Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения производного фенилаланина с хиназолиндионовым скелетом, которое представляет собой соединение, весьма применимое в качестве лекарственного средства, обладающего α-4-интегрин-ингибирующей активностью, и в качестве промежуточного соединения для получения такого производного.
Уровень техники
В последнее время были проведены специальные исследования воспалительных заболеваний, в патологию которых вовлечен процесс α-4-интегрин-зависимой адгезии, таких как ревматоидный артрит, воспалительный энтерит, системный эритематоз, множественный склероз, синдром Шегрена (Sjögren), астма, псориаз, аллергия, диабет, сердечно-сосудистые заболевания, артериосклероз, рестеноз, онкогенные гиперплазии, метастазирование опухоли и отторжение имплантата, и соответственно ожидается, что должно разрабатываться и применяться в качестве терапевтического или профилактического средства соединение, обладающее α-4-интегрин-ингибирующей активностью.
Заявителем настоящего изобретения было предложено изобретение, относящееся к новому производному фенилаланина, которое обладает α-4-интегрин-ингибирующей активностью и которое можно было бы рассматривать в качестве весьма применимого средства для лечения или профилактики воспалительного заболевания, в патологию которого вовлечен процесс α-4-интегрин-зависимой адгезии, и ранее заявителем подана патентная заявка (см. указанный ниже патентный документ 1). В данной патентной заявке сообщалось о способе получения такого производного фенилаланина с хиназолиндионовым скелетом, который включает в себя стадии нанесения производного фенилаланина на твердую фазу из полимерной смолы для создания при этом хиназолиндионового скелета благодаря применению промежуточного амидного продукта (см. указанный ниже патентный документ 1).
Кроме того, аналогично этому заявителем настоящего изобретения обнаружен способ получения такого производного, подходящий для применения в промышленном масштабе, и ранее подана патентная заявка (см. указанный ниже патентный документ 2). В данной патентной заявке сообщалось о способе, включающем в себя стадии взаимодействия производного антраниловой кислоты, чья карбоксильная группа защищена посредством сложноэфирной связи, с производным фенилаланина и получения в конечном итоге производного фенилаланина с хиназолиндионовым скелетом.
Кроме того, заявителем настоящего изобретения обнаружено производное фенилаланина с новым хиназолиндионовым скелетом, которое обладает α-4-интегрин-ингибирующей активностью, и ранее подана патентная заявка (указанный ниже патентный документ 3). В данной патентной заявке сообщалось о прямом (линейном) способе жидкофазного синтеза для последовательного формирования его скелета как способа получения.
Однако все еще существует потребность в разработке способа получения производного фенилаланина с хиназолиндионовым скелетом, который является подходящим для применения в промышленном масштабе, а также дополнительном синтетическом подходе, который еще не известен. Более конкретно, существует потребность в разработке способа получения того же самого продукта наряду с достижением ряда преимуществ, например в разработке более конвергентного синтетического подхода; в разработке способа, требующего для получения конечного соединения небольшого числа стадий, исходя из исходного материала; способа, требующего для образования конечного продукта более короткого периода времени; и способа, позволяющего производить конечное соединение с более высоким выходом; и получать в результате конечное соединение с высокой степенью чистоты.
Патентный документ 1: WO 02/16329
Патентный документ 2: WO 2004/74264
Патентный документ 3: WO 2005/61466
Описание изобретения
Соответственно целью настоящего изобретения является разработка способа получения производного фенилаланина с хиназолиндионовым скелетом, который позволяет подходящим образом получать данное соединение в промышленном масштабе с более высоким выходом.
Еще одной целью настоящего изобретения является разработка способа получения производного фенилаланина с хиназолиндионовым скелетом, который до сих пор неизвестен и который можно успешно осуществлять в промышленном масштабе.
Еще одной дополнительной целью настоящего изобретения является получение промежуточного соединения, применяемого при получении производного фенилаланина с хиназолиндионовым скелетом.
Таким образом, в настоящем изобретении предлагается способ (первый способ получения) получения производного фенилаланина с хиназолиндионовым циклом, представленного следующей формулой (1), или его фармацевтически приемлемой соли, который включает в себя следующие стадии (a), (b) и (c):

в которой R1 представляет собой либо фенильную группу, которая может содержать заместитель, либо пиридильную группу, которая может содержать заместитель; R2 представляет собой алкильную группу, которая может содержать заместитель; R3 представляет собой алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, или алкильную группу, замещенную аминогруппой; и R4 представляет собой атом водорода, алкильную группу или бензильную группу, которая может содержать заместитель;
(a) взаимодействие производного ацилфенилаланина, представленного следующей формулой (2), или его химически приемлемой соли:
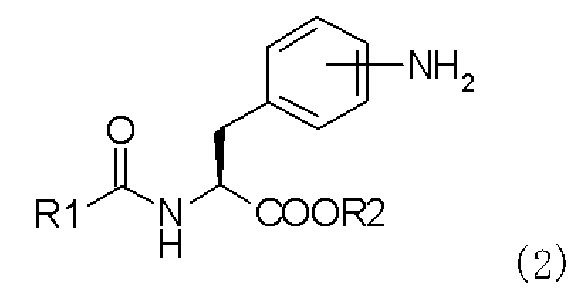 ,
,
в которой R1 и R2 имеют те же самые значения, которые указаны выше,
с реагентом, вводящим карбонильную группу, и производным антраниловой кислоты, представленным следующей формулой (3), или его химически приемлемой солью:
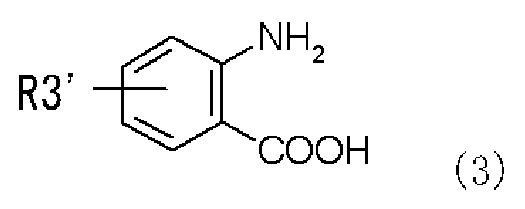 ,
,
в которой R3' представляет собой алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, которая может иметь защитную группу, или алкильную группу, замещенную аминогруппой, которая может иметь защитную группу,
с образованием при этом производного мочевины с асимметрично расположенными карбоксигруппами, представленного следующей формулой (4), или его химически приемлемой соли:
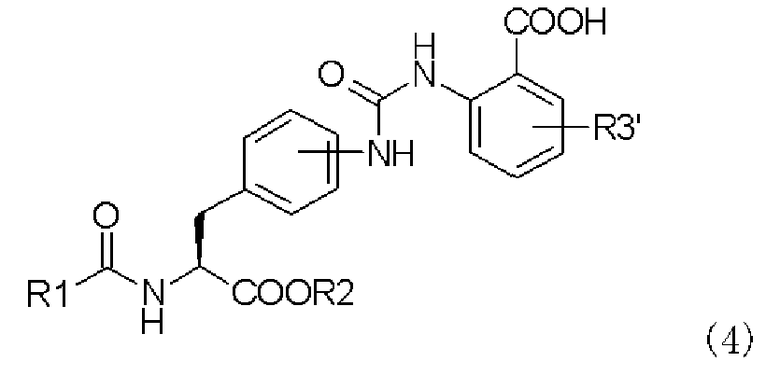 ,
,
в которой R1, R2 и R3' имеют те же самые значения, которые указаны выше;
(b) превращение производного мочевины с асимметрично расположенными карбоксигруппами формулы (4) в хиназолиндионовое производное, представленное следующей формулой (5), или его фармацевтически приемлемую соль в присутствии средства, активирующего карбоксильную группу:
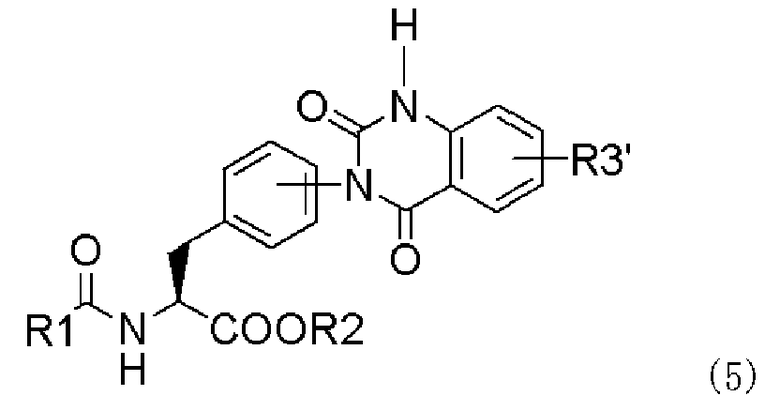 ,
,
в которой заместители R1-R3' имеют те же самые значения, которые указаны выше; и
(c) если требуется, замещение атома водорода, присоединенного к атому азота, присутствующему в хиназолиндионовом цикле хиназолиндионового производного формулы (5), N-алкильной группой с помощью N-алкилирующего средства и затем снятие защитных групп в полученном продукте, когда заместитель R3' защищен.
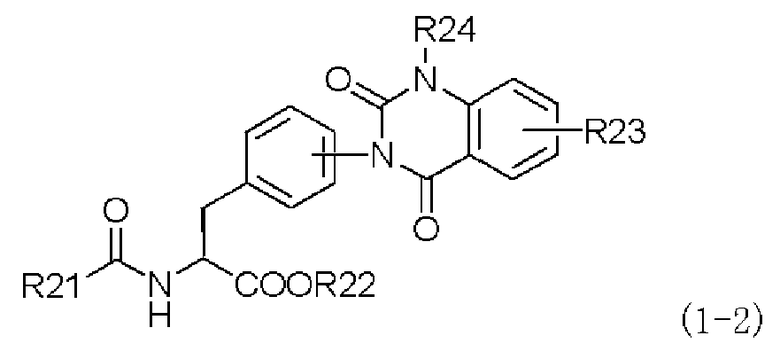
Кроме того, в настоящем изобретении дополнительно предлагается способ (второй способ получения) получения производного фенилаланина с хиназолиндионовым циклом, представленного следующей формулой (1-2), или его фармацевтически приемлемой соли, который включает в себя следующие стадии (a), (b), (c) и (d):
 ,
,
в которой R21 представляет собой либо фенильную группу, которая может содержать заместитель, либо пиридильную группу, которая может содержать заместитель; заместитель R22 представляет собой алкильную группу, которая может содержать заместитель; заместитель R23 представляет собой диалкиламиногруппу, моноалкиламиногруппу, аминогруппу, атом водорода, атом галогена, алкильную группу, перфторалкильную группу, алкоксигруппу, нитрогруппу, алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, алкильную группу, замещенную аминогруппой, алкильную группу, замещенную алкенильной группой, алкильную группу, замещенную алкинильной группой, карбоксильную группу, алкоксикарбонильную группу, алкилтиогруппу или арилтиогруппу; заместитель R24 представляет собой атом водорода, алкильную группу или бензильную группу, которая может содержать заместитель;
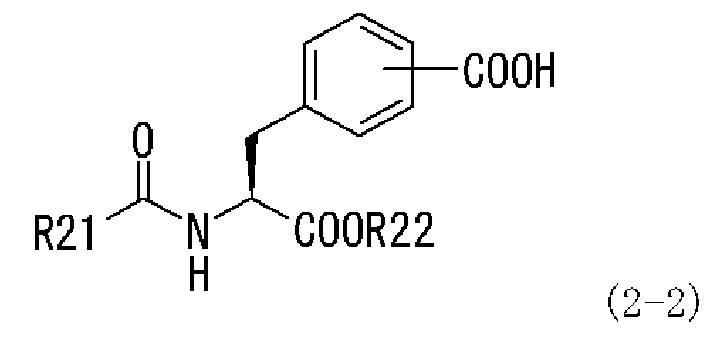
(a) превращение производного ацилфенилаланина, представленного следующей формулой (2-2), или его химически приемлемой соли:
 ,
,
в которой заместители R21 и R22 имеют те же самые значения, которые указаны выше,
в изоцианатное производное и затем превращение его карбоксильной группы в изоцианильную группу;
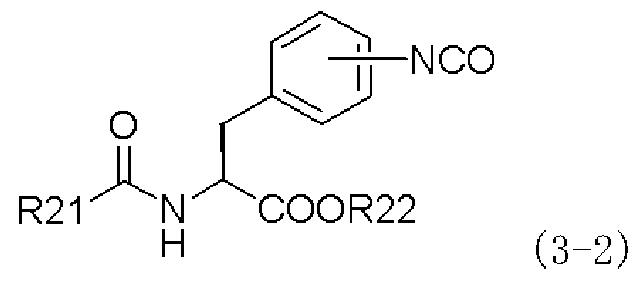
(b) взаимодействие полученного соединения, представленного следующей формулой (3-2), или его химически приемлемой соли:
 ,
,
в которой заместители R21 и R22 имеют те же самые значения, которые указаны выше,
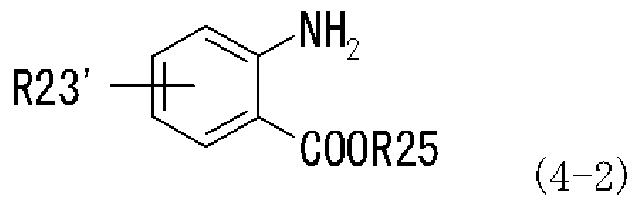
с производным антраниловой кислоты, представленным следующей формулой (4-2), или его химически приемлемой солью:
 ,
,
в которой R23' представляет собой диалкиламиногруппу, моноалкиламиногруппу, аминогруппу, которая может иметь защитную группу, атом водорода, атом галогена, алкильную группу, перфторалкильную группу, алкоксигруппу, нитрогруппу, алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, которая может иметь защитную группу, алкильную группу, замещенную аминогруппой, которая может иметь защитную группу, алкильную группу, замещенную алкенильной группой, алкильную группу, замещенную алкинильной группой, карбоксильную группу, алкоксикарбонильную группу, алкилтиогруппу или арилтиогруппу; и R25 представляет собой атом водорода или алкильную группу, которая может содержать заместитель;
(c) превращение полученного асимметричного производного мочевины, представленного следующей формулой (5-2), или его химически приемлемой соли:
 ,
,
в которой заместители R21, R22, R23' и R25 имеют те же самые значения, которые указаны выше,
в хиназолиндионовое производное, представленное следующей формулой (6-2), или его химически приемлемую соль:
 ,
,
в которой заместители R21-R23' имеют те же самые значения, которые указаны выше,
в присутствии средства, активирующего карбоксильную группу, когда R25 представляет собой атом водорода, или в присутствии основания, когда R25 представляет собой алкильную группу;
(d) если требуется, замещение атома водорода, присоединенного к атому азота, присутствующему в хиназолиндионовом цикле хиназолиндионового производного формулы (6-2), N-алкильной группой с помощью N-алкилирующего средства и затем снятие защитных групп в полученном продукте, когда заместитель R23' защищен.
Согласно настоящему изобретению дополнительно предлагается способ (третий способ получения) получения производного фенилаланина с хиназолиндионовым циклом, представленного следующей формулой (1-3), или его фармацевтически приемлемой соли, который включает в себя следующие стадии (a), (b) и (c):
 ,
,
в которой заместитель R31 представляет собой либо фенильную группу, которая может содержать заместитель, либо пиридильную группу, которая может содержать заместитель; R32 представляет собой алкильную группу, которая может содержать заместитель; R33 представляет собой диалкиламиногруппу, моноалкиламиногруппу, аминогруппу, атом водорода, атом галогена, алкильную группу, перфторалкильную группу, алкоксигруппу, нитрогруппу, алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, алкильную группу, замещенную аминогруппой, алкильную группу, замещенную алкенильной группой, алкильную группу, замещенную алкинильной группой, карбоксильную группу, алкоксикарбонильную группу, алкилтиогруппу или арилтиогруппу; R34 представляет собой атом водорода, алкильную группу или бензильную группу, которая может содержать заместитель;
(a) взаимодействие производного изатового ангидрида, представленного следующей формулой (2-3), или его химически приемлемой соли:
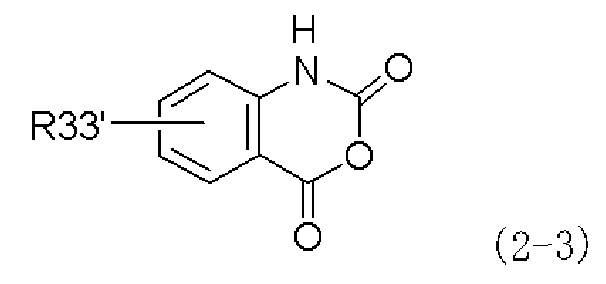 ,
,
в которой R33' представляет собой диалкиламиногруппу, моноалкиламиногруппу, аминогруппу, которая может иметь защитную группу, атом водорода, атом галогена, алкильную группу, перфторалкильную группу, алкоксигруппу, нитрогруппу, алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, которая может иметь защитную группу, алкильную группу, замещенную аминогруппой, которая может иметь защитную группу, алкильную группу, замещенную алкенильной группой, алкильную группу, замещенную алкинильной группой, карбоксильную группу, алкоксикарбонильную группу, алкилтиогруппу или арилтиогруппу,
с производным ацилфенилаланина, представленным следующей формулой (3-3), или его химически приемлемой солью:
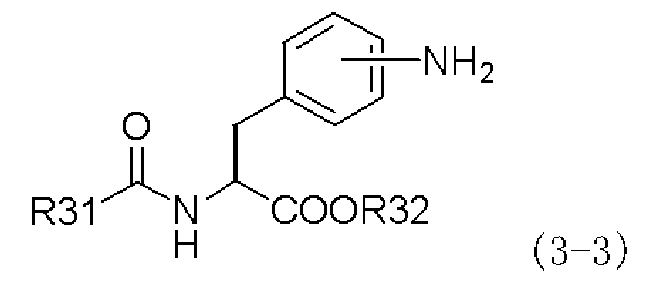 ,
,
в которой заместители R31 и R32 имеют те же самые значения, которые указаны выше;
(b) взаимодействие полученного амидного производного, представленного следующей формулой (4-3):
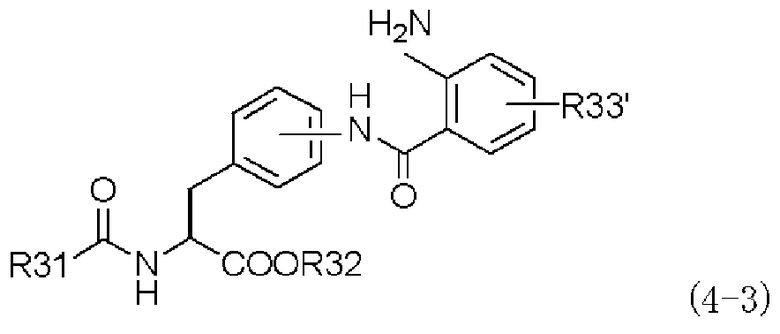 ,
,
в которой R31, R32 и R33' имеют те же самые значения, которые указаны выше,
с реагентом, вводящим карбонильную группу, с образованием при этом хиназолиндионового производного, представленного следующей формулой (5-3):
 ,
,
в которой заместители R31-R33' имеют те же самые значения, которые указаны выше; и
(c) если требуется, замещение атома водорода, присоединенного к атому азота, присутствующему в хиназолиндионовом цикле хиназолиндионового производного формулы (5-3), N-алкильной группой с помощью N-алкилирующего средства и затем снятие защитных групп в полученном продукте, когда заместитель R33' защищен.
Согласно настоящему изобретению дополнительно предлагается способ (четвертый способ получения) получения производного фенилаланина с хиназолиндионовым циклом, представленного следующей формулой (1-4), или его фармацевтически приемлемой соли, который включает в себя следующие стадии (a), (b) и (c):
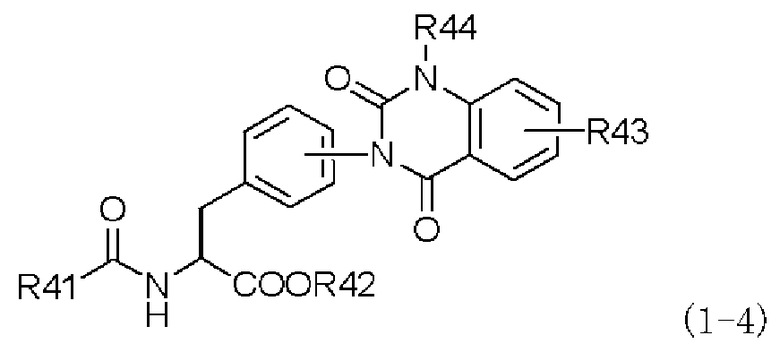 ,
,
в которой заместитель R41 представляет собой либо фенильную группу, которая может содержать заместитель, либо пиридильную группу, которая может содержать заместитель; заместитель R42 представляет собой алкильную группу, которая может содержать заместитель; R43 представляет собой диалкиламиногруппу, моноалкиламиногруппу, аминогруппу, атом водорода, атом галогена, алкильную группу, перфторалкильную группу, алкоксигруппу, нитрогруппу, алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, алкильную группу, замещенную аминогруппой, алкильную группу, замещенную алкенильной группой, алкильную группу, замещенную алкинильной группой, карбоксильную группу, алкоксикарбонильную группу, алкилтиогруппу или арилтиогруппу; R44 представляет собой атом водорода, алкильную группу или бензильную группу, которая может содержать заместитель;
(a) взаимодействие производного бензоксазина, представленного следующей формулой (2-4):
 ,
,
в которой заместитель R43' представляет собой диалкиламиногруппу, моноалкиламиногруппу, аминогруппу, которая может иметь защитную группу, атом водорода, атом галогена, алкильную группу, перфторалкильную группу, алкоксигруппу, нитрогруппу, алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, которая может иметь защитную группу, алкильную группу, замещенную аминогруппой, которая может иметь защитную группу, алкильную группу, замещенную алкенильной группой, алкильную группу, замещенную алкинильной группой, карбоксильную группу, алкоксикарбонильную группу, алкилтиогруппу или арилтиогруппу; и заместитель R45 представляет собой алкильную группу или фенильную группу, которая может содержать заместитель,
с производным ацилфенилаланина, представленным следующей формулой (3-4), или его химически приемлемой солью:
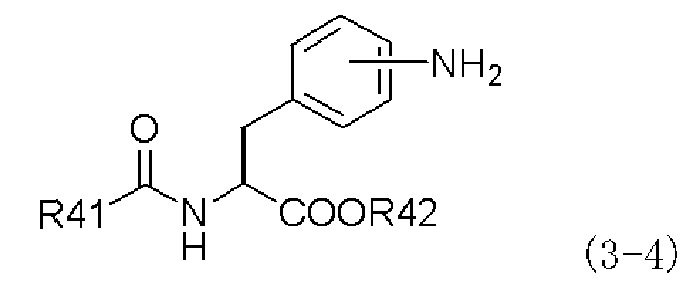 ,
,
в которой заместители R41 и R42 имеют те же самые значения, которые указаны выше;
(b) превращение полученного амидкарбаматного производного, представленного следующей формулой (4-4), или его химически приемлемой соли:
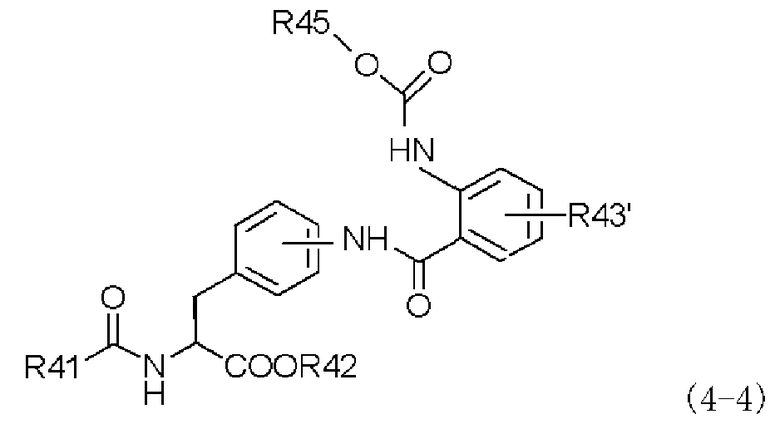 ,
,
в которой заместители R41, R42, R43' и R45 имеют те же самые значения, которые указаны выше;
в хиназолиндионовое производное, представленное следующей формулой (5-4), в присутствии основания:
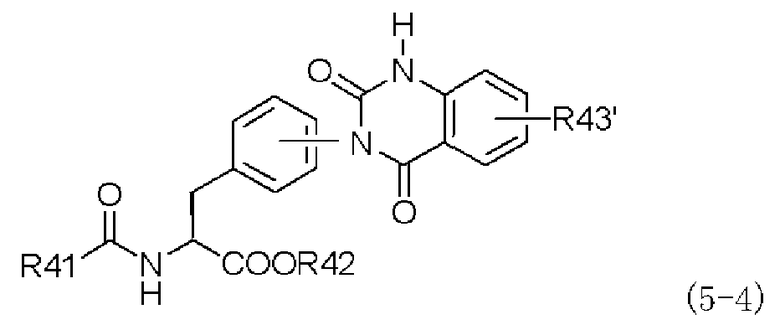 ,
,
в которой заместители R41-R43' имеют те же самые значения, которые указаны выше; и
(c) если требуется, замещение атома водорода, присоединенного к атому азота, присутствующему в хиназолиндионовом цикле хиназолиндионового производного формулы (5-4), N-алкильной группой с помощью N-алкилирующего средства и затем снятие защитных групп в полученном продукте, когда заместитель R43' защищен.
Наилучший способ осуществления изобретения
В настоящем изобретении хиназолиндионовая циклическая группа, представленная в формулах (1), (1-2), (1-3) и (1-4), может быть расположена на бензольном кольце фенилаланина в о-, м- или п-положении, однако циклическая группа в числе прочих предпочтительно находится в п-положении.
В настоящем изобретении заместители фенильной группы, которая может содержать заместитель, и заместители пиридильной группы, которая может содержать заместитель, представленные обозначениями R1, R21, R31 и R41, могут представлять собой, например, атомы галогенов, алкильные группы, галогеналкильные группы (включая перфторалкильные группы), алкоксигруппы, галогеналкоксигруппы (включая перфторалкоксигруппы), алкилтиогруппы, нитрогруппу, алкилсульфониламиногруппы и тетразолильную группу. При таком аспекте алкильные группы в качестве компонентов указанных заместителей предпочтительно представляют собой группы, содержащие от 1 до 6 атомов углерода и особенно предпочтительно - от 1 до 3 атомов углерода. Дополнительно число упомянутых выше заместителей находится в диапазоне от 1 до 5, предпочтительно в диапазоне от 1 до 3, и они могут быть одинаковыми или разными. Предпочтительно применяемые здесь в качестве заместителей R1, R21, R31 и R41 представляют собой фенильные группы, замещенные атомами галогенов, и/или алкильные группы, и конкретные их примеры включают в себя 2,6-дихлорфенильную группу, 2,6-диметилфенильную группу, 2-хлор-6-метилфенильную группу, 2-хлорфенильную группу, 2-метилфенильную группу, 2,4,6-трихлорфенильную группу, 2,4,6-триметилфенильную группу и 2,6-дихлор-4-метилфенильную группу.
В настоящем изобретении алкильные группы упомянутых выше замещенных или незамещенных алкильных групп, представленных обозначениями R2, R22, R32 и R42, предпочтительно представляют собой алкильные группы, содержащие от 1 до 6 атомов углерода, и особенно предпочтительно - алкильные группы, содержащие от 1 до 3 атомов углерода.
В каждой группе R2, R22, R32 и R42, содержащей заместитель, заместители могут представлять собой, например, замещенные или незамещенные низшие алкилкарбонилоксигруппы, замещенные или незамещенные низшие алкоксикарбонилоксигруппы, замещенные или незамещенные аминогруппы, низшие алкоксигруппы, атомы галогенов, замещенные или незамещенные арильные группы, замещенные или незамещенные гетероарильные группы и замещенные или незамещенные карбамоильные группы.
При таком аспекте, когда заместители представляют собой низшие алкилкарбонилоксигруппы, низшие алкоксикарбонилоксигруппы и низшие алкоксигруппы, их алкильные и алкоксигруппы предпочтительно представляют собой группы, содержащие от 1 до 6 атомов углерода, и они включают в себя цепи, подобные циклическим, линейным и разветвленным цепям.
Кроме того, когда заместители представляют собой арильные группы, арильная группа может представлять собой группу от моноциклической до бициклической ароматической углеводородной группы, содержащую от 6 до 10 атомов углерода. Их примеры включают в себя фенильные группы и нафтильные группы. Кроме того, когда заместители представляют собой гетероарильные группы, они могут представлять собой 5-8-членные группы, от моноциклических до трициклических ароматических гетероциклических групп, включающие в себя от 1 до 4 гетероатомов в качестве атомов, образующих цикл, выбранных из атома кислорода, атома серы и атома азота. Их конкретные примеры включают в себя пиридильную группу, пиридазинильную группу, пиримидильную группу, пиразинильную группу, фурильную группу, тиенильную группу, пирролильную группу, изоксазолильную группу, оксазолильную группу, изотиазолильную группу, тиазолильную группу, пиразолильную группу, имидазолильную группу, тетразолильную группу, индолильную группу, бензимидазолильную группу, хинолильную группу и изохинолильную группу. При этом заместителями для арильных и гетероарильных групп могут служить, например, атомы галогенов, алкоксигруппы, алкильные группы, гидроксильная группа, галогеналкильные группы и галогеналкоксигруппы. Среди них предпочтительно применяемыми здесь группами являются пиридильная группа, фурильная группа и тиенильная группа.
Между тем, если заместителями на R2, R22, R32 и R42 являются низшие алкилкарбонилоксигруппы или низшие алкоксикарбонилоксигруппы, их заместителями могут являться, например, низшие алкильные группы, низшие алкенильные группы, низшие алкоксигруппы, гидроксильная группа, аминогруппы и аминогруппы, каждая из которых замещена низшей алкильной группой (включая моно- и дизамещенные группы). Предпочтительно применяемыми в настоящем изобретении являются в числе прочих метильная группа и этильная группа.
Кроме того, если заместителями на R2, R22, R32 и R42 являются аминогруппы, их заместители могут представлять собой, например, низшие алкильные группы, низшие алкоксикарбонильные группы и низшие алкилсульфонильные группы. Среди них предпочтительными является метильная группа и этильная группа. Альтернативно два заместителя могут соединяться с образованием цикла, и такие две группы, когда они образуют цикл, могут перебиваться атомами кислорода, азота и/или серы. Например, замещенная аминогруппа включает в себя кольцеобразную аминогруппу, такую как 1-пиперидинильная группа или 4-морфолинильная группа; кольцеобразную амидную группу, такую как 2-оксо-1-пирролидинильная группа; и кольцеобразную мочевинную группу, такую как 2-оксоимидазолин-1-ильная группа или 2-оксоимидазолидин-1-ильная группа.
Кроме того, если заместителями на R2, R22, R32 и R42 являются арильные или гетероарильные группы, их заместители могут представлять собой, например, атомы галогенов, алкоксигруппы, алкильные группы, гидроксильную группу, галогеналкильные группы и галогеналкоксигруппы.
Дополнительно, если заместителями на R2, R22, R32 и R42 являются карбамоильные группы, их заместители могут представлять собой, например, низшие алкильные группы и фенильную группу, и они включают в себя моно- и дизамещенные группы.
Кроме того, если R2, R22, R32 и R42 содержат заместители, такие предпочтительно применяемые здесь заместители могут представлять собой, например, низшие алкилкарбонилоксигруппы, атом хлора, пиридильную группу, фурильную группу, тиенильную группу и низшие диалкилкарбамоильные группы.
В настоящем изобретении R3 предпочтительно представляет собой моноалкиламиноалкильную группу; R23, R33 и R43 предпочтительно представляют собой диалкиламиногруппы, атом водорода, атомы галогенов, моноалкиламиногруппы, алкильные группы, замещенные диалкиламиногруппами, алкильные группы, замещенные моноалкиламиногруппами, алкильные группы, замещенные алкинильными группами, алкильные группы, замещенные аминогруппами, карбоксильную группу, алкоксикарбонильные группы и алкилтиогруппы, и особенно предпочтительно R23, R33 и R43 представляют собой диалкиламиногруппы, моноалкиламиногруппы, алкильные группы, замещенные диалкиламиногруппами, алкильные группы, замещенные моноалкиламиногруппами, алкильные группы, замещенные аминогруппами, алкильные группы, замещенные алкинильными группами, карбоксильную группу, алкоксикарбонильные группы и алкилтиогруппы.
Кроме того, в настоящем изобретении R3', R23', R33' и R43' аналогичны группам, представленным соответственно обозначениями R3, R23, R33 и R43, или представляют собой группы, способные подвергаться превращению в аналогичные группы, представленные последними обозначениями, во время процессов производства соответственно. Защитные группы, присутствующие в R3', R23', R33' и R43', предпочтительно представляют собой группы, применяемые в настоящее время для защиты аминогрупп (Protective Groups in Organic Syntheses (3-е издание), T.W. Green, P.G.M. Wootz, JHON WILEY & SONS, INC., 1999), формильные группы, алкилкарбонильные группы и алкоксикарбонильные группы, в которых алкильные группы и алкоксигруппы предпочтительно представляют собой группы, содержащие от 1 до 3 атомов углерода. Предпочтительно применяемые здесь группы включают в себя группы, устойчивые к основным условиям и способные легко удаляться в кислых условиях, и более конкретно, предпочтительно применяемыми здесь группами являются формильные группы и трет-бутоксикарбонильная группа.
Диалкиламиногруппа означает здесь аминогруппу, замещенную двумя алкильными группами, каждая из которых содержит от 1 до 6 атомов углерода (включая циклические аминогруппы), предпочтительно аминогруппу или циклическую аминогруппу, содержащую от 2 до 6 атомов углерода, которая замещена двумя алкильными группами, каждая из которых содержит от 1 до 3 атомов углерода, и их конкретными примерами являются диметиламиногруппа, диэтиламиногруппа, метилэтиламиногруппа, пирролидинильная группа, пиперидинильная группа, дипропиламиногруппа, метилпропиламиногруппа и этилпропиламиногруппа.
Моноалкиламиногруппа означает здесь аминогруппу, замещенную одной алкильной группой, содержащей от 1 до 6 атомов углерода (включая аминогруппы, содержащие циклическую алкильную группу), предпочтительно аминогруппу, замещенную одной алкильной группой, содержащей от 1 до 4 атомов углерода, и их конкретные примеры включают в себя метиламиногруппу, этиламиногруппу, пропиламиногруппу, изопропиламиногруппу, бутиламиногруппу и циклопропилметиламиногруппу.
Применяемая здесь алкильная группа, замещенная диалкиламиногруппой, означает алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную диалкиламиногруппой, аналогичной описанной выше, предпочтительно означает алкильную группу, содержащую от 1 до 3 атомов углерода и замещенную диалкиламиногруппой, аналогичной описанной выше, и ее конкретными примерами являются метильная, этильная и пропильная группы, каждая из которых замещена диметиламиногруппой, диэтиламиногруппой, метилэтиламиногруппой, пирролидинильной группой, пиперидинильной группой, дипропиламиногруппой, метилпропиламиногруппой или этилпропиламиногруппой. Особенно предпочтительно применяемыми здесь являются диметиламинометильная группа, диэтиламинометильная группа и метилэтиламинометильная группа.
Применяемая здесь алкильная группа, замещенная моноалкиламиногруппой, означает алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную моноалкиламиногруппой, аналогичной описанной выше, предпочтительно означает алкильную группу, содержащую от 1 до 3 атомов углерода и замещенную моноалкиламиногруппой, аналогичной описанной выше, и ее конкретными примерами являются метильная, этильная и пропильная группы, каждая из которых замещена метиламиногруппой, этиламиногруппой, пропиламиногруппой, изопропиламиногруппой, бутиламиногруппой или циклопропилметиламиногруппой. Особенно предпочтительно применяемыми здесь являются метиламинометильная группа, этиламинометильная группа, метиламиноэтильная группа и этиламиноэтильная группа.
Применяемая здесь алкильная группа, замещенная аминогруппой, означает алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную аминогруппой, предпочтительно означает алкильную группу, содержащую от 1 до 3 атомов углерода и замещенную аминогруппой, и ее конкретными примерами являются аминометильная группа, аминоэтильная группа и аминопропильная группа.
Применяемая здесь алкильная группа, замещенная алкенильной группой, означает алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную алкенильной группой, содержащей от 2 до 6 атомов углерода, предпочтительно означает алкильную группу, содержащую от 1 до 3 атомов углерода и замещенную алкенильной группой, содержащей от 2 до 4 атомов углерода, и ее конкретными примерами являются -CH2CH=CH2 и -CH2CH2CH=CH2.
Применяемая здесь алкильная группа, замещенная алкинильной группой, означает алкильную группу, содержащую от 1 до 6 атомов углерода и замещенную алкинильной группой, содержащей от 2 до 6 атомов углерода, предпочтительно означает алкильную группу, содержащую от 1 до 3 атомов углерода и замещенную алкинильной группой, содержащей от 2 до 4 атомов углерода, и ее конкретными примерами являются -CH2C≡CH и -CH2CH2C≡CH.
Применяемая здесь алкоксикарбонильная группа означает алкоксикарбонильную группу, содержащую от 2 до 7 атомов углерода, предпочтительно означает алкоксикарбонильную группу, содержащую от 2 до 4 атомов углерода, и ее конкретные примеры включают в себя метоксикарбонильную группу, этоксикарбонильную группу и пропилоксикарбонильную группу.
Применяемая здесь алкилтиогруппа означает тиогруппу, замещенную алкильной группой, содержащей от 1 до 6 атомов углерода, предпочтительно означает тиогруппу, замещенную алкильной группой, содержащей от 1 до 3 атомов углерода, и ее конкретные примеры включают в себя метилтиогруппу, этилтиогруппу и пропилтиогруппу.
Применяемая здесь арилтиогруппа включает в себя, например, фенилтио- и нафтилтиогруппы.
Более конкретно, R3 предпочтительно представляет собой метиламинометильную и этиламинометильную группы; R23, R33 и R43 в числе прочих предпочтительно представляют собой диметиламино-, диэтиламино-, метилэтиламино-, пирролидинильную, пиперидинильную, метиламино-, этиламино-, пропиламино-, циклопропилметиламино-, диметиламинометильную, диэтиламинометильную, диметиламиноэтильную, диэтиламиноэтильную, метиламинометильную, этиламинометильную, пропиламинометильную, метиламиноэтильную, этиламиноэтильную, пропиламиноэтильную, HC≡CCH2, карбоксильную, метоксикарбонильную, этоксикарбонильную, метилтио- и этилтиогруппы.
Кроме того, в настоящем изобретении R3, R23, R33, R43, R3', R23', R33' и R43' предпочтительно расположены в п-положении относительно атома азота, соответствующего аминогруппе антраниловой кислоты.
В настоящем изобретении каждый из R4, R24, R34 и R44 предпочтительно представляет собой атом водорода или алкильную группу. При таком аспекте алкильная группа предпочтительно представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода. Дополнительно заместителями бензильной группы могут, например, являться алкильные группы, алкоксигруппы и атомы галогенов, однако предпочтительно бензильная группа не содержит никакого заместителя.
Заместитель R25 в числе прочих предпочтительно представляет собой алкильную группу. В данном контексте алкильная группа предпочтительно представляет собой алкильную группу, содержащую от 1 до 3 атомов углерода.
Заместитель R45 может представлять собой, например, алкильную группу и фенильную группу, которая может содержать заместитель, однако предпочтительно применяемые здесь группы в числе прочих включают в себя алкильные группы, каждая из которых содержит от 1 до 3 атомов углерода. При таком аспекте заместители, присутствующие на фенильной группе, могут представлять собой, например, алкильные группы, алкоксигруппы и атомы галогенов, однако предпочтительно фенильная группа не содержит никакого заместителя.
Согласно настоящему изобретению в первом способе получения в качестве промежуточного соединения, применяемого при получении производного фенилаланина с хиназолиндионовым скелетом, предлагается соединение, представленное следующей формулой (3-1):
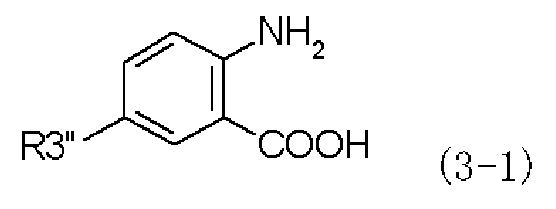 ,
,
в которой R3'' представляет собой член, выбранный из группы, состоящей из диалкиламиногрупп, алкильных групп, каждая из которых замещена моноалкиламиногруппой, N-алкил-N-формиламиноалкильных групп, N-алкил-N-алкилкарбониламиноалкильных групп и N-алкил-N-алкоксикарбониламиноалкильных групп.
Кроме того, в первом способе получения в качестве промежуточных соединений, применяемых при получении производных фенилаланина, каждое из которых имеет хиназолиндионовый скелет, предлагаются соединения, представленные следующими формулами, или их химически приемлемые соли:
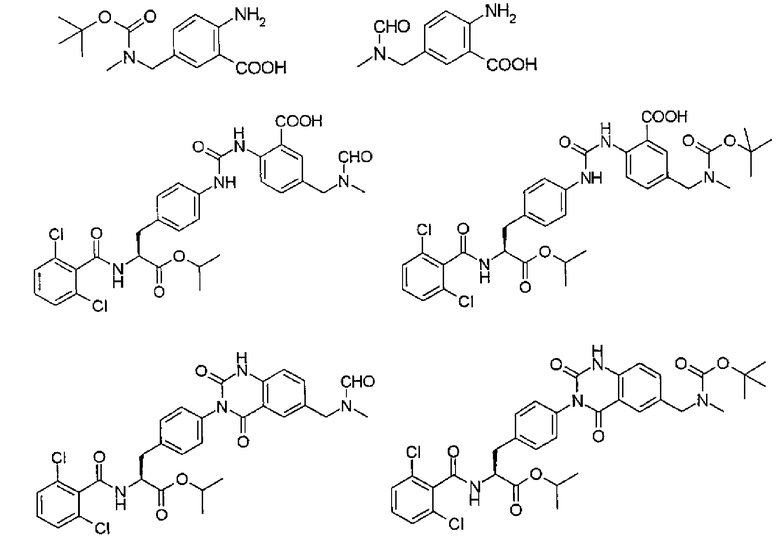
 .
.
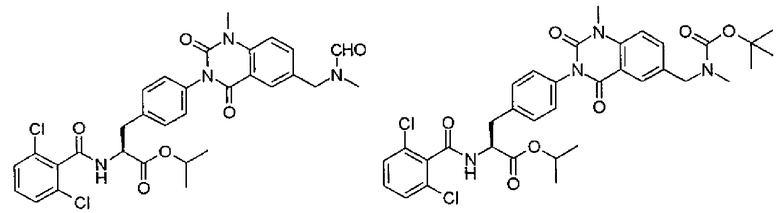
Кроме того, согласно настоящему изобретению во втором-четвертом способах получения в качестве промежуточных соединений, применяемых при получении производных фенилаланина, каждое из которых имеет хиназолиндионовый скелет, предлагаются соединения, представленные группой следующих формул, или их химически приемлемые соли:
 .
.
Далее здесь будет более подробно описан первый способ получения согласно настоящему изобретению.
Первый способ получения согласно настоящему изобретению выполнен на основе обнаружения следующего факта: если в качестве исходного материала для получения производного антраниловой кислоты, представленного формулой (3), или его химически приемлемой соли, в которой R3' представляет собой алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, которая может иметь защитную группу, или алкильную группу, замещенную аминогруппой, которая может иметь защитную группу, применяют соединение с карбоксильной группой (COOH-группу), не участвующей в образовании какой-либо сложноэфирной связи, выход реакции можно значительно повысить по сравнению с реакцией, проводимой с применением в качестве исходного материала соединения, у которого концевая карбоксильная группа образует сложноэфирную связь.
Согласно первому способу получения на стадии (a) производное ацилфенилаланина, представленное формулой (2), или его химически приемлемую соль сначала подвергают взаимодействию с реагентом, вводящим карбонильную группу, и производным антраниловой кислоты, представленным формулой (3), или его химически приемлемой солью. При таком аспекте производное ацилфенилаланина, представленное формулой (2), или его химически приемлемую соль можно получать, например, согласно способу, описанному в патентном документе 2 или 3, в то время как производное антраниловой кислоты, представленное формулой (3), или его химически приемлемую соль можно получать аналогичным образом, например, гидролизацией сложноэфирного производного, описанного в патентном документе 2.
В данном контексте термин «реагент, вводящий карбонильную группу» означает реагент, из которого в атомную группировку хиназолиндионового цикла извлекается только карбонильная группа. Примеры таких реагентов включают в себя 1,1'-карбонилдиимидазол (пример его синтеза описан в публикации «Organic Syntheses Collective» т. V, стр. 201-204, Wiley, New York, 1973) и сложный эфир хлормуравьиной кислоты. Указанные соединения не только известны, но и предлагаются на рынке и соответственно коммерчески доступны.
Кроме того, также можно применять реагенты, получаемые замещением имидазолильной группы 1,1'-карбонилдиимидазола другой гетероарильной удаляемой группой, такие как 1,1'-карбонил-ди-(1,2,4-триазол) (в данном реагенте имидазолильная группа замещена триазоилом, и реагент может быть коммерчески доступен). Заместители не ограничиваются имидазолильной и триазоильной группами, и можно применять любую гетероарильную удаляемую группу, отличающуюся от указанных выше групп.
Кроме того, реагенты, подходящие для применения в настоящем изобретении, включают N,N'-дисукцинимидилкарбонат (ДСК) (данный реагент является реагентом, вводящим карбонильную группу, в котором N-гидроксисукцинимидная группа выполняет функцию удаляемой группы, и который может быть коммерчески доступен).
Примеры таких сложных эфиров хлормуравьиной кислоты включают, но не ограничиваются перечисленным, сложные эфиры, содержащие от 2 до 10 атомов углерода, такие как фенилхлорформиат, нитрофенилхлорформиат, метоксифенилхлорформиат, метилхлорформиат, этилхлорформиат, изобутилхлорформиат, октилхлорформиат и бензилхлорформиат.
Кроме того, соединения, подходящие здесь для применения в качестве реагентов, вводящих карбонильную группу, также включают, например, фосген и аналоги фосгена (такие как трифосген).
Особенно предпочтительным для применения здесь реагентом, вводящим карбонильную группу, является 1,1'-карбонилдиимидазол. В частности, когда применяется 1,1'-карбонилдиимидазол, во время реакции образуется только небольшое количество побочных продуктов, и его применение отличается тем, что в результате реакции можно получить заданное асимметричное производное мочевины с высоким выходом.
Между тем реагент, вводящий карбонильную группу, предпочтительно применяется в количестве, находящемся в диапазоне от 0,8 до 1,5 мольных эквивалентов на один моль соединения формулы (2).
Концентрация реагента в упомянутой выше реакции является подходящей концентрацией для применения в промышленном процессе и находится, например, в диапазоне от 0,1 до 10M и желательно - около 1,3M, когда в качестве реакционного растворителя применяют DMF, в то же время принимая во внимание реологические свойства реакционного раствора и то, что при перемешивании раствора при перекристаллизации сталкиваются с трудностями.
Реакцию соединения формулы (2) с реагентом, вводящим карбонильную группу (таким как 1,1'-карбонилдиимидазол), с образованием имидазокарбонильного производного проводят при температуре, предпочтительно находящейся в диапазоне от приблизительно -40°С до точки кипения применяемого реакционного растворителя, и с промышленной точки зрения более предпочтительно температура реакции находится в диапазоне приблизительно от 0°С до 20°С. Кроме того, при образовании алкоксикарбонильного производного с помощью сложного эфира хлормуравьиной кислоты реакцию предпочтительно проводят при температуре в диапазоне от приблизительно -40°С до точки кипения применяемого реакционного растворителя, а с промышленной точки зрения более предпочтительно реакцию проводят при температуре в диапазоне приблизительно от 0°С до 40°С. В таком случае количество применяемого реагента предпочтительно находится в диапазоне от 1,0 до 1,1 эквивалентов.
Кроме того, соединение формулы (3) предпочтительно применяют в количестве, находящемся в диапазоне от 0,8 до 1,2 мольных эквивалентов на один моль соединения формулы (2).
Дополнительно, когда в качестве реагента, вводящего карбонильную группу, применяют сложный эфир хлормуравьиной кислоты, реакцию предпочтительно проводят в присутствии органического основания. Примерами таких органических оснований, предпочтительно применяемых здесь, являются триэтиламин, диизопропилэтиламин и пиридин.
Реакционные растворители, применяемые в данной реакции, представляют собой, например, органические растворители, которые обладают соответствующей способностью растворять соединение формулы (2) (например, сложный метиловый эфир Nα-(2,6-дихлорбензоил)-4-амино-L-фенилаланина), и их примеры, подходящие для применения здесь, включают в себя N,N-диметилформамид (DMF), диметоксиэтан (DME), диметилсульфоксид (DMSO), диметилацетамид (DMA), ацетонитрил, тетрагидрофуран (ТГФ) или смеси таких растворителей. Особенно предпочтительным является применение N,N-диметилформамида. Время реакции предпочтительно составляет приблизительно от 1 до 5 часов.
Последующую реакцию конденсации соединения формулы (2), превращенного в имидазокарбонильное производное или алкоксикарбонильное производное, с замещенной 2-аминобензойной кислотой формулы (3) проводят при температуре в предпочтительном диапазоне от 0°С до точки кипения применяемого реакционного растворителя. В частности, реакцию предпочтительно проводят при температуре около 50°С, учитывая, что реакцию образования мочевины можно завершить в течение времени реакции приблизительно от 2 до 3 часов, получая при этом производное мочевины с асимметрично расположенными карбоксильными группами формулы (4) с высоким выходом.
Однако при таком аспекте температура реакции и время проведения реакции не ограничиваются указанными выше конкретными диапазонами, время проведения реакции определяется в зависимости от температуры реакции, поэтому с промышленной точки зрения предпочтительно регулировать или контролировать реакционный раствор аналитическими средствами, такими как метод ВЭЖХ.
В упомянутой выше реакции порядок внесения исходного материала и реагента не ограничивается каким-либо конкретным порядком, однако предпочтительно, чтобы сначала соединение формулы (2) подвергалось взаимодействию с реагентом, вводящим карбонильную группу, для превращения первого в его имидазокарбонильное производное или алкоксикарбонильное производное, чтобы затем последнее подвергалось взаимодействию с соединением формулы (3), поскольку такой способ обеспечивает достижение более высокого выхода заданного продукта и уменьшение количества побочных продуктов по сравнению со способом, при котором соединение формулы (3) сначала превращается в свое имидазокарбонильное производное или свое алкоксикарбонильное производное. Однако в способе получения согласно настоящему изобретению соединение формулы (3) может сначала превращаться в свое имидазокарбонильное производное, или соединение формулы (2), реагент, вводящий карбонильную группу и соединение формулы (3) могут подвергаться взаимодействию одновременно.
На следующей стадии (b) полученное производное мочевины формулы (4) с асимметрично расположенными карбоксигруппами обрабатывают средством, активирующим карбоксильную группу, в подходящем реакционном растворителе с образованием при этом хиназолиндионового цикла и хиназолиндионового производного формулы (5).
Применяемый здесь термин «средство, активирующее карбоксильную группу» означает средство для активации карбоксильной группы, полученной из антраниловой кислоты, до такой степени или более высокой степени, что карбоксильная группа может взаимодействовать с внутримолекулярным атомом азота мочевины с образованием при этом хиназолиндионового цикла. Более конкретно примеры таких «средств, активирующих карбоксильную группу» включают в себя перечисленные выше реагенты с предпочтительно применяемым здесь 1,1'-карбонилдиимидазолом.
Дополнительно упомянутую выше стадию (b) можно аналогичным образом проводить сразу после выделения производного мочевины с асимметрично расположенными карбоксигруппами, представленного формулой (4), из реакционного раствора с помощью традиционного метода выделения, такого как кристаллизация, однако с промышленной точки зрения предпочтительно непрерывное проведение указанных стадий (или стадию (b) проводят без выделения промежуточного соединения формулы (4)).
После завершения упомянутой выше реакции образования хиназолиндионового цикла в реакционную систему можно добавлять спиртовой растворитель, чтобы разложить избыток 1,1'-карбонилдиимидазола (CDI) или сложного эфира хлормуравьиной кислоты или т.п. Фактически в качестве добавляемого спиртового растворителя предпочтительно применяют метанол и изопропиловый спирт. Кроме того, в случае продукта, содержащего в молекуле сложноэфирный фрагмент, добавление спиртового растворителя может вызвать протекание реакции транс-этерификации, и, следовательно, спиртовой растворитель предпочтительно должен выбираться с учетом характера сложного эфира.
Затем на стадии (c) атом водорода, связанный с атомом азота, присутствующим в хиназолиндионовом цикле хиназолиндионового производного формулы (5), если требуется, замещают N-алкильной группой с помощью N-алкилирующего средства и удаляют защитную группу, когда заместитель R3' защищен.
Другими словами, хиназолиндионовое производное, в котором R4 в формуле (1) представляет собой алкильную группу или замещенную или незамещенную бензильную группу, можно получать путем воздействия N-алкилирующего средства на хиназолиндионовое производное формулы (5) в присутствии основания.
В данном описании термин «N-алкилирующее средство» означает реагент, с помощью которого можно вводить в соединение алкильную группу на его атом азота, и примеры таких средств включают в себя галогеналкан, алкилсульфонат, бензилгалогенид, который может содержать заместитель.
При таком аспекте такими галогеналканами и алкилсульфонатами предпочтительно являются те, которые содержат от 1 до 10 атомов углерода. Дополнительно более предпочтительными являются те, которые содержат от 1 до 6 атомов углерода, и, в частности, предпочтительными являются те, которые содержат от 1 до 3 атомов углерода. Примерами галогеналканов являются метилйодид и этилйодид; а примерами алкилсульфонатов являются метилметансульфонат, этилметансульфонат, метилэтансульфонат, этилэтансульфонат, метил-п-толуолсульфонат и этил-п-толуолсульфонат. Кроме того, примерами бензилгалогенидов являются бензилхлорид и бензилбромид, и их заместителями могут быть, например, алкильные группы, алкоксигруппы и атомы галогенов. Более предпочтительно N-алкилирующее средство представляет собой член, выбранный из группы, состоящей из метил-п-толуолсульфоната, метилметансульфоната, метилйодида, метилбромида и метилхлорида.
Например, с промышленной точки зрения при получении соединения формулы (1), в котором R4 представляет собой метильную группу, в качестве N-алкилирующего средства подходящим образом применяется метил-п-толуолсульфонат. Другими словами, метил-п-толуолсульфонат по сравнению с летучим метилйодидом имеет высокую точку кипения и соответственно с ним легко обращаться при комнатной температуре. Кроме того, по сравнению с метилметансульфонатом метил-п-толуолсульфонат является предпочтительным с точки зрения реологических свойств полученного реакционного раствора и соответственно является подходящим для промышленных способов, которые связаны с операциями переноса жидкости.
Достаточно, чтобы реакционный растворитель, применяемый на данной стадии, мог растворять соединение, представленное формулой (5), и чтобы он представлял собой органический растворитель, который является стабильным во время реакции. Примеры таких растворителей включают N,N-диметилформамид и системы смешанных растворителей, содержащие N,N-диметилформамид и спирты, с предпочтительно применяемым здесь N,N-диметилформамидом.
Подходящее количество применяемого N-алкилирующего средства находится в диапазоне от 1 до 2 мольных эквивалентов и предпочтительно от 1,0 до 1,2 мольных эквивалентов, исходя из соединения формулы (4) или формулы (5), однако количество добавляемого реагента может увеличиваться или уменьшаться в зависимости от протекания реакции.
Примеры упомянутых выше оснований включают неорганические основания и органические основания. В данном контексте такими неорганическими основаниями могут являться, например, соли щелочных металлов (такие как карбонат калия, карбонат натрия, карбонат цезия, метилат натрия и этилат натрия) и соли щелочноземельных металлов (такие как карбонат кальция и карбонат магния). Кроме того, примеры органических оснований включают триэтиламин, этаноламин, морфолин, пиперидин, дициклогексиламин, 1,8-диазабицикло[5.4.0]ундека-7-ен (DBU) и N,N-диизопропил-N-этиламин (DIPEA). Предпочтительно основание является неорганическим основанием, с предпочтительно применяемым здесь в числе прочих карбонатом калия.
Количество применяемого основания предпочтительно находится в диапазоне от 1 до 2 мольных эквивалентов и более предпочтительно составляет приблизительно 1,5 мольных эквивалента, однако его количество не ограничивается таким конкретным диапазоном и может увеличиваться или уменьшаться в зависимости от протекания реакции.
Кроме того, если заместитель R3' защищен, удаление его защиты можно легко проводить согласно традиционному способу, такому как способ, при котором применяются кислые условия, или метод каталитического восстановления.
Если защитная группа представляет собой формильную группу, трет-бутоксикарбонильную группу или ацильную группу (такую как ацетильная или бензоильная группа), такую защитную группу можно удалять в кислых условиях. Формильную группу и трет-бутоксикарбонильную группу можно непосредственно удалять в кислых условиях, и соответственно они предпочтительно применяются в качестве защитных групп. Примеры таких кислот, подходящих для применения здесь, включают хлористоводородную кислоту, хлористый водород, серную кислоту, метансульфоновую кислоту, п-толуолсульфоновую кислоту, бромистоводородную кислоту и бромистый водород. В зависимости от соли соединения, получаемой в конечном счете, можно выбрать любую подходящую кислоту. Например, если солью заданного конечного соединения является гидрохлорид, в качестве такой кислоты, предпочтительной для применения, является хлористоводородная кислота или хлористый водород. Реакционные растворители, подходящие для применения здесь, включают, например, диметилформамид, изопропилацетат, метилацетат, изопропиловый спирт и метанол, однако на сложноэфирном фрагменте исходного лекарственного средства может быть вызвана реакция транс-этерификации, и, следовательно, предпочтительно применяют соответствующий спиртовой или алкилацетатный растворитель.
Кроме того, в случае таких групп, как бензилоксигруппа, бензильная группа и бензилоксикарбонильная группа, снятие защиты можно проводить, например, согласно методу каталитического восстановления.
Полученное производное фенилаланина, представленное формулой (1), или его фармацевтически приемлемую соль можно выделять с помощью традиционного метода выделения.
В первом способе получения заместитель R3, представленный в формуле (1), предпочтительно представляет собой метиламинометильную группу, этиламинометильную группу, диметиламинометильную группу или диэтиламинометильную группу, а R3', представленный в формулах (3)-(5), предпочтительно представляет собой метильную группу, замещенную метиламиногруппой, несущей защитную группу, метильную группу, замещенную этиламиногруппой, несущей защитную группу, диметиламинометильную группу или диэтиламинометильную группу.
Кроме того, также предпочтительно, чтобы заместитель R1, представленный в формулах (1)-(5), представлял собой 2,6-дихлорфенильную группу, 2-хлор-6-метилфенильную группу, 2-хлор-6-фторфенильную группу, 2,6-дифторфенильную группу или 2-фтор-6-метилфенильную группу; R4 представляет собой метильную группу или этильную группу; и заместитель на бензольном кольце фенилаланина расположен в п-положении.
Кроме того, предпочтительно, чтобы соединение формулы (2), в котором R1 в формуле (2) представляет собой 2,6-дихлорфенильную группу, и R2 представляет собой изопропильную группу, подвергалось взаимодействию с 1,1'-карбонилдиимидазолом в качестве агента, вводящего карбонильную группу, и соединением формулы (3), в котором R3' представляет собой N-формил-N-метиламинометильную группу, образуя при этом 2-(3-{4-[2(S)-2- (2,6-дихлорбензоиламино)-2-изопропоксикарбонилэтил]фенил}уреидо)-5-(N-формил-N-метиламинометил)бензойную кислоту формулы (4), в которой R1 представляет собой 2,6-дихлорфенильную группу, R2 представляет собой изопропильную группу, и R3' представляет собой N-формил-N-метиламинометильную группу; затем полученное соединение подвергают взаимодействию с 1,1'-карбонилдиимидазолом в качестве средства, активирующего карбоксильную группу, превращая при этом первый в сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (5), в которой R1 представляет собой 2,6-дихлорфенильную группу, R2 представляет собой изопропильную группу, и R3' представляет собой N-формил-N-метиламинометильную группу; затем с помощью метил-п-толуолсульфоната проводят N-алкилирование полученного продукта, превращая при этом продукт в сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина; и, в конечном счете, N-алкилированный продукт обрабатывают хлористым водородом для удаления его защитной формильной группы, получая при этом сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (1) или его гидрохлорид, в которых R1 представляет собой 2,6-дихлорфенильную группу, R2 представляет собой изопропильную группу, R3' представляет собой N-метиламинометильную группу, и R4 представляет собой метильную группу.
При таком аспекте упомянутое выше N-алкилирование с помощью метил-п-толуолсульфоната предпочтительно проводят в оснόвных условиях.
Далее будет подробно описан второй способ получения по настоящему изобретению.
Второй способ получения производного фенилаланина с хиназолиндионовым циклом, представленного формулой (1-2), или его фармацевтически приемлемой соли по настоящему изобретению осуществлен на основе обнаружения того факта, что асимметричное производное мочевины можно легко и просто получать без применения какого-либо реагента, вводящего карбонильную группу, путем превращения в изоцианат производного ацилфенилаланина, в котором карбоксильная группа связана с его фенильной группой, представленного формулой (2-2), или его химически приемлемой соли в качестве исходного материала вместо производного ацилфенилаланина, содержащего аминогруппу, связанную с его фенильной группой, с последующим превращением карбоксильной группы в изоцианильную группу и взаимодействием полученного изоцианильного производного с производным антраниловой кислоты, содержащим сложноэфирную группу алкилового эфира и представленного формулой (4-2), или его химически приемлемой солью.
Во втором способе получения производное ацилфенилаланина, представленное формулой (2-2), или его химически приемлемую соль сначала на стадии (a) подвергают реакции превращения в изоцианат для превращения карбоксильной группы в его изоцианильную группу с образованием при этом соединения формулы (3-2) или его химически приемлемой соли; затем полученное соединение формулы (3-2) или его химически приемлемую соль на стадии (b) подвергают взаимодействию с производным антраниловой кислоты формулы (4-2) или с его химически приемлемой солью, получая при этом асимметричное производное мочевины формулы (5-2) или его химически приемлемую соль; дополнительно на стадии (c) полученное асимметричное производное мочевины формулы (5-2) или его химически приемлемую соль обрабатывают средством, активирующим карбоксильную группу, когда R25 представляет собой атом водорода, или основанием, когда R25 представляет собой алкильную группу, получая при этом хиназолиндионовое производное формулы (6-2) или его химически приемлемую соль; затем на стадии (d) атом водорода, связанный с атомом азота в хиназолиндионовом цикле хиназолиндионового производного формулы (6-2), если требуется, замещают N-алкильной группой с помощью N-алкилирующего реагента; и в том случае, когда заместитель R23' защищен, с него удаляют защиту.
В таком контексте упомянутое выше N-алкилирование с помощью N-алкилирующего реагента предпочтительно проводят в оснόвных условиях.
При таком аспекте производное ацилфенилаланина, представленное формулой (2-2), или его химически приемлемую соль легко можно синтезировать, например, при взаимодействии галогенированного фенилаланина (предпочтительно 4-йод-L-фенилаланина), например, с 2,6-дихлорбензоилхлоридом в щелочных условиях с образованием при этом Nα-(2,6-дихлорбензоил)галогенированного фенилаланина, с последующим превращением в сложный алкиловый эфир традиционным способом и затем превращением галогена в галогенированном фенилаланине в карбоксильную группу традиционным способом. Затем полученное соединение можно подвергать взаимодействию со средством для превращения в изоцианат, таким как азидное соединение (предпочтительно дифенилфосфорилазид (DPPA)) для превращения карбоксильной группы соединения формулы (2-2) в изоцианильную группу, получая при этом соединение формулы (3-2) или его химически приемлемую соль. На данной стадии предпочтительно, чтобы средство для превращения в изоцианат применялось в количестве от 1 до 2 моль на один моль производного ацилфенилаланина, представленного формулой (2-2), или его химически приемлемой соли, и последнее соединение подвергают реакции превращения в изоцианат в органическом растворителе, таком как 1,2-диметоксиэтан в присутствии амина, такого как триэтиламин, при температуре в диапазоне от 70 до 95°С в течение приблизительно от одного до 5 часов, превращая при этом карбоксильную группу соединения формулы (2-2) в изоцианильную группу. Кроме того, вместо упомянутого способа также можно внедрить такой способ, при котором карбоксильную группу, присутствующую в соединении формулы (2-2), превращают в хлорангидрид и затем хлорангидрид подвергают взаимодействию с азидом натрия, осуществляя тем самым требуемую реакцию превращения в изоцианат.
С другой стороны, соединение, применяемое здесь в качестве производного антраниловой кислоты, представленное формулой (4-2), или его химически приемлемая соль включают в себя, например, сложноэфирные производные, такие как производные, описанные в патентном документе 2.
В настоящем изобретении соединение формулы (3-2) или его химически приемлемую соль затем подвергают взаимодействию с производным антраниловой кислоты, представленным формулой (4-2), или его химически приемлемой солью. На данной стадии соединение формулы (4-2) желательно применять в количестве, находящимся в диапазоне от 0,8 до 1,2 мольного эквивалента на один моль соединения формулы (3-2).
Указанную реакцию предпочтительно проводят в органическом растворителе, обладающем соответствующей способностью растворять соединение формулы (3-2), таком как N,N-диметилформамид (DMF), диметоксиэтан (DME), диметилсульфоксид (DMSO), диметилацетамид (DMA), ацетонитрил, тетрагидрофуран (ТГФ) или смесь из указанных растворителей. В качестве такого органического растворителя особенно предпочтительным является диметоксиэтан. Упомянутую реакцию предпочтительно проводят в присутствии органического основания, такого как триэтиламин, диизопропилэтиламин или пиридин, или азидного соединения (предпочтительно дифенилфосфорилазида (DPPA)). Реакцию предпочтительно проводят в течение времени реакции в диапазоне приблизительно от 1 до 5 часов при температуре реакции в диапазоне от 70 до 95°С.
В настоящем изобретении стадию (b) можно проводить сразу после выделения соединения формулы (3-2) из реакционного раствора согласно принятому в настоящее время методу выделения, такому как кристаллизация, однако с промышленной точки зрения предпочтительно непрерывное проведение указанных стадий (или стадию (b) проводят без выделения промежуточного соединения формулы (3-2)).
Затем на стадии (c) полученное асимметричное производное мочевины формулы (5-2) или его химически приемлемую соль обрабатывают в присутствии средства, активирующего карбоксильную группу, в случае, когда R25 представляет собой атом водорода, или в присутствии основания, в случае, когда R25 представляет собой алкильную группу, с образованием хиназолиндионового цикла, получая при этом хиназолиндионовое производное формулы (6-2) или его химически приемлемую соль. При таком аспекте средство, активирующее карбоксильную группу, и основание, подходящее для применения на упомянутой реакционной стадии, могут быть аналогичны средствам, перечисленным выше в связи с первым способом получения. Среди них средства, предпочтительно применяемые в качестве средств, активирующих карбоксильную группу, включают, например, 1,1'-карбонилдиимидазол, сложные эфиры хлормуравьиной кислоты, хлорангидриды алкилсульфоновых кислот, N,N'-дициклогексилкарбодиимид (DCC) и 1-этил-3-(3-диметиламинопропил)карбодиимид. Кроме того, соединения, предпочтительно применяемые здесь в качестве оснований, включают, например, карбонат калия и метилат натрия. Количества средства, активирующего карбоксильную группу, и основания, а также условия для их применения, описанные выше в связи с первым способом получения, можно аналогичным образом внедрить в упомянутом втором способе получения.
В настоящем изобретении стадию (c) можно проводить сразу после выделения соединения формулы (5-2) из реакционного раствора согласно принятому в настоящее время методу выделения, такому как кристаллизация, однако с промышленной точки зрения предпочтительно непрерывное проведение упомянутых стадий (или стадию (c) проводят без выделения промежуточного соединения формулы (5-2)).
После упомянутой выше реакции образования хиназолиндионового цикла в реакционную систему можно добавлять спиртовой растворитель, чтобы разложить присутствующий в ней избыток средства, активирующего карбоксильную группу. Примеры предпочтительно добавляемого спиртового растворителя включают, например, метанол или изопропиловый спирт. Кроме того, в случае продукта, содержащего в молекуле сложноэфирный фрагмент, добавление спиртового растворителя может вызвать протекание реакции транс-этерификации, и, следовательно, спиртовой растворитель предпочтительно следует выбирать с учетом характера сложного эфира.
Затем на стадии (d) атом водорода, связанный с атомом азота, присутствующим в хиназолиндионовом цикле хиназолиндионового производного формулы (6-2), если требуется, можно замещать N-алкильной группой с помощью N-алкилирующего средства, и когда заместитель R23' защищен, можно удалять защитную группу. Указанные стадии можно проводить согласно аналогичным процедурам, применяемым на стадии (c) первого способа получения. На данной стадии средства, предпочтительно применяемые здесь в качестве N-алкилирующего средства, включают метил-п-толуолсульфонат, метилметансульфонат, метилйодид, метилбромид и метилхлорид.
При этом N-алкильное замещение с помощью N-алкилирующего средства предпочтительно проводят в оснόвных условиях.
Во втором способе получения заместитель R23, представленный в формуле (1-2), предпочтительно представляет собой либо диметиламиногруппу, либо метиламинометильную группу; и заместитель R23', представленный в формулах (4-2)-(6-2), предпочтительно представляет собой одну из следующих групп: N-формил-N-метиламинометильную группу, N-(трет-бутоксикарбонил)-N-метиламинометильную группу, N-ацетил-N-метиламинометильную группу и диметиламиногруппу.
Кроме того, во втором способе получения предпочтительно, чтобы 2-(3-{4-[2(S)-2-(2,6-дихлорбензоиламино)-2-изопропоксикарбонилэтил]фенил}уреидо)-5-(N-формил-N-метиламинометил)бензойную кислоту формулы (5-2), в которой заместитель R21 представляет собой 2,6-дихлорфенильную группу, R22 представляет собой изопропильную группу, R23' представляет собой N-формил-N-метиламинометильную группу, и R25 представляет собой атом водорода, получали взаимодействием соединения формулы (3-2), в которой R21 представляет собой 2,6-дихлорфенильную группу, и R22 представляет собой изопропильную группу, с соединением формулы (4-2), в которой R23' представляет собой N-формил-N-метиламинометильную группу, и R25 представляет собой атом водорода; полученную бензойную кислоту подвергают взаимодействию с 1,1'-карбонилдиимидазолом в качестве средства, активирующего карбоксильную группу, превращая при этом бензойную кислоту в сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина, представленный формулой (6-2), в которой R21 представляет собой 2,6-дихлорфенильную группу, R22 представляет собой изопропильную группу, и R23' представляет собой N-формил-N-метиламинометильную группу; затем с помощью метил-п-толуолсульфоната сложный изопропиловый эфир подвергают N-алкилированию, превращая его при этом в сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина; и удаляют из сложного изопропилового эфира фенилаланина защитную формильную группу в кислых условиях с помощью хлористоводородной кислоты, получая при этом сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (1-2), в которой R21 представляет собой 2,6-дихлорфенильную группу, R22 представляет собой изопропильную группу, R23 представляет собой N-метиламинометильную группу, и R24 представляет собой метильную группу, или его гидрохлорид.
При таком аспекте упомянутое выше N-алкилирование с помощью метил-п-толуолсульфоната предпочтительно проводят в оснόвных условиях.
Кроме того, во втором способе получения предпочтительно, чтобы сложный метиловый эфир 2-(3-{4-[2(S)-2-(2,6-дихлорбензоиламино)-2-метилкарбонилэтил]фенил}уреидо)-5-(диметиламино)бензойной кислоты формулы (5-2), в которой R21 представляет собой 2,6-дихлорфенильную группу, R22 представляет собой метильную группу, R23' представляет собой диметиламиногруппу, и R25 представляет собой метильную группу, получали взаимодействием соединения формулы (3-2), в которой R21 представляет собой 2,6-дихлорфенильную группу, и R22 представляет собой метильную группу, с соединением формулы (4-2), в которой R23' представляет собой диметиламиногруппу, и R25 представляет собой метильную группу; полученный сложный метиловый эфир бензойной кислоты подвергают взаимодействию с карбонатом калия, превращая при этом сложный метиловый эфир бензойной кислоты в сложный метиловый эфир Nα-(2,6-дихлорбензоил)-4-{6-диметиламинохиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (6-2), в которой R21 представляет собой 2,6-дихлорфенильную группу, R22 представляет собой метильную группу, R23 представляет собой диметиламиногруппу; затем с помощью метил-п-толуолсульфоната проводят N-алкилирование полученного сложного метилового эфира, получая при этом сложный метиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-диметиламинохиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (1-2), в которой R21 представляет собой 2,6-дихлорфенильную группу, R22 представляет собой метильную группу, R23 представляет собой диметиламиногруппу, и R24 представляет собой метильную группу.
При таком аспекте упомянутое выше N-алкилирование с помощью метил-п-толуолсульфоната предпочтительно проводят в оснόвных условиях.
Далее будет более подробно описан третий способ получения согласно настоящему изобретению.
Третий способ получения согласно настоящему изобретению осуществлен на основе обнаружения того факта, что количество побочного продукта, присущее и иногда производимое при первом способе получения, можно уменьшать, когда в качестве исходного материала производное изатового ангидрида, представленное формулой (2-3), или его химически приемлемую соль подвергают взаимодействию с производным ацилфенилаланина, представленным формулой (3-3), или его химически приемлемой солью, получая при этом амидное производное формулы (4-3), с последующим получением производного фенилаланина с хиназолиндионовым циклом, представленного формулой (1-3), или его фармацевтически приемлемой соли в качестве конечного целевого соединения через амидное производное.
Широко известно, что реакция конденсации между изатовым ангидридом и амином приводит к образованию его соответствующего амидного производного (см. Science of Synthesis, т. 16, стр. 658). Однако в настоящем изобретении такое амидное производное формулы (4-3) синтезируют при взаимодействии конкретного производного изатового ангидрида, представленного формулой (2-3), или его химически приемлемой соли в качестве исходного материала с производным ацилфенилаланина, представленного формулой (3-3), или его химически приемлемой солью.
При таком аспекте производное изатового ангидрида, представленное формулой (2-3), или его химически приемлемую соль можно получать путем взаимодействия производного антраниловой кислоты, представленного следующей формулой (6-3) и несущего незащищенную карбоксильную группу и аминогруппу, или его химически приемлемой соли с реагентом, вводящим карбонильную группу:
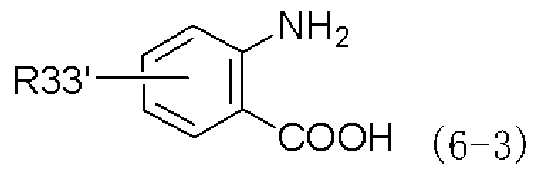 ,
,
в которой R33' имеет такое же значение, как указано выше.
При таком аспекте изатовый ангидрид, представленный формулой (2-3), можно получать путем взаимодействия производного антраниловой кислоты формулы (6-3) или его химически приемлемой соли с N,N'-карбонилдиимидазолом (CDI) или его эквивалентом (представленным формулой: R-CO-R', в которой R и R' представляют собой удаляемые группы и могут быть одинаковыми или разными, и каждый из R и R' представляет собой, например, имидазол, триазол, сукцинимидильную группу или атом галогена). На данной стадии более предпочтительным для применения является N,N'-карбонилдиимидазол (CDI).
В таком контексте в третий способ получения можно внедрить средство, активирующее карбоксильную группу, и условия его применения, аналогичные описанным выше в связи с первым способом получения, однако в третьем способе предпочтительным является применение 1,1'-карбонилдиимидазола, 1,1'-карбонилтриазола или сложного эфира хлормуравьиной кислоты.
Защитными группами для заместителя R33', представленного в формуле (6-3), желательно являются группы, способные выдерживать оснόвные условия и способные удаляться в кислых условиях, и более предпочтительно применяемыми здесь группами являются, например, формильная группа и трет-бутоксикарбонильная группа. Кроме того, диалкиламиногруппа, подходящая здесь для применения, более предпочтительно представляет собой диметиламиногруппу.
Органический растворитель, подходящий для применения в третьем способе получения, включает, например, N,N-диметилформамид (DMF), диметоксиэтан (DME), диметилсульфоксид (DMSO), диметилацетамид (DMA), ацетонитрил и тетрагидрофуран (ТГФ), и выбирается из органических растворителей, которые очень слабо взаимодействуют в числе прочих с N,N'-карбонилдиимидазолом (CDI) и более предпочтительно - с DMF.
Реакцию образования изатового ангидрида формулы (2-3) желательно проводить при температуре в диапазоне от 0 до 40°С и особенно предпочтительно - при температуре от 0 до 20°С.
Изатовый ангидрид формулы (2-3) можно выделять из реакционного раствора, и в таком случае изатовый ангидрид можно выделять согласно традиционному методу выделения, например, путем экстракции органическим растворителем с последующей концентрацией экстракта досуха. Реакционный раствор можно применять непосредственно в следующей реакции конденсации без выделения изатового ангидрида, например, к реакционному раствору, содержащему изатовый ангидрид формулы (2-3), добавляют производное ацилфенилаланина, представленное формулой (3-3), и затем полученную смесь нагревают, получая при этом амидное производное, представленное формулой (4-3).
С промышленной точки зрения указанные стадии предпочтительно проводить непрерывно, без выделения изатового ангидрида, тем самым уменьшая число необходимых стадий, однако настоящее изобретение вовсе не ограничивается таким конкретным вариантом осуществления.
Амидное производное, представленное формулой (4-3), также можно выделять из реакционного раствора согласно принятому в настоящее время методу выделения, однако реакционный раствор, содержащий амидное производное формулы (4-3), непосредственно подвергают взаимодействию с реагентом, вводящим карбонильную группу, получая при этом хиназолиндионовое производное, представленное формулой (5-3). С промышленной точки зрения указанные стадии предпочтительно проводить непрерывно, однако настоящее изобретение вовсе не ограничивается таким конкретным вариантом осуществления.
В реакции конденсации изатового ангидрида формулы (2-3) с производным ацилфенилаланина формулы (3-3) примерами применяемых в ней реакционных растворителей являются N,N-диметилформамид (DMF), диметоксиэтан (DME), диметилсульфоксид (DMSO), диметилацетамид (DMA), ацетонитрил и тетрагидрофуран (ТГФ), как описано выше, и растворитель выбирают из органических растворителей, которые очень слабо взаимодействуют в числе прочих с N,N'-карбонилдиимидазолом (CDI), более предпочтительно с DMF. Желательно, чтобы температура реакции находилась в диапазоне от 10 до 100°С и особенно предпочтительно - от 50 до 80°С. Однако при таком аспекте температуру реакции следует определять, принимая во внимание скорость образования целевого продукта (скорость исчезновения исходного материала) и образование побочных продуктов, и соответственно температура реакции не ограничивается упомянутым выше конкретным диапазоном. Время проведения реакции в общем случае находится в диапазоне приблизительно от 1 до 12 часов, однако степень протекания реакции контролируют или регулируют традиционно принятым методом с помощью ВЭЖХ, тем самым определяя время проведения реакции с учетом скорости производства продукта конденсации и скорости расходования исходного материала, и соответственно время проведения реакции не ограничивается упомянутым выше конкретным диапазоном.
При таком аспекте, который описан выше в связи с первым способом получения, реагент, вводящий карбонильную группу, предпочтительно представляет собой 1,1'-карбонилдиимидазол (CDI), метилхлорформиат, этилхлорформиат или фенилхлорформиат, и особенно предпочтительно реагент, вводящий карбонильную группу, представляет собой 1,1'-карбонилдиимидазол (CDI).
Затем на стадии (c) атом водорода, присоединенный к атому азота, присутствующему в хиназолиндионовом цикле полученного хиназолиндионового производного формулы (5-3), если требуется, можно заместить N-алкильной группой с помощью N-алкилирующего средства, и когда заместитель R33' защищен, можно удалять защитную группу. Указанные стадии можно проводить согласно процедурам, аналогичным применяемым на стадии (c) первого способа получения. На данной стадии средства, предпочтительно применяемые здесь в качестве N-алкилирующих средств, включают в себя метил-п-толуолсульфонат, метилметансульфонат, метилйодид, метилбромид и метилхлорид.
В третьем способе получения заместитель R33, представленный в формуле (1-3), предпочтительно представляет собой либо метиламинометильную группу, либо диметиламиногруппу; и заместитель R33', представленный в формулах (2-3), (4-3) и (5-3), предпочтительно представляет собой одну из групп: N-формил-N-метиламинометильную группу, N-(трет-бутоксикарбонил)-N-метиламинометильную группу, N-ацетил-N-метиламинометильную группу и диметиламиногруппу.
Кроме того, в изобретении предпочтительно, чтобы соединение формулы (3-3), в которой R31, представленный в формуле (3-3), представляет собой 2,6-дихлорфенильную группу, и R32 представляет собой изопропильную группу, подвергали взаимодействию с соединением формулы (2-3), в которой R33' представляет собой N-формил-N-метиламинометильную группу, образуя при этом сложный изопропиловый эфир 4-{2-амино-5-(N-формил-N-метиламинометил)бензоиламино}-Nα-(2,6-дихлорбензоил)-L-фенилаланина формулы (4-3), в которой R31 представляет собой 2,6-дихлорфенильную группу, R32 представляет собой изопропильную группу, и R33' представляет собой N-формил-N-метиламинометильную группу; полученный сложный изопропиловый эфир подвергают взаимодействию с 1,1'-карбонилдиимидазолом в качестве реагента, вводящего карбонильную группу, превращая при этом сложный изопропиловый эфир в сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (5-3), в которой R31 представляет собой 2,6-дихлорфенильную группу, R32 представляет собой изопропильную группу, и R33' представляет собой N-формил-N-метиламинометильную группу; затем сложный изопропиловый эфир формулы (5-3) с помощью метил-п-толуолсульфоната подвергают N-алкилированию, превращая при этом сложный эфир в сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина; затем с помощью хлористого водорода удаляют защитную формильную группу, получая при этом сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (1-3) или его гидрохлорид, в которой R31 представляет собой 2,6-дихлорфенильную группу, R32 представляет собой изопропильную группу, и R33 представляет собой N-метиламинометильную группу, и R34 представляет собой метильную группу.
При таком аспекте упомянутое выше N-алкилирование с помощью метил-п-толуолсульфоната предпочтительно проводят в оснόвных условиях.
Кроме того, в настоящем изобретении предпочтительно, чтобы соединение формулы (3-3), в которой R31, представленный в формуле (3-3), представляет собой 2,6-дихлорфенильную группу, и R32 представляет собой метильную группу, подвергали взаимодействию с соединением формулы (2-3), в которой R33' представляет собой диметиламиногруппу, получая при этом сложный метиловый эфир 4-{2-амино-5-диметиламинобензоиламино}-Nα-(2,6-дихлорбензоил)-L-фенилаланина формулы (4-3), в которой R31 представляет собой 2,6-дихлорфенильную группу, R32 представляет собой метильную группу, и R33' представляет собой диметиламиногруппу; полученный сложный метиловый эфир подвергают взаимодействию с 1,1'-карбонилдиимидазолом в качестве реагента, вводящего карбонильную группу, превращая при этом сложный метиловый эфир в сложный метиловый эфир Nα-(2,6-дихлорбензоил)-4-{6-диметиламинохиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (5-3), в которой R31 представляет собой 2,6-дихлорфенильную группу, R32 представляет собой метильную группу, и R33' представляет собой диметиламиногруппу; затем полученный сложный метиловый эфир формулы (5-3) подвергают N-алкилированию с помощью метил-п-толуолсульфоната, получая при этом сложный метиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-диметиламинохиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (1-3), в которой R31 представляет собой 2,6-дихлорфенильную группу, R32 представляет собой метильную группу, R33 представляет собой диметиламиногруппу, и R34 представляет собой метильную группу.
При таком аспекте упомянутое выше N-алкилирование с помощью метил-п-толуолсульфоната предпочтительно проводят в оснόвных условиях.
Далее более подробно будет описан четвертый способ получения согласно настоящему изобретению.
Четвертый способ получения согласно настоящему изобретению выполнен на основе обнаружения того факта, что количество побочного продукта, присущее и иногда производимое в первом способе получения, можно уменьшить, если в качестве исходного материала производное бензоксазина, представленное формулой (2-4), подвергают взаимодействию с производным ацилфенилаланина, представленным формулой (3-4), или его химически приемлемой солью, затем на полученное амидкарбаматное производное формулы (4-4) или его химически приемлемую соль действуют основанием, получая при этом хиназолиндионовое производное, представленное формулой (5-4), с последующим получением через посредство хиназолиндионового производного формулы (5-4) производного фенилаланина с хиназолиндионовым циклом, представленного формулой (1-4), или его фармацевтически приемлемой соли в качестве конечного целевого соединения.
Применяемое в качестве исходного материала производное бензоксазина, представленное формулой (2-4), можно легко получать путем взаимодействия производного антраниловой кислоты, представленного следующей формулой (6-4), или его химически приемлемой соли с алкилгалогенформиатом или фенилгалогенформиатом, который может содержать заместитель согласно способу, описанному в литературе (Heterocycle, 1999, 51(7): 1543-1561):
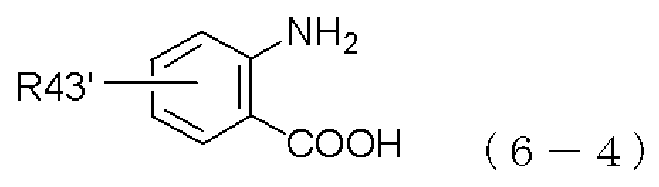 ,
,
в которой R43' имеет то же самое значение, которое указано выше.
Предпочтительно применяемый здесь в качестве такого реагента алкилгалогенформиат представляет собой сложные эфиры хлормуравьиной кислоты, и его конкретными примерами, подходящими здесь для применения, являются метилхлорформиат, этилхлорформиат, фенилхлорформиат, изопропилхлорформиат и бензилхлорформиат, с более предпочтительно применяемым здесь этилхлорформиатом. Кроме того, предпочтительно применяемыми здесь являются сложные эфиры хлормуравьиной кислоты, которые никогда не связаны с какой-либо побочной реакцией, такой как транс-этерификация. В качестве заместителя на фенильной группе фенилгалогенформиата, который может содержать заместитель, можно перечислить, например, алкильные группы, алкоксигруппы и атомы галогенов, однако предпочтительно применяемой здесь является фенильная группа, не содержащая никакого заместителя. Фенилгалогенформиат, который может содержать заместитель, предпочтительно представляет собой фенилхлорформиат.
Реакционные растворители, подходящие здесь для применения, включают, например, пиридин, N-метилморфолин, N,N-диметилформамид (DMF) и ацетонитрил, и более предпочтительно - пиридиновый растворитель. Кроме того, когда в качестве такого растворителя применяют DMF или ацетонитрил, также можно применять смешанный растворитель, к которому в качестве основания добавляют органическое основание или пиридин.
Производное бензоксазина формулы (2-4), полученное при взаимодействии производного антраниловой кислоты формулы (6-4) или его химически приемлемой соли с алкилгалогенформиатом, можно применять на следующей стадии без выделения производного бензоксазина из реакционного раствора, однако производное можно выделять согласно традиционному методу выделения. С промышленной точки зрения указанные стадии преимущественно проводят непрерывно.
При реакции конденсации производного бензоксазина формулы (2-4) с производным ацилфенилаланина формулы (3-4) производное ацилфенилаланина формулы (3-4) можно добавлять в реакционный раствор, содержащий производное бензоксазина формулы (2-4), образуя при этом соответствующее амидкарбаматное производное формулы (4-4).
Подходящими здесь для применения в качестве реакционных растворителей являются, например, DMF, ацетонитрил и пиридин. Температура реакции предпочтительно находится в диапазоне от 0 до 80°С и более предпочтительно - приблизительно от 10 до 30°С, однако в настоящем изобретении она не ограничивается указанным выше диапазоном. После того, как протекание реакции подтверждено, например, с помощью ВЭЖХ, продукт или амидкарбаматное производное формулы (4-4) выделяют согласно широко принятому методу выделения, такому как добавление слабого растворителя.
Хиназолиндионовый цикл можно образовывать путем обработки амидкарбаматного производного формулы (4-4) в оснόвных условиях, получая при этом хиназолиндионовое производное формулы (5-4). Реакционные растворители, подходящие здесь для применения, включают, например, DMF, N-метилпирролидон, ацетонитрил и смешанный растворитель DMF/спирт. В качестве таких оснований, подходящих здесь для применения, можно перечислить, например, органические основания, такие как триэтиламин и диизопропилэтиламин, углекислые соли щелочных металлов, такие как карбонат калия, карбонат натрия, гидрокарбонат калия, гидрокарбонат натрия. Из них предпочтительно применяемые здесь в качестве основания включают в себя карбонат калия или метилат натрия.
Температура реакции предпочтительно находится в диапазоне от 10 до 80°С и более предпочтительно - приблизительно от 20 до 40°С, однако температуру реакции следует определять, принимая во внимание скорость образования целевого продукта (скорость исчезновения исходного материала) и образование побочных продуктов, и соответственно температура реакции не ограничивается упомянутым выше конкретным диапазоном.
Затем на стадии (c) атом водорода, связанный с атомом азота, присутствующим в хиназолиндионовом цикле хиназолиндионового производного формулы (5-4), если требуется, замещают N-алкильной группой с помощью N-алкилирующего средства, и когда заместитель R43' защищен, можно удалять защитную группу. Указанные стадии можно проводить согласно аналогичным процедурам, применяемым на стадии (c) первого способа получения. На указанной стадии средства, предпочтительно применяемые здесь в качестве N-алкилирующих средств, включают в себя метил-п-толуолсульфонат, метилметансульфонат, метилйодид, метилбромид и метилхлорид.
В четвертом способе получения согласно настоящему изобретению предпочтительно, чтобы заместитель R43, представленный в формуле (1-4), представлял собой либо метиламинометильную группу, либо диметиламиногруппу, и заместитель R43', представленный в формулах (2-4), (4-4) и (5-4), представлял собой N-формил-N-метиламинометильную группу, N-(трет-бутоксикарбонил)-N-метиламинометильную группу, N-ацетил-N-метиламинометильную группу или диметиламиногруппу. При таком аспекте упомянутое выше N-алкилзамещение с помощью метил-п-толуолсульфоната предпочтительно проводят в основных условиях.
Кроме того, предпочтительно, чтобы соединение формулы (3-4), в которой R41 представляет собой 2,6-дихлорфенильную группу, и R42 представляет собой изопропильную группу, подвергалось взаимодействию с соединением формулы (2-4), в которой R43' представляет собой N-формил-N-метиламинометильную группу, и R45 представляет собой этильную группу, образуя при этом сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{2-этоксикарбониламино-5-(N-формил-N-метиламинометил)бензоиламино}-L-фенилаланина формулы (4-4), в которой R41 представляет собой 2,6-дихлорфенильную группу, R42 представляет собой изопропильную группу, R43' представляет собой N-формил-N-метиламинометильную группу, и R45 представляет собой этильную группу; затем сложный изопропиловый эфир формулы (4-4) подвергают взаимодействию с карбонатом калия, превращая при этом сложный эфир в сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (5-4), в которой R41 представляет собой 2,6-дихлорфенильную группу, R42 представляет собой изопропильную группу, и R43' представляет собой N-формил-N-метиламинометильную группу; затем сложный изопропиловый эфир с помощью метил-п-толуолсульфоната подвергают N-алкилированию, превращая при этом сложный эфир в сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина; и, в конечном счете, из полученного сложного изопропилового эфира с помощью хлористого водорода удаляют защитную формильную группу, получая при этом сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина или его гидрохлорид, в котором R41 представляет собой 2,6-дихлорфенильную группу, R42 представляет собой изопропильную группу, R43 представляет собой N-метиламинометильную группу, и R44 представляет собой метильную группу.
При таком аспекте упомянутое выше N-алкилирование с помощью метил-п-толуолсульфоната предпочтительно проводят в оснόвных условиях.
В изобретении также предпочтительно, чтобы соединение формулы (3-4), в которой R41 представляет собой 2,6-дихлорфенильную группу, и R42 представляет собой метильную группу, подвергалось взаимодействию с соединением формулы (2-4), в которой R43' представляет собой диметиламиногруппу, и R45 представляет собой этильную группу, образуя при этом сложный метиловый эфир Nα-(2,6-дихлорбензоил)-4-{2-этоксикарбониламино-4-диметиламинобензоиламино}-L-фенилаланина формулы (4-4), в которой R41 представляет собой 2,6-дихлорфенильную группу, R42 представляет собой метильную группу, R43' представляет собой диметиламиногруппу, и R45 представляет собой этильную группу; полученный сложный метиловый эфир подвергают взаимодействию с карбонатом калия, превращая при этом сложный эфир в сложный метиловый эфир Nα-(2,6-дихлорбензоил)-4-{6-диметиламинохиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (4-5), в которой R41 представляет собой 2,6-дихлорфенильную группу, R42 представляет собой метильную группу, и R43' представляет собой диметиламиногруппу; и затем сложный метиловый эфир с помощью метил-п-толуолсульфоната подвергают N-алкилированию, получая при этом сложный метиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-диметиламинохиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина формулы (1-4), в которой R41 представляет собой 2,6-дихлорфенильную группу, R42 представляет собой метильную группу, R43 представляет собой диметиламинометильную группу, и R44 представляет собой метильную группу.
При таком аспекте упомянутое выше N-алкилирование с помощью метил-п-толуолсульфоната предпочтительно проводят в оснόвных условиях.
Настоящее изобретение будет описано ниже более подробно со ссылкой на следующие примеры.
ПРИМЕРЫ
Прежде всего, далее следует объяснение методов анализа, применяемых в следующих примерах, и применяемых в них исходных материалов.
1. Условия анализа или тому подобное
(1) 1H и 13C ЯМР-спектроскопические анализы проводили с помощью прибора для ЯМР-анализа AVANCE 400 МГц, предоставленного компанией Bruker Company, или прибора для ЯМР-анализа GEMINI 300 МГц, предоставленного компанией Varian Company, в то же время применяя при измерении в качестве стандарта пик, обнаруживаемый для применяемого растворителя.
(2) ВЭЖХ-анализ проводили с помощью прибора серии LC, предоставленного компанией Shimadzu Corporation, и обрабатывали данные с помощью программного обеспечения для хроматографических анализов. Условия проведения ВЭЖХ-анализа следующие:
Элюирующий раствор: Раствор A: водный раствор, содержащий 0,1% трифторуксусной кислоты; Раствор B: ацетонитрильный раствор, содержащий 0,1% трифторуксусной кислоты;
Градиентные условия: (раствор A/раствор B) = первоначально (90/10) - спустя 25 минут (10/90) - спустя 30 минут (10/90);
Скорость потока: 1,0 мл/мин;
Применяемая колонка: колонка ODS с силикагелем с обращенной фазой (ODS-2 предоставлена компанией GL Science Company); размер колонки: 4,6 мм (внутренний диаметр)×150 мм (в длину);
Температура колонки: 40°С; количество впрыскиваемого образца: 10 мкл.
(3) Ионообменную хроматографию проводили с помощью прибора DIONEX (DX-120), предоставленного компанией Dionex Company, и анализировали данные с помощью программного обеспечения для хроматографического анализа. Применяемый элюирующий раствор: 1M Na2CO3/1M NaHCO3 = смешанный раствор 9/1.
(4) Температуру плавления определяли с помощью дифференциального сканирующего калориметрического анализатора (ДСК) (DSC6200, предоставленного компанией Seiko-Epolead Company). Применяемый контейнер представлял собой герметизированный SUS-контейнер или облегченный герметизированный контейнер алюминиевой кюветы.
(5) При масс-спектрометрическом (МС) исследовании применяли масс-спектрометр Thermo Quest TSQ700, предоставленный компанией Seiko-Epolead Company.
(6) Масс-спектрометрический анализ высокого разрешения (HRMS) проводили на приборе MS700V, предоставленном компанией JEOL Ltd.
2. Исходные материалы или тому подобное:
Растворители и исходные материалы, применяемые в следующих примерах, являются коммерчески доступными и применялись без какой-либо предварительной обработки, за исключением особых случаев. Сложный метиловый эфир 4-амино-Nα-(2,6-дихлорбензоил)-L-фенилаланина синтезировали согласно способу, описанному в патентном документе 2.
Пример 1
Синтез 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты
Стадия 1: Синтез 5-(N-метиламинометил)-2-нитробензойной кислоты
Способ 1: Синтез 5-(N-метиламинометил)-2-нитробензойной кислоты с применением DBH/AIBN и выделение ее путем кристаллизации при добавлении 2-пропанола
К 100 мл хлорбензола добавляли 50 г 5-метил-2-нитробензойной кислоты и 36,66 г 1,3-дибром-5,5-диметилгидантоина (DBH), получая при этом суспензию. Полученную суспензию нагревали до 40°С в атмосфере газообразного аргона, добавляли к суспензии 1,263 г азобис-(2-метилпропионитрила) (AIBN) и затем нагревали смесь до 80°С. По истечении одного часа реакционный раствор сразу охлаждали до 40°С, к реакционному раствору снова добавляли 18,33 г 1,3-дибром-5,5-диметилгидантоина и 1,263 г азобис-(2-метилпропионитрила) и снова нагревали полученную смесь до 80°С. Спустя 2 часа реакционный раствор охлаждали до 10°С по мере перемешивания его в течение ночи, выделяющееся при этом нерастворимое твердое вещество отфильтровывали и промывали последнее 40 мл хлорбензола. Фильтрат и промывную жидкость объединяли и полученную смесь по каплям добавляли к 260 мл 40% раствора метиламина/метанола, предварительно охлажденного до 10°С в течение более 80 минут, в то же время, принимая меры по предотвращению возможности избыточного образования тепла. Затем температуру реакционного раствора повышали до 25°С с последующим перемешиванием. После подтверждения с помощью ВЭЖХ протекания реакции полученный реакционный раствор подвергали перегонке при пониженном давлении (температура водяной бани: 50°С), концентрируя при этом раствор наполовину, и добавляли к концентрированному раствору 150 мл 2-пропанола и затравочные кристаллы. Смесь непрерывно перемешивали при 50°С и после выделения требуемого продукта добавляли к ней 100 мл 2-пропанола. Полученную взвесь снова подвергали перегонке при пониженном давлении, концентрируя при этом взвесь, концентрированную взвесь перемешивали при 50°С в течение одного часа, охлаждали до 9°С и затем выдерживали в течение 4 часов. Образовавшиеся при этом кристаллы отфильтровывали из взвеси и промывали кристаллы во влажном состоянии 100 мл 2-пропанола. Затем кристаллы во влажном состоянии сушили при 60°С и пониженном давлении, получая при этом 42,18 г указанного в заголовке соединения в виде твердого кристаллического вещества белого цвета (выход: 52%).
1H-ЯМР (400 МГц, D2O): δ 7,98 (1H, д, J=8,4 Гц), 7,50 (1H, дд, J=8,4, 1,9 Гц), 7,44 (д, 1H, J=1,7 Гц), 4,20 (с, 2H), 2,66 (с, 3H); 13C-ЯМР (100 МГц, D2O): δ 173,89, 145,56, 137,84, 136,83, 130,64, 129,07, 125,37, 51,56, 32,92.
Температура плавления (ДСК): Какой-либо точки плавления не обнаружено, но соединение разлагается (температура начала разложения: 239,6°С); МС (ESI+): m/z 210,8 (MH+), 421,1 (2MH+); HRMS (FAB): рассчитанное для C9H11N2O4 m/z 211,0719 (MH+); найденное m/z: 211,0723 (MH+).
Способ 2: Синтез 5-(N-метиламинометил)-2-нитробензойной кислоты путем бромирования с помощью NBS/BPO и выделение кристаллов при добавлении хлористого водорода/этилацетата
Получали суспензию из 6,12 г 5-метил-2-нитробензойной кислоты в 25 мл бензола и затем добавляли к суспензии 6,72 г N-бромсукцинимида (NBS) и 280 мг бензоилпероксида (BPO), содержащего 25% воды. Атмосферу реакционной системы заменяли атмосферой газообразного аргона и затем перемешивали полученную смесь при 80°С в течение 3 часов. Затем реакционный раствор охлаждали до 10°С, по каплям добавляли реакционный раствор к 32 мл 40% раствора метиламина в метаноле и перемешивали смесь при комнатной температуре в течение ночи. Реакционный раствор подвергали перегонке при пониженном давлении, чтобы приблизительно наполовину уменьшить объем реакционного раствора, и затем добавляли к реакционному раствору 23,5 мл раствора хлористый водород/этилацетат (3M). Выпавшее в осадок твердое вещество отфильтровывали и промывали 50 мл этилацетата. Затем твердое вещество сушили при пониженном давлении, получая при этом 8,08 г указанного в заголовке соединения в виде твердого кристаллического вещества (в виде смеси с гидрохлоридом метиламина). Обнаруженные физические свойства почти полностью идентичны свойствам, обнаруженным для аналогичного соединения, полученного в упомянутом выше примере синтеза.
Стадия 2: Синтез 5-(N-формил-N-метиламинометил)-2-нитробензойной кислоты
В колбу вытянутой формы (баклажаноподобной) объемом 100 мл добавляли 4,0 мл муравьиной кислоты и 2,7 мл уксусного ангидрида и перемешивали смесь при комнатной температуре. После этого к упомянутой выше смеси в виде двух порций добавляли 0,65 г формиата натрия и 5,58 г (содержание: 71,7% мас.) 5-(N-метиламинометил)-2-нитробензойной кислоты, затем перемешивали полученную смесь при комнатной температуре в течение одного часа. После подтверждения с помощью ВЭЖХ протекания реакции к содержимому добавляли 32 мл воды для выделения кристаллов и образования взвеси и затем охлаждали до 9°С и перемешивали в течение ночи. Твердое вещество, выделенное из реакционного раствора, отфильтровывали, промывали водой, сушили при пониженном давлении, получая при этом 3,95 г указанного в заголовке соединения в виде твердого кристаллического вещества белого цвета (выход: 87%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 8,32 и 8,19 (два с, 1H), 8,02 и 7,99 (два д, 1H, J=8,3 Гц), 7,74 и 7,67 (два д, 1H, J=1,8 Гц), 7,65 и 7,59 (два дд, 1H, J=8,3, 1,9 Гц), 4,62 и 4,58 (два с, 2H), 2,90 и 2,66 (два с, 3H); 13C-ЯМР (100 МГц, ДМСО-d6): δ 166,11, 166,05, 163,39, 163,32, 147,63, 147,33, 143,18, 143,01, 131,37, 131,23, 128,99, 128,85, 128,08, 128,06, 124,50, 124,41, 51,31, 46,26, 34,18, 29,18.
Температура плавления (ДСК): 174,8°С; МС (ESI+): m/z 239 (MH+), 477,1 (2MH+); HRMS (FAB): рассчитанное для C10H11N2O5 m/z 239,0688 (MH+); найденное m/z: 239,0667 (MH+).
Стадия 3: Синтез 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты
В двугорлую колбу вытянутой формы (баклажаноподобной формы) объемом 100 мл при комнатной температуре добавляли 6,98 г (содержание: 85,9%) 5-(N-формил-N-метиламинометил)-2-нитробензойной кислоты и 48 мл метанола, получая при этом суспензию, и затем к суспензии добавляли 3,5 мл 6M водного раствора гидроксида натрия с образованием при этом раствора (pH: 5,6). После добавления к раствору 1,028 г (0,9% мол. относительно исходного материала) 5% палладия на углероде (влажный продукт) в атмосфере газообразного аргона газовую фазу заменяли газообразным водородом и нагревали смесь до 40°С. Спустя 5 часов подтверждали протекание реакции с помощью ВЭЖХ (по исчезновению исходного материала), катализатор отфильтровывали, и оставшийся на фильтре катализатор промывали 18 мл метанола. Полученный фильтрат и промывочную жидкость объединяли, растворитель отгоняли при 40°С и пониженном давлении до тех пор, пока концентрация не достигла 2 л/кг, и затем добавляли 48 мл воды. К полученному концентрату при комнатной температуре добавляли 11,0 мл 2M водного раствора хлористоводородной кислоты, выделяя при этом кристаллы (pH: от 2 до 3). Полученную взвесь охлаждали до 9°С, после перемешивания взвеси в течение ночи кристаллы отфильтровывали и затем промывали кристаллы 24 мл воды. Полученные кристаллы сушили при 60°С и пониженном давлении, получая при этом 4,84 г указанного в заголовке соединения в виде твердого кристаллического вещества белого цвета (выход: 92%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 8,49 (ушир.с, 2H), 8,25 и 8,08 (два с, 1H), 7,60 и 7,59 (два д, 1H, J=2,1 Гц), 7,14 и 7,10 (два дд, 1H, J=8,5, 2,3 Гц), 6,76 и 6,72 (два д, 1H, J=8,4 Гц), 4,27 (с, 2H), 2,77 и 2,58 (два с, 3H); 13C-ЯМР (100 МГц, ДМСО-d6): δ 169,55, 169,52, 162,67, 162,62, 151,24, 151,00, 134,03, 133,65, 131,01, 130,76, 122,60, 122,54, 117,01, 116,88, 109,49, 109,48, 51,75, 46,14, 33,48, 28,60.
Температура плавления (ДСК): 174,2°С; МС (ESI+): m/z 231,3 (MH+), 417,2 (2MH+); HRMS (FAB): рассчитанное для C10H13N2O3 m/z 209,0926 (MH+); найденное m/z: 209,0950 (MH+).
Пример 2
Синтез 2-амино-5-диметиламинобензойной кислоты
Способ 1: Синтез указанного в заголовке соединения, исходя из 5-диметиламино-2-нитробензойной кислоты, в основных условиях, соответствующих каталитическому восстановлению

К 1,0 г 5-диметиламино-2-нитробензойной кислоты добавляли 16 мл метанола и 0,79 мл 6M водного раствора гидроксида натрия, затем нагревали смесь до 40°С до получения однородного раствора. Затем к раствору в атмосфере газообразного аргона добавляли 0,21 г 5% палладия на углероде (влажный продукт) и вводили в смесь газообразный водород для взаимодействия с ним. По истечении 5 часов подтверждали протекание реакции с помощью ВЭЖХ, затем отфильтровывали катализатор при пониженном давлении и промывали 10 мл метанола. Объединенные фильтрат и промывочную жидкость нейтрализовали добавлением 0,79 мл 6M водного раствора хлористоводородной кислоты (pH: 5,5), и затем отгоняли растворитель при пониженном давлении с осаждением при этом твердого вещества. К полученной суспензии добавляли 10 мл воды и перемешивали смесь при 10°С в течение одного часа. Образовавшиеся при этом кристаллы отделяли фильтрованием при пониженном давлении, промывали 10 мл воды и затем сушили при 60°С в течение 12 часов при пониженном давлении, получая при этом 0,56 г указанного в заголовке соединения в виде твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6): δ 8,21 (ушир.с, 3H), 7,10 (д, 1H, J=2,8 Гц), 6,97 (дд, 1H, J=9,1, 2,8 Гц), 6,70 (д, 1H, J=9,1 Гц), 2,72 (с, 6H); 13C-ЯМР (100 МГц, ДМСО-d6): δ 168,89, 144,55, 141,61, 123,29, 117,90, 114,78, 110,11, 41,95.
MС (ESI+): m/z 181,3 (MH+), (ESI-): m/z 179,2 (M-H-).
Способ 2: Указанное в заголовке соединение получали, исходя из дигидрохлорида сложного метилового эфира 2-амино-5-диметиламинобензойной кислоты, с помощью гидролиза в оснόвных условиях
К 5,0 г дигидрохлорида сложного метилового эфира 2-амино-5-диметиламинобензойной кислоты добавляли 15 мл воды и 15,6 мл 6M водного раствора гидроксида натрия и нагревали полученную смесь до 40°С в течение 2 часов. После подтверждения с помощью ВЭЖХ протекания реакции реакционную систему охлаждали до комнатной температуры, к реакционной системе по каплям добавляли 6M водный раствор хлористоводородной кислоты, нейтрализуя при этом систему и выделяя кристаллы (pH 4,9), и затем перемешивали реакционную систему при 10°С в течение 2 часов. Полученное при этом твердое вещество отделяли фильтрованием при пониженном давлении, промывали 30 мл воды и затем сушили при 60°С и пониженном давлении в течение 14 часов, получая при этом 3,14 г указанного в заголовке соединения в виде твердого вещества, окрашенного в серый цвет. Обнаруженные физические свойства почти полностью идентичны свойствам, обнаруженным для аналогичного соединения, полученного в упомянутом выше примере синтеза.
Пример 3
Синтез 2-амино-5-(N-трет-бутоксикарбонил-N-метиламинометил)бензойной кислоты
Стадия 1: Синтез гидрохлорида 5-метиламинометил-2-нитробензойной кислоты
К 260 мл бензола добавляли 29,52 г 5-метил-2-нитробензойной кислоты, 31,10 г N-бромсукцинимида и 1,00 г бензоилпероксида и нагревали полученную смесь при 70°С при перемешивании в течение ночи. После подтверждения с помощью ВЭЖХ протекания реакции реакционный раствор охлаждали до температуры, близкой к комнатной температуре, и затем концентрировали с помощью испарителя. К остатку, оставшемуся после концентрирования, добавляли 300 мл воды и 300 мл этилацетата, экстрагируя при этом продукт в органическую фазу. Органическую фазу три раза промывали 200 мл воды, к органической фазе добавляли небольшое количество сульфата магния для ее обезвоживания, и затем концентрировали органическую фазу с помощью испарителя. Полученный маслянистый продукт растворяли в 200 мл ацетонитрила и ацетонитрильный раствор по каплям добавляли к 400 мл 2M раствора метиламина в ТГФ на ледяной бане. После завершения добавления капель смесь концентрировали с помощью испарителя, к полученному остатку добавляли 300 мл этилацетата и затем к этому добавляли 250 мл 4M раствора хлористого водорода в этилацетате. Полученные кристаллы отфильтровывали и затем сушили при пониженном давлении, получая при этом 33,57 г смеси указанного в заголовке соединения и гидрохлорида метиламина (отношение = 1:3). Полученную смесь добавляли к 500 мл 2-пропанола, нагревали до 70°С при перемешивании, затем постепенно охлаждали от комнатной температуры до 10°С и собирали полученные осадки.
Стадия 2: Синтез 5-(N-трет-бутоксикарбонил-N-метиламинометил)-2-нитробензойной кислоты
К 25 мл ацетонитрила добавляли 2,46 г гидрохлорида 5-метиламинометил-2-нитробензойной кислоты, полученного на упомянутой выше стадии 1, и 2,40 г ди-трет-бутилдикарбоната, затем добавляли 3,5 мл триэтиламина и перемешивали полученную смесь в течение 3 часов при комнатной температуре. После подтверждения протекания реакции смесь концентрировали с помощью испарителя, полученный остаток разбавляли при добавлении 100 мл этилацетата и затем добавляли к разбавленному остатку 100 мл воды и 10 мл 2M раствора гидроксида натрия. Предполагаемый продукт экстрагировали водой, водную фазу слегка подкисляли при добавлении этилацетата и 2M раствора хлористоводородной кислоты, экстрагируя при этом требуемый продукт в органическую фазу. Затем органическую фазу промывали насыщенным водным раствором обычной соли, органическую фазу подвергали обезвоживанию при обработке сульфатом натрия, затем концентрировали досуха с помощью испарителя и сушили при пониженном давлении, получая при этом 2,214 г указанного в заголовке соединения в виде твердого вещества светло-желтого цвета (выход: 71%).
1H-ЯМР (300 МГц, ДМСО-d6): δ 10,76 (ушир.с, 1H), 7,89 (д, 1H, J=8,4 Гц), 7,68 (с, 1H), 7,51 (д, 1H, J=7,5 Гц), 4,55 (с, 2H), 2,90 (ушир.с, 3H), 1,48 (с, 9H).
Стадия 3: Синтез 2-амино-5-(N-трет-бутоксикарбонил-N-метиламинометил)бензойной кислоты
К 80 мл метанола добавляли 8,3 г 5-(N-трет-бутоксикарбонил-N-метиламинометил)-2-нитробензойной кислоты, полученной на упомянутой выше стадии 2, и 500 мг 10% Pd/C (влажный продукт), затем в полученную дисперсию вводили газообразный водород и перемешивали смесь при комнатной температуре. Спустя 1,5 часа к смеси дополнительно добавляли дополнительные 500 мг 10% Pd/C и перемешивали последнюю в течение более 7 часов в атмосфере газообразного водорода. Катализатор удаляли фильтрованием через целит и промывали целит метанолом. Фильтрат концентрировали досуха, получая при этом указанное в заголовке соединение.
1H-ЯМР (300 МГц, ДМСО-d6): δ 7,62 (ушир.с, 1H), 7,11 (д, 1H, J=6,6 Гц), 6,80 (д, 1H, J=8,1 Гц), 4,18 (с, 2H), 2,69 (с, 3H), 1,41 (с, 9H).
МС (ES+): m/z 281,0 (MH+).
Пример 4
Синтез сложного изопропилового эфира 4-амино-N α -(2,6-дихлорбензоил)-L-фенилаланина

Стадия 1: Синтез N α -(2,6-дихлорбензоил)-4-нитро-L-фенилаланина
К 13 мл ацетона добавляли 10,04 г п-нитро-L-фенилаланина и 28 мл воды, образуя при этом суспензию, и охлаждали последнюю на ледяной бане. Альтернативно к полученной суспензии по каплям добавляли 18,4 мл 6M раствора гидроксида натрия и 10,05 г 2,6-дихлорбензоилхлорида таким образом, что значение pH суспензии поддерживалось на уровне не менее 13 при температуре не более 15°С. После завершения добавления капель суспензию дополнительно перемешивали в течение 2 часов при охлаждении на ледяной бане, затем с помощью ВЭЖХ подтверждали протекание реакции и добавляли по каплям 36 мл 2M раствора хлористоводородной кислоты, в то же время принимая меры по предотвращению избыточного образования тепла. Суспензию, содержащую выделенное твердое вещество, перемешивали при 10°С в течение ночи, осажденные вещества отделяли фильтрованием и затем сушили при 60°С и пониженном давлении, получая при этом 17,32 г указанного в заголовке соединения в виде твердого кристаллического вещества белого цвета (выход: 95%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,13 (д, 1H, J=8,3 Гц), 8,16 (м, 2H), 7,59 (д, 2H, J=8,4 Гц), 7,42 (м, 3H), 4,79 (м, 1H), 3,30 (м, 1H), 3,06 (м, 1H); 13C-ЯМР (100 МГц, ДМСО-d6): δ 172,08, 163,56, 146,50, 146,00, 136,18, 131,33, 131,19, 130,88, 128,19, 123,37, 93,39, 52,98, 36,55.
Температура плавления (ДСК): 220,6°С; МС (ESI+, m/z): 383,0 (MH+), 385,1 (MH+).
Стадия 2: Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-нитро-L-фенилаланина
Способ 1: Реакция этерификации с метансульфоновой кислотой и выделение/сушка
Готовили суспензию из 359 мл 2-пропанола и 35,90 г Nα-(2,6-дихлорбензоил)-4-нитро-L-фенилаланина, полученного по упомянутому выше способу, и добавляли к суспензии 6,0 мл метансульфоновой кислоты. После перемешивания суспензии при 60°С в течение 19 часов ее охлаждали до комнатной температуры и извлекали полученные кристаллы фильтрованием. Полученные влажные кристаллы сушили при 60°С и пониженном давлении, получая при этом 38,29 г указанного в заголовке соединения в виде твердого кристаллического вещества белого цвета (выход: 96%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,21 (д, 1H, J=8,1 Гц), 8,16 (д, 2H, J=8,7 Гц), 7,60 (д, 2H, J=8,7 Гц), 7,46-7,38 (м, 3H), 4,95 (м, 1H), 4,82 (м, 1H), 3,27 (м, 1H), 3,08 (м, 1H), 1,22 (д, 3H, J=6,2 Гц), 1,18 (д, 3H, J=6,2 Гц); 13C-ЯМР (100 МГц, ДМСО-d6): δ 170,05, 163,50, 146,54, 145,63, 136,03, 131,31, 131,28, 130,91, 128,22, 123,38, 68,67, 53,13, 36,38, 21,71, 21,65.
Температура плавления (ДСК): 188,2°С; МС (ESI+, m/z): 425,1 (MH+), 427,0 (MH+).
Способ 2: Соединение реакции этерификации с метансульфоновой кислотой с последующей стадией путем исключения стадии сушки
К 150 мл 2-пропанола добавляли 14,92 г Nα-(2,6-дихлорбензоил)-4-нитро-L-фенилаланина при комнатной температуре, получая при этом суспензию. К суспензии добавляли 2,5 мл метансульфоновой кислоты, полученную смесь нагревали при перемешивании до 65°С в течение более 16 часов. После подтверждения с помощью ВЭЖХ протекания реакции реакционный раствор охлаждали до комнатной температуры и выпавшее в осадок твердое вещество выделяли фильтрованием. Кристаллы промывали 75 мл 2-пропанола, получая при этом указанное в заголовке соединение в виде влажных кристаллов белого цвета. Влажные кристаллы применяли на следующей стадии (реакция восстановления нитрогруппы) без их сушки.
Стадия 3: Синтез сложного изопропилового эфира 4-амино-N α -(2,6-дихлорбензоил)-L-фенилаланина
К 80 мл тетрагидрофурана в атмосфере газообразного азота при комнатной температуре добавляли 31,3 г (содержание: приблизительно 50% масс.) влажных кристаллов сложного изопропилового эфира Nα-(2,6-дихлорбензоил)-4-нитро-L-фенилаланина и затем перемешивали смесь. К смеси добавляли 80 мл 2-пропанола и 0,86 г 3% Pt-S/C (влажного продукта, полученного нанесением платины на углерод и последующим протравливанием катализатора серой), затем в реакционную систему вводили газообразный водород и перемешивали реакционную систему при обычном давлении. Реакционную систему, как она есть, перемешивали при комнатной температуре в течение ночи, и затем после подтверждения с помощью ВЭЖХ протекания реакции удаляли катализатор из реакционного раствора фильтрованием через целит. Полученный фильтрат (первоначальное количество 178 г) концентрировали отгонкой растворителя при пониженном давлении до тех пор, пока количество фильтрата не достигало 91 г, и добавляли к концентрату 40 мл 2-пропанола, регулируя при этом концентрацию конденсата. К полученной жидкости, концентрация которой была отрегулирована, при комнатной температуре по каплям добавляли 150 мл воды, выделяя при этом твердое кристаллическое вещество, и поддерживали полученную суспензию при температуре не выше 10°С в течение ночи. Полученные кристаллы отфильтровывали, промывали 100 мл воды и сушили полученные влажные кристаллы при 60°С и пониженном давлении, получая при этом 13,16 г указанного в заголовке соединения в виде твердого кристаллического вещества белого цвета (суммарный выход по двум указанным стадиям: 86%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,10 (д, 1H, J=7,7 Гц), 7,47-7,38 (м, 3H), 6,90 (д, 2H, J=8,2 Гц), 6,46 (д, 2H, J=8,3 Гц), 4,95-4,80 (м, 3H), 4,52 (м, 1H), 2,88 (м, 1H), 2,78 (м, 1H), 1,19 (д, 3H, J=6,2 Гц), 1,12 (д, 3H, J=6,2 Гц); 13C-ЯМР (100 МГц, ДМСО-d6): δ 170,63, 163,54, 147,33, 136,33, 131,47, 131,12, 129,77, 128,15, 123,76, 113,90, 68,09, 54,57, 36,39, 21,75, 21,61.
Температура плавления (ДСК): 115,3°С; МС (ESI+, m/z): 395,0 (MH+), 397,1 (MH+).
Пример 5
Синтез гидрохлорида сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{1-метил-6-(N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина с помощью промежуточного соединения с применением формильной группы в качестве защитной группы
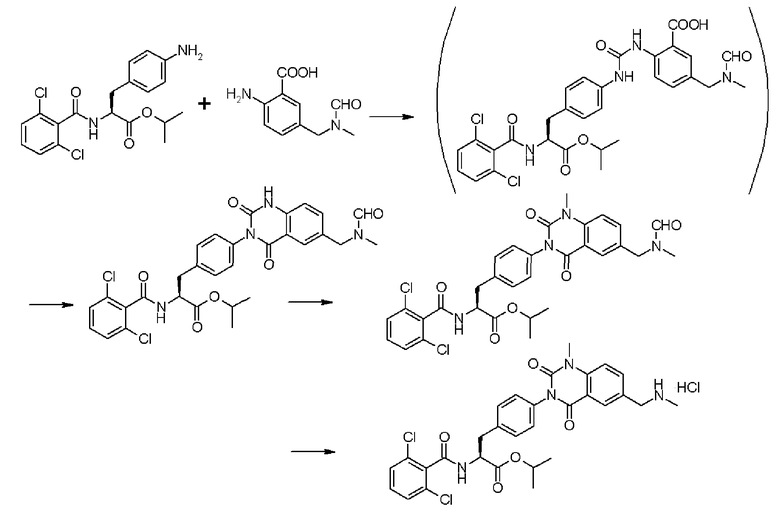
Стадия 1: Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4- [1H,3H]-дион-3-ил}-L-фенилаланина
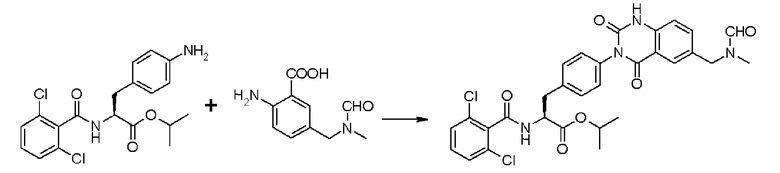
К 7,6 л N,N-диметилформамида в потоке газообразного азота добавляли 1,74 кг 1,1'-карбонилдиимидазола (CDI) и охлаждали смесь до 5°С. В полученный раствор в виде трех порций вводили 4,00 кг сложного изопропилового эфира 4-амино-Nα-(2,6-дихлорбензоил)-L-фенилаланина, в то же время принимая меры по предотвращению избыточного образования тепла. Затем полученную смесь перемешивали при температуре в диапазоне от 5 до 10°С в течение 1,5 часов, затем к смеси добавляли 2,11 кг 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты с последующим нагреванием при 60°С при перемешивании в течение одного часа. После подтверждения с помощью ВЭЖХ протекания реакции образования мочевинной связи реакционный раствор охлаждали до 15°С, в виде трех порций добавляли 1,99 кг 1,1'-карбонилдиимидазола (CDI), в то же время принимая меры по предотвращению появления вспенивания, и перемешивали полученную смесь при 60°С. После подтверждения с помощью ВЭЖХ протекания реакции образования хиназолиндионового цикла в реакционный раствор при 50°С по каплям добавляли 32 л 2-пропанола и затем добавляли к нему 2 г затравочных кристаллов. Полученную суспензию, содержащую продукт, охлаждали до 25°С и дополнительно по каплям добавляли дополнительные 40,01 л 2-пропанола.
Затем суспензию выдерживали в течение ночи при температуре в диапазоне от 10 до 15°С, образовавшееся при этом твердое кристаллическое вещество выделяли фильтрованием с применением центрифуги, и полученный осадок с фильтра промывали 8 л 2-пропанола. Осадок с фильтра сушили при 70°С и пониженном давлении, получая при этом 5,45 кг указанного в заголовке соединения в виде твердого кристаллического вещества белого цвета (выход: 88%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 11,53 (ушир.с, 1H), 9,24 (д, 1H, J=7,9 Гц), 8,33 и 8,15 (два с, 1H), 7,86 и 7,81 (два д, 1H, J=1,8 Гц), 7,61 и 7,56 (два дд, 1H, J=8,3, 1,9 Гц), 7,48-7,36 (м, 5H), 7,26-7,18 (м, 3H), 4,95 (септет, 1H, J=6,2 Гц), 4,76 (ддд, 1H, J=9,8, 8,2, 5,4 Гц), 4,50 и 4,48 (два с, 2H), 3,18 (дд, 1H, J=5,2, 14,0 Гц), 3,01 (дд, 1H, J=14,0, 9,9 Гц), 2,83 и 2,62 (два с, 3H), 1,23 (д, 3H, J=6,2 Гц), 1,19 (д, 3H, J=6,2 Гц); 13C-ЯМР (100 МГц, ДМСО-d6): δ 170,51, 163,67, 162,99, 162,92, 162,21, 150,29, 139,52, 139,25, 137,12, 136,25, 135,14, 134,95, 134,27, 131,61, 131,32, 131,23, 131,14, 129,75, 128,93, 128,13, 126,98, 126,96, 115,92, 115,82, 114,53, 114,48, 68,51, 53,77, 51,52, 46,12, 36,44, 33,71, 28,80, 21,76, 21,68.
Температура плавления (ДСК): 185,8°С; МС (ESI+): m/z 611 (MH+) и 633,1 (M+Na); (ESI-): m/z 609,1 (M-H-); МС (FAB): m/z 611,0 (MH+); HRMS (FAB): рассчитанное для C30H29Cl2N4O6 m/z 611,1464 (MH+), найденное m/z: 611,1461 (MH+).
При таком аспекте асимметричное производное мочевины, которое не выделяют на данной стадии, можно выделить согласно способу, описанному в другом примере.
Стадия 2: Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{1-метил-6-(N-формил-N-метиламинометил)хиназолин-2,4[1H,3H]-дион-3-ил}-L-фенилаланина

К 21,26 л N,N-диметилформамида (DMF) добавляли 5,445 кг сложного изопропилового эфира Nα-(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина, полученную смесь перемешивали при 23°С в потоке газообразного азота, чтобы растворить соединение сложного изопропилового эфира. Затем к полученному раствору добавляли 2,46 кг карбоната калия и 2,491 кг метил-п-толуолсульфоната и перемешивали смесь при нагревании до температуры от 55 до 65°С. После подтверждения с помощью ВЭЖХ протекания реакции N-метилирования к смеси добавляли 2,14 кг уксусной кислоты и затем перемешивали смесь при нагревании до 50°С в течение одного часа. Затем к реакционному раствору по каплям добавляли 10,9 л воды с последующим добавлением 6 г затравочных кристаллов и перемешиванием смеси при 50°С в течение 2 часов. К полученной взвеси дополнительно добавляли 10,9 л воды, смесь охлаждали до 25°С и перемешивали в течение ночи. Полученные при этом осадки извлекали фильтрованием с применением центрифуги, и полученный осадок с фильтра промывали 21,8 кг воды. Полученные влажные кристаллы сушили при 60°С и пониженном давлении, получая при этом 5,235 кг указанного в заголовке соединения в виде кристаллического твердого вещества белого цвета (выход: 94%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,25 (д, 1H, J=8,0 Гц), 8,36 и 8,16 (два с, 1H), 7,97 и 7,92 (два д, 1H, J=2,2 Гц), 7,73 и 7,67 (два дд, 1H, J=8,6, 2,2 Гц), 7,53 и 7,50 (два д, 1H, J=8,7 Гц), 7,47-7,37 (м, 5H), 7,20 (дд, 2H, J=8,4, 2,4 Гц), 4,95 (септет, 1H, J=6,2 Гц), 4,77 (ддд, 1H, J=9,9, 8,1, 5,2 Гц), 4,55 и 4,53 (два с, 2H), 3,53 и 3,52 (два с, 3H), 3,19 (дд, 1H, J=14,1, 5,2 Гц), 3,02 (дд, 1H, J=14,2, 10,0 Гц), 2,84 и 2,63 (два с, 3H), 1,23 (д, 3H, J=6,2 Гц), 1,19 (д, 3H, J=6,2 Гц); 13C-ЯМР (100 МГц, ДМСО-d6): δ 170,54, 163,71, 163,02, 162,94, 161,38, 150,57, 140,47, 140,20, 137,20, 136,27, 135,30, 135,13, 134,82, 131,64, 131,61, 131,54, 131,12, 129,84, 128,75, 128,10, 127,34, 127,30, 115,65, 115,59, 115,40, 115,28, 68,54, 53,75, 51,31, 45,97, 36,47, 33,77, 30,93, 30,90, 28,84, 21,75, 21,68.
Температура плавления (ДСК): 118,1°С; МС (ESI): m/z 625,1 (MH+); МС (FAB): m/z 625,1 (MH+); HRMS (FAB): рассчитанное для C31H31Cl2N4O6 m/z 625,1635 (MH+); найденное m/z: 625,1621 (MH+).
Стадия 3: Синтез гидрохлорида сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{1-метил-6-(N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
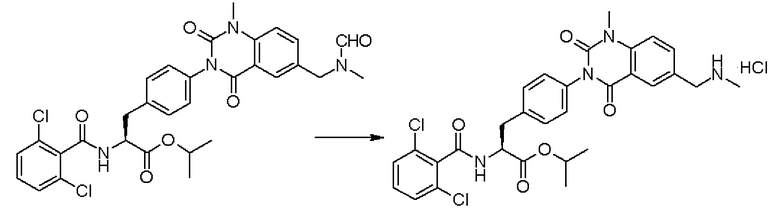
К 61,67 л 2-пропанола добавляли 5,226 кг сложного изопропилового эфира Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина, образуя при этом суспензию, и полученную суспензию охлаждали до 10°С и перемешивали в потоке газообразного азота. К полученной суспензии по каплям добавляли ацетилхлорид (10,00 кг) в течение более одного часа, в то же время принимая меры по предотвращению избыточного образования тепла, затем к суспензии добавляли 37,67 л изопропилацетата, и полученную смесь нагревали при перемешивании при 70°С в течение 24 часов. После подтверждения с помощью ВЭЖХ протекания реакции взвесь охлаждали до 25°С и перемешивали более 19 часов. Полученные осадки извлекали фильтрованием с применением центрифуги и полученный осадок с фильтра промывали 20,90 л 2-пропанола. Полученные влажные кристаллы сушили при 75°С и пониженном давлении, получая при этом 5,159 кг указанного в заголовке соединения в виде твердого кристаллического вещества белого цвета (выход: 95%).
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,45 (ушир.с, 2H), 9,26 (д, 1H, J=8,0 Гц), 8,23 (д, 1H, J=2,1 Гц), 8,02 (дд, 1H, J=2,0, 8,7 Гц), 7,58 (д, 1H, J=8,8 Гц), 7,38-7,47 (м, 5H), 7,21 (д, 2H, J=8,3 Гц), 4,95 (м, 1H), 4,77 (м, 1H), 4,21 (ушир.с, 2H), 3,55 (с, 3H), 3,19 (дд, 1H, J=5,2, 14,1 Гц), 3,02 (дд, 1H, J=10,0, 14,0 Гц), 2,52 (с, 3H), 1,23 (д, 3H, J=6,2 Гц), 1,19 (д, 3H, J=6,2 Гц); 13C-ЯМР (100 МГц, ДМСО-d6): δ 170,54, 163,71, 161,26, 150,59, 141,17, 137,45, 137,25, 136,22, 134,75, 131,60, 131,19, 130,09, 129,88, 128,73, 128,13, 126,68, 115,47, 115,27, 68,55, 53,76, 50,18, 36,44, 31,94, 31,05, 21,77, 21,70.
Температура плавления (ДСК): 254-258°С; МС (ESI): m/z 597,2 (MH+); МС (FAB): m/z 597,1 (MH+); HRMS (FAB): рассчитанное для C30H31Cl2N4O5 m/z 597,1672 (MH+); найденное m/z: 597,1691 (MH+).
Пример для сравнения 1
Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина (реакция с применением сложного эфира замещенной антраниловой кислоты)
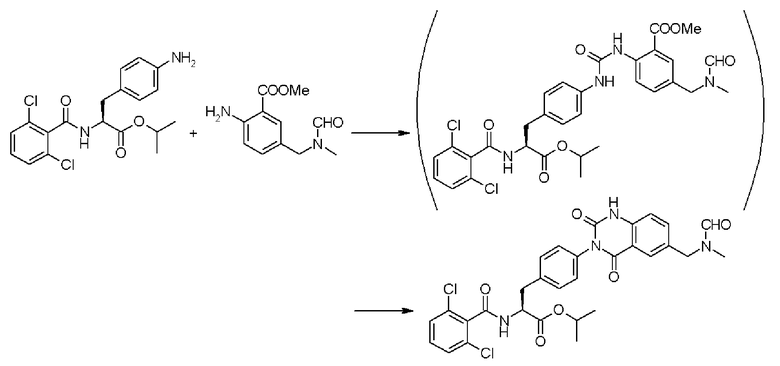
К 3,6 мл N,N-диметилформамида в потоке газообразного азота добавляли 0,79 г N,N'-карбонилдиимидазола (CDI) с образованием взвеси и охлаждали последнюю до 5°С. К полученной жидкости добавляли 1,8 г сложного изопропилового эфира Nα-(2,6-дихлорбензоил)-4-амино-L-фенилаланина двумя порциями, в то же время принимая меры по предотвращению избыточного образования тепла. Затем полученную смесь перемешивали при 10°С в течение одного часа, добавляли к ней 1,01 г метил-2-амино-5-(N-формил-N-метиламинометил)бензоата и перемешивали смесь при 60°С в течение 6 часов. После подтверждения исчезновения исходного материала с помощью ВЭЖХ реакционный раствор охлаждали до 50°С, добавляли к нему 14,4 мл 2-пропанола, также добавляли к нему затравочные кристаллы, осаждая при этом требуемый продукт. К полученной взвеси затем по каплям добавляли 18 мл 2-пропанола, смесь охлаждали до 10°С и перемешивали в течение ночи.
Образовавшееся при этом твердое вещество отделяли фильтрованием при пониженном давлении, промывали 3,6 мл 2-пропанола и затем сушили при 60°С и пониженном давлении, получая при этом 1,52 г (выход: 55%) указанного в заголовке соединения в виде твердого кристаллического вещества белого цвета. Было найдено, что физические свойства полученного соединения находятся в хорошем соответствии со свойствами, обнаруженными для соединения, полученного на стадии 1 примера 5.
Пример 6
Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
Стадия 1: Синтез 2-(3-{4-[2(S)-2-(2,6-дихлорбензоиламино)-2-изопропоксикарбонилэтил]фенил}уреидо)-5-(N-формил-N-метиламинометил)бензойной кислоты
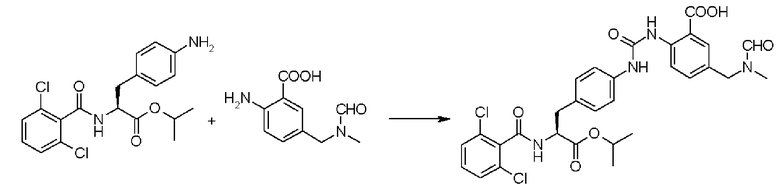
Готовили суспензию из 0,86 г 1,1'-карбонилдиимидазола (CDI) в 3,8 мл N,N-диметилформамида и охлаждали полученную взвесь до 10°С. В полученную взвесь вводили 1,90 г сложного изопропилового эфира 4-амино-Nα-(2,6-дихлорбензоил)-L-фенилаланина; полученную смесь перемешивали при 10°С в течение одного часа, к смеси добавляли 1,0 г 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты с последующим перемешиванием при 60°С в течение 2 часов. Затем к смеси при комнатной температуре добавляли 7,6 мл воды, 1,0 мл 6M водного раствора хлористоводородной кислоты и 1,0 мл 6M водного раствора гидроксида натрия. Полученный раствор постепенно, по каплям добавляли в другую колбу вытянутой (баклажаноподобной) формы, наполненную 3,8 мл воды и 1,1 мл 6M водного раствора хлористоводородной кислоты, выделяя при этом твердое вещество, и образованное таким образом твердое вещество выделяли фильтрованием с отсасыванием. Затем твердое вещество промывали 10 мл воды, сушили при 70°С и пониженном давлении в течение 2 часов, получая при этом 3,02 г указанного в заголовке соединения в виде твердого кристаллического вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6): δ10,41 и 10,37 (два с, 1H), 9,76 и 9,74 (два с, 1H), 9,17 (д, 1H, J=7,9 Гц), 8,46-8,31 (м, 1H), 8,31 и 8,14 (два с, 1H), 7,84 и 7,82 (два д, 1H, J=2,1 Гц), 7,48-7,37 (м, 6H), 7,19 (д, 2H, J=8,6 Гц), 4,92 (септет, 1H, J=6,2 Гц), 4,69-4,61 (м, 1H), 4,43 и 4,41 (два с, 2H), 3,04 (дд, 1H, J=5,7 и 14,2 Гц), 2,90 (дд, 1H, J=9,2, 14,0 Гц), 2,83 и 2,63 (два с, 3H), 1,21 (д, 3H, J=6,2 Гц), 1,15 (д, 3H, J=6,2 Гц); MS (ESI-): m/z 629,2 (M-H-).
МС (ESI-): m/z 629,2 (M-H-).
Стадия 2: Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
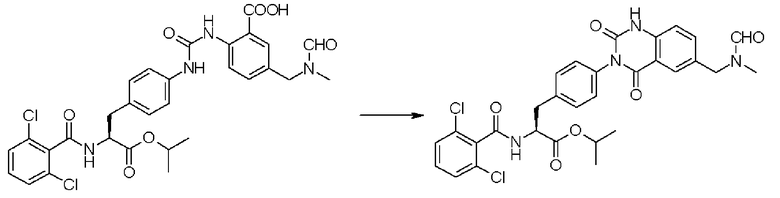
К 500 мг 2-(3-{4-[2(S)-2-(2,6-дихлорбензоиламино)-2-изопропоксикарбонилэтил]фенил}уреидо)-5-(N-формил-N-метиламинометил)бензойной кислоты добавляли 0,63 мл N,N-диметилформамида, получая при этом однородный раствор; затем к раствору добавляли 287 мг 1,1'-карбонилдиимидазола (CDI); полученную смесь нагревали до 60°С и перемешивали в течение 3 часов. После охлаждения реакционного раствора до 50°С добавляли 3,15 мл 2-пропанола и затем к нему добавляли затравочные кристаллы, выделяя при этом твердое вещество. Кроме того, к взвеси по каплям добавляли 2,52 мл 2-пропанола с последующим охлаждением взвеси до 10°С при перемешивании в течение ночи. Выделившееся при этом твердое вещество удаляли фильтрованием при пониженном давлении; полученный осадок с фильтра промывали 2 мл 2-пропанола и затем сушили при 70°С и пониженном давлении, получая при этом 375 мг требуемого продукта в виде твердого вещества белого цвета. Было найдено, что физические свойства полученного соединения находятся в хорошем соответствии со свойствами, обнаруженными для соединения, полученного на стадии 1 примера 5.
Пример 7
Синтез сложного метилового эфира 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты
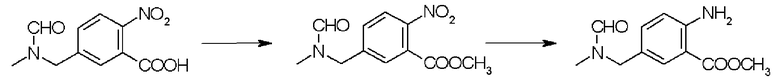
Стадия 1: Синтез сложного метилового эфира 5-(N-формил-N-метиламинометил)-2-нитробензойной кислоты
3,40 г 5-(N-формил-N-метиламинометил)-2-нитробензойной кислоты растворяли в 6,8 мл N,N-диметилформамида; затем добавляли к раствору 3,94 г карбоната калия и 1,77 мл метилйодида и перемешивали раствор при температуре в диапазоне от 20 до 30°С в течение 4 часов. После подтверждения с помощью ВЭЖХ исчезновения исходного материала к реакционному раствору добавляли 20,5 мл воды и 34 мл этилацетата. Органическую фазу отделяли от водной фазы, с последующей ее концентрацией, получая при этом 4,47 г заданного соединения в виде неочищенного продукта (маслянистое состояние).
1H-ЯМР (400 МГц, ДМСО-d6): δ 8,31 и 8,19 (два с, 1H), 8,11 и 8,08 (два д, 1H, J=8,2 Гц), 7,78-7,61 (м, 2H), 4,63 и 4,58 (два с, 2H), 3,86 и 3,85 (два с, 3H), 2,89 и 2,65 (два с, 3H).
Стадия 2: Синтез сложного метилового эфира 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты
4,47 г неочищенного сложного метилового эфира 5-(N-формил-N-метиламинометил)-2-нитробензойной кислоты, полученного на стадии 1, растворяли в 34 мл метанола; добавляли к раствору в атмосфере газообразного аргона 5% Pd/C (влажный продукт) и затем вводили в реакционный контейнер газообразный водород. Реакционную систему перемешивали при температуре реакции 40°С в течение 8 часов; газообразный водород заменяли газообразным аргоном; затем в реакционную систему добавляли 14 мл метанола и повышали ее температуру до 55°С. Катализатор Pd/C удаляли фильтрованием при пониженном давлении; катализатор промывали 25 мл метанола, а полученный фильтрат и промывочную жидкость поддерживали при температуре 40°С. Добавляли к ним по каплям воду (31 мл), выделяя при этом твердое вещество, и полученную взвесь перемешивали при 10°С в течение ночи. Полученное твердое вещество выделяли из взвеси фильтрованием при пониженном давлении, промывали 7 мл воды и затем сушили при 70°С и пониженном давлении в течение 5 часов, получая при этом 2,53 г указанного в заголовке соединения в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6): δ 8,26 и 8,10 (два с, 1H), 7,62 и 7,60 (два д, 1H, J=2,2 Гц), 7,17 и 7,14 (два дд, 1H, J=2,2, 8,6 Гц), 6,80 и 6,77 (два д, 1H, J=8,6 Гц), 6,71 и 6,67 (два ушир.с, 2H), 4,28 (с, 2H), 3,80 и 3,79 (два с, 3H), 2,78 и 2,59 (два с, 3H).
МС (ESI+): m/z 223,3 (MH+), 245,2 (M+Na).
Пример 8
Синтез сложного метилового эфира 2-амино-5-(N-трет-бутоксикарбонил-N-метиламинометил)бензойной кислоты

Стадия 1: Синтез сложного метилового эфира 5-метил-2-нитробензойной кислоты
В 200 мл N,N-диметилформамида растворяли 20,0 г 5-метил-2-нитробензойной кислоты, к полученному раствору добавляли 22,9 г карбоната калия и затем 8,2 мл метилйодида и перемешивали смесь при комнатной температуре в течение 4 часов. В реакционный раствор добавляли 400 мл воды и 200 мл 1M раствора хлористоводородной кислоты, затем экстрагировали 900 мл этилацетата; экстракт промывали 200 мл насыщенного раствора гидрокарбоната натрия и 200 мл насыщенного раствора обычной соли и затем отгоняли растворитель при пониженном давлении, получая при этом 20,6 г указанного в заголовке соединения в виде неочищенного продукта.
МС (ESI+): m/z 196 (MH+).
Стадия 2: Синтез сложного метилового эфира 5-(N-трет-бутоксикарбонил-N-метиламинометил)-2-нитробензойной кислоты
К 1,0 г неочищенного сложного метилового эфира 5-метил-2-нитробензойной кислоты, синтезированного на упомянутой выше стадии 1, добавляли 1,4 г N-бромсукцинимида, 0,5 г бензоилпероксида и 30 мл бензола; полученную смесь кипятили с обратным холодильником при перемешивании в течение 4 часов и отгоняли растворитель при пониженном давлении. Полученный остаток растворяли в 30 мл ацетонитрила, полученный раствор добавляли к 36 мл 2M раствора метиламина в ТГФ и дополнительно перемешивали смесь при комнатной температуре в течение 10 минут. Растворитель отгоняли при пониженном давлении, затем добавляли этилацетат с последующим четырехкратным промыванием его 1M раствором хлористоводородной кислоты. Полученную этилацетатную фазу промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором обычной соли и отгоняли растворитель при пониженном давлении. Полученный при этом неочищенный продукт растворяли в 30 мл ацетонитрила, добавляли к раствору 1,1 г ди-трет-бутилдикарбоната и 1,1 мл триэтиламина и перемешивали полученную смесь при комнатной температуре в течение ночи. Растворитель отгоняли при пониженном давлении, добавляли к нему этилацетат, затем удаляли нерастворимое вещество фильтрованием и полученный при этом фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на силикагеле (гексан:этилацетат = 90:10→80:20), получая при этом 0,71 г указанного в заголовке соединения. МС (ESI+): m/z 325 (MH+).
Стадия 3: Синтез сложного метилового эфира 2-амино-5-(N-трет-бутоксикарбонил-N-метиламинометил)бензойной кислоты
В 50 мл метанола растворяли 2,5 г сложного метилового эфира 5-(N-трет-бутоксикарбонил-N-метиламинометил)-2-нитробензойной кислоты, полученного на упомянутой выше стадии 2; добавляли к раствору 230 мг 10% Pd/C и перемешивали полученную смесь в атмосфере газообразного водорода при комнатной температуре в течение 3 часов. Реакционный раствор фильтровали через целит, промывали метанолом и затем отгоняли растворитель при пониженном давлении, получая при этом 2,26 г указанного в заголовке соединения.
МС (ESI+): m/z 295 (MH+).
Пример 9
Синтез сложного метилового эфира N α -(2,6-дихлорбензоил)-4-изоцианат-L-фенилаланина
Стадия 1: Синтез N α -(2,6-дихлорбензоил)-4-йод-L-фенилаланина
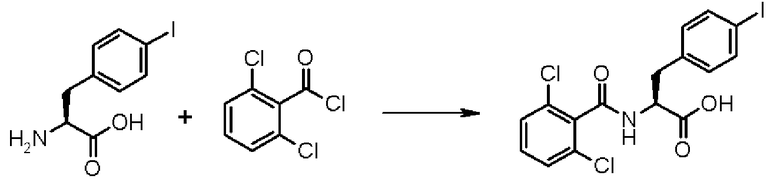
К 7,50 г 4-йод-L-фенилаланина добавляли воду (21 мл), ацетон (9,80 мл) и 6M водный раствор гидроксида натрия (5,20 мл) и перемешивали полученную смесь при 10°С, получая при этом однородный раствор. Альтернативно к полученному раствору добавляли 4,05 мл 2,6-дихлорбензоилхлорида и 4,50 мл 6M водного раствора гидроксида натрия, в то же время принимая меры по поддержанию значения pH не менее 13 и температуры не выше 15°С. После подтверждения завершения реакции в реакционную систему добавляли 6,50 мл 6M водного раствора хлористоводородной кислоты, осаждая при этом требуемый продукт. Выделенное таким образом твердое вещество выделяли фильтрованием при пониженном давлении, промывали 15 мл воды и сушили при 70°С в течение 4 часов при пониженном давлении, получая при этом 12,42 г заданного продукта.
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,06 (д, 1H, J=8,3 Гц), δ 7,63 (д, 2H, J=8,2 Гц), δ 7,48-7,35 (м, 3H), δ 7,11 (д, 2H, J=8,2 Гц), δ 4,68 (ддд, 1H, J=10,0, 8,4, 4,8 Гц), δ 3,11 (дд, 1H, J=14,0, 4,8 Гц), δ 2,87 (дд, 1H, J=14,0, 10,0 Гц).
MC (ESI+): m/z 463,9 (MH+) и 485,9 (M+Na), (ESI-): m/z 461,8 (M-H-).
Стадия 2: Синтез сложного метилового эфира N α -(2,6-дихлорбензоил)-4-иод-L-фенилаланина
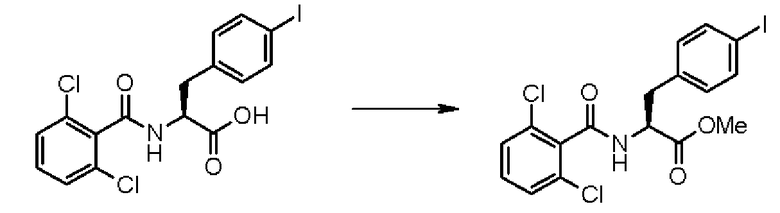
К 8,0 г Nα-(2,6-дихлорбензоил)-4-йод-L-фенилаланина добавляли метанол (40 мл) и 0,98 мл концентрированной серной кислоты и проводили реакцию в течение 4 часов, нагревая реакционную систему при 40°С. После подтверждения завершения реакции реакционную систему охлаждали до 10°С и по каплям добавляли 20 мл воды, выделяя при этом твердое вещество. Выпадающее в осадок твердое вещество выделяли фильтрованием при пониженном давлении, промывали 40 мл воды и затем сушили при 70°С и пониженном давлении, получая при этом 7,40 г требуемого продукта в виде твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,18 (д, 1H, J=8,1 Гц), 7,64 (д, 2H, J=8,1 Гц), 7,48-7,35 (м, 3H), 7,11 (д, 2H, J=8,2 Гц), 4,75 (ддд, 1H, J=10,0, 8,1, 4,9 Гц), 3,67 (с, 3H), 3,11 (дд, 1H, J=14,1, 5,1 Гц), 2,90 (дд, 1H, J=14,0, 10,2 Гц).
МС (ESI+): m/z 478,0 (MH+), (ESI-): m/z 475,9 (M-H-).
Стадия 3: Синтез сложного метилового эфира 4-карбоксил-N α -(2,6-дихлорбензоил)-L-фенилаланина
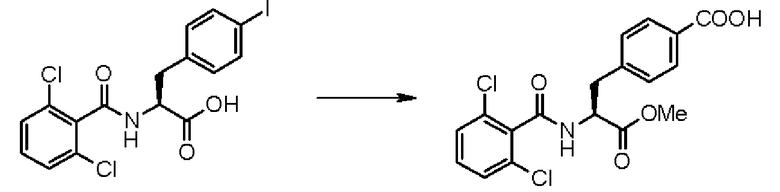
В пробирке, снабженной навинчивающейся крышкой, смешивали 0,5 мл DMF, 122 мг формиата натрия, 206 мкл диизопропилэтиламина и 113 мкл уксусного ангидрида и перемешивали смесь при комнатной температуре в течение 10 минут. Затем к полученной смеси добавляли 288 мг сложного метилового эфира Nα-(2,6-дихлорбензоил)-4-йод-L-фенилаланина, 32 мг 10% Pd/C, 76 мг хлорида лития и 0,4 мл DMF; полученную смесь дополнительно перемешивали, в то же время повышая температуру до 80°С; к смеси дополнительно добавляли 41 мг формиата натрия и 57 мкл уксусного ангидрида с последующим дополнительным перемешиванием. После подтверждения завершения реакции Pd/C удаляли фильтрованием и промывали 0,6 мл 6M раствора хлористоводородной кислоты, 0,3 мл воды и 1,3 мл DMF. К реакционному раствору при комнатной температуре по каплям добавляли воду (3,5 мл); к реакционному раствору дополнительно добавляли затравочные кристаллы, выделяя при этом твердое вещество, и охлаждали полученную суспензию до 10°С. Полученные осадки выделяли фильтрованием при пониженном давлении, промывали 3 мл воды и затем сушили при 60°С и пониженном давлении, получая при этом 217 мг грубо очищенного продукта в виде твердого вещества. Затем неочищенный продукт перекристаллизовывали из ацетонитрила, получая при этом 71 мг требуемого очищенного продукта.
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,22 (д, 1H, J=8,1 Гц), 7,86 (д, 2H, J=8,4 Гц), 7,48-7,35 (м, 5H), 4,81 (ддд, 1H, J=10,2, 8,3, 5,1 Гц), 3,67 (с, 3H), 3,23 (дд, 1H, J=14,0, 4,9 Гц), 3,02 (дд, 1H, J=14,0, 10,2 Гц).
МС (ESI+): m/z 396,1 (MH+), 418,1 (M+Na); МС (ESI-): m/z 394,1 (M-H-).
Стадия 4: Синтез сложного метилового эфира N α -(2,6-дихлорбензоил)-4-изоцианат-L-фенилаланина
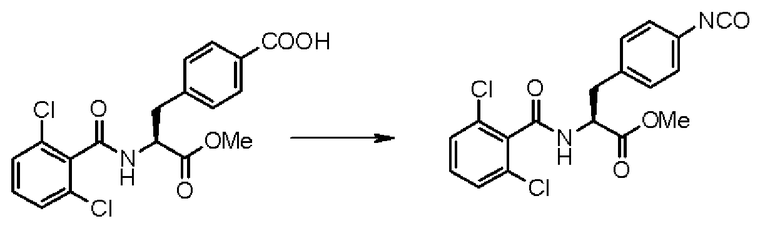
К 100 мг сложного метилового эфира 4-карбоксил-Nα-(2,6-дихлорбензоил)-L-фенилаланина добавляли 2 мл 1,2-диметоксиэтана (обезвоженного), 54 мкл триэтиламина и 55 мкл дифенилфосфорилазида (DPPA) и перемешивали полученную смесь при комнатной температуре в течение одного часа. Затем температуру смеси повышали до уровня в диапазоне от 80 до 90°С, перемешивали в течение 3 часов и затем удаляли растворитель перегонкой при пониженном давлении. Полученный таким образом маслянистый неочищенный продукт очищали с помощью колоночной флэш-хроматографии на силикагеле (простой диэтиловый эфир/н-гексан = 1/1→чистый диэтиловый эфир) и отгоняли растворитель при пониженном давлении, получая при этом требуемый продукт.
1H-ЯМР (400 МГц, CDCl3): δ 7,35-7,23 (м, 3H), 7,20-7,15 (м, 2H), 7,04-6,98 (м, 2H), 6,31 (ушир.д, 1H, J=7,3 Гц), 5,16 (дт, 1H, J=8,0, 5,8 Гц), 3,76 (с, 3H), 3,27 (дд, 1H, J=14,2, 5,7 Гц), 3,23 (дд, 1H, J=14,2, 5,7 Гц).
MC (ESI+): m/z 393,0, 415,0 (M+Na); MC (ESI-): m/z 391,1 (M-H-); IR (KBr), см-1: 2268,1(с), 1743,5(м), 1654,8(м), 1581,5(м), 1539,1(м), 1519,8(м).
Пример 10
Синтез сложного метилового эфира N α -(2,6-дихлорбензоил)-4-{6-диметиламинохиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
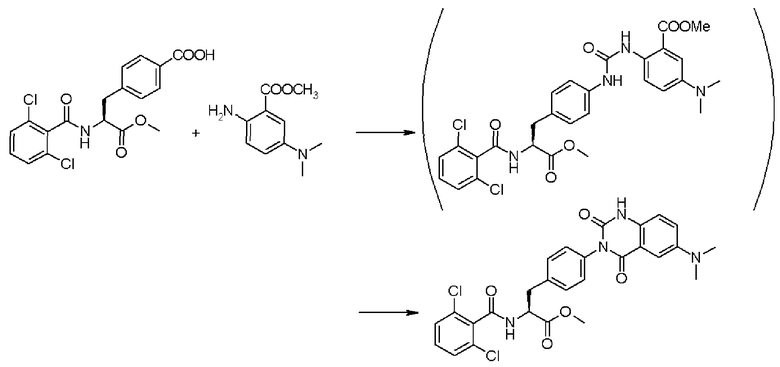
К 100 мг сложного метилового эфира 4-карбоксил-Nα-(2,6-дихлорбензоил)-L-фенилаланина добавляли диметоксиэтан (DME, 1 мл), триэтиламин (53 мкл) и дифенилфосфорилазид (DPPA, 55 мкл), полученную смесь нагревали до 40°С и перемешивали, по меньшей мере, более одного часа. К полученному реакционному раствору добавляли 63,7 мг дигидрохлорида сложного метилового эфира 2-амино-5-диметиламинобензойной кислоты, 71 мкл триэтиламина и 0,5 мл DME с образованием взвеси, и перемешивали полученную взвесь в течение не менее 3 часов при нагревании смеси при температуре в диапазоне от 80 до 90°С. Реакционный раствор концентрировали, затем добавляли к нему 5 мл воды, и дважды экстрагировали реакционный раствор этилацетатом (каждый раз по 5 мл). Полученную органическую фазу концентрировали досуха, получая при этом 0,4 мл N,N-диметилформамида в виде однородной дисперсии, затем к дисперсии добавляли 2 мл метанола, выделяя при этом твердое вещество, и перемешивали смесь при 10°С в течение ночи. Выделившееся твердое вещество извлекали фильтрованием при пониженном давлении, промывали 5 мл метанола и затем сушили при 70°С и пониженном давлении в течение 6 часов, получая при этом 75 мг требуемого соединения в виде твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, ДМСО-d6): δ 11,22 (с, 1H), 9,28 (д, 1H, J=8,1 Гц), 7,48-7,35 (м, 5H), 7,27 (дд, 1H, J=9,0, 2,8 Гц), 7,21-7,09 (м, 4H), 4,80 (ддд, 1H, J=10,0, 8,1, 4,9 Гц), 3,69 (с, 3H), 3,22 (дд, 1H, J=4,6, 14,2 Гц), 3,02 (дд, 1H, J=10,2, 13,8 Гц), 2,91 (с, 6H); 13C-ЯМР (400 МГц, ДМСО-d6): δ 171,70, 163,99, 162,75, 150,18, 146,83, 137,15, 136,34, 134,80, 131,78, 131,36, 131,15, 129,84, 129,18, 128,32, 122,05, 116,48, 115,03, 108,50, 53,70, 52,29, 40,93, 36,36.
МС (ESI+): m/z 555,1 (MH+) и 577,2 (M+Na); МС (ESI-): m/z 553,2 (M-H-).
В данном контексте полученное здесь соединение можно подвергать N-метилированию согласно способу, описанному в патентном документе 2 (WO 2004/74264), получая при этом соединение, представленное следующей структурной формулой:
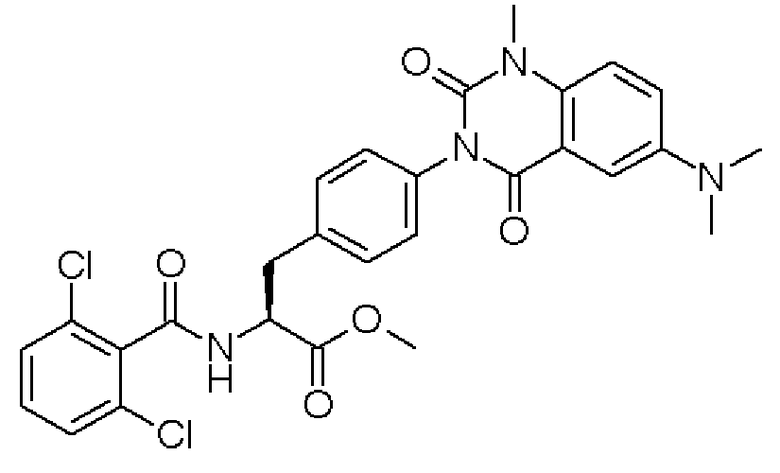
Пример 11
Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
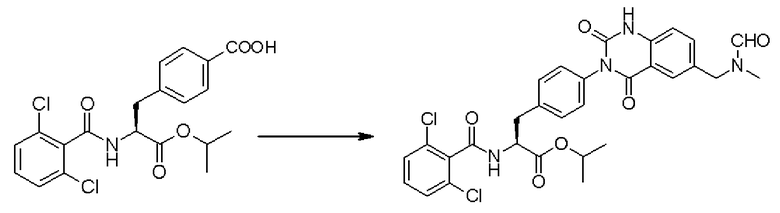
К 3,0 г сложного изопропилового эфира 4-карбоксил-Nα-(2,6-дихлорбензоил)-L-фенилаланина добавляли ацетонитрил (80 мл), триэтиламин (4,93 мл) и дифенилфосфорилазид (DPPA, 1,52 мл) и перемешивали полученную смесь в течение одного часа при нагревании ее до 60°С. К полученному реакционному раствору добавляли 1,47 г 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты и затем перемешивали смесь в течение не менее 12 часов при нагреванием смеси до 90°С. Затем добавляли к смеси 2,29 г 1,1'-карбонилдиимидазола (CDI) с последующим перемешиванием смеси в течение не менее 1,5 часов. После завершения реакции реакционный раствор концентрировали с последующим добавлением 500 мл этилацетата и 500 мл 1M раствора хлористоводородной кислоты и выделением с помощью экстракции. Полученную таким образом органическую фазу последовательно промывали смешанной жидкостью, содержащей 500 мл воды и 100 мл насыщенного раствора бикарбоната натрия, 300 мл насыщенного раствора бикарбоната натрия и затем 300 мл насыщенного водного раствора хлорида натрия и сушили над безводным сульфатом натрия. Затем органическую фазу концентрировали досуха, полученную взвесь промывали простым диэтиловым эфиром, полученное твердое вещество выделяли фильтрованием при пониженном давлении и затем промывали простым диэтиловым эфиром. Затем сушили твердое вещество при пониженном давлении, получая при этом 3,25 г заданного продукта в виде твердого вещества белого цвета. Было найдено, что физические свойства полученного соединения находятся в хорошем соответствии со свойствами, обнаруженными для аналогичного соединения, полученного в упомянутом выше примере синтеза.
Пример 12
Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{6-(N-трет-бутоксикарбонил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина

К 250 мг сложного изопропилового эфира Nα-(2,6-дихлорбензоил)-4-карбоксил-L-фенилаланина добавляли 20 мл ацетонитрила, 411 мкл триэтиламина и 127 мкл дифенилфосфорилазида (DPPA) и перемешивали полученную смесь в течение одного часа при нагревании ее до 60°С. К реакционному раствору добавляли 165 мг 2-амино-5-(N-трет-бутоксикарбонил-N-метиламинометил)бензойной кислоты и затем перемешивали смесь в течение 2 часов при нагревании смеси до 90°С. Затем добавляли 96 мг 1,1'-карбонилдиимидазола (CDI) с последующим перемешиванием смеси в течение ночи. После завершения реакции реакционный раствор подвергали выделению и очистке колоночной хроматографией с обращенной фазой, получая при этом 229 мг заданного продукта в виде твердого вещества. МС (ESI): m/z 683 (MH+).
Пример 13: Синтез 5-(N-формил-N-метиламинометил)изатового ангидрида
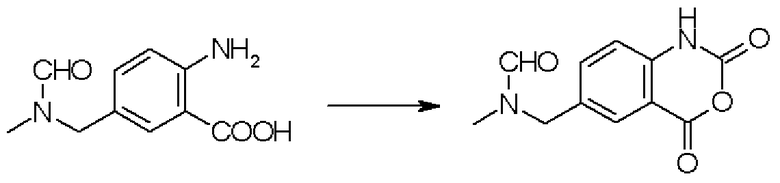
Готовили суспензию из 3,82 г 1,1'-карбонилдиимидазола (CDI) в 15,2 мл N,N-диметилформамида и охлаждали полученную суспензию до 5°С. К полученной взвеси двумя порциями добавляли 4,0 г 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты с последующим перемешиванием взвеси в течение одного часа. После подтверждения с помощью ВЭЖХ исчезновения исходного материала к взвеси добавляли затравочные кристаллы, в то же время добавляя по каплям 40,3 мл 1M водного раствора хлористоводородной кислоты для выделения твердого вещества, и полученное твердое вещество выделяли фильтрованием при пониженном давлении. После промывания твердого вещества 120 мл воды его сушили при 70°С в течение 13 часов при пониженном давлении, получая при этом 3,20 г указанного в заголовке соединения в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6): δ 11,82 (ушир.с, 1H), 8,32 и 8,15 (два с, 1H), 7,84 и 7,78 (два д, 1H, J=1,7 Гц), 7,65 и 7,60 (два дд, 1H, J=2,0, 8,4 Гц), 7,17 и 7,15 (два д, 1H, J=8,4 Гц), 4,49 и 4,47 (два с, 2H), 2,84 и 2,62 (два с, 3H).
МС (ESI+): m/z 235 (MH+), (ESI-): m/z 233,1 (M-H-).
Пример 14
Синтез 5-диметиламиноизатового ангидрида

Готовили суспензию из 144 мг 1,1'-карбонилдиимидазола (CDI) в 0,76 мл N,N-диметилформамида и охлаждали полученную суспензию до 5°С. К полученной взвеси добавляли 200 мг гидрохлорида 2-амино-5-диметиламинобензойной кислоты с последующим перемешиванием взвеси в течение одного часа или более, дополнительным добавлением 0,76 мл N,N-диметилформамида и 66 мг 1,1'-карбонилдиимидазола (CDI) и перемешиванием смеси в течение одного дополнительного часа. Выделившееся из суспензии твердое вещество выделяли фильтрованием при пониженном давлении, промывали 2 мл метанола и затем сушили при 60°С и пониженном давлении, получая при этом 91 мг требуемого продукта в виде твердого вещества желтого цвета.
1H-ЯМР (400 МГц, ДМСО-d6): δ 11,42 (ушир.с, 1H), δ 7,28 (дд, 1H, J=9,1, 3,0 Гц), 7,05 (д, 1H, J=8,7 Гц), 7,04 (д, 1H, J=3,2 Гц), 2,91 (с, 6H).
МС (ESI+): m/z 207,2 (M+H+) и 248,2 (M+MeCN) и 270,2 (M+MeCN+Na); (ESI-): m/z 205,1 (M-H-).
Пример 15
Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
Способ 1: Непрерывный способ осуществляют без выделения промежуточного амидного продукта

Готовили суспензию из 1,64 г 1,1'-карбонилдиимидазола (CDI) в 7,6 мл N,N-диметилформамида и охлаждали суспензию до 10°С. К полученной суспензии добавляли 2,0 г 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты и перемешивали смесь при 10°С. После подтверждения с помощью ВЭЖХ образования изатового ангидрида добавляли 3,80 г сложного изопропилового эфира 4-амино-Nα-(2,6-дихлорбензоил)-L-фенилаланина, проводя при этом амидирование при 60°С. После подтверждения с помощью ВЭЖХ завершения реакции реакционную систему охлаждали до 25°С, дополнительно добавляли 1,96 г 1,1'-карбонилдиимидазола (CDI) и снова нагревали смесь до 60°С. По истечении 2 часов и после подтверждения с помощью ВЭЖХ завершения реакции реакционную систему охлаждали до температуры в диапазоне от 50 до 40°С, добавляли к ней затравочные кристаллы, в тоже время добавляя по каплям 30,4 мл 2-пропанола, осаждая при этом заданный продукт. Дополнительно в реакционную систему добавляли по каплям дополнительные 38,0 мл 2-пропанола, полученную смесь перемешивали при 9°С в течение ночи, выделившееся твердое вещество выделяли фильтрованием при пониженном давлении и промывали 11,4 мл 2-пропанола.
Затем твердое вещество сушили при 60°С в течение 3 часов, получая при этом 3,51 г указанного в заголовке соединения в виде твердого кристаллического вещества белого цвета. Было найдено, что физические свойства полученного соединения находятся в хорошем соответствии со свойствами, обнаруженными для аналогичного соединения, полученного в упомянутом выше примере синтеза.
При таком аспекте промежуточный амидный продукт, который не выделен на данной стадии, можно выделять аналогичным образом согласно способу, отдельно описанному в другом примере.
Пример 16
Синтез сложного метилового эфира N α -(2,6-дихлорбензоил)-4-{6-диметиламинохиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
Способ 1: Непрерывный способ осуществляют без выделения промежуточного амида
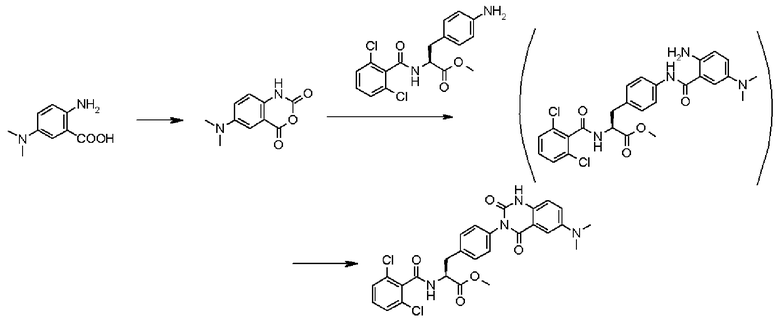
Готовили суспензию из 1,89 г 1,1'-карбонилдиимидазола (CDI)в 32 мл N,N-диметилформамида и охлаждали суспензию до 10°С. К полученной взвеси добавляли 1,96 г 2-амино-5-диметиламинобензойной кислоты и затем перемешивали ее при температуре в диапазоне от 10 до 25°С в течение 2 часов. Затем к смеси добавляли 4,00 г сложного метилового эфира Nα-(2,6-дихлорбензоил)-4-амино-L-фенилаланина и перемешивали смесь при 60°С в течение 2 часов. К полученному раствору добавляли 2,16 г 1,1'-карбонилдиимидазола (CDI), перемешивали смесь при 60°С в течение ночи и добавляли к смеси затравочные кристаллы при температуре в диапазоне от 60 до 10°С, в то же время добавляя к ней по каплям 160 мл метанола для выделения при этом твердого вещества. Полученную взвесь перемешивали при 10°С в течение ночи, твердое вещество выделяли фильтрованием при пониженном давлении, промывали 16 мл метанола и затем сушили при 70°С в течение 5 часов при пониженном давлении, получая при этом 1,84 г заданного соединения в виде твердого вещества светло-желтого цвета. Было найдено, что физические свойства полученного соединения находятся в хорошем соответствии со свойствами, обнаруженными для аналогичного соединения, полученного в упомянутом выше примере синтеза.
Пример 17
Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина (Способ 2, при котором выделяют промежуточный амидный продукт)
Стадия 1: Синтез сложного изопропилового эфира 4-{2-амино-5-(N-формил-N-метиламинометил)бензоиламино}-N α -(2,6-дихлорбензоил)-L-фенилаланина
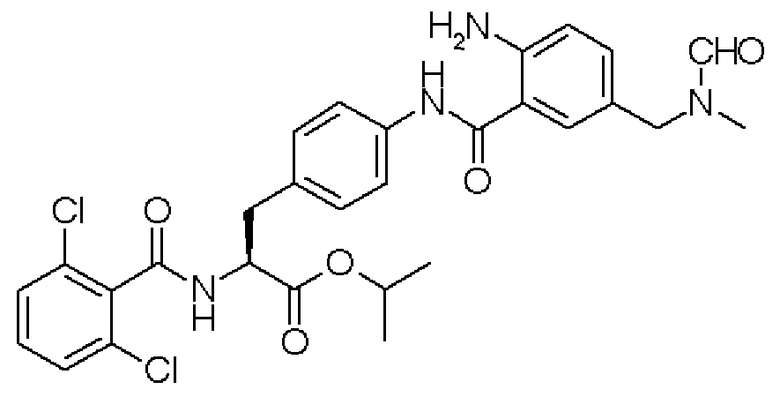
Готовили суспензию из 1,67 г 1,1'-карбонилдиимидазола (CDI) в 7,6 мл N,N-диметилформамида и охлаждали суспензию до 5°С. К полученной взвеси добавляли 2,0 г 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты для проведения при этом реакции образования производного изатового ангидрида при 5°С. После подтверждения с помощью ВЭЖХ завершения реакции в реакционную систему добавляли 3,80 г сложного изопропилового эфира 4-амино-Nα-(2,6-дихлорбензоил)-L-фенилаланина, проводя при этом амидирование при 60°С в течение ночи. После подтверждения с помощью ВЭЖХ завершения реакции реакционную систему охлаждали до температуры в диапазоне от 25 до 50°С с последующим добавлением 3 мл воды. Полученный раствор медленно, по каплям добавляли в другую колбу вытянутой формы (баклажаноподобной), в которую предварительно было добавлено 29 мл воды, для образования твердого материала, и затем выделившееся твердое вещество выделяли фильтрованием при пониженном давлении. После промывания твердого вещества 20 мл воды его сушили при пониженном давлении, получая при этом 5,50 г заданного соединения в виде неочищенного продукта. К неочищенному продукту добавляли 5,5 мл ацетонитрила для получения однородной дисперсии, добавляли к ней 137,5 мл 2-пропанола и затравочные кристаллы для осаждения твердого материала и перемешивали взвесь при 20°С в течение 2 часов. Выделившееся твердое вещество выделяли фильтрованием при пониженном давлении, промывали 20 мл 2-пропанола и затем сушили при пониженном давлении, получая при этом 2,60 г заданного продукта в виде твердого кристаллического вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,97 и 9,95 (два с, 1H), 9,19 (д, 1H, J=8,0 Гц), 8,27 и 8,10 (два с, 1H), 7,65-7,35 (м, 6H), 7,29-7,22 (м, 2H), 7,14-7,03 (м, 1H), 6,79-6,71 (м, 1H), 6,35 и 6,27 (два ушир.с, 2H), 4,92 (септет, 1H, J=6,2 Гц), 4,72-4,63 (м, 1H), 4,32 и 4,29 (два с, 2H), 3,07 (дд, 1H, J=14,0, 5,7 Гц), 2,94 (дд, 1H, J=14,0, 9,2 Гц), 2,81 и 2,64 (два с, 3H), 1,21 (д, 3H, J=6,4 Гц), 1,16 (д, 3H, J=6,4 Гц).
МС (ESI+): m/z 585,0 (MH+), (ESI-): m/z 583,2 (M-H-).
Стадия 2: Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
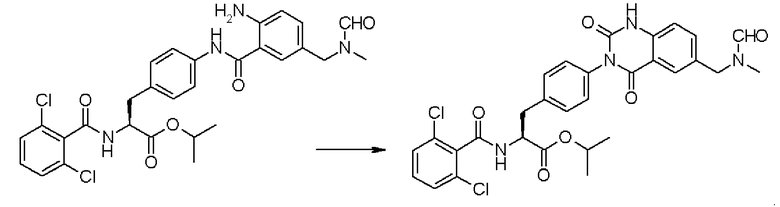
К 1,0 г сложного изопропилового эфира 4-{2-амино-5-(N-формил-N-метиламинометил)бензоиламино}-Nα-(2,6-дихлорбензоил)-L-фенилаланина добавляли 1,34 мл N,N-диметилформамида для образования при этом однородной дисперсии, затем к дисперсии добавляли 0,34 г 1,1'-карбонилдиимидазола (CDI) и полученную смесь нагревали при перемешивании при 60°С в течение не менее одного часа.
После охлаждения смеси до 50°С добавляли 6,7 мл 2-пропанола и затравочные кристаллы для выделения твердого материала. Дополнительно добавляли по каплям 5,36 мл 2-пропанола, получая при этом взвесь, и затем охлаждали до 10°С при перемешивании в течение ночи. Выделившееся твердое вещество выделяли фильтрованием при пониженном давлении, промывали 2 мл 2-пропанола и затем сушили при 70°С и пониженном давлении, получая при этом 0,85 г указанного в заголовке соединения в виде твердого вещества белого цвета. Было найдено, что физические свойства полученного соединения находятся в хорошем соответствии со свойствами, обнаруженными для аналогичного соединения, полученного в упомянутом выше примере синтеза.
Пример 18
Синтез сложного метилового эфира 4-{2-амино-6-диметиламинобензоиламино}-N α -(2,6-дихлорбензоил)-L-фенилаланина (Способ 2, при котором выделяют промежуточный амидный продукт):
Стадия 1: Синтез сложного метилового эфира 4-{2-амино-5-диметиламинобензоиламино}-N α -(2,6-дихлорбензоил)-L-фенилаланина

Указанное в заголовке соединение синтезировали путем взаимодействия сложного метилового эфира 4-амино-Nα-(2,6-дихлорбензоил)-L-фенилаланина с изатовым ангидридом, образованным с помощью реакции 2-амино-5-диметиламинобензойной кислоты с 1,1'-карбонилдиимидазолом (CDI).
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,96 (с, 1H), 9,22 (д, 1H, J=7,9 Гц), 7,61 (д, 2H, J=8,5 Гц), 7,48-7,38 (м, 3H), 7,23 (д, 2H, J=8,5 Гц), 6,95 (д, 1H, J=2,7 Гц), 6,88-6,84 (м, 1H), 6,69 (д, 1H, J=8,8 Гц), 5,62 (ушир.с, 2H), 4,74-4,68 (м, 1H), 3,66 (с, 3H), 3,12-3,06 (м, 1H), 2,98-2,91 (м, 1H), 2,79 (с, 6H); 13C-ЯМР (100 МГц, ДМСО-d6): δ 36,46, 42,13, 52,20, 54,11, 113,86, 117,09, 118,04, 120,32, 120,78, 128,37, 129,55, 131,39, 131,65, 132,21, 136,36, 138,17, 141,80, 142,25, 163,90, 168,33, 171,65.
Стадия 2: Синтез сложного метилового эфира N α -(2,6-дихлорбензоил)-4-{6-диметиламинохиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
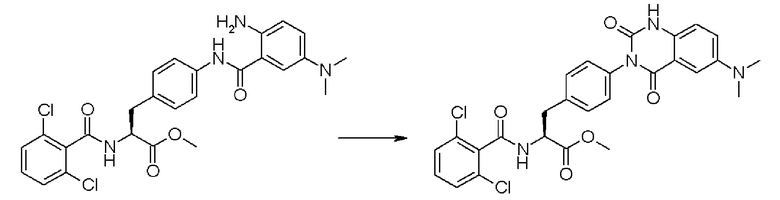
Готовили суспензию из 47,1 г сложного метилового эфира 4-{2-амино-5-диметиламинобензоиламино}-Nα-(2,6-дихлорбензоил)-L-фенилаланина, синтезированного на упомянутой выше стадии 1, в 480 мл ацетонитрила, затем к суспензии добавляли 15,9 г 1,1'-карбонилдиимидазола (CDI) и нагревали смесь при температуре в диапазоне от 55 до 60°С в течение 2,5 часов при перемешивании. После завершения реакции смесь охлаждали до 5°С и отфильтровывали полученные кристаллы, получая при этом 35,6 г первичных кристаллов. Фильтрат и промывочную жидкость концентрировали наполовину, затем добавляли к этому 200 мл воды, выделяя при этом кристаллы, полученную взвесь фильтровали и полученные кристаллы сушили, получая при этом 6,35 г дополнительных кристаллов. Было найдено, что физические свойства полученного соединения находятся в хорошем соответствии со свойствами, обнаруженными для аналогичного соединения, полученного в упомянутом выше примере синтеза.
Пример 19
Синтез 2-этокси-6-(N-формил-N-метиламинометил)-3,1-бензоксазин-4-она
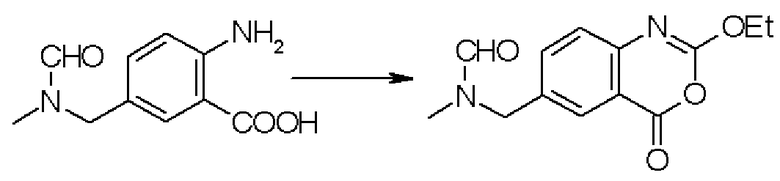
К 6,0 г 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты добавляли 30 мл пиридина и затем к смеси добавляли по каплям 11,1 мл этилхлорформиата, в то же время охлаждая смесь льдом. После истечения ночи к смеси по каплям добавляли 20 мл воды для выделения твердого вещества, затем твердое вещество выделяли фильтрованием при пониженном давлении и промывали 20 мл воды. Затем выделенное таким образом твердое вещество сушили при пониженном давлении, получая при этом 3,97 г заданного соединения в виде твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3): δ 8,33 и 8,19 (два с, 1H), 7,98 и 7,96 (два ушир.с, 1H), 7,65 и 7,57 (два дд, 1H, J=2,2, 8,4 Гц), 7,44 и 7,40 (два д, 1H, J=8,2 Гц), 4,59 и 4,47 (два с, 2H), 4,56-4,49 (м, 2H), 2,90 и 2,79 (два с, 3H), 1,50-1,43 (м, 3H).
МС (ESI+): m/z 263 (MH+) и 285,1 (M+Na).
Пример 20
Синтез 6-диметиламино-2-этокси-3,1-бензоксазин-4-она
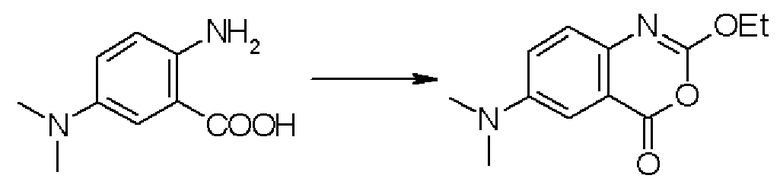
К 2,0 г 2-амино-5-диметиламинобензойной кислоты добавляли 10 мл ацетонитрила и 4,49 мл пиридина и нагревали раствор до 20°С. К полученному раствору постепенно добавляли 3,19 мл этилхлорформиата и затем перемешивали в течение не менее одного часа. После подтверждения завершения реакции к раствору по каплям добавляли 20 мл воды, обеспечивая выделение твердого материала, и охлаждали смесь до 10°С. После выделения твердого вещества, выделенного, таким образом, фильтрованием при пониженном давлении, его промывали 20 мл воды и затем сушили при 70°С и пониженном давлении, получая при этом 2,35 г заданного продукта в виде твердого вещества желтого цвета.
1H-ЯМР (400 МГц, ДМСО-d6): δ 7,31 (м, 2H), 7,12-7,07 (м, 1H), 4,40 (кв, 2H, J=7,1 Гц), 2,96 (с, 6H), 1,35 (т, 3H, J=7,2 Гц).
МС (ESI+): m/z 235,1 (M+H+), 276,2 (M+MeCN) и 298,2 (M+MeCN+Na).
Пример 21
Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{1-метил-5-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
Стадия 1: Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{2-этоксикарбониламино-5-(N-формил-N-метиламинометил)бензоиламино}-L-фенилаланина:
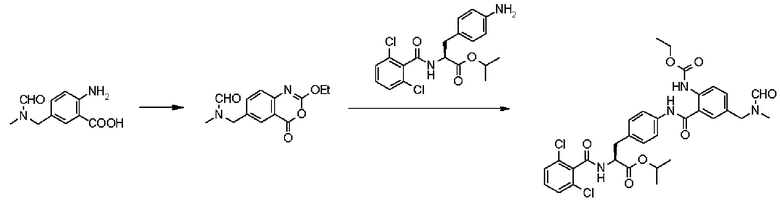
К 2,0 г 2-амино-5-(N-формил-N-метиламинометил)бензойной кислоты добавляли 10 мл ацетонитрила и 3,9 мл пиридина с образованием взвеси и охлаждали последнюю до 10°С. К полученной взвеси по каплям добавляли 2,76 мл этилхлорформиата более 5 минут и затем перемешивали в течение одного часа. После подтверждения с помощью ВЭЖХ исчезновения исходного материала к реакционному раствору добавляли 1,46 мл 2-пропанола для того, чтобы не вступивший в реакцию этилхлорформиат разложился при 25°С. К полученному реакционному раствору добавляли 3,80 г сложного изопропилового эфира 4-амино-Nα-(2,6-дихлорбензоил)-L-фенилаланина и перемешивали полученную смесь в течение ночи. Затем к смеси по каплям добавляли 30 мл 2-пропанола с последующим перемешиванием смеси при 10°С в течение 3 часов. Выделившееся твердое вещество выделяли фильтрованием при пониженном давлении, промывали 10 мл 2-пропанола и затем сушили при 60°С в течение 18 часов при пониженном давлении, получая при этом 4,35 г заданного продукта в виде твердого кристаллического вещества белого цвета.
1H-ЯМР (400 МГц, ДМСО-d6): δ 10,42 (с, 1H), 10,11 и 10,02 (два с, 1H), 9,20 (д, 1H, J=7,9 Гц), 8,31 и 8,15 (два с, 1H), 8,06 и 8,02 (два д, 1H, J=8,4 Гц), 7,72 и 7,66 (два д, 1H, J=1,7 Гц), 7,63-7,58 (м, 2H), 7,49-7,35 (м, 4H), 7,32-7,26 (м, 2H), 4,93 (септет, 1H, J=6,4 Гц), 4,73-4,65 (м, 1H), 4,47 и 4,46 (два с, 2H), 4,11 и 4,11 (два кв, 2H, J=7,2 Гц), 3,10 (дд, 1H, J=14,1, 5,9 Гц), 2,96 (дд, 1H, J=9,2, 14,0 Гц), 2,86 и 2,67 (два с, 3H), 1,22 и 1,21 (два т, 3H, J=7,2 Гц), 1,22 (д, 3H, J=6,0 Гц), 1,17 (д, 3H, J=6,4 Гц).
МС (ESI+): m/z 657,1 (MH+), (ESI-): m/z 655,2.
Стадия 2: Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{1-метил-6-(N-формил-N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
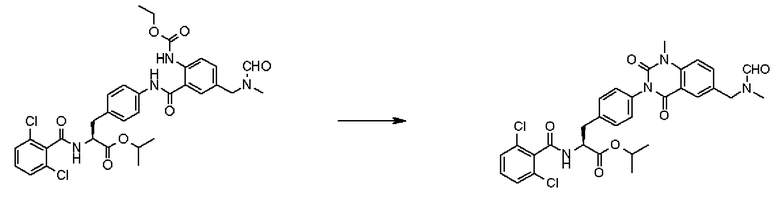
К 17,1 г сложного изопропилового эфира Nα-(2,6-дихлорбензоил)-4-{2-этоксикарбониламино-5-(N-формил-N-метиламинометил)бензоиламино}-L-фенилаланина, полученного на упомянутой выше стадии 1, добавляли 68 мл N,N-диметилформамида, 6,8 мл 2-пропанола и 7,55 г карбоната калия, и затем к полученной смеси добавляли 5,89 мл метил-п-толуолсульфоната с последующим перемешиванием смеси при комнатной температуре в течение ночи. После подтверждения с помощью ВЭЖХ завершения реакции добавляли 6,25 мл уксусной кислоты, чтобы погасить смесь, и затем к смеси по каплям добавляли 84 мл воды для выделения твердого материала. Выделившееся твердое вещество выделяли фильтрованием при пониженном давлении, промывали 82 мл воды и затем сушили при пониженном давлении, получая при этом 15,80 г заданного соединения в виде твердого кристаллического вещества белого цвета. Было найдено, что физические свойства полученного соединения находятся в хорошем соответствии со свойствами, обнаруженными для аналогичного соединения, полученного в упомянутом выше примере синтеза.
В таком контексте указанное соединение можно подвергать деформилированию согласно способу, подробно описанному на стадии 3 примера 5, тем самым превращая его в гидрохлорид сложного изопропилового эфира Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-метиламинометил)хиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина.
Пример 22
Синтез сложного изопропилового эфира N α -(2,6-дихлорбензоил)-4-{1-метил-6-диметиламинохиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина
Стадия 1: Синтез сложного метилового эфира N α -(2,6-дихлорбензоил)-4-{2-этоксикарбониламино-5-диметиламинобензоиламино}-L-фенилаланина
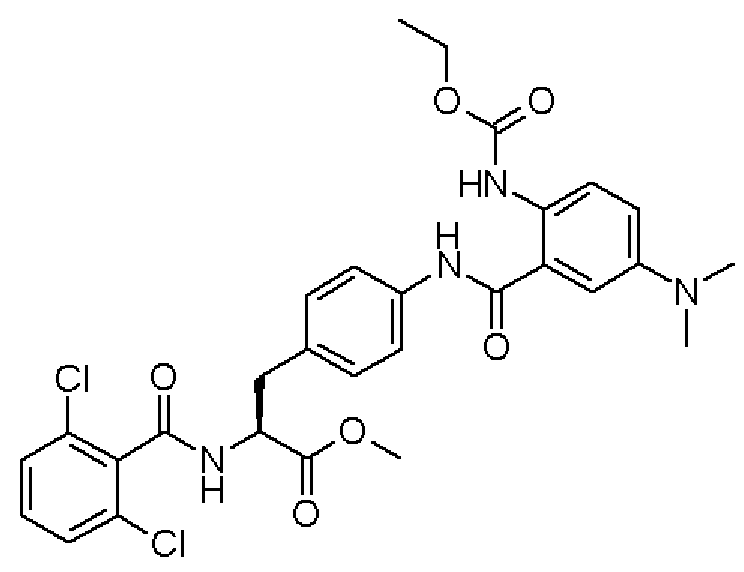
К 1,96 г 2-амино-5-диметиламинобензойной кислоты добавляли 12 мл ацетонитрила и 5,29 мл пиридина с образованием суспензии и затем охлаждали полученную суспензию до 4°С. К полученной суспензии добавляли по каплям 4,17 мл этилхлорформиата более 5 минут и затем перемешивали смесь при 25°С в течение одного часа. После подтверждения с помощью ВЭЖХ исчезновения исходного материала к смеси добавляли 0,7 мл этанола, чтобы разложить при этом избыток этилхлорформиата, и дополнительно перемешивали смесь в течение одного дополнительного часа. К реакционному раствору добавляли 4,0 г сложного метилового эфира 4-амино-Nα-(2,6-дихлорбензоил)-L-фенилаланина и 12 мл N,N-диметилформамида и перемешивали полученную смесь в течение ночи. Затем по каплям добавляли 48 мл метанола, полученную смесь перемешивали при 10°С в течение ночи и затем выделившееся из смеси твердое вещество выделяли фильтрованием при пониженном давлении. Затем твердое вещество промывали 8 мл метанола и сушили при 70°С в течение 5 часов при пониженном давлении, получая при этом 5,50 г указанного в заголовке соединения в виде твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, ДМСО-d6): δ 10,29 (с, 1H), 9,42 (ушир.с, 1H), 9,24 (д, 1H, J=7,9 Гц), 7,73 (ушир.с, 1H), 7,62 (д, 2H, J=8,4 Гц), 7,48-7,44 (м, 2H), 7,41 (дд, 1H, J=9,5, 6,2 Гц), 7,27 (д, 2H, J=8,4 Гц), 7,01 (д, 1H, J=2,7 Гц), 6,93 (дд, 1H, J=9,1, 2,9 Гц), 4,71 (ддд, 1H, J=9,2, 8,1, 5,7 Гц), 4,05 (кв, 2H, J=7,0 Гц), 3,66 (с, 3H), 3,10 (дд, 1H, J=14,0, 5,6 Гц), 2,96 (дд, 1H, J=14,0, 9,2 Гц), 2,93 (с, 6H), 1,18 (т, 3H, J=7,2 Гц).
МС (ESI+): m/z 601,2 (MH+) и 623,2 (M+Na), (ESI-): m/z 599,1 (M-H-).
Стадия 2: Синтез сложного метилового эфира N α -(2,6-дихлорбензоил)-4-{6-диметиламино-1-метилхиназолин-2,4-[1H,3H]-дион-3-ил}-L-фенилаланина

К 2,0 г сложного метилового эфира Nα-(2,6-дихлорбензоил)-4-{2-этоксикарбониламино-5-диметиламинобензоиламино}-L-фенилаланина, полученного на упомянутой выше стадии 1, добавляли 16 мл N,N-диметилформамида, 0,8 мл метанола и 0,91 г карбоната калия с последующим перемешиванием полученной смеси при 25°С в течение ночи. К полученному реакционному раствору добавляли 0,75 мл метил-п-толуолсульфоната, подвергая сложный метиловый эфир алкилированию при температуре в диапазоне от 25 до 40°С. После подтверждения с помощью ВЭЖХ исчезновения исходного материала добавляли 0,75 мл уксусной кислоты, чтобы погасить реакционный раствор, и затем к нему по каплям добавляли 16 мл воды, чтобы выделить твердый материал. Дополнительно к полученной смеси добавляли 8 мл смешанного раствора N,N-диметилформамид/вода (1/1) с последующим перемешиванием смеси при 25°С. Затем выделившееся при этом твердое вещество выделяли фильтрованием при пониженном давлении и промывали 8 мл воды. Затем выделенное твердое вещество сушили при 70°С в течение 4 часов при пониженном давлении, получая при этом 1,77 г заданного соединения в виде твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,28 (д, 1H, J=8,1 Гц), 7,48-7,36 (м, 6H), 7,31 (дд, 1H, J=3,0, 9,0 Гц), 7,24 (д, 1H, J=3,0 Гц), 7,20-7,15 (м, 2H), 4,18 (ддд, 1H, J=10,2, 8,1, 4,8 Гц), 3,69 (с, 3H), 3,49 (с, 3H), 3,22 (дд, 1H, J=14,1, 4,8 Гц), 3,02 (дд, 1H, J=14,2, 10,5 Гц), 2,94 (с, 6H).
МС (ESI+): m/z 569,2 (MH+) и 591,1 (M+Na), (ESI-): m/z 567,2 (M-H-).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ФЕНИЛАЛАНИНА | 2004 |
|
RU2390520C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2158739C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ТИОФЕНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2150470C1 |
| ПРОИЗВОДНОЕ АМИНОКИСЛОТЫ | 2009 |
|
RU2499790C2 |
| СУЛЬФОНАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2607081C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ФЕНИЛАЛАНИНА | 2001 |
|
RU2286340C2 |
| ПРОИЗВОДНОЕ СУЛЬФОНАМИДА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2014 |
|
RU2667520C9 |
| ПРОИЗВОДНЫЕ НИПЕКОТИНОВОЙ КИСЛОТЫ КАК ВЕЩЕСТВА, ПРЕПЯТСТВУЮЩИЕ ТРОМБООБРАЗОВАНИЮ | 1995 |
|
RU2135470C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, МЕДИЦИНСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ, ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2369613C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИНДАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВООПУХОЛЕВЫМ ДЕЙСТВИЕМ | 1995 |
|
RU2124017C1 |
Изобретение относится к улучшенному способу получения фенилаланина с хиназолиндионовым циклом, формулы (1), или его фармацевтически приемлемой соли, которое может быть использовано для получения лекарственного средства, для лечения воспалительных заболеваний, в патологию которых вовлечен процесс α-4-интегрин-зависимой адгезии, таких как артрит, воспалительный энтерит, рассеянный склероз и др. В формуле (1)
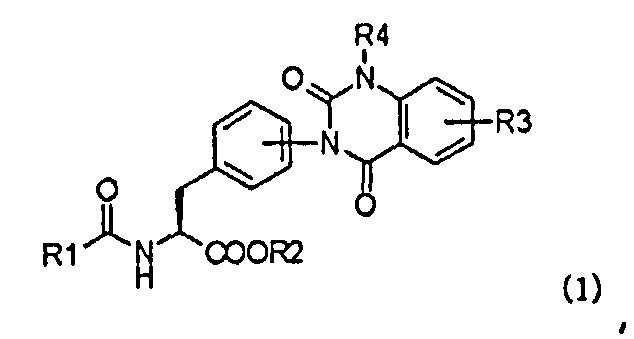
R1 представляет собой фенильную группу, которая может содержать заместитель; R2 представляет собой алкильную группу, которая может содержать заместитель; R3 представляет собой алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, или алкильную группу, замещенную аминогруппой; и R4 представляет собой атом водорода, алкильную группу или бензильную группу, которая может содержать заместитель. Способ включает стадии: (а) взаимодействие производного ацилфенилаланина формулы
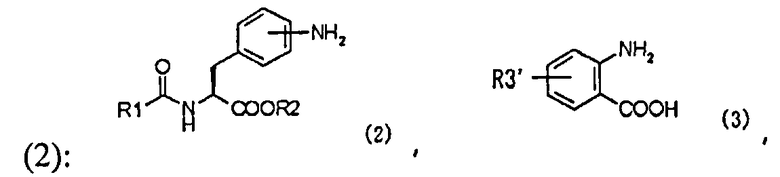
с реагентом, вводящим карбонильную группу и производным антраниловой кислоты, формулы (3), где R1, R2 имеют вышеуказанные значения, а R3' имеет значения R3, которые могут иметь защитную группу, с образованием производного мочевины с асимметрично расположенными карбоксигруппами, формулы (4), или его химически приемлемой соли:
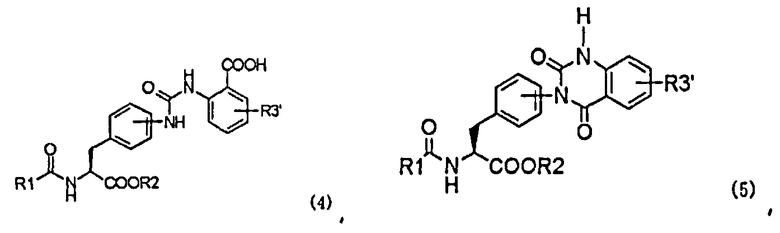
(b) превращение производного мочевины с асимметрично расположенными карбоксигруппами формулы (4) в хиназолиндионовое производное, представленное формулой (5), или его фармацевтически приемлемую соль, в присутствии средства, активирующего карбоксильную группу, где R1-R3' имеют те же самые значения, которые указаны выше; и (с)последующее N алкилирование соединения формулы (5) и снятие защитной группы, если это необходимо. Изобретение также относится к новым промежуточным продуктам. Способ позволяет упростить процесс за счет сокращения стадий и получить продукты с высоким выходом. 3 н. и 6 з.п. ф-лы, 22 пр.
1. Способ получения производного фенилаланина с хиназолиндионовым циклом, представленного следующей формулой (1), или его фармацевтически приемлемой соли, включающий следующие стадии (а), (b) и (с):

где R1 представляет собой фенильную группу, которая может содержать заместитель;
R2 представляет собой алкильную группу, которая может содержать заместитель; R3 представляет собой алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, или алкильную группу, замещенную аминогруппой; и
R4 представляет собой атом водорода, алкильную группу или бензильную группу, которая может содержать заместитель;
(а) взаимодействие производного ацилфенилаланина, представленного следующей формулой (2):

где R1 и R2 имеют те же значения, которые указаны выше,
с реагентом, вводящим карбонильную группу, выбранным из группы, состоящей из 1,1'-карбонилдиимидазола, 1,1'-карбонилдитриазола, сложного эфира хлормуравьиной кислоты, фосгена и трифосгена, и производным антраниловой кислоты, представленным следующей формулой (3):

где R3' представляет собой алкильную группу, замещенную диалкиламиногруппой, алкильную группу, замещенную моноалкиламиногруппой, которая может иметь защитную группу, или алкильную группу, замещенную аминогруппой, которая может иметь защитную группу,
с образованием при этом производного мочевины с асимметрично расположенными карбоксигруппами, представленного следующей формулой (4), или его химически приемлемой соли:
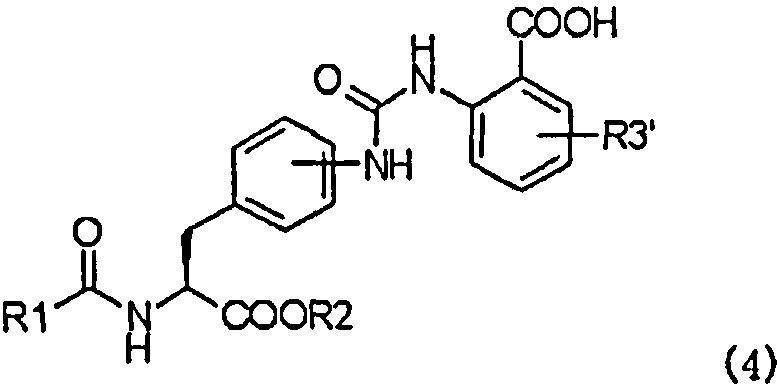
где R1, R2 и R3' имеют те же значения, которые указаны выше;
(b) превращение производного мочевины с асимметрично расположенными карбоксигруппами формулы (4) в хиназолиндионовое производное, представленное следующей формулой (5), или его фармацевтически приемлемую соль, в присутствии средства, активирующего карбоксильную группу, выбранного из группы, состоящей из 1,1'-карбонилдиимидазола, сложного эфира хлормуравьиной кислоты, хлорангидрида алкилсульфоновой кислоты, N,N'-дициклогексилкарбодиимида (DCC) и 1-этил-3-(3-диметиламинопропил)карбодиимида:
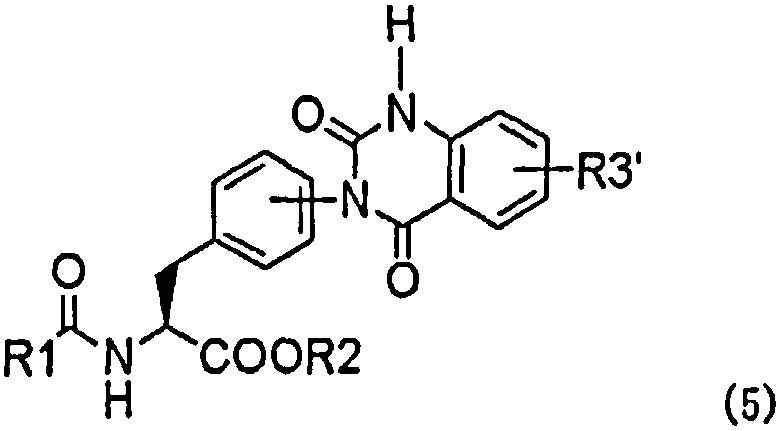
где R1-R3' имеют те же значения, которые указаны выше; и,
(с) если требуется, замещение атома водорода, связанного с атомом азота, присутствующим в хиназолиндионовом цикле хиназолиндионового производного формулы (5), N-алкильной группой с помощью N-алкилирующего средства и затем снятие защитных групп в полученном продукте, когда заместитель R3' защищен.
2. Способ по п.1, в котором R3, представленный в формуле (1), представляет собой метиламинометильную группу, этиламинометильную группу или диметиламинометильную группу; R3', представленный в формулах (3)-(5), представляет собой метильную группу, замещенную метиламиногруппой, имеющей защитную группу, метильную группу, замещенную этиламиногруппой, имеющей защитную группу, диметиламинометильную группу или диэтиламинометильную группу.
3. Способ по п.1, в котором R1 в формулах (1)-(5) представляет собой 2,6-дихлорфенильную группу, 2-хлор-6-метилфенильную группу, 2-хлор-6-фторфенильную группу, 2,6-дифторфенильную группу или 2-фтор-6-металфенильную группу; R4 представляет собой либо метильную группу, либо этильную группу; и заместитель, присутствующий на бензольном кольце фенилаланина, находится в п-положении кольца.
4. Способ по п.1, в котором N-алкилирующее средство представляет собой метил-п-толуолсульфонат, метилметансульфонат, метилйодид, метилбромид или металхлорид.
5. Способ по п.1, в котором соединение формулы (2) сначала подвергают взаимодействию с реагентом, вводящим карбонильную группу, а затем полученный продукт подвергают взаимодействию с соединением формулы (3).
6. Способ по п.1, в котором следующую стадию проводят без выделения соединения формулы (4).
7. Способ по п.1, в котором соединение формулы (2), в которой R1 в формуле (2) представляет собой 2,6-дихлорфенильную группу и R2 представляет собой изопропильную группу, подвергают взаимодействию с 1,1-карбонилдиимидазолом в качестве реагента, вводящего карбонильную группу, и соединением формулы (3), в которой R3' представляет собой N-формил-N-метиламинометильную группу, с образованием при этом 2-(3-{4-[2(S)-2-(2,6-дихлорбензоиламино)-2-изопропоксикарбонилэтил]фенил}уреидо)-5-(N-формил-N-метиламинометил)бензойной кислоты формулы (4), в которой R1 представляет собой 2,6-дихлорфенильную группу, R2 представляет собой изопропильную группу и R3' представляет собой N-формил-N-метиламинометильную группу; затем полученное соединение подвергают взаимодействию с 1,1-карбонилдиимидазолом в качестве средства, активирующего карбоксильную группу, превращая при этом первое соединение в сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{6-(N-формил-N-метиламинометил)хиназолин-2,4-[1Н,3Н]-дион-3-ил}-L-фенилаланина формулы (5), в которой R1 представляет собой 2,6-дихлорфенильную группу, R2 представляет собой изопропильную группу и R3' представляет собой N-формил-N-метиламинометильную группу; затем с помощью метил-п-толуолсульфоната проводят N-алкилирование полученного продукта, превращая при этом продукт в сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-формил-N-метиламинометил)хиназолин-2,4-[1Н,3Н]-дион-3-ил}-L-фенилаланина; и, в конечном счете, N-алкилированный продукт обрабатывают хлористым водородом для удаления его формильной группы, получая при этом сложный изопропиловый эфир Nα-(2,6-дихлорбензоил)-4-{1-метил-6-(N-метиламинометил)хиназолин-2,4-[1Н,3Н]-дион-3-ил}-L-фенилаланина формулы (1) или его гидрохлорид, в котором R1 представляет собой 2,6-дихлорфенильную группу, R2 представляет собой изопропильную группу, R3' представляет собой N-метиламинометильную группу и R4 представляет собой метильную группу.
8. Соединение, представленное следующей формулой (3-1):
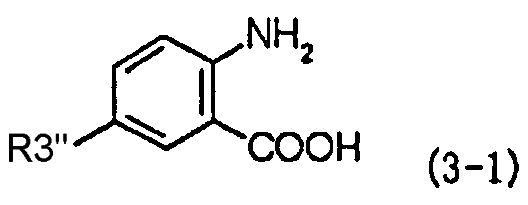
где R3'' представляет собой член, выбранный из группы, состоящей из N-(С1-С3алкил)-N-формиламинометильных групп и N-(С1-С3алкил)-N-алкоксикарбониламинометильных групп.
9. Соединение, представленное следующими формулами:

| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| US 4665070 A, 12.05.1887 | |||
| CA 1061356 A, 12.04.1977 | |||
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Реагент для минерализованных бентонитовых буровых растворов | 1988 |
|
SU1595870A1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ФЕНИЛАЛАНИНА | 2001 |
|
RU2286340C2 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |

