси-1-метилэтил]анилино]метиленмалонат
Соединение (300 мг), полученное в примере 51, диэтил этоксиметиленмалонат (632 мг) и хлорид тетрагексиламмония (57 мг) растворяли в ацетоне (3 мл). После добавления карбоната калия (445 мг), смесь перемешивали при комнатной температуре в течение 4,5 часов. По завершении реакции растворитель выпаривали. Затем полученный таким образом остаток подвергали колоночной хроматографии на силикагеле, получая таким образом 338 мг (84%) указанного в заголовке соединения в виде бесцветного твердого вещества. 1H-ЯМР (CDCl3, 270 МГц) δ: 1,13 (т, J=7,26 Гц, 3Н), 1,23 (т, J=7,26 Гц, 3Н), 2,34 (ушир.с, 1Н), 3,62-3,81 (м, 5Н), 4,16 (кв, J=7,26, 2Н), 6,87-7,11 (м, 2Н), 7,70 (с, 1Н).
ИК спектр (KBr): 3451, 3093, 2989, 1706, 1678 см-1
Масс-спектр; m/z: 375 (М+)
Пример 64: Диэтил [2,3,4-трифтор[(1S)-2-гидрокси-1-метилэтил]анилино]метиленмалонат
Соединение (103 мг), полученное в примере 51, диэтилэтоксиметиленмалонат (108 мг) и хлорид тетрагексиламмония (29 мг) растворяли в дихлорметане (1 мл). После добавления карбоната калия (138 мг) смесь перемешивали при комнатной температуре в течение 22 часов. По завершении реакции остаток отфильтровывали и растворитель выпаривали. Затем полученный таким образом остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 147 мг (78%) указанного в заголовке соединения в виде бесцветного твердого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 63.
Пример 65: Диэтил [2,3,4-трифтор[(1S)-2-гидрокси-1-метилэтил]анилино]метиленмалонат
трет-Бутоксид калия (62 мг) добавляли к ДМФ (2 мл) и охлаждали до 0°С. Затем по каплям добавляли раствор соединения (100 мг), полученного в примере 51, в ДМФ (200 мкл). После перемешивания в течение 15 минут добавляли по каплям диэтилэтоксиметиленмалонат и полученную смесь перемешивали в течение 8 часов при комнатной температуре. После обработки обычным образом смесь подвергали колоночной хроматографии на силикагеле, получая, таким образом, 137 мг (75%) указанного в заголовке соединения в виде бесцветного твердого вещества. 1H-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 63.
Пример 66: Диэтил [2,3,4-трифтор[(1S)-2-гидрокси-1-метилэтил]анилино]метиленмалонат
К соединению (103 мг), полученному в примере 51, добавляли диэтилэтоксиметиленмалонат (127 мг). Затем полученную смесь перемешивали в течение 1 часа при нагревании до 100°С при атмосферном давлении. Далее ее перемешивали при той же температуре в течение 1,5 часов при пониженном давлении и затем при атмосферном давлении дополнительно в течение 16 часов. По данным анализа ВЭЖХ с обращенной фазой с использованием соединения примера 63 в качестве образца полученный продукт соответствовал 142 мг (78%) указанного в заголовке соединения.
Пример 67: Диэтил [2,3,4-трифтор[(1S)-2-гидрокси-1-метилэтил]анилино]метиленмалонат
Соединение (103 мг), полученное в примере 51, и диметилэтоксиметиленмалонат (87 мг) растворяли в толуоле (3 мл) и смесь нагревали при кипении с обратным холодильником в течение 21 часа. Затем остаток фильтровали и растворитель выпаривали. Полученный остаток подвергали колоночной хроматографии на силикагеле, получая 125 мг (72%) указанного в заголовке соединения в виде бесцветных кристаллов.
1H-ЯМР (CDCl3, 270 МГц) δ: 1,22-1,25 (м, 3Н), 3,27 (с, 1Н), 3,57-3,82 (м, 8Н), 6,96-7,10 (м, 2Н), 7,76 (с, 1Н)
ИК спектр (KBr): 3452, 2954, 1722 см-1
Масс-спектр; m/z: 347(М+), 316, 284
Пример 68: Диэтил [2,3,4-трифтор[(1S)-2-гидрокси-1-метилэтил]анилино]метиленмалонат
При комнатной температуре гидроксид калия (330 мг) и хлорид тетрагексиламмония (190,1 мг) растворяли в ДМФ (15 мл). После добавления раствора (5 мл) (2S)-2-(2,3,4-трифторанилино)пропанола (1 г, 99,8% эи) и диэтилэтоксиметиленмалоната (2,09 г) в ДМФ полученную смесь перемешивали в течение 1 часа. После добавления воды жидкую реакционную смесь экстрагировали смесью растворителей, состоящей из этилацетата и н-гексана (3:2). Органический слой промывали водой и сушили над безводным сульфатом магния. После упаривания растворителя к полученному остатку добавляли IPE и смесь перемешивали при 0°С в течение 1 часа. Выпавшие при этом в осадок кристаллы отфильтровывали и полученный таким образом влажный продукт сушили при пониженном давлении. Таким образом получали указанное в заголовке соединение (1,65 г, 99,8% эи) в виде бесцветных кристаллов. 1Н-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 63.
Пример 69: Диэтил [2,3,4-трифтор[(1S)-2-гидрокси-1-метилэтил]анилино]метиленмалонат
При комнатной температуре гидроксид калия (330 мг) и гидросульфат тетрабутиламмония (82,7 мг) растворяли в ДМФ (15 мл). После добавления раствора (5 мл) (2S)-2-(2,3,4-трифторанилино)пропанола (1 г, 99,8% эи) и диэтилэтоксиметиленмалоната (2,09 г) в ДМФ полученную смесь перемешивали в течение 1 часа. После добавления воды жидкую реакционную смесь экстрагировали смесью растворителей, состоящей из этилацетата и н-гексана (3:2). Органический слой промывали водой и сушили над безводным сульфатом магния. После упаривания растворителя к полученному остатку добавляли IPE и смесь перемешивали при 0°С в течение 1 часа. Выпавшие при этом в осадок кристаллы отфильтровывали и полученный таким образом влажный продукт сушили при пониженном давлении. Таким образом получали указанное в заголовке соединение (1,7 г, 99,8% эи) в виде бесцветных кристаллов. 1Н-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 63.
Пример 70: Диэтил [2,3,4-трифтор[(1S)-2-гидрокси-1-метилэтил]анилино]метиленмалонат
При комнатной температуре гидроксид калия (330 мг) и гидросульфат тетрабутиламмония (82,7 мг) растворяли в ДМФ (15 мл). После добавления раствора (5 мл) (2S)-2-(2,3,4-трифторанилино)пропанола (1 г, 99,8% эи) и диэтилэтоксиметиленмалоната (2,09 г) в ДМФ, полученную смесь перемешивали в течение 1 часа. После добавления воды жидкую реакционную смесь экстрагировали смесью растворителей, состоящей из этилацетата и н-гексана (3:2). Органический слой промывали водой и сушили над безводным сульфатом магния. После упаривания растворителя к полученному остатку добавляли IPE и смесь перемешивали при 0°С в течение 1 часа. Выпавшие при этом в осадок кристаллы отфильтровывали и полученный таким образом влажный продукт сушили при пониженном давлении. Таким образом, получали указанное в заголовке соединение (1,65 г, 99,8% эи) в виде бесцветных кристаллов. 1Н-ЯМР и ИК спектральные данные продукта были идентичны данным соединения, полученного в примере 63.
Пример 71: Диэтил [(3S)-7,8-дифтор-3-метил-2,3-дигидро-4Н-[1,4]бензоксазин-4-ил]метиленмалонат
К ДМФ (5 мл) добавляли трет-бутоксид калия (74 мг) при охлаждении льдом. После добавления по каплям раствора соединения (200 мг), полученного в примере 63, в ДМФ (1 мл) полученную смесь перемешивали при 60°С в течение 18 часов. После обработки обычным образом полученный остаток подвергали колоночной хроматографии на силикагеле, получая, таким образом, 149 мг (79%) указанного в заголовке соединения. Физические константы полученных соединений были идентичны описанным в патенте Японии №2769174.
Пример 72: Диэтил [(3S)-7,8-дифтор-3-метил-2,3-дигидро-4Н-[1,4]бензоксазин-4-ил]метиленмалонат
К ДМФ (2 мл) добавляли трет-бутоксид калия (226 мг) при охлаждении льдом. После добавления по каплям раствора соединения (100 мг), полученного в примере 51, и диэтилэтоксиметиленмалоната (293 мг) в ДМФ (0,5 мл) полученную смесь перемешивали при комнатной температуре в течение 18 часов. После обработки обычным образом полученный остаток подвергали колоночной хроматографии на силикагеле, получая таким образом 113 мг (65%) указанного в заголовке соединения. Физические константы полученных соединений были идентичны описанным в патенте Японии №2769174.
Пример 73: Диэтил [(3S)-7,8-дифтор-3,4-дигидро-3-метил-2Н-[1,4]бензоксазин-4-ил]метиленмалонат
Гидроксид калия (180 мг) и гидросульфат тетрабутиламмония (90,4 мг) растворяли в ДМФ (15 мл) при нагревании до 60°С и добавляли раствор диэтил [2,3,4-трифтор[(1S)-2-гидрокси-1-метилэтил]анилино]метиленмалоната (1 г, 99,8% эи) и диэтилэтоксиметиленмалоната (120 мг) в ДМФ (85 мл). Полученную смесь перемешивали при той же температуре в течение 2 часов. После добавления воды жидкую реакционную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле. Таким образом получали 852 мг (99,8% эи) указанного в заголовке соединения в виде желтого маслянистого вещества. Различные спектральные данные были идентичны описанным в патенте Японии No. 2769174.
Пример 74: Диэтил [(3S)-7,8-дифтор-3,4-дигидро-3-метил-2Н-[1,4]бензоксазин-4-ил]метиленмалонат
Гидроксид калия (180 мг) и хлорид бензилтриметиламмония (49,5 мг) растворяли в ДМФ (15 мл) при нагревании до 70°С и добавляли раствор диэтил [2,3,4-трифтор[(1S)-2-гидрокси-1-метилэтил]анилино]метиленмалоната (1 г, 99,8% эи) и диэтилэтоксиметиленмалоната (120 мг) в ДМФ (5 мл). Полученную смесь перемешивали при той же температуре в течение 4 часов. После добавления воды жидкую реакционную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле. Таким образом получали 871 мг (99,8% эи) указанного в заголовке соединения в виде желтого маслянистого вещества. Различные спектральные данные были идентичны описанным в патенте Японии No. 2769174.
Пример 75: Диэтил [(3S)-7,8-дифтор-3,4-дигидро-3-метил-2Н-[1,4]бензоксазин-4-ил]метиленмалонат
Гидроксид калия (180 мг) и хлорид бензилтриметиламмония (60,7 мг) растворяли в ДМФ (15 мл) нагреванием до 60°С и добавляли раствор (5 мл) диэтил [2,3,4-трифтор[(1S)-2-гидрокси-1-метилэтил]анилино]метиленмалоната (1 г, 99,8% эи) и диэтилэтоксиметиленмалоната (120 мг) в ДМФ. Полученную смесь перемешивали при той же температуре в течение 7 часов. После добавления воды жидкую реакционную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле. Таким образом получали 899 мг (99,8% эи) указанного в заголовке соединения в виде желтого маслянистого вещества. Различные спектральные данные были идентичны описанным в патенте Японии No. 2769174.
Пример 76: Диэтил [(3S)-7,8-дифтор-3,4-дигидро-3-метил-2Н-[1,4]бензоксазин-4-ил]метиленмалонат
При комнатной температуре КОН (330 мг) и хлорид тетрагексиламмония (190,1 мг) растворяли в ДМФ (15 мл). После добавления раствора (5 мл) (2S)-2-(2,3,4-трифторанилино)-пропанола (1 г, 99,8% эи) и диэтилэтоксиметиленмалоната (2,09 г) в ДМФ смесь перемешивали в течение 1 часа. Затем ее нагревали до 60°С и добавляли раствор (5 мл) КОН (330 мг) и диэтилэтоксиметиленмалоната (120 мг) в ДМФ. Полученную смесь перемешивали при той же температуре в течение 5 часов. После добавления воды жидкую реакционную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния. После упаривания растворителя, полученный остаток подвергали колоночной хроматографии на силикагеле. Таким образом, получали 1,37 г (99,8% эи) указанного в заголовке соединения в виде желтого маслянистого вещества. Различные спектральные данные были идентичны описанным в патенте Японии No. 2769174.
Пример 77: Диэтил [(3S)-7,8-дифтор-3,4-дигидро-3-метил-2Н-[1,4]бензоксазин-4-ил]метиленмалонат
При комнатной температуре КОН (330 мг) и гидросульфат тетрабутиламмония (82,7 мг) растворяли в ДМФ (15 мл). После добавления раствора (5 мл) (2S)-2-(2,3,4-трифторанилино)-пропанола (1 г, 99,8% эи) и диэтилэтоксиметиленмалоната (2,09 г) в ДМФ смесь перемешивали в течение 1 часа. Затем ее нагревали до 60°С и добавляли раствор (5 мл) КОН (330 мг) и диэтилэтоксиметиленмалоната (120 мг) в ДМФ. Полученную смесь перемешивали при той же температуре в течение 5 часов. После добавления воды жидкую реакционную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния. После упаривания растворителя полученный остаток подвергали колоночной хроматографии на силикагеле. Таким образом, получали 1,3 г (99,8% эи) указанного в заголовке соединения в виде желтого маслянистого вещества. Различные спектральные данные были идентичны описанным в патенте Японии No. 2769174.
Пример 78: (3S)-(+)-7,8-Дифтор-3,4-дигидро-3-метил-2Н-[1,4]бензоксазин
К ДМФ (2 мл) добавляли гидрид натрия (39 мг) и смесь нагревали до 60°С на масляной бане. Затем добавляли по каплям раствор соединения (100 мг), полученного в примере 51, в ДМФ и полученную смесь перемешивали в течение 1 часа. После обработки обычным образом смесь подвергали колоночной хроматографии на силикагеле, получая 60 мг (66%) указанного в заголовке соединения. Оптическая чистота, определенная с использованием ВЭЖХ, составляла >94% эи. Различные спектральные данные были идентичны данным для образца, синтезированного отдельно.
Пример 79: (3S)-(+)-7,8-Дифтор-3,4-дигидро-3-метил-2Н-[1,4]бензоксазин
К ДМФ (2 мл) добавляли трет-бутоксид калия (110 мг) при охлаждении льдом. Затем добавляли по каплям раствор соединения (100 мг), полученного в примере 51, в ДМФ и полученную смесь перемешивали в течение 30 минут. После обработки обычным образом смесь подвергали колоночной хроматографии на силикагеле, получая 72 мг (79%) указанного в заголовке соединения. Оптическая чистота, определенная с использованием ВЭЖХ, составляла >94% эи. Различные спектральные данные были идентичны данным для образца, синтезированного отдельно.
Пример 80: (3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазин-п-толуолсульфонат
К трет-бутоксиду натрия (трет-BuONa; 748 мг) добавляли ДМА (8 мл). После растворения при нагревании при 80°С добавляли при той же температуре раствор (2S)-2-(2,3,4-трифторанилино)-пропанола (1,0 г; 99,8% эи) в ДМА (2 мл). После перемешивания в течение 30 минут и самопроизвольного охлаждения, добавляли при комнатной температуре воду (30 мл). Полученную смесь экстрагировали трижды этилацетатом (AcOEt; 20 мл). Экстрагированный таким образом органический слой концентрировали при пониженном давлении. Полученный раствор добавляли по каплям в раствор моногидрата п-толуолсульфоновой кислоты (927,5 мг) в AcOEt (10 мл). После перемешивания при комнатной температуре дополнительно в течение 1 часа кристаллы отфильтровывали, промывая AcOEt (7 мл). Полученный таким образом влажный продукт сушили при пониженном давлении, получая указанное в заголовке соединение (1,6 г) в виде бесцветных кристаллов.
1Н-ЯМР (CD3OD) δ: 1,43 (д, 3Н, J=5,7Гц),2,34 (д, 3Н, J=12,2 Гц), 3,85-3,89 (м, 1Н), 4,09-4,17 (м, 1Н), 7,22-7,32 (м, 1Н), 6,77-6,89 (м, 1Н).
Температура плавления: от 131 до 133°С (разложение)
Элементный анализ для C16H17NO4S
Вычислено (%): С, 53,77;Н, 4,79; N, 3,92%
Найдено (%): С, 53,80; Н, 4,81; N, 3,86%
Пример 81: (3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4] бензоксазин-п-толуолсульфонат
К трет-бутоксиду калия (трет-BuOK; 1,24 г) добавляли ДМФ (18 мл). После растворения при нагревании при 80°С добавляли при той же температуре раствор (2S)-2-(2,3,4-трифторанилино)пропанола (1,0 г; 99,8% эи) в ДМФ (2 мл). После перемешивания в течение 30 минут и самопроизвольного охлаждения, добавляли при комнатной температуре воду (40 мл). Полученную смесь экстрагировали трижды AcOEt (20 мл). Экстрагированный таким образом органический слой концентрировали при пониженном давлении. Полученный раствор добавляли по каплям в раствор моногидрата п-толуолсульфоновой кислоты (927,5 мг) в AcOEt (10 мл). После перемешивания при комнатной температуре дополнительно в течение 1 часа кристаллы отфильтровывали, промывая AcOEt (7 мл). Полученные таким образом кристаллы сушили при пониженном давлении, получая указанное в заголовке соединение (1,39 г) в виде бесцветных кристаллов. Различные спектральные данные были идентичны данным, полученным в примере 80.
Пример 82: (3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазин-п-толуолсульфонат
К гидриду натрия (NaH; 262 мг) добавляли ДМФ (18 мл). После растворения при нагревании при 80°С добавляли при той же температуре раствор (2S)-2-(2,3,4-трифторанилино)пропанола (1,0 г; 99,8% эи) в ДМФ (2 мл). После перемешивания в течение 30 минут и самопроизвольного охлаждения, добавляли при комнатной температуре воду (40 мл). Полученную смесь экстрагировали трижды AcOEt (20 мл). Экстрагированный таким образом органический слой концентрировали при пониженном давлении. Полученный раствор добавляли по каплям в раствор моногидрата п-толуолсульфоновой кислоты (927,5 мг) в AcOEt (10 мл). После перемешивания при комнатной температуре дополнительно в течение 1 часа кристаллы отфильтровывали, промывая AcOEt (7 мл). Полученные таким образом кристаллы сушили при пониженном давлении, получая указанное в заголовке соединение (1,14 г) в виде бесцветных кристаллов. Различные спектральные данные были идентичны данным, полученным в примере 80.
Пример 83: (3S)-1,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазин
(3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазин-п-толуолсульфонат (1 г) суспендировали в AcOEt (10 мл) и затем добавляли к суспензии водный раствор гидрокарбоната натрия (NaHCO3; 10 мл). После перемешивания при комнатной температуре в течение 1 часа смесь экстрагировали AcOEt. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая, таким образом, указанное в заголовке соединение (516 мг, 99,8% эи) в виде желтого маслянистого вещества.
1H-ЯМР (270 МГц, CDCl3) δ: 2,16 (с,3Н), 4,60 (с, 2Н), 6,28 (ддд, 1Н, J=2,3, 4,7, 8,9 Гц); 6,50-6,80 (м, 1Н).
Пример 84: (3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазин-метансульфонат
К трет-BuONa (748 мг) добавляли ДМА (8 мл). После растворения при нагревании при 80°С добавляли при той же температуре раствор (2S)-2-(2,3,4-трифторанилино)пропанола (1,0 г; 99,8% эи) в ДМА (2 мл). После перемешивания в течение 30 минут и самопроизвольного охлаждения, добавляли при комнатной температуре воду (30 мл). Полученную смесь экстрагировали трижды AcOEt (20 мл). Органические слои объединяли и концентрировали при пониженном давлении. Полученный раствор добавляли по каплям в раствор метансульфоновой кислоты (468, 4 мг) в AcOEt (5 мл). После перемешивания при комнатной температуре дополнительно в течение 1 часа кристаллы отфильтровывали, промывая AcOEt (5 мл). Полученный таким образом влажный продукт сушили, получая указанное в заголовке соединение (960,4 мг) в виде бесцветных кристаллов.
1Н-ЯМР (270 МГц, CD3OD) δ: 1,45 (д, 3Н, J=6,8 Гц), 2,68 (с, 3Н), 3,89-3,93 (м, 1Н), 4,17 (дд, 1Н, J=8,9, 12,2 Гц), 4,57 (дд, 1Н, J=2,7, 11,9 Гц), 6,96-7,15 (м, 2Н).
Температура плавления: от 131 до 133°С (разложение)
Элементный анализ для C10H13F2NO4S
Вычислено (%): С, 42,70; Н, 4,66%; N, 4,98%
Найдено (%): С, 42,70; Н, 4,66%; N, 4,92%
Пример 85: (3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазинметансульфонат
К трет-BuOK (1,24 г) добавляли ДМФ (18 мл). После растворения при нагревании при 80°С добавляли при той же температуре раствор (2S)-2-(2,3,4-трифторанилино)пропанола (1,0 г; 99,8% эи) в ДМФ (2 мл). После перемешивания в течение 30 минут и самопроизвольного охлаждения, добавляли при комнатной температуре воду (30 мл). Полученную смесь экстрагировали трижды AcOEt (20 мл). Органические слои объединяли и концентрировали при пониженном давлении. Полученный раствор добавляли по каплям в раствор метансульфоновой кислоты (468,4 мг) в AcOEt (5 мл). После перемешивания при комнатной температуре дополнительно в течение 1 часа кристаллы отфильтровывали, промывая AcOEt (5 мл). Полученный таким образом влажный продукт сушили при пониженном давлении, получая указанное в заголовке соединение (875 мг) в виде бесцветных кристаллов. Различные спектральные данные были идентичны данным, полученным в примере 84.
Пример 86: (3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазинметансульфонат
К NaH (262 мг) добавляли ДМФ (18 мл). После растворения при нагревании при 80°С добавляли при той же температуре раствор (2S)-2-(2,3,4-трифторанилино)пропанола (1,0 г; 99,8% эи) в ДМФ (2 мл). После перемешивания в течение 30 минут и самопроизвольного охлаждения, добавляли при комнатной температуре воду (30 мл). Полученную смесь экстрагировали трижды AcOEt (20 мл). Органические слои объединяли и концентрировали при пониженном давлении. Полученный раствор добавляли по каплям в раствор метансульфоновой кислоты (468,4 мг) в AcOEt (5 мл). После перемешивания при комнатной температуре дополнительно в течение 1 часа кристаллы отфильтровывали, промывая AcOEt (5 мл). Полученный таким образом влажный продукт сушили при пониженном давлении, получая указанное в заголовке соединение (894 мг) в виде бесцветных кристаллов. Различные спектральные данные были идентичны данным, полученным в примере 84.
Пример 87: (3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4] бензоксазин
(3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазин-п-толуолсульфонат (1 г) суспендировали в AcOEt (10 мл) и затем добавляли к нему водный раствор NaHCO3 (10 мл). После перемешивания при комнатной температуре в течение 1 часа смесь экстрагировали AcOEt. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая, таким образом, указанное в заголовке соединение (645,2 мг, 99,8% эи) в виде желтого маслянистого вещества. Различные спектральные данные были идентичны данным, полученным в примере 83.
Пример 88: (3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазин-(±)-камфорсульфонат
К трет-BuONa (748 мг) добавляли ДМА (8 мл). После растворения при нагревании при 80°С добавляли при той же температуре раствор (2S)-2-(2,3,4-трифторанилино)пропанол (1,0 г; 99,8% эи) в ДМА (2 мл). После перемешивания в течение 30 минут и охлаждения, оставляя смесь стоять, добавляли при комнатной температуре воду (30 мл). Полученную смесь экстрагировали трижды AcOEt (20 мл). Органические слои объединяли и концентрировали при пониженном давлении. Полученный раствор добавляли по каплям в раствор (±)-камфорсульфоновой кислоты (1,137 г) в 5% EtOH (этанол)/AcOEt (7 мл). После перемешивания при комнатной температуре дополнительно в течение 1 часа кристаллы отфильтровывали, промывая AcOEt (7 мл). Полученный таким образом влажный продукт сушили при пониженном давлении, получая указанное в заголовке соединение (1,8 г) в виде бесцветных кристаллов.
1H-ЯМР (270 МГц, CD3OD): 0,613 (с, 3Н), 0,847 (с, 3Н), 1,36-1,46 (м, 1Н), 1,45 (д, 3Н, J=6,5 Гц), 1,55-1,65 (м, 1Н), 1,88 (д, 1Н, J=18,4 Гц), 1,98-2,06 (м, 2Н), 2,76 (д, 1Н, J=14,6 Гц), 3,27 (д, 1Н, J=14,6 Гц), 3,85-3,97 (м, 1Н), 4,18 (дд, 1Н, J=8,6, 12,2 Гц), 4,57 (дд, 1Н, J-2,7, 11,9 Гц), 6,49-7,19 (м, 2Н).
Температура плавления: от 232 до 236°С (разложение)
Элементный анализ для C19H25NO5S
Вычислено (%): С, 54,66; Н, 6,04; N, 3,36%
Найдено (%): С, 54,63; Н, 6,04; N, 3,29%
Пример 89: (3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4] бензоксазин-(±)-камфорсульфонат
К т-ВиОК (1,24 г) добавляли ДМФ (18 мл). После растворения при нагревании при 80°С к нему добавляли при той же температуре раствор (2S)-2-(2,3,4-трифторанилино)пропанола (1,0 г; 99,8% эи) в ДМФ (2 мл). После перемешивания в течение 30 минут и самопроизвольного охлаждения, добавляли воду (30 мл) при комнатной температуре. Полученную смесь экстрагировали трижды AcOEt (20 мл). Органические слои объединяли и концентрировали при пониженном давлении. Полученный раствор добавляли по каплям к раствору (±)-камфорсульфоновой кислоты (1,137 г) в 5% EtOH/AcOEt (7 мл). После перемешивания при комнатной температуре дополнительно в течение 1 часа кристаллы отфильтровывали, промывая AcOEt (7 мл). Полученный таким образом влажный продукт сушили при пониженном давлении, получая указанное в заголовке соединение (1,72 г) в виде бесцветных кристаллов. Различные спектральные данные были идентичны данным, полученным в примере 88.
Пример 90: (3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4] бензоксазин-(±)-камфорсульфонат
К NaH (242 мг) добавляли ДМФ (18 мл). После растворения при нагревании при 80°С добавляли при той же температуре раствор (2S)-2-(2,3,4-трифторанилино)пропанола (1,0 г; 99,8% эи) в ДМФ (2 мл). После перемешивания в течение 30 минут и самопроизвольного охлаждения, добавляли воду (30 мл) при комнатной температуре. Полученную смесь экстрагировали трижды AcOEt (20 мл). Экстрагированный таким образом органический слой концентрировали при пониженном давлении. Полученный раствор добавляли по каплям в раствор (±)-камфорсульфоновой кислоты (1,137 г) в 5% EtOH/AcOEt (7 мл). После перемешивания при комнатной температуре дополнительно в течение 1 часа кристаллы отфильтровывали, промывая AcOEt (7 мл). Полученный таким образом влажный продукт сушили при пониженном давлении, получая указанное в заголовке соединение (1,41 г) в виде бесцветных кристаллов. Различные спектральные данные были идентичны данным, полученным в примере 88.
Пример 91: (3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазин
(3S)-7,8-Дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазин-п-толуолсульфонат (1 г) суспендировали в AcOEt (10 мл) и затем добавляли водный раствор NaHCO3 (10 мл). После перемешивания при комнатной температуре в течение 1 часа смесь экстрагировали AcOEt. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая, таким образом, указанное в заголовке соединение (438,9 мг, 99,8% эи) в виде желтого маслянистого вещества. Различные спектральные данные были идентичны данным, полученным в примере 83.
Пример 92: Диэтил [(3S)-7,8-дифтор-3-метил-2,3-дигидро-4Н-[1,4]бензоксазин-4-ил]метиленмалонат
К ДМФ (2,5 мл) добавляли трет-бутоксид калия (75 мг) при охлаждении льдом. После добавления по каплям раствора соединения (100 мг), полученного в примере 78, и диэтилэтоксиметиленмалоната (233 мг) в ДМФ (0,5 мл) полученную смесь перемешивали в течение 2 часов. После обработки обычным образом полученный остаток подвергали колоночной хроматографии на силикагеле, получая таким образом 153 мг (88%) указанного в заголовке соединения в виде маслянистого продукта. Физические константы полученных соединений были идентичны описанным в патенте Японии No. 2769174.
Пример 93: Диэтил [(3S)-7,8-дифтор-3-метил-2,3-дигидро-4Н-[1,4]бензоксазин-4-ил]метиленмалонат
1,20 г (99,8% эи) (3S)-7,8-дифтор-3-метил-3,4-дигидро-2Н-[1,4]бензоксазина растворяли в толуоле (0,5 мл). После добавления диэтилэтоксиметиленмалоната (1,92 г) смесь перемешивали при 120°С в течение 30 минут, а затем при 140°С при пониженном давлении в течение 30 минут. Остаток подвергали колоночной хроматографии на силикагеле. Таким образом 2,19 г (99,8% эи) указанного в заголовке соединения получали в виде желтого маслянистого продукта. Физические константы полученных соединений были идентичны описанным в патенте Японии No. 2769174.
Пример 94: 2-(2,3,4-Трифторанилино)пропил 4-нитробензоат
2-Гидроксипропил-4-нитробензоат (225 мг) растворяли в дихлорметане (1 мл) при перемешивании. При -50°С добавляли раствор ангидрида трифторметансульфоновой кислоты (339 мг), растворенного в дихлорметане (1 мл). После перемешивания при той же температуре в течение 30 минут дихлорметан выпаривали при пониженном давлении при 0°С. После растворения остатка в дихлорметане (1 мл) к нему добавяли по каплям при 0°С раствор 2, 3,4-трифторанилина (147,1 мг), растворенного в дихлорметане (1 мл), и полученную смесь перемешивали при той же температуре в течение 30 минут. Затем, к раствору добавляли дихлорметан (10 мл) с последующей промывкой водой (10 мл). Слой дихлорметана концентрировали при пониженном давлении и полученный остаток отделяли и очищали колоночной хроматографией на силикагеле, получая таким образом 159,4 мг (45%) указанного в заголовке соединения в виде желтых кристаллов.
1H-ЯМР (270 МГц, CDCl3) δ: 1,38 (д, 6,6 Гц, 3Н), 3,76-3,92 (м, 2Н), 4,30 (дд, J=5,3, 11,2 Гц, 1Н), 4,49 (дд, J=5,3, 11,2 Гц, 1Н), 6,46-6,55 (м, 1Н), 6,77-6,88 (м, 1Н), 8,17 (дд, J=2,0, 6,9 Гц, 2Н), 8,29 (дд, J=2,0, 6,9 Гц, 1Н).
Пример 95: 2-(2,3,4-Трифторанилино)пропанол
2-(2,3,4-Трифторанилино)пропил-4-нитробензоат (50 мг) и гидроксид калия (11,8 мг) добавляли к метанолу (2 мл) и растворяли при перемешивании. Затем смесь перемешивали при комнатной температуре в течение 18 часов. После выпаривания метанола при пониженном давлении добавляли хлороформ (5 мл) и воду (5 мл) и полученную смесь разделяли. Слой хлороформа концентрировали и очищали колоночной хроматографией на силикагеле, получая, таким образом, 19,8 мг (69,1%) указанного в заголовке соединения в виде бесцветного маслянистого продукта.
1H-ЯМР (270 МГц, CDCl3) δ: 1,22 (д, J=5,9 Гц, 3Н), 3,55-3,74 (м, 4Н), 6,3-6,5 (м, 1Н), 6,76-6,87 (м, 1Н).
Пример 96: 2-Гидроксипропил 4-нитробензоат
2-Гидроксипропанол (4,57 г) растворяли в толуоле (80 мл) при перемешивании и добавляли по каплям при 0°С триэтиламин (6,68 г). После перемешивания при той же температуре в течение 30 минут медленно добавляли раствор п-нитробензоилхлорида (11,4 г), растворенного в толуоле (12 мл). Полученную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Затем добавляли дихлорметан (50 мл) и осажденные таким образом кристаллы растворяли. Раствор промывали разбавленным водным раствором гидрокарбоната натрия (100 мл) и затем водным раствором хлористоводородной кислоты (0,5 моль/л). Полученный таким образом органический слой концентрировали и остаток растворяли в толуоле (45 мл) при нагревании. Затем его охлаждали, оставляя стоять при комнатной температуре для кристаллизации. Осажденные таким образом кристаллы собирали фильтрованием и сушили при пониженном давлении, давая 6,90 г (51%) указанного в заголовке соединения в виде желтых кристаллов. 1H-ЯМР (270 МГц, CDCl3) δ: 1,32 (д, 3,6 Гц, 3Н), 4,24-4,42 (м, 3Н), 8,22-8,33 (м, 4Н).
Пример 97: Этиловый эфир (3S)-(-)-9,10-дифтор-3-метил-7-оксо-2,3-дигидро-7Н-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновой кислоты
К S-(-)-7,8-дифтор-3-метил-2,3-дигидро-4Н-[1,4]бензоксазину (15,8 г) добавляли диэтилэтоксиметиленмалонат (24,0 г) и смесь перемешивали при пониженном давлении при 130-140°С в течение 1 часа. После охлаждения жидкую реакционную смесь растворяли в уксусном ангидриде (50 мл). При охлаждении льдом и перемешивании к смеси добавляли порциями жидкую смесь (80 мл) уксусный ангидрид-концентрированная серная кислота (2:1, об/об). После перемешивания при комнатной температуре в течение 1 часа, смесь перемешивали при температуре бани 50-60°С в течение 30 минут. После добавления ледяной воды жидкую реакционную смесь нейтрализовали добавлением порошкообразного карбоната калия и экстрагировали хлороформом. Экстракт промывали последовательно насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия и сушили над мирабилитом. После выпаривания хлороформа к остатку добавляли диэтиловый эфир. Кристаллы отфильтровывали, получая 20,0 г указанного в заголовке соединения.
Температура плавления: от 257 до 258°С
[α]D=-68,1° (с=0,250, уксусная кислота)
Пример 98: (3S)-(-)-9,10-Дифтор-3-метил-7-оксо-2,3-дигидро-7Н-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота
Полученное выше сложноэфирное соединение (19,5 г) растворяли в уксусной кислоте (150 мл). После добавления конц. хлористоводородной кислоты (400 мл) смесь нагревали при кипении с обратным холодильником в течение 3 часов. После охлаждения выпавшие таким образом в осадок кристаллы отфильтровывали и промывали последовательно водой, этанолом и диэтиловым эфиром с последующей сушкой, получая 16,2 г указанной в заголовке карбоновой кислоты. Температура плавления >300°С.
[α]D=-65,6°(с=0,985, ДМСО)
Пример 99: (3S)-(-)-9-Фтор-3-метил-10-(4-метил-1-пиперазинил)-7-оксо-2,3-дигидро-7Н-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота (левофлоксацин)
Полученную выше карбоновую кислоту (14,3 г) суспендировали в диэтиловом эфире (600 мл). После добавления комплекса трехфтористого бора с диэтиловым эфиром (70 мл) смесь перемешивали при комнатной температуре в течение 5 часов. После отбрасывания надосадочной жидкости декантацией остаток отфильтровывали, добавляя диэтиловый эфир и промывая диэтиловым эфиром с последующим высушиванием. Затем вещество растворяли в диметилсульфоксиде (100 мл). После добавления триэтиламина (14,2 мл) и N-метилпиперазина (7,3 мл) смесь перемешивали при комнатной температуре в течение 18 часов. После выпаривания растворителя при пониженном давлении к остатку добавляли диэтиловый эфир. Отфильтрованный желтый порошок суспендировали в 95% метаноле (400 мл) и добавляли к раствору триэтиламин (25 мл). После нагревания при кипении с обратным холодильником в течение 25 часов растворитель выпаривали при пониженном давлении. Остаток растворяли в 10%-ной хлористоводородной кислоте (500 мл), промывали трижды хлороформом и затем доводили до рН 11 водным раствором гидроксида натрия (4 моль/л). Затем рН опять доводили до рН 7,3 хлористоводородной кислотой (1 моль/л), экстрагировали хлороформом (2000 мл ×3) и сушили над мирабилитом. После выпаривания хлороформа полученное таким образом кристаллическое вещество перекристаллизовывали из смеси этанол-диэтиловый эфир, получая, таким образом, 12,0 г указанного в заголовке соединения (левофлоксацин).
Температура плавления: от 226 до 230°С (разложение)
[α]D=-76,9° (с=0,655, NaOH (0,05 моль/л))
Пример 100: (3S)-(-)-9-Фтор-3-метил-10-(4-метил-1-пиперазинил)-7-оксо-2,3-дигидро-7Н-пиридо[1,2,3-de]-[1,4]бензоксазин-6-карбоновая кислота (левофлоксацин)
(S)-(-)-9,10-Дифтор-3-метил-7-оксо-2,3-дигидро-7Н-пиридо [1,2,3-de] [1,4]бензоксазин-6-карбоновую кислоту (281 мг) растворяли в диэтиловом эфире (30 мл). При перемешивании при комнатной температуре добавляли к раствору большой избыток комплекса трехфтористый бор-диэтиловый эфир и реакцию в смеси продолжали 45 минут. Осадок отфильтровывали, промывали диэтиловым эфиром и сушили при пониженном давлении, получая, таким образом, хелатное соединение бора.
Температура разложения >300°С
[α]D=-9,4° (с=0,490, ДМСО)
Элементный анализ C13H8BF4NO4
Вычислено (%): С, 47,46; Н, 2,46; N, 4,26
Найдено (%): С, 47,68; Н, 2,59; N, 4,32
Данное хелатное соединение (310 мг) растворяли в диметилсульфоксиде (6 мл) и добавляли триэтиламин (0,32 мл) и N-метилпиперазин (0,13 мл). Полученную смесь перемешивали при комнатной температуре в течение 17 часов и затем упаривали досуха при пониженном давлении. Остаток промывали диэтиловым эфиром и растворяли в 95% этаноле (20 мл), содержащем триэтиламин (0,5 мл), с последующим нагреванием при кипении с обратным холодильником в течение 8 часов. После охлаждения остаток, полученный упариванием досуха, растворяли в разбавленной хлористоводородной кислоте (5%) и разделяли, встряхивая с хлороформом. Водный слой доводили до рН 11 гидроксидом натрия (1 моль/л) и затем до рН 7,4 хлористоводородной кислотой (1 моль/л). Затем экстрагировали хлороформом (50 мл ×3) и сушили над мирабилитом. После выпаривания хлороформа полученный таким образом порошок перекристаллизовывали из смеси этанол-диэтиловый эфир, получая таким образом 120 мг указанного в заголовке соединения в виде прозрачных тонких игл.
Температура плавления: от 225 до 227°С (разложение)
Элементный анализ для C18H20FN3O4
Вычислено (%): С, 58,37; Н, 5,72; N, 11,35
Найдено (%): С, 58,17; Н, 5,58; N, 11,27
Пример 101: Хелатный комплекс дифторида бора и (3S)-9,10-дифтор-3-метил-7-оксо-2,3-дигидро-7Н-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновой кислоты
(S)-Диэтил (7,8-дифтор-3-метил-3,4-дигидро-2Н-[1,4] бензоксазин-4-ил)метиленмалонат (2 г) смешивали с уксусным ангидридом (2 мл). При 140°С добавляли к нему 47% комплекс трехфтористый бор/тетрагидрофуран (0,8 мл) и полученную смесь перемешивали при нагревании при той же температуре в течение 1 часа. После выпаривания образующихся таким образом низкокипящих веществ жидкую реакционную смесь охлаждали до комнатной температуры. После добавления ацетона (10 мл) жидкую реакционную смесь перемешивали при той же температуре в течение 30 минут. Выпавшие при этом в осадок кристаллы отделяли и промывали ацетоном, получая 1,55 г указанного в заголовке соединения.
Пример 102: (3S)-(-)-9-Фтор-3-метил-10-(4-метил-1-пиперазинил-7-оксо-2,3-дигидро-7Н-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота (левофлоксацин)
(S)-(-)-9,10-Дифтор-3-метил-7-оксо-2,3-дигидро-7Н-пиридо [1,2,3-de][1,4]бензоксазин-6-карбоновую кислоту (21 мг) и N-метилпиперазин (30 мг) растворяли в безводном диметилсульфоксиде (3 мл) и перемешивали при температуре 130-140°С в течение 1 часа. После выпаривания растворителя к остатку добавляли этанол (2 мл). Выпавшее таким образом твердое вещество отфильтровывали и промывали последовательно небольшим количеством этанола и эфира. 14 мг полученного порошка подвергали колоночной хроматографии на силикагеле с использованием 5 г силикагеля и элюировали нижним слоем раствора хлороформ-метанол-вода (7:3:1), получая таким образом (S)-(-)-9-фтор-3-метил-10-(4-метил-1-пиперазинил)-7-оксо-2,3-дигидро-7Н-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновую кислоту. Описанный выше маточный раствор, полученный при фильтровании, фракционировали и подвергали тонкослойной хроматографии (силикагель, 20×20 см, 0,5 мм), проводя таким образом очистку с помощью проявления нижним слоем раствора хлороформ-метанол-вода (15:3:1). Продукты объединяли, получая таким образом 14 мг кристаллов целевого соединения.
Температура плавления: от 220 до 228°С (разложение).
Элементный анализ для C18H20FN3O4
Вычислено (%): С, 59,82; Н, 5,58; N, 11,63
Найдено (%): С, 60,01; Н, 5,69; N, 11,53
Масс-спектр (m/e); 361 (М+)
1H-ЯМР (CDCl3) δ (м.д.): 1,63 (3Н, д, J=7 Гц), 2,38 (3Н, с), 2,54-2,60 (4Н, м), 3,40-3,44 (4Н, м), 4,35-4,52 (3Н, м), 7,76 (1Н, д), 8,64 (1Н, с).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ СТЕРИЧЕСКИХ СОЕДИНЕНИЙ | 2007 |
|
RU2481326C2 |
| ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ОБЛАДАЮЩИХ АНТИДИАБЕТИЧЕСКОЙ АКТИВНОСТЬЮ | 1995 |
|
RU2151145C1 |
| ПРОИЗВОДНЫЕ АМИНОСПИРТОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2001 |
|
RU2233839C1 |
| ТАКСАНЫ С БОКОВОЙ ЦЕПЬЮ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ | 1993 |
|
RU2125042C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ФЕНИЛАЛАНИНА С ХИНАЗОЛИНДИОНОВЫМ СКЕЛЕТОМ И ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ, ПРИМЕНЯЕМЫХ ПРИ ПОЛУЧЕНИИ ТАКИХ ПРОИЗВОДНЫХ | 2007 |
|
RU2469028C2 |
| ПРОИЗВОДНЫЕ АЗОЛА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2047605C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ МАКРОЛИДОВ И КЕТОЛИДОВ, И ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ ДЛЯ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2608390C2 |
| ГЛЮКОПИРАНОЗИЛОКСИБЕНЗИЛБЕНЗОЛЬНЫЕ ПРОИЗВОДНЫЕ, ЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭТИ ПРОИЗВОДНЫЕ, И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ УКАЗАННЫХ ПРОИЗВОДНЫХ | 2001 |
|
RU2254340C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2018 |
|
RU2780892C2 |
| ПРОИЗВОДНОЕ БЕНЗОИЛПИРИДИНА ИЛИ ЕГО СОЛЬ, СОДЕРЖАЩИЙ ЕГО В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА ФУНГИЦИД, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2001 |
|
RU2255088C2 |
Изобретение относится к способу получения производных бензоксазина. Описывается способ получения соединения, представленного следующей формулой:
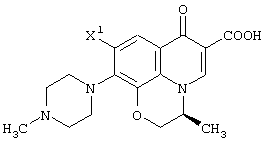 ,
,
который включает реакцию соединения, представленного формулой (I):
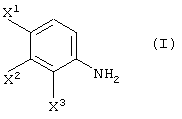 ,
,
с соединением, представленным формулой (II-1-а), в присутствии основания
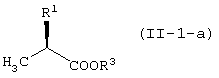 ,
,
давая соединение, представленное формулой (III-1-а)
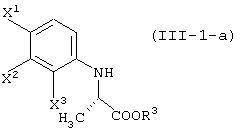 ,
,
восстановление данного соединения в соединение, представленное формулой (IV-а)
 ,
,
взаимодействие данного соединения с соединением, представленным следующей формулой:
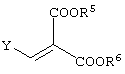 ,
,
давая соединение, представленное формулой (V-a)
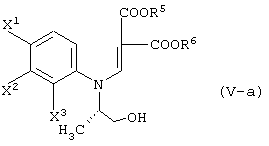 ,
,
а затем обработку данного соединения в присутствии основания с получением соединения, представленного формулой (VI-a)
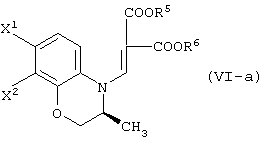 ,
,
обработку данного соединения соединением трехфтористого бора, с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
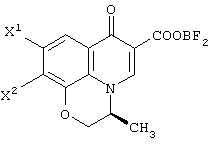 ,
,
реакцию данного соединения с 4-метилпиперазином с получением соединения, представленного следующей формулой:
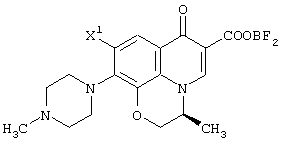 ,
,
затем расщепление и элиминирование хелата бора данного соединения; [в каждой из вышеуказанных формул, Х1, X2 и X3, каждый независимо, представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила; R5 и R6, каждый независимо, представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила; и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными, и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)]. Также описываются варианты вышеуказанного способа, способы получения промежуточных соединений и промежуточные соединения. Технический результат - предложены промышленно благоприятные способы получения промежуточных соединений, которые полезны при получения антибактериальных соединений. 30 н. и 66 з.п. ф-лы, 2 табл.
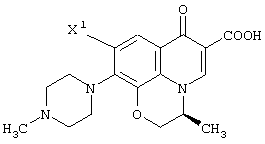
который включает реакцию соединения, представленного формулой (I)
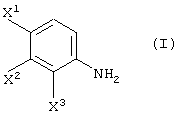
с соединением, представленным формулой (II-1-a), в присутствии основания
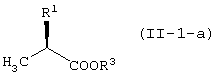
давая соединение, представленное формулой (III-1-a)
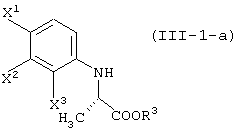
восстановление данного соединения в соединение, представленное формулой (IV-а)
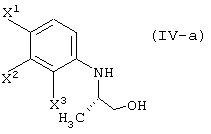
взаимодействие данного соединения с соединением, представленным следующей формулой:
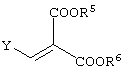
давая соединение, представленное формулой (V-a)
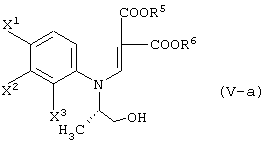
а затем обработку данного соединения в присутствии основания с получением соединения, представленного формулой (VI-a)
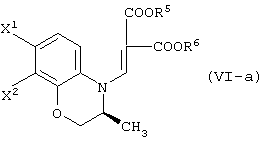
обработку данного соединения соединением трехфтористого бора с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
реакцию данного соединения с 4-метилпиперазином с получением соединения, представленного следующей формулой:
затем расщепление и элиминирование хелата бора данного соединения;
[в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила; R5 и R6 каждый независимо представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)].
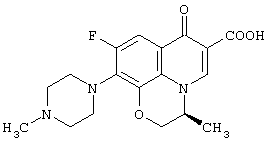
который включает реакцию соединения, представленного формулой (I)
с соединением, представленным формулой (II-2-a), в присутствии основания
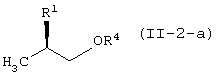
давая соединение, представленное формулой (III-2-a)
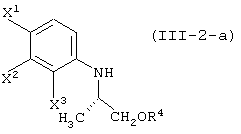
элиминирование защитной группы гидроксила данного соединения с получением соединения, представленного формулой (IV-a)
реакцию данного соединения с соединением, представленным следующей формулой:
давая соединение, представленное формулой (V-a)
а затем обработку данного соединения в присутствии основания с получением соединения, представленного формулой (VI-a)
обработку данного соединения соединением трехфтористого бора с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
реакцию данного соединения с 4-метилпиперазином с получением соединения, представленного следующей формулой:
затем расщепление и элиминирование хелата бора данного соединения;
[в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила; R5 и R6 каждый независимо представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)].
который включает реакцию соединения, представленного формулой (I)
с соединением, представленным формулой (II-1-a), в присутствии основания
давая соединение, представленное формулой (III-1-a)
восстановление данного соединения в соединение, представленное формулой (IV-a)
обработку данного соединения в присутствии основания с получением соединения, представленного формулой (VII-a)
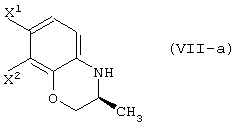
и реакцию данного соединения с соединением, представленным следующей формулой:
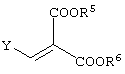
с получением соединения, представленного формулой (VI-a)
обработку данного соединения соединением трехфтористого бора с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
реакцию данного соединения с 4-метилпиперазином с получением соединения, представленного следующей формулой:
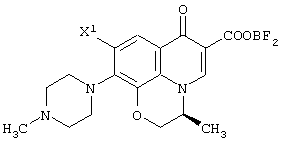
затем расщепление и элиминирование хелата бора данного соединения;
[в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила; R5 и R6 каждый независимо представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)].
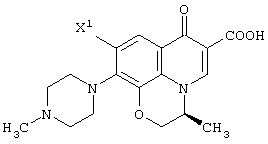
который включает реакцию соединения, представленного формулой (I)
с соединением, представленным формулой (II-2-а), в присутствии основания
давая соединение, представленное формулой (III-2-а)
элиминирование защитной группы гидроксила данного соединения с получением соединения, представленного формулой (IV-а)
обработку данного соединения в присутствии основания с получением соединения, представленного формулой (VII-a)
а затем реакцию данного соединения с соединением, представленным следующей формулой:
с получением соединения, представленного формулой (VI-a)
обработку данного соединения соединением трехфтористого бора с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
реакцию данного соединения с 4-метилпиперазином с получением соединения, представленного следующей формулой:

затем расщепление и элиминирование хелата бора данного соединения;
[в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила; R5 и R6 каждый независимо представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)].
который включает реакцию соединения, представленного формулой (I)
с соединением, представленным формулой (II-1), в присутствии основания
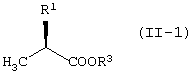
давая соединение, представленное формулой (III-1)
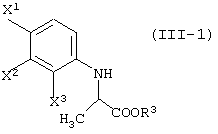
а затем данное соединение обрабатывают либо методом, который включает обработку данного соединения ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, после завершения данной обработки выделение продукта из обработанной жидкой смеси в случае соединения, представленного формулой (III-1), в которой R3 не является атомом водорода, либо методом, который включает оптическое разделение данного соединения по реакции с оптически активным органическим основанием, в случае соединения, представленного формулой (III-1), в которой R3 представляет атом водорода, с получением соединения карбоновой кислоты, представленного следующей формулой:
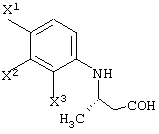
этерификацию данного соединения в присутствии спирта, представленного следующей формулой:
R7-OH
с получением сложноэфирного соединения, представленного следующей формулой:
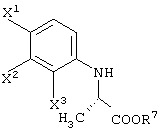
восстановление данного соединения в соединение, представленное формулой (IV-а)
реакцию данного соединения с соединением, представленным следующей формулой:
давая соединение, представленное формулой (V-a)
а затем обработку данного соединения в присутствии основания с получением соединения, представленного формулой (VI-a)
обработку данного соединения соединением трехфтористого бора с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
реакцию данного соединения с 4-метилпиперазином с получением соединения, представленного следующей формулой:
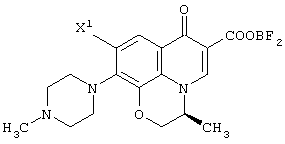
затем расщепление и элиминирование хелата бора данного соединения;
[в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила; R5 и R6 каждый независимо представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)].
который включает реакцию соединения, представленного формулой (I)
с соединением, представленным формулой (II-1), в присутствии основания
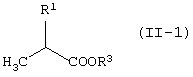
давая соединение, представленное формулой (III-1)
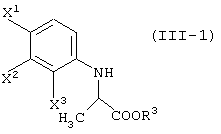
а затем данное соединение обрабатывают либо методом, который включает обработку данного соединения ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, после завершения данной обработки выделение продукта из обработанной жидкой смеси в случае соединения, представленного формулой (III-1), в которой R3 не является атомом водорода, либо методом, который включает оптическое разделение данного соединения по реакции с оптически активным органическим основанием, в случае соединения, представленного формулой (III-1), в которой R3 представляет атом водорода, с получением соединения карбоновой кислоты, представленного следующей формулой:
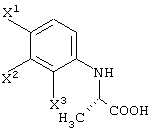
этерификацию данного соединения в присутствии спирта, представленного следующей формулой:
R7-OH
с получением сложноэфирного соединения, представленного следующей формулой:
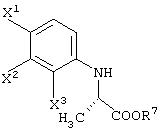
восстановление данного соединения в соединение, представленное формулой (IV-a)
обработку данного соединения в присутствии основания, давая соединение, представленное формулой (VII-a)
а затем реакцию данного соединения с соединением, представленным следующей формулой:
с получением соединения, представленного формулой (VI-a)
обработку данного соединения соединением трехфтористого бора с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
реакцию данного соединения с 4-метилпиперазином с получением соединения, представленного следующей формулой:
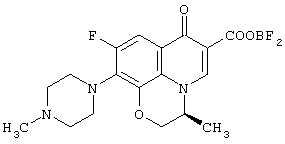
затем расщепление и элиминирование хелата бора данного соединения;
[в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила; R5 и R6, каждый независимо, представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)].
который включает реакцию соединения, представленного следующей формулой:
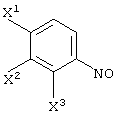
или следующей формулой:
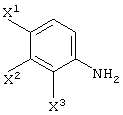
с соединением, представленным следующей формулой, в присутствии металлического катализатора в атмосфере газообразного водорода, необязательно в присутствии дегидратирующего агента или кислоты
СН3COCOOR3,
давая соединение, представленное формулой (III-1)
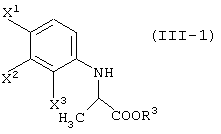
а затем данное соединение обрабатывают либо методом, который включает обработку данного соединения ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, после завершения данной обработки выделение продукта из обработанной жидкой смеси в случае соединения, представленного формулой (III-1), в которой R3 не является атомом водорода, либо методом, который включает оптическое разделение данного соединения по реакции с оптически активным органическим основанием, в случае соединения, представленного формулой (III-1), в которой R3 представляет атом водорода, с получением соединения карбоновой кислоты, представленного следующей формулой:
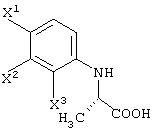
этерификацию данного соединения в присутствии спирта, представленного следующей формулой:
R7-OH
с получением сложноэфирного соединения, представленного следующей формулой:
восстановление данного соединения в соединение, представленное формулой (IV-a)
реакцию данного соединения с соединением, представленным следующей формулой:
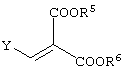
давая соединение, представленное формулой (V-a)
а затем обработку данного соединения в присутствии основания с получением соединения, представленного формулой (VI-a)
обработку данного соединения соединением трехфтористого бора с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
реакцию данного соединения с 4-метилпиперазином с получением соединения, представленного следующей формулой:

затем расщепление и элиминирование хелата бора данного соединения;
[в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила; R5 и R6 каждый независимо представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)].
который включает реакцию соединения, представленного следующей формулой:
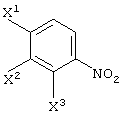
или следующей формулой:
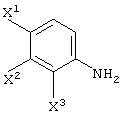
с соединением, представленным следующей формулой, в присутствии металлического катализатора в атмосфере газообразного водорода, необязательно в присутствии дегидратирующего агента или кислоты:
СН3COCOOR3,
давая соединение, представленное формулой (III-1)
а затем данное соединение обрабатывают либо методом, который включает обработку данного соединения ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, после завершения данной обработки выделение продукта из обработанной жидкой смеси в случае соединения, представленного формулой (III-1), в которой R3 не является атомом водорода, либо методом, который включает оптическое разделение данного соединения по реакции с оптически активным органическим основанием, в случае соединения, представленного формулой (III-1), в которой R3 представляет атом водорода, с получением соединения карбоновой кислоты, представленного следующей формулой:
этерификацию данного соединения в присутствии спирта, представленного следующей формулой:
R7-OH
с получением сложноэфирного соединения, представленного следующей формулой:
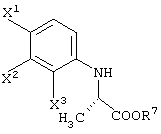
восстановление данного соединения в соединение, представленное формулой (IV-a)
обработку данного соединения в присутствии основания, давая соединение, представленное формулой (VII-a):
а затем реакцию данного соединения с соединением, представленным следующей формулой:
с получением соединения, представленного формулой (VI-a)
обработку данного соединения соединением трехфтористого бора с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
реакцию данного соединения с 4-метилпиперазином с получением соединения, представленного следующей формулой:
затем расщепление и элиминирование хелата бора данного соединения;
[в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила; R5 и R6, каждый независимо, представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода) ].
который включает реакцию соединения, представленного следующей формулой:
с соединением, представленным следующей формулой:
СН3COCOOR3
давая соединение, представленное следующей формулой:
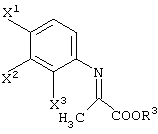
асимметрическое восстановление данного соединения в соединение, представленное формулой (III-1-a)
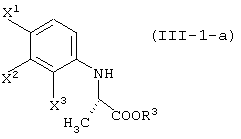
восстановление данного соединения в соединение, представленное формулой (IV-a)
реакцию данного соединения с соединением, представленным следующей формулой:
давая соединение, представленное формулой (V-a)
а затем обработку данного соединения в присутствии основания с получением соединения, представленного формулой (VI-a)
обработку данного соединения соединением трехфтористого бора с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
реакцию данного соединения с 4-метилпиперазином с получением соединения, представленного следующей формулой:
затем расщепление и элиминирование хелата бора данного соединения;
[в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила; R5 и R6, каждый независимо, представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)].
который включает реакцию соединения, представленного следующей формулой:
с соединением, представленным следующей формулой:
СН3COCOOR3
давая соединение, представленное следующей формулой:
асимметрическое восстановление данного соединения в соединение, представленное формулой (III-1-a)
восстановление данного соединения в соединение, представленное формулой (IV-а)
обработку данного соединения в присутствии основания, давая соединение, представленное формулой (VII-a)
а затем реакцию данного соединения с соединением, представленным следующей формулой:
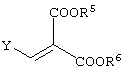
с получением соединения, представленного формулой (VI-a)
обработку данного соединения соединением трехфтористого бора с превращением его таким образом в хелатное соединение бора, представленное следующей формулой:
реакцию данного соединения с 4-метилпиперазином с получением соединения, представленного следующей формулой:
затем расщепление и элиминирование хелата бора данного соединения;
[в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R1 представляет уходящую группу; R3 представляет атом водорода или защитную группу карбоксила; R4 представляет защитную группу гидроксила; R5 и R6, каждый независимо, представляет алкильную группу, содержащую 1-6 атомов углерода; R7 представляет защитную группу карбоксила и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)].
который включает обработку соединения формулы (I-0)
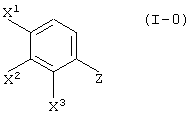
соединением следующей формулы:
СН3COCOOR3
в присутствии металлического катализатора в атмосфере газообразного водорода, необязательно в присутствии акцептора кислоты или кислоты;
(в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R3 представляет атом водорода или защитную группу карбоксила и Z представляет нитрогруппу или аминогруппу).
который включает обработку сложноэфирного соединения среди соединений, представленных формулой (III-1)
ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, а затем выделение и сбор продукта из обработанной жидкости;
(где Х1, X2 и X3 каждый независимо представляет атом галогена; R3 представляет атом водорода или защитную группу карбоксила).
который включает обработку сложноэфирного соединения среди соединений формулы (III-1)
в присутствии фермента, способного асимметрически гидролизовать сложный эфир, или жидкой культуральной среды микроорганизма, клеток данного микроорганизма или переработанных клеток данного микроорганизма, а затем отделение и удаление соединения формулы (III-1-b) из обработанной жидкости:
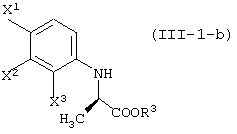
(где X1, X2 и X3 каждый независимо представляет атом галогена и R3 представляет атом водорода или защитную группу карбоксила).
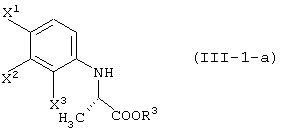
который включает обработку сложноэфирного соединения среди соединений, представленных формулой (III-1)
ферментом, способным асимметрически гидролизовать сложный эфир, или жидкой культуральной средой микроорганизма, клетками данного микроорганизма или переработанными клетками данного микроорганизма, а затем выделение и сбор продукта из обработанной жидкости;
(где X1, X2 и X3 каждый независимо представляет атом галогена; R3 представляет атом водорода или защитную группу карбоксила).
который включает обработку сложноэфирного соединения среди соединений, представленных формулой (III-1)
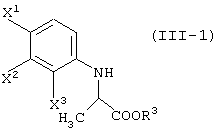
в присутствии фермента, способного асимметрически гидролизовать сложный эфир, или жидкой культуральной среды микроорганизма, клеток данного микроорганизма или переработанных клеток данного микроорганизма, а затем отделение и удаление соединения карбоновой кислоты, представленного следующей формулой из обработанной жидкости:
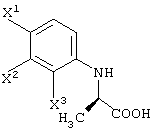
(где X1, X2 и X3 каждый независимо представляет атом галогена и R3 представляет атом водорода или защитную группу карбоксила).
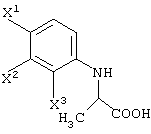
(где X1, X2 и X3 каждый независимо представляет атом галогена);
с использованием оптически активного органического основания;
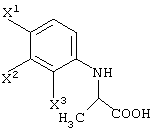
(где Х1, X2 и X3 каждый независимо представляет атом галогена);
оптически активным органическим основанием с получением диастереомерной соли одного из оптических изомеров 2-(2,3,4-тригалогенанилино)пропионовой кислоты и оптически активного органического основания, а затем обработку диастереомерной соли кислотой.
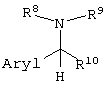
(в которой Aryl представляет арильную группу, необязательно имеющую атом галогена, нитрогруппу, цианогруппу, карбамоильную группу, алкильную группу, содержащую 1-6 атомов углерода, или алкоксигруппу, содержащую 1-6 атомов углерода; и
R8, R9 и R10 каждый независимо представляет
(1) фенильную группу, необязательно имеющую атом галогена, алкильную группу, содержащую 1-6 атомов углерода, галогеналкильную группу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, нитрогруппу, карбамоильную группу или цианогруппу;
(2) бензильную группу, необязательно содержащую атом галогена, алкильную группу, содержащую 1-6 атомов углерода, галогеналкильную группу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, нитрогруппу, карбамоильную группу или цианогруппу;
(3) алкильную группу, содержащую 1-6 атомов углерода; или
(4) атом водорода.
который включает обработку сложноэфирного соединения среди соединений, представленных формулой (III-1-b), в присутствии основания:
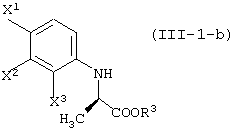
(где X1, X2 и X3 каждый независимо представляет атом галогена и R3 представляет атом водорода или защитную группу карбоксила).
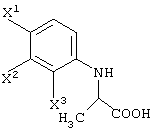
который включает рацемизирующую обработку сложноэфирного соединения среди соединений, представленных формулой (III-1-b), с помощью обработки в присутствии основания с последующим гидролизом:
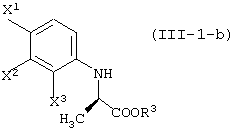
(где X1, X2 и X3 каждый независимо представляет атом галогена и R3 представляет атом водорода или защитную группу карбоксила).
который включает реакцию соединения, представленного формулой (III-1-a)
или соединения, представленного следующей формулой:
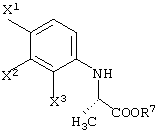
с соединением боргидрида металла в апротонном растворителе в присутствии спирта с получением соединения, представленного формулой (IV-a)
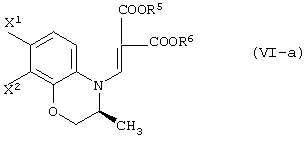
реакцию данного соединения с соединением, представленным следующей формулой в основных условиях:
давая соединение формулы (V-a)
а затем обработку данного соединения в основных условиях,
[в каждой из вышеуказанных формул X1, X2 и X3 каждый независимо представляет атом галогена; R3 представляет атом водорода или защитную группу карбоксила; R5, R6 и R7 каждый независимо, представляет алкильную группу, содержащую 1-6 атомов углерода, и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)].
который включает реакцию соединения, представленного формулой (IV-a)
с соединением, представленным следующей формулой в основных условиях:
[в каждой из вышеуказанных формул Х1, X2 и X3 каждый независимо представляет атом галогена; R3 представляет атом водорода или защитную группу карбоксила; R5 и R6 каждый независимо, представляет алкильную группу, содержащую 1-6 атомов углерода, и Y представляет алкоксигруппу, содержащую 1-6 атомов углерода, атом галогена или диалкиламиногруппу (где алкильные группы могут быть одинаковыми или различными и каждая представляет алкильную группу, содержащую 1-6 атомов углерода)].
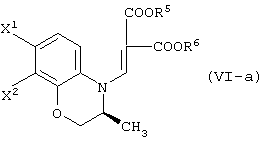
который включает реакцию соединения формулы (V-a) в основных условиях
(где X1, X2 и X3 каждый независимо представляет атом галогена и R5 и R6 каждый независимо представляет алкильную группу, содержащую 1-6 атомов углерода).
который включает обработку соединения, представленного формулой (III-1-a)
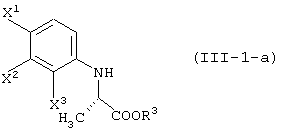
или соединения, представленного следующей формулой:
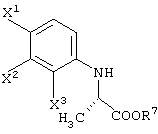
в апротонном растворителе соединением боргидрида металла и спиртом;
(где X1, X2 и X3 каждый независимо представляет атом галогена; R3 представляет атом водорода или защитную группу карбоксила и R7 представляет защитную группу карбоксила).
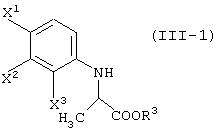
(в которой X1, X2 и X3 каждый независимо представляет атом галогена и R3 представляет атом водорода или алкильную группу).
(в которой X1, X2 и X3 каждый независимо представляет атом галогена и R3 представляет атом водорода или алкильную группу).
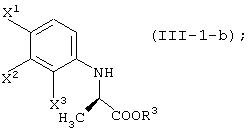
(в которой X1, X2 и X3 каждый независимо представляет атом галогена и R3 представляет атом водорода или алкильную группу).
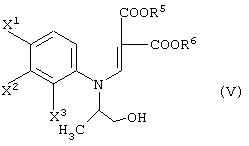
(в которой Х1, Х2 и Х3 каждый независимо представляет атом галогена и R5 и R6 каждый независимо представляет алкильную группу).
(в которой X1, X2 и X3 каждый независимо представляет атом галогена и R5 и R6 каждый независимо представляет алкильную группу).
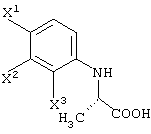
и оптически активного основания,
где Х1, X2 и X3 каждый независимо представляет атом галогена.
и оптически активного основания,
где X1, X2 и X3 каждый независимо представляет атом галогена.
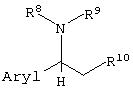
(в которой Aryl представляет арильную группу, необязательно имеющую атом галогена, нитрогруппу, цианогруппу, карбамоильную группу, алкильную группу, содержащую 1-6 атомов углерода, или алкоксигруппу, содержащую 1-6 атомов углерода; и R8, R9 и R10 каждый независимо представляет
(1) фенильную группу, необязательно имеющую атом галогена, алкильную группу, содержащую 1-6 атомов углерода, галогеналкильную группу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, нитрогруппу, карбамоильную группу или цианогруппу;
(2) бензильную группу, необязательно имеющую атом галогена, алкильную группу, содержащую 1-6 атомов углерода, галогеналкильную группу, содержащую 1-6 атомов углерода, алкоксигруппу, содержащую 1-6 атомов углерода, нитрогруппу, карбамоильную группу или цианогруппу;
(3) алкильную группу, содержащую 1-6 атомов углерода; или
(4) атом водорода).
| US 5644056 А, 01.07.1997 | |||
| СПОСОЁ ИЗГОТОВЛЕНИЯ АКТИВНОЙ МАССЫ | 0 |
|
SU322815A1 |
| 1,7-АНЕЛЛИРОВАННЫЕ ПРОИЗВОДНЫЕ 3-(ПИПЕРАЗИНОАЛКИЛ)-ИНДОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЙ ПРОДУКТ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2083580C1 |

